User login
Pediatric HM Highlights

It’s been a while since I went to a buffet and stuffed myself silly, but that’s how my mind felt on the flight home from Kansas City, Mo. After four incredibly packed days at Pediatric Hospital Medicine (PHM) 2011, I was one “wafer-thin mint” away from an explosion. What I wanted was some kind of a mental digestif. What I got instead was a light beer. It helped a little bit, but when I awoke during the landing in Austin, Texas, I realized that I remained in need of a better way to distill the thousand points of information from the conference into something more manageable.
Buoyed by Michelle Marks and Joel Tieder’s Top 10 Articles of the Year luncheon, specifically the piece on neurosurgeons and Kangaroo Care, I thought I might try my own version of a decompressive operation.
Without further delay, here are the top 10 things that I learned at Pediatric Hospital Medicine 2011:
10. We continue to grow as a field. Although the exact number of pediatric hospitalists in the U.S. remains somewhat unclear (but is probably between 1,000 and 2,000), what is known is that attendance at our annual meeting grows every year. Our tripartite meeting, sponsored by the American Academy of Pediatrics (AAP), SHM, and the Academic Pediatric Association (APA), hit a record 450 attendees this year. Beyond the physical numbers, it is quite clear that we are growing in many other domains as well.
9. It is time to re-evaluate the impact of CME on physician practice and outcomes. The literature on continuing professional education has been quite sobering to date, with nothing to show for the thousands of dollars spent per individual. But most of those studies were performed in the last century (think eight hours of lecture a day followed by dinners with big pharma) and I’ll bet that there was not the focus on learner-centered education that was evident in Kansas City.
With a dizzying array of workshops and interactive small group sessions spread amongst seven different tracks, it was difficult, if not impossible, to be a passive participant in the process. And since learning retention rates are generally proportionate to how active a role adults play in their own education, I am going to guess that many other attendees’ brains still have that “I’m thinking” hourglass icon over them. We have the conference planning committee to thank for this.
8. The JCPHM (Joint Council of Pediatric Hospital Medicine) will be increasingly important as we develop. Much like the constant stream of unfamiliar new vaccine names that have appeared in recent years, this proposed new committee comes with another long set of initials and an unfamiliar indication.
The JCPHM will function as a coordinating body, ensuring that work done through the AAP, SHM, and AAP are aligned to provide maximal benefit to pediatric hospitalists as a whole. And thus, similar to immunizations, the benefits will be most evident if we remain healthy as we grow in the context of an increasingly complex environment.
7. Our collective research accomplishments merit national recognition. It was not more than just a few years ago that we were in our research infancy. Posters at our meetings largely represented single-site descriptive studies, typically using survey methodology. This year we had four research breakout tracks, in addition to the plenary and three poster sessions.
Pediatric Research in Inpatient Settings (PRIS) leads the way with a Forbes-like listing of million-dollar grants and a partnership with Child Health Corporation of America’s (CHCA) uber-powerful Pediatric Health Information Systems (PHIS) database. Expect some landmark studies in the near future.
In the meantime, it is clear that the rest of the field is not languishing from smaller budgets. From clear outcome and process measurements of family-centered rounds to studying the spectrum of transitions of care to the impact of early warning systems, there was a predominant focus on quality and safety.
In fact, the tone was set at the opening keynote address, as Carolyn Clancy, MD, director of the Agency for Healthcare Research and Quality (AHRQ), described creative and innovative ways to study and translate work in this area to improved patient care.
6. We are poised to develop effective training systems for our future workforce. From the use of our core competencies in sessions to a full complement of workshops on education (from individual to team and from student to fellow), it is clear that thoughtful deliberation has paved the way for our future. We possess expertise in education that dovetails nicely with our need to grow and sustain an experienced, well-trained workforce.
Intrinsically, we know that we possess a unique body of skills, knowledge, and attitudes. The explicit articulation of this into longitudinal curricula will headline our evolution as a field.
5. Our new peer-reviewed journal has a bright future. Kudos to Shawn Ralston and the rest of the editorial board for publishing the first peer-reviewed edition of Hospital Pediatrics. Original research, evidence-based content, and practical commentary grace the pages with a little bit of something for everyone. The AAP has demonstrated a healthy level of support for this endeavor, as they sent out an introductory email announcing the journal to all of their membership over the weekend. I am confident that support from our pediatric hospitalist community will follow in the form of an exponential increase in quality submissions. Look no further than the PHM 2011 abstract book to get a preview of what our journal will highlight in the near future.
4. We connect with each other through a language of quality and value in our work. Quality spanned the continuum from conversation to collaboration, as like-minded souls shared ideas and passions amidst the sessions. The improved outcomes demonstrated by the Value in Inpatient Pediatrics (VIP) network are a testament to the positive change that can arise from such efforts. VIP, with its focus on inclusive and front-line collaboratives, also announced an upcoming merger with the AAP’s Quality Improvement and Innovation Network (QuIIN), approved by the AAP board in May.
Perhaps more impressive was that more than 12% of the attendees at PHM 2011 attended the annual VIP dinner and similar numbers signed up to participate in future efforts. At this pace, widespread improvement and value are easily within our sights.
3. Complex care is the new family-centered rounds. Atul Gawande’s recent New Yorker article about “Hot Spotters” could very well have been referring to the body of work that is represented by pediatric generalists (to include a fair number of hospitalists) over the past few years. Closing plenary speakers Robert Lyle and Patrick Casey wowed the audience as they described their medical home for medically complex children—and an estimated savings to Arkansas Medicaid of nearly $3 million per year.
As hospitals and hospital systems look to create accountable care organizations (ACOs), this kind of work will be increasingly prioritized, as it has the potential to generate the biggest gains in valued care.
2. STP, yeah, you know me. Chris Maloney and Suzanne Swanson Mendez brought down the house at the PHM Roundtable update as they presented preliminary results of their large and representative STP (strategic planning) Committee, which is mapping out future certification options for pediatric hospitalists. A lively debate ensued as questions surrounding how to best notify and involve pediatric hospitalists in these decisions came to the forefront. Are we a democracy? Are we a republic? Is there a better model for this decision?
Despite the lack of consensus on how to best move toward a decision, the discussion remained open and engaging. In contrast to recent certification decisions from other organizations, the audience clearly relished the opportunity to provide input, and the STP committee continues to look for able and willing participants.
1. A top-10 list is not enough to cover everything from PHM 2011. From clinical and practice conundrums galore to late nights at Spectators to Kevin Powell’s mad acting skills, a 1,500-word-top-10 list simply does not do the meeting justice! I place full blame on the planning committee for this overindulgent buffet and the unfortunate omission of many other meaningful lessons.
Thank you: Erin Stucky Fisher (chair), Brian Pate, Allison Ballantine, Matt Garber, Jeff Simmons, Doug Carlson and Tamara Simon.
If you’re feeling like you missed out, or have already digested and want more, PHM 2012 will be here soon enough to fill your appetite. Cincinnati, look out.
Dr. Shen is pediatric editor of The Hospitalist.
It’s been a while since I went to a buffet and stuffed myself silly, but that’s how my mind felt on the flight home from Kansas City, Mo. After four incredibly packed days at Pediatric Hospital Medicine (PHM) 2011, I was one “wafer-thin mint” away from an explosion. What I wanted was some kind of a mental digestif. What I got instead was a light beer. It helped a little bit, but when I awoke during the landing in Austin, Texas, I realized that I remained in need of a better way to distill the thousand points of information from the conference into something more manageable.
Buoyed by Michelle Marks and Joel Tieder’s Top 10 Articles of the Year luncheon, specifically the piece on neurosurgeons and Kangaroo Care, I thought I might try my own version of a decompressive operation.
Without further delay, here are the top 10 things that I learned at Pediatric Hospital Medicine 2011:
10. We continue to grow as a field. Although the exact number of pediatric hospitalists in the U.S. remains somewhat unclear (but is probably between 1,000 and 2,000), what is known is that attendance at our annual meeting grows every year. Our tripartite meeting, sponsored by the American Academy of Pediatrics (AAP), SHM, and the Academic Pediatric Association (APA), hit a record 450 attendees this year. Beyond the physical numbers, it is quite clear that we are growing in many other domains as well.
9. It is time to re-evaluate the impact of CME on physician practice and outcomes. The literature on continuing professional education has been quite sobering to date, with nothing to show for the thousands of dollars spent per individual. But most of those studies were performed in the last century (think eight hours of lecture a day followed by dinners with big pharma) and I’ll bet that there was not the focus on learner-centered education that was evident in Kansas City.
With a dizzying array of workshops and interactive small group sessions spread amongst seven different tracks, it was difficult, if not impossible, to be a passive participant in the process. And since learning retention rates are generally proportionate to how active a role adults play in their own education, I am going to guess that many other attendees’ brains still have that “I’m thinking” hourglass icon over them. We have the conference planning committee to thank for this.
8. The JCPHM (Joint Council of Pediatric Hospital Medicine) will be increasingly important as we develop. Much like the constant stream of unfamiliar new vaccine names that have appeared in recent years, this proposed new committee comes with another long set of initials and an unfamiliar indication.
The JCPHM will function as a coordinating body, ensuring that work done through the AAP, SHM, and AAP are aligned to provide maximal benefit to pediatric hospitalists as a whole. And thus, similar to immunizations, the benefits will be most evident if we remain healthy as we grow in the context of an increasingly complex environment.
7. Our collective research accomplishments merit national recognition. It was not more than just a few years ago that we were in our research infancy. Posters at our meetings largely represented single-site descriptive studies, typically using survey methodology. This year we had four research breakout tracks, in addition to the plenary and three poster sessions.
Pediatric Research in Inpatient Settings (PRIS) leads the way with a Forbes-like listing of million-dollar grants and a partnership with Child Health Corporation of America’s (CHCA) uber-powerful Pediatric Health Information Systems (PHIS) database. Expect some landmark studies in the near future.
In the meantime, it is clear that the rest of the field is not languishing from smaller budgets. From clear outcome and process measurements of family-centered rounds to studying the spectrum of transitions of care to the impact of early warning systems, there was a predominant focus on quality and safety.
In fact, the tone was set at the opening keynote address, as Carolyn Clancy, MD, director of the Agency for Healthcare Research and Quality (AHRQ), described creative and innovative ways to study and translate work in this area to improved patient care.
6. We are poised to develop effective training systems for our future workforce. From the use of our core competencies in sessions to a full complement of workshops on education (from individual to team and from student to fellow), it is clear that thoughtful deliberation has paved the way for our future. We possess expertise in education that dovetails nicely with our need to grow and sustain an experienced, well-trained workforce.
Intrinsically, we know that we possess a unique body of skills, knowledge, and attitudes. The explicit articulation of this into longitudinal curricula will headline our evolution as a field.
5. Our new peer-reviewed journal has a bright future. Kudos to Shawn Ralston and the rest of the editorial board for publishing the first peer-reviewed edition of Hospital Pediatrics. Original research, evidence-based content, and practical commentary grace the pages with a little bit of something for everyone. The AAP has demonstrated a healthy level of support for this endeavor, as they sent out an introductory email announcing the journal to all of their membership over the weekend. I am confident that support from our pediatric hospitalist community will follow in the form of an exponential increase in quality submissions. Look no further than the PHM 2011 abstract book to get a preview of what our journal will highlight in the near future.
4. We connect with each other through a language of quality and value in our work. Quality spanned the continuum from conversation to collaboration, as like-minded souls shared ideas and passions amidst the sessions. The improved outcomes demonstrated by the Value in Inpatient Pediatrics (VIP) network are a testament to the positive change that can arise from such efforts. VIP, with its focus on inclusive and front-line collaboratives, also announced an upcoming merger with the AAP’s Quality Improvement and Innovation Network (QuIIN), approved by the AAP board in May.
Perhaps more impressive was that more than 12% of the attendees at PHM 2011 attended the annual VIP dinner and similar numbers signed up to participate in future efforts. At this pace, widespread improvement and value are easily within our sights.
3. Complex care is the new family-centered rounds. Atul Gawande’s recent New Yorker article about “Hot Spotters” could very well have been referring to the body of work that is represented by pediatric generalists (to include a fair number of hospitalists) over the past few years. Closing plenary speakers Robert Lyle and Patrick Casey wowed the audience as they described their medical home for medically complex children—and an estimated savings to Arkansas Medicaid of nearly $3 million per year.
As hospitals and hospital systems look to create accountable care organizations (ACOs), this kind of work will be increasingly prioritized, as it has the potential to generate the biggest gains in valued care.
2. STP, yeah, you know me. Chris Maloney and Suzanne Swanson Mendez brought down the house at the PHM Roundtable update as they presented preliminary results of their large and representative STP (strategic planning) Committee, which is mapping out future certification options for pediatric hospitalists. A lively debate ensued as questions surrounding how to best notify and involve pediatric hospitalists in these decisions came to the forefront. Are we a democracy? Are we a republic? Is there a better model for this decision?
Despite the lack of consensus on how to best move toward a decision, the discussion remained open and engaging. In contrast to recent certification decisions from other organizations, the audience clearly relished the opportunity to provide input, and the STP committee continues to look for able and willing participants.
1. A top-10 list is not enough to cover everything from PHM 2011. From clinical and practice conundrums galore to late nights at Spectators to Kevin Powell’s mad acting skills, a 1,500-word-top-10 list simply does not do the meeting justice! I place full blame on the planning committee for this overindulgent buffet and the unfortunate omission of many other meaningful lessons.
Thank you: Erin Stucky Fisher (chair), Brian Pate, Allison Ballantine, Matt Garber, Jeff Simmons, Doug Carlson and Tamara Simon.
If you’re feeling like you missed out, or have already digested and want more, PHM 2012 will be here soon enough to fill your appetite. Cincinnati, look out.
Dr. Shen is pediatric editor of The Hospitalist.
It’s been a while since I went to a buffet and stuffed myself silly, but that’s how my mind felt on the flight home from Kansas City, Mo. After four incredibly packed days at Pediatric Hospital Medicine (PHM) 2011, I was one “wafer-thin mint” away from an explosion. What I wanted was some kind of a mental digestif. What I got instead was a light beer. It helped a little bit, but when I awoke during the landing in Austin, Texas, I realized that I remained in need of a better way to distill the thousand points of information from the conference into something more manageable.
Buoyed by Michelle Marks and Joel Tieder’s Top 10 Articles of the Year luncheon, specifically the piece on neurosurgeons and Kangaroo Care, I thought I might try my own version of a decompressive operation.
Without further delay, here are the top 10 things that I learned at Pediatric Hospital Medicine 2011:
10. We continue to grow as a field. Although the exact number of pediatric hospitalists in the U.S. remains somewhat unclear (but is probably between 1,000 and 2,000), what is known is that attendance at our annual meeting grows every year. Our tripartite meeting, sponsored by the American Academy of Pediatrics (AAP), SHM, and the Academic Pediatric Association (APA), hit a record 450 attendees this year. Beyond the physical numbers, it is quite clear that we are growing in many other domains as well.
9. It is time to re-evaluate the impact of CME on physician practice and outcomes. The literature on continuing professional education has been quite sobering to date, with nothing to show for the thousands of dollars spent per individual. But most of those studies were performed in the last century (think eight hours of lecture a day followed by dinners with big pharma) and I’ll bet that there was not the focus on learner-centered education that was evident in Kansas City.
With a dizzying array of workshops and interactive small group sessions spread amongst seven different tracks, it was difficult, if not impossible, to be a passive participant in the process. And since learning retention rates are generally proportionate to how active a role adults play in their own education, I am going to guess that many other attendees’ brains still have that “I’m thinking” hourglass icon over them. We have the conference planning committee to thank for this.
8. The JCPHM (Joint Council of Pediatric Hospital Medicine) will be increasingly important as we develop. Much like the constant stream of unfamiliar new vaccine names that have appeared in recent years, this proposed new committee comes with another long set of initials and an unfamiliar indication.
The JCPHM will function as a coordinating body, ensuring that work done through the AAP, SHM, and AAP are aligned to provide maximal benefit to pediatric hospitalists as a whole. And thus, similar to immunizations, the benefits will be most evident if we remain healthy as we grow in the context of an increasingly complex environment.
7. Our collective research accomplishments merit national recognition. It was not more than just a few years ago that we were in our research infancy. Posters at our meetings largely represented single-site descriptive studies, typically using survey methodology. This year we had four research breakout tracks, in addition to the plenary and three poster sessions.
Pediatric Research in Inpatient Settings (PRIS) leads the way with a Forbes-like listing of million-dollar grants and a partnership with Child Health Corporation of America’s (CHCA) uber-powerful Pediatric Health Information Systems (PHIS) database. Expect some landmark studies in the near future.
In the meantime, it is clear that the rest of the field is not languishing from smaller budgets. From clear outcome and process measurements of family-centered rounds to studying the spectrum of transitions of care to the impact of early warning systems, there was a predominant focus on quality and safety.
In fact, the tone was set at the opening keynote address, as Carolyn Clancy, MD, director of the Agency for Healthcare Research and Quality (AHRQ), described creative and innovative ways to study and translate work in this area to improved patient care.
6. We are poised to develop effective training systems for our future workforce. From the use of our core competencies in sessions to a full complement of workshops on education (from individual to team and from student to fellow), it is clear that thoughtful deliberation has paved the way for our future. We possess expertise in education that dovetails nicely with our need to grow and sustain an experienced, well-trained workforce.
Intrinsically, we know that we possess a unique body of skills, knowledge, and attitudes. The explicit articulation of this into longitudinal curricula will headline our evolution as a field.
5. Our new peer-reviewed journal has a bright future. Kudos to Shawn Ralston and the rest of the editorial board for publishing the first peer-reviewed edition of Hospital Pediatrics. Original research, evidence-based content, and practical commentary grace the pages with a little bit of something for everyone. The AAP has demonstrated a healthy level of support for this endeavor, as they sent out an introductory email announcing the journal to all of their membership over the weekend. I am confident that support from our pediatric hospitalist community will follow in the form of an exponential increase in quality submissions. Look no further than the PHM 2011 abstract book to get a preview of what our journal will highlight in the near future.
4. We connect with each other through a language of quality and value in our work. Quality spanned the continuum from conversation to collaboration, as like-minded souls shared ideas and passions amidst the sessions. The improved outcomes demonstrated by the Value in Inpatient Pediatrics (VIP) network are a testament to the positive change that can arise from such efforts. VIP, with its focus on inclusive and front-line collaboratives, also announced an upcoming merger with the AAP’s Quality Improvement and Innovation Network (QuIIN), approved by the AAP board in May.
