User login
AUDIO: Franchiser hopes to put dermatology ‘back in the hands of the dermatologist’
Dermatology has yet to conquer the cosmetic corner of the specialty. That’s according to Dr. Leslie S. Baumann of the Miami-based Skin Type Solutions, who explains a new franchise model she says will help “put dermatology back in the hands of dermatologists.”
In this interview, Dr. Baumann, who writes the Cosmeceutical Critique column for Skin & Allergy News, explains her new franchise method for selling skin care products in the dermatologist’s office, and why she thinks it will “disrupt” business as usual in the retail skin care marketplace, including for online retailers.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the cosmetic dermatology center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (McGraw-Hill, April 2002), and a book for consumers, “The Skin Type Solution” (Bantam, 2006). She has contributed to the Cosmeceutical Critique column in Skin & Allergy News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” will be published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy,Topix Pharmaceuticals, and Unilever.
On Twitter @whitneymcknight
Dermatology has yet to conquer the cosmetic corner of the specialty. That’s according to Dr. Leslie S. Baumann of the Miami-based Skin Type Solutions, who explains a new franchise model she says will help “put dermatology back in the hands of dermatologists.”
In this interview, Dr. Baumann, who writes the Cosmeceutical Critique column for Skin & Allergy News, explains her new franchise method for selling skin care products in the dermatologist’s office, and why she thinks it will “disrupt” business as usual in the retail skin care marketplace, including for online retailers.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the cosmetic dermatology center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (McGraw-Hill, April 2002), and a book for consumers, “The Skin Type Solution” (Bantam, 2006). She has contributed to the Cosmeceutical Critique column in Skin & Allergy News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” will be published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy,Topix Pharmaceuticals, and Unilever.
On Twitter @whitneymcknight
Dermatology has yet to conquer the cosmetic corner of the specialty. That’s according to Dr. Leslie S. Baumann of the Miami-based Skin Type Solutions, who explains a new franchise model she says will help “put dermatology back in the hands of dermatologists.”
In this interview, Dr. Baumann, who writes the Cosmeceutical Critique column for Skin & Allergy News, explains her new franchise method for selling skin care products in the dermatologist’s office, and why she thinks it will “disrupt” business as usual in the retail skin care marketplace, including for online retailers.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the cosmetic dermatology center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (McGraw-Hill, April 2002), and a book for consumers, “The Skin Type Solution” (Bantam, 2006). She has contributed to the Cosmeceutical Critique column in Skin & Allergy News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” will be published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy,Topix Pharmaceuticals, and Unilever.
On Twitter @whitneymcknight
Master Class: Office evaluation for incontinence
Ten years ago, urodynamics were widely viewed as the gold standard for evaluating urinary incontinence. We often turned to such testing to confirm or reject the findings of our basic evaluation before determining the best type of treatment – especially before proceeding with primary anti-incontinence surgery.
What has emerged in recent years is a body of evidence that tells us otherwise. We now know that urodynamics do not give us all the answers, and that we can be much more judicious with its use.

A good history followed by a thorough physical examination and some office tests often enables us to make sound treatment recommendations without costly and potentially uncomfortable urodynamic testing. The key lies in discerning complicated and uncomplicated cases. For patients deemed to have uncomplicated stress urinary incontinence (SUI) – especially those who have failed conservative management – we can comfortably recommend surgical repair without urodynamic testing.
Identifying uncomplicated SUI
The history is the most important part of the evaluation for incontinence. Every patient who answers “yes” to a basic opening question about whether she has any concerns about bladder control should be asked a series of questions that will enable the physician to fully understand her symptoms, their severity, and their impact on her life and daily activities.
It is critical to determine whether you are dealing with pure SUI, pure urge incontinence (UI), or SUI with a component of UI. Mixed incontinence is quite prevalent. An analysis of recent National Health and Nutrition Examination Survey (NHANES) data showed that of those women reporting incontinence symptoms, almost 50% reported pure SUI, and 34% reported mixed incontinence (J. Urol. 2008;179: 656-61). Other studies similarly have shown prevalence rates of mixed incontinence above 30%.
The International Urogynecological Association (IUGA) recommends the use of validated questionnaires to assess incontinence and the relative contribution of UI and SUI symptoms. Some physicians do find the organized and structured format of questionnaires helpful in their practices. Others have incorporated questions from various tools into history-taking templates on their electronic medical records. Still others have made them part of a mental checklist for history taking.

The short-form version of the Urogenital Distress Inventory (UDI-6), for instance, asks the patient whether she experiences – and how much she is bothered by – the following: frequent urination; leakage related to a feeling of urgency; leakage related to activity, coughing, or sneezing; small amount of leakage; difficulty emptying the bladder; and pain or discomfort in the lower abdominal or genital area.
The Incontinence Impact Questionnaire can be used to further assess the impact of symptoms. The short-form IIQ (the IIQ-7) asks, for instance, about the extent to which urine leakage has affected household chores, physical recreation, social activities, or emotional health.
Since the UDI and IIQ were developed about 20 years ago, at least several other urinary incontinence questionnaires have been developed and validated. Whether or not questionnaires are utilized as official tools, history taking should capture their essence and provide you with enough information to ascertain the type of incontinence, frequency of occurrence, severity, and effect on daily life.
The history also must assess the possibility of voiding dysfunction. Positive responses to questions about nocturia, hesitancy, and the need to immediately revoid, for instance, point toward complicated SUI and the need for further assessment before embarking on surgical treatment for SUI.
Patients who have uncomplicated SUI, on the other hand, will answer negatively to questions about symptoms of predominant urgency, functional impairment, continuous leakage, and/or incomplete emptying. They also will not have had recurrent urinary tract infections or medical conditions that can affect lower urinary tract function (such as neurologic disease and poorly controlled diabetes).
The physical exam
Along with the history, the physical exam is important for identifying complicated SUI and confirming which cases of SUI are truly uncomplicated. Evaluation should include a cough stress test to confirm leakage from the urethra under stress, an assessment of urethral mobility, and an assessment for pelvic organ prolapse.
The cough stress test is usually done with the patient in the supine or semirecumbent lithotomy position. If you strongly suspect stress incontinence but have a negative result, consider the following:
• Make sure the patient has a comfortably full bladder.
• Many women will contract their pelvic floor muscles when coughing to try to avoid leaking. You can apply pressure against the posterior vaginal wall either digitally or with half of the bivalve speculum to keep the patient from activating her muscles.
• The cough test can be performed in the standing position.
Assessing urethral mobility similarly involves simple observation while the patient is in a supine lithotomy position and straining. A Q-tip test or the Pelvic Organ Prolapse Quantification (POP-Q) system may be used, but visualization and palpation also are completely acceptable.
Just as the absence of urethral mobility is a red flag, so is prolapse beyond the hymen. This patient could potentially have urethral kinking, which can mask the severity of SUI or be a source of retention. Either finding the absence of urethral mobility or the presence of POP beyond the hymen moves the case from the uncomplicated to complicated category and signals the need for further evaluation with urodynamics or other tests.
These and other findings for uncomplicated versus complicated SUI are outlined in a committee opinion issued recently by the American College of Obstetricians and Gynecologists and the American Urogynecologic Society (Committee Opinion No. 603, Obstet .Gynecol. 2014;123:1403-7).
As the ACOG-AUGS recommendations point out, urinalysis is part of the minimum work-up for stress incontinence. Measurement of postvoid residual urine volume also becomes important when midurethral sling surgery is being contemplated for uncomplicated SUI. A normal volume rules out potential bladder-emptying abnormalities and provides final assurance that the patient is a good candidate for surgical repair.
Recent research on urodynamics
Evidence that a simple office-based incontinence evaluation without preoperative urodynamic testing is appropriate for uncomplicated predominant SUI comes largely from two recent randomized noninferiority trials.
One of these trials – a study from the Urinary Incontinence Treatment Network in the United States, known as the VALUE trial – randomized 630 women with uncomplicated SUI to pretreatment work-up with or without urodynamics. Treatment success at 12 months was similar for the two groups (approximately 77%).
This finding, the authors wrote, suggests that for women with uncomplicated SUI, a “basic office evaluation” (i.e., a positive provocative stress test, a normal postvoiding residual volume, an assessment or urethral mobility, and a negative urinalysis) is a “sufficient preoperative work-up” (N. Engl. J. Med. 2012;366:1987-97).
The diagnosis of SUI as made by office evaluation was confirmed in 97% of women who underwent urodynamic testing, and while there were some adjustments in diagnosis after urodynamics, there were no major changes in treatment decision making after the testing. Approximately 93% of women in both groups underwent midurethral sling surgery.
The second trial, a Dutch study, focused on women who had already undergone urodynamic testing and been shown to have discordant findings on urodynamics and their history and clinical exam. The women – all of whom had uncomplicated predominant SUI – were randomized to undergo immediate midurethral sling surgery or receive individually tailored treatment (including sling surgery, behavioral and physical therapy, pessary, and anticholinergics).
At 1 year, there was no clinically significant difference between the two groups in patients’ assessment of their symptoms as measured by the UDI. The authors concluded that “an immediate midurethral sling operation is not inferior to individually tailored treatment based on urodynamic findings” and that “urodynamics should no longer be advised routinely before primary surgery in these patients” (Obstet. Gynecol. 2013;121:999-1008).
When urge incontinence is involved
Urodynamic testing was never believed to be perfect, but these and other studies have highlighted its imperfections. Urodynamics creates an artificial condition in the bladder, in effect, and some of the findings will involve artifact. A systematic review of studies that compared diagnoses based on symptoms with diagnoses after urodynamic investigation was interesting in this regard; while the review did not assess impact on treatment, it showed that there is poor agreement between clinical symptoms and urodynamic-based diagnoses (Neurourol. Urodyn. 2011;30:495-502).
Certainly, women with complicated SUI – as well as women who have recurrent SUI after a prior surgical intervention – require further assessment, which likely includes multichannel urodynamic testing.
Urodynamics also can play a useful role in decision making and counseling for some patients whose incontinence is predominately SUI, but is believed to involve some degree of urinary urgency. Patients with mixed urinary incontinence fare worse after midurethral sling procedures compared with patients who have SUI alone, and I counsel my patients accordingly, emphasizing that the sling will not address aspects of their incontinence related to urgency. When I sense that a patient may have unreasonably high expectations for surgery, urodynamic testing can provide some perspective on possible postoperative outcomes.
Treatment for UI or overactive bladder often may be initiated after simple office-based evaluation, just as with SUI. The goal, similarly, is to discern relatively uncomplicated or straightforward cases from complicated ones. Urologic, medical, and neurologic histories should be obtained, for instance, and retention issues (which can aggravate UI) should be ruled out through the measurement of postvoid residual urine volume.
Just as with SUI, evaluation of suspected UI more often than not involves careful history taking and clinical probing. A voiding diary can sometimes be helpful; I send patients home with such a tool when the history is inconclusive or I suspect behavioral (excessive fluid intake) or functional issues as significant factors in bladder control.
It is important to keep in mind that patients with severe SUI may have urinary frequency as a learned response. Such patients appear to have overactive bladder in addition to SUI, but may actually be urinating frequently because they’ve learned that doing so results in less leakage. In our practice we’ve observed that patients with a learned response tend not to have nocturia, while those with overactive bladder do report nocturia.
Dr. Culbertson had no relevant financial disclosures.
Dr. Culbertson is a professor in the department of obstetrics and gynecology at the University of Chicago.
Ten years ago, urodynamics were widely viewed as the gold standard for evaluating urinary incontinence. We often turned to such testing to confirm or reject the findings of our basic evaluation before determining the best type of treatment – especially before proceeding with primary anti-incontinence surgery.
What has emerged in recent years is a body of evidence that tells us otherwise. We now know that urodynamics do not give us all the answers, and that we can be much more judicious with its use.
A good history followed by a thorough physical examination and some office tests often enables us to make sound treatment recommendations without costly and potentially uncomfortable urodynamic testing. The key lies in discerning complicated and uncomplicated cases. For patients deemed to have uncomplicated stress urinary incontinence (SUI) – especially those who have failed conservative management – we can comfortably recommend surgical repair without urodynamic testing.
Identifying uncomplicated SUI
The history is the most important part of the evaluation for incontinence. Every patient who answers “yes” to a basic opening question about whether she has any concerns about bladder control should be asked a series of questions that will enable the physician to fully understand her symptoms, their severity, and their impact on her life and daily activities.
It is critical to determine whether you are dealing with pure SUI, pure urge incontinence (UI), or SUI with a component of UI. Mixed incontinence is quite prevalent. An analysis of recent National Health and Nutrition Examination Survey (NHANES) data showed that of those women reporting incontinence symptoms, almost 50% reported pure SUI, and 34% reported mixed incontinence (J. Urol. 2008;179: 656-61). Other studies similarly have shown prevalence rates of mixed incontinence above 30%.
The International Urogynecological Association (IUGA) recommends the use of validated questionnaires to assess incontinence and the relative contribution of UI and SUI symptoms. Some physicians do find the organized and structured format of questionnaires helpful in their practices. Others have incorporated questions from various tools into history-taking templates on their electronic medical records. Still others have made them part of a mental checklist for history taking.
The short-form version of the Urogenital Distress Inventory (UDI-6), for instance, asks the patient whether she experiences – and how much she is bothered by – the following: frequent urination; leakage related to a feeling of urgency; leakage related to activity, coughing, or sneezing; small amount of leakage; difficulty emptying the bladder; and pain or discomfort in the lower abdominal or genital area.
The Incontinence Impact Questionnaire can be used to further assess the impact of symptoms. The short-form IIQ (the IIQ-7) asks, for instance, about the extent to which urine leakage has affected household chores, physical recreation, social activities, or emotional health.
Since the UDI and IIQ were developed about 20 years ago, at least several other urinary incontinence questionnaires have been developed and validated. Whether or not questionnaires are utilized as official tools, history taking should capture their essence and provide you with enough information to ascertain the type of incontinence, frequency of occurrence, severity, and effect on daily life.
The history also must assess the possibility of voiding dysfunction. Positive responses to questions about nocturia, hesitancy, and the need to immediately revoid, for instance, point toward complicated SUI and the need for further assessment before embarking on surgical treatment for SUI.
Patients who have uncomplicated SUI, on the other hand, will answer negatively to questions about symptoms of predominant urgency, functional impairment, continuous leakage, and/or incomplete emptying. They also will not have had recurrent urinary tract infections or medical conditions that can affect lower urinary tract function (such as neurologic disease and poorly controlled diabetes).
The physical exam
Along with the history, the physical exam is important for identifying complicated SUI and confirming which cases of SUI are truly uncomplicated. Evaluation should include a cough stress test to confirm leakage from the urethra under stress, an assessment of urethral mobility, and an assessment for pelvic organ prolapse.
The cough stress test is usually done with the patient in the supine or semirecumbent lithotomy position. If you strongly suspect stress incontinence but have a negative result, consider the following:
• Make sure the patient has a comfortably full bladder.
• Many women will contract their pelvic floor muscles when coughing to try to avoid leaking. You can apply pressure against the posterior vaginal wall either digitally or with half of the bivalve speculum to keep the patient from activating her muscles.
• The cough test can be performed in the standing position.
Assessing urethral mobility similarly involves simple observation while the patient is in a supine lithotomy position and straining. A Q-tip test or the Pelvic Organ Prolapse Quantification (POP-Q) system may be used, but visualization and palpation also are completely acceptable.
Just as the absence of urethral mobility is a red flag, so is prolapse beyond the hymen. This patient could potentially have urethral kinking, which can mask the severity of SUI or be a source of retention. Either finding the absence of urethral mobility or the presence of POP beyond the hymen moves the case from the uncomplicated to complicated category and signals the need for further evaluation with urodynamics or other tests.
These and other findings for uncomplicated versus complicated SUI are outlined in a committee opinion issued recently by the American College of Obstetricians and Gynecologists and the American Urogynecologic Society (Committee Opinion No. 603, Obstet .Gynecol. 2014;123:1403-7).
As the ACOG-AUGS recommendations point out, urinalysis is part of the minimum work-up for stress incontinence. Measurement of postvoid residual urine volume also becomes important when midurethral sling surgery is being contemplated for uncomplicated SUI. A normal volume rules out potential bladder-emptying abnormalities and provides final assurance that the patient is a good candidate for surgical repair.
Recent research on urodynamics
Evidence that a simple office-based incontinence evaluation without preoperative urodynamic testing is appropriate for uncomplicated predominant SUI comes largely from two recent randomized noninferiority trials.
One of these trials – a study from the Urinary Incontinence Treatment Network in the United States, known as the VALUE trial – randomized 630 women with uncomplicated SUI to pretreatment work-up with or without urodynamics. Treatment success at 12 months was similar for the two groups (approximately 77%).
This finding, the authors wrote, suggests that for women with uncomplicated SUI, a “basic office evaluation” (i.e., a positive provocative stress test, a normal postvoiding residual volume, an assessment or urethral mobility, and a negative urinalysis) is a “sufficient preoperative work-up” (N. Engl. J. Med. 2012;366:1987-97).
The diagnosis of SUI as made by office evaluation was confirmed in 97% of women who underwent urodynamic testing, and while there were some adjustments in diagnosis after urodynamics, there were no major changes in treatment decision making after the testing. Approximately 93% of women in both groups underwent midurethral sling surgery.
The second trial, a Dutch study, focused on women who had already undergone urodynamic testing and been shown to have discordant findings on urodynamics and their history and clinical exam. The women – all of whom had uncomplicated predominant SUI – were randomized to undergo immediate midurethral sling surgery or receive individually tailored treatment (including sling surgery, behavioral and physical therapy, pessary, and anticholinergics).
At 1 year, there was no clinically significant difference between the two groups in patients’ assessment of their symptoms as measured by the UDI. The authors concluded that “an immediate midurethral sling operation is not inferior to individually tailored treatment based on urodynamic findings” and that “urodynamics should no longer be advised routinely before primary surgery in these patients” (Obstet. Gynecol. 2013;121:999-1008).
When urge incontinence is involved
Urodynamic testing was never believed to be perfect, but these and other studies have highlighted its imperfections. Urodynamics creates an artificial condition in the bladder, in effect, and some of the findings will involve artifact. A systematic review of studies that compared diagnoses based on symptoms with diagnoses after urodynamic investigation was interesting in this regard; while the review did not assess impact on treatment, it showed that there is poor agreement between clinical symptoms and urodynamic-based diagnoses (Neurourol. Urodyn. 2011;30:495-502).
Certainly, women with complicated SUI – as well as women who have recurrent SUI after a prior surgical intervention – require further assessment, which likely includes multichannel urodynamic testing.
Urodynamics also can play a useful role in decision making and counseling for some patients whose incontinence is predominately SUI, but is believed to involve some degree of urinary urgency. Patients with mixed urinary incontinence fare worse after midurethral sling procedures compared with patients who have SUI alone, and I counsel my patients accordingly, emphasizing that the sling will not address aspects of their incontinence related to urgency. When I sense that a patient may have unreasonably high expectations for surgery, urodynamic testing can provide some perspective on possible postoperative outcomes.
Treatment for UI or overactive bladder often may be initiated after simple office-based evaluation, just as with SUI. The goal, similarly, is to discern relatively uncomplicated or straightforward cases from complicated ones. Urologic, medical, and neurologic histories should be obtained, for instance, and retention issues (which can aggravate UI) should be ruled out through the measurement of postvoid residual urine volume.
Just as with SUI, evaluation of suspected UI more often than not involves careful history taking and clinical probing. A voiding diary can sometimes be helpful; I send patients home with such a tool when the history is inconclusive or I suspect behavioral (excessive fluid intake) or functional issues as significant factors in bladder control.
