User login
EULAR: Panel targets six rheumatic disease comorbidities
ROME – Clinicians who care for patients with chronic inflammatory rheumatic diseases should consider regularly assessing six potential comorbidities these patients might develop, according to a set of “points to consider” developed by a task force of the European League Against Rheumatism.
The six comorbidities the working group’s report cites are ischemic cardiovascular disease, malignancies, infections, peptic ulcer, osteoporosis, and depression, Dr. Maxime Dougados said at the European Congress of Rheumatology.

This is the “minimum list of comorbidities to systematically check” for patients with inflammatory rheumatic diseases, said Dr. Dougados, professor and chief of rheumatology at Cochin Hospital in Paris.
The task force he heads plans to soon make available on the EULAR Website screening questionnaires for assessing the status of each of these six comorbidities. “We hope you will consider this initiative and implement these points to consider in your practice,” he said.
A seventh comorbidity to potentially add to the list for regular assessment is hypertension, said Dr. Deborah P.M. Symmons, professor of rheumatology and musculoskeletal epidemiology at the University of Manchester (England), in a separate talk at the meeting. Roughly 80% of patients with rheumatoid arthritis (RA) have at least one comorbidity, she noted.
Recent study results have documented the prevalence of comorbidities in patients with RA, Dr. Symmons said. For example, an analysis of data collected during 2011 and 2012 from 3,920 RA patients in 17 countries, including 400 U.S. patients, showed that depression was the most common comorbidity, affecting 15% of patients; other comorbidities included ischemic cardiovascular disease in 6%, malignancy in 5%, and hypertension in 11% (Ann. Rheum. Dis. 2014;73:62-8). A separate survey of 9,874 RA patients from 34 countries also published last year found patients had a median of two comorbidities each. The most common were hypertension in 32% of patients, osteoporosis in 18%, and osteoarthritis in 16% (Clin. Exp. Rheum. 2014;32:869-77).

“Chronic diseases cluster together, more than you would expect by chance, perhaps because of shared risk factors such as genetic or environmental, the direct impact of inflammation, and because of treatment” patients receive for their rheumatic disease, Dr. Symmons said.
The consequence is that clinicians who manage patients with RA or other rheumatic disease must be on the lookout for comorbidities and take them into consideration when planning management strategies. A rheumatologist might be most concerned about how comobidities will affect the rheumatic disease, but for patients the overriding concern is how all their chronic diseases, not just their rheumatic disease, will affect their quality of life and physical function, she noted. “We must constantly ask ourselves whether treatment of the RA will worsen the comorbidities, or will treatment of the comorbidities worsen the RA?”
Knowledge of how RA treatments will affect comorbidities is often lacking because patients with comorbidities are usually not enrolled in clinical trials, Dr. Symmons said.
She recommended that rheumatologists systematically screen patients annually for comorbidities and discuss with each patient and with clinicians from other relevant specialties appropriate treatment based on the patient’s global status. Steroid treatment should be minimized because of the risk it poses for causing or exacerbating hypertension, hyperlipidemia, diabetes, osteoporosis, and infection.
The rheumatologist does not necessarily need to be the clinician who manages all of a patient’s comorbidities, which might be better done by a primary care physician, but the rheumatologist should know that a patient’s comorbidities are being managed by someone, and this fact should be documented in the rheumatologist’s records for each patient, she said.
Dr. Dougados and Dr. Symmons said they had no relevant financial disclosures.
On Twitter@mitchelzoler
ROME – Clinicians who care for patients with chronic inflammatory rheumatic diseases should consider regularly assessing six potential comorbidities these patients might develop, according to a set of “points to consider” developed by a task force of the European League Against Rheumatism.
The six comorbidities the working group’s report cites are ischemic cardiovascular disease, malignancies, infections, peptic ulcer, osteoporosis, and depression, Dr. Maxime Dougados said at the European Congress of Rheumatology.
This is the “minimum list of comorbidities to systematically check” for patients with inflammatory rheumatic diseases, said Dr. Dougados, professor and chief of rheumatology at Cochin Hospital in Paris.
The task force he heads plans to soon make available on the EULAR Website screening questionnaires for assessing the status of each of these six comorbidities. “We hope you will consider this initiative and implement these points to consider in your practice,” he said.
A seventh comorbidity to potentially add to the list for regular assessment is hypertension, said Dr. Deborah P.M. Symmons, professor of rheumatology and musculoskeletal epidemiology at the University of Manchester (England), in a separate talk at the meeting. Roughly 80% of patients with rheumatoid arthritis (RA) have at least one comorbidity, she noted.
Recent study results have documented the prevalence of comorbidities in patients with RA, Dr. Symmons said. For example, an analysis of data collected during 2011 and 2012 from 3,920 RA patients in 17 countries, including 400 U.S. patients, showed that depression was the most common comorbidity, affecting 15% of patients; other comorbidities included ischemic cardiovascular disease in 6%, malignancy in 5%, and hypertension in 11% (Ann. Rheum. Dis. 2014;73:62-8). A separate survey of 9,874 RA patients from 34 countries also published last year found patients had a median of two comorbidities each. The most common were hypertension in 32% of patients, osteoporosis in 18%, and osteoarthritis in 16% (Clin. Exp. Rheum. 2014;32:869-77).
“Chronic diseases cluster together, more than you would expect by chance, perhaps because of shared risk factors such as genetic or environmental, the direct impact of inflammation, and because of treatment” patients receive for their rheumatic disease, Dr. Symmons said.
The consequence is that clinicians who manage patients with RA or other rheumatic disease must be on the lookout for comorbidities and take them into consideration when planning management strategies. A rheumatologist might be most concerned about how comobidities will affect the rheumatic disease, but for patients the overriding concern is how all their chronic diseases, not just their rheumatic disease, will affect their quality of life and physical function, she noted. “We must constantly ask ourselves whether treatment of the RA will worsen the comorbidities, or will treatment of the comorbidities worsen the RA?”
Knowledge of how RA treatments will affect comorbidities is often lacking because patients with comorbidities are usually not enrolled in clinical trials, Dr. Symmons said.
She recommended that rheumatologists systematically screen patients annually for comorbidities and discuss with each patient and with clinicians from other relevant specialties appropriate treatment based on the patient’s global status. Steroid treatment should be minimized because of the risk it poses for causing or exacerbating hypertension, hyperlipidemia, diabetes, osteoporosis, and infection.
The rheumatologist does not necessarily need to be the clinician who manages all of a patient’s comorbidities, which might be better done by a primary care physician, but the rheumatologist should know that a patient’s comorbidities are being managed by someone, and this fact should be documented in the rheumatologist’s records for each patient, she said.
Dr. Dougados and Dr. Symmons said they had no relevant financial disclosures.
On Twitter@mitchelzoler
ROME – Clinicians who care for patients with chronic inflammatory rheumatic diseases should consider regularly assessing six potential comorbidities these patients might develop, according to a set of “points to consider” developed by a task force of the European League Against Rheumatism.
The six comorbidities the working group’s report cites are ischemic cardiovascular disease, malignancies, infections, peptic ulcer, osteoporosis, and depression, Dr. Maxime Dougados said at the European Congress of Rheumatology.
This is the “minimum list of comorbidities to systematically check” for patients with inflammatory rheumatic diseases, said Dr. Dougados, professor and chief of rheumatology at Cochin Hospital in Paris.
The task force he heads plans to soon make available on the EULAR Website screening questionnaires for assessing the status of each of these six comorbidities. “We hope you will consider this initiative and implement these points to consider in your practice,” he said.
A seventh comorbidity to potentially add to the list for regular assessment is hypertension, said Dr. Deborah P.M. Symmons, professor of rheumatology and musculoskeletal epidemiology at the University of Manchester (England), in a separate talk at the meeting. Roughly 80% of patients with rheumatoid arthritis (RA) have at least one comorbidity, she noted.
Recent study results have documented the prevalence of comorbidities in patients with RA, Dr. Symmons said. For example, an analysis of data collected during 2011 and 2012 from 3,920 RA patients in 17 countries, including 400 U.S. patients, showed that depression was the most common comorbidity, affecting 15% of patients; other comorbidities included ischemic cardiovascular disease in 6%, malignancy in 5%, and hypertension in 11% (Ann. Rheum. Dis. 2014;73:62-8). A separate survey of 9,874 RA patients from 34 countries also published last year found patients had a median of two comorbidities each. The most common were hypertension in 32% of patients, osteoporosis in 18%, and osteoarthritis in 16% (Clin. Exp. Rheum. 2014;32:869-77).
“Chronic diseases cluster together, more than you would expect by chance, perhaps because of shared risk factors such as genetic or environmental, the direct impact of inflammation, and because of treatment” patients receive for their rheumatic disease, Dr. Symmons said.
The consequence is that clinicians who manage patients with RA or other rheumatic disease must be on the lookout for comorbidities and take them into consideration when planning management strategies. A rheumatologist might be most concerned about how comobidities will affect the rheumatic disease, but for patients the overriding concern is how all their chronic diseases, not just their rheumatic disease, will affect their quality of life and physical function, she noted. “We must constantly ask ourselves whether treatment of the RA will worsen the comorbidities, or will treatment of the comorbidities worsen the RA?”
Knowledge of how RA treatments will affect comorbidities is often lacking because patients with comorbidities are usually not enrolled in clinical trials, Dr. Symmons said.
She recommended that rheumatologists systematically screen patients annually for comorbidities and discuss with each patient and with clinicians from other relevant specialties appropriate treatment based on the patient’s global status. Steroid treatment should be minimized because of the risk it poses for causing or exacerbating hypertension, hyperlipidemia, diabetes, osteoporosis, and infection.
The rheumatologist does not necessarily need to be the clinician who manages all of a patient’s comorbidities, which might be better done by a primary care physician, but the rheumatologist should know that a patient’s comorbidities are being managed by someone, and this fact should be documented in the rheumatologist’s records for each patient, she said.
Dr. Dougados and Dr. Symmons said they had no relevant financial disclosures.
On Twitter@mitchelzoler
AT THE EULAR 2015 CONGRESS

Foot Rash + Gnarly Toenails = Man in Need of a Diagnosis
ANSWER
The correct answer is to perform a KOH examination (choice “b”), which takes just five minutes and offers the chance to establish the fungal origin of the rash. Although the patient’s skin is quite dry, the use of a moisturizer (choice “a”) is unlikely to address the overall problem. A punch biopsy (choice “c”) would be a logical choice if the KOH failed to solve the mystery. The use of combination creams (choice “d”) that contain a steroid (triamcinolone) and an antifungal (nystatin) is essentially an admission of the lack of a definitive diagnosis. For reasons discussed below, this strategy has almost no chance of helping.
DISCUSSION
In this case, the KOH prep showed numerous hyphal elements, confirming suspicions of a fungal origin. One potential source of these organisms was the patient’s feet, where fungal infection had been present for years (“more than 30,” questioning revealed).
A common scenario is one in which the patient applies a steroid cream to a bit of dry skin just above the feet, which allows the fungi to gain a “foothold” from which to spread upward onto the leg; this progress is assisted through scratching and additional steroid application. If no firm diagnosis is ever established, definitive treatment cannot be undertaken and the problem never resolves.
In my opinion, there is never a reason to prescribe a product containing nystatin. In 1950, when it was discovered by researchers working in New York State laboratories (after which it was named), its efficacy against Candida species represented a notable advance, given the limited drug choices available for that purpose. But it has little, if any, activity against the dermatophytes causing our patient’s problems. And the steroid (triamcinolone) in this combination product, far from adding any therapeutic benefit, effectively diminishes any natural immune response.
The other reason to refrain from prescribing nystatin is that, since its discovery, at least three generations of products that treat both fungi and yeast (the azoles, such as clotrimazole, econazole, and fluconazole) have become available and have been found to be very effective.
The more important issue in this case, however, is finally having an accurate diagnosis: tinea corporis, probably caused by the most common dermatophyte, Trichophyton rubrum. The patient’s body is obviously a very happy home for this ubiquitous organism, to the extent that our chances of eliminating it are quite small. But we can at least make the patient more comfortable.
Treatment entailed ketoconazole foam (applied bid to his legs) and a two-month course of oral terbinafine (250 mg/d), which cleared up most of the skin problem. For his overgrown toenails, the patient was advised to establish care with a podiatrist for regular trimming.
In terms of a differential, this patient might have had psoriasis or eczema—and may still have one or both, since there’s no law against having more than one condition in the same location. In time, we may have to reconsider our solitary diagnosis.
ANSWER
The correct answer is to perform a KOH examination (choice “b”), which takes just five minutes and offers the chance to establish the fungal origin of the rash. Although the patient’s skin is quite dry, the use of a moisturizer (choice “a”) is unlikely to address the overall problem. A punch biopsy (choice “c”) would be a logical choice if the KOH failed to solve the mystery. The use of combination creams (choice “d”) that contain a steroid (triamcinolone) and an antifungal (nystatin) is essentially an admission of the lack of a definitive diagnosis. For reasons discussed below, this strategy has almost no chance of helping.
DISCUSSION
In this case, the KOH prep showed numerous hyphal elements, confirming suspicions of a fungal origin. One potential source of these organisms was the patient’s feet, where fungal infection had been present for years (“more than 30,” questioning revealed).
A common scenario is one in which the patient applies a steroid cream to a bit of dry skin just above the feet, which allows the fungi to gain a “foothold” from which to spread upward onto the leg; this progress is assisted through scratching and additional steroid application. If no firm diagnosis is ever established, definitive treatment cannot be undertaken and the problem never resolves.
In my opinion, there is never a reason to prescribe a product containing nystatin. In 1950, when it was discovered by researchers working in New York State laboratories (after which it was named), its efficacy against Candida species represented a notable advance, given the limited drug choices available for that purpose. But it has little, if any, activity against the dermatophytes causing our patient’s problems. And the steroid (triamcinolone) in this combination product, far from adding any therapeutic benefit, effectively diminishes any natural immune response.
The other reason to refrain from prescribing nystatin is that, since its discovery, at least three generations of products that treat both fungi and yeast (the azoles, such as clotrimazole, econazole, and fluconazole) have become available and have been found to be very effective.
The more important issue in this case, however, is finally having an accurate diagnosis: tinea corporis, probably caused by the most common dermatophyte, Trichophyton rubrum. The patient’s body is obviously a very happy home for this ubiquitous organism, to the extent that our chances of eliminating it are quite small. But we can at least make the patient more comfortable.
Treatment entailed ketoconazole foam (applied bid to his legs) and a two-month course of oral terbinafine (250 mg/d), which cleared up most of the skin problem. For his overgrown toenails, the patient was advised to establish care with a podiatrist for regular trimming.
In terms of a differential, this patient might have had psoriasis or eczema—and may still have one or both, since there’s no law against having more than one condition in the same location. In time, we may have to reconsider our solitary diagnosis.
ANSWER
The correct answer is to perform a KOH examination (choice “b”), which takes just five minutes and offers the chance to establish the fungal origin of the rash. Although the patient’s skin is quite dry, the use of a moisturizer (choice “a”) is unlikely to address the overall problem. A punch biopsy (choice “c”) would be a logical choice if the KOH failed to solve the mystery. The use of combination creams (choice “d”) that contain a steroid (triamcinolone) and an antifungal (nystatin) is essentially an admission of the lack of a definitive diagnosis. For reasons discussed below, this strategy has almost no chance of helping.
DISCUSSION
In this case, the KOH prep showed numerous hyphal elements, confirming suspicions of a fungal origin. One potential source of these organisms was the patient’s feet, where fungal infection had been present for years (“more than 30,” questioning revealed).
A common scenario is one in which the patient applies a steroid cream to a bit of dry skin just above the feet, which allows the fungi to gain a “foothold” from which to spread upward onto the leg; this progress is assisted through scratching and additional steroid application. If no firm diagnosis is ever established, definitive treatment cannot be undertaken and the problem never resolves.
In my opinion, there is never a reason to prescribe a product containing nystatin. In 1950, when it was discovered by researchers working in New York State laboratories (after which it was named), its efficacy against Candida species represented a notable advance, given the limited drug choices available for that purpose. But it has little, if any, activity against the dermatophytes causing our patient’s problems. And the steroid (triamcinolone) in this combination product, far from adding any therapeutic benefit, effectively diminishes any natural immune response.
The other reason to refrain from prescribing nystatin is that, since its discovery, at least three generations of products that treat both fungi and yeast (the azoles, such as clotrimazole, econazole, and fluconazole) have become available and have been found to be very effective.
The more important issue in this case, however, is finally having an accurate diagnosis: tinea corporis, probably caused by the most common dermatophyte, Trichophyton rubrum. The patient’s body is obviously a very happy home for this ubiquitous organism, to the extent that our chances of eliminating it are quite small. But we can at least make the patient more comfortable.
Treatment entailed ketoconazole foam (applied bid to his legs) and a two-month course of oral terbinafine (250 mg/d), which cleared up most of the skin problem. For his overgrown toenails, the patient was advised to establish care with a podiatrist for regular trimming.
In terms of a differential, this patient might have had psoriasis or eczema—and may still have one or both, since there’s no law against having more than one condition in the same location. In time, we may have to reconsider our solitary diagnosis.
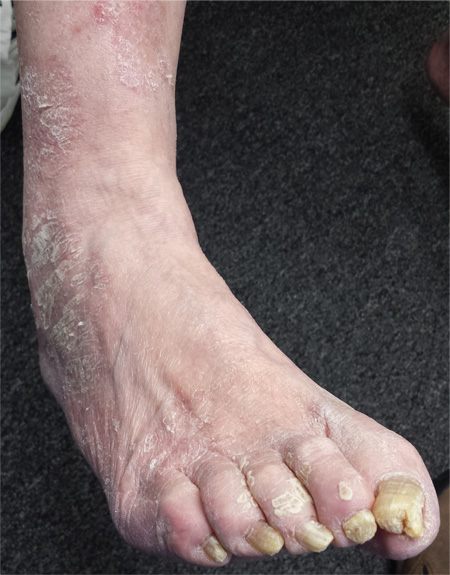
For several years, a 66-year-old man has had an itchy rash on his right leg; recently, it has become more bothersome. In general, he has noticed that when cold weather arrives, the rash improves slightly, but it inevitably worsens again as winter progresses. Over the years, the providers he has consulted have prescribed a number of topical products—among them, antifungal and steroid creams. Each of these products seems to help for a short period, then stops; at that point, the patient switches to a different product, with similar mixed results. The patient says he doesn’t have any other skin problems. Examination reveals patches of dry skin scattered from the knee to the top of the patient’s foot. Most have a faintly erythematous surface and arciform borders. These patches blend into a similar rash that covers the sides of both feet. All 10 toenails are grossly dystrophic, yellowed, and overgrown. The skin on the patient’s other leg is somewhat dry but otherwise unaffected.

Understanding Hematuria: IgA Nephropathy
Q) My hematuria patient had more significant proteinuria recently, so the nephrologist sent him for kidney biopsy. It was read as IgA nephropathy: “classic mesangial staining on IF with moderate-advanced chronic injury (15/32 gloms globally sclerosed, 40% IFTA, mild arteriosclerosis).” What exactly does this mean, and what is IgA nephropathy?
Immunoglobulin A (IgA) nephropathy is the most common type of glomerulonephritis; up to 40% of patients with IgA nephropathy develop end-stage renal disease within 20 years of diagnosis. More common in men, IgA nephropathy is usually diagnosed in people in their second or third decades of life.2,3
Considered an autoimmune disease, IgA nephropathy typically presents with macroscopic or gross hematuria that occurs within 24 hours of the onset of an upper respiratory infection (URI). The hematuria typically resolves quickly, in one to three days. An individual bacterial or viral element has not yet been identified.
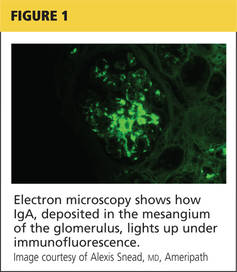
IgA nephropathy is an immune response to the URI. IgA is secreted from mucosal surfaces at the back of the mouth and then deposited in the glomerular mesangium, a “stalk of cells” associated with the capillaries of the renal glomerulus.1 It is speculated that genetics, environment, and/or hypersensitivity to food antigens may play a part in IgA nephropathy. Results from biopsies of blood relatives of patients with confirmed IgA nephropathy suggest a familial role.1
IgA nephropathy is prevalent in persons who live in the Pacific Rim and Southern Europe. However, this association may be the result of a sampling error due to investigation of all microscopic hematuria in these areas. In all, 90% of IgA is sporadic.4 It is often asymptomatic, aside from occasional back and flank pain secondary to inflammation of the renal capsule. Unfortunately, many patients develop renal impairment and hypertension by the time they are diagnosed.
Renal biopsy is used to confirm/diagnose IgA nephropathy. IgA, deposited in the mesangium of the glomerulus, lights up under immunofluorescence (IF; see Figure 1). In some patients, this mesangial deposition results in sclerosis, scarring, and/or inflammation of the glomerulus (see Figure 2).
An international panel of experts created guidelines (the Oxford classification system) for reporting IgA kidney biopsies. Six adverse pathologic features have been identified:
• Mesangial cellularity score
• Percentage of segmental sclerosis
• Endocapillary hypercellularity
• Cellular and/or fibrocellular crescents
• Percentage of interstitial fibrosis/tubular atrophy (IFTA)
• Arteriosclerosis score5,6
Interstitial fibrosis, crescents, and as little as 25% glomerular sclerosis found on biopsy increases the likelihood of disease progression.5 Clinically, hypertension, a reduced glomerular filtration rate, increasing age, and proteinuria of > 1g/24h have been identified as risk factors for progression of IgA nephropathy. Up to 30% of patients diagnosed will require renal replacement therapy within 20 years.1
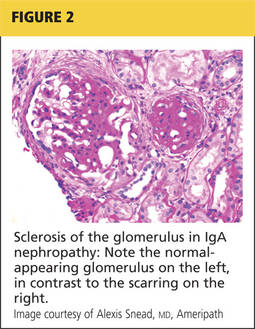
The case patient’s findings include the typical IF staining of IgA in the glomerulus. The biopsy report also indicates that 40% of the glomeruli (less than half) have interstitial fibrosis and that the structural integrity of the tubules has been affected secondary to IgA accumulation in the mesangium. These findings are suggestive of progressive disease.
There is no known way to stop IgA deposition in the mesangium. Tonsillectomy to reduce mucosal IgA release has been suggested but is controversial.
Treatment of IgA nephropathy focuses on preserving renal function by reducing proteinuria through the use of ACE inhibitors and/or angiotensin receptor blockers. Aggressive blood pressure management is achieved by blocking the renin-angiotensin-aldosterone system (RAAS).
Other methods for decreasing progression of renal disease are directed at reducing the immune and inflammatory response via immunosuppressant medications.3 The use of immunosuppressive agents, though controversial, is recommended for those who have progressive disease and/or proteinuria despite achieving target blood pressure with full RAAS blockade.1
Amy L. Hazel, RN, MSN, CNP
Kidney & Hypertension Consultants, Canton, Ohio
REFERENCES
1. Greenberg A. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2005.
2. Barratt J, Feehally J. IgA nephropathy [disease of the month]. J Am Soc Nephrol. 2005;16(7): 2088-2097.
3. Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int. 2006;69(11):1934-1938.
4. Johnson R, Feehally J. Comprehensive Clinical Nephrology. 2nd ed. London: Mosby; 2000.
5. Walsh M, Sar A, Lee D, et al. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clin J Am Soc Nephrol. 2010; 5(3):425-430.
6. Roberts I. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009; 76(5):546-556.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Amy L. Hazel, RN, MSN, CNP, who practices at Kidney & Hypertension Consultants in Canton, Ohio.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Amy L. Hazel, RN, MSN, CNP, who practices at Kidney & Hypertension Consultants in Canton, Ohio.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Amy L. Hazel, RN, MSN, CNP, who practices at Kidney & Hypertension Consultants in Canton, Ohio.
Q) My hematuria patient had more significant proteinuria recently, so the nephrologist sent him for kidney biopsy. It was read as IgA nephropathy: “classic mesangial staining on IF with moderate-advanced chronic injury (15/32 gloms globally sclerosed, 40% IFTA, mild arteriosclerosis).” What exactly does this mean, and what is IgA nephropathy?
Immunoglobulin A (IgA) nephropathy is the most common type of glomerulonephritis; up to 40% of patients with IgA nephropathy develop end-stage renal disease within 20 years of diagnosis. More common in men, IgA nephropathy is usually diagnosed in people in their second or third decades of life.2,3
Considered an autoimmune disease, IgA nephropathy typically presents with macroscopic or gross hematuria that occurs within 24 hours of the onset of an upper respiratory infection (URI). The hematuria typically resolves quickly, in one to three days. An individual bacterial or viral element has not yet been identified.
IgA nephropathy is an immune response to the URI. IgA is secreted from mucosal surfaces at the back of the mouth and then deposited in the glomerular mesangium, a “stalk of cells” associated with the capillaries of the renal glomerulus.1 It is speculated that genetics, environment, and/or hypersensitivity to food antigens may play a part in IgA nephropathy. Results from biopsies of blood relatives of patients with confirmed IgA nephropathy suggest a familial role.1
IgA nephropathy is prevalent in persons who live in the Pacific Rim and Southern Europe. However, this association may be the result of a sampling error due to investigation of all microscopic hematuria in these areas. In all, 90% of IgA is sporadic.4 It is often asymptomatic, aside from occasional back and flank pain secondary to inflammation of the renal capsule. Unfortunately, many patients develop renal impairment and hypertension by the time they are diagnosed.
Renal biopsy is used to confirm/diagnose IgA nephropathy. IgA, deposited in the mesangium of the glomerulus, lights up under immunofluorescence (IF; see Figure 1). In some patients, this mesangial deposition results in sclerosis, scarring, and/or inflammation of the glomerulus (see Figure 2).
An international panel of experts created guidelines (the Oxford classification system) for reporting IgA kidney biopsies. Six adverse pathologic features have been identified:
• Mesangial cellularity score
• Percentage of segmental sclerosis
• Endocapillary hypercellularity
• Cellular and/or fibrocellular crescents
• Percentage of interstitial fibrosis/tubular atrophy (IFTA)
• Arteriosclerosis score5,6
Interstitial fibrosis, crescents, and as little as 25% glomerular sclerosis found on biopsy increases the likelihood of disease progression.5 Clinically, hypertension, a reduced glomerular filtration rate, increasing age, and proteinuria of > 1g/24h have been identified as risk factors for progression of IgA nephropathy. Up to 30% of patients diagnosed will require renal replacement therapy within 20 years.1
The case patient’s findings include the typical IF staining of IgA in the glomerulus. The biopsy report also indicates that 40% of the glomeruli (less than half) have interstitial fibrosis and that the structural integrity of the tubules has been affected secondary to IgA accumulation in the mesangium. These findings are suggestive of progressive disease.
There is no known way to stop IgA deposition in the mesangium. Tonsillectomy to reduce mucosal IgA release has been suggested but is controversial.
Treatment of IgA nephropathy focuses on preserving renal function by reducing proteinuria through the use of ACE inhibitors and/or angiotensin receptor blockers. Aggressive blood pressure management is achieved by blocking the renin-angiotensin-aldosterone system (RAAS).
Other methods for decreasing progression of renal disease are directed at reducing the immune and inflammatory response via immunosuppressant medications.3 The use of immunosuppressive agents, though controversial, is recommended for those who have progressive disease and/or proteinuria despite achieving target blood pressure with full RAAS blockade.1
Amy L. Hazel, RN, MSN, CNP
Kidney & Hypertension Consultants, Canton, Ohio
REFERENCES
1. Greenberg A. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2005.
2. Barratt J, Feehally J. IgA nephropathy [disease of the month]. J Am Soc Nephrol. 2005;16(7): 2088-2097.
3. Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int. 2006;69(11):1934-1938.
4. Johnson R, Feehally J. Comprehensive Clinical Nephrology. 2nd ed. London: Mosby; 2000.
5. Walsh M, Sar A, Lee D, et al. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clin J Am Soc Nephrol. 2010; 5(3):425-430.
6. Roberts I. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009; 76(5):546-556.
Q) My hematuria patient had more significant proteinuria recently, so the nephrologist sent him for kidney biopsy. It was read as IgA nephropathy: “classic mesangial staining on IF with moderate-advanced chronic injury (15/32 gloms globally sclerosed, 40% IFTA, mild arteriosclerosis).” What exactly does this mean, and what is IgA nephropathy?
