User login
Addressing Alzheimer’s: A pragmatic approach
› Refer patients for formal neuropsychological testing when dementia is suspected but the history, clinical interview, and brief cognitive tests do not result in a definitive diagnosis. C
› Use non-drug therapies as first-line treatment for behavioral symptoms of Alzheimer’s disease (AD), as the adverse effects of drug therapy generally offset any benefit. B
› Recommend against feeding tubes for patients with late-stage AD as they are more apt to cause discomfort than to provide benefit. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Alzheimer’s disease (AD), the most common form of dementia, affects more than 5 million Americans.1 Estimates suggest that by 2050, the prevalence could triple, reaching 13 to 16 million.1 To effectively care for patients with AD and their families, family physicians need to be familiar with the latest evidence on all facets of care, from initial detection to patient management and end-of-life care.
This evidence-based review will help you toward that end by answering common questions regarding Alzheimer’s care, including whether routine screening is advisable, what tests should be ordered, which interventions (including nonpharmacologic options) are worth considering, and how best to counsel patients and families about end-of-life care.
Routine screening? Still subject to debate
In considering routine dementia screening in primary care, the key question is whether screening improves outcomes. Advocates note that individuals with dementia may appear unimpaired during office visits and may not report symptoms due to lack of insight; they point out, too, that waiting for an event that makes cognitive impairment obvious, such as a driving mishap, is risky.2 Those who advocate routine screening also note that only about half of those who have dementia are ever diagnosed.3
Others, including the US Preventive Services Task Force (USPSTF), disagree. In its 2014 evidence review, the USPSTF indicated that there is “insufficient evidence to assess the balance of benefits and harms of screening for cognitive impairment in older adults.”4
Mixed messages
The dearth of evidence is also reflected in the conflicting recommendations of the Affordable Care Act (ACA) and the Centers for Medicare and Medicaid Services (CMS). The ACA requires physicians to assess the cognitive function of Medicare patients during their annual wellness visits. CMS, however, instructs providers to screen for dementia only if observation or concerns raised by the patient or family suggest the possibility of impairment, and does not recommend any particular test.5
Cost-effectiveness analyses raise questions about the value of routine screening, as well. Evidence suggests that if a primary care physician screens 300 older patients, 39 will have a positive screen. But only about half of those 39 will agree to a diagnostic evaluation, and no more than 9 will ultimately be diagnosed with dementia. The estimated cost of identifying 9 cases is nearly $40,000—all in the absence of a treatment to cure or stop the progression of the disorder.6
The bottom line: Evidence does not support routine dementia screening of older adults. When cognitive impairment is suspected, however, physicians should conduct a diagnostic evaluation—and consider educating patients and families about the Alzheimer’s Association (AA)’s 10 warning signs of AD.7 (See “Is it Alzheimer’s? 10 warning signs”7 below.) A longer version, available at http://www.alz.org/national/documents/checklist_10signs.pdf, outlines the cognitive changes that are characteristic of healthy aging and compares them to changes suggestive of early dementia.7
Is it Alzheimer’s? 10 warning signs7 1. Memory loss that disrupts daily life |
How to proceed when you suspect AD
Step 1: Screening instrument. The first step in the diagnostic evaluation of a patient with suspected AD is to determine if, in fact, cognitive impairment is present. This can be done by screening with in-office screening instruments, such as the Mini-Cog (available at alz.org/documents_custom/minicog.pdf) or Mini-Mental State Examination (MMSE; health.gov.bc.ca/pharmacare/adti/clinician/pdf/ADTI%20SMMSE-GDS%20Reference%20Card.pdf), among others.8
Step 2: Clinical evaluation. If observation and test results suggest cognitive impairment, the next step is to determine whether clinical findings are consistent with the diagnostic criteria for AD (TABLE 1)9 developed by workgroups from the National Institute on Aging (NIA)/AA in 2011. A work-up is necessary to identify conditions that can mimic dementia (eg, depression) and behaviors that suggest another type of dementia, such as frontotemporal or Lewy body dementia.10 Lab testing should be included to rule out potentially reversible causes of cognitive dysfunction (eg, hypothyroidism, vitamin D deficiency).
Step 3: Neuropsychological evaluation. The NIA/AA recommends neuropsychological testing when the brief cognitive tests, history, and clinical work-up are not sufficient for a definitive diagnosis of dementia.9This generally involves a referral to a neuropsychologist, who conducts a battery of standardized tests to evaluate attention, memory, language, visual-spatial abilities, and executive functions, among others. Neuropsychological testing can confirm the presence of cognitive impairment and aid in the differential diagnosis by comparing the patient’s performance in these domains with characteristic features of different dementia syndromes.
Step 4: Brain imaging with either computed tomography or magnetic resonance imaging can be included in the work-up for patients with suspected AD to rule out abnormalities—eg, metastatic cancer, hydrocephalus, or occult chronic subdural hematoma—that could be causing cognitive impairment.9,10 Clinical features that generally warrant brain imaging include onset of cognitive impairment before age 60; unexplained focal neurologic signs or symptoms; abrupt onset or rapid decline; and/or predisposing conditions, such as cancer or anticoagulant treatment.10

The role of biomarkers and advanced brain imaging
Biomarkers that might provide confirmation of AD in patients who exhibit early symptoms of dementia have been studied extensively.11 The NIA/AA identified 2 categories of AD biomarkers:
- tests for β-amyloid deposition in the brain, including spinal fluid assays for β-amyloid (Aβ42) and positron emission tomography (PET) scans after intravenous injection of florbetapir or flutemetamol, which bind to amyloid in the brain; and
- tests for neuronal degeneration, which would include spinal fluid assays for tau protein and PET scans after injection of fluorodeoxyglucose (FDG), which shows decreased uptake in patients with AD.9
Research reveals the promise of these biomarkers as diagnostic tools, particularly in patients with an atypical presentation of dementia or mild cognitive impairment (MCI) that may be associated with early AD.12 (More on MCI in a moment.) However, the NIA/AA concluded that additional research is needed to validate these tests for routine diagnostic purposes. Medicare covers PET scans with FDG only for the differential diagnosis of AD vs frontotemporal dementia.13
Mild cognitive impairment: How likely that it will progress?
Along with diagnostic criteria for AD, the NIA/AA developed criteria for a symptomatic predementia phase of AD—often referred to as MCI.14 According to the workgroup, MCI is diagnosed when:
2. there is evidence of cognitive impairment, ideally through psychometric testing, revealing performance below expectation based on the patient’s age and education;
3. the patient is able to maintain independent functioning in daily life, despite mild problems or the need for minimal assistance; and
4. there is no significant impairment in social or occupational functioning.14
Progression: Less likely than you might think
Patients with MCI are at risk for progression to overt dementia, with an overall annual conversion rate from MCI to dementia estimated at 10% to 15%.15,16 This estimate must be interpreted with caution, however, because most studies were conducted prior to the 2011 guidelines, when different diagnostic criteria were used. Observers have noted, too, that the numbers largely reflect data collected in specialty clinics and that community-based studies reveal substantially lower conversion rates (3%-6% per year).16 In addition, evidence suggests that many patients with MCI demonstrate long-term stability or even reversal of deficits.17
While there is some consideration of the use of biomarkers and amyloid imaging tests to help determine which patients with MCI will progress to AD, practice guidelines do not currently recommend such testing and it is not covered by Medicare.
When evidence indicates an AD diagnosis
When faced with the need to communicate an AD diagnosis, follow the general recommendations for delivering any bad news or discouraging prognosis:
Prioritize and limit the information you provide, determining not only what the patient and family want to hear, but also how much they are able to comprehend.
Confirm that the patient and family understand the information you’ve provided.
Offer emotional support and recommend additional resources18 (TABLE 2).
Given the progressive cognitive decline that characterizes AD, it is important to address the primary caregiver’s understanding of, and ability to cope with, the disease. It is also important to explore beliefs and attitudes regarding AD. Keep in mind that different cultural groups tend to differ in their beliefs about the nature, cause, and appropriate management of AD, as well as the role of spirituality, help-seeking, and stigma.19,20
The progressive and ultimately fatal nature of AD also makes planning for the future a priority. Ideally, patients should be engaged in discussions regarding end-of-life care as early as possible, while they are still able to make informed decisions and express their preferences. Discussing end-of-life care can be overwhelming for newly diagnosed patients and their families, however, so it is important that you address issues—medical, financial, and legal planning, for example—that families should be considering.
TABLE 2
| Resources for newly diagnosed patients and families | |
| Issue | Resources |
| Education | Alzheimer’s and Dementia Caregiver Center http://www.alz.org/care/overview.asp NIA Alzheimer’s Disease Education and Referral Center http://www.nia.nih.gov/alzheimers/ |
| Planning (medical, financial, legal)/benefits | AARP Caregiving Resource Center http://www.aarp.org/home-family/caregiving Alzheimer’s Association Alzheimer’s Navigator https://www.alzheimersnavigator.org/ National Council on Aging Benefits Checkup https://www.benefitscheckup.org/ |
| Safety | Association for Driver Rehabilitation Specialists: Driving and Alzheimer’s/Dementia https://c.ymcdn.com/sites/www.aded.net/resource/resmgr/fact_Sheets/ADED_alzheimers-Dementia_fac.pdf NIA’s Home Safety for People with Alzheimer’s booklet http://www.nia.nih.gov/alzheimers/publication/home-safety-people-alzheimers-disease |
| Support | Caregiver Action Network http://caregiveraction.org/ |
Drugs address cognitive and behavioral function
No currently available treatments can cure or significantly alter the progression of AD, but 2 classes of medications are used in an attempt to improve cognitive function. One is cholinesterase inhibitors (ChEIs), which potentiate acetylcholine synaptic transmission. The other is N-methyl-D-aspartate (NMDA) glutamate receptor blockers. Other classes of drugs are sometimes used to treat behavioral symptoms of dementia, such as agitation, aggression, mood disorders, and psychosis (eg, delusions and hallucinations).
Cognitive function. Results from studies of pharmacologic management of MCI vary widely, but recent reviews have found no convincing evidence that either ChEIs or NMDA receptor blockers have an effect on progression from MCI to dementia.21,22 Neither class is FDA-approved for treating MCI.
In patients with dementia, the effects of ChEIs and NMDA receptor blockers on cognition are statistically significant but modest, and often of questionable clinical relevance.23 Nonetheless, among ChEIs, donepezil is approved by the US Food and Drug Administration (FDA) for mild, moderate, and severe dementia and galantamine and rivastigmine are approved for mild and moderate dementia. There is no evidence that any one ChEI is more effective than any other,24 and the choice of drugs is often guided by cost, adverse effects, and health plan formularies. Memantine, the only FDA-approved NMDA receptor blocker, is approved for moderate to severe dementia and can be used alone or in combination with a ChEI.
If these drugs are used in an attempt to improve cognition in AD, guidelines recommend the following approach for initial therapy: Prescribe a ChEI for the mild stage, a ChEI plus memantine for the moderate stage, and memantine (with or without a ChEI) for the severe stage.25 The recommendations also include monitoring every 6 months.
There is no consensus about when to discontinue medication. Various published recommendations call for continuing treatment until the patient has “lost all cognitive and functional abilities;”22 until the patient’s MMSE score falls below 10 and there is no indication that the drug is having a “worthwhile effect;”21 or until he or she has reached stage 7 on the Reisberg Functional Assessment Staging scale, indicating nonambulatory status with speech limited to one to 5 words a day.10
Behavioral function. A variety of drugs are used to treat behavioral symptoms in AD. While not FDA-approved for this use, the most widely prescribed agents are second-generation antipsychotics (aripiprazole, olanzapine, quetiapine, and risperidone). The main effect of these drugs is often nothing more than sedation, and one large multisite clinical trial concluded that the adverse effects offset the benefits for patients with AD.26 Indeed, the FDA has issued an advisory on the use of second-generation antipsychotics in AD patients, stating that they are associated with an increased risk of death.27 The recently updated Beers Criteria strongly recommend avoiding these drugs for treating behavioral disturbances in AD unless nonpharmacologic options have failed and the patient is a threat to self or others.28
Because of the black-box warning that antipsychotics increase the risk of death, some physicians have advocated obtaining informed consent prior to prescribing such medications.29 At the very least, when family or guardians are involved, a conversation about risks vs benefits should take place and be documented in the medical record.
Other drug classes are also sometimes used in an attempt to improve behavioral function, including anti-seizure medications (valproic acid, carbamazepine), antidepressants (trazodone and selective serotonin reuptake inhibitors), and anxiolytics (benzodiazepines and buspirone). Other than their sedating effects, there is no strong evidence that these drugs are effective for treating dementia-related behavioral disorders. If used, caution is required due to potential adverse effects.
Nonpharmacologic management is “promising”
A recent systematic review of nonpharmacologic interventions for MCI evaluated exercise, training in compensatory strategies, and engagement in cognitively stimulating activities and found “promising but inconclusive” results. The researchers found that studies show mostly positive effects on cognition but have significant methodologic limitations.30 Importantly, there is no evidence of delayed or reduced conversion to dementia.
For patients who already have mild-to-moderate dementia, cognitive stimulation seems to help in the short term.31 There is also some evidence that exercise and occupational therapy may slow functional decline,32 but the effects are small to modest and their actual clinical significance (eg, the ability to delay institutionalization) is unclear. There is promising but preliminary evidence that cognitive rehabilitation (helping patients devise strategies to complete daily activities) may improve functioning in everyday life.33
While behavioral symptoms are often due to the dementia itself, it is important to identify and treat medical and environmental causes that may be contributing, such as infection, pain, and loud or unsafe environments. As noted before, nonpharmacologic treatments are generally preferred for behavioral problems and should be considered prior to drug therapy. Approaches that identify and modify both the antecedents and consequences of problem behaviors and increase pleasant events have empirical support for the management of behavioral symptoms.34 Interventions including massage therapy, aromatherapy, exercise, and music therapy may also be effective in the short term for agitated behavior.35
Caregivers should be encouraged to receive training in these strategies through organizations like AA. Caregiver education and support can reduce caregivers’ distress and increase their self-efficacy and coping skills.36
End-of-life care must be addressed
Perhaps the most important aspect of end-of-life care in AD is assuring that families (or health care proxies) understand that AD is a fatal illness, with most patients dying within 4 to 8 years of diagnosis.1 Evidence indicates that patients whose proxies have a clear recognition of this are less likely to experience “burdensome” interventions such as parenteral therapy, emergency department visits, hospital admissions, and tube feedings in their last 3 months of life.37
Overall, decisions regarding discontinuing medical treatments in advanced AD should be made by balancing the likelihood of benefit with the potential for adverse effects.38 For example, the American Geriatrics Society recently recommended against feeding tubes because they often result in discomfort due to agitation, use of restraints, and worsening pressure ulcers.39
Unfortunately, only a minority of families receives straightforward information on the course and prognosis of AD, including the fact that patients eventually stop eating and that the natural cause of death is often an acute infection. Studies also show that patients with dementia are at risk for inadequate treatment of pain.40 Assuring adequate pain control is an essential component of end-of-life care.
Hospice. End-of-life care can often be improved with hospice care. This service is underused by patients with dementia, even though hospice care is available at no cost through Medicare. Hospice eligibility criteria for patients with AD are shown in
TABLE 3.41,42

Finally, a word about prevention
Numerous risk factors have been associated with an increased risk of AD (TABLE 4)2,3. Some, like age and genetics, are nonmodifiable, while others—particularly cardiovascular risk factors—can be modified.1 There are also factors associated with decreased risk, most notably, physical exercise and participating in cognitively stimulating activities.3 Identification of these factors has led to the hope that addressing them can prevent AD.
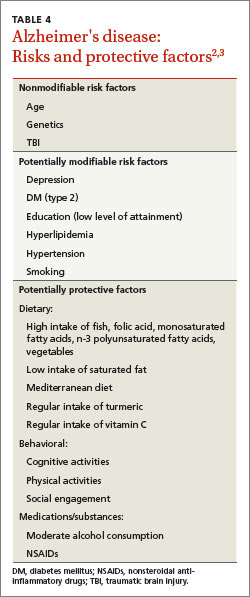
But association does not equal causation. In 2010, a report from the National Institutes of Health concluded that, although there are modifiable factors associated with AD, there is insufficient evidence that addressing any of them will actually prevent AD.43 In fact, there is good evidence that some of these factors (eg, statin therapy) are not effective in reducing the incidence of dementia, and that others (eg, vitamin E and estrogen therapy) are potentially harmful.44
The absence of empirically supported preventive interventions does not mean, however, that we should disregard these risks and protective factors. Encouraging social engagement, for example, may improve both emotional health and quality of life. Addressing cardiovascular risk factors can reduce the rate of coronary and cerebrovascular disease, potentially including vascular dementia, even if it does not reduce the rate of AD.
Studies are evaluating the use of monoclonal antibodies with anti-amyloid properties for preventing AD in individuals who have APOE ε4 genotypes or high amyloid loads on neuroimaging.45 It will be several years before results are available, however, and the outcome of these studies is uncertain as the use of anti-amyloid agents for treating established dementia has not been effective.46,47
CORRESPONDENCE
Marisa Menchola, PhD, Department of Psychiatry, University of Arizona College of Medicine, 1501 N. Campbell Ave., 7OPC. Tucson, AZ 85724; [email protected]
1. Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimer’s Association Web site. Available at: http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed December 2, 2014.
2. Román GC, Nash DT, Fillit H. Translating current knowledge into dementia prevention. Alzheimer Dis Assoc Disord. 2012;26:295-299.
3. Jak, AJ. The impact of physical and mental activity on cognitive aging. Curr Top Behav Neurosci. 2012;10:273-291.
4. US Preventive Services Task Force. Cognitive impairment in older adults: Screening. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/cognitive-impairment-in-older-adults-screening. Accessed November 28, 2014.
5. Centers for Medicare & Medicaid Services. The guide to Medicare preventive services. 4th ed. 2011. Available at: http://www.curemd.com/fqhc/The%20Guide%20to%20Medicare%20Preventative%20Services%20for%20Physicans,%20Providers%20and%20Suppliers.pdf. Accessed December 2, 2014.
6. Boustani, M. Dementia screening in primary care: not too fast! J Amer Geriatr Soc. 2013;61:1205-1207.
7. Alzheimer’s Association. Know the 10 signs: Early detection matters. Alzheimer’s Association Web site. Available at: http://www.alz.org/national/documents/checklist_10signs.pdf. Accessed December 2, 2014.
8. Cordell CB, Borson S, Boustani M, et al; Medicare Detection of Cognitive Impairment Workgroup. Alzheimer’s Association recommendations for operationalizing the detection of cognitive impairment during the Medicare Annual Wellness Visit in a primary care setting. Alzheimers Dement. 2013;9:141-150.
9. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263-269.
10. The American Geriatrics Society. A guide to dementia diagnosis and treatment. The American Geriatrics Society Web site. Available at: http://dementia.americangeriatrics.org/documents/AGS_PC_Dementia_Sheet_2010v2.pdf. Accessed December
2, 2014.
11. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207-216.
12. Johnson KA, Minoshima S, Bohnen NI, et al; Alzheimer’s Association; Society of Nuclear Medicine and Molecular Imaging; Amyloid Imaging Taskforce. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;9:e1-e16.
13. Centers for Medicare and Medicaid Services. National coverage determination (NCD) for FDG PET for dementia and neurodegenerative diseases (220.6.13). Centers for Medicare and Medicaid Services Web site. Available at: http://www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=288&ncdver=3&bc=BAABAAAAAAAA&. Accessed December 2, 2014.
14. Albert MS, DeKosky ST, Ruckson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270-179.
15. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia—meta-analysis of 41 robust inception studies. Acta Psychiatr Scand. 2009;119:252-265.
16. Farias ST, Mungas D, Reed BR, et al. Progression of mild cognitive impairment to dementia in clinic- vs community-based cohorts. Arch Neurol. 2009;66:1151-1157.
17. Bensadon BA, Odenheimer GL. Current management decisions in mild cognitive impairment. Clin Geriatr Med. 2013;29:847-871.
18. Ngo-Metzger Q, August KJ, Srinivasan M, et al. End-of-life care: guidelines for patient-centered communication. Am Fam Physician. 2008;77:167-174.
19. Sayegh P, Knight BG. Cross-cultural differences in dementia: the Sociocultural Health Belief Model. Int Psychogeriatr. 2013;25:517-530.
20. McDaniel SH, Campbell TL, Hepworth J, et al. Family-Oriented Primary Care. 2nd ed. New York, NY: Springer; 2005.
21. Bensadon BA, Odenheimer GL. Current management decisions in mild cognitive impairment. Clin Geriatr Med. 2013:29;847-871.
22. Russ TC, Morling JR. Cholinesterase inhibitors for mild cognitive impairment. Cochrane Database Syst Rev. 2012;9:CD009132.
23. Sadowsky CH, Galvin JE. Guidelines for the management of cognitive and behavioral problems in dementia. J Am Board Fam Med. 2012;25:350-366.
24. Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD005593.
25. Fillit HM, Doody RS, Binaso K, et al. Recommendations for best practices in the treatment of Alzheimer’s disease in managed care. Am J Geriatr Pharmacother. 2006;4(suppl A):S9-S24;quiz S25-S28.
26. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355:1525-1538.
27. US Food and Drug Administration. Public health advisory: Deaths with antipsychotics in elderly patients with behavioral disturbances. US Food and Drug Administration Web site. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm053171.htm. Published April 11, 2005. Updated August 16, 2013. Accessed December 2, 2014.
28. The American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60:616-631.
29. Brummel-Smith K. It’s time to require written informed consent when using antipsychotics in dementia. Br J Med Pract. 2008;1:4-6.
30. Huckans M, Hutson L, Twamley E, et al. Efficacy of cognitive rehabilitation therapies for mild cognitive impairment (MCI) in older adults: working toward a theoretical model and evidence-based interventions. Neuropsychol Rev. 2013;23:63-80.
31. Woods B, Aguirre E, Spector AE, et al. Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst Rev. 2012;2:CD005562.
32. McLaren AN, Lamantia MA, Callahan CM. Systematic review of non-pharmacologic interventions to delay functional decline in community-dwelling patients with dementia. Aging Ment Health. 2013;17:655-666.
33. Bahar-Fuchs A, Clare L, Woods B. Cognitive training and cognitive rehabilitation for mild to moderate Alzheimer’s disease and vascular dementia. Cochrane Database Syst Rev. 2013;6:CD003260.
34. Logsdon RG, McCurry SM, Teri L. Evidence-based psychological treatments for disruptive behaviors in individuals with dementia. Psychol Aging. 2007;22:28-36.
35. Raetz J. A nondrug approach to dementia. J Fam Pract. 2013;62:548-557.
36. Gallagher-Thompson D, Coon DW. Evidence-based psychological treatments for distress in family caregivers of older adults. Psychol Aging. 2007;22:37-51.
37. Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009:361:1529-1538.
38. Parsons C, Hughes CM, Passmore AP, et al. Withholding, discontinuing and withdrawing medications in dementia patients at the end of life: a neglected problem in the disadvantaged dying? Drugs Aging. 2010;27:435-449.
39. The American Geriatrics Society. Feeding tubes in advanced dementia position statement. The American Geriatrics Society Web site. Available at: http://www.americangeriatrics.org/files/documents/feeding.tubes.advanced.dementia.pdf. Accessed November 19, 2013.
40. Goodman C, Evans C, Wilcock J, et al. End of life care for community dwelling older people with dementia: an integrated review. Int J Geriatr Psychiatry. 2010;25:329-337.
41. Storey CP. A quick-reference guide to the hospice and palliative care training for physicians: UNIPAC self-study program. American Academy of Hospice and Palliative Medicine. Chicago; 2009.
42. Kaszniak AW, Kligman EW. Hospice care for patients with dementia. Elder Care. 2013. Arizona Alzheimer's Consortium Web site. Available at: http://azalz.org/wp-content/uploads/2013/07/Hospice-Care-for-Pts-with-Dementia.pdf. Accessed December 2, 2014.