It is important to keep in mind that patients with severe SUI may have urinary frequency as a learned response. Such patients appear to have overactive bladder in addition to SUI, but may actually be urinating frequently because they’ve learned that doing so results in less leakage. In our practice we’ve observed that patients with a learned response tend not to have nocturia, while those with overactive bladder do report nocturia.
Dr. Culbertson had no relevant financial disclosures.
Dr. Culbertson is a professor in the department of obstetrics and gynecology at the University of Chicago.
Ten years ago, urodynamics were widely viewed as the gold standard for evaluating urinary incontinence. We often turned to such testing to confirm or reject the findings of our basic evaluation before determining the best type of treatment – especially before proceeding with primary anti-incontinence surgery.
What has emerged in recent years is a body of evidence that tells us otherwise. We now know that urodynamics do not give us all the answers, and that we can be much more judicious with its use.
A good history followed by a thorough physical examination and some office tests often enables us to make sound treatment recommendations without costly and potentially uncomfortable urodynamic testing. The key lies in discerning complicated and uncomplicated cases. For patients deemed to have uncomplicated stress urinary incontinence (SUI) – especially those who have failed conservative management – we can comfortably recommend surgical repair without urodynamic testing.
Identifying uncomplicated SUI
The history is the most important part of the evaluation for incontinence. Every patient who answers “yes” to a basic opening question about whether she has any concerns about bladder control should be asked a series of questions that will enable the physician to fully understand her symptoms, their severity, and their impact on her life and daily activities.
It is critical to determine whether you are dealing with pure SUI, pure urge incontinence (UI), or SUI with a component of UI. Mixed incontinence is quite prevalent. An analysis of recent National Health and Nutrition Examination Survey (NHANES) data showed that of those women reporting incontinence symptoms, almost 50% reported pure SUI, and 34% reported mixed incontinence (J. Urol. 2008;179: 656-61). Other studies similarly have shown prevalence rates of mixed incontinence above 30%.
The International Urogynecological Association (IUGA) recommends the use of validated questionnaires to assess incontinence and the relative contribution of UI and SUI symptoms. Some physicians do find the organized and structured format of questionnaires helpful in their practices. Others have incorporated questions from various tools into history-taking templates on their electronic medical records. Still others have made them part of a mental checklist for history taking.
The short-form version of the Urogenital Distress Inventory (UDI-6), for instance, asks the patient whether she experiences – and how much she is bothered by – the following: frequent urination; leakage related to a feeling of urgency; leakage related to activity, coughing, or sneezing; small amount of leakage; difficulty emptying the bladder; and pain or discomfort in the lower abdominal or genital area.
The Incontinence Impact Questionnaire can be used to further assess the impact of symptoms. The short-form IIQ (the IIQ-7) asks, for instance, about the extent to which urine leakage has affected household chores, physical recreation, social activities, or emotional health.
Since the UDI and IIQ were developed about 20 years ago, at least several other urinary incontinence questionnaires have been developed and validated. Whether or not questionnaires are utilized as official tools, history taking should capture their essence and provide you with enough information to ascertain the type of incontinence, frequency of occurrence, severity, and effect on daily life.
The history also must assess the possibility of voiding dysfunction. Positive responses to questions about nocturia, hesitancy, and the need to immediately revoid, for instance, point toward complicated SUI and the need for further assessment before embarking on surgical treatment for SUI.
Patients who have uncomplicated SUI, on the other hand, will answer negatively to questions about symptoms of predominant urgency, functional impairment, continuous leakage, and/or incomplete emptying. They also will not have had recurrent urinary tract infections or medical conditions that can affect lower urinary tract function (such as neurologic disease and poorly controlled diabetes).
The physical exam
Along with the history, the physical exam is important for identifying complicated SUI and confirming which cases of SUI are truly uncomplicated. Evaluation should include a cough stress test to confirm leakage from the urethra under stress, an assessment of urethral mobility, and an assessment for pelvic organ prolapse.
The cough stress test is usually done with the patient in the supine or semirecumbent lithotomy position. If you strongly suspect stress incontinence but have a negative result, consider the following:
• Make sure the patient has a comfortably full bladder.
• Many women will contract their pelvic floor muscles when coughing to try to avoid leaking. You can apply pressure against the posterior vaginal wall either digitally or with half of the bivalve speculum to keep the patient from activating her muscles.
• The cough test can be performed in the standing position.
Assessing urethral mobility similarly involves simple observation while the patient is in a supine lithotomy position and straining. A Q-tip test or the Pelvic Organ Prolapse Quantification (POP-Q) system may be used, but visualization and palpation also are completely acceptable.
Just as the absence of urethral mobility is a red flag, so is prolapse beyond the hymen. This patient could potentially have urethral kinking, which can mask the severity of SUI or be a source of retention. Either finding the absence of urethral mobility or the presence of POP beyond the hymen moves the case from the uncomplicated to complicated category and signals the need for further evaluation with urodynamics or other tests.
These and other findings for uncomplicated versus complicated SUI are outlined in a committee opinion issued recently by the American College of Obstetricians and Gynecologists and the American Urogynecologic Society (Committee Opinion No. 603, Obstet .Gynecol. 2014;123:1403-7).
As the ACOG-AUGS recommendations point out, urinalysis is part of the minimum work-up for stress incontinence. Measurement of postvoid residual urine volume also becomes important when midurethral sling surgery is being contemplated for uncomplicated SUI. A normal volume rules out potential bladder-emptying abnormalities and provides final assurance that the patient is a good candidate for surgical repair.
Recent research on urodynamics
Evidence that a simple office-based incontinence evaluation without preoperative urodynamic testing is appropriate for uncomplicated predominant SUI comes largely from two recent randomized noninferiority trials.
One of these trials – a study from the Urinary Incontinence Treatment Network in the United States, known as the VALUE trial – randomized 630 women with uncomplicated SUI to pretreatment work-up with or without urodynamics. Treatment success at 12 months was similar for the two groups (approximately 77%).
This finding, the authors wrote, suggests that for women with uncomplicated SUI, a “basic office evaluation” (i.e., a positive provocative stress test, a normal postvoiding residual volume, an assessment or urethral mobility, and a negative urinalysis) is a “sufficient preoperative work-up” (N. Engl. J. Med. 2012;366:1987-97).
The diagnosis of SUI as made by office evaluation was confirmed in 97% of women who underwent urodynamic testing, and while there were some adjustments in diagnosis after urodynamics, there were no major changes in treatment decision making after the testing. Approximately 93% of women in both groups underwent midurethral sling surgery.
The second trial, a Dutch study, focused on women who had already undergone urodynamic testing and been shown to have discordant findings on urodynamics and their history and clinical exam. The women – all of whom had uncomplicated predominant SUI – were randomized to undergo immediate midurethral sling surgery or receive individually tailored treatment (including sling surgery, behavioral and physical therapy, pessary, and anticholinergics).
At 1 year, there was no clinically significant difference between the two groups in patients’ assessment of their symptoms as measured by the UDI. The authors concluded that “an immediate midurethral sling operation is not inferior to individually tailored treatment based on urodynamic findings” and that “urodynamics should no longer be advised routinely before primary surgery in these patients” (Obstet. Gynecol. 2013;121:999-1008).
When urge incontinence is involved
Urodynamic testing was never believed to be perfect, but these and other studies have highlighted its imperfections. Urodynamics creates an artificial condition in the bladder, in effect, and some of the findings will involve artifact. A systematic review of studies that compared diagnoses based on symptoms with diagnoses after urodynamic investigation was interesting in this regard; while the review did not assess impact on treatment, it showed that there is poor agreement between clinical symptoms and urodynamic-based diagnoses (Neurourol. Urodyn. 2011;30:495-502).
Certainly, women with complicated SUI – as well as women who have recurrent SUI after a prior surgical intervention – require further assessment, which likely includes multichannel urodynamic testing.
Urodynamics also can play a useful role in decision making and counseling for some patients whose incontinence is predominately SUI, but is believed to involve some degree of urinary urgency. Patients with mixed urinary incontinence fare worse after midurethral sling procedures compared with patients who have SUI alone, and I counsel my patients accordingly, emphasizing that the sling will not address aspects of their incontinence related to urgency. When I sense that a patient may have unreasonably high expectations for surgery, urodynamic testing can provide some perspective on possible postoperative outcomes.
Treatment for UI or overactive bladder often may be initiated after simple office-based evaluation, just as with SUI. The goal, similarly, is to discern relatively uncomplicated or straightforward cases from complicated ones. Urologic, medical, and neurologic histories should be obtained, for instance, and retention issues (which can aggravate UI) should be ruled out through the measurement of postvoid residual urine volume.
Just as with SUI, evaluation of suspected UI more often than not involves careful history taking and clinical probing. A voiding diary can sometimes be helpful; I send patients home with such a tool when the history is inconclusive or I suspect behavioral (excessive fluid intake) or functional issues as significant factors in bladder control.
It is important to keep in mind that patients with severe SUI may have urinary frequency as a learned response. Such patients appear to have overactive bladder in addition to SUI, but may actually be urinating frequently because they’ve learned that doing so results in less leakage. In our practice we’ve observed that patients with a learned response tend not to have nocturia, while those with overactive bladder do report nocturia.
Dr. Culbertson had no relevant financial disclosures.
Dr. Culbertson is a professor in the department of obstetrics and gynecology at the University of Chicago.

Gemcitabine: Best Alone or in Combination?
Gemcitabine (GEM) is the standard treatment for patients with locally advanced/metastatic pancreatic cancer (LA/MPC). Many studies have focused on finding combinations that might extend the efficacy of GEM. However, most studies have not found an improvement in overall survival (OS) for patients using GEM combination therapy, say researchers from Beijing Friendship Hospital in China. That is until recently, when researchers found that compared with GEM monotherapy, nanoparticle albumin-bound paclitaxel plus GEM significantly improved OS (8.5 months vs 6.7 months; P < .001) and progression-free survival (5.5 months vs 3.7 months; P < .001).
Other research has found that combining GEM with platinum, fluoropyrimidine, irinotecan, biotherapy, and others, for example, marginally but significantly affects OS, again compared with GEM monotherapy (P = .001). The combinations were associated with increased toxicity.
According to the researchers, the use of targeted therapies in cancer treatment is a “significant focus” of cancer research and has brought great clinical benefits in treating a variety of solid tumors. Studies that combined drugs to target epidermal growth factor receptor (EGFR), which is overexpressed in pancreatic tumors and associated with poor prognosis, had mixed results for patients with LA/MPC. This prompted researchers in this study to conduct a systematic survey of 10 randomized controlled trials: 3 phase 2 trials and 7 phase 3 trials. Of 3,899 patients, 1,989 received GEM + targeted agents and 1,910 received GEM as monotherapy or combined with placebo (PLC). In a subgroup of GEM + antiangiogenic agents, 733 patients received GEM + axitinib or bevacizumab and cilengitide, and 693 received GEM ± PLC.
No significant difference was seen in the OS rate between the GEM + targeted agents and GEM ± PLC groups (P = .85), and only a marginal difference was found in the 1-year survival rate (P = .05). In the subgroup analysis, the researchers found a significant increase in objective response rate with GEM + antiangiogenic agents vs GEM ± PLC (95% CI, 0.42-0.98; P = .04). However, they found no significant difference in OS, 1-year survival, or progression-free survival between those 2 groups.
The researchers advise further research concentrated on clarifying the “concrete targets” involved in the occurrence and progression of pancreatic cancer. They add that personalized therapy based on a patient’s stratification, tumor stage, and genetic background should also be considered.
Source
Li Q, Yuan Z, Yan H, Wen Z, Zhang R, Cao B. Clin Ther. 2014;36(7):1054-1063.
doi: 10.1016/j.clinthera.2014.05.066.
Gemcitabine (GEM) is the standard treatment for patients with locally advanced/metastatic pancreatic cancer (LA/MPC). Many studies have focused on finding combinations that might extend the efficacy of GEM. However, most studies have not found an improvement in overall survival (OS) for patients using GEM combination therapy, say researchers from Beijing Friendship Hospital in China. That is until recently, when researchers found that compared with GEM monotherapy, nanoparticle albumin-bound paclitaxel plus GEM significantly improved OS (8.5 months vs 6.7 months; P < .001) and progression-free survival (5.5 months vs 3.7 months; P < .001).
Other research has found that combining GEM with platinum, fluoropyrimidine, irinotecan, biotherapy, and others, for example, marginally but significantly affects OS, again compared with GEM monotherapy (P = .001). The combinations were associated with increased toxicity.
According to the researchers, the use of targeted therapies in cancer treatment is a “significant focus” of cancer research and has brought great clinical benefits in treating a variety of solid tumors. Studies that combined drugs to target epidermal growth factor receptor (EGFR), which is overexpressed in pancreatic tumors and associated with poor prognosis, had mixed results for patients with LA/MPC. This prompted researchers in this study to conduct a systematic survey of 10 randomized controlled trials: 3 phase 2 trials and 7 phase 3 trials. Of 3,899 patients, 1,989 received GEM + targeted agents and 1,910 received GEM as monotherapy or combined with placebo (PLC). In a subgroup of GEM + antiangiogenic agents, 733 patients received GEM + axitinib or bevacizumab and cilengitide, and 693 received GEM ± PLC.
No significant difference was seen in the OS rate between the GEM + targeted agents and GEM ± PLC groups (P = .85), and only a marginal difference was found in the 1-year survival rate (P = .05). In the subgroup analysis, the researchers found a significant increase in objective response rate with GEM + antiangiogenic agents vs GEM ± PLC (95% CI, 0.42-0.98; P = .04). However, they found no significant difference in OS, 1-year survival, or progression-free survival between those 2 groups.
The researchers advise further research concentrated on clarifying the “concrete targets” involved in the occurrence and progression of pancreatic cancer. They add that personalized therapy based on a patient’s stratification, tumor stage, and genetic background should also be considered.
Source
Li Q, Yuan Z, Yan H, Wen Z, Zhang R, Cao B. Clin Ther. 2014;36(7):1054-1063.
doi: 10.1016/j.clinthera.2014.05.066.
Gemcitabine (GEM) is the standard treatment for patients with locally advanced/metastatic pancreatic cancer (LA/MPC). Many studies have focused on finding combinations that might extend the efficacy of GEM. However, most studies have not found an improvement in overall survival (OS) for patients using GEM combination therapy, say researchers from Beijing Friendship Hospital in China. That is until recently, when researchers found that compared with GEM monotherapy, nanoparticle albumin-bound paclitaxel plus GEM significantly improved OS (8.5 months vs 6.7 months; P < .001) and progression-free survival (5.5 months vs 3.7 months; P < .001).
Other research has found that combining GEM with platinum, fluoropyrimidine, irinotecan, biotherapy, and others, for example, marginally but significantly affects OS, again compared with GEM monotherapy (P = .001). The combinations were associated with increased toxicity.
According to the researchers, the use of targeted therapies in cancer treatment is a “significant focus” of cancer research and has brought great clinical benefits in treating a variety of solid tumors. Studies that combined drugs to target epidermal growth factor receptor (EGFR), which is overexpressed in pancreatic tumors and associated with poor prognosis, had mixed results for patients with LA/MPC. This prompted researchers in this study to conduct a systematic survey of 10 randomized controlled trials: 3 phase 2 trials and 7 phase 3 trials. Of 3,899 patients, 1,989 received GEM + targeted agents and 1,910 received GEM as monotherapy or combined with placebo (PLC). In a subgroup of GEM + antiangiogenic agents, 733 patients received GEM + axitinib or bevacizumab and cilengitide, and 693 received GEM ± PLC.
No significant difference was seen in the OS rate between the GEM + targeted agents and GEM ± PLC groups (P = .85), and only a marginal difference was found in the 1-year survival rate (P = .05). In the subgroup analysis, the researchers found a significant increase in objective response rate with GEM + antiangiogenic agents vs GEM ± PLC (95% CI, 0.42-0.98; P = .04). However, they found no significant difference in OS, 1-year survival, or progression-free survival between those 2 groups.
The researchers advise further research concentrated on clarifying the “concrete targets” involved in the occurrence and progression of pancreatic cancer. They add that personalized therapy based on a patient’s stratification, tumor stage, and genetic background should also be considered.
Source
Li Q, Yuan Z, Yan H, Wen Z, Zhang R, Cao B. Clin Ther. 2014;36(7):1054-1063.
doi: 10.1016/j.clinthera.2014.05.066.
Dendritic cells promote Myc-driven lymphoma
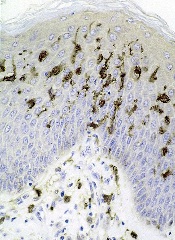
Studies have shown that dendritic cells (DCs) can contribute to tumor growth and help shield the tumor from the immune system in colon, stomach, breast, and prostate cancer.
Now, researchers have found evidence suggesting this phenomenon also occurs in Myc-driven lymphomas.
The team has also identified the molecular mechanism that induces the immune cells to promote tumor growth.
They reported these findings in Nature Communications.
Uta Höpken, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany, and her colleagues investigated how DCs drive tumor in mouse models of Eµ-Myc lymphoma.
The team began by depleting DCs in these mice and found that tumor growth was delayed—the first clue that DCs are indeed associated with lymphoma growth.
Next, the researchers found that, after contact with lymphoma cells, the DCs increasingly secrete immunomodulatory cytokines and growth factors. The cytokine secretion takes place in the spleen and lymph nodes.
Dr Höpken and her colleagues previously demonstrated that various forms of lymphoma cells settle in the lymph nodes and in the spleen, where they create their own survival niche. This process is regulated by selective cytokines and growth factors the researchers identified a few years ago.
“In these niches, almost everything is already there that the lymphoma cells as malignant B cells need to survive, including blood vessels and connective tissue cells [stromal cells],” Dr Höpken said. “The survival substances secreted by the DCs optimize the niche so that the tumors can grow better.”
This also means the DCs prevent the T lymphocytes from exercising their defensive function. Normally, healthy B or T cells settle in the respective B- or T-cell niches of the spleen and the lymph nodes to be made fit for immune defense.
“What is paradoxical is that the mouse lymphoma cells we studied—malignant B cells—found their survival niche in the T-cell zones of the lymph nodes and the spleen and not in the B-cell zones,” Dr Höpken said.
After making contact with the lymphoma cells, the DCs increasingly upregulate C/EBPβ, a transcription factor that promotes the production of cytokines that mediate inflammation.
The researchers found that C/EBPβ regulates DCs. Without it, the cells could not secrete inflammatory cytokines. C/EBPβ also indirectly blocks apoptosis in the lymphoma cells, allowing the cancer cells to grow unchecked.
The team pointed out that, even if their model of Eµ-Myc lymphoma is not entirely comparable to B-cell lymphomas in humans, it shows that lymphoma cells and DCs interact—a previously unknown molecular mechanism.
Furthermore, these findings may have clinical applications. The researchers noted that the immunomodulatory agent lenalidomide induces downregulation of C/EBPβ, which is secreted by cancer cells.
So Dr Höpken and her colleagues believe it might be appropriate to approve the use of lenalidomide for patients with Myc B-cell lymphoma, as an addition to their existing treatment, to strengthen their immune defense. ![]()
Studies have shown that dendritic cells (DCs) can contribute to tumor growth and help shield the tumor from the immune system in colon, stomach, breast, and prostate cancer.
Now, researchers have found evidence suggesting this phenomenon also occurs in Myc-driven lymphomas.
The team has also identified the molecular mechanism that induces the immune cells to promote tumor growth.
They reported these findings in Nature Communications.
