User login
Multifaceted Intervention Improves Anticoagulant Use in Patients With Atrial Fibrillation
A customized educational intervention significantly increases the use of oral anticoagulants among patients with atrial fibrillation at risk for stroke, according to data published online ahead of print August 28 in Lancet. The intervention also appears to reduce the risk of stroke.
“If this intervention could be broadly applied, which we believe is possible, the public health implications would be substantial,” said Christopher B. Granger, MD, Professor of Medicine at Duke University School of Medicine in Durham, North Carolina. “More than 33 million people worldwide have atrial fibrillation, which is a leading cause of stoke. Improving adherence to anticoagulation therapy would be a lifesaver.”

Intervention Included Education and Monitoring
Dr. Granger and colleagues conducted IMPACT-AF—a prospective, cluster-randomized, controlled trial—to evaluate the effect of a multifaceted educational intervention on the use of oral anticoagulation in patients with atrial fibrillation, compared with usual care. The study was conducted in Argentina, Brazil, China, India, and Romania. Eligible patients were 18 or older, had atrial fibrillation not resulting from reversible causes, and had an indication for oral anticoagulation. Patients with an absolute contraindication to oral anticoagulation, those with a mechanical prosthetic valve, and those who were not able to have one year of follow-up were excluded.
Clusters (ie, sites) in each country were paired and randomized 1:1 to receive an educational intervention or usual care. The intervention’s two components were education and regular monitoring. Education was provided through various media (eg, web-based materials, videos, and guideline recommendations) to patients, their families, and healthcare providers. It was customized for each country and described the benefits, risks, and costs of anticoagulant therapies. The monitoring component, which included feedback, was designed to promote anticoagulant initiation among appropriate candidates who were not being treated, prevent discontinuation among treated patients, and improve adherence. The investigators collected data at baseline, six months, and 12 months at all sites. The intervention sites had additional telephone calls or patient visits at one month, three months, and nine months.
The primary end point was the change in the proportion of patients treated with oral anticoagulants from baseline to one year. Key secondary end points included the proportion of patients who were on oral anticoagulation at baseline, six months, and 12 months; and the proportion of patients who were not on oral anticoagulation at baseline, but were on this therapy at six months and 12 months. Other secondary clinical outcomes included all-cause death, stroke, transient ischemic attack, and major bleeding.
Anticoagulant Use Increased
In all, researchers enrolled 2,281 participants at 48 clusters. Mean age was approximately 70, and about 47% of participants were women. Five patients (three in the intervention group) were lost to follow-up after baseline. The median follow-up duration was 12 months. Age, sex, educational level, and socioeconomic factors were well balanced between the two groups. The intervention group, however, had a higher proportion of patients with permanent atrial fibrillation, history of major bleeding, systemic embolism, and uncontrolled hypertension, and a lower proportion of patients with rheumatic valvular heart disease, heart failure or left ventricular dysfunction, vascular disease, and previous myocardial infarction, than the control group.
The proportion of patients on oral anticoagulation increased from 68% at baseline to 80% at one year in the intervention group and from 64% to 67% in the control group. The absolute difference between groups in the change of oral anticoagulation use was 9.1%. This result yielded an odds ratio of 3.28, representing the proportional increase in anticoagulation use from baseline to one year in the intervention group, compared with the control group. The effect size in favor of the intervention was consistent across prespecified subgroups. The primary outcome result was consistent in all five countries.
Furthermore, 95% of patients who were on oral anticoagulants at baseline in the intervention group and 94% of patients in the control group continued taking oral anticoagulants at one year. For patients who were not on oral anticoagulants at baseline, 48% in the intervention group and 18% in the control group were on oral anticoagulants at one year. This result yielded an odds ratio of 4.60, representing the proportional increase in anticoagulation use from baseline to one year in the intervention group, compared with the control group.
“Our study also found a reduction in strokes in the intervention group, compared with the control group,” said Renato D. Lopes, MD, PhD, Professor of Medicine at Duke University School of Medicine and principal investigator for Brazil. The hazard ratio of stroke was 0.48 for the intervention group, compared with the control group. “While this was a secondary outcome, it highlights the potential benefit of improved anticoagulation care,” he added.
The number needed to treat was 100 patients exposed to intervention to prevent one stroke event over one year. The rates of all-cause death and the composite of stroke, systemic embolism, or major bleeding did not differ between the intervention and control groups.
Intervention Might Help Patients Worldwide
The majority of participants were using vitamin K antagonists at baseline, and data suggest that non-vitamin K antagonists have advantages (eg, reduced intracerebral bleeds) over this class of oral anticoagulants. Approximately 8% of participants in the intervention group had switched from vitamin K antagonists to non-vitamin K antagonists at one year, while participants in the control group did not change their medication. “A similar educational approach could be used for all anticoagulant classes with even greater benefit to patients,” said Michael D. Ezekowitz, MD, a cardiologist at Lankenau Medical Center in Wynnewood, Pennsylvania, and Anthony P. Kent, MD, a resident at Bridgeport Hospital in Bridgeport, Connecticut, in an accompanying editorial.
The study was limited to five countries and relied on a technological intervention, which might hinder the generalization of the results to broader clinical practice, said Drs. Ezekowitz and Kent. Nevertheless, “we are confident that the impact of IMPACT-AF will benefit patients with atrial fibrillation worldwide,” they concluded.
—Erik Greb
Suggested Reading
Ezekowitz MD, Kent AP. The impact of IMPACT-AF. Lancet. 2017 Aug 28 [Epub ahead of print].
Vinereanu D, Lopes RD, Bahit MC, et al. A multifaceted intervention to improve treatment with oral anticoagulants in atrial fibrillation (IMPACT-AF): an international, cluster-randomised trial. Lancet. 2017 Aug 28 [Epub ahead of print].
A customized educational intervention significantly increases the use of oral anticoagulants among patients with atrial fibrillation at risk for stroke, according to data published online ahead of print August 28 in Lancet. The intervention also appears to reduce the risk of stroke.
“If this intervention could be broadly applied, which we believe is possible, the public health implications would be substantial,” said Christopher B. Granger, MD, Professor of Medicine at Duke University School of Medicine in Durham, North Carolina. “More than 33 million people worldwide have atrial fibrillation, which is a leading cause of stoke. Improving adherence to anticoagulation therapy would be a lifesaver.”
Intervention Included Education and Monitoring
Dr. Granger and colleagues conducted IMPACT-AF—a prospective, cluster-randomized, controlled trial—to evaluate the effect of a multifaceted educational intervention on the use of oral anticoagulation in patients with atrial fibrillation, compared with usual care. The study was conducted in Argentina, Brazil, China, India, and Romania. Eligible patients were 18 or older, had atrial fibrillation not resulting from reversible causes, and had an indication for oral anticoagulation. Patients with an absolute contraindication to oral anticoagulation, those with a mechanical prosthetic valve, and those who were not able to have one year of follow-up were excluded.
Clusters (ie, sites) in each country were paired and randomized 1:1 to receive an educational intervention or usual care. The intervention’s two components were education and regular monitoring. Education was provided through various media (eg, web-based materials, videos, and guideline recommendations) to patients, their families, and healthcare providers. It was customized for each country and described the benefits, risks, and costs of anticoagulant therapies. The monitoring component, which included feedback, was designed to promote anticoagulant initiation among appropriate candidates who were not being treated, prevent discontinuation among treated patients, and improve adherence. The investigators collected data at baseline, six months, and 12 months at all sites. The intervention sites had additional telephone calls or patient visits at one month, three months, and nine months.
The primary end point was the change in the proportion of patients treated with oral anticoagulants from baseline to one year. Key secondary end points included the proportion of patients who were on oral anticoagulation at baseline, six months, and 12 months; and the proportion of patients who were not on oral anticoagulation at baseline, but were on this therapy at six months and 12 months. Other secondary clinical outcomes included all-cause death, stroke, transient ischemic attack, and major bleeding.
Anticoagulant Use Increased
In all, researchers enrolled 2,281 participants at 48 clusters. Mean age was approximately 70, and about 47% of participants were women. Five patients (three in the intervention group) were lost to follow-up after baseline. The median follow-up duration was 12 months. Age, sex, educational level, and socioeconomic factors were well balanced between the two groups. The intervention group, however, had a higher proportion of patients with permanent atrial fibrillation, history of major bleeding, systemic embolism, and uncontrolled hypertension, and a lower proportion of patients with rheumatic valvular heart disease, heart failure or left ventricular dysfunction, vascular disease, and previous myocardial infarction, than the control group.
The proportion of patients on oral anticoagulation increased from 68% at baseline to 80% at one year in the intervention group and from 64% to 67% in the control group. The absolute difference between groups in the change of oral anticoagulation use was 9.1%. This result yielded an odds ratio of 3.28, representing the proportional increase in anticoagulation use from baseline to one year in the intervention group, compared with the control group. The effect size in favor of the intervention was consistent across prespecified subgroups. The primary outcome result was consistent in all five countries.
Furthermore, 95% of patients who were on oral anticoagulants at baseline in the intervention group and 94% of patients in the control group continued taking oral anticoagulants at one year. For patients who were not on oral anticoagulants at baseline, 48% in the intervention group and 18% in the control group were on oral anticoagulants at one year. This result yielded an odds ratio of 4.60, representing the proportional increase in anticoagulation use from baseline to one year in the intervention group, compared with the control group.
“Our study also found a reduction in strokes in the intervention group, compared with the control group,” said Renato D. Lopes, MD, PhD, Professor of Medicine at Duke University School of Medicine and principal investigator for Brazil. The hazard ratio of stroke was 0.48 for the intervention group, compared with the control group. “While this was a secondary outcome, it highlights the potential benefit of improved anticoagulation care,” he added.
The number needed to treat was 100 patients exposed to intervention to prevent one stroke event over one year. The rates of all-cause death and the composite of stroke, systemic embolism, or major bleeding did not differ between the intervention and control groups.
Intervention Might Help Patients Worldwide
The majority of participants were using vitamin K antagonists at baseline, and data suggest that non-vitamin K antagonists have advantages (eg, reduced intracerebral bleeds) over this class of oral anticoagulants. Approximately 8% of participants in the intervention group had switched from vitamin K antagonists to non-vitamin K antagonists at one year, while participants in the control group did not change their medication. “A similar educational approach could be used for all anticoagulant classes with even greater benefit to patients,” said Michael D. Ezekowitz, MD, a cardiologist at Lankenau Medical Center in Wynnewood, Pennsylvania, and Anthony P. Kent, MD, a resident at Bridgeport Hospital in Bridgeport, Connecticut, in an accompanying editorial.
The study was limited to five countries and relied on a technological intervention, which might hinder the generalization of the results to broader clinical practice, said Drs. Ezekowitz and Kent. Nevertheless, “we are confident that the impact of IMPACT-AF will benefit patients with atrial fibrillation worldwide,” they concluded.
—Erik Greb
Suggested Reading
Ezekowitz MD, Kent AP. The impact of IMPACT-AF. Lancet. 2017 Aug 28 [Epub ahead of print].
Vinereanu D, Lopes RD, Bahit MC, et al. A multifaceted intervention to improve treatment with oral anticoagulants in atrial fibrillation (IMPACT-AF): an international, cluster-randomised trial. Lancet. 2017 Aug 28 [Epub ahead of print].
A customized educational intervention significantly increases the use of oral anticoagulants among patients with atrial fibrillation at risk for stroke, according to data published online ahead of print August 28 in Lancet. The intervention also appears to reduce the risk of stroke.
“If this intervention could be broadly applied, which we believe is possible, the public health implications would be substantial,” said Christopher B. Granger, MD, Professor of Medicine at Duke University School of Medicine in Durham, North Carolina. “More than 33 million people worldwide have atrial fibrillation, which is a leading cause of stoke. Improving adherence to anticoagulation therapy would be a lifesaver.”
Intervention Included Education and Monitoring
Dr. Granger and colleagues conducted IMPACT-AF—a prospective, cluster-randomized, controlled trial—to evaluate the effect of a multifaceted educational intervention on the use of oral anticoagulation in patients with atrial fibrillation, compared with usual care. The study was conducted in Argentina, Brazil, China, India, and Romania. Eligible patients were 18 or older, had atrial fibrillation not resulting from reversible causes, and had an indication for oral anticoagulation. Patients with an absolute contraindication to oral anticoagulation, those with a mechanical prosthetic valve, and those who were not able to have one year of follow-up were excluded.
Clusters (ie, sites) in each country were paired and randomized 1:1 to receive an educational intervention or usual care. The intervention’s two components were education and regular monitoring. Education was provided through various media (eg, web-based materials, videos, and guideline recommendations) to patients, their families, and healthcare providers. It was customized for each country and described the benefits, risks, and costs of anticoagulant therapies. The monitoring component, which included feedback, was designed to promote anticoagulant initiation among appropriate candidates who were not being treated, prevent discontinuation among treated patients, and improve adherence. The investigators collected data at baseline, six months, and 12 months at all sites. The intervention sites had additional telephone calls or patient visits at one month, three months, and nine months.
The primary end point was the change in the proportion of patients treated with oral anticoagulants from baseline to one year. Key secondary end points included the proportion of patients who were on oral anticoagulation at baseline, six months, and 12 months; and the proportion of patients who were not on oral anticoagulation at baseline, but were on this therapy at six months and 12 months. Other secondary clinical outcomes included all-cause death, stroke, transient ischemic attack, and major bleeding.
Anticoagulant Use Increased
In all, researchers enrolled 2,281 participants at 48 clusters. Mean age was approximately 70, and about 47% of participants were women. Five patients (three in the intervention group) were lost to follow-up after baseline. The median follow-up duration was 12 months. Age, sex, educational level, and socioeconomic factors were well balanced between the two groups. The intervention group, however, had a higher proportion of patients with permanent atrial fibrillation, history of major bleeding, systemic embolism, and uncontrolled hypertension, and a lower proportion of patients with rheumatic valvular heart disease, heart failure or left ventricular dysfunction, vascular disease, and previous myocardial infarction, than the control group.
The proportion of patients on oral anticoagulation increased from 68% at baseline to 80% at one year in the intervention group and from 64% to 67% in the control group. The absolute difference between groups in the change of oral anticoagulation use was 9.1%. This result yielded an odds ratio of 3.28, representing the proportional increase in anticoagulation use from baseline to one year in the intervention group, compared with the control group. The effect size in favor of the intervention was consistent across prespecified subgroups. The primary outcome result was consistent in all five countries.
Furthermore, 95% of patients who were on oral anticoagulants at baseline in the intervention group and 94% of patients in the control group continued taking oral anticoagulants at one year. For patients who were not on oral anticoagulants at baseline, 48% in the intervention group and 18% in the control group were on oral anticoagulants at one year. This result yielded an odds ratio of 4.60, representing the proportional increase in anticoagulation use from baseline to one year in the intervention group, compared with the control group.
“Our study also found a reduction in strokes in the intervention group, compared with the control group,” said Renato D. Lopes, MD, PhD, Professor of Medicine at Duke University School of Medicine and principal investigator for Brazil. The hazard ratio of stroke was 0.48 for the intervention group, compared with the control group. “While this was a secondary outcome, it highlights the potential benefit of improved anticoagulation care,” he added.
The number needed to treat was 100 patients exposed to intervention to prevent one stroke event over one year. The rates of all-cause death and the composite of stroke, systemic embolism, or major bleeding did not differ between the intervention and control groups.
Intervention Might Help Patients Worldwide
The majority of participants were using vitamin K antagonists at baseline, and data suggest that non-vitamin K antagonists have advantages (eg, reduced intracerebral bleeds) over this class of oral anticoagulants. Approximately 8% of participants in the intervention group had switched from vitamin K antagonists to non-vitamin K antagonists at one year, while participants in the control group did not change their medication. “A similar educational approach could be used for all anticoagulant classes with even greater benefit to patients,” said Michael D. Ezekowitz, MD, a cardiologist at Lankenau Medical Center in Wynnewood, Pennsylvania, and Anthony P. Kent, MD, a resident at Bridgeport Hospital in Bridgeport, Connecticut, in an accompanying editorial.
The study was limited to five countries and relied on a technological intervention, which might hinder the generalization of the results to broader clinical practice, said Drs. Ezekowitz and Kent. Nevertheless, “we are confident that the impact of IMPACT-AF will benefit patients with atrial fibrillation worldwide,” they concluded.
—Erik Greb
Suggested Reading
Ezekowitz MD, Kent AP. The impact of IMPACT-AF. Lancet. 2017 Aug 28 [Epub ahead of print].
Vinereanu D, Lopes RD, Bahit MC, et al. A multifaceted intervention to improve treatment with oral anticoagulants in atrial fibrillation (IMPACT-AF): an international, cluster-randomised trial. Lancet. 2017 Aug 28 [Epub ahead of print].

Reengineering your office to be perfect for your patients
Independent of the Affordable Care Act or any upcoming changes in health care, the focus of an ObGyn practice remains paramount: the patient comes first.
The “recipe” for creating patient satisfaction and service excellence is predicated upon the mission of your practice and creating a shared vision with your employees. An action plan that is created and “visited/revisited”on a regular basis will serve to keep all abreast of the latest information to enhance the quality of patient care. It goes without saying, the ObGyn must first “lead by example” and always strive for satisfied patients who will tell their friends about your practice.
Start with the right tools
To organize a practice well, you need the right tools, which ideally include mission and vision statements and an action plan with goals and objectives.
Mission statement
A mission statement can be developed by the ObGyn(s) in your office or in concert with your staff. It should include:
- the “here and now” focus on the current approach to patient care
- why the practice exists (Develop a brief description of your practice, including the desired patient population.)
- the products and services offered and why and how those services are provided.
Here is an example of a mission statement for an ObGyn practice: “Our mission is to provide excellent, exceptional, personalized care for women of all ages in a warm and friendly environment. We incorporate leading-edge technology in our practice and continue to be a leader in obstetrics and gynecology.”
Vision statement
A vision statement should be developed in concert with your staff. It should include:
- the “then and there” focus on the historic perspective of your practice
- the ObGyn(s) and staff vision of the future
- what the ObGyn(s) and staff want to create.
The vision statement should energize and excite your personnel, create a shared and meaningful purpose, inspire passion and interest, and convey the values you want to share in your practice.
Here is an example of a vision statement for an ObGyn practice: “We aim to become the premier obstetrics and gynecology pro-vider to residents of (location) community.”
Action plan: Setting goals
To succeed, an ObGyn practice needs to:
- develop targets and challenges reflecting periodic (quarterly) meetings with staff and new entity development in the practice
- establish benchmarks and measurable parameters (How do you compare with other local practices? Set criteria/metrics to assess your progress.)
- ensure that the objectives support the goals (Develop goals and objectives over a defined period of time.)
- revisit the goals (Have they have been met? Do they need revision?)
Goals and objectives are essential for the continued health of your practice. This is all predicated upon developing a competitive advantage and then maintaining it.
Read about how to make a positive first impression on a new patient.
Is the environment welcoming?
When we examine a practice from the patient’s point of view, a good starting place is with the front desk. Have you looked at your front desk “from the outside in?” In one sense, this is the showcase of your practice.
Related article:
Four pillars of a successful practice: 2. Attract new patients
The first impression: Appointment scheduling
The first impression a patient receives about your practice occurs when she attempts to set up an appointment. Perhaps you might ask someone to call in to schedule an appointment. Is the caller immediately put on hold? Are your personnel courteous on the phone? Can she be seen quickly if she has a problem? How long is the wait for an annual exam? A test run can be very revealing.
Walk in the front door
When a patient walks in the door, does the physical office space radiate a friendly, relaxed atmosphere? Walk through the waiting room, then consultation and exam rooms as if you are a patient seeing it for the first time. Have you created an environment in which patients sense a well-organized office and the esprit de corps of the personnel? Does it look and smell fresh and clean? This all sends a loud and clear positive message about your practice.1–3
Here are some suggestions for making a waiting room more inviting:
- Provide a seating arrangement that is “patient centered.” For example, semi- circular arrangements allow easy viewing of any monitors in the waiting room.
- WiFi is a great addition. Post several signs with the user name and password.
- Offer computers for patients to use to complete registration
- Set up a fish tank. If well-maintained, it can be soothing to many people.
- Display medical information pamphlets, even if they are rarely taken.
- Provide a big screen television that offers information about your practice, including personnel and procedures.
Streaming ads for physician offices are available. One platform, Outcome Health (https://www.outcomehealth.com), provides flat-screen TVs and tablets that show patient education videos.4 Another vendor, Patient Point (http://patientpoint.com), offers waiting room networks, editorials, and other communications designed to support “the goals of improving healthcare.”5 Other available media include channel news and music programming to relax patients.6
Wait times. A patient’s perceived wait time and the actual wait time are often quite different. How long she waits to see the ObGyn is “numero uno” with regard to patient satisfaction and can be a key source of annoyance, irritability, stress, and anger.
Does someone inform waiting patients that the ObGyn is running late? Does staff at the front desk or perhaps your medical assistant inquire, “Can I get you anything? The doctor is running late,” or “Dr. Jones has just finished delivering a baby. He’ll be here in 10 minutes. He’ll see you first.”
Consultation and exam rooms
Suggestions to develop a relaxing environment in your consultation and exam rooms are7:
- decorate the walls with soft, pastel colors
- use “spa aesthetics” to create a colorful atmosphere with appropriate lighting, artwork, and modern furnishings
- present a few magazines neatly and update them periodically
- stock and appropriately maintain the patients rooms with medical supplies
- remember, “Subjects perceive people more positively in beautiful rooms than in ugly rooms.”5
Read about how to keep your patients satisfied and your business stable.
Set the lead example
The need for open and supportive communication between you and your office staff cannot be overly emphasized. An ideal office staff member understands and shares in the vision, is aware of stated goals and objectives, is responsive to patient needs, and wants to create a win-win environment.
Frequently discuss your expectations with your staff. Expect them to be responsive, courteous, competent, have good communication skills, and be influenced by the appearance of the physical environ-ment. Provide support and educational tools to help them successfully perform their work.
Related article:
Four pillars of a successful practice: 1. Keep your current patients happy
Discover your patients’ vision of customer service
Formal measurement of patient satisfaction began with Professor Irwin Press at the University of Notre Dame. Rod Ganey, a sociologist and statistician, then developed the Press Ganey Patient Satisfaction Survey. These points earlier conveyed by Maslow and Mintz8 addressed the “effects of esthetic surroundings.” Color and art proved to be preferences in an esthetically pleasing environment. Additional historical information has been provided by Siegrist, who addressed “the patient experience.”9 He cites the myth that patients do not fill out satisfaction surveys. Indeed they do. Patient satisfaction is not a personality contest but rather a reflection of the health care provider’s investment of time and effort to offer patient-centered care. Siegrist also notes that the patient’s family plays a key role in how a patient perceives her experience with her health care professional.9
The federal government has been actively involved in assessing patient satisfaction in the hospital setting since 2002. This is reflected in the Centers for Medicare and Medicaid Services, the Agency for Healthcare Research and Quality, and Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) surveys. The HCAHPS is a 27-question survey randomly administered to adult inpatients after discharge.10–12
The following metrics are often included in patient satisfaction surveys9,10:
- rating of hospital care from 0 (lowest) to 10 (highest)
- percentage of patients who would recommend a practice to family and friends
- number of patients who say their health care providers always communicate well
- the number of patients who report that the office is always clean and friendly.
Use of search engines focused on health care patient surveys can provide a number of options for clinicians to use in their practice.
Tips on patient satisfaction
Several interesting tips from the busi-ness world can be applied to an ObGyn’s practice14:
- You will only hear from 4% of unhappy customers.
- One dissatisfied customer tells 9.
- 95% of customers with resolved issues will do business with you again.
- If a problem is not addressed, that patient will tell 10 others.
- Resolve the problem and 5 people will know about it.
- It costs 5 times as much effort to gain 1 new customer.
- Loyal customers in 1 area of service are good prospects for other (new) services.
Related article:
Using the Internet in your practice. Part 2: Generating new patients using social media
Tell stories about good, satisfied patients
Sharing the stories of satisfied patients motivates others to consider coming to your practice. To develop these stories, offer a “suggestion box” where patients can leave compliments or comments about their experiences. Ask patients to record their positive reviews (be sure to obtain written consent before recording and publishing). Show the videos on the big-screen TVs in your waiting room and include patient reviews (written, audio, and video) on your website.15
Related article:
Four pillars of a successful practice: 4. Motivate your staff
Reevaluate periodically
Encouraging team spirit makes good business sense. Offer staff members bonuses for coming up with improved processes. Provide educational programs for staff on patient care, technology, etc. If a difficult experience occurs, discuss it openly with staff members without accusing, asking them for suggestions to improve the situation.16
To assess the monetary value of your practice, you need to know what contributes to your profit margin and overhead. What investments are the most profitable? Then monitor each segment of the office practice.
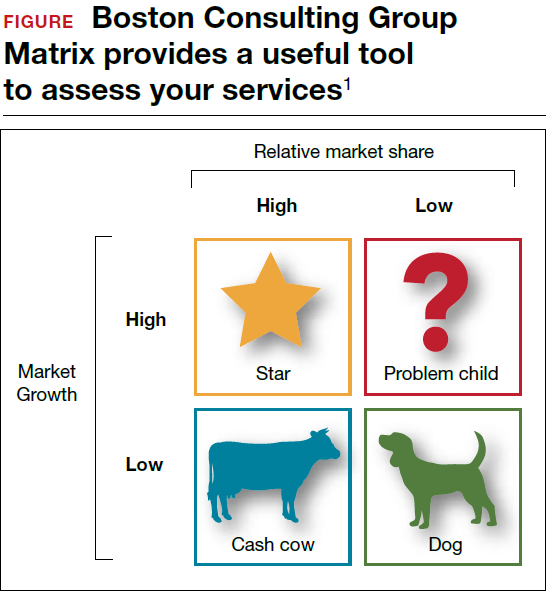
Should you proceed with a purchase? Should you take on a new hire? Let's look at one excellent model from the Boston Consulting Group (FIGURE) that provides insight into "low and high performance" aspects of business or practice.1
In the matrix, Stars use large amounts of cash and are leaders in cash generation. Stars lead to development of a Cash Cow, which are entities that generate profits and cash with low investment prerequisites. Dogs are segments of product and service line(s) that should be carefully reevaluated. A decision must be made to liquidate if the problem cannot be corrected. Question Marks have the worst cash characteristics of all and are associated with high demands and low profit margin(s).1
SWOT analysis
A SWOT analysis is most helpful when assessing a practice in real time. The basic tenets are2:
Strengths:
- prestigious reputation
- technological expertise
Weaknesses:
- antiquated computer system
- lack of experience in specific areas
Opportunities:
- growing market demand for a specific product or procedure
- provision of unique services
Threats:
- changing demographics
- competitive practices
- changes in health care third-party payers.
The American College of Obstetricians and Gynecologists (ACOG) has developed an "ACOG Medical Home Toolkit" to allow ObGyns to assess how significant the changes regarding payers will be to their practice. Sections include the patient/practice partnership support; clinical care information; community resources; care delivery management; performance measurement and improvement; and payment and finance.3 The toolkit is available for download from the ACOG website.
References
- Morrison A, Wensley R. Boxing up or boxed in? A short history of the Boston Consulting Group Share/Growth Matrix. J Market Manag. 1993;7(2):105-129. http://www.tandfonline.com/doi/abs/10.1080/0267257X.1991.9964145.
- Klasko SK, Toub DB. It's not a plan without a business plan. In: Sanfilippo JS, Nolan TE, Whiteside BH, eds. MBA Handbook for Healthcare Professionals. New York, NY: Parthenon Publishing Group; 2002:36-37.
- American Congress of Obstetricians and Gynecologists. ACOG Medical Home Toolkit. https://www.acog.org/About-ACOG/ACOG-Departments/Practice-Management-and-Managed-Care/ACOG-Medical-Home-Toolkit. Accessed August 14, 2017.
Bottom line
Ensuring that your patients have an outstanding experience is a smart business strategy. A unified approach that includes team members’ involvement to create a patient-centered environment will provide a quality experience and encourage patients to recommend your ObGyn practice to others.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Ulrich RS. Evidence-based environmental design for improving medical outcomes: Paper Delivered at a Conference Entitled Healing by Design: Building for Health Care in the 21st Century. Montreal: McGill University Health Centre; 2000. http://www.brikbase.org/sites/default/files/Evidence%20Based%20Environmental%20Design%20for%20Improving%20Medical.pdf. Accessed August 15, 2017.
- Becker F, Douglass S. The ecology of the patient visit: physical attractiveness, waiting times and perceived quality of care. J Ambul Care Manag. 2008;31(2):128–141.
- Becker F, Sweeney B, Parsons K. Ambulatory facility design and patients’ perceptions of healthcare quality. HERD. 2008;1(4):35–54.
- Outcome Health Website. https://www.outcomehealth.com/. Accessed August 14, 2017.
- Mazer SE. The waiting room: Where suffering begins. Healing Healthcare Systems website. http://www.healinghealth.com/waiting-room-suffering-begins/. Published November 7, 2014. Accessed August 14, 2017.
- Patient Point Programs Website. http://patientpoint.com/. Accessed August 14, 2017.
- Almquist J, Kelly C, Bromberg J, Bryant S, Christianson T, Montori V. Consultation room design and the clinical encounter: the space and interaction randomized trial. Health Environ Res Design. 2009;3(1):41–78.
- Maslow A, Mintz N. Effects of esthetic surroundings: I. Initial effects of three esthetic conditions upon perceiving “energy” and “well-being” in faces. J Psychology. 1956;41(2):247–254.
- Siegrist RB. The patient experience. In: Sanfilippo JS, Bieber E, Javich D, Siegrist R, eds. MBA for Healthcare. New York, NY: Oxford Press;2016:227–236.
- Press I. Patient satisfaction: Understanding and managing the experience of care. 2nd ed. Chicago, IL: Health Administration Press; 2005:66–78.
- Piper L, Tallman E. Hospital consumer assessment of healthcare providers and systems: An ethical leadership dilemma to satisfy patients. Health Care Manag (Frederick). 2016;35(2):151–155.
- Giordano L, Elliott M, Goldstein E, Lehrman W, Spencer P. Development, implementation and public reporting of HCAHPS survey. Med Care Res Rev. 2010;67(1):27–37.
- Jones KE. Helping the health profession help others: Applying business principles to the medical world. University of Tennessee, Knoxville Honors Thesis Projects. http://trace.tennessee.edu/cgi/viewcontent.cgi?article=1560&context=utk_chanhonoproj. Published 2002. Accessed August 14, 2017.
- Baum N. Marketing your practice: ethically, effectively and economically. In: Sanfilippo JS, Nolan TE, Whiteside BH, eds. MBA Handbook for Healthcare Professionals. New York, NY: Parthenon Publishing Group; 2002:123–154.
- Baum NH. Four pillars of a successful practice: 1. Keep your current patients happy. OBG Manag. 2013;25(3):49–56.
- Baum NH. Four pillars of a successful practice: 4. Motivate your staff. OBG Manag. 2013;25(8):29–33.

Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Academic Division Director, Reproductive Endocrinology and Infertility, Magee-Women’s Hospital, Pittsburgh, Pennsylvania. Dr. Sanfilippo is a member of the
The author reports no financial relationships relevant to this article.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Academic Division Director, Reproductive Endocrinology and Infertility, Magee-Women’s Hospital, Pittsburgh, Pennsylvania. Dr. Sanfilippo is a member of the
The author reports no financial relationships relevant to this article.
Dr. Sanfilippo is Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Academic Division Director, Reproductive Endocrinology and Infertility, Magee-Women’s Hospital, Pittsburgh, Pennsylvania. Dr. Sanfilippo is a member of the
The author reports no financial relationships relevant to this article.
