User login
The jealous insomniac
CASE Anxious and jealous
Mrs. H, age 28, presents to the emergency department (ED) with pressured speech, emotional lability, loose associations, and echolalia. On physical examination, Mrs. H is noted to have hand tremors. Mrs. H says she has not slept for the past 5 days and is experiencing anxiety and heart palpitations.
She also says that for the past 2 years she has believed that her husband is having an affair with her best friend. However, her current presentation—which she attributes to the alleged affair—began a week before she came to the ED. According to her husband, Mrs. H was “perfectly fine until a week ago” and her symptoms “appeared out of nowhere.” He reports that this has never happened before.
Mrs. H is admitted to the psychiatry unit. The nursing team reports that on the first night, Mrs. H was “running and screaming on the unit, out of control,” and was “tearful, manicky, and dysphoric.”
Mrs. H has no significant medical or psychiatric history. Her family history is significant for hyperthyroidism in her mother and maternal grandmother. Mrs. H says she smokes cigarettes (1 pack/d) but denies alcohol or illicit drug use.

EVALUATION A telling thyroid panel
Mrs. H undergoes laboratory testing, including a complete blood count, comprehensive metabolic panel, and thyroid panel due to her family history of thyroid-related disorders. The thyroid panel shows the presence of the thyroid-stimulating hormone (TSH) receptor antibody; a low TSH level; elevated triiodothyronine (T3) and thyroxine (T4) levels, with T3 > T4; elevated thyroid peroxidase (TPO) antibody; and elevated thyroglobulin antibody (Table 1). A scan shows the thyroid gland to be normal/top-normal size and is read by radiology to be indicative of a resolving thyroiditis vs Graves’ disease. An electrocardiogram indicates a heart rate of 139 beats per minute.
[polldaddy:10352133]
The authors’ observations
Mrs. H fits the presentation of psychosis secondary to Graves’ disease. However, our differential consisted of thyroiditis, brief psychotic disorder, delusional disorder (jealous type), and bipolar mania.
Brief psychotic disorder, bipolar mania, and delusional disorder were better explained by Graves’ disease, and Mrs. H’s jealous delusion resulted in functional impairment, which eliminated delusional disorder. Her family history of hyperthyroidism, as well as her sex and history of tobacco use, supported the diagnosis of Graves’ disease. Although Mrs. H did not experience goiter, ophthalmopathy, or dermopathy, which are common signs and symptoms of Graves’ disease (Table 2), she did present with irritability, insomnia, tachycardia, and a hand tremor. Her psychiatric symptoms included anxiety, emotional lability and, most importantly, psychosis. Her laboratory results included the presence of the TSH-receptor antibody, a low TSH level, and elevated T3 and T4 levels (T3>T4), confirming the diagnosis of early-onset Graves’ disease.
Continue to: Graves' disease
Graves’ disease
Graves’ disease is the most common cause of hyperthyroidism, representing approximately 50% to 80% of cases.1 Graves’ disease occurs most often in women, smokers, and those with a personal or family history of autoimmune disease; although patients of any age may be affected, the peak incidence occurs between age 40 and 60.1
Graves’ disease results from the production of immunoglobulin G (IgG) antibodies that activate the TSH receptor on the surface of thyroid follicular cells.1 The presence of the TSH-receptor antibody, in addition to a low TSH and elevated T3 and T4 levels (T3>T4), are common laboratory findings in patients with this disease. A thyroid scan will also show increased radiotracer accumulation.

Patients with Graves’ disease, as well as those with hyperthyroidism, tend to report weight loss, increased appetite, heat intolerance, irritability, insomnia, and palpitations. In addition to the above symptoms, the identifying signs and symptoms of Graves’ disease include a goiter, ophthalmopathy, and dermopathy (Table 2). Rarely, patients with Graves’ disease can present with psychosis, which is often complicated by thyrotoxicosis.2
[polldaddy:10352135]
TREATMENT Antipsychotic and a beta blocker
Based on her signs, symptoms, and laboratory findings, Mrs. H receives risperidone, 1 mg twice daily, for psychosis, and atenolol, 25 mg twice daily, for heart palpitations. Over 4 days, her symptoms decrease; she experiences more linear thought and decreased flight-of-ideas, and becomes unsure about the truth of her husband’s alleged affair. Her impulsive behaviors and severe mood lability cease. Her tachycardia remains controlled with atenolol.

The authors’ observations
Rapid initiation of treatment is important when managing patients with Graves’ disease, because untreated patients have a higher risk of psychiatric illness, cardiac disease, arrhythmia, and sudden cardiac death.1 Patients with Graves’ disease typically are treated with thionamides, radioactive iodine, and/or surgery. When a patient presents with psychosis as a result of thyrotoxicosis, treatment focuses on improving the thyrotoxicosis through anti-thyroid medications and beta blockers (Table 33). Psychotropic medications, such as antipsychotics, are not indicated for primary treatment, but are given to patients who have severe psychosis until symptoms have resolved.3 For Mrs. H, the severity of her psychosis necessitated risperidone in addition to atenolol.
OUTCOME Continuous medical management; no ablation
Mrs. H is discharged with immediate outpatient follow-up with an endocrinology team to discuss the best long-term management of her thyroiditis. Mrs. H opts for continuous medical management (as opposed to ablation) and is administered methimazole, 15 mg/d, to treat Graves’ disease.
The authors’ observations
This case provides useful information regarding recognizing psychosis as the initial sign of Graves’ disease. Although Graves’ disease represents 50% to 80% of cases of hyperthyroidism,1 psychosis as the first clinical presentation of this disease is extremely rare. Several case reports, however, have described this phenomenon,2,3 and further studies would be helpful to determine its true prevalence.
Continue to: Bottom Line
Bottom Line
Although extremely rare, psychosis as the initial clinical presentation of Graves’ disease can occur. The early diagnosis of Graves’ disease is critical to prevent cardiovascular implications and death.
Related Resources
- Abraham P, Acharya S. Current and emerging treatment options for Graves’ hyperthyroidism. Ther Clin Risk Manag. 2010;6:29-40.
- Bunevicius R, Prange AJ Jr. Psychiatric manifestations of Graves’ hyperthyroidism: pathophysiology and treatment options. CNS Drugs. 2006;20(11):897-909.
- Ginsberg J. Diagnosis and management of Graves’ disease. CMAJ. 2003;168(5):575-585.
Drug Brand Names
Atenolol • Tenormin
Methimazole • Tapazole
Risperidone • Risperdal
1. Girgis C, Champion B, Wall J. Current concepts in Graves’ disease. Ther Adv Endocrinol Metab. 2011;2(3):135-144.
2. Urias-Uribe L, Valdez-Solis E, González-Milán C, et al. Psychosis crisis associated with thyrotoxicosis due to Graves’ disease. Case Rep Psychiatry. 2017;2017:6803682. doi: 10.1155/2017/6803682.
3. Ugwu ET, Maluze J, Onyebueke GC. Graves’ thyrotoxicosis presenting as schizophreniform psychosis: a case report and literature review. Int J Endocrinol Metab. 2017;15(1):e41977. doi: 10.5812/ijem.41977.
CASE Anxious and jealous
Mrs. H, age 28, presents to the emergency department (ED) with pressured speech, emotional lability, loose associations, and echolalia. On physical examination, Mrs. H is noted to have hand tremors. Mrs. H says she has not slept for the past 5 days and is experiencing anxiety and heart palpitations.
She also says that for the past 2 years she has believed that her husband is having an affair with her best friend. However, her current presentation—which she attributes to the alleged affair—began a week before she came to the ED. According to her husband, Mrs. H was “perfectly fine until a week ago” and her symptoms “appeared out of nowhere.” He reports that this has never happened before.
Mrs. H is admitted to the psychiatry unit. The nursing team reports that on the first night, Mrs. H was “running and screaming on the unit, out of control,” and was “tearful, manicky, and dysphoric.”
Mrs. H has no significant medical or psychiatric history. Her family history is significant for hyperthyroidism in her mother and maternal grandmother. Mrs. H says she smokes cigarettes (1 pack/d) but denies alcohol or illicit drug use.
EVALUATION A telling thyroid panel
Mrs. H undergoes laboratory testing, including a complete blood count, comprehensive metabolic panel, and thyroid panel due to her family history of thyroid-related disorders. The thyroid panel shows the presence of the thyroid-stimulating hormone (TSH) receptor antibody; a low TSH level; elevated triiodothyronine (T3) and thyroxine (T4) levels, with T3 > T4; elevated thyroid peroxidase (TPO) antibody; and elevated thyroglobulin antibody (Table 1). A scan shows the thyroid gland to be normal/top-normal size and is read by radiology to be indicative of a resolving thyroiditis vs Graves’ disease. An electrocardiogram indicates a heart rate of 139 beats per minute.
[polldaddy:10352133]
The authors’ observations
Mrs. H fits the presentation of psychosis secondary to Graves’ disease. However, our differential consisted of thyroiditis, brief psychotic disorder, delusional disorder (jealous type), and bipolar mania.
Brief psychotic disorder, bipolar mania, and delusional disorder were better explained by Graves’ disease, and Mrs. H’s jealous delusion resulted in functional impairment, which eliminated delusional disorder. Her family history of hyperthyroidism, as well as her sex and history of tobacco use, supported the diagnosis of Graves’ disease. Although Mrs. H did not experience goiter, ophthalmopathy, or dermopathy, which are common signs and symptoms of Graves’ disease (Table 2), she did present with irritability, insomnia, tachycardia, and a hand tremor. Her psychiatric symptoms included anxiety, emotional lability and, most importantly, psychosis. Her laboratory results included the presence of the TSH-receptor antibody, a low TSH level, and elevated T3 and T4 levels (T3>T4), confirming the diagnosis of early-onset Graves’ disease.
Continue to: Graves' disease
Graves’ disease
Graves’ disease is the most common cause of hyperthyroidism, representing approximately 50% to 80% of cases.1 Graves’ disease occurs most often in women, smokers, and those with a personal or family history of autoimmune disease; although patients of any age may be affected, the peak incidence occurs between age 40 and 60.1
Graves’ disease results from the production of immunoglobulin G (IgG) antibodies that activate the TSH receptor on the surface of thyroid follicular cells.1 The presence of the TSH-receptor antibody, in addition to a low TSH and elevated T3 and T4 levels (T3>T4), are common laboratory findings in patients with this disease. A thyroid scan will also show increased radiotracer accumulation.
Patients with Graves’ disease, as well as those with hyperthyroidism, tend to report weight loss, increased appetite, heat intolerance, irritability, insomnia, and palpitations. In addition to the above symptoms, the identifying signs and symptoms of Graves’ disease include a goiter, ophthalmopathy, and dermopathy (Table 2). Rarely, patients with Graves’ disease can present with psychosis, which is often complicated by thyrotoxicosis.2
[polldaddy:10352135]
TREATMENT Antipsychotic and a beta blocker
Based on her signs, symptoms, and laboratory findings, Mrs. H receives risperidone, 1 mg twice daily, for psychosis, and atenolol, 25 mg twice daily, for heart palpitations. Over 4 days, her symptoms decrease; she experiences more linear thought and decreased flight-of-ideas, and becomes unsure about the truth of her husband’s alleged affair. Her impulsive behaviors and severe mood lability cease. Her tachycardia remains controlled with atenolol.
The authors’ observations
Rapid initiation of treatment is important when managing patients with Graves’ disease, because untreated patients have a higher risk of psychiatric illness, cardiac disease, arrhythmia, and sudden cardiac death.1 Patients with Graves’ disease typically are treated with thionamides, radioactive iodine, and/or surgery. When a patient presents with psychosis as a result of thyrotoxicosis, treatment focuses on improving the thyrotoxicosis through anti-thyroid medications and beta blockers (Table 33). Psychotropic medications, such as antipsychotics, are not indicated for primary treatment, but are given to patients who have severe psychosis until symptoms have resolved.3 For Mrs. H, the severity of her psychosis necessitated risperidone in addition to atenolol.
OUTCOME Continuous medical management; no ablation
Mrs. H is discharged with immediate outpatient follow-up with an endocrinology team to discuss the best long-term management of her thyroiditis. Mrs. H opts for continuous medical management (as opposed to ablation) and is administered methimazole, 15 mg/d, to treat Graves’ disease.
The authors’ observations
This case provides useful information regarding recognizing psychosis as the initial sign of Graves’ disease. Although Graves’ disease represents 50% to 80% of cases of hyperthyroidism,1 psychosis as the first clinical presentation of this disease is extremely rare. Several case reports, however, have described this phenomenon,2,3 and further studies would be helpful to determine its true prevalence.
Continue to: Bottom Line
Bottom Line
Although extremely rare, psychosis as the initial clinical presentation of Graves’ disease can occur. The early diagnosis of Graves’ disease is critical to prevent cardiovascular implications and death.
Related Resources
- Abraham P, Acharya S. Current and emerging treatment options for Graves’ hyperthyroidism. Ther Clin Risk Manag. 2010;6:29-40.
- Bunevicius R, Prange AJ Jr. Psychiatric manifestations of Graves’ hyperthyroidism: pathophysiology and treatment options. CNS Drugs. 2006;20(11):897-909.
- Ginsberg J. Diagnosis and management of Graves’ disease. CMAJ. 2003;168(5):575-585.
Drug Brand Names
Atenolol • Tenormin
Methimazole • Tapazole
Risperidone • Risperdal
CASE Anxious and jealous
Mrs. H, age 28, presents to the emergency department (ED) with pressured speech, emotional lability, loose associations, and echolalia. On physical examination, Mrs. H is noted to have hand tremors. Mrs. H says she has not slept for the past 5 days and is experiencing anxiety and heart palpitations.
She also says that for the past 2 years she has believed that her husband is having an affair with her best friend. However, her current presentation—which she attributes to the alleged affair—began a week before she came to the ED. According to her husband, Mrs. H was “perfectly fine until a week ago” and her symptoms “appeared out of nowhere.” He reports that this has never happened before.
Mrs. H is admitted to the psychiatry unit. The nursing team reports that on the first night, Mrs. H was “running and screaming on the unit, out of control,” and was “tearful, manicky, and dysphoric.”
Mrs. H has no significant medical or psychiatric history. Her family history is significant for hyperthyroidism in her mother and maternal grandmother. Mrs. H says she smokes cigarettes (1 pack/d) but denies alcohol or illicit drug use.
EVALUATION A telling thyroid panel
Mrs. H undergoes laboratory testing, including a complete blood count, comprehensive metabolic panel, and thyroid panel due to her family history of thyroid-related disorders. The thyroid panel shows the presence of the thyroid-stimulating hormone (TSH) receptor antibody; a low TSH level; elevated triiodothyronine (T3) and thyroxine (T4) levels, with T3 > T4; elevated thyroid peroxidase (TPO) antibody; and elevated thyroglobulin antibody (Table 1). A scan shows the thyroid gland to be normal/top-normal size and is read by radiology to be indicative of a resolving thyroiditis vs Graves’ disease. An electrocardiogram indicates a heart rate of 139 beats per minute.
[polldaddy:10352133]
The authors’ observations
Mrs. H fits the presentation of psychosis secondary to Graves’ disease. However, our differential consisted of thyroiditis, brief psychotic disorder, delusional disorder (jealous type), and bipolar mania.
Brief psychotic disorder, bipolar mania, and delusional disorder were better explained by Graves’ disease, and Mrs. H’s jealous delusion resulted in functional impairment, which eliminated delusional disorder. Her family history of hyperthyroidism, as well as her sex and history of tobacco use, supported the diagnosis of Graves’ disease. Although Mrs. H did not experience goiter, ophthalmopathy, or dermopathy, which are common signs and symptoms of Graves’ disease (Table 2), she did present with irritability, insomnia, tachycardia, and a hand tremor. Her psychiatric symptoms included anxiety, emotional lability and, most importantly, psychosis. Her laboratory results included the presence of the TSH-receptor antibody, a low TSH level, and elevated T3 and T4 levels (T3>T4), confirming the diagnosis of early-onset Graves’ disease.
Continue to: Graves' disease
Graves’ disease
Graves’ disease is the most common cause of hyperthyroidism, representing approximately 50% to 80% of cases.1 Graves’ disease occurs most often in women, smokers, and those with a personal or family history of autoimmune disease; although patients of any age may be affected, the peak incidence occurs between age 40 and 60.1
Graves’ disease results from the production of immunoglobulin G (IgG) antibodies that activate the TSH receptor on the surface of thyroid follicular cells.1 The presence of the TSH-receptor antibody, in addition to a low TSH and elevated T3 and T4 levels (T3>T4), are common laboratory findings in patients with this disease. A thyroid scan will also show increased radiotracer accumulation.
Patients with Graves’ disease, as well as those with hyperthyroidism, tend to report weight loss, increased appetite, heat intolerance, irritability, insomnia, and palpitations. In addition to the above symptoms, the identifying signs and symptoms of Graves’ disease include a goiter, ophthalmopathy, and dermopathy (Table 2). Rarely, patients with Graves’ disease can present with psychosis, which is often complicated by thyrotoxicosis.2
[polldaddy:10352135]
TREATMENT Antipsychotic and a beta blocker
Based on her signs, symptoms, and laboratory findings, Mrs. H receives risperidone, 1 mg twice daily, for psychosis, and atenolol, 25 mg twice daily, for heart palpitations. Over 4 days, her symptoms decrease; she experiences more linear thought and decreased flight-of-ideas, and becomes unsure about the truth of her husband’s alleged affair. Her impulsive behaviors and severe mood lability cease. Her tachycardia remains controlled with atenolol.
The authors’ observations
Rapid initiation of treatment is important when managing patients with Graves’ disease, because untreated patients have a higher risk of psychiatric illness, cardiac disease, arrhythmia, and sudden cardiac death.1 Patients with Graves’ disease typically are treated with thionamides, radioactive iodine, and/or surgery. When a patient presents with psychosis as a result of thyrotoxicosis, treatment focuses on improving the thyrotoxicosis through anti-thyroid medications and beta blockers (Table 33). Psychotropic medications, such as antipsychotics, are not indicated for primary treatment, but are given to patients who have severe psychosis until symptoms have resolved.3 For Mrs. H, the severity of her psychosis necessitated risperidone in addition to atenolol.
OUTCOME Continuous medical management; no ablation
Mrs. H is discharged with immediate outpatient follow-up with an endocrinology team to discuss the best long-term management of her thyroiditis. Mrs. H opts for continuous medical management (as opposed to ablation) and is administered methimazole, 15 mg/d, to treat Graves’ disease.
The authors’ observations
This case provides useful information regarding recognizing psychosis as the initial sign of Graves’ disease. Although Graves’ disease represents 50% to 80% of cases of hyperthyroidism,1 psychosis as the first clinical presentation of this disease is extremely rare. Several case reports, however, have described this phenomenon,2,3 and further studies would be helpful to determine its true prevalence.
Continue to: Bottom Line
Bottom Line
Although extremely rare, psychosis as the initial clinical presentation of Graves’ disease can occur. The early diagnosis of Graves’ disease is critical to prevent cardiovascular implications and death.
Related Resources
- Abraham P, Acharya S. Current and emerging treatment options for Graves’ hyperthyroidism. Ther Clin Risk Manag. 2010;6:29-40.
- Bunevicius R, Prange AJ Jr. Psychiatric manifestations of Graves’ hyperthyroidism: pathophysiology and treatment options. CNS Drugs. 2006;20(11):897-909.
- Ginsberg J. Diagnosis and management of Graves’ disease. CMAJ. 2003;168(5):575-585.
Drug Brand Names
Atenolol • Tenormin
Methimazole • Tapazole
Risperidone • Risperdal
1. Girgis C, Champion B, Wall J. Current concepts in Graves’ disease. Ther Adv Endocrinol Metab. 2011;2(3):135-144.
2. Urias-Uribe L, Valdez-Solis E, González-Milán C, et al. Psychosis crisis associated with thyrotoxicosis due to Graves’ disease. Case Rep Psychiatry. 2017;2017:6803682. doi: 10.1155/2017/6803682.
3. Ugwu ET, Maluze J, Onyebueke GC. Graves’ thyrotoxicosis presenting as schizophreniform psychosis: a case report and literature review. Int J Endocrinol Metab. 2017;15(1):e41977. doi: 10.5812/ijem.41977.
1. Girgis C, Champion B, Wall J. Current concepts in Graves’ disease. Ther Adv Endocrinol Metab. 2011;2(3):135-144.
2. Urias-Uribe L, Valdez-Solis E, González-Milán C, et al. Psychosis crisis associated with thyrotoxicosis due to Graves’ disease. Case Rep Psychiatry. 2017;2017:6803682. doi: 10.1155/2017/6803682.
3. Ugwu ET, Maluze J, Onyebueke GC. Graves’ thyrotoxicosis presenting as schizophreniform psychosis: a case report and literature review. Int J Endocrinol Metab. 2017;15(1):e41977. doi: 10.5812/ijem.41977.

Serotonin syndrome: How to keep your patients safe

Mr. S, age 55, comes to your clinic as a walk-in for management of major depressive disorder, insomnia, and migraines. He also has tobacco use disorder and hypertension. Several days ago, Mr. S had visited the clinic because he was continuing to experience depressive symptoms, so his sertraline was increased from 100 to 200 mg/d. His current medication regimen includes sertraline 200 mg/d, trazodone 100 mg/d, lisinopril 10 mg/d, and sumatriptan, 100 mg as needed for migraine. He says last week he used 4 or 5 doses of sumatriptan because he experienced several migraines. Mr. S also reports occasionally taking 2 tablets of trazodone instead of 1 on nights that he has trouble falling asleep.
Today, Mr. S presents with a low-grade fever, diarrhea, internal restlessness, and a racing heartbeat that started shortly after his last visit. During physical examination, he exhibits slow, continuous lateral eye movements. His vital signs are markedly elevated: blood pressure, 175/85 mm Hg; heart rate, 110 beats per minute; and temperature, 39°C (102.2°F). Based on his presentation, the treatment team decides to send Mr. S to urgent care for closer monitoring.
Serotonin syndrome is a drug-induced syndrome caused by overstimulation of serotonin receptors. The syndrome is characterized by a classic clinical triad consisting of mental status changes, autonomic hyperactivity, and neuromuscular abnormalities. The clinical presentation is highly variable, and the severity ranges from mild to life-threatening.1-3 The incidence and prevalence of serotonin syndrome has not been well defined.3 Serotonin syndrome may be underreported because mild cases are often overlooked due to nonspecific symptoms. In addition, lack of physician awareness of drug–drug interactions, signs and symptoms, and differential diagnoses may result in underdiagnosis or misdiagnosis.1-3
What causes it?
Serotonin syndrome is usually a consequence of a drug–drug interaction between 2 or more serotonergic agents.4 Serotonin syndrome may result following medication misuse, overdose, initiation of a serotonergic agent, or increase in the dose of a currently prescribed serotonergic agent.3,4 In addition to medication classes and specific agents, Table 12-5 lists the drug mechanisms associated with serotonin syndrome:
- inhibition of serotonin reuptake
- inhibition of serotonin metabolism
- increased serotonin synthesis
- agonism of the serotonin receptor.

The amount of serotonergic activity most likely to cause serotonin syndrome is unclear.4
Pathophysiology. Serotonin, also known as 5-hydroxytryptamine (5-HT), is a metabolite of the amino acid tryptophan. This neurotransmitter is located in both the CNS and the periphery. Regulation of the serotonergic system begins in the presynaptic neurons with decarboxylation and hydroxylation of tryptophan resulting in serotonin synthesis. Once serotonin is produced, it is released into the synaptic cleft, where it binds to serotonin receptors.1,4,5 After receptor binding, serotonin reuptake occurs in the presynaptic neurons, where it can be metabolized by the monoamine oxidase enzyme. Finally, the metabolites are excreted in the urine. Serotonin syndrome results when this regulatory system is disrupted due to hyperstimulation of the postsynaptic serotonin receptors, mainly via agonism of the 5-HT2A and 5-HT1A receptors.1,4,5
Continue to: A nonspecific presentation
A nonspecific presentation
Unfortunately, many of the symptoms of serotonin syndrome are nonspecific, and the severity varies among patients.2,3 The onset of symptoms usually occurs within 6 to 8 hours after ingestion of a serotonergic agent.5 It is important to immediately recognize the symptoms (Table 22-5) and formulate a differential diagnosis because sudden progression of symptoms is common and may lead to life-threatening circumstances.1,3

In mild cases of serotonin syndrome, patients may have a low-grade fever or be afebrile. Hyperthermia tends to be present in moderate and severe cases, with temperatures >41°C (105.8°F) during life-threatening cases. Diaphoresis and tachycardia may be present regardless of severity. Additional autonomic irregularities include hypertension, tachypnea, nausea, vomiting, diarrhea, and hyperactive bowel sounds. In terms of neuromuscular abnormalities, hyperreflexia is a primary concern, as well as myoclonus. As the severity progresses to life-threatening, the clonus may convert from inducible to spontaneous and slow, continuous lateral eye movements may be present. Additional neuromuscular symptoms include tremor, akathisia, and muscle rigidity.1,3-5
Common mental status changes during mild cases include restlessness and anxiety. Abnormal mentation during moderate cases may present as increased hypervigilance and agitation, and this may advance to delirium or coma in severe cases. As the severity intensifies, the risk of developing additional physiological complications also increases. Rhabdomyolysis may occur due to muscle damage and myoglobinuria secondary to hyperreflexia, myoclonus, hypertonicity, and muscle rigidity. Muscle breakdown may then progress to further complications, such as renal failure. In rare instances, serotonin syndrome can result in seizures or death.1,3-5
Medication history tips off the diagnosis
The first step in diagnosing serotonin syndrome is to conduct a thorough review of the patient’s medication history, specifically taking into account any recent exposure to serotonergic agents.3,5 It is important to ask about prescription medications as well as over-the-counter products, herbal supplements, and illicit substances.1,4 When reviewing the medication history, investigate whether there may have been a recent change in therapy with serotonergic agents. Also, determine when the patient’s symptoms began in relation to exposure to serotonergic agents.4