Uta Höpken, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany, and her colleagues investigated how DCs drive tumor in mouse models of Eµ-Myc lymphoma.
The team began by depleting DCs in these mice and found that tumor growth was delayed—the first clue that DCs are indeed associated with lymphoma growth.
Next, the researchers found that, after contact with lymphoma cells, the DCs increasingly secrete immunomodulatory cytokines and growth factors. The cytokine secretion takes place in the spleen and lymph nodes.
Dr Höpken and her colleagues previously demonstrated that various forms of lymphoma cells settle in the lymph nodes and in the spleen, where they create their own survival niche. This process is regulated by selective cytokines and growth factors the researchers identified a few years ago.
“In these niches, almost everything is already there that the lymphoma cells as malignant B cells need to survive, including blood vessels and connective tissue cells [stromal cells],” Dr Höpken said. “The survival substances secreted by the DCs optimize the niche so that the tumors can grow better.”
This also means the DCs prevent the T lymphocytes from exercising their defensive function. Normally, healthy B or T cells settle in the respective B- or T-cell niches of the spleen and the lymph nodes to be made fit for immune defense.
“What is paradoxical is that the mouse lymphoma cells we studied—malignant B cells—found their survival niche in the T-cell zones of the lymph nodes and the spleen and not in the B-cell zones,” Dr Höpken said.
After making contact with the lymphoma cells, the DCs increasingly upregulate C/EBPβ, a transcription factor that promotes the production of cytokines that mediate inflammation.
The researchers found that C/EBPβ regulates DCs. Without it, the cells could not secrete inflammatory cytokines. C/EBPβ also indirectly blocks apoptosis in the lymphoma cells, allowing the cancer cells to grow unchecked.
The team pointed out that, even if their model of Eµ-Myc lymphoma is not entirely comparable to B-cell lymphomas in humans, it shows that lymphoma cells and DCs interact—a previously unknown molecular mechanism.
Furthermore, these findings may have clinical applications. The researchers noted that the immunomodulatory agent lenalidomide induces downregulation of C/EBPβ, which is secreted by cancer cells.
So Dr Höpken and her colleagues believe it might be appropriate to approve the use of lenalidomide for patients with Myc B-cell lymphoma, as an addition to their existing treatment, to strengthen their immune defense. ![]()
Studies have shown that dendritic cells (DCs) can contribute to tumor growth and help shield the tumor from the immune system in colon, stomach, breast, and prostate cancer.
Now, researchers have found evidence suggesting this phenomenon also occurs in Myc-driven lymphomas.
The team has also identified the molecular mechanism that induces the immune cells to promote tumor growth.
They reported these findings in Nature Communications.
Uta Höpken, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany, and her colleagues investigated how DCs drive tumor in mouse models of Eµ-Myc lymphoma.
The team began by depleting DCs in these mice and found that tumor growth was delayed—the first clue that DCs are indeed associated with lymphoma growth.
Next, the researchers found that, after contact with lymphoma cells, the DCs increasingly secrete immunomodulatory cytokines and growth factors. The cytokine secretion takes place in the spleen and lymph nodes.
Dr Höpken and her colleagues previously demonstrated that various forms of lymphoma cells settle in the lymph nodes and in the spleen, where they create their own survival niche. This process is regulated by selective cytokines and growth factors the researchers identified a few years ago.
“In these niches, almost everything is already there that the lymphoma cells as malignant B cells need to survive, including blood vessels and connective tissue cells [stromal cells],” Dr Höpken said. “The survival substances secreted by the DCs optimize the niche so that the tumors can grow better.”
This also means the DCs prevent the T lymphocytes from exercising their defensive function. Normally, healthy B or T cells settle in the respective B- or T-cell niches of the spleen and the lymph nodes to be made fit for immune defense.
“What is paradoxical is that the mouse lymphoma cells we studied—malignant B cells—found their survival niche in the T-cell zones of the lymph nodes and the spleen and not in the B-cell zones,” Dr Höpken said.
After making contact with the lymphoma cells, the DCs increasingly upregulate C/EBPβ, a transcription factor that promotes the production of cytokines that mediate inflammation.
The researchers found that C/EBPβ regulates DCs. Without it, the cells could not secrete inflammatory cytokines. C/EBPβ also indirectly blocks apoptosis in the lymphoma cells, allowing the cancer cells to grow unchecked.
The team pointed out that, even if their model of Eµ-Myc lymphoma is not entirely comparable to B-cell lymphomas in humans, it shows that lymphoma cells and DCs interact—a previously unknown molecular mechanism.
Furthermore, these findings may have clinical applications. The researchers noted that the immunomodulatory agent lenalidomide induces downregulation of C/EBPβ, which is secreted by cancer cells.
So Dr Höpken and her colleagues believe it might be appropriate to approve the use of lenalidomide for patients with Myc B-cell lymphoma, as an addition to their existing treatment, to strengthen their immune defense. ![]()
Product appears safe, effective in rel/ref ALL

Credit: Bill Branson
Eryaspase, a product consisting of L-asparaginase encapsulated in red blood cells, may be safer and more effective than native E coli L-asparaginase, results of a phase 2/3 study suggest.
In patients with relapsed or refractory acute lymphoblastic leukemia (ALL), eryaspase given in combination with chemotherapy produced a higher complete response rate and fewer allergic reactions than native E coli L-asparaginase in combination with chemotherapy.
Eryaspase was also well-tolerated in patients who had previously experienced allergic reactions to L-asparaginase.
ERYTECH Pharma, the company developing eryaspase, recently announced these results from the GRASPIVOTALL trial.
The study included 80 children and adults with relapsed or refractory ALL, who were randomized to 1 of 3 treatment arms.
In the first 2 arms, researchers compared eryaspase to native E coli L-asparaginase, both in combination with standard chemotherapy (COOPRALL), in patients without prior allergies to L-asparaginase. In the third arm, the team evaluated eryaspase for patients who have experienced allergic reactions related to asparaginase in their first-line treatment.
The primary endpoint of the study consisted of 2 objectives: (a) superior safety, expressed as a significant reduction of the incidence of allergic reactions with eryaspase compared to native L-asparaginase, and (b) noninferior duration of asparaginase activity above the threshold of 100 IU/L during the induction phase in the nonallergic patients. Both endpoints needed to be met for the study to be considered positive.
The main secondary efficacy endpoints included complete response, minimal residual disease, event-free survival, and overall survival.
At 1 year of follow-up, both primary endpoints were met. There was a significant reduction of allergic reactions in the eryaspase arm compared to the native L-asparaginase arm—0% (0/26) and 42.9% (12/28), respectively (P<0.001).
And there was a significant increase in the duration of circulating asparaginase activity in the eryaspase arm compared to the native L-asparaginase arm. Asparaginase levels were maintained above 100 IU/L for an average of 20.5 days with up to 2 injections during the first month of treatment with eryaspase, compared to 9.2 days in the native L-asparaginase arm, with up to 8 injections of L-asparaginase (P<0.001).
In addition, the complete response rate was higher with eryaspase. At the end of the induction phase, 71.4% of patients (n=15) in the eryaspase arm had a complete response, compared to 42.3% of patients (n=11) in the native L-asparaginase arm.
Finally, the study showed that eryaspase is well-tolerated by patients with previous allergies to L-asparaginase. Two of the 26 patients with prior allergies to L-asparaginase experienced mild allergic reactions to eryaspase.
“The results of this study are an important step forward for the treatment of ALL patients that are at risk to receive L-asparaginase, which remains an important unmet medical need,” said Yves Betrand, MD, of the Institute for Pediatric Hematology and Oncology in Lyon, France.
“The virtual absence of allergic reactions, also in patients with prior allergies to L-asparaginase, is very encouraging.”
The analysis of additional secondary and exploratory endpoints for this study is ongoing. ERYTECH said results will be available later this year and are set to be presented at an upcoming scientific conference.
Based on the results of this and earlier studies of eryaspase, ERYTECH intends to submit its application for European Marketing Authorization in the first half of 2015.
The company also plans to accelerate the product’s development in ALL in the US and to launch phase 2 clinical trials in additional oncology indications with high unmet medical need. A phase 2b study of eryaspase in acute myeloid leukemia is already underway, with more than half of the patients enrolled. ![]()
Credit: Bill Branson
Eryaspase, a product consisting of L-asparaginase encapsulated in red blood cells, may be safer and more effective than native E coli L-asparaginase, results of a phase 2/3 study suggest.
In patients with relapsed or refractory acute lymphoblastic leukemia (ALL), eryaspase given in combination with chemotherapy produced a higher complete response rate and fewer allergic reactions than native E coli L-asparaginase in combination with chemotherapy.
Eryaspase was also well-tolerated in patients who had previously experienced allergic reactions to L-asparaginase.
ERYTECH Pharma, the company developing eryaspase, recently announced these results from the GRASPIVOTALL trial.
The study included 80 children and adults with relapsed or refractory ALL, who were randomized to 1 of 3 treatment arms.
In the first 2 arms, researchers compared eryaspase to native E coli L-asparaginase, both in combination with standard chemotherapy (COOPRALL), in patients without prior allergies to L-asparaginase. In the third arm, the team evaluated eryaspase for patients who have experienced allergic reactions related to asparaginase in their first-line treatment.
The primary endpoint of the study consisted of 2 objectives: (a) superior safety, expressed as a significant reduction of the incidence of allergic reactions with eryaspase compared to native L-asparaginase, and (b) noninferior duration of asparaginase activity above the threshold of 100 IU/L during the induction phase in the nonallergic patients. Both endpoints needed to be met for the study to be considered positive.
The main secondary efficacy endpoints included complete response, minimal residual disease, event-free survival, and overall survival.
At 1 year of follow-up, both primary endpoints were met. There was a significant reduction of allergic reactions in the eryaspase arm compared to the native L-asparaginase arm—0% (0/26) and 42.9% (12/28), respectively (P<0.001).
And there was a significant increase in the duration of circulating asparaginase activity in the eryaspase arm compared to the native L-asparaginase arm. Asparaginase levels were maintained above 100 IU/L for an average of 20.5 days with up to 2 injections during the first month of treatment with eryaspase, compared to 9.2 days in the native L-asparaginase arm, with up to 8 injections of L-asparaginase (P<0.001).
In addition, the complete response rate was higher with eryaspase. At the end of the induction phase, 71.4% of patients (n=15) in the eryaspase arm had a complete response, compared to 42.3% of patients (n=11) in the native L-asparaginase arm.
Finally, the study showed that eryaspase is well-tolerated by patients with previous allergies to L-asparaginase. Two of the 26 patients with prior allergies to L-asparaginase experienced mild allergic reactions to eryaspase.
“The results of this study are an important step forward for the treatment of ALL patients that are at risk to receive L-asparaginase, which remains an important unmet medical need,” said Yves Betrand, MD, of the Institute for Pediatric Hematology and Oncology in Lyon, France.
“The virtual absence of allergic reactions, also in patients with prior allergies to L-asparaginase, is very encouraging.”
The analysis of additional secondary and exploratory endpoints for this study is ongoing. ERYTECH said results will be available later this year and are set to be presented at an upcoming scientific conference.
Based on the results of this and earlier studies of eryaspase, ERYTECH intends to submit its application for European Marketing Authorization in the first half of 2015.
The company also plans to accelerate the product’s development in ALL in the US and to launch phase 2 clinical trials in additional oncology indications with high unmet medical need. A phase 2b study of eryaspase in acute myeloid leukemia is already underway, with more than half of the patients enrolled. ![]()
Credit: Bill Branson
Eryaspase, a product consisting of L-asparaginase encapsulated in red blood cells, may be safer and more effective than native E coli L-asparaginase, results of a phase 2/3 study suggest.
In patients with relapsed or refractory acute lymphoblastic leukemia (ALL), eryaspase given in combination with chemotherapy produced a higher complete response rate and fewer allergic reactions than native E coli L-asparaginase in combination with chemotherapy.
Eryaspase was also well-tolerated in patients who had previously experienced allergic reactions to L-asparaginase.
ERYTECH Pharma, the company developing eryaspase, recently announced these results from the GRASPIVOTALL trial.
The study included 80 children and adults with relapsed or refractory ALL, who were randomized to 1 of 3 treatment arms.
In the first 2 arms, researchers compared eryaspase to native E coli L-asparaginase, both in combination with standard chemotherapy (COOPRALL), in patients without prior allergies to L-asparaginase. In the third arm, the team evaluated eryaspase for patients who have experienced allergic reactions related to asparaginase in their first-line treatment.
The primary endpoint of the study consisted of 2 objectives: (a) superior safety, expressed as a significant reduction of the incidence of allergic reactions with eryaspase compared to native L-asparaginase, and (b) noninferior duration of asparaginase activity above the threshold of 100 IU/L during the induction phase in the nonallergic patients. Both endpoints needed to be met for the study to be considered positive.
The main secondary efficacy endpoints included complete response, minimal residual disease, event-free survival, and overall survival.
At 1 year of follow-up, both primary endpoints were met. There was a significant reduction of allergic reactions in the eryaspase arm compared to the native L-asparaginase arm—0% (0/26) and 42.9% (12/28), respectively (P<0.001).
And there was a significant increase in the duration of circulating asparaginase activity in the eryaspase arm compared to the native L-asparaginase arm. Asparaginase levels were maintained above 100 IU/L for an average of 20.5 days with up to 2 injections during the first month of treatment with eryaspase, compared to 9.2 days in the native L-asparaginase arm, with up to 8 injections of L-asparaginase (P<0.001).
In addition, the complete response rate was higher with eryaspase. At the end of the induction phase, 71.4% of patients (n=15) in the eryaspase arm had a complete response, compared to 42.3% of patients (n=11) in the native L-asparaginase arm.
Finally, the study showed that eryaspase is well-tolerated by patients with previous allergies to L-asparaginase. Two of the 26 patients with prior allergies to L-asparaginase experienced mild allergic reactions to eryaspase.
“The results of this study are an important step forward for the treatment of ALL patients that are at risk to receive L-asparaginase, which remains an important unmet medical need,” said Yves Betrand, MD, of the Institute for Pediatric Hematology and Oncology in Lyon, France.
“The virtual absence of allergic reactions, also in patients with prior allergies to L-asparaginase, is very encouraging.”
The analysis of additional secondary and exploratory endpoints for this study is ongoing. ERYTECH said results will be available later this year and are set to be presented at an upcoming scientific conference.
Based on the results of this and earlier studies of eryaspase, ERYTECH intends to submit its application for European Marketing Authorization in the first half of 2015.
The company also plans to accelerate the product’s development in ALL in the US and to launch phase 2 clinical trials in additional oncology indications with high unmet medical need. A phase 2b study of eryaspase in acute myeloid leukemia is already underway, with more than half of the patients enrolled. ![]()
Cancer treatment during pregnancy can be safe
MADRID—Two new studies suggest that children exposed to chemotherapy or radiotherapy in the womb can be spared negative long-term effects.
At a median age of 6, most of the children exposed to radiation in utero had neuropsychological, behavioral, and general health outcomes that were within normal ranges.
And children who were exposed to chemotherapy in the womb had normal mental and cardiac health at a median age of 2.
“When chemotherapy is administered after the first trimester of pregnancy, we cannot discern any problems in the children,” said Frederic Amant, MD, PhD, of University Hospitals Leuven in Belgium.
“Fear about the risks of chemotherapy administration should not be a reason to terminate a pregnancy, delay cancer treatment for the mother, or to deliver a baby prematurely.”
Dr Amant and his colleagues presented these findings (abstract 267PD_PR) and their findings on radiation (abstract 49LBA_PR) at the ESMO 2014 Congress.
Outcomes with chemotherapy
In the first study, the researchers recruited 38 children from the International Network for Cancer, Infertility and Pregnancy registry who were prenatally exposed to chemotherapy.
Most of the mothers had breast (61%) or hematologic (22%) cancers. Most were treated with anthracyclines (61%) and had received an average of 4 cycles (range, 1-7) of treatment.
The researchers assessed mental development and cardiac health in the children of these subjects, comparing them to 38 control children who were not exposed to chemotherapy.
At a median age of almost 2 years, mental development was in the normal range for both groups of children, and development was not significantly different between the groups. The mean Mental Development Index score was 99.13 for exposed children and 101.47 for controls.
Cardiac dimensions and functions were within normal ranges for both groups as well. Mean fractional shortening was 36% (range, 32-42) for exposed children and 39% (range, 32-51) for controls. Although the difference was signficant (P=0.004), the researchers said it was not clinically relevant.
“This paper points to the very important issue of long-term safety of prenatal exposure to chemotherapy and reinforces the notion that chemotherapy during gestation does not endanger the fetus and her or his subsequent development,” said Fedro Alessandro Peccatori, MD, PhD, of the European Institute of Oncology in Milan, Italy, who was not involved in this study.
“To further ameliorate neonatal outcome, a special effort should be made to prolong pregnancy duration, and stringent long-term follow-up should be pursued to confirm these findings. Meanwhile, specific measures to support prematurely delivered babies and their families should be implemented.”
Dr Amant said future studies will explore the effects of specific chemotherapy types in detail and include longer-term follow-up.
Outcomes with radiation
In a second study, Dr Amant and his colleagues explored the impact of radiotherapy on the children of women with cancer.
The study included 16 children, with a median age of 6 years, who had been exposed to radiotherapy in utero. The median maternal irradiation was 48 Gy (range, 12-70), and the median estimated fetal irradiation was 91 mGy (range, 0-1690).
Neuropsychological, behavioral, and general health outcomes were within normal ranges for most of the children. And the researchers found no linear relationship between the fetal dose of radiation and cognitive outcome.
However, there was a negative linear relationship between the gestational age at radiotherapy exposure and verbal intelligence, based on results in 8 children. Two of these children were exposed to radiotherapy in the third trimester and had verbal intelligence scores outside the normal range.
One of the 2 children had low scores on all variables of cognitive development, but other pregnancy-related complications are confounding factors. The child’s mother suffered from an aggressive non-Hodgkin tumor of the brain that impacted her general state, and she had a preterm delivery.
Dr Amant noted that this is based on a very small number of children, so the results should be interpreted with caution.
“We cannot exclude that there might be an impact of prenatal radiotherapy exposure,” he said, “but larger series are needed to further investigate this relationship.” ![]()
MADRID—Two new studies suggest that children exposed to chemotherapy or radiotherapy in the womb can be spared negative long-term effects.
At a median age of 6, most of the children exposed to radiation in utero had neuropsychological, behavioral, and general health outcomes that were within normal ranges.
And children who were exposed to chemotherapy in the womb had normal mental and cardiac health at a median age of 2.
“When chemotherapy is administered after the first trimester of pregnancy, we cannot discern any problems in the children,” said Frederic Amant, MD, PhD, of University Hospitals Leuven in Belgium.
“Fear about the risks of chemotherapy administration should not be a reason to terminate a pregnancy, delay cancer treatment for the mother, or to deliver a baby prematurely.”
Dr Amant and his colleagues presented these findings (abstract 267PD_PR) and their findings on radiation (abstract 49LBA_PR) at the ESMO 2014 Congress.
Outcomes with chemotherapy
In the first study, the researchers recruited 38 children from the International Network for Cancer, Infertility and Pregnancy registry who were prenatally exposed to chemotherapy.
Most of the mothers had breast (61%) or hematologic (22%) cancers. Most were treated with anthracyclines (61%) and had received an average of 4 cycles (range, 1-7) of treatment.
The researchers assessed mental development and cardiac health in the children of these subjects, comparing them to 38 control children who were not exposed to chemotherapy.
At a median age of almost 2 years, mental development was in the normal range for both groups of children, and development was not significantly different between the groups. The mean Mental Development Index score was 99.13 for exposed children and 101.47 for controls.
