User login
Monitoring effectively identifies seizures in postbypass neonates
In the first report evaluating the impact of a clinical guideline that calls for the use of postoperative continuous electroencephalography (CEEG) on infants after they’ve had cardiopulmonary bypass surgery, investigators at Children’s Hospital of Philadelphia and the University of Pennsylvania validated the clinical utility of routine CEEG monitoring and found that clinical assessment for seizures without CEEG is not a reliable marker for diagnosis and treatment.
In a report online in the Journal of Thoracic and Cardiovascular Surgery (J. Thorac. Cardiovasc. Surg. 2015 [doi:10.1016/j.jtcvs.2015.03.045]), Dr. Maryam Naim and colleagues said that CEEG identified electroencephalographic seizures in 8% of newborns after cardiopulmonary bypass surgery. The study, conducted over 18 months, evaluated 172 newborns, none older than 1 month, with 161 (94%) having undergone postoperative CEEG. They had CEEG within 6 hours of their return to the cardiac intensive care unit.
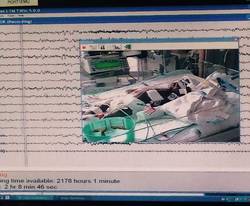
The study classified electroencephalographic seizures as EEG-only (also termed nonconvulsive seizures, with no observable clinical signs either at bedside or via video) or electroclinical seizures. Dr. Naim and colleagues said the majority of seizures they identified with CEEG would not have been noticed otherwise as they had no clinically obvious signs or symptoms.
The American Clinical Neurophysiology Society (ACNS) recommends that cardiac surgeons consider continuous CEEG monitoring in high-risk neonates with congenital heart disease (CHD) after bypass surgery, but Dr. Naim and coauthors raised the question of whether seizure incidence would justify routine CEEG for all neonates with CHD who’ve had bypass surgery, especially as health systems place greater emphasis on quality improvement programs and cost-effective strategies. The authors said that neonates with all types of congenital heart disease had seizures.
“In adult populations, CEEG has not been shown to significantly increase hospital costs, but cost-effectiveness analyses have not been performed in neonates with CHD,” the authors said.
So they attempted to identify at-risk populations of newborns who would benefit most from routine CEEG monitoring. In a multivariable model that the investigators used, both delayed sternal closure and longer deep hypothermic circulatory arrest (DHCA) during surgery seemed predictive of seizures, but the odds ratios for both were low, “suggesting the statistically significant findings may not be very useful in focusing CEEG implementation on a high-risk group.”
Previous studies have reported that identifying and treating seizures in newborns who have had bypass surgery may reduce secondary brain injury and improve outcomes (Pediatrics 2008;121:e759-67), and the Boston Circulatory Arrest Study showed an association between postoperative seizures and lower reading and math scores and lower cognitive and functional skills later in life (Circulation 2011;124:1361-1369). The authors cited other studies that showed older, critically ill children with “high seizure burdens” have had worse outcomes. (Critical Care Medicine 2013;31:215-23; Neurology 2014;82:396-404; Brain 2014;137:1429-38). They also pointed out increased risk if the seizure is not treated. “While occurrence of a seizure is a marker of brain injury, there may also be secondary injury if the seizure activity is not terminated,” Dr. Naim and coauthors said.
The investigators concluded that postoperative CEEG to identify seizures “is warranted,” and while they found some newborns may be at greater risk of postbypass seizures than others, they advocated for “widespread” monitoring strategies.
Their work also questioned the effectiveness of non-CEEG assessment. In the study, clinicians identified bedside events indicative of seizures – what the study termed “push-button events” – in 32 newborns, or about 18% of patients, but none of the events had an EEG correlate, so they were considered nonepileptic. When the authors looked more closely at those “push-button” events, they found they ranged from abnormal body movement in 14 and hypertension in 7 to tachycardia and abnormal face movements, among other characterizations, in lesser numbers.
“Furthermore, push-button events by bedside clinicians, including abnormal movements and hypertensive episodes concerning for possible seizures, did not have any EEG correlate, indicating that bedside clinical assessment for seizures without CEEG monitoring is unreliable,” Dr. Naim and colleagues said.
As to whether identifying and treating postbypass seizures in young newborns with CHD will improve long-term neurodevelopment in these children, the authors acknowledged that further study is needed.
They reported having no financial disclosures.
The findings of Dr. Maryam Naim and coauthors show that relying on physical examination alone is no longer adequate to rule out postoperative neurologic complications, Dr. Carl L. Backer and Dr. Bradley S. Marino said in their invited commentary on the study (J. Thorac. Cardiovasc. Surg. 2015 [doi:10.1016/j.jtcvs.2015.04.028]).
However, they noted that the level of “sophisticated monitoring” the investigators had at their disposal – 24-hour availability of EEG technologists, comprehensive 12-scalp electrode monitoring – is not available at all institutions. “What we need is a screening tool that is not as labor intensive,” Dr. Backer and Dr. Marino said – a screening CEEG monitor that would allow care teams to identify seizure activity at a minimal expense and serve as a basis for a full EEG for evaluation and avoid the expense and manpower for the vast majority of patients who do not have seizures.
Nonetheless, prevention of seizures in this newborn population is “critically important,” but that can only be achieved if the care team monitors for seizures and then assesses strategies, both during and after surgery, to eliminate development of seizures, the commentary authors said.
But the recent study points to the need for a multicenter, observational cross-sectional study using CEEG monitoring, Dr. Backer and Dr. Marino said.
Dr. Backer is a cardiovascular-thoracic surgeon and Dr. Marino is a cardiac surgeon at the Ann and Robert H. Lurie Children’s Hospital of Chicago.
The findings of Dr. Maryam Naim and coauthors show that relying on physical examination alone is no longer adequate to rule out postoperative neurologic complications, Dr. Carl L. Backer and Dr. Bradley S. Marino said in their invited commentary on the study (J. Thorac. Cardiovasc. Surg. 2015 [doi:10.1016/j.jtcvs.2015.04.028]).
However, they noted that the level of “sophisticated monitoring” the investigators had at their disposal – 24-hour availability of EEG technologists, comprehensive 12-scalp electrode monitoring – is not available at all institutions. “What we need is a screening tool that is not as labor intensive,” Dr. Backer and Dr. Marino said – a screening CEEG monitor that would allow care teams to identify seizure activity at a minimal expense and serve as a basis for a full EEG for evaluation and avoid the expense and manpower for the vast majority of patients who do not have seizures.
Nonetheless, prevention of seizures in this newborn population is “critically important,” but that can only be achieved if the care team monitors for seizures and then assesses strategies, both during and after surgery, to eliminate development of seizures, the commentary authors said.
But the recent study points to the need for a multicenter, observational cross-sectional study using CEEG monitoring, Dr. Backer and Dr. Marino said.
Dr. Backer is a cardiovascular-thoracic surgeon and Dr. Marino is a cardiac surgeon at the Ann and Robert H. Lurie Children’s Hospital of Chicago.
The findings of Dr. Maryam Naim and coauthors show that relying on physical examination alone is no longer adequate to rule out postoperative neurologic complications, Dr. Carl L. Backer and Dr. Bradley S. Marino said in their invited commentary on the study (J. Thorac. Cardiovasc. Surg. 2015 [doi:10.1016/j.jtcvs.2015.04.028]).
However, they noted that the level of “sophisticated monitoring” the investigators had at their disposal – 24-hour availability of EEG technologists, comprehensive 12-scalp electrode monitoring – is not available at all institutions. “What we need is a screening tool that is not as labor intensive,” Dr. Backer and Dr. Marino said – a screening CEEG monitor that would allow care teams to identify seizure activity at a minimal expense and serve as a basis for a full EEG for evaluation and avoid the expense and manpower for the vast majority of patients who do not have seizures.
Nonetheless, prevention of seizures in this newborn population is “critically important,” but that can only be achieved if the care team monitors for seizures and then assesses strategies, both during and after surgery, to eliminate development of seizures, the commentary authors said.
But the recent study points to the need for a multicenter, observational cross-sectional study using CEEG monitoring, Dr. Backer and Dr. Marino said.
Dr. Backer is a cardiovascular-thoracic surgeon and Dr. Marino is a cardiac surgeon at the Ann and Robert H. Lurie Children’s Hospital of Chicago.
In the first report evaluating the impact of a clinical guideline that calls for the use of postoperative continuous electroencephalography (CEEG) on infants after they’ve had cardiopulmonary bypass surgery, investigators at Children’s Hospital of Philadelphia and the University of Pennsylvania validated the clinical utility of routine CEEG monitoring and found that clinical assessment for seizures without CEEG is not a reliable marker for diagnosis and treatment.
In a report online in the Journal of Thoracic and Cardiovascular Surgery (J. Thorac. Cardiovasc. Surg. 2015 [doi:10.1016/j.jtcvs.2015.03.045]), Dr. Maryam Naim and colleagues said that CEEG identified electroencephalographic seizures in 8% of newborns after cardiopulmonary bypass surgery. The study, conducted over 18 months, evaluated 172 newborns, none older than 1 month, with 161 (94%) having undergone postoperative CEEG. They had CEEG within 6 hours of their return to the cardiac intensive care unit.
The study classified electroencephalographic seizures as EEG-only (also termed nonconvulsive seizures, with no observable clinical signs either at bedside or via video) or electroclinical seizures. Dr. Naim and colleagues said the majority of seizures they identified with CEEG would not have been noticed otherwise as they had no clinically obvious signs or symptoms.
The American Clinical Neurophysiology Society (ACNS) recommends that cardiac surgeons consider continuous CEEG monitoring in high-risk neonates with congenital heart disease (CHD) after bypass surgery, but Dr. Naim and coauthors raised the question of whether seizure incidence would justify routine CEEG for all neonates with CHD who’ve had bypass surgery, especially as health systems place greater emphasis on quality improvement programs and cost-effective strategies. The authors said that neonates with all types of congenital heart disease had seizures.
“In adult populations, CEEG has not been shown to significantly increase hospital costs, but cost-effectiveness analyses have not been performed in neonates with CHD,” the authors said.
So they attempted to identify at-risk populations of newborns who would benefit most from routine CEEG monitoring. In a multivariable model that the investigators used, both delayed sternal closure and longer deep hypothermic circulatory arrest (DHCA) during surgery seemed predictive of seizures, but the odds ratios for both were low, “suggesting the statistically significant findings may not be very useful in focusing CEEG implementation on a high-risk group.”
Previous studies have reported that identifying and treating seizures in newborns who have had bypass surgery may reduce secondary brain injury and improve outcomes (Pediatrics 2008;121:e759-67), and the Boston Circulatory Arrest Study showed an association between postoperative seizures and lower reading and math scores and lower cognitive and functional skills later in life (Circulation 2011;124:1361-1369). The authors cited other studies that showed older, critically ill children with “high seizure burdens” have had worse outcomes. (Critical Care Medicine 2013;31:215-23; Neurology 2014;82:396-404; Brain 2014;137:1429-38). They also pointed out increased risk if the seizure is not treated. “While occurrence of a seizure is a marker of brain injury, there may also be secondary injury if the seizure activity is not terminated,” Dr. Naim and coauthors said.
The investigators concluded that postoperative CEEG to identify seizures “is warranted,” and while they found some newborns may be at greater risk of postbypass seizures than others, they advocated for “widespread” monitoring strategies.
Their work also questioned the effectiveness of non-CEEG assessment. In the study, clinicians identified bedside events indicative of seizures – what the study termed “push-button events” – in 32 newborns, or about 18% of patients, but none of the events had an EEG correlate, so they were considered nonepileptic. When the authors looked more closely at those “push-button” events, they found they ranged from abnormal body movement in 14 and hypertension in 7 to tachycardia and abnormal face movements, among other characterizations, in lesser numbers.
“Furthermore, push-button events by bedside clinicians, including abnormal movements and hypertensive episodes concerning for possible seizures, did not have any EEG correlate, indicating that bedside clinical assessment for seizures without CEEG monitoring is unreliable,” Dr. Naim and colleagues said.
As to whether identifying and treating postbypass seizures in young newborns with CHD will improve long-term neurodevelopment in these children, the authors acknowledged that further study is needed.
They reported having no financial disclosures.
In the first report evaluating the impact of a clinical guideline that calls for the use of postoperative continuous electroencephalography (CEEG) on infants after they’ve had cardiopulmonary bypass surgery, investigators at Children’s Hospital of Philadelphia and the University of Pennsylvania validated the clinical utility of routine CEEG monitoring and found that clinical assessment for seizures without CEEG is not a reliable marker for diagnosis and treatment.
In a report online in the Journal of Thoracic and Cardiovascular Surgery (J. Thorac. Cardiovasc. Surg. 2015 [doi:10.1016/j.jtcvs.2015.03.045]), Dr. Maryam Naim and colleagues said that CEEG identified electroencephalographic seizures in 8% of newborns after cardiopulmonary bypass surgery. The study, conducted over 18 months, evaluated 172 newborns, none older than 1 month, with 161 (94%) having undergone postoperative CEEG. They had CEEG within 6 hours of their return to the cardiac intensive care unit.
The study classified electroencephalographic seizures as EEG-only (also termed nonconvulsive seizures, with no observable clinical signs either at bedside or via video) or electroclinical seizures. Dr. Naim and colleagues said the majority of seizures they identified with CEEG would not have been noticed otherwise as they had no clinically obvious signs or symptoms.
The American Clinical Neurophysiology Society (ACNS) recommends that cardiac surgeons consider continuous CEEG monitoring in high-risk neonates with congenital heart disease (CHD) after bypass surgery, but Dr. Naim and coauthors raised the question of whether seizure incidence would justify routine CEEG for all neonates with CHD who’ve had bypass surgery, especially as health systems place greater emphasis on quality improvement programs and cost-effective strategies. The authors said that neonates with all types of congenital heart disease had seizures.
“In adult populations, CEEG has not been shown to significantly increase hospital costs, but cost-effectiveness analyses have not been performed in neonates with CHD,” the authors said.
So they attempted to identify at-risk populations of newborns who would benefit most from routine CEEG monitoring. In a multivariable model that the investigators used, both delayed sternal closure and longer deep hypothermic circulatory arrest (DHCA) during surgery seemed predictive of seizures, but the odds ratios for both were low, “suggesting the statistically significant findings may not be very useful in focusing CEEG implementation on a high-risk group.”
Previous studies have reported that identifying and treating seizures in newborns who have had bypass surgery may reduce secondary brain injury and improve outcomes (Pediatrics 2008;121:e759-67), and the Boston Circulatory Arrest Study showed an association between postoperative seizures and lower reading and math scores and lower cognitive and functional skills later in life (Circulation 2011;124:1361-1369). The authors cited other studies that showed older, critically ill children with “high seizure burdens” have had worse outcomes. (Critical Care Medicine 2013;31:215-23; Neurology 2014;82:396-404; Brain 2014;137:1429-38). They also pointed out increased risk if the seizure is not treated. “While occurrence of a seizure is a marker of brain injury, there may also be secondary injury if the seizure activity is not terminated,” Dr. Naim and coauthors said.
The investigators concluded that postoperative CEEG to identify seizures “is warranted,” and while they found some newborns may be at greater risk of postbypass seizures than others, they advocated for “widespread” monitoring strategies.
Their work also questioned the effectiveness of non-CEEG assessment. In the study, clinicians identified bedside events indicative of seizures – what the study termed “push-button events” – in 32 newborns, or about 18% of patients, but none of the events had an EEG correlate, so they were considered nonepileptic. When the authors looked more closely at those “push-button” events, they found they ranged from abnormal body movement in 14 and hypertension in 7 to tachycardia and abnormal face movements, among other characterizations, in lesser numbers.
“Furthermore, push-button events by bedside clinicians, including abnormal movements and hypertensive episodes concerning for possible seizures, did not have any EEG correlate, indicating that bedside clinical assessment for seizures without CEEG monitoring is unreliable,” Dr. Naim and colleagues said.
As to whether identifying and treating postbypass seizures in young newborns with CHD will improve long-term neurodevelopment in these children, the authors acknowledged that further study is needed.
They reported having no financial disclosures.
FROM THE JOURNAL OF THORACIC AND CARDIOVASCULAR SURGERY
Key clinical point: Electroencephalography is more effective than clinical observation in identifying seizures in infants immediately after they’ve had cardiopulmonary bypass surgery.
Major finding: Postoperative CEEG identified seizures in 8% of newborns with congenital heart disease after coronary bypass surgery.
Data source: Chart review involved 172 neonates from a single center. Multiple logistic regression analysis assessed seizures and clinical and predictive factors.
Disclosures: The authors reported having no financial disclosures.
Care of the aging HIV patient
In the 1980s, human immunodeficiency virus (HIV) infection was considered untreatable and predictably lethal. Today, with highly effective antiretroviral therapy, it has become a chronic condition in which patients have a life expectancy comparable to that in the general population.
This change has led to new challenges for primary care physicians, many of whom now find themselves either the sole medical provider for or the comanager of aging HIV-infected patients. Given that about one-fifth of new HIV diagnoses are now in people over the age of 50, it is crucial that primary care providers be able to recognize and diagnose the disease in this population. In addition, they need to effectively manage the polypharmacy and subsequent drug interactions prevalent in older HIV-infected patients. Finally, the clinician must address comorbid diseases common in the elderly, specifically neurologic, cardiovascular, metabolic, and endocrine disorders, as well as performing routine cancer screening.
Take-home point
- As the number of people age 50 and older with HIV infection increases, primary care providers must be able to both recognize and manage the condition.
RISING PREVALENCE OF HIV IN THE ELDERLY
Globally, about 2.5 million people received a new diagnosis of HIV infection in 2011, and about 35 million people worldwide are currently living with it.1 An estimated 1.1 million Americans are living with HIV, and of these, about 16% do not know they are infected.2
Antiretroviral therapy has greatly improved the life expectancy of HIV-infected patients, and the number of HIV-infected people over age 50 continues to rise. A successfully treated HIV-positive person with a CD4 count higher than 350 × 106/L and a suppressed viral load now has a normal life expectancy.3 In 2011, nearly 20% of newly diagnosed HIV-infected people in the United States were over age 50, as were nearly 25% of those with a new diagnosis of acquired immune deficiency syndrome (AIDS).4 This year (2015), we expect that more than half of all HIV-infected people in the United States will be over age 50.5
The rising prevalence of HIV infection in this age group has prompted reevaluation of screening guidelines. The US Preventive Services Task Force recommends screening for HIV in all people ages 15 to 65, and also after age 65 in people at ongoing risk of infection.6 The American College of Physicians has suggested that the range for routine HIV screening be expanded to age 75.7 The cost-effectiveness of expanded and more frequent HIV testing appears to justify it.8
Take-home points
- An HIV-infected patient who is compliant with an appropriate antiretroviral regimen and has a CD4 count higher than 350 × 106/L and a suppressed viral load now has a normal life expectancy.
- Today, nearly 20% of newly diagnosed HIV-infected people and more than 50% of all HIV-infected people in the United States are over the age of 50.
- The age range for routine screening for HIV infection should be expanded.
HIGH-RISK GROUPS AMONG THE ELDERLY
Early in the HIV epidemic, older patients acquired HIV from blood transfusions received because of hemophilia and other disorders. However, this rapidly ceased after blood banks began screening blood products. Today, people over age 50 who acquire HIV have many of the same risk factors as younger people.
Men who have sex with men are the largest subgroup of HIV-infected people in the United States, even among those over age 50. In particular, white men who have sex with men now constitute the largest demographic group among the HIV-infected elderly.4
Intravenous drug users make up about 15% of older people with HIV.
Women who have sex with infected men or with men at risk of HIV infection make up the largest group of older women with HIV.4
Sex and the older person
Many older HIV-infected people remain sexually active and continue to engage in unprotected sexual intercourse far into advanced age. According to one survey, 53% of Americans ages 65 to 74 are engaging in sexual activity regularly; however, they are not using protective measures with up to 91% of casual partners and 70% of new partners.9,10 Many widowed and divorced people are dating again, and they may be unfamiliar with condom use or may be reluctant to use condoms because condoms can often make it difficult to maintain an erection.
Drugs for erectile dysfunction are making it easier for the elderly to engage in both vaginal and anal intercourse, but often without a condom.9 Older women who no longer worry about getting pregnant may be less likely to insist their partners use a condom and to practice safe sex. In addition, age-related thinning and dryness can cause vaginal tears, increasing the risk of HIV transmission.11
Take-home points
- People older than 50 have risk factors for HIV similar to those in younger people.
- Men who have sex with men compose the largest group of HIV-infected individuals in the elderly population.
- Unprotected sexual intercourse is common in the elderly for several reasons: unfamiliarity with condom use, difficulty maintaining an erection, lack of concern about possible pregnancy, and vaginal thinning and dryness in women.
UNDERDIAGNOSIS AND LATE DIAGNOSIS IN THE ELDERLY
The cumulative number of AIDS cases in adults age 50 and older increased nearly ninefold from 1990 to the end of 2009. Even more worrisome, one-half of HIV-positive adults over age 50 are diagnosed with AIDS simultaneously or within 1 year of their HIV diagnosis.4 This late diagnosis—and therefore late initiation of treatment—is associated with poorer health outcomes and more rapid disease progression.12
HIV infection in older adults often goes undiagnosed, for several reasons.
Providers may underestimate the risk in this population and therefore may not discuss HIV transmission or perform testing. Despite a US Centers for Disease Control and Prevention recommendation that people ages 13 to 64 be tested at least once, and more often if sexually active, only 35% of adults ages 45 to 64 have ever been tested for HIV infection.13
Older patients may not perceive themselves to be at risk of HIV infection because of lack of insight and information about its prevention and transmission. They are also less likely than younger adults to discuss their sexual habits or drug use with providers.14 In addition, compared with the young sexually active population, very little HIV prevention education is targeted to older people.15 Social stigmatization is also a concern for many HIV-infected elderly, as a perceived negative reputation within their community may prevent them from seeking care and disclosing their HIV status.
Take-home points
Reasons that HIV infection is underdiagnosed in the elderly include lack of:
- Provider recognition
- Insight and information about HIV prevention and transmission
- HIV-prevention education targeting the elderly
- Disclosure because of the social stigma of HIV infection.
HIV ACCELERATES AGING, AGING REDUCES IMMUNITY
Many HIV-positive people can expect to live as long as people in the general population, but those who are diagnosed late and thus are started on antiretroviral therapy later in the course of their infection have a reduced life expectancy. Longevity depends on both restoring the CD4 count to near-normal and suppressing the viral load to undetectable levels.3,16 This is especially important for older adults, as HIV may accelerate aging, and aging itself may speed the progression of HIV disease, so that therapy may result in delayed or only partial restoration of immunity.
Older age at the time of HIV infection is a strong predictor of accelerated HIV disease progression in the absence of therapy.17 Left untreated, older patients with HIV lose CD4 cells and progress to AIDS and death faster than younger patients. The deleterious effects of chronic immune activation in the course of HIV infection, combined with the immune senescence of aging, are thought to promote this accelerated course.18
Recent data indicate that starting antiretroviral therapy early can help prevent the CD4-cell impairment that occurs with aging.19 However, in adults over age 50, the capacity to restore the CD4 count with antiretroviral therapy apears to be reduced, despite demonstrated viral load suppression and better adherence.20 Although mean adherence rates appear higher in older HIV-infected patients, they are worse in those with neurocognitive impairment, highlighting the importance of evaluating neurocognition in this population.21
Decreased immune recovery and the subsequent increased risk of serious AIDS events are factors that now favor starting antiretroviral therapy in all HIV patients over age 50, regardless of CD4 count.
Take-home points
- Without treatment, HIV infection in older patients progresses more rapidly to AIDS and death than in younger patients.
- HIV-positive people over age 50 who have never received antiretroviral therapy should be strongly considered for it, regardless of the CD4 count.
SO MANY DRUGS, SO MANY INTERACTIONS
Since HIV patients are now living longer thanks to antiretroviral therapy, they are now experiencing more disease- and treatment-related problems. This has led to an increased likelihood of polypharmacy, defined here as the use of six or more medications.
In general, polypharmacy in the elderly is associated with adverse drug events, drug interactions, inappropriate medication use, delirium, falls, fractures, and poor medication adherence.22,23 But it becomes even more of a problem in HIV-infected elderly patients, as various drug interactions can alter the effectiveness of the antiretroviral regimen and can result in drug toxicity.
The most common classes of medications used in the elderly are antihypertensives, lipid-lowering agents, antiplatelet medications, antidepressants, anxiolytics, sedatives, and analgesics, and many of these have notable interactions with current antiretroviral regimens.24,25 Most medications, including antiretrovirals, are cleared by the liver or kidneys, and the function of these organs often decreases with age, resulting in impaired elimination and in drug accumulation.
Information on drug interactions is readily available from the US Department of Health and Human Services,26 drug interaction databases,27,28 and drug interaction software. The combination of antiretroviral therapy and preexisting polypharmacy significantly increases the risk of serious interactions, which can lead to drug toxicity, poorer adherence with antiretroviral therapy, loss of efficacy of the coadministered medication, or resurgence of HIV infection due to drug-drug interactions affecting the metabolism and ultimate efficacy of the antiretroviral therapy. An increased awareness of common drug-drug interactions can prevent coadministration of potentially harmful medications in elderly HIV patients.
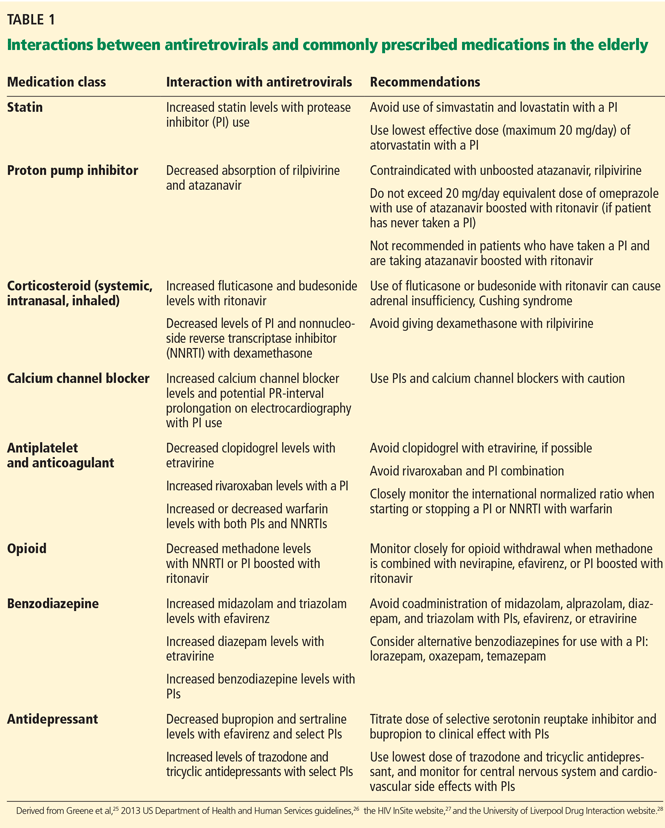
Important interactions between antiretroviral drugs and other drug classes are summarized in Table 1.25–28 Most notably:
- Simvastatin and lovastatin are contraindicated with any protease inhibitor.
- Proton pump inhibitors are not recommended for patients taking ritonavir-boosted atazanavir. If a proton pump inhibitor is necessary, the daily dose should not exceed 20 mg of omeprazole or its equivalent in patients who have never taken a protease inhibitor, and it should be taken 12 hours before boosted atazanavir.26
- Corticosteroids, whether systemic, inhaled, or intranasal (eg, fluticasone, budesonide), should be avoided in combination with any protease inhibitor, as they can cause iatrogenic Cushing syndrome and also pose the risk of adrenal crisis during acute illness.27
Take-home points
- In cases of preexisting polypharmacy, antiretroviral therapy can lead to significant drug toxicity, poor adherence to medications, and resurgence of HIV infection.
- Increased provider awareness of common drug-drug interactions can prevent the prescribing of potentially harmful drug combinations to HIV-infected elderly patients.
COMORBIDITIES
In recent years, more than half of the deaths in HIV patients on antiretroviral therapy have been from noninfectious comorbidities such as cardiovascular disease, bone disease, and renal failure, which often coexist and are associated with advanced age.29 In fact, both older age and each additional year of antiretroviral therapy are independent predictors of polypathology (simultaneous occurrence of two or more defined diseases).30 The Antiretroviral Therapy Cohort Collaboration found that age greater than 50 was strongly associated with increasing rates of non–AIDS-related malignancy and cardiovascular disease.31
CARDIOVASCULAR DISEASE
With the increasing life expectancy of HIV-infected adults on antiretroviral therapy, cardiovascular disease has become an important concern. HIV-infected adults appear to have a significantly greater risk of myocardial infarction and coronary artery disease than age-matched HIV-negative individuals.32 Strikingly, being older than 50 itself increases the risk of hospitalization for cardiovascular disease fivefold (incidence rate ratio 5.01, 95% confidence interval 3.41–7.38).33 In addition, HIV infection is associated with a risk of acute myocardial infarction 50% higher than that explained by recognized risk factors.34
This high prevalence of coronary artery disease is likely from a combination of factors, including increasing age and the chronic inflammation and immune activation associated with HIV infection.35 An association between untreated HIV disease and markers of risk for cardiovascular disease has been identified.36,37
In addition, antiretroviral therapy is associated with dyslipidemia, which is most pronounced with protease inhibitor regimens. Whether specific lipid changes associated with individual antiretroviral drugs affect cardiovascular risk remains uncertain. In the Data Collection on Adverse Events of Anti-HIV Drugs studies,38 only cumulative exposure to indinavir, lopinavir-ritonavir, and didanosine was associated with an increased risk of myocardial infarction.38
Traditional risk factors such as obesity, tobacco use, and genetic predisposition also apply to HIV-infected people.39 In fact, the prevalence of traditional risk factors such as smoking and dyslipidemia is generally higher in HIV-infected people than in the general population, although this situation may be improving.40
Science needs to elucidate the relationship between traditional and nontraditional risk factors for cardiovascular disease in older HIV-infected adults. In the meantime, older patients with HIV require aggressive management of modifiable risk factors.
Tools for assessing cardiovascular risk include the Framingham risk score41 and the Data Collection on Adverse Events of Anti-HIV Drugs 5-year risk calculator.42 The European AIDS Clinical Society guidelines recommend considering changing the antiretroviral regimen if the patient’s 10-year risk of cardiovascular disease is more than 20%.43 Recommended strategies for reducing cardiovascular risk in elderly patients with HIV infection include counseling about smoking cessation and weight loss at every clinic visit and optimally controlling dyslipidemia and hypertension using nationally accepted standardized guidelines.44
Take-home points
- HIV infection is associated with a 50% higher risk of acute myocardial infarction beyond that explained by traditional risk factors.
- Chronic inflammation, immune activation, and dyslipidemia associated with antiretroviral therapy all contribute to cardiovascular disease in HIV-infected patients.
- HIV-infected elderly patients require aggressive management of modifiable risk factors for cardiovascular disease.
ENDOCRINE DISEASE
Diabetes mellitus
The estimated prevalence of diabetes mellitus is 3% in HIV-infected people who have never received antiretroviral therapy, but glucose intolerance increases to the range of 10% to 25% in those who have started it.45 Glucose disorders are associated with traditional risk factors as well as with HIV-associated factors such as lipodystrophy and antiretroviral therapy, specifically long-term use of protease inhibitors.46 Although increasing age and obesity clearly play a role in the development of diabetes mellitus in this population, HIV-specific factors may also allow diabetes to develop at a lower level of adiposity than in people without HIV infection.47
Strategies for preventing type 2 diabetes mellitus in HIV-infected patients focus on avoiding excessive weight gain, especially after starting antiretroviral therapy; regularly screening for diabetes using hemoglobin A1c, both before and after starting antiretroviral therapy; and continuing to check hemoglobin A1c every 6 months. The target hemoglobin A1c should be less than 7.0%. This threshold should be increased to 8% in frail elderly adults if their anticipated life expectancy is less than 5 years, given their higher risk of hypoglycemia, polypharmacy, and drug interactions.48 In addition, as in HIV-negative patients, diabetes screening should be performed if systolic blood pressure exceeds 135/80 mm Hg.
Insulin sensitizers such as metformin and thiazolidinediones should be considered for treating diabetes in HIV-infected patients if no contraindications exist. Consideration may also be given to switching the antiretroviral regimen from a protease inhibitor-based regimen to a nonnucleoside reverse transcriptase inhibitor-based regimen.48
Take-home points
- Glucose intolerance has been associated with HIV-specific factors, including lipodystrophy and antiretroviral therapy.
- Avoiding excessive weight gain, use of insulin-sensitizing medications, and alteration in antiretroviral regimens should be considered for the treatment of diabetes mellitus in HIV infection.
Osteoporosis
Osteoporotic bone disease disproportionately affects patients with advanced HIV infection compared with patients of similar age.49 Bone mineral density is lower and the fracture rate is higher in HIV-infected individuals.
The pathogenesis of bone disease appears to be multifactorial. Traditional risk factors include hypogonadism, smoking, alcohol use, and low body weight, while HIV-related risk factors include chronic immune activation and antiretroviral therapy.50
Several antiretroviral regimens have been linked to clinically significant bone loss, including both tenofovir-based and protease inhibitor-based regimens.51 Most studies have shown that bone mineral density decreases by 2% to 6% in the first 2 years after starting these regimens52; however, long-term effects on bone loss are unknown.
Questions remain. For example, what are the exact mechanisms that lead to the acute decrease in bone mineral density after starting antiretroviral therapy? And why is vitamin D deficiency is so prevalent in HIV infection, with low vitamin D levels seen in up to 60% to 75% of elderly HIV-infected patients?53
Both the Work Group for the HIV and Aging Consensus Project54 and the European AIDS Clinical Society43 recommend screening for and treating causes of secondary low bone mineral density in HIV-infected men over age 50 and postmenopausal HIV-infected women. These causes include vitamin D deficiency. As of 2013, the National Osteoporosis Foundation guidelines include HIV infection and antiretroviral therapy as osteoporosis risk factors that should trigger screening for low bone mineral density with dual-energy x-ray absorptiometry (DXA).55
As in the general population, the preferred treatment for low bone mineral density in people with HIV is a bisphosphonate, in addition to ensuring adequate calcium and vitamin D intake. It is important to repeat DXA imaging every 2 years and to reassess the need for continued bisphosphonate therapy after 3 to 5 years because of a possible increased risk of fracture with prolonged use.
Take-home points
- Osteoporosis and vitamin D deficiency both appear to be more prevalent with HIV infection.
- HIV infection and antiretroviral therapy are risk factors that should prompt DXA screening to evaluate for osteoporosis.
NEUROCOGNITIVE DISORDERS
HIV-associated neurocognitive disorders are common, with an estimated 50% of HIV-infected patients experiencing some degree of cognitive loss and some progressing to dementia.56 Unfortunately, studies suggest that cognitive disorders can occur despite good HIV control with antiretroviral therapy, with one report demonstrating that 84% of patients with cognitive complaints and 64% without complaints were affected by an HIV-associated neurocognitive disorder.57
HIV-associated dementia is often subcortical, with fluctuating symptoms such as psychomotor retardation, difficulty multitasking, and apathy. In contrast to dementia syndromes such as Alzheimer disease, relentless progression is less common in HIV-infected patients who receive antiretroviral therapy.
The Mini-Mental State Examination should not be used to screen for HIV-associated neurocognitive disorders, as it does not assess the domains that are typically impaired. The Montreal Cognitive Assessment has been suggested as the best screening instrument in elderly HIV-infected patients; it is available at no cost at www.mocatest.org.58
As HIV-associated neurocognitive disorder is a diagnosis of exclusion, an evaluation for alternative diagnoses such as syphilis, hypothyroidism, and depression is recommended. If an HIV-associated neurocognitive disorder is diagnosed, referral to specialty care should be considered, as interventions such as lumbar puncture to assess cerebrospinal fluid viral escape and changing the antiretroviral regimen to improve central nervous system penetration are possible options under study.
Patients with poorly controlled HIV and a depressed CD4 count are at risk of a number of central nervous system complications in addition to HIV-associated neurocognitive disorders, eg, central nervous system toxoplasmosis, cryptococcal meningitis, progressive multifocal leukoencephalopathy, and primary central nervous system lymphoma. Adherence to an effective antiretroviral regimen is the primary prevention strategy.
Take-home points
- HIV-associated neurocognitive disorders and dementia can occur despite appropriate HIV control and adherence to antiretroviral therapy.
- Adherence to antiretroviral therapy is the primary prevention against most central nervous system complications in HIV infection.
GERIATRIC SYNDROMES
The aging HIV-infected adult may also be at increased risk of geriatric syndromes.
In particular, a frailty-related phenotype of weight loss, exhaustion, slowness, and low physical activity was more common in HIV-infected elderly than in noninfected elderly.59 HIV-infected men are 4.5 to 10 times more likely than age-matched controls to be frail, and the likelihood of frailty increases with age, duration of HIV infection, having a CD4 count lower than 350 × 106/L, and having uncontrolled HIV replication.60,61
Other geriatric syndromes such as falls, urinary incontinence, and functional impairment have been identified in 25% to 56% of older HIV-infected patients.62 Indeed, the combination of HIV and older age may adversely affect performance of instrumental activities of daily living.63 Also, as previously mentioned, nondisclosure, fear of HIV-related social stigmatization, and a desire to be self-reliant are all factors that perpetuate the social isolation that is common among the HIV-infected elderly.
For these reasons, a comprehensive approach involving a geriatrician, an infectious disease specialist, and community social workers is needed to manage the care of this aging population.
Take-home point
- Geriatric syndromes have an important impact on health in aging HIV patients.
CANCER SCREENING IN HIV PATIENTS
People with HIV have an elevated risk of cancer. Specifically, compared with the general population, their risk is:
- 3,640 times higher for Kaposi sarcoma
- 77 times higher for non-Hodgkin lymphomas
- 6 times higher for cervical cancer.64,65
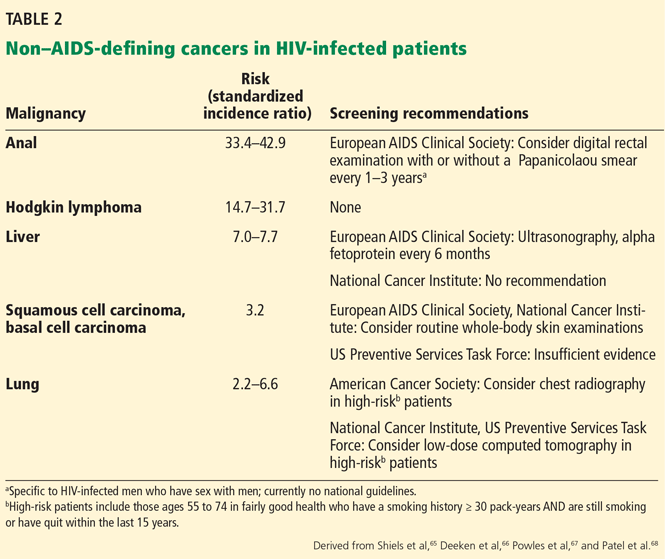
These cancers are considered “AIDS-defining,” and fortunately, the development of effective antiretroviral therapy in the 1990s has led to a marked reduction in their incidence. However, the aging HIV population is now experiencing a rise in the incidence of non–AIDS-defining cancers, such as cancers of the lung, liver, kidney, anus, head and neck, and skin, as well as Hodgkin lymphoma.66 Table 2 shows the standardized incidence ratio of selected non–AIDS-defining cancers in HIV-infected patients as reported in several large international studies.65,67,68 The etiology for the increased risk of non–AIDS-defining cancers in the HIV-infected population is not clear, but possible explanations include the virus itself, antiretroviral therapy, and co-infection with other viruses such as hepatitis B, hepatitis C, and Epstein-Barr virus.