Independent of the Affordable Care Act or any upcoming changes in health care, the focus of an ObGyn practice remains paramount: the patient comes first.
The “recipe” for creating patient satisfaction and service excellence is predicated upon the mission of your practice and creating a shared vision with your employees. An action plan that is created and “visited/revisited”on a regular basis will serve to keep all abreast of the latest information to enhance the quality of patient care. It goes without saying, the ObGyn must first “lead by example” and always strive for satisfied patients who will tell their friends about your practice.
Start with the right tools
To organize a practice well, you need the right tools, which ideally include mission and vision statements and an action plan with goals and objectives.
Mission statement
A mission statement can be developed by the ObGyn(s) in your office or in concert with your staff. It should include:
- the “here and now” focus on the current approach to patient care
- why the practice exists (Develop a brief description of your practice, including the desired patient population.)
- the products and services offered and why and how those services are provided.
Here is an example of a mission statement for an ObGyn practice: “Our mission is to provide excellent, exceptional, personalized care for women of all ages in a warm and friendly environment. We incorporate leading-edge technology in our practice and continue to be a leader in obstetrics and gynecology.”
Vision statement
A vision statement should be developed in concert with your staff. It should include:
- the “then and there” focus on the historic perspective of your practice
- the ObGyn(s) and staff vision of the future
- what the ObGyn(s) and staff want to create.
The vision statement should energize and excite your personnel, create a shared and meaningful purpose, inspire passion and interest, and convey the values you want to share in your practice.
Here is an example of a vision statement for an ObGyn practice: “We aim to become the premier obstetrics and gynecology pro-vider to residents of (location) community.”
Action plan: Setting goals
To succeed, an ObGyn practice needs to:
- develop targets and challenges reflecting periodic (quarterly) meetings with staff and new entity development in the practice
- establish benchmarks and measurable parameters (How do you compare with other local practices? Set criteria/metrics to assess your progress.)
- ensure that the objectives support the goals (Develop goals and objectives over a defined period of time.)
- revisit the goals (Have they have been met? Do they need revision?)
Goals and objectives are essential for the continued health of your practice. This is all predicated upon developing a competitive advantage and then maintaining it.
Read about how to make a positive first impression on a new patient.
Is the environment welcoming?
When we examine a practice from the patient’s point of view, a good starting place is with the front desk. Have you looked at your front desk “from the outside in?” In one sense, this is the showcase of your practice.
Related article:
Four pillars of a successful practice: 2. Attract new patients
The first impression: Appointment scheduling
The first impression a patient receives about your practice occurs when she attempts to set up an appointment. Perhaps you might ask someone to call in to schedule an appointment. Is the caller immediately put on hold? Are your personnel courteous on the phone? Can she be seen quickly if she has a problem? How long is the wait for an annual exam? A test run can be very revealing.
Walk in the front door
When a patient walks in the door, does the physical office space radiate a friendly, relaxed atmosphere? Walk through the waiting room, then consultation and exam rooms as if you are a patient seeing it for the first time. Have you created an environment in which patients sense a well-organized office and the esprit de corps of the personnel? Does it look and smell fresh and clean? This all sends a loud and clear positive message about your practice.1–3
Here are some suggestions for making a waiting room more inviting:
- Provide a seating arrangement that is “patient centered.” For example, semi- circular arrangements allow easy viewing of any monitors in the waiting room.
- WiFi is a great addition. Post several signs with the user name and password.
- Offer computers for patients to use to complete registration
- Set up a fish tank. If well-maintained, it can be soothing to many people.
- Display medical information pamphlets, even if they are rarely taken.
- Provide a big screen television that offers information about your practice, including personnel and procedures.
Streaming ads for physician offices are available. One platform, Outcome Health (https://www.outcomehealth.com), provides flat-screen TVs and tablets that show patient education videos.4 Another vendor, Patient Point (http://patientpoint.com), offers waiting room networks, editorials, and other communications designed to support “the goals of improving healthcare.”5 Other available media include channel news and music programming to relax patients.6
Wait times. A patient’s perceived wait time and the actual wait time are often quite different. How long she waits to see the ObGyn is “numero uno” with regard to patient satisfaction and can be a key source of annoyance, irritability, stress, and anger.
Does someone inform waiting patients that the ObGyn is running late? Does staff at the front desk or perhaps your medical assistant inquire, “Can I get you anything? The doctor is running late,” or “Dr. Jones has just finished delivering a baby. He’ll be here in 10 minutes. He’ll see you first.”
Consultation and exam rooms
Suggestions to develop a relaxing environment in your consultation and exam rooms are7:
- decorate the walls with soft, pastel colors
- use “spa aesthetics” to create a colorful atmosphere with appropriate lighting, artwork, and modern furnishings
- present a few magazines neatly and update them periodically
- stock and appropriately maintain the patients rooms with medical supplies
- remember, “Subjects perceive people more positively in beautiful rooms than in ugly rooms.”5
Read about how to keep your patients satisfied and your business stable.
Set the lead example
The need for open and supportive communication between you and your office staff cannot be overly emphasized. An ideal office staff member understands and shares in the vision, is aware of stated goals and objectives, is responsive to patient needs, and wants to create a win-win environment.
Frequently discuss your expectations with your staff. Expect them to be responsive, courteous, competent, have good communication skills, and be influenced by the appearance of the physical environ-ment. Provide support and educational tools to help them successfully perform their work.
Related article:
Four pillars of a successful practice: 1. Keep your current patients happy
Discover your patients’ vision of customer service
Formal measurement of patient satisfaction began with Professor Irwin Press at the University of Notre Dame. Rod Ganey, a sociologist and statistician, then developed the Press Ganey Patient Satisfaction Survey. These points earlier conveyed by Maslow and Mintz8 addressed the “effects of esthetic surroundings.” Color and art proved to be preferences in an esthetically pleasing environment. Additional historical information has been provided by Siegrist, who addressed “the patient experience.”9 He cites the myth that patients do not fill out satisfaction surveys. Indeed they do. Patient satisfaction is not a personality contest but rather a reflection of the health care provider’s investment of time and effort to offer patient-centered care. Siegrist also notes that the patient’s family plays a key role in how a patient perceives her experience with her health care professional.9
The federal government has been actively involved in assessing patient satisfaction in the hospital setting since 2002. This is reflected in the Centers for Medicare and Medicaid Services, the Agency for Healthcare Research and Quality, and Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) surveys. The HCAHPS is a 27-question survey randomly administered to adult inpatients after discharge.10–12
The following metrics are often included in patient satisfaction surveys9,10:
- rating of hospital care from 0 (lowest) to 10 (highest)
- percentage of patients who would recommend a practice to family and friends
- number of patients who say their health care providers always communicate well
- the number of patients who report that the office is always clean and friendly.
Use of search engines focused on health care patient surveys can provide a number of options for clinicians to use in their practice.
Tips on patient satisfaction
Several interesting tips from the busi-ness world can be applied to an ObGyn’s practice14:
- You will only hear from 4% of unhappy customers.
- One dissatisfied customer tells 9.
- 95% of customers with resolved issues will do business with you again.
- If a problem is not addressed, that patient will tell 10 others.
- Resolve the problem and 5 people will know about it.
- It costs 5 times as much effort to gain 1 new customer.
- Loyal customers in 1 area of service are good prospects for other (new) services.
Related article:
Using the Internet in your practice. Part 2: Generating new patients using social media
Tell stories about good, satisfied patients
Sharing the stories of satisfied patients motivates others to consider coming to your practice. To develop these stories, offer a “suggestion box” where patients can leave compliments or comments about their experiences. Ask patients to record their positive reviews (be sure to obtain written consent before recording and publishing). Show the videos on the big-screen TVs in your waiting room and include patient reviews (written, audio, and video) on your website.15
Related article:
Four pillars of a successful practice: 4. Motivate your staff
Reevaluate periodically
Encouraging team spirit makes good business sense. Offer staff members bonuses for coming up with improved processes. Provide educational programs for staff on patient care, technology, etc. If a difficult experience occurs, discuss it openly with staff members without accusing, asking them for suggestions to improve the situation.16
To assess the monetary value of your practice, you need to know what contributes to your profit margin and overhead. What investments are the most profitable? Then monitor each segment of the office practice.
Should you proceed with a purchase? Should you take on a new hire? Let's look at one excellent model from the Boston Consulting Group (FIGURE) that provides insight into "low and high performance" aspects of business or practice.1
In the matrix, Stars use large amounts of cash and are leaders in cash generation. Stars lead to development of a Cash Cow, which are entities that generate profits and cash with low investment prerequisites. Dogs are segments of product and service line(s) that should be carefully reevaluated. A decision must be made to liquidate if the problem cannot be corrected. Question Marks have the worst cash characteristics of all and are associated with high demands and low profit margin(s).1
SWOT analysis
A SWOT analysis is most helpful when assessing a practice in real time. The basic tenets are2:
Strengths:
- prestigious reputation
- technological expertise
Weaknesses:
- antiquated computer system
- lack of experience in specific areas
Opportunities:
- growing market demand for a specific product or procedure
- provision of unique services
Threats:
- changing demographics
- competitive practices
- changes in health care third-party payers.
The American College of Obstetricians and Gynecologists (ACOG) has developed an "ACOG Medical Home Toolkit" to allow ObGyns to assess how significant the changes regarding payers will be to their practice. Sections include the patient/practice partnership support; clinical care information; community resources; care delivery management; performance measurement and improvement; and payment and finance.3 The toolkit is available for download from the ACOG website.
References
- Morrison A, Wensley R. Boxing up or boxed in? A short history of the Boston Consulting Group Share/Growth Matrix. J Market Manag. 1993;7(2):105-129. http://www.tandfonline.com/doi/abs/10.1080/0267257X.1991.9964145.
- Klasko SK, Toub DB. It's not a plan without a business plan. In: Sanfilippo JS, Nolan TE, Whiteside BH, eds. MBA Handbook for Healthcare Professionals. New York, NY: Parthenon Publishing Group; 2002:36-37.
- American Congress of Obstetricians and Gynecologists. ACOG Medical Home Toolkit. https://www.acog.org/About-ACOG/ACOG-Departments/Practice-Management-and-Managed-Care/ACOG-Medical-Home-Toolkit. Accessed August 14, 2017.
Bottom line
Ensuring that your patients have an outstanding experience is a smart business strategy. A unified approach that includes team members’ involvement to create a patient-centered environment will provide a quality experience and encourage patients to recommend your ObGyn practice to others.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Independent of the Affordable Care Act or any upcoming changes in health care, the focus of an ObGyn practice remains paramount: the patient comes first.
The “recipe” for creating patient satisfaction and service excellence is predicated upon the mission of your practice and creating a shared vision with your employees. An action plan that is created and “visited/revisited”on a regular basis will serve to keep all abreast of the latest information to enhance the quality of patient care. It goes without saying, the ObGyn must first “lead by example” and always strive for satisfied patients who will tell their friends about your practice.
Start with the right tools
To organize a practice well, you need the right tools, which ideally include mission and vision statements and an action plan with goals and objectives.
Mission statement
A mission statement can be developed by the ObGyn(s) in your office or in concert with your staff. It should include:
- the “here and now” focus on the current approach to patient care
- why the practice exists (Develop a brief description of your practice, including the desired patient population.)
- the products and services offered and why and how those services are provided.
Here is an example of a mission statement for an ObGyn practice: “Our mission is to provide excellent, exceptional, personalized care for women of all ages in a warm and friendly environment. We incorporate leading-edge technology in our practice and continue to be a leader in obstetrics and gynecology.”
Vision statement
A vision statement should be developed in concert with your staff. It should include:
- the “then and there” focus on the historic perspective of your practice
- the ObGyn(s) and staff vision of the future
- what the ObGyn(s) and staff want to create.
The vision statement should energize and excite your personnel, create a shared and meaningful purpose, inspire passion and interest, and convey the values you want to share in your practice.
Here is an example of a vision statement for an ObGyn practice: “We aim to become the premier obstetrics and gynecology pro-vider to residents of (location) community.”
Action plan: Setting goals
To succeed, an ObGyn practice needs to:
- develop targets and challenges reflecting periodic (quarterly) meetings with staff and new entity development in the practice
- establish benchmarks and measurable parameters (How do you compare with other local practices? Set criteria/metrics to assess your progress.)
- ensure that the objectives support the goals (Develop goals and objectives over a defined period of time.)
- revisit the goals (Have they have been met? Do they need revision?)
Goals and objectives are essential for the continued health of your practice. This is all predicated upon developing a competitive advantage and then maintaining it.
Read about how to make a positive first impression on a new patient.
Is the environment welcoming?
When we examine a practice from the patient’s point of view, a good starting place is with the front desk. Have you looked at your front desk “from the outside in?” In one sense, this is the showcase of your practice.
Related article:
Four pillars of a successful practice: 2. Attract new patients
The first impression: Appointment scheduling
The first impression a patient receives about your practice occurs when she attempts to set up an appointment. Perhaps you might ask someone to call in to schedule an appointment. Is the caller immediately put on hold? Are your personnel courteous on the phone? Can she be seen quickly if she has a problem? How long is the wait for an annual exam? A test run can be very revealing.
Walk in the front door
When a patient walks in the door, does the physical office space radiate a friendly, relaxed atmosphere? Walk through the waiting room, then consultation and exam rooms as if you are a patient seeing it for the first time. Have you created an environment in which patients sense a well-organized office and the esprit de corps of the personnel? Does it look and smell fresh and clean? This all sends a loud and clear positive message about your practice.1–3
Here are some suggestions for making a waiting room more inviting:
- Provide a seating arrangement that is “patient centered.” For example, semi- circular arrangements allow easy viewing of any monitors in the waiting room.
- WiFi is a great addition. Post several signs with the user name and password.
- Offer computers for patients to use to complete registration
- Set up a fish tank. If well-maintained, it can be soothing to many people.
- Display medical information pamphlets, even if they are rarely taken.
- Provide a big screen television that offers information about your practice, including personnel and procedures.
Streaming ads for physician offices are available. One platform, Outcome Health (https://www.outcomehealth.com), provides flat-screen TVs and tablets that show patient education videos.4 Another vendor, Patient Point (http://patientpoint.com), offers waiting room networks, editorials, and other communications designed to support “the goals of improving healthcare.”5 Other available media include channel news and music programming to relax patients.6
Wait times. A patient’s perceived wait time and the actual wait time are often quite different. How long she waits to see the ObGyn is “numero uno” with regard to patient satisfaction and can be a key source of annoyance, irritability, stress, and anger.
Does someone inform waiting patients that the ObGyn is running late? Does staff at the front desk or perhaps your medical assistant inquire, “Can I get you anything? The doctor is running late,” or “Dr. Jones has just finished delivering a baby. He’ll be here in 10 minutes. He’ll see you first.”
Consultation and exam rooms
Suggestions to develop a relaxing environment in your consultation and exam rooms are7:
- decorate the walls with soft, pastel colors
- use “spa aesthetics” to create a colorful atmosphere with appropriate lighting, artwork, and modern furnishings
- present a few magazines neatly and update them periodically
- stock and appropriately maintain the patients rooms with medical supplies
- remember, “Subjects perceive people more positively in beautiful rooms than in ugly rooms.”5
Read about how to keep your patients satisfied and your business stable.
Set the lead example
The need for open and supportive communication between you and your office staff cannot be overly emphasized. An ideal office staff member understands and shares in the vision, is aware of stated goals and objectives, is responsive to patient needs, and wants to create a win-win environment.
Frequently discuss your expectations with your staff. Expect them to be responsive, courteous, competent, have good communication skills, and be influenced by the appearance of the physical environ-ment. Provide support and educational tools to help them successfully perform their work.
Related article:
Four pillars of a successful practice: 1. Keep your current patients happy
Discover your patients’ vision of customer service
Formal measurement of patient satisfaction began with Professor Irwin Press at the University of Notre Dame. Rod Ganey, a sociologist and statistician, then developed the Press Ganey Patient Satisfaction Survey. These points earlier conveyed by Maslow and Mintz8 addressed the “effects of esthetic surroundings.” Color and art proved to be preferences in an esthetically pleasing environment. Additional historical information has been provided by Siegrist, who addressed “the patient experience.”9 He cites the myth that patients do not fill out satisfaction surveys. Indeed they do. Patient satisfaction is not a personality contest but rather a reflection of the health care provider’s investment of time and effort to offer patient-centered care. Siegrist also notes that the patient’s family plays a key role in how a patient perceives her experience with her health care professional.9
The federal government has been actively involved in assessing patient satisfaction in the hospital setting since 2002. This is reflected in the Centers for Medicare and Medicaid Services, the Agency for Healthcare Research and Quality, and Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) surveys. The HCAHPS is a 27-question survey randomly administered to adult inpatients after discharge.10–12
The following metrics are often included in patient satisfaction surveys9,10:
- rating of hospital care from 0 (lowest) to 10 (highest)
- percentage of patients who would recommend a practice to family and friends
- number of patients who say their health care providers always communicate well
- the number of patients who report that the office is always clean and friendly.
Use of search engines focused on health care patient surveys can provide a number of options for clinicians to use in their practice.
Tips on patient satisfaction
Several interesting tips from the busi-ness world can be applied to an ObGyn’s practice14:
- You will only hear from 4% of unhappy customers.
- One dissatisfied customer tells 9.
- 95% of customers with resolved issues will do business with you again.
- If a problem is not addressed, that patient will tell 10 others.
- Resolve the problem and 5 people will know about it.
- It costs 5 times as much effort to gain 1 new customer.
- Loyal customers in 1 area of service are good prospects for other (new) services.
Related article:
Using the Internet in your practice. Part 2: Generating new patients using social media
Tell stories about good, satisfied patients
Sharing the stories of satisfied patients motivates others to consider coming to your practice. To develop these stories, offer a “suggestion box” where patients can leave compliments or comments about their experiences. Ask patients to record their positive reviews (be sure to obtain written consent before recording and publishing). Show the videos on the big-screen TVs in your waiting room and include patient reviews (written, audio, and video) on your website.15
Related article:
Four pillars of a successful practice: 4. Motivate your staff
Reevaluate periodically
Encouraging team spirit makes good business sense. Offer staff members bonuses for coming up with improved processes. Provide educational programs for staff on patient care, technology, etc. If a difficult experience occurs, discuss it openly with staff members without accusing, asking them for suggestions to improve the situation.16
To assess the monetary value of your practice, you need to know what contributes to your profit margin and overhead. What investments are the most profitable? Then monitor each segment of the office practice.
Should you proceed with a purchase? Should you take on a new hire? Let's look at one excellent model from the Boston Consulting Group (FIGURE) that provides insight into "low and high performance" aspects of business or practice.1
In the matrix, Stars use large amounts of cash and are leaders in cash generation. Stars lead to development of a Cash Cow, which are entities that generate profits and cash with low investment prerequisites. Dogs are segments of product and service line(s) that should be carefully reevaluated. A decision must be made to liquidate if the problem cannot be corrected. Question Marks have the worst cash characteristics of all and are associated with high demands and low profit margin(s).1
SWOT analysis
A SWOT analysis is most helpful when assessing a practice in real time. The basic tenets are2:
Strengths:
- prestigious reputation
- technological expertise
Weaknesses:
- antiquated computer system
- lack of experience in specific areas
Opportunities:
- growing market demand for a specific product or procedure
- provision of unique services
Threats:
- changing demographics
- competitive practices
- changes in health care third-party payers.
The American College of Obstetricians and Gynecologists (ACOG) has developed an "ACOG Medical Home Toolkit" to allow ObGyns to assess how significant the changes regarding payers will be to their practice. Sections include the patient/practice partnership support; clinical care information; community resources; care delivery management; performance measurement and improvement; and payment and finance.3 The toolkit is available for download from the ACOG website.
References
- Morrison A, Wensley R. Boxing up or boxed in? A short history of the Boston Consulting Group Share/Growth Matrix. J Market Manag. 1993;7(2):105-129. http://www.tandfonline.com/doi/abs/10.1080/0267257X.1991.9964145.
- Klasko SK, Toub DB. It's not a plan without a business plan. In: Sanfilippo JS, Nolan TE, Whiteside BH, eds. MBA Handbook for Healthcare Professionals. New York, NY: Parthenon Publishing Group; 2002:36-37.
- American Congress of Obstetricians and Gynecologists. ACOG Medical Home Toolkit. https://www.acog.org/About-ACOG/ACOG-Departments/Practice-Management-and-Managed-Care/ACOG-Medical-Home-Toolkit. Accessed August 14, 2017.
Bottom line
Ensuring that your patients have an outstanding experience is a smart business strategy. A unified approach that includes team members’ involvement to create a patient-centered environment will provide a quality experience and encourage patients to recommend your ObGyn practice to others.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Ulrich RS. Evidence-based environmental design for improving medical outcomes: Paper Delivered at a Conference Entitled Healing by Design: Building for Health Care in the 21st Century. Montreal: McGill University Health Centre; 2000. http://www.brikbase.org/sites/default/files/Evidence%20Based%20Environmental%20Design%20for%20Improving%20Medical.pdf. Accessed August 15, 2017.
- Becker F, Douglass S. The ecology of the patient visit: physical attractiveness, waiting times and perceived quality of care. J Ambul Care Manag. 2008;31(2):128–141.
- Becker F, Sweeney B, Parsons K. Ambulatory facility design and patients’ perceptions of healthcare quality. HERD. 2008;1(4):35–54.
- Outcome Health Website. https://www.outcomehealth.com/. Accessed August 14, 2017.
- Mazer SE. The waiting room: Where suffering begins. Healing Healthcare Systems website. http://www.healinghealth.com/waiting-room-suffering-begins/. Published November 7, 2014. Accessed August 14, 2017.
- Patient Point Programs Website. http://patientpoint.com/. Accessed August 14, 2017.
- Almquist J, Kelly C, Bromberg J, Bryant S, Christianson T, Montori V. Consultation room design and the clinical encounter: the space and interaction randomized trial. Health Environ Res Design. 2009;3(1):41–78.
- Maslow A, Mintz N. Effects of esthetic surroundings: I. Initial effects of three esthetic conditions upon perceiving “energy” and “well-being” in faces. J Psychology. 1956;41(2):247–254.
- Siegrist RB. The patient experience. In: Sanfilippo JS, Bieber E, Javich D, Siegrist R, eds. MBA for Healthcare. New York, NY: Oxford Press;2016:227–236.
- Press I. Patient satisfaction: Understanding and managing the experience of care. 2nd ed. Chicago, IL: Health Administration Press; 2005:66–78.
- Piper L, Tallman E. Hospital consumer assessment of healthcare providers and systems: An ethical leadership dilemma to satisfy patients. Health Care Manag (Frederick). 2016;35(2):151–155.
- Giordano L, Elliott M, Goldstein E, Lehrman W, Spencer P. Development, implementation and public reporting of HCAHPS survey. Med Care Res Rev. 2010;67(1):27–37.
- Jones KE. Helping the health profession help others: Applying business principles to the medical world. University of Tennessee, Knoxville Honors Thesis Projects. http://trace.tennessee.edu/cgi/viewcontent.cgi?article=1560&context=utk_chanhonoproj. Published 2002. Accessed August 14, 2017.
- Baum N. Marketing your practice: ethically, effectively and economically. In: Sanfilippo JS, Nolan TE, Whiteside BH, eds. MBA Handbook for Healthcare Professionals. New York, NY: Parthenon Publishing Group; 2002:123–154.
- Baum NH. Four pillars of a successful practice: 1. Keep your current patients happy. OBG Manag. 2013;25(3):49–56.
- Baum NH. Four pillars of a successful practice: 4. Motivate your staff. OBG Manag. 2013;25(8):29–33.
- Ulrich RS. Evidence-based environmental design for improving medical outcomes: Paper Delivered at a Conference Entitled Healing by Design: Building for Health Care in the 21st Century. Montreal: McGill University Health Centre; 2000. http://www.brikbase.org/sites/default/files/Evidence%20Based%20Environmental%20Design%20for%20Improving%20Medical.pdf. Accessed August 15, 2017.
- Becker F, Douglass S. The ecology of the patient visit: physical attractiveness, waiting times and perceived quality of care. J Ambul Care Manag. 2008;31(2):128–141.
- Becker F, Sweeney B, Parsons K. Ambulatory facility design and patients’ perceptions of healthcare quality. HERD. 2008;1(4):35–54.
- Outcome Health Website. https://www.outcomehealth.com/. Accessed August 14, 2017.
- Mazer SE. The waiting room: Where suffering begins. Healing Healthcare Systems website. http://www.healinghealth.com/waiting-room-suffering-begins/. Published November 7, 2014. Accessed August 14, 2017.
- Patient Point Programs Website. http://patientpoint.com/. Accessed August 14, 2017.
- Almquist J, Kelly C, Bromberg J, Bryant S, Christianson T, Montori V. Consultation room design and the clinical encounter: the space and interaction randomized trial. Health Environ Res Design. 2009;3(1):41–78.
- Maslow A, Mintz N. Effects of esthetic surroundings: I. Initial effects of three esthetic conditions upon perceiving “energy” and “well-being” in faces. J Psychology. 1956;41(2):247–254.
- Siegrist RB. The patient experience. In: Sanfilippo JS, Bieber E, Javich D, Siegrist R, eds. MBA for Healthcare. New York, NY: Oxford Press;2016:227–236.
- Press I. Patient satisfaction: Understanding and managing the experience of care. 2nd ed. Chicago, IL: Health Administration Press; 2005:66–78.
- Piper L, Tallman E. Hospital consumer assessment of healthcare providers and systems: An ethical leadership dilemma to satisfy patients. Health Care Manag (Frederick). 2016;35(2):151–155.
- Giordano L, Elliott M, Goldstein E, Lehrman W, Spencer P. Development, implementation and public reporting of HCAHPS survey. Med Care Res Rev. 2010;67(1):27–37.
- Jones KE. Helping the health profession help others: Applying business principles to the medical world. University of Tennessee, Knoxville Honors Thesis Projects. http://trace.tennessee.edu/cgi/viewcontent.cgi?article=1560&context=utk_chanhonoproj. Published 2002. Accessed August 14, 2017.
- Baum N. Marketing your practice: ethically, effectively and economically. In: Sanfilippo JS, Nolan TE, Whiteside BH, eds. MBA Handbook for Healthcare Professionals. New York, NY: Parthenon Publishing Group; 2002:123–154.
- Baum NH. Four pillars of a successful practice: 1. Keep your current patients happy. OBG Manag. 2013;25(3):49–56.
- Baum NH. Four pillars of a successful practice: 4. Motivate your staff. OBG Manag. 2013;25(8):29–33.
Sneak Peek: The Hospital Leader blog – Sept. 2017
Wrongful Life
There have been recent discussions in the lay media about a growing trend of litigation cases focused not on the “right to live,” but rather on the “right to die.” These cases have involved patients who received aggressive treatment, despite having documentation of their wishes not to receive such aggressive treatment. Although unsettling, it is not surprising that this issue has arisen, given the national conversations about the exorbitant cost of care at the end of life in the United States, and the frequency with which patients do not receive end-of-life care that is concordant with their wishes.
These conversations have spurred providers and patients to discuss and document their wishes, via advanced care directives and/or POLST orders (Physicians Orders for Life Sustaining Treatment). There is now even a national day devoted to advanced care decision making (National Healthcare Decisions Day).

But for situations where the paperwork is clear, and the patient actually does receive undesired aggressive care, more plaintiff attorneys are taking on these cases of the “right to die,” since now more people are recognizing and accepting that unwanted life is a type of harm.
This brings to light two important considerations in how we use advanced care planning documentation:
1. These documents should be treated as dynamic decision-making documents, not static documents that are filled out and filed at a single point in time. Patient wishes can and do change due to a variety of factors; any changes should be repeatedly sought to ensure consistency with care plans.
2. These documents should be the start of a conversation, not the end of a conversation. Written documentation can still be wrought with ambiguity; a conversation about the document can help clarify desires and ensure that wishes and care plans match.
In our ongoing desire to “do no harm,” overtreatment is increasingly being recognized by patients and families as a type of harm. To avoid these potentially catastrophic situations, we should all use advanced care documentation as the start of a careful conversation about goals of care and treatment choices. Hospitalists should work with their interprofessional team members (for example, case managers, social workers, nurse navigators, and so on) to make sure every patient has, or is at least working on, advance care directives, and guide the patient and family in decision-making that puts them at ease. With our patients, we can help ensure concordance between their end-of-life wishes and our care plans.
Read the full post at hospitalleader.org.
Also on The Hospital Leader…
Follow You, Follow Me by Tracy Cardin, ACNP-BC, SFHM
SHM Movers & Shakers, Hospital Silos & JHM Research in HM News by Felicia Steele
Wrongful Life
There have been recent discussions in the lay media about a growing trend of litigation cases focused not on the “right to live,” but rather on the “right to die.” These cases have involved patients who received aggressive treatment, despite having documentation of their wishes not to receive such aggressive treatment. Although unsettling, it is not surprising that this issue has arisen, given the national conversations about the exorbitant cost of care at the end of life in the United States, and the frequency with which patients do not receive end-of-life care that is concordant with their wishes.
These conversations have spurred providers and patients to discuss and document their wishes, via advanced care directives and/or POLST orders (Physicians Orders for Life Sustaining Treatment). There is now even a national day devoted to advanced care decision making (National Healthcare Decisions Day).
But for situations where the paperwork is clear, and the patient actually does receive undesired aggressive care, more plaintiff attorneys are taking on these cases of the “right to die,” since now more people are recognizing and accepting that unwanted life is a type of harm.
This brings to light two important considerations in how we use advanced care planning documentation:
1. These documents should be treated as dynamic decision-making documents, not static documents that are filled out and filed at a single point in time. Patient wishes can and do change due to a variety of factors; any changes should be repeatedly sought to ensure consistency with care plans.
2. These documents should be the start of a conversation, not the end of a conversation. Written documentation can still be wrought with ambiguity; a conversation about the document can help clarify desires and ensure that wishes and care plans match.
In our ongoing desire to “do no harm,” overtreatment is increasingly being recognized by patients and families as a type of harm. To avoid these potentially catastrophic situations, we should all use advanced care documentation as the start of a careful conversation about goals of care and treatment choices. Hospitalists should work with their interprofessional team members (for example, case managers, social workers, nurse navigators, and so on) to make sure every patient has, or is at least working on, advance care directives, and guide the patient and family in decision-making that puts them at ease. With our patients, we can help ensure concordance between their end-of-life wishes and our care plans.
Read the full post at hospitalleader.org.
Also on The Hospital Leader…
Follow You, Follow Me by Tracy Cardin, ACNP-BC, SFHM
SHM Movers & Shakers, Hospital Silos & JHM Research in HM News by Felicia Steele
Wrongful Life
There have been recent discussions in the lay media about a growing trend of litigation cases focused not on the “right to live,” but rather on the “right to die.” These cases have involved patients who received aggressive treatment, despite having documentation of their wishes not to receive such aggressive treatment. Although unsettling, it is not surprising that this issue has arisen, given the national conversations about the exorbitant cost of care at the end of life in the United States, and the frequency with which patients do not receive end-of-life care that is concordant with their wishes.
These conversations have spurred providers and patients to discuss and document their wishes, via advanced care directives and/or POLST orders (Physicians Orders for Life Sustaining Treatment). There is now even a national day devoted to advanced care decision making (National Healthcare Decisions Day).
But for situations where the paperwork is clear, and the patient actually does receive undesired aggressive care, more plaintiff attorneys are taking on these cases of the “right to die,” since now more people are recognizing and accepting that unwanted life is a type of harm.
This brings to light two important considerations in how we use advanced care planning documentation:
1. These documents should be treated as dynamic decision-making documents, not static documents that are filled out and filed at a single point in time. Patient wishes can and do change due to a variety of factors; any changes should be repeatedly sought to ensure consistency with care plans.
