User login
Helping patients with insomnia to help themselves
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
A patient handout from the health services department of the University of California, Berkley titled “Insomnia Self-Care Guide” teaches patients how their behaviors might be contributing to their sleep difficulties. In addition to providing general information about sleep, the handout, which is available at http://www.uhs.berkeley.edu/home/healthtopics/PDF%20Handouts/Insomnia.pdf, also includes a checklist of questions about behaviors tied to insomnia, such as “Do you drink alcohol in the evenings?” and suggestions for inducing sleep, such as exercising daily.
Teaching patients about Hepatitis C
The Liver Foundation offers a brief educational guide that teaches patients about the hepatitis C virus. The brochure, which is available at http://www.liverfoundation.org/downloads/alf_download_24.pdf, uses a Q&A format to explain what hepatitis C is, how it is spread, who is at risk, and how it is diagnosed and treated.
The Liver Foundation offers a brief educational guide that teaches patients about the hepatitis C virus. The brochure, which is available at http://www.liverfoundation.org/downloads/alf_download_24.pdf, uses a Q&A format to explain what hepatitis C is, how it is spread, who is at risk, and how it is diagnosed and treated.
The Liver Foundation offers a brief educational guide that teaches patients about the hepatitis C virus. The brochure, which is available at http://www.liverfoundation.org/downloads/alf_download_24.pdf, uses a Q&A format to explain what hepatitis C is, how it is spread, who is at risk, and how it is diagnosed and treated.
Umbilical hernia in a patient with cirrhosis
Ascites is the major predisposing factor, since it causes muscle wasting and increases intra-abdominal pressure.
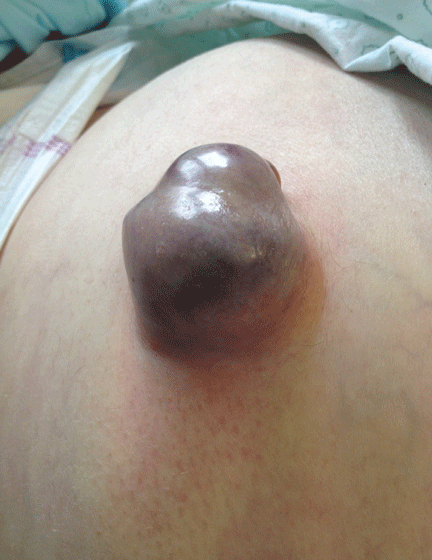
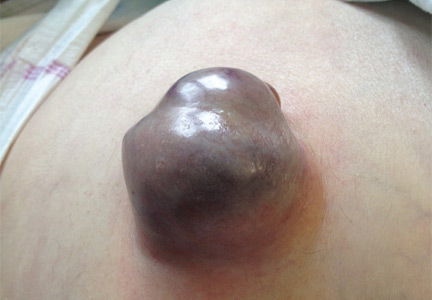
A 62-YEAR-OLD MAN was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
Ascites is the major predisposing factor, since it causes muscle wasting and increases intra-abdominal pressure.
Ascites is the major predisposing factor, since it causes muscle wasting and increases intra-abdominal pressure.
A 62-YEAR-OLD MAN was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
A 62-YEAR-OLD MAN was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
Online recommendations provide constantly updated HCV management guidelines
The newest guidelines for testing, managing, and treating hepatitis C infections in adults are part of a “living document” – a constantly updated online resource that reflects the ever-changing world of hepatitis research.
A joint venture of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), the document puts cutting-edge science in the hands of clinicians, Dr. Gary L. Davis wrote (Hepatology 2015 June 25 [doi:10.1002/hep.27950]). The continuously updated version may be accessed at any time at www.hcvguidelines.org.
“The pace of hepatitis C virus (HCV) drug development in recent years has accelerated dramatically,” wrote Dr. Davis, cochair for the AASLD/IDSA HCV Guidance writing group. “Such information and advice can be difficult to access readily given the diverse sources from which information is available, and the sometimes lengthy time needed for publication of original articles and scholarly perspectives. Traditional practice guidelines for more established areas of medicine and care often take years to develop and bring to publication. In the new era in hepatitis C treatment, such a process would not be nimble or timely enough to address the needs of patients with HCV infection, practitioners caring for these patients, or payers approving therapies for use.”
The online guidelines “will undergo real-time revisions as the field evolves,” Dr. Davis noted. A panel of 26 hepatologists and infectious diseases specialists and a patient advocate developed the original consensus recommendations.
The new update contains recommendations for direct antiviral drug regimens in treatment-naive patients and for all six HCV genotypes. A second section examines the recommended regimens for patients who have failed treatment with PEG-interferon and ribavirin, with or without a direct antiviral agent.
The document also gives guidance for managing patients with and without a sustained viral response and concludes with a section on treating special patient populations (decompensated cirrhosis, post-transplant HCV infections, renal impairment, and coinfection with HIV).
On Twitter @Alz_Gal
The newest guidelines for testing, managing, and treating hepatitis C infections in adults are part of a “living document” – a constantly updated online resource that reflects the ever-changing world of hepatitis research.
A joint venture of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), the document puts cutting-edge science in the hands of clinicians, Dr. Gary L. Davis wrote (Hepatology 2015 June 25 [doi:10.1002/hep.27950]). The continuously updated version may be accessed at any time at www.hcvguidelines.org.
“The pace of hepatitis C virus (HCV) drug development in recent years has accelerated dramatically,” wrote Dr. Davis, cochair for the AASLD/IDSA HCV Guidance writing group. “Such information and advice can be difficult to access readily given the diverse sources from which information is available, and the sometimes lengthy time needed for publication of original articles and scholarly perspectives. Traditional practice guidelines for more established areas of medicine and care often take years to develop and bring to publication. In the new era in hepatitis C treatment, such a process would not be nimble or timely enough to address the needs of patients with HCV infection, practitioners caring for these patients, or payers approving therapies for use.”
The online guidelines “will undergo real-time revisions as the field evolves,” Dr. Davis noted. A panel of 26 hepatologists and infectious diseases specialists and a patient advocate developed the original consensus recommendations.
The new update contains recommendations for direct antiviral drug regimens in treatment-naive patients and for all six HCV genotypes. A second section examines the recommended regimens for patients who have failed treatment with PEG-interferon and ribavirin, with or without a direct antiviral agent.
The document also gives guidance for managing patients with and without a sustained viral response and concludes with a section on treating special patient populations (decompensated cirrhosis, post-transplant HCV infections, renal impairment, and coinfection with HIV).
On Twitter @Alz_Gal
The newest guidelines for testing, managing, and treating hepatitis C infections in adults are part of a “living document” – a constantly updated online resource that reflects the ever-changing world of hepatitis research.
A joint venture of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), the document puts cutting-edge science in the hands of clinicians, Dr. Gary L. Davis wrote (Hepatology 2015 June 25 [doi:10.1002/hep.27950]). The continuously updated version may be accessed at any time at www.hcvguidelines.org.
“The pace of hepatitis C virus (HCV) drug development in recent years has accelerated dramatically,” wrote Dr. Davis, cochair for the AASLD/IDSA HCV Guidance writing group. “Such information and advice can be difficult to access readily given the diverse sources from which information is available, and the sometimes lengthy time needed for publication of original articles and scholarly perspectives. Traditional practice guidelines for more established areas of medicine and care often take years to develop and bring to publication. In the new era in hepatitis C treatment, such a process would not be nimble or timely enough to address the needs of patients with HCV infection, practitioners caring for these patients, or payers approving therapies for use.”
The online guidelines “will undergo real-time revisions as the field evolves,” Dr. Davis noted. A panel of 26 hepatologists and infectious diseases specialists and a patient advocate developed the original consensus recommendations.
The new update contains recommendations for direct antiviral drug regimens in treatment-naive patients and for all six HCV genotypes. A second section examines the recommended regimens for patients who have failed treatment with PEG-interferon and ribavirin, with or without a direct antiviral agent.
The document also gives guidance for managing patients with and without a sustained viral response and concludes with a section on treating special patient populations (decompensated cirrhosis, post-transplant HCV infections, renal impairment, and coinfection with HIV).
On Twitter @Alz_Gal
Stroke Centers More Common Where Laws Encourage Them
State laws have played a big part in boosting the number of hospitals where specialized stroke care is available, a new study shows.
During the study, the increase in the number of hospitals certified as primary stroke centers was more than twice as high in states with stroke legislation as in states without similar laws.
At these hospitals, a dedicated stroke-focused program staffed by professionals with special training delivers emergency therapy rapidly and reliably.
All hospitals should be able to see patients with stroke, but PSC certification attests to quality of care, said lead author Dr. Ken Uchino of the Cleveland Clinic.
"It takes money and effort to organize quality care," he told Reuters Health by email. "Sometimes a hospital is so small that the facility does not expect many patients with stroke to arrive. Sometimes the resources to provide quality care are not available, such as radiology technicians on call to run a CT scanner 24 hours a day or a specialist physician in the community."