Guidelines for cancer screening vary by organization, and the American Cancer Society, the National Cancer Institute, and the US Preventive Services Task Force do not have formal screening guidelines for the most common non–AIDS-defining cancers. The European AIDS Clinical Society, however, has proposed some screening recommendations for selected malignancies.43
In general, screening recommendations are similar to those for HIV-negative patients. A specific difference for HIV-infected patients is in cervical cancer screening. HIV-infected women should undergo a Papanicolaou smear at 6-month intervals during the first year after diagnosis of HIV infection and, if the results are normal, annually thereafter. There is no consensus as to whether human papillomavirus testing should be performed routinely on HIV-infected women.
At the time of this writing, there are no recommendations for routine screening for anal cancer, although some specialists recommend anal cytologic screening for HIV-positive men and women, and an annual digital anal examination may be useful to detect masses that could be anal cancer.69
Take-home points
- The incidence of non–AIDS-defining cancers is rising in the aging HIV population.
- There are currently no formal recommendations for routine screening for anal cancer.
FINAL WORD
Because patients with HIV are living longer as a result of newer effective combination antiretroviral therapies, physicians face a new challenge of managing conditions in these patients that are traditionally associated with aging. Providers will need to improve their understanding of drug-drug interactions and polypharmacy issues and be able to address the complex medical and psychosocial issues in this growing population. As patients with HIV on effective antiretroviral therapy grow older, the burden of comorbid medical disease will continue to increase.
- World Health Organization. HIV/AIDS: fact sheet. www.who.int/mediacentre/factsheets/fs360/en/. Accessed April 16, 2015.
- Centers for Disease Control and Prevention (CDC). HIV/AIDS: basic statistics. www.cdc.gov/hiv/basics/statistics.html. Accessed April 16, 2015.
- May MT, Gompels M, Delpech V, et al; UK Collaborative HIV Cohort (UK CHIC) Study. Impact on life expectancy of HIV-1 positive individuals of CD4+ cell count and viral load response to antiretroviral therapy. AIDS 2014; 28:1193–1202.
- Centers for Disease Control and Prevention (CDC). Diagnoses of HIV infection in the United States and dependent areas: HIV surveillance report. www.cdc.gov/hiv/library/reports/surveillance/2011/surveillance_Report_vol_23.html. Accessed April 16, 2015.
- Effros RB, Fletcher CV, Gebo K, et al. Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis 2008; 47:542–553.
- Moyer VA; US Preventive Services Task Force. Screening for HIV: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:51–60.
- Qaseem A, Snow V, Shekelle P, Hopkins R Jr, Owens DK; Clinical Efficacy Assessment Subcommittee, American College of Physicians. Screening for HIV in health care settings: a guidance statement from the American College of Physicians and HIV Medicine Association. Ann Intern Med 2009; 150:125–131.
- Lucas A, Armbruster B. The cost-effectiveness of expanded HIV screening in the United States. AIDS 2013; 27:795–801.
- Lindau ST, Schumm LP, Laumann EO, Levinson W, O’Muircheartaigh CA, Waite LJ. A study of sexuality and health among older adults in the United States. N Engl J Med 2007; 357:762–774.
- Schick V, Herbenick D, Reece M, et al. Sexual behaviors, condom use, and sexual health of Americans over 50: implications for sexual health promotion for older adults. J Sex Med 2010; 7(suppl 5):315–329.
- US Department of Health and Human Services, HIV/AIDS Bureau. The Ryan White HIV/AIDS program: population fact sheet: August 2010. Older adults. http://hab.hrsa.gov/abouthab/populations/olderadultsfacts.pdf. Accessed April 16, 2015.
- May M, Gompels M, Delpech V, et al. Impact of late diagnosis and treatment on life expectancy in people with HIV-1: UK Collaborative HIV Cohort (UK CHIC) Study. BMJ 2011; 343:d6016.
- Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention (CDC). Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep 2006; 55:1–17.
- Health Resources and Services Administration (HRSA); HIV/AIDS Bureau. HRSA CAREAction. The graying of HIV. http://hab.hrsa.gov/newspublications/careactionnewsletter/february2009.pdf. Accessed April 16, 2015.
- AIDS InfoNet. Fact sheet number 616: Older people and HIV. http://aidsinfonet.org/fact_sheets/view/616. Accessed April 16, 2015.
- Rodger AJ, Lodwick R, Schechter M, et al; INSIGHT SMART, ESPRIT Study Groups. Mortality in well controlled HIV in the continuous antiretroviral therapy arms of the SMART and ESPRIT trials compared with the general population. AIDS 2013; 27:973–979.
- Kitahata MM, Gange SJ, Abraham AG, et al; NA-ACCORD Investigators. Effect of early versus deferred antiretroviral therapy for HIV on survival. N Engl J Med 2009; 360:1815–1826.
- Cao W, Jamieson BD, Hultin LE, Hultin PM, Effros RB, Detels R. Premature aging of T cells is associated with faster HIV-1 disease progression. J Acquir Immune Defic Syndr 2009; 50:137–147.
- Allers K, Bösel D, Epple HJ, et al. Effect of age on the CD4+ T-cell impairment in HIV-infected persons without and with cART. J Acquir Immune Defic Syndr 2014; 66:7–15.
- Kalayjian RC, Spritzler J, Matining RM, et al. Older HIV-infected patients on antiretroviral therapy have B-cell expansion and attenuated CD4 cell increases with immune activation reduction. AIDS 2013; 27:1563–1571.
- Althoff KN, Gebo KA, Gange SJ, et al; North American AIDS Cohort Collaboration on Research and Design. CD4 count at presentation for HIV care in the United States and Canada: are those over 50 years more likely to have a delayed presentation? AIDS Res Ther 2010; 7:45.
- Freeland KN, Thompson AN, Zhao Y, Leal JE, Mauldin PD, Moran WP. Medication use and associated risk of falling in a geriatric outpatient population. Ann Pharmacother 2012; 46:1188–1192.
- Steinman MA, Hanlon JT. Managing medications in clinically complex elders: “There’s got to be a happy medium.” JAMA 2010; 304:1592–1601.
- Marzolini C, Elzi L, Gibbons S, et al; Swiss HIV Cohort Study. Prevalence of comedications and effect of potential drug-drug interactions in the Swiss HIV Cohort Study. Antivir Ther 2010; 15:413–423.
- Greene M, Justice AC, Lampiris HW, Valcour V. Management of human immunodeficiency virus infection in advanced age. JAMA 2013; 309:1397–1405.
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed April 16, 2015.
- UCSF Center for HIV Information. HIVInSite. Comprehensive, up-to-date information on HIV/AIDS treatment, prevention, and policy from the University of California San Francisco: database of antiretroviral drug interactions. http://hivinsite.ucsf.edu/. Accessed April 16, 2015.
- The University of Liverpool. Drug interaction charts. www.hiv-druginteractions.org. Accessed April 16, 2015.
- Vance DE, Mugavero M, Willig J, Raper JL, Saag MS. Aging with HIV: a cross-sectional study of comorbidity prevalence and clinical characteristics across decades of life. J Assoc Nurses AIDS Care 2011; 22:17–25.
- Guaraldi G, Orlando G, Zona S, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis 2011; 53:1120–1126.
- Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996-2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 2010; 50:1387–1396.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- Berry SA, Fleishman JA, Moore RD, Gebo KA; HIV Research Network. Trends in reasons for hospitalization in a multisite United States cohort of persons living with HIV, 2001-2008. J Acquir Immune Defic Syndr 2012; 59:368–375.
- Freiberg MS, Chang CC, Kuller LH, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med 2013; 173:614–622.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Worm SW, Sabin C, Weber R, et al. Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. J Infect Dis 2010; 201:318–330.
- Lake JE, Currier JS. Metabolic disease in HIV infection. Lancet Infect Dis 2013; 13:964–975.
- Data Collection on Adverse Events of Anti-HIV Drugs Study Group; Sabin CA, d’Arminio Monforte A, Friis-Moller N, et al. Changes over time in risk factors for cardiovascular disease and use of lipid-lowering drugs in HIV-infected individuals and impact on myocardial infarction. Clin Infect Dis 2008; 46:1101–1110.
- Falcone EL, Mangili A, Skinner S, Alam A, Polak JF, Wanke CA. Framingham risk score and early markers of atherosclerosis in a cohort of adults infected with HIV. Antivir Ther 2011; 16:1–8.
- Friis-Møller N, Thiébaut R, Reiss P, et al; DAD study group. Predicting the risk of cardiovascular disease in HIV-infected patients: the Data Collection on Adverse Effects of Anti-HIV Drugs study. Eur J Cardiovasc Prev Rehabil 2010; 17:491–501.
- European AIDS Clinical Society Guidelines (EACS). www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. Accessed April 16, 2015.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Samaras K. The burden of diabetes and hyperlipidemia in treated HIV infection and approaches for cardiometabolic care. Curr HIV/AIDS Rep 2012; 9:206–217.
- Rasmussen LD, Mathiesen ER, Kronborg G, Pedersen C, Gerstoft J, Obel N. Risk of diabetes mellitus in persons with and without HIV: a Danish nationwide population-based cohort study. PLoS One 2012; 7:e44575.
- Capeau J, Bouteloup V, Katlama C, et al; ANRS CO8 APROCO-COPILOTE Cohort Study Group. Ten-year diabetes incidence in 1,046 HIV-infected patients started on a combination antiretroviral treatment. AIDS 2012; 26:303–314.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006; 20:2165–2174.
- Rothman MS, Bessesen MT. HIV infection and osteoporosis: pathophysiology, diagnosis, and treatment options. Curr Osteoporos Rep 2012; 10:270–277.
- Bedimo R, Maalouf NM, Zhang S, Drechsler H, Tebas P. Osteoporotic fracture risk associated with cumulative exposure to tenofovir and other antiretroviral agents. AIDS 2012; 26:825–831.
- Brown TT, McComsey GA, King MS, Qaqish RB, Bernstein BM, da Silva BA. Loss of bone mineral density after antiretroviral therapy initiation, independent of antiretroviral regimen. J Acquir Immune Defic Syndr 2009; 51:554–561.
- Rodríguez M, Daniels B, Gunawardene S, Robbins GK. High frequency of vitamin D deficiency in ambulatory HIV-positive patients. AIDS Res Hum Retroviruses 2009; 25:9–14.
- Work Group for HIV and Aging Consensus Project. Summary report from the Human Immunodeficiency Virus and Aging Consensus Project: treatment strategies for clinicians managing older individuals with the human immunodeficiency virus. J Am Geriatr Soc 2012; 60:974–979.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. 2013 Issue, Version 3. http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 16, 2015.
- Heaton RK, Clifford DB, Franklin DR Jr, et al; CHARTER Group. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010; 75:2087–2096.
- Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010; 24:1243–1250.
- Valcour VG. Evaluating cognitive impairment in the clinical setting: practical screening and assessment tools. Top Antivir Med 2011; 19:175–180.
- Desquilbet L, Jacobson LP, Fried LP, et al; Multicenter AIDS Cohort Study. HIV-1 infection is associated with an earlier occurrence of a phenotype related to frailty. J Gerontol A Biol Sci Med Sci 2007; 62:1279–1286.
- Desquilbet L, Jacobson LP, Fried LP, et al. A frailty-related phenotype before HAART initiation as an independent risk factor for AIDS or death after HAART among HIV-infected men. J Gerontol A Biol Sci Med Sci 2011; 66:1030–1038.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Greene M, Valcour V, Miao Y, et al. Geriatric syndromes are common among older HIV-infected adults. 21st Conference on Retroviruses and Opportunistic Infections (CROI) 2014 March 3-6, Boston MA.
- Morgan EE, Iudicello JE, Weber E, et al; HIV Neurobehavioral Research Program (HNRP) Group. Synergistic effects of HIV infection and older age on daily functioning. J Acquir Immune Defic Syndr 2012; 61:341–348.
- Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 2007; 370:59–67.
- Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst 2011; 103:753–762.
- Deeken JF, Tjen-A-Looi A, Rudek MA, et al. The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin Infect Dis 2012; 55:1228–1235.
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol 2009; 27:884–890.
- Patel P, Hanson DL, Sullivan PS, et al; Adult and Adolescent Spectrum of Disease Project and HIV Outpatient Study Investigators. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992-2003. Ann Intern Med 2008; 148:728–736.
- Kaplan JE, Benson C, Holmes KK, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC); National Institutes of Health; HIV Medicine Association of the Infectious Diseases Society of America. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
In the 1980s, human immunodeficiency virus (HIV) infection was considered untreatable and predictably lethal. Today, with highly effective antiretroviral therapy, it has become a chronic condition in which patients have a life expectancy comparable to that in the general population.
This change has led to new challenges for primary care physicians, many of whom now find themselves either the sole medical provider for or the comanager of aging HIV-infected patients. Given that about one-fifth of new HIV diagnoses are now in people over the age of 50, it is crucial that primary care providers be able to recognize and diagnose the disease in this population. In addition, they need to effectively manage the polypharmacy and subsequent drug interactions prevalent in older HIV-infected patients. Finally, the clinician must address comorbid diseases common in the elderly, specifically neurologic, cardiovascular, metabolic, and endocrine disorders, as well as performing routine cancer screening.
Take-home point
- As the number of people age 50 and older with HIV infection increases, primary care providers must be able to both recognize and manage the condition.
RISING PREVALENCE OF HIV IN THE ELDERLY
Globally, about 2.5 million people received a new diagnosis of HIV infection in 2011, and about 35 million people worldwide are currently living with it.1 An estimated 1.1 million Americans are living with HIV, and of these, about 16% do not know they are infected.2
Antiretroviral therapy has greatly improved the life expectancy of HIV-infected patients, and the number of HIV-infected people over age 50 continues to rise. A successfully treated HIV-positive person with a CD4 count higher than 350 × 106/L and a suppressed viral load now has a normal life expectancy.3 In 2011, nearly 20% of newly diagnosed HIV-infected people in the United States were over age 50, as were nearly 25% of those with a new diagnosis of acquired immune deficiency syndrome (AIDS).4 This year (2015), we expect that more than half of all HIV-infected people in the United States will be over age 50.5
The rising prevalence of HIV infection in this age group has prompted reevaluation of screening guidelines. The US Preventive Services Task Force recommends screening for HIV in all people ages 15 to 65, and also after age 65 in people at ongoing risk of infection.6 The American College of Physicians has suggested that the range for routine HIV screening be expanded to age 75.7 The cost-effectiveness of expanded and more frequent HIV testing appears to justify it.8
Take-home points
- An HIV-infected patient who is compliant with an appropriate antiretroviral regimen and has a CD4 count higher than 350 × 106/L and a suppressed viral load now has a normal life expectancy.
- Today, nearly 20% of newly diagnosed HIV-infected people and more than 50% of all HIV-infected people in the United States are over the age of 50.
- The age range for routine screening for HIV infection should be expanded.
HIGH-RISK GROUPS AMONG THE ELDERLY
Early in the HIV epidemic, older patients acquired HIV from blood transfusions received because of hemophilia and other disorders. However, this rapidly ceased after blood banks began screening blood products. Today, people over age 50 who acquire HIV have many of the same risk factors as younger people.
Men who have sex with men are the largest subgroup of HIV-infected people in the United States, even among those over age 50. In particular, white men who have sex with men now constitute the largest demographic group among the HIV-infected elderly.4
Intravenous drug users make up about 15% of older people with HIV.
Women who have sex with infected men or with men at risk of HIV infection make up the largest group of older women with HIV.4
Sex and the older person
Many older HIV-infected people remain sexually active and continue to engage in unprotected sexual intercourse far into advanced age. According to one survey, 53% of Americans ages 65 to 74 are engaging in sexual activity regularly; however, they are not using protective measures with up to 91% of casual partners and 70% of new partners.9,10 Many widowed and divorced people are dating again, and they may be unfamiliar with condom use or may be reluctant to use condoms because condoms can often make it difficult to maintain an erection.
Drugs for erectile dysfunction are making it easier for the elderly to engage in both vaginal and anal intercourse, but often without a condom.9 Older women who no longer worry about getting pregnant may be less likely to insist their partners use a condom and to practice safe sex. In addition, age-related thinning and dryness can cause vaginal tears, increasing the risk of HIV transmission.11
Take-home points
- People older than 50 have risk factors for HIV similar to those in younger people.
- Men who have sex with men compose the largest group of HIV-infected individuals in the elderly population.
- Unprotected sexual intercourse is common in the elderly for several reasons: unfamiliarity with condom use, difficulty maintaining an erection, lack of concern about possible pregnancy, and vaginal thinning and dryness in women.
UNDERDIAGNOSIS AND LATE DIAGNOSIS IN THE ELDERLY
The cumulative number of AIDS cases in adults age 50 and older increased nearly ninefold from 1990 to the end of 2009. Even more worrisome, one-half of HIV-positive adults over age 50 are diagnosed with AIDS simultaneously or within 1 year of their HIV diagnosis.4 This late diagnosis—and therefore late initiation of treatment—is associated with poorer health outcomes and more rapid disease progression.12
HIV infection in older adults often goes undiagnosed, for several reasons.
Providers may underestimate the risk in this population and therefore may not discuss HIV transmission or perform testing. Despite a US Centers for Disease Control and Prevention recommendation that people ages 13 to 64 be tested at least once, and more often if sexually active, only 35% of adults ages 45 to 64 have ever been tested for HIV infection.13
Older patients may not perceive themselves to be at risk of HIV infection because of lack of insight and information about its prevention and transmission. They are also less likely than younger adults to discuss their sexual habits or drug use with providers.14 In addition, compared with the young sexually active population, very little HIV prevention education is targeted to older people.15 Social stigmatization is also a concern for many HIV-infected elderly, as a perceived negative reputation within their community may prevent them from seeking care and disclosing their HIV status.
Take-home points
Reasons that HIV infection is underdiagnosed in the elderly include lack of:
- Provider recognition
- Insight and information about HIV prevention and transmission
- HIV-prevention education targeting the elderly
- Disclosure because of the social stigma of HIV infection.
HIV ACCELERATES AGING, AGING REDUCES IMMUNITY
Many HIV-positive people can expect to live as long as people in the general population, but those who are diagnosed late and thus are started on antiretroviral therapy later in the course of their infection have a reduced life expectancy. Longevity depends on both restoring the CD4 count to near-normal and suppressing the viral load to undetectable levels.3,16 This is especially important for older adults, as HIV may accelerate aging, and aging itself may speed the progression of HIV disease, so that therapy may result in delayed or only partial restoration of immunity.
Older age at the time of HIV infection is a strong predictor of accelerated HIV disease progression in the absence of therapy.17 Left untreated, older patients with HIV lose CD4 cells and progress to AIDS and death faster than younger patients. The deleterious effects of chronic immune activation in the course of HIV infection, combined with the immune senescence of aging, are thought to promote this accelerated course.18
Recent data indicate that starting antiretroviral therapy early can help prevent the CD4-cell impairment that occurs with aging.19 However, in adults over age 50, the capacity to restore the CD4 count with antiretroviral therapy apears to be reduced, despite demonstrated viral load suppression and better adherence.20 Although mean adherence rates appear higher in older HIV-infected patients, they are worse in those with neurocognitive impairment, highlighting the importance of evaluating neurocognition in this population.21
Decreased immune recovery and the subsequent increased risk of serious AIDS events are factors that now favor starting antiretroviral therapy in all HIV patients over age 50, regardless of CD4 count.
Take-home points
- Without treatment, HIV infection in older patients progresses more rapidly to AIDS and death than in younger patients.
- HIV-positive people over age 50 who have never received antiretroviral therapy should be strongly considered for it, regardless of the CD4 count.
SO MANY DRUGS, SO MANY INTERACTIONS
Since HIV patients are now living longer thanks to antiretroviral therapy, they are now experiencing more disease- and treatment-related problems. This has led to an increased likelihood of polypharmacy, defined here as the use of six or more medications.
In general, polypharmacy in the elderly is associated with adverse drug events, drug interactions, inappropriate medication use, delirium, falls, fractures, and poor medication adherence.22,23 But it becomes even more of a problem in HIV-infected elderly patients, as various drug interactions can alter the effectiveness of the antiretroviral regimen and can result in drug toxicity.
The most common classes of medications used in the elderly are antihypertensives, lipid-lowering agents, antiplatelet medications, antidepressants, anxiolytics, sedatives, and analgesics, and many of these have notable interactions with current antiretroviral regimens.24,25 Most medications, including antiretrovirals, are cleared by the liver or kidneys, and the function of these organs often decreases with age, resulting in impaired elimination and in drug accumulation.
Information on drug interactions is readily available from the US Department of Health and Human Services,26 drug interaction databases,27,28 and drug interaction software. The combination of antiretroviral therapy and preexisting polypharmacy significantly increases the risk of serious interactions, which can lead to drug toxicity, poorer adherence with antiretroviral therapy, loss of efficacy of the coadministered medication, or resurgence of HIV infection due to drug-drug interactions affecting the metabolism and ultimate efficacy of the antiretroviral therapy. An increased awareness of common drug-drug interactions can prevent coadministration of potentially harmful medications in elderly HIV patients.
Important interactions between antiretroviral drugs and other drug classes are summarized in Table 1.25–28 Most notably:
- Simvastatin and lovastatin are contraindicated with any protease inhibitor.
- Proton pump inhibitors are not recommended for patients taking ritonavir-boosted atazanavir. If a proton pump inhibitor is necessary, the daily dose should not exceed 20 mg of omeprazole or its equivalent in patients who have never taken a protease inhibitor, and it should be taken 12 hours before boosted atazanavir.26
- Corticosteroids, whether systemic, inhaled, or intranasal (eg, fluticasone, budesonide), should be avoided in combination with any protease inhibitor, as they can cause iatrogenic Cushing syndrome and also pose the risk of adrenal crisis during acute illness.27
Take-home points
- In cases of preexisting polypharmacy, antiretroviral therapy can lead to significant drug toxicity, poor adherence to medications, and resurgence of HIV infection.
- Increased provider awareness of common drug-drug interactions can prevent the prescribing of potentially harmful drug combinations to HIV-infected elderly patients.
COMORBIDITIES
In recent years, more than half of the deaths in HIV patients on antiretroviral therapy have been from noninfectious comorbidities such as cardiovascular disease, bone disease, and renal failure, which often coexist and are associated with advanced age.29 In fact, both older age and each additional year of antiretroviral therapy are independent predictors of polypathology (simultaneous occurrence of two or more defined diseases).30 The Antiretroviral Therapy Cohort Collaboration found that age greater than 50 was strongly associated with increasing rates of non–AIDS-related malignancy and cardiovascular disease.31
CARDIOVASCULAR DISEASE
With the increasing life expectancy of HIV-infected adults on antiretroviral therapy, cardiovascular disease has become an important concern. HIV-infected adults appear to have a significantly greater risk of myocardial infarction and coronary artery disease than age-matched HIV-negative individuals.32 Strikingly, being older than 50 itself increases the risk of hospitalization for cardiovascular disease fivefold (incidence rate ratio 5.01, 95% confidence interval 3.41–7.38).33 In addition, HIV infection is associated with a risk of acute myocardial infarction 50% higher than that explained by recognized risk factors.34
This high prevalence of coronary artery disease is likely from a combination of factors, including increasing age and the chronic inflammation and immune activation associated with HIV infection.35 An association between untreated HIV disease and markers of risk for cardiovascular disease has been identified.36,37
In addition, antiretroviral therapy is associated with dyslipidemia, which is most pronounced with protease inhibitor regimens. Whether specific lipid changes associated with individual antiretroviral drugs affect cardiovascular risk remains uncertain. In the Data Collection on Adverse Events of Anti-HIV Drugs studies,38 only cumulative exposure to indinavir, lopinavir-ritonavir, and didanosine was associated with an increased risk of myocardial infarction.38
Traditional risk factors such as obesity, tobacco use, and genetic predisposition also apply to HIV-infected people.39 In fact, the prevalence of traditional risk factors such as smoking and dyslipidemia is generally higher in HIV-infected people than in the general population, although this situation may be improving.40
Science needs to elucidate the relationship between traditional and nontraditional risk factors for cardiovascular disease in older HIV-infected adults. In the meantime, older patients with HIV require aggressive management of modifiable risk factors.
Tools for assessing cardiovascular risk include the Framingham risk score41 and the Data Collection on Adverse Events of Anti-HIV Drugs 5-year risk calculator.42 The European AIDS Clinical Society guidelines recommend considering changing the antiretroviral regimen if the patient’s 10-year risk of cardiovascular disease is more than 20%.43 Recommended strategies for reducing cardiovascular risk in elderly patients with HIV infection include counseling about smoking cessation and weight loss at every clinic visit and optimally controlling dyslipidemia and hypertension using nationally accepted standardized guidelines.44
Take-home points
- HIV infection is associated with a 50% higher risk of acute myocardial infarction beyond that explained by traditional risk factors.
- Chronic inflammation, immune activation, and dyslipidemia associated with antiretroviral therapy all contribute to cardiovascular disease in HIV-infected patients.
- HIV-infected elderly patients require aggressive management of modifiable risk factors for cardiovascular disease.
ENDOCRINE DISEASE
Diabetes mellitus
The estimated prevalence of diabetes mellitus is 3% in HIV-infected people who have never received antiretroviral therapy, but glucose intolerance increases to the range of 10% to 25% in those who have started it.45 Glucose disorders are associated with traditional risk factors as well as with HIV-associated factors such as lipodystrophy and antiretroviral therapy, specifically long-term use of protease inhibitors.46 Although increasing age and obesity clearly play a role in the development of diabetes mellitus in this population, HIV-specific factors may also allow diabetes to develop at a lower level of adiposity than in people without HIV infection.47
Strategies for preventing type 2 diabetes mellitus in HIV-infected patients focus on avoiding excessive weight gain, especially after starting antiretroviral therapy; regularly screening for diabetes using hemoglobin A1c, both before and after starting antiretroviral therapy; and continuing to check hemoglobin A1c every 6 months. The target hemoglobin A1c should be less than 7.0%. This threshold should be increased to 8% in frail elderly adults if their anticipated life expectancy is less than 5 years, given their higher risk of hypoglycemia, polypharmacy, and drug interactions.48 In addition, as in HIV-negative patients, diabetes screening should be performed if systolic blood pressure exceeds 135/80 mm Hg.
Insulin sensitizers such as metformin and thiazolidinediones should be considered for treating diabetes in HIV-infected patients if no contraindications exist. Consideration may also be given to switching the antiretroviral regimen from a protease inhibitor-based regimen to a nonnucleoside reverse transcriptase inhibitor-based regimen.48
Take-home points
- Glucose intolerance has been associated with HIV-specific factors, including lipodystrophy and antiretroviral therapy.
- Avoiding excessive weight gain, use of insulin-sensitizing medications, and alteration in antiretroviral regimens should be considered for the treatment of diabetes mellitus in HIV infection.
Osteoporosis
Osteoporotic bone disease disproportionately affects patients with advanced HIV infection compared with patients of similar age.49 Bone mineral density is lower and the fracture rate is higher in HIV-infected individuals.
The pathogenesis of bone disease appears to be multifactorial. Traditional risk factors include hypogonadism, smoking, alcohol use, and low body weight, while HIV-related risk factors include chronic immune activation and antiretroviral therapy.50
Several antiretroviral regimens have been linked to clinically significant bone loss, including both tenofovir-based and protease inhibitor-based regimens.51 Most studies have shown that bone mineral density decreases by 2% to 6% in the first 2 years after starting these regimens52; however, long-term effects on bone loss are unknown.
Questions remain. For example, what are the exact mechanisms that lead to the acute decrease in bone mineral density after starting antiretroviral therapy? And why is vitamin D deficiency is so prevalent in HIV infection, with low vitamin D levels seen in up to 60% to 75% of elderly HIV-infected patients?53
Both the Work Group for the HIV and Aging Consensus Project54 and the European AIDS Clinical Society43 recommend screening for and treating causes of secondary low bone mineral density in HIV-infected men over age 50 and postmenopausal HIV-infected women. These causes include vitamin D deficiency. As of 2013, the National Osteoporosis Foundation guidelines include HIV infection and antiretroviral therapy as osteoporosis risk factors that should trigger screening for low bone mineral density with dual-energy x-ray absorptiometry (DXA).55
As in the general population, the preferred treatment for low bone mineral density in people with HIV is a bisphosphonate, in addition to ensuring adequate calcium and vitamin D intake. It is important to repeat DXA imaging every 2 years and to reassess the need for continued bisphosphonate therapy after 3 to 5 years because of a possible increased risk of fracture with prolonged use.
Take-home points
- Osteoporosis and vitamin D deficiency both appear to be more prevalent with HIV infection.
- HIV infection and antiretroviral therapy are risk factors that should prompt DXA screening to evaluate for osteoporosis.
NEUROCOGNITIVE DISORDERS
HIV-associated neurocognitive disorders are common, with an estimated 50% of HIV-infected patients experiencing some degree of cognitive loss and some progressing to dementia.56 Unfortunately, studies suggest that cognitive disorders can occur despite good HIV control with antiretroviral therapy, with one report demonstrating that 84% of patients with cognitive complaints and 64% without complaints were affected by an HIV-associated neurocognitive disorder.57
HIV-associated dementia is often subcortical, with fluctuating symptoms such as psychomotor retardation, difficulty multitasking, and apathy. In contrast to dementia syndromes such as Alzheimer disease, relentless progression is less common in HIV-infected patients who receive antiretroviral therapy.
The Mini-Mental State Examination should not be used to screen for HIV-associated neurocognitive disorders, as it does not assess the domains that are typically impaired. The Montreal Cognitive Assessment has been suggested as the best screening instrument in elderly HIV-infected patients; it is available at no cost at www.mocatest.org.58
As HIV-associated neurocognitive disorder is a diagnosis of exclusion, an evaluation for alternative diagnoses such as syphilis, hypothyroidism, and depression is recommended. If an HIV-associated neurocognitive disorder is diagnosed, referral to specialty care should be considered, as interventions such as lumbar puncture to assess cerebrospinal fluid viral escape and changing the antiretroviral regimen to improve central nervous system penetration are possible options under study.
Patients with poorly controlled HIV and a depressed CD4 count are at risk of a number of central nervous system complications in addition to HIV-associated neurocognitive disorders, eg, central nervous system toxoplasmosis, cryptococcal meningitis, progressive multifocal leukoencephalopathy, and primary central nervous system lymphoma. Adherence to an effective antiretroviral regimen is the primary prevention strategy.
Take-home points
- HIV-associated neurocognitive disorders and dementia can occur despite appropriate HIV control and adherence to antiretroviral therapy.
- Adherence to antiretroviral therapy is the primary prevention against most central nervous system complications in HIV infection.
GERIATRIC SYNDROMES
The aging HIV-infected adult may also be at increased risk of geriatric syndromes.
In particular, a frailty-related phenotype of weight loss, exhaustion, slowness, and low physical activity was more common in HIV-infected elderly than in noninfected elderly.59 HIV-infected men are 4.5 to 10 times more likely than age-matched controls to be frail, and the likelihood of frailty increases with age, duration of HIV infection, having a CD4 count lower than 350 × 106/L, and having uncontrolled HIV replication.60,61
Other geriatric syndromes such as falls, urinary incontinence, and functional impairment have been identified in 25% to 56% of older HIV-infected patients.62 Indeed, the combination of HIV and older age may adversely affect performance of instrumental activities of daily living.63 Also, as previously mentioned, nondisclosure, fear of HIV-related social stigmatization, and a desire to be self-reliant are all factors that perpetuate the social isolation that is common among the HIV-infected elderly.
For these reasons, a comprehensive approach involving a geriatrician, an infectious disease specialist, and community social workers is needed to manage the care of this aging population.
Take-home point
- Geriatric syndromes have an important impact on health in aging HIV patients.
CANCER SCREENING IN HIV PATIENTS
People with HIV have an elevated risk of cancer. Specifically, compared with the general population, their risk is:
- 3,640 times higher for Kaposi sarcoma
- 77 times higher for non-Hodgkin lymphomas
- 6 times higher for cervical cancer.64,65
These cancers are considered “AIDS-defining,” and fortunately, the development of effective antiretroviral therapy in the 1990s has led to a marked reduction in their incidence. However, the aging HIV population is now experiencing a rise in the incidence of non–AIDS-defining cancers, such as cancers of the lung, liver, kidney, anus, head and neck, and skin, as well as Hodgkin lymphoma.66 Table 2 shows the standardized incidence ratio of selected non–AIDS-defining cancers in HIV-infected patients as reported in several large international studies.65,67,68 The etiology for the increased risk of non–AIDS-defining cancers in the HIV-infected population is not clear, but possible explanations include the virus itself, antiretroviral therapy, and co-infection with other viruses such as hepatitis B, hepatitis C, and Epstein-Barr virus.
Guidelines for cancer screening vary by organization, and the American Cancer Society, the National Cancer Institute, and the US Preventive Services Task Force do not have formal screening guidelines for the most common non–AIDS-defining cancers. The European AIDS Clinical Society, however, has proposed some screening recommendations for selected malignancies.43
In general, screening recommendations are similar to those for HIV-negative patients. A specific difference for HIV-infected patients is in cervical cancer screening. HIV-infected women should undergo a Papanicolaou smear at 6-month intervals during the first year after diagnosis of HIV infection and, if the results are normal, annually thereafter. There is no consensus as to whether human papillomavirus testing should be performed routinely on HIV-infected women.
At the time of this writing, there are no recommendations for routine screening for anal cancer, although some specialists recommend anal cytologic screening for HIV-positive men and women, and an annual digital anal examination may be useful to detect masses that could be anal cancer.69
Take-home points
- The incidence of non–AIDS-defining cancers is rising in the aging HIV population.
- There are currently no formal recommendations for routine screening for anal cancer.
FINAL WORD
Because patients with HIV are living longer as a result of newer effective combination antiretroviral therapies, physicians face a new challenge of managing conditions in these patients that are traditionally associated with aging. Providers will need to improve their understanding of drug-drug interactions and polypharmacy issues and be able to address the complex medical and psychosocial issues in this growing population. As patients with HIV on effective antiretroviral therapy grow older, the burden of comorbid medical disease will continue to increase.
In the 1980s, human immunodeficiency virus (HIV) infection was considered untreatable and predictably lethal. Today, with highly effective antiretroviral therapy, it has become a chronic condition in which patients have a life expectancy comparable to that in the general population.
This change has led to new challenges for primary care physicians, many of whom now find themselves either the sole medical provider for or the comanager of aging HIV-infected patients. Given that about one-fifth of new HIV diagnoses are now in people over the age of 50, it is crucial that primary care providers be able to recognize and diagnose the disease in this population. In addition, they need to effectively manage the polypharmacy and subsequent drug interactions prevalent in older HIV-infected patients. Finally, the clinician must address comorbid diseases common in the elderly, specifically neurologic, cardiovascular, metabolic, and endocrine disorders, as well as performing routine cancer screening.
Take-home point
- As the number of people age 50 and older with HIV infection increases, primary care providers must be able to both recognize and manage the condition.
RISING PREVALENCE OF HIV IN THE ELDERLY
Globally, about 2.5 million people received a new diagnosis of HIV infection in 2011, and about 35 million people worldwide are currently living with it.1 An estimated 1.1 million Americans are living with HIV, and of these, about 16% do not know they are infected.2
Antiretroviral therapy has greatly improved the life expectancy of HIV-infected patients, and the number of HIV-infected people over age 50 continues to rise. A successfully treated HIV-positive person with a CD4 count higher than 350 × 106/L and a suppressed viral load now has a normal life expectancy.3 In 2011, nearly 20% of newly diagnosed HIV-infected people in the United States were over age 50, as were nearly 25% of those with a new diagnosis of acquired immune deficiency syndrome (AIDS).4 This year (2015), we expect that more than half of all HIV-infected people in the United States will be over age 50.5
The rising prevalence of HIV infection in this age group has prompted reevaluation of screening guidelines. The US Preventive Services Task Force recommends screening for HIV in all people ages 15 to 65, and also after age 65 in people at ongoing risk of infection.6 The American College of Physicians has suggested that the range for routine HIV screening be expanded to age 75.7 The cost-effectiveness of expanded and more frequent HIV testing appears to justify it.8
Take-home points
- An HIV-infected patient who is compliant with an appropriate antiretroviral regimen and has a CD4 count higher than 350 × 106/L and a suppressed viral load now has a normal life expectancy.
- Today, nearly 20% of newly diagnosed HIV-infected people and more than 50% of all HIV-infected people in the United States are over the age of 50.
- The age range for routine screening for HIV infection should be expanded.
HIGH-RISK GROUPS AMONG THE ELDERLY
Early in the HIV epidemic, older patients acquired HIV from blood transfusions received because of hemophilia and other disorders. However, this rapidly ceased after blood banks began screening blood products. Today, people over age 50 who acquire HIV have many of the same risk factors as younger people.
Men who have sex with men are the largest subgroup of HIV-infected people in the United States, even among those over age 50. In particular, white men who have sex with men now constitute the largest demographic group among the HIV-infected elderly.4
Intravenous drug users make up about 15% of older people with HIV.
Women who have sex with infected men or with men at risk of HIV infection make up the largest group of older women with HIV.4
Sex and the older person
Many older HIV-infected people remain sexually active and continue to engage in unprotected sexual intercourse far into advanced age. According to one survey, 53% of Americans ages 65 to 74 are engaging in sexual activity regularly; however, they are not using protective measures with up to 91% of casual partners and 70% of new partners.9,10 Many widowed and divorced people are dating again, and they may be unfamiliar with condom use or may be reluctant to use condoms because condoms can often make it difficult to maintain an erection.
Drugs for erectile dysfunction are making it easier for the elderly to engage in both vaginal and anal intercourse, but often without a condom.9 Older women who no longer worry about getting pregnant may be less likely to insist their partners use a condom and to practice safe sex. In addition, age-related thinning and dryness can cause vaginal tears, increasing the risk of HIV transmission.11
Take-home points
- People older than 50 have risk factors for HIV similar to those in younger people.
- Men who have sex with men compose the largest group of HIV-infected individuals in the elderly population.
- Unprotected sexual intercourse is common in the elderly for several reasons: unfamiliarity with condom use, difficulty maintaining an erection, lack of concern about possible pregnancy, and vaginal thinning and dryness in women.
UNDERDIAGNOSIS AND LATE DIAGNOSIS IN THE ELDERLY
The cumulative number of AIDS cases in adults age 50 and older increased nearly ninefold from 1990 to the end of 2009. Even more worrisome, one-half of HIV-positive adults over age 50 are diagnosed with AIDS simultaneously or within 1 year of their HIV diagnosis.4 This late diagnosis—and therefore late initiation of treatment—is associated with poorer health outcomes and more rapid disease progression.12
HIV infection in older adults often goes undiagnosed, for several reasons.
Providers may underestimate the risk in this population and therefore may not discuss HIV transmission or perform testing. Despite a US Centers for Disease Control and Prevention recommendation that people ages 13 to 64 be tested at least once, and more often if sexually active, only 35% of adults ages 45 to 64 have ever been tested for HIV infection.13
Older patients may not perceive themselves to be at risk of HIV infection because of lack of insight and information about its prevention and transmission. They are also less likely than younger adults to discuss their sexual habits or drug use with providers.14 In addition, compared with the young sexually active population, very little HIV prevention education is targeted to older people.15 Social stigmatization is also a concern for many HIV-infected elderly, as a perceived negative reputation within their community may prevent them from seeking care and disclosing their HIV status.
Take-home points
Reasons that HIV infection is underdiagnosed in the elderly include lack of:
- Provider recognition
- Insight and information about HIV prevention and transmission
- HIV-prevention education targeting the elderly
- Disclosure because of the social stigma of HIV infection.
