User login
The road to weight loss is paved with collusion and sabotage
Three big bumps on the weight-loss journey
The search for the Holy Grail. The destruction of the One Ring. The never-ending struggle to Lose Weight.
Like most legendary quests, weight loss is a journey, and we need support to help us achieve our goal. Maybe it’s gaining a new workout partner or finding a similarly-goaled Facebook Group. For a lot of people, it’s as simple as your friends and family. A recent study, however, suggests that the people closest to you may be your worst weight-loss enemies, and they might not even know it.
Researchers at the University of Surrey reviewed the literature on the positives and negatives of social support when it comes to weight loss and identified three types of negative effects: acts of sabotage, feeding behavior, and collusion.
Let’s start with the softest of intentions and work our way up. Collusion is the least negative. Friends and family may just go with the flow, even if it doesn’t agree with the goals of the person who’s trying to lose weight. It can even happen when health care professionals try to help their patients navigate or avoid obesity, ultimately killing with kindness, so to speak.
Next up, feeding behavior. Maybe you know someone whose love language is cooking. There are also people who share food because they don’t want to waste it or because they’re trying to be polite. They act out of the goodness of their hearts, but they’re putting up roadblocks to someone’s goals. These types of acts are usually one-sided, the researchers found. Remember, it’s okay to say, “No thanks.”
The last method, sabotage, is the most sinister. The saboteur may discourage others from eating healthy, undermine their efforts to be physically active, or take jabs at their confidence or self-esteem. Something as simple as criticizing someone for eating a salad or refusing to go on a walk with them can cause a setback.
“We need to explore this area further to develop interventions which could target family and friends and help them be more supportive in helping those they are close to lose weight,” said lead author Jane Odgen, PhD, of the University of Surrey, Guildford, England.
Like we said before, weight loss is a journey. The right support can only improve the odds of success.
Robots vs. mosquitoes
If there’s one thing robots are bad at, it’s giving solid mental health advice to people in crisis. If there’s one thing robots are very, very good at, it’s causing apocalypses. And joyous day for humanity, this time we’re not the ones being apocalypsed.
Yet.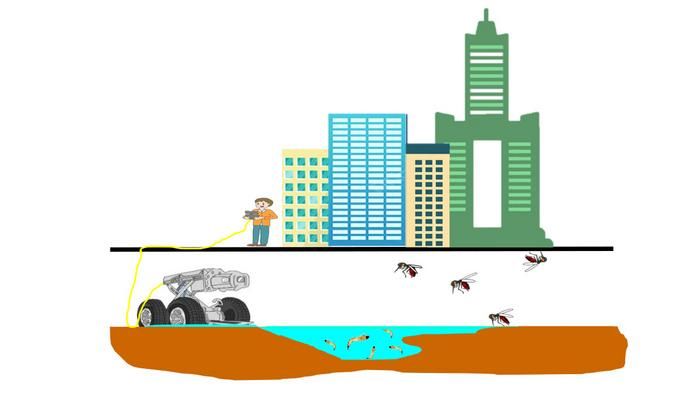
Taiwan has a big mosquito problem. Not only do the mosquitoes in Taiwan carry dengue – among other dangerous diseases – but they’ve urbanized. Not urbanized in the sense that they’ve acquired a taste for organic coffee and avocado toast (that would be the millennial mosquito, a separate but even more terrifying creature), but more that they’ve adapted to reproduce literally anywhere and everywhere. Taiwanese mosquitoes like to breed in roadside sewer ditches, and this is where our genocidal robot comes in.
To combat the new, dangerous form of street-savvy mosquito, researchers built a robot armed with both insecticide and high-temperature, high-pressure water jets and sent it into the sewers of Kaohsiung City. The robot’s goal was simple: Whenever it came across signs of heavy mosquito breeding – eggs, larvae, pupae, and so on – the robot went to work. Utilizing both its primary weapons, the robot scrubbed numerous breeding sites across the city clean.
The researchers could just sit back and wait to see how effective their robot was. In the immediate aftermath, at various monitoring sites placed alongside the ditches, adult mosquito density fell by two-thirds in areas targeted by the robot. That’s nothing to sniff at, and it does make sense. After all, mosquitoes are quite difficult to kill in their adult stage, why not target them when they’re young and basically immobile?
The researchers saw promise with their mosquito-killing robot, but we’ve noticed a rather large issue. Killing two-thirds of mosquitoes is fine, but the third that’s left will be very angry. Very angry indeed. After all, we’re targeting the mosquito equivalent of children. Let’s hope our mosquito Terminator managed to kill mosquito Sarah Connor, or we’re going to have a big problem on our hands a bit later down the line.
This is knot what you were expecting
Physicians who aren’t surgeons probably don’t realize it, but the big thing that’s been getting between the knot-tying specialists and perfect suturing technique all these years is a lack of physics. Don’t believe us? Well, maybe you’ll believe plastic surgeon Samia Guerid, MD, of Lausanne, Switzerland: “The lack of physics-based analysis has been a limitation.” Nuff said.