U.S. organizations first began certifying stroke centers in 2003. Some states developed their own certification programs, and many passed laws requiring ambulance personnel to take an acute stroke patient directly to a certified center, bypassing hospitals that are not certified.
These laws seem to have encouraged more hospitals to get certification, according to a paper online now in the journal Stroke.
Between 2009 and 2013, states with stroke legislation had a 16% increase in PSC certification, compared to a 6% increase in states without similar legislation.
"I think if a hospital administrator realizes that an ambulance might bypass his or her hospital because it is not stroke-certified, there is an incentive to organize stroke care in the hospital and have stroke center certification," Uchino said.
By 2013, about a third of short-term adult general hospitals with emergency departments in the U.S. were certified as primary stroke centers, he said. But growth rates have varied by state, and by 2013 there were still three states with only one certified center, he said.
Out of 4,640 general hospitals with emergency rooms in the country, 1,505 have been certified as primary stroke centers following action by state legislatures. But the proportion of stroke centers by state still varied from as low as 4% in Wyoming, which has no stroke legislation, to 100% in Delaware, which does have stroke laws.
"Massachusetts, Florida, and New Jersey, which passed stroke legislation in 2004, had 74% to 97% of the hospitals certified as stroke centers by 2013," Uchino said.
Larger, more urban hospitals in states with higher economic output are most likely to be certified as primary stroke centers, the researchers found.
Patients brought to a certified stroke center have a better chance of survival than those brought elsewhere, Uchino said.
Almost all large hospitals can and should be stroke centers, and small hospitals still need help to improve, he said.
"Small hospitals still can become stroke centers, but they had to be creative with how they pulled resources together," said Dr. Lee H. Schwamm of Massachusetts General Hospital and Harvard Medical School in Boston.
"Every community should have at least one" primary stroke center, Schwamm, who was not part of the new study, told Reuters Health by phone. "The real challenge is how do I ensure equitable access for people all over the country."
State laws have played a big part in boosting the number of hospitals where specialized stroke care is available, a new study shows.
During the study, the increase in the number of hospitals certified as primary stroke centers was more than twice as high in states with stroke legislation as in states without similar laws.
At these hospitals, a dedicated stroke-focused program staffed by professionals with special training delivers emergency therapy rapidly and reliably.
All hospitals should be able to see patients with stroke, but PSC certification attests to quality of care, said lead author Dr. Ken Uchino of the Cleveland Clinic.
"It takes money and effort to organize quality care," he told Reuters Health by email. "Sometimes a hospital is so small that the facility does not expect many patients with stroke to arrive. Sometimes the resources to provide quality care are not available, such as radiology technicians on call to run a CT scanner 24 hours a day or a specialist physician in the community."
U.S. organizations first began certifying stroke centers in 2003. Some states developed their own certification programs, and many passed laws requiring ambulance personnel to take an acute stroke patient directly to a certified center, bypassing hospitals that are not certified.
These laws seem to have encouraged more hospitals to get certification, according to a paper online now in the journal Stroke.
Between 2009 and 2013, states with stroke legislation had a 16% increase in PSC certification, compared to a 6% increase in states without similar legislation.
"I think if a hospital administrator realizes that an ambulance might bypass his or her hospital because it is not stroke-certified, there is an incentive to organize stroke care in the hospital and have stroke center certification," Uchino said.
By 2013, about a third of short-term adult general hospitals with emergency departments in the U.S. were certified as primary stroke centers, he said. But growth rates have varied by state, and by 2013 there were still three states with only one certified center, he said.
Out of 4,640 general hospitals with emergency rooms in the country, 1,505 have been certified as primary stroke centers following action by state legislatures. But the proportion of stroke centers by state still varied from as low as 4% in Wyoming, which has no stroke legislation, to 100% in Delaware, which does have stroke laws.
"Massachusetts, Florida, and New Jersey, which passed stroke legislation in 2004, had 74% to 97% of the hospitals certified as stroke centers by 2013," Uchino said.
Larger, more urban hospitals in states with higher economic output are most likely to be certified as primary stroke centers, the researchers found.
Patients brought to a certified stroke center have a better chance of survival than those brought elsewhere, Uchino said.
Almost all large hospitals can and should be stroke centers, and small hospitals still need help to improve, he said.
"Small hospitals still can become stroke centers, but they had to be creative with how they pulled resources together," said Dr. Lee H. Schwamm of Massachusetts General Hospital and Harvard Medical School in Boston.
"Every community should have at least one" primary stroke center, Schwamm, who was not part of the new study, told Reuters Health by phone. "The real challenge is how do I ensure equitable access for people all over the country."
State laws have played a big part in boosting the number of hospitals where specialized stroke care is available, a new study shows.
During the study, the increase in the number of hospitals certified as primary stroke centers was more than twice as high in states with stroke legislation as in states without similar laws.
At these hospitals, a dedicated stroke-focused program staffed by professionals with special training delivers emergency therapy rapidly and reliably.
All hospitals should be able to see patients with stroke, but PSC certification attests to quality of care, said lead author Dr. Ken Uchino of the Cleveland Clinic.
"It takes money and effort to organize quality care," he told Reuters Health by email. "Sometimes a hospital is so small that the facility does not expect many patients with stroke to arrive. Sometimes the resources to provide quality care are not available, such as radiology technicians on call to run a CT scanner 24 hours a day or a specialist physician in the community."
U.S. organizations first began certifying stroke centers in 2003. Some states developed their own certification programs, and many passed laws requiring ambulance personnel to take an acute stroke patient directly to a certified center, bypassing hospitals that are not certified.
These laws seem to have encouraged more hospitals to get certification, according to a paper online now in the journal Stroke.
Between 2009 and 2013, states with stroke legislation had a 16% increase in PSC certification, compared to a 6% increase in states without similar legislation.
"I think if a hospital administrator realizes that an ambulance might bypass his or her hospital because it is not stroke-certified, there is an incentive to organize stroke care in the hospital and have stroke center certification," Uchino said.
By 2013, about a third of short-term adult general hospitals with emergency departments in the U.S. were certified as primary stroke centers, he said. But growth rates have varied by state, and by 2013 there were still three states with only one certified center, he said.
Out of 4,640 general hospitals with emergency rooms in the country, 1,505 have been certified as primary stroke centers following action by state legislatures. But the proportion of stroke centers by state still varied from as low as 4% in Wyoming, which has no stroke legislation, to 100% in Delaware, which does have stroke laws.
"Massachusetts, Florida, and New Jersey, which passed stroke legislation in 2004, had 74% to 97% of the hospitals certified as stroke centers by 2013," Uchino said.
Larger, more urban hospitals in states with higher economic output are most likely to be certified as primary stroke centers, the researchers found.
Patients brought to a certified stroke center have a better chance of survival than those brought elsewhere, Uchino said.
Almost all large hospitals can and should be stroke centers, and small hospitals still need help to improve, he said.
"Small hospitals still can become stroke centers, but they had to be creative with how they pulled resources together," said Dr. Lee H. Schwamm of Massachusetts General Hospital and Harvard Medical School in Boston.
"Every community should have at least one" primary stroke center, Schwamm, who was not part of the new study, told Reuters Health by phone. "The real challenge is how do I ensure equitable access for people all over the country."
Approach could build bridge to HSCT in T-PLL
![]()
Photo by Chad McNeeley
A treatment approach that combines epigenetics and immunotherapy could provide a bridge to transplant in patients with T-cell prolymphocytic leukemia (T-PLL), according to researchers.
In a pilot study, the team found that combining alemtuzumab with epigenetic agents could override alemtuzumab resistance in T-PLL.
The approach produced complete responses (CRs) in 7 of 8 patients. The eighth patient achieved a partial response (PR).
However, 5 of the patients ultimately died of persistent disease, and 2 are alive in early relapse. One patient remains in remission after allogeneic hematopoietic stem cell transplant (HSCT).
“There’s been a revolution in the last few years seeing success with immunotherapy, and people speculated that, perhaps, if you combined epigenetic and immunotherapy, that might be even more spectacular,” said Thomas P. Loughran Jr, MD, of the University of Virginia Cancer Center in Charlottesville. “This is proof of principle that this might be true.”
Dr Loughran and his colleagues described this research in Science Translational Medicine.
The researchers evaluated 8 patients with T-PLL, 4 of whom were previously untreated (patients 3, 4, 5, and 6).
Patient 2 presented with alemtuzumab-resistant disease but achieved a CR after receiving the drug along with cladribine and vorinostat. He achieved PRs with the same regimen after a second and third relapse but ultimately progressed and died.
“It was unbelievable, really, seeing a patient who had already failed [alemtuzumab] literally going back into remission,” Dr Loughran said.
Patients 1, 4, 6, and 8 achieved a CR with cladribine and alemtuzumab, and patient 3 achieved a PR on the same regimen.
Patient 7 received cladribine and alemtuzumab as well but only achieved a CR with the addition of valproic acid. Patient 5 achieved a CR with cladribine, alemtuzumab, and vorinostat.
Patients 3, 4, and 5 ultimately died. Patient 4 experienced a grade 5 intracranial hemorrhage that may have been treatment-related.