HIV ACCELERATES AGING, AGING REDUCES IMMUNITY
Many HIV-positive people can expect to live as long as people in the general population, but those who are diagnosed late and thus are started on antiretroviral therapy later in the course of their infection have a reduced life expectancy. Longevity depends on both restoring the CD4 count to near-normal and suppressing the viral load to undetectable levels.3,16 This is especially important for older adults, as HIV may accelerate aging, and aging itself may speed the progression of HIV disease, so that therapy may result in delayed or only partial restoration of immunity.
Older age at the time of HIV infection is a strong predictor of accelerated HIV disease progression in the absence of therapy.17 Left untreated, older patients with HIV lose CD4 cells and progress to AIDS and death faster than younger patients. The deleterious effects of chronic immune activation in the course of HIV infection, combined with the immune senescence of aging, are thought to promote this accelerated course.18
Recent data indicate that starting antiretroviral therapy early can help prevent the CD4-cell impairment that occurs with aging.19 However, in adults over age 50, the capacity to restore the CD4 count with antiretroviral therapy apears to be reduced, despite demonstrated viral load suppression and better adherence.20 Although mean adherence rates appear higher in older HIV-infected patients, they are worse in those with neurocognitive impairment, highlighting the importance of evaluating neurocognition in this population.21
Decreased immune recovery and the subsequent increased risk of serious AIDS events are factors that now favor starting antiretroviral therapy in all HIV patients over age 50, regardless of CD4 count.
Take-home points
- Without treatment, HIV infection in older patients progresses more rapidly to AIDS and death than in younger patients.
- HIV-positive people over age 50 who have never received antiretroviral therapy should be strongly considered for it, regardless of the CD4 count.
SO MANY DRUGS, SO MANY INTERACTIONS
Since HIV patients are now living longer thanks to antiretroviral therapy, they are now experiencing more disease- and treatment-related problems. This has led to an increased likelihood of polypharmacy, defined here as the use of six or more medications.
In general, polypharmacy in the elderly is associated with adverse drug events, drug interactions, inappropriate medication use, delirium, falls, fractures, and poor medication adherence.22,23 But it becomes even more of a problem in HIV-infected elderly patients, as various drug interactions can alter the effectiveness of the antiretroviral regimen and can result in drug toxicity.
The most common classes of medications used in the elderly are antihypertensives, lipid-lowering agents, antiplatelet medications, antidepressants, anxiolytics, sedatives, and analgesics, and many of these have notable interactions with current antiretroviral regimens.24,25 Most medications, including antiretrovirals, are cleared by the liver or kidneys, and the function of these organs often decreases with age, resulting in impaired elimination and in drug accumulation.
Information on drug interactions is readily available from the US Department of Health and Human Services,26 drug interaction databases,27,28 and drug interaction software. The combination of antiretroviral therapy and preexisting polypharmacy significantly increases the risk of serious interactions, which can lead to drug toxicity, poorer adherence with antiretroviral therapy, loss of efficacy of the coadministered medication, or resurgence of HIV infection due to drug-drug interactions affecting the metabolism and ultimate efficacy of the antiretroviral therapy. An increased awareness of common drug-drug interactions can prevent coadministration of potentially harmful medications in elderly HIV patients.
Important interactions between antiretroviral drugs and other drug classes are summarized in Table 1.25–28 Most notably:
- Simvastatin and lovastatin are contraindicated with any protease inhibitor.
- Proton pump inhibitors are not recommended for patients taking ritonavir-boosted atazanavir. If a proton pump inhibitor is necessary, the daily dose should not exceed 20 mg of omeprazole or its equivalent in patients who have never taken a protease inhibitor, and it should be taken 12 hours before boosted atazanavir.26
- Corticosteroids, whether systemic, inhaled, or intranasal (eg, fluticasone, budesonide), should be avoided in combination with any protease inhibitor, as they can cause iatrogenic Cushing syndrome and also pose the risk of adrenal crisis during acute illness.27
Take-home points
- In cases of preexisting polypharmacy, antiretroviral therapy can lead to significant drug toxicity, poor adherence to medications, and resurgence of HIV infection.
- Increased provider awareness of common drug-drug interactions can prevent the prescribing of potentially harmful drug combinations to HIV-infected elderly patients.
COMORBIDITIES
In recent years, more than half of the deaths in HIV patients on antiretroviral therapy have been from noninfectious comorbidities such as cardiovascular disease, bone disease, and renal failure, which often coexist and are associated with advanced age.29 In fact, both older age and each additional year of antiretroviral therapy are independent predictors of polypathology (simultaneous occurrence of two or more defined diseases).30 The Antiretroviral Therapy Cohort Collaboration found that age greater than 50 was strongly associated with increasing rates of non–AIDS-related malignancy and cardiovascular disease.31
CARDIOVASCULAR DISEASE
With the increasing life expectancy of HIV-infected adults on antiretroviral therapy, cardiovascular disease has become an important concern. HIV-infected adults appear to have a significantly greater risk of myocardial infarction and coronary artery disease than age-matched HIV-negative individuals.32 Strikingly, being older than 50 itself increases the risk of hospitalization for cardiovascular disease fivefold (incidence rate ratio 5.01, 95% confidence interval 3.41–7.38).33 In addition, HIV infection is associated with a risk of acute myocardial infarction 50% higher than that explained by recognized risk factors.34
This high prevalence of coronary artery disease is likely from a combination of factors, including increasing age and the chronic inflammation and immune activation associated with HIV infection.35 An association between untreated HIV disease and markers of risk for cardiovascular disease has been identified.36,37
In addition, antiretroviral therapy is associated with dyslipidemia, which is most pronounced with protease inhibitor regimens. Whether specific lipid changes associated with individual antiretroviral drugs affect cardiovascular risk remains uncertain. In the Data Collection on Adverse Events of Anti-HIV Drugs studies,38 only cumulative exposure to indinavir, lopinavir-ritonavir, and didanosine was associated with an increased risk of myocardial infarction.38
Traditional risk factors such as obesity, tobacco use, and genetic predisposition also apply to HIV-infected people.39 In fact, the prevalence of traditional risk factors such as smoking and dyslipidemia is generally higher in HIV-infected people than in the general population, although this situation may be improving.40
Science needs to elucidate the relationship between traditional and nontraditional risk factors for cardiovascular disease in older HIV-infected adults. In the meantime, older patients with HIV require aggressive management of modifiable risk factors.
Tools for assessing cardiovascular risk include the Framingham risk score41 and the Data Collection on Adverse Events of Anti-HIV Drugs 5-year risk calculator.42 The European AIDS Clinical Society guidelines recommend considering changing the antiretroviral regimen if the patient’s 10-year risk of cardiovascular disease is more than 20%.43 Recommended strategies for reducing cardiovascular risk in elderly patients with HIV infection include counseling about smoking cessation and weight loss at every clinic visit and optimally controlling dyslipidemia and hypertension using nationally accepted standardized guidelines.44
Take-home points
- HIV infection is associated with a 50% higher risk of acute myocardial infarction beyond that explained by traditional risk factors.
- Chronic inflammation, immune activation, and dyslipidemia associated with antiretroviral therapy all contribute to cardiovascular disease in HIV-infected patients.
- HIV-infected elderly patients require aggressive management of modifiable risk factors for cardiovascular disease.
ENDOCRINE DISEASE
Diabetes mellitus
The estimated prevalence of diabetes mellitus is 3% in HIV-infected people who have never received antiretroviral therapy, but glucose intolerance increases to the range of 10% to 25% in those who have started it.45 Glucose disorders are associated with traditional risk factors as well as with HIV-associated factors such as lipodystrophy and antiretroviral therapy, specifically long-term use of protease inhibitors.46 Although increasing age and obesity clearly play a role in the development of diabetes mellitus in this population, HIV-specific factors may also allow diabetes to develop at a lower level of adiposity than in people without HIV infection.47
Strategies for preventing type 2 diabetes mellitus in HIV-infected patients focus on avoiding excessive weight gain, especially after starting antiretroviral therapy; regularly screening for diabetes using hemoglobin A1c, both before and after starting antiretroviral therapy; and continuing to check hemoglobin A1c every 6 months. The target hemoglobin A1c should be less than 7.0%. This threshold should be increased to 8% in frail elderly adults if their anticipated life expectancy is less than 5 years, given their higher risk of hypoglycemia, polypharmacy, and drug interactions.48 In addition, as in HIV-negative patients, diabetes screening should be performed if systolic blood pressure exceeds 135/80 mm Hg.
Insulin sensitizers such as metformin and thiazolidinediones should be considered for treating diabetes in HIV-infected patients if no contraindications exist. Consideration may also be given to switching the antiretroviral regimen from a protease inhibitor-based regimen to a nonnucleoside reverse transcriptase inhibitor-based regimen.48
Take-home points
- Glucose intolerance has been associated with HIV-specific factors, including lipodystrophy and antiretroviral therapy.
- Avoiding excessive weight gain, use of insulin-sensitizing medications, and alteration in antiretroviral regimens should be considered for the treatment of diabetes mellitus in HIV infection.
Osteoporosis
Osteoporotic bone disease disproportionately affects patients with advanced HIV infection compared with patients of similar age.49 Bone mineral density is lower and the fracture rate is higher in HIV-infected individuals.
The pathogenesis of bone disease appears to be multifactorial. Traditional risk factors include hypogonadism, smoking, alcohol use, and low body weight, while HIV-related risk factors include chronic immune activation and antiretroviral therapy.50
Several antiretroviral regimens have been linked to clinically significant bone loss, including both tenofovir-based and protease inhibitor-based regimens.51 Most studies have shown that bone mineral density decreases by 2% to 6% in the first 2 years after starting these regimens52; however, long-term effects on bone loss are unknown.
Questions remain. For example, what are the exact mechanisms that lead to the acute decrease in bone mineral density after starting antiretroviral therapy? And why is vitamin D deficiency is so prevalent in HIV infection, with low vitamin D levels seen in up to 60% to 75% of elderly HIV-infected patients?53
Both the Work Group for the HIV and Aging Consensus Project54 and the European AIDS Clinical Society43 recommend screening for and treating causes of secondary low bone mineral density in HIV-infected men over age 50 and postmenopausal HIV-infected women. These causes include vitamin D deficiency. As of 2013, the National Osteoporosis Foundation guidelines include HIV infection and antiretroviral therapy as osteoporosis risk factors that should trigger screening for low bone mineral density with dual-energy x-ray absorptiometry (DXA).55
As in the general population, the preferred treatment for low bone mineral density in people with HIV is a bisphosphonate, in addition to ensuring adequate calcium and vitamin D intake. It is important to repeat DXA imaging every 2 years and to reassess the need for continued bisphosphonate therapy after 3 to 5 years because of a possible increased risk of fracture with prolonged use.
Take-home points
- Osteoporosis and vitamin D deficiency both appear to be more prevalent with HIV infection.
- HIV infection and antiretroviral therapy are risk factors that should prompt DXA screening to evaluate for osteoporosis.
NEUROCOGNITIVE DISORDERS
HIV-associated neurocognitive disorders are common, with an estimated 50% of HIV-infected patients experiencing some degree of cognitive loss and some progressing to dementia.56 Unfortunately, studies suggest that cognitive disorders can occur despite good HIV control with antiretroviral therapy, with one report demonstrating that 84% of patients with cognitive complaints and 64% without complaints were affected by an HIV-associated neurocognitive disorder.57
HIV-associated dementia is often subcortical, with fluctuating symptoms such as psychomotor retardation, difficulty multitasking, and apathy. In contrast to dementia syndromes such as Alzheimer disease, relentless progression is less common in HIV-infected patients who receive antiretroviral therapy.
The Mini-Mental State Examination should not be used to screen for HIV-associated neurocognitive disorders, as it does not assess the domains that are typically impaired. The Montreal Cognitive Assessment has been suggested as the best screening instrument in elderly HIV-infected patients; it is available at no cost at www.mocatest.org.58
As HIV-associated neurocognitive disorder is a diagnosis of exclusion, an evaluation for alternative diagnoses such as syphilis, hypothyroidism, and depression is recommended. If an HIV-associated neurocognitive disorder is diagnosed, referral to specialty care should be considered, as interventions such as lumbar puncture to assess cerebrospinal fluid viral escape and changing the antiretroviral regimen to improve central nervous system penetration are possible options under study.
Patients with poorly controlled HIV and a depressed CD4 count are at risk of a number of central nervous system complications in addition to HIV-associated neurocognitive disorders, eg, central nervous system toxoplasmosis, cryptococcal meningitis, progressive multifocal leukoencephalopathy, and primary central nervous system lymphoma. Adherence to an effective antiretroviral regimen is the primary prevention strategy.
Take-home points
- HIV-associated neurocognitive disorders and dementia can occur despite appropriate HIV control and adherence to antiretroviral therapy.
- Adherence to antiretroviral therapy is the primary prevention against most central nervous system complications in HIV infection.
GERIATRIC SYNDROMES
The aging HIV-infected adult may also be at increased risk of geriatric syndromes.
In particular, a frailty-related phenotype of weight loss, exhaustion, slowness, and low physical activity was more common in HIV-infected elderly than in noninfected elderly.59 HIV-infected men are 4.5 to 10 times more likely than age-matched controls to be frail, and the likelihood of frailty increases with age, duration of HIV infection, having a CD4 count lower than 350 × 106/L, and having uncontrolled HIV replication.60,61
Other geriatric syndromes such as falls, urinary incontinence, and functional impairment have been identified in 25% to 56% of older HIV-infected patients.62 Indeed, the combination of HIV and older age may adversely affect performance of instrumental activities of daily living.63 Also, as previously mentioned, nondisclosure, fear of HIV-related social stigmatization, and a desire to be self-reliant are all factors that perpetuate the social isolation that is common among the HIV-infected elderly.
For these reasons, a comprehensive approach involving a geriatrician, an infectious disease specialist, and community social workers is needed to manage the care of this aging population.
Take-home point
- Geriatric syndromes have an important impact on health in aging HIV patients.
CANCER SCREENING IN HIV PATIENTS
People with HIV have an elevated risk of cancer. Specifically, compared with the general population, their risk is:
- 3,640 times higher for Kaposi sarcoma
- 77 times higher for non-Hodgkin lymphomas
- 6 times higher for cervical cancer.64,65
These cancers are considered “AIDS-defining,” and fortunately, the development of effective antiretroviral therapy in the 1990s has led to a marked reduction in their incidence. However, the aging HIV population is now experiencing a rise in the incidence of non–AIDS-defining cancers, such as cancers of the lung, liver, kidney, anus, head and neck, and skin, as well as Hodgkin lymphoma.66 Table 2 shows the standardized incidence ratio of selected non–AIDS-defining cancers in HIV-infected patients as reported in several large international studies.65,67,68 The etiology for the increased risk of non–AIDS-defining cancers in the HIV-infected population is not clear, but possible explanations include the virus itself, antiretroviral therapy, and co-infection with other viruses such as hepatitis B, hepatitis C, and Epstein-Barr virus.
Guidelines for cancer screening vary by organization, and the American Cancer Society, the National Cancer Institute, and the US Preventive Services Task Force do not have formal screening guidelines for the most common non–AIDS-defining cancers. The European AIDS Clinical Society, however, has proposed some screening recommendations for selected malignancies.43
In general, screening recommendations are similar to those for HIV-negative patients. A specific difference for HIV-infected patients is in cervical cancer screening. HIV-infected women should undergo a Papanicolaou smear at 6-month intervals during the first year after diagnosis of HIV infection and, if the results are normal, annually thereafter. There is no consensus as to whether human papillomavirus testing should be performed routinely on HIV-infected women.
At the time of this writing, there are no recommendations for routine screening for anal cancer, although some specialists recommend anal cytologic screening for HIV-positive men and women, and an annual digital anal examination may be useful to detect masses that could be anal cancer.69
Take-home points
- The incidence of non–AIDS-defining cancers is rising in the aging HIV population.
- There are currently no formal recommendations for routine screening for anal cancer.
FINAL WORD
Because patients with HIV are living longer as a result of newer effective combination antiretroviral therapies, physicians face a new challenge of managing conditions in these patients that are traditionally associated with aging. Providers will need to improve their understanding of drug-drug interactions and polypharmacy issues and be able to address the complex medical and psychosocial issues in this growing population. As patients with HIV on effective antiretroviral therapy grow older, the burden of comorbid medical disease will continue to increase.
- World Health Organization. HIV/AIDS: fact sheet. www.who.int/mediacentre/factsheets/fs360/en/. Accessed April 16, 2015.
- Centers for Disease Control and Prevention (CDC). HIV/AIDS: basic statistics. www.cdc.gov/hiv/basics/statistics.html. Accessed April 16, 2015.
- May MT, Gompels M, Delpech V, et al; UK Collaborative HIV Cohort (UK CHIC) Study. Impact on life expectancy of HIV-1 positive individuals of CD4+ cell count and viral load response to antiretroviral therapy. AIDS 2014; 28:1193–1202.
- Centers for Disease Control and Prevention (CDC). Diagnoses of HIV infection in the United States and dependent areas: HIV surveillance report. www.cdc.gov/hiv/library/reports/surveillance/2011/surveillance_Report_vol_23.html. Accessed April 16, 2015.
- Effros RB, Fletcher CV, Gebo K, et al. Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis 2008; 47:542–553.
- Moyer VA; US Preventive Services Task Force. Screening for HIV: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:51–60.
- Qaseem A, Snow V, Shekelle P, Hopkins R Jr, Owens DK; Clinical Efficacy Assessment Subcommittee, American College of Physicians. Screening for HIV in health care settings: a guidance statement from the American College of Physicians and HIV Medicine Association. Ann Intern Med 2009; 150:125–131.
- Lucas A, Armbruster B. The cost-effectiveness of expanded HIV screening in the United States. AIDS 2013; 27:795–801.
- Lindau ST, Schumm LP, Laumann EO, Levinson W, O’Muircheartaigh CA, Waite LJ. A study of sexuality and health among older adults in the United States. N Engl J Med 2007; 357:762–774.
- Schick V, Herbenick D, Reece M, et al. Sexual behaviors, condom use, and sexual health of Americans over 50: implications for sexual health promotion for older adults. J Sex Med 2010; 7(suppl 5):315–329.
- US Department of Health and Human Services, HIV/AIDS Bureau. The Ryan White HIV/AIDS program: population fact sheet: August 2010. Older adults. http://hab.hrsa.gov/abouthab/populations/olderadultsfacts.pdf. Accessed April 16, 2015.
- May M, Gompels M, Delpech V, et al. Impact of late diagnosis and treatment on life expectancy in people with HIV-1: UK Collaborative HIV Cohort (UK CHIC) Study. BMJ 2011; 343:d6016.
- Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention (CDC). Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep 2006; 55:1–17.
- Health Resources and Services Administration (HRSA); HIV/AIDS Bureau. HRSA CAREAction. The graying of HIV. http://hab.hrsa.gov/newspublications/careactionnewsletter/february2009.pdf. Accessed April 16, 2015.
- AIDS InfoNet. Fact sheet number 616: Older people and HIV. http://aidsinfonet.org/fact_sheets/view/616. Accessed April 16, 2015.
- Rodger AJ, Lodwick R, Schechter M, et al; INSIGHT SMART, ESPRIT Study Groups. Mortality in well controlled HIV in the continuous antiretroviral therapy arms of the SMART and ESPRIT trials compared with the general population. AIDS 2013; 27:973–979.
- Kitahata MM, Gange SJ, Abraham AG, et al; NA-ACCORD Investigators. Effect of early versus deferred antiretroviral therapy for HIV on survival. N Engl J Med 2009; 360:1815–1826.
- Cao W, Jamieson BD, Hultin LE, Hultin PM, Effros RB, Detels R. Premature aging of T cells is associated with faster HIV-1 disease progression. J Acquir Immune Defic Syndr 2009; 50:137–147.
- Allers K, Bösel D, Epple HJ, et al. Effect of age on the CD4+ T-cell impairment in HIV-infected persons without and with cART. J Acquir Immune Defic Syndr 2014; 66:7–15.
- Kalayjian RC, Spritzler J, Matining RM, et al. Older HIV-infected patients on antiretroviral therapy have B-cell expansion and attenuated CD4 cell increases with immune activation reduction. AIDS 2013; 27:1563–1571.
- Althoff KN, Gebo KA, Gange SJ, et al; North American AIDS Cohort Collaboration on Research and Design. CD4 count at presentation for HIV care in the United States and Canada: are those over 50 years more likely to have a delayed presentation? AIDS Res Ther 2010; 7:45.
- Freeland KN, Thompson AN, Zhao Y, Leal JE, Mauldin PD, Moran WP. Medication use and associated risk of falling in a geriatric outpatient population. Ann Pharmacother 2012; 46:1188–1192.
- Steinman MA, Hanlon JT. Managing medications in clinically complex elders: “There’s got to be a happy medium.” JAMA 2010; 304:1592–1601.
- Marzolini C, Elzi L, Gibbons S, et al; Swiss HIV Cohort Study. Prevalence of comedications and effect of potential drug-drug interactions in the Swiss HIV Cohort Study. Antivir Ther 2010; 15:413–423.
- Greene M, Justice AC, Lampiris HW, Valcour V. Management of human immunodeficiency virus infection in advanced age. JAMA 2013; 309:1397–1405.
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed April 16, 2015.
- UCSF Center for HIV Information. HIVInSite. Comprehensive, up-to-date information on HIV/AIDS treatment, prevention, and policy from the University of California San Francisco: database of antiretroviral drug interactions. http://hivinsite.ucsf.edu/. Accessed April 16, 2015.
- The University of Liverpool. Drug interaction charts. www.hiv-druginteractions.org. Accessed April 16, 2015.
- Vance DE, Mugavero M, Willig J, Raper JL, Saag MS. Aging with HIV: a cross-sectional study of comorbidity prevalence and clinical characteristics across decades of life. J Assoc Nurses AIDS Care 2011; 22:17–25.
- Guaraldi G, Orlando G, Zona S, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis 2011; 53:1120–1126.
- Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996-2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 2010; 50:1387–1396.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- Berry SA, Fleishman JA, Moore RD, Gebo KA; HIV Research Network. Trends in reasons for hospitalization in a multisite United States cohort of persons living with HIV, 2001-2008. J Acquir Immune Defic Syndr 2012; 59:368–375.
- Freiberg MS, Chang CC, Kuller LH, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med 2013; 173:614–622.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Worm SW, Sabin C, Weber R, et al. Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. J Infect Dis 2010; 201:318–330.
- Lake JE, Currier JS. Metabolic disease in HIV infection. Lancet Infect Dis 2013; 13:964–975.
- Data Collection on Adverse Events of Anti-HIV Drugs Study Group; Sabin CA, d’Arminio Monforte A, Friis-Moller N, et al. Changes over time in risk factors for cardiovascular disease and use of lipid-lowering drugs in HIV-infected individuals and impact on myocardial infarction. Clin Infect Dis 2008; 46:1101–1110.
- Falcone EL, Mangili A, Skinner S, Alam A, Polak JF, Wanke CA. Framingham risk score and early markers of atherosclerosis in a cohort of adults infected with HIV. Antivir Ther 2011; 16:1–8.
- Friis-Møller N, Thiébaut R, Reiss P, et al; DAD study group. Predicting the risk of cardiovascular disease in HIV-infected patients: the Data Collection on Adverse Effects of Anti-HIV Drugs study. Eur J Cardiovasc Prev Rehabil 2010; 17:491–501.
- European AIDS Clinical Society Guidelines (EACS). www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. Accessed April 16, 2015.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Samaras K. The burden of diabetes and hyperlipidemia in treated HIV infection and approaches for cardiometabolic care. Curr HIV/AIDS Rep 2012; 9:206–217.
- Rasmussen LD, Mathiesen ER, Kronborg G, Pedersen C, Gerstoft J, Obel N. Risk of diabetes mellitus in persons with and without HIV: a Danish nationwide population-based cohort study. PLoS One 2012; 7:e44575.
- Capeau J, Bouteloup V, Katlama C, et al; ANRS CO8 APROCO-COPILOTE Cohort Study Group. Ten-year diabetes incidence in 1,046 HIV-infected patients started on a combination antiretroviral treatment. AIDS 2012; 26:303–314.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006; 20:2165–2174.
- Rothman MS, Bessesen MT. HIV infection and osteoporosis: pathophysiology, diagnosis, and treatment options. Curr Osteoporos Rep 2012; 10:270–277.
- Bedimo R, Maalouf NM, Zhang S, Drechsler H, Tebas P. Osteoporotic fracture risk associated with cumulative exposure to tenofovir and other antiretroviral agents. AIDS 2012; 26:825–831.
- Brown TT, McComsey GA, King MS, Qaqish RB, Bernstein BM, da Silva BA. Loss of bone mineral density after antiretroviral therapy initiation, independent of antiretroviral regimen. J Acquir Immune Defic Syndr 2009; 51:554–561.
- Rodríguez M, Daniels B, Gunawardene S, Robbins GK. High frequency of vitamin D deficiency in ambulatory HIV-positive patients. AIDS Res Hum Retroviruses 2009; 25:9–14.
- Work Group for HIV and Aging Consensus Project. Summary report from the Human Immunodeficiency Virus and Aging Consensus Project: treatment strategies for clinicians managing older individuals with the human immunodeficiency virus. J Am Geriatr Soc 2012; 60:974–979.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. 2013 Issue, Version 3. http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 16, 2015.
- Heaton RK, Clifford DB, Franklin DR Jr, et al; CHARTER Group. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010; 75:2087–2096.
- Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010; 24:1243–1250.
- Valcour VG. Evaluating cognitive impairment in the clinical setting: practical screening and assessment tools. Top Antivir Med 2011; 19:175–180.
- Desquilbet L, Jacobson LP, Fried LP, et al; Multicenter AIDS Cohort Study. HIV-1 infection is associated with an earlier occurrence of a phenotype related to frailty. J Gerontol A Biol Sci Med Sci 2007; 62:1279–1286.
- Desquilbet L, Jacobson LP, Fried LP, et al. A frailty-related phenotype before HAART initiation as an independent risk factor for AIDS or death after HAART among HIV-infected men. J Gerontol A Biol Sci Med Sci 2011; 66:1030–1038.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Greene M, Valcour V, Miao Y, et al. Geriatric syndromes are common among older HIV-infected adults. 21st Conference on Retroviruses and Opportunistic Infections (CROI) 2014 March 3-6, Boston MA.
- Morgan EE, Iudicello JE, Weber E, et al; HIV Neurobehavioral Research Program (HNRP) Group. Synergistic effects of HIV infection and older age on daily functioning. J Acquir Immune Defic Syndr 2012; 61:341–348.
- Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 2007; 370:59–67.
- Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst 2011; 103:753–762.
- Deeken JF, Tjen-A-Looi A, Rudek MA, et al. The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin Infect Dis 2012; 55:1228–1235.
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol 2009; 27:884–890.
- Patel P, Hanson DL, Sullivan PS, et al; Adult and Adolescent Spectrum of Disease Project and HIV Outpatient Study Investigators. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992-2003. Ann Intern Med 2008; 148:728–736.
- Kaplan JE, Benson C, Holmes KK, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC); National Institutes of Health; HIV Medicine Association of the Infectious Diseases Society of America. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
- World Health Organization. HIV/AIDS: fact sheet. www.who.int/mediacentre/factsheets/fs360/en/. Accessed April 16, 2015.
- Centers for Disease Control and Prevention (CDC). HIV/AIDS: basic statistics. www.cdc.gov/hiv/basics/statistics.html. Accessed April 16, 2015.
- May MT, Gompels M, Delpech V, et al; UK Collaborative HIV Cohort (UK CHIC) Study. Impact on life expectancy of HIV-1 positive individuals of CD4+ cell count and viral load response to antiretroviral therapy. AIDS 2014; 28:1193–1202.
- Centers for Disease Control and Prevention (CDC). Diagnoses of HIV infection in the United States and dependent areas: HIV surveillance report. www.cdc.gov/hiv/library/reports/surveillance/2011/surveillance_Report_vol_23.html. Accessed April 16, 2015.
- Effros RB, Fletcher CV, Gebo K, et al. Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis 2008; 47:542–553.
- Moyer VA; US Preventive Services Task Force. Screening for HIV: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:51–60.
- Qaseem A, Snow V, Shekelle P, Hopkins R Jr, Owens DK; Clinical Efficacy Assessment Subcommittee, American College of Physicians. Screening for HIV in health care settings: a guidance statement from the American College of Physicians and HIV Medicine Association. Ann Intern Med 2009; 150:125–131.
- Lucas A, Armbruster B. The cost-effectiveness of expanded HIV screening in the United States. AIDS 2013; 27:795–801.
- Lindau ST, Schumm LP, Laumann EO, Levinson W, O’Muircheartaigh CA, Waite LJ. A study of sexuality and health among older adults in the United States. N Engl J Med 2007; 357:762–774.
- Schick V, Herbenick D, Reece M, et al. Sexual behaviors, condom use, and sexual health of Americans over 50: implications for sexual health promotion for older adults. J Sex Med 2010; 7(suppl 5):315–329.
- US Department of Health and Human Services, HIV/AIDS Bureau. The Ryan White HIV/AIDS program: population fact sheet: August 2010. Older adults. http://hab.hrsa.gov/abouthab/populations/olderadultsfacts.pdf. Accessed April 16, 2015.
- May M, Gompels M, Delpech V, et al. Impact of late diagnosis and treatment on life expectancy in people with HIV-1: UK Collaborative HIV Cohort (UK CHIC) Study. BMJ 2011; 343:d6016.
- Branson BM, Handsfield HH, Lampe MA, et al; Centers for Disease Control and Prevention (CDC). Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. MMWR Recomm Rep 2006; 55:1–17.
- Health Resources and Services Administration (HRSA); HIV/AIDS Bureau. HRSA CAREAction. The graying of HIV. http://hab.hrsa.gov/newspublications/careactionnewsletter/february2009.pdf. Accessed April 16, 2015.
- AIDS InfoNet. Fact sheet number 616: Older people and HIV. http://aidsinfonet.org/fact_sheets/view/616. Accessed April 16, 2015.
- Rodger AJ, Lodwick R, Schechter M, et al; INSIGHT SMART, ESPRIT Study Groups. Mortality in well controlled HIV in the continuous antiretroviral therapy arms of the SMART and ESPRIT trials compared with the general population. AIDS 2013; 27:973–979.
- Kitahata MM, Gange SJ, Abraham AG, et al; NA-ACCORD Investigators. Effect of early versus deferred antiretroviral therapy for HIV on survival. N Engl J Med 2009; 360:1815–1826.
- Cao W, Jamieson BD, Hultin LE, Hultin PM, Effros RB, Detels R. Premature aging of T cells is associated with faster HIV-1 disease progression. J Acquir Immune Defic Syndr 2009; 50:137–147.
- Allers K, Bösel D, Epple HJ, et al. Effect of age on the CD4+ T-cell impairment in HIV-infected persons without and with cART. J Acquir Immune Defic Syndr 2014; 66:7–15.
- Kalayjian RC, Spritzler J, Matining RM, et al. Older HIV-infected patients on antiretroviral therapy have B-cell expansion and attenuated CD4 cell increases with immune activation reduction. AIDS 2013; 27:1563–1571.
- Althoff KN, Gebo KA, Gange SJ, et al; North American AIDS Cohort Collaboration on Research and Design. CD4 count at presentation for HIV care in the United States and Canada: are those over 50 years more likely to have a delayed presentation? AIDS Res Ther 2010; 7:45.
- Freeland KN, Thompson AN, Zhao Y, Leal JE, Mauldin PD, Moran WP. Medication use and associated risk of falling in a geriatric outpatient population. Ann Pharmacother 2012; 46:1188–1192.
- Steinman MA, Hanlon JT. Managing medications in clinically complex elders: “There’s got to be a happy medium.” JAMA 2010; 304:1592–1601.
- Marzolini C, Elzi L, Gibbons S, et al; Swiss HIV Cohort Study. Prevalence of comedications and effect of potential drug-drug interactions in the Swiss HIV Cohort Study. Antivir Ther 2010; 15:413–423.
- Greene M, Justice AC, Lampiris HW, Valcour V. Management of human immunodeficiency virus infection in advanced age. JAMA 2013; 309:1397–1405.
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Department of Health and Human Services. http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed April 16, 2015.
- UCSF Center for HIV Information. HIVInSite. Comprehensive, up-to-date information on HIV/AIDS treatment, prevention, and policy from the University of California San Francisco: database of antiretroviral drug interactions. http://hivinsite.ucsf.edu/. Accessed April 16, 2015.
- The University of Liverpool. Drug interaction charts. www.hiv-druginteractions.org. Accessed April 16, 2015.
- Vance DE, Mugavero M, Willig J, Raper JL, Saag MS. Aging with HIV: a cross-sectional study of comorbidity prevalence and clinical characteristics across decades of life. J Assoc Nurses AIDS Care 2011; 22:17–25.
- Guaraldi G, Orlando G, Zona S, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis 2011; 53:1120–1126.
- Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996-2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 2010; 50:1387–1396.
- Currier JS, Taylor A, Boyd F, et al. Coronary heart disease in HIV-infected individuals. J Acquir Immune Defic Syndr 2003; 33:506–512.
- Berry SA, Fleishman JA, Moore RD, Gebo KA; HIV Research Network. Trends in reasons for hospitalization in a multisite United States cohort of persons living with HIV, 2001-2008. J Acquir Immune Defic Syndr 2012; 59:368–375.
- Freiberg MS, Chang CC, Kuller LH, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med 2013; 173:614–622.
- Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab 2007; 92:2506–2512.
- Calmy A, Gayet-Ageron A, Montecucco F, et al; STACCATO Study Group. HIV increases markers of cardiovascular risk: results from a randomized, treatment interruption trial. AIDS 2009; 23:929–939.
- Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: exploratory analyses from the SMART trial. Antivir Ther 2008; 13:177–187.
- Worm SW, Sabin C, Weber R, et al. Risk of myocardial infarction in patients with HIV infection exposed to specific individual antiretroviral drugs from the 3 major drug classes: the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study. J Infect Dis 2010; 201:318–330.
- Lake JE, Currier JS. Metabolic disease in HIV infection. Lancet Infect Dis 2013; 13:964–975.
- Data Collection on Adverse Events of Anti-HIV Drugs Study Group; Sabin CA, d’Arminio Monforte A, Friis-Moller N, et al. Changes over time in risk factors for cardiovascular disease and use of lipid-lowering drugs in HIV-infected individuals and impact on myocardial infarction. Clin Infect Dis 2008; 46:1101–1110.
- Falcone EL, Mangili A, Skinner S, Alam A, Polak JF, Wanke CA. Framingham risk score and early markers of atherosclerosis in a cohort of adults infected with HIV. Antivir Ther 2011; 16:1–8.
- Friis-Møller N, Thiébaut R, Reiss P, et al; DAD study group. Predicting the risk of cardiovascular disease in HIV-infected patients: the Data Collection on Adverse Effects of Anti-HIV Drugs study. Eur J Cardiovasc Prev Rehabil 2010; 17:491–501.
- European AIDS Clinical Society Guidelines (EACS). www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. Accessed April 16, 2015.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Samaras K. The burden of diabetes and hyperlipidemia in treated HIV infection and approaches for cardiometabolic care. Curr HIV/AIDS Rep 2012; 9:206–217.
- Rasmussen LD, Mathiesen ER, Kronborg G, Pedersen C, Gerstoft J, Obel N. Risk of diabetes mellitus in persons with and without HIV: a Danish nationwide population-based cohort study. PLoS One 2012; 7:e44575.
- Capeau J, Bouteloup V, Katlama C, et al; ANRS CO8 APROCO-COPILOTE Cohort Study Group. Ten-year diabetes incidence in 1,046 HIV-infected patients started on a combination antiretroviral treatment. AIDS 2012; 26:303–314.
- American Diabetes Association. Standards of medical care in diabetes—2013. Diabetes Care 2013; 36(suppl 1):S11–S66.
- Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS 2006; 20:2165–2174.
- Rothman MS, Bessesen MT. HIV infection and osteoporosis: pathophysiology, diagnosis, and treatment options. Curr Osteoporos Rep 2012; 10:270–277.
- Bedimo R, Maalouf NM, Zhang S, Drechsler H, Tebas P. Osteoporotic fracture risk associated with cumulative exposure to tenofovir and other antiretroviral agents. AIDS 2012; 26:825–831.
- Brown TT, McComsey GA, King MS, Qaqish RB, Bernstein BM, da Silva BA. Loss of bone mineral density after antiretroviral therapy initiation, independent of antiretroviral regimen. J Acquir Immune Defic Syndr 2009; 51:554–561.
- Rodríguez M, Daniels B, Gunawardene S, Robbins GK. High frequency of vitamin D deficiency in ambulatory HIV-positive patients. AIDS Res Hum Retroviruses 2009; 25:9–14.
- Work Group for HIV and Aging Consensus Project. Summary report from the Human Immunodeficiency Virus and Aging Consensus Project: treatment strategies for clinicians managing older individuals with the human immunodeficiency virus. J Am Geriatr Soc 2012; 60:974–979.
- National Osteoporosis Foundation. Clinician’s guide to prevention and treatment of osteoporosis. 2013 Issue, Version 3. http://nof.org/files/nof/public/content/file/2791/upload/919.pdf. Accessed April 16, 2015.
- Heaton RK, Clifford DB, Franklin DR Jr, et al; CHARTER Group. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010; 75:2087–2096.
- Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010; 24:1243–1250.
- Valcour VG. Evaluating cognitive impairment in the clinical setting: practical screening and assessment tools. Top Antivir Med 2011; 19:175–180.
- Desquilbet L, Jacobson LP, Fried LP, et al; Multicenter AIDS Cohort Study. HIV-1 infection is associated with an earlier occurrence of a phenotype related to frailty. J Gerontol A Biol Sci Med Sci 2007; 62:1279–1286.
- Desquilbet L, Jacobson LP, Fried LP, et al. A frailty-related phenotype before HAART initiation as an independent risk factor for AIDS or death after HAART among HIV-infected men. J Gerontol A Biol Sci Med Sci 2011; 66:1030–1038.
- Fried LP, Tangen CM, Walston J, et al; Cardiovascular Health Study Collaborative Research Group. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 2001; 56:M146–M156.
- Greene M, Valcour V, Miao Y, et al. Geriatric syndromes are common among older HIV-infected adults. 21st Conference on Retroviruses and Opportunistic Infections (CROI) 2014 March 3-6, Boston MA.
- Morgan EE, Iudicello JE, Weber E, et al; HIV Neurobehavioral Research Program (HNRP) Group. Synergistic effects of HIV infection and older age on daily functioning. J Acquir Immune Defic Syndr 2012; 61:341–348.
- Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet 2007; 370:59–67.
- Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst 2011; 103:753–762.
- Deeken JF, Tjen-A-Looi A, Rudek MA, et al. The rising challenge of non-AIDS-defining cancers in HIV-infected patients. Clin Infect Dis 2012; 55:1228–1235.
- Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J Clin Oncol 2009; 27:884–890.
- Patel P, Hanson DL, Sullivan PS, et al; Adult and Adolescent Spectrum of Disease Project and HIV Outpatient Study Investigators. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992-2003. Ann Intern Med 2008; 148:728–736.
- Kaplan JE, Benson C, Holmes KK, Brooks JT, Pau A, Masur H; Centers for Disease Control and Prevention (CDC); National Institutes of Health; HIV Medicine Association of the Infectious Diseases Society of America. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep 2009; 58:1–207.
KEY POINTS
- Today, nearly 20% of newly diagnosed HIV-infected people and more than 50% of all HIV-infected people in the United States are over age 50.