After the medication review, conduct a thorough physical and neurologic examination to identify current symptoms and severity.1,3 No specific laboratory test is available to definitively confirm the diagnosis of serotonin syndrome.1,4 Monitoring of serum serotonin is not recommended because the levels do not correlate with symptom severity.3 The recommended diagnostic tool is the Hunter Serotonin Toxicity Criteria (Figure1,3).3,4 Historically, the Sternbach’s Diagnostic Criteria for serotonin syndrome were used for diagnosis; however, the Hunter Serotonin Toxicity Criteria are more sensitive (96% vs 75%) and more specific (97% vs 84%) than the Sternbach’s Diagnostic Criteria for serotonin syndrome.1,3-5
Continue to: In addition to using the proper diagnostic tool...
In addition to using the proper diagnostic tool, conduct a differential diagnosis to rule out other drug-induced syndromes, such as anticholinergic toxidrome, neuroleptic malignant syndrome, or malignant hyperthermia.1,3,5 Autonomic instability, including hypertension, tachycardia, tachypnea, and hyperthermia, may be present in all of the aforementioned drug-induced syndromes.1 As a result, the clinician must monitor for other symptoms that may differentiate the disease states to establish a clear diagnosis.
Discontinue agents, offer supportive care
There are no official published guidelines for managing serotonin syndrome.5 Regardless of the severity of a patient’s presentation, all serotonergic agents should be discontinued immediately. In addition, supportive care should be initiated for symptom management. Intravenous fluid replacement is recommended for hydration and to treat hyperthermia. External cooling may also be warranted to reduce body temperatures. Vital signs should be stabilized with appropriate pharmacotherapy.1,3-5
Benzodiazepines are considered a mainstay for relief of agitation during serotonin syndrome of any severity. In life-threatening cases—which are characterized by hyperthermia >41°C (105.8°F)—sedation, paralysis, and intubation may be necessary to maintain the airway, breathing, and circulation.1,3-5 Because treatment of hyperthermia requires elimination of hyperreflexia, paralysis is recommended.1 Nondepolarizing neuromuscular blocking agents, such as vecuronium, are preferred over depolarizing agents due to their decreased potential for rhabdomyolysis.1,3
Cyproheptadine, a histamine-1 receptor antagonist and a 5-HT2A receptor antagonist, is recommended for off-label treatment of serotonin syndrome to help decrease the intensity of symptoms. This should be initiated as a single dose of 12 mg followed by 2 mg every 2 hours until symptoms improve.1,3,5 After stabilization, a maintenance dose of 8 mg every 6 hours is recommended. Doses should not exceed the maximum recommended dose of 0.5 mg/kg/d.1,3,6 The most common adverse reactions associated with cyproheptadine are sedation and anticholinergic adverse effects.1,4,6
Antipsychotics, such as olanzapine and chlorpromazine, have been considered treatment alternatives due to their associated 5-HT2A receptor antagonism. However, there is limited data supporting such use.1,4 Antipsychotics should be used with caution because neuroleptic malignant syndrome may be mistaken for serotonin syndrome. Use of antipyretics is not recommended for treating fever and hyperthermia because the increase in body temperature is secondary to excessive muscle activity rather than dysfunction of the hypothalamic temperature set point.1,3,5 Physical restraints are also not recommended because their use may provoke further hyperthermia and increase the risk of rhabdomyolysis.3,5
Continue to: Ultimately, the duration of treatment...
Ultimately, the duration of treatment will be influenced by the pharmacokinetics of the serotonergic agents that induced the serotonin syndrome. Following resolution, retrial of the offending serotonergic agents should be carefully assessed. A retrial should only be considered after an adequate washout period has been observed, and clinicians should consider utilizing lower doses.2,5
Take steps for prevention
Patients at highest risk of developing serotonin syndrome are those who have multiple comorbidities that result in treatment with multiple serotonergic agents.3 Clinicians and patients alike need to be educated about the signs and symptoms of serotonin syndrome to promote early recognition. Also consider modifying your prescribing practices to minimize the use of multiple serotonergic agents. When switching between serotonergic agents, institute safe washout periods. Encourage patients to adhere to their prescribed medication regimens. Using electronic ordering systems can help detect drug–drug interactions.1,3 Prophylaxis with cyproheptadine may be considered in high-risk patients; however, no clinical trials have been conducted to evaluate using cyproheptadine to prevent serotonin syndrome.7
CASE CONTINUED
Upon further assessment in urgent care, Mr. S is found to have muscle rigidity in addition to ocular clonus and a temperature >38°C (100.4°F). Because Mr. S’s symptoms coincide with a recent increase of sertraline and increased use of both trazodone and sumatriptan, he meets Hunter Serotonin Toxicity Criteria. Therefore, his symptoms are likely related to excessive increase in serotonergic activity. Mr. S is admitted to the hospital for closer monitoring, and his sertraline, trazodone, and sumatriptan are held. He receives IV fluids for several days as well as cyproheptadine, 8 mg every 6 hours after stabilization, until his symptoms resolve. On Day 4, Mr. S no longer experiences diarrhea and internal restlessness. His vital signs return to normal, and as a result of symptom resolution, he is discharged from the hospital. The treatment team discusses changing his medication regimen to avoid multiple serotonergic agents. Mr. S is switched from sertraline to bupropion XL, 150 mg/d. Sumatriptan, 100 mg/d as needed, is continued for acute migraine treatment. Trazodone is discontinued and replaced with melatonin, 3 mg/d. The team also counsels Mr. S on the importance of proper adherence to his medication regimen. He is advised to return to the clinic in 2 weeks for reassessment of safety and efficacy.
Related Resource
- Turner AH, Kim JJ, McCarron RM. Differentiating serotonin syndrome and neuroleptic malignant syndrome. Current Psychiatry. 2019;18(2):30-36.
Drug Brand Names
Almotriptan • Axert
Buprenorphine • Subutex
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cyproheptadine • Periactin
Eletriptan • Relpax
Frovatriptan • Frova
Granisetron • Kytril
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Methadone • Dolophine, Methadose
Metoclopramide • Reglan
Mirtazapine • Remeron
Naratriptan • Amerge
Olanzapine • Zyprexa
Ondansetron • Zofran
Rizatriptan • Maxalt
Sertraline • Zoloft
Sumatriptan • Imitrex tablets
Tapentadol • Nucynta
Tramadol • Conzip
Trazodone • Desyrel, Oleptro
Valproic acid • Depakene, Depakote
Vecuronium • Norcuron
Zolmitriptan • Zomig
1. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
2. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395-400.
3. Wang RZ, Vashistha V, Kaur S, et al. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
4. Bartlett D. Drug-induced serotonin syndrome. Crit Care Nurse. 2017;37(1):49-54.
5. Frank C. Recognition and treatment of serotonin syndrome. Can Fam Physician. 2008;54(7):988-992.
6. Cyproheptadine hydrochloride tablets [package insert]. Hayward, CA: Impax Generics; 2017.
7. Deardorff OG, Khan T, Kulkarni G, et al. Serotonin syndrome: prophylactic treatment with cyproheptadine. Prim Care Companion CNS Disord. 2016;18(4). doi: 10.4088/PCC.16br01966.
Mr. S, age 55, comes to your clinic as a walk-in for management of major depressive disorder, insomnia, and migraines. He also has tobacco use disorder and hypertension. Several days ago, Mr. S had visited the clinic because he was continuing to experience depressive symptoms, so his sertraline was increased from 100 to 200 mg/d. His current medication regimen includes sertraline 200 mg/d, trazodone 100 mg/d, lisinopril 10 mg/d, and sumatriptan, 100 mg as needed for migraine. He says last week he used 4 or 5 doses of sumatriptan because he experienced several migraines. Mr. S also reports occasionally taking 2 tablets of trazodone instead of 1 on nights that he has trouble falling asleep.
Today, Mr. S presents with a low-grade fever, diarrhea, internal restlessness, and a racing heartbeat that started shortly after his last visit. During physical examination, he exhibits slow, continuous lateral eye movements. His vital signs are markedly elevated: blood pressure, 175/85 mm Hg; heart rate, 110 beats per minute; and temperature, 39°C (102.2°F). Based on his presentation, the treatment team decides to send Mr. S to urgent care for closer monitoring.
Serotonin syndrome is a drug-induced syndrome caused by overstimulation of serotonin receptors. The syndrome is characterized by a classic clinical triad consisting of mental status changes, autonomic hyperactivity, and neuromuscular abnormalities. The clinical presentation is highly variable, and the severity ranges from mild to life-threatening.1-3 The incidence and prevalence of serotonin syndrome has not been well defined.3 Serotonin syndrome may be underreported because mild cases are often overlooked due to nonspecific symptoms. In addition, lack of physician awareness of drug–drug interactions, signs and symptoms, and differential diagnoses may result in underdiagnosis or misdiagnosis.1-3
What causes it?
Serotonin syndrome is usually a consequence of a drug–drug interaction between 2 or more serotonergic agents.4 Serotonin syndrome may result following medication misuse, overdose, initiation of a serotonergic agent, or increase in the dose of a currently prescribed serotonergic agent.3,4 In addition to medication classes and specific agents, Table 12-5 lists the drug mechanisms associated with serotonin syndrome:
- inhibition of serotonin reuptake
- inhibition of serotonin metabolism
- increased serotonin synthesis
- agonism of the serotonin receptor.
The amount of serotonergic activity most likely to cause serotonin syndrome is unclear.4
Pathophysiology. Serotonin, also known as 5-hydroxytryptamine (5-HT), is a metabolite of the amino acid tryptophan. This neurotransmitter is located in both the CNS and the periphery. Regulation of the serotonergic system begins in the presynaptic neurons with decarboxylation and hydroxylation of tryptophan resulting in serotonin synthesis. Once serotonin is produced, it is released into the synaptic cleft, where it binds to serotonin receptors.1,4,5 After receptor binding, serotonin reuptake occurs in the presynaptic neurons, where it can be metabolized by the monoamine oxidase enzyme. Finally, the metabolites are excreted in the urine. Serotonin syndrome results when this regulatory system is disrupted due to hyperstimulation of the postsynaptic serotonin receptors, mainly via agonism of the 5-HT2A and 5-HT1A receptors.1,4,5
Continue to: A nonspecific presentation
A nonspecific presentation
Unfortunately, many of the symptoms of serotonin syndrome are nonspecific, and the severity varies among patients.2,3 The onset of symptoms usually occurs within 6 to 8 hours after ingestion of a serotonergic agent.5 It is important to immediately recognize the symptoms (Table 22-5) and formulate a differential diagnosis because sudden progression of symptoms is common and may lead to life-threatening circumstances.1,3
In mild cases of serotonin syndrome, patients may have a low-grade fever or be afebrile. Hyperthermia tends to be present in moderate and severe cases, with temperatures >41°C (105.8°F) during life-threatening cases. Diaphoresis and tachycardia may be present regardless of severity. Additional autonomic irregularities include hypertension, tachypnea, nausea, vomiting, diarrhea, and hyperactive bowel sounds. In terms of neuromuscular abnormalities, hyperreflexia is a primary concern, as well as myoclonus. As the severity progresses to life-threatening, the clonus may convert from inducible to spontaneous and slow, continuous lateral eye movements may be present. Additional neuromuscular symptoms include tremor, akathisia, and muscle rigidity.1,3-5
Common mental status changes during mild cases include restlessness and anxiety. Abnormal mentation during moderate cases may present as increased hypervigilance and agitation, and this may advance to delirium or coma in severe cases. As the severity intensifies, the risk of developing additional physiological complications also increases. Rhabdomyolysis may occur due to muscle damage and myoglobinuria secondary to hyperreflexia, myoclonus, hypertonicity, and muscle rigidity. Muscle breakdown may then progress to further complications, such as renal failure. In rare instances, serotonin syndrome can result in seizures or death.1,3-5
Medication history tips off the diagnosis
The first step in diagnosing serotonin syndrome is to conduct a thorough review of the patient’s medication history, specifically taking into account any recent exposure to serotonergic agents.3,5 It is important to ask about prescription medications as well as over-the-counter products, herbal supplements, and illicit substances.1,4 When reviewing the medication history, investigate whether there may have been a recent change in therapy with serotonergic agents. Also, determine when the patient’s symptoms began in relation to exposure to serotonergic agents.4
After the medication review, conduct a thorough physical and neurologic examination to identify current symptoms and severity.1,3 No specific laboratory test is available to definitively confirm the diagnosis of serotonin syndrome.1,4 Monitoring of serum serotonin is not recommended because the levels do not correlate with symptom severity.3 The recommended diagnostic tool is the Hunter Serotonin Toxicity Criteria (Figure1,3).3,4 Historically, the Sternbach’s Diagnostic Criteria for serotonin syndrome were used for diagnosis; however, the Hunter Serotonin Toxicity Criteria are more sensitive (96% vs 75%) and more specific (97% vs 84%) than the Sternbach’s Diagnostic Criteria for serotonin syndrome.1,3-5
Continue to: In addition to using the proper diagnostic tool...
In addition to using the proper diagnostic tool, conduct a differential diagnosis to rule out other drug-induced syndromes, such as anticholinergic toxidrome, neuroleptic malignant syndrome, or malignant hyperthermia.1,3,5 Autonomic instability, including hypertension, tachycardia, tachypnea, and hyperthermia, may be present in all of the aforementioned drug-induced syndromes.1 As a result, the clinician must monitor for other symptoms that may differentiate the disease states to establish a clear diagnosis.
Discontinue agents, offer supportive care
There are no official published guidelines for managing serotonin syndrome.5 Regardless of the severity of a patient’s presentation, all serotonergic agents should be discontinued immediately. In addition, supportive care should be initiated for symptom management. Intravenous fluid replacement is recommended for hydration and to treat hyperthermia. External cooling may also be warranted to reduce body temperatures. Vital signs should be stabilized with appropriate pharmacotherapy.1,3-5
Benzodiazepines are considered a mainstay for relief of agitation during serotonin syndrome of any severity. In life-threatening cases—which are characterized by hyperthermia >41°C (105.8°F)—sedation, paralysis, and intubation may be necessary to maintain the airway, breathing, and circulation.1,3-5 Because treatment of hyperthermia requires elimination of hyperreflexia, paralysis is recommended.1 Nondepolarizing neuromuscular blocking agents, such as vecuronium, are preferred over depolarizing agents due to their decreased potential for rhabdomyolysis.1,3
Cyproheptadine, a histamine-1 receptor antagonist and a 5-HT2A receptor antagonist, is recommended for off-label treatment of serotonin syndrome to help decrease the intensity of symptoms. This should be initiated as a single dose of 12 mg followed by 2 mg every 2 hours until symptoms improve.1,3,5 After stabilization, a maintenance dose of 8 mg every 6 hours is recommended. Doses should not exceed the maximum recommended dose of 0.5 mg/kg/d.1,3,6 The most common adverse reactions associated with cyproheptadine are sedation and anticholinergic adverse effects.1,4,6
Antipsychotics, such as olanzapine and chlorpromazine, have been considered treatment alternatives due to their associated 5-HT2A receptor antagonism. However, there is limited data supporting such use.1,4 Antipsychotics should be used with caution because neuroleptic malignant syndrome may be mistaken for serotonin syndrome. Use of antipyretics is not recommended for treating fever and hyperthermia because the increase in body temperature is secondary to excessive muscle activity rather than dysfunction of the hypothalamic temperature set point.1,3,5 Physical restraints are also not recommended because their use may provoke further hyperthermia and increase the risk of rhabdomyolysis.3,5
Continue to: Ultimately, the duration of treatment...
Ultimately, the duration of treatment will be influenced by the pharmacokinetics of the serotonergic agents that induced the serotonin syndrome. Following resolution, retrial of the offending serotonergic agents should be carefully assessed. A retrial should only be considered after an adequate washout period has been observed, and clinicians should consider utilizing lower doses.2,5
Take steps for prevention
Patients at highest risk of developing serotonin syndrome are those who have multiple comorbidities that result in treatment with multiple serotonergic agents.3 Clinicians and patients alike need to be educated about the signs and symptoms of serotonin syndrome to promote early recognition. Also consider modifying your prescribing practices to minimize the use of multiple serotonergic agents. When switching between serotonergic agents, institute safe washout periods. Encourage patients to adhere to their prescribed medication regimens. Using electronic ordering systems can help detect drug–drug interactions.1,3 Prophylaxis with cyproheptadine may be considered in high-risk patients; however, no clinical trials have been conducted to evaluate using cyproheptadine to prevent serotonin syndrome.7
CASE CONTINUED
Upon further assessment in urgent care, Mr. S is found to have muscle rigidity in addition to ocular clonus and a temperature >38°C (100.4°F). Because Mr. S’s symptoms coincide with a recent increase of sertraline and increased use of both trazodone and sumatriptan, he meets Hunter Serotonin Toxicity Criteria. Therefore, his symptoms are likely related to excessive increase in serotonergic activity. Mr. S is admitted to the hospital for closer monitoring, and his sertraline, trazodone, and sumatriptan are held. He receives IV fluids for several days as well as cyproheptadine, 8 mg every 6 hours after stabilization, until his symptoms resolve. On Day 4, Mr. S no longer experiences diarrhea and internal restlessness. His vital signs return to normal, and as a result of symptom resolution, he is discharged from the hospital. The treatment team discusses changing his medication regimen to avoid multiple serotonergic agents. Mr. S is switched from sertraline to bupropion XL, 150 mg/d. Sumatriptan, 100 mg/d as needed, is continued for acute migraine treatment. Trazodone is discontinued and replaced with melatonin, 3 mg/d. The team also counsels Mr. S on the importance of proper adherence to his medication regimen. He is advised to return to the clinic in 2 weeks for reassessment of safety and efficacy.
Related Resource
- Turner AH, Kim JJ, McCarron RM. Differentiating serotonin syndrome and neuroleptic malignant syndrome. Current Psychiatry. 2019;18(2):30-36.
Drug Brand Names
Almotriptan • Axert
Buprenorphine • Subutex
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cyproheptadine • Periactin
Eletriptan • Relpax
Frovatriptan • Frova
Granisetron • Kytril
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Methadone • Dolophine, Methadose
Metoclopramide • Reglan
Mirtazapine • Remeron
Naratriptan • Amerge
Olanzapine • Zyprexa
Ondansetron • Zofran
Rizatriptan • Maxalt
Sertraline • Zoloft
Sumatriptan • Imitrex tablets
Tapentadol • Nucynta
Tramadol • Conzip
Trazodone • Desyrel, Oleptro
Valproic acid • Depakene, Depakote
Vecuronium • Norcuron
Zolmitriptan • Zomig
Mr. S, age 55, comes to your clinic as a walk-in for management of major depressive disorder, insomnia, and migraines. He also has tobacco use disorder and hypertension. Several days ago, Mr. S had visited the clinic because he was continuing to experience depressive symptoms, so his sertraline was increased from 100 to 200 mg/d. His current medication regimen includes sertraline 200 mg/d, trazodone 100 mg/d, lisinopril 10 mg/d, and sumatriptan, 100 mg as needed for migraine. He says last week he used 4 or 5 doses of sumatriptan because he experienced several migraines. Mr. S also reports occasionally taking 2 tablets of trazodone instead of 1 on nights that he has trouble falling asleep.
Today, Mr. S presents with a low-grade fever, diarrhea, internal restlessness, and a racing heartbeat that started shortly after his last visit. During physical examination, he exhibits slow, continuous lateral eye movements. His vital signs are markedly elevated: blood pressure, 175/85 mm Hg; heart rate, 110 beats per minute; and temperature, 39°C (102.2°F). Based on his presentation, the treatment team decides to send Mr. S to urgent care for closer monitoring.
Serotonin syndrome is a drug-induced syndrome caused by overstimulation of serotonin receptors. The syndrome is characterized by a classic clinical triad consisting of mental status changes, autonomic hyperactivity, and neuromuscular abnormalities. The clinical presentation is highly variable, and the severity ranges from mild to life-threatening.1-3 The incidence and prevalence of serotonin syndrome has not been well defined.3 Serotonin syndrome may be underreported because mild cases are often overlooked due to nonspecific symptoms. In addition, lack of physician awareness of drug–drug interactions, signs and symptoms, and differential diagnoses may result in underdiagnosis or misdiagnosis.1-3
What causes it?
Serotonin syndrome is usually a consequence of a drug–drug interaction between 2 or more serotonergic agents.4 Serotonin syndrome may result following medication misuse, overdose, initiation of a serotonergic agent, or increase in the dose of a currently prescribed serotonergic agent.3,4 In addition to medication classes and specific agents, Table 12-5 lists the drug mechanisms associated with serotonin syndrome:
- inhibition of serotonin reuptake
- inhibition of serotonin metabolism
- increased serotonin synthesis
- agonism of the serotonin receptor.
The amount of serotonergic activity most likely to cause serotonin syndrome is unclear.4
Pathophysiology. Serotonin, also known as 5-hydroxytryptamine (5-HT), is a metabolite of the amino acid tryptophan. This neurotransmitter is located in both the CNS and the periphery. Regulation of the serotonergic system begins in the presynaptic neurons with decarboxylation and hydroxylation of tryptophan resulting in serotonin synthesis. Once serotonin is produced, it is released into the synaptic cleft, where it binds to serotonin receptors.1,4,5 After receptor binding, serotonin reuptake occurs in the presynaptic neurons, where it can be metabolized by the monoamine oxidase enzyme. Finally, the metabolites are excreted in the urine. Serotonin syndrome results when this regulatory system is disrupted due to hyperstimulation of the postsynaptic serotonin receptors, mainly via agonism of the 5-HT2A and 5-HT1A receptors.1,4,5
Continue to: A nonspecific presentation
A nonspecific presentation
Unfortunately, many of the symptoms of serotonin syndrome are nonspecific, and the severity varies among patients.2,3 The onset of symptoms usually occurs within 6 to 8 hours after ingestion of a serotonergic agent.5 It is important to immediately recognize the symptoms (Table 22-5) and formulate a differential diagnosis because sudden progression of symptoms is common and may lead to life-threatening circumstances.1,3
In mild cases of serotonin syndrome, patients may have a low-grade fever or be afebrile. Hyperthermia tends to be present in moderate and severe cases, with temperatures >41°C (105.8°F) during life-threatening cases. Diaphoresis and tachycardia may be present regardless of severity. Additional autonomic irregularities include hypertension, tachypnea, nausea, vomiting, diarrhea, and hyperactive bowel sounds. In terms of neuromuscular abnormalities, hyperreflexia is a primary concern, as well as myoclonus. As the severity progresses to life-threatening, the clonus may convert from inducible to spontaneous and slow, continuous lateral eye movements may be present. Additional neuromuscular symptoms include tremor, akathisia, and muscle rigidity.1,3-5
Common mental status changes during mild cases include restlessness and anxiety. Abnormal mentation during moderate cases may present as increased hypervigilance and agitation, and this may advance to delirium or coma in severe cases. As the severity intensifies, the risk of developing additional physiological complications also increases. Rhabdomyolysis may occur due to muscle damage and myoglobinuria secondary to hyperreflexia, myoclonus, hypertonicity, and muscle rigidity. Muscle breakdown may then progress to further complications, such as renal failure. In rare instances, serotonin syndrome can result in seizures or death.1,3-5
Medication history tips off the diagnosis
The first step in diagnosing serotonin syndrome is to conduct a thorough review of the patient’s medication history, specifically taking into account any recent exposure to serotonergic agents.3,5 It is important to ask about prescription medications as well as over-the-counter products, herbal supplements, and illicit substances.1,4 When reviewing the medication history, investigate whether there may have been a recent change in therapy with serotonergic agents. Also, determine when the patient’s symptoms began in relation to exposure to serotonergic agents.4
After the medication review, conduct a thorough physical and neurologic examination to identify current symptoms and severity.1,3 No specific laboratory test is available to definitively confirm the diagnosis of serotonin syndrome.1,4 Monitoring of serum serotonin is not recommended because the levels do not correlate with symptom severity.3 The recommended diagnostic tool is the Hunter Serotonin Toxicity Criteria (Figure1,3).3,4 Historically, the Sternbach’s Diagnostic Criteria for serotonin syndrome were used for diagnosis; however, the Hunter Serotonin Toxicity Criteria are more sensitive (96% vs 75%) and more specific (97% vs 84%) than the Sternbach’s Diagnostic Criteria for serotonin syndrome.1,3-5
Continue to: In addition to using the proper diagnostic tool...
In addition to using the proper diagnostic tool, conduct a differential diagnosis to rule out other drug-induced syndromes, such as anticholinergic toxidrome, neuroleptic malignant syndrome, or malignant hyperthermia.1,3,5 Autonomic instability, including hypertension, tachycardia, tachypnea, and hyperthermia, may be present in all of the aforementioned drug-induced syndromes.1 As a result, the clinician must monitor for other symptoms that may differentiate the disease states to establish a clear diagnosis.
Discontinue agents, offer supportive care
There are no official published guidelines for managing serotonin syndrome.5 Regardless of the severity of a patient’s presentation, all serotonergic agents should be discontinued immediately. In addition, supportive care should be initiated for symptom management. Intravenous fluid replacement is recommended for hydration and to treat hyperthermia. External cooling may also be warranted to reduce body temperatures. Vital signs should be stabilized with appropriate pharmacotherapy.1,3-5
Benzodiazepines are considered a mainstay for relief of agitation during serotonin syndrome of any severity. In life-threatening cases—which are characterized by hyperthermia >41°C (105.8°F)—sedation, paralysis, and intubation may be necessary to maintain the airway, breathing, and circulation.1,3-5 Because treatment of hyperthermia requires elimination of hyperreflexia, paralysis is recommended.1 Nondepolarizing neuromuscular blocking agents, such as vecuronium, are preferred over depolarizing agents due to their decreased potential for rhabdomyolysis.1,3
Cyproheptadine, a histamine-1 receptor antagonist and a 5-HT2A receptor antagonist, is recommended for off-label treatment of serotonin syndrome to help decrease the intensity of symptoms. This should be initiated as a single dose of 12 mg followed by 2 mg every 2 hours until symptoms improve.1,3,5 After stabilization, a maintenance dose of 8 mg every 6 hours is recommended. Doses should not exceed the maximum recommended dose of 0.5 mg/kg/d.1,3,6 The most common adverse reactions associated with cyproheptadine are sedation and anticholinergic adverse effects.1,4,6
Antipsychotics, such as olanzapine and chlorpromazine, have been considered treatment alternatives due to their associated 5-HT2A receptor antagonism. However, there is limited data supporting such use.1,4 Antipsychotics should be used with caution because neuroleptic malignant syndrome may be mistaken for serotonin syndrome. Use of antipyretics is not recommended for treating fever and hyperthermia because the increase in body temperature is secondary to excessive muscle activity rather than dysfunction of the hypothalamic temperature set point.1,3,5 Physical restraints are also not recommended because their use may provoke further hyperthermia and increase the risk of rhabdomyolysis.3,5
Continue to: Ultimately, the duration of treatment...
Ultimately, the duration of treatment will be influenced by the pharmacokinetics of the serotonergic agents that induced the serotonin syndrome. Following resolution, retrial of the offending serotonergic agents should be carefully assessed. A retrial should only be considered after an adequate washout period has been observed, and clinicians should consider utilizing lower doses.2,5
Take steps for prevention
Patients at highest risk of developing serotonin syndrome are those who have multiple comorbidities that result in treatment with multiple serotonergic agents.3 Clinicians and patients alike need to be educated about the signs and symptoms of serotonin syndrome to promote early recognition. Also consider modifying your prescribing practices to minimize the use of multiple serotonergic agents. When switching between serotonergic agents, institute safe washout periods. Encourage patients to adhere to their prescribed medication regimens. Using electronic ordering systems can help detect drug–drug interactions.1,3 Prophylaxis with cyproheptadine may be considered in high-risk patients; however, no clinical trials have been conducted to evaluate using cyproheptadine to prevent serotonin syndrome.7
CASE CONTINUED
Upon further assessment in urgent care, Mr. S is found to have muscle rigidity in addition to ocular clonus and a temperature >38°C (100.4°F). Because Mr. S’s symptoms coincide with a recent increase of sertraline and increased use of both trazodone and sumatriptan, he meets Hunter Serotonin Toxicity Criteria. Therefore, his symptoms are likely related to excessive increase in serotonergic activity. Mr. S is admitted to the hospital for closer monitoring, and his sertraline, trazodone, and sumatriptan are held. He receives IV fluids for several days as well as cyproheptadine, 8 mg every 6 hours after stabilization, until his symptoms resolve. On Day 4, Mr. S no longer experiences diarrhea and internal restlessness. His vital signs return to normal, and as a result of symptom resolution, he is discharged from the hospital. The treatment team discusses changing his medication regimen to avoid multiple serotonergic agents. Mr. S is switched from sertraline to bupropion XL, 150 mg/d. Sumatriptan, 100 mg/d as needed, is continued for acute migraine treatment. Trazodone is discontinued and replaced with melatonin, 3 mg/d. The team also counsels Mr. S on the importance of proper adherence to his medication regimen. He is advised to return to the clinic in 2 weeks for reassessment of safety and efficacy.
Related Resource
- Turner AH, Kim JJ, McCarron RM. Differentiating serotonin syndrome and neuroleptic malignant syndrome. Current Psychiatry. 2019;18(2):30-36.
Drug Brand Names
Almotriptan • Axert
Buprenorphine • Subutex
Bupropion • Wellbutrin, Zyban
Buspirone • BuSpar
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Cyproheptadine • Periactin
Eletriptan • Relpax
Frovatriptan • Frova
Granisetron • Kytril
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Methadone • Dolophine, Methadose
Metoclopramide • Reglan
Mirtazapine • Remeron
Naratriptan • Amerge
Olanzapine • Zyprexa
Ondansetron • Zofran
Rizatriptan • Maxalt
Sertraline • Zoloft
Sumatriptan • Imitrex tablets
Tapentadol • Nucynta
Tramadol • Conzip
Trazodone • Desyrel, Oleptro
Valproic acid • Depakene, Depakote
Vecuronium • Norcuron
Zolmitriptan • Zomig
1. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
2. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395-400.
3. Wang RZ, Vashistha V, Kaur S, et al. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
4. Bartlett D. Drug-induced serotonin syndrome. Crit Care Nurse. 2017;37(1):49-54.
5. Frank C. Recognition and treatment of serotonin syndrome. Can Fam Physician. 2008;54(7):988-992.
6. Cyproheptadine hydrochloride tablets [package insert]. Hayward, CA: Impax Generics; 2017.
7. Deardorff OG, Khan T, Kulkarni G, et al. Serotonin syndrome: prophylactic treatment with cyproheptadine. Prim Care Companion CNS Disord. 2016;18(4). doi: 10.4088/PCC.16br01966.
1. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352(11):1112-1120.
2. Beakley BD, Kaye AM, Kaye AD. Tramadol, pharmacology, side effects, and serotonin syndrome: a review. Pain Physician. 2015;18(4):395-400.
3. Wang RZ, Vashistha V, Kaur S, et al. Serotonin syndrome: preventing, recognizing, and treating it. Cleve Clin J Med. 2016;83(11):810-817.
4. Bartlett D. Drug-induced serotonin syndrome. Crit Care Nurse. 2017;37(1):49-54.
5. Frank C. Recognition and treatment of serotonin syndrome. Can Fam Physician. 2008;54(7):988-992.
6. Cyproheptadine hydrochloride tablets [package insert]. Hayward, CA: Impax Generics; 2017.
7. Deardorff OG, Khan T, Kulkarni G, et al. Serotonin syndrome: prophylactic treatment with cyproheptadine. Prim Care Companion CNS Disord. 2016;18(4). doi: 10.4088/PCC.16br01966.

Acute Graft-vs-host Disease Following Liver Transplantation
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
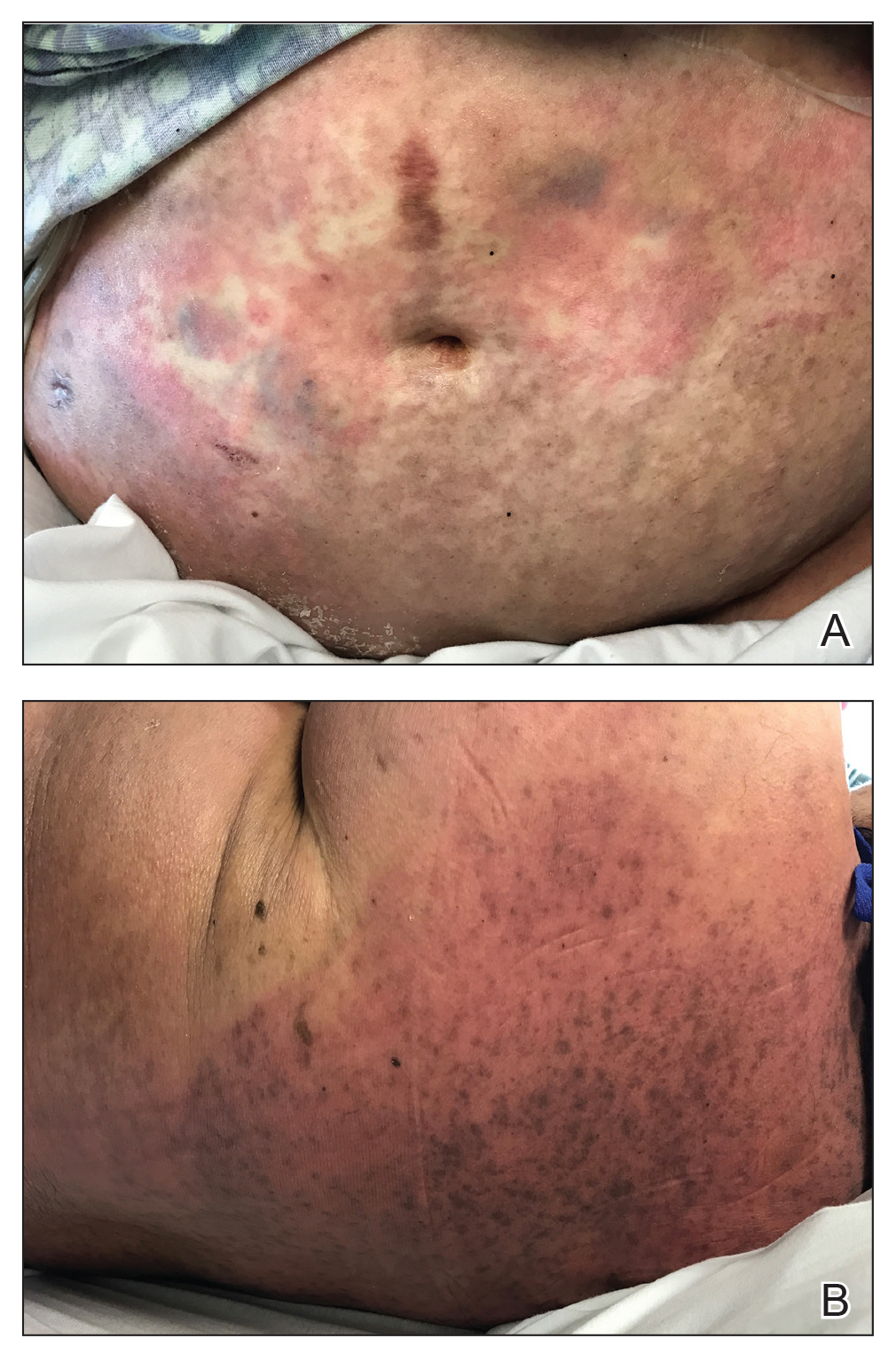
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
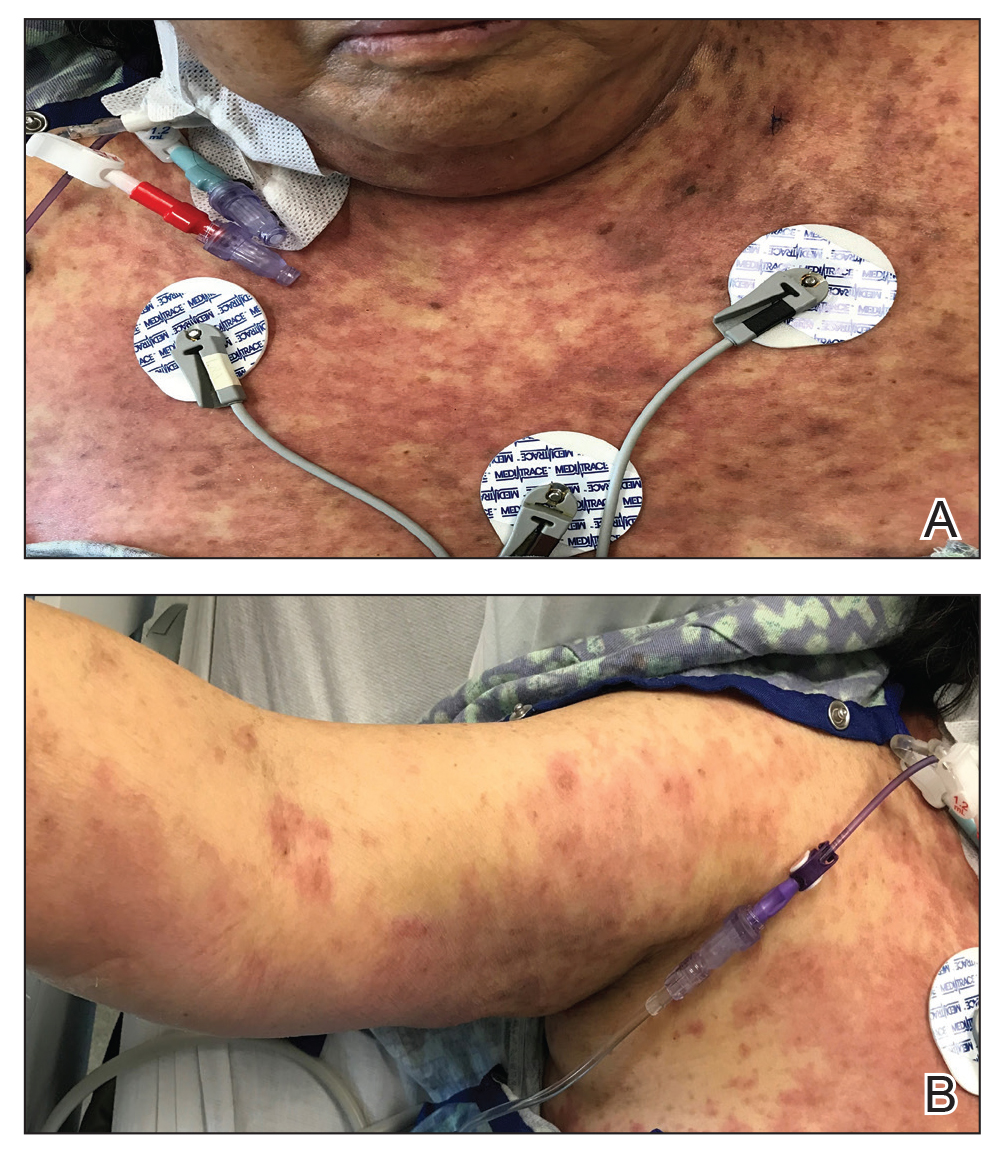
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
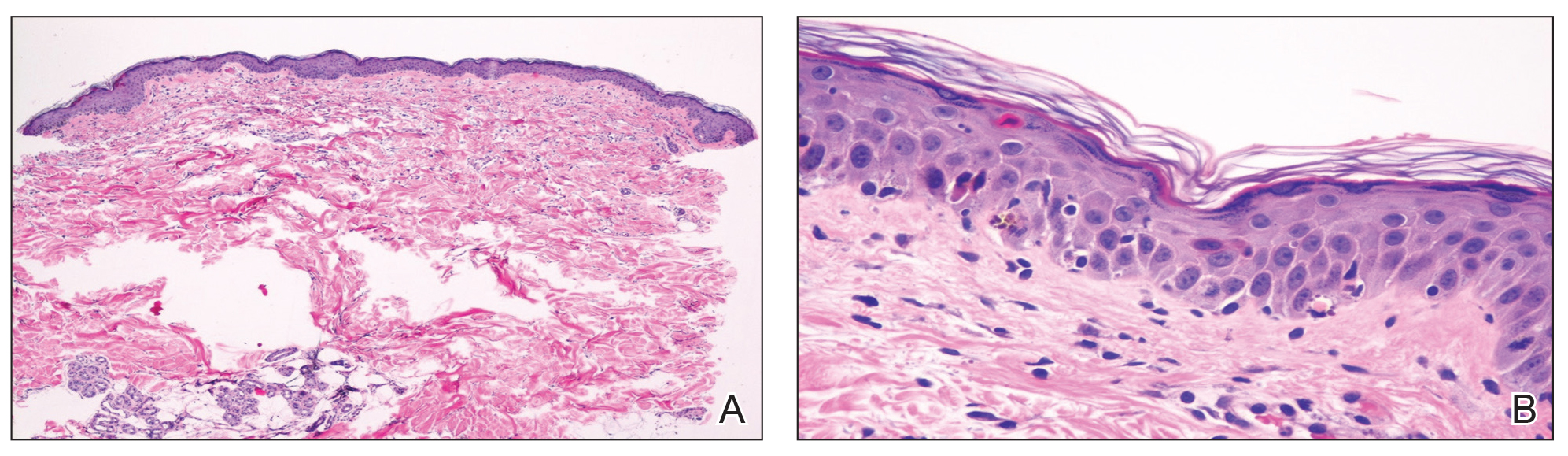
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
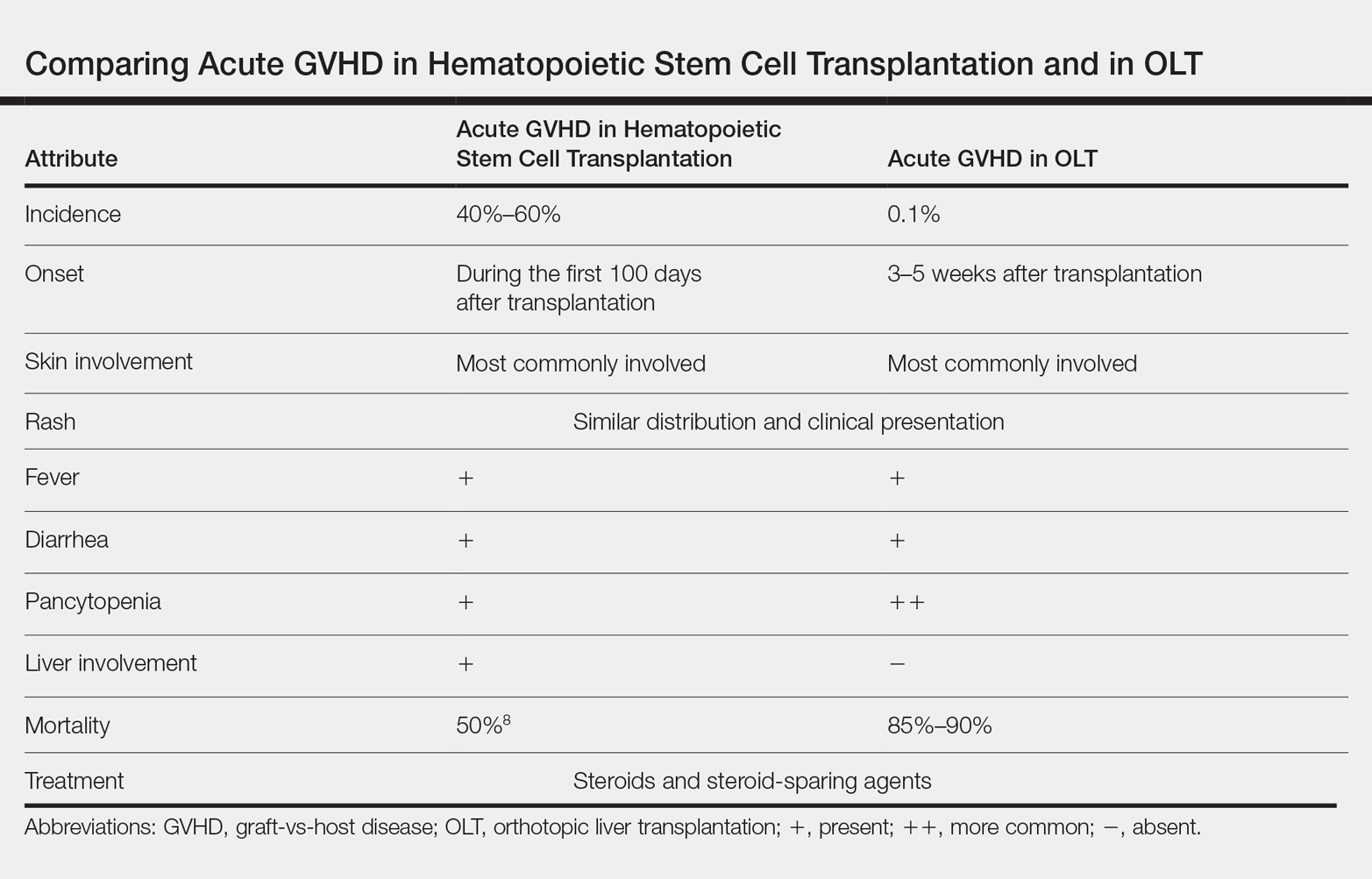
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Practice Points
- Acute graft-vs-host disease (GVHD) is a T cell–mediated reaction in which donor T lymphocytes attack host tissue in the setting of immunosuppression.
- Acute GVHD is more common in allogeneic stem cell transplantation but can occur in the setting of solid organ transplantation.
- Symptoms of acute GVHD include rash with or without pruritus, fever, pancytopenia, and diarrhea.
- Early recognition and treatment with systemic steroids can improve mortality.
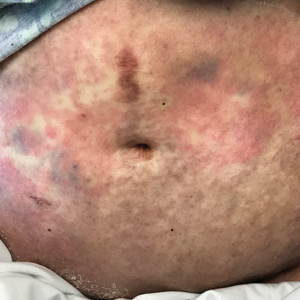
Trends in Nail Services May Cause Dermatitis: Not Your Mother’s Nail Polish
In 2017, consumers spent an average of $8.53 billion on nail services.1 This booming industry is set to grow to more than $15.5 billion by 2024.2 Nail polishes and other nail cosmetic trends can present new exposures for consumers, including chemicals that can elicit allergic contact dermatitis. In this article, we discuss new nail trends and their associated allergens, the acrylates.
Tosylamide/Formaldehyde Resin
Traditionally, the most widely recognized nail polish allergen has been tosylamide/formaldehyde resin (TSFR). However, there now are many touted TSFR-free nail polishes on the market, and the rate of positive reactions to this chemical has been declining in recent years. The North American Contact Dermatitis Group reported a positive reaction rate of 1.3% from 2005 through 2006,3 and rates decreased to 0.9% from 2015 through 2016.4 An Australian study demonstrated a similar reduction in positive reaction rates to nail polish chemicals, with only 0.7% of patients reacting to TSFR from 2014 to 2016 and 0% in 2017. It is theorized that this reduction occurred from replacing TSFR in traditional nail polishes with other chemicals such as polyester resins and cellulose acetate butyrate.5
Acrylate-Based Nail Treatments
Consumers recently have been gravitating toward acrylate-based nail treatments vs traditional nail polishes for a variety of reasons. Often referred to as gels, dips, or shellac, acrylate-based nail treatments represent a hot new trend in nail cosmetics. These manicures are resistant to chipping and scratches, creating a like-new look that lasts for weeks after application. The long-lasting nature of acrylate-based nail polishes has made them wildly popular with consumers.
Traditional acrylic nails consist of a powder polymer mixed with a liquid monomer, which polymerizes when a catalyst is added.6 The procedure is time consuming and can take up to 2 hours for application. In contrast, the newer gel manicure can be completed faster and includes application of acrylate-based nail polish, including a base coat, 2 coats of color, and a top coat. Exposure to either a light-emitting diode (30–60 seconds) or UVA (2 minutes) lamp is necessary after each coat is applied for polymerization (Figure 1).6 This long-lasting, semipermanent manicure typically is what patients are referring to when they say they have “gel nails.”
Gel dipping powders (referred to as dips) are another long-lasting acrylate-based nail treatment. This type of polish uses ethyl cyanoacrylate, a slightly different acrylate (yes, that IS super glue). After the nail is prepared, a base polish is applied to three-quarters of the nail and it is dipped into a natural color dip powder. The base polish is then applied to the entire nail, followed by a dip into the polish color of choice. This process is completed twice, followed by shaping and application of a top coat (Figure 2).
base coat. B, Application of dip powder to gel polish. Note the entire
distal finger and nail are dipped into the powder. C, Shaping of the
nail after the second coat of color is applied.
Finally, there are nail wraps, which are similar to stickers placed over or extending the nail plate. The wraps can be made from linen, silk, vinyl, or other material. Ethyl cyanoacrylate and isopropyl-2-cyanoacrylates have been identified in nail wrap adhesive.7 The heated product is directly applied to the prepared nail, and the excess wrap is filed off. Additional nail polish and a top coat usually are applied to finish the nail. Many of these products are available for in-salon use as well as online purchase and home application by consumers.
Acrylate Allergy
Patients who are allergic to acrylates can present with different patterns of dermatitis. Although the majority of patients present with dermatitis on the hands, fingers, or wrists, up to 10% may only have facial and neck dermatitis.8 Less commonly, the abdomen and thighs can be involved.6,8 Nail technicians most commonly present with pulpitis with cutaneous fissures.8 Other symptoms can include subungual hyperkeratosis, onycholysis, and nail dystrophy. Paresthesia, urticaria, and upper respiratory tract symptoms can occur but are less common.6,8
Acrylate allergy typically is the result of sensitization to the acrylate monomers. In theory, gel nail acrylate materials are polymerized following exposure to a light-emitting diode or UVA lamp; however, there likely is some incomplete polymerization, which can increase the risk for development of allergy. Allergen exposure can occur due to incorrect application of the light source; inadvertent monomer exposure, which occurs when nail technicians wipe extra acrylate off of a client’s finger(s); or inadvertent application of acrylate monomers to objects in the nail technician’s work environment.6,8
Several acrylate nail allergens have been reported. Many studies have identified 2-hydroxyethyl methacrylate (HEMA) as the most common nail acrylate allergen.8,9 At least one study identified 2-hydroxypropyl methacrylate as the most common, with HEMA in second place.6 Other reported acrylate allergens have included ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, methyl methacrylate, ethyl cyanoacrylate, 1,4-butanediol diacrylate, hydroxypropyl acrylate, and 2-hydroxyethyl acrylate.8,9
The American Contact Dermatitis Society Core Allergen Series and the North American Contact Dermatitis Group screening series currently include HEMA, methyl methacrylate, ethyl acrylate, ethyl cyanoacrylate, and TSFR.4,10 Of note, acrylates are not included in the thin-layer rapid use epicutaneous (T.R.U.E.) patch test (SmartPractice), so they will be missed if this series is used.11 In the setting of suspected nail acrylate allergy, some authors recommend initial screening with HEMA and ethyl cyanoacrylate, with extended acrylate testing if both are negative.8
Upon patch testing with an acrylate series, patients frequently react to 2 or more acrylates and the reactions can be strong (++) or extreme (+++), which may represent cosensitization or cross-sensitization.8 The likelihood of cross-reactivity between acrylates is not clear, though it has been postulated that it is theoretically possible.6
An important pearl for patch testers using the chamber method is proper storage of acrylate allergens and assembly of trays prior to patch testing. Similar to all haptens, manufacturers recommend that acrylates should be stored in a refrigerator, but some authors suggest that acrylates should be stored in the freezer.12 Acrylates are volatile chemicals and rapidly degrade when exposed to air. A methyl methacrylate preparation loaded into an inert quadrate (IQ) chamber and stored at room temperature showed a nearly undetectable amount of any residual methyl methacrylate 24 hours later. Refrigeration of allergens in chambers slowed but did not stop eventual degradation, with nearly all acrylate preparations reaching an undetectable level of allergen by day 8.13 Acrylates, along with other volatile allergens, should only be loaded into chambers immediately prior to placement on the patient.
Allergy Prevention
Prevention of nail acrylate allergy among consumers is simple: avoid contact with the offending allergen. Acrylate spillover (ie, applying the acrylate onto the skin) and direct contact with objects and working surfaces contaminated with acrylate-based nail products should be avoided.8 Avoidance is more complicated for nail technicians, but it is thought that nitrile gloves allow for the best dexterity and allergen avoidance when acrylate exposure is brief.14 Allowable exposure times with nitrile gloves may be 15 to 30 minutes. After this times passes, a glove change is required to avoid exposure.14 Wearing nitrile gloves for longer than 15 to 30 minutes will result in cutaneous exposure and risk for dermatitis in sensitized patients. If longer wear is desired, one option includes cutting the fingertips off of Silver Shield/4H gloves (Honeywell Safety Products USA, Inc), applying them to the distal fingers, and wearing a standard nitrile glove over top, known as the finger stall technique.6 In one study, this technique was recommended to nail technicians with acrylate allergy. A telephone survey conducted 4 to 43 months later confirmed that 36% (8/22) of participants were using the technique without symptoms. In this same study, 73% (16/22) had continued working as nail technicians.6
Acrylates are used for other medical purposes, including dental procedures, orthopedic procedures, surgical glues, wound dressings, and contact and intraocular lenses. They also have additional cosmetic applications, including eyelash and hair extensions.8 Therefore, it is vital that patients disclose any history of acrylate allergy to both their medical and cosmetic providers.
Our Final Interpretation
Acrylate allergy has become increasingly common, and long-lasting nail treatments often are the culprit. Whether through gels, dips, or shellac, repeated exposure to acrylates through nail treatments can increase the risk for allergy. The T.R.U.E. test alone will not make the diagnosis, as acrylates are not present in this patch test system. It is important to remind your allergic patients that acrylates are present in other compounds used for medical and cosmetic purposes. Avoidance is key, and for allergic patients who love to bedazzle their nails, we suggest less-permanent, acrylate-free nail polishes as alternatives.
- 2017-2018 industry statistics highlights. Nails Magazine. http://files.nailsmag.com/handouts/nabb2017-18stats-lr.pdf. Accessed May 17, 2019.
- Nail polish market size worth $15.55 billion by 2024. Grand View Research website. https://www.grandviewresearch.com/press-release/global-nail-polish-market. Published October 2017. Accessed May 17, 2019.
- Zug KA, Warshaw EM, Fowler JF, et al. Patch-test results of the North American Contact Dermatitis Group 2005-2006. Dermatitis. 2009;20:149-160.
- DeKoven J, Warshaw EM, Zug KA, et al. North American Contact Dermatitis Group patch test results: 2015-2016. Dermatitis. 2018;29:297-309.
- Lee S, Maor D, Palmer A, et al. Declining prevalence of allergic contact dermatitis caused by tosylamide/formaldehyde in nail polish. Contact Dermatitis. 2018;79:184-185.
- Gatica-Ortega ME, Pastor-Nieto MA, Mercader-García P, et al. Allergic contact dermatitis caused by (meth)acrylates in long-lasting nail polish: are we facing a new epidemic in the beauty industry? Contact Dermatitis. 2017;7:360-366.
- Fitzgerald DA, Bhaggoe R, English JS. Contact sensitivity to cyanoacrylate nail-adhesive with dermatitis at remote sites. Contact Dermatitis. 1995;32:175-176.
- Goncalo M, Pinho A, Agner T et al. Allergic contact dermatitis caused by nail acrylates in Europe. an EECDRG study. Contact Dermatitis. 2017;78:254-260.
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians—a 10-year study [published online January 14, 2019]. Contact Dermatitis. doi:10.1111/cod.13216.
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society core allergen series: 2017 update. Dermatitis. 2017;28:141-143.
- T.R.U.E. TEST ready-to-use patch test panels. Smart Practice website. https://www.smartpractice.com/shop/wa/category?cn=T.R.U.E.-TEST%C2%AE-Ready-to-Use-Patch-Test-Panels&id=508222&m=SPA. Accessed May 17, 2019.
- Good AT, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methylacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Goon A, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians—thoughts on a rising concern [published online January 17, 2019]. Contact Dermatitis. doi:10.1111/cod.13222.
In 2017, consumers spent an average of $8.53 billion on nail services.1 This booming industry is set to grow to more than $15.5 billion by 2024.2 Nail polishes and other nail cosmetic trends can present new exposures for consumers, including chemicals that can elicit allergic contact dermatitis. In this article, we discuss new nail trends and their associated allergens, the acrylates.
Tosylamide/Formaldehyde Resin
Traditionally, the most widely recognized nail polish allergen has been tosylamide/formaldehyde resin (TSFR). However, there now are many touted TSFR-free nail polishes on the market, and the rate of positive reactions to this chemical has been declining in recent years. The North American Contact Dermatitis Group reported a positive reaction rate of 1.3% from 2005 through 2006,3 and rates decreased to 0.9% from 2015 through 2016.4 An Australian study demonstrated a similar reduction in positive reaction rates to nail polish chemicals, with only 0.7% of patients reacting to TSFR from 2014 to 2016 and 0% in 2017. It is theorized that this reduction occurred from replacing TSFR in traditional nail polishes with other chemicals such as polyester resins and cellulose acetate butyrate.5
Acrylate-Based Nail Treatments
Consumers recently have been gravitating toward acrylate-based nail treatments vs traditional nail polishes for a variety of reasons. Often referred to as gels, dips, or shellac, acrylate-based nail treatments represent a hot new trend in nail cosmetics. These manicures are resistant to chipping and scratches, creating a like-new look that lasts for weeks after application. The long-lasting nature of acrylate-based nail polishes has made them wildly popular with consumers.
Traditional acrylic nails consist of a powder polymer mixed with a liquid monomer, which polymerizes when a catalyst is added.6 The procedure is time consuming and can take up to 2 hours for application. In contrast, the newer gel manicure can be completed faster and includes application of acrylate-based nail polish, including a base coat, 2 coats of color, and a top coat. Exposure to either a light-emitting diode (30–60 seconds) or UVA (2 minutes) lamp is necessary after each coat is applied for polymerization (Figure 1).6 This long-lasting, semipermanent manicure typically is what patients are referring to when they say they have “gel nails.”
Gel dipping powders (referred to as dips) are another long-lasting acrylate-based nail treatment. This type of polish uses ethyl cyanoacrylate, a slightly different acrylate (yes, that IS super glue). After the nail is prepared, a base polish is applied to three-quarters of the nail and it is dipped into a natural color dip powder. The base polish is then applied to the entire nail, followed by a dip into the polish color of choice. This process is completed twice, followed by shaping and application of a top coat (Figure 2).
base coat. B, Application of dip powder to gel polish. Note the entire
distal finger and nail are dipped into the powder. C, Shaping of the
nail after the second coat of color is applied.
Finally, there are nail wraps, which are similar to stickers placed over or extending the nail plate. The wraps can be made from linen, silk, vinyl, or other material. Ethyl cyanoacrylate and isopropyl-2-cyanoacrylates have been identified in nail wrap adhesive.7 The heated product is directly applied to the prepared nail, and the excess wrap is filed off. Additional nail polish and a top coat usually are applied to finish the nail. Many of these products are available for in-salon use as well as online purchase and home application by consumers.
Acrylate Allergy
Patients who are allergic to acrylates can present with different patterns of dermatitis. Although the majority of patients present with dermatitis on the hands, fingers, or wrists, up to 10% may only have facial and neck dermatitis.8 Less commonly, the abdomen and thighs can be involved.6,8 Nail technicians most commonly present with pulpitis with cutaneous fissures.8 Other symptoms can include subungual hyperkeratosis, onycholysis, and nail dystrophy. Paresthesia, urticaria, and upper respiratory tract symptoms can occur but are less common.6,8
Acrylate allergy typically is the result of sensitization to the acrylate monomers. In theory, gel nail acrylate materials are polymerized following exposure to a light-emitting diode or UVA lamp; however, there likely is some incomplete polymerization, which can increase the risk for development of allergy. Allergen exposure can occur due to incorrect application of the light source; inadvertent monomer exposure, which occurs when nail technicians wipe extra acrylate off of a client’s finger(s); or inadvertent application of acrylate monomers to objects in the nail technician’s work environment.6,8
Several acrylate nail allergens have been reported. Many studies have identified 2-hydroxyethyl methacrylate (HEMA) as the most common nail acrylate allergen.8,9 At least one study identified 2-hydroxypropyl methacrylate as the most common, with HEMA in second place.6 Other reported acrylate allergens have included ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, methyl methacrylate, ethyl cyanoacrylate, 1,4-butanediol diacrylate, hydroxypropyl acrylate, and 2-hydroxyethyl acrylate.8,9
The American Contact Dermatitis Society Core Allergen Series and the North American Contact Dermatitis Group screening series currently include HEMA, methyl methacrylate, ethyl acrylate, ethyl cyanoacrylate, and TSFR.4,10 Of note, acrylates are not included in the thin-layer rapid use epicutaneous (T.R.U.E.) patch test (SmartPractice), so they will be missed if this series is used.11 In the setting of suspected nail acrylate allergy, some authors recommend initial screening with HEMA and ethyl cyanoacrylate, with extended acrylate testing if both are negative.8
Upon patch testing with an acrylate series, patients frequently react to 2 or more acrylates and the reactions can be strong (++) or extreme (+++), which may represent cosensitization or cross-sensitization.8 The likelihood of cross-reactivity between acrylates is not clear, though it has been postulated that it is theoretically possible.6
An important pearl for patch testers using the chamber method is proper storage of acrylate allergens and assembly of trays prior to patch testing. Similar to all haptens, manufacturers recommend that acrylates should be stored in a refrigerator, but some authors suggest that acrylates should be stored in the freezer.12 Acrylates are volatile chemicals and rapidly degrade when exposed to air. A methyl methacrylate preparation loaded into an inert quadrate (IQ) chamber and stored at room temperature showed a nearly undetectable amount of any residual methyl methacrylate 24 hours later. Refrigeration of allergens in chambers slowed but did not stop eventual degradation, with nearly all acrylate preparations reaching an undetectable level of allergen by day 8.13 Acrylates, along with other volatile allergens, should only be loaded into chambers immediately prior to placement on the patient.
Allergy Prevention
Prevention of nail acrylate allergy among consumers is simple: avoid contact with the offending allergen. Acrylate spillover (ie, applying the acrylate onto the skin) and direct contact with objects and working surfaces contaminated with acrylate-based nail products should be avoided.8 Avoidance is more complicated for nail technicians, but it is thought that nitrile gloves allow for the best dexterity and allergen avoidance when acrylate exposure is brief.14 Allowable exposure times with nitrile gloves may be 15 to 30 minutes. After this times passes, a glove change is required to avoid exposure.14 Wearing nitrile gloves for longer than 15 to 30 minutes will result in cutaneous exposure and risk for dermatitis in sensitized patients. If longer wear is desired, one option includes cutting the fingertips off of Silver Shield/4H gloves (Honeywell Safety Products USA, Inc), applying them to the distal fingers, and wearing a standard nitrile glove over top, known as the finger stall technique.6 In one study, this technique was recommended to nail technicians with acrylate allergy. A telephone survey conducted 4 to 43 months later confirmed that 36% (8/22) of participants were using the technique without symptoms. In this same study, 73% (16/22) had continued working as nail technicians.6
Acrylates are used for other medical purposes, including dental procedures, orthopedic procedures, surgical glues, wound dressings, and contact and intraocular lenses. They also have additional cosmetic applications, including eyelash and hair extensions.8 Therefore, it is vital that patients disclose any history of acrylate allergy to both their medical and cosmetic providers.
Our Final Interpretation
Acrylate allergy has become increasingly common, and long-lasting nail treatments often are the culprit. Whether through gels, dips, or shellac, repeated exposure to acrylates through nail treatments can increase the risk for allergy. The T.R.U.E. test alone will not make the diagnosis, as acrylates are not present in this patch test system. It is important to remind your allergic patients that acrylates are present in other compounds used for medical and cosmetic purposes. Avoidance is key, and for allergic patients who love to bedazzle their nails, we suggest less-permanent, acrylate-free nail polishes as alternatives.
In 2017, consumers spent an average of $8.53 billion on nail services.1 This booming industry is set to grow to more than $15.5 billion by 2024.2 Nail polishes and other nail cosmetic trends can present new exposures for consumers, including chemicals that can elicit allergic contact dermatitis. In this article, we discuss new nail trends and their associated allergens, the acrylates.
Tosylamide/Formaldehyde Resin
Traditionally, the most widely recognized nail polish allergen has been tosylamide/formaldehyde resin (TSFR). However, there now are many touted TSFR-free nail polishes on the market, and the rate of positive reactions to this chemical has been declining in recent years. The North American Contact Dermatitis Group reported a positive reaction rate of 1.3% from 2005 through 2006,3 and rates decreased to 0.9% from 2015 through 2016.4 An Australian study demonstrated a similar reduction in positive reaction rates to nail polish chemicals, with only 0.7% of patients reacting to TSFR from 2014 to 2016 and 0% in 2017. It is theorized that this reduction occurred from replacing TSFR in traditional nail polishes with other chemicals such as polyester resins and cellulose acetate butyrate.5
Acrylate-Based Nail Treatments
Consumers recently have been gravitating toward acrylate-based nail treatments vs traditional nail polishes for a variety of reasons. Often referred to as gels, dips, or shellac, acrylate-based nail treatments represent a hot new trend in nail cosmetics. These manicures are resistant to chipping and scratches, creating a like-new look that lasts for weeks after application. The long-lasting nature of acrylate-based nail polishes has made them wildly popular with consumers.
Traditional acrylic nails consist of a powder polymer mixed with a liquid monomer, which polymerizes when a catalyst is added.6 The procedure is time consuming and can take up to 2 hours for application. In contrast, the newer gel manicure can be completed faster and includes application of acrylate-based nail polish, including a base coat, 2 coats of color, and a top coat. Exposure to either a light-emitting diode (30–60 seconds) or UVA (2 minutes) lamp is necessary after each coat is applied for polymerization (Figure 1).6 This long-lasting, semipermanent manicure typically is what patients are referring to when they say they have “gel nails.”
Gel dipping powders (referred to as dips) are another long-lasting acrylate-based nail treatment. This type of polish uses ethyl cyanoacrylate, a slightly different acrylate (yes, that IS super glue). After the nail is prepared, a base polish is applied to three-quarters of the nail and it is dipped into a natural color dip powder. The base polish is then applied to the entire nail, followed by a dip into the polish color of choice. This process is completed twice, followed by shaping and application of a top coat (Figure 2).
base coat. B, Application of dip powder to gel polish. Note the entire
distal finger and nail are dipped into the powder. C, Shaping of the
nail after the second coat of color is applied.
Finally, there are nail wraps, which are similar to stickers placed over or extending the nail plate. The wraps can be made from linen, silk, vinyl, or other material. Ethyl cyanoacrylate and isopropyl-2-cyanoacrylates have been identified in nail wrap adhesive.7 The heated product is directly applied to the prepared nail, and the excess wrap is filed off. Additional nail polish and a top coat usually are applied to finish the nail. Many of these products are available for in-salon use as well as online purchase and home application by consumers.
Acrylate Allergy
Patients who are allergic to acrylates can present with different patterns of dermatitis. Although the majority of patients present with dermatitis on the hands, fingers, or wrists, up to 10% may only have facial and neck dermatitis.8 Less commonly, the abdomen and thighs can be involved.6,8 Nail technicians most commonly present with pulpitis with cutaneous fissures.8 Other symptoms can include subungual hyperkeratosis, onycholysis, and nail dystrophy. Paresthesia, urticaria, and upper respiratory tract symptoms can occur but are less common.6,8
Acrylate allergy typically is the result of sensitization to the acrylate monomers. In theory, gel nail acrylate materials are polymerized following exposure to a light-emitting diode or UVA lamp; however, there likely is some incomplete polymerization, which can increase the risk for development of allergy. Allergen exposure can occur due to incorrect application of the light source; inadvertent monomer exposure, which occurs when nail technicians wipe extra acrylate off of a client’s finger(s); or inadvertent application of acrylate monomers to objects in the nail technician’s work environment.6,8
Several acrylate nail allergens have been reported. Many studies have identified 2-hydroxyethyl methacrylate (HEMA) as the most common nail acrylate allergen.8,9 At least one study identified 2-hydroxypropyl methacrylate as the most common, with HEMA in second place.6 Other reported acrylate allergens have included ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, methyl methacrylate, ethyl cyanoacrylate, 1,4-butanediol diacrylate, hydroxypropyl acrylate, and 2-hydroxyethyl acrylate.8,9
The American Contact Dermatitis Society Core Allergen Series and the North American Contact Dermatitis Group screening series currently include HEMA, methyl methacrylate, ethyl acrylate, ethyl cyanoacrylate, and TSFR.4,10 Of note, acrylates are not included in the thin-layer rapid use epicutaneous (T.R.U.E.) patch test (SmartPractice), so they will be missed if this series is used.11 In the setting of suspected nail acrylate allergy, some authors recommend initial screening with HEMA and ethyl cyanoacrylate, with extended acrylate testing if both are negative.8
Upon patch testing with an acrylate series, patients frequently react to 2 or more acrylates and the reactions can be strong (++) or extreme (+++), which may represent cosensitization or cross-sensitization.8 The likelihood of cross-reactivity between acrylates is not clear, though it has been postulated that it is theoretically possible.6
An important pearl for patch testers using the chamber method is proper storage of acrylate allergens and assembly of trays prior to patch testing. Similar to all haptens, manufacturers recommend that acrylates should be stored in a refrigerator, but some authors suggest that acrylates should be stored in the freezer.12 Acrylates are volatile chemicals and rapidly degrade when exposed to air. A methyl methacrylate preparation loaded into an inert quadrate (IQ) chamber and stored at room temperature showed a nearly undetectable amount of any residual methyl methacrylate 24 hours later. Refrigeration of allergens in chambers slowed but did not stop eventual degradation, with nearly all acrylate preparations reaching an undetectable level of allergen by day 8.13 Acrylates, along with other volatile allergens, should only be loaded into chambers immediately prior to placement on the patient.
Allergy Prevention
Prevention of nail acrylate allergy among consumers is simple: avoid contact with the offending allergen. Acrylate spillover (ie, applying the acrylate onto the skin) and direct contact with objects and working surfaces contaminated with acrylate-based nail products should be avoided.8 Avoidance is more complicated for nail technicians, but it is thought that nitrile gloves allow for the best dexterity and allergen avoidance when acrylate exposure is brief.14 Allowable exposure times with nitrile gloves may be 15 to 30 minutes. After this times passes, a glove change is required to avoid exposure.14 Wearing nitrile gloves for longer than 15 to 30 minutes will result in cutaneous exposure and risk for dermatitis in sensitized patients. If longer wear is desired, one option includes cutting the fingertips off of Silver Shield/4H gloves (Honeywell Safety Products USA, Inc), applying them to the distal fingers, and wearing a standard nitrile glove over top, known as the finger stall technique.6 In one study, this technique was recommended to nail technicians with acrylate allergy. A telephone survey conducted 4 to 43 months later confirmed that 36% (8/22) of participants were using the technique without symptoms. In this same study, 73% (16/22) had continued working as nail technicians.6
Acrylates are used for other medical purposes, including dental procedures, orthopedic procedures, surgical glues, wound dressings, and contact and intraocular lenses. They also have additional cosmetic applications, including eyelash and hair extensions.8 Therefore, it is vital that patients disclose any history of acrylate allergy to both their medical and cosmetic providers.
Our Final Interpretation
Acrylate allergy has become increasingly common, and long-lasting nail treatments often are the culprit. Whether through gels, dips, or shellac, repeated exposure to acrylates through nail treatments can increase the risk for allergy. The T.R.U.E. test alone will not make the diagnosis, as acrylates are not present in this patch test system. It is important to remind your allergic patients that acrylates are present in other compounds used for medical and cosmetic purposes. Avoidance is key, and for allergic patients who love to bedazzle their nails, we suggest less-permanent, acrylate-free nail polishes as alternatives.
- 2017-2018 industry statistics highlights. Nails Magazine. http://files.nailsmag.com/handouts/nabb2017-18stats-lr.pdf. Accessed May 17, 2019.
- Nail polish market size worth $15.55 billion by 2024. Grand View Research website. https://www.grandviewresearch.com/press-release/global-nail-polish-market. Published October 2017. Accessed May 17, 2019.
- Zug KA, Warshaw EM, Fowler JF, et al. Patch-test results of the North American Contact Dermatitis Group 2005-2006. Dermatitis. 2009;20:149-160.
- DeKoven J, Warshaw EM, Zug KA, et al. North American Contact Dermatitis Group patch test results: 2015-2016. Dermatitis. 2018;29:297-309.
- Lee S, Maor D, Palmer A, et al. Declining prevalence of allergic contact dermatitis caused by tosylamide/formaldehyde in nail polish. Contact Dermatitis. 2018;79:184-185.
- Gatica-Ortega ME, Pastor-Nieto MA, Mercader-García P, et al. Allergic contact dermatitis caused by (meth)acrylates in long-lasting nail polish: are we facing a new epidemic in the beauty industry? Contact Dermatitis. 2017;7:360-366.
- Fitzgerald DA, Bhaggoe R, English JS. Contact sensitivity to cyanoacrylate nail-adhesive with dermatitis at remote sites. Contact Dermatitis. 1995;32:175-176.
- Goncalo M, Pinho A, Agner T et al. Allergic contact dermatitis caused by nail acrylates in Europe. an EECDRG study. Contact Dermatitis. 2017;78:254-260.
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians—a 10-year study [published online January 14, 2019]. Contact Dermatitis. doi:10.1111/cod.13216.
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society core allergen series: 2017 update. Dermatitis. 2017;28:141-143.
- T.R.U.E. TEST ready-to-use patch test panels. Smart Practice website. https://www.smartpractice.com/shop/wa/category?cn=T.R.U.E.-TEST%C2%AE-Ready-to-Use-Patch-Test-Panels&id=508222&m=SPA. Accessed May 17, 2019.
- Good AT, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methylacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Goon A, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians—thoughts on a rising concern [published online January 17, 2019]. Contact Dermatitis. doi:10.1111/cod.13222.
- 2017-2018 industry statistics highlights. Nails Magazine. http://files.nailsmag.com/handouts/nabb2017-18stats-lr.pdf. Accessed May 17, 2019.
- Nail polish market size worth $15.55 billion by 2024. Grand View Research website. https://www.grandviewresearch.com/press-release/global-nail-polish-market. Published October 2017. Accessed May 17, 2019.
- Zug KA, Warshaw EM, Fowler JF, et al. Patch-test results of the North American Contact Dermatitis Group 2005-2006. Dermatitis. 2009;20:149-160.
- DeKoven J, Warshaw EM, Zug KA, et al. North American Contact Dermatitis Group patch test results: 2015-2016. Dermatitis. 2018;29:297-309.
- Lee S, Maor D, Palmer A, et al. Declining prevalence of allergic contact dermatitis caused by tosylamide/formaldehyde in nail polish. Contact Dermatitis. 2018;79:184-185.
- Gatica-Ortega ME, Pastor-Nieto MA, Mercader-García P, et al. Allergic contact dermatitis caused by (meth)acrylates in long-lasting nail polish: are we facing a new epidemic in the beauty industry? Contact Dermatitis. 2017;7:360-366.
- Fitzgerald DA, Bhaggoe R, English JS. Contact sensitivity to cyanoacrylate nail-adhesive with dermatitis at remote sites. Contact Dermatitis. 1995;32:175-176.
- Goncalo M, Pinho A, Agner T et al. Allergic contact dermatitis caused by nail acrylates in Europe. an EECDRG study. Contact Dermatitis. 2017;78:254-260.
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians—a 10-year study [published online January 14, 2019]. Contact Dermatitis. doi:10.1111/cod.13216.
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society core allergen series: 2017 update. Dermatitis. 2017;28:141-143.
- T.R.U.E. TEST ready-to-use patch test panels. Smart Practice website. https://www.smartpractice.com/shop/wa/category?cn=T.R.U.E.-TEST%C2%AE-Ready-to-Use-Patch-Test-Panels&id=508222&m=SPA. Accessed May 17, 2019.
- Good AT, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methylacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Goon A, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians—thoughts on a rising concern [published online January 17, 2019]. Contact Dermatitis. doi:10.1111/cod.13222.
Practice Points
- Changing trends in nail services mean new exposures for consumers. Traditional nail polish has been replaced by semipermanent nail polish, which contains acrylates.
- Acrylates are a common cause of allergic contact dermatitis from nail polish. Acrylates can be found in gel, dip, and shellac nail polishes, among others.
- Patch testing with 2-hydroxyethyl methacrylate and ethyl cyanoacrylate can screen many patients for allergy due to nail services.