43. Daviglus ML, Bell CC, Berrettini W, et al. NIH state-of-the-science conference statement: Preventing Alzheimer’s disease and cognitive decline. NIH Consens State Sci Statements. 2010;27:1-30.
44. Patterson C, Feightner JW, Garcia A, et al. Diagnosis and treatment of dementia: 1. Risk assessment and primary prevention of Alzheimer disease. CMAJ. 2008;178:548-556.
45. Carrillo MC, Brashear HR, Logovinsky V, et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimers Dement. 2013;9:123-131.e1.
46. Salloway S, Sperling R, Fox NC, et al; Bapineuzumab 301 and 302 Clinical Trial Investigators. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s Disease. N Engl J Med. 2014;370:322-333.
47. Doody RS, Thomas RG, Farlow M, et al; Alheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s Disease. N Engl J Med. 2014:370:311-321.
› Refer patients for formal neuropsychological testing when dementia is suspected but the history, clinical interview, and brief cognitive tests do not result in a definitive diagnosis. C
› Use non-drug therapies as first-line treatment for behavioral symptoms of Alzheimer’s disease (AD), as the adverse effects of drug therapy generally offset any benefit. B
› Recommend against feeding tubes for patients with late-stage AD as they are more apt to cause discomfort than to provide benefit. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Alzheimer’s disease (AD), the most common form of dementia, affects more than 5 million Americans.1 Estimates suggest that by 2050, the prevalence could triple, reaching 13 to 16 million.1 To effectively care for patients with AD and their families, family physicians need to be familiar with the latest evidence on all facets of care, from initial detection to patient management and end-of-life care.
This evidence-based review will help you toward that end by answering common questions regarding Alzheimer’s care, including whether routine screening is advisable, what tests should be ordered, which interventions (including nonpharmacologic options) are worth considering, and how best to counsel patients and families about end-of-life care.
Routine screening? Still subject to debate
In considering routine dementia screening in primary care, the key question is whether screening improves outcomes. Advocates note that individuals with dementia may appear unimpaired during office visits and may not report symptoms due to lack of insight; they point out, too, that waiting for an event that makes cognitive impairment obvious, such as a driving mishap, is risky.2 Those who advocate routine screening also note that only about half of those who have dementia are ever diagnosed.3
Others, including the US Preventive Services Task Force (USPSTF), disagree. In its 2014 evidence review, the USPSTF indicated that there is “insufficient evidence to assess the balance of benefits and harms of screening for cognitive impairment in older adults.”4
Mixed messages
The dearth of evidence is also reflected in the conflicting recommendations of the Affordable Care Act (ACA) and the Centers for Medicare and Medicaid Services (CMS). The ACA requires physicians to assess the cognitive function of Medicare patients during their annual wellness visits. CMS, however, instructs providers to screen for dementia only if observation or concerns raised by the patient or family suggest the possibility of impairment, and does not recommend any particular test.5
Cost-effectiveness analyses raise questions about the value of routine screening, as well. Evidence suggests that if a primary care physician screens 300 older patients, 39 will have a positive screen. But only about half of those 39 will agree to a diagnostic evaluation, and no more than 9 will ultimately be diagnosed with dementia. The estimated cost of identifying 9 cases is nearly $40,000—all in the absence of a treatment to cure or stop the progression of the disorder.6
The bottom line: Evidence does not support routine dementia screening of older adults. When cognitive impairment is suspected, however, physicians should conduct a diagnostic evaluation—and consider educating patients and families about the Alzheimer’s Association (AA)’s 10 warning signs of AD.7 (See “Is it Alzheimer’s? 10 warning signs”7 below.) A longer version, available at http://www.alz.org/national/documents/checklist_10signs.pdf, outlines the cognitive changes that are characteristic of healthy aging and compares them to changes suggestive of early dementia.7
Is it Alzheimer’s? 10 warning signs7 1. Memory loss that disrupts daily life |
How to proceed when you suspect AD
Step 1: Screening instrument. The first step in the diagnostic evaluation of a patient with suspected AD is to determine if, in fact, cognitive impairment is present. This can be done by screening with in-office screening instruments, such as the Mini-Cog (available at alz.org/documents_custom/minicog.pdf) or Mini-Mental State Examination (MMSE; health.gov.bc.ca/pharmacare/adti/clinician/pdf/ADTI%20SMMSE-GDS%20Reference%20Card.pdf), among others.8
Step 2: Clinical evaluation. If observation and test results suggest cognitive impairment, the next step is to determine whether clinical findings are consistent with the diagnostic criteria for AD (TABLE 1)9 developed by workgroups from the National Institute on Aging (NIA)/AA in 2011. A work-up is necessary to identify conditions that can mimic dementia (eg, depression) and behaviors that suggest another type of dementia, such as frontotemporal or Lewy body dementia.10 Lab testing should be included to rule out potentially reversible causes of cognitive dysfunction (eg, hypothyroidism, vitamin D deficiency).
Step 3: Neuropsychological evaluation. The NIA/AA recommends neuropsychological testing when the brief cognitive tests, history, and clinical work-up are not sufficient for a definitive diagnosis of dementia.9This generally involves a referral to a neuropsychologist, who conducts a battery of standardized tests to evaluate attention, memory, language, visual-spatial abilities, and executive functions, among others. Neuropsychological testing can confirm the presence of cognitive impairment and aid in the differential diagnosis by comparing the patient’s performance in these domains with characteristic features of different dementia syndromes.
Step 4: Brain imaging with either computed tomography or magnetic resonance imaging can be included in the work-up for patients with suspected AD to rule out abnormalities—eg, metastatic cancer, hydrocephalus, or occult chronic subdural hematoma—that could be causing cognitive impairment.9,10 Clinical features that generally warrant brain imaging include onset of cognitive impairment before age 60; unexplained focal neurologic signs or symptoms; abrupt onset or rapid decline; and/or predisposing conditions, such as cancer or anticoagulant treatment.10
The role of biomarkers and advanced brain imaging
Biomarkers that might provide confirmation of AD in patients who exhibit early symptoms of dementia have been studied extensively.11 The NIA/AA identified 2 categories of AD biomarkers:
- tests for β-amyloid deposition in the brain, including spinal fluid assays for β-amyloid (Aβ42) and positron emission tomography (PET) scans after intravenous injection of florbetapir or flutemetamol, which bind to amyloid in the brain; and
- tests for neuronal degeneration, which would include spinal fluid assays for tau protein and PET scans after injection of fluorodeoxyglucose (FDG), which shows decreased uptake in patients with AD.9
Research reveals the promise of these biomarkers as diagnostic tools, particularly in patients with an atypical presentation of dementia or mild cognitive impairment (MCI) that may be associated with early AD.12 (More on MCI in a moment.) However, the NIA/AA concluded that additional research is needed to validate these tests for routine diagnostic purposes. Medicare covers PET scans with FDG only for the differential diagnosis of AD vs frontotemporal dementia.13
Mild cognitive impairment: How likely that it will progress?
Along with diagnostic criteria for AD, the NIA/AA developed criteria for a symptomatic predementia phase of AD—often referred to as MCI.14 According to the workgroup, MCI is diagnosed when:
2. there is evidence of cognitive impairment, ideally through psychometric testing, revealing performance below expectation based on the patient’s age and education;
3. the patient is able to maintain independent functioning in daily life, despite mild problems or the need for minimal assistance; and
4. there is no significant impairment in social or occupational functioning.14
Progression: Less likely than you might think
Patients with MCI are at risk for progression to overt dementia, with an overall annual conversion rate from MCI to dementia estimated at 10% to 15%.15,16 This estimate must be interpreted with caution, however, because most studies were conducted prior to the 2011 guidelines, when different diagnostic criteria were used. Observers have noted, too, that the numbers largely reflect data collected in specialty clinics and that community-based studies reveal substantially lower conversion rates (3%-6% per year).16 In addition, evidence suggests that many patients with MCI demonstrate long-term stability or even reversal of deficits.17
While there is some consideration of the use of biomarkers and amyloid imaging tests to help determine which patients with MCI will progress to AD, practice guidelines do not currently recommend such testing and it is not covered by Medicare.
When evidence indicates an AD diagnosis
When faced with the need to communicate an AD diagnosis, follow the general recommendations for delivering any bad news or discouraging prognosis:
Prioritize and limit the information you provide, determining not only what the patient and family want to hear, but also how much they are able to comprehend.
Confirm that the patient and family understand the information you’ve provided.
Offer emotional support and recommend additional resources18 (TABLE 2).
Given the progressive cognitive decline that characterizes AD, it is important to address the primary caregiver’s understanding of, and ability to cope with, the disease. It is also important to explore beliefs and attitudes regarding AD. Keep in mind that different cultural groups tend to differ in their beliefs about the nature, cause, and appropriate management of AD, as well as the role of spirituality, help-seeking, and stigma.19,20
The progressive and ultimately fatal nature of AD also makes planning for the future a priority. Ideally, patients should be engaged in discussions regarding end-of-life care as early as possible, while they are still able to make informed decisions and express their preferences. Discussing end-of-life care can be overwhelming for newly diagnosed patients and their families, however, so it is important that you address issues—medical, financial, and legal planning, for example—that families should be considering.
TABLE 2
| Resources for newly diagnosed patients and families | |
| Issue | Resources |
| Education | Alzheimer’s and Dementia Caregiver Center http://www.alz.org/care/overview.asp NIA Alzheimer’s Disease Education and Referral Center http://www.nia.nih.gov/alzheimers/ |
| Planning (medical, financial, legal)/benefits | AARP Caregiving Resource Center http://www.aarp.org/home-family/caregiving Alzheimer’s Association Alzheimer’s Navigator https://www.alzheimersnavigator.org/ National Council on Aging Benefits Checkup https://www.benefitscheckup.org/ |
| Safety | Association for Driver Rehabilitation Specialists: Driving and Alzheimer’s/Dementia https://c.ymcdn.com/sites/www.aded.net/resource/resmgr/fact_Sheets/ADED_alzheimers-Dementia_fac.pdf NIA’s Home Safety for People with Alzheimer’s booklet http://www.nia.nih.gov/alzheimers/publication/home-safety-people-alzheimers-disease |
| Support | Caregiver Action Network http://caregiveraction.org/ |
Drugs address cognitive and behavioral function
No currently available treatments can cure or significantly alter the progression of AD, but 2 classes of medications are used in an attempt to improve cognitive function. One is cholinesterase inhibitors (ChEIs), which potentiate acetylcholine synaptic transmission. The other is N-methyl-D-aspartate (NMDA) glutamate receptor blockers. Other classes of drugs are sometimes used to treat behavioral symptoms of dementia, such as agitation, aggression, mood disorders, and psychosis (eg, delusions and hallucinations).
Cognitive function. Results from studies of pharmacologic management of MCI vary widely, but recent reviews have found no convincing evidence that either ChEIs or NMDA receptor blockers have an effect on progression from MCI to dementia.21,22 Neither class is FDA-approved for treating MCI.
In patients with dementia, the effects of ChEIs and NMDA receptor blockers on cognition are statistically significant but modest, and often of questionable clinical relevance.23 Nonetheless, among ChEIs, donepezil is approved by the US Food and Drug Administration (FDA) for mild, moderate, and severe dementia and galantamine and rivastigmine are approved for mild and moderate dementia. There is no evidence that any one ChEI is more effective than any other,24 and the choice of drugs is often guided by cost, adverse effects, and health plan formularies. Memantine, the only FDA-approved NMDA receptor blocker, is approved for moderate to severe dementia and can be used alone or in combination with a ChEI.
If these drugs are used in an attempt to improve cognition in AD, guidelines recommend the following approach for initial therapy: Prescribe a ChEI for the mild stage, a ChEI plus memantine for the moderate stage, and memantine (with or without a ChEI) for the severe stage.25 The recommendations also include monitoring every 6 months.
There is no consensus about when to discontinue medication. Various published recommendations call for continuing treatment until the patient has “lost all cognitive and functional abilities;”22 until the patient’s MMSE score falls below 10 and there is no indication that the drug is having a “worthwhile effect;”21 or until he or she has reached stage 7 on the Reisberg Functional Assessment Staging scale, indicating nonambulatory status with speech limited to one to 5 words a day.10
Behavioral function. A variety of drugs are used to treat behavioral symptoms in AD. While not FDA-approved for this use, the most widely prescribed agents are second-generation antipsychotics (aripiprazole, olanzapine, quetiapine, and risperidone). The main effect of these drugs is often nothing more than sedation, and one large multisite clinical trial concluded that the adverse effects offset the benefits for patients with AD.26 Indeed, the FDA has issued an advisory on the use of second-generation antipsychotics in AD patients, stating that they are associated with an increased risk of death.27 The recently updated Beers Criteria strongly recommend avoiding these drugs for treating behavioral disturbances in AD unless nonpharmacologic options have failed and the patient is a threat to self or others.28
Because of the black-box warning that antipsychotics increase the risk of death, some physicians have advocated obtaining informed consent prior to prescribing such medications.29 At the very least, when family or guardians are involved, a conversation about risks vs benefits should take place and be documented in the medical record.
Other drug classes are also sometimes used in an attempt to improve behavioral function, including anti-seizure medications (valproic acid, carbamazepine), antidepressants (trazodone and selective serotonin reuptake inhibitors), and anxiolytics (benzodiazepines and buspirone). Other than their sedating effects, there is no strong evidence that these drugs are effective for treating dementia-related behavioral disorders. If used, caution is required due to potential adverse effects.
Nonpharmacologic management is “promising”
A recent systematic review of nonpharmacologic interventions for MCI evaluated exercise, training in compensatory strategies, and engagement in cognitively stimulating activities and found “promising but inconclusive” results. The researchers found that studies show mostly positive effects on cognition but have significant methodologic limitations.30 Importantly, there is no evidence of delayed or reduced conversion to dementia.
For patients who already have mild-to-moderate dementia, cognitive stimulation seems to help in the short term.31 There is also some evidence that exercise and occupational therapy may slow functional decline,32 but the effects are small to modest and their actual clinical significance (eg, the ability to delay institutionalization) is unclear. There is promising but preliminary evidence that cognitive rehabilitation (helping patients devise strategies to complete daily activities) may improve functioning in everyday life.33
While behavioral symptoms are often due to the dementia itself, it is important to identify and treat medical and environmental causes that may be contributing, such as infection, pain, and loud or unsafe environments. As noted before, nonpharmacologic treatments are generally preferred for behavioral problems and should be considered prior to drug therapy. Approaches that identify and modify both the antecedents and consequences of problem behaviors and increase pleasant events have empirical support for the management of behavioral symptoms.34 Interventions including massage therapy, aromatherapy, exercise, and music therapy may also be effective in the short term for agitated behavior.35
Caregivers should be encouraged to receive training in these strategies through organizations like AA. Caregiver education and support can reduce caregivers’ distress and increase their self-efficacy and coping skills.36
End-of-life care must be addressed
Perhaps the most important aspect of end-of-life care in AD is assuring that families (or health care proxies) understand that AD is a fatal illness, with most patients dying within 4 to 8 years of diagnosis.1 Evidence indicates that patients whose proxies have a clear recognition of this are less likely to experience “burdensome” interventions such as parenteral therapy, emergency department visits, hospital admissions, and tube feedings in their last 3 months of life.37
Overall, decisions regarding discontinuing medical treatments in advanced AD should be made by balancing the likelihood of benefit with the potential for adverse effects.38 For example, the American Geriatrics Society recently recommended against feeding tubes because they often result in discomfort due to agitation, use of restraints, and worsening pressure ulcers.39
Unfortunately, only a minority of families receives straightforward information on the course and prognosis of AD, including the fact that patients eventually stop eating and that the natural cause of death is often an acute infection. Studies also show that patients with dementia are at risk for inadequate treatment of pain.40 Assuring adequate pain control is an essential component of end-of-life care.
Hospice. End-of-life care can often be improved with hospice care. This service is underused by patients with dementia, even though hospice care is available at no cost through Medicare. Hospice eligibility criteria for patients with AD are shown in
TABLE 3.41,42
Finally, a word about prevention
Numerous risk factors have been associated with an increased risk of AD (TABLE 4)2,3. Some, like age and genetics, are nonmodifiable, while others—particularly cardiovascular risk factors—can be modified.1 There are also factors associated with decreased risk, most notably, physical exercise and participating in cognitively stimulating activities.3 Identification of these factors has led to the hope that addressing them can prevent AD.
But association does not equal causation. In 2010, a report from the National Institutes of Health concluded that, although there are modifiable factors associated with AD, there is insufficient evidence that addressing any of them will actually prevent AD.43 In fact, there is good evidence that some of these factors (eg, statin therapy) are not effective in reducing the incidence of dementia, and that others (eg, vitamin E and estrogen therapy) are potentially harmful.44
The absence of empirically supported preventive interventions does not mean, however, that we should disregard these risks and protective factors. Encouraging social engagement, for example, may improve both emotional health and quality of life. Addressing cardiovascular risk factors can reduce the rate of coronary and cerebrovascular disease, potentially including vascular dementia, even if it does not reduce the rate of AD.
Studies are evaluating the use of monoclonal antibodies with anti-amyloid properties for preventing AD in individuals who have APOE ε4 genotypes or high amyloid loads on neuroimaging.45 It will be several years before results are available, however, and the outcome of these studies is uncertain as the use of anti-amyloid agents for treating established dementia has not been effective.46,47
CORRESPONDENCE
Marisa Menchola, PhD, Department of Psychiatry, University of Arizona College of Medicine, 1501 N. Campbell Ave., 7OPC. Tucson, AZ 85724; [email protected]
› Refer patients for formal neuropsychological testing when dementia is suspected but the history, clinical interview, and brief cognitive tests do not result in a definitive diagnosis. C
› Use non-drug therapies as first-line treatment for behavioral symptoms of Alzheimer’s disease (AD), as the adverse effects of drug therapy generally offset any benefit. B
› Recommend against feeding tubes for patients with late-stage AD as they are more apt to cause discomfort than to provide benefit. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Alzheimer’s disease (AD), the most common form of dementia, affects more than 5 million Americans.1 Estimates suggest that by 2050, the prevalence could triple, reaching 13 to 16 million.1 To effectively care for patients with AD and their families, family physicians need to be familiar with the latest evidence on all facets of care, from initial detection to patient management and end-of-life care.
This evidence-based review will help you toward that end by answering common questions regarding Alzheimer’s care, including whether routine screening is advisable, what tests should be ordered, which interventions (including nonpharmacologic options) are worth considering, and how best to counsel patients and families about end-of-life care.
Routine screening? Still subject to debate
In considering routine dementia screening in primary care, the key question is whether screening improves outcomes. Advocates note that individuals with dementia may appear unimpaired during office visits and may not report symptoms due to lack of insight; they point out, too, that waiting for an event that makes cognitive impairment obvious, such as a driving mishap, is risky.2 Those who advocate routine screening also note that only about half of those who have dementia are ever diagnosed.3
Others, including the US Preventive Services Task Force (USPSTF), disagree. In its 2014 evidence review, the USPSTF indicated that there is “insufficient evidence to assess the balance of benefits and harms of screening for cognitive impairment in older adults.”4
Mixed messages
The dearth of evidence is also reflected in the conflicting recommendations of the Affordable Care Act (ACA) and the Centers for Medicare and Medicaid Services (CMS). The ACA requires physicians to assess the cognitive function of Medicare patients during their annual wellness visits. CMS, however, instructs providers to screen for dementia only if observation or concerns raised by the patient or family suggest the possibility of impairment, and does not recommend any particular test.5
Cost-effectiveness analyses raise questions about the value of routine screening, as well. Evidence suggests that if a primary care physician screens 300 older patients, 39 will have a positive screen. But only about half of those 39 will agree to a diagnostic evaluation, and no more than 9 will ultimately be diagnosed with dementia. The estimated cost of identifying 9 cases is nearly $40,000—all in the absence of a treatment to cure or stop the progression of the disorder.6
The bottom line: Evidence does not support routine dementia screening of older adults. When cognitive impairment is suspected, however, physicians should conduct a diagnostic evaluation—and consider educating patients and families about the Alzheimer’s Association (AA)’s 10 warning signs of AD.7 (See “Is it Alzheimer’s? 10 warning signs”7 below.) A longer version, available at http://www.alz.org/national/documents/checklist_10signs.pdf, outlines the cognitive changes that are characteristic of healthy aging and compares them to changes suggestive of early dementia.7
Is it Alzheimer’s? 10 warning signs7 1. Memory loss that disrupts daily life |
How to proceed when you suspect AD
Step 1: Screening instrument. The first step in the diagnostic evaluation of a patient with suspected AD is to determine if, in fact, cognitive impairment is present. This can be done by screening with in-office screening instruments, such as the Mini-Cog (available at alz.org/documents_custom/minicog.pdf) or Mini-Mental State Examination (MMSE; health.gov.bc.ca/pharmacare/adti/clinician/pdf/ADTI%20SMMSE-GDS%20Reference%20Card.pdf), among others.8
Step 2: Clinical evaluation. If observation and test results suggest cognitive impairment, the next step is to determine whether clinical findings are consistent with the diagnostic criteria for AD (TABLE 1)9 developed by workgroups from the National Institute on Aging (NIA)/AA in 2011. A work-up is necessary to identify conditions that can mimic dementia (eg, depression) and behaviors that suggest another type of dementia, such as frontotemporal or Lewy body dementia.10 Lab testing should be included to rule out potentially reversible causes of cognitive dysfunction (eg, hypothyroidism, vitamin D deficiency).
Step 3: Neuropsychological evaluation. The NIA/AA recommends neuropsychological testing when the brief cognitive tests, history, and clinical work-up are not sufficient for a definitive diagnosis of dementia.9This generally involves a referral to a neuropsychologist, who conducts a battery of standardized tests to evaluate attention, memory, language, visual-spatial abilities, and executive functions, among others. Neuropsychological testing can confirm the presence of cognitive impairment and aid in the differential diagnosis by comparing the patient’s performance in these domains with characteristic features of different dementia syndromes.
Step 4: Brain imaging with either computed tomography or magnetic resonance imaging can be included in the work-up for patients with suspected AD to rule out abnormalities—eg, metastatic cancer, hydrocephalus, or occult chronic subdural hematoma—that could be causing cognitive impairment.9,10 Clinical features that generally warrant brain imaging include onset of cognitive impairment before age 60; unexplained focal neurologic signs or symptoms; abrupt onset or rapid decline; and/or predisposing conditions, such as cancer or anticoagulant treatment.10
The role of biomarkers and advanced brain imaging
Biomarkers that might provide confirmation of AD in patients who exhibit early symptoms of dementia have been studied extensively.11 The NIA/AA identified 2 categories of AD biomarkers:
- tests for β-amyloid deposition in the brain, including spinal fluid assays for β-amyloid (Aβ42) and positron emission tomography (PET) scans after intravenous injection of florbetapir or flutemetamol, which bind to amyloid in the brain; and
- tests for neuronal degeneration, which would include spinal fluid assays for tau protein and PET scans after injection of fluorodeoxyglucose (FDG), which shows decreased uptake in patients with AD.9
Research reveals the promise of these biomarkers as diagnostic tools, particularly in patients with an atypical presentation of dementia or mild cognitive impairment (MCI) that may be associated with early AD.12 (More on MCI in a moment.) However, the NIA/AA concluded that additional research is needed to validate these tests for routine diagnostic purposes. Medicare covers PET scans with FDG only for the differential diagnosis of AD vs frontotemporal dementia.13
Mild cognitive impairment: How likely that it will progress?
