User login
Interpreting SPRINT: How low should you go?
In treating hypertension, lower systolic pressure is better than higher—with caveats. This is the message of the Systolic Blood Pressure Intervention Trial (SPRINT),1 a large, federally funded study that was halted early when patients at high cardiovascular risk who were randomized to a goal systolic pressure of less than 120 mm Hg were found to have better outcomes, including lower rates of heart failure, death from cardiovascular causes, and death from any cause, than patients randomized to a goal of less than 140 mm Hg.
The caveats: the benefit came at a price of more adverse events. Also, the trial excluded patients who had diabetes mellitus or previous strokes, so it is uncertain if these subgroups would also benefit from intensive lowering of systolic pressure—and in earlier trials they did not.
This article reviews the trial design and protocol, summarizes the results, and briefly discusses the implications of these results.
BEFORE SPRINT
Hypertension is very common in adults in the United States, and is a risk factor for heart disease, stroke, heart failure, and kidney disease. The estimated prevalence of hypertension in the 2011–2014 National Health and Nutrition Examination Survey (NHANES) was 29%, and the prevalence increases with age (7.3% in those ages 18 to 39, 32.2% in those ages 40 to 59, and 64.9% in those ages 60 and older).2 Isolated systolic hypertension (ie, systolic blood pressure > 140 mm Hg with diastolic pressure < 90 mm Hg) is the most common form of hypertension after age 50.3
Clinical trials have provided substantial evidence that treating hypertension reduces the incidence of stroke, myocardial infarction, and heart failure.4,5 Although observational studies show a progressive and linear rise in cardiovascular risk as systolic blood pressure rises above 115 mm Hg,6 clinical trials in the general population have not documented benefits of lowering systolic pressure to this level.7–11 However, clinical trials that directly evaluated two different blood pressure goals in the general population showed benefit with achieving systolic blood pressure less than 150 mm Hg,7,9 with limited data on lower blood pressure targets.10–12
No benefit found in intensive systolic lowering in diabetes or after stroke
The Action to Control Cardiovascular Risk in Diabetes-Blood Pressure (ACCORD BP) trial13 in patients with type 2 diabetes found no benefit in lowering systolic pressure to less than 120 mm Hg compared with less than 140 mm Hg in terms of the trial’s primary composite cardiovascular outcome (ie, nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes). However, the intensively treated group in this trial did enjoy a benefit in terms of fewer stroke events.
The Secondary Prevention of Small Subcortical Strokes (SPS3) trial14 in patients with stroke found no significant benefit in lowering systolic pressure to less than 130 mm Hg compared with less than 150 mm Hg for overall risk of another stroke, but a significant benefit was noted in reduced risk of intracerebral hemorrhage.
Current guidelines, based on available evidence, advocate treatment to a systolic goal of less than 140 mm Hg in most patients, and recommend relaxing this goal to less than 150 mm Hg in the elderly.15,16
Given the uncertainty surrounding optimal systolic targets, SPRINT was designed to test the hypothesis that a goal of less than 120 mm Hg would reduce the risk of cardiovascular events more than the generally accepted systolic goal of less than 140 mm Hg.17 Patients with diabetes and stroke were excluded because a similar hypothesis was tested in the ACCORD BP and SPS3 trials, which included patients with these conditions.
SPRINT DESIGN
SPRINT was a randomized, controlled, open-label trial sponsored by the National Institutes of Health and conducted at 102 US sites.
Inclusion criteria. Participants had to be at least 50 years old, with systolic pressure of 130 to 180 mm Hg, and had to have at least one cardiovascular risk factor, eg:
- Clinical or subclinical cardiovascular disease (other than stroke)
- Chronic kidney disease, defined as estimated glomerular filtration rate (eGFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation, of 20 to less than 60 mL/min/1.73 m2
- Framingham risk score of 15% of more
- Age 75 or older.
Major exclusion criteria included:
- Diabetes
- Stroke
- Polycystic kidney disease
- Chronic kidney disease with an eGFR less than 20 mL/min/1.73 m2
- Proteinuria (excretion > 1 g/day).
Intensive vs standard treatment
Participants were randomized to receive intensive treatment (systolic goal < 120 mm Hg) or standard treatment (systolic goal < 140 mm Hg). Baseline antihypertensive medications were adjusted to achieve blood pressure goals based on randomization assignment.
Doses of medications were adjusted on the basis of an average of three seated office blood pressure measurements after a 5-minute period of rest, taken with an automated monitor (Omron Healthcare Model 907); the same monitor was used and the same protocol was followed at all participating sites. Blood pressure was also measured after standing for 1 minute to assess orthostatic change.
Lifestyle modifications were encouraged in both groups. There was no restriction on using any antihypertensive medication, and this was at the discretion of individual investigators. Thiazide-type diuretics were encouraged as first-line agents (with chlorthalidone encouraged as the primary thiazide-type diuretic).
Outcomes measured
The primary outcome was a composite of myocardial infarction, acute coronary syndrome not resulting in myocardial infarction, stroke, acute decompensated heart failure, and cardiovascular mortality.
Secondary outcomes included individual components of the primary composite outcome, all-cause mortality, and the composite of primary outcome and all-cause mortality.
Renal outcomes were assessed as:
- Incident albuminuria (doubling of the urinary albumin-to-creatinine ratio from less than 10 mg/g to more than 10 mg/g)
- Composite of a 50% decrease in eGFR or development of end-stage renal disease requiring long-term dialysis or kidney transplantation (in those with baseline chronic kidney disease)
- A 30% decrease in eGFR (in those without chronic kidney disease).1,17
SPRINT also recruited participants to two nested substudies: SPRINT MIND and SPRINT MIND MRI, to study differences in cognitive outcomes and small-vessel ischemic disease between intensive treatment and standard treatment.
STUDY RESULTS
Older patients at risk, but without diabetes
Of 14,692 participants screened, 9,361 were enrolled in the study between 2010 and 2013. Baseline characteristics were comparable in both groups.
Demographics. The mean age of the participants was 67.9, and about 28% were 75 or older. About 36% were women, 58% white, 30% black, and 11% Hispanic.
Cardiovascular risk. The mean Framingham risk score was 20% (ie, they had a 20% risk of having a cardiovascular event within 10 years), and 61% of the participants had a risk score of at least 15%. Twenty percent already had cardiovascular disease.
Blood pressure. The average baseline blood pressure was 139.7/78.2 mm Hg. One-third of the participants had baseline systolic pressures of 132 mm Hg or less, another third had pressures in the range of 132 to 145, and the rest had 145 mm Hg or higher.
Renal function. The mean serum creatinine level was about 1.1 mg/dL. The mean eGFR was about 71 mL/min/1.73 m2 as calculated by the MDRD equation, and about 28% had eGFRs less than 60. The mean ratio of urinary albumin to creatinine was 44.1 mg/g in the intensive treatment group and 41.1 in the standard treatment group.
Other. The mean total cholesterol level was 190 mg/dL, fasting plasma glucose 99 mg/dL, and body mass index nearly 30 kg/m2.
Blood pressure during treatment
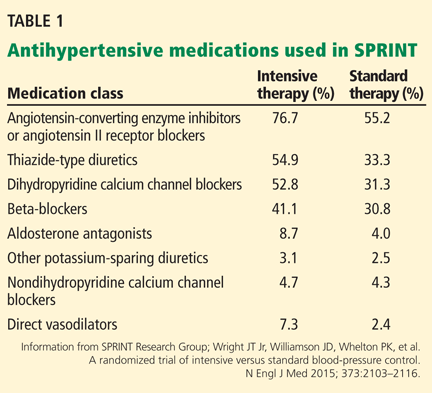
People in the intensive treatment group were taking a mean of 2.8 antihypertensive medications, and those in the standard treatment group were taking 1.8. Patients in the intensive group required greater use of all classes of medications to achieve goal systolic pressure (Table 1).
Study halted early due to efficacy
Throughout the 3.26 years of follow-up, the average difference in systolic pressure between the two groups was 13.1 mm Hg, with a mean systolic pressure of 121.5 mm Hg in the intensive treatment group and 134.6 mm Hg in the standard treatment group. The mean diastolic blood pressure was 68.7 mm Hg in the intensive treatment group and 76.3 mm Hg in the standard treatment group.
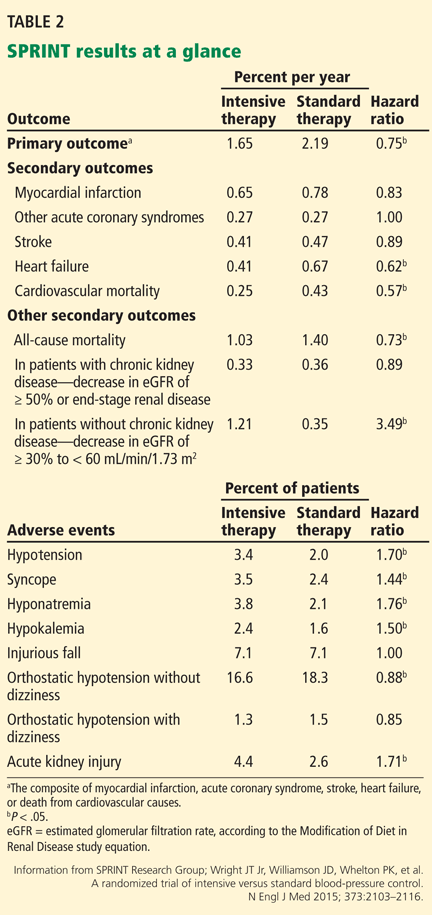
Although the study was planned to run for an average follow-up of 5 years, the National Heart, Lung, and Blood Institute terminated it early at a median of 3.26 years in view of lower rates of the primary outcome and of heart failure and death in the intensive treatment group (Table 2).
The effects on the primary outcome and mortality were consistent across the prespecified subgroups of age (< 75 vs ≥ 75), sex (female vs male), race (black vs nonblack), cardiovascular disease (presence or absence at baseline), prior chronic kidney disease (presence or absence at baseline), and across blood pressure tertiles (≤ 132 mm Hg, > 132 to < 145 mm Hg, ≥ 145 mm Hg).
Follow-up for assessment of cognitive outcomes (SPRINT MIND) and small-vessel ischemic disease (SPRINT MIND MRI) is ongoing.
WHAT DOES THIS MEAN?
SPRINT is the first large, adequately powered, randomized trial to demonstrate cardiovascular and mortality benefit from lowering the systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk but without a history of diabetes mellitus or stroke.1
Most SPRINT patients had reasonably controlled blood pressure at baseline (the mean systolic pressure was 139.7 mm Hg, and two-thirds of participants had systolic pressure < 145 mm Hg). Of note, however, this trial excluded patients with systolic pressure higher than 180 mm Hg. There was excellent separation of systolic pressure between the two groups beginning at 1 year, which was consistent through the course of the trial.
The cardiovascular benefit in the intensive treatment group was predominantly driven by lower rates of heart failure (a 38% reduction in the intensive treatment group, P = .0002) and cardiovascular mortality (a 43% reduction in the intensive treatment group, P = .005), while there was no significant difference between the two groups in myocardial infarction or stroke. The beneficial effect on heart failure events is consistent with results from other trials including the Systolic Hypertension in the Elderly Program,7 Systolic Hypertension in Europe,8 and Hypertension in the Very Elderly Trial,9 all of which showed greatest risk reduction for heart failure events with systolic pressure-lowering (although to higher systolic levels than SPRINT).7–9 It is unclear why there was no beneficial effect on stroke events. The reduction in all-cause mortality in the intensive treatment group in SPRINT was greater than the reduction in cardiovascular deaths, which is also unexplained.
Although the study was terminated early due to efficacy (which introduces the possible bias that the estimated effect size will be too high), the number of primary end points reached was large (562 in the two groups combined), providing reassurance that the findings are valid. There was no blinding in the study (both participants and study investigators were aware of treatment assignment and study medications), but there was a structured assessment of outcomes and adverse events, with adjudication done by blinded reviewers.
SPRINT used an automated device for blood pressure measurement, which is known to reduce the “white coat” effect and correlates tightly with average daytime blood pressure done by ambulatory blood pressure monitoring.18 However, in clinical practice automated devices may not be available and a strict protocol for correct measurement may not be followed, with the possible result that blood pressure may be overestimated and overtreated.
What about diastolic pressure?
The trial, by design, focused on lowering systolic pressure (given the greater prevalence of isolated systolic hypertension with age), and the implications of lowering diastolic pressure are unclear. The issue of a J-shaped relationship between diastolic pressure and cardiovascular risk is debated in the literature: patients with a diastolic pressure of 60 to 65 mm Hg, especially those with existing coronary artery disease, may not tolerate aggressive blood pressure-lowering.19,20 Further analysis of this association (if any) from SPRINT will be helpful.
What about patients with diabetes?
Patients were excluded from SPRINT if they were under age 50, were at low cardiovascular risk, or had diabetes, raising the question of whether the results apply to these groups as well.

The question is particularly relevant in diabetes, as the ACCORD BP study, which used the same blood pressure targets as SPRINT, did not show a significant difference in the primary cardiovascular outcome between the intensive and standard treatments in patients with diabetes (Table 3).13 In ACCORD BP, the rate of the primary outcome was 12% lower in the intensive treatment group than in the standard treatment group, but the 95% confidence interval was –27% to +6%, so the finding was not statistically significant. However, the wide confidence interval does not exclude the possibility of a benefit that was comparable to that observed in SPRINT.
It has been speculated that ACCORD BP was underpowered to detect significant differences in the primary outcome.21 An analysis combining data from both trials indicated that effects on individual outcomes were generally consistent in both trials (with no significant heterogeneity noted).22 Also, the primary composite outcome in ACCORD did not include heart failure, which is particularly sensitive to blood pressure reduction.
Additionally, ACCORD BP had a 2 × 2 factorial design involving a simultaneous comparison of intensive vs standard glycemic control, which may have influenced the effects due to blood pressure. Indeed, a post hoc analysis showed that there was a significant 26% lower risk of the primary outcome in ACCORD BP patients who received intensive systolic pressure control plus standard glycemic control than in those receiving standard systolic control plus standard glycemic control.23
Are more adverse events an acceptable trade-off?
Adverse events, including acute kidney injury, were more frequent in the intensive therapy group in SPRINT.
Acute kidney injury was coded as an adverse event on the basis of this diagnosis being included in the hospital discharge summary (as a primary or main secondary diagnosis) and if considered by the safety officer to be one of the top three reasons for admission or continued hospitalization. Further analysis of renal events should be forthcoming.
People in the intensive treatment group, on average, needed one more medication than those in the standard treatment group. Some of the adverse events may be related to the antihypertensive medications taken (eg, electrolyte abnormalities such as hyponatremia and hypokalemia due to diuretic use), and others may be related to blood pressure-lowering (eg, acute kidney injury due to renal hypoperfusion).
At this point, the long-term effects of these adverse events, especially on kidney function, are not known. Patients enrolled in clinical trials tend to be healthier than patients seen in clinical practice; thus, the rate of adverse events reported in the trial may be lower than one would see in the real world.
Does lower systolic pressure protect or harm the kidneys?
SPRINT included patients with stage 3 and 4 chronic kidney disease (ie, with eGFR 20–50 mL/min/1.73 m2), but it was designed to assess cardiovascular outcomes, not the progression of chronic kidney disease. The trial excluded patients with diabetic nephropathy or high degrees of proteinuria.
Earlier randomized trials that focused on chronic kidney disease progression, including the MDRD24 and the African American Study of Kidney Disease and Hypertension,25 did not show benefit with more aggressive blood pressure-lowering (except in patients with higher degrees of proteinuria), and these trials were not powered to assess effects on cardiovascular outcomes.24,25
The Irbesartan Diabetic Nephropathy Trial,26,27 which was done in patients with overt diabetic nephropathy, showed that a progressively lower achieved systolic pressure down to 120 mm Hg predicted lower rates of heart failure, cardiovascular mortality, and renal events (although the trial target was ≤ 130/85 mm Hg and few participants achieved systolic pressure lower than 120 mm Hg).
IMPLICATIONS FOR MANAGEMENT
The recent estimates of hypertension prevalence and control from NHANES show that only about 53% of hypertensive adults have their blood pressure under control (defined as systolic pressure < 140 mm Hg and diastolic pressure < 90 mm Hg).2 Analysis of the NHANES 2007–2012 data showed that 16.7% or 8.2 million US adults with treated hypertension meet the eligibility criteria for SPRINT.28
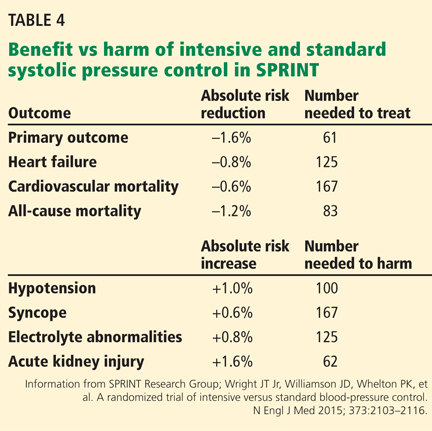
Although the SPRINT results support the notion that “lower is better,” the risks and benefits of intensive control will need to be balanced in individual patients. Table 4 shows the number needed to treat and number needed to harm in the trial.
More aggressive management of hypertension is challenging. The median systolic pressure achieved in the intensive group in SPRINT was just over 120 mm Hg, which implies that at least half of the participants in the intensive group did not achieve the goal of less than 120 mm Hg. While it may be reasonable to aim for systolic pressure of less than 120 or 125 mm Hg in patients who fit the SPRINT criteria and can tolerate intensive blood pressure lowering, it would be prudent to aim for a more conservative goal in elderly patients who are frail and at risk for falls, considering the higher incidence of specified adverse events in the intensive group.
Results of cognitive outcomes, as well as data related to quality of life, are still awaited. Long-term renal outcomes are also unclear.
As noted above, the question of generalizability of SPRINT results to patients with diabetes is open to debate. In our opinion, with currently available evidence, it is difficult to conclusively answer the question of whether a lower systolic target provides cardiovascular benefit in diabetes. It is also unclear whether similar beneficial results would be seen with intensive treatment in a population at low cardiovascular risk. The American Heart Association and the American College of Cardiology are in the process of formulating new hypertension guidelines, and evidence from SPRINT will inform any new recommendations.
As more medications will likely be needed for intensive systolic blood pressure control, side effects and tolerability of medications with polypharmacy and potential nonadherence with increasing complexity of medication regimens should be kept in mind. Lifestyle modifications will need to be emphasized and reinforced, with greater use of combination antihypertensive therapy.
The data from SPRINT indicate that lower systolic pressure is better, as long as untoward clinical events can be monitored and avoided or easily managed. Careful monitoring will likely entail more frequent clinic visits and more frequent assessment of renal function and electrolyte levels (participants in the intensive group in the trial were seen every month until goal was achieved). A team approach that includes pharmacists and nurse practitioners, along with optimal use of best practice algorithms and remote monitoring technology, will need to be implemented for efficient and effective care.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS data brief, no. 220. Hyattsville, MD: National Center for Health Statistics. 2015.
- Franklin SS, Jacobs MJ, Wong ND, L’Italien GJ, Lapuerta P. Predominance of isolated systolic hypertension among middle-aged and elderly US hypertensives: analysis based on National Health and Nutrition Examination Survey (NHANES) III. Hypertension 2001; 37:869–874.
- Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000; 356:1955–1964.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: Valsartan in Elderly Isolated Systolic Hypertension study. Hypertension 2010; 56:196–202.
- Liu L, Zhang Y, Liu G, Li W, Zhang X, Zanchetti A; FEVER Study Group. The Felodipine Event Reduction (FEVER) Study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23:2157–2172.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520. Erratum in: JAMA. 2014; 311:1809.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Ambrosius WT, Sink KM, Foy CG, et al; SPRINT Study Research Group. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014; 11:532–546.
- Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Conventional versus automated measurement of blood pressure in the office (CAMBO) trial. Fam Pract 2012; 29:376–382.
- Messerli FH, Mancia G, Conti CR, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893.
- Boutitie F, Gueyffier F, Pocock S, Fagard R, Boissel JP; INDANA Project Steering Committee; INdividual Data ANalysis of Antihypertensive intervention. J-shaped relationship between blood pressure and mortality in hypertensive patients: new insights from a meta-analysis of individual-patient data. Ann Intern Med 2002; 136:438–448.
- Mancia G. Effects of intensive blood pressure control in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 2010; 122:847–849.
- Perkovic V, Rodgers A. Redefining blood-pressure targets—SPRINT starts the marathon. N Engl J Med 2015; 373:2175–2178.
- Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care 2014; 37:1721–1728.
- Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease study. Ann Intern Med 1995; 123:754–762.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Berl T, Hunsicker LG, Lewis JB, et al; Collaborative Study Group. Impact of achieved blood pressure on cardiovascular outcomes in the Irbesartan Diabetic Nephropathy Trial. J Am Soc Nephrol 2005; 16:2170–2179.
- Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the Irbesartan Diabetic Nephropathy Trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16:3027–3037.
- Bress AP, Tanner RM, Hess R, Colantonio LD, Shimbo D, Muntner P. Generalizability of results from the Systolic Blood Pressure Intervention Trial (SPRINT) to the US adult population. J Am Coll Cardiol 2015 Oct 31. doi: 10.1016/j.jacc.2015.10.037. Epub ahead of print.
In treating hypertension, lower systolic pressure is better than higher—with caveats. This is the message of the Systolic Blood Pressure Intervention Trial (SPRINT),1 a large, federally funded study that was halted early when patients at high cardiovascular risk who were randomized to a goal systolic pressure of less than 120 mm Hg were found to have better outcomes, including lower rates of heart failure, death from cardiovascular causes, and death from any cause, than patients randomized to a goal of less than 140 mm Hg.
The caveats: the benefit came at a price of more adverse events. Also, the trial excluded patients who had diabetes mellitus or previous strokes, so it is uncertain if these subgroups would also benefit from intensive lowering of systolic pressure—and in earlier trials they did not.
This article reviews the trial design and protocol, summarizes the results, and briefly discusses the implications of these results.
BEFORE SPRINT
Hypertension is very common in adults in the United States, and is a risk factor for heart disease, stroke, heart failure, and kidney disease. The estimated prevalence of hypertension in the 2011–2014 National Health and Nutrition Examination Survey (NHANES) was 29%, and the prevalence increases with age (7.3% in those ages 18 to 39, 32.2% in those ages 40 to 59, and 64.9% in those ages 60 and older).2 Isolated systolic hypertension (ie, systolic blood pressure > 140 mm Hg with diastolic pressure < 90 mm Hg) is the most common form of hypertension after age 50.3
Clinical trials have provided substantial evidence that treating hypertension reduces the incidence of stroke, myocardial infarction, and heart failure.4,5 Although observational studies show a progressive and linear rise in cardiovascular risk as systolic blood pressure rises above 115 mm Hg,6 clinical trials in the general population have not documented benefits of lowering systolic pressure to this level.7–11 However, clinical trials that directly evaluated two different blood pressure goals in the general population showed benefit with achieving systolic blood pressure less than 150 mm Hg,7,9 with limited data on lower blood pressure targets.10–12
No benefit found in intensive systolic lowering in diabetes or after stroke
The Action to Control Cardiovascular Risk in Diabetes-Blood Pressure (ACCORD BP) trial13 in patients with type 2 diabetes found no benefit in lowering systolic pressure to less than 120 mm Hg compared with less than 140 mm Hg in terms of the trial’s primary composite cardiovascular outcome (ie, nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes). However, the intensively treated group in this trial did enjoy a benefit in terms of fewer stroke events.