Between a rock and a hard place
CASE Irritable and short of breath
Mr. B, age 75, who lives alone, is brought to the emergency department (ED) for evaluation of shortness of breath. Mr. B is normally highly independent, and is able to drive, manage his own finances, attend to activities of daily living, and participate in social functions at church. On the day before he was taken to the ED, his home nurse had come to his home to dispense medications and found Mr. B was irritable, verbally rude, and repeatedly scratching the right side of his head. The nurse was unsure if Mr. B had taken his medications over the weekend. She called for emergency services, but Mr. B refused to go to the ED, and he was able to decline care because he was not in an acute medical emergency (95% oxygen on pulse oximetry).
The next day, when Mr. B’s nurse returned to his home, she found him to be tachypneic and verbigerating the phrase “I don’t know.” She contacted emergency services again, and Mr. B was taken to the ED.
In the ED, Mr. B has tachycardia, tachypnea, increased work of breathing, and diffuse rhonchi. He continues to repeat the phrase “I don’t know” and scratches the right side of his head repeatedly. The ED clinicians consult Psychiatry due to Mr. B’s confusion and because his nurse reports that his presentation is similar to a previous psychiatric hospitalization 9 years earlier.
[polldaddy:10332862]
EVALUATION Complex comorbidities
Mr. B has a lengthy history of schizophrenia, chronic right-sided heart failure secondary to pulmonary hypertension, moderate chronic obstructive pulmonary disease, hypertension, type 2 diabetes mellitus, and prostatic adenocarcinoma after external beam radiation therapy.
His symptoms of schizophrenia had been stable on his long-standing outpatient psychotropic regimen of haloperidol, 5 mg nightly; mirtazapine, 15 mg nightly, for appetite stimulation and insomnia; and trazodone, 100 mg nightly for insomnia. Mr. B has been receiving assertive community treatment (ACT) psychiatric services for schizophrenia; a nurse refills his pill box with his medications weekly. He does not have a history of medication nonadherence, and his nurse did not think he had missed any doses before the weekend.
He has acute changes in depressed mood, perseveration, and a Mini-Mental State Examination (MMSE) score of 26 (missing points for delayed recall and inability to construct a sentence), which indicates a cognitive assessment score on the low end of the normal range for people with at least an eighth grade education.
At the hospital, the psychiatrist diagnoses hypoactive delirium due to Mr. B’s fluctuating attention and disorientation. She also recommends that Mr. B continue his outpatient psychotropic regimen, and adds oral haloperidol, 5 mg, as needed for agitation (his QTc interval is 451 ms; reference range for men <430 ms, borderline prolonged 431 to 450 ms, prolonged >450 ms).
Continue to: An initial laboratory workup...
An initial laboratory workup and electrocardiogram reveal that Mr. B has an elevated troponin level (0.21 ng/mL; reference range <0.04; 0.04 to 0.39 ng/mL is elevated above the 99th percentile of a healthy population), non-ST-elevation myocardial infarction type II, Q waves in lead III, arteriovenous fistula with right axis deviation, acute on chronic kidney failure (creatinine level of 2.1 mg/dL, up from baseline of 1.4 mg/dL; reference range 0.84 to 1.21 mg/dL), elevated brain natriuretic peptide (111 pg/mL; reference range <125 pg/mL), and an elevated lactate level of 5.51 mmol/L (reference range 0.5 to 1 mmol/L). He also has a mixed respiratory alkalosis and metabolic acidosis with increased anion gap, transaminitis (aspartate aminotransferase 149 U/L; reference range 10 to 40 U/L), and elevated alkaline phosphatase (151 IU/L; reference range 44 to 147 IU/L). Urinalysis shows moderate ketones and is negative for nitrite or leukocyte esterase.
A brain CT rules out stroke. A chest X-ray shows subtle left basilar reticular opacity with a follow-up lateral view showing no consolidation and prominent pulmonary vasculature without overt edema.
In the ED, Mr. B is determined to have decision-making capacity and is able to authorize all treatment. Cardiology is also consulted, and Mr. B is admitted to the cardiac intensive care unit (CCU) for cardiogenic shock with close cardiac monitoring.
The Psychiatry and Cardiology teams discuss the risks and benefits of continuing antipsychotics. Due to the imminent risk of harm to Mr. B because of his significant agitation in the ED, which required treatment with one dose of IM haloperidol, 5 mg, and lorazepam, 2 mg, and close monitoring, the teams agree that the benefits of continuing haloperidol outweigh the risks.
On hospital Day 2, Mr. B’s repetitive scratching resolves. He is moved from the CCU to a general medical unit, where he begins to have episodes of mutism and negativism. By hospital Day 6, catatonia is suspected due to a MMSE of 6/30 and a Bush- Francis Catatonia Rating Scale (BFCRS) score of 14 for predominant stereotypy, perseveration, and withdrawal (Table 1). The teams determine that Mr. B lacks decisionmaking capacity due to his inability to rationally manipulate information. His brother is contacted and authorizes all treatment, deferring decision-making to the medical teams caring for Mr. B.
 on hospital Day 6")
Continue to: Mr. B undergoes an EEG...
Mr. B undergoes an EEG, which rules out nonconvulsive status epilepticus and is consistent with encephalopathy/delirium. Neuroleptic malignant syndrome (NMS) is considered but is less likely because Mr. B had been receiving a stable dose of haloperidol for several years, is afebrile, has stable vital signs, has no muscle rigidity, and no evidence of leukocytosis, creatine kinase elevation, myoglobinuria, hyperkalemia, hyperphosphatemia, thrombocytosis, or hypocalcemia.
Based on these clinical findings, Mr. B is diagnosed with catatonia and delirium.
The authors’ observations
Delirium, characterized by inattention and changes in mental status, is a syndrome due to acute brain dysfunction. It can be subclassified as hyperactive or hypoactive based on the change of activity. Simple catatonia is characterized by changes in behavior, affect, and motor function (with hyper- or hypoactivity). It may arise from gammaaminobutyric acid hypoactivity, dopamine (D2) hypoactivity, and possibly glutamate N-methyl-d-aspartate (NMDA) hyperactivity.1 Malignant catatonia is simple catatonia combined with autonomic instability and hyperthermia, which is a life-threatening condition. The BFCRS is commonly used to assess symptoms.2
Both catatonia and delirium result in significant morbidity and mortality. The 2 conditions share signs and symptoms yet rarely are diagnosed at the same time. DSM-IV, DSM-IV-TR, and DSM-5 state that a diagnosis of catatonia due to another medical condition cannot be made exclusively in the presence of delirium.3,4 DSM-IV and DSM-IV-TR required at least 2 criteria from 5 areas, including motoric immobility, excessive motor activity, extreme negativism or mutism, peculiarities of voluntary movement, and echolalia or echopraxia. Instead of grouping symptoms into clusters, DSM-5 requires 3 criteria of 12 individual symptoms.3,4 A co-occurrence with a medical illness precludes using the DSM-5 “catatonia associated with another mental disorder (catatonia specifier)” with the “unspecified catatonia” diagnosis category.4
However, a growing body of literature suggests that delirium and catatonia can cooccur.5,6 In 2017, Wilson et al6 found that of 136 critically ill patients in the ICU, 43% (58 patients) had only delirium, 3% (4 patients) had only catatonia, 31% (42 patients) had both, and 24% (32 patients) had neither. In patients with both catatonia and delirium, the most common signs of catatonia were autonomic abnormalities (96%), immobility/ stupor (87%), staring (77%), mutism (60%), and posturing (60%).
Continue to: The differential diagnosis...
The differential diagnosis of catatonia is extensive and varied.3,4 The most common psychiatric causes are mood disorders (13% to 31%) and psychotic disorders (7% to 17%).7 Neuromedical etiologies account for 4% to 46% of cases.7 The most common medical and neurologic causes are seizure disorder, acute intermittent porphyria, systemic lupus erythematosus, and drugrelated adverse effects (particularly due to clozapine withdrawal, risperidone, and phencyclidine).7
A workup that includes physical examination, laboratory testing, and neuroimaging can be helpful to identify delirium and catatonia, but there is limited literature to guide identifying coexisting delirium and catatonia other than a blend of physical exam findings of delirium and catatonia. Electroencephalogram may be normal in primary catatonia or may show nonspecific changes in secondary catatonia.8 Additionally, discharges in the frontal lobes and anterior limbic systems with diffuse background slowing and dysrhythmic patterns may be seen.7 Neuroimaging with MRI can help to evaluate catatonia.9 Laboratory testing such as creatine phosphokinase levels can be high in simple catatonia and are often elevated in malignant catatonia.7 Considering the possible co-occurrence of delirium and catatonia is critical to providing good patient care because the 2 conditions are treated differently.
[polldaddy:10332867]
TREATMENT A balancing act
Over the next month, Mr. B alternates between appearing catatonic or delirious. When he appears more catatonic, the dose of lorazepam is increased, which results in increased impulsivity and agitation and leads to multiple interventions from the behavioral emergency response team. At times, the team must use restraints and haloperidol because Mr. B pulls out IV lines and is considered at high risk for falls. When Mr. B appears more delirious and the dose of lorazepam is decreased, he becomes more catatonic.
Following the diagnosis of catatonia on Day 6, oral haloperidol is discontinued to further mitigate Mr. B’s risk of developing NMS. On hospital Day 6, Mr. B improves significantly after a 2-mg IV lorazepam challenge, with a BFCRS score of 6. At this point, he is started on lorazepam, 1 mg IV 3 times a day.
On Day 7, based on the complicated nature of Mr. B’s medical and psychiatric comorbidities, the treatment team considers ECT to minimize medication adverse effects, but Mr. B’s medical condition is too tenuous.
Continue to: On Day 7...
On Day 7, lorazepam is decreased to 0.5 mg/0.5 mg/1 mg IV. On Day 9, it is further decreased to 0.5 mg IV 3 times a day because Mr. B appears to be more delirious. On Day 10, lorazepam is increased to 1 mg IV 3 times a day, and oral haloperidol, 2 mg as needed for agitation, is restarted after multiple nights when Mr. B had behavioral emergencies and was treated with IM haloperidol and lorazepam. On Day 11, lorazepam is decreased and switched from IV formulation to oral, 0.5 mg 3 times a day. On Day 13, oral haloperidol is increased to 2 mg twice a day because of overnight behavioral emergencies requiring treatment with IV haloperidol, 4 mg. On Day 17, oral haloperidol is increased to 2 mg in the morning and 3 mg every night at bedtime because Mr. B has increased morning agitation. On Day 19, oral lorazepam is increased to 1 mg 3 times a day because Mr. B appears more catatonic. On Day 21, oral haloperidol is consolidated to 5 mg every night at bedtime. On Day 31, oral lorazepam is increased to 2 mg/1 mg/1 mg because he appears more catatonic with increased stuttering and mannerisms. On Day 33, oral haloperidol is increased to 6 mg every night at bedtime because Mr. B has morning agitation.
Multiple lorazepam and haloperidol dose adjustments are needed to balance the situation: combating catatonia, addressing delirium, managing schizophrenia symptoms, and improving Mr. B’s cardiac status. Finally, Mr. B is stabilized on oral lorazepam, 2 mg every morning, 1 mg every day at noon, and 1 mg every day at bedtime, and oral haloperidol, 6 mg every day at bedtime. This regimen, Mr. B has a BFCRS score of 1 (Table 2) and returns to his baseline mental status.