Cardiac dimensions and functions were within normal ranges for both groups as well. Mean fractional shortening was 36% (range, 32-42) for exposed children and 39% (range, 32-51) for controls. Although the difference was signficant (P=0.004), the researchers said it was not clinically relevant.
“This paper points to the very important issue of long-term safety of prenatal exposure to chemotherapy and reinforces the notion that chemotherapy during gestation does not endanger the fetus and her or his subsequent development,” said Fedro Alessandro Peccatori, MD, PhD, of the European Institute of Oncology in Milan, Italy, who was not involved in this study.
“To further ameliorate neonatal outcome, a special effort should be made to prolong pregnancy duration, and stringent long-term follow-up should be pursued to confirm these findings. Meanwhile, specific measures to support prematurely delivered babies and their families should be implemented.”
Dr Amant said future studies will explore the effects of specific chemotherapy types in detail and include longer-term follow-up.
Outcomes with radiation
In a second study, Dr Amant and his colleagues explored the impact of radiotherapy on the children of women with cancer.
The study included 16 children, with a median age of 6 years, who had been exposed to radiotherapy in utero. The median maternal irradiation was 48 Gy (range, 12-70), and the median estimated fetal irradiation was 91 mGy (range, 0-1690).
Neuropsychological, behavioral, and general health outcomes were within normal ranges for most of the children. And the researchers found no linear relationship between the fetal dose of radiation and cognitive outcome.
However, there was a negative linear relationship between the gestational age at radiotherapy exposure and verbal intelligence, based on results in 8 children. Two of these children were exposed to radiotherapy in the third trimester and had verbal intelligence scores outside the normal range.
One of the 2 children had low scores on all variables of cognitive development, but other pregnancy-related complications are confounding factors. The child’s mother suffered from an aggressive non-Hodgkin tumor of the brain that impacted her general state, and she had a preterm delivery.
Dr Amant noted that this is based on a very small number of children, so the results should be interpreted with caution.
“We cannot exclude that there might be an impact of prenatal radiotherapy exposure,” he said, “but larger series are needed to further investigate this relationship.” ![]()
MADRID—Two new studies suggest that children exposed to chemotherapy or radiotherapy in the womb can be spared negative long-term effects.
At a median age of 6, most of the children exposed to radiation in utero had neuropsychological, behavioral, and general health outcomes that were within normal ranges.
And children who were exposed to chemotherapy in the womb had normal mental and cardiac health at a median age of 2.
“When chemotherapy is administered after the first trimester of pregnancy, we cannot discern any problems in the children,” said Frederic Amant, MD, PhD, of University Hospitals Leuven in Belgium.
“Fear about the risks of chemotherapy administration should not be a reason to terminate a pregnancy, delay cancer treatment for the mother, or to deliver a baby prematurely.”
Dr Amant and his colleagues presented these findings (abstract 267PD_PR) and their findings on radiation (abstract 49LBA_PR) at the ESMO 2014 Congress.
Outcomes with chemotherapy
In the first study, the researchers recruited 38 children from the International Network for Cancer, Infertility and Pregnancy registry who were prenatally exposed to chemotherapy.
Most of the mothers had breast (61%) or hematologic (22%) cancers. Most were treated with anthracyclines (61%) and had received an average of 4 cycles (range, 1-7) of treatment.
The researchers assessed mental development and cardiac health in the children of these subjects, comparing them to 38 control children who were not exposed to chemotherapy.
At a median age of almost 2 years, mental development was in the normal range for both groups of children, and development was not significantly different between the groups. The mean Mental Development Index score was 99.13 for exposed children and 101.47 for controls.
Cardiac dimensions and functions were within normal ranges for both groups as well. Mean fractional shortening was 36% (range, 32-42) for exposed children and 39% (range, 32-51) for controls. Although the difference was signficant (P=0.004), the researchers said it was not clinically relevant.
“This paper points to the very important issue of long-term safety of prenatal exposure to chemotherapy and reinforces the notion that chemotherapy during gestation does not endanger the fetus and her or his subsequent development,” said Fedro Alessandro Peccatori, MD, PhD, of the European Institute of Oncology in Milan, Italy, who was not involved in this study.
“To further ameliorate neonatal outcome, a special effort should be made to prolong pregnancy duration, and stringent long-term follow-up should be pursued to confirm these findings. Meanwhile, specific measures to support prematurely delivered babies and their families should be implemented.”
Dr Amant said future studies will explore the effects of specific chemotherapy types in detail and include longer-term follow-up.
Outcomes with radiation
In a second study, Dr Amant and his colleagues explored the impact of radiotherapy on the children of women with cancer.
The study included 16 children, with a median age of 6 years, who had been exposed to radiotherapy in utero. The median maternal irradiation was 48 Gy (range, 12-70), and the median estimated fetal irradiation was 91 mGy (range, 0-1690).
Neuropsychological, behavioral, and general health outcomes were within normal ranges for most of the children. And the researchers found no linear relationship between the fetal dose of radiation and cognitive outcome.
However, there was a negative linear relationship between the gestational age at radiotherapy exposure and verbal intelligence, based on results in 8 children. Two of these children were exposed to radiotherapy in the third trimester and had verbal intelligence scores outside the normal range.
One of the 2 children had low scores on all variables of cognitive development, but other pregnancy-related complications are confounding factors. The child’s mother suffered from an aggressive non-Hodgkin tumor of the brain that impacted her general state, and she had a preterm delivery.
Dr Amant noted that this is based on a very small number of children, so the results should be interpreted with caution.
“We cannot exclude that there might be an impact of prenatal radiotherapy exposure,” he said, “but larger series are needed to further investigate this relationship.” ![]()
Consolidation can improve PFS in HL
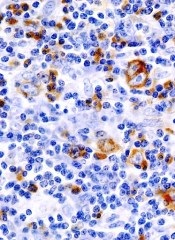
Consolidation therapy with brentuximab vedotin can improve progression-free survival (PFS) for Hodgkin lymphoma (HL) patients who have undergone a transplant, according to a phase 3 study.
The trial, known as AETHERA, is a comparison of single-agent brentuximab vedotin to placebo in patients with HL who were at risk of relapse following autologous stem cell transplant (ASCT).
Brentuximab vedotin conferred a 75% improvement in PFS over placebo.
However, there was no significant difference in overall survival between the 2 treatment arms.
These results were recently announced by Seattle Genetics Inc. and Takeda Pharmaceutical Company Limited, the companies developing brentuximab vedotin (Adcetris).
The companies said more complete results from AETHERA will be presented at the 2014 ASH Annual Meeting in December.
AETHERA is a randomized, double-blind, placebo-controlled study designed to evaluate the potential of brentuximab vedotin to extend PFS post-ASCT in patients with HL who have at least one risk factor for progression. In addition to the primary endpoint of PFS, secondary endpoints included overall survival, safety, and tolerability.
Patients were eligible if they had risk factors for residual HL, defined as a history of refractory HL, those who relapse or progress within a year of receiving frontline chemotherapy, and/or those who have disease outside of the lymph nodes at the time of pre-ASCT relapse.
The study included 329 patients who received brentuximab vedotin or placebo every 3 weeks for up to a year.
The researchers assessed PFS a minimum of 2 years after the initiation of treatment for all patients. There was a significant improvement in PFS with brentuximab vedotin compared to placebo (hazard ratio=0.57; P=0.001).
However, a prespecified interim analysis of overall survival showed no significant difference between the treatment arms.
Patients in both arms who experienced progression received a variety of subsequent therapies. Most patients on the placebo arm received brentuximab vedotin after progression.
A further analysis of overall survival is planned in 2016. The safety profile of brentuximab vedotin in the AETHERA trial was generally consistent with the existing prescribing information.
“We anticipate reporting more complete AETHERA data at the ASH Annual Meeting in December and intend to submit a supplemental Biologics License Application to the FDA in 2015 for approval in this setting,” said Clay B. Siegall, PhD, President and Chief Executive Officer of Seattle Genetics.
The FDA has already granted brentuximab vedotin accelerated approval to treat HL patients after ASCT failure or after the failure of at least 2 prior multiagent chemotherapy regimens in patients who are not ASCT candidates. The FDA also granted the drug accelerated approval to treat systemic anaplastic large cell lymphoma after the failure of at least 1 prior multiagent chemotherapy regimen.
The European Commission granted brentuximab vedotin conditional marketing authorization for the same indications. In both cases, the drug can gain full, traditional approval once studies have shown it confers a clinical benefit.
Brentuximab vedotin has a boxed warning detailing the risk of progressive multifocal leukoencephalopathy associated with use of the drug. The drug has also been shown to pose a risk of pulmonary toxicity when combined with bleomycin. ![]()
Consolidation therapy with brentuximab vedotin can improve progression-free survival (PFS) for Hodgkin lymphoma (HL) patients who have undergone a transplant, according to a phase 3 study.
The trial, known as AETHERA, is a comparison of single-agent brentuximab vedotin to placebo in patients with HL who were at risk of relapse following autologous stem cell transplant (ASCT).
Brentuximab vedotin conferred a 75% improvement in PFS over placebo.
However, there was no significant difference in overall survival between the 2 treatment arms.
These results were recently announced by Seattle Genetics Inc. and Takeda Pharmaceutical Company Limited, the companies developing brentuximab vedotin (Adcetris).
The companies said more complete results from AETHERA will be presented at the 2014 ASH Annual Meeting in December.
AETHERA is a randomized, double-blind, placebo-controlled study designed to evaluate the potential of brentuximab vedotin to extend PFS post-ASCT in patients with HL who have at least one risk factor for progression. In addition to the primary endpoint of PFS, secondary endpoints included overall survival, safety, and tolerability.
Patients were eligible if they had risk factors for residual HL, defined as a history of refractory HL, those who relapse or progress within a year of receiving frontline chemotherapy, and/or those who have disease outside of the lymph nodes at the time of pre-ASCT relapse.
The study included 329 patients who received brentuximab vedotin or placebo every 3 weeks for up to a year.
The researchers assessed PFS a minimum of 2 years after the initiation of treatment for all patients. There was a significant improvement in PFS with brentuximab vedotin compared to placebo (hazard ratio=0.57; P=0.001).
However, a prespecified interim analysis of overall survival showed no significant difference between the treatment arms.
Patients in both arms who experienced progression received a variety of subsequent therapies. Most patients on the placebo arm received brentuximab vedotin after progression.
A further analysis of overall survival is planned in 2016. The safety profile of brentuximab vedotin in the AETHERA trial was generally consistent with the existing prescribing information.
“We anticipate reporting more complete AETHERA data at the ASH Annual Meeting in December and intend to submit a supplemental Biologics License Application to the FDA in 2015 for approval in this setting,” said Clay B. Siegall, PhD, President and Chief Executive Officer of Seattle Genetics.
The FDA has already granted brentuximab vedotin accelerated approval to treat HL patients after ASCT failure or after the failure of at least 2 prior multiagent chemotherapy regimens in patients who are not ASCT candidates. The FDA also granted the drug accelerated approval to treat systemic anaplastic large cell lymphoma after the failure of at least 1 prior multiagent chemotherapy regimen.
The European Commission granted brentuximab vedotin conditional marketing authorization for the same indications. In both cases, the drug can gain full, traditional approval once studies have shown it confers a clinical benefit.
Brentuximab vedotin has a boxed warning detailing the risk of progressive multifocal leukoencephalopathy associated with use of the drug. The drug has also been shown to pose a risk of pulmonary toxicity when combined with bleomycin. ![]()
Consolidation therapy with brentuximab vedotin can improve progression-free survival (PFS) for Hodgkin lymphoma (HL) patients who have undergone a transplant, according to a phase 3 study.
The trial, known as AETHERA, is a comparison of single-agent brentuximab vedotin to placebo in patients with HL who were at risk of relapse following autologous stem cell transplant (ASCT).
Brentuximab vedotin conferred a 75% improvement in PFS over placebo.
However, there was no significant difference in overall survival between the 2 treatment arms.
These results were recently announced by Seattle Genetics Inc. and Takeda Pharmaceutical Company Limited, the companies developing brentuximab vedotin (Adcetris).
The companies said more complete results from AETHERA will be presented at the 2014 ASH Annual Meeting in December.
AETHERA is a randomized, double-blind, placebo-controlled study designed to evaluate the potential of brentuximab vedotin to extend PFS post-ASCT in patients with HL who have at least one risk factor for progression. In addition to the primary endpoint of PFS, secondary endpoints included overall survival, safety, and tolerability.
Patients were eligible if they had risk factors for residual HL, defined as a history of refractory HL, those who relapse or progress within a year of receiving frontline chemotherapy, and/or those who have disease outside of the lymph nodes at the time of pre-ASCT relapse.
The study included 329 patients who received brentuximab vedotin or placebo every 3 weeks for up to a year.
The researchers assessed PFS a minimum of 2 years after the initiation of treatment for all patients. There was a significant improvement in PFS with brentuximab vedotin compared to placebo (hazard ratio=0.57; P=0.001).
However, a prespecified interim analysis of overall survival showed no significant difference between the treatment arms.
Patients in both arms who experienced progression received a variety of subsequent therapies. Most patients on the placebo arm received brentuximab vedotin after progression.
A further analysis of overall survival is planned in 2016. The safety profile of brentuximab vedotin in the AETHERA trial was generally consistent with the existing prescribing information.
“We anticipate reporting more complete AETHERA data at the ASH Annual Meeting in December and intend to submit a supplemental Biologics License Application to the FDA in 2015 for approval in this setting,” said Clay B. Siegall, PhD, President and Chief Executive Officer of Seattle Genetics.
The FDA has already granted brentuximab vedotin accelerated approval to treat HL patients after ASCT failure or after the failure of at least 2 prior multiagent chemotherapy regimens in patients who are not ASCT candidates. The FDA also granted the drug accelerated approval to treat systemic anaplastic large cell lymphoma after the failure of at least 1 prior multiagent chemotherapy regimen.
The European Commission granted brentuximab vedotin conditional marketing authorization for the same indications. In both cases, the drug can gain full, traditional approval once studies have shown it confers a clinical benefit.
Brentuximab vedotin has a boxed warning detailing the risk of progressive multifocal leukoencephalopathy associated with use of the drug. The drug has also been shown to pose a risk of pulmonary toxicity when combined with bleomycin. ![]()
The five “I’s” of electronic health records
It is clear at this point that physicians are not friends of electronic health records (EHRs). The predominant sentiment is that EHRs are costly (1/3 of providers are buying their second EHR system) and are poorly functional. Much has been said about the failures of EHRs. The shortcomings discussed have ranged from lack of cost benefits to interoperability with medical devices and security of interoperability with medical devices. Decreasing provider productivity and direct patient interaction time are also of concern. These opinions were raised in a ‘Medical Economics’ survey as well as a study by Rand Corporation. Interestingly, physicians do not desire to return to paper records. I will discuss what I call “The Five Important ‘I’s” of EHRs.
1. EHRs are not INTUITIVE. Navigating an EHR is akin to guessing what is behind Door Number 2 of “Let’s Make a Deal.” Documentation does not follow a provider’s thought process or the interaction workflow. It is built to meet regulatory and billing requirements. Many physicians are required to learn and be facile with different EHRs if they go to different hospitals. The AMA has called for a design overhaul of EHRs.
2. EHRs are IMPOSING. Providers are spending an inordinate amount of time with the EHR and less with patients. Most providers do not receive adequate training time, which is inversely related to privacy and security breaches. EHRs are inflexible and progressive practices with ambitious patient quality initiatives cannot implement them because of IT issues.
3. EHRs have limited INTEROPERABILITY. At a recent session of the Office of the National Coordinator at the annual conference of the American Health Information Management Association, Chief Science Officer Doug Fridsma laid out an ambitious vision of what he calls the “Learning Healthcare System” which comprises the building blocks of health IT systems. This will result in improved interoperability by way of the system adapting to change with encounters.
4. EHRs need INSIGHTFUL analytics. Data without good analytics is almost useless. Clinical decision support tools make EHRs pertinent insomuch as they can incorporate accepted practice guidelines as well as customized “best practice” decision support. These follow provider workflows and make the tool more intuitive. Add to this proscribing analytics that actually recommend (not prescribe) tests or treatment plans, and one ends up with a physician’s friend.
5. EHRs must INCLUDE robust portals. Robust patient portals will be critical in creating a true patient-centric health care system. Most portals used today are proprietary to the customer’s EHR vendor because of its low cost. There are some excellent third-party portals that have the ability to corral data from different providers who might have different EHR vendors. In addition, they are places to communicate multimedia content including video consultations.
While this list is not inclusive of all issues regarding EHRs, it serves as a focal point for discussion by clinicians about them. A 6th ‘I’ might be ‘IMMOBILE.’ A physician running back and forth to computer stations from patient beds creates self-evident inefficiencies. Presently available (not offered by most all vendors) mobile versions of EHRs have their own drawbacks. The small screen on a smartphone is a severe limitation, though many physicians do use tablets. That being said, in a 2013 cited survey by Black Book, only 8% of physician responders used the mobile EHR for purposes of ePrescribing, accessing records, ordering tests, or viewing results. As a champion of digital health technologies, I can only be frustrated about the vision I and many others have for their use. However, as with most technologies (and few have been as disruptive as EHRs) adoption in health care is slow. I look forward to leaders like Doug Fridsma and organizations like HIMSS, which has excellent representation by clinicians to help bring about necessary changes.
It is clear at this point that physicians are not friends of electronic health records (EHRs). The predominant sentiment is that EHRs are costly (1/3 of providers are buying their second EHR system) and are poorly functional. Much has been said about the failures of EHRs. The shortcomings discussed have ranged from lack of cost benefits to interoperability with medical devices and security of interoperability with medical devices. Decreasing provider productivity and direct patient interaction time are also of concern. These opinions were raised in a ‘Medical Economics’ survey as well as a study by Rand Corporation. Interestingly, physicians do not desire to return to paper records. I will discuss what I call “The Five Important ‘I’s” of EHRs.
1. EHRs are not INTUITIVE. Navigating an EHR is akin to guessing what is behind Door Number 2 of “Let’s Make a Deal.” Documentation does not follow a provider’s thought process or the interaction workflow. It is built to meet regulatory and billing requirements. Many physicians are required to learn and be facile with different EHRs if they go to different hospitals. The AMA has called for a design overhaul of EHRs.
2. EHRs are IMPOSING. Providers are spending an inordinate amount of time with the EHR and less with patients. Most providers do not receive adequate training time, which is inversely related to privacy and security breaches. EHRs are inflexible and progressive practices with ambitious patient quality initiatives cannot implement them because of IT issues.
3. EHRs have limited INTEROPERABILITY. At a recent session of the Office of the National Coordinator at the annual conference of the American Health Information Management Association, Chief Science Officer Doug Fridsma laid out an ambitious vision of what he calls the “Learning Healthcare System” which comprises the building blocks of health IT systems. This will result in improved interoperability by way of the system adapting to change with encounters.
4. EHRs need INSIGHTFUL analytics. Data without good analytics is almost useless. Clinical decision support tools make EHRs pertinent insomuch as they can incorporate accepted practice guidelines as well as customized “best practice” decision support. These follow provider workflows and make the tool more intuitive. Add to this proscribing analytics that actually recommend (not prescribe) tests or treatment plans, and one ends up with a physician’s friend.
5. EHRs must INCLUDE robust portals. Robust patient portals will be critical in creating a true patient-centric health care system. Most portals used today are proprietary to the customer’s EHR vendor because of its low cost. There are some excellent third-party portals that have the ability to corral data from different providers who might have different EHR vendors. In addition, they are places to communicate multimedia content including video consultations.