Perhaps more impressive was that more than 12% of the attendees at PHM 2011 attended the annual VIP dinner and similar numbers signed up to participate in future efforts. At this pace, widespread improvement and value are easily within our sights.
3. Complex care is the new family-centered rounds. Atul Gawande’s recent New Yorker article about “Hot Spotters” could very well have been referring to the body of work that is represented by pediatric generalists (to include a fair number of hospitalists) over the past few years. Closing plenary speakers Robert Lyle and Patrick Casey wowed the audience as they described their medical home for medically complex children—and an estimated savings to Arkansas Medicaid of nearly $3 million per year.
As hospitals and hospital systems look to create accountable care organizations (ACOs), this kind of work will be increasingly prioritized, as it has the potential to generate the biggest gains in valued care.
2. STP, yeah, you know me. Chris Maloney and Suzanne Swanson Mendez brought down the house at the PHM Roundtable update as they presented preliminary results of their large and representative STP (strategic planning) Committee, which is mapping out future certification options for pediatric hospitalists. A lively debate ensued as questions surrounding how to best notify and involve pediatric hospitalists in these decisions came to the forefront. Are we a democracy? Are we a republic? Is there a better model for this decision?
Despite the lack of consensus on how to best move toward a decision, the discussion remained open and engaging. In contrast to recent certification decisions from other organizations, the audience clearly relished the opportunity to provide input, and the STP committee continues to look for able and willing participants.
1. A top-10 list is not enough to cover everything from PHM 2011. From clinical and practice conundrums galore to late nights at Spectators to Kevin Powell’s mad acting skills, a 1,500-word-top-10 list simply does not do the meeting justice! I place full blame on the planning committee for this overindulgent buffet and the unfortunate omission of many other meaningful lessons.
Thank you: Erin Stucky Fisher (chair), Brian Pate, Allison Ballantine, Matt Garber, Jeff Simmons, Doug Carlson and Tamara Simon.
If you’re feeling like you missed out, or have already digested and want more, PHM 2012 will be here soon enough to fill your appetite. Cincinnati, look out.
Dr. Shen is pediatric editor of The Hospitalist.
Dr. Hospitalist

What I am curious about are the current issues around using length of stay (LOS) or cost/case or the like as part of compensation packages. I have had discussions with several other folks, and I think I am getting the picture. However, it sounds like there has been some new interpretation of the laws around gainsharing, and that is what I am curious about.
K.S., Ohio
Dr. Hospitalist responds:
Tough question, and let me start by saying that I’m not a healthcare lawyer: This stuff is tricky. I’ll do my best to explain the current situation as I understand it, but I’m no expert on this.
So gainsharing, as generally defined in healthcare, is where a hospital and a group of physicians design a contract around services for which the two sides can share in any savings. Physicians are paid fee-for-service by Medicare, thus they are reimbursed per unit of work, with no incentive for cost control. Hospitals are paid on a per-case (or per-procedure) basis, so cost control means a lot to them: Because they get a set payment, any savings generated, they get to keep. Ideally, this means that better performance leads to more efficient care, less waste, and better outcomes. Unfortunately, that’s not always what happens, especially in the view of the federal government (you know, the guys who issue the bright orange jumpsuits for you to wear when you break the law).
Gainsharing has an interesting history as interpreted by the Office of the Inspector General (OIG) and the Centers for Medicare & Medicaid Services (CMS). Back in 1999, the OIG explicitly stopped any gainsharing models between physicians and hospitals based on concerns that these contracts might reduce the care provided to patients. The opinion was that there might be a “race to the bottom” in terms of cutting expenses (read: services).
Since then, there has been only incremental movement forward in the form of demonstration projects. One project looked at two very specific procedures: cardiac catheterization and coronary artery bypass grafting (CABG). The results showed that gainsharing could be beneficial to the hospital-physician relationship, and, more important, not harmful to the patient. There has since been some movement toward gainsharing, but only in the context of specific procedures, with very clear safeguards around it, including an independent auditor. Nothing to this point has suggested that a cost per case or adjusted LOS gainsharing agreement would pass muster with the OIG.
So, at this point in time, I would caution against any contract that contained explicit references connecting compensation to a change in hospital costs, such as reducing LOS or cost per case. The new accountable-care organization (ACO) model might be a different prism through which to view this, but it’s a world apart from an individual physician or hospitalist group contract (see “A Chilly Reception,” August 2011, p. 23).
For contractual compensation, I think that quality metrics can fill a need, and there are lots of ways to be creative here. You could set a target around something measurable (appropriate DVT prophylaxis is just one example) and tie dollars to that specific performance. The key is avoiding any language that would imply additional physician compensation for a reduction in patient services.
Might things change in the future? Your guess is as good as mine.
What I am curious about are the current issues around using length of stay (LOS) or cost/case or the like as part of compensation packages. I have had discussions with several other folks, and I think I am getting the picture. However, it sounds like there has been some new interpretation of the laws around gainsharing, and that is what I am curious about.
K.S., Ohio
Dr. Hospitalist responds:
Tough question, and let me start by saying that I’m not a healthcare lawyer: This stuff is tricky. I’ll do my best to explain the current situation as I understand it, but I’m no expert on this.
So gainsharing, as generally defined in healthcare, is where a hospital and a group of physicians design a contract around services for which the two sides can share in any savings. Physicians are paid fee-for-service by Medicare, thus they are reimbursed per unit of work, with no incentive for cost control. Hospitals are paid on a per-case (or per-procedure) basis, so cost control means a lot to them: Because they get a set payment, any savings generated, they get to keep. Ideally, this means that better performance leads to more efficient care, less waste, and better outcomes. Unfortunately, that’s not always what happens, especially in the view of the federal government (you know, the guys who issue the bright orange jumpsuits for you to wear when you break the law).
Gainsharing has an interesting history as interpreted by the Office of the Inspector General (OIG) and the Centers for Medicare & Medicaid Services (CMS). Back in 1999, the OIG explicitly stopped any gainsharing models between physicians and hospitals based on concerns that these contracts might reduce the care provided to patients. The opinion was that there might be a “race to the bottom” in terms of cutting expenses (read: services).
Since then, there has been only incremental movement forward in the form of demonstration projects. One project looked at two very specific procedures: cardiac catheterization and coronary artery bypass grafting (CABG). The results showed that gainsharing could be beneficial to the hospital-physician relationship, and, more important, not harmful to the patient. There has since been some movement toward gainsharing, but only in the context of specific procedures, with very clear safeguards around it, including an independent auditor. Nothing to this point has suggested that a cost per case or adjusted LOS gainsharing agreement would pass muster with the OIG.
So, at this point in time, I would caution against any contract that contained explicit references connecting compensation to a change in hospital costs, such as reducing LOS or cost per case. The new accountable-care organization (ACO) model might be a different prism through which to view this, but it’s a world apart from an individual physician or hospitalist group contract (see “A Chilly Reception,” August 2011, p. 23).
For contractual compensation, I think that quality metrics can fill a need, and there are lots of ways to be creative here. You could set a target around something measurable (appropriate DVT prophylaxis is just one example) and tie dollars to that specific performance. The key is avoiding any language that would imply additional physician compensation for a reduction in patient services.
Might things change in the future? Your guess is as good as mine.
What I am curious about are the current issues around using length of stay (LOS) or cost/case or the like as part of compensation packages. I have had discussions with several other folks, and I think I am getting the picture. However, it sounds like there has been some new interpretation of the laws around gainsharing, and that is what I am curious about.
K.S., Ohio
Dr. Hospitalist responds:
Tough question, and let me start by saying that I’m not a healthcare lawyer: This stuff is tricky. I’ll do my best to explain the current situation as I understand it, but I’m no expert on this.
So gainsharing, as generally defined in healthcare, is where a hospital and a group of physicians design a contract around services for which the two sides can share in any savings. Physicians are paid fee-for-service by Medicare, thus they are reimbursed per unit of work, with no incentive for cost control. Hospitals are paid on a per-case (or per-procedure) basis, so cost control means a lot to them: Because they get a set payment, any savings generated, they get to keep. Ideally, this means that better performance leads to more efficient care, less waste, and better outcomes. Unfortunately, that’s not always what happens, especially in the view of the federal government (you know, the guys who issue the bright orange jumpsuits for you to wear when you break the law).
Gainsharing has an interesting history as interpreted by the Office of the Inspector General (OIG) and the Centers for Medicare & Medicaid Services (CMS). Back in 1999, the OIG explicitly stopped any gainsharing models between physicians and hospitals based on concerns that these contracts might reduce the care provided to patients. The opinion was that there might be a “race to the bottom” in terms of cutting expenses (read: services).
Since then, there has been only incremental movement forward in the form of demonstration projects. One project looked at two very specific procedures: cardiac catheterization and coronary artery bypass grafting (CABG). The results showed that gainsharing could be beneficial to the hospital-physician relationship, and, more important, not harmful to the patient. There has since been some movement toward gainsharing, but only in the context of specific procedures, with very clear safeguards around it, including an independent auditor. Nothing to this point has suggested that a cost per case or adjusted LOS gainsharing agreement would pass muster with the OIG.
So, at this point in time, I would caution against any contract that contained explicit references connecting compensation to a change in hospital costs, such as reducing LOS or cost per case. The new accountable-care organization (ACO) model might be a different prism through which to view this, but it’s a world apart from an individual physician or hospitalist group contract (see “A Chilly Reception,” August 2011, p. 23).
For contractual compensation, I think that quality metrics can fill a need, and there are lots of ways to be creative here. You could set a target around something measurable (appropriate DVT prophylaxis is just one example) and tie dollars to that specific performance. The key is avoiding any language that would imply additional physician compensation for a reduction in patient services.
Might things change in the future? Your guess is as good as mine.
ONLINE EXCLUSIVE: Emergency Medicine Companies Venture into Hospital Medicine
Hollywood, Fla.-based Hospital Physician Partners (HPP) was an ED business when opportunity came knocking: Hospital administrators started asking, “Can you provide us with some hospitalists to go with our emergency-room doctors?”
Today, HPP is firmly in the HM business—and all signs point toward more hospitals hiring companies to handle both emergency care and inpatient care.
“In many ways, we expanded our efforts into hospitalist medicine as a result of requests from our hospital partners,” says Ed Weinberg, HPP’s chief operating officer. “Their needs were such that they asked us to provide hospital medicine services. So from that, it became clear that it was an area that was really growing. And that is something we are pursuing as vigorously as we are emergency medicine.”
HPP handling both emergency care and hospital medicine can help with the transition of patients from the ED to a hospital bed upstairs, he says.
“That’s where our efficiencies are, because we have physicians working who are carrying out the same philosophy,” he says.
Out of HPP’s 120 contracts, 15 are in hospital medicine. But the HM contract numbers are growing quickly, Weinberg notes.
EmCare has about 400 emergency-medicine programs and more than 50 HM programs, according to Mark Hamm, CEO of EmCare Inpatient Services. He says that it can be much more cost effective to contract with one company for both hospitalist and ED services, something hospitals find attractive.
EmCare service agreements range from completely separate emergency and HM staffs to small, rural hospitals where ED physicians also do rounds. Some hospitals “just don’t have the money for a full-time hospitalist and don’t really need one,” Hamm says.
The patient transitions tend to go more smoothly when both types of care are provided by EmCare, he adds. If they’re not, there can be slowdowns.
“Our goal is to quickly and appropriately move patients through the system,” he says. “If we have a hospitalist provider that’s not really on the same page, that can create bottlenecks. But it’s a blip. Our goal is to sit down, even if it’s not an EmCare hospitalist, to sit down with that director and say, ‘Hey look, let’s be the leader here, let’s work together and appropriately expedite these patients.’ We do the same thing on the hospitalist side.”
Inpatient care promises to be a big part of their future business, the executives agreed.
“Hospital medicine,” Weinberg says, “is growing by leaps and bounds.”
Hollywood, Fla.-based Hospital Physician Partners (HPP) was an ED business when opportunity came knocking: Hospital administrators started asking, “Can you provide us with some hospitalists to go with our emergency-room doctors?”
Today, HPP is firmly in the HM business—and all signs point toward more hospitals hiring companies to handle both emergency care and inpatient care.
“In many ways, we expanded our efforts into hospitalist medicine as a result of requests from our hospital partners,” says Ed Weinberg, HPP’s chief operating officer. “Their needs were such that they asked us to provide hospital medicine services. So from that, it became clear that it was an area that was really growing. And that is something we are pursuing as vigorously as we are emergency medicine.”
HPP handling both emergency care and hospital medicine can help with the transition of patients from the ED to a hospital bed upstairs, he says.
“That’s where our efficiencies are, because we have physicians working who are carrying out the same philosophy,” he says.
Out of HPP’s 120 contracts, 15 are in hospital medicine. But the HM contract numbers are growing quickly, Weinberg notes.
EmCare has about 400 emergency-medicine programs and more than 50 HM programs, according to Mark Hamm, CEO of EmCare Inpatient Services. He says that it can be much more cost effective to contract with one company for both hospitalist and ED services, something hospitals find attractive.
EmCare service agreements range from completely separate emergency and HM staffs to small, rural hospitals where ED physicians also do rounds. Some hospitals “just don’t have the money for a full-time hospitalist and don’t really need one,” Hamm says.
The patient transitions tend to go more smoothly when both types of care are provided by EmCare, he adds. If they’re not, there can be slowdowns.
“Our goal is to quickly and appropriately move patients through the system,” he says. “If we have a hospitalist provider that’s not really on the same page, that can create bottlenecks. But it’s a blip. Our goal is to sit down, even if it’s not an EmCare hospitalist, to sit down with that director and say, ‘Hey look, let’s be the leader here, let’s work together and appropriately expedite these patients.’ We do the same thing on the hospitalist side.”
Inpatient care promises to be a big part of their future business, the executives agreed.
“Hospital medicine,” Weinberg says, “is growing by leaps and bounds.”
Hollywood, Fla.-based Hospital Physician Partners (HPP) was an ED business when opportunity came knocking: Hospital administrators started asking, “Can you provide us with some hospitalists to go with our emergency-room doctors?”
Today, HPP is firmly in the HM business—and all signs point toward more hospitals hiring companies to handle both emergency care and inpatient care.
“In many ways, we expanded our efforts into hospitalist medicine as a result of requests from our hospital partners,” says Ed Weinberg, HPP’s chief operating officer. “Their needs were such that they asked us to provide hospital medicine services. So from that, it became clear that it was an area that was really growing. And that is something we are pursuing as vigorously as we are emergency medicine.”
HPP handling both emergency care and hospital medicine can help with the transition of patients from the ED to a hospital bed upstairs, he says.
“That’s where our efficiencies are, because we have physicians working who are carrying out the same philosophy,” he says.
Out of HPP’s 120 contracts, 15 are in hospital medicine. But the HM contract numbers are growing quickly, Weinberg notes.
EmCare has about 400 emergency-medicine programs and more than 50 HM programs, according to Mark Hamm, CEO of EmCare Inpatient Services. He says that it can be much more cost effective to contract with one company for both hospitalist and ED services, something hospitals find attractive.
EmCare service agreements range from completely separate emergency and HM staffs to small, rural hospitals where ED physicians also do rounds. Some hospitals “just don’t have the money for a full-time hospitalist and don’t really need one,” Hamm says.
The patient transitions tend to go more smoothly when both types of care are provided by EmCare, he adds. If they’re not, there can be slowdowns.
“Our goal is to quickly and appropriately move patients through the system,” he says. “If we have a hospitalist provider that’s not really on the same page, that can create bottlenecks. But it’s a blip. Our goal is to sit down, even if it’s not an EmCare hospitalist, to sit down with that director and say, ‘Hey look, let’s be the leader here, let’s work together and appropriately expedite these patients.’ We do the same thing on the hospitalist side.”
Inpatient care promises to be a big part of their future business, the executives agreed.
“Hospital medicine,” Weinberg says, “is growing by leaps and bounds.”
ONLINE EXCLUSIVE: Weighing the Costs of Palliative Care
Hospitalist David Mitchell, MD, PhD, was moonlighting in an Ohio hospital when a nurse called him about a gravely ill older patient who was experiencing shortness of breath. Should she administer the diuretic Lasix to help clear his lung congestion?
Dr. Mitchell, now a hospitalist at Sibley Memorial Hospital in Washington, D.C., and a member of SHM’s Performance Standards Committee, decided to see the patient in person and review his charts. He found that the patient had severe dementia, hadn’t walked in months, and was declining despite more than two weeks in the hospital and daily visits by three specialists.
Dr. Mitchell called the patient’s son and explained the situation, then asked whether the son thought his father would want to continue receiving aggressive therapy. “The son said, ‘Oh, no. He would never want to continue like this.’ So we stopped all the treatments, and he died by the next day,” Dr. Mitchell says.
To him, the anecdote highlights how far medicine has to go in providing personalized palliative care that honors the wishes of patients and their families. It also demonstrates how ignoring those wishes and failing to communicate can contribute to the huge costs associated with end-of-life medical care. Every day, the three specialists seeing the patient were recommending the same course of therapy. “But nobody was being the quarterback and saying, ‘Hey, listen. This is not working,’ ” Dr. Mitchell says.
Hospitalists, he says, are in an ideal position to step up and play a pivotal role in providing the kind of patient-centered care that could improve both quality and cost. So far, however, Dr. Mitchell says he’s seen wide variation in how hospitalists communicate with a patient’s family about end-of-life decisions. “For the ones who do have these conversations, the family is almost always glad that somebody finally said, ‘Do we have to do these tests? Do we have to continue to try to save his life?’ ” Dr. Mitchell says.
Time constraints, he says, are the main reason why hospitalists don’t have such conversations more often. “The communication dies when you’re busy.” And the remedy? Dr. Mitchell says the only thing that will help shift the focus from seeing as many patients as possible to making sure every encounter is a high-quality, efficient one is payment reform in the form of bundled payments to hospitals and physicians. In theory, professional standards can encourage more uniformity, he says. “But when it hits the trenches, it’s the payment that speaks.”
Hospitalist David Mitchell, MD, PhD, was moonlighting in an Ohio hospital when a nurse called him about a gravely ill older patient who was experiencing shortness of breath. Should she administer the diuretic Lasix to help clear his lung congestion?