- The diagnosis and treatment of HIV tends to be delayed in elderly patients, with deleterious effects.
- Antiretroviral drugs have a number of interactions with drugs commonly used in elderly patients.
- Several diseases are more common in HIV-positive patients, including cardiovascular disease, diabetes mellitus, osteoporosis, dementia, and various malignant diseases. These merit aggressive screening and preventive measures.

Cannabinoid hyperemesis syndrome: Marijuana is both antiemetic and proemetic
With the growing use of marijuana, reports have appeared of a newly recognized condition in long-term heavy users termed cannabinoid hyperemesis syndrome.1
This syndrome is interesting for at least two reasons. First, paradoxically, marijuana appears to have an emetic effect with chronic use, whereas it usually has the opposite effect and is used as an antiemetic in patients undergoing chemotherapy. Second, patients develop a compulsion to bathe or shower in extremely hot water to relieve the symptoms.
In this article, we review the pathophysiology, clinical presentation, diagnosis, and management of this emerging condition.
MARIJUANA USE ON THE RISE
Marijuana is the most widely used illicit drug worldwide. Although statistics on its use vary, a report from the Pew Research Center2 stated that 49% of Americans say they have tried it. Several states now allow the use of marijuana for medicinal purposes, and Colorado and Washington have legalized it for recreational use. This marks a major turning point and may accelerate the slow-growing acceptance of marijuana use in the United States.
Marijuana has been used to treat HIV-associated anorexia and wasting, convulsions, glaucoma, headache, and chemotherapy-induced nausea and vomiting.3–5
Cannabinoid hyperemesis syndrome was first described in 2004 in South Australia.1 Since its recognition, an increasing number of cases have been identified worldwide. However, there are still no population-based studies to estimate its exact prevalence.
THC PREVENTS VOMITING—AND CAUSES IT
Delta-9-tetrahydrocannabinol (THC) is the principal psychoactive component in marijuana.6,7 There are two types of cannabinoid receptors in humans: CB1 and CB2. Both are found in the central nervous system and autonomic nervous system. Activation of CB1 receptors is responsible for the psychoactive effects of cannabinoids such as altered consciousness, euphoria, relaxation, perceptual disturbances, intensified sensory experiences, cognitive impairment, and increased reaction time. The physiologic role of CB2 is not known.
THC as an antiemetic
The antiemetic property of THC is not well understood but has been linked to activation of CB1 receptors found on the enteric plexus, presynaptic parasympathetic system, and central nervous system, particularly the cerebellum, hypothalamus, and vomiting center in the medulla.1,8–12 Stimulation and blockade of CB1 receptors can inhibit and induce vomiting in a dose-dependent manner, implicating endogenous cannabinoids in emetic circuits.12
THC as a proemetic
The mechanism of the paradoxical hyperemetic effect of THC is unknown, but several concepts have been proposed.
Chronic cannabis use can lead to down-regulation of CB1 receptors.13 Simonetto et al10 suggested that the central effects of long-term cannabis use on the hypothalamic-pituitary-adrenal axis may play a central role in the development of hyperemesis.10
Cannabinoids have a long half-life and are lipophilic.1 When used infrequently, they prevent vomiting. But with chronic use, high concentrations of THC can accumulate in the body, including cerebral fat, and can cause severe nausea and vomiting.8,9 This paradoxic hyperemesis was observed in people using intravenous crude marijuana extract.7 The same response was also noted in ferrets injected with 2-arachidonoylglycerol, a potent cannabinoid agonist.11
Patients who experience hyperemesis from chronic cannabis use may also have a genetic variation in their hepatic drug-transforming enzymes that results in excessive levels of cannabis metabolites that promote emesis.1,14
Delayed gastric emptying has also been linked to the proemetic effect of THC. However, this association became controversial when a large case series study showed that only 30% of patients with cannabinoid hyperemesis syndrome had delayed emptying on gastric scintigraphy.10
It is also possible that excessive stimulation of cannabinoid receptors in the gut can cause diffuse splanchnic vasodilation and contribute to the abdominal pain.13
DIAGNOSING CANNABINOID HYPEREMESIS SYNDROME
Cannabinoid hyperemesis syndrome is a clinical diagnosis typically seen in young patients (under age 50) with a long history of marijuana use. They present with severe, cyclic nausea and vomiting and admit to compulsively taking extremely hot showers or baths. Most patients report using marijuana for more than a year before developing episodes of severe vomiting. However, one study found that as many as 32% of patients had used it less than 1 year before experiencing symptoms.10
Other associated nonspecific symptoms are diaphoresis, bloating, abdominal discomfort, flushing, and weight loss. Symptoms are relieved with long, hot showers or baths and cessation of marijuana use. Taking a complete history is key to making the diagnosis.

In 2004, Allen et al1 first defined cannabinoid hyperemesis as excessive marijuana use associated with cyclical vomiting and abdominal pain.1 In 2012, Simonetto et al10 proposed diagnostic criteria (Table 1). Although not yet validated, these criteria are based on the largest series of cases of cannabinoid hyperemesis syndrome to date (98 patients).10
THE THREE PHASES OF CANNABINOID HYPEREMESIS
The clinical presentation of cannabinoid hyperemesis syndrome can be divided into three phases: prodromal, vomiting, and resolution.
Prodromal phase
During this phase, patients often appear anxious and agitated and display a spectrum of autonomic symptoms such as sweating, flushing, and constantly sipping water due to thirst. They may sometimes have abdominal pain that is usually epigastric but may also be diffuse. Their symptoms are associated with severe nausea, usually early in the morning or when they see or smell food. Appetite and eating patterns remain normal. Compulsive hot bathing or showering is minimal at this phase.
Vomiting phase
In this next phase, patients experience incapacitating nausea and vomiting that may occur without warning and are resistant to conventional antiemetics such as ondansetron and promethazine.14 However, patients eventually learn that hot baths or showers relieve the symptoms, and this behavior eventually becomes a compulsion. The higher the temperature of the water, the better the effect on symptoms.1 Low-grade pyrexia, excessive thirst, orthostasis, abdominal tenderness, weight loss, and sometimes even superficial skin burns have been reported.1,9,15–18
Recovery phase
During the final phase of cannabinoid hyperemesis syndrome, most patients experience marked resolution of symptoms after 24 to 48 hours of conservative management (bowel rest until symptoms resolve, slowly advancing diet as tolerated, intravenous fluids, and electrolyte monitoring and repletion as necessary), and most importantly, cessation of cannabis use. However, the time from cessation of marijuana use to resolution of symptoms may be as long as 1 week to 1 month.1,10,14 Patients begin to resume their normal diet and daily activities. The bathing-showering compulsion subsides, and patients regain lost weight after 3 to 6 months.1
In all case series and reports, resumption of cannabis use causes the symptoms to recur. This recurrence is compelling evidence that cannabis is the cause of the hyperemesis and should be part of the essential criteria for the diagnosis of cannabinoid hyperemesis syndrome.
WHY COMPULSIVE HOT BATHING?
The mechanism behind this unique characteristic of cannabinoid hyperemesis syndrome is not known. Several theories have been suggested, but no study has identified the exact explanation for this phenomenon.1,9,10,13–15,17–31
One suggested mechanism is a response by the thermoregulatory center of the brain to the dose-dependent hypothermic effects of THC, or even a direct effect of CB1 receptor activation in the hypothalamus.9 Cannabis toxicity could disrupt the equilibrium of satiety, thirst, digestive, and thermoregulatory systems of the hypothalamus, and this interference could resolve with hot bathing.1
The so-called “cutaneous steal” syndrome has also been proposed, in which cutaneous vasodilation caused by hot water decreases the blood volume available for the splanchnic circulation thought to be responsible for the abdominal pain and vomiting.13 The compulsive hot bathing may also be a response by the brain to the anxiety or psychological stress induced by severe nausea and vomiting.14
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of cannabinoid hyperemesis syndrome includes mainly cyclic vomiting syndrome and psychogenic vomiting. A careful history is useful, as is ruling out medication-induced reactions, toxins, pregnancy, and gastrointestinal, neurologic, metabolic, and endocrine causes. All three of these vomiting syndromes can present with a cyclic pattern of nausea and vomiting. Cannabis use is common in all three and so is not helpful in differentiating them. But the characteristic compulsive hot bathing and showering is unique and pathognomonic of cannabinoid hyperemesis syndrome.32
Endoscopic examination may reveal esophagitis and gastritis from severe bouts of retching.26
Cyclic vomiting syndrome
The Rome III criteria for the diagnosis of cyclic vomiting syndrome include three or more stereotypic episodes of acute-onset nausea and vomiting lasting less than 1 week, alternating with intervals of completely normal health. The criteria should be fulfilled for the previous 3 months with symptom onset at least 6 months before diagnosis.33
In a series of 17 patients with adult-onset cyclic vomiting syndrome,18 the average age at onset was 30, and 13 (76%) of the patients were women. Fifteen (88%) of the patients experienced a prodrome or aura of abdominal pain or headache, and in this group, a trigger such as emotional stress and infection could also be identified in 9 (60%).
Unlike in cannabinoid hyperemesis syndrome, most patients with cyclic vomiting syndrome have a family history of migraine headache, and the prevalence of psychological stressors is high.31 Also, patients with cannabinoid hyperemesis syndrome do not respond to medications that usually abort migraine episodes,15 whereas patients with cyclic vomiting syndrome, especially those who have a family history of migraines, may respond to antimigraine medications such as triptans. There is evidence of clinical psychological overlap between cyclic vomiting syndrome, abdominal migraine, and migraine headaches. Some authors recommend antimigraine therapy even in the absence of a family or personal history of migraine if, after a careful history and physical examination, the diagnosis of cyclic vomiting syndrome seems likely. Moreover, nonmedical management such as sleep, dark rooms, and quiet environment are not as effective in cannabinoid hyperemesis syndrome as they are in cyclic vomiting syndrome.18
Psychogenic vomiting
Psychogenic vomiting is classically defined as vomiting caused by psychological mechanisms without any obvious organic cause.13 It occurs most commonly in patients with major depressive disorder or conversion disorder.34 The mechanism appears to be a combination of past organic or gastrointestinal functional abnormalities and emotional problems, and multiple patterns of vomiting can occur. Most of these patients can be treated with behavioral therapy, antidepressant drug therapy, and supportive psychotherapy.34,35
ASKING A SERIES OF QUESTIONS
Most patients with cannabinoid hyperemesis syndrome have a history of frequent visits to emergency departments or clinics for persistent nausea and vomiting, and they may have undergone extensive diagnostic workups to exclude structural, inflammatory, infectious, and functional diseases of the bowel.23,24
To prevent unnecessary testing and use of healthcare resources, Wallace et al32 proposed an algorithm to help guide clinicians in diagnosing and treating patients with suspected cannabinoid hyperemesis syndrome. A patient presenting with severe nausea and vomiting should prompt a series of questions:
Do the signs and symptoms suggest a severe underlying medical cause? If so, this should be pursued.
Do symptoms improve while taking a hot shower or bath? If not, pursue an appropriate diagnostic evaluation and treatment for conditions other than cannabinoid hyperemesis syndrome.
Is the bathing compulsive? If not, consider other diagnoses, but remain suspicious about cannabinoid hyperemesis syndrome.
Does the patient currently use cannabis daily or almost daily, and has the patient done so for at least the past year? If the patient denies using cannabis, a urine drug screen for THC may be useful. If the patient admits to use, a presumptive diagnosis of cannabinoid hyperemesis syndrome can be made.
Does the patient have signs or symptoms of volume depletion, or is the patient unable to tolerate oral hydration? Encourage oral hydration or provide intravenous hydration, and provide cannabis cessation counseling.
Do the symptoms improve? If yes, great! Provide cessation counseling, resources, and follow-up. If not:
Is the patient still using cannabis? If not, it is time to rethink the diagnosis.
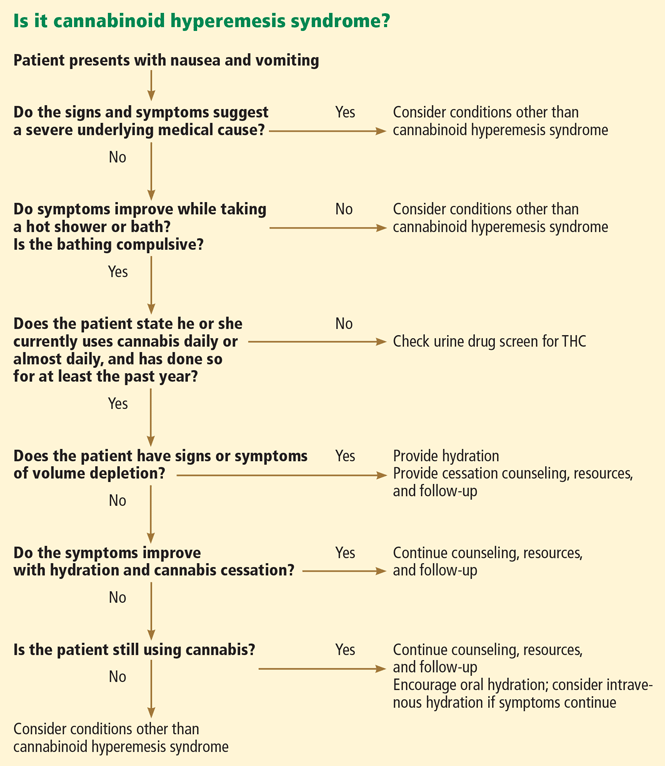
Treatment in the acute setting is supportive and includes intravenous hydration and correction of electrolytes. Conventional antiemetics such as ondansetron, metoclopramide, prochlorperazine, and promethazine have not been effective in relieving hyperemesis.9,12,14 This implies that the mechanism of emesis likely does not involve dopaminergic and serotonin pathways in the central and autonomic nervous systems.
Cessation of cannabis use is key for long-term resolution of symptoms. Efforts should be made to provide counseling and encourage patients to stop using the drug entirely (Figure 1).
SOMETHING TO THINK ABOUT
With the high prevalence of chronic cannabis abuse and the recent legalization of recreational marijuana use, we will all likely encounter a patient with cannabinoid hyperemesis. With adequate knowledge of this phenomenon, we can avoid unnecessary workups and inappropriate medical and surgical treatment in patients presenting with recurrent vomiting of unknown cause. The diagnosis can easily be made by simply asking for a history of chronic marijuana use and symptoms related to cannabinoid hyperemesis syndrome, such as relief of symptoms with hot baths or showers and with marijuana cessation.
Conservative management and fluid resuscitation is important in the acute setting, but cessation of marijuana use and follow-up counseling are the key components for treating patients with cannabinoid hyperemesis syndrome and for preventing recurrence.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Motel S. 6 facts about marijuana. Factank. News in the Numbers Pew Research Center. www.pewresearch.org/fact-tank/2015/04/14/6-facts-about-marijuana/. Accessed June 2, 2015.
- Walsh D, Nelson KA, Mahmoud FA. Established and potential therapeutic applications of cannabinoids in oncology. Support Care Cancer 2003; 11:137–143.
- Tramèr MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ 2001; 323:16–21.
- Davis M, Maida V, Daeninck P, Pergolizzi J. The emerging role of cannabinoid neuromodulators in symptom management. Support Care Cancer 2007; 15:63–71.
- National Institutes of Health (NIH). National Institute on Drug Abuse. Drug facts: marijuana. www.nida.nih.gov/infofacts/marijuana. Accessed April 29, 2015.
- Vaziri ND, Thomas R, Sterling M, et al. Toxicity with intravenous injection of crude marijuana extract. Clin Toxicol 1981; 18:353–366.
- Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992; 258:1946–1949.
- Chang YH, Windish DM. Cannabinoid hyperemesis relieved by compulsive bathing. Mayo Clin Proc 2009; 84:76–78.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Darmani NA. The potent emetogenic effects of the endocannabinoid, 2-AG (2-arachidonoylglycerol) are blocked by delta(9)-tetrahydrocannabinol and other cannnabinoids. J Pharmacol Exp Ther 2002; 300:34–42.
- Darmani NA, Sim-Selley LJ, Martin BR, et al. Antiemetic and motor-depressive actions of CP55,940: cannabinoid CB1 receptor characterization, distribution, and G-protein activation. Eur J Pharmacol 2003; 459:83–95.
- Leibovich MA. Psychogenic vomiting. Psychotherapeutic considerations. Psychother Psychosom 1973; 22:263–268.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Sontineni SP, Chaudhary S, Sontineni V, Lanspa SJ. Cannabinoid hyperemesis syndrome: clinical diagnosis of an underrecognised manifestation of chronic cannabis abuse. World J Gastroenterol 2009; 15:1264–1266.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Price SL, Fisher C, Kumar R, Hilgerson A. Cannabinoid hyperemesis syndrome as the underlying cause of intractable nausea and vomiting. J Am Osteopath Assoc 2011; 111:166–169.
- Lee LY, Abbott L, Moodie S, Anderson S. Cyclic vomiting syndrome in 28 patients: demographics, features and outcomes. Eur J Gastroenterol Hepatol 2012; 24:939–943.
- Wallace D, Martin AL, Park B. Cannabinoid hyperemesis: marijuana puts patients in hot water. Australas Psychiatry 2007; 15:156–158.
- Ashton CH. Adverse effects of cannabis and cannabinoids. Br J Anaesth 1999; 83:637–649.
- Cota D, Steiner MA, Marsicano G, et al. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 2007; 148:1574–1581.
- McCallum RW, Soykan I, Sridhar KR, Ricci DA, Lange RC, Plankey MW. Delta-9-tetrahydrocannabinol delays the gastric emptying of solid food in humans: a double-blind, randomized study. Aliment Pharmacol Ther 1999; 13:77–80.
- Donnino MW, Cocchi MN, Miller J, Fisher J. Cannabinoid hyperemesis: a case series. J Emerg Med 2011; 40:e63–e66.
- Singh E, Coyle W. Cannabinoid hyperemesis. Am J Gastroenterol 2008; 103:1048–1049.
- Carnett JB. Intercostal neuralgia as a cause of abdominal pain and tenderness. Surg Gynecol Obstet 1926; 42:625–632.
- Patterson DA, Smith E, Monahan M, et al. Cannabinoid hyperemesis and compulsive bathing: a case series and paradoxical pathophysiological explanation. J Am Board Fam Med 2010; 23:790–793.
- Izzo AA, Camilleri M. Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut 2008; 57:1140–1155.
- Pertwee RG. Cannabinoids and the gastrointestinal tract. Gut 2001; 48:859–867.
- Choung RS, Locke GR 3rd, Lee RM, Schleck CD, Zinsmeister AR, Talley NJ. Cyclic vomiting syndrome and functional vomiting in adults: association with cannabinoid use in males. Neurogastroenterol Motil 2012; 24:20–26,e1.
- Nicolson SE, Denysenko L, Mulcare JL, Vito JP, Chabon B. Cannabinoid hyperemesis syndrome: a case series and review of previous reports. Psychosomatics 2012; 53:212–219.
- Miller JB, Walsh M, Patel PA, et al. Pediatric cannabinoid hyperemesis: two cases. Pediatr Emerg Care 2010; 26:919–920.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Tack J, Taley NJ, Camilleri M, et al. Functional gastroduodenal disorders. Gastroenterology 2006; 130:1466–1479.
- Muraoka M, Mine K, Matsumoto K, Nakai Y, Nakagawa T. Psychogenic vomiting: the relation between patterns of vomiting and psychiatric diagnoses. Gut 1990; 31:526–528.
- Stravynski A. Behavioral treatment of psychogenic vomiting in the context of social phobia. J Nerv Ment Dis 1983; 171:448–451.
With the growing use of marijuana, reports have appeared of a newly recognized condition in long-term heavy users termed cannabinoid hyperemesis syndrome.1
This syndrome is interesting for at least two reasons. First, paradoxically, marijuana appears to have an emetic effect with chronic use, whereas it usually has the opposite effect and is used as an antiemetic in patients undergoing chemotherapy. Second, patients develop a compulsion to bathe or shower in extremely hot water to relieve the symptoms.
In this article, we review the pathophysiology, clinical presentation, diagnosis, and management of this emerging condition.
MARIJUANA USE ON THE RISE
Marijuana is the most widely used illicit drug worldwide. Although statistics on its use vary, a report from the Pew Research Center2 stated that 49% of Americans say they have tried it. Several states now allow the use of marijuana for medicinal purposes, and Colorado and Washington have legalized it for recreational use. This marks a major turning point and may accelerate the slow-growing acceptance of marijuana use in the United States.
Marijuana has been used to treat HIV-associated anorexia and wasting, convulsions, glaucoma, headache, and chemotherapy-induced nausea and vomiting.3–5
Cannabinoid hyperemesis syndrome was first described in 2004 in South Australia.1 Since its recognition, an increasing number of cases have been identified worldwide. However, there are still no population-based studies to estimate its exact prevalence.
THC PREVENTS VOMITING—AND CAUSES IT
Delta-9-tetrahydrocannabinol (THC) is the principal psychoactive component in marijuana.6,7 There are two types of cannabinoid receptors in humans: CB1 and CB2. Both are found in the central nervous system and autonomic nervous system. Activation of CB1 receptors is responsible for the psychoactive effects of cannabinoids such as altered consciousness, euphoria, relaxation, perceptual disturbances, intensified sensory experiences, cognitive impairment, and increased reaction time. The physiologic role of CB2 is not known.
THC as an antiemetic
The antiemetic property of THC is not well understood but has been linked to activation of CB1 receptors found on the enteric plexus, presynaptic parasympathetic system, and central nervous system, particularly the cerebellum, hypothalamus, and vomiting center in the medulla.1,8–12 Stimulation and blockade of CB1 receptors can inhibit and induce vomiting in a dose-dependent manner, implicating endogenous cannabinoids in emetic circuits.12
THC as a proemetic
The mechanism of the paradoxical hyperemetic effect of THC is unknown, but several concepts have been proposed.
Chronic cannabis use can lead to down-regulation of CB1 receptors.13 Simonetto et al10 suggested that the central effects of long-term cannabis use on the hypothalamic-pituitary-adrenal axis may play a central role in the development of hyperemesis.10
Cannabinoids have a long half-life and are lipophilic.1 When used infrequently, they prevent vomiting. But with chronic use, high concentrations of THC can accumulate in the body, including cerebral fat, and can cause severe nausea and vomiting.8,9 This paradoxic hyperemesis was observed in people using intravenous crude marijuana extract.7 The same response was also noted in ferrets injected with 2-arachidonoylglycerol, a potent cannabinoid agonist.11
Patients who experience hyperemesis from chronic cannabis use may also have a genetic variation in their hepatic drug-transforming enzymes that results in excessive levels of cannabis metabolites that promote emesis.1,14
Delayed gastric emptying has also been linked to the proemetic effect of THC. However, this association became controversial when a large case series study showed that only 30% of patients with cannabinoid hyperemesis syndrome had delayed emptying on gastric scintigraphy.10
It is also possible that excessive stimulation of cannabinoid receptors in the gut can cause diffuse splanchnic vasodilation and contribute to the abdominal pain.13
DIAGNOSING CANNABINOID HYPEREMESIS SYNDROME
Cannabinoid hyperemesis syndrome is a clinical diagnosis typically seen in young patients (under age 50) with a long history of marijuana use. They present with severe, cyclic nausea and vomiting and admit to compulsively taking extremely hot showers or baths. Most patients report using marijuana for more than a year before developing episodes of severe vomiting. However, one study found that as many as 32% of patients had used it less than 1 year before experiencing symptoms.10
Other associated nonspecific symptoms are diaphoresis, bloating, abdominal discomfort, flushing, and weight loss. Symptoms are relieved with long, hot showers or baths and cessation of marijuana use. Taking a complete history is key to making the diagnosis.
In 2004, Allen et al1 first defined cannabinoid hyperemesis as excessive marijuana use associated with cyclical vomiting and abdominal pain.1 In 2012, Simonetto et al10 proposed diagnostic criteria (Table 1). Although not yet validated, these criteria are based on the largest series of cases of cannabinoid hyperemesis syndrome to date (98 patients).10
THE THREE PHASES OF CANNABINOID HYPEREMESIS
The clinical presentation of cannabinoid hyperemesis syndrome can be divided into three phases: prodromal, vomiting, and resolution.
Prodromal phase
During this phase, patients often appear anxious and agitated and display a spectrum of autonomic symptoms such as sweating, flushing, and constantly sipping water due to thirst. They may sometimes have abdominal pain that is usually epigastric but may also be diffuse. Their symptoms are associated with severe nausea, usually early in the morning or when they see or smell food. Appetite and eating patterns remain normal. Compulsive hot bathing or showering is minimal at this phase.
Vomiting phase
In this next phase, patients experience incapacitating nausea and vomiting that may occur without warning and are resistant to conventional antiemetics such as ondansetron and promethazine.14 However, patients eventually learn that hot baths or showers relieve the symptoms, and this behavior eventually becomes a compulsion. The higher the temperature of the water, the better the effect on symptoms.1 Low-grade pyrexia, excessive thirst, orthostasis, abdominal tenderness, weight loss, and sometimes even superficial skin burns have been reported.1,9,15–18
Recovery phase
During the final phase of cannabinoid hyperemesis syndrome, most patients experience marked resolution of symptoms after 24 to 48 hours of conservative management (bowel rest until symptoms resolve, slowly advancing diet as tolerated, intravenous fluids, and electrolyte monitoring and repletion as necessary), and most importantly, cessation of cannabis use. However, the time from cessation of marijuana use to resolution of symptoms may be as long as 1 week to 1 month.1,10,14 Patients begin to resume their normal diet and daily activities. The bathing-showering compulsion subsides, and patients regain lost weight after 3 to 6 months.1
In all case series and reports, resumption of cannabis use causes the symptoms to recur. This recurrence is compelling evidence that cannabis is the cause of the hyperemesis and should be part of the essential criteria for the diagnosis of cannabinoid hyperemesis syndrome.
WHY COMPULSIVE HOT BATHING?
The mechanism behind this unique characteristic of cannabinoid hyperemesis syndrome is not known. Several theories have been suggested, but no study has identified the exact explanation for this phenomenon.1,9,10,13–15,17–31
One suggested mechanism is a response by the thermoregulatory center of the brain to the dose-dependent hypothermic effects of THC, or even a direct effect of CB1 receptor activation in the hypothalamus.9 Cannabis toxicity could disrupt the equilibrium of satiety, thirst, digestive, and thermoregulatory systems of the hypothalamus, and this interference could resolve with hot bathing.1
The so-called “cutaneous steal” syndrome has also been proposed, in which cutaneous vasodilation caused by hot water decreases the blood volume available for the splanchnic circulation thought to be responsible for the abdominal pain and vomiting.13 The compulsive hot bathing may also be a response by the brain to the anxiety or psychological stress induced by severe nausea and vomiting.14
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of cannabinoid hyperemesis syndrome includes mainly cyclic vomiting syndrome and psychogenic vomiting. A careful history is useful, as is ruling out medication-induced reactions, toxins, pregnancy, and gastrointestinal, neurologic, metabolic, and endocrine causes. All three of these vomiting syndromes can present with a cyclic pattern of nausea and vomiting. Cannabis use is common in all three and so is not helpful in differentiating them. But the characteristic compulsive hot bathing and showering is unique and pathognomonic of cannabinoid hyperemesis syndrome.32
Endoscopic examination may reveal esophagitis and gastritis from severe bouts of retching.26
Cyclic vomiting syndrome
The Rome III criteria for the diagnosis of cyclic vomiting syndrome include three or more stereotypic episodes of acute-onset nausea and vomiting lasting less than 1 week, alternating with intervals of completely normal health. The criteria should be fulfilled for the previous 3 months with symptom onset at least 6 months before diagnosis.33
In a series of 17 patients with adult-onset cyclic vomiting syndrome,18 the average age at onset was 30, and 13 (76%) of the patients were women. Fifteen (88%) of the patients experienced a prodrome or aura of abdominal pain or headache, and in this group, a trigger such as emotional stress and infection could also be identified in 9 (60%).
Unlike in cannabinoid hyperemesis syndrome, most patients with cyclic vomiting syndrome have a family history of migraine headache, and the prevalence of psychological stressors is high.31 Also, patients with cannabinoid hyperemesis syndrome do not respond to medications that usually abort migraine episodes,15 whereas patients with cyclic vomiting syndrome, especially those who have a family history of migraines, may respond to antimigraine medications such as triptans. There is evidence of clinical psychological overlap between cyclic vomiting syndrome, abdominal migraine, and migraine headaches. Some authors recommend antimigraine therapy even in the absence of a family or personal history of migraine if, after a careful history and physical examination, the diagnosis of cyclic vomiting syndrome seems likely. Moreover, nonmedical management such as sleep, dark rooms, and quiet environment are not as effective in cannabinoid hyperemesis syndrome as they are in cyclic vomiting syndrome.18
Psychogenic vomiting
Psychogenic vomiting is classically defined as vomiting caused by psychological mechanisms without any obvious organic cause.13 It occurs most commonly in patients with major depressive disorder or conversion disorder.34 The mechanism appears to be a combination of past organic or gastrointestinal functional abnormalities and emotional problems, and multiple patterns of vomiting can occur. Most of these patients can be treated with behavioral therapy, antidepressant drug therapy, and supportive psychotherapy.34,35
ASKING A SERIES OF QUESTIONS
Most patients with cannabinoid hyperemesis syndrome have a history of frequent visits to emergency departments or clinics for persistent nausea and vomiting, and they may have undergone extensive diagnostic workups to exclude structural, inflammatory, infectious, and functional diseases of the bowel.23,24
To prevent unnecessary testing and use of healthcare resources, Wallace et al32 proposed an algorithm to help guide clinicians in diagnosing and treating patients with suspected cannabinoid hyperemesis syndrome. A patient presenting with severe nausea and vomiting should prompt a series of questions:
Do the signs and symptoms suggest a severe underlying medical cause? If so, this should be pursued.
Do symptoms improve while taking a hot shower or bath? If not, pursue an appropriate diagnostic evaluation and treatment for conditions other than cannabinoid hyperemesis syndrome.
Is the bathing compulsive? If not, consider other diagnoses, but remain suspicious about cannabinoid hyperemesis syndrome.
Does the patient currently use cannabis daily or almost daily, and has the patient done so for at least the past year? If the patient denies using cannabis, a urine drug screen for THC may be useful. If the patient admits to use, a presumptive diagnosis of cannabinoid hyperemesis syndrome can be made.
Does the patient have signs or symptoms of volume depletion, or is the patient unable to tolerate oral hydration? Encourage oral hydration or provide intravenous hydration, and provide cannabis cessation counseling.
Do the symptoms improve? If yes, great! Provide cessation counseling, resources, and follow-up. If not:
Is the patient still using cannabis? If not, it is time to rethink the diagnosis.
Treatment in the acute setting is supportive and includes intravenous hydration and correction of electrolytes. Conventional antiemetics such as ondansetron, metoclopramide, prochlorperazine, and promethazine have not been effective in relieving hyperemesis.9,12,14 This implies that the mechanism of emesis likely does not involve dopaminergic and serotonin pathways in the central and autonomic nervous systems.
Cessation of cannabis use is key for long-term resolution of symptoms. Efforts should be made to provide counseling and encourage patients to stop using the drug entirely (Figure 1).
SOMETHING TO THINK ABOUT
With the high prevalence of chronic cannabis abuse and the recent legalization of recreational marijuana use, we will all likely encounter a patient with cannabinoid hyperemesis. With adequate knowledge of this phenomenon, we can avoid unnecessary workups and inappropriate medical and surgical treatment in patients presenting with recurrent vomiting of unknown cause. The diagnosis can easily be made by simply asking for a history of chronic marijuana use and symptoms related to cannabinoid hyperemesis syndrome, such as relief of symptoms with hot baths or showers and with marijuana cessation.
Conservative management and fluid resuscitation is important in the acute setting, but cessation of marijuana use and follow-up counseling are the key components for treating patients with cannabinoid hyperemesis syndrome and for preventing recurrence.
With the growing use of marijuana, reports have appeared of a newly recognized condition in long-term heavy users termed cannabinoid hyperemesis syndrome.1
This syndrome is interesting for at least two reasons. First, paradoxically, marijuana appears to have an emetic effect with chronic use, whereas it usually has the opposite effect and is used as an antiemetic in patients undergoing chemotherapy. Second, patients develop a compulsion to bathe or shower in extremely hot water to relieve the symptoms.
In this article, we review the pathophysiology, clinical presentation, diagnosis, and management of this emerging condition.
MARIJUANA USE ON THE RISE
Marijuana is the most widely used illicit drug worldwide. Although statistics on its use vary, a report from the Pew Research Center2 stated that 49% of Americans say they have tried it. Several states now allow the use of marijuana for medicinal purposes, and Colorado and Washington have legalized it for recreational use. This marks a major turning point and may accelerate the slow-growing acceptance of marijuana use in the United States.
Marijuana has been used to treat HIV-associated anorexia and wasting, convulsions, glaucoma, headache, and chemotherapy-induced nausea and vomiting.3–5
Cannabinoid hyperemesis syndrome was first described in 2004 in South Australia.1 Since its recognition, an increasing number of cases have been identified worldwide. However, there are still no population-based studies to estimate its exact prevalence.
THC PREVENTS VOMITING—AND CAUSES IT
Delta-9-tetrahydrocannabinol (THC) is the principal psychoactive component in marijuana.6,7 There are two types of cannabinoid receptors in humans: CB1 and CB2. Both are found in the central nervous system and autonomic nervous system. Activation of CB1 receptors is responsible for the psychoactive effects of cannabinoids such as altered consciousness, euphoria, relaxation, perceptual disturbances, intensified sensory experiences, cognitive impairment, and increased reaction time. The physiologic role of CB2 is not known.
THC as an antiemetic
The antiemetic property of THC is not well understood but has been linked to activation of CB1 receptors found on the enteric plexus, presynaptic parasympathetic system, and central nervous system, particularly the cerebellum, hypothalamus, and vomiting center in the medulla.1,8–12 Stimulation and blockade of CB1 receptors can inhibit and induce vomiting in a dose-dependent manner, implicating endogenous cannabinoids in emetic circuits.12
THC as a proemetic
The mechanism of the paradoxical hyperemetic effect of THC is unknown, but several concepts have been proposed.
Chronic cannabis use can lead to down-regulation of CB1 receptors.13 Simonetto et al10 suggested that the central effects of long-term cannabis use on the hypothalamic-pituitary-adrenal axis may play a central role in the development of hyperemesis.10
Cannabinoids have a long half-life and are lipophilic.1 When used infrequently, they prevent vomiting. But with chronic use, high concentrations of THC can accumulate in the body, including cerebral fat, and can cause severe nausea and vomiting.8,9 This paradoxic hyperemesis was observed in people using intravenous crude marijuana extract.7 The same response was also noted in ferrets injected with 2-arachidonoylglycerol, a potent cannabinoid agonist.11
Patients who experience hyperemesis from chronic cannabis use may also have a genetic variation in their hepatic drug-transforming enzymes that results in excessive levels of cannabis metabolites that promote emesis.1,14
Delayed gastric emptying has also been linked to the proemetic effect of THC. However, this association became controversial when a large case series study showed that only 30% of patients with cannabinoid hyperemesis syndrome had delayed emptying on gastric scintigraphy.10
It is also possible that excessive stimulation of cannabinoid receptors in the gut can cause diffuse splanchnic vasodilation and contribute to the abdominal pain.13
DIAGNOSING CANNABINOID HYPEREMESIS SYNDROME
Cannabinoid hyperemesis syndrome is a clinical diagnosis typically seen in young patients (under age 50) with a long history of marijuana use. They present with severe, cyclic nausea and vomiting and admit to compulsively taking extremely hot showers or baths. Most patients report using marijuana for more than a year before developing episodes of severe vomiting. However, one study found that as many as 32% of patients had used it less than 1 year before experiencing symptoms.10
Other associated nonspecific symptoms are diaphoresis, bloating, abdominal discomfort, flushing, and weight loss. Symptoms are relieved with long, hot showers or baths and cessation of marijuana use. Taking a complete history is key to making the diagnosis.
In 2004, Allen et al1 first defined cannabinoid hyperemesis as excessive marijuana use associated with cyclical vomiting and abdominal pain.1 In 2012, Simonetto et al10 proposed diagnostic criteria (Table 1). Although not yet validated, these criteria are based on the largest series of cases of cannabinoid hyperemesis syndrome to date (98 patients).10
THE THREE PHASES OF CANNABINOID HYPEREMESIS
The clinical presentation of cannabinoid hyperemesis syndrome can be divided into three phases: prodromal, vomiting, and resolution.
Prodromal phase
During this phase, patients often appear anxious and agitated and display a spectrum of autonomic symptoms such as sweating, flushing, and constantly sipping water due to thirst. They may sometimes have abdominal pain that is usually epigastric but may also be diffuse. Their symptoms are associated with severe nausea, usually early in the morning or when they see or smell food. Appetite and eating patterns remain normal. Compulsive hot bathing or showering is minimal at this phase.
Vomiting phase
In this next phase, patients experience incapacitating nausea and vomiting that may occur without warning and are resistant to conventional antiemetics such as ondansetron and promethazine.14 However, patients eventually learn that hot baths or showers relieve the symptoms, and this behavior eventually becomes a compulsion. The higher the temperature of the water, the better the effect on symptoms.1 Low-grade pyrexia, excessive thirst, orthostasis, abdominal tenderness, weight loss, and sometimes even superficial skin burns have been reported.1,9,15–18
Recovery phase
During the final phase of cannabinoid hyperemesis syndrome, most patients experience marked resolution of symptoms after 24 to 48 hours of conservative management (bowel rest until symptoms resolve, slowly advancing diet as tolerated, intravenous fluids, and electrolyte monitoring and repletion as necessary), and most importantly, cessation of cannabis use. However, the time from cessation of marijuana use to resolution of symptoms may be as long as 1 week to 1 month.1,10,14 Patients begin to resume their normal diet and daily activities. The bathing-showering compulsion subsides, and patients regain lost weight after 3 to 6 months.1
In all case series and reports, resumption of cannabis use causes the symptoms to recur. This recurrence is compelling evidence that cannabis is the cause of the hyperemesis and should be part of the essential criteria for the diagnosis of cannabinoid hyperemesis syndrome.
WHY COMPULSIVE HOT BATHING?
The mechanism behind this unique characteristic of cannabinoid hyperemesis syndrome is not known. Several theories have been suggested, but no study has identified the exact explanation for this phenomenon.1,9,10,13–15,17–31
One suggested mechanism is a response by the thermoregulatory center of the brain to the dose-dependent hypothermic effects of THC, or even a direct effect of CB1 receptor activation in the hypothalamus.9 Cannabis toxicity could disrupt the equilibrium of satiety, thirst, digestive, and thermoregulatory systems of the hypothalamus, and this interference could resolve with hot bathing.1
The so-called “cutaneous steal” syndrome has also been proposed, in which cutaneous vasodilation caused by hot water decreases the blood volume available for the splanchnic circulation thought to be responsible for the abdominal pain and vomiting.13 The compulsive hot bathing may also be a response by the brain to the anxiety or psychological stress induced by severe nausea and vomiting.14
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of cannabinoid hyperemesis syndrome includes mainly cyclic vomiting syndrome and psychogenic vomiting. A careful history is useful, as is ruling out medication-induced reactions, toxins, pregnancy, and gastrointestinal, neurologic, metabolic, and endocrine causes. All three of these vomiting syndromes can present with a cyclic pattern of nausea and vomiting. Cannabis use is common in all three and so is not helpful in differentiating them. But the characteristic compulsive hot bathing and showering is unique and pathognomonic of cannabinoid hyperemesis syndrome.32
Endoscopic examination may reveal esophagitis and gastritis from severe bouts of retching.26
Cyclic vomiting syndrome
The Rome III criteria for the diagnosis of cyclic vomiting syndrome include three or more stereotypic episodes of acute-onset nausea and vomiting lasting less than 1 week, alternating with intervals of completely normal health. The criteria should be fulfilled for the previous 3 months with symptom onset at least 6 months before diagnosis.33
In a series of 17 patients with adult-onset cyclic vomiting syndrome,18 the average age at onset was 30, and 13 (76%) of the patients were women. Fifteen (88%) of the patients experienced a prodrome or aura of abdominal pain or headache, and in this group, a trigger such as emotional stress and infection could also be identified in 9 (60%).
