User login
Fine-Tuning the Discharge Process
The first metrics from SHM's Project BOOST mentorship program won't be ready until later this year, but the recent addition of more intervention sites comes as pilot institutions are reporting success in changing the discharge culture.
SHM recently announced 24 new sites for Project BOOST (Better Outcomes for Older Adults through Safe Transitions), bringing the number of participating institutions to 30. Each site features SHM mentors working with hospitalists to improve transitional care via a discharge planning toolkit.
Emmanuel King, MD, director of the Nurse Practitioner Hospitalist Service at the Hospital of the University of Pennsylvania in Philadelphia, says a major shift is implementing the "7P Risk Scale," a transitional-care checklist. Dr. King says some of his staff initially balked at depression screening and questions about health literacy, but when the tools were introduced and the checklist items were embraced, hospitalists felt "included in and comfortable with the process."
"Tweaking it to meet the needs of the team was a great idea," says Dr. King, assistant professor of clinical at UPenn's School of Medicine. "We've been able to get the team to buy in."
Tina Budnitz, MPH, SHM senior advisor for quality initiatives, says some early responses to Project BOOST have been better than expected, especially in the area of follow-up tasks.
"I was expecting people to say they were incredibly time-intensive," Budnitz says. "Some of the hospitalists got back to us and said, 'We think it's a good idea to call every patient, regardless of their risk status.' "
The first metrics from SHM's Project BOOST mentorship program won't be ready until later this year, but the recent addition of more intervention sites comes as pilot institutions are reporting success in changing the discharge culture.
SHM recently announced 24 new sites for Project BOOST (Better Outcomes for Older Adults through Safe Transitions), bringing the number of participating institutions to 30. Each site features SHM mentors working with hospitalists to improve transitional care via a discharge planning toolkit.
Emmanuel King, MD, director of the Nurse Practitioner Hospitalist Service at the Hospital of the University of Pennsylvania in Philadelphia, says a major shift is implementing the "7P Risk Scale," a transitional-care checklist. Dr. King says some of his staff initially balked at depression screening and questions about health literacy, but when the tools were introduced and the checklist items were embraced, hospitalists felt "included in and comfortable with the process."
"Tweaking it to meet the needs of the team was a great idea," says Dr. King, assistant professor of clinical at UPenn's School of Medicine. "We've been able to get the team to buy in."
Tina Budnitz, MPH, SHM senior advisor for quality initiatives, says some early responses to Project BOOST have been better than expected, especially in the area of follow-up tasks.
"I was expecting people to say they were incredibly time-intensive," Budnitz says. "Some of the hospitalists got back to us and said, 'We think it's a good idea to call every patient, regardless of their risk status.' "
The first metrics from SHM's Project BOOST mentorship program won't be ready until later this year, but the recent addition of more intervention sites comes as pilot institutions are reporting success in changing the discharge culture.
SHM recently announced 24 new sites for Project BOOST (Better Outcomes for Older Adults through Safe Transitions), bringing the number of participating institutions to 30. Each site features SHM mentors working with hospitalists to improve transitional care via a discharge planning toolkit.
Emmanuel King, MD, director of the Nurse Practitioner Hospitalist Service at the Hospital of the University of Pennsylvania in Philadelphia, says a major shift is implementing the "7P Risk Scale," a transitional-care checklist. Dr. King says some of his staff initially balked at depression screening and questions about health literacy, but when the tools were introduced and the checklist items were embraced, hospitalists felt "included in and comfortable with the process."
"Tweaking it to meet the needs of the team was a great idea," says Dr. King, assistant professor of clinical at UPenn's School of Medicine. "We've been able to get the team to buy in."
Tina Budnitz, MPH, SHM senior advisor for quality initiatives, says some early responses to Project BOOST have been better than expected, especially in the area of follow-up tasks.
"I was expecting people to say they were incredibly time-intensive," Budnitz says. "Some of the hospitalists got back to us and said, 'We think it's a good idea to call every patient, regardless of their risk status.' "
HM Spreads Its Wings
Hospitalists are not just general internists anymore, having successfully branched out into such subspecialties as cardiology, pulmonology, and gastroenterology, according to a March 12 study in the New England Journal of Medicine (2009;360:1102-12).
In the first quantitative national review to study hospitalists based on Medicare payment data, a team of researchers at the University of Texas Medical Branch at Galveston calculated that the percentage of internal medicine physicians practicing as hospitalists jumped to 19% in 2006 from 5.9% in 1995.
Perhaps more interesting is the number of cardiologists, pulmonologists, gastroenterologists, family physicians, and general practitioners who work as hospitalists totaled roughly 20% in 2006. The study defined hospitalists as those who generated more than 90% of their E/M claims from hospitalized patients.
HM appears to have even more room to grow, as more physicians move toward the HM model and away from primary care, according to an editorial accompanying the NEJM study. The editorial debated the value-adds and the complications caused by the presence of hospitalists in all phases of the care continuum. The authors also acknowledged the model is widely accepted as beneficial.
"The economic and practical forces that promoted the growth in the care of patients by hospitalists are intensifying, not lessening, and hospitalists are here to stay," according to the editorial, written by a trio of NEJM editors, including editor-in-chief Jeffrey M. Drazen, MD. "It is time to focus on how to enhance the value."
Hospitalists are not just general internists anymore, having successfully branched out into such subspecialties as cardiology, pulmonology, and gastroenterology, according to a March 12 study in the New England Journal of Medicine (2009;360:1102-12).
In the first quantitative national review to study hospitalists based on Medicare payment data, a team of researchers at the University of Texas Medical Branch at Galveston calculated that the percentage of internal medicine physicians practicing as hospitalists jumped to 19% in 2006 from 5.9% in 1995.
Perhaps more interesting is the number of cardiologists, pulmonologists, gastroenterologists, family physicians, and general practitioners who work as hospitalists totaled roughly 20% in 2006. The study defined hospitalists as those who generated more than 90% of their E/M claims from hospitalized patients.
HM appears to have even more room to grow, as more physicians move toward the HM model and away from primary care, according to an editorial accompanying the NEJM study. The editorial debated the value-adds and the complications caused by the presence of hospitalists in all phases of the care continuum. The authors also acknowledged the model is widely accepted as beneficial.
"The economic and practical forces that promoted the growth in the care of patients by hospitalists are intensifying, not lessening, and hospitalists are here to stay," according to the editorial, written by a trio of NEJM editors, including editor-in-chief Jeffrey M. Drazen, MD. "It is time to focus on how to enhance the value."
Hospitalists are not just general internists anymore, having successfully branched out into such subspecialties as cardiology, pulmonology, and gastroenterology, according to a March 12 study in the New England Journal of Medicine (2009;360:1102-12).
In the first quantitative national review to study hospitalists based on Medicare payment data, a team of researchers at the University of Texas Medical Branch at Galveston calculated that the percentage of internal medicine physicians practicing as hospitalists jumped to 19% in 2006 from 5.9% in 1995.
Perhaps more interesting is the number of cardiologists, pulmonologists, gastroenterologists, family physicians, and general practitioners who work as hospitalists totaled roughly 20% in 2006. The study defined hospitalists as those who generated more than 90% of their E/M claims from hospitalized patients.
HM appears to have even more room to grow, as more physicians move toward the HM model and away from primary care, according to an editorial accompanying the NEJM study. The editorial debated the value-adds and the complications caused by the presence of hospitalists in all phases of the care continuum. The authors also acknowledged the model is widely accepted as beneficial.
"The economic and practical forces that promoted the growth in the care of patients by hospitalists are intensifying, not lessening, and hospitalists are here to stay," according to the editorial, written by a trio of NEJM editors, including editor-in-chief Jeffrey M. Drazen, MD. "It is time to focus on how to enhance the value."
Interhospital Transfer of Children
Interhospital transfer of critically ill and injured children is necessitated by variation in resource availability between hospitals. Critically ill children judged in need of clinical services or expertise not locally available undergo transfer to hospitals with more appropriate resource capabilities and expertise, with the expectation that clinical outcomes of transfer will be better than nontransfer.
Significant variation both in the availability of pediatric critical care services across US hospitals1 and in child mortality among hospitals without pediatric critical care services2 suggests that interhospital transfer will remain an integral part of healthcare delivery for critically ill and injured children. Timely provision of definitive care for acute life‐threatening disease is associated with good clinical outcomes.3, 4 While prior studies have examined clinical outcomes and resource consumption among critically ill adults who underwent interhospital transfer for intensive care,59 there is scarce information regarding clinical characteristics and outcomes of interhospital transfer for critically ill and injured children.
This study was conducted to test the hypothesis that, among critically ill and injured children who undergo interhospital transfer for intensive care, children transferred after an initial hospitalization at the referring facility will have higher mortality, longer length of stay (LOS), and higher resource consumption than children transferred directly from the emergency department (ED) of the referring hospitals.
METHODS
Study Design
We conducted a secondary analysis of administrative claims data from the Michigan Medicaid program for the period January 1, 2002, to December 31, 2004. The data included all paid claims for health services rendered to enrollees in the Medicaid program. The Institutional Review Board of the University of Michigan Medical School approved the study.
Study Sample and Variable Identification
A 3‐step approach was employed to identify interhospital transfer admissions for intensive care of children. Initially, the Medicaid claims were queried to identify all hospitalizations for children 018 years who received intensive care services, using Medicare revenue codes.10 Admissions for neonatal intensive care were excluded from the analysis. The American Hospital Association Guide to the Health Care Field, a compendium of US healthcare facilities, was used to verify the presence of intensive care facilities.11, 12 Subsequently, to identify the subset of children who underwent interhospital transfer, data were queried for the presence of claims from another hospital, and the date of discharge from the referring hospital had to be the same as the date of admission to the receiving hospital intensive care unit (ICU). Finally, to ascertain the source of interhospital transfer, Medicare revenue codes and current procedural terminology (CPT) codes were used to identify claims for receipt of services at specific sites within the referring hospital; namely, the ED, ward, or the ICU. This information was used to categorize admissions into 1 of 3 pathways of interhospital transfer:
ED transferFrom the ED of the referring hospital to the ICU of the receiving hospital.
Ward transferFrom the wards of the referring hospital to the ICU of the receiving hospital.
Inter‐ICU transferFrom the ICU of the referring hospital to the ICU of the receiving hospital.
Dependent Variables
Mortality at the Receiving Hospital
This is determined by linkage to vital statistics records maintained by the Michigan Department of Community Health, Division of Vital Records and Health Statistics.
LOS at the Receiving Hospital
This is determined as the count of days of hospitalization at the receiving hospital. Of note, this includes ICU days and non‐ICU days at the receiving hospital.
Independent Variables
Source of Interhospital Transfer
The main (exposure) independent variable. Categorized into ED, ward, or inter‐ICU transfers, as described.
Patient Demographics
Age and gender.
Comorbid Illness
Determined using International Classification of Diseases, ninth revision (ICD‐9) diagnosis codes, applying methodology as described.13
Organ Dysfunction at the Referring and Receiving Hospitals
Determined using ICD‐9 diagnosis codes, applying methodology as described.14
Patient Diagnostic Categories
Eleven diagnostic categories were created based on primary admission diagnoses (Appendix A).
LOS at the Referring Hospital
Determined as the count of days of hospitalization at the referring hospital.
Receipt of Cardiopulmonary Resuscitation (CPR) on the Date of Interhospital Transfer
Determined using procedure codes.
Receipt of Medical‐Surgical Procedures at the Receiving Hospital
Identified through the use of ICD‐9 procedure codes, CPT codes, and Healthcare Common Procedure Coding System codes. The procedures are listed in Appendix B.
Statistical Analysis
Descriptive statistics were used to characterize the study sample. According to the 3 sources of interhospital transfer, patient characteristics (age, gender, presence of organ dysfunction, and comorbid illness), median LOS at the referring hospital, and receipt of CPR on the date of interhospital transfer were compared using chi‐square tests for categorical variables, and Kruskal‐Wallis tests for continuous variables. Similarly, outcome variables of in‐hospital mortality and median LOS at the receiving hospital were compared across the 3 sources of interhospital transfer. Analysis of variance was used to compare mean LOS at the receiving hospital across the 3 sources of interhospital transfer. Median (with interquartile range [IQR]) and mean (with standard deviation [SD]) values are presented to describe LOS, given skew in LOS data.
To account for potential confounding of LOS and mortality at the receiving hospital by the presence of organ dysfunction and comorbid illness1316 at the referring hospital, multivariate logistic regression and multiple linear regression analyses were conducted to estimate the odds of in‐hospital mortality and the incremental LOS, respectively, for ward and inter‐ICU transfers, compared with ED transfers. Statistical analyses were conducted using Stata 8 for windows (Stata Corporation, College Station, TX). A 2‐tailed level of 0.05 was used as the threshold for statistical significance.
RESULTS
Patient Characteristics
Of 1,643 transfer admissions for intensive care during the study period, 1022 (62%) were ED transfers, 512 (31%) were ward transfers, and 109 (7%) were inter‐ICU transfers. The average age was 2 years, with male gender (57%) predominance. Comorbid illness was present in 19% of admissions, while 11% had evidence of organ dysfunction at the referring hospital. Table 1 presents key patient demographic and clinical characteristics at the referring hospitals, by transfer source. Inter‐ICU and ward transfers were younger than ED transfers, and had a higher preponderance of comorbid illness and organ dysfunction. At the time of interhospital transfer, compared with ED transfers, the proportion of admissions with organ dysfunction (a marker of illness severity) was 3‐fold and 8‐fold higher among ward and inter‐ICU transfers, respectively.
| Transfer Source | P | |||
|---|---|---|---|---|
| Characteristics | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | |
| ||||
| Median age in years (IQR) | 2 (09) | 1 (07) | 1 (010) | <0.01 |
| Male (%) | 57.8 | 56.2 | 47.6 | 0.13 |
| Comorbid illness (% ) | 13.1 | 25.0 | 50.5 | <0.01 |
| Pretransfer hospital length of stay (days) | ||||
| Median (IQR) | 0 | 1 (02) | 3 (18) | <0.01 |
| Mean (SD) | 0.2 (5.2) | 1.6 (4.8) | 9.7 (18.0) | <0.01 |
| Pretransfer organ dysfunction (%) | 5.5 | 14.5 | 40.4 | <0.01 |
Patterns of Transfer
The leading diagnoses among all children were respiratory disease, trauma, and neurological disease (Table 2), with some variation in diagnoses by source of interhospital transfer. For example, cardiovascular disease was the second leading diagnosis after respiratory disease among the inter‐ICU transfers, while more children with endocrine disease (predominantly diabetic ketoacidosis), traumatic injury, or drug poisoning were transferred directly from the ED, than from the ward or the ICU settings. For burn care, 80% (45/56) of all transfer admissions were direct from the ED (Table 3). The majority (78%) of children with traumatic injuries were directly transferred from the ED to the ICU, while the remainder were transferred after initial care delivered on the ward (18%) or ICU (4%) settings prior to interhospital transfer for definitive intensive trauma care. Importantly, among the inter‐ICU transfers, 104 (95%) were transferred to pediatric ICUs from referring hospitals with adult and pediatric ICU facilities, suggesting uptransfer for specialized care. Five children were transferred between hospitals with adult ICU facilities.
| Transfer Source | ||||
|---|---|---|---|---|
| Diagnostic Category (%) | Overall* (n = 1639) | ED* (n = 1018) | Ward (n = 512) | Inter‐ICU (n = 109) |
| ||||
| Respiratory disease | 35.1 | 32.8 | 41.0 | 28.4 |
| Trauma | 16.2 | 20.5 | 9.2 | 9.1 |
| Neurological disease | 12.4 | 12.5 | 12.3 | 11.9 |
| Gastrointestinal disease | 6.7 | 5.4 | 7.4 | 11.9 |
| Infectious disease | 5.8 | 4.0 | 8.4 | 10.0 |
| Endocrine disease | 5.5 | 7.9 | 1.8 | 0 |
| Drug overdose/poisoning | 5.0 | 6.4 | 2.9 | 1.8 |
| Cardiovascular disease | 4.8 | 2.8 | 6.3 | 16.5 |
| Hematologic/oncologic disease | 2.0 | 1.6 | 2.9 | 1.8 |
| Cardiac arrest | 0.2 | 0 | 0.6 | 0.9 |
| Other diagnoses | 6.2 | 5.4 | 7.2 | 7.7 |
| Transfer Source | |||||
|---|---|---|---|---|---|
| Characteristics (%) | Overall (n = 1643) | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | P |
| Respiratory | 26.8 | 19.0 | 36.7 | 54.1 | <0.01 |
| Radiological | 21.2 | 19.5 | 20.5 | 41.3 | <0.01 |
| Vascular access | 20.0 | 15.2 | 27.0 | 33.0 | <0.01 |
| Gastrointestinal | 3.9 | 3.0 | 3.7 | 12.8 | <0.01 |
| Neurological | 3.8 | 3.2 | 3.7 | 10.1 | <0.01 |
| Cardiovascular | 3.6 | 1.8 | 4.1 | 18.4 | <0.01 |
| Burn care | 3.4 | 4.5 | 2.0 | 0 | <0.01 |
| General surgery | 3.2 | 2.1 | 4.3 | 8.3 | <0.01 |
| Dialysis | 2.6 | 2.0 | 2.5 | 8.3 | <0.01 |
| ECMO | 2.1 | 1.3 | 2.2 | 9.2 | <0.01 |
CPR was performed on the date of interhospital transfer for 23 patients (1.4% of the sample), of whom 13 (56.5%) were ward transfers, 8 (34.8%) were inter‐ICU transfers, and 2 (8.7%) were ED transfers (P < 0.02). Two‐thirds of these children did not survive subsequent hospitalization at the receiving hospitals.
Clinical Outcomes and Resource Utilization at the Receiving Hospitals
At the receiving hospitals, other than burn care, medical‐surgical procedures were performed most often among the inter‐ICU transfers. Ward transfers also had higher receipt of procedures compared with ED transfers (Table 3). The inter‐ICU and ward transfers had a higher preponderance of organ dysfunction at the receiving hospitals, compared to the ED transfers (38.5% and 29.3% versus 20.8%, P < 0.01).
Clinical outcomes at the receiving hospitals varied significantly according to the source of interhospital transfer (Table 4). Sixty‐six (4%) of patients died at the receiving hospitals. In comparison with ED transfers, unadjusted in‐hospital mortality was 2‐fold and 3‐fold higher among the ward and inter‐ICU transfers, respectively. Also, hospital LOS was significantly longer among the ward and inter‐ICU transfers than for the ED transfers.
| Transfer Source | ||||
|---|---|---|---|---|
| Characteristics | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | P |
| ||||
| Mortality (%) | 2.8 | 5.5 | 8.3 | <0.01 |
| Length of stay (days) | ||||
| Median (IQR) | 3 (27) | 5 (312) | 13 (724) | <0.01 |
| Mean (SD) | 6.7 (10.4) | 8.5 (9.2) | 21.4 (22.9) | <0.01 |
In multivariate analyses adjusting for patient age, and the presence of comorbid illness and organ dysfunction at the referring hospital, compared with ED transfers, odds of mortality were significantly higher (odds ratio [OR], 1.76; 95% confidence interval [CI], 1.023.03) for ward transfers. Inter‐ICU transfers also had higher odds of mortality (OR, 2.07; 95% CI, 0.884.86), without achieving statistical significance. Similarly, compared with ED transfers, LOS at the receiving hospital was longer by 1.5 days (95% CI, 0.32.7 days) for ward transfers, and by 13.5 days (95% CI, 11.115.8 days) for inter‐ICU transfers.
DISCUSSION
This study is the first to highlight significant variation in clinical outcomes and resource consumption after interhospital transfer of critically ill and injured children, depending on the source of transfer. In comparison with children transferred directly from the referring hospitals' ED settings, children transferred from the referring hospitals' wards had higher mortality, while those who underwent inter‐ICU transfer had significantly higher resource consumption. In addition, ward transfers had the highest proportion of children who underwent CPR on the date of interhospital transfer, highlighting elevated severity of disease prior to transfer and an urgent need for improved understanding of pretransfer clinical care and medical decision‐making. The findings raise the possibility that more timely transfer of some patients directly from community hospital EDs to regional ICUs might improve survival and reduce resource consumption.
Although interhospital transfers are common in everyday clinical practice, there has been a knowledge gap in pediatric acute and critical care medicine regarding the clinical outcomes and resource consumption among children who undergo such transfers. Findings from the current study narrow this gap by relating triage at the referring hospitals to clinical outcomes and resource utilization at the receiving hospitals.
Certain distinct transfer patterns were observed. Most children with burn injury underwent direct transfer from the ED to the ICU; this transfer pattern may be related both to the limited availability of ICUs with burn care capability in Michigan and to the acuity of burn injuries, which often mandates immediate triage to hospitals with intensive burn care facilities. Conversely, while the majority of children with traumatic injuries were directly transferred from emergency to intensive care, over one‐fifth were transferred after initial care delivered on the ward or ICU settings prior to interhospital transfer for definitive intensive trauma care. Such imperfect regionalization of trauma care suggests further study of clinical outcomes and resource utilization among injured children is warranted. Likewise, cardiovascular disease was prominent among the inter‐ICU transfers, suggesting a clinical practice pattern of stabilization and resuscitation at the initial ICU prior to interhospital vertical or uptransfer for definitive cardiac care at the receiving hospitals.
It remains unknown whether the timing of interhospital transfer of critically ill children is a determinant of clinical outcomes. Prior studies among adults have reported higher mortality with prolonged duration of pre‐ICU care on the ward.4, 17 In the current study, ward and inter‐ICU transfers were initially hospitalized for a median of 1 and 3 days, respectively, prior to transfer. While we could not determine from administrative data what the precise triggers for interhospital transfer in this study were, it is instructive to note that ward transfers comprised more than one‐half of all children who received CPR on the date of transfer. For children who received CPR, severe clinical deterioration likely triggered transfer to hospitals with ICU facilities, but because only a minority of children received CPR overall, other triggers of transfer warrant investigation. For most of the children transferred, it seems plausible that the precipitant of transfer was likely a mismatch of their clinical status with the clinical capacities of the facilities where they were initially hospitalized. Future work should investigate if there is an association between clinical outcomes at the receiving hospitals, and both the timing of interhospital transfer and the clinical status of patients at transfer.
Importantly, compared with ED transfers, ward transfers demonstrated elevated odds of mortality after adjustment for coexisting comorbid illness, patient age, and pretransfer organ dysfunction at the referring hospital. Some possible explanations for this finding include the progression of disease while receiving care on the ward, or suboptimal access to ICU facilities due to barriers to transfer at either the referring or receiving hospitals. Importantly, progression of disease in ward settings may be detected by early identification of children at high risk of clinical deterioration on the wards of hospitals without ICU facilities, prior to cardiopulmonary arrest, because death after CPR may not be averted with subsequent ICU care.18
Various approaches to facilitate rapid and appropriate triage and reassessment of children in hospitals without ICU facilities, prior to severe clinical deterioration or need for CPR, must be investigated. These approaches might include in‐hospital measures such as the establishment of medical emergency teams to respond to clinical deterioration on the wards19 or collaborative interhospital measures such as the use of telemedicine20 or similar remote communication/triage systems to enhance communication between clinical caregivers at hospitals with ICU facilities and those in hospitals without ICU facilities. Furthermore, interhospital transfer agreements may facilitate expeditious and appropriate transfer of severely ill patients to hospitals with ICU facilities.
Access to hospitals with ICU facilities might also influence outcomes for critically ill children admitted initially to wards of hospitals without ICU facilities. Kanter2 reported significant variation in mortality among children who received care at New York hospitals without ICU facilities. Of note, 27% of statewide pediatric inpatient deaths occurred in those hospitals without ICU facilities. It appeared that, while some pediatric deaths in hospitals without ICU facilities were expected, regional variation in such mortality might have been associated with reduced access to, or poor utilization of, hospitals with ICU facilities. Barriers to interhospital transfers might include underrecognition of mismatch between patient illness severity and hospital capability at referring hospitals, or lack of capacity to accept transfers at the receiving hospitals. Further study is warranted to investigate clinical decision‐making underlying the initiation of the interhospital transfer processes, and procedural or institutional barriers that might hinder the transfer of critically ill children from hospitals without ICU facilities.
Resource consumption at the receiving hospitals, measured by hospital LOS and receipt of medical‐surgical procedures, was highest among the inter‐ICU transfers. This was an expected finding, given the high frequency of organ dysfunction among the inter‐ICU transfers, before and after interhospital transfer. These patients had the highest use of advanced and resource‐intensive technology, including continuous renal replacement therapy, extracorporeal membrane oxygenation, and cardiovascular procedures such as open‐heart surgery. In addition, the duration of hospitalization at the receiving hospital was 2 weeks longer among the inter‐ICU transfers when compared with the ED transfers. Such prolonged hospitalization has been previously associated with significantly increased resource consumption.4, 6 In the absence of physiologic data pertaining to illness severity, however, it is unknown whether this observed differential LOS by source of interhospital transfer might be attributable to both unobserved illness severity and/or extensive in‐hospital post‐ICU multidisciplinary rehabilitative care for inter‐ICU transfer patients, compared with ED transfer patients.
Our study findings need to be interpreted in light of certain limitations. Administrative claims data do not allow for assessment of the quality of hospital care, a factor that might play an important role in patient clinical outcomes. The data lacked any physiologic information that might enhance the ability to estimate patient severity of illness; the analysis used the presence of organ dysfunction at the referring hospitals as a proxy for illness severity. The use of diagnosis codebased measures of severity adjustment, as employed in the current study, however, has been reported to be comparable with clinical severity measures because of the relatively complete capture of diagnosis codes for life‐threatening conditions occurring late in the hospitalization, such as prior to interhospital transfer in the current study.2123
The absence of clinical information prevented assessment of the likelihood of in‐hospital morbidity, transport complications, and need for various therapeutic interventions after ICU care, which are also highly relevant outcomes of interhospital transfers. It is unknown if the small sample size among inter‐ICU transfers limited the ability to demonstrate a statistically significant difference in odds of mortality among inter‐ICU transfers compared with ED transfers.
Also, the identification of diagnoses and procedures was made using multiple coding instruments and is therefore susceptible to inaccuracies of detection and attribution that may have biased the findings. Study findings did not include cost, because cost data were not available for children enrolled in Medicaid managed care plans under capitated arrangements. Finally, it is unknown how generalizable the current study findings might be to children with private insurance, or to children who are uninsured.
The study findings highlight potential opportunities for future research. Further studies are warranted to identify key characteristics that differentiate children admitted to nonpediatric hospitals who are subsequently transferred to pediatric hospitals with ICU facilities versus the children who are not transferred. Also, in‐depth study of the decision‐making that underlies interhospital transfer of critically ill or injured children to hospitals with ICU facilities for advanced care after initial hospitalization is vital to improved understanding of factors that might contribute to the extensive resource consumption and mortality burden borne by these children. The existence and effectiveness of interhospital transfer agreements at the state level needs to be examined specifically as it relates to patterns and clinical outcomes of interhospital transfer of critically ill and injured children in the US.
In conclusion, in this multiyear, statewide sample among critically ill and injured children enrolled by a statewide public payer, clinical outcomes were worse and resource consumption higher, among children who underwent interhospital transfer after initial hospitalization compared with those transferred directly from referring EDs. The findings raise the possibility that more timely transfer of some patients directly from community hospital EDs to regional ICUs might improve survival and reduce resource consumption.
Efforts to improve the care of critically ill and injured children may be enhanced by improved understanding of the medical decision‐making underlying interhospital transfer; application of innovative methods to identify and ensure rapid access to clinical expertise for children initially admitted to hospitals without pediatric intensive care facilities who might subsequently require intensive care; and routine reassessment of hospitalized children to ensure effective and efficient triage and re‐triage at the ED, ward, and ICU levels of referring hospitals.
- ,,,,.A national survey of pediatric critical care resources in the United States.Pediatrics.2005;115:e382–386.
- .Regional variation in child mortality at hospitals lacking a pediatric intensive care unit.Crit Care Med.2002;30:94–99.
- ,,,,,.Direct transport to tertiary trauma centers versus transfer from lower level facilities: impact on mortality and morbidity among patients with major trauma.J Trauma.1997;43:288–296.
- ,,,.Timing of intensive care unit admission in relation to ICU outcome.Crit Care Med.1990;18:1231–1235.
- ,:Admission source to the medical intensive care unit predicts hospital death independent of APACHE II score.JAMA.1990;264:2389–2394.
- ,,,,.Accepting critically ill transfer patients: adverse effect on a referral center's outcome and benchmark measures.Ann Intern Med.2003;138:882–890.
- ,,,,,.Elective intrahospital admissions versus acute interhospital transfers to a surgical intensive care unit: cost and outcome prediction.J Trauma.1991;31:915–918.
- ,,,,.Adverse effect on a referral intensive care unit's performance of accepting patients transferred from another intensive care unit.Crit Care Med.2005;33:705–710.
- ,,,.Prognostic factors for mortality following interhospital transfers to the medical intensive care unit of a tertiary referral center.Crit Care Med.2003;31:1981–1986.
- National Government Services. Medicare UB‐04 Revenue Codes. Available at http://www.ngsmedicare.com/NGSMedicare/PartA/EducationandSupport/ToolsandMaterials/0908ub‐04.pdf. Accessed April 7,2008.
- American Hospital Association.AHA Guide to the Health Care Field.2002 ed.Chicago:American Hospital Association;2002.
- American Hospital Association.AHA Guide to the Health Care Field.2003 ed.Chicago:American Hospital Association;2003.
- ,,.Pediatric deaths attributable to complex chronic conditions: a population‐based study of Washington State, 1980–1997.Pediatrics.2000;106:205–209.
- ,,,.Importance of organ dysfunction in determining hospital outcomes in children.J Pediatr.2004;144:595–601.
- ,,, et al.Cumulative influence of organ dysfunctions and septic state on mortality of critically ill children.Am J Respir Crit Care Med.2005;171:348–353.
- ,,,,,.The epidemiology of severe sepsis in children in the United States.Am J Respir Crit Care Med.2003;167:695–701.
- ,,,.The longer patients are in hospital before intensive care admission the higher their mortality.Intensive Care Med.2004;30:1908–1913.
- ,.A prospective study of outcome of in‐patient pediatric cardiopulmonary arrest.Resuscitation.2006;71:310–318.
- ,,, et al.Effect of a rapid response team on hospital‐wide mortality and code rates outside the ICU in a children's hospital.JAMA.2007;298:2267–2274.
- ,,,,,.Use of telemedicine to provide pediatric critical care consultations to underserved rural northern California.J Pediatr.2004;144:375–380.
- ,.Risk‐adjusting acute myocardial infarction mortality: are APR‐DRGs the right tool?Health Serv Res.2000;34:1469–1489.
- ,,,,.Predicting in‐hospital deaths from coronary artery bypass graft surgery: do different severity measures give different predictions?Med Care.1998;36:28–39.
- ,,.Patient and hospital correlates of clinical outcomes and resource‐utilization in severe pediatric sepsis.Pediatrics.2007;119:487–494.
Interhospital transfer of critically ill and injured children is necessitated by variation in resource availability between hospitals. Critically ill children judged in need of clinical services or expertise not locally available undergo transfer to hospitals with more appropriate resource capabilities and expertise, with the expectation that clinical outcomes of transfer will be better than nontransfer.
Significant variation both in the availability of pediatric critical care services across US hospitals1 and in child mortality among hospitals without pediatric critical care services2 suggests that interhospital transfer will remain an integral part of healthcare delivery for critically ill and injured children. Timely provision of definitive care for acute life‐threatening disease is associated with good clinical outcomes.3, 4 While prior studies have examined clinical outcomes and resource consumption among critically ill adults who underwent interhospital transfer for intensive care,59 there is scarce information regarding clinical characteristics and outcomes of interhospital transfer for critically ill and injured children.
This study was conducted to test the hypothesis that, among critically ill and injured children who undergo interhospital transfer for intensive care, children transferred after an initial hospitalization at the referring facility will have higher mortality, longer length of stay (LOS), and higher resource consumption than children transferred directly from the emergency department (ED) of the referring hospitals.
METHODS
Study Design
We conducted a secondary analysis of administrative claims data from the Michigan Medicaid program for the period January 1, 2002, to December 31, 2004. The data included all paid claims for health services rendered to enrollees in the Medicaid program. The Institutional Review Board of the University of Michigan Medical School approved the study.
Study Sample and Variable Identification
A 3‐step approach was employed to identify interhospital transfer admissions for intensive care of children. Initially, the Medicaid claims were queried to identify all hospitalizations for children 018 years who received intensive care services, using Medicare revenue codes.10 Admissions for neonatal intensive care were excluded from the analysis. The American Hospital Association Guide to the Health Care Field, a compendium of US healthcare facilities, was used to verify the presence of intensive care facilities.11, 12 Subsequently, to identify the subset of children who underwent interhospital transfer, data were queried for the presence of claims from another hospital, and the date of discharge from the referring hospital had to be the same as the date of admission to the receiving hospital intensive care unit (ICU). Finally, to ascertain the source of interhospital transfer, Medicare revenue codes and current procedural terminology (CPT) codes were used to identify claims for receipt of services at specific sites within the referring hospital; namely, the ED, ward, or the ICU. This information was used to categorize admissions into 1 of 3 pathways of interhospital transfer:
ED transferFrom the ED of the referring hospital to the ICU of the receiving hospital.
Ward transferFrom the wards of the referring hospital to the ICU of the receiving hospital.
Inter‐ICU transferFrom the ICU of the referring hospital to the ICU of the receiving hospital.
Dependent Variables
Mortality at the Receiving Hospital
This is determined by linkage to vital statistics records maintained by the Michigan Department of Community Health, Division of Vital Records and Health Statistics.
LOS at the Receiving Hospital
This is determined as the count of days of hospitalization at the receiving hospital. Of note, this includes ICU days and non‐ICU days at the receiving hospital.
Independent Variables
Source of Interhospital Transfer
The main (exposure) independent variable. Categorized into ED, ward, or inter‐ICU transfers, as described.
Patient Demographics
Age and gender.
Comorbid Illness
Determined using International Classification of Diseases, ninth revision (ICD‐9) diagnosis codes, applying methodology as described.13
Organ Dysfunction at the Referring and Receiving Hospitals
Determined using ICD‐9 diagnosis codes, applying methodology as described.14
Patient Diagnostic Categories
Eleven diagnostic categories were created based on primary admission diagnoses (Appendix A).
LOS at the Referring Hospital
Determined as the count of days of hospitalization at the referring hospital.
Receipt of Cardiopulmonary Resuscitation (CPR) on the Date of Interhospital Transfer
Determined using procedure codes.
Receipt of Medical‐Surgical Procedures at the Receiving Hospital
Identified through the use of ICD‐9 procedure codes, CPT codes, and Healthcare Common Procedure Coding System codes. The procedures are listed in Appendix B.
Statistical Analysis
Descriptive statistics were used to characterize the study sample. According to the 3 sources of interhospital transfer, patient characteristics (age, gender, presence of organ dysfunction, and comorbid illness), median LOS at the referring hospital, and receipt of CPR on the date of interhospital transfer were compared using chi‐square tests for categorical variables, and Kruskal‐Wallis tests for continuous variables. Similarly, outcome variables of in‐hospital mortality and median LOS at the receiving hospital were compared across the 3 sources of interhospital transfer. Analysis of variance was used to compare mean LOS at the receiving hospital across the 3 sources of interhospital transfer. Median (with interquartile range [IQR]) and mean (with standard deviation [SD]) values are presented to describe LOS, given skew in LOS data.
To account for potential confounding of LOS and mortality at the receiving hospital by the presence of organ dysfunction and comorbid illness1316 at the referring hospital, multivariate logistic regression and multiple linear regression analyses were conducted to estimate the odds of in‐hospital mortality and the incremental LOS, respectively, for ward and inter‐ICU transfers, compared with ED transfers. Statistical analyses were conducted using Stata 8 for windows (Stata Corporation, College Station, TX). A 2‐tailed level of 0.05 was used as the threshold for statistical significance.
RESULTS
Patient Characteristics
Of 1,643 transfer admissions for intensive care during the study period, 1022 (62%) were ED transfers, 512 (31%) were ward transfers, and 109 (7%) were inter‐ICU transfers. The average age was 2 years, with male gender (57%) predominance. Comorbid illness was present in 19% of admissions, while 11% had evidence of organ dysfunction at the referring hospital. Table 1 presents key patient demographic and clinical characteristics at the referring hospitals, by transfer source. Inter‐ICU and ward transfers were younger than ED transfers, and had a higher preponderance of comorbid illness and organ dysfunction. At the time of interhospital transfer, compared with ED transfers, the proportion of admissions with organ dysfunction (a marker of illness severity) was 3‐fold and 8‐fold higher among ward and inter‐ICU transfers, respectively.
| Transfer Source | P | |||
|---|---|---|---|---|
| Characteristics | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | |
| ||||
| Median age in years (IQR) | 2 (09) | 1 (07) | 1 (010) | <0.01 |
| Male (%) | 57.8 | 56.2 | 47.6 | 0.13 |
| Comorbid illness (% ) | 13.1 | 25.0 | 50.5 | <0.01 |
| Pretransfer hospital length of stay (days) | ||||
| Median (IQR) | 0 | 1 (02) | 3 (18) | <0.01 |
| Mean (SD) | 0.2 (5.2) | 1.6 (4.8) | 9.7 (18.0) | <0.01 |
| Pretransfer organ dysfunction (%) | 5.5 | 14.5 | 40.4 | <0.01 |
Patterns of Transfer
The leading diagnoses among all children were respiratory disease, trauma, and neurological disease (Table 2), with some variation in diagnoses by source of interhospital transfer. For example, cardiovascular disease was the second leading diagnosis after respiratory disease among the inter‐ICU transfers, while more children with endocrine disease (predominantly diabetic ketoacidosis), traumatic injury, or drug poisoning were transferred directly from the ED, than from the ward or the ICU settings. For burn care, 80% (45/56) of all transfer admissions were direct from the ED (Table 3). The majority (78%) of children with traumatic injuries were directly transferred from the ED to the ICU, while the remainder were transferred after initial care delivered on the ward (18%) or ICU (4%) settings prior to interhospital transfer for definitive intensive trauma care. Importantly, among the inter‐ICU transfers, 104 (95%) were transferred to pediatric ICUs from referring hospitals with adult and pediatric ICU facilities, suggesting uptransfer for specialized care. Five children were transferred between hospitals with adult ICU facilities.
| Transfer Source | ||||
|---|---|---|---|---|
| Diagnostic Category (%) | Overall* (n = 1639) | ED* (n = 1018) | Ward (n = 512) | Inter‐ICU (n = 109) |
| ||||
| Respiratory disease | 35.1 | 32.8 | 41.0 | 28.4 |
| Trauma | 16.2 | 20.5 | 9.2 | 9.1 |
| Neurological disease | 12.4 | 12.5 | 12.3 | 11.9 |
| Gastrointestinal disease | 6.7 | 5.4 | 7.4 | 11.9 |
| Infectious disease | 5.8 | 4.0 | 8.4 | 10.0 |
| Endocrine disease | 5.5 | 7.9 | 1.8 | 0 |
| Drug overdose/poisoning | 5.0 | 6.4 | 2.9 | 1.8 |
| Cardiovascular disease | 4.8 | 2.8 | 6.3 | 16.5 |
| Hematologic/oncologic disease | 2.0 | 1.6 | 2.9 | 1.8 |
| Cardiac arrest | 0.2 | 0 | 0.6 | 0.9 |
| Other diagnoses | 6.2 | 5.4 | 7.2 | 7.7 |
| Transfer Source | |||||
|---|---|---|---|---|---|
| Characteristics (%) | Overall (n = 1643) | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | P |
| Respiratory | 26.8 | 19.0 | 36.7 | 54.1 | <0.01 |
| Radiological | 21.2 | 19.5 | 20.5 | 41.3 | <0.01 |
| Vascular access | 20.0 | 15.2 | 27.0 | 33.0 | <0.01 |
| Gastrointestinal | 3.9 | 3.0 | 3.7 | 12.8 | <0.01 |
| Neurological | 3.8 | 3.2 | 3.7 | 10.1 | <0.01 |
| Cardiovascular | 3.6 | 1.8 | 4.1 | 18.4 | <0.01 |
| Burn care | 3.4 | 4.5 | 2.0 | 0 | <0.01 |
| General surgery | 3.2 | 2.1 | 4.3 | 8.3 | <0.01 |
| Dialysis | 2.6 | 2.0 | 2.5 | 8.3 | <0.01 |
| ECMO | 2.1 | 1.3 | 2.2 | 9.2 | <0.01 |
CPR was performed on the date of interhospital transfer for 23 patients (1.4% of the sample), of whom 13 (56.5%) were ward transfers, 8 (34.8%) were inter‐ICU transfers, and 2 (8.7%) were ED transfers (P < 0.02). Two‐thirds of these children did not survive subsequent hospitalization at the receiving hospitals.
Clinical Outcomes and Resource Utilization at the Receiving Hospitals
At the receiving hospitals, other than burn care, medical‐surgical procedures were performed most often among the inter‐ICU transfers. Ward transfers also had higher receipt of procedures compared with ED transfers (Table 3). The inter‐ICU and ward transfers had a higher preponderance of organ dysfunction at the receiving hospitals, compared to the ED transfers (38.5% and 29.3% versus 20.8%, P < 0.01).
Clinical outcomes at the receiving hospitals varied significantly according to the source of interhospital transfer (Table 4). Sixty‐six (4%) of patients died at the receiving hospitals. In comparison with ED transfers, unadjusted in‐hospital mortality was 2‐fold and 3‐fold higher among the ward and inter‐ICU transfers, respectively. Also, hospital LOS was significantly longer among the ward and inter‐ICU transfers than for the ED transfers.
| Transfer Source | ||||
|---|---|---|---|---|
| Characteristics | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | P |
| ||||
| Mortality (%) | 2.8 | 5.5 | 8.3 | <0.01 |
| Length of stay (days) | ||||
| Median (IQR) | 3 (27) | 5 (312) | 13 (724) | <0.01 |
| Mean (SD) | 6.7 (10.4) | 8.5 (9.2) | 21.4 (22.9) | <0.01 |
In multivariate analyses adjusting for patient age, and the presence of comorbid illness and organ dysfunction at the referring hospital, compared with ED transfers, odds of mortality were significantly higher (odds ratio [OR], 1.76; 95% confidence interval [CI], 1.023.03) for ward transfers. Inter‐ICU transfers also had higher odds of mortality (OR, 2.07; 95% CI, 0.884.86), without achieving statistical significance. Similarly, compared with ED transfers, LOS at the receiving hospital was longer by 1.5 days (95% CI, 0.32.7 days) for ward transfers, and by 13.5 days (95% CI, 11.115.8 days) for inter‐ICU transfers.
DISCUSSION
This study is the first to highlight significant variation in clinical outcomes and resource consumption after interhospital transfer of critically ill and injured children, depending on the source of transfer. In comparison with children transferred directly from the referring hospitals' ED settings, children transferred from the referring hospitals' wards had higher mortality, while those who underwent inter‐ICU transfer had significantly higher resource consumption. In addition, ward transfers had the highest proportion of children who underwent CPR on the date of interhospital transfer, highlighting elevated severity of disease prior to transfer and an urgent need for improved understanding of pretransfer clinical care and medical decision‐making. The findings raise the possibility that more timely transfer of some patients directly from community hospital EDs to regional ICUs might improve survival and reduce resource consumption.
Although interhospital transfers are common in everyday clinical practice, there has been a knowledge gap in pediatric acute and critical care medicine regarding the clinical outcomes and resource consumption among children who undergo such transfers. Findings from the current study narrow this gap by relating triage at the referring hospitals to clinical outcomes and resource utilization at the receiving hospitals.