2. These documents should be the start of a conversation, not the end of a conversation. Written documentation can still be wrought with ambiguity; a conversation about the document can help clarify desires and ensure that wishes and care plans match.
In our ongoing desire to “do no harm,” overtreatment is increasingly being recognized by patients and families as a type of harm. To avoid these potentially catastrophic situations, we should all use advanced care documentation as the start of a careful conversation about goals of care and treatment choices. Hospitalists should work with their interprofessional team members (for example, case managers, social workers, nurse navigators, and so on) to make sure every patient has, or is at least working on, advance care directives, and guide the patient and family in decision-making that puts them at ease. With our patients, we can help ensure concordance between their end-of-life wishes and our care plans.
Read the full post at hospitalleader.org.
Also on The Hospital Leader…
Follow You, Follow Me by Tracy Cardin, ACNP-BC, SFHM
SHM Movers & Shakers, Hospital Silos & JHM Research in HM News by Felicia Steele
Current Approaches to Measuring Functional Status Among Older Adults in VA Primary Care Clinics
The ability to perform activities of daily living (ADLs), commonly called functional status, is central to older adults’ quality of life (QOL) and independence.1,2 Understanding functional status is key to improving outcomes for older adults. In community-dwelling older adults with difficulty performing basic ADLs, practical interventions, including physical and occupational therapy, can improve functioning and prevent functional decline.3,4 Understanding function also is important for delivering patient-centered care, including individualizing cancer screening,5 evaluating how patients will tolerate interventions,6-9 and helping patients and families determine the need for long-term services and supports.
For these reasons, assessing functional status is a cornerstone of geriatrics practice. However, most older adults are cared for in primary care settings where routine measurement of functional status is uncommon.10,11 Although policy leaders have long noted this gap and the obstacle it poses to improving the quality and outcomes of care for older adults, many health care systems have been slow to incorporate measurement of functional status into routine patient care.12-14
Over the past several years, the VA has been a leader in the efforts to address this barrier by implementing routine, standardized measurement of functional status in primary care clinics. Initially, the VA encouraged, but did not require, measurement of functional status among older adults, but the implementation barriers and facilitators were not formally assessed.15 In a postimplementation evaluation, the authors found that a relatively small number of medical centers implemented functional measures. Moreover, the level of implementation seemed to vary across sites. Some sites were collecting complete measures on all eligible older patients, while other sites were collecting measures less consistently.15
As part of a national VA initiative to learn how best to implement standardized functional status measurement, the authors are conducting a qualitative study, including a formal assessment of barriers and facilitators to implementing functional assessments in VA primary care clinics. In the current project, which serves as formative work for this larger ongoing study, the authors identified and described current processes for measuring functional status in VA primary care patient aligned care team (PACT) and Geriatric (GeriPACT) clinics.
Methods
A rapid qualitative analysis approach was used, which included semistructured interviews with primary care stakeholders and rapid data analysis to summarize each clinic’s approach to measuring functional status and develop process maps for each clinic (eFigures 1, 2, 3, and 4 ). Interviews and analyses were conducted by a team consisting of a geriatrician clinician-researcher, a medical anthropologist, and a research coordinator. The institutional review boards of the San Francisco VAMC and the University of California, San Francisco approved the study.
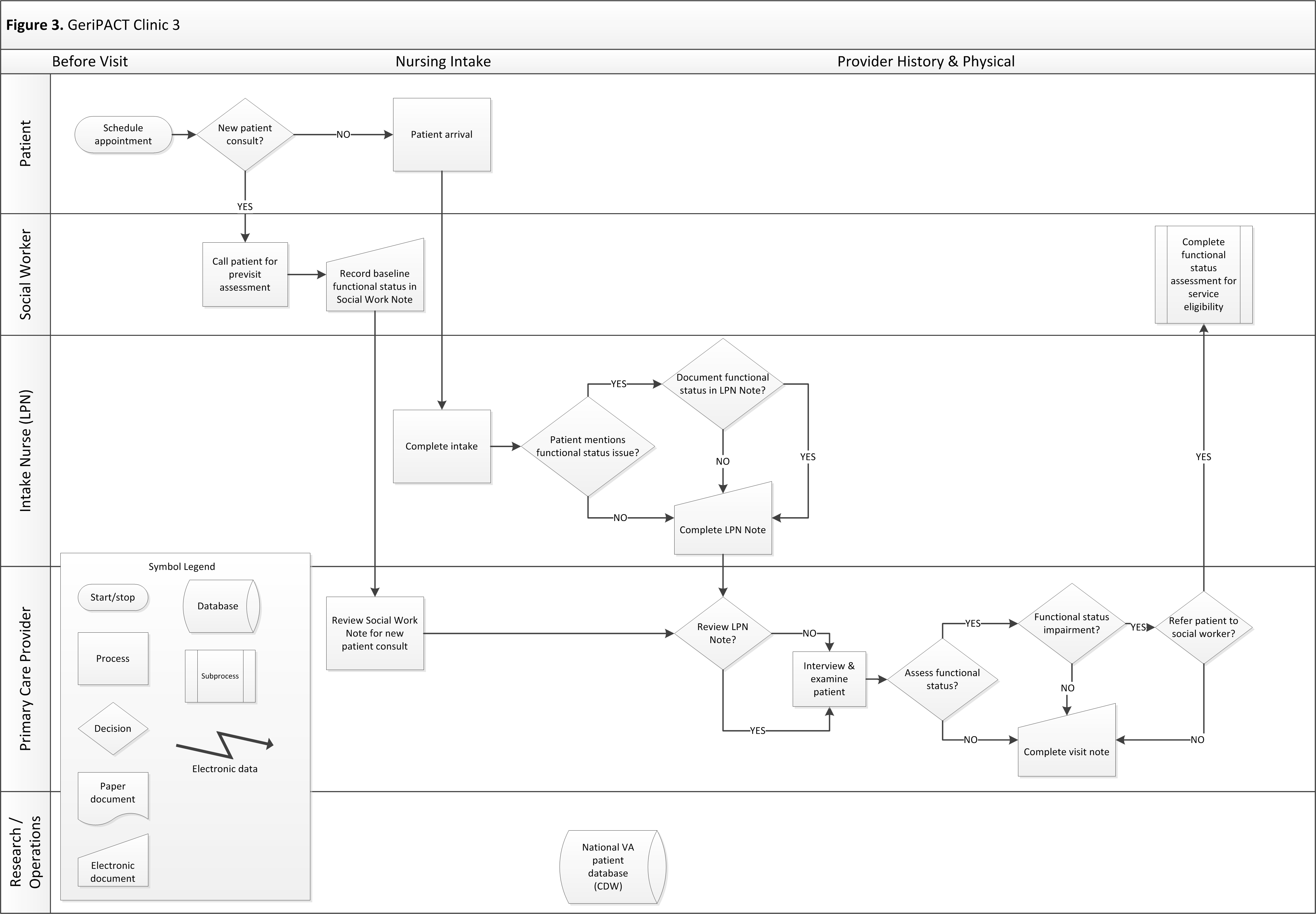
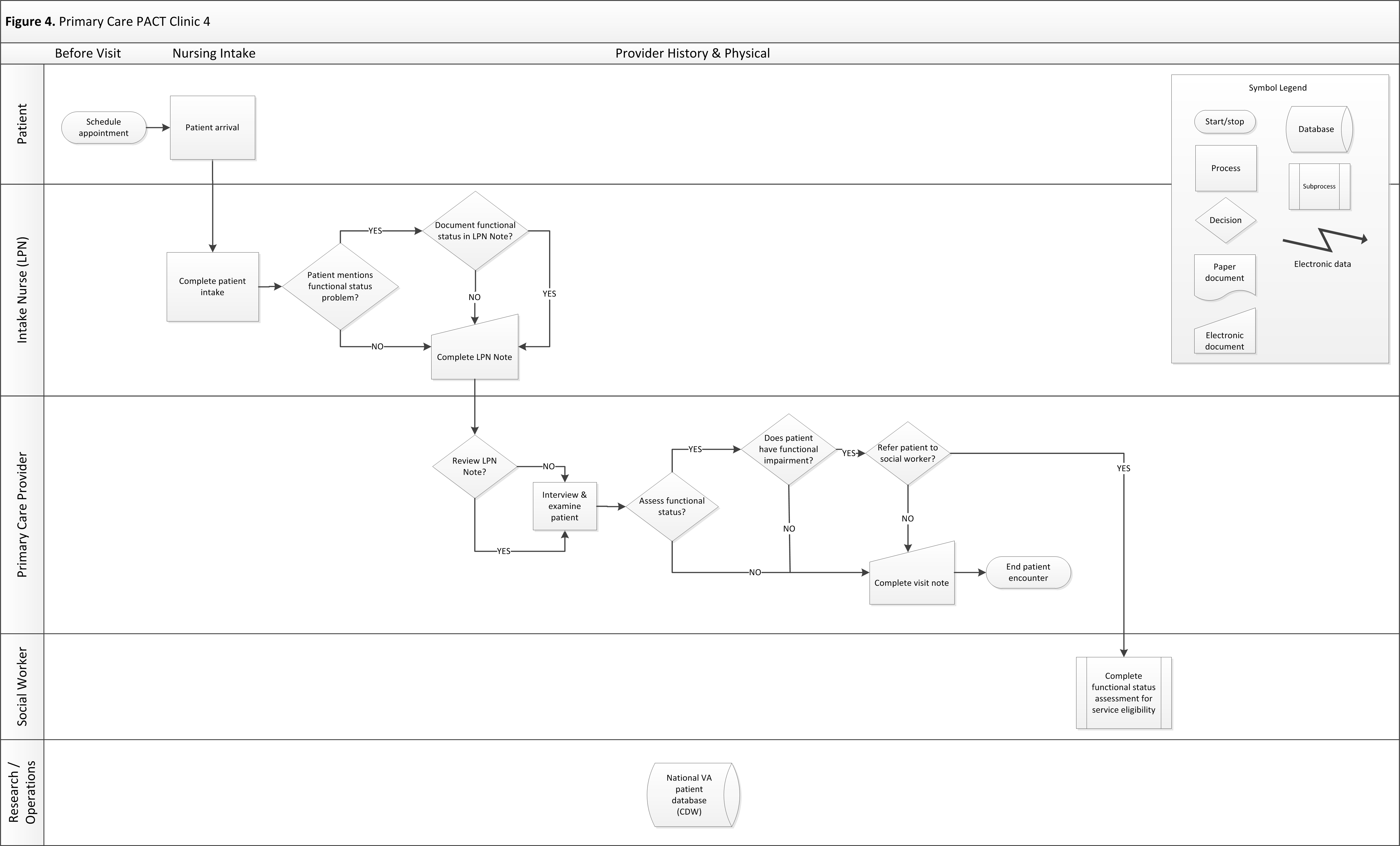
Sampling Strategy
In order to identify VAMCs with varying approaches to assessing functional status in older patients who attended primary care appointments, the study used a criterion sampling approach.16,17 First, national “health factors” data were extracted from the VA Corporate Data Warehouse (CDW). Health factors are patient data collected through screening tools called clinical reminders, which prompt clinic staff and providers to enter data into checkbox-formatted templates. The study then identified medical centers that collected health factors data from patients aged ≥ 65 years (157 of 165 medical centers). A keyword search identified health factors related to the Katz ADL (bathing, dressing, transferring, toileting, and eating), and Lawton Instrumental ADL (IADL) Scale (using the telephone, shopping, preparing food, housekeeping, doing laundry, using transportation, managing medications, and managing finances).18,19 Health factors that were not collected during a primary care appointment were excluded.
Of the original 157 medical centers, 139 met these initial inclusion criteria. Among these 139 medical centers, 66 centers did not collect complete data on these 5 ADLs and 8 IADLs (eg, only ADLs or only IADLs, or only certain ADLs or IADLs).
Two medical centers were selected in each of the following 3 categories: (1) routinely used clinical reminders to collect standardized data on the Katz ADL and the Lawton IADL Scale; (2) routinely used clinical reminders to collect functional status data but collected partial information; and (3) did not use a clinical reminder to collect functional status data. To ensure that these 6 medical centers were geographically representative, the sample included at least 1 site from each of the 5 VA regions: 1 North Atlantic, 1 Southeast, 1 Midwest, 2 Continental (1 from the northern Continental region and 1 from the southern), and 1 Pacific. Three sites that included GeriPACTs also were sampled.
Primary care PACT and GeriPACT members from these 6 medical centers were recruited to participate. These PACT members included individuals who can assess function or use functional status information to inform patient care, including front-line nursing staff (licensed practical nurses [LPNs], and registered nurses [RNs]), primary care providers (medical doctors [MDs] and nurse practitioners [NPs]), and social workers (SWs).
Local bargaining units, nurse managers, and clinic directors provided lists of all clinic staff. All members of each group then received recruitment e-mails. Phone interviews were scheduled with interested participants. In several cases, a snowball sampling approach was used to increase enrollment numbers by asking interview participants to recommend colleagues who might be interested in participating.17
Data Collection
Telephone interviews were conducted between March 2016 and October 2016 using semistructured guides developed from the project aims and from related literature in implementation science.20,21 Interview domains included clinic structure, team member roles and responsibilities, current practices for collecting functional status data, and opinions on barriers and facilitators to assessing and recording functional status (Appendix:
Data Analysis
Rapid analysis, a team-based qualitative approach was used to engage efficiently and systematically with the data.22,23 This approach allowed results to be analyzed more quickly than in traditional qualitative analysis in order to inform intervention design and develop implementation strategies.23 Rapid analysis typically includes organization of interview data into summary templates, followed by a matrix analysis, which was used to create process maps.24
Summary Templates
Summary templates were developed from the interview guides by shortening each question into a representative code. The project team then read the transcripts and summarized key points in the appropriate section of the template. This process, known as data reduction, is used to organize and highlight material so conclusions can be drawn from the data easily.22 In order to maintain rigor and trustworthiness, one team member conducted the interview, and a different team member created the interview summary. All team members reviewed each summary and met regularly to discuss results.
The summary templates were converted into matrix analyses, a method of displaying data to identify relationships, including commonalities and differences.24 The matrixes were organized by stakeholder group and clinic in order to compare functional status assessment and documentation workflows across clinics.
Process Maps
Finally, the team used the matrix data to create process maps for each clinic of when, where, and by whom functional status information was assessed and documented. These maps were created using Microsoft Visio (Redmond, WA). The maps integrated perspectives from all participants to give an overview of the process for collecting functional status data in each clinic setting. To ensure accuracy, participants at each site received process maps to solicit feedback and validation.
Results
Forty-six participants at 6 medical centers (20 MDs and NPs, 19 RNs and LPNs, and 7 SWs) from 9 primary care clinics provided samples and interviews. The study team identified 3 general approaches to functional status assessment: (1) Routine collection of functional status data via a standardized clinical reminder; (2) Routine collection of functional status data via methods other than a clinical reminder (eg, a previsit telephone screen or electronic note template); and (3) Ad hoc approaches to measuring functional status (ie, no standard or routine approach to assessing or documenting functional status). The study team selected 4 clinics (2 PACTs and 2 GeriPACTs) clinics to serve as examples of the 3 identified approaches.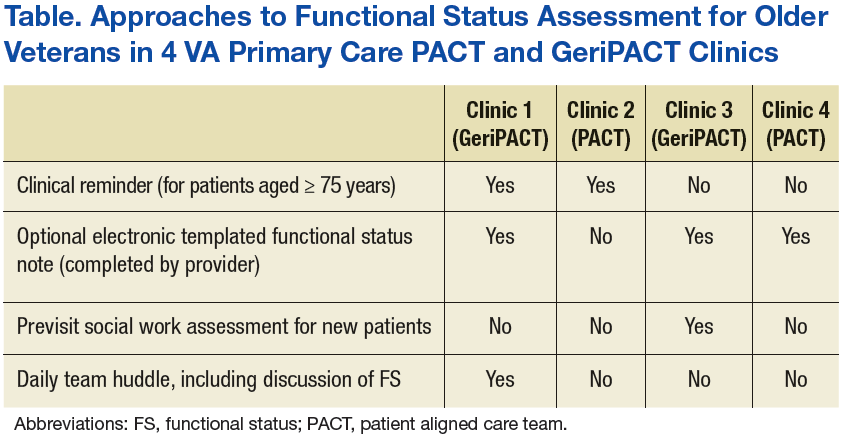
The processes for functional status assessment in each of 4 clinics are summarized in the following detailed descriptions (Table).
Clinic 1
Clinic 1 is a GeriPACT clinic that routinely assesses and documents functional status for all patients (efigure 1, available at feprac.com). The clinic’s current process includes 4 elements: (1) a patient questionnaire; (2) an annual clinical reminder administered by an RN; (3) a primary care provider (PCP) assessment; and (4) a postvisit SW assessment if referred by the PCP.
All newly referred patients are mailed a paper questionnaire that includes questions about their medical history and functional status. The patient is asked to bring the completed questionnaire to the first appointment. The clinic RN completes this form for returning patients at every visit during patient intake.
Second, the clinic uses an annual functional status clinical reminder for patients aged ≥ 75 years. The reminder includes questions about a patient’s ability to perform ADLs and IADLs with 3 to 4 response options for each question. If the clinical reminder is due at the time of a patient appointment, the RN fills out the reminder using information from the paper questionnaire. The RN also records this functional status in the nursing intake note. The RN may elect to designate the PCP as a cosigner for the nursing intake note especially if there are concerns about or changes in the patient’s functional status.
Third, the RN brings the paper form to the PCP, who often uses the questionnaire to guide the patient history. The PCP then uses the questionnaire and patient history to complete a functional status template within their visit note. The PCP also may use this information to inform patient care (eg, to make referrals to physical or occupational therapy).
Finally, the PCP might refer the patient to SW. The SW may be able to see the patient immediately after the PCP appointment, but if not, the SW follows up with a phone call to complete further functional status assessment and eligibility forms.
In addition to the above assessments by individual team members, the PACT has an interdisciplinary team huddle at the end of each clinic to discuss any issues or concerns about specific patients. The huddles often focus on issues related to functional status.
Clinic 2
Clinic 2 is a primary care PACT clinic that routinely assesses and documents functional status (eFigure 2, available at fedprac.com). The clinic process includes 3 steps: an annual clinical reminder for patients aged ≥ 75 years; a PCP assessment; and a postvisit SW assessment if referred by the PCP.
First, patients see an LPN for the intake process. During intake, the LPN records vitals and completes relevant clinical reminders. Similar to Clinic 1, Clinic 2 requires an annual functional status clinical reminder that includes ADLs and IADLs for patients aged ≥ 75 years. Patient information from the intake and clinical reminders are recorded by the LPN in a preventative medicine note in the electronic health record. This note is printed and handed to the PCP.
The PCP may review the preventative medicine note prior to completing the patient history and physical, including the functional status clinical reminder when applicable. If the PCP follows up on any functional issues identified by the LPN or completes further assessment of patient function, he or she may use this information to refer the patient to services or to place a SW consult; the PCP’s functional assessment is documented in a free-form visit note.
When the SW receives a consult, a chart review for social history, demographic information, and previous functional status assessments is conducted. The SW then calls the patient to administer functional and cognitive assessments over the phone and refers the patient to appropriate services based on eligibility.
Clinic 3
Clinic 3 is a GeriPACT clinic where functional status information is routinely collected for all new patients but may or may not be collected for returning patients (eFigure 3, available at fedprac.com). The process for new patients includes a previsit SW assessment; an informal LPN screening (ie, not based on a standardized clinical reminder); a PCP assessment; and a postvisit SW assessment if referred by the provider. The process for returning patients is similar but omits the previsit social work assessment. New patients complete a comprehensive questionnaire with a SW before their first clinic visit. The questionnaire is completed by phone and involves an extensive social and medical history, including an assessment of ADLs and IADLs. This assessment is recorded in a free-form social work note.
Next, both new and returning patients see an LPN who completes the intake process, including vitals and clinical reminders. Clinic 3 does not have a clinical reminder for functional status. However, the LPN could elect to ask about ADLs or IADLs if the patient brings up a functional issue related to the chief symptom or if the LPN observes something that indicates possible functional impairment, such as difficulty walking or a disheveled appearance. If discussed, this information is recorded in the LPN intake note, and the LPN also could verbally inform the PCP of the patient’s functional status. The RN is not formally involved in intake or functional status assessment in this clinic.
Finally, the patient sees the PCP, who may or may not have reviewed the LPN note. The PCP may assess functional status at his or her discretion, but there was no required assessment. The PCP could complete an optional functional status assessment template included in the PCP visit note. The PCP can refer the patient to services or to SW for further evaluation.
Clinic 4
Clinic 4 is a primary care PACT clinic that does not routinely measure functional status (eFigure 4, available at fedprac.com). The approach includes an informal LPN screening (ie, not based on a standardized clinical reminder); a PCP assessment; and a postvisit social worker assessment if referred by the provider. These steps are very similar to those of clinic 3, but they do not include a previsit SW assessment for new patients.
Although not represented within the 4 clinics described in this article, the content of functional status clinical reminders differed across the 9 clinics in the larger sample. Clinical reminders differed across several domains, including the type of question stems (scripted questions for each ADL vs categories for each activity); response options (eg, dichotomous vs ≥ 3 options), and the presence of free-text boxes to allow staff to enter any additional notes.
Discussion
Approaches to assessing and documenting functional status varied widely. Whereas some clinics primarily used informal approaches to assessing and documenting functional status (ie, neither routine nor standardized), others used a routine, standardized clinical reminder, and some combined several standardized approaches to measuring function. The study team identified variability across several domains of the functional status assessment process, including documentation, workflow, and clinical reminder content.
Approaches to functional assessment differed between GeriPACT and PACT clinics. Consistent with the central role that functional status assessment plays in geriatrics practice, GeriPACTs tended to employ a routine, multidisciplinary approach to measuring functional status. This approach included standardized functional assessments by multiple primary care team members, including LPNs, SWs, and PCPs. In contrast, when PACTs completed standardized functional status assessment, it was generally carried out by a single team member (typically an LPN). The PCPs in PACTs used a nonroutine approach to assess functional status in which they performed detailed functional assessments for certain high-risk patients and referred a subset for further SW evaluation.
These processes are consistent with research showing that standardized functional status data are seldom collected routinely in nongeriatric primary care settings.11 Reports by PCPs that they did not always assess functional status also are consistent with previous research demonstrating that clinicians are not always aware of their patients’ functional ability.10
In addition to highlighting differences between GeriPACT and PACTs, the identified processes illustrate the variability in documentation, clinic workflow, and clinical reminder content across all clinics. Approaches to documentation included checkbox-formatted clinical reminders with and without associated nursing notes, patient questionnaires, and templated PCP and SW notes. Clinics employed varying approaches to collect functional status information and to ensure that those data were shared with the team. Clinic staff assessed functional status at different times during the clinical encounter. Clinics used several approaches to share this information with team members, including warm handoffs from LPNs to PCPs, interdisciplinary team huddles, and electronic signoffs. Finally, clinical reminder content varied between clinics, with differences in the wording of ADL and IADL questions as well as in the number and type of response options.
This variability highlights the challenges inherent in developing a routine, standardized approach to measuring functional status that can be adapted across primary care settings. Such an approach must be both flexible enough to accommodate variation in workflow and structured enough to capture accurate data that can be used to guide clinical decisions. Capturing accurate, standardized data in CDW also will inform efforts to improve population health by allowing VHA leaders to understand the scope of disability among older veterans and plan for service needs and interventions.
Whereas the larger qualitative study will identify the specific barriers and facilitators to developing and implementing such an approach, current clinic processes present here offer hints as to which features may be important. For example, several clinics collected functional status information before the visit by telephone or questionnaire. Therefore, it will be important to choose a functional status assessment instrument that is validated for both telephone and in-person use. Similarly, some clinics had structured clinical reminders with categoric response options, whereas others included free-text boxes. Incorporating both categoric responses (to ensure accurate data) as well as free-text (to allow for additional notes about a patient’s specific circumstances that may influence service needs) may be one approach.
Limitations
This study’s approach to identifying clinic processes had several limitations. First, the authors did not send process maps to clinic directors for verification. However, speaking with PACT members who carry out clinic processes is likely the most accurate way to identify practice. Second, the results may not be generalizable to all VA primary care settings. Due to resource limitations and project scope, community-based outpatient clinics (CBOCs) were not included. Compared with clinics based in medical centers, CBOCs may have different staffing levels, practice models, and needs regarding implementation of functional status assessment.
Although 46 participants from 9 clinics were interviewed, there are likely additional approaches to measuring functional status that are not represented within this sample. In addition, 3 of the 4 clinics included are affiliated with academic institutions, and all 4 are located in large cities. Efforts to include rural VAMCs were not successful. Finally, clinic-level characteristics were not reported, which may impact clinic processes. Although study participants were asked about clinic characteristics, they were often unsure or only able to provide rough estimates. In the ongoing qualitative study, the authors will attempt to collect more reliable data about these clinic-level characteristics and to examine the potential role these characteristics may play as barriers or facilitators to implementing routine assessment of functional status in primary care settings.
Conclusion
VA primary care clinics had widely varying approaches for assessing and documenting functional status. This work along with a larger ongoing qualitative study that includes interviews with veterans will directly inform the design and implementation of a standardized, patient-centered approach to functional assessment that can be adapted across varied primary care settings. Implementing standardized functional status measurement will allow the VA to serve veterans better by using functional status information to refer patients to appropriate services and to deliver patient-centered care with the potential to improve patient function and quality of life.
1.Covinsky KE, Wu AW, Landefeld CS, et al. Health status versus quality of life in older patients: does the distinction matter? Am J Med. 1999;106(4):435-440.
2. Fried TR, McGraw S, Agostini JV, Tinetti ME. Views of older persons with multiple morbidities on competing outcomes and clinical decision-making. J Am Geriatr Soc. 2008;56(10):1839-1844.
3. Beswick AD, Rees K, Dieppe P, et al. Complex interventions to improve physical function and maintain independent living in elderly people: a systematic review and meta-analysis. Lancet. 2008;371(9614):725-735.
4. Szanton SL, Leff B, Wolff JL, Roberts L, Gitlin LN. Home-based care program reduces disability and promotes aging in place. Health Aff (Millwood). 2016;35(9):1558-1563.
5. Walter LC, Covinsky KE. Cancer screening in elderly patients: a framework for individualized decision making. JAMA. 2001;285(21):2750-2756.
6. Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med. 2009;361(16):1539-1547.
7. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29(25):3457-3465.
8. Crawford RS, Cambria RP, Abularrage CJ, et al. Preoperative functional status predicts perioperative outcomes after infrainguinal bypass surgery. J Vasc Surg. 2010;51(2):351-358; discussion 358-359.
9. Arnold SV, Reynolds MR, Lei Y, et al; PARTNER Investigators. Predictors of poor outcomes after transcatheter aortic valve replacement: results from the PARTNER (Placement of Aortic Transcatheter Valve) trial. Circulation. 2014;129(25):2682-2690.
10. Calkins DR, Rubenstein LV, Cleary PD, et al. Failure of physicians to recognize functional disability in ambulatory patients. Ann Intern Med. 1991;114(6):451-454.
11. Bogardus ST Jr, Towle V, Williams CS, Desai MM, Inouye SK. What does the medical record reveal about functional status? A comparison of medical record and interview data. J Gen Intern Med. 2001;16(11):728-736.
12. Bierman AS. Functional status: the six vital sign. J Gen Intern Med. 2001;16(11):785-786.
13. Iezzoni LI, Greenberg MS. Capturing and classifying functional status information in administrative databases. Health Care Financ Rev. 2003;24(3):61-76.
14. Clauser SB, Bierman AS. Significance of functional status data for payment and quality. Health Care Financ Rev. 2003;24(3):1-12.
15. Brown RT, Komaiko KD, Shi Y, et al. Bringing functional status into a big data world: validation of national Veterans Affairs functional status data. PloS One. 2017;12(6):e0178726.
16. Palinkas LA, Horwitz SM, Green CA, Wisdom JP, Duan N, Hoagwood K. Purposeful sampling for qualitative data collection and analysis in mixed method implementation research. Adm Policy Ment Health. 2015;42(5):533-544.
17. Patton MQ. Qualitative Research Evaluation and Methods. 4th ed. Thousand Oaks, CA: Sage; 2015.
18. Katz S, Ford AB, Moskowitz RW, Jackson BA, Jaffe MW. Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychosocial function. JAMA. 1963;185(12):914-919.
19. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3):179-186.
20. Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4(1):50.
21. Saleem JJ, Patterson ES, Militello L, Render ML, Orshansky G, Asch SM. Exploring barriers and facilitators to the use of computerized clinical reminders. J Am Med Inform Assoc. 2005;12(4):438-447.
22. Miles MB, Huberman AM, Saldana J. Qualitative Data Analysis: A Methods Sourcebook. 3rd ed. Thousand Oaks, CA: Sage; 2014.
23. Hamilton AB. Qualitative methods in rapid turn-around health services research. https://www.hsrd .research.va.gov/for_researchers/cyber_seminars /archives/video_archive.cfm?SessionID=780. Published December 11, 2013. Accessed August 9, 2017.
24. Averill JB. Matrix analysis as a complementary analytic strategy in qualitative inquiry. Qual Health Res. 2002;12(6):855-866.
The ability to perform activities of daily living (ADLs), commonly called functional status, is central to older adults’ quality of life (QOL) and independence.1,2 Understanding functional status is key to improving outcomes for older adults. In community-dwelling older adults with difficulty performing basic ADLs, practical interventions, including physical and occupational therapy, can improve functioning and prevent functional decline.3,4 Understanding function also is important for delivering patient-centered care, including individualizing cancer screening,5 evaluating how patients will tolerate interventions,6-9 and helping patients and families determine the need for long-term services and supports.
For these reasons, assessing functional status is a cornerstone of geriatrics practice. However, most older adults are cared for in primary care settings where routine measurement of functional status is uncommon.10,11 Although policy leaders have long noted this gap and the obstacle it poses to improving the quality and outcomes of care for older adults, many health care systems have been slow to incorporate measurement of functional status into routine patient care.12-14
Over the past several years, the VA has been a leader in the efforts to address this barrier by implementing routine, standardized measurement of functional status in primary care clinics. Initially, the VA encouraged, but did not require, measurement of functional status among older adults, but the implementation barriers and facilitators were not formally assessed.15 In a postimplementation evaluation, the authors found that a relatively small number of medical centers implemented functional measures. Moreover, the level of implementation seemed to vary across sites. Some sites were collecting complete measures on all eligible older patients, while other sites were collecting measures less consistently.15
As part of a national VA initiative to learn how best to implement standardized functional status measurement, the authors are conducting a qualitative study, including a formal assessment of barriers and facilitators to implementing functional assessments in VA primary care clinics. In the current project, which serves as formative work for this larger ongoing study, the authors identified and described current processes for measuring functional status in VA primary care patient aligned care team (PACT) and Geriatric (GeriPACT) clinics.
Methods
A rapid qualitative analysis approach was used, which included semistructured interviews with primary care stakeholders and rapid data analysis to summarize each clinic’s approach to measuring functional status and develop process maps for each clinic (eFigures 1, 2, 3, and 4 ). Interviews and analyses were conducted by a team consisting of a geriatrician clinician-researcher, a medical anthropologist, and a research coordinator. The institutional review boards of the San Francisco VAMC and the University of California, San Francisco approved the study.
Sampling Strategy
In order to identify VAMCs with varying approaches to assessing functional status in older patients who attended primary care appointments, the study used a criterion sampling approach.16,17 First, national “health factors” data were extracted from the VA Corporate Data Warehouse (CDW). Health factors are patient data collected through screening tools called clinical reminders, which prompt clinic staff and providers to enter data into checkbox-formatted templates. The study then identified medical centers that collected health factors data from patients aged ≥ 65 years (157 of 165 medical centers). A keyword search identified health factors related to the Katz ADL (bathing, dressing, transferring, toileting, and eating), and Lawton Instrumental ADL (IADL) Scale (using the telephone, shopping, preparing food, housekeeping, doing laundry, using transportation, managing medications, and managing finances).18,19 Health factors that were not collected during a primary care appointment were excluded.