Along with diagnostic criteria for AD, the NIA/AA developed criteria for a symptomatic predementia phase of AD—often referred to as MCI.14 According to the workgroup, MCI is diagnosed when:
2. there is evidence of cognitive impairment, ideally through psychometric testing, revealing performance below expectation based on the patient’s age and education;
3. the patient is able to maintain independent functioning in daily life, despite mild problems or the need for minimal assistance; and
4. there is no significant impairment in social or occupational functioning.14
Progression: Less likely than you might think
Patients with MCI are at risk for progression to overt dementia, with an overall annual conversion rate from MCI to dementia estimated at 10% to 15%.15,16 This estimate must be interpreted with caution, however, because most studies were conducted prior to the 2011 guidelines, when different diagnostic criteria were used. Observers have noted, too, that the numbers largely reflect data collected in specialty clinics and that community-based studies reveal substantially lower conversion rates (3%-6% per year).16 In addition, evidence suggests that many patients with MCI demonstrate long-term stability or even reversal of deficits.17
While there is some consideration of the use of biomarkers and amyloid imaging tests to help determine which patients with MCI will progress to AD, practice guidelines do not currently recommend such testing and it is not covered by Medicare.
When evidence indicates an AD diagnosis
When faced with the need to communicate an AD diagnosis, follow the general recommendations for delivering any bad news or discouraging prognosis:
Prioritize and limit the information you provide, determining not only what the patient and family want to hear, but also how much they are able to comprehend.
Confirm that the patient and family understand the information you’ve provided.
Offer emotional support and recommend additional resources18 (TABLE 2).
Given the progressive cognitive decline that characterizes AD, it is important to address the primary caregiver’s understanding of, and ability to cope with, the disease. It is also important to explore beliefs and attitudes regarding AD. Keep in mind that different cultural groups tend to differ in their beliefs about the nature, cause, and appropriate management of AD, as well as the role of spirituality, help-seeking, and stigma.19,20
The progressive and ultimately fatal nature of AD also makes planning for the future a priority. Ideally, patients should be engaged in discussions regarding end-of-life care as early as possible, while they are still able to make informed decisions and express their preferences. Discussing end-of-life care can be overwhelming for newly diagnosed patients and their families, however, so it is important that you address issues—medical, financial, and legal planning, for example—that families should be considering.
TABLE 2
| Resources for newly diagnosed patients and families | |
| Issue | Resources |
| Education | Alzheimer’s and Dementia Caregiver Center http://www.alz.org/care/overview.asp NIA Alzheimer’s Disease Education and Referral Center http://www.nia.nih.gov/alzheimers/ |
| Planning (medical, financial, legal)/benefits | AARP Caregiving Resource Center http://www.aarp.org/home-family/caregiving Alzheimer’s Association Alzheimer’s Navigator https://www.alzheimersnavigator.org/ National Council on Aging Benefits Checkup https://www.benefitscheckup.org/ |
| Safety | Association for Driver Rehabilitation Specialists: Driving and Alzheimer’s/Dementia https://c.ymcdn.com/sites/www.aded.net/resource/resmgr/fact_Sheets/ADED_alzheimers-Dementia_fac.pdf NIA’s Home Safety for People with Alzheimer’s booklet http://www.nia.nih.gov/alzheimers/publication/home-safety-people-alzheimers-disease |
| Support | Caregiver Action Network http://caregiveraction.org/ |
Drugs address cognitive and behavioral function
No currently available treatments can cure or significantly alter the progression of AD, but 2 classes of medications are used in an attempt to improve cognitive function. One is cholinesterase inhibitors (ChEIs), which potentiate acetylcholine synaptic transmission. The other is N-methyl-D-aspartate (NMDA) glutamate receptor blockers. Other classes of drugs are sometimes used to treat behavioral symptoms of dementia, such as agitation, aggression, mood disorders, and psychosis (eg, delusions and hallucinations).
Cognitive function. Results from studies of pharmacologic management of MCI vary widely, but recent reviews have found no convincing evidence that either ChEIs or NMDA receptor blockers have an effect on progression from MCI to dementia.21,22 Neither class is FDA-approved for treating MCI.
In patients with dementia, the effects of ChEIs and NMDA receptor blockers on cognition are statistically significant but modest, and often of questionable clinical relevance.23 Nonetheless, among ChEIs, donepezil is approved by the US Food and Drug Administration (FDA) for mild, moderate, and severe dementia and galantamine and rivastigmine are approved for mild and moderate dementia. There is no evidence that any one ChEI is more effective than any other,24 and the choice of drugs is often guided by cost, adverse effects, and health plan formularies. Memantine, the only FDA-approved NMDA receptor blocker, is approved for moderate to severe dementia and can be used alone or in combination with a ChEI.
If these drugs are used in an attempt to improve cognition in AD, guidelines recommend the following approach for initial therapy: Prescribe a ChEI for the mild stage, a ChEI plus memantine for the moderate stage, and memantine (with or without a ChEI) for the severe stage.25 The recommendations also include monitoring every 6 months.
There is no consensus about when to discontinue medication. Various published recommendations call for continuing treatment until the patient has “lost all cognitive and functional abilities;”22 until the patient’s MMSE score falls below 10 and there is no indication that the drug is having a “worthwhile effect;”21 or until he or she has reached stage 7 on the Reisberg Functional Assessment Staging scale, indicating nonambulatory status with speech limited to one to 5 words a day.10
Behavioral function. A variety of drugs are used to treat behavioral symptoms in AD. While not FDA-approved for this use, the most widely prescribed agents are second-generation antipsychotics (aripiprazole, olanzapine, quetiapine, and risperidone). The main effect of these drugs is often nothing more than sedation, and one large multisite clinical trial concluded that the adverse effects offset the benefits for patients with AD.26 Indeed, the FDA has issued an advisory on the use of second-generation antipsychotics in AD patients, stating that they are associated with an increased risk of death.27 The recently updated Beers Criteria strongly recommend avoiding these drugs for treating behavioral disturbances in AD unless nonpharmacologic options have failed and the patient is a threat to self or others.28
Because of the black-box warning that antipsychotics increase the risk of death, some physicians have advocated obtaining informed consent prior to prescribing such medications.29 At the very least, when family or guardians are involved, a conversation about risks vs benefits should take place and be documented in the medical record.
Other drug classes are also sometimes used in an attempt to improve behavioral function, including anti-seizure medications (valproic acid, carbamazepine), antidepressants (trazodone and selective serotonin reuptake inhibitors), and anxiolytics (benzodiazepines and buspirone). Other than their sedating effects, there is no strong evidence that these drugs are effective for treating dementia-related behavioral disorders. If used, caution is required due to potential adverse effects.
Nonpharmacologic management is “promising”
A recent systematic review of nonpharmacologic interventions for MCI evaluated exercise, training in compensatory strategies, and engagement in cognitively stimulating activities and found “promising but inconclusive” results. The researchers found that studies show mostly positive effects on cognition but have significant methodologic limitations.30 Importantly, there is no evidence of delayed or reduced conversion to dementia.
For patients who already have mild-to-moderate dementia, cognitive stimulation seems to help in the short term.31 There is also some evidence that exercise and occupational therapy may slow functional decline,32 but the effects are small to modest and their actual clinical significance (eg, the ability to delay institutionalization) is unclear. There is promising but preliminary evidence that cognitive rehabilitation (helping patients devise strategies to complete daily activities) may improve functioning in everyday life.33
While behavioral symptoms are often due to the dementia itself, it is important to identify and treat medical and environmental causes that may be contributing, such as infection, pain, and loud or unsafe environments. As noted before, nonpharmacologic treatments are generally preferred for behavioral problems and should be considered prior to drug therapy. Approaches that identify and modify both the antecedents and consequences of problem behaviors and increase pleasant events have empirical support for the management of behavioral symptoms.34 Interventions including massage therapy, aromatherapy, exercise, and music therapy may also be effective in the short term for agitated behavior.35
Caregivers should be encouraged to receive training in these strategies through organizations like AA. Caregiver education and support can reduce caregivers’ distress and increase their self-efficacy and coping skills.36
End-of-life care must be addressed
Perhaps the most important aspect of end-of-life care in AD is assuring that families (or health care proxies) understand that AD is a fatal illness, with most patients dying within 4 to 8 years of diagnosis.1 Evidence indicates that patients whose proxies have a clear recognition of this are less likely to experience “burdensome” interventions such as parenteral therapy, emergency department visits, hospital admissions, and tube feedings in their last 3 months of life.37
Overall, decisions regarding discontinuing medical treatments in advanced AD should be made by balancing the likelihood of benefit with the potential for adverse effects.38 For example, the American Geriatrics Society recently recommended against feeding tubes because they often result in discomfort due to agitation, use of restraints, and worsening pressure ulcers.39
Unfortunately, only a minority of families receives straightforward information on the course and prognosis of AD, including the fact that patients eventually stop eating and that the natural cause of death is often an acute infection. Studies also show that patients with dementia are at risk for inadequate treatment of pain.40 Assuring adequate pain control is an essential component of end-of-life care.
Hospice. End-of-life care can often be improved with hospice care. This service is underused by patients with dementia, even though hospice care is available at no cost through Medicare. Hospice eligibility criteria for patients with AD are shown in
TABLE 3.41,42
Finally, a word about prevention
Numerous risk factors have been associated with an increased risk of AD (TABLE 4)2,3. Some, like age and genetics, are nonmodifiable, while others—particularly cardiovascular risk factors—can be modified.1 There are also factors associated with decreased risk, most notably, physical exercise and participating in cognitively stimulating activities.3 Identification of these factors has led to the hope that addressing them can prevent AD.
But association does not equal causation. In 2010, a report from the National Institutes of Health concluded that, although there are modifiable factors associated with AD, there is insufficient evidence that addressing any of them will actually prevent AD.43 In fact, there is good evidence that some of these factors (eg, statin therapy) are not effective in reducing the incidence of dementia, and that others (eg, vitamin E and estrogen therapy) are potentially harmful.44
The absence of empirically supported preventive interventions does not mean, however, that we should disregard these risks and protective factors. Encouraging social engagement, for example, may improve both emotional health and quality of life. Addressing cardiovascular risk factors can reduce the rate of coronary and cerebrovascular disease, potentially including vascular dementia, even if it does not reduce the rate of AD.
Studies are evaluating the use of monoclonal antibodies with anti-amyloid properties for preventing AD in individuals who have APOE ε4 genotypes or high amyloid loads on neuroimaging.45 It will be several years before results are available, however, and the outcome of these studies is uncertain as the use of anti-amyloid agents for treating established dementia has not been effective.46,47
CORRESPONDENCE
Marisa Menchola, PhD, Department of Psychiatry, University of Arizona College of Medicine, 1501 N. Campbell Ave., 7OPC. Tucson, AZ 85724; [email protected]
1. Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimer’s Association Web site. Available at: http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed December 2, 2014.
2. Román GC, Nash DT, Fillit H. Translating current knowledge into dementia prevention. Alzheimer Dis Assoc Disord. 2012;26:295-299.
3. Jak, AJ. The impact of physical and mental activity on cognitive aging. Curr Top Behav Neurosci. 2012;10:273-291.
4. US Preventive Services Task Force. Cognitive impairment in older adults: Screening. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/cognitive-impairment-in-older-adults-screening. Accessed November 28, 2014.
5. Centers for Medicare & Medicaid Services. The guide to Medicare preventive services. 4th ed. 2011. Available at: http://www.curemd.com/fqhc/The%20Guide%20to%20Medicare%20Preventative%20Services%20for%20Physicans,%20Providers%20and%20Suppliers.pdf. Accessed December 2, 2014.
6. Boustani, M. Dementia screening in primary care: not too fast! J Amer Geriatr Soc. 2013;61:1205-1207.
7. Alzheimer’s Association. Know the 10 signs: Early detection matters. Alzheimer’s Association Web site. Available at: http://www.alz.org/national/documents/checklist_10signs.pdf. Accessed December 2, 2014.
8. Cordell CB, Borson S, Boustani M, et al; Medicare Detection of Cognitive Impairment Workgroup. Alzheimer’s Association recommendations for operationalizing the detection of cognitive impairment during the Medicare Annual Wellness Visit in a primary care setting. Alzheimers Dement. 2013;9:141-150.
9. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263-269.
10. The American Geriatrics Society. A guide to dementia diagnosis and treatment. The American Geriatrics Society Web site. Available at: http://dementia.americangeriatrics.org/documents/AGS_PC_Dementia_Sheet_2010v2.pdf. Accessed December
2, 2014.
11. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207-216.
12. Johnson KA, Minoshima S, Bohnen NI, et al; Alzheimer’s Association; Society of Nuclear Medicine and Molecular Imaging; Amyloid Imaging Taskforce. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;9:e1-e16.
13. Centers for Medicare and Medicaid Services. National coverage determination (NCD) for FDG PET for dementia and neurodegenerative diseases (220.6.13). Centers for Medicare and Medicaid Services Web site. Available at: http://www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=288&ncdver=3&bc=BAABAAAAAAAA&. Accessed December 2, 2014.
14. Albert MS, DeKosky ST, Ruckson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270-179.
15. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia—meta-analysis of 41 robust inception studies. Acta Psychiatr Scand. 2009;119:252-265.
16. Farias ST, Mungas D, Reed BR, et al. Progression of mild cognitive impairment to dementia in clinic- vs community-based cohorts. Arch Neurol. 2009;66:1151-1157.
17. Bensadon BA, Odenheimer GL. Current management decisions in mild cognitive impairment. Clin Geriatr Med. 2013;29:847-871.
18. Ngo-Metzger Q, August KJ, Srinivasan M, et al. End-of-life care: guidelines for patient-centered communication. Am Fam Physician. 2008;77:167-174.
19. Sayegh P, Knight BG. Cross-cultural differences in dementia: the Sociocultural Health Belief Model. Int Psychogeriatr. 2013;25:517-530.
20. McDaniel SH, Campbell TL, Hepworth J, et al. Family-Oriented Primary Care. 2nd ed. New York, NY: Springer; 2005.
21. Bensadon BA, Odenheimer GL. Current management decisions in mild cognitive impairment. Clin Geriatr Med. 2013:29;847-871.
22. Russ TC, Morling JR. Cholinesterase inhibitors for mild cognitive impairment. Cochrane Database Syst Rev. 2012;9:CD009132.
23. Sadowsky CH, Galvin JE. Guidelines for the management of cognitive and behavioral problems in dementia. J Am Board Fam Med. 2012;25:350-366.
24. Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD005593.
25. Fillit HM, Doody RS, Binaso K, et al. Recommendations for best practices in the treatment of Alzheimer’s disease in managed care. Am J Geriatr Pharmacother. 2006;4(suppl A):S9-S24;quiz S25-S28.
26. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355:1525-1538.
27. US Food and Drug Administration. Public health advisory: Deaths with antipsychotics in elderly patients with behavioral disturbances. US Food and Drug Administration Web site. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm053171.htm. Published April 11, 2005. Updated August 16, 2013. Accessed December 2, 2014.
28. The American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60:616-631.
29. Brummel-Smith K. It’s time to require written informed consent when using antipsychotics in dementia. Br J Med Pract. 2008;1:4-6.
30. Huckans M, Hutson L, Twamley E, et al. Efficacy of cognitive rehabilitation therapies for mild cognitive impairment (MCI) in older adults: working toward a theoretical model and evidence-based interventions. Neuropsychol Rev. 2013;23:63-80.
31. Woods B, Aguirre E, Spector AE, et al. Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst Rev. 2012;2:CD005562.
32. McLaren AN, Lamantia MA, Callahan CM. Systematic review of non-pharmacologic interventions to delay functional decline in community-dwelling patients with dementia. Aging Ment Health. 2013;17:655-666.
33. Bahar-Fuchs A, Clare L, Woods B. Cognitive training and cognitive rehabilitation for mild to moderate Alzheimer’s disease and vascular dementia. Cochrane Database Syst Rev. 2013;6:CD003260.
34. Logsdon RG, McCurry SM, Teri L. Evidence-based psychological treatments for disruptive behaviors in individuals with dementia. Psychol Aging. 2007;22:28-36.
35. Raetz J. A nondrug approach to dementia. J Fam Pract. 2013;62:548-557.
36. Gallagher-Thompson D, Coon DW. Evidence-based psychological treatments for distress in family caregivers of older adults. Psychol Aging. 2007;22:37-51.
37. Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009:361:1529-1538.
38. Parsons C, Hughes CM, Passmore AP, et al. Withholding, discontinuing and withdrawing medications in dementia patients at the end of life: a neglected problem in the disadvantaged dying? Drugs Aging. 2010;27:435-449.
39. The American Geriatrics Society. Feeding tubes in advanced dementia position statement. The American Geriatrics Society Web site. Available at: http://www.americangeriatrics.org/files/documents/feeding.tubes.advanced.dementia.pdf. Accessed November 19, 2013.
40. Goodman C, Evans C, Wilcock J, et al. End of life care for community dwelling older people with dementia: an integrated review. Int J Geriatr Psychiatry. 2010;25:329-337.
41. Storey CP. A quick-reference guide to the hospice and palliative care training for physicians: UNIPAC self-study program. American Academy of Hospice and Palliative Medicine. Chicago; 2009.
42. Kaszniak AW, Kligman EW. Hospice care for patients with dementia. Elder Care. 2013. Arizona Alzheimer's Consortium Web site. Available at: http://azalz.org/wp-content/uploads/2013/07/Hospice-Care-for-Pts-with-Dementia.pdf. Accessed December 2, 2014.
43. Daviglus ML, Bell CC, Berrettini W, et al. NIH state-of-the-science conference statement: Preventing Alzheimer’s disease and cognitive decline. NIH Consens State Sci Statements. 2010;27:1-30.
44. Patterson C, Feightner JW, Garcia A, et al. Diagnosis and treatment of dementia: 1. Risk assessment and primary prevention of Alzheimer disease. CMAJ. 2008;178:548-556.
45. Carrillo MC, Brashear HR, Logovinsky V, et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimers Dement. 2013;9:123-131.e1.
46. Salloway S, Sperling R, Fox NC, et al; Bapineuzumab 301 and 302 Clinical Trial Investigators. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s Disease. N Engl J Med. 2014;370:322-333.
47. Doody RS, Thomas RG, Farlow M, et al; Alheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s Disease. N Engl J Med. 2014:370:311-321.
1. Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimer’s Association Web site. Available at: http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed December 2, 2014.
2. Román GC, Nash DT, Fillit H. Translating current knowledge into dementia prevention. Alzheimer Dis Assoc Disord. 2012;26:295-299.
3. Jak, AJ. The impact of physical and mental activity on cognitive aging. Curr Top Behav Neurosci. 2012;10:273-291.
4. US Preventive Services Task Force. Cognitive impairment in older adults: Screening. US Preventive Services Task Force Web site. Available at: http://www.uspreventiveservicestaskforce.org/Page/Topic/recommendation-summary/cognitive-impairment-in-older-adults-screening. Accessed November 28, 2014.
5. Centers for Medicare & Medicaid Services. The guide to Medicare preventive services. 4th ed. 2011. Available at: http://www.curemd.com/fqhc/The%20Guide%20to%20Medicare%20Preventative%20Services%20for%20Physicans,%20Providers%20and%20Suppliers.pdf. Accessed December 2, 2014.
6. Boustani, M. Dementia screening in primary care: not too fast! J Amer Geriatr Soc. 2013;61:1205-1207.
7. Alzheimer’s Association. Know the 10 signs: Early detection matters. Alzheimer’s Association Web site. Available at: http://www.alz.org/national/documents/checklist_10signs.pdf. Accessed December 2, 2014.
8. Cordell CB, Borson S, Boustani M, et al; Medicare Detection of Cognitive Impairment Workgroup. Alzheimer’s Association recommendations for operationalizing the detection of cognitive impairment during the Medicare Annual Wellness Visit in a primary care setting. Alzheimers Dement. 2013;9:141-150.
9. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263-269.
10. The American Geriatrics Society. A guide to dementia diagnosis and treatment. The American Geriatrics Society Web site. Available at: http://dementia.americangeriatrics.org/documents/AGS_PC_Dementia_Sheet_2010v2.pdf. Accessed December
2, 2014.
11. Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207-216.
12. Johnson KA, Minoshima S, Bohnen NI, et al; Alzheimer’s Association; Society of Nuclear Medicine and Molecular Imaging; Amyloid Imaging Taskforce. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;9:e1-e16.
13. Centers for Medicare and Medicaid Services. National coverage determination (NCD) for FDG PET for dementia and neurodegenerative diseases (220.6.13). Centers for Medicare and Medicaid Services Web site. Available at: http://www.cms.gov/medicare-coverage-database/details/ncd-details.aspx?NCDId=288&ncdver=3&bc=BAABAAAAAAAA&. Accessed December 2, 2014.
14. Albert MS, DeKosky ST, Ruckson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270-179.
15. Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia—meta-analysis of 41 robust inception studies. Acta Psychiatr Scand. 2009;119:252-265.
16. Farias ST, Mungas D, Reed BR, et al. Progression of mild cognitive impairment to dementia in clinic- vs community-based cohorts. Arch Neurol. 2009;66:1151-1157.
17. Bensadon BA, Odenheimer GL. Current management decisions in mild cognitive impairment. Clin Geriatr Med. 2013;29:847-871.
18. Ngo-Metzger Q, August KJ, Srinivasan M, et al. End-of-life care: guidelines for patient-centered communication. Am Fam Physician. 2008;77:167-174.
19. Sayegh P, Knight BG. Cross-cultural differences in dementia: the Sociocultural Health Belief Model. Int Psychogeriatr. 2013;25:517-530.
20. McDaniel SH, Campbell TL, Hepworth J, et al. Family-Oriented Primary Care. 2nd ed. New York, NY: Springer; 2005.
21. Bensadon BA, Odenheimer GL. Current management decisions in mild cognitive impairment. Clin Geriatr Med. 2013:29;847-871.
22. Russ TC, Morling JR. Cholinesterase inhibitors for mild cognitive impairment. Cochrane Database Syst Rev. 2012;9:CD009132.
23. Sadowsky CH, Galvin JE. Guidelines for the management of cognitive and behavioral problems in dementia. J Am Board Fam Med. 2012;25:350-366.
24. Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006;(1):CD005593.
25. Fillit HM, Doody RS, Binaso K, et al. Recommendations for best practices in the treatment of Alzheimer’s disease in managed care. Am J Geriatr Pharmacother. 2006;4(suppl A):S9-S24;quiz S25-S28.
26. Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med. 2006;355:1525-1538.
27. US Food and Drug Administration. Public health advisory: Deaths with antipsychotics in elderly patients with behavioral disturbances. US Food and Drug Administration Web site. Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm053171.htm. Published April 11, 2005. Updated August 16, 2013. Accessed December 2, 2014.
28. The American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2012;60:616-631.
29. Brummel-Smith K. It’s time to require written informed consent when using antipsychotics in dementia. Br J Med Pract. 2008;1:4-6.
30. Huckans M, Hutson L, Twamley E, et al. Efficacy of cognitive rehabilitation therapies for mild cognitive impairment (MCI) in older adults: working toward a theoretical model and evidence-based interventions. Neuropsychol Rev. 2013;23:63-80.
31. Woods B, Aguirre E, Spector AE, et al. Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst Rev. 2012;2:CD005562.
32. McLaren AN, Lamantia MA, Callahan CM. Systematic review of non-pharmacologic interventions to delay functional decline in community-dwelling patients with dementia. Aging Ment Health. 2013;17:655-666.
33. Bahar-Fuchs A, Clare L, Woods B. Cognitive training and cognitive rehabilitation for mild to moderate Alzheimer’s disease and vascular dementia. Cochrane Database Syst Rev. 2013;6:CD003260.
34. Logsdon RG, McCurry SM, Teri L. Evidence-based psychological treatments for disruptive behaviors in individuals with dementia. Psychol Aging. 2007;22:28-36.
35. Raetz J. A nondrug approach to dementia. J Fam Pract. 2013;62:548-557.
36. Gallagher-Thompson D, Coon DW. Evidence-based psychological treatments for distress in family caregivers of older adults. Psychol Aging. 2007;22:37-51.
37. Mitchell SL, Teno JM, Kiely DK, et al. The clinical course of advanced dementia. N Engl J Med. 2009:361:1529-1538.
38. Parsons C, Hughes CM, Passmore AP, et al. Withholding, discontinuing and withdrawing medications in dementia patients at the end of life: a neglected problem in the disadvantaged dying? Drugs Aging. 2010;27:435-449.
