User login
Product Update: Hologic Aptima HSV Assay, Cianna SAVI SCOUT, Olympus Hystero-Resectoscope, and Clarius Ultrasound Scanners
DIAGNOSE HERPES SIMPLEX VIRUS

According to the CDC, infections with HSV-2 affect more than 24 million Americans. Patients with HSV-2 strain are at increased risk for contracting and transmitting HIV. Pregnant women infected with HSV-2 are at risk of transmitting the virus to their babies, with increased risk for neurologic complications in the child.
FOR MORE INFORMATION, VISIT: http://www.hologic.com/search/site/aptima%20hsv
PRECISELY TARGET TISSUE DURING LUMPECTOMY OR BIOPSY
Cianna Medical reported recent data showing that, when compared with wire localization, the SCOUT reduces breast surgery operating room (OR) delay times by 72.5%, resulted in an average 29- minute reduction in OR waiting time, and significantly improved workflow efficiency.
FOR MORE INFORMATION, VISIT: https://www.ciannamedical.com/savi-scout/
PLASMA HYSTEROSCOPIC RESECTION

During gynecologic procedures, the Olympus 8.5-mm hystero-resectoscope uses a combination of radio frequency, energy, and saline to create plasma, an electrically conductive gas cloud of vapor and charged particles. Due to its conductivity, plasma allows energy to cross into targeted tissue at lower energy levels than with more traditional approaches. This effect leads to lower operating temperatures and therefore less thermal spread.
FOR MORE INFORMATION, VISIT: http://olympusmedical.com.sg
APP-BASED HANDHELD ULTRASOUND

High-resolution images can be saved, reviewed, and managed on the secure Clarius Cloud. Built with a durable magnesium shell, each device has an IPX7 immersion rating so it can be sterilized. Power is obtained from a rechargeable battery that will last for more than 45 minutes of scanning; 2 batteries come with each Clarius device.
FOR MORE INFORMATION, VISIT: https://www.clarius.me/
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
DIAGNOSE HERPES SIMPLEX VIRUS
According to the CDC, infections with HSV-2 affect more than 24 million Americans. Patients with HSV-2 strain are at increased risk for contracting and transmitting HIV. Pregnant women infected with HSV-2 are at risk of transmitting the virus to their babies, with increased risk for neurologic complications in the child.
FOR MORE INFORMATION, VISIT: http://www.hologic.com/search/site/aptima%20hsv
PRECISELY TARGET TISSUE DURING LUMPECTOMY OR BIOPSY
Cianna Medical reported recent data showing that, when compared with wire localization, the SCOUT reduces breast surgery operating room (OR) delay times by 72.5%, resulted in an average 29- minute reduction in OR waiting time, and significantly improved workflow efficiency.
FOR MORE INFORMATION, VISIT: https://www.ciannamedical.com/savi-scout/
PLASMA HYSTEROSCOPIC RESECTION
During gynecologic procedures, the Olympus 8.5-mm hystero-resectoscope uses a combination of radio frequency, energy, and saline to create plasma, an electrically conductive gas cloud of vapor and charged particles. Due to its conductivity, plasma allows energy to cross into targeted tissue at lower energy levels than with more traditional approaches. This effect leads to lower operating temperatures and therefore less thermal spread.
FOR MORE INFORMATION, VISIT: http://olympusmedical.com.sg
APP-BASED HANDHELD ULTRASOUND
High-resolution images can be saved, reviewed, and managed on the secure Clarius Cloud. Built with a durable magnesium shell, each device has an IPX7 immersion rating so it can be sterilized. Power is obtained from a rechargeable battery that will last for more than 45 minutes of scanning; 2 batteries come with each Clarius device.
FOR MORE INFORMATION, VISIT: https://www.clarius.me/
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
DIAGNOSE HERPES SIMPLEX VIRUS
According to the CDC, infections with HSV-2 affect more than 24 million Americans. Patients with HSV-2 strain are at increased risk for contracting and transmitting HIV. Pregnant women infected with HSV-2 are at risk of transmitting the virus to their babies, with increased risk for neurologic complications in the child.
FOR MORE INFORMATION, VISIT: http://www.hologic.com/search/site/aptima%20hsv
PRECISELY TARGET TISSUE DURING LUMPECTOMY OR BIOPSY
Cianna Medical reported recent data showing that, when compared with wire localization, the SCOUT reduces breast surgery operating room (OR) delay times by 72.5%, resulted in an average 29- minute reduction in OR waiting time, and significantly improved workflow efficiency.
FOR MORE INFORMATION, VISIT: https://www.ciannamedical.com/savi-scout/
PLASMA HYSTEROSCOPIC RESECTION
During gynecologic procedures, the Olympus 8.5-mm hystero-resectoscope uses a combination of radio frequency, energy, and saline to create plasma, an electrically conductive gas cloud of vapor and charged particles. Due to its conductivity, plasma allows energy to cross into targeted tissue at lower energy levels than with more traditional approaches. This effect leads to lower operating temperatures and therefore less thermal spread.
FOR MORE INFORMATION, VISIT: http://olympusmedical.com.sg
APP-BASED HANDHELD ULTRASOUND
High-resolution images can be saved, reviewed, and managed on the secure Clarius Cloud. Built with a durable magnesium shell, each device has an IPX7 immersion rating so it can be sterilized. Power is obtained from a rechargeable battery that will last for more than 45 minutes of scanning; 2 batteries come with each Clarius device.
FOR MORE INFORMATION, VISIT: https://www.clarius.me/
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Comparison of Methods to Define High Use of Inpatient Services Using Population-Based Data
As healthcare system use and costs continue to rise, increased importance has been placed on identifying the small subgroup of patients that drive this trend.1 It is estimated that 5% of healthcare users account for over 60% of healthcare spending.2-6 Furthermore, care for these “high users” is expensive due to an over-reliance on inpatient services. Approximately 40% of all health spending is for inpatient care, the largest single category of health spending, which is similarly skewed toward high users.1,3,5 Improving our understanding of this population may provide an opportunity to direct improvement efforts to a select group of patients with a potentially high benefit, as well as move care away from the costly inpatient setting.
However, the development of effective interventions to improve patient experience and outcomes while decreasing costs (referred to as the “Triple Aim” by the Institute for Health Improvement) for high users of inpatient services hinges on the methodology used to identify this high-risk population.7 There is substantial variability in definitions of high users; the most common definitions are based on the number of hospital encounters, days spent in the hospital, and hospital costs.8-15 Definitions have intrinsic differences in their implications around appropriateness, efficiency, and financial sustainability of inpatient resource use. Though the constructs underlying these definitions are highly variable, direct comparisons of differences in patient capture are limited.
A recent study from a single US center explored the clinical characteristics of hospital patients based on definitions of use vs cost and observed important differences in patients’ profiles and outcomes.12 While this suggests that the choice of definition may have major implications for whom to target (and the efficacy of any proposed interventions), this concept has not been explored at the population level. Therefore, we used population-based administrative data from a single-payer healthcare system to compare 3 common definitions of high inpatient service use and their influence on patient capture, health outcomes, and inpatient system burden.
METHODS
Data Sources and Study Population
We conducted a retrospective population-based study using administrative and clinical data for the province of Alberta, including the discharge abstracts database, physician claims, ambulatory care records, population health registry file, and aggregated data from the Canadian census.16 We identified all adults who had 1 or more hospitalizations with a discharge date between April 1, 2012, and March 31, 2013, though the admission date could be prior to April 1, 2012.
Definition of High-Inpatient Use
High-inpatient use was defined using 3 metrics: number of inpatient episodes, length of stay, and cost. As in prior studies, for each definition, individuals in the upper5th percentile of the relevant distribution were designated “high users,”2,15 while patients in the lower 95th percentile were considered “nonhigh users.” Patients could be defined as a high user in more than 1 definition.
Patients with 3 or more hospital episodes were defined as high users for the “number of inpatient episodes” definition. A hospital episode of care was defined as an event that resulted in discharge (or death) from an inpatient facility. If an individual was admitted to a hospital and transferred to another facility within 1 day of discharge, the hospitalizations were considered part of the same episode of care.
The “length of stay” definition refers to the cumulative number of days spent in an inpatient facility for all eligible episodes of care. Patients with 56 or more days in hospital during the study period were considered high users. Day of admission and discharge were considered full inpatient days, regardless of the time of admission and discharge.
The “cost” definition considered the cumulative estimated cost of every eligible episode of care. We estimated costs for each hospitalization using resource intensity weights (RIW). This is a relative weighted value for the average inpatient case after taking factors such as age, comorbidity, and procedures into account. The RIW for each episode was multiplied by the national average inpatient cost.17 Based on this definition, patients with a cumulative hospital cost of ≥ $63,597 were deemed high users. All costs were calculated in Canadian Dollars (CAD, $) and adjusted to 2013 dollars based on Statistics Canada’s Consumer Price Index.18
Demographic, Clinical, and Encounter Characteristics
Individual characteristics were measured using a combination of provincial administrative data sources. All measures were recorded as of the admission date of the first eligible hospitalization. Demographic characteristics included age, sex, First Nations status, urban/rural status (based on the individual’s residential postal code), and median neighborhood income quintile. Clinical characteristics included 28 comorbid conditions defined based on separate validated International Statistical Classification of Disease and Health Related Problems, Tenth Revision, Canada (ICD-10-CA) coding algorithms reported individually and cumulatively (categorized as 0, 1, 2–3, and 4+).19 Primary care attachment was defined as the percentage of all outpatient primary care visits made to a single practitioner in the 2-year period prior to their first hospitalization (among those with ≥3 visits). Attachment was categorized as 75%-100% (good attachment), 50%-74% (moderate attachment), or <50% (low attachment).20,21
We also identified hospital encounter-level characteristics. These included the most responsible diagnosis, admission category (elective or urgent/emergent), and discharge disposition for each hospital episode. Reported health outcomes included the proportion of patients with in-hospital mortality and those with at least one 30-day, all-cause readmission to hospital.
Analysis
Patient characteristics were described using proportions and means (standard deviation) as appropriate for high users and nonhigh users within and across each definition. Encounter characteristics were also described and stratified by age category (18-64 or 65+ years). Comparison of patient capture was then analyzed among patients who were high use by at least 1 definition. The overlap and agreement of the 3 definitions were compared using a Venn diagram and kappa statistic. The 10 most responsible diagnoses (based on frequency) were also compared across definitions and stratified by age.
Finally, the percentage of system burden accounted for by each measure was calculated as the amount used by high users divided by the total amount used by the entire study population (x 100). To assess the potential modifying effect of age, results were stratified by age category for each definition.
All analyses were conducted using Stata 11.2 (StataCorp LP, College Station, TX).22 The Conjoint Health Research Ethics Board of the University of Calgary approved this study and granted waiver of patient consent. This manuscript is written in accordance with reporting guidelines for studies conducted using observational routinely collected health data (RECORD statement).23
RESULTS
Comparison of Patient and Encounter-level Characterist
")
ics
")
A total of 219,106 adults had 283,204 inpatient episodes of care within the study timeframe. There were 12,707 (5.8%), 11,095 (5.1%), and 10,956 (5.0%) patients defined as high users based on number of inpatient episodes, length of stay, and cost, respectively (supplementary Figure 1). Regardless of definition, when compared to their non–high use counterparts, patients classified as high use were more likely to be male, older, in a lower median neighborhood income quintile, and have a higher level of comorbidity. Comparing across definitions of high use, those defined by number of inpatient episodes were more likely to be younger, live in rural areas, have better primary care attachment, and have fewer comorbidities, compared to the other definitions. High users by length of stay were more likely to be older and had a higher proportion of mental health–related comorbidities, including dementia and depression, as compared with the other definitions. Results were largely similar for those defined by cost (Table 1).
Encounter-level analyses
Comparison of Patient Capture and Inpatient Burden

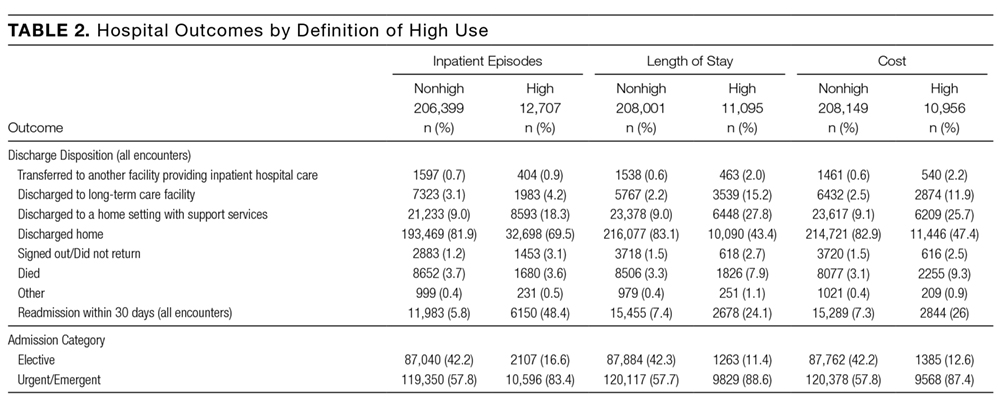
Of the 22,691 individuals who were defined as high use by at least 1 definition, 2,331 (10.3%) were consistently high use across all 3 definitions (kappa = 0.38; Figure 1). Of the 13,682 individuals classified as high use by at least 1 of length of stay or cost, 8369 (61.2%) were defined as high use by both definitions (kappa = 0.75). However, of the 12,707 defined as high use by the number of inpatient episodes, only 3698 (29.1%) were also defined as high use by another definition. Exploration of the most responsible diagnoses across definitions showed that congestive heart failure (2.8%-3.5%), chronic obstructive pulmonary disease (1.6%-3.2%), and dementia (0.6%-2.2%) were the most frequent. Acute medical conditions (eg, pneumonia [1.8%] or gastroenteritis [0.7%]) that may result in multiple shorter hospitalizations were observed at higher frequencies among high users defined by inpatient episodes, while conditions commonly requiring rehabilitation (eg, fracture [1.8%] and stroke [1.7%]) were more common among high users defined by length of stay and cost (supplementary Table 2). Stratification by age showed marked differences in the diagnoses across high-use definitions. Among hi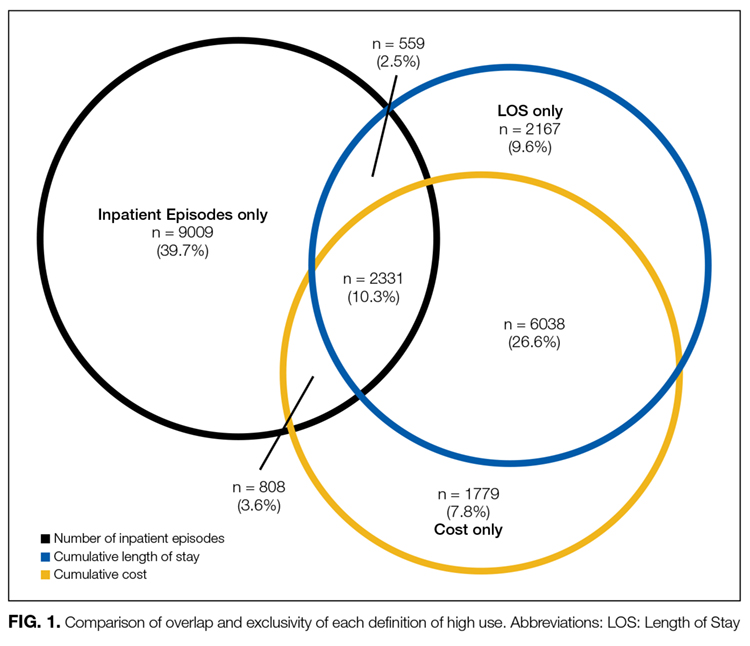
When assessing inpatient system burden, high users by number of inpatient episodes accounted for 47,044 (16.6%) of the 283,204 episodes. High users defined by length of stay accounted for 1,286,539 (46.4%) days of 2,773,561 total days, while high users defined by cost accumulated $1.4 billion (38.9%) of the estimated $3.7 billion in inpatient expenditures. High users defined by cost and length of stay each accounted for comparatively few episode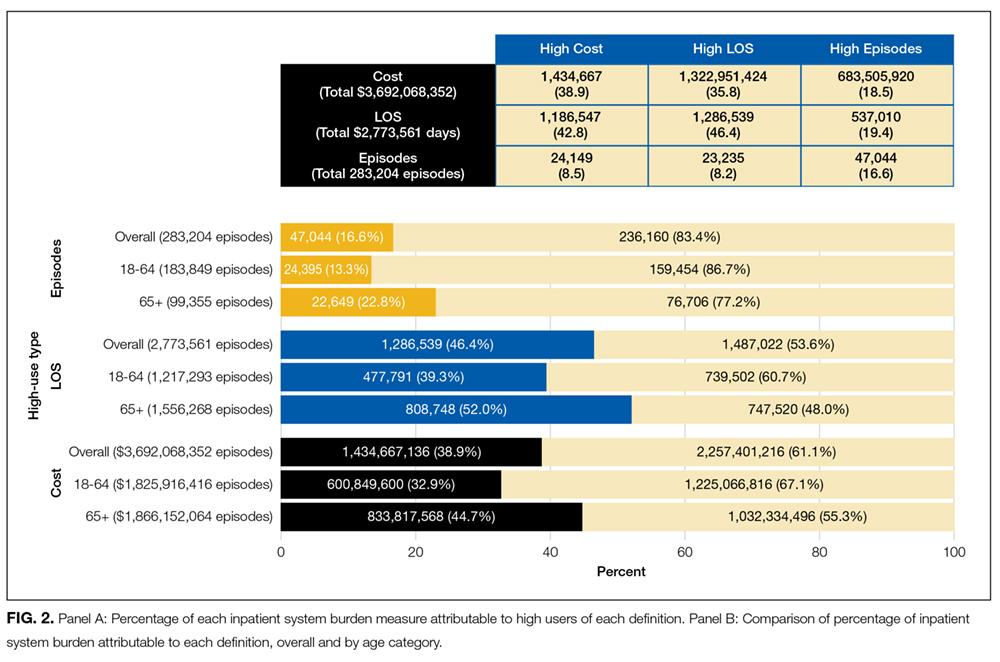
DISCUSSION
Using a large population-based cohort of all adults with at least 1 hospitalization in the province of Alberta, Canada, within a 12-month period, we compared 3 commonly used definitions of high inpatient use. The choice of definition had a substantial influence on the types of patients categorized as high use, as well as the proportion of total inpatient utilization that was associated with high users. The definition based on number of inpatient episodes captured a distinct population of high users, while the populations identified using cumulative length of stay or cost were similar.
Differences within and between definitions were especially apparent in age-stratified analyses: Greater length of stay or higher cost among patients aged 18-64 years identifies a large proportion of psychological conditions, while a greater number of inpatient episodes identifies acute conditions and childbirth or labor-related complications. Conversely, definitions based on length of stay and cost in the elderly (65+) identified groups with chronic conditions that result in progressive functional decline (often requiring increasing supportive services upon discharge) or conditions that require significant rehabilitation prior to discharge. Regarding inpatient system burden, high users defined by number of inpatient episodes accounted for a small proportion of total inpatient episodes, while high users defined by length of stay and cost accounted for nearly half of the accumulated hospital days and cost for each. These findings highlight the need for careful consideration of how high use is defined when studying high-user populations and implications for targeting subpopulations for intervention.