Dr. Mitchell, now a hospitalist at Sibley Memorial Hospital in Washington, D.C., and a member of SHM’s Performance Standards Committee, decided to see the patient in person and review his charts. He found that the patient had severe dementia, hadn’t walked in months, and was declining despite more than two weeks in the hospital and daily visits by three specialists.
Dr. Mitchell called the patient’s son and explained the situation, then asked whether the son thought his father would want to continue receiving aggressive therapy. “The son said, ‘Oh, no. He would never want to continue like this.’ So we stopped all the treatments, and he died by the next day,” Dr. Mitchell says.
To him, the anecdote highlights how far medicine has to go in providing personalized palliative care that honors the wishes of patients and their families. It also demonstrates how ignoring those wishes and failing to communicate can contribute to the huge costs associated with end-of-life medical care. Every day, the three specialists seeing the patient were recommending the same course of therapy. “But nobody was being the quarterback and saying, ‘Hey, listen. This is not working,’ ” Dr. Mitchell says.
Hospitalists, he says, are in an ideal position to step up and play a pivotal role in providing the kind of patient-centered care that could improve both quality and cost. So far, however, Dr. Mitchell says he’s seen wide variation in how hospitalists communicate with a patient’s family about end-of-life decisions. “For the ones who do have these conversations, the family is almost always glad that somebody finally said, ‘Do we have to do these tests? Do we have to continue to try to save his life?’ ” Dr. Mitchell says.
Time constraints, he says, are the main reason why hospitalists don’t have such conversations more often. “The communication dies when you’re busy.” And the remedy? Dr. Mitchell says the only thing that will help shift the focus from seeing as many patients as possible to making sure every encounter is a high-quality, efficient one is payment reform in the form of bundled payments to hospitals and physicians. In theory, professional standards can encourage more uniformity, he says. “But when it hits the trenches, it’s the payment that speaks.”
Hospitalist David Mitchell, MD, PhD, was moonlighting in an Ohio hospital when a nurse called him about a gravely ill older patient who was experiencing shortness of breath. Should she administer the diuretic Lasix to help clear his lung congestion?
Dr. Mitchell, now a hospitalist at Sibley Memorial Hospital in Washington, D.C., and a member of SHM’s Performance Standards Committee, decided to see the patient in person and review his charts. He found that the patient had severe dementia, hadn’t walked in months, and was declining despite more than two weeks in the hospital and daily visits by three specialists.
Dr. Mitchell called the patient’s son and explained the situation, then asked whether the son thought his father would want to continue receiving aggressive therapy. “The son said, ‘Oh, no. He would never want to continue like this.’ So we stopped all the treatments, and he died by the next day,” Dr. Mitchell says.
To him, the anecdote highlights how far medicine has to go in providing personalized palliative care that honors the wishes of patients and their families. It also demonstrates how ignoring those wishes and failing to communicate can contribute to the huge costs associated with end-of-life medical care. Every day, the three specialists seeing the patient were recommending the same course of therapy. “But nobody was being the quarterback and saying, ‘Hey, listen. This is not working,’ ” Dr. Mitchell says.
Hospitalists, he says, are in an ideal position to step up and play a pivotal role in providing the kind of patient-centered care that could improve both quality and cost. So far, however, Dr. Mitchell says he’s seen wide variation in how hospitalists communicate with a patient’s family about end-of-life decisions. “For the ones who do have these conversations, the family is almost always glad that somebody finally said, ‘Do we have to do these tests? Do we have to continue to try to save his life?’ ” Dr. Mitchell says.
Time constraints, he says, are the main reason why hospitalists don’t have such conversations more often. “The communication dies when you’re busy.” And the remedy? Dr. Mitchell says the only thing that will help shift the focus from seeing as many patients as possible to making sure every encounter is a high-quality, efficient one is payment reform in the form of bundled payments to hospitals and physicians. In theory, professional standards can encourage more uniformity, he says. “But when it hits the trenches, it’s the payment that speaks.”
ONLINE EXCLUSIVE: Listen to Russell Holman and Ed Weinberg discuss companies' acquisition strategies
ONLINE EXCLUSIVE: Listen to Tosha Wetterneck and Keiki Hinami discuss burnout and career satisfaction
ONLINE EXCLUSIVE: Listen to David Meltzer and Scott Lundberg talk about HM efficiency
How proposed changes to personality disorders in DSM-5 will affect researchers
Crizotinib Approval Personalizes Lung Cancer Therapy
The swift approval of crizotinib capsules by the Food and Drug Administration as the first and only targeted therapy for locally advanced or metastatic ALK-positive non–small cell lung cancer represents another milestone in biomarker-driven, personalized medicine.
Crizotinib was approved, along with a companion diagnostic test, Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit, which identifies the anaplastic lymphoma kinase (ALK) fusion gene that the drug targets. Pfizer is authorized to market crizotinib as Xalkori for use in patients who test positive for the abnormality.
The ALK fusion gene – comprising portions of the EML4 (echinoderm microtubule–associated proteinlike 4) gene and the ALK gene – is present in about 3%-5% of all patients with NSCLC. Although the percentage is small, it translates to approximately 6,000-11,000 new patients annually in the United States, which is more than the numbers being diagnosed with Hodgkin’s disease, cancer of the testis, or chronic myelogenous leukemia.

"Having this number of people we can affect is really an important development in oncology," commented Dr. Mark G. Kris, chief of the thoracic oncology service at Memorial Sloan-Kettering Cancer Center in New York, during a press briefing sponsored by Pfizer.
The approval "is a delivery on the promise of personalized medicine and genomic medicine. Many folks have wondered how these concepts translate into reality. This is an example of exactly that," said Dr. Kris, a professor of medicine at Cornell University in New York.
Crizotinib is part of a paradigm shift in lung cancer management, observed Dr. Paul A. Bunn Jr., professor of medicine and the James Dudley Chair in Cancer Research at the University of Colorado at Denver. Increasingly, patients are tested for specific mutations and if they are positive, they can be treated with targeted therapies, usually pills that are safer and typically more effective than the conventional chemotherapy and radiation.
The American Society of Clinical Oncology and the National Comprehensive Cancer Network have each issued guidelines recommending that NSCLC patients be tested for mutation of the EGFR (epidermal growth factor receptor) to identify those who might benefit from tyrosine kinase inhibitors targeting EGFR. Gefitinib (Iressa) and erlotinib (Tarceva) have proved effective in clinical trials in this population, but only erlotinib is widely available in the United States.
The experimental BATTLE (Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination) trials program at the University of Texas M.D. Anderson Cancer Center in Houston has shown that it is feasible to biopsy late-stage NSCLC patients and base the choice of therapies on the results of molecular tests for abnormal KRAS, EGFR, and other genes.
Currently, the Lung Cancer Mutation Consortium is engaged in a collaborative project profiling 10 genes, including KRAS, ALK, and EGFR, in 1,000 patients at participating cancer centers. Investigators have reported that 280 (54%) of the first 516 patients were found to have at least one known driver mutation. Patients are being directed to approved targeted therapies where available, or to clinical trials of novel therapies targeting their specific mutations.
"The message is, lung cancers are not all the same. They have different molecular changes," Dr. Bunn said at the crizotinib briefing. "At the time of diagnosis, physicians need to be testing their patients for these molecular changes, and patients who have one of these changes are likely to have prolonged benefit from a pill that is more active than multiagent chemotherapy."
Indeed, Dr. Kris pointed out, in clinical trials crizotinib benefited nearly all patients with the ALK fusion gene, although the degree of benefit varied. On the other hand, withholding crizotinib from patients who test negative means they will be spared the side effects and the waste of time and resources associated with a treatment that won’t work for them. "Having a tablet makes the administration so much easier than intravenous chemotherapy," he added.
How Crizotinib Received FDA Approval
The FDA approval was based on two multicenter, single-arm studies that enrolled 255 late-stage NSCLC patients who tested positive for the ALK fusion gene before enrollment. In most cases, the patients had prior chemotherapy. Objective response rates were 50% (median duration, 42 weeks) in one study, and 61% (median duration, 48 weeks) in the other.
Among the most common side effects in the trials, the FDA listed vision disorders (visual impairment, flashes of light, blurred vision, floaters, double vision, sensitivity to light, and visual field defects), nausea, diarrhea, vomiting, edema, and constipation. Pfizer noted that grade 3 or 4 adverse events occurring in 4% or more of patients included increased ALT and neutropenia; it said that QT prolongation has also been observed.
The FDA also warned that crizotinib has been associated with potentially life-threatening pneumonitis (4 of 255 patients; 1.6%); the drug should be stopped permanently in patients with treatment-related pneumonitis, the agency said. Pregnancy is also a contraindication.
Consideration of crizotinib was expedited under the FDA’s priority review program for drugs with the potential to provide major advances in diseases for which no effective therapy exists. No survival data were reported, and the agency granted accelerated approval based on the objective response rates being reasonably likely to predict clinical benefit. Confirmatory trials are required.
Pfizer is already conducting two randomized, open-label, postmarketing phase III studies: one comparing the safety and efficacy of crizotinib with standard of care chemotherapy (pemetrexed [Alimta] or docetaxel [Taxotere]) in patients with previously treated, advanced, ALK-positive NSCLC, and the other comparing efficacy and safety of the agent to pemetrexed/cisplatin or pemetrexed/carboplatin in previously untreated patients with advanced, ALK-positive, nonsquamous NSCLC, according to a Pfizer statement.
Dr. Kris predicted that the addition of the required ALK fusion diagnostic test – at a cost of $250 – won’t be a large burden. "I think it will be very quick to add ALK testing as part of that," he said, noting the ASCO and NCCN recommendations for EGFR testing.
Because the fusion gene has been seen in patients with lung tumors other than adenocarcinomas, testing should ideally be done in all lung cancer patients, Dr. Kris added. "Based on broader experience with EGFR mutations, I think we’ve learned that there is no clinical profile that comes anywhere near the accuracy of the testing. ... I would caution against any particular kind of profile that would make one select who to test and who not to test," he said.
Recommended dosing is 250 mg taken orally twice daily with or without food. In some patients, a dosing interruption and/or dose reduction to 200 mg taken orally twice daily may be required; if further reduction is necessary, the label recommends 250 mg taken orally once daily.
Monthly treatment with crizotinib will cost $9,600. And because the tumor is driven by the mutation, the treatment is indefinite. Several patients from the trials have now been taking the drug for more than a year. "There is no stop time as long as they’re benefiting," said Dr. Mace Rothenberg, Pfizer senior vice president of clinical development and medical affairs in the oncology business unit.
Pfizer has two financial assistance programs for patients. The Pfizer First Resource Program connects eligible insured patients to specialty pharmacies to get reimbursement-support services and to obtain the medications. For uninsured and underinsured patients, the program will provide eligible patients with free medication. There is also a copay assistance program for eligible privately insured patients. Information about eligibility can be obtained by calling First Resource (1-877-744-5675), or by visiting XALKORI.com.
Dr. Kris is a consultant for Pfizer. Dr. Bunn has served as a consultant or on an advisory board for several companies in the past 2 years, but noted that he received no funding for any press discussions or research on crizotinib and has no Pfizer stock.
The swift approval of crizotinib capsules by the Food and Drug Administration as the first and only targeted therapy for locally advanced or metastatic ALK-positive non–small cell lung cancer represents another milestone in biomarker-driven, personalized medicine.
Crizotinib was approved, along with a companion diagnostic test, Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit, which identifies the anaplastic lymphoma kinase (ALK) fusion gene that the drug targets. Pfizer is authorized to market crizotinib as Xalkori for use in patients who test positive for the abnormality.
The ALK fusion gene – comprising portions of the EML4 (echinoderm microtubule–associated proteinlike 4) gene and the ALK gene – is present in about 3%-5% of all patients with NSCLC. Although the percentage is small, it translates to approximately 6,000-11,000 new patients annually in the United States, which is more than the numbers being diagnosed with Hodgkin’s disease, cancer of the testis, or chronic myelogenous leukemia.
"Having this number of people we can affect is really an important development in oncology," commented Dr. Mark G. Kris, chief of the thoracic oncology service at Memorial Sloan-Kettering Cancer Center in New York, during a press briefing sponsored by Pfizer.
The approval "is a delivery on the promise of personalized medicine and genomic medicine. Many folks have wondered how these concepts translate into reality. This is an example of exactly that," said Dr. Kris, a professor of medicine at Cornell University in New York.
Crizotinib is part of a paradigm shift in lung cancer management, observed Dr. Paul A. Bunn Jr., professor of medicine and the James Dudley Chair in Cancer Research at the University of Colorado at Denver. Increasingly, patients are tested for specific mutations and if they are positive, they can be treated with targeted therapies, usually pills that are safer and typically more effective than the conventional chemotherapy and radiation.
The American Society of Clinical Oncology and the National Comprehensive Cancer Network have each issued guidelines recommending that NSCLC patients be tested for mutation of the EGFR (epidermal growth factor receptor) to identify those who might benefit from tyrosine kinase inhibitors targeting EGFR. Gefitinib (Iressa) and erlotinib (Tarceva) have proved effective in clinical trials in this population, but only erlotinib is widely available in the United States.
The experimental BATTLE (Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination) trials program at the University of Texas M.D. Anderson Cancer Center in Houston has shown that it is feasible to biopsy late-stage NSCLC patients and base the choice of therapies on the results of molecular tests for abnormal KRAS, EGFR, and other genes.
Currently, the Lung Cancer Mutation Consortium is engaged in a collaborative project profiling 10 genes, including KRAS, ALK, and EGFR, in 1,000 patients at participating cancer centers. Investigators have reported that 280 (54%) of the first 516 patients were found to have at least one known driver mutation. Patients are being directed to approved targeted therapies where available, or to clinical trials of novel therapies targeting their specific mutations.
"The message is, lung cancers are not all the same. They have different molecular changes," Dr. Bunn said at the crizotinib briefing. "At the time of diagnosis, physicians need to be testing their patients for these molecular changes, and patients who have one of these changes are likely to have prolonged benefit from a pill that is more active than multiagent chemotherapy."
Indeed, Dr. Kris pointed out, in clinical trials crizotinib benefited nearly all patients with the ALK fusion gene, although the degree of benefit varied. On the other hand, withholding crizotinib from patients who test negative means they will be spared the side effects and the waste of time and resources associated with a treatment that won’t work for them. "Having a tablet makes the administration so much easier than intravenous chemotherapy," he added.
How Crizotinib Received FDA Approval
The FDA approval was based on two multicenter, single-arm studies that enrolled 255 late-stage NSCLC patients who tested positive for the ALK fusion gene before enrollment. In most cases, the patients had prior chemotherapy. Objective response rates were 50% (median duration, 42 weeks) in one study, and 61% (median duration, 48 weeks) in the other.
Among the most common side effects in the trials, the FDA listed vision disorders (visual impairment, flashes of light, blurred vision, floaters, double vision, sensitivity to light, and visual field defects), nausea, diarrhea, vomiting, edema, and constipation. Pfizer noted that grade 3 or 4 adverse events occurring in 4% or more of patients included increased ALT and neutropenia; it said that QT prolongation has also been observed.
The FDA also warned that crizotinib has been associated with potentially life-threatening pneumonitis (4 of 255 patients; 1.6%); the drug should be stopped permanently in patients with treatment-related pneumonitis, the agency said. Pregnancy is also a contraindication.
Consideration of crizotinib was expedited under the FDA’s priority review program for drugs with the potential to provide major advances in diseases for which no effective therapy exists. No survival data were reported, and the agency granted accelerated approval based on the objective response rates being reasonably likely to predict clinical benefit. Confirmatory trials are required.
Pfizer is already conducting two randomized, open-label, postmarketing phase III studies: one comparing the safety and efficacy of crizotinib with standard of care chemotherapy (pemetrexed [Alimta] or docetaxel [Taxotere]) in patients with previously treated, advanced, ALK-positive NSCLC, and the other comparing efficacy and safety of the agent to pemetrexed/cisplatin or pemetrexed/carboplatin in previously untreated patients with advanced, ALK-positive, nonsquamous NSCLC, according to a Pfizer statement.
Dr. Kris predicted that the addition of the required ALK fusion diagnostic test – at a cost of $250 – won’t be a large burden. "I think it will be very quick to add ALK testing as part of that," he said, noting the ASCO and NCCN recommendations for EGFR testing.
Because the fusion gene has been seen in patients with lung tumors other than adenocarcinomas, testing should ideally be done in all lung cancer patients, Dr. Kris added. "Based on broader experience with EGFR mutations, I think we’ve learned that there is no clinical profile that comes anywhere near the accuracy of the testing. ... I would caution against any particular kind of profile that would make one select who to test and who not to test," he said.
Recommended dosing is 250 mg taken orally twice daily with or without food. In some patients, a dosing interruption and/or dose reduction to 200 mg taken orally twice daily may be required; if further reduction is necessary, the label recommends 250 mg taken orally once daily.
Monthly treatment with crizotinib will cost $9,600. And because the tumor is driven by the mutation, the treatment is indefinite. Several patients from the trials have now been taking the drug for more than a year. "There is no stop time as long as they’re benefiting," said Dr. Mace Rothenberg, Pfizer senior vice president of clinical development and medical affairs in the oncology business unit.
Pfizer has two financial assistance programs for patients. The Pfizer First Resource Program connects eligible insured patients to specialty pharmacies to get reimbursement-support services and to obtain the medications. For uninsured and underinsured patients, the program will provide eligible patients with free medication. There is also a copay assistance program for eligible privately insured patients. Information about eligibility can be obtained by calling First Resource (1-877-744-5675), or by visiting XALKORI.com.
Dr. Kris is a consultant for Pfizer. Dr. Bunn has served as a consultant or on an advisory board for several companies in the past 2 years, but noted that he received no funding for any press discussions or research on crizotinib and has no Pfizer stock.
The swift approval of crizotinib capsules by the Food and Drug Administration as the first and only targeted therapy for locally advanced or metastatic ALK-positive non–small cell lung cancer represents another milestone in biomarker-driven, personalized medicine.
Crizotinib was approved, along with a companion diagnostic test, Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit, which identifies the anaplastic lymphoma kinase (ALK) fusion gene that the drug targets. Pfizer is authorized to market crizotinib as Xalkori for use in patients who test positive for the abnormality.
The ALK fusion gene – comprising portions of the EML4 (echinoderm microtubule–associated proteinlike 4) gene and the ALK gene – is present in about 3%-5% of all patients with NSCLC. Although the percentage is small, it translates to approximately 6,000-11,000 new patients annually in the United States, which is more than the numbers being diagnosed with Hodgkin’s disease, cancer of the testis, or chronic myelogenous leukemia.