Unlike in cannabinoid hyperemesis syndrome, most patients with cyclic vomiting syndrome have a family history of migraine headache, and the prevalence of psychological stressors is high.31 Also, patients with cannabinoid hyperemesis syndrome do not respond to medications that usually abort migraine episodes,15 whereas patients with cyclic vomiting syndrome, especially those who have a family history of migraines, may respond to antimigraine medications such as triptans. There is evidence of clinical psychological overlap between cyclic vomiting syndrome, abdominal migraine, and migraine headaches. Some authors recommend antimigraine therapy even in the absence of a family or personal history of migraine if, after a careful history and physical examination, the diagnosis of cyclic vomiting syndrome seems likely. Moreover, nonmedical management such as sleep, dark rooms, and quiet environment are not as effective in cannabinoid hyperemesis syndrome as they are in cyclic vomiting syndrome.18
Psychogenic vomiting
Psychogenic vomiting is classically defined as vomiting caused by psychological mechanisms without any obvious organic cause.13 It occurs most commonly in patients with major depressive disorder or conversion disorder.34 The mechanism appears to be a combination of past organic or gastrointestinal functional abnormalities and emotional problems, and multiple patterns of vomiting can occur. Most of these patients can be treated with behavioral therapy, antidepressant drug therapy, and supportive psychotherapy.34,35
ASKING A SERIES OF QUESTIONS
Most patients with cannabinoid hyperemesis syndrome have a history of frequent visits to emergency departments or clinics for persistent nausea and vomiting, and they may have undergone extensive diagnostic workups to exclude structural, inflammatory, infectious, and functional diseases of the bowel.23,24
To prevent unnecessary testing and use of healthcare resources, Wallace et al32 proposed an algorithm to help guide clinicians in diagnosing and treating patients with suspected cannabinoid hyperemesis syndrome. A patient presenting with severe nausea and vomiting should prompt a series of questions:
Do the signs and symptoms suggest a severe underlying medical cause? If so, this should be pursued.
Do symptoms improve while taking a hot shower or bath? If not, pursue an appropriate diagnostic evaluation and treatment for conditions other than cannabinoid hyperemesis syndrome.
Is the bathing compulsive? If not, consider other diagnoses, but remain suspicious about cannabinoid hyperemesis syndrome.
Does the patient currently use cannabis daily or almost daily, and has the patient done so for at least the past year? If the patient denies using cannabis, a urine drug screen for THC may be useful. If the patient admits to use, a presumptive diagnosis of cannabinoid hyperemesis syndrome can be made.
Does the patient have signs or symptoms of volume depletion, or is the patient unable to tolerate oral hydration? Encourage oral hydration or provide intravenous hydration, and provide cannabis cessation counseling.
Do the symptoms improve? If yes, great! Provide cessation counseling, resources, and follow-up. If not:
Is the patient still using cannabis? If not, it is time to rethink the diagnosis.
Treatment in the acute setting is supportive and includes intravenous hydration and correction of electrolytes. Conventional antiemetics such as ondansetron, metoclopramide, prochlorperazine, and promethazine have not been effective in relieving hyperemesis.9,12,14 This implies that the mechanism of emesis likely does not involve dopaminergic and serotonin pathways in the central and autonomic nervous systems.
Cessation of cannabis use is key for long-term resolution of symptoms. Efforts should be made to provide counseling and encourage patients to stop using the drug entirely (Figure 1).
SOMETHING TO THINK ABOUT
With the high prevalence of chronic cannabis abuse and the recent legalization of recreational marijuana use, we will all likely encounter a patient with cannabinoid hyperemesis. With adequate knowledge of this phenomenon, we can avoid unnecessary workups and inappropriate medical and surgical treatment in patients presenting with recurrent vomiting of unknown cause. The diagnosis can easily be made by simply asking for a history of chronic marijuana use and symptoms related to cannabinoid hyperemesis syndrome, such as relief of symptoms with hot baths or showers and with marijuana cessation.
Conservative management and fluid resuscitation is important in the acute setting, but cessation of marijuana use and follow-up counseling are the key components for treating patients with cannabinoid hyperemesis syndrome and for preventing recurrence.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Motel S. 6 facts about marijuana. Factank. News in the Numbers Pew Research Center. www.pewresearch.org/fact-tank/2015/04/14/6-facts-about-marijuana/. Accessed June 2, 2015.
- Walsh D, Nelson KA, Mahmoud FA. Established and potential therapeutic applications of cannabinoids in oncology. Support Care Cancer 2003; 11:137–143.
- Tramèr MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ 2001; 323:16–21.
- Davis M, Maida V, Daeninck P, Pergolizzi J. The emerging role of cannabinoid neuromodulators in symptom management. Support Care Cancer 2007; 15:63–71.
- National Institutes of Health (NIH). National Institute on Drug Abuse. Drug facts: marijuana. www.nida.nih.gov/infofacts/marijuana. Accessed April 29, 2015.
- Vaziri ND, Thomas R, Sterling M, et al. Toxicity with intravenous injection of crude marijuana extract. Clin Toxicol 1981; 18:353–366.
- Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992; 258:1946–1949.
- Chang YH, Windish DM. Cannabinoid hyperemesis relieved by compulsive bathing. Mayo Clin Proc 2009; 84:76–78.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Darmani NA. The potent emetogenic effects of the endocannabinoid, 2-AG (2-arachidonoylglycerol) are blocked by delta(9)-tetrahydrocannabinol and other cannnabinoids. J Pharmacol Exp Ther 2002; 300:34–42.
- Darmani NA, Sim-Selley LJ, Martin BR, et al. Antiemetic and motor-depressive actions of CP55,940: cannabinoid CB1 receptor characterization, distribution, and G-protein activation. Eur J Pharmacol 2003; 459:83–95.
- Leibovich MA. Psychogenic vomiting. Psychotherapeutic considerations. Psychother Psychosom 1973; 22:263–268.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Sontineni SP, Chaudhary S, Sontineni V, Lanspa SJ. Cannabinoid hyperemesis syndrome: clinical diagnosis of an underrecognised manifestation of chronic cannabis abuse. World J Gastroenterol 2009; 15:1264–1266.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Price SL, Fisher C, Kumar R, Hilgerson A. Cannabinoid hyperemesis syndrome as the underlying cause of intractable nausea and vomiting. J Am Osteopath Assoc 2011; 111:166–169.
- Lee LY, Abbott L, Moodie S, Anderson S. Cyclic vomiting syndrome in 28 patients: demographics, features and outcomes. Eur J Gastroenterol Hepatol 2012; 24:939–943.
- Wallace D, Martin AL, Park B. Cannabinoid hyperemesis: marijuana puts patients in hot water. Australas Psychiatry 2007; 15:156–158.
- Ashton CH. Adverse effects of cannabis and cannabinoids. Br J Anaesth 1999; 83:637–649.
- Cota D, Steiner MA, Marsicano G, et al. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 2007; 148:1574–1581.
- McCallum RW, Soykan I, Sridhar KR, Ricci DA, Lange RC, Plankey MW. Delta-9-tetrahydrocannabinol delays the gastric emptying of solid food in humans: a double-blind, randomized study. Aliment Pharmacol Ther 1999; 13:77–80.
- Donnino MW, Cocchi MN, Miller J, Fisher J. Cannabinoid hyperemesis: a case series. J Emerg Med 2011; 40:e63–e66.
- Singh E, Coyle W. Cannabinoid hyperemesis. Am J Gastroenterol 2008; 103:1048–1049.
- Carnett JB. Intercostal neuralgia as a cause of abdominal pain and tenderness. Surg Gynecol Obstet 1926; 42:625–632.
- Patterson DA, Smith E, Monahan M, et al. Cannabinoid hyperemesis and compulsive bathing: a case series and paradoxical pathophysiological explanation. J Am Board Fam Med 2010; 23:790–793.
- Izzo AA, Camilleri M. Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut 2008; 57:1140–1155.
- Pertwee RG. Cannabinoids and the gastrointestinal tract. Gut 2001; 48:859–867.
- Choung RS, Locke GR 3rd, Lee RM, Schleck CD, Zinsmeister AR, Talley NJ. Cyclic vomiting syndrome and functional vomiting in adults: association with cannabinoid use in males. Neurogastroenterol Motil 2012; 24:20–26,e1.
- Nicolson SE, Denysenko L, Mulcare JL, Vito JP, Chabon B. Cannabinoid hyperemesis syndrome: a case series and review of previous reports. Psychosomatics 2012; 53:212–219.
- Miller JB, Walsh M, Patel PA, et al. Pediatric cannabinoid hyperemesis: two cases. Pediatr Emerg Care 2010; 26:919–920.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Tack J, Taley NJ, Camilleri M, et al. Functional gastroduodenal disorders. Gastroenterology 2006; 130:1466–1479.
- Muraoka M, Mine K, Matsumoto K, Nakai Y, Nakagawa T. Psychogenic vomiting: the relation between patterns of vomiting and psychiatric diagnoses. Gut 1990; 31:526–528.
- Stravynski A. Behavioral treatment of psychogenic vomiting in the context of social phobia. J Nerv Ment Dis 1983; 171:448–451.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Motel S. 6 facts about marijuana. Factank. News in the Numbers Pew Research Center. www.pewresearch.org/fact-tank/2015/04/14/6-facts-about-marijuana/. Accessed June 2, 2015.
- Walsh D, Nelson KA, Mahmoud FA. Established and potential therapeutic applications of cannabinoids in oncology. Support Care Cancer 2003; 11:137–143.
- Tramèr MR, Carroll D, Campbell FA, Reynolds DJ, Moore RA, McQuay HJ. Cannabinoids for control of chemotherapy induced nausea and vomiting: quantitative systematic review. BMJ 2001; 323:16–21.
- Davis M, Maida V, Daeninck P, Pergolizzi J. The emerging role of cannabinoid neuromodulators in symptom management. Support Care Cancer 2007; 15:63–71.
- National Institutes of Health (NIH). National Institute on Drug Abuse. Drug facts: marijuana. www.nida.nih.gov/infofacts/marijuana. Accessed April 29, 2015.
- Vaziri ND, Thomas R, Sterling M, et al. Toxicity with intravenous injection of crude marijuana extract. Clin Toxicol 1981; 18:353–366.
- Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992; 258:1946–1949.
- Chang YH, Windish DM. Cannabinoid hyperemesis relieved by compulsive bathing. Mayo Clin Proc 2009; 84:76–78.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Darmani NA. The potent emetogenic effects of the endocannabinoid, 2-AG (2-arachidonoylglycerol) are blocked by delta(9)-tetrahydrocannabinol and other cannnabinoids. J Pharmacol Exp Ther 2002; 300:34–42.
- Darmani NA, Sim-Selley LJ, Martin BR, et al. Antiemetic and motor-depressive actions of CP55,940: cannabinoid CB1 receptor characterization, distribution, and G-protein activation. Eur J Pharmacol 2003; 459:83–95.
- Leibovich MA. Psychogenic vomiting. Psychotherapeutic considerations. Psychother Psychosom 1973; 22:263–268.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Sontineni SP, Chaudhary S, Sontineni V, Lanspa SJ. Cannabinoid hyperemesis syndrome: clinical diagnosis of an underrecognised manifestation of chronic cannabis abuse. World J Gastroenterol 2009; 15:1264–1266.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Price SL, Fisher C, Kumar R, Hilgerson A. Cannabinoid hyperemesis syndrome as the underlying cause of intractable nausea and vomiting. J Am Osteopath Assoc 2011; 111:166–169.
- Lee LY, Abbott L, Moodie S, Anderson S. Cyclic vomiting syndrome in 28 patients: demographics, features and outcomes. Eur J Gastroenterol Hepatol 2012; 24:939–943.
- Wallace D, Martin AL, Park B. Cannabinoid hyperemesis: marijuana puts patients in hot water. Australas Psychiatry 2007; 15:156–158.
- Ashton CH. Adverse effects of cannabis and cannabinoids. Br J Anaesth 1999; 83:637–649.
- Cota D, Steiner MA, Marsicano G, et al. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 2007; 148:1574–1581.
- McCallum RW, Soykan I, Sridhar KR, Ricci DA, Lange RC, Plankey MW. Delta-9-tetrahydrocannabinol delays the gastric emptying of solid food in humans: a double-blind, randomized study. Aliment Pharmacol Ther 1999; 13:77–80.
- Donnino MW, Cocchi MN, Miller J, Fisher J. Cannabinoid hyperemesis: a case series. J Emerg Med 2011; 40:e63–e66.
- Singh E, Coyle W. Cannabinoid hyperemesis. Am J Gastroenterol 2008; 103:1048–1049.
- Carnett JB. Intercostal neuralgia as a cause of abdominal pain and tenderness. Surg Gynecol Obstet 1926; 42:625–632.
- Patterson DA, Smith E, Monahan M, et al. Cannabinoid hyperemesis and compulsive bathing: a case series and paradoxical pathophysiological explanation. J Am Board Fam Med 2010; 23:790–793.
- Izzo AA, Camilleri M. Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut 2008; 57:1140–1155.
- Pertwee RG. Cannabinoids and the gastrointestinal tract. Gut 2001; 48:859–867.
- Choung RS, Locke GR 3rd, Lee RM, Schleck CD, Zinsmeister AR, Talley NJ. Cyclic vomiting syndrome and functional vomiting in adults: association with cannabinoid use in males. Neurogastroenterol Motil 2012; 24:20–26,e1.
- Nicolson SE, Denysenko L, Mulcare JL, Vito JP, Chabon B. Cannabinoid hyperemesis syndrome: a case series and review of previous reports. Psychosomatics 2012; 53:212–219.
- Miller JB, Walsh M, Patel PA, et al. Pediatric cannabinoid hyperemesis: two cases. Pediatr Emerg Care 2010; 26:919–920.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Tack J, Taley NJ, Camilleri M, et al. Functional gastroduodenal disorders. Gastroenterology 2006; 130:1466–1479.
- Muraoka M, Mine K, Matsumoto K, Nakai Y, Nakagawa T. Psychogenic vomiting: the relation between patterns of vomiting and psychiatric diagnoses. Gut 1990; 31:526–528.
- Stravynski A. Behavioral treatment of psychogenic vomiting in the context of social phobia. J Nerv Ment Dis 1983; 171:448–451.
KEY POINTS
- The prodromal phase is characterized by severe anxiety and agitation. Patients display a spectrum of autonomic symptoms such as sweating, flushing, constantly sipping water due to thirst, and colicky abdominal pain.
- In the second phase, patients develop incapacitating nausea and vomiting that may occur without warning and is usually resistant to conventional antiemetics such as ondansetron and promethazine. During this phase, patients learn the immediate relieving effects of taking hot baths.
- After 24 to 48 hours of conservative management, intravenous fluid replacement, and, most importantly, cessation of cannabis use, patients experience marked resolution of symptoms. The compulsive hot-bathing behavior subsides. However, eventually, patients go back to using marijuana, and the cycle of symptoms recurs.

Bell palsy: Clinical examination and management
Bell palsy is an idiopathic peripheral nerve disorder involving the facial nerve (ie, cranial nerve VII) and manifesting as acute, ipsilateral facial muscle weakness. It is named after Sir Charles Bell, who in 1821 first described the anatomy of the facial nerve.1 Although the disorder is clinically benign, patients can be devastated by its disfigurement.
The annual incidence of Bell palsy is 20 per 100,000, with no predilection for sex or ethnicity. It can affect people at any age, but the incidence is slightly higher after age 40.2,3 Risk factors include diabetes, pregnancy, severe preeclampsia, obesity, and hypertension.4–7
THE FACIAL NERVE IS VULNERABLE TO TRAUMA AND COMPRESSION
A basic understanding of the neuroanatomy of the facial nerve provides clues for distinguishing a central lesion from a peripheral lesion. This differentiation is important because the causes and management differ.
The facial nerve is a mixed sensory and motor nerve, carrying fibers involved in facial expression, taste, lacrimation, salivation, and sensation of the ear. It originates in the lower pons and exits the brainstem ventrally at the pontomedullary junction. After entering the internal acoustic meatus, it travels 20 to 30 mm in the facial canal, the longest bony course of any cranial nerve, making it highly susceptible to trauma and compression by edema.8
In the facial canal, it makes a posterior and inferior turn, forming a bend (ie, the genu of the facial nerve). The genu is proximal to the geniculate ganglion, which contains the facial nerve’s primary sensory neurons for taste and sensation. The motor branch of the facial nerve then exits the cranium via the stylomastoid foramen and passes through the parotid gland, where it divides into temporofacial and cervicofacial trunks.9
The facial nerve has five terminal branches that innervate the muscles of facial expression:
- The temporal branch (muscles of the forehead and superior part of the orbicularis oculi)
- The zygomatic branch (muscles of the nasolabial fold and cheek, eg, nasalis and zygomaticus).
- The buccal branch (the buccinators and inferior part of the orbicularis oculi)
- The marginal mandibular branch (the depressors of the mouth, eg, depressor anguli and mentalis)
- The cervical branch (the platysma muscle).
INFLAMMATION IS BELIEVED TO BE RESPONSIBLE
Although the precise cause of Bell palsy is not known, one theory is that inflammation of the nerve causes focal edema, demyelination, and ischemia. Several studies have suggested that herpes virus simplex type 1 infection may be involved.10
FACIAL DROOPING, EYELID WEAKNESS, OTHER SYMPTOMS
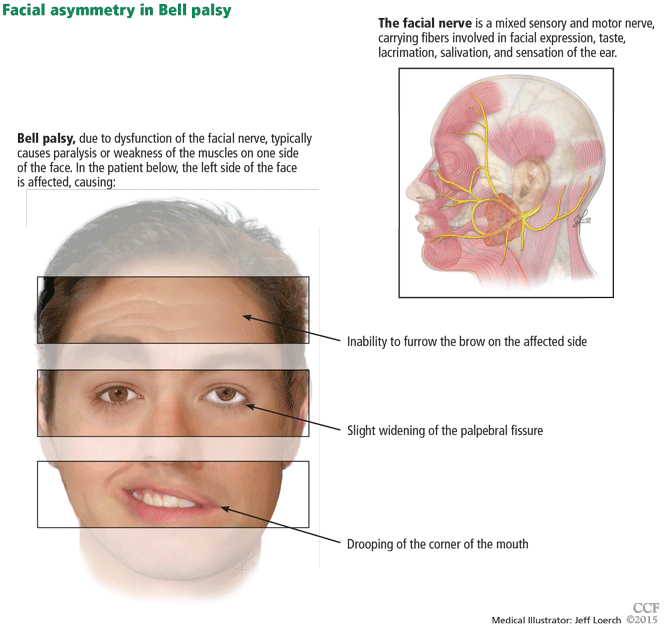
Symptoms of Bell palsy include ipsilateral sagging of the eyebrow, drooping of the face, flattening of the nasolabial fold, and inability to fully close the eye, pucker the lips, or raise the corner of the mouth (Figure 1). Symptoms develop within hours and are maximal by 3 days.
About 70% of patients have associated ipsilateral pain around the ear. If facial pain is present with sensory and hearing loss, a tumor of the parotid gland or viral otitis must be considered.11 Other complaints may include hyperacusis due to disruption of nerve fibers to the stapedius muscle, changes in taste, and dry eye from parasympathetic dysfunction. Some patients report paresthesias over the face, which most often represent motor symptoms misconstrued as sensory changes.
PHYSICAL EXAMINATION
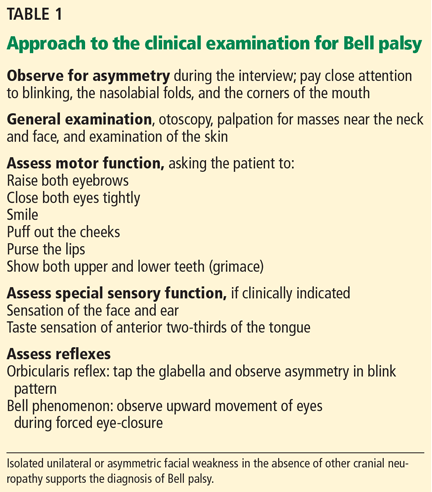
The clinical examination should include a complete neurologic and general examination, including otoscopy and attention to the skin and parotid gland. Vesicles or scabbing around the ear should prompt testing for herpes zoster. Careful observation during the interview while the patient is talking may reveal subtle signs of weakness and provide additional clues.
A systematic approach to the assessment of a patient with suspected Bell palsy is recommended (Table 1) and outlined below:
Does the patient have peripheral facial palsy?
In Bell palsy, wrinkling of the forehead on the affected side when raising the eyebrows is either asymmetrical or absent.
If the forehead muscles are spared and the lower face is weak, this signifies a central lesion such as a stroke or other structural abnormality and not a peripheral lesion of the facial nerve (eg, Bell palsy).
Can the patient close the eyes tightly?
Normally, the patient should be able to close both eyes tightly, and the eyelashes should be buried between the eyelids. In Bell palsy, when the patient attempts to close the eyes, the affected side shows incomplete closure and the eye may remain partly open.
Assess the strength of the orbicularis oculi by trying to open the eyes. The patient who is attempting to close the eyelids tightly but cannot will demonstrate the Bell phenomenon, ie, the examiner is able to force open the eyelids, and the eyes are deviated upward and laterally.
Closely observe the blink pattern, as the involved side in Bell palsy may slightly lag behind the normal eye, and the patient may be unable to close the eye completely.
Is the smile symmetric?
Note flattening of the nasolabial fold on one side, which indicates facial weakness.
Can the patient puff out the cheeks?
Ask the patient to hold air in the mouth against resistance. This assesses the strength of the buccinator muscle.
Can the patient purse the lips?
Ask the patient to pucker or purse the lips and observe for asymmetry or weakness on the affected side.
Test the orbicularis oris muscle by trying to spread the lips apart while the patient resists, and observe for weakness on one side.
Is there a symmetric grimace?
This will test the muscles involved in depressing the angles of the mouth and platysma.
Are taste, sensation, and hearing intact?
Other testable functions of the facial nerve, including taste, sensation, and hearing, do not always need to be assessed but can be in patients with specific sensory deficits.
Abnormalities in taste can support localization of the problem either proximal or distal to the branch point of fibers mediating taste. The facial nerve supplies taste fibers to the anterior two-thirds of the tongue. Sweet and salty taste can be screened with sugar and salt. Tell the patient to close the eyes, and using a tongue blade, apply a small amount of sugar or salt on the side of the tongue. Ask the patient to identify the taste and repeat with the other sample after he or she has rinsed the mouth.
Somatic sensory fibers supplied by the facial nerve innervate the inner ear and a small area behind the ear, but these may be difficult to assess objectively. Formal audiologic testing may be needed if hearing is impaired.
Facial nerve reflexes
A number of facial reflexes can be tested, including the orbicularis oculi, palpebral-oculogyric, and corneal reflexes.12
The orbicularis oculi reflex is tested by gentle finger percussion of the glabella while observing for involuntary blinking with each stimulus. The afferent branch of this reflex is carried by the trigeminal nerve, while the efferent response is carried by the facial nerve. In peripheral facial nerve palsy, this reflex is weakened or absent on the affected side.
The palpebral-oculogyric reflex, or Bell phenomenon, produces upward and lateral deviation of the eyes when attempting forceful eyelid closure. In this reflex, the afferent fibers are carried by the facial nerve and the efferent fibers travel in the oculomotor nerve to the superior rectus muscle. In Bell palsy, this reflex is visible because of failure of adequate eyelid closure.
The corneal reflex is elicited by stimulating the cornea with a wisp of cotton, causing reflexive closure of the both eyes. The affected side may show slowed or absent lid closure when tested on either side. The sensory afferent fibers are carried by the trigeminal nerve, and the motor efferent fibers are carried by the facial nerve.
Grading of facial paralysis
The House-Brackmann scale is the most widely used tool for grading the degree of facial paralysis and for predicting recovery. Grades are I to VI, with grade I indicating normal function, and grade VI, complete paralysis.
Patients with some preserved motor function generally have good recovery, but those with complete paralysis may have long-term residual deficits.13
A DIAGNOSIS OF EXCLUSION

The diagnosis of Bell palsy is made by excluding other causes of unilateral facial paralysis, and 30% to 60% of cases of facial palsy are caused by an underlying disorder that mimics Bell palsy, including central nervous system lesion (eg, stroke, demyelinating disease), parotid gland tumor, Lyme disease, Ramsay Hunt syndrome, granulomatous disease, otitis media, cholesteatoma, diabetes, trauma, and Guillain-Barré syndrome (Table 2).14,15 Many of these conditions have associated features that help distinguish them from Bell palsy. Facial palsy that does not improve after 3 weeks should prompt referral to a neurologist.
Brain lesions
It is uncommon to have isolated facial palsy with a cortical or subcortical brain lesion, since the corticobulbar and corticospinal tracts travel in close proximity. Cortical signs such as hemiparesis, hemisensory loss, neglect, and dysarthria suggest a lesion of the cerebral cortex. Additionally, forehead muscle sparing is expected in supranuclear lesions.
Brainstem lesions can manifest with multiple ipsilateral cranial nerve palsies and contralateral limb weakness. Sarcoidosis and leptomeningeal carcinomatosis tend to involve the skull base and present with multiple cranial neuropathies.
Tumors of the brain or parotid gland have an insidious onset and may cause systemic signs such as fevers, chills, and weight loss. Headache, seizures, and hearing loss indicate an intracranial lesion. A palpable mass near the ear, neck, or parotid gland requires imaging of the face to look for a parotid gland tumor.
Infection
A number of infections can cause acute facial paralysis. The most common is herpes simplex virus, and the next most common is varicella zoster.14 Herpes simplex virus, Ramsay Hunt syndrome, and Lyme disease may have associated pain and skin changes. Erythema of the tympanic membrane suggests otitis media, especially in the setting of ear pain and hearing loss.
Ramsay Hunt syndrome is caused by reactivation of the herpes zoster virus from the geniculate ganglion, affecting the facial nerve. Careful examination of the ear canal and the oropharynx may show vesicles.
In Lyme disease, facial palsy is the most common cranial neuropathy, seen in 50% to 63% of patients with Borrelia burgdorferi meningitis.16,17 In people with a history of rash, arthralgia, tick bite, or travel to an endemic region, Lyme titers should be checked before starting the patient on corticosteroids.
Bilateral facial palsy is rare and occurs in fewer than 1% of patients. It has been reported in patients with Lyme disease, Guillain-Barré syndrome, sarcoidosis, diabetes mellitus, viral infection, and pontine glioma.18
DIAGNOSTIC EVALUATION
Serologic testing, electrodiagnostic studies, and imaging are not routinely necessary to diagnose Bell palsy. However, referral to the appropriate specialist (neurologist, otolaryngologist, optometrist, ophthalmologist) is advised if the patient has sparing of the forehead muscle, multiple cranial neuropathies, signs of infection, or persistent weakness without significant improvement at 3 weeks.
Laboratory testing
A complete blood cell count with differential may point to infection or a lymphoproliferative disorder. When indicated, screening for diabetes mellitus with fasting blood glucose or hemoglobin A1c may be helpful. In Lyme-endemic regions, patients should undergo an enzyme-linked immunosorbent assay or an indirect fluorescent antibody test to screen for the disease. If positive, the diagnosis of Lyme disease should be confirmed by Western blot. If vesicles are present on examination, check serum antibodies for herpes zoster. In the appropriate clinical setting, angiotensin-converting enzyme, human immunodeficiency virus, and inflammatory markers can be tested.
Cerebrospinal fluid analysis is generally not helpful in diagnosing Bell palsy but can differentiate it from Guillain-Barré syndrome, leptomeningeal carcinomatosis, and infection involving the central nervous system.
Imaging
Imaging is not recommended in the initial evaluation of Bell palsy unless symptoms and the examination are atypical. From 5% to 7% of cases of facial palsy are caused by a tumor (eg, facial neuroma, cholesteatoma, hemangioma, meningioma), whether benign or malignant.14,15 Therefore, in patients with insidious onset of symptoms that do not improve in about 3 weeks, contrast-enhanced computed tomography or gadolinium-enhanced magnetic resonance imaging of the internal auditory canal and face is warranted.
Electrodiagnostic studies
Electrodiagnostic testing is typically not part of the evaluation of acute Bell palsy, but in patients with complete paralysis, it may help assess the degree of nerve injury and the chances of recovery, especially since patients with complete paralysis have a higher risk of incomplete recovery.19 Electrodiagnostic studies should be performed at least 1 week after symptom onset to avoid false-negative results.
TREATMENT
The treatment of Bell palsy focuses on maximizing recovery and minimizing associated complications.
Protect the eyes
Patients who cannot completely close their eyes should be given instructions on ocular protective care to prevent exposure keratopathy. Frequent application of lubricant eyedrops with artificial tears during the day or ophthalmic ointment at bedtime is recommended. The physician should also recommend protective eyewear such as sunglasses during the day. Eye patching or taping at night may be useful but could be harmful if applied too loosely or too tightly. Patients with vision loss or eye irritation should be referred to an ophthalmologist.19
Corticosteroids are recommended in the first 72 hours
In two randomized clinical trials (conducted by Sullivan et al20 in 511 patients and Engström et al21 in 829 patients), prednisolone was found to be beneficial if started within 72 hours of symptom onset.
In a double-blind, randomized, placebo-controlled study of prednisone in 58 patients, those who received the drug recovered faster, although long-term outcomes in these patients were not significantly different than those in the control group.22 The American Academy of Neurology23 rated this study as class II, ie, not meeting all of its criteria for the highest level of evidence, class I. Nevertheless, although prednisone lacks class I evidence, its use is recommended because it is a precursor to its active metabolite, prednisolone, which has been studied extensively.
The current guidelines of the American Academy of Neurology, updated in 2012, state, “For patients with new-onset Bell palsy, steroids are highly likely to be effective and should be offered to increase the probability of recovery of facial nerve function”23 (level A evidence, ie, established as effective). They also concluded that adverse effects of corticosteroids were generally minor and temporary.

Similarly, the guidelines of the American Academy of Otolaryngology–Head and Neck Surgery, published in 2013, recommend oral corticosteroids within 72 hours of onset of symptoms of Bell palsy for patients age 16 and older.19 The recommendation is for a 10-day course of corticosteroids with at least 5 days at a high dose (prednisolone 50 mg orally daily for 10 days, or prednisone 60 mg orally daily for 5 days, followed by a 5-day taper). The benefit of corticosteroids after 72 hours is unclear (Table 3).19
Even though the guidelines recommend corticosteroids, the decision to use them in diabetic patients and pregnant women should be individualized. Discretion is advised, as not all patients with Bell palsy need to be treated. Most recover spontaneously, especially those with mild symptoms.
Antiviral therapy may offer modest benefit
Antiviral therapy has not been shown to be beneficial in Bell palsy, and current guidelines do not recommend oral antiviral therapy alone.19 However, an antiviral combined with a corticosteroid may offer modest benefit if started within 72 hours of symptom onset (level C evidence, ie, possibly effective).23 Patients starting antiviral therapy should understand that its benefit has not been established.
Surgical decompression remains controversial
A Cochrane systematic review in 2011 found insufficient evidence regarding the safety and efficacy of surgical intervention in Bell palsy.24 Surgery should be considered only for patients with complete paralysis with a greater than 90% reduction in motor amplitude on a nerve conduction study compared with the unaffected side, and absent volitional activity on needle examination.19,25
Acupuncture: No recommendation
Currently, there is no recommendation for acupuncture in the treatment of Bell palsy.19 A recent randomized clinical trial suggests benefit from acupuncture combined with corticosteroids,26 but high-quality studies to support its use are lacking.26
Physical therapy: Insufficient evidence
There is insufficient evidence to show that physical therapy has benefit—or harm—in Bell palsy. However, some low-quality studies indicated that facial exercises and mime therapy may improve function in patients with moderate paralysis.27
Follow-up
Patients should be instructed to call at 2 weeks to report progress of symptoms and to be reevaluated within or at 1 month, with close attention to facial weakness and eye irritation. Further evaluation is needed if there has been no improvement, if symptoms have worsened, or if new symptoms have appeared.
The psychosocial impact of Bell palsy cannot be discounted, as the disfigurement can have negative implications for self-esteem and social relationships. Appropriate referral to an ophthalmologist, neurologist, otolaryngologist, social worker, or a plastic surgeon may be necessary.
COMPLICATIONS AND PROGNOSIS
Most patients with Bell palsy recover completely, but up to 30% have residual symptoms at 6 months.14,20 Furthermore, although Bell palsy usually has a monophasic course, 7% to 12% of patients have a recurrence.3,15
Long-term complications can include residual facial weakness, facial synkinesis, facial contracture, and facial spasm.14,28 Incomplete eye closure may benefit from surgery (tarsorrhaphy or gold-weight implantation) to prevent corneal ulceration. Facial synkinesis is due to aberrant nerve regeneration and occurs in 15% to 20% of patients after recovery from Bell palsy.29 Patients may describe tearing while chewing (“crocodile tears”), involuntary movement of the corners of the mouth with blinking, or ipsilateral eye-closing when the jaw opens (“jaw-winking”). Facial contracture, facial synkinesis, and facial spasm can be treated with botulinum toxin injection.30
- Grzybowski A, Kaufman MH. Sir Charles Bell (1774-1842): contributions to neuro-ophthalmology. Acta Ophthalmol Scand 2007; 85:897–901.
- De Diego-Sastre JI, Prim-Espada MP, Fernández-García F. The epidemiology of Bell’s palsy. Rev Neurol 2005; 41:287–290. In Spanish.
- Morris AM, Deeks SL, Hill MD, et al. Annualized incidence and spectrum of illness from an outbreak investigation of Bell’s palsy. Neuroepidemiology 2002; 21:255–261.
- Bosco D, Plastino M, Bosco F, et al. Bell’s palsy: a manifestation of prediabetes? Acta Neurol Scand 2011; 123:68–72.
- Riga M, Kefalidis G, Danielides V. The role of diabetes mellitus in the clinical presentation and prognosis of Bell palsy. J Am Board Fam Med 2012; 25:819–826.
- Hilsinger RL Jr, Adour KK, Doty HE. Idiopathic facial paralysis, pregnancy, and the menstrual cycle. Ann Otol Rhinol Laryngol 1975; 84:433–442.
- Savadi-Oskouei D, Abedi A, Sadeghi-Bazargani H. Independent role of hypertension in Bell’s palsy: a case-control study. Eur Neurol 2008; 60:253–257.
- Murai A, Kariya S, Tamura K, et al. The facial nerve canal in patients with Bell’s palsy: an investigation by high-resolution computed tomography with multiplanar reconstruction. Eur Arch Otorhinolaryngol 2013; 270:2035–2038.
- Blumenfeld H. Neuroanatomy Through Clinical Cases. 1st ed. Sunderland, MA: Sinauer; 2002:479–484.
- Murakami S, Mizobuchi M, Nakashiro Y, Doi T, Hato N, Yanagihara N. Bell palsy and herpes simplex virus: identification of viral DNA in endoneurial fluid and muscle. Ann Intern Med 1996; 124:27–30.
- Boahene DO, Olsen KD, Driscoll C, Lewis JE, McDonald TJ. Facial nerve paralysis secondary to occult malignant neoplasms. Otolaryngol Head Neck Surg 2004; 130:459–465.
- DeJong RN. The Neurologic Examination: Incorporating the fundamentals of neuroanatomy and neurophysiology. 4th ed. New York, NY: Harper & Row; 1979:178–198.
- House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg 1985; 93:146–147.
- Peitersen E. Bell’s palsy: the spontaneous course of 2,500 peripheral facial nerve palsies of different etiologies. Acta Otolaryngol Suppl 2002; 549:4–30.
- Hohman MH, Hadlock TA. Etiology, diagnosis, and management of facial palsy: 2000 patients at a facial nerve center. Laryngoscope 2014; 124:E283–E293.
- Ackermann R, Hörstrup P, Schmidt R. Tick-borne meningopolyneuritis (Garin-Bujadoux, Bannwarth). Yale J Biol Med 1984; 57:485–490.
- Pachner AR, Steere AC. The triad of neurologic manifestations of Lyme disease: meningitis, cranial neuritis, and radiculoneuritis. Neurology 1985; 35:47–53.
- Keane JR. Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature. Neurology 1994; 44:1198–1202.
- Baugh RF, Basura GJ, Ishii LE, et al. Clinical practice guideline: Bell’s palsy. Otolaryngol Head Neck Surg 2013; 149(suppl 3):S1–S27.
- Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007; 357:1598–1607.
- Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol 2008; 7:993–1000.
- Lagalla G, Logullo F, Di Bella P, Provinciali L, Ceravolo MG. Influence of early high-dose steroid treatment on Bell’s palsy evolution. Neurol Sci 2002; 23:107–112.
- Gronseth GS, Paduga R; American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012; 79:2209–2213.
- McAllister K, Walker D, Donnan PT, Swan I. Surgical interventions for the early management of Bell’s palsy. Cochrane Database Syst Rev 2011; 2:CD007468.
- Gantz BJ, Rubinstein JT, Gidley P, Woodworth GG. Surgical management of Bell’s palsy. Laryngoscope 1999; 109:1177–1188.
- Xu SB, Huang B, Zhang CY, et al. Effectiveness of strengthened stimulation during acupuncture for the treatment of Bell palsy: a randomized controlled trial. CMAJ 2013; 185:473–479.
- Teixeira LJ, Valbuza JS, Prado GF. Physical therapy for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev 2011; 12:CD006283.
- Yaltho TC, Jankovic J. The many faces of hemifacial spasm: differential diagnosis of unilateral facial spasms. Mov Disord 2011; 26:1582–1592.
- Celik M, Forta H, Vural C. The development of synkinesis after facial nerve paralysis. Eur Neurol 2000; 43:147–151.
- Chua CN, Quhill F, Jones E, Voon LW, Ahad M, Rowson N. Treatment of aberrant facial nerve regeneration with botulinum toxin A. Orbit 2004; 23:213–218.
Bell palsy is an idiopathic peripheral nerve disorder involving the facial nerve (ie, cranial nerve VII) and manifesting as acute, ipsilateral facial muscle weakness. It is named after Sir Charles Bell, who in 1821 first described the anatomy of the facial nerve.1 Although the disorder is clinically benign, patients can be devastated by its disfigurement.
The annual incidence of Bell palsy is 20 per 100,000, with no predilection for sex or ethnicity. It can affect people at any age, but the incidence is slightly higher after age 40.2,3 Risk factors include diabetes, pregnancy, severe preeclampsia, obesity, and hypertension.4–7
THE FACIAL NERVE IS VULNERABLE TO TRAUMA AND COMPRESSION
A basic understanding of the neuroanatomy of the facial nerve provides clues for distinguishing a central lesion from a peripheral lesion. This differentiation is important because the causes and management differ.