The Secondary Prevention of Small Subcortical Strokes (SPS3) trial14 in patients with stroke found no significant benefit in lowering systolic pressure to less than 130 mm Hg compared with less than 150 mm Hg for overall risk of another stroke, but a significant benefit was noted in reduced risk of intracerebral hemorrhage.
Current guidelines, based on available evidence, advocate treatment to a systolic goal of less than 140 mm Hg in most patients, and recommend relaxing this goal to less than 150 mm Hg in the elderly.15,16
Given the uncertainty surrounding optimal systolic targets, SPRINT was designed to test the hypothesis that a goal of less than 120 mm Hg would reduce the risk of cardiovascular events more than the generally accepted systolic goal of less than 140 mm Hg.17 Patients with diabetes and stroke were excluded because a similar hypothesis was tested in the ACCORD BP and SPS3 trials, which included patients with these conditions.
SPRINT DESIGN
SPRINT was a randomized, controlled, open-label trial sponsored by the National Institutes of Health and conducted at 102 US sites.
Inclusion criteria. Participants had to be at least 50 years old, with systolic pressure of 130 to 180 mm Hg, and had to have at least one cardiovascular risk factor, eg:
- Clinical or subclinical cardiovascular disease (other than stroke)
- Chronic kidney disease, defined as estimated glomerular filtration rate (eGFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation, of 20 to less than 60 mL/min/1.73 m2
- Framingham risk score of 15% of more
- Age 75 or older.
Major exclusion criteria included:
- Diabetes
- Stroke
- Polycystic kidney disease
- Chronic kidney disease with an eGFR less than 20 mL/min/1.73 m2
- Proteinuria (excretion > 1 g/day).
Intensive vs standard treatment
Participants were randomized to receive intensive treatment (systolic goal < 120 mm Hg) or standard treatment (systolic goal < 140 mm Hg). Baseline antihypertensive medications were adjusted to achieve blood pressure goals based on randomization assignment.
Doses of medications were adjusted on the basis of an average of three seated office blood pressure measurements after a 5-minute period of rest, taken with an automated monitor (Omron Healthcare Model 907); the same monitor was used and the same protocol was followed at all participating sites. Blood pressure was also measured after standing for 1 minute to assess orthostatic change.
Lifestyle modifications were encouraged in both groups. There was no restriction on using any antihypertensive medication, and this was at the discretion of individual investigators. Thiazide-type diuretics were encouraged as first-line agents (with chlorthalidone encouraged as the primary thiazide-type diuretic).
Outcomes measured
The primary outcome was a composite of myocardial infarction, acute coronary syndrome not resulting in myocardial infarction, stroke, acute decompensated heart failure, and cardiovascular mortality.
Secondary outcomes included individual components of the primary composite outcome, all-cause mortality, and the composite of primary outcome and all-cause mortality.
Renal outcomes were assessed as:
- Incident albuminuria (doubling of the urinary albumin-to-creatinine ratio from less than 10 mg/g to more than 10 mg/g)
- Composite of a 50% decrease in eGFR or development of end-stage renal disease requiring long-term dialysis or kidney transplantation (in those with baseline chronic kidney disease)
- A 30% decrease in eGFR (in those without chronic kidney disease).1,17
SPRINT also recruited participants to two nested substudies: SPRINT MIND and SPRINT MIND MRI, to study differences in cognitive outcomes and small-vessel ischemic disease between intensive treatment and standard treatment.
STUDY RESULTS
Older patients at risk, but without diabetes
Of 14,692 participants screened, 9,361 were enrolled in the study between 2010 and 2013. Baseline characteristics were comparable in both groups.
Demographics. The mean age of the participants was 67.9, and about 28% were 75 or older. About 36% were women, 58% white, 30% black, and 11% Hispanic.
Cardiovascular risk. The mean Framingham risk score was 20% (ie, they had a 20% risk of having a cardiovascular event within 10 years), and 61% of the participants had a risk score of at least 15%. Twenty percent already had cardiovascular disease.
Blood pressure. The average baseline blood pressure was 139.7/78.2 mm Hg. One-third of the participants had baseline systolic pressures of 132 mm Hg or less, another third had pressures in the range of 132 to 145, and the rest had 145 mm Hg or higher.
Renal function. The mean serum creatinine level was about 1.1 mg/dL. The mean eGFR was about 71 mL/min/1.73 m2 as calculated by the MDRD equation, and about 28% had eGFRs less than 60. The mean ratio of urinary albumin to creatinine was 44.1 mg/g in the intensive treatment group and 41.1 in the standard treatment group.
Other. The mean total cholesterol level was 190 mg/dL, fasting plasma glucose 99 mg/dL, and body mass index nearly 30 kg/m2.
Blood pressure during treatment
People in the intensive treatment group were taking a mean of 2.8 antihypertensive medications, and those in the standard treatment group were taking 1.8. Patients in the intensive group required greater use of all classes of medications to achieve goal systolic pressure (Table 1).
Study halted early due to efficacy
Throughout the 3.26 years of follow-up, the average difference in systolic pressure between the two groups was 13.1 mm Hg, with a mean systolic pressure of 121.5 mm Hg in the intensive treatment group and 134.6 mm Hg in the standard treatment group. The mean diastolic blood pressure was 68.7 mm Hg in the intensive treatment group and 76.3 mm Hg in the standard treatment group.
Although the study was planned to run for an average follow-up of 5 years, the National Heart, Lung, and Blood Institute terminated it early at a median of 3.26 years in view of lower rates of the primary outcome and of heart failure and death in the intensive treatment group (Table 2).
The effects on the primary outcome and mortality were consistent across the prespecified subgroups of age (< 75 vs ≥ 75), sex (female vs male), race (black vs nonblack), cardiovascular disease (presence or absence at baseline), prior chronic kidney disease (presence or absence at baseline), and across blood pressure tertiles (≤ 132 mm Hg, > 132 to < 145 mm Hg, ≥ 145 mm Hg).
Follow-up for assessment of cognitive outcomes (SPRINT MIND) and small-vessel ischemic disease (SPRINT MIND MRI) is ongoing.
WHAT DOES THIS MEAN?
SPRINT is the first large, adequately powered, randomized trial to demonstrate cardiovascular and mortality benefit from lowering the systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk but without a history of diabetes mellitus or stroke.1
Most SPRINT patients had reasonably controlled blood pressure at baseline (the mean systolic pressure was 139.7 mm Hg, and two-thirds of participants had systolic pressure < 145 mm Hg). Of note, however, this trial excluded patients with systolic pressure higher than 180 mm Hg. There was excellent separation of systolic pressure between the two groups beginning at 1 year, which was consistent through the course of the trial.
The cardiovascular benefit in the intensive treatment group was predominantly driven by lower rates of heart failure (a 38% reduction in the intensive treatment group, P = .0002) and cardiovascular mortality (a 43% reduction in the intensive treatment group, P = .005), while there was no significant difference between the two groups in myocardial infarction or stroke. The beneficial effect on heart failure events is consistent with results from other trials including the Systolic Hypertension in the Elderly Program,7 Systolic Hypertension in Europe,8 and Hypertension in the Very Elderly Trial,9 all of which showed greatest risk reduction for heart failure events with systolic pressure-lowering (although to higher systolic levels than SPRINT).7–9 It is unclear why there was no beneficial effect on stroke events. The reduction in all-cause mortality in the intensive treatment group in SPRINT was greater than the reduction in cardiovascular deaths, which is also unexplained.
Although the study was terminated early due to efficacy (which introduces the possible bias that the estimated effect size will be too high), the number of primary end points reached was large (562 in the two groups combined), providing reassurance that the findings are valid. There was no blinding in the study (both participants and study investigators were aware of treatment assignment and study medications), but there was a structured assessment of outcomes and adverse events, with adjudication done by blinded reviewers.
SPRINT used an automated device for blood pressure measurement, which is known to reduce the “white coat” effect and correlates tightly with average daytime blood pressure done by ambulatory blood pressure monitoring.18 However, in clinical practice automated devices may not be available and a strict protocol for correct measurement may not be followed, with the possible result that blood pressure may be overestimated and overtreated.
What about diastolic pressure?
The trial, by design, focused on lowering systolic pressure (given the greater prevalence of isolated systolic hypertension with age), and the implications of lowering diastolic pressure are unclear. The issue of a J-shaped relationship between diastolic pressure and cardiovascular risk is debated in the literature: patients with a diastolic pressure of 60 to 65 mm Hg, especially those with existing coronary artery disease, may not tolerate aggressive blood pressure-lowering.19,20 Further analysis of this association (if any) from SPRINT will be helpful.
What about patients with diabetes?
Patients were excluded from SPRINT if they were under age 50, were at low cardiovascular risk, or had diabetes, raising the question of whether the results apply to these groups as well.
The question is particularly relevant in diabetes, as the ACCORD BP study, which used the same blood pressure targets as SPRINT, did not show a significant difference in the primary cardiovascular outcome between the intensive and standard treatments in patients with diabetes (Table 3).13 In ACCORD BP, the rate of the primary outcome was 12% lower in the intensive treatment group than in the standard treatment group, but the 95% confidence interval was –27% to +6%, so the finding was not statistically significant. However, the wide confidence interval does not exclude the possibility of a benefit that was comparable to that observed in SPRINT.
It has been speculated that ACCORD BP was underpowered to detect significant differences in the primary outcome.21 An analysis combining data from both trials indicated that effects on individual outcomes were generally consistent in both trials (with no significant heterogeneity noted).22 Also, the primary composite outcome in ACCORD did not include heart failure, which is particularly sensitive to blood pressure reduction.
Additionally, ACCORD BP had a 2 × 2 factorial design involving a simultaneous comparison of intensive vs standard glycemic control, which may have influenced the effects due to blood pressure. Indeed, a post hoc analysis showed that there was a significant 26% lower risk of the primary outcome in ACCORD BP patients who received intensive systolic pressure control plus standard glycemic control than in those receiving standard systolic control plus standard glycemic control.23
Are more adverse events an acceptable trade-off?
Adverse events, including acute kidney injury, were more frequent in the intensive therapy group in SPRINT.
Acute kidney injury was coded as an adverse event on the basis of this diagnosis being included in the hospital discharge summary (as a primary or main secondary diagnosis) and if considered by the safety officer to be one of the top three reasons for admission or continued hospitalization. Further analysis of renal events should be forthcoming.
People in the intensive treatment group, on average, needed one more medication than those in the standard treatment group. Some of the adverse events may be related to the antihypertensive medications taken (eg, electrolyte abnormalities such as hyponatremia and hypokalemia due to diuretic use), and others may be related to blood pressure-lowering (eg, acute kidney injury due to renal hypoperfusion).
At this point, the long-term effects of these adverse events, especially on kidney function, are not known. Patients enrolled in clinical trials tend to be healthier than patients seen in clinical practice; thus, the rate of adverse events reported in the trial may be lower than one would see in the real world.
Does lower systolic pressure protect or harm the kidneys?
SPRINT included patients with stage 3 and 4 chronic kidney disease (ie, with eGFR 20–50 mL/min/1.73 m2), but it was designed to assess cardiovascular outcomes, not the progression of chronic kidney disease. The trial excluded patients with diabetic nephropathy or high degrees of proteinuria.
Earlier randomized trials that focused on chronic kidney disease progression, including the MDRD24 and the African American Study of Kidney Disease and Hypertension,25 did not show benefit with more aggressive blood pressure-lowering (except in patients with higher degrees of proteinuria), and these trials were not powered to assess effects on cardiovascular outcomes.24,25
The Irbesartan Diabetic Nephropathy Trial,26,27 which was done in patients with overt diabetic nephropathy, showed that a progressively lower achieved systolic pressure down to 120 mm Hg predicted lower rates of heart failure, cardiovascular mortality, and renal events (although the trial target was ≤ 130/85 mm Hg and few participants achieved systolic pressure lower than 120 mm Hg).
IMPLICATIONS FOR MANAGEMENT
The recent estimates of hypertension prevalence and control from NHANES show that only about 53% of hypertensive adults have their blood pressure under control (defined as systolic pressure < 140 mm Hg and diastolic pressure < 90 mm Hg).2 Analysis of the NHANES 2007–2012 data showed that 16.7% or 8.2 million US adults with treated hypertension meet the eligibility criteria for SPRINT.28
Although the SPRINT results support the notion that “lower is better,” the risks and benefits of intensive control will need to be balanced in individual patients. Table 4 shows the number needed to treat and number needed to harm in the trial.
More aggressive management of hypertension is challenging. The median systolic pressure achieved in the intensive group in SPRINT was just over 120 mm Hg, which implies that at least half of the participants in the intensive group did not achieve the goal of less than 120 mm Hg. While it may be reasonable to aim for systolic pressure of less than 120 or 125 mm Hg in patients who fit the SPRINT criteria and can tolerate intensive blood pressure lowering, it would be prudent to aim for a more conservative goal in elderly patients who are frail and at risk for falls, considering the higher incidence of specified adverse events in the intensive group.
Results of cognitive outcomes, as well as data related to quality of life, are still awaited. Long-term renal outcomes are also unclear.
As noted above, the question of generalizability of SPRINT results to patients with diabetes is open to debate. In our opinion, with currently available evidence, it is difficult to conclusively answer the question of whether a lower systolic target provides cardiovascular benefit in diabetes. It is also unclear whether similar beneficial results would be seen with intensive treatment in a population at low cardiovascular risk. The American Heart Association and the American College of Cardiology are in the process of formulating new hypertension guidelines, and evidence from SPRINT will inform any new recommendations.
As more medications will likely be needed for intensive systolic blood pressure control, side effects and tolerability of medications with polypharmacy and potential nonadherence with increasing complexity of medication regimens should be kept in mind. Lifestyle modifications will need to be emphasized and reinforced, with greater use of combination antihypertensive therapy.
The data from SPRINT indicate that lower systolic pressure is better, as long as untoward clinical events can be monitored and avoided or easily managed. Careful monitoring will likely entail more frequent clinic visits and more frequent assessment of renal function and electrolyte levels (participants in the intensive group in the trial were seen every month until goal was achieved). A team approach that includes pharmacists and nurse practitioners, along with optimal use of best practice algorithms and remote monitoring technology, will need to be implemented for efficient and effective care.
In treating hypertension, lower systolic pressure is better than higher—with caveats. This is the message of the Systolic Blood Pressure Intervention Trial (SPRINT),1 a large, federally funded study that was halted early when patients at high cardiovascular risk who were randomized to a goal systolic pressure of less than 120 mm Hg were found to have better outcomes, including lower rates of heart failure, death from cardiovascular causes, and death from any cause, than patients randomized to a goal of less than 140 mm Hg.
The caveats: the benefit came at a price of more adverse events. Also, the trial excluded patients who had diabetes mellitus or previous strokes, so it is uncertain if these subgroups would also benefit from intensive lowering of systolic pressure—and in earlier trials they did not.
This article reviews the trial design and protocol, summarizes the results, and briefly discusses the implications of these results.
BEFORE SPRINT
Hypertension is very common in adults in the United States, and is a risk factor for heart disease, stroke, heart failure, and kidney disease. The estimated prevalence of hypertension in the 2011–2014 National Health and Nutrition Examination Survey (NHANES) was 29%, and the prevalence increases with age (7.3% in those ages 18 to 39, 32.2% in those ages 40 to 59, and 64.9% in those ages 60 and older).2 Isolated systolic hypertension (ie, systolic blood pressure > 140 mm Hg with diastolic pressure < 90 mm Hg) is the most common form of hypertension after age 50.3
Clinical trials have provided substantial evidence that treating hypertension reduces the incidence of stroke, myocardial infarction, and heart failure.4,5 Although observational studies show a progressive and linear rise in cardiovascular risk as systolic blood pressure rises above 115 mm Hg,6 clinical trials in the general population have not documented benefits of lowering systolic pressure to this level.7–11 However, clinical trials that directly evaluated two different blood pressure goals in the general population showed benefit with achieving systolic blood pressure less than 150 mm Hg,7,9 with limited data on lower blood pressure targets.10–12
No benefit found in intensive systolic lowering in diabetes or after stroke
The Action to Control Cardiovascular Risk in Diabetes-Blood Pressure (ACCORD BP) trial13 in patients with type 2 diabetes found no benefit in lowering systolic pressure to less than 120 mm Hg compared with less than 140 mm Hg in terms of the trial’s primary composite cardiovascular outcome (ie, nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes). However, the intensively treated group in this trial did enjoy a benefit in terms of fewer stroke events.
The Secondary Prevention of Small Subcortical Strokes (SPS3) trial14 in patients with stroke found no significant benefit in lowering systolic pressure to less than 130 mm Hg compared with less than 150 mm Hg for overall risk of another stroke, but a significant benefit was noted in reduced risk of intracerebral hemorrhage.
Current guidelines, based on available evidence, advocate treatment to a systolic goal of less than 140 mm Hg in most patients, and recommend relaxing this goal to less than 150 mm Hg in the elderly.15,16
Given the uncertainty surrounding optimal systolic targets, SPRINT was designed to test the hypothesis that a goal of less than 120 mm Hg would reduce the risk of cardiovascular events more than the generally accepted systolic goal of less than 140 mm Hg.17 Patients with diabetes and stroke were excluded because a similar hypothesis was tested in the ACCORD BP and SPS3 trials, which included patients with these conditions.
SPRINT DESIGN
SPRINT was a randomized, controlled, open-label trial sponsored by the National Institutes of Health and conducted at 102 US sites.
Inclusion criteria. Participants had to be at least 50 years old, with systolic pressure of 130 to 180 mm Hg, and had to have at least one cardiovascular risk factor, eg:
- Clinical or subclinical cardiovascular disease (other than stroke)
- Chronic kidney disease, defined as estimated glomerular filtration rate (eGFR), calculated by the Modification of Diet in Renal Disease (MDRD) study equation, of 20 to less than 60 mL/min/1.73 m2
- Framingham risk score of 15% of more
- Age 75 or older.
Major exclusion criteria included:
- Diabetes
- Stroke
- Polycystic kidney disease
- Chronic kidney disease with an eGFR less than 20 mL/min/1.73 m2
- Proteinuria (excretion > 1 g/day).
Intensive vs standard treatment
Participants were randomized to receive intensive treatment (systolic goal < 120 mm Hg) or standard treatment (systolic goal < 140 mm Hg). Baseline antihypertensive medications were adjusted to achieve blood pressure goals based on randomization assignment.
Doses of medications were adjusted on the basis of an average of three seated office blood pressure measurements after a 5-minute period of rest, taken with an automated monitor (Omron Healthcare Model 907); the same monitor was used and the same protocol was followed at all participating sites. Blood pressure was also measured after standing for 1 minute to assess orthostatic change.
Lifestyle modifications were encouraged in both groups. There was no restriction on using any antihypertensive medication, and this was at the discretion of individual investigators. Thiazide-type diuretics were encouraged as first-line agents (with chlorthalidone encouraged as the primary thiazide-type diuretic).
Outcomes measured
The primary outcome was a composite of myocardial infarction, acute coronary syndrome not resulting in myocardial infarction, stroke, acute decompensated heart failure, and cardiovascular mortality.
Secondary outcomes included individual components of the primary composite outcome, all-cause mortality, and the composite of primary outcome and all-cause mortality.
Renal outcomes were assessed as:
- Incident albuminuria (doubling of the urinary albumin-to-creatinine ratio from less than 10 mg/g to more than 10 mg/g)
- Composite of a 50% decrease in eGFR or development of end-stage renal disease requiring long-term dialysis or kidney transplantation (in those with baseline chronic kidney disease)
- A 30% decrease in eGFR (in those without chronic kidney disease).1,17
SPRINT also recruited participants to two nested substudies: SPRINT MIND and SPRINT MIND MRI, to study differences in cognitive outcomes and small-vessel ischemic disease between intensive treatment and standard treatment.
STUDY RESULTS
Older patients at risk, but without diabetes
Of 14,692 participants screened, 9,361 were enrolled in the study between 2010 and 2013. Baseline characteristics were comparable in both groups.
Demographics. The mean age of the participants was 67.9, and about 28% were 75 or older. About 36% were women, 58% white, 30% black, and 11% Hispanic.
Cardiovascular risk. The mean Framingham risk score was 20% (ie, they had a 20% risk of having a cardiovascular event within 10 years), and 61% of the participants had a risk score of at least 15%. Twenty percent already had cardiovascular disease.
Blood pressure. The average baseline blood pressure was 139.7/78.2 mm Hg. One-third of the participants had baseline systolic pressures of 132 mm Hg or less, another third had pressures in the range of 132 to 145, and the rest had 145 mm Hg or higher.
Renal function. The mean serum creatinine level was about 1.1 mg/dL. The mean eGFR was about 71 mL/min/1.73 m2 as calculated by the MDRD equation, and about 28% had eGFRs less than 60. The mean ratio of urinary albumin to creatinine was 44.1 mg/g in the intensive treatment group and 41.1 in the standard treatment group.
Other. The mean total cholesterol level was 190 mg/dL, fasting plasma glucose 99 mg/dL, and body mass index nearly 30 kg/m2.
Blood pressure during treatment
People in the intensive treatment group were taking a mean of 2.8 antihypertensive medications, and those in the standard treatment group were taking 1.8. Patients in the intensive group required greater use of all classes of medications to achieve goal systolic pressure (Table 1).
Study halted early due to efficacy
Throughout the 3.26 years of follow-up, the average difference in systolic pressure between the two groups was 13.1 mm Hg, with a mean systolic pressure of 121.5 mm Hg in the intensive treatment group and 134.6 mm Hg in the standard treatment group. The mean diastolic blood pressure was 68.7 mm Hg in the intensive treatment group and 76.3 mm Hg in the standard treatment group.
Although the study was planned to run for an average follow-up of 5 years, the National Heart, Lung, and Blood Institute terminated it early at a median of 3.26 years in view of lower rates of the primary outcome and of heart failure and death in the intensive treatment group (Table 2).
The effects on the primary outcome and mortality were consistent across the prespecified subgroups of age (< 75 vs ≥ 75), sex (female vs male), race (black vs nonblack), cardiovascular disease (presence or absence at baseline), prior chronic kidney disease (presence or absence at baseline), and across blood pressure tertiles (≤ 132 mm Hg, > 132 to < 145 mm Hg, ≥ 145 mm Hg).
Follow-up for assessment of cognitive outcomes (SPRINT MIND) and small-vessel ischemic disease (SPRINT MIND MRI) is ongoing.
WHAT DOES THIS MEAN?
SPRINT is the first large, adequately powered, randomized trial to demonstrate cardiovascular and mortality benefit from lowering the systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk but without a history of diabetes mellitus or stroke.1
Most SPRINT patients had reasonably controlled blood pressure at baseline (the mean systolic pressure was 139.7 mm Hg, and two-thirds of participants had systolic pressure < 145 mm Hg). Of note, however, this trial excluded patients with systolic pressure higher than 180 mm Hg. There was excellent separation of systolic pressure between the two groups beginning at 1 year, which was consistent through the course of the trial.