39. The American Geriatrics Society. Feeding tubes in advanced dementia position statement. The American Geriatrics Society Web site. Available at: http://www.americangeriatrics.org/files/documents/feeding.tubes.advanced.dementia.pdf. Accessed November 19, 2013.
40. Goodman C, Evans C, Wilcock J, et al. End of life care for community dwelling older people with dementia: an integrated review. Int J Geriatr Psychiatry. 2010;25:329-337.
41. Storey CP. A quick-reference guide to the hospice and palliative care training for physicians: UNIPAC self-study program. American Academy of Hospice and Palliative Medicine. Chicago; 2009.
42. Kaszniak AW, Kligman EW. Hospice care for patients with dementia. Elder Care. 2013. Arizona Alzheimer's Consortium Web site. Available at: http://azalz.org/wp-content/uploads/2013/07/Hospice-Care-for-Pts-with-Dementia.pdf. Accessed December 2, 2014.
43. Daviglus ML, Bell CC, Berrettini W, et al. NIH state-of-the-science conference statement: Preventing Alzheimer’s disease and cognitive decline. NIH Consens State Sci Statements. 2010;27:1-30.
44. Patterson C, Feightner JW, Garcia A, et al. Diagnosis and treatment of dementia: 1. Risk assessment and primary prevention of Alzheimer disease. CMAJ. 2008;178:548-556.
45. Carrillo MC, Brashear HR, Logovinsky V, et al. Can we prevent Alzheimer’s disease? Secondary “prevention” trials in Alzheimer’s disease. Alzheimers Dement. 2013;9:123-131.e1.
46. Salloway S, Sperling R, Fox NC, et al; Bapineuzumab 301 and 302 Clinical Trial Investigators. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s Disease. N Engl J Med. 2014;370:322-333.
47. Doody RS, Thomas RG, Farlow M, et al; Alheimer’s Disease Cooperative Study Steering Committee; Solanezumab Study Group. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s Disease. N Engl J Med. 2014:370:311-321.

Do annual pelvic exams benefit asymptomatic women who receive regular Pap smears?
No evidence exists to support a clinical benefit from annual pelvic examinations for asymptomatic women who receive Pap smears every 3 to 5 years. However, the American College of Obstetricians and Gynecologists (ACOG) committee on gynecologic practice recommends annual pelvic exams (strength of recommendation [SOR]: C, expert opinion).
Urine testing alone reliably diagnoses gonorrhea and chlamydia (SOR: A, systematic review of cohort studies).
Pelvic examinations unreliably detect adnexal masses (SOR: B, single cohort study); pelvic exams accompanied by ultrasound fail to affect outcomes in ovarian cancer screening (SOR: B, cohort studies).
Pelvic exams aren’t necessary before prescribing oral contraceptive pills (OCPs) (SOR: C, expert opinion).
Vulvar carcinoma has a low prevalence and is usually symptomatic (SOR: B, ecologic study and a case series).
EVIDENCE SUMMARY
A systematic review and meta-analysis included 29 studies that compared the sensitivity and specificity of nucleic acid amplification tests on specimens collected invasively from the cervix or urethra with noninvasively collected urine specimens.1 Studies included both asymptomatic and symptomatic patients. Reference standards varied and included cervical culture, enzyme immunoassay, direct fluorescent antibody, ligase chain reaction, and positive results on 2 of 3 nucleic acid amplification assays.
The sensitivity and specificity of chlamydia and gonorrhea detection didn’t differ between urine and cervical specimens. The pooled sensitivity and specificity for polymerase chain reaction urine samples were 83.3% (95% confidence interval [CI], 77.7%-88.9%) and 99.5% (CI, 99.3%-99.8%), respectively, and for cervical samples 85.5% (CI, 80.3%-90.6%) and 99.6% (CI, 99.4%-99.8%), respectively.1
Pelvic exams detect adnexal masses, but not reliably
A prospective cohort of 127 women undergoing pelvic surgery had preoperative bimanual exams under anesthesia to detect an adnexal mass.2 The gold standard for detection was findings at surgery. The woman had a high prevalence (20%) of ovarian masses. Indications for surgery included diagnosis, sterilization, and suspected malignancy.
When the preoperative bimanual examination detected a left adnexal mass, the odds of finding one at surgery increased 2.8 times, whereas when the exam was normal the odds decreased by 0.8 (positive predictive value [PPV]=0.64; 95% CI, 0.45-0.83). Conversely, the preoperative examination failed to correctly predict a right adnexal mass regardless of the result; the likelihood ratio for both normal and abnormal right adnexal examinations was 1 (PPV=0.26; 95% CI, 0.12-0.47).
What about pelvic exams with ultrasound?
An investigation of transvaginal ultrasonography (TVUS) from November 1987 to January 1991 screened a cohort of 1300 asymptomatic postmenopausal women for an ovarian tumor.3 To be eligible for the study, subjects had to have been without menses for at least 6 months and have no history of a pelvic tumor. Each woman underwent both a pelvic exam and TVUS.
TVUS found that 33 of the women had abnormal ovarian size and morphology when compared with normal standards. Twenty-seven of the 33, who had abnormalities that persisted longer than 1 month, underwent exploratory laparotomy. Ovarian enlargement also was apparent on clinical examination in 10 patients.
Of the 27 patients who underwent surgery, 2 had primary ovarian carcinomas. Significantly, both women had documented normal pelvic examinations on screening.
Another cohort trial conducted between October 1984 and July 1987 studied 801 women ages 40 to 70 years who were at high risk for ovarian cancer.4 Risk factors included nulliparity; symptoms such as abdominal pain, urinary frequency, or irregular bleeding; a personal history of cancer; and a family history of ovarian, breast, or endometrial cancer.
The women underwent both pelvic examination and abdominal ultrasound scanning. Fifty-one patients had abnormal pelvic examinations but normal sonograms. None of the 51 patients, who were followed to the end of the study, developed evidence of ovarian carcinoma. Abnormal abdominal ultrasound scans in 163 patients resulted in 3 diagnoses of malignancy. The 3 patients had normal pelvic examinations.
A pelvic exam isn’t needed before prescribing hormonal contraception
A 2001 JAMA literature review addressed pelvic exams as a prerequisite for administering hormonal contraceptives.5 Investigators identified consensus statements, policy statements, and reviews on the subject and contacted major health associations such as the World Health Organization for their recommendations.
Despite a lack of evidence, these expert sources concluded that a pelvic exam isn’t necessary to identify conditions in which OCPs are contraindicated (pregnancy, breast cancer, hypertension, and thromboembolic disease). Medical history and blood pressure measurement provide adequate screening.
Vulvar cancer is rare and usually symptomatic
Vulvar disease is uncommon and almost always symptomatic. The United Kingdom national cancer registry found an incidence of 3.7 per 100,000.6 A prospective study of 102 women presenting with squamous cell carcinoma of the vulva showed that 94% reported a history of symptomatic vulvar irritation.7 Eighty-eight percent had had symptoms for longer than 6 months.
RECOMMENDATIONS
Regarding screening for gonorrhea and chlamydia, the United States Preventive Services Task Force (USPSTF) states that newer tests, including nucleic acid amplification tests of urine, have improved sensitivity and comparable specificity when compared with cervical culture.8,9
The USPSTF recommends against screening for ovarian cancer in general, (Grade D recommendation: no net benefit or the harms outweigh the benefits). The Task Force states that the sensitivity of pelvic examination in detecting ovarian cancer is unknown based on several ultrasound studies.10
A 2012 ACOG committee opinion recommends that an annual pelvic examination remain a part of the well-woman visit even though the committee found no evidence in support of an annual exam for asymptomatic, low-risk patients.11 The committee notes that patients and providers should discuss the decision to perform a pelvic exam annually.
1. Cook RL, Hutchison SL, østergarrd L, et al. Systemic review: noninvasive testing for Chlamydia trachomatis and Neisseria gonorrheoeae. Ann Intern Med. 2005;142:914-925.
2. Padilla LA, Radosevich DM, Milad MP. Accuracy of the pelvic examination in detecting adnexal masses. Obset Gynecol. 2000;96:593-598.
3. Van Nagell JR Jr, DePriest PD, Puls LE, et al. Ovarian cancer screening in asymptomatic postmenopausal women by transvaginal sonography. Cancer. 1991;68:458-462.
4. Andolf E, Jørgensen C, Astedt B. Ultrasound examination for detection of ovarian carcinoma in risk groups. Obstet Gyenocol. 1990;75:106-109.
5. Stewart FH, Harper CC, Ellerston CE, et al. Clinical breast and pelvic examination requirements for hormonal contraception: current practice vs evidence. JAMA. 2001;285:2232-2239.
6. CancerResearchUK. Vulval cancer incidence statistics. Cancer Research UK Web site. Available at: http://info.cancerresearchuk.org/cancerstats/types/vulva/incidence/. Accessed October 30, 2013.
7. Jones RW, Joura EA. Analyzing prior clinical events at presentation in 102 women with vulvar carcinoma. Evidence of diagnostic delays. J Reprod Med. 1999;44:766-768.
8. US Preventive Services Task Force. Screening for gonorrhea: recommendation statement. Ann Fam Med. 2005;3:263-267.
9. US Preventive Services Task Force. Screening for chlamydial infection: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2007;147:128-134.
10. US Preventive Services Task Force. Screening for ovarian cancer: recommendation statement. Ann Fam Med. 2004;2:260-262.
11. Committee on Gyencologic Practice. Committee opinion no. 534: well-woman visit. Obstet Gynecol. 2012;120:421-424.
No evidence exists to support a clinical benefit from annual pelvic examinations for asymptomatic women who receive Pap smears every 3 to 5 years. However, the American College of Obstetricians and Gynecologists (ACOG) committee on gynecologic practice recommends annual pelvic exams (strength of recommendation [SOR]: C, expert opinion).
Urine testing alone reliably diagnoses gonorrhea and chlamydia (SOR: A, systematic review of cohort studies).
Pelvic examinations unreliably detect adnexal masses (SOR: B, single cohort study); pelvic exams accompanied by ultrasound fail to affect outcomes in ovarian cancer screening (SOR: B, cohort studies).
Pelvic exams aren’t necessary before prescribing oral contraceptive pills (OCPs) (SOR: C, expert opinion).
Vulvar carcinoma has a low prevalence and is usually symptomatic (SOR: B, ecologic study and a case series).
EVIDENCE SUMMARY
A systematic review and meta-analysis included 29 studies that compared the sensitivity and specificity of nucleic acid amplification tests on specimens collected invasively from the cervix or urethra with noninvasively collected urine specimens.1 Studies included both asymptomatic and symptomatic patients. Reference standards varied and included cervical culture, enzyme immunoassay, direct fluorescent antibody, ligase chain reaction, and positive results on 2 of 3 nucleic acid amplification assays.
The sensitivity and specificity of chlamydia and gonorrhea detection didn’t differ between urine and cervical specimens. The pooled sensitivity and specificity for polymerase chain reaction urine samples were 83.3% (95% confidence interval [CI], 77.7%-88.9%) and 99.5% (CI, 99.3%-99.8%), respectively, and for cervical samples 85.5% (CI, 80.3%-90.6%) and 99.6% (CI, 99.4%-99.8%), respectively.1
Pelvic exams detect adnexal masses, but not reliably
A prospective cohort of 127 women undergoing pelvic surgery had preoperative bimanual exams under anesthesia to detect an adnexal mass.2 The gold standard for detection was findings at surgery. The woman had a high prevalence (20%) of ovarian masses. Indications for surgery included diagnosis, sterilization, and suspected malignancy.
When the preoperative bimanual examination detected a left adnexal mass, the odds of finding one at surgery increased 2.8 times, whereas when the exam was normal the odds decreased by 0.8 (positive predictive value [PPV]=0.64; 95% CI, 0.45-0.83). Conversely, the preoperative examination failed to correctly predict a right adnexal mass regardless of the result; the likelihood ratio for both normal and abnormal right adnexal examinations was 1 (PPV=0.26; 95% CI, 0.12-0.47).
What about pelvic exams with ultrasound?
An investigation of transvaginal ultrasonography (TVUS) from November 1987 to January 1991 screened a cohort of 1300 asymptomatic postmenopausal women for an ovarian tumor.3 To be eligible for the study, subjects had to have been without menses for at least 6 months and have no history of a pelvic tumor. Each woman underwent both a pelvic exam and TVUS.
TVUS found that 33 of the women had abnormal ovarian size and morphology when compared with normal standards. Twenty-seven of the 33, who had abnormalities that persisted longer than 1 month, underwent exploratory laparotomy. Ovarian enlargement also was apparent on clinical examination in 10 patients.
Of the 27 patients who underwent surgery, 2 had primary ovarian carcinomas. Significantly, both women had documented normal pelvic examinations on screening.
Another cohort trial conducted between October 1984 and July 1987 studied 801 women ages 40 to 70 years who were at high risk for ovarian cancer.4 Risk factors included nulliparity; symptoms such as abdominal pain, urinary frequency, or irregular bleeding; a personal history of cancer; and a family history of ovarian, breast, or endometrial cancer.
The women underwent both pelvic examination and abdominal ultrasound scanning. Fifty-one patients had abnormal pelvic examinations but normal sonograms. None of the 51 patients, who were followed to the end of the study, developed evidence of ovarian carcinoma. Abnormal abdominal ultrasound scans in 163 patients resulted in 3 diagnoses of malignancy. The 3 patients had normal pelvic examinations.
A pelvic exam isn’t needed before prescribing hormonal contraception
A 2001 JAMA literature review addressed pelvic exams as a prerequisite for administering hormonal contraceptives.5 Investigators identified consensus statements, policy statements, and reviews on the subject and contacted major health associations such as the World Health Organization for their recommendations.
Despite a lack of evidence, these expert sources concluded that a pelvic exam isn’t necessary to identify conditions in which OCPs are contraindicated (pregnancy, breast cancer, hypertension, and thromboembolic disease). Medical history and blood pressure measurement provide adequate screening.
Vulvar cancer is rare and usually symptomatic
Vulvar disease is uncommon and almost always symptomatic. The United Kingdom national cancer registry found an incidence of 3.7 per 100,000.6 A prospective study of 102 women presenting with squamous cell carcinoma of the vulva showed that 94% reported a history of symptomatic vulvar irritation.7 Eighty-eight percent had had symptoms for longer than 6 months.
RECOMMENDATIONS
Regarding screening for gonorrhea and chlamydia, the United States Preventive Services Task Force (USPSTF) states that newer tests, including nucleic acid amplification tests of urine, have improved sensitivity and comparable specificity when compared with cervical culture.8,9
The USPSTF recommends against screening for ovarian cancer in general, (Grade D recommendation: no net benefit or the harms outweigh the benefits). The Task Force states that the sensitivity of pelvic examination in detecting ovarian cancer is unknown based on several ultrasound studies.10
A 2012 ACOG committee opinion recommends that an annual pelvic examination remain a part of the well-woman visit even though the committee found no evidence in support of an annual exam for asymptomatic, low-risk patients.11 The committee notes that patients and providers should discuss the decision to perform a pelvic exam annually.
No evidence exists to support a clinical benefit from annual pelvic examinations for asymptomatic women who receive Pap smears every 3 to 5 years. However, the American College of Obstetricians and Gynecologists (ACOG) committee on gynecologic practice recommends annual pelvic exams (strength of recommendation [SOR]: C, expert opinion).
Urine testing alone reliably diagnoses gonorrhea and chlamydia (SOR: A, systematic review of cohort studies).
Pelvic examinations unreliably detect adnexal masses (SOR: B, single cohort study); pelvic exams accompanied by ultrasound fail to affect outcomes in ovarian cancer screening (SOR: B, cohort studies).
Pelvic exams aren’t necessary before prescribing oral contraceptive pills (OCPs) (SOR: C, expert opinion).
Vulvar carcinoma has a low prevalence and is usually symptomatic (SOR: B, ecologic study and a case series).
EVIDENCE SUMMARY
A systematic review and meta-analysis included 29 studies that compared the sensitivity and specificity of nucleic acid amplification tests on specimens collected invasively from the cervix or urethra with noninvasively collected urine specimens.1 Studies included both asymptomatic and symptomatic patients. Reference standards varied and included cervical culture, enzyme immunoassay, direct fluorescent antibody, ligase chain reaction, and positive results on 2 of 3 nucleic acid amplification assays.
The sensitivity and specificity of chlamydia and gonorrhea detection didn’t differ between urine and cervical specimens. The pooled sensitivity and specificity for polymerase chain reaction urine samples were 83.3% (95% confidence interval [CI], 77.7%-88.9%) and 99.5% (CI, 99.3%-99.8%), respectively, and for cervical samples 85.5% (CI, 80.3%-90.6%) and 99.6% (CI, 99.4%-99.8%), respectively.1
Pelvic exams detect adnexal masses, but not reliably
A prospective cohort of 127 women undergoing pelvic surgery had preoperative bimanual exams under anesthesia to detect an adnexal mass.2 The gold standard for detection was findings at surgery. The woman had a high prevalence (20%) of ovarian masses. Indications for surgery included diagnosis, sterilization, and suspected malignancy.
When the preoperative bimanual examination detected a left adnexal mass, the odds of finding one at surgery increased 2.8 times, whereas when the exam was normal the odds decreased by 0.8 (positive predictive value [PPV]=0.64; 95% CI, 0.45-0.83). Conversely, the preoperative examination failed to correctly predict a right adnexal mass regardless of the result; the likelihood ratio for both normal and abnormal right adnexal examinations was 1 (PPV=0.26; 95% CI, 0.12-0.47).
What about pelvic exams with ultrasound?
An investigation of transvaginal ultrasonography (TVUS) from November 1987 to January 1991 screened a cohort of 1300 asymptomatic postmenopausal women for an ovarian tumor.3 To be eligible for the study, subjects had to have been without menses for at least 6 months and have no history of a pelvic tumor. Each woman underwent both a pelvic exam and TVUS.
TVUS found that 33 of the women had abnormal ovarian size and morphology when compared with normal standards. Twenty-seven of the 33, who had abnormalities that persisted longer than 1 month, underwent exploratory laparotomy. Ovarian enlargement also was apparent on clinical examination in 10 patients.
Of the 27 patients who underwent surgery, 2 had primary ovarian carcinomas. Significantly, both women had documented normal pelvic examinations on screening.
Another cohort trial conducted between October 1984 and July 1987 studied 801 women ages 40 to 70 years who were at high risk for ovarian cancer.4 Risk factors included nulliparity; symptoms such as abdominal pain, urinary frequency, or irregular bleeding; a personal history of cancer; and a family history of ovarian, breast, or endometrial cancer.
The women underwent both pelvic examination and abdominal ultrasound scanning. Fifty-one patients had abnormal pelvic examinations but normal sonograms. None of the 51 patients, who were followed to the end of the study, developed evidence of ovarian carcinoma. Abnormal abdominal ultrasound scans in 163 patients resulted in 3 diagnoses of malignancy. The 3 patients had normal pelvic examinations.
A pelvic exam isn’t needed before prescribing hormonal contraception
A 2001 JAMA literature review addressed pelvic exams as a prerequisite for administering hormonal contraceptives.5 Investigators identified consensus statements, policy statements, and reviews on the subject and contacted major health associations such as the World Health Organization for their recommendations.
Despite a lack of evidence, these expert sources concluded that a pelvic exam isn’t necessary to identify conditions in which OCPs are contraindicated (pregnancy, breast cancer, hypertension, and thromboembolic disease). Medical history and blood pressure measurement provide adequate screening.
Vulvar cancer is rare and usually symptomatic
Vulvar disease is uncommon and almost always symptomatic. The United Kingdom national cancer registry found an incidence of 3.7 per 100,000.6 A prospective study of 102 women presenting with squamous cell carcinoma of the vulva showed that 94% reported a history of symptomatic vulvar irritation.7 Eighty-eight percent had had symptoms for longer than 6 months.
RECOMMENDATIONS
Regarding screening for gonorrhea and chlamydia, the United States Preventive Services Task Force (USPSTF) states that newer tests, including nucleic acid amplification tests of urine, have improved sensitivity and comparable specificity when compared with cervical culture.8,9
The USPSTF recommends against screening for ovarian cancer in general, (Grade D recommendation: no net benefit or the harms outweigh the benefits). The Task Force states that the sensitivity of pelvic examination in detecting ovarian cancer is unknown based on several ultrasound studies.10
A 2012 ACOG committee opinion recommends that an annual pelvic examination remain a part of the well-woman visit even though the committee found no evidence in support of an annual exam for asymptomatic, low-risk patients.11 The committee notes that patients and providers should discuss the decision to perform a pelvic exam annually.
1. Cook RL, Hutchison SL, østergarrd L, et al. Systemic review: noninvasive testing for Chlamydia trachomatis and Neisseria gonorrheoeae. Ann Intern Med. 2005;142:914-925.
2. Padilla LA, Radosevich DM, Milad MP. Accuracy of the pelvic examination in detecting adnexal masses. Obset Gynecol. 2000;96:593-598.
3. Van Nagell JR Jr, DePriest PD, Puls LE, et al. Ovarian cancer screening in asymptomatic postmenopausal women by transvaginal sonography. Cancer. 1991;68:458-462.
4. Andolf E, Jørgensen C, Astedt B. Ultrasound examination for detection of ovarian carcinoma in risk groups. Obstet Gyenocol. 1990;75:106-109.
5. Stewart FH, Harper CC, Ellerston CE, et al. Clinical breast and pelvic examination requirements for hormonal contraception: current practice vs evidence. JAMA. 2001;285:2232-2239.
6. CancerResearchUK. Vulval cancer incidence statistics. Cancer Research UK Web site. Available at: http://info.cancerresearchuk.org/cancerstats/types/vulva/incidence/. Accessed October 30, 2013.
7. Jones RW, Joura EA. Analyzing prior clinical events at presentation in 102 women with vulvar carcinoma. Evidence of diagnostic delays. J Reprod Med. 1999;44:766-768.
8. US Preventive Services Task Force. Screening for gonorrhea: recommendation statement. Ann Fam Med. 2005;3:263-267.
9. US Preventive Services Task Force. Screening for chlamydial infection: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2007;147:128-134.
10. US Preventive Services Task Force. Screening for ovarian cancer: recommendation statement. Ann Fam Med. 2004;2:260-262.
11. Committee on Gyencologic Practice. Committee opinion no. 534: well-woman visit. Obstet Gynecol. 2012;120:421-424.
1. Cook RL, Hutchison SL, østergarrd L, et al. Systemic review: noninvasive testing for Chlamydia trachomatis and Neisseria gonorrheoeae. Ann Intern Med. 2005;142:914-925.
2. Padilla LA, Radosevich DM, Milad MP. Accuracy of the pelvic examination in detecting adnexal masses. Obset Gynecol. 2000;96:593-598.
3. Van Nagell JR Jr, DePriest PD, Puls LE, et al. Ovarian cancer screening in asymptomatic postmenopausal women by transvaginal sonography. Cancer. 1991;68:458-462.
4. Andolf E, Jørgensen C, Astedt B. Ultrasound examination for detection of ovarian carcinoma in risk groups. Obstet Gyenocol. 1990;75:106-109.
5. Stewart FH, Harper CC, Ellerston CE, et al. Clinical breast and pelvic examination requirements for hormonal contraception: current practice vs evidence. JAMA. 2001;285:2232-2239.
6. CancerResearchUK. Vulval cancer incidence statistics. Cancer Research UK Web site. Available at: http://info.cancerresearchuk.org/cancerstats/types/vulva/incidence/. Accessed October 30, 2013.
7. Jones RW, Joura EA. Analyzing prior clinical events at presentation in 102 women with vulvar carcinoma. Evidence of diagnostic delays. J Reprod Med. 1999;44:766-768.
8. US Preventive Services Task Force. Screening for gonorrhea: recommendation statement. Ann Fam Med. 2005;3:263-267.
9. US Preventive Services Task Force. Screening for chlamydial infection: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2007;147:128-134.