Certain distinct transfer patterns were observed. Most children with burn injury underwent direct transfer from the ED to the ICU; this transfer pattern may be related both to the limited availability of ICUs with burn care capability in Michigan and to the acuity of burn injuries, which often mandates immediate triage to hospitals with intensive burn care facilities. Conversely, while the majority of children with traumatic injuries were directly transferred from emergency to intensive care, over one‐fifth were transferred after initial care delivered on the ward or ICU settings prior to interhospital transfer for definitive intensive trauma care. Such imperfect regionalization of trauma care suggests further study of clinical outcomes and resource utilization among injured children is warranted. Likewise, cardiovascular disease was prominent among the inter‐ICU transfers, suggesting a clinical practice pattern of stabilization and resuscitation at the initial ICU prior to interhospital vertical or uptransfer for definitive cardiac care at the receiving hospitals.
It remains unknown whether the timing of interhospital transfer of critically ill children is a determinant of clinical outcomes. Prior studies among adults have reported higher mortality with prolonged duration of pre‐ICU care on the ward.4, 17 In the current study, ward and inter‐ICU transfers were initially hospitalized for a median of 1 and 3 days, respectively, prior to transfer. While we could not determine from administrative data what the precise triggers for interhospital transfer in this study were, it is instructive to note that ward transfers comprised more than one‐half of all children who received CPR on the date of transfer. For children who received CPR, severe clinical deterioration likely triggered transfer to hospitals with ICU facilities, but because only a minority of children received CPR overall, other triggers of transfer warrant investigation. For most of the children transferred, it seems plausible that the precipitant of transfer was likely a mismatch of their clinical status with the clinical capacities of the facilities where they were initially hospitalized. Future work should investigate if there is an association between clinical outcomes at the receiving hospitals, and both the timing of interhospital transfer and the clinical status of patients at transfer.
Importantly, compared with ED transfers, ward transfers demonstrated elevated odds of mortality after adjustment for coexisting comorbid illness, patient age, and pretransfer organ dysfunction at the referring hospital. Some possible explanations for this finding include the progression of disease while receiving care on the ward, or suboptimal access to ICU facilities due to barriers to transfer at either the referring or receiving hospitals. Importantly, progression of disease in ward settings may be detected by early identification of children at high risk of clinical deterioration on the wards of hospitals without ICU facilities, prior to cardiopulmonary arrest, because death after CPR may not be averted with subsequent ICU care.18
Various approaches to facilitate rapid and appropriate triage and reassessment of children in hospitals without ICU facilities, prior to severe clinical deterioration or need for CPR, must be investigated. These approaches might include in‐hospital measures such as the establishment of medical emergency teams to respond to clinical deterioration on the wards19 or collaborative interhospital measures such as the use of telemedicine20 or similar remote communication/triage systems to enhance communication between clinical caregivers at hospitals with ICU facilities and those in hospitals without ICU facilities. Furthermore, interhospital transfer agreements may facilitate expeditious and appropriate transfer of severely ill patients to hospitals with ICU facilities.
Access to hospitals with ICU facilities might also influence outcomes for critically ill children admitted initially to wards of hospitals without ICU facilities. Kanter2 reported significant variation in mortality among children who received care at New York hospitals without ICU facilities. Of note, 27% of statewide pediatric inpatient deaths occurred in those hospitals without ICU facilities. It appeared that, while some pediatric deaths in hospitals without ICU facilities were expected, regional variation in such mortality might have been associated with reduced access to, or poor utilization of, hospitals with ICU facilities. Barriers to interhospital transfers might include underrecognition of mismatch between patient illness severity and hospital capability at referring hospitals, or lack of capacity to accept transfers at the receiving hospitals. Further study is warranted to investigate clinical decision‐making underlying the initiation of the interhospital transfer processes, and procedural or institutional barriers that might hinder the transfer of critically ill children from hospitals without ICU facilities.
Resource consumption at the receiving hospitals, measured by hospital LOS and receipt of medical‐surgical procedures, was highest among the inter‐ICU transfers. This was an expected finding, given the high frequency of organ dysfunction among the inter‐ICU transfers, before and after interhospital transfer. These patients had the highest use of advanced and resource‐intensive technology, including continuous renal replacement therapy, extracorporeal membrane oxygenation, and cardiovascular procedures such as open‐heart surgery. In addition, the duration of hospitalization at the receiving hospital was 2 weeks longer among the inter‐ICU transfers when compared with the ED transfers. Such prolonged hospitalization has been previously associated with significantly increased resource consumption.4, 6 In the absence of physiologic data pertaining to illness severity, however, it is unknown whether this observed differential LOS by source of interhospital transfer might be attributable to both unobserved illness severity and/or extensive in‐hospital post‐ICU multidisciplinary rehabilitative care for inter‐ICU transfer patients, compared with ED transfer patients.
Our study findings need to be interpreted in light of certain limitations. Administrative claims data do not allow for assessment of the quality of hospital care, a factor that might play an important role in patient clinical outcomes. The data lacked any physiologic information that might enhance the ability to estimate patient severity of illness; the analysis used the presence of organ dysfunction at the referring hospitals as a proxy for illness severity. The use of diagnosis codebased measures of severity adjustment, as employed in the current study, however, has been reported to be comparable with clinical severity measures because of the relatively complete capture of diagnosis codes for life‐threatening conditions occurring late in the hospitalization, such as prior to interhospital transfer in the current study.2123
The absence of clinical information prevented assessment of the likelihood of in‐hospital morbidity, transport complications, and need for various therapeutic interventions after ICU care, which are also highly relevant outcomes of interhospital transfers. It is unknown if the small sample size among inter‐ICU transfers limited the ability to demonstrate a statistically significant difference in odds of mortality among inter‐ICU transfers compared with ED transfers.
Also, the identification of diagnoses and procedures was made using multiple coding instruments and is therefore susceptible to inaccuracies of detection and attribution that may have biased the findings. Study findings did not include cost, because cost data were not available for children enrolled in Medicaid managed care plans under capitated arrangements. Finally, it is unknown how generalizable the current study findings might be to children with private insurance, or to children who are uninsured.
The study findings highlight potential opportunities for future research. Further studies are warranted to identify key characteristics that differentiate children admitted to nonpediatric hospitals who are subsequently transferred to pediatric hospitals with ICU facilities versus the children who are not transferred. Also, in‐depth study of the decision‐making that underlies interhospital transfer of critically ill or injured children to hospitals with ICU facilities for advanced care after initial hospitalization is vital to improved understanding of factors that might contribute to the extensive resource consumption and mortality burden borne by these children. The existence and effectiveness of interhospital transfer agreements at the state level needs to be examined specifically as it relates to patterns and clinical outcomes of interhospital transfer of critically ill and injured children in the US.
In conclusion, in this multiyear, statewide sample among critically ill and injured children enrolled by a statewide public payer, clinical outcomes were worse and resource consumption higher, among children who underwent interhospital transfer after initial hospitalization compared with those transferred directly from referring EDs. The findings raise the possibility that more timely transfer of some patients directly from community hospital EDs to regional ICUs might improve survival and reduce resource consumption.
Efforts to improve the care of critically ill and injured children may be enhanced by improved understanding of the medical decision‐making underlying interhospital transfer; application of innovative methods to identify and ensure rapid access to clinical expertise for children initially admitted to hospitals without pediatric intensive care facilities who might subsequently require intensive care; and routine reassessment of hospitalized children to ensure effective and efficient triage and re‐triage at the ED, ward, and ICU levels of referring hospitals.
Interhospital transfer of critically ill and injured children is necessitated by variation in resource availability between hospitals. Critically ill children judged in need of clinical services or expertise not locally available undergo transfer to hospitals with more appropriate resource capabilities and expertise, with the expectation that clinical outcomes of transfer will be better than nontransfer.
Significant variation both in the availability of pediatric critical care services across US hospitals1 and in child mortality among hospitals without pediatric critical care services2 suggests that interhospital transfer will remain an integral part of healthcare delivery for critically ill and injured children. Timely provision of definitive care for acute life‐threatening disease is associated with good clinical outcomes.3, 4 While prior studies have examined clinical outcomes and resource consumption among critically ill adults who underwent interhospital transfer for intensive care,59 there is scarce information regarding clinical characteristics and outcomes of interhospital transfer for critically ill and injured children.
This study was conducted to test the hypothesis that, among critically ill and injured children who undergo interhospital transfer for intensive care, children transferred after an initial hospitalization at the referring facility will have higher mortality, longer length of stay (LOS), and higher resource consumption than children transferred directly from the emergency department (ED) of the referring hospitals.
METHODS
Study Design
We conducted a secondary analysis of administrative claims data from the Michigan Medicaid program for the period January 1, 2002, to December 31, 2004. The data included all paid claims for health services rendered to enrollees in the Medicaid program. The Institutional Review Board of the University of Michigan Medical School approved the study.
Study Sample and Variable Identification
A 3‐step approach was employed to identify interhospital transfer admissions for intensive care of children. Initially, the Medicaid claims were queried to identify all hospitalizations for children 018 years who received intensive care services, using Medicare revenue codes.10 Admissions for neonatal intensive care were excluded from the analysis. The American Hospital Association Guide to the Health Care Field, a compendium of US healthcare facilities, was used to verify the presence of intensive care facilities.11, 12 Subsequently, to identify the subset of children who underwent interhospital transfer, data were queried for the presence of claims from another hospital, and the date of discharge from the referring hospital had to be the same as the date of admission to the receiving hospital intensive care unit (ICU). Finally, to ascertain the source of interhospital transfer, Medicare revenue codes and current procedural terminology (CPT) codes were used to identify claims for receipt of services at specific sites within the referring hospital; namely, the ED, ward, or the ICU. This information was used to categorize admissions into 1 of 3 pathways of interhospital transfer:
ED transferFrom the ED of the referring hospital to the ICU of the receiving hospital.
Ward transferFrom the wards of the referring hospital to the ICU of the receiving hospital.
Inter‐ICU transferFrom the ICU of the referring hospital to the ICU of the receiving hospital.
Dependent Variables
Mortality at the Receiving Hospital
This is determined by linkage to vital statistics records maintained by the Michigan Department of Community Health, Division of Vital Records and Health Statistics.
LOS at the Receiving Hospital
This is determined as the count of days of hospitalization at the receiving hospital. Of note, this includes ICU days and non‐ICU days at the receiving hospital.
Independent Variables
Source of Interhospital Transfer
The main (exposure) independent variable. Categorized into ED, ward, or inter‐ICU transfers, as described.
Patient Demographics
Age and gender.
Comorbid Illness
Determined using International Classification of Diseases, ninth revision (ICD‐9) diagnosis codes, applying methodology as described.13
Organ Dysfunction at the Referring and Receiving Hospitals
Determined using ICD‐9 diagnosis codes, applying methodology as described.14
Patient Diagnostic Categories
Eleven diagnostic categories were created based on primary admission diagnoses (Appendix A).
LOS at the Referring Hospital
Determined as the count of days of hospitalization at the referring hospital.
Receipt of Cardiopulmonary Resuscitation (CPR) on the Date of Interhospital Transfer
Determined using procedure codes.
Receipt of Medical‐Surgical Procedures at the Receiving Hospital
Identified through the use of ICD‐9 procedure codes, CPT codes, and Healthcare Common Procedure Coding System codes. The procedures are listed in Appendix B.
Statistical Analysis
Descriptive statistics were used to characterize the study sample. According to the 3 sources of interhospital transfer, patient characteristics (age, gender, presence of organ dysfunction, and comorbid illness), median LOS at the referring hospital, and receipt of CPR on the date of interhospital transfer were compared using chi‐square tests for categorical variables, and Kruskal‐Wallis tests for continuous variables. Similarly, outcome variables of in‐hospital mortality and median LOS at the receiving hospital were compared across the 3 sources of interhospital transfer. Analysis of variance was used to compare mean LOS at the receiving hospital across the 3 sources of interhospital transfer. Median (with interquartile range [IQR]) and mean (with standard deviation [SD]) values are presented to describe LOS, given skew in LOS data.
To account for potential confounding of LOS and mortality at the receiving hospital by the presence of organ dysfunction and comorbid illness1316 at the referring hospital, multivariate logistic regression and multiple linear regression analyses were conducted to estimate the odds of in‐hospital mortality and the incremental LOS, respectively, for ward and inter‐ICU transfers, compared with ED transfers. Statistical analyses were conducted using Stata 8 for windows (Stata Corporation, College Station, TX). A 2‐tailed level of 0.05 was used as the threshold for statistical significance.
RESULTS
Patient Characteristics
Of 1,643 transfer admissions for intensive care during the study period, 1022 (62%) were ED transfers, 512 (31%) were ward transfers, and 109 (7%) were inter‐ICU transfers. The average age was 2 years, with male gender (57%) predominance. Comorbid illness was present in 19% of admissions, while 11% had evidence of organ dysfunction at the referring hospital. Table 1 presents key patient demographic and clinical characteristics at the referring hospitals, by transfer source. Inter‐ICU and ward transfers were younger than ED transfers, and had a higher preponderance of comorbid illness and organ dysfunction. At the time of interhospital transfer, compared with ED transfers, the proportion of admissions with organ dysfunction (a marker of illness severity) was 3‐fold and 8‐fold higher among ward and inter‐ICU transfers, respectively.
| Transfer Source | P | |||
|---|---|---|---|---|
| Characteristics | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | |
| ||||
| Median age in years (IQR) | 2 (09) | 1 (07) | 1 (010) | <0.01 |
| Male (%) | 57.8 | 56.2 | 47.6 | 0.13 |
| Comorbid illness (% ) | 13.1 | 25.0 | 50.5 | <0.01 |
| Pretransfer hospital length of stay (days) | ||||
| Median (IQR) | 0 | 1 (02) | 3 (18) | <0.01 |
| Mean (SD) | 0.2 (5.2) | 1.6 (4.8) | 9.7 (18.0) | <0.01 |
| Pretransfer organ dysfunction (%) | 5.5 | 14.5 | 40.4 | <0.01 |
Patterns of Transfer
The leading diagnoses among all children were respiratory disease, trauma, and neurological disease (Table 2), with some variation in diagnoses by source of interhospital transfer. For example, cardiovascular disease was the second leading diagnosis after respiratory disease among the inter‐ICU transfers, while more children with endocrine disease (predominantly diabetic ketoacidosis), traumatic injury, or drug poisoning were transferred directly from the ED, than from the ward or the ICU settings. For burn care, 80% (45/56) of all transfer admissions were direct from the ED (Table 3). The majority (78%) of children with traumatic injuries were directly transferred from the ED to the ICU, while the remainder were transferred after initial care delivered on the ward (18%) or ICU (4%) settings prior to interhospital transfer for definitive intensive trauma care. Importantly, among the inter‐ICU transfers, 104 (95%) were transferred to pediatric ICUs from referring hospitals with adult and pediatric ICU facilities, suggesting uptransfer for specialized care. Five children were transferred between hospitals with adult ICU facilities.
| Transfer Source | ||||
|---|---|---|---|---|
| Diagnostic Category (%) | Overall* (n = 1639) | ED* (n = 1018) | Ward (n = 512) | Inter‐ICU (n = 109) |
| ||||
| Respiratory disease | 35.1 | 32.8 | 41.0 | 28.4 |
| Trauma | 16.2 | 20.5 | 9.2 | 9.1 |
| Neurological disease | 12.4 | 12.5 | 12.3 | 11.9 |
| Gastrointestinal disease | 6.7 | 5.4 | 7.4 | 11.9 |
| Infectious disease | 5.8 | 4.0 | 8.4 | 10.0 |
| Endocrine disease | 5.5 | 7.9 | 1.8 | 0 |
| Drug overdose/poisoning | 5.0 | 6.4 | 2.9 | 1.8 |
| Cardiovascular disease | 4.8 | 2.8 | 6.3 | 16.5 |
| Hematologic/oncologic disease | 2.0 | 1.6 | 2.9 | 1.8 |
| Cardiac arrest | 0.2 | 0 | 0.6 | 0.9 |
| Other diagnoses | 6.2 | 5.4 | 7.2 | 7.7 |
| Transfer Source | |||||
|---|---|---|---|---|---|
| Characteristics (%) | Overall (n = 1643) | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | P |
| Respiratory | 26.8 | 19.0 | 36.7 | 54.1 | <0.01 |
| Radiological | 21.2 | 19.5 | 20.5 | 41.3 | <0.01 |
| Vascular access | 20.0 | 15.2 | 27.0 | 33.0 | <0.01 |
| Gastrointestinal | 3.9 | 3.0 | 3.7 | 12.8 | <0.01 |
| Neurological | 3.8 | 3.2 | 3.7 | 10.1 | <0.01 |
| Cardiovascular | 3.6 | 1.8 | 4.1 | 18.4 | <0.01 |
| Burn care | 3.4 | 4.5 | 2.0 | 0 | <0.01 |
| General surgery | 3.2 | 2.1 | 4.3 | 8.3 | <0.01 |
| Dialysis | 2.6 | 2.0 | 2.5 | 8.3 | <0.01 |
| ECMO | 2.1 | 1.3 | 2.2 | 9.2 | <0.01 |
CPR was performed on the date of interhospital transfer for 23 patients (1.4% of the sample), of whom 13 (56.5%) were ward transfers, 8 (34.8%) were inter‐ICU transfers, and 2 (8.7%) were ED transfers (P < 0.02). Two‐thirds of these children did not survive subsequent hospitalization at the receiving hospitals.
Clinical Outcomes and Resource Utilization at the Receiving Hospitals
At the receiving hospitals, other than burn care, medical‐surgical procedures were performed most often among the inter‐ICU transfers. Ward transfers also had higher receipt of procedures compared with ED transfers (Table 3). The inter‐ICU and ward transfers had a higher preponderance of organ dysfunction at the receiving hospitals, compared to the ED transfers (38.5% and 29.3% versus 20.8%, P < 0.01).
Clinical outcomes at the receiving hospitals varied significantly according to the source of interhospital transfer (Table 4). Sixty‐six (4%) of patients died at the receiving hospitals. In comparison with ED transfers, unadjusted in‐hospital mortality was 2‐fold and 3‐fold higher among the ward and inter‐ICU transfers, respectively. Also, hospital LOS was significantly longer among the ward and inter‐ICU transfers than for the ED transfers.
| Transfer Source | ||||
|---|---|---|---|---|
| Characteristics | ED (n = 1022) | Ward (n = 512) | Inter‐ICU (n = 109) | P |
| ||||
| Mortality (%) | 2.8 | 5.5 | 8.3 | <0.01 |
| Length of stay (days) | ||||
| Median (IQR) | 3 (27) | 5 (312) | 13 (724) | <0.01 |
| Mean (SD) | 6.7 (10.4) | 8.5 (9.2) | 21.4 (22.9) | <0.01 |
In multivariate analyses adjusting for patient age, and the presence of comorbid illness and organ dysfunction at the referring hospital, compared with ED transfers, odds of mortality were significantly higher (odds ratio [OR], 1.76; 95% confidence interval [CI], 1.023.03) for ward transfers. Inter‐ICU transfers also had higher odds of mortality (OR, 2.07; 95% CI, 0.884.86), without achieving statistical significance. Similarly, compared with ED transfers, LOS at the receiving hospital was longer by 1.5 days (95% CI, 0.32.7 days) for ward transfers, and by 13.5 days (95% CI, 11.115.8 days) for inter‐ICU transfers.
DISCUSSION
This study is the first to highlight significant variation in clinical outcomes and resource consumption after interhospital transfer of critically ill and injured children, depending on the source of transfer. In comparison with children transferred directly from the referring hospitals' ED settings, children transferred from the referring hospitals' wards had higher mortality, while those who underwent inter‐ICU transfer had significantly higher resource consumption. In addition, ward transfers had the highest proportion of children who underwent CPR on the date of interhospital transfer, highlighting elevated severity of disease prior to transfer and an urgent need for improved understanding of pretransfer clinical care and medical decision‐making. The findings raise the possibility that more timely transfer of some patients directly from community hospital EDs to regional ICUs might improve survival and reduce resource consumption.
Although interhospital transfers are common in everyday clinical practice, there has been a knowledge gap in pediatric acute and critical care medicine regarding the clinical outcomes and resource consumption among children who undergo such transfers. Findings from the current study narrow this gap by relating triage at the referring hospitals to clinical outcomes and resource utilization at the receiving hospitals.
Certain distinct transfer patterns were observed. Most children with burn injury underwent direct transfer from the ED to the ICU; this transfer pattern may be related both to the limited availability of ICUs with burn care capability in Michigan and to the acuity of burn injuries, which often mandates immediate triage to hospitals with intensive burn care facilities. Conversely, while the majority of children with traumatic injuries were directly transferred from emergency to intensive care, over one‐fifth were transferred after initial care delivered on the ward or ICU settings prior to interhospital transfer for definitive intensive trauma care. Such imperfect regionalization of trauma care suggests further study of clinical outcomes and resource utilization among injured children is warranted. Likewise, cardiovascular disease was prominent among the inter‐ICU transfers, suggesting a clinical practice pattern of stabilization and resuscitation at the initial ICU prior to interhospital vertical or uptransfer for definitive cardiac care at the receiving hospitals.
It remains unknown whether the timing of interhospital transfer of critically ill children is a determinant of clinical outcomes. Prior studies among adults have reported higher mortality with prolonged duration of pre‐ICU care on the ward.4, 17 In the current study, ward and inter‐ICU transfers were initially hospitalized for a median of 1 and 3 days, respectively, prior to transfer. While we could not determine from administrative data what the precise triggers for interhospital transfer in this study were, it is instructive to note that ward transfers comprised more than one‐half of all children who received CPR on the date of transfer. For children who received CPR, severe clinical deterioration likely triggered transfer to hospitals with ICU facilities, but because only a minority of children received CPR overall, other triggers of transfer warrant investigation. For most of the children transferred, it seems plausible that the precipitant of transfer was likely a mismatch of their clinical status with the clinical capacities of the facilities where they were initially hospitalized. Future work should investigate if there is an association between clinical outcomes at the receiving hospitals, and both the timing of interhospital transfer and the clinical status of patients at transfer.
Importantly, compared with ED transfers, ward transfers demonstrated elevated odds of mortality after adjustment for coexisting comorbid illness, patient age, and pretransfer organ dysfunction at the referring hospital. Some possible explanations for this finding include the progression of disease while receiving care on the ward, or suboptimal access to ICU facilities due to barriers to transfer at either the referring or receiving hospitals. Importantly, progression of disease in ward settings may be detected by early identification of children at high risk of clinical deterioration on the wards of hospitals without ICU facilities, prior to cardiopulmonary arrest, because death after CPR may not be averted with subsequent ICU care.18
Various approaches to facilitate rapid and appropriate triage and reassessment of children in hospitals without ICU facilities, prior to severe clinical deterioration or need for CPR, must be investigated. These approaches might include in‐hospital measures such as the establishment of medical emergency teams to respond to clinical deterioration on the wards19 or collaborative interhospital measures such as the use of telemedicine20 or similar remote communication/triage systems to enhance communication between clinical caregivers at hospitals with ICU facilities and those in hospitals without ICU facilities. Furthermore, interhospital transfer agreements may facilitate expeditious and appropriate transfer of severely ill patients to hospitals with ICU facilities.
Access to hospitals with ICU facilities might also influence outcomes for critically ill children admitted initially to wards of hospitals without ICU facilities. Kanter2 reported significant variation in mortality among children who received care at New York hospitals without ICU facilities. Of note, 27% of statewide pediatric inpatient deaths occurred in those hospitals without ICU facilities. It appeared that, while some pediatric deaths in hospitals without ICU facilities were expected, regional variation in such mortality might have been associated with reduced access to, or poor utilization of, hospitals with ICU facilities. Barriers to interhospital transfers might include underrecognition of mismatch between patient illness severity and hospital capability at referring hospitals, or lack of capacity to accept transfers at the receiving hospitals. Further study is warranted to investigate clinical decision‐making underlying the initiation of the interhospital transfer processes, and procedural or institutional barriers that might hinder the transfer of critically ill children from hospitals without ICU facilities.
Resource consumption at the receiving hospitals, measured by hospital LOS and receipt of medical‐surgical procedures, was highest among the inter‐ICU transfers. This was an expected finding, given the high frequency of organ dysfunction among the inter‐ICU transfers, before and after interhospital transfer. These patients had the highest use of advanced and resource‐intensive technology, including continuous renal replacement therapy, extracorporeal membrane oxygenation, and cardiovascular procedures such as open‐heart surgery. In addition, the duration of hospitalization at the receiving hospital was 2 weeks longer among the inter‐ICU transfers when compared with the ED transfers. Such prolonged hospitalization has been previously associated with significantly increased resource consumption.4, 6 In the absence of physiologic data pertaining to illness severity, however, it is unknown whether this observed differential LOS by source of interhospital transfer might be attributable to both unobserved illness severity and/or extensive in‐hospital post‐ICU multidisciplinary rehabilitative care for inter‐ICU transfer patients, compared with ED transfer patients.
Our study findings need to be interpreted in light of certain limitations. Administrative claims data do not allow for assessment of the quality of hospital care, a factor that might play an important role in patient clinical outcomes. The data lacked any physiologic information that might enhance the ability to estimate patient severity of illness; the analysis used the presence of organ dysfunction at the referring hospitals as a proxy for illness severity. The use of diagnosis codebased measures of severity adjustment, as employed in the current study, however, has been reported to be comparable with clinical severity measures because of the relatively complete capture of diagnosis codes for life‐threatening conditions occurring late in the hospitalization, such as prior to interhospital transfer in the current study.2123
The absence of clinical information prevented assessment of the likelihood of in‐hospital morbidity, transport complications, and need for various therapeutic interventions after ICU care, which are also highly relevant outcomes of interhospital transfers. It is unknown if the small sample size among inter‐ICU transfers limited the ability to demonstrate a statistically significant difference in odds of mortality among inter‐ICU transfers compared with ED transfers.
Also, the identification of diagnoses and procedures was made using multiple coding instruments and is therefore susceptible to inaccuracies of detection and attribution that may have biased the findings. Study findings did not include cost, because cost data were not available for children enrolled in Medicaid managed care plans under capitated arrangements. Finally, it is unknown how generalizable the current study findings might be to children with private insurance, or to children who are uninsured.
The study findings highlight potential opportunities for future research. Further studies are warranted to identify key characteristics that differentiate children admitted to nonpediatric hospitals who are subsequently transferred to pediatric hospitals with ICU facilities versus the children who are not transferred. Also, in‐depth study of the decision‐making that underlies interhospital transfer of critically ill or injured children to hospitals with ICU facilities for advanced care after initial hospitalization is vital to improved understanding of factors that might contribute to the extensive resource consumption and mortality burden borne by these children. The existence and effectiveness of interhospital transfer agreements at the state level needs to be examined specifically as it relates to patterns and clinical outcomes of interhospital transfer of critically ill and injured children in the US.
In conclusion, in this multiyear, statewide sample among critically ill and injured children enrolled by a statewide public payer, clinical outcomes were worse and resource consumption higher, among children who underwent interhospital transfer after initial hospitalization compared with those transferred directly from referring EDs. The findings raise the possibility that more timely transfer of some patients directly from community hospital EDs to regional ICUs might improve survival and reduce resource consumption.
Efforts to improve the care of critically ill and injured children may be enhanced by improved understanding of the medical decision‐making underlying interhospital transfer; application of innovative methods to identify and ensure rapid access to clinical expertise for children initially admitted to hospitals without pediatric intensive care facilities who might subsequently require intensive care; and routine reassessment of hospitalized children to ensure effective and efficient triage and re‐triage at the ED, ward, and ICU levels of referring hospitals.
- ,,,,.A national survey of pediatric critical care resources in the United States.Pediatrics.2005;115:e382–386.
- .Regional variation in child mortality at hospitals lacking a pediatric intensive care unit.Crit Care Med.2002;30:94–99.
- ,,,,,.Direct transport to tertiary trauma centers versus transfer from lower level facilities: impact on mortality and morbidity among patients with major trauma.J Trauma.1997;43:288–296.
- ,,,.Timing of intensive care unit admission in relation to ICU outcome.Crit Care Med.1990;18:1231–1235.
- ,:Admission source to the medical intensive care unit predicts hospital death independent of APACHE II score.JAMA.1990;264:2389–2394.
- ,,,,.Accepting critically ill transfer patients: adverse effect on a referral center's outcome and benchmark measures.Ann Intern Med.2003;138:882–890.
- ,,,,,.Elective intrahospital admissions versus acute interhospital transfers to a surgical intensive care unit: cost and outcome prediction.J Trauma.1991;31:915–918.
- ,,,,.Adverse effect on a referral intensive care unit's performance of accepting patients transferred from another intensive care unit.Crit Care Med.2005;33:705–710.
- ,,,.Prognostic factors for mortality following interhospital transfers to the medical intensive care unit of a tertiary referral center.Crit Care Med.2003;31:1981–1986.
- National Government Services. Medicare UB‐04 Revenue Codes. Available at http://www.ngsmedicare.com/NGSMedicare/PartA/EducationandSupport/ToolsandMaterials/0908ub‐04.pdf. Accessed April 7,2008.
- American Hospital Association.AHA Guide to the Health Care Field.2002 ed.Chicago:American Hospital Association;2002.
- American Hospital Association.AHA Guide to the Health Care Field.2003 ed.Chicago:American Hospital Association;2003.
- ,,.Pediatric deaths attributable to complex chronic conditions: a population‐based study of Washington State, 1980–1997.Pediatrics.2000;106:205–209.
- ,,,.Importance of organ dysfunction in determining hospital outcomes in children.J Pediatr.2004;144:595–601.
- ,,, et al.Cumulative influence of organ dysfunctions and septic state on mortality of critically ill children.Am J Respir Crit Care Med.2005;171:348–353.
- ,,,,,.The epidemiology of severe sepsis in children in the United States.Am J Respir Crit Care Med.2003;167:695–701.
- ,,,.The longer patients are in hospital before intensive care admission the higher their mortality.Intensive Care Med.2004;30:1908–1913.
- ,.A prospective study of outcome of in‐patient pediatric cardiopulmonary arrest.Resuscitation.2006;71:310–318.
- ,,, et al.Effect of a rapid response team on hospital‐wide mortality and code rates outside the ICU in a children's hospital.JAMA.2007;298:2267–2274.
- ,,,,,.Use of telemedicine to provide pediatric critical care consultations to underserved rural northern California.J Pediatr.2004;144:375–380.
- ,.Risk‐adjusting acute myocardial infarction mortality: are APR‐DRGs the right tool?Health Serv Res.2000;34:1469–1489.
- ,,,,.Predicting in‐hospital deaths from coronary artery bypass graft surgery: do different severity measures give different predictions?Med Care.1998;36:28–39.
- ,,.Patient and hospital correlates of clinical outcomes and resource‐utilization in severe pediatric sepsis.Pediatrics.2007;119:487–494.
- ,,,,.A national survey of pediatric critical care resources in the United States.Pediatrics.2005;115:e382–386.
- .Regional variation in child mortality at hospitals lacking a pediatric intensive care unit.Crit Care Med.2002;30:94–99.
- ,,,,,.Direct transport to tertiary trauma centers versus transfer from lower level facilities: impact on mortality and morbidity among patients with major trauma.J Trauma.1997;43:288–296.
- ,,,.Timing of intensive care unit admission in relation to ICU outcome.Crit Care Med.1990;18:1231–1235.
- ,:Admission source to the medical intensive care unit predicts hospital death independent of APACHE II score.JAMA.1990;264:2389–2394.
- ,,,,.Accepting critically ill transfer patients: adverse effect on a referral center's outcome and benchmark measures.Ann Intern Med.2003;138:882–890.
- ,,,,,.Elective intrahospital admissions versus acute interhospital transfers to a surgical intensive care unit: cost and outcome prediction.J Trauma.1991;31:915–918.
- ,,,,.Adverse effect on a referral intensive care unit's performance of accepting patients transferred from another intensive care unit.Crit Care Med.2005;33:705–710.
- ,,,.Prognostic factors for mortality following interhospital transfers to the medical intensive care unit of a tertiary referral center.Crit Care Med.2003;31:1981–1986.
- National Government Services. Medicare UB‐04 Revenue Codes. Available at http://www.ngsmedicare.com/NGSMedicare/PartA/EducationandSupport/ToolsandMaterials/0908ub‐04.pdf. Accessed April 7,2008.
- American Hospital Association.AHA Guide to the Health Care Field.2002 ed.Chicago:American Hospital Association;2002.
- American Hospital Association.AHA Guide to the Health Care Field.2003 ed.Chicago:American Hospital Association;2003.
- ,,.Pediatric deaths attributable to complex chronic conditions: a population‐based study of Washington State, 1980–1997.Pediatrics.2000;106:205–209.
- ,,,.Importance of organ dysfunction in determining hospital outcomes in children.J Pediatr.2004;144:595–601.
- ,,, et al.Cumulative influence of organ dysfunctions and septic state on mortality of critically ill children.Am J Respir Crit Care Med.2005;171:348–353.
- ,,,,,.The epidemiology of severe sepsis in children in the United States.Am J Respir Crit Care Med.2003;167:695–701.
- ,,,.The longer patients are in hospital before intensive care admission the higher their mortality.Intensive Care Med.2004;30:1908–1913.
- ,.A prospective study of outcome of in‐patient pediatric cardiopulmonary arrest.Resuscitation.2006;71:310–318.
- ,,, et al.Effect of a rapid response team on hospital‐wide mortality and code rates outside the ICU in a children's hospital.JAMA.2007;298:2267–2274.
- ,,,,,.Use of telemedicine to provide pediatric critical care consultations to underserved rural northern California.J Pediatr.2004;144:375–380.
- ,.Risk‐adjusting acute myocardial infarction mortality: are APR‐DRGs the right tool?Health Serv Res.2000;34:1469–1489.
- ,,,,.Predicting in‐hospital deaths from coronary artery bypass graft surgery: do different severity measures give different predictions?Med Care.1998;36:28–39.
- ,,.Patient and hospital correlates of clinical outcomes and resource‐utilization in severe pediatric sepsis.Pediatrics.2007;119:487–494.
Copyright © 2009 Society of Hospital Medicine
I Don't Think We're in the Adult Inpatient Unit
The March issue of the Journal of Hospital Medicine represents a landmark for pediatric hospital medicine (PHM), with 100% of the original research content devoted to pediatrics. Since the days of the National Association of Inpatient Physicians, pediatric hospitalists have consistently constituted 8% to 10% of the membership of the Society of Hospital Medicine (SHM). SHM has always welcomed pediatrics and pediatricians into the community of hospital medicine. A pediatrician has sat on the board since the founding of the National Association of Inpatient Physicians, and for the past 3 years, there has been a formal pediatric board seat. The Hospitalist has consistently included pediatric content with program descriptions and literature reviews. This past July, more than 325 pediatric hospitalists gathered in Denver for the largest PHM meeting ever, a 4‐day event trisponsored by SHM, the American Academy of Pediatrics (AAP), and the Academic Pediatric Association (APA).
As pediatric hospitalists, we have prospered by following the successes of adult hospitalists. We have flattered/emmitated our adult colleagues with pediatric voluntary referral policies, core competencies, salary surveys, fellowship programs, and quality improvement projects. In other areas, pediatrics has set trends for (adult) hospital medicine. Pediatrics developed the medical home concept. We zealously advocate for family‐centered rounds. (Imagine actually rounding in the room with the patient, family, nurse, and physician. It certainly beats flipping cards in the conference room)! Pediatricians have developed global fee codes for evaluation and management services (albeit limited to neonatal and pediatric critical care). As evidenced by the trisponsored meeting mentioned previously and the Pediatric Research in Inpatient Settings Network, we have created collaborative relationships among the pediatric academic (APA), professional (AAP), and hospitalist organizations (SHM) that serve as models for other disciplines and their respective sandboxes.
Research and publications are where we most lag behind our adult colleagues and where the most work needs to be done for us to achieve legitimacy as practitioners and as a discipline. This issue of the Journal of Hospital Medicine is a harbinger of more pediatric content to come, with topics that run the gamut of PHM. Woolford et al.1 highlight clinical, public health, and public policy issues with their analysis of the increased costs and morbidity associated with obesity and inpatient hospitalizations. Wilkes et al.2 explore the logistic issues surrounding influenza testing. As is frequently true for hospitalists, our expertise is not purely clinical: Is oseltamvir effective and, if so, in what age groups? That question is probably best left to the infectious disease community. Rather, Wilkes et al. highlight both the provider and system issues involved in reliably and expeditiously obtaining, reporting, and communicating flu antigen test results so that clinicians and families have the opportunity to consider oseltamvir use within the first 48 hours of disease. Odetola et al.'s3 analysis of a Michigan administrative data set suggests that morbidity, length of stay, and resource utilization are decreased for patients who ultimately require pediatric critical care when these patients are directly transferred from the emergency room to a facility with a pediatric intensive care unit (PICU) in comparison with the morbidity, length of stay, and resource utilization of patients who are initially admitted to the ward from the emergency room and then transferred to a facility with a PICU. This study lacks the rigor of prospectively collected physiological data and would probably never receive institutional review board approval for randomization, but it certainly raises key questions about appropriate transfer criteria for patients cared for in hospitals without a PICU. This is a key quality concern for pediatric hospitalists practicing in smaller, community hospital settings.
The 2 most controversial articles in this pediatric inpatient potpourri are the studies conducted by Freed and Kelly examining pediatric hospitalist training, practice, and career goals4 and PHM fellowship programs.5 These studies are part of a 6‐perspective analysis of pediatric hospitalists/PHM requested by the American Board of Pediatrics (ABP) to provide background to the ABP as it begins to grapple with its role in certifying pediatricians whose primary practice is inpatient pediatrics. A previously published study analyzed the perspective of PHM group leaders.6 The remaining studies assess the perspectives of residency program directors, department chairs, and hospital leaders.
Not surprisingly, these 3 articles46 tend to be more critical of the PHM movement and its current state than are articles and commentaries written by those of us who are practicing hospitalists. As a hospitalist, my initial reaction was to focus on the studies' shortcomings. The methods seemed flawed, the criticisms seemed unwarranted, and the study limitations seemed underappreciated. Aside from the fellowship study, which surveyed the entire n = 8 universe of PHM fellowship programs, the group leader and hospitalist surveys suffer from a selection bias. Sampling for these studies was based on hospital size and type. Although this sampling strategy is appropriate for comparing programs across hospitals, it fails to account for programs of different sizes in different settings. It is not the best sampling strategy for a denominator of all pediatric hospitalists. For example, community hospital programs without residents are often much bigger than academic programs with residents. Community pediatric hospitalists are likely underrepresented in Freed's survey.4 From a study design standpoint, it does not appear that specific a priori hypotheses were generated when subgroups were compared. Rather, one suspects that every possible comparison was analyzed. Thus, the percent differences from one group to another are best considered descriptive rather than rigorously statistically significant at a p 0.05 level. Some criticisms addressed to hospitalists apply to all pediatricians. Given the current emphasis on quality assessment, wouldn't most office‐based pediatricians (and particularly group leaders) believe that they need extra training in this field? When less than 50% of hospitals require practitioners in established subboarded specialties to be board‐certified to maintain hospital privileges,7 is it surprising to see that privileging standards vary for pediatric hospitalists?
However, nitpicking these studies is a defensive response that does a disservice both to the reports and more importantly to the PHM community as a whole and to the children, parents, and colleagues that we serve. There is no denying that we are a young, evolving field with significant inter‐institutional and at times intra‐institutional variability. All of us in the PHM community, leaders and lurkers, need to rise to the challenges offered by comprehensive analysis. Freed's sample of 431 hospitalists4 is significantly larger than the sample of 265 hospitalist participants in the latest Pediatric Research in Inpatient Settings survey.8 The perceptions of external observers are crucial; it would be a mistake to dismiss their findings or to ignore their interpretations and criticisms.
Certainly none would challenge the variability of practice revealed in Freed's analyses.46 Remember, if you've seen one pediatric hospital medicine program, you've seen ONE pediatric hospital medicine program. Some may see this variability as a weakness; others may see it as a strength. We must be equally receptive to other less‐flattering observations, data, and conclusions included in these reports to the ABP. All programs target seamless communication with referring physicians, but hospitalists and referring physicians alike agree that we do not achieve it, as evidenced by the work of Harlan et al.9 in this issue. SHM is taking the lead in developing performance standards for transitions of care and has created best discharge practices for the geriatric population.10 Similarly, we in the PHM community would do well to ramp up our self‐assessment and quality improvement activities. Our recusal from Centers for Medicare and Medicaid Services reporting requirements for (adult) inpatient quality metrics does not excuse us from pursuing voluntary, rigorous, transparent, public reporting on pediatric quality indicators. As Freed et al.6 clearly implied, the public and payers expect this of us. No doubt, if we do not first propose and implement our own standards, external standards will be imposed upon us.
Aside from the question of mandatory fellowship training for hospitalists, does the vision implied in the studies commissioned by the ABP vary significantly from the challenges to PHM that Sandy Melzer11 presented at his keynote address at the Denver meeting? Melzer used strategic planning principles to outline a future vision for PHM, including the following:
-
Harm is eliminated from the inpatient setting.
-
Inpatient care is evidence‐based for all conditions treated.
-
Hospital care is highly coordinated, especially for children with chronic conditions.
-
A robust research agenda supports all aspects of inpatient care.
Is not the work done by the SHM and APA to develop core competencies for PHM an effort to define our field and identify (uniform) expectations? Do not the criteria for designation as a fellow of hospital medicine (5 years as a practicing hospitalist; 2 national meetings; and a minimum combination of leadership, teamwork, and quality improvement activities)12 serve to recognize the commitment and accomplishments that distinguish a true hospitalist practicing systems‐based hospital medicine from a physician who simply works in the hospital?
There is no need for pediatric hospitalists to respond defensively to the hospitalist studies commissioned by the ABP. In fact, Freed46 has done us a favor by adding dimension and texture to the preliminary outlines of what it means for PHM to be ultimately successful. Both Freed and Melzer11 are describing the same path. As hospitalists, we tend to take pride in how far we have already come along this adventure. External observers such as Freed remind of us of how far we still need to go. Either way, Dorothy Gale, MD, pediatric hospitalist, has a relatively well‐identified yellow brick road to follow with specific challenges and charges to meet. What is unclear is whether formal acknowledgment will be awarded at the end of this journey and, if so, what form it will take. Options include (1) recognition of focused practice in hospital medicine with maintenance of certification, (2) SHM fellowship, (3) a traditionally boarded subspecialty, or (4) all of the above.
Any formal designation will be of secondary importance. Remember, the wizard did not change anything when he bestowed the diploma, the heart‐shaped testimonial, and the medal of valor. Like the scarecrow, tin man, and lion, all the qualities that we need for success as pediatric hospitalists are already within us. No wizard's pronouncements will help us provide better care to our patients. Change will come from working together on shared goals with mutual support along our common path. Look to the Journal of Hospital Medicine for frequent updates on the journey. See you in the Emerald City.
The March issue of the Journal of Hospital Medicine represents a landmark for pediatric hospital medicine (PHM), with 100% of the original research content devoted to pediatrics. Since the days of the National Association of Inpatient Physicians, pediatric hospitalists have consistently constituted 8% to 10% of the membership of the Society of Hospital Medicine (SHM). SHM has always welcomed pediatrics and pediatricians into the community of hospital medicine. A pediatrician has sat on the board since the founding of the National Association of Inpatient Physicians, and for the past 3 years, there has been a formal pediatric board seat. The Hospitalist has consistently included pediatric content with program descriptions and literature reviews. This past July, more than 325 pediatric hospitalists gathered in Denver for the largest PHM meeting ever, a 4‐day event trisponsored by SHM, the American Academy of Pediatrics (AAP), and the Academic Pediatric Association (APA).
As pediatric hospitalists, we have prospered by following the successes of adult hospitalists. We have flattered/emmitated our adult colleagues with pediatric voluntary referral policies, core competencies, salary surveys, fellowship programs, and quality improvement projects. In other areas, pediatrics has set trends for (adult) hospital medicine. Pediatrics developed the medical home concept. We zealously advocate for family‐centered rounds. (Imagine actually rounding in the room with the patient, family, nurse, and physician. It certainly beats flipping cards in the conference room)! Pediatricians have developed global fee codes for evaluation and management services (albeit limited to neonatal and pediatric critical care). As evidenced by the trisponsored meeting mentioned previously and the Pediatric Research in Inpatient Settings Network, we have created collaborative relationships among the pediatric academic (APA), professional (AAP), and hospitalist organizations (SHM) that serve as models for other disciplines and their respective sandboxes.