"Having this number of people we can affect is really an important development in oncology," commented Dr. Mark G. Kris, chief of the thoracic oncology service at Memorial Sloan-Kettering Cancer Center in New York, during a press briefing sponsored by Pfizer.
The approval "is a delivery on the promise of personalized medicine and genomic medicine. Many folks have wondered how these concepts translate into reality. This is an example of exactly that," said Dr. Kris, a professor of medicine at Cornell University in New York.
Crizotinib is part of a paradigm shift in lung cancer management, observed Dr. Paul A. Bunn Jr., professor of medicine and the James Dudley Chair in Cancer Research at the University of Colorado at Denver. Increasingly, patients are tested for specific mutations and if they are positive, they can be treated with targeted therapies, usually pills that are safer and typically more effective than the conventional chemotherapy and radiation.
The American Society of Clinical Oncology and the National Comprehensive Cancer Network have each issued guidelines recommending that NSCLC patients be tested for mutation of the EGFR (epidermal growth factor receptor) to identify those who might benefit from tyrosine kinase inhibitors targeting EGFR. Gefitinib (Iressa) and erlotinib (Tarceva) have proved effective in clinical trials in this population, but only erlotinib is widely available in the United States.
The experimental BATTLE (Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination) trials program at the University of Texas M.D. Anderson Cancer Center in Houston has shown that it is feasible to biopsy late-stage NSCLC patients and base the choice of therapies on the results of molecular tests for abnormal KRAS, EGFR, and other genes.
Currently, the Lung Cancer Mutation Consortium is engaged in a collaborative project profiling 10 genes, including KRAS, ALK, and EGFR, in 1,000 patients at participating cancer centers. Investigators have reported that 280 (54%) of the first 516 patients were found to have at least one known driver mutation. Patients are being directed to approved targeted therapies where available, or to clinical trials of novel therapies targeting their specific mutations.
"The message is, lung cancers are not all the same. They have different molecular changes," Dr. Bunn said at the crizotinib briefing. "At the time of diagnosis, physicians need to be testing their patients for these molecular changes, and patients who have one of these changes are likely to have prolonged benefit from a pill that is more active than multiagent chemotherapy."
Indeed, Dr. Kris pointed out, in clinical trials crizotinib benefited nearly all patients with the ALK fusion gene, although the degree of benefit varied. On the other hand, withholding crizotinib from patients who test negative means they will be spared the side effects and the waste of time and resources associated with a treatment that won’t work for them. "Having a tablet makes the administration so much easier than intravenous chemotherapy," he added.
How Crizotinib Received FDA Approval
The FDA approval was based on two multicenter, single-arm studies that enrolled 255 late-stage NSCLC patients who tested positive for the ALK fusion gene before enrollment. In most cases, the patients had prior chemotherapy. Objective response rates were 50% (median duration, 42 weeks) in one study, and 61% (median duration, 48 weeks) in the other.
Among the most common side effects in the trials, the FDA listed vision disorders (visual impairment, flashes of light, blurred vision, floaters, double vision, sensitivity to light, and visual field defects), nausea, diarrhea, vomiting, edema, and constipation. Pfizer noted that grade 3 or 4 adverse events occurring in 4% or more of patients included increased ALT and neutropenia; it said that QT prolongation has also been observed.
The FDA also warned that crizotinib has been associated with potentially life-threatening pneumonitis (4 of 255 patients; 1.6%); the drug should be stopped permanently in patients with treatment-related pneumonitis, the agency said. Pregnancy is also a contraindication.
Consideration of crizotinib was expedited under the FDA’s priority review program for drugs with the potential to provide major advances in diseases for which no effective therapy exists. No survival data were reported, and the agency granted accelerated approval based on the objective response rates being reasonably likely to predict clinical benefit. Confirmatory trials are required.
Pfizer is already conducting two randomized, open-label, postmarketing phase III studies: one comparing the safety and efficacy of crizotinib with standard of care chemotherapy (pemetrexed [Alimta] or docetaxel [Taxotere]) in patients with previously treated, advanced, ALK-positive NSCLC, and the other comparing efficacy and safety of the agent to pemetrexed/cisplatin or pemetrexed/carboplatin in previously untreated patients with advanced, ALK-positive, nonsquamous NSCLC, according to a Pfizer statement.
Dr. Kris predicted that the addition of the required ALK fusion diagnostic test – at a cost of $250 – won’t be a large burden. "I think it will be very quick to add ALK testing as part of that," he said, noting the ASCO and NCCN recommendations for EGFR testing.
Because the fusion gene has been seen in patients with lung tumors other than adenocarcinomas, testing should ideally be done in all lung cancer patients, Dr. Kris added. "Based on broader experience with EGFR mutations, I think we’ve learned that there is no clinical profile that comes anywhere near the accuracy of the testing. ... I would caution against any particular kind of profile that would make one select who to test and who not to test," he said.
Recommended dosing is 250 mg taken orally twice daily with or without food. In some patients, a dosing interruption and/or dose reduction to 200 mg taken orally twice daily may be required; if further reduction is necessary, the label recommends 250 mg taken orally once daily.
Monthly treatment with crizotinib will cost $9,600. And because the tumor is driven by the mutation, the treatment is indefinite. Several patients from the trials have now been taking the drug for more than a year. "There is no stop time as long as they’re benefiting," said Dr. Mace Rothenberg, Pfizer senior vice president of clinical development and medical affairs in the oncology business unit.
Pfizer has two financial assistance programs for patients. The Pfizer First Resource Program connects eligible insured patients to specialty pharmacies to get reimbursement-support services and to obtain the medications. For uninsured and underinsured patients, the program will provide eligible patients with free medication. There is also a copay assistance program for eligible privately insured patients. Information about eligibility can be obtained by calling First Resource (1-877-744-5675), or by visiting XALKORI.com.
Dr. Kris is a consultant for Pfizer. Dr. Bunn has served as a consultant or on an advisory board for several companies in the past 2 years, but noted that he received no funding for any press discussions or research on crizotinib and has no Pfizer stock.
Venous thromboembolism: What to do after anticoagulation is started
Deep vein thrombosis and pulmonary embolism are collectively referred to as venous thromboembolic (VTE) disease. They affect approximately 100,000 to 300,000 patients per year in the United States.1 Although patients with deep vein thrombosis can be treated as outpatients, many are admitted for the initiation of anticoagulation. Initial anticoagulation usually requires the overlap of a parenteral anticoagulant (unfractionated heparin, low-molecular-weight heparin [LMWH] or fondaparinux) with warfarin for a minimum of 5 days and until the international normalized ratio (INR) of the prothrombin time is above 2.0 for at least 24 hours.2
Three clinical issues need to be addressed after the initiation of anticoagulation for VTE:
- Determination of the length of anticoagulation with the correct anticoagulant
- Prevention of postthrombotic syndrome
- Appropriate screening for occult malignancy.
HOW LONG SHOULD VTE BE TREATED?
The duration of anticoagulation has been a matter of debate.
The risk of recurrent VTE appears related to clinical risk factors that a patient has at the time of the initial thrombotic event. An epidemiologic study3 found that patients with VTE treated for approximately 6 months had a low rate of recurrence (0% at 2 years of follow-up) if surgery was the risk factor. The risk climbed to 9% if the risk factor was nonsurgical and to 19% if there were no provoking risk factors.
The likelihood of VTE recurrence and therefore the recommended duration of treatment depend on whether the VTE event was provoked, cancer-related, recurrent, thrombophilia-related, or idiopathic. We address each of these scenarios below.
HOW LONG TO TREAT PROVOKED VTE
A VTE event is considered provoked if the patient had a clear inciting risk factor. As defined in various clinical trials, these risk factors include:
- Hospitalization with confinement to bed for 3 or more consecutive days in the last 3 months
- Surgery or general anesthesia in the last 3 months
- Immobilization for more than 7 days, regardless of the cause
- Trauma in the last 3 months
- Pregnancy
- Use of an oral contraceptive, regardless of which estrogen or progesterone analogue it contains
- Travel for more than 4 hours
- Recent childbirth.
However, the trials that tested different lengths of anticoagulation have varied markedly in how they defined provoked deep vein thrombosis.4–7
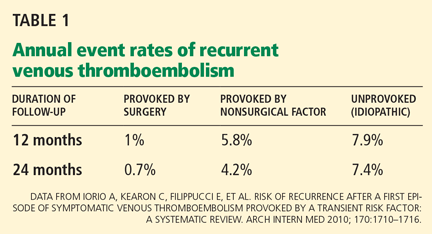
Recommendation: Warfarin or equivalent for 3 months
The American College of Chest Physicians (ACCP) recommends 3 months of anticoagulation with warfarin or another vitamin K antagonist for patients with VTE secondary to a transient (reversible) risk factor,2 and we agree.
HOW LONG TO TREAT CANCER-RELATED VTE
Patients with cancer are at higher risk of developing VTE. Furthermore, in one study,9 compared with other patients with VTE, patients with cancer were three times more likely to have another episode of VTE, with a cumulative rate of recurrence at 1 year of 21% vs 7%. Cancer patients were also twice as likely to suffer major bleeding complications while on anticoagulation.9
Warfarin is a difficult drug to manage because it has many interactions with foods, diseases, and other drugs. These difficulties are amplified in many cancer patients during chemotherapy.
Warfarin was compared with a LMWH in four randomized trials in cancer patients, and a meta-analysis10 found a 50% relative reduction in the rates of recurrent deep vein thrombosis and pulmonary embolism with LMWH treatment. These results were driven primarily by the CLOT trial (Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer),11 which showed an 8% absolute risk reduction (number needed to treat 13) without an increase in major bleeding when cancer-related VTE was treated with an LMWH—ie, dalteparin (Fragmin)—for 6 months compared with warfarin.
Current thinking suggests that VTE should be treated until the cancer is resolved. However, this hypothesis has not been adequately tested, and consequently, the ACCP gives it only a level 1C recommendation.2 The largest of the four trials comparing warfarin and an LMWH lasted only 6 months. The safety of extending LMWH treatment beyond 6 months is currently unknown but is under investigation (clinicaltrials.gov identifier NCT00942968).
Recommendation: LMWH therapy for at least 6 months
The ACCP guidelines recommend LMWH therapy for 3 to 6 months, followed by warfarin or another vitamin K antagonist or continued LMWH treatment until the cancer is resolved.2
The National Comprehensive Cancer Network guidelines recommend an LMWH for 6 months as monotherapy and indefinite anticoagulation if the cancer is still active.12
The American Society of Clinical Oncology guidelines recommend an LMWH for at least 6 months and indefinite anticoagulant therapy for selected patients with active cancer.13
We agree that patients with active cancer should receive an LMWH for at least 6 months and indefinite anticoagulation until the cancer is resolved.
In our experience, many patients are reluctant to give themselves the daily injections that LMWH therapy requires, and so they need to be well-informed about the marked decrease in VTE recurrence with this less-convenient and more-expensive therapy. Many patients face insurance barriers to cover the cost of LMWH therapy; however, careful attention to preauthorization can usually overcome this obstacle.
HOW LONG TO TREAT RECURRENT VTE
It makes clinical sense that patients who have a second VTE event should be treated indefinitely. This theory was tested in a randomized clinical trial14 in which patients with provoked or unprovoked VTE were randomized after their second event to receive anticoagulation for 6 months vs indefinitely.
After 4 years of follow-up, the recurrence rate was 21% in patients assigned to 6 months of treatment and only 3% in patients who continued anticoagulation throughout the trial. On the other hand, major hemorrhage occurred in 3% of patients treated for 6 months and in 9% in patients who continued anticoagulation indefinitely.
Of note, most of the patients in this trial had unprovoked (idiopathic) VTE, so the results should not be extrapolated to patients with provoked VTE, who accounted for only 20% of the study population.14
Recommendation: Long-term anticoagulation
We agree with the ACCP recommendation2 that patients who have had a second episode of unprovoked VTE should receive long-term anticoagulation. Because of a lack of data, the duration of therapy for patients with a second episode of provoked VTE should be individualized.
HOW LONG TO TREAT THROMBOPHILIA-RELATED VTE
Inherited thrombophilias
Patients with VTE that is not related to a clear provoking risk factor or cancer frequently have testing to evaluate for a hypercoagulable state. This workup traditionally includes the most common inherited thrombophilias for gene mutations for factor V and prothrombin as well as for deficiencies in protein C, protein S, antithrombin and the acquired antiphospholipid syndrome.
The key questions that should be asked prior to embarking on this workup are:
- Will the results change the length of therapy for the patient?
- Will testing the patient help with genetic counseling and possible testing of family members?
- Will the results change the targeted INR range for warfarin or other vitamin K antagonist therapy?
Patients with inherited thrombophilia have a greater risk of developing an initial VTE event; however, these tests do not help predict the recurrence of VTE in patients with established disease more than clinical risk factors do. A prospective study demonstrated this by looking at the effect of thrombophilia and clinical factors on the recurrence of venous thrombosis and found that inherited prothrombotic abnormalities do not appear to play an important role in the risk of a recurrent event.15 On the other hand, clinical factors, such as whether the first event was idiopathic or provoked, appear more important in determining the duration of anticoagulation therapy.15 A systematic review of the common inherited thrombophilias showed the VTE recurrence rate of patients with factor V Leiden was higher than in patients without the mutation; however, the absolute rates of recurrence were not much different than what would be expected in patients with idiopathic VTE.16
A retrospective study involving a large cohort of families of patients who already had experienced a first episode of either idiopathic or provoked VTE showed high annual risks of recurrent VTE associated with hereditary deficiencies of protein S (8.4%), protein C (6.0%), and antithrombin (10%).17 However, for the more commonly occurring genetic thrombophilias, the factor V Leiden and prothrombin G20210A mutations, family members with either gene abnormality had low rates of VTE, suggesting that testing of relatives of probands is not clinically useful.16
Antiphospholipid syndrome
Antiphospholipid syndrome is an acquired thrombophilia. A patient has thrombotic antiphospholipid syndrome when there is a history of vascular thrombosis in the presence of persistently positive tests (at least 12 weeks apart) for lupus anticoagulants, anticardiolipin antibodies, or anti-beta-2 glycoprotein I. A prospective study of 412 patients with a first episode of VTE found that 15% were positive for anticardiolipin antibody at the end of 6 months of anticoagulation. The risk of recurrent VTE after 4 years was 29% in patients with antibodies and 14% in those without antibodies (relative risk 2.1; 95% confidence interval [CI] 1.3–3.3; P =.0013).18
Recent reviews advise indefinite warfarin anticoagulation in patients with VTE and persistence of antiphospholipid antibodies.19 However, the optimal duration of anticoagulation is uncertain. Until well-designed clinical trials are done, the current general consensus is to anticoagulate these patients indefinitely.20,21 Retrospective studies had suggested that patients with antiphospholipid antibodies required a higher therapeutic INR range; however, this observation was tested in two trials that found no difference in thromboembolic rates when patients were randomized to an INR of 2.0–3.0 vs 3.1–4.0,22 or 2.0–3.0 vs 3.0–4.5.23
No formal recommendations
In the absence of strong evidence, the ACCP guidelines do not include a recommendation on the duration of anticoagulation treatment specific to inherited thrombophilias. We believe that clinical factors are more important than inherited thrombophilias for deciding the duration of anticoagulation, and that testing is almost never indicated or useful. However, patients with antiphospholipid syndrome are at high risk of recurrence, and it is our practice to anticoagulate these patients indefinitely.
HOW LONG TO TREAT UNPROVOKED (IDIOPATHIC) VTE
A VTE event is thought to be idiopathic if it occurs without a clearly identified provoking factor.
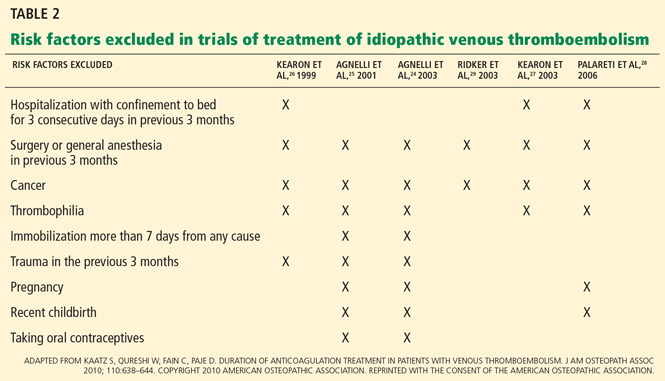
In an observational study,3 patients with oral contraceptive use, transient illness, immobilization, or a history of travel had an 8.8% risk of recurrence vs 19.4% in patients with unprovoked VTE. The meta-analysis discussed above (Table 1)8 also shows that patients with these nonsurgical risk factors have a lower rate of recurrence than patients with idiopathic VTE.
The high rate of recurrence of idiopathic VTE (4% to 27% after 3 months of anticoagulation24–26) suggests that a longer duration of treatment is reasonable. However, increasing the length of therapy from 3 to 12 months delays but does not prevent recurrence, the risk of which begins to accumulate once anticoagulation is stopped.24,25
Three promising strategies to identify subgroups of patients with idiopathic VTE who are at highest risk of recurrence and who would benefit the most from prolonged anticoagulation are d-dimer testing, evaluation for residual vein thrombosis in patients who present with a deep vein thrombosis, and clinical prediction rules.
d-dimer testing
d-dimer is a degradation product of fibrin and is an indirect marker of residual thrombosis.30
In a systematic review of patients with a first episode of unprovoked VTE,31 a normal d-dimer concentration at the end of at least 3 months of anticoagulation was associated with a 3.5% annual risk of recurrence, whereas an elevated d-dimer level at that time was associated with an annual risk of 8.9%. These results were confirmed in a systematic review of individual patient data.32
In a randomized trial,28 patients with an idiopathic VTE event who received anticoagulation for at least 3 months had their d-dimer level measured 1 month after cessation of treatment. Those with an elevated level were randomized to either resume anticoagulation or not. Patients who resumed anticoagulation had an annual recurrence rate of 2%; however, those who were allocated not to restart anticoagulation had a recurrence rate of 10.9% per year. There was no difference in the rate of major bleeding between the two groups. Patients in this clinical trial who had a normal d-dimer level did not restart anticoagulation and had an annual recurrence rate of 4.4%.