Immunoglobulin A (IgA) nephropathy is the most common type of glomerulonephritis; up to 40% of patients with IgA nephropathy develop end-stage renal disease within 20 years of diagnosis. More common in men, IgA nephropathy is usually diagnosed in people in their second or third decades of life.2,3
Considered an autoimmune disease, IgA nephropathy typically presents with macroscopic or gross hematuria that occurs within 24 hours of the onset of an upper respiratory infection (URI). The hematuria typically resolves quickly, in one to three days. An individual bacterial or viral element has not yet been identified.
IgA nephropathy is an immune response to the URI. IgA is secreted from mucosal surfaces at the back of the mouth and then deposited in the glomerular mesangium, a “stalk of cells” associated with the capillaries of the renal glomerulus.1 It is speculated that genetics, environment, and/or hypersensitivity to food antigens may play a part in IgA nephropathy. Results from biopsies of blood relatives of patients with confirmed IgA nephropathy suggest a familial role.1
IgA nephropathy is prevalent in persons who live in the Pacific Rim and Southern Europe. However, this association may be the result of a sampling error due to investigation of all microscopic hematuria in these areas. In all, 90% of IgA is sporadic.4 It is often asymptomatic, aside from occasional back and flank pain secondary to inflammation of the renal capsule. Unfortunately, many patients develop renal impairment and hypertension by the time they are diagnosed.
Renal biopsy is used to confirm/diagnose IgA nephropathy. IgA, deposited in the mesangium of the glomerulus, lights up under immunofluorescence (IF; see Figure 1). In some patients, this mesangial deposition results in sclerosis, scarring, and/or inflammation of the glomerulus (see Figure 2).
An international panel of experts created guidelines (the Oxford classification system) for reporting IgA kidney biopsies. Six adverse pathologic features have been identified:
• Mesangial cellularity score
• Percentage of segmental sclerosis
• Endocapillary hypercellularity
• Cellular and/or fibrocellular crescents
• Percentage of interstitial fibrosis/tubular atrophy (IFTA)
• Arteriosclerosis score5,6
Interstitial fibrosis, crescents, and as little as 25% glomerular sclerosis found on biopsy increases the likelihood of disease progression.5 Clinically, hypertension, a reduced glomerular filtration rate, increasing age, and proteinuria of > 1g/24h have been identified as risk factors for progression of IgA nephropathy. Up to 30% of patients diagnosed will require renal replacement therapy within 20 years.1
The case patient’s findings include the typical IF staining of IgA in the glomerulus. The biopsy report also indicates that 40% of the glomeruli (less than half) have interstitial fibrosis and that the structural integrity of the tubules has been affected secondary to IgA accumulation in the mesangium. These findings are suggestive of progressive disease.
There is no known way to stop IgA deposition in the mesangium. Tonsillectomy to reduce mucosal IgA release has been suggested but is controversial.
Treatment of IgA nephropathy focuses on preserving renal function by reducing proteinuria through the use of ACE inhibitors and/or angiotensin receptor blockers. Aggressive blood pressure management is achieved by blocking the renin-angiotensin-aldosterone system (RAAS).
Other methods for decreasing progression of renal disease are directed at reducing the immune and inflammatory response via immunosuppressant medications.3 The use of immunosuppressive agents, though controversial, is recommended for those who have progressive disease and/or proteinuria despite achieving target blood pressure with full RAAS blockade.1
Amy L. Hazel, RN, MSN, CNP
Kidney & Hypertension Consultants, Canton, Ohio
REFERENCES
1. Greenberg A. Primer on Kidney Diseases. 5th ed. Philadelphia, PA: Elsevier Saunders; 2005.
2. Barratt J, Feehally J. IgA nephropathy [disease of the month]. J Am Soc Nephrol. 2005;16(7): 2088-2097.
3. Barratt J, Feehally J. Treatment of IgA nephropathy. Kidney Int. 2006;69(11):1934-1938.
4. Johnson R, Feehally J. Comprehensive Clinical Nephrology. 2nd ed. London: Mosby; 2000.
5. Walsh M, Sar A, Lee D, et al. Histopathologic features aid in predicting risk for progression of IgA nephropathy. Clin J Am Soc Nephrol. 2010; 5(3):425-430.
6. Roberts I. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009; 76(5):546-556.
Extended-interval dosing appears safe, effective

Photo courtesy of Biogen
TORONTO—Full results of a phase 3 study support extended-interval dosing with a recombinant factor IX Fc fusion protein (rFIXFc) over FIX products with a standard half-life, according to a speaker at the 2015 ISTH Congress.
Kathelijn Fischer, MD, PhD, of the University Medical Center Utrecht in The Netherlands, reported results with rFIXFc (also known as eftrenonacog alfa and Alprolix), in children with severe hemophilia B who were enrolled on the KIDS B-LONG study.
rFIXFc was successful in preventing and treating bleeding episodes in these patients. Furthermore, the patients did not develop inhibitors, and there were no serious adverse events related to treatment.
Dr Fischer presented these results as abstract LB009. Interim results of this study helped support the US approval of rFIXFc for use in children. The trial was sponsored by Sobi and Biogen, the companies developing rFIXFc.
KIDS B-LONG included 30 boys younger than 12 who had severe hemophilia B. The patients had at least 50 prior exposure days to FIX therapies and no history of inhibitors.
At baseline, all patients were receiving FIX prophylaxis. Seventy-seven percent of patients were receiving 2 or more doses a week.
On day 1 of the study, patients received rFIXFc at 50 IU/kg. They then received weekly prophylaxis at an initial dose of 50 IU/kg to 60 IU/kg. Doses were adjusted throughout the study, but the maximum was 100 IU/kg. The minimum dosing frequency was once a week, and the maximum was twice a week.
Twenty-seven patients (90%) completed the study. The median time spent on study was 49.4 weeks, and 24 patients (80%) received rFIXFc injections on at least 50 separate days.
Safety
None of the patients developed inhibitors or non-neutralizing anti-rFIXFc antibodies. There were no anaphylactic reactions, hypersensitivity reactions, thrombotic events, or deaths.
Adverse events occurred in 86.7% of patients. The most frequent were nanopharyngitis (23.3%) and falls (20%). Eleven serious adverse events occurred in 4 patients. None were considered related to treatment, and none led to study discontinuation.
One adverse event was considered related to rFIXFc. A 3-year-old child experienced decreased appetite.
Efficacy
The median annualized bleeding rate (ABR) was 2.0 overall, 1.1 in children under 6, and 2.1 in children ages 6 to 11.
For spontaneous bleeds, the median ABR was 0, both overall and in the 2 age groups. For joint bleeds, the median ABR was 0 overall and in the younger age group, but it was 1.1 for the older children.
Thirty-three percent of patients had no bleeding episodes while on study, and 63% had no joint bleeds.
Ninety-seven percent of patients receiving rFIXFc prophylaxis had no change in their dosing interval.
For patients under 6, the median prophylactic dose was 59.4 IU/kg/week (range, 53.0-64.8). For patients ages 6 to 11, the median dose was 57.8 IU/kg/week (range, 51.7-65.0)
When patients received rFIXFc to treat bleeding, 75% of bleeds were controlled with 1 infusion, and 91.7% were controlled with 1 or 2 infusions. The median dose per infusion was 63.5 IU/kg (range, 48.9-99.4).
Pharmacokinetics
The terminal half-life of rFIXFc was 66.5 hours for children under 6 and 70.3 hours for children ages 6 to 11. The clearance was 4.4 mL/hour/kg and 3.5 mL/hour/kg, respectively. The incremental recovery (IR) was 0.59 IU/dL per IU/kg and 0.72 IU/dL per IU/kg, respectively.
Compared to pre-study treatment with BeneFIX (recombinant FIX) at 50 IU/kg, rFIXFc at 50 IU/kg had a significantly longer half-life. In children younger than 6, the half-life was 66.5 hours for rFIXFc and 18.2 hours for BeneFIX (P<0.001). In children ages 6 to 11, the half-lives were 71.1 and 19.2 hours, respectively (P<0.001).
There was no significant difference between the treatments with regard to IR for children under 6. IR was 0.59 with rFIXFc and 0.52 with BeneFIX (P=0.109). However, there was a significant difference in IR for children ages 6 to 11—0.70 for rFIXFc and 0.54 for BeneFIX (P=0.003). ![]()
Photo courtesy of Biogen
TORONTO—Full results of a phase 3 study support extended-interval dosing with a recombinant factor IX Fc fusion protein (rFIXFc) over FIX products with a standard half-life, according to a speaker at the 2015 ISTH Congress.
Kathelijn Fischer, MD, PhD, of the University Medical Center Utrecht in The Netherlands, reported results with rFIXFc (also known as eftrenonacog alfa and Alprolix), in children with severe hemophilia B who were enrolled on the KIDS B-LONG study.
rFIXFc was successful in preventing and treating bleeding episodes in these patients. Furthermore, the patients did not develop inhibitors, and there were no serious adverse events related to treatment.
Dr Fischer presented these results as abstract LB009. Interim results of this study helped support the US approval of rFIXFc for use in children. The trial was sponsored by Sobi and Biogen, the companies developing rFIXFc.
KIDS B-LONG included 30 boys younger than 12 who had severe hemophilia B. The patients had at least 50 prior exposure days to FIX therapies and no history of inhibitors.
At baseline, all patients were receiving FIX prophylaxis. Seventy-seven percent of patients were receiving 2 or more doses a week.
On day 1 of the study, patients received rFIXFc at 50 IU/kg. They then received weekly prophylaxis at an initial dose of 50 IU/kg to 60 IU/kg. Doses were adjusted throughout the study, but the maximum was 100 IU/kg. The minimum dosing frequency was once a week, and the maximum was twice a week.
Twenty-seven patients (90%) completed the study. The median time spent on study was 49.4 weeks, and 24 patients (80%) received rFIXFc injections on at least 50 separate days.
Safety
None of the patients developed inhibitors or non-neutralizing anti-rFIXFc antibodies. There were no anaphylactic reactions, hypersensitivity reactions, thrombotic events, or deaths.
Adverse events occurred in 86.7% of patients. The most frequent were nanopharyngitis (23.3%) and falls (20%). Eleven serious adverse events occurred in 4 patients. None were considered related to treatment, and none led to study discontinuation.
One adverse event was considered related to rFIXFc. A 3-year-old child experienced decreased appetite.
Efficacy
The median annualized bleeding rate (ABR) was 2.0 overall, 1.1 in children under 6, and 2.1 in children ages 6 to 11.
For spontaneous bleeds, the median ABR was 0, both overall and in the 2 age groups. For joint bleeds, the median ABR was 0 overall and in the younger age group, but it was 1.1 for the older children.
Thirty-three percent of patients had no bleeding episodes while on study, and 63% had no joint bleeds.
Ninety-seven percent of patients receiving rFIXFc prophylaxis had no change in their dosing interval.
For patients under 6, the median prophylactic dose was 59.4 IU/kg/week (range, 53.0-64.8). For patients ages 6 to 11, the median dose was 57.8 IU/kg/week (range, 51.7-65.0)
When patients received rFIXFc to treat bleeding, 75% of bleeds were controlled with 1 infusion, and 91.7% were controlled with 1 or 2 infusions. The median dose per infusion was 63.5 IU/kg (range, 48.9-99.4).
Pharmacokinetics
The terminal half-life of rFIXFc was 66.5 hours for children under 6 and 70.3 hours for children ages 6 to 11. The clearance was 4.4 mL/hour/kg and 3.5 mL/hour/kg, respectively. The incremental recovery (IR) was 0.59 IU/dL per IU/kg and 0.72 IU/dL per IU/kg, respectively.
Compared to pre-study treatment with BeneFIX (recombinant FIX) at 50 IU/kg, rFIXFc at 50 IU/kg had a significantly longer half-life. In children younger than 6, the half-life was 66.5 hours for rFIXFc and 18.2 hours for BeneFIX (P<0.001). In children ages 6 to 11, the half-lives were 71.1 and 19.2 hours, respectively (P<0.001).
There was no significant difference between the treatments with regard to IR for children under 6. IR was 0.59 with rFIXFc and 0.52 with BeneFIX (P=0.109). However, there was a significant difference in IR for children ages 6 to 11—0.70 for rFIXFc and 0.54 for BeneFIX (P=0.003). ![]()
Photo courtesy of Biogen
TORONTO—Full results of a phase 3 study support extended-interval dosing with a recombinant factor IX Fc fusion protein (rFIXFc) over FIX products with a standard half-life, according to a speaker at the 2015 ISTH Congress.
Kathelijn Fischer, MD, PhD, of the University Medical Center Utrecht in The Netherlands, reported results with rFIXFc (also known as eftrenonacog alfa and Alprolix), in children with severe hemophilia B who were enrolled on the KIDS B-LONG study.
rFIXFc was successful in preventing and treating bleeding episodes in these patients. Furthermore, the patients did not develop inhibitors, and there were no serious adverse events related to treatment.
Dr Fischer presented these results as abstract LB009. Interim results of this study helped support the US approval of rFIXFc for use in children. The trial was sponsored by Sobi and Biogen, the companies developing rFIXFc.
KIDS B-LONG included 30 boys younger than 12 who had severe hemophilia B. The patients had at least 50 prior exposure days to FIX therapies and no history of inhibitors.
At baseline, all patients were receiving FIX prophylaxis. Seventy-seven percent of patients were receiving 2 or more doses a week.
On day 1 of the study, patients received rFIXFc at 50 IU/kg. They then received weekly prophylaxis at an initial dose of 50 IU/kg to 60 IU/kg. Doses were adjusted throughout the study, but the maximum was 100 IU/kg. The minimum dosing frequency was once a week, and the maximum was twice a week.
Twenty-seven patients (90%) completed the study. The median time spent on study was 49.4 weeks, and 24 patients (80%) received rFIXFc injections on at least 50 separate days.
Safety
None of the patients developed inhibitors or non-neutralizing anti-rFIXFc antibodies. There were no anaphylactic reactions, hypersensitivity reactions, thrombotic events, or deaths.
Adverse events occurred in 86.7% of patients. The most frequent were nanopharyngitis (23.3%) and falls (20%). Eleven serious adverse events occurred in 4 patients. None were considered related to treatment, and none led to study discontinuation.
One adverse event was considered related to rFIXFc. A 3-year-old child experienced decreased appetite.
Efficacy
The median annualized bleeding rate (ABR) was 2.0 overall, 1.1 in children under 6, and 2.1 in children ages 6 to 11.
For spontaneous bleeds, the median ABR was 0, both overall and in the 2 age groups. For joint bleeds, the median ABR was 0 overall and in the younger age group, but it was 1.1 for the older children.
Thirty-three percent of patients had no bleeding episodes while on study, and 63% had no joint bleeds.
Ninety-seven percent of patients receiving rFIXFc prophylaxis had no change in their dosing interval.
For patients under 6, the median prophylactic dose was 59.4 IU/kg/week (range, 53.0-64.8). For patients ages 6 to 11, the median dose was 57.8 IU/kg/week (range, 51.7-65.0)
When patients received rFIXFc to treat bleeding, 75% of bleeds were controlled with 1 infusion, and 91.7% were controlled with 1 or 2 infusions. The median dose per infusion was 63.5 IU/kg (range, 48.9-99.4).
Pharmacokinetics
The terminal half-life of rFIXFc was 66.5 hours for children under 6 and 70.3 hours for children ages 6 to 11. The clearance was 4.4 mL/hour/kg and 3.5 mL/hour/kg, respectively. The incremental recovery (IR) was 0.59 IU/dL per IU/kg and 0.72 IU/dL per IU/kg, respectively.
Compared to pre-study treatment with BeneFIX (recombinant FIX) at 50 IU/kg, rFIXFc at 50 IU/kg had a significantly longer half-life. In children younger than 6, the half-life was 66.5 hours for rFIXFc and 18.2 hours for BeneFIX (P<0.001). In children ages 6 to 11, the half-lives were 71.1 and 19.2 hours, respectively (P<0.001).
There was no significant difference between the treatments with regard to IR for children under 6. IR was 0.59 with rFIXFc and 0.52 with BeneFIX (P=0.109). However, there was a significant difference in IR for children ages 6 to 11—0.70 for rFIXFc and 0.54 for BeneFIX (P=0.003). ![]()
Drug can produce durable responses in rel/ref PTCL
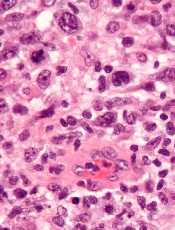
anaplastic large cell lymphoma
The histone deacetylase inhibitor belinostat can produce durable responses in patients with relapsed/refractory peripheral T-cell lymphoma (PTCL), results of the BELIEF study suggest.
The overall response rate (ORR) was low in this heavily pretreated population, at about 26%.
But responses occurred across PTCL subtypes and irrespective of a patient’s prior treatment, and the median duration of response was 13.6 months.
The researchers said toxicity was manageable, and the rate of grade 3/4 thrombocytopenia was low.
“This is a very exciting time in the treatment of patients with PTCL,” said Owen O’Connor, MD, PhD, of Columbia University Medical Center in New York, New York.
“At long last, we finally have tools in the therapeutic armamentarium to help our patients. Belinostat represents the latest drug approved for patients with [relapsed/refractory] PTCL that has relatively few side effects and produces long durations of benefit, even in patients who have received multiple conventional treatments in the past.”
Dr O’Connor and his colleagues reported results with belinostat in the Journal of Clinical Oncology. The research was sponsored by Spectrum Pharmaceuticals, Inc., the company developing belinostat (as Beleodaq).
BELIEF was a single-arm, phase 2 trial that enrolled 129 patients with relapsed/refractory PTCL. One hundred and twenty patients were evaluable. They had a median age of 64 (range, 29-81), and roughly half of patients were female.
PTCL subtypes included PTCL-not otherwise specified (n=77), angioimmunoblastic T-cell lymphoma (n=22), ALK-negative anaplastic large cell lymphoma (n=13), ALK-positive anaplastic large cell lymphoma (n=2), enteropathy-associated T-cell lymphoma (n=2), nasal type extranodal natural killer T-cell lymphoma (n=2), and hepatosplenic T-cell lymphoma (n=2).
The patients had received a median of 2 prior therapies (range, 1-8), including multi-agent and single-agent regimens, as well as transplant.
For this study, the patients received belinostat (1000 mg/m2) as daily, 30-minute infusions on days 1-5 every 21 days until disease progression or unacceptable toxicity.
Response and survival
The study’s primary endpoint was ORR, as assessed by an independent review committee using the International Working Group criteria. The ORR was 25.8% (n=31), including 13 complete responses (10.8%) and 18 partial responses (15%).
The median time to response was 5.6 weeks, and the median duration of response was 13.6 months by International Working Group criteria. The median duration of response based on the date of first response to progressive disease or death was 8.4 months.
Among patients who achieved a complete response, the median duration of response was not reached and exceeded 29 months. The longest ongoing patient response is more than 36 months.
The median progression-free survival was 1.6 months, and the median overall survival was 7.9 months.
Forty-six patients were censored for overall survival because they were alive at the data cutoff point. Seven of these patients continued to receive belinostat. Five were in complete response, 1 had a partial response, and 1 had stable disease.
Twelve patients underwent stem cell transplant after belinostat, and 10 of these patients were still alive at the data cutoff. Their overall survival ranged from 9.4 months to 22.9 months.
Adverse events
Treatment-emergent adverse events (AEs) occurred in 96.9% of patients, and treatment-related AEs occurred in 83.7%. The most common treatment-related AEs were nausea (38.0%), fatigue (28.7%), and vomiting (24.0%). Serious AEs occurred in 47.3% of patients.
Grade 3/4 related AEs were reported in 61.2% of patients, and the most common were anemia (10.9%), thrombocytopenia (7.0%), dyspnea (6.2%), neutropenia (6.2%), fatigue (5.4%), and pneumonia (5.4%).
About 12% of patients underwent a dose reduction due to AEs, 19.4% discontinued treatment due to AEs, and 10.9% of these AEs were considered treatment-related.
Twenty-two patients (17.1%) died, 12 (9.3%) of progressive disease and 10 (7.8%) of AEs. One death was considered related to belinostat. This patient had tolerated 9 cycles of the drug without complications but had elevated liver function tests at the start of cycle 10 that subsequently led to death from toxic liver failure. ![]()
anaplastic large cell lymphoma
The histone deacetylase inhibitor belinostat can produce durable responses in patients with relapsed/refractory peripheral T-cell lymphoma (PTCL), results of the BELIEF study suggest.
The overall response rate (ORR) was low in this heavily pretreated population, at about 26%.
But responses occurred across PTCL subtypes and irrespective of a patient’s prior treatment, and the median duration of response was 13.6 months.
The researchers said toxicity was manageable, and the rate of grade 3/4 thrombocytopenia was low.
“This is a very exciting time in the treatment of patients with PTCL,” said Owen O’Connor, MD, PhD, of Columbia University Medical Center in New York, New York.
“At long last, we finally have tools in the therapeutic armamentarium to help our patients. Belinostat represents the latest drug approved for patients with [relapsed/refractory] PTCL that has relatively few side effects and produces long durations of benefit, even in patients who have received multiple conventional treatments in the past.”
Dr O’Connor and his colleagues reported results with belinostat in the Journal of Clinical Oncology. The research was sponsored by Spectrum Pharmaceuticals, Inc., the company developing belinostat (as Beleodaq).
BELIEF was a single-arm, phase 2 trial that enrolled 129 patients with relapsed/refractory PTCL. One hundred and twenty patients were evaluable. They had a median age of 64 (range, 29-81), and roughly half of patients were female.
PTCL subtypes included PTCL-not otherwise specified (n=77), angioimmunoblastic T-cell lymphoma (n=22), ALK-negative anaplastic large cell lymphoma (n=13), ALK-positive anaplastic large cell lymphoma (n=2), enteropathy-associated T-cell lymphoma (n=2), nasal type extranodal natural killer T-cell lymphoma (n=2), and hepatosplenic T-cell lymphoma (n=2).
The patients had received a median of 2 prior therapies (range, 1-8), including multi-agent and single-agent regimens, as well as transplant.
For this study, the patients received belinostat (1000 mg/m2) as daily, 30-minute infusions on days 1-5 every 21 days until disease progression or unacceptable toxicity.
Response and survival
The study’s primary endpoint was ORR, as assessed by an independent review committee using the International Working Group criteria. The ORR was 25.8% (n=31), including 13 complete responses (10.8%) and 18 partial responses (15%).
The median time to response was 5.6 weeks, and the median duration of response was 13.6 months by International Working Group criteria. The median duration of response based on the date of first response to progressive disease or death was 8.4 months.
Among patients who achieved a complete response, the median duration of response was not reached and exceeded 29 months. The longest ongoing patient response is more than 36 months.
The median progression-free survival was 1.6 months, and the median overall survival was 7.9 months.
Forty-six patients were censored for overall survival because they were alive at the data cutoff point. Seven of these patients continued to receive belinostat. Five were in complete response, 1 had a partial response, and 1 had stable disease.
Twelve patients underwent stem cell transplant after belinostat, and 10 of these patients were still alive at the data cutoff. Their overall survival ranged from 9.4 months to 22.9 months.
Adverse events
Treatment-emergent adverse events (AEs) occurred in 96.9% of patients, and treatment-related AEs occurred in 83.7%. The most common treatment-related AEs were nausea (38.0%), fatigue (28.7%), and vomiting (24.0%). Serious AEs occurred in 47.3% of patients.
Grade 3/4 related AEs were reported in 61.2% of patients, and the most common were anemia (10.9%), thrombocytopenia (7.0%), dyspnea (6.2%), neutropenia (6.2%), fatigue (5.4%), and pneumonia (5.4%).
About 12% of patients underwent a dose reduction due to AEs, 19.4% discontinued treatment due to AEs, and 10.9% of these AEs were considered treatment-related.
Twenty-two patients (17.1%) died, 12 (9.3%) of progressive disease and 10 (7.8%) of AEs. One death was considered related to belinostat. This patient had tolerated 9 cycles of the drug without complications but had elevated liver function tests at the start of cycle 10 that subsequently led to death from toxic liver failure. ![]()
anaplastic large cell lymphoma
The histone deacetylase inhibitor belinostat can produce durable responses in patients with relapsed/refractory peripheral T-cell lymphoma (PTCL), results of the BELIEF study suggest.
The overall response rate (ORR) was low in this heavily pretreated population, at about 26%.
But responses occurred across PTCL subtypes and irrespective of a patient’s prior treatment, and the median duration of response was 13.6 months.
The researchers said toxicity was manageable, and the rate of grade 3/4 thrombocytopenia was low.
“This is a very exciting time in the treatment of patients with PTCL,” said Owen O’Connor, MD, PhD, of Columbia University Medical Center in New York, New York.
“At long last, we finally have tools in the therapeutic armamentarium to help our patients. Belinostat represents the latest drug approved for patients with [relapsed/refractory] PTCL that has relatively few side effects and produces long durations of benefit, even in patients who have received multiple conventional treatments in the past.”
Dr O’Connor and his colleagues reported results with belinostat in the Journal of Clinical Oncology. The research was sponsored by Spectrum Pharmaceuticals, Inc., the company developing belinostat (as Beleodaq).
BELIEF was a single-arm, phase 2 trial that enrolled 129 patients with relapsed/refractory PTCL. One hundred and twenty patients were evaluable. They had a median age of 64 (range, 29-81), and roughly half of patients were female.
PTCL subtypes included PTCL-not otherwise specified (n=77), angioimmunoblastic T-cell lymphoma (n=22), ALK-negative anaplastic large cell lymphoma (n=13), ALK-positive anaplastic large cell lymphoma (n=2), enteropathy-associated T-cell lymphoma (n=2), nasal type extranodal natural killer T-cell lymphoma (n=2), and hepatosplenic T-cell lymphoma (n=2).
The patients had received a median of 2 prior therapies (range, 1-8), including multi-agent and single-agent regimens, as well as transplant.
For this study, the patients received belinostat (1000 mg/m2) as daily, 30-minute infusions on days 1-5 every 21 days until disease progression or unacceptable toxicity.
Response and survival
The study’s primary endpoint was ORR, as assessed by an independent review committee using the International Working Group criteria. The ORR was 25.8% (n=31), including 13 complete responses (10.8%) and 18 partial responses (15%).
The median time to response was 5.6 weeks, and the median duration of response was 13.6 months by International Working Group criteria. The median duration of response based on the date of first response to progressive disease or death was 8.4 months.
Among patients who achieved a complete response, the median duration of response was not reached and exceeded 29 months. The longest ongoing patient response is more than 36 months.
The median progression-free survival was 1.6 months, and the median overall survival was 7.9 months.
Forty-six patients were censored for overall survival because they were alive at the data cutoff point. Seven of these patients continued to receive belinostat. Five were in complete response, 1 had a partial response, and 1 had stable disease.
Twelve patients underwent stem cell transplant after belinostat, and 10 of these patients were still alive at the data cutoff. Their overall survival ranged from 9.4 months to 22.9 months.
Adverse events
Treatment-emergent adverse events (AEs) occurred in 96.9% of patients, and treatment-related AEs occurred in 83.7%. The most common treatment-related AEs were nausea (38.0%), fatigue (28.7%), and vomiting (24.0%). Serious AEs occurred in 47.3% of patients.
Grade 3/4 related AEs were reported in 61.2% of patients, and the most common were anemia (10.9%), thrombocytopenia (7.0%), dyspnea (6.2%), neutropenia (6.2%), fatigue (5.4%), and pneumonia (5.4%).
About 12% of patients underwent a dose reduction due to AEs, 19.4% discontinued treatment due to AEs, and 10.9% of these AEs were considered treatment-related.
Twenty-two patients (17.1%) died, 12 (9.3%) of progressive disease and 10 (7.8%) of AEs. One death was considered related to belinostat. This patient had tolerated 9 cycles of the drug without complications but had elevated liver function tests at the start of cycle 10 that subsequently led to death from toxic liver failure. ![]()
Intervention reduces CLABSIs in pediatric patients
Staphylococcus infection
Photo by Bill Branson
NASHVILLE—A single-center study has shown that incorporating antimicrobial cloths into an infection-prevention protocol can reduce the incidence of central line-associated bloodstream infections (CLABSIs) in pediatric patients.
After the hospital implemented daily “baths” with disposable cloths containing 2% chlorhexidine gluconate (CHG), its CLABSI incidence fell 59% over a 6-month period.
The details of this experience were presented at the APIC 2015 Annual Conference (abstract 013).
The study was conducted at Riley Hospital for Children at Indiana University Health in Indianapolis. The hospital previously used CHG for daily bathing in the hematology/oncology unit and found it successfully reduced CLABSIs there.
This prompted infection preventionists to consider implementing the practice hospital-wide, regardless of whether patients had central-line catheters.