Research and publications are where we most lag behind our adult colleagues and where the most work needs to be done for us to achieve legitimacy as practitioners and as a discipline. This issue of the Journal of Hospital Medicine is a harbinger of more pediatric content to come, with topics that run the gamut of PHM. Woolford et al.1 highlight clinical, public health, and public policy issues with their analysis of the increased costs and morbidity associated with obesity and inpatient hospitalizations. Wilkes et al.2 explore the logistic issues surrounding influenza testing. As is frequently true for hospitalists, our expertise is not purely clinical: Is oseltamvir effective and, if so, in what age groups? That question is probably best left to the infectious disease community. Rather, Wilkes et al. highlight both the provider and system issues involved in reliably and expeditiously obtaining, reporting, and communicating flu antigen test results so that clinicians and families have the opportunity to consider oseltamvir use within the first 48 hours of disease. Odetola et al.'s3 analysis of a Michigan administrative data set suggests that morbidity, length of stay, and resource utilization are decreased for patients who ultimately require pediatric critical care when these patients are directly transferred from the emergency room to a facility with a pediatric intensive care unit (PICU) in comparison with the morbidity, length of stay, and resource utilization of patients who are initially admitted to the ward from the emergency room and then transferred to a facility with a PICU. This study lacks the rigor of prospectively collected physiological data and would probably never receive institutional review board approval for randomization, but it certainly raises key questions about appropriate transfer criteria for patients cared for in hospitals without a PICU. This is a key quality concern for pediatric hospitalists practicing in smaller, community hospital settings.
The 2 most controversial articles in this pediatric inpatient potpourri are the studies conducted by Freed and Kelly examining pediatric hospitalist training, practice, and career goals4 and PHM fellowship programs.5 These studies are part of a 6‐perspective analysis of pediatric hospitalists/PHM requested by the American Board of Pediatrics (ABP) to provide background to the ABP as it begins to grapple with its role in certifying pediatricians whose primary practice is inpatient pediatrics. A previously published study analyzed the perspective of PHM group leaders.6 The remaining studies assess the perspectives of residency program directors, department chairs, and hospital leaders.
Not surprisingly, these 3 articles46 tend to be more critical of the PHM movement and its current state than are articles and commentaries written by those of us who are practicing hospitalists. As a hospitalist, my initial reaction was to focus on the studies' shortcomings. The methods seemed flawed, the criticisms seemed unwarranted, and the study limitations seemed underappreciated. Aside from the fellowship study, which surveyed the entire n = 8 universe of PHM fellowship programs, the group leader and hospitalist surveys suffer from a selection bias. Sampling for these studies was based on hospital size and type. Although this sampling strategy is appropriate for comparing programs across hospitals, it fails to account for programs of different sizes in different settings. It is not the best sampling strategy for a denominator of all pediatric hospitalists. For example, community hospital programs without residents are often much bigger than academic programs with residents. Community pediatric hospitalists are likely underrepresented in Freed's survey.4 From a study design standpoint, it does not appear that specific a priori hypotheses were generated when subgroups were compared. Rather, one suspects that every possible comparison was analyzed. Thus, the percent differences from one group to another are best considered descriptive rather than rigorously statistically significant at a p 0.05 level. Some criticisms addressed to hospitalists apply to all pediatricians. Given the current emphasis on quality assessment, wouldn't most office‐based pediatricians (and particularly group leaders) believe that they need extra training in this field? When less than 50% of hospitals require practitioners in established subboarded specialties to be board‐certified to maintain hospital privileges,7 is it surprising to see that privileging standards vary for pediatric hospitalists?
However, nitpicking these studies is a defensive response that does a disservice both to the reports and more importantly to the PHM community as a whole and to the children, parents, and colleagues that we serve. There is no denying that we are a young, evolving field with significant inter‐institutional and at times intra‐institutional variability. All of us in the PHM community, leaders and lurkers, need to rise to the challenges offered by comprehensive analysis. Freed's sample of 431 hospitalists4 is significantly larger than the sample of 265 hospitalist participants in the latest Pediatric Research in Inpatient Settings survey.8 The perceptions of external observers are crucial; it would be a mistake to dismiss their findings or to ignore their interpretations and criticisms.
Certainly none would challenge the variability of practice revealed in Freed's analyses.46 Remember, if you've seen one pediatric hospital medicine program, you've seen ONE pediatric hospital medicine program. Some may see this variability as a weakness; others may see it as a strength. We must be equally receptive to other less‐flattering observations, data, and conclusions included in these reports to the ABP. All programs target seamless communication with referring physicians, but hospitalists and referring physicians alike agree that we do not achieve it, as evidenced by the work of Harlan et al.9 in this issue. SHM is taking the lead in developing performance standards for transitions of care and has created best discharge practices for the geriatric population.10 Similarly, we in the PHM community would do well to ramp up our self‐assessment and quality improvement activities. Our recusal from Centers for Medicare and Medicaid Services reporting requirements for (adult) inpatient quality metrics does not excuse us from pursuing voluntary, rigorous, transparent, public reporting on pediatric quality indicators. As Freed et al.6 clearly implied, the public and payers expect this of us. No doubt, if we do not first propose and implement our own standards, external standards will be imposed upon us.
Aside from the question of mandatory fellowship training for hospitalists, does the vision implied in the studies commissioned by the ABP vary significantly from the challenges to PHM that Sandy Melzer11 presented at his keynote address at the Denver meeting? Melzer used strategic planning principles to outline a future vision for PHM, including the following:
-
Harm is eliminated from the inpatient setting.
-
Inpatient care is evidence‐based for all conditions treated.
-
Hospital care is highly coordinated, especially for children with chronic conditions.
-
A robust research agenda supports all aspects of inpatient care.
Is not the work done by the SHM and APA to develop core competencies for PHM an effort to define our field and identify (uniform) expectations? Do not the criteria for designation as a fellow of hospital medicine (5 years as a practicing hospitalist; 2 national meetings; and a minimum combination of leadership, teamwork, and quality improvement activities)12 serve to recognize the commitment and accomplishments that distinguish a true hospitalist practicing systems‐based hospital medicine from a physician who simply works in the hospital?
There is no need for pediatric hospitalists to respond defensively to the hospitalist studies commissioned by the ABP. In fact, Freed46 has done us a favor by adding dimension and texture to the preliminary outlines of what it means for PHM to be ultimately successful. Both Freed and Melzer11 are describing the same path. As hospitalists, we tend to take pride in how far we have already come along this adventure. External observers such as Freed remind of us of how far we still need to go. Either way, Dorothy Gale, MD, pediatric hospitalist, has a relatively well‐identified yellow brick road to follow with specific challenges and charges to meet. What is unclear is whether formal acknowledgment will be awarded at the end of this journey and, if so, what form it will take. Options include (1) recognition of focused practice in hospital medicine with maintenance of certification, (2) SHM fellowship, (3) a traditionally boarded subspecialty, or (4) all of the above.
Any formal designation will be of secondary importance. Remember, the wizard did not change anything when he bestowed the diploma, the heart‐shaped testimonial, and the medal of valor. Like the scarecrow, tin man, and lion, all the qualities that we need for success as pediatric hospitalists are already within us. No wizard's pronouncements will help us provide better care to our patients. Change will come from working together on shared goals with mutual support along our common path. Look to the Journal of Hospital Medicine for frequent updates on the journey. See you in the Emerald City.
The March issue of the Journal of Hospital Medicine represents a landmark for pediatric hospital medicine (PHM), with 100% of the original research content devoted to pediatrics. Since the days of the National Association of Inpatient Physicians, pediatric hospitalists have consistently constituted 8% to 10% of the membership of the Society of Hospital Medicine (SHM). SHM has always welcomed pediatrics and pediatricians into the community of hospital medicine. A pediatrician has sat on the board since the founding of the National Association of Inpatient Physicians, and for the past 3 years, there has been a formal pediatric board seat. The Hospitalist has consistently included pediatric content with program descriptions and literature reviews. This past July, more than 325 pediatric hospitalists gathered in Denver for the largest PHM meeting ever, a 4‐day event trisponsored by SHM, the American Academy of Pediatrics (AAP), and the Academic Pediatric Association (APA).
As pediatric hospitalists, we have prospered by following the successes of adult hospitalists. We have flattered/emmitated our adult colleagues with pediatric voluntary referral policies, core competencies, salary surveys, fellowship programs, and quality improvement projects. In other areas, pediatrics has set trends for (adult) hospital medicine. Pediatrics developed the medical home concept. We zealously advocate for family‐centered rounds. (Imagine actually rounding in the room with the patient, family, nurse, and physician. It certainly beats flipping cards in the conference room)! Pediatricians have developed global fee codes for evaluation and management services (albeit limited to neonatal and pediatric critical care). As evidenced by the trisponsored meeting mentioned previously and the Pediatric Research in Inpatient Settings Network, we have created collaborative relationships among the pediatric academic (APA), professional (AAP), and hospitalist organizations (SHM) that serve as models for other disciplines and their respective sandboxes.
Research and publications are where we most lag behind our adult colleagues and where the most work needs to be done for us to achieve legitimacy as practitioners and as a discipline. This issue of the Journal of Hospital Medicine is a harbinger of more pediatric content to come, with topics that run the gamut of PHM. Woolford et al.1 highlight clinical, public health, and public policy issues with their analysis of the increased costs and morbidity associated with obesity and inpatient hospitalizations. Wilkes et al.2 explore the logistic issues surrounding influenza testing. As is frequently true for hospitalists, our expertise is not purely clinical: Is oseltamvir effective and, if so, in what age groups? That question is probably best left to the infectious disease community. Rather, Wilkes et al. highlight both the provider and system issues involved in reliably and expeditiously obtaining, reporting, and communicating flu antigen test results so that clinicians and families have the opportunity to consider oseltamvir use within the first 48 hours of disease. Odetola et al.'s3 analysis of a Michigan administrative data set suggests that morbidity, length of stay, and resource utilization are decreased for patients who ultimately require pediatric critical care when these patients are directly transferred from the emergency room to a facility with a pediatric intensive care unit (PICU) in comparison with the morbidity, length of stay, and resource utilization of patients who are initially admitted to the ward from the emergency room and then transferred to a facility with a PICU. This study lacks the rigor of prospectively collected physiological data and would probably never receive institutional review board approval for randomization, but it certainly raises key questions about appropriate transfer criteria for patients cared for in hospitals without a PICU. This is a key quality concern for pediatric hospitalists practicing in smaller, community hospital settings.
The 2 most controversial articles in this pediatric inpatient potpourri are the studies conducted by Freed and Kelly examining pediatric hospitalist training, practice, and career goals4 and PHM fellowship programs.5 These studies are part of a 6‐perspective analysis of pediatric hospitalists/PHM requested by the American Board of Pediatrics (ABP) to provide background to the ABP as it begins to grapple with its role in certifying pediatricians whose primary practice is inpatient pediatrics. A previously published study analyzed the perspective of PHM group leaders.6 The remaining studies assess the perspectives of residency program directors, department chairs, and hospital leaders.
Not surprisingly, these 3 articles46 tend to be more critical of the PHM movement and its current state than are articles and commentaries written by those of us who are practicing hospitalists. As a hospitalist, my initial reaction was to focus on the studies' shortcomings. The methods seemed flawed, the criticisms seemed unwarranted, and the study limitations seemed underappreciated. Aside from the fellowship study, which surveyed the entire n = 8 universe of PHM fellowship programs, the group leader and hospitalist surveys suffer from a selection bias. Sampling for these studies was based on hospital size and type. Although this sampling strategy is appropriate for comparing programs across hospitals, it fails to account for programs of different sizes in different settings. It is not the best sampling strategy for a denominator of all pediatric hospitalists. For example, community hospital programs without residents are often much bigger than academic programs with residents. Community pediatric hospitalists are likely underrepresented in Freed's survey.4 From a study design standpoint, it does not appear that specific a priori hypotheses were generated when subgroups were compared. Rather, one suspects that every possible comparison was analyzed. Thus, the percent differences from one group to another are best considered descriptive rather than rigorously statistically significant at a p 0.05 level. Some criticisms addressed to hospitalists apply to all pediatricians. Given the current emphasis on quality assessment, wouldn't most office‐based pediatricians (and particularly group leaders) believe that they need extra training in this field? When less than 50% of hospitals require practitioners in established subboarded specialties to be board‐certified to maintain hospital privileges,7 is it surprising to see that privileging standards vary for pediatric hospitalists?
However, nitpicking these studies is a defensive response that does a disservice both to the reports and more importantly to the PHM community as a whole and to the children, parents, and colleagues that we serve. There is no denying that we are a young, evolving field with significant inter‐institutional and at times intra‐institutional variability. All of us in the PHM community, leaders and lurkers, need to rise to the challenges offered by comprehensive analysis. Freed's sample of 431 hospitalists4 is significantly larger than the sample of 265 hospitalist participants in the latest Pediatric Research in Inpatient Settings survey.8 The perceptions of external observers are crucial; it would be a mistake to dismiss their findings or to ignore their interpretations and criticisms.
Certainly none would challenge the variability of practice revealed in Freed's analyses.46 Remember, if you've seen one pediatric hospital medicine program, you've seen ONE pediatric hospital medicine program. Some may see this variability as a weakness; others may see it as a strength. We must be equally receptive to other less‐flattering observations, data, and conclusions included in these reports to the ABP. All programs target seamless communication with referring physicians, but hospitalists and referring physicians alike agree that we do not achieve it, as evidenced by the work of Harlan et al.9 in this issue. SHM is taking the lead in developing performance standards for transitions of care and has created best discharge practices for the geriatric population.10 Similarly, we in the PHM community would do well to ramp up our self‐assessment and quality improvement activities. Our recusal from Centers for Medicare and Medicaid Services reporting requirements for (adult) inpatient quality metrics does not excuse us from pursuing voluntary, rigorous, transparent, public reporting on pediatric quality indicators. As Freed et al.6 clearly implied, the public and payers expect this of us. No doubt, if we do not first propose and implement our own standards, external standards will be imposed upon us.
Aside from the question of mandatory fellowship training for hospitalists, does the vision implied in the studies commissioned by the ABP vary significantly from the challenges to PHM that Sandy Melzer11 presented at his keynote address at the Denver meeting? Melzer used strategic planning principles to outline a future vision for PHM, including the following:
-
Harm is eliminated from the inpatient setting.
-
Inpatient care is evidence‐based for all conditions treated.
-
Hospital care is highly coordinated, especially for children with chronic conditions.
-
A robust research agenda supports all aspects of inpatient care.
Is not the work done by the SHM and APA to develop core competencies for PHM an effort to define our field and identify (uniform) expectations? Do not the criteria for designation as a fellow of hospital medicine (5 years as a practicing hospitalist; 2 national meetings; and a minimum combination of leadership, teamwork, and quality improvement activities)12 serve to recognize the commitment and accomplishments that distinguish a true hospitalist practicing systems‐based hospital medicine from a physician who simply works in the hospital?
There is no need for pediatric hospitalists to respond defensively to the hospitalist studies commissioned by the ABP. In fact, Freed46 has done us a favor by adding dimension and texture to the preliminary outlines of what it means for PHM to be ultimately successful. Both Freed and Melzer11 are describing the same path. As hospitalists, we tend to take pride in how far we have already come along this adventure. External observers such as Freed remind of us of how far we still need to go. Either way, Dorothy Gale, MD, pediatric hospitalist, has a relatively well‐identified yellow brick road to follow with specific challenges and charges to meet. What is unclear is whether formal acknowledgment will be awarded at the end of this journey and, if so, what form it will take. Options include (1) recognition of focused practice in hospital medicine with maintenance of certification, (2) SHM fellowship, (3) a traditionally boarded subspecialty, or (4) all of the above.
Any formal designation will be of secondary importance. Remember, the wizard did not change anything when he bestowed the diploma, the heart‐shaped testimonial, and the medal of valor. Like the scarecrow, tin man, and lion, all the qualities that we need for success as pediatric hospitalists are already within us. No wizard's pronouncements will help us provide better care to our patients. Change will come from working together on shared goals with mutual support along our common path. Look to the Journal of Hospital Medicine for frequent updates on the journey. See you in the Emerald City.
Predictors of Smoking and Relapse
Tobacco use in the United States is the chief avoidable cause of death in the United States.1 The health benefits of smoking cessation are widely known, including reductions in the risk for lung cancer, chronic obstructive pulmonary disease, and heart disease.2, 3 Particularly for patients with symptomatic coronary artery disease, smoking cessation reduces the risk of mortality by 30% to 50%.4, 5
Being hospitalized for a major cardiac event spurs many smokers to stop smoking. Acute and chronic health events are associated with a much lower likelihood of continued smoking, both immediately and over time. Cessation rates among smokers hospitalized for a cardiac condition, such as acute coronary syndrome (ACS), range from 31% without intervention to 60% with sustained intervention posthospitalization, at 1‐year follow‐up.610
Various studies have examined predictors of continued smoking among patients with heart disease. However, few studies have focused on prognostic factors in patients hospitalized for their heart condition, illustrating a gap in the literature. Factors found to affect smoking cessation rates have included: mood disorders, such as current or history of depression,6, 1113 a high level of state‐anxiety,13 and hostility or tensions;12 severity of disease, such as history of previous cardiac event,6, 9 history of smoking‐related pulmonary disease,6 severity of the cardiac disease,6, 12 having 1 or more risk factors for coronary artery disease other than smoking,14 or unstable angina;14 greater nicotine dependence or heavy smoking at index hospitalization;6, 9, 14, 15 and the presence of other smokers in the home/work environment.16
Data from a recently completed randomized controlled trial of a health behavior intervention within the context of hospital quality improvement provided the opportunity to study factors predictive of successfully quitting smoking in hospitalized cardiac patients. The description and results of that trial, called the Heart After Hospitalization Recovery Program (HARP), are reported elsewhere.17, 18 In summary, the health behavior intervention program studied in the trial was not successful in improving the smoking cessation rates above the control group receiving only the hospital quality improvement (QI) approach. Results of the QI intervention, the ACS Guidelines Applied to Practice (GAP) program, showed gains in survival that appeared to be due to better adherence to guidelines, which included a patient contract for behavior change.19, 20 Therefore, the purpose of this work is to describe all the preadmission smoking patients in the study, regardless of trial group assignment, and examine predictive factors for smoking cessation and relapse to smoking after their hospital discharge for ACS.
PATIENTS AND METHODS
The institutional review boards of the authors' university and each of the 5 participating hospitals approved the HARP study.
Settings and Subjects
Patients were recruited from 5 hospitals located in 2 adjacent counties in a Midwestern state. The 2 counties were similar: each had 1 major city surrounded by suburbs and outlying rural and farming areas, diverse populations with a minority population higher than the state average (20% versus 14.5%), a high unemployment rate (above 8%), and an industrial/manufacturing economic base.
Patient eligibility criteria included: admission to 1 of 5 participating study hospitals, a documented serum troponin I level greater than the upper limits of normal observed in each hospital, and a working diagnosis of ACS. Exclusion criteria included: discharge to any nonhome setting, possession of any significant mental/cognitive impairments, lack of a home telephone, or non‐English speaking. Trained nurse recruiters approached hospitalized patients, providing information on study participation and attempting to obtain consent. Recruitment occurred between January 14, 2002 and April 13, 2003. A mean number of 2.29 standard deviation (SD) 1.82 contacts were made with patients having elevated troponin levels to determine their actual eligibility.
Measures
Interview data were collected from patients at the following time points: shortly after hospital discharge (baseline), and 3 and 8 months postdischarge. Survey telephone calls lasted approximately 30 to 40 minutes and were conducted by trained survey researchers at the university's Institute for Public Policy and Social Research. Surveyors were blinded to the group assignment of the participants they were interviewing.
At the baseline interview, conducted between 1 and 4 weeks after discharge from the hospital (mean 14.11 9.6 days), patients answered questions of survey interviewers including demographic and background information, comorbid conditions (Charlson comorbidity index [CCI] method),21 history of depression, current depressive symptoms (Center for Epidemiological Studies‐Depression [CES‐D] tool)22, and tobacco use.
Smoking status at the time of hospitalization was established based on 2 sources of information: the medical record chart audits, and the baseline interview. The baseline interview asked Have you ever smoked tobacco? Respondents answering yes were asked: Do you smoke every day, some days, or not at all?; How many cigarettes do you now smoke per day on average?; and On average, when you smoked in the past 30 days, how many cigarettes did you smoke? Respondents who reported that they quit smoking were asked when they had quit. Comparing the time between the baseline interview and the original hospital admission, the research team could establish who quit by the time of the baseline interview, but had still been a smoker at the time of hospitalization. Therefore, this group of patients was considered smokers for the purpose of this study; defined as smoking at the time of hospitalization for this heart event. All other patients were treated as nonsmokers. Smoking status at hospitalization was then confirmed with the hospital medical record, which provided information on whether the patient was a current, ever, or never smoker, the years smoked, and the number of packs currently smoked per day.
In the subsequent 3‐month and 8‐month interviews, each patient's reported smoking status and frequency was reassessed through the items, Have there been any changes in your tobacco use in the past 3 months? and During the last month, have you smoked everyday, some days, or not at all? If the patient reported current smoking, then he/she was asked On the average, when you smoked during the past 30 days, about how many cigarettes did you smoke a day? If the patient reported quitting, he/she was asked How long ago did you quit smoking? Household smoking was assessed by the question In the past 30 days, has anyone, besides yourself, smoked cigarettes, cigars, or pipes anywhere inside your home?
Data Analysis
In addition to descriptive statistics characterizing the analysis sample, the analysis relies on multinomial logit regression models to predict who among the smoking ACS patients discharged would continue to smoke, would quit for the observation period, or would relapse.23, 24 Multinomial logit regression represents an extension of the more familiar binary logistic regression25 involving comparisons of all possible pairs of outcomes. Specifically, instead of employing separate logistic regression models to compare permanent quitters to continuing smokers, relapsers to continuing smoker, and permanent quitters to relapsers, these comparisons are contrasts in the multinomial model.
Thus, in addition to obtaining adjusted odds ratio (OR)s that take account of the simultaneous influence of all predictors, this analysis produces overall goodness‐of‐fit indicators and multivariate significance tests, which test whether coefficients associated with a particular independent variable have a simultaneous effect on the outcome across all categories. For instance, a multinomial P‐value of less than 0.05 for a subject's history of depression would indicate that such a history has a significant effect on whether or not subjects continue to smoke, quit for good, or relapse. The specific ORs and their associated significance levels then indicate if and to what degree prior depression affects each 2‐way contrast (quitters versus smokers, relapsers versus smokers, quitters versus relapsers). The analysis was conducted using the mlogit procedure of the STATA, 9.0 software package.26
RESULTS
Subjects
Of 719 consenting patients, 166 (23.1%) were initially assessed to be smokers based on their medical record documentation. Smoking status at hospitalization could not be verified and smoking status after hospitalization could not be determined for 15 patients who did not participate in any of the follow‐up interviews, therefore they were excluded. An additional 15 patients were excluded because of the lack of a second follow‐up interview, which would have allowed us to determine whether they quit smoking for at least 3 months or relapsed. Therefore, we include only the 136 cases with generally complete interview data in the current analysis. Table 1 depicts the demographic, medical, and smoking‐related characteristics of these patients.
| Variable | Number or Mean SD | Percentage |
|---|---|---|
| ||
| Age (years) | 53.32 9.52 | |
| Gender | ||
| Male | 83 | 61 |
| Female | 53 | 39 |
| White/nonwhite race | ||
| White | 112 | 82.4 |
| Nonwhite/multiracial/other | 24 | 17.6 |
| Marital status | ||
| Married | 80 | 59.3 |
| Divorced/separated/widowed | 56 | 40.7 |
| Work for pay or profit | ||
| Yes | 71 | 52.2 |
| No | 64 | 47.1 |
| Missing | 1 | 0.7 |
| Highest education | ||
| High‐school diploma or less | 79 | 58.1 |
| Some college or more | 57 | 41.9 |
| Family income | ||
| Less than $15,000 per year | 36 | 26.5 |
| $15,000 or more per year | 90 | 66.2 |
| No information | 10 | 7.4 |
| Ejection fraction (EF) | ||
| EF 35% | 14 | 10.3 |
| EF > 35% | 110 | 80.9 |
| Unmeasured | 12 | 8.8 |
| Number of comorbid conditions | 1.79 1.81 | |
| Number of persons living in household | 2.63 1.41 | |
| Past diagnosis of depression | ||
| Yes | 40 | 29.4 |
| No | 96 | 70.6 |
Significant Predictors of Cessation and Relapse
Of the 136 smokers who were interviewed at baseline and completed the follow‐up surveys, 45 continued to smoke at baseline and at subsequent interviews in which they participated (33.1%) were defined as continuing smokers. Sixty‐five patients quit smoking (quitters) and remained nonsmokers for the time of observationat least 1 more interview wave or an additional 3 months (47.8% of respondents). Twenty‐six of the 136 (19.1%) were relapsers. They reported quitting smoking shortly after their hospitalization, but reported smoking again at either the 3‐month or 8‐month interview. Smoking again was defined as answering every day or some days to the question During the last month, have you smoked every day, some days, or not at all?).
Table 2 shows the cross‐tabulations and Table 3 shows the result from the multinominal regression analysis identifying significant predictors of quitting and relapsing as compared to continued smoking, relapsing versus quitting. Initially, the model included patient age (continuous variable), sex, years of education, race/ethnicity (other versus Caucasian) and insurance status (Medicare, Medicaid, employer‐based private insurance, other private insurance, no insurance) among the predictor variables, but they were eliminated from the model based on the nonsignificance of the likelihood ratio chi square test associated with each of them. In the final model, we retained only significant predictor variables, except for 1: membership in the study groups (intervention group with coaching, intervention group without coaching, control group). To ease interpretation, the table displays 3 columns of the adjusted ORs, comparing all 2‐way comparisons of outcomes. The third column, comparing relapsers and quitters, is technically redundant, as its ORs represent the ratios of the other 2 columns, but the values may be of interest to readers. The multinomial P‐values in the right‐hand column confirm that, with the sole exception of study group membership, every remaining independent variable is a significant predictor of smoking status after hospital discharge.
| Independent Variables | Smokers [n (%)]* | Relapsers [n (%)] | Quitters [n (%)] | P‐Value |
|---|---|---|---|---|
| ||||
| Household income | ||||
| <$15,000 | 21 (58) | 6 (17) | 9 (25) | |
| $15,000+ | 22 (24) | 19 (21) | 59 (54) | 0.003 |
| Study group | ||||
| QI only | 22 (37) | 13 (21) | 25 (42) | |
| QI‐plus HARP (coached) | 14 (27) | 10 (20) | 27 (53) | |
| QI‐plus HARP (not coached) | 9 (36) | 3 (12) | 13 (52) | 0.644 |
| History of depression | ||||
| No | 30 (31) | 12 (13) | 54 (56) | |
| Yes | 14 (36) | 14 (36) | 11 (28) | 0.002 |
| Smokers in household | ||||
| No | 19 (23) | 11 (14) | 51 (63) | |
| Yes | 26 (47) | 15 (27) | 14 (25) | 0.000 |
| Intensity of smoking | ||||
| Moderate/heavy | 26 (29) | 13 (15) | 49 (56) | |
| Light | 19 (40) | 13 (27) | 16 (33) | 0.034 |
| Independent Variables | Successful Quitters* versus Smokers OR (95% CI) | Relapsers versus Smokers OR (95% CI) | Relapsers versus Successful Quitters* OR (95% CI) | Multinomial P Value |
|---|---|---|---|---|
| ||||
| Household income ($15,000)∥ | 0.007 | |||
| $15,000+ | 4.72 (1.69‐12.87) | 3.38 (1.09‐9.97) | 1.72 (0.85‐3.46) | 0.005 |
| Study group (intervention)∥ | 0.56 | |||
| QI‐plus HARP (uncoached) | 0.86 (0.24‐3.05) | 0.46 (0.10‐2.65) | 0.53 (0.26‐1.09) | 0.65 |
| QI only | 0.52 (0.19‐1.39) | 0.81 (0.28‐2.70) | 1.56 (0.85‐2.84) | 0.42 |
| History of depression (yes) | 0.42 (0.16‐1.41) | 2.66 (1.02‐7.49) | 6.38 (2.34‐17.34) | 0.007 |
| Smokers in household (yes) | 0.20 (0.08‐0.55) | 0.97 (0.32‐2.61) | 4.74 (1.57‐14.24) | 0.001 |
| Intensity of smoking (moderate to heavy)∥ | ||||
| Light | 0.20 (0.04‐0.99) | 1.29 (0.44‐3.56) | 0.16 (0.05‐0.57) | 0.08 |
As the data show, patients with higher household incomes have substantially higher odds of quitting than low‐income patients (OR = 4.72; P = 0.001); yet they also have greater odds of relapsing (OR = 3.38; P = 0.04). Patients with a history/past diagnosis of depression are not more likely to quit than those without a depression history; however, they have larger odds of relapsing back to smoking (ie, the OR for the comparison of relapsers versus smokers (OR = 2.66; P = 0.05) almost reaches the conventional significance level, while the OR for the contrast of relapsers versus quitters (OR = 6.38; P = 0.002) is significant and of substantial magnitude. By comparison, the presence of other smokers in a patient's household both lowers the odds of becoming a successful quitter (OR = 0.20; P = 0.001), and raises the odds of relapsing after initial quitting (OR = 4.74; P = 0.005). While lighter smokers (defined as <10 cigarettes a day) before hospitalization do not appear to be more successful in quitting than heavier smokers (defined as >20 cigarettes a day) (OR = 1.29; P = 0.62), they are less likely to relapse to smoking if they quit (OR = 0.16; P = 0.03).
Specific results relating to the telephone counseling intervention are found elsewhere.17, 18 However, we did include in Table 2 the specific results for the study groups to illustrate that the intervention program was not a factor predicting cessation. Analysis of the data using intention‐to‐treat (assuming all losses to follow‐up were continuing smokers) resulted in similar findings at the 8‐month follow‐up (2 = 2.635; degrees of freedom [df] = 2; P = 0.268).
DISCUSSION
The smoking cessation rate of 56.8% (n = 111; only those with 8‐month follow‐up) in this study population at 8 months compares favorably with the range of 31% to 60% shown in earlier studies of cardiac populations.610, 27 Assuming more conservatively that the survey nonresponders were all smokers yields a 46.3% quit rate (n = 136; all those with at least 1 follow‐up), which is within the range reported in the literature.
The intervention program was not a factor predicting cessation. Most posthospital follow‐up counseling is associated with increased smoking abstinence at follow‐up.28, 29 It is possible that the GAP in‐hospital QI initiative in these hospitals contributed to improving the cessation of smokers in both trial arms, thereby negating the effect of the counseling‐only option, although we did not specifically study the effect of the GAP intervention. It is also possible that we were underpowered to detect a statistically significant difference given our sample size of smokers.
Several characteristics were associated with successful smoking cessation in posthospitalized ACS patients. These included higher incomes, no other smokers in the household, and being a lighter smoker. We also found, however, that those with a history of depression, and heavier smokers also had higher rates of relapsing. As with previous research, our results support the evidence that heavier smokers have greater difficulty quitting smoking.6, 9, 14, 15 Heavier smoking indicates a greater nicotine addiction.27 However, 1 study of smoking cessation of smokers at a tertiary referral, cardiothoracic hospital found that smokers with greater pack years (eg, number of years smoked at an equivalent of 1 pack per day), had a higher likelihood of abstinence at a 12‐month follow‐up.30 More intense efforts are likely needed to assist smokers with a more significant addiction. Perhaps studies are needed to better understand the physiological and genetic mechanisms of nicotine addiction and effective treatment options for this group.
Our results also demonstrate that those with a history of depression were more likely to relapse. Several researchers have demonstrated that in patients with a history of depression, return of depressive symptoms upon a cessation attempt may precipitate relapse.28, 29 Current depressive symptoms, as measured by the CES‐D, were not associated with decreased rates of quitting or relapsing. After controlling for history of depression, the CES‐D score was no longer a predictor of quitting or relapsing in our data.
Similar to other studies, smokers in this study who reported having other smokers in the household had a more difficult time both quitting and remaining abstinent.16, 31 A related controversy concerns the efficacy of including (smoking and nonsmoking) family members in interventions to sustain longer‐term abstinence. Including family members has demonstrated efficacy in some research,3234 although the optimal means of involving family members in smoking cessation interventions has not yet been identified. Severity of cardiac disease (as measured by ejection fraction) and the presence of comorbid conditions were not found to be associated with smoking continuation or cessation. We did not find in this sample of ACS patients that smoking cessation rates increased with age during the follow‐up survey time points.
There are several limitations to our study. First, we did not biochemically validate self‐reported smoking cessation rates. However, it is generally found that self‐reports of cessation are accurate in research studies.35 Also participants may have incorrectly stated their quit rates due to recall bias. We were unable to fully capture use of smoking cessation pharmacotherapy (such as bupropion or nicotine replacement), which may have better explained success with cessation. Unfortunately, this is also not usually captured in the literature on studies of this nature. Last, since this study enrolled only cardiac patients in 2 similar community populations, these results may not be fully generalizable to other communities.
For smokers suffering from cardiac disease, there are few better ways to prevent a second heart event than quitting smoking. Judging from these results, there still remain a great number of hospitalized smokers who either choose to, or are unable to, successfully quit smoking, even after hospitalization for a serious cardiac event. Further research is needed to understand what individual motivating or household mechanisms may be best considered when encouraging this group of smokers to quit permanently.
Acknowledgements
Special thanks to Dr. Azfar Siddiqi for database management, Chrystal Price for data entry, and Camille Proden for chart abstraction and study recruitment. Supported by Agency for Health Research and Quality (AHRQ) grant number R01 HS 10531 (to M.H.‐R.).
- ,,,.Actual causes of death in the United States 2000.JAMA.2004;291(10):1238–1245.
- ,.Methods to enhance smoking cessation after myocardial infarction.Med Clin North Am.2000;84(1):63–80.
- .The problem of tobacco smoking.BMJ.2004;328:217–219.
- .Effects of cessation of smoking after myocardial infarction.J Cardiovasc Risk.1998;5(3):173–176.
- ,,,.Effect of smoking cessation on mortality after myocardial infarction.Ann Intern Med.2000;160:939–944.
- ,,,,,.Smoking habits and predictors of continued smoking in patients with acute coronary syndromes.JAdv Nurs.2004;46(6):614–623.
- ,,.Brief intervention during hospital admission to help patients to give up smoking after myocardial infarction and bypass surgery: randomised controlled trial.BMJ.2002;324(7329):87–89.
- ,,.The effects of counseling on smoking cessation among patients hospitalized with chronic obstructive pulmonary disease: a randomized clinical trial.Int J Addict.1991;26(1):107–119.
- ,.Randomised controlled trial of smoking cessation intervention after admission for coronary heart disease.BMJ.2003;327(7426):1254–1257.
- ,,,,.Registration and management of smoking behaviour in patients with coronary heart disease. The EUROASPIRE Survey.Eur Heart J.1999;20(22):1630–1637.
- ,,, et al.Depression and anxiety as predictors of outcome after myocardial infarction.Psychosom Med.2000;62(2):212–219.
- ,,, et al.Predictors of smoking cessation in patients with a diagnosis of coronary artery disease.J Cardiopulm Rehabil.2002;22(3):143–147.
- ,,, et al.Modification of smoking habits five months after myocardial infarction: relationship with personality characteristics.J Psychosom Res.1996;40(4):369–378.
- ,,, et al.Predictors of smoking cessation after percutaneous coronary revascularization.Mayo Clin Proc.1998;73(3):205–209.
- ,,,.Predictors of success in smoking cessation among hospitalized patients.Respirology.2005;10(1):63–69.
- ,,.Socio‐demographic predictors of quitting smoking: how important are household factors.Addiction.2004;99(6):770–777.
- ,,,,,.Health behavior goals of cardiac patients after hospitalization.Am J Health Behav.2006;30(4):387–399.
- ,,, et al.Does outpatient telephone coaching add to hospital quality improvement following hospitalization for acute coronary syndrome?J Gen Intern Med.2008;23(9):1464–1470.
- ,,, et al.Improving quality of care for acute myocardial infarction. The guidelines applied in practice (GAP) initiative.JAMA.2002;287(10):1269–1276.
- ,,, et al.Improving quality of care for acute myocardial infarction.JAMA.2002;287(10):1269–1276.
- ,,,.A new method of classifying prognostic comorbidity in longitudinal studies: development and validation.J Chronic Dis.1987;40(5):373–383.
- ,.Center for Epidemiologic Studies Depression Scale. In:Keyser DJ,Sweetland RC, eds.Test Critiques. Vol2.Kansas City, MO:Test Corporation;1985:144–160.
- .Categorical Data Analysis.New York, NY:Wiley 1990.
- .Regression Models for Categorical and Limited Dependent Variables.Thousand Oaks, CA:Sage;1997.
- ,.Applied Logistic Regression.New York, NY:John Wiley 1989.
- Stata Statistical Software: Release 9.College Station, TX:StataCorp LP;2005.
- ,,,,.Registration and management of smoking behaviour in patients with coronary heart disease. The EUROASPIRE Survey.Eur Heart J.1999;20(22):1630–1637.
- ,,,.Interventions for smoking cessation in hospitalized patients.Cochrane Database Syst Rev.2003;(1):CD001837.
- ,,.Smoking cessation interventions among hospitalized patients: what have we learned.Prev Med.2001;32(4):376–388.
- ,,, et al.Effectiveness of hospital‐based smoking cessation.Chest.2005;128(1):216–223.
- ,,.Reducing cardiovascular risk: identifying predictors of smoking relapse.Can J Cardiovasc Nurs.2003;13(3):7–12.
- ,,, et al.A randomized trial of a family‐based smoking prevention intervention in managed care.Prev Med.2003;37:617–626.
- ,,,,.Enhancing partner support to improve smoking cessation.Cochrane Database Syst Rev.2004;(3):CD002928.
- ,,,,,.Couple dynamics of change‐resistant smoking: toward a family consultation model.Fam Process.2001;40:115–131.
- ,,,.Factors associated with discrepancies between self‐reports on cigarette smoking and measured serum cotinine levels among persons aged 17 years or older: Third National Health and Nutrition Examination Survey, 1988–1994.Am J Epidemiol.2001;153(8):807–814.
Tobacco use in the United States is the chief avoidable cause of death in the United States.1 The health benefits of smoking cessation are widely known, including reductions in the risk for lung cancer, chronic obstructive pulmonary disease, and heart disease.2, 3 Particularly for patients with symptomatic coronary artery disease, smoking cessation reduces the risk of mortality by 30% to 50%.4, 5
Being hospitalized for a major cardiac event spurs many smokers to stop smoking. Acute and chronic health events are associated with a much lower likelihood of continued smoking, both immediately and over time. Cessation rates among smokers hospitalized for a cardiac condition, such as acute coronary syndrome (ACS), range from 31% without intervention to 60% with sustained intervention posthospitalization, at 1‐year follow‐up.610
Various studies have examined predictors of continued smoking among patients with heart disease. However, few studies have focused on prognostic factors in patients hospitalized for their heart condition, illustrating a gap in the literature. Factors found to affect smoking cessation rates have included: mood disorders, such as current or history of depression,6, 1113 a high level of state‐anxiety,13 and hostility or tensions;12 severity of disease, such as history of previous cardiac event,6, 9 history of smoking‐related pulmonary disease,6 severity of the cardiac disease,6, 12 having 1 or more risk factors for coronary artery disease other than smoking,14 or unstable angina;14 greater nicotine dependence or heavy smoking at index hospitalization;6, 9, 14, 15 and the presence of other smokers in the home/work environment.16
Data from a recently completed randomized controlled trial of a health behavior intervention within the context of hospital quality improvement provided the opportunity to study factors predictive of successfully quitting smoking in hospitalized cardiac patients. The description and results of that trial, called the Heart After Hospitalization Recovery Program (HARP), are reported elsewhere.17, 18 In summary, the health behavior intervention program studied in the trial was not successful in improving the smoking cessation rates above the control group receiving only the hospital quality improvement (QI) approach. Results of the QI intervention, the ACS Guidelines Applied to Practice (GAP) program, showed gains in survival that appeared to be due to better adherence to guidelines, which included a patient contract for behavior change.19, 20 Therefore, the purpose of this work is to describe all the preadmission smoking patients in the study, regardless of trial group assignment, and examine predictive factors for smoking cessation and relapse to smoking after their hospital discharge for ACS.
PATIENTS AND METHODS
The institutional review boards of the authors' university and each of the 5 participating hospitals approved the HARP study.
Settings and Subjects
Patients were recruited from 5 hospitals located in 2 adjacent counties in a Midwestern state. The 2 counties were similar: each had 1 major city surrounded by suburbs and outlying rural and farming areas, diverse populations with a minority population higher than the state average (20% versus 14.5%), a high unemployment rate (above 8%), and an industrial/manufacturing economic base.
Patient eligibility criteria included: admission to 1 of 5 participating study hospitals, a documented serum troponin I level greater than the upper limits of normal observed in each hospital, and a working diagnosis of ACS. Exclusion criteria included: discharge to any nonhome setting, possession of any significant mental/cognitive impairments, lack of a home telephone, or non‐English speaking. Trained nurse recruiters approached hospitalized patients, providing information on study participation and attempting to obtain consent. Recruitment occurred between January 14, 2002 and April 13, 2003. A mean number of 2.29 standard deviation (SD) 1.82 contacts were made with patients having elevated troponin levels to determine their actual eligibility.
Measures
Interview data were collected from patients at the following time points: shortly after hospital discharge (baseline), and 3 and 8 months postdischarge. Survey telephone calls lasted approximately 30 to 40 minutes and were conducted by trained survey researchers at the university's Institute for Public Policy and Social Research. Surveyors were blinded to the group assignment of the participants they were interviewing.
At the baseline interview, conducted between 1 and 4 weeks after discharge from the hospital (mean 14.11 9.6 days), patients answered questions of survey interviewers including demographic and background information, comorbid conditions (Charlson comorbidity index [CCI] method),21 history of depression, current depressive symptoms (Center for Epidemiological Studies‐Depression [CES‐D] tool)22, and tobacco use.
Smoking status at the time of hospitalization was established based on 2 sources of information: the medical record chart audits, and the baseline interview. The baseline interview asked Have you ever smoked tobacco? Respondents answering yes were asked: Do you smoke every day, some days, or not at all?; How many cigarettes do you now smoke per day on average?; and On average, when you smoked in the past 30 days, how many cigarettes did you smoke? Respondents who reported that they quit smoking were asked when they had quit. Comparing the time between the baseline interview and the original hospital admission, the research team could establish who quit by the time of the baseline interview, but had still been a smoker at the time of hospitalization. Therefore, this group of patients was considered smokers for the purpose of this study; defined as smoking at the time of hospitalization for this heart event. All other patients were treated as nonsmokers. Smoking status at hospitalization was then confirmed with the hospital medical record, which provided information on whether the patient was a current, ever, or never smoker, the years smoked, and the number of packs currently smoked per day.
In the subsequent 3‐month and 8‐month interviews, each patient's reported smoking status and frequency was reassessed through the items, Have there been any changes in your tobacco use in the past 3 months? and During the last month, have you smoked everyday, some days, or not at all? If the patient reported current smoking, then he/she was asked On the average, when you smoked during the past 30 days, about how many cigarettes did you smoke a day? If the patient reported quitting, he/she was asked How long ago did you quit smoking? Household smoking was assessed by the question In the past 30 days, has anyone, besides yourself, smoked cigarettes, cigars, or pipes anywhere inside your home?
Data Analysis
In addition to descriptive statistics characterizing the analysis sample, the analysis relies on multinomial logit regression models to predict who among the smoking ACS patients discharged would continue to smoke, would quit for the observation period, or would relapse.23, 24 Multinomial logit regression represents an extension of the more familiar binary logistic regression25 involving comparisons of all possible pairs of outcomes. Specifically, instead of employing separate logistic regression models to compare permanent quitters to continuing smokers, relapsers to continuing smoker, and permanent quitters to relapsers, these comparisons are contrasts in the multinomial model.