That’s not enough for you, is it? Fine, we were warned.
Any surgical knot, Dr. Guerid and associates explained in a written statement, involves the “complex interplay” between six key factors: topology, geometry, elasticity, contact, friction, and polymer plasticity of the suturing filament. The strength of a suture “depends on the tension applied during the tying of the knot, [which] permanently deforms, or stretches the filament, creating a holding force.” Not enough tension and the knot comes undone, while too much snaps the filament.
For the experiment, Dr. Guerid tied a few dozen surgical knots, which were then scanned using x-ray micro–computed tomography to facilitate finite element modeling with a “3D continuum-level constitutive model for elastic-viscoplastic mechanical behavior” – no, we have no idea what that means, either – developed by the research team.
That model, and a great deal of math – so much math – allowed the researchers to define a threshold between loose and tight knots and uncover “relationships between knot strength and pretension, friction, and number of throws,” they said.
But what about the big question? The one about the ideal amount of tension? You may want to sit down. The answer to the ultimate question of the relationship between knot pretension and strength is … Did we mention that the team had its own mathematician? Their predictive model for safe knot-tying is … You’re not going to like this. The best way to teach safe knot-tying to both trainees and robots is … not ready yet.
The secret to targeting the knot tension sweet spot, for now, anyway, is still intuition gained from years of experience. Nobody ever said science was perfect … or easy … or quick.
Three big bumps on the weight-loss journey
The search for the Holy Grail. The destruction of the One Ring. The never-ending struggle to Lose Weight.
Like most legendary quests, weight loss is a journey, and we need support to help us achieve our goal. Maybe it’s gaining a new workout partner or finding a similarly-goaled Facebook Group. For a lot of people, it’s as simple as your friends and family. A recent study, however, suggests that the people closest to you may be your worst weight-loss enemies, and they might not even know it.
Researchers at the University of Surrey reviewed the literature on the positives and negatives of social support when it comes to weight loss and identified three types of negative effects: acts of sabotage, feeding behavior, and collusion.
Let’s start with the softest of intentions and work our way up. Collusion is the least negative. Friends and family may just go with the flow, even if it doesn’t agree with the goals of the person who’s trying to lose weight. It can even happen when health care professionals try to help their patients navigate or avoid obesity, ultimately killing with kindness, so to speak.
Next up, feeding behavior. Maybe you know someone whose love language is cooking. There are also people who share food because they don’t want to waste it or because they’re trying to be polite. They act out of the goodness of their hearts, but they’re putting up roadblocks to someone’s goals. These types of acts are usually one-sided, the researchers found. Remember, it’s okay to say, “No thanks.”
The last method, sabotage, is the most sinister. The saboteur may discourage others from eating healthy, undermine their efforts to be physically active, or take jabs at their confidence or self-esteem. Something as simple as criticizing someone for eating a salad or refusing to go on a walk with them can cause a setback.
“We need to explore this area further to develop interventions which could target family and friends and help them be more supportive in helping those they are close to lose weight,” said lead author Jane Odgen, PhD, of the University of Surrey, Guildford, England.
Like we said before, weight loss is a journey. The right support can only improve the odds of success.
Robots vs. mosquitoes
If there’s one thing robots are bad at, it’s giving solid mental health advice to people in crisis. If there’s one thing robots are very, very good at, it’s causing apocalypses. And joyous day for humanity, this time we’re not the ones being apocalypsed.
Yet.
Taiwan has a big mosquito problem. Not only do the mosquitoes in Taiwan carry dengue – among other dangerous diseases – but they’ve urbanized. Not urbanized in the sense that they’ve acquired a taste for organic coffee and avocado toast (that would be the millennial mosquito, a separate but even more terrifying creature), but more that they’ve adapted to reproduce literally anywhere and everywhere. Taiwanese mosquitoes like to breed in roadside sewer ditches, and this is where our genocidal robot comes in.
To combat the new, dangerous form of street-savvy mosquito, researchers built a robot armed with both insecticide and high-temperature, high-pressure water jets and sent it into the sewers of Kaohsiung City. The robot’s goal was simple: Whenever it came across signs of heavy mosquito breeding – eggs, larvae, pupae, and so on – the robot went to work. Utilizing both its primary weapons, the robot scrubbed numerous breeding sites across the city clean.
The researchers could just sit back and wait to see how effective their robot was. In the immediate aftermath, at various monitoring sites placed alongside the ditches, adult mosquito density fell by two-thirds in areas targeted by the robot. That’s nothing to sniff at, and it does make sense. After all, mosquitoes are quite difficult to kill in their adult stage, why not target them when they’re young and basically immobile?
The researchers saw promise with their mosquito-killing robot, but we’ve noticed a rather large issue. Killing two-thirds of mosquitoes is fine, but the third that’s left will be very angry. Very angry indeed. After all, we’re targeting the mosquito equivalent of children. Let’s hope our mosquito Terminator managed to kill mosquito Sarah Connor, or we’re going to have a big problem on our hands a bit later down the line.
This is knot what you were expecting
Physicians who aren’t surgeons probably don’t realize it, but the big thing that’s been getting between the knot-tying specialists and perfect suturing technique all these years is a lack of physics. Don’t believe us? Well, maybe you’ll believe plastic surgeon Samia Guerid, MD, of Lausanne, Switzerland: “The lack of physics-based analysis has been a limitation.” Nuff said.
That’s not enough for you, is it? Fine, we were warned.
Any surgical knot, Dr. Guerid and associates explained in a written statement, involves the “complex interplay” between six key factors: topology, geometry, elasticity, contact, friction, and polymer plasticity of the suturing filament. The strength of a suture “depends on the tension applied during the tying of the knot, [which] permanently deforms, or stretches the filament, creating a holding force.” Not enough tension and the knot comes undone, while too much snaps the filament.
For the experiment, Dr. Guerid tied a few dozen surgical knots, which were then scanned using x-ray micro–computed tomography to facilitate finite element modeling with a “3D continuum-level constitutive model for elastic-viscoplastic mechanical behavior” – no, we have no idea what that means, either – developed by the research team.
That model, and a great deal of math – so much math – allowed the researchers to define a threshold between loose and tight knots and uncover “relationships between knot strength and pretension, friction, and number of throws,” they said.
But what about the big question? The one about the ideal amount of tension? You may want to sit down. The answer to the ultimate question of the relationship between knot pretension and strength is … Did we mention that the team had its own mathematician? Their predictive model for safe knot-tying is … You’re not going to like this. The best way to teach safe knot-tying to both trainees and robots is … not ready yet.
The secret to targeting the knot tension sweet spot, for now, anyway, is still intuition gained from years of experience. Nobody ever said science was perfect … or easy … or quick.
Three big bumps on the weight-loss journey
The search for the Holy Grail. The destruction of the One Ring. The never-ending struggle to Lose Weight.
Like most legendary quests, weight loss is a journey, and we need support to help us achieve our goal. Maybe it’s gaining a new workout partner or finding a similarly-goaled Facebook Group. For a lot of people, it’s as simple as your friends and family. A recent study, however, suggests that the people closest to you may be your worst weight-loss enemies, and they might not even know it.
Researchers at the University of Surrey reviewed the literature on the positives and negatives of social support when it comes to weight loss and identified three types of negative effects: acts of sabotage, feeding behavior, and collusion.
Let’s start with the softest of intentions and work our way up. Collusion is the least negative. Friends and family may just go with the flow, even if it doesn’t agree with the goals of the person who’s trying to lose weight. It can even happen when health care professionals try to help their patients navigate or avoid obesity, ultimately killing with kindness, so to speak.
Next up, feeding behavior. Maybe you know someone whose love language is cooking. There are also people who share food because they don’t want to waste it or because they’re trying to be polite. They act out of the goodness of their hearts, but they’re putting up roadblocks to someone’s goals. These types of acts are usually one-sided, the researchers found. Remember, it’s okay to say, “No thanks.”
The last method, sabotage, is the most sinister. The saboteur may discourage others from eating healthy, undermine their efforts to be physically active, or take jabs at their confidence or self-esteem. Something as simple as criticizing someone for eating a salad or refusing to go on a walk with them can cause a setback.
“We need to explore this area further to develop interventions which could target family and friends and help them be more supportive in helping those they are close to lose weight,” said lead author Jane Odgen, PhD, of the University of Surrey, Guildford, England.
Like we said before, weight loss is a journey. The right support can only improve the odds of success.
Robots vs. mosquitoes
If there’s one thing robots are bad at, it’s giving solid mental health advice to people in crisis. If there’s one thing robots are very, very good at, it’s causing apocalypses. And joyous day for humanity, this time we’re not the ones being apocalypsed.
Yet.
Taiwan has a big mosquito problem. Not only do the mosquitoes in Taiwan carry dengue – among other dangerous diseases – but they’ve urbanized. Not urbanized in the sense that they’ve acquired a taste for organic coffee and avocado toast (that would be the millennial mosquito, a separate but even more terrifying creature), but more that they’ve adapted to reproduce literally anywhere and everywhere. Taiwanese mosquitoes like to breed in roadside sewer ditches, and this is where our genocidal robot comes in.
To combat the new, dangerous form of street-savvy mosquito, researchers built a robot armed with both insecticide and high-temperature, high-pressure water jets and sent it into the sewers of Kaohsiung City. The robot’s goal was simple: Whenever it came across signs of heavy mosquito breeding – eggs, larvae, pupae, and so on – the robot went to work. Utilizing both its primary weapons, the robot scrubbed numerous breeding sites across the city clean.
The researchers could just sit back and wait to see how effective their robot was. In the immediate aftermath, at various monitoring sites placed alongside the ditches, adult mosquito density fell by two-thirds in areas targeted by the robot. That’s nothing to sniff at, and it does make sense. After all, mosquitoes are quite difficult to kill in their adult stage, why not target them when they’re young and basically immobile?
The researchers saw promise with their mosquito-killing robot, but we’ve noticed a rather large issue. Killing two-thirds of mosquitoes is fine, but the third that’s left will be very angry. Very angry indeed. After all, we’re targeting the mosquito equivalent of children. Let’s hope our mosquito Terminator managed to kill mosquito Sarah Connor, or we’re going to have a big problem on our hands a bit later down the line.
This is knot what you were expecting
Physicians who aren’t surgeons probably don’t realize it, but the big thing that’s been getting between the knot-tying specialists and perfect suturing technique all these years is a lack of physics. Don’t believe us? Well, maybe you’ll believe plastic surgeon Samia Guerid, MD, of Lausanne, Switzerland: “The lack of physics-based analysis has been a limitation.” Nuff said.
That’s not enough for you, is it? Fine, we were warned.
Any surgical knot, Dr. Guerid and associates explained in a written statement, involves the “complex interplay” between six key factors: topology, geometry, elasticity, contact, friction, and polymer plasticity of the suturing filament. The strength of a suture “depends on the tension applied during the tying of the knot, [which] permanently deforms, or stretches the filament, creating a holding force.” Not enough tension and the knot comes undone, while too much snaps the filament.
For the experiment, Dr. Guerid tied a few dozen surgical knots, which were then scanned using x-ray micro–computed tomography to facilitate finite element modeling with a “3D continuum-level constitutive model for elastic-viscoplastic mechanical behavior” – no, we have no idea what that means, either – developed by the research team.
That model, and a great deal of math – so much math – allowed the researchers to define a threshold between loose and tight knots and uncover “relationships between knot strength and pretension, friction, and number of throws,” they said.
But what about the big question? The one about the ideal amount of tension? You may want to sit down. The answer to the ultimate question of the relationship between knot pretension and strength is … Did we mention that the team had its own mathematician? Their predictive model for safe knot-tying is … You’re not going to like this. The best way to teach safe knot-tying to both trainees and robots is … not ready yet.
The secret to targeting the knot tension sweet spot, for now, anyway, is still intuition gained from years of experience. Nobody ever said science was perfect … or easy … or quick.
New bill would provide greater length of time to sue doctors
A bill in the Maine legislature would have the medical malpractice statute of limitations clock start running when a patient discovers the negligence, which could be years after treatment took place. And other states could follow suit with similar bills. What danger does this pose for doctors?
As it stands, the time limit for patients to be able to bring a medical malpractice lawsuit varies by state. For physicians, this extends their period of liability and could potentially increase the number of lawsuits against them.
“The theory behind a statute of limitations is that states want to provide a reasonable, but not indefinite, amount of time for someone to bring a case to court,” says Patrick T. O’Rourke, Esq., adjunct professor at University of Colorado School of Law, Boulder.
Without a statute of limitations, people could bring claims many years after the fact, which makes it harder to obtain and preserve evidence, Mr. O’Rourke says.
In most cases, it isn’t necessary for a patient to know the full extent of their injury or that their physician acted wrongfully or negligently for the statute of limitations to begin running.
Time of injury versus time of discovery
Most states’ laws dictate that the statute of limitations begins at a set time “after the cause of action accrues.” That means that the clock starts ticking from the date of the procedure, surgery, or treatment. In most states, that time is 2 or 3 years.
This can bar some patients from taking any action at all because the statute of limitations ran out. Because of these hurdles, the proposed bill in Maine would extend the statute of limitations.
Proponents of the bill say that patients would still have 3 years to file suit; it just changes when the clock starts. But opponents feel it could open the door to a limitless system in which people have an indefinite time to sue.
Many states already have discovery rules that extend the statute of limitations when the harm was not immediately obvious to the patient. The legal expectation is that patients who have significant pain or unexpected health conditions will seek medical treatment to investigate what’s wrong. Patients who don’t address the situation promptly are not protected by the discovery rule.
“It is the injured person’s obligation, once learning of the injury, to take action to protect their rights,” says Mr. O’Rourke.
Some states have also enacted other claims requirements in medical malpractice cases that are prerequisites for bringing lawsuits that have periods attached to them. For instance, in Florida, parties have 10 days to provide relevant medical records during the investigation period for a malpractice suit, and in Maine, before filing any malpractice action, a plaintiff must file a complaint with a prelitigation screening panel.
Medical malpractice statutes of limitations by state
Although each state has a basic statute of limitations, many states also include clauses for discovery rules. For example, in Vermont, in addition to the 3-year statute of limitations, a patient can pursue legal recourse “2 years from the date the injury is or reasonably should have been discovered, whichever occurs later, but not later than 7 years from the date of the incident.”
In some states, such as Virginia, special extensions apply in cases in which fraud, concealment, or intentional misrepresentation prevented discovery of the injury within the statute of limitations. And in most states, the statute of limitations is much longer for cases in which medical malpractice involves a child, usually at least until the child turns 18.
Statutes of limitations by state
1 Year: California, Kentucky, Louisiana, Ohio, Tennessee
2 Years: Alabama, Alaska, Arizona, Arkansas, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Illinois, Indiana, Iowa, Kansas, Michigan, Mississippi, Missouri, Nebraska, New Hampshire, New Jersey, North Dakota, Oklahoma, Oregon, Pennsylvania, South Dakota, Texas, Utah, Virginia, West Virginia, Wyoming
2.5 Years: New York
3 Years: Washington D.C., Maine, Maryland, Massachusetts, Montana, Nevada, New Mexico, North Carolina, Rhode Island, South Carolina, Vermont, Washington, Wisconsin
4 Years: Minnesota
To protect yourself
Mr. O’Rourke says that if your state enacts a law that extends the statute of limitations for medical malpractice, there aren’t any proactive changes you need to make in terms of your day-to-day practice of medicine.
“Physicians should continue to provide care that is consistent with the standards of care for their specialty and ensure that the documentation accurately reflects the care they rendered,” he says.
Always be candid and up-front about a patient’s condition, Mr. O’Rourke says, especially if malpractice is on the table.
“If a physician misleads a patient about the nature or extent of an injury, that could prevent the statute of limitations from beginning to run,” he says. “Being open and honest about an injury doesn’t mean that a physician must admit any fault. The patient is owed timely, accurate, and candid information about their condition.”
Keep good records
If the statute of limitations increases, you’ll need to have access to the medical records for as long as the statute is in place, but this shouldn’t have an effect on your records keeping if you’re up to date with HIPAA compliance, says Mr. O’Rourke.
“I don’t think an extension of the statute should cause physicians to change their practices, particularly with the retention of medical records, which should be maintained consistently with HIPAA requirements irrespective of the limitations period in a particular state,” he adds.
Keep an eye on malpractice insurance rates
It’s possible that your malpractice insurance could go up as a result of laws that increase the statute of limitations. But Mr. O’Rourke thinks it likely won’t be a significant amount.
He says it’s “theoretically possible” that an increase in a limitations period could result in an increase in your malpractice insurance, since some claims that would otherwise have been barred because of time could then proceed, but the increase would be nominal.
“I would expect any increase to be fairly marginal because the majority of claims will already be accounted for on an actuarial basis,” he says. “I also don’t think that the extension of a limitations period would increase the award of damages in a particular case. The injuries should be the same under either limitations period, so the compensable loss should not increase.”
Anything that makes it easier for patients to recover should increase the cost of professional liability insurance, and vice versa, says Charles Silver, McDonald Endowed Chair in Civil Procedure at University of Texas at Austin School of Law and coauthor of “Medical Malpractice Litigation: How It Works – Why Tort Reform Hasn’t Helped.” But the long-term trend across the country is toward declining rates of liability and declining payouts on claims.
“The likelihood of being sued successfully by a former patient is low, as is the risk of having to pay out of pocket to settle a claim,” he says. In 2022, the number of adverse reports nationally was 38,938, and out of those, 10,807 resulted in a payout.
In his research on medical malpractice in Texas, Mr. Silver says physicians who carried $1 million in coverage essentially never faced any personal liability on medical malpractice claims. “[This means] that they never had to write a check to a victim,” he says. “Insurers provided all the money. I suspect that the same is true nationwide.”
Key takeaways
Ultimately, to protect yourself and your practice, you can do the following:
- Know the statute of limitations and discovery rules for your state.
- Review your coverage with your insurer to better understand your liability.
- Keep accurate records for as long as your statute requires.
- Notify your insurer or risk management department as soon as possible in the event of an adverse outcome with a patient, Mr. O’Rourke advises.
“The most important thing a physician can do to avoid being sued, even when negligent, is to treat patients with kindness and respect,” says Mr. Silver. “Patients don’t expect doctors to be perfect, and they rarely sue doctors they like.”
A version of this article first appeared on Medscape.com.
A bill in the Maine legislature would have the medical malpractice statute of limitations clock start running when a patient discovers the negligence, which could be years after treatment took place. And other states could follow suit with similar bills. What danger does this pose for doctors?
As it stands, the time limit for patients to be able to bring a medical malpractice lawsuit varies by state. For physicians, this extends their period of liability and could potentially increase the number of lawsuits against them.
“The theory behind a statute of limitations is that states want to provide a reasonable, but not indefinite, amount of time for someone to bring a case to court,” says Patrick T. O’Rourke, Esq., adjunct professor at University of Colorado School of Law, Boulder.
Without a statute of limitations, people could bring claims many years after the fact, which makes it harder to obtain and preserve evidence, Mr. O’Rourke says.
In most cases, it isn’t necessary for a patient to know the full extent of their injury or that their physician acted wrongfully or negligently for the statute of limitations to begin running.
Time of injury versus time of discovery
Most states’ laws dictate that the statute of limitations begins at a set time “after the cause of action accrues.” That means that the clock starts ticking from the date of the procedure, surgery, or treatment. In most states, that time is 2 or 3 years.
This can bar some patients from taking any action at all because the statute of limitations ran out. Because of these hurdles, the proposed bill in Maine would extend the statute of limitations.
Proponents of the bill say that patients would still have 3 years to file suit; it just changes when the clock starts. But opponents feel it could open the door to a limitless system in which people have an indefinite time to sue.
Many states already have discovery rules that extend the statute of limitations when the harm was not immediately obvious to the patient. The legal expectation is that patients who have significant pain or unexpected health conditions will seek medical treatment to investigate what’s wrong. Patients who don’t address the situation promptly are not protected by the discovery rule.
“It is the injured person’s obligation, once learning of the injury, to take action to protect their rights,” says Mr. O’Rourke.
Some states have also enacted other claims requirements in medical malpractice cases that are prerequisites for bringing lawsuits that have periods attached to them. For instance, in Florida, parties have 10 days to provide relevant medical records during the investigation period for a malpractice suit, and in Maine, before filing any malpractice action, a plaintiff must file a complaint with a prelitigation screening panel.
Medical malpractice statutes of limitations by state
Although each state has a basic statute of limitations, many states also include clauses for discovery rules. For example, in Vermont, in addition to the 3-year statute of limitations, a patient can pursue legal recourse “2 years from the date the injury is or reasonably should have been discovered, whichever occurs later, but not later than 7 years from the date of the incident.”
In some states, such as Virginia, special extensions apply in cases in which fraud, concealment, or intentional misrepresentation prevented discovery of the injury within the statute of limitations. And in most states, the statute of limitations is much longer for cases in which medical malpractice involves a child, usually at least until the child turns 18.
Statutes of limitations by state
1 Year: California, Kentucky, Louisiana, Ohio, Tennessee
2 Years: Alabama, Alaska, Arizona, Arkansas, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Illinois, Indiana, Iowa, Kansas, Michigan, Mississippi, Missouri, Nebraska, New Hampshire, New Jersey, North Dakota, Oklahoma, Oregon, Pennsylvania, South Dakota, Texas, Utah, Virginia, West Virginia, Wyoming
2.5 Years: New York
3 Years: Washington D.C., Maine, Maryland, Massachusetts, Montana, Nevada, New Mexico, North Carolina, Rhode Island, South Carolina, Vermont, Washington, Wisconsin
4 Years: Minnesota
To protect yourself
Mr. O’Rourke says that if your state enacts a law that extends the statute of limitations for medical malpractice, there aren’t any proactive changes you need to make in terms of your day-to-day practice of medicine.
“Physicians should continue to provide care that is consistent with the standards of care for their specialty and ensure that the documentation accurately reflects the care they rendered,” he says.
Always be candid and up-front about a patient’s condition, Mr. O’Rourke says, especially if malpractice is on the table.
“If a physician misleads a patient about the nature or extent of an injury, that could prevent the statute of limitations from beginning to run,” he says. “Being open and honest about an injury doesn’t mean that a physician must admit any fault. The patient is owed timely, accurate, and candid information about their condition.”
Keep good records
If the statute of limitations increases, you’ll need to have access to the medical records for as long as the statute is in place, but this shouldn’t have an effect on your records keeping if you’re up to date with HIPAA compliance, says Mr. O’Rourke.
“I don’t think an extension of the statute should cause physicians to change their practices, particularly with the retention of medical records, which should be maintained consistently with HIPAA requirements irrespective of the limitations period in a particular state,” he adds.
Keep an eye on malpractice insurance rates
It’s possible that your malpractice insurance could go up as a result of laws that increase the statute of limitations. But Mr. O’Rourke thinks it likely won’t be a significant amount.
He says it’s “theoretically possible” that an increase in a limitations period could result in an increase in your malpractice insurance, since some claims that would otherwise have been barred because of time could then proceed, but the increase would be nominal.
“I would expect any increase to be fairly marginal because the majority of claims will already be accounted for on an actuarial basis,” he says. “I also don’t think that the extension of a limitations period would increase the award of damages in a particular case. The injuries should be the same under either limitations period, so the compensable loss should not increase.”
Anything that makes it easier for patients to recover should increase the cost of professional liability insurance, and vice versa, says Charles Silver, McDonald Endowed Chair in Civil Procedure at University of Texas at Austin School of Law and coauthor of “Medical Malpractice Litigation: How It Works – Why Tort Reform Hasn’t Helped.” But the long-term trend across the country is toward declining rates of liability and declining payouts on claims.
“The likelihood of being sued successfully by a former patient is low, as is the risk of having to pay out of pocket to settle a claim,” he says. In 2022, the number of adverse reports nationally was 38,938, and out of those, 10,807 resulted in a payout.
In his research on medical malpractice in Texas, Mr. Silver says physicians who carried $1 million in coverage essentially never faced any personal liability on medical malpractice claims. “[This means] that they never had to write a check to a victim,” he says. “Insurers provided all the money. I suspect that the same is true nationwide.”
Key takeaways
Ultimately, to protect yourself and your practice, you can do the following:
- Know the statute of limitations and discovery rules for your state.
- Review your coverage with your insurer to better understand your liability.
- Keep accurate records for as long as your statute requires.
- Notify your insurer or risk management department as soon as possible in the event of an adverse outcome with a patient, Mr. O’Rourke advises.
“The most important thing a physician can do to avoid being sued, even when negligent, is to treat patients with kindness and respect,” says Mr. Silver. “Patients don’t expect doctors to be perfect, and they rarely sue doctors they like.”
A version of this article first appeared on Medscape.com.
A bill in the Maine legislature would have the medical malpractice statute of limitations clock start running when a patient discovers the negligence, which could be years after treatment took place. And other states could follow suit with similar bills. What danger does this pose for doctors?
As it stands, the time limit for patients to be able to bring a medical malpractice lawsuit varies by state. For physicians, this extends their period of liability and could potentially increase the number of lawsuits against them.
“The theory behind a statute of limitations is that states want to provide a reasonable, but not indefinite, amount of time for someone to bring a case to court,” says Patrick T. O’Rourke, Esq., adjunct professor at University of Colorado School of Law, Boulder.
Without a statute of limitations, people could bring claims many years after the fact, which makes it harder to obtain and preserve evidence, Mr. O’Rourke says.
In most cases, it isn’t necessary for a patient to know the full extent of their injury or that their physician acted wrongfully or negligently for the statute of limitations to begin running.
Time of injury versus time of discovery
Most states’ laws dictate that the statute of limitations begins at a set time “after the cause of action accrues.” That means that the clock starts ticking from the date of the procedure, surgery, or treatment. In most states, that time is 2 or 3 years.
This can bar some patients from taking any action at all because the statute of limitations ran out. Because of these hurdles, the proposed bill in Maine would extend the statute of limitations.
Proponents of the bill say that patients would still have 3 years to file suit; it just changes when the clock starts. But opponents feel it could open the door to a limitless system in which people have an indefinite time to sue.
Many states already have discovery rules that extend the statute of limitations when the harm was not immediately obvious to the patient. The legal expectation is that patients who have significant pain or unexpected health conditions will seek medical treatment to investigate what’s wrong. Patients who don’t address the situation promptly are not protected by the discovery rule.
“It is the injured person’s obligation, once learning of the injury, to take action to protect their rights,” says Mr. O’Rourke.
Some states have also enacted other claims requirements in medical malpractice cases that are prerequisites for bringing lawsuits that have periods attached to them. For instance, in Florida, parties have 10 days to provide relevant medical records during the investigation period for a malpractice suit, and in Maine, before filing any malpractice action, a plaintiff must file a complaint with a prelitigation screening panel.
Medical malpractice statutes of limitations by state
Although each state has a basic statute of limitations, many states also include clauses for discovery rules. For example, in Vermont, in addition to the 3-year statute of limitations, a patient can pursue legal recourse “2 years from the date the injury is or reasonably should have been discovered, whichever occurs later, but not later than 7 years from the date of the incident.”
In some states, such as Virginia, special extensions apply in cases in which fraud, concealment, or intentional misrepresentation prevented discovery of the injury within the statute of limitations. And in most states, the statute of limitations is much longer for cases in which medical malpractice involves a child, usually at least until the child turns 18.
Statutes of limitations by state
1 Year: California, Kentucky, Louisiana, Ohio, Tennessee
2 Years: Alabama, Alaska, Arizona, Arkansas, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Illinois, Indiana, Iowa, Kansas, Michigan, Mississippi, Missouri, Nebraska, New Hampshire, New Jersey, North Dakota, Oklahoma, Oregon, Pennsylvania, South Dakota, Texas, Utah, Virginia, West Virginia, Wyoming
2.5 Years: New York
3 Years: Washington D.C., Maine, Maryland, Massachusetts, Montana, Nevada, New Mexico, North Carolina, Rhode Island, South Carolina, Vermont, Washington, Wisconsin
4 Years: Minnesota
To protect yourself
Mr. O’Rourke says that if your state enacts a law that extends the statute of limitations for medical malpractice, there aren’t any proactive changes you need to make in terms of your day-to-day practice of medicine.
“Physicians should continue to provide care that is consistent with the standards of care for their specialty and ensure that the documentation accurately reflects the care they rendered,” he says.
Always be candid and up-front about a patient’s condition, Mr. O’Rourke says, especially if malpractice is on the table.
“If a physician misleads a patient about the nature or extent of an injury, that could prevent the statute of limitations from beginning to run,” he says. “Being open and honest about an injury doesn’t mean that a physician must admit any fault. The patient is owed timely, accurate, and candid information about their condition.”
Keep good records
If the statute of limitations increases, you’ll need to have access to the medical records for as long as the statute is in place, but this shouldn’t have an effect on your records keeping if you’re up to date with HIPAA compliance, says Mr. O’Rourke.
“I don’t think an extension of the statute should cause physicians to change their practices, particularly with the retention of medical records, which should be maintained consistently with HIPAA requirements irrespective of the limitations period in a particular state,” he adds.
Keep an eye on malpractice insurance rates
It’s possible that your malpractice insurance could go up as a result of laws that increase the statute of limitations. But Mr. O’Rourke thinks it likely won’t be a significant amount.
He says it’s “theoretically possible” that an increase in a limitations period could result in an increase in your malpractice insurance, since some claims that would otherwise have been barred because of time could then proceed, but the increase would be nominal.
“I would expect any increase to be fairly marginal because the majority of claims will already be accounted for on an actuarial basis,” he says. “I also don’t think that the extension of a limitations period would increase the award of damages in a particular case. The injuries should be the same under either limitations period, so the compensable loss should not increase.”
Anything that makes it easier for patients to recover should increase the cost of professional liability insurance, and vice versa, says Charles Silver, McDonald Endowed Chair in Civil Procedure at University of Texas at Austin School of Law and coauthor of “Medical Malpractice Litigation: How It Works – Why Tort Reform Hasn’t Helped.” But the long-term trend across the country is toward declining rates of liability and declining payouts on claims.
“The likelihood of being sued successfully by a former patient is low, as is the risk of having to pay out of pocket to settle a claim,” he says. In 2022, the number of adverse reports nationally was 38,938, and out of those, 10,807 resulted in a payout.
In his research on medical malpractice in Texas, Mr. Silver says physicians who carried $1 million in coverage essentially never faced any personal liability on medical malpractice claims. “[This means] that they never had to write a check to a victim,” he says. “Insurers provided all the money. I suspect that the same is true nationwide.”
Key takeaways
Ultimately, to protect yourself and your practice, you can do the following:
- Know the statute of limitations and discovery rules for your state.
- Review your coverage with your insurer to better understand your liability.
- Keep accurate records for as long as your statute requires.
- Notify your insurer or risk management department as soon as possible in the event of an adverse outcome with a patient, Mr. O’Rourke advises.
“The most important thing a physician can do to avoid being sued, even when negligent, is to treat patients with kindness and respect,” says Mr. Silver. “Patients don’t expect doctors to be perfect, and they rarely sue doctors they like.”
A version of this article first appeared on Medscape.com.
Protecting your practice data
While data protection is important in any industry, it is particularly critical in health care because in addition to the usual financial records, trade secrets, and other valuable data, confidential patient information is also at risk.
You may think that your computer vendor is responsible for safeguarding your data, but third parties can only do so much. And if your data is compromised, the ultimate responsibility is yours – not to mention the financial loss, and the damage to your practice’s reputation.