Evaluation for residual thrombosis

A randomized trial34 in patients with both provoked and idiopathic deep vein thrombosis showed a reduction in recurrence when those who had residual vein thrombosis were given extended anticoagulation. In the subset of patients whose deep vein thrombosis was idiopathic, the recurrence rate was 17% per year when treatment lasted only 3 months and 10% when it was extended for up to 1 year.
Another trial35 randomized patients with provoked and idiopathic deep vein thrombosis to receive anticoagulation for the usual duration or to continue treatment until recanalization of the residual thrombus was demonstrated on follow-up Doppler ultrasonography. Patients who received this ultrasonography-tailored treatment had a lower rate of recurrence of VTE; however, the absolute reductions in recurrence rates cannot be calculated from this report for patients with idiopathic deep vein thrombosis.
A prospective observational study36 of the predictive value of d-dimer status and residual vein thrombus found that only d-dimer was an independent risk factor for recurrent VTE after vitamin K antagonist withdrawal.
A clinical prediction rule: ‘Men and HERDOO2’
A promising tool for predicting if a patient is at low risk of recurrent VTE after the first episode of proximal deep vein thrombosis or pulmonary embolism is known by the mnemonic device “Men and HERDOO2.” It is based on data prospectively derived by Rodger et al37 to identify patients with less than a 3% annual risk of recurrent VTE after their first event of idiopathic proximal deep vein thrombosis or pulmonary embolism. Risk factors for recurrent VTE were male sex (the “men” of “Men and HERDOO2”), signs of postthrombotic syndrome, including hyperpigmentation of the lower extremities, edema or redness of either leg, a d-dimer level > 250 μg/L, obesity (body mass index > 30 kg/m2, and older age (> 65 years).
Overall, one-fourth of the population were women with no risk factors or one risk factor, and their risk of recurrence was 1.6% per year. Men and women who had two or more risk factors for postthrombotic syndrome (hyperpigmentation, edema, or redness), elevated d-dimer, obesity, or older age were predicted to be at higher risk of recurrent VTE. Patients such as this should be considered for indefinite anticoagulation.
Ideally, clinical prediction rules should be validated in a separate group of patients before they are used routinely in practice,38 and this clinical prediction rule is currently being tested in the REVERSE II study. If the results are consistent, this will be an easy-to-use tool to help identify patients who likely can safely stop anticoagulation therapy after 3 to 6 months (clinicaltrials.gov Identifier: NCT00967304).
The location of the thrombosis also influences the likelihood of recurrence. Patients with isolated distal (calf) deep vein thrombosis are less likely to suffer recurrent VTE than those who present with proximal deep vein thrombosis. However, trials focusing specifically on the precise subset of idiopathic isolated distal deep vein thrombosis are lacking. In a randomized trial39 comparing 6 vs 12 weeks of anticoagulation for isolated distal deep vein thrombosis and 12 vs 24 weeks for proximal deep vein thrombosis, the annual rates of recurrence after 12 weeks of treatment were approximately 3.4% for isolated distal and 8.1% for proximal deep vein thrombosis.39
Recommendation: At least 3 months of warfarin or equivalent
We agree with the ACCP recommendation2 that patients with unprovoked VTE should receive at least 3 months of anticoagulation with a vitamin K antagonist.
If the patient has no risk factors for bleeding and good anticoagulant monitoring is achievable, we agree with long-term anticoagulation for proximal unprovoked deep vein thrombosis or pulmonary embolism, and 3 months of therapy for isolated distal unprovoked deep vein thrombosis.
Patient preferences and the risk of recurrence vs the risk of bleeding should be discussed with patients when contemplating indefinite anticoagulation.
If testing is being considered to assist in the decision to prescribe indefinite anticoagulation, we prefer using d-dimer levels rather than ultrasonography to detect residual venous thrombosis because of its ease of use and the strength of the current evidence.
PREVENTING POSTTHROMBOTIC SYNDROME
The postthrombotic (postphlebitic) syndrome is a chronic and burdensome consequence of deep vein thrombosis that occurs despite anticoagulation therapy. It is estimated to affect 23% to 60% of patients and typically manifests in the first 2 years.40 It is not only costly in clinical terms, with decreased quality of life for the patient, but health care expenditures have been estimated to range from $400 per year in a Brazilian study to $7,000 per year in a US study.40
Typical symptoms include leg pain, heaviness, swelling, and cramping. In severe cases, chronic venous ulcers can occur and are difficult to treat.41
The definition of postthrombotic syndrome has been unclear over the years, and six different scales that measure signs and symptoms have been reported.42
The Villalta scale has been proposed by the International Society of Thrombosis and Hemostasis as a diagnostic standard to define postthrombotic syndrome.42 This validated scale is based on five clinical symptoms, six clinical signs, and the presence or absence of venous ulcers. Each clinical symptom and sign is scored as mild (1 point), moderate (2 points), or severe (3 points). Symptoms include pain, cramps, heaviness, paresthesia, and pruritus; the six clinical signs are pretibial edema, skin induration, hyperpigmentation, redness, venous ectasia, and pain on calf compression.
According to the International Society of Thrombosis and Hemostasis, postthrombotic syndrome is present if the Villalta score is 5 or greater or if a venous ulcer is present in a leg with previous deep vein thrombosis. Further, using the Villalta scale, postthrombotic syndrome can be categorized as mild (score 5–9), moderate (10–14), or severe (≥ 15).
A limitation of the Villalta scale is that the presence or absence of a venous ulcer has not been assigned a score. Since a venous ulcer requires more aggressive measures, the society defines postthrombotic syndrome as severe if venous ulcers are present.42
Acute symptoms of deep vein thrombosis may take months to resolve and, indeed, acute symptoms may transition to chronic symptoms without a symptom-free interval. It is recommended that postthrombotic syndrome not be diagnosed before 3 months to avoid inappropriately attributing acute symptoms and signs of deep vein thrombosis to the postthrombotic syndrome.42
Studies of stockings
A systematic review of three randomized trials44 concluded that elastic compression stockings reduce the risk of postthrombotic syndrome (any severity) from 43% to 20% and severe postthrombotic syndrome from 15% to 7%.43
The first of these trials44 randomized patients soon after the diagnosis of deep vein thrombosis to receive made-to-order compression stockings that were rated at 30 to 40 mm Hg or to be in a control group that did not receive stockings. The second trial45 randomized patients 1 year after the index event of deep vein thrombosis to receive 20- to 30-mm Hg stockings or stockings that were two sizes too large (the control group). The third study46 randomly allocated patients to receive “off-the-shelf” stockings (30–40 mm Hg) or no stockings. Each study used its own definition of postthrombotic syndrome.
Although these studies strongly suggest compression stockings prevent postthrombotic syndrome, several methodologic issues remain:
- A standard definition of postthrombotic syndrome was not used
- The amount of compression varied between studies
- The studies were not blinded.
Lack of blinding becomes most significant when an outcome is based on subjective findings, like the symptoms that make up a large part of the diagnosis of postthrombotic syndrome.
The SOX trial, currently under way, is designed to address these methodologic issues and should be completed in 2012 (clinicaltrials.gov Identifier: NCT00143598).
Recommendation: Stockings for at least 2 years
We agree with the ACCP recommendation that a patient who has had a symptomatic proximal deep vein thrombosis should wear an elastic compression stocking with an ankle pressure gradient of 30 to 40 mm Hg as soon as possible after starting anticoagulant therapy and continuing for a minimum of 2 years.2
SCREENING FOR OCCULT MALIGNANCY
VTE can be the first manifestation of cancer.
French physician Armand Trousseau, in the 1860s, was the first to describe disseminated intravascular coagulation closely associated with adenocarcinoma. Ironically, several years later, after suffering for weeks from abdominal pain, he declared to one of his students that he had developed thrombosis, and he died of gastric cancer shortly thereafter.47
Since cancer is a well-known risk factor for VTE, it is logical to screen for cancer as an explanation for an idiopathic VTE event.48 To make an informed decision, one needs to understand the rate of occult cancer at the time VTE is diagnosed, the risk of future development of cancer, and the utility of extensive cancer screening.
The clinical efficacy, side effects, and cost-effectiveness of cancer screening in patients with idiopathic VTE are unknown. However, a systematic review47 of 34 studies found that, in patients with idiopathic VTE, cancer was diagnosed within 1 month in 6.1%, within 6 months in 8.6%, and within 1 year in 10.0% (95% CI 8.6–11.3).
A subset of studies compared two strategies for screening soon after the diagnosis of idiopathic VTE: a strategy limited to the history, physical examination, basic blood work, and chest radiography vs an extensive screening strategy that also included serum tumor markers or abdominal ultrasonography or computed tomography. Limited screening detected 49% of the prevalent cancers; extensive screening increased this rate to 70%. Stated another way, the detection rate for prevalent cancers was 5% with limited screening and 7% with extensive screening soon after the diagnosis of idiopathic VTE.47
Patients with idiopathic VTE had higher rates of cancer within 1 month of diagnosis than patients with provoked VTE (6.1% vs 1.9%), and this difference persisted at 1 year (10.0% vs 2.6%).47
Recommendation: Individualized cancer screening
Patients with idiopathic VTE have a significant risk of occult cancer within the first year after diagnosis, and cancer screening should be considered. Our practice for patients with idiopathic VTE is to perform a history and physical examination and ensure that the patient is up to date on age- and sex-specific cancer screening.
The use of additional imaging or biomarkers should be discussed with patients so they can balance the risks (radiation and potential false-positive results with their downstream consequences), costs, and potential benefits, given the lack of proven survival benefit or cost-effectiveness.
ORAL ANTICOAGULANT MANAGEMENT
Warfarin’s multiple interactions, along with the need for INR monitoring, make it a difficult medication to manage.
The Joint Commission, the US organization for health service accreditation and certification, has defined National Patient Safety Goals and quality measures for the management of anticoagulation.49 Organized anticoagulation management services, dosing algorithms, and patient self-testing using capillary INR meters or patient self-management of warfarin were recommended as tools to improve the time patients spend in the therapeutic INR range.50
Two new oral anticoagulants
The limitations of warfarin have stimulated the search for newer oral anticoagulants that do not require laboratory monitoring or have as many diet and drug interactions.
Two trials have been published with experimental oral anticoagulants that had similar efficacy and safety as warfarin in the treatment of VTE.
The study of dabigatran (Pradaxa) vs warfarin in the treatment of acute VTE (the RECOVER trial)51 randomized 2,539 patients with acute VTE to receive the oral direct thrombin inhibitor dabigatran or warfarin for approximately 6 months. Of note, each treatment group received a median of 6 days of heparin, LMWH, or fondaparinux at the beginning of blinded therapy. The rates of recurrent VTE and major bleeding were similar between the treatment arms, and overall bleeding was less with dabigatran. Dabigatran was approved in the United States in October 2010 for stroke prevention in atrial fibrillation but has yet to be approved for the treatment of VTE pending further study (clinicaltrials.gov Identifier: NCT00680186).
A study of oral rivaroxaban (Xarelto) for symptomatic VTE (the EINSTEIN-DVT trial) 52 randomized 3,449 patients with acute deep vein thrombosis to rivaroxaban or enoxaparin (Lovenox) overlapped with warfarin or another vitamin K antagonist in the usual manner. No difference was noted between the treatments in the rate of recurrence of VTE or of major bleeding. Of note, patients randomized to rivaroxaban received 15 mg twice a day for the first 3 weeks of treatment and then 20 mg per day for the remainder of their therapy and did not require parenteral anticoagulant overlap.
The long-awaited promise of easier-to-use oral anticoagulants for the treatment of VTE is drawing near and has the potential to revolutionize the treatment of this common disorder. In the meantime, close monitoring of warfarin and careful patient education regarding its use are essential. And even with the development of new drugs in the future, it is still imperative that patients with acute VTE receive the correct length of anticoagulation treatment, are prescribed stockings to prevent postthrombotic syndrome, and are updated on routine cancer screening.
- Spencer FA, Emery C, Lessard D, et al. The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 2006; 21:722–727.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523–526.
- Schulman S, Lockner D, Juhlin-Dannfelt A. The duration of oral anticoagulation after deep vein thrombosis. A randomized study. Acta Med Scand 1985; 217:547–552.
- Optimum duration of anticoagulation for deep-vein thrombosis and pulmonary embolism. Research Committee of the British Thoracic Society. Lancet 1992; 340:873–876.
- Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. Duration of Anticoagulation Trial Study Group. N Engl J Med 1995; 332:1661–1665.
- Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743–749.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med 2010; 170:1710–1716.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hull RD, Pineo GF, Brant RF, et al; LITE Trial Investigators. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology, Venous Thromboembolic Disease. http://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed August 3, 2011.
- Lyman GH, Khorana AA, Falanga A, et al; American Society of Clinical Oncology. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Schulman S, Granqvist S, Holmström M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–2485.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost 2009; 101:93–99.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med 1998; 104:332–338.
- Derksen RH, de Groot PG. Towards evidence-based treatment of thrombotic antiphospholipid syndrome. Lupus 2010; 19:470–474.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Fonseca AG, D’Cruz DP. Controversies in the antiphospholipid syndrome: can we ever stop warfarin? J Autoimmune Dis 2008; 5:6.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3:848–853.
- Agnelli G, Prandoni P, Becattini C, et al; Warfarin Optimal Duration Italian Trial Investigators. Extended oral anticoagulant therapy after a first episode of pulmonary embolism. Ann Intern Med 2003; 139:19–25.
- Agnelli G, Prandoni P, Santamaria MG, et al; Warfarin Optimal Duration Italian Trial Investigators. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Kearon C, Ginsberg JS, Kovacs MJ, et al; Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–639.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Ridker PM, Goldhaber SZ, Glynn RJ. Low-intensity versus conventional-intensity warfarin for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:2164–2167.
- Bockenstedt P. D-dimer in venous thromboembolism. N Engl J Med 2003; 349:1203–1204.
- Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008; 149:481–490,W94.
- Douketis J, Tosetto A, Marcucci M, et al. Patient-level metaanalysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med 2010; 153:523–531.
- Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med 2002; 137:955–960.
- Siragusa S, Malato A, Anastasio R, et al. Residual vein thrombosis to establish duration of anticoagulation after a first episode of deep vein thrombosis: the Duration of Anticoagulation based on Compression UltraSonography (DACUS) study. Blood 2008; 112:511–515.
- Prandoni P, Prins MH, Lensing AW, et al; AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep venous thrombosis: a randomized trial. Ann Intern Med 2009; 150:577–585.
- Cosmi B, Legnani C, Cini M, Guazzaloca G, Palareti G. D-dimer levels in combination with residual venous obstruction and the risk of recurrence after anticoagulation withdrawal for a first idiopathic deep vein thrombosis. Thromb Haemost 2005; 94:969–974.
- Rodger MA, Kahn SR, Wells PS, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ 2008; 179:417–426.
- McGinn TG, Guyatt GH, Wyer PC, Naylor CD, Stiell IG, Richardson WS. Users’ guides to the medical literature: XXII: how to use articles about clinical decision rules. Evidence-Based Medicine Working Group. JAMA 2000; 284:79–84.
- Pinede L, Ninet J, Duhaut P, et al; Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103:2453–2460.
- Ashrani AA, Heit JA. Incidence and cost burden of postthrombotic syndrome. J Thromb Thrombolysis 2009; 28:465–476.
- Kahn SR, Shrier I, Julian JA, et al. Determinants and time course of the postthrombotic syndrome after acute deep venous thrombosis. Ann Intern Med 2008; 149:698–707.
- Kahn SR, Partsch H, Vedantham S, Prandoni P, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of post-thrombotic syndrome of the leg for use in clinical investigations: a recommendation for standardization. J Thromb Haemost 2009; 7:879–883.
- Kolbach DN, Sandbrink MW, Hamulyak K, Neumann HA, Prins MH. Non-pharmaceutical measures for prevention of post-thrombotic syndrome. Cochrane Database Syst Rev 2004;CD004174.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet 1997; 349:759–762.
- Ginsberg JS, Hirsh J, Julian J, et al. Prevention and treatment of postphlebitic syndrome: results of a 3-part study. Arch Intern Med 2001; 161:2105–2109.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the post-thrombotic syndrome: a randomized, controlled trial. Ann Intern Med 2004; 141:249–256.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008; 149:323–333.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Kaatz S. Impact on patient care: patient case through the continuum of care. J Thromb Thrombolysis 2010; 29:167–170.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Schulman S, Kearon C, Kakkar AK, et al; for the RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2452.
- The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363;2499–2510.
Deep vein thrombosis and pulmonary embolism are collectively referred to as venous thromboembolic (VTE) disease. They affect approximately 100,000 to 300,000 patients per year in the United States.1 Although patients with deep vein thrombosis can be treated as outpatients, many are admitted for the initiation of anticoagulation. Initial anticoagulation usually requires the overlap of a parenteral anticoagulant (unfractionated heparin, low-molecular-weight heparin [LMWH] or fondaparinux) with warfarin for a minimum of 5 days and until the international normalized ratio (INR) of the prothrombin time is above 2.0 for at least 24 hours.2
Three clinical issues need to be addressed after the initiation of anticoagulation for VTE:
- Determination of the length of anticoagulation with the correct anticoagulant
- Prevention of postthrombotic syndrome
- Appropriate screening for occult malignancy.
HOW LONG SHOULD VTE BE TREATED?
The duration of anticoagulation has been a matter of debate.
The risk of recurrent VTE appears related to clinical risk factors that a patient has at the time of the initial thrombotic event. An epidemiologic study3 found that patients with VTE treated for approximately 6 months had a low rate of recurrence (0% at 2 years of follow-up) if surgery was the risk factor. The risk climbed to 9% if the risk factor was nonsurgical and to 19% if there were no provoking risk factors.
The likelihood of VTE recurrence and therefore the recommended duration of treatment depend on whether the VTE event was provoked, cancer-related, recurrent, thrombophilia-related, or idiopathic. We address each of these scenarios below.
HOW LONG TO TREAT PROVOKED VTE
A VTE event is considered provoked if the patient had a clear inciting risk factor. As defined in various clinical trials, these risk factors include:
- Hospitalization with confinement to bed for 3 or more consecutive days in the last 3 months
- Surgery or general anesthesia in the last 3 months
- Immobilization for more than 7 days, regardless of the cause
- Trauma in the last 3 months
- Pregnancy
- Use of an oral contraceptive, regardless of which estrogen or progesterone analogue it contains
- Travel for more than 4 hours
- Recent childbirth.