Our results add to those from previous studies. A US-based, single-center study of 2566 individuals compared definitions of high inpatient use based on cost and frequency of admission and found that patients defined by cost were predominantly hospitalized for surgical conditions, while those fulfilling the episode-based definition were often hospitalized for medical conditions.12 The most responsible diagnoses for patient hospitalizations in our study reflect this. We extended this comparison to consider the impact of age on outcomes and inpatient system burden and found that older age was also linked to poorer outcomes and increased burden. We also considered a third definition (cumulative length of stay), which provided another opportunity for comparison. The presence of chronic conditions requiring rehabilitation and possible alternate level of care days within our cohort highlights the utility of this length of stay-based approach when considering definitions. Although there were similarities between patients defined by length of stay and cost, partly due to cost being largely a function of length of stay, there were also important differences in their patient profiles. Those defined by cost tended to have conditions requiring surgical procedures not requiring extended in-hospital rehabilitation. Furthermore, the higher proportion of in-hospital mortality among those defined by cost may also reflect the fact that patients tend to accrue the majority of their healthcare expenditures during the final 120 days of life.24
Each definition of high use identified complex patients; however, the differences between the various types of high users identified by these definitions suggest that they are not interchangeable. Arguably, selection of the most appropriate definition should depend on the objective of measuring high users, particularly if an intervention is planned. Interventions for high users are complex, requiring both medical and nonmedical components. The current literature in this area has often focused on case management programs, collaboration with community-based social support programs, and improving coordination and transitions of care.25-27 While many of these approaches require considerable involvement outside of the inpatient setting, these interventions can be informed by defining who high users of inpatient services are. Our findings show several possible subgroups of high users, which could be targeted for intervention. For example, an inpatient episode-based definition, which identifies patients with frequent encounters for acute conditions (eg, pneumonia and urinary tract infections), would be informative if an intervention targeted reductions in inpatient use and readmission rates. Alternatively, an intervention designed to improve community-based mental health programs would best be informed by a definition based on length of stay in which high users with underlying mental health conditions were prevalent. Such interventions are rarely mutually exclusive and require multiple perspectives to inform their objectives. A well-designed intervention will not only address the medical characteristics of high users but also the social determinants of health that place patients at risk of high inpatient use.
Our study should be interpreted in light of its limitations. First, measures of disease severity were not available to further characterize similarities and differences across high-use groups. Furthermore, we were unable to account for other social determinants of health that may be relevant to inpatient system usage. Second, direct cost of hospitalizations was estimated based on RIW and is thus reflective of expected rather than actual costs. However, this will have minimal impact on capture, as patients defined by this metric require substantial costs to be included in the top fifth percentile, and thus deviations in individual hospitalization costs will have minimal influence on the cumulative cost. Finally, while inpatient spending makes up a large proportion of healthcare spending, there is likely a number of different high-use profiles found outside of the acute care setting. Despite these limitations, our study includes several key strengths. The use of population-level data allows for analysis that is robust and more generalizable than studies from single centers. Additionally, the comparison of 3 independent definitions allows for a greater comparison of the nuances of each definition. Our study also considers the important impact of age as an effect modifier of inpatient use in the general population and identifies distinct patient profiles that exist across each definition.
CONCLUSIONS
Definitions of high use of inpatient services based on number of inpatient episodes, days spent in hospital, and total hospital costs identify patient populations with different characteristics and differ substantially in their impact on health outcomes and inpatient burden. These results highlight the need for careful consideration of the context of the study or intervention and the implications of selecting a specific definition of high inpatient use at study conception. Ultimately, the performance of an intervention in high-use populations is likely to be conditional on the fit of the patient population generated by the chosen definition of high inpatient use to the objectives of a study.
Acknowledgments
This study is based in part on data provided by Alberta Health and Alberta Health Services. The interpretation and conclusions are those of the researchers and do not represent the views of the Government of Alberta. Neither the Government of Alberta nor Alberta Health express any opinion in relation to this study.
Disclosure
Dr. Hemmelgarn is supported by the Roy and Vi Baay Chair in Kidney Research. Dr. Manns is supported by the Svare Professorship in Health Economics and by a Health Scholar Award by Alberta Innovates Health Solutions (AIHS). Dr. Tonelli is supported by the David Freeze chair in Health Services Research. The Interdisciplinary Chronic Disease Collaboration is funded by AIHS—Collaborative Research and Innovation Opportunity (CRIO) Team Grants Program.
1. National Health Expenditure Trends, 1975 to 2015. Canadian Institute for Health Information. 2015. https://secure.cihi.ca/free_products/nhex_trends_narrative_report_2015_en.pdf. Accessed on June 23, 2016.
2. Berk ML, Monheit AC. The concentration of health care expenditures, revisited. Health Aff (Millwood). 2001;20:9-18. PubMed
3. Wodchis WP, Austin PC, Henry DA. A 3-year study of high-cost users of health care. CMAJ. 2016;188(3):182-188. PubMed
4. Forget EL, Roos LL, Deber RB, Wald R. Variations in Lifetime Healthcare Costs across a Population. Healthc Policy. 2008;4:e148-e167. PubMed
5. Joynt KE, Gawande AA, Orav EJ, Jha AK. Contribution of preventable acute care spending to total spending for high-cost Medicare patients. JAMA. 2013;309:2572-2578. PubMed
6. Riley GF. Long-term trends in the concentration of Medicare spending. Health Aff (Millwood). 2007;26:808-816. PubMed
7. IHI Triple Aim Initiative. Institute for Healthcare Improvement. 2015. http://www.ihi.org/engage/initiatives/TripleAim/Pages/default.aspx. Accessed on June 17, 2016.
8. Johansen H, Nair C, Bond J. Who goes to the hospital? An investigation of high users of hospital days. Health Reports. 1994;6(2):253-277. PubMed
9. Conwell LJ, Cohen JW. Characteristics of persons with high medical expenditures in the US civilian noninstitutionalized population. MEPS Statistical Brief# 73. 2002.
10. Lemstra M, Mackenbach J, Neudorf C, Nannapaneni U. High health care utilization and costs associated with lower socio-economic status: Results from a linked dataset. CJPH. 2009;100(3):180-183. PubMed
11. Macnee CL, McCabe S, Clarke PN, Fiske M, Campbell S. Typology of high users of health services among a rural medicaid population. Pub Health Nurs. 2009;26(5):396-404. PubMed
12. Nguyen OK, Tang N, Hillman JM, Gonzales R. What’s cost got to do with it? Association between hospital costs and frequency of admissions among “high users” of hospital care. J. Hosp Med. 2013;8(12):665-671. PubMed
13. Rosella LC, Fitzpatrick T, Wodchis WP, Calzavara A, Manson H, Goel V. High-cost health care users in Ontario, Canada: Demographic, socio-economic, and health status characteristics. BMC Health Serv Res. 2014;14(1):532. PubMed
14. Cohen SB. The Concentration of Health Care Expenditures and Related Expenses for Costly Medical Conditions, 2009. Agency for Healthcare Research and Quality Statistical Brief #359; 2012.
15. Ronksley PE, McKay JA, Kobewka DM, Mulpuru S, Forster AJ. Patterns of health care use in a high-cost inpatient population in Ottawa, Ontario: A retrospective observational study. CMAJ Open. 2015; 3:E111-E118. PubMed
16. Hemmelgarn BR, Clement F, Manns BJ, et al. Overview of the Alberta Kidney Disease Network. BMC Nephrol. 2009;10:30. PubMed
17. DAD Resource Intensity Weights and Expected Length of Stay. Canadian Institute for Health Information. 2016. https://www.cihi.ca/en/data-and-standards/standards/case-mix/resource-indicators-dad-resource-intensity-weights-and. Accessed on June 24, 2016.
18. Statistics Canada. The Canadian Consumer Price Index Reference Paper, Statistics Canada Catalogue no. 62-553-X.
19. Tonelli M, Wiebe N, Fortin M, et al. Methods for identifying 30 chronic conditions: Application to administrative data. BMC Med Inform Decis Mak. 2015;17:15(1):1. PubMed
20. Jaakkimainen RL, Klein-Geltink J, Guttmann A, Barnsley J, Jagorski B, Kopp A. Indicators of primary care based on administrative data. In Primary Care in Ontario: ICES Atlas. Toronto, Ontario: Institute for Clinical Evaluative Sciences; 2006.
21. Jee SH, Cabana MD. Indices for continuity of care: A systematic review of the literature. Med Care Res Rev. 2006;63:158-188. PubMed
22. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP. 2009.
23. Benchimol EI, Smeeth L, Guttmann A, et al. The REporting of studies Conducted using Observational Routinely-collected health Data (RECORD) statement. PLoS Med. 2015;12(10):e1001885. PubMed
24. Tanuseputro P, Wodchis WP, Fowler R, et al. The health care cost of dying: A population-based retrospective cohort study of the last year of life in ontario, canada. PLoS One. 2015;10(3):e0121759. PubMed
25. Hong CS, Siegel AL, Ferris TG. Caring for high-need, high-cost patients: What makes for a successful care management program? Issue Brief (Commonw Fund). 2014;19:1-19. PubMed
26. Birnbaum M, Halper DE. Rethinking service delivery for high-cost Medicaid patients. Medicaid Institute. 2009. http://shnny.org/research/rethinking-service-delivery-for-high-cost-medicaid-patients/. Accessed on Jan 11, 2017.
27. Pan-Canadian forum on high users of health care. Canadian Institute for Health Information. 2014. https://secure.cihi.ca/free_products/highusers_summary_report_revised_EN_web.pdf. Accessed on Jan 11, 2017.
As healthcare system use and costs continue to rise, increased importance has been placed on identifying the small subgroup of patients that drive this trend.1 It is estimated that 5% of healthcare users account for over 60% of healthcare spending.2-6 Furthermore, care for these “high users” is expensive due to an over-reliance on inpatient services. Approximately 40% of all health spending is for inpatient care, the largest single category of health spending, which is similarly skewed toward high users.1,3,5 Improving our understanding of this population may provide an opportunity to direct improvement efforts to a select group of patients with a potentially high benefit, as well as move care away from the costly inpatient setting.
However, the development of effective interventions to improve patient experience and outcomes while decreasing costs (referred to as the “Triple Aim” by the Institute for Health Improvement) for high users of inpatient services hinges on the methodology used to identify this high-risk population.7 There is substantial variability in definitions of high users; the most common definitions are based on the number of hospital encounters, days spent in the hospital, and hospital costs.8-15 Definitions have intrinsic differences in their implications around appropriateness, efficiency, and financial sustainability of inpatient resource use. Though the constructs underlying these definitions are highly variable, direct comparisons of differences in patient capture are limited.
A recent study from a single US center explored the clinical characteristics of hospital patients based on definitions of use vs cost and observed important differences in patients’ profiles and outcomes.12 While this suggests that the choice of definition may have major implications for whom to target (and the efficacy of any proposed interventions), this concept has not been explored at the population level. Therefore, we used population-based administrative data from a single-payer healthcare system to compare 3 common definitions of high inpatient service use and their influence on patient capture, health outcomes, and inpatient system burden.
METHODS
Data Sources and Study Population
We conducted a retrospective population-based study using administrative and clinical data for the province of Alberta, including the discharge abstracts database, physician claims, ambulatory care records, population health registry file, and aggregated data from the Canadian census.16 We identified all adults who had 1 or more hospitalizations with a discharge date between April 1, 2012, and March 31, 2013, though the admission date could be prior to April 1, 2012.
Definition of High-Inpatient Use
High-inpatient use was defined using 3 metrics: number of inpatient episodes, length of stay, and cost. As in prior studies, for each definition, individuals in the upper5th percentile of the relevant distribution were designated “high users,”2,15 while patients in the lower 95th percentile were considered “nonhigh users.” Patients could be defined as a high user in more than 1 definition.
Patients with 3 or more hospital episodes were defined as high users for the “number of inpatient episodes” definition. A hospital episode of care was defined as an event that resulted in discharge (or death) from an inpatient facility. If an individual was admitted to a hospital and transferred to another facility within 1 day of discharge, the hospitalizations were considered part of the same episode of care.
The “length of stay” definition refers to the cumulative number of days spent in an inpatient facility for all eligible episodes of care. Patients with 56 or more days in hospital during the study period were considered high users. Day of admission and discharge were considered full inpatient days, regardless of the time of admission and discharge.
The “cost” definition considered the cumulative estimated cost of every eligible episode of care. We estimated costs for each hospitalization using resource intensity weights (RIW). This is a relative weighted value for the average inpatient case after taking factors such as age, comorbidity, and procedures into account. The RIW for each episode was multiplied by the national average inpatient cost.17 Based on this definition, patients with a cumulative hospital cost of ≥ $63,597 were deemed high users. All costs were calculated in Canadian Dollars (CAD, $) and adjusted to 2013 dollars based on Statistics Canada’s Consumer Price Index.18
Demographic, Clinical, and Encounter Characteristics
Individual characteristics were measured using a combination of provincial administrative data sources. All measures were recorded as of the admission date of the first eligible hospitalization. Demographic characteristics included age, sex, First Nations status, urban/rural status (based on the individual’s residential postal code), and median neighborhood income quintile. Clinical characteristics included 28 comorbid conditions defined based on separate validated International Statistical Classification of Disease and Health Related Problems, Tenth Revision, Canada (ICD-10-CA) coding algorithms reported individually and cumulatively (categorized as 0, 1, 2–3, and 4+).19 Primary care attachment was defined as the percentage of all outpatient primary care visits made to a single practitioner in the 2-year period prior to their first hospitalization (among those with ≥3 visits). Attachment was categorized as 75%-100% (good attachment), 50%-74% (moderate attachment), or <50% (low attachment).20,21
We also identified hospital encounter-level characteristics. These included the most responsible diagnosis, admission category (elective or urgent/emergent), and discharge disposition for each hospital episode. Reported health outcomes included the proportion of patients with in-hospital mortality and those with at least one 30-day, all-cause readmission to hospital.
Analysis
Patient characteristics were described using proportions and means (standard deviation) as appropriate for high users and nonhigh users within and across each definition. Encounter characteristics were also described and stratified by age category (18-64 or 65+ years). Comparison of patient capture was then analyzed among patients who were high use by at least 1 definition. The overlap and agreement of the 3 definitions were compared using a Venn diagram and kappa statistic. The 10 most responsible diagnoses (based on frequency) were also compared across definitions and stratified by age.
Finally, the percentage of system burden accounted for by each measure was calculated as the amount used by high users divided by the total amount used by the entire study population (x 100). To assess the potential modifying effect of age, results were stratified by age category for each definition.
All analyses were conducted using Stata 11.2 (StataCorp LP, College Station, TX).22 The Conjoint Health Research Ethics Board of the University of Calgary approved this study and granted waiver of patient consent. This manuscript is written in accordance with reporting guidelines for studies conducted using observational routinely collected health data (RECORD statement).23
RESULTS
Comparison of Patient and Encounter-level Characterist
ics
A total of 219,106 adults had 283,204 inpatient episodes of care within the study timeframe. There were 12,707 (5.8%), 11,095 (5.1%), and 10,956 (5.0%) patients defined as high users based on number of inpatient episodes, length of stay, and cost, respectively (supplementary Figure 1). Regardless of definition, when compared to their non–high use counterparts, patients classified as high use were more likely to be male, older, in a lower median neighborhood income quintile, and have a higher level of comorbidity. Comparing across definitions of high use, those defined by number of inpatient episodes were more likely to be younger, live in rural areas, have better primary care attachment, and have fewer comorbidities, compared to the other definitions. High users by length of stay were more likely to be older and had a higher proportion of mental health–related comorbidities, including dementia and depression, as compared with the other definitions. Results were largely similar for those defined by cost (Table 1).
Encounter-level analyses
Comparison of Patient Capture and Inpatient Burden
Of the 22,691 individuals who were defined as high use by at least 1 definition, 2,331 (10.3%) were consistently high use across all 3 definitions (kappa = 0.38; Figure 1). Of the 13,682 individuals classified as high use by at least 1 of length of stay or cost, 8369 (61.2%) were defined as high use by both definitions (kappa = 0.75). However, of the 12,707 defined as high use by the number of inpatient episodes, only 3698 (29.1%) were also defined as high use by another definition. Exploration of the most responsible diagnoses across definitions showed that congestive heart failure (2.8%-3.5%), chronic obstructive pulmonary disease (1.6%-3.2%), and dementia (0.6%-2.2%) were the most frequent. Acute medical conditions (eg, pneumonia [1.8%] or gastroenteritis [0.7%]) that may result in multiple shorter hospitalizations were observed at higher frequencies among high users defined by inpatient episodes, while conditions commonly requiring rehabilitation (eg, fracture [1.8%] and stroke [1.7%]) were more common among high users defined by length of stay and cost (supplementary Table 2). Stratification by age showed marked differences in the diagnoses across high-use definitions. Among hi
When assessing inpatient system burden, high users by number of inpatient episodes accounted for 47,044 (16.6%) of the 283,204 episodes. High users defined by length of stay accounted for 1,286,539 (46.4%) days of 2,773,561 total days, while high users defined by cost accumulated $1.4 billion (38.9%) of the estimated $3.7 billion in inpatient expenditures. High users defined by cost and length of stay each accounted for comparatively few episode
DISCUSSION
Using a large population-based cohort of all adults with at least 1 hospitalization in the province of Alberta, Canada, within a 12-month period, we compared 3 commonly used definitions of high inpatient use. The choice of definition had a substantial influence on the types of patients categorized as high use, as well as the proportion of total inpatient utilization that was associated with high users. The definition based on number of inpatient episodes captured a distinct population of high users, while the populations identified using cumulative length of stay or cost were similar.
Differences within and between definitions were especially apparent in age-stratified analyses: Greater length of stay or higher cost among patients aged 18-64 years identifies a large proportion of psychological conditions, while a greater number of inpatient episodes identifies acute conditions and childbirth or labor-related complications. Conversely, definitions based on length of stay and cost in the elderly (65+) identified groups with chronic conditions that result in progressive functional decline (often requiring increasing supportive services upon discharge) or conditions that require significant rehabilitation prior to discharge. Regarding inpatient system burden, high users defined by number of inpatient episodes accounted for a small proportion of total inpatient episodes, while high users defined by length of stay and cost accounted for nearly half of the accumulated hospital days and cost for each. These findings highlight the need for careful consideration of how high use is defined when studying high-user populations and implications for targeting subpopulations for intervention.
Our results add to those from previous studies. A US-based, single-center study of 2566 individuals compared definitions of high inpatient use based on cost and frequency of admission and found that patients defined by cost were predominantly hospitalized for surgical conditions, while those fulfilling the episode-based definition were often hospitalized for medical conditions.12 The most responsible diagnoses for patient hospitalizations in our study reflect this. We extended this comparison to consider the impact of age on outcomes and inpatient system burden and found that older age was also linked to poorer outcomes and increased burden. We also considered a third definition (cumulative length of stay), which provided another opportunity for comparison. The presence of chronic conditions requiring rehabilitation and possible alternate level of care days within our cohort highlights the utility of this length of stay-based approach when considering definitions. Although there were similarities between patients defined by length of stay and cost, partly due to cost being largely a function of length of stay, there were also important differences in their patient profiles. Those defined by cost tended to have conditions requiring surgical procedures not requiring extended in-hospital rehabilitation. Furthermore, the higher proportion of in-hospital mortality among those defined by cost may also reflect the fact that patients tend to accrue the majority of their healthcare expenditures during the final 120 days of life.24
Each definition of high use identified complex patients; however, the differences between the various types of high users identified by these definitions suggest that they are not interchangeable. Arguably, selection of the most appropriate definition should depend on the objective of measuring high users, particularly if an intervention is planned. Interventions for high users are complex, requiring both medical and nonmedical components. The current literature in this area has often focused on case management programs, collaboration with community-based social support programs, and improving coordination and transitions of care.25-27 While many of these approaches require considerable involvement outside of the inpatient setting, these interventions can be informed by defining who high users of inpatient services are. Our findings show several possible subgroups of high users, which could be targeted for intervention. For example, an inpatient episode-based definition, which identifies patients with frequent encounters for acute conditions (eg, pneumonia and urinary tract infections), would be informative if an intervention targeted reductions in inpatient use and readmission rates. Alternatively, an intervention designed to improve community-based mental health programs would best be informed by a definition based on length of stay in which high users with underlying mental health conditions were prevalent. Such interventions are rarely mutually exclusive and require multiple perspectives to inform their objectives. A well-designed intervention will not only address the medical characteristics of high users but also the social determinants of health that place patients at risk of high inpatient use.
Our study should be interpreted in light of its limitations. First, measures of disease severity were not available to further characterize similarities and differences across high-use groups. Furthermore, we were unable to account for other social determinants of health that may be relevant to inpatient system usage. Second, direct cost of hospitalizations was estimated based on RIW and is thus reflective of expected rather than actual costs. However, this will have minimal impact on capture, as patients defined by this metric require substantial costs to be included in the top fifth percentile, and thus deviations in individual hospitalization costs will have minimal influence on the cumulative cost. Finally, while inpatient spending makes up a large proportion of healthcare spending, there is likely a number of different high-use profiles found outside of the acute care setting. Despite these limitations, our study includes several key strengths. The use of population-level data allows for analysis that is robust and more generalizable than studies from single centers. Additionally, the comparison of 3 independent definitions allows for a greater comparison of the nuances of each definition. Our study also considers the important impact of age as an effect modifier of inpatient use in the general population and identifies distinct patient profiles that exist across each definition.