The infection-prevention team worked with nursing staff, parents, and hospital leadership to adopt daily CHG bathing for all patients and to strengthen adherence to a bundle of prevention practices already in place for patients with central lines.
In addition to daily bathing with CHG-impregnated wipes, the strategies included daily linen changes, assessment of central-line dressings, ensuring use of the appropriate technique for giving medications, and regular tubing and cap changes on the lines.
“We took great care to ensure successful implementation of the new bathing regimen,” said Adam N. Karcz, an infection preventionist at the hospital.
“By educating everyone on the care team, including parents, and standardizing bathing procedures, we were able to dramatically reduce infections and save healthcare dollars in just 6 months.”
Bathing compliance increased from 45% to 81% during the 6-month study period. During the control period—6 months prior to implementation—the 269-bed hospital had 22 CLABSIs. During the implementation period, there were 9 CLABSIs.
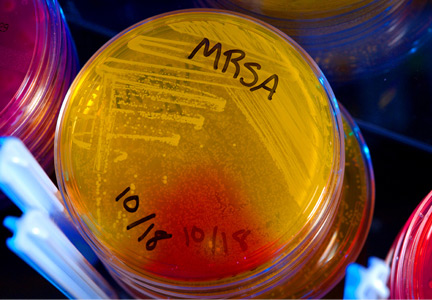
The hospital also experienced a 56% drop in the number of methicillin-resistant Staphylococcus aureus (MRSA) infections during this time period.
The reduction in healthcare-associated infections during the implementation period represents a potential cost savings of $297,999. ![]()
Staphylococcus infection
Photo by Bill Branson
NASHVILLE—A single-center study has shown that incorporating antimicrobial cloths into an infection-prevention protocol can reduce the incidence of central line-associated bloodstream infections (CLABSIs) in pediatric patients.
After the hospital implemented daily “baths” with disposable cloths containing 2% chlorhexidine gluconate (CHG), its CLABSI incidence fell 59% over a 6-month period.
The details of this experience were presented at the APIC 2015 Annual Conference (abstract 013).
The study was conducted at Riley Hospital for Children at Indiana University Health in Indianapolis. The hospital previously used CHG for daily bathing in the hematology/oncology unit and found it successfully reduced CLABSIs there.
This prompted infection preventionists to consider implementing the practice hospital-wide, regardless of whether patients had central-line catheters.
The infection-prevention team worked with nursing staff, parents, and hospital leadership to adopt daily CHG bathing for all patients and to strengthen adherence to a bundle of prevention practices already in place for patients with central lines.
In addition to daily bathing with CHG-impregnated wipes, the strategies included daily linen changes, assessment of central-line dressings, ensuring use of the appropriate technique for giving medications, and regular tubing and cap changes on the lines.
“We took great care to ensure successful implementation of the new bathing regimen,” said Adam N. Karcz, an infection preventionist at the hospital.
“By educating everyone on the care team, including parents, and standardizing bathing procedures, we were able to dramatically reduce infections and save healthcare dollars in just 6 months.”
Bathing compliance increased from 45% to 81% during the 6-month study period. During the control period—6 months prior to implementation—the 269-bed hospital had 22 CLABSIs. During the implementation period, there were 9 CLABSIs.
The hospital also experienced a 56% drop in the number of methicillin-resistant Staphylococcus aureus (MRSA) infections during this time period.
The reduction in healthcare-associated infections during the implementation period represents a potential cost savings of $297,999. ![]()
Staphylococcus infection
Photo by Bill Branson
NASHVILLE—A single-center study has shown that incorporating antimicrobial cloths into an infection-prevention protocol can reduce the incidence of central line-associated bloodstream infections (CLABSIs) in pediatric patients.
After the hospital implemented daily “baths” with disposable cloths containing 2% chlorhexidine gluconate (CHG), its CLABSI incidence fell 59% over a 6-month period.
The details of this experience were presented at the APIC 2015 Annual Conference (abstract 013).
The study was conducted at Riley Hospital for Children at Indiana University Health in Indianapolis. The hospital previously used CHG for daily bathing in the hematology/oncology unit and found it successfully reduced CLABSIs there.
This prompted infection preventionists to consider implementing the practice hospital-wide, regardless of whether patients had central-line catheters.
The infection-prevention team worked with nursing staff, parents, and hospital leadership to adopt daily CHG bathing for all patients and to strengthen adherence to a bundle of prevention practices already in place for patients with central lines.
In addition to daily bathing with CHG-impregnated wipes, the strategies included daily linen changes, assessment of central-line dressings, ensuring use of the appropriate technique for giving medications, and regular tubing and cap changes on the lines.
“We took great care to ensure successful implementation of the new bathing regimen,” said Adam N. Karcz, an infection preventionist at the hospital.
“By educating everyone on the care team, including parents, and standardizing bathing procedures, we were able to dramatically reduce infections and save healthcare dollars in just 6 months.”
Bathing compliance increased from 45% to 81% during the 6-month study period. During the control period—6 months prior to implementation—the 269-bed hospital had 22 CLABSIs. During the implementation period, there were 9 CLABSIs.
The hospital also experienced a 56% drop in the number of methicillin-resistant Staphylococcus aureus (MRSA) infections during this time period.
The reduction in healthcare-associated infections during the implementation period represents a potential cost savings of $297,999. ![]()
Malpractice Counsel
Acute Coronary Syndromes
A 53-year-old obese man presented to the ED complaining of pain in his chest, bilateral jaw, and back. He stated that his symptoms had started the previous evening and had increased in severity the morning of presentation. He denied any change in breathing, or any nausea or vomiting. The patient’s medical history was significant for hypertension and chronic back pain. Regarding his social history, the patient noted that he smoked one pack of cigarettes per day; he denied alcohol use.
On physical examination, the patient’s weight was 350 lb, and he was profusely diaphoretic. Vital signs were remarkable for an elevated blood pressure (BP) of 214/106 mm Hg; respiratory rate (RR), heart rate (HR), temperature (T), and oxygen saturation were normal. The head, eyes, ears, nose, and throat examination was normal, and there was no jugular venous distention. The lung and heart examinations were also normal, and the abdominal examination was unremarkable. The patient had 2+ pitting edema in his lower extremities, which he said had been present for the past few weeks. The back examination was unremarkable, and the neurological examination was completely normal, including deep tendon reflexes.

Because of the presence of chest and back pain and history of hypertension, the EP ordered a computed tomography (CT) scan of the chest with intravenous (IV) contrast to rule out aortic dissection. He also administered 0.2 mg of clonidine orally for the elevated BP. Approximately 20 minutes later, the patient was given 2 mg morphine IV for the back pain and another 0.2 mg of clonidine orally. The elevated BP responded to the clonidine, and the patient stated he was feeling better.
The CT scan of the chest was interpreted by radiology services as normal. The patient was then administered 325 mg of aspirin by mouth. Since the EP’s hospital did not have facilities for cardiac catheterization, the EP consulted with a physician at another facility regarding a possible transfer. The consulted physician did not accept the patient for transfer, but instead recommended keeping the patient at the EP’s institution for observation and continuing treatment for the elevated BP and pain. The EP agreed, and diagnosed the patient with a hypertensive emergency and a flare-up of his chronic back pain.
In the ED, the patient’s BP decreased to near normal levels, and he was feeling much improved. Approximately 5.5 hours after his arrival to the ED, he was admitted to a monitored bed under the care of a hospitalist.
A few hours later, the patient began to complain of burning in the epigastric area; analgesics and nitroglycerin were administered and a repeat ECG was ordered. A second troponin level, drawn approximately 6 hours after the original, was found to be significantly elevated. The repeat ECG demonstrated sinus tachycardia with ST-segment depression.
The hospitalist was concerned about an acute coronary syndrome (ACS) and attempted to make contact with the other facility to transfer the patient for an emergent cardiac catheterization. The consulted physician agreed to accept the patient and recommended starting an IV heparin drip and giving clopidogrel bisulfate (Plavix). While arranging for the transfer, the patient suffered a cardiac arrest; resuscitation attempts were unsuccessful.
The family of the patient sued the hospital, the EP, and the hospitalist, alleging the EP failed to recognize that the initial ECG and elevated troponin level were suggestive of an ACS. They also complained that the morphine, oxygen, nitroglycerin, and aspirin were not started in a timely manner. In addition, the family claimed the decedent should have been immediately transferred to another facility because the defendant’s hospital could not perform cardiac catheterization. They further alleged that the hospitalist failed to perform an independent evaluation of the patient and also failed to obtain a repeat 12-lead ECG sooner. Lastly, the plaintiffs claimed that the hospital’s nursing staff was negligent in failing to provide nursing care for 3 hours prior to the patient being found unresponsive.
The defendant EP asserted that the initial ECG was nondiagnostic and that the initial troponin level, while elevated, was nonspecific. He argued the ED evaluation and care provided was appropriate. Following trial, a defense verdict was returned.
Discussion
Fortunately, the jury ruled correctly in this case. Acute coronary sydromes can be some of the most challenging medical conditions to evaluate and manage in the ED. The EP’s initial cardiac workup and evaluation for a possible acute thoracic aortic dissection were appropriate—an acute thoracic aortic dissection is a true cardiovascular emergency. After interpreting the initial ECG as nondiagnostic (specifically, to rule out evidence of ST-segment elevation, myocardial infarction [MI], or STEMI), obtaining the contrast CT scan of the chest emergently was critically important. This patient had multiple risk factors for aortic dissection: he was a male between the ages of 50 and 55 years old (the mean age for proximal thoracic aortic dissection); he had a history of hypertension; and he was experiencing chest and back pain.1
Once an acute aortic dissection was excluded, focusing on a cardiac etiology, as the EP did, was appropriate. The only criticism is that this patient probably should have been managed with an IV antihypertensive agent to allow for a more controlled BP reduction; this, however, does not seem to have played any role in the patient’s ultimate outcome.
Acute coronary syndromes are a dynamic process and progress over time. The EP was clearly concerned about an ACS very early in the case, as evidenced by his attempt to transfer the patient to a facility with specialized cardiac capabilities. After not being able to do so, the most appropriate next step was his admission of the patient to a monitored bed with serial cardiac enzymes and ECGs. It is well known that initial evaluation of both ECG and cardiac enzymes can be normal early on in an ACS. Patients with a normal or nonspecific ECG have a 1% to 5% incidence of MI and a 4% to 23% incidence of unstable angina.2
This patient ultimately experienced a non-ST-segment elevation myocardial infarction (NSTEMI). However, this diagnosis did not become evident until several hours after the patient’s admission to the hospital. It is unfortunate the physician consulted by the EP at the onset did not agree to accept this patient. This patient’s best chance for survival was at a facility capable of percutaneous coronary intervention.
Serotonin Syndrome
| A 20-year-old man was brought to the ED by his friends for concerns of an overdose. Just prior to arrival, the patient reportedly drank the entire contents of a bottle of cough medicine containing dextromethorphan. His friends reported the patient had been depressed lately, but was otherwise in good health. The patient was not known to abuse alcohol or use illicit drugs. The EP was unable to obtain any history from the patient, who was extremely agitated and yelling frequently. A review of the hospital records revealed the patient had been admitted a few months prior for a suicide attempt. On physical examination, the patient’s vital signs were: pulse, 126 beats/minute; BP, 144/92 mm Hg, RR 22 breaths/minute; and T, 100.6˚F. Oxygen saturation was 99% on room air. The patient was diaphoretic, agitated, and only able to provide one-word answers between screaming episodes. His pupils were mildly dilated but reactive. The cardiac examination revealed a tachycardic rate with a normal rhythm, and no murmurs, rubs, or gallops. The lungs were clear to auscultation bilaterally. The abdomen was soft and nontender, without guarding or rebound. The patient would not cooperate for a neurological examination, but was found to be moving all four extremities with good strength. He was noted to have myoclonus. The EP immediately called the Poison Control Center for advice about treatment. In the meantime, laboratory studies were drawn, including an alcohol level, acetaminophen level, salicylate level, and a urine drug screen. A 12-lead ECG demonstrated a sinus tachycardia with a normal axis. The patient was given IV lorazepam to treat the agitation. The patient’s alcohol, acetaminophen, and salicylate levels were all negative. The EP attempted to transfer the patient to another facility with a higher level of care, but unfortunately, the patient went into cardiac arrest and died in the ED. An autopsy showed that the patient died from serotonin syndrome as a result of acute dextromethorphan and selegiline toxicity. It was later discovered that the patient had been prescribed selegiline as an antidepressant following his recent hospitalization for the suicide attempt. Unfortunately, this information was not available in the records from his previous presentation or from the patient or his friends during the history taking. The patient’s family sued the EP for failing to diagnose serotonin syndrome. They argued the patient did not die from a suicide, but rather from serotonin syndrome. The EP contended the patient had deliberately combined the two drugs to commit suicide. Both parties argued application of the state’s “dead man’s statute” (also known as a “dead man’s act” or “dead man’s rule”). Following trial, a defense verdict was returned. Discussion Serotonin syndrome (or serotonin toxicity) is a drug-induced syndrome characterized by a cluster of dose-related adverse effects due to increased serotonin concentrations in the central nervous system.1 Severe toxicity, as seen in this case, usually occurs only when two or more serotonergic drugs (even when each is at therapeutic dose) are combined. One of the drugs is usually a monoamine oxidase inhibitor (MAOI).1 While selegiline is used primarily as an adjunct treatment for Parkinson disease, it is also used to treat depression, attention deficit and hyperactivity disorder, and Alzheimer disease. Its primary mechanism of action is as an irreversible inhibitor of MAO. Dextromethorphan is used primarily as an antitussive (cough suppressant). It is also used recreationally for its reported effects as a hallucinogen. Its mechanism of action occurs through several effects, one of which is as a nonselective serotonin reuptake inhibitor (NSRI). Although the label on all NSRIs clearly states this medication should not be taken with MAOIs (ie, selegiline), few lay people know the mechanism of action of their medications. The patient in this case took a combination of medications that are known to cause severe serotonin toxicity. It is unclear whether or not he was aware of the dangers associated with combining these two medications. The classic triad of clinical features of serotonin syndrome are neuromuscular excitation (eg, clonus, hyperreflexia, myoclonus, rigidity); autonomic nervous system excitation (eg, hyperthermia, tachycardia); and altered mental status (eg, agitation, confusion).1 The onset of symptoms typically occurs within a few hours of ingestion. Serotonin syndrome can be confused with neuroleptic malignant syndrome (NMS), but there are three key differentiating features: (1) In NMS, symptom onset is slow, usually over days, not hours; (2) extrapyramidal features and rigidity are much more prominent in NMS; and (3) clonus is usually pronounced and easily elicited (especially with ankle dorsiflexion) in serotonin syndrome, but minimal to absent in NMS.1 The initial treatment of serotonin syndrome involves symptomatic care and discontinuation of all serotonergic drugs.2 Benzodiazepines can be used for muscle relaxation and treatment of agitation. All patients with serotonin syndrome require hospital admission, and those with severe toxicity should be admitted to an intensive care unit. Cyproheptadine is the most effective antiserotonergic agent, but it is only available in oral formulation. Chlorpromazine IV has also been used to treat serotonin syndrome, but resulting hypotension is a drawback.1 Approximately 25% of patients with severe serotonin toxicity require intubation and mechanical ventilation. Most patients show dramatic improvement within 24 hours of symptom onset.2 Regarding the dead man statute, according to Cornell University Law School, this statute states that in a civil action, a party with an interest in the litigation may not testify against a dead party about communications with the dead party. This is a state statute and therefore the exact wording varies from state to state. The Federal Rules of Evidence does not contain a dead man’s statute. |
Reference - Acute Coronary Syndromes
- Pacini D, Di Marco L, Fortuna D, et al. Acute aortic dissection: epidemiology and outcomes. Int J Cardiol. 2013;167(6):2806-2812.
- Hollander JE, Diercks DB. Acute coronary syndromes: acute myocardial infarction and unstable angina. In: Tintinalli JE, Stapczynski JS, Cline DM, Ma OJ, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine—A Comprehensive Study Guide. 7th ed. New York; McGraw Hill Medical; 2011:367.
Reference - Serotonin Syndrome
- Buckley NA, Dawson AH, Isbister GK. Serotonin Syndrome. BMJ. 2014;348:g1626.
- Mills KC, Bora KM. Atypical antidepressants, serotonin reuptake inhibitors, and serotonin syndrome. In: Tintinalli JE, Stapczynski JS, Cline DM, Ma OJ, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine—A Comprehensive Study Guide. 7th ed. New York; McGraw Hill Medical; 2011:1202.
Acute Coronary Syndromes
A 53-year-old obese man presented to the ED complaining of pain in his chest, bilateral jaw, and back. He stated that his symptoms had started the previous evening and had increased in severity the morning of presentation. He denied any change in breathing, or any nausea or vomiting. The patient’s medical history was significant for hypertension and chronic back pain. Regarding his social history, the patient noted that he smoked one pack of cigarettes per day; he denied alcohol use.
On physical examination, the patient’s weight was 350 lb, and he was profusely diaphoretic. Vital signs were remarkable for an elevated blood pressure (BP) of 214/106 mm Hg; respiratory rate (RR), heart rate (HR), temperature (T), and oxygen saturation were normal. The head, eyes, ears, nose, and throat examination was normal, and there was no jugular venous distention. The lung and heart examinations were also normal, and the abdominal examination was unremarkable. The patient had 2+ pitting edema in his lower extremities, which he said had been present for the past few weeks. The back examination was unremarkable, and the neurological examination was completely normal, including deep tendon reflexes.
Because of the presence of chest and back pain and history of hypertension, the EP ordered a computed tomography (CT) scan of the chest with intravenous (IV) contrast to rule out aortic dissection. He also administered 0.2 mg of clonidine orally for the elevated BP. Approximately 20 minutes later, the patient was given 2 mg morphine IV for the back pain and another 0.2 mg of clonidine orally. The elevated BP responded to the clonidine, and the patient stated he was feeling better.
The CT scan of the chest was interpreted by radiology services as normal. The patient was then administered 325 mg of aspirin by mouth. Since the EP’s hospital did not have facilities for cardiac catheterization, the EP consulted with a physician at another facility regarding a possible transfer. The consulted physician did not accept the patient for transfer, but instead recommended keeping the patient at the EP’s institution for observation and continuing treatment for the elevated BP and pain. The EP agreed, and diagnosed the patient with a hypertensive emergency and a flare-up of his chronic back pain.
In the ED, the patient’s BP decreased to near normal levels, and he was feeling much improved. Approximately 5.5 hours after his arrival to the ED, he was admitted to a monitored bed under the care of a hospitalist.
A few hours later, the patient began to complain of burning in the epigastric area; analgesics and nitroglycerin were administered and a repeat ECG was ordered. A second troponin level, drawn approximately 6 hours after the original, was found to be significantly elevated. The repeat ECG demonstrated sinus tachycardia with ST-segment depression.
The hospitalist was concerned about an acute coronary syndrome (ACS) and attempted to make contact with the other facility to transfer the patient for an emergent cardiac catheterization. The consulted physician agreed to accept the patient and recommended starting an IV heparin drip and giving clopidogrel bisulfate (Plavix). While arranging for the transfer, the patient suffered a cardiac arrest; resuscitation attempts were unsuccessful.
The family of the patient sued the hospital, the EP, and the hospitalist, alleging the EP failed to recognize that the initial ECG and elevated troponin level were suggestive of an ACS. They also complained that the morphine, oxygen, nitroglycerin, and aspirin were not started in a timely manner. In addition, the family claimed the decedent should have been immediately transferred to another facility because the defendant’s hospital could not perform cardiac catheterization. They further alleged that the hospitalist failed to perform an independent evaluation of the patient and also failed to obtain a repeat 12-lead ECG sooner. Lastly, the plaintiffs claimed that the hospital’s nursing staff was negligent in failing to provide nursing care for 3 hours prior to the patient being found unresponsive.
The defendant EP asserted that the initial ECG was nondiagnostic and that the initial troponin level, while elevated, was nonspecific. He argued the ED evaluation and care provided was appropriate. Following trial, a defense verdict was returned.
Discussion
Fortunately, the jury ruled correctly in this case. Acute coronary sydromes can be some of the most challenging medical conditions to evaluate and manage in the ED. The EP’s initial cardiac workup and evaluation for a possible acute thoracic aortic dissection were appropriate—an acute thoracic aortic dissection is a true cardiovascular emergency. After interpreting the initial ECG as nondiagnostic (specifically, to rule out evidence of ST-segment elevation, myocardial infarction [MI], or STEMI), obtaining the contrast CT scan of the chest emergently was critically important. This patient had multiple risk factors for aortic dissection: he was a male between the ages of 50 and 55 years old (the mean age for proximal thoracic aortic dissection); he had a history of hypertension; and he was experiencing chest and back pain.1
Once an acute aortic dissection was excluded, focusing on a cardiac etiology, as the EP did, was appropriate. The only criticism is that this patient probably should have been managed with an IV antihypertensive agent to allow for a more controlled BP reduction; this, however, does not seem to have played any role in the patient’s ultimate outcome.
Acute coronary syndromes are a dynamic process and progress over time. The EP was clearly concerned about an ACS very early in the case, as evidenced by his attempt to transfer the patient to a facility with specialized cardiac capabilities. After not being able to do so, the most appropriate next step was his admission of the patient to a monitored bed with serial cardiac enzymes and ECGs. It is well known that initial evaluation of both ECG and cardiac enzymes can be normal early on in an ACS. Patients with a normal or nonspecific ECG have a 1% to 5% incidence of MI and a 4% to 23% incidence of unstable angina.2
This patient ultimately experienced a non-ST-segment elevation myocardial infarction (NSTEMI). However, this diagnosis did not become evident until several hours after the patient’s admission to the hospital. It is unfortunate the physician consulted by the EP at the onset did not agree to accept this patient. This patient’s best chance for survival was at a facility capable of percutaneous coronary intervention.
Serotonin Syndrome
| A 20-year-old man was brought to the ED by his friends for concerns of an overdose. Just prior to arrival, the patient reportedly drank the entire contents of a bottle of cough medicine containing dextromethorphan. His friends reported the patient had been depressed lately, but was otherwise in good health. The patient was not known to abuse alcohol or use illicit drugs. The EP was unable to obtain any history from the patient, who was extremely agitated and yelling frequently. A review of the hospital records revealed the patient had been admitted a few months prior for a suicide attempt. On physical examination, the patient’s vital signs were: pulse, 126 beats/minute; BP, 144/92 mm Hg, RR 22 breaths/minute; and T, 100.6˚F. Oxygen saturation was 99% on room air. The patient was diaphoretic, agitated, and only able to provide one-word answers between screaming episodes. His pupils were mildly dilated but reactive. The cardiac examination revealed a tachycardic rate with a normal rhythm, and no murmurs, rubs, or gallops. The lungs were clear to auscultation bilaterally. The abdomen was soft and nontender, without guarding or rebound. The patient would not cooperate for a neurological examination, but was found to be moving all four extremities with good strength. He was noted to have myoclonus. The EP immediately called the Poison Control Center for advice about treatment. In the meantime, laboratory studies were drawn, including an alcohol level, acetaminophen level, salicylate level, and a urine drug screen. A 12-lead ECG demonstrated a sinus tachycardia with a normal axis. The patient was given IV lorazepam to treat the agitation. The patient’s alcohol, acetaminophen, and salicylate levels were all negative. The EP attempted to transfer the patient to another facility with a higher level of care, but unfortunately, the patient went into cardiac arrest and died in the ED. An autopsy showed that the patient died from serotonin syndrome as a result of acute dextromethorphan and selegiline toxicity. It was later discovered that the patient had been prescribed selegiline as an antidepressant following his recent hospitalization for the suicide attempt. Unfortunately, this information was not available in the records from his previous presentation or from the patient or his friends during the history taking. The patient’s family sued the EP for failing to diagnose serotonin syndrome. They argued the patient did not die from a suicide, but rather from serotonin syndrome. The EP contended the patient had deliberately combined the two drugs to commit suicide. Both parties argued application of the state’s “dead man’s statute” (also known as a “dead man’s act” or “dead man’s rule”). Following trial, a defense verdict was returned. Discussion Serotonin syndrome (or serotonin toxicity) is a drug-induced syndrome characterized by a cluster of dose-related adverse effects due to increased serotonin concentrations in the central nervous system.1 Severe toxicity, as seen in this case, usually occurs only when two or more serotonergic drugs (even when each is at therapeutic dose) are combined. One of the drugs is usually a monoamine oxidase inhibitor (MAOI).1 While selegiline is used primarily as an adjunct treatment for Parkinson disease, it is also used to treat depression, attention deficit and hyperactivity disorder, and Alzheimer disease. Its primary mechanism of action is as an irreversible inhibitor of MAO. Dextromethorphan is used primarily as an antitussive (cough suppressant). It is also used recreationally for its reported effects as a hallucinogen. Its mechanism of action occurs through several effects, one of which is as a nonselective serotonin reuptake inhibitor (NSRI). Although the label on all NSRIs clearly states this medication should not be taken with MAOIs (ie, selegiline), few lay people know the mechanism of action of their medications. The patient in this case took a combination of medications that are known to cause severe serotonin toxicity. It is unclear whether or not he was aware of the dangers associated with combining these two medications. The classic triad of clinical features of serotonin syndrome are neuromuscular excitation (eg, clonus, hyperreflexia, myoclonus, rigidity); autonomic nervous system excitation (eg, hyperthermia, tachycardia); and altered mental status (eg, agitation, confusion).1 The onset of symptoms typically occurs within a few hours of ingestion. Serotonin syndrome can be confused with neuroleptic malignant syndrome (NMS), but there are three key differentiating features: (1) In NMS, symptom onset is slow, usually over days, not hours; (2) extrapyramidal features and rigidity are much more prominent in NMS; and (3) clonus is usually pronounced and easily elicited (especially with ankle dorsiflexion) in serotonin syndrome, but minimal to absent in NMS.1 The initial treatment of serotonin syndrome involves symptomatic care and discontinuation of all serotonergic drugs.2 Benzodiazepines can be used for muscle relaxation and treatment of agitation. All patients with serotonin syndrome require hospital admission, and those with severe toxicity should be admitted to an intensive care unit. Cyproheptadine is the most effective antiserotonergic agent, but it is only available in oral formulation. Chlorpromazine IV has also been used to treat serotonin syndrome, but resulting hypotension is a drawback.1 Approximately 25% of patients with severe serotonin toxicity require intubation and mechanical ventilation. Most patients show dramatic improvement within 24 hours of symptom onset.2 Regarding the dead man statute, according to Cornell University Law School, this statute states that in a civil action, a party with an interest in the litigation may not testify against a dead party about communications with the dead party. This is a state statute and therefore the exact wording varies from state to state. The Federal Rules of Evidence does not contain a dead man’s statute. |
Acute Coronary Syndromes
A 53-year-old obese man presented to the ED complaining of pain in his chest, bilateral jaw, and back. He stated that his symptoms had started the previous evening and had increased in severity the morning of presentation. He denied any change in breathing, or any nausea or vomiting. The patient’s medical history was significant for hypertension and chronic back pain. Regarding his social history, the patient noted that he smoked one pack of cigarettes per day; he denied alcohol use.
On physical examination, the patient’s weight was 350 lb, and he was profusely diaphoretic. Vital signs were remarkable for an elevated blood pressure (BP) of 214/106 mm Hg; respiratory rate (RR), heart rate (HR), temperature (T), and oxygen saturation were normal. The head, eyes, ears, nose, and throat examination was normal, and there was no jugular venous distention. The lung and heart examinations were also normal, and the abdominal examination was unremarkable. The patient had 2+ pitting edema in his lower extremities, which he said had been present for the past few weeks. The back examination was unremarkable, and the neurological examination was completely normal, including deep tendon reflexes.
Because of the presence of chest and back pain and history of hypertension, the EP ordered a computed tomography (CT) scan of the chest with intravenous (IV) contrast to rule out aortic dissection. He also administered 0.2 mg of clonidine orally for the elevated BP. Approximately 20 minutes later, the patient was given 2 mg morphine IV for the back pain and another 0.2 mg of clonidine orally. The elevated BP responded to the clonidine, and the patient stated he was feeling better.
The CT scan of the chest was interpreted by radiology services as normal. The patient was then administered 325 mg of aspirin by mouth. Since the EP’s hospital did not have facilities for cardiac catheterization, the EP consulted with a physician at another facility regarding a possible transfer. The consulted physician did not accept the patient for transfer, but instead recommended keeping the patient at the EP’s institution for observation and continuing treatment for the elevated BP and pain. The EP agreed, and diagnosed the patient with a hypertensive emergency and a flare-up of his chronic back pain.