Patients 7 and 8 are in relapse, but they are receiving treatment and are under consideration for HSCT. Patient 6 went on to HSCT after achieving a CR and has been in remission for 4 years.
Major adverse events in this study include neutropenia (5 grade 4, 3 grade 3), thrombocytopenia (2 grade 4, 1 grade 3), anemia (1 grade 2, 2 grade 3), grade 3 febrile neutropenia (n=2), grade 2 lung infection (n=2), grade 1 CMV reactivation (n=1), grade 3 oral mucositis (n=1), grade 3 skin infection (n=1), and grade 5 intracranial hemorrhage (n=1). ![]()
![]()
Photo by Chad McNeeley
A treatment approach that combines epigenetics and immunotherapy could provide a bridge to transplant in patients with T-cell prolymphocytic leukemia (T-PLL), according to researchers.
In a pilot study, the team found that combining alemtuzumab with epigenetic agents could override alemtuzumab resistance in T-PLL.
The approach produced complete responses (CRs) in 7 of 8 patients. The eighth patient achieved a partial response (PR).
However, 5 of the patients ultimately died of persistent disease, and 2 are alive in early relapse. One patient remains in remission after allogeneic hematopoietic stem cell transplant (HSCT).
“There’s been a revolution in the last few years seeing success with immunotherapy, and people speculated that, perhaps, if you combined epigenetic and immunotherapy, that might be even more spectacular,” said Thomas P. Loughran Jr, MD, of the University of Virginia Cancer Center in Charlottesville. “This is proof of principle that this might be true.”
Dr Loughran and his colleagues described this research in Science Translational Medicine.
The researchers evaluated 8 patients with T-PLL, 4 of whom were previously untreated (patients 3, 4, 5, and 6).
Patient 2 presented with alemtuzumab-resistant disease but achieved a CR after receiving the drug along with cladribine and vorinostat. He achieved PRs with the same regimen after a second and third relapse but ultimately progressed and died.
“It was unbelievable, really, seeing a patient who had already failed [alemtuzumab] literally going back into remission,” Dr Loughran said.
Patients 1, 4, 6, and 8 achieved a CR with cladribine and alemtuzumab, and patient 3 achieved a PR on the same regimen.
Patient 7 received cladribine and alemtuzumab as well but only achieved a CR with the addition of valproic acid. Patient 5 achieved a CR with cladribine, alemtuzumab, and vorinostat.
Patients 3, 4, and 5 ultimately died. Patient 4 experienced a grade 5 intracranial hemorrhage that may have been treatment-related.
Patients 7 and 8 are in relapse, but they are receiving treatment and are under consideration for HSCT. Patient 6 went on to HSCT after achieving a CR and has been in remission for 4 years.
Major adverse events in this study include neutropenia (5 grade 4, 3 grade 3), thrombocytopenia (2 grade 4, 1 grade 3), anemia (1 grade 2, 2 grade 3), grade 3 febrile neutropenia (n=2), grade 2 lung infection (n=2), grade 1 CMV reactivation (n=1), grade 3 oral mucositis (n=1), grade 3 skin infection (n=1), and grade 5 intracranial hemorrhage (n=1). ![]()
![]()
Photo by Chad McNeeley
A treatment approach that combines epigenetics and immunotherapy could provide a bridge to transplant in patients with T-cell prolymphocytic leukemia (T-PLL), according to researchers.
In a pilot study, the team found that combining alemtuzumab with epigenetic agents could override alemtuzumab resistance in T-PLL.
The approach produced complete responses (CRs) in 7 of 8 patients. The eighth patient achieved a partial response (PR).
However, 5 of the patients ultimately died of persistent disease, and 2 are alive in early relapse. One patient remains in remission after allogeneic hematopoietic stem cell transplant (HSCT).
“There’s been a revolution in the last few years seeing success with immunotherapy, and people speculated that, perhaps, if you combined epigenetic and immunotherapy, that might be even more spectacular,” said Thomas P. Loughran Jr, MD, of the University of Virginia Cancer Center in Charlottesville. “This is proof of principle that this might be true.”
Dr Loughran and his colleagues described this research in Science Translational Medicine.
The researchers evaluated 8 patients with T-PLL, 4 of whom were previously untreated (patients 3, 4, 5, and 6).
Patient 2 presented with alemtuzumab-resistant disease but achieved a CR after receiving the drug along with cladribine and vorinostat. He achieved PRs with the same regimen after a second and third relapse but ultimately progressed and died.
“It was unbelievable, really, seeing a patient who had already failed [alemtuzumab] literally going back into remission,” Dr Loughran said.
Patients 1, 4, 6, and 8 achieved a CR with cladribine and alemtuzumab, and patient 3 achieved a PR on the same regimen.
Patient 7 received cladribine and alemtuzumab as well but only achieved a CR with the addition of valproic acid. Patient 5 achieved a CR with cladribine, alemtuzumab, and vorinostat.
Patients 3, 4, and 5 ultimately died. Patient 4 experienced a grade 5 intracranial hemorrhage that may have been treatment-related.
Patients 7 and 8 are in relapse, but they are receiving treatment and are under consideration for HSCT. Patient 6 went on to HSCT after achieving a CR and has been in remission for 4 years.
Major adverse events in this study include neutropenia (5 grade 4, 3 grade 3), thrombocytopenia (2 grade 4, 1 grade 3), anemia (1 grade 2, 2 grade 3), grade 3 febrile neutropenia (n=2), grade 2 lung infection (n=2), grade 1 CMV reactivation (n=1), grade 3 oral mucositis (n=1), grade 3 skin infection (n=1), and grade 5 intracranial hemorrhage (n=1). ![]()
Agent reduces fibrosis, improves platelet counts in MF

VIENNA—The immunotherapeutic agent PRM-151, when given alone or in combination with ruxolitinib, can reduce bone marrow fibrosis and improve platelet counts in patients with myelofibrosis (MF), results of a phase 2 trial suggest.
Using a novel assessment technique known as computer-assisted image analysis (CIA), researchers found that PRM-151, with or without ruxolitinib, prompted fibrosis responses in nearly three-quarters of patients studied.
And nearly 60% of thrombocytopenic patients saw improvements in their platelet counts.
“Thrombocytopenia remains a significant problem for many patients with myelofibrosis, and a new treatment that is not myelosuppressive and actually increases platelet counts would be of great benefit to patients,” said Srdan Verstovsek, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
He and his colleagues presented this research—the first part of a 2-stage study—at the 20th Congress of the European Hematology Association (abstract P677*). The trial is sponsored by Promedior, the company developing PRM-151.
Stage 1 of the study included 27 patients with a median age of 67 (range, 51-85). They had been diagnosed with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The patients received PRM-151 at 10 mg/kg IV dosed weekly (n=8) or monthly (n=7), or ruxolitinib plus PRM-151 at 10 mg/kg IV dosed weekly (n=6) or monthly (n=6). The patients who received ruxolitinib were already taking the drug, and doses varied. The mean duration of ruxolitinib treatment was 1.6 years (range, 0.6-3 years).
Patients were set to receive study treatment for 24 weeks but could continue beyond that if they experienced clinical improvement.
Response assessment: Using CIA
Hematopathologists who were blinded to the patient, treatment, and time point performed morphologic analysis on bone marrow specimens taken at baseline and at 12, 24, and 36 weeks (if available).
The pathologists also performed CIA on whole-slide scans from specimens taken at the same time points. The idea to use CIA came after morphologic analyses revealed some surprising findings.
“[The pathologists] were really struck by the fact that, in the patients who had reductions in fibrosis, there were other elements in the bone marrow that showed improvements,” said Beth Trehu, MD, Chief Medical Officer at Promedior. “There were trends toward normalization of the red cells and of the megakaryocytes.”
“They also remarked that, whereas the baseline samples were totally homogeneous—a grade 3 was a grade 3 throughout the bone marrow—after treatment, the samples became very heterogeneous. There were areas of grade 3, grade 2, grade 1, and 0, all in one sample.”
To solve this problem, the pathologists decided that a sample’s WHO grade would be defined as whatever grade was present in at least 50% of the sample. But the team still thought these grades weren’t accurately quantifying the effects of PRM-151.
So they turned to CIA, which allowed them to quantify the volume of collagen or reticulin fibers in the bone marrow.
Fibrosis and platelet responses
According to CIA, 73% of evaluable patients (19/26) experienced reductions in bone marrow fibrosis at any time during the trial.
Of the 23 evaluable patients who had grade 2 or 3 fibrosis at baseline, 11 patients had a reduction of 1 grade or more during the study period, according to morphologic analysis. Nine patients had a fibrosis response by morphology at the last time point they were assessed.
Reductions in fibrosis correlated with increased platelet counts in thrombocytopenic patients. Fifty-seven percent of thrombocytopenic patients (8/14) saw an improvement in platelet counts after treatment.