10. US Preventive Services Task Force. Screening for ovarian cancer: recommendation statement. Ann Fam Med. 2004;2:260-262.
11. Committee on Gyencologic Practice. Committee opinion no. 534: well-woman visit. Obstet Gynecol. 2012;120:421-424.
Evidence-based answers from the Family Physicians Inquiries Network
Tuberculosis: Which drug regimen and when
› Obtain a problem-focused history and physical, as well as chest radiography, to rule out active pulmonary tuberculosis (TB) before initiating treatment for latent tuberculosis infection (LTBI). B
› Prescribe isoniazid 5 mg/kg/d (10 mg/kg/d in children) up to a maximum dose of 300 mg/d for 9 months for most patients with LTBI. B
› Ensure that directly observed therapy is used for all patients with active TB, as well as for select high-risk cases of LTBI. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mitchell J, age 62, comes to see you because he’s had a cough with increasing dyspnea for a month. Mr. J has never smoked but has type 2 diabetes mellitus. He also tells you that over the past month, he’s had occasional night sweats and has lost 8 pounds, although he’s not changed his diet. During the past week, he’s noticed blood-tinged sputum. Physical examination reveals a thin, chronically ill appearing man with an oral temperature of 100.6°F and mild tachypnea. You order a complete blood count, chest x-ray, and metabolic profile, administer a tuberculin skin test (TST), and initiate levofloxacin 500 mg/d for a presumed bacterial pneumonia. His lab work reveals mild leukocytosis and hyperglycemia, and the chest x-ray shows a left upper lobe infiltrate. The TST reaction—4 mm 50 hours after placement—was negative.
Mr. J returns a week later and says he feels worse. Your examination reveals worsened tachypnea, with tachycardia and crackles over the left upper lung fields.
How would you proceed with his care?
More people die of tuberculosis (TB) each year than any other infectious disease except human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. In 2013, an estimated 9 million people worldwide developed active TB and 1.5 million died of the disease.1 Many of these deaths could have been prevented if patients had received a diagnosis and treatment during the latent phase (when the patient was infected, but had no active disease), or as soon as the patient developed active disease. In this article we describe treatment for both latent and active TB.
Before treating latent TB infection, first rule out active TB
Patients with latent tuberculosis infection (LTBI) have a 5% to 10% lifetime risk of developing active TB disease.2 Treatment of LTBI can reduce this risk to 1% to 2%.3
Although not the focus of this article, diagnosis of LTBI is made by using either a TST, in which the patient receives an intradermal injection of purified protein derivative and the size of the skin induration is measured 48 to 72 hours after administration, or an interferon-gamma release assay (IGRA), which requires a blood draw. After receiving a positive test result for LTBI, the next step is to rule out active TB.4 This is necessary because the primary treatment regimen for LTBI involves only one drug, whereas treating active TB with one drug is strongly associated with treatment failure and future resistance to that drug.5
To rule out active TB, perform a brief, problem-focused history and physical, and obtain a chest x-ray.4 Pertinent findings that suggest active disease include:
- any history of recent weight loss, unexplained fever, night sweats, cough or hemoptysis
- fever or any unexpected lung findings on physical exam
- any parenchymal infiltrates on chest x-ray. (Granulomas and scarring may be signs of previously healed TB infection, but do not indicate active TB.)
Any of these findings should prompt a further investigation to either confirm or definitively rule out active TB disease. In the absence of these findings, the physician may proceed with treatment for LTBI.
Latent TB infection treatment: Isoniazid alone, or another regimen?
The current preferred regimen for most patients with LTBI is 9 months of isoniazid (INH) 5 mg/kg/d (10 mg/kg/d in children) up to a maximum of 300 mg/d. This regimen has been recommended by the Centers for Disease Control and Prevention (CDC), the American Thoracic Society, and the Infectious Diseases Society of America.3 However, there are 3 other CDC-recommended LTBI treatment regimens that include INH, INH plus rifapentine (RPT), or rifampin (RIF) for 6, 3, or 4 months, respectively (TABLE 1).6 These other regimens may be considered under certain circumstances. For example, INH and rifapentine might be used to treat an otherwise healthy patient who has had recent exposure to an individual with active, contagious TB.
If the patient is pregnant. INH is a pregnancy category C drug. Treatment for LTBI during pregnancy is generally regarded as safe and should be strongly considered if the patient has risk factors for progression to active TB, such as a recent exposure to someone with active TB.7 In otherwise healthy patients, treatment for LTBI may be deferred until after delivery.
Take steps to avoid complications of drug therapy
Drug-induced hepatitis is the primary adverse effect of INH treatment. Risk increases with age, previous hepatic injury, or concomitant use of other hepatotoxic medications. The risk is very small (<0.1%) for healthy children but may be over 10% for adults with multiple risk factors.8 Hepatitis is generally preceded by asymptomatic elevation of liver function tests (LFTs), which is much more common than clinical hepatitis.
Baseline LFTs should be obtained in patients who:
- have underlying liver disease, such as hepatitis B or C9
- consume ≥2 alcoholic drinks daily or >5 drinks at a time on any occasion
- take other medications with potential hepatotoxicity, such as statins
- have HIV infection10
- are pregnant or postpartum.
If a patient being considered for INH treatment has not had serologic testing for HIV, hepatitis B, or hepatitis C, these tests should be done prior to initiating INH. LFTs should be monitored every 1 to 2 months during INH therapy for patients who have ≥1 of these conditions and normal baseline LFTs. If baseline transaminases are >3 times the upper limit of normal, treatment for LTBI should probably be withheld, though might be considered in those whose LFTs return to normal after withdrawal of a modifiable risk factor, such as alcohol or a statin medication.
After beginning LTBI treatment, patients should be monitored regularly for signs and symptoms of hepatitis, including anorexia, nausea, abdominal pain, icterus, and dark urine, and LFTs performed if these develop. If during treatment transaminases increase to >3 times normal in a symptomatic patient (or >5 times normal in an asymptomatic patient), INH should be stopped and generally not resumed, even after LFTs return to normal. (Such patients would be considered to have partially treated LTBI, and their physicians should be alert to signs and symptoms of active TB, such as unexplained fever, weight loss, or blood-tinged sputum, during subsequent patient encounters.)
Peripheral neuropathy is a less common adverse effect of INH. It occurs in up to 2% of patients and is caused by interference with vitamin B6 (pyridoxine) metabolism. It can be prevented by supplementation with pyridoxine 25 to 50 mg/d. Vitamin B6, however, does not prevent INH-induced hepatotoxicity.
Noncompliance is a concern with INH therapy because treatment typically requires a 9-month course of daily medication.11 Patients for whom compliance is likely to be an issue might be considered for a 3-month, 12-dose course of once-weekly, directly-observed therapy (DOT) with INH and RPT administered by a public health agency. (See “Which patients with TB should receive directly observed therapy?” on page 32.12-14) A randomized, open-label trial involving nearly 8000 patients in 4 low-risk countries found this regimen was as effective as 9 months of self-administered INH.15 The CDC has published recommendations for using this regimen.16
Suspect active TB? Don’t wait for cultures to begin Tx
Unlike LTBI, for which the results of diagnostic testing are available within a few days, active TB is diagnosed by culture, which may take as long as 6 to 8 weeks. However, if you suspect your patient has active TB, do not delay treatment while waiting for culture results, or defer treatment for a patient who has a negative acid-fast bacilli (AFB) smear or rapid nucleic acid amplification test.17 These 2 tests, which are routinely performed during TB cultures, look for other evidence of the presence of TB bacilli; they are not as accurate as cultures, but results are available within days. Likewise, a negative TST or IGRA should not prevent empiric treatment for active TB. Treatment for active TB should be begun empirically based on risk factors and clinical presentation, and can be modified or stopped if cultures are negative, the patient fails to improve, or an alternative diagnosis is found to explain the patient’s symptoms.
Rapid testing for evidence of active TB disease—as well as resistance to medications commonly used to treat TB—can be performed using newer modalities such as MODS (Microscopic-Observation Drug-Susceptibility)18,19 or Xpert MTB/RIF20 testing. However, these tests are not available in many hospitals, and culture and drug sensitivity testing remain the gold standard.21
CASE › Mr. J’s clinical history and chest x-ray findings are highly suggestive of active TB. It was not unreasonable to initially treat him for a bacterial pneumonia, although fluoroquinolones should be used cautiously in this setting, because they are one of the most effective second-line drugs for TB, and using them as a single agent will often invoke drug resistance. Because he failed to respond to treatment for bacterial pneumonia and his presentation suggests TB or another serious cause of nonresponsiveness to standard treatment for community-acquired pneumonia (CAP), you admit him to the hospital.

Treatment for active TB requires multiple drugs in 2 phases
While all family physicians should suspect active TB in appropriate clinical situations and be comfortable with obtaining cultures and initiating empiric treatment, most will want to seek consultation with an infectious disease (ID) specialist especially in the scenarios listed in TABLE 2.5,22 Delayed or inappropriate treatment of active TB remains a major public health problem and cause of multidrug-resistant TB. Inappropriate treatment has been shown to be associated with a 27-fold increase in treatment failure.23 TB treatment guidelines are available from the CDC,24 World Health Organization,25 and International Union Against Tuberculosis and Lung Disease.26
Appropriate treatment requires the use of multiple medications administered in 2 phases. In the initial phase, a patient with suspected TB should begin 4 drugs—usually INH, RIF, ethambutol (EMB), and pyrazinamide (PZA)—for 2 months.1,2,27 The daily pediatric and adult doses and common adverse effects of these medications are summarized in TABLE 3.28 Although most cases of TB can be adequately treated with 2 drugs to which the organism is susceptible, 4 drugs are used initially while awaiting drug sensitivity test results because of the risk of inadequately treating a strain of drug-resistant TB. Before beginning these medications, a chest x-ray, LFTs, HIV antibody test, hepatitis B and C serologies, a serum creatinine, and complete blood count should be obtained in all patients.5 If EMB is prescribed, the patient should also undergo testing for red-green color discrimination, because red-green color vision disturbance is a potential adverse effect of this medication.
All 4 drugs may be administered as a single daily dose, and may be taken together.29 They are ordinarily given either daily for 8 weeks, or daily for 2 weeks followed by a twice-weekly schedule for the remaining 6 weeks in higher doses, although the twice-weekly dose of RIF is the same as the daily dose. All are pregnancy category C, although for active TB, the benefit of treatment is almost always greater than the potential harm.
The continuation phase of treatment starts at 8 weeks, when the results of initial cultures and drug sensitivity tests should be available to guide therapy. A second set of cultures and AFB smears is obtained at 8 weeks to document clearing of the initial infection and guide duration of the continuation phase. If the initial culture was positive for Mycobacterium tuberculosis and the organism was sensitive to both INH and RIF, these 2 drugs should be continued for another 4 months (for a total of 6 months of treatment). PZA and EMB may be stopped at 2 months if the organism is sensitive to both INH and RIF. Thus, for most patients with active TB, the standard regimen will be 4 drugs for 2 months, then 2 drugs for 4 months.2
When should the standard treatment regimen be modified?
If the second set of cultures obtained 2 months after beginning drug treatment is positive and there was cavitary disease on the initial chest x-ray, the continuation phase should be extended by 7 months (for a total of 9 months of treatment).30 If a patient has either cavitary disease or persistently positive cultures (but not both), then the length of therapy is determined on an individual basis in consultation with an ID specialist.
Should a patient’s cultures show resistance to any of the first-line drugs, obtain consultation with an ID specialist. Treatment of multidrug-resistant TB (resistant to INH and RIF) and its subset, extensively drug-resistant TB (resistant to INH and RIF, plus any fluoroquinolone, plus either an aminoglycoside or capreomycin) requires prolonged courses of therapy with multiple drugs administered by DOT.31,32
If at any point during treatment a patient shows clinical deterioration that’s believed to be due to a resurgence of his or her TB disease, obtain a new set of cultures and, in consultation with an ID specialist, add at least 2 drugs to which the patient has not been exposed. Never add only one drug to a failing regimen; active TB always requires 2 drugs to cure, and the patient may have developed resistance to all of the drugs he or she is currently receiving.
If initial cultures are negative for Mycobacterium tuberculosis but the patient responds to treatment, he or she is considered to have “culture-negative TB,” and should generally be continued on INH and RIF for 2 more months after completion of the initial treatment phase (for a total of 4 months of INH and RIF).33
Remember to report. In the United States, active TB must be reported to your local health department, which can be invaluable in coordinating care and administering DOT.
Directly observed therapy (DOT) is preferred for certain high-risk patients with latent tuberculosis infection (LTBI), including those who are younger than 5 years of age, test positive for human immunodeficiency virus, are receiving immunosuppressive therapy, have chest radiography evidence of healed TB, have recently converted to active TB status while receiving serial TB testing, or have recently been exposed to active TB.12
Treatment for active TB should always be given by DOT.13 Because DOT is labor-intensive, twice-weekly dosing is usually preferred.14
CASE › In the hospital, Mr. J was placed in respiratory isolation, had prompt sputum cultures for TB, and was started on empiric treatment for active TB with INH, RIF, PZA, and EMB in standard doses. A search for other causes of nonresponsiveness to CAP showed no evidence of malignancy or HIV infection. He improved steadily and was discharged from the hospital after 2 weeks to complete 2 months of 4-drug therapy, with follow-up care coordinated by the local health department, including a home health nurse experienced in administering DOT. Cultures were positive for Mycobacterium tuberculosis sensitive to all drugs tested. After his initial 2 months of 4-drug therapy, he completed 4 months of additional treatment with INH and RIF, given by DOT, and recovered completely.
CORRESPONDENCE
Jeff Hall, MD, University of South Carolina Department of Family and Preventive Medicine, 3209 Colonial Drive, Columbia, SC 29203; [email protected]
1. World Health Organization. Global tuberculosis report 2014. World Health Organization Web site. Available at: http://www.who.int/tb/publications/global_report/en/. Accessed December 15, 2014.
2. Zumla AI, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745-755.
3. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr4906a1.htm. Accessed December 15, 2014.
4. Hauck FR, Neese BH, Panchal AS, et al. Identification and management of latent tuberculosis infection. Am Fam Physician. 2009;79:879-886.
5. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77.
6. Centers for Disease Control and Prevention. Latent tuberculosis infection: A guide for primary health care providers. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/tb/publications/LTBI/default.htm. Accessed December 11, 2014.
7. Centers for Disease Control and Prevention. Fact sheet: tuberculosis and pregnancy. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/factsheets/specpop/pregnancy.htm. Accessed September 6, 2014.
8. Kunst H, Khan KS. Age-related risk of hepatotoxicity in the treatment of latent tuberculosis infection: a systematic review. Int J Tuberc Lung Dis. 2010;14:1374-1381.
9. Bliven EE, Podewils LJ. The role of chronic hepatitis in isoniazid hepatotoxicity during treatment for latent tuberculosis infection. Int J Tuberc Lung Dis. 2009;13:1054-1060.
10. Akolo C, Adetifa I, Shepperd S, et al. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;1:CD000171.
11. Horsburgh CR Jr, Goldberg S, Bethel J, et al; Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401-409.
12. Horsburgh CR Jr. Priorities for the treatment of latent tuberculosis infection in the United States. N Engl J Med. 2004;350:2060-2070.
13. Potter B, Rindfleisch K, Kraus CK. Management of active tuberculosis. Am Fam Physician. 2005;72:2225-2232.
14. Volmink J, Garner P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. 2007;4:CD003343.
15. Sterling TR, Villarina ME, Borisov AS, et al; TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155-2166.
16. Centers for Disease Control and Prevention (CDC). Recommendations for use of an isoniazid-rifapentine regimen with direct observation to treat latent Mycobacterium tuberculosis infection. MMWR Morb Mortal Wkly Rep. 2011;60:1650-1653.
17. Inge LD, Wilson JW. Update on the treatment of tuberculosis. Am Fam Physician. 2008;78:457-465.
18. Moore DA, Evans CA, Gilman RH, et al. Microscopic-observation drug-susceptibility assay for the diagnosis of TB. N Engl J Med. 2006;355:1539-1550.
19. Minion J, Leung E, Menzies D, et al. Microscopic-observation drug susceptibility and thin layer agar assays for the detection of drug resistant tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:688-698.
20. Boehme CC, Nabeta P, Hilleman D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363:1005-1015.
21. Arentz M, Sorensen B, Horne DJ, et al. Systematic review of the performance of rapid rifampicin resistance testing for drug-resistant tuberculosis. PLoS One. 2013;8:e76533.
22. Sia IG, Wieland ML. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348-361.
23. van der Werf MJ, Langendam MW, Huitric E, et al. Multidrug resistance after inappropriate tuberculosis treatment: a meta-analysis. Eur Respir J. 2012;39:1511-1519.
24. Centers for Disease Control and Prevention. Tuberculosis (TB). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/guidelines/default.htm. Accessed September 6, 2014.
25. World Health Organization. Treatment of tuberculosis guidelines. 4th ed. World Health Organization Web site. Available at: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf?ua=1. Accessed September 6, 2014.
26. International Union Against Tuberculosis and Lung Disease. Management of tuberculosis: A guide to the essentials of good clinical practice. 6th ed. 2010. International Union Against Tuberculosis and Lung Disease Web site. Available at: http://www.theunion.org/what-we-do/publications/technical/management-of-tuberculosis-a-guide-to-the-essentials-of-good-clinical-practice. Accessed September 6, 2014.
27. Combs DL, O’Brien RJ, Geiter LJ. USPHS Tuberculosis Short-Course Chemotherapy Trial 21: effectiveness, toxicity and acceptability. The report of the final results. Ann Intern Med. 1990;112:397-406.
28. Drugs for tuberculosis. Treat Guidel Med Lett. 2012;10:29-36.
29. Chang KC, Leung CC, Grosset J, et al. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66:997-1007.
30. Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention and the Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
31. Lynch JB. Multidrug-resistant tuberculosis. Med Clin North Am. 2013;97:553-579,ix-x.
32. Keshavjee S, Farmer PE. Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med. 2012;367:931-936.
33. Dutt AK, Moers D, Stead WW. Smear- and culture-negative pulmonary tuberculosis: four-month short-course chemotherapy. Am Rev Respir Dis. 1989;139:867-870.
› Obtain a problem-focused history and physical, as well as chest radiography, to rule out active pulmonary tuberculosis (TB) before initiating treatment for latent tuberculosis infection (LTBI). B
› Prescribe isoniazid 5 mg/kg/d (10 mg/kg/d in children) up to a maximum dose of 300 mg/d for 9 months for most patients with LTBI. B
› Ensure that directly observed therapy is used for all patients with active TB, as well as for select high-risk cases of LTBI. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mitchell J, age 62, comes to see you because he’s had a cough with increasing dyspnea for a month. Mr. J has never smoked but has type 2 diabetes mellitus. He also tells you that over the past month, he’s had occasional night sweats and has lost 8 pounds, although he’s not changed his diet. During the past week, he’s noticed blood-tinged sputum. Physical examination reveals a thin, chronically ill appearing man with an oral temperature of 100.6°F and mild tachypnea. You order a complete blood count, chest x-ray, and metabolic profile, administer a tuberculin skin test (TST), and initiate levofloxacin 500 mg/d for a presumed bacterial pneumonia. His lab work reveals mild leukocytosis and hyperglycemia, and the chest x-ray shows a left upper lobe infiltrate. The TST reaction—4 mm 50 hours after placement—was negative.
Mr. J returns a week later and says he feels worse. Your examination reveals worsened tachypnea, with tachycardia and crackles over the left upper lung fields.
How would you proceed with his care?
More people die of tuberculosis (TB) each year than any other infectious disease except human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. In 2013, an estimated 9 million people worldwide developed active TB and 1.5 million died of the disease.1 Many of these deaths could have been prevented if patients had received a diagnosis and treatment during the latent phase (when the patient was infected, but had no active disease), or as soon as the patient developed active disease. In this article we describe treatment for both latent and active TB.
Before treating latent TB infection, first rule out active TB
Patients with latent tuberculosis infection (LTBI) have a 5% to 10% lifetime risk of developing active TB disease.2 Treatment of LTBI can reduce this risk to 1% to 2%.3
Although not the focus of this article, diagnosis of LTBI is made by using either a TST, in which the patient receives an intradermal injection of purified protein derivative and the size of the skin induration is measured 48 to 72 hours after administration, or an interferon-gamma release assay (IGRA), which requires a blood draw. After receiving a positive test result for LTBI, the next step is to rule out active TB.4 This is necessary because the primary treatment regimen for LTBI involves only one drug, whereas treating active TB with one drug is strongly associated with treatment failure and future resistance to that drug.5
To rule out active TB, perform a brief, problem-focused history and physical, and obtain a chest x-ray.4 Pertinent findings that suggest active disease include:
- any history of recent weight loss, unexplained fever, night sweats, cough or hemoptysis
- fever or any unexpected lung findings on physical exam
- any parenchymal infiltrates on chest x-ray. (Granulomas and scarring may be signs of previously healed TB infection, but do not indicate active TB.)
Any of these findings should prompt a further investigation to either confirm or definitively rule out active TB disease. In the absence of these findings, the physician may proceed with treatment for LTBI.
Latent TB infection treatment: Isoniazid alone, or another regimen?
The current preferred regimen for most patients with LTBI is 9 months of isoniazid (INH) 5 mg/kg/d (10 mg/kg/d in children) up to a maximum of 300 mg/d. This regimen has been recommended by the Centers for Disease Control and Prevention (CDC), the American Thoracic Society, and the Infectious Diseases Society of America.3 However, there are 3 other CDC-recommended LTBI treatment regimens that include INH, INH plus rifapentine (RPT), or rifampin (RIF) for 6, 3, or 4 months, respectively (TABLE 1).6 These other regimens may be considered under certain circumstances. For example, INH and rifapentine might be used to treat an otherwise healthy patient who has had recent exposure to an individual with active, contagious TB.
If the patient is pregnant. INH is a pregnancy category C drug. Treatment for LTBI during pregnancy is generally regarded as safe and should be strongly considered if the patient has risk factors for progression to active TB, such as a recent exposure to someone with active TB.7 In otherwise healthy patients, treatment for LTBI may be deferred until after delivery.
Take steps to avoid complications of drug therapy
Drug-induced hepatitis is the primary adverse effect of INH treatment. Risk increases with age, previous hepatic injury, or concomitant use of other hepatotoxic medications. The risk is very small (<0.1%) for healthy children but may be over 10% for adults with multiple risk factors.8 Hepatitis is generally preceded by asymptomatic elevation of liver function tests (LFTs), which is much more common than clinical hepatitis.
Baseline LFTs should be obtained in patients who:
- have underlying liver disease, such as hepatitis B or C9
- consume ≥2 alcoholic drinks daily or >5 drinks at a time on any occasion
- take other medications with potential hepatotoxicity, such as statins
- have HIV infection10
- are pregnant or postpartum.
If a patient being considered for INH treatment has not had serologic testing for HIV, hepatitis B, or hepatitis C, these tests should be done prior to initiating INH. LFTs should be monitored every 1 to 2 months during INH therapy for patients who have ≥1 of these conditions and normal baseline LFTs. If baseline transaminases are >3 times the upper limit of normal, treatment for LTBI should probably be withheld, though might be considered in those whose LFTs return to normal after withdrawal of a modifiable risk factor, such as alcohol or a statin medication.
After beginning LTBI treatment, patients should be monitored regularly for signs and symptoms of hepatitis, including anorexia, nausea, abdominal pain, icterus, and dark urine, and LFTs performed if these develop. If during treatment transaminases increase to >3 times normal in a symptomatic patient (or >5 times normal in an asymptomatic patient), INH should be stopped and generally not resumed, even after LFTs return to normal. (Such patients would be considered to have partially treated LTBI, and their physicians should be alert to signs and symptoms of active TB, such as unexplained fever, weight loss, or blood-tinged sputum, during subsequent patient encounters.)
Peripheral neuropathy is a less common adverse effect of INH. It occurs in up to 2% of patients and is caused by interference with vitamin B6 (pyridoxine) metabolism. It can be prevented by supplementation with pyridoxine 25 to 50 mg/d. Vitamin B6, however, does not prevent INH-induced hepatotoxicity.