CONCLUSIONS
Definitions of high use of inpatient services based on number of inpatient episodes, days spent in hospital, and total hospital costs identify patient populations with different characteristics and differ substantially in their impact on health outcomes and inpatient burden. These results highlight the need for careful consideration of the context of the study or intervention and the implications of selecting a specific definition of high inpatient use at study conception. Ultimately, the performance of an intervention in high-use populations is likely to be conditional on the fit of the patient population generated by the chosen definition of high inpatient use to the objectives of a study.
Acknowledgments
This study is based in part on data provided by Alberta Health and Alberta Health Services. The interpretation and conclusions are those of the researchers and do not represent the views of the Government of Alberta. Neither the Government of Alberta nor Alberta Health express any opinion in relation to this study.
Disclosure
Dr. Hemmelgarn is supported by the Roy and Vi Baay Chair in Kidney Research. Dr. Manns is supported by the Svare Professorship in Health Economics and by a Health Scholar Award by Alberta Innovates Health Solutions (AIHS). Dr. Tonelli is supported by the David Freeze chair in Health Services Research. The Interdisciplinary Chronic Disease Collaboration is funded by AIHS—Collaborative Research and Innovation Opportunity (CRIO) Team Grants Program.
As healthcare system use and costs continue to rise, increased importance has been placed on identifying the small subgroup of patients that drive this trend.1 It is estimated that 5% of healthcare users account for over 60% of healthcare spending.2-6 Furthermore, care for these “high users” is expensive due to an over-reliance on inpatient services. Approximately 40% of all health spending is for inpatient care, the largest single category of health spending, which is similarly skewed toward high users.1,3,5 Improving our understanding of this population may provide an opportunity to direct improvement efforts to a select group of patients with a potentially high benefit, as well as move care away from the costly inpatient setting.
However, the development of effective interventions to improve patient experience and outcomes while decreasing costs (referred to as the “Triple Aim” by the Institute for Health Improvement) for high users of inpatient services hinges on the methodology used to identify this high-risk population.7 There is substantial variability in definitions of high users; the most common definitions are based on the number of hospital encounters, days spent in the hospital, and hospital costs.8-15 Definitions have intrinsic differences in their implications around appropriateness, efficiency, and financial sustainability of inpatient resource use. Though the constructs underlying these definitions are highly variable, direct comparisons of differences in patient capture are limited.
A recent study from a single US center explored the clinical characteristics of hospital patients based on definitions of use vs cost and observed important differences in patients’ profiles and outcomes.12 While this suggests that the choice of definition may have major implications for whom to target (and the efficacy of any proposed interventions), this concept has not been explored at the population level. Therefore, we used population-based administrative data from a single-payer healthcare system to compare 3 common definitions of high inpatient service use and their influence on patient capture, health outcomes, and inpatient system burden.
METHODS
Data Sources and Study Population
We conducted a retrospective population-based study using administrative and clinical data for the province of Alberta, including the discharge abstracts database, physician claims, ambulatory care records, population health registry file, and aggregated data from the Canadian census.16 We identified all adults who had 1 or more hospitalizations with a discharge date between April 1, 2012, and March 31, 2013, though the admission date could be prior to April 1, 2012.
Definition of High-Inpatient Use
High-inpatient use was defined using 3 metrics: number of inpatient episodes, length of stay, and cost. As in prior studies, for each definition, individuals in the upper5th percentile of the relevant distribution were designated “high users,”2,15 while patients in the lower 95th percentile were considered “nonhigh users.” Patients could be defined as a high user in more than 1 definition.
Patients with 3 or more hospital episodes were defined as high users for the “number of inpatient episodes” definition. A hospital episode of care was defined as an event that resulted in discharge (or death) from an inpatient facility. If an individual was admitted to a hospital and transferred to another facility within 1 day of discharge, the hospitalizations were considered part of the same episode of care.
The “length of stay” definition refers to the cumulative number of days spent in an inpatient facility for all eligible episodes of care. Patients with 56 or more days in hospital during the study period were considered high users. Day of admission and discharge were considered full inpatient days, regardless of the time of admission and discharge.
The “cost” definition considered the cumulative estimated cost of every eligible episode of care. We estimated costs for each hospitalization using resource intensity weights (RIW). This is a relative weighted value for the average inpatient case after taking factors such as age, comorbidity, and procedures into account. The RIW for each episode was multiplied by the national average inpatient cost.17 Based on this definition, patients with a cumulative hospital cost of ≥ $63,597 were deemed high users. All costs were calculated in Canadian Dollars (CAD, $) and adjusted to 2013 dollars based on Statistics Canada’s Consumer Price Index.18
Demographic, Clinical, and Encounter Characteristics
Individual characteristics were measured using a combination of provincial administrative data sources. All measures were recorded as of the admission date of the first eligible hospitalization. Demographic characteristics included age, sex, First Nations status, urban/rural status (based on the individual’s residential postal code), and median neighborhood income quintile. Clinical characteristics included 28 comorbid conditions defined based on separate validated International Statistical Classification of Disease and Health Related Problems, Tenth Revision, Canada (ICD-10-CA) coding algorithms reported individually and cumulatively (categorized as 0, 1, 2–3, and 4+).19 Primary care attachment was defined as the percentage of all outpatient primary care visits made to a single practitioner in the 2-year period prior to their first hospitalization (among those with ≥3 visits). Attachment was categorized as 75%-100% (good attachment), 50%-74% (moderate attachment), or <50% (low attachment).20,21
We also identified hospital encounter-level characteristics. These included the most responsible diagnosis, admission category (elective or urgent/emergent), and discharge disposition for each hospital episode. Reported health outcomes included the proportion of patients with in-hospital mortality and those with at least one 30-day, all-cause readmission to hospital.
Analysis
Patient characteristics were described using proportions and means (standard deviation) as appropriate for high users and nonhigh users within and across each definition. Encounter characteristics were also described and stratified by age category (18-64 or 65+ years). Comparison of patient capture was then analyzed among patients who were high use by at least 1 definition. The overlap and agreement of the 3 definitions were compared using a Venn diagram and kappa statistic. The 10 most responsible diagnoses (based on frequency) were also compared across definitions and stratified by age.
Finally, the percentage of system burden accounted for by each measure was calculated as the amount used by high users divided by the total amount used by the entire study population (x 100). To assess the potential modifying effect of age, results were stratified by age category for each definition.
All analyses were conducted using Stata 11.2 (StataCorp LP, College Station, TX).22 The Conjoint Health Research Ethics Board of the University of Calgary approved this study and granted waiver of patient consent. This manuscript is written in accordance with reporting guidelines for studies conducted using observational routinely collected health data (RECORD statement).23
RESULTS
Comparison of Patient and Encounter-level Characterist
ics
A total of 219,106 adults had 283,204 inpatient episodes of care within the study timeframe. There were 12,707 (5.8%), 11,095 (5.1%), and 10,956 (5.0%) patients defined as high users based on number of inpatient episodes, length of stay, and cost, respectively (supplementary Figure 1). Regardless of definition, when compared to their non–high use counterparts, patients classified as high use were more likely to be male, older, in a lower median neighborhood income quintile, and have a higher level of comorbidity. Comparing across definitions of high use, those defined by number of inpatient episodes were more likely to be younger, live in rural areas, have better primary care attachment, and have fewer comorbidities, compared to the other definitions. High users by length of stay were more likely to be older and had a higher proportion of mental health–related comorbidities, including dementia and depression, as compared with the other definitions. Results were largely similar for those defined by cost (Table 1).
Encounter-level analyses
Comparison of Patient Capture and Inpatient Burden
Of the 22,691 individuals who were defined as high use by at least 1 definition, 2,331 (10.3%) were consistently high use across all 3 definitions (kappa = 0.38; Figure 1). Of the 13,682 individuals classified as high use by at least 1 of length of stay or cost, 8369 (61.2%) were defined as high use by both definitions (kappa = 0.75). However, of the 12,707 defined as high use by the number of inpatient episodes, only 3698 (29.1%) were also defined as high use by another definition. Exploration of the most responsible diagnoses across definitions showed that congestive heart failure (2.8%-3.5%), chronic obstructive pulmonary disease (1.6%-3.2%), and dementia (0.6%-2.2%) were the most frequent. Acute medical conditions (eg, pneumonia [1.8%] or gastroenteritis [0.7%]) that may result in multiple shorter hospitalizations were observed at higher frequencies among high users defined by inpatient episodes, while conditions commonly requiring rehabilitation (eg, fracture [1.8%] and stroke [1.7%]) were more common among high users defined by length of stay and cost (supplementary Table 2). Stratification by age showed marked differences in the diagnoses across high-use definitions. Among hi
When assessing inpatient system burden, high users by number of inpatient episodes accounted for 47,044 (16.6%) of the 283,204 episodes. High users defined by length of stay accounted for 1,286,539 (46.4%) days of 2,773,561 total days, while high users defined by cost accumulated $1.4 billion (38.9%) of the estimated $3.7 billion in inpatient expenditures. High users defined by cost and length of stay each accounted for comparatively few episode
DISCUSSION
Using a large population-based cohort of all adults with at least 1 hospitalization in the province of Alberta, Canada, within a 12-month period, we compared 3 commonly used definitions of high inpatient use. The choice of definition had a substantial influence on the types of patients categorized as high use, as well as the proportion of total inpatient utilization that was associated with high users. The definition based on number of inpatient episodes captured a distinct population of high users, while the populations identified using cumulative length of stay or cost were similar.
Differences within and between definitions were especially apparent in age-stratified analyses: Greater length of stay or higher cost among patients aged 18-64 years identifies a large proportion of psychological conditions, while a greater number of inpatient episodes identifies acute conditions and childbirth or labor-related complications. Conversely, definitions based on length of stay and cost in the elderly (65+) identified groups with chronic conditions that result in progressive functional decline (often requiring increasing supportive services upon discharge) or conditions that require significant rehabilitation prior to discharge. Regarding inpatient system burden, high users defined by number of inpatient episodes accounted for a small proportion of total inpatient episodes, while high users defined by length of stay and cost accounted for nearly half of the accumulated hospital days and cost for each. These findings highlight the need for careful consideration of how high use is defined when studying high-user populations and implications for targeting subpopulations for intervention.
Our results add to those from previous studies. A US-based, single-center study of 2566 individuals compared definitions of high inpatient use based on cost and frequency of admission and found that patients defined by cost were predominantly hospitalized for surgical conditions, while those fulfilling the episode-based definition were often hospitalized for medical conditions.12 The most responsible diagnoses for patient hospitalizations in our study reflect this. We extended this comparison to consider the impact of age on outcomes and inpatient system burden and found that older age was also linked to poorer outcomes and increased burden. We also considered a third definition (cumulative length of stay), which provided another opportunity for comparison. The presence of chronic conditions requiring rehabilitation and possible alternate level of care days within our cohort highlights the utility of this length of stay-based approach when considering definitions. Although there were similarities between patients defined by length of stay and cost, partly due to cost being largely a function of length of stay, there were also important differences in their patient profiles. Those defined by cost tended to have conditions requiring surgical procedures not requiring extended in-hospital rehabilitation. Furthermore, the higher proportion of in-hospital mortality among those defined by cost may also reflect the fact that patients tend to accrue the majority of their healthcare expenditures during the final 120 days of life.24
Each definition of high use identified complex patients; however, the differences between the various types of high users identified by these definitions suggest that they are not interchangeable. Arguably, selection of the most appropriate definition should depend on the objective of measuring high users, particularly if an intervention is planned. Interventions for high users are complex, requiring both medical and nonmedical components. The current literature in this area has often focused on case management programs, collaboration with community-based social support programs, and improving coordination and transitions of care.25-27 While many of these approaches require considerable involvement outside of the inpatient setting, these interventions can be informed by defining who high users of inpatient services are. Our findings show several possible subgroups of high users, which could be targeted for intervention. For example, an inpatient episode-based definition, which identifies patients with frequent encounters for acute conditions (eg, pneumonia and urinary tract infections), would be informative if an intervention targeted reductions in inpatient use and readmission rates. Alternatively, an intervention designed to improve community-based mental health programs would best be informed by a definition based on length of stay in which high users with underlying mental health conditions were prevalent. Such interventions are rarely mutually exclusive and require multiple perspectives to inform their objectives. A well-designed intervention will not only address the medical characteristics of high users but also the social determinants of health that place patients at risk of high inpatient use.
Our study should be interpreted in light of its limitations. First, measures of disease severity were not available to further characterize similarities and differences across high-use groups. Furthermore, we were unable to account for other social determinants of health that may be relevant to inpatient system usage. Second, direct cost of hospitalizations was estimated based on RIW and is thus reflective of expected rather than actual costs. However, this will have minimal impact on capture, as patients defined by this metric require substantial costs to be included in the top fifth percentile, and thus deviations in individual hospitalization costs will have minimal influence on the cumulative cost. Finally, while inpatient spending makes up a large proportion of healthcare spending, there is likely a number of different high-use profiles found outside of the acute care setting. Despite these limitations, our study includes several key strengths. The use of population-level data allows for analysis that is robust and more generalizable than studies from single centers. Additionally, the comparison of 3 independent definitions allows for a greater comparison of the nuances of each definition. Our study also considers the important impact of age as an effect modifier of inpatient use in the general population and identifies distinct patient profiles that exist across each definition.
CONCLUSIONS
Definitions of high use of inpatient services based on number of inpatient episodes, days spent in hospital, and total hospital costs identify patient populations with different characteristics and differ substantially in their impact on health outcomes and inpatient burden. These results highlight the need for careful consideration of the context of the study or intervention and the implications of selecting a specific definition of high inpatient use at study conception. Ultimately, the performance of an intervention in high-use populations is likely to be conditional on the fit of the patient population generated by the chosen definition of high inpatient use to the objectives of a study.
Acknowledgments
This study is based in part on data provided by Alberta Health and Alberta Health Services. The interpretation and conclusions are those of the researchers and do not represent the views of the Government of Alberta. Neither the Government of Alberta nor Alberta Health express any opinion in relation to this study.
Disclosure
Dr. Hemmelgarn is supported by the Roy and Vi Baay Chair in Kidney Research. Dr. Manns is supported by the Svare Professorship in Health Economics and by a Health Scholar Award by Alberta Innovates Health Solutions (AIHS). Dr. Tonelli is supported by the David Freeze chair in Health Services Research. The Interdisciplinary Chronic Disease Collaboration is funded by AIHS—Collaborative Research and Innovation Opportunity (CRIO) Team Grants Program.
1. National Health Expenditure Trends, 1975 to 2015. Canadian Institute for Health Information. 2015. https://secure.cihi.ca/free_products/nhex_trends_narrative_report_2015_en.pdf. Accessed on June 23, 2016.
2. Berk ML, Monheit AC. The concentration of health care expenditures, revisited. Health Aff (Millwood). 2001;20:9-18. PubMed
3. Wodchis WP, Austin PC, Henry DA. A 3-year study of high-cost users of health care. CMAJ. 2016;188(3):182-188. PubMed
4. Forget EL, Roos LL, Deber RB, Wald R. Variations in Lifetime Healthcare Costs across a Population. Healthc Policy. 2008;4:e148-e167. PubMed
5. Joynt KE, Gawande AA, Orav EJ, Jha AK. Contribution of preventable acute care spending to total spending for high-cost Medicare patients. JAMA. 2013;309:2572-2578. PubMed
6. Riley GF. Long-term trends in the concentration of Medicare spending. Health Aff (Millwood). 2007;26:808-816. PubMed
7. IHI Triple Aim Initiative. Institute for Healthcare Improvement. 2015. http://www.ihi.org/engage/initiatives/TripleAim/Pages/default.aspx. Accessed on June 17, 2016.
8. Johansen H, Nair C, Bond J. Who goes to the hospital? An investigation of high users of hospital days. Health Reports. 1994;6(2):253-277. PubMed
9. Conwell LJ, Cohen JW. Characteristics of persons with high medical expenditures in the US civilian noninstitutionalized population. MEPS Statistical Brief# 73. 2002.
10. Lemstra M, Mackenbach J, Neudorf C, Nannapaneni U. High health care utilization and costs associated with lower socio-economic status: Results from a linked dataset. CJPH. 2009;100(3):180-183. PubMed
11. Macnee CL, McCabe S, Clarke PN, Fiske M, Campbell S. Typology of high users of health services among a rural medicaid population. Pub Health Nurs. 2009;26(5):396-404. PubMed
12. Nguyen OK, Tang N, Hillman JM, Gonzales R. What’s cost got to do with it? Association between hospital costs and frequency of admissions among “high users” of hospital care. J. Hosp Med. 2013;8(12):665-671. PubMed
13. Rosella LC, Fitzpatrick T, Wodchis WP, Calzavara A, Manson H, Goel V. High-cost health care users in Ontario, Canada: Demographic, socio-economic, and health status characteristics. BMC Health Serv Res. 2014;14(1):532. PubMed
14. Cohen SB. The Concentration of Health Care Expenditures and Related Expenses for Costly Medical Conditions, 2009. Agency for Healthcare Research and Quality Statistical Brief #359; 2012.
15. Ronksley PE, McKay JA, Kobewka DM, Mulpuru S, Forster AJ. Patterns of health care use in a high-cost inpatient population in Ottawa, Ontario: A retrospective observational study. CMAJ Open. 2015; 3:E111-E118. PubMed
16. Hemmelgarn BR, Clement F, Manns BJ, et al. Overview of the Alberta Kidney Disease Network. BMC Nephrol. 2009;10:30. PubMed
17. DAD Resource Intensity Weights and Expected Length of Stay. Canadian Institute for Health Information. 2016. https://www.cihi.ca/en/data-and-standards/standards/case-mix/resource-indicators-dad-resource-intensity-weights-and. Accessed on June 24, 2016.
18. Statistics Canada. The Canadian Consumer Price Index Reference Paper, Statistics Canada Catalogue no. 62-553-X.
19. Tonelli M, Wiebe N, Fortin M, et al. Methods for identifying 30 chronic conditions: Application to administrative data. BMC Med Inform Decis Mak. 2015;17:15(1):1. PubMed
20. Jaakkimainen RL, Klein-Geltink J, Guttmann A, Barnsley J, Jagorski B, Kopp A. Indicators of primary care based on administrative data. In Primary Care in Ontario: ICES Atlas. Toronto, Ontario: Institute for Clinical Evaluative Sciences; 2006.
21. Jee SH, Cabana MD. Indices for continuity of care: A systematic review of the literature. Med Care Res Rev. 2006;63:158-188. PubMed
22. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP. 2009.
23. Benchimol EI, Smeeth L, Guttmann A, et al. The REporting of studies Conducted using Observational Routinely-collected health Data (RECORD) statement. PLoS Med. 2015;12(10):e1001885. PubMed
24. Tanuseputro P, Wodchis WP, Fowler R, et al. The health care cost of dying: A population-based retrospective cohort study of the last year of life in ontario, canada. PLoS One. 2015;10(3):e0121759. PubMed
25. Hong CS, Siegel AL, Ferris TG. Caring for high-need, high-cost patients: What makes for a successful care management program? Issue Brief (Commonw Fund). 2014;19:1-19. PubMed
26. Birnbaum M, Halper DE. Rethinking service delivery for high-cost Medicaid patients. Medicaid Institute. 2009. http://shnny.org/research/rethinking-service-delivery-for-high-cost-medicaid-patients/. Accessed on Jan 11, 2017.
27. Pan-Canadian forum on high users of health care. Canadian Institute for Health Information. 2014. https://secure.cihi.ca/free_products/highusers_summary_report_revised_EN_web.pdf. Accessed on Jan 11, 2017.
1. National Health Expenditure Trends, 1975 to 2015. Canadian Institute for Health Information. 2015. https://secure.cihi.ca/free_products/nhex_trends_narrative_report_2015_en.pdf. Accessed on June 23, 2016.
2. Berk ML, Monheit AC. The concentration of health care expenditures, revisited. Health Aff (Millwood). 2001;20:9-18. PubMed
3. Wodchis WP, Austin PC, Henry DA. A 3-year study of high-cost users of health care. CMAJ. 2016;188(3):182-188. PubMed
4. Forget EL, Roos LL, Deber RB, Wald R. Variations in Lifetime Healthcare Costs across a Population. Healthc Policy. 2008;4:e148-e167. PubMed
5. Joynt KE, Gawande AA, Orav EJ, Jha AK. Contribution of preventable acute care spending to total spending for high-cost Medicare patients. JAMA. 2013;309:2572-2578. PubMed
6. Riley GF. Long-term trends in the concentration of Medicare spending. Health Aff (Millwood). 2007;26:808-816. PubMed
7. IHI Triple Aim Initiative. Institute for Healthcare Improvement. 2015. http://www.ihi.org/engage/initiatives/TripleAim/Pages/default.aspx. Accessed on June 17, 2016.