Of the original 157 medical centers, 139 met these initial inclusion criteria. Among these 139 medical centers, 66 centers did not collect complete data on these 5 ADLs and 8 IADLs (eg, only ADLs or only IADLs, or only certain ADLs or IADLs).
Two medical centers were selected in each of the following 3 categories: (1) routinely used clinical reminders to collect standardized data on the Katz ADL and the Lawton IADL Scale; (2) routinely used clinical reminders to collect functional status data but collected partial information; and (3) did not use a clinical reminder to collect functional status data. To ensure that these 6 medical centers were geographically representative, the sample included at least 1 site from each of the 5 VA regions: 1 North Atlantic, 1 Southeast, 1 Midwest, 2 Continental (1 from the northern Continental region and 1 from the southern), and 1 Pacific. Three sites that included GeriPACTs also were sampled.
Primary care PACT and GeriPACT members from these 6 medical centers were recruited to participate. These PACT members included individuals who can assess function or use functional status information to inform patient care, including front-line nursing staff (licensed practical nurses [LPNs], and registered nurses [RNs]), primary care providers (medical doctors [MDs] and nurse practitioners [NPs]), and social workers (SWs).
Local bargaining units, nurse managers, and clinic directors provided lists of all clinic staff. All members of each group then received recruitment e-mails. Phone interviews were scheduled with interested participants. In several cases, a snowball sampling approach was used to increase enrollment numbers by asking interview participants to recommend colleagues who might be interested in participating.17
Data Collection
Telephone interviews were conducted between March 2016 and October 2016 using semistructured guides developed from the project aims and from related literature in implementation science.20,21 Interview domains included clinic structure, team member roles and responsibilities, current practices for collecting functional status data, and opinions on barriers and facilitators to assessing and recording functional status (Appendix:
Data Analysis
Rapid analysis, a team-based qualitative approach was used to engage efficiently and systematically with the data.22,23 This approach allowed results to be analyzed more quickly than in traditional qualitative analysis in order to inform intervention design and develop implementation strategies.23 Rapid analysis typically includes organization of interview data into summary templates, followed by a matrix analysis, which was used to create process maps.24
Summary Templates
Summary templates were developed from the interview guides by shortening each question into a representative code. The project team then read the transcripts and summarized key points in the appropriate section of the template. This process, known as data reduction, is used to organize and highlight material so conclusions can be drawn from the data easily.22 In order to maintain rigor and trustworthiness, one team member conducted the interview, and a different team member created the interview summary. All team members reviewed each summary and met regularly to discuss results.
The summary templates were converted into matrix analyses, a method of displaying data to identify relationships, including commonalities and differences.24 The matrixes were organized by stakeholder group and clinic in order to compare functional status assessment and documentation workflows across clinics.
Process Maps
Finally, the team used the matrix data to create process maps for each clinic of when, where, and by whom functional status information was assessed and documented. These maps were created using Microsoft Visio (Redmond, WA). The maps integrated perspectives from all participants to give an overview of the process for collecting functional status data in each clinic setting. To ensure accuracy, participants at each site received process maps to solicit feedback and validation.
Results
Forty-six participants at 6 medical centers (20 MDs and NPs, 19 RNs and LPNs, and 7 SWs) from 9 primary care clinics provided samples and interviews. The study team identified 3 general approaches to functional status assessment: (1) Routine collection of functional status data via a standardized clinical reminder; (2) Routine collection of functional status data via methods other than a clinical reminder (eg, a previsit telephone screen or electronic note template); and (3) Ad hoc approaches to measuring functional status (ie, no standard or routine approach to assessing or documenting functional status). The study team selected 4 clinics (2 PACTs and 2 GeriPACTs) clinics to serve as examples of the 3 identified approaches.
The processes for functional status assessment in each of 4 clinics are summarized in the following detailed descriptions (Table).
Clinic 1
Clinic 1 is a GeriPACT clinic that routinely assesses and documents functional status for all patients (efigure 1, available at feprac.com). The clinic’s current process includes 4 elements: (1) a patient questionnaire; (2) an annual clinical reminder administered by an RN; (3) a primary care provider (PCP) assessment; and (4) a postvisit SW assessment if referred by the PCP.
All newly referred patients are mailed a paper questionnaire that includes questions about their medical history and functional status. The patient is asked to bring the completed questionnaire to the first appointment. The clinic RN completes this form for returning patients at every visit during patient intake.
Second, the clinic uses an annual functional status clinical reminder for patients aged ≥ 75 years. The reminder includes questions about a patient’s ability to perform ADLs and IADLs with 3 to 4 response options for each question. If the clinical reminder is due at the time of a patient appointment, the RN fills out the reminder using information from the paper questionnaire. The RN also records this functional status in the nursing intake note. The RN may elect to designate the PCP as a cosigner for the nursing intake note especially if there are concerns about or changes in the patient’s functional status.
Third, the RN brings the paper form to the PCP, who often uses the questionnaire to guide the patient history. The PCP then uses the questionnaire and patient history to complete a functional status template within their visit note. The PCP also may use this information to inform patient care (eg, to make referrals to physical or occupational therapy).
Finally, the PCP might refer the patient to SW. The SW may be able to see the patient immediately after the PCP appointment, but if not, the SW follows up with a phone call to complete further functional status assessment and eligibility forms.
In addition to the above assessments by individual team members, the PACT has an interdisciplinary team huddle at the end of each clinic to discuss any issues or concerns about specific patients. The huddles often focus on issues related to functional status.
Clinic 2
Clinic 2 is a primary care PACT clinic that routinely assesses and documents functional status (eFigure 2, available at fedprac.com). The clinic process includes 3 steps: an annual clinical reminder for patients aged ≥ 75 years; a PCP assessment; and a postvisit SW assessment if referred by the PCP.
First, patients see an LPN for the intake process. During intake, the LPN records vitals and completes relevant clinical reminders. Similar to Clinic 1, Clinic 2 requires an annual functional status clinical reminder that includes ADLs and IADLs for patients aged ≥ 75 years. Patient information from the intake and clinical reminders are recorded by the LPN in a preventative medicine note in the electronic health record. This note is printed and handed to the PCP.
The PCP may review the preventative medicine note prior to completing the patient history and physical, including the functional status clinical reminder when applicable. If the PCP follows up on any functional issues identified by the LPN or completes further assessment of patient function, he or she may use this information to refer the patient to services or to place a SW consult; the PCP’s functional assessment is documented in a free-form visit note.
When the SW receives a consult, a chart review for social history, demographic information, and previous functional status assessments is conducted. The SW then calls the patient to administer functional and cognitive assessments over the phone and refers the patient to appropriate services based on eligibility.
Clinic 3
Clinic 3 is a GeriPACT clinic where functional status information is routinely collected for all new patients but may or may not be collected for returning patients (eFigure 3, available at fedprac.com). The process for new patients includes a previsit SW assessment; an informal LPN screening (ie, not based on a standardized clinical reminder); a PCP assessment; and a postvisit SW assessment if referred by the provider. The process for returning patients is similar but omits the previsit social work assessment. New patients complete a comprehensive questionnaire with a SW before their first clinic visit. The questionnaire is completed by phone and involves an extensive social and medical history, including an assessment of ADLs and IADLs. This assessment is recorded in a free-form social work note.
Next, both new and returning patients see an LPN who completes the intake process, including vitals and clinical reminders. Clinic 3 does not have a clinical reminder for functional status. However, the LPN could elect to ask about ADLs or IADLs if the patient brings up a functional issue related to the chief symptom or if the LPN observes something that indicates possible functional impairment, such as difficulty walking or a disheveled appearance. If discussed, this information is recorded in the LPN intake note, and the LPN also could verbally inform the PCP of the patient’s functional status. The RN is not formally involved in intake or functional status assessment in this clinic.
Finally, the patient sees the PCP, who may or may not have reviewed the LPN note. The PCP may assess functional status at his or her discretion, but there was no required assessment. The PCP could complete an optional functional status assessment template included in the PCP visit note. The PCP can refer the patient to services or to SW for further evaluation.
Clinic 4
Clinic 4 is a primary care PACT clinic that does not routinely measure functional status (eFigure 4, available at fedprac.com). The approach includes an informal LPN screening (ie, not based on a standardized clinical reminder); a PCP assessment; and a postvisit social worker assessment if referred by the provider. These steps are very similar to those of clinic 3, but they do not include a previsit SW assessment for new patients.
Although not represented within the 4 clinics described in this article, the content of functional status clinical reminders differed across the 9 clinics in the larger sample. Clinical reminders differed across several domains, including the type of question stems (scripted questions for each ADL vs categories for each activity); response options (eg, dichotomous vs ≥ 3 options), and the presence of free-text boxes to allow staff to enter any additional notes.
Discussion
Approaches to assessing and documenting functional status varied widely. Whereas some clinics primarily used informal approaches to assessing and documenting functional status (ie, neither routine nor standardized), others used a routine, standardized clinical reminder, and some combined several standardized approaches to measuring function. The study team identified variability across several domains of the functional status assessment process, including documentation, workflow, and clinical reminder content.
Approaches to functional assessment differed between GeriPACT and PACT clinics. Consistent with the central role that functional status assessment plays in geriatrics practice, GeriPACTs tended to employ a routine, multidisciplinary approach to measuring functional status. This approach included standardized functional assessments by multiple primary care team members, including LPNs, SWs, and PCPs. In contrast, when PACTs completed standardized functional status assessment, it was generally carried out by a single team member (typically an LPN). The PCPs in PACTs used a nonroutine approach to assess functional status in which they performed detailed functional assessments for certain high-risk patients and referred a subset for further SW evaluation.
These processes are consistent with research showing that standardized functional status data are seldom collected routinely in nongeriatric primary care settings.11 Reports by PCPs that they did not always assess functional status also are consistent with previous research demonstrating that clinicians are not always aware of their patients’ functional ability.10
In addition to highlighting differences between GeriPACT and PACTs, the identified processes illustrate the variability in documentation, clinic workflow, and clinical reminder content across all clinics. Approaches to documentation included checkbox-formatted clinical reminders with and without associated nursing notes, patient questionnaires, and templated PCP and SW notes. Clinics employed varying approaches to collect functional status information and to ensure that those data were shared with the team. Clinic staff assessed functional status at different times during the clinical encounter. Clinics used several approaches to share this information with team members, including warm handoffs from LPNs to PCPs, interdisciplinary team huddles, and electronic signoffs. Finally, clinical reminder content varied between clinics, with differences in the wording of ADL and IADL questions as well as in the number and type of response options.
This variability highlights the challenges inherent in developing a routine, standardized approach to measuring functional status that can be adapted across primary care settings. Such an approach must be both flexible enough to accommodate variation in workflow and structured enough to capture accurate data that can be used to guide clinical decisions. Capturing accurate, standardized data in CDW also will inform efforts to improve population health by allowing VHA leaders to understand the scope of disability among older veterans and plan for service needs and interventions.
Whereas the larger qualitative study will identify the specific barriers and facilitators to developing and implementing such an approach, current clinic processes present here offer hints as to which features may be important. For example, several clinics collected functional status information before the visit by telephone or questionnaire. Therefore, it will be important to choose a functional status assessment instrument that is validated for both telephone and in-person use. Similarly, some clinics had structured clinical reminders with categoric response options, whereas others included free-text boxes. Incorporating both categoric responses (to ensure accurate data) as well as free-text (to allow for additional notes about a patient’s specific circumstances that may influence service needs) may be one approach.
Limitations
This study’s approach to identifying clinic processes had several limitations. First, the authors did not send process maps to clinic directors for verification. However, speaking with PACT members who carry out clinic processes is likely the most accurate way to identify practice. Second, the results may not be generalizable to all VA primary care settings. Due to resource limitations and project scope, community-based outpatient clinics (CBOCs) were not included. Compared with clinics based in medical centers, CBOCs may have different staffing levels, practice models, and needs regarding implementation of functional status assessment.
Although 46 participants from 9 clinics were interviewed, there are likely additional approaches to measuring functional status that are not represented within this sample. In addition, 3 of the 4 clinics included are affiliated with academic institutions, and all 4 are located in large cities. Efforts to include rural VAMCs were not successful. Finally, clinic-level characteristics were not reported, which may impact clinic processes. Although study participants were asked about clinic characteristics, they were often unsure or only able to provide rough estimates. In the ongoing qualitative study, the authors will attempt to collect more reliable data about these clinic-level characteristics and to examine the potential role these characteristics may play as barriers or facilitators to implementing routine assessment of functional status in primary care settings.
Conclusion
VA primary care clinics had widely varying approaches for assessing and documenting functional status. This work along with a larger ongoing qualitative study that includes interviews with veterans will directly inform the design and implementation of a standardized, patient-centered approach to functional assessment that can be adapted across varied primary care settings. Implementing standardized functional status measurement will allow the VA to serve veterans better by using functional status information to refer patients to appropriate services and to deliver patient-centered care with the potential to improve patient function and quality of life.
The ability to perform activities of daily living (ADLs), commonly called functional status, is central to older adults’ quality of life (QOL) and independence.1,2 Understanding functional status is key to improving outcomes for older adults. In community-dwelling older adults with difficulty performing basic ADLs, practical interventions, including physical and occupational therapy, can improve functioning and prevent functional decline.3,4 Understanding function also is important for delivering patient-centered care, including individualizing cancer screening,5 evaluating how patients will tolerate interventions,6-9 and helping patients and families determine the need for long-term services and supports.
For these reasons, assessing functional status is a cornerstone of geriatrics practice. However, most older adults are cared for in primary care settings where routine measurement of functional status is uncommon.10,11 Although policy leaders have long noted this gap and the obstacle it poses to improving the quality and outcomes of care for older adults, many health care systems have been slow to incorporate measurement of functional status into routine patient care.12-14
Over the past several years, the VA has been a leader in the efforts to address this barrier by implementing routine, standardized measurement of functional status in primary care clinics. Initially, the VA encouraged, but did not require, measurement of functional status among older adults, but the implementation barriers and facilitators were not formally assessed.15 In a postimplementation evaluation, the authors found that a relatively small number of medical centers implemented functional measures. Moreover, the level of implementation seemed to vary across sites. Some sites were collecting complete measures on all eligible older patients, while other sites were collecting measures less consistently.15
As part of a national VA initiative to learn how best to implement standardized functional status measurement, the authors are conducting a qualitative study, including a formal assessment of barriers and facilitators to implementing functional assessments in VA primary care clinics. In the current project, which serves as formative work for this larger ongoing study, the authors identified and described current processes for measuring functional status in VA primary care patient aligned care team (PACT) and Geriatric (GeriPACT) clinics.
Methods
A rapid qualitative analysis approach was used, which included semistructured interviews with primary care stakeholders and rapid data analysis to summarize each clinic’s approach to measuring functional status and develop process maps for each clinic (eFigures 1, 2, 3, and 4 ). Interviews and analyses were conducted by a team consisting of a geriatrician clinician-researcher, a medical anthropologist, and a research coordinator. The institutional review boards of the San Francisco VAMC and the University of California, San Francisco approved the study.
Sampling Strategy
In order to identify VAMCs with varying approaches to assessing functional status in older patients who attended primary care appointments, the study used a criterion sampling approach.16,17 First, national “health factors” data were extracted from the VA Corporate Data Warehouse (CDW). Health factors are patient data collected through screening tools called clinical reminders, which prompt clinic staff and providers to enter data into checkbox-formatted templates. The study then identified medical centers that collected health factors data from patients aged ≥ 65 years (157 of 165 medical centers). A keyword search identified health factors related to the Katz ADL (bathing, dressing, transferring, toileting, and eating), and Lawton Instrumental ADL (IADL) Scale (using the telephone, shopping, preparing food, housekeeping, doing laundry, using transportation, managing medications, and managing finances).18,19 Health factors that were not collected during a primary care appointment were excluded.
Of the original 157 medical centers, 139 met these initial inclusion criteria. Among these 139 medical centers, 66 centers did not collect complete data on these 5 ADLs and 8 IADLs (eg, only ADLs or only IADLs, or only certain ADLs or IADLs).
Two medical centers were selected in each of the following 3 categories: (1) routinely used clinical reminders to collect standardized data on the Katz ADL and the Lawton IADL Scale; (2) routinely used clinical reminders to collect functional status data but collected partial information; and (3) did not use a clinical reminder to collect functional status data. To ensure that these 6 medical centers were geographically representative, the sample included at least 1 site from each of the 5 VA regions: 1 North Atlantic, 1 Southeast, 1 Midwest, 2 Continental (1 from the northern Continental region and 1 from the southern), and 1 Pacific. Three sites that included GeriPACTs also were sampled.
Primary care PACT and GeriPACT members from these 6 medical centers were recruited to participate. These PACT members included individuals who can assess function or use functional status information to inform patient care, including front-line nursing staff (licensed practical nurses [LPNs], and registered nurses [RNs]), primary care providers (medical doctors [MDs] and nurse practitioners [NPs]), and social workers (SWs).
Local bargaining units, nurse managers, and clinic directors provided lists of all clinic staff. All members of each group then received recruitment e-mails. Phone interviews were scheduled with interested participants. In several cases, a snowball sampling approach was used to increase enrollment numbers by asking interview participants to recommend colleagues who might be interested in participating.17
Data Collection
Telephone interviews were conducted between March 2016 and October 2016 using semistructured guides developed from the project aims and from related literature in implementation science.20,21 Interview domains included clinic structure, team member roles and responsibilities, current practices for collecting functional status data, and opinions on barriers and facilitators to assessing and recording functional status (Appendix:
Data Analysis
Rapid analysis, a team-based qualitative approach was used to engage efficiently and systematically with the data.22,23 This approach allowed results to be analyzed more quickly than in traditional qualitative analysis in order to inform intervention design and develop implementation strategies.23 Rapid analysis typically includes organization of interview data into summary templates, followed by a matrix analysis, which was used to create process maps.24
Summary Templates
Summary templates were developed from the interview guides by shortening each question into a representative code. The project team then read the transcripts and summarized key points in the appropriate section of the template. This process, known as data reduction, is used to organize and highlight material so conclusions can be drawn from the data easily.22 In order to maintain rigor and trustworthiness, one team member conducted the interview, and a different team member created the interview summary. All team members reviewed each summary and met regularly to discuss results.
The summary templates were converted into matrix analyses, a method of displaying data to identify relationships, including commonalities and differences.24 The matrixes were organized by stakeholder group and clinic in order to compare functional status assessment and documentation workflows across clinics.
Process Maps
Finally, the team used the matrix data to create process maps for each clinic of when, where, and by whom functional status information was assessed and documented. These maps were created using Microsoft Visio (Redmond, WA). The maps integrated perspectives from all participants to give an overview of the process for collecting functional status data in each clinic setting. To ensure accuracy, participants at each site received process maps to solicit feedback and validation.
Results
Forty-six participants at 6 medical centers (20 MDs and NPs, 19 RNs and LPNs, and 7 SWs) from 9 primary care clinics provided samples and interviews. The study team identified 3 general approaches to functional status assessment: (1) Routine collection of functional status data via a standardized clinical reminder; (2) Routine collection of functional status data via methods other than a clinical reminder (eg, a previsit telephone screen or electronic note template); and (3) Ad hoc approaches to measuring functional status (ie, no standard or routine approach to assessing or documenting functional status). The study team selected 4 clinics (2 PACTs and 2 GeriPACTs) clinics to serve as examples of the 3 identified approaches.
The processes for functional status assessment in each of 4 clinics are summarized in the following detailed descriptions (Table).
Clinic 1
Clinic 1 is a GeriPACT clinic that routinely assesses and documents functional status for all patients (efigure 1, available at feprac.com). The clinic’s current process includes 4 elements: (1) a patient questionnaire; (2) an annual clinical reminder administered by an RN; (3) a primary care provider (PCP) assessment; and (4) a postvisit SW assessment if referred by the PCP.
All newly referred patients are mailed a paper questionnaire that includes questions about their medical history and functional status. The patient is asked to bring the completed questionnaire to the first appointment. The clinic RN completes this form for returning patients at every visit during patient intake.
Second, the clinic uses an annual functional status clinical reminder for patients aged ≥ 75 years. The reminder includes questions about a patient’s ability to perform ADLs and IADLs with 3 to 4 response options for each question. If the clinical reminder is due at the time of a patient appointment, the RN fills out the reminder using information from the paper questionnaire. The RN also records this functional status in the nursing intake note. The RN may elect to designate the PCP as a cosigner for the nursing intake note especially if there are concerns about or changes in the patient’s functional status.
Third, the RN brings the paper form to the PCP, who often uses the questionnaire to guide the patient history. The PCP then uses the questionnaire and patient history to complete a functional status template within their visit note. The PCP also may use this information to inform patient care (eg, to make referrals to physical or occupational therapy).
Finally, the PCP might refer the patient to SW. The SW may be able to see the patient immediately after the PCP appointment, but if not, the SW follows up with a phone call to complete further functional status assessment and eligibility forms.
In addition to the above assessments by individual team members, the PACT has an interdisciplinary team huddle at the end of each clinic to discuss any issues or concerns about specific patients. The huddles often focus on issues related to functional status.
Clinic 2
Clinic 2 is a primary care PACT clinic that routinely assesses and documents functional status (eFigure 2, available at fedprac.com). The clinic process includes 3 steps: an annual clinical reminder for patients aged ≥ 75 years; a PCP assessment; and a postvisit SW assessment if referred by the PCP.
First, patients see an LPN for the intake process. During intake, the LPN records vitals and completes relevant clinical reminders. Similar to Clinic 1, Clinic 2 requires an annual functional status clinical reminder that includes ADLs and IADLs for patients aged ≥ 75 years. Patient information from the intake and clinical reminders are recorded by the LPN in a preventative medicine note in the electronic health record. This note is printed and handed to the PCP.
The PCP may review the preventative medicine note prior to completing the patient history and physical, including the functional status clinical reminder when applicable. If the PCP follows up on any functional issues identified by the LPN or completes further assessment of patient function, he or she may use this information to refer the patient to services or to place a SW consult; the PCP’s functional assessment is documented in a free-form visit note.
When the SW receives a consult, a chart review for social history, demographic information, and previous functional status assessments is conducted. The SW then calls the patient to administer functional and cognitive assessments over the phone and refers the patient to appropriate services based on eligibility.
Clinic 3
Clinic 3 is a GeriPACT clinic where functional status information is routinely collected for all new patients but may or may not be collected for returning patients (eFigure 3, available at fedprac.com). The process for new patients includes a previsit SW assessment; an informal LPN screening (ie, not based on a standardized clinical reminder); a PCP assessment; and a postvisit SW assessment if referred by the provider. The process for returning patients is similar but omits the previsit social work assessment. New patients complete a comprehensive questionnaire with a SW before their first clinic visit. The questionnaire is completed by phone and involves an extensive social and medical history, including an assessment of ADLs and IADLs. This assessment is recorded in a free-form social work note.
Next, both new and returning patients see an LPN who completes the intake process, including vitals and clinical reminders. Clinic 3 does not have a clinical reminder for functional status. However, the LPN could elect to ask about ADLs or IADLs if the patient brings up a functional issue related to the chief symptom or if the LPN observes something that indicates possible functional impairment, such as difficulty walking or a disheveled appearance. If discussed, this information is recorded in the LPN intake note, and the LPN also could verbally inform the PCP of the patient’s functional status. The RN is not formally involved in intake or functional status assessment in this clinic.
Finally, the patient sees the PCP, who may or may not have reviewed the LPN note. The PCP may assess functional status at his or her discretion, but there was no required assessment. The PCP could complete an optional functional status assessment template included in the PCP visit note. The PCP can refer the patient to services or to SW for further evaluation.
Clinic 4
Clinic 4 is a primary care PACT clinic that does not routinely measure functional status (eFigure 4, available at fedprac.com). The approach includes an informal LPN screening (ie, not based on a standardized clinical reminder); a PCP assessment; and a postvisit social worker assessment if referred by the provider. These steps are very similar to those of clinic 3, but they do not include a previsit SW assessment for new patients.
Although not represented within the 4 clinics described in this article, the content of functional status clinical reminders differed across the 9 clinics in the larger sample. Clinical reminders differed across several domains, including the type of question stems (scripted questions for each ADL vs categories for each activity); response options (eg, dichotomous vs ≥ 3 options), and the presence of free-text boxes to allow staff to enter any additional notes.
Discussion
Approaches to assessing and documenting functional status varied widely. Whereas some clinics primarily used informal approaches to assessing and documenting functional status (ie, neither routine nor standardized), others used a routine, standardized clinical reminder, and some combined several standardized approaches to measuring function. The study team identified variability across several domains of the functional status assessment process, including documentation, workflow, and clinical reminder content.
Approaches to functional assessment differed between GeriPACT and PACT clinics. Consistent with the central role that functional status assessment plays in geriatrics practice, GeriPACTs tended to employ a routine, multidisciplinary approach to measuring functional status. This approach included standardized functional assessments by multiple primary care team members, including LPNs, SWs, and PCPs. In contrast, when PACTs completed standardized functional status assessment, it was generally carried out by a single team member (typically an LPN). The PCPs in PACTs used a nonroutine approach to assess functional status in which they performed detailed functional assessments for certain high-risk patients and referred a subset for further SW evaluation.
These processes are consistent with research showing that standardized functional status data are seldom collected routinely in nongeriatric primary care settings.11 Reports by PCPs that they did not always assess functional status also are consistent with previous research demonstrating that clinicians are not always aware of their patients’ functional ability.10
In addition to highlighting differences between GeriPACT and PACTs, the identified processes illustrate the variability in documentation, clinic workflow, and clinical reminder content across all clinics. Approaches to documentation included checkbox-formatted clinical reminders with and without associated nursing notes, patient questionnaires, and templated PCP and SW notes. Clinics employed varying approaches to collect functional status information and to ensure that those data were shared with the team. Clinic staff assessed functional status at different times during the clinical encounter. Clinics used several approaches to share this information with team members, including warm handoffs from LPNs to PCPs, interdisciplinary team huddles, and electronic signoffs. Finally, clinical reminder content varied between clinics, with differences in the wording of ADL and IADL questions as well as in the number and type of response options.
This variability highlights the challenges inherent in developing a routine, standardized approach to measuring functional status that can be adapted across primary care settings. Such an approach must be both flexible enough to accommodate variation in workflow and structured enough to capture accurate data that can be used to guide clinical decisions. Capturing accurate, standardized data in CDW also will inform efforts to improve population health by allowing VHA leaders to understand the scope of disability among older veterans and plan for service needs and interventions.
Whereas the larger qualitative study will identify the specific barriers and facilitators to developing and implementing such an approach, current clinic processes present here offer hints as to which features may be important. For example, several clinics collected functional status information before the visit by telephone or questionnaire. Therefore, it will be important to choose a functional status assessment instrument that is validated for both telephone and in-person use. Similarly, some clinics had structured clinical reminders with categoric response options, whereas others included free-text boxes. Incorporating both categoric responses (to ensure accurate data) as well as free-text (to allow for additional notes about a patient’s specific circumstances that may influence service needs) may be one approach.
Limitations
This study’s approach to identifying clinic processes had several limitations. First, the authors did not send process maps to clinic directors for verification. However, speaking with PACT members who carry out clinic processes is likely the most accurate way to identify practice. Second, the results may not be generalizable to all VA primary care settings. Due to resource limitations and project scope, community-based outpatient clinics (CBOCs) were not included. Compared with clinics based in medical centers, CBOCs may have different staffing levels, practice models, and needs regarding implementation of functional status assessment.
Although 46 participants from 9 clinics were interviewed, there are likely additional approaches to measuring functional status that are not represented within this sample. In addition, 3 of the 4 clinics included are affiliated with academic institutions, and all 4 are located in large cities. Efforts to include rural VAMCs were not successful. Finally, clinic-level characteristics were not reported, which may impact clinic processes. Although study participants were asked about clinic characteristics, they were often unsure or only able to provide rough estimates. In the ongoing qualitative study, the authors will attempt to collect more reliable data about these clinic-level characteristics and to examine the potential role these characteristics may play as barriers or facilitators to implementing routine assessment of functional status in primary care settings.
Conclusion
VA primary care clinics had widely varying approaches for assessing and documenting functional status. This work along with a larger ongoing qualitative study that includes interviews with veterans will directly inform the design and implementation of a standardized, patient-centered approach to functional assessment that can be adapted across varied primary care settings. Implementing standardized functional status measurement will allow the VA to serve veterans better by using functional status information to refer patients to appropriate services and to deliver patient-centered care with the potential to improve patient function and quality of life.
1.Covinsky KE, Wu AW, Landefeld CS, et al. Health status versus quality of life in older patients: does the distinction matter? Am J Med. 1999;106(4):435-440.
2. Fried TR, McGraw S, Agostini JV, Tinetti ME. Views of older persons with multiple morbidities on competing outcomes and clinical decision-making. J Am Geriatr Soc. 2008;56(10):1839-1844.
3. Beswick AD, Rees K, Dieppe P, et al. Complex interventions to improve physical function and maintain independent living in elderly people: a systematic review and meta-analysis. Lancet. 2008;371(9614):725-735.
4. Szanton SL, Leff B, Wolff JL, Roberts L, Gitlin LN. Home-based care program reduces disability and promotes aging in place. Health Aff (Millwood). 2016;35(9):1558-1563.
5. Walter LC, Covinsky KE. Cancer screening in elderly patients: a framework for individualized decision making. JAMA. 2001;285(21):2750-2756.
6. Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med. 2009;361(16):1539-1547.
7. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29(25):3457-3465.
8. Crawford RS, Cambria RP, Abularrage CJ, et al. Preoperative functional status predicts perioperative outcomes after infrainguinal bypass surgery. J Vasc Surg. 2010;51(2):351-358; discussion 358-359.
9. Arnold SV, Reynolds MR, Lei Y, et al; PARTNER Investigators. Predictors of poor outcomes after transcatheter aortic valve replacement: results from the PARTNER (Placement of Aortic Transcatheter Valve) trial. Circulation. 2014;129(25):2682-2690.
10. Calkins DR, Rubenstein LV, Cleary PD, et al. Failure of physicians to recognize functional disability in ambulatory patients. Ann Intern Med. 1991;114(6):451-454.
11. Bogardus ST Jr, Towle V, Williams CS, Desai MM, Inouye SK. What does the medical record reveal about functional status? A comparison of medical record and interview data. J Gen Intern Med. 2001;16(11):728-736.
12. Bierman AS. Functional status: the six vital sign. J Gen Intern Med. 2001;16(11):785-786.
13. Iezzoni LI, Greenberg MS. Capturing and classifying functional status information in administrative databases. Health Care Financ Rev. 2003;24(3):61-76.
14. Clauser SB, Bierman AS. Significance of functional status data for payment and quality. Health Care Financ Rev. 2003;24(3):1-12.
15. Brown RT, Komaiko KD, Shi Y, et al. Bringing functional status into a big data world: validation of national Veterans Affairs functional status data. PloS One. 2017;12(6):e0178726.
16. Palinkas LA, Horwitz SM, Green CA, Wisdom JP, Duan N, Hoagwood K. Purposeful sampling for qualitative data collection and analysis in mixed method implementation research. Adm Policy Ment Health. 2015;42(5):533-544.
17. Patton MQ. Qualitative Research Evaluation and Methods. 4th ed. Thousand Oaks, CA: Sage; 2015.
18. Katz S, Ford AB, Moskowitz RW, Jackson BA, Jaffe MW. Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychosocial function. JAMA. 1963;185(12):914-919.
19. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3):179-186.
20. Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4(1):50.
21. Saleem JJ, Patterson ES, Militello L, Render ML, Orshansky G, Asch SM. Exploring barriers and facilitators to the use of computerized clinical reminders. J Am Med Inform Assoc. 2005;12(4):438-447.
22. Miles MB, Huberman AM, Saldana J. Qualitative Data Analysis: A Methods Sourcebook. 3rd ed. Thousand Oaks, CA: Sage; 2014.
23. Hamilton AB. Qualitative methods in rapid turn-around health services research. https://www.hsrd .research.va.gov/for_researchers/cyber_seminars /archives/video_archive.cfm?SessionID=780. Published December 11, 2013. Accessed August 9, 2017.
24. Averill JB. Matrix analysis as a complementary analytic strategy in qualitative inquiry. Qual Health Res. 2002;12(6):855-866.
1.Covinsky KE, Wu AW, Landefeld CS, et al. Health status versus quality of life in older patients: does the distinction matter? Am J Med. 1999;106(4):435-440.
2. Fried TR, McGraw S, Agostini JV, Tinetti ME. Views of older persons with multiple morbidities on competing outcomes and clinical decision-making. J Am Geriatr Soc. 2008;56(10):1839-1844.
3. Beswick AD, Rees K, Dieppe P, et al. Complex interventions to improve physical function and maintain independent living in elderly people: a systematic review and meta-analysis. Lancet. 2008;371(9614):725-735.
4. Szanton SL, Leff B, Wolff JL, Roberts L, Gitlin LN. Home-based care program reduces disability and promotes aging in place. Health Aff (Millwood). 2016;35(9):1558-1563.
5. Walter LC, Covinsky KE. Cancer screening in elderly patients: a framework for individualized decision making. JAMA. 2001;285(21):2750-2756.
6. Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med. 2009;361(16):1539-1547.
7. Hurria A, Togawa K, Mohile SG, et al. Predicting chemotherapy toxicity in older adults with cancer: a prospective multicenter study. J Clin Oncol. 2011;29(25):3457-3465.
8. Crawford RS, Cambria RP, Abularrage CJ, et al. Preoperative functional status predicts perioperative outcomes after infrainguinal bypass surgery. J Vasc Surg. 2010;51(2):351-358; discussion 358-359.
9. Arnold SV, Reynolds MR, Lei Y, et al; PARTNER Investigators. Predictors of poor outcomes after transcatheter aortic valve replacement: results from the PARTNER (Placement of Aortic Transcatheter Valve) trial. Circulation. 2014;129(25):2682-2690.
10. Calkins DR, Rubenstein LV, Cleary PD, et al. Failure of physicians to recognize functional disability in ambulatory patients. Ann Intern Med. 1991;114(6):451-454.
11. Bogardus ST Jr, Towle V, Williams CS, Desai MM, Inouye SK. What does the medical record reveal about functional status? A comparison of medical record and interview data. J Gen Intern Med. 2001;16(11):728-736.
12. Bierman AS. Functional status: the six vital sign. J Gen Intern Med. 2001;16(11):785-786.
13. Iezzoni LI, Greenberg MS. Capturing and classifying functional status information in administrative databases. Health Care Financ Rev. 2003;24(3):61-76.
14. Clauser SB, Bierman AS. Significance of functional status data for payment and quality. Health Care Financ Rev. 2003;24(3):1-12.
15. Brown RT, Komaiko KD, Shi Y, et al. Bringing functional status into a big data world: validation of national Veterans Affairs functional status data. PloS One. 2017;12(6):e0178726.
16. Palinkas LA, Horwitz SM, Green CA, Wisdom JP, Duan N, Hoagwood K. Purposeful sampling for qualitative data collection and analysis in mixed method implementation research. Adm Policy Ment Health. 2015;42(5):533-544.
17. Patton MQ. Qualitative Research Evaluation and Methods. 4th ed. Thousand Oaks, CA: Sage; 2015.
18. Katz S, Ford AB, Moskowitz RW, Jackson BA, Jaffe MW. Studies of illness in the aged. The index of ADL: a standardized measure of biological and psychosocial function. JAMA. 1963;185(12):914-919.
19. Lawton MP, Brody EM. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9(3):179-186.
20. Damschroder LJ, Aron DC, Keith RE, Kirsh SR, Alexander JA, Lowery JC. Fostering implementation of health services research findings into practice: a consolidated framework for advancing implementation science. Implement Sci. 2009;4(1):50.
21. Saleem JJ, Patterson ES, Militello L, Render ML, Orshansky G, Asch SM. Exploring barriers and facilitators to the use of computerized clinical reminders. J Am Med Inform Assoc. 2005;12(4):438-447.
22. Miles MB, Huberman AM, Saldana J. Qualitative Data Analysis: A Methods Sourcebook. 3rd ed. Thousand Oaks, CA: Sage; 2014.
23. Hamilton AB. Qualitative methods in rapid turn-around health services research. https://www.hsrd .research.va.gov/for_researchers/cyber_seminars /archives/video_archive.cfm?SessionID=780. Published December 11, 2013. Accessed August 9, 2017.
24. Averill JB. Matrix analysis as a complementary analytic strategy in qualitative inquiry. Qual Health Res. 2002;12(6):855-866.
Woman dies following cervical cone biopsy: $4.25M award
Woman dies following cervical cone biopsy: $4.25M award
A 46-year-old woman underwent a cervical cone biopsy at a Veterans Administration (VA) hospital on July 18. Following the test, significant bleeding occurred. The gynecologic surgeon attempted to control the hemorrhage by injecting fe
ESTATE'S CLAIM:
The surgeon’s actions were negligent. She removed too much tissue during the biopsy, injured the vaginal and uterine walls, and failed to timely diagnose and appropriately treat the injuries. The ferric subsulfate solution entered the abdominal cavity via the perforation, causing peritonitis and bowel injuries. A pathology report from the bowel resection surgery informed the surgeon that the bowel was not properly reconnected after the damaged portion was removed, but this condition was neither detected intraoperatively nor treated postoperatively.
DEFENDANTS' DEFENSE:
The surgeon moved for summary judgment, countering that, as a federal employee, she was exempt from personal liability for the services performed as an employee of the VA. That motion was denied. She then argued that injury to the vaginal/uterine wall is a known complication of the biopsy procedure.
VERDICT:
A $4.25 million Illinois verdict was returned in federal court.
Related article:
Reducing maternal mortality in the United States—Let’s get organized!
Needle stick not reported to patient
A woman delivered a baby assisted by an on-call ObGyn. When the baby developed fetal tachycardia, the ObGyn recommended expediting delivery and discussed various options and the risks of each option. The mother chose a vaginal forceps delivery. During the procedure, the mother experienced a 3rd-degree perineal laceration and a few minor lacerations, which were repaired. The mother was in pain, so the ObGyn performed a revision repair. During the procedure, the ObGyn accidentally stuck himself with a clean needle. He replaced the needle and changed his glove. The mother reported instant pain relief following revision and was discharged. After the needle incident, the ObGyn’s thumb became red and swollen, so he took antibiotics.
Two days after discharge, the patient reported to the ObGyn’s office with fever, pain, and a foul odor emanating from the surgery site. She was given the diagnosis of pelvic incisional cellulitis and was taken to the operating room for exploration and debridement. The patient developed septic shock and necrotizing fasciitis. She was placed on a ventilator and underwent 13 surgeries.
PATIENTS' CLAIM:
The ObGyn was negligent. The patient claimed breach of duty: the ObGyn did not disclose that his thumb was swollen and that he took antibiotics.
PHYSICIANS' DEFENSE:
There was no breach of duty. He did not feel the need to concern the patient about an injury to himself that did not affect her.
VERDICT:
A Kansas defense verdict was returned.
Related article:
2017 Update on infectious disease
Catheter removal, air embolism: $3.5M settlement
A 44-year-old woman underwent gynecologic surgery on April 22. She developed a rectovaginal fistula and other complications. Intravenous antibiotics were required and parenteral nutrition was delivered through a central venous catheter. On May 22, after a hospital nurse removed the catheter, an air embolism developed, causing a brain injury. The patient has a mental disability and residual leg tremors.
PATIENTS' CLAIM:
Because of the surgeon’s negligence during surgery, a fistula developed. The nurse negligently removed the catheter, causing the embolism.
DEFENDANTS' DEFENSE:
The case settled during the trial.
VERDICT:
A $3.5 million Illinois settlement was reached, including payments of $1 million from the surgeon and $2.5 million from the hospital.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Woman dies following cervical cone biopsy: $4.25M award
A 46-year-old woman underwent a cervical cone biopsy at a Veterans Administration (VA) hospital on July 18. Following the test, significant bleeding occurred. The gynecologic surgeon attempted to control the hemorrhage by injecting fe
ESTATE'S CLAIM:
The surgeon’s actions were negligent. She removed too much tissue during the biopsy, injured the vaginal and uterine walls, and failed to timely diagnose and appropriately treat the injuries. The ferric subsulfate solution entered the abdominal cavity via the perforation, causing peritonitis and bowel injuries. A pathology report from the bowel resection surgery informed the surgeon that the bowel was not properly reconnected after the damaged portion was removed, but this condition was neither detected intraoperatively nor treated postoperatively.
DEFENDANTS' DEFENSE:
The surgeon moved for summary judgment, countering that, as a federal employee, she was exempt from personal liability for the services performed as an employee of the VA. That motion was denied. She then argued that injury to the vaginal/uterine wall is a known complication of the biopsy procedure.
VERDICT:
A $4.25 million Illinois verdict was returned in federal court.
Related article:
Reducing maternal mortality in the United States—Let’s get organized!
Needle stick not reported to patient
A woman delivered a baby assisted by an on-call ObGyn. When the baby developed fetal tachycardia, the ObGyn recommended expediting delivery and discussed various options and the risks of each option. The mother chose a vaginal forceps delivery. During the procedure, the mother experienced a 3rd-degree perineal laceration and a few minor lacerations, which were repaired. The mother was in pain, so the ObGyn performed a revision repair. During the procedure, the ObGyn accidentally stuck himself with a clean needle. He replaced the needle and changed his glove. The mother reported instant pain relief following revision and was discharged. After the needle incident, the ObGyn’s thumb became red and swollen, so he took antibiotics.
Two days after discharge, the patient reported to the ObGyn’s office with fever, pain, and a foul odor emanating from the surgery site. She was given the diagnosis of pelvic incisional cellulitis and was taken to the operating room for exploration and debridement. The patient developed septic shock and necrotizing fasciitis. She was placed on a ventilator and underwent 13 surgeries.
PATIENTS' CLAIM:
The ObGyn was negligent. The patient claimed breach of duty: the ObGyn did not disclose that his thumb was swollen and that he took antibiotics.
PHYSICIANS' DEFENSE:
There was no breach of duty. He did not feel the need to concern the patient about an injury to himself that did not affect her.
VERDICT:
A Kansas defense verdict was returned.
Related article:
2017 Update on infectious disease
Catheter removal, air embolism: $3.5M settlement
A 44-year-old woman underwent gynecologic surgery on April 22. She developed a rectovaginal fistula and other complications. Intravenous antibiotics were required and parenteral nutrition was delivered through a central venous catheter. On May 22, after a hospital nurse removed the catheter, an air embolism developed, causing a brain injury. The patient has a mental disability and residual leg tremors.
PATIENTS' CLAIM:
Because of the surgeon’s negligence during surgery, a fistula developed. The nurse negligently removed the catheter, causing the embolism.
DEFENDANTS' DEFENSE:
The case settled during the trial.
VERDICT:
A $3.5 million Illinois settlement was reached, including payments of $1 million from the surgeon and $2.5 million from the hospital.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Woman dies following cervical cone biopsy: $4.25M award
A 46-year-old woman underwent a cervical cone biopsy at a Veterans Administration (VA) hospital on July 18. Following the test, significant bleeding occurred. The gynecologic surgeon attempted to control the hemorrhage by injecting fe
ESTATE'S CLAIM:
The surgeon’s actions were negligent. She removed too much tissue during the biopsy, injured the vaginal and uterine walls, and failed to timely diagnose and appropriately treat the injuries. The ferric subsulfate solution entered the abdominal cavity via the perforation, causing peritonitis and bowel injuries. A pathology report from the bowel resection surgery informed the surgeon that the bowel was not properly reconnected after the damaged portion was removed, but this condition was neither detected intraoperatively nor treated postoperatively.
DEFENDANTS' DEFENSE:
The surgeon moved for summary judgment, countering that, as a federal employee, she was exempt from personal liability for the services performed as an employee of the VA. That motion was denied. She then argued that injury to the vaginal/uterine wall is a known complication of the biopsy procedure.
VERDICT:
A $4.25 million Illinois verdict was returned in federal court.
Related article:
Reducing maternal mortality in the United States—Let’s get organized!
Needle stick not reported to patient
A woman delivered a baby assisted by an on-call ObGyn. When the baby developed fetal tachycardia, the ObGyn recommended expediting delivery and discussed various options and the risks of each option. The mother chose a vaginal forceps delivery. During the procedure, the mother experienced a 3rd-degree perineal laceration and a few minor lacerations, which were repaired. The mother was in pain, so the ObGyn performed a revision repair. During the procedure, the ObGyn accidentally stuck himself with a clean needle. He replaced the needle and changed his glove. The mother reported instant pain relief following revision and was discharged. After the needle incident, the ObGyn’s thumb became red and swollen, so he took antibiotics.
Two days after discharge, the patient reported to the ObGyn’s office with fever, pain, and a foul odor emanating from the surgery site. She was given the diagnosis of pelvic incisional cellulitis and was taken to the operating room for exploration and debridement. The patient developed septic shock and necrotizing fasciitis. She was placed on a ventilator and underwent 13 surgeries.
PATIENTS' CLAIM:
The ObGyn was negligent. The patient claimed breach of duty: the ObGyn did not disclose that his thumb was swollen and that he took antibiotics.
PHYSICIANS' DEFENSE:
There was no breach of duty. He did not feel the need to concern the patient about an injury to himself that did not affect her.
VERDICT:
A Kansas defense verdict was returned.
Related article:
2017 Update on infectious disease
Catheter removal, air embolism: $3.5M settlement
A 44-year-old woman underwent gynecologic surgery on April 22. She developed a rectovaginal fistula and other complications. Intravenous antibiotics were required and parenteral nutrition was delivered through a central venous catheter. On May 22, after a hospital nurse removed the catheter, an air embolism developed, causing a brain injury. The patient has a mental disability and residual leg tremors.
PATIENTS' CLAIM:
Because of the surgeon’s negligence during surgery, a fistula developed. The nurse negligently removed the catheter, causing the embolism.
DEFENDANTS' DEFENSE:
The case settled during the trial.
VERDICT:
A $3.5 million Illinois settlement was reached, including payments of $1 million from the surgeon and $2.5 million from the hospital.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Salmonella infections: The source may be as close as your patient’s backyard
I recently received a group text from a friend voicing her frustration that her neighbor had acquired chickens, and she shared a photo of some roaming freely in the front yard. Naturally, my response was related to the potential infectious disease exposure and infections. Another friend chimed in “fresh eggs, and these are free range chickens. They don’t get sick. ... Many people in my area have chickens.” Unbeknownst to my friends, they had helped me select the ID Consult topic for this month.
Nontyphoidal Salmonella bacteria are associated with a wide spectrum of infections which range from asymptomatic gastrointestinal carriage to bacteremia, meningitis, osteomyelitis, and focal infections. Invasive disease is seen most often in children younger than 5 years of age, persons aged 65 years or older, and individuals with hemoglobinopathies including sickle cell disease and those with immunodeficiencies. Annually, the Centers for Disease Control and Prevention estimates that nontyphoidal salmonellosis is responsible for 1.2 million illnesses, 23,000 hospitalizations, and 450 deaths in the United States. Gastroenteritis is the most common manifestation of the disease and is characterized by abdominal cramps, diarrhea, and fever that develops 12-72 hours after exposure. It is usually self-limited. As previously reported in this column (June, 2017), Salmonella is one of the top two foodborne pathogens in the United States, and most outbreaks have been associated with consumption of contaminated food. But wait, contaminated food is not the only cause of some of our most recent outbreaks.
Live poultry-associated salmonellosis (LPAS)
LPAS was first reported in the 1950s. More recent epidemiologic data was published by C. Basler et al. (Emerging Infect Dis. 2016;22[10]:1705-11). LPAS was defined as two or more culture confirmed human Salmonella infections with a combination of epidemiologic, laboratory, or traceback evidence linking illnesses to contact with live poultry. The median outbreak size involved 26 cases (range, 4-363) and 77% (41 of 53) were multistate. The median age of the patients was 9 years (range, less than 1 to 92 years), and 31% were aged 5 years or younger. Exposure to chicks and ducklings was reported in 85% and 38%, respectively. High-risk practices included keeping poultry inside of the home (46%), snuggling baby birds (49%), and kissing baby birds (13%). The median time from purchase of poultry to onset of illness was 17 days (range, 1-672), and 66% reported onset of illness less than 30 days after purchase. Almost 52% reported owning poultry for less than 1 year.
The number of outbreaks continued to increase. From 1990 to 2005, there were a total of 17 outbreaks, compared with 36 between 2006 and 2014. Historically, outbreaks occurred in children around Easter when brightly colored dyed chicks were purchased. In the above review, 80% of outbreaks began in February, March, or April with an average duration of 4.9 months (range, 1-12).
Salmonella isolates

Backyard flocks and LPAS
More recently outbreaks have been associated with backyard flocks occurring year round and affecting both adults and children in contrast to seasonal peaks. The first multistate backyard flock outbreak was documented in 2007. Currently, the CDC is investigating 10 separate multistate outbreaks that began on Jan. 4, 2017. It involves 48 states, 961 infected individuals, 215 hospitalizations, and 1 death. At least 5 salmonella serotypes have been isolated.
What about the hatcheries?
It’s estimated that 50 million live poultry are sold annually. Birds are shipped within 24 hours after hatching via the U.S. Postal Service in boxes containing up to 100 chicks. Delivery occurs within 72 hours of hatching. Approximately 20 mail order hatcheries provide the majority of poultry sold to the general public. The National Poultry Improvement Plan (NPIP) is a voluntary state and federal testing and certification program whose goal is to eliminate poultry disease from breeder flocks to prevent egg-transmitted and hatchery-disseminated diseases. All hatcheries may participate. They also may participate in the voluntary Salmonella monitoring program. Note participation is not mandatory.
Preventing future outbreaks: patient/parental education is mandatory
1. Make sure your parents know about the association of Salmonella and live poultry. Reinforce these are farm animals, not pets. Purchase birds from hatcheries that participate in NPIP and the Salmonella monitoring programs.
2. Chicks, ducklings, or other live poultry should not be taken to schools, day care facilities, or nursing homes. Poultry should not be allowed in the home or in areas where food or drink is being prepared or consumed.
3. Poultry should not be snuggled, kissed, or allowed to touch one’s mouth. Hand washing with soap and water should occur after touching live poultry or any object touched in areas where they live or roam.
4. Contact with live poultry should be avoided in those at risk for developing serious infections including persons aged 5 years or younger, 65 years or older, immunocompromised individuals, and those with hemoglobinopathies.
5. All equipment used to care for live birds should be washed outdoors. Owners should have designated shoes when caring for poultry which should never be worn inside the home.
Hopefully, the next time you see a patient with fever and diarrhea you will recall this topic and ask about their contact with live poultry.
Additional resources to facilitate discussions can be found at www.cdc.gov/salmonella.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Email her at [email protected].
I recently received a group text from a friend voicing her frustration that her neighbor had acquired chickens, and she shared a photo of some roaming freely in the front yard. Naturally, my response was related to the potential infectious disease exposure and infections. Another friend chimed in “fresh eggs, and these are free range chickens. They don’t get sick. ... Many people in my area have chickens.” Unbeknownst to my friends, they had helped me select the ID Consult topic for this month.
Nontyphoidal Salmonella bacteria are associated with a wide spectrum of infections which range from asymptomatic gastrointestinal carriage to bacteremia, meningitis, osteomyelitis, and focal infections. Invasive disease is seen most often in children younger than 5 years of age, persons aged 65 years or older, and individuals with hemoglobinopathies including sickle cell disease and those with immunodeficiencies. Annually, the Centers for Disease Control and Prevention estimates that nontyphoidal salmonellosis is responsible for 1.2 million illnesses, 23,000 hospitalizations, and 450 deaths in the United States. Gastroenteritis is the most common manifestation of the disease and is characterized by abdominal cramps, diarrhea, and fever that develops 12-72 hours after exposure. It is usually self-limited. As previously reported in this column (June, 2017), Salmonella is one of the top two foodborne pathogens in the United States, and most outbreaks have been associated with consumption of contaminated food. But wait, contaminated food is not the only cause of some of our most recent outbreaks.
Live poultry-associated salmonellosis (LPAS)
LPAS was first reported in the 1950s. More recent epidemiologic data was published by C. Basler et al. (Emerging Infect Dis. 2016;22[10]:1705-11). LPAS was defined as two or more culture confirmed human Salmonella infections with a combination of epidemiologic, laboratory, or traceback evidence linking illnesses to contact with live poultry. The median outbreak size involved 26 cases (range, 4-363) and 77% (41 of 53) were multistate. The median age of the patients was 9 years (range, less than 1 to 92 years), and 31% were aged 5 years or younger. Exposure to chicks and ducklings was reported in 85% and 38%, respectively. High-risk practices included keeping poultry inside of the home (46%), snuggling baby birds (49%), and kissing baby birds (13%). The median time from purchase of poultry to onset of illness was 17 days (range, 1-672), and 66% reported onset of illness less than 30 days after purchase. Almost 52% reported owning poultry for less than 1 year.
The number of outbreaks continued to increase. From 1990 to 2005, there were a total of 17 outbreaks, compared with 36 between 2006 and 2014. Historically, outbreaks occurred in children around Easter when brightly colored dyed chicks were purchased. In the above review, 80% of outbreaks began in February, March, or April with an average duration of 4.9 months (range, 1-12).
Salmonella isolates
Backyard flocks and LPAS
More recently outbreaks have been associated with backyard flocks occurring year round and affecting both adults and children in contrast to seasonal peaks. The first multistate backyard flock outbreak was documented in 2007. Currently, the CDC is investigating 10 separate multistate outbreaks that began on Jan. 4, 2017. It involves 48 states, 961 infected individuals, 215 hospitalizations, and 1 death. At least 5 salmonella serotypes have been isolated.
What about the hatcheries?
It’s estimated that 50 million live poultry are sold annually. Birds are shipped within 24 hours after hatching via the U.S. Postal Service in boxes containing up to 100 chicks. Delivery occurs within 72 hours of hatching. Approximately 20 mail order hatcheries provide the majority of poultry sold to the general public. The National Poultry Improvement Plan (NPIP) is a voluntary state and federal testing and certification program whose goal is to eliminate poultry disease from breeder flocks to prevent egg-transmitted and hatchery-disseminated diseases. All hatcheries may participate. They also may participate in the voluntary Salmonella monitoring program. Note participation is not mandatory.
Preventing future outbreaks: patient/parental education is mandatory
1. Make sure your parents know about the association of Salmonella and live poultry. Reinforce these are farm animals, not pets. Purchase birds from hatcheries that participate in NPIP and the Salmonella monitoring programs.
2. Chicks, ducklings, or other live poultry should not be taken to schools, day care facilities, or nursing homes. Poultry should not be allowed in the home or in areas where food or drink is being prepared or consumed.
3. Poultry should not be snuggled, kissed, or allowed to touch one’s mouth. Hand washing with soap and water should occur after touching live poultry or any object touched in areas where they live or roam.
4. Contact with live poultry should be avoided in those at risk for developing serious infections including persons aged 5 years or younger, 65 years or older, immunocompromised individuals, and those with hemoglobinopathies.
5. All equipment used to care for live birds should be washed outdoors. Owners should have designated shoes when caring for poultry which should never be worn inside the home.
Hopefully, the next time you see a patient with fever and diarrhea you will recall this topic and ask about their contact with live poultry.
Additional resources to facilitate discussions can be found at www.cdc.gov/salmonella.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Email her at [email protected].
I recently received a group text from a friend voicing her frustration that her neighbor had acquired chickens, and she shared a photo of some roaming freely in the front yard. Naturally, my response was related to the potential infectious disease exposure and infections. Another friend chimed in “fresh eggs, and these are free range chickens. They don’t get sick. ... Many people in my area have chickens.” Unbeknownst to my friends, they had helped me select the ID Consult topic for this month.
Nontyphoidal Salmonella bacteria are associated with a wide spectrum of infections which range from asymptomatic gastrointestinal carriage to bacteremia, meningitis, osteomyelitis, and focal infections. Invasive disease is seen most often in children younger than 5 years of age, persons aged 65 years or older, and individuals with hemoglobinopathies including sickle cell disease and those with immunodeficiencies. Annually, the Centers for Disease Control and Prevention estimates that nontyphoidal salmonellosis is responsible for 1.2 million illnesses, 23,000 hospitalizations, and 450 deaths in the United States. Gastroenteritis is the most common manifestation of the disease and is characterized by abdominal cramps, diarrhea, and fever that develops 12-72 hours after exposure. It is usually self-limited. As previously reported in this column (June, 2017), Salmonella is one of the top two foodborne pathogens in the United States, and most outbreaks have been associated with consumption of contaminated food. But wait, contaminated food is not the only cause of some of our most recent outbreaks.
Live poultry-associated salmonellosis (LPAS)
LPAS was first reported in the 1950s. More recent epidemiologic data was published by C. Basler et al. (Emerging Infect Dis. 2016;22[10]:1705-11). LPAS was defined as two or more culture confirmed human Salmonella infections with a combination of epidemiologic, laboratory, or traceback evidence linking illnesses to contact with live poultry. The median outbreak size involved 26 cases (range, 4-363) and 77% (41 of 53) were multistate. The median age of the patients was 9 years (range, less than 1 to 92 years), and 31% were aged 5 years or younger. Exposure to chicks and ducklings was reported in 85% and 38%, respectively. High-risk practices included keeping poultry inside of the home (46%), snuggling baby birds (49%), and kissing baby birds (13%). The median time from purchase of poultry to onset of illness was 17 days (range, 1-672), and 66% reported onset of illness less than 30 days after purchase. Almost 52% reported owning poultry for less than 1 year.
The number of outbreaks continued to increase. From 1990 to 2005, there were a total of 17 outbreaks, compared with 36 between 2006 and 2014. Historically, outbreaks occurred in children around Easter when brightly colored dyed chicks were purchased. In the above review, 80% of outbreaks began in February, March, or April with an average duration of 4.9 months (range, 1-12).
Salmonella isolates
Backyard flocks and LPAS
More recently outbreaks have been associated with backyard flocks occurring year round and affecting both adults and children in contrast to seasonal peaks. The first multistate backyard flock outbreak was documented in 2007. Currently, the CDC is investigating 10 separate multistate outbreaks that began on Jan. 4, 2017. It involves 48 states, 961 infected individuals, 215 hospitalizations, and 1 death. At least 5 salmonella serotypes have been isolated.
What about the hatcheries?
It’s estimated that 50 million live poultry are sold annually. Birds are shipped within 24 hours after hatching via the U.S. Postal Service in boxes containing up to 100 chicks. Delivery occurs within 72 hours of hatching. Approximately 20 mail order hatcheries provide the majority of poultry sold to the general public. The National Poultry Improvement Plan (NPIP) is a voluntary state and federal testing and certification program whose goal is to eliminate poultry disease from breeder flocks to prevent egg-transmitted and hatchery-disseminated diseases. All hatcheries may participate. They also may participate in the voluntary Salmonella monitoring program. Note participation is not mandatory.
Preventing future outbreaks: patient/parental education is mandatory
1. Make sure your parents know about the association of Salmonella and live poultry. Reinforce these are farm animals, not pets. Purchase birds from hatcheries that participate in NPIP and the Salmonella monitoring programs.
2. Chicks, ducklings, or other live poultry should not be taken to schools, day care facilities, or nursing homes. Poultry should not be allowed in the home or in areas where food or drink is being prepared or consumed.
3. Poultry should not be snuggled, kissed, or allowed to touch one’s mouth. Hand washing with soap and water should occur after touching live poultry or any object touched in areas where they live or roam.
4. Contact with live poultry should be avoided in those at risk for developing serious infections including persons aged 5 years or younger, 65 years or older, immunocompromised individuals, and those with hemoglobinopathies.
5. All equipment used to care for live birds should be washed outdoors. Owners should have designated shoes when caring for poultry which should never be worn inside the home.
Hopefully, the next time you see a patient with fever and diarrhea you will recall this topic and ask about their contact with live poultry.
Additional resources to facilitate discussions can be found at www.cdc.gov/salmonella.
Dr. Word is a pediatric infectious disease specialist and director of the Houston Travel Medicine Clinic. She said she had no relevant financial disclosures. Email her at [email protected].
Consider adding chemotherapy after GI surgery
Adjuvant chemotherapy was associated with improved overall survival rates at 3 years in patients who had surgery for gastroesophageal cancer, based on retrospective data from more than 10,000 adults.
Preoperative chemoradiotherapy and resection has shown benefits in patients with gastroesophageal adenocarcinoma, but the potential benefits of adjuvant chemotherapy (AC) after surgery in these patients has not been well studied, wrote Ali A. El Mokdad, MD, of the University of Texas Southwestern Medical Center, Dallas, and his colleagues (JAMA Oncol. 2017 Sep 21. doi: 10.1001/jamaoncol.2017.2805).
The researchers reviewed data from 10,086 patients in the National Cancer Database during 2006-2013. Of these, 814 (8%) received adjuvant chemotherapy after surgery and 9,272 (94%) received no additional intervention beyond postoperative observation. The average age of the patients was 61 years, and 88% were men.
The average survival rates at 3 years after surgery were 40 months for the adjuvant group and 34 months for the observation group (hazard ratio, 0.79). The overall survival rates in the adjuvant group were 94%, 54%, and 38% at 1,3, and 5 years, respectively, compared with rates of 88%, 47%, and 34%, in the observation group.
The findings were limited in part by the retrospective nature of the study, the researchers said. In addition, “the estimated effect of AC on overall survival is subject to selection bias and immortal time bias given that the study was observational,” they noted.
However, the results support the addition of chemotherapy for gastroesophageal surgery patients, and “provide compelling motivation to explore the potential benefit of adjuvant chemotherapy in a randomized clinical trial,” they said. “A two-arm phase 2 trial design using recurrence-free survival as a primary endpoint is an appealing first step,” they added.
The researchers had no financial conflicts to disclose.
The study findings “seem to indicate that additional systemic chemotherapy could be advantageous for patients treated with neoadjuvant chemoradiotherapy for resectable gastroesophageal cancer,” wrote David Cunningham, MD, FMedSci, and Elizabeth C. Smyth, MB, BCh., MSc., in an accompanying editorial.
“The small percentage of patients treated with adjuvant chemotherapy is reassuring; neoadjuvant chemoradiotherapy and surgery followed by adjuvant chemotherapy is not a treatment approach endorsed by current national or international guidelines,” they noted. The findings suggest that the 4% increase in overall survival at 3 years is promising because most gastroesophageal cancer recurrences arise within 3 years of surgery, they said. “However, these results require validation in the form of a randomized clinical trial,” they emphasized (JAMA Oncol. 2017 Sep 21. doi: 10.1001/jamaoncol.2017.2792).
Dr. Cunningham and Ms. Smyth are affiliated with the department of gastrointestinal oncology and lymphoma at the Royal Marsden Hospital, London. Dr. Cunningham disclosed institutional research funding from Amgen, AstraZeneca, Bayer, Celgene, MedImmune, Merck Serono, Merrimack, and Sanofi. Ms. Smyth disclosed honoraria for advisory roles with Five Prime Therapeutics, Bristol-Myers Squibb, and Gritstone Oncology.