In addition to the security vulnerabilities inherent in any system, there are external vulnerabilities, such as weak passwords, viruses, and hacking (either externally or internally). And as hardware becomes more and more portable, there is the increasing risk of theft of platforms and storage media containing confidential data.
A close and ongoing relationship with your hardware and software vendors is essential to good data protection. Your office should have a permanent contact at each company, and you should talk to them regularly. Ask them what sort of firewalls, antivirus software, and other safeguards are in place to protect your system. Whenever they identify a bug or other vulnerability, you should know about it. They should tell you about each software update, what improvements it makes, and what defects it fixes. You should also know about any changes to your data encryption.
Encryption has become an essential component of data protection. It is especially important if you use portable devices such as laptops, pads, or smart phones to store and transport patient information. If you lose one of these devices, or a thumb drive or other storage media, HIPAA will probably not consider it a breach if the data it contains is encrypted.
Encryption isn’t perfect, of course. Log-in credentials can be stolen; and data that is stored in house is can be hacked with malware and phishing techniques, especially if the key to decryption is located on that server. And make sure that employees are not putting any medical data on their own private (unencrypted) devices.
Each employee should have his or her own password, and sharing should be strictly prohibited. Multifactor authentication is becoming increasingly popular for an extra level of security.
Your vendor should require you to change your passwords every few months. If it doesn’t, you need to establish a timetable to do it yourself. All passwords should be strong (no birthdays, pet names, etc.), and they shouldn’t be the same or similar to old passwords.
In some offices, I’ve been surprised to see that every employee has unrestricted access to all practice data. The vulnerabilities of such an arrangement are obvious. There is no reason why receptionists, for example, should have access to medical histories, and insurance people don’t need to know what medications a patient is on. Your vendor can help you design partitions that restrict each employee to only the information they need access to.
Ask if your vendor provides security training for employees. If not, look into hiring a security firm to do it. Regular security training can help employees to recognize data security attacks like phishing, and instills a heightened sense of security awareness and vigilance among staff. They will also gain a better understanding of the role they play in maintaining the overall security of your office.
It goes without saying that third parties, such as business vendors, payers, and managed care providers, should never have access to patient records or other personal health information.
Backing up data
I have written many times about the importance of regularly backing up your data. Industry statistics show that fully 10% of hard drives fail in any given year, and 43% of computer users lose one or more files every year in the form of clinical data, financial records, photos, email, documents, and other important information. Recovery of lost data, when it’s possible at all, can be very expensive.
Even if your EHR vendor backs up your data, you should consider making a separate backup of your own. Backup drives have been known to fail too; and if you decide to switch computer vendors, you don’t want to be at the mercy of the old company that might be reluctant to transfer your data without a hefty payment.
The first rule of backing up is to store your backup drives in a different location from your computers. Unfortunately, that’s a pain; and external drives can be lost or stolen, creating a HIPAA nightmare. So an increasingly popular alternative is automatic remote backup. Several companies offer that service, and the cost is very reasonable for individual computers. Backing up an entire office costs more, depending on how many computers and/or servers you have, but it’s still very reasonable and includes other services, such as operating system and network share support.
The procedure is simple: You create an account and tell the service which files you want copied. Your first backup can take a long time, often days, depending on how much data you are sending and how fast your Internet connection runs. After that the program runs in the background, copying only those files that have changed since the previous backup. Files are encrypted before leaving your computer, and they remain encrypted at the service’s data center, making them HIPAA compliant and, theoretically, only accessible by you.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].
While data protection is important in any industry, it is particularly critical in health care because in addition to the usual financial records, trade secrets, and other valuable data, confidential patient information is also at risk.
You may think that your computer vendor is responsible for safeguarding your data, but third parties can only do so much. And if your data is compromised, the ultimate responsibility is yours – not to mention the financial loss, and the damage to your practice’s reputation.
In addition to the security vulnerabilities inherent in any system, there are external vulnerabilities, such as weak passwords, viruses, and hacking (either externally or internally). And as hardware becomes more and more portable, there is the increasing risk of theft of platforms and storage media containing confidential data.
A close and ongoing relationship with your hardware and software vendors is essential to good data protection. Your office should have a permanent contact at each company, and you should talk to them regularly. Ask them what sort of firewalls, antivirus software, and other safeguards are in place to protect your system. Whenever they identify a bug or other vulnerability, you should know about it. They should tell you about each software update, what improvements it makes, and what defects it fixes. You should also know about any changes to your data encryption.
Encryption has become an essential component of data protection. It is especially important if you use portable devices such as laptops, pads, or smart phones to store and transport patient information. If you lose one of these devices, or a thumb drive or other storage media, HIPAA will probably not consider it a breach if the data it contains is encrypted.
Encryption isn’t perfect, of course. Log-in credentials can be stolen; and data that is stored in house is can be hacked with malware and phishing techniques, especially if the key to decryption is located on that server. And make sure that employees are not putting any medical data on their own private (unencrypted) devices.
Each employee should have his or her own password, and sharing should be strictly prohibited. Multifactor authentication is becoming increasingly popular for an extra level of security.
Your vendor should require you to change your passwords every few months. If it doesn’t, you need to establish a timetable to do it yourself. All passwords should be strong (no birthdays, pet names, etc.), and they shouldn’t be the same or similar to old passwords.
In some offices, I’ve been surprised to see that every employee has unrestricted access to all practice data. The vulnerabilities of such an arrangement are obvious. There is no reason why receptionists, for example, should have access to medical histories, and insurance people don’t need to know what medications a patient is on. Your vendor can help you design partitions that restrict each employee to only the information they need access to.
Ask if your vendor provides security training for employees. If not, look into hiring a security firm to do it. Regular security training can help employees to recognize data security attacks like phishing, and instills a heightened sense of security awareness and vigilance among staff. They will also gain a better understanding of the role they play in maintaining the overall security of your office.
It goes without saying that third parties, such as business vendors, payers, and managed care providers, should never have access to patient records or other personal health information.
Backing up data
I have written many times about the importance of regularly backing up your data. Industry statistics show that fully 10% of hard drives fail in any given year, and 43% of computer users lose one or more files every year in the form of clinical data, financial records, photos, email, documents, and other important information. Recovery of lost data, when it’s possible at all, can be very expensive.
Even if your EHR vendor backs up your data, you should consider making a separate backup of your own. Backup drives have been known to fail too; and if you decide to switch computer vendors, you don’t want to be at the mercy of the old company that might be reluctant to transfer your data without a hefty payment.
The first rule of backing up is to store your backup drives in a different location from your computers. Unfortunately, that’s a pain; and external drives can be lost or stolen, creating a HIPAA nightmare. So an increasingly popular alternative is automatic remote backup. Several companies offer that service, and the cost is very reasonable for individual computers. Backing up an entire office costs more, depending on how many computers and/or servers you have, but it’s still very reasonable and includes other services, such as operating system and network share support.
The procedure is simple: You create an account and tell the service which files you want copied. Your first backup can take a long time, often days, depending on how much data you are sending and how fast your Internet connection runs. After that the program runs in the background, copying only those files that have changed since the previous backup. Files are encrypted before leaving your computer, and they remain encrypted at the service’s data center, making them HIPAA compliant and, theoretically, only accessible by you.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].
While data protection is important in any industry, it is particularly critical in health care because in addition to the usual financial records, trade secrets, and other valuable data, confidential patient information is also at risk.
You may think that your computer vendor is responsible for safeguarding your data, but third parties can only do so much. And if your data is compromised, the ultimate responsibility is yours – not to mention the financial loss, and the damage to your practice’s reputation.
In addition to the security vulnerabilities inherent in any system, there are external vulnerabilities, such as weak passwords, viruses, and hacking (either externally or internally). And as hardware becomes more and more portable, there is the increasing risk of theft of platforms and storage media containing confidential data.
A close and ongoing relationship with your hardware and software vendors is essential to good data protection. Your office should have a permanent contact at each company, and you should talk to them regularly. Ask them what sort of firewalls, antivirus software, and other safeguards are in place to protect your system. Whenever they identify a bug or other vulnerability, you should know about it. They should tell you about each software update, what improvements it makes, and what defects it fixes. You should also know about any changes to your data encryption.
Encryption has become an essential component of data protection. It is especially important if you use portable devices such as laptops, pads, or smart phones to store and transport patient information. If you lose one of these devices, or a thumb drive or other storage media, HIPAA will probably not consider it a breach if the data it contains is encrypted.
Encryption isn’t perfect, of course. Log-in credentials can be stolen; and data that is stored in house is can be hacked with malware and phishing techniques, especially if the key to decryption is located on that server. And make sure that employees are not putting any medical data on their own private (unencrypted) devices.
Each employee should have his or her own password, and sharing should be strictly prohibited. Multifactor authentication is becoming increasingly popular for an extra level of security.
Your vendor should require you to change your passwords every few months. If it doesn’t, you need to establish a timetable to do it yourself. All passwords should be strong (no birthdays, pet names, etc.), and they shouldn’t be the same or similar to old passwords.
In some offices, I’ve been surprised to see that every employee has unrestricted access to all practice data. The vulnerabilities of such an arrangement are obvious. There is no reason why receptionists, for example, should have access to medical histories, and insurance people don’t need to know what medications a patient is on. Your vendor can help you design partitions that restrict each employee to only the information they need access to.
Ask if your vendor provides security training for employees. If not, look into hiring a security firm to do it. Regular security training can help employees to recognize data security attacks like phishing, and instills a heightened sense of security awareness and vigilance among staff. They will also gain a better understanding of the role they play in maintaining the overall security of your office.
It goes without saying that third parties, such as business vendors, payers, and managed care providers, should never have access to patient records or other personal health information.
Backing up data
I have written many times about the importance of regularly backing up your data. Industry statistics show that fully 10% of hard drives fail in any given year, and 43% of computer users lose one or more files every year in the form of clinical data, financial records, photos, email, documents, and other important information. Recovery of lost data, when it’s possible at all, can be very expensive.
Even if your EHR vendor backs up your data, you should consider making a separate backup of your own. Backup drives have been known to fail too; and if you decide to switch computer vendors, you don’t want to be at the mercy of the old company that might be reluctant to transfer your data without a hefty payment.
The first rule of backing up is to store your backup drives in a different location from your computers. Unfortunately, that’s a pain; and external drives can be lost or stolen, creating a HIPAA nightmare. So an increasingly popular alternative is automatic remote backup. Several companies offer that service, and the cost is very reasonable for individual computers. Backing up an entire office costs more, depending on how many computers and/or servers you have, but it’s still very reasonable and includes other services, such as operating system and network share support.
The procedure is simple: You create an account and tell the service which files you want copied. Your first backup can take a long time, often days, depending on how much data you are sending and how fast your Internet connection runs. After that the program runs in the background, copying only those files that have changed since the previous backup. Files are encrypted before leaving your computer, and they remain encrypted at the service’s data center, making them HIPAA compliant and, theoretically, only accessible by you.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News. Write to him at [email protected].