While this list is not inclusive of all issues regarding EHRs, it serves as a focal point for discussion by clinicians about them. A 6th ‘I’ might be ‘IMMOBILE.’ A physician running back and forth to computer stations from patient beds creates self-evident inefficiencies. Presently available (not offered by most all vendors) mobile versions of EHRs have their own drawbacks. The small screen on a smartphone is a severe limitation, though many physicians do use tablets. That being said, in a 2013 cited survey by Black Book, only 8% of physician responders used the mobile EHR for purposes of ePrescribing, accessing records, ordering tests, or viewing results. As a champion of digital health technologies, I can only be frustrated about the vision I and many others have for their use. However, as with most technologies (and few have been as disruptive as EHRs) adoption in health care is slow. I look forward to leaders like Doug Fridsma and organizations like HIMSS, which has excellent representation by clinicians to help bring about necessary changes.
It is clear at this point that physicians are not friends of electronic health records (EHRs). The predominant sentiment is that EHRs are costly (1/3 of providers are buying their second EHR system) and are poorly functional. Much has been said about the failures of EHRs. The shortcomings discussed have ranged from lack of cost benefits to interoperability with medical devices and security of interoperability with medical devices. Decreasing provider productivity and direct patient interaction time are also of concern. These opinions were raised in a ‘Medical Economics’ survey as well as a study by Rand Corporation. Interestingly, physicians do not desire to return to paper records. I will discuss what I call “The Five Important ‘I’s” of EHRs.
1. EHRs are not INTUITIVE. Navigating an EHR is akin to guessing what is behind Door Number 2 of “Let’s Make a Deal.” Documentation does not follow a provider’s thought process or the interaction workflow. It is built to meet regulatory and billing requirements. Many physicians are required to learn and be facile with different EHRs if they go to different hospitals. The AMA has called for a design overhaul of EHRs.
2. EHRs are IMPOSING. Providers are spending an inordinate amount of time with the EHR and less with patients. Most providers do not receive adequate training time, which is inversely related to privacy and security breaches. EHRs are inflexible and progressive practices with ambitious patient quality initiatives cannot implement them because of IT issues.
3. EHRs have limited INTEROPERABILITY. At a recent session of the Office of the National Coordinator at the annual conference of the American Health Information Management Association, Chief Science Officer Doug Fridsma laid out an ambitious vision of what he calls the “Learning Healthcare System” which comprises the building blocks of health IT systems. This will result in improved interoperability by way of the system adapting to change with encounters.
4. EHRs need INSIGHTFUL analytics. Data without good analytics is almost useless. Clinical decision support tools make EHRs pertinent insomuch as they can incorporate accepted practice guidelines as well as customized “best practice” decision support. These follow provider workflows and make the tool more intuitive. Add to this proscribing analytics that actually recommend (not prescribe) tests or treatment plans, and one ends up with a physician’s friend.
5. EHRs must INCLUDE robust portals. Robust patient portals will be critical in creating a true patient-centric health care system. Most portals used today are proprietary to the customer’s EHR vendor because of its low cost. There are some excellent third-party portals that have the ability to corral data from different providers who might have different EHR vendors. In addition, they are places to communicate multimedia content including video consultations.
While this list is not inclusive of all issues regarding EHRs, it serves as a focal point for discussion by clinicians about them. A 6th ‘I’ might be ‘IMMOBILE.’ A physician running back and forth to computer stations from patient beds creates self-evident inefficiencies. Presently available (not offered by most all vendors) mobile versions of EHRs have their own drawbacks. The small screen on a smartphone is a severe limitation, though many physicians do use tablets. That being said, in a 2013 cited survey by Black Book, only 8% of physician responders used the mobile EHR for purposes of ePrescribing, accessing records, ordering tests, or viewing results. As a champion of digital health technologies, I can only be frustrated about the vision I and many others have for their use. However, as with most technologies (and few have been as disruptive as EHRs) adoption in health care is slow. I look forward to leaders like Doug Fridsma and organizations like HIMSS, which has excellent representation by clinicians to help bring about necessary changes.

What are cancer survivors’ needs and how well are they being met?
ABSTRACT
Purpose This study sought to identify the needs and unmet needs of the growing number of adult cancer survivors.
Methods Vermont survivor advocates partnered with academic researchers to create a survivor registry and conduct a cross-sectional survey of cancer-related needs and unmet needs of adult survivors. The mailed survey addressed 53 specific needs in 5 domains based on prior research, contributions from the research partners, and pilot testing. Results were summarized by computing proportions who reported having needs met or unmet.
Results Survey participants included 1668 of 2005 individuals invited from the survivor registry (83%); 65.7% were ages 60 or older and 61.9% were women. These participants had received their diagnosis 2 to 16 years earlier; 77.5% had been diagnosed ≥5 years previously; 30.2% had at least one unmet need in the emotional, social, and spiritual (e) domain; just 14.4% had at least one unmet need in the economic and legal domain. The most commonly identified individual unmet needs were in the e and the information (i) domains and included “help reducing stress” (14.8% of all respondents) and “information about possible after effects of treatment” (14.4%).
Conclusions Most needs of these longer-term survivors were met, but substantial proportions of survivors identified unmet needs. Unmet needs such as information about late and long-term adverse effects of treatment could be met within clinical care with a cancer survivor care plan, but some survivors may require referral to services focused on stress and coping.
Following a successful course of treatment for cancer, many patients return to or remain in the care of their primary care physician (PCP). What often goes unrecognized, however, are these cancer survivors’ unique needs—physical, psychological, social, spiritual, economic, and legal—and the informational and professional services available to address them.1,2
Increased cancer survival creates new needs. There are already >12 million cancer survivors in the United States and >30 million worldwide.3 As baby boomers age, the number of cancers diagnosed over the next 45 years will double4 and improved diagnosis and treatments are already prolonging survivors’ lives. With the greater number of cancer survivors and longer survival time, a cancer survivorship advocacy community has developed to help identify and address the concerns, needs, and benefits of having lived with, through, and beyond a cancer diagnosis.
The purpose of our study. Some of these areas of need have been studied extensively with childhood survivors, breast cancer survivors, and, more recently, prostate cancer survivors. However, few studies have examined adult survivors from all cancer types5-9 or have had cohorts large enough to yield meaningful information.5,7-9 The aim of this study was to describe the needs of adult survivors of all cancer types in a general population from Vermont and to determine whether these needs were met. The results of this study can help identify the services needed by cancer survivors.
METHODS
Population and sample
In November 2009, we invited all survivors listed in a cancer survivor registry to complete a 12-page survey. The registry10 was created as part of the Cancer Survivor Community Study, a community-based participatory research project funded by the National Cancer Institute. The study’s Steering Committee was comprised of cancer survivors, cancer registrars, and researchers. We identified and invited cancer survivors from 4 hospital registries in northwest and central Vermont to participate. Registry participants who indicated willingness to enroll in research studies received an invitation letter and informed consent form, the 12-page survey, and an addressed and stamped return envelope. We obtained Institutional Review Board (IRB) approval for these procedures at the University of Vermont and at local hospital IRBs.
Instrument development
A working group from the Steering Committee reviewed a range of available instruments to assess cancer survivors’ needs.9,11-15 We determined that the survey most relevant to our objectives was the Cancer Survivors’ Unmet Needs (CaSun) instrument.13 Because CaSun was developed in Australia, we carefully examined each question for appropriateness to our target audience. We eliminated several questions that we thought less important, added questions from other instruments, and simplified the survey format. Survivors from the Steering Committee pilot tested the draft questionnaire to identify awkward wording or concepts.
We piloted the revised draft using a standardized feedback form with cancer survivors who were not connected to our project and not enrolled in the survivor registry, and with residents at a senior center. Students and a teacher from an Adult Basic Education program helped to ensure easy readability. Our final instrument had 53 questions about needs in 5 domains. Questions within each domain completed the lead-in, “Since your cancer diagnosis, did you need....” We asked participants to check only 1 of the 3 boxes to the right of each question to indicate that there was no need in that area, that there was a need and it was met, or that there was a need and it was not met. We obtained self-reported demographic data during enrollment in the registry.
Data analysis
We summarized data by computing the percent of survivors who reported having each need (either met or unmet) and the percent for whom the need was unmet. The latter was computed both as a percent of all survivors and as a percent of those who had the need. We also calculated the percentage of survivors that had at least one need and at least one unmet need in each domain, as well as the average number of needs per survivor in each domain. We used SPSS for Unix, Release 6.1 (AIX 3.2)(IBM, Armonk, New York).
RESULTS
Of the 2005 cancer survivors invited into the study, 1668 responded, yielding a participation rate of 83%. TABLE 1 describes the self-reported demographic and cancer characteristics of participants in this study. Most respondents were female, ≥60 years old, urban dwellers, married or with a partner, well educated, and had household incomes of ≥$50,000. There were more breast cancer survivors than survivors of other cancers, and 14.6% of all survivors reported being diagnosed with more than one cancer. Cancer was diagnosed at stages 1 or 2 for 78.3% of the participants; 61.9% reported having undergone ≥2 treatment regimens.
The survey addressed needs in 5 domains: access to care and services (A); information (I); emotional, social, and spiritual (E); physical (P); and economic and legal (L). More than 80% of respondents reported having at least one need in the A, I, and E domains. The E domain had the most survivors with at least one unmet need (N=503), followed by the I (N=410) and P (N=375) domains.
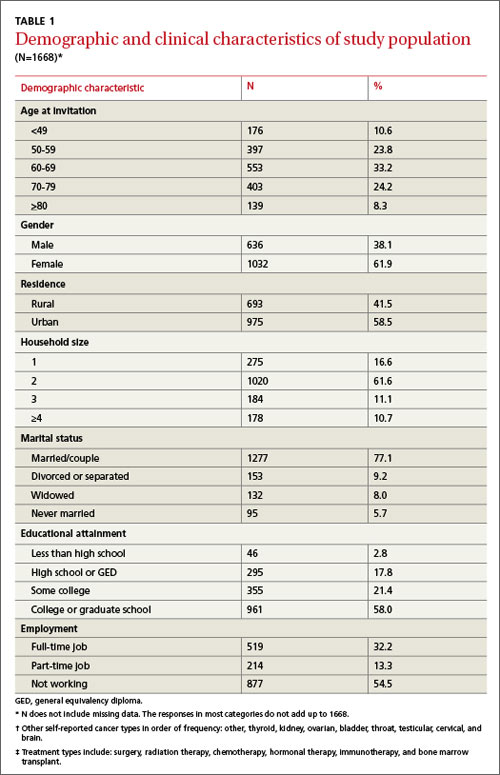
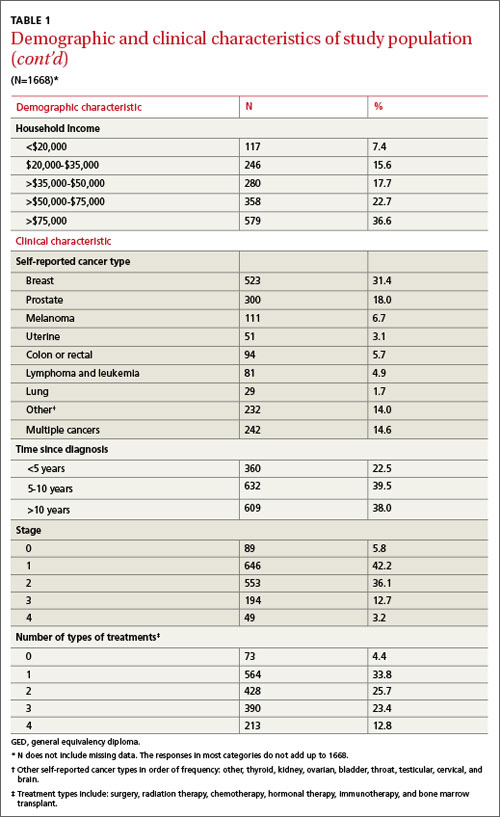
Identifying unmet needs. TABLE 2 shows results for the specific questions within the domains in the order they were asked. Most participants who had a need also had it met. However, some needs that were not commonly reported were deemed unmet by a large proportion of those who expressed the need. For example, the A need for “A case manager to whom you could go to find out about services whenever they were needed” (A5) was reported by only 29.1% of survivors. But 32.1% of those reporting the need said it was unmet, which corresponds to 9.4% of all study participants having the need unmet. Similarly, the need for “More information about complementary and alternative medicine” (I3) was reported by about a quarter of the study population, 41.4% of whom (9.8% of all participants) reported it as unmet. In the P domain, the need for “Help to address problems with your sex life” (P4) was reported by only 26.5% of the respondents; yet 40.7% of those reporting the need had it unmet. Similarly, in the L domain, “Help with life insurance concerns as a result of your cancer” (L3) was only reported by 10.9% of the participants but was unmet for 46.4% of those who reported the need, or 5% of all study participants.
Most commonly expressed needs. TABLE 2 also identifies 12 needs reported by ≥50% of participants. Three of these needs were in the A domain, 6 in the I domain, and 3 in the E domain. The 2 most common needs related to A: the need “To feel like you were managing your health together with the medical team” (A3) was reported by 68.6% and was viewed as unmet by 5.2% of all respondents; the need for “Access to screening for recurrence or other cancers” (A7) was reported by 63.8% of the survivors but was deemed unmet by only 3.1% of all the respondents. “More information about possible after effects of your treatment” (I5) was a need for 63.2% that went unmet in 22.9% (14.4% of all participants). “Help managing your concerns about the cancer coming back” (E13) was reported as a need by 54.1% and as unmet by 11.8% of all participants.
The rank order of 7 unmet needs reported by ≥10% of the participants is shown in TABLE 3. Four of the 7 unmet needs were in the E domain. The most common unmet need in this domain was “Help reducing stress in your life” (E19).
Only 3 needs were both commonly reported and also unmet for at least 10% of the participants: “More information about possible after effects of your treatment” (I5), “More information about possible side effects of your treatment” (I4), and “Help managing your concerns about the cancer coming back” (E13).
DISCUSSION
The survey instrument we used to assess the needs of cancer survivors in a large community-based registry included a detailed list of potential needs generated, in part, by representatives of the survivor community. Most cancer survivor needs mentioned in this survey were met. However, some needs were not met for substantial proportions of respondents and should be examined carefully to determine whether services could be improved to better address them. This study was planned and implemented by researchers and cancer survivors using community-based participatory principles to learn about local needs. The results of this study may be generalizable to similar populations of survivors and will inform the survivorship goals for the Vermont State Cancer Plan and future Vermont Cancer Survivor Network activities.
Acting on patients’ expressed needs. Over 80% of participants had needs in the A, I, and E domains. The most commonly reported need was in the A domain, “To feel like you were managing your health together with the medical team” (A3). It was also a top need in other studies that asked this question.16,17 A cancer diagnosis may cause patients to feel out of control. Participation in the management of their health may help them gain a greater sense of control. PCP accommodation of expressed patient preferences may be an important part of a cancer survivor’s long-term adaptation to the disease.
Six of the 12 most frequently reported needs and 2 frequently reported unmet needs were in the I domain. Communication of information increases patients’ involvement in decision-making and enables them to cope better during diagnosis, treatment, and follow-up.18 “More information about possible after effects of your treatment” and “More information about possible side effects of your treatment” were reported by a high proportion of participants, and many also reported these needs as unmet. In another study about health-related information needs of survivors, 52% wanted more information about “What late and long-term side effects of cancer treatment are expected”19; and in a 2005 review of information needs, 12% of survivors reported similar needs.20 Two recent articles also noted such needs in adolescent and young adult cancer survivors.21,22 Based on current evidence, it would be advisable to discuss anticipated effects of treatment with patients not only at the outset but also at the end of treatment, and to write it in a cancer survivor care plan.
Individual needs that were not met for at least 10% of respondents, regardless of how common the need (TABLE 3), provided additional insights. Among these 7 needs, 3 also were reported as a need by more than 50% of respondents (TABLE 2), and 4 by <50%, indicating that some less common needs are not being met adequately. Among these 7 prominent unmet needs, 4 were E Issues (TABLE 3) and 2 were I Issues.
Unmet needs are an opportunity to improve care. In our study and in others, E needs were most likely to be unmet.17,23-26 Among the 4 common unmet E needs, 2 (E19 and E11) focused on generalized stress and worry, and one (E15) focused on concern about illness impact on family members or partners. Although these issues may be challenging to address successfully in a typical clinical environment, others have confirmed the importance of these needs and proposed ways to meet them.27 The fourth most common unmet E need focused on concern about cancer recurrence, also a prominent need found in other studies.15-17 These needs might be addressed more adequately in the course of usual clinical care by PCPs or specialists. In fact, the American College of Surgeons’ Commission on Cancer 2012 standards now require psychosocial distress screening and the provision of referral for psychosocial services.24 Our results are consistent in many respects with prior studies of needs reported by cancer survivors in other countries. The CaSUN survey developed by Hodgkinson et al13 has been applied to several survivor populations in Australia. In a diverse survivor sample, specific E, I, and A issues were frequently reported as unmet needs.13 The most prominent unmet needs in a gynecologic cancer sample using CaSUN focused on emotional and social issues such as worry, stress, coping, and relationships with, and expectations of, others.25
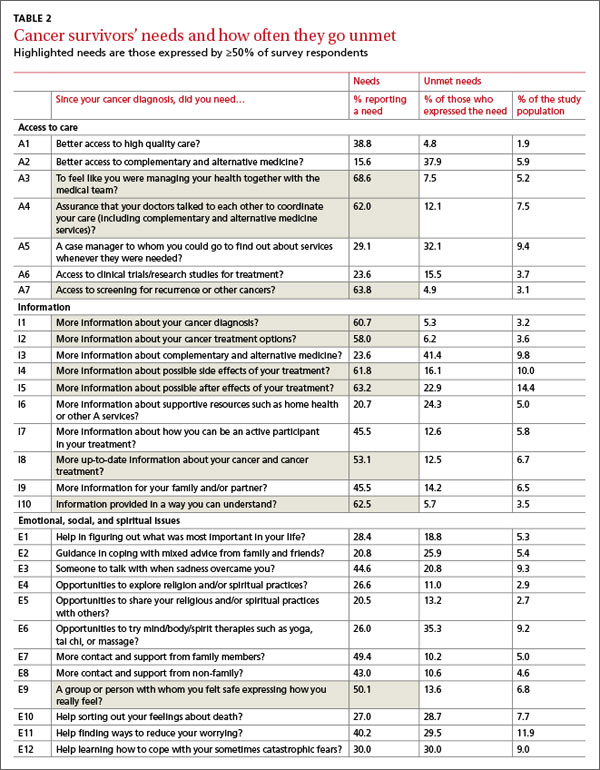
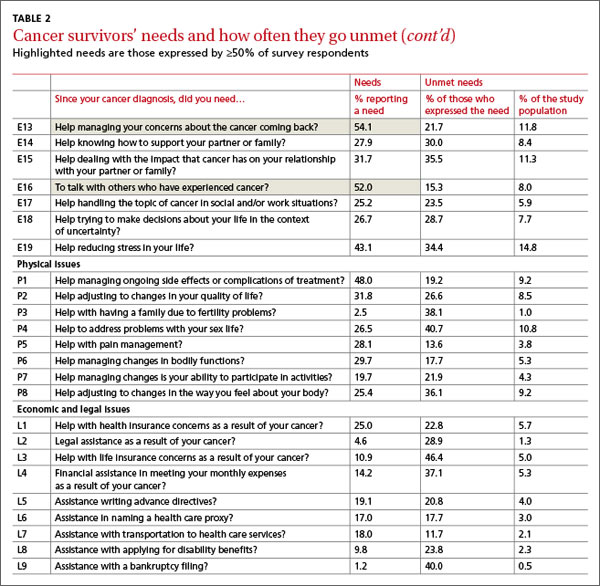
Barg et al23 conducted a survey of unmet needs in the United States using a detailed list based on prior survivor research and targeting individuals in a cancer registry. The most prominent area of need expressed was “emotional,” similar to the high rank of E needs in our study. In contrast to our study, however, physical and financial issues also were prominent. The latter variances might be explained by differences in access to care, or perhaps the study’s low response rate (23.8%). A similar survey reported by Campbell et al12 identified needs in the emotional domain as the most cited unmet survivor needs based on psychometrically developed subscales of a 152-item survey (29% response rate).