The study findings “seem to indicate that additional systemic chemotherapy could be advantageous for patients treated with neoadjuvant chemoradiotherapy for resectable gastroesophageal cancer,” wrote David Cunningham, MD, FMedSci, and Elizabeth C. Smyth, MB, BCh., MSc., in an accompanying editorial.
“The small percentage of patients treated with adjuvant chemotherapy is reassuring; neoadjuvant chemoradiotherapy and surgery followed by adjuvant chemotherapy is not a treatment approach endorsed by current national or international guidelines,” they noted. The findings suggest that the 4% increase in overall survival at 3 years is promising because most gastroesophageal cancer recurrences arise within 3 years of surgery, they said. “However, these results require validation in the form of a randomized clinical trial,” they emphasized (JAMA Oncol. 2017 Sep 21. doi: 10.1001/jamaoncol.2017.2792).
Dr. Cunningham and Ms. Smyth are affiliated with the department of gastrointestinal oncology and lymphoma at the Royal Marsden Hospital, London. Dr. Cunningham disclosed institutional research funding from Amgen, AstraZeneca, Bayer, Celgene, MedImmune, Merck Serono, Merrimack, and Sanofi. Ms. Smyth disclosed honoraria for advisory roles with Five Prime Therapeutics, Bristol-Myers Squibb, and Gritstone Oncology.
The study findings “seem to indicate that additional systemic chemotherapy could be advantageous for patients treated with neoadjuvant chemoradiotherapy for resectable gastroesophageal cancer,” wrote David Cunningham, MD, FMedSci, and Elizabeth C. Smyth, MB, BCh., MSc., in an accompanying editorial.
“The small percentage of patients treated with adjuvant chemotherapy is reassuring; neoadjuvant chemoradiotherapy and surgery followed by adjuvant chemotherapy is not a treatment approach endorsed by current national or international guidelines,” they noted. The findings suggest that the 4% increase in overall survival at 3 years is promising because most gastroesophageal cancer recurrences arise within 3 years of surgery, they said. “However, these results require validation in the form of a randomized clinical trial,” they emphasized (JAMA Oncol. 2017 Sep 21. doi: 10.1001/jamaoncol.2017.2792).
Dr. Cunningham and Ms. Smyth are affiliated with the department of gastrointestinal oncology and lymphoma at the Royal Marsden Hospital, London. Dr. Cunningham disclosed institutional research funding from Amgen, AstraZeneca, Bayer, Celgene, MedImmune, Merck Serono, Merrimack, and Sanofi. Ms. Smyth disclosed honoraria for advisory roles with Five Prime Therapeutics, Bristol-Myers Squibb, and Gritstone Oncology.
Adjuvant chemotherapy was associated with improved overall survival rates at 3 years in patients who had surgery for gastroesophageal cancer, based on retrospective data from more than 10,000 adults.
Preoperative chemoradiotherapy and resection has shown benefits in patients with gastroesophageal adenocarcinoma, but the potential benefits of adjuvant chemotherapy (AC) after surgery in these patients has not been well studied, wrote Ali A. El Mokdad, MD, of the University of Texas Southwestern Medical Center, Dallas, and his colleagues (JAMA Oncol. 2017 Sep 21. doi: 10.1001/jamaoncol.2017.2805).
The researchers reviewed data from 10,086 patients in the National Cancer Database during 2006-2013. Of these, 814 (8%) received adjuvant chemotherapy after surgery and 9,272 (94%) received no additional intervention beyond postoperative observation. The average age of the patients was 61 years, and 88% were men.
The average survival rates at 3 years after surgery were 40 months for the adjuvant group and 34 months for the observation group (hazard ratio, 0.79). The overall survival rates in the adjuvant group were 94%, 54%, and 38% at 1,3, and 5 years, respectively, compared with rates of 88%, 47%, and 34%, in the observation group.
The findings were limited in part by the retrospective nature of the study, the researchers said. In addition, “the estimated effect of AC on overall survival is subject to selection bias and immortal time bias given that the study was observational,” they noted.
However, the results support the addition of chemotherapy for gastroesophageal surgery patients, and “provide compelling motivation to explore the potential benefit of adjuvant chemotherapy in a randomized clinical trial,” they said. “A two-arm phase 2 trial design using recurrence-free survival as a primary endpoint is an appealing first step,” they added.
The researchers had no financial conflicts to disclose.
Adjuvant chemotherapy was associated with improved overall survival rates at 3 years in patients who had surgery for gastroesophageal cancer, based on retrospective data from more than 10,000 adults.
Preoperative chemoradiotherapy and resection has shown benefits in patients with gastroesophageal adenocarcinoma, but the potential benefits of adjuvant chemotherapy (AC) after surgery in these patients has not been well studied, wrote Ali A. El Mokdad, MD, of the University of Texas Southwestern Medical Center, Dallas, and his colleagues (JAMA Oncol. 2017 Sep 21. doi: 10.1001/jamaoncol.2017.2805).
The researchers reviewed data from 10,086 patients in the National Cancer Database during 2006-2013. Of these, 814 (8%) received adjuvant chemotherapy after surgery and 9,272 (94%) received no additional intervention beyond postoperative observation. The average age of the patients was 61 years, and 88% were men.
The average survival rates at 3 years after surgery were 40 months for the adjuvant group and 34 months for the observation group (hazard ratio, 0.79). The overall survival rates in the adjuvant group were 94%, 54%, and 38% at 1,3, and 5 years, respectively, compared with rates of 88%, 47%, and 34%, in the observation group.
The findings were limited in part by the retrospective nature of the study, the researchers said. In addition, “the estimated effect of AC on overall survival is subject to selection bias and immortal time bias given that the study was observational,” they noted.
However, the results support the addition of chemotherapy for gastroesophageal surgery patients, and “provide compelling motivation to explore the potential benefit of adjuvant chemotherapy in a randomized clinical trial,” they said. “A two-arm phase 2 trial design using recurrence-free survival as a primary endpoint is an appealing first step,” they added.
The researchers had no financial conflicts to disclose.
FROM JAMA ONCOLOGY
Key clinical point: Patients undergoing surgery for gastroesophageal cancer may benefit from additional chemotherapy.
Major finding: Overall survival rates improved in patients who received adjuvant chemotherapy, compared with those who did not (40 months vs. 34 months, respectively).
Data source: A review of 10,086 adults in the National Cancer Database who underwent gastroesophageal cancer surgery during 2006-2013.
Disclosures: The researchers had no financial conflicts to disclose.
Hospital Privileging Practices for Bedside Procedures: A Survey of Hospitalist Experts
Performance of 6 bedside procedures (paracentesis, thoracentesis, lumbar puncture, arthrocentesis, central venous catheter [CVC] placement, and arterial line placement) are considered core competencies for hospitalists.1 Yet, the American Board of Internal Medicine (ABIM) no longer requires demonstration of manual competency for bedside procedures, and graduates may enter the workforce with minimal or no experience performing such procedures.2 As such, the burden falls on hospital privileging committees to ensure providers have the necessary training and experience to competently perform invasive procedures before granting institutional privileges to perform them.3 Although recommendations for privileging to perform certain surgical procedures have been proposed,4,5 there are no widely accepted guidelines for initial or ongoing privileging of common invasive bedside procedures performed by hospitalists, and current privileging practices vary significantly.
In 2015, the Society of Hospital Medicine (SHM) set up a Point-of-Care Ultrasound (POCUS) Task Force to draft evidence-based guidelines on the use of ultrasound to perform bedside procedures. The recommendations for certification of competency in ultrasound-guided procedures may guide institutional privileging. The purpose of this study was to better understand current hospital privileging practices for invasive bedside procedures both with and without ultrasound guidance and how current practices are perceived by experts.
METHODS
Study Design, Setting, and Participants
After approval by the University of Texas Health Science Center at San Antonio Institutional Review Board, we conducted a survey of hospital privileging processes for bedside procedures from a convenience sample of hospitalist procedure experts on the SHM POCUS Task Force. All 21 hospitalists on the task force were invited to participate, including the authors of this article. These hospitalists represent 21 unique institutions, and all have clinical, educational, and/or research expertise in ultrasound-guided bedside procedures.
Survey Design
A 26-question, electronic survey on privileging for bedside procedures was conducted (Appendix A). Twenty questions addressed procedures in general, such as minimum numbers of procedures required and use of simulation. Six questions focused on the use of ultrasound guidance. To provide context, many questions were framed to assess a privileging process being drafted by the task force. Answers were either multiple choice or free text.
Data Collection and Analysis
All members of the task force were invited to complete the survey by e-mail during November 2016. A reminder e-mail was sent on the day after initial distribution. No compensation was offered, and participation was not required. Survey results were compiled electronically through Research Electronic Data Capture, or “REDCap”TM (Nashville, Tennessee), and data analysis was performed with Stata version 14 (College Station, Texas). Means of current and recommended minimum thresholds were calculated by excluding responses of “I don’t know,” and responses of “no minimum number threshold” were coded as 0.
RESULTS
The survey response rate was 100% (21 of 21). All experts were hospitalists, but 2 also identified themselves as intensivists. Experts practiced in a variety of hospital settings, including private university hospitals (43%), public university hospitals (19%), Veterans Affairs teaching hospitals (14%), community teaching hospitals (14%), and community nonteaching hospitals (10%). Most hospitals (90%) were teaching hospitals for internal medicine trainees. All experts have personally performed bedside procedures on a regular basis, and most (86%) had leadership roles in teaching procedures to students, residents, fellows, physician assistants, nurse practitioners, and/or physicians. Approximately half (57%) were involved in granting privileges for bedside procedures at their institutions.
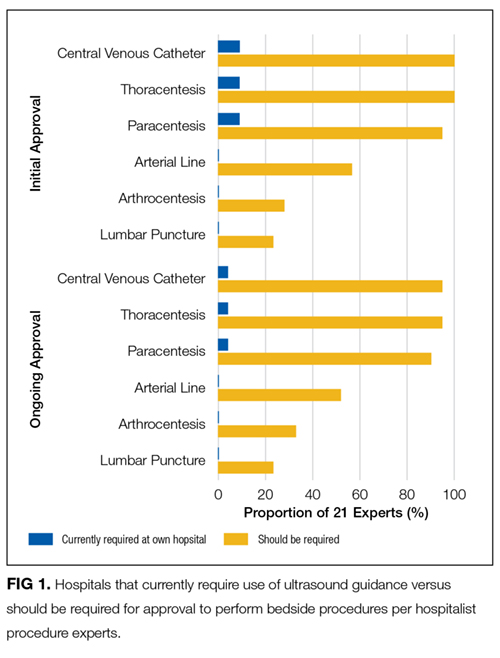
Only half of the experts reported that their hospitals required a minimum number of procedures to earn initial (48%) or ongoing (52%) privileges to perform bedside procedures. Nevertheless, most experts thought there ought to be minimum numbers of procedures for initial (81%) and ongoing (81%) privileging, recommending higher minimums for both initial and ongoing privileging than are currently required at their hospitals (Figure 2).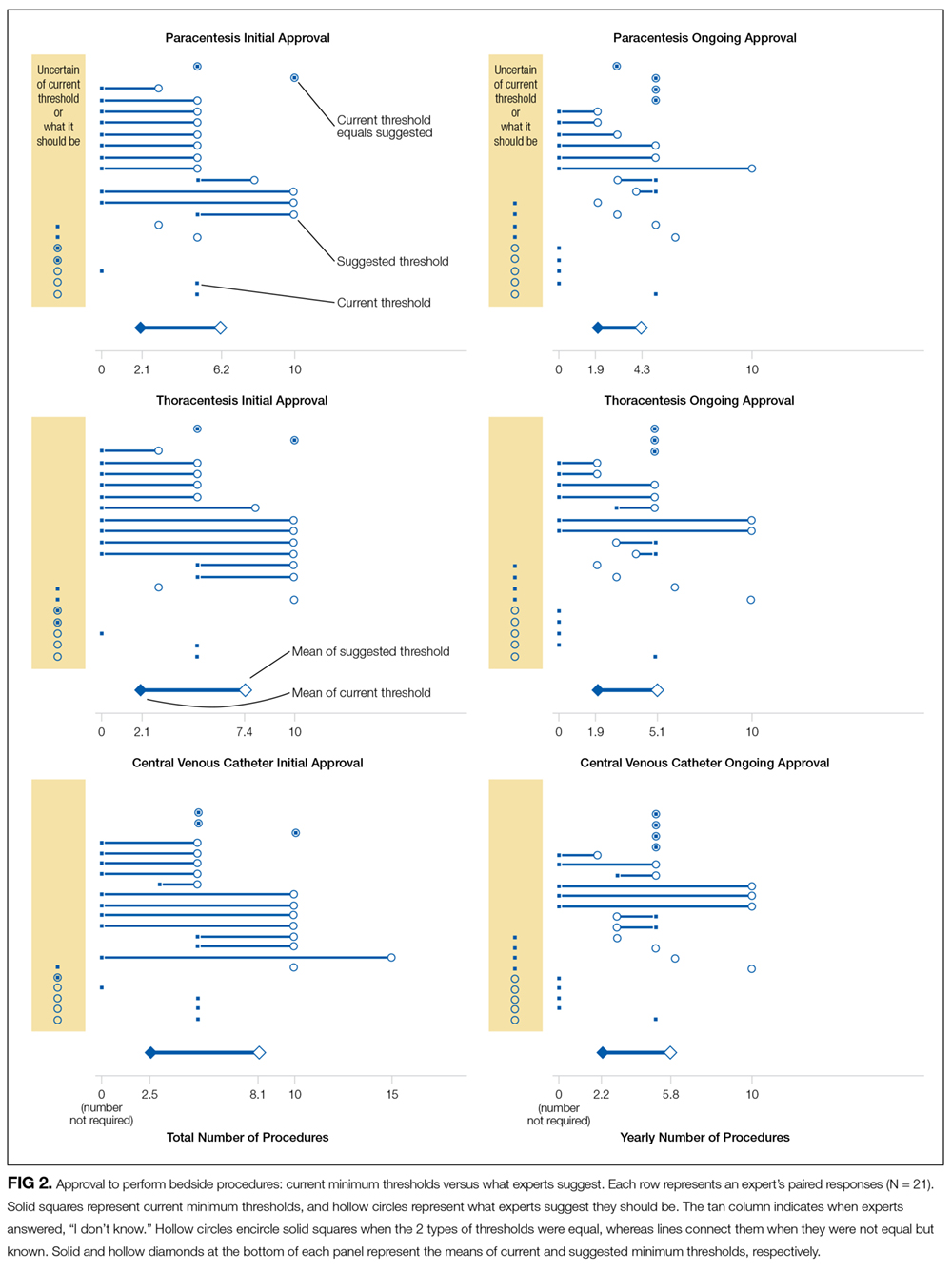
Most hospitalist procedure experts thought that simulation training (67%) and direct observation of procedural skills (71%) should be core components of an initial privileging process. Many of the experts who did not agree with direct observation or simulation training as core components of initial privileging had concerns about feasibility with respect to manpower, availability of simulation equipment, and costs. In contrast, the majority (67%) did not think it was necessary to directly observe providers for ongoing privileging when routine monitoring was in place for periprocedural complications, which all experts (100%) agreed should be in place.
DISCUSSION
Our survey identified 3 distinct differences between hospitalist procedure experts’ recommendations and their own hospitals’ current privileging practices. First, whereas experts recommended ultrasound guidance for thoracentesis, paracentesis, and CVC placement, it is rarely a current requirement. Second, experts recommend requiring minimum numbers of procedures for both initial and ongoing privileging even though such minimums are not currently required at half of their hospitals. Third, recommended minimum numbers were generally higher than those currently in place.
The routine use of ultrasound guidance for thoracentesis, paracentesis, and CVC placement is likely a result of increased adoption based on the literature showing clinical benefits.6-9 Thus, the expert recommendations for required use of ultrasound guidance for these procedures seems both appropriate and feasible. The procedure minimums identified in our study are similar to prior ABIM guidelines when manual competency was required for board certification in internal medicine and are comparable to recent minimums proposed by the Society of Critical Care Medicine, both of which recommended a minimum of 5 to 10 per procedure.10,11 Nevertheless, no commonly agreed-upon minimum number of procedures currently exists for certification of competency, and the variability seen in the experts’ responses further supports the idea that no specific number will guarantee competence. Thus, while requiring minimum numbers of procedures was generally considered necessary by our experts, minimums alone were also considered insufficient for initial privileging because most recommended that direct observation and simulation should be part of an initial privileging process.
These findings encourage more rigorous requirements for both initial and ongoing privileging of procedures. Nevertheless, our findings were rarely unanimous. The most frequently cited reason for disagreement on our findings was feasibility and capacity for direct observation, and the absence of ultrasound equipment or simulators, particularly in resource-limited clinical environments.
Our study has several strengths and limitations. One strength is the recruitment of study experts specifically composed of hospitalist procedure experts from diverse geographic and hospital settings. Yet, we acknowledge that our findings may not be generalizable to other specialties. Another strength is we obtained 100% participation from the experts surveyed. Weaknesses of this study include the relatively small number of experts who are likely to be biased in favor of both the use of ultrasound guidance and higher standards for privileging. We also relied on self-reported data about privileging processes rather than direct observation of those practices. Finally, questions were framed in the context of only 1 possible privileging pathway, and experts may respond differently to a different framing.
CONCLUSION
Our findings may guide the development of more standardized frameworks for initial and ongoing privileging of hospitalists for invasive bedside procedures. In particular, additional privileging requirements may include the routine use of ultrasound guidance for paracentesis, thoracentesis, and CVC insertion; simulation preceding direct observation of manual skills if possible; and higher required minimums of procedures for both initial and ongoing privileging. The goal of a standardized framework for privileging should be directed at improving the quality and safety of bedside procedures but must consider feasibility in diverse clinical settings where hospitalists work.
Acknowledgments
The authors thank the hospitalists on the SHM POCUS Task Force who provided data about their institutions’ privileging processes and requirements. They are also grateful to Loretta M. Grikis, MLS, AHIP, at the White River Junction Veterans Affairs Medical Center for her assistance as a medical librarian.
Disclosure
B
1. Nichani S, Jonathan Crocker, MD, Nick Fitterman, MD, Michael Lukela, MD, Updating the core competencies in hospital medicine—2017 revision: Introduction and methodology. J Hosp Med 2017;12(4);283-287. PubMed
2. American Board of Internal Medicine. Policies and Procedures for Certification. http://www.abim.org/certification/policies/imss/im.aspx - procedures. Published July 2016. Accessed on November 8, 2016.
3. Department of Health & Human Services. Centers for Medicare & Medicaid Services (CMS) Requirements for Hospital Medical Staff Privileging. Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/downloads/SCLetter05-04.pdf. Published November 12, 2004. Accessed on November 8, 2016.
4. Blackmon SH, Cooke DT, Whyte R, et al. The Society of Thoracic Surgeons Expert Consensus Statement: A tool kit to assist thoracic surgeons seeking privileging to use new technology and perform advanced procedures in general thoracic surgery. Ann Thorac Surg. 2016;101(3):1230-1237. PubMed
5. Bhora FY, Al-Ayoubi AM, Rehmani SS, Forleiter CM, Raad WN, Belsley SG. Robotically assisted thoracic surgery: proposed guidelines for privileging and credentialing. Innovations (Phila). 2016;11(6):386-389. PubMed
6. Mercaldi CJ, Lanes SF. Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143:532-538. PubMed
7. Patel PA, Ernst FR, Gunnarsson CL. Ultrasonography guidance reduces complications and costs associated with thoracentesis procedures. J Clin Ultrasound. 2012;40:135-141. PubMed
8. Brass P, Hellmich M, Kolodziej L, Schick G, Smith AF. Ultrasound guidance versus anatomical landmarks for internal jugular vein catheterization. Cochrane Database Syst Rev. 2015;1:CD006962. DOI: 10.1002/14651858.CD006962.pub2. PubMed
9. Brass P, Hellmich M, Kolodziej L, Schick G, Smith AF. Ultrasound guidance versus anatomical landmarks for subclavian or femoral vein catheterization. Cochrane Database Syst Rev. 2015;1:CD011447. DOI: 10.1002/14651858.CD011447. PubMed
10. American Board of Internal Medicine. Policies and Procedures. Philadelphia, PA; July 1990.
11. Society of Critical Care Medicine Ultrasound Certification Task Force. Recommendations for Achieving and Maintaining Competence and Credentialing in Critical Care Ultrasound with Focused Cardiac Ultrasound and Advanced Critical Care Echocardiography. http://journals.lww.com/ccmjournal/Documents/Critical%20Care%20Ultrasound.pdf. Published 2013. Accessed November 8, 2016.
Performance of 6 bedside procedures (paracentesis, thoracentesis, lumbar puncture, arthrocentesis, central venous catheter [CVC] placement, and arterial line placement) are considered core competencies for hospitalists.1 Yet, the American Board of Internal Medicine (ABIM) no longer requires demonstration of manual competency for bedside procedures, and graduates may enter the workforce with minimal or no experience performing such procedures.2 As such, the burden falls on hospital privileging committees to ensure providers have the necessary training and experience to competently perform invasive procedures before granting institutional privileges to perform them.3 Although recommendations for privileging to perform certain surgical procedures have been proposed,4,5 there are no widely accepted guidelines for initial or ongoing privileging of common invasive bedside procedures performed by hospitalists, and current privileging practices vary significantly.
In 2015, the Society of Hospital Medicine (SHM) set up a Point-of-Care Ultrasound (POCUS) Task Force to draft evidence-based guidelines on the use of ultrasound to perform bedside procedures. The recommendations for certification of competency in ultrasound-guided procedures may guide institutional privileging. The purpose of this study was to better understand current hospital privileging practices for invasive bedside procedures both with and without ultrasound guidance and how current practices are perceived by experts.
METHODS
Study Design, Setting, and Participants
After approval by the University of Texas Health Science Center at San Antonio Institutional Review Board, we conducted a survey of hospital privileging processes for bedside procedures from a convenience sample of hospitalist procedure experts on the SHM POCUS Task Force. All 21 hospitalists on the task force were invited to participate, including the authors of this article. These hospitalists represent 21 unique institutions, and all have clinical, educational, and/or research expertise in ultrasound-guided bedside procedures.
Survey Design
A 26-question, electronic survey on privileging for bedside procedures was conducted (Appendix A). Twenty questions addressed procedures in general, such as minimum numbers of procedures required and use of simulation. Six questions focused on the use of ultrasound guidance. To provide context, many questions were framed to assess a privileging process being drafted by the task force. Answers were either multiple choice or free text.
Data Collection and Analysis
All members of the task force were invited to complete the survey by e-mail during November 2016. A reminder e-mail was sent on the day after initial distribution. No compensation was offered, and participation was not required. Survey results were compiled electronically through Research Electronic Data Capture, or “REDCap”TM (Nashville, Tennessee), and data analysis was performed with Stata version 14 (College Station, Texas). Means of current and recommended minimum thresholds were calculated by excluding responses of “I don’t know,” and responses of “no minimum number threshold” were coded as 0.
RESULTS
The survey response rate was 100% (21 of 21). All experts were hospitalists, but 2 also identified themselves as intensivists. Experts practiced in a variety of hospital settings, including private university hospitals (43%), public university hospitals (19%), Veterans Affairs teaching hospitals (14%), community teaching hospitals (14%), and community nonteaching hospitals (10%). Most hospitals (90%) were teaching hospitals for internal medicine trainees. All experts have personally performed bedside procedures on a regular basis, and most (86%) had leadership roles in teaching procedures to students, residents, fellows, physician assistants, nurse practitioners, and/or physicians. Approximately half (57%) were involved in granting privileges for bedside procedures at their institutions.
Only half of the experts reported that their hospitals required a minimum number of procedures to earn initial (48%) or ongoing (52%) privileges to perform bedside procedures. Nevertheless, most experts thought there ought to be minimum numbers of procedures for initial (81%) and ongoing (81%) privileging, recommending higher minimums for both initial and ongoing privileging than are currently required at their hospitals (Figure 2).
Most hospitalist procedure experts thought that simulation training (67%) and direct observation of procedural skills (71%) should be core components of an initial privileging process. Many of the experts who did not agree with direct observation or simulation training as core components of initial privileging had concerns about feasibility with respect to manpower, availability of simulation equipment, and costs. In contrast, the majority (67%) did not think it was necessary to directly observe providers for ongoing privileging when routine monitoring was in place for periprocedural complications, which all experts (100%) agreed should be in place.
DISCUSSION
Our survey identified 3 distinct differences between hospitalist procedure experts’ recommendations and their own hospitals’ current privileging practices. First, whereas experts recommended ultrasound guidance for thoracentesis, paracentesis, and CVC placement, it is rarely a current requirement. Second, experts recommend requiring minimum numbers of procedures for both initial and ongoing privileging even though such minimums are not currently required at half of their hospitals. Third, recommended minimum numbers were generally higher than those currently in place.
The routine use of ultrasound guidance for thoracentesis, paracentesis, and CVC placement is likely a result of increased adoption based on the literature showing clinical benefits.6-9 Thus, the expert recommendations for required use of ultrasound guidance for these procedures seems both appropriate and feasible. The procedure minimums identified in our study are similar to prior ABIM guidelines when manual competency was required for board certification in internal medicine and are comparable to recent minimums proposed by the Society of Critical Care Medicine, both of which recommended a minimum of 5 to 10 per procedure.10,11 Nevertheless, no commonly agreed-upon minimum number of procedures currently exists for certification of competency, and the variability seen in the experts’ responses further supports the idea that no specific number will guarantee competence. Thus, while requiring minimum numbers of procedures was generally considered necessary by our experts, minimums alone were also considered insufficient for initial privileging because most recommended that direct observation and simulation should be part of an initial privileging process.
These findings encourage more rigorous requirements for both initial and ongoing privileging of procedures. Nevertheless, our findings were rarely unanimous. The most frequently cited reason for disagreement on our findings was feasibility and capacity for direct observation, and the absence of ultrasound equipment or simulators, particularly in resource-limited clinical environments.
Our study has several strengths and limitations. One strength is the recruitment of study experts specifically composed of hospitalist procedure experts from diverse geographic and hospital settings. Yet, we acknowledge that our findings may not be generalizable to other specialties. Another strength is we obtained 100% participation from the experts surveyed. Weaknesses of this study include the relatively small number of experts who are likely to be biased in favor of both the use of ultrasound guidance and higher standards for privileging. We also relied on self-reported data about privileging processes rather than direct observation of those practices. Finally, questions were framed in the context of only 1 possible privileging pathway, and experts may respond differently to a different framing.
CONCLUSION
Our findings may guide the development of more standardized frameworks for initial and ongoing privileging of hospitalists for invasive bedside procedures. In particular, additional privileging requirements may include the routine use of ultrasound guidance for paracentesis, thoracentesis, and CVC insertion; simulation preceding direct observation of manual skills if possible; and higher required minimums of procedures for both initial and ongoing privileging. The goal of a standardized framework for privileging should be directed at improving the quality and safety of bedside procedures but must consider feasibility in diverse clinical settings where hospitalists work.
Acknowledgments
The authors thank the hospitalists on the SHM POCUS Task Force who provided data about their institutions’ privileging processes and requirements. They are also grateful to Loretta M. Grikis, MLS, AHIP, at the White River Junction Veterans Affairs Medical Center for her assistance as a medical librarian.
Disclosure
B
Performance of 6 bedside procedures (paracentesis, thoracentesis, lumbar puncture, arthrocentesis, central venous catheter [CVC] placement, and arterial line placement) are considered core competencies for hospitalists.1 Yet, the American Board of Internal Medicine (ABIM) no longer requires demonstration of manual competency for bedside procedures, and graduates may enter the workforce with minimal or no experience performing such procedures.2 As such, the burden falls on hospital privileging committees to ensure providers have the necessary training and experience to competently perform invasive procedures before granting institutional privileges to perform them.3 Although recommendations for privileging to perform certain surgical procedures have been proposed,4,5 there are no widely accepted guidelines for initial or ongoing privileging of common invasive bedside procedures performed by hospitalists, and current privileging practices vary significantly.
In 2015, the Society of Hospital Medicine (SHM) set up a Point-of-Care Ultrasound (POCUS) Task Force to draft evidence-based guidelines on the use of ultrasound to perform bedside procedures. The recommendations for certification of competency in ultrasound-guided procedures may guide institutional privileging. The purpose of this study was to better understand current hospital privileging practices for invasive bedside procedures both with and without ultrasound guidance and how current practices are perceived by experts.
METHODS
Study Design, Setting, and Participants
After approval by the University of Texas Health Science Center at San Antonio Institutional Review Board, we conducted a survey of hospital privileging processes for bedside procedures from a convenience sample of hospitalist procedure experts on the SHM POCUS Task Force. All 21 hospitalists on the task force were invited to participate, including the authors of this article. These hospitalists represent 21 unique institutions, and all have clinical, educational, and/or research expertise in ultrasound-guided bedside procedures.
Survey Design
A 26-question, electronic survey on privileging for bedside procedures was conducted (Appendix A). Twenty questions addressed procedures in general, such as minimum numbers of procedures required and use of simulation. Six questions focused on the use of ultrasound guidance. To provide context, many questions were framed to assess a privileging process being drafted by the task force. Answers were either multiple choice or free text.
Data Collection and Analysis
All members of the task force were invited to complete the survey by e-mail during November 2016. A reminder e-mail was sent on the day after initial distribution. No compensation was offered, and participation was not required. Survey results were compiled electronically through Research Electronic Data Capture, or “REDCap”TM (Nashville, Tennessee), and data analysis was performed with Stata version 14 (College Station, Texas). Means of current and recommended minimum thresholds were calculated by excluding responses of “I don’t know,” and responses of “no minimum number threshold” were coded as 0.
RESULTS
The survey response rate was 100% (21 of 21). All experts were hospitalists, but 2 also identified themselves as intensivists. Experts practiced in a variety of hospital settings, including private university hospitals (43%), public university hospitals (19%), Veterans Affairs teaching hospitals (14%), community teaching hospitals (14%), and community nonteaching hospitals (10%). Most hospitals (90%) were teaching hospitals for internal medicine trainees. All experts have personally performed bedside procedures on a regular basis, and most (86%) had leadership roles in teaching procedures to students, residents, fellows, physician assistants, nurse practitioners, and/or physicians. Approximately half (57%) were involved in granting privileges for bedside procedures at their institutions.
Only half of the experts reported that their hospitals required a minimum number of procedures to earn initial (48%) or ongoing (52%) privileges to perform bedside procedures. Nevertheless, most experts thought there ought to be minimum numbers of procedures for initial (81%) and ongoing (81%) privileging, recommending higher minimums for both initial and ongoing privileging than are currently required at their hospitals (Figure 2).
Most hospitalist procedure experts thought that simulation training (67%) and direct observation of procedural skills (71%) should be core components of an initial privileging process. Many of the experts who did not agree with direct observation or simulation training as core components of initial privileging had concerns about feasibility with respect to manpower, availability of simulation equipment, and costs. In contrast, the majority (67%) did not think it was necessary to directly observe providers for ongoing privileging when routine monitoring was in place for periprocedural complications, which all experts (100%) agreed should be in place.
DISCUSSION
Our survey identified 3 distinct differences between hospitalist procedure experts’ recommendations and their own hospitals’ current privileging practices. First, whereas experts recommended ultrasound guidance for thoracentesis, paracentesis, and CVC placement, it is rarely a current requirement. Second, experts recommend requiring minimum numbers of procedures for both initial and ongoing privileging even though such minimums are not currently required at half of their hospitals. Third, recommended minimum numbers were generally higher than those currently in place.