The cardiovascular benefit in the intensive treatment group was predominantly driven by lower rates of heart failure (a 38% reduction in the intensive treatment group, P = .0002) and cardiovascular mortality (a 43% reduction in the intensive treatment group, P = .005), while there was no significant difference between the two groups in myocardial infarction or stroke. The beneficial effect on heart failure events is consistent with results from other trials including the Systolic Hypertension in the Elderly Program,7 Systolic Hypertension in Europe,8 and Hypertension in the Very Elderly Trial,9 all of which showed greatest risk reduction for heart failure events with systolic pressure-lowering (although to higher systolic levels than SPRINT).7–9 It is unclear why there was no beneficial effect on stroke events. The reduction in all-cause mortality in the intensive treatment group in SPRINT was greater than the reduction in cardiovascular deaths, which is also unexplained.
Although the study was terminated early due to efficacy (which introduces the possible bias that the estimated effect size will be too high), the number of primary end points reached was large (562 in the two groups combined), providing reassurance that the findings are valid. There was no blinding in the study (both participants and study investigators were aware of treatment assignment and study medications), but there was a structured assessment of outcomes and adverse events, with adjudication done by blinded reviewers.
SPRINT used an automated device for blood pressure measurement, which is known to reduce the “white coat” effect and correlates tightly with average daytime blood pressure done by ambulatory blood pressure monitoring.18 However, in clinical practice automated devices may not be available and a strict protocol for correct measurement may not be followed, with the possible result that blood pressure may be overestimated and overtreated.
What about diastolic pressure?
The trial, by design, focused on lowering systolic pressure (given the greater prevalence of isolated systolic hypertension with age), and the implications of lowering diastolic pressure are unclear. The issue of a J-shaped relationship between diastolic pressure and cardiovascular risk is debated in the literature: patients with a diastolic pressure of 60 to 65 mm Hg, especially those with existing coronary artery disease, may not tolerate aggressive blood pressure-lowering.19,20 Further analysis of this association (if any) from SPRINT will be helpful.
What about patients with diabetes?
Patients were excluded from SPRINT if they were under age 50, were at low cardiovascular risk, or had diabetes, raising the question of whether the results apply to these groups as well.
The question is particularly relevant in diabetes, as the ACCORD BP study, which used the same blood pressure targets as SPRINT, did not show a significant difference in the primary cardiovascular outcome between the intensive and standard treatments in patients with diabetes (Table 3).13 In ACCORD BP, the rate of the primary outcome was 12% lower in the intensive treatment group than in the standard treatment group, but the 95% confidence interval was –27% to +6%, so the finding was not statistically significant. However, the wide confidence interval does not exclude the possibility of a benefit that was comparable to that observed in SPRINT.
It has been speculated that ACCORD BP was underpowered to detect significant differences in the primary outcome.21 An analysis combining data from both trials indicated that effects on individual outcomes were generally consistent in both trials (with no significant heterogeneity noted).22 Also, the primary composite outcome in ACCORD did not include heart failure, which is particularly sensitive to blood pressure reduction.
Additionally, ACCORD BP had a 2 × 2 factorial design involving a simultaneous comparison of intensive vs standard glycemic control, which may have influenced the effects due to blood pressure. Indeed, a post hoc analysis showed that there was a significant 26% lower risk of the primary outcome in ACCORD BP patients who received intensive systolic pressure control plus standard glycemic control than in those receiving standard systolic control plus standard glycemic control.23
Are more adverse events an acceptable trade-off?
Adverse events, including acute kidney injury, were more frequent in the intensive therapy group in SPRINT.
Acute kidney injury was coded as an adverse event on the basis of this diagnosis being included in the hospital discharge summary (as a primary or main secondary diagnosis) and if considered by the safety officer to be one of the top three reasons for admission or continued hospitalization. Further analysis of renal events should be forthcoming.
People in the intensive treatment group, on average, needed one more medication than those in the standard treatment group. Some of the adverse events may be related to the antihypertensive medications taken (eg, electrolyte abnormalities such as hyponatremia and hypokalemia due to diuretic use), and others may be related to blood pressure-lowering (eg, acute kidney injury due to renal hypoperfusion).
At this point, the long-term effects of these adverse events, especially on kidney function, are not known. Patients enrolled in clinical trials tend to be healthier than patients seen in clinical practice; thus, the rate of adverse events reported in the trial may be lower than one would see in the real world.
Does lower systolic pressure protect or harm the kidneys?
SPRINT included patients with stage 3 and 4 chronic kidney disease (ie, with eGFR 20–50 mL/min/1.73 m2), but it was designed to assess cardiovascular outcomes, not the progression of chronic kidney disease. The trial excluded patients with diabetic nephropathy or high degrees of proteinuria.
Earlier randomized trials that focused on chronic kidney disease progression, including the MDRD24 and the African American Study of Kidney Disease and Hypertension,25 did not show benefit with more aggressive blood pressure-lowering (except in patients with higher degrees of proteinuria), and these trials were not powered to assess effects on cardiovascular outcomes.24,25
The Irbesartan Diabetic Nephropathy Trial,26,27 which was done in patients with overt diabetic nephropathy, showed that a progressively lower achieved systolic pressure down to 120 mm Hg predicted lower rates of heart failure, cardiovascular mortality, and renal events (although the trial target was ≤ 130/85 mm Hg and few participants achieved systolic pressure lower than 120 mm Hg).
IMPLICATIONS FOR MANAGEMENT
The recent estimates of hypertension prevalence and control from NHANES show that only about 53% of hypertensive adults have their blood pressure under control (defined as systolic pressure < 140 mm Hg and diastolic pressure < 90 mm Hg).2 Analysis of the NHANES 2007–2012 data showed that 16.7% or 8.2 million US adults with treated hypertension meet the eligibility criteria for SPRINT.28
Although the SPRINT results support the notion that “lower is better,” the risks and benefits of intensive control will need to be balanced in individual patients. Table 4 shows the number needed to treat and number needed to harm in the trial.
More aggressive management of hypertension is challenging. The median systolic pressure achieved in the intensive group in SPRINT was just over 120 mm Hg, which implies that at least half of the participants in the intensive group did not achieve the goal of less than 120 mm Hg. While it may be reasonable to aim for systolic pressure of less than 120 or 125 mm Hg in patients who fit the SPRINT criteria and can tolerate intensive blood pressure lowering, it would be prudent to aim for a more conservative goal in elderly patients who are frail and at risk for falls, considering the higher incidence of specified adverse events in the intensive group.
Results of cognitive outcomes, as well as data related to quality of life, are still awaited. Long-term renal outcomes are also unclear.
As noted above, the question of generalizability of SPRINT results to patients with diabetes is open to debate. In our opinion, with currently available evidence, it is difficult to conclusively answer the question of whether a lower systolic target provides cardiovascular benefit in diabetes. It is also unclear whether similar beneficial results would be seen with intensive treatment in a population at low cardiovascular risk. The American Heart Association and the American College of Cardiology are in the process of formulating new hypertension guidelines, and evidence from SPRINT will inform any new recommendations.
As more medications will likely be needed for intensive systolic blood pressure control, side effects and tolerability of medications with polypharmacy and potential nonadherence with increasing complexity of medication regimens should be kept in mind. Lifestyle modifications will need to be emphasized and reinforced, with greater use of combination antihypertensive therapy.
The data from SPRINT indicate that lower systolic pressure is better, as long as untoward clinical events can be monitored and avoided or easily managed. Careful monitoring will likely entail more frequent clinic visits and more frequent assessment of renal function and electrolyte levels (participants in the intensive group in the trial were seen every month until goal was achieved). A team approach that includes pharmacists and nurse practitioners, along with optimal use of best practice algorithms and remote monitoring technology, will need to be implemented for efficient and effective care.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS data brief, no. 220. Hyattsville, MD: National Center for Health Statistics. 2015.
- Franklin SS, Jacobs MJ, Wong ND, L’Italien GJ, Lapuerta P. Predominance of isolated systolic hypertension among middle-aged and elderly US hypertensives: analysis based on National Health and Nutrition Examination Survey (NHANES) III. Hypertension 2001; 37:869–874.
- Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000; 356:1955–1964.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: Valsartan in Elderly Isolated Systolic Hypertension study. Hypertension 2010; 56:196–202.
- Liu L, Zhang Y, Liu G, Li W, Zhang X, Zanchetti A; FEVER Study Group. The Felodipine Event Reduction (FEVER) Study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23:2157–2172.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520. Erratum in: JAMA. 2014; 311:1809.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Ambrosius WT, Sink KM, Foy CG, et al; SPRINT Study Research Group. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014; 11:532–546.
- Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Conventional versus automated measurement of blood pressure in the office (CAMBO) trial. Fam Pract 2012; 29:376–382.
- Messerli FH, Mancia G, Conti CR, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893.
- Boutitie F, Gueyffier F, Pocock S, Fagard R, Boissel JP; INDANA Project Steering Committee; INdividual Data ANalysis of Antihypertensive intervention. J-shaped relationship between blood pressure and mortality in hypertensive patients: new insights from a meta-analysis of individual-patient data. Ann Intern Med 2002; 136:438–448.
- Mancia G. Effects of intensive blood pressure control in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 2010; 122:847–849.
- Perkovic V, Rodgers A. Redefining blood-pressure targets—SPRINT starts the marathon. N Engl J Med 2015; 373:2175–2178.
- Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care 2014; 37:1721–1728.
- Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease study. Ann Intern Med 1995; 123:754–762.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Berl T, Hunsicker LG, Lewis JB, et al; Collaborative Study Group. Impact of achieved blood pressure on cardiovascular outcomes in the Irbesartan Diabetic Nephropathy Trial. J Am Soc Nephrol 2005; 16:2170–2179.
- Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the Irbesartan Diabetic Nephropathy Trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16:3027–3037.
- Bress AP, Tanner RM, Hess R, Colantonio LD, Shimbo D, Muntner P. Generalizability of results from the Systolic Blood Pressure Intervention Trial (SPRINT) to the US adult population. J Am Coll Cardiol 2015 Oct 31. doi: 10.1016/j.jacc.2015.10.037. Epub ahead of print.
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015; 373:2103–2116.
- Yoon SS, Fryar CD, Carroll MD. Hypertension prevalence and control among adults: United States, 2011–2014. NCHS data brief, no. 220. Hyattsville, MD: National Center for Health Statistics. 2015.
- Franklin SS, Jacobs MJ, Wong ND, L’Italien GJ, Lapuerta P. Predominance of isolated systolic hypertension among middle-aged and elderly US hypertensives: analysis based on National Health and Nutrition Examination Survey (NHANES) III. Hypertension 2001; 37:869–874.
- Neal B, MacMahon S, Chapman N; Blood Pressure Lowering Treatment Trialists’ Collaboration. Effects of ACE inhibitors, calcium antagonists, and other blood-pressure-lowering drugs: results of prospectively designed overviews of randomised trials. Blood Pressure Lowering Treatment Trialists’ Collaboration. Lancet 2000; 356:1955–1964.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R; Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360:1903–1913.
- SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991; 265:3255–3264.
- Staessen JA, Fagard R, Thijs L, et al. Randomised double-blind comparison of placebo and active treatment for older patients with isolated systolic hypertension. The Systolic Hypertension in Europe (Syst-Eur) Trial Investigators. Lancet 1997; 350:757–764.
- Beckett NS, Peters R, Fletcher AE, et al; HYVET Study Group. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358:1887–1898.
- JATOS Study Group. Principal results of the Japanese trial to assess optimal systolic blood pressure in elderly hypertensive patients (JATOS). Hypertens Res 2008; 31:2115–2127.
- Ogihara T, Saruta T, Rakugi H, et al; Valsartan in Elderly Isolated Systolic Hypertension Study Group. Target blood pressure for treatment of isolated systolic hypertension in the elderly: Valsartan in Elderly Isolated Systolic Hypertension study. Hypertension 2010; 56:196–202.
- Liu L, Zhang Y, Liu G, Li W, Zhang X, Zanchetti A; FEVER Study Group. The Felodipine Event Reduction (FEVER) Study: a randomized long-term placebo-controlled trial in Chinese hypertensive patients. J Hypertens 2005; 23:2157–2172.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- SPS3 Study Group; Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013; 382:507–515.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520. Erratum in: JAMA. 2014; 311:1809.
- Weber MA, Schiffrin EL, White WB, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens (Greenwich) 2014; 16:14–26.
- Ambrosius WT, Sink KM, Foy CG, et al; SPRINT Study Research Group. The design and rationale of a multicenter clinical trial comparing two strategies for control of systolic blood pressure: the Systolic Blood Pressure Intervention Trial (SPRINT). Clin Trials 2014; 11:532–546.
- Myers MG, Godwin M, Dawes M, Kiss A, Tobe SW, Kaczorowski J. Conventional versus automated measurement of blood pressure in the office (CAMBO) trial. Fam Pract 2012; 29:376–382.
- Messerli FH, Mancia G, Conti CR, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893.
- Boutitie F, Gueyffier F, Pocock S, Fagard R, Boissel JP; INDANA Project Steering Committee; INdividual Data ANalysis of Antihypertensive intervention. J-shaped relationship between blood pressure and mortality in hypertensive patients: new insights from a meta-analysis of individual-patient data. Ann Intern Med 2002; 136:438–448.
- Mancia G. Effects of intensive blood pressure control in the management of patients with type 2 diabetes mellitus in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Circulation 2010; 122:847–849.
- Perkovic V, Rodgers A. Redefining blood-pressure targets—SPRINT starts the marathon. N Engl J Med 2015; 373:2175–2178.
- Margolis KL, O’Connor PJ, Morgan TM, et al. Outcomes of combined cardiovascular risk factor management strategies in type 2 diabetes: the ACCORD randomized trial. Diabetes Care 2014; 37:1721–1728.
- Peterson JC, Adler S, Burkart JM, et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease study. Ann Intern Med 1995; 123:754–762.
- Agodoa LY, Appel L, Bakris GL, et al; African American Study of Kidney Disease and Hypertension (AASK) Study Group. Effect of ramipril vs amlodipine on renal outcomes in hypertensive nephrosclerosis: a randomized controlled trial. JAMA 2001; 285:2719–2728.
- Berl T, Hunsicker LG, Lewis JB, et al; Collaborative Study Group. Impact of achieved blood pressure on cardiovascular outcomes in the Irbesartan Diabetic Nephropathy Trial. J Am Soc Nephrol 2005; 16:2170–2179.
- Pohl MA, Blumenthal S, Cordonnier DJ, et al. Independent and additive impact of blood pressure control and angiotensin II receptor blockade on renal outcomes in the Irbesartan Diabetic Nephropathy Trial: clinical implications and limitations. J Am Soc Nephrol 2005; 16:3027–3037.
- Bress AP, Tanner RM, Hess R, Colantonio LD, Shimbo D, Muntner P. Generalizability of results from the Systolic Blood Pressure Intervention Trial (SPRINT) to the US adult population. J Am Coll Cardiol 2015 Oct 31. doi: 10.1016/j.jacc.2015.10.037. Epub ahead of print.
KEY POINTS
- SPRINT is the first large prospective randomized trial to show evidence of cardiovascular and mortality benefit for intensive lowering of systolic blood pressure (goal < 120 mm Hg) in older patients at cardiovascular risk, but without a history of diabetes mellitus or stroke.
- A similar trial in patients with type 2 diabetes mellitus did not show significant benefit of intensive treatment.
- Intensive treatment was associated with more adverse events, including hypotension, syncope, electrolyte abnormalities, and acute kidney injury.
- It is unclear if these results can be extrapolated to patients with a history of diabetes or stroke, younger patients, or those with low cardiovascular risk.
- Healthcare providers should engage patients in a shared decision-making process, with discussion of the benefits and risks associated with intensive lowering of blood pressure.
Advances in the treatment of dyslipidemia
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:

They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:
They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
The 2013 joint guidelines of the American College of Cardiology and American Heart Association (ACC/AHA)1 on the treatment of blood cholesterol to reduce cardiovascular risk recommend high-intensity statin therapy for secondary prevention of cardiovascular events. The question of primary prevention is not so straightforward, and the recommended strategy has come under fire. In addition, the guidelines focus on statins and not on LDL-C levels, and the role of nonstatin lipid-lowering drugs and the value of reducing LDL-C levels to well below levels currently regarded as “normal” remain unclear.
This article comments on the 2013 ACC/AHA guidelines, reviews the data on optimal LDL-C levels, and discusses new nonstatin agents.
ACC/AHA GUIDELINES: A MIXED MESSAGE
The 2013 ACC/AHA cholesterol guidelines1 can be characterized by the title from the famous Western film “The Good, the Bad, and the Ugly.”
The good: A clear message to treat
The guidelines deliver an unambiguous message to treat patients at high risk with high-intensity statin therapy. This mandate is very helpful as it should reduce the undertreatment of patients.
The seemingly bad
Two common misconceptions regarding the guidelines:
They abandon LDL-C targets. Actually, the guidelines do not argue for or against targets; they simply remain silent, citing that randomized trials have not been conducted with LDL-C targets as specific goals. Technically, this statement is true. However, it seems contrived to argue, for example, that the benefit of atorvastatin 80 mg over 10 mg in the Treating to New Targets trial could not be reliably ascribed to the lower LDL-C achieved with the higher dose, but rather to some undefined benefit of high-intensity statin therapy, especially as the guidelines define the intensity of statins by the degree of LDL-C lowering. In fact, by correlating the incidence of coronary heart disease events with the levels of LDL-C achieved in those trials, conclusions can reasonably be drawn from such data (Figure 1).2
The guidelines do not recommend nonstatin drugs. Actually, the guidelines note that clinicians are free to consider other therapies, especially those proven to reduce the risk of cardiovascular events, a central principle of medicine. Since the guidelines were published, data have emerged indicating that the role of nonstatin drugs also needs consideration.
The ugly: Risk calculator untested
The guidelines promote the use of a risk calculator developed by the ACC/AHA to estimate the 10-year risk of an atherosclerotic event for people whose LDL-C levels are between 70 and 189 mg/dL to help decide whether to initiate statin therapy for primary prevention of atherosclerotic cardiovascular disease. Such an approach is reasonable, although the risk score was promulgated without evidence to support its utility.
Media coverage of the risk calculator was fierce. Some physicians found imperfections in the risk score (as is true for all risk scores), resulting in public mistrust of the guidelines and of the medical community as a whole. This needless controversy may have compromised the main message—that LDL-C should be lowered in many people—a message backed by strong evidence.
Alternative strategies proposed
Ridker et al3 have proposed a hybrid strategy to guide statin use for apparently healthy people that combines the ACC/AHA guideline approach with entry criteria for randomized clinical trials that showed statin efficacy for primary prevention.
Genetic analysis may offer another approach. Mega et al4 stratified more than 48,000 people by a genetic risk score based on 27 genetic variants and found a significant association with risk of coronary events. Targeting therapy to people found to be at higher risk on this basis offers greater risk reduction than expected for the general population. Biomarkers and imaging tests are other potentially useful risk determinants.
LDL-C: LOWER IS BETTER
Although no clinical trial has yet targeted specific LDL-C levels, there is plenty of evidence that lower LDL-C levels offer greater benefit (Figure 1).2
In 1994, the Scandinavian Simvastatin Survival Study5 established the benefit of statins in patients with known vascular disease. The mean LDL-C level achieved in the active treatment group was 120 mg/dL. More trials followed supporting the benefits of statins and of reducing LDL-C from average levels in the 120s down to 100 mg/dL.
In 2004, the Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis in Myocardial Infarction 22 trial6 observed an even greater risk reduction in patients with known risk by treating with statins; the mean LDL-C level achieved in the group randomized to an intensive regimen of atorvastatin 80 mg per day was 62 mg/dL. The same year, the Adult Treatment Panel III of the National Cholesterol Education Program7 issued updated guidelines including an optional goal of LDL-C less than 70 mg/dL for patients at very high risk.
In 2008, the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER)8 found a significantly lower incidence of major cardiovascular events at 2 years in apparently healthy men and women with baseline LDL-C levels of less than 130 mg/dL after treatment with rosuvastatin 20 mg daily, with an achieved median LDL-C of 55 mg/dL.
How low should LDL-C go?
Evidence from clinical trials indicates a 20% to 25% reduction in the risk of cardiovascular events for every 39-mg/dL decrease in LDL-C. Extrapolating the data, cardiovascular disease risk would be reduced to zero if LDL-C were brought down below 40 mg/dL.
Brown and Goldstein,9 who won the 1985 Nobel Prize in medicine for their work in cholesterol metabolism, estimated that a plasma level of LDL-C of only 25 mg/dL would be sufficient to nourish cells with cholesterol. Cells can synthesize all the cholesterol they need, underscoring that LDL-C is simply the final end-product that the liver removes from circulation.
Other evidence that lower LDL-C does not have adverse effects comes from non-Western populations as well as from other mammals. Total cholesterol levels range in the low 100s mg/dL in Native American and African tribal populations, with LDL-C estimated to be about 50 to 75 mg/dL. Elephants, baboons, and foxes have even lower levels.10
Clinical trial data also support that LDL-C levels below the current “normal” are better. The Cholesterol Treatment Trialists’ Collaboration11 analyzed data from more than 160,000 patients in 26 trials that evaluated either more- vs less-intensive statin regimens or statin treatment vs control. No baseline level below which lowering LDL-C further was not beneficial was found. Patients who started out with an LDL-C level of less than 77 mg/dL had the same risk reduction of major vascular events when the level was dropped to 50 mg/dL as those who started at higher levels and reduced their LDL-C by the same amount. In the JUPITER trial, even those with a baseline LDL-C of less than 60 mg/dL benefited from statin therapy.12
BEYOND STATINS
Ezetimibe further lowers risk
Ezetimibe is a nonstatin drug that reduces LDL-C by about 15% to 20%. The Improved Reduction of Outcomes: Vytorin Efficacy International Trial13 registered more than 18,000 patients with a baseline LDL-C level of less than 125 mg/dL (or 100 mg/dL if already on lipid-lowering therapy) who had been stabilized shortly after an acute cardiovascular event. They were randomized to receive either simvastatin 40 mg or combined simvastatin 40 mg and ezetimibe 10 mg. The study intended to determine two things: whether ezetimibe could further lower LDL-C when combined with a statin, and whether risk could be reduced further by driving the LDL-C below 70 mg/dL and down to the mid-50s.
After 1 year, the average LDL-C level was 70 mg/dL in the simvastatin group and 53 mg/dL in the combined simvastatin and ezetimibe group. At 7 years, for the primary end point (cardiovascular death, myocardial infarction, unstable angina requiring hospitalization, coronary revascularization, or stroke), there was a 6% reduction of events in the combined drug treatment group, with the number of people needed to treat being 50 to prevent one event. For the narrower end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke, there was a 10% risk reduction in the combined drug treatment arm.14
The amount of risk reduction is exactly what was predicted by the Cholesterol Treatment Trialists’ Collaboration’s plot of reduction in events vs reduction in LDL-C based on the analysis of 26 trials, adding further evidence that it is the LDL-C reduction itself, rather than the means by which LDL-C is reduced, that is critical for benefit.
PCSK9 inhibitors: A new approach
Mutations in the gene for proprotein convertase subtilisin kexin type 9 (PCSK9) have become a new focus of interest for reducing LDL-C and cardiovascular risk.15 PCSK9 binds to the LDL-C receptor on the surface of hepatocytes and escorts it to its destruction in the lysosomes, rather than allowing it to return to the cell surface to take more LDL-C out of circulation.