8. Johansen H, Nair C, Bond J. Who goes to the hospital? An investigation of high users of hospital days. Health Reports. 1994;6(2):253-277. PubMed
9. Conwell LJ, Cohen JW. Characteristics of persons with high medical expenditures in the US civilian noninstitutionalized population. MEPS Statistical Brief# 73. 2002.
10. Lemstra M, Mackenbach J, Neudorf C, Nannapaneni U. High health care utilization and costs associated with lower socio-economic status: Results from a linked dataset. CJPH. 2009;100(3):180-183. PubMed
11. Macnee CL, McCabe S, Clarke PN, Fiske M, Campbell S. Typology of high users of health services among a rural medicaid population. Pub Health Nurs. 2009;26(5):396-404. PubMed
12. Nguyen OK, Tang N, Hillman JM, Gonzales R. What’s cost got to do with it? Association between hospital costs and frequency of admissions among “high users” of hospital care. J. Hosp Med. 2013;8(12):665-671. PubMed
13. Rosella LC, Fitzpatrick T, Wodchis WP, Calzavara A, Manson H, Goel V. High-cost health care users in Ontario, Canada: Demographic, socio-economic, and health status characteristics. BMC Health Serv Res. 2014;14(1):532. PubMed
14. Cohen SB. The Concentration of Health Care Expenditures and Related Expenses for Costly Medical Conditions, 2009. Agency for Healthcare Research and Quality Statistical Brief #359; 2012.
15. Ronksley PE, McKay JA, Kobewka DM, Mulpuru S, Forster AJ. Patterns of health care use in a high-cost inpatient population in Ottawa, Ontario: A retrospective observational study. CMAJ Open. 2015; 3:E111-E118. PubMed
16. Hemmelgarn BR, Clement F, Manns BJ, et al. Overview of the Alberta Kidney Disease Network. BMC Nephrol. 2009;10:30. PubMed
17. DAD Resource Intensity Weights and Expected Length of Stay. Canadian Institute for Health Information. 2016. https://www.cihi.ca/en/data-and-standards/standards/case-mix/resource-indicators-dad-resource-intensity-weights-and. Accessed on June 24, 2016.
18. Statistics Canada. The Canadian Consumer Price Index Reference Paper, Statistics Canada Catalogue no. 62-553-X.
19. Tonelli M, Wiebe N, Fortin M, et al. Methods for identifying 30 chronic conditions: Application to administrative data. BMC Med Inform Decis Mak. 2015;17:15(1):1. PubMed
20. Jaakkimainen RL, Klein-Geltink J, Guttmann A, Barnsley J, Jagorski B, Kopp A. Indicators of primary care based on administrative data. In Primary Care in Ontario: ICES Atlas. Toronto, Ontario: Institute for Clinical Evaluative Sciences; 2006.
21. Jee SH, Cabana MD. Indices for continuity of care: A systematic review of the literature. Med Care Res Rev. 2006;63:158-188. PubMed
22. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP. 2009.
23. Benchimol EI, Smeeth L, Guttmann A, et al. The REporting of studies Conducted using Observational Routinely-collected health Data (RECORD) statement. PLoS Med. 2015;12(10):e1001885. PubMed
24. Tanuseputro P, Wodchis WP, Fowler R, et al. The health care cost of dying: A population-based retrospective cohort study of the last year of life in ontario, canada. PLoS One. 2015;10(3):e0121759. PubMed
25. Hong CS, Siegel AL, Ferris TG. Caring for high-need, high-cost patients: What makes for a successful care management program? Issue Brief (Commonw Fund). 2014;19:1-19. PubMed
26. Birnbaum M, Halper DE. Rethinking service delivery for high-cost Medicaid patients. Medicaid Institute. 2009. http://shnny.org/research/rethinking-service-delivery-for-high-cost-medicaid-patients/. Accessed on Jan 11, 2017.
27. Pan-Canadian forum on high users of health care. Canadian Institute for Health Information. 2014. https://secure.cihi.ca/free_products/highusers_summary_report_revised_EN_web.pdf. Accessed on Jan 11, 2017.
© 2017 Society of Hospital Medicine
Trans-Scaphoid Transcapitate Perilunate Fracture-Dislocation
Take-Home Points
- TSTC-PLFD is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.
- TSTC-PLFD is associated by a complex ligamentous injury of the wrist.
- Impaction of the wrist in extension seems to be the most important predictor of this injury.
- Optimal treatment for TSTC-PLFD is open reduction, anatomical alignment, and ligamentous and osseous stabilization.
- The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.
Trans-scaphoid transcapitate (TSTC) perilunate fracture-dislocation (PLFD) is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.1 Isolated capitate fractures with or without rotation of its proximal fragment have been well described.2,3 Obviously, this specific type of injury represents just the osseous part of a more complex ligamentous wrist injury.2,3
TSTC-PLFD was first described by Nicholson4 in 1940. In 1956, Fenton5 coined the term scaphocapitate syndrome, which became widely known. With PLFD, accurate diagnosis may be delayed. Usually, only the scaphoid fracture is identified by radiologic examination, and thus the severity of the injury is underestimated and appropriate treatment delayed.3,6,7 The English literature includes only case reports and small series on this rare perilunate injury.6-9 In this article, we report the case of an adult with TSTC-PLFD. We describe the radiographic and intraoperative findings, review the current surgical principles for reduction and stabilization of this injury, and assess the clinical and radiologic outcomes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 32-year-old man sustained an isolated injury of his right (dominant) hand after falling from a height of 6 feet and landing on his outstretched right arm with the wrist in extension. 

With the patient under general anesthesia and a humerus tourniquet applied, an external fixator was placed for spanning of the wrist joint. The dorsal aspect of the wrist joint was approached through a midline longitudinal 5-cm incision, centered over the Lister tubercle. For adequate exposure of the dorsal wrist, a flap of the dorsal capsule was raised with the apex at the triquetrum and a radial broad base, as previously described.9 An avulsion fracture at the insertion of the dorsal capsule to the triquetrum was observed. The dorsal surface of the hamate and lunate showed a small area of bone contusion with hemorrhagic infiltration. The scapholunate and lunotriquetral ligaments were intact. The proximal fragment of the capitate was identified deep into the space between the lunate and distal capitate fragment; the articular surface of the bone fragment was rotated 180° distally (Figure 3). 

Skin sutures were removed 2 weeks after surgery, K-wires 6 weeks after surgery, and the external fixator 8 weeks after surgery. At 8 weeks, radiographs showed healing of both fractures, scaphoid and capitate. The patient was allowed gradual passive and active-assisted range-of-motion exercises of the wrist at 8 weeks, and he returned to work 3 months after surgery. At 12-month follow-up, all fractures were completely healed, and the wrist was stable and pain-free. 
Discussion
The exact biomechanism of TSTC-PLFD is unclear. Impaction of the wrist in extension seems to be the most important predictor of this injury.5,7,9-11 According to Stein and Siegel,10 scaphoid fractures first allow hyperextension of the wrist; the lunate and the capitate rotate dorsally, and the dorsal surface of the capitate impacts the dorsal edge of the distal radius, causing a fracture of the neck of the capitate. If the wrist continues to rotate into further hyperextension, the unsupported, proximal part of the capitate rotates 90° around itself.9,10 When the carpus returns to neutral position, the bone fragment of the capitate rotates further, reaching a position of 180°, with its proximal articular surface facing distally. In this type of injury, the axis of rotation is transverse (radioulnar), in contrast to the perpendicular (anteroposterior) axis of rotation suggested by the initial report by Fenton.5 The scaphoid is fractured by impaction of the radial styloid process. Monahan and Galasko11 reported a case of capitate fracture with palmar displacement and 90° rotation of the proximal bone fragment; the fragmented surface was facing dorsally. A transverse axis of rotation, as in our patient’s case, could explain this type of displacement supporting the mechanism of injury proposed by Stein and Siegel.10 Vance and colleagues7 described various patterns of scaphocapitate fractures and concluded that no single mechanism of injury accounts for these types of injuries. Other authors have considered scaphocapitate syndrome as a specific type of TSTC-PLFD, one that reduces either spontaneously or with manipulation.1,3,12 Detailed evaluation of standard anteroposterior and lateral wrist radiographs can provide enough evidence for the diagnosis of this injury. Computed tomography may define further the type and extent of injury.7 In our patient’s case, wrist impaction caused the scaphoid and capitate fractures and the avulsion of the capsule attachment to the triquetrum. The distal fragment of the capitate subluxated dorsally in relation to the lunate. The lateral radiograph of the wrist showed its position in the lunate fossa. According to the classification of Herzberg and colleagues12 and Mayfield and colleagues,13 this represents a dorsal PLFD of the greater carpal bones arc.
Conservative treatment is not recommended for PLFD because closed reduction usually is not possible, and poor functional outcomes are common. Instead, optimal treatment is open reduction, anatomical alignment, and ligamentous and osseous stabilization.7,12,14,15 Dorsal, palmar, and combined approaches have been used in surgery for perilunate injuries. A dorsal approach through a radius-based capsular flap allows excellent exposure of the dorsal wrist and facilitates reduction of fractures.9 Capitate reduction should precede scaphoid reduction because scaphoid reduction cannot be easily maintained, especially when the fracture interface is comminuted.7 In addition, scaphoid reduction may be guided from the radial surface of the capitate. Moreover, when the scaphoid is fixated first, reduction of the rotated head of the capitate usually is difficult. In our patient’s case, traction applied through the external fixator facilitated reduction and K-wire fixation of the capitate fracture. After scaphoid fixation, the K-wires were advanced through the capitate to the lunate to stabilize the capitolunate joint. The wrist must be immobilized for 6 to 8 weeks after surgical repair of PLFD. A cast can be used, but, as with our patient, an external fixator facilitates fracture reduction and wrist stability during osteosynthesis. During immobilization, the wrist should be maintained in neutral position to avoid stretching the dorsal and palmar wrist capsule and ligaments.16The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.17-20 Similar to scaphoid fractures, capitate fractures proximal to the waist of the capitate are associated with increased risk of osteonecrosis. Therefore, anatomical reduction and stabilization favor revascularization of the proximal bone fragment. Moreover, any osteonecrosis that occurs in the proximal part of the capitate is not an indication for further surgery as long as wrist height is maintained. Nonunion is not common after open reduction and internal fixation of PLFD (eg, our patient’s fractures healed completely).17 Radiographically, nonunion is characterized by bone absorption and sclerosis of the ends of the bone. Treatment of capitate nonunion depends on symptom severity, bone fragment size, and radiographic evidence of arthritic changes.3,7,21-23 Treatment options include resection of sclerotic edges, bone grafting, and stabilization21 and removal of the proximal capitate fragment and limited arthrodesis,22 as arthritic changes likely are inevitable.22,23TSTC-PLFD is a rare wrist injury. Careful radiographic evaluation of the carpal bones and their relationships on both anteroposterior and lateral views is mandatory in making the correct diagnosis. Open reduction (preferably with use of an external fixator) and internal fixation are recommended for optimal healing and functional outcomes.
Am J Orthop. 2017;46(4):E230-E234. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
2. Volk AG, Schnall SB, Merkle P, Stevanovic M. Unusual capitate fracture: a case report. J Hand Surg Am. 1995;20(4):581-582.
3. Apergis E, Darmanis S, Kastanis G, Papanikolaou A. Does the term scaphocapitate syndrome need to be revised? A report of 6 cases. J Hand Surg Br. 2001;26(5):441-445.
4. Nicholson CB. Fracture dislocation of the os magnum. J Roy Navy Med Serv. 1940;26:289-291.
5. Fenton RL. The naviculo-capitate fracture syndrome. J Bone Joint Surg Am. 1956;38(3):681-684.
6. Strohm PC, Laier P, Müller CA, Gutorski S, Pfister U. Scaphocapitate fracture syndrome of both hands—first description of a bilateral occurrence of a rare carpal injury [in German]. Unfallchirurg. 2003;106(4):339-342.
7. Vance RM, Gelberman R, Evans EF. Scaphocapitate fractures. Patterns of dislocation, mechanisms of injury, and preliminary results of treatment. J Bone Joint Surg Am. 1980;62(2):271-276.
8. Apostolides JG, Lifchez SD, Christy MR. Complex and rare fracture patterns in perilunate dislocations. Hand. 2011;6(3):287-294.
9. Berger RA, Bishop AT, Bettinger PC. New dorsal capsulotomy for the surgical exposure of the wrist. Ann Plast Surg. 1995;35(1):54-59.
10. Stein F, Siegel MW. Naviculocapitate fracture syndrome. A case report: new thoughts on the mechanism of injury. J Bone Joint Surg Am. 1969;51(2):391-395.
11. Monahan PR, Galasko CS. The scapho-capitate fracture syndrome. A mechanism of injury. J Bone Joint Surg Br. 1972;54(1):122-124.
12. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
13. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
14. Moneim MS, Hofammann KE 3rd, Omer GE. Transscaphoid perilunate fracture-dislocation. Result of open reduction and pin fixation. Clin Orthop Relat Res. 1984;(190):227-235.
15. Andreasi A, Coppo M, Danda F. Trans-scapho-capitate perilunar dislocation of the carpus. Ital J Orthop Traumatol. 1986;12(4):461-466.
16. Song D, Goodman S, Gilula LA, Wollstein R. Ulnocarpal translation in perilunate dislocations. J Hand Surg Eur. 2009;34(3):388-390.
17. Rand JA, Linscheid RL, Dobyns JH. Capitate fractures: a long-term follow-up. Clin Orthop Relat Res. 1982;(165):209-216.
18. Panagis JS, Gelberman RH, Taleisnik J, Baumgaertner M. The arterial anatomy of the human carpus. Part II: the intraosseous vascularity. J Hand Surg Am. 1983;8(4):375-382.
19. Freedman DM, Botte MJ, Gelberman RH. Vascularity of the carpus. Clin Orthop Relat Res. 2001;(383):47-59.
20. Vander Grend R, Dell PC, Glowczewskie F, Leslie B, Ruby LK. Intraosseous blood supply of the capitate and its correlation with aseptic necrosis. J Hand Surg Am. 1984;9(5):677-683.
21. Rico AA, Holguin PH, Martin JG. Pseudarthrosis of the capitate. J Hand Surg Br. 1999;24(3):382-384.
22. Kumar A, Olney DB. Multiple carpometacarpal dislocations. J Accid Emerg Med. 1994;11(4):257-258.
23. Kohut GN. Extra-articular fractures of the distal radius in young adults. A technique of closed reposition and stabilisation by mono-segmental, radio-radial external fixator. Ann Chir Main Memb Super. 1995;14(1):14-19.
Take-Home Points
- TSTC-PLFD is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.
- TSTC-PLFD is associated by a complex ligamentous injury of the wrist.
- Impaction of the wrist in extension seems to be the most important predictor of this injury.
- Optimal treatment for TSTC-PLFD is open reduction, anatomical alignment, and ligamentous and osseous stabilization.
- The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.
Trans-scaphoid transcapitate (TSTC) perilunate fracture-dislocation (PLFD) is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.1 Isolated capitate fractures with or without rotation of its proximal fragment have been well described.2,3 Obviously, this specific type of injury represents just the osseous part of a more complex ligamentous wrist injury.2,3
TSTC-PLFD was first described by Nicholson4 in 1940. In 1956, Fenton5 coined the term scaphocapitate syndrome, which became widely known. With PLFD, accurate diagnosis may be delayed. Usually, only the scaphoid fracture is identified by radiologic examination, and thus the severity of the injury is underestimated and appropriate treatment delayed.3,6,7 The English literature includes only case reports and small series on this rare perilunate injury.6-9 In this article, we report the case of an adult with TSTC-PLFD. We describe the radiographic and intraoperative findings, review the current surgical principles for reduction and stabilization of this injury, and assess the clinical and radiologic outcomes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 32-year-old man sustained an isolated injury of his right (dominant) hand after falling from a height of 6 feet and landing on his outstretched right arm with the wrist in extension.
With the patient under general anesthesia and a humerus tourniquet applied, an external fixator was placed for spanning of the wrist joint. The dorsal aspect of the wrist joint was approached through a midline longitudinal 5-cm incision, centered over the Lister tubercle. For adequate exposure of the dorsal wrist, a flap of the dorsal capsule was raised with the apex at the triquetrum and a radial broad base, as previously described.9 An avulsion fracture at the insertion of the dorsal capsule to the triquetrum was observed. The dorsal surface of the hamate and lunate showed a small area of bone contusion with hemorrhagic infiltration. The scapholunate and lunotriquetral ligaments were intact. The proximal fragment of the capitate was identified deep into the space between the lunate and distal capitate fragment; the articular surface of the bone fragment was rotated 180° distally (Figure 3).
Skin sutures were removed 2 weeks after surgery, K-wires 6 weeks after surgery, and the external fixator 8 weeks after surgery. At 8 weeks, radiographs showed healing of both fractures, scaphoid and capitate. The patient was allowed gradual passive and active-assisted range-of-motion exercises of the wrist at 8 weeks, and he returned to work 3 months after surgery. At 12-month follow-up, all fractures were completely healed, and the wrist was stable and pain-free.
Discussion
The exact biomechanism of TSTC-PLFD is unclear. Impaction of the wrist in extension seems to be the most important predictor of this injury.5,7,9-11 According to Stein and Siegel,10 scaphoid fractures first allow hyperextension of the wrist; the lunate and the capitate rotate dorsally, and the dorsal surface of the capitate impacts the dorsal edge of the distal radius, causing a fracture of the neck of the capitate. If the wrist continues to rotate into further hyperextension, the unsupported, proximal part of the capitate rotates 90° around itself.9,10 When the carpus returns to neutral position, the bone fragment of the capitate rotates further, reaching a position of 180°, with its proximal articular surface facing distally. In this type of injury, the axis of rotation is transverse (radioulnar), in contrast to the perpendicular (anteroposterior) axis of rotation suggested by the initial report by Fenton.5 The scaphoid is fractured by impaction of the radial styloid process. Monahan and Galasko11 reported a case of capitate fracture with palmar displacement and 90° rotation of the proximal bone fragment; the fragmented surface was facing dorsally. A transverse axis of rotation, as in our patient’s case, could explain this type of displacement supporting the mechanism of injury proposed by Stein and Siegel.10 Vance and colleagues7 described various patterns of scaphocapitate fractures and concluded that no single mechanism of injury accounts for these types of injuries. Other authors have considered scaphocapitate syndrome as a specific type of TSTC-PLFD, one that reduces either spontaneously or with manipulation.1,3,12 Detailed evaluation of standard anteroposterior and lateral wrist radiographs can provide enough evidence for the diagnosis of this injury. Computed tomography may define further the type and extent of injury.7 In our patient’s case, wrist impaction caused the scaphoid and capitate fractures and the avulsion of the capsule attachment to the triquetrum. The distal fragment of the capitate subluxated dorsally in relation to the lunate. The lateral radiograph of the wrist showed its position in the lunate fossa. According to the classification of Herzberg and colleagues12 and Mayfield and colleagues,13 this represents a dorsal PLFD of the greater carpal bones arc.