Results across treatment groups were as follows:
| Treatment group (n) | WHO fibrosis
response at any time |
CIA fibrosis
response at any time |
Platelet
improvement |
| PRM-151 QW (7) | 3 | 4 | 2 |
| PRM-151 Q4W (7) | 3 | 6 | 4 |
| PRM-151 QW + RUX (6) | 2 | 4 | 1 |
| PRM-151 Q4W + RUX (6) | 3 | 5 | 1 |
“The responses we saw with single-agent PRM-151, in particular, give us a lot of confidence that this drug is absolutely reversing fibrosis in the bone marrow,” Dr Trehu said. “And that is very nicely correlated with improvements in platelets, which are much harder to see in patients who are getting ruxolitinib because it’s a myelosuppressive agent.”
Dr Trehu added that these data suggest a monthly dose of PRM-151 is sufficient to treat MF patients, and there is a path forward for PRM-151 both alone and in combination with ruxolitinib.
Hemoglobin and spleen responses
The researchers also observed some improvements in hemoglobin levels and spleen size after treatment.
Of the 15 patients who had hemoglobin levels below 100 g/L at baseline, 3 met criteria for hemoglobin improvement. This included becoming transfusion independent, having a 50% reduction in the need for transfusion, or experiencing a 2 g/L increase in hemoglobin.
“Other patients had a nice trend toward hemoglobin improvement,” Dr Trehu noted.
Of the 20 patients with palpable spleen at baseline, reductions occurred in all but 1 patient. Four patients had a 50% or greater reduction in spleen size, but this response did not last beyond 12 weeks.
Adverse events
Forty-eight percent of patients (13/27) had at least 1 treatment-related adverse event (AE). Grade 1 AEs included diarrhea (n=3), fatigue (n=2), bruising at the infusion site (n=2), oral herpes (n=1), joint swelling (n=2), and headache (n=2). One patient had grade 2 oral herpes.
There were 5 serious AEs that were possibly treatment-related. Three events, from which patients recovered, were abdominal pain, sialadenitis, and pneumonia. The other 2 serious AEs were gastroenteritis and pneumonia, which resulted in death in an 85-year-old patient.
There were 2 deaths unrelated to treatment. One was due to pneumonia, and the other was a result of multi-organ failure and cardiac arrest. ![]()
*Information in the abstract differs from that presented at the meeting.
VIENNA—The immunotherapeutic agent PRM-151, when given alone or in combination with ruxolitinib, can reduce bone marrow fibrosis and improve platelet counts in patients with myelofibrosis (MF), results of a phase 2 trial suggest.
Using a novel assessment technique known as computer-assisted image analysis (CIA), researchers found that PRM-151, with or without ruxolitinib, prompted fibrosis responses in nearly three-quarters of patients studied.
And nearly 60% of thrombocytopenic patients saw improvements in their platelet counts.
“Thrombocytopenia remains a significant problem for many patients with myelofibrosis, and a new treatment that is not myelosuppressive and actually increases platelet counts would be of great benefit to patients,” said Srdan Verstovsek, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
He and his colleagues presented this research—the first part of a 2-stage study—at the 20th Congress of the European Hematology Association (abstract P677*). The trial is sponsored by Promedior, the company developing PRM-151.
Stage 1 of the study included 27 patients with a median age of 67 (range, 51-85). They had been diagnosed with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The patients received PRM-151 at 10 mg/kg IV dosed weekly (n=8) or monthly (n=7), or ruxolitinib plus PRM-151 at 10 mg/kg IV dosed weekly (n=6) or monthly (n=6). The patients who received ruxolitinib were already taking the drug, and doses varied. The mean duration of ruxolitinib treatment was 1.6 years (range, 0.6-3 years).
Patients were set to receive study treatment for 24 weeks but could continue beyond that if they experienced clinical improvement.
Response assessment: Using CIA
Hematopathologists who were blinded to the patient, treatment, and time point performed morphologic analysis on bone marrow specimens taken at baseline and at 12, 24, and 36 weeks (if available).
The pathologists also performed CIA on whole-slide scans from specimens taken at the same time points. The idea to use CIA came after morphologic analyses revealed some surprising findings.
“[The pathologists] were really struck by the fact that, in the patients who had reductions in fibrosis, there were other elements in the bone marrow that showed improvements,” said Beth Trehu, MD, Chief Medical Officer at Promedior. “There were trends toward normalization of the red cells and of the megakaryocytes.”
“They also remarked that, whereas the baseline samples were totally homogeneous—a grade 3 was a grade 3 throughout the bone marrow—after treatment, the samples became very heterogeneous. There were areas of grade 3, grade 2, grade 1, and 0, all in one sample.”
To solve this problem, the pathologists decided that a sample’s WHO grade would be defined as whatever grade was present in at least 50% of the sample. But the team still thought these grades weren’t accurately quantifying the effects of PRM-151.
So they turned to CIA, which allowed them to quantify the volume of collagen or reticulin fibers in the bone marrow.
Fibrosis and platelet responses
According to CIA, 73% of evaluable patients (19/26) experienced reductions in bone marrow fibrosis at any time during the trial.
Of the 23 evaluable patients who had grade 2 or 3 fibrosis at baseline, 11 patients had a reduction of 1 grade or more during the study period, according to morphologic analysis. Nine patients had a fibrosis response by morphology at the last time point they were assessed.
Reductions in fibrosis correlated with increased platelet counts in thrombocytopenic patients. Fifty-seven percent of thrombocytopenic patients (8/14) saw an improvement in platelet counts after treatment.
Results across treatment groups were as follows:
| Treatment group (n) | WHO fibrosis
response at any time |
CIA fibrosis
response at any time |
Platelet
improvement |
| PRM-151 QW (7) | 3 | 4 | 2 |
| PRM-151 Q4W (7) | 3 | 6 | 4 |
| PRM-151 QW + RUX (6) | 2 | 4 | 1 |
| PRM-151 Q4W + RUX (6) | 3 | 5 | 1 |
“The responses we saw with single-agent PRM-151, in particular, give us a lot of confidence that this drug is absolutely reversing fibrosis in the bone marrow,” Dr Trehu said. “And that is very nicely correlated with improvements in platelets, which are much harder to see in patients who are getting ruxolitinib because it’s a myelosuppressive agent.”
Dr Trehu added that these data suggest a monthly dose of PRM-151 is sufficient to treat MF patients, and there is a path forward for PRM-151 both alone and in combination with ruxolitinib.
Hemoglobin and spleen responses
The researchers also observed some improvements in hemoglobin levels and spleen size after treatment.
Of the 15 patients who had hemoglobin levels below 100 g/L at baseline, 3 met criteria for hemoglobin improvement. This included becoming transfusion independent, having a 50% reduction in the need for transfusion, or experiencing a 2 g/L increase in hemoglobin.
“Other patients had a nice trend toward hemoglobin improvement,” Dr Trehu noted.
Of the 20 patients with palpable spleen at baseline, reductions occurred in all but 1 patient. Four patients had a 50% or greater reduction in spleen size, but this response did not last beyond 12 weeks.
Adverse events
Forty-eight percent of patients (13/27) had at least 1 treatment-related adverse event (AE). Grade 1 AEs included diarrhea (n=3), fatigue (n=2), bruising at the infusion site (n=2), oral herpes (n=1), joint swelling (n=2), and headache (n=2). One patient had grade 2 oral herpes.
There were 5 serious AEs that were possibly treatment-related. Three events, from which patients recovered, were abdominal pain, sialadenitis, and pneumonia. The other 2 serious AEs were gastroenteritis and pneumonia, which resulted in death in an 85-year-old patient.
There were 2 deaths unrelated to treatment. One was due to pneumonia, and the other was a result of multi-organ failure and cardiac arrest. ![]()
*Information in the abstract differs from that presented at the meeting.
VIENNA—The immunotherapeutic agent PRM-151, when given alone or in combination with ruxolitinib, can reduce bone marrow fibrosis and improve platelet counts in patients with myelofibrosis (MF), results of a phase 2 trial suggest.
Using a novel assessment technique known as computer-assisted image analysis (CIA), researchers found that PRM-151, with or without ruxolitinib, prompted fibrosis responses in nearly three-quarters of patients studied.
And nearly 60% of thrombocytopenic patients saw improvements in their platelet counts.
“Thrombocytopenia remains a significant problem for many patients with myelofibrosis, and a new treatment that is not myelosuppressive and actually increases platelet counts would be of great benefit to patients,” said Srdan Verstovsek, MD, PhD, of the University of Texas MD Anderson Cancer Center in Houston.
He and his colleagues presented this research—the first part of a 2-stage study—at the 20th Congress of the European Hematology Association (abstract P677*). The trial is sponsored by Promedior, the company developing PRM-151.
Stage 1 of the study included 27 patients with a median age of 67 (range, 51-85). They had been diagnosed with primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The patients received PRM-151 at 10 mg/kg IV dosed weekly (n=8) or monthly (n=7), or ruxolitinib plus PRM-151 at 10 mg/kg IV dosed weekly (n=6) or monthly (n=6). The patients who received ruxolitinib were already taking the drug, and doses varied. The mean duration of ruxolitinib treatment was 1.6 years (range, 0.6-3 years).