Esophageal diseases: Key new concepts
CHICAGO – These include novel care approaches for esophageal diseases that were published in recent AGA best practice updates on gastroesophageal reflux disease (GERD), extraesophageal reflux, and Barrett’s esophagus, as well as randomized clinical trial data examining therapeutic approaches for erosive esophagitis and eosinophilic esophagitis.

Here are a few highlights: Complications of chronic gastroesophageal reflux include erosive esophagitis for which healing and maintenance of healing is crucial to reduce further erosive sequelae. Healing is typically achieved with pump inhibitor (PPI) therapy. Potassium competitive acid blockers are active prodrugs that bind to the H+/K+ ATPase and have been demonstrated to have a more potent and faster onset in suppressing gastric acid secretion, compared with PPIs.
In a recent phase 3 randomized trial of more than 1,000 adults with erosive esophagitis, the potassium competitive acid blocker vonoprazan was found to be noninferior to lansoprazole in inducing and maintaining healing of erosive esophagitis. Overall, the proportions of subjects that achieved healing by week 8 and maintained healing up to 24 weeks were higher with vonoprazan, when compared with lansoprazole, with a greater treatment effect seen in subjects with severe erosive esophagitis (Los Angeles grade C or D) (Laine L et al. Gastroenterology. Jan 2023;164[1]:61-71).
Screening patients at risk of Barrett’s esophagus (BE), another erosive sequelae of chronic GERD, is critical for early detection and prevention of esophageal cancer. Upper GI endoscopy is standard for Barrett’s screening; however, screening rates of at-risk populations are suboptimal.
In a recent retrospective analysis of a multipractice health care network, only 39% of a screen-eligible population were noted to have undergone upper GI endoscopy. These findings highlight the critical need to improve screening for Barrett’s, including potential of the newer nonendoscopic screening modalities such as swallowable capsule devices combined with a biomarker or cell-collection devices, as well as the need for risk stratification/prediction tools and collaboration with primary care physicians (Eluri S et al. Am J Gastroenterol. Nov 2022;117[11]:1764-71).
Therapeutic options for eosinophilic esophagitis (EoE) have expanded over the past year. Randomized trials demonstrate the efficacy of varied therapeutic approaches including the monoclonal antibody dupilumab as well as topical corticosteroids such as fluticasone propionate orally disintegrated tablet and budesonide oral suspension.
In terms of food elimination diets, a recent multicenter randomized open-label trial identified comparable rates of partial histologic remission with both a traditional six-food elimination diet and a one-food animal milk elimination diet in patients with EoE, though those treated with a six-food elimination were more likely to achieve complete remission (< 1 eosinophil/high power field). Results suggest elimination of animal milk alone is an acceptable initial dietary therapy for EoE, with potential to convert to six-food elimination or alternative therapy when histologic response is not achieved (Kliewer K. Lancet Gastroenterol Hepatol. [published online Feb 2023]).
Dr. Yadlapati is an associate professor in gastroenterology at the University of California, San Diego. She disclosed relationships with Medtronic (Institutional), Ironwood Pharmaceuticals (Institutional), Phathom Pharmaceuticals, and Ironwood Pharmaceuticals. She serves on the advisory board with stock options for RJS Mediagnostix.
These remarks were made during one of the AGA Postgraduate Course sessions held at DDW 2023.
DDW is sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA), the American Society for Gastrointestinal Endoscopy (ASGE) and The Society for Surgery of the Alimentary Tract (SSAT).
CHICAGO – These include novel care approaches for esophageal diseases that were published in recent AGA best practice updates on gastroesophageal reflux disease (GERD), extraesophageal reflux, and Barrett’s esophagus, as well as randomized clinical trial data examining therapeutic approaches for erosive esophagitis and eosinophilic esophagitis.
Here are a few highlights: Complications of chronic gastroesophageal reflux include erosive esophagitis for which healing and maintenance of healing is crucial to reduce further erosive sequelae. Healing is typically achieved with pump inhibitor (PPI) therapy. Potassium competitive acid blockers are active prodrugs that bind to the H+/K+ ATPase and have been demonstrated to have a more potent and faster onset in suppressing gastric acid secretion, compared with PPIs.
In a recent phase 3 randomized trial of more than 1,000 adults with erosive esophagitis, the potassium competitive acid blocker vonoprazan was found to be noninferior to lansoprazole in inducing and maintaining healing of erosive esophagitis. Overall, the proportions of subjects that achieved healing by week 8 and maintained healing up to 24 weeks were higher with vonoprazan, when compared with lansoprazole, with a greater treatment effect seen in subjects with severe erosive esophagitis (Los Angeles grade C or D) (Laine L et al. Gastroenterology. Jan 2023;164[1]:61-71).
Screening patients at risk of Barrett’s esophagus (BE), another erosive sequelae of chronic GERD, is critical for early detection and prevention of esophageal cancer. Upper GI endoscopy is standard for Barrett’s screening; however, screening rates of at-risk populations are suboptimal.
In a recent retrospective analysis of a multipractice health care network, only 39% of a screen-eligible population were noted to have undergone upper GI endoscopy. These findings highlight the critical need to improve screening for Barrett’s, including potential of the newer nonendoscopic screening modalities such as swallowable capsule devices combined with a biomarker or cell-collection devices, as well as the need for risk stratification/prediction tools and collaboration with primary care physicians (Eluri S et al. Am J Gastroenterol. Nov 2022;117[11]:1764-71).
Therapeutic options for eosinophilic esophagitis (EoE) have expanded over the past year. Randomized trials demonstrate the efficacy of varied therapeutic approaches including the monoclonal antibody dupilumab as well as topical corticosteroids such as fluticasone propionate orally disintegrated tablet and budesonide oral suspension.
In terms of food elimination diets, a recent multicenter randomized open-label trial identified comparable rates of partial histologic remission with both a traditional six-food elimination diet and a one-food animal milk elimination diet in patients with EoE, though those treated with a six-food elimination were more likely to achieve complete remission (< 1 eosinophil/high power field). Results suggest elimination of animal milk alone is an acceptable initial dietary therapy for EoE, with potential to convert to six-food elimination or alternative therapy when histologic response is not achieved (Kliewer K. Lancet Gastroenterol Hepatol. [published online Feb 2023]).
Dr. Yadlapati is an associate professor in gastroenterology at the University of California, San Diego. She disclosed relationships with Medtronic (Institutional), Ironwood Pharmaceuticals (Institutional), Phathom Pharmaceuticals, and Ironwood Pharmaceuticals. She serves on the advisory board with stock options for RJS Mediagnostix.
These remarks were made during one of the AGA Postgraduate Course sessions held at DDW 2023.
DDW is sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA), the American Society for Gastrointestinal Endoscopy (ASGE) and The Society for Surgery of the Alimentary Tract (SSAT).
CHICAGO – These include novel care approaches for esophageal diseases that were published in recent AGA best practice updates on gastroesophageal reflux disease (GERD), extraesophageal reflux, and Barrett’s esophagus, as well as randomized clinical trial data examining therapeutic approaches for erosive esophagitis and eosinophilic esophagitis.
Here are a few highlights: Complications of chronic gastroesophageal reflux include erosive esophagitis for which healing and maintenance of healing is crucial to reduce further erosive sequelae. Healing is typically achieved with pump inhibitor (PPI) therapy. Potassium competitive acid blockers are active prodrugs that bind to the H+/K+ ATPase and have been demonstrated to have a more potent and faster onset in suppressing gastric acid secretion, compared with PPIs.
In a recent phase 3 randomized trial of more than 1,000 adults with erosive esophagitis, the potassium competitive acid blocker vonoprazan was found to be noninferior to lansoprazole in inducing and maintaining healing of erosive esophagitis. Overall, the proportions of subjects that achieved healing by week 8 and maintained healing up to 24 weeks were higher with vonoprazan, when compared with lansoprazole, with a greater treatment effect seen in subjects with severe erosive esophagitis (Los Angeles grade C or D) (Laine L et al. Gastroenterology. Jan 2023;164[1]:61-71).
Screening patients at risk of Barrett’s esophagus (BE), another erosive sequelae of chronic GERD, is critical for early detection and prevention of esophageal cancer. Upper GI endoscopy is standard for Barrett’s screening; however, screening rates of at-risk populations are suboptimal.
In a recent retrospective analysis of a multipractice health care network, only 39% of a screen-eligible population were noted to have undergone upper GI endoscopy. These findings highlight the critical need to improve screening for Barrett’s, including potential of the newer nonendoscopic screening modalities such as swallowable capsule devices combined with a biomarker or cell-collection devices, as well as the need for risk stratification/prediction tools and collaboration with primary care physicians (Eluri S et al. Am J Gastroenterol. Nov 2022;117[11]:1764-71).
Therapeutic options for eosinophilic esophagitis (EoE) have expanded over the past year. Randomized trials demonstrate the efficacy of varied therapeutic approaches including the monoclonal antibody dupilumab as well as topical corticosteroids such as fluticasone propionate orally disintegrated tablet and budesonide oral suspension.
In terms of food elimination diets, a recent multicenter randomized open-label trial identified comparable rates of partial histologic remission with both a traditional six-food elimination diet and a one-food animal milk elimination diet in patients with EoE, though those treated with a six-food elimination were more likely to achieve complete remission (< 1 eosinophil/high power field). Results suggest elimination of animal milk alone is an acceptable initial dietary therapy for EoE, with potential to convert to six-food elimination or alternative therapy when histologic response is not achieved (Kliewer K. Lancet Gastroenterol Hepatol. [published online Feb 2023]).
Dr. Yadlapati is an associate professor in gastroenterology at the University of California, San Diego. She disclosed relationships with Medtronic (Institutional), Ironwood Pharmaceuticals (Institutional), Phathom Pharmaceuticals, and Ironwood Pharmaceuticals. She serves on the advisory board with stock options for RJS Mediagnostix.
These remarks were made during one of the AGA Postgraduate Course sessions held at DDW 2023.
DDW is sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA), the American Society for Gastrointestinal Endoscopy (ASGE) and The Society for Surgery of the Alimentary Tract (SSAT).
AT DDW 2023
Smart-bed technology reveals insomnia, flu risk link
The study of smart-bed sleepers found that there was a statistically significant correlation between a higher number of episodes of influenza-like illnesses (ILI) per year with longer duration compared to people without insomnia.
However, more research is needed to determine causality and whether insomnia may predispose to ILI or whether ILI affects long-term sleep behavior, the researchers noted.
“Several lines of evidence make me think that it’s more likely that insomnia makes one more vulnerable to influenza through pathways that involve decreased immune function,” study investigator Gary Garcia-Molina, PhD, with Sleep Number Labs, San Jose, Calif., said in an interview.
Sleep disorders, including insomnia, can dampen immune function and an individual’s ability to fight off illness, he noted.
The findings were presented at the annual meeting of the Associated Professional Sleep Societies.
Smart, connected devices
Pathophysiological responses to respiratory viral infection affect sleep duration and quality in addition to breathing function. “Smart” and “connected” devices that monitor biosignals over time have shown promise for monitoring infectious disease.
In an earlier study presented at SLEEP 2021, Dr. Garcia-Molina and colleagues found that real-world biometric data obtained from a smart bed can help predict and track symptoms of COVID-19 and other respiratory infections. They showed that worsening of COVID-19 symptoms correlated with an increase in sleep duration, breathing rate, and heart rate and a decrease in sleep quality.
In the new study, the researchers evaluated vulnerability to ILI in people with insomnia.
They quantified insomnia over time using the insomnia severity index (ISI). They quantified ILI vulnerability using an established artificial intelligence model they developed that estimates the daily probability of ILI symptoms from a Sleep Number smart bed using ballistocardiograph sensors.
Smart bed data – including daily and restful sleep duration, sleep latency, sleep quality, heart rate, breathing rate and motion level – were queried from 2019 (pre-COVID) and 2021.
A total of 1,680 smart sleepers had nearly constant ISI scores over the study period, with 249 having insomnia and 1,431 not having insomnia.
Data from both 2019 and 2021 show that smart sleepers with insomnia had significantly more and longer ILI episodes per year, compared with peers without insomnia.
For 2019, individuals without insomnia had 1.2 ILI episodes on average, which was significantly less (P < .01) than individuals with insomnia, at 1.5 episodes. The average ILI episode duration for those without insomnia was 4.3 days, which was significantly lower (P < .01) in those with insomnia group, at 6.1 days.
The data for 2021 show similar results, with the no-insomnia group having significantly fewer (P < .01) ILI episodes (about 1.2), compared with the insomnia group (about 1.5).
The average ILI episode duration for the no-insomnia group was 5 days, which was significantly less (P < .01) than the insomnia group, at 6.1 days.
The researchers said their study adds to other data on the relationship between sleep and overall health and well-being. It also highlights the potential health risk of insomnia and the importance of identifying and treating sleep disorders.
“Sleep has such a profound influence on health and wellness and the ability to capture these data unobtrusively in such an easy way and with such a large number of participants paves the way to investigate different aspects of health and disease,” Dr. Garcia-Molina said.
Rich data source
In a comment, Adam C. Powell, PhD, president of Payer+Provider Syndicate, a management advisory and operational consulting firm, said “smart beds provide a new data source for passively monitoring the health of individuals.”
“Unlike active monitoring methods requiring self-report, passive monitoring enables data to be captured without an individual taking any action. This data can be potentially integrated with data from other sources, such as pedometers, smart scales, and smart blood pressure cuffs, to gain a more holistic understanding of how an individual’s activities and behaviors impact their well-being,” said Dr. Powell, who wasn’t involved in the study.
There are some methodological limitations to the study, he noted.
“While the dependent variables examined were the duration and presence of episodes of influenza-like illness, they did not directly measure these episodes. Instead, they calculated the daily probability of influenza-like illness symptoms using a model that received input from the ballistocardiograph sensors in the smart beds,” Dr. Powell noted.
“The model used to calculate daily probability of influenza-like illness was created by examining associations between individuals’ smart-bed sensor data and population-level trends in influenza-like illness reported by the Centers for Disease Control and Prevention,” he explained.
Nonetheless, the findings are “consistent with the literature. It has been established by other researchers that impaired sleep is associated with greater risk of influenza, as well as other illnesses,” Dr. Powell said.
Funding for the study was provided by Sleep Number. Dr. Garcia-Molina and five coauthors are employed by Sleep Number. Dr. Powell reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The study of smart-bed sleepers found that there was a statistically significant correlation between a higher number of episodes of influenza-like illnesses (ILI) per year with longer duration compared to people without insomnia.
However, more research is needed to determine causality and whether insomnia may predispose to ILI or whether ILI affects long-term sleep behavior, the researchers noted.
“Several lines of evidence make me think that it’s more likely that insomnia makes one more vulnerable to influenza through pathways that involve decreased immune function,” study investigator Gary Garcia-Molina, PhD, with Sleep Number Labs, San Jose, Calif., said in an interview.
Sleep disorders, including insomnia, can dampen immune function and an individual’s ability to fight off illness, he noted.
The findings were presented at the annual meeting of the Associated Professional Sleep Societies.
Smart, connected devices
Pathophysiological responses to respiratory viral infection affect sleep duration and quality in addition to breathing function. “Smart” and “connected” devices that monitor biosignals over time have shown promise for monitoring infectious disease.
In an earlier study presented at SLEEP 2021, Dr. Garcia-Molina and colleagues found that real-world biometric data obtained from a smart bed can help predict and track symptoms of COVID-19 and other respiratory infections. They showed that worsening of COVID-19 symptoms correlated with an increase in sleep duration, breathing rate, and heart rate and a decrease in sleep quality.
In the new study, the researchers evaluated vulnerability to ILI in people with insomnia.
They quantified insomnia over time using the insomnia severity index (ISI). They quantified ILI vulnerability using an established artificial intelligence model they developed that estimates the daily probability of ILI symptoms from a Sleep Number smart bed using ballistocardiograph sensors.
Smart bed data – including daily and restful sleep duration, sleep latency, sleep quality, heart rate, breathing rate and motion level – were queried from 2019 (pre-COVID) and 2021.
A total of 1,680 smart sleepers had nearly constant ISI scores over the study period, with 249 having insomnia and 1,431 not having insomnia.
Data from both 2019 and 2021 show that smart sleepers with insomnia had significantly more and longer ILI episodes per year, compared with peers without insomnia.
For 2019, individuals without insomnia had 1.2 ILI episodes on average, which was significantly less (P < .01) than individuals with insomnia, at 1.5 episodes. The average ILI episode duration for those without insomnia was 4.3 days, which was significantly lower (P < .01) in those with insomnia group, at 6.1 days.
The data for 2021 show similar results, with the no-insomnia group having significantly fewer (P < .01) ILI episodes (about 1.2), compared with the insomnia group (about 1.5).
The average ILI episode duration for the no-insomnia group was 5 days, which was significantly less (P < .01) than the insomnia group, at 6.1 days.
The researchers said their study adds to other data on the relationship between sleep and overall health and well-being. It also highlights the potential health risk of insomnia and the importance of identifying and treating sleep disorders.
“Sleep has such a profound influence on health and wellness and the ability to capture these data unobtrusively in such an easy way and with such a large number of participants paves the way to investigate different aspects of health and disease,” Dr. Garcia-Molina said.
Rich data source
In a comment, Adam C. Powell, PhD, president of Payer+Provider Syndicate, a management advisory and operational consulting firm, said “smart beds provide a new data source for passively monitoring the health of individuals.”
“Unlike active monitoring methods requiring self-report, passive monitoring enables data to be captured without an individual taking any action. This data can be potentially integrated with data from other sources, such as pedometers, smart scales, and smart blood pressure cuffs, to gain a more holistic understanding of how an individual’s activities and behaviors impact their well-being,” said Dr. Powell, who wasn’t involved in the study.
There are some methodological limitations to the study, he noted.
“While the dependent variables examined were the duration and presence of episodes of influenza-like illness, they did not directly measure these episodes. Instead, they calculated the daily probability of influenza-like illness symptoms using a model that received input from the ballistocardiograph sensors in the smart beds,” Dr. Powell noted.
“The model used to calculate daily probability of influenza-like illness was created by examining associations between individuals’ smart-bed sensor data and population-level trends in influenza-like illness reported by the Centers for Disease Control and Prevention,” he explained.
Nonetheless, the findings are “consistent with the literature. It has been established by other researchers that impaired sleep is associated with greater risk of influenza, as well as other illnesses,” Dr. Powell said.
Funding for the study was provided by Sleep Number. Dr. Garcia-Molina and five coauthors are employed by Sleep Number. Dr. Powell reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The study of smart-bed sleepers found that there was a statistically significant correlation between a higher number of episodes of influenza-like illnesses (ILI) per year with longer duration compared to people without insomnia.
However, more research is needed to determine causality and whether insomnia may predispose to ILI or whether ILI affects long-term sleep behavior, the researchers noted.
“Several lines of evidence make me think that it’s more likely that insomnia makes one more vulnerable to influenza through pathways that involve decreased immune function,” study investigator Gary Garcia-Molina, PhD, with Sleep Number Labs, San Jose, Calif., said in an interview.
Sleep disorders, including insomnia, can dampen immune function and an individual’s ability to fight off illness, he noted.
The findings were presented at the annual meeting of the Associated Professional Sleep Societies.
Smart, connected devices
Pathophysiological responses to respiratory viral infection affect sleep duration and quality in addition to breathing function. “Smart” and “connected” devices that monitor biosignals over time have shown promise for monitoring infectious disease.
In an earlier study presented at SLEEP 2021, Dr. Garcia-Molina and colleagues found that real-world biometric data obtained from a smart bed can help predict and track symptoms of COVID-19 and other respiratory infections. They showed that worsening of COVID-19 symptoms correlated with an increase in sleep duration, breathing rate, and heart rate and a decrease in sleep quality.
In the new study, the researchers evaluated vulnerability to ILI in people with insomnia.
They quantified insomnia over time using the insomnia severity index (ISI). They quantified ILI vulnerability using an established artificial intelligence model they developed that estimates the daily probability of ILI symptoms from a Sleep Number smart bed using ballistocardiograph sensors.
Smart bed data – including daily and restful sleep duration, sleep latency, sleep quality, heart rate, breathing rate and motion level – were queried from 2019 (pre-COVID) and 2021.
A total of 1,680 smart sleepers had nearly constant ISI scores over the study period, with 249 having insomnia and 1,431 not having insomnia.
Data from both 2019 and 2021 show that smart sleepers with insomnia had significantly more and longer ILI episodes per year, compared with peers without insomnia.
For 2019, individuals without insomnia had 1.2 ILI episodes on average, which was significantly less (P < .01) than individuals with insomnia, at 1.5 episodes. The average ILI episode duration for those without insomnia was 4.3 days, which was significantly lower (P < .01) in those with insomnia group, at 6.1 days.
The data for 2021 show similar results, with the no-insomnia group having significantly fewer (P < .01) ILI episodes (about 1.2), compared with the insomnia group (about 1.5).
The average ILI episode duration for the no-insomnia group was 5 days, which was significantly less (P < .01) than the insomnia group, at 6.1 days.
The researchers said their study adds to other data on the relationship between sleep and overall health and well-being. It also highlights the potential health risk of insomnia and the importance of identifying and treating sleep disorders.
“Sleep has such a profound influence on health and wellness and the ability to capture these data unobtrusively in such an easy way and with such a large number of participants paves the way to investigate different aspects of health and disease,” Dr. Garcia-Molina said.
Rich data source
In a comment, Adam C. Powell, PhD, president of Payer+Provider Syndicate, a management advisory and operational consulting firm, said “smart beds provide a new data source for passively monitoring the health of individuals.”
“Unlike active monitoring methods requiring self-report, passive monitoring enables data to be captured without an individual taking any action. This data can be potentially integrated with data from other sources, such as pedometers, smart scales, and smart blood pressure cuffs, to gain a more holistic understanding of how an individual’s activities and behaviors impact their well-being,” said Dr. Powell, who wasn’t involved in the study.
There are some methodological limitations to the study, he noted.
“While the dependent variables examined were the duration and presence of episodes of influenza-like illness, they did not directly measure these episodes. Instead, they calculated the daily probability of influenza-like illness symptoms using a model that received input from the ballistocardiograph sensors in the smart beds,” Dr. Powell noted.
“The model used to calculate daily probability of influenza-like illness was created by examining associations between individuals’ smart-bed sensor data and population-level trends in influenza-like illness reported by the Centers for Disease Control and Prevention,” he explained.
Nonetheless, the findings are “consistent with the literature. It has been established by other researchers that impaired sleep is associated with greater risk of influenza, as well as other illnesses,” Dr. Powell said.
Funding for the study was provided by Sleep Number. Dr. Garcia-Molina and five coauthors are employed by Sleep Number. Dr. Powell reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM SLEEP 2023
Investing in the future of GI
This leads to promising investigators walking away from GI research frustrated by a lack of support. Investigators in the early stages of their careers are particularly hard hit.
Decades of research have revolutionized the care of many digestive disease patients. These patients, as well as everyone in the GI field – clinicians and researchers alike – have benefited from discoveries made by dedicated investigators, past and present.
Creative young researchers are poised to make groundbreaking discoveries that will shape the future of gastroenterology. Unfortunately, declining government funding for biomedical research puts this potential in jeopardy. We’re at risk of losing an entire generation of researchers if we don’t act now.
To fill this gap, the AGA Research Foundation invites you to support young investigators’ research careers, allowing them to make discoveries that could ultimately improve patient care and even cure diseases.
“We are at the threshold of key research advances that will cure digestive diseases. We have the manpower, we have trained the people, now we need to have the security that they can stay in research and advance these cures,” said Kim Elaine Barrett, PhD, AGAF, AGA legacy society donor and AGA governing board member.