Conservative treatment is not recommended for PLFD because closed reduction usually is not possible, and poor functional outcomes are common. Instead, optimal treatment is open reduction, anatomical alignment, and ligamentous and osseous stabilization.7,12,14,15 Dorsal, palmar, and combined approaches have been used in surgery for perilunate injuries. A dorsal approach through a radius-based capsular flap allows excellent exposure of the dorsal wrist and facilitates reduction of fractures.9 Capitate reduction should precede scaphoid reduction because scaphoid reduction cannot be easily maintained, especially when the fracture interface is comminuted.7 In addition, scaphoid reduction may be guided from the radial surface of the capitate. Moreover, when the scaphoid is fixated first, reduction of the rotated head of the capitate usually is difficult. In our patient’s case, traction applied through the external fixator facilitated reduction and K-wire fixation of the capitate fracture. After scaphoid fixation, the K-wires were advanced through the capitate to the lunate to stabilize the capitolunate joint. The wrist must be immobilized for 6 to 8 weeks after surgical repair of PLFD. A cast can be used, but, as with our patient, an external fixator facilitates fracture reduction and wrist stability during osteosynthesis. During immobilization, the wrist should be maintained in neutral position to avoid stretching the dorsal and palmar wrist capsule and ligaments.16The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.17-20 Similar to scaphoid fractures, capitate fractures proximal to the waist of the capitate are associated with increased risk of osteonecrosis. Therefore, anatomical reduction and stabilization favor revascularization of the proximal bone fragment. Moreover, any osteonecrosis that occurs in the proximal part of the capitate is not an indication for further surgery as long as wrist height is maintained. Nonunion is not common after open reduction and internal fixation of PLFD (eg, our patient’s fractures healed completely).17 Radiographically, nonunion is characterized by bone absorption and sclerosis of the ends of the bone. Treatment of capitate nonunion depends on symptom severity, bone fragment size, and radiographic evidence of arthritic changes.3,7,21-23 Treatment options include resection of sclerotic edges, bone grafting, and stabilization21 and removal of the proximal capitate fragment and limited arthrodesis,22 as arthritic changes likely are inevitable.22,23TSTC-PLFD is a rare wrist injury. Careful radiographic evaluation of the carpal bones and their relationships on both anteroposterior and lateral views is mandatory in making the correct diagnosis. Open reduction (preferably with use of an external fixator) and internal fixation are recommended for optimal healing and functional outcomes.
Am J Orthop. 2017;46(4):E230-E234. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- TSTC-PLFD is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.
- TSTC-PLFD is associated by a complex ligamentous injury of the wrist.
- Impaction of the wrist in extension seems to be the most important predictor of this injury.
- Optimal treatment for TSTC-PLFD is open reduction, anatomical alignment, and ligamentous and osseous stabilization.
- The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.
Trans-scaphoid transcapitate (TSTC) perilunate fracture-dislocation (PLFD) is a rare hyperextension wrist injury characterized by fracture of both the scaphoid and the capitate and rotation of the proximal bone fragment of the capitate.1 Isolated capitate fractures with or without rotation of its proximal fragment have been well described.2,3 Obviously, this specific type of injury represents just the osseous part of a more complex ligamentous wrist injury.2,3
TSTC-PLFD was first described by Nicholson4 in 1940. In 1956, Fenton5 coined the term scaphocapitate syndrome, which became widely known. With PLFD, accurate diagnosis may be delayed. Usually, only the scaphoid fracture is identified by radiologic examination, and thus the severity of the injury is underestimated and appropriate treatment delayed.3,6,7 The English literature includes only case reports and small series on this rare perilunate injury.6-9 In this article, we report the case of an adult with TSTC-PLFD. We describe the radiographic and intraoperative findings, review the current surgical principles for reduction and stabilization of this injury, and assess the clinical and radiologic outcomes. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 32-year-old man sustained an isolated injury of his right (dominant) hand after falling from a height of 6 feet and landing on his outstretched right arm with the wrist in extension.
With the patient under general anesthesia and a humerus tourniquet applied, an external fixator was placed for spanning of the wrist joint. The dorsal aspect of the wrist joint was approached through a midline longitudinal 5-cm incision, centered over the Lister tubercle. For adequate exposure of the dorsal wrist, a flap of the dorsal capsule was raised with the apex at the triquetrum and a radial broad base, as previously described.9 An avulsion fracture at the insertion of the dorsal capsule to the triquetrum was observed. The dorsal surface of the hamate and lunate showed a small area of bone contusion with hemorrhagic infiltration. The scapholunate and lunotriquetral ligaments were intact. The proximal fragment of the capitate was identified deep into the space between the lunate and distal capitate fragment; the articular surface of the bone fragment was rotated 180° distally (Figure 3).
Skin sutures were removed 2 weeks after surgery, K-wires 6 weeks after surgery, and the external fixator 8 weeks after surgery. At 8 weeks, radiographs showed healing of both fractures, scaphoid and capitate. The patient was allowed gradual passive and active-assisted range-of-motion exercises of the wrist at 8 weeks, and he returned to work 3 months after surgery. At 12-month follow-up, all fractures were completely healed, and the wrist was stable and pain-free.
Discussion
The exact biomechanism of TSTC-PLFD is unclear. Impaction of the wrist in extension seems to be the most important predictor of this injury.5,7,9-11 According to Stein and Siegel,10 scaphoid fractures first allow hyperextension of the wrist; the lunate and the capitate rotate dorsally, and the dorsal surface of the capitate impacts the dorsal edge of the distal radius, causing a fracture of the neck of the capitate. If the wrist continues to rotate into further hyperextension, the unsupported, proximal part of the capitate rotates 90° around itself.9,10 When the carpus returns to neutral position, the bone fragment of the capitate rotates further, reaching a position of 180°, with its proximal articular surface facing distally. In this type of injury, the axis of rotation is transverse (radioulnar), in contrast to the perpendicular (anteroposterior) axis of rotation suggested by the initial report by Fenton.5 The scaphoid is fractured by impaction of the radial styloid process. Monahan and Galasko11 reported a case of capitate fracture with palmar displacement and 90° rotation of the proximal bone fragment; the fragmented surface was facing dorsally. A transverse axis of rotation, as in our patient’s case, could explain this type of displacement supporting the mechanism of injury proposed by Stein and Siegel.10 Vance and colleagues7 described various patterns of scaphocapitate fractures and concluded that no single mechanism of injury accounts for these types of injuries. Other authors have considered scaphocapitate syndrome as a specific type of TSTC-PLFD, one that reduces either spontaneously or with manipulation.1,3,12 Detailed evaluation of standard anteroposterior and lateral wrist radiographs can provide enough evidence for the diagnosis of this injury. Computed tomography may define further the type and extent of injury.7 In our patient’s case, wrist impaction caused the scaphoid and capitate fractures and the avulsion of the capsule attachment to the triquetrum. The distal fragment of the capitate subluxated dorsally in relation to the lunate. The lateral radiograph of the wrist showed its position in the lunate fossa. According to the classification of Herzberg and colleagues12 and Mayfield and colleagues,13 this represents a dorsal PLFD of the greater carpal bones arc.
Conservative treatment is not recommended for PLFD because closed reduction usually is not possible, and poor functional outcomes are common. Instead, optimal treatment is open reduction, anatomical alignment, and ligamentous and osseous stabilization.7,12,14,15 Dorsal, palmar, and combined approaches have been used in surgery for perilunate injuries. A dorsal approach through a radius-based capsular flap allows excellent exposure of the dorsal wrist and facilitates reduction of fractures.9 Capitate reduction should precede scaphoid reduction because scaphoid reduction cannot be easily maintained, especially when the fracture interface is comminuted.7 In addition, scaphoid reduction may be guided from the radial surface of the capitate. Moreover, when the scaphoid is fixated first, reduction of the rotated head of the capitate usually is difficult. In our patient’s case, traction applied through the external fixator facilitated reduction and K-wire fixation of the capitate fracture. After scaphoid fixation, the K-wires were advanced through the capitate to the lunate to stabilize the capitolunate joint. The wrist must be immobilized for 6 to 8 weeks after surgical repair of PLFD. A cast can be used, but, as with our patient, an external fixator facilitates fracture reduction and wrist stability during osteosynthesis. During immobilization, the wrist should be maintained in neutral position to avoid stretching the dorsal and palmar wrist capsule and ligaments.16The most important complications of scaphoid and capitate fractures and PLFD are osteonecrosis and nonunion.17-20 Similar to scaphoid fractures, capitate fractures proximal to the waist of the capitate are associated with increased risk of osteonecrosis. Therefore, anatomical reduction and stabilization favor revascularization of the proximal bone fragment. Moreover, any osteonecrosis that occurs in the proximal part of the capitate is not an indication for further surgery as long as wrist height is maintained. Nonunion is not common after open reduction and internal fixation of PLFD (eg, our patient’s fractures healed completely).17 Radiographically, nonunion is characterized by bone absorption and sclerosis of the ends of the bone. Treatment of capitate nonunion depends on symptom severity, bone fragment size, and radiographic evidence of arthritic changes.3,7,21-23 Treatment options include resection of sclerotic edges, bone grafting, and stabilization21 and removal of the proximal capitate fragment and limited arthrodesis,22 as arthritic changes likely are inevitable.22,23TSTC-PLFD is a rare wrist injury. Careful radiographic evaluation of the carpal bones and their relationships on both anteroposterior and lateral views is mandatory in making the correct diagnosis. Open reduction (preferably with use of an external fixator) and internal fixation are recommended for optimal healing and functional outcomes.
Am J Orthop. 2017;46(4):E230-E234. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
2. Volk AG, Schnall SB, Merkle P, Stevanovic M. Unusual capitate fracture: a case report. J Hand Surg Am. 1995;20(4):581-582.
3. Apergis E, Darmanis S, Kastanis G, Papanikolaou A. Does the term scaphocapitate syndrome need to be revised? A report of 6 cases. J Hand Surg Br. 2001;26(5):441-445.
4. Nicholson CB. Fracture dislocation of the os magnum. J Roy Navy Med Serv. 1940;26:289-291.
5. Fenton RL. The naviculo-capitate fracture syndrome. J Bone Joint Surg Am. 1956;38(3):681-684.
6. Strohm PC, Laier P, Müller CA, Gutorski S, Pfister U. Scaphocapitate fracture syndrome of both hands—first description of a bilateral occurrence of a rare carpal injury [in German]. Unfallchirurg. 2003;106(4):339-342.
7. Vance RM, Gelberman R, Evans EF. Scaphocapitate fractures. Patterns of dislocation, mechanisms of injury, and preliminary results of treatment. J Bone Joint Surg Am. 1980;62(2):271-276.
8. Apostolides JG, Lifchez SD, Christy MR. Complex and rare fracture patterns in perilunate dislocations. Hand. 2011;6(3):287-294.
9. Berger RA, Bishop AT, Bettinger PC. New dorsal capsulotomy for the surgical exposure of the wrist. Ann Plast Surg. 1995;35(1):54-59.
10. Stein F, Siegel MW. Naviculocapitate fracture syndrome. A case report: new thoughts on the mechanism of injury. J Bone Joint Surg Am. 1969;51(2):391-395.
11. Monahan PR, Galasko CS. The scapho-capitate fracture syndrome. A mechanism of injury. J Bone Joint Surg Br. 1972;54(1):122-124.
12. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
13. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
14. Moneim MS, Hofammann KE 3rd, Omer GE. Transscaphoid perilunate fracture-dislocation. Result of open reduction and pin fixation. Clin Orthop Relat Res. 1984;(190):227-235.
15. Andreasi A, Coppo M, Danda F. Trans-scapho-capitate perilunar dislocation of the carpus. Ital J Orthop Traumatol. 1986;12(4):461-466.
16. Song D, Goodman S, Gilula LA, Wollstein R. Ulnocarpal translation in perilunate dislocations. J Hand Surg Eur. 2009;34(3):388-390.
17. Rand JA, Linscheid RL, Dobyns JH. Capitate fractures: a long-term follow-up. Clin Orthop Relat Res. 1982;(165):209-216.
18. Panagis JS, Gelberman RH, Taleisnik J, Baumgaertner M. The arterial anatomy of the human carpus. Part II: the intraosseous vascularity. J Hand Surg Am. 1983;8(4):375-382.
19. Freedman DM, Botte MJ, Gelberman RH. Vascularity of the carpus. Clin Orthop Relat Res. 2001;(383):47-59.
20. Vander Grend R, Dell PC, Glowczewskie F, Leslie B, Ruby LK. Intraosseous blood supply of the capitate and its correlation with aseptic necrosis. J Hand Surg Am. 1984;9(5):677-683.
21. Rico AA, Holguin PH, Martin JG. Pseudarthrosis of the capitate. J Hand Surg Br. 1999;24(3):382-384.
22. Kumar A, Olney DB. Multiple carpometacarpal dislocations. J Accid Emerg Med. 1994;11(4):257-258.
23. Kohut GN. Extra-articular fractures of the distal radius in young adults. A technique of closed reposition and stabilisation by mono-segmental, radio-radial external fixator. Ann Chir Main Memb Super. 1995;14(1):14-19.
1. Johnson RP. The acutely injured wrist and its residuals. Clin Orthop Relat Res. 1980;(149):33-44.
2. Volk AG, Schnall SB, Merkle P, Stevanovic M. Unusual capitate fracture: a case report. J Hand Surg Am. 1995;20(4):581-582.
3. Apergis E, Darmanis S, Kastanis G, Papanikolaou A. Does the term scaphocapitate syndrome need to be revised? A report of 6 cases. J Hand Surg Br. 2001;26(5):441-445.
4. Nicholson CB. Fracture dislocation of the os magnum. J Roy Navy Med Serv. 1940;26:289-291.
5. Fenton RL. The naviculo-capitate fracture syndrome. J Bone Joint Surg Am. 1956;38(3):681-684.
6. Strohm PC, Laier P, Müller CA, Gutorski S, Pfister U. Scaphocapitate fracture syndrome of both hands—first description of a bilateral occurrence of a rare carpal injury [in German]. Unfallchirurg. 2003;106(4):339-342.
7. Vance RM, Gelberman R, Evans EF. Scaphocapitate fractures. Patterns of dislocation, mechanisms of injury, and preliminary results of treatment. J Bone Joint Surg Am. 1980;62(2):271-276.
8. Apostolides JG, Lifchez SD, Christy MR. Complex and rare fracture patterns in perilunate dislocations. Hand. 2011;6(3):287-294.
9. Berger RA, Bishop AT, Bettinger PC. New dorsal capsulotomy for the surgical exposure of the wrist. Ann Plast Surg. 1995;35(1):54-59.
10. Stein F, Siegel MW. Naviculocapitate fracture syndrome. A case report: new thoughts on the mechanism of injury. J Bone Joint Surg Am. 1969;51(2):391-395.
11. Monahan PR, Galasko CS. The scapho-capitate fracture syndrome. A mechanism of injury. J Bone Joint Surg Br. 1972;54(1):122-124.
12. Herzberg G, Comtet JJ, Linscheid RL, Amadio PC, Cooney WP, Stalder J. Perilunate dislocations and fracture-dislocations: a multicenter study. J Hand Surg Am. 1993;18(5):768-779.
13. Mayfield JK, Johnson RP, Kilcoyne RK. Carpal dislocations: pathomechanics and progressive perilunar instability. J Hand Surg Am. 1980;5(3):226-241.
14. Moneim MS, Hofammann KE 3rd, Omer GE. Transscaphoid perilunate fracture-dislocation. Result of open reduction and pin fixation. Clin Orthop Relat Res. 1984;(190):227-235.
15. Andreasi A, Coppo M, Danda F. Trans-scapho-capitate perilunar dislocation of the carpus. Ital J Orthop Traumatol. 1986;12(4):461-466.
16. Song D, Goodman S, Gilula LA, Wollstein R. Ulnocarpal translation in perilunate dislocations. J Hand Surg Eur. 2009;34(3):388-390.
17. Rand JA, Linscheid RL, Dobyns JH. Capitate fractures: a long-term follow-up. Clin Orthop Relat Res. 1982;(165):209-216.
18. Panagis JS, Gelberman RH, Taleisnik J, Baumgaertner M. The arterial anatomy of the human carpus. Part II: the intraosseous vascularity. J Hand Surg Am. 1983;8(4):375-382.
19. Freedman DM, Botte MJ, Gelberman RH. Vascularity of the carpus. Clin Orthop Relat Res. 2001;(383):47-59.
20. Vander Grend R, Dell PC, Glowczewskie F, Leslie B, Ruby LK. Intraosseous blood supply of the capitate and its correlation with aseptic necrosis. J Hand Surg Am. 1984;9(5):677-683.
21. Rico AA, Holguin PH, Martin JG. Pseudarthrosis of the capitate. J Hand Surg Br. 1999;24(3):382-384.
22. Kumar A, Olney DB. Multiple carpometacarpal dislocations. J Accid Emerg Med. 1994;11(4):257-258.
23. Kohut GN. Extra-articular fractures of the distal radius in young adults. A technique of closed reposition and stabilisation by mono-segmental, radio-radial external fixator. Ann Chir Main Memb Super. 1995;14(1):14-19.
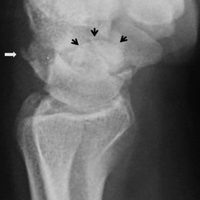
Models provide new understanding of sickle cell disease
Computer models have revealed new details of what happens inside a red blood cell affected by sickle cell disease (SCD), according to research published in Biophysical Journal.
In patients with SCD, mutated hemoglobin can polymerize, assembling into long fibers that push against the membranes of red blood cells and force them out of shape.
“The goal of our work is to model both how these sickle hemoglobin fibers form as well as the mechanical properties of those fibers,” said study author Lu Lu, a PhD student at Brown University in Providence, Rhode Island.
“There had been separate models for each of these things individually developed by us, but this brings those together into one comprehensive model.”
The model uses detailed biomechanical data on how sickle hemoglobin molecules behave and bind with each other to simulate the assembly of a polymer fiber.
Prior to this work, the problem had been that, as the fiber grows, so does the amount of data the model must crunch. Modeling an entire polymer fiber at a cellular scale using the details of each molecule was simply too computationally expensive.
“Even the world’s fastest supercomputers wouldn’t be able to handle it,” said study author George Karniadakis, PhD, of Brown University.
“There’s just too much happening and no way to capture it all computationally. That’s what we were able to overcome with this work.”
The researchers’ solution was to apply what they call a mesoscopic adaptive resolution scheme (MARS).
The MARS model calculates the detailed dynamics of each individual hemoglobin molecule only at the end of polymer fibers, where new molecules are being recruited into the fiber.
Once 4 layers of a fiber have been established, the model automatically dials back the resolution at which it represents that section. The model retains the important information about how the fiber behaves mechanically but glosses over the fine details of each constituent molecule.
“By eliminating the fine details where we don’t need them, we develop a model that can simulate this whole process and its effects on a red blood cell,” Dr Karniadakis said.
Using the new MARS simulations, the researchers were able to show how different configurations of growing polymer fibers are able to produce cells with different shapes.
“We are able to produce a polymerization profile for each of the cell types associated with the disease,” Dr Karniadakis said. “Now, the goal is to use these models to look for ways of preventing the disease onset.”
Using these new models, Dr Karniadakis and his colleagues can run simulations that include fetal hemoglobin. Those simulations could be used to confirm the theory that fetal hemoglobin disrupts polymerization, as well as determine how much fetal hemoglobin is necessary.
That could help in establishing better dosage guidelines for hydroxyurea or in developing new and more effective drugs for SCD, according to the researchers.
“The models give us a way to do preliminary testing on new approaches to stopping this disease,” Dr Karniadakis said. “Now that we can simulate the entire polymerization process, we think the models will be much more useful.” ![]()
Computer models have revealed new details of what happens inside a red blood cell affected by sickle cell disease (SCD), according to research published in Biophysical Journal.
In patients with SCD, mutated hemoglobin can polymerize, assembling into long fibers that push against the membranes of red blood cells and force them out of shape.
“The goal of our work is to model both how these sickle hemoglobin fibers form as well as the mechanical properties of those fibers,” said study author Lu Lu, a PhD student at Brown University in Providence, Rhode Island.
“There had been separate models for each of these things individually developed by us, but this brings those together into one comprehensive model.”
The model uses detailed biomechanical data on how sickle hemoglobin molecules behave and bind with each other to simulate the assembly of a polymer fiber.
Prior to this work, the problem had been that, as the fiber grows, so does the amount of data the model must crunch. Modeling an entire polymer fiber at a cellular scale using the details of each molecule was simply too computationally expensive.
“Even the world’s fastest supercomputers wouldn’t be able to handle it,” said study author George Karniadakis, PhD, of Brown University.
“There’s just too much happening and no way to capture it all computationally. That’s what we were able to overcome with this work.”
The researchers’ solution was to apply what they call a mesoscopic adaptive resolution scheme (MARS).
The MARS model calculates the detailed dynamics of each individual hemoglobin molecule only at the end of polymer fibers, where new molecules are being recruited into the fiber.
Once 4 layers of a fiber have been established, the model automatically dials back the resolution at which it represents that section. The model retains the important information about how the fiber behaves mechanically but glosses over the fine details of each constituent molecule.
“By eliminating the fine details where we don’t need them, we develop a model that can simulate this whole process and its effects on a red blood cell,” Dr Karniadakis said.
Using the new MARS simulations, the researchers were able to show how different configurations of growing polymer fibers are able to produce cells with different shapes.