Thus, in addition to obtaining adjusted odds ratio (OR)s that take account of the simultaneous influence of all predictors, this analysis produces overall goodness‐of‐fit indicators and multivariate significance tests, which test whether coefficients associated with a particular independent variable have a simultaneous effect on the outcome across all categories. For instance, a multinomial P‐value of less than 0.05 for a subject's history of depression would indicate that such a history has a significant effect on whether or not subjects continue to smoke, quit for good, or relapse. The specific ORs and their associated significance levels then indicate if and to what degree prior depression affects each 2‐way contrast (quitters versus smokers, relapsers versus smokers, quitters versus relapsers). The analysis was conducted using the mlogit procedure of the STATA, 9.0 software package.26
RESULTS
Subjects
Of 719 consenting patients, 166 (23.1%) were initially assessed to be smokers based on their medical record documentation. Smoking status at hospitalization could not be verified and smoking status after hospitalization could not be determined for 15 patients who did not participate in any of the follow‐up interviews, therefore they were excluded. An additional 15 patients were excluded because of the lack of a second follow‐up interview, which would have allowed us to determine whether they quit smoking for at least 3 months or relapsed. Therefore, we include only the 136 cases with generally complete interview data in the current analysis. Table 1 depicts the demographic, medical, and smoking‐related characteristics of these patients.
| Variable | Number or Mean SD | Percentage |
|---|---|---|
| ||
| Age (years) | 53.32 9.52 | |
| Gender | ||
| Male | 83 | 61 |
| Female | 53 | 39 |
| White/nonwhite race | ||
| White | 112 | 82.4 |
| Nonwhite/multiracial/other | 24 | 17.6 |
| Marital status | ||
| Married | 80 | 59.3 |
| Divorced/separated/widowed | 56 | 40.7 |
| Work for pay or profit | ||
| Yes | 71 | 52.2 |
| No | 64 | 47.1 |
| Missing | 1 | 0.7 |
| Highest education | ||
| High‐school diploma or less | 79 | 58.1 |
| Some college or more | 57 | 41.9 |
| Family income | ||
| Less than $15,000 per year | 36 | 26.5 |
| $15,000 or more per year | 90 | 66.2 |
| No information | 10 | 7.4 |
| Ejection fraction (EF) | ||
| EF 35% | 14 | 10.3 |
| EF > 35% | 110 | 80.9 |
| Unmeasured | 12 | 8.8 |
| Number of comorbid conditions | 1.79 1.81 | |
| Number of persons living in household | 2.63 1.41 | |
| Past diagnosis of depression | ||
| Yes | 40 | 29.4 |
| No | 96 | 70.6 |
Significant Predictors of Cessation and Relapse
Of the 136 smokers who were interviewed at baseline and completed the follow‐up surveys, 45 continued to smoke at baseline and at subsequent interviews in which they participated (33.1%) were defined as continuing smokers. Sixty‐five patients quit smoking (quitters) and remained nonsmokers for the time of observationat least 1 more interview wave or an additional 3 months (47.8% of respondents). Twenty‐six of the 136 (19.1%) were relapsers. They reported quitting smoking shortly after their hospitalization, but reported smoking again at either the 3‐month or 8‐month interview. Smoking again was defined as answering every day or some days to the question During the last month, have you smoked every day, some days, or not at all?).
Table 2 shows the cross‐tabulations and Table 3 shows the result from the multinominal regression analysis identifying significant predictors of quitting and relapsing as compared to continued smoking, relapsing versus quitting. Initially, the model included patient age (continuous variable), sex, years of education, race/ethnicity (other versus Caucasian) and insurance status (Medicare, Medicaid, employer‐based private insurance, other private insurance, no insurance) among the predictor variables, but they were eliminated from the model based on the nonsignificance of the likelihood ratio chi square test associated with each of them. In the final model, we retained only significant predictor variables, except for 1: membership in the study groups (intervention group with coaching, intervention group without coaching, control group). To ease interpretation, the table displays 3 columns of the adjusted ORs, comparing all 2‐way comparisons of outcomes. The third column, comparing relapsers and quitters, is technically redundant, as its ORs represent the ratios of the other 2 columns, but the values may be of interest to readers. The multinomial P‐values in the right‐hand column confirm that, with the sole exception of study group membership, every remaining independent variable is a significant predictor of smoking status after hospital discharge.
| Independent Variables | Smokers [n (%)]* | Relapsers [n (%)] | Quitters [n (%)] | P‐Value |
|---|---|---|---|---|
| ||||
| Household income | ||||
| <$15,000 | 21 (58) | 6 (17) | 9 (25) | |
| $15,000+ | 22 (24) | 19 (21) | 59 (54) | 0.003 |
| Study group | ||||
| QI only | 22 (37) | 13 (21) | 25 (42) | |
| QI‐plus HARP (coached) | 14 (27) | 10 (20) | 27 (53) | |
| QI‐plus HARP (not coached) | 9 (36) | 3 (12) | 13 (52) | 0.644 |
| History of depression | ||||
| No | 30 (31) | 12 (13) | 54 (56) | |
| Yes | 14 (36) | 14 (36) | 11 (28) | 0.002 |
| Smokers in household | ||||
| No | 19 (23) | 11 (14) | 51 (63) | |
| Yes | 26 (47) | 15 (27) | 14 (25) | 0.000 |
| Intensity of smoking | ||||
| Moderate/heavy | 26 (29) | 13 (15) | 49 (56) | |
| Light | 19 (40) | 13 (27) | 16 (33) | 0.034 |
| Independent Variables | Successful Quitters* versus Smokers OR (95% CI) | Relapsers versus Smokers OR (95% CI) | Relapsers versus Successful Quitters* OR (95% CI) | Multinomial P Value |
|---|---|---|---|---|
| ||||
| Household income ($15,000)∥ | 0.007 | |||
| $15,000+ | 4.72 (1.69‐12.87) | 3.38 (1.09‐9.97) | 1.72 (0.85‐3.46) | 0.005 |
| Study group (intervention)∥ | 0.56 | |||
| QI‐plus HARP (uncoached) | 0.86 (0.24‐3.05) | 0.46 (0.10‐2.65) | 0.53 (0.26‐1.09) | 0.65 |
| QI only | 0.52 (0.19‐1.39) | 0.81 (0.28‐2.70) | 1.56 (0.85‐2.84) | 0.42 |
| History of depression (yes) | 0.42 (0.16‐1.41) | 2.66 (1.02‐7.49) | 6.38 (2.34‐17.34) | 0.007 |
| Smokers in household (yes) | 0.20 (0.08‐0.55) | 0.97 (0.32‐2.61) | 4.74 (1.57‐14.24) | 0.001 |
| Intensity of smoking (moderate to heavy)∥ | ||||
| Light | 0.20 (0.04‐0.99) | 1.29 (0.44‐3.56) | 0.16 (0.05‐0.57) | 0.08 |
As the data show, patients with higher household incomes have substantially higher odds of quitting than low‐income patients (OR = 4.72; P = 0.001); yet they also have greater odds of relapsing (OR = 3.38; P = 0.04). Patients with a history/past diagnosis of depression are not more likely to quit than those without a depression history; however, they have larger odds of relapsing back to smoking (ie, the OR for the comparison of relapsers versus smokers (OR = 2.66; P = 0.05) almost reaches the conventional significance level, while the OR for the contrast of relapsers versus quitters (OR = 6.38; P = 0.002) is significant and of substantial magnitude. By comparison, the presence of other smokers in a patient's household both lowers the odds of becoming a successful quitter (OR = 0.20; P = 0.001), and raises the odds of relapsing after initial quitting (OR = 4.74; P = 0.005). While lighter smokers (defined as <10 cigarettes a day) before hospitalization do not appear to be more successful in quitting than heavier smokers (defined as >20 cigarettes a day) (OR = 1.29; P = 0.62), they are less likely to relapse to smoking if they quit (OR = 0.16; P = 0.03).
Specific results relating to the telephone counseling intervention are found elsewhere.17, 18 However, we did include in Table 2 the specific results for the study groups to illustrate that the intervention program was not a factor predicting cessation. Analysis of the data using intention‐to‐treat (assuming all losses to follow‐up were continuing smokers) resulted in similar findings at the 8‐month follow‐up (2 = 2.635; degrees of freedom [df] = 2; P = 0.268).
DISCUSSION
The smoking cessation rate of 56.8% (n = 111; only those with 8‐month follow‐up) in this study population at 8 months compares favorably with the range of 31% to 60% shown in earlier studies of cardiac populations.610, 27 Assuming more conservatively that the survey nonresponders were all smokers yields a 46.3% quit rate (n = 136; all those with at least 1 follow‐up), which is within the range reported in the literature.
The intervention program was not a factor predicting cessation. Most posthospital follow‐up counseling is associated with increased smoking abstinence at follow‐up.28, 29 It is possible that the GAP in‐hospital QI initiative in these hospitals contributed to improving the cessation of smokers in both trial arms, thereby negating the effect of the counseling‐only option, although we did not specifically study the effect of the GAP intervention. It is also possible that we were underpowered to detect a statistically significant difference given our sample size of smokers.
Several characteristics were associated with successful smoking cessation in posthospitalized ACS patients. These included higher incomes, no other smokers in the household, and being a lighter smoker. We also found, however, that those with a history of depression, and heavier smokers also had higher rates of relapsing. As with previous research, our results support the evidence that heavier smokers have greater difficulty quitting smoking.6, 9, 14, 15 Heavier smoking indicates a greater nicotine addiction.27 However, 1 study of smoking cessation of smokers at a tertiary referral, cardiothoracic hospital found that smokers with greater pack years (eg, number of years smoked at an equivalent of 1 pack per day), had a higher likelihood of abstinence at a 12‐month follow‐up.30 More intense efforts are likely needed to assist smokers with a more significant addiction. Perhaps studies are needed to better understand the physiological and genetic mechanisms of nicotine addiction and effective treatment options for this group.
Our results also demonstrate that those with a history of depression were more likely to relapse. Several researchers have demonstrated that in patients with a history of depression, return of depressive symptoms upon a cessation attempt may precipitate relapse.28, 29 Current depressive symptoms, as measured by the CES‐D, were not associated with decreased rates of quitting or relapsing. After controlling for history of depression, the CES‐D score was no longer a predictor of quitting or relapsing in our data.
Similar to other studies, smokers in this study who reported having other smokers in the household had a more difficult time both quitting and remaining abstinent.16, 31 A related controversy concerns the efficacy of including (smoking and nonsmoking) family members in interventions to sustain longer‐term abstinence. Including family members has demonstrated efficacy in some research,3234 although the optimal means of involving family members in smoking cessation interventions has not yet been identified. Severity of cardiac disease (as measured by ejection fraction) and the presence of comorbid conditions were not found to be associated with smoking continuation or cessation. We did not find in this sample of ACS patients that smoking cessation rates increased with age during the follow‐up survey time points.
There are several limitations to our study. First, we did not biochemically validate self‐reported smoking cessation rates. However, it is generally found that self‐reports of cessation are accurate in research studies.35 Also participants may have incorrectly stated their quit rates due to recall bias. We were unable to fully capture use of smoking cessation pharmacotherapy (such as bupropion or nicotine replacement), which may have better explained success with cessation. Unfortunately, this is also not usually captured in the literature on studies of this nature. Last, since this study enrolled only cardiac patients in 2 similar community populations, these results may not be fully generalizable to other communities.
For smokers suffering from cardiac disease, there are few better ways to prevent a second heart event than quitting smoking. Judging from these results, there still remain a great number of hospitalized smokers who either choose to, or are unable to, successfully quit smoking, even after hospitalization for a serious cardiac event. Further research is needed to understand what individual motivating or household mechanisms may be best considered when encouraging this group of smokers to quit permanently.
Acknowledgements
Special thanks to Dr. Azfar Siddiqi for database management, Chrystal Price for data entry, and Camille Proden for chart abstraction and study recruitment. Supported by Agency for Health Research and Quality (AHRQ) grant number R01 HS 10531 (to M.H.‐R.).
Tobacco use in the United States is the chief avoidable cause of death in the United States.1 The health benefits of smoking cessation are widely known, including reductions in the risk for lung cancer, chronic obstructive pulmonary disease, and heart disease.2, 3 Particularly for patients with symptomatic coronary artery disease, smoking cessation reduces the risk of mortality by 30% to 50%.4, 5
Being hospitalized for a major cardiac event spurs many smokers to stop smoking. Acute and chronic health events are associated with a much lower likelihood of continued smoking, both immediately and over time. Cessation rates among smokers hospitalized for a cardiac condition, such as acute coronary syndrome (ACS), range from 31% without intervention to 60% with sustained intervention posthospitalization, at 1‐year follow‐up.610
Various studies have examined predictors of continued smoking among patients with heart disease. However, few studies have focused on prognostic factors in patients hospitalized for their heart condition, illustrating a gap in the literature. Factors found to affect smoking cessation rates have included: mood disorders, such as current or history of depression,6, 1113 a high level of state‐anxiety,13 and hostility or tensions;12 severity of disease, such as history of previous cardiac event,6, 9 history of smoking‐related pulmonary disease,6 severity of the cardiac disease,6, 12 having 1 or more risk factors for coronary artery disease other than smoking,14 or unstable angina;14 greater nicotine dependence or heavy smoking at index hospitalization;6, 9, 14, 15 and the presence of other smokers in the home/work environment.16
Data from a recently completed randomized controlled trial of a health behavior intervention within the context of hospital quality improvement provided the opportunity to study factors predictive of successfully quitting smoking in hospitalized cardiac patients. The description and results of that trial, called the Heart After Hospitalization Recovery Program (HARP), are reported elsewhere.17, 18 In summary, the health behavior intervention program studied in the trial was not successful in improving the smoking cessation rates above the control group receiving only the hospital quality improvement (QI) approach. Results of the QI intervention, the ACS Guidelines Applied to Practice (GAP) program, showed gains in survival that appeared to be due to better adherence to guidelines, which included a patient contract for behavior change.19, 20 Therefore, the purpose of this work is to describe all the preadmission smoking patients in the study, regardless of trial group assignment, and examine predictive factors for smoking cessation and relapse to smoking after their hospital discharge for ACS.
PATIENTS AND METHODS
The institutional review boards of the authors' university and each of the 5 participating hospitals approved the HARP study.
Settings and Subjects
Patients were recruited from 5 hospitals located in 2 adjacent counties in a Midwestern state. The 2 counties were similar: each had 1 major city surrounded by suburbs and outlying rural and farming areas, diverse populations with a minority population higher than the state average (20% versus 14.5%), a high unemployment rate (above 8%), and an industrial/manufacturing economic base.
Patient eligibility criteria included: admission to 1 of 5 participating study hospitals, a documented serum troponin I level greater than the upper limits of normal observed in each hospital, and a working diagnosis of ACS. Exclusion criteria included: discharge to any nonhome setting, possession of any significant mental/cognitive impairments, lack of a home telephone, or non‐English speaking. Trained nurse recruiters approached hospitalized patients, providing information on study participation and attempting to obtain consent. Recruitment occurred between January 14, 2002 and April 13, 2003. A mean number of 2.29 standard deviation (SD) 1.82 contacts were made with patients having elevated troponin levels to determine their actual eligibility.
Measures
Interview data were collected from patients at the following time points: shortly after hospital discharge (baseline), and 3 and 8 months postdischarge. Survey telephone calls lasted approximately 30 to 40 minutes and were conducted by trained survey researchers at the university's Institute for Public Policy and Social Research. Surveyors were blinded to the group assignment of the participants they were interviewing.
At the baseline interview, conducted between 1 and 4 weeks after discharge from the hospital (mean 14.11 9.6 days), patients answered questions of survey interviewers including demographic and background information, comorbid conditions (Charlson comorbidity index [CCI] method),21 history of depression, current depressive symptoms (Center for Epidemiological Studies‐Depression [CES‐D] tool)22, and tobacco use.
Smoking status at the time of hospitalization was established based on 2 sources of information: the medical record chart audits, and the baseline interview. The baseline interview asked Have you ever smoked tobacco? Respondents answering yes were asked: Do you smoke every day, some days, or not at all?; How many cigarettes do you now smoke per day on average?; and On average, when you smoked in the past 30 days, how many cigarettes did you smoke? Respondents who reported that they quit smoking were asked when they had quit. Comparing the time between the baseline interview and the original hospital admission, the research team could establish who quit by the time of the baseline interview, but had still been a smoker at the time of hospitalization. Therefore, this group of patients was considered smokers for the purpose of this study; defined as smoking at the time of hospitalization for this heart event. All other patients were treated as nonsmokers. Smoking status at hospitalization was then confirmed with the hospital medical record, which provided information on whether the patient was a current, ever, or never smoker, the years smoked, and the number of packs currently smoked per day.
In the subsequent 3‐month and 8‐month interviews, each patient's reported smoking status and frequency was reassessed through the items, Have there been any changes in your tobacco use in the past 3 months? and During the last month, have you smoked everyday, some days, or not at all? If the patient reported current smoking, then he/she was asked On the average, when you smoked during the past 30 days, about how many cigarettes did you smoke a day? If the patient reported quitting, he/she was asked How long ago did you quit smoking? Household smoking was assessed by the question In the past 30 days, has anyone, besides yourself, smoked cigarettes, cigars, or pipes anywhere inside your home?
Data Analysis
In addition to descriptive statistics characterizing the analysis sample, the analysis relies on multinomial logit regression models to predict who among the smoking ACS patients discharged would continue to smoke, would quit for the observation period, or would relapse.23, 24 Multinomial logit regression represents an extension of the more familiar binary logistic regression25 involving comparisons of all possible pairs of outcomes. Specifically, instead of employing separate logistic regression models to compare permanent quitters to continuing smokers, relapsers to continuing smoker, and permanent quitters to relapsers, these comparisons are contrasts in the multinomial model.
Thus, in addition to obtaining adjusted odds ratio (OR)s that take account of the simultaneous influence of all predictors, this analysis produces overall goodness‐of‐fit indicators and multivariate significance tests, which test whether coefficients associated with a particular independent variable have a simultaneous effect on the outcome across all categories. For instance, a multinomial P‐value of less than 0.05 for a subject's history of depression would indicate that such a history has a significant effect on whether or not subjects continue to smoke, quit for good, or relapse. The specific ORs and their associated significance levels then indicate if and to what degree prior depression affects each 2‐way contrast (quitters versus smokers, relapsers versus smokers, quitters versus relapsers). The analysis was conducted using the mlogit procedure of the STATA, 9.0 software package.26
RESULTS
Subjects
Of 719 consenting patients, 166 (23.1%) were initially assessed to be smokers based on their medical record documentation. Smoking status at hospitalization could not be verified and smoking status after hospitalization could not be determined for 15 patients who did not participate in any of the follow‐up interviews, therefore they were excluded. An additional 15 patients were excluded because of the lack of a second follow‐up interview, which would have allowed us to determine whether they quit smoking for at least 3 months or relapsed. Therefore, we include only the 136 cases with generally complete interview data in the current analysis. Table 1 depicts the demographic, medical, and smoking‐related characteristics of these patients.
| Variable | Number or Mean SD | Percentage |
|---|---|---|
| ||
| Age (years) | 53.32 9.52 | |
| Gender | ||
| Male | 83 | 61 |
| Female | 53 | 39 |
| White/nonwhite race | ||
| White | 112 | 82.4 |
| Nonwhite/multiracial/other | 24 | 17.6 |
| Marital status | ||
| Married | 80 | 59.3 |
| Divorced/separated/widowed | 56 | 40.7 |
| Work for pay or profit | ||
| Yes | 71 | 52.2 |
| No | 64 | 47.1 |
| Missing | 1 | 0.7 |
| Highest education | ||
| High‐school diploma or less | 79 | 58.1 |
| Some college or more | 57 | 41.9 |
| Family income | ||
| Less than $15,000 per year | 36 | 26.5 |
| $15,000 or more per year | 90 | 66.2 |
| No information | 10 | 7.4 |
| Ejection fraction (EF) | ||
| EF 35% | 14 | 10.3 |
| EF > 35% | 110 | 80.9 |
| Unmeasured | 12 | 8.8 |
| Number of comorbid conditions | 1.79 1.81 | |
| Number of persons living in household | 2.63 1.41 | |
| Past diagnosis of depression | ||
| Yes | 40 | 29.4 |
| No | 96 | 70.6 |
Significant Predictors of Cessation and Relapse
Of the 136 smokers who were interviewed at baseline and completed the follow‐up surveys, 45 continued to smoke at baseline and at subsequent interviews in which they participated (33.1%) were defined as continuing smokers. Sixty‐five patients quit smoking (quitters) and remained nonsmokers for the time of observationat least 1 more interview wave or an additional 3 months (47.8% of respondents). Twenty‐six of the 136 (19.1%) were relapsers. They reported quitting smoking shortly after their hospitalization, but reported smoking again at either the 3‐month or 8‐month interview. Smoking again was defined as answering every day or some days to the question During the last month, have you smoked every day, some days, or not at all?).
Table 2 shows the cross‐tabulations and Table 3 shows the result from the multinominal regression analysis identifying significant predictors of quitting and relapsing as compared to continued smoking, relapsing versus quitting. Initially, the model included patient age (continuous variable), sex, years of education, race/ethnicity (other versus Caucasian) and insurance status (Medicare, Medicaid, employer‐based private insurance, other private insurance, no insurance) among the predictor variables, but they were eliminated from the model based on the nonsignificance of the likelihood ratio chi square test associated with each of them. In the final model, we retained only significant predictor variables, except for 1: membership in the study groups (intervention group with coaching, intervention group without coaching, control group). To ease interpretation, the table displays 3 columns of the adjusted ORs, comparing all 2‐way comparisons of outcomes. The third column, comparing relapsers and quitters, is technically redundant, as its ORs represent the ratios of the other 2 columns, but the values may be of interest to readers. The multinomial P‐values in the right‐hand column confirm that, with the sole exception of study group membership, every remaining independent variable is a significant predictor of smoking status after hospital discharge.
| Independent Variables | Smokers [n (%)]* | Relapsers [n (%)] | Quitters [n (%)] | P‐Value |
|---|---|---|---|---|
| ||||
| Household income | ||||
| <$15,000 | 21 (58) | 6 (17) | 9 (25) | |
| $15,000+ | 22 (24) | 19 (21) | 59 (54) | 0.003 |
| Study group | ||||
| QI only | 22 (37) | 13 (21) | 25 (42) | |
| QI‐plus HARP (coached) | 14 (27) | 10 (20) | 27 (53) | |
| QI‐plus HARP (not coached) | 9 (36) | 3 (12) | 13 (52) | 0.644 |
| History of depression | ||||
| No | 30 (31) | 12 (13) | 54 (56) | |
| Yes | 14 (36) | 14 (36) | 11 (28) | 0.002 |
| Smokers in household | ||||
| No | 19 (23) | 11 (14) | 51 (63) | |
| Yes | 26 (47) | 15 (27) | 14 (25) | 0.000 |
| Intensity of smoking | ||||
| Moderate/heavy | 26 (29) | 13 (15) | 49 (56) | |
| Light | 19 (40) | 13 (27) | 16 (33) | 0.034 |
| Independent Variables | Successful Quitters* versus Smokers OR (95% CI) | Relapsers versus Smokers OR (95% CI) | Relapsers versus Successful Quitters* OR (95% CI) | Multinomial P Value |
|---|---|---|---|---|
| ||||
| Household income ($15,000)∥ | 0.007 | |||
| $15,000+ | 4.72 (1.69‐12.87) | 3.38 (1.09‐9.97) | 1.72 (0.85‐3.46) | 0.005 |
| Study group (intervention)∥ | 0.56 | |||
| QI‐plus HARP (uncoached) | 0.86 (0.24‐3.05) | 0.46 (0.10‐2.65) | 0.53 (0.26‐1.09) | 0.65 |
| QI only | 0.52 (0.19‐1.39) | 0.81 (0.28‐2.70) | 1.56 (0.85‐2.84) | 0.42 |
| History of depression (yes) | 0.42 (0.16‐1.41) | 2.66 (1.02‐7.49) | 6.38 (2.34‐17.34) | 0.007 |
| Smokers in household (yes) | 0.20 (0.08‐0.55) | 0.97 (0.32‐2.61) | 4.74 (1.57‐14.24) | 0.001 |
| Intensity of smoking (moderate to heavy)∥ | ||||
| Light | 0.20 (0.04‐0.99) | 1.29 (0.44‐3.56) | 0.16 (0.05‐0.57) | 0.08 |
As the data show, patients with higher household incomes have substantially higher odds of quitting than low‐income patients (OR = 4.72; P = 0.001); yet they also have greater odds of relapsing (OR = 3.38; P = 0.04). Patients with a history/past diagnosis of depression are not more likely to quit than those without a depression history; however, they have larger odds of relapsing back to smoking (ie, the OR for the comparison of relapsers versus smokers (OR = 2.66; P = 0.05) almost reaches the conventional significance level, while the OR for the contrast of relapsers versus quitters (OR = 6.38; P = 0.002) is significant and of substantial magnitude. By comparison, the presence of other smokers in a patient's household both lowers the odds of becoming a successful quitter (OR = 0.20; P = 0.001), and raises the odds of relapsing after initial quitting (OR = 4.74; P = 0.005). While lighter smokers (defined as <10 cigarettes a day) before hospitalization do not appear to be more successful in quitting than heavier smokers (defined as >20 cigarettes a day) (OR = 1.29; P = 0.62), they are less likely to relapse to smoking if they quit (OR = 0.16; P = 0.03).
Specific results relating to the telephone counseling intervention are found elsewhere.17, 18 However, we did include in Table 2 the specific results for the study groups to illustrate that the intervention program was not a factor predicting cessation. Analysis of the data using intention‐to‐treat (assuming all losses to follow‐up were continuing smokers) resulted in similar findings at the 8‐month follow‐up (2 = 2.635; degrees of freedom [df] = 2; P = 0.268).
DISCUSSION
The smoking cessation rate of 56.8% (n = 111; only those with 8‐month follow‐up) in this study population at 8 months compares favorably with the range of 31% to 60% shown in earlier studies of cardiac populations.610, 27 Assuming more conservatively that the survey nonresponders were all smokers yields a 46.3% quit rate (n = 136; all those with at least 1 follow‐up), which is within the range reported in the literature.
The intervention program was not a factor predicting cessation. Most posthospital follow‐up counseling is associated with increased smoking abstinence at follow‐up.28, 29 It is possible that the GAP in‐hospital QI initiative in these hospitals contributed to improving the cessation of smokers in both trial arms, thereby negating the effect of the counseling‐only option, although we did not specifically study the effect of the GAP intervention. It is also possible that we were underpowered to detect a statistically significant difference given our sample size of smokers.
Several characteristics were associated with successful smoking cessation in posthospitalized ACS patients. These included higher incomes, no other smokers in the household, and being a lighter smoker. We also found, however, that those with a history of depression, and heavier smokers also had higher rates of relapsing. As with previous research, our results support the evidence that heavier smokers have greater difficulty quitting smoking.6, 9, 14, 15 Heavier smoking indicates a greater nicotine addiction.27 However, 1 study of smoking cessation of smokers at a tertiary referral, cardiothoracic hospital found that smokers with greater pack years (eg, number of years smoked at an equivalent of 1 pack per day), had a higher likelihood of abstinence at a 12‐month follow‐up.30 More intense efforts are likely needed to assist smokers with a more significant addiction. Perhaps studies are needed to better understand the physiological and genetic mechanisms of nicotine addiction and effective treatment options for this group.
Our results also demonstrate that those with a history of depression were more likely to relapse. Several researchers have demonstrated that in patients with a history of depression, return of depressive symptoms upon a cessation attempt may precipitate relapse.28, 29 Current depressive symptoms, as measured by the CES‐D, were not associated with decreased rates of quitting or relapsing. After controlling for history of depression, the CES‐D score was no longer a predictor of quitting or relapsing in our data.
Similar to other studies, smokers in this study who reported having other smokers in the household had a more difficult time both quitting and remaining abstinent.16, 31 A related controversy concerns the efficacy of including (smoking and nonsmoking) family members in interventions to sustain longer‐term abstinence. Including family members has demonstrated efficacy in some research,3234 although the optimal means of involving family members in smoking cessation interventions has not yet been identified. Severity of cardiac disease (as measured by ejection fraction) and the presence of comorbid conditions were not found to be associated with smoking continuation or cessation. We did not find in this sample of ACS patients that smoking cessation rates increased with age during the follow‐up survey time points.
There are several limitations to our study. First, we did not biochemically validate self‐reported smoking cessation rates. However, it is generally found that self‐reports of cessation are accurate in research studies.35 Also participants may have incorrectly stated their quit rates due to recall bias. We were unable to fully capture use of smoking cessation pharmacotherapy (such as bupropion or nicotine replacement), which may have better explained success with cessation. Unfortunately, this is also not usually captured in the literature on studies of this nature. Last, since this study enrolled only cardiac patients in 2 similar community populations, these results may not be fully generalizable to other communities.
For smokers suffering from cardiac disease, there are few better ways to prevent a second heart event than quitting smoking. Judging from these results, there still remain a great number of hospitalized smokers who either choose to, or are unable to, successfully quit smoking, even after hospitalization for a serious cardiac event. Further research is needed to understand what individual motivating or household mechanisms may be best considered when encouraging this group of smokers to quit permanently.
Acknowledgements
Special thanks to Dr. Azfar Siddiqi for database management, Chrystal Price for data entry, and Camille Proden for chart abstraction and study recruitment. Supported by Agency for Health Research and Quality (AHRQ) grant number R01 HS 10531 (to M.H.‐R.).
- ,,,.Actual causes of death in the United States 2000.JAMA.2004;291(10):1238–1245.
- ,.Methods to enhance smoking cessation after myocardial infarction.Med Clin North Am.2000;84(1):63–80.
- .The problem of tobacco smoking.BMJ.2004;328:217–219.
- .Effects of cessation of smoking after myocardial infarction.J Cardiovasc Risk.1998;5(3):173–176.
- ,,,.Effect of smoking cessation on mortality after myocardial infarction.Ann Intern Med.2000;160:939–944.
- ,,,,,.Smoking habits and predictors of continued smoking in patients with acute coronary syndromes.JAdv Nurs.2004;46(6):614–623.
- ,,.Brief intervention during hospital admission to help patients to give up smoking after myocardial infarction and bypass surgery: randomised controlled trial.BMJ.2002;324(7329):87–89.
- ,,.The effects of counseling on smoking cessation among patients hospitalized with chronic obstructive pulmonary disease: a randomized clinical trial.Int J Addict.1991;26(1):107–119.
- ,.Randomised controlled trial of smoking cessation intervention after admission for coronary heart disease.BMJ.2003;327(7426):1254–1257.
- ,,,,.Registration and management of smoking behaviour in patients with coronary heart disease. The EUROASPIRE Survey.Eur Heart J.1999;20(22):1630–1637.
- ,,, et al.Depression and anxiety as predictors of outcome after myocardial infarction.Psychosom Med.2000;62(2):212–219.
- ,,, et al.Predictors of smoking cessation in patients with a diagnosis of coronary artery disease.J Cardiopulm Rehabil.2002;22(3):143–147.
- ,,, et al.Modification of smoking habits five months after myocardial infarction: relationship with personality characteristics.J Psychosom Res.1996;40(4):369–378.
- ,,, et al.Predictors of smoking cessation after percutaneous coronary revascularization.Mayo Clin Proc.1998;73(3):205–209.
- ,,,.Predictors of success in smoking cessation among hospitalized patients.Respirology.2005;10(1):63–69.
- ,,.Socio‐demographic predictors of quitting smoking: how important are household factors.Addiction.2004;99(6):770–777.
- ,,,,,.Health behavior goals of cardiac patients after hospitalization.Am J Health Behav.2006;30(4):387–399.
- ,,, et al.Does outpatient telephone coaching add to hospital quality improvement following hospitalization for acute coronary syndrome?J Gen Intern Med.2008;23(9):1464–1470.
- ,,, et al.Improving quality of care for acute myocardial infarction. The guidelines applied in practice (GAP) initiative.JAMA.2002;287(10):1269–1276.
- ,,, et al.Improving quality of care for acute myocardial infarction.JAMA.2002;287(10):1269–1276.
- ,,,.A new method of classifying prognostic comorbidity in longitudinal studies: development and validation.J Chronic Dis.1987;40(5):373–383.
- ,.Center for Epidemiologic Studies Depression Scale. In:Keyser DJ,Sweetland RC, eds.Test Critiques. Vol2.Kansas City, MO:Test Corporation;1985:144–160.
- .Categorical Data Analysis.New York, NY:Wiley 1990.
- .Regression Models for Categorical and Limited Dependent Variables.Thousand Oaks, CA:Sage;1997.
- ,.Applied Logistic Regression.New York, NY:John Wiley 1989.
- Stata Statistical Software: Release 9.College Station, TX:StataCorp LP;2005.
- ,,,,.Registration and management of smoking behaviour in patients with coronary heart disease. The EUROASPIRE Survey.Eur Heart J.1999;20(22):1630–1637.
- ,,,.Interventions for smoking cessation in hospitalized patients.Cochrane Database Syst Rev.2003;(1):CD001837.
- ,,.Smoking cessation interventions among hospitalized patients: what have we learned.Prev Med.2001;32(4):376–388.
- ,,, et al.Effectiveness of hospital‐based smoking cessation.Chest.2005;128(1):216–223.
- ,,.Reducing cardiovascular risk: identifying predictors of smoking relapse.Can J Cardiovasc Nurs.2003;13(3):7–12.
- ,,, et al.A randomized trial of a family‐based smoking prevention intervention in managed care.Prev Med.2003;37:617–626.
- ,,,,.Enhancing partner support to improve smoking cessation.Cochrane Database Syst Rev.2004;(3):CD002928.
- ,,,,,.Couple dynamics of change‐resistant smoking: toward a family consultation model.Fam Process.2001;40:115–131.
- ,,,.Factors associated with discrepancies between self‐reports on cigarette smoking and measured serum cotinine levels among persons aged 17 years or older: Third National Health and Nutrition Examination Survey, 1988–1994.Am J Epidemiol.2001;153(8):807–814.
- ,,,.Actual causes of death in the United States 2000.JAMA.2004;291(10):1238–1245.
- ,.Methods to enhance smoking cessation after myocardial infarction.Med Clin North Am.2000;84(1):63–80.
- .The problem of tobacco smoking.BMJ.2004;328:217–219.
- .Effects of cessation of smoking after myocardial infarction.J Cardiovasc Risk.1998;5(3):173–176.
- ,,,.Effect of smoking cessation on mortality after myocardial infarction.Ann Intern Med.2000;160:939–944.
- ,,,,,.Smoking habits and predictors of continued smoking in patients with acute coronary syndromes.JAdv Nurs.2004;46(6):614–623.
- ,,.Brief intervention during hospital admission to help patients to give up smoking after myocardial infarction and bypass surgery: randomised controlled trial.BMJ.2002;324(7329):87–89.
- ,,.The effects of counseling on smoking cessation among patients hospitalized with chronic obstructive pulmonary disease: a randomized clinical trial.Int J Addict.1991;26(1):107–119.
- ,.Randomised controlled trial of smoking cessation intervention after admission for coronary heart disease.BMJ.2003;327(7426):1254–1257.
- ,,,,.Registration and management of smoking behaviour in patients with coronary heart disease. The EUROASPIRE Survey.Eur Heart J.1999;20(22):1630–1637.
- ,,, et al.Depression and anxiety as predictors of outcome after myocardial infarction.Psychosom Med.2000;62(2):212–219.
- ,,, et al.Predictors of smoking cessation in patients with a diagnosis of coronary artery disease.J Cardiopulm Rehabil.2002;22(3):143–147.
- ,,, et al.Modification of smoking habits five months after myocardial infarction: relationship with personality characteristics.J Psychosom Res.1996;40(4):369–378.
- ,,, et al.Predictors of smoking cessation after percutaneous coronary revascularization.Mayo Clin Proc.1998;73(3):205–209.
- ,,,.Predictors of success in smoking cessation among hospitalized patients.Respirology.2005;10(1):63–69.
- ,,.Socio‐demographic predictors of quitting smoking: how important are household factors.Addiction.2004;99(6):770–777.
- ,,,,,.Health behavior goals of cardiac patients after hospitalization.Am J Health Behav.2006;30(4):387–399.
- ,,, et al.Does outpatient telephone coaching add to hospital quality improvement following hospitalization for acute coronary syndrome?J Gen Intern Med.2008;23(9):1464–1470.
- ,,, et al.Improving quality of care for acute myocardial infarction. The guidelines applied in practice (GAP) initiative.JAMA.2002;287(10):1269–1276.
- ,,, et al.Improving quality of care for acute myocardial infarction.JAMA.2002;287(10):1269–1276.
- ,,,.A new method of classifying prognostic comorbidity in longitudinal studies: development and validation.J Chronic Dis.1987;40(5):373–383.
- ,.Center for Epidemiologic Studies Depression Scale. In:Keyser DJ,Sweetland RC, eds.Test Critiques. Vol2.Kansas City, MO:Test Corporation;1985:144–160.
- .Categorical Data Analysis.New York, NY:Wiley 1990.
- .Regression Models for Categorical and Limited Dependent Variables.Thousand Oaks, CA:Sage;1997.
- ,.Applied Logistic Regression.New York, NY:John Wiley 1989.
- Stata Statistical Software: Release 9.College Station, TX:StataCorp LP;2005.
- ,,,,.Registration and management of smoking behaviour in patients with coronary heart disease. The EUROASPIRE Survey.Eur Heart J.1999;20(22):1630–1637.
- ,,,.Interventions for smoking cessation in hospitalized patients.Cochrane Database Syst Rev.2003;(1):CD001837.
- ,,.Smoking cessation interventions among hospitalized patients: what have we learned.Prev Med.2001;32(4):376–388.
- ,,, et al.Effectiveness of hospital‐based smoking cessation.Chest.2005;128(1):216–223.
- ,,.Reducing cardiovascular risk: identifying predictors of smoking relapse.Can J Cardiovasc Nurs.2003;13(3):7–12.
- ,,, et al.A randomized trial of a family‐based smoking prevention intervention in managed care.Prev Med.2003;37:617–626.
- ,,,,.Enhancing partner support to improve smoking cessation.Cochrane Database Syst Rev.2004;(3):CD002928.
- ,,,,,.Couple dynamics of change‐resistant smoking: toward a family consultation model.Fam Process.2001;40:115–131.
- ,,,.Factors associated with discrepancies between self‐reports on cigarette smoking and measured serum cotinine levels among persons aged 17 years or older: Third National Health and Nutrition Examination Survey, 1988–1994.Am J Epidemiol.2001;153(8):807–814.
Copyright © 2009 Society of Hospital Medicine
Hospital Charges for Childhood Obesity
With increases in the prevalence of obesity among children and adults over the past 3 decades in the United States,13 healthcare expenditures attributed to obesity have climbed steadily, to over $100 billion in excess expenditures annually.4 Several studies have examined healthcare costs associated with obesity in adults,48 but these studies have not attempted to distinguish between excess expenditures in inpatient versus outpatient settings. In contrast, the only 2 national economic analyses of childhood obesity have focused exclusively on the inpatient setting, because the health and economic consequences of obesity among children may be most apparent in cases in which obesity is causally linked to other diagnoses (eg, type 2 diabetes mellitus, gall bladder disease) or is a comorbidity that complicates hospitalizations.9, 10
In our previous study of obesity as a comorbidity for hospitalized children, we examined the incremental charges and length‐of‐stay (LOS) for hospitalizations for the most common nonpregnancy/nonchildbirth pediatric diagnoses, comparing those coded with obesity as a secondary diagnosis versus those without.10 Using data from the Agency for Healthcare Research and Quality (AHRQ) Kid's Inpatient Database (KID) for the year 2000, we found that obesity was associated with higher charges and longer LOS for all 4 of the conditions studied (asthma, pneumonia, affective disorders, and appendicitis). Our findings regarding asthma and affective disorders echoed earlier analyses of hospitalizations for conditions clinically linked to obesity.9 However, our study was the first to demonstrate that childhood obesity is a clinically and economically significant complicating factor for conditions not thought to be linked to obesity (pneumonia, appendicitis).
For this current study, our objective was to use more recent child hospitalization data from 2003 to determine whether our prior findings of incremental charges and LOS associated with hospitalizations where obesity was coded as a secondary diagnosis compared to those where it was not, were stable over time, and whether the magnitude of differences was consistent over a period of 4 years. We hypothesized that incremental differences in hospital charges and LOS between discharges with and without obesity would be seen in the 2003 data and that the magnitude of these differences in 2003 would be similar to those in 2000. Because obesity prevalence among children increased from 2000 to 2003,11 we also hypothesized that there would be a corresponding increase in the proportion of hospitalizations with obesity as a comorbidity.
METHODS
Data Source and Sample
We analyzed data from the AHRQ KID. The KID is a nationally representative sample of annual pediatric hospital discharges. Analysis using the KID allows for improved estimates due to the discharges from community, nonrehabilitation hospitals.12 It provides data found in standard hospital discharge abstracts for more than 2 million pediatric discharges, including International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes, LOS, total hospital charges, and patient demographic information.12 Our original analysis (published in Obesity, July 2007)10 utilized data from the 2000 KID. For this analysis we utilized data from the 2003 KID (the most recent version available), and for comparison we converted the results from the 2000 study into 2003 dollars using the Consumer Price Index for Medical Care.
Using ICD‐9‐CM codes, the KID provides the principal diagnosis for each discharge, along with up to 14 secondary diagnoses. It also provides Clinical Classification Software (CCS) codes, a diagnostic categorization scheme that permits grouping of related conditions. ICD‐9‐CM codes are collapsed into a smaller number of categories that are sometimes more useful for presenting descriptive statistics than are individual ICD‐9‐CM codes or the much broader categories of Diagnosis Related Groups (DRGs). For example, all ICD‐9‐CM codes for specific types of pneumonia would be grouped together under 1 CCS code but would exclude other respiratory conditions such as pneumothorax which would generally be included in the respiratory condition DRG.12
The 2000 KID contained 2,516,833 unweighted discharges, representing 7,291,038 discharges in the population. In the 2003 KID, there were 2,984,129 unweighted discharges representing 7,409,162 discharges in the population. Our sample included all discharges for nonpregnancy‐related conditions, in children 2 years of age (due to the Centers for Disease Control definition for overweight based on body mass index [BMI] that starts at age 2 years)13, 14 to 18 years of age (2000 weighted n = 1,527,309; 2003 weighted n = 1,613,258); these numbers exclude discharges with obesity as a primary diagnosis.
Key Variables
Our outcome variables were LOS and total charges for each of the common nonpregnancy‐related principal discharge diagnoses studied. For the 2000 and 2003 KID, total charges included all hospital fees with the exception of professional fees.15
The main independent variable of interest was presence of obesity as a secondary diagnosis. Discharges were classified as either with or without obesity based on the presence of the ICD‐9‐CM code 278.0x as a secondary diagnosis (1 if yes, 0 if no). This code captures obesity unspecified (278.00), overweight (278.01), and morbid obesity (278.02). Of note, the distribution of these codes did not vary significantly between the 2 study years.
Other independent variables included sex, age (2‐5 years, 6‐10 years, 11‐14 years, and 15‐18 years), race/ethnicity (white, black, Hispanic, and other), region (Northeast, Midwest, South, and West), hospital type (based on classification by the National Association of Children's Hospitals and Related Institutions [NACHRI] as general hospital, children's unit in a general hospital, and children's hospital) and expected primary payer (Medicaid, private, and other). We chose independent variables due to their established association with our outcomes and for their patterns of association with childhood obesity.
We did not include LOS as a covariate in models of charges because obesity may have been associated with the outcome indirectly through LOS as well as directly as a main effect. To fully interpret the combination of these effects would require analyses beyond the scope of this work.