The facial nerve is a mixed sensory and motor nerve, carrying fibers involved in facial expression, taste, lacrimation, salivation, and sensation of the ear. It originates in the lower pons and exits the brainstem ventrally at the pontomedullary junction. After entering the internal acoustic meatus, it travels 20 to 30 mm in the facial canal, the longest bony course of any cranial nerve, making it highly susceptible to trauma and compression by edema.8
In the facial canal, it makes a posterior and inferior turn, forming a bend (ie, the genu of the facial nerve). The genu is proximal to the geniculate ganglion, which contains the facial nerve’s primary sensory neurons for taste and sensation. The motor branch of the facial nerve then exits the cranium via the stylomastoid foramen and passes through the parotid gland, where it divides into temporofacial and cervicofacial trunks.9
The facial nerve has five terminal branches that innervate the muscles of facial expression:
- The temporal branch (muscles of the forehead and superior part of the orbicularis oculi)
- The zygomatic branch (muscles of the nasolabial fold and cheek, eg, nasalis and zygomaticus).
- The buccal branch (the buccinators and inferior part of the orbicularis oculi)
- The marginal mandibular branch (the depressors of the mouth, eg, depressor anguli and mentalis)
- The cervical branch (the platysma muscle).
INFLAMMATION IS BELIEVED TO BE RESPONSIBLE
Although the precise cause of Bell palsy is not known, one theory is that inflammation of the nerve causes focal edema, demyelination, and ischemia. Several studies have suggested that herpes virus simplex type 1 infection may be involved.10
FACIAL DROOPING, EYELID WEAKNESS, OTHER SYMPTOMS
Symptoms of Bell palsy include ipsilateral sagging of the eyebrow, drooping of the face, flattening of the nasolabial fold, and inability to fully close the eye, pucker the lips, or raise the corner of the mouth (Figure 1). Symptoms develop within hours and are maximal by 3 days.
About 70% of patients have associated ipsilateral pain around the ear. If facial pain is present with sensory and hearing loss, a tumor of the parotid gland or viral otitis must be considered.11 Other complaints may include hyperacusis due to disruption of nerve fibers to the stapedius muscle, changes in taste, and dry eye from parasympathetic dysfunction. Some patients report paresthesias over the face, which most often represent motor symptoms misconstrued as sensory changes.
PHYSICAL EXAMINATION
The clinical examination should include a complete neurologic and general examination, including otoscopy and attention to the skin and parotid gland. Vesicles or scabbing around the ear should prompt testing for herpes zoster. Careful observation during the interview while the patient is talking may reveal subtle signs of weakness and provide additional clues.
A systematic approach to the assessment of a patient with suspected Bell palsy is recommended (Table 1) and outlined below:
Does the patient have peripheral facial palsy?
In Bell palsy, wrinkling of the forehead on the affected side when raising the eyebrows is either asymmetrical or absent.
If the forehead muscles are spared and the lower face is weak, this signifies a central lesion such as a stroke or other structural abnormality and not a peripheral lesion of the facial nerve (eg, Bell palsy).
Can the patient close the eyes tightly?
Normally, the patient should be able to close both eyes tightly, and the eyelashes should be buried between the eyelids. In Bell palsy, when the patient attempts to close the eyes, the affected side shows incomplete closure and the eye may remain partly open.
Assess the strength of the orbicularis oculi by trying to open the eyes. The patient who is attempting to close the eyelids tightly but cannot will demonstrate the Bell phenomenon, ie, the examiner is able to force open the eyelids, and the eyes are deviated upward and laterally.
Closely observe the blink pattern, as the involved side in Bell palsy may slightly lag behind the normal eye, and the patient may be unable to close the eye completely.
Is the smile symmetric?
Note flattening of the nasolabial fold on one side, which indicates facial weakness.
Can the patient puff out the cheeks?
Ask the patient to hold air in the mouth against resistance. This assesses the strength of the buccinator muscle.
Can the patient purse the lips?
Ask the patient to pucker or purse the lips and observe for asymmetry or weakness on the affected side.
Test the orbicularis oris muscle by trying to spread the lips apart while the patient resists, and observe for weakness on one side.
Is there a symmetric grimace?
This will test the muscles involved in depressing the angles of the mouth and platysma.
Are taste, sensation, and hearing intact?
Other testable functions of the facial nerve, including taste, sensation, and hearing, do not always need to be assessed but can be in patients with specific sensory deficits.
Abnormalities in taste can support localization of the problem either proximal or distal to the branch point of fibers mediating taste. The facial nerve supplies taste fibers to the anterior two-thirds of the tongue. Sweet and salty taste can be screened with sugar and salt. Tell the patient to close the eyes, and using a tongue blade, apply a small amount of sugar or salt on the side of the tongue. Ask the patient to identify the taste and repeat with the other sample after he or she has rinsed the mouth.
Somatic sensory fibers supplied by the facial nerve innervate the inner ear and a small area behind the ear, but these may be difficult to assess objectively. Formal audiologic testing may be needed if hearing is impaired.
Facial nerve reflexes
A number of facial reflexes can be tested, including the orbicularis oculi, palpebral-oculogyric, and corneal reflexes.12
The orbicularis oculi reflex is tested by gentle finger percussion of the glabella while observing for involuntary blinking with each stimulus. The afferent branch of this reflex is carried by the trigeminal nerve, while the efferent response is carried by the facial nerve. In peripheral facial nerve palsy, this reflex is weakened or absent on the affected side.
The palpebral-oculogyric reflex, or Bell phenomenon, produces upward and lateral deviation of the eyes when attempting forceful eyelid closure. In this reflex, the afferent fibers are carried by the facial nerve and the efferent fibers travel in the oculomotor nerve to the superior rectus muscle. In Bell palsy, this reflex is visible because of failure of adequate eyelid closure.
The corneal reflex is elicited by stimulating the cornea with a wisp of cotton, causing reflexive closure of the both eyes. The affected side may show slowed or absent lid closure when tested on either side. The sensory afferent fibers are carried by the trigeminal nerve, and the motor efferent fibers are carried by the facial nerve.
Grading of facial paralysis
The House-Brackmann scale is the most widely used tool for grading the degree of facial paralysis and for predicting recovery. Grades are I to VI, with grade I indicating normal function, and grade VI, complete paralysis.
Patients with some preserved motor function generally have good recovery, but those with complete paralysis may have long-term residual deficits.13
A DIAGNOSIS OF EXCLUSION
The diagnosis of Bell palsy is made by excluding other causes of unilateral facial paralysis, and 30% to 60% of cases of facial palsy are caused by an underlying disorder that mimics Bell palsy, including central nervous system lesion (eg, stroke, demyelinating disease), parotid gland tumor, Lyme disease, Ramsay Hunt syndrome, granulomatous disease, otitis media, cholesteatoma, diabetes, trauma, and Guillain-Barré syndrome (Table 2).14,15 Many of these conditions have associated features that help distinguish them from Bell palsy. Facial palsy that does not improve after 3 weeks should prompt referral to a neurologist.
Brain lesions
It is uncommon to have isolated facial palsy with a cortical or subcortical brain lesion, since the corticobulbar and corticospinal tracts travel in close proximity. Cortical signs such as hemiparesis, hemisensory loss, neglect, and dysarthria suggest a lesion of the cerebral cortex. Additionally, forehead muscle sparing is expected in supranuclear lesions.
Brainstem lesions can manifest with multiple ipsilateral cranial nerve palsies and contralateral limb weakness. Sarcoidosis and leptomeningeal carcinomatosis tend to involve the skull base and present with multiple cranial neuropathies.
Tumors of the brain or parotid gland have an insidious onset and may cause systemic signs such as fevers, chills, and weight loss. Headache, seizures, and hearing loss indicate an intracranial lesion. A palpable mass near the ear, neck, or parotid gland requires imaging of the face to look for a parotid gland tumor.
Infection
A number of infections can cause acute facial paralysis. The most common is herpes simplex virus, and the next most common is varicella zoster.14 Herpes simplex virus, Ramsay Hunt syndrome, and Lyme disease may have associated pain and skin changes. Erythema of the tympanic membrane suggests otitis media, especially in the setting of ear pain and hearing loss.
Ramsay Hunt syndrome is caused by reactivation of the herpes zoster virus from the geniculate ganglion, affecting the facial nerve. Careful examination of the ear canal and the oropharynx may show vesicles.
In Lyme disease, facial palsy is the most common cranial neuropathy, seen in 50% to 63% of patients with Borrelia burgdorferi meningitis.16,17 In people with a history of rash, arthralgia, tick bite, or travel to an endemic region, Lyme titers should be checked before starting the patient on corticosteroids.
Bilateral facial palsy is rare and occurs in fewer than 1% of patients. It has been reported in patients with Lyme disease, Guillain-Barré syndrome, sarcoidosis, diabetes mellitus, viral infection, and pontine glioma.18
DIAGNOSTIC EVALUATION
Serologic testing, electrodiagnostic studies, and imaging are not routinely necessary to diagnose Bell palsy. However, referral to the appropriate specialist (neurologist, otolaryngologist, optometrist, ophthalmologist) is advised if the patient has sparing of the forehead muscle, multiple cranial neuropathies, signs of infection, or persistent weakness without significant improvement at 3 weeks.
Laboratory testing
A complete blood cell count with differential may point to infection or a lymphoproliferative disorder. When indicated, screening for diabetes mellitus with fasting blood glucose or hemoglobin A1c may be helpful. In Lyme-endemic regions, patients should undergo an enzyme-linked immunosorbent assay or an indirect fluorescent antibody test to screen for the disease. If positive, the diagnosis of Lyme disease should be confirmed by Western blot. If vesicles are present on examination, check serum antibodies for herpes zoster. In the appropriate clinical setting, angiotensin-converting enzyme, human immunodeficiency virus, and inflammatory markers can be tested.
Cerebrospinal fluid analysis is generally not helpful in diagnosing Bell palsy but can differentiate it from Guillain-Barré syndrome, leptomeningeal carcinomatosis, and infection involving the central nervous system.
Imaging
Imaging is not recommended in the initial evaluation of Bell palsy unless symptoms and the examination are atypical. From 5% to 7% of cases of facial palsy are caused by a tumor (eg, facial neuroma, cholesteatoma, hemangioma, meningioma), whether benign or malignant.14,15 Therefore, in patients with insidious onset of symptoms that do not improve in about 3 weeks, contrast-enhanced computed tomography or gadolinium-enhanced magnetic resonance imaging of the internal auditory canal and face is warranted.
Electrodiagnostic studies
Electrodiagnostic testing is typically not part of the evaluation of acute Bell palsy, but in patients with complete paralysis, it may help assess the degree of nerve injury and the chances of recovery, especially since patients with complete paralysis have a higher risk of incomplete recovery.19 Electrodiagnostic studies should be performed at least 1 week after symptom onset to avoid false-negative results.
TREATMENT
The treatment of Bell palsy focuses on maximizing recovery and minimizing associated complications.
Protect the eyes
Patients who cannot completely close their eyes should be given instructions on ocular protective care to prevent exposure keratopathy. Frequent application of lubricant eyedrops with artificial tears during the day or ophthalmic ointment at bedtime is recommended. The physician should also recommend protective eyewear such as sunglasses during the day. Eye patching or taping at night may be useful but could be harmful if applied too loosely or too tightly. Patients with vision loss or eye irritation should be referred to an ophthalmologist.19
Corticosteroids are recommended in the first 72 hours
In two randomized clinical trials (conducted by Sullivan et al20 in 511 patients and Engström et al21 in 829 patients), prednisolone was found to be beneficial if started within 72 hours of symptom onset.
In a double-blind, randomized, placebo-controlled study of prednisone in 58 patients, those who received the drug recovered faster, although long-term outcomes in these patients were not significantly different than those in the control group.22 The American Academy of Neurology23 rated this study as class II, ie, not meeting all of its criteria for the highest level of evidence, class I. Nevertheless, although prednisone lacks class I evidence, its use is recommended because it is a precursor to its active metabolite, prednisolone, which has been studied extensively.
The current guidelines of the American Academy of Neurology, updated in 2012, state, “For patients with new-onset Bell palsy, steroids are highly likely to be effective and should be offered to increase the probability of recovery of facial nerve function”23 (level A evidence, ie, established as effective). They also concluded that adverse effects of corticosteroids were generally minor and temporary.
Similarly, the guidelines of the American Academy of Otolaryngology–Head and Neck Surgery, published in 2013, recommend oral corticosteroids within 72 hours of onset of symptoms of Bell palsy for patients age 16 and older.19 The recommendation is for a 10-day course of corticosteroids with at least 5 days at a high dose (prednisolone 50 mg orally daily for 10 days, or prednisone 60 mg orally daily for 5 days, followed by a 5-day taper). The benefit of corticosteroids after 72 hours is unclear (Table 3).19
Even though the guidelines recommend corticosteroids, the decision to use them in diabetic patients and pregnant women should be individualized. Discretion is advised, as not all patients with Bell palsy need to be treated. Most recover spontaneously, especially those with mild symptoms.
Antiviral therapy may offer modest benefit
Antiviral therapy has not been shown to be beneficial in Bell palsy, and current guidelines do not recommend oral antiviral therapy alone.19 However, an antiviral combined with a corticosteroid may offer modest benefit if started within 72 hours of symptom onset (level C evidence, ie, possibly effective).23 Patients starting antiviral therapy should understand that its benefit has not been established.
Surgical decompression remains controversial
A Cochrane systematic review in 2011 found insufficient evidence regarding the safety and efficacy of surgical intervention in Bell palsy.24 Surgery should be considered only for patients with complete paralysis with a greater than 90% reduction in motor amplitude on a nerve conduction study compared with the unaffected side, and absent volitional activity on needle examination.19,25
Acupuncture: No recommendation
Currently, there is no recommendation for acupuncture in the treatment of Bell palsy.19 A recent randomized clinical trial suggests benefit from acupuncture combined with corticosteroids,26 but high-quality studies to support its use are lacking.26
Physical therapy: Insufficient evidence
There is insufficient evidence to show that physical therapy has benefit—or harm—in Bell palsy. However, some low-quality studies indicated that facial exercises and mime therapy may improve function in patients with moderate paralysis.27
Follow-up
Patients should be instructed to call at 2 weeks to report progress of symptoms and to be reevaluated within or at 1 month, with close attention to facial weakness and eye irritation. Further evaluation is needed if there has been no improvement, if symptoms have worsened, or if new symptoms have appeared.
The psychosocial impact of Bell palsy cannot be discounted, as the disfigurement can have negative implications for self-esteem and social relationships. Appropriate referral to an ophthalmologist, neurologist, otolaryngologist, social worker, or a plastic surgeon may be necessary.
COMPLICATIONS AND PROGNOSIS
Most patients with Bell palsy recover completely, but up to 30% have residual symptoms at 6 months.14,20 Furthermore, although Bell palsy usually has a monophasic course, 7% to 12% of patients have a recurrence.3,15
Long-term complications can include residual facial weakness, facial synkinesis, facial contracture, and facial spasm.14,28 Incomplete eye closure may benefit from surgery (tarsorrhaphy or gold-weight implantation) to prevent corneal ulceration. Facial synkinesis is due to aberrant nerve regeneration and occurs in 15% to 20% of patients after recovery from Bell palsy.29 Patients may describe tearing while chewing (“crocodile tears”), involuntary movement of the corners of the mouth with blinking, or ipsilateral eye-closing when the jaw opens (“jaw-winking”). Facial contracture, facial synkinesis, and facial spasm can be treated with botulinum toxin injection.30
Bell palsy is an idiopathic peripheral nerve disorder involving the facial nerve (ie, cranial nerve VII) and manifesting as acute, ipsilateral facial muscle weakness. It is named after Sir Charles Bell, who in 1821 first described the anatomy of the facial nerve.1 Although the disorder is clinically benign, patients can be devastated by its disfigurement.
The annual incidence of Bell palsy is 20 per 100,000, with no predilection for sex or ethnicity. It can affect people at any age, but the incidence is slightly higher after age 40.2,3 Risk factors include diabetes, pregnancy, severe preeclampsia, obesity, and hypertension.4–7
THE FACIAL NERVE IS VULNERABLE TO TRAUMA AND COMPRESSION
A basic understanding of the neuroanatomy of the facial nerve provides clues for distinguishing a central lesion from a peripheral lesion. This differentiation is important because the causes and management differ.
The facial nerve is a mixed sensory and motor nerve, carrying fibers involved in facial expression, taste, lacrimation, salivation, and sensation of the ear. It originates in the lower pons and exits the brainstem ventrally at the pontomedullary junction. After entering the internal acoustic meatus, it travels 20 to 30 mm in the facial canal, the longest bony course of any cranial nerve, making it highly susceptible to trauma and compression by edema.8
In the facial canal, it makes a posterior and inferior turn, forming a bend (ie, the genu of the facial nerve). The genu is proximal to the geniculate ganglion, which contains the facial nerve’s primary sensory neurons for taste and sensation. The motor branch of the facial nerve then exits the cranium via the stylomastoid foramen and passes through the parotid gland, where it divides into temporofacial and cervicofacial trunks.9
The facial nerve has five terminal branches that innervate the muscles of facial expression:
- The temporal branch (muscles of the forehead and superior part of the orbicularis oculi)
- The zygomatic branch (muscles of the nasolabial fold and cheek, eg, nasalis and zygomaticus).
- The buccal branch (the buccinators and inferior part of the orbicularis oculi)
- The marginal mandibular branch (the depressors of the mouth, eg, depressor anguli and mentalis)
- The cervical branch (the platysma muscle).
INFLAMMATION IS BELIEVED TO BE RESPONSIBLE
Although the precise cause of Bell palsy is not known, one theory is that inflammation of the nerve causes focal edema, demyelination, and ischemia. Several studies have suggested that herpes virus simplex type 1 infection may be involved.10
FACIAL DROOPING, EYELID WEAKNESS, OTHER SYMPTOMS
Symptoms of Bell palsy include ipsilateral sagging of the eyebrow, drooping of the face, flattening of the nasolabial fold, and inability to fully close the eye, pucker the lips, or raise the corner of the mouth (Figure 1). Symptoms develop within hours and are maximal by 3 days.
About 70% of patients have associated ipsilateral pain around the ear. If facial pain is present with sensory and hearing loss, a tumor of the parotid gland or viral otitis must be considered.11 Other complaints may include hyperacusis due to disruption of nerve fibers to the stapedius muscle, changes in taste, and dry eye from parasympathetic dysfunction. Some patients report paresthesias over the face, which most often represent motor symptoms misconstrued as sensory changes.
PHYSICAL EXAMINATION
The clinical examination should include a complete neurologic and general examination, including otoscopy and attention to the skin and parotid gland. Vesicles or scabbing around the ear should prompt testing for herpes zoster. Careful observation during the interview while the patient is talking may reveal subtle signs of weakness and provide additional clues.
A systematic approach to the assessment of a patient with suspected Bell palsy is recommended (Table 1) and outlined below:
Does the patient have peripheral facial palsy?
In Bell palsy, wrinkling of the forehead on the affected side when raising the eyebrows is either asymmetrical or absent.
If the forehead muscles are spared and the lower face is weak, this signifies a central lesion such as a stroke or other structural abnormality and not a peripheral lesion of the facial nerve (eg, Bell palsy).
Can the patient close the eyes tightly?
Normally, the patient should be able to close both eyes tightly, and the eyelashes should be buried between the eyelids. In Bell palsy, when the patient attempts to close the eyes, the affected side shows incomplete closure and the eye may remain partly open.
Assess the strength of the orbicularis oculi by trying to open the eyes. The patient who is attempting to close the eyelids tightly but cannot will demonstrate the Bell phenomenon, ie, the examiner is able to force open the eyelids, and the eyes are deviated upward and laterally.
Closely observe the blink pattern, as the involved side in Bell palsy may slightly lag behind the normal eye, and the patient may be unable to close the eye completely.
Is the smile symmetric?
Note flattening of the nasolabial fold on one side, which indicates facial weakness.
Can the patient puff out the cheeks?
Ask the patient to hold air in the mouth against resistance. This assesses the strength of the buccinator muscle.
Can the patient purse the lips?
Ask the patient to pucker or purse the lips and observe for asymmetry or weakness on the affected side.
Test the orbicularis oris muscle by trying to spread the lips apart while the patient resists, and observe for weakness on one side.
Is there a symmetric grimace?
This will test the muscles involved in depressing the angles of the mouth and platysma.
Are taste, sensation, and hearing intact?
Other testable functions of the facial nerve, including taste, sensation, and hearing, do not always need to be assessed but can be in patients with specific sensory deficits.
Abnormalities in taste can support localization of the problem either proximal or distal to the branch point of fibers mediating taste. The facial nerve supplies taste fibers to the anterior two-thirds of the tongue. Sweet and salty taste can be screened with sugar and salt. Tell the patient to close the eyes, and using a tongue blade, apply a small amount of sugar or salt on the side of the tongue. Ask the patient to identify the taste and repeat with the other sample after he or she has rinsed the mouth.
Somatic sensory fibers supplied by the facial nerve innervate the inner ear and a small area behind the ear, but these may be difficult to assess objectively. Formal audiologic testing may be needed if hearing is impaired.
Facial nerve reflexes
A number of facial reflexes can be tested, including the orbicularis oculi, palpebral-oculogyric, and corneal reflexes.12
The orbicularis oculi reflex is tested by gentle finger percussion of the glabella while observing for involuntary blinking with each stimulus. The afferent branch of this reflex is carried by the trigeminal nerve, while the efferent response is carried by the facial nerve. In peripheral facial nerve palsy, this reflex is weakened or absent on the affected side.
The palpebral-oculogyric reflex, or Bell phenomenon, produces upward and lateral deviation of the eyes when attempting forceful eyelid closure. In this reflex, the afferent fibers are carried by the facial nerve and the efferent fibers travel in the oculomotor nerve to the superior rectus muscle. In Bell palsy, this reflex is visible because of failure of adequate eyelid closure.
The corneal reflex is elicited by stimulating the cornea with a wisp of cotton, causing reflexive closure of the both eyes. The affected side may show slowed or absent lid closure when tested on either side. The sensory afferent fibers are carried by the trigeminal nerve, and the motor efferent fibers are carried by the facial nerve.
Grading of facial paralysis
The House-Brackmann scale is the most widely used tool for grading the degree of facial paralysis and for predicting recovery. Grades are I to VI, with grade I indicating normal function, and grade VI, complete paralysis.
Patients with some preserved motor function generally have good recovery, but those with complete paralysis may have long-term residual deficits.13
A DIAGNOSIS OF EXCLUSION
The diagnosis of Bell palsy is made by excluding other causes of unilateral facial paralysis, and 30% to 60% of cases of facial palsy are caused by an underlying disorder that mimics Bell palsy, including central nervous system lesion (eg, stroke, demyelinating disease), parotid gland tumor, Lyme disease, Ramsay Hunt syndrome, granulomatous disease, otitis media, cholesteatoma, diabetes, trauma, and Guillain-Barré syndrome (Table 2).14,15 Many of these conditions have associated features that help distinguish them from Bell palsy. Facial palsy that does not improve after 3 weeks should prompt referral to a neurologist.
Brain lesions
It is uncommon to have isolated facial palsy with a cortical or subcortical brain lesion, since the corticobulbar and corticospinal tracts travel in close proximity. Cortical signs such as hemiparesis, hemisensory loss, neglect, and dysarthria suggest a lesion of the cerebral cortex. Additionally, forehead muscle sparing is expected in supranuclear lesions.
Brainstem lesions can manifest with multiple ipsilateral cranial nerve palsies and contralateral limb weakness. Sarcoidosis and leptomeningeal carcinomatosis tend to involve the skull base and present with multiple cranial neuropathies.
Tumors of the brain or parotid gland have an insidious onset and may cause systemic signs such as fevers, chills, and weight loss. Headache, seizures, and hearing loss indicate an intracranial lesion. A palpable mass near the ear, neck, or parotid gland requires imaging of the face to look for a parotid gland tumor.
Infection
A number of infections can cause acute facial paralysis. The most common is herpes simplex virus, and the next most common is varicella zoster.14 Herpes simplex virus, Ramsay Hunt syndrome, and Lyme disease may have associated pain and skin changes. Erythema of the tympanic membrane suggests otitis media, especially in the setting of ear pain and hearing loss.
Ramsay Hunt syndrome is caused by reactivation of the herpes zoster virus from the geniculate ganglion, affecting the facial nerve. Careful examination of the ear canal and the oropharynx may show vesicles.
In Lyme disease, facial palsy is the most common cranial neuropathy, seen in 50% to 63% of patients with Borrelia burgdorferi meningitis.16,17 In people with a history of rash, arthralgia, tick bite, or travel to an endemic region, Lyme titers should be checked before starting the patient on corticosteroids.
Bilateral facial palsy is rare and occurs in fewer than 1% of patients. It has been reported in patients with Lyme disease, Guillain-Barré syndrome, sarcoidosis, diabetes mellitus, viral infection, and pontine glioma.18
DIAGNOSTIC EVALUATION
Serologic testing, electrodiagnostic studies, and imaging are not routinely necessary to diagnose Bell palsy. However, referral to the appropriate specialist (neurologist, otolaryngologist, optometrist, ophthalmologist) is advised if the patient has sparing of the forehead muscle, multiple cranial neuropathies, signs of infection, or persistent weakness without significant improvement at 3 weeks.
Laboratory testing
A complete blood cell count with differential may point to infection or a lymphoproliferative disorder. When indicated, screening for diabetes mellitus with fasting blood glucose or hemoglobin A1c may be helpful. In Lyme-endemic regions, patients should undergo an enzyme-linked immunosorbent assay or an indirect fluorescent antibody test to screen for the disease. If positive, the diagnosis of Lyme disease should be confirmed by Western blot. If vesicles are present on examination, check serum antibodies for herpes zoster. In the appropriate clinical setting, angiotensin-converting enzyme, human immunodeficiency virus, and inflammatory markers can be tested.
Cerebrospinal fluid analysis is generally not helpful in diagnosing Bell palsy but can differentiate it from Guillain-Barré syndrome, leptomeningeal carcinomatosis, and infection involving the central nervous system.
Imaging
Imaging is not recommended in the initial evaluation of Bell palsy unless symptoms and the examination are atypical. From 5% to 7% of cases of facial palsy are caused by a tumor (eg, facial neuroma, cholesteatoma, hemangioma, meningioma), whether benign or malignant.14,15 Therefore, in patients with insidious onset of symptoms that do not improve in about 3 weeks, contrast-enhanced computed tomography or gadolinium-enhanced magnetic resonance imaging of the internal auditory canal and face is warranted.
Electrodiagnostic studies
Electrodiagnostic testing is typically not part of the evaluation of acute Bell palsy, but in patients with complete paralysis, it may help assess the degree of nerve injury and the chances of recovery, especially since patients with complete paralysis have a higher risk of incomplete recovery.19 Electrodiagnostic studies should be performed at least 1 week after symptom onset to avoid false-negative results.
TREATMENT
The treatment of Bell palsy focuses on maximizing recovery and minimizing associated complications.
Protect the eyes
Patients who cannot completely close their eyes should be given instructions on ocular protective care to prevent exposure keratopathy. Frequent application of lubricant eyedrops with artificial tears during the day or ophthalmic ointment at bedtime is recommended. The physician should also recommend protective eyewear such as sunglasses during the day. Eye patching or taping at night may be useful but could be harmful if applied too loosely or too tightly. Patients with vision loss or eye irritation should be referred to an ophthalmologist.19
Corticosteroids are recommended in the first 72 hours
In two randomized clinical trials (conducted by Sullivan et al20 in 511 patients and Engström et al21 in 829 patients), prednisolone was found to be beneficial if started within 72 hours of symptom onset.
In a double-blind, randomized, placebo-controlled study of prednisone in 58 patients, those who received the drug recovered faster, although long-term outcomes in these patients were not significantly different than those in the control group.22 The American Academy of Neurology23 rated this study as class II, ie, not meeting all of its criteria for the highest level of evidence, class I. Nevertheless, although prednisone lacks class I evidence, its use is recommended because it is a precursor to its active metabolite, prednisolone, which has been studied extensively.
The current guidelines of the American Academy of Neurology, updated in 2012, state, “For patients with new-onset Bell palsy, steroids are highly likely to be effective and should be offered to increase the probability of recovery of facial nerve function”23 (level A evidence, ie, established as effective). They also concluded that adverse effects of corticosteroids were generally minor and temporary.
Similarly, the guidelines of the American Academy of Otolaryngology–Head and Neck Surgery, published in 2013, recommend oral corticosteroids within 72 hours of onset of symptoms of Bell palsy for patients age 16 and older.19 The recommendation is for a 10-day course of corticosteroids with at least 5 days at a high dose (prednisolone 50 mg orally daily for 10 days, or prednisone 60 mg orally daily for 5 days, followed by a 5-day taper). The benefit of corticosteroids after 72 hours is unclear (Table 3).19
Even though the guidelines recommend corticosteroids, the decision to use them in diabetic patients and pregnant women should be individualized. Discretion is advised, as not all patients with Bell palsy need to be treated. Most recover spontaneously, especially those with mild symptoms.
Antiviral therapy may offer modest benefit
Antiviral therapy has not been shown to be beneficial in Bell palsy, and current guidelines do not recommend oral antiviral therapy alone.19 However, an antiviral combined with a corticosteroid may offer modest benefit if started within 72 hours of symptom onset (level C evidence, ie, possibly effective).23 Patients starting antiviral therapy should understand that its benefit has not been established.
Surgical decompression remains controversial
A Cochrane systematic review in 2011 found insufficient evidence regarding the safety and efficacy of surgical intervention in Bell palsy.24 Surgery should be considered only for patients with complete paralysis with a greater than 90% reduction in motor amplitude on a nerve conduction study compared with the unaffected side, and absent volitional activity on needle examination.19,25
Acupuncture: No recommendation
Currently, there is no recommendation for acupuncture in the treatment of Bell palsy.19 A recent randomized clinical trial suggests benefit from acupuncture combined with corticosteroids,26 but high-quality studies to support its use are lacking.26
Physical therapy: Insufficient evidence
There is insufficient evidence to show that physical therapy has benefit—or harm—in Bell palsy. However, some low-quality studies indicated that facial exercises and mime therapy may improve function in patients with moderate paralysis.27
Follow-up
Patients should be instructed to call at 2 weeks to report progress of symptoms and to be reevaluated within or at 1 month, with close attention to facial weakness and eye irritation. Further evaluation is needed if there has been no improvement, if symptoms have worsened, or if new symptoms have appeared.
The psychosocial impact of Bell palsy cannot be discounted, as the disfigurement can have negative implications for self-esteem and social relationships. Appropriate referral to an ophthalmologist, neurologist, otolaryngologist, social worker, or a plastic surgeon may be necessary.
COMPLICATIONS AND PROGNOSIS
Most patients with Bell palsy recover completely, but up to 30% have residual symptoms at 6 months.14,20 Furthermore, although Bell palsy usually has a monophasic course, 7% to 12% of patients have a recurrence.3,15
Long-term complications can include residual facial weakness, facial synkinesis, facial contracture, and facial spasm.14,28 Incomplete eye closure may benefit from surgery (tarsorrhaphy or gold-weight implantation) to prevent corneal ulceration. Facial synkinesis is due to aberrant nerve regeneration and occurs in 15% to 20% of patients after recovery from Bell palsy.29 Patients may describe tearing while chewing (“crocodile tears”), involuntary movement of the corners of the mouth with blinking, or ipsilateral eye-closing when the jaw opens (“jaw-winking”). Facial contracture, facial synkinesis, and facial spasm can be treated with botulinum toxin injection.30
- Grzybowski A, Kaufman MH. Sir Charles Bell (1774-1842): contributions to neuro-ophthalmology. Acta Ophthalmol Scand 2007; 85:897–901.
- De Diego-Sastre JI, Prim-Espada MP, Fernández-García F. The epidemiology of Bell’s palsy. Rev Neurol 2005; 41:287–290. In Spanish.
- Morris AM, Deeks SL, Hill MD, et al. Annualized incidence and spectrum of illness from an outbreak investigation of Bell’s palsy. Neuroepidemiology 2002; 21:255–261.
- Bosco D, Plastino M, Bosco F, et al. Bell’s palsy: a manifestation of prediabetes? Acta Neurol Scand 2011; 123:68–72.
- Riga M, Kefalidis G, Danielides V. The role of diabetes mellitus in the clinical presentation and prognosis of Bell palsy. J Am Board Fam Med 2012; 25:819–826.
- Hilsinger RL Jr, Adour KK, Doty HE. Idiopathic facial paralysis, pregnancy, and the menstrual cycle. Ann Otol Rhinol Laryngol 1975; 84:433–442.
- Savadi-Oskouei D, Abedi A, Sadeghi-Bazargani H. Independent role of hypertension in Bell’s palsy: a case-control study. Eur Neurol 2008; 60:253–257.
- Murai A, Kariya S, Tamura K, et al. The facial nerve canal in patients with Bell’s palsy: an investigation by high-resolution computed tomography with multiplanar reconstruction. Eur Arch Otorhinolaryngol 2013; 270:2035–2038.
- Blumenfeld H. Neuroanatomy Through Clinical Cases. 1st ed. Sunderland, MA: Sinauer; 2002:479–484.
- Murakami S, Mizobuchi M, Nakashiro Y, Doi T, Hato N, Yanagihara N. Bell palsy and herpes simplex virus: identification of viral DNA in endoneurial fluid and muscle. Ann Intern Med 1996; 124:27–30.
- Boahene DO, Olsen KD, Driscoll C, Lewis JE, McDonald TJ. Facial nerve paralysis secondary to occult malignant neoplasms. Otolaryngol Head Neck Surg 2004; 130:459–465.
- DeJong RN. The Neurologic Examination: Incorporating the fundamentals of neuroanatomy and neurophysiology. 4th ed. New York, NY: Harper & Row; 1979:178–198.
- House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg 1985; 93:146–147.
- Peitersen E. Bell’s palsy: the spontaneous course of 2,500 peripheral facial nerve palsies of different etiologies. Acta Otolaryngol Suppl 2002; 549:4–30.
- Hohman MH, Hadlock TA. Etiology, diagnosis, and management of facial palsy: 2000 patients at a facial nerve center. Laryngoscope 2014; 124:E283–E293.
- Ackermann R, Hörstrup P, Schmidt R. Tick-borne meningopolyneuritis (Garin-Bujadoux, Bannwarth). Yale J Biol Med 1984; 57:485–490.
- Pachner AR, Steere AC. The triad of neurologic manifestations of Lyme disease: meningitis, cranial neuritis, and radiculoneuritis. Neurology 1985; 35:47–53.
- Keane JR. Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature. Neurology 1994; 44:1198–1202.
- Baugh RF, Basura GJ, Ishii LE, et al. Clinical practice guideline: Bell’s palsy. Otolaryngol Head Neck Surg 2013; 149(suppl 3):S1–S27.
- Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007; 357:1598–1607.
- Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol 2008; 7:993–1000.
- Lagalla G, Logullo F, Di Bella P, Provinciali L, Ceravolo MG. Influence of early high-dose steroid treatment on Bell’s palsy evolution. Neurol Sci 2002; 23:107–112.
- Gronseth GS, Paduga R; American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012; 79:2209–2213.
- McAllister K, Walker D, Donnan PT, Swan I. Surgical interventions for the early management of Bell’s palsy. Cochrane Database Syst Rev 2011; 2:CD007468.
- Gantz BJ, Rubinstein JT, Gidley P, Woodworth GG. Surgical management of Bell’s palsy. Laryngoscope 1999; 109:1177–1188.
- Xu SB, Huang B, Zhang CY, et al. Effectiveness of strengthened stimulation during acupuncture for the treatment of Bell palsy: a randomized controlled trial. CMAJ 2013; 185:473–479.
- Teixeira LJ, Valbuza JS, Prado GF. Physical therapy for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev 2011; 12:CD006283.
- Yaltho TC, Jankovic J. The many faces of hemifacial spasm: differential diagnosis of unilateral facial spasms. Mov Disord 2011; 26:1582–1592.
- Celik M, Forta H, Vural C. The development of synkinesis after facial nerve paralysis. Eur Neurol 2000; 43:147–151.
- Chua CN, Quhill F, Jones E, Voon LW, Ahad M, Rowson N. Treatment of aberrant facial nerve regeneration with botulinum toxin A. Orbit 2004; 23:213–218.
- Grzybowski A, Kaufman MH. Sir Charles Bell (1774-1842): contributions to neuro-ophthalmology. Acta Ophthalmol Scand 2007; 85:897–901.
- De Diego-Sastre JI, Prim-Espada MP, Fernández-García F. The epidemiology of Bell’s palsy. Rev Neurol 2005; 41:287–290. In Spanish.
- Morris AM, Deeks SL, Hill MD, et al. Annualized incidence and spectrum of illness from an outbreak investigation of Bell’s palsy. Neuroepidemiology 2002; 21:255–261.
- Bosco D, Plastino M, Bosco F, et al. Bell’s palsy: a manifestation of prediabetes? Acta Neurol Scand 2011; 123:68–72.
- Riga M, Kefalidis G, Danielides V. The role of diabetes mellitus in the clinical presentation and prognosis of Bell palsy. J Am Board Fam Med 2012; 25:819–826.
- Hilsinger RL Jr, Adour KK, Doty HE. Idiopathic facial paralysis, pregnancy, and the menstrual cycle. Ann Otol Rhinol Laryngol 1975; 84:433–442.
- Savadi-Oskouei D, Abedi A, Sadeghi-Bazargani H. Independent role of hypertension in Bell’s palsy: a case-control study. Eur Neurol 2008; 60:253–257.
- Murai A, Kariya S, Tamura K, et al. The facial nerve canal in patients with Bell’s palsy: an investigation by high-resolution computed tomography with multiplanar reconstruction. Eur Arch Otorhinolaryngol 2013; 270:2035–2038.
- Blumenfeld H. Neuroanatomy Through Clinical Cases. 1st ed. Sunderland, MA: Sinauer; 2002:479–484.
- Murakami S, Mizobuchi M, Nakashiro Y, Doi T, Hato N, Yanagihara N. Bell palsy and herpes simplex virus: identification of viral DNA in endoneurial fluid and muscle. Ann Intern Med 1996; 124:27–30.
- Boahene DO, Olsen KD, Driscoll C, Lewis JE, McDonald TJ. Facial nerve paralysis secondary to occult malignant neoplasms. Otolaryngol Head Neck Surg 2004; 130:459–465.
- DeJong RN. The Neurologic Examination: Incorporating the fundamentals of neuroanatomy and neurophysiology. 4th ed. New York, NY: Harper & Row; 1979:178–198.
- House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg 1985; 93:146–147.
- Peitersen E. Bell’s palsy: the spontaneous course of 2,500 peripheral facial nerve palsies of different etiologies. Acta Otolaryngol Suppl 2002; 549:4–30.
- Hohman MH, Hadlock TA. Etiology, diagnosis, and management of facial palsy: 2000 patients at a facial nerve center. Laryngoscope 2014; 124:E283–E293.
- Ackermann R, Hörstrup P, Schmidt R. Tick-borne meningopolyneuritis (Garin-Bujadoux, Bannwarth). Yale J Biol Med 1984; 57:485–490.
- Pachner AR, Steere AC. The triad of neurologic manifestations of Lyme disease: meningitis, cranial neuritis, and radiculoneuritis. Neurology 1985; 35:47–53.
- Keane JR. Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature. Neurology 1994; 44:1198–1202.
- Baugh RF, Basura GJ, Ishii LE, et al. Clinical practice guideline: Bell’s palsy. Otolaryngol Head Neck Surg 2013; 149(suppl 3):S1–S27.
- Sullivan FM, Swan IR, Donnan PT, et al. Early treatment with prednisolone or acyclovir in Bell’s palsy. N Engl J Med 2007; 357:1598–1607.
- Engström M, Berg T, Stjernquist-Desatnik A, et al. Prednisolone and valaciclovir in Bell’s palsy: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet Neurol 2008; 7:993–1000.