“We are able to produce a polymerization profile for each of the cell types associated with the disease,” Dr Karniadakis said. “Now, the goal is to use these models to look for ways of preventing the disease onset.”
Using these new models, Dr Karniadakis and his colleagues can run simulations that include fetal hemoglobin. Those simulations could be used to confirm the theory that fetal hemoglobin disrupts polymerization, as well as determine how much fetal hemoglobin is necessary.
That could help in establishing better dosage guidelines for hydroxyurea or in developing new and more effective drugs for SCD, according to the researchers.
“The models give us a way to do preliminary testing on new approaches to stopping this disease,” Dr Karniadakis said. “Now that we can simulate the entire polymerization process, we think the models will be much more useful.” ![]()
Computer models have revealed new details of what happens inside a red blood cell affected by sickle cell disease (SCD), according to research published in Biophysical Journal.
In patients with SCD, mutated hemoglobin can polymerize, assembling into long fibers that push against the membranes of red blood cells and force them out of shape.
“The goal of our work is to model both how these sickle hemoglobin fibers form as well as the mechanical properties of those fibers,” said study author Lu Lu, a PhD student at Brown University in Providence, Rhode Island.
“There had been separate models for each of these things individually developed by us, but this brings those together into one comprehensive model.”
The model uses detailed biomechanical data on how sickle hemoglobin molecules behave and bind with each other to simulate the assembly of a polymer fiber.
Prior to this work, the problem had been that, as the fiber grows, so does the amount of data the model must crunch. Modeling an entire polymer fiber at a cellular scale using the details of each molecule was simply too computationally expensive.
“Even the world’s fastest supercomputers wouldn’t be able to handle it,” said study author George Karniadakis, PhD, of Brown University.
“There’s just too much happening and no way to capture it all computationally. That’s what we were able to overcome with this work.”
The researchers’ solution was to apply what they call a mesoscopic adaptive resolution scheme (MARS).
The MARS model calculates the detailed dynamics of each individual hemoglobin molecule only at the end of polymer fibers, where new molecules are being recruited into the fiber.
Once 4 layers of a fiber have been established, the model automatically dials back the resolution at which it represents that section. The model retains the important information about how the fiber behaves mechanically but glosses over the fine details of each constituent molecule.
“By eliminating the fine details where we don’t need them, we develop a model that can simulate this whole process and its effects on a red blood cell,” Dr Karniadakis said.
Using the new MARS simulations, the researchers were able to show how different configurations of growing polymer fibers are able to produce cells with different shapes.
“We are able to produce a polymerization profile for each of the cell types associated with the disease,” Dr Karniadakis said. “Now, the goal is to use these models to look for ways of preventing the disease onset.”
Using these new models, Dr Karniadakis and his colleagues can run simulations that include fetal hemoglobin. Those simulations could be used to confirm the theory that fetal hemoglobin disrupts polymerization, as well as determine how much fetal hemoglobin is necessary.
That could help in establishing better dosage guidelines for hydroxyurea or in developing new and more effective drugs for SCD, according to the researchers.
“The models give us a way to do preliminary testing on new approaches to stopping this disease,” Dr Karniadakis said. “Now that we can simulate the entire polymerization process, we think the models will be much more useful.” ![]()
FDA grants drug breakthrough designation for AML

The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to venetoclax (Venclexta®).
The designation is for venetoclax in combination with low-dose cytarabine to treat elderly patients with previously untreated acute myeloid leukemia (AML) who are ineligible for intensive chemotherapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About venetoclax
Venetoclax is a small molecule designed to selectively bind and inhibit the BCL-2 protein. The drug is being developed by AbbVie and Roche.
Last year, the FDA granted venetoclax accelerated approval to treat patients with chronic lymphocytic leukemia who have 17p deletion and have received at least one prior therapy. Continued approval of venetoclax for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA granted venetoclax breakthrough therapy designation for the AML indication based on data from an ongoing phase 1/2 study. Preliminary data from the study were presented at the 22nd European Hematology Association (EHA) Annual Congress.
The presentation included data on 61 elderly patients (older than 65) with previously untreated AML who were ineligible for intensive chemotherapy.
They received venetoclax in combination with low-dose cytarabine (as well as prophylaxis for tumor lysis syndrome). The patients’ median time on treatment was 6 months (range, <1 to 19 months), and 72% of patients discontinued treatment.
The overall response rate was 65%. Twenty-five percent of patients achieved a complete response, 38% had a complete response with incomplete blood count recovery, and 2% had a partial response.
The 30-day death rate was 3%, the 60-day death rate was 15%, and the median overall survival was approximately 12 months.
The most common adverse events of any grade (occurring in at least 30% of patients) were nausea (74%), hypokalemia (46%), diarrhea (46%), fatigue (44%), decreased appetite (41%), constipation (34%), hypomagnesemia (34%), vomiting (31%), thrombocytopenia (44%), febrile neutropenia (38%), and neutropenia (33%).
Grade 3/4 adverse events (occurring in at least 10% of patients) included thrombocytopenia (44%), febrile neutropenia (36%), neutropenia (33%), anemia (28%), hypokalemia (16%), hypophosphatemia (13%), and hypertension (12%). ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to venetoclax (Venclexta®).
The designation is for venetoclax in combination with low-dose cytarabine to treat elderly patients with previously untreated acute myeloid leukemia (AML) who are ineligible for intensive chemotherapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About venetoclax
Venetoclax is a small molecule designed to selectively bind and inhibit the BCL-2 protein. The drug is being developed by AbbVie and Roche.
Last year, the FDA granted venetoclax accelerated approval to treat patients with chronic lymphocytic leukemia who have 17p deletion and have received at least one prior therapy. Continued approval of venetoclax for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA granted venetoclax breakthrough therapy designation for the AML indication based on data from an ongoing phase 1/2 study. Preliminary data from the study were presented at the 22nd European Hematology Association (EHA) Annual Congress.
The presentation included data on 61 elderly patients (older than 65) with previously untreated AML who were ineligible for intensive chemotherapy.
They received venetoclax in combination with low-dose cytarabine (as well as prophylaxis for tumor lysis syndrome). The patients’ median time on treatment was 6 months (range, <1 to 19 months), and 72% of patients discontinued treatment.
The overall response rate was 65%. Twenty-five percent of patients achieved a complete response, 38% had a complete response with incomplete blood count recovery, and 2% had a partial response.
The 30-day death rate was 3%, the 60-day death rate was 15%, and the median overall survival was approximately 12 months.
The most common adverse events of any grade (occurring in at least 30% of patients) were nausea (74%), hypokalemia (46%), diarrhea (46%), fatigue (44%), decreased appetite (41%), constipation (34%), hypomagnesemia (34%), vomiting (31%), thrombocytopenia (44%), febrile neutropenia (38%), and neutropenia (33%).
Grade 3/4 adverse events (occurring in at least 10% of patients) included thrombocytopenia (44%), febrile neutropenia (36%), neutropenia (33%), anemia (28%), hypokalemia (16%), hypophosphatemia (13%), and hypertension (12%). ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation to venetoclax (Venclexta®).
The designation is for venetoclax in combination with low-dose cytarabine to treat elderly patients with previously untreated acute myeloid leukemia (AML) who are ineligible for intensive chemotherapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
Breakthrough designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as a rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About venetoclax
Venetoclax is a small molecule designed to selectively bind and inhibit the BCL-2 protein. The drug is being developed by AbbVie and Roche.
Last year, the FDA granted venetoclax accelerated approval to treat patients with chronic lymphocytic leukemia who have 17p deletion and have received at least one prior therapy. Continued approval of venetoclax for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA granted venetoclax breakthrough therapy designation for the AML indication based on data from an ongoing phase 1/2 study. Preliminary data from the study were presented at the 22nd European Hematology Association (EHA) Annual Congress.
The presentation included data on 61 elderly patients (older than 65) with previously untreated AML who were ineligible for intensive chemotherapy.
They received venetoclax in combination with low-dose cytarabine (as well as prophylaxis for tumor lysis syndrome). The patients’ median time on treatment was 6 months (range, <1 to 19 months), and 72% of patients discontinued treatment.
The overall response rate was 65%. Twenty-five percent of patients achieved a complete response, 38% had a complete response with incomplete blood count recovery, and 2% had a partial response.
The 30-day death rate was 3%, the 60-day death rate was 15%, and the median overall survival was approximately 12 months.
The most common adverse events of any grade (occurring in at least 30% of patients) were nausea (74%), hypokalemia (46%), diarrhea (46%), fatigue (44%), decreased appetite (41%), constipation (34%), hypomagnesemia (34%), vomiting (31%), thrombocytopenia (44%), febrile neutropenia (38%), and neutropenia (33%).
Grade 3/4 adverse events (occurring in at least 10% of patients) included thrombocytopenia (44%), febrile neutropenia (36%), neutropenia (33%), anemia (28%), hypokalemia (16%), hypophosphatemia (13%), and hypertension (12%). ![]()
FDA clears new plasmapheresis system

The US Food and Drug Administration (FDA) has granted 510(k) clearance for Haemonetics Corporation’s NexSys PCS™ plasmapheresis system.
The open architecture of NexSys PCS facilitates bi-directional connectivity to donor management systems, enabling automated collection procedure programming and automated end-of-procedure documentation.
The guided operation, large touch screen, and on-screen troubleshooting assistance on NexSys PCS are designed to improve plasma center efficiency. The goal is to reduce donors’ time in centers and improve the centers’ collection capacity.
“NexSys PCS is designed to increase productivity and improve quality and compliance in plasma collection centers,” said Christopher Simon, CEO of Haemonetics.
“Each of these benefits is noteworthy and, when combined, we believe will unlock meaningful value for our customers.”
Haemonetics plans to begin limited production of NexSys PCS devices immediately and to pursue further regulatory clearances for additional enhancements to the overall product offering.
NexSys PCS was previously referred to as PCS 300. ![]()
The US Food and Drug Administration (FDA) has granted 510(k) clearance for Haemonetics Corporation’s NexSys PCS™ plasmapheresis system.
The open architecture of NexSys PCS facilitates bi-directional connectivity to donor management systems, enabling automated collection procedure programming and automated end-of-procedure documentation.
The guided operation, large touch screen, and on-screen troubleshooting assistance on NexSys PCS are designed to improve plasma center efficiency. The goal is to reduce donors’ time in centers and improve the centers’ collection capacity.
“NexSys PCS is designed to increase productivity and improve quality and compliance in plasma collection centers,” said Christopher Simon, CEO of Haemonetics.
“Each of these benefits is noteworthy and, when combined, we believe will unlock meaningful value for our customers.”
Haemonetics plans to begin limited production of NexSys PCS devices immediately and to pursue further regulatory clearances for additional enhancements to the overall product offering.
NexSys PCS was previously referred to as PCS 300. ![]()
The US Food and Drug Administration (FDA) has granted 510(k) clearance for Haemonetics Corporation’s NexSys PCS™ plasmapheresis system.
The open architecture of NexSys PCS facilitates bi-directional connectivity to donor management systems, enabling automated collection procedure programming and automated end-of-procedure documentation.
The guided operation, large touch screen, and on-screen troubleshooting assistance on NexSys PCS are designed to improve plasma center efficiency. The goal is to reduce donors’ time in centers and improve the centers’ collection capacity.
“NexSys PCS is designed to increase productivity and improve quality and compliance in plasma collection centers,” said Christopher Simon, CEO of Haemonetics.
“Each of these benefits is noteworthy and, when combined, we believe will unlock meaningful value for our customers.”
Haemonetics plans to begin limited production of NexSys PCS devices immediately and to pursue further regulatory clearances for additional enhancements to the overall product offering.
NexSys PCS was previously referred to as PCS 300. ![]()
Glioblastoma: Prognosis is poor, but new therapies are emerging
The questioning of former FBI director James B. Comey by Sen. John McCain (R-Ariz.) during a June 8 Senate Intelligence Committee hearing raised more than a few eyebrows; Sen. McCain seemed confused and disoriented, at one point referring to Mr. Comey as “President Comey,” but a possible medical explanation emerged soon after.
On July 14, Sen. McCain, 80, underwent surgery to remove a 5-cm blood clot that had been discovered above his left eye during a physical, and on July 19, the Mayo Clinic in Phoenix, where he had undergone the procedure, announced at his request that, “subsequent tissue pathology revealed that a primary brain tumor known as a glioblastoma was associated with the blood clot.”
Glioblastoma features
While Sen. McCain’s symptoms can’t necessarily be attributed to the glioblastoma, it is not unusual for glioblastoma patients to present with some sort of neurologic deficit, such as speech issues, unilateral weakness, or confusion, according to Eudocia Quant Lee, MD, a neuro-oncologist at Dana-Farber Cancer Institute, Boston.

Neuro-oncologist Manmeet Singh Ahluwalia, MD, of the Cleveland Clinic said seizures, persistent headaches, double or blurred vision, and changes in ability to think and learn can also be presenting symptoms.
Glioblastoma is the most common malignant primary brain tumor diagnosed in adults, with an estimated 12,000-13,000 new cases occurring each year in the United States. It is more common among older adults but can occur in younger patients. It arises in the brain and generally stays within the central nervous system, Dr. Lee explained, noting that it is much less common than lung cancer, breast cancer, and melanoma.

This is particularly true for older patients.
Prognosis and age
“We know, in general – as with most cancers – that the older you’re diagnosed with your cancer, the poorer your prognosis is,” she said, adding that other health issues and the ability to tolerate treatment can affect outcomes.
Outcomes also can be affected by type of surgery, functional status, extent of treatment, and molecular subtypes of the glioblastoma, Dr. Ahluwalia said.
Survival generally ranges about 14-18 months, although about 10% of patients live 5 years or longer.

The study, presented in a poster by Michael Weller, MD, of University Hospital and University of Zürich and his colleagues, also showed that, compared with the 398 older patients who survived less than 2 years (median, 6.2 months), those who survived longer had “more intensive up-front treatment and a trend toward higher initial Karnofsky performance scores as distinguishing clinical factors.”
In addition, molecular analyses showed more frequent O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation in those with longer survival, while isocitrate dehydrogenase (IDH) mutations were restricted to single patients.
“Collectively, our findings confirm that LTS is rare in elderly patients with glioblastoma and that clinical and tumor-associated molecular factors linked to LTS resemble those in standard-age patients, except for less common IDH mutation,” the investigators wrote.
Another abstract published online in conjunction with the ASCO annual meeting looked at outcomes, based on age and MGMT analysis, and similarly found that aggressive treatment with chemoradiation is associated with better outcomes in both younger and older patients.
In that study by Suryanarayan Mohapatra, MD, of Cleveland Clinic–Fairview Hospital, and his colleagues – including Dr. Ahluwalia – 567 of 1,165 patients were aged 65 years or older. The benefits of chemoradiation therapy, which was associated with a significantly lower risk of death vs. radiation therapy alone in the study, were more pronounced among the older patients (hazard ratio, 0.45 vs. 0.61 for those under age 65 years), but the difference did not reach statistical significance.
Dr. Mohapatra and his colleagues also showed that more aggressive therapy resulted in better overall and progression-free survival regardless of MGMT methylation status, but that there was no difference between the age groups on this measure. Overall and progression-free survival also were significantly better with gross-total resection and subtotal resection vs. biopsy only, and with diagnosis during 2009 and later vs. during 2007-2008. However, a difference between the two age groups was seen with respect to overall survival only among those diagnosed during 2009 or later, with a more prominent impact among the younger group, the investigators reported.
“Older individuals often get less aggressive treatment. However, based on the research, active and functional older patients should get aggressive treatment,” Dr. Ahluwalia said. “We advocate tailor-made treatment that takes into account patient condition, location of tumor, functional status, etc., in addition to patient age.”
Standard treatment approaches
The first step in the treatment of glioblastomas is maximal safe therapy, Dr. Lee said.
“You want to achieve as much of a resection as possible without leaving the patient with some sort of permanent neurologic deficit that could severely compromise the quality of their life,” she explained.
Sen. McCain’s tumor was “completely resected by imaging criteria,” according to the Mayo Clinic statement, which also noted that treatment options might include a combination of chemotherapy and radiation.
Indeed, the standard of care for glioblastomas after surgery is combined chemotherapy and radiation – typically given as approximately 6 weeks of radiation combined with oral temozolomide chemotherapy – followed by 6 monthly cycles of temozolomide, she explained.
Radiation is sometimes given for only 3 weeks, but this option is mainly reserved for elderly patients, she said, adding that trials in patients aged 65-70 years have shown that this shorter course of radiation can be equally effective but potentially less toxic.
Emerging treatment approaches
Another treatment that has shown promise involves the use of tumor-treating fields (TTFields) – a locoregionally delivered antimitotic treatment that disrupts cell division and organelle assembly.
A 2015 phase 3 trial showed that adding TTFields to maintenance temozolomide significantly prolonged progression-free and overall survival, Dr. Ahluwalia said.
Other studies, including two presented during poster sessions at the ASCO annual meeting, have shown a progression-free survival benefit with the addition of bevacizumab to the treatment regimen. One open-label phase 2 study showed that hypofractionated radiotherapy in combination with IV bevacizumab every 2 weeks vs. radiotherapy alone in newly diagnosed patients over age 65 years improved progression-free survival (median, 7.6 vs. 4.8 months), but not overall survival (median, 12.1 vs. 12.2 months).
Another phase 2 study presented by Phioanh (Leia) Nghiemphu, MD, of the University of California, Los Angeles, showed that in newly diagnosed patients aged 70 years and older, upfront treatment with bevacizumab and temozolomide was associated with promising survival benefits (overall survival, 12.3 months; progression-free survival, 5.1 months) and tolerable side effects. The best survival in multivariate analysis was in patients who received radiotherapy at progression; it was unclear whether the addition of bevacizumab led to a survival advantage, but it may have allowed delay of radiotherapy treatment, she noted.
“Although we have no cure for glioblastoma, treatments can control tumor growth for a period of time, and there are additional promising therapies emerging every day to treat this deadly cancer, she said in an interview. “There has been increasing interest in developing better therapies for the older patients with glioblastoma with less toxicity and still-robust survival, such as the addition of bevacizumab or a short course of radiotherapy with temozolomide chemotherapy.”
Dr. Ahluwalia encourages clinical trial participation for patients diagnosed with glioblastoma and noted that he is particularly excited about immunotherapy and targeted therapy trials.
Dr. Lee has served as consultant to Eli Lilly. Dr. Ahluwalia disclosed a financial relationship with multiple companies, including Novocure, which markets a TTFields device. Dr. Weller disclosed a financial relationships with multiple companies, including Novocure; Merck Sharp & Dohme, which markets temozolomide; and Roche, which markets bevacizumab. Dr. Nghiemphu has received research funding from Genentech/Roche and Novartis. Dr. Mohapatra reporting having no disclosures.
The questioning of former FBI director James B. Comey by Sen. John McCain (R-Ariz.) during a June 8 Senate Intelligence Committee hearing raised more than a few eyebrows; Sen. McCain seemed confused and disoriented, at one point referring to Mr. Comey as “President Comey,” but a possible medical explanation emerged soon after.
On July 14, Sen. McCain, 80, underwent surgery to remove a 5-cm blood clot that had been discovered above his left eye during a physical, and on July 19, the Mayo Clinic in Phoenix, where he had undergone the procedure, announced at his request that, “subsequent tissue pathology revealed that a primary brain tumor known as a glioblastoma was associated with the blood clot.”
Glioblastoma features
While Sen. McCain’s symptoms can’t necessarily be attributed to the glioblastoma, it is not unusual for glioblastoma patients to present with some sort of neurologic deficit, such as speech issues, unilateral weakness, or confusion, according to Eudocia Quant Lee, MD, a neuro-oncologist at Dana-Farber Cancer Institute, Boston.
Neuro-oncologist Manmeet Singh Ahluwalia, MD, of the Cleveland Clinic said seizures, persistent headaches, double or blurred vision, and changes in ability to think and learn can also be presenting symptoms.
Glioblastoma is the most common malignant primary brain tumor diagnosed in adults, with an estimated 12,000-13,000 new cases occurring each year in the United States. It is more common among older adults but can occur in younger patients. It arises in the brain and generally stays within the central nervous system, Dr. Lee explained, noting that it is much less common than lung cancer, breast cancer, and melanoma.
This is particularly true for older patients.
Prognosis and age
“We know, in general – as with most cancers – that the older you’re diagnosed with your cancer, the poorer your prognosis is,” she said, adding that other health issues and the ability to tolerate treatment can affect outcomes.