Patients were set to receive study treatment for 24 weeks but could continue beyond that if they experienced clinical improvement.
Response assessment: Using CIA
Hematopathologists who were blinded to the patient, treatment, and time point performed morphologic analysis on bone marrow specimens taken at baseline and at 12, 24, and 36 weeks (if available).
The pathologists also performed CIA on whole-slide scans from specimens taken at the same time points. The idea to use CIA came after morphologic analyses revealed some surprising findings.
“[The pathologists] were really struck by the fact that, in the patients who had reductions in fibrosis, there were other elements in the bone marrow that showed improvements,” said Beth Trehu, MD, Chief Medical Officer at Promedior. “There were trends toward normalization of the red cells and of the megakaryocytes.”
“They also remarked that, whereas the baseline samples were totally homogeneous—a grade 3 was a grade 3 throughout the bone marrow—after treatment, the samples became very heterogeneous. There were areas of grade 3, grade 2, grade 1, and 0, all in one sample.”
To solve this problem, the pathologists decided that a sample’s WHO grade would be defined as whatever grade was present in at least 50% of the sample. But the team still thought these grades weren’t accurately quantifying the effects of PRM-151.
So they turned to CIA, which allowed them to quantify the volume of collagen or reticulin fibers in the bone marrow.
Fibrosis and platelet responses
According to CIA, 73% of evaluable patients (19/26) experienced reductions in bone marrow fibrosis at any time during the trial.
Of the 23 evaluable patients who had grade 2 or 3 fibrosis at baseline, 11 patients had a reduction of 1 grade or more during the study period, according to morphologic analysis. Nine patients had a fibrosis response by morphology at the last time point they were assessed.
Reductions in fibrosis correlated with increased platelet counts in thrombocytopenic patients. Fifty-seven percent of thrombocytopenic patients (8/14) saw an improvement in platelet counts after treatment.
Results across treatment groups were as follows:
| Treatment group (n) | WHO fibrosis
response at any time |
CIA fibrosis
response at any time |
Platelet
improvement |
| PRM-151 QW (7) | 3 | 4 | 2 |
| PRM-151 Q4W (7) | 3 | 6 | 4 |
| PRM-151 QW + RUX (6) | 2 | 4 | 1 |
| PRM-151 Q4W + RUX (6) | 3 | 5 | 1 |
“The responses we saw with single-agent PRM-151, in particular, give us a lot of confidence that this drug is absolutely reversing fibrosis in the bone marrow,” Dr Trehu said. “And that is very nicely correlated with improvements in platelets, which are much harder to see in patients who are getting ruxolitinib because it’s a myelosuppressive agent.”
Dr Trehu added that these data suggest a monthly dose of PRM-151 is sufficient to treat MF patients, and there is a path forward for PRM-151 both alone and in combination with ruxolitinib.
Hemoglobin and spleen responses
The researchers also observed some improvements in hemoglobin levels and spleen size after treatment.
Of the 15 patients who had hemoglobin levels below 100 g/L at baseline, 3 met criteria for hemoglobin improvement. This included becoming transfusion independent, having a 50% reduction in the need for transfusion, or experiencing a 2 g/L increase in hemoglobin.
“Other patients had a nice trend toward hemoglobin improvement,” Dr Trehu noted.
Of the 20 patients with palpable spleen at baseline, reductions occurred in all but 1 patient. Four patients had a 50% or greater reduction in spleen size, but this response did not last beyond 12 weeks.
Adverse events
Forty-eight percent of patients (13/27) had at least 1 treatment-related adverse event (AE). Grade 1 AEs included diarrhea (n=3), fatigue (n=2), bruising at the infusion site (n=2), oral herpes (n=1), joint swelling (n=2), and headache (n=2). One patient had grade 2 oral herpes.
There were 5 serious AEs that were possibly treatment-related. Three events, from which patients recovered, were abdominal pain, sialadenitis, and pneumonia. The other 2 serious AEs were gastroenteritis and pneumonia, which resulted in death in an 85-year-old patient.
There were 2 deaths unrelated to treatment. One was due to pneumonia, and the other was a result of multi-organ failure and cardiac arrest. ![]()
*Information in the abstract differs from that presented at the meeting.
Nanobody can help treat acquired TTP
Image by Erhabor Osaro
TORONTO—Adding the anti-von Willebrand factor nanobody caplacizumab to standard therapy can provide clinical benefits for patients with
acquired thrombotic thrombocytopenic purpura (TTP), according to research presented at the 2015 ISTH Congress.
In the phase 2 TITAN trial, patients who received caplacizumab and standard therapy had a significantly shorter time to platelet normalization than patients who received standard therapy and placebo.
Caplacizumab was also associated with a shorter time to troponin, creatinine, and lactate dehydrogenase (LDH) normalization, as well as a reduced need for plasma exchange (PE).
However, some patients relapsed after stopping caplacizumab, and the majority of these relapses could be linked to unresolved disease activity. The drug also increased the risk of mild-to-moderate bleeding compared to placebo.
Flora Peyvandi, MD, PhD, of the University of Milan in Italy, presented data from the TITAN trial at ISTH as abstract LB006. Information on using ADAMTS13 activity for treatment monitoring in the TITAN trial was also presented at the meeting, as abstract OR363.
TITAN was sponsored by Ablynx, the company developing caplacizumab.
The study enrolled 75 patients with acquired TTP from January 2011 to January 2014. Patients were randomized to receive caplacizumab (n=36) or placebo (n=39).
All patients also received the current standard of care, which is, primarily, multiple PEs. The protocol for the study was adapted in September 2013 to allow 1 day of PE prior to study enrollment.
Patients in the caplacizumab arm immediately received an intravenous bolus dose of 10 mg and then a 10 mg subcutaneous dose of the drug daily until 30 days had elapsed after the final PE. Patients in the control arm received placebo at the same time points.
Platelet responses and relapse
The study’s primary endpoint was the time to platelet normalization. And patients in the caplacizumab arm achieved platelet normalization at more than twice the rate of patients in the placebo arm (overall hazard rate ratio=2.2, P=0.005).
Among patients with no prior PE, the median time to platelet normalization was 3 days in caplacizumab-treated patients (n=34) and 4.9 days in controls (n=35). Among patients with 1 prior PE, the median time to platelet normalization was 2.4 days in the caplacizumab arm (n=2) and 4.3 days in the placebo arm (n=4).
In the caplacizumab arm, a higher proportion of patients achieved a complete remission, and fewer patients had exacerbations of TTP. The complete remission rates were 81% and 46%, respectively. And the exacerbation rates were 8% and 28%, respectively.
However, the rate of exacerbation and/or relapse at up to 1 month of follow-up was the same in both arms, at 28%.
Seven patients relapsed within 10 days of stopping caplacizumab. All of these patients had continuous low ADAMTS13 activity (<10%) during and near the end of treatment.
Cardiac, renal, and tissue responses
Additional analyses revealed that patients in the caplacizumab arm had a faster time to troponin normalization than those in the placebo arm. At baseline, 53% of caplacizumab-treated patients and 46% of controls had elevated troponin. The median time to normalization was 9 days and 27 days, respectively.
Patients in the caplacizumab arm also had a faster time to creatinine normalization than those in the placebo arm. At baseline, 31% of caplacizumab-treated patients and 41% of controls had elevated creatinine. The median time to normalization was 4 days and 6 days, respectively.
And patients in the caplacizumab arm had a faster time to LDH normalization than those in the placebo arm. At baseline, 91% of caplacizumab-treated patients and 87% of controls had elevated LDH. The median time to normalization was 3 days and 4 days, respectively.
However, the investigators said they could not rule out a possible effect of PE therapy on LDH, creatinine, and troponin levels.
Need for PE
Results showed that caplacizumab could reduce the need for PE, at least while patients continued to receive the drug.
Patients in the caplacizumab arm required fewer PEs during the daily PE treatment period and the overall treatment period. But this benefit was lost during the 1-month follow-up period, which is reflective of the increased number of relapses, according to the investigators.
During the daily PE treatment period, the mean number of PE days was 5.9 in the caplacizumab arm and 7.9 in the placebo arm. During the overall treatment period, it was 7.7 and 11.7 days, respectively. And during the overall treatment and follow-up period, it was 10.2 and 11.7 days, respectively.
Adverse events
The investigators said caplacizumab increased the risk of bleeding events, but these were readily managed.
Treatment-emergent adverse events (AEs) occurred in 97% of patients in the caplacizumab arm and 100% of patients in the placebo arm. Eight percent of caplacizumab-treated patients discontinued treatment due to AEs, but none of the controls did.
Fifty-four percent of patients in the caplacizumab arm had a bleeding event, as did 38% of patients in the placebo arm. Eighty percent of the bleeding events in the caplacizumab arm were mild. Three patients required drug treatment. None required von Willebrand factor or factor VIII substitution.