By joining others in supporting the AGA Research Foundation, you will ensure that young researchers have opportunities to continue their life-saving work.
Learn more or make a contribution at www.foundation.gastro.org.
This leads to promising investigators walking away from GI research frustrated by a lack of support. Investigators in the early stages of their careers are particularly hard hit.
Decades of research have revolutionized the care of many digestive disease patients. These patients, as well as everyone in the GI field – clinicians and researchers alike – have benefited from discoveries made by dedicated investigators, past and present.
Creative young researchers are poised to make groundbreaking discoveries that will shape the future of gastroenterology. Unfortunately, declining government funding for biomedical research puts this potential in jeopardy. We’re at risk of losing an entire generation of researchers if we don’t act now.
To fill this gap, the AGA Research Foundation invites you to support young investigators’ research careers, allowing them to make discoveries that could ultimately improve patient care and even cure diseases.
“We are at the threshold of key research advances that will cure digestive diseases. We have the manpower, we have trained the people, now we need to have the security that they can stay in research and advance these cures,” said Kim Elaine Barrett, PhD, AGAF, AGA legacy society donor and AGA governing board member.
By joining others in supporting the AGA Research Foundation, you will ensure that young researchers have opportunities to continue their life-saving work.
Learn more or make a contribution at www.foundation.gastro.org.
This leads to promising investigators walking away from GI research frustrated by a lack of support. Investigators in the early stages of their careers are particularly hard hit.
Decades of research have revolutionized the care of many digestive disease patients. These patients, as well as everyone in the GI field – clinicians and researchers alike – have benefited from discoveries made by dedicated investigators, past and present.
Creative young researchers are poised to make groundbreaking discoveries that will shape the future of gastroenterology. Unfortunately, declining government funding for biomedical research puts this potential in jeopardy. We’re at risk of losing an entire generation of researchers if we don’t act now.
To fill this gap, the AGA Research Foundation invites you to support young investigators’ research careers, allowing them to make discoveries that could ultimately improve patient care and even cure diseases.
“We are at the threshold of key research advances that will cure digestive diseases. We have the manpower, we have trained the people, now we need to have the security that they can stay in research and advance these cures,” said Kim Elaine Barrett, PhD, AGAF, AGA legacy society donor and AGA governing board member.
By joining others in supporting the AGA Research Foundation, you will ensure that young researchers have opportunities to continue their life-saving work.
Learn more or make a contribution at www.foundation.gastro.org.
Long-term Remission of Pyoderma Gangrenosum, Acne, and Hidradenitis Suppurativa Syndrome
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.

Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.

After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.

The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.

Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.

PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.
Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.
After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.
The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.
Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.
PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.
Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.
After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.
The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.
Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.
PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
Practice Points
- Despite phenotypic similarities among pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (PASH) syndrome; pyogenic arthritis, PG, and acne syndrome; and pyogenic arthritis–PASH syndrome, there are genotypic differences that contribute to unique inflammatory cytokine patterns and the need for distinct pharmacologic considerations within each entity.
- When formulating therapeutic regimens for patients with PASH syndrome, it is essential for dermatologists to consider the likelihood of hyperactivity in multiple pathways of the innate immune system and utilize a combination of multimodal antiinflammatory therapies.

Trailblazer for women in gastroenterology, Dr. Barbara H. Jung takes over as AGA president
She currently serves as the first woman Robert G. Petersdorf professor and chair of internal medicine at the University of Washington, Seattle, and is the fourth woman to lead the American Gastroenterological Association as its president.
Dr. Jung is an international expert in the field of transforming growth factor–beta superfamily signaling in colon cancer and has made significant contributions at AGA prior to becoming president, most recently as a member of the finance and operations committee, chair-elect of the audit committee and vice chair of the AGA Research Foundation.

Born in Portland, Ore., and raised in Munich, Germany, Dr. Jung’s parents provided unconditional support for her career choice in medicine and nurtured her leadership skills throughout her childhood.
Her academic career began at Ludwig Maximilians University of Munich followed by postdoctoral studies in colon cancer at the Sidney Kimmel Cancer Center in San Diego and eventually culminating in an internal medicine residency at the University of California, San Diego.
Dr. Jung joined the AGA Governing Board in June 2021 as vice president and served as president-elect prior to assuming the top leadership role. Over her time as an AGA member (which started during fellowship), Dr. Jung has also served on the AGA Audit Committee, AGA Registry Research and Publications Committee, AGA Research Policy Committee, and AGA Innovation and Technology Task Force. In 2017, she co-organized the AGA Academic Skills Workshop to train the next generation of gastroenterologists.
She currently serves as the first woman Robert G. Petersdorf professor and chair of internal medicine at the University of Washington, Seattle, and is the fourth woman to lead the American Gastroenterological Association as its president.
Dr. Jung is an international expert in the field of transforming growth factor–beta superfamily signaling in colon cancer and has made significant contributions at AGA prior to becoming president, most recently as a member of the finance and operations committee, chair-elect of the audit committee and vice chair of the AGA Research Foundation.
Born in Portland, Ore., and raised in Munich, Germany, Dr. Jung’s parents provided unconditional support for her career choice in medicine and nurtured her leadership skills throughout her childhood.
Her academic career began at Ludwig Maximilians University of Munich followed by postdoctoral studies in colon cancer at the Sidney Kimmel Cancer Center in San Diego and eventually culminating in an internal medicine residency at the University of California, San Diego.
Dr. Jung joined the AGA Governing Board in June 2021 as vice president and served as president-elect prior to assuming the top leadership role. Over her time as an AGA member (which started during fellowship), Dr. Jung has also served on the AGA Audit Committee, AGA Registry Research and Publications Committee, AGA Research Policy Committee, and AGA Innovation and Technology Task Force. In 2017, she co-organized the AGA Academic Skills Workshop to train the next generation of gastroenterologists.
She currently serves as the first woman Robert G. Petersdorf professor and chair of internal medicine at the University of Washington, Seattle, and is the fourth woman to lead the American Gastroenterological Association as its president.
Dr. Jung is an international expert in the field of transforming growth factor–beta superfamily signaling in colon cancer and has made significant contributions at AGA prior to becoming president, most recently as a member of the finance and operations committee, chair-elect of the audit committee and vice chair of the AGA Research Foundation.
Born in Portland, Ore., and raised in Munich, Germany, Dr. Jung’s parents provided unconditional support for her career choice in medicine and nurtured her leadership skills throughout her childhood.
Her academic career began at Ludwig Maximilians University of Munich followed by postdoctoral studies in colon cancer at the Sidney Kimmel Cancer Center in San Diego and eventually culminating in an internal medicine residency at the University of California, San Diego.
Dr. Jung joined the AGA Governing Board in June 2021 as vice president and served as president-elect prior to assuming the top leadership role. Over her time as an AGA member (which started during fellowship), Dr. Jung has also served on the AGA Audit Committee, AGA Registry Research and Publications Committee, AGA Research Policy Committee, and AGA Innovation and Technology Task Force. In 2017, she co-organized the AGA Academic Skills Workshop to train the next generation of gastroenterologists.
What is new in hepatology in 2023?
CHICAGO – Two landmark phase 2 trials of the glucagon-like peptide 1 (GLP-1) receptor agonist, semaglutide, Food and Drug Administration–approved for type 2 diabetes or obesity, demonstrated improvements in nonalcoholic steatohepatitis (NASH) activity and resolution in stage 2 or 3 fibrosis (F2/F3), but not F4 fibrosis.1,2 The first randomized trial to compare the efficacy and safety of bariatric surgery with lifestyle intervention plus best medical care for histologically confirmed NASH demonstrated the superiority of bariatric surgery.3
There has been a paradigm shift in risk stratification of cirrhosis and targets for prevention and treatment of decompensation. A newer term, advanced chronic liver disease (ACLD), represents a shift away from needing a histologic or radiologic diagnosis of cirrhosis while reduction in portal pressure is a therapeutic goal. Evidence supports noninvasive liver stiffness measurement (LSM) using transient elastography to identify those with clinically significant portal hypertension (CSPH) (e.g., hepatic venous pressure gradient ≥ 10 mm Hg) who may benefit from therapy to lower portal pressure. Accordingly, based on landmark studies, the American Association for the Study of Liver Diseases (AASLD) recommends early utilization of nonselective beta-blocker therapy, with carvedilol as a preferred agent, in CSPH to decrease the risk of decompensation and mortality.4

The definition of hepatorenal syndrome has also substantially evolved.5 Terminology now acknowledges significant renal impairment at lower creatinine levels than previously used and recognizes the contribution of chronic kidney disease. The FDA approved terlipressin, a potent vasoconstrictor, for the treatment of hepatorenal syndrome with acute kidney injury (HRS-AKI, formerly HRS-Type I).