In the ED, the patient’s BP decreased to near normal levels, and he was feeling much improved. Approximately 5.5 hours after his arrival to the ED, he was admitted to a monitored bed under the care of a hospitalist.
A few hours later, the patient began to complain of burning in the epigastric area; analgesics and nitroglycerin were administered and a repeat ECG was ordered. A second troponin level, drawn approximately 6 hours after the original, was found to be significantly elevated. The repeat ECG demonstrated sinus tachycardia with ST-segment depression.
The hospitalist was concerned about an acute coronary syndrome (ACS) and attempted to make contact with the other facility to transfer the patient for an emergent cardiac catheterization. The consulted physician agreed to accept the patient and recommended starting an IV heparin drip and giving clopidogrel bisulfate (Plavix). While arranging for the transfer, the patient suffered a cardiac arrest; resuscitation attempts were unsuccessful.
The family of the patient sued the hospital, the EP, and the hospitalist, alleging the EP failed to recognize that the initial ECG and elevated troponin level were suggestive of an ACS. They also complained that the morphine, oxygen, nitroglycerin, and aspirin were not started in a timely manner. In addition, the family claimed the decedent should have been immediately transferred to another facility because the defendant’s hospital could not perform cardiac catheterization. They further alleged that the hospitalist failed to perform an independent evaluation of the patient and also failed to obtain a repeat 12-lead ECG sooner. Lastly, the plaintiffs claimed that the hospital’s nursing staff was negligent in failing to provide nursing care for 3 hours prior to the patient being found unresponsive.
The defendant EP asserted that the initial ECG was nondiagnostic and that the initial troponin level, while elevated, was nonspecific. He argued the ED evaluation and care provided was appropriate. Following trial, a defense verdict was returned.
Discussion
Fortunately, the jury ruled correctly in this case. Acute coronary sydromes can be some of the most challenging medical conditions to evaluate and manage in the ED. The EP’s initial cardiac workup and evaluation for a possible acute thoracic aortic dissection were appropriate—an acute thoracic aortic dissection is a true cardiovascular emergency. After interpreting the initial ECG as nondiagnostic (specifically, to rule out evidence of ST-segment elevation, myocardial infarction [MI], or STEMI), obtaining the contrast CT scan of the chest emergently was critically important. This patient had multiple risk factors for aortic dissection: he was a male between the ages of 50 and 55 years old (the mean age for proximal thoracic aortic dissection); he had a history of hypertension; and he was experiencing chest and back pain.1
Once an acute aortic dissection was excluded, focusing on a cardiac etiology, as the EP did, was appropriate. The only criticism is that this patient probably should have been managed with an IV antihypertensive agent to allow for a more controlled BP reduction; this, however, does not seem to have played any role in the patient’s ultimate outcome.
Acute coronary syndromes are a dynamic process and progress over time. The EP was clearly concerned about an ACS very early in the case, as evidenced by his attempt to transfer the patient to a facility with specialized cardiac capabilities. After not being able to do so, the most appropriate next step was his admission of the patient to a monitored bed with serial cardiac enzymes and ECGs. It is well known that initial evaluation of both ECG and cardiac enzymes can be normal early on in an ACS. Patients with a normal or nonspecific ECG have a 1% to 5% incidence of MI and a 4% to 23% incidence of unstable angina.2
This patient ultimately experienced a non-ST-segment elevation myocardial infarction (NSTEMI). However, this diagnosis did not become evident until several hours after the patient’s admission to the hospital. It is unfortunate the physician consulted by the EP at the onset did not agree to accept this patient. This patient’s best chance for survival was at a facility capable of percutaneous coronary intervention.
Serotonin Syndrome
| A 20-year-old man was brought to the ED by his friends for concerns of an overdose. Just prior to arrival, the patient reportedly drank the entire contents of a bottle of cough medicine containing dextromethorphan. His friends reported the patient had been depressed lately, but was otherwise in good health. The patient was not known to abuse alcohol or use illicit drugs. The EP was unable to obtain any history from the patient, who was extremely agitated and yelling frequently. A review of the hospital records revealed the patient had been admitted a few months prior for a suicide attempt. On physical examination, the patient’s vital signs were: pulse, 126 beats/minute; BP, 144/92 mm Hg, RR 22 breaths/minute; and T, 100.6˚F. Oxygen saturation was 99% on room air. The patient was diaphoretic, agitated, and only able to provide one-word answers between screaming episodes. His pupils were mildly dilated but reactive. The cardiac examination revealed a tachycardic rate with a normal rhythm, and no murmurs, rubs, or gallops. The lungs were clear to auscultation bilaterally. The abdomen was soft and nontender, without guarding or rebound. The patient would not cooperate for a neurological examination, but was found to be moving all four extremities with good strength. He was noted to have myoclonus. The EP immediately called the Poison Control Center for advice about treatment. In the meantime, laboratory studies were drawn, including an alcohol level, acetaminophen level, salicylate level, and a urine drug screen. A 12-lead ECG demonstrated a sinus tachycardia with a normal axis. The patient was given IV lorazepam to treat the agitation. The patient’s alcohol, acetaminophen, and salicylate levels were all negative. The EP attempted to transfer the patient to another facility with a higher level of care, but unfortunately, the patient went into cardiac arrest and died in the ED. An autopsy showed that the patient died from serotonin syndrome as a result of acute dextromethorphan and selegiline toxicity. It was later discovered that the patient had been prescribed selegiline as an antidepressant following his recent hospitalization for the suicide attempt. Unfortunately, this information was not available in the records from his previous presentation or from the patient or his friends during the history taking. The patient’s family sued the EP for failing to diagnose serotonin syndrome. They argued the patient did not die from a suicide, but rather from serotonin syndrome. The EP contended the patient had deliberately combined the two drugs to commit suicide. Both parties argued application of the state’s “dead man’s statute” (also known as a “dead man’s act” or “dead man’s rule”). Following trial, a defense verdict was returned. Discussion Serotonin syndrome (or serotonin toxicity) is a drug-induced syndrome characterized by a cluster of dose-related adverse effects due to increased serotonin concentrations in the central nervous system.1 Severe toxicity, as seen in this case, usually occurs only when two or more serotonergic drugs (even when each is at therapeutic dose) are combined. One of the drugs is usually a monoamine oxidase inhibitor (MAOI).1 While selegiline is used primarily as an adjunct treatment for Parkinson disease, it is also used to treat depression, attention deficit and hyperactivity disorder, and Alzheimer disease. Its primary mechanism of action is as an irreversible inhibitor of MAO. Dextromethorphan is used primarily as an antitussive (cough suppressant). It is also used recreationally for its reported effects as a hallucinogen. Its mechanism of action occurs through several effects, one of which is as a nonselective serotonin reuptake inhibitor (NSRI). Although the label on all NSRIs clearly states this medication should not be taken with MAOIs (ie, selegiline), few lay people know the mechanism of action of their medications. The patient in this case took a combination of medications that are known to cause severe serotonin toxicity. It is unclear whether or not he was aware of the dangers associated with combining these two medications. The classic triad of clinical features of serotonin syndrome are neuromuscular excitation (eg, clonus, hyperreflexia, myoclonus, rigidity); autonomic nervous system excitation (eg, hyperthermia, tachycardia); and altered mental status (eg, agitation, confusion).1 The onset of symptoms typically occurs within a few hours of ingestion. Serotonin syndrome can be confused with neuroleptic malignant syndrome (NMS), but there are three key differentiating features: (1) In NMS, symptom onset is slow, usually over days, not hours; (2) extrapyramidal features and rigidity are much more prominent in NMS; and (3) clonus is usually pronounced and easily elicited (especially with ankle dorsiflexion) in serotonin syndrome, but minimal to absent in NMS.1 The initial treatment of serotonin syndrome involves symptomatic care and discontinuation of all serotonergic drugs.2 Benzodiazepines can be used for muscle relaxation and treatment of agitation. All patients with serotonin syndrome require hospital admission, and those with severe toxicity should be admitted to an intensive care unit. Cyproheptadine is the most effective antiserotonergic agent, but it is only available in oral formulation. Chlorpromazine IV has also been used to treat serotonin syndrome, but resulting hypotension is a drawback.1 Approximately 25% of patients with severe serotonin toxicity require intubation and mechanical ventilation. Most patients show dramatic improvement within 24 hours of symptom onset.2 Regarding the dead man statute, according to Cornell University Law School, this statute states that in a civil action, a party with an interest in the litigation may not testify against a dead party about communications with the dead party. This is a state statute and therefore the exact wording varies from state to state. The Federal Rules of Evidence does not contain a dead man’s statute. |
Reference - Acute Coronary Syndromes
- Pacini D, Di Marco L, Fortuna D, et al. Acute aortic dissection: epidemiology and outcomes. Int J Cardiol. 2013;167(6):2806-2812.
- Hollander JE, Diercks DB. Acute coronary syndromes: acute myocardial infarction and unstable angina. In: Tintinalli JE, Stapczynski JS, Cline DM, Ma OJ, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine—A Comprehensive Study Guide. 7th ed. New York; McGraw Hill Medical; 2011:367.
Reference - Serotonin Syndrome
- Buckley NA, Dawson AH, Isbister GK. Serotonin Syndrome. BMJ. 2014;348:g1626.
- Mills KC, Bora KM. Atypical antidepressants, serotonin reuptake inhibitors, and serotonin syndrome. In: Tintinalli JE, Stapczynski JS, Cline DM, Ma OJ, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine—A Comprehensive Study Guide. 7th ed. New York; McGraw Hill Medical; 2011:1202.
Reference - Acute Coronary Syndromes
- Pacini D, Di Marco L, Fortuna D, et al. Acute aortic dissection: epidemiology and outcomes. Int J Cardiol. 2013;167(6):2806-2812.
- Hollander JE, Diercks DB. Acute coronary syndromes: acute myocardial infarction and unstable angina. In: Tintinalli JE, Stapczynski JS, Cline DM, Ma OJ, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine—A Comprehensive Study Guide. 7th ed. New York; McGraw Hill Medical; 2011:367.
Reference - Serotonin Syndrome
- Buckley NA, Dawson AH, Isbister GK. Serotonin Syndrome. BMJ. 2014;348:g1626.
- Mills KC, Bora KM. Atypical antidepressants, serotonin reuptake inhibitors, and serotonin syndrome. In: Tintinalli JE, Stapczynski JS, Cline DM, Ma OJ, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine—A Comprehensive Study Guide. 7th ed. New York; McGraw Hill Medical; 2011:1202.

Case Studies in Toxicology: When Doing More for the Sake of Better Health Goes Wrong
Case
A 62-year-old man with a history of hypercholesterolemia and HIV infection presented to the ED for evaluation of diffuse myalgia and tea-colored urine. His medication history included lopinavir/ritonavir (Kaletra) and simvastatin. A week prior to presentation, the patient’s primary care physician had instructed him to increase his daily dose of simvastatin from 40 mg to 80 mg. The patient stated that he had taken simvastatin 80 mg daily for approximately 5 days and then, 2 days prior to presentation, had independently further increased the dose to 160 mg daily.
In the ED, the patient reported feeling fatigued. His initial vital signs were: blood pressure, 129/86 mm Hg; heart rate, 93 beats/minute; respiratory rate, 17 breaths/minute; and temperature, 98.5˚F. Oxygen saturation was 98% on room air. His physical examination was unremarkable. Initial laboratory testing revealed the following: creatine kinase (CK) 350,000 U/L; blood urea nitrogen, 27 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase (AST), 2,950 U/L; and alanine aminotransferase (ALT), 1,305 U/L.
What can cause tea-colored/cola-colored urine and myalgia?
Numerous medications can result in dark-colored urine. These include antimalarial drugs such as chloroquine and primaquine; antibiotics such as metronidazole or nitrofurantoin; and the muscle relaxant methocarbamol. Myalgia and tea-colored urine are the hallmarks of rhabdomyolysis. Rhabdomyolysis involves the destruction of myocytes, which can occur as a result of a long list of processes, including crush injuries, poor oxygenation or perfusion, hypermetabolic states, and direct (or indirect) toxin-mediated myocyte damage.1 The list of toxic substances that can cause rhabdomyolysis is extensive, and statins are one of the most common drug-induced causes (Table).
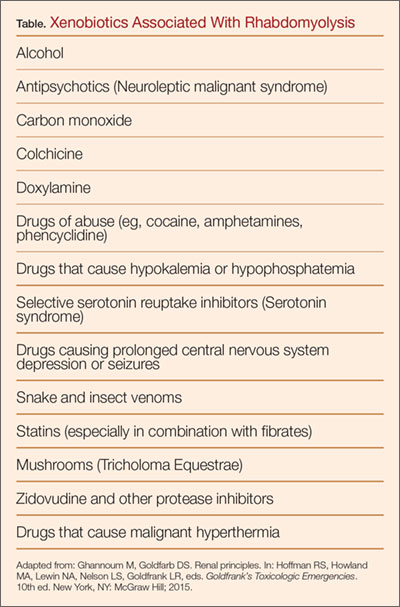
Simvastatin is one of seven currently available 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (ie, statins) that are commonly used to treat hypercholesterolemia. Because simvastatin is lipophilic, it can more readily cross cell membranes than nonlipophilic statins such as pravastatin. Simvastatin, therefore, has a propensity to disrupt the cellular integrity of myocytes and hepatocytes.What is the likely cause of this patient’s rhabdomyolysis?
At doses greater than 40 mg daily, simvastatin is associated with myalgia, myositis, and rhabdomyolysis. In December 2011, the US Food and Drug Administration (FDA) released a drug safety announcement recommending the originally approved maximum daily dose of simvastatin 80 mg be limited to patients who have already tolerated that dose for at least 12 months without evidence of muscular injury. The FDA further recommended no new patients be escalated to this dose. According to the FDA, patients taking 80 mg of simvastatin daily are also at increased risk of myopathy.
The metabolism of simvastatin, in addition to increased dosage of the drug, contributes to its potential for adverse effects. Of the seven available statins, only atorvastatin, lovastatin, and simvastatin are metabolized by the cytochrome P450 3A4 (CYP3A4). Lovastatin and simvastatin appear to have the highest potential for drug-drug interactions when coadministered with drugs that inhibit this enzyme (eg, ritonavir).2 The resulting elevation in blood concentration of simvastatin increases the risk of rhabdomyolysis. Other nonlipophilic statins, such as pravastatin, which are mostly eliminated unchanged in the urine and bile, would be preferable for patients taking CYP3A4 inhibitors.
How should patients with rhabdomyolysis be monitored?
Statins interfere with the myocyte’s ability to produce adenosine triphosphate, most likely by depleting coenzyme Q—one of the complexes found in the electron transport chain of the mitochondria. Under conditions of a high-energy requirement, myocytes incapable of producing sufficient energy ultimately fail and lyse, releasing cellular contents such as CK and myoglobin.1 The serum CK activity serves as a marker of muscle injury and should be monitored closely in patients with rhabdomyolysis. Although values above 5,000 U/L has been associated with renal injury,4 in healthy patients with access to hydration, renal injury is relatively uncommon with CK activities less than 50,000 U/L. Even though the prediction of renal failure is difficult, a validated nephrotoxicity prediction instrument using the patient’s age, gender, and initial laboratory data (serum creatinine, calcium, CK, phosphate, and bicarbonate) is available.5
Although the association between rhabdomyolysis and acute renal injury is well established, the mechanism remains unclear. Myoglobin from skeletal myocytes passes through the glomerulus without causing damage and is reabsorbed in the proximal renal tubular cell. Iron is subsequently released from the porphyrin ring and, in large concentrations, exceeds the binding capacity of the tissue ferritin. Because it is a transition metal, the free iron ion participates in oxidant stress reactions causing direct injury to the renal tubular cells.6 Furthermore, myoglobin also combines with renal tubular proteins, a process enhanced by an environment with lower pH, to form casts and cause renal tubular obstruction.
Patients with rhabdomyolysis may also be at risk for aminotransferase elevation, as occurred in the patient presented here. This elevation is most likely due to myocyte injury. In addition, potassium release due to myocyte destruction may cause life-threatening hyperkalemia, and phosphate liberation from these myocytes may cause hypocalcemia. Laboratory monitoring along with an electrocardiogram should be performed as required.
What is the treatment for rhabdomyolysis?
No adequate randomized controlled trials exist to guide the treatment of patients with rhabdomyolysis. As a result, recommendations for management come from retrospective observational studies, animal studies, case reports, and expert opinion.7
Once airway, breathing, and circulation have been addressed, patients with statin-induced rhabdomyolysis should be immediately treated with intravenous (IV) fluids to maintain renal perfusion, which helps to limit acute renal injury. Normal saline appears to be the most recommended fluid type, with a goal of maintaining a urine output of approximately 3 to 5 mL/kg/h.4,7
Some recommendations include the use of a sodium bicarbonate infusion to raise the urine pH, which may help limit the formation of renal casts from myoglobin. The data to support the benefit of sodium bicarbonate, however, is weak.3 A 2013 systematic review indicated that sodium bicarbonate should only be used to treat severe metabolic acidosis in patients with rhabdomyolysis.4
In addition to sodium bicarbonate, the use of diuretics is also discouraged by current recommendations. In patients with refractory electrolyte abnormalities or renal failure, hemodialysis may be required. Before disposition of a patient, his or her medication list should be reconciled to reflect statin discontinuation.
Case Conclusion
The patient received IV normal saline to maintain his urine output at 2 to 3 cc/kg/h. His repeat creatinine was 0.8 mg/dL and remained stable on repeat testing. His CK and AST concentrations trended down during his hospitalization. On hospital day 4, laboratory values were CK, less than 10,000 U/L; AST, 56 U/L; and ALT, 23 U/L. He had normal serum potassium levels and no dysrhythmia on electrocardiogram. His symptoms resolved on hospital day 2, and he was discharged on hospital day 4 with instructions to discontinue simvastatin.
Dr Fernandez is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
- Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52(10):815-831.
- Brown CV, Rhee P, Chan L, Evans K, Demetriades D, Velmahos GC. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? J Trauma. 2004;56(6):1191-1196.
- Scharman EJ, Troutman WG. Prevention of kidney injury following rhabdomyolysis: a systematic review. Ann Pharmacother. 2013;47(1):90-105.
- McMahon GM, Zeng X, Waikar SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
- Visweswaran P, Guntupalli J. Rhabdomyolysis. Crit Care Clin. 1999;15(2):415-428, ix-x.
- Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013;144(3):1058-1065.
Case
A 62-year-old man with a history of hypercholesterolemia and HIV infection presented to the ED for evaluation of diffuse myalgia and tea-colored urine. His medication history included lopinavir/ritonavir (Kaletra) and simvastatin. A week prior to presentation, the patient’s primary care physician had instructed him to increase his daily dose of simvastatin from 40 mg to 80 mg. The patient stated that he had taken simvastatin 80 mg daily for approximately 5 days and then, 2 days prior to presentation, had independently further increased the dose to 160 mg daily.
In the ED, the patient reported feeling fatigued. His initial vital signs were: blood pressure, 129/86 mm Hg; heart rate, 93 beats/minute; respiratory rate, 17 breaths/minute; and temperature, 98.5˚F. Oxygen saturation was 98% on room air. His physical examination was unremarkable. Initial laboratory testing revealed the following: creatine kinase (CK) 350,000 U/L; blood urea nitrogen, 27 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase (AST), 2,950 U/L; and alanine aminotransferase (ALT), 1,305 U/L.
What can cause tea-colored/cola-colored urine and myalgia?
Numerous medications can result in dark-colored urine. These include antimalarial drugs such as chloroquine and primaquine; antibiotics such as metronidazole or nitrofurantoin; and the muscle relaxant methocarbamol. Myalgia and tea-colored urine are the hallmarks of rhabdomyolysis. Rhabdomyolysis involves the destruction of myocytes, which can occur as a result of a long list of processes, including crush injuries, poor oxygenation or perfusion, hypermetabolic states, and direct (or indirect) toxin-mediated myocyte damage.1 The list of toxic substances that can cause rhabdomyolysis is extensive, and statins are one of the most common drug-induced causes (Table).
Simvastatin is one of seven currently available 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (ie, statins) that are commonly used to treat hypercholesterolemia. Because simvastatin is lipophilic, it can more readily cross cell membranes than nonlipophilic statins such as pravastatin. Simvastatin, therefore, has a propensity to disrupt the cellular integrity of myocytes and hepatocytes.What is the likely cause of this patient’s rhabdomyolysis?
At doses greater than 40 mg daily, simvastatin is associated with myalgia, myositis, and rhabdomyolysis. In December 2011, the US Food and Drug Administration (FDA) released a drug safety announcement recommending the originally approved maximum daily dose of simvastatin 80 mg be limited to patients who have already tolerated that dose for at least 12 months without evidence of muscular injury. The FDA further recommended no new patients be escalated to this dose. According to the FDA, patients taking 80 mg of simvastatin daily are also at increased risk of myopathy.
The metabolism of simvastatin, in addition to increased dosage of the drug, contributes to its potential for adverse effects. Of the seven available statins, only atorvastatin, lovastatin, and simvastatin are metabolized by the cytochrome P450 3A4 (CYP3A4). Lovastatin and simvastatin appear to have the highest potential for drug-drug interactions when coadministered with drugs that inhibit this enzyme (eg, ritonavir).2 The resulting elevation in blood concentration of simvastatin increases the risk of rhabdomyolysis. Other nonlipophilic statins, such as pravastatin, which are mostly eliminated unchanged in the urine and bile, would be preferable for patients taking CYP3A4 inhibitors.
How should patients with rhabdomyolysis be monitored?
Statins interfere with the myocyte’s ability to produce adenosine triphosphate, most likely by depleting coenzyme Q—one of the complexes found in the electron transport chain of the mitochondria. Under conditions of a high-energy requirement, myocytes incapable of producing sufficient energy ultimately fail and lyse, releasing cellular contents such as CK and myoglobin.1 The serum CK activity serves as a marker of muscle injury and should be monitored closely in patients with rhabdomyolysis. Although values above 5,000 U/L has been associated with renal injury,4 in healthy patients with access to hydration, renal injury is relatively uncommon with CK activities less than 50,000 U/L. Even though the prediction of renal failure is difficult, a validated nephrotoxicity prediction instrument using the patient’s age, gender, and initial laboratory data (serum creatinine, calcium, CK, phosphate, and bicarbonate) is available.5
Although the association between rhabdomyolysis and acute renal injury is well established, the mechanism remains unclear. Myoglobin from skeletal myocytes passes through the glomerulus without causing damage and is reabsorbed in the proximal renal tubular cell. Iron is subsequently released from the porphyrin ring and, in large concentrations, exceeds the binding capacity of the tissue ferritin. Because it is a transition metal, the free iron ion participates in oxidant stress reactions causing direct injury to the renal tubular cells.6 Furthermore, myoglobin also combines with renal tubular proteins, a process enhanced by an environment with lower pH, to form casts and cause renal tubular obstruction.
Patients with rhabdomyolysis may also be at risk for aminotransferase elevation, as occurred in the patient presented here. This elevation is most likely due to myocyte injury. In addition, potassium release due to myocyte destruction may cause life-threatening hyperkalemia, and phosphate liberation from these myocytes may cause hypocalcemia. Laboratory monitoring along with an electrocardiogram should be performed as required.
What is the treatment for rhabdomyolysis?
No adequate randomized controlled trials exist to guide the treatment of patients with rhabdomyolysis. As a result, recommendations for management come from retrospective observational studies, animal studies, case reports, and expert opinion.7
Once airway, breathing, and circulation have been addressed, patients with statin-induced rhabdomyolysis should be immediately treated with intravenous (IV) fluids to maintain renal perfusion, which helps to limit acute renal injury. Normal saline appears to be the most recommended fluid type, with a goal of maintaining a urine output of approximately 3 to 5 mL/kg/h.4,7
Some recommendations include the use of a sodium bicarbonate infusion to raise the urine pH, which may help limit the formation of renal casts from myoglobin. The data to support the benefit of sodium bicarbonate, however, is weak.3 A 2013 systematic review indicated that sodium bicarbonate should only be used to treat severe metabolic acidosis in patients with rhabdomyolysis.4
In addition to sodium bicarbonate, the use of diuretics is also discouraged by current recommendations. In patients with refractory electrolyte abnormalities or renal failure, hemodialysis may be required. Before disposition of a patient, his or her medication list should be reconciled to reflect statin discontinuation.
Case Conclusion
The patient received IV normal saline to maintain his urine output at 2 to 3 cc/kg/h. His repeat creatinine was 0.8 mg/dL and remained stable on repeat testing. His CK and AST concentrations trended down during his hospitalization. On hospital day 4, laboratory values were CK, less than 10,000 U/L; AST, 56 U/L; and ALT, 23 U/L. He had normal serum potassium levels and no dysrhythmia on electrocardiogram. His symptoms resolved on hospital day 2, and he was discharged on hospital day 4 with instructions to discontinue simvastatin.
Dr Fernandez is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 62-year-old man with a history of hypercholesterolemia and HIV infection presented to the ED for evaluation of diffuse myalgia and tea-colored urine. His medication history included lopinavir/ritonavir (Kaletra) and simvastatin. A week prior to presentation, the patient’s primary care physician had instructed him to increase his daily dose of simvastatin from 40 mg to 80 mg. The patient stated that he had taken simvastatin 80 mg daily for approximately 5 days and then, 2 days prior to presentation, had independently further increased the dose to 160 mg daily.
In the ED, the patient reported feeling fatigued. His initial vital signs were: blood pressure, 129/86 mm Hg; heart rate, 93 beats/minute; respiratory rate, 17 breaths/minute; and temperature, 98.5˚F. Oxygen saturation was 98% on room air. His physical examination was unremarkable. Initial laboratory testing revealed the following: creatine kinase (CK) 350,000 U/L; blood urea nitrogen, 27 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase (AST), 2,950 U/L; and alanine aminotransferase (ALT), 1,305 U/L.
What can cause tea-colored/cola-colored urine and myalgia?
Numerous medications can result in dark-colored urine. These include antimalarial drugs such as chloroquine and primaquine; antibiotics such as metronidazole or nitrofurantoin; and the muscle relaxant methocarbamol. Myalgia and tea-colored urine are the hallmarks of rhabdomyolysis. Rhabdomyolysis involves the destruction of myocytes, which can occur as a result of a long list of processes, including crush injuries, poor oxygenation or perfusion, hypermetabolic states, and direct (or indirect) toxin-mediated myocyte damage.1 The list of toxic substances that can cause rhabdomyolysis is extensive, and statins are one of the most common drug-induced causes (Table).
Simvastatin is one of seven currently available 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (ie, statins) that are commonly used to treat hypercholesterolemia. Because simvastatin is lipophilic, it can more readily cross cell membranes than nonlipophilic statins such as pravastatin. Simvastatin, therefore, has a propensity to disrupt the cellular integrity of myocytes and hepatocytes.What is the likely cause of this patient’s rhabdomyolysis?
At doses greater than 40 mg daily, simvastatin is associated with myalgia, myositis, and rhabdomyolysis. In December 2011, the US Food and Drug Administration (FDA) released a drug safety announcement recommending the originally approved maximum daily dose of simvastatin 80 mg be limited to patients who have already tolerated that dose for at least 12 months without evidence of muscular injury. The FDA further recommended no new patients be escalated to this dose. According to the FDA, patients taking 80 mg of simvastatin daily are also at increased risk of myopathy.
The metabolism of simvastatin, in addition to increased dosage of the drug, contributes to its potential for adverse effects. Of the seven available statins, only atorvastatin, lovastatin, and simvastatin are metabolized by the cytochrome P450 3A4 (CYP3A4). Lovastatin and simvastatin appear to have the highest potential for drug-drug interactions when coadministered with drugs that inhibit this enzyme (eg, ritonavir).2 The resulting elevation in blood concentration of simvastatin increases the risk of rhabdomyolysis. Other nonlipophilic statins, such as pravastatin, which are mostly eliminated unchanged in the urine and bile, would be preferable for patients taking CYP3A4 inhibitors.
How should patients with rhabdomyolysis be monitored?