Analyses
Using CCS codes, we identified the 4 most common principal nonpregnancy‐related discharge diagnoses for children 2‐18 years old. For both 2000 and 2003 these were asthma (CCS 128), pneumonia (CCS 122), affective disorders (eg, depression and bipolar disorder) (CCS 69), and appendicitis (CCS 142). Importantly, this group of diagnoses included conditions clinically associated with obesity (asthma and affective disorders)16 and conditions not associated with obesity (pneumonia and appendicitis). Given this distinction we analyzed the 4 conditions separately.
For all discharges with these common principal diagnoses, we calculated mean LOS and mean total charges. Bivariate and multivariable analyses were conducted using simple and multiple linear regression, respectively. These analyses were designed to test the study hypothesis that obesity as a secondary diagnosis is associated with incremental economic charges and LOS. All analyses were performed on log‐transformed LOS and charge data. We included in our models those characteristics that we hypothesized were potentially related to hospital charges and LOS, based on published literature,4, 17, 18 and for that reason all covariates were retained in the final models regardless of bivariate findings. Differences in the incremental LOS and charges for 2003 versus 2000 were compared using t‐tests.
For each independent variable with missing data, we included in the analyses a category for unreported values. The KID is known to have a large number of missing data for race/ethnicity; therefore, in keeping with other studies utilizing the KID,10, 19 we also conducted multivariate analyses excluding those discharges with unreported race as a sensitivity analysis. Of note, analyses of these data excluding children of unreported race were not substantively different than those in which the unreported group was included. We present our findings including the unreported race category.
Predicted values on the log scale, for those with obesity and without obesity as a secondary diagnosis adjusted for the listed covariates, were obtained. We then back‐transformed these to their original scales and units using methods developed by Duan.20 For each principal diagnosis category, we analyzed the differences in predicted mean LOS and predicted mean total charges between discharges with and without obesity as a secondary diagnosis, adjusted for the listed covariates, using the P values obtained from the regression analyses. All results are presented in 2003 dollars.
In keeping with our earlier analysis we considered the potential influence of comorbidities other than obesity on LOS and charges. Therefore, we examined whether other comorbidities were coded more frequently among those discharges with obesity as a secondary diagnosis than those discharges without obesity. As seen in the 2000 KID, the 2003 data revealed that diabetes was more commonly seen with obesity‐related hospitalizations than with those hospitalizations without obesity. However, the proportion of discharges with obesity and diabetes was low for all of the principal diagnostic categories studied (asthma 4.8%, pneumonia 6.0%, affective disorders 4.2%, and appendicitis 1.9%). Thus, we judged the co‐occurrence of diabetes and obesity as secondary diagnoses too infrequent to be an explanatory factor for the overall incremental differences in LOS and charges.
All analyses were weighted to account for the complex probability sampling of the dataset and permit inferences regarding national hospital discharge patterns. The same sample of discharges was used to analyze LOS and charges, with the discharge weighting variable (DISCWT) used for all analyses. All results are presented as weighted data unless otherwise noted. Analyses were conducted using STATA 8.0 (Stata Corporation, College Station, TX) and SUDAAN version 9 (Research Triangle Institute, Research Triangle Park, NC).
This study was approved by the Institutional Review Board of the University of Michigan Medical School.
RESULTS
Sample Characteristics
The characteristics of the study population are presented in Table 1. In 2003, the overall proportion of nonpregnancy‐related discharges for children 2‐18 years old coded with obesity as a secondary diagnosis was 1.6%, an increase from 1.1% in 2000. Within the 4 most common nonpregnancy‐related CCS category diagnoses (asthma, pneumonia, affective disorders, and appendicitis) the proportion of discharges with obesity coded as a secondary diagnosis increased from 2000 to 2003 (Figure 1)
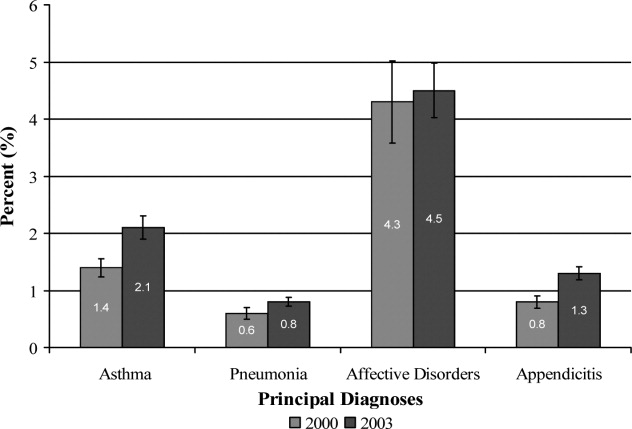
| Variables | Discharges | |||
|---|---|---|---|---|
| With Obesity as Secondary Diagnosis | Without Obesity | |||
| 2000 | 2003 | 2000 | 2003 | |
| Unweighted, n | 8,696 | 15,546 | 762,407 | 943,182 |
| Weighted population size | 17,672 | 25,709 | 1,509,637 | 1,587,549 |
| Age | ||||
| 2‐5 years (%) | 4.8 | 4.8 | 27.4 | 29.0 |
| 6‐10 years (%) | 14.7 | 15.9 | 22.4 | 22.3 |
| 11‐14 years (%) | 34.4 | 34.1 | 20.9 | 20.9 |
| 15‐18 years (%) | 46.1 | 45.2 | 29.3 | 27.8 |
| Sex | ||||
| Male (%) | 45.5 | 46.8 | 53.8 | 53.4 |
| Race/Ethnicity | ||||
| White (%) | 46.8 | 32.2 | 50.9 | 39.3 |
| Black (%) | 20.7 | 20.4 | 14.3 | 12.6 |
| Hispanic (%) | 14.9 | 16.8 | 14.3 | 14.4 |
| Other (%) | 4.8 | 5.0 | 5.6 | 5.6 |
| Unreported (%) | 12.8 | 25.6 | 14.9 | 28.1 |
| Payer | ||||
| Medicaid (%) | 42.6 | 46.5 | 32.1 | 36.9 |
| Private (%) | 47.1 | 42.6 | 58.0 | 53.3 |
| Other (%) | 9.4 | 10.6 | 9.4 | 9.6 |
| Unreported (%) | 0.9 | 0.3 | 0.5 | 0.2 |
| Hospital Region | ||||
| Northeast (%) | 17.3 | 15.9 | 21.7 | 18.7 |
| Midwest (%) | 24.6 | 24.8 | 20.0 | 23.3 |
| South (%) | 37.6 | 38.4 | 36.3 | 37.0 |
| West (%) | 20.5 | 20.9 | 22.0 | 21.0 |
| Hospital type | ||||
| General hospital (%) | 68.2 | 61.4 | 61.0 | 57.1 |
| Children's unit in general hospital (%) | 15.2 | 17.8 | 18.6 | 19.2 |
| Children's hospitals (%) | 14.3 | 14.9 | 17.9 | 17.6 |
| Unreported (%) | 2.3% | 5.9 | 2.5 | 6.1 |
Incremental Differences in Mean Charges Associated with Obesity
In Table 2, we present results for analyses of mean charges. Following the pattern in 2000 for all 4 of these common conditions, in 2003 the adjusted mean total hospital charges were statistically significantly higher for discharges in which obesity was listed as a secondary diagnosis, compared with those in which it was not. Moreover, the magnitude of these differences was somewhat greater in 2003 than in 2000, although it did not achieve statistical significance (P > 0.05) (Figure 2). Specifically, the difference in charges among asthma discharges with and without obesity as a comorbidity was 9% greater in 2003 than in 2000, 17% greater among pneumonia discharges, 121% greater among affective disorders, and 3% greater among appendicitis discharges.
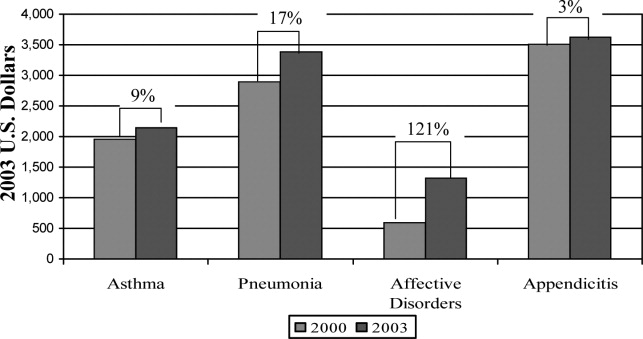
| Adjusted Mean Charges ($) | ||||||
|---|---|---|---|---|---|---|
| 2000 | 2003 | |||||
| With Obesity | Without Obesity | Difference | With Obesity | Without Obesity | Difference | |
| ||||||
| Asthma | 8,847 | 6,884 | 1,963* | 10,589 | 8,444 | 2,145 |
| Pneumonia | 13,930 | 11,036 | 2,894* | 16,609 | 13,219 | 3,390 |
| Affective disorders | 9,446 | 8,850 | 596 | 11,942 | 10,619 | 1,323 |
| Appendicitis | 16,101 | 12,587 | 3,514 | 19,213 | 15,586 | 3,627 |
Incremental Differences in Mean LOS Associated with Obesity
Compared with those discharges without obesity coded, obesity as a secondary diagnosis was associated with a statistically significantly longer mean LOS for all four diagnoses in 2003 (Table 3). In addition, for all diagnoses except asthma, the magnitude of the difference was somewhat greater in 2003 than in 2000, although it did not reach statistical significance (P > 0.05). The greatest increase was seen with appendicitis, with the incremental difference in LOS between those with obesity and those without going from 0.17 days in 2000 to 0.83 days in 2003; an increase of over 300%.
| Adjusted Mean LOS | ||||||
|---|---|---|---|---|---|---|
| 2000 | 2003 | |||||
| With Obesity | Without Obesity | Difference | With Obesity | Without Obesity | Difference | |
| ||||||
| Asthma | 3.04 | 2.45 | 0.59* | 2.88 | 2.44 | 0.44* |
| Pneumonia | 4.26 | 3.89 | 0.37 | 4.39 | 3.83 | 0.56 |
| Affective disorders | 7.72 | 7.11 | 0.61* | 8.23 | 7.42 | 0.81* |
| Appendicitis | 3.33 | 3.16 | 0.17 | 3.91 | 3.08 | 0.83* |
DISCUSSION
Prior studies have explored the resource utilization and expenditures associated with obesity in adult populations and among obese children in the outpatient setting.48, 21 Few, however, have examined charges related to inpatient care of obese children. Our studies are the first to utilize actual charge data from a nationally representative sample to explore the economic implications of obesity among children hospitalized for common pediatric illnesses.
Our findings from this national analysis support the hypothesis that, for the 4 conditions studied, statistically significantly higher mean total hospital charges and longer mean LOS for those discharges with obesity coded as a secondary diagnosis versus those without obesity coded occurred for both 2000 and 2003, even when controlling for sex, age, race/ethnicity, region, payer, and hospital type. Our analyses also suggested that the magnitude of the incremental differences in charges from 2000 to 2003 increased somewhat, and the magnitude of the incremental differences in LOS for all of these common conditions (except asthma) is also increasing.
While these findings serve to confirm higher incremental charges and LOS associated with obesity for hospitalized children, they raise the question of why charges and LOS for children with obesity might increase at a greater rate than for those without obesity. Higher hospital charges and LOS for children with obesity coded as a secondary diagnosis may be explained by greater resource utilization due to obesity that: 1) increases the technical complexity of procedures such as surgical interventions or intravenous catheter (IV) placement;22 2) leads to greater illness severity, as has been suggested in studies of adult patients;23, 24 or 3) leads to more complications such as secondary infections.22 One explanation for the possible widening of the gap in charges during the time period studied might be that discharges coded with obesity in 2003 reflect children who were more severely obese and had a greater severity of illness, leading to higher resource utilization than those with obesity in the 2000 dataset. However, the ICD‐9‐CM code for morbid obesity (278.02) was not used more often in 2003 than in 2000. Alternatively, we suspect that between 2000 and 2003, due to an increasing awareness of the problem of childhood obesity, physicians may have become more likely to order tests or consultations specifically related to the treatment or evaluation of obesity. Other than the increase in the proportion of discharges with obesity as a comorbidity from 2000 to 2003, we were unable to explore these possible explanations with the KID datasets.
In this sample of discharges, with only 1.1% and 1.6% coded with obesity as a secondary diagnosis in 2000 and 2003, respectively, it is important to note that these discharges should not be interpreted as the prevalence of obesity in hospitalized children. Indeed, in a recent study of children hospitalized for surgical procedures at a large Midwestern tertiary care hospital, 31.6% were found to be overweight or obese.25 However, we posit that the cases coded with obesity as a secondary diagnosis in this sample represent the cases in which obesity presents a recognized factor that complicated the clinical course. Further work should explore the mechanisms by which obesity impacts the care of children hospitalized for common conditions. For example, children with obesity may require more procedures and may experience more treatment complications. These specific interventions should be the target of clinically focused analyses.
Limitations
Analyses utilizing discharge data are potentially limited by the accuracy and consistency of coding. Whether the discharges coded with obesity reflect all cases in which obesity was a complicating factor is not known. Based on the national prevalence of childhood obesity it is likely than more than 1.6% of the children hospitalized in 2003 were obese. However, whether obesity impacted the hospital course sufficiently to be included among the secondary diagnoses (as stipulated by ICD‐9‐CM guidelines for official recording)26 in more than 1.6% of cases is unknown.
This study is also limited by the inability to address the processes that might account for the consistently higher charges and longer LOS seen for those discharges coded with obesity versus those without obesity coded. The KID provides some information regarding procedures performed but cannot be reliably used to examine this aspect of patient care.27 An additional limitation of the KID data set is that it contains information about deidentified discharges. This leads to the possibility of having individual patients in the dataset with multiple hospitalizations. In this study we examined the relationship between obesity as a secondary diagnosis and incremental charges and LOS for the 4 most common clinical categories for which children are hospitalized. Findings may differ for other conditions not evaluated here. Finally, information regarding costs can only be inferred from the charge data provided by the KID. However, the ratio of charges to costs would not be expected to vary by obesity status.
CONCLUSIONS
These results extend our earlier findings of higher charges and longer LOS for pediatric discharges coded with obesity versus those without. In addition, this analysis suggests a widening gap of incremental hospital charges and LOS associated with obesity as a comorbidity for common pediatric conditions. These findings present a heightening financial imperative for further research to evaluate factors associated with greater resource utilization among obese pediatric patients.
- ,,,.Prevalence and trends in overweight among US children and adolescents, 1999–2000.JAMA.2002;288(14):1728–1732.
- ,.Epidemic increase in childhood overweight, 1986–1998.JAMA.2001;286(22):2845–2848.
- ,.Overweight children and adolescents: description, epidemiology, and demographics.Pediatrics.1998;101(Pt 2):497–504.
- ,,.National medical spending attributable to overweight and obesity: how much, and who's paying?Health Aff (Millwood).2003; (Suppl Web Exclusives):W3‐219–26.
- ,,,.The impact of obesity on rising medical spending.Health Aff (Millwood).2004;(Suppl Web Exclusives):W4‐480–6.
- ,.Current estimates of the economic cost of obesity in the United States.Obes Res.1998;6(2):97–106.
- ,,,,.Lifetime health and economic benefits of weight loss among obese persons.Am J Public Health.1999;89(10):1536–1542.
- ,,.The health and cost consequences of obesity among the future elderly.Health Aff (Millwood).2005;24(Suppl 2):W5R30–W5R41.
- ,.Economic burden of obesity in youths aged 6 to 17 years: 1979–1999.Pediatrics.2002;109(5):E81–E81.
- ,,,.Incremental hospital charges associated with obesity as a secondary diagnosis.Obesity (Silver Spring).2007;15:1895–1901.
- ,,,,,.Prevalence of overweight and obesity in the United States, 1999–2004.JAMA.2006;295(13):1549–1555.
- Healthcare Cost and Utilization Project.2005. Overview of the Kid's Inpatient Database. Available at:http://www.hcup‐us.ahrq.gov/kidoverview.jsp. Accessed December 2008.
- CDC Body Mass Index: BMI for Children and Teens. Available at: http://www.cdc.gov/nccdphp/dnpa/bmi. Accessed December2008.
- ,,,,,.Prevalence of overweight among preschool children in the United States, 1971 through 1994.Pediatrics.1997;99(4):E1.
- Healthcare Cost and Utilization Project, 2002 and 2004. Description of data elements: inpatient core file. Available at: http://www.hcup‐us.ahrq.gov/db/nation/kid/DataElements_KID_Core_2000.pdf; http://www.hcup‐us.ahrq.gov/db/nation/kid/KID_2003_CORE_Volume1_A‐L.pdf;http://www.hcup‐us. ahrq.gov/db/nation/kid/KID_2003_CORE_Volume2_M‐Z.pdf. Accessed December2008.
- .Health consequences of obesity in youth: childhood predictors of adult disease.Pediatrics.1998;101:518–525.
- ,,.Lengths of stay and costs associated with children's hospitals.Pediatrics.2005;115(4):839–844.
- ,,, et al.Health care expenditures associated with overweight and obesity among US adults: importance of age and race.Am J Public Health.2005;95(1):159–165.
- ,,,.Effects of race, insurance status, and hospital volume on perforated appendicitis in children.Pediatrics.2005;115(4):920–925.
- .Smearing estimate: a nonparametric retransformation method.J Am Stat Assoc.1983;78:605–610.
- ,,,.Resource utilization and expenditures for overweight and obese children.Arch Pediatr Adolesc Med.2007 Jan;161(1):11–14.
- ,.Appendicitis in the obese child.J Pediatr Surg.2007;42(5):857–861.
- ,.Management of the obese critically ill patient.Crit Care Clin.2001;17:187–200.
- ,,,,.Total respiratory system, lung, and chest wall mechanics in sedated‐paralyzed postoperative morbidly obese patients.Chest.1996;109:144–151.
- ,,,,.Prevalence of overweight and obesity in a U.S. pediatric surgical population.J Natl Med Assoc.2007;99(1):46–48, 50–51.
- Centers for Disease Control and Prevention. ICD‐9‐CM. Official Guidelines for Coding and Reporting. Effective April 1, 2005. Available at: http://www.cdc.gov/nchs/data/icd9/icdguide.pdf. Accessed December2008.
- ,,,.Predictors of hospital charges for children admitted with asthma.Ambul Pediatr.2006;6(1):15–20.
With increases in the prevalence of obesity among children and adults over the past 3 decades in the United States,13 healthcare expenditures attributed to obesity have climbed steadily, to over $100 billion in excess expenditures annually.4 Several studies have examined healthcare costs associated with obesity in adults,48 but these studies have not attempted to distinguish between excess expenditures in inpatient versus outpatient settings. In contrast, the only 2 national economic analyses of childhood obesity have focused exclusively on the inpatient setting, because the health and economic consequences of obesity among children may be most apparent in cases in which obesity is causally linked to other diagnoses (eg, type 2 diabetes mellitus, gall bladder disease) or is a comorbidity that complicates hospitalizations.9, 10
In our previous study of obesity as a comorbidity for hospitalized children, we examined the incremental charges and length‐of‐stay (LOS) for hospitalizations for the most common nonpregnancy/nonchildbirth pediatric diagnoses, comparing those coded with obesity as a secondary diagnosis versus those without.10 Using data from the Agency for Healthcare Research and Quality (AHRQ) Kid's Inpatient Database (KID) for the year 2000, we found that obesity was associated with higher charges and longer LOS for all 4 of the conditions studied (asthma, pneumonia, affective disorders, and appendicitis). Our findings regarding asthma and affective disorders echoed earlier analyses of hospitalizations for conditions clinically linked to obesity.9 However, our study was the first to demonstrate that childhood obesity is a clinically and economically significant complicating factor for conditions not thought to be linked to obesity (pneumonia, appendicitis).
For this current study, our objective was to use more recent child hospitalization data from 2003 to determine whether our prior findings of incremental charges and LOS associated with hospitalizations where obesity was coded as a secondary diagnosis compared to those where it was not, were stable over time, and whether the magnitude of differences was consistent over a period of 4 years. We hypothesized that incremental differences in hospital charges and LOS between discharges with and without obesity would be seen in the 2003 data and that the magnitude of these differences in 2003 would be similar to those in 2000. Because obesity prevalence among children increased from 2000 to 2003,11 we also hypothesized that there would be a corresponding increase in the proportion of hospitalizations with obesity as a comorbidity.
METHODS
Data Source and Sample
We analyzed data from the AHRQ KID. The KID is a nationally representative sample of annual pediatric hospital discharges. Analysis using the KID allows for improved estimates due to the discharges from community, nonrehabilitation hospitals.12 It provides data found in standard hospital discharge abstracts for more than 2 million pediatric discharges, including International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes, LOS, total hospital charges, and patient demographic information.12 Our original analysis (published in Obesity, July 2007)10 utilized data from the 2000 KID. For this analysis we utilized data from the 2003 KID (the most recent version available), and for comparison we converted the results from the 2000 study into 2003 dollars using the Consumer Price Index for Medical Care.
Using ICD‐9‐CM codes, the KID provides the principal diagnosis for each discharge, along with up to 14 secondary diagnoses. It also provides Clinical Classification Software (CCS) codes, a diagnostic categorization scheme that permits grouping of related conditions. ICD‐9‐CM codes are collapsed into a smaller number of categories that are sometimes more useful for presenting descriptive statistics than are individual ICD‐9‐CM codes or the much broader categories of Diagnosis Related Groups (DRGs). For example, all ICD‐9‐CM codes for specific types of pneumonia would be grouped together under 1 CCS code but would exclude other respiratory conditions such as pneumothorax which would generally be included in the respiratory condition DRG.12
The 2000 KID contained 2,516,833 unweighted discharges, representing 7,291,038 discharges in the population. In the 2003 KID, there were 2,984,129 unweighted discharges representing 7,409,162 discharges in the population. Our sample included all discharges for nonpregnancy‐related conditions, in children 2 years of age (due to the Centers for Disease Control definition for overweight based on body mass index [BMI] that starts at age 2 years)13, 14 to 18 years of age (2000 weighted n = 1,527,309; 2003 weighted n = 1,613,258); these numbers exclude discharges with obesity as a primary diagnosis.
Key Variables
Our outcome variables were LOS and total charges for each of the common nonpregnancy‐related principal discharge diagnoses studied. For the 2000 and 2003 KID, total charges included all hospital fees with the exception of professional fees.15
The main independent variable of interest was presence of obesity as a secondary diagnosis. Discharges were classified as either with or without obesity based on the presence of the ICD‐9‐CM code 278.0x as a secondary diagnosis (1 if yes, 0 if no). This code captures obesity unspecified (278.00), overweight (278.01), and morbid obesity (278.02). Of note, the distribution of these codes did not vary significantly between the 2 study years.
Other independent variables included sex, age (2‐5 years, 6‐10 years, 11‐14 years, and 15‐18 years), race/ethnicity (white, black, Hispanic, and other), region (Northeast, Midwest, South, and West), hospital type (based on classification by the National Association of Children's Hospitals and Related Institutions [NACHRI] as general hospital, children's unit in a general hospital, and children's hospital) and expected primary payer (Medicaid, private, and other). We chose independent variables due to their established association with our outcomes and for their patterns of association with childhood obesity.
We did not include LOS as a covariate in models of charges because obesity may have been associated with the outcome indirectly through LOS as well as directly as a main effect. To fully interpret the combination of these effects would require analyses beyond the scope of this work.
Analyses
Using CCS codes, we identified the 4 most common principal nonpregnancy‐related discharge diagnoses for children 2‐18 years old. For both 2000 and 2003 these were asthma (CCS 128), pneumonia (CCS 122), affective disorders (eg, depression and bipolar disorder) (CCS 69), and appendicitis (CCS 142). Importantly, this group of diagnoses included conditions clinically associated with obesity (asthma and affective disorders)16 and conditions not associated with obesity (pneumonia and appendicitis). Given this distinction we analyzed the 4 conditions separately.
For all discharges with these common principal diagnoses, we calculated mean LOS and mean total charges. Bivariate and multivariable analyses were conducted using simple and multiple linear regression, respectively. These analyses were designed to test the study hypothesis that obesity as a secondary diagnosis is associated with incremental economic charges and LOS. All analyses were performed on log‐transformed LOS and charge data. We included in our models those characteristics that we hypothesized were potentially related to hospital charges and LOS, based on published literature,4, 17, 18 and for that reason all covariates were retained in the final models regardless of bivariate findings. Differences in the incremental LOS and charges for 2003 versus 2000 were compared using t‐tests.
For each independent variable with missing data, we included in the analyses a category for unreported values. The KID is known to have a large number of missing data for race/ethnicity; therefore, in keeping with other studies utilizing the KID,10, 19 we also conducted multivariate analyses excluding those discharges with unreported race as a sensitivity analysis. Of note, analyses of these data excluding children of unreported race were not substantively different than those in which the unreported group was included. We present our findings including the unreported race category.
Predicted values on the log scale, for those with obesity and without obesity as a secondary diagnosis adjusted for the listed covariates, were obtained. We then back‐transformed these to their original scales and units using methods developed by Duan.20 For each principal diagnosis category, we analyzed the differences in predicted mean LOS and predicted mean total charges between discharges with and without obesity as a secondary diagnosis, adjusted for the listed covariates, using the P values obtained from the regression analyses. All results are presented in 2003 dollars.
In keeping with our earlier analysis we considered the potential influence of comorbidities other than obesity on LOS and charges. Therefore, we examined whether other comorbidities were coded more frequently among those discharges with obesity as a secondary diagnosis than those discharges without obesity. As seen in the 2000 KID, the 2003 data revealed that diabetes was more commonly seen with obesity‐related hospitalizations than with those hospitalizations without obesity. However, the proportion of discharges with obesity and diabetes was low for all of the principal diagnostic categories studied (asthma 4.8%, pneumonia 6.0%, affective disorders 4.2%, and appendicitis 1.9%). Thus, we judged the co‐occurrence of diabetes and obesity as secondary diagnoses too infrequent to be an explanatory factor for the overall incremental differences in LOS and charges.
All analyses were weighted to account for the complex probability sampling of the dataset and permit inferences regarding national hospital discharge patterns. The same sample of discharges was used to analyze LOS and charges, with the discharge weighting variable (DISCWT) used for all analyses. All results are presented as weighted data unless otherwise noted. Analyses were conducted using STATA 8.0 (Stata Corporation, College Station, TX) and SUDAAN version 9 (Research Triangle Institute, Research Triangle Park, NC).
This study was approved by the Institutional Review Board of the University of Michigan Medical School.
RESULTS
Sample Characteristics
The characteristics of the study population are presented in Table 1. In 2003, the overall proportion of nonpregnancy‐related discharges for children 2‐18 years old coded with obesity as a secondary diagnosis was 1.6%, an increase from 1.1% in 2000. Within the 4 most common nonpregnancy‐related CCS category diagnoses (asthma, pneumonia, affective disorders, and appendicitis) the proportion of discharges with obesity coded as a secondary diagnosis increased from 2000 to 2003 (Figure 1)
| Variables | Discharges | |||
|---|---|---|---|---|
| With Obesity as Secondary Diagnosis | Without Obesity | |||
| 2000 | 2003 | 2000 | 2003 | |
| Unweighted, n | 8,696 | 15,546 | 762,407 | 943,182 |
| Weighted population size | 17,672 | 25,709 | 1,509,637 | 1,587,549 |
| Age | ||||
| 2‐5 years (%) | 4.8 | 4.8 | 27.4 | 29.0 |
| 6‐10 years (%) | 14.7 | 15.9 | 22.4 | 22.3 |
| 11‐14 years (%) | 34.4 | 34.1 | 20.9 | 20.9 |
| 15‐18 years (%) | 46.1 | 45.2 | 29.3 | 27.8 |
| Sex | ||||
| Male (%) | 45.5 | 46.8 | 53.8 | 53.4 |
| Race/Ethnicity | ||||
| White (%) | 46.8 | 32.2 | 50.9 | 39.3 |
| Black (%) | 20.7 | 20.4 | 14.3 | 12.6 |
| Hispanic (%) | 14.9 | 16.8 | 14.3 | 14.4 |
| Other (%) | 4.8 | 5.0 | 5.6 | 5.6 |
| Unreported (%) | 12.8 | 25.6 | 14.9 | 28.1 |
| Payer | ||||
| Medicaid (%) | 42.6 | 46.5 | 32.1 | 36.9 |
| Private (%) | 47.1 | 42.6 | 58.0 | 53.3 |
| Other (%) | 9.4 | 10.6 | 9.4 | 9.6 |
| Unreported (%) | 0.9 | 0.3 | 0.5 | 0.2 |
| Hospital Region | ||||
| Northeast (%) | 17.3 | 15.9 | 21.7 | 18.7 |
| Midwest (%) | 24.6 | 24.8 | 20.0 | 23.3 |
| South (%) | 37.6 | 38.4 | 36.3 | 37.0 |
| West (%) | 20.5 | 20.9 | 22.0 | 21.0 |
| Hospital type | ||||
| General hospital (%) | 68.2 | 61.4 | 61.0 | 57.1 |
| Children's unit in general hospital (%) | 15.2 | 17.8 | 18.6 | 19.2 |
| Children's hospitals (%) | 14.3 | 14.9 | 17.9 | 17.6 |
| Unreported (%) | 2.3% | 5.9 | 2.5 | 6.1 |
Incremental Differences in Mean Charges Associated with Obesity
In Table 2, we present results for analyses of mean charges. Following the pattern in 2000 for all 4 of these common conditions, in 2003 the adjusted mean total hospital charges were statistically significantly higher for discharges in which obesity was listed as a secondary diagnosis, compared with those in which it was not. Moreover, the magnitude of these differences was somewhat greater in 2003 than in 2000, although it did not achieve statistical significance (P > 0.05) (Figure 2). Specifically, the difference in charges among asthma discharges with and without obesity as a comorbidity was 9% greater in 2003 than in 2000, 17% greater among pneumonia discharges, 121% greater among affective disorders, and 3% greater among appendicitis discharges.
| Adjusted Mean Charges ($) | ||||||
|---|---|---|---|---|---|---|
| 2000 | 2003 | |||||
| With Obesity | Without Obesity | Difference | With Obesity | Without Obesity | Difference | |
| ||||||
| Asthma | 8,847 | 6,884 | 1,963* | 10,589 | 8,444 | 2,145 |
| Pneumonia | 13,930 | 11,036 | 2,894* | 16,609 | 13,219 | 3,390 |
| Affective disorders | 9,446 | 8,850 | 596 | 11,942 | 10,619 | 1,323 |
| Appendicitis | 16,101 | 12,587 | 3,514 | 19,213 | 15,586 | 3,627 |
Incremental Differences in Mean LOS Associated with Obesity
Compared with those discharges without obesity coded, obesity as a secondary diagnosis was associated with a statistically significantly longer mean LOS for all four diagnoses in 2003 (Table 3). In addition, for all diagnoses except asthma, the magnitude of the difference was somewhat greater in 2003 than in 2000, although it did not reach statistical significance (P > 0.05). The greatest increase was seen with appendicitis, with the incremental difference in LOS between those with obesity and those without going from 0.17 days in 2000 to 0.83 days in 2003; an increase of over 300%.
| Adjusted Mean LOS | ||||||
|---|---|---|---|---|---|---|
| 2000 | 2003 | |||||
| With Obesity | Without Obesity | Difference | With Obesity | Without Obesity | Difference | |
| ||||||
| Asthma | 3.04 | 2.45 | 0.59* | 2.88 | 2.44 | 0.44* |
| Pneumonia | 4.26 | 3.89 | 0.37 | 4.39 | 3.83 | 0.56 |
| Affective disorders | 7.72 | 7.11 | 0.61* | 8.23 | 7.42 | 0.81* |
| Appendicitis | 3.33 | 3.16 | 0.17 | 3.91 | 3.08 | 0.83* |
DISCUSSION
Prior studies have explored the resource utilization and expenditures associated with obesity in adult populations and among obese children in the outpatient setting.48, 21 Few, however, have examined charges related to inpatient care of obese children. Our studies are the first to utilize actual charge data from a nationally representative sample to explore the economic implications of obesity among children hospitalized for common pediatric illnesses.
Our findings from this national analysis support the hypothesis that, for the 4 conditions studied, statistically significantly higher mean total hospital charges and longer mean LOS for those discharges with obesity coded as a secondary diagnosis versus those without obesity coded occurred for both 2000 and 2003, even when controlling for sex, age, race/ethnicity, region, payer, and hospital type. Our analyses also suggested that the magnitude of the incremental differences in charges from 2000 to 2003 increased somewhat, and the magnitude of the incremental differences in LOS for all of these common conditions (except asthma) is also increasing.
While these findings serve to confirm higher incremental charges and LOS associated with obesity for hospitalized children, they raise the question of why charges and LOS for children with obesity might increase at a greater rate than for those without obesity. Higher hospital charges and LOS for children with obesity coded as a secondary diagnosis may be explained by greater resource utilization due to obesity that: 1) increases the technical complexity of procedures such as surgical interventions or intravenous catheter (IV) placement;22 2) leads to greater illness severity, as has been suggested in studies of adult patients;23, 24 or 3) leads to more complications such as secondary infections.22 One explanation for the possible widening of the gap in charges during the time period studied might be that discharges coded with obesity in 2003 reflect children who were more severely obese and had a greater severity of illness, leading to higher resource utilization than those with obesity in the 2000 dataset. However, the ICD‐9‐CM code for morbid obesity (278.02) was not used more often in 2003 than in 2000. Alternatively, we suspect that between 2000 and 2003, due to an increasing awareness of the problem of childhood obesity, physicians may have become more likely to order tests or consultations specifically related to the treatment or evaluation of obesity. Other than the increase in the proportion of discharges with obesity as a comorbidity from 2000 to 2003, we were unable to explore these possible explanations with the KID datasets.
In this sample of discharges, with only 1.1% and 1.6% coded with obesity as a secondary diagnosis in 2000 and 2003, respectively, it is important to note that these discharges should not be interpreted as the prevalence of obesity in hospitalized children. Indeed, in a recent study of children hospitalized for surgical procedures at a large Midwestern tertiary care hospital, 31.6% were found to be overweight or obese.25 However, we posit that the cases coded with obesity as a secondary diagnosis in this sample represent the cases in which obesity presents a recognized factor that complicated the clinical course. Further work should explore the mechanisms by which obesity impacts the care of children hospitalized for common conditions. For example, children with obesity may require more procedures and may experience more treatment complications. These specific interventions should be the target of clinically focused analyses.
Limitations
Analyses utilizing discharge data are potentially limited by the accuracy and consistency of coding. Whether the discharges coded with obesity reflect all cases in which obesity was a complicating factor is not known. Based on the national prevalence of childhood obesity it is likely than more than 1.6% of the children hospitalized in 2003 were obese. However, whether obesity impacted the hospital course sufficiently to be included among the secondary diagnoses (as stipulated by ICD‐9‐CM guidelines for official recording)26 in more than 1.6% of cases is unknown.
This study is also limited by the inability to address the processes that might account for the consistently higher charges and longer LOS seen for those discharges coded with obesity versus those without obesity coded. The KID provides some information regarding procedures performed but cannot be reliably used to examine this aspect of patient care.27 An additional limitation of the KID data set is that it contains information about deidentified discharges. This leads to the possibility of having individual patients in the dataset with multiple hospitalizations. In this study we examined the relationship between obesity as a secondary diagnosis and incremental charges and LOS for the 4 most common clinical categories for which children are hospitalized. Findings may differ for other conditions not evaluated here. Finally, information regarding costs can only be inferred from the charge data provided by the KID. However, the ratio of charges to costs would not be expected to vary by obesity status.
CONCLUSIONS
These results extend our earlier findings of higher charges and longer LOS for pediatric discharges coded with obesity versus those without. In addition, this analysis suggests a widening gap of incremental hospital charges and LOS associated with obesity as a comorbidity for common pediatric conditions. These findings present a heightening financial imperative for further research to evaluate factors associated with greater resource utilization among obese pediatric patients.
With increases in the prevalence of obesity among children and adults over the past 3 decades in the United States,13 healthcare expenditures attributed to obesity have climbed steadily, to over $100 billion in excess expenditures annually.4 Several studies have examined healthcare costs associated with obesity in adults,48 but these studies have not attempted to distinguish between excess expenditures in inpatient versus outpatient settings. In contrast, the only 2 national economic analyses of childhood obesity have focused exclusively on the inpatient setting, because the health and economic consequences of obesity among children may be most apparent in cases in which obesity is causally linked to other diagnoses (eg, type 2 diabetes mellitus, gall bladder disease) or is a comorbidity that complicates hospitalizations.9, 10
In our previous study of obesity as a comorbidity for hospitalized children, we examined the incremental charges and length‐of‐stay (LOS) for hospitalizations for the most common nonpregnancy/nonchildbirth pediatric diagnoses, comparing those coded with obesity as a secondary diagnosis versus those without.10 Using data from the Agency for Healthcare Research and Quality (AHRQ) Kid's Inpatient Database (KID) for the year 2000, we found that obesity was associated with higher charges and longer LOS for all 4 of the conditions studied (asthma, pneumonia, affective disorders, and appendicitis). Our findings regarding asthma and affective disorders echoed earlier analyses of hospitalizations for conditions clinically linked to obesity.9 However, our study was the first to demonstrate that childhood obesity is a clinically and economically significant complicating factor for conditions not thought to be linked to obesity (pneumonia, appendicitis).
For this current study, our objective was to use more recent child hospitalization data from 2003 to determine whether our prior findings of incremental charges and LOS associated with hospitalizations where obesity was coded as a secondary diagnosis compared to those where it was not, were stable over time, and whether the magnitude of differences was consistent over a period of 4 years. We hypothesized that incremental differences in hospital charges and LOS between discharges with and without obesity would be seen in the 2003 data and that the magnitude of these differences in 2003 would be similar to those in 2000. Because obesity prevalence among children increased from 2000 to 2003,11 we also hypothesized that there would be a corresponding increase in the proportion of hospitalizations with obesity as a comorbidity.
METHODS
Data Source and Sample
We analyzed data from the AHRQ KID. The KID is a nationally representative sample of annual pediatric hospital discharges. Analysis using the KID allows for improved estimates due to the discharges from community, nonrehabilitation hospitals.12 It provides data found in standard hospital discharge abstracts for more than 2 million pediatric discharges, including International Classification of Diseases, Ninth Revision, Clinical Modification (ICD‐9‐CM) codes, LOS, total hospital charges, and patient demographic information.12 Our original analysis (published in Obesity, July 2007)10 utilized data from the 2000 KID. For this analysis we utilized data from the 2003 KID (the most recent version available), and for comparison we converted the results from the 2000 study into 2003 dollars using the Consumer Price Index for Medical Care.
Using ICD‐9‐CM codes, the KID provides the principal diagnosis for each discharge, along with up to 14 secondary diagnoses. It also provides Clinical Classification Software (CCS) codes, a diagnostic categorization scheme that permits grouping of related conditions. ICD‐9‐CM codes are collapsed into a smaller number of categories that are sometimes more useful for presenting descriptive statistics than are individual ICD‐9‐CM codes or the much broader categories of Diagnosis Related Groups (DRGs). For example, all ICD‐9‐CM codes for specific types of pneumonia would be grouped together under 1 CCS code but would exclude other respiratory conditions such as pneumothorax which would generally be included in the respiratory condition DRG.12
The 2000 KID contained 2,516,833 unweighted discharges, representing 7,291,038 discharges in the population. In the 2003 KID, there were 2,984,129 unweighted discharges representing 7,409,162 discharges in the population. Our sample included all discharges for nonpregnancy‐related conditions, in children 2 years of age (due to the Centers for Disease Control definition for overweight based on body mass index [BMI] that starts at age 2 years)13, 14 to 18 years of age (2000 weighted n = 1,527,309; 2003 weighted n = 1,613,258); these numbers exclude discharges with obesity as a primary diagnosis.
Key Variables
Our outcome variables were LOS and total charges for each of the common nonpregnancy‐related principal discharge diagnoses studied. For the 2000 and 2003 KID, total charges included all hospital fees with the exception of professional fees.15
The main independent variable of interest was presence of obesity as a secondary diagnosis. Discharges were classified as either with or without obesity based on the presence of the ICD‐9‐CM code 278.0x as a secondary diagnosis (1 if yes, 0 if no). This code captures obesity unspecified (278.00), overweight (278.01), and morbid obesity (278.02). Of note, the distribution of these codes did not vary significantly between the 2 study years.
Other independent variables included sex, age (2‐5 years, 6‐10 years, 11‐14 years, and 15‐18 years), race/ethnicity (white, black, Hispanic, and other), region (Northeast, Midwest, South, and West), hospital type (based on classification by the National Association of Children's Hospitals and Related Institutions [NACHRI] as general hospital, children's unit in a general hospital, and children's hospital) and expected primary payer (Medicaid, private, and other). We chose independent variables due to their established association with our outcomes and for their patterns of association with childhood obesity.
We did not include LOS as a covariate in models of charges because obesity may have been associated with the outcome indirectly through LOS as well as directly as a main effect. To fully interpret the combination of these effects would require analyses beyond the scope of this work.
Analyses
Using CCS codes, we identified the 4 most common principal nonpregnancy‐related discharge diagnoses for children 2‐18 years old. For both 2000 and 2003 these were asthma (CCS 128), pneumonia (CCS 122), affective disorders (eg, depression and bipolar disorder) (CCS 69), and appendicitis (CCS 142). Importantly, this group of diagnoses included conditions clinically associated with obesity (asthma and affective disorders)16 and conditions not associated with obesity (pneumonia and appendicitis). Given this distinction we analyzed the 4 conditions separately.
For all discharges with these common principal diagnoses, we calculated mean LOS and mean total charges. Bivariate and multivariable analyses were conducted using simple and multiple linear regression, respectively. These analyses were designed to test the study hypothesis that obesity as a secondary diagnosis is associated with incremental economic charges and LOS. All analyses were performed on log‐transformed LOS and charge data. We included in our models those characteristics that we hypothesized were potentially related to hospital charges and LOS, based on published literature,4, 17, 18 and for that reason all covariates were retained in the final models regardless of bivariate findings. Differences in the incremental LOS and charges for 2003 versus 2000 were compared using t‐tests.
For each independent variable with missing data, we included in the analyses a category for unreported values. The KID is known to have a large number of missing data for race/ethnicity; therefore, in keeping with other studies utilizing the KID,10, 19 we also conducted multivariate analyses excluding those discharges with unreported race as a sensitivity analysis. Of note, analyses of these data excluding children of unreported race were not substantively different than those in which the unreported group was included. We present our findings including the unreported race category.
Predicted values on the log scale, for those with obesity and without obesity as a secondary diagnosis adjusted for the listed covariates, were obtained. We then back‐transformed these to their original scales and units using methods developed by Duan.20 For each principal diagnosis category, we analyzed the differences in predicted mean LOS and predicted mean total charges between discharges with and without obesity as a secondary diagnosis, adjusted for the listed covariates, using the P values obtained from the regression analyses. All results are presented in 2003 dollars.
In keeping with our earlier analysis we considered the potential influence of comorbidities other than obesity on LOS and charges. Therefore, we examined whether other comorbidities were coded more frequently among those discharges with obesity as a secondary diagnosis than those discharges without obesity. As seen in the 2000 KID, the 2003 data revealed that diabetes was more commonly seen with obesity‐related hospitalizations than with those hospitalizations without obesity. However, the proportion of discharges with obesity and diabetes was low for all of the principal diagnostic categories studied (asthma 4.8%, pneumonia 6.0%, affective disorders 4.2%, and appendicitis 1.9%). Thus, we judged the co‐occurrence of diabetes and obesity as secondary diagnoses too infrequent to be an explanatory factor for the overall incremental differences in LOS and charges.
All analyses were weighted to account for the complex probability sampling of the dataset and permit inferences regarding national hospital discharge patterns. The same sample of discharges was used to analyze LOS and charges, with the discharge weighting variable (DISCWT) used for all analyses. All results are presented as weighted data unless otherwise noted. Analyses were conducted using STATA 8.0 (Stata Corporation, College Station, TX) and SUDAAN version 9 (Research Triangle Institute, Research Triangle Park, NC).
This study was approved by the Institutional Review Board of the University of Michigan Medical School.