The AASLD published updates in advanced management of variceal bleeding highlighting intravascular interventions for esophageal, gastric, and ectopic varices. Data support early transjugular intrahepatic portosystemic shunt (TIPS) creation in patients with Child-Turcotte-Pugh (CTP) B or CTP C (< 14 points) within 72 hours of initial bleeding. However, hepatic encephalopathy remains a common complication. A randomized controlled trial demonstrated that rifaximin started 14 days prior to elective TIPS may reduce post-TIPS hepatic encephalopathy by 52%.6

Surveillance and treatment approaches for hepatocellular carcinoma (HCC) continue to evolve as reflected in new AASLD guidance in June 2023. Screening among high-risk patients should include ultrasound and serum alpha-fetoprotein every 6 months and novel screening approaches including imaging and blood-based biomarkers are on the horizon. There also have been significant advances in systemic therapies for HCC. First-line therapy for advanced HCC with CTP A cirrhosis now includes either atezolizumab/bevacizumab or durvalumab/tremelimumab. There is an evolving role for systemic therapy in the adjuvant setting as well, with emerging data supporting checkpoint inhibitors after resection or ablation among patients at high risk of recurrence.

Allocation policies continue to evolve for liver transplantation as waitlist prioritization transitions to MELD 3.0. The new score improves predictive accuracy for short-term mortality and addresses sex disparity in waitlist outcomes by lowering the prioritization of creatinine (and its threshold) and adding albumin to the current MELD-Na formula. MELD 3.0 will be adopted by the United Network for Organ Sharing in July 2023.
It has been a whirlwind 2023 for the liver community and we are excited and hopeful for new breakthroughs to come.
Dr. VanWagner is with the division of digestive and liver diseases, University of Texas Southwestern Medical Center; Dr. Serper is with the division of gastroenterology and hepatology, University of Pennsylvania; Dr. Goldberg is with the division of digestive health and liver diseases, University of Miami; and Dr. Verna is with the division of digestive and liver diseases, Columbia University. Dr. VanWagner is an adviser for Numares and Novo Nordisk and received a research grant from W.L. Gore & Associates. Dr. Serper disclosed research funding from Grifols. Dr. Verna disclosed research support from Salix. Dr. Goldberg had no disclosures. These remarks were made during one of the AGA Postgraduate Course sessions held at DDW 2023. DDW is sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA), the American Society for Gastrointestinal Endoscopy (ASGE) and The Society for Surgery of the Alimentary Tract (SSAT).
References
1. Loomba R et al. Semaglutide 2.4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:511-22.
2. Newsome PN et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113-24.
3. Verrastro O et al. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): A multicentre, open-label, randomised trial. Lancet 2023;401:1786-97.
4. Rowe IA et al. Quantifying the benefit of nonselective beta-blockers in the prevention of hepatic decompensation: A Bayesian reanalysis of the PREDESCI trial. Hepatology 2023 Mar 13. doi: 10.1097/HEP.0000000000000342.
5. Nadim MK and Garcia-Tsao G. Acute kidney injury in patients with cirrhosis. N Engl J Med. 2023;388:733-45.
6. Bureau C et al. The use of rifaximin in the prevention of overt hepatic encephalopathy after transjugular intrahepatic portosystemic shunt: A randomized controlled trial. Ann Intern Med. 2021;174:633-40.
CHICAGO – Two landmark phase 2 trials of the glucagon-like peptide 1 (GLP-1) receptor agonist, semaglutide, Food and Drug Administration–approved for type 2 diabetes or obesity, demonstrated improvements in nonalcoholic steatohepatitis (NASH) activity and resolution in stage 2 or 3 fibrosis (F2/F3), but not F4 fibrosis.1,2 The first randomized trial to compare the efficacy and safety of bariatric surgery with lifestyle intervention plus best medical care for histologically confirmed NASH demonstrated the superiority of bariatric surgery.3
There has been a paradigm shift in risk stratification of cirrhosis and targets for prevention and treatment of decompensation. A newer term, advanced chronic liver disease (ACLD), represents a shift away from needing a histologic or radiologic diagnosis of cirrhosis while reduction in portal pressure is a therapeutic goal. Evidence supports noninvasive liver stiffness measurement (LSM) using transient elastography to identify those with clinically significant portal hypertension (CSPH) (e.g., hepatic venous pressure gradient ≥ 10 mm Hg) who may benefit from therapy to lower portal pressure. Accordingly, based on landmark studies, the American Association for the Study of Liver Diseases (AASLD) recommends early utilization of nonselective beta-blocker therapy, with carvedilol as a preferred agent, in CSPH to decrease the risk of decompensation and mortality.4
The definition of hepatorenal syndrome has also substantially evolved.5 Terminology now acknowledges significant renal impairment at lower creatinine levels than previously used and recognizes the contribution of chronic kidney disease. The FDA approved terlipressin, a potent vasoconstrictor, for the treatment of hepatorenal syndrome with acute kidney injury (HRS-AKI, formerly HRS-Type I).
The AASLD published updates in advanced management of variceal bleeding highlighting intravascular interventions for esophageal, gastric, and ectopic varices. Data support early transjugular intrahepatic portosystemic shunt (TIPS) creation in patients with Child-Turcotte-Pugh (CTP) B or CTP C (< 14 points) within 72 hours of initial bleeding. However, hepatic encephalopathy remains a common complication. A randomized controlled trial demonstrated that rifaximin started 14 days prior to elective TIPS may reduce post-TIPS hepatic encephalopathy by 52%.6
Surveillance and treatment approaches for hepatocellular carcinoma (HCC) continue to evolve as reflected in new AASLD guidance in June 2023. Screening among high-risk patients should include ultrasound and serum alpha-fetoprotein every 6 months and novel screening approaches including imaging and blood-based biomarkers are on the horizon. There also have been significant advances in systemic therapies for HCC. First-line therapy for advanced HCC with CTP A cirrhosis now includes either atezolizumab/bevacizumab or durvalumab/tremelimumab. There is an evolving role for systemic therapy in the adjuvant setting as well, with emerging data supporting checkpoint inhibitors after resection or ablation among patients at high risk of recurrence.
Allocation policies continue to evolve for liver transplantation as waitlist prioritization transitions to MELD 3.0. The new score improves predictive accuracy for short-term mortality and addresses sex disparity in waitlist outcomes by lowering the prioritization of creatinine (and its threshold) and adding albumin to the current MELD-Na formula. MELD 3.0 will be adopted by the United Network for Organ Sharing in July 2023.
It has been a whirlwind 2023 for the liver community and we are excited and hopeful for new breakthroughs to come.
Dr. VanWagner is with the division of digestive and liver diseases, University of Texas Southwestern Medical Center; Dr. Serper is with the division of gastroenterology and hepatology, University of Pennsylvania; Dr. Goldberg is with the division of digestive health and liver diseases, University of Miami; and Dr. Verna is with the division of digestive and liver diseases, Columbia University. Dr. VanWagner is an adviser for Numares and Novo Nordisk and received a research grant from W.L. Gore & Associates. Dr. Serper disclosed research funding from Grifols. Dr. Verna disclosed research support from Salix. Dr. Goldberg had no disclosures. These remarks were made during one of the AGA Postgraduate Course sessions held at DDW 2023. DDW is sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA), the American Society for Gastrointestinal Endoscopy (ASGE) and The Society for Surgery of the Alimentary Tract (SSAT).
References
1. Loomba R et al. Semaglutide 2.4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:511-22.
2. Newsome PN et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113-24.
3. Verrastro O et al. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): A multicentre, open-label, randomised trial. Lancet 2023;401:1786-97.
4. Rowe IA et al. Quantifying the benefit of nonselective beta-blockers in the prevention of hepatic decompensation: A Bayesian reanalysis of the PREDESCI trial. Hepatology 2023 Mar 13. doi: 10.1097/HEP.0000000000000342.
5. Nadim MK and Garcia-Tsao G. Acute kidney injury in patients with cirrhosis. N Engl J Med. 2023;388:733-45.
6. Bureau C et al. The use of rifaximin in the prevention of overt hepatic encephalopathy after transjugular intrahepatic portosystemic shunt: A randomized controlled trial. Ann Intern Med. 2021;174:633-40.
CHICAGO – Two landmark phase 2 trials of the glucagon-like peptide 1 (GLP-1) receptor agonist, semaglutide, Food and Drug Administration–approved for type 2 diabetes or obesity, demonstrated improvements in nonalcoholic steatohepatitis (NASH) activity and resolution in stage 2 or 3 fibrosis (F2/F3), but not F4 fibrosis.1,2 The first randomized trial to compare the efficacy and safety of bariatric surgery with lifestyle intervention plus best medical care for histologically confirmed NASH demonstrated the superiority of bariatric surgery.3
There has been a paradigm shift in risk stratification of cirrhosis and targets for prevention and treatment of decompensation. A newer term, advanced chronic liver disease (ACLD), represents a shift away from needing a histologic or radiologic diagnosis of cirrhosis while reduction in portal pressure is a therapeutic goal. Evidence supports noninvasive liver stiffness measurement (LSM) using transient elastography to identify those with clinically significant portal hypertension (CSPH) (e.g., hepatic venous pressure gradient ≥ 10 mm Hg) who may benefit from therapy to lower portal pressure. Accordingly, based on landmark studies, the American Association for the Study of Liver Diseases (AASLD) recommends early utilization of nonselective beta-blocker therapy, with carvedilol as a preferred agent, in CSPH to decrease the risk of decompensation and mortality.4
The definition of hepatorenal syndrome has also substantially evolved.5 Terminology now acknowledges significant renal impairment at lower creatinine levels than previously used and recognizes the contribution of chronic kidney disease. The FDA approved terlipressin, a potent vasoconstrictor, for the treatment of hepatorenal syndrome with acute kidney injury (HRS-AKI, formerly HRS-Type I).
The AASLD published updates in advanced management of variceal bleeding highlighting intravascular interventions for esophageal, gastric, and ectopic varices. Data support early transjugular intrahepatic portosystemic shunt (TIPS) creation in patients with Child-Turcotte-Pugh (CTP) B or CTP C (< 14 points) within 72 hours of initial bleeding. However, hepatic encephalopathy remains a common complication. A randomized controlled trial demonstrated that rifaximin started 14 days prior to elective TIPS may reduce post-TIPS hepatic encephalopathy by 52%.6
Surveillance and treatment approaches for hepatocellular carcinoma (HCC) continue to evolve as reflected in new AASLD guidance in June 2023. Screening among high-risk patients should include ultrasound and serum alpha-fetoprotein every 6 months and novel screening approaches including imaging and blood-based biomarkers are on the horizon. There also have been significant advances in systemic therapies for HCC. First-line therapy for advanced HCC with CTP A cirrhosis now includes either atezolizumab/bevacizumab or durvalumab/tremelimumab. There is an evolving role for systemic therapy in the adjuvant setting as well, with emerging data supporting checkpoint inhibitors after resection or ablation among patients at high risk of recurrence.
Allocation policies continue to evolve for liver transplantation as waitlist prioritization transitions to MELD 3.0. The new score improves predictive accuracy for short-term mortality and addresses sex disparity in waitlist outcomes by lowering the prioritization of creatinine (and its threshold) and adding albumin to the current MELD-Na formula. MELD 3.0 will be adopted by the United Network for Organ Sharing in July 2023.
It has been a whirlwind 2023 for the liver community and we are excited and hopeful for new breakthroughs to come.
Dr. VanWagner is with the division of digestive and liver diseases, University of Texas Southwestern Medical Center; Dr. Serper is with the division of gastroenterology and hepatology, University of Pennsylvania; Dr. Goldberg is with the division of digestive health and liver diseases, University of Miami; and Dr. Verna is with the division of digestive and liver diseases, Columbia University. Dr. VanWagner is an adviser for Numares and Novo Nordisk and received a research grant from W.L. Gore & Associates. Dr. Serper disclosed research funding from Grifols. Dr. Verna disclosed research support from Salix. Dr. Goldberg had no disclosures. These remarks were made during one of the AGA Postgraduate Course sessions held at DDW 2023. DDW is sponsored by the American Association for the Study of Liver Diseases (AASLD), the American Gastroenterological Association (AGA), the American Society for Gastrointestinal Endoscopy (ASGE) and The Society for Surgery of the Alimentary Tract (SSAT).
References
1. Loomba R et al. Semaglutide 2.4 mg once weekly in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2023;8:511-22.
2. Newsome PN et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113-24.
3. Verrastro O et al. Bariatric-metabolic surgery versus lifestyle intervention plus best medical care in non-alcoholic steatohepatitis (BRAVES): A multicentre, open-label, randomised trial. Lancet 2023;401:1786-97.
4. Rowe IA et al. Quantifying the benefit of nonselective beta-blockers in the prevention of hepatic decompensation: A Bayesian reanalysis of the PREDESCI trial. Hepatology 2023 Mar 13. doi: 10.1097/HEP.0000000000000342.
5. Nadim MK and Garcia-Tsao G. Acute kidney injury in patients with cirrhosis. N Engl J Med. 2023;388:733-45.
6. Bureau C et al. The use of rifaximin in the prevention of overt hepatic encephalopathy after transjugular intrahepatic portosystemic shunt: A randomized controlled trial. Ann Intern Med. 2021;174:633-40.
AT DDW 2023
The evolving pulmonary landscape in HIV
Chronic pulmonary disease continues to be a major cause of morbidity and mortality in individuals living with the human immunodeficiency virus, even with optimal HIV control. And this is independent, as seen in many studies, of age, smoking, and pulmonary infections.
Both chronic pulmonary obstructive disease (COPD) and lung cancer occur more frequently in people living with HIV than in the general population, and at earlier ages, and with worse outcomes. The risk for emphysema and interstitial lung abnormalities also appears to be higher, research has shown. And asthma has also recently emerged as another important lung disease in people with HIV (PWH).