Outcomes also can be affected by type of surgery, functional status, extent of treatment, and molecular subtypes of the glioblastoma, Dr. Ahluwalia said.
Survival generally ranges about 14-18 months, although about 10% of patients live 5 years or longer.
The study, presented in a poster by Michael Weller, MD, of University Hospital and University of Zürich and his colleagues, also showed that, compared with the 398 older patients who survived less than 2 years (median, 6.2 months), those who survived longer had “more intensive up-front treatment and a trend toward higher initial Karnofsky performance scores as distinguishing clinical factors.”
In addition, molecular analyses showed more frequent O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation in those with longer survival, while isocitrate dehydrogenase (IDH) mutations were restricted to single patients.
“Collectively, our findings confirm that LTS is rare in elderly patients with glioblastoma and that clinical and tumor-associated molecular factors linked to LTS resemble those in standard-age patients, except for less common IDH mutation,” the investigators wrote.
Another abstract published online in conjunction with the ASCO annual meeting looked at outcomes, based on age and MGMT analysis, and similarly found that aggressive treatment with chemoradiation is associated with better outcomes in both younger and older patients.
In that study by Suryanarayan Mohapatra, MD, of Cleveland Clinic–Fairview Hospital, and his colleagues – including Dr. Ahluwalia – 567 of 1,165 patients were aged 65 years or older. The benefits of chemoradiation therapy, which was associated with a significantly lower risk of death vs. radiation therapy alone in the study, were more pronounced among the older patients (hazard ratio, 0.45 vs. 0.61 for those under age 65 years), but the difference did not reach statistical significance.
Dr. Mohapatra and his colleagues also showed that more aggressive therapy resulted in better overall and progression-free survival regardless of MGMT methylation status, but that there was no difference between the age groups on this measure. Overall and progression-free survival also were significantly better with gross-total resection and subtotal resection vs. biopsy only, and with diagnosis during 2009 and later vs. during 2007-2008. However, a difference between the two age groups was seen with respect to overall survival only among those diagnosed during 2009 or later, with a more prominent impact among the younger group, the investigators reported.
“Older individuals often get less aggressive treatment. However, based on the research, active and functional older patients should get aggressive treatment,” Dr. Ahluwalia said. “We advocate tailor-made treatment that takes into account patient condition, location of tumor, functional status, etc., in addition to patient age.”
Standard treatment approaches
The first step in the treatment of glioblastomas is maximal safe therapy, Dr. Lee said.
“You want to achieve as much of a resection as possible without leaving the patient with some sort of permanent neurologic deficit that could severely compromise the quality of their life,” she explained.
Sen. McCain’s tumor was “completely resected by imaging criteria,” according to the Mayo Clinic statement, which also noted that treatment options might include a combination of chemotherapy and radiation.
Indeed, the standard of care for glioblastomas after surgery is combined chemotherapy and radiation – typically given as approximately 6 weeks of radiation combined with oral temozolomide chemotherapy – followed by 6 monthly cycles of temozolomide, she explained.
Radiation is sometimes given for only 3 weeks, but this option is mainly reserved for elderly patients, she said, adding that trials in patients aged 65-70 years have shown that this shorter course of radiation can be equally effective but potentially less toxic.
Emerging treatment approaches
Another treatment that has shown promise involves the use of tumor-treating fields (TTFields) – a locoregionally delivered antimitotic treatment that disrupts cell division and organelle assembly.
A 2015 phase 3 trial showed that adding TTFields to maintenance temozolomide significantly prolonged progression-free and overall survival, Dr. Ahluwalia said.
Other studies, including two presented during poster sessions at the ASCO annual meeting, have shown a progression-free survival benefit with the addition of bevacizumab to the treatment regimen. One open-label phase 2 study showed that hypofractionated radiotherapy in combination with IV bevacizumab every 2 weeks vs. radiotherapy alone in newly diagnosed patients over age 65 years improved progression-free survival (median, 7.6 vs. 4.8 months), but not overall survival (median, 12.1 vs. 12.2 months).
Another phase 2 study presented by Phioanh (Leia) Nghiemphu, MD, of the University of California, Los Angeles, showed that in newly diagnosed patients aged 70 years and older, upfront treatment with bevacizumab and temozolomide was associated with promising survival benefits (overall survival, 12.3 months; progression-free survival, 5.1 months) and tolerable side effects. The best survival in multivariate analysis was in patients who received radiotherapy at progression; it was unclear whether the addition of bevacizumab led to a survival advantage, but it may have allowed delay of radiotherapy treatment, she noted.
“Although we have no cure for glioblastoma, treatments can control tumor growth for a period of time, and there are additional promising therapies emerging every day to treat this deadly cancer, she said in an interview. “There has been increasing interest in developing better therapies for the older patients with glioblastoma with less toxicity and still-robust survival, such as the addition of bevacizumab or a short course of radiotherapy with temozolomide chemotherapy.”
Dr. Ahluwalia encourages clinical trial participation for patients diagnosed with glioblastoma and noted that he is particularly excited about immunotherapy and targeted therapy trials.
Dr. Lee has served as consultant to Eli Lilly. Dr. Ahluwalia disclosed a financial relationship with multiple companies, including Novocure, which markets a TTFields device. Dr. Weller disclosed a financial relationships with multiple companies, including Novocure; Merck Sharp & Dohme, which markets temozolomide; and Roche, which markets bevacizumab. Dr. Nghiemphu has received research funding from Genentech/Roche and Novartis. Dr. Mohapatra reporting having no disclosures.
The questioning of former FBI director James B. Comey by Sen. John McCain (R-Ariz.) during a June 8 Senate Intelligence Committee hearing raised more than a few eyebrows; Sen. McCain seemed confused and disoriented, at one point referring to Mr. Comey as “President Comey,” but a possible medical explanation emerged soon after.
On July 14, Sen. McCain, 80, underwent surgery to remove a 5-cm blood clot that had been discovered above his left eye during a physical, and on July 19, the Mayo Clinic in Phoenix, where he had undergone the procedure, announced at his request that, “subsequent tissue pathology revealed that a primary brain tumor known as a glioblastoma was associated with the blood clot.”
Glioblastoma features
While Sen. McCain’s symptoms can’t necessarily be attributed to the glioblastoma, it is not unusual for glioblastoma patients to present with some sort of neurologic deficit, such as speech issues, unilateral weakness, or confusion, according to Eudocia Quant Lee, MD, a neuro-oncologist at Dana-Farber Cancer Institute, Boston.
Neuro-oncologist Manmeet Singh Ahluwalia, MD, of the Cleveland Clinic said seizures, persistent headaches, double or blurred vision, and changes in ability to think and learn can also be presenting symptoms.
Glioblastoma is the most common malignant primary brain tumor diagnosed in adults, with an estimated 12,000-13,000 new cases occurring each year in the United States. It is more common among older adults but can occur in younger patients. It arises in the brain and generally stays within the central nervous system, Dr. Lee explained, noting that it is much less common than lung cancer, breast cancer, and melanoma.
This is particularly true for older patients.
Prognosis and age
“We know, in general – as with most cancers – that the older you’re diagnosed with your cancer, the poorer your prognosis is,” she said, adding that other health issues and the ability to tolerate treatment can affect outcomes.
Outcomes also can be affected by type of surgery, functional status, extent of treatment, and molecular subtypes of the glioblastoma, Dr. Ahluwalia said.
Survival generally ranges about 14-18 months, although about 10% of patients live 5 years or longer.
The study, presented in a poster by Michael Weller, MD, of University Hospital and University of Zürich and his colleagues, also showed that, compared with the 398 older patients who survived less than 2 years (median, 6.2 months), those who survived longer had “more intensive up-front treatment and a trend toward higher initial Karnofsky performance scores as distinguishing clinical factors.”
In addition, molecular analyses showed more frequent O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation in those with longer survival, while isocitrate dehydrogenase (IDH) mutations were restricted to single patients.
“Collectively, our findings confirm that LTS is rare in elderly patients with glioblastoma and that clinical and tumor-associated molecular factors linked to LTS resemble those in standard-age patients, except for less common IDH mutation,” the investigators wrote.
Another abstract published online in conjunction with the ASCO annual meeting looked at outcomes, based on age and MGMT analysis, and similarly found that aggressive treatment with chemoradiation is associated with better outcomes in both younger and older patients.
In that study by Suryanarayan Mohapatra, MD, of Cleveland Clinic–Fairview Hospital, and his colleagues – including Dr. Ahluwalia – 567 of 1,165 patients were aged 65 years or older. The benefits of chemoradiation therapy, which was associated with a significantly lower risk of death vs. radiation therapy alone in the study, were more pronounced among the older patients (hazard ratio, 0.45 vs. 0.61 for those under age 65 years), but the difference did not reach statistical significance.
Dr. Mohapatra and his colleagues also showed that more aggressive therapy resulted in better overall and progression-free survival regardless of MGMT methylation status, but that there was no difference between the age groups on this measure. Overall and progression-free survival also were significantly better with gross-total resection and subtotal resection vs. biopsy only, and with diagnosis during 2009 and later vs. during 2007-2008. However, a difference between the two age groups was seen with respect to overall survival only among those diagnosed during 2009 or later, with a more prominent impact among the younger group, the investigators reported.
“Older individuals often get less aggressive treatment. However, based on the research, active and functional older patients should get aggressive treatment,” Dr. Ahluwalia said. “We advocate tailor-made treatment that takes into account patient condition, location of tumor, functional status, etc., in addition to patient age.”
Standard treatment approaches
The first step in the treatment of glioblastomas is maximal safe therapy, Dr. Lee said.
“You want to achieve as much of a resection as possible without leaving the patient with some sort of permanent neurologic deficit that could severely compromise the quality of their life,” she explained.
Sen. McCain’s tumor was “completely resected by imaging criteria,” according to the Mayo Clinic statement, which also noted that treatment options might include a combination of chemotherapy and radiation.
Indeed, the standard of care for glioblastomas after surgery is combined chemotherapy and radiation – typically given as approximately 6 weeks of radiation combined with oral temozolomide chemotherapy – followed by 6 monthly cycles of temozolomide, she explained.
Radiation is sometimes given for only 3 weeks, but this option is mainly reserved for elderly patients, she said, adding that trials in patients aged 65-70 years have shown that this shorter course of radiation can be equally effective but potentially less toxic.
Emerging treatment approaches
Another treatment that has shown promise involves the use of tumor-treating fields (TTFields) – a locoregionally delivered antimitotic treatment that disrupts cell division and organelle assembly.
A 2015 phase 3 trial showed that adding TTFields to maintenance temozolomide significantly prolonged progression-free and overall survival, Dr. Ahluwalia said.
Other studies, including two presented during poster sessions at the ASCO annual meeting, have shown a progression-free survival benefit with the addition of bevacizumab to the treatment regimen. One open-label phase 2 study showed that hypofractionated radiotherapy in combination with IV bevacizumab every 2 weeks vs. radiotherapy alone in newly diagnosed patients over age 65 years improved progression-free survival (median, 7.6 vs. 4.8 months), but not overall survival (median, 12.1 vs. 12.2 months).
Another phase 2 study presented by Phioanh (Leia) Nghiemphu, MD, of the University of California, Los Angeles, showed that in newly diagnosed patients aged 70 years and older, upfront treatment with bevacizumab and temozolomide was associated with promising survival benefits (overall survival, 12.3 months; progression-free survival, 5.1 months) and tolerable side effects. The best survival in multivariate analysis was in patients who received radiotherapy at progression; it was unclear whether the addition of bevacizumab led to a survival advantage, but it may have allowed delay of radiotherapy treatment, she noted.
“Although we have no cure for glioblastoma, treatments can control tumor growth for a period of time, and there are additional promising therapies emerging every day to treat this deadly cancer, she said in an interview. “There has been increasing interest in developing better therapies for the older patients with glioblastoma with less toxicity and still-robust survival, such as the addition of bevacizumab or a short course of radiotherapy with temozolomide chemotherapy.”
Dr. Ahluwalia encourages clinical trial participation for patients diagnosed with glioblastoma and noted that he is particularly excited about immunotherapy and targeted therapy trials.
Dr. Lee has served as consultant to Eli Lilly. Dr. Ahluwalia disclosed a financial relationship with multiple companies, including Novocure, which markets a TTFields device. Dr. Weller disclosed a financial relationships with multiple companies, including Novocure; Merck Sharp & Dohme, which markets temozolomide; and Roche, which markets bevacizumab. Dr. Nghiemphu has received research funding from Genentech/Roche and Novartis. Dr. Mohapatra reporting having no disclosures.
When the painful ‘bumps’ are calciphylaxis, what’s next?
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.

Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
Six Steps to Reduce Taxes on Investments: Minimizing What You Pay in a Tough Environment
Orthopedic physicians in the highest income tax brackets may have been presented with an unpleasant surprise in recent years when they learned of their investment tax liability. A prolonged period of strong domestic stock performance from 2009 to 2016, combined with the implementation of The American Taxpayer Relief Act of 2012, may have resulted in significantly higher taxes for many of you.
The top ordinary income tax rates increased by 24% when including the Net Investment Income surtax, while the top capital gains rate was increased by more than 58%. Writing a large check to the Internal Revenue Service serves as a harsh reminder that tax planning requires attention throughout the year, and is not a technique you can properly manage a few weeks before an April 15 deadline.
Proper tax planning became more critical as we moved into an era of higher taxes. A multi-year bull market for domestic stocks has caused many traditional investment vehicles to hold large amounts of unrealized gains, which can become realized gains if you are not careful. Most major equity indices took a breath in 2015 and finished the year in the red, which created a planning opportunity for astute investors and their advisors. Stocks in the US and emerging market countries quickly bounced back in 2016; however, European stocks struggled and continue to trade well below peak levels reached nearly a decade ago. Investors who missed the opportunity to offset gains of the prior 2 years may have an opportunity to reduce their tax bill in 2017.
In this article, we will provide you with 6 suggestions that could save you thousands of dollars in investment taxes over the next several years.
1. Account Registration Matters: A common mistake investors make is the failure to implement a tax diversification strategy. Brokerage accounts, Roth IRAs, and qualified plans are subject to various forms of taxation. It is important to utilize the tax advantages of these tools to ensure they work for you in the most productive manner possible. A properly integrated approach is critical during your accumulation phase. Further, it is just as important when you enter the distribution period of your investment life cycle (ie, retirement).
Master Limited Partnerships offer a potentially advantageous income stream for a brokerage account, while it is generally preferable for qualified accounts to own high yield bonds and corporate debt, as they are taxed at ordinary income rates. There are countless additional examples we could discuss, but the lesson is simple: it is important to review the pieces of your plan with an advisor who will consider both tax diversification and security diversification as they relate to your specific circumstances.
2. Consider Owning Municipal Bonds in Taxable Accounts: Most municipal bonds are exempt from federal taxation. Certain issues may also be exempt from state and local taxes. If you are in the highest federal tax bracket, you may be paying tax on investment income at a rate of 43.4%. Under these circumstances, a municipal bond yielding 3% will provide a superior after tax return in comparison to a corporate bond yielding 5% in an individual or joint registration, a pass-through LLC, or in many trust accounts. Therefore, it is important in many circumstances to make certain your long-term plan utilizes the advantages of owning certain municipal bonds in taxable accounts.
3. Be Cognizant of Holding Periods: Long-term capital gains rates are much more favorable than short-term rates. Holding a security for a period of 12 months presents an opportunity to save nearly 20% on the taxation of your appreciated position. For example, an initial investment of $50,000 which grows to $100,000 represents a $50,000 unrealized gain. If an investor in the highest tax bracket simply delays liquidation of the position (assuming the security price does not change) the tax savings in this scenario would be $9,800. Although an awareness of the holding period of a security would appear to be a basic principal of investing, many mutual funds and managed accounts are not designed for tax sensitivity. High income investors should be aware that the average client of most advisors is not in the highest federal tax bracket. Therefore, it is generally advantageous to seek the advice of a financial professional with experience executing an appropriate exit strategy that is aware of holding periods.
4. Proactively Realize Losses to Offset Gains: As mentioned in the opening paragraphs of the article, 2015 presented investors with an opportunity to realize losses in domestic stocks for the first time in 4 years. Clients with a diversified portfolio may still have an opportunity to offset gains in domestic stocks by selling foreign equities. One benefit of diversifying across asset classes is that if the portfolio is structured properly, the securities typically will not move in tandem. This divergence of returns among asset classes not only reduces portfolio volatility, but it creates a tax planning opportunity. Domestic equities experienced tremendous appreciation over a 5-year period through 2014; however, international stocks, commodities, and multiple fixed income investments experienced down years. Astute advisors were presented with the opportunity to save clients thousands of dollars in taxes by performing strategic tax swaps prior to year-end. It is important to understand the rules relating to wash sales when executing such tactics. The laws are confusing, and if a mistake is made your loss could be disallowed. Make certain your advisor is well-versed in utilizing tax offsets.
5. Think Twice About Gifting Cash: This is not to discourage your charitable intentions. Quite the opposite is true. However, a successful investor can occasionally find themselves in a precarious position. You may have allocated 5% of your portfolio to a growth stock with significant upside. Several years have passed, the security has experienced explosive growth, and it now represents 15% of your investable assets. Suddenly your portfolio has a concentrated position with significant gains, and the level of risk is no longer consistent with your long-term objectives. The sound practice of rebalancing your portfolio then becomes very costly, because liquidation of the stock could create a taxable event that may negatively impact your net return.
By planning ahead of time, you may be able to gift a portion of the appreciated security to a charitable organization able to accept this type of donation. The value of your gift can be replaced with the cash you originally intended to donate to the charitable organization and, in this scenario, your cash will create a new cost basis. The charity can liquidate the stock without paying tax, and you have removed a future tax liability from your portfolio. Implementing the aforementioned gifting strategy offers the potential to save thousands of dollars in taxes over the life of your portfolio.
6. Understand your Mutual Fund’s Tax Cost Ratio: The technical detail behind a mutual fund’s tax cost ratio is beyond the scope of this article. Our intent is to simply bring this topic to your attention. Tax cost ratio represents the percentage of an investor’s assets that are lost to taxes. Mutual funds avoid double taxation, provided they pay at least 90% of net investment income and realized capital gains to shareholders at the end of the calendar year. But all mutual funds are not created equally, and proper research will allow you to identify funds that are tax efficient.
A well-managed mutual fund will add diversification to a portfolio while creating the opportunity to outperform asset classes with inefficient markets. You do need to be aware of funds with excessive turnover. An understanding of when a fund pays its capital gains distributions is a critical component of successful investing. A poorly timed fund purchase can result in acquiring another investor’s tax liability. It is not unusual for an investor to experience a negative return in a calendar year, yet find himself on the receiving end of a capital gains distribution. Understanding the tax cost ratios of the funds that make up portions of your investment plan will enable you to take advantage of the many benefits of owning mutual funds.
The above steps are by no means the only tax strategies experienced advisors can execute on behalf of their clients. This article highlights several strategies you should discuss with your advisor to determine if implementation is appropriate for your unique portfolio and overall financial situation. Successful investing requires discipline that extends beyond proper security selection. While gross returns are important and should not be ignored, the percentage return you see on your statements does not tell the full story.
In today’s tax environment, successful investors must choose an advisor who will help them look beyond portfolio earnings and focus on strategic after-tax asset growth.
To receive a free hardcopy of Wealth Protection Planning for Orthopaedic Surgeons, please call 877-656-4362. Visit www.ojmbookstore.com and enter promotional code AJO30 for a free ebook download of Wealth Protection Planning or one of our other ebooks for your Kindle or iPad.
Orthopedic physicians in the highest income tax brackets may have been presented with an unpleasant surprise in recent years when they learned of their investment tax liability. A prolonged period of strong domestic stock performance from 2009 to 2016, combined with the implementation of The American Taxpayer Relief Act of 2012, may have resulted in significantly higher taxes for many of you.
The top ordinary income tax rates increased by 24% when including the Net Investment Income surtax, while the top capital gains rate was increased by more than 58%. Writing a large check to the Internal Revenue Service serves as a harsh reminder that tax planning requires attention throughout the year, and is not a technique you can properly manage a few weeks before an April 15 deadline.
Proper tax planning became more critical as we moved into an era of higher taxes. A multi-year bull market for domestic stocks has caused many traditional investment vehicles to hold large amounts of unrealized gains, which can become realized gains if you are not careful. Most major equity indices took a breath in 2015 and finished the year in the red, which created a planning opportunity for astute investors and their advisors. Stocks in the US and emerging market countries quickly bounced back in 2016; however, European stocks struggled and continue to trade well below peak levels reached nearly a decade ago. Investors who missed the opportunity to offset gains of the prior 2 years may have an opportunity to reduce their tax bill in 2017.