- Lagalla G, Logullo F, Di Bella P, Provinciali L, Ceravolo MG. Influence of early high-dose steroid treatment on Bell’s palsy evolution. Neurol Sci 2002; 23:107–112.
- Gronseth GS, Paduga R; American Academy of Neurology. Evidence-based guideline update: steroids and antivirals for Bell palsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012; 79:2209–2213.
- McAllister K, Walker D, Donnan PT, Swan I. Surgical interventions for the early management of Bell’s palsy. Cochrane Database Syst Rev 2011; 2:CD007468.
- Gantz BJ, Rubinstein JT, Gidley P, Woodworth GG. Surgical management of Bell’s palsy. Laryngoscope 1999; 109:1177–1188.
- Xu SB, Huang B, Zhang CY, et al. Effectiveness of strengthened stimulation during acupuncture for the treatment of Bell palsy: a randomized controlled trial. CMAJ 2013; 185:473–479.
- Teixeira LJ, Valbuza JS, Prado GF. Physical therapy for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev 2011; 12:CD006283.
- Yaltho TC, Jankovic J. The many faces of hemifacial spasm: differential diagnosis of unilateral facial spasms. Mov Disord 2011; 26:1582–1592.
- Celik M, Forta H, Vural C. The development of synkinesis after facial nerve paralysis. Eur Neurol 2000; 43:147–151.
- Chua CN, Quhill F, Jones E, Voon LW, Ahad M, Rowson N. Treatment of aberrant facial nerve regeneration with botulinum toxin A. Orbit 2004; 23:213–218.
KEY POINTS
- Bell palsy is an acute disorder of the facial nerve causing unilateral facial weakness, pain, abnormal taste, and reduced tearing.
- Although herpes simplex virus reactivation is suspected in the pathogenesis, the exact cause is unknown.
- An additional workup is warranted for abnormalities beyond isolated facial nerve palsy.
- Guidelines recommend starting corticosteroids for patients who present within 3 days of symptom onset. There is no compelling evidence to support antiviral therapy, physical therapy, acupuncture, or surgical decompression.
A continuous cardiac murmur
A 45-year-old woman presents with shortness of breath that has been progressively worsening for 3 weeks. She has no history of medical conditions and is taking no medications. Her blood pressure is 132/68 mm Hg, pulse 90 beats per minute, respirations 14 per minute, and oxygen saturation 95% on room air by pulse oximetry.
Physical examination reveals clear lung fields and no jugular venous distention or peripheral edema. However, she has a grade 3 of 6 continuous murmur audible over the entire precordium that does not change in intensity with respiration.
1. Which of the following is the likely cause of this patient’s cardiac murmur?
- Ventricular septal defect
- Atrial septal defect
- Ruptured sinus of Valsalva aneurysm
- Aortic regurgitation
- Patent ductus arteriosus
- Pulmonic stenosis
Table 1 summarizes the characteristics of the murmurs caused by these various cardiac defects.
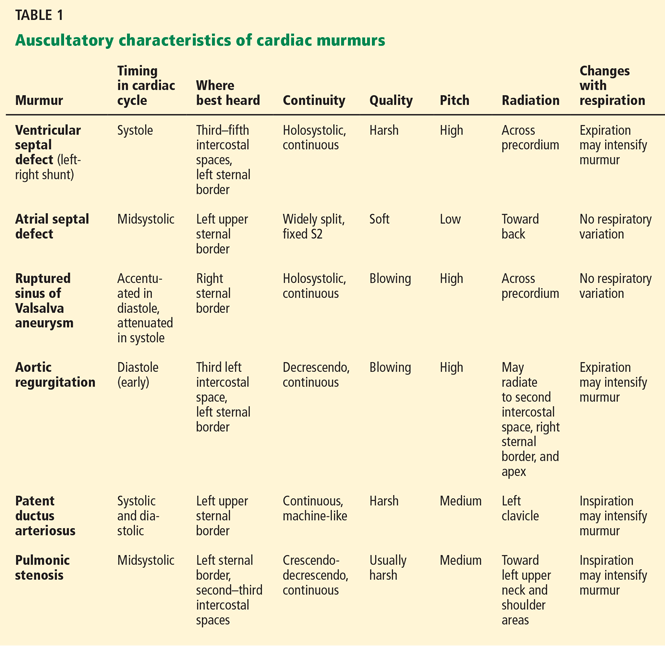
Ventricular septal defect causes murmurs that are characteristically holosystolic and heard best at the lower left sternal border with radiation to the right lower sternal border, which overlies the defect.
The murmur of restrictive ventricular septal defect is most often holosystolic because the pressure difference between the ventricles is generated almost instantly at the onset of systole with a left-to-right shunt continuing throughout ventricular contraction. In contrast, nonrestrictive ventricular septal defects generally do not generate a murmur, since pressure is equalized across the defect. This left-to-right shunting may lead to right ventricular volume overload, resulting in delayed closure of the pulmonary valve and a widely split S2. Irreversible pulmonary hypertension with shunt reversal may occur if the defect remains untreated.1
Atrial septal defect. The most characteristic feature of atrial septal defect is a fixed split S2 resulting from right ventricular volume overload due to left-to-right atrial shunting of blood flow. As flow is shunted from the left to the right atrium and subsequently into the right ventricle, ejection of excess blood through the pulmonary valve produces a midsystolic flow murmur, heard best over the left upper sternal border, that may radiate to the back.
Ruptured sinus of Valsalva aneurysm. The pressure is higher in the aorta than in the right atrium throughout the cardiac cycle, and if a shunt is created between the two structures by a ruptured sinus of Valsalva aneurysm, the blood flow across this shunt throughout the cardiac cycle produces a continuous murmur. In contrast, if a sinus of Valsalva aneurysm ruptures into the right ventricle, the murmur is accentuated in diastole and attenuated in systole, and is often associated with pounding pulses and a thrill along either the left or right sternal border.1
Aortic regurgitation causes a diastolic murmur as blood flows retrograde into the left ventricle through the incompetent aortic valve. This murmur is usually described as a blowing, decrescendo murmur heard best at the third left intercostal space.
Patent ductus arteriosus is a communication between the descending thoracic aorta and the pulmonary artery that fails to close at birth. The hallmark murmur associated with this defect is a continuous “machine-like” murmur located at the upper left sternal border, often radiating down the left side of the sternum into the back. Of note, increasing the systemic pressure by the Valsalva maneuver or handgrip exercise will increase the diastolic component of the continuous murmur associated with ruptured sinus of Valsalva aneurysm, helping to differentiate it from patent ductus arteriosus.2
Pulmonic stenosis causes a systolic murmur heard best at the second intercostal space along the left sternal border and having a crescendo-decrescendo intensity and harsh quality. As the right ventricle takes longer to eject its blood volume through the stenotic pulmonary valve, the delay in closure between the aortic and pulmonary valve is widened, resulting in a significant splitting of the S2. In addition, any maneuver that increases preload will also increase the intensity of the murmur.3
Our patient has a murmur that is continuous, is heard across the entire precordium, and has no respiratory variation. These features are most consistent with a sinus of Valsalva aneurysm that has ruptured into the right atrium.
The 2008 update of the joint American College of Cardiology and American Heart Association guidelines4 recommends further evaluation of diastolic or continuous murmurs with echocardiography, as these murmurs are most often signs of a pathologic condition. In addition, echocardiography is warranted to evaluate grade 3 or higher systolic murmurs and those that are holosystolic.4
SINUS OF VALSALVA ANEURYSM
Sinus of Valsalva aneurysm is rare, with an incidence of 0.09% to 0.15%. From 65% to 85% are in the right coronary cusp, 10% to 30% are in the noncoronary cusp, and fewer than 5% are in the left coronary cusp.5
This condition is most often congenital, accounting for up to 3.5% of congenital cardiac anomalies, though it can be acquired. Formation of the aneurysm is generally related to weakening of elastic fibers and muscular tissues that progresses over time.
Many cases of sinus of Valsalva aneurysm are associated with additional cardiac defects.1 Ventricular septal defect is the most common coexisting congenital anomaly, occurring in up to 53% of patients and frequently associated with aneurysms involving the right coronary cusp and with sinus of Valsalva aneurysm.6 Other congenital anomalies often accompanying sinus of Valsalva aneurysm include pulmonary stenosis, atrial septal defect, bicuspid aortic valve, tetralogy of Fallot, patent ductus arteriosus, coarctation of the aorta, and subaortic stenosis. Another associated condition is aortic regurgitation, for which more than half of affected patients eventually require aortic valve replacement.2
Acquired sinus of Valsalva aneurysm can be the result of endocarditis, trauma, surgery, cardiac catheterization, or inflammatory or degenerative processes including, rarely, tertiary syphilis.3
Sinus of Valsalva aneurysm often remains asymptomatic, but symptoms may arise if the aneurysm ruptures, resulting in intracardiac shunting or aneurysm-associated compression of adjacent cardiac structures such as coronary arteries. Rupture may be spontaneous, secondary to chest trauma or excess exertion, or iatrogenic.
Imaging studies such as echocardiography, cardiac computed tomography, and cardiac magnetic resonance imaging are essential in diagnosing and managing sinus of Valsalva aneurysm and identifying coexisting cardiac anomalies.
Rupture occurs most commonly into the right ventricle, followed in frequency by the right atrium or left atrium. Once rupture occurs, median survival is 1 to 2 years if left untreated, with death often secondary to congestive heart failure or infective endocarditis.7
Surgery remains the preferred approach to the treatment of ruptured sinus of Valsalva aneurysm. Operative risk is reasonably low and long-term outcomes are good. The appropriate therapy for unruptured and asymptomatic sinus of Valsalva aneurysm remains less clear.
Successful transcatheter closure of ruptured sinus of Valsalva aneurysm has been described using Amplatzer devices, a procedure that avoids sternotomy and cardiopulmonary bypass. Despite advances in percutaneous techniques, open surgery with or without aortic valve replacement remains the current standard of care.8
BACK TO OUR PATIENT
In the case described above, the initial diagnostic study done to evaluate the patient’s dyspnea and murmur was transthoracic echocardiography, which demonstrated a relatively preserved ejection fraction with mild aortic regurgitation and an aneurysmal structure extending from the aortic root toward the right atrium.
Transesophageal echocardiography confirmed this finding (Figure 1). Cross-sectional imaging of the aortic valve (Figure 2) showed the aneurysm arising from the noncoronary cusp and communicating with the right atrium. Color flow Doppler (Figure 3) confirmed continuous flow between the aneurysmal sinus and right atrium throughout the cardiac cycle, consistent with the continuous murmur noted on physical examination.
The aneurysm was also noted on aortography (Figure 4) obtained before the patient underwent surgery to correct it. The surgery was successful, no complications occurred, and the murmur and associated dyspnea had completely resolved at subsequent follow-up.
This case highlights the importance of imaging studies such as echocardiography in diagnosing and managing sinus of Valsalva aneurysm, and also the importance of physical examination in guiding the diagnostic evaluation and differentiating this condition from other cardiac disorders.
- Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011:1411–1468.
- Topi B, John J, Agarwal A, et al. An uncommon cause of a continuous murmur. Exp Clin Cardiol 2012; 17:148–149.
- Constant J. Bedside Cardiology. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999:268–320.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Jung SH, Yun TJ, Im YM, et al. Ruptured sinus of Valsalva aneurysm: transaortic repair may cause sinus of Valsalva distortion and aortic regurgitation. J Thorac Cardiovasc Surg 2008; 135:1153–1158.
- Post MC, Braam RL, Groenemeijer BE, Nicastia D, Rensing BJ, Schepens MA. Rupture of right coronary sinus of Valsalva aneurysm into right ventricle. Neth Heart J 2010; 18:209–211.
- Moustafa S, Mookadam F, Cooper L, et al. Sinus of Valsalva aneurysms—47 years of a single center experience and systematic overview of published reports. Am J Cardiol 2007; 99:1159–1164.
- Zhao SH, Yan CW, Zhu XY, et al. Transcatheter occlusion of the ruptured sinus of Valsalva aneurysm with an Amplatzer duct occluder. Int J Cardiol 2008; 129:81–85.
A 45-year-old woman presents with shortness of breath that has been progressively worsening for 3 weeks. She has no history of medical conditions and is taking no medications. Her blood pressure is 132/68 mm Hg, pulse 90 beats per minute, respirations 14 per minute, and oxygen saturation 95% on room air by pulse oximetry.
Physical examination reveals clear lung fields and no jugular venous distention or peripheral edema. However, she has a grade 3 of 6 continuous murmur audible over the entire precordium that does not change in intensity with respiration.
1. Which of the following is the likely cause of this patient’s cardiac murmur?
- Ventricular septal defect
- Atrial septal defect
- Ruptured sinus of Valsalva aneurysm
- Aortic regurgitation
- Patent ductus arteriosus
- Pulmonic stenosis
Table 1 summarizes the characteristics of the murmurs caused by these various cardiac defects.
Ventricular septal defect causes murmurs that are characteristically holosystolic and heard best at the lower left sternal border with radiation to the right lower sternal border, which overlies the defect.
The murmur of restrictive ventricular septal defect is most often holosystolic because the pressure difference between the ventricles is generated almost instantly at the onset of systole with a left-to-right shunt continuing throughout ventricular contraction. In contrast, nonrestrictive ventricular septal defects generally do not generate a murmur, since pressure is equalized across the defect. This left-to-right shunting may lead to right ventricular volume overload, resulting in delayed closure of the pulmonary valve and a widely split S2. Irreversible pulmonary hypertension with shunt reversal may occur if the defect remains untreated.1
Atrial septal defect. The most characteristic feature of atrial septal defect is a fixed split S2 resulting from right ventricular volume overload due to left-to-right atrial shunting of blood flow. As flow is shunted from the left to the right atrium and subsequently into the right ventricle, ejection of excess blood through the pulmonary valve produces a midsystolic flow murmur, heard best over the left upper sternal border, that may radiate to the back.
Ruptured sinus of Valsalva aneurysm. The pressure is higher in the aorta than in the right atrium throughout the cardiac cycle, and if a shunt is created between the two structures by a ruptured sinus of Valsalva aneurysm, the blood flow across this shunt throughout the cardiac cycle produces a continuous murmur. In contrast, if a sinus of Valsalva aneurysm ruptures into the right ventricle, the murmur is accentuated in diastole and attenuated in systole, and is often associated with pounding pulses and a thrill along either the left or right sternal border.1
Aortic regurgitation causes a diastolic murmur as blood flows retrograde into the left ventricle through the incompetent aortic valve. This murmur is usually described as a blowing, decrescendo murmur heard best at the third left intercostal space.
Patent ductus arteriosus is a communication between the descending thoracic aorta and the pulmonary artery that fails to close at birth. The hallmark murmur associated with this defect is a continuous “machine-like” murmur located at the upper left sternal border, often radiating down the left side of the sternum into the back. Of note, increasing the systemic pressure by the Valsalva maneuver or handgrip exercise will increase the diastolic component of the continuous murmur associated with ruptured sinus of Valsalva aneurysm, helping to differentiate it from patent ductus arteriosus.2
Pulmonic stenosis causes a systolic murmur heard best at the second intercostal space along the left sternal border and having a crescendo-decrescendo intensity and harsh quality. As the right ventricle takes longer to eject its blood volume through the stenotic pulmonary valve, the delay in closure between the aortic and pulmonary valve is widened, resulting in a significant splitting of the S2. In addition, any maneuver that increases preload will also increase the intensity of the murmur.3
Our patient has a murmur that is continuous, is heard across the entire precordium, and has no respiratory variation. These features are most consistent with a sinus of Valsalva aneurysm that has ruptured into the right atrium.
The 2008 update of the joint American College of Cardiology and American Heart Association guidelines4 recommends further evaluation of diastolic or continuous murmurs with echocardiography, as these murmurs are most often signs of a pathologic condition. In addition, echocardiography is warranted to evaluate grade 3 or higher systolic murmurs and those that are holosystolic.4
SINUS OF VALSALVA ANEURYSM
Sinus of Valsalva aneurysm is rare, with an incidence of 0.09% to 0.15%. From 65% to 85% are in the right coronary cusp, 10% to 30% are in the noncoronary cusp, and fewer than 5% are in the left coronary cusp.5
This condition is most often congenital, accounting for up to 3.5% of congenital cardiac anomalies, though it can be acquired. Formation of the aneurysm is generally related to weakening of elastic fibers and muscular tissues that progresses over time.
Many cases of sinus of Valsalva aneurysm are associated with additional cardiac defects.1 Ventricular septal defect is the most common coexisting congenital anomaly, occurring in up to 53% of patients and frequently associated with aneurysms involving the right coronary cusp and with sinus of Valsalva aneurysm.6 Other congenital anomalies often accompanying sinus of Valsalva aneurysm include pulmonary stenosis, atrial septal defect, bicuspid aortic valve, tetralogy of Fallot, patent ductus arteriosus, coarctation of the aorta, and subaortic stenosis. Another associated condition is aortic regurgitation, for which more than half of affected patients eventually require aortic valve replacement.2
Acquired sinus of Valsalva aneurysm can be the result of endocarditis, trauma, surgery, cardiac catheterization, or inflammatory or degenerative processes including, rarely, tertiary syphilis.3
Sinus of Valsalva aneurysm often remains asymptomatic, but symptoms may arise if the aneurysm ruptures, resulting in intracardiac shunting or aneurysm-associated compression of adjacent cardiac structures such as coronary arteries. Rupture may be spontaneous, secondary to chest trauma or excess exertion, or iatrogenic.
Imaging studies such as echocardiography, cardiac computed tomography, and cardiac magnetic resonance imaging are essential in diagnosing and managing sinus of Valsalva aneurysm and identifying coexisting cardiac anomalies.
Rupture occurs most commonly into the right ventricle, followed in frequency by the right atrium or left atrium. Once rupture occurs, median survival is 1 to 2 years if left untreated, with death often secondary to congestive heart failure or infective endocarditis.7
Surgery remains the preferred approach to the treatment of ruptured sinus of Valsalva aneurysm. Operative risk is reasonably low and long-term outcomes are good. The appropriate therapy for unruptured and asymptomatic sinus of Valsalva aneurysm remains less clear.
Successful transcatheter closure of ruptured sinus of Valsalva aneurysm has been described using Amplatzer devices, a procedure that avoids sternotomy and cardiopulmonary bypass. Despite advances in percutaneous techniques, open surgery with or without aortic valve replacement remains the current standard of care.8
BACK TO OUR PATIENT
In the case described above, the initial diagnostic study done to evaluate the patient’s dyspnea and murmur was transthoracic echocardiography, which demonstrated a relatively preserved ejection fraction with mild aortic regurgitation and an aneurysmal structure extending from the aortic root toward the right atrium.
Transesophageal echocardiography confirmed this finding (Figure 1). Cross-sectional imaging of the aortic valve (Figure 2) showed the aneurysm arising from the noncoronary cusp and communicating with the right atrium. Color flow Doppler (Figure 3) confirmed continuous flow between the aneurysmal sinus and right atrium throughout the cardiac cycle, consistent with the continuous murmur noted on physical examination.
The aneurysm was also noted on aortography (Figure 4) obtained before the patient underwent surgery to correct it. The surgery was successful, no complications occurred, and the murmur and associated dyspnea had completely resolved at subsequent follow-up.
This case highlights the importance of imaging studies such as echocardiography in diagnosing and managing sinus of Valsalva aneurysm, and also the importance of physical examination in guiding the diagnostic evaluation and differentiating this condition from other cardiac disorders.
A 45-year-old woman presents with shortness of breath that has been progressively worsening for 3 weeks. She has no history of medical conditions and is taking no medications. Her blood pressure is 132/68 mm Hg, pulse 90 beats per minute, respirations 14 per minute, and oxygen saturation 95% on room air by pulse oximetry.
Physical examination reveals clear lung fields and no jugular venous distention or peripheral edema. However, she has a grade 3 of 6 continuous murmur audible over the entire precordium that does not change in intensity with respiration.
1. Which of the following is the likely cause of this patient’s cardiac murmur?
- Ventricular septal defect
- Atrial septal defect
- Ruptured sinus of Valsalva aneurysm
- Aortic regurgitation
- Patent ductus arteriosus
- Pulmonic stenosis
Table 1 summarizes the characteristics of the murmurs caused by these various cardiac defects.
Ventricular septal defect causes murmurs that are characteristically holosystolic and heard best at the lower left sternal border with radiation to the right lower sternal border, which overlies the defect.
The murmur of restrictive ventricular septal defect is most often holosystolic because the pressure difference between the ventricles is generated almost instantly at the onset of systole with a left-to-right shunt continuing throughout ventricular contraction. In contrast, nonrestrictive ventricular septal defects generally do not generate a murmur, since pressure is equalized across the defect. This left-to-right shunting may lead to right ventricular volume overload, resulting in delayed closure of the pulmonary valve and a widely split S2. Irreversible pulmonary hypertension with shunt reversal may occur if the defect remains untreated.1
Atrial septal defect. The most characteristic feature of atrial septal defect is a fixed split S2 resulting from right ventricular volume overload due to left-to-right atrial shunting of blood flow. As flow is shunted from the left to the right atrium and subsequently into the right ventricle, ejection of excess blood through the pulmonary valve produces a midsystolic flow murmur, heard best over the left upper sternal border, that may radiate to the back.
Ruptured sinus of Valsalva aneurysm. The pressure is higher in the aorta than in the right atrium throughout the cardiac cycle, and if a shunt is created between the two structures by a ruptured sinus of Valsalva aneurysm, the blood flow across this shunt throughout the cardiac cycle produces a continuous murmur. In contrast, if a sinus of Valsalva aneurysm ruptures into the right ventricle, the murmur is accentuated in diastole and attenuated in systole, and is often associated with pounding pulses and a thrill along either the left or right sternal border.1
Aortic regurgitation causes a diastolic murmur as blood flows retrograde into the left ventricle through the incompetent aortic valve. This murmur is usually described as a blowing, decrescendo murmur heard best at the third left intercostal space.
Patent ductus arteriosus is a communication between the descending thoracic aorta and the pulmonary artery that fails to close at birth. The hallmark murmur associated with this defect is a continuous “machine-like” murmur located at the upper left sternal border, often radiating down the left side of the sternum into the back. Of note, increasing the systemic pressure by the Valsalva maneuver or handgrip exercise will increase the diastolic component of the continuous murmur associated with ruptured sinus of Valsalva aneurysm, helping to differentiate it from patent ductus arteriosus.2
Pulmonic stenosis causes a systolic murmur heard best at the second intercostal space along the left sternal border and having a crescendo-decrescendo intensity and harsh quality. As the right ventricle takes longer to eject its blood volume through the stenotic pulmonary valve, the delay in closure between the aortic and pulmonary valve is widened, resulting in a significant splitting of the S2. In addition, any maneuver that increases preload will also increase the intensity of the murmur.3
Our patient has a murmur that is continuous, is heard across the entire precordium, and has no respiratory variation. These features are most consistent with a sinus of Valsalva aneurysm that has ruptured into the right atrium.
The 2008 update of the joint American College of Cardiology and American Heart Association guidelines4 recommends further evaluation of diastolic or continuous murmurs with echocardiography, as these murmurs are most often signs of a pathologic condition. In addition, echocardiography is warranted to evaluate grade 3 or higher systolic murmurs and those that are holosystolic.4
SINUS OF VALSALVA ANEURYSM
Sinus of Valsalva aneurysm is rare, with an incidence of 0.09% to 0.15%. From 65% to 85% are in the right coronary cusp, 10% to 30% are in the noncoronary cusp, and fewer than 5% are in the left coronary cusp.5
This condition is most often congenital, accounting for up to 3.5% of congenital cardiac anomalies, though it can be acquired. Formation of the aneurysm is generally related to weakening of elastic fibers and muscular tissues that progresses over time.
Many cases of sinus of Valsalva aneurysm are associated with additional cardiac defects.1 Ventricular septal defect is the most common coexisting congenital anomaly, occurring in up to 53% of patients and frequently associated with aneurysms involving the right coronary cusp and with sinus of Valsalva aneurysm.6 Other congenital anomalies often accompanying sinus of Valsalva aneurysm include pulmonary stenosis, atrial septal defect, bicuspid aortic valve, tetralogy of Fallot, patent ductus arteriosus, coarctation of the aorta, and subaortic stenosis. Another associated condition is aortic regurgitation, for which more than half of affected patients eventually require aortic valve replacement.2
Acquired sinus of Valsalva aneurysm can be the result of endocarditis, trauma, surgery, cardiac catheterization, or inflammatory or degenerative processes including, rarely, tertiary syphilis.3
Sinus of Valsalva aneurysm often remains asymptomatic, but symptoms may arise if the aneurysm ruptures, resulting in intracardiac shunting or aneurysm-associated compression of adjacent cardiac structures such as coronary arteries. Rupture may be spontaneous, secondary to chest trauma or excess exertion, or iatrogenic.
Imaging studies such as echocardiography, cardiac computed tomography, and cardiac magnetic resonance imaging are essential in diagnosing and managing sinus of Valsalva aneurysm and identifying coexisting cardiac anomalies.
Rupture occurs most commonly into the right ventricle, followed in frequency by the right atrium or left atrium. Once rupture occurs, median survival is 1 to 2 years if left untreated, with death often secondary to congestive heart failure or infective endocarditis.7
Surgery remains the preferred approach to the treatment of ruptured sinus of Valsalva aneurysm. Operative risk is reasonably low and long-term outcomes are good. The appropriate therapy for unruptured and asymptomatic sinus of Valsalva aneurysm remains less clear.
Successful transcatheter closure of ruptured sinus of Valsalva aneurysm has been described using Amplatzer devices, a procedure that avoids sternotomy and cardiopulmonary bypass. Despite advances in percutaneous techniques, open surgery with or without aortic valve replacement remains the current standard of care.8
BACK TO OUR PATIENT
In the case described above, the initial diagnostic study done to evaluate the patient’s dyspnea and murmur was transthoracic echocardiography, which demonstrated a relatively preserved ejection fraction with mild aortic regurgitation and an aneurysmal structure extending from the aortic root toward the right atrium.
Transesophageal echocardiography confirmed this finding (Figure 1). Cross-sectional imaging of the aortic valve (Figure 2) showed the aneurysm arising from the noncoronary cusp and communicating with the right atrium. Color flow Doppler (Figure 3) confirmed continuous flow between the aneurysmal sinus and right atrium throughout the cardiac cycle, consistent with the continuous murmur noted on physical examination.
The aneurysm was also noted on aortography (Figure 4) obtained before the patient underwent surgery to correct it. The surgery was successful, no complications occurred, and the murmur and associated dyspnea had completely resolved at subsequent follow-up.
This case highlights the importance of imaging studies such as echocardiography in diagnosing and managing sinus of Valsalva aneurysm, and also the importance of physical examination in guiding the diagnostic evaluation and differentiating this condition from other cardiac disorders.
- Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011:1411–1468.
- Topi B, John J, Agarwal A, et al. An uncommon cause of a continuous murmur. Exp Clin Cardiol 2012; 17:148–149.
- Constant J. Bedside Cardiology. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999:268–320.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Jung SH, Yun TJ, Im YM, et al. Ruptured sinus of Valsalva aneurysm: transaortic repair may cause sinus of Valsalva distortion and aortic regurgitation. J Thorac Cardiovasc Surg 2008; 135:1153–1158.
- Post MC, Braam RL, Groenemeijer BE, Nicastia D, Rensing BJ, Schepens MA. Rupture of right coronary sinus of Valsalva aneurysm into right ventricle. Neth Heart J 2010; 18:209–211.
- Moustafa S, Mookadam F, Cooper L, et al. Sinus of Valsalva aneurysms—47 years of a single center experience and systematic overview of published reports. Am J Cardiol 2007; 99:1159–1164.
- Zhao SH, Yan CW, Zhu XY, et al. Transcatheter occlusion of the ruptured sinus of Valsalva aneurysm with an Amplatzer duct occluder. Int J Cardiol 2008; 129:81–85.
- Bonow RO, Mann DL, Zipes DP, Libby P. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2011:1411–1468.
- Topi B, John J, Agarwal A, et al. An uncommon cause of a continuous murmur. Exp Clin Cardiol 2012; 17:148–149.
- Constant J. Bedside Cardiology. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999:268–320.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Jung SH, Yun TJ, Im YM, et al. Ruptured sinus of Valsalva aneurysm: transaortic repair may cause sinus of Valsalva distortion and aortic regurgitation. J Thorac Cardiovasc Surg 2008; 135:1153–1158.
- Post MC, Braam RL, Groenemeijer BE, Nicastia D, Rensing BJ, Schepens MA. Rupture of right coronary sinus of Valsalva aneurysm into right ventricle. Neth Heart J 2010; 18:209–211.
- Moustafa S, Mookadam F, Cooper L, et al. Sinus of Valsalva aneurysms—47 years of a single center experience and systematic overview of published reports. Am J Cardiol 2007; 99:1159–1164.
- Zhao SH, Yan CW, Zhu XY, et al. Transcatheter occlusion of the ruptured sinus of Valsalva aneurysm with an Amplatzer duct occluder. Int J Cardiol 2008; 129:81–85.
An unusual cause of vitamin B12 and iron deficiency
A 76-year-old woman visiting from Ethiopia presented for further evaluation of concomitant iron and vitamin B12 deficiency anemia that had developed over the previous 6 months. During that time, she had complained of ongoing fatigue and increasing paresthesias in the hands and feet.
At presentation, her hemoglobin concentration was 7.8 g/dL (reference range 11.5–15), with a mean corpuscular volume of 81.8 fL (81.5–97.0). These values were down from her baseline hemoglobin of 12 g/dL and corpuscular volume of 85.8 recorded more than 1 year ago. Serum studies showed an iron concentration of 21 µg/dL (37–170), ferritin 3 ng/mL (10–107), and percent saturation of transferrin 5% (20%–55%). Also noted was a low vitamin B12 level of 108 pg/mL (180–1,241 pg/mL). She had no overt signs of gastrointestinal blood loss. She did not report altered bowel habits or use of nonsteroidal anti-inflammatory medications.
Given her country of origin, she was sent for initial stool testing for ova and parasites, which was unrevealing.
She underwent esophagogastroduodenoscopy and colonoscopy, which revealed no underlying cause of her iron deficiency or vitamin B12 insufficiency. But further evaluation with capsule endoscopy showed evidence of a tapeworm in the distal duodenum (Figure 1).
She was given praziquantel in a single oral dose of 10 mg/kg. Repeat stool culture 1 month later showed no evidence of tapeworm infection, and at follow-up 3 months later, her hemoglobin had recovered to 13.2 g/dL with a corpuscular volume of 87.6 fL and no residual vitamin B12 or iron deficiency. She reported complete resolution of fatigue and of paresthesias of the hands and feet.
DIPHYLLOBOTHRIUM LATUM
The appearance on capsule endoscopy indicated Diphyllobothrium latum as the likely parasite. This tapeworm is acquired by ingesting undercooked or raw fish. Infection is most common in Northern Europe but has been reported in Africa.1
As it grows, the tapeworm develops chains of segments and can reach a length of 1 to 15 meters.1 In humans, it typically resides in the small intestine. Most patients are asymptomatic or have moderate nonspecific symptoms such as abdominal pain and diarrhea. A key differentiating aspect of D latum infection is vitamin B12 deficiency caused by consumption of the vitamin by the parasite, as well as by parasite-mediated dissociation of the vitamin B12-intrinsic factor complex, thus making the vitamin unavailable to the host.
Up to 40% of people infected with D latum develop low levels of vitamin B12, and 2% develop symptomatic megaloblastic anemia.2 Iron deficiency anemia is uncommon but has been reported.3 In our patient, the concomitant iron deficiency was probably secondary to involvement of the duodenum, where a significant amount of dietary iron is absorbed.
The diagnosis is typically established by stool testing for ova and parasites. When stool samples do not reveal a cause of the symptoms, as in this patient, endoscopy can be used. Capsule endoscopy has not been widely used in the diagnosis of intestinal helminth infection, although reports exist describing the use of capsule endoscopy to detect intestinal parasites. Notably, as in this case, intestinal parasite infection is occasionally found during investigations of anemia and vitamin deficiencies of unknown cause.4
As in our patient, treatment of infection with this species of tapeworm typically involves a single oral dose of praziquantel; this off-label use has been shown to lead to resolution of symptoms in nearly all patients treated.5
- Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am 1996; 25:637–653.
- Scholz T, Garcia HH, Kuchta R, Wicht B. Update on the human broad tapeworm (genus Diphyllobothrium), including clinical relevance. Clin Microbiol Rev 2009; 22:146–160,
- Stanciu C, Trifan A, Singeap AM, Sfarti C, Cojocariu C, Luca M. Diphyllobothrium latum identified by capsule endoscopy—an unusual cause of iron-deficiency anemia. J Gastrointestin Liver Dis 2009; 18:142.
- Soga K, Handa O, Yamada M, et al. In vivo imaging of intestinal helminths by capsule endoscopy. Parasitol Int 2014; 63:221–228.
- Drugs for Parasitic Infections. 3rd edition. Treatment guidelines from the Medical Letter 2010. The Medical Letter, Inc., New Rochelle, NY.
A 76-year-old woman visiting from Ethiopia presented for further evaluation of concomitant iron and vitamin B12 deficiency anemia that had developed over the previous 6 months. During that time, she had complained of ongoing fatigue and increasing paresthesias in the hands and feet.
At presentation, her hemoglobin concentration was 7.8 g/dL (reference range 11.5–15), with a mean corpuscular volume of 81.8 fL (81.5–97.0). These values were down from her baseline hemoglobin of 12 g/dL and corpuscular volume of 85.8 recorded more than 1 year ago. Serum studies showed an iron concentration of 21 µg/dL (37–170), ferritin 3 ng/mL (10–107), and percent saturation of transferrin 5% (20%–55%). Also noted was a low vitamin B12 level of 108 pg/mL (180–1,241 pg/mL). She had no overt signs of gastrointestinal blood loss. She did not report altered bowel habits or use of nonsteroidal anti-inflammatory medications.
Given her country of origin, she was sent for initial stool testing for ova and parasites, which was unrevealing.
She underwent esophagogastroduodenoscopy and colonoscopy, which revealed no underlying cause of her iron deficiency or vitamin B12 insufficiency. But further evaluation with capsule endoscopy showed evidence of a tapeworm in the distal duodenum (Figure 1).
She was given praziquantel in a single oral dose of 10 mg/kg. Repeat stool culture 1 month later showed no evidence of tapeworm infection, and at follow-up 3 months later, her hemoglobin had recovered to 13.2 g/dL with a corpuscular volume of 87.6 fL and no residual vitamin B12 or iron deficiency. She reported complete resolution of fatigue and of paresthesias of the hands and feet.
DIPHYLLOBOTHRIUM LATUM
The appearance on capsule endoscopy indicated Diphyllobothrium latum as the likely parasite. This tapeworm is acquired by ingesting undercooked or raw fish. Infection is most common in Northern Europe but has been reported in Africa.1
As it grows, the tapeworm develops chains of segments and can reach a length of 1 to 15 meters.1 In humans, it typically resides in the small intestine. Most patients are asymptomatic or have moderate nonspecific symptoms such as abdominal pain and diarrhea. A key differentiating aspect of D latum infection is vitamin B12 deficiency caused by consumption of the vitamin by the parasite, as well as by parasite-mediated dissociation of the vitamin B12-intrinsic factor complex, thus making the vitamin unavailable to the host.
Up to 40% of people infected with D latum develop low levels of vitamin B12, and 2% develop symptomatic megaloblastic anemia.2 Iron deficiency anemia is uncommon but has been reported.3 In our patient, the concomitant iron deficiency was probably secondary to involvement of the duodenum, where a significant amount of dietary iron is absorbed.
The diagnosis is typically established by stool testing for ova and parasites. When stool samples do not reveal a cause of the symptoms, as in this patient, endoscopy can be used. Capsule endoscopy has not been widely used in the diagnosis of intestinal helminth infection, although reports exist describing the use of capsule endoscopy to detect intestinal parasites. Notably, as in this case, intestinal parasite infection is occasionally found during investigations of anemia and vitamin deficiencies of unknown cause.4
As in our patient, treatment of infection with this species of tapeworm typically involves a single oral dose of praziquantel; this off-label use has been shown to lead to resolution of symptoms in nearly all patients treated.5
A 76-year-old woman visiting from Ethiopia presented for further evaluation of concomitant iron and vitamin B12 deficiency anemia that had developed over the previous 6 months. During that time, she had complained of ongoing fatigue and increasing paresthesias in the hands and feet.
At presentation, her hemoglobin concentration was 7.8 g/dL (reference range 11.5–15), with a mean corpuscular volume of 81.8 fL (81.5–97.0). These values were down from her baseline hemoglobin of 12 g/dL and corpuscular volume of 85.8 recorded more than 1 year ago. Serum studies showed an iron concentration of 21 µg/dL (37–170), ferritin 3 ng/mL (10–107), and percent saturation of transferrin 5% (20%–55%). Also noted was a low vitamin B12 level of 108 pg/mL (180–1,241 pg/mL). She had no overt signs of gastrointestinal blood loss. She did not report altered bowel habits or use of nonsteroidal anti-inflammatory medications.
Given her country of origin, she was sent for initial stool testing for ova and parasites, which was unrevealing.
She underwent esophagogastroduodenoscopy and colonoscopy, which revealed no underlying cause of her iron deficiency or vitamin B12 insufficiency. But further evaluation with capsule endoscopy showed evidence of a tapeworm in the distal duodenum (Figure 1).
She was given praziquantel in a single oral dose of 10 mg/kg. Repeat stool culture 1 month later showed no evidence of tapeworm infection, and at follow-up 3 months later, her hemoglobin had recovered to 13.2 g/dL with a corpuscular volume of 87.6 fL and no residual vitamin B12 or iron deficiency. She reported complete resolution of fatigue and of paresthesias of the hands and feet.
DIPHYLLOBOTHRIUM LATUM
The appearance on capsule endoscopy indicated Diphyllobothrium latum as the likely parasite. This tapeworm is acquired by ingesting undercooked or raw fish. Infection is most common in Northern Europe but has been reported in Africa.1
As it grows, the tapeworm develops chains of segments and can reach a length of 1 to 15 meters.1 In humans, it typically resides in the small intestine. Most patients are asymptomatic or have moderate nonspecific symptoms such as abdominal pain and diarrhea. A key differentiating aspect of D latum infection is vitamin B12 deficiency caused by consumption of the vitamin by the parasite, as well as by parasite-mediated dissociation of the vitamin B12-intrinsic factor complex, thus making the vitamin unavailable to the host.
Up to 40% of people infected with D latum develop low levels of vitamin B12, and 2% develop symptomatic megaloblastic anemia.2 Iron deficiency anemia is uncommon but has been reported.3 In our patient, the concomitant iron deficiency was probably secondary to involvement of the duodenum, where a significant amount of dietary iron is absorbed.
The diagnosis is typically established by stool testing for ova and parasites. When stool samples do not reveal a cause of the symptoms, as in this patient, endoscopy can be used. Capsule endoscopy has not been widely used in the diagnosis of intestinal helminth infection, although reports exist describing the use of capsule endoscopy to detect intestinal parasites. Notably, as in this case, intestinal parasite infection is occasionally found during investigations of anemia and vitamin deficiencies of unknown cause.4
As in our patient, treatment of infection with this species of tapeworm typically involves a single oral dose of praziquantel; this off-label use has been shown to lead to resolution of symptoms in nearly all patients treated.5
- Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am 1996; 25:637–653.