Statins interfere with the myocyte’s ability to produce adenosine triphosphate, most likely by depleting coenzyme Q—one of the complexes found in the electron transport chain of the mitochondria. Under conditions of a high-energy requirement, myocytes incapable of producing sufficient energy ultimately fail and lyse, releasing cellular contents such as CK and myoglobin.1 The serum CK activity serves as a marker of muscle injury and should be monitored closely in patients with rhabdomyolysis. Although values above 5,000 U/L has been associated with renal injury,4 in healthy patients with access to hydration, renal injury is relatively uncommon with CK activities less than 50,000 U/L. Even though the prediction of renal failure is difficult, a validated nephrotoxicity prediction instrument using the patient’s age, gender, and initial laboratory data (serum creatinine, calcium, CK, phosphate, and bicarbonate) is available.5
Although the association between rhabdomyolysis and acute renal injury is well established, the mechanism remains unclear. Myoglobin from skeletal myocytes passes through the glomerulus without causing damage and is reabsorbed in the proximal renal tubular cell. Iron is subsequently released from the porphyrin ring and, in large concentrations, exceeds the binding capacity of the tissue ferritin. Because it is a transition metal, the free iron ion participates in oxidant stress reactions causing direct injury to the renal tubular cells.6 Furthermore, myoglobin also combines with renal tubular proteins, a process enhanced by an environment with lower pH, to form casts and cause renal tubular obstruction.
Patients with rhabdomyolysis may also be at risk for aminotransferase elevation, as occurred in the patient presented here. This elevation is most likely due to myocyte injury. In addition, potassium release due to myocyte destruction may cause life-threatening hyperkalemia, and phosphate liberation from these myocytes may cause hypocalcemia. Laboratory monitoring along with an electrocardiogram should be performed as required.
What is the treatment for rhabdomyolysis?
No adequate randomized controlled trials exist to guide the treatment of patients with rhabdomyolysis. As a result, recommendations for management come from retrospective observational studies, animal studies, case reports, and expert opinion.7
Once airway, breathing, and circulation have been addressed, patients with statin-induced rhabdomyolysis should be immediately treated with intravenous (IV) fluids to maintain renal perfusion, which helps to limit acute renal injury. Normal saline appears to be the most recommended fluid type, with a goal of maintaining a urine output of approximately 3 to 5 mL/kg/h.4,7
Some recommendations include the use of a sodium bicarbonate infusion to raise the urine pH, which may help limit the formation of renal casts from myoglobin. The data to support the benefit of sodium bicarbonate, however, is weak.3 A 2013 systematic review indicated that sodium bicarbonate should only be used to treat severe metabolic acidosis in patients with rhabdomyolysis.4
In addition to sodium bicarbonate, the use of diuretics is also discouraged by current recommendations. In patients with refractory electrolyte abnormalities or renal failure, hemodialysis may be required. Before disposition of a patient, his or her medication list should be reconciled to reflect statin discontinuation.
Case Conclusion
The patient received IV normal saline to maintain his urine output at 2 to 3 cc/kg/h. His repeat creatinine was 0.8 mg/dL and remained stable on repeat testing. His CK and AST concentrations trended down during his hospitalization. On hospital day 4, laboratory values were CK, less than 10,000 U/L; AST, 56 U/L; and ALT, 23 U/L. He had normal serum potassium levels and no dysrhythmia on electrocardiogram. His symptoms resolved on hospital day 2, and he was discharged on hospital day 4 with instructions to discontinue simvastatin.
Dr Fernandez is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
- Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52(10):815-831.
- Brown CV, Rhee P, Chan L, Evans K, Demetriades D, Velmahos GC. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? J Trauma. 2004;56(6):1191-1196.
- Scharman EJ, Troutman WG. Prevention of kidney injury following rhabdomyolysis: a systematic review. Ann Pharmacother. 2013;47(1):90-105.
- McMahon GM, Zeng X, Waikar SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
- Visweswaran P, Guntupalli J. Rhabdomyolysis. Crit Care Clin. 1999;15(2):415-428, ix-x.
- Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013;144(3):1058-1065.
- Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
- Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52(10):815-831.
- Brown CV, Rhee P, Chan L, Evans K, Demetriades D, Velmahos GC. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? J Trauma. 2004;56(6):1191-1196.
- Scharman EJ, Troutman WG. Prevention of kidney injury following rhabdomyolysis: a systematic review. Ann Pharmacother. 2013;47(1):90-105.
- McMahon GM, Zeng X, Waikar SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
- Visweswaran P, Guntupalli J. Rhabdomyolysis. Crit Care Clin. 1999;15(2):415-428, ix-x.
- Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013;144(3):1058-1065.
Urologic applications of botulinum toxin
Patients with loss of bladder control experience discomfort, embarrassment, personal care and health issues, and, often, significant pain, all with a decidedly negative impact on quality of life. Although some patients may find lifestyle modifications, drug therapy, and self-catheterization acceptable and effective, there is a clear need for more options.
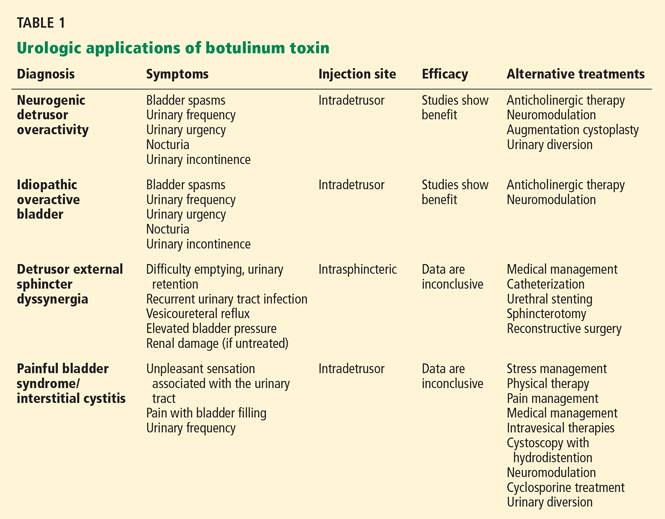
Botulinum toxin, or onabotulinumtoxinA, is currently approved by the US Food and Drug Administration (FDA) for neurogenic detrusor overactivity and overactive bladder refractory to drug therapy. Studies so far have shown botulinum toxin injection to be safe and effective for these conditions, and these results have led to interest in off-label uses, eg, for detrusor external sphincter dyssynergia (DESD), motor and sensory urgency, and painful bladder syndrome/interstitial cystitis (Table 1).
Although more data from clinical trials are needed, botulinum toxin injection offers patients a much-needed treatment option.
HOW BOTULINUM TOXIN WORKS
Seven serotypes identified
Discovered in 1897, botulinum toxin is a neurotoxin produced by the gram-positive, rod-shaped anaerobic bacterium Clostridium botulinum1 and is the most poisonous naturally occurring toxin known.2 Seven immunologically distinct antigenic serotypes have been identified (A, B, C1, D, E, F, and G),1 but only types A and B are available for clinical use.
Most research into potential therapeutic uses has focused on type A, which has the longest duration of action, a clinical advantage.3 Recently, work has been done to further characterize other serotypes and to isolate additional variants of botulinum toxin. For example, serotype E, the predominant serotype associated with foodborne botulism, is being studied in an effort to prevent future outbreaks.4
Our discussion focuses on clinical uses of the serotype A botulinum toxin preparation, which we will refer to simply as botulinum toxin.
Studies exploring how it works
Botulinum toxin exerts its effects by binding to peripheral cholinergic terminals, inhibiting release of acetylcholine at the neuromuscular junction. Flaccid paralysis ensues as a result.
Results of animal studies have shed additional light on the specific actions of botulinum toxin A:
- It may alter levels of nerve growth factor and transient receptor potential vanilloid 1 in rats, and this may provide an additional mechanism of reducing bladder detrusor overactivity.5
- In addition to blocking acetylcholine release from motor neurons, it inhibits the release of neurotransmitters involved in bladder sensory afferent pathways.6
- It inhibits the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6,7
- It promotes apoptosis in prostatic tissue; however, this effect has not been shown in the bladder.3
The time necessary to recover function after botulinum toxin paralysis depends on the subtype of botulinum toxin as well as on the type of nerve terminal. Chemodenervation lasts from 3 to 6 months when the toxin is injected into the neuromuscular junction of skeletal muscle, and considerably longer (up to 1 year) when injected into the autonomic neurons of smooth muscle.2,6
TREATMENT OF NEUROGENIC DETRUSOR OVERACTIVITY
Neurogenic detrusor overactivity involves involuntary contractions of the bladder resulting from spinal cord injury, multiple sclerosis, and other neurologic conditions. An estimated 273,000 people in the United States have a spinal cord injury, and 81% of them have urologic symptoms ranging from areflexia to overactivity.8 From 75% to 100% of patients with multiple sclerosis have urologic symptoms, and detrusor overactivity is the most common.9
Detrusor overactivity can cause urinary urgency, urinary frequency, and urgency incontinence, significantly affecting quality of life and leading to skin breakdown, sacral ulcerations, and challenges with personal care.
Anticholinergic drugs have been the mainstay of therapy. If drug therapy failed, the next option was reconstructive surgery, often augmentation cystoplasty. Thus, botulinum toxin injection is an important advance in treatment options.
Studies that showed effectiveness
Botulinum toxin for neurogenic detrusor overactivity was first studied by Schurch et al.10 In their study, 200 U or 300 U was injected into the trigone of 21 patients with spinal cord injury and urgency incontinence managed with intermittent self-catheterization.10 At 6 weeks after injection, 17 of the 19 patients seen at follow-up visits were completely continent. Urodynamic evaluation revealed significant increases in maximum cystometric capacity and in volume at first involuntary detrusor contraction, and a decrease in detrusor voiding pressure. Of the 11 patients available for follow-up at 16 and 36 weeks, improvements in measures of incontinence and urodynamic function persisted.
In addition, two small randomized controlled trials11,12 showed significant increases in cystometric bladder capacity, significant improvement in quality-of-life measures, and reduction in episodes of urgency incontinence.
In 2011 and 2012, two multicenter double-blind randomized controlled trials reported on patients with multiple sclerosis and spinal cord injury with neurogenic detrusor overactivity inadequately managed with drug therapy. The patients were randomized to botulinum toxin injection (200 U or 300 U) or placebo injection.13,14 The primary end point for both studies was the change from baseline in episodes of urinary incontinence per week at week 6. Secondary end points were maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and score on the Incontinence Quality of Life scale.15
In both studies, the mean number of urinary incontinence episodes per week was 33 at baseline. At week 6, Cruz et al14 found that patients who received botulinum toxin injection had significantly fewer episodes per week (21.8 fewer with 200 U, 19.4 fewer with 300 U) than those in the placebo group, who had 13.2 fewer episodes per week (P < .01). Ginsberg et al13 reported decreases in the mean number of episodes of urinary incontinence of 21, 23, and 9 episodes per week in the 200 U, 300 U, and placebo groups, respectively (P < .001). The patients who received botulinum toxin had statistically significant improvements in maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and Incontinence Quality of Life scores compared with placebo (P < .001). Thirty-eight percent of patients in the treatment group were fully continent.13,14
Safety and adverse effects
The most frequently reported adverse events were urinary tract infection (24% of patients)13,14 and urinary retention requiring initiation of clean intermittent catheterization. In the study by Cruz et al,14 these were reported in 30% with 200 U, 42% with 300 U, and 12% with placebo, while in the study by Ginsberg et al13 they were reported in 35% with 200 U, 42% with 300 U, and 10% with placebo.
In a study of long-term safety and efficacy of botulinum toxin injection in patients with neurogenic detrusor overactivity, Kennelly et al16 found that patients undergoing repeat injections had sustained reductions in episodes of incontinence and increases in the maximum cystometric capacity and quality of life scores, with no increase in adverse events over time.16
But is it cost-effective?
While botulinum toxin injection may be safe and effective for neurogenic detrusor overactivity, is it cost-effective?
Carlson et al17 used a Markov State Transition model to assess the cost of refractory neurogenic detrusor overactivity in patients receiving botulinum toxin vs best supportive care (incontinence pads, medications, intermittent self-catheterization).17 They found that the injections were more expensive than supportive care but were cost-effective when considering the reduction in episodes of incontinence, the reduced need for incontinence products, and improvement in measures of quality of life.
What the evidence indicates
Trials of botulinum toxin injection for neurogenic detrusor overactivity have shown that it improves continence, maximum cystometric capacity, detrusor pressures, and quality of life. The main adverse effects are urinary tract infection and urinary retention requiring intermittent self-catheterization.
Although many patients with this condition are already self-catheterizing, the physician must discuss this before botulinum toxin therapy to ensure that the patient or a family member is able to perform catheterization. Studies have shown that patients have an increase in urinary tract infections after botulinum injections. But in these studies, a urinary tract infection was defined as 100,000 colony-forming units or the presence of leukocytosis with or without symptoms. It is important to remember that patients on intermittent catheterization have bacteriuria and should be treated only for symptomatic, not asymptomatic, bacteriuria.
TREATMENT OF IDIOPATHIC OVERACTIVE BLADDER
Patients with idiopathic overactive bladder have urinary urgency accompanied by urgency incontinence, nocturia, or urinary frequency.18 The prevalence of this condition has been reported to range from 1.7% to 13.3% in men age 30 and older and 7% to 30.3% in women of similar ages. About one-third of women with overactive bladder also have detrusor overactivity.19 Overactive bladder presents a significant economic and medical burden on the healthcare system, as well as having a negative impact on quality of life.
The FDA approved botulinum toxin injection for treatment of idiopathic overactive bladder in January 2013.
Evidence of effectiveness
Two multicenter randomized controlled trials20,21 of botulinum toxin 100 U enrolled patients age 18 and older who had more than three episodes of urinary urgency incontinence in a 3-day period or more than eight micturitions per day inadequately managed by anticholinergic drug therapy. Primary end points were the change from baseline in the number of episodes of urinary incontinence per day and the proportion of patients with a positive response on the Treatment Benefit Scale22 at week 12. Secondary end points included episodes of urinary urgency incontinence, micturition, urgency, and nocturia, and scores on health-related quality of life questionnaires (Incontinence Quality of Life scale, King’s Health Questionnaire).
In both studies, patients receiving botulinum toxin had significantly fewer episodes of incontinence compared with placebo (−2.65 vs −0.87; P < .001 and −2.95 vs −1.03; P < .001).20,21 Reductions from baseline in all other symptoms of overactive bladder, a positive treatment response on the treatment benefit scale, and improvements in quality-of-life scores were also significantly greater with botulinum toxin injection than with placebo (P ≤ .01).
As in the studies of neurogenic detrusor overactivity, the most common adverse effects were urinary tract infection (occurring in 15.5%20 and 24.1%21 of patients) and urinary retention requiring self-catheterization (5.4%20 and 6.9%21).
The largest study to date of anticholinergic therapy vs botulinum toxin injection23 in women with urinary urgency incontinence, published in 2012, studied nearly 250 women who had five or more episodes of idiopathic urgency incontinence in a 3-day period. They were randomized either to daily oral therapy (solifenacin 5 mg with possible escalation to 10 mg and, if necessary, a subsequent switch to extended-release trospium 60 mg) plus one intradetrusor injection of saline, or to a daily oral placebo plus one injection of botulinum toxin 100 U.23
The dropout rate was low in both groups, with 93% of patients in both groups completing the 6-month protocol. Women experienced a mean reduction in urgency incontinence episodes of 3.4 per day (baseline 5) in the anticholinergic group vs 3.3 episodes in the botulinum toxin group (P = .81). However, more patients achieved complete resolution of urinary urgency incontinence in the botulinum toxin group than in the anticholinergic therapy group (27% vs 13%; P = .003). Quality of life improved in both groups without a significant difference between the groups. The botulinum toxin group had higher rates of initiation of self-catheterization (5% vs 0%, P = .01) and urinary tract infection (33% vs 13%, P < .001).23
Botulinum toxin as a third-line therapy
In May 2014, the American Urological Association updated its guidelines on idiopathic overactive bladder24 to include botulinum toxin injection as standard third-line therapy for patients in whom behavioral and medical management (ie, anticholinergics and beta-3-agonists) failed.
Interpreting the evidence to date
Overall, studies in idiopathic overactive bladder have shown a reduction in episodes of urgency incontinence and other symptoms, with some data also demonstrating a corresponding improvement in quality of life.
As in neurogenic detrusor overactivity, the main risks associated with botulinum toxin injection are urinary tract infection and the need to initiate self-catheterization. Although 94% of patients studied did not require self-catheterization after injection, the patient’s ability to perform self-catheterization should be determined before proceeding with botulinum toxin injections.
DETRUSOR EXTERNAL SPHINCTER DYSSYNERGIA
Botulinum toxin has been used not only to improve bladder storage but also to facilitate bladder emptying, as in patients with DESD, a lack of coordination between the bladder and the urinary sphincter. Normal voiding involves relaxation of the urinary sphincter and contraction of the bladder; in DESD the sphincter contracts and works against the bladder’s ability to empty. This leads not only to difficulty emptying the bladder but also to elevated bladder pressure, which can cause renal damage if untreated.
DESD can be seen after injury between the pontine micturition center, which coordinates activity between the bladder and the sphincter, and the caudal spinal cord. This can occur in spinal cord injury, multiple sclerosis, myelomeningocele, and transverse myelitis and can cause significant morbidity for the patient.
Treatment options include drug therapy, injection of botulinum toxin into the sphincter, clean intermittent catheterization, indwelling catheterization, urethral stenting, sphincterotomy, and reconstructive surgery such as urinary diversion.25
The goals of therapy are to avoid the need for clean intermittent catheterization in patients who have difficulty with manual dexterity, and to avoid the need for surgical procedures such as sphincterotomy and urinary diversion. The efficacy of urethral stenting is low, and medical management can be limited.26
In the first published report on botulinum toxin for DESD (in 1988),27 of 11 patients with spinal cord injury and DESD who received botulinum toxin injected into the external urethral sphincter, 10 showed signs of sphincter denervation on electromyography and reductions in urethral pressure profiles and postvoid residual volumes. Schurch et al28 and de Sèze et al29 also reported reductions in postvoid residual volume and maximal urethral pressures in patients with spinal cord injury and DESD.
In 2005, Gallien et al30 reported what is still the largest multicenter randomized controlled trial of botulinum toxin injection in DESD. Eighty-six patients with multiple sclerosis, DESD, and chronic urinary retention were randomized to receive either a single transperineal botulinum toxin injection of 100 U plus the alpha-1-blocker alfuzosin, or a placebo injection plus alfuzosin. Botulinum toxin treatment was associated with significantly increased voided volumes and reduced premicturition and maximal detrusor pressures, but no significant decrease in postvoid residual volume.30
More study needed
Despite these findings, a Cochrane Review concluded that, given the limited experience with intrasphincteric injection of botulinum toxin, data from larger randomized controlled trials are needed before making definitive recommendations.25 In the meantime, the clinician must weigh the low morbidity of the procedure against the limited options in the treatment of these patients.
OFF-LABEL UROLOGIC INDICATIONS
Botulinum toxin injection has been studied off-label for painful bladder syndrome/interstitial cystitis and for chronic prostatic pain. Patients with these conditions often describe pain with filling of the bladder, which leads to urinary frequency in an attempt to relieve the pain.
These pain syndromes can be difficult to treat and can have a devastating impact on quality of life. Treatment options include pain management, stress management, physical therapy, intravesical therapies, cystoscopy with hydrodistention, neuromodulation, cyclosporine, urinary diversion surgery, and botulinum toxin injection (an off-label use).31
In painful bladder syndrome/interstitial cystitis, botulinum toxin is thought to act on sensory afferent pathways, as well as to inhibit the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6 In animal studies,32 botulinum toxin was found to inhibit the afferent neural response by inhibiting mechanoreceptor-mediated release of adenosine triphosphate and by causing a decrease in calcitonin gene-related peptide, which helps regulate micturition and mediates painful bladder sensation.
Clinical studies to date in pelvic pain syndromes
Data from clinical studies of botulinum toxin injection for pelvic pain syndromes are limited. Zermann et al33 performed transurethral perisphincteric injection in 11 men with chronic prostatic pain, 9 of whom reported subjective pain relief, with an average decrease from 7.2 to 1.6 on a visual analogue scale. Postinjection urodynamic studies showed a decrease in functional urethral length, urethral closure pressure, and postvoid residual volume, and an increase in the peak and average flow rates.33
Abbott et al34 evaluated the effect of botulinum toxin injection into the levator ani in 12 women with chronic pelvic pain and pelvic floor hypertonicity. Pelvic floor manometry showed significant reduction in resting muscle pressures and improvements in dyspareunia and nonmenstrual pain. There were also improvements in quality of life and dyschezia, but these were not statistically significant.
Smith et al35 injected botulinum toxin into the detrusor of 13 women with refractory painful bladder syndrome and interstitial cystitis,35 and 9 women (69%) noted statistically significant improvements in the Interstitial Cystitis Symptom Index and Interstitial Cystitis Problem Index, daytime frequency, nocturia, pain, and urodynamic parameters (volume at first desire to void, and maximum cystometric capacity).
In a prospective randomized study of patients with refractory painful bladder syndrome and interstitial cystitis, Kuo and Chancellor36 compared suburothelial injection of 200 U or 100 U of botulinum toxin plus hydrodistention against hydrodistention alone.Patients who received botulinum toxin had increased bladder capacity and improved long-term pain relief, but no difference was noted between 200 U and 100 U, and more adverse effects were seen with the higher dose.36
Pinto et al37 treated 16 women with refractory painful bladder syndrome and interstitial cystitis with intratrigonal injections of botulinum toxin and reported improvements in pain scores, symptom scores, urinary frequency, and quality-of-life measures. The effect lasted 9.9 months (± 2.4 months) and persisted with successive injections.37
More study needed
Although these studies show that botulinum toxin injection for pelvic pain syndromes has the potential to improve pain, urinary frequency, bladder sensation, bladder capacity, and quality of life, larger randomized controlled trials are needed.
Again, the treatment options are limited for refractory painful bladder syndrome and interstitial cystitis. Patients may be desperate for relief from their symptoms. Practitioners must manage expectations and properly inform patients of the potential risks of treatments, especially with patients who will easily agree to further treatment with the smallest hope of relief.
INJECTION TECHNIQUES

For general points about the procedure to discuss with patients, see “What to tell patients.”
Cystoscopic detrusor injection
This procedure is usually done on an outpatient basis (eg, office, ambulatory surgery center). With the patient in the lithotomy position, 100 mL of 2% lidocaine is instilled into the bladder and is allowed 15 to 20 minutes to take effect. A flexible or rigid cystoscope can be used. Depending on the indication, the bladder is injected with 100 U to 300 U of botulinum toxin. The ideal depth of injection is 2 mm in the detrusor muscle, with each injection spaced about 1 cm apart. The recommended administration for 100 U is to inject 20 sites with 0.5 U per mL of saline and, for 200 U, to inject 30 sites with about 0.67 U per mL of saline.38 The location of the injections into the detrusor can vary, as long as adequate spacing is assured.
Injection sites vary. Proponents of injecting the trigone argue that as it is an area of greater nerve density, patients will have a better clinical response. Opponents argue that trigonal injection could result in distal ureteral paralysis and subsequent ureteral reflux. However, this theoretical concern has not been observed clinically.
Urethral injection (off-label use)
The urethra can be injected cystoscopically or periurethrally. Cystoscopic injection involves localization of the external sphincter using the rigid cystoscope and collagen needle; a total of 100 U is injected into the sphincter under direct vision, typically at the 3 o’clock and 9 o’clock positions.35 The periurethral technique is an option in women and involves a spinal needle with 100 U to 200 U of botulinum toxin injected into the external sphincter muscle at the 2 o’clock and 10 o’clock positions.
ADVERSE EFFECTS AND CONTRAINDICATIONS
Adverse effects are rare for urologic applications. The injections are localized, with little systemic absorption, and the doses are 1/1,000th of the theorized lethal dose in a 70-kg male.2 The maximum recommended dose for a 3-month period is 360 U.
Generalized muscle weakness has been reported in a paraplegic patient and in a tetraplegic patient after detrusor injections.2 Interestingly, both patients had return of bladder spasticity within 2 months, prompting speculation about diffusion of botulinum toxin through the bladder wall.2
Repeat injections can cause an immune response in up to 5% of patients.6 Patients undergoing repeat injections are at risk of forming neutralizing antibodies that can interfere with the efficacy of botulinum toxin.6 In a study by Schulte-Baukloh et al, all patients with antibodies to botulinum toxin had a history of recurrent urinary tract infection.39
Botulinum toxin injection is contraindicated in patients with preexisting neuromuscular disease, such as myasthenia gravis, Eaton-Lambert syndrome, and amyotrophic lateral sclerosis. It should also be avoided in patients who are breastfeeding, pregnant, or using agents that potentiate neuromuscular weakness, such as aminoglycosides.
Patients should be informed that some formulations of botulinum toxin include a stabilizer such as albumin derived from human blood, as this may be of religious or cultural significance.
- Leippold T, Reitz A, Schurch B. Botulinum toxin as a new therapy option for voiding disorders: current state of the art. Eur Urol 2003; 44:165–174.
- Sahai A, Khan M, Fowler CJ, Dasgupta P. Botulinum toxin for the treatment of lower urinary tract symptoms: a review. Neurourol Urodyn 2005; 24:2–12.
- Cruz F. Targets for botulinum toxin in the lower urinary tract. Neurourol Urodyn 2014; 33:31–38.
- Weedmark KA, Lambert DL, Mabon P, et al. Two novel toxin variants revealed by whole-genome sequencing of 175 Clostridium botulinum type E strains. Appl Environ Microbiol 2014; 80:6334–6345.
- Ha US, Park EY, Kim JC. Effect of botulinum toxin on expression of nerve growth factor and transient receptor potential vanilloid 1 in urothelium and detrusor muscle of rats with bladder outlet obstruction-induced detrusor overactivity. Urology 2011; 78:721.e1–721.e6
- Frenkl TL, Rackley RR. Injectable neuromodulatory agents: botulinum toxin therapy. Urol Clin North Am 2005; 32:89–99.
- Ikeda Y, Zabbarova IV, Birder LA, et al. Botulinum neurotoxin serotype A suppresses neurotransmitter release from afferent as well as efferent nerves in the urinary bladder. Eur Urol 2012; 62:1157–1164.
- Goldmark E, Niver B, Ginsberg DA. Neurogenic bladder: from diagnosis to management. Curr Urol Rep 2014; 15:448.
- Andersson KE. Current and future drugs for treatment of MS-associated bladder dysfunction. Ann Phys Rehabil Med 2014; 57:321–328.
- Schurch B, Stöhrer M, Kramer G, Schmid DM, Gaul G, Hauri D. Botulinum-A toxin for treating detrusor hyperreflexia in spinal cord injured patients: a new alternative to anticholinergic drugs? Preliminary results. J Urol 2000; 164:692–697.
- Schurch B, de Sèze M, Denys P, et al; Botox Detrusor Hyperreflexia Study Team. Botulinum toxin type a is a safe and effective treatment for neurogenic urinary incontinence: results of a single treatment, randomized, placebo controlled 6-month study. J Urol 2005; 174:196–200.
- Ehren I, Volz D, Farrelly E, et al. Efficacy and impact of botulinum toxin A on quality of life in patients with neurogenic detrusor overactivity: a randomised, placebo-controlled, double-blind study. Scand J Urol Nephrol 2007; 41:335–340.
- Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol 2012; 187:2131–2139.
- Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol 2011; 60:742–750.
- Wagner TH, Patrick DL, Bavendam TG, Martin ML, Buesching DP. Quality of life of persons with urinary incontinence: development of a new measure. Urology 1996: 47:67–71.
- Kennelly M, Dmochowski R, Ethans K, et al. Long-term efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: an interim analysis. Urology 2013; 81:491–497.
- Carlson JJ, Hansen RN, Dmochowski RR, Globe DR, Colayco DC, Sullivan SD. Estimating the cost-effectiveness of onabotulinumtoxinA for neurogenic detrusor overactivity in the United States. Clin Ther 2013; 35:414–424.
- Abrams P, Cardozo L, Fall M, et al; Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology 2003; 61:37–49.
- Milsom I, Coyne KS, Nicholson S, Kvasz M, Chen CI, Wein AJ. Global prevalence and economic burden of urgency urinary incontinence: a systematic review. Eur Urol 2014; 65:79–95.
- Nitti VW, Dmochowski R, Herschorn S, et al; EMBARK Study Group. OnabotulinumtoxinA for the treatment of patients with overactive bladder and urinary incontinence: results of a phase 3, randomized, placebo controlled trial. J Urol 2013; 189:2186–2193.
- Chapple C, Sievert KD, MacDiarmid S, et al. OnabotulinumtoxinA 100 U significantly improves all idiopathic overactive bladder symptoms and quality of life in patients with overactive bladder and urinary incontinence: a randomised, double-blind, placebo-controlled trial. Eur Urol 2013; 64:249–256.