However, the trials that tested different lengths of anticoagulation have varied markedly in how they defined provoked deep vein thrombosis.4–7
Recommendation: Warfarin or equivalent for 3 months
The American College of Chest Physicians (ACCP) recommends 3 months of anticoagulation with warfarin or another vitamin K antagonist for patients with VTE secondary to a transient (reversible) risk factor,2 and we agree.
HOW LONG TO TREAT CANCER-RELATED VTE
Patients with cancer are at higher risk of developing VTE. Furthermore, in one study,9 compared with other patients with VTE, patients with cancer were three times more likely to have another episode of VTE, with a cumulative rate of recurrence at 1 year of 21% vs 7%. Cancer patients were also twice as likely to suffer major bleeding complications while on anticoagulation.9
Warfarin is a difficult drug to manage because it has many interactions with foods, diseases, and other drugs. These difficulties are amplified in many cancer patients during chemotherapy.
Warfarin was compared with a LMWH in four randomized trials in cancer patients, and a meta-analysis10 found a 50% relative reduction in the rates of recurrent deep vein thrombosis and pulmonary embolism with LMWH treatment. These results were driven primarily by the CLOT trial (Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer),11 which showed an 8% absolute risk reduction (number needed to treat 13) without an increase in major bleeding when cancer-related VTE was treated with an LMWH—ie, dalteparin (Fragmin)—for 6 months compared with warfarin.
Current thinking suggests that VTE should be treated until the cancer is resolved. However, this hypothesis has not been adequately tested, and consequently, the ACCP gives it only a level 1C recommendation.2 The largest of the four trials comparing warfarin and an LMWH lasted only 6 months. The safety of extending LMWH treatment beyond 6 months is currently unknown but is under investigation (clinicaltrials.gov identifier NCT00942968).
Recommendation: LMWH therapy for at least 6 months
The ACCP guidelines recommend LMWH therapy for 3 to 6 months, followed by warfarin or another vitamin K antagonist or continued LMWH treatment until the cancer is resolved.2
The National Comprehensive Cancer Network guidelines recommend an LMWH for 6 months as monotherapy and indefinite anticoagulation if the cancer is still active.12
The American Society of Clinical Oncology guidelines recommend an LMWH for at least 6 months and indefinite anticoagulant therapy for selected patients with active cancer.13
We agree that patients with active cancer should receive an LMWH for at least 6 months and indefinite anticoagulation until the cancer is resolved.
In our experience, many patients are reluctant to give themselves the daily injections that LMWH therapy requires, and so they need to be well-informed about the marked decrease in VTE recurrence with this less-convenient and more-expensive therapy. Many patients face insurance barriers to cover the cost of LMWH therapy; however, careful attention to preauthorization can usually overcome this obstacle.
HOW LONG TO TREAT RECURRENT VTE
It makes clinical sense that patients who have a second VTE event should be treated indefinitely. This theory was tested in a randomized clinical trial14 in which patients with provoked or unprovoked VTE were randomized after their second event to receive anticoagulation for 6 months vs indefinitely.
After 4 years of follow-up, the recurrence rate was 21% in patients assigned to 6 months of treatment and only 3% in patients who continued anticoagulation throughout the trial. On the other hand, major hemorrhage occurred in 3% of patients treated for 6 months and in 9% in patients who continued anticoagulation indefinitely.
Of note, most of the patients in this trial had unprovoked (idiopathic) VTE, so the results should not be extrapolated to patients with provoked VTE, who accounted for only 20% of the study population.14
Recommendation: Long-term anticoagulation
We agree with the ACCP recommendation2 that patients who have had a second episode of unprovoked VTE should receive long-term anticoagulation. Because of a lack of data, the duration of therapy for patients with a second episode of provoked VTE should be individualized.
HOW LONG TO TREAT THROMBOPHILIA-RELATED VTE
Inherited thrombophilias
Patients with VTE that is not related to a clear provoking risk factor or cancer frequently have testing to evaluate for a hypercoagulable state. This workup traditionally includes the most common inherited thrombophilias for gene mutations for factor V and prothrombin as well as for deficiencies in protein C, protein S, antithrombin and the acquired antiphospholipid syndrome.
The key questions that should be asked prior to embarking on this workup are:
- Will the results change the length of therapy for the patient?
- Will testing the patient help with genetic counseling and possible testing of family members?
- Will the results change the targeted INR range for warfarin or other vitamin K antagonist therapy?
Patients with inherited thrombophilia have a greater risk of developing an initial VTE event; however, these tests do not help predict the recurrence of VTE in patients with established disease more than clinical risk factors do. A prospective study demonstrated this by looking at the effect of thrombophilia and clinical factors on the recurrence of venous thrombosis and found that inherited prothrombotic abnormalities do not appear to play an important role in the risk of a recurrent event.15 On the other hand, clinical factors, such as whether the first event was idiopathic or provoked, appear more important in determining the duration of anticoagulation therapy.15 A systematic review of the common inherited thrombophilias showed the VTE recurrence rate of patients with factor V Leiden was higher than in patients without the mutation; however, the absolute rates of recurrence were not much different than what would be expected in patients with idiopathic VTE.16
A retrospective study involving a large cohort of families of patients who already had experienced a first episode of either idiopathic or provoked VTE showed high annual risks of recurrent VTE associated with hereditary deficiencies of protein S (8.4%), protein C (6.0%), and antithrombin (10%).17 However, for the more commonly occurring genetic thrombophilias, the factor V Leiden and prothrombin G20210A mutations, family members with either gene abnormality had low rates of VTE, suggesting that testing of relatives of probands is not clinically useful.16
Antiphospholipid syndrome
Antiphospholipid syndrome is an acquired thrombophilia. A patient has thrombotic antiphospholipid syndrome when there is a history of vascular thrombosis in the presence of persistently positive tests (at least 12 weeks apart) for lupus anticoagulants, anticardiolipin antibodies, or anti-beta-2 glycoprotein I. A prospective study of 412 patients with a first episode of VTE found that 15% were positive for anticardiolipin antibody at the end of 6 months of anticoagulation. The risk of recurrent VTE after 4 years was 29% in patients with antibodies and 14% in those without antibodies (relative risk 2.1; 95% confidence interval [CI] 1.3–3.3; P =.0013).18
Recent reviews advise indefinite warfarin anticoagulation in patients with VTE and persistence of antiphospholipid antibodies.19 However, the optimal duration of anticoagulation is uncertain. Until well-designed clinical trials are done, the current general consensus is to anticoagulate these patients indefinitely.20,21 Retrospective studies had suggested that patients with antiphospholipid antibodies required a higher therapeutic INR range; however, this observation was tested in two trials that found no difference in thromboembolic rates when patients were randomized to an INR of 2.0–3.0 vs 3.1–4.0,22 or 2.0–3.0 vs 3.0–4.5.23
No formal recommendations
In the absence of strong evidence, the ACCP guidelines do not include a recommendation on the duration of anticoagulation treatment specific to inherited thrombophilias. We believe that clinical factors are more important than inherited thrombophilias for deciding the duration of anticoagulation, and that testing is almost never indicated or useful. However, patients with antiphospholipid syndrome are at high risk of recurrence, and it is our practice to anticoagulate these patients indefinitely.
HOW LONG TO TREAT UNPROVOKED (IDIOPATHIC) VTE
A VTE event is thought to be idiopathic if it occurs without a clearly identified provoking factor.
In an observational study,3 patients with oral contraceptive use, transient illness, immobilization, or a history of travel had an 8.8% risk of recurrence vs 19.4% in patients with unprovoked VTE. The meta-analysis discussed above (Table 1)8 also shows that patients with these nonsurgical risk factors have a lower rate of recurrence than patients with idiopathic VTE.
The high rate of recurrence of idiopathic VTE (4% to 27% after 3 months of anticoagulation24–26) suggests that a longer duration of treatment is reasonable. However, increasing the length of therapy from 3 to 12 months delays but does not prevent recurrence, the risk of which begins to accumulate once anticoagulation is stopped.24,25
Three promising strategies to identify subgroups of patients with idiopathic VTE who are at highest risk of recurrence and who would benefit the most from prolonged anticoagulation are d-dimer testing, evaluation for residual vein thrombosis in patients who present with a deep vein thrombosis, and clinical prediction rules.
d-dimer testing
d-dimer is a degradation product of fibrin and is an indirect marker of residual thrombosis.30
In a systematic review of patients with a first episode of unprovoked VTE,31 a normal d-dimer concentration at the end of at least 3 months of anticoagulation was associated with a 3.5% annual risk of recurrence, whereas an elevated d-dimer level at that time was associated with an annual risk of 8.9%. These results were confirmed in a systematic review of individual patient data.32
In a randomized trial,28 patients with an idiopathic VTE event who received anticoagulation for at least 3 months had their d-dimer level measured 1 month after cessation of treatment. Those with an elevated level were randomized to either resume anticoagulation or not. Patients who resumed anticoagulation had an annual recurrence rate of 2%; however, those who were allocated not to restart anticoagulation had a recurrence rate of 10.9% per year. There was no difference in the rate of major bleeding between the two groups. Patients in this clinical trial who had a normal d-dimer level did not restart anticoagulation and had an annual recurrence rate of 4.4%.
Evaluation for residual thrombosis
A randomized trial34 in patients with both provoked and idiopathic deep vein thrombosis showed a reduction in recurrence when those who had residual vein thrombosis were given extended anticoagulation. In the subset of patients whose deep vein thrombosis was idiopathic, the recurrence rate was 17% per year when treatment lasted only 3 months and 10% when it was extended for up to 1 year.
Another trial35 randomized patients with provoked and idiopathic deep vein thrombosis to receive anticoagulation for the usual duration or to continue treatment until recanalization of the residual thrombus was demonstrated on follow-up Doppler ultrasonography. Patients who received this ultrasonography-tailored treatment had a lower rate of recurrence of VTE; however, the absolute reductions in recurrence rates cannot be calculated from this report for patients with idiopathic deep vein thrombosis.
A prospective observational study36 of the predictive value of d-dimer status and residual vein thrombus found that only d-dimer was an independent risk factor for recurrent VTE after vitamin K antagonist withdrawal.
A clinical prediction rule: ‘Men and HERDOO2’
A promising tool for predicting if a patient is at low risk of recurrent VTE after the first episode of proximal deep vein thrombosis or pulmonary embolism is known by the mnemonic device “Men and HERDOO2.” It is based on data prospectively derived by Rodger et al37 to identify patients with less than a 3% annual risk of recurrent VTE after their first event of idiopathic proximal deep vein thrombosis or pulmonary embolism. Risk factors for recurrent VTE were male sex (the “men” of “Men and HERDOO2”), signs of postthrombotic syndrome, including hyperpigmentation of the lower extremities, edema or redness of either leg, a d-dimer level > 250 μg/L, obesity (body mass index > 30 kg/m2, and older age (> 65 years).
Overall, one-fourth of the population were women with no risk factors or one risk factor, and their risk of recurrence was 1.6% per year. Men and women who had two or more risk factors for postthrombotic syndrome (hyperpigmentation, edema, or redness), elevated d-dimer, obesity, or older age were predicted to be at higher risk of recurrent VTE. Patients such as this should be considered for indefinite anticoagulation.
Ideally, clinical prediction rules should be validated in a separate group of patients before they are used routinely in practice,38 and this clinical prediction rule is currently being tested in the REVERSE II study. If the results are consistent, this will be an easy-to-use tool to help identify patients who likely can safely stop anticoagulation therapy after 3 to 6 months (clinicaltrials.gov Identifier: NCT00967304).
The location of the thrombosis also influences the likelihood of recurrence. Patients with isolated distal (calf) deep vein thrombosis are less likely to suffer recurrent VTE than those who present with proximal deep vein thrombosis. However, trials focusing specifically on the precise subset of idiopathic isolated distal deep vein thrombosis are lacking. In a randomized trial39 comparing 6 vs 12 weeks of anticoagulation for isolated distal deep vein thrombosis and 12 vs 24 weeks for proximal deep vein thrombosis, the annual rates of recurrence after 12 weeks of treatment were approximately 3.4% for isolated distal and 8.1% for proximal deep vein thrombosis.39
Recommendation: At least 3 months of warfarin or equivalent
We agree with the ACCP recommendation2 that patients with unprovoked VTE should receive at least 3 months of anticoagulation with a vitamin K antagonist.
If the patient has no risk factors for bleeding and good anticoagulant monitoring is achievable, we agree with long-term anticoagulation for proximal unprovoked deep vein thrombosis or pulmonary embolism, and 3 months of therapy for isolated distal unprovoked deep vein thrombosis.
Patient preferences and the risk of recurrence vs the risk of bleeding should be discussed with patients when contemplating indefinite anticoagulation.
If testing is being considered to assist in the decision to prescribe indefinite anticoagulation, we prefer using d-dimer levels rather than ultrasonography to detect residual venous thrombosis because of its ease of use and the strength of the current evidence.
PREVENTING POSTTHROMBOTIC SYNDROME
The postthrombotic (postphlebitic) syndrome is a chronic and burdensome consequence of deep vein thrombosis that occurs despite anticoagulation therapy. It is estimated to affect 23% to 60% of patients and typically manifests in the first 2 years.40 It is not only costly in clinical terms, with decreased quality of life for the patient, but health care expenditures have been estimated to range from $400 per year in a Brazilian study to $7,000 per year in a US study.40
Typical symptoms include leg pain, heaviness, swelling, and cramping. In severe cases, chronic venous ulcers can occur and are difficult to treat.41
The definition of postthrombotic syndrome has been unclear over the years, and six different scales that measure signs and symptoms have been reported.42
The Villalta scale has been proposed by the International Society of Thrombosis and Hemostasis as a diagnostic standard to define postthrombotic syndrome.42 This validated scale is based on five clinical symptoms, six clinical signs, and the presence or absence of venous ulcers. Each clinical symptom and sign is scored as mild (1 point), moderate (2 points), or severe (3 points). Symptoms include pain, cramps, heaviness, paresthesia, and pruritus; the six clinical signs are pretibial edema, skin induration, hyperpigmentation, redness, venous ectasia, and pain on calf compression.
According to the International Society of Thrombosis and Hemostasis, postthrombotic syndrome is present if the Villalta score is 5 or greater or if a venous ulcer is present in a leg with previous deep vein thrombosis. Further, using the Villalta scale, postthrombotic syndrome can be categorized as mild (score 5–9), moderate (10–14), or severe (≥ 15).
A limitation of the Villalta scale is that the presence or absence of a venous ulcer has not been assigned a score. Since a venous ulcer requires more aggressive measures, the society defines postthrombotic syndrome as severe if venous ulcers are present.42
Acute symptoms of deep vein thrombosis may take months to resolve and, indeed, acute symptoms may transition to chronic symptoms without a symptom-free interval. It is recommended that postthrombotic syndrome not be diagnosed before 3 months to avoid inappropriately attributing acute symptoms and signs of deep vein thrombosis to the postthrombotic syndrome.42
Studies of stockings
A systematic review of three randomized trials44 concluded that elastic compression stockings reduce the risk of postthrombotic syndrome (any severity) from 43% to 20% and severe postthrombotic syndrome from 15% to 7%.43
The first of these trials44 randomized patients soon after the diagnosis of deep vein thrombosis to receive made-to-order compression stockings that were rated at 30 to 40 mm Hg or to be in a control group that did not receive stockings. The second trial45 randomized patients 1 year after the index event of deep vein thrombosis to receive 20- to 30-mm Hg stockings or stockings that were two sizes too large (the control group). The third study46 randomly allocated patients to receive “off-the-shelf” stockings (30–40 mm Hg) or no stockings. Each study used its own definition of postthrombotic syndrome.
Although these studies strongly suggest compression stockings prevent postthrombotic syndrome, several methodologic issues remain:
- A standard definition of postthrombotic syndrome was not used
- The amount of compression varied between studies
- The studies were not blinded.
Lack of blinding becomes most significant when an outcome is based on subjective findings, like the symptoms that make up a large part of the diagnosis of postthrombotic syndrome.
The SOX trial, currently under way, is designed to address these methodologic issues and should be completed in 2012 (clinicaltrials.gov Identifier: NCT00143598).
Recommendation: Stockings for at least 2 years
We agree with the ACCP recommendation that a patient who has had a symptomatic proximal deep vein thrombosis should wear an elastic compression stocking with an ankle pressure gradient of 30 to 40 mm Hg as soon as possible after starting anticoagulant therapy and continuing for a minimum of 2 years.2
SCREENING FOR OCCULT MALIGNANCY
VTE can be the first manifestation of cancer.
French physician Armand Trousseau, in the 1860s, was the first to describe disseminated intravascular coagulation closely associated with adenocarcinoma. Ironically, several years later, after suffering for weeks from abdominal pain, he declared to one of his students that he had developed thrombosis, and he died of gastric cancer shortly thereafter.47
Since cancer is a well-known risk factor for VTE, it is logical to screen for cancer as an explanation for an idiopathic VTE event.48 To make an informed decision, one needs to understand the rate of occult cancer at the time VTE is diagnosed, the risk of future development of cancer, and the utility of extensive cancer screening.
The clinical efficacy, side effects, and cost-effectiveness of cancer screening in patients with idiopathic VTE are unknown. However, a systematic review47 of 34 studies found that, in patients with idiopathic VTE, cancer was diagnosed within 1 month in 6.1%, within 6 months in 8.6%, and within 1 year in 10.0% (95% CI 8.6–11.3).
A subset of studies compared two strategies for screening soon after the diagnosis of idiopathic VTE: a strategy limited to the history, physical examination, basic blood work, and chest radiography vs an extensive screening strategy that also included serum tumor markers or abdominal ultrasonography or computed tomography. Limited screening detected 49% of the prevalent cancers; extensive screening increased this rate to 70%. Stated another way, the detection rate for prevalent cancers was 5% with limited screening and 7% with extensive screening soon after the diagnosis of idiopathic VTE.47
Patients with idiopathic VTE had higher rates of cancer within 1 month of diagnosis than patients with provoked VTE (6.1% vs 1.9%), and this difference persisted at 1 year (10.0% vs 2.6%).47
Recommendation: Individualized cancer screening
Patients with idiopathic VTE have a significant risk of occult cancer within the first year after diagnosis, and cancer screening should be considered. Our practice for patients with idiopathic VTE is to perform a history and physical examination and ensure that the patient is up to date on age- and sex-specific cancer screening.
The use of additional imaging or biomarkers should be discussed with patients so they can balance the risks (radiation and potential false-positive results with their downstream consequences), costs, and potential benefits, given the lack of proven survival benefit or cost-effectiveness.