These results from several studies, including ours, call for more detailed exploration of the E needs of long-term cancer survivors. A useful framework developed by Stein et al28 accounts for factors contributing to cancer stress and burden as well as resources available to survivors (intrapersonal, social, informational, and tangible services), with the interactions between these 2 domains determining how well a survivor will be able to cope. There clearly is a role for development of more effective communication channels and focused services to meet survivor needs.
The list of most common unmet needs in TABLE 3 also includes a focus on “problems with your sex life” (P4). This is an area that may be difficult to address in a cancer care setting because of the focus on disease management. Primary care providers might be better prepared to address this issue because they likely encounter similar issues among the wide range of patients they serve. However, a recent study reported that only 46% of internists were somewhat or likely to initiate a discussion about sexuality with cancer survivors.29 Some additional preparation for physicians to address this need might be warranted.
The proportion in this sample reporting needs for access to, or information about, complementary and alternative medicine services fell below the thresholds chosen to designate common needs in this study. Although reported use is relatively common among cancer survivor in several studies,30-32 it appears that in our survivor sample, those who were interested in these approaches encountered only moderate barriers.
Study limitations. We invited participants from a registry unlikely to include cancer survivors with lower educational attainment or from rural locations9—that is, our participants were less likely to have challenges in obtaining appropriate services and information. This sample limitation therefore likely underestimates the overall level of needs among cancer survivors.
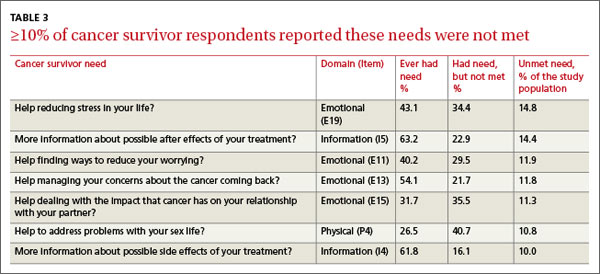
This was a cross-sectional assessment of perceived needs among a diverse group of survivors, which may have overlooked needs that were met but only after considerable effort on the part of survivors. Longitudinal studies would provide more complete accounts of how readily needs are met and the changes in needs at different times in the continuum of care.
The Vermont population is less diverse racially and ethnically, but not with respect to household income or educational attainment, than the overall US population. Access to health care also is relatively high in Vermont compared with many other states. According to a 2009 Vermont Household Health Insurance Survey, only 7.6% of Vermonters are uninsured.33
CORRESPONDENCE
Berta Geller, EdD, University of Vermont, Health Promotion Research/Family Medicine, 1 South Prospect Street, Burlington, VT 05401-3444; [email protected]
ACKNOWLEDGEMENTS
We would like to thank Anne Dorwaldt, Kathy Howe, Mark Bowman and John Mace at the University of Vermont, and the Cancer Survivor Community Study Steering Committee for their contributions to the successful completion of this study. We also thank the cancer survivors who participated in the pilot testing and the overall survey.
1. Potosky A, Han PK, Rowland J, et al. Differences between primary care physicians’ and oncologists’ knowledge, attitudes and practices regarding the care of cancer survivors. J Gen Intern Med. 2011;26:1403-1410.
2. Rowland J. Survivorship research: past, present, and future. In: Chang AE, Ganz PA, Hayes DF, et al, eds. Oncology: An Evidence-Based Approach. New York, NY: Springer; 2005:1753-1767.
3. Jemal A, Center MM, DeSantis C, et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-1907.
4. Hewitt ME, Greenfield S, Stovall E, eds. From Cancer Patient to Cancer Survivor: Lost in Transition. Washington, DC: National Academies Press; 2005.
5. Hawkins NA, Smith T, Zhao L, et al. Health-related behavior change after cancer: results of the American Cancer Society’s studies of cancer survivors (SCS). J Cancer Surviv. 2010;4:20-32.
6. Katz ML, Reiter PL, Corbin S, et al. Are rural Ohio Appalachia cancer survivors needs different than urban cancer survivors? J Cancer Surviv. 2010;4:140-148.
7. Yabroff KR, Lawrence WF, Clauser S, et al. Burden of illness in cancer survivors: findings from a population-based national sample. J Natl Cancer Inst. 2004;96:1322-1330.
8. Sanson-Fisher R, Girgis A, Boyes A, et al; Supportive Care Review Group. The unmet supportive care needs of patients with cancer. Cancer. 2000;88:226-237.
9. Smith T, Stein KD, Mehta CC, et al. The rationale, design, and implementation of the American Cancer Society’s studies of cancer survivors. Cancer. 2007;109:1-12.
10. Geller BM, Mace J, Vacek P, et al. Are cancer survivors willing to participate in research? J Community Health. 2011;36: 772-778.
11. Crespi CM, Ganz PA, Petersen L, et al. Refinement and psychometric evaluation of the impact of cancer scale. J Natl Cancer Inst. 2008;100:1530-1541.
12. Campbell HS, Sanson-Fisher R, Turner D, et al. Psychometric properties of cancer survivors’ unmet needs survey. Support Care Cancer. 2010;19:221-230.
13. Hodgkinson K, Butow P, Hunt GE, et al. The development and evaluation of a measure to assess cancer survivors’ unmet supportive care needs: the CaSUN (Cancer Survivors’ Unmet Needs measure). Psychooncology. 2007;16:796-804.
14. Zebrack BJ, Ganz PA, Bernaards CA, et al. Assessing the impact of cancer: development of a new instrument for long-term survivors. Psychooncology. 2006;15:407-421.
15. Arora NK, Hesse BW, Rimer BK, et al. Frustrated and confused: the American public rates its cancer-related information-seeking experiences. J Gen Intern Med. 2008;23:223-228.
16. Hodgkinson K, Butow P, Hunt GE, et al. Breast cancer survivors’ supportive care needs 2-10 years after diagnosis. Support Care Cancer. 2007;15:515-523.
17. Harrison SE, Watson EK, Ward AM, et al. Primary health and supportive care needs of long-term cancer survivors: a questionnaire study. J Clin Oncol. 2011;29:2091-2098.
18. Rutten LJ, Squiers L, Treiman K. Requests for information by family and friends of cancer patients calling the National Cancer Institute’s Cancer Information Service. Psychooncology. 2006;15:664-672.
19. Beckjord EB, Arora NK, McLaughlin W, et al. Health-related information needs in a large and diverse sample of adult cancer survivors: implications for cancer care. J Cancer Surviv. 2008;2:179-189.
20. Rutten LJ, Arora NK, Bakos AD, et al. Information needs and sources of information among cancer patients: a systematic review of research (1980-2003). Patient Educ Couns. 2005;57:250-261.
21. Zebrack BJ, Block R, Hayes-Lattin B, et al. Psychosocial service use and unmet need among recently diagnosed adolescent and youg adult patients. Cancer. 2013;119:201-214.
22. Keegan TH, Lichtensztajn DY, Kato I, et al. Unmet adolescent and young adult cancer survivors information and service needs: a population-based cancer registry study. J Cancer Surviv. 2012;6:239-250.
23. Barg KF, Cronholm PF, Straton JB, et al. Unmet psychosocial needs of Pennsylvanians with cancer: 1996-2005. Cancer. 2007;110:631-639.
24. American College of Surgeons. Cancer Program Standards 2012: Ensuring Patient-Centered Care. Chicago, IL: American College of Surgeons; 2011.
25. Hodgkinson K, Butow P, Fuchs A, et al. Long-term survival from gynecologic cancer: psychosocial outcomes, supportive care needs and positive outcomes. Gynecol Oncol. 2007;104:381-389.
26. Houts PS, Yasko JM, Kahn SB, et al. Unmet psychological, social, and economic needs of persons with cancer in Pennsylvania. Cancer. 1986;58:2355-2361.
27. Holland JC, Reznik I. Pathways for psychosocial care of cancer survivors. Cancer. 2005;104(11 suppl):2624-2637.
28. Stein KD, Syrjala KL, Andrykowski MA. Physical and psychological long-term and late effects of cancer. Cancer. 2008;112(11 suppl):2577-2592.
29. Park ER, Bober SL, Campbell EG, et al. General: Internist communication about sexual function with cancer survivors. J Gen Intern Med. 2009;24(suppl 2):S407-S411.
30. Girgis A, Adams J, Sibbritt D. The use of complementary and alternative therapies by patients with cancer. Oncol Res. 2005;15:281-289.
31. Gansler T, Kaw C, Crammer C, et al. A population-based study of prevalence of complementary methods use by cancer survivors: a report from the American Cancer Society’s studies of cancer survivors. Cancer. 2008;113:1048-1057.
32. Fouladbakhsh JM, Stommel M. Gender, symptom experience, and use of complementary and alternative medicine practices among cancer survivors in the U.S. cancer population. Oncol Nurs Forum. 2010;37:E7-E15.
33. Vermont Department of Financial Regulation. Vermont Household Health Insurance Survey (VHHIS). Vermont Department of Financial Regulation Web site. Available at: http://www.dfr.vermont.gov/insurance/health-insurance/vermont-household-health-insurance-survey-vhhis. Accessed July 15, 2014.
ABSTRACT
Purpose This study sought to identify the needs and unmet needs of the growing number of adult cancer survivors.
Methods Vermont survivor advocates partnered with academic researchers to create a survivor registry and conduct a cross-sectional survey of cancer-related needs and unmet needs of adult survivors. The mailed survey addressed 53 specific needs in 5 domains based on prior research, contributions from the research partners, and pilot testing. Results were summarized by computing proportions who reported having needs met or unmet.
Results Survey participants included 1668 of 2005 individuals invited from the survivor registry (83%); 65.7% were ages 60 or older and 61.9% were women. These participants had received their diagnosis 2 to 16 years earlier; 77.5% had been diagnosed ≥5 years previously; 30.2% had at least one unmet need in the emotional, social, and spiritual (e) domain; just 14.4% had at least one unmet need in the economic and legal domain. The most commonly identified individual unmet needs were in the e and the information (i) domains and included “help reducing stress” (14.8% of all respondents) and “information about possible after effects of treatment” (14.4%).
Conclusions Most needs of these longer-term survivors were met, but substantial proportions of survivors identified unmet needs. Unmet needs such as information about late and long-term adverse effects of treatment could be met within clinical care with a cancer survivor care plan, but some survivors may require referral to services focused on stress and coping.
Following a successful course of treatment for cancer, many patients return to or remain in the care of their primary care physician (PCP). What often goes unrecognized, however, are these cancer survivors’ unique needs—physical, psychological, social, spiritual, economic, and legal—and the informational and professional services available to address them.1,2
Increased cancer survival creates new needs. There are already >12 million cancer survivors in the United States and >30 million worldwide.3 As baby boomers age, the number of cancers diagnosed over the next 45 years will double4 and improved diagnosis and treatments are already prolonging survivors’ lives. With the greater number of cancer survivors and longer survival time, a cancer survivorship advocacy community has developed to help identify and address the concerns, needs, and benefits of having lived with, through, and beyond a cancer diagnosis.
The purpose of our study. Some of these areas of need have been studied extensively with childhood survivors, breast cancer survivors, and, more recently, prostate cancer survivors. However, few studies have examined adult survivors from all cancer types5-9 or have had cohorts large enough to yield meaningful information.5,7-9 The aim of this study was to describe the needs of adult survivors of all cancer types in a general population from Vermont and to determine whether these needs were met. The results of this study can help identify the services needed by cancer survivors.
METHODS
Population and sample
In November 2009, we invited all survivors listed in a cancer survivor registry to complete a 12-page survey. The registry10 was created as part of the Cancer Survivor Community Study, a community-based participatory research project funded by the National Cancer Institute. The study’s Steering Committee was comprised of cancer survivors, cancer registrars, and researchers. We identified and invited cancer survivors from 4 hospital registries in northwest and central Vermont to participate. Registry participants who indicated willingness to enroll in research studies received an invitation letter and informed consent form, the 12-page survey, and an addressed and stamped return envelope. We obtained Institutional Review Board (IRB) approval for these procedures at the University of Vermont and at local hospital IRBs.
Instrument development
A working group from the Steering Committee reviewed a range of available instruments to assess cancer survivors’ needs.9,11-15 We determined that the survey most relevant to our objectives was the Cancer Survivors’ Unmet Needs (CaSun) instrument.13 Because CaSun was developed in Australia, we carefully examined each question for appropriateness to our target audience. We eliminated several questions that we thought less important, added questions from other instruments, and simplified the survey format. Survivors from the Steering Committee pilot tested the draft questionnaire to identify awkward wording or concepts.
We piloted the revised draft using a standardized feedback form with cancer survivors who were not connected to our project and not enrolled in the survivor registry, and with residents at a senior center. Students and a teacher from an Adult Basic Education program helped to ensure easy readability. Our final instrument had 53 questions about needs in 5 domains. Questions within each domain completed the lead-in, “Since your cancer diagnosis, did you need....” We asked participants to check only 1 of the 3 boxes to the right of each question to indicate that there was no need in that area, that there was a need and it was met, or that there was a need and it was not met. We obtained self-reported demographic data during enrollment in the registry.
Data analysis
We summarized data by computing the percent of survivors who reported having each need (either met or unmet) and the percent for whom the need was unmet. The latter was computed both as a percent of all survivors and as a percent of those who had the need. We also calculated the percentage of survivors that had at least one need and at least one unmet need in each domain, as well as the average number of needs per survivor in each domain. We used SPSS for Unix, Release 6.1 (AIX 3.2)(IBM, Armonk, New York).
RESULTS
Of the 2005 cancer survivors invited into the study, 1668 responded, yielding a participation rate of 83%. TABLE 1 describes the self-reported demographic and cancer characteristics of participants in this study. Most respondents were female, ≥60 years old, urban dwellers, married or with a partner, well educated, and had household incomes of ≥$50,000. There were more breast cancer survivors than survivors of other cancers, and 14.6% of all survivors reported being diagnosed with more than one cancer. Cancer was diagnosed at stages 1 or 2 for 78.3% of the participants; 61.9% reported having undergone ≥2 treatment regimens.
The survey addressed needs in 5 domains: access to care and services (A); information (I); emotional, social, and spiritual (E); physical (P); and economic and legal (L). More than 80% of respondents reported having at least one need in the A, I, and E domains. The E domain had the most survivors with at least one unmet need (N=503), followed by the I (N=410) and P (N=375) domains.
Identifying unmet needs. TABLE 2 shows results for the specific questions within the domains in the order they were asked. Most participants who had a need also had it met. However, some needs that were not commonly reported were deemed unmet by a large proportion of those who expressed the need. For example, the A need for “A case manager to whom you could go to find out about services whenever they were needed” (A5) was reported by only 29.1% of survivors. But 32.1% of those reporting the need said it was unmet, which corresponds to 9.4% of all study participants having the need unmet. Similarly, the need for “More information about complementary and alternative medicine” (I3) was reported by about a quarter of the study population, 41.4% of whom (9.8% of all participants) reported it as unmet. In the P domain, the need for “Help to address problems with your sex life” (P4) was reported by only 26.5% of the respondents; yet 40.7% of those reporting the need had it unmet. Similarly, in the L domain, “Help with life insurance concerns as a result of your cancer” (L3) was only reported by 10.9% of the participants but was unmet for 46.4% of those who reported the need, or 5% of all study participants.
Most commonly expressed needs. TABLE 2 also identifies 12 needs reported by ≥50% of participants. Three of these needs were in the A domain, 6 in the I domain, and 3 in the E domain. The 2 most common needs related to A: the need “To feel like you were managing your health together with the medical team” (A3) was reported by 68.6% and was viewed as unmet by 5.2% of all respondents; the need for “Access to screening for recurrence or other cancers” (A7) was reported by 63.8% of the survivors but was deemed unmet by only 3.1% of all the respondents. “More information about possible after effects of your treatment” (I5) was a need for 63.2% that went unmet in 22.9% (14.4% of all participants). “Help managing your concerns about the cancer coming back” (E13) was reported as a need by 54.1% and as unmet by 11.8% of all participants.
The rank order of 7 unmet needs reported by ≥10% of the participants is shown in TABLE 3. Four of the 7 unmet needs were in the E domain. The most common unmet need in this domain was “Help reducing stress in your life” (E19).
Only 3 needs were both commonly reported and also unmet for at least 10% of the participants: “More information about possible after effects of your treatment” (I5), “More information about possible side effects of your treatment” (I4), and “Help managing your concerns about the cancer coming back” (E13).
DISCUSSION
The survey instrument we used to assess the needs of cancer survivors in a large community-based registry included a detailed list of potential needs generated, in part, by representatives of the survivor community. Most cancer survivor needs mentioned in this survey were met. However, some needs were not met for substantial proportions of respondents and should be examined carefully to determine whether services could be improved to better address them. This study was planned and implemented by researchers and cancer survivors using community-based participatory principles to learn about local needs. The results of this study may be generalizable to similar populations of survivors and will inform the survivorship goals for the Vermont State Cancer Plan and future Vermont Cancer Survivor Network activities.
Acting on patients’ expressed needs. Over 80% of participants had needs in the A, I, and E domains. The most commonly reported need was in the A domain, “To feel like you were managing your health together with the medical team” (A3). It was also a top need in other studies that asked this question.16,17 A cancer diagnosis may cause patients to feel out of control. Participation in the management of their health may help them gain a greater sense of control. PCP accommodation of expressed patient preferences may be an important part of a cancer survivor’s long-term adaptation to the disease.
Six of the 12 most frequently reported needs and 2 frequently reported unmet needs were in the I domain. Communication of information increases patients’ involvement in decision-making and enables them to cope better during diagnosis, treatment, and follow-up.18 “More information about possible after effects of your treatment” and “More information about possible side effects of your treatment” were reported by a high proportion of participants, and many also reported these needs as unmet. In another study about health-related information needs of survivors, 52% wanted more information about “What late and long-term side effects of cancer treatment are expected”19; and in a 2005 review of information needs, 12% of survivors reported similar needs.20 Two recent articles also noted such needs in adolescent and young adult cancer survivors.21,22 Based on current evidence, it would be advisable to discuss anticipated effects of treatment with patients not only at the outset but also at the end of treatment, and to write it in a cancer survivor care plan.
Individual needs that were not met for at least 10% of respondents, regardless of how common the need (TABLE 3), provided additional insights. Among these 7 needs, 3 also were reported as a need by more than 50% of respondents (TABLE 2), and 4 by <50%, indicating that some less common needs are not being met adequately. Among these 7 prominent unmet needs, 4 were E Issues (TABLE 3) and 2 were I Issues.