RESULTS
Sample Characteristics
The characteristics of the study population are presented in Table 1. In 2003, the overall proportion of nonpregnancy‐related discharges for children 2‐18 years old coded with obesity as a secondary diagnosis was 1.6%, an increase from 1.1% in 2000. Within the 4 most common nonpregnancy‐related CCS category diagnoses (asthma, pneumonia, affective disorders, and appendicitis) the proportion of discharges with obesity coded as a secondary diagnosis increased from 2000 to 2003 (Figure 1)
| Variables | Discharges | |||
|---|---|---|---|---|
| With Obesity as Secondary Diagnosis | Without Obesity | |||
| 2000 | 2003 | 2000 | 2003 | |
| Unweighted, n | 8,696 | 15,546 | 762,407 | 943,182 |
| Weighted population size | 17,672 | 25,709 | 1,509,637 | 1,587,549 |
| Age | ||||
| 2‐5 years (%) | 4.8 | 4.8 | 27.4 | 29.0 |
| 6‐10 years (%) | 14.7 | 15.9 | 22.4 | 22.3 |
| 11‐14 years (%) | 34.4 | 34.1 | 20.9 | 20.9 |
| 15‐18 years (%) | 46.1 | 45.2 | 29.3 | 27.8 |
| Sex | ||||
| Male (%) | 45.5 | 46.8 | 53.8 | 53.4 |
| Race/Ethnicity | ||||
| White (%) | 46.8 | 32.2 | 50.9 | 39.3 |
| Black (%) | 20.7 | 20.4 | 14.3 | 12.6 |
| Hispanic (%) | 14.9 | 16.8 | 14.3 | 14.4 |
| Other (%) | 4.8 | 5.0 | 5.6 | 5.6 |
| Unreported (%) | 12.8 | 25.6 | 14.9 | 28.1 |
| Payer | ||||
| Medicaid (%) | 42.6 | 46.5 | 32.1 | 36.9 |
| Private (%) | 47.1 | 42.6 | 58.0 | 53.3 |
| Other (%) | 9.4 | 10.6 | 9.4 | 9.6 |
| Unreported (%) | 0.9 | 0.3 | 0.5 | 0.2 |
| Hospital Region | ||||
| Northeast (%) | 17.3 | 15.9 | 21.7 | 18.7 |
| Midwest (%) | 24.6 | 24.8 | 20.0 | 23.3 |
| South (%) | 37.6 | 38.4 | 36.3 | 37.0 |
| West (%) | 20.5 | 20.9 | 22.0 | 21.0 |
| Hospital type | ||||
| General hospital (%) | 68.2 | 61.4 | 61.0 | 57.1 |
| Children's unit in general hospital (%) | 15.2 | 17.8 | 18.6 | 19.2 |
| Children's hospitals (%) | 14.3 | 14.9 | 17.9 | 17.6 |
| Unreported (%) | 2.3% | 5.9 | 2.5 | 6.1 |
Incremental Differences in Mean Charges Associated with Obesity
In Table 2, we present results for analyses of mean charges. Following the pattern in 2000 for all 4 of these common conditions, in 2003 the adjusted mean total hospital charges were statistically significantly higher for discharges in which obesity was listed as a secondary diagnosis, compared with those in which it was not. Moreover, the magnitude of these differences was somewhat greater in 2003 than in 2000, although it did not achieve statistical significance (P > 0.05) (Figure 2). Specifically, the difference in charges among asthma discharges with and without obesity as a comorbidity was 9% greater in 2003 than in 2000, 17% greater among pneumonia discharges, 121% greater among affective disorders, and 3% greater among appendicitis discharges.
| Adjusted Mean Charges ($) | ||||||
|---|---|---|---|---|---|---|
| 2000 | 2003 | |||||
| With Obesity | Without Obesity | Difference | With Obesity | Without Obesity | Difference | |
| ||||||
| Asthma | 8,847 | 6,884 | 1,963* | 10,589 | 8,444 | 2,145 |
| Pneumonia | 13,930 | 11,036 | 2,894* | 16,609 | 13,219 | 3,390 |
| Affective disorders | 9,446 | 8,850 | 596 | 11,942 | 10,619 | 1,323 |
| Appendicitis | 16,101 | 12,587 | 3,514 | 19,213 | 15,586 | 3,627 |
Incremental Differences in Mean LOS Associated with Obesity
Compared with those discharges without obesity coded, obesity as a secondary diagnosis was associated with a statistically significantly longer mean LOS for all four diagnoses in 2003 (Table 3). In addition, for all diagnoses except asthma, the magnitude of the difference was somewhat greater in 2003 than in 2000, although it did not reach statistical significance (P > 0.05). The greatest increase was seen with appendicitis, with the incremental difference in LOS between those with obesity and those without going from 0.17 days in 2000 to 0.83 days in 2003; an increase of over 300%.
| Adjusted Mean LOS | ||||||
|---|---|---|---|---|---|---|
| 2000 | 2003 | |||||
| With Obesity | Without Obesity | Difference | With Obesity | Without Obesity | Difference | |
| ||||||
| Asthma | 3.04 | 2.45 | 0.59* | 2.88 | 2.44 | 0.44* |
| Pneumonia | 4.26 | 3.89 | 0.37 | 4.39 | 3.83 | 0.56 |
| Affective disorders | 7.72 | 7.11 | 0.61* | 8.23 | 7.42 | 0.81* |
| Appendicitis | 3.33 | 3.16 | 0.17 | 3.91 | 3.08 | 0.83* |
DISCUSSION
Prior studies have explored the resource utilization and expenditures associated with obesity in adult populations and among obese children in the outpatient setting.48, 21 Few, however, have examined charges related to inpatient care of obese children. Our studies are the first to utilize actual charge data from a nationally representative sample to explore the economic implications of obesity among children hospitalized for common pediatric illnesses.
Our findings from this national analysis support the hypothesis that, for the 4 conditions studied, statistically significantly higher mean total hospital charges and longer mean LOS for those discharges with obesity coded as a secondary diagnosis versus those without obesity coded occurred for both 2000 and 2003, even when controlling for sex, age, race/ethnicity, region, payer, and hospital type. Our analyses also suggested that the magnitude of the incremental differences in charges from 2000 to 2003 increased somewhat, and the magnitude of the incremental differences in LOS for all of these common conditions (except asthma) is also increasing.
While these findings serve to confirm higher incremental charges and LOS associated with obesity for hospitalized children, they raise the question of why charges and LOS for children with obesity might increase at a greater rate than for those without obesity. Higher hospital charges and LOS for children with obesity coded as a secondary diagnosis may be explained by greater resource utilization due to obesity that: 1) increases the technical complexity of procedures such as surgical interventions or intravenous catheter (IV) placement;22 2) leads to greater illness severity, as has been suggested in studies of adult patients;23, 24 or 3) leads to more complications such as secondary infections.22 One explanation for the possible widening of the gap in charges during the time period studied might be that discharges coded with obesity in 2003 reflect children who were more severely obese and had a greater severity of illness, leading to higher resource utilization than those with obesity in the 2000 dataset. However, the ICD‐9‐CM code for morbid obesity (278.02) was not used more often in 2003 than in 2000. Alternatively, we suspect that between 2000 and 2003, due to an increasing awareness of the problem of childhood obesity, physicians may have become more likely to order tests or consultations specifically related to the treatment or evaluation of obesity. Other than the increase in the proportion of discharges with obesity as a comorbidity from 2000 to 2003, we were unable to explore these possible explanations with the KID datasets.
In this sample of discharges, with only 1.1% and 1.6% coded with obesity as a secondary diagnosis in 2000 and 2003, respectively, it is important to note that these discharges should not be interpreted as the prevalence of obesity in hospitalized children. Indeed, in a recent study of children hospitalized for surgical procedures at a large Midwestern tertiary care hospital, 31.6% were found to be overweight or obese.25 However, we posit that the cases coded with obesity as a secondary diagnosis in this sample represent the cases in which obesity presents a recognized factor that complicated the clinical course. Further work should explore the mechanisms by which obesity impacts the care of children hospitalized for common conditions. For example, children with obesity may require more procedures and may experience more treatment complications. These specific interventions should be the target of clinically focused analyses.
Limitations
Analyses utilizing discharge data are potentially limited by the accuracy and consistency of coding. Whether the discharges coded with obesity reflect all cases in which obesity was a complicating factor is not known. Based on the national prevalence of childhood obesity it is likely than more than 1.6% of the children hospitalized in 2003 were obese. However, whether obesity impacted the hospital course sufficiently to be included among the secondary diagnoses (as stipulated by ICD‐9‐CM guidelines for official recording)26 in more than 1.6% of cases is unknown.
This study is also limited by the inability to address the processes that might account for the consistently higher charges and longer LOS seen for those discharges coded with obesity versus those without obesity coded. The KID provides some information regarding procedures performed but cannot be reliably used to examine this aspect of patient care.27 An additional limitation of the KID data set is that it contains information about deidentified discharges. This leads to the possibility of having individual patients in the dataset with multiple hospitalizations. In this study we examined the relationship between obesity as a secondary diagnosis and incremental charges and LOS for the 4 most common clinical categories for which children are hospitalized. Findings may differ for other conditions not evaluated here. Finally, information regarding costs can only be inferred from the charge data provided by the KID. However, the ratio of charges to costs would not be expected to vary by obesity status.
CONCLUSIONS
These results extend our earlier findings of higher charges and longer LOS for pediatric discharges coded with obesity versus those without. In addition, this analysis suggests a widening gap of incremental hospital charges and LOS associated with obesity as a comorbidity for common pediatric conditions. These findings present a heightening financial imperative for further research to evaluate factors associated with greater resource utilization among obese pediatric patients.
- ,,,.Prevalence and trends in overweight among US children and adolescents, 1999–2000.JAMA.2002;288(14):1728–1732.
- ,.Epidemic increase in childhood overweight, 1986–1998.JAMA.2001;286(22):2845–2848.
- ,.Overweight children and adolescents: description, epidemiology, and demographics.Pediatrics.1998;101(Pt 2):497–504.
- ,,.National medical spending attributable to overweight and obesity: how much, and who's paying?Health Aff (Millwood).2003; (Suppl Web Exclusives):W3‐219–26.
- ,,,.The impact of obesity on rising medical spending.Health Aff (Millwood).2004;(Suppl Web Exclusives):W4‐480–6.
- ,.Current estimates of the economic cost of obesity in the United States.Obes Res.1998;6(2):97–106.
- ,,,,.Lifetime health and economic benefits of weight loss among obese persons.Am J Public Health.1999;89(10):1536–1542.
- ,,.The health and cost consequences of obesity among the future elderly.Health Aff (Millwood).2005;24(Suppl 2):W5R30–W5R41.
- ,.Economic burden of obesity in youths aged 6 to 17 years: 1979–1999.Pediatrics.2002;109(5):E81–E81.
- ,,,.Incremental hospital charges associated with obesity as a secondary diagnosis.Obesity (Silver Spring).2007;15:1895–1901.
- ,,,,,.Prevalence of overweight and obesity in the United States, 1999–2004.JAMA.2006;295(13):1549–1555.
- Healthcare Cost and Utilization Project.2005. Overview of the Kid's Inpatient Database. Available at:http://www.hcup‐us.ahrq.gov/kidoverview.jsp. Accessed December 2008.
- CDC Body Mass Index: BMI for Children and Teens. Available at: http://www.cdc.gov/nccdphp/dnpa/bmi. Accessed December2008.
- ,,,,,.Prevalence of overweight among preschool children in the United States, 1971 through 1994.Pediatrics.1997;99(4):E1.
- Healthcare Cost and Utilization Project, 2002 and 2004. Description of data elements: inpatient core file. Available at: http://www.hcup‐us.ahrq.gov/db/nation/kid/DataElements_KID_Core_2000.pdf; http://www.hcup‐us.ahrq.gov/db/nation/kid/KID_2003_CORE_Volume1_A‐L.pdf;http://www.hcup‐us. ahrq.gov/db/nation/kid/KID_2003_CORE_Volume2_M‐Z.pdf. Accessed December2008.
- .Health consequences of obesity in youth: childhood predictors of adult disease.Pediatrics.1998;101:518–525.
- ,,.Lengths of stay and costs associated with children's hospitals.Pediatrics.2005;115(4):839–844.
- ,,, et al.Health care expenditures associated with overweight and obesity among US adults: importance of age and race.Am J Public Health.2005;95(1):159–165.
- ,,,.Effects of race, insurance status, and hospital volume on perforated appendicitis in children.Pediatrics.2005;115(4):920–925.
- .Smearing estimate: a nonparametric retransformation method.J Am Stat Assoc.1983;78:605–610.
- ,,,.Resource utilization and expenditures for overweight and obese children.Arch Pediatr Adolesc Med.2007 Jan;161(1):11–14.
- ,.Appendicitis in the obese child.J Pediatr Surg.2007;42(5):857–861.
- ,.Management of the obese critically ill patient.Crit Care Clin.2001;17:187–200.
- ,,,,.Total respiratory system, lung, and chest wall mechanics in sedated‐paralyzed postoperative morbidly obese patients.Chest.1996;109:144–151.
- ,,,,.Prevalence of overweight and obesity in a U.S. pediatric surgical population.J Natl Med Assoc.2007;99(1):46–48, 50–51.
- Centers for Disease Control and Prevention. ICD‐9‐CM. Official Guidelines for Coding and Reporting. Effective April 1, 2005. Available at: http://www.cdc.gov/nchs/data/icd9/icdguide.pdf. Accessed December2008.
- ,,,.Predictors of hospital charges for children admitted with asthma.Ambul Pediatr.2006;6(1):15–20.
- ,,,.Prevalence and trends in overweight among US children and adolescents, 1999–2000.JAMA.2002;288(14):1728–1732.
- ,.Epidemic increase in childhood overweight, 1986–1998.JAMA.2001;286(22):2845–2848.
- ,.Overweight children and adolescents: description, epidemiology, and demographics.Pediatrics.1998;101(Pt 2):497–504.
- ,,.National medical spending attributable to overweight and obesity: how much, and who's paying?Health Aff (Millwood).2003; (Suppl Web Exclusives):W3‐219–26.
- ,,,.The impact of obesity on rising medical spending.Health Aff (Millwood).2004;(Suppl Web Exclusives):W4‐480–6.
- ,.Current estimates of the economic cost of obesity in the United States.Obes Res.1998;6(2):97–106.
- ,,,,.Lifetime health and economic benefits of weight loss among obese persons.Am J Public Health.1999;89(10):1536–1542.
- ,,.The health and cost consequences of obesity among the future elderly.Health Aff (Millwood).2005;24(Suppl 2):W5R30–W5R41.
- ,.Economic burden of obesity in youths aged 6 to 17 years: 1979–1999.Pediatrics.2002;109(5):E81–E81.
- ,,,.Incremental hospital charges associated with obesity as a secondary diagnosis.Obesity (Silver Spring).2007;15:1895–1901.
- ,,,,,.Prevalence of overweight and obesity in the United States, 1999–2004.JAMA.2006;295(13):1549–1555.
- Healthcare Cost and Utilization Project.2005. Overview of the Kid's Inpatient Database. Available at:http://www.hcup‐us.ahrq.gov/kidoverview.jsp. Accessed December 2008.
- CDC Body Mass Index: BMI for Children and Teens. Available at: http://www.cdc.gov/nccdphp/dnpa/bmi. Accessed December2008.
- ,,,,,.Prevalence of overweight among preschool children in the United States, 1971 through 1994.Pediatrics.1997;99(4):E1.
- Healthcare Cost and Utilization Project, 2002 and 2004. Description of data elements: inpatient core file. Available at: http://www.hcup‐us.ahrq.gov/db/nation/kid/DataElements_KID_Core_2000.pdf; http://www.hcup‐us.ahrq.gov/db/nation/kid/KID_2003_CORE_Volume1_A‐L.pdf;http://www.hcup‐us. ahrq.gov/db/nation/kid/KID_2003_CORE_Volume2_M‐Z.pdf. Accessed December2008.
- .Health consequences of obesity in youth: childhood predictors of adult disease.Pediatrics.1998;101:518–525.
- ,,.Lengths of stay and costs associated with children's hospitals.Pediatrics.2005;115(4):839–844.
- ,,, et al.Health care expenditures associated with overweight and obesity among US adults: importance of age and race.Am J Public Health.2005;95(1):159–165.
- ,,,.Effects of race, insurance status, and hospital volume on perforated appendicitis in children.Pediatrics.2005;115(4):920–925.
- .Smearing estimate: a nonparametric retransformation method.J Am Stat Assoc.1983;78:605–610.
- ,,,.Resource utilization and expenditures for overweight and obese children.Arch Pediatr Adolesc Med.2007 Jan;161(1):11–14.
- ,.Appendicitis in the obese child.J Pediatr Surg.2007;42(5):857–861.
- ,.Management of the obese critically ill patient.Crit Care Clin.2001;17:187–200.
- ,,,,.Total respiratory system, lung, and chest wall mechanics in sedated‐paralyzed postoperative morbidly obese patients.Chest.1996;109:144–151.
- ,,,,.Prevalence of overweight and obesity in a U.S. pediatric surgical population.J Natl Med Assoc.2007;99(1):46–48, 50–51.
- Centers for Disease Control and Prevention. ICD‐9‐CM. Official Guidelines for Coding and Reporting. Effective April 1, 2005. Available at: http://www.cdc.gov/nchs/data/icd9/icdguide.pdf. Accessed December2008.
- ,,,.Predictors of hospital charges for children admitted with asthma.Ambul Pediatr.2006;6(1):15–20.
Copyright © 2009 Society of Hospital Medicine
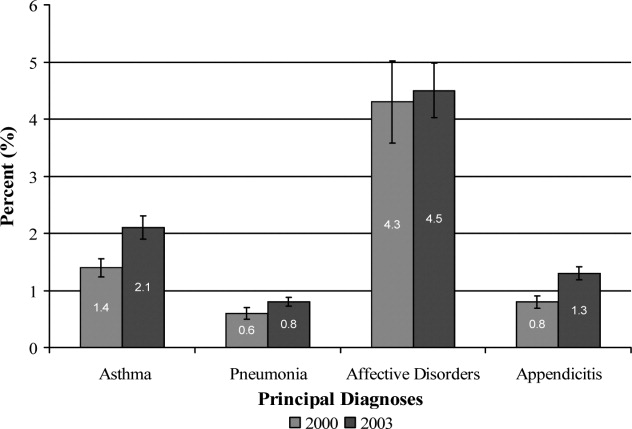
Symmetrical drug‐related intertriginous and flexural exanthema after coronary artery angiography
A 57‐year‐old woman developed a pruritic rash 6 hours after undergoing coronary angiography. On exam, symmetrical, eczematous plaques were noted in her bilateral groin (Figure 1), buttocks, axillae (Figure 2), and the intertriginous folds of her breasts. No palmar, plantar, or mucosal lesions were noted and laboratory tests were normal. This patient presents with symmetrical drug‐related intertriginous and flexural exanthema (SDRIFE) secondary to iodine‐based contrast dye. It is a type IV hypersensitivity reaction most often reported to nickel, mercury, and systemic antibiotics, although previous sensitization is often unknown. Also called baboon syndrome because its distribution mimics the pink bottom of a baboon, SDRIFE appears hours to days after exposure to the offending agent. The unusual distribution may be explained by high concentrations of the allergen in sweat. Resolution is typical with discontinuation of the offending drug, although antihistamines, topical steroids, and possibly oral steroids may be useful adjuncts.
A 57‐year‐old woman developed a pruritic rash 6 hours after undergoing coronary angiography. On exam, symmetrical, eczematous plaques were noted in her bilateral groin (Figure 1), buttocks, axillae (Figure 2), and the intertriginous folds of her breasts. No palmar, plantar, or mucosal lesions were noted and laboratory tests were normal. This patient presents with symmetrical drug‐related intertriginous and flexural exanthema (SDRIFE) secondary to iodine‐based contrast dye. It is a type IV hypersensitivity reaction most often reported to nickel, mercury, and systemic antibiotics, although previous sensitization is often unknown. Also called baboon syndrome because its distribution mimics the pink bottom of a baboon, SDRIFE appears hours to days after exposure to the offending agent. The unusual distribution may be explained by high concentrations of the allergen in sweat. Resolution is typical with discontinuation of the offending drug, although antihistamines, topical steroids, and possibly oral steroids may be useful adjuncts.
A 57‐year‐old woman developed a pruritic rash 6 hours after undergoing coronary angiography. On exam, symmetrical, eczematous plaques were noted in her bilateral groin (Figure 1), buttocks, axillae (Figure 2), and the intertriginous folds of her breasts. No palmar, plantar, or mucosal lesions were noted and laboratory tests were normal. This patient presents with symmetrical drug‐related intertriginous and flexural exanthema (SDRIFE) secondary to iodine‐based contrast dye. It is a type IV hypersensitivity reaction most often reported to nickel, mercury, and systemic antibiotics, although previous sensitization is often unknown. Also called baboon syndrome because its distribution mimics the pink bottom of a baboon, SDRIFE appears hours to days after exposure to the offending agent. The unusual distribution may be explained by high concentrations of the allergen in sweat. Resolution is typical with discontinuation of the offending drug, although antihistamines, topical steroids, and possibly oral steroids may be useful adjuncts.
Out of Africa
I knew that he was going to die. I do not remember when it became evident to me, and I was not sure how to tell the family. I thought that I could arrange a family meeting and inform them of the sad reality in a calm, sympathetic manner. The patient had chronic lymphocytic leukemia, and his case was advanced. The only medication available to him was chlorambucil. As the days passed, I could not bring myself to call the family meeting because they had so much hope. Every day as we got results and I shared them, I would sandwich the bad news with some optimism to ease their pain. Well, his white blood cell count has come down, but his platelet count and red blood cell counts are very low, and this puts him in danger of bleeding. The medicine is bringing the white cell count down but has not yet brought the other cell counts up. What we can do is give him some blood. I tried not to allow despair to creep into my thoughts or my voice. I knew that the blood bank had no platelets or packed red blood cells. He was not eating or drinking, and we had placed a nasogastric tube through which his family fed him wheat or millet porridge (manufactured tube feeds are not widely available in Uganda). I tried not to think about the time that he had almost died a few weeks before.
I had been called to the bedside because the patient was in respiratory distress. The doctor on call was in his office when I arrived, and I wondered why he was not at the bedside. I took one look at the patient and had to step away for a moment to compose myself. I felt the tears threatening to come, but I had to stop them. This was not the time for emotions. I had to assess the patient and make some quick decisions. The doctor on call seemed to have given up. He was a young trainee in a system in which you treat when you can and, if the situation is hopeless, you move on to the next patient. There are no resources for perpetuating hope. This is so different from my practice in the United States, where if a patient wants everything done, we will do it. We are not taught when to give up hope, and futility does not figure into the allocation of resources. I looked at the patient struggling to breathe and felt that I had to do all that I could for him. I asked the doctor on call to place the patient on oxygen and hoped that the tanks were not empty. I was worried about a lot of things, such as pulmonary embolus, myocardial infarction, and pneumonia. Diagnosing any of these would not be easy (the hospital did not have a computed tomography scanner, and obtaining cardiac enzymes was not as simple as clicking a button on a computer). First things first: the chest X‐ray. I thanked God that we were in a private hospital, one of the best in the city of Kampala, so we were able to get a chest X‐ray right away. As we transported the patient (portable X‐rays are nonexistent), the resident told me that he had called the consultant (the equivalent of an attending physician in the United States), who happened to be out of town. The consultant instructed us to transfer the patient to Mulago Hospital (the largest tertiary center in Uganda with well over 1000 beds and some of the equipment that you might find in an American hospital). I wondered how an attending physician could be out of town and leave a resident in charge. The thought was disturbing, but I had no time to ponder it. I later learned that physicians are so poorly paid that many have their own private clinics. My patient got the X‐ray, and I reviewed it with the resident. Tuberculosis, he said. Tuberculosis was this resident's reality. Many patients who need chest X‐rays in Uganda have tuberculosis. As I reviewed the X‐ray, though, I was certain that this was congestive heart failure. However, in Uganda, congestive heart failure is rarely diagnosed in the hospital. Patients with an ejection fraction low enough to cause congestion generally die before they get to a hospital. I knew that some furosemide would work for this patient, but I could not get the resident to listen to me. He had orders from the consultant to transfer the patient immediately, and the ambulance was ready. I tried to convince the resident to administer furosemide before transferring the patient, but he feared administering a drug not approved by his superior. As the patient was loaded onto the ambulance, I reflected for a second on how different things would be if we were in the United States. We arrived at Mulago in record time, and I tried to get the intake doctors to understand what the problem was; however, they did not want to hear from the US doctor. I stared in frustration as they wasted valuable time. I wondered how long the patient would survive in respiratory distress with nothing being done. I called the patient's son and asked him to come to Mulago immediately. Miraculously, he had already been on his way. As I held the patient's hand, sure that he would die right then and there in a waiting area as nobody did anything, I saw the patient's son. I knew that he was a pharmacist, and I asked him to go to the pharmacy and buy furosemide and some syringes. In Uganda, one can buy any medication without a prescription. Luckily, the hospital pharmacy had the drug. We treated the patient, and in no time, his breathing had returned to normal.
I was jolted back to reality. He was dying, and I knew it. He had had many close calls. There was the time that he got the wrong blood during a blood transfusion. I informed the doctor on call as the blood was being administered that I thought the patient was getting a transfusion reaction because he had rigors. The physician on call suggested covering him in blankets, and I suggested stopping the infusion and administering steroids. The pack of blood showed that he was getting his blood type. The patient was typed and crossed again, and to our surprise, we got a different result. I went to the laboratory to perform a third, tie‐breaking cross match and was surprised to note that the reagents had passed their expiration date. However, I knew that these were small battles we were winning and that there was no winning the war.
I recognized that the challenges of practicing medicine in the developing world were many. I wondered how the patients of families with fewer resources survived. The answer was obvious: they didn't. I personally picked up blood when it was available from the blood bank and vividly remember walking from the blood bank at night to the private hospital with units of blood in each hand. Once we arrived at the hospital, I had to warm the blood to room temperature by holding it close to my own skin. Many tests that we perform routinely on a hospitalized patient in the United States are not available.
There was still the problem of breaking the news to the family. Despite everything that had been done and the many near misses that the patient had survived, he was still going to die. It turns out that the family was more intuitive than I thought. One day, the son came to me and asked how long his dad had. Not long, I said quietly. I thought about all that I could potentially do if I had the patient in the hospital at which I worked in the United States. Would it have made a difference? I do not know. It was impossible doctoring this patient, and I suspect doing it in a resource‐rich environment would not have made it any easier. You see this patient, perhaps the most important patient of my life, certainly a patient that I will never forget, was my father.
It had been 15 years since I had traveled to the United States for an education. I knew that my father was so incredibly proud of me. I think that he was the happiest I had ever seen him when he attended my graduation from medical school in Minnesota. I had been looking forward to this visit back home because it had been 3 years since I had last seen my family. I was somewhat concerned because my father had told me a week before I traveled that he was not feeling well. When I arrived, there seemed to be relief on my brother's face when he met me at the airport. We drove straight to the hospital, and along with the joy of seeing me, I could sense that my father was glad that I was home at this particular point in time. They had just received the diagnosis. He had leukemia, and they were glad that their doctor was home. They had particular faith in the daughter (sister) sent abroad for an education. Things would now be okay. Initially, I never got to choose the role of doctor that I played in the final chapter of my father's life. The decision was made for me out of my family's desperation to make sure that they had left no stone unturned to help my father, and I accepted it out of necessity. As my father became my father when I entered this world, I became his doctor when he was leaving it; there was never any question in my mind, as there never was in his. As it became clear that my father would not survive, I chose to continue the role of doctor. I have watched many patients die as a physician and have done my best to make sure that their passing is comfortable, peaceful, and dignified. The doctor could help this patient die, but the daughter could not watch her father go. When it was evident that he had only days to live and did not need this doctor or know his daughter, I flew back to the United States. Three days later my father died. I was not physically at his bedside, but my spirit was. I have no regrets. Although the head knows that he passed on, in my mind's eye, he is laughing and has a twinkle in his eye. I could not bear to see him without life. A piece of my heart is buried with him, and for this reason, I will never be out of Africa.
Acknowledgements
The author is indebted to J.B. Kisuule and seeks to honor his life of service. Thank you to Dr. Roy Ziegelstein for his help with this article.
I knew that he was going to die. I do not remember when it became evident to me, and I was not sure how to tell the family. I thought that I could arrange a family meeting and inform them of the sad reality in a calm, sympathetic manner. The patient had chronic lymphocytic leukemia, and his case was advanced. The only medication available to him was chlorambucil. As the days passed, I could not bring myself to call the family meeting because they had so much hope. Every day as we got results and I shared them, I would sandwich the bad news with some optimism to ease their pain. Well, his white blood cell count has come down, but his platelet count and red blood cell counts are very low, and this puts him in danger of bleeding. The medicine is bringing the white cell count down but has not yet brought the other cell counts up. What we can do is give him some blood. I tried not to allow despair to creep into my thoughts or my voice. I knew that the blood bank had no platelets or packed red blood cells. He was not eating or drinking, and we had placed a nasogastric tube through which his family fed him wheat or millet porridge (manufactured tube feeds are not widely available in Uganda). I tried not to think about the time that he had almost died a few weeks before.
I had been called to the bedside because the patient was in respiratory distress. The doctor on call was in his office when I arrived, and I wondered why he was not at the bedside. I took one look at the patient and had to step away for a moment to compose myself. I felt the tears threatening to come, but I had to stop them. This was not the time for emotions. I had to assess the patient and make some quick decisions. The doctor on call seemed to have given up. He was a young trainee in a system in which you treat when you can and, if the situation is hopeless, you move on to the next patient. There are no resources for perpetuating hope. This is so different from my practice in the United States, where if a patient wants everything done, we will do it. We are not taught when to give up hope, and futility does not figure into the allocation of resources. I looked at the patient struggling to breathe and felt that I had to do all that I could for him. I asked the doctor on call to place the patient on oxygen and hoped that the tanks were not empty. I was worried about a lot of things, such as pulmonary embolus, myocardial infarction, and pneumonia. Diagnosing any of these would not be easy (the hospital did not have a computed tomography scanner, and obtaining cardiac enzymes was not as simple as clicking a button on a computer). First things first: the chest X‐ray. I thanked God that we were in a private hospital, one of the best in the city of Kampala, so we were able to get a chest X‐ray right away. As we transported the patient (portable X‐rays are nonexistent), the resident told me that he had called the consultant (the equivalent of an attending physician in the United States), who happened to be out of town. The consultant instructed us to transfer the patient to Mulago Hospital (the largest tertiary center in Uganda with well over 1000 beds and some of the equipment that you might find in an American hospital). I wondered how an attending physician could be out of town and leave a resident in charge. The thought was disturbing, but I had no time to ponder it. I later learned that physicians are so poorly paid that many have their own private clinics. My patient got the X‐ray, and I reviewed it with the resident. Tuberculosis, he said. Tuberculosis was this resident's reality. Many patients who need chest X‐rays in Uganda have tuberculosis. As I reviewed the X‐ray, though, I was certain that this was congestive heart failure. However, in Uganda, congestive heart failure is rarely diagnosed in the hospital. Patients with an ejection fraction low enough to cause congestion generally die before they get to a hospital. I knew that some furosemide would work for this patient, but I could not get the resident to listen to me. He had orders from the consultant to transfer the patient immediately, and the ambulance was ready. I tried to convince the resident to administer furosemide before transferring the patient, but he feared administering a drug not approved by his superior. As the patient was loaded onto the ambulance, I reflected for a second on how different things would be if we were in the United States. We arrived at Mulago in record time, and I tried to get the intake doctors to understand what the problem was; however, they did not want to hear from the US doctor. I stared in frustration as they wasted valuable time. I wondered how long the patient would survive in respiratory distress with nothing being done. I called the patient's son and asked him to come to Mulago immediately. Miraculously, he had already been on his way. As I held the patient's hand, sure that he would die right then and there in a waiting area as nobody did anything, I saw the patient's son. I knew that he was a pharmacist, and I asked him to go to the pharmacy and buy furosemide and some syringes. In Uganda, one can buy any medication without a prescription. Luckily, the hospital pharmacy had the drug. We treated the patient, and in no time, his breathing had returned to normal.
I was jolted back to reality. He was dying, and I knew it. He had had many close calls. There was the time that he got the wrong blood during a blood transfusion. I informed the doctor on call as the blood was being administered that I thought the patient was getting a transfusion reaction because he had rigors. The physician on call suggested covering him in blankets, and I suggested stopping the infusion and administering steroids. The pack of blood showed that he was getting his blood type. The patient was typed and crossed again, and to our surprise, we got a different result. I went to the laboratory to perform a third, tie‐breaking cross match and was surprised to note that the reagents had passed their expiration date. However, I knew that these were small battles we were winning and that there was no winning the war.
I recognized that the challenges of practicing medicine in the developing world were many. I wondered how the patients of families with fewer resources survived. The answer was obvious: they didn't. I personally picked up blood when it was available from the blood bank and vividly remember walking from the blood bank at night to the private hospital with units of blood in each hand. Once we arrived at the hospital, I had to warm the blood to room temperature by holding it close to my own skin. Many tests that we perform routinely on a hospitalized patient in the United States are not available.
There was still the problem of breaking the news to the family. Despite everything that had been done and the many near misses that the patient had survived, he was still going to die. It turns out that the family was more intuitive than I thought. One day, the son came to me and asked how long his dad had. Not long, I said quietly. I thought about all that I could potentially do if I had the patient in the hospital at which I worked in the United States. Would it have made a difference? I do not know. It was impossible doctoring this patient, and I suspect doing it in a resource‐rich environment would not have made it any easier. You see this patient, perhaps the most important patient of my life, certainly a patient that I will never forget, was my father.
It had been 15 years since I had traveled to the United States for an education. I knew that my father was so incredibly proud of me. I think that he was the happiest I had ever seen him when he attended my graduation from medical school in Minnesota. I had been looking forward to this visit back home because it had been 3 years since I had last seen my family. I was somewhat concerned because my father had told me a week before I traveled that he was not feeling well. When I arrived, there seemed to be relief on my brother's face when he met me at the airport. We drove straight to the hospital, and along with the joy of seeing me, I could sense that my father was glad that I was home at this particular point in time. They had just received the diagnosis. He had leukemia, and they were glad that their doctor was home. They had particular faith in the daughter (sister) sent abroad for an education. Things would now be okay. Initially, I never got to choose the role of doctor that I played in the final chapter of my father's life. The decision was made for me out of my family's desperation to make sure that they had left no stone unturned to help my father, and I accepted it out of necessity. As my father became my father when I entered this world, I became his doctor when he was leaving it; there was never any question in my mind, as there never was in his. As it became clear that my father would not survive, I chose to continue the role of doctor. I have watched many patients die as a physician and have done my best to make sure that their passing is comfortable, peaceful, and dignified. The doctor could help this patient die, but the daughter could not watch her father go. When it was evident that he had only days to live and did not need this doctor or know his daughter, I flew back to the United States. Three days later my father died. I was not physically at his bedside, but my spirit was. I have no regrets. Although the head knows that he passed on, in my mind's eye, he is laughing and has a twinkle in his eye. I could not bear to see him without life. A piece of my heart is buried with him, and for this reason, I will never be out of Africa.
Acknowledgements
The author is indebted to J.B. Kisuule and seeks to honor his life of service. Thank you to Dr. Roy Ziegelstein for his help with this article.
I knew that he was going to die. I do not remember when it became evident to me, and I was not sure how to tell the family. I thought that I could arrange a family meeting and inform them of the sad reality in a calm, sympathetic manner. The patient had chronic lymphocytic leukemia, and his case was advanced. The only medication available to him was chlorambucil. As the days passed, I could not bring myself to call the family meeting because they had so much hope. Every day as we got results and I shared them, I would sandwich the bad news with some optimism to ease their pain. Well, his white blood cell count has come down, but his platelet count and red blood cell counts are very low, and this puts him in danger of bleeding. The medicine is bringing the white cell count down but has not yet brought the other cell counts up. What we can do is give him some blood. I tried not to allow despair to creep into my thoughts or my voice. I knew that the blood bank had no platelets or packed red blood cells. He was not eating or drinking, and we had placed a nasogastric tube through which his family fed him wheat or millet porridge (manufactured tube feeds are not widely available in Uganda). I tried not to think about the time that he had almost died a few weeks before.
I had been called to the bedside because the patient was in respiratory distress. The doctor on call was in his office when I arrived, and I wondered why he was not at the bedside. I took one look at the patient and had to step away for a moment to compose myself. I felt the tears threatening to come, but I had to stop them. This was not the time for emotions. I had to assess the patient and make some quick decisions. The doctor on call seemed to have given up. He was a young trainee in a system in which you treat when you can and, if the situation is hopeless, you move on to the next patient. There are no resources for perpetuating hope. This is so different from my practice in the United States, where if a patient wants everything done, we will do it. We are not taught when to give up hope, and futility does not figure into the allocation of resources. I looked at the patient struggling to breathe and felt that I had to do all that I could for him. I asked the doctor on call to place the patient on oxygen and hoped that the tanks were not empty. I was worried about a lot of things, such as pulmonary embolus, myocardial infarction, and pneumonia. Diagnosing any of these would not be easy (the hospital did not have a computed tomography scanner, and obtaining cardiac enzymes was not as simple as clicking a button on a computer). First things first: the chest X‐ray. I thanked God that we were in a private hospital, one of the best in the city of Kampala, so we were able to get a chest X‐ray right away. As we transported the patient (portable X‐rays are nonexistent), the resident told me that he had called the consultant (the equivalent of an attending physician in the United States), who happened to be out of town. The consultant instructed us to transfer the patient to Mulago Hospital (the largest tertiary center in Uganda with well over 1000 beds and some of the equipment that you might find in an American hospital). I wondered how an attending physician could be out of town and leave a resident in charge. The thought was disturbing, but I had no time to ponder it. I later learned that physicians are so poorly paid that many have their own private clinics. My patient got the X‐ray, and I reviewed it with the resident. Tuberculosis, he said. Tuberculosis was this resident's reality. Many patients who need chest X‐rays in Uganda have tuberculosis. As I reviewed the X‐ray, though, I was certain that this was congestive heart failure. However, in Uganda, congestive heart failure is rarely diagnosed in the hospital. Patients with an ejection fraction low enough to cause congestion generally die before they get to a hospital. I knew that some furosemide would work for this patient, but I could not get the resident to listen to me. He had orders from the consultant to transfer the patient immediately, and the ambulance was ready. I tried to convince the resident to administer furosemide before transferring the patient, but he feared administering a drug not approved by his superior. As the patient was loaded onto the ambulance, I reflected for a second on how different things would be if we were in the United States. We arrived at Mulago in record time, and I tried to get the intake doctors to understand what the problem was; however, they did not want to hear from the US doctor. I stared in frustration as they wasted valuable time. I wondered how long the patient would survive in respiratory distress with nothing being done. I called the patient's son and asked him to come to Mulago immediately. Miraculously, he had already been on his way. As I held the patient's hand, sure that he would die right then and there in a waiting area as nobody did anything, I saw the patient's son. I knew that he was a pharmacist, and I asked him to go to the pharmacy and buy furosemide and some syringes. In Uganda, one can buy any medication without a prescription. Luckily, the hospital pharmacy had the drug. We treated the patient, and in no time, his breathing had returned to normal.
I was jolted back to reality. He was dying, and I knew it. He had had many close calls. There was the time that he got the wrong blood during a blood transfusion. I informed the doctor on call as the blood was being administered that I thought the patient was getting a transfusion reaction because he had rigors. The physician on call suggested covering him in blankets, and I suggested stopping the infusion and administering steroids. The pack of blood showed that he was getting his blood type. The patient was typed and crossed again, and to our surprise, we got a different result. I went to the laboratory to perform a third, tie‐breaking cross match and was surprised to note that the reagents had passed their expiration date. However, I knew that these were small battles we were winning and that there was no winning the war.
I recognized that the challenges of practicing medicine in the developing world were many. I wondered how the patients of families with fewer resources survived. The answer was obvious: they didn't. I personally picked up blood when it was available from the blood bank and vividly remember walking from the blood bank at night to the private hospital with units of blood in each hand. Once we arrived at the hospital, I had to warm the blood to room temperature by holding it close to my own skin. Many tests that we perform routinely on a hospitalized patient in the United States are not available.
There was still the problem of breaking the news to the family. Despite everything that had been done and the many near misses that the patient had survived, he was still going to die. It turns out that the family was more intuitive than I thought. One day, the son came to me and asked how long his dad had. Not long, I said quietly. I thought about all that I could potentially do if I had the patient in the hospital at which I worked in the United States. Would it have made a difference? I do not know. It was impossible doctoring this patient, and I suspect doing it in a resource‐rich environment would not have made it any easier. You see this patient, perhaps the most important patient of my life, certainly a patient that I will never forget, was my father.
It had been 15 years since I had traveled to the United States for an education. I knew that my father was so incredibly proud of me. I think that he was the happiest I had ever seen him when he attended my graduation from medical school in Minnesota. I had been looking forward to this visit back home because it had been 3 years since I had last seen my family. I was somewhat concerned because my father had told me a week before I traveled that he was not feeling well. When I arrived, there seemed to be relief on my brother's face when he met me at the airport. We drove straight to the hospital, and along with the joy of seeing me, I could sense that my father was glad that I was home at this particular point in time. They had just received the diagnosis. He had leukemia, and they were glad that their doctor was home. They had particular faith in the daughter (sister) sent abroad for an education. Things would now be okay. Initially, I never got to choose the role of doctor that I played in the final chapter of my father's life. The decision was made for me out of my family's desperation to make sure that they had left no stone unturned to help my father, and I accepted it out of necessity. As my father became my father when I entered this world, I became his doctor when he was leaving it; there was never any question in my mind, as there never was in his. As it became clear that my father would not survive, I chose to continue the role of doctor. I have watched many patients die as a physician and have done my best to make sure that their passing is comfortable, peaceful, and dignified. The doctor could help this patient die, but the daughter could not watch her father go. When it was evident that he had only days to live and did not need this doctor or know his daughter, I flew back to the United States. Three days later my father died. I was not physically at his bedside, but my spirit was. I have no regrets. Although the head knows that he passed on, in my mind's eye, he is laughing and has a twinkle in his eye. I could not bear to see him without life. A piece of my heart is buried with him, and for this reason, I will never be out of Africa.
Acknowledgements
The author is indebted to J.B. Kisuule and seeks to honor his life of service. Thank you to Dr. Roy Ziegelstein for his help with this article.
Pediatric Hospitalists
There has been marked recent growth in the employment and utilization of both pediatric and adult hospitalists. Recent data demonstrate that approximately 25% of current pediatric hospitalist programs are less than 2 years old.1 Some have posited that this growth is due to increasing pressure from the public and payors to deliver cost‐effective and high‐quality care.2 However, little is known about the mechanisms by which those who deliver care in this framework are trained, nor the scope of clinical practice they provide.37 One study has shown that among those who direct pediatric hospitalist services there is a great degree of variability in the description of the roles, work patterns, and employment characteristics of hospitalists.1 That study provided only 1 perspective on the roles and career trajectories of those in the field. To better understand both the range and frequency of experiences, clinical and nonclinical roles, training, work expectations, and career plans, we conducted a national survey study of practicing pediatric hospitalists.