ORAL ANTICOAGULANT MANAGEMENT
Warfarin’s multiple interactions, along with the need for INR monitoring, make it a difficult medication to manage.
The Joint Commission, the US organization for health service accreditation and certification, has defined National Patient Safety Goals and quality measures for the management of anticoagulation.49 Organized anticoagulation management services, dosing algorithms, and patient self-testing using capillary INR meters or patient self-management of warfarin were recommended as tools to improve the time patients spend in the therapeutic INR range.50
Two new oral anticoagulants
The limitations of warfarin have stimulated the search for newer oral anticoagulants that do not require laboratory monitoring or have as many diet and drug interactions.
Two trials have been published with experimental oral anticoagulants that had similar efficacy and safety as warfarin in the treatment of VTE.
The study of dabigatran (Pradaxa) vs warfarin in the treatment of acute VTE (the RECOVER trial)51 randomized 2,539 patients with acute VTE to receive the oral direct thrombin inhibitor dabigatran or warfarin for approximately 6 months. Of note, each treatment group received a median of 6 days of heparin, LMWH, or fondaparinux at the beginning of blinded therapy. The rates of recurrent VTE and major bleeding were similar between the treatment arms, and overall bleeding was less with dabigatran. Dabigatran was approved in the United States in October 2010 for stroke prevention in atrial fibrillation but has yet to be approved for the treatment of VTE pending further study (clinicaltrials.gov Identifier: NCT00680186).
A study of oral rivaroxaban (Xarelto) for symptomatic VTE (the EINSTEIN-DVT trial) 52 randomized 3,449 patients with acute deep vein thrombosis to rivaroxaban or enoxaparin (Lovenox) overlapped with warfarin or another vitamin K antagonist in the usual manner. No difference was noted between the treatments in the rate of recurrence of VTE or of major bleeding. Of note, patients randomized to rivaroxaban received 15 mg twice a day for the first 3 weeks of treatment and then 20 mg per day for the remainder of their therapy and did not require parenteral anticoagulant overlap.
The long-awaited promise of easier-to-use oral anticoagulants for the treatment of VTE is drawing near and has the potential to revolutionize the treatment of this common disorder. In the meantime, close monitoring of warfarin and careful patient education regarding its use are essential. And even with the development of new drugs in the future, it is still imperative that patients with acute VTE receive the correct length of anticoagulation treatment, are prescribed stockings to prevent postthrombotic syndrome, and are updated on routine cancer screening.
Deep vein thrombosis and pulmonary embolism are collectively referred to as venous thromboembolic (VTE) disease. They affect approximately 100,000 to 300,000 patients per year in the United States.1 Although patients with deep vein thrombosis can be treated as outpatients, many are admitted for the initiation of anticoagulation. Initial anticoagulation usually requires the overlap of a parenteral anticoagulant (unfractionated heparin, low-molecular-weight heparin [LMWH] or fondaparinux) with warfarin for a minimum of 5 days and until the international normalized ratio (INR) of the prothrombin time is above 2.0 for at least 24 hours.2
Three clinical issues need to be addressed after the initiation of anticoagulation for VTE:
- Determination of the length of anticoagulation with the correct anticoagulant
- Prevention of postthrombotic syndrome
- Appropriate screening for occult malignancy.
HOW LONG SHOULD VTE BE TREATED?
The duration of anticoagulation has been a matter of debate.
The risk of recurrent VTE appears related to clinical risk factors that a patient has at the time of the initial thrombotic event. An epidemiologic study3 found that patients with VTE treated for approximately 6 months had a low rate of recurrence (0% at 2 years of follow-up) if surgery was the risk factor. The risk climbed to 9% if the risk factor was nonsurgical and to 19% if there were no provoking risk factors.
The likelihood of VTE recurrence and therefore the recommended duration of treatment depend on whether the VTE event was provoked, cancer-related, recurrent, thrombophilia-related, or idiopathic. We address each of these scenarios below.
HOW LONG TO TREAT PROVOKED VTE
A VTE event is considered provoked if the patient had a clear inciting risk factor. As defined in various clinical trials, these risk factors include:
- Hospitalization with confinement to bed for 3 or more consecutive days in the last 3 months
- Surgery or general anesthesia in the last 3 months
- Immobilization for more than 7 days, regardless of the cause
- Trauma in the last 3 months
- Pregnancy
- Use of an oral contraceptive, regardless of which estrogen or progesterone analogue it contains
- Travel for more than 4 hours
- Recent childbirth.
However, the trials that tested different lengths of anticoagulation have varied markedly in how they defined provoked deep vein thrombosis.4–7
Recommendation: Warfarin or equivalent for 3 months
The American College of Chest Physicians (ACCP) recommends 3 months of anticoagulation with warfarin or another vitamin K antagonist for patients with VTE secondary to a transient (reversible) risk factor,2 and we agree.
HOW LONG TO TREAT CANCER-RELATED VTE
Patients with cancer are at higher risk of developing VTE. Furthermore, in one study,9 compared with other patients with VTE, patients with cancer were three times more likely to have another episode of VTE, with a cumulative rate of recurrence at 1 year of 21% vs 7%. Cancer patients were also twice as likely to suffer major bleeding complications while on anticoagulation.9
Warfarin is a difficult drug to manage because it has many interactions with foods, diseases, and other drugs. These difficulties are amplified in many cancer patients during chemotherapy.
Warfarin was compared with a LMWH in four randomized trials in cancer patients, and a meta-analysis10 found a 50% relative reduction in the rates of recurrent deep vein thrombosis and pulmonary embolism with LMWH treatment. These results were driven primarily by the CLOT trial (Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer),11 which showed an 8% absolute risk reduction (number needed to treat 13) without an increase in major bleeding when cancer-related VTE was treated with an LMWH—ie, dalteparin (Fragmin)—for 6 months compared with warfarin.
Current thinking suggests that VTE should be treated until the cancer is resolved. However, this hypothesis has not been adequately tested, and consequently, the ACCP gives it only a level 1C recommendation.2 The largest of the four trials comparing warfarin and an LMWH lasted only 6 months. The safety of extending LMWH treatment beyond 6 months is currently unknown but is under investigation (clinicaltrials.gov identifier NCT00942968).
Recommendation: LMWH therapy for at least 6 months
The ACCP guidelines recommend LMWH therapy for 3 to 6 months, followed by warfarin or another vitamin K antagonist or continued LMWH treatment until the cancer is resolved.2
The National Comprehensive Cancer Network guidelines recommend an LMWH for 6 months as monotherapy and indefinite anticoagulation if the cancer is still active.12
The American Society of Clinical Oncology guidelines recommend an LMWH for at least 6 months and indefinite anticoagulant therapy for selected patients with active cancer.13
We agree that patients with active cancer should receive an LMWH for at least 6 months and indefinite anticoagulation until the cancer is resolved.
In our experience, many patients are reluctant to give themselves the daily injections that LMWH therapy requires, and so they need to be well-informed about the marked decrease in VTE recurrence with this less-convenient and more-expensive therapy. Many patients face insurance barriers to cover the cost of LMWH therapy; however, careful attention to preauthorization can usually overcome this obstacle.
HOW LONG TO TREAT RECURRENT VTE
It makes clinical sense that patients who have a second VTE event should be treated indefinitely. This theory was tested in a randomized clinical trial14 in which patients with provoked or unprovoked VTE were randomized after their second event to receive anticoagulation for 6 months vs indefinitely.
After 4 years of follow-up, the recurrence rate was 21% in patients assigned to 6 months of treatment and only 3% in patients who continued anticoagulation throughout the trial. On the other hand, major hemorrhage occurred in 3% of patients treated for 6 months and in 9% in patients who continued anticoagulation indefinitely.
Of note, most of the patients in this trial had unprovoked (idiopathic) VTE, so the results should not be extrapolated to patients with provoked VTE, who accounted for only 20% of the study population.14
Recommendation: Long-term anticoagulation
We agree with the ACCP recommendation2 that patients who have had a second episode of unprovoked VTE should receive long-term anticoagulation. Because of a lack of data, the duration of therapy for patients with a second episode of provoked VTE should be individualized.
HOW LONG TO TREAT THROMBOPHILIA-RELATED VTE
Inherited thrombophilias
Patients with VTE that is not related to a clear provoking risk factor or cancer frequently have testing to evaluate for a hypercoagulable state. This workup traditionally includes the most common inherited thrombophilias for gene mutations for factor V and prothrombin as well as for deficiencies in protein C, protein S, antithrombin and the acquired antiphospholipid syndrome.
The key questions that should be asked prior to embarking on this workup are:
- Will the results change the length of therapy for the patient?
- Will testing the patient help with genetic counseling and possible testing of family members?
- Will the results change the targeted INR range for warfarin or other vitamin K antagonist therapy?
Patients with inherited thrombophilia have a greater risk of developing an initial VTE event; however, these tests do not help predict the recurrence of VTE in patients with established disease more than clinical risk factors do. A prospective study demonstrated this by looking at the effect of thrombophilia and clinical factors on the recurrence of venous thrombosis and found that inherited prothrombotic abnormalities do not appear to play an important role in the risk of a recurrent event.15 On the other hand, clinical factors, such as whether the first event was idiopathic or provoked, appear more important in determining the duration of anticoagulation therapy.15 A systematic review of the common inherited thrombophilias showed the VTE recurrence rate of patients with factor V Leiden was higher than in patients without the mutation; however, the absolute rates of recurrence were not much different than what would be expected in patients with idiopathic VTE.16
A retrospective study involving a large cohort of families of patients who already had experienced a first episode of either idiopathic or provoked VTE showed high annual risks of recurrent VTE associated with hereditary deficiencies of protein S (8.4%), protein C (6.0%), and antithrombin (10%).17 However, for the more commonly occurring genetic thrombophilias, the factor V Leiden and prothrombin G20210A mutations, family members with either gene abnormality had low rates of VTE, suggesting that testing of relatives of probands is not clinically useful.16
Antiphospholipid syndrome
Antiphospholipid syndrome is an acquired thrombophilia. A patient has thrombotic antiphospholipid syndrome when there is a history of vascular thrombosis in the presence of persistently positive tests (at least 12 weeks apart) for lupus anticoagulants, anticardiolipin antibodies, or anti-beta-2 glycoprotein I. A prospective study of 412 patients with a first episode of VTE found that 15% were positive for anticardiolipin antibody at the end of 6 months of anticoagulation. The risk of recurrent VTE after 4 years was 29% in patients with antibodies and 14% in those without antibodies (relative risk 2.1; 95% confidence interval [CI] 1.3–3.3; P =.0013).18
Recent reviews advise indefinite warfarin anticoagulation in patients with VTE and persistence of antiphospholipid antibodies.19 However, the optimal duration of anticoagulation is uncertain. Until well-designed clinical trials are done, the current general consensus is to anticoagulate these patients indefinitely.20,21 Retrospective studies had suggested that patients with antiphospholipid antibodies required a higher therapeutic INR range; however, this observation was tested in two trials that found no difference in thromboembolic rates when patients were randomized to an INR of 2.0–3.0 vs 3.1–4.0,22 or 2.0–3.0 vs 3.0–4.5.23
No formal recommendations
In the absence of strong evidence, the ACCP guidelines do not include a recommendation on the duration of anticoagulation treatment specific to inherited thrombophilias. We believe that clinical factors are more important than inherited thrombophilias for deciding the duration of anticoagulation, and that testing is almost never indicated or useful. However, patients with antiphospholipid syndrome are at high risk of recurrence, and it is our practice to anticoagulate these patients indefinitely.
HOW LONG TO TREAT UNPROVOKED (IDIOPATHIC) VTE
A VTE event is thought to be idiopathic if it occurs without a clearly identified provoking factor.
In an observational study,3 patients with oral contraceptive use, transient illness, immobilization, or a history of travel had an 8.8% risk of recurrence vs 19.4% in patients with unprovoked VTE. The meta-analysis discussed above (Table 1)8 also shows that patients with these nonsurgical risk factors have a lower rate of recurrence than patients with idiopathic VTE.
The high rate of recurrence of idiopathic VTE (4% to 27% after 3 months of anticoagulation24–26) suggests that a longer duration of treatment is reasonable. However, increasing the length of therapy from 3 to 12 months delays but does not prevent recurrence, the risk of which begins to accumulate once anticoagulation is stopped.24,25
Three promising strategies to identify subgroups of patients with idiopathic VTE who are at highest risk of recurrence and who would benefit the most from prolonged anticoagulation are d-dimer testing, evaluation for residual vein thrombosis in patients who present with a deep vein thrombosis, and clinical prediction rules.
d-dimer testing
d-dimer is a degradation product of fibrin and is an indirect marker of residual thrombosis.30
In a systematic review of patients with a first episode of unprovoked VTE,31 a normal d-dimer concentration at the end of at least 3 months of anticoagulation was associated with a 3.5% annual risk of recurrence, whereas an elevated d-dimer level at that time was associated with an annual risk of 8.9%. These results were confirmed in a systematic review of individual patient data.32
In a randomized trial,28 patients with an idiopathic VTE event who received anticoagulation for at least 3 months had their d-dimer level measured 1 month after cessation of treatment. Those with an elevated level were randomized to either resume anticoagulation or not. Patients who resumed anticoagulation had an annual recurrence rate of 2%; however, those who were allocated not to restart anticoagulation had a recurrence rate of 10.9% per year. There was no difference in the rate of major bleeding between the two groups. Patients in this clinical trial who had a normal d-dimer level did not restart anticoagulation and had an annual recurrence rate of 4.4%.
Evaluation for residual thrombosis
A randomized trial34 in patients with both provoked and idiopathic deep vein thrombosis showed a reduction in recurrence when those who had residual vein thrombosis were given extended anticoagulation. In the subset of patients whose deep vein thrombosis was idiopathic, the recurrence rate was 17% per year when treatment lasted only 3 months and 10% when it was extended for up to 1 year.
Another trial35 randomized patients with provoked and idiopathic deep vein thrombosis to receive anticoagulation for the usual duration or to continue treatment until recanalization of the residual thrombus was demonstrated on follow-up Doppler ultrasonography. Patients who received this ultrasonography-tailored treatment had a lower rate of recurrence of VTE; however, the absolute reductions in recurrence rates cannot be calculated from this report for patients with idiopathic deep vein thrombosis.
A prospective observational study36 of the predictive value of d-dimer status and residual vein thrombus found that only d-dimer was an independent risk factor for recurrent VTE after vitamin K antagonist withdrawal.
A clinical prediction rule: ‘Men and HERDOO2’
A promising tool for predicting if a patient is at low risk of recurrent VTE after the first episode of proximal deep vein thrombosis or pulmonary embolism is known by the mnemonic device “Men and HERDOO2.” It is based on data prospectively derived by Rodger et al37 to identify patients with less than a 3% annual risk of recurrent VTE after their first event of idiopathic proximal deep vein thrombosis or pulmonary embolism. Risk factors for recurrent VTE were male sex (the “men” of “Men and HERDOO2”), signs of postthrombotic syndrome, including hyperpigmentation of the lower extremities, edema or redness of either leg, a d-dimer level > 250 μg/L, obesity (body mass index > 30 kg/m2, and older age (> 65 years).
Overall, one-fourth of the population were women with no risk factors or one risk factor, and their risk of recurrence was 1.6% per year. Men and women who had two or more risk factors for postthrombotic syndrome (hyperpigmentation, edema, or redness), elevated d-dimer, obesity, or older age were predicted to be at higher risk of recurrent VTE. Patients such as this should be considered for indefinite anticoagulation.
Ideally, clinical prediction rules should be validated in a separate group of patients before they are used routinely in practice,38 and this clinical prediction rule is currently being tested in the REVERSE II study. If the results are consistent, this will be an easy-to-use tool to help identify patients who likely can safely stop anticoagulation therapy after 3 to 6 months (clinicaltrials.gov Identifier: NCT00967304).
The location of the thrombosis also influences the likelihood of recurrence. Patients with isolated distal (calf) deep vein thrombosis are less likely to suffer recurrent VTE than those who present with proximal deep vein thrombosis. However, trials focusing specifically on the precise subset of idiopathic isolated distal deep vein thrombosis are lacking. In a randomized trial39 comparing 6 vs 12 weeks of anticoagulation for isolated distal deep vein thrombosis and 12 vs 24 weeks for proximal deep vein thrombosis, the annual rates of recurrence after 12 weeks of treatment were approximately 3.4% for isolated distal and 8.1% for proximal deep vein thrombosis.39
Recommendation: At least 3 months of warfarin or equivalent
We agree with the ACCP recommendation2 that patients with unprovoked VTE should receive at least 3 months of anticoagulation with a vitamin K antagonist.
If the patient has no risk factors for bleeding and good anticoagulant monitoring is achievable, we agree with long-term anticoagulation for proximal unprovoked deep vein thrombosis or pulmonary embolism, and 3 months of therapy for isolated distal unprovoked deep vein thrombosis.
Patient preferences and the risk of recurrence vs the risk of bleeding should be discussed with patients when contemplating indefinite anticoagulation.
If testing is being considered to assist in the decision to prescribe indefinite anticoagulation, we prefer using d-dimer levels rather than ultrasonography to detect residual venous thrombosis because of its ease of use and the strength of the current evidence.
PREVENTING POSTTHROMBOTIC SYNDROME
The postthrombotic (postphlebitic) syndrome is a chronic and burdensome consequence of deep vein thrombosis that occurs despite anticoagulation therapy. It is estimated to affect 23% to 60% of patients and typically manifests in the first 2 years.40 It is not only costly in clinical terms, with decreased quality of life for the patient, but health care expenditures have been estimated to range from $400 per year in a Brazilian study to $7,000 per year in a US study.40
Typical symptoms include leg pain, heaviness, swelling, and cramping. In severe cases, chronic venous ulcers can occur and are difficult to treat.41
The definition of postthrombotic syndrome has been unclear over the years, and six different scales that measure signs and symptoms have been reported.42
The Villalta scale has been proposed by the International Society of Thrombosis and Hemostasis as a diagnostic standard to define postthrombotic syndrome.42 This validated scale is based on five clinical symptoms, six clinical signs, and the presence or absence of venous ulcers. Each clinical symptom and sign is scored as mild (1 point), moderate (2 points), or severe (3 points). Symptoms include pain, cramps, heaviness, paresthesia, and pruritus; the six clinical signs are pretibial edema, skin induration, hyperpigmentation, redness, venous ectasia, and pain on calf compression.