People with a gain-of-function mutation (conferring too much PCSK9, resulting in fewer LDL-C receptors and more LDL-C in circulation) are a more recently recognized subset of those with autosomal-dominant familial hypercholesterolemia. They have total cholesterol levels in the 90th percentile, tendon xanthomas, and a high risk of myocardial infarction and stroke at a young age.
Conversely, those with a nonsense mutation in PCSK9—leading to loss of function—have a 28% reduction in mean LDL-C and 88% reduction in risk of coronary heart disease compared with those without the mutation.16 Two women (ages 32 and 21, fertile) have been found who have inactivating mutations in both PCSK9 alleles, and both are in apparent good health, with LDL-C levels of 14 mg/dL and 15 mg/dL, respectively.17,18
Dramatic reduction in LDL-C
Monoclonal antibodies have been developed that bind PCSK9 and block its action with the goal of developing new LDL-C–lowering treatments. Phase 2 clinical trials of varying doses of evolocumab (Repatha), a drug in this class, combined with standard therapy (a statin with or without ezetimibe), found a 66% reduction of LDL-C at high doses at 12 weeks compared with standard therapy alone, with concomitant reductions in other atherogenic lipoproteins.19 Patients who could not tolerate statins because of myalgia responded well to evolocumab.20
Patients with heterozygous familial hypercholesterolemia also had a substantial reduction in LDL-C (55% at the highest dosage), even though they have fewer LDL-C receptors for the drug to act upon.21 People with homozygous familial hypercholesterolemia and no LDL-C receptors had a lesser relative reduction in LDL-C that depended on the type of mutations they had. Nonetheless, given how high LDL-C levels are in this population, the absolute decreases in LDL-C level were quite impressive.
Cardiovascular risk reduced
Data at nearly 1 year showed continued reduction of LDL-C by about 60% (absolute reduction: 73 mg/dL), as well as a lower incidence of cardiovascular events starting at just 3 months, much sooner than observed in some statin trials.22 Benefits were found regardless of subgroup (sex, age, statin use, baseline LDL-C level, or known vascular disease). No difference was found in the safety profile between the evolocumab and control arms. Only 2.4% of participants discontinued evolocumab because of adverse events, and the incidence of adverse effects did not correlate with LDL-C level achieved.
Neurocognitive effects occurred in 0.9% of the evolocumab arm vs 0.3% in the control arm. This difference has not been explained: although there is cholesterol in the central nervous system, it is generated locally, and lipoproteins—and evolocumab—are not thought to cross the blood-brain barrier.
Long-term trials of evolocumab are currently under way for patients with cardiovascular disease, as are trials of two other PCSK9 inhibitors, alirocumab and bococizumab, in addition to standard statin therapy.
On July 24, 2015, the US Food and Drug Administration (FDA) approved the first PCSK9 inhibitor, alirocumab (Praluent) for patients with heterozygous familial hypercholesterolemia or those with clinical atherosclerotic cardiovascular disease who require additional lowering of LDL-C. The starting dosage is 75 mg subcutaneously every 2 weeks, which can be increased up to 150 mg every 2 weeks.
Evolocumab was approved by the FDA on August 27, 2015, for the same indications. The dosage is 140 mg subcutaneously every 2 weeks or 420 mg every month.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
- Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-S45. Erratum in: Circulation 2014; 129:S46–S48.
- Raymond C, Cho L, Rocco M, Hazen SL. New cholesterol guidelines: worth the wait? Cleve Clin J Med 2014; 81:11–19.
- Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol 2015; 65:942–948.
- Mega JL, Stitziel NO, Smith JG, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet 2015; 385:2264–2271.
- Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 1994; 344:1383–1389.
- Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- Grundy SM, Cleeman JI, Merz CN, et al; National Heart, Lung, and Blood Institute; American College of Cardiology Foundation; American Heart Association. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239. Erratum in Circulation 2004; 110:763.
- Ridker PM, Danielson E, Fonseca FAH, et al; JUPITER Study Group. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science 1986; 232:34–47.
- Hochholzer W, Giugliano RP. Lipid lowering goals: back to nature? Ther Adv Cardiovasc Dis 2010; 4:185–191.
- Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010; 376:1670–1681.
- Hsia J, MacFadyen JG, Monyak J, Ridker PM. Cardiovascular event reduction and adverse events among subjects attaining low-density lipoprotein cholesterol <50 mg/dl with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol 2011; 57:1666–1675.
- Cannon CP, Giugliano RP, Blazing MA, et al; IMPROVE-IT Investigators. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndromes. Am Heart J 2008; 156:826–832.
- Cannon CP, Blazing MA, Giugliano RP, et al for the IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med 2015; 372:2387–2397.
- Giugliano RP, Sabatine MS. Are PCSK9 Inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol 2015; 65:2638–2651.
- Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354:1264–1272.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79:514-523.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007; 193:445–448.
- Giugliano RP, Desai NR, Kohli P, et al; LAPLACE-TIMI 57 Investigators. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012; 380:2007–2017.
- Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA 2012; 308:2497–2506.
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012; 126:2408–2417.
- Sabatine MS, Giugliano RP, Wiviott SD, et al; Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372:1500–1509.
KEY POINTS
- Patients at high risk of atherosclerotic cardiovascular disease should be treated with high-intensity statin therapy.
- To date, no baseline level has been identified beneath which lowering LDL-C does not provide clinical benefit.
- The benefits of lower LDL-C are seen with a variety of pharmacologic interventions and in people who have naturally low levels due to genetic variants.
- Clinical trial evidence supports that ezetimibe reduces the risk of cardiovascular events.
- Proprotein convertase subtilisin kexin type 9 (PCSK9) inhibitors reduce LDL-C by approximately 60%, and preliminary data show that they reduce the risk of cardiovascular events.

When we need to remember that it is more than a job

“I am forever humbled.” So said a heart failure specialist on rounds when I was a resident in the intensive care unit several decades ago. He was talking about the perpetual mismatch between a physician’s level of knowledge and the unpredictability inherent in the management and outcome of critically ill patients. His words ring true for me nearly every day. We should never think we are so smart that we are truly in control of our patients’ outcomes or that we don’t make mistakes—but we also cannot become so paralyzed by the awareness of our limitations that we don’t make decisions.
I have spoken those same powerful words many times on teaching rounds. I also frequently push them to the back of my mind. As a consultant at a major medical center, I am supposed to know. It is a fine line we walk.
I know I am not alone in harboring these self-doubts. Ready access to online information does little to assuage the concern that we can never know enough. Have I ordered enough diagnostic tests to be sure? Have I ordered too many tests and thus will be penalized for providing cost-ineffective care? Should we follow generic guidelines, or deviate from the guidelines based on our clinical instincts, our own interpretation of the literature, and the patient’s unique circumstances and desires?
And then what happens when we make wrong decisions, or even the right decisions that result in a poor patient outcome, which of course is at some point inevitable? We are told to be open about errors, to be honest and transparent about our limitations, to throw down our elaborate emotional and intellectual defensive shields and expose our vulnerability.
But what do we experience emotionally when we are named in a malpractice suit? We may have done all that we thought we could do: we responsibly explored the diagnostic and therapeutic options, provided empathetic care, and listened to the voice of the patient. Yet an adverse outcome still occurred. The practice of medicine is indeed humbling. We feel crushed. We revisit the patient’s care in a vivid perpetual replay loop in our head. Maybe we didn’t evaluate all the options as we should have. If we had been a bit smarter, a bit more efficient, maybe the outcome would have been different.
Then during a deposition, the plaintiff’s counsel points out the temporal and documentary inconsistencies in the electronic medical record: “Doctor, you say you saw the patient at 2:00 pm, but there was no note finalized until 10:00 pm…and why was your documented physical exam exactly the same as the one from the day before and exactly the same as that of the resident who saw the patient that afternoon?” We now feel crushed, totally vulnerable, emotionally trampled, and often isolated and disconnected from our patients and peers. The intellectualized humility becomes transformed into a sense of inadequacy. Why should I keep doing this?
In this issue of the Journal, experienced malpractice attorney Kevin Giordano explores aspects of the malpractice process as they relate to the physicians he defends. He notes how the electronic medical record, a tool ostensibly in place to improve communication and the sharing of medical information between caregivers and patients, can be our worst enemy in a courtroom. He discusses the pressures of our complicated healthcare system that promote documentation errors that he must try to explain away to the jury in our defense, demonstrating that these documentation errors do not necessarily mirror the care and caring of the named physicians. This is critically important information for us to understand and to act on for our personal protection, but it is not his most important message to us.
Mr. Giordano is a sincere, empathetic, and proficient professional. He has spoken for and to many physicians. He has listened to us and observed our behaviors. And as he has defended many of us in a court of law, he has learned to diagnose in his clients the damage that can persist following involvement in a malpractice case and the emotional scars the malpractice experience leaves on physicians. He emphasizes that we must not let the event of a malpractice suit force us to withdraw and strip us of our connection and engagement to patients. If anything, he and Drs. Susan Rehm and Bradford Borden, in an accompanying editorial, urge us to keep in mind that our personal engagement with patients and the mindful practice of medicine is our raison d’être as physicians.
I am continuously humbled by the breadth of the pathology, clinical medicine, and social challenges that I encounter on a daily basis, armed with limited knowledge and experience. It is intellectually rewarding to make an arcane diagnosis or to see an individualized therapy work as I had hoped. But I agree with Mr. Giordano that it is the genuine engagement with patients that provides us with the real joy in the practice of medicine and pushes us to deliver care at a consistently proficient level. We must not forget that, even in the face of significant and emotionally challenging events such as being named in a malpractice suit. It is the nature of our engagement with our patients and our colleagues that make what we do more than a job.
As more physicians in the United States become employed by health systems, I hope that the administrative leaders within these systems truly recognize these issues. As they struggle to balance the provision of safe high-quality care to patients with their increasingly complex financial spreadsheets, I hope that the emotional health of their physician employees is not forgotten. And not just after a malpractice suit.
“I am forever humbled.” So said a heart failure specialist on rounds when I was a resident in the intensive care unit several decades ago. He was talking about the perpetual mismatch between a physician’s level of knowledge and the unpredictability inherent in the management and outcome of critically ill patients. His words ring true for me nearly every day. We should never think we are so smart that we are truly in control of our patients’ outcomes or that we don’t make mistakes—but we also cannot become so paralyzed by the awareness of our limitations that we don’t make decisions.
I have spoken those same powerful words many times on teaching rounds. I also frequently push them to the back of my mind. As a consultant at a major medical center, I am supposed to know. It is a fine line we walk.
I know I am not alone in harboring these self-doubts. Ready access to online information does little to assuage the concern that we can never know enough. Have I ordered enough diagnostic tests to be sure? Have I ordered too many tests and thus will be penalized for providing cost-ineffective care? Should we follow generic guidelines, or deviate from the guidelines based on our clinical instincts, our own interpretation of the literature, and the patient’s unique circumstances and desires?
And then what happens when we make wrong decisions, or even the right decisions that result in a poor patient outcome, which of course is at some point inevitable? We are told to be open about errors, to be honest and transparent about our limitations, to throw down our elaborate emotional and intellectual defensive shields and expose our vulnerability.
But what do we experience emotionally when we are named in a malpractice suit? We may have done all that we thought we could do: we responsibly explored the diagnostic and therapeutic options, provided empathetic care, and listened to the voice of the patient. Yet an adverse outcome still occurred. The practice of medicine is indeed humbling. We feel crushed. We revisit the patient’s care in a vivid perpetual replay loop in our head. Maybe we didn’t evaluate all the options as we should have. If we had been a bit smarter, a bit more efficient, maybe the outcome would have been different.
Then during a deposition, the plaintiff’s counsel points out the temporal and documentary inconsistencies in the electronic medical record: “Doctor, you say you saw the patient at 2:00 pm, but there was no note finalized until 10:00 pm…and why was your documented physical exam exactly the same as the one from the day before and exactly the same as that of the resident who saw the patient that afternoon?” We now feel crushed, totally vulnerable, emotionally trampled, and often isolated and disconnected from our patients and peers. The intellectualized humility becomes transformed into a sense of inadequacy. Why should I keep doing this?
In this issue of the Journal, experienced malpractice attorney Kevin Giordano explores aspects of the malpractice process as they relate to the physicians he defends. He notes how the electronic medical record, a tool ostensibly in place to improve communication and the sharing of medical information between caregivers and patients, can be our worst enemy in a courtroom. He discusses the pressures of our complicated healthcare system that promote documentation errors that he must try to explain away to the jury in our defense, demonstrating that these documentation errors do not necessarily mirror the care and caring of the named physicians. This is critically important information for us to understand and to act on for our personal protection, but it is not his most important message to us.
Mr. Giordano is a sincere, empathetic, and proficient professional. He has spoken for and to many physicians. He has listened to us and observed our behaviors. And as he has defended many of us in a court of law, he has learned to diagnose in his clients the damage that can persist following involvement in a malpractice case and the emotional scars the malpractice experience leaves on physicians. He emphasizes that we must not let the event of a malpractice suit force us to withdraw and strip us of our connection and engagement to patients. If anything, he and Drs. Susan Rehm and Bradford Borden, in an accompanying editorial, urge us to keep in mind that our personal engagement with patients and the mindful practice of medicine is our raison d’être as physicians.
I am continuously humbled by the breadth of the pathology, clinical medicine, and social challenges that I encounter on a daily basis, armed with limited knowledge and experience. It is intellectually rewarding to make an arcane diagnosis or to see an individualized therapy work as I had hoped. But I agree with Mr. Giordano that it is the genuine engagement with patients that provides us with the real joy in the practice of medicine and pushes us to deliver care at a consistently proficient level. We must not forget that, even in the face of significant and emotionally challenging events such as being named in a malpractice suit. It is the nature of our engagement with our patients and our colleagues that make what we do more than a job.
As more physicians in the United States become employed by health systems, I hope that the administrative leaders within these systems truly recognize these issues. As they struggle to balance the provision of safe high-quality care to patients with their increasingly complex financial spreadsheets, I hope that the emotional health of their physician employees is not forgotten. And not just after a malpractice suit.
“I am forever humbled.” So said a heart failure specialist on rounds when I was a resident in the intensive care unit several decades ago. He was talking about the perpetual mismatch between a physician’s level of knowledge and the unpredictability inherent in the management and outcome of critically ill patients. His words ring true for me nearly every day. We should never think we are so smart that we are truly in control of our patients’ outcomes or that we don’t make mistakes—but we also cannot become so paralyzed by the awareness of our limitations that we don’t make decisions.
I have spoken those same powerful words many times on teaching rounds. I also frequently push them to the back of my mind. As a consultant at a major medical center, I am supposed to know. It is a fine line we walk.
I know I am not alone in harboring these self-doubts. Ready access to online information does little to assuage the concern that we can never know enough. Have I ordered enough diagnostic tests to be sure? Have I ordered too many tests and thus will be penalized for providing cost-ineffective care? Should we follow generic guidelines, or deviate from the guidelines based on our clinical instincts, our own interpretation of the literature, and the patient’s unique circumstances and desires?
And then what happens when we make wrong decisions, or even the right decisions that result in a poor patient outcome, which of course is at some point inevitable? We are told to be open about errors, to be honest and transparent about our limitations, to throw down our elaborate emotional and intellectual defensive shields and expose our vulnerability.
But what do we experience emotionally when we are named in a malpractice suit? We may have done all that we thought we could do: we responsibly explored the diagnostic and therapeutic options, provided empathetic care, and listened to the voice of the patient. Yet an adverse outcome still occurred. The practice of medicine is indeed humbling. We feel crushed. We revisit the patient’s care in a vivid perpetual replay loop in our head. Maybe we didn’t evaluate all the options as we should have. If we had been a bit smarter, a bit more efficient, maybe the outcome would have been different.
Then during a deposition, the plaintiff’s counsel points out the temporal and documentary inconsistencies in the electronic medical record: “Doctor, you say you saw the patient at 2:00 pm, but there was no note finalized until 10:00 pm…and why was your documented physical exam exactly the same as the one from the day before and exactly the same as that of the resident who saw the patient that afternoon?” We now feel crushed, totally vulnerable, emotionally trampled, and often isolated and disconnected from our patients and peers. The intellectualized humility becomes transformed into a sense of inadequacy. Why should I keep doing this?
In this issue of the Journal, experienced malpractice attorney Kevin Giordano explores aspects of the malpractice process as they relate to the physicians he defends. He notes how the electronic medical record, a tool ostensibly in place to improve communication and the sharing of medical information between caregivers and patients, can be our worst enemy in a courtroom. He discusses the pressures of our complicated healthcare system that promote documentation errors that he must try to explain away to the jury in our defense, demonstrating that these documentation errors do not necessarily mirror the care and caring of the named physicians. This is critically important information for us to understand and to act on for our personal protection, but it is not his most important message to us.
Mr. Giordano is a sincere, empathetic, and proficient professional. He has spoken for and to many physicians. He has listened to us and observed our behaviors. And as he has defended many of us in a court of law, he has learned to diagnose in his clients the damage that can persist following involvement in a malpractice case and the emotional scars the malpractice experience leaves on physicians. He emphasizes that we must not let the event of a malpractice suit force us to withdraw and strip us of our connection and engagement to patients. If anything, he and Drs. Susan Rehm and Bradford Borden, in an accompanying editorial, urge us to keep in mind that our personal engagement with patients and the mindful practice of medicine is our raison d’être as physicians.
I am continuously humbled by the breadth of the pathology, clinical medicine, and social challenges that I encounter on a daily basis, armed with limited knowledge and experience. It is intellectually rewarding to make an arcane diagnosis or to see an individualized therapy work as I had hoped. But I agree with Mr. Giordano that it is the genuine engagement with patients that provides us with the real joy in the practice of medicine and pushes us to deliver care at a consistently proficient level. We must not forget that, even in the face of significant and emotionally challenging events such as being named in a malpractice suit. It is the nature of our engagement with our patients and our colleagues that make what we do more than a job.
As more physicians in the United States become employed by health systems, I hope that the administrative leaders within these systems truly recognize these issues. As they struggle to balance the provision of safe high-quality care to patients with their increasingly complex financial spreadsheets, I hope that the emotional health of their physician employees is not forgotten. And not just after a malpractice suit.
The emotional impact of a malpractice suit on physicians: Maintaining resilience
Physicians who have been involved in malpractice actions are all too familiar with the range of emotions they experience during the process. Anxiety, fear, frustration, remorse, self-doubt, shame, betrayal, anger…no pleasant feelings here. Add malpractice stress to the high level of pressure experienced at home and at work, and crisis looms.
In his commentary in this issue, Kevin Giordano states, “it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team toward disconnection.”1
Because of the nature of our work as physicians, we are isolated, and malpractice isolates us further. Because of embarrassment, we avoid talking with our colleagues and managers. Legal counsel reminds us to correspond with no one about the details of the case. Spouses and friends may offer support, but it is difficult— perhaps impossible—to be reassured.
Isolation fuels our self-doubt and erodes our confidence, leading us to focus on what may go wrong, rather than on healing. Every decision is fraught with anxiety, and efficiency evaporates. Paralysis may set in, leading to disengagement from patient care and increasing the chance of further problems.
IT TAKES RESILIENCE TO THRIVE
It takes resilience to thrive in today’s pressure-cooker healthcare environment, let alone in the setting of malpractice stress. Resilient people are able to face reality and see a better future, put things into perspective, and bounce back from adversity.2 Resilience, a trait that protects against stress and burnout, is relevant at the personal, managerial, and system levels. Though this definition is not specific to caregiver or malpractice stress, it is applicable. It is an essential component of wellness and requires perpetual attention to self-care for successful maintenance.
Studies of physicians who have avoided burnout reveal remarkably consistent qualities, including finding gratification related to work, maintaining useful habits and practices, and developing attitudes that keep them resilient.3 Rather than adding activities to their full schedules, these physicians stayed resilient through mindfulness of various aspects of their daily lives. Interactions with colleagues—discussing cases, treatments, and outcomes (including errors)—proved vital. Professional development, encompassing activities such as continuing education, coaching, mentoring, and counseling, was recognized as an important self-directed resilience measure. Maintenance of relationships with family and friends, cultivation of leisure-time activities, and appreciation of the need for personal reflection time were traits often found in resilient physicians.
FOSTERING RESILIENCE
As part of the Mayo Clinic’s biannual survey of its physicians, Shanafelt et al4 studied relationships between qualities of physician leaders and burnout and satisfaction among the physicians they supervised. Many of the desirable leadership traits were related to building relationships through respectful communication, along with provision of opportunities for personal and professional development. The acknowledgment that resilient, healthy physicians are satisfied, productive, and able to provide safer and higher-quality patient care should lead to the establishment of physician wellness as a “dashboard metric.” This makes priorities clear by rewarding managers who foster self-care and resilience among their staff.
Likewise, at the healthcare system level, Beckman5 recognized that organizations can provide opportunities to promote resilience among caregivers. Organizational initiatives that set the stage for resilience include:
- Curricula to enhance communication with patients, coworkers, and family
- “Best practices” for efficient and effective patient care
- Self-care through health insurance incentives and educational sessions
- Accessible, affordable, and confidential behavioral health support
- Time for self-care activities during the workday
- Coaching and mentoring programs
- Narrative-and-reflection groups and mindfulness training.5
Through an atmosphere of support for resilience, organizations provide a place for physicians to maintain a sense of meaning and purpose in their work. For individuals facing malpractice action, this infrastructure can be used to weather the storm. As Mr. Giordano writes, staying engaged “may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.”1 We must pay attention to developing individual physicians, educating managers, and building systems so that caregivers can remain engaged and resilient. It may help those affected by malpractice stress, and perhaps as importantly, it may reduce the chance of future “disconnection” leading to recourse in the legal system.
- Giordano KC. It is not the critic’s voice that should count. Cleve Clin J Med 2016; 83:174–176.
- Coutu DL. How resilience works. Harv Bus Rev 2002; 80(5):46–55.
- Zwack J, Schweitzer J. If every fifth physician is affected by burnout, what about the other four? Acad Med 2013; 88:382–389.
- Shanafelt TD, Gorringe G, Menaker R, et al. Impact of organizational leadership on physician burnout and satisfaction. Mayo Clin Proc 2015; 90:432–440.
- Beckman H. The role of medical culture in the journey to resilience. Acad Med 2015; 90:710–712.
Physicians who have been involved in malpractice actions are all too familiar with the range of emotions they experience during the process. Anxiety, fear, frustration, remorse, self-doubt, shame, betrayal, anger…no pleasant feelings here. Add malpractice stress to the high level of pressure experienced at home and at work, and crisis looms.
In his commentary in this issue, Kevin Giordano states, “it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team toward disconnection.”1
Because of the nature of our work as physicians, we are isolated, and malpractice isolates us further. Because of embarrassment, we avoid talking with our colleagues and managers. Legal counsel reminds us to correspond with no one about the details of the case. Spouses and friends may offer support, but it is difficult— perhaps impossible—to be reassured.
Isolation fuels our self-doubt and erodes our confidence, leading us to focus on what may go wrong, rather than on healing. Every decision is fraught with anxiety, and efficiency evaporates. Paralysis may set in, leading to disengagement from patient care and increasing the chance of further problems.