Serious AEs occurred in 57% of caplacizumab-treated patients and 51% of controls. Serious bleeding events occurred in 6% and 5%, respectively. ![]()
Image by Erhabor Osaro
TORONTO—Adding the anti-von Willebrand factor nanobody caplacizumab to standard therapy can provide clinical benefits for patients with
acquired thrombotic thrombocytopenic purpura (TTP), according to research presented at the 2015 ISTH Congress.
In the phase 2 TITAN trial, patients who received caplacizumab and standard therapy had a significantly shorter time to platelet normalization than patients who received standard therapy and placebo.
Caplacizumab was also associated with a shorter time to troponin, creatinine, and lactate dehydrogenase (LDH) normalization, as well as a reduced need for plasma exchange (PE).
However, some patients relapsed after stopping caplacizumab, and the majority of these relapses could be linked to unresolved disease activity. The drug also increased the risk of mild-to-moderate bleeding compared to placebo.
Flora Peyvandi, MD, PhD, of the University of Milan in Italy, presented data from the TITAN trial at ISTH as abstract LB006. Information on using ADAMTS13 activity for treatment monitoring in the TITAN trial was also presented at the meeting, as abstract OR363.
TITAN was sponsored by Ablynx, the company developing caplacizumab.
The study enrolled 75 patients with acquired TTP from January 2011 to January 2014. Patients were randomized to receive caplacizumab (n=36) or placebo (n=39).
All patients also received the current standard of care, which is, primarily, multiple PEs. The protocol for the study was adapted in September 2013 to allow 1 day of PE prior to study enrollment.
Patients in the caplacizumab arm immediately received an intravenous bolus dose of 10 mg and then a 10 mg subcutaneous dose of the drug daily until 30 days had elapsed after the final PE. Patients in the control arm received placebo at the same time points.
Platelet responses and relapse
The study’s primary endpoint was the time to platelet normalization. And patients in the caplacizumab arm achieved platelet normalization at more than twice the rate of patients in the placebo arm (overall hazard rate ratio=2.2, P=0.005).
Among patients with no prior PE, the median time to platelet normalization was 3 days in caplacizumab-treated patients (n=34) and 4.9 days in controls (n=35). Among patients with 1 prior PE, the median time to platelet normalization was 2.4 days in the caplacizumab arm (n=2) and 4.3 days in the placebo arm (n=4).
In the caplacizumab arm, a higher proportion of patients achieved a complete remission, and fewer patients had exacerbations of TTP. The complete remission rates were 81% and 46%, respectively. And the exacerbation rates were 8% and 28%, respectively.
However, the rate of exacerbation and/or relapse at up to 1 month of follow-up was the same in both arms, at 28%.
Seven patients relapsed within 10 days of stopping caplacizumab. All of these patients had continuous low ADAMTS13 activity (<10%) during and near the end of treatment.
Cardiac, renal, and tissue responses
Additional analyses revealed that patients in the caplacizumab arm had a faster time to troponin normalization than those in the placebo arm. At baseline, 53% of caplacizumab-treated patients and 46% of controls had elevated troponin. The median time to normalization was 9 days and 27 days, respectively.
Patients in the caplacizumab arm also had a faster time to creatinine normalization than those in the placebo arm. At baseline, 31% of caplacizumab-treated patients and 41% of controls had elevated creatinine. The median time to normalization was 4 days and 6 days, respectively.
And patients in the caplacizumab arm had a faster time to LDH normalization than those in the placebo arm. At baseline, 91% of caplacizumab-treated patients and 87% of controls had elevated LDH. The median time to normalization was 3 days and 4 days, respectively.
However, the investigators said they could not rule out a possible effect of PE therapy on LDH, creatinine, and troponin levels.
Need for PE
Results showed that caplacizumab could reduce the need for PE, at least while patients continued to receive the drug.
Patients in the caplacizumab arm required fewer PEs during the daily PE treatment period and the overall treatment period. But this benefit was lost during the 1-month follow-up period, which is reflective of the increased number of relapses, according to the investigators.
During the daily PE treatment period, the mean number of PE days was 5.9 in the caplacizumab arm and 7.9 in the placebo arm. During the overall treatment period, it was 7.7 and 11.7 days, respectively. And during the overall treatment and follow-up period, it was 10.2 and 11.7 days, respectively.
Adverse events
The investigators said caplacizumab increased the risk of bleeding events, but these were readily managed.
Treatment-emergent adverse events (AEs) occurred in 97% of patients in the caplacizumab arm and 100% of patients in the placebo arm. Eight percent of caplacizumab-treated patients discontinued treatment due to AEs, but none of the controls did.
Fifty-four percent of patients in the caplacizumab arm had a bleeding event, as did 38% of patients in the placebo arm. Eighty percent of the bleeding events in the caplacizumab arm were mild. Three patients required drug treatment. None required von Willebrand factor or factor VIII substitution.
Serious AEs occurred in 57% of caplacizumab-treated patients and 51% of controls. Serious bleeding events occurred in 6% and 5%, respectively. ![]()
Image by Erhabor Osaro
TORONTO—Adding the anti-von Willebrand factor nanobody caplacizumab to standard therapy can provide clinical benefits for patients with
acquired thrombotic thrombocytopenic purpura (TTP), according to research presented at the 2015 ISTH Congress.
In the phase 2 TITAN trial, patients who received caplacizumab and standard therapy had a significantly shorter time to platelet normalization than patients who received standard therapy and placebo.
Caplacizumab was also associated with a shorter time to troponin, creatinine, and lactate dehydrogenase (LDH) normalization, as well as a reduced need for plasma exchange (PE).
However, some patients relapsed after stopping caplacizumab, and the majority of these relapses could be linked to unresolved disease activity. The drug also increased the risk of mild-to-moderate bleeding compared to placebo.
Flora Peyvandi, MD, PhD, of the University of Milan in Italy, presented data from the TITAN trial at ISTH as abstract LB006. Information on using ADAMTS13 activity for treatment monitoring in the TITAN trial was also presented at the meeting, as abstract OR363.
TITAN was sponsored by Ablynx, the company developing caplacizumab.
The study enrolled 75 patients with acquired TTP from January 2011 to January 2014. Patients were randomized to receive caplacizumab (n=36) or placebo (n=39).
All patients also received the current standard of care, which is, primarily, multiple PEs. The protocol for the study was adapted in September 2013 to allow 1 day of PE prior to study enrollment.
Patients in the caplacizumab arm immediately received an intravenous bolus dose of 10 mg and then a 10 mg subcutaneous dose of the drug daily until 30 days had elapsed after the final PE. Patients in the control arm received placebo at the same time points.
Platelet responses and relapse
The study’s primary endpoint was the time to platelet normalization. And patients in the caplacizumab arm achieved platelet normalization at more than twice the rate of patients in the placebo arm (overall hazard rate ratio=2.2, P=0.005).
Among patients with no prior PE, the median time to platelet normalization was 3 days in caplacizumab-treated patients (n=34) and 4.9 days in controls (n=35). Among patients with 1 prior PE, the median time to platelet normalization was 2.4 days in the caplacizumab arm (n=2) and 4.3 days in the placebo arm (n=4).
In the caplacizumab arm, a higher proportion of patients achieved a complete remission, and fewer patients had exacerbations of TTP. The complete remission rates were 81% and 46%, respectively. And the exacerbation rates were 8% and 28%, respectively.
However, the rate of exacerbation and/or relapse at up to 1 month of follow-up was the same in both arms, at 28%.
Seven patients relapsed within 10 days of stopping caplacizumab. All of these patients had continuous low ADAMTS13 activity (<10%) during and near the end of treatment.
Cardiac, renal, and tissue responses
Additional analyses revealed that patients in the caplacizumab arm had a faster time to troponin normalization than those in the placebo arm. At baseline, 53% of caplacizumab-treated patients and 46% of controls had elevated troponin. The median time to normalization was 9 days and 27 days, respectively.
Patients in the caplacizumab arm also had a faster time to creatinine normalization than those in the placebo arm. At baseline, 31% of caplacizumab-treated patients and 41% of controls had elevated creatinine. The median time to normalization was 4 days and 6 days, respectively.
And patients in the caplacizumab arm had a faster time to LDH normalization than those in the placebo arm. At baseline, 91% of caplacizumab-treated patients and 87% of controls had elevated LDH. The median time to normalization was 3 days and 4 days, respectively.
However, the investigators said they could not rule out a possible effect of PE therapy on LDH, creatinine, and troponin levels.
Need for PE
Results showed that caplacizumab could reduce the need for PE, at least while patients continued to receive the drug.
Patients in the caplacizumab arm required fewer PEs during the daily PE treatment period and the overall treatment period. But this benefit was lost during the 1-month follow-up period, which is reflective of the increased number of relapses, according to the investigators.
During the daily PE treatment period, the mean number of PE days was 5.9 in the caplacizumab arm and 7.9 in the placebo arm. During the overall treatment period, it was 7.7 and 11.7 days, respectively. And during the overall treatment and follow-up period, it was 10.2 and 11.7 days, respectively.
Adverse events
The investigators said caplacizumab increased the risk of bleeding events, but these were readily managed.