- Colman S, Chapple C, Nitti V, Haag-Molkenteller C, Hastedt C, Massow U. Validation of Treatment Benefit Scale for assessing subjective outcomes in treatment of overactive bladder. Urology 2008; 72:803–807.
- Visco AG, Brubaker L, Richter HE, et al; Pelvic Floor Disorders Network. Anticholinergic therapy vs onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med 2012; 367:1803–1813.
- Gormley EA, Lightner DJ, Burgio KL, et al. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline. www.auanet.org/education/guidelines/overactive-bladder.cfm. Accessed June 11, 2015.
- Utomo E, Groen J, Blok BF. Surgical management of functional bladder outlet obstruction in adults with neurogenic bladder dysfunction. Cochrane Database Syst Rev 2014; 5:CD004927.
- Mahfouz W, Corcos J. Management of detrusor external sphincter dyssynergia in neurogenic bladder. Eur J Phys Rehabil Med 2011; 47:639–650.
- Dykstra DD, Sidi AA, Scott AB, Pagel JM, Goldish GD. Effects of botulinum A toxin on detrusor-sphincter dyssynergia in spinal cord injury patients. J Urol 1988; 139:919–922.
- Schurch B, Hauri D, Rodic B, Curt A, Meyer M, Rossier AB. Botulinum-A toxin as a treatment of detrusor-sphincter dyssynergia: a prospective study in 24 spinal cord injury patients. J Urol 1996; 155:1023–1029.
- de Sèze M, Petit H, Gallien, de Sèze MP, Joseph PA, Mazaux JM, Barat M. Botulinum a toxin and detrusor sphincter dyssynergia: a double-blind lidocaine-controlled study in 13 patients with spinal cord disease. Eur Urol 2002; 42:56–62.
- Gallien P, Reymann JM, Amarenco G, Nicolas B, de Sèze M, Bellissant E. Placebo controlled, randomised, double blind study of the effects of botulinum A toxin on detrusor sphincter dyssynergia in multiple sclerosis patients. J Neurol Neurosurg Psychiatry 2005; 76:1670–1676.
- Hanno PM, Burks DA, Clemens JQ, et al; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc. AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 2011; 185:2162–2170.
- Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol 2004; 172:1529–1532.
- Zermann DH, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol 2000; 38:393–399.
- Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol 2006; 108:915–923.
- Smith CP, Radziszewski P, Borkowski A, Somogyi GT, Boone TB, Chancellor MB. Botulinum toxin A has antinociceptive effects in treating interstitial cystitis. Urology 2004; 64:871–875.
- Kuo HC, Chancellor MB. Comparison of intravesical botulinum toxin type A injections plus hydrodistention with hydrodistention alone for the treatment of refractory interstitial cystitis/painful bladder syndrome. BJU Int 2009: 104:657–661.
- Pinto R, Lopes T, Silva J, Silva C, Dinis P, Cruz F. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol 2013; 189:548–553.
- Rovner E. Chapter 6: Practical aspects of administration of onabotulinumtoxinA. Neurourol Urodyn 2014; 33(suppl 3):S32–S37.
- Schulte-Baukloh H, Herholz J, Bigalke H, Miller K, Knispel HH. Results of a BoNT/A antibody study in children and adolescents after onabotulinumtoxin A (Botox®) detrusor injection. Urol Int 2011; 87:434–438.
Patients with loss of bladder control experience discomfort, embarrassment, personal care and health issues, and, often, significant pain, all with a decidedly negative impact on quality of life. Although some patients may find lifestyle modifications, drug therapy, and self-catheterization acceptable and effective, there is a clear need for more options.
Botulinum toxin, or onabotulinumtoxinA, is currently approved by the US Food and Drug Administration (FDA) for neurogenic detrusor overactivity and overactive bladder refractory to drug therapy. Studies so far have shown botulinum toxin injection to be safe and effective for these conditions, and these results have led to interest in off-label uses, eg, for detrusor external sphincter dyssynergia (DESD), motor and sensory urgency, and painful bladder syndrome/interstitial cystitis (Table 1).
Although more data from clinical trials are needed, botulinum toxin injection offers patients a much-needed treatment option.
HOW BOTULINUM TOXIN WORKS
Seven serotypes identified
Discovered in 1897, botulinum toxin is a neurotoxin produced by the gram-positive, rod-shaped anaerobic bacterium Clostridium botulinum1 and is the most poisonous naturally occurring toxin known.2 Seven immunologically distinct antigenic serotypes have been identified (A, B, C1, D, E, F, and G),1 but only types A and B are available for clinical use.
Most research into potential therapeutic uses has focused on type A, which has the longest duration of action, a clinical advantage.3 Recently, work has been done to further characterize other serotypes and to isolate additional variants of botulinum toxin. For example, serotype E, the predominant serotype associated with foodborne botulism, is being studied in an effort to prevent future outbreaks.4
Our discussion focuses on clinical uses of the serotype A botulinum toxin preparation, which we will refer to simply as botulinum toxin.
Studies exploring how it works
Botulinum toxin exerts its effects by binding to peripheral cholinergic terminals, inhibiting release of acetylcholine at the neuromuscular junction. Flaccid paralysis ensues as a result.
Results of animal studies have shed additional light on the specific actions of botulinum toxin A:
- It may alter levels of nerve growth factor and transient receptor potential vanilloid 1 in rats, and this may provide an additional mechanism of reducing bladder detrusor overactivity.5
- In addition to blocking acetylcholine release from motor neurons, it inhibits the release of neurotransmitters involved in bladder sensory afferent pathways.6
- It inhibits the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6,7
- It promotes apoptosis in prostatic tissue; however, this effect has not been shown in the bladder.3
The time necessary to recover function after botulinum toxin paralysis depends on the subtype of botulinum toxin as well as on the type of nerve terminal. Chemodenervation lasts from 3 to 6 months when the toxin is injected into the neuromuscular junction of skeletal muscle, and considerably longer (up to 1 year) when injected into the autonomic neurons of smooth muscle.2,6
TREATMENT OF NEUROGENIC DETRUSOR OVERACTIVITY
Neurogenic detrusor overactivity involves involuntary contractions of the bladder resulting from spinal cord injury, multiple sclerosis, and other neurologic conditions. An estimated 273,000 people in the United States have a spinal cord injury, and 81% of them have urologic symptoms ranging from areflexia to overactivity.8 From 75% to 100% of patients with multiple sclerosis have urologic symptoms, and detrusor overactivity is the most common.9
Detrusor overactivity can cause urinary urgency, urinary frequency, and urgency incontinence, significantly affecting quality of life and leading to skin breakdown, sacral ulcerations, and challenges with personal care.
Anticholinergic drugs have been the mainstay of therapy. If drug therapy failed, the next option was reconstructive surgery, often augmentation cystoplasty. Thus, botulinum toxin injection is an important advance in treatment options.
Studies that showed effectiveness
Botulinum toxin for neurogenic detrusor overactivity was first studied by Schurch et al.10 In their study, 200 U or 300 U was injected into the trigone of 21 patients with spinal cord injury and urgency incontinence managed with intermittent self-catheterization.10 At 6 weeks after injection, 17 of the 19 patients seen at follow-up visits were completely continent. Urodynamic evaluation revealed significant increases in maximum cystometric capacity and in volume at first involuntary detrusor contraction, and a decrease in detrusor voiding pressure. Of the 11 patients available for follow-up at 16 and 36 weeks, improvements in measures of incontinence and urodynamic function persisted.
In addition, two small randomized controlled trials11,12 showed significant increases in cystometric bladder capacity, significant improvement in quality-of-life measures, and reduction in episodes of urgency incontinence.
In 2011 and 2012, two multicenter double-blind randomized controlled trials reported on patients with multiple sclerosis and spinal cord injury with neurogenic detrusor overactivity inadequately managed with drug therapy. The patients were randomized to botulinum toxin injection (200 U or 300 U) or placebo injection.13,14 The primary end point for both studies was the change from baseline in episodes of urinary incontinence per week at week 6. Secondary end points were maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and score on the Incontinence Quality of Life scale.15
In both studies, the mean number of urinary incontinence episodes per week was 33 at baseline. At week 6, Cruz et al14 found that patients who received botulinum toxin injection had significantly fewer episodes per week (21.8 fewer with 200 U, 19.4 fewer with 300 U) than those in the placebo group, who had 13.2 fewer episodes per week (P < .01). Ginsberg et al13 reported decreases in the mean number of episodes of urinary incontinence of 21, 23, and 9 episodes per week in the 200 U, 300 U, and placebo groups, respectively (P < .001). The patients who received botulinum toxin had statistically significant improvements in maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and Incontinence Quality of Life scores compared with placebo (P < .001). Thirty-eight percent of patients in the treatment group were fully continent.13,14
Safety and adverse effects
The most frequently reported adverse events were urinary tract infection (24% of patients)13,14 and urinary retention requiring initiation of clean intermittent catheterization. In the study by Cruz et al,14 these were reported in 30% with 200 U, 42% with 300 U, and 12% with placebo, while in the study by Ginsberg et al13 they were reported in 35% with 200 U, 42% with 300 U, and 10% with placebo.
In a study of long-term safety and efficacy of botulinum toxin injection in patients with neurogenic detrusor overactivity, Kennelly et al16 found that patients undergoing repeat injections had sustained reductions in episodes of incontinence and increases in the maximum cystometric capacity and quality of life scores, with no increase in adverse events over time.16
But is it cost-effective?
While botulinum toxin injection may be safe and effective for neurogenic detrusor overactivity, is it cost-effective?
Carlson et al17 used a Markov State Transition model to assess the cost of refractory neurogenic detrusor overactivity in patients receiving botulinum toxin vs best supportive care (incontinence pads, medications, intermittent self-catheterization).17 They found that the injections were more expensive than supportive care but were cost-effective when considering the reduction in episodes of incontinence, the reduced need for incontinence products, and improvement in measures of quality of life.
What the evidence indicates
Trials of botulinum toxin injection for neurogenic detrusor overactivity have shown that it improves continence, maximum cystometric capacity, detrusor pressures, and quality of life. The main adverse effects are urinary tract infection and urinary retention requiring intermittent self-catheterization.
Although many patients with this condition are already self-catheterizing, the physician must discuss this before botulinum toxin therapy to ensure that the patient or a family member is able to perform catheterization. Studies have shown that patients have an increase in urinary tract infections after botulinum injections. But in these studies, a urinary tract infection was defined as 100,000 colony-forming units or the presence of leukocytosis with or without symptoms. It is important to remember that patients on intermittent catheterization have bacteriuria and should be treated only for symptomatic, not asymptomatic, bacteriuria.
TREATMENT OF IDIOPATHIC OVERACTIVE BLADDER
Patients with idiopathic overactive bladder have urinary urgency accompanied by urgency incontinence, nocturia, or urinary frequency.18 The prevalence of this condition has been reported to range from 1.7% to 13.3% in men age 30 and older and 7% to 30.3% in women of similar ages. About one-third of women with overactive bladder also have detrusor overactivity.19 Overactive bladder presents a significant economic and medical burden on the healthcare system, as well as having a negative impact on quality of life.
The FDA approved botulinum toxin injection for treatment of idiopathic overactive bladder in January 2013.
Evidence of effectiveness
Two multicenter randomized controlled trials20,21 of botulinum toxin 100 U enrolled patients age 18 and older who had more than three episodes of urinary urgency incontinence in a 3-day period or more than eight micturitions per day inadequately managed by anticholinergic drug therapy. Primary end points were the change from baseline in the number of episodes of urinary incontinence per day and the proportion of patients with a positive response on the Treatment Benefit Scale22 at week 12. Secondary end points included episodes of urinary urgency incontinence, micturition, urgency, and nocturia, and scores on health-related quality of life questionnaires (Incontinence Quality of Life scale, King’s Health Questionnaire).
In both studies, patients receiving botulinum toxin had significantly fewer episodes of incontinence compared with placebo (−2.65 vs −0.87; P < .001 and −2.95 vs −1.03; P < .001).20,21 Reductions from baseline in all other symptoms of overactive bladder, a positive treatment response on the treatment benefit scale, and improvements in quality-of-life scores were also significantly greater with botulinum toxin injection than with placebo (P ≤ .01).
As in the studies of neurogenic detrusor overactivity, the most common adverse effects were urinary tract infection (occurring in 15.5%20 and 24.1%21 of patients) and urinary retention requiring self-catheterization (5.4%20 and 6.9%21).
The largest study to date of anticholinergic therapy vs botulinum toxin injection23 in women with urinary urgency incontinence, published in 2012, studied nearly 250 women who had five or more episodes of idiopathic urgency incontinence in a 3-day period. They were randomized either to daily oral therapy (solifenacin 5 mg with possible escalation to 10 mg and, if necessary, a subsequent switch to extended-release trospium 60 mg) plus one intradetrusor injection of saline, or to a daily oral placebo plus one injection of botulinum toxin 100 U.23
The dropout rate was low in both groups, with 93% of patients in both groups completing the 6-month protocol. Women experienced a mean reduction in urgency incontinence episodes of 3.4 per day (baseline 5) in the anticholinergic group vs 3.3 episodes in the botulinum toxin group (P = .81). However, more patients achieved complete resolution of urinary urgency incontinence in the botulinum toxin group than in the anticholinergic therapy group (27% vs 13%; P = .003). Quality of life improved in both groups without a significant difference between the groups. The botulinum toxin group had higher rates of initiation of self-catheterization (5% vs 0%, P = .01) and urinary tract infection (33% vs 13%, P < .001).23
Botulinum toxin as a third-line therapy
In May 2014, the American Urological Association updated its guidelines on idiopathic overactive bladder24 to include botulinum toxin injection as standard third-line therapy for patients in whom behavioral and medical management (ie, anticholinergics and beta-3-agonists) failed.
Interpreting the evidence to date
Overall, studies in idiopathic overactive bladder have shown a reduction in episodes of urgency incontinence and other symptoms, with some data also demonstrating a corresponding improvement in quality of life.
As in neurogenic detrusor overactivity, the main risks associated with botulinum toxin injection are urinary tract infection and the need to initiate self-catheterization. Although 94% of patients studied did not require self-catheterization after injection, the patient’s ability to perform self-catheterization should be determined before proceeding with botulinum toxin injections.
DETRUSOR EXTERNAL SPHINCTER DYSSYNERGIA
Botulinum toxin has been used not only to improve bladder storage but also to facilitate bladder emptying, as in patients with DESD, a lack of coordination between the bladder and the urinary sphincter. Normal voiding involves relaxation of the urinary sphincter and contraction of the bladder; in DESD the sphincter contracts and works against the bladder’s ability to empty. This leads not only to difficulty emptying the bladder but also to elevated bladder pressure, which can cause renal damage if untreated.
DESD can be seen after injury between the pontine micturition center, which coordinates activity between the bladder and the sphincter, and the caudal spinal cord. This can occur in spinal cord injury, multiple sclerosis, myelomeningocele, and transverse myelitis and can cause significant morbidity for the patient.
Treatment options include drug therapy, injection of botulinum toxin into the sphincter, clean intermittent catheterization, indwelling catheterization, urethral stenting, sphincterotomy, and reconstructive surgery such as urinary diversion.25
The goals of therapy are to avoid the need for clean intermittent catheterization in patients who have difficulty with manual dexterity, and to avoid the need for surgical procedures such as sphincterotomy and urinary diversion. The efficacy of urethral stenting is low, and medical management can be limited.26
In the first published report on botulinum toxin for DESD (in 1988),27 of 11 patients with spinal cord injury and DESD who received botulinum toxin injected into the external urethral sphincter, 10 showed signs of sphincter denervation on electromyography and reductions in urethral pressure profiles and postvoid residual volumes. Schurch et al28 and de Sèze et al29 also reported reductions in postvoid residual volume and maximal urethral pressures in patients with spinal cord injury and DESD.
In 2005, Gallien et al30 reported what is still the largest multicenter randomized controlled trial of botulinum toxin injection in DESD. Eighty-six patients with multiple sclerosis, DESD, and chronic urinary retention were randomized to receive either a single transperineal botulinum toxin injection of 100 U plus the alpha-1-blocker alfuzosin, or a placebo injection plus alfuzosin. Botulinum toxin treatment was associated with significantly increased voided volumes and reduced premicturition and maximal detrusor pressures, but no significant decrease in postvoid residual volume.30
More study needed
Despite these findings, a Cochrane Review concluded that, given the limited experience with intrasphincteric injection of botulinum toxin, data from larger randomized controlled trials are needed before making definitive recommendations.25 In the meantime, the clinician must weigh the low morbidity of the procedure against the limited options in the treatment of these patients.
OFF-LABEL UROLOGIC INDICATIONS
Botulinum toxin injection has been studied off-label for painful bladder syndrome/interstitial cystitis and for chronic prostatic pain. Patients with these conditions often describe pain with filling of the bladder, which leads to urinary frequency in an attempt to relieve the pain.
These pain syndromes can be difficult to treat and can have a devastating impact on quality of life. Treatment options include pain management, stress management, physical therapy, intravesical therapies, cystoscopy with hydrodistention, neuromodulation, cyclosporine, urinary diversion surgery, and botulinum toxin injection (an off-label use).31
In painful bladder syndrome/interstitial cystitis, botulinum toxin is thought to act on sensory afferent pathways, as well as to inhibit the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6 In animal studies,32 botulinum toxin was found to inhibit the afferent neural response by inhibiting mechanoreceptor-mediated release of adenosine triphosphate and by causing a decrease in calcitonin gene-related peptide, which helps regulate micturition and mediates painful bladder sensation.
Clinical studies to date in pelvic pain syndromes
Data from clinical studies of botulinum toxin injection for pelvic pain syndromes are limited. Zermann et al33 performed transurethral perisphincteric injection in 11 men with chronic prostatic pain, 9 of whom reported subjective pain relief, with an average decrease from 7.2 to 1.6 on a visual analogue scale. Postinjection urodynamic studies showed a decrease in functional urethral length, urethral closure pressure, and postvoid residual volume, and an increase in the peak and average flow rates.33
Abbott et al34 evaluated the effect of botulinum toxin injection into the levator ani in 12 women with chronic pelvic pain and pelvic floor hypertonicity. Pelvic floor manometry showed significant reduction in resting muscle pressures and improvements in dyspareunia and nonmenstrual pain. There were also improvements in quality of life and dyschezia, but these were not statistically significant.
Smith et al35 injected botulinum toxin into the detrusor of 13 women with refractory painful bladder syndrome and interstitial cystitis,35 and 9 women (69%) noted statistically significant improvements in the Interstitial Cystitis Symptom Index and Interstitial Cystitis Problem Index, daytime frequency, nocturia, pain, and urodynamic parameters (volume at first desire to void, and maximum cystometric capacity).
In a prospective randomized study of patients with refractory painful bladder syndrome and interstitial cystitis, Kuo and Chancellor36 compared suburothelial injection of 200 U or 100 U of botulinum toxin plus hydrodistention against hydrodistention alone.Patients who received botulinum toxin had increased bladder capacity and improved long-term pain relief, but no difference was noted between 200 U and 100 U, and more adverse effects were seen with the higher dose.36
Pinto et al37 treated 16 women with refractory painful bladder syndrome and interstitial cystitis with intratrigonal injections of botulinum toxin and reported improvements in pain scores, symptom scores, urinary frequency, and quality-of-life measures. The effect lasted 9.9 months (± 2.4 months) and persisted with successive injections.37
More study needed
Although these studies show that botulinum toxin injection for pelvic pain syndromes has the potential to improve pain, urinary frequency, bladder sensation, bladder capacity, and quality of life, larger randomized controlled trials are needed.
Again, the treatment options are limited for refractory painful bladder syndrome and interstitial cystitis. Patients may be desperate for relief from their symptoms. Practitioners must manage expectations and properly inform patients of the potential risks of treatments, especially with patients who will easily agree to further treatment with the smallest hope of relief.
INJECTION TECHNIQUES
For general points about the procedure to discuss with patients, see “What to tell patients.”
Cystoscopic detrusor injection
This procedure is usually done on an outpatient basis (eg, office, ambulatory surgery center). With the patient in the lithotomy position, 100 mL of 2% lidocaine is instilled into the bladder and is allowed 15 to 20 minutes to take effect. A flexible or rigid cystoscope can be used. Depending on the indication, the bladder is injected with 100 U to 300 U of botulinum toxin. The ideal depth of injection is 2 mm in the detrusor muscle, with each injection spaced about 1 cm apart. The recommended administration for 100 U is to inject 20 sites with 0.5 U per mL of saline and, for 200 U, to inject 30 sites with about 0.67 U per mL of saline.38 The location of the injections into the detrusor can vary, as long as adequate spacing is assured.
Injection sites vary. Proponents of injecting the trigone argue that as it is an area of greater nerve density, patients will have a better clinical response. Opponents argue that trigonal injection could result in distal ureteral paralysis and subsequent ureteral reflux. However, this theoretical concern has not been observed clinically.
Urethral injection (off-label use)
The urethra can be injected cystoscopically or periurethrally. Cystoscopic injection involves localization of the external sphincter using the rigid cystoscope and collagen needle; a total of 100 U is injected into the sphincter under direct vision, typically at the 3 o’clock and 9 o’clock positions.35 The periurethral technique is an option in women and involves a spinal needle with 100 U to 200 U of botulinum toxin injected into the external sphincter muscle at the 2 o’clock and 10 o’clock positions.
ADVERSE EFFECTS AND CONTRAINDICATIONS
Adverse effects are rare for urologic applications. The injections are localized, with little systemic absorption, and the doses are 1/1,000th of the theorized lethal dose in a 70-kg male.2 The maximum recommended dose for a 3-month period is 360 U.
Generalized muscle weakness has been reported in a paraplegic patient and in a tetraplegic patient after detrusor injections.2 Interestingly, both patients had return of bladder spasticity within 2 months, prompting speculation about diffusion of botulinum toxin through the bladder wall.2
Repeat injections can cause an immune response in up to 5% of patients.6 Patients undergoing repeat injections are at risk of forming neutralizing antibodies that can interfere with the efficacy of botulinum toxin.6 In a study by Schulte-Baukloh et al, all patients with antibodies to botulinum toxin had a history of recurrent urinary tract infection.39
Botulinum toxin injection is contraindicated in patients with preexisting neuromuscular disease, such as myasthenia gravis, Eaton-Lambert syndrome, and amyotrophic lateral sclerosis. It should also be avoided in patients who are breastfeeding, pregnant, or using agents that potentiate neuromuscular weakness, such as aminoglycosides.
Patients should be informed that some formulations of botulinum toxin include a stabilizer such as albumin derived from human blood, as this may be of religious or cultural significance.
Patients with loss of bladder control experience discomfort, embarrassment, personal care and health issues, and, often, significant pain, all with a decidedly negative impact on quality of life. Although some patients may find lifestyle modifications, drug therapy, and self-catheterization acceptable and effective, there is a clear need for more options.
Botulinum toxin, or onabotulinumtoxinA, is currently approved by the US Food and Drug Administration (FDA) for neurogenic detrusor overactivity and overactive bladder refractory to drug therapy. Studies so far have shown botulinum toxin injection to be safe and effective for these conditions, and these results have led to interest in off-label uses, eg, for detrusor external sphincter dyssynergia (DESD), motor and sensory urgency, and painful bladder syndrome/interstitial cystitis (Table 1).
Although more data from clinical trials are needed, botulinum toxin injection offers patients a much-needed treatment option.
HOW BOTULINUM TOXIN WORKS
Seven serotypes identified
Discovered in 1897, botulinum toxin is a neurotoxin produced by the gram-positive, rod-shaped anaerobic bacterium Clostridium botulinum1 and is the most poisonous naturally occurring toxin known.2 Seven immunologically distinct antigenic serotypes have been identified (A, B, C1, D, E, F, and G),1 but only types A and B are available for clinical use.
Most research into potential therapeutic uses has focused on type A, which has the longest duration of action, a clinical advantage.3 Recently, work has been done to further characterize other serotypes and to isolate additional variants of botulinum toxin. For example, serotype E, the predominant serotype associated with foodborne botulism, is being studied in an effort to prevent future outbreaks.4
Our discussion focuses on clinical uses of the serotype A botulinum toxin preparation, which we will refer to simply as botulinum toxin.
Studies exploring how it works
Botulinum toxin exerts its effects by binding to peripheral cholinergic terminals, inhibiting release of acetylcholine at the neuromuscular junction. Flaccid paralysis ensues as a result.
Results of animal studies have shed additional light on the specific actions of botulinum toxin A:
- It may alter levels of nerve growth factor and transient receptor potential vanilloid 1 in rats, and this may provide an additional mechanism of reducing bladder detrusor overactivity.5
- In addition to blocking acetylcholine release from motor neurons, it inhibits the release of neurotransmitters involved in bladder sensory afferent pathways.6
- It inhibits the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6,7
- It promotes apoptosis in prostatic tissue; however, this effect has not been shown in the bladder.3
The time necessary to recover function after botulinum toxin paralysis depends on the subtype of botulinum toxin as well as on the type of nerve terminal. Chemodenervation lasts from 3 to 6 months when the toxin is injected into the neuromuscular junction of skeletal muscle, and considerably longer (up to 1 year) when injected into the autonomic neurons of smooth muscle.2,6
TREATMENT OF NEUROGENIC DETRUSOR OVERACTIVITY
Neurogenic detrusor overactivity involves involuntary contractions of the bladder resulting from spinal cord injury, multiple sclerosis, and other neurologic conditions. An estimated 273,000 people in the United States have a spinal cord injury, and 81% of them have urologic symptoms ranging from areflexia to overactivity.8 From 75% to 100% of patients with multiple sclerosis have urologic symptoms, and detrusor overactivity is the most common.9
Detrusor overactivity can cause urinary urgency, urinary frequency, and urgency incontinence, significantly affecting quality of life and leading to skin breakdown, sacral ulcerations, and challenges with personal care.
Anticholinergic drugs have been the mainstay of therapy. If drug therapy failed, the next option was reconstructive surgery, often augmentation cystoplasty. Thus, botulinum toxin injection is an important advance in treatment options.
Studies that showed effectiveness
Botulinum toxin for neurogenic detrusor overactivity was first studied by Schurch et al.10 In their study, 200 U or 300 U was injected into the trigone of 21 patients with spinal cord injury and urgency incontinence managed with intermittent self-catheterization.10 At 6 weeks after injection, 17 of the 19 patients seen at follow-up visits were completely continent. Urodynamic evaluation revealed significant increases in maximum cystometric capacity and in volume at first involuntary detrusor contraction, and a decrease in detrusor voiding pressure. Of the 11 patients available for follow-up at 16 and 36 weeks, improvements in measures of incontinence and urodynamic function persisted.
In addition, two small randomized controlled trials11,12 showed significant increases in cystometric bladder capacity, significant improvement in quality-of-life measures, and reduction in episodes of urgency incontinence.
In 2011 and 2012, two multicenter double-blind randomized controlled trials reported on patients with multiple sclerosis and spinal cord injury with neurogenic detrusor overactivity inadequately managed with drug therapy. The patients were randomized to botulinum toxin injection (200 U or 300 U) or placebo injection.13,14 The primary end point for both studies was the change from baseline in episodes of urinary incontinence per week at week 6. Secondary end points were maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and score on the Incontinence Quality of Life scale.15
In both studies, the mean number of urinary incontinence episodes per week was 33 at baseline. At week 6, Cruz et al14 found that patients who received botulinum toxin injection had significantly fewer episodes per week (21.8 fewer with 200 U, 19.4 fewer with 300 U) than those in the placebo group, who had 13.2 fewer episodes per week (P < .01). Ginsberg et al13 reported decreases in the mean number of episodes of urinary incontinence of 21, 23, and 9 episodes per week in the 200 U, 300 U, and placebo groups, respectively (P < .001). The patients who received botulinum toxin had statistically significant improvements in maximum cystometric capacity, maximum detrusor pressure during first involuntary detrusor contraction, and Incontinence Quality of Life scores compared with placebo (P < .001). Thirty-eight percent of patients in the treatment group were fully continent.13,14
Safety and adverse effects
The most frequently reported adverse events were urinary tract infection (24% of patients)13,14 and urinary retention requiring initiation of clean intermittent catheterization. In the study by Cruz et al,14 these were reported in 30% with 200 U, 42% with 300 U, and 12% with placebo, while in the study by Ginsberg et al13 they were reported in 35% with 200 U, 42% with 300 U, and 10% with placebo.
In a study of long-term safety and efficacy of botulinum toxin injection in patients with neurogenic detrusor overactivity, Kennelly et al16 found that patients undergoing repeat injections had sustained reductions in episodes of incontinence and increases in the maximum cystometric capacity and quality of life scores, with no increase in adverse events over time.16
But is it cost-effective?