Noncompliance is a concern with INH therapy because treatment typically requires a 9-month course of daily medication.11 Patients for whom compliance is likely to be an issue might be considered for a 3-month, 12-dose course of once-weekly, directly-observed therapy (DOT) with INH and RPT administered by a public health agency. (See “Which patients with TB should receive directly observed therapy?” on page 32.12-14) A randomized, open-label trial involving nearly 8000 patients in 4 low-risk countries found this regimen was as effective as 9 months of self-administered INH.15 The CDC has published recommendations for using this regimen.16
Suspect active TB? Don’t wait for cultures to begin Tx
Unlike LTBI, for which the results of diagnostic testing are available within a few days, active TB is diagnosed by culture, which may take as long as 6 to 8 weeks. However, if you suspect your patient has active TB, do not delay treatment while waiting for culture results, or defer treatment for a patient who has a negative acid-fast bacilli (AFB) smear or rapid nucleic acid amplification test.17 These 2 tests, which are routinely performed during TB cultures, look for other evidence of the presence of TB bacilli; they are not as accurate as cultures, but results are available within days. Likewise, a negative TST or IGRA should not prevent empiric treatment for active TB. Treatment for active TB should be begun empirically based on risk factors and clinical presentation, and can be modified or stopped if cultures are negative, the patient fails to improve, or an alternative diagnosis is found to explain the patient’s symptoms.
Rapid testing for evidence of active TB disease—as well as resistance to medications commonly used to treat TB—can be performed using newer modalities such as MODS (Microscopic-Observation Drug-Susceptibility)18,19 or Xpert MTB/RIF20 testing. However, these tests are not available in many hospitals, and culture and drug sensitivity testing remain the gold standard.21
CASE › Mr. J’s clinical history and chest x-ray findings are highly suggestive of active TB. It was not unreasonable to initially treat him for a bacterial pneumonia, although fluoroquinolones should be used cautiously in this setting, because they are one of the most effective second-line drugs for TB, and using them as a single agent will often invoke drug resistance. Because he failed to respond to treatment for bacterial pneumonia and his presentation suggests TB or another serious cause of nonresponsiveness to standard treatment for community-acquired pneumonia (CAP), you admit him to the hospital.
Treatment for active TB requires multiple drugs in 2 phases
While all family physicians should suspect active TB in appropriate clinical situations and be comfortable with obtaining cultures and initiating empiric treatment, most will want to seek consultation with an infectious disease (ID) specialist especially in the scenarios listed in TABLE 2.5,22 Delayed or inappropriate treatment of active TB remains a major public health problem and cause of multidrug-resistant TB. Inappropriate treatment has been shown to be associated with a 27-fold increase in treatment failure.23 TB treatment guidelines are available from the CDC,24 World Health Organization,25 and International Union Against Tuberculosis and Lung Disease.26
Appropriate treatment requires the use of multiple medications administered in 2 phases. In the initial phase, a patient with suspected TB should begin 4 drugs—usually INH, RIF, ethambutol (EMB), and pyrazinamide (PZA)—for 2 months.1,2,27 The daily pediatric and adult doses and common adverse effects of these medications are summarized in TABLE 3.28 Although most cases of TB can be adequately treated with 2 drugs to which the organism is susceptible, 4 drugs are used initially while awaiting drug sensitivity test results because of the risk of inadequately treating a strain of drug-resistant TB. Before beginning these medications, a chest x-ray, LFTs, HIV antibody test, hepatitis B and C serologies, a serum creatinine, and complete blood count should be obtained in all patients.5 If EMB is prescribed, the patient should also undergo testing for red-green color discrimination, because red-green color vision disturbance is a potential adverse effect of this medication.
All 4 drugs may be administered as a single daily dose, and may be taken together.29 They are ordinarily given either daily for 8 weeks, or daily for 2 weeks followed by a twice-weekly schedule for the remaining 6 weeks in higher doses, although the twice-weekly dose of RIF is the same as the daily dose. All are pregnancy category C, although for active TB, the benefit of treatment is almost always greater than the potential harm.
The continuation phase of treatment starts at 8 weeks, when the results of initial cultures and drug sensitivity tests should be available to guide therapy. A second set of cultures and AFB smears is obtained at 8 weeks to document clearing of the initial infection and guide duration of the continuation phase. If the initial culture was positive for Mycobacterium tuberculosis and the organism was sensitive to both INH and RIF, these 2 drugs should be continued for another 4 months (for a total of 6 months of treatment). PZA and EMB may be stopped at 2 months if the organism is sensitive to both INH and RIF. Thus, for most patients with active TB, the standard regimen will be 4 drugs for 2 months, then 2 drugs for 4 months.2
When should the standard treatment regimen be modified?
If the second set of cultures obtained 2 months after beginning drug treatment is positive and there was cavitary disease on the initial chest x-ray, the continuation phase should be extended by 7 months (for a total of 9 months of treatment).30 If a patient has either cavitary disease or persistently positive cultures (but not both), then the length of therapy is determined on an individual basis in consultation with an ID specialist.
Should a patient’s cultures show resistance to any of the first-line drugs, obtain consultation with an ID specialist. Treatment of multidrug-resistant TB (resistant to INH and RIF) and its subset, extensively drug-resistant TB (resistant to INH and RIF, plus any fluoroquinolone, plus either an aminoglycoside or capreomycin) requires prolonged courses of therapy with multiple drugs administered by DOT.31,32
If at any point during treatment a patient shows clinical deterioration that’s believed to be due to a resurgence of his or her TB disease, obtain a new set of cultures and, in consultation with an ID specialist, add at least 2 drugs to which the patient has not been exposed. Never add only one drug to a failing regimen; active TB always requires 2 drugs to cure, and the patient may have developed resistance to all of the drugs he or she is currently receiving.
If initial cultures are negative for Mycobacterium tuberculosis but the patient responds to treatment, he or she is considered to have “culture-negative TB,” and should generally be continued on INH and RIF for 2 more months after completion of the initial treatment phase (for a total of 4 months of INH and RIF).33
Remember to report. In the United States, active TB must be reported to your local health department, which can be invaluable in coordinating care and administering DOT.
Directly observed therapy (DOT) is preferred for certain high-risk patients with latent tuberculosis infection (LTBI), including those who are younger than 5 years of age, test positive for human immunodeficiency virus, are receiving immunosuppressive therapy, have chest radiography evidence of healed TB, have recently converted to active TB status while receiving serial TB testing, or have recently been exposed to active TB.12
Treatment for active TB should always be given by DOT.13 Because DOT is labor-intensive, twice-weekly dosing is usually preferred.14
CASE › In the hospital, Mr. J was placed in respiratory isolation, had prompt sputum cultures for TB, and was started on empiric treatment for active TB with INH, RIF, PZA, and EMB in standard doses. A search for other causes of nonresponsiveness to CAP showed no evidence of malignancy or HIV infection. He improved steadily and was discharged from the hospital after 2 weeks to complete 2 months of 4-drug therapy, with follow-up care coordinated by the local health department, including a home health nurse experienced in administering DOT. Cultures were positive for Mycobacterium tuberculosis sensitive to all drugs tested. After his initial 2 months of 4-drug therapy, he completed 4 months of additional treatment with INH and RIF, given by DOT, and recovered completely.
CORRESPONDENCE
Jeff Hall, MD, University of South Carolina Department of Family and Preventive Medicine, 3209 Colonial Drive, Columbia, SC 29203; [email protected]
› Obtain a problem-focused history and physical, as well as chest radiography, to rule out active pulmonary tuberculosis (TB) before initiating treatment for latent tuberculosis infection (LTBI). B
› Prescribe isoniazid 5 mg/kg/d (10 mg/kg/d in children) up to a maximum dose of 300 mg/d for 9 months for most patients with LTBI. B
› Ensure that directly observed therapy is used for all patients with active TB, as well as for select high-risk cases of LTBI. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mitchell J, age 62, comes to see you because he’s had a cough with increasing dyspnea for a month. Mr. J has never smoked but has type 2 diabetes mellitus. He also tells you that over the past month, he’s had occasional night sweats and has lost 8 pounds, although he’s not changed his diet. During the past week, he’s noticed blood-tinged sputum. Physical examination reveals a thin, chronically ill appearing man with an oral temperature of 100.6°F and mild tachypnea. You order a complete blood count, chest x-ray, and metabolic profile, administer a tuberculin skin test (TST), and initiate levofloxacin 500 mg/d for a presumed bacterial pneumonia. His lab work reveals mild leukocytosis and hyperglycemia, and the chest x-ray shows a left upper lobe infiltrate. The TST reaction—4 mm 50 hours after placement—was negative.
Mr. J returns a week later and says he feels worse. Your examination reveals worsened tachypnea, with tachycardia and crackles over the left upper lung fields.
How would you proceed with his care?
More people die of tuberculosis (TB) each year than any other infectious disease except human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. In 2013, an estimated 9 million people worldwide developed active TB and 1.5 million died of the disease.1 Many of these deaths could have been prevented if patients had received a diagnosis and treatment during the latent phase (when the patient was infected, but had no active disease), or as soon as the patient developed active disease. In this article we describe treatment for both latent and active TB.
Before treating latent TB infection, first rule out active TB
Patients with latent tuberculosis infection (LTBI) have a 5% to 10% lifetime risk of developing active TB disease.2 Treatment of LTBI can reduce this risk to 1% to 2%.3
Although not the focus of this article, diagnosis of LTBI is made by using either a TST, in which the patient receives an intradermal injection of purified protein derivative and the size of the skin induration is measured 48 to 72 hours after administration, or an interferon-gamma release assay (IGRA), which requires a blood draw. After receiving a positive test result for LTBI, the next step is to rule out active TB.4 This is necessary because the primary treatment regimen for LTBI involves only one drug, whereas treating active TB with one drug is strongly associated with treatment failure and future resistance to that drug.5
To rule out active TB, perform a brief, problem-focused history and physical, and obtain a chest x-ray.4 Pertinent findings that suggest active disease include:
- any history of recent weight loss, unexplained fever, night sweats, cough or hemoptysis
- fever or any unexpected lung findings on physical exam
- any parenchymal infiltrates on chest x-ray. (Granulomas and scarring may be signs of previously healed TB infection, but do not indicate active TB.)
Any of these findings should prompt a further investigation to either confirm or definitively rule out active TB disease. In the absence of these findings, the physician may proceed with treatment for LTBI.
Latent TB infection treatment: Isoniazid alone, or another regimen?
The current preferred regimen for most patients with LTBI is 9 months of isoniazid (INH) 5 mg/kg/d (10 mg/kg/d in children) up to a maximum of 300 mg/d. This regimen has been recommended by the Centers for Disease Control and Prevention (CDC), the American Thoracic Society, and the Infectious Diseases Society of America.3 However, there are 3 other CDC-recommended LTBI treatment regimens that include INH, INH plus rifapentine (RPT), or rifampin (RIF) for 6, 3, or 4 months, respectively (TABLE 1).6 These other regimens may be considered under certain circumstances. For example, INH and rifapentine might be used to treat an otherwise healthy patient who has had recent exposure to an individual with active, contagious TB.
If the patient is pregnant. INH is a pregnancy category C drug. Treatment for LTBI during pregnancy is generally regarded as safe and should be strongly considered if the patient has risk factors for progression to active TB, such as a recent exposure to someone with active TB.7 In otherwise healthy patients, treatment for LTBI may be deferred until after delivery.
Take steps to avoid complications of drug therapy
Drug-induced hepatitis is the primary adverse effect of INH treatment. Risk increases with age, previous hepatic injury, or concomitant use of other hepatotoxic medications. The risk is very small (<0.1%) for healthy children but may be over 10% for adults with multiple risk factors.8 Hepatitis is generally preceded by asymptomatic elevation of liver function tests (LFTs), which is much more common than clinical hepatitis.
Baseline LFTs should be obtained in patients who:
- have underlying liver disease, such as hepatitis B or C9
- consume ≥2 alcoholic drinks daily or >5 drinks at a time on any occasion
- take other medications with potential hepatotoxicity, such as statins
- have HIV infection10
- are pregnant or postpartum.
If a patient being considered for INH treatment has not had serologic testing for HIV, hepatitis B, or hepatitis C, these tests should be done prior to initiating INH. LFTs should be monitored every 1 to 2 months during INH therapy for patients who have ≥1 of these conditions and normal baseline LFTs. If baseline transaminases are >3 times the upper limit of normal, treatment for LTBI should probably be withheld, though might be considered in those whose LFTs return to normal after withdrawal of a modifiable risk factor, such as alcohol or a statin medication.
After beginning LTBI treatment, patients should be monitored regularly for signs and symptoms of hepatitis, including anorexia, nausea, abdominal pain, icterus, and dark urine, and LFTs performed if these develop. If during treatment transaminases increase to >3 times normal in a symptomatic patient (or >5 times normal in an asymptomatic patient), INH should be stopped and generally not resumed, even after LFTs return to normal. (Such patients would be considered to have partially treated LTBI, and their physicians should be alert to signs and symptoms of active TB, such as unexplained fever, weight loss, or blood-tinged sputum, during subsequent patient encounters.)
Peripheral neuropathy is a less common adverse effect of INH. It occurs in up to 2% of patients and is caused by interference with vitamin B6 (pyridoxine) metabolism. It can be prevented by supplementation with pyridoxine 25 to 50 mg/d. Vitamin B6, however, does not prevent INH-induced hepatotoxicity.
Noncompliance is a concern with INH therapy because treatment typically requires a 9-month course of daily medication.11 Patients for whom compliance is likely to be an issue might be considered for a 3-month, 12-dose course of once-weekly, directly-observed therapy (DOT) with INH and RPT administered by a public health agency. (See “Which patients with TB should receive directly observed therapy?” on page 32.12-14) A randomized, open-label trial involving nearly 8000 patients in 4 low-risk countries found this regimen was as effective as 9 months of self-administered INH.15 The CDC has published recommendations for using this regimen.16
Suspect active TB? Don’t wait for cultures to begin Tx
Unlike LTBI, for which the results of diagnostic testing are available within a few days, active TB is diagnosed by culture, which may take as long as 6 to 8 weeks. However, if you suspect your patient has active TB, do not delay treatment while waiting for culture results, or defer treatment for a patient who has a negative acid-fast bacilli (AFB) smear or rapid nucleic acid amplification test.17 These 2 tests, which are routinely performed during TB cultures, look for other evidence of the presence of TB bacilli; they are not as accurate as cultures, but results are available within days. Likewise, a negative TST or IGRA should not prevent empiric treatment for active TB. Treatment for active TB should be begun empirically based on risk factors and clinical presentation, and can be modified or stopped if cultures are negative, the patient fails to improve, or an alternative diagnosis is found to explain the patient’s symptoms.
Rapid testing for evidence of active TB disease—as well as resistance to medications commonly used to treat TB—can be performed using newer modalities such as MODS (Microscopic-Observation Drug-Susceptibility)18,19 or Xpert MTB/RIF20 testing. However, these tests are not available in many hospitals, and culture and drug sensitivity testing remain the gold standard.21
CASE › Mr. J’s clinical history and chest x-ray findings are highly suggestive of active TB. It was not unreasonable to initially treat him for a bacterial pneumonia, although fluoroquinolones should be used cautiously in this setting, because they are one of the most effective second-line drugs for TB, and using them as a single agent will often invoke drug resistance. Because he failed to respond to treatment for bacterial pneumonia and his presentation suggests TB or another serious cause of nonresponsiveness to standard treatment for community-acquired pneumonia (CAP), you admit him to the hospital.
Treatment for active TB requires multiple drugs in 2 phases
While all family physicians should suspect active TB in appropriate clinical situations and be comfortable with obtaining cultures and initiating empiric treatment, most will want to seek consultation with an infectious disease (ID) specialist especially in the scenarios listed in TABLE 2.5,22 Delayed or inappropriate treatment of active TB remains a major public health problem and cause of multidrug-resistant TB. Inappropriate treatment has been shown to be associated with a 27-fold increase in treatment failure.23 TB treatment guidelines are available from the CDC,24 World Health Organization,25 and International Union Against Tuberculosis and Lung Disease.26
Appropriate treatment requires the use of multiple medications administered in 2 phases. In the initial phase, a patient with suspected TB should begin 4 drugs—usually INH, RIF, ethambutol (EMB), and pyrazinamide (PZA)—for 2 months.1,2,27 The daily pediatric and adult doses and common adverse effects of these medications are summarized in TABLE 3.28 Although most cases of TB can be adequately treated with 2 drugs to which the organism is susceptible, 4 drugs are used initially while awaiting drug sensitivity test results because of the risk of inadequately treating a strain of drug-resistant TB. Before beginning these medications, a chest x-ray, LFTs, HIV antibody test, hepatitis B and C serologies, a serum creatinine, and complete blood count should be obtained in all patients.5 If EMB is prescribed, the patient should also undergo testing for red-green color discrimination, because red-green color vision disturbance is a potential adverse effect of this medication.
All 4 drugs may be administered as a single daily dose, and may be taken together.29 They are ordinarily given either daily for 8 weeks, or daily for 2 weeks followed by a twice-weekly schedule for the remaining 6 weeks in higher doses, although the twice-weekly dose of RIF is the same as the daily dose. All are pregnancy category C, although for active TB, the benefit of treatment is almost always greater than the potential harm.
The continuation phase of treatment starts at 8 weeks, when the results of initial cultures and drug sensitivity tests should be available to guide therapy. A second set of cultures and AFB smears is obtained at 8 weeks to document clearing of the initial infection and guide duration of the continuation phase. If the initial culture was positive for Mycobacterium tuberculosis and the organism was sensitive to both INH and RIF, these 2 drugs should be continued for another 4 months (for a total of 6 months of treatment). PZA and EMB may be stopped at 2 months if the organism is sensitive to both INH and RIF. Thus, for most patients with active TB, the standard regimen will be 4 drugs for 2 months, then 2 drugs for 4 months.2
When should the standard treatment regimen be modified?
If the second set of cultures obtained 2 months after beginning drug treatment is positive and there was cavitary disease on the initial chest x-ray, the continuation phase should be extended by 7 months (for a total of 9 months of treatment).30 If a patient has either cavitary disease or persistently positive cultures (but not both), then the length of therapy is determined on an individual basis in consultation with an ID specialist.
Should a patient’s cultures show resistance to any of the first-line drugs, obtain consultation with an ID specialist. Treatment of multidrug-resistant TB (resistant to INH and RIF) and its subset, extensively drug-resistant TB (resistant to INH and RIF, plus any fluoroquinolone, plus either an aminoglycoside or capreomycin) requires prolonged courses of therapy with multiple drugs administered by DOT.31,32
If at any point during treatment a patient shows clinical deterioration that’s believed to be due to a resurgence of his or her TB disease, obtain a new set of cultures and, in consultation with an ID specialist, add at least 2 drugs to which the patient has not been exposed. Never add only one drug to a failing regimen; active TB always requires 2 drugs to cure, and the patient may have developed resistance to all of the drugs he or she is currently receiving.
If initial cultures are negative for Mycobacterium tuberculosis but the patient responds to treatment, he or she is considered to have “culture-negative TB,” and should generally be continued on INH and RIF for 2 more months after completion of the initial treatment phase (for a total of 4 months of INH and RIF).33
Remember to report. In the United States, active TB must be reported to your local health department, which can be invaluable in coordinating care and administering DOT.
Directly observed therapy (DOT) is preferred for certain high-risk patients with latent tuberculosis infection (LTBI), including those who are younger than 5 years of age, test positive for human immunodeficiency virus, are receiving immunosuppressive therapy, have chest radiography evidence of healed TB, have recently converted to active TB status while receiving serial TB testing, or have recently been exposed to active TB.12
Treatment for active TB should always be given by DOT.13 Because DOT is labor-intensive, twice-weekly dosing is usually preferred.14
CASE › In the hospital, Mr. J was placed in respiratory isolation, had prompt sputum cultures for TB, and was started on empiric treatment for active TB with INH, RIF, PZA, and EMB in standard doses. A search for other causes of nonresponsiveness to CAP showed no evidence of malignancy or HIV infection. He improved steadily and was discharged from the hospital after 2 weeks to complete 2 months of 4-drug therapy, with follow-up care coordinated by the local health department, including a home health nurse experienced in administering DOT. Cultures were positive for Mycobacterium tuberculosis sensitive to all drugs tested. After his initial 2 months of 4-drug therapy, he completed 4 months of additional treatment with INH and RIF, given by DOT, and recovered completely.
CORRESPONDENCE
Jeff Hall, MD, University of South Carolina Department of Family and Preventive Medicine, 3209 Colonial Drive, Columbia, SC 29203; [email protected]
1. World Health Organization. Global tuberculosis report 2014. World Health Organization Web site. Available at: http://www.who.int/tb/publications/global_report/en/. Accessed December 15, 2014.
2. Zumla AI, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745-755.
3. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr4906a1.htm. Accessed December 15, 2014.
4. Hauck FR, Neese BH, Panchal AS, et al. Identification and management of latent tuberculosis infection. Am Fam Physician. 2009;79:879-886.
5. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77.
6. Centers for Disease Control and Prevention. Latent tuberculosis infection: A guide for primary health care providers. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/tb/publications/LTBI/default.htm. Accessed December 11, 2014.
7. Centers for Disease Control and Prevention. Fact sheet: tuberculosis and pregnancy. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/factsheets/specpop/pregnancy.htm. Accessed September 6, 2014.
8. Kunst H, Khan KS. Age-related risk of hepatotoxicity in the treatment of latent tuberculosis infection: a systematic review. Int J Tuberc Lung Dis. 2010;14:1374-1381.
9. Bliven EE, Podewils LJ. The role of chronic hepatitis in isoniazid hepatotoxicity during treatment for latent tuberculosis infection. Int J Tuberc Lung Dis. 2009;13:1054-1060.
10. Akolo C, Adetifa I, Shepperd S, et al. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;1:CD000171.
11. Horsburgh CR Jr, Goldberg S, Bethel J, et al; Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401-409.
12. Horsburgh CR Jr. Priorities for the treatment of latent tuberculosis infection in the United States. N Engl J Med. 2004;350:2060-2070.
13. Potter B, Rindfleisch K, Kraus CK. Management of active tuberculosis. Am Fam Physician. 2005;72:2225-2232.
14. Volmink J, Garner P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. 2007;4:CD003343.
15. Sterling TR, Villarina ME, Borisov AS, et al; TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155-2166.
16. Centers for Disease Control and Prevention (CDC). Recommendations for use of an isoniazid-rifapentine regimen with direct observation to treat latent Mycobacterium tuberculosis infection. MMWR Morb Mortal Wkly Rep. 2011;60:1650-1653.
17. Inge LD, Wilson JW. Update on the treatment of tuberculosis. Am Fam Physician. 2008;78:457-465.
18. Moore DA, Evans CA, Gilman RH, et al. Microscopic-observation drug-susceptibility assay for the diagnosis of TB. N Engl J Med. 2006;355:1539-1550.
19. Minion J, Leung E, Menzies D, et al. Microscopic-observation drug susceptibility and thin layer agar assays for the detection of drug resistant tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:688-698.
20. Boehme CC, Nabeta P, Hilleman D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363:1005-1015.
21. Arentz M, Sorensen B, Horne DJ, et al. Systematic review of the performance of rapid rifampicin resistance testing for drug-resistant tuberculosis. PLoS One. 2013;8:e76533.
22. Sia IG, Wieland ML. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348-361.
23. van der Werf MJ, Langendam MW, Huitric E, et al. Multidrug resistance after inappropriate tuberculosis treatment: a meta-analysis. Eur Respir J. 2012;39:1511-1519.
24. Centers for Disease Control and Prevention. Tuberculosis (TB). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/guidelines/default.htm. Accessed September 6, 2014.
25. World Health Organization. Treatment of tuberculosis guidelines. 4th ed. World Health Organization Web site. Available at: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf?ua=1. Accessed September 6, 2014.