The authors’ observations
Delirium and catatonia typically have different treatments. Delirium is routinely treated by addressing the underlying medical and environmental factors, and managing comorbid symptoms such as agitation and disturbing hallucinations by prescribing antipsychotics, restoring the sleep-wake cycle with melatonin, initiating nonpharmacologic behavioral management, and avoiding deliriogenic medications such as benzodiazepines, opioids, and steroids.10 Catatonia is managed by prescribing benzodiazepines (with or without ECT) and by avoiding dopamine antagonists such as antipsychotics and metoclopramide (which may worsen catatonia or precipitate malignant catatonia).
The first-line treatment for catatonia is benzodiazepines, with IV preferred over IM, sublingual, or oral formulations. Electroconvulsive therapy is commonly used with benzodiazepines and is effective in 85% to 90% of patients. For ECT, bitemporal placement and daily treatment with brief pulses are frequently used. It is also effective in 60% of patients who fail to respond to benzodiazepines. Thus, ECT should be considered within the first 48 to 72 hours of benzodiazepine failure.7
Amantadine, a NMDA antagonist, may be a possible treatment for catatonia. A case report published in 1986 described a patient who developed catatonia after the abrupt withdrawal of amantadine during neuroleptic therapy.11 Memantine also may serve as a treatment for catatonia through glutamate antagonism. A review identified 25 cases of patients with catatonia who were treated with amantadine or memantine.12 Oral amantadine was administered at 100 to 400 mg/d in divided doses, with lower doses for patients with diminished renal function.12 Memantine was administered at 5 to 20 mg/d.12 All patients showed improvement after 1 to 7 days of treatment.12 Thus, memantine may be considered for patients with catatonic schizophrenia or comorbid catatonia and delirium. Although memantine was not considered in Mr. B’s case, he would have been a good candidate for treatment with this agent.
Continue to: There are also case reports of...
There are also case reports of aripiprazole being used for catatonia in the context of psychosis or delirium in both adults and adolescents.13-15 Other medications used in case reports for treating catatonia include carbamazepine, valproate, and secondgeneration antipsychotics.7
Because most of the literature on pharmacotherapy for catatonia consists of case reports or small case series, further research on medication management of catatonia and delirium is needed to guide treatment.
OUTCOME Multiple rehospitalizations
On Day 57, Mr. B is discharged to a skilled nursing facility due to significant deconditioning. He is discharged with continued follow-up with his ACT psychiatrist and nurse. Mr. B’s catatonia remains resolved; however, he is unable to be safely managed at the skilled nursing facility.
During the next 7 months, he is readmitted to the ICU for acute on chronic hypoxic respiratory failure 5 times; his rehospitalizations are complicated by delirium due to cardiogenic shock and urosepsis. Mild hyperactive delirium re-emerges after worsening respiratory failure and contributes to falls in the skilled nursing facility.
Six months later, Mr. B continues to receive the initial hospital discharge lorazepam regimen of 2 mg every morning, 1 mg every day at noon, and 1 mg every night at bedtime. The Psychiatry team slowly tapers this to 0.5 mg twice daily.
Continue to: On Day 5...
On Day 5 of Mr. B’s fifth hospital readmission, based on his advance directive, Mr. B’s family implements the do-not-resuscitate and do-not-intubate orders. He is transitioned to comfort measures, and dies on Day 6 with his brother and the hospital chaplain present.
Bottom Line
Delirium and catatonia share signs and symptoms, yet rarely are diagnosed at the same time. Both conditions result in significant morbidity and mortality. An emerging literature supports the concurrence of these 2 syndromes and aids in their diagnosis and treatment. Comorbidity with other medical conditions, common with both delirium and catatonia, substantially complicates treatment; thus, additional research into new treatment approaches is critical.
Related Resources
- Wilson JE, Carlson R, Duggan MC, et al. Delirium and catatonia in critically ill patients: the delirium and catatonia prospective cohort investigation. Crit Care Med. 2017;45(11):1837-1844.
- Catatonia Information Center. Penn State University. http://catatonia.org/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Clozapine • Clozaril
Haloperidol • Haldol
Lorazepam • Ativan
Memantine • Namenda
Metoclopramide • Reglan
Mirtazapine • Remeron
Risperidone • Risperdal
Topiramate • Topamax
Trazodone • Desyrel
Valproate • Depacon, Depakene, Depakote
1. Northoff G. What catatonia can tell us about “top-down modulation”: a neuropsychiatric hypothesis. Behav Brain Sci. 2002;25(5):555-577; discussion 578-604.
2. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
3. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Oldham MA, Lee HB. Catatonia vis-à-vis delirium: the significance of recognizing catatonia in altered mental status. Gen Hosp Psychiatry. 2015;37(6):554-559.
6. Wilson JE, Carlson R, Duggan MC. Delirium and catatonia in critically ill patients: the delirium and catatonia prospective cohort investigation. Crit Care Med. 2017;45(11):1837-1844.
7. Fricchione GL, Gross AF, Huffman JC, et al. Chapter 21: Catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fricchione GL, Cassem NH, et al. Massachusetts General Hospital Handbook of General Hospital Psychiatry, 6th Ed. Philadelphia, PA: Saunders Elsevier; 2010:273-288.
8. Van der Kooi AW, Zaal IJ, Klijn FA, et al. Delirium detection using EEG: what and how to measure. Chest. 2015;147(1):94-101.
9. Wilson JE, Niu K, Nicolson SE, et al. The diagnostic criteria and structure of catatonia. Schizophr Res. 2015;164 (1-3):256-262.
10. Maldonado JR. Acute brain failure: pathophysiology, diagnosis, management, and sequelae of delirium. Crit Care Clin. 2017;33(3):461-519.
11. Brown CS, Wittkowsky AK, Bryant SG. Neurolepticinduced catatonia after abrupt withdrawal of amantadine during neuroleptic therapy. Pharmacotherapy. 1986;6(4):193-195.
12. Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406-412.
13. Huffman JC, Fricchione GL. Catatonia and psychosis in a patient with AIDS: treatment with lorazepam and aripiprazole. J Clin Psychopharmacol. 2005;25(5):508-510.
14. Roberto AJ, Pinnaka S, Mohan A, et al. Adolescent catatonia successfully treated with lorazepam and aripiprazole. Case Rep Psychiatry. 2014;2014:309517.
15. Voros V, Kovacs A, Herold R, et al. Effectiveness of intramuscular aripiprazole injection in patients with catatonia: report on three cases. Pharmacopsychiatry. 2009;42(6):286-287.
CASE Irritable and short of breath
Mr. B, age 75, who lives alone, is brought to the emergency department (ED) for evaluation of shortness of breath. Mr. B is normally highly independent, and is able to drive, manage his own finances, attend to activities of daily living, and participate in social functions at church. On the day before he was taken to the ED, his home nurse had come to his home to dispense medications and found Mr. B was irritable, verbally rude, and repeatedly scratching the right side of his head. The nurse was unsure if Mr. B had taken his medications over the weekend. She called for emergency services, but Mr. B refused to go to the ED, and he was able to decline care because he was not in an acute medical emergency (95% oxygen on pulse oximetry).
The next day, when Mr. B’s nurse returned to his home, she found him to be tachypneic and verbigerating the phrase “I don’t know.” She contacted emergency services again, and Mr. B was taken to the ED.
In the ED, Mr. B has tachycardia, tachypnea, increased work of breathing, and diffuse rhonchi. He continues to repeat the phrase “I don’t know” and scratches the right side of his head repeatedly. The ED clinicians consult Psychiatry due to Mr. B’s confusion and because his nurse reports that his presentation is similar to a previous psychiatric hospitalization 9 years earlier.
[polldaddy:10332862]
EVALUATION Complex comorbidities
Mr. B has a lengthy history of schizophrenia, chronic right-sided heart failure secondary to pulmonary hypertension, moderate chronic obstructive pulmonary disease, hypertension, type 2 diabetes mellitus, and prostatic adenocarcinoma after external beam radiation therapy.
His symptoms of schizophrenia had been stable on his long-standing outpatient psychotropic regimen of haloperidol, 5 mg nightly; mirtazapine, 15 mg nightly, for appetite stimulation and insomnia; and trazodone, 100 mg nightly for insomnia. Mr. B has been receiving assertive community treatment (ACT) psychiatric services for schizophrenia; a nurse refills his pill box with his medications weekly. He does not have a history of medication nonadherence, and his nurse did not think he had missed any doses before the weekend.
He has acute changes in depressed mood, perseveration, and a Mini-Mental State Examination (MMSE) score of 26 (missing points for delayed recall and inability to construct a sentence), which indicates a cognitive assessment score on the low end of the normal range for people with at least an eighth grade education.
At the hospital, the psychiatrist diagnoses hypoactive delirium due to Mr. B’s fluctuating attention and disorientation. She also recommends that Mr. B continue his outpatient psychotropic regimen, and adds oral haloperidol, 5 mg, as needed for agitation (his QTc interval is 451 ms; reference range for men <430 ms, borderline prolonged 431 to 450 ms, prolonged >450 ms).
Continue to: An initial laboratory workup...
An initial laboratory workup and electrocardiogram reveal that Mr. B has an elevated troponin level (0.21 ng/mL; reference range <0.04; 0.04 to 0.39 ng/mL is elevated above the 99th percentile of a healthy population), non-ST-elevation myocardial infarction type II, Q waves in lead III, arteriovenous fistula with right axis deviation, acute on chronic kidney failure (creatinine level of 2.1 mg/dL, up from baseline of 1.4 mg/dL; reference range 0.84 to 1.21 mg/dL), elevated brain natriuretic peptide (111 pg/mL; reference range <125 pg/mL), and an elevated lactate level of 5.51 mmol/L (reference range 0.5 to 1 mmol/L). He also has a mixed respiratory alkalosis and metabolic acidosis with increased anion gap, transaminitis (aspartate aminotransferase 149 U/L; reference range 10 to 40 U/L), and elevated alkaline phosphatase (151 IU/L; reference range 44 to 147 IU/L). Urinalysis shows moderate ketones and is negative for nitrite or leukocyte esterase.
A brain CT rules out stroke. A chest X-ray shows subtle left basilar reticular opacity with a follow-up lateral view showing no consolidation and prominent pulmonary vasculature without overt edema.
In the ED, Mr. B is determined to have decision-making capacity and is able to authorize all treatment. Cardiology is also consulted, and Mr. B is admitted to the cardiac intensive care unit (CCU) for cardiogenic shock with close cardiac monitoring.
The Psychiatry and Cardiology teams discuss the risks and benefits of continuing antipsychotics. Due to the imminent risk of harm to Mr. B because of his significant agitation in the ED, which required treatment with one dose of IM haloperidol, 5 mg, and lorazepam, 2 mg, and close monitoring, the teams agree that the benefits of continuing haloperidol outweigh the risks.
On hospital Day 2, Mr. B’s repetitive scratching resolves. He is moved from the CCU to a general medical unit, where he begins to have episodes of mutism and negativism. By hospital Day 6, catatonia is suspected due to a MMSE of 6/30 and a Bush- Francis Catatonia Rating Scale (BFCRS) score of 14 for predominant stereotypy, perseveration, and withdrawal (Table 1). The teams determine that Mr. B lacks decisionmaking capacity due to his inability to rationally manipulate information. His brother is contacted and authorizes all treatment, deferring decision-making to the medical teams caring for Mr. B.
Continue to: Mr. B undergoes an EEG...
Mr. B undergoes an EEG, which rules out nonconvulsive status epilepticus and is consistent with encephalopathy/delirium. Neuroleptic malignant syndrome (NMS) is considered but is less likely because Mr. B had been receiving a stable dose of haloperidol for several years, is afebrile, has stable vital signs, has no muscle rigidity, and no evidence of leukocytosis, creatine kinase elevation, myoglobinuria, hyperkalemia, hyperphosphatemia, thrombocytosis, or hypocalcemia.
Based on these clinical findings, Mr. B is diagnosed with catatonia and delirium.
The authors’ observations
Delirium, characterized by inattention and changes in mental status, is a syndrome due to acute brain dysfunction. It can be subclassified as hyperactive or hypoactive based on the change of activity. Simple catatonia is characterized by changes in behavior, affect, and motor function (with hyper- or hypoactivity). It may arise from gammaaminobutyric acid hypoactivity, dopamine (D2) hypoactivity, and possibly glutamate N-methyl-d-aspartate (NMDA) hyperactivity.1 Malignant catatonia is simple catatonia combined with autonomic instability and hyperthermia, which is a life-threatening condition. The BFCRS is commonly used to assess symptoms.2
Both catatonia and delirium result in significant morbidity and mortality. The 2 conditions share signs and symptoms yet rarely are diagnosed at the same time. DSM-IV, DSM-IV-TR, and DSM-5 state that a diagnosis of catatonia due to another medical condition cannot be made exclusively in the presence of delirium.3,4 DSM-IV and DSM-IV-TR required at least 2 criteria from 5 areas, including motoric immobility, excessive motor activity, extreme negativism or mutism, peculiarities of voluntary movement, and echolalia or echopraxia. Instead of grouping symptoms into clusters, DSM-5 requires 3 criteria of 12 individual symptoms.3,4 A co-occurrence with a medical illness precludes using the DSM-5 “catatonia associated with another mental disorder (catatonia specifier)” with the “unspecified catatonia” diagnosis category.4
However, a growing body of literature suggests that delirium and catatonia can cooccur.5,6 In 2017, Wilson et al6 found that of 136 critically ill patients in the ICU, 43% (58 patients) had only delirium, 3% (4 patients) had only catatonia, 31% (42 patients) had both, and 24% (32 patients) had neither. In patients with both catatonia and delirium, the most common signs of catatonia were autonomic abnormalities (96%), immobility/ stupor (87%), staring (77%), mutism (60%), and posturing (60%).
Continue to: The differential diagnosis...
The differential diagnosis of catatonia is extensive and varied.3,4 The most common psychiatric causes are mood disorders (13% to 31%) and psychotic disorders (7% to 17%).7 Neuromedical etiologies account for 4% to 46% of cases.7 The most common medical and neurologic causes are seizure disorder, acute intermittent porphyria, systemic lupus erythematosus, and drugrelated adverse effects (particularly due to clozapine withdrawal, risperidone, and phencyclidine).7
A workup that includes physical examination, laboratory testing, and neuroimaging can be helpful to identify delirium and catatonia, but there is limited literature to guide identifying coexisting delirium and catatonia other than a blend of physical exam findings of delirium and catatonia. Electroencephalogram may be normal in primary catatonia or may show nonspecific changes in secondary catatonia.8 Additionally, discharges in the frontal lobes and anterior limbic systems with diffuse background slowing and dysrhythmic patterns may be seen.7 Neuroimaging with MRI can help to evaluate catatonia.9 Laboratory testing such as creatine phosphokinase levels can be high in simple catatonia and are often elevated in malignant catatonia.7 Considering the possible co-occurrence of delirium and catatonia is critical to providing good patient care because the 2 conditions are treated differently.
[polldaddy:10332867]
TREATMENT A balancing act
Over the next month, Mr. B alternates between appearing catatonic or delirious. When he appears more catatonic, the dose of lorazepam is increased, which results in increased impulsivity and agitation and leads to multiple interventions from the behavioral emergency response team. At times, the team must use restraints and haloperidol because Mr. B pulls out IV lines and is considered at high risk for falls. When Mr. B appears more delirious and the dose of lorazepam is decreased, he becomes more catatonic.
Following the diagnosis of catatonia on Day 6, oral haloperidol is discontinued to further mitigate Mr. B’s risk of developing NMS. On hospital Day 6, Mr. B improves significantly after a 2-mg IV lorazepam challenge, with a BFCRS score of 6. At this point, he is started on lorazepam, 1 mg IV 3 times a day.
On Day 7, based on the complicated nature of Mr. B’s medical and psychiatric comorbidities, the treatment team considers ECT to minimize medication adverse effects, but Mr. B’s medical condition is too tenuous.
Continue to: On Day 7...
On Day 7, lorazepam is decreased to 0.5 mg/0.5 mg/1 mg IV. On Day 9, it is further decreased to 0.5 mg IV 3 times a day because Mr. B appears to be more delirious. On Day 10, lorazepam is increased to 1 mg IV 3 times a day, and oral haloperidol, 2 mg as needed for agitation, is restarted after multiple nights when Mr. B had behavioral emergencies and was treated with IM haloperidol and lorazepam. On Day 11, lorazepam is decreased and switched from IV formulation to oral, 0.5 mg 3 times a day. On Day 13, oral haloperidol is increased to 2 mg twice a day because of overnight behavioral emergencies requiring treatment with IV haloperidol, 4 mg. On Day 17, oral haloperidol is increased to 2 mg in the morning and 3 mg every night at bedtime because Mr. B has increased morning agitation. On Day 19, oral lorazepam is increased to 1 mg 3 times a day because Mr. B appears more catatonic. On Day 21, oral haloperidol is consolidated to 5 mg every night at bedtime. On Day 31, oral lorazepam is increased to 2 mg/1 mg/1 mg because he appears more catatonic with increased stuttering and mannerisms. On Day 33, oral haloperidol is increased to 6 mg every night at bedtime because Mr. B has morning agitation.
Multiple lorazepam and haloperidol dose adjustments are needed to balance the situation: combating catatonia, addressing delirium, managing schizophrenia symptoms, and improving Mr. B’s cardiac status. Finally, Mr. B is stabilized on oral lorazepam, 2 mg every morning, 1 mg every day at noon, and 1 mg every day at bedtime, and oral haloperidol, 6 mg every day at bedtime. This regimen, Mr. B has a BFCRS score of 1 (Table 2) and returns to his baseline mental status.
The authors’ observations
Delirium and catatonia typically have different treatments. Delirium is routinely treated by addressing the underlying medical and environmental factors, and managing comorbid symptoms such as agitation and disturbing hallucinations by prescribing antipsychotics, restoring the sleep-wake cycle with melatonin, initiating nonpharmacologic behavioral management, and avoiding deliriogenic medications such as benzodiazepines, opioids, and steroids.10 Catatonia is managed by prescribing benzodiazepines (with or without ECT) and by avoiding dopamine antagonists such as antipsychotics and metoclopramide (which may worsen catatonia or precipitate malignant catatonia).
The first-line treatment for catatonia is benzodiazepines, with IV preferred over IM, sublingual, or oral formulations. Electroconvulsive therapy is commonly used with benzodiazepines and is effective in 85% to 90% of patients. For ECT, bitemporal placement and daily treatment with brief pulses are frequently used. It is also effective in 60% of patients who fail to respond to benzodiazepines. Thus, ECT should be considered within the first 48 to 72 hours of benzodiazepine failure.7
Amantadine, a NMDA antagonist, may be a possible treatment for catatonia. A case report published in 1986 described a patient who developed catatonia after the abrupt withdrawal of amantadine during neuroleptic therapy.11 Memantine also may serve as a treatment for catatonia through glutamate antagonism. A review identified 25 cases of patients with catatonia who were treated with amantadine or memantine.12 Oral amantadine was administered at 100 to 400 mg/d in divided doses, with lower doses for patients with diminished renal function.12 Memantine was administered at 5 to 20 mg/d.12 All patients showed improvement after 1 to 7 days of treatment.12 Thus, memantine may be considered for patients with catatonic schizophrenia or comorbid catatonia and delirium. Although memantine was not considered in Mr. B’s case, he would have been a good candidate for treatment with this agent.
Continue to: There are also case reports of...
There are also case reports of aripiprazole being used for catatonia in the context of psychosis or delirium in both adults and adolescents.13-15 Other medications used in case reports for treating catatonia include carbamazepine, valproate, and secondgeneration antipsychotics.7
Because most of the literature on pharmacotherapy for catatonia consists of case reports or small case series, further research on medication management of catatonia and delirium is needed to guide treatment.
OUTCOME Multiple rehospitalizations
On Day 57, Mr. B is discharged to a skilled nursing facility due to significant deconditioning. He is discharged with continued follow-up with his ACT psychiatrist and nurse. Mr. B’s catatonia remains resolved; however, he is unable to be safely managed at the skilled nursing facility.
During the next 7 months, he is readmitted to the ICU for acute on chronic hypoxic respiratory failure 5 times; his rehospitalizations are complicated by delirium due to cardiogenic shock and urosepsis. Mild hyperactive delirium re-emerges after worsening respiratory failure and contributes to falls in the skilled nursing facility.
Six months later, Mr. B continues to receive the initial hospital discharge lorazepam regimen of 2 mg every morning, 1 mg every day at noon, and 1 mg every night at bedtime. The Psychiatry team slowly tapers this to 0.5 mg twice daily.
Continue to: On Day 5...
On Day 5 of Mr. B’s fifth hospital readmission, based on his advance directive, Mr. B’s family implements the do-not-resuscitate and do-not-intubate orders. He is transitioned to comfort measures, and dies on Day 6 with his brother and the hospital chaplain present.
Bottom Line
Delirium and catatonia share signs and symptoms, yet rarely are diagnosed at the same time. Both conditions result in significant morbidity and mortality. An emerging literature supports the concurrence of these 2 syndromes and aids in their diagnosis and treatment. Comorbidity with other medical conditions, common with both delirium and catatonia, substantially complicates treatment; thus, additional research into new treatment approaches is critical.
Related Resources
- Wilson JE, Carlson R, Duggan MC, et al. Delirium and catatonia in critically ill patients: the delirium and catatonia prospective cohort investigation. Crit Care Med. 2017;45(11):1837-1844.
- Catatonia Information Center. Penn State University. http://catatonia.org/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Clozapine • Clozaril
Haloperidol • Haldol
Lorazepam • Ativan
Memantine • Namenda
Metoclopramide • Reglan
Mirtazapine • Remeron
Risperidone • Risperdal
Topiramate • Topamax
Trazodone • Desyrel
Valproate • Depacon, Depakene, Depakote
CASE Irritable and short of breath
Mr. B, age 75, who lives alone, is brought to the emergency department (ED) for evaluation of shortness of breath. Mr. B is normally highly independent, and is able to drive, manage his own finances, attend to activities of daily living, and participate in social functions at church. On the day before he was taken to the ED, his home nurse had come to his home to dispense medications and found Mr. B was irritable, verbally rude, and repeatedly scratching the right side of his head. The nurse was unsure if Mr. B had taken his medications over the weekend. She called for emergency services, but Mr. B refused to go to the ED, and he was able to decline care because he was not in an acute medical emergency (95% oxygen on pulse oximetry).
The next day, when Mr. B’s nurse returned to his home, she found him to be tachypneic and verbigerating the phrase “I don’t know.” She contacted emergency services again, and Mr. B was taken to the ED.
In the ED, Mr. B has tachycardia, tachypnea, increased work of breathing, and diffuse rhonchi. He continues to repeat the phrase “I don’t know” and scratches the right side of his head repeatedly. The ED clinicians consult Psychiatry due to Mr. B’s confusion and because his nurse reports that his presentation is similar to a previous psychiatric hospitalization 9 years earlier.
[polldaddy:10332862]
EVALUATION Complex comorbidities
Mr. B has a lengthy history of schizophrenia, chronic right-sided heart failure secondary to pulmonary hypertension, moderate chronic obstructive pulmonary disease, hypertension, type 2 diabetes mellitus, and prostatic adenocarcinoma after external beam radiation therapy.
His symptoms of schizophrenia had been stable on his long-standing outpatient psychotropic regimen of haloperidol, 5 mg nightly; mirtazapine, 15 mg nightly, for appetite stimulation and insomnia; and trazodone, 100 mg nightly for insomnia. Mr. B has been receiving assertive community treatment (ACT) psychiatric services for schizophrenia; a nurse refills his pill box with his medications weekly. He does not have a history of medication nonadherence, and his nurse did not think he had missed any doses before the weekend.
He has acute changes in depressed mood, perseveration, and a Mini-Mental State Examination (MMSE) score of 26 (missing points for delayed recall and inability to construct a sentence), which indicates a cognitive assessment score on the low end of the normal range for people with at least an eighth grade education.
At the hospital, the psychiatrist diagnoses hypoactive delirium due to Mr. B’s fluctuating attention and disorientation. She also recommends that Mr. B continue his outpatient psychotropic regimen, and adds oral haloperidol, 5 mg, as needed for agitation (his QTc interval is 451 ms; reference range for men <430 ms, borderline prolonged 431 to 450 ms, prolonged >450 ms).
Continue to: An initial laboratory workup...
An initial laboratory workup and electrocardiogram reveal that Mr. B has an elevated troponin level (0.21 ng/mL; reference range <0.04; 0.04 to 0.39 ng/mL is elevated above the 99th percentile of a healthy population), non-ST-elevation myocardial infarction type II, Q waves in lead III, arteriovenous fistula with right axis deviation, acute on chronic kidney failure (creatinine level of 2.1 mg/dL, up from baseline of 1.4 mg/dL; reference range 0.84 to 1.21 mg/dL), elevated brain natriuretic peptide (111 pg/mL; reference range <125 pg/mL), and an elevated lactate level of 5.51 mmol/L (reference range 0.5 to 1 mmol/L). He also has a mixed respiratory alkalosis and metabolic acidosis with increased anion gap, transaminitis (aspartate aminotransferase 149 U/L; reference range 10 to 40 U/L), and elevated alkaline phosphatase (151 IU/L; reference range 44 to 147 IU/L). Urinalysis shows moderate ketones and is negative for nitrite or leukocyte esterase.
A brain CT rules out stroke. A chest X-ray shows subtle left basilar reticular opacity with a follow-up lateral view showing no consolidation and prominent pulmonary vasculature without overt edema.
In the ED, Mr. B is determined to have decision-making capacity and is able to authorize all treatment. Cardiology is also consulted, and Mr. B is admitted to the cardiac intensive care unit (CCU) for cardiogenic shock with close cardiac monitoring.
The Psychiatry and Cardiology teams discuss the risks and benefits of continuing antipsychotics. Due to the imminent risk of harm to Mr. B because of his significant agitation in the ED, which required treatment with one dose of IM haloperidol, 5 mg, and lorazepam, 2 mg, and close monitoring, the teams agree that the benefits of continuing haloperidol outweigh the risks.
On hospital Day 2, Mr. B’s repetitive scratching resolves. He is moved from the CCU to a general medical unit, where he begins to have episodes of mutism and negativism. By hospital Day 6, catatonia is suspected due to a MMSE of 6/30 and a Bush- Francis Catatonia Rating Scale (BFCRS) score of 14 for predominant stereotypy, perseveration, and withdrawal (Table 1). The teams determine that Mr. B lacks decisionmaking capacity due to his inability to rationally manipulate information. His brother is contacted and authorizes all treatment, deferring decision-making to the medical teams caring for Mr. B.
Continue to: Mr. B undergoes an EEG...
Mr. B undergoes an EEG, which rules out nonconvulsive status epilepticus and is consistent with encephalopathy/delirium. Neuroleptic malignant syndrome (NMS) is considered but is less likely because Mr. B had been receiving a stable dose of haloperidol for several years, is afebrile, has stable vital signs, has no muscle rigidity, and no evidence of leukocytosis, creatine kinase elevation, myoglobinuria, hyperkalemia, hyperphosphatemia, thrombocytosis, or hypocalcemia.
Based on these clinical findings, Mr. B is diagnosed with catatonia and delirium.
The authors’ observations
Delirium, characterized by inattention and changes in mental status, is a syndrome due to acute brain dysfunction. It can be subclassified as hyperactive or hypoactive based on the change of activity. Simple catatonia is characterized by changes in behavior, affect, and motor function (with hyper- or hypoactivity). It may arise from gammaaminobutyric acid hypoactivity, dopamine (D2) hypoactivity, and possibly glutamate N-methyl-d-aspartate (NMDA) hyperactivity.1 Malignant catatonia is simple catatonia combined with autonomic instability and hyperthermia, which is a life-threatening condition. The BFCRS is commonly used to assess symptoms.2
Both catatonia and delirium result in significant morbidity and mortality. The 2 conditions share signs and symptoms yet rarely are diagnosed at the same time. DSM-IV, DSM-IV-TR, and DSM-5 state that a diagnosis of catatonia due to another medical condition cannot be made exclusively in the presence of delirium.3,4 DSM-IV and DSM-IV-TR required at least 2 criteria from 5 areas, including motoric immobility, excessive motor activity, extreme negativism or mutism, peculiarities of voluntary movement, and echolalia or echopraxia. Instead of grouping symptoms into clusters, DSM-5 requires 3 criteria of 12 individual symptoms.3,4 A co-occurrence with a medical illness precludes using the DSM-5 “catatonia associated with another mental disorder (catatonia specifier)” with the “unspecified catatonia” diagnosis category.4
However, a growing body of literature suggests that delirium and catatonia can cooccur.5,6 In 2017, Wilson et al6 found that of 136 critically ill patients in the ICU, 43% (58 patients) had only delirium, 3% (4 patients) had only catatonia, 31% (42 patients) had both, and 24% (32 patients) had neither. In patients with both catatonia and delirium, the most common signs of catatonia were autonomic abnormalities (96%), immobility/ stupor (87%), staring (77%), mutism (60%), and posturing (60%).
Continue to: The differential diagnosis...
The differential diagnosis of catatonia is extensive and varied.3,4 The most common psychiatric causes are mood disorders (13% to 31%) and psychotic disorders (7% to 17%).7 Neuromedical etiologies account for 4% to 46% of cases.7 The most common medical and neurologic causes are seizure disorder, acute intermittent porphyria, systemic lupus erythematosus, and drugrelated adverse effects (particularly due to clozapine withdrawal, risperidone, and phencyclidine).7
A workup that includes physical examination, laboratory testing, and neuroimaging can be helpful to identify delirium and catatonia, but there is limited literature to guide identifying coexisting delirium and catatonia other than a blend of physical exam findings of delirium and catatonia. Electroencephalogram may be normal in primary catatonia or may show nonspecific changes in secondary catatonia.8 Additionally, discharges in the frontal lobes and anterior limbic systems with diffuse background slowing and dysrhythmic patterns may be seen.7 Neuroimaging with MRI can help to evaluate catatonia.9 Laboratory testing such as creatine phosphokinase levels can be high in simple catatonia and are often elevated in malignant catatonia.7 Considering the possible co-occurrence of delirium and catatonia is critical to providing good patient care because the 2 conditions are treated differently.
[polldaddy:10332867]
TREATMENT A balancing act
Over the next month, Mr. B alternates between appearing catatonic or delirious. When he appears more catatonic, the dose of lorazepam is increased, which results in increased impulsivity and agitation and leads to multiple interventions from the behavioral emergency response team. At times, the team must use restraints and haloperidol because Mr. B pulls out IV lines and is considered at high risk for falls. When Mr. B appears more delirious and the dose of lorazepam is decreased, he becomes more catatonic.
Following the diagnosis of catatonia on Day 6, oral haloperidol is discontinued to further mitigate Mr. B’s risk of developing NMS. On hospital Day 6, Mr. B improves significantly after a 2-mg IV lorazepam challenge, with a BFCRS score of 6. At this point, he is started on lorazepam, 1 mg IV 3 times a day.
On Day 7, based on the complicated nature of Mr. B’s medical and psychiatric comorbidities, the treatment team considers ECT to minimize medication adverse effects, but Mr. B’s medical condition is too tenuous.
Continue to: On Day 7...
On Day 7, lorazepam is decreased to 0.5 mg/0.5 mg/1 mg IV. On Day 9, it is further decreased to 0.5 mg IV 3 times a day because Mr. B appears to be more delirious. On Day 10, lorazepam is increased to 1 mg IV 3 times a day, and oral haloperidol, 2 mg as needed for agitation, is restarted after multiple nights when Mr. B had behavioral emergencies and was treated with IM haloperidol and lorazepam. On Day 11, lorazepam is decreased and switched from IV formulation to oral, 0.5 mg 3 times a day. On Day 13, oral haloperidol is increased to 2 mg twice a day because of overnight behavioral emergencies requiring treatment with IV haloperidol, 4 mg. On Day 17, oral haloperidol is increased to 2 mg in the morning and 3 mg every night at bedtime because Mr. B has increased morning agitation. On Day 19, oral lorazepam is increased to 1 mg 3 times a day because Mr. B appears more catatonic. On Day 21, oral haloperidol is consolidated to 5 mg every night at bedtime. On Day 31, oral lorazepam is increased to 2 mg/1 mg/1 mg because he appears more catatonic with increased stuttering and mannerisms. On Day 33, oral haloperidol is increased to 6 mg every night at bedtime because Mr. B has morning agitation.
Multiple lorazepam and haloperidol dose adjustments are needed to balance the situation: combating catatonia, addressing delirium, managing schizophrenia symptoms, and improving Mr. B’s cardiac status. Finally, Mr. B is stabilized on oral lorazepam, 2 mg every morning, 1 mg every day at noon, and 1 mg every day at bedtime, and oral haloperidol, 6 mg every day at bedtime. This regimen, Mr. B has a BFCRS score of 1 (Table 2) and returns to his baseline mental status.
The authors’ observations
Delirium and catatonia typically have different treatments. Delirium is routinely treated by addressing the underlying medical and environmental factors, and managing comorbid symptoms such as agitation and disturbing hallucinations by prescribing antipsychotics, restoring the sleep-wake cycle with melatonin, initiating nonpharmacologic behavioral management, and avoiding deliriogenic medications such as benzodiazepines, opioids, and steroids.10 Catatonia is managed by prescribing benzodiazepines (with or without ECT) and by avoiding dopamine antagonists such as antipsychotics and metoclopramide (which may worsen catatonia or precipitate malignant catatonia).
The first-line treatment for catatonia is benzodiazepines, with IV preferred over IM, sublingual, or oral formulations. Electroconvulsive therapy is commonly used with benzodiazepines and is effective in 85% to 90% of patients. For ECT, bitemporal placement and daily treatment with brief pulses are frequently used. It is also effective in 60% of patients who fail to respond to benzodiazepines. Thus, ECT should be considered within the first 48 to 72 hours of benzodiazepine failure.7
Amantadine, a NMDA antagonist, may be a possible treatment for catatonia. A case report published in 1986 described a patient who developed catatonia after the abrupt withdrawal of amantadine during neuroleptic therapy.11 Memantine also may serve as a treatment for catatonia through glutamate antagonism. A review identified 25 cases of patients with catatonia who were treated with amantadine or memantine.12 Oral amantadine was administered at 100 to 400 mg/d in divided doses, with lower doses for patients with diminished renal function.12 Memantine was administered at 5 to 20 mg/d.12 All patients showed improvement after 1 to 7 days of treatment.12 Thus, memantine may be considered for patients with catatonic schizophrenia or comorbid catatonia and delirium. Although memantine was not considered in Mr. B’s case, he would have been a good candidate for treatment with this agent.
Continue to: There are also case reports of...
There are also case reports of aripiprazole being used for catatonia in the context of psychosis or delirium in both adults and adolescents.13-15 Other medications used in case reports for treating catatonia include carbamazepine, valproate, and secondgeneration antipsychotics.7
Because most of the literature on pharmacotherapy for catatonia consists of case reports or small case series, further research on medication management of catatonia and delirium is needed to guide treatment.
OUTCOME Multiple rehospitalizations
On Day 57, Mr. B is discharged to a skilled nursing facility due to significant deconditioning. He is discharged with continued follow-up with his ACT psychiatrist and nurse. Mr. B’s catatonia remains resolved; however, he is unable to be safely managed at the skilled nursing facility.
During the next 7 months, he is readmitted to the ICU for acute on chronic hypoxic respiratory failure 5 times; his rehospitalizations are complicated by delirium due to cardiogenic shock and urosepsis. Mild hyperactive delirium re-emerges after worsening respiratory failure and contributes to falls in the skilled nursing facility.
Six months later, Mr. B continues to receive the initial hospital discharge lorazepam regimen of 2 mg every morning, 1 mg every day at noon, and 1 mg every night at bedtime. The Psychiatry team slowly tapers this to 0.5 mg twice daily.
Continue to: On Day 5...
On Day 5 of Mr. B’s fifth hospital readmission, based on his advance directive, Mr. B’s family implements the do-not-resuscitate and do-not-intubate orders. He is transitioned to comfort measures, and dies on Day 6 with his brother and the hospital chaplain present.
Bottom Line
Delirium and catatonia share signs and symptoms, yet rarely are diagnosed at the same time. Both conditions result in significant morbidity and mortality. An emerging literature supports the concurrence of these 2 syndromes and aids in their diagnosis and treatment. Comorbidity with other medical conditions, common with both delirium and catatonia, substantially complicates treatment; thus, additional research into new treatment approaches is critical.
Related Resources
- Wilson JE, Carlson R, Duggan MC, et al. Delirium and catatonia in critically ill patients: the delirium and catatonia prospective cohort investigation. Crit Care Med. 2017;45(11):1837-1844.
- Catatonia Information Center. Penn State University. http://catatonia.org/.
Drug Brand Names
Amantadine • Symmetrel
Aripiprazole • Abilify
Carbamazepine • Carbatrol, Tegretol
Clozapine • Clozaril
Haloperidol • Haldol
Lorazepam • Ativan
Memantine • Namenda
Metoclopramide • Reglan
Mirtazapine • Remeron
Risperidone • Risperdal
Topiramate • Topamax
Trazodone • Desyrel
Valproate • Depacon, Depakene, Depakote
1. Northoff G. What catatonia can tell us about “top-down modulation”: a neuropsychiatric hypothesis. Behav Brain Sci. 2002;25(5):555-577; discussion 578-604.
2. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
3. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Oldham MA, Lee HB. Catatonia vis-à-vis delirium: the significance of recognizing catatonia in altered mental status. Gen Hosp Psychiatry. 2015;37(6):554-559.
6. Wilson JE, Carlson R, Duggan MC. Delirium and catatonia in critically ill patients: the delirium and catatonia prospective cohort investigation. Crit Care Med. 2017;45(11):1837-1844.
7. Fricchione GL, Gross AF, Huffman JC, et al. Chapter 21: Catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fricchione GL, Cassem NH, et al. Massachusetts General Hospital Handbook of General Hospital Psychiatry, 6th Ed. Philadelphia, PA: Saunders Elsevier; 2010:273-288.
8. Van der Kooi AW, Zaal IJ, Klijn FA, et al. Delirium detection using EEG: what and how to measure. Chest. 2015;147(1):94-101.
9. Wilson JE, Niu K, Nicolson SE, et al. The diagnostic criteria and structure of catatonia. Schizophr Res. 2015;164 (1-3):256-262.
10. Maldonado JR. Acute brain failure: pathophysiology, diagnosis, management, and sequelae of delirium. Crit Care Clin. 2017;33(3):461-519.
11. Brown CS, Wittkowsky AK, Bryant SG. Neurolepticinduced catatonia after abrupt withdrawal of amantadine during neuroleptic therapy. Pharmacotherapy. 1986;6(4):193-195.
12. Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406-412.
13. Huffman JC, Fricchione GL. Catatonia and psychosis in a patient with AIDS: treatment with lorazepam and aripiprazole. J Clin Psychopharmacol. 2005;25(5):508-510.
14. Roberto AJ, Pinnaka S, Mohan A, et al. Adolescent catatonia successfully treated with lorazepam and aripiprazole. Case Rep Psychiatry. 2014;2014:309517.
15. Voros V, Kovacs A, Herold R, et al. Effectiveness of intramuscular aripiprazole injection in patients with catatonia: report on three cases. Pharmacopsychiatry. 2009;42(6):286-287.
1. Northoff G. What catatonia can tell us about “top-down modulation”: a neuropsychiatric hypothesis. Behav Brain Sci. 2002;25(5):555-577; discussion 578-604.
2. Bush G, Fink M, Petrides G, et al. Catatonia. I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129-136.
3. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Diagnostic and Statistical Manual of Mental Disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
5. Oldham MA, Lee HB. Catatonia vis-à-vis delirium: the significance of recognizing catatonia in altered mental status. Gen Hosp Psychiatry. 2015;37(6):554-559.
6. Wilson JE, Carlson R, Duggan MC. Delirium and catatonia in critically ill patients: the delirium and catatonia prospective cohort investigation. Crit Care Med. 2017;45(11):1837-1844.
7. Fricchione GL, Gross AF, Huffman JC, et al. Chapter 21: Catatonia, neuroleptic malignant syndrome, and serotonin syndrome. In: Stern TA, Fricchione GL, Cassem NH, et al. Massachusetts General Hospital Handbook of General Hospital Psychiatry, 6th Ed. Philadelphia, PA: Saunders Elsevier; 2010:273-288.
8. Van der Kooi AW, Zaal IJ, Klijn FA, et al. Delirium detection using EEG: what and how to measure. Chest. 2015;147(1):94-101.
9. Wilson JE, Niu K, Nicolson SE, et al. The diagnostic criteria and structure of catatonia. Schizophr Res. 2015;164 (1-3):256-262.
10. Maldonado JR. Acute brain failure: pathophysiology, diagnosis, management, and sequelae of delirium. Crit Care Clin. 2017;33(3):461-519.
11. Brown CS, Wittkowsky AK, Bryant SG. Neurolepticinduced catatonia after abrupt withdrawal of amantadine during neuroleptic therapy. Pharmacotherapy. 1986;6(4):193-195.
12. Carroll BT, Goforth HW, Thomas C, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406-412.
13. Huffman JC, Fricchione GL. Catatonia and psychosis in a patient with AIDS: treatment with lorazepam and aripiprazole. J Clin Psychopharmacol. 2005;25(5):508-510.
14. Roberto AJ, Pinnaka S, Mohan A, et al. Adolescent catatonia successfully treated with lorazepam and aripiprazole. Case Rep Psychiatry. 2014;2014:309517.
15. Voros V, Kovacs A, Herold R, et al. Effectiveness of intramuscular aripiprazole injection in patients with catatonia: report on three cases. Pharmacopsychiatry. 2009;42(6):286-287.

From sweet to belligerent in the blink of an eye
CASE Combative and agitated
Ms. P, age 87, presents to the emergency department (ED) with her caregiver, who says Ms. P has new-onset altered mental status, agitation, and combativeness.
Ms. P resides at a long-term care (LTC) facility, where according to the nurses she normally is pleasant, well-oriented, and cooperative. Ms. P’s medical history includes major depressive disorder, generalized anxiety disorder, hypertension, chronic kidney disease (CKD) stage III, peptic ulcer disease, gastroesophageal reflux disease, coronary artery disease with 2 past myocardial infarctions requiring stents, chronic obstructive pulmonary disease, hyperlipidemia, bradycardia requiring a pacemaker, paroxysmal atrial fibrillation, asthma, aortic stenosis, peripheral vascular disease, esophageal stricture requiring dilation, deep vein thrombosis, and migraines.
Mr. P’s medication list includes acetaminophen, 650 mg every 6 hours; ipratropium/albuterol nebulized solution, 3 mL 4 times a day; aspirin, 81 mg/d; atorvastatin, 40 mg/d; calcitonin, 1 spray nasally at bedtime; clopidogrel, 75 mg/d; ezetimibe, 10 mg/d; fluoxetine, 20 mg/d; furosemide, 20 mg/d; isosorbide dinitrate, 120 mg/d; lisinopril, 15 mg/d; risperidone, 0.5 mg/d; magnesium oxide, 800 mg/d; pantoprazole, 40 mg/d; polyethylene glycol, 17 g/d; sotalol, 160 mg/d; olanzapine, 5 mg IM every 6 hours as needed for agitation; and tramadol, 50 mg every 8 hours as needed for headache.
Seven days before coming to the ED, Ms. P was started on ceftriaxone, 1 g/d, for suspected community-acquired pneumonia. At that time, the nursing staff noticed behavioral changes. Soon after, Ms. P began refusing all her medications. Two days before presenting to the ED, Ms. P was started on nitrofurantoin, 200 mg/d, for a suspected urinary tract infection, but it was discontinued because of an allergy.
Her caregiver reports that while at the LTC facility, Ms. P’s behavioral changes worsened. Ms. P claimed to be Jesus Christ and said she was talking to the devil; she chased other residents around the facility and slapped medications away from the nursing staff. According to caregivers, this behavior was out of character.
Shortly after arriving in the ED, Ms. P is admitted to the psychiatric unit.
[polldaddy:10332748]

The authors’ observations
Delirium is a complex, acute alteration in a patient’s mental status compared with his/her baseline functioning1 (Table 12). The onset of delirium is quick, happening within hours to days, with fluctuations in mental function. Patients might present with hyperactive, hypoactive, or mixed delirium.3 Patients with hyperactive delirium often have delusions and hallucinations; these patients might be agitated and could become violent with family and caregivers.3 Patients with hypoactive delirium are less likely to experience hallucinations and more likely to show symptoms of sedation.3 Patients with hypoactive delirium can be difficult to diagnose because it is challenging to interview them and understand what might be the cause of their sedated state. Patients also can exhibit a mixed delirium in which they fluctuate between periods of hyperactivity and hypoactivity.3
Continue to: Suspected delirium...
Suspected delirium should be considered a medical emergency because the outcome could be fatal.1 It is important to uncover and treat the underlying cause(s) of delirium rather than solely administering antipsychotics, which might mask the presenting symptoms. In an older study, Francis and Kapoor4 reported that 56% of geriatric patients with delirium had a single definite or probable etiology, while the other 44% had about 2.8 etiologies per patient on average. Delirium risk factors, causes, and factors to consider during patient evaluation are listed in Table 21,3,5-7 and Table 3.1,3,5-7

A synergistic relationship between comorbidities, environment, and medications can induce delirium.5 Identifying irreversible and reversible causes is the key to treating delirium. After the cause has been identified, it can be addressed and the patient could return to his/her previous level of functioning. If the delirium is the result of multiple irreversible causes, it could become chronic.