While botulinum toxin injection may be safe and effective for neurogenic detrusor overactivity, is it cost-effective?
Carlson et al17 used a Markov State Transition model to assess the cost of refractory neurogenic detrusor overactivity in patients receiving botulinum toxin vs best supportive care (incontinence pads, medications, intermittent self-catheterization).17 They found that the injections were more expensive than supportive care but were cost-effective when considering the reduction in episodes of incontinence, the reduced need for incontinence products, and improvement in measures of quality of life.
What the evidence indicates
Trials of botulinum toxin injection for neurogenic detrusor overactivity have shown that it improves continence, maximum cystometric capacity, detrusor pressures, and quality of life. The main adverse effects are urinary tract infection and urinary retention requiring intermittent self-catheterization.
Although many patients with this condition are already self-catheterizing, the physician must discuss this before botulinum toxin therapy to ensure that the patient or a family member is able to perform catheterization. Studies have shown that patients have an increase in urinary tract infections after botulinum injections. But in these studies, a urinary tract infection was defined as 100,000 colony-forming units or the presence of leukocytosis with or without symptoms. It is important to remember that patients on intermittent catheterization have bacteriuria and should be treated only for symptomatic, not asymptomatic, bacteriuria.
TREATMENT OF IDIOPATHIC OVERACTIVE BLADDER
Patients with idiopathic overactive bladder have urinary urgency accompanied by urgency incontinence, nocturia, or urinary frequency.18 The prevalence of this condition has been reported to range from 1.7% to 13.3% in men age 30 and older and 7% to 30.3% in women of similar ages. About one-third of women with overactive bladder also have detrusor overactivity.19 Overactive bladder presents a significant economic and medical burden on the healthcare system, as well as having a negative impact on quality of life.
The FDA approved botulinum toxin injection for treatment of idiopathic overactive bladder in January 2013.
Evidence of effectiveness
Two multicenter randomized controlled trials20,21 of botulinum toxin 100 U enrolled patients age 18 and older who had more than three episodes of urinary urgency incontinence in a 3-day period or more than eight micturitions per day inadequately managed by anticholinergic drug therapy. Primary end points were the change from baseline in the number of episodes of urinary incontinence per day and the proportion of patients with a positive response on the Treatment Benefit Scale22 at week 12. Secondary end points included episodes of urinary urgency incontinence, micturition, urgency, and nocturia, and scores on health-related quality of life questionnaires (Incontinence Quality of Life scale, King’s Health Questionnaire).
In both studies, patients receiving botulinum toxin had significantly fewer episodes of incontinence compared with placebo (−2.65 vs −0.87; P < .001 and −2.95 vs −1.03; P < .001).20,21 Reductions from baseline in all other symptoms of overactive bladder, a positive treatment response on the treatment benefit scale, and improvements in quality-of-life scores were also significantly greater with botulinum toxin injection than with placebo (P ≤ .01).
As in the studies of neurogenic detrusor overactivity, the most common adverse effects were urinary tract infection (occurring in 15.5%20 and 24.1%21 of patients) and urinary retention requiring self-catheterization (5.4%20 and 6.9%21).
The largest study to date of anticholinergic therapy vs botulinum toxin injection23 in women with urinary urgency incontinence, published in 2012, studied nearly 250 women who had five or more episodes of idiopathic urgency incontinence in a 3-day period. They were randomized either to daily oral therapy (solifenacin 5 mg with possible escalation to 10 mg and, if necessary, a subsequent switch to extended-release trospium 60 mg) plus one intradetrusor injection of saline, or to a daily oral placebo plus one injection of botulinum toxin 100 U.23
The dropout rate was low in both groups, with 93% of patients in both groups completing the 6-month protocol. Women experienced a mean reduction in urgency incontinence episodes of 3.4 per day (baseline 5) in the anticholinergic group vs 3.3 episodes in the botulinum toxin group (P = .81). However, more patients achieved complete resolution of urinary urgency incontinence in the botulinum toxin group than in the anticholinergic therapy group (27% vs 13%; P = .003). Quality of life improved in both groups without a significant difference between the groups. The botulinum toxin group had higher rates of initiation of self-catheterization (5% vs 0%, P = .01) and urinary tract infection (33% vs 13%, P < .001).23
Botulinum toxin as a third-line therapy
In May 2014, the American Urological Association updated its guidelines on idiopathic overactive bladder24 to include botulinum toxin injection as standard third-line therapy for patients in whom behavioral and medical management (ie, anticholinergics and beta-3-agonists) failed.
Interpreting the evidence to date
Overall, studies in idiopathic overactive bladder have shown a reduction in episodes of urgency incontinence and other symptoms, with some data also demonstrating a corresponding improvement in quality of life.
As in neurogenic detrusor overactivity, the main risks associated with botulinum toxin injection are urinary tract infection and the need to initiate self-catheterization. Although 94% of patients studied did not require self-catheterization after injection, the patient’s ability to perform self-catheterization should be determined before proceeding with botulinum toxin injections.
DETRUSOR EXTERNAL SPHINCTER DYSSYNERGIA
Botulinum toxin has been used not only to improve bladder storage but also to facilitate bladder emptying, as in patients with DESD, a lack of coordination between the bladder and the urinary sphincter. Normal voiding involves relaxation of the urinary sphincter and contraction of the bladder; in DESD the sphincter contracts and works against the bladder’s ability to empty. This leads not only to difficulty emptying the bladder but also to elevated bladder pressure, which can cause renal damage if untreated.
DESD can be seen after injury between the pontine micturition center, which coordinates activity between the bladder and the sphincter, and the caudal spinal cord. This can occur in spinal cord injury, multiple sclerosis, myelomeningocele, and transverse myelitis and can cause significant morbidity for the patient.
Treatment options include drug therapy, injection of botulinum toxin into the sphincter, clean intermittent catheterization, indwelling catheterization, urethral stenting, sphincterotomy, and reconstructive surgery such as urinary diversion.25
The goals of therapy are to avoid the need for clean intermittent catheterization in patients who have difficulty with manual dexterity, and to avoid the need for surgical procedures such as sphincterotomy and urinary diversion. The efficacy of urethral stenting is low, and medical management can be limited.26
In the first published report on botulinum toxin for DESD (in 1988),27 of 11 patients with spinal cord injury and DESD who received botulinum toxin injected into the external urethral sphincter, 10 showed signs of sphincter denervation on electromyography and reductions in urethral pressure profiles and postvoid residual volumes. Schurch et al28 and de Sèze et al29 also reported reductions in postvoid residual volume and maximal urethral pressures in patients with spinal cord injury and DESD.
In 2005, Gallien et al30 reported what is still the largest multicenter randomized controlled trial of botulinum toxin injection in DESD. Eighty-six patients with multiple sclerosis, DESD, and chronic urinary retention were randomized to receive either a single transperineal botulinum toxin injection of 100 U plus the alpha-1-blocker alfuzosin, or a placebo injection plus alfuzosin. Botulinum toxin treatment was associated with significantly increased voided volumes and reduced premicturition and maximal detrusor pressures, but no significant decrease in postvoid residual volume.30
More study needed
Despite these findings, a Cochrane Review concluded that, given the limited experience with intrasphincteric injection of botulinum toxin, data from larger randomized controlled trials are needed before making definitive recommendations.25 In the meantime, the clinician must weigh the low morbidity of the procedure against the limited options in the treatment of these patients.
OFF-LABEL UROLOGIC INDICATIONS
Botulinum toxin injection has been studied off-label for painful bladder syndrome/interstitial cystitis and for chronic prostatic pain. Patients with these conditions often describe pain with filling of the bladder, which leads to urinary frequency in an attempt to relieve the pain.
These pain syndromes can be difficult to treat and can have a devastating impact on quality of life. Treatment options include pain management, stress management, physical therapy, intravesical therapies, cystoscopy with hydrodistention, neuromodulation, cyclosporine, urinary diversion surgery, and botulinum toxin injection (an off-label use).31
In painful bladder syndrome/interstitial cystitis, botulinum toxin is thought to act on sensory afferent pathways, as well as to inhibit the release of substance P and glutamate, neuropeptides involved in sensory and nociceptive pathways.6 In animal studies,32 botulinum toxin was found to inhibit the afferent neural response by inhibiting mechanoreceptor-mediated release of adenosine triphosphate and by causing a decrease in calcitonin gene-related peptide, which helps regulate micturition and mediates painful bladder sensation.
Clinical studies to date in pelvic pain syndromes
Data from clinical studies of botulinum toxin injection for pelvic pain syndromes are limited. Zermann et al33 performed transurethral perisphincteric injection in 11 men with chronic prostatic pain, 9 of whom reported subjective pain relief, with an average decrease from 7.2 to 1.6 on a visual analogue scale. Postinjection urodynamic studies showed a decrease in functional urethral length, urethral closure pressure, and postvoid residual volume, and an increase in the peak and average flow rates.33
Abbott et al34 evaluated the effect of botulinum toxin injection into the levator ani in 12 women with chronic pelvic pain and pelvic floor hypertonicity. Pelvic floor manometry showed significant reduction in resting muscle pressures and improvements in dyspareunia and nonmenstrual pain. There were also improvements in quality of life and dyschezia, but these were not statistically significant.
Smith et al35 injected botulinum toxin into the detrusor of 13 women with refractory painful bladder syndrome and interstitial cystitis,35 and 9 women (69%) noted statistically significant improvements in the Interstitial Cystitis Symptom Index and Interstitial Cystitis Problem Index, daytime frequency, nocturia, pain, and urodynamic parameters (volume at first desire to void, and maximum cystometric capacity).
In a prospective randomized study of patients with refractory painful bladder syndrome and interstitial cystitis, Kuo and Chancellor36 compared suburothelial injection of 200 U or 100 U of botulinum toxin plus hydrodistention against hydrodistention alone.Patients who received botulinum toxin had increased bladder capacity and improved long-term pain relief, but no difference was noted between 200 U and 100 U, and more adverse effects were seen with the higher dose.36
Pinto et al37 treated 16 women with refractory painful bladder syndrome and interstitial cystitis with intratrigonal injections of botulinum toxin and reported improvements in pain scores, symptom scores, urinary frequency, and quality-of-life measures. The effect lasted 9.9 months (± 2.4 months) and persisted with successive injections.37
More study needed
Although these studies show that botulinum toxin injection for pelvic pain syndromes has the potential to improve pain, urinary frequency, bladder sensation, bladder capacity, and quality of life, larger randomized controlled trials are needed.
Again, the treatment options are limited for refractory painful bladder syndrome and interstitial cystitis. Patients may be desperate for relief from their symptoms. Practitioners must manage expectations and properly inform patients of the potential risks of treatments, especially with patients who will easily agree to further treatment with the smallest hope of relief.
INJECTION TECHNIQUES
For general points about the procedure to discuss with patients, see “What to tell patients.”
Cystoscopic detrusor injection
This procedure is usually done on an outpatient basis (eg, office, ambulatory surgery center). With the patient in the lithotomy position, 100 mL of 2% lidocaine is instilled into the bladder and is allowed 15 to 20 minutes to take effect. A flexible or rigid cystoscope can be used. Depending on the indication, the bladder is injected with 100 U to 300 U of botulinum toxin. The ideal depth of injection is 2 mm in the detrusor muscle, with each injection spaced about 1 cm apart. The recommended administration for 100 U is to inject 20 sites with 0.5 U per mL of saline and, for 200 U, to inject 30 sites with about 0.67 U per mL of saline.38 The location of the injections into the detrusor can vary, as long as adequate spacing is assured.
Injection sites vary. Proponents of injecting the trigone argue that as it is an area of greater nerve density, patients will have a better clinical response. Opponents argue that trigonal injection could result in distal ureteral paralysis and subsequent ureteral reflux. However, this theoretical concern has not been observed clinically.
Urethral injection (off-label use)
The urethra can be injected cystoscopically or periurethrally. Cystoscopic injection involves localization of the external sphincter using the rigid cystoscope and collagen needle; a total of 100 U is injected into the sphincter under direct vision, typically at the 3 o’clock and 9 o’clock positions.35 The periurethral technique is an option in women and involves a spinal needle with 100 U to 200 U of botulinum toxin injected into the external sphincter muscle at the 2 o’clock and 10 o’clock positions.
ADVERSE EFFECTS AND CONTRAINDICATIONS
Adverse effects are rare for urologic applications. The injections are localized, with little systemic absorption, and the doses are 1/1,000th of the theorized lethal dose in a 70-kg male.2 The maximum recommended dose for a 3-month period is 360 U.
Generalized muscle weakness has been reported in a paraplegic patient and in a tetraplegic patient after detrusor injections.2 Interestingly, both patients had return of bladder spasticity within 2 months, prompting speculation about diffusion of botulinum toxin through the bladder wall.2
Repeat injections can cause an immune response in up to 5% of patients.6 Patients undergoing repeat injections are at risk of forming neutralizing antibodies that can interfere with the efficacy of botulinum toxin.6 In a study by Schulte-Baukloh et al, all patients with antibodies to botulinum toxin had a history of recurrent urinary tract infection.39
Botulinum toxin injection is contraindicated in patients with preexisting neuromuscular disease, such as myasthenia gravis, Eaton-Lambert syndrome, and amyotrophic lateral sclerosis. It should also be avoided in patients who are breastfeeding, pregnant, or using agents that potentiate neuromuscular weakness, such as aminoglycosides.
Patients should be informed that some formulations of botulinum toxin include a stabilizer such as albumin derived from human blood, as this may be of religious or cultural significance.
- Leippold T, Reitz A, Schurch B. Botulinum toxin as a new therapy option for voiding disorders: current state of the art. Eur Urol 2003; 44:165–174.
- Sahai A, Khan M, Fowler CJ, Dasgupta P. Botulinum toxin for the treatment of lower urinary tract symptoms: a review. Neurourol Urodyn 2005; 24:2–12.
- Cruz F. Targets for botulinum toxin in the lower urinary tract. Neurourol Urodyn 2014; 33:31–38.
- Weedmark KA, Lambert DL, Mabon P, et al. Two novel toxin variants revealed by whole-genome sequencing of 175 Clostridium botulinum type E strains. Appl Environ Microbiol 2014; 80:6334–6345.
- Ha US, Park EY, Kim JC. Effect of botulinum toxin on expression of nerve growth factor and transient receptor potential vanilloid 1 in urothelium and detrusor muscle of rats with bladder outlet obstruction-induced detrusor overactivity. Urology 2011; 78:721.e1–721.e6
- Frenkl TL, Rackley RR. Injectable neuromodulatory agents: botulinum toxin therapy. Urol Clin North Am 2005; 32:89–99.
- Ikeda Y, Zabbarova IV, Birder LA, et al. Botulinum neurotoxin serotype A suppresses neurotransmitter release from afferent as well as efferent nerves in the urinary bladder. Eur Urol 2012; 62:1157–1164.
- Goldmark E, Niver B, Ginsberg DA. Neurogenic bladder: from diagnosis to management. Curr Urol Rep 2014; 15:448.
- Andersson KE. Current and future drugs for treatment of MS-associated bladder dysfunction. Ann Phys Rehabil Med 2014; 57:321–328.
- Schurch B, Stöhrer M, Kramer G, Schmid DM, Gaul G, Hauri D. Botulinum-A toxin for treating detrusor hyperreflexia in spinal cord injured patients: a new alternative to anticholinergic drugs? Preliminary results. J Urol 2000; 164:692–697.
- Schurch B, de Sèze M, Denys P, et al; Botox Detrusor Hyperreflexia Study Team. Botulinum toxin type a is a safe and effective treatment for neurogenic urinary incontinence: results of a single treatment, randomized, placebo controlled 6-month study. J Urol 2005; 174:196–200.
- Ehren I, Volz D, Farrelly E, et al. Efficacy and impact of botulinum toxin A on quality of life in patients with neurogenic detrusor overactivity: a randomised, placebo-controlled, double-blind study. Scand J Urol Nephrol 2007; 41:335–340.
- Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol 2012; 187:2131–2139.
- Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol 2011; 60:742–750.
- Wagner TH, Patrick DL, Bavendam TG, Martin ML, Buesching DP. Quality of life of persons with urinary incontinence: development of a new measure. Urology 1996: 47:67–71.
- Kennelly M, Dmochowski R, Ethans K, et al. Long-term efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: an interim analysis. Urology 2013; 81:491–497.
- Carlson JJ, Hansen RN, Dmochowski RR, Globe DR, Colayco DC, Sullivan SD. Estimating the cost-effectiveness of onabotulinumtoxinA for neurogenic detrusor overactivity in the United States. Clin Ther 2013; 35:414–424.
- Abrams P, Cardozo L, Fall M, et al; Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology 2003; 61:37–49.
- Milsom I, Coyne KS, Nicholson S, Kvasz M, Chen CI, Wein AJ. Global prevalence and economic burden of urgency urinary incontinence: a systematic review. Eur Urol 2014; 65:79–95.
- Nitti VW, Dmochowski R, Herschorn S, et al; EMBARK Study Group. OnabotulinumtoxinA for the treatment of patients with overactive bladder and urinary incontinence: results of a phase 3, randomized, placebo controlled trial. J Urol 2013; 189:2186–2193.
- Chapple C, Sievert KD, MacDiarmid S, et al. OnabotulinumtoxinA 100 U significantly improves all idiopathic overactive bladder symptoms and quality of life in patients with overactive bladder and urinary incontinence: a randomised, double-blind, placebo-controlled trial. Eur Urol 2013; 64:249–256.
- Colman S, Chapple C, Nitti V, Haag-Molkenteller C, Hastedt C, Massow U. Validation of Treatment Benefit Scale for assessing subjective outcomes in treatment of overactive bladder. Urology 2008; 72:803–807.
- Visco AG, Brubaker L, Richter HE, et al; Pelvic Floor Disorders Network. Anticholinergic therapy vs onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med 2012; 367:1803–1813.
- Gormley EA, Lightner DJ, Burgio KL, et al. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline. www.auanet.org/education/guidelines/overactive-bladder.cfm. Accessed June 11, 2015.
- Utomo E, Groen J, Blok BF. Surgical management of functional bladder outlet obstruction in adults with neurogenic bladder dysfunction. Cochrane Database Syst Rev 2014; 5:CD004927.
- Mahfouz W, Corcos J. Management of detrusor external sphincter dyssynergia in neurogenic bladder. Eur J Phys Rehabil Med 2011; 47:639–650.
- Dykstra DD, Sidi AA, Scott AB, Pagel JM, Goldish GD. Effects of botulinum A toxin on detrusor-sphincter dyssynergia in spinal cord injury patients. J Urol 1988; 139:919–922.
- Schurch B, Hauri D, Rodic B, Curt A, Meyer M, Rossier AB. Botulinum-A toxin as a treatment of detrusor-sphincter dyssynergia: a prospective study in 24 spinal cord injury patients. J Urol 1996; 155:1023–1029.
- de Sèze M, Petit H, Gallien, de Sèze MP, Joseph PA, Mazaux JM, Barat M. Botulinum a toxin and detrusor sphincter dyssynergia: a double-blind lidocaine-controlled study in 13 patients with spinal cord disease. Eur Urol 2002; 42:56–62.
- Gallien P, Reymann JM, Amarenco G, Nicolas B, de Sèze M, Bellissant E. Placebo controlled, randomised, double blind study of the effects of botulinum A toxin on detrusor sphincter dyssynergia in multiple sclerosis patients. J Neurol Neurosurg Psychiatry 2005; 76:1670–1676.
- Hanno PM, Burks DA, Clemens JQ, et al; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc. AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 2011; 185:2162–2170.
- Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol 2004; 172:1529–1532.
- Zermann DH, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol 2000; 38:393–399.
- Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol 2006; 108:915–923.
- Smith CP, Radziszewski P, Borkowski A, Somogyi GT, Boone TB, Chancellor MB. Botulinum toxin A has antinociceptive effects in treating interstitial cystitis. Urology 2004; 64:871–875.
- Kuo HC, Chancellor MB. Comparison of intravesical botulinum toxin type A injections plus hydrodistention with hydrodistention alone for the treatment of refractory interstitial cystitis/painful bladder syndrome. BJU Int 2009: 104:657–661.
- Pinto R, Lopes T, Silva J, Silva C, Dinis P, Cruz F. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol 2013; 189:548–553.
- Rovner E. Chapter 6: Practical aspects of administration of onabotulinumtoxinA. Neurourol Urodyn 2014; 33(suppl 3):S32–S37.
- Schulte-Baukloh H, Herholz J, Bigalke H, Miller K, Knispel HH. Results of a BoNT/A antibody study in children and adolescents after onabotulinumtoxin A (Botox®) detrusor injection. Urol Int 2011; 87:434–438.
- Leippold T, Reitz A, Schurch B. Botulinum toxin as a new therapy option for voiding disorders: current state of the art. Eur Urol 2003; 44:165–174.
- Sahai A, Khan M, Fowler CJ, Dasgupta P. Botulinum toxin for the treatment of lower urinary tract symptoms: a review. Neurourol Urodyn 2005; 24:2–12.
- Cruz F. Targets for botulinum toxin in the lower urinary tract. Neurourol Urodyn 2014; 33:31–38.
- Weedmark KA, Lambert DL, Mabon P, et al. Two novel toxin variants revealed by whole-genome sequencing of 175 Clostridium botulinum type E strains. Appl Environ Microbiol 2014; 80:6334–6345.
- Ha US, Park EY, Kim JC. Effect of botulinum toxin on expression of nerve growth factor and transient receptor potential vanilloid 1 in urothelium and detrusor muscle of rats with bladder outlet obstruction-induced detrusor overactivity. Urology 2011; 78:721.e1–721.e6
- Frenkl TL, Rackley RR. Injectable neuromodulatory agents: botulinum toxin therapy. Urol Clin North Am 2005; 32:89–99.
- Ikeda Y, Zabbarova IV, Birder LA, et al. Botulinum neurotoxin serotype A suppresses neurotransmitter release from afferent as well as efferent nerves in the urinary bladder. Eur Urol 2012; 62:1157–1164.
- Goldmark E, Niver B, Ginsberg DA. Neurogenic bladder: from diagnosis to management. Curr Urol Rep 2014; 15:448.
- Andersson KE. Current and future drugs for treatment of MS-associated bladder dysfunction. Ann Phys Rehabil Med 2014; 57:321–328.
- Schurch B, Stöhrer M, Kramer G, Schmid DM, Gaul G, Hauri D. Botulinum-A toxin for treating detrusor hyperreflexia in spinal cord injured patients: a new alternative to anticholinergic drugs? Preliminary results. J Urol 2000; 164:692–697.
- Schurch B, de Sèze M, Denys P, et al; Botox Detrusor Hyperreflexia Study Team. Botulinum toxin type a is a safe and effective treatment for neurogenic urinary incontinence: results of a single treatment, randomized, placebo controlled 6-month study. J Urol 2005; 174:196–200.
- Ehren I, Volz D, Farrelly E, et al. Efficacy and impact of botulinum toxin A on quality of life in patients with neurogenic detrusor overactivity: a randomised, placebo-controlled, double-blind study. Scand J Urol Nephrol 2007; 41:335–340.
- Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol 2012; 187:2131–2139.
- Cruz F, Herschorn S, Aliotta P, et al. Efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: a randomised, double-blind, placebo-controlled trial. Eur Urol 2011; 60:742–750.
- Wagner TH, Patrick DL, Bavendam TG, Martin ML, Buesching DP. Quality of life of persons with urinary incontinence: development of a new measure. Urology 1996: 47:67–71.
- Kennelly M, Dmochowski R, Ethans K, et al. Long-term efficacy and safety of onabotulinumtoxinA in patients with urinary incontinence due to neurogenic detrusor overactivity: an interim analysis. Urology 2013; 81:491–497.
- Carlson JJ, Hansen RN, Dmochowski RR, Globe DR, Colayco DC, Sullivan SD. Estimating the cost-effectiveness of onabotulinumtoxinA for neurogenic detrusor overactivity in the United States. Clin Ther 2013; 35:414–424.
- Abrams P, Cardozo L, Fall M, et al; Standardisation Sub-Committee of the International Continence Society. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the International Continence Society. Urology 2003; 61:37–49.
- Milsom I, Coyne KS, Nicholson S, Kvasz M, Chen CI, Wein AJ. Global prevalence and economic burden of urgency urinary incontinence: a systematic review. Eur Urol 2014; 65:79–95.
- Nitti VW, Dmochowski R, Herschorn S, et al; EMBARK Study Group. OnabotulinumtoxinA for the treatment of patients with overactive bladder and urinary incontinence: results of a phase 3, randomized, placebo controlled trial. J Urol 2013; 189:2186–2193.
- Chapple C, Sievert KD, MacDiarmid S, et al. OnabotulinumtoxinA 100 U significantly improves all idiopathic overactive bladder symptoms and quality of life in patients with overactive bladder and urinary incontinence: a randomised, double-blind, placebo-controlled trial. Eur Urol 2013; 64:249–256.
- Colman S, Chapple C, Nitti V, Haag-Molkenteller C, Hastedt C, Massow U. Validation of Treatment Benefit Scale for assessing subjective outcomes in treatment of overactive bladder. Urology 2008; 72:803–807.
- Visco AG, Brubaker L, Richter HE, et al; Pelvic Floor Disorders Network. Anticholinergic therapy vs onabotulinumtoxinA for urgency urinary incontinence. N Engl J Med 2012; 367:1803–1813.
- Gormley EA, Lightner DJ, Burgio KL, et al. Diagnosis and treatment of overactive bladder (non-neurogenic) in adults: AUA/SUFU Guideline. www.auanet.org/education/guidelines/overactive-bladder.cfm. Accessed June 11, 2015.
- Utomo E, Groen J, Blok BF. Surgical management of functional bladder outlet obstruction in adults with neurogenic bladder dysfunction. Cochrane Database Syst Rev 2014; 5:CD004927.
- Mahfouz W, Corcos J. Management of detrusor external sphincter dyssynergia in neurogenic bladder. Eur J Phys Rehabil Med 2011; 47:639–650.
- Dykstra DD, Sidi AA, Scott AB, Pagel JM, Goldish GD. Effects of botulinum A toxin on detrusor-sphincter dyssynergia in spinal cord injury patients. J Urol 1988; 139:919–922.
- Schurch B, Hauri D, Rodic B, Curt A, Meyer M, Rossier AB. Botulinum-A toxin as a treatment of detrusor-sphincter dyssynergia: a prospective study in 24 spinal cord injury patients. J Urol 1996; 155:1023–1029.
- de Sèze M, Petit H, Gallien, de Sèze MP, Joseph PA, Mazaux JM, Barat M. Botulinum a toxin and detrusor sphincter dyssynergia: a double-blind lidocaine-controlled study in 13 patients with spinal cord disease. Eur Urol 2002; 42:56–62.
- Gallien P, Reymann JM, Amarenco G, Nicolas B, de Sèze M, Bellissant E. Placebo controlled, randomised, double blind study of the effects of botulinum A toxin on detrusor sphincter dyssynergia in multiple sclerosis patients. J Neurol Neurosurg Psychiatry 2005; 76:1670–1676.
- Hanno PM, Burks DA, Clemens JQ, et al; Interstitial Cystitis Guidelines Panel of the American Urological Association Education and Research, Inc. AUA guideline for the diagnosis and treatment of interstitial cystitis/bladder pain syndrome. J Urol 2011; 185:2162–2170.
- Chuang YC, Yoshimura N, Huang CC, Chiang PH, Chancellor MB. Intravesical botulinum toxin a administration produces analgesia against acetic acid induced bladder pain responses in rats. J Urol 2004; 172:1529–1532.
- Zermann DH, Ishigooka M, Schubert J, Schmidt RA. Perisphincteric injection of botulinum toxin type A. A treatment option for patients with chronic prostatic pain? Eur Urol 2000; 38:393–399.
- Abbott JA, Jarvis SK, Lyons SD, Thomson A, Vancaille TG. Botulinum toxin type A for chronic pain and pelvic floor spasm in women: a randomized controlled trial. Obstet Gynecol 2006; 108:915–923.
- Smith CP, Radziszewski P, Borkowski A, Somogyi GT, Boone TB, Chancellor MB. Botulinum toxin A has antinociceptive effects in treating interstitial cystitis. Urology 2004; 64:871–875.
- Kuo HC, Chancellor MB. Comparison of intravesical botulinum toxin type A injections plus hydrodistention with hydrodistention alone for the treatment of refractory interstitial cystitis/painful bladder syndrome. BJU Int 2009: 104:657–661.