- Scholz T, Garcia HH, Kuchta R, Wicht B. Update on the human broad tapeworm (genus Diphyllobothrium), including clinical relevance. Clin Microbiol Rev 2009; 22:146–160,
- Stanciu C, Trifan A, Singeap AM, Sfarti C, Cojocariu C, Luca M. Diphyllobothrium latum identified by capsule endoscopy—an unusual cause of iron-deficiency anemia. J Gastrointestin Liver Dis 2009; 18:142.
- Soga K, Handa O, Yamada M, et al. In vivo imaging of intestinal helminths by capsule endoscopy. Parasitol Int 2014; 63:221–228.
- Drugs for Parasitic Infections. 3rd edition. Treatment guidelines from the Medical Letter 2010. The Medical Letter, Inc., New Rochelle, NY.
- Schantz PM. Tapeworms (cestodiasis). Gastroenterol Clin North Am 1996; 25:637–653.
- Scholz T, Garcia HH, Kuchta R, Wicht B. Update on the human broad tapeworm (genus Diphyllobothrium), including clinical relevance. Clin Microbiol Rev 2009; 22:146–160,
- Stanciu C, Trifan A, Singeap AM, Sfarti C, Cojocariu C, Luca M. Diphyllobothrium latum identified by capsule endoscopy—an unusual cause of iron-deficiency anemia. J Gastrointestin Liver Dis 2009; 18:142.
- Soga K, Handa O, Yamada M, et al. In vivo imaging of intestinal helminths by capsule endoscopy. Parasitol Int 2014; 63:221–228.
- Drugs for Parasitic Infections. 3rd edition. Treatment guidelines from the Medical Letter 2010. The Medical Letter, Inc., New Rochelle, NY.
Umbilical hernia in a patient with cirrhosis
A 62-year-old man was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
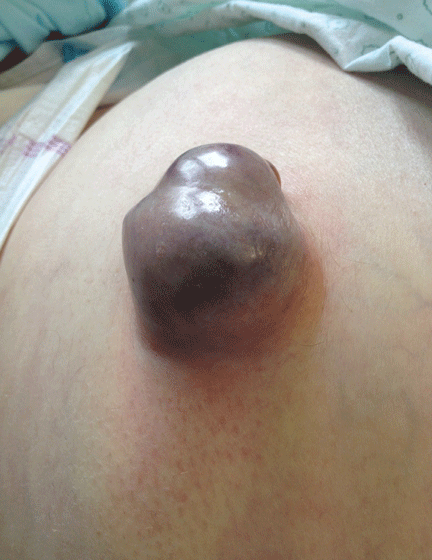
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
A 62-year-old man was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
A 62-year-old man was admitted to the intensive care unit with esophageal variceal bleeding. He had a long history of alcohol abuse with secondary cirrhosis, with a Child-Pugh score of 11 on a scale of 15 (class C—the most severe) at presentation. He also had a history of uncomplicated umbilical hernia, 6 cm in diameter without overlying trophic skin alterations.
Treatment with somatostatin, endoscopic band ligation, and prophylactic antibiotics was initiated for the variceal bleeding. The next day, he was transferred to the hepatology floor. His condition stabilized during the next week, but then he abruptly became diaphoretic and less talkative. Physical examination revealed a painful and irreducible umbilical hernia (Figure 1). He was rushed for umbilical hernia repair with resection of a necrotic segment of small bowel. His recovery after surgery was uneventful, and he was eventually discharged.
UMBILICAL HERNIA AND CIRRHOSIS
Umbilical hernia is common in cirrhotic patients suffering from ascites, with a prevalence up to 20%, which is 10 times higher than in the general population.1 Ascites is the major predisposing factor since it causes muscle wasting and increases intra-abdominal pressure.
A unique feature of cirrhosis is low physiologic reserve, which increases the risk of death from complications of umbilical hernia and makes the patient more vulnerable to perioperative complications during repair. Because of the high operative risk, umbilical hernia repair has traditionally been reserved for the most complicated cases, such as strangulation of the bowel or rupture of the skin with leakage of ascitic fluid.2,3 Many patients are thus managed conservatively, with watchful waiting.
However, the natural course of umbilical hernia tends toward complications (eg, bowel incarceration, rupture of the overlying skin), which necessitate urgent repair.4 The risk of death with hernia repair in this urgent setting is seven times higher than for elective hernia repair in cirrhotic patients.5 More recent data indicate that elective repair in patients with well-compensated cirrhosis carries complication and mortality rates similar to those in noncirrhotic patients.5–8 Therefore, patients who should undergo umbilical hernia repair are not only those with complicated umbilical hernia (strangulation or ascites leak), but also those with well-compensated cirrhosis at risk of complications.
Factors that pose a particularly high risk of complications of repair are large hernia (> 5 cm), hernia associated with pain, intermittent incarceration, and trophic alterations of the overlying skin.1 In these patients, elective repair should be considered if hepatic function is preserved, if ascites is well managed (sodium restriction, diuretics, and sometimes even preoperative transjugular intrahepatic portosystemic shunt placement), and if the patient is not expected to undergo liver transplantation in the near future. If liver transplantation is anticipated in the short term, umbilical hernia can be managed concomitantly. Management of ascites after umbilical hernia repair is essential for prevention of recurrence.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
- Dokmak S, Aussilhou B, Belghiti J. Umbilical hernias and cirrhose. J Visc Surg 2012; 149(suppl 5):e32–e39.
- Baron HC. Umbilical hernia secondary to cirrhosis of the liver. Complications of surgical correction. N Engl J Med 1960; 263:824–828.
- Hansen JB, Thulstrup AM, Vilstup H, Sørensen HT. Danish nationwide cohort study of postoperative death in patients with liver cirrhosis undergoing hernia repair. Br J Surg 2002; 89:805–806.
- Marsman HA, Heisterkamp J, Halm JA, Tilanus HW, Metselaar HJ, Kazemier G. Management in patients with liver cirrhosis and an umbilical hernia. Surgery 2007; 142:372–375.
- Carbonell AM, Wolfe LG, DeMaria EJ. Poor outcomes in cirrhosis-associated hernia repair: a nationwide cohort study of 32,033 patients. Hernia 2005; 9:353–357.
- Eker HH, van Ramshorst GH, de Goede B, et al. A prospective study on elective umbilical hernia repair in patients with liver cirrhosis and ascites. Surgery 2011; 150:542–546.
- Gray SH, Vick CC, Graham LA, Finan KR, Neumayer LA, Hawn MT. Umbilical herniorrhapy in cirrhosis: improved outcomes with elective repair. J Gastrointest Surg 2008; 12:675–681.
- McKay A, Dixon E, Bathe O, Sutherland F. Umbilical hernia repair in the presence of cirrhosis and ascites: results of a survey and review of the literature. Hernia 2009; 13:461–468.
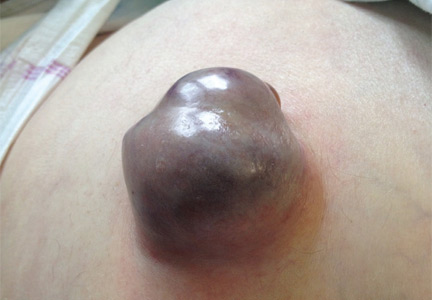
Electrocardiographic changes in amitriptyline overdose
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
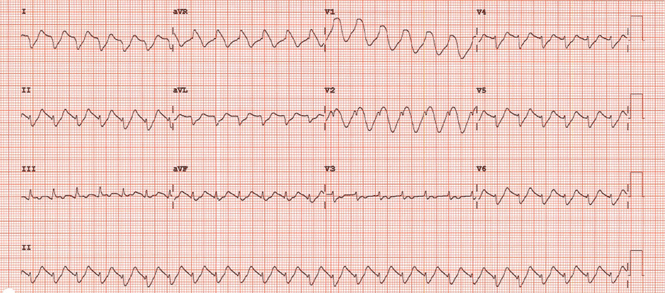
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
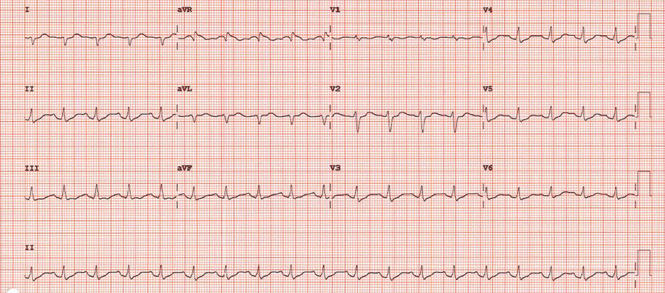
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
A 49-year-old woman with a history of depression, bipolar disorder, and chronic back pain was brought to the emergency department unresponsive after having taken an unknown quantity of amitriptyline tablets.
On arrival, she was comatose, with a score of 3 (the lowest possible score) on the 15-point Glasgow Coma Scale. Her blood pressure was 65/22 mm Hg, heart rate 121 beats per minute, respiratory rate 14 per minute, and oxygen saturation 88% on room air. The rest of the initial physical examination was normal.
She was immediately intubated, put on mechanical ventilation, and given an infusion of a 1-L bolus of normal saline and 50 mmol (1 mmol/kg) of sodium bicarbonate. Norepinephrine infusion was started. Gastric lavage was not done.
Results of initial laboratory testing showed a serum potassium of 2.9 mmol/L (reference range 3.5–5.0) and a serum magnesium of 1.6 mmol/L (1.7–2.6), which were corrected with infusion of 60 mmol of potassium chloride and 2 g of magnesium sulfate. The serum amitriptyline measurement was ordered at the time of her presentation to the emergency department.
Arterial blood gas analysis showed:
- pH 7.15 (normal range 7.35–7.45)
- Paco2 66 mm Hg (34–46)
- Pao2 229 mm Hg (85–95)
- Bicarbonate 22 mmol/L (22–26).
The initial electrocardiogram (ECG) (Figure 1) showed regular wide-complex tachycardia with no definite right or left bundle branch block morphology, no discernible P waves, a QRS duration of 198 msec, right axis deviation, and no Brugada criteria to suggest ventricular tachycardia.
She remained hypotensive, with regular wide-complex tachycardia on the ECG. She was given an additional 1-L bolus of normal saline and 100 mmol (2 mmol/kg) of sodium bicarbonate, and within 1 minute the wide-complex tachycardia resolved to narrow-complex sinus tachycardia (Figure 2). At this point, an infusion of 150 mmol/L of sodium bicarbonate in dextrose 5% in water was started, with serial ECGs to monitor the QRS duration and serial arterial blood gas monitoring to maintain the pH between 7.45 and 7.55.
TRANSFER TO THE ICU
She was then transferred to the intensive care unit (ICU), where she remained for 2 weeks. While in the ICU, she had a single recurrence of wide-complex tachycardia that resolved immediately with an infusion of 100 mmol of sodium bicarbonate. A urine toxicology screen was negative, and the serum amitriptyline measurement, returned from the laboratory 48 hours after her initial presentation, was 594 ng/mL (reference range 100–250 ng/mL). She was eventually weaned off the norepinephrine infusion after 20 hours, the sodium bicarbonate infusion was discontinued after 4 days, and she was taken off mechanical ventilation after 10 days. Also during her ICU stay, she had seizures on day 3 and developed aspiration pneumonia.
From the ICU, she was transferred to a regular floor, where she stayed for another week and then was transferred to a rehabilitation center. This patient was known to have clinical depression and to have attempted suicide once before. She had recently been under additional psychosocial stresses, which likely prompted this second attempt.
She reportedly had no neurologic or cardiovascular sequelae after her discharge from the hospital.
AMITRIPTYLINE OVERDOSE
Amitriptyline causes a relatively high number of fatal overdoses, at 34 per 1 million prescriptions.1 Death is usually from hypotension and ventricular arrhythmia caused by blockage of cardiac fast sodium channels leading to disturbances of cardiac conduction such as wide-complex tachycardia.
Other manifestations of amitriptyline overdose include seizures, sedation, and anticholinergic toxicity from variable blockade of gamma-aminobutyric acid receptors, histamine 1 receptors, and alpha receptors.2
Of the various changes on ECG described with amitriptyline overdose, sinus tachycardia is the most common. A QRS duration greater than 100 msec, right to extreme-right axis deviation with negative QRS complexes in leads I and aVL, and an R-wave amplitude greater than 3 mm in lead aVR are indications for sodium bicarbonate infusion, especially in hemodynamically unstable patients.3 Sodium bicarbonate increases the serum concentration of sodium and thereby overcomes the sodium channel blockade. It also alkalinizes the serum, favoring an electrically neutral form of amitriptyline that binds less to receptors and binds more to alpha-1-acid glycoprotein, decreasing the fraction of free drug available for toxicity.4
In patients with amitriptyline overdose, wide-complex tachycardia and hypotension refractory to sodium bicarbonate infusion can be treated with lidocaine, magnesium sulfate, direct-current cardioversion, and lipid resuscitation.5,6 Treatment with class IA, IC, and III antiarrhythmics is contraindicated, as they block sodium channels and thus can worsen conduction disturbances.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
- Henry JA, Alexander CA, Sener EK. Relative mortality from overdose of antidepressants. BMJ 1995; 310:221–224.
- Shannon M, Merola J, Lovejoy FH Jr. Hypotension in severe tricyclic antidepressant overdose. Am J Emerg Med 1988; 6:439–442.
- Liebelt EL, Francis PD, Woolf AD. ECG lead aVR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med 1995; 26:195–201.
- Sayniuk BI, Jhamandas V. Mechanism of reversal of toxic effects of amitriptyline on cardiac Purkinje fibres by sodium bicarbonate. J Pharmacol Exp Ther 1984; 231:387.
- Kiberd MB, Minor SF. Lipid therapy for the treatment of a refractory amitriptyline overdose. CJEM 2012; 14:193–197.
- Harvey M, Cave G. Case report: successful lipid resuscitation in multidrug overdose with predominant tricyclic antidepressant toxidrome. Int J Emerg Med 2012; 5:8.
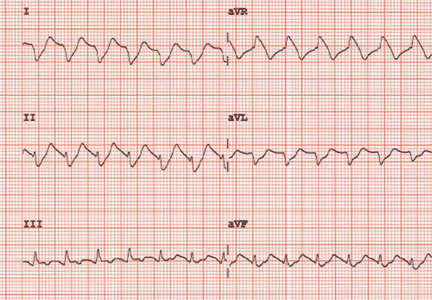
The cohabitation of art and genomic science

The art of medicine includes picking the right drug for the right patient, especially when we can choose between different classes of efficacious therapies. But, in view of our growing understanding of the human genome, can science replace art?
That question is part of the promise of pharmacogenetics, the study of how inter-individual genetic differences influence a patient’s response to a specific drug. A patient’s genome dictates the expression of specific enzymes that metabolize a drug with various efficiencies: variant alleles may result in slightly different proteins that express different enzymatic activity, ie, different substrate affinities for a drug resulting in more or less efficient metabolism. Genomic differences may also dictate whether a specific biochemical pathway is dominant in generating a specific pathophysiologic response, in which case drugs that affect that pathway may be strikingly effective. This may partly explain the various responses to different antihypertensive drugs.
Another less well-understood example of pharmacogenetics is the link between specific HLA haplotypes and a dramatic increase in allergic reactions to specific medications, such as the link between HLA-B*57:01 and abacavir hypersensitivity.
In this issue of the Journal, DiPiero et al discuss thiopurine methyltransferase (TPMT), an enzyme responsible for the degradation of azathioprine, and how knowing the genetically determined relative activity of this enzyme should influence our initial dosing of this and related drugs. Patients with certain variant alleles of TPMT degrade azathioprine more slowly, and these patients are at higher risk of myelosuppressive toxicity from the drug when it is given at the full weight-based dose. The TPMT test is expensive but not prohibitively so, and it would seem that genomic testing is a reasonable clinical and cost-effective option.
As in the abacavir scenario noted above, genomic-based dosing of azathioprine makes scientific sense and offers proof of principle for the validity of pharmacogenomics. But is it truly a clinical game-changer?
The answer depends in part on how the prescribing physician doses the drug, which depends in part on what disease is being treated, how fast the drug needs to be at full dose, and whether there are equally effective alternatives. Recommendations have been offered that state if TPMT activity is normal, we can start at the usual maintenance dose of 1.5 to 2 mg/kg/day (or occasionally more). But if the patient is heterozygous for the wild-type gene and thus is a slower drug metabolizer, then initial dosing “should” be reduced to 25 to 50 mg/day, with close observation of the white blood cell count as the dose is slowly increased to the target. The very rare patient who is homozygous for a non–wild-type allele should not be given the drug.
My usual practice has been to start patients on 50 mg or less daily and slowly titrate up, asking them how they are tolerating the drug and watching the white count—notably, the same approach to be taken if I had done genotyping before starting the drug and had found the patient to be heterozygous for the TPMT gene.
Interestingly, one pragmatic clinical trial tested whether genotyping patients before starting azathioprine—with subsequent suggested dosing of the drug based on the genotype as above—was safer and cheaper than letting physicians dose as they chose.1 It turned out that physicians participating in this study still dosed their patients conservatively. Even knowing that they might be able to give full doses from the start in patients with normal TPMT activity, many chose not to. I assume that many of those physicians felt as I do that there was no urgency in reaching the presumed-to-be-effective full weight-based therapeutic dose. (We don’t have a good clinical marker of azathioprine’s efficacy). At 4 months, the maintenance dose was about the same in all groups.
We have robust evidence to support the role of pharmacogenetics in informing the dosing of several medications, more than just the ones I have mentioned here. And in the right settings, we should use pharmacogenetic testing to limit toxicity and perhaps enhance efficacy in our drug selection. As the field moves rapidly forward, we will have many opportunities to improve clinical care by using our patients’ genomic information.
But like it or bemoan it, even when we have science in the house, the art of medicine still plays a role in our clinical decisions.
- Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014; 17:22–33.
The art of medicine includes picking the right drug for the right patient, especially when we can choose between different classes of efficacious therapies. But, in view of our growing understanding of the human genome, can science replace art?
That question is part of the promise of pharmacogenetics, the study of how inter-individual genetic differences influence a patient’s response to a specific drug. A patient’s genome dictates the expression of specific enzymes that metabolize a drug with various efficiencies: variant alleles may result in slightly different proteins that express different enzymatic activity, ie, different substrate affinities for a drug resulting in more or less efficient metabolism. Genomic differences may also dictate whether a specific biochemical pathway is dominant in generating a specific pathophysiologic response, in which case drugs that affect that pathway may be strikingly effective. This may partly explain the various responses to different antihypertensive drugs.
Another less well-understood example of pharmacogenetics is the link between specific HLA haplotypes and a dramatic increase in allergic reactions to specific medications, such as the link between HLA-B*57:01 and abacavir hypersensitivity.
In this issue of the Journal, DiPiero et al discuss thiopurine methyltransferase (TPMT), an enzyme responsible for the degradation of azathioprine, and how knowing the genetically determined relative activity of this enzyme should influence our initial dosing of this and related drugs. Patients with certain variant alleles of TPMT degrade azathioprine more slowly, and these patients are at higher risk of myelosuppressive toxicity from the drug when it is given at the full weight-based dose. The TPMT test is expensive but not prohibitively so, and it would seem that genomic testing is a reasonable clinical and cost-effective option.
As in the abacavir scenario noted above, genomic-based dosing of azathioprine makes scientific sense and offers proof of principle for the validity of pharmacogenomics. But is it truly a clinical game-changer?
The answer depends in part on how the prescribing physician doses the drug, which depends in part on what disease is being treated, how fast the drug needs to be at full dose, and whether there are equally effective alternatives. Recommendations have been offered that state if TPMT activity is normal, we can start at the usual maintenance dose of 1.5 to 2 mg/kg/day (or occasionally more). But if the patient is heterozygous for the wild-type gene and thus is a slower drug metabolizer, then initial dosing “should” be reduced to 25 to 50 mg/day, with close observation of the white blood cell count as the dose is slowly increased to the target. The very rare patient who is homozygous for a non–wild-type allele should not be given the drug.
My usual practice has been to start patients on 50 mg or less daily and slowly titrate up, asking them how they are tolerating the drug and watching the white count—notably, the same approach to be taken if I had done genotyping before starting the drug and had found the patient to be heterozygous for the TPMT gene.
Interestingly, one pragmatic clinical trial tested whether genotyping patients before starting azathioprine—with subsequent suggested dosing of the drug based on the genotype as above—was safer and cheaper than letting physicians dose as they chose.1 It turned out that physicians participating in this study still dosed their patients conservatively. Even knowing that they might be able to give full doses from the start in patients with normal TPMT activity, many chose not to. I assume that many of those physicians felt as I do that there was no urgency in reaching the presumed-to-be-effective full weight-based therapeutic dose. (We don’t have a good clinical marker of azathioprine’s efficacy). At 4 months, the maintenance dose was about the same in all groups.
We have robust evidence to support the role of pharmacogenetics in informing the dosing of several medications, more than just the ones I have mentioned here. And in the right settings, we should use pharmacogenetic testing to limit toxicity and perhaps enhance efficacy in our drug selection. As the field moves rapidly forward, we will have many opportunities to improve clinical care by using our patients’ genomic information.
But like it or bemoan it, even when we have science in the house, the art of medicine still plays a role in our clinical decisions.
The art of medicine includes picking the right drug for the right patient, especially when we can choose between different classes of efficacious therapies. But, in view of our growing understanding of the human genome, can science replace art?
That question is part of the promise of pharmacogenetics, the study of how inter-individual genetic differences influence a patient’s response to a specific drug. A patient’s genome dictates the expression of specific enzymes that metabolize a drug with various efficiencies: variant alleles may result in slightly different proteins that express different enzymatic activity, ie, different substrate affinities for a drug resulting in more or less efficient metabolism. Genomic differences may also dictate whether a specific biochemical pathway is dominant in generating a specific pathophysiologic response, in which case drugs that affect that pathway may be strikingly effective. This may partly explain the various responses to different antihypertensive drugs.
Another less well-understood example of pharmacogenetics is the link between specific HLA haplotypes and a dramatic increase in allergic reactions to specific medications, such as the link between HLA-B*57:01 and abacavir hypersensitivity.
In this issue of the Journal, DiPiero et al discuss thiopurine methyltransferase (TPMT), an enzyme responsible for the degradation of azathioprine, and how knowing the genetically determined relative activity of this enzyme should influence our initial dosing of this and related drugs. Patients with certain variant alleles of TPMT degrade azathioprine more slowly, and these patients are at higher risk of myelosuppressive toxicity from the drug when it is given at the full weight-based dose. The TPMT test is expensive but not prohibitively so, and it would seem that genomic testing is a reasonable clinical and cost-effective option.
As in the abacavir scenario noted above, genomic-based dosing of azathioprine makes scientific sense and offers proof of principle for the validity of pharmacogenomics. But is it truly a clinical game-changer?
The answer depends in part on how the prescribing physician doses the drug, which depends in part on what disease is being treated, how fast the drug needs to be at full dose, and whether there are equally effective alternatives. Recommendations have been offered that state if TPMT activity is normal, we can start at the usual maintenance dose of 1.5 to 2 mg/kg/day (or occasionally more). But if the patient is heterozygous for the wild-type gene and thus is a slower drug metabolizer, then initial dosing “should” be reduced to 25 to 50 mg/day, with close observation of the white blood cell count as the dose is slowly increased to the target. The very rare patient who is homozygous for a non–wild-type allele should not be given the drug.
My usual practice has been to start patients on 50 mg or less daily and slowly titrate up, asking them how they are tolerating the drug and watching the white count—notably, the same approach to be taken if I had done genotyping before starting the drug and had found the patient to be heterozygous for the TPMT gene.
Interestingly, one pragmatic clinical trial tested whether genotyping patients before starting azathioprine—with subsequent suggested dosing of the drug based on the genotype as above—was safer and cheaper than letting physicians dose as they chose.1 It turned out that physicians participating in this study still dosed their patients conservatively. Even knowing that they might be able to give full doses from the start in patients with normal TPMT activity, many chose not to. I assume that many of those physicians felt as I do that there was no urgency in reaching the presumed-to-be-effective full weight-based therapeutic dose. (We don’t have a good clinical marker of azathioprine’s efficacy). At 4 months, the maintenance dose was about the same in all groups.
We have robust evidence to support the role of pharmacogenetics in informing the dosing of several medications, more than just the ones I have mentioned here. And in the right settings, we should use pharmacogenetic testing to limit toxicity and perhaps enhance efficacy in our drug selection. As the field moves rapidly forward, we will have many opportunities to improve clinical care by using our patients’ genomic information.
But like it or bemoan it, even when we have science in the house, the art of medicine still plays a role in our clinical decisions.
- Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014; 17:22–33.
- Thompson AJ, Newman WG, Elliott RA, Roberts SA, Tricker K, Payne K. The cost-effectiveness of a pharmacogenetic test: a trial-based evaluation of TPMT genotyping for azathioprine. Value Health 2014; 17:22–33.
Should thiopurine methyltransferase (TPMT) activity be determined before prescribing azathioprine, mercaptopurine, or thioguanine?
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
The thiopurines azathioprine, mercaptopurine, and thioguanine are prodrugs that are converted to active thioguanine nucleotide metabolites or methylated by thiopurine methyltransferase (TPMT) to compounds with less pharmacologic activity. In the absence of TPMT activity, patients are likely to have higher concentrations of thioguanine nucleotides, which can pose an increased risk of severe life-threatening myelosuppression. Determining TPMT activity, either directly by phenotyping or indirectly by determining the specific genetic allele (different alleles have different enzymatic activity), can help identify patients at greater risk of severe myelosuppression. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
THIOPURINES AND TPMT
Azathioprine, mercaptopurine, and thioguanine are used for treating autoimmune and inflammatory diseases1–3 and certain types of cancer such as leukemias and lymphomas.1,4–6 Typically, azathioprine is used to treat nonmalignant conditions, thioguanine is used to treat malignancies, and mercaptopurine can be used to treat both malignant and nonmalignant conditions.
Although the exact mechanism of action of these drugs has not been completely elucidated, the active thioguanine nucleotide metabolites are thought to be incorporated into the DNA of leukocytes, resulting in DNA damage that subsequently leads to cell death and myelosuppression.7–9
Variants of the TPMT gene may alter the activity of the TPMT enzyme, resulting in individual variability in thiopurine metabolism. Compared with people with normal (high) TPMT activity, those with intermediate or low TPMT activity metabolize the drugs more slowly, and are likely to have higher thioguanine nucleotide concentrations and therefore an increased risk of myelosuppression.
One of the earliest correlations between TPMT activity and thiopurine-induced myelosuppression was described in a pediatric patient with acute lymphocytic leukemia.10 After being prescribed a conventional mercaptopurine dosage (75 mg/m2 daily), the patient developed severe myelosuppression and was observed to have a thioguanine nucleotide metabolite concentration seven times the observed population median. TPMT phenotyping demonstrated that the patient had low TPMT activity. Reducing the mercaptopurine dose by approximately 90% resulted in normalization of thioguanine nucleotide metabolite concentrations, and the myelosuppression subsequently resolved.
Approximately 10% of the population has intermediate TPMT activity and 0.3% has low or absent TPMT activity, though these percentages vary depending on ancestry.1 Research has demonstrated that approximately 30% to 60% of those with intermediate TPMT activity cannot tolerate a full thiopurine dose (eg, azathioprine 2–3 mg/kg/day or mercaptopurine 1.5 mg/kg/day).1 Almost all patients with low TPMT activity will develop life-threating myelosuppression if prescribed a full thiopurine dose.1
SHOULD TPMT ACTIVITY BE DETERMINED FOR EVERY PATIENT PRESCRIBED A THIOPURINE?
Although determining TPMT activity in thiopurine-naïve patients will assist clinicians in selecting a thiopurine starting dose or in deciding if an alternative agent is warranted, there are instances when a clinician may elect to not perform a TPMT genotype or phenotype test. For example, determining TPMT activity is not recommended for patients who previously tolerated thiopurine therapy at full steady-state doses.
The required starting dose of a thiopurine can influence the decision on whether or not to test for TPMT activity. TPMT genotyping or phenotyping may be of most benefit for patients requiring immediate full doses of a thiopurine.11 Ideally, TPMT activity should be determined before prescribing immediate full doses of a thiopurine. This could be achieved by preemptively ordering a TPMT test in patients likely to require immunosuppression—for example, in patients diagnosed with inflammatory or autoimmune diseases. If therapy cannot be delayed and TPMT activity is unknown, ordering a TPMT test at the time of prescribing a full thiopurine dose is still of benefit. Depending on the clinical laboratory utilized for testing, TPMT phenotype results are usually reported in 3 to 5 days, and TPMT genotype results are usually reported in 5 to 7 days. Because most patients will not reach steady-state concentrations for 2 to 6 weeks, clinicians could initiate immediate full doses of a thiopurine and modify therapy based on TPMT test results before accumulation of thioguanine nucleotide metabolites occurs. Caution should be used with this approach, particularly in situations where the clinical laboratory may not return results in a timely manner.
For patients who are candidates for an initial low dose of a thiopurine, clinicians may choose to slowly titrate doses based on response and tolerability instead of determining TPMT activity.11 Depending on the starting dose and how slowly titration occurs, initiating a thiopurine at a low dose and titrating based on response can be a feasible approach for patients with intermediate TPMT activity. Because drastic thiopurine dose reductions of approximately 10-fold are required for patients with low TPMT activity, which is a much smaller dosage than most clinicians will initially prescribe, the starting dosage will likely not be low enough to prevent myelosuppression in patients with low TPMT activity.1,10
Determining TPMT activity can help clinicians establish an appropriate titration schedule. Patients with normal TPMT activity will usually reach thiopurine steady-state concentrations in 2 weeks, and the dosage can be titrated based on response.1 Alterations in TPMT activity influence the pharmacokinetic parameters of thiopurines, and the time to reach steady-state is extended to 4 or 6 weeks for those with intermediate or low TPMT activity.1 Increasing the thiopurine dosage before reaching steady state can lead to the prescribing of doses that will not be tolerated, resulting in myelosuppression.
Factors to consider when deciding if TPMT activity should be assessed include the disease state being treated and corresponding starting dose, the need for immediate full doses, and previous documented tolerance of thiopurines at steady-state doses. As with many aspects of medicine that have multiple options, coupled with an increase in patient access to healthcare information, the decision to test for TPMT activity may include shared decision-making between patients and providers. Although TPMT genotyping or phenotyping can help identify those at greatest risk of severe myelosuppression, such assays do not replace routine monitoring for myelosuppression, hepatotoxicity, or pancreatitis that may be caused by thiopurines.
WHAT TESTS ARE AVAILABLE TO DETERMINE TPMT ACTIVITY?
Patients with intermediate or low TPMT activity can be identified by either genotyping or phenotyping. There are considerations, though, that clinicians should be aware of before selecting a particular test.
TPMT genotyping
![]()
Four TPMT alleles, TPMT*2, *3A, *3B, and *3C, account for over 90% of inactivating polymorphisms.12 Therefore, most reference laboratories only analyze for those genetic variants. Based on the reported test result, a predicted phenotype (eg, normal, intermediate, or low TPMT activity) can be assigned. Table 1 lists the predicted phenotypes for select genotyping results.
TPMT phenotyping
Phenotyping quantitates TPMT enzyme activity in erythrocytes, and based on the result, patients are classified as having normal, intermediate, or low TPMT activity. Because internal standards and other testing conditions may differ between reference laboratories, test results must be interpreted in the context of the laboratory that performed the assay.
Which test is right for my patient?
In most cases, either the genotype or the phenotype test provides sufficient information to guide thiopurine therapy. There are certain circumstances, though, in which the genotype or phenotype test is less informative.
TPMT genotyping, when performed using a blood specimen, is not recommended in those with a history of allogeneic bone marrow transplantation, as the result would reflect the donor’s genotype, not the patient’s. In such instances, monitoring of white blood cell counts and thiopurine metabolites may be more beneficial.
TPMT phenotyping may be inaccurate if performed within 30 to 90 days of an erythrocyte transfusion, as the test result may be influenced by donor erythrocytes. If a patient is receiving erythrocyte transfusions, TPMT genotyping is preferable to phenotyping.
Test cost may also be a consideration when determining if the genotype or phenotype test is best for your patient. Costs vary by laboratory, but phenotyping is generally less expensive than genotyping. The cost of genotyping, though, continues to decrease.13 The approximate commercial cost is $200 for phenotyping and $450 for genotyping, but laboratory fees may be substantially higher. Several insurance plans, including Medicare, cover TPMT testing, but reimbursement and copayments vary, depending on the patient’s specific plan.
There are conflicting data as to whether determining TPMT status is11,14–18 or is not19 cost-effective. Multiple studies suggest that the cost of genotyping a sufficient number of patients to identify a single individual at high risk of myelosuppression is cheaper than the costs associated with treating an adverse event. Additional cost-benefit studies are needed, particularly studies that consider how bundled payments and outcomes-based reimbursement influence cost-effectiveness.
MODIFYING THIOPURINE THERAPY BASED ON TPMT ACTIVITY
![]()
There is a strong correlation between TPMT activity and tolerated thiopurine doses, with those having intermediate or low TPMT activity requiring lower doses.10,20–23 Adjusting mercaptopurine doses based on TPMT activity to prevent hematopoietic toxicity has been successfully demonstrated in pediatric patients with acute lymphoblastic leukemia.24 Furthermore, reducing initial thiopurine doses to avoid myelosuppression and titrating based on response has been shown to not compromise outcomes.1,25,26 The Clinical Pharmacogenetic Implementation Consortium (CPIC) has developed an evidence-based guideline on how to adjust thiopurine doses based on TPMT activity,1 summarized in Table 2. These dosing recommendations are classified as “strong.”
Patients with normal TPMT activity should be prescribed the usual thiopurine starting dose as indicated by disease-specific guidelines.
For those with intermediate TPMT activity, the CPIC guideline recommends reducing the initial targeted full dose of azathioprine and mercaptopurine by 30% to 70% and reducing the targeted full dose of thioguanine by 30% to 50%. The percentage of dose reduction depends on the targeted full dose. Siegel and Sands27 suggested that for those who are diagnosed with inflammatory bowel disease and have intermediate TPMT activity, azathioprine should be initiated at a low dose and titrated to 1.25 mg/kg and mercaptopurine should be initiated at a low dose and titrated to 0.75 mg/kg. Based on these titration goals, if the targeted full dose for mercaptopurine is 1 mg/kg, then a dose reduction of approximately 30% would be more appropriate. If the targeted full dose is 1.5 mg/kg, a dose reduction of approximately 50% would be more appropriate. Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 2 to 4 weeks to reach steady state before dose titration.
For those with low TPMT activity, alternative therapy should be considered for nonmalignant conditions because of the risk of severe myelosuppression. For malignancy, or if a thiopurine is warranted for a nonmalignant condition, consider a 90% dose reduction and give the drug three times per week instead of daily. For example, acute lymphoblastic leukemia patients with low TPMT activity can be started on mercaptopurine 10 mg/m2 three times per week instead of the usual starting dose.10 Thiopurine doses should be titrated based on response and disease-specific guidelines, allowing 4 to 6 weeks to reach steady state before dose titration.
RECOMMENDATIONS
Individuals with intermediate or low TPMT activity have an increased risk of myelosuppression. Because of the elevated risk for morbidity and death, especially for patients with low TPMT activity, multiple guidelines and regulatory agencies recommend TPMT genotyping or phenotyping if a thiopurine is prescribed.25,28–32 Although additional cost-benefit analysis studies are needed, evidence suggests testing for TPMT activity may be cheaper than the costs associated with treating myelosuppression.
In view of treatment guidelines, the recommendations of regulatory agencies, cost-benefit analyses, and the availability of gene-based dosing recommendations, we consider the benefits of testing for TPMT activity to greatly outweigh any associated risks. Therefore, we recommend that TPMT testing be strongly considered before initiating therapy with a thiopurine.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Ansari A, Arenas M, Greenfield SM, et al. Prospective evaluation of the pharmacogenetics of azathioprine in the treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2008; 28:973–983.
- Beswick L, Friedman AB, Sparrow MP. The role of thiopurine metabolite monitoring in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol 2014; 8:383–392.
- Gervasini G, Vagace JM. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012; 3:249.
- Levinsen M, Rotevatn EØ, Rosthøj S, et al; Nordic Society of Paediatric Haematology, Oncology. Pharmacogenetically based dosing of thiopurines in childhood acute lymphoblastic leukemia: influence on cure rates and risk of second cancer. Pediatr Blood Cancer 2014; 61:797–802.
- Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Pharmacol 2012; 68:1233–1242.
- Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:715–729.
- Fairchild CR, Maybaum J, Kennedy KA. Concurrent unilateral chromatid damage and DNA strand breakage in response to 6-thioguanine treatment. Biochem Pharmacol 1986; 35:3533–3541.
- Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull 2006; 79–80:153–170.
- Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr 1991; 119:985–989.
- Gardiner SJ, Gearry RB, Barclay ML, Begg EJ. Two cases of thiopurine methyltransferase (TPMT) deficiency—a lucky save and a near miss with azathioprine. Br J Clin Pharmacol 2006; 62:473–476.
- Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther 2013; 93:324–325.
- Altman RB. Pharmacogenomics: “noninferiority” is sufficient for initial implementation. Clin Pharmacol Ther 2011; 89:348–350.
- van den Akker-van Marle ME, Gurwitz D, Detmar SB, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics 2006; 7:783–792.
- Clunie GP, Lennard L. Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology (Oxford) 2004; 43:13–18.
- Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn’s disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol 2005; 100:2239–2247.
- Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther 2004; 20:593–599.
- Marra CA, Esdaile JM, Anis AH. Practical pharmacogenetics: the cost effectiveness of screening for thiopurine s-methyltransferase polymorphisms in patients with rheumatological conditions treated with azathioprine. J Rheumatol 2002; 29:2507–2512.
- Donnan JR, Ungar WJ, Mathews M, Hancock-Howard RL, Rahman P. A cost effectiveness analysis of thiopurine methyltransferase testing for guiding 6-mercaptopurine dosing in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 2011; 57:231–239.
- Lennard L, Gibson BE, Nicole T, Lilleyman JS. Congenital thiopurine methyltransferase deficiency and 6-mercaptopurine toxicity during treatment for acute lymphoblastic leukaemia. Arch Dis Child 1993; 69:577–579.
- Hindorf U, Lindqvist M, Hildebrand H, Fagerberg U, Almer S. Adverse events leading to modification of therapy in a large cohort of patients with inflammatory bowel disease. Aliment Pharmacol Ther 2006; 24:331–342.
- Relling MV, Hancock ML, Rivera GK, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 1999; 91:2001–2008.
- Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93:2817–2823.
- Pui CH, Pei D, Sandlund JT, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010; 24:371–382.
- Ford LT, Berg JD. Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol 2010; 63:288–295.
- Schmiegelow K, Forestier E, Hellebostad M, et al; Nordic Society of Paediatric Haematology and Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010; 24:345–354.
- Siegel CA, Sands BE. Review article: practical management of inflammatory bowel disease patients taking immunomodulators. Aliment Pharmacol Ther 2005; 22:1–16.
- Mayberry JF, Lobo A, Ford AC, Thomas A. NICE clinical guideline (CG152): the management of Crohn’s disease in adults, children and young people. Aliment Pharmacol Ther 2013; 37:195–203
- Mowat C, Cole A, Windsor A, et al; IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011; 60:571–607.
- Turner D, Levine A, Escher JC, et al; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr 2012; 55:340–361.
- Bernstein CN, Fried M, Krabshuis JH, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis 2010; 16:112–124.
- Becquemont L, Alfirevic A, Amstutz U, et al. Practical recommendations for pharmacogenomics-based prescription: 2010 ESF-UB Conference on Pharmacogenetics and Pharmacogenomics. Pharmacogenomics 2011; 12:113–124.