IT TAKES RESILIENCE TO THRIVE
It takes resilience to thrive in today’s pressure-cooker healthcare environment, let alone in the setting of malpractice stress. Resilient people are able to face reality and see a better future, put things into perspective, and bounce back from adversity.2 Resilience, a trait that protects against stress and burnout, is relevant at the personal, managerial, and system levels. Though this definition is not specific to caregiver or malpractice stress, it is applicable. It is an essential component of wellness and requires perpetual attention to self-care for successful maintenance.
Studies of physicians who have avoided burnout reveal remarkably consistent qualities, including finding gratification related to work, maintaining useful habits and practices, and developing attitudes that keep them resilient.3 Rather than adding activities to their full schedules, these physicians stayed resilient through mindfulness of various aspects of their daily lives. Interactions with colleagues—discussing cases, treatments, and outcomes (including errors)—proved vital. Professional development, encompassing activities such as continuing education, coaching, mentoring, and counseling, was recognized as an important self-directed resilience measure. Maintenance of relationships with family and friends, cultivation of leisure-time activities, and appreciation of the need for personal reflection time were traits often found in resilient physicians.
FOSTERING RESILIENCE
As part of the Mayo Clinic’s biannual survey of its physicians, Shanafelt et al4 studied relationships between qualities of physician leaders and burnout and satisfaction among the physicians they supervised. Many of the desirable leadership traits were related to building relationships through respectful communication, along with provision of opportunities for personal and professional development. The acknowledgment that resilient, healthy physicians are satisfied, productive, and able to provide safer and higher-quality patient care should lead to the establishment of physician wellness as a “dashboard metric.” This makes priorities clear by rewarding managers who foster self-care and resilience among their staff.
Likewise, at the healthcare system level, Beckman5 recognized that organizations can provide opportunities to promote resilience among caregivers. Organizational initiatives that set the stage for resilience include:
- Curricula to enhance communication with patients, coworkers, and family
- “Best practices” for efficient and effective patient care
- Self-care through health insurance incentives and educational sessions
- Accessible, affordable, and confidential behavioral health support
- Time for self-care activities during the workday
- Coaching and mentoring programs
- Narrative-and-reflection groups and mindfulness training.5
Through an atmosphere of support for resilience, organizations provide a place for physicians to maintain a sense of meaning and purpose in their work. For individuals facing malpractice action, this infrastructure can be used to weather the storm. As Mr. Giordano writes, staying engaged “may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.”1 We must pay attention to developing individual physicians, educating managers, and building systems so that caregivers can remain engaged and resilient. It may help those affected by malpractice stress, and perhaps as importantly, it may reduce the chance of future “disconnection” leading to recourse in the legal system.
Physicians who have been involved in malpractice actions are all too familiar with the range of emotions they experience during the process. Anxiety, fear, frustration, remorse, self-doubt, shame, betrayal, anger…no pleasant feelings here. Add malpractice stress to the high level of pressure experienced at home and at work, and crisis looms.
In his commentary in this issue, Kevin Giordano states, “it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team toward disconnection.”1
Because of the nature of our work as physicians, we are isolated, and malpractice isolates us further. Because of embarrassment, we avoid talking with our colleagues and managers. Legal counsel reminds us to correspond with no one about the details of the case. Spouses and friends may offer support, but it is difficult— perhaps impossible—to be reassured.
Isolation fuels our self-doubt and erodes our confidence, leading us to focus on what may go wrong, rather than on healing. Every decision is fraught with anxiety, and efficiency evaporates. Paralysis may set in, leading to disengagement from patient care and increasing the chance of further problems.
IT TAKES RESILIENCE TO THRIVE
It takes resilience to thrive in today’s pressure-cooker healthcare environment, let alone in the setting of malpractice stress. Resilient people are able to face reality and see a better future, put things into perspective, and bounce back from adversity.2 Resilience, a trait that protects against stress and burnout, is relevant at the personal, managerial, and system levels. Though this definition is not specific to caregiver or malpractice stress, it is applicable. It is an essential component of wellness and requires perpetual attention to self-care for successful maintenance.
Studies of physicians who have avoided burnout reveal remarkably consistent qualities, including finding gratification related to work, maintaining useful habits and practices, and developing attitudes that keep them resilient.3 Rather than adding activities to their full schedules, these physicians stayed resilient through mindfulness of various aspects of their daily lives. Interactions with colleagues—discussing cases, treatments, and outcomes (including errors)—proved vital. Professional development, encompassing activities such as continuing education, coaching, mentoring, and counseling, was recognized as an important self-directed resilience measure. Maintenance of relationships with family and friends, cultivation of leisure-time activities, and appreciation of the need for personal reflection time were traits often found in resilient physicians.
FOSTERING RESILIENCE
As part of the Mayo Clinic’s biannual survey of its physicians, Shanafelt et al4 studied relationships between qualities of physician leaders and burnout and satisfaction among the physicians they supervised. Many of the desirable leadership traits were related to building relationships through respectful communication, along with provision of opportunities for personal and professional development. The acknowledgment that resilient, healthy physicians are satisfied, productive, and able to provide safer and higher-quality patient care should lead to the establishment of physician wellness as a “dashboard metric.” This makes priorities clear by rewarding managers who foster self-care and resilience among their staff.
Likewise, at the healthcare system level, Beckman5 recognized that organizations can provide opportunities to promote resilience among caregivers. Organizational initiatives that set the stage for resilience include:
- Curricula to enhance communication with patients, coworkers, and family
- “Best practices” for efficient and effective patient care
- Self-care through health insurance incentives and educational sessions
- Accessible, affordable, and confidential behavioral health support
- Time for self-care activities during the workday
- Coaching and mentoring programs
- Narrative-and-reflection groups and mindfulness training.5
Through an atmosphere of support for resilience, organizations provide a place for physicians to maintain a sense of meaning and purpose in their work. For individuals facing malpractice action, this infrastructure can be used to weather the storm. As Mr. Giordano writes, staying engaged “may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.”1 We must pay attention to developing individual physicians, educating managers, and building systems so that caregivers can remain engaged and resilient. It may help those affected by malpractice stress, and perhaps as importantly, it may reduce the chance of future “disconnection” leading to recourse in the legal system.
- Giordano KC. It is not the critic’s voice that should count. Cleve Clin J Med 2016; 83:174–176.
- Coutu DL. How resilience works. Harv Bus Rev 2002; 80(5):46–55.
- Zwack J, Schweitzer J. If every fifth physician is affected by burnout, what about the other four? Acad Med 2013; 88:382–389.
- Shanafelt TD, Gorringe G, Menaker R, et al. Impact of organizational leadership on physician burnout and satisfaction. Mayo Clin Proc 2015; 90:432–440.
- Beckman H. The role of medical culture in the journey to resilience. Acad Med 2015; 90:710–712.
- Giordano KC. It is not the critic’s voice that should count. Cleve Clin J Med 2016; 83:174–176.
- Coutu DL. How resilience works. Harv Bus Rev 2002; 80(5):46–55.
- Zwack J, Schweitzer J. If every fifth physician is affected by burnout, what about the other four? Acad Med 2013; 88:382–389.
- Shanafelt TD, Gorringe G, Menaker R, et al. Impact of organizational leadership on physician burnout and satisfaction. Mayo Clin Proc 2015; 90:432–440.
- Beckman H. The role of medical culture in the journey to resilience. Acad Med 2015; 90:710–712.

It is not the critic’s voice that should count
During my 25 years as a defense attorney, I have seen the traumatic impact that the allegation of medical malpractice can have on healthcare providers. And I have seen many times that in the aftermath of a case it remains difficult, if not impossible, for the practitioner to return to the clinical setting unscarred by the process. Although vindication by the jury provides some solace, by itself it does not create healing. Instead, the critic’s voice continues to resonate long after the trial.
During a lawsuit, physicians and other providers are commonly confronted with incidental imperfections in the care they provided, errors in their documentation, or both. Consequently, a provider’s perception of events and ultimately the meaning derived from the experience is shaped less by the valid defenses and opinions of the supportive defense experts than by the inconsequential flaws and errors that can often be found in any medical record.
A RECENT CASE
Recently, I defended a hospital team consisting of a hospitalist, trauma surgeon, three residents, and a nurse. The case involved a 74-year-old man who was admitted to the hospital with pancreatitis of unknown cause. Six days after admission, he died of complications of acute respiratory distress syndrome. The team was accused of causing the patient’s death. Specifically, the plaintiff alleged that although the patient’s liver enzyme levels were improving, his condition was deteriorating, and he ultimately developed hemorrhagic pancreatitis. It was the plaintiff’s contention that proper ongoing evaluation, including computed tomographic imaging, would have led to treatment that would have avoided the worsening of pancreatitis, development of an ileus, and ultimately the insult to his bowel and lungs that they claim caused acute respiratory distress syndrome and death. The patient was survived by his wife and their three children. After his death, hospital representatives and the hospitalist met with her in an effort to explain the events that led to her husband’s death. Unfortunately, these discussions did not ameliorate her feelings of loss and anger. She filed a lawsuit, and 4 years later, the case went to trial.
During the trial, the plaintiff’s attorney highlighted errors in the electronic medical record. Entries had been cut and pasted, saving time, but without updating information that had changed in the interim. The inaccuracies included “assessment: worsening pancreatitis” on a day it was considered to have improved. Another entry contained “persistent fever” on a day when no fever was present. Other mistakes involved notes that contained care plans made after morning rounds that were not revised later in the day after changes in the patient’s condition necessitated a change in the plan. In fact, most references to medication dosing in the progress notes on the last 2 days did not match the medication dosing documented in the medication administration record.
In the end, the plaintiff’s counsel did not convince the jury that the healthcare team had been negligent, but unfortunately, she planted doubt in the minds of the caregivers themselves. Perhaps in part, these doubts were the result of having to defend a bad outcome in the face of criticism that was based solely in retrospect. But the providers’ doubts seemed mostly to emanate from the inadequacies in their documentation as they observed how every entry in a far-from-perfect medical record was scrutinized and then manipulated to challenge its textual integrity—and to portray the healthcare team as unengaged and substandard clinicians.
Despite the team’s high level of engagement and the quality of care they provided, any imperfection—whether a documentation error or a minor omission in some aspect of the care provided to this complex patient—became a source of self-doubt and self-criticism.
THE ELECTRONIC MEDICAL RECORD: A MIXED BLESSING
Documentation failures have long been used to “prove” that physicians are disconnected from the clinical situation. The electronic medical record has not proved to be a strong shield against malpractice allegations. In fact, because the electronic medical record absorbs more of the physician’s time and that of the care team’s members, efforts to save time through work-arounds and shortcuts have increased the risk of errors in entering information.
For instance, drop-down menus have led to wrong selections. Cutting and pasting has led to entries that contain data superseded by clinical events, thus creating contradictions within the record itself, and worse, with the physician’s own testimony pertaining to the basis of the clinical decision-making. And boilerplate language has created difficulties when the language does not completely fit the context or when inapplicable verbiage that fills itself in automatically goes unedited. An emergency department physician I represented at trial had to awkwardly explain that some of the data reported in his physical exam findings were inaccurate because of programmed language and should have been deleted; he had no explanation for his oversight.
But my experience has been that juries can forgive imperfections in documentation and even incidental aspects of care. They want to trust that the clinician was there for, and there with, the patient. This emphasis is what allowed us to defend the case involving the patient with pancreatitis. Clinical judgment means being engaged enough to choose what you pay attention to and to process the data you receive.
Unfortunately, the electronic medical record seems designed more for billing and for guarding against claims of fraud than for communication among clinicians or documenting clinically significant events. Many clinicians believe that redundancy and standardized phraseology have weakened the meaningful use of the medical record, as the clinical information is now of questionable reliability or value or is simply hard to find. Consequently, the electronic medical record has become less effective as a communication tool for providing continuity of care.
More importantly, the electronic medical record too often places the physician in front of a computer, so that the computer becomes the focus, not the patient. Studies suggest that the way the electronic medical record is currently used in the examination room affects the quality of physician-patient communication as well as the physician’s cognitive processing of information. Unless the physician is alert and attuned, the electronic medical record can be a barrier to connection. This not only creates the potential for mistakes, but it can also cause patients to question the quality of care they are getting and to distrust the level of the provider’s engagement. In this context, the likelihood that the patient retains an attorney increases when a bad outcome occurs, avoidable or not.
WHAT PATIENTS WANT FROM PHYSICIANS
When I first began seeing my own primary care physician, her office was 5 minutes from my home. Then she relocated to a practice 15 minutes away. And then, because of office consolidation and acquisition, her office was relocated 40 minutes away.
So why do I still go to her? Her training is not better than that of most internists, and my medical history is not so complex that I require more care than most 55-year-old men. I am only speculating, but I would guess that she is not the most financially productive physician in her group. I know that her transition to the electronic medical record has been difficult. Recently, I asked her about it. Except in some situations, she does not type while taking a history, and she stays totally away from the computer while in the examination room with me. She sits a couple of feet from me, and it feels like the days before the electronic medical record. She is clearly more comfortable listening and taking notes first and worrying about the electronic record later. I imagine she stays later to do her notes than most of the other physicians, or she finishes them at home.
The reason I continue to see her as my primary care physician is that she remains totally engaged during my office visit. What tells me that is not just her avoidance of the computer or her body language, but the depth of questions she asks. My responses often prompt her to look back at an earlier office note, and she will then ask follow-up questions to confirm what she had previously recorded. Her examination is thorough, with testing to confirm and retesting to be sure. Doing this may mean that she has difficulty meeting financial or administrative benchmarks established by her practice. I don’t know. But I have no doubt that the likelihood of her missing something in her clinical care is small, and what I suspect is even smaller is the risk that one of her patients would bring a lawsuit against her, given the time she takes to listen and remain connected throughout the office visit.
STAYING CONNECTED, IN SPITE OF EVERYTHING
My point is not to suggest that everyone must conform to the same practice philosophy, particularly with the economic pressures in the medical field. What I am suggesting is that it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team towards disconnection. Quality healthcare means making every effort to remain engaged at all times with your patient’s care, which will reduce the likelihood of a bad outcome and may preserve the physician-patient relationship even when a bad outcome occurs.
In the end, perhaps it is not possible to avoid being named as a defendant in a malpractice case, just as it is not possible to avoid all bad medical outcomes despite exceptional care. In law, as in medicine, there are always factors beyond your control. My aspiration is to find a pathway to get providers through the system unbroken—also not an easy task. But one thing I know is true: the more you can stay engaged in the care you provide and in your documentation, the more you will preclude a plaintiff’s attorney from exploiting the effects of the forces within the system that drive providers toward disconnection. As long as you stay engaged and supported by the knowledge that the care provided was appropriate, it is my hope that the voice of the critic will not count as much in the aftermath of a malpractice case. But more importantly, it may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.
During my 25 years as a defense attorney, I have seen the traumatic impact that the allegation of medical malpractice can have on healthcare providers. And I have seen many times that in the aftermath of a case it remains difficult, if not impossible, for the practitioner to return to the clinical setting unscarred by the process. Although vindication by the jury provides some solace, by itself it does not create healing. Instead, the critic’s voice continues to resonate long after the trial.
During a lawsuit, physicians and other providers are commonly confronted with incidental imperfections in the care they provided, errors in their documentation, or both. Consequently, a provider’s perception of events and ultimately the meaning derived from the experience is shaped less by the valid defenses and opinions of the supportive defense experts than by the inconsequential flaws and errors that can often be found in any medical record.
A RECENT CASE
Recently, I defended a hospital team consisting of a hospitalist, trauma surgeon, three residents, and a nurse. The case involved a 74-year-old man who was admitted to the hospital with pancreatitis of unknown cause. Six days after admission, he died of complications of acute respiratory distress syndrome. The team was accused of causing the patient’s death. Specifically, the plaintiff alleged that although the patient’s liver enzyme levels were improving, his condition was deteriorating, and he ultimately developed hemorrhagic pancreatitis. It was the plaintiff’s contention that proper ongoing evaluation, including computed tomographic imaging, would have led to treatment that would have avoided the worsening of pancreatitis, development of an ileus, and ultimately the insult to his bowel and lungs that they claim caused acute respiratory distress syndrome and death. The patient was survived by his wife and their three children. After his death, hospital representatives and the hospitalist met with her in an effort to explain the events that led to her husband’s death. Unfortunately, these discussions did not ameliorate her feelings of loss and anger. She filed a lawsuit, and 4 years later, the case went to trial.
During the trial, the plaintiff’s attorney highlighted errors in the electronic medical record. Entries had been cut and pasted, saving time, but without updating information that had changed in the interim. The inaccuracies included “assessment: worsening pancreatitis” on a day it was considered to have improved. Another entry contained “persistent fever” on a day when no fever was present. Other mistakes involved notes that contained care plans made after morning rounds that were not revised later in the day after changes in the patient’s condition necessitated a change in the plan. In fact, most references to medication dosing in the progress notes on the last 2 days did not match the medication dosing documented in the medication administration record.
In the end, the plaintiff’s counsel did not convince the jury that the healthcare team had been negligent, but unfortunately, she planted doubt in the minds of the caregivers themselves. Perhaps in part, these doubts were the result of having to defend a bad outcome in the face of criticism that was based solely in retrospect. But the providers’ doubts seemed mostly to emanate from the inadequacies in their documentation as they observed how every entry in a far-from-perfect medical record was scrutinized and then manipulated to challenge its textual integrity—and to portray the healthcare team as unengaged and substandard clinicians.
Despite the team’s high level of engagement and the quality of care they provided, any imperfection—whether a documentation error or a minor omission in some aspect of the care provided to this complex patient—became a source of self-doubt and self-criticism.
THE ELECTRONIC MEDICAL RECORD: A MIXED BLESSING
Documentation failures have long been used to “prove” that physicians are disconnected from the clinical situation. The electronic medical record has not proved to be a strong shield against malpractice allegations. In fact, because the electronic medical record absorbs more of the physician’s time and that of the care team’s members, efforts to save time through work-arounds and shortcuts have increased the risk of errors in entering information.
For instance, drop-down menus have led to wrong selections. Cutting and pasting has led to entries that contain data superseded by clinical events, thus creating contradictions within the record itself, and worse, with the physician’s own testimony pertaining to the basis of the clinical decision-making. And boilerplate language has created difficulties when the language does not completely fit the context or when inapplicable verbiage that fills itself in automatically goes unedited. An emergency department physician I represented at trial had to awkwardly explain that some of the data reported in his physical exam findings were inaccurate because of programmed language and should have been deleted; he had no explanation for his oversight.
But my experience has been that juries can forgive imperfections in documentation and even incidental aspects of care. They want to trust that the clinician was there for, and there with, the patient. This emphasis is what allowed us to defend the case involving the patient with pancreatitis. Clinical judgment means being engaged enough to choose what you pay attention to and to process the data you receive.
Unfortunately, the electronic medical record seems designed more for billing and for guarding against claims of fraud than for communication among clinicians or documenting clinically significant events. Many clinicians believe that redundancy and standardized phraseology have weakened the meaningful use of the medical record, as the clinical information is now of questionable reliability or value or is simply hard to find. Consequently, the electronic medical record has become less effective as a communication tool for providing continuity of care.
More importantly, the electronic medical record too often places the physician in front of a computer, so that the computer becomes the focus, not the patient. Studies suggest that the way the electronic medical record is currently used in the examination room affects the quality of physician-patient communication as well as the physician’s cognitive processing of information. Unless the physician is alert and attuned, the electronic medical record can be a barrier to connection. This not only creates the potential for mistakes, but it can also cause patients to question the quality of care they are getting and to distrust the level of the provider’s engagement. In this context, the likelihood that the patient retains an attorney increases when a bad outcome occurs, avoidable or not.
WHAT PATIENTS WANT FROM PHYSICIANS
When I first began seeing my own primary care physician, her office was 5 minutes from my home. Then she relocated to a practice 15 minutes away. And then, because of office consolidation and acquisition, her office was relocated 40 minutes away.
So why do I still go to her? Her training is not better than that of most internists, and my medical history is not so complex that I require more care than most 55-year-old men. I am only speculating, but I would guess that she is not the most financially productive physician in her group. I know that her transition to the electronic medical record has been difficult. Recently, I asked her about it. Except in some situations, she does not type while taking a history, and she stays totally away from the computer while in the examination room with me. She sits a couple of feet from me, and it feels like the days before the electronic medical record. She is clearly more comfortable listening and taking notes first and worrying about the electronic record later. I imagine she stays later to do her notes than most of the other physicians, or she finishes them at home.
The reason I continue to see her as my primary care physician is that she remains totally engaged during my office visit. What tells me that is not just her avoidance of the computer or her body language, but the depth of questions she asks. My responses often prompt her to look back at an earlier office note, and she will then ask follow-up questions to confirm what she had previously recorded. Her examination is thorough, with testing to confirm and retesting to be sure. Doing this may mean that she has difficulty meeting financial or administrative benchmarks established by her practice. I don’t know. But I have no doubt that the likelihood of her missing something in her clinical care is small, and what I suspect is even smaller is the risk that one of her patients would bring a lawsuit against her, given the time she takes to listen and remain connected throughout the office visit.
STAYING CONNECTED, IN SPITE OF EVERYTHING
My point is not to suggest that everyone must conform to the same practice philosophy, particularly with the economic pressures in the medical field. What I am suggesting is that it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team towards disconnection. Quality healthcare means making every effort to remain engaged at all times with your patient’s care, which will reduce the likelihood of a bad outcome and may preserve the physician-patient relationship even when a bad outcome occurs.
In the end, perhaps it is not possible to avoid being named as a defendant in a malpractice case, just as it is not possible to avoid all bad medical outcomes despite exceptional care. In law, as in medicine, there are always factors beyond your control. My aspiration is to find a pathway to get providers through the system unbroken—also not an easy task. But one thing I know is true: the more you can stay engaged in the care you provide and in your documentation, the more you will preclude a plaintiff’s attorney from exploiting the effects of the forces within the system that drive providers toward disconnection. As long as you stay engaged and supported by the knowledge that the care provided was appropriate, it is my hope that the voice of the critic will not count as much in the aftermath of a malpractice case. But more importantly, it may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.
During my 25 years as a defense attorney, I have seen the traumatic impact that the allegation of medical malpractice can have on healthcare providers. And I have seen many times that in the aftermath of a case it remains difficult, if not impossible, for the practitioner to return to the clinical setting unscarred by the process. Although vindication by the jury provides some solace, by itself it does not create healing. Instead, the critic’s voice continues to resonate long after the trial.
During a lawsuit, physicians and other providers are commonly confronted with incidental imperfections in the care they provided, errors in their documentation, or both. Consequently, a provider’s perception of events and ultimately the meaning derived from the experience is shaped less by the valid defenses and opinions of the supportive defense experts than by the inconsequential flaws and errors that can often be found in any medical record.