According to the International Society of Thrombosis and Hemostasis, postthrombotic syndrome is present if the Villalta score is 5 or greater or if a venous ulcer is present in a leg with previous deep vein thrombosis. Further, using the Villalta scale, postthrombotic syndrome can be categorized as mild (score 5–9), moderate (10–14), or severe (≥ 15).
A limitation of the Villalta scale is that the presence or absence of a venous ulcer has not been assigned a score. Since a venous ulcer requires more aggressive measures, the society defines postthrombotic syndrome as severe if venous ulcers are present.42
Acute symptoms of deep vein thrombosis may take months to resolve and, indeed, acute symptoms may transition to chronic symptoms without a symptom-free interval. It is recommended that postthrombotic syndrome not be diagnosed before 3 months to avoid inappropriately attributing acute symptoms and signs of deep vein thrombosis to the postthrombotic syndrome.42
Studies of stockings
A systematic review of three randomized trials44 concluded that elastic compression stockings reduce the risk of postthrombotic syndrome (any severity) from 43% to 20% and severe postthrombotic syndrome from 15% to 7%.43
The first of these trials44 randomized patients soon after the diagnosis of deep vein thrombosis to receive made-to-order compression stockings that were rated at 30 to 40 mm Hg or to be in a control group that did not receive stockings. The second trial45 randomized patients 1 year after the index event of deep vein thrombosis to receive 20- to 30-mm Hg stockings or stockings that were two sizes too large (the control group). The third study46 randomly allocated patients to receive “off-the-shelf” stockings (30–40 mm Hg) or no stockings. Each study used its own definition of postthrombotic syndrome.
Although these studies strongly suggest compression stockings prevent postthrombotic syndrome, several methodologic issues remain:
- A standard definition of postthrombotic syndrome was not used
- The amount of compression varied between studies
- The studies were not blinded.
Lack of blinding becomes most significant when an outcome is based on subjective findings, like the symptoms that make up a large part of the diagnosis of postthrombotic syndrome.
The SOX trial, currently under way, is designed to address these methodologic issues and should be completed in 2012 (clinicaltrials.gov Identifier: NCT00143598).
Recommendation: Stockings for at least 2 years
We agree with the ACCP recommendation that a patient who has had a symptomatic proximal deep vein thrombosis should wear an elastic compression stocking with an ankle pressure gradient of 30 to 40 mm Hg as soon as possible after starting anticoagulant therapy and continuing for a minimum of 2 years.2
SCREENING FOR OCCULT MALIGNANCY
VTE can be the first manifestation of cancer.
French physician Armand Trousseau, in the 1860s, was the first to describe disseminated intravascular coagulation closely associated with adenocarcinoma. Ironically, several years later, after suffering for weeks from abdominal pain, he declared to one of his students that he had developed thrombosis, and he died of gastric cancer shortly thereafter.47
Since cancer is a well-known risk factor for VTE, it is logical to screen for cancer as an explanation for an idiopathic VTE event.48 To make an informed decision, one needs to understand the rate of occult cancer at the time VTE is diagnosed, the risk of future development of cancer, and the utility of extensive cancer screening.
The clinical efficacy, side effects, and cost-effectiveness of cancer screening in patients with idiopathic VTE are unknown. However, a systematic review47 of 34 studies found that, in patients with idiopathic VTE, cancer was diagnosed within 1 month in 6.1%, within 6 months in 8.6%, and within 1 year in 10.0% (95% CI 8.6–11.3).
A subset of studies compared two strategies for screening soon after the diagnosis of idiopathic VTE: a strategy limited to the history, physical examination, basic blood work, and chest radiography vs an extensive screening strategy that also included serum tumor markers or abdominal ultrasonography or computed tomography. Limited screening detected 49% of the prevalent cancers; extensive screening increased this rate to 70%. Stated another way, the detection rate for prevalent cancers was 5% with limited screening and 7% with extensive screening soon after the diagnosis of idiopathic VTE.47
Patients with idiopathic VTE had higher rates of cancer within 1 month of diagnosis than patients with provoked VTE (6.1% vs 1.9%), and this difference persisted at 1 year (10.0% vs 2.6%).47
Recommendation: Individualized cancer screening
Patients with idiopathic VTE have a significant risk of occult cancer within the first year after diagnosis, and cancer screening should be considered. Our practice for patients with idiopathic VTE is to perform a history and physical examination and ensure that the patient is up to date on age- and sex-specific cancer screening.
The use of additional imaging or biomarkers should be discussed with patients so they can balance the risks (radiation and potential false-positive results with their downstream consequences), costs, and potential benefits, given the lack of proven survival benefit or cost-effectiveness.
ORAL ANTICOAGULANT MANAGEMENT
Warfarin’s multiple interactions, along with the need for INR monitoring, make it a difficult medication to manage.
The Joint Commission, the US organization for health service accreditation and certification, has defined National Patient Safety Goals and quality measures for the management of anticoagulation.49 Organized anticoagulation management services, dosing algorithms, and patient self-testing using capillary INR meters or patient self-management of warfarin were recommended as tools to improve the time patients spend in the therapeutic INR range.50
Two new oral anticoagulants
The limitations of warfarin have stimulated the search for newer oral anticoagulants that do not require laboratory monitoring or have as many diet and drug interactions.
Two trials have been published with experimental oral anticoagulants that had similar efficacy and safety as warfarin in the treatment of VTE.
The study of dabigatran (Pradaxa) vs warfarin in the treatment of acute VTE (the RECOVER trial)51 randomized 2,539 patients with acute VTE to receive the oral direct thrombin inhibitor dabigatran or warfarin for approximately 6 months. Of note, each treatment group received a median of 6 days of heparin, LMWH, or fondaparinux at the beginning of blinded therapy. The rates of recurrent VTE and major bleeding were similar between the treatment arms, and overall bleeding was less with dabigatran. Dabigatran was approved in the United States in October 2010 for stroke prevention in atrial fibrillation but has yet to be approved for the treatment of VTE pending further study (clinicaltrials.gov Identifier: NCT00680186).
A study of oral rivaroxaban (Xarelto) for symptomatic VTE (the EINSTEIN-DVT trial) 52 randomized 3,449 patients with acute deep vein thrombosis to rivaroxaban or enoxaparin (Lovenox) overlapped with warfarin or another vitamin K antagonist in the usual manner. No difference was noted between the treatments in the rate of recurrence of VTE or of major bleeding. Of note, patients randomized to rivaroxaban received 15 mg twice a day for the first 3 weeks of treatment and then 20 mg per day for the remainder of their therapy and did not require parenteral anticoagulant overlap.
The long-awaited promise of easier-to-use oral anticoagulants for the treatment of VTE is drawing near and has the potential to revolutionize the treatment of this common disorder. In the meantime, close monitoring of warfarin and careful patient education regarding its use are essential. And even with the development of new drugs in the future, it is still imperative that patients with acute VTE receive the correct length of anticoagulation treatment, are prescribed stockings to prevent postthrombotic syndrome, and are updated on routine cancer screening.
- Spencer FA, Emery C, Lessard D, et al. The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 2006; 21:722–727.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523–526.
- Schulman S, Lockner D, Juhlin-Dannfelt A. The duration of oral anticoagulation after deep vein thrombosis. A randomized study. Acta Med Scand 1985; 217:547–552.
- Optimum duration of anticoagulation for deep-vein thrombosis and pulmonary embolism. Research Committee of the British Thoracic Society. Lancet 1992; 340:873–876.
- Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. Duration of Anticoagulation Trial Study Group. N Engl J Med 1995; 332:1661–1665.
- Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743–749.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med 2010; 170:1710–1716.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hull RD, Pineo GF, Brant RF, et al; LITE Trial Investigators. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology, Venous Thromboembolic Disease. http://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed August 3, 2011.
- Lyman GH, Khorana AA, Falanga A, et al; American Society of Clinical Oncology. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Schulman S, Granqvist S, Holmström M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–2485.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost 2009; 101:93–99.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med 1998; 104:332–338.
- Derksen RH, de Groot PG. Towards evidence-based treatment of thrombotic antiphospholipid syndrome. Lupus 2010; 19:470–474.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Fonseca AG, D’Cruz DP. Controversies in the antiphospholipid syndrome: can we ever stop warfarin? J Autoimmune Dis 2008; 5:6.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3:848–853.
- Agnelli G, Prandoni P, Becattini C, et al; Warfarin Optimal Duration Italian Trial Investigators. Extended oral anticoagulant therapy after a first episode of pulmonary embolism. Ann Intern Med 2003; 139:19–25.
- Agnelli G, Prandoni P, Santamaria MG, et al; Warfarin Optimal Duration Italian Trial Investigators. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Kearon C, Ginsberg JS, Kovacs MJ, et al; Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–639.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Ridker PM, Goldhaber SZ, Glynn RJ. Low-intensity versus conventional-intensity warfarin for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:2164–2167.
- Bockenstedt P. D-dimer in venous thromboembolism. N Engl J Med 2003; 349:1203–1204.
- Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008; 149:481–490,W94.
- Douketis J, Tosetto A, Marcucci M, et al. Patient-level metaanalysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med 2010; 153:523–531.
- Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med 2002; 137:955–960.
- Siragusa S, Malato A, Anastasio R, et al. Residual vein thrombosis to establish duration of anticoagulation after a first episode of deep vein thrombosis: the Duration of Anticoagulation based on Compression UltraSonography (DACUS) study. Blood 2008; 112:511–515.
- Prandoni P, Prins MH, Lensing AW, et al; AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep venous thrombosis: a randomized trial. Ann Intern Med 2009; 150:577–585.
- Cosmi B, Legnani C, Cini M, Guazzaloca G, Palareti G. D-dimer levels in combination with residual venous obstruction and the risk of recurrence after anticoagulation withdrawal for a first idiopathic deep vein thrombosis. Thromb Haemost 2005; 94:969–974.
- Rodger MA, Kahn SR, Wells PS, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ 2008; 179:417–426.
- McGinn TG, Guyatt GH, Wyer PC, Naylor CD, Stiell IG, Richardson WS. Users’ guides to the medical literature: XXII: how to use articles about clinical decision rules. Evidence-Based Medicine Working Group. JAMA 2000; 284:79–84.
- Pinede L, Ninet J, Duhaut P, et al; Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103:2453–2460.
- Ashrani AA, Heit JA. Incidence and cost burden of postthrombotic syndrome. J Thromb Thrombolysis 2009; 28:465–476.
- Kahn SR, Shrier I, Julian JA, et al. Determinants and time course of the postthrombotic syndrome after acute deep venous thrombosis. Ann Intern Med 2008; 149:698–707.
- Kahn SR, Partsch H, Vedantham S, Prandoni P, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of post-thrombotic syndrome of the leg for use in clinical investigations: a recommendation for standardization. J Thromb Haemost 2009; 7:879–883.
- Kolbach DN, Sandbrink MW, Hamulyak K, Neumann HA, Prins MH. Non-pharmaceutical measures for prevention of post-thrombotic syndrome. Cochrane Database Syst Rev 2004;CD004174.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet 1997; 349:759–762.
- Ginsberg JS, Hirsh J, Julian J, et al. Prevention and treatment of postphlebitic syndrome: results of a 3-part study. Arch Intern Med 2001; 161:2105–2109.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the post-thrombotic syndrome: a randomized, controlled trial. Ann Intern Med 2004; 141:249–256.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008; 149:323–333.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Kaatz S. Impact on patient care: patient case through the continuum of care. J Thromb Thrombolysis 2010; 29:167–170.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Schulman S, Kearon C, Kakkar AK, et al; for the RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2452.
- The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363;2499–2510.
- Spencer FA, Emery C, Lessard D, et al. The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 2006; 21:722–727.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ; American College of Chest Physicians. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):454S–545S.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523–526.
- Schulman S, Lockner D, Juhlin-Dannfelt A. The duration of oral anticoagulation after deep vein thrombosis. A randomized study. Acta Med Scand 1985; 217:547–552.
- Optimum duration of anticoagulation for deep-vein thrombosis and pulmonary embolism. Research Committee of the British Thoracic Society. Lancet 1992; 340:873–876.
- Schulman S, Rhedin AS, Lindmarker P, et al. A comparison of six weeks with six months of oral anticoagulant therapy after a first episode of venous thromboembolism. Duration of Anticoagulation Trial Study Group. N Engl J Med 1995; 332:1661–1665.
- Kearon C, Ginsberg JS, Anderson DR, et al. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743–749.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med 2010; 170:1710–1716.
- Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002; 100:3484–3488.
- Hull RD, Pineo GF, Brant RF, et al; LITE Trial Investigators. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Lee AY, Levine MN, Baker RI, et al; Randomized Comparison of Low-Molecular-Weight Heparin versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients with Cancer (CLOT) Investigators. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology, Venous Thromboembolic Disease. http://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed August 3, 2011.
- Lyman GH, Khorana AA, Falanga A, et al; American Society of Clinical Oncology. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Schulman S, Granqvist S, Holmström M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA 2005; 293:2352–2361.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA 2009; 301:2472–2485.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost 2009; 101:93–99.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med 1998; 104:332–338.
- Derksen RH, de Groot PG. Towards evidence-based treatment of thrombotic antiphospholipid syndrome. Lupus 2010; 19:470–474.
- Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006; 295:1050–1057.
- Fonseca AG, D’Cruz DP. Controversies in the antiphospholipid syndrome: can we ever stop warfarin? J Autoimmune Dis 2008; 5:6.
- Crowther MA, Ginsberg JS, Julian J, et al. A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome. N Engl J Med 2003; 349:1133–1138.
- Finazzi G, Marchioli R, Brancaccio V, et al. A randomized clinical trial of high-intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS). J Thromb Haemost 2005; 3:848–853.
- Agnelli G, Prandoni P, Becattini C, et al; Warfarin Optimal Duration Italian Trial Investigators. Extended oral anticoagulant therapy after a first episode of pulmonary embolism. Ann Intern Med 2003; 139:19–25.
- Agnelli G, Prandoni P, Santamaria MG, et al; Warfarin Optimal Duration Italian Trial Investigators. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Kearon C, Ginsberg JS, Kovacs MJ, et al; Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity warfarin therapy with conventional-intensity warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631–639.
- Palareti G, Cosmi B, Legnani C, et al; PROLONG Investigators. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med 2006; 355:1780–1789.
- Ridker PM, Goldhaber SZ, Glynn RJ. Low-intensity versus conventional-intensity warfarin for prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:2164–2167.
- Bockenstedt P. D-dimer in venous thromboembolism. N Engl J Med 2003; 349:1203–1204.
- Verhovsek M, Douketis JD, Yi Q, et al. Systematic review: D-dimer to predict recurrent disease after stopping anticoagulant therapy for unprovoked venous thromboembolism. Ann Intern Med 2008; 149:481–490,W94.
- Douketis J, Tosetto A, Marcucci M, et al. Patient-level metaanalysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med 2010; 153:523–531.
- Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med 2002; 137:955–960.
- Siragusa S, Malato A, Anastasio R, et al. Residual vein thrombosis to establish duration of anticoagulation after a first episode of deep vein thrombosis: the Duration of Anticoagulation based on Compression UltraSonography (DACUS) study. Blood 2008; 112:511–515.
- Prandoni P, Prins MH, Lensing AW, et al; AESOPUS Investigators. Residual thrombosis on ultrasonography to guide the duration of anticoagulation in patients with deep venous thrombosis: a randomized trial. Ann Intern Med 2009; 150:577–585.
- Cosmi B, Legnani C, Cini M, Guazzaloca G, Palareti G. D-dimer levels in combination with residual venous obstruction and the risk of recurrence after anticoagulation withdrawal for a first idiopathic deep vein thrombosis. Thromb Haemost 2005; 94:969–974.
- Rodger MA, Kahn SR, Wells PS, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ 2008; 179:417–426.
- McGinn TG, Guyatt GH, Wyer PC, Naylor CD, Stiell IG, Richardson WS. Users’ guides to the medical literature: XXII: how to use articles about clinical decision rules. Evidence-Based Medicine Working Group. JAMA 2000; 284:79–84.
- Pinede L, Ninet J, Duhaut P, et al; Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103:2453–2460.
- Ashrani AA, Heit JA. Incidence and cost burden of postthrombotic syndrome. J Thromb Thrombolysis 2009; 28:465–476.
- Kahn SR, Shrier I, Julian JA, et al. Determinants and time course of the postthrombotic syndrome after acute deep venous thrombosis. Ann Intern Med 2008; 149:698–707.
- Kahn SR, Partsch H, Vedantham S, Prandoni P, Kearon C; Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definition of post-thrombotic syndrome of the leg for use in clinical investigations: a recommendation for standardization. J Thromb Haemost 2009; 7:879–883.
- Kolbach DN, Sandbrink MW, Hamulyak K, Neumann HA, Prins MH. Non-pharmaceutical measures for prevention of post-thrombotic syndrome. Cochrane Database Syst Rev 2004;CD004174.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet 1997; 349:759–762.
- Ginsberg JS, Hirsh J, Julian J, et al. Prevention and treatment of postphlebitic syndrome: results of a 3-part study. Arch Intern Med 2001; 161:2105–2109.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the post-thrombotic syndrome: a randomized, controlled trial. Ann Intern Med 2004; 141:249–256.
- Carrier M, Le Gal G, Wells PS, Fergusson D, Ramsay T, Rodger MA. Systematic review: the Trousseau syndrome revisited: should we screen extensively for cancer in patients with venous thromboembolism? Ann Intern Med 2008; 149:323–333.
- Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005; 293:715–722.
- Kaatz S. Impact on patient care: patient case through the continuum of care. J Thromb Thrombolysis 2010; 29:167–170.
- Ansell J, Hirsh J, Hylek E, Jacobson A, Crowther M, Palareti G; American College of Chest Physicians. Pharmacology and management of the vitamin K antagonists: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):160S–198S.
- Schulman S, Kearon C, Kakkar AK, et al; for the RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2452.
- The EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363;2499–2510.
KEY POINTS
- A low-molecular-weight heparin for at least 6 months is the treatment of choice for cancer-related VTE.
- We recommend 3 months of anticoagulation for VTE caused by a reversible risk factor and indefinite treatment for idiopathic VTE in patients without risk factors for bleeding who can get anticoagulation monitoring.
- Clinical factors are more important in deciding the duration of anticoagulation therapy than evidence of an inherited thrombophilic state.
- Elastic compression stockings reduce the risk of postthrombotic syndrome substantially.
- Patients with idiopathic VTE should have a basic screening for malignancy.