Unmet needs are an opportunity to improve care. In our study and in others, E needs were most likely to be unmet.17,23-26 Among the 4 common unmet E needs, 2 (E19 and E11) focused on generalized stress and worry, and one (E15) focused on concern about illness impact on family members or partners. Although these issues may be challenging to address successfully in a typical clinical environment, others have confirmed the importance of these needs and proposed ways to meet them.27 The fourth most common unmet E need focused on concern about cancer recurrence, also a prominent need found in other studies.15-17 These needs might be addressed more adequately in the course of usual clinical care by PCPs or specialists. In fact, the American College of Surgeons’ Commission on Cancer 2012 standards now require psychosocial distress screening and the provision of referral for psychosocial services.24 Our results are consistent in many respects with prior studies of needs reported by cancer survivors in other countries. The CaSUN survey developed by Hodgkinson et al13 has been applied to several survivor populations in Australia. In a diverse survivor sample, specific E, I, and A issues were frequently reported as unmet needs.13 The most prominent unmet needs in a gynecologic cancer sample using CaSUN focused on emotional and social issues such as worry, stress, coping, and relationships with, and expectations of, others.25
Barg et al23 conducted a survey of unmet needs in the United States using a detailed list based on prior survivor research and targeting individuals in a cancer registry. The most prominent area of need expressed was “emotional,” similar to the high rank of E needs in our study. In contrast to our study, however, physical and financial issues also were prominent. The latter variances might be explained by differences in access to care, or perhaps the study’s low response rate (23.8%). A similar survey reported by Campbell et al12 identified needs in the emotional domain as the most cited unmet survivor needs based on psychometrically developed subscales of a 152-item survey (29% response rate).
These results from several studies, including ours, call for more detailed exploration of the E needs of long-term cancer survivors. A useful framework developed by Stein et al28 accounts for factors contributing to cancer stress and burden as well as resources available to survivors (intrapersonal, social, informational, and tangible services), with the interactions between these 2 domains determining how well a survivor will be able to cope. There clearly is a role for development of more effective communication channels and focused services to meet survivor needs.
The list of most common unmet needs in TABLE 3 also includes a focus on “problems with your sex life” (P4). This is an area that may be difficult to address in a cancer care setting because of the focus on disease management. Primary care providers might be better prepared to address this issue because they likely encounter similar issues among the wide range of patients they serve. However, a recent study reported that only 46% of internists were somewhat or likely to initiate a discussion about sexuality with cancer survivors.29 Some additional preparation for physicians to address this need might be warranted.
The proportion in this sample reporting needs for access to, or information about, complementary and alternative medicine services fell below the thresholds chosen to designate common needs in this study. Although reported use is relatively common among cancer survivor in several studies,30-32 it appears that in our survivor sample, those who were interested in these approaches encountered only moderate barriers.
Study limitations. We invited participants from a registry unlikely to include cancer survivors with lower educational attainment or from rural locations9—that is, our participants were less likely to have challenges in obtaining appropriate services and information. This sample limitation therefore likely underestimates the overall level of needs among cancer survivors.
This was a cross-sectional assessment of perceived needs among a diverse group of survivors, which may have overlooked needs that were met but only after considerable effort on the part of survivors. Longitudinal studies would provide more complete accounts of how readily needs are met and the changes in needs at different times in the continuum of care.
The Vermont population is less diverse racially and ethnically, but not with respect to household income or educational attainment, than the overall US population. Access to health care also is relatively high in Vermont compared with many other states. According to a 2009 Vermont Household Health Insurance Survey, only 7.6% of Vermonters are uninsured.33
CORRESPONDENCE
Berta Geller, EdD, University of Vermont, Health Promotion Research/Family Medicine, 1 South Prospect Street, Burlington, VT 05401-3444; [email protected]
ACKNOWLEDGEMENTS
We would like to thank Anne Dorwaldt, Kathy Howe, Mark Bowman and John Mace at the University of Vermont, and the Cancer Survivor Community Study Steering Committee for their contributions to the successful completion of this study. We also thank the cancer survivors who participated in the pilot testing and the overall survey.
ABSTRACT
Purpose This study sought to identify the needs and unmet needs of the growing number of adult cancer survivors.
Methods Vermont survivor advocates partnered with academic researchers to create a survivor registry and conduct a cross-sectional survey of cancer-related needs and unmet needs of adult survivors. The mailed survey addressed 53 specific needs in 5 domains based on prior research, contributions from the research partners, and pilot testing. Results were summarized by computing proportions who reported having needs met or unmet.
Results Survey participants included 1668 of 2005 individuals invited from the survivor registry (83%); 65.7% were ages 60 or older and 61.9% were women. These participants had received their diagnosis 2 to 16 years earlier; 77.5% had been diagnosed ≥5 years previously; 30.2% had at least one unmet need in the emotional, social, and spiritual (e) domain; just 14.4% had at least one unmet need in the economic and legal domain. The most commonly identified individual unmet needs were in the e and the information (i) domains and included “help reducing stress” (14.8% of all respondents) and “information about possible after effects of treatment” (14.4%).
Conclusions Most needs of these longer-term survivors were met, but substantial proportions of survivors identified unmet needs. Unmet needs such as information about late and long-term adverse effects of treatment could be met within clinical care with a cancer survivor care plan, but some survivors may require referral to services focused on stress and coping.
Following a successful course of treatment for cancer, many patients return to or remain in the care of their primary care physician (PCP). What often goes unrecognized, however, are these cancer survivors’ unique needs—physical, psychological, social, spiritual, economic, and legal—and the informational and professional services available to address them.1,2
Increased cancer survival creates new needs. There are already >12 million cancer survivors in the United States and >30 million worldwide.3 As baby boomers age, the number of cancers diagnosed over the next 45 years will double4 and improved diagnosis and treatments are already prolonging survivors’ lives. With the greater number of cancer survivors and longer survival time, a cancer survivorship advocacy community has developed to help identify and address the concerns, needs, and benefits of having lived with, through, and beyond a cancer diagnosis.
The purpose of our study. Some of these areas of need have been studied extensively with childhood survivors, breast cancer survivors, and, more recently, prostate cancer survivors. However, few studies have examined adult survivors from all cancer types5-9 or have had cohorts large enough to yield meaningful information.5,7-9 The aim of this study was to describe the needs of adult survivors of all cancer types in a general population from Vermont and to determine whether these needs were met. The results of this study can help identify the services needed by cancer survivors.
METHODS
Population and sample
In November 2009, we invited all survivors listed in a cancer survivor registry to complete a 12-page survey. The registry10 was created as part of the Cancer Survivor Community Study, a community-based participatory research project funded by the National Cancer Institute. The study’s Steering Committee was comprised of cancer survivors, cancer registrars, and researchers. We identified and invited cancer survivors from 4 hospital registries in northwest and central Vermont to participate. Registry participants who indicated willingness to enroll in research studies received an invitation letter and informed consent form, the 12-page survey, and an addressed and stamped return envelope. We obtained Institutional Review Board (IRB) approval for these procedures at the University of Vermont and at local hospital IRBs.
Instrument development
A working group from the Steering Committee reviewed a range of available instruments to assess cancer survivors’ needs.9,11-15 We determined that the survey most relevant to our objectives was the Cancer Survivors’ Unmet Needs (CaSun) instrument.13 Because CaSun was developed in Australia, we carefully examined each question for appropriateness to our target audience. We eliminated several questions that we thought less important, added questions from other instruments, and simplified the survey format. Survivors from the Steering Committee pilot tested the draft questionnaire to identify awkward wording or concepts.
We piloted the revised draft using a standardized feedback form with cancer survivors who were not connected to our project and not enrolled in the survivor registry, and with residents at a senior center. Students and a teacher from an Adult Basic Education program helped to ensure easy readability. Our final instrument had 53 questions about needs in 5 domains. Questions within each domain completed the lead-in, “Since your cancer diagnosis, did you need....” We asked participants to check only 1 of the 3 boxes to the right of each question to indicate that there was no need in that area, that there was a need and it was met, or that there was a need and it was not met. We obtained self-reported demographic data during enrollment in the registry.
Data analysis
We summarized data by computing the percent of survivors who reported having each need (either met or unmet) and the percent for whom the need was unmet. The latter was computed both as a percent of all survivors and as a percent of those who had the need. We also calculated the percentage of survivors that had at least one need and at least one unmet need in each domain, as well as the average number of needs per survivor in each domain. We used SPSS for Unix, Release 6.1 (AIX 3.2)(IBM, Armonk, New York).
RESULTS
Of the 2005 cancer survivors invited into the study, 1668 responded, yielding a participation rate of 83%. TABLE 1 describes the self-reported demographic and cancer characteristics of participants in this study. Most respondents were female, ≥60 years old, urban dwellers, married or with a partner, well educated, and had household incomes of ≥$50,000. There were more breast cancer survivors than survivors of other cancers, and 14.6% of all survivors reported being diagnosed with more than one cancer. Cancer was diagnosed at stages 1 or 2 for 78.3% of the participants; 61.9% reported having undergone ≥2 treatment regimens.
The survey addressed needs in 5 domains: access to care and services (A); information (I); emotional, social, and spiritual (E); physical (P); and economic and legal (L). More than 80% of respondents reported having at least one need in the A, I, and E domains. The E domain had the most survivors with at least one unmet need (N=503), followed by the I (N=410) and P (N=375) domains.
Identifying unmet needs. TABLE 2 shows results for the specific questions within the domains in the order they were asked. Most participants who had a need also had it met. However, some needs that were not commonly reported were deemed unmet by a large proportion of those who expressed the need. For example, the A need for “A case manager to whom you could go to find out about services whenever they were needed” (A5) was reported by only 29.1% of survivors. But 32.1% of those reporting the need said it was unmet, which corresponds to 9.4% of all study participants having the need unmet. Similarly, the need for “More information about complementary and alternative medicine” (I3) was reported by about a quarter of the study population, 41.4% of whom (9.8% of all participants) reported it as unmet. In the P domain, the need for “Help to address problems with your sex life” (P4) was reported by only 26.5% of the respondents; yet 40.7% of those reporting the need had it unmet. Similarly, in the L domain, “Help with life insurance concerns as a result of your cancer” (L3) was only reported by 10.9% of the participants but was unmet for 46.4% of those who reported the need, or 5% of all study participants.
Most commonly expressed needs. TABLE 2 also identifies 12 needs reported by ≥50% of participants. Three of these needs were in the A domain, 6 in the I domain, and 3 in the E domain. The 2 most common needs related to A: the need “To feel like you were managing your health together with the medical team” (A3) was reported by 68.6% and was viewed as unmet by 5.2% of all respondents; the need for “Access to screening for recurrence or other cancers” (A7) was reported by 63.8% of the survivors but was deemed unmet by only 3.1% of all the respondents. “More information about possible after effects of your treatment” (I5) was a need for 63.2% that went unmet in 22.9% (14.4% of all participants). “Help managing your concerns about the cancer coming back” (E13) was reported as a need by 54.1% and as unmet by 11.8% of all participants.
The rank order of 7 unmet needs reported by ≥10% of the participants is shown in TABLE 3. Four of the 7 unmet needs were in the E domain. The most common unmet need in this domain was “Help reducing stress in your life” (E19).
Only 3 needs were both commonly reported and also unmet for at least 10% of the participants: “More information about possible after effects of your treatment” (I5), “More information about possible side effects of your treatment” (I4), and “Help managing your concerns about the cancer coming back” (E13).
DISCUSSION
The survey instrument we used to assess the needs of cancer survivors in a large community-based registry included a detailed list of potential needs generated, in part, by representatives of the survivor community. Most cancer survivor needs mentioned in this survey were met. However, some needs were not met for substantial proportions of respondents and should be examined carefully to determine whether services could be improved to better address them. This study was planned and implemented by researchers and cancer survivors using community-based participatory principles to learn about local needs. The results of this study may be generalizable to similar populations of survivors and will inform the survivorship goals for the Vermont State Cancer Plan and future Vermont Cancer Survivor Network activities.
Acting on patients’ expressed needs. Over 80% of participants had needs in the A, I, and E domains. The most commonly reported need was in the A domain, “To feel like you were managing your health together with the medical team” (A3). It was also a top need in other studies that asked this question.16,17 A cancer diagnosis may cause patients to feel out of control. Participation in the management of their health may help them gain a greater sense of control. PCP accommodation of expressed patient preferences may be an important part of a cancer survivor’s long-term adaptation to the disease.
Six of the 12 most frequently reported needs and 2 frequently reported unmet needs were in the I domain. Communication of information increases patients’ involvement in decision-making and enables them to cope better during diagnosis, treatment, and follow-up.18 “More information about possible after effects of your treatment” and “More information about possible side effects of your treatment” were reported by a high proportion of participants, and many also reported these needs as unmet. In another study about health-related information needs of survivors, 52% wanted more information about “What late and long-term side effects of cancer treatment are expected”19; and in a 2005 review of information needs, 12% of survivors reported similar needs.20 Two recent articles also noted such needs in adolescent and young adult cancer survivors.21,22 Based on current evidence, it would be advisable to discuss anticipated effects of treatment with patients not only at the outset but also at the end of treatment, and to write it in a cancer survivor care plan.
Individual needs that were not met for at least 10% of respondents, regardless of how common the need (TABLE 3), provided additional insights. Among these 7 needs, 3 also were reported as a need by more than 50% of respondents (TABLE 2), and 4 by <50%, indicating that some less common needs are not being met adequately. Among these 7 prominent unmet needs, 4 were E Issues (TABLE 3) and 2 were I Issues.
Unmet needs are an opportunity to improve care. In our study and in others, E needs were most likely to be unmet.17,23-26 Among the 4 common unmet E needs, 2 (E19 and E11) focused on generalized stress and worry, and one (E15) focused on concern about illness impact on family members or partners. Although these issues may be challenging to address successfully in a typical clinical environment, others have confirmed the importance of these needs and proposed ways to meet them.27 The fourth most common unmet E need focused on concern about cancer recurrence, also a prominent need found in other studies.15-17 These needs might be addressed more adequately in the course of usual clinical care by PCPs or specialists. In fact, the American College of Surgeons’ Commission on Cancer 2012 standards now require psychosocial distress screening and the provision of referral for psychosocial services.24 Our results are consistent in many respects with prior studies of needs reported by cancer survivors in other countries. The CaSUN survey developed by Hodgkinson et al13 has been applied to several survivor populations in Australia. In a diverse survivor sample, specific E, I, and A issues were frequently reported as unmet needs.13 The most prominent unmet needs in a gynecologic cancer sample using CaSUN focused on emotional and social issues such as worry, stress, coping, and relationships with, and expectations of, others.25
Barg et al23 conducted a survey of unmet needs in the United States using a detailed list based on prior survivor research and targeting individuals in a cancer registry. The most prominent area of need expressed was “emotional,” similar to the high rank of E needs in our study. In contrast to our study, however, physical and financial issues also were prominent. The latter variances might be explained by differences in access to care, or perhaps the study’s low response rate (23.8%). A similar survey reported by Campbell et al12 identified needs in the emotional domain as the most cited unmet survivor needs based on psychometrically developed subscales of a 152-item survey (29% response rate).
These results from several studies, including ours, call for more detailed exploration of the E needs of long-term cancer survivors. A useful framework developed by Stein et al28 accounts for factors contributing to cancer stress and burden as well as resources available to survivors (intrapersonal, social, informational, and tangible services), with the interactions between these 2 domains determining how well a survivor will be able to cope. There clearly is a role for development of more effective communication channels and focused services to meet survivor needs.
The list of most common unmet needs in TABLE 3 also includes a focus on “problems with your sex life” (P4). This is an area that may be difficult to address in a cancer care setting because of the focus on disease management. Primary care providers might be better prepared to address this issue because they likely encounter similar issues among the wide range of patients they serve. However, a recent study reported that only 46% of internists were somewhat or likely to initiate a discussion about sexuality with cancer survivors.29 Some additional preparation for physicians to address this need might be warranted.
The proportion in this sample reporting needs for access to, or information about, complementary and alternative medicine services fell below the thresholds chosen to designate common needs in this study. Although reported use is relatively common among cancer survivor in several studies,30-32 it appears that in our survivor sample, those who were interested in these approaches encountered only moderate barriers.
Study limitations. We invited participants from a registry unlikely to include cancer survivors with lower educational attainment or from rural locations9—that is, our participants were less likely to have challenges in obtaining appropriate services and information. This sample limitation therefore likely underestimates the overall level of needs among cancer survivors.
This was a cross-sectional assessment of perceived needs among a diverse group of survivors, which may have overlooked needs that were met but only after considerable effort on the part of survivors. Longitudinal studies would provide more complete accounts of how readily needs are met and the changes in needs at different times in the continuum of care.
The Vermont population is less diverse racially and ethnically, but not with respect to household income or educational attainment, than the overall US population. Access to health care also is relatively high in Vermont compared with many other states. According to a 2009 Vermont Household Health Insurance Survey, only 7.6% of Vermonters are uninsured.33
CORRESPONDENCE
Berta Geller, EdD, University of Vermont, Health Promotion Research/Family Medicine, 1 South Prospect Street, Burlington, VT 05401-3444; [email protected]
ACKNOWLEDGEMENTS
We would like to thank Anne Dorwaldt, Kathy Howe, Mark Bowman and John Mace at the University of Vermont, and the Cancer Survivor Community Study Steering Committee for their contributions to the successful completion of this study. We also thank the cancer survivors who participated in the pilot testing and the overall survey.
1. Potosky A, Han PK, Rowland J, et al. Differences between primary care physicians’ and oncologists’ knowledge, attitudes and practices regarding the care of cancer survivors. J Gen Intern Med. 2011;26:1403-1410.
2. Rowland J. Survivorship research: past, present, and future. In: Chang AE, Ganz PA, Hayes DF, et al, eds. Oncology: An Evidence-Based Approach. New York, NY: Springer; 2005:1753-1767.
3. Jemal A, Center MM, DeSantis C, et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-1907.
4. Hewitt ME, Greenfield S, Stovall E, eds. From Cancer Patient to Cancer Survivor: Lost in Transition. Washington, DC: National Academies Press; 2005.
5. Hawkins NA, Smith T, Zhao L, et al. Health-related behavior change after cancer: results of the American Cancer Society’s studies of cancer survivors (SCS). J Cancer Surviv. 2010;4:20-32.
6. Katz ML, Reiter PL, Corbin S, et al. Are rural Ohio Appalachia cancer survivors needs different than urban cancer survivors? J Cancer Surviv. 2010;4:140-148.
7. Yabroff KR, Lawrence WF, Clauser S, et al. Burden of illness in cancer survivors: findings from a population-based national sample. J Natl Cancer Inst. 2004;96:1322-1330.
8. Sanson-Fisher R, Girgis A, Boyes A, et al; Supportive Care Review Group. The unmet supportive care needs of patients with cancer. Cancer. 2000;88:226-237.
9. Smith T, Stein KD, Mehta CC, et al. The rationale, design, and implementation of the American Cancer Society’s studies of cancer survivors. Cancer. 2007;109:1-12.
10. Geller BM, Mace J, Vacek P, et al. Are cancer survivors willing to participate in research? J Community Health. 2011;36: 772-778.
11. Crespi CM, Ganz PA, Petersen L, et al. Refinement and psychometric evaluation of the impact of cancer scale. J Natl Cancer Inst. 2008;100:1530-1541.
12. Campbell HS, Sanson-Fisher R, Turner D, et al. Psychometric properties of cancer survivors’ unmet needs survey. Support Care Cancer. 2010;19:221-230.
13. Hodgkinson K, Butow P, Hunt GE, et al. The development and evaluation of a measure to assess cancer survivors’ unmet supportive care needs: the CaSUN (Cancer Survivors’ Unmet Needs measure). Psychooncology. 2007;16:796-804.
14. Zebrack BJ, Ganz PA, Bernaards CA, et al. Assessing the impact of cancer: development of a new instrument for long-term survivors. Psychooncology. 2006;15:407-421.