A RECENT CASE
Recently, I defended a hospital team consisting of a hospitalist, trauma surgeon, three residents, and a nurse. The case involved a 74-year-old man who was admitted to the hospital with pancreatitis of unknown cause. Six days after admission, he died of complications of acute respiratory distress syndrome. The team was accused of causing the patient’s death. Specifically, the plaintiff alleged that although the patient’s liver enzyme levels were improving, his condition was deteriorating, and he ultimately developed hemorrhagic pancreatitis. It was the plaintiff’s contention that proper ongoing evaluation, including computed tomographic imaging, would have led to treatment that would have avoided the worsening of pancreatitis, development of an ileus, and ultimately the insult to his bowel and lungs that they claim caused acute respiratory distress syndrome and death. The patient was survived by his wife and their three children. After his death, hospital representatives and the hospitalist met with her in an effort to explain the events that led to her husband’s death. Unfortunately, these discussions did not ameliorate her feelings of loss and anger. She filed a lawsuit, and 4 years later, the case went to trial.
During the trial, the plaintiff’s attorney highlighted errors in the electronic medical record. Entries had been cut and pasted, saving time, but without updating information that had changed in the interim. The inaccuracies included “assessment: worsening pancreatitis” on a day it was considered to have improved. Another entry contained “persistent fever” on a day when no fever was present. Other mistakes involved notes that contained care plans made after morning rounds that were not revised later in the day after changes in the patient’s condition necessitated a change in the plan. In fact, most references to medication dosing in the progress notes on the last 2 days did not match the medication dosing documented in the medication administration record.
In the end, the plaintiff’s counsel did not convince the jury that the healthcare team had been negligent, but unfortunately, she planted doubt in the minds of the caregivers themselves. Perhaps in part, these doubts were the result of having to defend a bad outcome in the face of criticism that was based solely in retrospect. But the providers’ doubts seemed mostly to emanate from the inadequacies in their documentation as they observed how every entry in a far-from-perfect medical record was scrutinized and then manipulated to challenge its textual integrity—and to portray the healthcare team as unengaged and substandard clinicians.
Despite the team’s high level of engagement and the quality of care they provided, any imperfection—whether a documentation error or a minor omission in some aspect of the care provided to this complex patient—became a source of self-doubt and self-criticism.
THE ELECTRONIC MEDICAL RECORD: A MIXED BLESSING
Documentation failures have long been used to “prove” that physicians are disconnected from the clinical situation. The electronic medical record has not proved to be a strong shield against malpractice allegations. In fact, because the electronic medical record absorbs more of the physician’s time and that of the care team’s members, efforts to save time through work-arounds and shortcuts have increased the risk of errors in entering information.
For instance, drop-down menus have led to wrong selections. Cutting and pasting has led to entries that contain data superseded by clinical events, thus creating contradictions within the record itself, and worse, with the physician’s own testimony pertaining to the basis of the clinical decision-making. And boilerplate language has created difficulties when the language does not completely fit the context or when inapplicable verbiage that fills itself in automatically goes unedited. An emergency department physician I represented at trial had to awkwardly explain that some of the data reported in his physical exam findings were inaccurate because of programmed language and should have been deleted; he had no explanation for his oversight.
But my experience has been that juries can forgive imperfections in documentation and even incidental aspects of care. They want to trust that the clinician was there for, and there with, the patient. This emphasis is what allowed us to defend the case involving the patient with pancreatitis. Clinical judgment means being engaged enough to choose what you pay attention to and to process the data you receive.
Unfortunately, the electronic medical record seems designed more for billing and for guarding against claims of fraud than for communication among clinicians or documenting clinically significant events. Many clinicians believe that redundancy and standardized phraseology have weakened the meaningful use of the medical record, as the clinical information is now of questionable reliability or value or is simply hard to find. Consequently, the electronic medical record has become less effective as a communication tool for providing continuity of care.
More importantly, the electronic medical record too often places the physician in front of a computer, so that the computer becomes the focus, not the patient. Studies suggest that the way the electronic medical record is currently used in the examination room affects the quality of physician-patient communication as well as the physician’s cognitive processing of information. Unless the physician is alert and attuned, the electronic medical record can be a barrier to connection. This not only creates the potential for mistakes, but it can also cause patients to question the quality of care they are getting and to distrust the level of the provider’s engagement. In this context, the likelihood that the patient retains an attorney increases when a bad outcome occurs, avoidable or not.
WHAT PATIENTS WANT FROM PHYSICIANS
When I first began seeing my own primary care physician, her office was 5 minutes from my home. Then she relocated to a practice 15 minutes away. And then, because of office consolidation and acquisition, her office was relocated 40 minutes away.
So why do I still go to her? Her training is not better than that of most internists, and my medical history is not so complex that I require more care than most 55-year-old men. I am only speculating, but I would guess that she is not the most financially productive physician in her group. I know that her transition to the electronic medical record has been difficult. Recently, I asked her about it. Except in some situations, she does not type while taking a history, and she stays totally away from the computer while in the examination room with me. She sits a couple of feet from me, and it feels like the days before the electronic medical record. She is clearly more comfortable listening and taking notes first and worrying about the electronic record later. I imagine she stays later to do her notes than most of the other physicians, or she finishes them at home.
The reason I continue to see her as my primary care physician is that she remains totally engaged during my office visit. What tells me that is not just her avoidance of the computer or her body language, but the depth of questions she asks. My responses often prompt her to look back at an earlier office note, and she will then ask follow-up questions to confirm what she had previously recorded. Her examination is thorough, with testing to confirm and retesting to be sure. Doing this may mean that she has difficulty meeting financial or administrative benchmarks established by her practice. I don’t know. But I have no doubt that the likelihood of her missing something in her clinical care is small, and what I suspect is even smaller is the risk that one of her patients would bring a lawsuit against her, given the time she takes to listen and remain connected throughout the office visit.
STAYING CONNECTED, IN SPITE OF EVERYTHING
My point is not to suggest that everyone must conform to the same practice philosophy, particularly with the economic pressures in the medical field. What I am suggesting is that it is not easy to stay connected in a healthcare system in which the system’s structure is driving physicians and other members of the healthcare team towards disconnection. Quality healthcare means making every effort to remain engaged at all times with your patient’s care, which will reduce the likelihood of a bad outcome and may preserve the physician-patient relationship even when a bad outcome occurs.
In the end, perhaps it is not possible to avoid being named as a defendant in a malpractice case, just as it is not possible to avoid all bad medical outcomes despite exceptional care. In law, as in medicine, there are always factors beyond your control. My aspiration is to find a pathway to get providers through the system unbroken—also not an easy task. But one thing I know is true: the more you can stay engaged in the care you provide and in your documentation, the more you will preclude a plaintiff’s attorney from exploiting the effects of the forces within the system that drive providers toward disconnection. As long as you stay engaged and supported by the knowledge that the care provided was appropriate, it is my hope that the voice of the critic will not count as much in the aftermath of a malpractice case. But more importantly, it may allow you to draw meaning and reconciliation from the fact that throughout the patient’s illness, undeterred by the complexities of today’s healthcare system, you remained the attentive and compassionate healer you hoped to be when you first became a healthcare professional.

Fungal folliculitis masquerading as acute exanthematous pustulosis
THE DIFFERENTIAL DIAGNOSIS
The appearance of sterile pustules after starting a new antibiotic raised suspicion for acute localized exanthematous pustulosis, a variant of acute generalized exanthematous pustulosis. It is a serious but uncommon adverse drug reaction, with a frequency of one to five cases per million per year. The eruption of erythematous plaques studded with sterile pustules classically appears 1 to 5 days after starting a drug.1 Piperacillin-tazobactam has been infrequently reported in association with acute exanthematous pustulosis, but antibiotics in general are among the most commonly reported culprits.2–4
Clues to the correct diagnosis
Although our concern for acute localized exanthematous pustulosis was warranted, the morphology and distribution of this patient’s exanthem also raised suspicion of fungal folliculitis, which is more common.
Malassezia folliculitis appears as a monomorphic papular and pustular eruption on the chest, back, and face,5 as in our patient. Differentiating fungal folliculitis from pustulosis is important, as each condition is treated differently: Malassezia folliculitis is treated with antifungals,5 and acute localized exanthematous pustulosis is managed with cessation of the offending drug, supportive care, and systemic or topical steroids.4
Take-home point
Our experience with this patient was a reminder to consider fungal folliculitis in the differential diagnosis of a pustular eruption, so as to allow appropriate management and to avert discontinuation of potentially life-saving medications.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol 2012; 53:87–92.
- Talati S, Lala M, Kapupara H, Thet Z. Acute generalized exanthematous pustulosis: a rare clinical entity with use of piperacillin/tazobactam. Am J Ther 2009; 16:591–592.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR). Br J Dermatol 2007; 157:989–996.
- Huilaja L, Kallioinen M, Soronen M, Riekki R, Tasanen K. Acute localized exanthematous pustulosis on inguinal area secondary to piperacillin/tazobactam. Acta Derm Venereol 2014; 94:106–107.
- Rubenstein RM, Malerich SA. Malassezia (pityrosporum) folliculitis. J Clin Aesthet Dermatol 2014; 73:37–41.
THE DIFFERENTIAL DIAGNOSIS
The appearance of sterile pustules after starting a new antibiotic raised suspicion for acute localized exanthematous pustulosis, a variant of acute generalized exanthematous pustulosis. It is a serious but uncommon adverse drug reaction, with a frequency of one to five cases per million per year. The eruption of erythematous plaques studded with sterile pustules classically appears 1 to 5 days after starting a drug.1 Piperacillin-tazobactam has been infrequently reported in association with acute exanthematous pustulosis, but antibiotics in general are among the most commonly reported culprits.2–4
Clues to the correct diagnosis
Although our concern for acute localized exanthematous pustulosis was warranted, the morphology and distribution of this patient’s exanthem also raised suspicion of fungal folliculitis, which is more common.
Malassezia folliculitis appears as a monomorphic papular and pustular eruption on the chest, back, and face,5 as in our patient. Differentiating fungal folliculitis from pustulosis is important, as each condition is treated differently: Malassezia folliculitis is treated with antifungals,5 and acute localized exanthematous pustulosis is managed with cessation of the offending drug, supportive care, and systemic or topical steroids.4
Take-home point
Our experience with this patient was a reminder to consider fungal folliculitis in the differential diagnosis of a pustular eruption, so as to allow appropriate management and to avert discontinuation of potentially life-saving medications.
THE DIFFERENTIAL DIAGNOSIS
The appearance of sterile pustules after starting a new antibiotic raised suspicion for acute localized exanthematous pustulosis, a variant of acute generalized exanthematous pustulosis. It is a serious but uncommon adverse drug reaction, with a frequency of one to five cases per million per year. The eruption of erythematous plaques studded with sterile pustules classically appears 1 to 5 days after starting a drug.1 Piperacillin-tazobactam has been infrequently reported in association with acute exanthematous pustulosis, but antibiotics in general are among the most commonly reported culprits.2–4
Clues to the correct diagnosis
Although our concern for acute localized exanthematous pustulosis was warranted, the morphology and distribution of this patient’s exanthem also raised suspicion of fungal folliculitis, which is more common.
Malassezia folliculitis appears as a monomorphic papular and pustular eruption on the chest, back, and face,5 as in our patient. Differentiating fungal folliculitis from pustulosis is important, as each condition is treated differently: Malassezia folliculitis is treated with antifungals,5 and acute localized exanthematous pustulosis is managed with cessation of the offending drug, supportive care, and systemic or topical steroids.4
Take-home point
Our experience with this patient was a reminder to consider fungal folliculitis in the differential diagnosis of a pustular eruption, so as to allow appropriate management and to avert discontinuation of potentially life-saving medications.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol 2012; 53:87–92.
- Talati S, Lala M, Kapupara H, Thet Z. Acute generalized exanthematous pustulosis: a rare clinical entity with use of piperacillin/tazobactam. Am J Ther 2009; 16:591–592.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR). Br J Dermatol 2007; 157:989–996.
- Huilaja L, Kallioinen M, Soronen M, Riekki R, Tasanen K. Acute localized exanthematous pustulosis on inguinal area secondary to piperacillin/tazobactam. Acta Derm Venereol 2014; 94:106–107.
- Rubenstein RM, Malerich SA. Malassezia (pityrosporum) folliculitis. J Clin Aesthet Dermatol 2014; 73:37–41.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol 2012; 53:87–92.
- Talati S, Lala M, Kapupara H, Thet Z. Acute generalized exanthematous pustulosis: a rare clinical entity with use of piperacillin/tazobactam. Am J Ther 2009; 16:591–592.
- Sidoroff A, Dunant A, Viboud C, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP)-results of a multinational case-control study (EuroSCAR). Br J Dermatol 2007; 157:989–996.
- Huilaja L, Kallioinen M, Soronen M, Riekki R, Tasanen K. Acute localized exanthematous pustulosis on inguinal area secondary to piperacillin/tazobactam. Acta Derm Venereol 2014; 94:106–107.
- Rubenstein RM, Malerich SA. Malassezia (pityrosporum) folliculitis. J Clin Aesthet Dermatol 2014; 73:37–41.
FDA calls for more safety data, patient counseling for Essure
The Food and Drug Administration has ordered Bayer to conduct a 2,000-patient postmarketing study of the Essure implantable birth control device, to explore the risks it may pose to some women and examine how it is being employed in clinical practice.
Bayer, which manufactures the device, will also be required to add a boxed warning to the label, describing potential adverse events, and a “Patient Decision Checklist” to help guide preimplantation discussions, under draft guidance issued by the FDA on Feb. 29.
The 3-year observational study will compare data on complications, pregnancy, and pregnancy loss in Essure patients and in women who undergo bilateral tubal ligation, according to Dr. William Maisel, chief scientist at the FDA’s Center for Devices and Radiological Health.
“This will be a large observational study,” he said during an FDA press briefing on Feb. 29. “The specific questions will relate to overall complications rates; perforation, migration, and expulsion; chronic pelvic pain; abnormal uterine bleeding; allergy and hypersensitivity,” and obstetric outcomes.
Because FDA is requiring such a large patient cohort with long-term follow-up, final study results will be years away. Therefore, Bayer will be required to release data intermittently to keep the public well informed as research progresses, Dr. Maisel said.
The requested study will also examine why some patients don’t have a confirmation test to ensure that Essure has been properly placed 3 months after insertion – a key area that seems related to a number of reported adverse events, including pregnancy and device migration.
Despite the push for additional data, the FDA still believes that Essure is an appropriate and safe permanent option for the majority of women who want permanent birth control, Dr. Maisel said.
“It’s the only nonincisional form of permanent birth control. It requires no general anesthetic to insert, and most women go back to work in a day,” he said. “It is highly effective at preventing pregnancy, and it contains no drugs or hormones. Essure should remain an option for women seeking permanent birth control who are informed of its risks.”
Bayer officials said they will continue to work closely with the FDA to support the safe and effective use of the device.
“Patient safety and appropriate use of Essure are our greatest priorities,” Dr. Dario Mirski, senior vice president and head of medical affairs Americas at Bayer said in a statement. “A woman’s decision to choose a birth control method is a very important and personal one, and Bayer is committed to providing physicians with resources, tools, and information to help them counsel women about Essure.”
The boxed warning announced by the FDA will outline the adverse events that may be associated with Essure, including those that might occur during insertion and removal. The “Patient Decision Checklist” will be designed to help doctors stress the importance of the 3-month confirmation test to determine that it is correctly placed and that sufficient scar tissue has formed to prevent pregnancy. Both patient and physician will have to sign off on the checklist before the device is employed.
The FDA is seeking public comments on the proposed language for the warnings. The docket will be open for 60 days.
“The actions we are taking today will encourage important conversations between women and their doctors to help patients make more informed decisions about whether or not Essure is right for them,” said Dr. Maisel. “They also reflect our recognition that more rigorous research is needed to better understand if certain women are at heightened risk of complications.”
The draft guidance solidifies discussions that occurred last fall during a meeting of the FDA Obstetrics and Gynecology Devices Panel. During that meeting, the 19-member panel heard testimony from dozens of women who developed pain and other serious problems, including autoimmune diseases, after receiving Essure.
Since the device was approved in 2002, the FDA has received more than 5,000 complaints of such adverse reactions. These include 631 pregnancies and 294 pregnancy losses.
Between the September meeting and the FDA’s draft guidance announcement, the agency has received even more information from patients. A 22,000-person support group called Essure Problems collected and submitted a large amount of personal and clinical data. The package was submitted to the FDA on Feb. 22 and includes surgical notes and photos; letters from doctors and surgeons; pregnancy records; data on fetal death, ectopic pregnancies and miscarriage; and a series of electromicrographic images purporting to show defects of the coils’ metal ribbons.
The administrators of the Essure Problems groups blasted the FDA for requiring studies, rather than removing the device from the market. “These studies could take several years, and leaving the device on the market will only put more women’s lives at risk,” they wrote.
Rep. Mike Fitzpatrick (R-Pa.), who has been critical of Essure, said that the FDA’s actions are inadequate and said he will push for congressional action, including blocking government agencies from purchasing the device and revoking the FDA’s approval of Essure.
The Food and Drug Administration has ordered Bayer to conduct a 2,000-patient postmarketing study of the Essure implantable birth control device, to explore the risks it may pose to some women and examine how it is being employed in clinical practice.
Bayer, which manufactures the device, will also be required to add a boxed warning to the label, describing potential adverse events, and a “Patient Decision Checklist” to help guide preimplantation discussions, under draft guidance issued by the FDA on Feb. 29.
The 3-year observational study will compare data on complications, pregnancy, and pregnancy loss in Essure patients and in women who undergo bilateral tubal ligation, according to Dr. William Maisel, chief scientist at the FDA’s Center for Devices and Radiological Health.
“This will be a large observational study,” he said during an FDA press briefing on Feb. 29. “The specific questions will relate to overall complications rates; perforation, migration, and expulsion; chronic pelvic pain; abnormal uterine bleeding; allergy and hypersensitivity,” and obstetric outcomes.
Because FDA is requiring such a large patient cohort with long-term follow-up, final study results will be years away. Therefore, Bayer will be required to release data intermittently to keep the public well informed as research progresses, Dr. Maisel said.
The requested study will also examine why some patients don’t have a confirmation test to ensure that Essure has been properly placed 3 months after insertion – a key area that seems related to a number of reported adverse events, including pregnancy and device migration.
Despite the push for additional data, the FDA still believes that Essure is an appropriate and safe permanent option for the majority of women who want permanent birth control, Dr. Maisel said.
“It’s the only nonincisional form of permanent birth control. It requires no general anesthetic to insert, and most women go back to work in a day,” he said. “It is highly effective at preventing pregnancy, and it contains no drugs or hormones. Essure should remain an option for women seeking permanent birth control who are informed of its risks.”
Bayer officials said they will continue to work closely with the FDA to support the safe and effective use of the device.
“Patient safety and appropriate use of Essure are our greatest priorities,” Dr. Dario Mirski, senior vice president and head of medical affairs Americas at Bayer said in a statement. “A woman’s decision to choose a birth control method is a very important and personal one, and Bayer is committed to providing physicians with resources, tools, and information to help them counsel women about Essure.”
The boxed warning announced by the FDA will outline the adverse events that may be associated with Essure, including those that might occur during insertion and removal. The “Patient Decision Checklist” will be designed to help doctors stress the importance of the 3-month confirmation test to determine that it is correctly placed and that sufficient scar tissue has formed to prevent pregnancy. Both patient and physician will have to sign off on the checklist before the device is employed.
The FDA is seeking public comments on the proposed language for the warnings. The docket will be open for 60 days.
“The actions we are taking today will encourage important conversations between women and their doctors to help patients make more informed decisions about whether or not Essure is right for them,” said Dr. Maisel. “They also reflect our recognition that more rigorous research is needed to better understand if certain women are at heightened risk of complications.”
The draft guidance solidifies discussions that occurred last fall during a meeting of the FDA Obstetrics and Gynecology Devices Panel. During that meeting, the 19-member panel heard testimony from dozens of women who developed pain and other serious problems, including autoimmune diseases, after receiving Essure.
Since the device was approved in 2002, the FDA has received more than 5,000 complaints of such adverse reactions. These include 631 pregnancies and 294 pregnancy losses.
Between the September meeting and the FDA’s draft guidance announcement, the agency has received even more information from patients. A 22,000-person support group called Essure Problems collected and submitted a large amount of personal and clinical data. The package was submitted to the FDA on Feb. 22 and includes surgical notes and photos; letters from doctors and surgeons; pregnancy records; data on fetal death, ectopic pregnancies and miscarriage; and a series of electromicrographic images purporting to show defects of the coils’ metal ribbons.
The administrators of the Essure Problems groups blasted the FDA for requiring studies, rather than removing the device from the market. “These studies could take several years, and leaving the device on the market will only put more women’s lives at risk,” they wrote.
Rep. Mike Fitzpatrick (R-Pa.), who has been critical of Essure, said that the FDA’s actions are inadequate and said he will push for congressional action, including blocking government agencies from purchasing the device and revoking the FDA’s approval of Essure.
The Food and Drug Administration has ordered Bayer to conduct a 2,000-patient postmarketing study of the Essure implantable birth control device, to explore the risks it may pose to some women and examine how it is being employed in clinical practice.
Bayer, which manufactures the device, will also be required to add a boxed warning to the label, describing potential adverse events, and a “Patient Decision Checklist” to help guide preimplantation discussions, under draft guidance issued by the FDA on Feb. 29.
The 3-year observational study will compare data on complications, pregnancy, and pregnancy loss in Essure patients and in women who undergo bilateral tubal ligation, according to Dr. William Maisel, chief scientist at the FDA’s Center for Devices and Radiological Health.
“This will be a large observational study,” he said during an FDA press briefing on Feb. 29. “The specific questions will relate to overall complications rates; perforation, migration, and expulsion; chronic pelvic pain; abnormal uterine bleeding; allergy and hypersensitivity,” and obstetric outcomes.
Because FDA is requiring such a large patient cohort with long-term follow-up, final study results will be years away. Therefore, Bayer will be required to release data intermittently to keep the public well informed as research progresses, Dr. Maisel said.
The requested study will also examine why some patients don’t have a confirmation test to ensure that Essure has been properly placed 3 months after insertion – a key area that seems related to a number of reported adverse events, including pregnancy and device migration.
Despite the push for additional data, the FDA still believes that Essure is an appropriate and safe permanent option for the majority of women who want permanent birth control, Dr. Maisel said.
“It’s the only nonincisional form of permanent birth control. It requires no general anesthetic to insert, and most women go back to work in a day,” he said. “It is highly effective at preventing pregnancy, and it contains no drugs or hormones. Essure should remain an option for women seeking permanent birth control who are informed of its risks.”
Bayer officials said they will continue to work closely with the FDA to support the safe and effective use of the device.
“Patient safety and appropriate use of Essure are our greatest priorities,” Dr. Dario Mirski, senior vice president and head of medical affairs Americas at Bayer said in a statement. “A woman’s decision to choose a birth control method is a very important and personal one, and Bayer is committed to providing physicians with resources, tools, and information to help them counsel women about Essure.”
The boxed warning announced by the FDA will outline the adverse events that may be associated with Essure, including those that might occur during insertion and removal. The “Patient Decision Checklist” will be designed to help doctors stress the importance of the 3-month confirmation test to determine that it is correctly placed and that sufficient scar tissue has formed to prevent pregnancy. Both patient and physician will have to sign off on the checklist before the device is employed.
The FDA is seeking public comments on the proposed language for the warnings. The docket will be open for 60 days.
“The actions we are taking today will encourage important conversations between women and their doctors to help patients make more informed decisions about whether or not Essure is right for them,” said Dr. Maisel. “They also reflect our recognition that more rigorous research is needed to better understand if certain women are at heightened risk of complications.”