Treatment-emergent adverse events (AEs) occurred in 97% of patients in the caplacizumab arm and 100% of patients in the placebo arm. Eight percent of caplacizumab-treated patients discontinued treatment due to AEs, but none of the controls did.
Fifty-four percent of patients in the caplacizumab arm had a bleeding event, as did 38% of patients in the placebo arm. Eighty percent of the bleeding events in the caplacizumab arm were mild. Three patients required drug treatment. None required von Willebrand factor or factor VIII substitution.
Serious AEs occurred in 57% of caplacizumab-treated patients and 51% of controls. Serious bleeding events occurred in 6% and 5%, respectively. ![]()
Bivalirudin provides no net benefit in PCI, doc says

Photo by Frank C. Muller
Results of a large meta-analysis indicate that bivalirudin may not be appropriate thromboprophylaxis for patients undergoing percutaneous coronary intervention (PCI).
Investigators found that bivalirudin was associated with an increased risk of stent thrombosis, when compared to an active control group.
Bivalirudin did confer a lower risk of major bleeding and cardiac death than the other anticoagulants studied.
But the drug had “no discernible impact” on all-cause mortality, according to the investigators.
“The take-home message is that, essentially, we didn’t find a net benefit with bivalirudin,” said Behnood Bikdeli, MD, of Yale-New Haven Hospital in Connecticut.
He and his colleagues detailed this discovery in Thrombosis Research.
The team analyzed data from 25 trials involving 41,243 PCI patients. The trials compared bivalirudin to other anticoagulants—largely unfractionated heparin, with or without other agents.
Compared with the active control group, bivalirudin use was associated with a significantly increased risk of definite stent thrombosis (risk ratio[RR]=1.73, P<0.001) and a significantly decreased risk of major bleeding (RR=0.59, P<0.001) and cardiac death (RR=0.72, P=0.05).
The risk of acute myocardial infarction was similar between the 2 groups (RR=1.00, P=0.96), as was the risk of all-cause mortality (RR=0.96, P=0.69).
The investigators said these results were consistent across a wide set of subgroup and sensitivity analyses.
“In our extensive analyses, we could not identify any patient subgroups that had less bleeding but no increase in the risk of clots, or stent thrombosis [with bivalirudin],” Dr Bikdeli said.
“This expensive medication, which is being used in a widespread manner, is not doing a slam-dunk better job. We might need more studies to see if a select minority of patients could potentially fare better with it.” ![]()
Photo by Frank C. Muller
Results of a large meta-analysis indicate that bivalirudin may not be appropriate thromboprophylaxis for patients undergoing percutaneous coronary intervention (PCI).
Investigators found that bivalirudin was associated with an increased risk of stent thrombosis, when compared to an active control group.
Bivalirudin did confer a lower risk of major bleeding and cardiac death than the other anticoagulants studied.
But the drug had “no discernible impact” on all-cause mortality, according to the investigators.
“The take-home message is that, essentially, we didn’t find a net benefit with bivalirudin,” said Behnood Bikdeli, MD, of Yale-New Haven Hospital in Connecticut.
He and his colleagues detailed this discovery in Thrombosis Research.
The team analyzed data from 25 trials involving 41,243 PCI patients. The trials compared bivalirudin to other anticoagulants—largely unfractionated heparin, with or without other agents.
Compared with the active control group, bivalirudin use was associated with a significantly increased risk of definite stent thrombosis (risk ratio[RR]=1.73, P<0.001) and a significantly decreased risk of major bleeding (RR=0.59, P<0.001) and cardiac death (RR=0.72, P=0.05).
The risk of acute myocardial infarction was similar between the 2 groups (RR=1.00, P=0.96), as was the risk of all-cause mortality (RR=0.96, P=0.69).
The investigators said these results were consistent across a wide set of subgroup and sensitivity analyses.
“In our extensive analyses, we could not identify any patient subgroups that had less bleeding but no increase in the risk of clots, or stent thrombosis [with bivalirudin],” Dr Bikdeli said.
“This expensive medication, which is being used in a widespread manner, is not doing a slam-dunk better job. We might need more studies to see if a select minority of patients could potentially fare better with it.” ![]()
Photo by Frank C. Muller
Results of a large meta-analysis indicate that bivalirudin may not be appropriate thromboprophylaxis for patients undergoing percutaneous coronary intervention (PCI).
Investigators found that bivalirudin was associated with an increased risk of stent thrombosis, when compared to an active control group.
Bivalirudin did confer a lower risk of major bleeding and cardiac death than the other anticoagulants studied.
But the drug had “no discernible impact” on all-cause mortality, according to the investigators.
“The take-home message is that, essentially, we didn’t find a net benefit with bivalirudin,” said Behnood Bikdeli, MD, of Yale-New Haven Hospital in Connecticut.
He and his colleagues detailed this discovery in Thrombosis Research.
The team analyzed data from 25 trials involving 41,243 PCI patients. The trials compared bivalirudin to other anticoagulants—largely unfractionated heparin, with or without other agents.
Compared with the active control group, bivalirudin use was associated with a significantly increased risk of definite stent thrombosis (risk ratio[RR]=1.73, P<0.001) and a significantly decreased risk of major bleeding (RR=0.59, P<0.001) and cardiac death (RR=0.72, P=0.05).
The risk of acute myocardial infarction was similar between the 2 groups (RR=1.00, P=0.96), as was the risk of all-cause mortality (RR=0.96, P=0.69).
The investigators said these results were consistent across a wide set of subgroup and sensitivity analyses.
“In our extensive analyses, we could not identify any patient subgroups that had less bleeding but no increase in the risk of clots, or stent thrombosis [with bivalirudin],” Dr Bikdeli said.
“This expensive medication, which is being used in a widespread manner, is not doing a slam-dunk better job. We might need more studies to see if a select minority of patients could potentially fare better with it.” ![]()
Product Update: LILETTA, myHDL, KleenSpec, STEPS Forward
NEW, SMALL IUD NOW AVAILABLE
LILETTA™ (levonorgestrel-releasing intrauterine system) 52 mg is now available for use by women to prevent pregnancy for up to 3 years. LILETTA is a small, flexible plastic T-shaped system 32 mm x 32 mm in size. It works to prevent pregnancy by slowly releasing levonorgestrel (LNG), a progestin, at an initial release rate of 18.6 µg/day with an average in vivo release rate of LNG of approximately 15.6 µg/day over a period of 3 years. Generally, LILETTA can be inserted at any time if the provider is reasonably certain that the woman is not pregnant. While LILETTA is intended for use up to 3 years, according to a press release from Actavis, it can be removed by a clinician at any time and can be replaced at the time of removal with a new LILETTA if continued contraceptive protection is desired.
The February 2015 FDA approval of LILETTA was based on the largest hormonal IUD trial conducted in the United States, designed to reflect the US population, says the manufacturer. The IUD was studied in women aged 16 to 45 years who were nulliparous or parous with a BMI of 15.8 kg/m2 to 61.6 kg/m2.
Through a partnership between Actavis and Medicines360 described at www.liletta.com, IUD-appropriate women, regardless of income and insurance coverage, now have access to this IUD at their doctor’s offices and at public health clinics enrolled in the 340B Drug Pricing Program.
FOR MORE INFORMATION, VISIT www.liletta.com

MYHDL APP ON APPLE WATCH, iPAD, iPHONE
The myHDL Physician App from Health Diagnostic Laboratory, Inc. (HDL) is newly available on the Apple Watch, iPhone, and iPad. The app is designed to allow users to view and manage patient cases from their wrist on all models of Apple Watch. HDL says that the myHDL app helps clinicians offer effective, personalized care to their patients, and features include the ability to manage multiple patients’ results, ease of use through color coding and categorized test results, and security and privacy. HDL provides comprehensive biomarker testing and clinical health consulting for earlier disease detection and targeted disease management. The myHDL app is available on iTunes App Store and can be downloaded at www.myhdlapp.com.
FOR MORE INFORMATION, VISIT www.hdlabinc.com
SINGLE-USE LED SPECULA IN 3 SIZES
Welch Allyn, Inc. recently launched the KleenSpec® Single Use LED Vaginal Specula intended for use in hospital emergency departments, labor and delivery units, urgent care centers, ambulatory care surgery centers, clinics, and other women’s health treatment centers.
Welch Allyn says that the KleenSpec Single Use LED Vaginal Specula are 100% acrylic and designed with smooth, molded edges to deliver maximum patient comfort. Wide handles are meant to provide comfortable ergonomics, ease of use, and good balance. According to Welch Allyn, the cordless device’s LED light source provides enhanced visualization of the examination area by supplying uniform white light for more than 30 minutes. The sealed LED light source and Lithium-primary battery are placed in the handle to reduce patient risk. The LED and battery can be removed for disposal or recycling. The device is ready to use out of the package and has a 5-year shelf life. The KleenSpec Single Use LED Vaginal Specula are available in extra small, small, and medium sizes in a distinctive color scheme to help clinicians identify various sizes.