The routine use of ultrasound guidance for thoracentesis, paracentesis, and CVC placement is likely a result of increased adoption based on the literature showing clinical benefits.6-9 Thus, the expert recommendations for required use of ultrasound guidance for these procedures seems both appropriate and feasible. The procedure minimums identified in our study are similar to prior ABIM guidelines when manual competency was required for board certification in internal medicine and are comparable to recent minimums proposed by the Society of Critical Care Medicine, both of which recommended a minimum of 5 to 10 per procedure.10,11 Nevertheless, no commonly agreed-upon minimum number of procedures currently exists for certification of competency, and the variability seen in the experts’ responses further supports the idea that no specific number will guarantee competence. Thus, while requiring minimum numbers of procedures was generally considered necessary by our experts, minimums alone were also considered insufficient for initial privileging because most recommended that direct observation and simulation should be part of an initial privileging process.
These findings encourage more rigorous requirements for both initial and ongoing privileging of procedures. Nevertheless, our findings were rarely unanimous. The most frequently cited reason for disagreement on our findings was feasibility and capacity for direct observation, and the absence of ultrasound equipment or simulators, particularly in resource-limited clinical environments.
Our study has several strengths and limitations. One strength is the recruitment of study experts specifically composed of hospitalist procedure experts from diverse geographic and hospital settings. Yet, we acknowledge that our findings may not be generalizable to other specialties. Another strength is we obtained 100% participation from the experts surveyed. Weaknesses of this study include the relatively small number of experts who are likely to be biased in favor of both the use of ultrasound guidance and higher standards for privileging. We also relied on self-reported data about privileging processes rather than direct observation of those practices. Finally, questions were framed in the context of only 1 possible privileging pathway, and experts may respond differently to a different framing.
CONCLUSION
Our findings may guide the development of more standardized frameworks for initial and ongoing privileging of hospitalists for invasive bedside procedures. In particular, additional privileging requirements may include the routine use of ultrasound guidance for paracentesis, thoracentesis, and CVC insertion; simulation preceding direct observation of manual skills if possible; and higher required minimums of procedures for both initial and ongoing privileging. The goal of a standardized framework for privileging should be directed at improving the quality and safety of bedside procedures but must consider feasibility in diverse clinical settings where hospitalists work.
Acknowledgments
The authors thank the hospitalists on the SHM POCUS Task Force who provided data about their institutions’ privileging processes and requirements. They are also grateful to Loretta M. Grikis, MLS, AHIP, at the White River Junction Veterans Affairs Medical Center for her assistance as a medical librarian.
Disclosure
B
1. Nichani S, Jonathan Crocker, MD, Nick Fitterman, MD, Michael Lukela, MD, Updating the core competencies in hospital medicine—2017 revision: Introduction and methodology. J Hosp Med 2017;12(4);283-287. PubMed
2. American Board of Internal Medicine. Policies and Procedures for Certification. http://www.abim.org/certification/policies/imss/im.aspx - procedures. Published July 2016. Accessed on November 8, 2016.
3. Department of Health & Human Services. Centers for Medicare & Medicaid Services (CMS) Requirements for Hospital Medical Staff Privileging. Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/downloads/SCLetter05-04.pdf. Published November 12, 2004. Accessed on November 8, 2016.
4. Blackmon SH, Cooke DT, Whyte R, et al. The Society of Thoracic Surgeons Expert Consensus Statement: A tool kit to assist thoracic surgeons seeking privileging to use new technology and perform advanced procedures in general thoracic surgery. Ann Thorac Surg. 2016;101(3):1230-1237. PubMed
5. Bhora FY, Al-Ayoubi AM, Rehmani SS, Forleiter CM, Raad WN, Belsley SG. Robotically assisted thoracic surgery: proposed guidelines for privileging and credentialing. Innovations (Phila). 2016;11(6):386-389. PubMed
6. Mercaldi CJ, Lanes SF. Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143:532-538. PubMed
7. Patel PA, Ernst FR, Gunnarsson CL. Ultrasonography guidance reduces complications and costs associated with thoracentesis procedures. J Clin Ultrasound. 2012;40:135-141. PubMed
8. Brass P, Hellmich M, Kolodziej L, Schick G, Smith AF. Ultrasound guidance versus anatomical landmarks for internal jugular vein catheterization. Cochrane Database Syst Rev. 2015;1:CD006962. DOI: 10.1002/14651858.CD006962.pub2. PubMed
9. Brass P, Hellmich M, Kolodziej L, Schick G, Smith AF. Ultrasound guidance versus anatomical landmarks for subclavian or femoral vein catheterization. Cochrane Database Syst Rev. 2015;1:CD011447. DOI: 10.1002/14651858.CD011447. PubMed
10. American Board of Internal Medicine. Policies and Procedures. Philadelphia, PA; July 1990.
11. Society of Critical Care Medicine Ultrasound Certification Task Force. Recommendations for Achieving and Maintaining Competence and Credentialing in Critical Care Ultrasound with Focused Cardiac Ultrasound and Advanced Critical Care Echocardiography. http://journals.lww.com/ccmjournal/Documents/Critical%20Care%20Ultrasound.pdf. Published 2013. Accessed November 8, 2016.
1. Nichani S, Jonathan Crocker, MD, Nick Fitterman, MD, Michael Lukela, MD, Updating the core competencies in hospital medicine—2017 revision: Introduction and methodology. J Hosp Med 2017;12(4);283-287. PubMed
2. American Board of Internal Medicine. Policies and Procedures for Certification. http://www.abim.org/certification/policies/imss/im.aspx - procedures. Published July 2016. Accessed on November 8, 2016.
3. Department of Health & Human Services. Centers for Medicare & Medicaid Services (CMS) Requirements for Hospital Medical Staff Privileging. Centers for Medicare and Medicaid Services website. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/downloads/SCLetter05-04.pdf. Published November 12, 2004. Accessed on November 8, 2016.
4. Blackmon SH, Cooke DT, Whyte R, et al. The Society of Thoracic Surgeons Expert Consensus Statement: A tool kit to assist thoracic surgeons seeking privileging to use new technology and perform advanced procedures in general thoracic surgery. Ann Thorac Surg. 2016;101(3):1230-1237. PubMed
5. Bhora FY, Al-Ayoubi AM, Rehmani SS, Forleiter CM, Raad WN, Belsley SG. Robotically assisted thoracic surgery: proposed guidelines for privileging and credentialing. Innovations (Phila). 2016;11(6):386-389. PubMed
6. Mercaldi CJ, Lanes SF. Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143:532-538. PubMed
7. Patel PA, Ernst FR, Gunnarsson CL. Ultrasonography guidance reduces complications and costs associated with thoracentesis procedures. J Clin Ultrasound. 2012;40:135-141. PubMed
8. Brass P, Hellmich M, Kolodziej L, Schick G, Smith AF. Ultrasound guidance versus anatomical landmarks for internal jugular vein catheterization. Cochrane Database Syst Rev. 2015;1:CD006962. DOI: 10.1002/14651858.CD006962.pub2. PubMed
9. Brass P, Hellmich M, Kolodziej L, Schick G, Smith AF. Ultrasound guidance versus anatomical landmarks for subclavian or femoral vein catheterization. Cochrane Database Syst Rev. 2015;1:CD011447. DOI: 10.1002/14651858.CD011447. PubMed
10. American Board of Internal Medicine. Policies and Procedures. Philadelphia, PA; July 1990.
11. Society of Critical Care Medicine Ultrasound Certification Task Force. Recommendations for Achieving and Maintaining Competence and Credentialing in Critical Care Ultrasound with Focused Cardiac Ultrasound and Advanced Critical Care Echocardiography. http://journals.lww.com/ccmjournal/Documents/Critical%20Care%20Ultrasound.pdf. Published 2013. Accessed November 8, 2016.
© 2017 Society of Hospital Medicine
Planned, Related or Preventable: Defining Readmissions to Capture Quality of Care
In this issue of the Journal of Hospital Medicine, Ellimoottil and colleagues examine characteristics of readmissions identified as planned by the planned readmission algorithm developed for the Center for Medicare & Medicaid Services (CMS) by using Medicare claims data from 131 hospitals in Michigan.1 They found that a substantial portion of readmissions currently classified as planned by the algorithm appear to be nonelective, as defined by the presence of a charge by an emergency medicine physician or an admission type of emergent or urgent, making those hospitalizations unlikely to be planned. They suggest that the algorithm could be modified to exclude such cases from the planned designation.
To determine whether modifying the algorithm as recommended is a good idea, it is helpful to examine the origins of the existing planned readmission algorithm. The algorithm originated as a consequence of hospital accountability measures for readmissions and was developed by this author in collaboration with colleagues at Yale University and elsewhere.2 Readmission measures have been controversial in part because clearly some (undetermined) fraction of readmissions is unavoidable. Many commentators have asked that readmission measures therefore capture only avoidable or related readmissions. Avoidable readmissions are those that could have been prevented by members of the healthcare system through actions taken during or after hospitalization, such as patient counseling, communication among team members, and guideline-concordant medical care. Related readmissions are those directly stemming from the index admission. However, reliably and accurately defining such events has proven elusive. One study, for instance, found the rate of physician-assessed preventability in published studies ranged from 9% to 48%.3 The challenge is even greater in trying to determine preventability using just claims data, without physician review of charts. Imagine, for instance, a patient with heart failure who is readmitted with heart failure exacerbation. The readmission preceded by a large fast-food meal is likely preventable; although even in this case, some would argue the healthcare system should not be held accountable for a readmission if the patient had been properly counseled about avoiding salty food. The one preceded by progressively worsening systolic function in a patient who reliably takes medications, weighs herself daily, and watches her diet is likely not. But both appear identical in claims. Related is also a difficult concept to operationalize. A recently hospitalized patient readmitted with pneumonia might have acquired it in the hospital (related) or from her grandchild 2 weeks later (unrelated). Again, both appear identical in claims.
In the ideal world, clinicians would be held accountable only for preventable readmissions. In practice, that has not proven to be possible.
Instead, the CMS readmission measures omit readmissions that are thought to be planned in advance: necessary and intentional readmissions. Defining a planned readmission is conceptually easier than defining a preventable readmission, yet even this is not always straightforward. The clearest case might be a person with a longstanding plan to have an elective surgery (say, a hip replacement) who is briefly admitted with something minor enough not to delay a subsequent admission for the scheduled surgery. Other patients are admitted with acute problems that require follow-up hospitalization (for instance, an acute myocardial infarction that requires a coronary artery bypass graft 2 weeks later).4 More ambiguous are patients who are sent home on a course of treatment with a plan for rehospitalization if it fails; for instance, a patient with gangrene is sent home on intravenous antibiotics but fails to improve and is rehospitalized for an amputation. Is that readmission planned or unplanned? Reasonable people might disagree.
Nonetheless, assuming it is desirable to at least try to identify and remove planned readmissions from measures, there are a number of ways in which one might do so. Perhaps the simplest would be to classify each hospitalization as planned or not on the UB-04 claim form. Such a process would be very feasible but also subject to gaming or coding variability. Given that there is some ambiguity and no standard about what types of readmissions are planned and that current policy provides incentives to reduce unplanned readmission rates, hospitals might vary in the cases to which they would apply such a code. This approach, therefore, has not been favored by payers to date. An alternative is to prospectively flag admissions that are expected to result in planned readmissions. In fiscal year 2014, the CMS implemented this option for newborns and patients with acute myocardial infarction by creating new discharge status codes of “discharged to [location] with a planned acute care hospital inpatient readmission.” Institutions can flag discharges that they know at the time of discharge will be followed by a readmission, such as a newborn who requires a repeat hospitalization for repair of a congenital anomaly.5 There is no time span required for the planned readmission to qualify. However, the difficulty in broadening the applicability of this option to all discharges lies in identification and matching; there also remains a possibility for gaming. The code does not specify when the readmission is expected nor for what diagnosis or procedure. How, then, do we know if the subsequent readmission is the one anticipated? Unexpected readmissions may still occur in the interim. Conversely, what if the discharging clinicians don’t know about an anticipated planned procedure? What would stop hospitals from labeling every discharge as expected to be followed by a planned readmission? These considerations have largely prevented the CMS from asking hospitals to apply the new code widely or from applying the code to identify planned readmissions.
Instead, the existing algorithm attempts to identify procedures that might be done on an elective basis and assumes readmissions with these procedures are planned if paired with a nonurgent diagnosis. Ellimoottil and colleagues attempt to verify whether this is accurate using a creative approach of seeking emergency department (ED) charges and admission type of emergent or urgent, and they found that roughly half of planned readmissions are, in fact, likely unplanned. This figure agrees closely with the original chart review validation of the algorithm. In particular, they found that some procedures, such as percutaneous cardiac interventions, appear to be paired regularly with a nonurgent principal diagnosis, such as coronary artery disease, even when done on an urgent basis.
This validation was performed prior to the availability of version 4.0 of the planned readmission algorithm, which removes several high-frequency procedures from the potentially planned readmission list (including cardiac devices and diagnostic cardiac catheterizations) that were very frequently mischaracterized as planned in the original chart validation.6 At least 8 such cases were also identified in this validation according to the table. Therefore, the misclassification rate of the current algorithm version is probably less than that reported in this article. Nonetheless, percutaneous transluminal coronary angioplasty remains on the planned procedure list in version 4.0 and appears to account for a substantial error rate, and it is likely that the authors’ approach would improve the accuracy even of the newer version of the algorithm.
The advantages of the suggested modifications are that they do not require chart review and could be readily adopted by the CMS. Although seeking ED charges for Medicare is somewhat cumbersome in that they are recorded in a different data set than the inpatient hospitalizations, there is no absolute barrier to adding this step to the algorithm, and doing so has substantial face validity. That said, identifying ED visits is not straightforward because nonemergency services can be provided in the ED (ie, critical care or observation care) and because facilities and providers have different billing requirements, producing different estimates depending on the data set used.7 Including admission type would be easier, but it would be less conservative and likely less accurate, as this field has not been validated and is not typically audited. Nonetheless, adding the presence of ED charges seems likely to improve the accuracy of the algorithm. As the CMS continues to refine the planned readmission algorithm, these proposed changes would be very reasonable to study with chart validation and, if valid, to consider adopting.
Disclosure
Dr. Horwitz reports grants from Center for Medicare & Medicaid Services, grants from Agency for Healthcare Research and Quality, during the conduct of the study.
1. Ellimoottil C, Khouri R, Dhir A, Hou H, Miller D, Dupree J. An opportunity to improve Medicare’s planned readmissions measure. J Hosp Med. 2017;12(10):840-842.
2. Horwitz LI, Grady JN, Cohen DB, et al. Development and validation of an algorithm to identify planned readmissions from claims data. J Hosp Med. 2015;10(10):670-677. PubMed
3. Benbassat J, Taragin M. Hospital readmissions as a measure of quality of health care: advantages and limitations. Arch Intern Med. 2000;160(8):1074-1081. PubMed
4. Assmann A, Boeken U, Akhyari P, Lichtenberg A. Appropriate timing of coronary artery bypass grafting after acute myocardial infarction. Thorac Cardiovasc Surg. 2012;60(7):446-451. PubMed
5. Inpatient Prospective Payment System/Long-Term Care Hospital (IPPS/LTCH) Final Rule, 78 Fed. Reg. 27520 (Aug 19, 2013) (to be codified at 42 C.F.R. Parts 424, 414, 419, 424, 482, 485 and 489). http://www.gpo.gov/fdsys/pkg/FR-2013-08-19/pdf/2013-18956.pdf. Accessed on May 4, 2017.
6. Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation. 2016 Condition-Specific Measures Updates and Specifications Report: Hospital-Level 30-Day Risk-Standardized Readmission Measures. March 2016.
7. Venkatesh AK, Mei H, Kocher KE, et al. Identification of emergency department visits in Medicare administrative claims: approaches and implications. Acad Emerg Med. 2017;24(4):422-431. PubMed
In this issue of the Journal of Hospital Medicine, Ellimoottil and colleagues examine characteristics of readmissions identified as planned by the planned readmission algorithm developed for the Center for Medicare & Medicaid Services (CMS) by using Medicare claims data from 131 hospitals in Michigan.1 They found that a substantial portion of readmissions currently classified as planned by the algorithm appear to be nonelective, as defined by the presence of a charge by an emergency medicine physician or an admission type of emergent or urgent, making those hospitalizations unlikely to be planned. They suggest that the algorithm could be modified to exclude such cases from the planned designation.
To determine whether modifying the algorithm as recommended is a good idea, it is helpful to examine the origins of the existing planned readmission algorithm. The algorithm originated as a consequence of hospital accountability measures for readmissions and was developed by this author in collaboration with colleagues at Yale University and elsewhere.2 Readmission measures have been controversial in part because clearly some (undetermined) fraction of readmissions is unavoidable. Many commentators have asked that readmission measures therefore capture only avoidable or related readmissions. Avoidable readmissions are those that could have been prevented by members of the healthcare system through actions taken during or after hospitalization, such as patient counseling, communication among team members, and guideline-concordant medical care. Related readmissions are those directly stemming from the index admission. However, reliably and accurately defining such events has proven elusive. One study, for instance, found the rate of physician-assessed preventability in published studies ranged from 9% to 48%.3 The challenge is even greater in trying to determine preventability using just claims data, without physician review of charts. Imagine, for instance, a patient with heart failure who is readmitted with heart failure exacerbation. The readmission preceded by a large fast-food meal is likely preventable; although even in this case, some would argue the healthcare system should not be held accountable for a readmission if the patient had been properly counseled about avoiding salty food. The one preceded by progressively worsening systolic function in a patient who reliably takes medications, weighs herself daily, and watches her diet is likely not. But both appear identical in claims. Related is also a difficult concept to operationalize. A recently hospitalized patient readmitted with pneumonia might have acquired it in the hospital (related) or from her grandchild 2 weeks later (unrelated). Again, both appear identical in claims.
In the ideal world, clinicians would be held accountable only for preventable readmissions. In practice, that has not proven to be possible.
Instead, the CMS readmission measures omit readmissions that are thought to be planned in advance: necessary and intentional readmissions. Defining a planned readmission is conceptually easier than defining a preventable readmission, yet even this is not always straightforward. The clearest case might be a person with a longstanding plan to have an elective surgery (say, a hip replacement) who is briefly admitted with something minor enough not to delay a subsequent admission for the scheduled surgery. Other patients are admitted with acute problems that require follow-up hospitalization (for instance, an acute myocardial infarction that requires a coronary artery bypass graft 2 weeks later).4 More ambiguous are patients who are sent home on a course of treatment with a plan for rehospitalization if it fails; for instance, a patient with gangrene is sent home on intravenous antibiotics but fails to improve and is rehospitalized for an amputation. Is that readmission planned or unplanned? Reasonable people might disagree.
Nonetheless, assuming it is desirable to at least try to identify and remove planned readmissions from measures, there are a number of ways in which one might do so. Perhaps the simplest would be to classify each hospitalization as planned or not on the UB-04 claim form. Such a process would be very feasible but also subject to gaming or coding variability. Given that there is some ambiguity and no standard about what types of readmissions are planned and that current policy provides incentives to reduce unplanned readmission rates, hospitals might vary in the cases to which they would apply such a code. This approach, therefore, has not been favored by payers to date. An alternative is to prospectively flag admissions that are expected to result in planned readmissions. In fiscal year 2014, the CMS implemented this option for newborns and patients with acute myocardial infarction by creating new discharge status codes of “discharged to [location] with a planned acute care hospital inpatient readmission.” Institutions can flag discharges that they know at the time of discharge will be followed by a readmission, such as a newborn who requires a repeat hospitalization for repair of a congenital anomaly.5 There is no time span required for the planned readmission to qualify. However, the difficulty in broadening the applicability of this option to all discharges lies in identification and matching; there also remains a possibility for gaming. The code does not specify when the readmission is expected nor for what diagnosis or procedure. How, then, do we know if the subsequent readmission is the one anticipated? Unexpected readmissions may still occur in the interim. Conversely, what if the discharging clinicians don’t know about an anticipated planned procedure? What would stop hospitals from labeling every discharge as expected to be followed by a planned readmission? These considerations have largely prevented the CMS from asking hospitals to apply the new code widely or from applying the code to identify planned readmissions.
Instead, the existing algorithm attempts to identify procedures that might be done on an elective basis and assumes readmissions with these procedures are planned if paired with a nonurgent diagnosis. Ellimoottil and colleagues attempt to verify whether this is accurate using a creative approach of seeking emergency department (ED) charges and admission type of emergent or urgent, and they found that roughly half of planned readmissions are, in fact, likely unplanned. This figure agrees closely with the original chart review validation of the algorithm. In particular, they found that some procedures, such as percutaneous cardiac interventions, appear to be paired regularly with a nonurgent principal diagnosis, such as coronary artery disease, even when done on an urgent basis.
This validation was performed prior to the availability of version 4.0 of the planned readmission algorithm, which removes several high-frequency procedures from the potentially planned readmission list (including cardiac devices and diagnostic cardiac catheterizations) that were very frequently mischaracterized as planned in the original chart validation.6 At least 8 such cases were also identified in this validation according to the table. Therefore, the misclassification rate of the current algorithm version is probably less than that reported in this article. Nonetheless, percutaneous transluminal coronary angioplasty remains on the planned procedure list in version 4.0 and appears to account for a substantial error rate, and it is likely that the authors’ approach would improve the accuracy even of the newer version of the algorithm.
The advantages of the suggested modifications are that they do not require chart review and could be readily adopted by the CMS. Although seeking ED charges for Medicare is somewhat cumbersome in that they are recorded in a different data set than the inpatient hospitalizations, there is no absolute barrier to adding this step to the algorithm, and doing so has substantial face validity. That said, identifying ED visits is not straightforward because nonemergency services can be provided in the ED (ie, critical care or observation care) and because facilities and providers have different billing requirements, producing different estimates depending on the data set used.7 Including admission type would be easier, but it would be less conservative and likely less accurate, as this field has not been validated and is not typically audited. Nonetheless, adding the presence of ED charges seems likely to improve the accuracy of the algorithm. As the CMS continues to refine the planned readmission algorithm, these proposed changes would be very reasonable to study with chart validation and, if valid, to consider adopting.
Disclosure
Dr. Horwitz reports grants from Center for Medicare & Medicaid Services, grants from Agency for Healthcare Research and Quality, during the conduct of the study.
In this issue of the Journal of Hospital Medicine, Ellimoottil and colleagues examine characteristics of readmissions identified as planned by the planned readmission algorithm developed for the Center for Medicare & Medicaid Services (CMS) by using Medicare claims data from 131 hospitals in Michigan.1 They found that a substantial portion of readmissions currently classified as planned by the algorithm appear to be nonelective, as defined by the presence of a charge by an emergency medicine physician or an admission type of emergent or urgent, making those hospitalizations unlikely to be planned. They suggest that the algorithm could be modified to exclude such cases from the planned designation.
To determine whether modifying the algorithm as recommended is a good idea, it is helpful to examine the origins of the existing planned readmission algorithm. The algorithm originated as a consequence of hospital accountability measures for readmissions and was developed by this author in collaboration with colleagues at Yale University and elsewhere.2 Readmission measures have been controversial in part because clearly some (undetermined) fraction of readmissions is unavoidable. Many commentators have asked that readmission measures therefore capture only avoidable or related readmissions. Avoidable readmissions are those that could have been prevented by members of the healthcare system through actions taken during or after hospitalization, such as patient counseling, communication among team members, and guideline-concordant medical care. Related readmissions are those directly stemming from the index admission. However, reliably and accurately defining such events has proven elusive. One study, for instance, found the rate of physician-assessed preventability in published studies ranged from 9% to 48%.3 The challenge is even greater in trying to determine preventability using just claims data, without physician review of charts. Imagine, for instance, a patient with heart failure who is readmitted with heart failure exacerbation. The readmission preceded by a large fast-food meal is likely preventable; although even in this case, some would argue the healthcare system should not be held accountable for a readmission if the patient had been properly counseled about avoiding salty food. The one preceded by progressively worsening systolic function in a patient who reliably takes medications, weighs herself daily, and watches her diet is likely not. But both appear identical in claims. Related is also a difficult concept to operationalize. A recently hospitalized patient readmitted with pneumonia might have acquired it in the hospital (related) or from her grandchild 2 weeks later (unrelated). Again, both appear identical in claims.
In the ideal world, clinicians would be held accountable only for preventable readmissions. In practice, that has not proven to be possible.
Instead, the CMS readmission measures omit readmissions that are thought to be planned in advance: necessary and intentional readmissions. Defining a planned readmission is conceptually easier than defining a preventable readmission, yet even this is not always straightforward. The clearest case might be a person with a longstanding plan to have an elective surgery (say, a hip replacement) who is briefly admitted with something minor enough not to delay a subsequent admission for the scheduled surgery. Other patients are admitted with acute problems that require follow-up hospitalization (for instance, an acute myocardial infarction that requires a coronary artery bypass graft 2 weeks later).4 More ambiguous are patients who are sent home on a course of treatment with a plan for rehospitalization if it fails; for instance, a patient with gangrene is sent home on intravenous antibiotics but fails to improve and is rehospitalized for an amputation. Is that readmission planned or unplanned? Reasonable people might disagree.
Nonetheless, assuming it is desirable to at least try to identify and remove planned readmissions from measures, there are a number of ways in which one might do so. Perhaps the simplest would be to classify each hospitalization as planned or not on the UB-04 claim form. Such a process would be very feasible but also subject to gaming or coding variability. Given that there is some ambiguity and no standard about what types of readmissions are planned and that current policy provides incentives to reduce unplanned readmission rates, hospitals might vary in the cases to which they would apply such a code. This approach, therefore, has not been favored by payers to date. An alternative is to prospectively flag admissions that are expected to result in planned readmissions. In fiscal year 2014, the CMS implemented this option for newborns and patients with acute myocardial infarction by creating new discharge status codes of “discharged to [location] with a planned acute care hospital inpatient readmission.” Institutions can flag discharges that they know at the time of discharge will be followed by a readmission, such as a newborn who requires a repeat hospitalization for repair of a congenital anomaly.5 There is no time span required for the planned readmission to qualify. However, the difficulty in broadening the applicability of this option to all discharges lies in identification and matching; there also remains a possibility for gaming. The code does not specify when the readmission is expected nor for what diagnosis or procedure. How, then, do we know if the subsequent readmission is the one anticipated? Unexpected readmissions may still occur in the interim. Conversely, what if the discharging clinicians don’t know about an anticipated planned procedure? What would stop hospitals from labeling every discharge as expected to be followed by a planned readmission? These considerations have largely prevented the CMS from asking hospitals to apply the new code widely or from applying the code to identify planned readmissions.
Instead, the existing algorithm attempts to identify procedures that might be done on an elective basis and assumes readmissions with these procedures are planned if paired with a nonurgent diagnosis. Ellimoottil and colleagues attempt to verify whether this is accurate using a creative approach of seeking emergency department (ED) charges and admission type of emergent or urgent, and they found that roughly half of planned readmissions are, in fact, likely unplanned. This figure agrees closely with the original chart review validation of the algorithm. In particular, they found that some procedures, such as percutaneous cardiac interventions, appear to be paired regularly with a nonurgent principal diagnosis, such as coronary artery disease, even when done on an urgent basis.
This validation was performed prior to the availability of version 4.0 of the planned readmission algorithm, which removes several high-frequency procedures from the potentially planned readmission list (including cardiac devices and diagnostic cardiac catheterizations) that were very frequently mischaracterized as planned in the original chart validation.6 At least 8 such cases were also identified in this validation according to the table. Therefore, the misclassification rate of the current algorithm version is probably less than that reported in this article. Nonetheless, percutaneous transluminal coronary angioplasty remains on the planned procedure list in version 4.0 and appears to account for a substantial error rate, and it is likely that the authors’ approach would improve the accuracy even of the newer version of the algorithm.
The advantages of the suggested modifications are that they do not require chart review and could be readily adopted by the CMS. Although seeking ED charges for Medicare is somewhat cumbersome in that they are recorded in a different data set than the inpatient hospitalizations, there is no absolute barrier to adding this step to the algorithm, and doing so has substantial face validity. That said, identifying ED visits is not straightforward because nonemergency services can be provided in the ED (ie, critical care or observation care) and because facilities and providers have different billing requirements, producing different estimates depending on the data set used.7 Including admission type would be easier, but it would be less conservative and likely less accurate, as this field has not been validated and is not typically audited. Nonetheless, adding the presence of ED charges seems likely to improve the accuracy of the algorithm. As the CMS continues to refine the planned readmission algorithm, these proposed changes would be very reasonable to study with chart validation and, if valid, to consider adopting.
Disclosure
Dr. Horwitz reports grants from Center for Medicare & Medicaid Services, grants from Agency for Healthcare Research and Quality, during the conduct of the study.
1. Ellimoottil C, Khouri R, Dhir A, Hou H, Miller D, Dupree J. An opportunity to improve Medicare’s planned readmissions measure. J Hosp Med. 2017;12(10):840-842.
2. Horwitz LI, Grady JN, Cohen DB, et al. Development and validation of an algorithm to identify planned readmissions from claims data. J Hosp Med. 2015;10(10):670-677. PubMed
3. Benbassat J, Taragin M. Hospital readmissions as a measure of quality of health care: advantages and limitations. Arch Intern Med. 2000;160(8):1074-1081. PubMed
4. Assmann A, Boeken U, Akhyari P, Lichtenberg A. Appropriate timing of coronary artery bypass grafting after acute myocardial infarction. Thorac Cardiovasc Surg. 2012;60(7):446-451. PubMed
5. Inpatient Prospective Payment System/Long-Term Care Hospital (IPPS/LTCH) Final Rule, 78 Fed. Reg. 27520 (Aug 19, 2013) (to be codified at 42 C.F.R. Parts 424, 414, 419, 424, 482, 485 and 489). http://www.gpo.gov/fdsys/pkg/FR-2013-08-19/pdf/2013-18956.pdf. Accessed on May 4, 2017.
6. Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation. 2016 Condition-Specific Measures Updates and Specifications Report: Hospital-Level 30-Day Risk-Standardized Readmission Measures. March 2016.
7. Venkatesh AK, Mei H, Kocher KE, et al. Identification of emergency department visits in Medicare administrative claims: approaches and implications. Acad Emerg Med. 2017;24(4):422-431. PubMed
1. Ellimoottil C, Khouri R, Dhir A, Hou H, Miller D, Dupree J. An opportunity to improve Medicare’s planned readmissions measure. J Hosp Med. 2017;12(10):840-842.
2. Horwitz LI, Grady JN, Cohen DB, et al. Development and validation of an algorithm to identify planned readmissions from claims data. J Hosp Med. 2015;10(10):670-677. PubMed
3. Benbassat J, Taragin M. Hospital readmissions as a measure of quality of health care: advantages and limitations. Arch Intern Med. 2000;160(8):1074-1081. PubMed
4. Assmann A, Boeken U, Akhyari P, Lichtenberg A. Appropriate timing of coronary artery bypass grafting after acute myocardial infarction. Thorac Cardiovasc Surg. 2012;60(7):446-451. PubMed
5. Inpatient Prospective Payment System/Long-Term Care Hospital (IPPS/LTCH) Final Rule, 78 Fed. Reg. 27520 (Aug 19, 2013) (to be codified at 42 C.F.R. Parts 424, 414, 419, 424, 482, 485 and 489). http://www.gpo.gov/fdsys/pkg/FR-2013-08-19/pdf/2013-18956.pdf. Accessed on May 4, 2017.
6. Yale New Haven Health Services Corporation Center for Outcomes Research and Evaluation. 2016 Condition-Specific Measures Updates and Specifications Report: Hospital-Level 30-Day Risk-Standardized Readmission Measures. March 2016.
7. Venkatesh AK, Mei H, Kocher KE, et al. Identification of emergency department visits in Medicare administrative claims: approaches and implications. Acad Emerg Med. 2017;24(4):422-431. PubMed
Noise and Light Pollution in the Hospital: A Call for Action
“Unnecessary noise is the most cruel abuse of care which can be inflicted on either the sick or the well.”
–Florence Nightingale1
Motivated by the “unsustainable” rise in noise pollution and its “direct, as well as cumulative, adverse health effects,” an expert World Health Organization (WHO) task force composed the Guidelines for Community Noise, outlining specific noise recommendations for public settings, including hospitals.2 In ward settings, these guidelines mandate that background noise (which is defined as unwanted sound) levels average <35 decibels (dB; ie, a typical library) during the day, average <30 dB at night, and peak no higher than 40 dB (ie, a normal conversation), a level sufficient to awaken someone from sleep.
Since the publication of these guidelines in 1999, substantial new research has added to our understanding of hospital noise levels. Recent research has demonstrated that few, if any, hospitals comply with WHO noise recommendations.3 Moreover, since 1960, hospital sound levels have risen ~4 dB per decade; based on the logarithmic decibel scale, if this trend continues, this translates to a 528% increase in loudness by 2020.3
The overwhelming majority of research on hospital noise has focused on the intensive care unit (ICU), where beeping machines and busy staff often push peak nighttime noise levels over 80 dB (ie, a kitchen blender).4 When evaluated during sleep, noise in the ICU causes frequent arousals and awakenings. When noise is combined with other factors, such as bright light and patient care interactions, poor sleep quality invariably results.4
While it has been known for years that critically ill patients experience markedly fragmented and nonrestorative sleep,5 poor sleep has recently gained attention due to its potential role as a modifiable risk factor for delirium and its associated consequences, including prolonged length of stay and long-lasting neuropsychological and physical impairments.6 Due to this interest, numerous interventions have been attempted,7 including multicomponent bundles to promote sleep,8 which have been shown to reduce delirium in the ICU.9-12 Therefore, efforts to promote sleep in the ICU, including interventions to minimize nighttime noise, are recommended in Society of Critical Care Medicine clinical practice guidelines13 and are listed as a top 5 research priority by an expert panel of ICU delirium researchers.14
In contrast to the ICU, there has been little attention paid to noise in other patient care areas. Existing studies in non-ICU ward settings suggest that excessive noise is common,3 similar to the ICU, and that patients experience poor sleep, with noise being a significant disruptor of sleep.5,15,16 Such poor sleep is thought to contribute to uncontrolled pain, labile blood pressure, and dissatisfaction with care.16,17
In this issue of the Journal of Hospital Medicine, Jaiswal and colleagues18 report on an important study evaluating sound and light levels in both non-ICU and ICU settings within a busy tertiary-care hospital. In 8 general ward, 8 telemetry, and 8 ICU patient rooms, the investigators used meters to record sound and light levels for 24 to 72 hours. In each of these locations, they detected average hourly sound levels ranging from 45 to 54 dB, 47 to 55 dB, and 56 to 60 dB, respectively, with ICUs consistently registering the highest hourly sound levels. Notably, all locations exceeded WHO noise limits at all hours of the day. As a novel measure, the investigators evaluated sound level changes (SLCs), or the difference between peak and background sound levels, based on research suggesting that dramatic SLCs (≥17.5 dB) are more disruptive than constant loud noise.19 The authors observed that SLCs ≥17.5 dB occur predominantly during daytime hours and, interestingly, at a similar rate in the wards versus the ICU.
Importantly, the authors do not link their findings with patient sleep or other patient outcomes but instead focus on employing rigorous methods to gather continuous recordings. By measuring light levels, the authors bring attention to an issue often considered less disruptive to sleep than noise.6,10,20 Similar to prior research,21 Jaiswal and colleagues demonstrate low levels of light at night, with no substantial difference between non-ICU and ICU settings. As a key finding, the authors bring attention to low levels of light during daytime hours, particularly in the morning, when levels range from 22 to 101 lux in the wards and 16 to 39 lux in the ICU. While the optimal timing and brightness of light exposure remains unknown, it is well established that ambient light is the most potent cue for circadian rhythms, with levels >100 lux necessary to suppress melatonin, the key hormone involved in circadian entrainment. Hence, the levels of morning light observed in this study were likely insufficient to maintain healthy circadian rhythms. When exposed to abnormal light levels and factors such as noise, stress, and medications, hospitalized patients are at risk for circadian rhythm misalignment, which can disrupt sleep and trigger a complex molecular cascade, leading to end-organ dysfunction including depressed immunity, glucose dysregulation, arrhythmias, and delirium.22-24
What are the major takeaway messages from this study? First, it confirms that sound levels are not only high in the ICU but also in non-ICU wards. As hospital ratings and reimbursements now rely on favorable patient ratings, future noise-reduction efforts will surely expand more vigorously across patient care areas.25 Second, SLCs and daytime recordings must be included in efforts to understand and improve sleep and circadian rhythms in hospitalized patients. Finally, this study provides a sobering reminder of the challenge of meeting WHO guidelines and facilitating an optimal healing environment for patients. Sadly, hospital sound levels continue to rise, and quiet-time interventions consistently fail to lower noise to levels anywhere near WHO limits.26 Hence, to make any progress, hospitals of the future must entertain novel design modifications (eg, sound-absorbing walls and alternative room layouts), fix common sources of noise pollution (eg, ventilation systems and alarms), and critically evaluate and update interventions aimed at improving sleep and aligning circadian rhythms for hospitalized patients.27
Acknowledgments
B.B.K. is currently supported by a grant through the University of California, Los Angeles Clinical Translational Research Institute and the National Institutes of Health’s National Center for Advancing Translational Sciences (UL1TR000124).
Disclosure
The authors have nothing to disclose.
1. Nightingale F. Notes on Nursing: What It Is, and What It Is Not. Harrison; 1860. PubMed
2. Berglund B, Lindvall T, Schwela DH. Guidelines for Community Noise. Geneva, Switzerland: World Health Organization, 1999. http://www.who.int/docstore/peh/noise/guidelines2.html. Accessed on June 23, 2017.
3. Busch-Vishniac IJ, West JE, Barnhill C, Hunter T, Orellana D, Chivukula R. Noise levels in Johns Hopkins Hospital. J Acoust Soc Am. 2005;118(6):3629-3645. PubMed
4. Kamdar BB, Needham DM, Collop NA. Sleep deprivation in critical illness: its role in physical and psychological recovery. J Intensive Care Med. 2012;27(2):97-111. PubMed
5. Knauert MP, Malik V, Kamdar BB. Sleep and sleep disordered breathing in hospitalized patients. Semin Respir Crit Care Med. 2014;35(5):582-592. PubMed
6. Kamdar BB, Knauert MP, Jones SF, et al. Perceptions and practices regarding sleep in the intensive care unit. A survey of 1,223 critical care providers. Ann Am Thorac Soc. 2016;13(8):1370-1377. PubMed
7. DuBose JR, Hadi K. Improving inpatient environments to support patient sleep. Int J Qual Health Care. 2016;28(5):540-553. PubMed
8. Kamdar BB, Kamdar BB, Needham DM. Bundling sleep promotion with delirium prevention: ready for prime time? Anaesthesia. 2014;69(6):527-531. PubMed
9. Patel J, Baldwin J, Bunting P, Laha S. The effect of a multicomponent multidisciplinary bundle of interventions on sleep and delirium in medical and surgical intensive care patients. Anaesthesia. 2014;69(6):540-549. PubMed
10. Kamdar BB, King LM, Collop NA, et al. The effect of a quality improvement intervention on perceived sleep quality and cognition in a medical ICU. Crit Care Med. 2013;41(3):800-809. PubMed
11. van de Pol I, van Iterson M, Maaskant J. Effect of nocturnal sound reduction on the incidence of delirium in intensive care unit patients: An interrupted time series analysis. Intensive Crit Care Nurs. 2017;41:18-25. PubMed
12. Flannery AH, Oyler DR, Weinhouse GL. The impact of interventions to improve sleep on delirium in the ICU: a systematic review and research framework. Crit Care Med. 2016;44(12):2231-2240. PubMed
13. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41(1):263-306. PubMed
14. Pandharipande PP, Ely EW, Arora RC, et al. The intensive care delirium research agenda: a multinational, interprofessional perspective [published online ahead of print June 13, 2017]. Intensive Care Med. PubMed
15. Topf M, Thompson S. Interactive relationships between hospital patients’ noise-induced stress and other stress with sleep. Heart Lung. 2001;30(4):237-243. PubMed
16. Tamrat R, Huynh-Le MP, Goyal M. Non-pharmacologic interventions to improve the sleep of hospitalized patients: a systematic review. J Gen Intern Med. 2014;29(5):788-795. PubMed
17. Fillary J, Chaplin H, Jones G, Thompson A, Holme A, Wilson P. Noise at night in hospital general wards: a mapping of the literature. Br J Nurs. 2015;24(10):536-540. PubMed
18. Jaiswal SJ, Garcia S, Owens RL. Sound and light levels are similarly disruptive in ICU and non-ICU wards. J Hosp Med. 2017;12(10):798-804. https://doi.org/10.12788/jhm.2826.
19. Stanchina ML, Abu-Hijleh M, Chaudhry BK, Carlisle CC, Millman RP. The influence of white noise on sleep in subjects exposed to ICU noise. Sleep Med. 2005;6(5):423-428. PubMed
20. Freedman NS, Kotzer N, Schwab RJ. Patient perception of sleep quality and etiology of sleep disruption in the intensive care unit. Am J Respir Crit Care Med. 1999;159(4, Pt 1):1155-1162. PubMed
21. Meyer TJ, Eveloff SE, Bauer MS, Schwartz WA, Hill NS, Millman RP. Adverse environmental conditions in the respiratory and medical ICU settings. Chest. 1994;105(4):1211-1216. PubMed
22. Castro R, Angus DC, Rosengart MR. The effect of light on critical illness. Crit Care. 2011;15(2):218. PubMed
23. Brainard J, Gobel M, Scott B, Koeppen M, Eckle T. Health implications of disrupted circadian rhythms and the potential for daylight as therapy. Anesthesiology. 2015;122(5):1170-1175. PubMed
24. Fitzgerald JM, Adamis D, Trzepacz PT, et al. Delirium: a disturbance of circadian integrity? Med Hypotheses. 2013;81(4):568-576. PubMed
25. Stafford A, Haverland A, Bridges E. Noise in the ICU. Am J Nurs. 2014;114(5):57-63. PubMed
26. Tainter CR, Levine AR, Quraishi SA, et al. Noise levels in surgical ICUs are consistently above recommended standards. Crit Care Med. 2016;44(1):147-152. PubMed
27. Ulrich RS, Zimring C, Zhu X, et al. A review of the research literature on evidence-based healthcare design. HERD. 2008;1(3):61-125. PubMed
“Unnecessary noise is the most cruel abuse of care which can be inflicted on either the sick or the well.”
–Florence Nightingale1
Motivated by the “unsustainable” rise in noise pollution and its “direct, as well as cumulative, adverse health effects,” an expert World Health Organization (WHO) task force composed the Guidelines for Community Noise, outlining specific noise recommendations for public settings, including hospitals.2 In ward settings, these guidelines mandate that background noise (which is defined as unwanted sound) levels average <35 decibels (dB; ie, a typical library) during the day, average <30 dB at night, and peak no higher than 40 dB (ie, a normal conversation), a level sufficient to awaken someone from sleep.
Since the publication of these guidelines in 1999, substantial new research has added to our understanding of hospital noise levels. Recent research has demonstrated that few, if any, hospitals comply with WHO noise recommendations.3 Moreover, since 1960, hospital sound levels have risen ~4 dB per decade; based on the logarithmic decibel scale, if this trend continues, this translates to a 528% increase in loudness by 2020.3
The overwhelming majority of research on hospital noise has focused on the intensive care unit (ICU), where beeping machines and busy staff often push peak nighttime noise levels over 80 dB (ie, a kitchen blender).4 When evaluated during sleep, noise in the ICU causes frequent arousals and awakenings. When noise is combined with other factors, such as bright light and patient care interactions, poor sleep quality invariably results.4
While it has been known for years that critically ill patients experience markedly fragmented and nonrestorative sleep,5 poor sleep has recently gained attention due to its potential role as a modifiable risk factor for delirium and its associated consequences, including prolonged length of stay and long-lasting neuropsychological and physical impairments.6 Due to this interest, numerous interventions have been attempted,7 including multicomponent bundles to promote sleep,8 which have been shown to reduce delirium in the ICU.9-12 Therefore, efforts to promote sleep in the ICU, including interventions to minimize nighttime noise, are recommended in Society of Critical Care Medicine clinical practice guidelines13 and are listed as a top 5 research priority by an expert panel of ICU delirium researchers.14
In contrast to the ICU, there has been little attention paid to noise in other patient care areas. Existing studies in non-ICU ward settings suggest that excessive noise is common,3 similar to the ICU, and that patients experience poor sleep, with noise being a significant disruptor of sleep.5,15,16 Such poor sleep is thought to contribute to uncontrolled pain, labile blood pressure, and dissatisfaction with care.16,17
In this issue of the Journal of Hospital Medicine, Jaiswal and colleagues18 report on an important study evaluating sound and light levels in both non-ICU and ICU settings within a busy tertiary-care hospital. In 8 general ward, 8 telemetry, and 8 ICU patient rooms, the investigators used meters to record sound and light levels for 24 to 72 hours. In each of these locations, they detected average hourly sound levels ranging from 45 to 54 dB, 47 to 55 dB, and 56 to 60 dB, respectively, with ICUs consistently registering the highest hourly sound levels. Notably, all locations exceeded WHO noise limits at all hours of the day. As a novel measure, the investigators evaluated sound level changes (SLCs), or the difference between peak and background sound levels, based on research suggesting that dramatic SLCs (≥17.5 dB) are more disruptive than constant loud noise.19 The authors observed that SLCs ≥17.5 dB occur predominantly during daytime hours and, interestingly, at a similar rate in the wards versus the ICU.
Importantly, the authors do not link their findings with patient sleep or other patient outcomes but instead focus on employing rigorous methods to gather continuous recordings. By measuring light levels, the authors bring attention to an issue often considered less disruptive to sleep than noise.6,10,20 Similar to prior research,21 Jaiswal and colleagues demonstrate low levels of light at night, with no substantial difference between non-ICU and ICU settings. As a key finding, the authors bring attention to low levels of light during daytime hours, particularly in the morning, when levels range from 22 to 101 lux in the wards and 16 to 39 lux in the ICU. While the optimal timing and brightness of light exposure remains unknown, it is well established that ambient light is the most potent cue for circadian rhythms, with levels >100 lux necessary to suppress melatonin, the key hormone involved in circadian entrainment. Hence, the levels of morning light observed in this study were likely insufficient to maintain healthy circadian rhythms. When exposed to abnormal light levels and factors such as noise, stress, and medications, hospitalized patients are at risk for circadian rhythm misalignment, which can disrupt sleep and trigger a complex molecular cascade, leading to end-organ dysfunction including depressed immunity, glucose dysregulation, arrhythmias, and delirium.22-24
What are the major takeaway messages from this study? First, it confirms that sound levels are not only high in the ICU but also in non-ICU wards. As hospital ratings and reimbursements now rely on favorable patient ratings, future noise-reduction efforts will surely expand more vigorously across patient care areas.25 Second, SLCs and daytime recordings must be included in efforts to understand and improve sleep and circadian rhythms in hospitalized patients. Finally, this study provides a sobering reminder of the challenge of meeting WHO guidelines and facilitating an optimal healing environment for patients. Sadly, hospital sound levels continue to rise, and quiet-time interventions consistently fail to lower noise to levels anywhere near WHO limits.26 Hence, to make any progress, hospitals of the future must entertain novel design modifications (eg, sound-absorbing walls and alternative room layouts), fix common sources of noise pollution (eg, ventilation systems and alarms), and critically evaluate and update interventions aimed at improving sleep and aligning circadian rhythms for hospitalized patients.27
Acknowledgments
B.B.K. is currently supported by a grant through the University of California, Los Angeles Clinical Translational Research Institute and the National Institutes of Health’s National Center for Advancing Translational Sciences (UL1TR000124).
Disclosure
The authors have nothing to disclose.
“Unnecessary noise is the most cruel abuse of care which can be inflicted on either the sick or the well.”
–Florence Nightingale1
Motivated by the “unsustainable” rise in noise pollution and its “direct, as well as cumulative, adverse health effects,” an expert World Health Organization (WHO) task force composed the Guidelines for Community Noise, outlining specific noise recommendations for public settings, including hospitals.2 In ward settings, these guidelines mandate that background noise (which is defined as unwanted sound) levels average <35 decibels (dB; ie, a typical library) during the day, average <30 dB at night, and peak no higher than 40 dB (ie, a normal conversation), a level sufficient to awaken someone from sleep.
Since the publication of these guidelines in 1999, substantial new research has added to our understanding of hospital noise levels. Recent research has demonstrated that few, if any, hospitals comply with WHO noise recommendations.3 Moreover, since 1960, hospital sound levels have risen ~4 dB per decade; based on the logarithmic decibel scale, if this trend continues, this translates to a 528% increase in loudness by 2020.3
The overwhelming majority of research on hospital noise has focused on the intensive care unit (ICU), where beeping machines and busy staff often push peak nighttime noise levels over 80 dB (ie, a kitchen blender).4 When evaluated during sleep, noise in the ICU causes frequent arousals and awakenings. When noise is combined with other factors, such as bright light and patient care interactions, poor sleep quality invariably results.4
While it has been known for years that critically ill patients experience markedly fragmented and nonrestorative sleep,5 poor sleep has recently gained attention due to its potential role as a modifiable risk factor for delirium and its associated consequences, including prolonged length of stay and long-lasting neuropsychological and physical impairments.6 Due to this interest, numerous interventions have been attempted,7 including multicomponent bundles to promote sleep,8 which have been shown to reduce delirium in the ICU.9-12 Therefore, efforts to promote sleep in the ICU, including interventions to minimize nighttime noise, are recommended in Society of Critical Care Medicine clinical practice guidelines13 and are listed as a top 5 research priority by an expert panel of ICU delirium researchers.14
In contrast to the ICU, there has been little attention paid to noise in other patient care areas. Existing studies in non-ICU ward settings suggest that excessive noise is common,3 similar to the ICU, and that patients experience poor sleep, with noise being a significant disruptor of sleep.5,15,16 Such poor sleep is thought to contribute to uncontrolled pain, labile blood pressure, and dissatisfaction with care.16,17
In this issue of the Journal of Hospital Medicine, Jaiswal and colleagues18 report on an important study evaluating sound and light levels in both non-ICU and ICU settings within a busy tertiary-care hospital. In 8 general ward, 8 telemetry, and 8 ICU patient rooms, the investigators used meters to record sound and light levels for 24 to 72 hours. In each of these locations, they detected average hourly sound levels ranging from 45 to 54 dB, 47 to 55 dB, and 56 to 60 dB, respectively, with ICUs consistently registering the highest hourly sound levels. Notably, all locations exceeded WHO noise limits at all hours of the day. As a novel measure, the investigators evaluated sound level changes (SLCs), or the difference between peak and background sound levels, based on research suggesting that dramatic SLCs (≥17.5 dB) are more disruptive than constant loud noise.19 The authors observed that SLCs ≥17.5 dB occur predominantly during daytime hours and, interestingly, at a similar rate in the wards versus the ICU.
Importantly, the authors do not link their findings with patient sleep or other patient outcomes but instead focus on employing rigorous methods to gather continuous recordings. By measuring light levels, the authors bring attention to an issue often considered less disruptive to sleep than noise.6,10,20 Similar to prior research,21 Jaiswal and colleagues demonstrate low levels of light at night, with no substantial difference between non-ICU and ICU settings. As a key finding, the authors bring attention to low levels of light during daytime hours, particularly in the morning, when levels range from 22 to 101 lux in the wards and 16 to 39 lux in the ICU. While the optimal timing and brightness of light exposure remains unknown, it is well established that ambient light is the most potent cue for circadian rhythms, with levels >100 lux necessary to suppress melatonin, the key hormone involved in circadian entrainment. Hence, the levels of morning light observed in this study were likely insufficient to maintain healthy circadian rhythms. When exposed to abnormal light levels and factors such as noise, stress, and medications, hospitalized patients are at risk for circadian rhythm misalignment, which can disrupt sleep and trigger a complex molecular cascade, leading to end-organ dysfunction including depressed immunity, glucose dysregulation, arrhythmias, and delirium.22-24
What are the major takeaway messages from this study? First, it confirms that sound levels are not only high in the ICU but also in non-ICU wards. As hospital ratings and reimbursements now rely on favorable patient ratings, future noise-reduction efforts will surely expand more vigorously across patient care areas.25 Second, SLCs and daytime recordings must be included in efforts to understand and improve sleep and circadian rhythms in hospitalized patients. Finally, this study provides a sobering reminder of the challenge of meeting WHO guidelines and facilitating an optimal healing environment for patients. Sadly, hospital sound levels continue to rise, and quiet-time interventions consistently fail to lower noise to levels anywhere near WHO limits.26 Hence, to make any progress, hospitals of the future must entertain novel design modifications (eg, sound-absorbing walls and alternative room layouts), fix common sources of noise pollution (eg, ventilation systems and alarms), and critically evaluate and update interventions aimed at improving sleep and aligning circadian rhythms for hospitalized patients.27
Acknowledgments
B.B.K. is currently supported by a grant through the University of California, Los Angeles Clinical Translational Research Institute and the National Institutes of Health’s National Center for Advancing Translational Sciences (UL1TR000124).
Disclosure
The authors have nothing to disclose.
1. Nightingale F. Notes on Nursing: What It Is, and What It Is Not. Harrison; 1860. PubMed
2. Berglund B, Lindvall T, Schwela DH. Guidelines for Community Noise. Geneva, Switzerland: World Health Organization, 1999. http://www.who.int/docstore/peh/noise/guidelines2.html. Accessed on June 23, 2017.
3. Busch-Vishniac IJ, West JE, Barnhill C, Hunter T, Orellana D, Chivukula R. Noise levels in Johns Hopkins Hospital. J Acoust Soc Am. 2005;118(6):3629-3645. PubMed
4. Kamdar BB, Needham DM, Collop NA. Sleep deprivation in critical illness: its role in physical and psychological recovery. J Intensive Care Med. 2012;27(2):97-111. PubMed
5. Knauert MP, Malik V, Kamdar BB. Sleep and sleep disordered breathing in hospitalized patients. Semin Respir Crit Care Med. 2014;35(5):582-592. PubMed
6. Kamdar BB, Knauert MP, Jones SF, et al. Perceptions and practices regarding sleep in the intensive care unit. A survey of 1,223 critical care providers. Ann Am Thorac Soc. 2016;13(8):1370-1377. PubMed
7. DuBose JR, Hadi K. Improving inpatient environments to support patient sleep. Int J Qual Health Care. 2016;28(5):540-553. PubMed
8. Kamdar BB, Kamdar BB, Needham DM. Bundling sleep promotion with delirium prevention: ready for prime time? Anaesthesia. 2014;69(6):527-531. PubMed
9. Patel J, Baldwin J, Bunting P, Laha S. The effect of a multicomponent multidisciplinary bundle of interventions on sleep and delirium in medical and surgical intensive care patients. Anaesthesia. 2014;69(6):540-549. PubMed
10. Kamdar BB, King LM, Collop NA, et al. The effect of a quality improvement intervention on perceived sleep quality and cognition in a medical ICU. Crit Care Med. 2013;41(3):800-809. PubMed
11. van de Pol I, van Iterson M, Maaskant J. Effect of nocturnal sound reduction on the incidence of delirium in intensive care unit patients: An interrupted time series analysis. Intensive Crit Care Nurs. 2017;41:18-25. PubMed
12. Flannery AH, Oyler DR, Weinhouse GL. The impact of interventions to improve sleep on delirium in the ICU: a systematic review and research framework. Crit Care Med. 2016;44(12):2231-2240. PubMed
13. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41(1):263-306. PubMed
14. Pandharipande PP, Ely EW, Arora RC, et al. The intensive care delirium research agenda: a multinational, interprofessional perspective [published online ahead of print June 13, 2017]. Intensive Care Med. PubMed
15. Topf M, Thompson S. Interactive relationships between hospital patients’ noise-induced stress and other stress with sleep. Heart Lung. 2001;30(4):237-243. PubMed
16. Tamrat R, Huynh-Le MP, Goyal M. Non-pharmacologic interventions to improve the sleep of hospitalized patients: a systematic review. J Gen Intern Med. 2014;29(5):788-795. PubMed
17. Fillary J, Chaplin H, Jones G, Thompson A, Holme A, Wilson P. Noise at night in hospital general wards: a mapping of the literature. Br J Nurs. 2015;24(10):536-540. PubMed
18. Jaiswal SJ, Garcia S, Owens RL. Sound and light levels are similarly disruptive in ICU and non-ICU wards. J Hosp Med. 2017;12(10):798-804. https://doi.org/10.12788/jhm.2826.
19. Stanchina ML, Abu-Hijleh M, Chaudhry BK, Carlisle CC, Millman RP. The influence of white noise on sleep in subjects exposed to ICU noise. Sleep Med. 2005;6(5):423-428. PubMed
20. Freedman NS, Kotzer N, Schwab RJ. Patient perception of sleep quality and etiology of sleep disruption in the intensive care unit. Am J Respir Crit Care Med. 1999;159(4, Pt 1):1155-1162. PubMed
21. Meyer TJ, Eveloff SE, Bauer MS, Schwartz WA, Hill NS, Millman RP. Adverse environmental conditions in the respiratory and medical ICU settings. Chest. 1994;105(4):1211-1216. PubMed
22. Castro R, Angus DC, Rosengart MR. The effect of light on critical illness. Crit Care. 2011;15(2):218. PubMed
23. Brainard J, Gobel M, Scott B, Koeppen M, Eckle T. Health implications of disrupted circadian rhythms and the potential for daylight as therapy. Anesthesiology. 2015;122(5):1170-1175. PubMed
24. Fitzgerald JM, Adamis D, Trzepacz PT, et al. Delirium: a disturbance of circadian integrity? Med Hypotheses. 2013;81(4):568-576. PubMed
25. Stafford A, Haverland A, Bridges E. Noise in the ICU. Am J Nurs. 2014;114(5):57-63. PubMed
26. Tainter CR, Levine AR, Quraishi SA, et al. Noise levels in surgical ICUs are consistently above recommended standards. Crit Care Med. 2016;44(1):147-152. PubMed
27. Ulrich RS, Zimring C, Zhu X, et al. A review of the research literature on evidence-based healthcare design. HERD. 2008;1(3):61-125. PubMed
1. Nightingale F. Notes on Nursing: What It Is, and What It Is Not. Harrison; 1860. PubMed
2. Berglund B, Lindvall T, Schwela DH. Guidelines for Community Noise. Geneva, Switzerland: World Health Organization, 1999. http://www.who.int/docstore/peh/noise/guidelines2.html. Accessed on June 23, 2017.
3. Busch-Vishniac IJ, West JE, Barnhill C, Hunter T, Orellana D, Chivukula R. Noise levels in Johns Hopkins Hospital. J Acoust Soc Am. 2005;118(6):3629-3645. PubMed
4. Kamdar BB, Needham DM, Collop NA. Sleep deprivation in critical illness: its role in physical and psychological recovery. J Intensive Care Med. 2012;27(2):97-111. PubMed
5. Knauert MP, Malik V, Kamdar BB. Sleep and sleep disordered breathing in hospitalized patients. Semin Respir Crit Care Med. 2014;35(5):582-592. PubMed
6. Kamdar BB, Knauert MP, Jones SF, et al. Perceptions and practices regarding sleep in the intensive care unit. A survey of 1,223 critical care providers. Ann Am Thorac Soc. 2016;13(8):1370-1377. PubMed
7. DuBose JR, Hadi K. Improving inpatient environments to support patient sleep. Int J Qual Health Care. 2016;28(5):540-553. PubMed
8. Kamdar BB, Kamdar BB, Needham DM. Bundling sleep promotion with delirium prevention: ready for prime time? Anaesthesia. 2014;69(6):527-531. PubMed
9. Patel J, Baldwin J, Bunting P, Laha S. The effect of a multicomponent multidisciplinary bundle of interventions on sleep and delirium in medical and surgical intensive care patients. Anaesthesia. 2014;69(6):540-549. PubMed
10. Kamdar BB, King LM, Collop NA, et al. The effect of a quality improvement intervention on perceived sleep quality and cognition in a medical ICU. Crit Care Med. 2013;41(3):800-809. PubMed
11. van de Pol I, van Iterson M, Maaskant J. Effect of nocturnal sound reduction on the incidence of delirium in intensive care unit patients: An interrupted time series analysis. Intensive Crit Care Nurs. 2017;41:18-25. PubMed
12. Flannery AH, Oyler DR, Weinhouse GL. The impact of interventions to improve sleep on delirium in the ICU: a systematic review and research framework. Crit Care Med. 2016;44(12):2231-2240. PubMed
13. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41(1):263-306. PubMed
14. Pandharipande PP, Ely EW, Arora RC, et al. The intensive care delirium research agenda: a multinational, interprofessional perspective [published online ahead of print June 13, 2017]. Intensive Care Med. PubMed
15. Topf M, Thompson S. Interactive relationships between hospital patients’ noise-induced stress and other stress with sleep. Heart Lung. 2001;30(4):237-243. PubMed
16. Tamrat R, Huynh-Le MP, Goyal M. Non-pharmacologic interventions to improve the sleep of hospitalized patients: a systematic review. J Gen Intern Med. 2014;29(5):788-795. PubMed
17. Fillary J, Chaplin H, Jones G, Thompson A, Holme A, Wilson P. Noise at night in hospital general wards: a mapping of the literature. Br J Nurs. 2015;24(10):536-540. PubMed
18. Jaiswal SJ, Garcia S, Owens RL. Sound and light levels are similarly disruptive in ICU and non-ICU wards. J Hosp Med. 2017;12(10):798-804. https://doi.org/10.12788/jhm.2826.
19. Stanchina ML, Abu-Hijleh M, Chaudhry BK, Carlisle CC, Millman RP. The influence of white noise on sleep in subjects exposed to ICU noise. Sleep Med. 2005;6(5):423-428. PubMed
20. Freedman NS, Kotzer N, Schwab RJ. Patient perception of sleep quality and etiology of sleep disruption in the intensive care unit. Am J Respir Crit Care Med. 1999;159(4, Pt 1):1155-1162. PubMed
21. Meyer TJ, Eveloff SE, Bauer MS, Schwartz WA, Hill NS, Millman RP. Adverse environmental conditions in the respiratory and medical ICU settings. Chest. 1994;105(4):1211-1216. PubMed
22. Castro R, Angus DC, Rosengart MR. The effect of light on critical illness. Crit Care. 2011;15(2):218. PubMed
23. Brainard J, Gobel M, Scott B, Koeppen M, Eckle T. Health implications of disrupted circadian rhythms and the potential for daylight as therapy. Anesthesiology. 2015;122(5):1170-1175. PubMed
24. Fitzgerald JM, Adamis D, Trzepacz PT, et al. Delirium: a disturbance of circadian integrity? Med Hypotheses. 2013;81(4):568-576. PubMed
25. Stafford A, Haverland A, Bridges E. Noise in the ICU. Am J Nurs. 2014;114(5):57-63. PubMed
26. Tainter CR, Levine AR, Quraishi SA, et al. Noise levels in surgical ICUs are consistently above recommended standards. Crit Care Med. 2016;44(1):147-152. PubMed
27. Ulrich RS, Zimring C, Zhu X, et al. A review of the research literature on evidence-based healthcare design. HERD. 2008;1(3):61-125. PubMed
© 2017 Society of Hospital Medicine