[polldaddy:10332749]
EVALUATION Cardiac dysfunction
Ms. P undergoes laboratory testing. The results include: white blood cell count, 5.9/µL; hemoglobin, 13.6 g/dL; hematocrit, 42.6%; platelets, 304 × 103/µL; sodium,143 mEq/L; potassium, 3.2 mEq/L; chloride, 96 mEq/L; carbon dioxide, 23 mEq/L; blood glucose, 87 mg/dL; creatinine, 1.2 mg/dL; estimated creatinine clearance (eCrCl) level of 33 mL/min/1.73 m2; calcium, 9.5 mg/dL; albumin, 3.6 g/dL; liver enzymes within normal limits; thyroid-stimulating hormone, 0.78 mIU/L; vitamin B12, 995 pg/mL; folic acid, 16.6 ng/mL; vitamin D, 31 pg/mL; and rapid plasma reagin: nonreactive. Urinalysis is unremarkable, and no culture is performed. Urine drug screening/toxicology is positive for the benzodiazepines that she received in the ED (oral alprazolam 0.25 mg given once and oral lorazepam 0.5 mg given once).
Electrocardiogram (ECG) shows atrial flutter/tachycardia with rapid ventricular response, marked left axis deviation, nonspecific ST- and T-wave abnormality, QT/QTC of 301/387 ms, and ventricular rate 151 beats per minute. A CT scan of the head and brain without contrast shows mild atrophy and chronic white matter changes and no acute intracranial abnormality. A two-view chest radiography shows no acute cardiopulmonary findings. Her temperature is 98.4°F; heart rate is 122 beats per minute; respiratory rate is 20 breaths per minute; blood pressure is 161/98 mm Hg; and oxygen saturation is 86% on room air.
Based on this data, Ms. P’s cardiac condition seems to be worsening, which is thought to be caused by her refusal of furosemide, lisinopril, isosorbide, sotalol, clopidogrel, and aspirin. The treatment team plans to work on compliance to resolve these cardiac issues and places Ms. P on 1:1 observation with a sitter and music in attempt to calm her.
Continue to: The authors' observations
The authors’ observations
Many factors can contribute to behavioral or cognitive changes in geriatric patients. Often, a major change noted in an older patient can be attributed to new-onset dementia, dementia with behavioral disturbances, delirium, depression, or acute psychosis. These potential causes should be considered and ruled out in a step-by-step progression. Because patients are unreliable historians during acute distress, a complete history from family or caregivers and exhaustive workup is paramount.
TREATMENT Medication adjustments
In an attempt to resolve Ms. P’s disruptive behaviors, her risperidone dosage is changed to 0.5 mg twice daily. Ms. P is encouraged to use the provided oxygen to raise her saturation level.
On hospital Day 3, a loose stool prompts a Clostridium difficile test as a possible source of delirium; however, the results are negative.
On hospital Day 4, Ms. P is confused and irritable overnight, yelling profanities at staff, refusing care, inappropriately disrobing, and having difficulty falling asleep and staying asleep. Risperidone is discontinued because it appears to have had little or no effect on Ms. P’s disruptive behaviors. Olanzapine, 10 mg/d, is initiated with mirtazapine, 7.5 mg/d, to help with mood, appetite, and sleep. Fluoxetine is also discontinued because of a possible interaction with clopidogrel.
On hospital Days 6 to 8, Ms. P remains upset and unable to follow instructions. Melatonin is initiated to improve her sleep cycle. On Day 9, she continues to decline and is cursing at hospital staff; haloperidol is initiated at 5 mg every morning, 10 mg at bedtime, and 5 mg IM as needed for agitation. Her sleep improves with melatonin and mirtazapine. IV hydration also is initiated. Ms. P has a slight improvement in medication compliance. On Day 11, haloperidol is increased to 5 mg in the morning, 5 mg in the afternoon, and 10 mg at bedtime. On Day 12, haloperidol is changed to 7.5 mg twice daily; a slight improvement in Ms. P’s behavior is noted.
Continue to: On hospital Day 13...
On hospital Day 13, Ms. P’s behavior declines again. She screams profanities at staff and does not recognize the clinicians who have been providing care to her. The physician initiates valproic acid, 125 mg, 3 times a day, to target Ms. P’s behavioral disturbances. A pharmacist notes that the patient’s sotalol could be contributing to Ms. P’s psychiatric presentation, and that based on her eCrCl level of 33 mL/min/1.73 m2, a dosage adjustment or medication change might be warranted.
On Day 14, Ms. P displays erratic behavior and intermittent tachycardia. A cardiac consultation is ordered. A repeat ECG reveals atrial fibrillation with rapid rate and a QT/QTc of 409/432 ms. Ms. P is transferred to the telemetry unit, where the cardiologist discontinues sotalol because the dosage is not properly renally adjusted. Sotalol hydrochloride has been associated with life-threatening ventricular tachycardia.8 Diltiazem, 30 mg every 6 hours is initiated to replace sotalol.
By Day 16, the treatment team notes improved cognition and behavior. On Day 17, the cardiologist reports that Ms. P’s atrial fibrillation is controlled. An ECG reveals mild left ventricular hypertrophy, an ejection fraction of 50% to 55%, no stenosis in the mitral or tricuspid valves, no valvular pulmonic stenosis, and moderate aortic sclerosis. Cardiac markers also are evaluated (creatinine phosphokinase: 105 U/L; creatinine kinase–MB fraction: 2.6 ng/mL; troponin: 0.01 ng/mL; pro-B-type natriuretic peptide: 2,073 pg/mL); and myocardial infarction is ruled out.
On Day 19, Ms. P’s diltiazem is consolidated to a controlled-delivery formulation, 180 mg/d, along with the addition of metoprolol, 12.5 mg twice daily. Ms. P is transferred back to the psychiatric unit.
OUTCOME Gradual improvement
On Days 20 to 23, Ms. P shows remarkable progress, and her mental status, cognition, and behavior slowly return to baseline. Haloperidol and valproic acid are tapered and discontinued. Ms. P is observed to be healthy and oriented to person, place, and time.
Continue to: On Day 25...
On Day 25, she is discharged from the hospital, and returns to the LTC facility.
The authors’ observations
Ms. P’s delirium was a combination of her older age, non-renally adjusted sotalol, and CKD. At admission, the hospital treatment team first thought that pneumonia or antibiotic use could have caused delirium. However, Ms. P’s condition did not improve after antibiotics were stopped. In addition, several chest radiographs found no evidence of pneumonia. It is important to check for any source of infection because infection is a common source of delirium in older patients.1 Urine samples revealed no pathogens, a C. difficile test was negative, and the patient’s white blood cell counts remained within normal limits. Physicians began looking elsewhere for potential causes of Ms. P’s delirium.
Ms. P’s vital signs ruled out a temperature irregularity or hypertension as the cause of her delirium. She has a slightly low oxygen saturation when she first presented, but this quickly returned to normal with administration of oxygen, which ruled out hypoxemia. Laboratory results concluded that Ms. P’s glucose levels were within a normal range and she had no electrolyte imbalances. A head CT scan showed slight atrophy of white matter that is consistent with Ms. P’s age. The head CT scan also showed that Ms. P had no acute condition or head trauma.
In terms of organ function, Ms. P was in relatively healthy condition other than paroxysmal atrial fibrillation and CKD. Chronic kidney disease can interrupt the normal pharmacokinetics of medications. Reviewing Ms. P’s medication list, several agents could have induced delirium, including antidepressants, antipsychotics, cardiovascular medications (beta blocker/antiarrhythmic [sotalol]), and opioid analgesics such as tramadol.5 Ms. P’s condition did not improve after discontinuing fluoxetine, risperidone, or olanzapine, although haloperidol was started in their place. Ms. P scored an 8 on the Naranjo Adverse Drug Reaction Probability Scale, indicating this event was a probable adverse drug reaction.9
Identifying a cause
This was a unique case where sotalol was identified as the culprit for inducing Ms. P’s delirium, because her age and CKD are irreversible. It is important to note that antiarrhythmics can induce arrhythmias when present in high concentrations or administered without appropriate renal dose adjustments. Although Ms. P’s serum levels of sotalol were not evaluated, because of her renal impairment, it is possible that toxic levels of sotalol accumulated and lead to arrhythmias and delirium. Of note, a cardiologist was consulted to safely change Ms. P to a calcium channel blocker so she could undergo cardiac monitoring. With the addition of diltiazem and metoprolol, the patient’s delirium subsided and her arrhythmia was controlled. Once the source of Ms. P’s delirium had been identified, antipsychotics were no longer needed.
Continue to: Bottom Line
Bottom Line
Delirium is a complex disorder that often has multiple causes, both reversible and irreversible. A “process of elimination” approach should be used to accurately identify and manage delirium. If a patient with delirium has little to no response to antipsychotic medications, the underlying cause or causes likely has not yet been addressed, and the evaluation should continue.
Related Resources
- Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456-1466.
- Inouye SK, Westendorp RGJ, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
Drug Brand Names
Acyclovir • Zovirax
Alprazolam • Niravam, Xanax
Amantadine • Symmetrel
Amphotericin B • Abelcet
Atorvastatin • Lipitor
Atropine • Atropen
Baclofen • EnovaRX-Baclofen
Benztropine • Cogentin
Bromocriptine • Cycloset
Calcitonin • Miacalcin
Carbamazepine • Tegretol
Carbidopa-levodopa • Duopa
Ceftriaxone • Rocephin
Chlorpromazine • Thorazine
Clonidine • Catapres
Clopidogrel • Plavix
Cyclobenzaprine • Amrix
Digoxin • Lanoxin
Diltiazem • Cardizem
Disulfiram • Antabuse
Ezetimibe • Zetia
Fluoxetine • Prozac
Fluphenazine • Prolixin
Furosemide • Lasix
Haloperidol • Haldol
Ipratropium/albuterol nebulized solution • Combivent Respimat
Isoniazid • Isotamine
Isosorbide nitrate • Dilatrate
Levetiracetam • Keppra
Levodopa • Stalevo
Linezolid • Zyvox
Lisinopril • Zestril
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Magnesium Oxide • Mag-200
Meperidine • Demerol
Methyldopa • Aldomet
Metoprolol • Lopressor
Metronidazole • Flagyl
Mirtazapine • Remeron
Nitrofurantoin • Macrobid
Olanzapine • Zyprexa
Pantoprazole • Protonix
Phenytoin • Dilantin
Pramipexole • Mirapex
Rifampin • Rifadin
Risperidone • Risperdal
Ropinirole • Requip
Sotalol hydrochloride • Betapace AF
Tramadol • Ultram
Trihexyphenidyl • Trihexane
Valproic acid • Depakote
1. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention, and treatment. Nat Rev Neurol. 2009;5(4):210-220.
2. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
3. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
4. Francis J, Kapoor WN. Delirium in hospitalized elderly. J Gen Intern Med. 1990;5(1):65-79.
5. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
6. Cook IA. Guideline watch: practice guideline for the treatment of patients with delirium. Arlington, VA: American Psychiatric Publishing; 2004.
7. Bourgeois J, Ategan A, Losier B. Delirium in the hospital: emphasis on the management of geriatric patients. Current Psychiatry. 2014;13(8):29,36-42.
8. Betapace AF [package insert]. Zug, Switzerland: Covis Pharma; 2016.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
CASE Combative and agitated
Ms. P, age 87, presents to the emergency department (ED) with her caregiver, who says Ms. P has new-onset altered mental status, agitation, and combativeness.
Ms. P resides at a long-term care (LTC) facility, where according to the nurses she normally is pleasant, well-oriented, and cooperative. Ms. P’s medical history includes major depressive disorder, generalized anxiety disorder, hypertension, chronic kidney disease (CKD) stage III, peptic ulcer disease, gastroesophageal reflux disease, coronary artery disease with 2 past myocardial infarctions requiring stents, chronic obstructive pulmonary disease, hyperlipidemia, bradycardia requiring a pacemaker, paroxysmal atrial fibrillation, asthma, aortic stenosis, peripheral vascular disease, esophageal stricture requiring dilation, deep vein thrombosis, and migraines.
Mr. P’s medication list includes acetaminophen, 650 mg every 6 hours; ipratropium/albuterol nebulized solution, 3 mL 4 times a day; aspirin, 81 mg/d; atorvastatin, 40 mg/d; calcitonin, 1 spray nasally at bedtime; clopidogrel, 75 mg/d; ezetimibe, 10 mg/d; fluoxetine, 20 mg/d; furosemide, 20 mg/d; isosorbide dinitrate, 120 mg/d; lisinopril, 15 mg/d; risperidone, 0.5 mg/d; magnesium oxide, 800 mg/d; pantoprazole, 40 mg/d; polyethylene glycol, 17 g/d; sotalol, 160 mg/d; olanzapine, 5 mg IM every 6 hours as needed for agitation; and tramadol, 50 mg every 8 hours as needed for headache.
Seven days before coming to the ED, Ms. P was started on ceftriaxone, 1 g/d, for suspected community-acquired pneumonia. At that time, the nursing staff noticed behavioral changes. Soon after, Ms. P began refusing all her medications. Two days before presenting to the ED, Ms. P was started on nitrofurantoin, 200 mg/d, for a suspected urinary tract infection, but it was discontinued because of an allergy.
Her caregiver reports that while at the LTC facility, Ms. P’s behavioral changes worsened. Ms. P claimed to be Jesus Christ and said she was talking to the devil; she chased other residents around the facility and slapped medications away from the nursing staff. According to caregivers, this behavior was out of character.
Shortly after arriving in the ED, Ms. P is admitted to the psychiatric unit.
[polldaddy:10332748]
The authors’ observations
Delirium is a complex, acute alteration in a patient’s mental status compared with his/her baseline functioning1 (Table 12). The onset of delirium is quick, happening within hours to days, with fluctuations in mental function. Patients might present with hyperactive, hypoactive, or mixed delirium.3 Patients with hyperactive delirium often have delusions and hallucinations; these patients might be agitated and could become violent with family and caregivers.3 Patients with hypoactive delirium are less likely to experience hallucinations and more likely to show symptoms of sedation.3 Patients with hypoactive delirium can be difficult to diagnose because it is challenging to interview them and understand what might be the cause of their sedated state. Patients also can exhibit a mixed delirium in which they fluctuate between periods of hyperactivity and hypoactivity.3
Continue to: Suspected delirium...
Suspected delirium should be considered a medical emergency because the outcome could be fatal.1 It is important to uncover and treat the underlying cause(s) of delirium rather than solely administering antipsychotics, which might mask the presenting symptoms. In an older study, Francis and Kapoor4 reported that 56% of geriatric patients with delirium had a single definite or probable etiology, while the other 44% had about 2.8 etiologies per patient on average. Delirium risk factors, causes, and factors to consider during patient evaluation are listed in Table 21,3,5-7 and Table 3.1,3,5-7
A synergistic relationship between comorbidities, environment, and medications can induce delirium.5 Identifying irreversible and reversible causes is the key to treating delirium. After the cause has been identified, it can be addressed and the patient could return to his/her previous level of functioning. If the delirium is the result of multiple irreversible causes, it could become chronic.
[polldaddy:10332749]
EVALUATION Cardiac dysfunction
Ms. P undergoes laboratory testing. The results include: white blood cell count, 5.9/µL; hemoglobin, 13.6 g/dL; hematocrit, 42.6%; platelets, 304 × 103/µL; sodium,143 mEq/L; potassium, 3.2 mEq/L; chloride, 96 mEq/L; carbon dioxide, 23 mEq/L; blood glucose, 87 mg/dL; creatinine, 1.2 mg/dL; estimated creatinine clearance (eCrCl) level of 33 mL/min/1.73 m2; calcium, 9.5 mg/dL; albumin, 3.6 g/dL; liver enzymes within normal limits; thyroid-stimulating hormone, 0.78 mIU/L; vitamin B12, 995 pg/mL; folic acid, 16.6 ng/mL; vitamin D, 31 pg/mL; and rapid plasma reagin: nonreactive. Urinalysis is unremarkable, and no culture is performed. Urine drug screening/toxicology is positive for the benzodiazepines that she received in the ED (oral alprazolam 0.25 mg given once and oral lorazepam 0.5 mg given once).
Electrocardiogram (ECG) shows atrial flutter/tachycardia with rapid ventricular response, marked left axis deviation, nonspecific ST- and T-wave abnormality, QT/QTC of 301/387 ms, and ventricular rate 151 beats per minute. A CT scan of the head and brain without contrast shows mild atrophy and chronic white matter changes and no acute intracranial abnormality. A two-view chest radiography shows no acute cardiopulmonary findings. Her temperature is 98.4°F; heart rate is 122 beats per minute; respiratory rate is 20 breaths per minute; blood pressure is 161/98 mm Hg; and oxygen saturation is 86% on room air.
Based on this data, Ms. P’s cardiac condition seems to be worsening, which is thought to be caused by her refusal of furosemide, lisinopril, isosorbide, sotalol, clopidogrel, and aspirin. The treatment team plans to work on compliance to resolve these cardiac issues and places Ms. P on 1:1 observation with a sitter and music in attempt to calm her.
Continue to: The authors' observations
The authors’ observations
Many factors can contribute to behavioral or cognitive changes in geriatric patients. Often, a major change noted in an older patient can be attributed to new-onset dementia, dementia with behavioral disturbances, delirium, depression, or acute psychosis. These potential causes should be considered and ruled out in a step-by-step progression. Because patients are unreliable historians during acute distress, a complete history from family or caregivers and exhaustive workup is paramount.
TREATMENT Medication adjustments
In an attempt to resolve Ms. P’s disruptive behaviors, her risperidone dosage is changed to 0.5 mg twice daily. Ms. P is encouraged to use the provided oxygen to raise her saturation level.
On hospital Day 3, a loose stool prompts a Clostridium difficile test as a possible source of delirium; however, the results are negative.
On hospital Day 4, Ms. P is confused and irritable overnight, yelling profanities at staff, refusing care, inappropriately disrobing, and having difficulty falling asleep and staying asleep. Risperidone is discontinued because it appears to have had little or no effect on Ms. P’s disruptive behaviors. Olanzapine, 10 mg/d, is initiated with mirtazapine, 7.5 mg/d, to help with mood, appetite, and sleep. Fluoxetine is also discontinued because of a possible interaction with clopidogrel.
On hospital Days 6 to 8, Ms. P remains upset and unable to follow instructions. Melatonin is initiated to improve her sleep cycle. On Day 9, she continues to decline and is cursing at hospital staff; haloperidol is initiated at 5 mg every morning, 10 mg at bedtime, and 5 mg IM as needed for agitation. Her sleep improves with melatonin and mirtazapine. IV hydration also is initiated. Ms. P has a slight improvement in medication compliance. On Day 11, haloperidol is increased to 5 mg in the morning, 5 mg in the afternoon, and 10 mg at bedtime. On Day 12, haloperidol is changed to 7.5 mg twice daily; a slight improvement in Ms. P’s behavior is noted.
Continue to: On hospital Day 13...
On hospital Day 13, Ms. P’s behavior declines again. She screams profanities at staff and does not recognize the clinicians who have been providing care to her. The physician initiates valproic acid, 125 mg, 3 times a day, to target Ms. P’s behavioral disturbances. A pharmacist notes that the patient’s sotalol could be contributing to Ms. P’s psychiatric presentation, and that based on her eCrCl level of 33 mL/min/1.73 m2, a dosage adjustment or medication change might be warranted.
On Day 14, Ms. P displays erratic behavior and intermittent tachycardia. A cardiac consultation is ordered. A repeat ECG reveals atrial fibrillation with rapid rate and a QT/QTc of 409/432 ms. Ms. P is transferred to the telemetry unit, where the cardiologist discontinues sotalol because the dosage is not properly renally adjusted. Sotalol hydrochloride has been associated with life-threatening ventricular tachycardia.8 Diltiazem, 30 mg every 6 hours is initiated to replace sotalol.
By Day 16, the treatment team notes improved cognition and behavior. On Day 17, the cardiologist reports that Ms. P’s atrial fibrillation is controlled. An ECG reveals mild left ventricular hypertrophy, an ejection fraction of 50% to 55%, no stenosis in the mitral or tricuspid valves, no valvular pulmonic stenosis, and moderate aortic sclerosis. Cardiac markers also are evaluated (creatinine phosphokinase: 105 U/L; creatinine kinase–MB fraction: 2.6 ng/mL; troponin: 0.01 ng/mL; pro-B-type natriuretic peptide: 2,073 pg/mL); and myocardial infarction is ruled out.
On Day 19, Ms. P’s diltiazem is consolidated to a controlled-delivery formulation, 180 mg/d, along with the addition of metoprolol, 12.5 mg twice daily. Ms. P is transferred back to the psychiatric unit.
OUTCOME Gradual improvement
On Days 20 to 23, Ms. P shows remarkable progress, and her mental status, cognition, and behavior slowly return to baseline. Haloperidol and valproic acid are tapered and discontinued. Ms. P is observed to be healthy and oriented to person, place, and time.
Continue to: On Day 25...
On Day 25, she is discharged from the hospital, and returns to the LTC facility.
The authors’ observations
Ms. P’s delirium was a combination of her older age, non-renally adjusted sotalol, and CKD. At admission, the hospital treatment team first thought that pneumonia or antibiotic use could have caused delirium. However, Ms. P’s condition did not improve after antibiotics were stopped. In addition, several chest radiographs found no evidence of pneumonia. It is important to check for any source of infection because infection is a common source of delirium in older patients.1 Urine samples revealed no pathogens, a C. difficile test was negative, and the patient’s white blood cell counts remained within normal limits. Physicians began looking elsewhere for potential causes of Ms. P’s delirium.
Ms. P’s vital signs ruled out a temperature irregularity or hypertension as the cause of her delirium. She has a slightly low oxygen saturation when she first presented, but this quickly returned to normal with administration of oxygen, which ruled out hypoxemia. Laboratory results concluded that Ms. P’s glucose levels were within a normal range and she had no electrolyte imbalances. A head CT scan showed slight atrophy of white matter that is consistent with Ms. P’s age. The head CT scan also showed that Ms. P had no acute condition or head trauma.
In terms of organ function, Ms. P was in relatively healthy condition other than paroxysmal atrial fibrillation and CKD. Chronic kidney disease can interrupt the normal pharmacokinetics of medications. Reviewing Ms. P’s medication list, several agents could have induced delirium, including antidepressants, antipsychotics, cardiovascular medications (beta blocker/antiarrhythmic [sotalol]), and opioid analgesics such as tramadol.5 Ms. P’s condition did not improve after discontinuing fluoxetine, risperidone, or olanzapine, although haloperidol was started in their place. Ms. P scored an 8 on the Naranjo Adverse Drug Reaction Probability Scale, indicating this event was a probable adverse drug reaction.9
Identifying a cause
This was a unique case where sotalol was identified as the culprit for inducing Ms. P’s delirium, because her age and CKD are irreversible. It is important to note that antiarrhythmics can induce arrhythmias when present in high concentrations or administered without appropriate renal dose adjustments. Although Ms. P’s serum levels of sotalol were not evaluated, because of her renal impairment, it is possible that toxic levels of sotalol accumulated and lead to arrhythmias and delirium. Of note, a cardiologist was consulted to safely change Ms. P to a calcium channel blocker so she could undergo cardiac monitoring. With the addition of diltiazem and metoprolol, the patient’s delirium subsided and her arrhythmia was controlled. Once the source of Ms. P’s delirium had been identified, antipsychotics were no longer needed.
Continue to: Bottom Line
Bottom Line
Delirium is a complex disorder that often has multiple causes, both reversible and irreversible. A “process of elimination” approach should be used to accurately identify and manage delirium. If a patient with delirium has little to no response to antipsychotic medications, the underlying cause or causes likely has not yet been addressed, and the evaluation should continue.
Related Resources
- Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456-1466.
- Inouye SK, Westendorp RGJ, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
Drug Brand Names
Acyclovir • Zovirax
Alprazolam • Niravam, Xanax
Amantadine • Symmetrel
Amphotericin B • Abelcet
Atorvastatin • Lipitor
Atropine • Atropen
Baclofen • EnovaRX-Baclofen
Benztropine • Cogentin
Bromocriptine • Cycloset
Calcitonin • Miacalcin
Carbamazepine • Tegretol
Carbidopa-levodopa • Duopa
Ceftriaxone • Rocephin
Chlorpromazine • Thorazine
Clonidine • Catapres
Clopidogrel • Plavix
Cyclobenzaprine • Amrix
Digoxin • Lanoxin
Diltiazem • Cardizem
Disulfiram • Antabuse
Ezetimibe • Zetia
Fluoxetine • Prozac
Fluphenazine • Prolixin
Furosemide • Lasix
Haloperidol • Haldol
Ipratropium/albuterol nebulized solution • Combivent Respimat
Isoniazid • Isotamine
Isosorbide nitrate • Dilatrate
Levetiracetam • Keppra
Levodopa • Stalevo
Linezolid • Zyvox
Lisinopril • Zestril
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Magnesium Oxide • Mag-200
Meperidine • Demerol
Methyldopa • Aldomet
Metoprolol • Lopressor
Metronidazole • Flagyl
Mirtazapine • Remeron
Nitrofurantoin • Macrobid
Olanzapine • Zyprexa
Pantoprazole • Protonix
Phenytoin • Dilantin
Pramipexole • Mirapex
Rifampin • Rifadin
Risperidone • Risperdal
Ropinirole • Requip
Sotalol hydrochloride • Betapace AF
Tramadol • Ultram
Trihexyphenidyl • Trihexane
Valproic acid • Depakote
CASE Combative and agitated
Ms. P, age 87, presents to the emergency department (ED) with her caregiver, who says Ms. P has new-onset altered mental status, agitation, and combativeness.
Ms. P resides at a long-term care (LTC) facility, where according to the nurses she normally is pleasant, well-oriented, and cooperative. Ms. P’s medical history includes major depressive disorder, generalized anxiety disorder, hypertension, chronic kidney disease (CKD) stage III, peptic ulcer disease, gastroesophageal reflux disease, coronary artery disease with 2 past myocardial infarctions requiring stents, chronic obstructive pulmonary disease, hyperlipidemia, bradycardia requiring a pacemaker, paroxysmal atrial fibrillation, asthma, aortic stenosis, peripheral vascular disease, esophageal stricture requiring dilation, deep vein thrombosis, and migraines.
Mr. P’s medication list includes acetaminophen, 650 mg every 6 hours; ipratropium/albuterol nebulized solution, 3 mL 4 times a day; aspirin, 81 mg/d; atorvastatin, 40 mg/d; calcitonin, 1 spray nasally at bedtime; clopidogrel, 75 mg/d; ezetimibe, 10 mg/d; fluoxetine, 20 mg/d; furosemide, 20 mg/d; isosorbide dinitrate, 120 mg/d; lisinopril, 15 mg/d; risperidone, 0.5 mg/d; magnesium oxide, 800 mg/d; pantoprazole, 40 mg/d; polyethylene glycol, 17 g/d; sotalol, 160 mg/d; olanzapine, 5 mg IM every 6 hours as needed for agitation; and tramadol, 50 mg every 8 hours as needed for headache.
Seven days before coming to the ED, Ms. P was started on ceftriaxone, 1 g/d, for suspected community-acquired pneumonia. At that time, the nursing staff noticed behavioral changes. Soon after, Ms. P began refusing all her medications. Two days before presenting to the ED, Ms. P was started on nitrofurantoin, 200 mg/d, for a suspected urinary tract infection, but it was discontinued because of an allergy.
Her caregiver reports that while at the LTC facility, Ms. P’s behavioral changes worsened. Ms. P claimed to be Jesus Christ and said she was talking to the devil; she chased other residents around the facility and slapped medications away from the nursing staff. According to caregivers, this behavior was out of character.
Shortly after arriving in the ED, Ms. P is admitted to the psychiatric unit.
[polldaddy:10332748]
The authors’ observations
Delirium is a complex, acute alteration in a patient’s mental status compared with his/her baseline functioning1 (Table 12). The onset of delirium is quick, happening within hours to days, with fluctuations in mental function. Patients might present with hyperactive, hypoactive, or mixed delirium.3 Patients with hyperactive delirium often have delusions and hallucinations; these patients might be agitated and could become violent with family and caregivers.3 Patients with hypoactive delirium are less likely to experience hallucinations and more likely to show symptoms of sedation.3 Patients with hypoactive delirium can be difficult to diagnose because it is challenging to interview them and understand what might be the cause of their sedated state. Patients also can exhibit a mixed delirium in which they fluctuate between periods of hyperactivity and hypoactivity.3
Continue to: Suspected delirium...
Suspected delirium should be considered a medical emergency because the outcome could be fatal.1 It is important to uncover and treat the underlying cause(s) of delirium rather than solely administering antipsychotics, which might mask the presenting symptoms. In an older study, Francis and Kapoor4 reported that 56% of geriatric patients with delirium had a single definite or probable etiology, while the other 44% had about 2.8 etiologies per patient on average. Delirium risk factors, causes, and factors to consider during patient evaluation are listed in Table 21,3,5-7 and Table 3.1,3,5-7
A synergistic relationship between comorbidities, environment, and medications can induce delirium.5 Identifying irreversible and reversible causes is the key to treating delirium. After the cause has been identified, it can be addressed and the patient could return to his/her previous level of functioning. If the delirium is the result of multiple irreversible causes, it could become chronic.
[polldaddy:10332749]
EVALUATION Cardiac dysfunction
Ms. P undergoes laboratory testing. The results include: white blood cell count, 5.9/µL; hemoglobin, 13.6 g/dL; hematocrit, 42.6%; platelets, 304 × 103/µL; sodium,143 mEq/L; potassium, 3.2 mEq/L; chloride, 96 mEq/L; carbon dioxide, 23 mEq/L; blood glucose, 87 mg/dL; creatinine, 1.2 mg/dL; estimated creatinine clearance (eCrCl) level of 33 mL/min/1.73 m2; calcium, 9.5 mg/dL; albumin, 3.6 g/dL; liver enzymes within normal limits; thyroid-stimulating hormone, 0.78 mIU/L; vitamin B12, 995 pg/mL; folic acid, 16.6 ng/mL; vitamin D, 31 pg/mL; and rapid plasma reagin: nonreactive. Urinalysis is unremarkable, and no culture is performed. Urine drug screening/toxicology is positive for the benzodiazepines that she received in the ED (oral alprazolam 0.25 mg given once and oral lorazepam 0.5 mg given once).
Electrocardiogram (ECG) shows atrial flutter/tachycardia with rapid ventricular response, marked left axis deviation, nonspecific ST- and T-wave abnormality, QT/QTC of 301/387 ms, and ventricular rate 151 beats per minute. A CT scan of the head and brain without contrast shows mild atrophy and chronic white matter changes and no acute intracranial abnormality. A two-view chest radiography shows no acute cardiopulmonary findings. Her temperature is 98.4°F; heart rate is 122 beats per minute; respiratory rate is 20 breaths per minute; blood pressure is 161/98 mm Hg; and oxygen saturation is 86% on room air.
Based on this data, Ms. P’s cardiac condition seems to be worsening, which is thought to be caused by her refusal of furosemide, lisinopril, isosorbide, sotalol, clopidogrel, and aspirin. The treatment team plans to work on compliance to resolve these cardiac issues and places Ms. P on 1:1 observation with a sitter and music in attempt to calm her.
Continue to: The authors' observations
The authors’ observations
Many factors can contribute to behavioral or cognitive changes in geriatric patients. Often, a major change noted in an older patient can be attributed to new-onset dementia, dementia with behavioral disturbances, delirium, depression, or acute psychosis. These potential causes should be considered and ruled out in a step-by-step progression. Because patients are unreliable historians during acute distress, a complete history from family or caregivers and exhaustive workup is paramount.
TREATMENT Medication adjustments
In an attempt to resolve Ms. P’s disruptive behaviors, her risperidone dosage is changed to 0.5 mg twice daily. Ms. P is encouraged to use the provided oxygen to raise her saturation level.
On hospital Day 3, a loose stool prompts a Clostridium difficile test as a possible source of delirium; however, the results are negative.
On hospital Day 4, Ms. P is confused and irritable overnight, yelling profanities at staff, refusing care, inappropriately disrobing, and having difficulty falling asleep and staying asleep. Risperidone is discontinued because it appears to have had little or no effect on Ms. P’s disruptive behaviors. Olanzapine, 10 mg/d, is initiated with mirtazapine, 7.5 mg/d, to help with mood, appetite, and sleep. Fluoxetine is also discontinued because of a possible interaction with clopidogrel.
On hospital Days 6 to 8, Ms. P remains upset and unable to follow instructions. Melatonin is initiated to improve her sleep cycle. On Day 9, she continues to decline and is cursing at hospital staff; haloperidol is initiated at 5 mg every morning, 10 mg at bedtime, and 5 mg IM as needed for agitation. Her sleep improves with melatonin and mirtazapine. IV hydration also is initiated. Ms. P has a slight improvement in medication compliance. On Day 11, haloperidol is increased to 5 mg in the morning, 5 mg in the afternoon, and 10 mg at bedtime. On Day 12, haloperidol is changed to 7.5 mg twice daily; a slight improvement in Ms. P’s behavior is noted.
Continue to: On hospital Day 13...
On hospital Day 13, Ms. P’s behavior declines again. She screams profanities at staff and does not recognize the clinicians who have been providing care to her. The physician initiates valproic acid, 125 mg, 3 times a day, to target Ms. P’s behavioral disturbances. A pharmacist notes that the patient’s sotalol could be contributing to Ms. P’s psychiatric presentation, and that based on her eCrCl level of 33 mL/min/1.73 m2, a dosage adjustment or medication change might be warranted.
On Day 14, Ms. P displays erratic behavior and intermittent tachycardia. A cardiac consultation is ordered. A repeat ECG reveals atrial fibrillation with rapid rate and a QT/QTc of 409/432 ms. Ms. P is transferred to the telemetry unit, where the cardiologist discontinues sotalol because the dosage is not properly renally adjusted. Sotalol hydrochloride has been associated with life-threatening ventricular tachycardia.8 Diltiazem, 30 mg every 6 hours is initiated to replace sotalol.
By Day 16, the treatment team notes improved cognition and behavior. On Day 17, the cardiologist reports that Ms. P’s atrial fibrillation is controlled. An ECG reveals mild left ventricular hypertrophy, an ejection fraction of 50% to 55%, no stenosis in the mitral or tricuspid valves, no valvular pulmonic stenosis, and moderate aortic sclerosis. Cardiac markers also are evaluated (creatinine phosphokinase: 105 U/L; creatinine kinase–MB fraction: 2.6 ng/mL; troponin: 0.01 ng/mL; pro-B-type natriuretic peptide: 2,073 pg/mL); and myocardial infarction is ruled out.
On Day 19, Ms. P’s diltiazem is consolidated to a controlled-delivery formulation, 180 mg/d, along with the addition of metoprolol, 12.5 mg twice daily. Ms. P is transferred back to the psychiatric unit.
OUTCOME Gradual improvement
On Days 20 to 23, Ms. P shows remarkable progress, and her mental status, cognition, and behavior slowly return to baseline. Haloperidol and valproic acid are tapered and discontinued. Ms. P is observed to be healthy and oriented to person, place, and time.
Continue to: On Day 25...
On Day 25, she is discharged from the hospital, and returns to the LTC facility.
The authors’ observations
Ms. P’s delirium was a combination of her older age, non-renally adjusted sotalol, and CKD. At admission, the hospital treatment team first thought that pneumonia or antibiotic use could have caused delirium. However, Ms. P’s condition did not improve after antibiotics were stopped. In addition, several chest radiographs found no evidence of pneumonia. It is important to check for any source of infection because infection is a common source of delirium in older patients.1 Urine samples revealed no pathogens, a C. difficile test was negative, and the patient’s white blood cell counts remained within normal limits. Physicians began looking elsewhere for potential causes of Ms. P’s delirium.
Ms. P’s vital signs ruled out a temperature irregularity or hypertension as the cause of her delirium. She has a slightly low oxygen saturation when she first presented, but this quickly returned to normal with administration of oxygen, which ruled out hypoxemia. Laboratory results concluded that Ms. P’s glucose levels were within a normal range and she had no electrolyte imbalances. A head CT scan showed slight atrophy of white matter that is consistent with Ms. P’s age. The head CT scan also showed that Ms. P had no acute condition or head trauma.
In terms of organ function, Ms. P was in relatively healthy condition other than paroxysmal atrial fibrillation and CKD. Chronic kidney disease can interrupt the normal pharmacokinetics of medications. Reviewing Ms. P’s medication list, several agents could have induced delirium, including antidepressants, antipsychotics, cardiovascular medications (beta blocker/antiarrhythmic [sotalol]), and opioid analgesics such as tramadol.5 Ms. P’s condition did not improve after discontinuing fluoxetine, risperidone, or olanzapine, although haloperidol was started in their place. Ms. P scored an 8 on the Naranjo Adverse Drug Reaction Probability Scale, indicating this event was a probable adverse drug reaction.9
Identifying a cause
This was a unique case where sotalol was identified as the culprit for inducing Ms. P’s delirium, because her age and CKD are irreversible. It is important to note that antiarrhythmics can induce arrhythmias when present in high concentrations or administered without appropriate renal dose adjustments. Although Ms. P’s serum levels of sotalol were not evaluated, because of her renal impairment, it is possible that toxic levels of sotalol accumulated and lead to arrhythmias and delirium. Of note, a cardiologist was consulted to safely change Ms. P to a calcium channel blocker so she could undergo cardiac monitoring. With the addition of diltiazem and metoprolol, the patient’s delirium subsided and her arrhythmia was controlled. Once the source of Ms. P’s delirium had been identified, antipsychotics were no longer needed.
Continue to: Bottom Line
Bottom Line
Delirium is a complex disorder that often has multiple causes, both reversible and irreversible. A “process of elimination” approach should be used to accurately identify and manage delirium. If a patient with delirium has little to no response to antipsychotic medications, the underlying cause or causes likely has not yet been addressed, and the evaluation should continue.
Related Resources
- Marcantonio ER. Delirium in hospitalized older adults. N Engl J Med. 2017;377:1456-1466.
- Inouye SK, Westendorp RGJ, Saczynski JS. Delirium in elderly people. Lancet. 2014;383(9920):911-922.
Drug Brand Names
Acyclovir • Zovirax
Alprazolam • Niravam, Xanax
Amantadine • Symmetrel
Amphotericin B • Abelcet
Atorvastatin • Lipitor
Atropine • Atropen
Baclofen • EnovaRX-Baclofen
Benztropine • Cogentin
Bromocriptine • Cycloset
Calcitonin • Miacalcin
Carbamazepine • Tegretol
Carbidopa-levodopa • Duopa
Ceftriaxone • Rocephin
Chlorpromazine • Thorazine
Clonidine • Catapres
Clopidogrel • Plavix
Cyclobenzaprine • Amrix
Digoxin • Lanoxin
Diltiazem • Cardizem
Disulfiram • Antabuse
Ezetimibe • Zetia
Fluoxetine • Prozac
Fluphenazine • Prolixin
Furosemide • Lasix
Haloperidol • Haldol
Ipratropium/albuterol nebulized solution • Combivent Respimat
Isoniazid • Isotamine
Isosorbide nitrate • Dilatrate
Levetiracetam • Keppra
Levodopa • Stalevo
Linezolid • Zyvox
Lisinopril • Zestril
Lithium • Eskalith, Lithobid
Lorazepam • Ativan
Magnesium Oxide • Mag-200
Meperidine • Demerol
Methyldopa • Aldomet
Metoprolol • Lopressor
Metronidazole • Flagyl
Mirtazapine • Remeron
Nitrofurantoin • Macrobid
Olanzapine • Zyprexa
Pantoprazole • Protonix
Phenytoin • Dilantin
Pramipexole • Mirapex
Rifampin • Rifadin
Risperidone • Risperdal
Ropinirole • Requip
Sotalol hydrochloride • Betapace AF
Tramadol • Ultram
Trihexyphenidyl • Trihexane
Valproic acid • Depakote
1. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention, and treatment. Nat Rev Neurol. 2009;5(4):210-220.
2. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
3. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
4. Francis J, Kapoor WN. Delirium in hospitalized elderly. J Gen Intern Med. 1990;5(1):65-79.
5. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
6. Cook IA. Guideline watch: practice guideline for the treatment of patients with delirium. Arlington, VA: American Psychiatric Publishing; 2004.
7. Bourgeois J, Ategan A, Losier B. Delirium in the hospital: emphasis on the management of geriatric patients. Current Psychiatry. 2014;13(8):29,36-42.
8. Betapace AF [package insert]. Zug, Switzerland: Covis Pharma; 2016.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
1. Fong TG, Tulebaev SR, Inouye SK. Delirium in elderly adults: diagnosis, prevention, and treatment. Nat Rev Neurol. 2009;5(4):210-220.
2. Diagnostic and statistical manual of mental disorders, fifth edition. Washington, DC: American Psychiatric Association; 2013.
3. American Psychiatric Association. Practice guideline for the treatment of patients with delirium. Am J Psychiatry. 1999;156(suppl 5):1-20.
4. Francis J, Kapoor WN. Delirium in hospitalized elderly. J Gen Intern Med. 1990;5(1):65-79.
5. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
6. Cook IA. Guideline watch: practice guideline for the treatment of patients with delirium. Arlington, VA: American Psychiatric Publishing; 2004.
7. Bourgeois J, Ategan A, Losier B. Delirium in the hospital: emphasis on the management of geriatric patients. Current Psychiatry. 2014;13(8):29,36-42.
8. Betapace AF [package insert]. Zug, Switzerland: Covis Pharma; 2016.
9. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.