In this article, we will provide you with 6 suggestions that could save you thousands of dollars in investment taxes over the next several years.
1. Account Registration Matters: A common mistake investors make is the failure to implement a tax diversification strategy. Brokerage accounts, Roth IRAs, and qualified plans are subject to various forms of taxation. It is important to utilize the tax advantages of these tools to ensure they work for you in the most productive manner possible. A properly integrated approach is critical during your accumulation phase. Further, it is just as important when you enter the distribution period of your investment life cycle (ie, retirement).
Master Limited Partnerships offer a potentially advantageous income stream for a brokerage account, while it is generally preferable for qualified accounts to own high yield bonds and corporate debt, as they are taxed at ordinary income rates. There are countless additional examples we could discuss, but the lesson is simple: it is important to review the pieces of your plan with an advisor who will consider both tax diversification and security diversification as they relate to your specific circumstances.
2. Consider Owning Municipal Bonds in Taxable Accounts: Most municipal bonds are exempt from federal taxation. Certain issues may also be exempt from state and local taxes. If you are in the highest federal tax bracket, you may be paying tax on investment income at a rate of 43.4%. Under these circumstances, a municipal bond yielding 3% will provide a superior after tax return in comparison to a corporate bond yielding 5% in an individual or joint registration, a pass-through LLC, or in many trust accounts. Therefore, it is important in many circumstances to make certain your long-term plan utilizes the advantages of owning certain municipal bonds in taxable accounts.
3. Be Cognizant of Holding Periods: Long-term capital gains rates are much more favorable than short-term rates. Holding a security for a period of 12 months presents an opportunity to save nearly 20% on the taxation of your appreciated position. For example, an initial investment of $50,000 which grows to $100,000 represents a $50,000 unrealized gain. If an investor in the highest tax bracket simply delays liquidation of the position (assuming the security price does not change) the tax savings in this scenario would be $9,800. Although an awareness of the holding period of a security would appear to be a basic principal of investing, many mutual funds and managed accounts are not designed for tax sensitivity. High income investors should be aware that the average client of most advisors is not in the highest federal tax bracket. Therefore, it is generally advantageous to seek the advice of a financial professional with experience executing an appropriate exit strategy that is aware of holding periods.
4. Proactively Realize Losses to Offset Gains: As mentioned in the opening paragraphs of the article, 2015 presented investors with an opportunity to realize losses in domestic stocks for the first time in 4 years. Clients with a diversified portfolio may still have an opportunity to offset gains in domestic stocks by selling foreign equities. One benefit of diversifying across asset classes is that if the portfolio is structured properly, the securities typically will not move in tandem. This divergence of returns among asset classes not only reduces portfolio volatility, but it creates a tax planning opportunity. Domestic equities experienced tremendous appreciation over a 5-year period through 2014; however, international stocks, commodities, and multiple fixed income investments experienced down years. Astute advisors were presented with the opportunity to save clients thousands of dollars in taxes by performing strategic tax swaps prior to year-end. It is important to understand the rules relating to wash sales when executing such tactics. The laws are confusing, and if a mistake is made your loss could be disallowed. Make certain your advisor is well-versed in utilizing tax offsets.
5. Think Twice About Gifting Cash: This is not to discourage your charitable intentions. Quite the opposite is true. However, a successful investor can occasionally find themselves in a precarious position. You may have allocated 5% of your portfolio to a growth stock with significant upside. Several years have passed, the security has experienced explosive growth, and it now represents 15% of your investable assets. Suddenly your portfolio has a concentrated position with significant gains, and the level of risk is no longer consistent with your long-term objectives. The sound practice of rebalancing your portfolio then becomes very costly, because liquidation of the stock could create a taxable event that may negatively impact your net return.
By planning ahead of time, you may be able to gift a portion of the appreciated security to a charitable organization able to accept this type of donation. The value of your gift can be replaced with the cash you originally intended to donate to the charitable organization and, in this scenario, your cash will create a new cost basis. The charity can liquidate the stock without paying tax, and you have removed a future tax liability from your portfolio. Implementing the aforementioned gifting strategy offers the potential to save thousands of dollars in taxes over the life of your portfolio.
6. Understand your Mutual Fund’s Tax Cost Ratio: The technical detail behind a mutual fund’s tax cost ratio is beyond the scope of this article. Our intent is to simply bring this topic to your attention. Tax cost ratio represents the percentage of an investor’s assets that are lost to taxes. Mutual funds avoid double taxation, provided they pay at least 90% of net investment income and realized capital gains to shareholders at the end of the calendar year. But all mutual funds are not created equally, and proper research will allow you to identify funds that are tax efficient.
A well-managed mutual fund will add diversification to a portfolio while creating the opportunity to outperform asset classes with inefficient markets. You do need to be aware of funds with excessive turnover. An understanding of when a fund pays its capital gains distributions is a critical component of successful investing. A poorly timed fund purchase can result in acquiring another investor’s tax liability. It is not unusual for an investor to experience a negative return in a calendar year, yet find himself on the receiving end of a capital gains distribution. Understanding the tax cost ratios of the funds that make up portions of your investment plan will enable you to take advantage of the many benefits of owning mutual funds.
The above steps are by no means the only tax strategies experienced advisors can execute on behalf of their clients. This article highlights several strategies you should discuss with your advisor to determine if implementation is appropriate for your unique portfolio and overall financial situation. Successful investing requires discipline that extends beyond proper security selection. While gross returns are important and should not be ignored, the percentage return you see on your statements does not tell the full story.
In today’s tax environment, successful investors must choose an advisor who will help them look beyond portfolio earnings and focus on strategic after-tax asset growth.
To receive a free hardcopy of Wealth Protection Planning for Orthopaedic Surgeons, please call 877-656-4362. Visit www.ojmbookstore.com and enter promotional code AJO30 for a free ebook download of Wealth Protection Planning or one of our other ebooks for your Kindle or iPad.
Orthopedic physicians in the highest income tax brackets may have been presented with an unpleasant surprise in recent years when they learned of their investment tax liability. A prolonged period of strong domestic stock performance from 2009 to 2016, combined with the implementation of The American Taxpayer Relief Act of 2012, may have resulted in significantly higher taxes for many of you.
The top ordinary income tax rates increased by 24% when including the Net Investment Income surtax, while the top capital gains rate was increased by more than 58%. Writing a large check to the Internal Revenue Service serves as a harsh reminder that tax planning requires attention throughout the year, and is not a technique you can properly manage a few weeks before an April 15 deadline.
Proper tax planning became more critical as we moved into an era of higher taxes. A multi-year bull market for domestic stocks has caused many traditional investment vehicles to hold large amounts of unrealized gains, which can become realized gains if you are not careful. Most major equity indices took a breath in 2015 and finished the year in the red, which created a planning opportunity for astute investors and their advisors. Stocks in the US and emerging market countries quickly bounced back in 2016; however, European stocks struggled and continue to trade well below peak levels reached nearly a decade ago. Investors who missed the opportunity to offset gains of the prior 2 years may have an opportunity to reduce their tax bill in 2017.
In this article, we will provide you with 6 suggestions that could save you thousands of dollars in investment taxes over the next several years.
1. Account Registration Matters: A common mistake investors make is the failure to implement a tax diversification strategy. Brokerage accounts, Roth IRAs, and qualified plans are subject to various forms of taxation. It is important to utilize the tax advantages of these tools to ensure they work for you in the most productive manner possible. A properly integrated approach is critical during your accumulation phase. Further, it is just as important when you enter the distribution period of your investment life cycle (ie, retirement).
Master Limited Partnerships offer a potentially advantageous income stream for a brokerage account, while it is generally preferable for qualified accounts to own high yield bonds and corporate debt, as they are taxed at ordinary income rates. There are countless additional examples we could discuss, but the lesson is simple: it is important to review the pieces of your plan with an advisor who will consider both tax diversification and security diversification as they relate to your specific circumstances.
2. Consider Owning Municipal Bonds in Taxable Accounts: Most municipal bonds are exempt from federal taxation. Certain issues may also be exempt from state and local taxes. If you are in the highest federal tax bracket, you may be paying tax on investment income at a rate of 43.4%. Under these circumstances, a municipal bond yielding 3% will provide a superior after tax return in comparison to a corporate bond yielding 5% in an individual or joint registration, a pass-through LLC, or in many trust accounts. Therefore, it is important in many circumstances to make certain your long-term plan utilizes the advantages of owning certain municipal bonds in taxable accounts.
3. Be Cognizant of Holding Periods: Long-term capital gains rates are much more favorable than short-term rates. Holding a security for a period of 12 months presents an opportunity to save nearly 20% on the taxation of your appreciated position. For example, an initial investment of $50,000 which grows to $100,000 represents a $50,000 unrealized gain. If an investor in the highest tax bracket simply delays liquidation of the position (assuming the security price does not change) the tax savings in this scenario would be $9,800. Although an awareness of the holding period of a security would appear to be a basic principal of investing, many mutual funds and managed accounts are not designed for tax sensitivity. High income investors should be aware that the average client of most advisors is not in the highest federal tax bracket. Therefore, it is generally advantageous to seek the advice of a financial professional with experience executing an appropriate exit strategy that is aware of holding periods.
4. Proactively Realize Losses to Offset Gains: As mentioned in the opening paragraphs of the article, 2015 presented investors with an opportunity to realize losses in domestic stocks for the first time in 4 years. Clients with a diversified portfolio may still have an opportunity to offset gains in domestic stocks by selling foreign equities. One benefit of diversifying across asset classes is that if the portfolio is structured properly, the securities typically will not move in tandem. This divergence of returns among asset classes not only reduces portfolio volatility, but it creates a tax planning opportunity. Domestic equities experienced tremendous appreciation over a 5-year period through 2014; however, international stocks, commodities, and multiple fixed income investments experienced down years. Astute advisors were presented with the opportunity to save clients thousands of dollars in taxes by performing strategic tax swaps prior to year-end. It is important to understand the rules relating to wash sales when executing such tactics. The laws are confusing, and if a mistake is made your loss could be disallowed. Make certain your advisor is well-versed in utilizing tax offsets.
5. Think Twice About Gifting Cash: This is not to discourage your charitable intentions. Quite the opposite is true. However, a successful investor can occasionally find themselves in a precarious position. You may have allocated 5% of your portfolio to a growth stock with significant upside. Several years have passed, the security has experienced explosive growth, and it now represents 15% of your investable assets. Suddenly your portfolio has a concentrated position with significant gains, and the level of risk is no longer consistent with your long-term objectives. The sound practice of rebalancing your portfolio then becomes very costly, because liquidation of the stock could create a taxable event that may negatively impact your net return.
By planning ahead of time, you may be able to gift a portion of the appreciated security to a charitable organization able to accept this type of donation. The value of your gift can be replaced with the cash you originally intended to donate to the charitable organization and, in this scenario, your cash will create a new cost basis. The charity can liquidate the stock without paying tax, and you have removed a future tax liability from your portfolio. Implementing the aforementioned gifting strategy offers the potential to save thousands of dollars in taxes over the life of your portfolio.
6. Understand your Mutual Fund’s Tax Cost Ratio: The technical detail behind a mutual fund’s tax cost ratio is beyond the scope of this article. Our intent is to simply bring this topic to your attention. Tax cost ratio represents the percentage of an investor’s assets that are lost to taxes. Mutual funds avoid double taxation, provided they pay at least 90% of net investment income and realized capital gains to shareholders at the end of the calendar year. But all mutual funds are not created equally, and proper research will allow you to identify funds that are tax efficient.
A well-managed mutual fund will add diversification to a portfolio while creating the opportunity to outperform asset classes with inefficient markets. You do need to be aware of funds with excessive turnover. An understanding of when a fund pays its capital gains distributions is a critical component of successful investing. A poorly timed fund purchase can result in acquiring another investor’s tax liability. It is not unusual for an investor to experience a negative return in a calendar year, yet find himself on the receiving end of a capital gains distribution. Understanding the tax cost ratios of the funds that make up portions of your investment plan will enable you to take advantage of the many benefits of owning mutual funds.
The above steps are by no means the only tax strategies experienced advisors can execute on behalf of their clients. This article highlights several strategies you should discuss with your advisor to determine if implementation is appropriate for your unique portfolio and overall financial situation. Successful investing requires discipline that extends beyond proper security selection. While gross returns are important and should not be ignored, the percentage return you see on your statements does not tell the full story.
In today’s tax environment, successful investors must choose an advisor who will help them look beyond portfolio earnings and focus on strategic after-tax asset growth.
To receive a free hardcopy of Wealth Protection Planning for Orthopaedic Surgeons, please call 877-656-4362. Visit www.ojmbookstore.com and enter promotional code AJO30 for a free ebook download of Wealth Protection Planning or one of our other ebooks for your Kindle or iPad.
Bone marrow transplantation for epidermolysis bullosa continues to evolve
CHICAGO – Bone marrow transplantation is evolving as a promising treatment for patients with the most severe forms of epidermolysis bullosa.
“Is this a cure? It’s not,” Dr. Jakub Tolar, MD, PhD, said at the World Congress of Pediatric Dermatology. “It is, however, a path toward understanding how we can treat this grave disorder in a systemic way.”

The University of Minnesota BMT Team has also observed a correlation between the engraftment in the blood and engraftment in the skin. “We have skin engraftment as high as 50%, which is good,” Dr. Tolar said. “The more donor cells engrafted in the skin, the more types of collagen you express.”
The clinicians have also been able to reduce the amount of chemotherapy and radiation patients require prior to transplant, for the BMT to work and skin to heal. “We were able to make it so that the last 11 patients are surviving and having benefit from the transplant, with the exception of one,” Dr. Tolar said. “How does this work? We still don’t entirely know. This is not a shot in the dark, however, this is the continuation of a very long process where we were first able to show that bone marrow transplant is an efficient stem cell therapy for leukemia, and about 20 years ago for the lysosomal enzyme deficiencies.” Their hunt for the cell that travels from the bone marrow to skin and produces type 7 collagen is continuing. “What haunts me is that BMT, which works in recessive dystrophic EB, works only in some types of junctional EB, those with alpha-3 chain deficiency of laminin 322.” he continued. “There has been no benefit to bone marrow transplantation for children with mutations of beta-3 chain of laminin 322, so we have closed enrollment for this one form of junctional EB. Survival in this group was 40%. Other types of junctional EB continue to be eligible for the study.”
Dr. Tolar recommended keratinocyte-driven or thymic epithelium cell type–driven therapy for patients with mutations of beta-3. “The deficiency of thymic function seems to be key in the inability to benefit from BMT in this form of junctional EB,” he said. “We have seen children who have engrafted, their skin got better, and then they died of infection many months after transplant. When we look at the immune profile and the thymic epithelial cells, they are both deficient – very abnormal.”
Despite current challenges, Dr. Tolar expressed optimism about the future of BMT in EB patients. “We have the same approach that we have in cancer care: deep empathy for all patients, radical international collaboration, and rapid laboratory and clinical prototyping,” he said. “It’s time to move from two-dimensional science to three-dimensional science; we need to study all aspects of EB simultaneously, from gene to cell to tissue to individual to patient population, and to understand the properties of the whole EB pathobiology that emerge at each level of biological complexity. By connecting information from these layers of disease network, we can better understand EB and create comb

Dr. Tolar reported having no financial disclosures.
CHICAGO – Bone marrow transplantation is evolving as a promising treatment for patients with the most severe forms of epidermolysis bullosa.
“Is this a cure? It’s not,” Dr. Jakub Tolar, MD, PhD, said at the World Congress of Pediatric Dermatology. “It is, however, a path toward understanding how we can treat this grave disorder in a systemic way.”
The University of Minnesota BMT Team has also observed a correlation between the engraftment in the blood and engraftment in the skin. “We have skin engraftment as high as 50%, which is good,” Dr. Tolar said. “The more donor cells engrafted in the skin, the more types of collagen you express.”
The clinicians have also been able to reduce the amount of chemotherapy and radiation patients require prior to transplant, for the BMT to work and skin to heal. “We were able to make it so that the last 11 patients are surviving and having benefit from the transplant, with the exception of one,” Dr. Tolar said. “How does this work? We still don’t entirely know. This is not a shot in the dark, however, this is the continuation of a very long process where we were first able to show that bone marrow transplant is an efficient stem cell therapy for leukemia, and about 20 years ago for the lysosomal enzyme deficiencies.” Their hunt for the cell that travels from the bone marrow to skin and produces type 7 collagen is continuing. “What haunts me is that BMT, which works in recessive dystrophic EB, works only in some types of junctional EB, those with alpha-3 chain deficiency of laminin 322.” he continued. “There has been no benefit to bone marrow transplantation for children with mutations of beta-3 chain of laminin 322, so we have closed enrollment for this one form of junctional EB. Survival in this group was 40%. Other types of junctional EB continue to be eligible for the study.”
Dr. Tolar recommended keratinocyte-driven or thymic epithelium cell type–driven therapy for patients with mutations of beta-3. “The deficiency of thymic function seems to be key in the inability to benefit from BMT in this form of junctional EB,” he said. “We have seen children who have engrafted, their skin got better, and then they died of infection many months after transplant. When we look at the immune profile and the thymic epithelial cells, they are both deficient – very abnormal.”
Despite current challenges, Dr. Tolar expressed optimism about the future of BMT in EB patients. “We have the same approach that we have in cancer care: deep empathy for all patients, radical international collaboration, and rapid laboratory and clinical prototyping,” he said. “It’s time to move from two-dimensional science to three-dimensional science; we need to study all aspects of EB simultaneously, from gene to cell to tissue to individual to patient population, and to understand the properties of the whole EB pathobiology that emerge at each level of biological complexity. By connecting information from these layers of disease network, we can better understand EB and create comb
Dr. Tolar reported having no financial disclosures.
CHICAGO – Bone marrow transplantation is evolving as a promising treatment for patients with the most severe forms of epidermolysis bullosa.
“Is this a cure? It’s not,” Dr. Jakub Tolar, MD, PhD, said at the World Congress of Pediatric Dermatology. “It is, however, a path toward understanding how we can treat this grave disorder in a systemic way.”
The University of Minnesota BMT Team has also observed a correlation between the engraftment in the blood and engraftment in the skin. “We have skin engraftment as high as 50%, which is good,” Dr. Tolar said. “The more donor cells engrafted in the skin, the more types of collagen you express.”
The clinicians have also been able to reduce the amount of chemotherapy and radiation patients require prior to transplant, for the BMT to work and skin to heal. “We were able to make it so that the last 11 patients are surviving and having benefit from the transplant, with the exception of one,” Dr. Tolar said. “How does this work? We still don’t entirely know. This is not a shot in the dark, however, this is the continuation of a very long process where we were first able to show that bone marrow transplant is an efficient stem cell therapy for leukemia, and about 20 years ago for the lysosomal enzyme deficiencies.” Their hunt for the cell that travels from the bone marrow to skin and produces type 7 collagen is continuing. “What haunts me is that BMT, which works in recessive dystrophic EB, works only in some types of junctional EB, those with alpha-3 chain deficiency of laminin 322.” he continued. “There has been no benefit to bone marrow transplantation for children with mutations of beta-3 chain of laminin 322, so we have closed enrollment for this one form of junctional EB. Survival in this group was 40%. Other types of junctional EB continue to be eligible for the study.”
Dr. Tolar recommended keratinocyte-driven or thymic epithelium cell type–driven therapy for patients with mutations of beta-3. “The deficiency of thymic function seems to be key in the inability to benefit from BMT in this form of junctional EB,” he said. “We have seen children who have engrafted, their skin got better, and then they died of infection many months after transplant. When we look at the immune profile and the thymic epithelial cells, they are both deficient – very abnormal.”
Despite current challenges, Dr. Tolar expressed optimism about the future of BMT in EB patients. “We have the same approach that we have in cancer care: deep empathy for all patients, radical international collaboration, and rapid laboratory and clinical prototyping,” he said. “It’s time to move from two-dimensional science to three-dimensional science; we need to study all aspects of EB simultaneously, from gene to cell to tissue to individual to patient population, and to understand the properties of the whole EB pathobiology that emerge at each level of biological complexity. By connecting information from these layers of disease network, we can better understand EB and create comb
Dr. Tolar reported having no financial disclosures.
AT WCPD 2017