15. Arora NK, Hesse BW, Rimer BK, et al. Frustrated and confused: the American public rates its cancer-related information-seeking experiences. J Gen Intern Med. 2008;23:223-228.
16. Hodgkinson K, Butow P, Hunt GE, et al. Breast cancer survivors’ supportive care needs 2-10 years after diagnosis. Support Care Cancer. 2007;15:515-523.
17. Harrison SE, Watson EK, Ward AM, et al. Primary health and supportive care needs of long-term cancer survivors: a questionnaire study. J Clin Oncol. 2011;29:2091-2098.
18. Rutten LJ, Squiers L, Treiman K. Requests for information by family and friends of cancer patients calling the National Cancer Institute’s Cancer Information Service. Psychooncology. 2006;15:664-672.
19. Beckjord EB, Arora NK, McLaughlin W, et al. Health-related information needs in a large and diverse sample of adult cancer survivors: implications for cancer care. J Cancer Surviv. 2008;2:179-189.
20. Rutten LJ, Arora NK, Bakos AD, et al. Information needs and sources of information among cancer patients: a systematic review of research (1980-2003). Patient Educ Couns. 2005;57:250-261.
21. Zebrack BJ, Block R, Hayes-Lattin B, et al. Psychosocial service use and unmet need among recently diagnosed adolescent and youg adult patients. Cancer. 2013;119:201-214.
22. Keegan TH, Lichtensztajn DY, Kato I, et al. Unmet adolescent and young adult cancer survivors information and service needs: a population-based cancer registry study. J Cancer Surviv. 2012;6:239-250.
23. Barg KF, Cronholm PF, Straton JB, et al. Unmet psychosocial needs of Pennsylvanians with cancer: 1996-2005. Cancer. 2007;110:631-639.
24. American College of Surgeons. Cancer Program Standards 2012: Ensuring Patient-Centered Care. Chicago, IL: American College of Surgeons; 2011.
25. Hodgkinson K, Butow P, Fuchs A, et al. Long-term survival from gynecologic cancer: psychosocial outcomes, supportive care needs and positive outcomes. Gynecol Oncol. 2007;104:381-389.
26. Houts PS, Yasko JM, Kahn SB, et al. Unmet psychological, social, and economic needs of persons with cancer in Pennsylvania. Cancer. 1986;58:2355-2361.
27. Holland JC, Reznik I. Pathways for psychosocial care of cancer survivors. Cancer. 2005;104(11 suppl):2624-2637.
28. Stein KD, Syrjala KL, Andrykowski MA. Physical and psychological long-term and late effects of cancer. Cancer. 2008;112(11 suppl):2577-2592.
29. Park ER, Bober SL, Campbell EG, et al. General: Internist communication about sexual function with cancer survivors. J Gen Intern Med. 2009;24(suppl 2):S407-S411.
30. Girgis A, Adams J, Sibbritt D. The use of complementary and alternative therapies by patients with cancer. Oncol Res. 2005;15:281-289.
31. Gansler T, Kaw C, Crammer C, et al. A population-based study of prevalence of complementary methods use by cancer survivors: a report from the American Cancer Society’s studies of cancer survivors. Cancer. 2008;113:1048-1057.
32. Fouladbakhsh JM, Stommel M. Gender, symptom experience, and use of complementary and alternative medicine practices among cancer survivors in the U.S. cancer population. Oncol Nurs Forum. 2010;37:E7-E15.
33. Vermont Department of Financial Regulation. Vermont Household Health Insurance Survey (VHHIS). Vermont Department of Financial Regulation Web site. Available at: http://www.dfr.vermont.gov/insurance/health-insurance/vermont-household-health-insurance-survey-vhhis. Accessed July 15, 2014.
1. Potosky A, Han PK, Rowland J, et al. Differences between primary care physicians’ and oncologists’ knowledge, attitudes and practices regarding the care of cancer survivors. J Gen Intern Med. 2011;26:1403-1410.
2. Rowland J. Survivorship research: past, present, and future. In: Chang AE, Ganz PA, Hayes DF, et al, eds. Oncology: An Evidence-Based Approach. New York, NY: Springer; 2005:1753-1767.
3. Jemal A, Center MM, DeSantis C, et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-1907.
4. Hewitt ME, Greenfield S, Stovall E, eds. From Cancer Patient to Cancer Survivor: Lost in Transition. Washington, DC: National Academies Press; 2005.
5. Hawkins NA, Smith T, Zhao L, et al. Health-related behavior change after cancer: results of the American Cancer Society’s studies of cancer survivors (SCS). J Cancer Surviv. 2010;4:20-32.
6. Katz ML, Reiter PL, Corbin S, et al. Are rural Ohio Appalachia cancer survivors needs different than urban cancer survivors? J Cancer Surviv. 2010;4:140-148.
7. Yabroff KR, Lawrence WF, Clauser S, et al. Burden of illness in cancer survivors: findings from a population-based national sample. J Natl Cancer Inst. 2004;96:1322-1330.
8. Sanson-Fisher R, Girgis A, Boyes A, et al; Supportive Care Review Group. The unmet supportive care needs of patients with cancer. Cancer. 2000;88:226-237.
9. Smith T, Stein KD, Mehta CC, et al. The rationale, design, and implementation of the American Cancer Society’s studies of cancer survivors. Cancer. 2007;109:1-12.
10. Geller BM, Mace J, Vacek P, et al. Are cancer survivors willing to participate in research? J Community Health. 2011;36: 772-778.
11. Crespi CM, Ganz PA, Petersen L, et al. Refinement and psychometric evaluation of the impact of cancer scale. J Natl Cancer Inst. 2008;100:1530-1541.
12. Campbell HS, Sanson-Fisher R, Turner D, et al. Psychometric properties of cancer survivors’ unmet needs survey. Support Care Cancer. 2010;19:221-230.
13. Hodgkinson K, Butow P, Hunt GE, et al. The development and evaluation of a measure to assess cancer survivors’ unmet supportive care needs: the CaSUN (Cancer Survivors’ Unmet Needs measure). Psychooncology. 2007;16:796-804.
14. Zebrack BJ, Ganz PA, Bernaards CA, et al. Assessing the impact of cancer: development of a new instrument for long-term survivors. Psychooncology. 2006;15:407-421.
15. Arora NK, Hesse BW, Rimer BK, et al. Frustrated and confused: the American public rates its cancer-related information-seeking experiences. J Gen Intern Med. 2008;23:223-228.
16. Hodgkinson K, Butow P, Hunt GE, et al. Breast cancer survivors’ supportive care needs 2-10 years after diagnosis. Support Care Cancer. 2007;15:515-523.
17. Harrison SE, Watson EK, Ward AM, et al. Primary health and supportive care needs of long-term cancer survivors: a questionnaire study. J Clin Oncol. 2011;29:2091-2098.
18. Rutten LJ, Squiers L, Treiman K. Requests for information by family and friends of cancer patients calling the National Cancer Institute’s Cancer Information Service. Psychooncology. 2006;15:664-672.
19. Beckjord EB, Arora NK, McLaughlin W, et al. Health-related information needs in a large and diverse sample of adult cancer survivors: implications for cancer care. J Cancer Surviv. 2008;2:179-189.
20. Rutten LJ, Arora NK, Bakos AD, et al. Information needs and sources of information among cancer patients: a systematic review of research (1980-2003). Patient Educ Couns. 2005;57:250-261.
21. Zebrack BJ, Block R, Hayes-Lattin B, et al. Psychosocial service use and unmet need among recently diagnosed adolescent and youg adult patients. Cancer. 2013;119:201-214.
22. Keegan TH, Lichtensztajn DY, Kato I, et al. Unmet adolescent and young adult cancer survivors information and service needs: a population-based cancer registry study. J Cancer Surviv. 2012;6:239-250.
23. Barg KF, Cronholm PF, Straton JB, et al. Unmet psychosocial needs of Pennsylvanians with cancer: 1996-2005. Cancer. 2007;110:631-639.
24. American College of Surgeons. Cancer Program Standards 2012: Ensuring Patient-Centered Care. Chicago, IL: American College of Surgeons; 2011.
25. Hodgkinson K, Butow P, Fuchs A, et al. Long-term survival from gynecologic cancer: psychosocial outcomes, supportive care needs and positive outcomes. Gynecol Oncol. 2007;104:381-389.
26. Houts PS, Yasko JM, Kahn SB, et al. Unmet psychological, social, and economic needs of persons with cancer in Pennsylvania. Cancer. 1986;58:2355-2361.
27. Holland JC, Reznik I. Pathways for psychosocial care of cancer survivors. Cancer. 2005;104(11 suppl):2624-2637.
28. Stein KD, Syrjala KL, Andrykowski MA. Physical and psychological long-term and late effects of cancer. Cancer. 2008;112(11 suppl):2577-2592.
29. Park ER, Bober SL, Campbell EG, et al. General: Internist communication about sexual function with cancer survivors. J Gen Intern Med. 2009;24(suppl 2):S407-S411.
30. Girgis A, Adams J, Sibbritt D. The use of complementary and alternative therapies by patients with cancer. Oncol Res. 2005;15:281-289.
31. Gansler T, Kaw C, Crammer C, et al. A population-based study of prevalence of complementary methods use by cancer survivors: a report from the American Cancer Society’s studies of cancer survivors. Cancer. 2008;113:1048-1057.
32. Fouladbakhsh JM, Stommel M. Gender, symptom experience, and use of complementary and alternative medicine practices among cancer survivors in the U.S. cancer population. Oncol Nurs Forum. 2010;37:E7-E15.
33. Vermont Department of Financial Regulation. Vermont Household Health Insurance Survey (VHHIS). Vermont Department of Financial Regulation Web site. Available at: http://www.dfr.vermont.gov/insurance/health-insurance/vermont-household-health-insurance-survey-vhhis. Accessed July 15, 2014.
Failure to properly manage a patient’s hypertension
Failure to properly manage a patient’s hypertension
A 44-YEAR-OLD MAN WHO WEIGHED >450 POUNDS went to his internist for treatment of hypertension. At a work-related physical the previous day, his blood pressure had been 160/110 mm Hg. After examination, the internist wrote a 30-day prescription for amlodipine, 5 mg/d, with 3 refills. The patient saw the physician 2 weeks later but not again until 3 months later. At that visit, the internist prescribed amlodipine, 5 mg/d, for 90 days with 2 refills. The patient missed his next appointment, which was set for 4 months later, but when his medication was about to run out, he was able to get a prescription for 10 months’ worth of amlodipine by phone. The patient died 2 months before the prescription ran out.
PLAINTIFF’S CLAIM The physician failed to properly manage and monitor the patient’s hypertension. The dosage of amlodipine was insufficient.
THE DEFENSE The patient was noncompliant and failed to show for follow-up appointments. The dosage of amlodipine was sufficient. The cause of death was unknown because no autopsy was performed.
VERDICT $136,000 New Jersey verdict.
COMMENT If we accept a patient into our practice, we need to have reasonable policies for patients to show up for follow-up, and to consider having them find another physician if they do not.
Did the patient’s age discourage proper evaluation?
THREE MONTHS AFTER NOTICING BLOOD IN HER STOOL, a 19-year-old woman went to see her physician. Without ordering a flexible sigmoidoscopy or colonoscopy, the physician diagnosed a healing anal fissure. Approximately 4 years later, the patient developed bloody diarrhea and went to a gastroenterologist, who found a 2.6 cm lesion in her rectum during a flexible sigmoidoscopy. Biopsy confirmed a low-grade adenocarcinoma. Imaging studies revealed that the cancer had spread to her lungs and liver, and she was diagnosed with Stage IV rectal cancer. After 2 years of extensive treatment that included surgical resection, conventional and experimental chemotherapy, and radiation therapy, the patient died.
PLAINTIFF’S CLAIM If the physician had ordered endoscopy exams when the patient first presented for treatment, testing could have identified a polyp or early-stage cancer.
THE DEFENSE No information about the defense is available.
VERDICT $2.5 million Maryland verdict.
COMMENT Colon cancer in a 19-year-old is extraordinarily rare. I doubt that the patient didn’t experience any more rectal bleeding until 4 years after she first sought treatment. A lesson in this tragic case is to be sure to document when you tell patients to “come back to see me right away if this happens again.”
23-year-old dies when myocarditis is mistaken for bronchitis
A 23-YEAR-OLD MAN PRESENTED TO THE EMERGENCY DEPARTMENT (ED) with chest tightness, cough, and fever. After a chest x-ray, the ED physician diagnosed bronchitis and sent the patient home with prescriptions for hydrocodone/acetaminophen and antibiotics. He was found dead in his bed less than 24 hours later. An autopsy determined the cause of death was myocarditis.
PLAINTIFF’S CLAIM The physician didn’t perform an electrocardiogram (EKG), which is a routine evaluation for a patient with chest pain. The EKG would have detected myocarditis.
THE DEFENSE The patient was evaluated properly. An EKG was not necessary.
VERDICT $2.9 million Massachusetts verdict.
COMMENT I think the jury got this one wrong. I don’t think an EKG is necessary for every case of acute bronchitis. However, I do wonder if the chest x-ray showed a large heart shadow.
Failure to properly manage a patient’s hypertension
A 44-YEAR-OLD MAN WHO WEIGHED >450 POUNDS went to his internist for treatment of hypertension. At a work-related physical the previous day, his blood pressure had been 160/110 mm Hg. After examination, the internist wrote a 30-day prescription for amlodipine, 5 mg/d, with 3 refills. The patient saw the physician 2 weeks later but not again until 3 months later. At that visit, the internist prescribed amlodipine, 5 mg/d, for 90 days with 2 refills. The patient missed his next appointment, which was set for 4 months later, but when his medication was about to run out, he was able to get a prescription for 10 months’ worth of amlodipine by phone. The patient died 2 months before the prescription ran out.
PLAINTIFF’S CLAIM The physician failed to properly manage and monitor the patient’s hypertension. The dosage of amlodipine was insufficient.
THE DEFENSE The patient was noncompliant and failed to show for follow-up appointments. The dosage of amlodipine was sufficient. The cause of death was unknown because no autopsy was performed.
VERDICT $136,000 New Jersey verdict.
COMMENT If we accept a patient into our practice, we need to have reasonable policies for patients to show up for follow-up, and to consider having them find another physician if they do not.
Did the patient’s age discourage proper evaluation?
THREE MONTHS AFTER NOTICING BLOOD IN HER STOOL, a 19-year-old woman went to see her physician. Without ordering a flexible sigmoidoscopy or colonoscopy, the physician diagnosed a healing anal fissure. Approximately 4 years later, the patient developed bloody diarrhea and went to a gastroenterologist, who found a 2.6 cm lesion in her rectum during a flexible sigmoidoscopy. Biopsy confirmed a low-grade adenocarcinoma. Imaging studies revealed that the cancer had spread to her lungs and liver, and she was diagnosed with Stage IV rectal cancer. After 2 years of extensive treatment that included surgical resection, conventional and experimental chemotherapy, and radiation therapy, the patient died.
PLAINTIFF’S CLAIM If the physician had ordered endoscopy exams when the patient first presented for treatment, testing could have identified a polyp or early-stage cancer.
THE DEFENSE No information about the defense is available.
VERDICT $2.5 million Maryland verdict.
COMMENT Colon cancer in a 19-year-old is extraordinarily rare. I doubt that the patient didn’t experience any more rectal bleeding until 4 years after she first sought treatment. A lesson in this tragic case is to be sure to document when you tell patients to “come back to see me right away if this happens again.”
23-year-old dies when myocarditis is mistaken for bronchitis
A 23-YEAR-OLD MAN PRESENTED TO THE EMERGENCY DEPARTMENT (ED) with chest tightness, cough, and fever. After a chest x-ray, the ED physician diagnosed bronchitis and sent the patient home with prescriptions for hydrocodone/acetaminophen and antibiotics. He was found dead in his bed less than 24 hours later. An autopsy determined the cause of death was myocarditis.
PLAINTIFF’S CLAIM The physician didn’t perform an electrocardiogram (EKG), which is a routine evaluation for a patient with chest pain. The EKG would have detected myocarditis.
THE DEFENSE The patient was evaluated properly. An EKG was not necessary.
VERDICT $2.9 million Massachusetts verdict.
COMMENT I think the jury got this one wrong. I don’t think an EKG is necessary for every case of acute bronchitis. However, I do wonder if the chest x-ray showed a large heart shadow.
Failure to properly manage a patient’s hypertension
A 44-YEAR-OLD MAN WHO WEIGHED >450 POUNDS went to his internist for treatment of hypertension. At a work-related physical the previous day, his blood pressure had been 160/110 mm Hg. After examination, the internist wrote a 30-day prescription for amlodipine, 5 mg/d, with 3 refills. The patient saw the physician 2 weeks later but not again until 3 months later. At that visit, the internist prescribed amlodipine, 5 mg/d, for 90 days with 2 refills. The patient missed his next appointment, which was set for 4 months later, but when his medication was about to run out, he was able to get a prescription for 10 months’ worth of amlodipine by phone. The patient died 2 months before the prescription ran out.
PLAINTIFF’S CLAIM The physician failed to properly manage and monitor the patient’s hypertension. The dosage of amlodipine was insufficient.
THE DEFENSE The patient was noncompliant and failed to show for follow-up appointments. The dosage of amlodipine was sufficient. The cause of death was unknown because no autopsy was performed.
VERDICT $136,000 New Jersey verdict.
COMMENT If we accept a patient into our practice, we need to have reasonable policies for patients to show up for follow-up, and to consider having them find another physician if they do not.
Did the patient’s age discourage proper evaluation?
THREE MONTHS AFTER NOTICING BLOOD IN HER STOOL, a 19-year-old woman went to see her physician. Without ordering a flexible sigmoidoscopy or colonoscopy, the physician diagnosed a healing anal fissure. Approximately 4 years later, the patient developed bloody diarrhea and went to a gastroenterologist, who found a 2.6 cm lesion in her rectum during a flexible sigmoidoscopy. Biopsy confirmed a low-grade adenocarcinoma. Imaging studies revealed that the cancer had spread to her lungs and liver, and she was diagnosed with Stage IV rectal cancer. After 2 years of extensive treatment that included surgical resection, conventional and experimental chemotherapy, and radiation therapy, the patient died.
PLAINTIFF’S CLAIM If the physician had ordered endoscopy exams when the patient first presented for treatment, testing could have identified a polyp or early-stage cancer.
THE DEFENSE No information about the defense is available.
VERDICT $2.5 million Maryland verdict.
COMMENT Colon cancer in a 19-year-old is extraordinarily rare. I doubt that the patient didn’t experience any more rectal bleeding until 4 years after she first sought treatment. A lesson in this tragic case is to be sure to document when you tell patients to “come back to see me right away if this happens again.”
23-year-old dies when myocarditis is mistaken for bronchitis
A 23-YEAR-OLD MAN PRESENTED TO THE EMERGENCY DEPARTMENT (ED) with chest tightness, cough, and fever. After a chest x-ray, the ED physician diagnosed bronchitis and sent the patient home with prescriptions for hydrocodone/acetaminophen and antibiotics. He was found dead in his bed less than 24 hours later. An autopsy determined the cause of death was myocarditis.
PLAINTIFF’S CLAIM The physician didn’t perform an electrocardiogram (EKG), which is a routine evaluation for a patient with chest pain. The EKG would have detected myocarditis.
THE DEFENSE The patient was evaluated properly. An EKG was not necessary.
VERDICT $2.9 million Massachusetts verdict.
COMMENT I think the jury got this one wrong. I don’t think an EKG is necessary for every case of acute bronchitis. However, I do wonder if the chest x-ray showed a large heart shadow.