26. International Union Against Tuberculosis and Lung Disease. Management of tuberculosis: A guide to the essentials of good clinical practice. 6th ed. 2010. International Union Against Tuberculosis and Lung Disease Web site. Available at: http://www.theunion.org/what-we-do/publications/technical/management-of-tuberculosis-a-guide-to-the-essentials-of-good-clinical-practice. Accessed September 6, 2014.
27. Combs DL, O’Brien RJ, Geiter LJ. USPHS Tuberculosis Short-Course Chemotherapy Trial 21: effectiveness, toxicity and acceptability. The report of the final results. Ann Intern Med. 1990;112:397-406.
28. Drugs for tuberculosis. Treat Guidel Med Lett. 2012;10:29-36.
29. Chang KC, Leung CC, Grosset J, et al. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66:997-1007.
30. Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention and the Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
31. Lynch JB. Multidrug-resistant tuberculosis. Med Clin North Am. 2013;97:553-579,ix-x.
32. Keshavjee S, Farmer PE. Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med. 2012;367:931-936.
33. Dutt AK, Moers D, Stead WW. Smear- and culture-negative pulmonary tuberculosis: four-month short-course chemotherapy. Am Rev Respir Dis. 1989;139:867-870.
1. World Health Organization. Global tuberculosis report 2014. World Health Organization Web site. Available at: http://www.who.int/tb/publications/global_report/en/. Accessed December 15, 2014.
2. Zumla AI, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745-755.
3. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr4906a1.htm. Accessed December 15, 2014.
4. Hauck FR, Neese BH, Panchal AS, et al. Identification and management of latent tuberculosis infection. Am Fam Physician. 2009;79:879-886.
5. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77.
6. Centers for Disease Control and Prevention. Latent tuberculosis infection: A guide for primary health care providers. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/tb/publications/LTBI/default.htm. Accessed December 11, 2014.
7. Centers for Disease Control and Prevention. Fact sheet: tuberculosis and pregnancy. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/factsheets/specpop/pregnancy.htm. Accessed September 6, 2014.
8. Kunst H, Khan KS. Age-related risk of hepatotoxicity in the treatment of latent tuberculosis infection: a systematic review. Int J Tuberc Lung Dis. 2010;14:1374-1381.
9. Bliven EE, Podewils LJ. The role of chronic hepatitis in isoniazid hepatotoxicity during treatment for latent tuberculosis infection. Int J Tuberc Lung Dis. 2009;13:1054-1060.
10. Akolo C, Adetifa I, Shepperd S, et al. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;1:CD000171.
11. Horsburgh CR Jr, Goldberg S, Bethel J, et al; Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401-409.
12. Horsburgh CR Jr. Priorities for the treatment of latent tuberculosis infection in the United States. N Engl J Med. 2004;350:2060-2070.
13. Potter B, Rindfleisch K, Kraus CK. Management of active tuberculosis. Am Fam Physician. 2005;72:2225-2232.
14. Volmink J, Garner P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. 2007;4:CD003343.
15. Sterling TR, Villarina ME, Borisov AS, et al; TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155-2166.
16. Centers for Disease Control and Prevention (CDC). Recommendations for use of an isoniazid-rifapentine regimen with direct observation to treat latent Mycobacterium tuberculosis infection. MMWR Morb Mortal Wkly Rep. 2011;60:1650-1653.
17. Inge LD, Wilson JW. Update on the treatment of tuberculosis. Am Fam Physician. 2008;78:457-465.
18. Moore DA, Evans CA, Gilman RH, et al. Microscopic-observation drug-susceptibility assay for the diagnosis of TB. N Engl J Med. 2006;355:1539-1550.
19. Minion J, Leung E, Menzies D, et al. Microscopic-observation drug susceptibility and thin layer agar assays for the detection of drug resistant tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:688-698.
20. Boehme CC, Nabeta P, Hilleman D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363:1005-1015.
21. Arentz M, Sorensen B, Horne DJ, et al. Systematic review of the performance of rapid rifampicin resistance testing for drug-resistant tuberculosis. PLoS One. 2013;8:e76533.
22. Sia IG, Wieland ML. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348-361.
23. van der Werf MJ, Langendam MW, Huitric E, et al. Multidrug resistance after inappropriate tuberculosis treatment: a meta-analysis. Eur Respir J. 2012;39:1511-1519.
24. Centers for Disease Control and Prevention. Tuberculosis (TB). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/guidelines/default.htm. Accessed September 6, 2014.
25. World Health Organization. Treatment of tuberculosis guidelines. 4th ed. World Health Organization Web site. Available at: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf?ua=1. Accessed September 6, 2014.
26. International Union Against Tuberculosis and Lung Disease. Management of tuberculosis: A guide to the essentials of good clinical practice. 6th ed. 2010. International Union Against Tuberculosis and Lung Disease Web site. Available at: http://www.theunion.org/what-we-do/publications/technical/management-of-tuberculosis-a-guide-to-the-essentials-of-good-clinical-practice. Accessed September 6, 2014.
27. Combs DL, O’Brien RJ, Geiter LJ. USPHS Tuberculosis Short-Course Chemotherapy Trial 21: effectiveness, toxicity and acceptability. The report of the final results. Ann Intern Med. 1990;112:397-406.
28. Drugs for tuberculosis. Treat Guidel Med Lett. 2012;10:29-36.
29. Chang KC, Leung CC, Grosset J, et al. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66:997-1007.
30. Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention and the Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
31. Lynch JB. Multidrug-resistant tuberculosis. Med Clin North Am. 2013;97:553-579,ix-x.
32. Keshavjee S, Farmer PE. Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med. 2012;367:931-936.
33. Dutt AK, Moers D, Stead WW. Smear- and culture-negative pulmonary tuberculosis: four-month short-course chemotherapy. Am Rev Respir Dis. 1989;139:867-870.
January 2015 Quiz 1
ANSWER: D
Critique
The presentation is one of intermittent solid food dysphagia in the setting of frequent heartburn in a young white male. The differential diagnosis includes reflux esophagitis and peptic stricture, as well as eosinophilic esophagitis. Sometimes, the two conditions overlap. Empiric proton pump inhibitor therapy is of value in clarifying the diagnosis of gastroesophageal reflux disease in patients with typical symptoms of heartburn and regurgitation. This is because the likelihood of gastroesophageal reflux disease is very high in patients with typical reflux symptoms. However, in this setting eosinophilic esophagitis needs to be excluded. So the optimal approach is to initiate proton pump inhibitor therapy, and then inspect and biopsy the esophagus. Treatments specific for eosinophilic esophagitis (topical fluticasone, montelukast) will only be indicated if the diagnosis of eosinophilic esophagitis is confirmed.
Prednisone is not utilized often orally for the management of eosinophilic esophagitis. Baclofen, a GABA-B receptor agonist, has been demonstrated to reduce the frequency of transient lower esophageal sphincter relaxations and improve residual symptoms in patients on PPI therapy.
- Numans M.E., Lau J., de Wit N.J., et al. Short-term treatment with proton-pump inhibitors as a test for gastroesophageal reflux disease: a meta-analysis of diagnostic test characteristics. Ann. Intern. Med. 2004;140:518-27.
- Kapel R.C., Miller J.K., Torres C., Aksoy S., Lash R., Katzka D.A. Eosinophilic esophagitis: A prevalent disease in the United States that affects all age groups. Gastroenterology 2008;134:1316-21.
- Rothenberg M.E. Biology and treatment of eosinophilic esophagitis. Gastroenterology 2009;137:1238-49.
ANSWER: D
Critique
The presentation is one of intermittent solid food dysphagia in the setting of frequent heartburn in a young white male. The differential diagnosis includes reflux esophagitis and peptic stricture, as well as eosinophilic esophagitis. Sometimes, the two conditions overlap. Empiric proton pump inhibitor therapy is of value in clarifying the diagnosis of gastroesophageal reflux disease in patients with typical symptoms of heartburn and regurgitation. This is because the likelihood of gastroesophageal reflux disease is very high in patients with typical reflux symptoms. However, in this setting eosinophilic esophagitis needs to be excluded. So the optimal approach is to initiate proton pump inhibitor therapy, and then inspect and biopsy the esophagus. Treatments specific for eosinophilic esophagitis (topical fluticasone, montelukast) will only be indicated if the diagnosis of eosinophilic esophagitis is confirmed.
Prednisone is not utilized often orally for the management of eosinophilic esophagitis. Baclofen, a GABA-B receptor agonist, has been demonstrated to reduce the frequency of transient lower esophageal sphincter relaxations and improve residual symptoms in patients on PPI therapy.
ANSWER: D
Critique
The presentation is one of intermittent solid food dysphagia in the setting of frequent heartburn in a young white male. The differential diagnosis includes reflux esophagitis and peptic stricture, as well as eosinophilic esophagitis. Sometimes, the two conditions overlap. Empiric proton pump inhibitor therapy is of value in clarifying the diagnosis of gastroesophageal reflux disease in patients with typical symptoms of heartburn and regurgitation. This is because the likelihood of gastroesophageal reflux disease is very high in patients with typical reflux symptoms. However, in this setting eosinophilic esophagitis needs to be excluded. So the optimal approach is to initiate proton pump inhibitor therapy, and then inspect and biopsy the esophagus. Treatments specific for eosinophilic esophagitis (topical fluticasone, montelukast) will only be indicated if the diagnosis of eosinophilic esophagitis is confirmed.
Prednisone is not utilized often orally for the management of eosinophilic esophagitis. Baclofen, a GABA-B receptor agonist, has been demonstrated to reduce the frequency of transient lower esophageal sphincter relaxations and improve residual symptoms in patients on PPI therapy.
- Numans M.E., Lau J., de Wit N.J., et al. Short-term treatment with proton-pump inhibitors as a test for gastroesophageal reflux disease: a meta-analysis of diagnostic test characteristics. Ann. Intern. Med. 2004;140:518-27.
- Kapel R.C., Miller J.K., Torres C., Aksoy S., Lash R., Katzka D.A. Eosinophilic esophagitis: A prevalent disease in the United States that affects all age groups. Gastroenterology 2008;134:1316-21.
- Rothenberg M.E. Biology and treatment of eosinophilic esophagitis. Gastroenterology 2009;137:1238-49.
- Numans M.E., Lau J., de Wit N.J., et al. Short-term treatment with proton-pump inhibitors as a test for gastroesophageal reflux disease: a meta-analysis of diagnostic test characteristics. Ann. Intern. Med. 2004;140:518-27.
- Kapel R.C., Miller J.K., Torres C., Aksoy S., Lash R., Katzka D.A. Eosinophilic esophagitis: A prevalent disease in the United States that affects all age groups. Gastroenterology 2008;134:1316-21.
- Rothenberg M.E. Biology and treatment of eosinophilic esophagitis. Gastroenterology 2009;137:1238-49.
January 2015 Quiz 2
ANSWER: D
Critique
Villous blunting is not specific to celiac disease. In this case, the patient’s history supports a diagnosis of common variable immunodeficiency (CVID), which is characterized by low levels of two Ig classes and recurrent infections. The infections most commonly involve the upper and lower respiratory tract.
Chronic diarrhea is seen in 40%-60% of patients and may lead to malabsorption. Diarrhea can be the result of infections (most commonly Salmonella, Campylobacter, Clostridium difficile, and Giardia lamblia), inflammatory disorders, or malignancy. Biopsies reveal villous blunting similar to that seen in celiac disease. Unlike celiac disease, however, the biopsies lack plasma cells. These patients also differ from those with celiac disease in that their celiac serologies are negative.
- Shah V.H., Rotterdam H., Kotler D.P., et al. All that scallops is not celiac disease. Gastrointest. Endoscopy 2000;51:717-20.
- Sperber K.F., Mayer L. Gastrointestinal manifestations of common variable immunodeficiency. Immunol. Allergy Clin. North Am. 1988;8:423-34.
ANSWER: D
Critique
Villous blunting is not specific to celiac disease. In this case, the patient’s history supports a diagnosis of common variable immunodeficiency (CVID), which is characterized by low levels of two Ig classes and recurrent infections. The infections most commonly involve the upper and lower respiratory tract.
Chronic diarrhea is seen in 40%-60% of patients and may lead to malabsorption. Diarrhea can be the result of infections (most commonly Salmonella, Campylobacter, Clostridium difficile, and Giardia lamblia), inflammatory disorders, or malignancy. Biopsies reveal villous blunting similar to that seen in celiac disease. Unlike celiac disease, however, the biopsies lack plasma cells. These patients also differ from those with celiac disease in that their celiac serologies are negative.
ANSWER: D
Critique
Villous blunting is not specific to celiac disease. In this case, the patient’s history supports a diagnosis of common variable immunodeficiency (CVID), which is characterized by low levels of two Ig classes and recurrent infections. The infections most commonly involve the upper and lower respiratory tract.
Chronic diarrhea is seen in 40%-60% of patients and may lead to malabsorption. Diarrhea can be the result of infections (most commonly Salmonella, Campylobacter, Clostridium difficile, and Giardia lamblia), inflammatory disorders, or malignancy. Biopsies reveal villous blunting similar to that seen in celiac disease. Unlike celiac disease, however, the biopsies lack plasma cells. These patients also differ from those with celiac disease in that their celiac serologies are negative.
- Shah V.H., Rotterdam H., Kotler D.P., et al. All that scallops is not celiac disease. Gastrointest. Endoscopy 2000;51:717-20.
- Sperber K.F., Mayer L. Gastrointestinal manifestations of common variable immunodeficiency. Immunol. Allergy Clin. North Am. 1988;8:423-34.
- Shah V.H., Rotterdam H., Kotler D.P., et al. All that scallops is not celiac disease. Gastrointest. Endoscopy 2000;51:717-20.
- Sperber K.F., Mayer L. Gastrointestinal manifestations of common variable immunodeficiency. Immunol. Allergy Clin. North Am. 1988;8:423-34.
Easy bruising • low platelet count • recent cold-like illness • Dx?
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
THE CASE
A 6-year-old girl was brought to the emergency department (ED) by her mother after the child had bumped her head while playing. While the physician examined the child’s head, the mother remarked that her daughter had recently developed bruises that appeared suddenly and only after minor, if any, known trauma. The ED physician determined that the child’s bump to the head was nothing to worry about, attributed the bruising to the child being a “healthy, active 6-year-old,” and sent her home.
Two days later the child was brought to our office because the mother was still concerned about her daughter’s easy bruising. The mother pointed out ecchymosis scattered across her daughter’s extremities and torso. The child denied any pain or other complaints, including any active or recurrent bleeding. Upon further questioning, the mother mentioned that her daughter had recovered from a cold-like illness several weeks earlier.
THE DIAGNOSIS
We ordered a complete blood count (CBC) and peripheral smear, which were normal except for the platelet count, which was 7000/mcL (normal, 150,000-450,000/mcL). Based on the child’s easy bruising and isolated thrombocytopenia, we diagnosed immune thrombocytopenia, which is also known as idiopathic thrombocytopenic purpura (ITP).
DISCUSSION
In ITP, autoantibodies are directed against platelets, leading to their sequestration and destruction in the spleen and a resultant drop in platelet count.1 Children with ITP typically present between the ages of 2 and 10 years, with a peak incidence between 2 and 5 years.2 The incidence is estimated to be as high as 8 per 100,000 children.3 However, this estimate primarily reflects symptomatic children, and the true incidence of childhood ITP may be much higher because asymptomatic children may not be brought in to see a doctor. For the majority of patients, ITP resolves within 3 months. However, for 20% to 30% of patients, thrombocytopenia will last beyond 6 months, with or without treatment.4 In 1% of cases, patients will have a recurrence of ITP.3
In addition to easy bruising, nearly all patients who present with possible ITP will complain of cutaneous bleeding, typically a nose bleed or bleeding in the oral cavity.2 Upon questioning, 60% of patients will report a history of recent infection.4 Not surprisingly, bleeding severity correlates inversely with platelet count; severe bleeding is seen in patients with a platelet count <10,000/mcL.
While rare, the more worrisome complications include intracranial hemorrhage, with an incidence of 0.1% to 0.8%, and other serious hemorrhages that would require transfusion, with an estimated incidence of 2.9%.2
Vast differential seen in child bruising
When a child presents with bruising, perform a thorough history, including birth and prenatal course, as well as a physical to exclude other potential causes, such as physical abuse, use of herbal remedies or other natural supplements that may not be disclosed as medication, or even environmental exposure. When bruising is present in a child who has isolated thrombocytopenia, the diagnosis of ITP may be straightforward. However, many conditions may share thrombocytopenia in their disease process and should be considered in the differential diagnosis of a child who you suspect may have ITP.
Suspect physical abuse in a bruised child who does not have thrombocytopenia, whose mood is flat or depressed, or who has experienced recurrent injuries or bruising.
Leukemia, particularly acute lymphoblastic leukemia (ALL), the predominant leukemia found in children, should be ruled out, as well. Symptoms that may distinguish a child with ALL from one with ITP include fever, weight loss, and joint pain, as well as signs such as lymphadenopathy, hepatosplenomegaly, anemia, and leukocytosis. A peripheral smear may be ordered to help confirm or exclude a diagnosis of ALL should any of the above be present in a child with thrombocytopenia.5 It may show lymphoblasts and/or atypical cells in a patient with ALL.5
Infections should also be included in a differential when a patient is suspected of having ITP, particularly if he or she has systemic symptoms. Viral infections that may cause thrombocytopenia include mononucleosis, dengue virus, human herpesvirus-6, and human immunodeficiency virus.6,7
ITP often follows an infection, and the incidence of ITP may be higher during winter months, when infections are more common. However, infection may not always be the cause of ITP. Sepsis may also lead to thrombocytopenia, but a child with sepsis would present very differently from a child who has only ITP. A septic child would present acutely ill with signs and symptoms of severe systemic illness, such as high fever, altered mental status, tachycardia, pallor, diaphoresis, and hypotension.
Drug-induced thrombocytopenia (DIT) should be considered in any child who is taking or recently took a medication that may cause thrombocytopenia. Medications that can cause thrombocytopenia include heparin, quinine, vancomycin, trimethoprim-sulfamethoxazole, rifampin, carbamazepine, phenytoin, piperacillin, linezolid, and valproic acid.8 The measles, mumps, and rubella vaccine also can cause thrombocytopenia.8 A careful medication history may determine if the child is at risk for DIT.
To narrow the differential, obtain a CBC and peripheral smear when evaluating a patient you suspect may have ITP5 (strength of recommendation [SOR]: A). A CBC will determine the patient’s platelet count and a peripheral smear should be obtained to exclude other possible diagnoses.5
If there are any questions regarding the results of a peripheral smear, it may be necessary to perform a bone marrow aspiration. This, however, is not usually necessary in an otherwise typical case of ITP.9 Bone marrow aspiration may, however, be necessary to reevaluate the initial diagnosis for a child who does not respond to treatment for ITP.
Corticosteroids, IVIg are usually effective
The first step in treating a patient with ITP is to limit the risk of further injury or bleeding, by stopping nonsteroidal anti-inflammatory drugs or ending participation in contact sports2,9 (SOR: C). The next step is to determine if pharmacologic therapy is warranted.
Medication, if necessary, is the mainstay of treatment for patients with ITP, particularly those experiencing significant bleeding.2 Corticosteroids, intravenous (IV) immunoglobulin (IVIg), and IV Rho(D) immune globulin (also known as anti-D) are the medications typically used to treat a child with ITP, depending on availability of the drugs, bleeding or bleeding risk, as well as convenience of dosing. For example, corticosteroids can be used orally or IV, whereas IVIg and IV Rho(D) may not be readily available in some treatment settings.
Corticosteroids have been shown to more rapidly increase platelet count compared to placebo and appear to have a dose-related effect.10,11 Oral prednisone can be dosed at 1 to 2 mg/kg/d for 14 days and then tapered over the course of one week10,11 or one may prescribe 4 mg/kg/d for 4 days.10,11 IV methylprednisolone typically is given at 30 mg/kg/d for 3 to 4 days.9
IVIg may have greater efficacy than corticosteroids in treating ITP, but it may also cause adverse effects, including nausea, headache, and fever. IVIg can be administered as a single 800 to 1000 mg/kg dose, or as a daily 400 mg/kg dose for 5 days; higher doses should be reserved for patients with severe bleeding.12
If ITP persists despite the use of corticosteroids or IVIg, IV Rho(D) Ig may be used in patients with Rho(D)-positive blood at a single dose of 25 to 50 mcg/kg, with additional doses administered on separate days as required to elevate platelet count. However, only Rho(D)-positive patients are eligible for anti-D treatment.
The response rates/times and adverse effects of common treatments for ITP are summarized in the TABLE.9 A small randomized study found that oral methylprednisolone 30 mg/kg/d for 3 days followed by 20 mg/kg/d for an additional 4 days was comparable to IVIg 0.4 g/kg/d for 5 days.11 A different study that compared oral methylprednisolone (30 mg/kg/d or 50 mg/kg/d for 7 days) and IVIg (0.5 g/kg/d for 5 days) found no difference in outcomes among the 3 treatments.13 One advantage, though, of IVIg is that it can be administered as a single IV dose, rather than multiple doses over several weeks, as is the case with oral prednisone.9,11-13
Follow platelet counts closely. Patients with ITP should have their platelet counts monitored at least once weekly and as often as twice weekly. The frequency of monitoring may be tapered depending on an individual patient’s response to treatment and the severity of the thrombocytopenia.14
We referred our patient to a nearby children’s hospital, where a repeat CBC showed her platelets had decreased to 3000/mcL. She received a 6-hour infusion of IVIg and was discharged with instructions to have her CBC closely monitored. Her platelets remained stable until 4 weeks later, when they decreased from 102,000/mcL to 71,000/mcL. She received a second infusion of IVIg as an outpatient.
Soon after, she went to our ED with a headache, nausea, and fever of 102°F. A computed tomography scan of her head was normal; a repeat CBC showed no elevation in white blood cells but her hemoglobin had decreased from 11.9 g/dL to 9.7 g/dL. (Her platelets were 254,000/mcL.) The patient’s complaints were likely adverse effects of the IVIg. The CBC abnormalities, fever, headache, and malaise resolved shortly thereafter and the patient remains asymptomatic with no recurrence of ITP.
THE TAKEAWAY
Suspect ITP in a child who bruises easily and who also has thrombocytopenia. Order a CBC and peripheral blood smear to rule out other potential illnesses. Pharmacotherapy, if needed, typically consists of an oral or IV corticosteroid or IVIg; IV Rho(D) Ig may be used in patients who are Rho(D)-positive who don’t respond to other treatments. Patients with ITP should have their platelet count monitored at least once weekly until platelets have increased to 150,000/mcL or higher. Frequency of monitoring may be reduced as the clinical picture improves and the patient remains stable. More frequent monitoring may be necessary based on severity, complications, and response to treatment.
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
1. Johnsen J. Pathogenesis in immune thrombocytopenia: new insights. Hematology Am Soc Hematol Educ Program. 2012;2012:306-312.
2. Kühne T, Buchanan GR, Zimmerman S, et al; Intercontinental Childhood ITP Study Group. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143:605-608.
3. Kurtzberg J, Stockman JA 3rd. Idiopathic autoimmune thrombocytopenic purpura. Adv Pediatr. 1994;41:111-134.
4. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease. Acta Paediatr. 2005;94:178-184.
5. Margolin JF, Steuber CP, Poplack DG. Acute lymphoblastic leukemia. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2001: 317-321.
6. Hashimoto H, Maruyama H, Fujimoto K, et al. Hematologic findings associated with thrombocytopenia during the acute phase of exanthem subitum confirmed by primary human herpesvirus-6 infection. J Pediatr Hematol Oncol. 2002;24:211-214.
7. La Russa VF, Innis BL. Mechanisms of dengue virus-induced bone marrow suppression. Baillieres Clin Haematol. 1995;8:249-270.
8. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. Thromb Haemost. 2009;7:911-918.
9. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186.