“There is evidence that the severity of immunocompromise associated with HIV infection is linked with chronic lung diseases. People who have a lower CD4 cell count or a higher viral load do have an increased risk of COPD and emphysema as well as potentially lung cancer. ,” said Kristina Crothers, MD, professor in the division of pulmonary, critical care, and sleep medicine at the University of Washington, Seattle.
Research has evolved from a focus on the epidemiology of HIV-related chronic lung diseases to a current emphasis on “trying to understand further the mechanisms [behind the heightened risk] through more benchwork and corollary translational studies, and then to the next level of trying to understand what this means for how we should manage people with HIV who have chronic lung diseases,” Dr. Crothers said. “Should management be tailored for people with HIV infection?”
Impairments in immune pathways, local and systemic inflammation, oxidative stress, dysbiosis, and accelerated cellular senescence are among potential mechanisms, but until ongoing mechanistic research yields more answers, pulmonologists should simply – but importantly – be aware of the increased risk and have a low threshold for investigating respiratory symptoms, she and other experts said in interviews. Referral of eligible patients for lung cancer screening is also a priority, as is smoking cessation, they said.
Notably, while spirometry has been the most commonly studied lung function measure in PWH, another noninvasive measure, diffusing capacity for carbon monoxide (DLCO), has garnered attention in the past decade and thus far appears to be the more frequent lung function abnormality.
In an analysis published in 2020 from the longitudinal Multicenter AIDS Cohort Study (MACS) – a study of a subcohort of 591 men with HIV and 476 without HIV – those with HIV were found to have a 1.6-fold increased risk of mild DLCO impairment (< 80% of predicted normal) and a 3-fold higher risk of more severe DLCO impairment (< 60% of predicted normal). There was no significant difference in spirometry findings by HIV status.
Such findings on DLCO are worthy of consideration in clinical practice, even in the absence of HIV-specific screening guidelines for noncommunicable lung diseases, Dr. Crothers said. “In thinking about screening and diagnosing chronic lung diseases in these patients, I’d not only consider spirometry, but also diffusing capacity” when possible, she said. Impaired DLCO is seen with emphysema and pulmonary vascular diseases like pulmonary hypertension and also interstitial lung diseases.
Key chronic lung diseases
Ken M. Kunisaki, MD, MS, associate professor of medicine at the University of Minnesota, Minneapolis, and the first author of the MACS analysis of lung function – one of the most recent and largest reports of DLCO impairment – points out that studies of chest computed tomography (CT) have also documented higher rates of emphysema and interstitial lung abnormalities.

A chest CT analysis from a cohort in Denmark (the Copenhagen Comorbidity in HIV Infection [COCOMO] cohort) found interstitial lung abnormalities in 10.9% of more than 700 PWH which represented a 1.8-fold increased risk compared to HIV-negative controls. And a study from an Italian sample of never-smoking PWH and controls reported emphysema in 18% and 4%, respectively. These studies, which did not measure DLCO, are among those discussed in a 2021 review by Dr. Kunisaki of advances in HIV-associated chronic lung disease research.

COPD is the best studied and most commonly encountered chronic lung disease in PWH. “Particularly for COPD, what’s both interesting and unfortunate is that we haven’t really seen any changes in the epidemiology with ART (antiretroviral therapy) – we’re still seeing the same findings, like the association of HIV with worse COPD at younger ages,” said Alison Morris, MD, MS, professor of medicine, immunology, and clinical and translational research at the University of Pittsburgh. “It doesn’t seem to have improved.”
Its prevalence has varied widely from cohort to cohort, from as low as 3% (similar to the general population) to over 40%, Dr. Kunisaki said, emphasizing that many studies, including studies showing higher rates, have controlled for current and past smoking. In evaluating patients with low or no smoking burden, “don’t discount respiratory symptoms as possibly reflecting underlying lung disease because COPD can develop with low to no smoking history in those with HIV,” he advised.
A better understanding of how a chronic viral infection like HIV leads to heightened COPD risk will not only help those with HIV, he notes, but also people without HIV who have COPD but have never smoked – a woefully underappreciated and understudied population. Ongoing research, he said, “should help us understand COPD pathogenesis generally.”
Research on asthma is relatively limited thus far, but it does appear that PWH may be more prone to developing severe asthma, just as with COPD, said Dr. Kunisaki, also a staff physician at the Minneapolis Veterans Administration Health Care System. Research has shown, for instance, that people with HIV more frequently needed aggressive respiratory support when hospitalized for asthma exacerbations.
It’s unclear how much of this potentially increased severity is attributable to the biology of HIV’s impact on the body and how much relates to social factors like disparities in income and access to care, Dr. Kunisaki said, noting that the same questions apply to the more frequent COPD exacerbations documented in PWH.
Dr. Crothers points out that, while most studies do not suggest a difference in the incidence of asthma in PWH, “there is some data from researchers looking at asthma profiles [suggesting] that the biomarkers associated with asthma may be different in people with and without HIV,” signaling potentially different molecular or biologic underpinnings of the disease.
Incidence rates of lung cancer in PWH, meanwhile, have declined over the last 2 decades, but lung cancer remains the leading cause of cancer-related mortality in PWH and occurs at a rate that is 2-2.5 times higher than that of individuals not infected with HIV, according to