The draft guidance solidifies discussions that occurred last fall during a meeting of the FDA Obstetrics and Gynecology Devices Panel. During that meeting, the 19-member panel heard testimony from dozens of women who developed pain and other serious problems, including autoimmune diseases, after receiving Essure.
Since the device was approved in 2002, the FDA has received more than 5,000 complaints of such adverse reactions. These include 631 pregnancies and 294 pregnancy losses.
Between the September meeting and the FDA’s draft guidance announcement, the agency has received even more information from patients. A 22,000-person support group called Essure Problems collected and submitted a large amount of personal and clinical data. The package was submitted to the FDA on Feb. 22 and includes surgical notes and photos; letters from doctors and surgeons; pregnancy records; data on fetal death, ectopic pregnancies and miscarriage; and a series of electromicrographic images purporting to show defects of the coils’ metal ribbons.
The administrators of the Essure Problems groups blasted the FDA for requiring studies, rather than removing the device from the market. “These studies could take several years, and leaving the device on the market will only put more women’s lives at risk,” they wrote.
Rep. Mike Fitzpatrick (R-Pa.), who has been critical of Essure, said that the FDA’s actions are inadequate and said he will push for congressional action, including blocking government agencies from purchasing the device and revoking the FDA’s approval of Essure.
Stronger evidence links Zika to Guillain-Barré syndrome
Serological evidence from French Polynesia links an outbreak of Zika virus to a spike in cases of Guillain-Barré syndrome seen there in 2013-2014.
The research, published online Feb. 29 in The Lancet, is the first to use a case-control design to demonstrate that Zika, a mosquito-borne flavivirus, is associated with Guillain-Barré syndrome (Lancet. 2016 Feb 29. doi: 10.1016/S0140-6736(16)00562-6).
Guillain-Barré syndrome (GBS) is an immune-mediated flaccid paralysis that can follow viral or bacterial infections. Most patients with GBS recover with intensive care in hospitals, although the syndrome can be permanently debilitating or, in rare cases, fatal.

As a large outbreak of Zika continues in Central and South America, hospitals should be prepared for excess GBS cases, the authors of the study say, and assure adequate intensive-care capacity to treat them. Based on the 66% attack rate of Zika during the French Polynesia outbreak, investigators estimated the incidence of GBS at 0.24 per 1,000 Zika infections, but noted that it could be different in the current outbreak.
Dr. Van-Mai Cao-Lormeau of the Unit of Emerging Infectious Diseases at Institut Louis Malardé in Papeete, French Polynesia, alongside colleagues in France and French Polynesia, used a case-control design to compare serological samples from 42 patients (74% male) diagnosed at a Tahiti hospital with GBS with samples from age-and sex-matched patients who presented at the same hospital, also during the time of the outbreak, with a nonfebrile illness (n = 98) or with acute Zika disease without neurological symptoms (n = 70).
The investigators found that all but one patient with GBS had Zika virus antibodies, and all of them had neutralizing antibodies to Zika virus. By comparison, only 56% (n = 54) of the control group admitted with nonfebrile illness had neutralizing antibodies (P less than .0001).
Also, 93% of the GBS patients had Zika virus immunoglobulin M (IgM) and 88% reported symptoms consistent with Zika infection a mean of 6 days before onset of neurological symptoms. Acute Zika infection is usually characterized by rash, fever, and conjunctivitis.
Past dengue virus infection, which had been considered a possible risk factor for Zika-mediated GBS, did not differ significantly between patients in the control groups and those with GBS.
The investigators were also able to subtype the clinical characteristics of the GBS cases as consistent with acute motor axonal neuropathy, or AMAN, phenotype. However, the antibodies typically seen associated with AMAN were not seen in these patients, leading investigators to suspect that a different biological pathway was responsible.
More than a third of the GBS patients in the study required intensive care, most of these also with respiratory support, though none died.
The government of France, the European Union, and the Wellcome Trust funded the study. The researchers declared that they had no competing interests.
Zika virus can be added to our list of viruses that can cause Guillain-Barré syndrome, and investigation of these cases should include tests for Zika when there is a possibility of infection by that virus. Whether Zika will be proven to pose a greater threat in causing Guillain-Barré syndrome than its various flavivirus cousins remains to be determined. A little caution should be taken because the data are still scarce and we do not know whether the current Zika virus is identical to that in previous outbreaks, whether it will behave exactly the same in a different population with a different genetic and immunity background, or whether a cofactor or co-infection is responsible. Reassuringly, the investigators did not find any evidence that previous dengue infection enhanced the severity of the disease, which could substantially have increased the threat in areas of regular activity.
Dr. David W. Smith is a clinical professor of pathology and laboratory medicine at the University of Western Australia in Perth. John Mackenzie, Ph.D., is a professor of tropical and infectious diseases at Curtin University in Bentley, Australia. They had no competing interests to disclose.
Zika virus can be added to our list of viruses that can cause Guillain-Barré syndrome, and investigation of these cases should include tests for Zika when there is a possibility of infection by that virus. Whether Zika will be proven to pose a greater threat in causing Guillain-Barré syndrome than its various flavivirus cousins remains to be determined. A little caution should be taken because the data are still scarce and we do not know whether the current Zika virus is identical to that in previous outbreaks, whether it will behave exactly the same in a different population with a different genetic and immunity background, or whether a cofactor or co-infection is responsible. Reassuringly, the investigators did not find any evidence that previous dengue infection enhanced the severity of the disease, which could substantially have increased the threat in areas of regular activity.
Dr. David W. Smith is a clinical professor of pathology and laboratory medicine at the University of Western Australia in Perth. John Mackenzie, Ph.D., is a professor of tropical and infectious diseases at Curtin University in Bentley, Australia. They had no competing interests to disclose.
Zika virus can be added to our list of viruses that can cause Guillain-Barré syndrome, and investigation of these cases should include tests for Zika when there is a possibility of infection by that virus. Whether Zika will be proven to pose a greater threat in causing Guillain-Barré syndrome than its various flavivirus cousins remains to be determined. A little caution should be taken because the data are still scarce and we do not know whether the current Zika virus is identical to that in previous outbreaks, whether it will behave exactly the same in a different population with a different genetic and immunity background, or whether a cofactor or co-infection is responsible. Reassuringly, the investigators did not find any evidence that previous dengue infection enhanced the severity of the disease, which could substantially have increased the threat in areas of regular activity.
Dr. David W. Smith is a clinical professor of pathology and laboratory medicine at the University of Western Australia in Perth. John Mackenzie, Ph.D., is a professor of tropical and infectious diseases at Curtin University in Bentley, Australia. They had no competing interests to disclose.
Serological evidence from French Polynesia links an outbreak of Zika virus to a spike in cases of Guillain-Barré syndrome seen there in 2013-2014.
The research, published online Feb. 29 in The Lancet, is the first to use a case-control design to demonstrate that Zika, a mosquito-borne flavivirus, is associated with Guillain-Barré syndrome (Lancet. 2016 Feb 29. doi: 10.1016/S0140-6736(16)00562-6).
Guillain-Barré syndrome (GBS) is an immune-mediated flaccid paralysis that can follow viral or bacterial infections. Most patients with GBS recover with intensive care in hospitals, although the syndrome can be permanently debilitating or, in rare cases, fatal.
As a large outbreak of Zika continues in Central and South America, hospitals should be prepared for excess GBS cases, the authors of the study say, and assure adequate intensive-care capacity to treat them. Based on the 66% attack rate of Zika during the French Polynesia outbreak, investigators estimated the incidence of GBS at 0.24 per 1,000 Zika infections, but noted that it could be different in the current outbreak.
Dr. Van-Mai Cao-Lormeau of the Unit of Emerging Infectious Diseases at Institut Louis Malardé in Papeete, French Polynesia, alongside colleagues in France and French Polynesia, used a case-control design to compare serological samples from 42 patients (74% male) diagnosed at a Tahiti hospital with GBS with samples from age-and sex-matched patients who presented at the same hospital, also during the time of the outbreak, with a nonfebrile illness (n = 98) or with acute Zika disease without neurological symptoms (n = 70).
The investigators found that all but one patient with GBS had Zika virus antibodies, and all of them had neutralizing antibodies to Zika virus. By comparison, only 56% (n = 54) of the control group admitted with nonfebrile illness had neutralizing antibodies (P less than .0001).
Also, 93% of the GBS patients had Zika virus immunoglobulin M (IgM) and 88% reported symptoms consistent with Zika infection a mean of 6 days before onset of neurological symptoms. Acute Zika infection is usually characterized by rash, fever, and conjunctivitis.
Past dengue virus infection, which had been considered a possible risk factor for Zika-mediated GBS, did not differ significantly between patients in the control groups and those with GBS.
The investigators were also able to subtype the clinical characteristics of the GBS cases as consistent with acute motor axonal neuropathy, or AMAN, phenotype. However, the antibodies typically seen associated with AMAN were not seen in these patients, leading investigators to suspect that a different biological pathway was responsible.
More than a third of the GBS patients in the study required intensive care, most of these also with respiratory support, though none died.
The government of France, the European Union, and the Wellcome Trust funded the study. The researchers declared that they had no competing interests.
Serological evidence from French Polynesia links an outbreak of Zika virus to a spike in cases of Guillain-Barré syndrome seen there in 2013-2014.
The research, published online Feb. 29 in The Lancet, is the first to use a case-control design to demonstrate that Zika, a mosquito-borne flavivirus, is associated with Guillain-Barré syndrome (Lancet. 2016 Feb 29. doi: 10.1016/S0140-6736(16)00562-6).
Guillain-Barré syndrome (GBS) is an immune-mediated flaccid paralysis that can follow viral or bacterial infections. Most patients with GBS recover with intensive care in hospitals, although the syndrome can be permanently debilitating or, in rare cases, fatal.
As a large outbreak of Zika continues in Central and South America, hospitals should be prepared for excess GBS cases, the authors of the study say, and assure adequate intensive-care capacity to treat them. Based on the 66% attack rate of Zika during the French Polynesia outbreak, investigators estimated the incidence of GBS at 0.24 per 1,000 Zika infections, but noted that it could be different in the current outbreak.
Dr. Van-Mai Cao-Lormeau of the Unit of Emerging Infectious Diseases at Institut Louis Malardé in Papeete, French Polynesia, alongside colleagues in France and French Polynesia, used a case-control design to compare serological samples from 42 patients (74% male) diagnosed at a Tahiti hospital with GBS with samples from age-and sex-matched patients who presented at the same hospital, also during the time of the outbreak, with a nonfebrile illness (n = 98) or with acute Zika disease without neurological symptoms (n = 70).
The investigators found that all but one patient with GBS had Zika virus antibodies, and all of them had neutralizing antibodies to Zika virus. By comparison, only 56% (n = 54) of the control group admitted with nonfebrile illness had neutralizing antibodies (P less than .0001).
Also, 93% of the GBS patients had Zika virus immunoglobulin M (IgM) and 88% reported symptoms consistent with Zika infection a mean of 6 days before onset of neurological symptoms. Acute Zika infection is usually characterized by rash, fever, and conjunctivitis.
Past dengue virus infection, which had been considered a possible risk factor for Zika-mediated GBS, did not differ significantly between patients in the control groups and those with GBS.
The investigators were also able to subtype the clinical characteristics of the GBS cases as consistent with acute motor axonal neuropathy, or AMAN, phenotype. However, the antibodies typically seen associated with AMAN were not seen in these patients, leading investigators to suspect that a different biological pathway was responsible.
More than a third of the GBS patients in the study required intensive care, most of these also with respiratory support, though none died.
The government of France, the European Union, and the Wellcome Trust funded the study. The researchers declared that they had no competing interests.
FROM THE LANCET
Key clinical point: Acute infection with Zika virus in French Polynesia was associated with Guillain-Barré syndrome.
Major finding: Among GBS patients admitted to hospitals during an 2013-2014 outbreak of Zika virus, nearly all had antibodies or neutralizing antibodies to Zika, vs. 56% of age and sex-matched controls (P less than .0001).
Data source: A case-cohort study comparing blood results from 42 GBS cases and two cohorts of controls, one with acute Zika infection without GBS (n = 70) and another admitted during the outbreak for other illnesses (n = 98).
Disclosures: The French government, the European Union, and the Wellcome Trust sponsored the study. Investigators disclosed no conflicts of interest.

Increase in Blood Pressure May Improve Survival in TBI
SAN DIEGO—In the setting of traumatic brain injury (TBI), increases in systolic blood pressure after the blood pressure nadir are independently associated with improved survival in patients with hypotension. In addition, even substantial blood pressure increases do not seem to harm patients with normal blood pressure. These findings come from a subanalysis of the ongoing Excellence in Prehospital Injury Care (EPIC) TBI study.
“Little is known about the patterns of blood pressure in TBI in the field,” said Daniel W. Spaite, MD, Professor and Virginia Piper Endowed Chair of Emergency Medicine at the University of Arizona in Tucson, at the Annual Meeting of the National Association of EMS Physicians (NAEMSP). “For instance, nobody knows whether it’s better to have your blood pressure increasing, stable, or decreasing in the field with regard to outcome, especially mortality. Typical studies that do have EMS data linked only have a single blood pressure measurement documented, so there’s no knowledge of trends in EMS blood pressure in TBI.”

Dr. Spaite and his colleagues evaluated the association between mortality and increases in prehospital systolic blood pressure after the lowest recorded measurement in patients with major TBI who are part of the EPIC study, the statewide implementation of TBI guidelines from the Brain Trauma Foundation and the NAEMSP. Data sources include the Arizona State Trauma Registry, which has comprehensive hospital outcome data. “The cases are then linked, and the EMS patient care reports are carefully abstracted by the EPIC data team,” Dr. Spaite explained. “This included major TBI (which is, clinically, both moderate and severe) and all patients whose lowest systolic blood pressure was between 40 and 300 mmHg.”
The researchers used logistic regression to examine the association between the increase in EMS systolic blood pressure after the lowest EMS blood pressure recorded and its association with adjusted probability of death. They then separated the study population into four cohorts, based on each patient’s prehospital systolic blood pressure (ie, 40–89 mmHg, 90–139 mmHg, 140–159 mmHg, and 160–300 mmHg). In each cohort, they identified the independent association between the magnitude of increase in systolic blood pressure and the adjusted probability of death.
Dr. Spaite reported findings from 14,567 patients with TBI. More than two-thirds (68%) of participants were male, and their mean age was 45. The researchers observed that in the hypotensive cohort, mortality dropped significantly if the systolic blood pressure increased after the lowest recorded systolic blood pressure. “Improvements were dramatic with increases of 40–80 mmHg,” he said. In the normotensive group, increases in systolic blood pressure were associated with slight reductions in mortality. Large increases in systolic blood pressure, such as in the range of 70–90 mmHg, did not appear to be detrimental.In the mildly hypertensive group, large systolic increases were associated with higher mortality. “Interestingly, even if your lowest [systolic blood pressure] is between 140 and 159 mmHg, until you get above an increase of 40 mmHg above that, you don’t start seeing increases in mortality,” said Dr. Spaite. In the severely hypertensive group, mortality was higher with any subsequent increase in systolic blood pressure, “which doesn’t surprise any of us,” he said. “It’s dramatically higher if the increase is large.”
Dr. Spaite emphasized that the current analysis is based on observational data, “so this does not prove that treating hypotension improves outcome. … That direct question is part of the EPIC study itself and awaits the final analysis, hopefully in mid-2017. This is the first large report of blood pressure trends in the prehospital management of TBI.”
He concluded that the current findings in the hypotensive and normotensive cohorts “support guideline recommendations for restoring and optimizing cerebral perfusion in EMS TBI management. What is fascinating about the literature is that the focus in TBI has always been on hypotension, but there’s very little information about what’s the best or the optimal blood pressure.”
—Doug Brunk
SAN DIEGO—In the setting of traumatic brain injury (TBI), increases in systolic blood pressure after the blood pressure nadir are independently associated with improved survival in patients with hypotension. In addition, even substantial blood pressure increases do not seem to harm patients with normal blood pressure. These findings come from a subanalysis of the ongoing Excellence in Prehospital Injury Care (EPIC) TBI study.
“Little is known about the patterns of blood pressure in TBI in the field,” said Daniel W. Spaite, MD, Professor and Virginia Piper Endowed Chair of Emergency Medicine at the University of Arizona in Tucson, at the Annual Meeting of the National Association of EMS Physicians (NAEMSP). “For instance, nobody knows whether it’s better to have your blood pressure increasing, stable, or decreasing in the field with regard to outcome, especially mortality. Typical studies that do have EMS data linked only have a single blood pressure measurement documented, so there’s no knowledge of trends in EMS blood pressure in TBI.”
Dr. Spaite and his colleagues evaluated the association between mortality and increases in prehospital systolic blood pressure after the lowest recorded measurement in patients with major TBI who are part of the EPIC study, the statewide implementation of TBI guidelines from the Brain Trauma Foundation and the NAEMSP. Data sources include the Arizona State Trauma Registry, which has comprehensive hospital outcome data. “The cases are then linked, and the EMS patient care reports are carefully abstracted by the EPIC data team,” Dr. Spaite explained. “This included major TBI (which is, clinically, both moderate and severe) and all patients whose lowest systolic blood pressure was between 40 and 300 mmHg.”
The researchers used logistic regression to examine the association between the increase in EMS systolic blood pressure after the lowest EMS blood pressure recorded and its association with adjusted probability of death. They then separated the study population into four cohorts, based on each patient’s prehospital systolic blood pressure (ie, 40–89 mmHg, 90–139 mmHg, 140–159 mmHg, and 160–300 mmHg). In each cohort, they identified the independent association between the magnitude of increase in systolic blood pressure and the adjusted probability of death.
Dr. Spaite reported findings from 14,567 patients with TBI. More than two-thirds (68%) of participants were male, and their mean age was 45. The researchers observed that in the hypotensive cohort, mortality dropped significantly if the systolic blood pressure increased after the lowest recorded systolic blood pressure. “Improvements were dramatic with increases of 40–80 mmHg,” he said. In the normotensive group, increases in systolic blood pressure were associated with slight reductions in mortality. Large increases in systolic blood pressure, such as in the range of 70–90 mmHg, did not appear to be detrimental.In the mildly hypertensive group, large systolic increases were associated with higher mortality. “Interestingly, even if your lowest [systolic blood pressure] is between 140 and 159 mmHg, until you get above an increase of 40 mmHg above that, you don’t start seeing increases in mortality,” said Dr. Spaite. In the severely hypertensive group, mortality was higher with any subsequent increase in systolic blood pressure, “which doesn’t surprise any of us,” he said. “It’s dramatically higher if the increase is large.”
Dr. Spaite emphasized that the current analysis is based on observational data, “so this does not prove that treating hypotension improves outcome. … That direct question is part of the EPIC study itself and awaits the final analysis, hopefully in mid-2017. This is the first large report of blood pressure trends in the prehospital management of TBI.”
He concluded that the current findings in the hypotensive and normotensive cohorts “support guideline recommendations for restoring and optimizing cerebral perfusion in EMS TBI management. What is fascinating about the literature is that the focus in TBI has always been on hypotension, but there’s very little information about what’s the best or the optimal blood pressure.”
—Doug Brunk
SAN DIEGO—In the setting of traumatic brain injury (TBI), increases in systolic blood pressure after the blood pressure nadir are independently associated with improved survival in patients with hypotension. In addition, even substantial blood pressure increases do not seem to harm patients with normal blood pressure. These findings come from a subanalysis of the ongoing Excellence in Prehospital Injury Care (EPIC) TBI study.
“Little is known about the patterns of blood pressure in TBI in the field,” said Daniel W. Spaite, MD, Professor and Virginia Piper Endowed Chair of Emergency Medicine at the University of Arizona in Tucson, at the Annual Meeting of the National Association of EMS Physicians (NAEMSP). “For instance, nobody knows whether it’s better to have your blood pressure increasing, stable, or decreasing in the field with regard to outcome, especially mortality. Typical studies that do have EMS data linked only have a single blood pressure measurement documented, so there’s no knowledge of trends in EMS blood pressure in TBI.”
Dr. Spaite and his colleagues evaluated the association between mortality and increases in prehospital systolic blood pressure after the lowest recorded measurement in patients with major TBI who are part of the EPIC study, the statewide implementation of TBI guidelines from the Brain Trauma Foundation and the NAEMSP. Data sources include the Arizona State Trauma Registry, which has comprehensive hospital outcome data. “The cases are then linked, and the EMS patient care reports are carefully abstracted by the EPIC data team,” Dr. Spaite explained. “This included major TBI (which is, clinically, both moderate and severe) and all patients whose lowest systolic blood pressure was between 40 and 300 mmHg.”
The researchers used logistic regression to examine the association between the increase in EMS systolic blood pressure after the lowest EMS blood pressure recorded and its association with adjusted probability of death. They then separated the study population into four cohorts, based on each patient’s prehospital systolic blood pressure (ie, 40–89 mmHg, 90–139 mmHg, 140–159 mmHg, and 160–300 mmHg). In each cohort, they identified the independent association between the magnitude of increase in systolic blood pressure and the adjusted probability of death.
Dr. Spaite reported findings from 14,567 patients with TBI. More than two-thirds (68%) of participants were male, and their mean age was 45. The researchers observed that in the hypotensive cohort, mortality dropped significantly if the systolic blood pressure increased after the lowest recorded systolic blood pressure. “Improvements were dramatic with increases of 40–80 mmHg,” he said. In the normotensive group, increases in systolic blood pressure were associated with slight reductions in mortality. Large increases in systolic blood pressure, such as in the range of 70–90 mmHg, did not appear to be detrimental.In the mildly hypertensive group, large systolic increases were associated with higher mortality. “Interestingly, even if your lowest [systolic blood pressure] is between 140 and 159 mmHg, until you get above an increase of 40 mmHg above that, you don’t start seeing increases in mortality,” said Dr. Spaite. In the severely hypertensive group, mortality was higher with any subsequent increase in systolic blood pressure, “which doesn’t surprise any of us,” he said. “It’s dramatically higher if the increase is large.”
Dr. Spaite emphasized that the current analysis is based on observational data, “so this does not prove that treating hypotension improves outcome. … That direct question is part of the EPIC study itself and awaits the final analysis, hopefully in mid-2017. This is the first large report of blood pressure trends in the prehospital management of TBI.”
He concluded that the current findings in the hypotensive and normotensive cohorts “support guideline recommendations for restoring and optimizing cerebral perfusion in EMS TBI management. What is fascinating about the literature is that the focus in TBI has always been on hypotension, but there’s very little information about what’s the best or the optimal blood pressure.”
—Doug Brunk

The microbiome in celiac disease: Beyond diet-genetic interactions
Evidence points to the mix of bacteria that make the gut their home, collectively called the microbiome.
INHERITING THE WRONG GENES and eating the wrong food (ie, gluten) are necessary for celiac disease to develop, but are not enough by themselves. Something else must be contributing, and evidence is pointing to the mix of bacteria that make our guts their home, collectively called the microbiome.