- Pinto R, Lopes T, Silva J, Silva C, Dinis P, Cruz F. Persistent therapeutic effect of repeated injections of onabotulinum toxin a in refractory bladder pain syndrome/interstitial cystitis. J Urol 2013; 189:548–553.
- Rovner E. Chapter 6: Practical aspects of administration of onabotulinumtoxinA. Neurourol Urodyn 2014; 33(suppl 3):S32–S37.
- Schulte-Baukloh H, Herholz J, Bigalke H, Miller K, Knispel HH. Results of a BoNT/A antibody study in children and adolescents after onabotulinumtoxin A (Botox®) detrusor injection. Urol Int 2011; 87:434–438.
KEY POINTS
- Anticholinergic drugs have been the first-line therapy for neurogenic detrusor overactivity. If drug therapy failed, the next option was reconstructive surgery such as cystoplasty. Botulinum toxin injection may be an option in select patients.
- Urinary tract infection and urinary retention requiring intermittent self-catheterization are the most common adverse events of botulinum toxin injection in trials of patients with neurogenic detrusor overactivity or idiopathic overactive bladder.
- Small studies have shown that botulinum toxin injection for painful bladder syndrome/interstitial cystitis can improve pain, urinary frequency, and quality of life. But larger randomized controlled trials are needed.
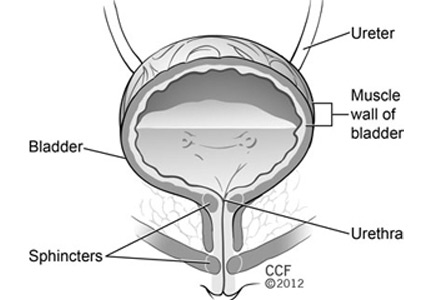
Ceftaroline fosamil: A super-cephalosporin?
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
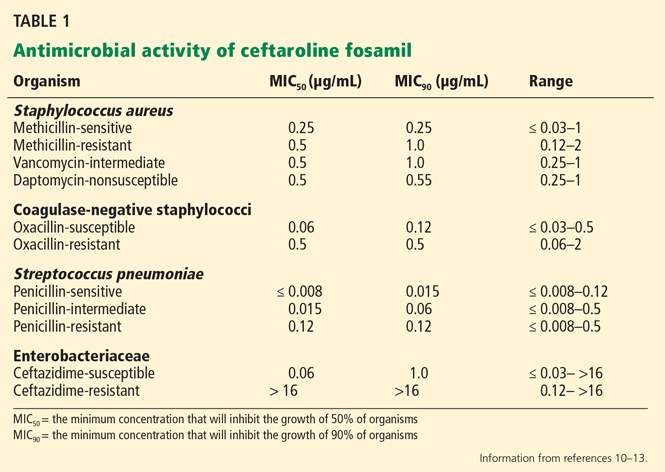
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.

Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19

Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.

Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
Ceftaroline fosamil (Teflaro), introduced to the US market in October 2010, is the first beta-lactam agent with clinically useful activity against methicillin-resistant Staphylococcus aureus (MRSA). Currently, it is approved by the US Food and Drug Administration (FDA) to treat acute bacterial skin and skin-structure infections and community-acquired bacterial pneumonia caused by susceptible microorganisms.
In an era of increasing drug resistance and limited numbers of antimicrobials in the drug-production pipeline, ceftaroline is a step forward in fulfilling the Infectious Diseases Society of America’s “10 × ’20 Initiative” to increase support for drug research and manufacturing, with the goal of producing 10 new antimicrobial drugs by the year 2020.1 Ceftaroline was the first of several antibiotics to receive FDA approval in response to this initiative. It was followed by dalbavancin (May 2014), tedizolid phosphate (June 2014), oritavancin (August 2014), ceftolozane-tazobactam (December 2014), and ceftazidime-avibactam (February 2015). These antibiotic agents are aimed at treating infections caused by drug-resistant gram-positive and gram-negative microorganisms. It is important to understand and optimize the use of these new antibiotic agents in order to decrease the risk of emerging antibiotic resistance and superinfections (eg, Clostridium difficile infection) caused by antibiotic overuse or misuse.
This article provides an overview of ceftaroline’s mechanisms of action and resistance, spectrum of activity, pharmacokinetic properties, adverse effects, and current place in therapy.
AN ERA OF MULTIDRUG-RESISTANT MICROORGANISMS
Increasing rates of antimicrobial resistance threaten the efficacy of antimicrobial drugs in the daily practice of medicine. The World Health Organization has labeled antimicrobial resistance one of the three greatest threats to human health. Global efforts are under way to stimulate development of new antimicrobial agents and to decrease rates of antimicrobial resistance.
Staphylococcus aureus: A threat, even with vancomycin
Between 1998 and 2005, S aureus was one of the most common inpatient and outpatient isolates reported by clinical laboratories throughout the United States.2
Treatment of S aureus infection is complicated by a variety of resistance mechanisms that have evolved over time. In fact, the first resistant isolate of S aureus emerged not long after penicillin’s debut into clinical practice, and now the majority of strains are resistant to penicillin.
Methicillin was designed to overcome this beta-lactamase resistance and became the treatment of choice for penicillin-resistant S aureus isolates. However, MRSA isolates soon emerged because of the organism’s acquisition of penicillin-binding protein PBP2a via the mecA gene, leading to decreased binding affinity of methicillin.3
Since then, several agents active against MRSA (vancomycin, daptomycin, linezolid, tigecycline) have been introduced and continue to be widely used. While vancomycin is considered the first-line option for a variety of MRSA infections, its use has been threatened because of the emergence of vancomycin-intermediate-resistant S aureus (VISA), S aureus strains displaying vancomycin heteroresistance (hVISA), and vancomycin-resistant S aureus (VRSA) strains.4
VISA and hVISA isolates emerged through sequential mutations that lead to autolytic activity and cell-wall thickening. In contrast, the mechanism of resistance in VRSA is by acquisition of the vanA resistance gene, which alters the binding site of vancomycin from d-alanine-d-alanine to d-alanine-d-lactate.5
Streptococcus pneumoniae resistance: A continuing problem
The prevalence of drug resistance in S pneumoniae has risen since the late 1990s. A 2013 report from the SENTRY Antimicrobial Surveillance Program stated that almost 20% of S pneumoniae isolates were resistant to amoxicillin-clavulanate, and similar trends have been observed for penicillin (14.8%) and ceftriaxone (11.7%).6
S pneumoniae resistance is acquired through modifications of the penicillin-binding proteins, namely PBP1a, PBP2b, PBP2x, and, less frequently, PBP2a. These modifications lead to decreased binding affinity for most beta-lactams.7
Clinical impact of multidrug-resistant S aureus and S pneumoniae
In 2011, the US Centers for Disease Control and Prevention reported an estimated 80,000 severe MRSA infections and 11,000 MRSA-related deaths in the United States.8 In the same report, drug-resistant S pneumoniae was estimated to be responsible for almost 1.2 million illnesses and 7,000 deaths per year, leading to upwards of $96 million in related medical costs.
While invasive drug-resistant S pneumoniae infections usually affect patients at the extremes of age (under age 5 and over age 65), they have had a serious impact on patients of all ages.8
In light of the increasing prevalence of multidrug-resistant organisms, newer antimicrobial agents with novel mechanisms of action are needed.
CEFTAROLINE: A BETA-LACTAM WITH ANTI-MRSA ACTIVITY
The cephalosporins, a class of beta-lactam antibiotics, were originally derived from the fungus Cephalosporium (now called Acremonium). There are now many agents in this class, each containing a nucleus consisting of a beta-lactam ring fused to a six-member dihydrothiazine ring, and two side chains that can be modified to affect antibacterial activity and pharmacokinetic properties.
Cephalosporins are typically categorized into “generations.” With some exceptions, the first- and second-generation agents have good activity against gram-positive microorganisms, including methicillin-susceptible S aureus—but not against MRSA. The third- and fourth-generation cephalosporins have better gram-negative activity, with many agents having activity against the gram-negative bacterium Pseudomonas aeruginosa.
Enterococcal isolates are intrinsically resistant to cephalosporins. Additionally, cephalosporins are not active against anaerobic bacteria, except for a subset of structurally unique second-generation cephalosporins, ie, cefotetan and cefoxitin.
Ceftaroline was synthesized with specific manipulations of the side chains to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae isolates, making it the first available beta-lactam with this ability.
Mechanism of action
Ceftaroline binds to penicillin-binding proteins, inhibiting transpeptidation. This interaction blocks the final stage of peptidoglycan synthesis and inhibits bacterial cell wall formation, ultimately leading to cellular autolysis and microorganism death. Ceftaroline binds with high affinity to PBP2a and PBP2x, expanding its activity to encompass MRSA and penicillin-resistant S pneumoniae isolates.9
Spectrum of activity
Ceftaroline has in vitro activity against many gram-positive and gram-negative bacteria,10–13 including (Table 1):
- Methicillin-susceptible and methicillin-resistant staphylococci
- VISA, VRSA, and hVISA
- Daptomycin-nonsusceptible S aureus
- Streptococcal species, including penicillin-resistant S pneumoniae
- Enterobacteriaceae, including Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Citrobacter koseri, Citrobacter freundii, Enterobacter cloacae, Enterobacter aerogenes, Moraxella catarrhalis, Morganella morganii, and Proteus mirabilis.
Of note, ceftaroline is not active against Pseudomonas species, Enterococcus species, or Bacteroides fragilis. In addition, it is not active against the “atypical” respiratory pathogens Mycoplasma pneumoniae, Chlamydophila pneumoniae, and Legionella pneumophila.
Ceftaroline resistance
Gram-negative organisms appear to develop resistance to ceftaroline at rates similar to those observed with the other oxyimino-cephalosporins (eg, ceftriaxone). Ceftaroline is inactive against gram-negative organisms producing extended-spectrum beta-lactamases, including K pneumoniae carbapenemase and metallo-beta-lactamases.14 In addition, it induces the expression of AmpC beta-lactamases.
Although currently uncommon, resistance to ceftaroline has also been reported in S aureus strains.15 The mechanism of resistance is decreased binding affinity for PBP2a due to amino acid substitutions on the nonpenicillin-binding domains.15
Pharmacokinetic profile
An understanding of pharmacokinetics is key in optimizing the dose of antimicrobials so that the drugs are used most effectively and pathogens do not develop resistance to them.
Ceftaroline fosamil is a prodrug that, upon intravenous administration, is rapidly converted by phosphatase enzymes to its active moiety, ceftaroline. Its pharmacokinetic profile is summarized in Table 2.16,17 Its volume of distribution is similar to that of the fourth-generation cephalosporin cefepime.
Ceftaroline is then hydrolyzed into its inactive metabolite, ceftaroline M-1. It undergoes little hepatic metabolism and lacks properties to make it a substrate, inhibitor, or inducer of the CYP450 enzyme system and therefore is not likely to cause notable CYP450-related drug-drug interactions.
Like most other beta-lactams, ceftaroline is primarily excreted by the kidneys. Furthermore, an estimated 21% of a dose is eliminated with each intermittent hemodialysis session. Therefore, renal and intermittent hemodialysis dose adjustments are necessary. The estimated elimination half-life is 2.6 hours, necessitating dosing two to three times daily, depending on the indication and infectious inoculum.
Ceftaroline dosing
Ceftaroline is available only in a parenteral preparation and is typically given at a dose of 600 mg every 12 hours.10 The intravenous infusion is given over 1 hour.
The current stability data require reconstituted ceftaroline to be used within 6 hours at room temperature and within 24 hours if refrigerated.10
Ceftaroline requires dosing adjustments for patients with renal insufficiency. Per the manufacturer, renal dosing adjustments are based on the creatinine clearance rate, as estimated by the Cockroft-Gault formula:
- Creatinine clearance > 50 mL/min: no dosage adjustment necessary
- Creatinine clearance > 30 to ≤ 50 mL/min: give 400 mg every 12 hours
- Creatinine clearance ≥ 15 to ≤ 30 mL/min: give 300 mg every 12 hours
- Creatinine clearance < 15 mL/min or on intermittent dialysis: give 200 mg every 12 hours.
Ongoing clinical trials are investigating a higher-dosing strategy of 600 mg every 8 hours for patients with community-acquired bacterial pneumonia at risk of MRSA bacteremia.18
CLINICAL TRIALS LEADING TO CEFTAROLINE’S APPROVAL
Ceftaroline was approved for the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections due to susceptible pathogens on the basis of phase 3 comparator trials.
Community-acquired bacterial pneumonia: The FOCUS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of community-acquired bacterial pneumonia was studied in two randomized, double-blind, noninferiority trials, known as Ceftaroline Community-acquired Pneumonia vs Ceftriaxone (FOCUS) 1 and FOCUS 2.19,20
Patients were adults and not critically ill, as was reflected by their being in Pneumonia Outcomes Research Team (PORT) risk class III or IV (with class V indicating the highest risk of death). Therefore, the results may not be completely applicable to critically ill patients or those not admitted to the hospital. Of note, patients were excluded from the trials if they had infections known or thought to be due to MRSA or to atypical organisms.21 Baseline characteristics and patient demographics were similar between study groups in both trials.
A bacterial pathogen was identified in 26.1% of the patients included in the modified intent-to-treat analysis of the pooled data of the trials; the most common pathogens were S pneumoniae, methicillin-sensitive S aureus, Haemophilus influenzae, K pneumoniae, and E coli.21
Treatment. Patients received either ceftaroline 600 mg every 12 hours (or a lower dose based on renal function) or ceftriaxone 1 g every 24 hours. In addition, in the FOCUS 1 trial, patients in both treatment groups received clarithromycin 500 mg every 12 hours for the first day.19
Results. In both trials and in the integrated analysis, ceftaroline was noninferior to ceftriaxone (Table 3).22 In the integrated analysis of both trials, compared with the ceftriaxone group, the ceftaroline group had a higher clinical cure rate among patients classified as PORT risk class III (86.8% vs 79.2%, weighted treatment difference 12.6%, 95% confidence interval [CI] 1.3–13.8) and among patients who had not received prior antibiotic treatment (85.5% vs 74.9%, weighted treatment difference 11.2%, 95% CI 4.5–18.0).21
Acute bacterial skin and skin-structure infections: The CANVAS 1 and 2 trials
The efficacy and safety of ceftaroline in the treatment of complicated acute bacterial skin and skin-structure infections was studied in two randomized, double-blind trials: Ceftaroline Versus Vancomycin in Skin and Skin Structure Infections (CANVAS) 1 and CANVAS 2.23,24
Patients. Adult patients with a diagnosis of community-acquired skin and skin-structure infections warranting at least 5 days of intravenous antimicrobial therapy were included in the trials. Important protocol exclusions were patients with diabetic foot ulcers, decubitus ulcers, burns, ulcers associated with peripheral vascular disease accompanied by osteomyelitis, and suspected P aeruginosa infections.25 This limits the external validity of ceftaroline use in the aforementioned excluded patient populations.
Patients in each treatment group of the trials had similar demographic characteristics. The most common infections were cellulitis, major abscess requiring surgical intervention, wound infection, and infected ulcer. Bacteremia was present in 4.2% of patients in the ceftaroline group and in 3.8% of patients in the vancomycin-aztreonam group. The most common pathogen was S aureus. Methicillin resistance was present in 40% of the ceftaroline group and 34% of the control group.
Treatment. Patients received either ceftaroline 600 mg every 12 hours or the combination of vancomycin 1 g plus aztreonam 1 g given 12 hours, for 5 to 14 days.
Results. As assessed at a “test-of-cure” visit 8 to 15 days after the last dose of study medication, the efficacy of ceftaroline was similar to that of vancomycin-aztreonam, meeting the set noninferiority goal (Table 4).25 Moreover, if assessed on day 2 or 3 (a new end point recommended by the FDA), the rate of cessation of erythema spread and absence of fever was higher in the ceftaroline group than in the vancomycin-aztreonam group.26 However, this end point was not in the original trial protocol.
CEFTAROLINE FOR OTHER INDICATIONS
As noted, ceftaroline has been approved for treating community-acquired bacterial pneumonia and acute bacterial skin and skin-structure infections. In addition, it has been used in several studies in animals, and case reports of non-FDA approved indications including endocarditis and osteomyelitis have been published. Clinical trials are evaluating its use in pediatric patients, as well as for community-acquired bacterial pneumonia with risk for MRSA and for MRSA bacteremia.
Endocarditis
Animal studies have demonstrated ceftaroline to have bactericidal activity against MRSA and hVISA in endocarditis.27
A few case series have been published describing ceftaroline’s use as salvage therapy for persistent MRSA bacteremia and endocarditis. For example, Ho et al28 reported using it in three patients who had endocarditis as a source of their persistent bacteremia. All three patients had resolution of their MRSA bloodstream infection following ceftaroline therapy. The dosage was 600 mg every 8 hours, which is higher than in the manufacturer’s prescribing information.
Lin et al29 reported using ceftaroline in five patients with either possible or probable endocarditis. Three of the five patients had clinical cure as defined by resolution or improvement of all signs and symptoms of infection, and not requiring further antimicrobial therapy.29
More data from clinical trials would be beneficial in defining ceftaroline’s role in treating endocarditis caused by susceptible microorganisms.
Osteomyelitis
In animal studies of osteomyelitis, ceftaroline exhibited activity against MRSA in infected bone and joint fluid. Compared with vancomycin and linezolid, ceftaroline was associated with more significant decreases in bacterial load in the infected joint fluid, bone marrow, and bone.30
Lin et al29 gave ceftaroline to two patients with bone and joint infections, both of whom had received other therapies that had failed. The doses of ceftaroline were higher than those recommended in the prescribing information; clinical cure was noted in both cases following the switch.
These data come from case series, and more study of ceftaroline’s role in the treatment of osteomyelitis infections is warranted.
Meningitis
The use of ceftaroline in meningitis has been studied in rabbits. While ceftaroline penetrated into the cerebrospinal fluid in only negligible amounts in healthy rabbits (3% penetration), its penetration improved to 15% in animals with inflamed meninges. Ceftaroline cerebrospinal fluid levels in inflamed meninges were sufficient to provide bactericidal activity against penicillin-sensitive and resistant S pneumoniae strains as well as K pneumoniae and E coli strains.31,32
REPORTED ADVERSE EFFECTS OF CEFTAROLINE
Overall, ceftaroline was well tolerated in clinical trials, and its safety profile was similar to those of the comparator agents (ceftriaxone and vancomycin-aztreonam).
As with the other cephalosporins, hypersensitivity reactions have been reported with ceftaroline. In the clinical trials, 3% of patients developed a rash with ceftaroline.33,34 Patients with a history of beta-lactam allergy were excluded from the trials, so the rate of cross-reactivity with penicillins and with other cephalosporins is unknown.
In the phase 3 clinical trials, gastrointestinal side effects including diarrhea (5%), nausea (4%), and vomiting (2%) were reported with ceftaroline. C difficile-associated diarrhea has also been reported.33
As with other cephalosporins, ceftaroline can cause a false-positive result on the Coombs test. Approximately 11% of ceftaroline-treated patients in phase 3 clinical trials had a positive Coombs test, but hemolytic anemia did not occur in any patients.33,34
Discontinuation of ceftaroline due to an adverse reaction was reported in 2.7% of patients receiving the drug during phase 3 trials, compared with 3.7% with comparator agents.
WHEN SHOULD CEFTAROLINE BE USED IN DAILY PRACTICE?
Ceftaroline has been shown to be at least as effective as ceftriaxone in treating community-acquired bacterial pneumonia, and at least as effective as vancomycin-aztreonam in treating acute bacterial skin and skin-structure infections. The 2014 Infectious Diseases Society of America’s guidelines for the diagnosis and management of skin and soft-tissue infections recommend ceftaroline as an option for empiric therapy for purulent skin and soft-tissue infections.35
The guidelines on community-acquired pneumonia have not been updated since 2007, which was before ceftaroline was approved. However, these guidelines are currently undergoing revision and may provide insight on ceftaroline’s place in the treatment of community-acquired bacterial pneumonia.36
Currently, ceftaroline’s routine use for these indications should be balanced by its higher cost ($150 for a 600-mg dose) compared with ceftriaxone ($5 for a 1-g dose) or vancomycin ($25 for a 1-g dose). The drug’s in vitro activity against drug-resistant pneumococci and S aureus, including MRSA, hVISA, and VISA may help fill an unmet need or provide a safer and more tolerable alternative to currently available therapies.
However, ceftaroline’s lack of activity against P aeruginosa and carbapenem-resistant Enterobacteriaceae does not meet the public health threat needs stemming from these multidrug-resistant microorganisms. Ongoing clinical trials in patients with more serious MRSA infections will provide important information about ceftaroline’s role as an anti-MRSA agent.
While the discovery of antimicrobials has had one of the greatest impacts on medicine, continued antibiotic use is threatened by the emergence of drug-resistant pathogens. Therefore, it is as important as ever to be good stewards of our currently available antimicrobials. Developing usage and dosing criteria for antimicrobials based on available data and literature is a step forward in optimizing the use of antibiotics—a precious medical resource.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
- Infectious Diseases Society of America. The 10 x ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 2010; 50:1081–1083.
- Styers D, Sheehan DJ, Hogan P, Sahm DF. Laboratory-based surveillance of current antimicrobial resistance patterns and trends among Staphylococcus aureus: 2005 status in the United States. Ann Clin Microbiol Antimicrob 2006; 5:2.
- Farrell DJ, Castanheira M, Mendes RE, Sader HS, Jones RN. In vitro activity of ceftaroline against multidrug-resistant Staphylococcus aureus and Streptococcus pneumoniae: a review of published studies and the AWARE Surveillance Program (2008-2010). Clin Infect Dis 2012; 55(suppl 3):S206–S214.
- Holmes NE, Johnson PD, Howden BP. Relationship between vancomycin-resistant Staphylococcus aureus, vancomycin-intermediate S. aureus, high vancomycin MIC, and outcome in serious S. aureus infections. J Clin Microbiol 2012; 50:2548–2552.
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111:1265–1273.
- Jones RN, Sader HS, Mendes RE, Flamm RK. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998-2011). Diag Microbiol Infect Dis 2013; 75:107–109.
- Zapun A, Contreras-Martel C, Vernet T. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 2008; 32:361–385.
- Centers for Disease Control and Prevention (CDC). Antibiotic resistance threats in the United States 2013. cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed June 1, 2015.
- Moisan H, Pruneau M, Malouin F. Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 2010; 65:713–716.
- Forest Laboratories, Inc. Teflaro® (ceftaroline fosamil): prescribing information. www.frx.com/pi/teflaro_pi.pdf. Accessed June 1, 2015.
- Richter SS, Heilmann KP, Dohrn CL, et al. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob Agents Chemother 2011; 55:4154–4160.
- Ge Y, Biek D, Talbot GH, Sahm DF. In vitro profiling of ceftaroline against a collection of recent bacterial clinical isolates from across the United States. Antimicrob Agents Chemother 2008; 52:3398–3407.
- Saravolatz L, Pawlak J, Johnson L. In vitro activity of ceftaroline against community-associated methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, and daptomycin-nonsusceptible Staphylococcus aureus isolates. Antimicrob Agents Chemother 2010; 54:3027–3030.
- Mushtaq S, Livermore DM. AmpC induction by ceftaroline. J Antimicrob Chemother 2010; 65:586–588.
- Mendes RE, Tsakris A, Sader HS, et al. Characterization of methicillin-resistant Staphylococcus aureus displaying increased MICs of ceftaroline. J Antimicrob Chemother 2012; 67:1321–1324.
- Lodise TP, Low DE. Ceftaroline fosamil in the treatment of community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections. Drugs 2012; 72:1473–1493.
- Riccobene TA, Su SF, Rank D. Single- and multiple-dose study to determine the safety, tolerability, and pharmacokinetics of ceftaroline fosamil in combination with avibactam in healthy subjects. Antimicrob Agents Chemother 2013; 57:1496–1504.
- US National Institutes of Health. ClinicalTrials.gov. Evaluation of ceftaroline fosamil versus a comparator in adult subjects with community-acquired bacterial pneumonia (CABP) with risk for methicillin-resistant Staphylococcus aureus. http://clinicaltrials.gov/ct2/show/NCT01645735. Accessed June 1, 2015.
- File TM Jr, Low DE, Eckburg PB, et al; FOCUS 1 investigators. FOCUS 1: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii19–iii32.
- Low DE, File TM Jr, Eckburg PB, et al; FOCUS 2 investigators. FOCUS 2: a randomized, double-blinded, multicentre, phase III trial of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii33–iii44.
- File TM Jr, Low DE, Eckburg PB, et al. Integrated analysis of FOCUS 1 and FOCUS 2: randomized, doubled-blinded, multicenter phase 3 trials of the efficacy and safety of ceftaroline fosamil versus ceftriaxone in patients with community-acquired pneumonia. Clin Infect Dis 2010; 51:1395–1405.
- Food and Drug Administration (FDA). Ceftaroline fosamil for the treatment of community-acquired bacterial pneumonia and complicated skin and skin structure infections. www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/anti-infectivedrugsadvisorycommittee/ucm224656.pdf. Accessed June 1, 2015.
- Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 1 investigators. CANVAS 1: the first phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv41–iv51.
- Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T; CANVAS 2 investigators. CANVAS 2: the second phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv53-iv65.
- Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis 2010; 51:641–650.
- Friedland HD, O’Neal T, Biek D, et al. CANVAS 1 and 2: analysis of clinical response at day 3 in two phase 3 trials of ceftaroline fosamil versus vancomycin plus aztreonam in treatment of acute bacterial skin and skin structure infections. Antimicrob Agents Chemother 2012; 56:2231–2236.
- Jacqueline C, Caillon J, Le Mabecque V, et al. In vivo efficacy of ceftaroline (PPI-0903), a new broad-spectrum cephalosporin, compared with linezolid and vancomycin against methicillin-resistant and vancomycin-intermediate Staphylococcus aureus in a rabbit endocarditis model. Antimicrob Agents Chemother 2007; 51:3397–3400.
- Ho TT, Cadena J, Childs LM, Gonzalez-Velez M, Lewis JS 2nd. Methicillin-resistant Staphylococcus aureus bacteraemia and endocarditis treated with ceftaroline salvage therapy. J Antimicrob Chemother 2012; 67:1267–1270.
- Lin JC, Aung G, Thomas A, Jahng M, Johns S, Fierer J. The use of ceftaroline fosamil in methicillin-resistant Staphylococcus aureus endocarditis and deep-seated MRSA infections: a retrospective case series of 10 patients. J Infect Chemother 2013; 19:42–49.
- Jacqueline C, Amador G, Caillon J, et al. Efficacy of the new cephalosporin ceftaroline in the treatment of experimental methicillin-resistant Staphylococcus aureus acute osteomyelitis. J Antimicrob Chemother 2010; 65:1749–1752.
- Stucki A, Acosta F, Cottagnoud M, Cottagnoud P. Efficacy of ceftaroline fosamil against Escherichia coli and Klebsiella pneumoniae strains in a rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:5808–5810.
- Cottagnoud P, Cottagnoud M, Acosta F, Stucki A. Efficacy of ceftaroline fosamil against penicillin-sensitive and -resistant Streptococcus pneumoniae in an experimental rabbit meningitis model. Antimicrob Agents Chemother 2013; 57:4653–4655.
- Corrado ML. Integrated safety summary of CANVAS 1 and 2 trials: phase III, randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother 2010; 65(suppl 4):iv67–iv71.
- Rank DR, Friedland HD, Laudano JB. Integrated safety summary of FOCUS 1 and FOCUS 2 trials: phase III randomized, double-blind studies evaluating ceftaroline fosamil for the treatment of patients with community-acquired pneumonia. J Antimicrob Chemother 2011; 66(suppl 3):iii53–iii59.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis 2014; 59:147–159.
- Mandell LA, Wunderink RG, Anzueto A, et al; Infectious Diseases Society of America; American Thoracic Society. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44:S27–S72.
KEY POINTS
- Resistance of S aureus and S pneumoniae to multiple antimicrobial drugs is on the rise, and new agents are urgently needed.
- Ceftaroline’s molecular structure was designed to provide enhanced activity against MRSA and multidrug-resistant S pneumoniae.
- In clinical trials leading to its approval, ceftaroline was found to be at least as effective as ceftriaxone in treating community-acquired pneumonia and at least as effective as vancomycin plus aztreonam in treating acute bacterial skin and skin-structure infections.
- The routine use of ceftaroline for these indications should be balanced by its higher cost compared with ceftriaxone or vancomycin. Ongoing studies should shed more light on its role in treatment.