METHODS
Sample
We identified all 761 hospitals in the American Hospital Association (AHA)'s 2005 Annual Survey of Hospitals that reported to have both a hospitalist service (adult and/or pediatric) and pediatric beds. From these 761 hospitals, we selected a random sample of 213, stratified by:
Council of Teaching Hospital (COTH) designation
National Association of Children's Hospitals & Related Institutions (NACHRI) membership
Freestanding children's hospitals
Metropolitan Statistical Area (MSA) (urban versus rural location)
Hospital size (small: <250 total beds versus large: 250 total beds)
Some hospitals are included in more than 1 category. Thus, there is some overlap of hospitals in the analysis. Of these 213 hospitals, 97 were removed from the sample because they did not have at least 1 pediatric hospitalist. In a separate study, we surveyed hospitalist program directors at 112 of the remaining 116 hospitals from June through September 2006. These results have been published.1
Pediatric hospitalist program directors at these 112 participating hospitals were asked to provide the names of all practicing pediatric hospitalists in their respective programs. Ninety‐five of these program directors provided a list of hospitalists at their institutions, representing 85% of the hospitals in our previous study. A total of 530 practicing pediatric hospitalists were identified to us in this manner. Of these 530 hospitalists, 67% (N = 338) were from teaching hospitals, 71% (N = 374) were from children's hospitals, 43% (N = 230) were from freestanding children's hospitals, and 69% (N = 354) were from hospitals with 250 beds. These are not mutually exclusive categories.
Survey Instrument
We developed a structured questionnaire to be administered by mail. The survey contained 25 items and was designed to be completed in 10 minutes or less. The survey focused on exploring the characteristics of hospitalist clinical and nonclinical practice, service schedule, training, and career goals. The questionnaire was comprised of a mixture of fixed‐choice, Likert‐scale, and open‐ended questions.
Questionnaire Administration
In October 2006, the first mailing of questionnaires was sent via priority mail. The survey packet contained a personalized cover letter signed by the principal investigator (G.L.F.), the instrument, a business reply mail envelope, and a $5 bill as an incentive. Two additional mailings were sent to nonrespondents in November 2006 and January 2007.
Data Analysis
First, frequency distributions were calculated for all survey items. Next, comparisons were made between respondents indicating they held an academic appointment and those who did not. For the purposes of this analysis, academic pediatric hospitalists were defined as those respondents holding a full‐time or part‐time academic appointment. Nonacademic pediatric hospitalists were defined as respondents holding an adjunct or volunteer faculty position, or no academic appointment. Finally, chi‐square statistics were used to compare pediatric hospitalist responses by hospital demographics such as teaching status, children's hospital status, NACHRI freestanding hospital designation, and hospital bed size.
The study was approved by the University of Michigan Medical Institutional Review Board.
RESULTS
Response Rate
Of the initial 530 survey packets mailed, 18 were returned as undeliverable by the postal service and 431 physicians returned the survey. This yielded an overall response rate of 84%. Of the 431 respondents, 40 physicians were ineligible because they no longer provided inpatient care to children or did not consider themselves to be hospitalists. Thus, the final sample for analysis was 391.
Hospitalist Employment Characteristics
Demographics of Hospital Worksite
Of the 391 respondents, 61% (N = 237) were from teaching hospitals, 73% (N = 287) from children's hospitals, 47% (N = 182) from freestanding children's hospitals, and 66% (N = 258) from hospitals with more than 250 beds.
Physician Demographics
The mean age of respondents was 39 years and 59% were female. The majority were employed by a hospital or health system (56%), 20% were employed by a university, and 4% were employed by both. Eight percent reported employment by a general physician medical group, 7% were employed by a hospitalist‐only group, and 4% reported other sources of employment. Half of respondents (N = 196) reported holding a full‐time (40%) or part‐time (10%) academic appointment. Approximately half the respondents (N = 194) were considered nonacademic hospitalists.
More than half of respondents (54%; N = 211) had been practicing as hospitalists for at least 3 years. Reported time as a practicing hospitalist ranged from <1 year to 26 years, while the average length of time was 63 months (Table 1). These figures may be skewed because those hospitalists with higher turnover rates might have left their position during the period of time from when they were selected into the sample until the time of survey administration.
| Length of Time as Hospitalist | % (N) |
|---|---|
| |
| 12 months | 13 (51) |
| 13‐24 months | 18 (71) |
| 25‐36 months | 14 (56) |
| 37‐60 months | 17 (67) |
| >61 months | 37 (144) |
Clinical Practice
Most respondents reported that the pediatric inpatient unit (94%) and inpatient consultation service (51%) were a part of their regular clinical assignment (Table 2). A majority did not provide service in the normal newborn nursery (58%), subspecialty inpatient service (52%), pediatric intensive care unit (ICU) (70%), neonatal ICU (77%), transports (85%), outpatient clinics (66%), or as part of an emergency response team (53%).
| Part of Regular Clinical Assignment % (N) | Occasionally % (N) | Never % (N) | |
|---|---|---|---|
| |||
| Pediatric inpatient unit | 94 (368) | 3 (13) | 2 (9) |
| Inpatient consultation service | 51 (199) | 40 (155) | 9 (35) |
| Normal newborn nursery | 29 (110) | 13 (50) | 58 (223) |
| Emergency department | 25 (95) | 28 (108) | 47 (178) |
| Subspecialty inpatient service | 25 (92) | 23 (86) | 52 (196) |
| Emergency response team | 23 (87) | 24 (91) | 53 (201) |
| Outpatient/outreach clinics | 18 (68) | 16 (61) | 66 (253) |
| Pediatric ICU | 14 (54) | 16 (59) | 70 (268) |
| Neonatal ICU | 12 (44) | 11 (42) | 77 (294) |
| Transports | 9 (33) | 6 (23) | 85 (319) |
With regard to procedures, many (53%) respondents reported that they routinely perform or supervise lumbar punctures. Several services are never performed or never supervised by the majority of pediatric hospitalists, including infusion services (57%), peripherally inserted central catheter (PICC) placement (76%), central line placement (67%), and circumcision (85%).
Professional Roles and Parameters
Respondents reported that they participate in a variety of nonclinical activities. Ninety‐four percent of hospitalists were involved in education, and 45% reported having a leadership role in that area. The majority of respondents participated in quality improvement (QI) initiatives (84%) and practice guideline development (81%), with one‐quarter of hospitalists reporting a leadership role in each of these activities. Slightly more than half of respondents reported involvement in hospital administration (52%) and utilization review (55%) (Table 3).
| Participation | No Involvement % (N) | ||
|---|---|---|---|
| Participation of Any Type % (N) | Leadership Role % (N) | ||
| |||
| Education (students, house staff) | 94 (368) | 45 (177) | 6 (22) |
| Quality improvement initiatives | 84 (330) | 25 (99) | 16 (61) |
| Practice guideline development | 81 (313) | 26 (101) | 19 (74) |
| Utilization review | 55 (213) | 11 (41) | 45 (172) |
| Hospital administration | 52 (202) | 16 (60) | 48 (184) |
On average, hospitalists reported spending 61% of their time providing inpatient care (excluding clinical teaching) and 16% of their time providing clinical teaching or supervising residents. More than one‐third of respondents (38%) spent more than 75% of their time providing direct inpatient care. Research (3%), administrative duties (8%), and nonclinical teaching (3%) were reported to be a small part of hospitalist professional time.
Pediatric Hospitalist Service Schedule
The majority of respondents reported that their assigned clinical schedule was a combination of shift and call (61%).
When on service, over half of responding pediatric hospitalists (58%) reported that they spend 40 to 60 hours onsite per week. Less than one‐fifth of respondents (19%) reported that they provide <40 hours of onsite coverage when on service. Most (97%) provide some type of night coverage, including taking calls from home or providing onsite coverage.
Hospitalist Training and Continuing Education
Only 51 of the 391 respondents (13%) had received some type of fellowship training, mostly in general pediatrics or the pediatric subspecialties. Only 5 respondents had received fellowship training in hospital medicine.
Fifty‐eight percent of respondents reported that they had received no hospitalist‐specific training. One‐fifth reported that they received training through a workshop at a professional meeting, while fewer respondents had received hospitalist training though a continuing medical education (CME) course (16%) or a mentoring program (17%).
Respondents were asked to rate the adequacy of their respective training in preparing them for their work as hospitalists. The vast majority rated their training in general clinical skills (94%) and communication (85%) as fully adequate. However, respondents found their training for some of the nonclinical aspects of their positions to be deficient. Many respondents rated training for QI projects (38%) and hospital administrative duties (46%) as inadequate (Table 4).
| Fully Adequate % (N) | Somewhat Adequate % (N) | Not Adequate % (N) | NA % (N) | |
|---|---|---|---|---|
| ||||
| General clinical skills | 94 (367) | 5 (21) | 0 (0) | 0 (1) |
| Communication skills | 85 (330) | 14 (53) | 1 (5) | 0 (1) |
| Coordination of care | 73 (284) | 23 (89) | 4 (15) | 0 (1) |
| Clinical procedure experience | 67 (258) | 32 (123) | 1 (5) | 1 (2) |
| Teaching skills (resident and medical student teaching) | 64 (248) | 31 (120) | 3 (13) | 2 (8) |
| Attending newborn deliveries | 60 (233) | 18 (70) | 4 (14) | 19 (72) |
| Running resuscitation (codes) | 45 (173) | 46 (177) | 5 (21) | 5 (18) |
| Quality improvement projects | 14 (55) | 42 (162) | 38 (148) | 6 (22) |
| Hospital administrative duties | 10 (37) | 37 (144) | 46 (177) | 8 (31) |
Survey respondents were asked to indicate the extent to which they agreed or disagreed with 3 statements regarding hospitalist training. The majority of respondents believed that hospitalists need training in QI methods (70%). However, most pediatric hospitalists (73%) did not believe that additional training beyond residency should be required. Only one‐third (36%) of respondents agreed that current CME offerings are adequate for their needs as a pediatric hospitalist.
Career Goals and Expectations
Respondents were asked to select 1 or more reasons why they became pediatric hospitalists. The top factors influencing respondents' decision to become a hospitalist were reported to be a preference for the inpatient setting (73%), clinical variety (72%), enjoyment of teaching in the inpatient setting (58%), and a flexible schedule (52%) (Table 5).
| Factor | % (N) |
|---|---|
| |
| Prefer inpatient setting | 73 (284) |
| Clinical variety | 72 (281) |
| Enjoy teaching in inpatient setting | 58 (225) |
| Flexible schedule | 52 (202) |
| Defined hours | 41 (161) |
| Attractive career opportunities | 21 (80) |
| Salary | 18 (70) |
| Unsure of long‐term career direction | 13 (51) |
| Other | 7 (28) |
| Needed short‐term employment | 4 (15) |
| Only position available | 3 (10) |
The majority (85%) were satisfied with their position as a pediatric hospitalist, with 37% reporting that they were extremely satisfied. Over one‐half (61%) expected to remain a hospitalist for the duration of their career.
RESULTS BY ACADEMIC STATUS
Only significant differences between academic and nonacademic hospitalists are presented.
Clinical Practice by Academic Status
Nonacademic respondents were more likely than academic respondents to report regular service in the normal newborn nursery, pediatric ICU, neonatal ICU, transports, emergency department, and as part of an emergency response team. Academic respondents were more likely to report regular service in outpatient clinics. Nonacademic respondents were more likely than academic respondents to perform or supervise lumbar punctures, sedation services, PICC or central line insertions, and circumcisions (Table 6).
| Academic* (N = 196) | Nonacademic (N = 194) | P Value | |
|---|---|---|---|
| |||
| Regularly provides service | |||
| Normal newborn nursery | 16% | 42% | <0.0001 |
| Pediatric ICU | 9% | 20% | 0.0065 |
| Neonatal ICU | 4% | 20% | <0.0001 |
| Transports | 3% | 15% | <0.0001 |
| Emergency department | 16% | 34% | <0.0001 |
| Emergency response team | 17% | 29% | <0.0001 |
| Outpatient clinic | 23% | 13% | 0.0168 |
| Performs or supervises procedures | |||
| Lumbar puncture | 84% | 92% | 0.0152 |
| Sedation services | 50% | 64% | 0.0055 |
| PICC insertion | 8% | 18% | 0.0031 |
| Central line insertion | 11% | 23% | 0.0018 |
| Circumcision | 5% | 16% | 0.0002 |
| Holds leadership roles | |||
| Education (student or house staff) | 63% | 27% | <0.0001 |
| Hospital administration | 21% | 10% | <0.0001 |
| Quality improvement initiatives | 33% | 18% | 0.0005 |
Professional Roles and Parameters by Academic Status
Responding academic pediatric hospitalists were twice as likely as nonacademic respondents to have a leadership role in the education of students and house staff and to hold a leadership position in hospital administration. The academic respondents were also more likely to report a leadership role in QI initiatives (Table 6).
Clinical and Educational Activities by Academic Status
Academic pediatric hospitalist respondents reported spending on average 52% of their time providing inpatient care (excluding teaching), in contrast to the nonacademic hospitalist respondents who reported 71% of their time was spent providing inpatient care (P < 0.0001). Academic respondents also reported that 19% of their time was spent providing inpatient teaching or supervising residents, compared to 12% of nonacademic respondents (P < 0.0001). Responding academic pediatric hospitalists reported spending a greater proportion of time participating in nonclinical teaching activities (5% versus 2%; P < 0.0001), administrative duties (11% versus 5%; P < 0.0001), and research (4% versus 1%; P < 0.0001) compared to the nonacademic respondents.
Nonacademic respondents were more likely than academic respondents to report no hospitalist‐specific training (64% versus 54%; P = 0.0324).
RESULTS BY HOSPITAL CHARACTERISTICS
For each hospital characteristic, only significant differences between dichotomized groups are presented.
Children's Hospitals versus Other Hospitals
Clinical Practice
Pediatric hospitalist respondents practicing in NACHRI hospitals were more likely to report that they provide regular service for general pediatric inpatients (98% versus 86%; P < 0.0001) as well as subspecialty inpatients (27% versus 17%; P = 0.044). Non‐NACHRI pediatric hospitalist respondents were twice as likely to report the provision of regular service in the normal newborn nursery (49% versus 22%; P < 0.0001), the neonatal ICU (21% versus 8%, P = 0.002), and the emergency department (38% versus 20%; P < 0.0001).
Among respondents, pediatric hospitalists who were not working at a children's hospital were more likely to report that they sometimes or routinely performed lumbar punctures (93% versus 85%; P = 0.037), infusion services (36% versus 21%; P = 0.003), and were twice as likely to perform circumcision (16% versus 8%; P = 0.041) compared to those working at children's hospitals.
Professional Roles and Parameters
Respondents working in children's hospitals were twice as likely to hold a leadership position in utilization review (12% versus 6%; P = 0.012), though respondents from non‐NACHRI hospitals were more likely to at least participate in utilization review (58% versus 40%; P = 0.004).
Hospitalist Training
Respondents from non‐NACHRI hospitals were more likely to report that they had received no hospitalist‐specific training (68% versus 56%; P = 0.029). Those at NACHRI hospitals were twice as likely to have received hospitalist training through a mentoring program (20% versus 9%; P = 0.009).
Freestanding versus Nonfreestanding Children's Hospitals
Clinical Practice
Pediatric hospitalist respondents employed at institutions that are not freestanding children's hospitals were more likely to report that they provided regular service in the normal newborn nursery (42% versus 14%; P < 0.0001), pediatric ICU (22% versus 5%), emergency department (32% versus 17%; P < 0.0001), and outpatient clinics (23% versus 12%; P = 0.0068). They were also more likely to perform or supervise sedation services (63% versus 50%; P = 0.0116), infusion services (32% versus 17%; P = 0.0006), PICC insertions (19% versus 6%; P = 0.0002), central line insertions (23% versus 11%; P = 0.0024), and circumcisions (16% versus 3%; P < 0.0001).
Professional Roles and Parameters
Among respondents, pediatric hospitalists employed by nonfreestanding children's hospitals were more likely to report participation in utilization review (51% versus 38%; P = 0.02).
Hospital Size
Clinical Practice
Pediatric hospitalist respondents working at large hospitals were twice as likely to report that they regularly provided service in the pediatric ICU (18% versus 7%; P = 0.0072) and were more likely to regularly perform circumcisions (13% versus 5%; P = 0.0069). Respondents from small hospitals were more likely to provide regular service in the neonatal ICU (20% versus 7%; P = 0.0013).
COTH Status: Teaching versus Nonteaching Hospitals
Clinical Practice
Among survey respondents, pediatric hospitalists employed by COTH hospitals were more likely to provide regular service in the neonatal ICU, compared to their peers in nonteaching hospitals (15% versus 6%; P = 0.0109). Those employed by non‐COTH hospitals were more likely to provide service in subspecialty inpatient service (38% versus 16%; P < 0.0001), transports (14% versus 6%; P = 0.0227), inpatient consultation (61% versus 45%; P = 0.0086), and the emergency response team (29% versus 19%; P = 0.0021).
Professional Roles and Parameters
Respondents from COTH hospitals were more likely to have no involvement in utilization review, compared to their peers at non‐COTH hospitals (49% versus 37%; P = 0.0220).
DISCUSSION
This study provides the most comprehensive information available regarding the clinical and nonclinical roles, training, work expectations, and career plans of pediatric hospitalists. Among the most important of our findings is the distribution of the length of time that pediatric hospitalists had served in their roles. While over one‐third (37%) reported having been practicing as hospitalists for over 5 years, 45% of our respondents had been in practice for fewer than 3 years. This is consistent with both the perceptions of rapid growth of the field and with significant turnover of hospitalists.1, 8 It is important to note that our findings may actually overestimate the proportion of hospitalists with longer durations of employment as our sampling strategy would have been less likely to include those who left the field within the first 12 to 18 months of practice. Nevertheless, over half (61%) of our respondents expected to remain a hospitalist for the duration of their career and few reported choosing to become a hospitalist as a short‐term employment option. This finding has important implications for the future stability of the hospitalist workforce and the potential development of specific expertise among this cadre of clinicians.6
The demographic profile of pediatric hospitalists was also consistent with these findings. The mean age of 39 years for our respondents is indicative of a significant proportion of this group of physicians recently having completed their residency training. Further, the gender distribution approximates that of current pediatric residency graduates, thus indicating that that this is not a clinical choice for which there would be a skewed distribution as is the case in some pediatric subspecialties.9
Our findings were similar to the 2004 Ottolini et al.10 findings on the roles of pediatric hospitalists. Respondents in our study reported spending less time providing inpatient care (61% versus 75%), providing clinical teaching or supervising residents (16% versus 26%), performing administrative duties (8% versus 19%), and conducting research (3% versus 9%) compared with the respondents in the Ottolini et al.10 survey.
At this point in time, fewer than half of our respondents reported any hospitalist‐specific training, including workshops at professional meetings or CME coursework. As there are a paucity of fellowships offering postresidency training in pediatric hospital medicine, and most of the existing programs are newly established, few in practice have completed such programs.11 In addition, most respondents reported that current CME offerings do not meet their needs, and that they could have used additional QI training to prepare them for their role as pediatric hospitalists. However, almost three‐quarters of respondents (73%) do not believe any additional training beyond residency should be required. As such, it is unclear if a defined, unique body of knowledge specific to hospitalists is either needed or desired by those currently in the field.
Although there are a broad range of potential clinical roles within hospital medicine, and this clinical variety influenced most respondents' decisions to become hospitalists, the current scope of an individual hospitalist tends to become somewhat focused.12, 13 While we found almost all provided service on the pediatric inpatient unit, many fewer provided inpatient consultation and normal newborn care, or were involved in interhospital transport or as part of an emergency response team. There is also wide variation in the types of procedures performed or supervised by hospitalists at different institutions. More than half never perform or supervise infusion services, PICC or central line placement, or circumcision. The variation seen among hospitalists practicing in different hospital settings likely is a result, at least in part, of different needs in teaching hospitals for both service and for clinical experience of trainees. For example, our results demonstrate that pediatric hospitalists in nonteaching and non‐children's hospitals are more likely to have a broader scope of clinical care provision. Another potential issue is that some hospitalists may be employed by institutions which have no pediatric ICU, neonatal ICU, or other specialty unit. As such, these hospitalists would not have the opportunity to work in such settings.
Further, those without academic appointments are also more likely to have expanded clinical roles compared with their academic counterparts. This may be due to the fact that there is likely a greater number of subspecialty‐trained pediatric providers in academic centers and thus the need for hospitalists to cover specific services or perform specific procedures is lessened. There may also be a desire to prevent competition among care providers within the same institution. In contrast, hospitalists with academic appointments are more likely (though still uncommonly) to have taken leadership roles in hospital administration and QI initiatives. Thus, the nature of their efforts appears to expand into nonclinical delivery areas.
Clearly, hospitalists report they have assumed a significant role in the clinical teaching of trainees at all levels, with 94% of our respondents maintaining at least some involvement in education. On average, they spend 16% of their time in educational efforts. However, there are few data on the impact of their work in this area.5, 13 Studies in pediatrics to date have been limited to a few institutions,3, 5 and have not addressed the issue from the perspective of residency program directors or those who are in charge of inpatient curricula.
This study, like the majority of studies related to pediatric hospitalists, is hampered by the difficulty of identifying pediatric hospitalists. Rather than utilizing a hospital medicine membership list, which would be potentially biased by self‐selection, we attempted to obtain a more representative sample through utilization of the AHA database.
CONCLUSIONS
Findings from this study provide an additional perspective regarding pediatric hospitalists to add to our previous study of hospitalist program directors.1 However, the field is currently a moving target. Our data demonstrate that there is significant flux in the hospitalist workforce, uncertainty regarding turnover, and variation in the roles of these professionals in their clinical and nonclinical work environment. Moreover, additional studies of the educational impact of hospitalists on residency and medical student education are needed. Questions regarding the nature and degree of resident autonomy and experience conducting procedures in the hospitalist environment have been raised. These must be assessed through studies of residency program directors, their expectations of residents, and the curricula they have developed.
As with any new phenomenon, it will take time to understand the impact of hospitalists in a variety of domains. Additional research will be helpful in following the development of this field and the manner in which it will interface with existing medical practice and educational programs.
- ,,,; The Research Advisory Committee of the American Board of Pediatrics.Characteristics of the pediatric hospitalist workforce: its roles and work environment.Pediatrics.2007;120:33–39.
- .The evolution of the hospitalist model in the United States.Med Clin North Am.2002;86:687–706.
- ,.Hospitalists in children's hospitals: what we know now and what we need to know.J Pediatr.2006;148:296–299.
- ,.Hospitalists: the new model of inpatient medical care in the United States.Eur J Intern Med.2003;14:65–70.
- ,,,,,.Effect of a pediatric hospitalist system on housestaff education and experience.Arch Pediatr Adolesc Med.2002;156:877–883.
- ,,,.Hospitalists' perceptions of their residency training needs: results of a national survey.Am J Med.2001;111:247–254.
- ,,,,,.Pediatric hospitalists in Canada and the United States: a survey of pediatric academic department chairs.Ambul Pediatr.2001;1:338–339.
- .Hospitalists in the United States: mission accomplished or work in progress?N Engl J Med.2004;350:1935–1936.
- ,.Pediatric workforce: a look at general pediatrics data from the American Board of Pediatrics.J Pediatr.2006;148:166–169.
- ,,,,PRIS survey: pediatric hospitalist roles and training needs [Abstr].Pediatr Res.2004;55:360A.
- ,,,.Hospital medicine fellowships: works in progress.Am J Med.2006;119:1.e1–1.e7.
- ,,.How hospitalists spend their time: insights on efficiency and safety.J Hosp Med.2006;1:88–93.
- ,,.Pediatric hospitalists fill varied roles in the care of newborns.Pediatr Ann.2003;32:802–810.
There has been marked recent growth in the employment and utilization of both pediatric and adult hospitalists. Recent data demonstrate that approximately 25% of current pediatric hospitalist programs are less than 2 years old.1 Some have posited that this growth is due to increasing pressure from the public and payors to deliver cost‐effective and high‐quality care.2 However, little is known about the mechanisms by which those who deliver care in this framework are trained, nor the scope of clinical practice they provide.37 One study has shown that among those who direct pediatric hospitalist services there is a great degree of variability in the description of the roles, work patterns, and employment characteristics of hospitalists.1 That study provided only 1 perspective on the roles and career trajectories of those in the field. To better understand both the range and frequency of experiences, clinical and nonclinical roles, training, work expectations, and career plans, we conducted a national survey study of practicing pediatric hospitalists.
METHODS
Sample
We identified all 761 hospitals in the American Hospital Association (AHA)'s 2005 Annual Survey of Hospitals that reported to have both a hospitalist service (adult and/or pediatric) and pediatric beds. From these 761 hospitals, we selected a random sample of 213, stratified by:
Council of Teaching Hospital (COTH) designation
National Association of Children's Hospitals & Related Institutions (NACHRI) membership
Freestanding children's hospitals
Metropolitan Statistical Area (MSA) (urban versus rural location)
Hospital size (small: <250 total beds versus large: 250 total beds)
Some hospitals are included in more than 1 category. Thus, there is some overlap of hospitals in the analysis. Of these 213 hospitals, 97 were removed from the sample because they did not have at least 1 pediatric hospitalist. In a separate study, we surveyed hospitalist program directors at 112 of the remaining 116 hospitals from June through September 2006. These results have been published.1
Pediatric hospitalist program directors at these 112 participating hospitals were asked to provide the names of all practicing pediatric hospitalists in their respective programs. Ninety‐five of these program directors provided a list of hospitalists at their institutions, representing 85% of the hospitals in our previous study. A total of 530 practicing pediatric hospitalists were identified to us in this manner. Of these 530 hospitalists, 67% (N = 338) were from teaching hospitals, 71% (N = 374) were from children's hospitals, 43% (N = 230) were from freestanding children's hospitals, and 69% (N = 354) were from hospitals with 250 beds. These are not mutually exclusive categories.
Survey Instrument
We developed a structured questionnaire to be administered by mail. The survey contained 25 items and was designed to be completed in 10 minutes or less. The survey focused on exploring the characteristics of hospitalist clinical and nonclinical practice, service schedule, training, and career goals. The questionnaire was comprised of a mixture of fixed‐choice, Likert‐scale, and open‐ended questions.
Questionnaire Administration
In October 2006, the first mailing of questionnaires was sent via priority mail. The survey packet contained a personalized cover letter signed by the principal investigator (G.L.F.), the instrument, a business reply mail envelope, and a $5 bill as an incentive. Two additional mailings were sent to nonrespondents in November 2006 and January 2007.
Data Analysis
First, frequency distributions were calculated for all survey items. Next, comparisons were made between respondents indicating they held an academic appointment and those who did not. For the purposes of this analysis, academic pediatric hospitalists were defined as those respondents holding a full‐time or part‐time academic appointment. Nonacademic pediatric hospitalists were defined as respondents holding an adjunct or volunteer faculty position, or no academic appointment. Finally, chi‐square statistics were used to compare pediatric hospitalist responses by hospital demographics such as teaching status, children's hospital status, NACHRI freestanding hospital designation, and hospital bed size.
The study was approved by the University of Michigan Medical Institutional Review Board.
RESULTS
Response Rate
Of the initial 530 survey packets mailed, 18 were returned as undeliverable by the postal service and 431 physicians returned the survey. This yielded an overall response rate of 84%. Of the 431 respondents, 40 physicians were ineligible because they no longer provided inpatient care to children or did not consider themselves to be hospitalists. Thus, the final sample for analysis was 391.
Hospitalist Employment Characteristics
Demographics of Hospital Worksite
Of the 391 respondents, 61% (N = 237) were from teaching hospitals, 73% (N = 287) from children's hospitals, 47% (N = 182) from freestanding children's hospitals, and 66% (N = 258) from hospitals with more than 250 beds.
Physician Demographics
The mean age of respondents was 39 years and 59% were female. The majority were employed by a hospital or health system (56%), 20% were employed by a university, and 4% were employed by both. Eight percent reported employment by a general physician medical group, 7% were employed by a hospitalist‐only group, and 4% reported other sources of employment. Half of respondents (N = 196) reported holding a full‐time (40%) or part‐time (10%) academic appointment. Approximately half the respondents (N = 194) were considered nonacademic hospitalists.
More than half of respondents (54%; N = 211) had been practicing as hospitalists for at least 3 years. Reported time as a practicing hospitalist ranged from <1 year to 26 years, while the average length of time was 63 months (Table 1). These figures may be skewed because those hospitalists with higher turnover rates might have left their position during the period of time from when they were selected into the sample until the time of survey administration.
| Length of Time as Hospitalist | % (N) |
|---|---|
| |
| 12 months | 13 (51) |
| 13‐24 months | 18 (71) |
| 25‐36 months | 14 (56) |
| 37‐60 months | 17 (67) |
| >61 months | 37 (144) |
Clinical Practice
Most respondents reported that the pediatric inpatient unit (94%) and inpatient consultation service (51%) were a part of their regular clinical assignment (Table 2). A majority did not provide service in the normal newborn nursery (58%), subspecialty inpatient service (52%), pediatric intensive care unit (ICU) (70%), neonatal ICU (77%), transports (85%), outpatient clinics (66%), or as part of an emergency response team (53%).
| Part of Regular Clinical Assignment % (N) | Occasionally % (N) | Never % (N) | |
|---|---|---|---|
| |||
| Pediatric inpatient unit | 94 (368) | 3 (13) | 2 (9) |
| Inpatient consultation service | 51 (199) | 40 (155) | 9 (35) |
| Normal newborn nursery | 29 (110) | 13 (50) | 58 (223) |
| Emergency department | 25 (95) | 28 (108) | 47 (178) |
| Subspecialty inpatient service | 25 (92) | 23 (86) | 52 (196) |
| Emergency response team | 23 (87) | 24 (91) | 53 (201) |
| Outpatient/outreach clinics | 18 (68) | 16 (61) | 66 (253) |
| Pediatric ICU | 14 (54) | 16 (59) | 70 (268) |
| Neonatal ICU | 12 (44) | 11 (42) | 77 (294) |
| Transports | 9 (33) | 6 (23) | 85 (319) |
With regard to procedures, many (53%) respondents reported that they routinely perform or supervise lumbar punctures. Several services are never performed or never supervised by the majority of pediatric hospitalists, including infusion services (57%), peripherally inserted central catheter (PICC) placement (76%), central line placement (67%), and circumcision (85%).
Professional Roles and Parameters
Respondents reported that they participate in a variety of nonclinical activities. Ninety‐four percent of hospitalists were involved in education, and 45% reported having a leadership role in that area. The majority of respondents participated in quality improvement (QI) initiatives (84%) and practice guideline development (81%), with one‐quarter of hospitalists reporting a leadership role in each of these activities. Slightly more than half of respondents reported involvement in hospital administration (52%) and utilization review (55%) (Table 3).
| Participation | No Involvement % (N) | ||
|---|---|---|---|
| Participation of Any Type % (N) | Leadership Role % (N) | ||
| |||
| Education (students, house staff) | 94 (368) | 45 (177) | 6 (22) |
| Quality improvement initiatives | 84 (330) | 25 (99) | 16 (61) |
| Practice guideline development | 81 (313) | 26 (101) | 19 (74) |
| Utilization review | 55 (213) | 11 (41) | 45 (172) |
| Hospital administration | 52 (202) | 16 (60) | 48 (184) |
On average, hospitalists reported spending 61% of their time providing inpatient care (excluding clinical teaching) and 16% of their time providing clinical teaching or supervising residents. More than one‐third of respondents (38%) spent more than 75% of their time providing direct inpatient care. Research (3%), administrative duties (8%), and nonclinical teaching (3%) were reported to be a small part of hospitalist professional time.
Pediatric Hospitalist Service Schedule
The majority of respondents reported that their assigned clinical schedule was a combination of shift and call (61%).
When on service, over half of responding pediatric hospitalists (58%) reported that they spend 40 to 60 hours onsite per week. Less than one‐fifth of respondents (19%) reported that they provide <40 hours of onsite coverage when on service. Most (97%) provide some type of night coverage, including taking calls from home or providing onsite coverage.
Hospitalist Training and Continuing Education
Only 51 of the 391 respondents (13%) had received some type of fellowship training, mostly in general pediatrics or the pediatric subspecialties. Only 5 respondents had received fellowship training in hospital medicine.
Fifty‐eight percent of respondents reported that they had received no hospitalist‐specific training. One‐fifth reported that they received training through a workshop at a professional meeting, while fewer respondents had received hospitalist training though a continuing medical education (CME) course (16%) or a mentoring program (17%).
Respondents were asked to rate the adequacy of their respective training in preparing them for their work as hospitalists. The vast majority rated their training in general clinical skills (94%) and communication (85%) as fully adequate. However, respondents found their training for some of the nonclinical aspects of their positions to be deficient. Many respondents rated training for QI projects (38%) and hospital administrative duties (46%) as inadequate (Table 4).
| Fully Adequate % (N) | Somewhat Adequate % (N) | Not Adequate % (N) | NA % (N) | |
|---|---|---|---|---|
| ||||
| General clinical skills | 94 (367) | 5 (21) | 0 (0) | 0 (1) |
| Communication skills | 85 (330) | 14 (53) | 1 (5) | 0 (1) |
| Coordination of care | 73 (284) | 23 (89) | 4 (15) | 0 (1) |
| Clinical procedure experience | 67 (258) | 32 (123) | 1 (5) | 1 (2) |
| Teaching skills (resident and medical student teaching) | 64 (248) | 31 (120) | 3 (13) | 2 (8) |
| Attending newborn deliveries | 60 (233) | 18 (70) | 4 (14) | 19 (72) |
| Running resuscitation (codes) | 45 (173) | 46 (177) | 5 (21) | 5 (18) |
| Quality improvement projects | 14 (55) | 42 (162) | 38 (148) | 6 (22) |
| Hospital administrative duties | 10 (37) | 37 (144) | 46 (177) | 8 (31) |
Survey respondents were asked to indicate the extent to which they agreed or disagreed with 3 statements regarding hospitalist training. The majority of respondents believed that hospitalists need training in QI methods (70%). However, most pediatric hospitalists (73%) did not believe that additional training beyond residency should be required. Only one‐third (36%) of respondents agreed that current CME offerings are adequate for their needs as a pediatric hospitalist.
Career Goals and Expectations
Respondents were asked to select 1 or more reasons why they became pediatric hospitalists. The top factors influencing respondents' decision to become a hospitalist were reported to be a preference for the inpatient setting (73%), clinical variety (72%), enjoyment of teaching in the inpatient setting (58%), and a flexible schedule (52%) (Table 5).
| Factor | % (N) |
|---|---|
| |
| Prefer inpatient setting | 73 (284) |
| Clinical variety | 72 (281) |
| Enjoy teaching in inpatient setting | 58 (225) |
| Flexible schedule | 52 (202) |
| Defined hours | 41 (161) |
| Attractive career opportunities | 21 (80) |
| Salary | 18 (70) |
| Unsure of long‐term career direction | 13 (51) |
| Other | 7 (28) |
| Needed short‐term employment | 4 (15) |
| Only position available | 3 (10) |
The majority (85%) were satisfied with their position as a pediatric hospitalist, with 37% reporting that they were extremely satisfied. Over one‐half (61%) expected to remain a hospitalist for the duration of their career.
RESULTS BY ACADEMIC STATUS
Only significant differences between academic and nonacademic hospitalists are presented.
Clinical Practice by Academic Status
Nonacademic respondents were more likely than academic respondents to report regular service in the normal newborn nursery, pediatric ICU, neonatal ICU, transports, emergency department, and as part of an emergency response team. Academic respondents were more likely to report regular service in outpatient clinics. Nonacademic respondents were more likely than academic respondents to perform or supervise lumbar punctures, sedation services, PICC or central line insertions, and circumcisions (Table 6).
| Academic* (N = 196) | Nonacademic (N = 194) | P Value | |
|---|---|---|---|
| |||
| Regularly provides service | |||
| Normal newborn nursery | 16% | 42% | <0.0001 |
| Pediatric ICU | 9% | 20% | 0.0065 |
| Neonatal ICU | 4% | 20% | <0.0001 |
| Transports | 3% | 15% | <0.0001 |
| Emergency department | 16% | 34% | <0.0001 |
| Emergency response team | 17% | 29% | <0.0001 |
| Outpatient clinic | 23% | 13% | 0.0168 |
| Performs or supervises procedures | |||
| Lumbar puncture | 84% | 92% | 0.0152 |
| Sedation services | 50% | 64% | 0.0055 |
| PICC insertion | 8% | 18% | 0.0031 |
| Central line insertion | 11% | 23% | 0.0018 |
| Circumcision | 5% | 16% | 0.0002 |
| Holds leadership roles | |||
| Education (student or house staff) | 63% | 27% | <0.0001 |
| Hospital administration | 21% | 10% | <0.0001 |
| Quality improvement initiatives | 33% | 18% | 0.0005 |
Professional Roles and Parameters by Academic Status
Responding academic pediatric hospitalists were twice as likely as nonacademic respondents to have a leadership role in the education of students and house staff and to hold a leadership position in hospital administration. The academic respondents were also more likely to report a leadership role in QI initiatives (Table 6).
Clinical and Educational Activities by Academic Status
Academic pediatric hospitalist respondents reported spending on average 52% of their time providing inpatient care (excluding teaching), in contrast to the nonacademic hospitalist respondents who reported 71% of their time was spent providing inpatient care (P < 0.0001). Academic respondents also reported that 19% of their time was spent providing inpatient teaching or supervising residents, compared to 12% of nonacademic respondents (P < 0.0001). Responding academic pediatric hospitalists reported spending a greater proportion of time participating in nonclinical teaching activities (5% versus 2%; P < 0.0001), administrative duties (11% versus 5%; P < 0.0001), and research (4% versus 1%; P < 0.0001) compared to the nonacademic respondents.
Nonacademic respondents were more likely than academic respondents to report no hospitalist‐specific training (64% versus 54%; P = 0.0324).
RESULTS BY HOSPITAL CHARACTERISTICS
For each hospital characteristic, only significant differences between dichotomized groups are presented.
Children's Hospitals versus Other Hospitals
Clinical Practice
Pediatric hospitalist respondents practicing in NACHRI hospitals were more likely to report that they provide regular service for general pediatric inpatients (98% versus 86%; P < 0.0001) as well as subspecialty inpatients (27% versus 17%; P = 0.044). Non‐NACHRI pediatric hospitalist respondents were twice as likely to report the provision of regular service in the normal newborn nursery (49% versus 22%; P < 0.0001), the neonatal ICU (21% versus 8%, P = 0.002), and the emergency department (38% versus 20%; P < 0.0001).
Among respondents, pediatric hospitalists who were not working at a children's hospital were more likely to report that they sometimes or routinely performed lumbar punctures (93% versus 85%; P = 0.037), infusion services (36% versus 21%; P = 0.003), and were twice as likely to perform circumcision (16% versus 8%; P = 0.041) compared to those working at children's hospitals.
Professional Roles and Parameters
Respondents working in children's hospitals were twice as likely to hold a leadership position in utilization review (12% versus 6%; P = 0.012), though respondents from non‐NACHRI hospitals were more likely to at least participate in utilization review (58% versus 40%; P = 0.004).
Hospitalist Training
Respondents from non‐NACHRI hospitals were more likely to report that they had received no hospitalist‐specific training (68% versus 56%; P = 0.029). Those at NACHRI hospitals were twice as likely to have received hospitalist training through a mentoring program (20% versus 9%; P = 0.009).
Freestanding versus Nonfreestanding Children's Hospitals
Clinical Practice
Pediatric hospitalist respondents employed at institutions that are not freestanding children's hospitals were more likely to report that they provided regular service in the normal newborn nursery (42% versus 14%; P < 0.0001), pediatric ICU (22% versus 5%), emergency department (32% versus 17%; P < 0.0001), and outpatient clinics (23% versus 12%; P = 0.0068). They were also more likely to perform or supervise sedation services (63% versus 50%; P = 0.0116), infusion services (32% versus 17%; P = 0.0006), PICC insertions (19% versus 6%; P = 0.0002), central line insertions (23% versus 11%; P = 0.0024), and circumcisions (16% versus 3%; P < 0.0001).
Professional Roles and Parameters
Among respondents, pediatric hospitalists employed by nonfreestanding children's hospitals were more likely to report participation in utilization review (51% versus 38%; P = 0.02).
Hospital Size
Clinical Practice
Pediatric hospitalist respondents working at large hospitals were twice as likely to report that they regularly provided service in the pediatric ICU (18% versus 7%; P = 0.0072) and were more likely to regularly perform circumcisions (13% versus 5%; P = 0.0069). Respondents from small hospitals were more likely to provide regular service in the neonatal ICU (20% versus 7%; P = 0.0013).
COTH Status: Teaching versus Nonteaching Hospitals
Clinical Practice
Among survey respondents, pediatric hospitalists employed by COTH hospitals were more likely to provide regular service in the neonatal ICU, compared to their peers in nonteaching hospitals (15% versus 6%; P = 0.0109). Those employed by non‐COTH hospitals were more likely to provide service in subspecialty inpatient service (38% versus 16%; P < 0.0001), transports (14% versus 6%; P = 0.0227), inpatient consultation (61% versus 45%; P = 0.0086), and the emergency response team (29% versus 19%; P = 0.0021).
Professional Roles and Parameters
Respondents from COTH hospitals were more likely to have no involvement in utilization review, compared to their peers at non‐COTH hospitals (49% versus 37%; P = 0.0220).
DISCUSSION
This study provides the most comprehensive information available regarding the clinical and nonclinical roles, training, work expectations, and career plans of pediatric hospitalists. Among the most important of our findings is the distribution of the length of time that pediatric hospitalists had served in their roles. While over one‐third (37%) reported having been practicing as hospitalists for over 5 years, 45% of our respondents had been in practice for fewer than 3 years. This is consistent with both the perceptions of rapid growth of the field and with significant turnover of hospitalists.1, 8 It is important to note that our findings may actually overestimate the proportion of hospitalists with longer durations of employment as our sampling strategy would have been less likely to include those who left the field within the first 12 to 18 months of practice. Nevertheless, over half (61%) of our respondents expected to remain a hospitalist for the duration of their career and few reported choosing to become a hospitalist as a short‐term employment option. This finding has important implications for the future stability of the hospitalist workforce and the potential development of specific expertise among this cadre of clinicians.6
The demographic profile of pediatric hospitalists was also consistent with these findings. The mean age of 39 years for our respondents is indicative of a significant proportion of this group of physicians recently having completed their residency training. Further, the gender distribution approximates that of current pediatric residency graduates, thus indicating that that this is not a clinical choice for which there would be a skewed distribution as is the case in some pediatric subspecialties.9
Our findings were similar to the 2004 Ottolini et al.10 findings on the roles of pediatric hospitalists. Respondents in our study reported spending less time providing inpatient care (61% versus 75%), providing clinical teaching or supervising residents (16% versus 26%), performing administrative duties (8% versus 19%), and conducting research (3% versus 9%) compared with the respondents in the Ottolini et al.10 survey.
At this point in time, fewer than half of our respondents reported any hospitalist‐specific training, including workshops at professional meetings or CME coursework. As there are a paucity of fellowships offering postresidency training in pediatric hospital medicine, and most of the existing programs are newly established, few in practice have completed such programs.11 In addition, most respondents reported that current CME offerings do not meet their needs, and that they could have used additional QI training to prepare them for their role as pediatric hospitalists. However, almost three‐quarters of respondents (73%) do not believe any additional training beyond residency should be required. As such, it is unclear if a defined, unique body of knowledge specific to hospitalists is either needed or desired by those currently in the field.
Although there are a broad range of potential clinical roles within hospital medicine, and this clinical variety influenced most respondents' decisions to become hospitalists, the current scope of an individual hospitalist tends to become somewhat focused.12, 13 While we found almost all provided service on the pediatric inpatient unit, many fewer provided inpatient consultation and normal newborn care, or were involved in interhospital transport or as part of an emergency response team. There is also wide variation in the types of procedures performed or supervised by hospitalists at different institutions. More than half never perform or supervise infusion services, PICC or central line placement, or circumcision. The variation seen among hospitalists practicing in different hospital settings likely is a result, at least in part, of different needs in teaching hospitals for both service and for clinical experience of trainees. For example, our results demonstrate that pediatric hospitalists in nonteaching and non‐children's hospitals are more likely to have a broader scope of clinical care provision. Another potential issue is that some hospitalists may be employed by institutions which have no pediatric ICU, neonatal ICU, or other specialty unit. As such, these hospitalists would not have the opportunity to work in such settings.