Unipolar vs bipolar depression: A clinician’s perspective
Mrs. W, age 36, who is married, has a history of military service, and is currently employed as a paralegal, is referred to our practice by her family physician. She complains of severe depression that impairs her ability to function at work. She had seen several other psychiatrists in both military and civilian settings, and had been treated with multiple antidepressants, including fluoxetine, sertraline, bupropion, and paroxetine.
At the time of her initial psychiatric evaluation, she is taking duloxetine, 90 mg/d, but still is experiencing depressive symptoms. She is tearful, sad, lacks energy, spends too much time in bed, and is experiencing thoughts of hopelessness, despair, and escape, verging on thoughts of suicide. As a result, she needs to scale back her work schedule to part-time. When asked about how long she had been suffering from depression, she responds “I’ve been depressed all my life.” She had been briefly hospitalized at age 16, when she made a suicide attempt by overdose. There had been no subsequent suicide attempts or psychiatric hospitalizations, although she acknowledges having intermittent suicidal thoughts.
Mrs. W’s clinical presentation is similar to that of many patients entering our practice—patients who have recurrent depression that began in early life and a history of failure to respond to multiple antidepressants. She and other patients with similar presentations are not suffering from treatment-resistant depression and in need of a trial of electroconvulsive therapy, transcranial magnetic stimulation, direct current stimulation, vagus nerve stimulation, or intranasal esketamine. She has bipolar disorder, and had been repeatedly misdiagnosed and treated inappropriately with antidepressant monotherapy.
In a previous article1 (“Controversies in bipolar disorder: Trust evidence or experience?,”
In this article, based on our more than 25 years of experience in diagnosing and treating psychiatric disorders in patients of all ages, we expand on those observations.
Misdiagnosis is common
Bipolar depression is frequently misdiagnosed as unipolar depression in outpatient2-8 and inpatient9 settings, and in children and adolescents.10 Mrs. W is typical of patients who have what we consider a bipolar spectrum disorder and receive an inaccurate diagnosis and treatment that is ineffective or may worsen the course of their illness.
Reliance on DSM-511 and its predecessor, DSM-IV, is a part of the problem of misdiagnosis because the diagnostic criteria for bipolar disorder fail to capture the clinical features of many patients with “softer” (less obvious manic and hypomanic) variants of the disorder.12,13 For example, DSM-5 criteria for a hypomanic episode (the mild high experienced by patients with a soft bipolar disorder) require that the episode lasts “at least 4 consecutive days” and is “present most of the day, nearly every day.” In our experience, the majority of hypomanic episodes are shorter—ranging from a half-day to 2 days, averaging perhaps 1.5 days.
Continue to: DSM-5 also requires...
DSM-5 also requires severity criteria for hypomania that patients with unequivocal hypomanic episodes often do not meet. For example, they may fail to experience flight of ideas or racing thoughts, or engage in activities such as “unrestrained buying sprees, sexual indiscretions, or foolish business investments.” These patients usually describe these mild highs as feeling normal and report a happier mood, more smiles and laughter, increased energy, less sleepiness, increased talkativeness, increased socialization, and improved motivation to complete tasks left undone and projects left unfinished because of the previous depressive episode. These softer (subthreshold) hypomanic episodes are authentic and, if clinicians do not identify them, may lead to misdiagnosis and inappropriate treatment.
Patients who present with depression often fail to report these brief, subthreshold hypomanic episodes or consider them to be irrelevant to their diagnosis and treatment.12,13 Probing questions can often elicit these unreported highs. For example, a patient with depression should be asked, “Have you had a single good day during the last month?” and “Where were you and what did you do during that day?” Eliciting a history of brief periods of improved mood is the key to differentiating between unipolar and bipolar depression. Screening instruments such as the Mood Disorders Questionnaire14 and the Bipolar Spectrum Diagnostic Scale15 may be helpful in distinguishing between unipolar and bipolar depression. However, we offer our thoughts on making that crucial distinction.
Distinguishing between these 2 types of depression
Although it may be difficult to distinguish between unipolar and bipolar depression, especially in the absence of a history of distinct manic or hypomanic episodes, we find the following criteria to be useful in making that determination.
Age of onset. Bipolar spectrum disorders typically begin earlier in life than unipolar depression.10,16-19 A typical presentation of bipolar disorder in children and adolescents is depression or agitated mixed states with features of both mania and depression, often accompanied by rapid mood cycling.20,21 Unipolar depression usually begins later in life, and patients do not have a history of significant depressive episodes or mood swings in childhood or adolescence. An important question to ask a patient with a chief complaint of depression is, “How old were you when you first experienced an episode of depression?”
Gender differences. Bipolar spectrum disorders with more subtle (softer) presentations, such as subthreshold highs, occur more often in women than men.22 However, overall rates of bipolar disorder may be slightly higher in men than in women.23 Unipolar melancholic depression occurs at approximately the same frequency in men and women.24
Continue to: Rapidity of onset
Rapidity of onset. Bipolar depressive episodes develop more rapidly than unipolar episodes. It is common for a patient with a bipolar spectrum disorder to transition from normal to very depressed virtually overnight, whereas in our clinical experience, unipolar episodes progress more slowly, often over several months.
Deliberate self-harm. Adolescents and young adults with a bipolar spectrum disorder frequently engage in self-injurious behavior, usually cutting with a knife, razor, or even sharp fingernails.25 Although these patients may also have thoughts of suicide and make suicide attempts, the individual usually perceives cutting as a means of gaining relief from tension and distress. These behaviors are often associated with a diagnosis of a personality disorder; in our opinion, however, they are hallmarks of a bipolar spectrum disorder.
ADHD. Bipolar disorder frequently co-occurs with attention-deficit/hyperactivity disorder (ADHD).26,27 Adults with bipolar disorder often have ADHD symptoms, which can complicate their treatment and cause functional impairment even after their mood disorder has been stabilized.28
Substance use disorders. Excessive use of alcohol and drugs is common among people with a wide range of psychiatric disorders, but patients with bipolar disorder have an unusually high rate of co-occurring substance use disorders—40% to 50%.29,30
Appetite and weight differences. Patients with unipolar depression usually experience loss of appetite and weight loss, whereas in our clinical experience, patients with bipolar depression often overeat, crave carbohydrates, and gain weight.
Continue to: Sleep problems
Sleep problems. Patients with bipolar depression have an increased need for sleep (the opposite of what they experience during highs), are sleepy during the day regardless of how many hours they sleep, and have difficulty getting up in the morning. Patients with unipolar depression also have a sleep disturbance: they may fall asleep easily, sleep for a few hours, and then awaken but are unable to fall back to sleep.31 Yet these patients usually do not complain of sleepiness during the day.
Diurnal variation of mood. Patients with unipolar depression often report that their depressive symptoms fluctuate in a circadian manner. For example, they may report that their depression is worse in the morning but improves toward evening.31 This regular alteration of circadian rhythm usually is not evident in patients with bipolar depression, whose mood may vary unpredictably or in response to stressors. Some patients with bipolar disorder, however, exhibit ultradian (ultra-rapid) mood cycling, which may be confused with the diurnal mood variation seen in patients with unipolar depression.
Tendency to recur. Although both unipolar and bipolar depressive episodes recur, a pattern of multiple recurring episodes beginning in early life is characteristic of bipolar spectrum disorders.
Behavioral history. Patients with bipolar depression are more likely than patients with unipolar depression to have a history of multiple marriages, multiple romantic relationships, episodes of promiscuity, legal problems, or financial extravagance.
Response to antidepressants. Patients with bipolar depression exhibit atypical responses to antidepressant monotherapy, such as worsening of depressive symptoms, initial improvement of mood with subsequent loss of effectiveness, premature response to an antidepressant (eg, improvement of mood within 1 to 2 days of beginning the antidepressant), fluctuation of depressive symptoms (mood cycling), or precipitation of a hypomanic or manic episode. We believe that a history of multiple failed antidepressant trials is compelling evidence of misdiagnosis of a bipolar spectrum disorder as unipolar depression.
Continue to: Genetics
Genetics. Bipolar disorder is one of the most heritable of illnesses.32 Family history is important, but affected relatives may have been misdiagnosed with unipolar depression or schizophrenia, or said to have experienced “nervous breakdowns.”
Consequences of misdiagnosis
Misdiagnosis of patients with bipolar disorder is not benign. We see patients who have suffered needlessly for years with severe depression and mood instability. After trying antidepressant after antidepressant without benefit, they begin to feel hopeless, believing they have tried everything and that nothing works for them. Often, these patients have dropped out of high school or college, or lost jobs, friends, and spouses due to their disabling but misdiagnosed psychiatric disorder. Patients with misdiagnosed bipolar disorder have an increased risk of suicide attempts and psychiatric hospitalization.5,8
Misdiagnosis of patients with bipolar disorder is not limited to nonpsychiatric physicians. The majority of patients with bipolar spectrum disorders are misdiagnosed by outpatient psychiatrists as having unipolar depression.2-7 At least 45% of patients hospitalized for depression have bipolar disorder—and most of these patients are treated inappropriately with antidepressants.9 The STAR*D study,33,34 a large randomized clinical trial of antidepressants, concluded that more than one-third of patients had not remitted from their depression after treatment with 3 different antidepressants. In our opinion, many of the nonresponding patients may have undiagnosed bipolar depression, which predictably leads to a failure to respond adequately to antidepressants. We believe that the customary inclusion and exclusion criteria used to select participants for these research studies miss subtle (subthreshold) hypomanic episodes that fall short of meeting DSM criteria for duration and severity. This phenomenon may account for the results of studies that conclude that antidepressants are, at best, minimally more effective than placebo.35
When a patient with a bipolar spectrum disorder is misdiagnosed and treated with an antidepressant, the usual result is mood destabilization. Reports of mood swings, increased crying, and suicidal thoughts and suicidal gestures in children, adolescents, and young adults treated with antidepressants led the FDA to issue a “black-box” warning.36 Because bipolar depression typically begins in youth,10,18,19 the behaviors cited in the warning may reflect misdiagnosis of bipolar depression as unipolar depression, and consequent mood destabilization as a result of treatment with an antidepressant in the absence of a mood stabilizer.
Depression and life stressors
Since many patients who are depressed present with a history of significant stressors, clinicians often face the problem of distinguishing between clinical depression and stress-induced depression. We believe that one typical symptom of depression—increased sensitivity to stressors—may help in making that distinction. A patient who is depressed will often attribute depression to stressors such as marital conflict, divorce, problems with a teenage child, work pressures, financial pressures, or the illness or death of a family member or pet. If clinical depression (unipolar or bipolar) is present, the symptoms are persistent, sometimes antedate the stressor by days or weeks, often outlast the stressor, increase in severity over time, and are disproportional to the stressor. Clinical depression can also cause the patient to become obsessed with traumatic events or losses that occurred many years earlier.
Continue to: Our approach to treatment
Our approach to treatment
Patients with mood disorders often benefit from a combination of pharmacologic management and psychotherapy. Psychotherapy is particularly important in addressing the functional impairment, diminished self-worth, and interpersonal conflicts that often accompany clinical depression. Several styles or systems of psychotherapy have been developed to benefit patients with mood disorders. Their effectiveness may depend on the patient’s ability to gain insight,37 but in our opinion, the most important attribute of helpful psychotherapy is the rapport established between the patient and the therapist, and the therapist’s ability to empathize with the patient and instill in the patient a sense of optimism and hope. We often recommend that patients attend meetings of the Depression and Bipolar Support Alliance (DBSA), a national support group with chapters throughout the country. Patients often find that attending these meetings is both educational and emotionally rewarding.
The foundational pharmacologic treatment for bipolar disorder is a mood stabilizer. The medications we consider to be effective mood stabilizers (some with an FDA indication for bipolar maintenance, some without) are lithium carbonate, divalproex sodium, carbamazepine, oxcarbazepine, and lamotrigine.
Each of these mood stabilizers has its advantages, disadvantages, risks, and adverse effects. For example, although divalproex is a reliable mood stabilizer, it has a significant risk of causing birth defects if taken during pregnancy and can cause increased appetite and weight gain. Carbamazepine has significant drug interactions and the potential to cause neurologic adverse effects, while oxcarbazepine, a derivative of carbamazepine, has fewer drug interactions but is more likely to cause hyponatremia. Lamotrigine must be titrated very slowly to reduce the risk of a potentially fatal skin rash (ie, Stevens-Johnson syndrome or toxic epidermal necrolysis). Lithium is effective but has a significant adverse-effect burden: impairment of renal function with long-term use, nephrogenic diabetes insipidus, hypothyroidism, hyperparathyroidism, acne, and weight gain. Lithium also has potential interactions with multiple commonly prescribed medications, including antihypertensives and diuretics, as well as over-the-counter pain relievers such as ibuprofen and naproxen.
Second-generation antipsychotics (SGAs) have mood stabilizing, antidepressant, and anti-manic properties and are often useful in managing bipolar disorder. In our experience, for patients with bipolar disorder, SGAs are best used in combination with a mood stabilizer. Although virtually all SGAs have demonstrated effectiveness in the treatment of psychosis and some phases of bipolar disorder, the newer agents (aripiprazole, brexpiprazole, lurasidone, and cariprazine) are relatively free of metabolic adverse effects such as weight gain, abnormal cholesterol levels, increased prolactin levels, insulin resistance, and increased risk of diabetes.
Antidepressants may be effective in treating unipolar depression, but when treating bipolar depression, they should be used cautiously and only in combination with a mood stabilizer.
Continue to: As we observed...
As we observed in our previous article,1 thyroid laboratory monitoring and supplementation are critical components of managing mood disorders (Box 138-41).
Box 1
Conventional laboratory reference ranges often indicate that thyroid-stimulating hormone (TSH) levels as high as 4.0, 4.5, or 5.0 mU/L are normal. A recent meta- analysis determined that treatment of subclinical hypothyroidism (elevated TSH with normal free thyroxine) does not benefit patients’ quality of life.38 Patients with mood disorders, however, often fail to respond to mood stabilizers and other psychiatric medications unless their TSH is <3.0 or even <2.5 mU/L.39,40 We typically augment with liothyronine because, unlike levothyroxine, it works quickly, does not require deiodination to be activated, and, contrary to some reports, its elimination and biologic half-life are sufficient for single daily dosing.41
Moving towards better diagnoses
The emergence of a criteria-based psychiatric system in 1980 with the publication of DSM-III, and its subsequent revisions and updates, constituted a major advance in psychiatric diagnosis. As we learn more about the pathophysiology, genetics, and epigenetics of psychiatric symptoms and syndromes, future diagnostic systems will improve problems of validity that have yet to be resolved. While we believe that, for the most part, DSM-5 was an advance over the previous diagnostic iteration, we have 2 issues with DSM-5 in terms of the diagnosis of bipolar disorder (Box 210,12,13,18,19,42).
Box 2
Based on our clinical experience treating thousands of patients over 25 years, we have 2 issues with DSM-5 regarding bipolar disorder:
1. The DSM-5 criteria for hypomania fail to reflect the features of clinical presentations commonly seen in our practice. The majority of patients with authentic bipolar syndromes do not have hypomanias that last for at least 4 days or reach the level of severity required for a DSM-5 diagnosis of hypomania. This results in misdiagnosis of patients with bipolar depression as suffering from unipolar depression, which leads to inappropriate treatment with antidepressant monotherapy.
2. Bipolar disorder frequently makes its first appearance in childhood and adolescence,10,18,19 and increasing numbers of young patients have been receiving this diagnosis.42 In our opinion, this increase reflects clinicians’ improved diagnostic skills. Perhaps alarmed by the increase in young people receiving a diagnosis of bipolar disorder, the authors of DSM-5 created a new diagnosis for children: disruptive mood dysregulation disorder. This diagnostic addition is based on the finding that children with these mood symptoms may not subsequently exhibit classic DSM-5 manic or hypomanic episodes. But the lack of such episodes does not preclude a diagnosis of bipolar disorder, because many adults with unequivocal bipolar spectrum disorders have subthreshold hypomanias and thus fail to exhibit classic manic or hypomanic episodes.12,13
A rose by any other name would smell as sweet. Children who exhibit symptoms of disruptive mood dysregulation disorder— chronic irritability and protracted temper outbursts—usually suffer from depression and mood instability. In our opinion, it is irrational and confusing to clinicians to separate out with a new diagnosis an arbitrarily defined group of children who exhibit substantially the same symptoms as those who receive a diagnosis of bipolar disorder.
Patients with a chief complaint of depression are often given a diagnosis of “major depression, rule out bipolar disorder.” We believe that this formula should be turned on its head. In our opinion, based on our clinical experience, we think that most patients who present to a clinician’s office or psychiatric hospital with depression have bipolar depression, not unipolar depression. We hope that our experience and observations derived from treating thousands of patients over more than 25 years may be helpful to clinicians who sometimes struggle to bring relief to their patients with mood disorders.
CASE CONTINUED
Return to work
Mrs. W is now doing well. She is taking a lower dosage of duloxetine, 60 mg/d, in combination with the mood stabilizer lamotrigine, 200 mg/d. She returns to work full-time as a paralegal and no longer is experiencing depressive episodes.
Bottom Line
Patients with bipolar depression are often misdiagnosed with unipolar depression and treated inappropriately with antidepressant monotherapy, which often results in mood destabilization. Based on our clinical experience, a careful assessment of select criteria, including age of onset, rapidity of onset, comorbidities, diurnal mood variations, and more, can be useful for distinguishing between unipolar and bipolar depression.
Related Resources
- Nasrallah HA. Misdiagnosing bipolar depression as major depressive disorder. Current Psychiatry. 2013;12(10):20-21,A.
- Ghaemi SN. Bipolar spectrum: a review of the concept and a vision for the future. Psychiatry Investig. 2013;10(3):218-224.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Bupropion • Wellbutrin
Carbamazepine • Tegretol, Equetro
Cariprazine • Vraylar
Divalproex • Depakote
Duloxetine • Cymbalta
Esketamine • Spravato
Fluoxetine • Prozac
Lamotrigine • Lamictal
Levothyroxine • Synthroid, Levoxyl
Liothyronine • Cytomel
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Oxcarbazepine • Oxtellar XR, Trileptal
Paroxetine • Paxil
Sertraline • Zoloft
1. Miller GE, Noel RL. Controversies in bipolar disorder: trust evidence or experience? Current Psychiatry. 2009;8(2):27-28,31-33,39.
2. Glick ID. Undiagnosed bipolar disorder: new syndromes and new treatments. Prim Care Companion J Clin Psychiatry. 2004;6(1):27-33.
3. Ghaemi SN, Sachs GS, Chiou AM, et al. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord. 1999;52(1-3):135-144.
4. Blanco C, Laje G, Olfson M, et al. Trends in the treatment of bipolar disorder by outpatient psychiatrists. Am J Psychiatry. 2002;159(6):1005-1010.
5. Shi L, Thiebaud P, McCombs JS. The impact of unrecognized bipolar disorders for patients treated with antidepressants in the fee-for-services California Medicaid (Medi-Cal) program. J Affect Disord. 2004;82(3):373-383.
6. Sidor MM, MacQueen GM. Antidepressants for the acute treatment of bipolar depression: a systematic review and meta-analysis. J Clin Psychiatry. 2011;72(2):156-167.
7. Hughes T, Cardno A, West R, et al. Unrecognized bipolar disorder among UK primary care patients prescribed antidepressants: an observational study. Br J Gen Pract. 2016;66(643):e71-e77.
8. Keck PE Jr, Kessler RC, Ross R. Clinical and economic effects of unrecognized or inadequately treated bipolar disorder. J Psychiatric Pract. 2008;14(Suppl 2):31-38.
9. Goldberg JF, Harrow M, Whiteside JF. Risk for bipolar illness in inpatients initially hospitalized for unipolar depression. Am J Psychiatry. 2001:158(8):1265-1270.
10. Chilakamarri JK, Filkowski MM, Ghaemi SN. Misdiagnosis of bipolar disorder in children and adolescents: a comparison with ADHD and major depressive disorder. Ann Clin Psychiatry. 2011;23(1):25-29.
11. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
12. Bowden CL. A different depression: clinical distinctions between bipolar and unipolar depression. J Affect Disord. 2005;84(2-3):117-125.
13. Baldassano C. Distinctions between bipolar I and bipolar II depression. Current Psychiatry. 2017;16(8):S7-S16.
14. Hirschfeld MA, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: The Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
15. Ghaemi SN, Miller CJ, Berv DA, et al. Sensitivity and specificity a new bipolar spectrum diagnostic scale. J Affect Disorder. 2005;84(2-3):273-277
16. Suppes T, Leverich G, Keck P, et al. The Stanley Foundation Continuing Bipolar Treatment Outcome Network. II. Demographics and illness characteristics of the first 261 patients. J Affect Disorder. 2001;67(1-3):45-49.
17. Perlis RH, Miyahara S, Marangell LB. Long-term implications of early onset in bipolar disorder: data from the first 1000 participants in the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP–BD). Biol Psychiatry. 2004;55(9):875-881.
18. Baldessarini RJ, Bolzani L, Kruz N, et al. Onset age of bipolar disorders at six international sites. J Affect Disord. 2010;121(1-2):143-146.
19. Post RM, Altshuler LL, Kupka R, et al. More childhood onset bipolar disorder in the United States than Canada or Europe: implications for treatment and prevention. Neurosci Biobehav Rev. 2017;74(Pt A):204-213.
20. Geller B, Luby J. Child and adolescent bipolar disorder: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1168-1176.
21. Findling RL, Gracious BL, McNamara NK, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord. 2001;3(4):202-210.
22. Arnold LM. Gender differences in bipolar disorder. Psychiatr Clin North Am. 2003;26(3):595-620.
23. Deflorio A, Jones I. Is sex important? Gender differences in bipolar disorder. Int Rev Psychiatry. 2010;22(5):437-452.
24. Bogren M, Brådvik L, Holmstrand C, et al. Gender differences in subtypes of depression by first incidence and age of onset: a follow-up of the Lunby population. Eur Arch Psychiatry Clin Neurosci. 2018;268(2):179-189.
25. Singhal A, Ross J, Seminog O, et al. Risks of self-harm and suicide in people with specific psychiatric and physical disorders: comparisons between disorders using English national record linkage. J R Soc Med. 2014;107(5):194-204.
26. Joshi G, Wilens T. Comorbidity in pediatric bipolar disorder. Child Adolesc Psychiatr Clin N Amer. 2009;18(2):291-319.
27. Youngtrom EA, Arnold LE, Frazier TW. Bipolar and ADHD comorbidity: both artifact and outgrowth of shared mechanisms. Clin Psychol (New York). 2010;17(4):350-359.
28. McIntyre RS, Kennedy SH, Soczynska JK, et al. Attention-deficit/hyperactivity disorder in adults with bipolar disorder or major depressive disorder: results from the International Mood Disorders Collaborative Project. Prime Care Companion J Clin Psychiatry. 2010;12(3). doi:10.4088/PCC.09m00861gry.
29. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiological Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
30. Hunt GE, Malhi GS, Cleary M, et al. Prevalence of comorbid bipolar and substance use disorders in clinical settings, 1990-2015: systematic review and meta-analysis. J Affect Disord. 2016;206:331-349.
31. Agargun MY, Besiroglu L, Cilli AS, et al. Nightmares, suicide attempts, and melancholic features in patients with unipolar major depression. J Affect Disord. 2007;98(3):267-270.
32. Birmaher B, Axelson D, Monk K, et al. Lifetime psychiatric disorders in school-aged offspring of parents with bipolar disorder: the Pittsburgh Bipolar Offspring Study. Arch Gen Psychiatry. 2009;66(3):287-296.
33. Rush AJ, Trivedi MH, Wisniewski SR, et al. Buproprion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231-1242.
34. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
35. Kirsch I. Antidepressants and the placebo effect. Z Psychol. 2014;222(3):128-134.
36. U.S. Food and Drug Administration. Suicidality in children and adolescents being treated with antidepressant medications. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161679.htm. Published February 5, 2018. Accessed May 10, 2019.
37. Jennissen S, Huber J, Ehrenthal JC, et al. Association between insight and outcome of psychotherapy; systematic review and meta-analysis. Am J Psychiatry. 2018;175(10):961-969.
38. Feller M, Snel M, Moutzouri E, et al. Association of thyroid hormone therapy with quality of life and thyroid-related symptoms in patients with subclinical hypothyroidism. JAMA. 2018;320(13):1349-1359.
39. Cole DP, Thase ME, Mallinger AG, et al. Slower treatment response in bipolar depression predicted by lower pretreatment thyroid function. Am J Psychiatry. 2002;159(1):116-121.
40. Parmentier T, Sienaert P. The use of triiodothyronine (T3) in the treatment of bipolar depression: a review of the literature. J Affect Disord. 2018;229:410-414.
41. Koda-Kimbe MA, Alldredge BK. Koda-Kimble and Young’s applied therapeutics: the clinical use of drugs (10th ed). Baltimore, MD: Walters Klower Health/Lippincott Williams & Wilkins; 2012.
42. Moreno C, Laje G, Blanco C, et al. National trends in the outpatient diagnosis and treatment of bipolar disorder in youth. Arch Gen Psychiatry. 2007;64(9):1032-1039.
Mrs. W, age 36, who is married, has a history of military service, and is currently employed as a paralegal, is referred to our practice by her family physician. She complains of severe depression that impairs her ability to function at work. She had seen several other psychiatrists in both military and civilian settings, and had been treated with multiple antidepressants, including fluoxetine, sertraline, bupropion, and paroxetine.
At the time of her initial psychiatric evaluation, she is taking duloxetine, 90 mg/d, but still is experiencing depressive symptoms. She is tearful, sad, lacks energy, spends too much time in bed, and is experiencing thoughts of hopelessness, despair, and escape, verging on thoughts of suicide. As a result, she needs to scale back her work schedule to part-time. When asked about how long she had been suffering from depression, she responds “I’ve been depressed all my life.” She had been briefly hospitalized at age 16, when she made a suicide attempt by overdose. There had been no subsequent suicide attempts or psychiatric hospitalizations, although she acknowledges having intermittent suicidal thoughts.
Mrs. W’s clinical presentation is similar to that of many patients entering our practice—patients who have recurrent depression that began in early life and a history of failure to respond to multiple antidepressants. She and other patients with similar presentations are not suffering from treatment-resistant depression and in need of a trial of electroconvulsive therapy, transcranial magnetic stimulation, direct current stimulation, vagus nerve stimulation, or intranasal esketamine. She has bipolar disorder, and had been repeatedly misdiagnosed and treated inappropriately with antidepressant monotherapy.
In a previous article1 (“Controversies in bipolar disorder: Trust evidence or experience?,”
In this article, based on our more than 25 years of experience in diagnosing and treating psychiatric disorders in patients of all ages, we expand on those observations.
Misdiagnosis is common
Bipolar depression is frequently misdiagnosed as unipolar depression in outpatient2-8 and inpatient9 settings, and in children and adolescents.10 Mrs. W is typical of patients who have what we consider a bipolar spectrum disorder and receive an inaccurate diagnosis and treatment that is ineffective or may worsen the course of their illness.
Reliance on DSM-511 and its predecessor, DSM-IV, is a part of the problem of misdiagnosis because the diagnostic criteria for bipolar disorder fail to capture the clinical features of many patients with “softer” (less obvious manic and hypomanic) variants of the disorder.12,13 For example, DSM-5 criteria for a hypomanic episode (the mild high experienced by patients with a soft bipolar disorder) require that the episode lasts “at least 4 consecutive days” and is “present most of the day, nearly every day.” In our experience, the majority of hypomanic episodes are shorter—ranging from a half-day to 2 days, averaging perhaps 1.5 days.
Continue to: DSM-5 also requires...
DSM-5 also requires severity criteria for hypomania that patients with unequivocal hypomanic episodes often do not meet. For example, they may fail to experience flight of ideas or racing thoughts, or engage in activities such as “unrestrained buying sprees, sexual indiscretions, or foolish business investments.” These patients usually describe these mild highs as feeling normal and report a happier mood, more smiles and laughter, increased energy, less sleepiness, increased talkativeness, increased socialization, and improved motivation to complete tasks left undone and projects left unfinished because of the previous depressive episode. These softer (subthreshold) hypomanic episodes are authentic and, if clinicians do not identify them, may lead to misdiagnosis and inappropriate treatment.
Patients who present with depression often fail to report these brief, subthreshold hypomanic episodes or consider them to be irrelevant to their diagnosis and treatment.12,13 Probing questions can often elicit these unreported highs. For example, a patient with depression should be asked, “Have you had a single good day during the last month?” and “Where were you and what did you do during that day?” Eliciting a history of brief periods of improved mood is the key to differentiating between unipolar and bipolar depression. Screening instruments such as the Mood Disorders Questionnaire14 and the Bipolar Spectrum Diagnostic Scale15 may be helpful in distinguishing between unipolar and bipolar depression. However, we offer our thoughts on making that crucial distinction.
Distinguishing between these 2 types of depression
Although it may be difficult to distinguish between unipolar and bipolar depression, especially in the absence of a history of distinct manic or hypomanic episodes, we find the following criteria to be useful in making that determination.
Age of onset. Bipolar spectrum disorders typically begin earlier in life than unipolar depression.10,16-19 A typical presentation of bipolar disorder in children and adolescents is depression or agitated mixed states with features of both mania and depression, often accompanied by rapid mood cycling.20,21 Unipolar depression usually begins later in life, and patients do not have a history of significant depressive episodes or mood swings in childhood or adolescence. An important question to ask a patient with a chief complaint of depression is, “How old were you when you first experienced an episode of depression?”
Gender differences. Bipolar spectrum disorders with more subtle (softer) presentations, such as subthreshold highs, occur more often in women than men.22 However, overall rates of bipolar disorder may be slightly higher in men than in women.23 Unipolar melancholic depression occurs at approximately the same frequency in men and women.24
Continue to: Rapidity of onset
Rapidity of onset. Bipolar depressive episodes develop more rapidly than unipolar episodes. It is common for a patient with a bipolar spectrum disorder to transition from normal to very depressed virtually overnight, whereas in our clinical experience, unipolar episodes progress more slowly, often over several months.
Deliberate self-harm. Adolescents and young adults with a bipolar spectrum disorder frequently engage in self-injurious behavior, usually cutting with a knife, razor, or even sharp fingernails.25 Although these patients may also have thoughts of suicide and make suicide attempts, the individual usually perceives cutting as a means of gaining relief from tension and distress. These behaviors are often associated with a diagnosis of a personality disorder; in our opinion, however, they are hallmarks of a bipolar spectrum disorder.
ADHD. Bipolar disorder frequently co-occurs with attention-deficit/hyperactivity disorder (ADHD).26,27 Adults with bipolar disorder often have ADHD symptoms, which can complicate their treatment and cause functional impairment even after their mood disorder has been stabilized.28
Substance use disorders. Excessive use of alcohol and drugs is common among people with a wide range of psychiatric disorders, but patients with bipolar disorder have an unusually high rate of co-occurring substance use disorders—40% to 50%.29,30
Appetite and weight differences. Patients with unipolar depression usually experience loss of appetite and weight loss, whereas in our clinical experience, patients with bipolar depression often overeat, crave carbohydrates, and gain weight.
Continue to: Sleep problems
Sleep problems. Patients with bipolar depression have an increased need for sleep (the opposite of what they experience during highs), are sleepy during the day regardless of how many hours they sleep, and have difficulty getting up in the morning. Patients with unipolar depression also have a sleep disturbance: they may fall asleep easily, sleep for a few hours, and then awaken but are unable to fall back to sleep.31 Yet these patients usually do not complain of sleepiness during the day.
Diurnal variation of mood. Patients with unipolar depression often report that their depressive symptoms fluctuate in a circadian manner. For example, they may report that their depression is worse in the morning but improves toward evening.31 This regular alteration of circadian rhythm usually is not evident in patients with bipolar depression, whose mood may vary unpredictably or in response to stressors. Some patients with bipolar disorder, however, exhibit ultradian (ultra-rapid) mood cycling, which may be confused with the diurnal mood variation seen in patients with unipolar depression.
Tendency to recur. Although both unipolar and bipolar depressive episodes recur, a pattern of multiple recurring episodes beginning in early life is characteristic of bipolar spectrum disorders.
Behavioral history. Patients with bipolar depression are more likely than patients with unipolar depression to have a history of multiple marriages, multiple romantic relationships, episodes of promiscuity, legal problems, or financial extravagance.
Response to antidepressants. Patients with bipolar depression exhibit atypical responses to antidepressant monotherapy, such as worsening of depressive symptoms, initial improvement of mood with subsequent loss of effectiveness, premature response to an antidepressant (eg, improvement of mood within 1 to 2 days of beginning the antidepressant), fluctuation of depressive symptoms (mood cycling), or precipitation of a hypomanic or manic episode. We believe that a history of multiple failed antidepressant trials is compelling evidence of misdiagnosis of a bipolar spectrum disorder as unipolar depression.
Continue to: Genetics
Genetics. Bipolar disorder is one of the most heritable of illnesses.32 Family history is important, but affected relatives may have been misdiagnosed with unipolar depression or schizophrenia, or said to have experienced “nervous breakdowns.”
Consequences of misdiagnosis
Misdiagnosis of patients with bipolar disorder is not benign. We see patients who have suffered needlessly for years with severe depression and mood instability. After trying antidepressant after antidepressant without benefit, they begin to feel hopeless, believing they have tried everything and that nothing works for them. Often, these patients have dropped out of high school or college, or lost jobs, friends, and spouses due to their disabling but misdiagnosed psychiatric disorder. Patients with misdiagnosed bipolar disorder have an increased risk of suicide attempts and psychiatric hospitalization.5,8
Misdiagnosis of patients with bipolar disorder is not limited to nonpsychiatric physicians. The majority of patients with bipolar spectrum disorders are misdiagnosed by outpatient psychiatrists as having unipolar depression.2-7 At least 45% of patients hospitalized for depression have bipolar disorder—and most of these patients are treated inappropriately with antidepressants.9 The STAR*D study,33,34 a large randomized clinical trial of antidepressants, concluded that more than one-third of patients had not remitted from their depression after treatment with 3 different antidepressants. In our opinion, many of the nonresponding patients may have undiagnosed bipolar depression, which predictably leads to a failure to respond adequately to antidepressants. We believe that the customary inclusion and exclusion criteria used to select participants for these research studies miss subtle (subthreshold) hypomanic episodes that fall short of meeting DSM criteria for duration and severity. This phenomenon may account for the results of studies that conclude that antidepressants are, at best, minimally more effective than placebo.35
When a patient with a bipolar spectrum disorder is misdiagnosed and treated with an antidepressant, the usual result is mood destabilization. Reports of mood swings, increased crying, and suicidal thoughts and suicidal gestures in children, adolescents, and young adults treated with antidepressants led the FDA to issue a “black-box” warning.36 Because bipolar depression typically begins in youth,10,18,19 the behaviors cited in the warning may reflect misdiagnosis of bipolar depression as unipolar depression, and consequent mood destabilization as a result of treatment with an antidepressant in the absence of a mood stabilizer.
Depression and life stressors
Since many patients who are depressed present with a history of significant stressors, clinicians often face the problem of distinguishing between clinical depression and stress-induced depression. We believe that one typical symptom of depression—increased sensitivity to stressors—may help in making that distinction. A patient who is depressed will often attribute depression to stressors such as marital conflict, divorce, problems with a teenage child, work pressures, financial pressures, or the illness or death of a family member or pet. If clinical depression (unipolar or bipolar) is present, the symptoms are persistent, sometimes antedate the stressor by days or weeks, often outlast the stressor, increase in severity over time, and are disproportional to the stressor. Clinical depression can also cause the patient to become obsessed with traumatic events or losses that occurred many years earlier.
Continue to: Our approach to treatment
Our approach to treatment
Patients with mood disorders often benefit from a combination of pharmacologic management and psychotherapy. Psychotherapy is particularly important in addressing the functional impairment, diminished self-worth, and interpersonal conflicts that often accompany clinical depression. Several styles or systems of psychotherapy have been developed to benefit patients with mood disorders. Their effectiveness may depend on the patient’s ability to gain insight,37 but in our opinion, the most important attribute of helpful psychotherapy is the rapport established between the patient and the therapist, and the therapist’s ability to empathize with the patient and instill in the patient a sense of optimism and hope. We often recommend that patients attend meetings of the Depression and Bipolar Support Alliance (DBSA), a national support group with chapters throughout the country. Patients often find that attending these meetings is both educational and emotionally rewarding.
The foundational pharmacologic treatment for bipolar disorder is a mood stabilizer. The medications we consider to be effective mood stabilizers (some with an FDA indication for bipolar maintenance, some without) are lithium carbonate, divalproex sodium, carbamazepine, oxcarbazepine, and lamotrigine.
Each of these mood stabilizers has its advantages, disadvantages, risks, and adverse effects. For example, although divalproex is a reliable mood stabilizer, it has a significant risk of causing birth defects if taken during pregnancy and can cause increased appetite and weight gain. Carbamazepine has significant drug interactions and the potential to cause neurologic adverse effects, while oxcarbazepine, a derivative of carbamazepine, has fewer drug interactions but is more likely to cause hyponatremia. Lamotrigine must be titrated very slowly to reduce the risk of a potentially fatal skin rash (ie, Stevens-Johnson syndrome or toxic epidermal necrolysis). Lithium is effective but has a significant adverse-effect burden: impairment of renal function with long-term use, nephrogenic diabetes insipidus, hypothyroidism, hyperparathyroidism, acne, and weight gain. Lithium also has potential interactions with multiple commonly prescribed medications, including antihypertensives and diuretics, as well as over-the-counter pain relievers such as ibuprofen and naproxen.
Second-generation antipsychotics (SGAs) have mood stabilizing, antidepressant, and anti-manic properties and are often useful in managing bipolar disorder. In our experience, for patients with bipolar disorder, SGAs are best used in combination with a mood stabilizer. Although virtually all SGAs have demonstrated effectiveness in the treatment of psychosis and some phases of bipolar disorder, the newer agents (aripiprazole, brexpiprazole, lurasidone, and cariprazine) are relatively free of metabolic adverse effects such as weight gain, abnormal cholesterol levels, increased prolactin levels, insulin resistance, and increased risk of diabetes.
Antidepressants may be effective in treating unipolar depression, but when treating bipolar depression, they should be used cautiously and only in combination with a mood stabilizer.
Continue to: As we observed...
As we observed in our previous article,1 thyroid laboratory monitoring and supplementation are critical components of managing mood disorders (Box 138-41).
Box 1
Conventional laboratory reference ranges often indicate that thyroid-stimulating hormone (TSH) levels as high as 4.0, 4.5, or 5.0 mU/L are normal. A recent meta- analysis determined that treatment of subclinical hypothyroidism (elevated TSH with normal free thyroxine) does not benefit patients’ quality of life.38 Patients with mood disorders, however, often fail to respond to mood stabilizers and other psychiatric medications unless their TSH is <3.0 or even <2.5 mU/L.39,40 We typically augment with liothyronine because, unlike levothyroxine, it works quickly, does not require deiodination to be activated, and, contrary to some reports, its elimination and biologic half-life are sufficient for single daily dosing.41
Moving towards better diagnoses
The emergence of a criteria-based psychiatric system in 1980 with the publication of DSM-III, and its subsequent revisions and updates, constituted a major advance in psychiatric diagnosis. As we learn more about the pathophysiology, genetics, and epigenetics of psychiatric symptoms and syndromes, future diagnostic systems will improve problems of validity that have yet to be resolved. While we believe that, for the most part, DSM-5 was an advance over the previous diagnostic iteration, we have 2 issues with DSM-5 in terms of the diagnosis of bipolar disorder (Box 210,12,13,18,19,42).
Box 2
Based on our clinical experience treating thousands of patients over 25 years, we have 2 issues with DSM-5 regarding bipolar disorder:
1. The DSM-5 criteria for hypomania fail to reflect the features of clinical presentations commonly seen in our practice. The majority of patients with authentic bipolar syndromes do not have hypomanias that last for at least 4 days or reach the level of severity required for a DSM-5 diagnosis of hypomania. This results in misdiagnosis of patients with bipolar depression as suffering from unipolar depression, which leads to inappropriate treatment with antidepressant monotherapy.
2. Bipolar disorder frequently makes its first appearance in childhood and adolescence,10,18,19 and increasing numbers of young patients have been receiving this diagnosis.42 In our opinion, this increase reflects clinicians’ improved diagnostic skills. Perhaps alarmed by the increase in young people receiving a diagnosis of bipolar disorder, the authors of DSM-5 created a new diagnosis for children: disruptive mood dysregulation disorder. This diagnostic addition is based on the finding that children with these mood symptoms may not subsequently exhibit classic DSM-5 manic or hypomanic episodes. But the lack of such episodes does not preclude a diagnosis of bipolar disorder, because many adults with unequivocal bipolar spectrum disorders have subthreshold hypomanias and thus fail to exhibit classic manic or hypomanic episodes.12,13
A rose by any other name would smell as sweet. Children who exhibit symptoms of disruptive mood dysregulation disorder— chronic irritability and protracted temper outbursts—usually suffer from depression and mood instability. In our opinion, it is irrational and confusing to clinicians to separate out with a new diagnosis an arbitrarily defined group of children who exhibit substantially the same symptoms as those who receive a diagnosis of bipolar disorder.
Patients with a chief complaint of depression are often given a diagnosis of “major depression, rule out bipolar disorder.” We believe that this formula should be turned on its head. In our opinion, based on our clinical experience, we think that most patients who present to a clinician’s office or psychiatric hospital with depression have bipolar depression, not unipolar depression. We hope that our experience and observations derived from treating thousands of patients over more than 25 years may be helpful to clinicians who sometimes struggle to bring relief to their patients with mood disorders.
CASE CONTINUED
Return to work
Mrs. W is now doing well. She is taking a lower dosage of duloxetine, 60 mg/d, in combination with the mood stabilizer lamotrigine, 200 mg/d. She returns to work full-time as a paralegal and no longer is experiencing depressive episodes.
Bottom Line
Patients with bipolar depression are often misdiagnosed with unipolar depression and treated inappropriately with antidepressant monotherapy, which often results in mood destabilization. Based on our clinical experience, a careful assessment of select criteria, including age of onset, rapidity of onset, comorbidities, diurnal mood variations, and more, can be useful for distinguishing between unipolar and bipolar depression.
Related Resources
- Nasrallah HA. Misdiagnosing bipolar depression as major depressive disorder. Current Psychiatry. 2013;12(10):20-21,A.
- Ghaemi SN. Bipolar spectrum: a review of the concept and a vision for the future. Psychiatry Investig. 2013;10(3):218-224.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Bupropion • Wellbutrin
Carbamazepine • Tegretol, Equetro
Cariprazine • Vraylar
Divalproex • Depakote
Duloxetine • Cymbalta
Esketamine • Spravato
Fluoxetine • Prozac
Lamotrigine • Lamictal
Levothyroxine • Synthroid, Levoxyl
Liothyronine • Cytomel
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Oxcarbazepine • Oxtellar XR, Trileptal
Paroxetine • Paxil
Sertraline • Zoloft
Mrs. W, age 36, who is married, has a history of military service, and is currently employed as a paralegal, is referred to our practice by her family physician. She complains of severe depression that impairs her ability to function at work. She had seen several other psychiatrists in both military and civilian settings, and had been treated with multiple antidepressants, including fluoxetine, sertraline, bupropion, and paroxetine.
At the time of her initial psychiatric evaluation, she is taking duloxetine, 90 mg/d, but still is experiencing depressive symptoms. She is tearful, sad, lacks energy, spends too much time in bed, and is experiencing thoughts of hopelessness, despair, and escape, verging on thoughts of suicide. As a result, she needs to scale back her work schedule to part-time. When asked about how long she had been suffering from depression, she responds “I’ve been depressed all my life.” She had been briefly hospitalized at age 16, when she made a suicide attempt by overdose. There had been no subsequent suicide attempts or psychiatric hospitalizations, although she acknowledges having intermittent suicidal thoughts.
Mrs. W’s clinical presentation is similar to that of many patients entering our practice—patients who have recurrent depression that began in early life and a history of failure to respond to multiple antidepressants. She and other patients with similar presentations are not suffering from treatment-resistant depression and in need of a trial of electroconvulsive therapy, transcranial magnetic stimulation, direct current stimulation, vagus nerve stimulation, or intranasal esketamine. She has bipolar disorder, and had been repeatedly misdiagnosed and treated inappropriately with antidepressant monotherapy.
In a previous article1 (“Controversies in bipolar disorder: Trust evidence or experience?,”
In this article, based on our more than 25 years of experience in diagnosing and treating psychiatric disorders in patients of all ages, we expand on those observations.
Misdiagnosis is common
Bipolar depression is frequently misdiagnosed as unipolar depression in outpatient2-8 and inpatient9 settings, and in children and adolescents.10 Mrs. W is typical of patients who have what we consider a bipolar spectrum disorder and receive an inaccurate diagnosis and treatment that is ineffective or may worsen the course of their illness.
Reliance on DSM-511 and its predecessor, DSM-IV, is a part of the problem of misdiagnosis because the diagnostic criteria for bipolar disorder fail to capture the clinical features of many patients with “softer” (less obvious manic and hypomanic) variants of the disorder.12,13 For example, DSM-5 criteria for a hypomanic episode (the mild high experienced by patients with a soft bipolar disorder) require that the episode lasts “at least 4 consecutive days” and is “present most of the day, nearly every day.” In our experience, the majority of hypomanic episodes are shorter—ranging from a half-day to 2 days, averaging perhaps 1.5 days.
Continue to: DSM-5 also requires...
DSM-5 also requires severity criteria for hypomania that patients with unequivocal hypomanic episodes often do not meet. For example, they may fail to experience flight of ideas or racing thoughts, or engage in activities such as “unrestrained buying sprees, sexual indiscretions, or foolish business investments.” These patients usually describe these mild highs as feeling normal and report a happier mood, more smiles and laughter, increased energy, less sleepiness, increased talkativeness, increased socialization, and improved motivation to complete tasks left undone and projects left unfinished because of the previous depressive episode. These softer (subthreshold) hypomanic episodes are authentic and, if clinicians do not identify them, may lead to misdiagnosis and inappropriate treatment.
Patients who present with depression often fail to report these brief, subthreshold hypomanic episodes or consider them to be irrelevant to their diagnosis and treatment.12,13 Probing questions can often elicit these unreported highs. For example, a patient with depression should be asked, “Have you had a single good day during the last month?” and “Where were you and what did you do during that day?” Eliciting a history of brief periods of improved mood is the key to differentiating between unipolar and bipolar depression. Screening instruments such as the Mood Disorders Questionnaire14 and the Bipolar Spectrum Diagnostic Scale15 may be helpful in distinguishing between unipolar and bipolar depression. However, we offer our thoughts on making that crucial distinction.
Distinguishing between these 2 types of depression
Although it may be difficult to distinguish between unipolar and bipolar depression, especially in the absence of a history of distinct manic or hypomanic episodes, we find the following criteria to be useful in making that determination.
Age of onset. Bipolar spectrum disorders typically begin earlier in life than unipolar depression.10,16-19 A typical presentation of bipolar disorder in children and adolescents is depression or agitated mixed states with features of both mania and depression, often accompanied by rapid mood cycling.20,21 Unipolar depression usually begins later in life, and patients do not have a history of significant depressive episodes or mood swings in childhood or adolescence. An important question to ask a patient with a chief complaint of depression is, “How old were you when you first experienced an episode of depression?”
Gender differences. Bipolar spectrum disorders with more subtle (softer) presentations, such as subthreshold highs, occur more often in women than men.22 However, overall rates of bipolar disorder may be slightly higher in men than in women.23 Unipolar melancholic depression occurs at approximately the same frequency in men and women.24
Continue to: Rapidity of onset
Rapidity of onset. Bipolar depressive episodes develop more rapidly than unipolar episodes. It is common for a patient with a bipolar spectrum disorder to transition from normal to very depressed virtually overnight, whereas in our clinical experience, unipolar episodes progress more slowly, often over several months.
Deliberate self-harm. Adolescents and young adults with a bipolar spectrum disorder frequently engage in self-injurious behavior, usually cutting with a knife, razor, or even sharp fingernails.25 Although these patients may also have thoughts of suicide and make suicide attempts, the individual usually perceives cutting as a means of gaining relief from tension and distress. These behaviors are often associated with a diagnosis of a personality disorder; in our opinion, however, they are hallmarks of a bipolar spectrum disorder.
ADHD. Bipolar disorder frequently co-occurs with attention-deficit/hyperactivity disorder (ADHD).26,27 Adults with bipolar disorder often have ADHD symptoms, which can complicate their treatment and cause functional impairment even after their mood disorder has been stabilized.28
Substance use disorders. Excessive use of alcohol and drugs is common among people with a wide range of psychiatric disorders, but patients with bipolar disorder have an unusually high rate of co-occurring substance use disorders—40% to 50%.29,30
Appetite and weight differences. Patients with unipolar depression usually experience loss of appetite and weight loss, whereas in our clinical experience, patients with bipolar depression often overeat, crave carbohydrates, and gain weight.
Continue to: Sleep problems
Sleep problems. Patients with bipolar depression have an increased need for sleep (the opposite of what they experience during highs), are sleepy during the day regardless of how many hours they sleep, and have difficulty getting up in the morning. Patients with unipolar depression also have a sleep disturbance: they may fall asleep easily, sleep for a few hours, and then awaken but are unable to fall back to sleep.31 Yet these patients usually do not complain of sleepiness during the day.
Diurnal variation of mood. Patients with unipolar depression often report that their depressive symptoms fluctuate in a circadian manner. For example, they may report that their depression is worse in the morning but improves toward evening.31 This regular alteration of circadian rhythm usually is not evident in patients with bipolar depression, whose mood may vary unpredictably or in response to stressors. Some patients with bipolar disorder, however, exhibit ultradian (ultra-rapid) mood cycling, which may be confused with the diurnal mood variation seen in patients with unipolar depression.
Tendency to recur. Although both unipolar and bipolar depressive episodes recur, a pattern of multiple recurring episodes beginning in early life is characteristic of bipolar spectrum disorders.
Behavioral history. Patients with bipolar depression are more likely than patients with unipolar depression to have a history of multiple marriages, multiple romantic relationships, episodes of promiscuity, legal problems, or financial extravagance.
Response to antidepressants. Patients with bipolar depression exhibit atypical responses to antidepressant monotherapy, such as worsening of depressive symptoms, initial improvement of mood with subsequent loss of effectiveness, premature response to an antidepressant (eg, improvement of mood within 1 to 2 days of beginning the antidepressant), fluctuation of depressive symptoms (mood cycling), or precipitation of a hypomanic or manic episode. We believe that a history of multiple failed antidepressant trials is compelling evidence of misdiagnosis of a bipolar spectrum disorder as unipolar depression.
Continue to: Genetics
Genetics. Bipolar disorder is one of the most heritable of illnesses.32 Family history is important, but affected relatives may have been misdiagnosed with unipolar depression or schizophrenia, or said to have experienced “nervous breakdowns.”
Consequences of misdiagnosis
Misdiagnosis of patients with bipolar disorder is not benign. We see patients who have suffered needlessly for years with severe depression and mood instability. After trying antidepressant after antidepressant without benefit, they begin to feel hopeless, believing they have tried everything and that nothing works for them. Often, these patients have dropped out of high school or college, or lost jobs, friends, and spouses due to their disabling but misdiagnosed psychiatric disorder. Patients with misdiagnosed bipolar disorder have an increased risk of suicide attempts and psychiatric hospitalization.5,8
Misdiagnosis of patients with bipolar disorder is not limited to nonpsychiatric physicians. The majority of patients with bipolar spectrum disorders are misdiagnosed by outpatient psychiatrists as having unipolar depression.2-7 At least 45% of patients hospitalized for depression have bipolar disorder—and most of these patients are treated inappropriately with antidepressants.9 The STAR*D study,33,34 a large randomized clinical trial of antidepressants, concluded that more than one-third of patients had not remitted from their depression after treatment with 3 different antidepressants. In our opinion, many of the nonresponding patients may have undiagnosed bipolar depression, which predictably leads to a failure to respond adequately to antidepressants. We believe that the customary inclusion and exclusion criteria used to select participants for these research studies miss subtle (subthreshold) hypomanic episodes that fall short of meeting DSM criteria for duration and severity. This phenomenon may account for the results of studies that conclude that antidepressants are, at best, minimally more effective than placebo.35
When a patient with a bipolar spectrum disorder is misdiagnosed and treated with an antidepressant, the usual result is mood destabilization. Reports of mood swings, increased crying, and suicidal thoughts and suicidal gestures in children, adolescents, and young adults treated with antidepressants led the FDA to issue a “black-box” warning.36 Because bipolar depression typically begins in youth,10,18,19 the behaviors cited in the warning may reflect misdiagnosis of bipolar depression as unipolar depression, and consequent mood destabilization as a result of treatment with an antidepressant in the absence of a mood stabilizer.
Depression and life stressors
Since many patients who are depressed present with a history of significant stressors, clinicians often face the problem of distinguishing between clinical depression and stress-induced depression. We believe that one typical symptom of depression—increased sensitivity to stressors—may help in making that distinction. A patient who is depressed will often attribute depression to stressors such as marital conflict, divorce, problems with a teenage child, work pressures, financial pressures, or the illness or death of a family member or pet. If clinical depression (unipolar or bipolar) is present, the symptoms are persistent, sometimes antedate the stressor by days or weeks, often outlast the stressor, increase in severity over time, and are disproportional to the stressor. Clinical depression can also cause the patient to become obsessed with traumatic events or losses that occurred many years earlier.
Continue to: Our approach to treatment
Our approach to treatment
Patients with mood disorders often benefit from a combination of pharmacologic management and psychotherapy. Psychotherapy is particularly important in addressing the functional impairment, diminished self-worth, and interpersonal conflicts that often accompany clinical depression. Several styles or systems of psychotherapy have been developed to benefit patients with mood disorders. Their effectiveness may depend on the patient’s ability to gain insight,37 but in our opinion, the most important attribute of helpful psychotherapy is the rapport established between the patient and the therapist, and the therapist’s ability to empathize with the patient and instill in the patient a sense of optimism and hope. We often recommend that patients attend meetings of the Depression and Bipolar Support Alliance (DBSA), a national support group with chapters throughout the country. Patients often find that attending these meetings is both educational and emotionally rewarding.
The foundational pharmacologic treatment for bipolar disorder is a mood stabilizer. The medications we consider to be effective mood stabilizers (some with an FDA indication for bipolar maintenance, some without) are lithium carbonate, divalproex sodium, carbamazepine, oxcarbazepine, and lamotrigine.
Each of these mood stabilizers has its advantages, disadvantages, risks, and adverse effects. For example, although divalproex is a reliable mood stabilizer, it has a significant risk of causing birth defects if taken during pregnancy and can cause increased appetite and weight gain. Carbamazepine has significant drug interactions and the potential to cause neurologic adverse effects, while oxcarbazepine, a derivative of carbamazepine, has fewer drug interactions but is more likely to cause hyponatremia. Lamotrigine must be titrated very slowly to reduce the risk of a potentially fatal skin rash (ie, Stevens-Johnson syndrome or toxic epidermal necrolysis). Lithium is effective but has a significant adverse-effect burden: impairment of renal function with long-term use, nephrogenic diabetes insipidus, hypothyroidism, hyperparathyroidism, acne, and weight gain. Lithium also has potential interactions with multiple commonly prescribed medications, including antihypertensives and diuretics, as well as over-the-counter pain relievers such as ibuprofen and naproxen.
Second-generation antipsychotics (SGAs) have mood stabilizing, antidepressant, and anti-manic properties and are often useful in managing bipolar disorder. In our experience, for patients with bipolar disorder, SGAs are best used in combination with a mood stabilizer. Although virtually all SGAs have demonstrated effectiveness in the treatment of psychosis and some phases of bipolar disorder, the newer agents (aripiprazole, brexpiprazole, lurasidone, and cariprazine) are relatively free of metabolic adverse effects such as weight gain, abnormal cholesterol levels, increased prolactin levels, insulin resistance, and increased risk of diabetes.
Antidepressants may be effective in treating unipolar depression, but when treating bipolar depression, they should be used cautiously and only in combination with a mood stabilizer.
Continue to: As we observed...
As we observed in our previous article,1 thyroid laboratory monitoring and supplementation are critical components of managing mood disorders (Box 138-41).
Box 1
Conventional laboratory reference ranges often indicate that thyroid-stimulating hormone (TSH) levels as high as 4.0, 4.5, or 5.0 mU/L are normal. A recent meta- analysis determined that treatment of subclinical hypothyroidism (elevated TSH with normal free thyroxine) does not benefit patients’ quality of life.38 Patients with mood disorders, however, often fail to respond to mood stabilizers and other psychiatric medications unless their TSH is <3.0 or even <2.5 mU/L.39,40 We typically augment with liothyronine because, unlike levothyroxine, it works quickly, does not require deiodination to be activated, and, contrary to some reports, its elimination and biologic half-life are sufficient for single daily dosing.41
Moving towards better diagnoses
The emergence of a criteria-based psychiatric system in 1980 with the publication of DSM-III, and its subsequent revisions and updates, constituted a major advance in psychiatric diagnosis. As we learn more about the pathophysiology, genetics, and epigenetics of psychiatric symptoms and syndromes, future diagnostic systems will improve problems of validity that have yet to be resolved. While we believe that, for the most part, DSM-5 was an advance over the previous diagnostic iteration, we have 2 issues with DSM-5 in terms of the diagnosis of bipolar disorder (Box 210,12,13,18,19,42).
Box 2
Based on our clinical experience treating thousands of patients over 25 years, we have 2 issues with DSM-5 regarding bipolar disorder:
1. The DSM-5 criteria for hypomania fail to reflect the features of clinical presentations commonly seen in our practice. The majority of patients with authentic bipolar syndromes do not have hypomanias that last for at least 4 days or reach the level of severity required for a DSM-5 diagnosis of hypomania. This results in misdiagnosis of patients with bipolar depression as suffering from unipolar depression, which leads to inappropriate treatment with antidepressant monotherapy.
2. Bipolar disorder frequently makes its first appearance in childhood and adolescence,10,18,19 and increasing numbers of young patients have been receiving this diagnosis.42 In our opinion, this increase reflects clinicians’ improved diagnostic skills. Perhaps alarmed by the increase in young people receiving a diagnosis of bipolar disorder, the authors of DSM-5 created a new diagnosis for children: disruptive mood dysregulation disorder. This diagnostic addition is based on the finding that children with these mood symptoms may not subsequently exhibit classic DSM-5 manic or hypomanic episodes. But the lack of such episodes does not preclude a diagnosis of bipolar disorder, because many adults with unequivocal bipolar spectrum disorders have subthreshold hypomanias and thus fail to exhibit classic manic or hypomanic episodes.12,13
A rose by any other name would smell as sweet. Children who exhibit symptoms of disruptive mood dysregulation disorder— chronic irritability and protracted temper outbursts—usually suffer from depression and mood instability. In our opinion, it is irrational and confusing to clinicians to separate out with a new diagnosis an arbitrarily defined group of children who exhibit substantially the same symptoms as those who receive a diagnosis of bipolar disorder.
Patients with a chief complaint of depression are often given a diagnosis of “major depression, rule out bipolar disorder.” We believe that this formula should be turned on its head. In our opinion, based on our clinical experience, we think that most patients who present to a clinician’s office or psychiatric hospital with depression have bipolar depression, not unipolar depression. We hope that our experience and observations derived from treating thousands of patients over more than 25 years may be helpful to clinicians who sometimes struggle to bring relief to their patients with mood disorders.
CASE CONTINUED
Return to work
Mrs. W is now doing well. She is taking a lower dosage of duloxetine, 60 mg/d, in combination with the mood stabilizer lamotrigine, 200 mg/d. She returns to work full-time as a paralegal and no longer is experiencing depressive episodes.
Bottom Line
Patients with bipolar depression are often misdiagnosed with unipolar depression and treated inappropriately with antidepressant monotherapy, which often results in mood destabilization. Based on our clinical experience, a careful assessment of select criteria, including age of onset, rapidity of onset, comorbidities, diurnal mood variations, and more, can be useful for distinguishing between unipolar and bipolar depression.
Related Resources
- Nasrallah HA. Misdiagnosing bipolar depression as major depressive disorder. Current Psychiatry. 2013;12(10):20-21,A.
- Ghaemi SN. Bipolar spectrum: a review of the concept and a vision for the future. Psychiatry Investig. 2013;10(3):218-224.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Bupropion • Wellbutrin
Carbamazepine • Tegretol, Equetro
Cariprazine • Vraylar
Divalproex • Depakote
Duloxetine • Cymbalta
Esketamine • Spravato
Fluoxetine • Prozac
Lamotrigine • Lamictal
Levothyroxine • Synthroid, Levoxyl
Liothyronine • Cytomel
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Oxcarbazepine • Oxtellar XR, Trileptal
Paroxetine • Paxil
Sertraline • Zoloft
1. Miller GE, Noel RL. Controversies in bipolar disorder: trust evidence or experience? Current Psychiatry. 2009;8(2):27-28,31-33,39.
2. Glick ID. Undiagnosed bipolar disorder: new syndromes and new treatments. Prim Care Companion J Clin Psychiatry. 2004;6(1):27-33.
3. Ghaemi SN, Sachs GS, Chiou AM, et al. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord. 1999;52(1-3):135-144.
4. Blanco C, Laje G, Olfson M, et al. Trends in the treatment of bipolar disorder by outpatient psychiatrists. Am J Psychiatry. 2002;159(6):1005-1010.
5. Shi L, Thiebaud P, McCombs JS. The impact of unrecognized bipolar disorders for patients treated with antidepressants in the fee-for-services California Medicaid (Medi-Cal) program. J Affect Disord. 2004;82(3):373-383.
6. Sidor MM, MacQueen GM. Antidepressants for the acute treatment of bipolar depression: a systematic review and meta-analysis. J Clin Psychiatry. 2011;72(2):156-167.
7. Hughes T, Cardno A, West R, et al. Unrecognized bipolar disorder among UK primary care patients prescribed antidepressants: an observational study. Br J Gen Pract. 2016;66(643):e71-e77.
8. Keck PE Jr, Kessler RC, Ross R. Clinical and economic effects of unrecognized or inadequately treated bipolar disorder. J Psychiatric Pract. 2008;14(Suppl 2):31-38.
9. Goldberg JF, Harrow M, Whiteside JF. Risk for bipolar illness in inpatients initially hospitalized for unipolar depression. Am J Psychiatry. 2001:158(8):1265-1270.
10. Chilakamarri JK, Filkowski MM, Ghaemi SN. Misdiagnosis of bipolar disorder in children and adolescents: a comparison with ADHD and major depressive disorder. Ann Clin Psychiatry. 2011;23(1):25-29.
11. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
12. Bowden CL. A different depression: clinical distinctions between bipolar and unipolar depression. J Affect Disord. 2005;84(2-3):117-125.
13. Baldassano C. Distinctions between bipolar I and bipolar II depression. Current Psychiatry. 2017;16(8):S7-S16.
14. Hirschfeld MA, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: The Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
15. Ghaemi SN, Miller CJ, Berv DA, et al. Sensitivity and specificity a new bipolar spectrum diagnostic scale. J Affect Disorder. 2005;84(2-3):273-277
16. Suppes T, Leverich G, Keck P, et al. The Stanley Foundation Continuing Bipolar Treatment Outcome Network. II. Demographics and illness characteristics of the first 261 patients. J Affect Disorder. 2001;67(1-3):45-49.
17. Perlis RH, Miyahara S, Marangell LB. Long-term implications of early onset in bipolar disorder: data from the first 1000 participants in the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP–BD). Biol Psychiatry. 2004;55(9):875-881.
18. Baldessarini RJ, Bolzani L, Kruz N, et al. Onset age of bipolar disorders at six international sites. J Affect Disord. 2010;121(1-2):143-146.
19. Post RM, Altshuler LL, Kupka R, et al. More childhood onset bipolar disorder in the United States than Canada or Europe: implications for treatment and prevention. Neurosci Biobehav Rev. 2017;74(Pt A):204-213.
20. Geller B, Luby J. Child and adolescent bipolar disorder: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1168-1176.
21. Findling RL, Gracious BL, McNamara NK, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord. 2001;3(4):202-210.
22. Arnold LM. Gender differences in bipolar disorder. Psychiatr Clin North Am. 2003;26(3):595-620.
23. Deflorio A, Jones I. Is sex important? Gender differences in bipolar disorder. Int Rev Psychiatry. 2010;22(5):437-452.
24. Bogren M, Brådvik L, Holmstrand C, et al. Gender differences in subtypes of depression by first incidence and age of onset: a follow-up of the Lunby population. Eur Arch Psychiatry Clin Neurosci. 2018;268(2):179-189.
25. Singhal A, Ross J, Seminog O, et al. Risks of self-harm and suicide in people with specific psychiatric and physical disorders: comparisons between disorders using English national record linkage. J R Soc Med. 2014;107(5):194-204.
26. Joshi G, Wilens T. Comorbidity in pediatric bipolar disorder. Child Adolesc Psychiatr Clin N Amer. 2009;18(2):291-319.
27. Youngtrom EA, Arnold LE, Frazier TW. Bipolar and ADHD comorbidity: both artifact and outgrowth of shared mechanisms. Clin Psychol (New York). 2010;17(4):350-359.
28. McIntyre RS, Kennedy SH, Soczynska JK, et al. Attention-deficit/hyperactivity disorder in adults with bipolar disorder or major depressive disorder: results from the International Mood Disorders Collaborative Project. Prime Care Companion J Clin Psychiatry. 2010;12(3). doi:10.4088/PCC.09m00861gry.
29. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiological Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
30. Hunt GE, Malhi GS, Cleary M, et al. Prevalence of comorbid bipolar and substance use disorders in clinical settings, 1990-2015: systematic review and meta-analysis. J Affect Disord. 2016;206:331-349.
31. Agargun MY, Besiroglu L, Cilli AS, et al. Nightmares, suicide attempts, and melancholic features in patients with unipolar major depression. J Affect Disord. 2007;98(3):267-270.
32. Birmaher B, Axelson D, Monk K, et al. Lifetime psychiatric disorders in school-aged offspring of parents with bipolar disorder: the Pittsburgh Bipolar Offspring Study. Arch Gen Psychiatry. 2009;66(3):287-296.
33. Rush AJ, Trivedi MH, Wisniewski SR, et al. Buproprion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231-1242.
34. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
35. Kirsch I. Antidepressants and the placebo effect. Z Psychol. 2014;222(3):128-134.
36. U.S. Food and Drug Administration. Suicidality in children and adolescents being treated with antidepressant medications. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161679.htm. Published February 5, 2018. Accessed May 10, 2019.
37. Jennissen S, Huber J, Ehrenthal JC, et al. Association between insight and outcome of psychotherapy; systematic review and meta-analysis. Am J Psychiatry. 2018;175(10):961-969.
38. Feller M, Snel M, Moutzouri E, et al. Association of thyroid hormone therapy with quality of life and thyroid-related symptoms in patients with subclinical hypothyroidism. JAMA. 2018;320(13):1349-1359.
39. Cole DP, Thase ME, Mallinger AG, et al. Slower treatment response in bipolar depression predicted by lower pretreatment thyroid function. Am J Psychiatry. 2002;159(1):116-121.
40. Parmentier T, Sienaert P. The use of triiodothyronine (T3) in the treatment of bipolar depression: a review of the literature. J Affect Disord. 2018;229:410-414.
41. Koda-Kimbe MA, Alldredge BK. Koda-Kimble and Young’s applied therapeutics: the clinical use of drugs (10th ed). Baltimore, MD: Walters Klower Health/Lippincott Williams & Wilkins; 2012.
42. Moreno C, Laje G, Blanco C, et al. National trends in the outpatient diagnosis and treatment of bipolar disorder in youth. Arch Gen Psychiatry. 2007;64(9):1032-1039.
1. Miller GE, Noel RL. Controversies in bipolar disorder: trust evidence or experience? Current Psychiatry. 2009;8(2):27-28,31-33,39.
2. Glick ID. Undiagnosed bipolar disorder: new syndromes and new treatments. Prim Care Companion J Clin Psychiatry. 2004;6(1):27-33.
3. Ghaemi SN, Sachs GS, Chiou AM, et al. Is bipolar disorder still underdiagnosed? Are antidepressants overutilized? J Affect Disord. 1999;52(1-3):135-144.
4. Blanco C, Laje G, Olfson M, et al. Trends in the treatment of bipolar disorder by outpatient psychiatrists. Am J Psychiatry. 2002;159(6):1005-1010.
5. Shi L, Thiebaud P, McCombs JS. The impact of unrecognized bipolar disorders for patients treated with antidepressants in the fee-for-services California Medicaid (Medi-Cal) program. J Affect Disord. 2004;82(3):373-383.
6. Sidor MM, MacQueen GM. Antidepressants for the acute treatment of bipolar depression: a systematic review and meta-analysis. J Clin Psychiatry. 2011;72(2):156-167.
7. Hughes T, Cardno A, West R, et al. Unrecognized bipolar disorder among UK primary care patients prescribed antidepressants: an observational study. Br J Gen Pract. 2016;66(643):e71-e77.
8. Keck PE Jr, Kessler RC, Ross R. Clinical and economic effects of unrecognized or inadequately treated bipolar disorder. J Psychiatric Pract. 2008;14(Suppl 2):31-38.
9. Goldberg JF, Harrow M, Whiteside JF. Risk for bipolar illness in inpatients initially hospitalized for unipolar depression. Am J Psychiatry. 2001:158(8):1265-1270.
10. Chilakamarri JK, Filkowski MM, Ghaemi SN. Misdiagnosis of bipolar disorder in children and adolescents: a comparison with ADHD and major depressive disorder. Ann Clin Psychiatry. 2011;23(1):25-29.
11. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
12. Bowden CL. A different depression: clinical distinctions between bipolar and unipolar depression. J Affect Disord. 2005;84(2-3):117-125.
13. Baldassano C. Distinctions between bipolar I and bipolar II depression. Current Psychiatry. 2017;16(8):S7-S16.
14. Hirschfeld MA, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: The Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
15. Ghaemi SN, Miller CJ, Berv DA, et al. Sensitivity and specificity a new bipolar spectrum diagnostic scale. J Affect Disorder. 2005;84(2-3):273-277
16. Suppes T, Leverich G, Keck P, et al. The Stanley Foundation Continuing Bipolar Treatment Outcome Network. II. Demographics and illness characteristics of the first 261 patients. J Affect Disorder. 2001;67(1-3):45-49.
17. Perlis RH, Miyahara S, Marangell LB. Long-term implications of early onset in bipolar disorder: data from the first 1000 participants in the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP–BD). Biol Psychiatry. 2004;55(9):875-881.
18. Baldessarini RJ, Bolzani L, Kruz N, et al. Onset age of bipolar disorders at six international sites. J Affect Disord. 2010;121(1-2):143-146.
19. Post RM, Altshuler LL, Kupka R, et al. More childhood onset bipolar disorder in the United States than Canada or Europe: implications for treatment and prevention. Neurosci Biobehav Rev. 2017;74(Pt A):204-213.
20. Geller B, Luby J. Child and adolescent bipolar disorder: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1168-1176.
21. Findling RL, Gracious BL, McNamara NK, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord. 2001;3(4):202-210.
22. Arnold LM. Gender differences in bipolar disorder. Psychiatr Clin North Am. 2003;26(3):595-620.
23. Deflorio A, Jones I. Is sex important? Gender differences in bipolar disorder. Int Rev Psychiatry. 2010;22(5):437-452.
24. Bogren M, Brådvik L, Holmstrand C, et al. Gender differences in subtypes of depression by first incidence and age of onset: a follow-up of the Lunby population. Eur Arch Psychiatry Clin Neurosci. 2018;268(2):179-189.
25. Singhal A, Ross J, Seminog O, et al. Risks of self-harm and suicide in people with specific psychiatric and physical disorders: comparisons between disorders using English national record linkage. J R Soc Med. 2014;107(5):194-204.
26. Joshi G, Wilens T. Comorbidity in pediatric bipolar disorder. Child Adolesc Psychiatr Clin N Amer. 2009;18(2):291-319.
27. Youngtrom EA, Arnold LE, Frazier TW. Bipolar and ADHD comorbidity: both artifact and outgrowth of shared mechanisms. Clin Psychol (New York). 2010;17(4):350-359.
28. McIntyre RS, Kennedy SH, Soczynska JK, et al. Attention-deficit/hyperactivity disorder in adults with bipolar disorder or major depressive disorder: results from the International Mood Disorders Collaborative Project. Prime Care Companion J Clin Psychiatry. 2010;12(3). doi:10.4088/PCC.09m00861gry.
29. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiological Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
30. Hunt GE, Malhi GS, Cleary M, et al. Prevalence of comorbid bipolar and substance use disorders in clinical settings, 1990-2015: systematic review and meta-analysis. J Affect Disord. 2016;206:331-349.
31. Agargun MY, Besiroglu L, Cilli AS, et al. Nightmares, suicide attempts, and melancholic features in patients with unipolar major depression. J Affect Disord. 2007;98(3):267-270.
32. Birmaher B, Axelson D, Monk K, et al. Lifetime psychiatric disorders in school-aged offspring of parents with bipolar disorder: the Pittsburgh Bipolar Offspring Study. Arch Gen Psychiatry. 2009;66(3):287-296.
33. Rush AJ, Trivedi MH, Wisniewski SR, et al. Buproprion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354(12):1231-1242.
34. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am J Psychiatry. 2006;163(11):1905-1917.
35. Kirsch I. Antidepressants and the placebo effect. Z Psychol. 2014;222(3):128-134.
36. U.S. Food and Drug Administration. Suicidality in children and adolescents being treated with antidepressant medications. https://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm161679.htm. Published February 5, 2018. Accessed May 10, 2019.
37. Jennissen S, Huber J, Ehrenthal JC, et al. Association between insight and outcome of psychotherapy; systematic review and meta-analysis. Am J Psychiatry. 2018;175(10):961-969.
38. Feller M, Snel M, Moutzouri E, et al. Association of thyroid hormone therapy with quality of life and thyroid-related symptoms in patients with subclinical hypothyroidism. JAMA. 2018;320(13):1349-1359.
39. Cole DP, Thase ME, Mallinger AG, et al. Slower treatment response in bipolar depression predicted by lower pretreatment thyroid function. Am J Psychiatry. 2002;159(1):116-121.
40. Parmentier T, Sienaert P. The use of triiodothyronine (T3) in the treatment of bipolar depression: a review of the literature. J Affect Disord. 2018;229:410-414.
41. Koda-Kimbe MA, Alldredge BK. Koda-Kimble and Young’s applied therapeutics: the clinical use of drugs (10th ed). Baltimore, MD: Walters Klower Health/Lippincott Williams & Wilkins; 2012.
42. Moreno C, Laje G, Blanco C, et al. National trends in the outpatient diagnosis and treatment of bipolar disorder in youth. Arch Gen Psychiatry. 2007;64(9):1032-1039.