Janice Leung, MD, of the division of respiratory medicine at the University of British Columbia and the Centre for Heart Lung Innovation at St. Paul’s Hospital in Vancouver.
Patients with HIV have “worse outcomes overall and a higher risk of mortality, even when presenting at the same stage,” said Dr. Leung, who reviewed trends in COPD and lung cancer in a recently published opinion piece.
Potential drivers
A bird’s eye view of potential – and likely interrelated – mechanisms for chronic lung disease includes chronic immune activation that impairs innate and adaptive immune pathways; chronic inflammation systemically and in the lung despite viral suppression; persistence of the virus in latent reservoirs in the lung, particularly in alveolar macrophages and T cells; HIV-related proteins contributing to oxidative stress; accelerated cellular aging; dysbiosis; and ongoing injury from inhaled toxins.
All are described in the literature and are being further explored. “It’s likely that multiple pathways are playing a role,” said Dr. Crothers, “and it could be that the balance of one to another leads to different manifestations of disease.”
Biomarkers that have been elevated and associated with different features of chronic lung disease – such as airflow obstruction, low DLCO, and emphysema – include markers of inflammation (e.g., C-reactive protein, interleukin-6), monocyte activation (e.g., soluble CD14), and markers of endothelial dysfunction, she noted in a 2021 commentary marking 40 years since the first reported cases of acquired immunodeficiency syndrome.
In her laboratory, Dr. Leung is using new epigenetic markers to look at the pathogenesis of accelerated aging in the lung. By profiling bronchial epithelial brushings for DNA methylation and gene expression, they have found that “people living with both HIV and COPD have the fastest epigenetic age acceleration in their airway epithelium,” she said. The findings “suggest that the HIV lung is aging faster.”
They reported their findings in 2022, describing methylation disruptions along age-related pathways such as cellular senescence, longevity regulation, and insulin signaling.
Dr. Leung and her team have also studied the lung microbiome and found lower microbial diversity in the airway epithelium in patients with HIV than those without, especially in those with HIV and COPD. The National Institutes of Health–sponsored Lung HIV Microbiome Project found that changes in the lung microbiome are most pronounced in patients who haven’t yet initiated ART, but research in her lab suggests ongoing suppression of microbial diversity even after ART, she said.
Dr. Morris is particularly interested in the oral microbiome, having found through her research that changes in the oral microbiome in PWH were more related to impaired lung function than alterations in the lung and gut microbiome. “That may be in part because of the way we measure things,” she said. “But we also think that the oral microbiome probably seeds the lung [through micro-aspiration].” A study published in 2020 from the Pittsburgh site of the MACS described alterations in oral microbial communities in PWH with abnormal lung function.
Preliminary research suggests that improved dental cleaning and periodontal work in PWH and COPD may influence the severity of COPD, she noted.
“We don’t see as much of a signal with the gut microbiome [and HIV status or lung function], though there could still be ways in which gut microbiome influences the lung,” through systemic inflammation, the release of metabolites into the bloodstream, or microbial translocation, for instance, she said.
The potential role of translocation of members of the microbiome, in fact, is an area of active research for Dr. Morris. Members of the microbiome – viruses and fungi in addition to bacteria – “can get into the bloodstream from the mouth, from the lung, from the gut, to stimulate inflammation and worsen lung disease,” she said.
Key questions in an evolving research landscape
Dr. Kunisaki looks forward to research providing a more longitudinal look at lung function decline– a move beyond a dominance of cross-sectional studies – as well as research that is more comprehensive, with simultaneous collection of various functional measures (eg., DLCO with chest imaging and fractional excretion of nitric oxide (FENO – a standardized breath measure of Th2 airway inflammation).
The several-year-old NIH-supported MACS/WIHS (Women’s Interagency HIV Study) Combined Cohort study, in which Dr. Kunisaki and Dr. Morris participate, aims in part to identity biomarkers of increased risk for chronic lung disease and other chronic disorders and to develop strategies for more effective interventions and treatments.
Researchers will also share biospecimens, “which will allow more mechanistic work,” Dr. Kunisaki noted. (The combined cohort study includes participants from the earlier, separate MACS and WIHS studies.)
Questions about treatment strategies include the risks versus benefits of inhaled corticosteroids, which may increase an already elevated risk of respiratory infections like bacterial pneumonia in PWH, Dr. Kunisaki said.
[An aside: Inhaled corticosteroids also have well-described interactions with ART regimens that contain CYP3A4 inhibitors (e.g., ritonavir and cobicistat) that can lead to hypercortisolism. In patients who require both types of drugs, he said, beclomethasone has the least interactions and is the preferred inhaled corticosteroid.]
For Dr. Crothers, unanswered critical questions include – as she wrote in her 2021 commentary – the question of how guidelines for the management of COPD and asthma should be adapted for PWH. Is COPD in PWH more or less responsive to inhaled corticosteroids, for instance? And are antifibrotic treatments for interstitial lung disease and immunotherapies for asthma or lung cancer similarly effective, and are there any increased risks for harms in people with HIV?
There’s also the question of whether PWH should be screened for lung cancer earlier and with a lower smoking exposure than is advised under current guidelines for the general population, she said in the interview. “And should the approach to shared decision-making be modified for people with HIV?” she said. “We’re doing some work on these questions” right now.
None of the researchers interviewed reported any conflicts of interest relevant to the story. Dr. Kunisaki reported that he has no relevant disclosures, and said that his comments are his personal views and not official views of the U.S. Government, Department of Veterans Affairs, the Minneapolis VA, or the University of Minnesota.
Chronic pulmonary disease continues to be a major cause of morbidity and mortality in individuals living with the human immunodeficiency virus, even with optimal HIV control. And this is independent, as seen in many studies, of age, smoking, and pulmonary infections.
Both chronic pulmonary obstructive disease (COPD) and lung cancer occur more frequently in people living with HIV than in the general population, and at earlier ages, and with worse outcomes. The risk for emphysema and interstitial lung abnormalities also appears to be higher, research has shown. And asthma has also recently emerged as another important lung disease in people with HIV (PWH).
“There is evidence that the severity of immunocompromise associated with HIV infection is linked with chronic lung diseases. People who have a lower CD4 cell count or a higher viral load do have an increased risk of COPD and emphysema as well as potentially lung cancer. ,” said Kristina Crothers, MD, professor in the division of pulmonary, critical care, and sleep medicine at the University of Washington, Seattle.
Research has evolved from a focus on the epidemiology of HIV-related chronic lung diseases to a current emphasis on “trying to understand further the mechanisms [behind the heightened risk] through more benchwork and corollary translational studies, and then to the next level of trying to understand what this means for how we should manage people with HIV who have chronic lung diseases,” Dr. Crothers said. “Should management be tailored for people with HIV infection?”
Impairments in immune pathways, local and systemic inflammation, oxidative stress, dysbiosis, and accelerated cellular senescence are among potential mechanisms, but until ongoing mechanistic research yields more answers, pulmonologists should simply – but importantly – be aware of the increased risk and have a low threshold for investigating respiratory symptoms, she and other experts said in interviews. Referral of eligible patients for lung cancer screening is also a priority, as is smoking cessation, they said.
Notably, while spirometry has been the most commonly studied lung function measure in PWH, another noninvasive measure, diffusing capacity for carbon monoxide (DLCO), has garnered attention in the past decade and thus far appears to be the more frequent lung function abnormality.
In an analysis published in 2020 from the longitudinal Multicenter AIDS Cohort Study (MACS) – a study of a subcohort of 591 men with HIV and 476 without HIV – those with HIV were found to have a 1.6-fold increased risk of mild DLCO impairment (< 80% of predicted normal) and a 3-fold higher risk of more severe DLCO impairment (< 60% of predicted normal). There was no significant difference in spirometry findings by HIV status.
Such findings on DLCO are worthy of consideration in clinical practice, even in the absence of HIV-specific screening guidelines for noncommunicable lung diseases, Dr. Crothers said. “In thinking about screening and diagnosing chronic lung diseases in these patients, I’d not only consider spirometry, but also diffusing capacity” when possible, she said. Impaired DLCO is seen with emphysema and pulmonary vascular diseases like pulmonary hypertension and also interstitial lung diseases.
Key chronic lung diseases
Ken M. Kunisaki, MD, MS, associate professor of medicine at the University of Minnesota, Minneapolis, and the first author of the MACS analysis of lung function – one of the most recent and largest reports of DLCO impairment – points out that studies of chest computed tomography (CT) have also documented higher rates of emphysema and interstitial lung abnormalities.
A chest CT analysis from a cohort in Denmark (the Copenhagen Comorbidity in HIV Infection [COCOMO] cohort) found interstitial lung abnormalities in 10.9% of more than 700 PWH which represented a 1.8-fold increased risk compared to HIV-negative controls. And a study from an Italian sample of never-smoking PWH and controls reported emphysema in 18% and 4%, respectively. These studies, which did not measure DLCO, are among those discussed in a 2021 review by Dr. Kunisaki of advances in HIV-associated chronic lung disease research.
COPD is the best studied and most commonly encountered chronic lung disease in PWH. “Particularly for COPD, what’s both interesting and unfortunate is that we haven’t really seen any changes in the epidemiology with ART (antiretroviral therapy) – we’re still seeing the same findings, like the association of HIV with worse COPD at younger ages,” said Alison Morris, MD, MS, professor of medicine, immunology, and clinical and translational research at the University of Pittsburgh. “It doesn’t seem to have improved.”
Its prevalence has varied widely from cohort to cohort, from as low as 3% (similar to the general population) to over 40%, Dr. Kunisaki said, emphasizing that many studies, including studies showing higher rates, have controlled for current and past smoking. In evaluating patients with low or no smoking burden, “don’t discount respiratory symptoms as possibly reflecting underlying lung disease because COPD can develop with low to no smoking history in those with HIV,” he advised.
A better understanding of how a chronic viral infection like HIV leads to heightened COPD risk will not only help those with HIV, he notes, but also people without HIV who have COPD but have never smoked – a woefully underappreciated and understudied population. Ongoing research, he said, “should help us understand COPD pathogenesis generally.”
Research on asthma is relatively limited thus far, but it does appear that PWH may be more prone to developing severe asthma, just as with COPD, said Dr. Kunisaki, also a staff physician at the Minneapolis Veterans Administration Health Care System. Research has shown, for instance, that people with HIV more frequently needed aggressive respiratory support when hospitalized for asthma exacerbations.
It’s unclear how much of this potentially increased severity is attributable to the biology of HIV’s impact on the body and how much relates to social factors like disparities in income and access to care, Dr. Kunisaki said, noting that the same questions apply to the more frequent COPD exacerbations documented in PWH.
Dr. Crothers points out that, while most studies do not suggest a difference in the incidence of asthma in PWH, “there is some data from researchers looking at asthma profiles [suggesting] that the biomarkers associated with asthma may be different in people with and without HIV,” signaling potentially different molecular or biologic underpinnings of the disease.
Incidence rates of lung cancer in PWH, meanwhile, have declined over the last 2 decades, but lung cancer remains the leading cause of cancer-related mortality in PWH and occurs at a rate that is 2-2.5 times higher than that of individuals not infected with HIV, according to
Janice Leung, MD, of the division of respiratory medicine at the University of British Columbia and the Centre for Heart Lung Innovation at St. Paul’s Hospital in Vancouver.
Patients with HIV have “worse outcomes overall and a higher risk of mortality, even when presenting at the same stage,” said Dr. Leung, who reviewed trends in COPD and lung cancer in a recently published opinion piece.
Potential drivers
A bird’s eye view of potential – and likely interrelated – mechanisms for chronic lung disease includes chronic immune activation that impairs innate and adaptive immune pathways; chronic inflammation systemically and in the lung despite viral suppression; persistence of the virus in latent reservoirs in the lung, particularly in alveolar macrophages and T cells; HIV-related proteins contributing to oxidative stress; accelerated cellular aging; dysbiosis; and ongoing injury from inhaled toxins.
All are described in the literature and are being further explored. “It’s likely that multiple pathways are playing a role,” said Dr. Crothers, “and it could be that the balance of one to another leads to different manifestations of disease.”
Biomarkers that have been elevated and associated with different features of chronic lung disease – such as airflow obstruction, low DLCO, and emphysema – include markers of inflammation (e.g., C-reactive protein, interleukin-6), monocyte activation (e.g., soluble CD14), and markers of endothelial dysfunction, she noted in a 2021 commentary marking 40 years since the first reported cases of acquired immunodeficiency syndrome.
In her laboratory, Dr. Leung is using new epigenetic markers to look at the pathogenesis of accelerated aging in the lung. By profiling bronchial epithelial brushings for DNA methylation and gene expression, they have found that “people living with both HIV and COPD have the fastest epigenetic age acceleration in their airway epithelium,” she said. The findings “suggest that the HIV lung is aging faster.”
They reported their findings in 2022, describing methylation disruptions along age-related pathways such as cellular senescence, longevity regulation, and insulin signaling.
Dr. Leung and her team have also studied the lung microbiome and found lower microbial diversity in the airway epithelium in patients with HIV than those without, especially in those with HIV and COPD. The National Institutes of Health–sponsored Lung HIV Microbiome Project found that changes in the lung microbiome are most pronounced in patients who haven’t yet initiated ART, but research in her lab suggests ongoing suppression of microbial diversity even after ART, she said.
Dr. Morris is particularly interested in the oral microbiome, having found through her research that changes in the oral microbiome in PWH were more related to impaired lung function than alterations in the lung and gut microbiome. “That may be in part because of the way we measure things,” she said. “But we also think that the oral microbiome probably seeds the lung [through micro-aspiration].” A study published in 2020 from the Pittsburgh site of the MACS described alterations in oral microbial communities in PWH with abnormal lung function.
Preliminary research suggests that improved dental cleaning and periodontal work in PWH and COPD may influence the severity of COPD, she noted.
“We don’t see as much of a signal with the gut microbiome [and HIV status or lung function], though there could still be ways in which gut microbiome influences the lung,” through systemic inflammation, the release of metabolites into the bloodstream, or microbial translocation, for instance, she said.
The potential role of translocation of members of the microbiome, in fact, is an area of active research for Dr. Morris. Members of the microbiome – viruses and fungi in addition to bacteria – “can get into the bloodstream from the mouth, from the lung, from the gut, to stimulate inflammation and worsen lung disease,” she said.
Key questions in an evolving research landscape
Dr. Kunisaki looks forward to research providing a more longitudinal look at lung function decline– a move beyond a dominance of cross-sectional studies – as well as research that is more comprehensive, with simultaneous collection of various functional measures (eg., DLCO with chest imaging and fractional excretion of nitric oxide (FENO – a standardized breath measure of Th2 airway inflammation).
The several-year-old NIH-supported MACS/WIHS (Women’s Interagency HIV Study) Combined Cohort study, in which Dr. Kunisaki and Dr. Morris participate, aims in part to identity biomarkers of increased risk for chronic lung disease and other chronic disorders and to develop strategies for more effective interventions and treatments.
Researchers will also share biospecimens, “which will allow more mechanistic work,” Dr. Kunisaki noted. (The combined cohort study includes participants from the earlier, separate MACS and WIHS studies.)
Questions about treatment strategies include the risks versus benefits of inhaled corticosteroids, which may increase an already elevated risk of respiratory infections like bacterial pneumonia in PWH, Dr. Kunisaki said.
[An aside: Inhaled corticosteroids also have well-described interactions with ART regimens that contain CYP3A4 inhibitors (e.g., ritonavir and cobicistat) that can lead to hypercortisolism. In patients who require both types of drugs, he said, beclomethasone has the least interactions and is the preferred inhaled corticosteroid.]
For Dr. Crothers, unanswered critical questions include – as she wrote in her 2021 commentary – the question of how guidelines for the management of COPD and asthma should be adapted for PWH. Is COPD in PWH more or less responsive to inhaled corticosteroids, for instance? And are antifibrotic treatments for interstitial lung disease and immunotherapies for asthma or lung cancer similarly effective, and are there any increased risks for harms in people with HIV?
There’s also the question of whether PWH should be screened for lung cancer earlier and with a lower smoking exposure than is advised under current guidelines for the general population, she said in the interview. “And should the approach to shared decision-making be modified for people with HIV?” she said. “We’re doing some work on these questions” right now.
None of the researchers interviewed reported any conflicts of interest relevant to the story. Dr. Kunisaki reported that he has no relevant disclosures, and said that his comments are his personal views and not official views of the U.S. Government, Department of Veterans Affairs, the Minneapolis VA, or the University of Minnesota.
Chronic pulmonary disease continues to be a major cause of morbidity and mortality in individuals living with the human immunodeficiency virus, even with optimal HIV control. And this is independent, as seen in many studies, of age, smoking, and pulmonary infections.
Both chronic pulmonary obstructive disease (COPD) and lung cancer occur more frequently in people living with HIV than in the general population, and at earlier ages, and with worse outcomes. The risk for emphysema and interstitial lung abnormalities also appears to be higher, research has shown. And asthma has also recently emerged as another important lung disease in people with HIV (PWH).
“There is evidence that the severity of immunocompromise associated with HIV infection is linked with chronic lung diseases. People who have a lower CD4 cell count or a higher viral load do have an increased risk of COPD and emphysema as well as potentially lung cancer. ,” said Kristina Crothers, MD, professor in the division of pulmonary, critical care, and sleep medicine at the University of Washington, Seattle.
Research has evolved from a focus on the epidemiology of HIV-related chronic lung diseases to a current emphasis on “trying to understand further the mechanisms [behind the heightened risk] through more benchwork and corollary translational studies, and then to the next level of trying to understand what this means for how we should manage people with HIV who have chronic lung diseases,” Dr. Crothers said. “Should management be tailored for people with HIV infection?”
Impairments in immune pathways, local and systemic inflammation, oxidative stress, dysbiosis, and accelerated cellular senescence are among potential mechanisms, but until ongoing mechanistic research yields more answers, pulmonologists should simply – but importantly – be aware of the increased risk and have a low threshold for investigating respiratory symptoms, she and other experts said in interviews. Referral of eligible patients for lung cancer screening is also a priority, as is smoking cessation, they said.
Notably, while spirometry has been the most commonly studied lung function measure in PWH, another noninvasive measure, diffusing capacity for carbon monoxide (DLCO), has garnered attention in the past decade and thus far appears to be the more frequent lung function abnormality.
In an analysis published in 2020 from the longitudinal Multicenter AIDS Cohort Study (MACS) – a study of a subcohort of 591 men with HIV and 476 without HIV – those with HIV were found to have a 1.6-fold increased risk of mild DLCO impairment (< 80% of predicted normal) and a 3-fold higher risk of more severe DLCO impairment (< 60% of predicted normal). There was no significant difference in spirometry findings by HIV status.
Such findings on DLCO are worthy of consideration in clinical practice, even in the absence of HIV-specific screening guidelines for noncommunicable lung diseases, Dr. Crothers said. “In thinking about screening and diagnosing chronic lung diseases in these patients, I’d not only consider spirometry, but also diffusing capacity” when possible, she said. Impaired DLCO is seen with emphysema and pulmonary vascular diseases like pulmonary hypertension and also interstitial lung diseases.
Key chronic lung diseases
Ken M. Kunisaki, MD, MS, associate professor of medicine at the University of Minnesota, Minneapolis, and the first author of the MACS analysis of lung function – one of the most recent and largest reports of DLCO impairment – points out that studies of chest computed tomography (CT) have also documented higher rates of emphysema and interstitial lung abnormalities.
A chest CT analysis from a cohort in Denmark (the Copenhagen Comorbidity in HIV Infection [COCOMO] cohort) found interstitial lung abnormalities in 10.9% of more than 700 PWH which represented a 1.8-fold increased risk compared to HIV-negative controls. And a study from an Italian sample of never-smoking PWH and controls reported emphysema in 18% and 4%, respectively. These studies, which did not measure DLCO, are among those discussed in a 2021 review by Dr. Kunisaki of advances in HIV-associated chronic lung disease research.
COPD is the best studied and most commonly encountered chronic lung disease in PWH. “Particularly for COPD, what’s both interesting and unfortunate is that we haven’t really seen any changes in the epidemiology with ART (antiretroviral therapy) – we’re still seeing the same findings, like the association of HIV with worse COPD at younger ages,” said Alison Morris, MD, MS, professor of medicine, immunology, and clinical and translational research at the University of Pittsburgh. “It doesn’t seem to have improved.”
Its prevalence has varied widely from cohort to cohort, from as low as 3% (similar to the general population) to over 40%, Dr. Kunisaki said, emphasizing that many studies, including studies showing higher rates, have controlled for current and past smoking. In evaluating patients with low or no smoking burden, “don’t discount respiratory symptoms as possibly reflecting underlying lung disease because COPD can develop with low to no smoking history in those with HIV,” he advised.
A better understanding of how a chronic viral infection like HIV leads to heightened COPD risk will not only help those with HIV, he notes, but also people without HIV who have COPD but have never smoked – a woefully underappreciated and understudied population. Ongoing research, he said, “should help us understand COPD pathogenesis generally.”
Research on asthma is relatively limited thus far, but it does appear that PWH may be more prone to developing severe asthma, just as with COPD, said Dr. Kunisaki, also a staff physician at the Minneapolis Veterans Administration Health Care System. Research has shown, for instance, that people with HIV more frequently needed aggressive respiratory support when hospitalized for asthma exacerbations.
It’s unclear how much of this potentially increased severity is attributable to the biology of HIV’s impact on the body and how much relates to social factors like disparities in income and access to care, Dr. Kunisaki said, noting that the same questions apply to the more frequent COPD exacerbations documented in PWH.
Dr. Crothers points out that, while most studies do not suggest a difference in the incidence of asthma in PWH, “there is some data from researchers looking at asthma profiles [suggesting] that the biomarkers associated with asthma may be different in people with and without HIV,” signaling potentially different molecular or biologic underpinnings of the disease.
Incidence rates of lung cancer in PWH, meanwhile, have declined over the last 2 decades, but lung cancer remains the leading cause of cancer-related mortality in PWH and occurs at a rate that is 2-2.5 times higher than that of individuals not infected with HIV, according to
Janice Leung, MD, of the division of respiratory medicine at the University of British Columbia and the Centre for Heart Lung Innovation at St. Paul’s Hospital in Vancouver.
Patients with HIV have “worse outcomes overall and a higher risk of mortality, even when presenting at the same stage,” said Dr. Leung, who reviewed trends in COPD and lung cancer in a recently published opinion piece.
Potential drivers
A bird’s eye view of potential – and likely interrelated – mechanisms for chronic lung disease includes chronic immune activation that impairs innate and adaptive immune pathways; chronic inflammation systemically and in the lung despite viral suppression; persistence of the virus in latent reservoirs in the lung, particularly in alveolar macrophages and T cells; HIV-related proteins contributing to oxidative stress; accelerated cellular aging; dysbiosis; and ongoing injury from inhaled toxins.
All are described in the literature and are being further explored. “It’s likely that multiple pathways are playing a role,” said Dr. Crothers, “and it could be that the balance of one to another leads to different manifestations of disease.”
Biomarkers that have been elevated and associated with different features of chronic lung disease – such as airflow obstruction, low DLCO, and emphysema – include markers of inflammation (e.g., C-reactive protein, interleukin-6), monocyte activation (e.g., soluble CD14), and markers of endothelial dysfunction, she noted in a 2021 commentary marking 40 years since the first reported cases of acquired immunodeficiency syndrome.
In her laboratory, Dr. Leung is using new epigenetic markers to look at the pathogenesis of accelerated aging in the lung. By profiling bronchial epithelial brushings for DNA methylation and gene expression, they have found that “people living with both HIV and COPD have the fastest epigenetic age acceleration in their airway epithelium,” she said. The findings “suggest that the HIV lung is aging faster.”
They reported their findings in 2022, describing methylation disruptions along age-related pathways such as cellular senescence, longevity regulation, and insulin signaling.
Dr. Leung and her team have also studied the lung microbiome and found lower microbial diversity in the airway epithelium in patients with HIV than those without, especially in those with HIV and COPD. The National Institutes of Health–sponsored Lung HIV Microbiome Project found that changes in the lung microbiome are most pronounced in patients who haven’t yet initiated ART, but research in her lab suggests ongoing suppression of microbial diversity even after ART, she said.
Dr. Morris is particularly interested in the oral microbiome, having found through her research that changes in the oral microbiome in PWH were more related to impaired lung function than alterations in the lung and gut microbiome. “That may be in part because of the way we measure things,” she said. “But we also think that the oral microbiome probably seeds the lung [through micro-aspiration].” A study published in 2020 from the Pittsburgh site of the MACS described alterations in oral microbial communities in PWH with abnormal lung function.
Preliminary research suggests that improved dental cleaning and periodontal work in PWH and COPD may influence the severity of COPD, she noted.
“We don’t see as much of a signal with the gut microbiome [and HIV status or lung function], though there could still be ways in which gut microbiome influences the lung,” through systemic inflammation, the release of metabolites into the bloodstream, or microbial translocation, for instance, she said.
The potential role of translocation of members of the microbiome, in fact, is an area of active research for Dr. Morris. Members of the microbiome – viruses and fungi in addition to bacteria – “can get into the bloodstream from the mouth, from the lung, from the gut, to stimulate inflammation and worsen lung disease,” she said.
Key questions in an evolving research landscape
Dr. Kunisaki looks forward to research providing a more longitudinal look at lung function decline– a move beyond a dominance of cross-sectional studies – as well as research that is more comprehensive, with simultaneous collection of various functional measures (eg., DLCO with chest imaging and fractional excretion of nitric oxide (FENO – a standardized breath measure of Th2 airway inflammation).
The several-year-old NIH-supported MACS/WIHS (Women’s Interagency HIV Study) Combined Cohort study, in which Dr. Kunisaki and Dr. Morris participate, aims in part to identity biomarkers of increased risk for chronic lung disease and other chronic disorders and to develop strategies for more effective interventions and treatments.
Researchers will also share biospecimens, “which will allow more mechanistic work,” Dr. Kunisaki noted. (The combined cohort study includes participants from the earlier, separate MACS and WIHS studies.)
Questions about treatment strategies include the risks versus benefits of inhaled corticosteroids, which may increase an already elevated risk of respiratory infections like bacterial pneumonia in PWH, Dr. Kunisaki said.
[An aside: Inhaled corticosteroids also have well-described interactions with ART regimens that contain CYP3A4 inhibitors (e.g., ritonavir and cobicistat) that can lead to hypercortisolism. In patients who require both types of drugs, he said, beclomethasone has the least interactions and is the preferred inhaled corticosteroid.]
For Dr. Crothers, unanswered critical questions include – as she wrote in her 2021 commentary – the question of how guidelines for the management of COPD and asthma should be adapted for PWH. Is COPD in PWH more or less responsive to inhaled corticosteroids, for instance? And are antifibrotic treatments for interstitial lung disease and immunotherapies for asthma or lung cancer similarly effective, and are there any increased risks for harms in people with HIV?
There’s also the question of whether PWH should be screened for lung cancer earlier and with a lower smoking exposure than is advised under current guidelines for the general population, she said in the interview. “And should the approach to shared decision-making be modified for people with HIV?” she said. “We’re doing some work on these questions” right now.
None of the researchers interviewed reported any conflicts of interest relevant to the story. Dr. Kunisaki reported that he has no relevant disclosures, and said that his comments are his personal views and not official views of the U.S. Government, Department of Veterans Affairs, the Minneapolis VA, or the University of Minnesota.