Further, those without academic appointments are also more likely to have expanded clinical roles compared with their academic counterparts. This may be due to the fact that there is likely a greater number of subspecialty‐trained pediatric providers in academic centers and thus the need for hospitalists to cover specific services or perform specific procedures is lessened. There may also be a desire to prevent competition among care providers within the same institution. In contrast, hospitalists with academic appointments are more likely (though still uncommonly) to have taken leadership roles in hospital administration and QI initiatives. Thus, the nature of their efforts appears to expand into nonclinical delivery areas.
Clearly, hospitalists report they have assumed a significant role in the clinical teaching of trainees at all levels, with 94% of our respondents maintaining at least some involvement in education. On average, they spend 16% of their time in educational efforts. However, there are few data on the impact of their work in this area.5, 13 Studies in pediatrics to date have been limited to a few institutions,3, 5 and have not addressed the issue from the perspective of residency program directors or those who are in charge of inpatient curricula.
This study, like the majority of studies related to pediatric hospitalists, is hampered by the difficulty of identifying pediatric hospitalists. Rather than utilizing a hospital medicine membership list, which would be potentially biased by self‐selection, we attempted to obtain a more representative sample through utilization of the AHA database.
CONCLUSIONS
Findings from this study provide an additional perspective regarding pediatric hospitalists to add to our previous study of hospitalist program directors.1 However, the field is currently a moving target. Our data demonstrate that there is significant flux in the hospitalist workforce, uncertainty regarding turnover, and variation in the roles of these professionals in their clinical and nonclinical work environment. Moreover, additional studies of the educational impact of hospitalists on residency and medical student education are needed. Questions regarding the nature and degree of resident autonomy and experience conducting procedures in the hospitalist environment have been raised. These must be assessed through studies of residency program directors, their expectations of residents, and the curricula they have developed.
As with any new phenomenon, it will take time to understand the impact of hospitalists in a variety of domains. Additional research will be helpful in following the development of this field and the manner in which it will interface with existing medical practice and educational programs.
There has been marked recent growth in the employment and utilization of both pediatric and adult hospitalists. Recent data demonstrate that approximately 25% of current pediatric hospitalist programs are less than 2 years old.1 Some have posited that this growth is due to increasing pressure from the public and payors to deliver cost‐effective and high‐quality care.2 However, little is known about the mechanisms by which those who deliver care in this framework are trained, nor the scope of clinical practice they provide.37 One study has shown that among those who direct pediatric hospitalist services there is a great degree of variability in the description of the roles, work patterns, and employment characteristics of hospitalists.1 That study provided only 1 perspective on the roles and career trajectories of those in the field. To better understand both the range and frequency of experiences, clinical and nonclinical roles, training, work expectations, and career plans, we conducted a national survey study of practicing pediatric hospitalists.
METHODS
Sample
We identified all 761 hospitals in the American Hospital Association (AHA)'s 2005 Annual Survey of Hospitals that reported to have both a hospitalist service (adult and/or pediatric) and pediatric beds. From these 761 hospitals, we selected a random sample of 213, stratified by:
Council of Teaching Hospital (COTH) designation
National Association of Children's Hospitals & Related Institutions (NACHRI) membership
Freestanding children's hospitals
Metropolitan Statistical Area (MSA) (urban versus rural location)
Hospital size (small: <250 total beds versus large: 250 total beds)
Some hospitals are included in more than 1 category. Thus, there is some overlap of hospitals in the analysis. Of these 213 hospitals, 97 were removed from the sample because they did not have at least 1 pediatric hospitalist. In a separate study, we surveyed hospitalist program directors at 112 of the remaining 116 hospitals from June through September 2006. These results have been published.1
Pediatric hospitalist program directors at these 112 participating hospitals were asked to provide the names of all practicing pediatric hospitalists in their respective programs. Ninety‐five of these program directors provided a list of hospitalists at their institutions, representing 85% of the hospitals in our previous study. A total of 530 practicing pediatric hospitalists were identified to us in this manner. Of these 530 hospitalists, 67% (N = 338) were from teaching hospitals, 71% (N = 374) were from children's hospitals, 43% (N = 230) were from freestanding children's hospitals, and 69% (N = 354) were from hospitals with 250 beds. These are not mutually exclusive categories.
Survey Instrument
We developed a structured questionnaire to be administered by mail. The survey contained 25 items and was designed to be completed in 10 minutes or less. The survey focused on exploring the characteristics of hospitalist clinical and nonclinical practice, service schedule, training, and career goals. The questionnaire was comprised of a mixture of fixed‐choice, Likert‐scale, and open‐ended questions.
Questionnaire Administration
In October 2006, the first mailing of questionnaires was sent via priority mail. The survey packet contained a personalized cover letter signed by the principal investigator (G.L.F.), the instrument, a business reply mail envelope, and a $5 bill as an incentive. Two additional mailings were sent to nonrespondents in November 2006 and January 2007.
Data Analysis
First, frequency distributions were calculated for all survey items. Next, comparisons were made between respondents indicating they held an academic appointment and those who did not. For the purposes of this analysis, academic pediatric hospitalists were defined as those respondents holding a full‐time or part‐time academic appointment. Nonacademic pediatric hospitalists were defined as respondents holding an adjunct or volunteer faculty position, or no academic appointment. Finally, chi‐square statistics were used to compare pediatric hospitalist responses by hospital demographics such as teaching status, children's hospital status, NACHRI freestanding hospital designation, and hospital bed size.
The study was approved by the University of Michigan Medical Institutional Review Board.
RESULTS
Response Rate
Of the initial 530 survey packets mailed, 18 were returned as undeliverable by the postal service and 431 physicians returned the survey. This yielded an overall response rate of 84%. Of the 431 respondents, 40 physicians were ineligible because they no longer provided inpatient care to children or did not consider themselves to be hospitalists. Thus, the final sample for analysis was 391.
Hospitalist Employment Characteristics
Demographics of Hospital Worksite
Of the 391 respondents, 61% (N = 237) were from teaching hospitals, 73% (N = 287) from children's hospitals, 47% (N = 182) from freestanding children's hospitals, and 66% (N = 258) from hospitals with more than 250 beds.
Physician Demographics
The mean age of respondents was 39 years and 59% were female. The majority were employed by a hospital or health system (56%), 20% were employed by a university, and 4% were employed by both. Eight percent reported employment by a general physician medical group, 7% were employed by a hospitalist‐only group, and 4% reported other sources of employment. Half of respondents (N = 196) reported holding a full‐time (40%) or part‐time (10%) academic appointment. Approximately half the respondents (N = 194) were considered nonacademic hospitalists.
More than half of respondents (54%; N = 211) had been practicing as hospitalists for at least 3 years. Reported time as a practicing hospitalist ranged from <1 year to 26 years, while the average length of time was 63 months (Table 1). These figures may be skewed because those hospitalists with higher turnover rates might have left their position during the period of time from when they were selected into the sample until the time of survey administration.
| Length of Time as Hospitalist | % (N) |
|---|---|
| |
| 12 months | 13 (51) |
| 13‐24 months | 18 (71) |
| 25‐36 months | 14 (56) |
| 37‐60 months | 17 (67) |
| >61 months | 37 (144) |
Clinical Practice
Most respondents reported that the pediatric inpatient unit (94%) and inpatient consultation service (51%) were a part of their regular clinical assignment (Table 2). A majority did not provide service in the normal newborn nursery (58%), subspecialty inpatient service (52%), pediatric intensive care unit (ICU) (70%), neonatal ICU (77%), transports (85%), outpatient clinics (66%), or as part of an emergency response team (53%).
| Part of Regular Clinical Assignment % (N) | Occasionally % (N) | Never % (N) | |
|---|---|---|---|
| |||
| Pediatric inpatient unit | 94 (368) | 3 (13) | 2 (9) |
| Inpatient consultation service | 51 (199) | 40 (155) | 9 (35) |
| Normal newborn nursery | 29 (110) | 13 (50) | 58 (223) |
| Emergency department | 25 (95) | 28 (108) | 47 (178) |
| Subspecialty inpatient service | 25 (92) | 23 (86) | 52 (196) |
| Emergency response team | 23 (87) | 24 (91) | 53 (201) |
| Outpatient/outreach clinics | 18 (68) | 16 (61) | 66 (253) |
| Pediatric ICU | 14 (54) | 16 (59) | 70 (268) |
| Neonatal ICU | 12 (44) | 11 (42) | 77 (294) |
| Transports | 9 (33) | 6 (23) | 85 (319) |
With regard to procedures, many (53%) respondents reported that they routinely perform or supervise lumbar punctures. Several services are never performed or never supervised by the majority of pediatric hospitalists, including infusion services (57%), peripherally inserted central catheter (PICC) placement (76%), central line placement (67%), and circumcision (85%).
Professional Roles and Parameters
Respondents reported that they participate in a variety of nonclinical activities. Ninety‐four percent of hospitalists were involved in education, and 45% reported having a leadership role in that area. The majority of respondents participated in quality improvement (QI) initiatives (84%) and practice guideline development (81%), with one‐quarter of hospitalists reporting a leadership role in each of these activities. Slightly more than half of respondents reported involvement in hospital administration (52%) and utilization review (55%) (Table 3).
| Participation | No Involvement % (N) | ||
|---|---|---|---|
| Participation of Any Type % (N) | Leadership Role % (N) | ||
| |||
| Education (students, house staff) | 94 (368) | 45 (177) | 6 (22) |
| Quality improvement initiatives | 84 (330) | 25 (99) | 16 (61) |
| Practice guideline development | 81 (313) | 26 (101) | 19 (74) |
| Utilization review | 55 (213) | 11 (41) | 45 (172) |
| Hospital administration | 52 (202) | 16 (60) | 48 (184) |
On average, hospitalists reported spending 61% of their time providing inpatient care (excluding clinical teaching) and 16% of their time providing clinical teaching or supervising residents. More than one‐third of respondents (38%) spent more than 75% of their time providing direct inpatient care. Research (3%), administrative duties (8%), and nonclinical teaching (3%) were reported to be a small part of hospitalist professional time.
Pediatric Hospitalist Service Schedule
The majority of respondents reported that their assigned clinical schedule was a combination of shift and call (61%).
When on service, over half of responding pediatric hospitalists (58%) reported that they spend 40 to 60 hours onsite per week. Less than one‐fifth of respondents (19%) reported that they provide <40 hours of onsite coverage when on service. Most (97%) provide some type of night coverage, including taking calls from home or providing onsite coverage.
Hospitalist Training and Continuing Education
Only 51 of the 391 respondents (13%) had received some type of fellowship training, mostly in general pediatrics or the pediatric subspecialties. Only 5 respondents had received fellowship training in hospital medicine.
Fifty‐eight percent of respondents reported that they had received no hospitalist‐specific training. One‐fifth reported that they received training through a workshop at a professional meeting, while fewer respondents had received hospitalist training though a continuing medical education (CME) course (16%) or a mentoring program (17%).
Respondents were asked to rate the adequacy of their respective training in preparing them for their work as hospitalists. The vast majority rated their training in general clinical skills (94%) and communication (85%) as fully adequate. However, respondents found their training for some of the nonclinical aspects of their positions to be deficient. Many respondents rated training for QI projects (38%) and hospital administrative duties (46%) as inadequate (Table 4).
| Fully Adequate % (N) | Somewhat Adequate % (N) | Not Adequate % (N) | NA % (N) | |
|---|---|---|---|---|
| ||||
| General clinical skills | 94 (367) | 5 (21) | 0 (0) | 0 (1) |
| Communication skills | 85 (330) | 14 (53) | 1 (5) | 0 (1) |
| Coordination of care | 73 (284) | 23 (89) | 4 (15) | 0 (1) |
| Clinical procedure experience | 67 (258) | 32 (123) | 1 (5) | 1 (2) |
| Teaching skills (resident and medical student teaching) | 64 (248) | 31 (120) | 3 (13) | 2 (8) |
| Attending newborn deliveries | 60 (233) | 18 (70) | 4 (14) | 19 (72) |
| Running resuscitation (codes) | 45 (173) | 46 (177) | 5 (21) | 5 (18) |
| Quality improvement projects | 14 (55) | 42 (162) | 38 (148) | 6 (22) |
| Hospital administrative duties | 10 (37) | 37 (144) | 46 (177) | 8 (31) |
Survey respondents were asked to indicate the extent to which they agreed or disagreed with 3 statements regarding hospitalist training. The majority of respondents believed that hospitalists need training in QI methods (70%). However, most pediatric hospitalists (73%) did not believe that additional training beyond residency should be required. Only one‐third (36%) of respondents agreed that current CME offerings are adequate for their needs as a pediatric hospitalist.
Career Goals and Expectations
Respondents were asked to select 1 or more reasons why they became pediatric hospitalists. The top factors influencing respondents' decision to become a hospitalist were reported to be a preference for the inpatient setting (73%), clinical variety (72%), enjoyment of teaching in the inpatient setting (58%), and a flexible schedule (52%) (Table 5).
| Factor | % (N) |
|---|---|
| |
| Prefer inpatient setting | 73 (284) |
| Clinical variety | 72 (281) |
| Enjoy teaching in inpatient setting | 58 (225) |
| Flexible schedule | 52 (202) |
| Defined hours | 41 (161) |
| Attractive career opportunities | 21 (80) |
| Salary | 18 (70) |
| Unsure of long‐term career direction | 13 (51) |
| Other | 7 (28) |
| Needed short‐term employment | 4 (15) |
| Only position available | 3 (10) |
The majority (85%) were satisfied with their position as a pediatric hospitalist, with 37% reporting that they were extremely satisfied. Over one‐half (61%) expected to remain a hospitalist for the duration of their career.
RESULTS BY ACADEMIC STATUS
Only significant differences between academic and nonacademic hospitalists are presented.
Clinical Practice by Academic Status
Nonacademic respondents were more likely than academic respondents to report regular service in the normal newborn nursery, pediatric ICU, neonatal ICU, transports, emergency department, and as part of an emergency response team. Academic respondents were more likely to report regular service in outpatient clinics. Nonacademic respondents were more likely than academic respondents to perform or supervise lumbar punctures, sedation services, PICC or central line insertions, and circumcisions (Table 6).
| Academic* (N = 196) | Nonacademic (N = 194) | P Value | |
|---|---|---|---|
| |||
| Regularly provides service | |||
| Normal newborn nursery | 16% | 42% | <0.0001 |
| Pediatric ICU | 9% | 20% | 0.0065 |
| Neonatal ICU | 4% | 20% | <0.0001 |
| Transports | 3% | 15% | <0.0001 |
| Emergency department | 16% | 34% | <0.0001 |
| Emergency response team | 17% | 29% | <0.0001 |
| Outpatient clinic | 23% | 13% | 0.0168 |
| Performs or supervises procedures | |||
| Lumbar puncture | 84% | 92% | 0.0152 |
| Sedation services | 50% | 64% | 0.0055 |
| PICC insertion | 8% | 18% | 0.0031 |
| Central line insertion | 11% | 23% | 0.0018 |
| Circumcision | 5% | 16% | 0.0002 |
| Holds leadership roles | |||
| Education (student or house staff) | 63% | 27% | <0.0001 |
| Hospital administration | 21% | 10% | <0.0001 |
| Quality improvement initiatives | 33% | 18% | 0.0005 |
Professional Roles and Parameters by Academic Status
Responding academic pediatric hospitalists were twice as likely as nonacademic respondents to have a leadership role in the education of students and house staff and to hold a leadership position in hospital administration. The academic respondents were also more likely to report a leadership role in QI initiatives (Table 6).
Clinical and Educational Activities by Academic Status
Academic pediatric hospitalist respondents reported spending on average 52% of their time providing inpatient care (excluding teaching), in contrast to the nonacademic hospitalist respondents who reported 71% of their time was spent providing inpatient care (P < 0.0001). Academic respondents also reported that 19% of their time was spent providing inpatient teaching or supervising residents, compared to 12% of nonacademic respondents (P < 0.0001). Responding academic pediatric hospitalists reported spending a greater proportion of time participating in nonclinical teaching activities (5% versus 2%; P < 0.0001), administrative duties (11% versus 5%; P < 0.0001), and research (4% versus 1%; P < 0.0001) compared to the nonacademic respondents.
Nonacademic respondents were more likely than academic respondents to report no hospitalist‐specific training (64% versus 54%; P = 0.0324).
RESULTS BY HOSPITAL CHARACTERISTICS
For each hospital characteristic, only significant differences between dichotomized groups are presented.
Children's Hospitals versus Other Hospitals
Clinical Practice
Pediatric hospitalist respondents practicing in NACHRI hospitals were more likely to report that they provide regular service for general pediatric inpatients (98% versus 86%; P < 0.0001) as well as subspecialty inpatients (27% versus 17%; P = 0.044). Non‐NACHRI pediatric hospitalist respondents were twice as likely to report the provision of regular service in the normal newborn nursery (49% versus 22%; P < 0.0001), the neonatal ICU (21% versus 8%, P = 0.002), and the emergency department (38% versus 20%; P < 0.0001).
Among respondents, pediatric hospitalists who were not working at a children's hospital were more likely to report that they sometimes or routinely performed lumbar punctures (93% versus 85%; P = 0.037), infusion services (36% versus 21%; P = 0.003), and were twice as likely to perform circumcision (16% versus 8%; P = 0.041) compared to those working at children's hospitals.
Professional Roles and Parameters
Respondents working in children's hospitals were twice as likely to hold a leadership position in utilization review (12% versus 6%; P = 0.012), though respondents from non‐NACHRI hospitals were more likely to at least participate in utilization review (58% versus 40%; P = 0.004).
Hospitalist Training
Respondents from non‐NACHRI hospitals were more likely to report that they had received no hospitalist‐specific training (68% versus 56%; P = 0.029). Those at NACHRI hospitals were twice as likely to have received hospitalist training through a mentoring program (20% versus 9%; P = 0.009).
Freestanding versus Nonfreestanding Children's Hospitals
Clinical Practice
Pediatric hospitalist respondents employed at institutions that are not freestanding children's hospitals were more likely to report that they provided regular service in the normal newborn nursery (42% versus 14%; P < 0.0001), pediatric ICU (22% versus 5%), emergency department (32% versus 17%; P < 0.0001), and outpatient clinics (23% versus 12%; P = 0.0068). They were also more likely to perform or supervise sedation services (63% versus 50%; P = 0.0116), infusion services (32% versus 17%; P = 0.0006), PICC insertions (19% versus 6%; P = 0.0002), central line insertions (23% versus 11%; P = 0.0024), and circumcisions (16% versus 3%; P < 0.0001).
Professional Roles and Parameters
Among respondents, pediatric hospitalists employed by nonfreestanding children's hospitals were more likely to report participation in utilization review (51% versus 38%; P = 0.02).
Hospital Size
Clinical Practice
Pediatric hospitalist respondents working at large hospitals were twice as likely to report that they regularly provided service in the pediatric ICU (18% versus 7%; P = 0.0072) and were more likely to regularly perform circumcisions (13% versus 5%; P = 0.0069). Respondents from small hospitals were more likely to provide regular service in the neonatal ICU (20% versus 7%; P = 0.0013).
COTH Status: Teaching versus Nonteaching Hospitals
Clinical Practice
Among survey respondents, pediatric hospitalists employed by COTH hospitals were more likely to provide regular service in the neonatal ICU, compared to their peers in nonteaching hospitals (15% versus 6%; P = 0.0109). Those employed by non‐COTH hospitals were more likely to provide service in subspecialty inpatient service (38% versus 16%; P < 0.0001), transports (14% versus 6%; P = 0.0227), inpatient consultation (61% versus 45%; P = 0.0086), and the emergency response team (29% versus 19%; P = 0.0021).
Professional Roles and Parameters
Respondents from COTH hospitals were more likely to have no involvement in utilization review, compared to their peers at non‐COTH hospitals (49% versus 37%; P = 0.0220).
DISCUSSION
This study provides the most comprehensive information available regarding the clinical and nonclinical roles, training, work expectations, and career plans of pediatric hospitalists. Among the most important of our findings is the distribution of the length of time that pediatric hospitalists had served in their roles. While over one‐third (37%) reported having been practicing as hospitalists for over 5 years, 45% of our respondents had been in practice for fewer than 3 years. This is consistent with both the perceptions of rapid growth of the field and with significant turnover of hospitalists.1, 8 It is important to note that our findings may actually overestimate the proportion of hospitalists with longer durations of employment as our sampling strategy would have been less likely to include those who left the field within the first 12 to 18 months of practice. Nevertheless, over half (61%) of our respondents expected to remain a hospitalist for the duration of their career and few reported choosing to become a hospitalist as a short‐term employment option. This finding has important implications for the future stability of the hospitalist workforce and the potential development of specific expertise among this cadre of clinicians.6
The demographic profile of pediatric hospitalists was also consistent with these findings. The mean age of 39 years for our respondents is indicative of a significant proportion of this group of physicians recently having completed their residency training. Further, the gender distribution approximates that of current pediatric residency graduates, thus indicating that that this is not a clinical choice for which there would be a skewed distribution as is the case in some pediatric subspecialties.9
Our findings were similar to the 2004 Ottolini et al.10 findings on the roles of pediatric hospitalists. Respondents in our study reported spending less time providing inpatient care (61% versus 75%), providing clinical teaching or supervising residents (16% versus 26%), performing administrative duties (8% versus 19%), and conducting research (3% versus 9%) compared with the respondents in the Ottolini et al.10 survey.
At this point in time, fewer than half of our respondents reported any hospitalist‐specific training, including workshops at professional meetings or CME coursework. As there are a paucity of fellowships offering postresidency training in pediatric hospital medicine, and most of the existing programs are newly established, few in practice have completed such programs.11 In addition, most respondents reported that current CME offerings do not meet their needs, and that they could have used additional QI training to prepare them for their role as pediatric hospitalists. However, almost three‐quarters of respondents (73%) do not believe any additional training beyond residency should be required. As such, it is unclear if a defined, unique body of knowledge specific to hospitalists is either needed or desired by those currently in the field.
Although there are a broad range of potential clinical roles within hospital medicine, and this clinical variety influenced most respondents' decisions to become hospitalists, the current scope of an individual hospitalist tends to become somewhat focused.12, 13 While we found almost all provided service on the pediatric inpatient unit, many fewer provided inpatient consultation and normal newborn care, or were involved in interhospital transport or as part of an emergency response team. There is also wide variation in the types of procedures performed or supervised by hospitalists at different institutions. More than half never perform or supervise infusion services, PICC or central line placement, or circumcision. The variation seen among hospitalists practicing in different hospital settings likely is a result, at least in part, of different needs in teaching hospitals for both service and for clinical experience of trainees. For example, our results demonstrate that pediatric hospitalists in nonteaching and non‐children's hospitals are more likely to have a broader scope of clinical care provision. Another potential issue is that some hospitalists may be employed by institutions which have no pediatric ICU, neonatal ICU, or other specialty unit. As such, these hospitalists would not have the opportunity to work in such settings.
Further, those without academic appointments are also more likely to have expanded clinical roles compared with their academic counterparts. This may be due to the fact that there is likely a greater number of subspecialty‐trained pediatric providers in academic centers and thus the need for hospitalists to cover specific services or perform specific procedures is lessened. There may also be a desire to prevent competition among care providers within the same institution. In contrast, hospitalists with academic appointments are more likely (though still uncommonly) to have taken leadership roles in hospital administration and QI initiatives. Thus, the nature of their efforts appears to expand into nonclinical delivery areas.
Clearly, hospitalists report they have assumed a significant role in the clinical teaching of trainees at all levels, with 94% of our respondents maintaining at least some involvement in education. On average, they spend 16% of their time in educational efforts. However, there are few data on the impact of their work in this area.5, 13 Studies in pediatrics to date have been limited to a few institutions,3, 5 and have not addressed the issue from the perspective of residency program directors or those who are in charge of inpatient curricula.
This study, like the majority of studies related to pediatric hospitalists, is hampered by the difficulty of identifying pediatric hospitalists. Rather than utilizing a hospital medicine membership list, which would be potentially biased by self‐selection, we attempted to obtain a more representative sample through utilization of the AHA database.
CONCLUSIONS
Findings from this study provide an additional perspective regarding pediatric hospitalists to add to our previous study of hospitalist program directors.1 However, the field is currently a moving target. Our data demonstrate that there is significant flux in the hospitalist workforce, uncertainty regarding turnover, and variation in the roles of these professionals in their clinical and nonclinical work environment. Moreover, additional studies of the educational impact of hospitalists on residency and medical student education are needed. Questions regarding the nature and degree of resident autonomy and experience conducting procedures in the hospitalist environment have been raised. These must be assessed through studies of residency program directors, their expectations of residents, and the curricula they have developed.
As with any new phenomenon, it will take time to understand the impact of hospitalists in a variety of domains. Additional research will be helpful in following the development of this field and the manner in which it will interface with existing medical practice and educational programs.
- ,,,; The Research Advisory Committee of the American Board of Pediatrics.Characteristics of the pediatric hospitalist workforce: its roles and work environment.Pediatrics.2007;120:33–39.
- .The evolution of the hospitalist model in the United States.Med Clin North Am.2002;86:687–706.
- ,.Hospitalists in children's hospitals: what we know now and what we need to know.J Pediatr.2006;148:296–299.
- ,.Hospitalists: the new model of inpatient medical care in the United States.Eur J Intern Med.2003;14:65–70.
- ,,,,,.Effect of a pediatric hospitalist system on housestaff education and experience.Arch Pediatr Adolesc Med.2002;156:877–883.
- ,,,.Hospitalists' perceptions of their residency training needs: results of a national survey.Am J Med.2001;111:247–254.
- ,,,,,.Pediatric hospitalists in Canada and the United States: a survey of pediatric academic department chairs.Ambul Pediatr.2001;1:338–339.
- .Hospitalists in the United States: mission accomplished or work in progress?N Engl J Med.2004;350:1935–1936.
- ,.Pediatric workforce: a look at general pediatrics data from the American Board of Pediatrics.J Pediatr.2006;148:166–169.
- ,,,,PRIS survey: pediatric hospitalist roles and training needs [Abstr].Pediatr Res.2004;55:360A.
- ,,,.Hospital medicine fellowships: works in progress.Am J Med.2006;119:1.e1–1.e7.
- ,,.How hospitalists spend their time: insights on efficiency and safety.J Hosp Med.2006;1:88–93.
- ,,.Pediatric hospitalists fill varied roles in the care of newborns.Pediatr Ann.2003;32:802–810.
- ,,,; The Research Advisory Committee of the American Board of Pediatrics.Characteristics of the pediatric hospitalist workforce: its roles and work environment.Pediatrics.2007;120:33–39.
- .The evolution of the hospitalist model in the United States.Med Clin North Am.2002;86:687–706.
- ,.Hospitalists in children's hospitals: what we know now and what we need to know.J Pediatr.2006;148:296–299.
- ,.Hospitalists: the new model of inpatient medical care in the United States.Eur J Intern Med.2003;14:65–70.
- ,,,,,.Effect of a pediatric hospitalist system on housestaff education and experience.Arch Pediatr Adolesc Med.2002;156:877–883.
- ,,,.Hospitalists' perceptions of their residency training needs: results of a national survey.Am J Med.2001;111:247–254.
- ,,,,,.Pediatric hospitalists in Canada and the United States: a survey of pediatric academic department chairs.Ambul Pediatr.2001;1:338–339.
- .Hospitalists in the United States: mission accomplished or work in progress?N Engl J Med.2004;350:1935–1936.
- ,.Pediatric workforce: a look at general pediatrics data from the American Board of Pediatrics.J Pediatr.2006;148:166–169.
- ,,,,PRIS survey: pediatric hospitalist roles and training needs [Abstr].Pediatr Res.2004;55:360A.
- ,,,.Hospital medicine fellowships: works in progress.Am J Med.2006;119:1.e1–1.e7.
- ,,.How hospitalists spend their time: insights on efficiency and safety.J Hosp Med.2006;1:88–93.
- ,,.Pediatric hospitalists fill varied roles in the care of newborns.Pediatr Ann.2003;32:802–810.
Copyright © 2009 Society of Hospital Medicine
Takotsubo Cardiomyopathy
Takotsubo cardiomyopathy, also known as transient left ventricular apical ballooning, stress cardiomyopathy, and broken heart syndrome, is a condition that mimics acute myocardial infarction. Patients typically present with chest pain, electrocardiographic changes consistent with acute ischemia or infarct, and elevated cardiac enzymes in the absence of significant coronary artery disease. Left ventriculography demonstrates a characteristic pattern of dysfunction: dyskinesis of the cardiac apex and hyperkinesis of the base. This resulting appearance of apical ballooning is reminiscent of the takotsubo, a Japanese octopus pot with a wide base and narrow top. The syndrome occurs almost exclusively in postmenopausal women and demonstrates a distinct temporal association with extreme emotional or physiological stress. The pathophysiology is poorly understood, but one theory suggests that the transient cardiomyopathy reflects myocardial stunning due to excessive sympathetic output.1 Treatment is supportive, and most patients rapidly recover normal systolic function.
A 57‐year‐old African American female with a past medical history significant only for chronic obstructive pulmonary disease presented with severe dyspnea that was progressive over several hours following the unexpected death of her son. She denied chest pain, palpitations, cough, or fever. On examination, she was afebrile with a blood pressure of 145/82 mm Hg, a pulse of 90 beats per minute, and a respiratory rate of 24 breaths per minute with oxygen saturations of 88% on room air. Lung examination revealed coarse breath sounds with a slightly prolonged expiration phase, but it was otherwise clear. Cardiac examination was unremarkable. Chest radiograph showed only emphysematous changes. Initial electrocardiogram and serial cardiac enzymes were negative. A computed tomography pulmonary angiogram showed no evidence of pulmonary embolism. The patient was admitted with the diagnosis of chronic obstructive pulmonary disease exacerbation and treated with supplemental oxygen, bronchodilators, and corticosteroids.
On the following day, the patient developed worsening dyspnea, hypoxia, and diffuse crackles on examination. Electrocardiogram at that time demonstrated ST‐segment elevations in leads V1 and V2 as well as T‐wave inversions in all precordial leads (Figure 1). The troponin‐I concentration was 1.92 ng/mL (0.05 nL), and the brain natriuretic peptide concentration was 1425 pg/mL. The patient underwent urgent cardiac catheterization with no evidence of coronary artery obstruction. Left ventriculogram revealed a hyperdynamic base and akinetic apex extending into the mid‐heart (Figure 2). Left ventricular systolic function was severely reduced, with an estimated ejection fraction of 10% to 15%. The normal diastolic ventriculogram image is shown for comparison (Figure 3). These findings were felt to be consistent with takotsubo syndrome. The patient required inotropic support briefly but experienced full clinical recovery by the sixth hospital day.

DISCUSSION
Takotsubo cardiomyopathy was first described in Japan in 1991.2 Although the condition made a relatively recent debut in the United States, with the first case series published in 2003, subsequent reports have suggested that the condition is not rare.3 Recent analyses of Western populations estimate the prevalence to be approximately 2% among patients with acute coronary syndrome.1, 4, 5 Because women compose the majority of patients with takotsubo cardiomyopathy, the prevalence among women as a subset of patients with acute coronary syndrome is likely much higher. The syndrome has been described as a clinical entity in Japanese, European, Caucasian, and African American patients.2, 3 Interesting differences appear to exist among different ethnic groups. For example, evidence suggests that the condition is more likely to be precipitated by emotional stress in Caucasians, whereas physiological stress is a more frequent trigger in Asians.6
Although chest pain is described as a cardinal feature in takotsubo cardiomyopathy, existing data suggest that African American patients may lack this typical symptom. The first African American female reported with takotsubo syndrome presented with heart failure and hypotension in the absence of chest pain.1 Subsequently, Patel et al.7 reported 5 African American women with takotsubo syndrome. Three patients presented with dyspnea, and 2 presented with nausea: none of the patients experienced chest pain. Our case adds to this evidence by describing an African American woman with takotsubo syndrome whose presenting symptom was severe dyspnea without chest pain. Unlike the majority of reported cases, electrocardiographic and biomarker abnormalities were not present in our patient at admission. As with our patient, the diagnosis of takotsubo cardiomyopathy may initially be overlooked in African Americans because of the atypical presentation. As takotsubo syndrome becomes increasingly recognized in the United States, clinicians are encouraged to consider the diagnosis in African American women who present with severe dyspnea in the setting of extreme emotional or physiological stress. Further research on the pathophysiology of takotsubo cardiomyopathy is needed to explain why such differences in presenting symptoms may exist.
- ,,, et al.Neurohormonal features of myocardial stunning due to sudden emotional stress.N Engl J Med.2005;352:540–548.
- ,,, et al.Myocardial stunning due to simultaneous multivessel coronary spasm: a review of 5 cases.J Cardiol.1991;21:203–214.
- ,,, et al.A syndrome of transient left ventricular apical wall motion abnormality in the absence of coronary disease: a perspective from the United States.Cardiology.2003;100:61–66.
- ,,, et al.Clinical characteristics and thrombolysis in myocardial infarction frame counts in women with transient left ventricular ballooning syndrome.Am J Cardiol.2004;94:343–346.
- ,,, et al.Left ventricular apical ballooning: not an uncommon variant of acute myocardial infarction in women.Clin Cardiol.2006;29:9–12.
- ,.Clinical characteristics, demographics and prognosis of transient left ventricular apical ballooning syndrome.Heart Fail Rev.2005;10:311–316.
- ,,, et al.Takotsubo syndrome in African‐American women with atypical presentations: a single‐center experience.Clin Cardiol2007;30:14–18.
Takotsubo cardiomyopathy, also known as transient left ventricular apical ballooning, stress cardiomyopathy, and broken heart syndrome, is a condition that mimics acute myocardial infarction. Patients typically present with chest pain, electrocardiographic changes consistent with acute ischemia or infarct, and elevated cardiac enzymes in the absence of significant coronary artery disease. Left ventriculography demonstrates a characteristic pattern of dysfunction: dyskinesis of the cardiac apex and hyperkinesis of the base. This resulting appearance of apical ballooning is reminiscent of the takotsubo, a Japanese octopus pot with a wide base and narrow top. The syndrome occurs almost exclusively in postmenopausal women and demonstrates a distinct temporal association with extreme emotional or physiological stress. The pathophysiology is poorly understood, but one theory suggests that the transient cardiomyopathy reflects myocardial stunning due to excessive sympathetic output.1 Treatment is supportive, and most patients rapidly recover normal systolic function.
A 57‐year‐old African American female with a past medical history significant only for chronic obstructive pulmonary disease presented with severe dyspnea that was progressive over several hours following the unexpected death of her son. She denied chest pain, palpitations, cough, or fever. On examination, she was afebrile with a blood pressure of 145/82 mm Hg, a pulse of 90 beats per minute, and a respiratory rate of 24 breaths per minute with oxygen saturations of 88% on room air. Lung examination revealed coarse breath sounds with a slightly prolonged expiration phase, but it was otherwise clear. Cardiac examination was unremarkable. Chest radiograph showed only emphysematous changes. Initial electrocardiogram and serial cardiac enzymes were negative. A computed tomography pulmonary angiogram showed no evidence of pulmonary embolism. The patient was admitted with the diagnosis of chronic obstructive pulmonary disease exacerbation and treated with supplemental oxygen, bronchodilators, and corticosteroids.
On the following day, the patient developed worsening dyspnea, hypoxia, and diffuse crackles on examination. Electrocardiogram at that time demonstrated ST‐segment elevations in leads V1 and V2 as well as T‐wave inversions in all precordial leads (Figure 1). The troponin‐I concentration was 1.92 ng/mL (0.05 nL), and the brain natriuretic peptide concentration was 1425 pg/mL. The patient underwent urgent cardiac catheterization with no evidence of coronary artery obstruction. Left ventriculogram revealed a hyperdynamic base and akinetic apex extending into the mid‐heart (Figure 2). Left ventricular systolic function was severely reduced, with an estimated ejection fraction of 10% to 15%. The normal diastolic ventriculogram image is shown for comparison (Figure 3). These findings were felt to be consistent with takotsubo syndrome. The patient required inotropic support briefly but experienced full clinical recovery by the sixth hospital day.
DISCUSSION
Takotsubo cardiomyopathy was first described in Japan in 1991.2 Although the condition made a relatively recent debut in the United States, with the first case series published in 2003, subsequent reports have suggested that the condition is not rare.3 Recent analyses of Western populations estimate the prevalence to be approximately 2% among patients with acute coronary syndrome.1, 4, 5 Because women compose the majority of patients with takotsubo cardiomyopathy, the prevalence among women as a subset of patients with acute coronary syndrome is likely much higher. The syndrome has been described as a clinical entity in Japanese, European, Caucasian, and African American patients.2, 3 Interesting differences appear to exist among different ethnic groups. For example, evidence suggests that the condition is more likely to be precipitated by emotional stress in Caucasians, whereas physiological stress is a more frequent trigger in Asians.6
Although chest pain is described as a cardinal feature in takotsubo cardiomyopathy, existing data suggest that African American patients may lack this typical symptom. The first African American female reported with takotsubo syndrome presented with heart failure and hypotension in the absence of chest pain.1 Subsequently, Patel et al.7 reported 5 African American women with takotsubo syndrome. Three patients presented with dyspnea, and 2 presented with nausea: none of the patients experienced chest pain. Our case adds to this evidence by describing an African American woman with takotsubo syndrome whose presenting symptom was severe dyspnea without chest pain. Unlike the majority of reported cases, electrocardiographic and biomarker abnormalities were not present in our patient at admission. As with our patient, the diagnosis of takotsubo cardiomyopathy may initially be overlooked in African Americans because of the atypical presentation. As takotsubo syndrome becomes increasingly recognized in the United States, clinicians are encouraged to consider the diagnosis in African American women who present with severe dyspnea in the setting of extreme emotional or physiological stress. Further research on the pathophysiology of takotsubo cardiomyopathy is needed to explain why such differences in presenting symptoms may exist.
Takotsubo cardiomyopathy, also known as transient left ventricular apical ballooning, stress cardiomyopathy, and broken heart syndrome, is a condition that mimics acute myocardial infarction. Patients typically present with chest pain, electrocardiographic changes consistent with acute ischemia or infarct, and elevated cardiac enzymes in the absence of significant coronary artery disease. Left ventriculography demonstrates a characteristic pattern of dysfunction: dyskinesis of the cardiac apex and hyperkinesis of the base. This resulting appearance of apical ballooning is reminiscent of the takotsubo, a Japanese octopus pot with a wide base and narrow top. The syndrome occurs almost exclusively in postmenopausal women and demonstrates a distinct temporal association with extreme emotional or physiological stress. The pathophysiology is poorly understood, but one theory suggests that the transient cardiomyopathy reflects myocardial stunning due to excessive sympathetic output.1 Treatment is supportive, and most patients rapidly recover normal systolic function.
A 57‐year‐old African American female with a past medical history significant only for chronic obstructive pulmonary disease presented with severe dyspnea that was progressive over several hours following the unexpected death of her son. She denied chest pain, palpitations, cough, or fever. On examination, she was afebrile with a blood pressure of 145/82 mm Hg, a pulse of 90 beats per minute, and a respiratory rate of 24 breaths per minute with oxygen saturations of 88% on room air. Lung examination revealed coarse breath sounds with a slightly prolonged expiration phase, but it was otherwise clear. Cardiac examination was unremarkable. Chest radiograph showed only emphysematous changes. Initial electrocardiogram and serial cardiac enzymes were negative. A computed tomography pulmonary angiogram showed no evidence of pulmonary embolism. The patient was admitted with the diagnosis of chronic obstructive pulmonary disease exacerbation and treated with supplemental oxygen, bronchodilators, and corticosteroids.
On the following day, the patient developed worsening dyspnea, hypoxia, and diffuse crackles on examination. Electrocardiogram at that time demonstrated ST‐segment elevations in leads V1 and V2 as well as T‐wave inversions in all precordial leads (Figure 1). The troponin‐I concentration was 1.92 ng/mL (0.05 nL), and the brain natriuretic peptide concentration was 1425 pg/mL. The patient underwent urgent cardiac catheterization with no evidence of coronary artery obstruction. Left ventriculogram revealed a hyperdynamic base and akinetic apex extending into the mid‐heart (Figure 2). Left ventricular systolic function was severely reduced, with an estimated ejection fraction of 10% to 15%. The normal diastolic ventriculogram image is shown for comparison (Figure 3). These findings were felt to be consistent with takotsubo syndrome. The patient required inotropic support briefly but experienced full clinical recovery by the sixth hospital day.
DISCUSSION
Takotsubo cardiomyopathy was first described in Japan in 1991.2 Although the condition made a relatively recent debut in the United States, with the first case series published in 2003, subsequent reports have suggested that the condition is not rare.3 Recent analyses of Western populations estimate the prevalence to be approximately 2% among patients with acute coronary syndrome.1, 4, 5 Because women compose the majority of patients with takotsubo cardiomyopathy, the prevalence among women as a subset of patients with acute coronary syndrome is likely much higher. The syndrome has been described as a clinical entity in Japanese, European, Caucasian, and African American patients.2, 3 Interesting differences appear to exist among different ethnic groups. For example, evidence suggests that the condition is more likely to be precipitated by emotional stress in Caucasians, whereas physiological stress is a more frequent trigger in Asians.6
Although chest pain is described as a cardinal feature in takotsubo cardiomyopathy, existing data suggest that African American patients may lack this typical symptom. The first African American female reported with takotsubo syndrome presented with heart failure and hypotension in the absence of chest pain.1 Subsequently, Patel et al.7 reported 5 African American women with takotsubo syndrome. Three patients presented with dyspnea, and 2 presented with nausea: none of the patients experienced chest pain. Our case adds to this evidence by describing an African American woman with takotsubo syndrome whose presenting symptom was severe dyspnea without chest pain. Unlike the majority of reported cases, electrocardiographic and biomarker abnormalities were not present in our patient at admission. As with our patient, the diagnosis of takotsubo cardiomyopathy may initially be overlooked in African Americans because of the atypical presentation. As takotsubo syndrome becomes increasingly recognized in the United States, clinicians are encouraged to consider the diagnosis in African American women who present with severe dyspnea in the setting of extreme emotional or physiological stress. Further research on the pathophysiology of takotsubo cardiomyopathy is needed to explain why such differences in presenting symptoms may exist.
- ,,, et al.Neurohormonal features of myocardial stunning due to sudden emotional stress.N Engl J Med.2005;352:540–548.
- ,,, et al.Myocardial stunning due to simultaneous multivessel coronary spasm: a review of 5 cases.J Cardiol.1991;21:203–214.
- ,,, et al.A syndrome of transient left ventricular apical wall motion abnormality in the absence of coronary disease: a perspective from the United States.Cardiology.2003;100:61–66.
- ,,, et al.Clinical characteristics and thrombolysis in myocardial infarction frame counts in women with transient left ventricular ballooning syndrome.Am J Cardiol.2004;94:343–346.
- ,,, et al.Left ventricular apical ballooning: not an uncommon variant of acute myocardial infarction in women.Clin Cardiol.2006;29:9–12.
- ,.Clinical characteristics, demographics and prognosis of transient left ventricular apical ballooning syndrome.Heart Fail Rev.2005;10:311–316.
- ,,, et al.Takotsubo syndrome in African‐American women with atypical presentations: a single‐center experience.Clin Cardiol2007;30:14–18.
- ,,, et al.Neurohormonal features of myocardial stunning due to sudden emotional stress.N Engl J Med.2005;352:540–548.
- ,,, et al.Myocardial stunning due to simultaneous multivessel coronary spasm: a review of 5 cases.J Cardiol.1991;21:203–214.
- ,,, et al.A syndrome of transient left ventricular apical wall motion abnormality in the absence of coronary disease: a perspective from the United States.Cardiology.2003;100:61–66.
- ,,, et al.Clinical characteristics and thrombolysis in myocardial infarction frame counts in women with transient left ventricular ballooning syndrome.Am J Cardiol.2004;94:343–346.
- ,,, et al.Left ventricular apical ballooning: not an uncommon variant of acute myocardial infarction in women.Clin Cardiol.2006;29:9–12.
- ,.Clinical characteristics, demographics and prognosis of transient left ventricular apical ballooning syndrome.Heart Fail Rev.2005;10:311–316.
- ,,, et al.Takotsubo syndrome in African‐American women with atypical presentations: a single‐center experience.Clin Cardiol2007;30:14–18.