Advanced Melanoma: Treatment After Progression on First-line Therapy
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).

For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28

Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).
For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28
Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved objective response rates (ORRs), progression-free survival (PFS), and overall survival (OS) for patients with metastatic melanoma. This article reviews current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy. The selection of first-line therapy for metastatic melanoma is reviewed in a separate article.
Case Presentation
A 62-year-old man was diagnosed with stage IIA melanoma after undergoing wide local excision of a right scalp lesion (final staging was consistent with pT3aN0M0). After 3.5 years of follow-up, he developed symptoms of vertigo, diplopia, and recurrent falls prompting medical attention. Magnetic resonance imaging (MRI) brain revealed multiple supratentorial and infratentorial lesions concerning for intracranial metastases and computed tomography (CT) chest/abdomen/pelvis revealed a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He was treated with intravenous dexamethasone and further evaluation with an endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass revealed metastatic melanoma. The patient underwent whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy. Testing showed the melanoma was positive for a BRAF V600K mutation. He was started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially did well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass.
After 3 months of therapy, surveillance PET-CT notes increasing size and FDG avidity of the right lower lobe mass. MRI brain reveals resolution of several previously noted metastases, but with interval development of a new left frontal lobe mass concerning for progressive disease.
What is the general approach to treatment of metastatic melanoma after progression on first-line therapy?
Based on the current evidence, there is no definitive algorithm for the treatment of metastatic melanoma after progression on first-line therapy. Enrollment in clinical trials is encouraged to further elucidate the best sequencing of treatment. The current practice is to typically switch class of agents after progression on front-line therapy to either immunotherapy that has not yet been tried or to molecularly targeted therapy in patients harboring a BRAF V600 mutation. After further progression of disease, retreatment with a previously received agent is possible, and this may be combined with investigational therapies.
Immune Checkpoint Inhibitors in Progressive Disease
The 2 major populations of patients to consider are those with BRAF wild-type melanomas who progress on first-line immunotherapy and those with BRAF V600 mutation–positive melanoma who progress on molecularly targeted therapy with BRAF and MEK inhibitors. There is relatively limited data on the efficacy of immune checkpoint inhibition after progression on anti-programmed cell death 1 (PD-1) monotherapy. A small retrospective study of patients who progressed on anti-PD-1 monotherapy were treated with ipilimumab, with a 10% ORR and another 8% having stable disease for more than 6 months; however, 35% of patients experienced grade 3 to 5 immune-related adverse events.1 The only prospective data supports the efficacy of anti-PD-1 therapy after progression on ipilimumab, as supported by the CheckMate 037 trial (nivolumab versus chemotherapy)2 and KEYNOTE-002 trial (pembrolizumab versus chemotherapy)3,4; however, this is no longer applicable as ipilimumab is no longer given in the first-line setting and has been replaced by anti-PD-1 monotherapy or combination immunotherapy.
Another interesting facet of PD-1 monotherapy is the idea of treatment beyond progression. The concept of pseudoprogression—whereby patients receiving PD-1 inhibitors initially meet Response Evaluation Criteria in Solid Tumors (RECIST) criteria for progression, but then later go on to demonstrate significant decreases in tumor burden on subsequent imaging studies—has been described in melanoma patients receiving such immunotherapies. It is thought that pseudoprogression occurs due to either an initial delay in anti-tumor response to the immunotherapy or from the measured target lesion appearing larger due to surrounding immune/inflammatory infiltrate. In an analysis of individual patient data pooled from 8 multicenter clinical trials, 19% of patients were treated beyond initially documented RECIST progression and had subsequent imaging to evaluate the tumor burden; in these patients, the same target lesion later met RECIST criteria for response, with a greater than 30% reduction in tumor size. Furthermore, of the evaluable cohort, the median OS in patients who did receive treatment beyond progression was 24.4 months compared to 11.2 months in those who did not receive treatment beyond progression.5 While further randomized studies are warranted to characterize the potential benefit, the existing data suggests that selected patients who are doing well clinically despite evidence of radiographic progressive disease may benefit from continued treatment with PD-1 inhibitors.
Combination immunotherapy with both PD-1 and CTLA-4 blockade has been studied retrospectively in the second-line setting. A retrospective analysis of patients who had progressive disease on PD-1 inhibitor monotherapy compared the outcomes of patients who received just ipilimumab to those of patients who received both ipilimumab and nivolumab. The ORR (16% ipilimumab vs 21% combination group) and 1-year OS (54% vs 55%) were similar in both groups,6 and this demonstrated significantly less efficacy with combination therapy when compared to use in the first-line setting, albeit in a separate prospective trial.7 A multicenter, retrospective study by Tétu and colleagues compared outcomes with ipilimumab plus nivolumab across 3 groups that included previously untreated patients, patients who had progressed on single-agent immunotherapy, and patients who had progressed on prior molecularly targeted therapy.8 Despite clearly inferior efficacy in previously treated patients, the results support combination immunotherapy as a viable treatment option in the second-line setting. Outcomes are reported in Table 1 below. Of note, there is an ongoing phase 2 trial to assess the use of combined PD-1 and CTLA-4 inhibitors versus CTLA-4 inhibition alone after progression on first-line PD-1 inhibitor monotherapy (NCT03033576).
For patients with BRAF V600–mutation positive melanoma who progress on front-line molecularly targeted therapy, immune checkpoint inhibitor therapy with either anti-PD-1 monotherapy or combination anti-PD-1 and ipilimumab should be considered. The KEYNOTE-006 trial that demonstrated superiority of pembrolizumab compared to ipilimumab included patients who had received up to 1 prior systemic therapy that was not a PD-1 or CTLA-4 inhibitor, and subgroup analysis demonstrated efficacy with pembrolizumab in patients who had received prior treatment with a BRAF inhibitor.9 The retrospective analysis by Tétu et al (Table 1) noted efficacy of combination nivolumab and ipilimumab in patients treated with prior molecularly targeted therapy, as evidenced by an ORR of 35% and median OS of 16.5 months.8
A retrospective trial by Ackerman et al analyzed ORR, median PFS, and median OS from the time of commencement of BRAF inhibitor therapy (with or without a MEK inhibitor), and the comparison was made between those who received ipilimumab before or after molecularly targeted therapy. While ipilimumab is no longer the first-line immunotherapy agent used in advanced melanoma, the study did highlight some important concepts. First, ORRs to BRAF inhibitors were similar between the 2 treatment groups. The conclusions of the analysis were that there was no significant difference in median PFS or OS in regard to which therapy was given first, but median OS after BRAF inhibitors were discontinued was very short and patients had poor responses to ipilimumab after stopping a BRAF inhibitor. This highlights the concern that patients who have progressive disease on molecularly targeted therapy often have a poor performance status and undergo too rapid of a clinical decline to derive benefit from immunotherapy, which can often take weeks to months to take effect.10
A more recent retrospective study by Johnson et al compared efficacy outcomes in patients who received single-agent anti-PD-1 therapy prior to molecularly targeted therapy (BRAF inhibitor with or without MEK inhibitor) to those who received molecularly targeted therapy prior to anti-PD-1 therapy. The difference in median OS was not statistically significant (27.5 months with PD-1 inhibitor first vs 40.3 months with molecularly targeted therapy first). Both treatments demonstrated second-line efficacy, but outcomes were inferior to those reported when either type of therapy was used in the first-line setting. Interestingly, patients who were maintained on molecularly targeted therapy for more than 6 months prior to progression demonstrated an improved ORR to subsequent anti-PD-1 therapy (34% vs 15%).11
Molecularly Targeted Therapy in Progressive Disease
When melanoma patients with a BRAF V600 mutation are treated initially with immunotherapy and demonstrate progressive disease, molecularly targeted therapy with combined BRAF and MEK inhibition should be considered for second-line therapy. While there are no dedicated prospective trial results with BRAF/MEK inhibitors after progression on immune checkpoint inhibitors, for practical purposes, it may be reasonable to extrapolate outcomes from the currently available first-line studies.12-16 An ongoing study (NCT02224781) in which patients are randomized to receive ipilimumab/nivolumab followed by dabrafenib/trametinib at progression versus the reverse order is designed to help answer the question of optimal sequencing and timing of therapy. Johnson et al’s retrospective analysis of patients receiving single-agent anti-PD-1 therapy prior to molecularly targeted therapy compared to the reverse order concluded that there was no statistically significant difference in median OS.11 Ackerman et al’s retrospective study of patients who had received ipilimumab before or after molecularly targeted therapy noted similar response rates to molecularly targeted therapy in each treatment group.10
The issue of re-treatment with a BRAF/MEK inhibitor in a patient already progressing on targeted therapy is a more challenging situation, and currently available data suggests there is limited benefit. However, select patients may be considered for this approach. The combination of dabrafenib/trametinib demonstrated an ORR of approximately 15% in a cohort of patients who progressed on single-agent BRAF inhibitor therapy, with a suggestion that those patients who had previously derived benefit for more than 6 months may have a more favorable outcome.17
Based on the hypothesis that acquired resistance to BRAF/MEK inhibition may be reversible if the selective pressure of the medication is held for a period of time, a phase 2 trial analyzed outcomes with retreatment. The study included patients with BRAF V600–mutant melanoma who had progressed on prior BRAF inhibition (with or without MEK inhibitor) and required that they had been off of therapy for at least 12 weeks. Of the 25 patients who received dabrafenib plus trametinib as retreatment, 32% demonstrated a partial response and 40% had stable disease.18 While further studies are warranted, retreatment with molecularly targeted therapy may be a viable option, especially in light of the multiple approved BRAF and MEK inhibitor combinations.
Another concept that has been studied is treatment beyond disease progression with molecularly targeted therapy. In a retrospective analysis of patients who had progressed on a single-agent BRAF inhibitor, 39% of those patients were continued on the same BRAF inhibitor and compared to patients who received no subsequent therapy or changed to an alternative systemic therapy. In the multivariable analysis adjusting for other prognostic factors, continued treatment with the BRAF inhibitor was associated with prolonged OS.19
Case Conclusion
The patient is started on second-line therapy with nivolumab and ipilimumab and demonstrates a partial response. One year later he continues to feel well with decreased size of the intracranial and right lower lobe lesions, and without any interval development of new areas of metastatic disease.
Special Considerations
Intralesional Therapies
Talimogene laherparepvec (T-VEC) is a genetically modified herpesvirus-1 oncolytic virus that is injected into melanoma skin lesions and leads to the expression of granulocyte-macrophage colony-stimulating factor. While T-VEC is currently approved for local treatment of unresectable cutaneous, subcutaneous, or nodal recurrences,20 it has also been investigated in combination with other therapies for patients with advanced disease. In patients with previously treated melanoma, T-VEC plus ipilimumab demonstrated superior ORR to ipilimumab alone (39% vs 18%), and the tumor response was not limited to the injected lesions. The observation of systemic response suggests synergy between T-VEC and immune checkpoint blockade in enhancing the anti-tumor immune response.21 The phase 1b MASTERKEY-265 trial combining pembrolizumab and T-VEC led to an ORR of 62% and CR of 33%.22 A phase 3 trial comparing pembrolizumab plus T-VEC to pembrolizumab alone is ongoing (NCT02263508).
Melanoma Brain Metastases
The presence of brain metastases is a common event in patients with metastatic melanoma, and often confers a poor prognosis.23 The approach to the management of brain metastases should be multidisciplinary among medical oncology, neurosurgery, and radiation oncology providers, as treatment algorithms continue to rapidly evolve. Historically, there has been little prospective clinical trial data regarding optimal systemic therapy, and local therapies such as surgery or stereotactic radiation have long been the mainstay of therapy for intracranial disease.24 However, recent data with both immunotherapy and molecularly targeted therapy has demonstrated efficacy with intracranial metastases.
A recent trial of combined nivolumab and ipilimumab as frontline therapy in patients with asymptomatic melanoma brain metastases demonstrated a complete response rate of 26% and partial response rate of 30% in patients with a median follow-up of 14 months.25 In a separate study, ipilimumab plus nivolumab demonstrated better intracranial ORR when compared to nivolumab alone in asymptomatic, previously untreated patients. Outcomes were better in patients presenting with asymptomatic versus symptomatic brain metastases.26 Collectively, these results suggest that systemic immunotherapy alone may be adequate for patients with asymptomatic, previously untreated brain metastases.
For molecularly targeted therapy in patients with BRAF mutations and brain metastases, the BREAK-MB trial demonstrated that an intracranial response was attainable with dabrafenib regardless of whether the patient had previously received local therapy in the form of surgery or radiation.27 The COMBI-MB trial enhanced the preexisting data by testing the intracranial efficacy of dabrafenib plus trametinib in 4 different cohorts of patients, further supporting that systemic molecularly targeted therapy can provide significant intracranial activity in patients with both symptomatic and asymptomatic brain lesions and regardless of prior local therapy (Table 2).28
Conclusion
The treatment of advanced melanoma has been drastically improved over the past decade by the development and study of immune checkpoint inhibitors and molecularly targeted agents. There is still much to learn regarding the optimal combination and sequencing of therapies. Many of these trials are ongoing and will provide additional evidence to guide treatment decisions moving forward.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
1. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer. 2016;114:1084-1089.
2. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-384.
3. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-918.
4. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
5. Beaver JA, Hazarika M, Mulkey F, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2018;19:229-239.
6. Zimmer L, Apuri S, Eroglu Z, et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer. 2017;75:47-55.
7. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
8. Tétu P, Mangana J, Dummer R, et al. Benefit of the nivolumab and ipilimumab combination in pretreated advanced melanoma. Eur J Cancer. 2018;93:147-149.
9. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
10. Ackerman A, Klein O, McDermott D, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120:1695-1701.
11. Johnson DB, Pectasides E, Feld E, et al. Sequencing treatment in BRAFV600 mutant melanoma: anti-pd-1 before and after BRAF inhibition. J Immunother. 2017;40:31-35.
12. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
13. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
14. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-1260.
15. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
16. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
17. Johnson DB, Flaherty KT, Weber, JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697-3704.
18. Schreuer M, Jansen Y, Planken S, et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017;18:464-472.
19. Chan MM, Haydu LE, Azer MW, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120:3142-3153.
20. Imlygic (talimogene laherparepvec) suspension for intralesional injection [package insert]. Thousand Oaks, CA: BioVex; 2015.
21. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase ii study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol. 2018;36:1658-1667.
22. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2018;174:1031-1032.
23. Sampson JH, Carter Jr. JH, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88:11-20.
24. Yamamoto M, Serizawa T, Shuto T, et al. Stereotactic radiosurgery for patients with multiple brain metastases (JLGK0901): a multi-institutional prospective observational study. Lancet Oncol. 2014;15:387-395.
25. Tawbi HA, Forsyth PA, Hamid O, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379:722-730.
26. Long GV, Atkinson V, La S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicenter randomised phase 2 study. Lancet Oncol. 2018;19:672-681.
27. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicenter, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087-1095.
28. Davies MA, Saiag P, Robert C, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicenter, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18:863-873.
Advanced Melanoma: First-line Therapy
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.

Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.

Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.
Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.
Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
Malignant melanoma is the most serious form of primary skin cancer and one of the only malignancies in which the incidence rate has been rising. It is estimated that in 2018 there were 91,270 newly diagnosed cases and 9320 deaths from advanced melanoma in the United States. Melanoma is the fifth most common cancer type in males and the sixth most common in females. Despite rising incidence rates, improvement in the treatment of advanced melanoma has resulted in declining death rates over the past decade.1 Although most melanoma is diagnosed at an early stage and can be cured with surgical excision, the prognosis for metastatic melanoma had been historically poor prior to recent advancements in treatment. Conventional chemotherapy treatment with dacarbazine or temozolomide resulted in response rates ranging from 7.5% to 12.1%, but without much impact on median overall survival (OS), with reported OS ranging from 6.4 to 7.8 months. Combination approaches with interferon alfa-2B and low-dose interleukin-2 resulted in improved response rates compared with traditional chemotherapy, but again without survival benefit.2
Immunotherapy in the form of high-dose interleukin-2 emerged as the first therapy to alter the natural history of advanced melanoma, with both improved response rates (objective response rate [ORR], 16%) and median OS (2 months), with some patients achieving durable responses lasting more than 30 months. However, significant systemic toxicity limited its application to carefully selected patients.3 The past decade has brought rapid advancements in treatment with immune checkpoint inhibitors and molecularly targeted agents, which have significantly improved ORRs, progression-free survival (PFS), and OS for patients with metastatic melanoma.4-8
This review is the first of 2 articles focusing on the treatment and sequencing of therapies in advanced melanoma. Here, we review the selection of first-line therapy for metastatic melanoma. Current evidence for immune checkpoint blockade and molecularly targeted agents in the treatment of metastatic melanoma after progression on first-line therapy is discussed in a separate article.
Pathogenesis
The incidence of melanoma is strongly associated with ultraviolet light–mediated DNA damage related to sun exposure. Specifically, melanoma is associated to a greater degree with intense intermittent sun exposure and sunburn, but not associated with higher occupational exposure.9 Ultraviolet radiation can induce DNA damage by a number of mechanisms, and deficient DNA repair leads to somatic mutations that drive the progression from normal melanocyte to melanoma.10
The most commonly identified genetic mutations in cutaneous melanomas are alterations in the mitogen-activated protein kinase (MAPK) pathway. Typically, an extracellular growth factor causes dimerization of the growth factor receptor, which activates the intracellular RAS GTPase protein. Subsequently BRAF is phosphorylated within the kinase domain, which leads to downstream activation of the MEK and ERK kinases through phosphorylation. Activated ERK leads to phosphorylation of various cytoplasmic and nuclear targets, and the downstream effects of these changes promote cellular proliferation. While activation of this pathway usually requires phosphorylation of BRAF by RAS, mutations placing an acidic amino acid near the kinase domain mimics phosphorylation and leads to constitutive activation of the BRAF serine/threonine kinase in the absence of upstream signaling from extracellular growth factors mediated through RAS.11 One study of tumor samples of 71 patients with cutaneous melanoma detected NRAS mutations in 30% and BRAF mutations in 59% of all tumors tested. Of the BRAF mutation–positive tumors, 88% harbored the Val599Glu mutation, now commonly referred to as the BRAF V600E mutation. The same study demonstrated that the vast majority of BRAF mutations were seen in the primary tumor and were preserved when metastases were analyzed. Additionally, both NRAS and BRAF mutations were detected in the radial growth phase of the melanoma tumor. These findings indicate that alterations in the MAPK pathway occur early in the pathogenesis of advanced melanoma.11 Another group demonstrated that 66% of malignant melanoma tumor samples harbored BRAF mutations, of which 80% were specifically the V600E mutation. In vitro assays showed that the BRAF V600E–mutated kinase had greater than 10-fold kinase activity compared to wild-type BRAF, and that this kinase enhanced cellular proliferation even when upstream NRAS signaling was inhibited.12
The Cancer Genome Atlas Network performed a large analysis of tumor samples from 331 different melanoma patients and studied variations at the DNA, RNA, and protein levels. The study established a framework of 4 notable genomic subtypes, including mutant BRAF (52%), mutant RAS (28%), mutant NF1 (14%), and triple wild-type (6%). Additionally, mRNA transcriptomic analysis of overexpressed genes identified 3 different subclasses, which were labeled as “immune,” “keratin,” and “MITF-low.” The immune subclass was characterized by increased expression of proteins found in immune cells, immune signaling molecules, immune checkpoint proteins, cytokines, and chemokines, and correlated with increased lymphocyte invasion within the tumor. Interestingly, in the post-accession survival analysis, the “immune” transcriptomic subclass was statistically correlated with an improved prognosis.13 Having an understanding of the molecular pathogenesis of advanced melanoma helps to create a framework for understanding the mechanisms of current standard of care therapies for the disease.
Case Presentation
A 62-year-old Caucasian man with a history of well-controlled type 2 diabetes mellitus and hypertension is being followed by his dermatologist for surveillance of melanocytic nevi. On follow-up he is noted to have an asymmetrical melanocytic lesion over the right scalp with irregular borders and variegated color. He is asymptomatic and the remainder of physical examination is unremarkable, as he has no other concerning skin lesions and no cervical, axillary, or inguinal lymphadenopathy.
How is melanoma diagnosed?
Detailed discussion about diagnosis and staging will be deferred in this review of treatment of advanced melanoma. In brief, melanoma is best diagnosed by excisional biopsy and histopathology. Staging of melanoma is done according to the American Joint Committee on Cancer’s (AJCC) Cancer Staging Manual, 8th edition, using a TNM staging system that incorporates tumor thickness (Breslow depth); ulceration; number of involved regional lymph nodes; presence of in-transit, satellite, and/or microsatellite metastases; distant metastases; and serum lactate dehydrogenase level.14
Case Continued
The patient undergoes a wide excisional biopsy of the right scalp lesion, which is consistent with malignant melanoma. Pathology demonstrates a Breslow depth of 2.6 mm, 2 mitotic figures/mm2, and no evidence of ulceration. He subsequently undergoes wide local excision with 0/3 sentinel lymph nodes positive for malignancy. His final staging is consistent with pT3aN0M0, stage IIA melanoma.
He is seen in follow-up with medical oncology for the next 3.5 years without any evidence of disease recurrence. He then develops symptoms of vertigo, diplopia, and recurrent falls, prompting medical attention. Magnetic resonance imaging (MRI) brain reveals multiple supratentorial and infratentorial lesions concerning for intracranial metastases. Further imaging with computed tomography (CT) chest/abdomen/pelvis reveals a right lower lobe pulmonary mass with right hilar and subcarinal lymphadenopathy. He is admitted for treatment with intravenous dexamethasone and further evaluation with endobronchial ultrasound-guided fine-needle aspiration of the right lower lobe mass, which reveals metastatic melanoma. Given the extent of his intracranial metastases, he is treated with whole brain radiation therapy for symptomatic relief prior to initiating systemic therapy.
What is the general approach to first-line treatment for metastatic melanoma?
The past decade has brought an abundance of data supporting the use of immunotherapy with immune checkpoint inhibitors or molecularly targeted therapy with combined BRAF/MEK inhibitors in the first-line setting.4-8 After the diagnosis of metastatic melanoma has been made, molecular testing is recommended to determine the BRAF status of the tumor. Immunotherapy is the clear choice for first-line therapy in the absence of an activating BRAF V600 mutation. When a BRAF V600 mutation is present, current evidence supports the use of either immunotherapy or molecularly targeted therapy as first-line therapy.
To date, there have been no prospective clinical trials comparing the sequencing of immunotherapy and molecularly targeted therapy in the first-line setting. An ongoing clinical trial (NCT02224781) is comparing dabrafenib and trametinib followed by ipilimumab and nivolumab at time of progression to ipilimumab and nivolumab followed by dabrafenib and trametinib in patients with newly diagnosed stage III/IV BRAF V600 mutation–positive melanoma. The primary outcome measure is 2-year OS. Until completion of that trial, current practice regarding which type of therapy to use in the first-line setting is based on a number of factors including clinical characteristics and provider preferences.
Data suggest that immunotherapies can produce durable responses, especially after treatment completion or discontinuation, albeit at the expense of taking a longer time to achieve clinical benefit and the risk of potentially serious immune-related adverse effects. This idea of a durable, off-treatment response is highlighted by a study that followed 105 patients who had achieved a complete response (CR) and found that 24-month disease-free survival from the time of CR was 90.9% in all patients and 89.9% in the 67 patients who had discontinued pembrolizumab after attaining CR.15 BRAF/MEK inhibition has the potential for rapid clinical responses, though concerns exist about the development of resistance to therapy. The following sections explore the evidence supporting the use of these therapies.
Immunotherapy with Immune Checkpoint Inhibitors
Immunotherapy via immune checkpoint blockade has revolutionized the treatment of many solid tumors over the past decade. The promise of immunotherapy revolves around the potential for achieving a dynamic and durable systemic response against cancer by augmenting the antitumor effects of the immune system. T-cells are central to mounting a systemic antitumor response, and, in addition to antigen recognition, their function depends heavily on fine tuning between co-stimulatory and co-inhibitory signaling. The cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expressed on T-cells was the first discovered co-inhibitory receptor of T-cell activation.16 Later, it was discovered that the programmed cell death 1 receptor (PD-1), expressed on T-cells, and its ligands PD-L1 and PD-L2, expressed on antigen presenting cells, tumor cells, or other cells in the tumor microenvironment, also served as a potent negative regulator of T-cell function.17
Together, these 2 signaling pathways help to maintain peripheral immune tolerance, whereby autoreactive T-cells that have escaped from the thymus are silenced to prevent autoimmunity. However, these pathways can also be utilized by cancer cells to escape immune surveillance. Monoclonal antibodies that inhibit the aforementioned co-inhibitory signaling pathways, and thus augment the immune response, have proven to be an effective anticancer therapy capable of producing profound and durable responses in certain malignancies.16,17
Ipilimumab
Ipilimumab is a monoclonal antibody that inhibits the function of the CTLA-4 co-inhibitory immune checkpoint. In a phase 3 randomized controlled trial of 676 patients with previously treated metastatic melanoma, ipilimumab at a dose of 3 mg/kg every 3 weeks for 4 cycles, with or without a gp100 peptide vaccine, resulted in an improved median OS of 10.0 and 10.1 months, respectively, compared to 6.4 months in those receiving the peptide vaccine alone, meeting the primary endpoint.4 Subsequently, a phase 3 trial of 502 patients with untreated metastatic melanoma compared ipilimumab at a dose of 10 mg/kg every 3 weeks for 4 cycles plus dacarbazine to dacarbazine plus placebo and found a significant increase in median OS (11.2 months vs 9.1 months), with no additive benefit of chemotherapy. There was a higher reported rate of grade 3 or 4 adverse events in this trial with ipilimumab dosed at 10 mg/kg, which was felt to be dose-related.18 These trials were the first to show improved OS with any systemic therapy in metastatic melanoma and led to US Food and Drug Administration approval of ipilimumab for this indication in 2011.
PD-1 Inhibitor Monotherapy
The PD-1 inhibitors nivolumab and pembrolizumab were initially approved for metastatic melanoma after progression on ipilimumab. In the phase 1 trial of patients with previously treated metastatic melanoma, nivolumab therapy resulted in an ORR of 28%.19 The subsequent phase 2 trial conducted in pretreated patients, including patients who had progressed on ipilimumab, confirmed a similar ORR of 31%, as well as a median PFS of 3.7 months and a median OS of 16.8 months. The estimated response duration in patients who did achieve a response to therapy was 2 years.20 A phase 3 trial (CheckMate 037) comparing nivolumab (n = 120) to investigator’s choice chemotherapy (n = 47) in those with melanoma refractory to ipilimumab demonstrated that nivolumab was superior for the primary endpoint of ORR (31.7% vs 10.6%), had less toxicity (5% rate of grade 3 or 4 adverse events versus 9%), and increased median duration of response (32 months vs 13 months).21
The phase 1 trial (KEYNOTE-001) testing the efficacy of pembrolizumab demonstrated an ORR of 33% in the total population of patients treated and an ORR of 45% in those who were treatment-naive. Additionally, the median OS was 23 months for the total population and 31 months for treatment-naive patients, with only 14% of patients experiencing a grade 3 or 4 adverse event.22 The KEYNOTE-002 phase 2 trial compared 2 different pembrolizumab doses (2 mg/kg and 10 mg/kg every 3 weeks) to investigator’s choice chemotherapy (paclitaxel plus carboplatin, paclitaxel, carboplatin, dacarbazine, or oral temozolomide) in 540 patients with advanced melanoma with documented progression on ipilimumab with or without prior progression on molecularly targeted therapy if positive for a BRAF V600 mutation. The final analysis demonstrated significantly improved ORR with pembrolizumab (22% at 2 mg/kg vs 26% at 10 mg/kg vs 4% chemotherapy) and significantly improved 24-month PFS (16% vs 22% vs 0.6%, respectively). There was a nonstatistically significant improvement in median OS (13.4 months vs 14.7 months vs 10 months), although 55% of the patients initially assigned to the chemotherapy arm crossed over and received pembrolizumab after documentation of progressive disease.23,24
Because PD-1 inhibition improved efficacy with less toxicity than chemotherapy when studied in progressive disease, subsequent studies focused on PD-1 inhibition in the frontline setting. CheckMate 066 was a phase 3 trial comparing nivolumab to dacarbazine as first-line therapy for 418 patients with untreated metastatic melanoma who did not have a BRAF mutation. For the primary end point of 1-year OS, nivolumab was superior to dacarbazine (72.9% vs 42.1%; hazard ratio [HR], 0.42; P < 0.001). Treatment with nivolumab also resulted in superior ORR (40% vs 14%) and PFS (5.1 months vs 2.2 months). Additionally, nivolumab therapy had a lower rate of grade 3 or 4 toxicity compared to dacarbazine (11.7% vs 17.6%).25
The KEYNOTE-006 trial compared 2 separate dosing schedules of pembrolizumab (10 mg/kg every 2 weeks versus every 3 weeks) to ipilimumab (3 mg/kg every 3 weeks for 4 cycles) in a 1:1:1 ratio in 834 patients with metastatic melanoma who had received up to 1 prior systemic therapy, but no prior CTLA-4 or PD-1 inhibitors. The first published data reported statistically significant outcomes for the co-primary end points of 6-month PFS (47.3% for pembrolizumab every 2 weeks vs 46.4% for pembrolizumab every 3 weeks vs 26.5% for ipilimumab; HR, 0.58 for both pembrolizumab groups compared to ipilimumab; P < 0.001) and 12-month OS (74.1% vs 68.4% vs 58.2%) with pembrolizumab compared to ipilimumab. Compared to ipilimumab, pembrolizumab every 2 weeks had a hazard ratio of 0.63 (P = 0.0005) and pembrolizumab every 3 weeks had a hazard ratio of 0.69 (P = 0.0036). The pembrolizumab groups was also had lower rates of grade 3 to 5 toxicity (13.3% vs 10.1% vs 19.9%).5 Updated outcomes demonstrated improved ORR compared to the first analysis (37% vs 36% vs 13%), and improved OS (median OS, not reached for the pembrolizumab groups vs 16.0 months for the ipilimumab group; HR, 0.68, P = 0.0009 for pembrolizumab every 2 weeks versus HR 0.68, P = 0.0008 for pembrolizumab every 3 weeks).26 In addition, 24-month OS was 55% in both pembrolizumab groups compared to 43% in the ipilimumab group. Grade 3 or 4 toxicity occurred less frequently with pembrolizumab (17% vs 17% vs 20%).
Further analysis from the KEYNOTE-006 trial data demonstrated improved ORR, PFS, and OS with pembrolizumab compared to ipilimumab in tumors positive for PD-L1 expression. For PD-L1-negative tumors, response rate was higher, and PFS and OS rates were similar with pembrolizumab compared to ipilimumab. Given that pembrolizumab was associated with similar survival outcomes in PD-L1-negative tumors and with less toxicity than ipilimumab, the superiority of PD-L1 inhibitors over ipilimumab was further supported, regardless of tumor PD-L1 status.27
In sum, PD-1 inhibition should be considered the first-line immunotherapy in advanced melanoma, either alone or in combination with ipilimumab, as discussed in the following section. There is no longer a role for ipilimumab monotherapy in the first-line setting, based on evidence from direct comparison to single-agent PD-1 inhibition in clinical trials that demonstrated superior efficacy and less serious toxicity with PD-1 inhibitors.5,26 The finding that ORR and OS outcomes with single-agent PD-1 inhibitors are higher in treatment-naive patients compared to those receiving prior therapies also supports this approach.22
Combination CTLA-4 and PD-1 Therapy
Despite the potential for durable responses, the majority of patients fail to respond to single-agent PD-1 therapy. Given that preclinical data had suggested the potential for synergy between dual inhibition of CTLA-4 and PD-1, clinical trials were designed to test this approach. The first randomized phase 2 trial that established superior efficacy with combination therapy was the CheckMate 069 trial comparing nivolumab plus ipilimumab to ipilimumab monotherapy. Combination therapy resulted in increased ORR (59% vs 11%), median PFS (not reached vs 3.0 months), 2-year PFS (51.3% vs 12.0%), and 2-year OS (63.8% vs 53.6%).28 Similarly, a phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and 1-year OS of 89%.29
The landmark phase 3 CheckMate 067 trial analyzed efficacy outcomes for 3 different treatment regimens including nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy in previously untreated patients with unresectable stage III or IV melanoma. The trial was powered to compare survival outcomes for both the combination therapy arm against ipilimumab and the nivolumab monotherapy arm against ipilimumab, but not to compare combination therapy to nivolumab monotherapy. The initial analysis demonstrated a median PFS of 11.5 months with combination therapy versus 6.9 months with nivolumab and 2.9 months with ipilimumab, as well as an ORR of 58% versus 44% and 19%, respectively (Table 1).6 The updated 3-year survival outcomes from CheckMate 067 were notable for superior median OS with combination therapy (not reached in combination vs 37.6 months for nivolumab vs 19.9 months ipilimumab), improved 3-year OS (58% vs 52% vs 34%), and improved 3-year PFS (39% vs 32% vs 10%).7 In the reported 4-year survival outcomes, median OS was not reached in the combination therapy group, and was 36.9 months in the nivolumab monotherapy group and 19.9 months in the ipilimumab monotherapy group. Rates of grade 3 or 4 adverse events were significantly higher in the combination therapy group, at 59% compared to 22% with nivolumab monotherapy and 28% with ipilimumab alone.30 The 3- and 4-year OS outcomes (58% and 54%, respectively) with combination therapy were the highest seen in any phase 3 trial for treatment of advanced melanoma, supporting its use as the best approved first-line therapy in those who can tolerate the potential toxicity of combination therapy7,30 The conclusions from this landmark trial were that both combination therapy and nivolumab monotherapy resulted in statistically significant improvement in OS compared to ipilimumab.
Toxicity Associated with Immune Checkpoint Inhibitors
While immune checkpoint inhibitors have revolutionized the treatment of many solid tumor malignancies, this new class of cancer therapy has brought about a new type of toxicity for clinicians to be aware of, termed immune-related adverse events (irAEs). As immune checkpoint inhibitors amplify the immune response against malignancy, they also increase the likelihood that autoreactive T-cells persist and proliferate within the circulation. Therefore, these therapies can result in almost any type of autoimmune side effect. The most commonly reported irAEs in large clinical trials studying CTLA-4 and PD-1 inhibitors include rash/pruritus, diarrhea/colitis, hepatitis, endocrinopathies (thyroiditis, hypophysitis, adrenalitis), and pneumonitis. Other more rare toxicities include pancreatitis, autoimmune hematologic toxicities, cardiac toxicity (myocarditis, heart failure), and neurologic toxicities (neuropathies, myasthenia gravis-like syndrome, Guillain-Barré syndrome). It has been observed that PD-1 inhibitors have a lower incidence of irAEs than CTLA-4 inhibitors, and that the combined use of PD-1 and CTLA-4 inhibitors is associated with a greater incidence of irAEs compared to monotherapy with either agent.31 Toxicities associated with ipilimumab have been noted to be dose dependent.18 Generally, these toxicities are treated with immunosuppression in the form of glucocorticoids and are often reversible.31 There are several published guidelines that include algorithms for the management of irAEs by organizations such as the National Comprehensive Cancer Network.32
For example, previously untreated patients treated with ipilimumab plus dacarbazine as compared to dacarbazine plus placebo had greater grade 3 or 4 adverse events (56.3% vs 27.5%), and 77.7% of patients experiencing an irAE of any grade.18 In the CheckMate 066 trial comparing frontline nivolumab to dacarbazine, nivolumab had a lower rate of grade 3 or 4 toxicity (11.7% vs 17.6%) and irAEs were relatively infrequent, with diarrhea and elevated alanine aminotransferase level each being the most prominent irAE (affecting 1.0% of patients).25 In the KEYNOTE-006 trial, irAEs seen in more than 1% of patients treated with pembrolizumab included colitis, hepatitis, hypothyroidism, and hyperthyroidism, whereas those occurring in more than 1% of patients treated with ipilimumab included colitis and hypophysitis. Overall, there were lower rates of grade 3 to 5 toxicity with the 2 pembrolizumab doses compared to ipilimumab (13.3% pembrolizumab every 2 weeks vs 10.1% pembrolizumab every 3 weeks vs 19.9% ipilimumab).5 In the CheckMate 067 trial comparing nivolumab plus ipilimumab, nivolumab monotherapy, and ipilimumab monotherapy, rates of treatment-related adverse events of any grade were higher in the combination group (96% combination vs 86% nivolumab vs 86% ipilimumab), as were rates of grade 3 or 4 adverse events (59% vs 21% vs 28%, respectively). The irAE profile was similar to that demonstrated in prior studies: rash/pruritus were the most common, and diarrhea/colitis, elevated aminotransferases, and endocrinopathies were among the more common irAEs.7
Alternative dosing strategies have been investigated in an effort to preserve efficacy and minimize toxicity. A phase 1b trial of pembrolizumab plus reduced-dose ipilimumab demonstrated an ORR of 61%, with a 1-year PFS of 69% and a 1-year OS of 89%. This combination led to 45% of patients having a grade 3 or 4 adverse event, 60% having irAEs of any grade, and only 27% having grade 3 or 4 irAEs.29 The CheckMate 067 trial studied the combination of nivolumab 1 mg/kg plus ipilimumab 3 mg/kg.6 The CheckMate 511 trial compared different combination dosing strategies (nivolumab 3 mg/kg + ipilimumab 1 mg/kg versus nivolumab 1 mg/kg + ipilimumab 3 mg/kg) to assess for safety benefit. In the results published in abstract form, the reduced ipilimumab dose (nivolumab 3 mg/kg + ipilimumab 1 mg/kg arm) resulted in significantly decreased grade 3 to 5 adverse events (33.9% vs 48.3%) without significant differences in ORR, PFS, or OS.33
The question about the efficacy of checkpoint inhibitors in patients who discontinue treatment due to irAEs has been raised, as one hypothesis suggests that such toxicities may also indicate that the antitumor immune response has been activated. In a retrospective pooled analysis of phase 2 and 3 trials where patients received combination therapy with ipilimumab and nivolumab and discontinued therapy during the induction phase due to irAEs, outcomes did not appear to be inferior. Median PFS was 8.4 months in those who discontinued therapy compared to 10.8 months in those who continued therapy, but this did not reach statistical significance. Median OS had not been reached in either group and ORR was actually higher in those who discontinued due to adverse events (58.3% vs 50.2%). While this retrospective analysis needs to be validated, it does suggest that patients likely derive antitumor benefit from immunotherapy even if they have to discontinue therapy due to irAEs. Of note, patients in this analysis were not trialed on nivolumab monotherapy after receiving immunosuppressive treatment for toxicity related to combination therapy, which has since been deemed a reasonable treatment option.34
Molecularly Targeted Therapy for Metastatic Melanoma
As previously mentioned, the MAPK pathway is frequently altered in metastatic melanoma and thus serves as a target for therapy. Mutations in BRAF can cause constitutive activation of the protein’s kinase function, which subsequently phosphorylates/activates MEK in the absence of extracellular growth signals and causes increased cellular proliferation. For the roughly half of patients diagnosed with metastatic melanoma who harbor a BRAF V600 mutation, molecularly targeted therapy with BRAF/MEK inhibitors has emerged as a standard of care treatment option. As such, all patients with advanced disease should be tested for BRAF mutations.
After early phase 1 studies of the BRAF inhibitor vemurafenib demonstrated successful inhibition of mutated BRAF,35 subsequent studies confirmed the benefit of BRAF targeted therapy. In the phase 3 randomized controlled BRIM-3 trial comparing vemurafenib with dacarbazine for treatment of 675 patients with previously untreated metastatic melanoma positive for a BRAF V600E mutation, the vemurafenib group had superior ORR and 6-month OS during the first analysis.36 In a subsequent analysis, median PFS and median OS were also superior with vemurafenib compared to dacarbazine, as vemurafenib had a median OS of 13.6 months compared to 9.7 months with dacarbazine (HR, 0.70; P = 0.0008).37 Dabrafenib was the next BRAF inhibitor to demonstrate clinical efficacy with superior PFS compared to dacarbazine.38
Despite tumor shrinkage in the majority of patients, the development of resistance to therapy was an issue early on. The development of acquired resistance emerged as a heterogeneous process, though many of the identified resistance mechanisms involved reactivation of the MAPK pathway.39 A phase 3 trial of 322 patients with metastatic melanoma comparing the MEK inhibitor trametinib as monotherapy against chemotherapy demonstrated a modest improvement in both median PFS and OS.40 As a result, subsequent efforts focused on a strategy of concurrent MEK inhibition as a means to overcome resistance to molecularly targeted monotherapy
At least 4 large phase 3 randomized controlled trials of combination therapy with BRAF plus MEK inhibitors showed an improved ORR, PFS, and OS when compared to BRAF inhibition alone. The COMBI-d trial comparing dabrafenib plus trametinib versus dabrafenib alone was the first to demonstrate the superiority of combined BRAF/MEK inhibition and made combination therapy the current standard of care for patients with metastatic melanoma and a BRAF V600 mutation. In the final analysis of this trial, 3-year PFS was 22% with combination therapy compared to 12% with dabrafenib alone, and 3-year OS was 44% compared to 32%.8,41,42 A second trial with the combination of dabrafenib and trametinib (COMBI-V) also demonstrated superior efficacy when compared to single-agent vemurafenib without increased toxicity.43 Subsequently, the combination of vemurafenib with MEK inhibitor cobimetinib demonstrated superiority compared to vemurafenib alone,44 followed by the newest combination encorafenib (BRAF inhibitor) and binimetinib (MEK inhibitor) proving superior to either vemurafenib or encorafenib alone.45,46
It is important to note that there have been no studies directly comparing the efficacy of the 3 approved BRAF/MEK inhibitor combinations, but the 3 different regimens have some differences in their toxicity profiles (Table 2). Of note, single-agent BRAF inhibition was associated with increased cutaneous toxicity, including secondary squamous cell carcinoma and keratoacanthoma,47 which was demonstrated to be driven by paradoxical activation of the MAPK pathway.48 The concerning cutaneous toxicities such as squamous cell carcinoma were substantially reduced by combination BRAF/MEK inhibitor therapy.47 Collectively, the higher efficacy along with manageable toxicity profile established combination BRAF/MEK inhibition as the preferred regimen for patients with BRAF-mutated metastatic melanoma who are being considered for molecularly targeted therapy. BRAF inhibitor monotherapy should only be used when there is a specific concern regarding the use of a MEK inhibitor in certain clinical circumstances.
Other driver mutations associated with metastatic melanoma such as NRAS-mutated tumors have proven more difficult to effectively treat with molecularly targeted therapy, with one study showing that the MEK inhibitor binimetinib resulted in a modest improvement in ORR and median PFS without OS benefit compared to dacarbazine.49 Several phase 2 trials involving metastatic melanoma harboring a c-Kit alteration have demonstrated some efficacy with the tyrosine kinase inhibitor imatinib. The largest phase 2 trial of 43 patients treated with imatinib resulted in a 53.5% disease control rate (23.3% partial response and 30.2% stable disease), with 9 of the 10 patients who achieved partial response having a mutation in either exon 11 or 13. Median PFS was 3.5 months and 1-year OS was 51.0%.50
Case Conclusion
Prior to initiation of systemic therapy, the patient’s melanoma is tested and is found to be positive for a BRAF V600K mutation. At his follow-up appointment, the patient continues to endorse generalized weakness, fatigue, issues with balance, and residual pulmonary symptoms after being treated for post-obstructive pneumonia. Given his current symptoms and extent of metastatic disease, immunotherapy is deferred and he is started on combination molecularly targeted therapy with dabrafenib and trametinib. He initially does well, with a partial response noted by resolution of symptoms and decreased size of his intracranial metastases and decreased size of the right lower lobe mass. Further follow-up of this patient is presented in the second article in this 2-part review of advanced melanoma.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7-30.
2. Ives NJ, Stowe RL, Lorigan P, Wheatley K. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2621 patients. J Clin Oncol. 2007;25:5426-34.
3. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-16.
4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23.
5. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2522-2532.
6. Larkin J, Chiarion-Sileni V, Gonazalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34.
7. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345-1356.
8. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
9. Elwood JM, Jopson J. Melanoma and sun exposure: an overview of published studies. Int J Cancer. 1997;73:198-203.
10. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 199;340:1341-1348.
11. Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483-8.
12. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54.
13. Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681-96.
14. Gershenwald JE, Scolyer RA, Hess KR, et al. Melanoma staging: evidence-based changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J Clin. 2017;67:472-492.
15. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. 2018;36:1668-1674.
16. Salama AKS, Hodi FS. Cytotoxic T-lymphocyte-associated antigen-4. Clin Cancer Res. 2011;17:4622-8.
17. Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767-1778.
18. Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517-2526.
19. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-2454.
20. Topalian S, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020-30.
21. Weber JS, D’Angelo SP, Minor D, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375-84.
22. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315:1600-1609.
23. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908-18.
24. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer. 2017;86:37-45.
25. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
26. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicenter, randomised, open-label phase 3 study (KEYNOTE-006). Lancet Oncol. 2017;390:1853-1862.
27. Carlino MS, Long GV, Schadendorf D, et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006. A randomised clinical trial. Eur J Cancer. 2018;101:236-243.
28. Hodi FS, Chesney J, Pavlick AC, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558-1568.
29. Long GV, Atkinson V, Cebon JS, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. 2017;18:1202-10.
30. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480-1492.
31. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol. 2016;2:1346-1353.
32. National Comprehensive Cancer Network. Management of immunotherapy-related toxicities (version 2.2019). www.nccn.org/professionals/physician_gls/pdf/immunotherapy.pdf. Accessed April 8, 2019.
33. Lebbé C, Meyer N, Mortier L, et al. Initial results from a phase IIIb/IV study evaluating two dosing regimens of nivolumab (NIVO) in combination with ipilimumab (IPI) in patients with advanced melanoma (CheckMate 511) [Abstract LBA47]. Ann Oncol. 2018;29:mdy424.057.
34. Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase ii and iii trials. J Clin Oncol. 2017;35:3807-3814.
35. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
36. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
37. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
38. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicenter, open-label, phase 3 randomised controlled trial. Lancet Oncol. 2012;380:358-365.
39. Rizos H, Menzies AM, Pupo GM, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965-1977.
40. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-114.
41. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicenter, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2015;386:444-451.
42. Long GV, Flaherty KT, Stroyakovskiy D, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28:1631-1639.
43. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
44. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-260.
45. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicenter, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19:603-615.
46. Dummer R, Ascierto PA, Gogas HJ, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2018;19:1315-1327.
47. Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA. Dermatol 2015;151:1103-1109.
48. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
49. Dummer R, Schadendorf D, Ascierto P, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicenter, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435-445.
50. Guo J, Si L, Kong Y, et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29:2904-2909.
Management of Late Pulmonary Complications After Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1. 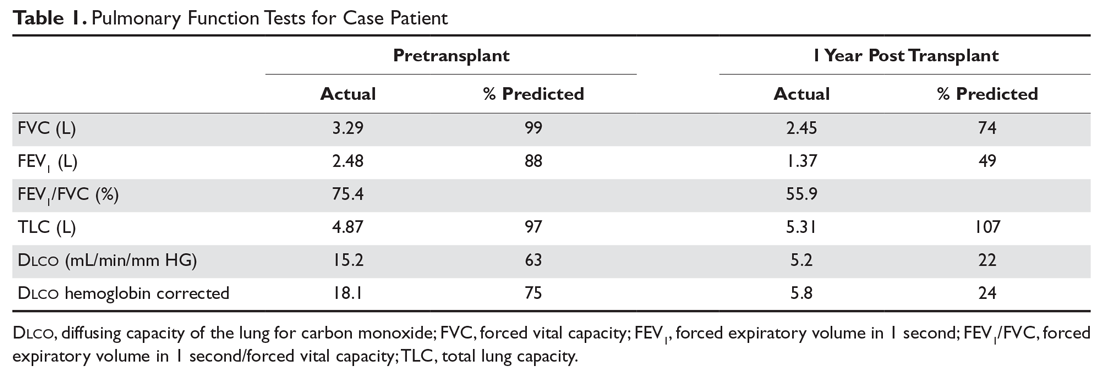
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2). 
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).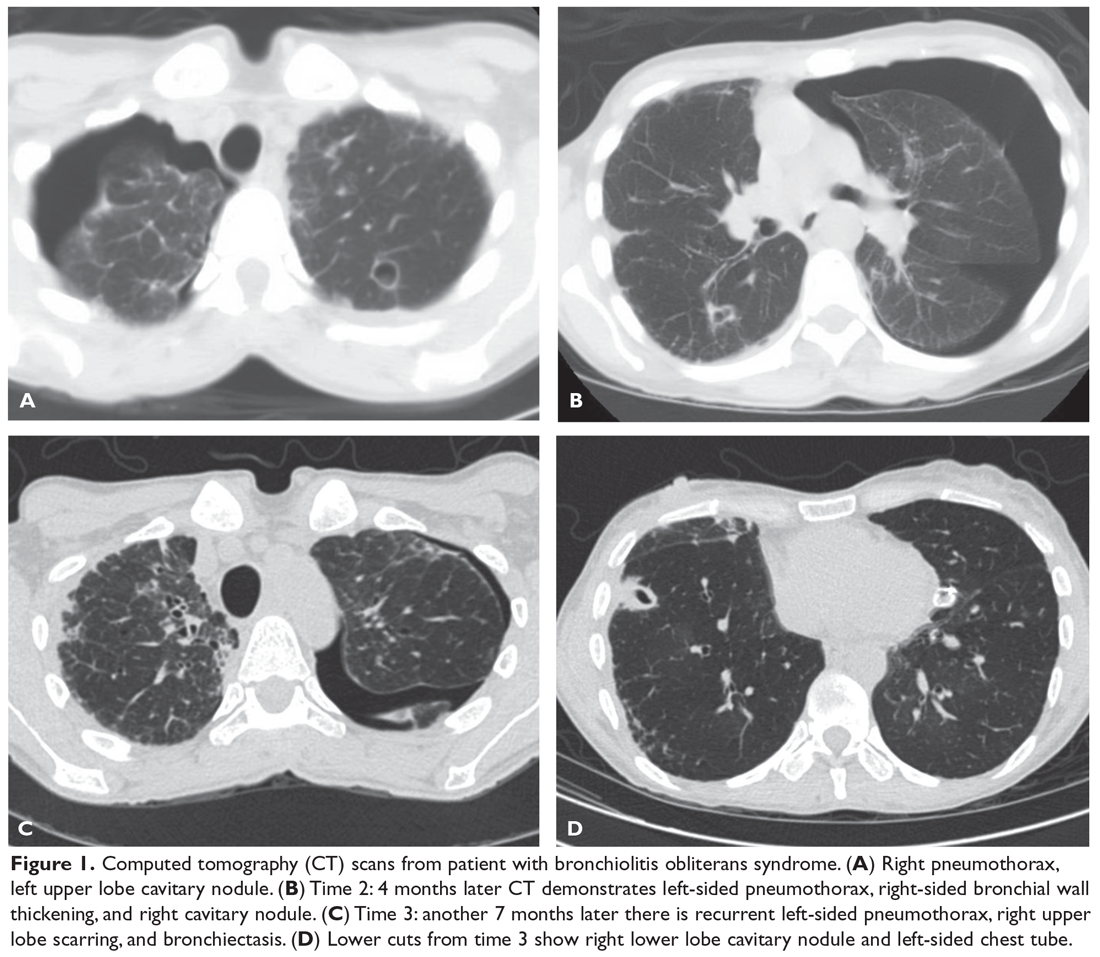
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.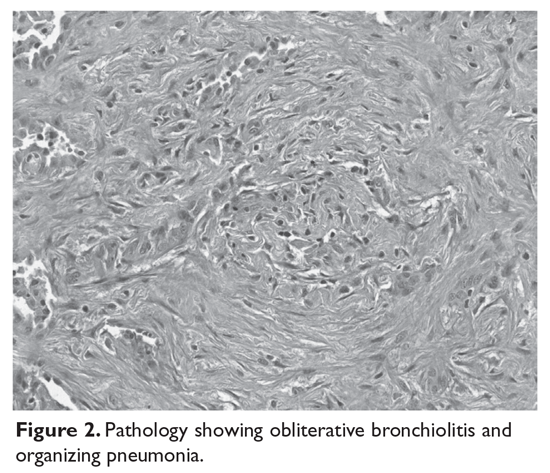
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.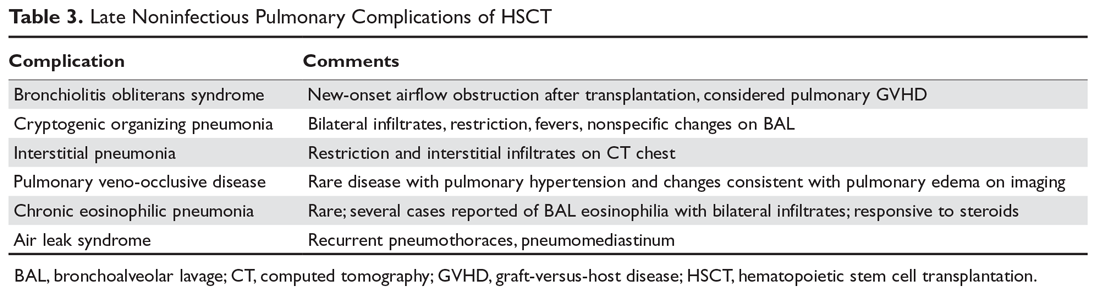
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1.
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2).
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1.
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2).
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.