FOR MORE INFORMATION, VISIT www.welchallyn.com
![]()
AMA’S PRACTICE TRANSFORMATION SERIES
AMA STEPS Forward™ is an online, interactive practice transformation series offering innovative strategies to help physicians and their staff refocus their practice. The American Medical Association (AMA) developed this initiative after a recent AMA-RAND report found that the satisfaction physicians derive from their work is eroding as they spend more and more time on administrative tasks. The AMA’s goal by offering STEPS Forward is to help clinicians achieve the “Quadruple Aim” to provide better patient experiences, better population health, lower overall costs, and improved professional satisfaction.
Physicians can access a collection of interactive educational modules to help deal with common practice challenges and also earn CME credit. Currently, 16 modules include steps for implementation, case studies, and downloadable videos, tools, and resources that address: practice efficiency and patient care, patient health, physician health, and technology and innovation. Additional modules are planned.
FOR MORE INFORMATION, VISIT www.stepsforward.org
NEW, SMALL IUD NOW AVAILABLE
LILETTA™ (levonorgestrel-releasing intrauterine system) 52 mg is now available for use by women to prevent pregnancy for up to 3 years. LILETTA is a small, flexible plastic T-shaped system 32 mm x 32 mm in size. It works to prevent pregnancy by slowly releasing levonorgestrel (LNG), a progestin, at an initial release rate of 18.6 µg/day with an average in vivo release rate of LNG of approximately 15.6 µg/day over a period of 3 years. Generally, LILETTA can be inserted at any time if the provider is reasonably certain that the woman is not pregnant. While LILETTA is intended for use up to 3 years, according to a press release from Actavis, it can be removed by a clinician at any time and can be replaced at the time of removal with a new LILETTA if continued contraceptive protection is desired.
The February 2015 FDA approval of LILETTA was based on the largest hormonal IUD trial conducted in the United States, designed to reflect the US population, says the manufacturer. The IUD was studied in women aged 16 to 45 years who were nulliparous or parous with a BMI of 15.8 kg/m2 to 61.6 kg/m2.
Through a partnership between Actavis and Medicines360 described at www.liletta.com, IUD-appropriate women, regardless of income and insurance coverage, now have access to this IUD at their doctor’s offices and at public health clinics enrolled in the 340B Drug Pricing Program.
FOR MORE INFORMATION, VISIT www.liletta.com
MYHDL APP ON APPLE WATCH, iPAD, iPHONE
The myHDL Physician App from Health Diagnostic Laboratory, Inc. (HDL) is newly available on the Apple Watch, iPhone, and iPad. The app is designed to allow users to view and manage patient cases from their wrist on all models of Apple Watch. HDL says that the myHDL app helps clinicians offer effective, personalized care to their patients, and features include the ability to manage multiple patients’ results, ease of use through color coding and categorized test results, and security and privacy. HDL provides comprehensive biomarker testing and clinical health consulting for earlier disease detection and targeted disease management. The myHDL app is available on iTunes App Store and can be downloaded at www.myhdlapp.com.
FOR MORE INFORMATION, VISIT www.hdlabinc.com
SINGLE-USE LED SPECULA IN 3 SIZES
Welch Allyn, Inc. recently launched the KleenSpec® Single Use LED Vaginal Specula intended for use in hospital emergency departments, labor and delivery units, urgent care centers, ambulatory care surgery centers, clinics, and other women’s health treatment centers.
Welch Allyn says that the KleenSpec Single Use LED Vaginal Specula are 100% acrylic and designed with smooth, molded edges to deliver maximum patient comfort. Wide handles are meant to provide comfortable ergonomics, ease of use, and good balance. According to Welch Allyn, the cordless device’s LED light source provides enhanced visualization of the examination area by supplying uniform white light for more than 30 minutes. The sealed LED light source and Lithium-primary battery are placed in the handle to reduce patient risk. The LED and battery can be removed for disposal or recycling. The device is ready to use out of the package and has a 5-year shelf life. The KleenSpec Single Use LED Vaginal Specula are available in extra small, small, and medium sizes in a distinctive color scheme to help clinicians identify various sizes.
FOR MORE INFORMATION, VISIT www.welchallyn.com
![]()
AMA’S PRACTICE TRANSFORMATION SERIES
AMA STEPS Forward™ is an online, interactive practice transformation series offering innovative strategies to help physicians and their staff refocus their practice. The American Medical Association (AMA) developed this initiative after a recent AMA-RAND report found that the satisfaction physicians derive from their work is eroding as they spend more and more time on administrative tasks. The AMA’s goal by offering STEPS Forward is to help clinicians achieve the “Quadruple Aim” to provide better patient experiences, better population health, lower overall costs, and improved professional satisfaction.
Physicians can access a collection of interactive educational modules to help deal with common practice challenges and also earn CME credit. Currently, 16 modules include steps for implementation, case studies, and downloadable videos, tools, and resources that address: practice efficiency and patient care, patient health, physician health, and technology and innovation. Additional modules are planned.
FOR MORE INFORMATION, VISIT www.stepsforward.org
NEW, SMALL IUD NOW AVAILABLE
LILETTA™ (levonorgestrel-releasing intrauterine system) 52 mg is now available for use by women to prevent pregnancy for up to 3 years. LILETTA is a small, flexible plastic T-shaped system 32 mm x 32 mm in size. It works to prevent pregnancy by slowly releasing levonorgestrel (LNG), a progestin, at an initial release rate of 18.6 µg/day with an average in vivo release rate of LNG of approximately 15.6 µg/day over a period of 3 years. Generally, LILETTA can be inserted at any time if the provider is reasonably certain that the woman is not pregnant. While LILETTA is intended for use up to 3 years, according to a press release from Actavis, it can be removed by a clinician at any time and can be replaced at the time of removal with a new LILETTA if continued contraceptive protection is desired.
The February 2015 FDA approval of LILETTA was based on the largest hormonal IUD trial conducted in the United States, designed to reflect the US population, says the manufacturer. The IUD was studied in women aged 16 to 45 years who were nulliparous or parous with a BMI of 15.8 kg/m2 to 61.6 kg/m2.
Through a partnership between Actavis and Medicines360 described at www.liletta.com, IUD-appropriate women, regardless of income and insurance coverage, now have access to this IUD at their doctor’s offices and at public health clinics enrolled in the 340B Drug Pricing Program.
FOR MORE INFORMATION, VISIT www.liletta.com
MYHDL APP ON APPLE WATCH, iPAD, iPHONE
The myHDL Physician App from Health Diagnostic Laboratory, Inc. (HDL) is newly available on the Apple Watch, iPhone, and iPad. The app is designed to allow users to view and manage patient cases from their wrist on all models of Apple Watch. HDL says that the myHDL app helps clinicians offer effective, personalized care to their patients, and features include the ability to manage multiple patients’ results, ease of use through color coding and categorized test results, and security and privacy. HDL provides comprehensive biomarker testing and clinical health consulting for earlier disease detection and targeted disease management. The myHDL app is available on iTunes App Store and can be downloaded at www.myhdlapp.com.
FOR MORE INFORMATION, VISIT www.hdlabinc.com
SINGLE-USE LED SPECULA IN 3 SIZES
Welch Allyn, Inc. recently launched the KleenSpec® Single Use LED Vaginal Specula intended for use in hospital emergency departments, labor and delivery units, urgent care centers, ambulatory care surgery centers, clinics, and other women’s health treatment centers.
Welch Allyn says that the KleenSpec Single Use LED Vaginal Specula are 100% acrylic and designed with smooth, molded edges to deliver maximum patient comfort. Wide handles are meant to provide comfortable ergonomics, ease of use, and good balance. According to Welch Allyn, the cordless device’s LED light source provides enhanced visualization of the examination area by supplying uniform white light for more than 30 minutes. The sealed LED light source and Lithium-primary battery are placed in the handle to reduce patient risk. The LED and battery can be removed for disposal or recycling. The device is ready to use out of the package and has a 5-year shelf life. The KleenSpec Single Use LED Vaginal Specula are available in extra small, small, and medium sizes in a distinctive color scheme to help clinicians identify various sizes.
FOR MORE INFORMATION, VISIT www.welchallyn.com
![]()
AMA’S PRACTICE TRANSFORMATION SERIES
AMA STEPS Forward™ is an online, interactive practice transformation series offering innovative strategies to help physicians and their staff refocus their practice. The American Medical Association (AMA) developed this initiative after a recent AMA-RAND report found that the satisfaction physicians derive from their work is eroding as they spend more and more time on administrative tasks. The AMA’s goal by offering STEPS Forward is to help clinicians achieve the “Quadruple Aim” to provide better patient experiences, better population health, lower overall costs, and improved professional satisfaction.
Physicians can access a collection of interactive educational modules to help deal with common practice challenges and also earn CME credit. Currently, 16 modules include steps for implementation, case studies, and downloadable videos, tools, and resources that address: practice efficiency and patient care, patient health, physician health, and technology and innovation. Additional modules are planned.
FOR MORE INFORMATION, VISIT www.stepsforward.org