Celiac disease is a highly prevalent, chronic, immune-mediated form of enteropathy.1 It affects 0.5% to 1% of the population, and although it is mostly seen in people of northern European descent, those in other populations can develop the disease as well. Historically, celiac disease was classified as an infant condition. However, it now commonly presents later in life (between ages 10 and 40) and often with extraintestinal manifestations.2
In this issue of Cleveland Clinic Journal of Medicine, Kochhar et al provide a comprehensive updated review of celiac disease.3
GENES AND GLUTEN ARE NECESSARY BUT NOT SUFFICIENT
Although genetic factors and exposure to gluten in the diet are proven to be necessary for celiac disease to develop, they are not sufficient. Evidence of this is in the numbers; although one-third of the general population carries the HLA susceptibility genes (specifically HLA-DQ2 and DQ8),4 only 2% to 5% of people with these genes develop clinically evident celiac disease.
Additional environmental factors must be contributing to disease development, but these other factors are poorly understood. Some of the possible culprits that might influence the risk of disease occurrence and the timing of its onset include5:
- The amount and quality of gluten ingested—the higher the concentration of gluten, the higher the risk, and different grains have gluten varieties with more or less immunogenic capabilities, ie, T-cell activation properties
- The pattern of infant feeding—the risk may be lower with breastfeeding than with formula
- The age at which gluten is introduced into the diet—the risk may be higher if gluten is introduced earlier.6
More recently, studies of the pathogenesis of celiac disease and gene-environmental interactions have expanded beyond host predisposition and dietary factors.
OUR BODIES, OUR MICROBIOMES: A SYMBIOTIC RELATIONSHIP
The role of the human microbiome in autoimmune disease is now being elucidated.7 Remarkably, the microorganisms living in our bodies outnumber our body cells by a factor of 10, and their genomes vastly exceed our own protein-coding genome capabilities by a factor of 100.
The gut microbiome is now considered a true bioreactor with enzymatic and immunologic capabilities beyond (and complementary to) those of its host. The commensal microbiome of the host intestine provides benefits that can be broken down into three broad categories:
- Nutritional—producing essential amino acids and vitamins
- Metabolic—degrading complex polysaccharides from dietary fibers
- Immunologic—shaping the host immune system while cooperating with it against pathogenic microorganisms.
The immunologic function is highly relevant. We have coevolved with our bacteria in a mutually beneficial, symbiotic relationship in which we maintain an active state of low inflammation so that a constant bacterial and dietary antigenic load can be tolerated.
Is there a core human microbiome shared by all individuals? And what is the impact of altering the relative microbial composition (dysbiosis) in physiologic and disease states? To find out, the National Institutes of Health launched the Human Microbiome Project8 in 2008. Important tools in this work include novel culture-independent approaches (high-throughput DNA sequencing and whole-microbiome “shotgun” sequencing with metagenomic analysis) and computational analytical tools.9
An accumulating body of evidence is now available from animal models and human studies correlating states of intestinal dysbiosis (disruption in homeostatic community composition) with various disease processes. These have ranged from inflammatory bowel disease to systemic autoimmune disorders such as psoriasis, inflammatory arthropathies, and demyelinating central nervous system diseases.10–14
RESEARCH INTO THE MICROBIOME IN CELIAC DISEASE
Celiac disease has also served as a unique model for studying this biologic relationship, and the microbiome has been postulated to have a role in its pathogenesis.15 Multiple clinical studies demonstrate that a state of intestinal dysbiosis is indeed associated with celiac disease.
Specifically, decreases in the abundance of Firmicutes spp and increases in Proteobacteria spp have been detected in both children and adults with active celiac disease.16,17 Intriguingly, overrepresentation of Proteobacteria was also correlated with disease activity. Other studies have reported decreases in the proportion of reportedly protective, anti-inflammatory bacteria such as Bifidobacterium and increases in the proportion of Bacteroides and Escherichia coli in patients with active disease.18,19 Altered diversity and altered metabolic function, ie, decreased concentration of protective short-chain fatty acids of the microbiota, have also been reported in patients with celiac disease.19,20
To move beyond correlative studies and mechanistically address the possibility of causation, multiple groups have used a gnotobiotic approach, ie, maintaining animals under germ-free conditions and incorporating microbes of interest. This approach is highly relevant in studying whether the bacterial community composition is capable of modulating loss of tolerance to gluten in genetically susceptible hosts. A few notable examples have been published.
In germ-free rats, long-term feeding of gliadin, but not albumin, from birth until 2 months of age induced moderate small-intestinal damage.21 Similarly, germ-free nonobese diabetic-DQ8 mice developed more severe gluten-induced disease than mice with normal intestinal bacteria.22
These findings suggest that the normal gut microbiome may have intrinsic beneficial properties capable of reducing the inflammatory effects associated with gluten ingestion. Notably, the specific composition of the intestinal microbiome can define the fate of gluten-induced pathology. Mice colonized with commensal microbiota are indeed protected from gluten-induced pathology, while mice colonized with Proteobacteria spp develop a moderate degree of gluten-induced disease. When Escherichia coli derived from patients with celiac disease is added to commensal colonization, the celiac disease-like phenotype develops.23
Taken together, these studies support the hypothesis that the intestinal microbiome may be another environmental factor involved in the development of celiac disease.
QUESTIONS AND CHALLENGES REMAIN
The results of clinical studies are not necessarily consistent at the taxonomy level. The fields of metagenomics, which investigates all genes and their enzymatic function in a given community, and metabolomics, which identifies bacterial end-products, characterizing their functional capabilities, are still in their infancy and will be required to further investigate functionality of the altered microbiome in celiac disease.
Second, the directionality—the causality or consequences of this dysbiosis—and timing—the moment at which changes occur, ie, after introducing gluten or at the time when symptoms appear—remain elusive, and prospective studies in humans will be essential.
Finally, more mechanistic studies in animal models are needed to dissect the host immune response to dietary gluten and perturbation of intestinal community composition. This may lead to the possibility of future interventions in the form of prebiotics, probiotics, or specific metabolites, complementary to gluten avoidance.
In the meantime, increasing disease awareness and rapid diagnosis and treatment continue to be of utmost importance to address the clinical consequences of celiac disease in both children and adults.
Supported by: Grant No. K23AR064318 from NIAMS to Dr. Scher; The Colton Center for Autoimmunity; The Riley Family Foundation.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014; 168:272–278.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Kochhar GS, Singh T, Gill A, Kirby DF. Celiac disease: an internist’s perspective. Cleve Clin J Med 2016; 83:217–227.
- Gutierrez-Achury J, Zhernakova A, Pulit SL, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet 2015; 47:577–578.
- Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med 2010; 42:530–538.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res 2009; 19:2317–2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65.
- Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015; 67:128–139.
- Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3:e2719.
- Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–1463.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn‘s disease. Cell Host Microbe 2014; 15:382–392.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12:497–506.
- Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79:5472–5479.
- Wacklin P, Kaukinen K, Tuovinen E, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19:934–941.
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62:264–269.
- Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11:219.
- Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10:175.
- Stepankova R, Tlaskalova-Hogenova H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scand J Gastroenterol 1996; 31:551–557.
- Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol 2011; 187:4338–4346.
- Galipeau HJ, Verdu EF. Gut microbes and adverse food reactions: focus on gluten related disorders. Gut Microbes 2014; 5:594–605.
Evidence points to the mix of bacteria that make the gut their home, collectively called the microbiome.
Evidence points to the mix of bacteria that make the gut their home, collectively called the microbiome.
INHERITING THE WRONG GENES and eating the wrong food (ie, gluten) are necessary for celiac disease to develop, but are not enough by themselves. Something else must be contributing, and evidence is pointing to the mix of bacteria that make our guts their home, collectively called the microbiome.
Celiac disease is a highly prevalent, chronic, immune-mediated form of enteropathy.1 It affects 0.5% to 1% of the population, and although it is mostly seen in people of northern European descent, those in other populations can develop the disease as well. Historically, celiac disease was classified as an infant condition. However, it now commonly presents later in life (between ages 10 and 40) and often with extraintestinal manifestations.2
In this issue of Cleveland Clinic Journal of Medicine, Kochhar et al provide a comprehensive updated review of celiac disease.3
GENES AND GLUTEN ARE NECESSARY BUT NOT SUFFICIENT
Although genetic factors and exposure to gluten in the diet are proven to be necessary for celiac disease to develop, they are not sufficient. Evidence of this is in the numbers; although one-third of the general population carries the HLA susceptibility genes (specifically HLA-DQ2 and DQ8),4 only 2% to 5% of people with these genes develop clinically evident celiac disease.
Additional environmental factors must be contributing to disease development, but these other factors are poorly understood. Some of the possible culprits that might influence the risk of disease occurrence and the timing of its onset include5:
- The amount and quality of gluten ingested—the higher the concentration of gluten, the higher the risk, and different grains have gluten varieties with more or less immunogenic capabilities, ie, T-cell activation properties
- The pattern of infant feeding—the risk may be lower with breastfeeding than with formula
- The age at which gluten is introduced into the diet—the risk may be higher if gluten is introduced earlier.6
More recently, studies of the pathogenesis of celiac disease and gene-environmental interactions have expanded beyond host predisposition and dietary factors.
OUR BODIES, OUR MICROBIOMES: A SYMBIOTIC RELATIONSHIP
The role of the human microbiome in autoimmune disease is now being elucidated.7 Remarkably, the microorganisms living in our bodies outnumber our body cells by a factor of 10, and their genomes vastly exceed our own protein-coding genome capabilities by a factor of 100.
The gut microbiome is now considered a true bioreactor with enzymatic and immunologic capabilities beyond (and complementary to) those of its host. The commensal microbiome of the host intestine provides benefits that can be broken down into three broad categories:
- Nutritional—producing essential amino acids and vitamins
- Metabolic—degrading complex polysaccharides from dietary fibers
- Immunologic—shaping the host immune system while cooperating with it against pathogenic microorganisms.
The immunologic function is highly relevant. We have coevolved with our bacteria in a mutually beneficial, symbiotic relationship in which we maintain an active state of low inflammation so that a constant bacterial and dietary antigenic load can be tolerated.
Is there a core human microbiome shared by all individuals? And what is the impact of altering the relative microbial composition (dysbiosis) in physiologic and disease states? To find out, the National Institutes of Health launched the Human Microbiome Project8 in 2008. Important tools in this work include novel culture-independent approaches (high-throughput DNA sequencing and whole-microbiome “shotgun” sequencing with metagenomic analysis) and computational analytical tools.9
An accumulating body of evidence is now available from animal models and human studies correlating states of intestinal dysbiosis (disruption in homeostatic community composition) with various disease processes. These have ranged from inflammatory bowel disease to systemic autoimmune disorders such as psoriasis, inflammatory arthropathies, and demyelinating central nervous system diseases.10–14
RESEARCH INTO THE MICROBIOME IN CELIAC DISEASE
Celiac disease has also served as a unique model for studying this biologic relationship, and the microbiome has been postulated to have a role in its pathogenesis.15 Multiple clinical studies demonstrate that a state of intestinal dysbiosis is indeed associated with celiac disease.
Specifically, decreases in the abundance of Firmicutes spp and increases in Proteobacteria spp have been detected in both children and adults with active celiac disease.16,17 Intriguingly, overrepresentation of Proteobacteria was also correlated with disease activity. Other studies have reported decreases in the proportion of reportedly protective, anti-inflammatory bacteria such as Bifidobacterium and increases in the proportion of Bacteroides and Escherichia coli in patients with active disease.18,19 Altered diversity and altered metabolic function, ie, decreased concentration of protective short-chain fatty acids of the microbiota, have also been reported in patients with celiac disease.19,20
To move beyond correlative studies and mechanistically address the possibility of causation, multiple groups have used a gnotobiotic approach, ie, maintaining animals under germ-free conditions and incorporating microbes of interest. This approach is highly relevant in studying whether the bacterial community composition is capable of modulating loss of tolerance to gluten in genetically susceptible hosts. A few notable examples have been published.
In germ-free rats, long-term feeding of gliadin, but not albumin, from birth until 2 months of age induced moderate small-intestinal damage.21 Similarly, germ-free nonobese diabetic-DQ8 mice developed more severe gluten-induced disease than mice with normal intestinal bacteria.22
These findings suggest that the normal gut microbiome may have intrinsic beneficial properties capable of reducing the inflammatory effects associated with gluten ingestion. Notably, the specific composition of the intestinal microbiome can define the fate of gluten-induced pathology. Mice colonized with commensal microbiota are indeed protected from gluten-induced pathology, while mice colonized with Proteobacteria spp develop a moderate degree of gluten-induced disease. When Escherichia coli derived from patients with celiac disease is added to commensal colonization, the celiac disease-like phenotype develops.23
Taken together, these studies support the hypothesis that the intestinal microbiome may be another environmental factor involved in the development of celiac disease.
QUESTIONS AND CHALLENGES REMAIN
The results of clinical studies are not necessarily consistent at the taxonomy level. The fields of metagenomics, which investigates all genes and their enzymatic function in a given community, and metabolomics, which identifies bacterial end-products, characterizing their functional capabilities, are still in their infancy and will be required to further investigate functionality of the altered microbiome in celiac disease.
Second, the directionality—the causality or consequences of this dysbiosis—and timing—the moment at which changes occur, ie, after introducing gluten or at the time when symptoms appear—remain elusive, and prospective studies in humans will be essential.
Finally, more mechanistic studies in animal models are needed to dissect the host immune response to dietary gluten and perturbation of intestinal community composition. This may lead to the possibility of future interventions in the form of prebiotics, probiotics, or specific metabolites, complementary to gluten avoidance.
In the meantime, increasing disease awareness and rapid diagnosis and treatment continue to be of utmost importance to address the clinical consequences of celiac disease in both children and adults.
Supported by: Grant No. K23AR064318 from NIAMS to Dr. Scher; The Colton Center for Autoimmunity; The Riley Family Foundation.
INHERITING THE WRONG GENES and eating the wrong food (ie, gluten) are necessary for celiac disease to develop, but are not enough by themselves. Something else must be contributing, and evidence is pointing to the mix of bacteria that make our guts their home, collectively called the microbiome.
Celiac disease is a highly prevalent, chronic, immune-mediated form of enteropathy.1 It affects 0.5% to 1% of the population, and although it is mostly seen in people of northern European descent, those in other populations can develop the disease as well. Historically, celiac disease was classified as an infant condition. However, it now commonly presents later in life (between ages 10 and 40) and often with extraintestinal manifestations.2
In this issue of Cleveland Clinic Journal of Medicine, Kochhar et al provide a comprehensive updated review of celiac disease.3
GENES AND GLUTEN ARE NECESSARY BUT NOT SUFFICIENT
Although genetic factors and exposure to gluten in the diet are proven to be necessary for celiac disease to develop, they are not sufficient. Evidence of this is in the numbers; although one-third of the general population carries the HLA susceptibility genes (specifically HLA-DQ2 and DQ8),4 only 2% to 5% of people with these genes develop clinically evident celiac disease.
Additional environmental factors must be contributing to disease development, but these other factors are poorly understood. Some of the possible culprits that might influence the risk of disease occurrence and the timing of its onset include5:
- The amount and quality of gluten ingested—the higher the concentration of gluten, the higher the risk, and different grains have gluten varieties with more or less immunogenic capabilities, ie, T-cell activation properties
- The pattern of infant feeding—the risk may be lower with breastfeeding than with formula
- The age at which gluten is introduced into the diet—the risk may be higher if gluten is introduced earlier.6
More recently, studies of the pathogenesis of celiac disease and gene-environmental interactions have expanded beyond host predisposition and dietary factors.
OUR BODIES, OUR MICROBIOMES: A SYMBIOTIC RELATIONSHIP
The role of the human microbiome in autoimmune disease is now being elucidated.7 Remarkably, the microorganisms living in our bodies outnumber our body cells by a factor of 10, and their genomes vastly exceed our own protein-coding genome capabilities by a factor of 100.
The gut microbiome is now considered a true bioreactor with enzymatic and immunologic capabilities beyond (and complementary to) those of its host. The commensal microbiome of the host intestine provides benefits that can be broken down into three broad categories:
- Nutritional—producing essential amino acids and vitamins
- Metabolic—degrading complex polysaccharides from dietary fibers
- Immunologic—shaping the host immune system while cooperating with it against pathogenic microorganisms.
The immunologic function is highly relevant. We have coevolved with our bacteria in a mutually beneficial, symbiotic relationship in which we maintain an active state of low inflammation so that a constant bacterial and dietary antigenic load can be tolerated.
Is there a core human microbiome shared by all individuals? And what is the impact of altering the relative microbial composition (dysbiosis) in physiologic and disease states? To find out, the National Institutes of Health launched the Human Microbiome Project8 in 2008. Important tools in this work include novel culture-independent approaches (high-throughput DNA sequencing and whole-microbiome “shotgun” sequencing with metagenomic analysis) and computational analytical tools.9
An accumulating body of evidence is now available from animal models and human studies correlating states of intestinal dysbiosis (disruption in homeostatic community composition) with various disease processes. These have ranged from inflammatory bowel disease to systemic autoimmune disorders such as psoriasis, inflammatory arthropathies, and demyelinating central nervous system diseases.10–14
RESEARCH INTO THE MICROBIOME IN CELIAC DISEASE
Celiac disease has also served as a unique model for studying this biologic relationship, and the microbiome has been postulated to have a role in its pathogenesis.15 Multiple clinical studies demonstrate that a state of intestinal dysbiosis is indeed associated with celiac disease.
Specifically, decreases in the abundance of Firmicutes spp and increases in Proteobacteria spp have been detected in both children and adults with active celiac disease.16,17 Intriguingly, overrepresentation of Proteobacteria was also correlated with disease activity. Other studies have reported decreases in the proportion of reportedly protective, anti-inflammatory bacteria such as Bifidobacterium and increases in the proportion of Bacteroides and Escherichia coli in patients with active disease.18,19 Altered diversity and altered metabolic function, ie, decreased concentration of protective short-chain fatty acids of the microbiota, have also been reported in patients with celiac disease.19,20
To move beyond correlative studies and mechanistically address the possibility of causation, multiple groups have used a gnotobiotic approach, ie, maintaining animals under germ-free conditions and incorporating microbes of interest. This approach is highly relevant in studying whether the bacterial community composition is capable of modulating loss of tolerance to gluten in genetically susceptible hosts. A few notable examples have been published.
In germ-free rats, long-term feeding of gliadin, but not albumin, from birth until 2 months of age induced moderate small-intestinal damage.21 Similarly, germ-free nonobese diabetic-DQ8 mice developed more severe gluten-induced disease than mice with normal intestinal bacteria.22
These findings suggest that the normal gut microbiome may have intrinsic beneficial properties capable of reducing the inflammatory effects associated with gluten ingestion. Notably, the specific composition of the intestinal microbiome can define the fate of gluten-induced pathology. Mice colonized with commensal microbiota are indeed protected from gluten-induced pathology, while mice colonized with Proteobacteria spp develop a moderate degree of gluten-induced disease. When Escherichia coli derived from patients with celiac disease is added to commensal colonization, the celiac disease-like phenotype develops.23
Taken together, these studies support the hypothesis that the intestinal microbiome may be another environmental factor involved in the development of celiac disease.
QUESTIONS AND CHALLENGES REMAIN
The results of clinical studies are not necessarily consistent at the taxonomy level. The fields of metagenomics, which investigates all genes and their enzymatic function in a given community, and metabolomics, which identifies bacterial end-products, characterizing their functional capabilities, are still in their infancy and will be required to further investigate functionality of the altered microbiome in celiac disease.
Second, the directionality—the causality or consequences of this dysbiosis—and timing—the moment at which changes occur, ie, after introducing gluten or at the time when symptoms appear—remain elusive, and prospective studies in humans will be essential.
Finally, more mechanistic studies in animal models are needed to dissect the host immune response to dietary gluten and perturbation of intestinal community composition. This may lead to the possibility of future interventions in the form of prebiotics, probiotics, or specific metabolites, complementary to gluten avoidance.
In the meantime, increasing disease awareness and rapid diagnosis and treatment continue to be of utmost importance to address the clinical consequences of celiac disease in both children and adults.
Supported by: Grant No. K23AR064318 from NIAMS to Dr. Scher; The Colton Center for Autoimmunity; The Riley Family Foundation.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014; 168:272–278.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Kochhar GS, Singh T, Gill A, Kirby DF. Celiac disease: an internist’s perspective. Cleve Clin J Med 2016; 83:217–227.
- Gutierrez-Achury J, Zhernakova A, Pulit SL, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet 2015; 47:577–578.
- Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med 2010; 42:530–538.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res 2009; 19:2317–2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65.
- Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015; 67:128–139.
- Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3:e2719.
- Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–1463.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn‘s disease. Cell Host Microbe 2014; 15:382–392.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12:497–506.
- Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79:5472–5479.
- Wacklin P, Kaukinen K, Tuovinen E, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19:934–941.
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62:264–269.
- Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11:219.
- Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10:175.
- Stepankova R, Tlaskalova-Hogenova H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scand J Gastroenterol 1996; 31:551–557.
- Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol 2011; 187:4338–4346.
- Galipeau HJ, Verdu EF. Gut microbes and adverse food reactions: focus on gluten related disorders. Gut Microbes 2014; 5:594–605.
- Guandalini S, Assiri A. Celiac disease: a review. JAMA Pediatr 2014; 168:272–278.
- Green PH, Cellier C. Celiac disease. N Engl J Med 2007; 357:1731–1743.
- Kochhar GS, Singh T, Gill A, Kirby DF. Celiac disease: an internist’s perspective. Cleve Clin J Med 2016; 83:217–227.
- Gutierrez-Achury J, Zhernakova A, Pulit SL, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet 2015; 47:577–578.
- Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med 2010; 42:530–538.
- Norris JM, Barriga K, Hoffenberg EJ, et al. Risk of celiac disease autoimmunity and timing of gluten introduction in the diet of infants at increased risk of disease. JAMA 2005; 293:2343–2351.
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007; 449:804–810.
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, et al. The NIH Human Microbiome Project. Genome Res 2009; 19:2317–2323.
- Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65.
- Scher JU, Sczesnak A, Longman RS, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013; 2:e01202.
- Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol 2015; 67:128–139.
- Gao Z, Tseng CH, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS One 2008; 3:e2719.
- Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155:1451–1463.
- Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn‘s disease. Cell Host Microbe 2014; 15:382–392.
- Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol 2015; 12:497–506.
- Sanchez E, Donat E, Ribes-Koninckx C, Fernandez-Murga ML, Sanz Y. Duodenal-mucosal bacteria associated with celiac disease in children. Appl Environ Microbiol 2013; 79:5472–5479.
- Wacklin P, Kaukinen K, Tuovinen E, et al. The duodenal microbiota composition of adult celiac disease patients is associated with the clinical manifestation of the disease. Inflamm Bowel Dis 2013; 19:934–941.
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol 2009; 62:264–269.
- Di Cagno R, De Angelis M, De Pasquale I, et al. Duodenal and faecal microbiota of celiac children: molecular, phenotype and metabolome characterization. BMC Microbiol 2011; 11:219.
- Schippa S, Iebba V, Barbato M, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC Microbiol 2010; 10:175.
- Stepankova R, Tlaskalova-Hogenova H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scand J Gastroenterol 1996; 31:551–557.
- Galipeau HJ, Rulli NE, Jury J, et al. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol 2011; 187:4338–4346.
- Galipeau HJ, Verdu EF. Gut microbes and adverse food reactions: focus on gluten related disorders. Gut Microbes 2014; 5:594–605.