10. Bellucci S, Charpak Y, Chastang C, et al. Low doses v conventional doses of corticoids in immune thrombocytopenic purpura (ITP): results of a randomized clinical trial in 160 children, 223 adults. Blood. 1988;71:1165-1169.
11. Ozsoylu S, Sayli TR, Oztürk G. Oral megadose methylprednisolone versus intravenous immunoglobulin for acute childhood idiopathic thrombocytopenic purpura. Pediatr Hematol Oncol. 1993;10:317-321.
12. Beck CE, Nathan PC, Parkin PC, et al. Corticosteroids versus intravenous immune globulin for the treatment of acute immune thrombocytopenic purpura in children: a systematic review and meta-analysis of randomized controlled trials. J Pediatr. 2005;147:521-527.
13. Albayrak D, Işlek I, Kalaycí AG, et al. Acute immune thrombocytopenic purpura: a comparative study of very high oral doses of methylprednisolone and intravenously administered immune globulin. J Pediatr. 1994;125(6 pt 1):1004-1007.
14. Tarantino MD, Madden RM, Fennewald DL, et al. Treatment of childhood acute immune thrombocytopenic purpura with anti-D immune globulin or pooled immune globulin. J Pediatr. 1999;134:21-26.
Hyperthyroidism • myalgia • rapidly progressing paralysis • Dx?
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
THE CASE
A 26-year-old Hispanic woman presented to the emergency department (ED) with myalgia and weakness. The work-up revealed profound hyperthyroidism, with a thyroid-stimulating hormone (TSH) <0.01 mIU/mL (normal, 0.4-4.2 mIU/L), potassium 2.4 mEq/L (normal, 3.7-5.2 mEq/L), hypophosphatemia, and low urinary potassium. There were no prior symptoms and family history was negative for endocrinopathies. She was admitted and started on methimazole 10 mg twice a day for thyroid suppression and given propranolol 10 mg twice a day for anticipated hyperadrenergic adverse effects. The remainder of her hospital stay was uneventful and she was discharged 6 days after admission. Soon after, an outpatient thyroid scan ordered by her primary care physician confirmed that the patient had Graves’ disease.
Eight months later, the patient returned to the ED with myalgia and rapidly progressing paralysis from the neck down; she was immediately intubated. Her potassium level was 1.2 mEq/L. An electrocardiogram (EKG) revealed conduction abnormalities consistent with hypokalemia.
THE DIAGNOSIS
Based on our patient’s paralysis, hyperthyroidism, and hypokalemia, we diagnosed thyrotoxic hypokalemic periodic paralysis (THPP), a rare endocrinopathy that causes electrolyte disturbances that can result in paralysis and lethal tachyarrhythmias.1-6
Patients with THPP typically have a history of myalgia, cramping, and stiffness followed by weakness or paralysis that tends to develop rapidly, most commonly in the late evening or early morning1-4,6,7 (TABLE1-9). Proximal muscles are predominantly affected symmetrically and the attacks usually resolve in a period of hours to several days. Ocular, bulbar, and respiratory muscles are usually spared, but these can be affected by the hypokalemia.1
DISCUSSION
Traditionally THPP has been seen primarily in Asia, with an incidence as high as 2%.1-6 The incidence in the United States is lower (0.1%-0.2%) and THPP occurs primarily in Asian, African, Hispanic, and Native American populations.1,4,6
Although thyrotoxicosis is more common in women, THPP has a predilection for men (20:1).1,3-6 THPP occurs in patients with hyperthyroidism, most commonly from Graves’ disease,1,6 who are exposed to certain precipitating factors, such as exercise, carbohydrate loading, high-salt diet, excessive alcohol consumption, trauma, cold exposure, infection, menstruation, or emotional stress.1,6 THPP can also occur in people taking medications such as corticosteroids, β2-adrenergic bronchodilators, epinephrine, acetazolamide, insulin, nonsteroidal anti-inflammatory drugs, thyroxine, amiodarone, and tiratricol.1,5,6 THPP is more common in the summer.1
A genetic basis for THPP. A Kir2.6 mutation results in a thyroid hormone-sensitive channelopathy involving the sodium-potassium-adenosine triphosphate (Na+,K+-ATPase) pump, which appears to be responsible for THPP.1-6,8,9 This mutation should not be confused with the pathogenesis of familial periodic paralysis (FPP)—a hereditary disorder resulting in abnormalities in calcium, sodium, and potassium channels on skeletal muscle cells that leads to multiple electrolyte derangements and paralysis identical to that observed in THPP.1
Hypokalemia may be exacerbated by catecholamine-induced potassium shifts.1,4,6 This is from the increased β2-adrenergic stimulation from the concurrent hyperadrenergic state caused by the underlying hyperthyroidism.1,4,6 Hyperinsulinemia from sympathetic stimulation of the insulin-releasing pancreatic beta cells also exacerbates hypokalemia.1,4,6
Focus treatment on correcting electrolytes
Initial evaluation of a patient suspected of having THPP should include a complete blood count, TSH and serum and urine electrolyte tests, and an EKG. Further work-up may require ultrasound and scan of the thyroid upon confirmation of thyrotoxicosis and hypokalemia. Physical examination may reveal thyromegaly. Exophthalmos and other hyperthyroidism symptoms often are absent.1
Diagnosis confirmed? Treat the hypokalemia first. Acute management of THPP centers on electrolyte correction. Total body stores of potassium in patients with THPP are usually normal, so the physician must use care to avoid excessive potassium administration.1-5 Rebound hyperkalemia can occur in patients who receive >90 mEq/L of potassium chloride within 24 hours.1
Definitive therapy may include antithyroid medication, radioactive iodine ablation (RIA), and/or thyroidectomy.1-5 All have the common goal of controlling the hyperthyroidism and preventing recurrent paralysis, which occurs in 62.2% of patients within the first 3 months following diagnosis.3 If antithyroid medications fail, then RIA is the next choice.1 Beta-blockers work by decreasing the Na+,K+-ATPase activity from the underlying hyperadrenergic state.1 Administration of acetazolamide—which is the primary treatment modality for FPP and idiopathic periodic paralysis—can precipitate THPP attacks and is contraindicated.1,5
If medical management is unsuccessful or the patient develops compression symptoms, then thyroidectomy should be considered.3 If the patient chooses thyroidectomy, medical optimization with antithyroid medications is indicated to mitigate the risks of anesthesia. When the thyroidectomy is performed by an experienced thyroid surgeon, the long-term results are excellent.
Our patient. Once our patient’s hypokalemia was corrected, she was successfully extubated. Despite appropriate medical therapy, her hyperthyroidism was poorly controlled. The endocrinologist believed that RIA was suboptimal for 3 reasons: 1) it might result in incomplete ablation, 2) it required a long treatment period to be effective, and 3) its prolonged course of treatment extended the time interval that the patient would be at risk for recurrent paralysis.
A surgeon was consulted for definitive treatment with thyroidectomy. Our patient’s medications were changed to propylthiouracil 150 mg every 8 hours and propranolol 10 mg twice a day until a euthyroid state was achieved and she could tolerate a general anesthetic without precipitating a thyroid storm. Two months later, she underwent total thyroidectomy without complication. Her postoperative course was normal.
THE TAKEAWAY
Thyrotoxic hypokalemic periodic paralysis is rare. Patients typically present with myalgia, cramping, and stiffness that progress to paralysis. Prompt electrolyte repletion is paramount for successful outcomes.1-5 Control of hyperthyroidism is the long-term goal.1-5 Definitive therapy can be achieved medically or surgically. Total thyroidectomy is a reasonable treatment option for medically refractory hyperthyroidism or when RIA is contraindicated. Long-term prognosis is excellent.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.
1. Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80:99-105.
2. Antonello IC, Antonello VS, de Los Santos CA, et al. Thyrotoxic hypokalemic periodic paralysis: a life-threatening syndrome. Eur J Emerg Med. 2009;16:43-44.
3. Lin YC, Wu CW, Chen HC, et al. Surgical treatment for thyrotoxic hypokalemic periodic paralysis: case report. World J Surg Oncol. 2012;10:21.
4. El-Hennawy AS, Nesa M, Mahmood AK. Thyrotoxic hypokalemic periodic paralysis triggered by high carbohydrate diet. Am J Ther. 2007;14:499-501.
5. Chang CC, Cheng CJ, Sung CC, et al. A 10-year analysis of thyrotoxic periodic paralysis in 135 patients: focus on symptomatology and precipitants. Eur J Endocrinol. 2013;169:529-536.
6. Vijayakumar A, Ashwath G, Thimmappa D. Thyrotoxic periodic paralysis: clinical challenges. J Thyroid Res. 2014;2014:649502.
7. Ray S, Kundu S, Goswami M, et al. An unusual cause of muscle weakness: a diagnostic challenge for clinicians. BMJ Case Rep. 2012;2012.
8. Dassau L, Conti LR, Radeke CM, et al. Kir2.6 regulates the surface expression of Kir2.x inward rectifier potassium channels. J Biol Chem. 2011;286:9526-9541.
9. Ryan DP, da Silva MR, Soong TW, et al. Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell. 2010;140:88-98.

Insulin therapy and cancer risk
To the Editor: We read with interest the article by Ching Sun et al1 on the relationship between diabetes therapy and cancer risk. We noted that there was no reference in the text to the long-acting insulins detemir and degludec, and we would like to add some relevant information.
With regard to detemir, a meta-analysis published in 2009 showed that patients treated with this insulin had a lower or similar rate of occurrence of a cancer compared with patients treated with neutral protamine Hagedorn insulin or insulin glargine.2 In addition, in a cohort study, no difference in cancer risk between insulin detemir users and nonusers was reported.3
Insulin detemir has a lower binding affinity for human insulin receptor isoform A (IR-A) relative to human insulin, and a much lower affinity for isoform B (IR-B). The binding affinity ratio of insulinlike growth factor-1 (IGF-1) receptor to insulin receptor for detemir is less than or equal to 1 relative to human insulin and displays a dissociation pattern from the insulin receptor that is similar to or faster than that of human insulin. Consequently, the relative mitogenic potency of detemir in cell types predominantly expressing either the IGF-1 receptor or the insulin receptor is low and corresponds to its IGF-1 receptor and insulin receptor affinities.4
Regarding insulin degludec, its affinity for both IR-A and IR-B, as well as for the IGF-1 receptor, has been found to be lower than human insulin. Its mitogenic response, in the absence of albumin, was reported to range from 4% to 14% relative to human insulin.5 Furthermore, in cellular assays, in which no albumin was added, the in vitro metabolic potency was determined to be in the range of 8% to 20%, resulting in a mitogenic-to-metabolic potency ratio of 1 or lower.5
It appears that insulins detemir and degludec have low mitogenic potential. However, additional studies are needed, especially with degludec, to further determine long-term safety.
- Ching Sun GE, Kashyap SR, Nasr C. Diabetes therapy and cancer risk: where do we stand when treating patients? Cleve Clin J Med 2014; 81:620–628.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Hansen BF, Glendorf T, Hegelund AC, et al. Molecular characterization of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X10. PLoS One 2012; 7:e34274.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
To the Editor: We read with interest the article by Ching Sun et al1 on the relationship between diabetes therapy and cancer risk. We noted that there was no reference in the text to the long-acting insulins detemir and degludec, and we would like to add some relevant information.
With regard to detemir, a meta-analysis published in 2009 showed that patients treated with this insulin had a lower or similar rate of occurrence of a cancer compared with patients treated with neutral protamine Hagedorn insulin or insulin glargine.2 In addition, in a cohort study, no difference in cancer risk between insulin detemir users and nonusers was reported.3
Insulin detemir has a lower binding affinity for human insulin receptor isoform A (IR-A) relative to human insulin, and a much lower affinity for isoform B (IR-B). The binding affinity ratio of insulinlike growth factor-1 (IGF-1) receptor to insulin receptor for detemir is less than or equal to 1 relative to human insulin and displays a dissociation pattern from the insulin receptor that is similar to or faster than that of human insulin. Consequently, the relative mitogenic potency of detemir in cell types predominantly expressing either the IGF-1 receptor or the insulin receptor is low and corresponds to its IGF-1 receptor and insulin receptor affinities.4
Regarding insulin degludec, its affinity for both IR-A and IR-B, as well as for the IGF-1 receptor, has been found to be lower than human insulin. Its mitogenic response, in the absence of albumin, was reported to range from 4% to 14% relative to human insulin.5 Furthermore, in cellular assays, in which no albumin was added, the in vitro metabolic potency was determined to be in the range of 8% to 20%, resulting in a mitogenic-to-metabolic potency ratio of 1 or lower.5
It appears that insulins detemir and degludec have low mitogenic potential. However, additional studies are needed, especially with degludec, to further determine long-term safety.
To the Editor: We read with interest the article by Ching Sun et al1 on the relationship between diabetes therapy and cancer risk. We noted that there was no reference in the text to the long-acting insulins detemir and degludec, and we would like to add some relevant information.
With regard to detemir, a meta-analysis published in 2009 showed that patients treated with this insulin had a lower or similar rate of occurrence of a cancer compared with patients treated with neutral protamine Hagedorn insulin or insulin glargine.2 In addition, in a cohort study, no difference in cancer risk between insulin detemir users and nonusers was reported.3
Insulin detemir has a lower binding affinity for human insulin receptor isoform A (IR-A) relative to human insulin, and a much lower affinity for isoform B (IR-B). The binding affinity ratio of insulinlike growth factor-1 (IGF-1) receptor to insulin receptor for detemir is less than or equal to 1 relative to human insulin and displays a dissociation pattern from the insulin receptor that is similar to or faster than that of human insulin. Consequently, the relative mitogenic potency of detemir in cell types predominantly expressing either the IGF-1 receptor or the insulin receptor is low and corresponds to its IGF-1 receptor and insulin receptor affinities.4
Regarding insulin degludec, its affinity for both IR-A and IR-B, as well as for the IGF-1 receptor, has been found to be lower than human insulin. Its mitogenic response, in the absence of albumin, was reported to range from 4% to 14% relative to human insulin.5 Furthermore, in cellular assays, in which no albumin was added, the in vitro metabolic potency was determined to be in the range of 8% to 20%, resulting in a mitogenic-to-metabolic potency ratio of 1 or lower.5
It appears that insulins detemir and degludec have low mitogenic potential. However, additional studies are needed, especially with degludec, to further determine long-term safety.
- Ching Sun GE, Kashyap SR, Nasr C. Diabetes therapy and cancer risk: where do we stand when treating patients? Cleve Clin J Med 2014; 81:620–628.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Hansen BF, Glendorf T, Hegelund AC, et al. Molecular characterization of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X10. PLoS One 2012; 7:e34274.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Ching Sun GE, Kashyap SR, Nasr C. Diabetes therapy and cancer risk: where do we stand when treating patients? Cleve Clin J Med 2014; 81:620–628.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Hansen BF, Glendorf T, Hegelund AC, et al. Molecular characterization of long-acting insulin analogues in comparison with human insulin, IGF-1 and insulin X10. PLoS One 2012; 7:e34274.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
In reply: Insulin therapy and cancer risk
In Reply: Dr. Fountas et al highlight further data on insulin therapy and cancer risk, specifically in regard to insulin detemir and insulin degludec. Detemir first gained US Food and Drug Administration (FDA) approval in 2005 as a basal insulin, dosed once or twice daily.1 Compared with regular human insulin, detemir has demonstrated proliferative and antiapoptotic activities in vitro in various cancer cell lines—eg, HCT-116 (colorectal cancer), PC-3 (prostate cancer), and MCF-7 (breast adenocarcinoma).2 But clinically, detemir has not demonstrated increased cancer risk compared with other basal insulins in randomized controlled trials or cohort studies.3–5
Degludec (U-200 insulin) is equal to twice the concentration of the usual U-100 insulin therapies presently available. In February 2013, the drug application for insulin degludec failed to obtain FDA approval, and the FDA requested additional data on cardiovascular safety. Thus, degludec is not currently available in the United States.6
Besides ameliorating nocturnal hypoglycemia,7 U-200 insulin may mitigate potential mitogenic effects.8 However, there are still very few data on degludec compared with the amount of data on insulin glargine. Insulin analogues with a decreased dissociation rate from the insulin receptor are associated with higher mitogenic potency than metabolic potency compared with human insulin.9,10 Degludec, like detemir, has an elevated dissociation rate from the insulin receptor, a low affinity for IGF-1 receptors, and a low mitogenic activity in vitro.8
At this juncture, neither detemir nor degludec has been associated with higher cancer risk, but these therapies are relatively new. And as Dr. Fountas et al indicated, their safety, particularly in regard to cancer risk in diabetes patients, should continue to be assessed.
- Levemir [package insert]. Plainsboro, NJ: Novo Nordisk Inc; 2013.
- Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 2009; 25:41–49.
- Simó R, Plana-Ripoll O, Puente D, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8:e79968.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Novo Nordisk. 2013. Novo Nordisk receives Complete Response Letter in the US for Tresiba® and Ryzodeg®. [Press release]. www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=83700060-0ce3-4577-a35a-f3e57801637d. Accessed December 1, 2014.
- Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379:1489–1497.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Hansen BF, Danielsen GM, Drejer K, et al. Sustained signaling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J 1996; 315:271–279.
- Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49:999–1005.
In Reply: Dr. Fountas et al highlight further data on insulin therapy and cancer risk, specifically in regard to insulin detemir and insulin degludec. Detemir first gained US Food and Drug Administration (FDA) approval in 2005 as a basal insulin, dosed once or twice daily.1 Compared with regular human insulin, detemir has demonstrated proliferative and antiapoptotic activities in vitro in various cancer cell lines—eg, HCT-116 (colorectal cancer), PC-3 (prostate cancer), and MCF-7 (breast adenocarcinoma).2 But clinically, detemir has not demonstrated increased cancer risk compared with other basal insulins in randomized controlled trials or cohort studies.3–5
Degludec (U-200 insulin) is equal to twice the concentration of the usual U-100 insulin therapies presently available. In February 2013, the drug application for insulin degludec failed to obtain FDA approval, and the FDA requested additional data on cardiovascular safety. Thus, degludec is not currently available in the United States.6
Besides ameliorating nocturnal hypoglycemia,7 U-200 insulin may mitigate potential mitogenic effects.8 However, there are still very few data on degludec compared with the amount of data on insulin glargine. Insulin analogues with a decreased dissociation rate from the insulin receptor are associated with higher mitogenic potency than metabolic potency compared with human insulin.9,10 Degludec, like detemir, has an elevated dissociation rate from the insulin receptor, a low affinity for IGF-1 receptors, and a low mitogenic activity in vitro.8
At this juncture, neither detemir nor degludec has been associated with higher cancer risk, but these therapies are relatively new. And as Dr. Fountas et al indicated, their safety, particularly in regard to cancer risk in diabetes patients, should continue to be assessed.
In Reply: Dr. Fountas et al highlight further data on insulin therapy and cancer risk, specifically in regard to insulin detemir and insulin degludec. Detemir first gained US Food and Drug Administration (FDA) approval in 2005 as a basal insulin, dosed once or twice daily.1 Compared with regular human insulin, detemir has demonstrated proliferative and antiapoptotic activities in vitro in various cancer cell lines—eg, HCT-116 (colorectal cancer), PC-3 (prostate cancer), and MCF-7 (breast adenocarcinoma).2 But clinically, detemir has not demonstrated increased cancer risk compared with other basal insulins in randomized controlled trials or cohort studies.3–5
Degludec (U-200 insulin) is equal to twice the concentration of the usual U-100 insulin therapies presently available. In February 2013, the drug application for insulin degludec failed to obtain FDA approval, and the FDA requested additional data on cardiovascular safety. Thus, degludec is not currently available in the United States.6
Besides ameliorating nocturnal hypoglycemia,7 U-200 insulin may mitigate potential mitogenic effects.8 However, there are still very few data on degludec compared with the amount of data on insulin glargine. Insulin analogues with a decreased dissociation rate from the insulin receptor are associated with higher mitogenic potency than metabolic potency compared with human insulin.9,10 Degludec, like detemir, has an elevated dissociation rate from the insulin receptor, a low affinity for IGF-1 receptors, and a low mitogenic activity in vitro.8
At this juncture, neither detemir nor degludec has been associated with higher cancer risk, but these therapies are relatively new. And as Dr. Fountas et al indicated, their safety, particularly in regard to cancer risk in diabetes patients, should continue to be assessed.
- Levemir [package insert]. Plainsboro, NJ: Novo Nordisk Inc; 2013.
- Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 2009; 25:41–49.
- Simó R, Plana-Ripoll O, Puente D, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8:e79968.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Novo Nordisk. 2013. Novo Nordisk receives Complete Response Letter in the US for Tresiba® and Ryzodeg®. [Press release]. www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=83700060-0ce3-4577-a35a-f3e57801637d. Accessed December 1, 2014.
- Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379:1489–1497.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Hansen BF, Danielsen GM, Drejer K, et al. Sustained signaling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J 1996; 315:271–279.
- Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49:999–1005.
- Levemir [package insert]. Plainsboro, NJ: Novo Nordisk Inc; 2013.
- Weinstein D, Simon M, Yehezkel E, Laron Z, Werner H. Insulin analogues display IGF-I-like mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab Res Rev 2009; 25:41–49.
- Simó R, Plana-Ripoll O, Puente D, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona case-control study. PLoS One. 2013; 8:e79968.
- Fagot JP, Blotière PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins? A French nationwide cohort study based on national administrative databases. Diabetes Care 2013; 36:294–301.
- Dejgaard A, Lynggaard H, Råstam J, Krogsgaard Thomsen M. No evidence of increased risk of malignancies in patients with diabetes treated with insulin detemir: a meta-analysis. Diabetologia 2009; 52:2507–2512.
- Novo Nordisk. 2013. Novo Nordisk receives Complete Response Letter in the US for Tresiba® and Ryzodeg®. [Press release]. www.novonordisk.com/include/asp/exe_news_attachment.asp?sAttachmentGUID=83700060-0ce3-4577-a35a-f3e57801637d. Accessed December 1, 2014.
- Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379:1489–1497.
- Nishimura E, Sørensen AR, Hansen BF, et al. Insulin degludec: a new ultra-long, basal insulin designed to maintain full metabolic effect while minimizing mitogenic potential. Diabetologia 2010; 53:S388–S389.
- Hansen BF, Danielsen GM, Drejer K, et al. Sustained signaling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochem J 1996; 315:271–279.
- Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes 2000; 49:999–1005.
Management of Plasma Cell Disorders
The plasma cell disorders are a spectrum of conditions that include asymptomatic precursor conditions—monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)—as well as symptomatic multiple myeloma (MM) and solitary plasmacytoma. Other plasma cell disorders include immunoglobulin light chain amyloidosis and POEMS syndrome, which are characterized by a unique set of end-organ manifestations. There are other related plasma cell and B-cell proliferations, such as light chain deposition disease and cryoglobulinemia, that are beyond the scope of this review but are relevant to the hematologist/oncologist and have been reviewed in detail elsewhere.
To read the full article in PDF:
The plasma cell disorders are a spectrum of conditions that include asymptomatic precursor conditions—monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)—as well as symptomatic multiple myeloma (MM) and solitary plasmacytoma. Other plasma cell disorders include immunoglobulin light chain amyloidosis and POEMS syndrome, which are characterized by a unique set of end-organ manifestations. There are other related plasma cell and B-cell proliferations, such as light chain deposition disease and cryoglobulinemia, that are beyond the scope of this review but are relevant to the hematologist/oncologist and have been reviewed in detail elsewhere.
To read the full article in PDF:
The plasma cell disorders are a spectrum of conditions that include asymptomatic precursor conditions—monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)—as well as symptomatic multiple myeloma (MM) and solitary plasmacytoma. Other plasma cell disorders include immunoglobulin light chain amyloidosis and POEMS syndrome, which are characterized by a unique set of end-organ manifestations. There are other related plasma cell and B-cell proliferations, such as light chain deposition disease and cryoglobulinemia, that are beyond the scope of this review but are relevant to the hematologist/oncologist and have been reviewed in detail elsewhere.
To read the full article in PDF: