User login
Outcomes After Peripheral Nerve Block in Hip Arthroscopy
ABSTRACT
Pain control following hip arthroscopy presents a significant clinical challenge, with postoperative pain requiring considerable opioid use. Peripheral nerve blocks (PNBs) have emerged as one option to improve pain and limit the consequences of opioid use. The purpose of this study is to provide a comprehensive review of outcomes associated with PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications. A systematic review of PubMed, Medline, Scopus, and Embase databases was conducted through January 2015 for English-language articles reporting outcome data, with 2 reviewers independently reviewing studies for inclusion. When available, similar outcomes were combined to generate frequency-weighted means. Six studies met the inclusion criteria for this review, reporting on 710 patients undergoing hip arthroscopy. The mean ages were 37.0 and 37.7 years for the PNB and comparator groups, respectively, with a reported total of 281 (40.5%) male and 412 (59.5%) female patients. Postoperative post-anesthesia care unit (PACU) pain was consistently reduced in the PNB group, with the use of a lower morphine equivalent dose and lower rates of inpatient admission, compared with that in the control groups. Postoperative nausea and/or vomiting as well as PACU discharge time showed mixed results. High satisfaction and few complications were reported. In conclusion, PNB is associated with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions, though similar rates of nausea/vomiting and time to discharge were reported. Current PNB techniques are varied, and future research efforts should focus on examining which of these methods provides the optimal risk-benefit profile in hip arthroscopy.
Continue to: Hip arthroscopy has emerged...
Hip arthroscopy has emerged as a useful procedure in the diagnosis and treatment of hip pathology,1-8 experiencing a substantial rise in popularity in recent years, with the number of procedures growing by a factor of 18 from 1999 to 20099 and 25 from 2006 to 2013.10 Though hip arthroscopy is beneficial in many cases, marked postoperative pain has presented a substantial challenge, with patients requiring considerable doses of opiate-based medications in the post-anesthesia care unit (PACU).11,12 Increased narcotic use carries increased side effects, including postoperative nausea and vomiting,13 and poorly managed pain leads to increased unplanned admissions.14 Furthermore, patients with chronic hip pain and long-term opioid use may experience heightened and prolonged pain following the procedure, owing to medication tolerance and reduced opioid efficacy in this setting.15
Several pain control strategies have been employed in patients undergoing hip arthroscopy. General anesthesia16,17 and combined spinal epidural (CSE)18 are commonly used. However, such techniques rely heavily on opioids for postoperative pain control,11 and epidural anesthesia commonly requires adjunctive treatments (eg, neuromuscular blockade) to ensure muscle relaxation for joint distraction.19 One technique that has been employed recently is peripheral nerve block (PNB), which has been associated with a significant decrease in postoperative opioid use and nausea and vomiting.13,20 This method has proven successful in other fields of arthroscopy, including shoulder arthroscopy, in which it resulted in faster recovery, reduced opioid consumption,21 and demonstrated cost-effectiveness22 compared with general anesthesia and knee arthroscopy.23-26 As it is a relatively new field, little is known about the use of PNB in hip arthroscopy.
The goal of this systematic review was to comprehensively review the studies reporting on PNB in hip arthroscopy. We specifically focused on outcomes, including postoperative pain; analgesic use; nausea, vomiting, and antiemetic use; discharge time; inpatient admission; and patient satisfaction, as well as the complications associated with the use of PNB. Our knowledge of outcomes associated with PNB in hip arthroscopy is based on a few individual studies that have reported on small groups of patients using a variety of outcome measures and other findings. Furthermore, each of these studies commonly reflects the experience of an individual surgeon at a single institution and, when taken alone, may not be an accurate representation of the more general outcomes associated with PNB. A comprehensive review of such studies will provide surgeons, anesthesiologists, and patients with a better understanding of the anticipated outcomes of using PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications.
MATERIALS AND METHODS
A systematic review of outcomes associated with PNB in hip arthroscopy was performed using the available English-language literature in accordance with the guidelines laid out by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement and included studies retrieved from the PubMed, Medline, Scopus, and Embase computerized literature databases. Searches were executed comprising all years from database inception through January 2015. Articles were retrieved by an electronic search of medical subject headings and keyword terms and their respective combinations (Table 1). The inclusion criteria for studies in this systematic review were studies that (1) were written in the English language and (2) reported explicit outcome data. The exclusion criteria were (1) review articles, meta-analyses, case reports, abstracts/conference papers, comments/letters, or technique articles without reported patient data and (2) basic research, biomechanics, or animal/cadaveric studies without reported patient data.
Table 1. Search Terms Entered to Identify English-Language Studies Through January 2015
Database | Search terms |
PubMed, Scopus | Keyword: (hip AND arthroscopy) AND (pain control OR pain management OR pain regimen OR nerve block OR spinal anesthesia OR regional anesthesia OR general anesthesia) |
Medline | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “Anesthesia, General” OR “Anesthesia” OR “Anesthesia, Inhalation”, OR “Balanced Anesthesia” OR “Anesthesia, Local” OR “Anesthesia, Spinal” OR “Anesthesia, Conduction” OR “Nerve Block”) |
Embase | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “General Anesthesia” OR “Anesthesia” OR “Inhalation Anesthesia”, OR “Balanced Anesthesia” OR “Local Anesthesia” OR “Spinal Anesthesia” OR “Regional Anesthesia” OR “Nerve Block”) |
The literature search strategy is outlined in the Figure. The initial title search yielded a subset of possible articles that were then further included or excluded on the basis of the contents of the article’s abstract, wherein articles were again selected on the basis of the aforementioned inclusion and exclusion criteria. Articles selected in both the title and abstract phases underwent full-text review, during which the full text of each qualifying article was reviewed. In addition, the reference sections from articles undergoing full-text review were scanned to identify any additional studies that had not been identified in the original literature search. Appropriate studies for final inclusion were then selected at this stage. The title, abstract, and full-text selection process were performed by 2 of the study authors (Dr. Steinhaus and Dr. Lynch), with any discrepancies being discussed and resolved by mutual agreement.
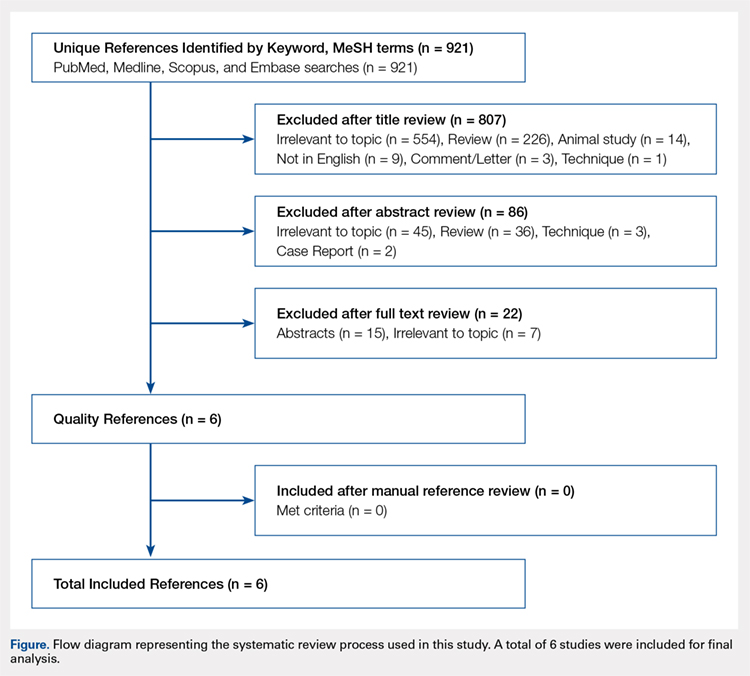
Continue to: For all 6 included studies...
For all 6 included studies,16-18,27-29 data were collected regarding the study specifics, patients included, and outcomes measured in the study. The journal of publication, type of study, level of evidence, and type of PNB, as well as the presence of a comparator group were noted (Table 2). Patient information included the number of patients at baseline and follow-up, mean age, gender, weight, height, body mass index, American Society of Anesthesiologists (ASA) status, and the specific procedures performed. In addition, data were collected on outcomes, including postoperative pain, as well as secondary outcomes and additional findings reported by the studies (Table 3). Where possible, weighted averages were calculated across all studies to obtain aggregate data.
RESULTS
STUDY INCLUSION
Six studies, all published between 2012 and 2014, were included in this systematic review (Table 2). Three studies involved lumbar plexus block, 2 studies involved femoral nerve block, and 1 study evaluated fascia iliaca block. Two studies used a control group of patients who received only general anesthesia (compared with the treatment group who received both general anesthesia and PNB); another study compared intravenous morphine with PNB; and 1 study compared CSE alone with PNB in addition to epidural.
DEMOGRAPHIC DATA
Demographic data from the included studies are presented in Table 2. In total, 710 and 549 patients were evaluated at baseline and final follow-up, respectively, which represents a follow-up rate of 77%. The frequency-weighted mean age of patients receiving PNB was 37.0 years compared with 37.7 years in the comparison groups, and the studies reported a total of 281 (40.5%) male and 412 (59.5%) female patients. The procedures performed were heterogeneously reported; therefore, totals were not tabulated, although the reported procedures included osteochondroplasty, labral débridement, labral and/or capsular repair, gluteus minimus repair, and synovectomy.
POSTOPERATIVE PAIN
Four studies reported on postoperative pain, and these data are presented in Table 3. In a retrospective study of patients receiving femoral nerve block in addition to general anesthesia, Dold and colleagues16 noted postoperative pain at 0, 15, 30, 45, and 60 minutes following arrival in the PACU, and discovered a statistically significantly lower level of pain at 60 minutes compared with inpatients receiving general anesthesia alone. YaDeau and colleagues18 found a significantly lower level of pain at rest in the PACU for those receiving CSE and lumbar plexus blockade compared with those receiving CSE alone. This significant difference did not persist at 24 hours or 6 months after the procedure, nor did it exist for pain with movement at any time point. Similarly, Schroeder and colleagues17 examined patients receiving general anesthesia and lumbar plexus block and found a significant reduction in pain immediately postoperatively in the PACU, though these effects disappeared the day following the procedure. Krych and colleagues27 also reported on postoperative pain in patients undergoing fascia iliaca blockade, although they did not include a comparator group. Outcome comparison between patients who received PNB and controls in the PACU and 1 day following the procedure are presented in Table 4.
ANALGESIC USE
Four studies reported on analgesic use after PNB, and these data are presented in Table 3. Dold and colleagues16 noted analgesic use intraoperatively, in the PACU, and in the surgical day care unit (SDCU). These authors found a significant reduction in morphine equivalent dose given in the operating room and in the PACU in the group receiving PNB, with a nonsignificant trend toward lower use of oxycodone in the SDCU. Schroeder and colleagues17 similarly reported significant reductions in morphine equivalent dose intraoperatively and in Phase I recovery for patients receiving PNB, and these differences disappeared in Phase II recovery as well as intraoperatively if the block dose was considered. In addition, these authors found a significant reduction in the use of fentanyl and hydromorphone in the operating room in the PNB group, as well as a significant reduction in the proportion of patients receiving ketorolac in the operating room or PACU. Finally, YaDeau and colleagues18 reported total analgesic usage in the PACU among PNB patients compared with those receiving CSE alone and showed a strong trend toward reduced use in the PNB group, although this difference was not significant (P = .051). PACU analgesic use is presented in Table 4.
Continue to: Postoperative nausea...
POSTOPERATIVE NAUSEA/VOMITING AND ANTIEMETIC USE
Five studies presented data on nausea, vomiting, or antiemetic use following PNB and are shown in Table 3. YaDeau and colleagues18 reported nausea among 34% of patients in the PNB group, compared with 20% in the control group, vomiting in 2% and 7%, respectively, and antiemetic use in 12% of both groups. Dold and colleagues16 identified a similar trend, with 41.1% of patients in the PNB group and 32.5% of patients in the control group experiencing postoperative nausea or vomiting, while Krych and colleagues27 noted only 10% of PNB patients with mild nausea and none requiring antiemetic use. In their study of patients receiving PNB, Schroeder and colleagues17 found a significant reduction in antiemetic use among PNB patients compared with those receiving general anesthesia alone. Similarly, Ward and colleagues29 noted a significant difference in postoperative nausea, with 10% of patients in the PNB group experiencing postoperative nausea compared with 75% of those in the comparator group who received intravenous morphine. The mean percentage of patients experiencing postoperative nausea and/or vomiting is shown in Table 4.
DISCHARGE TIME
Four studies presented data on discharge time from the PACU and are summarized in Table 3. Three of these studies included a comparator group. Both Dold and colleagues16 and YaDeau and colleagues18 reported an increase in the time to discharge for patients receiving PNB, although these differences were not significant. The study by Ward and colleagues,29 on the other hand, noted a significant reduction in the time to discharge for the PNB group. In addition to these studies, Krych and colleagues27 examined the time from skin closure to discharge for patients receiving PNB, noting a mean 199 minutes for the patients in their study. Mean times to discharge for the PNB and control groups are presented in Table 4.
INPATIENT ADMISSION
Four studies presented data on the proportion of study participants who were admitted as inpatients, and these data are shown in Table 3. Dold and colleagues16 reported no inpatient admissions in their PNB group compared with 5.0% for the control group (both cases of pain control), while YaDeau and colleagues18 found that 3 admissions occurred, with 2 in the control group (1 for oxygen desaturation and the other for intractable pain and nausea) and 1 from the PNB group (epidural spread and urinary retention). Two additional studies reported data on PNB groups alone. Krych and colleagues27 observed no overnight admissions in their study, while Nye and colleagues28 reported 1 readmission for bilateral leg numbness and weakness due to epidural spread, which resolved following discontinuation of the block. The mean proportion of inpatient admissions is presented in Table 4.
SATISFACTION
A total of 3 studies examined patient satisfaction, and these data are presented in Table 3. In their study, Ward and colleagues29 reported a significantly greater rate of satisfaction at 1 day postoperatively among the patients in the PNB group (90%) than among patients who received intravenous morphine (25%) (P < .0001). Similarly, YaDeau and colleagues18 noted greater satisfaction among the PNB group than among the control group, with PNB patients rating their satisfaction at a mean of 8.6 and control patients at a mean of 7.9 on a 10-point scale (0-10) 24 hours postoperatively, although this difference was not significant. Finally, Krych and colleagues27 found that 67% of patients were “very satisfied” and 33% were “satisfied”, based on a Likert scale.
COMPLICATIONS
Four studies presented data on complications, and these findings are summarized in Table 3. In their work, Nye and colleagues28 reported most extensively on complications associated with PNB. Overall, the authors found a rate of significant complications of 3.8%. In terms of specific complications, they noted local anesthetic systemic toxicity (0.9%), epidural spread (0.5%), sensory or motor deficits (9.4%), falls (0.5%), and catheter issues. In their study of patients receiving PNB and CSE, YaDeau and colleagues18 identified 1 patient in the PNB group with epidural spread and urinary retention, while they noted 1 case of oxygen desaturation and another case of intractable pain and nausea in the group receiving CSE alone, all 3 of which required inpatient admission. They found no permanent adverse events attributable to the PNB. In another study, Dold and colleagues16 observed no complications in patients receiving PNB compared with those in 2 admissions in the control group for inadequate pain control. Similarly, Krych and colleagues27 identified no complications in patients who received PNB in their study.
DISCUSSION
Hip arthroscopy has experienced a substantial gain in popularity in recent years, emerging as a beneficial technique for both the diagnosis and treatment of diverse hip pathologies in patients spanning a variety of demographics. Nevertheless, postoperative pain control, as well as medication side effects and unwanted patient admissions, present major challenges to the treating surgeon. As an adjuvant measure, peripheral nerve block represents one option to improve postoperative pain management, while at the same time addressing the adverse effects of considerable opioid use, which is commonly seen in these patients. Early experience with this method in hip arthroscopy was reported in a case series by Lee and colleagues.12 In an attempt to reduce postoperative pain, as well as limit the adverse effects and delay in discharge associated with considerable opioid use in the PACU, the authors used preoperative paravertebral blocks of L1 and L2 in 2 patients requiring hip arthroscopy with encouraging results. Since then, a number of studies have attempted the use of PNB in hip arthroscopy.16-18,27-29 However, we were unable to identify any prior reviews reporting on peripheral nerve blockade in hip arthroscopy, and thus this study is unique in providing a greater understanding of the outcomes associated with PNB use.
In general, we found that PNB was associated with improved outcomes. Based on the studies included in this review, there was a statistically significantly lower level of pain in the PACU for femoral nerve block (compared with general anesthesia alone)16 and lumbar plexus blockade (compared with general anesthesia17 and CSE18 alone). Nevertheless, these effects are likely short-lived, with differences disappearing the day following the procedure. In terms of analgesic use, 2 studies report significant reductions in analgesic use intraoperatively and in the PACU/Phase I recovery,16,17 with a third reporting a strong trend toward reduced analgesic use in the PACU (P = .051).18 Finally, we report fewer admissions for the PNB group, as well as high rates of satisfaction and few complications across these studies.
Continue to: Unlike these measures...
Unlike these measures, postoperative nausea, vomiting, and antiemetic use, as well as time to discharge, showed more mixed results. With regard to nausea/vomiting, 2 studies16,18 reported nonsignificantly increased rates in the PNB group, whereas others reported significant reductions in nausea/vomiting29 and in the proportion of patients receiving antiemetics.17 Similarly, mixed results were seen in terms of patient discharge time from the PACU. Two studies16,18 reported a nonsignificant increase in time to discharge for the PNB group, while another29 noted a significant reduction for the PNB group compared with those receiving intravenous morphine. These mixed results were surprising, as we expected reductions in opioid use to result in fewer instances of nausea/vomiting and a quicker time to discharge. The reasons underlying these findings are not clear, although it has been suggested that current discharge guidelines and clinical pathways limit the ability to take advantage of the accelerated timeline offered by regional anesthesia.16,30 As experience with PNB grows, our guidelines and pathways are likely to adapt to capitalize on these advantages, and future studies may show more reliable improvements in these measures.
While rare, the risk of bleeding requiring blood transfusion following hip arthroscopy is one of the most common complications of this procedure. Though the studies included in this review did not report on the need for transfusion, a recent study by Cvetanovich and colleagues10 used a national database and found that, of patients undergoing hip arthroscopy (n = 1338), 0.4% (n = 5) had bleeding requiring a transfusion, with 0.3% (n = 4) requiring return to the operating room, similar to an earlier study by Clarke and colleagues,31 who noted bleeding from the portal site in 0.4% of hip arthroscopy patients. In terms of risk factors, Cvetanovich and colleagues10 found that ASA class, older age, and prior cardiac surgery were significantly associated with minor and overall complications, whereas both regional anesthesia/monitored anesthesia care and alcohol consumption of >2 drinks a day were significantly associated with minor complications, including bleeding requiring transfusions. They noted, however, that these risk factors accounted for only 5% of the variance in complication rates, indicating that other unidentified variables better explained the variance in complication rates. These authors concluded that complications associated with hip arthroscopy are so rare that we may not be able to predict which risk factors or anesthesia types are more likely to cause them. Further characterization of bleeding following hip arthroscopy and its associated risk factors is a valuable area for future research.
LIMITATIONS
Our study contains a number of limitations. This review included studies whose level of evidence varied from I to IV; therefore, our study is limited by any bias or heterogeneity introduced in patient recruitment, selection, variability of technique, data collection, and analysis used in these studies. This heterogeneity is most apparent in the block types and comparator groups. Furthermore, several different outcome measures were reported across the 6 studies used in this review, which decreased the relevance of any one of these individual outcomes. Finally, given the limited data that currently exist for the use of PNB in hip arthroscopy, we are unable to note meaningful differences between various types of PNBs, such as differences in postoperative pain or other measures such as quadriceps weakness, which can accompany femoral nerve block.12 While it is important to read our work with these limitations in mind, this systematic review is, to our knowledge, the only comprehensive review to date of studies reporting on PNB in hip arthroscopy, providing clinicians and patients with a greater understanding of the associated outcomes across these studies.
CONCLUSION
This systematic review shows improved outcomes and few complications with PNB use in hip arthroscopy, with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions. Although opioid use was reduced in these studies, we found similar rates of postoperative nausea/vomiting as well as similar time to discharge from the PACU, which may reflect our continued reliance on outdated discharge guidelines and clinical pathways. Current attempts to provide peripheral nerve blockade are quite varied, with studies targeting femoral nerve, fascia iliaca, L1/L2 paravertebral, and lumbar plexus blockade. Future research efforts with a large prospective trial investigating these techniques should focus on which of these PNBs presents the optimal risk-benefit profile for hip arthroscopy patients and thus appropriately address the clinical questions at hand.
This paper will be judged for the Resident Writer’s Award.
- Baber YF, Robinson AH, Villar RN. Is diagnostic arthroscopy of the hip worthwhile? A prospective review of 328 adults investigated for hip pain. J Bone Joint Surg Br. 1999;81:600-603.
- Byrd JW, Jones KS. Arthroscopic management of femoroacetabular impingement: minimum 2-year follow-up. Arthroscopy. 2011;27:1379-1388.
- Larson CM, Giveans MR. Arthroscopic management of femoroacetabular impingement: early outcomes measures. Arthroscopy. 2008;24:540-546.
- O'Leary JA, Berend K, Vail TP. The relationship between diagnosis and outcome in arthroscopy of the hip. Arthroscopy. 2001;17:181-188.
- Philippon M, Schenker M, Briggs K, Kuppersmith D. Femoroacetabular impingement in 45 professional athletes: associated pathologies and return to sport following arthroscopic decompression. Knee Surg Sports Traumatol Arthrosc. 2007;15:908-914.
- Potter BK, Freedman BA, Andersen RC, Bojescul JA, Kuklo TR, Murphy KP. Correlation of Short Form-36 and disability status with outcomes of arthroscopic acetabular labral debridement. Am J Sports Med. 2005;33:864-870.
- Robertson WJ, Kadrmas WR, Kelly BT. Arthroscopic management of labral tears in the hip: a systematic review of the literature. Clin Orthop Relat Res. 2007;455:88-92.
- Yusaf MA, Hame SL. Arthroscopy of the hip. Curr Sports Med Rep. 2008;7:269-274.
- Colvin AC, Harrast J, Harner C. Trends in hip arthroscopy. J Bone Joint Surg Am. 2012;94:e23.
- Cvetanovich GL, Chalmers PN, Levy DM, et al. Hip arthroscopy surgical volume trends and 30-day postoperative complications. Arthroscopy. 2016 Apr 8. [Epub before print].
- Baker JF, Byrne DP, Hunter K, Mulhall KJ. Post-operative opiate requirements after hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2011;19:1399-1402.
- Lee EM, Murphy KP, Ben-David B. Postoperative analgesia for hip arthroscopy: combined L1 and L2 paravertebral blocks. J Clin Anesth. 2008;20:462-465.
- Ganesh A, Rose JB, Wells L, et al. Continuous peripheral nerve blockade for inpatient and outpatient postoperative analgesia in children. Anesth Analg. 2007;105:1234-1242.
- Williams BA, Kentor ML, Vogt MT, et al. Femoral-sciatic nerve blocks for complex outpatient knee surgery are associated with less postoperative pain before same-day discharge: a review of 1,200 consecutive cases from the period 1996-1999. Anesthesiology. 2003;98:1206-1213.
- Zywiel MG, Stroh DA, Lee SY, Bonutti PM, Mont MA. Chronic opioid use prior to total knee arthroplasty. J Bone Joint Surg Am. 2011;93:1988-1993.
- Dold AP, Murnaghan L, Xing J, Abdallah FW, Brull R, Whelan DB. Preoperative femoral nerve block in hip arthroscopic surgery: a retrospective review of 108 consecutive cases. Am J Sports Med. 2014;42:144-149.
- Schroeder KM, Donnelly MJ, Anderson BM, Ford MP, Keene JS. The analgesic impact of preoperative lumbar plexus blocks for hip arthroscopy. A retrospective review. Hip Int. 2013;23:93-98.
- YaDeau JT, Tedore T, Goytizolo EA, et al. Lumbar plexus blockade reduces pain after hip arthroscopy: a prospective randomized controlled trial. Anesth Analg. 2012;115:968-972.
- Smart LR, Oetgen M, Noonan B, Medvecky M. Beginning hip arthroscopy: indications, positioning, portals, basic techniques, and complications. Arthroscopy. 2007;23:1348-1353.
- Stevens M, Harrison G, McGrail M. A modified fascia iliaca compartment block has significant morphine-sparing effect after total hip arthroplasty. Anaesth Intensive Care. 2007;35:949-952.
- Lehmann LJ, Loosen G, Weiss C, Schmittner MD. Interscalene plexus block versus general anaesthesia for shoulder surgery: a randomized controlled study. Eur J Orthop Surg Traumatol. 2015;25:255-261.
- Gonano C, Kettner SC, Ernstbrunner M, Schebesta K, Chiari A, Marhofer P. Comparison of economical aspects of interscalene brachial plexus blockade and general anaesthesia for arthroscopic shoulder surgery. Br J Anaesth. 2009;103:428-433.
- Hadzic A, Karaca PE, Hobeika P, et al. Peripheral nerve blocks result in superior recovery profile compared with general anesthesia in outpatient knee arthroscopy. Anesth Analg. 2005;100:976-981.
- Hsu LP, Oh S, Nuber GW, et al. Nerve block of the infrapatellar branch of the saphenous nerve in knee arthroscopy: a prospective, double-blinded, randomized, placebo-controlled trial. J Bone Joint Surg Am. 2013;95:1465-1472.
- Montes FR, Zarate E, Grueso R, et al. Comparison of spinal anesthesia with combined sciatic-femoral nerve block for outpatient knee arthroscopy. J Clin Anesth. 2008;20:415-420.
- Wulf H, Lowe J, Gnutzmann KH, Steinfeldt T. Femoral nerve block with ropivacaine or bupivacaine in day case anterior crucial ligament reconstruction. Acta Anaesthesiol Scand. 2010;54:414-420.
- Krych AJ, Baran S, Kuzma SA, Smith HM, Johnson RL, Levy BA. Utility of multimodal analgesia with fascia iliaca blockade for acute pain management following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22:843-847.
- Nye ZB, Horn JL, Crittenden W, Abrahams MS, Aziz MF. Ambulatory continuous posterior lumbar plexus blocks following hip arthroscopy: a review of 213 cases. J Clin Anesth. 2013;25:268-274.
- Ward JP, Albert DB, Altman R, Goldstein RY, Cuff G, Youm T. Are femoral nerve blocks effective for early postoperative pain management after hip arthroscopy? Arthroscopy. 2012;28:1064-1069.
- Liu SS, Strodtbeck WM, Richman JM, Wu CL. A comparison of regional versus general anesthesia for ambulatory anesthesia: a meta-analysis of randomized controlled trials. Anesth Analg. 2005;101:1634-1642.
- Clarke MT, Arora A, Villar RN. Hip arthroscopy: complications in 1054 cases. Clin Orthop Relat Res. 2003;406:84-88.
ABSTRACT
Pain control following hip arthroscopy presents a significant clinical challenge, with postoperative pain requiring considerable opioid use. Peripheral nerve blocks (PNBs) have emerged as one option to improve pain and limit the consequences of opioid use. The purpose of this study is to provide a comprehensive review of outcomes associated with PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications. A systematic review of PubMed, Medline, Scopus, and Embase databases was conducted through January 2015 for English-language articles reporting outcome data, with 2 reviewers independently reviewing studies for inclusion. When available, similar outcomes were combined to generate frequency-weighted means. Six studies met the inclusion criteria for this review, reporting on 710 patients undergoing hip arthroscopy. The mean ages were 37.0 and 37.7 years for the PNB and comparator groups, respectively, with a reported total of 281 (40.5%) male and 412 (59.5%) female patients. Postoperative post-anesthesia care unit (PACU) pain was consistently reduced in the PNB group, with the use of a lower morphine equivalent dose and lower rates of inpatient admission, compared with that in the control groups. Postoperative nausea and/or vomiting as well as PACU discharge time showed mixed results. High satisfaction and few complications were reported. In conclusion, PNB is associated with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions, though similar rates of nausea/vomiting and time to discharge were reported. Current PNB techniques are varied, and future research efforts should focus on examining which of these methods provides the optimal risk-benefit profile in hip arthroscopy.
Continue to: Hip arthroscopy has emerged...
Hip arthroscopy has emerged as a useful procedure in the diagnosis and treatment of hip pathology,1-8 experiencing a substantial rise in popularity in recent years, with the number of procedures growing by a factor of 18 from 1999 to 20099 and 25 from 2006 to 2013.10 Though hip arthroscopy is beneficial in many cases, marked postoperative pain has presented a substantial challenge, with patients requiring considerable doses of opiate-based medications in the post-anesthesia care unit (PACU).11,12 Increased narcotic use carries increased side effects, including postoperative nausea and vomiting,13 and poorly managed pain leads to increased unplanned admissions.14 Furthermore, patients with chronic hip pain and long-term opioid use may experience heightened and prolonged pain following the procedure, owing to medication tolerance and reduced opioid efficacy in this setting.15
Several pain control strategies have been employed in patients undergoing hip arthroscopy. General anesthesia16,17 and combined spinal epidural (CSE)18 are commonly used. However, such techniques rely heavily on opioids for postoperative pain control,11 and epidural anesthesia commonly requires adjunctive treatments (eg, neuromuscular blockade) to ensure muscle relaxation for joint distraction.19 One technique that has been employed recently is peripheral nerve block (PNB), which has been associated with a significant decrease in postoperative opioid use and nausea and vomiting.13,20 This method has proven successful in other fields of arthroscopy, including shoulder arthroscopy, in which it resulted in faster recovery, reduced opioid consumption,21 and demonstrated cost-effectiveness22 compared with general anesthesia and knee arthroscopy.23-26 As it is a relatively new field, little is known about the use of PNB in hip arthroscopy.
The goal of this systematic review was to comprehensively review the studies reporting on PNB in hip arthroscopy. We specifically focused on outcomes, including postoperative pain; analgesic use; nausea, vomiting, and antiemetic use; discharge time; inpatient admission; and patient satisfaction, as well as the complications associated with the use of PNB. Our knowledge of outcomes associated with PNB in hip arthroscopy is based on a few individual studies that have reported on small groups of patients using a variety of outcome measures and other findings. Furthermore, each of these studies commonly reflects the experience of an individual surgeon at a single institution and, when taken alone, may not be an accurate representation of the more general outcomes associated with PNB. A comprehensive review of such studies will provide surgeons, anesthesiologists, and patients with a better understanding of the anticipated outcomes of using PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications.
MATERIALS AND METHODS
A systematic review of outcomes associated with PNB in hip arthroscopy was performed using the available English-language literature in accordance with the guidelines laid out by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement and included studies retrieved from the PubMed, Medline, Scopus, and Embase computerized literature databases. Searches were executed comprising all years from database inception through January 2015. Articles were retrieved by an electronic search of medical subject headings and keyword terms and their respective combinations (Table 1). The inclusion criteria for studies in this systematic review were studies that (1) were written in the English language and (2) reported explicit outcome data. The exclusion criteria were (1) review articles, meta-analyses, case reports, abstracts/conference papers, comments/letters, or technique articles without reported patient data and (2) basic research, biomechanics, or animal/cadaveric studies without reported patient data.
Table 1. Search Terms Entered to Identify English-Language Studies Through January 2015
Database | Search terms |
PubMed, Scopus | Keyword: (hip AND arthroscopy) AND (pain control OR pain management OR pain regimen OR nerve block OR spinal anesthesia OR regional anesthesia OR general anesthesia) |
Medline | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “Anesthesia, General” OR “Anesthesia” OR “Anesthesia, Inhalation”, OR “Balanced Anesthesia” OR “Anesthesia, Local” OR “Anesthesia, Spinal” OR “Anesthesia, Conduction” OR “Nerve Block”) |
Embase | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “General Anesthesia” OR “Anesthesia” OR “Inhalation Anesthesia”, OR “Balanced Anesthesia” OR “Local Anesthesia” OR “Spinal Anesthesia” OR “Regional Anesthesia” OR “Nerve Block”) |
The literature search strategy is outlined in the Figure. The initial title search yielded a subset of possible articles that were then further included or excluded on the basis of the contents of the article’s abstract, wherein articles were again selected on the basis of the aforementioned inclusion and exclusion criteria. Articles selected in both the title and abstract phases underwent full-text review, during which the full text of each qualifying article was reviewed. In addition, the reference sections from articles undergoing full-text review were scanned to identify any additional studies that had not been identified in the original literature search. Appropriate studies for final inclusion were then selected at this stage. The title, abstract, and full-text selection process were performed by 2 of the study authors (Dr. Steinhaus and Dr. Lynch), with any discrepancies being discussed and resolved by mutual agreement.
Continue to: For all 6 included studies...
For all 6 included studies,16-18,27-29 data were collected regarding the study specifics, patients included, and outcomes measured in the study. The journal of publication, type of study, level of evidence, and type of PNB, as well as the presence of a comparator group were noted (Table 2). Patient information included the number of patients at baseline and follow-up, mean age, gender, weight, height, body mass index, American Society of Anesthesiologists (ASA) status, and the specific procedures performed. In addition, data were collected on outcomes, including postoperative pain, as well as secondary outcomes and additional findings reported by the studies (Table 3). Where possible, weighted averages were calculated across all studies to obtain aggregate data.
RESULTS
STUDY INCLUSION
Six studies, all published between 2012 and 2014, were included in this systematic review (Table 2). Three studies involved lumbar plexus block, 2 studies involved femoral nerve block, and 1 study evaluated fascia iliaca block. Two studies used a control group of patients who received only general anesthesia (compared with the treatment group who received both general anesthesia and PNB); another study compared intravenous morphine with PNB; and 1 study compared CSE alone with PNB in addition to epidural.
DEMOGRAPHIC DATA
Demographic data from the included studies are presented in Table 2. In total, 710 and 549 patients were evaluated at baseline and final follow-up, respectively, which represents a follow-up rate of 77%. The frequency-weighted mean age of patients receiving PNB was 37.0 years compared with 37.7 years in the comparison groups, and the studies reported a total of 281 (40.5%) male and 412 (59.5%) female patients. The procedures performed were heterogeneously reported; therefore, totals were not tabulated, although the reported procedures included osteochondroplasty, labral débridement, labral and/or capsular repair, gluteus minimus repair, and synovectomy.
POSTOPERATIVE PAIN
Four studies reported on postoperative pain, and these data are presented in Table 3. In a retrospective study of patients receiving femoral nerve block in addition to general anesthesia, Dold and colleagues16 noted postoperative pain at 0, 15, 30, 45, and 60 minutes following arrival in the PACU, and discovered a statistically significantly lower level of pain at 60 minutes compared with inpatients receiving general anesthesia alone. YaDeau and colleagues18 found a significantly lower level of pain at rest in the PACU for those receiving CSE and lumbar plexus blockade compared with those receiving CSE alone. This significant difference did not persist at 24 hours or 6 months after the procedure, nor did it exist for pain with movement at any time point. Similarly, Schroeder and colleagues17 examined patients receiving general anesthesia and lumbar plexus block and found a significant reduction in pain immediately postoperatively in the PACU, though these effects disappeared the day following the procedure. Krych and colleagues27 also reported on postoperative pain in patients undergoing fascia iliaca blockade, although they did not include a comparator group. Outcome comparison between patients who received PNB and controls in the PACU and 1 day following the procedure are presented in Table 4.
ANALGESIC USE
Four studies reported on analgesic use after PNB, and these data are presented in Table 3. Dold and colleagues16 noted analgesic use intraoperatively, in the PACU, and in the surgical day care unit (SDCU). These authors found a significant reduction in morphine equivalent dose given in the operating room and in the PACU in the group receiving PNB, with a nonsignificant trend toward lower use of oxycodone in the SDCU. Schroeder and colleagues17 similarly reported significant reductions in morphine equivalent dose intraoperatively and in Phase I recovery for patients receiving PNB, and these differences disappeared in Phase II recovery as well as intraoperatively if the block dose was considered. In addition, these authors found a significant reduction in the use of fentanyl and hydromorphone in the operating room in the PNB group, as well as a significant reduction in the proportion of patients receiving ketorolac in the operating room or PACU. Finally, YaDeau and colleagues18 reported total analgesic usage in the PACU among PNB patients compared with those receiving CSE alone and showed a strong trend toward reduced use in the PNB group, although this difference was not significant (P = .051). PACU analgesic use is presented in Table 4.
Continue to: Postoperative nausea...
POSTOPERATIVE NAUSEA/VOMITING AND ANTIEMETIC USE
Five studies presented data on nausea, vomiting, or antiemetic use following PNB and are shown in Table 3. YaDeau and colleagues18 reported nausea among 34% of patients in the PNB group, compared with 20% in the control group, vomiting in 2% and 7%, respectively, and antiemetic use in 12% of both groups. Dold and colleagues16 identified a similar trend, with 41.1% of patients in the PNB group and 32.5% of patients in the control group experiencing postoperative nausea or vomiting, while Krych and colleagues27 noted only 10% of PNB patients with mild nausea and none requiring antiemetic use. In their study of patients receiving PNB, Schroeder and colleagues17 found a significant reduction in antiemetic use among PNB patients compared with those receiving general anesthesia alone. Similarly, Ward and colleagues29 noted a significant difference in postoperative nausea, with 10% of patients in the PNB group experiencing postoperative nausea compared with 75% of those in the comparator group who received intravenous morphine. The mean percentage of patients experiencing postoperative nausea and/or vomiting is shown in Table 4.
DISCHARGE TIME
Four studies presented data on discharge time from the PACU and are summarized in Table 3. Three of these studies included a comparator group. Both Dold and colleagues16 and YaDeau and colleagues18 reported an increase in the time to discharge for patients receiving PNB, although these differences were not significant. The study by Ward and colleagues,29 on the other hand, noted a significant reduction in the time to discharge for the PNB group. In addition to these studies, Krych and colleagues27 examined the time from skin closure to discharge for patients receiving PNB, noting a mean 199 minutes for the patients in their study. Mean times to discharge for the PNB and control groups are presented in Table 4.
INPATIENT ADMISSION
Four studies presented data on the proportion of study participants who were admitted as inpatients, and these data are shown in Table 3. Dold and colleagues16 reported no inpatient admissions in their PNB group compared with 5.0% for the control group (both cases of pain control), while YaDeau and colleagues18 found that 3 admissions occurred, with 2 in the control group (1 for oxygen desaturation and the other for intractable pain and nausea) and 1 from the PNB group (epidural spread and urinary retention). Two additional studies reported data on PNB groups alone. Krych and colleagues27 observed no overnight admissions in their study, while Nye and colleagues28 reported 1 readmission for bilateral leg numbness and weakness due to epidural spread, which resolved following discontinuation of the block. The mean proportion of inpatient admissions is presented in Table 4.
SATISFACTION
A total of 3 studies examined patient satisfaction, and these data are presented in Table 3. In their study, Ward and colleagues29 reported a significantly greater rate of satisfaction at 1 day postoperatively among the patients in the PNB group (90%) than among patients who received intravenous morphine (25%) (P < .0001). Similarly, YaDeau and colleagues18 noted greater satisfaction among the PNB group than among the control group, with PNB patients rating their satisfaction at a mean of 8.6 and control patients at a mean of 7.9 on a 10-point scale (0-10) 24 hours postoperatively, although this difference was not significant. Finally, Krych and colleagues27 found that 67% of patients were “very satisfied” and 33% were “satisfied”, based on a Likert scale.
COMPLICATIONS
Four studies presented data on complications, and these findings are summarized in Table 3. In their work, Nye and colleagues28 reported most extensively on complications associated with PNB. Overall, the authors found a rate of significant complications of 3.8%. In terms of specific complications, they noted local anesthetic systemic toxicity (0.9%), epidural spread (0.5%), sensory or motor deficits (9.4%), falls (0.5%), and catheter issues. In their study of patients receiving PNB and CSE, YaDeau and colleagues18 identified 1 patient in the PNB group with epidural spread and urinary retention, while they noted 1 case of oxygen desaturation and another case of intractable pain and nausea in the group receiving CSE alone, all 3 of which required inpatient admission. They found no permanent adverse events attributable to the PNB. In another study, Dold and colleagues16 observed no complications in patients receiving PNB compared with those in 2 admissions in the control group for inadequate pain control. Similarly, Krych and colleagues27 identified no complications in patients who received PNB in their study.
DISCUSSION
Hip arthroscopy has experienced a substantial gain in popularity in recent years, emerging as a beneficial technique for both the diagnosis and treatment of diverse hip pathologies in patients spanning a variety of demographics. Nevertheless, postoperative pain control, as well as medication side effects and unwanted patient admissions, present major challenges to the treating surgeon. As an adjuvant measure, peripheral nerve block represents one option to improve postoperative pain management, while at the same time addressing the adverse effects of considerable opioid use, which is commonly seen in these patients. Early experience with this method in hip arthroscopy was reported in a case series by Lee and colleagues.12 In an attempt to reduce postoperative pain, as well as limit the adverse effects and delay in discharge associated with considerable opioid use in the PACU, the authors used preoperative paravertebral blocks of L1 and L2 in 2 patients requiring hip arthroscopy with encouraging results. Since then, a number of studies have attempted the use of PNB in hip arthroscopy.16-18,27-29 However, we were unable to identify any prior reviews reporting on peripheral nerve blockade in hip arthroscopy, and thus this study is unique in providing a greater understanding of the outcomes associated with PNB use.
In general, we found that PNB was associated with improved outcomes. Based on the studies included in this review, there was a statistically significantly lower level of pain in the PACU for femoral nerve block (compared with general anesthesia alone)16 and lumbar plexus blockade (compared with general anesthesia17 and CSE18 alone). Nevertheless, these effects are likely short-lived, with differences disappearing the day following the procedure. In terms of analgesic use, 2 studies report significant reductions in analgesic use intraoperatively and in the PACU/Phase I recovery,16,17 with a third reporting a strong trend toward reduced analgesic use in the PACU (P = .051).18 Finally, we report fewer admissions for the PNB group, as well as high rates of satisfaction and few complications across these studies.
Continue to: Unlike these measures...
Unlike these measures, postoperative nausea, vomiting, and antiemetic use, as well as time to discharge, showed more mixed results. With regard to nausea/vomiting, 2 studies16,18 reported nonsignificantly increased rates in the PNB group, whereas others reported significant reductions in nausea/vomiting29 and in the proportion of patients receiving antiemetics.17 Similarly, mixed results were seen in terms of patient discharge time from the PACU. Two studies16,18 reported a nonsignificant increase in time to discharge for the PNB group, while another29 noted a significant reduction for the PNB group compared with those receiving intravenous morphine. These mixed results were surprising, as we expected reductions in opioid use to result in fewer instances of nausea/vomiting and a quicker time to discharge. The reasons underlying these findings are not clear, although it has been suggested that current discharge guidelines and clinical pathways limit the ability to take advantage of the accelerated timeline offered by regional anesthesia.16,30 As experience with PNB grows, our guidelines and pathways are likely to adapt to capitalize on these advantages, and future studies may show more reliable improvements in these measures.
While rare, the risk of bleeding requiring blood transfusion following hip arthroscopy is one of the most common complications of this procedure. Though the studies included in this review did not report on the need for transfusion, a recent study by Cvetanovich and colleagues10 used a national database and found that, of patients undergoing hip arthroscopy (n = 1338), 0.4% (n = 5) had bleeding requiring a transfusion, with 0.3% (n = 4) requiring return to the operating room, similar to an earlier study by Clarke and colleagues,31 who noted bleeding from the portal site in 0.4% of hip arthroscopy patients. In terms of risk factors, Cvetanovich and colleagues10 found that ASA class, older age, and prior cardiac surgery were significantly associated with minor and overall complications, whereas both regional anesthesia/monitored anesthesia care and alcohol consumption of >2 drinks a day were significantly associated with minor complications, including bleeding requiring transfusions. They noted, however, that these risk factors accounted for only 5% of the variance in complication rates, indicating that other unidentified variables better explained the variance in complication rates. These authors concluded that complications associated with hip arthroscopy are so rare that we may not be able to predict which risk factors or anesthesia types are more likely to cause them. Further characterization of bleeding following hip arthroscopy and its associated risk factors is a valuable area for future research.
LIMITATIONS
Our study contains a number of limitations. This review included studies whose level of evidence varied from I to IV; therefore, our study is limited by any bias or heterogeneity introduced in patient recruitment, selection, variability of technique, data collection, and analysis used in these studies. This heterogeneity is most apparent in the block types and comparator groups. Furthermore, several different outcome measures were reported across the 6 studies used in this review, which decreased the relevance of any one of these individual outcomes. Finally, given the limited data that currently exist for the use of PNB in hip arthroscopy, we are unable to note meaningful differences between various types of PNBs, such as differences in postoperative pain or other measures such as quadriceps weakness, which can accompany femoral nerve block.12 While it is important to read our work with these limitations in mind, this systematic review is, to our knowledge, the only comprehensive review to date of studies reporting on PNB in hip arthroscopy, providing clinicians and patients with a greater understanding of the associated outcomes across these studies.
CONCLUSION
This systematic review shows improved outcomes and few complications with PNB use in hip arthroscopy, with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions. Although opioid use was reduced in these studies, we found similar rates of postoperative nausea/vomiting as well as similar time to discharge from the PACU, which may reflect our continued reliance on outdated discharge guidelines and clinical pathways. Current attempts to provide peripheral nerve blockade are quite varied, with studies targeting femoral nerve, fascia iliaca, L1/L2 paravertebral, and lumbar plexus blockade. Future research efforts with a large prospective trial investigating these techniques should focus on which of these PNBs presents the optimal risk-benefit profile for hip arthroscopy patients and thus appropriately address the clinical questions at hand.
This paper will be judged for the Resident Writer’s Award.
ABSTRACT
Pain control following hip arthroscopy presents a significant clinical challenge, with postoperative pain requiring considerable opioid use. Peripheral nerve blocks (PNBs) have emerged as one option to improve pain and limit the consequences of opioid use. The purpose of this study is to provide a comprehensive review of outcomes associated with PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications. A systematic review of PubMed, Medline, Scopus, and Embase databases was conducted through January 2015 for English-language articles reporting outcome data, with 2 reviewers independently reviewing studies for inclusion. When available, similar outcomes were combined to generate frequency-weighted means. Six studies met the inclusion criteria for this review, reporting on 710 patients undergoing hip arthroscopy. The mean ages were 37.0 and 37.7 years for the PNB and comparator groups, respectively, with a reported total of 281 (40.5%) male and 412 (59.5%) female patients. Postoperative post-anesthesia care unit (PACU) pain was consistently reduced in the PNB group, with the use of a lower morphine equivalent dose and lower rates of inpatient admission, compared with that in the control groups. Postoperative nausea and/or vomiting as well as PACU discharge time showed mixed results. High satisfaction and few complications were reported. In conclusion, PNB is associated with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions, though similar rates of nausea/vomiting and time to discharge were reported. Current PNB techniques are varied, and future research efforts should focus on examining which of these methods provides the optimal risk-benefit profile in hip arthroscopy.
Continue to: Hip arthroscopy has emerged...
Hip arthroscopy has emerged as a useful procedure in the diagnosis and treatment of hip pathology,1-8 experiencing a substantial rise in popularity in recent years, with the number of procedures growing by a factor of 18 from 1999 to 20099 and 25 from 2006 to 2013.10 Though hip arthroscopy is beneficial in many cases, marked postoperative pain has presented a substantial challenge, with patients requiring considerable doses of opiate-based medications in the post-anesthesia care unit (PACU).11,12 Increased narcotic use carries increased side effects, including postoperative nausea and vomiting,13 and poorly managed pain leads to increased unplanned admissions.14 Furthermore, patients with chronic hip pain and long-term opioid use may experience heightened and prolonged pain following the procedure, owing to medication tolerance and reduced opioid efficacy in this setting.15
Several pain control strategies have been employed in patients undergoing hip arthroscopy. General anesthesia16,17 and combined spinal epidural (CSE)18 are commonly used. However, such techniques rely heavily on opioids for postoperative pain control,11 and epidural anesthesia commonly requires adjunctive treatments (eg, neuromuscular blockade) to ensure muscle relaxation for joint distraction.19 One technique that has been employed recently is peripheral nerve block (PNB), which has been associated with a significant decrease in postoperative opioid use and nausea and vomiting.13,20 This method has proven successful in other fields of arthroscopy, including shoulder arthroscopy, in which it resulted in faster recovery, reduced opioid consumption,21 and demonstrated cost-effectiveness22 compared with general anesthesia and knee arthroscopy.23-26 As it is a relatively new field, little is known about the use of PNB in hip arthroscopy.
The goal of this systematic review was to comprehensively review the studies reporting on PNB in hip arthroscopy. We specifically focused on outcomes, including postoperative pain; analgesic use; nausea, vomiting, and antiemetic use; discharge time; inpatient admission; and patient satisfaction, as well as the complications associated with the use of PNB. Our knowledge of outcomes associated with PNB in hip arthroscopy is based on a few individual studies that have reported on small groups of patients using a variety of outcome measures and other findings. Furthermore, each of these studies commonly reflects the experience of an individual surgeon at a single institution and, when taken alone, may not be an accurate representation of the more general outcomes associated with PNB. A comprehensive review of such studies will provide surgeons, anesthesiologists, and patients with a better understanding of the anticipated outcomes of using PNB in hip arthroscopy. We hypothesize that the use of PNB in hip arthroscopy leads to improved outcomes and is associated with few complications.
MATERIALS AND METHODS
A systematic review of outcomes associated with PNB in hip arthroscopy was performed using the available English-language literature in accordance with the guidelines laid out by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement and included studies retrieved from the PubMed, Medline, Scopus, and Embase computerized literature databases. Searches were executed comprising all years from database inception through January 2015. Articles were retrieved by an electronic search of medical subject headings and keyword terms and their respective combinations (Table 1). The inclusion criteria for studies in this systematic review were studies that (1) were written in the English language and (2) reported explicit outcome data. The exclusion criteria were (1) review articles, meta-analyses, case reports, abstracts/conference papers, comments/letters, or technique articles without reported patient data and (2) basic research, biomechanics, or animal/cadaveric studies without reported patient data.
Table 1. Search Terms Entered to Identify English-Language Studies Through January 2015
Database | Search terms |
PubMed, Scopus | Keyword: (hip AND arthroscopy) AND (pain control OR pain management OR pain regimen OR nerve block OR spinal anesthesia OR regional anesthesia OR general anesthesia) |
Medline | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “Anesthesia, General” OR “Anesthesia” OR “Anesthesia, Inhalation”, OR “Balanced Anesthesia” OR “Anesthesia, Local” OR “Anesthesia, Spinal” OR “Anesthesia, Conduction” OR “Nerve Block”) |
Embase | MeSH (includes both MeSH terms and keywords): (Hip) AND (Arthroscopy) AND (“Pain Management” OR “General Anesthesia” OR “Anesthesia” OR “Inhalation Anesthesia”, OR “Balanced Anesthesia” OR “Local Anesthesia” OR “Spinal Anesthesia” OR “Regional Anesthesia” OR “Nerve Block”) |
The literature search strategy is outlined in the Figure. The initial title search yielded a subset of possible articles that were then further included or excluded on the basis of the contents of the article’s abstract, wherein articles were again selected on the basis of the aforementioned inclusion and exclusion criteria. Articles selected in both the title and abstract phases underwent full-text review, during which the full text of each qualifying article was reviewed. In addition, the reference sections from articles undergoing full-text review were scanned to identify any additional studies that had not been identified in the original literature search. Appropriate studies for final inclusion were then selected at this stage. The title, abstract, and full-text selection process were performed by 2 of the study authors (Dr. Steinhaus and Dr. Lynch), with any discrepancies being discussed and resolved by mutual agreement.
Continue to: For all 6 included studies...
For all 6 included studies,16-18,27-29 data were collected regarding the study specifics, patients included, and outcomes measured in the study. The journal of publication, type of study, level of evidence, and type of PNB, as well as the presence of a comparator group were noted (Table 2). Patient information included the number of patients at baseline and follow-up, mean age, gender, weight, height, body mass index, American Society of Anesthesiologists (ASA) status, and the specific procedures performed. In addition, data were collected on outcomes, including postoperative pain, as well as secondary outcomes and additional findings reported by the studies (Table 3). Where possible, weighted averages were calculated across all studies to obtain aggregate data.
RESULTS
STUDY INCLUSION
Six studies, all published between 2012 and 2014, were included in this systematic review (Table 2). Three studies involved lumbar plexus block, 2 studies involved femoral nerve block, and 1 study evaluated fascia iliaca block. Two studies used a control group of patients who received only general anesthesia (compared with the treatment group who received both general anesthesia and PNB); another study compared intravenous morphine with PNB; and 1 study compared CSE alone with PNB in addition to epidural.
DEMOGRAPHIC DATA
Demographic data from the included studies are presented in Table 2. In total, 710 and 549 patients were evaluated at baseline and final follow-up, respectively, which represents a follow-up rate of 77%. The frequency-weighted mean age of patients receiving PNB was 37.0 years compared with 37.7 years in the comparison groups, and the studies reported a total of 281 (40.5%) male and 412 (59.5%) female patients. The procedures performed were heterogeneously reported; therefore, totals were not tabulated, although the reported procedures included osteochondroplasty, labral débridement, labral and/or capsular repair, gluteus minimus repair, and synovectomy.
POSTOPERATIVE PAIN
Four studies reported on postoperative pain, and these data are presented in Table 3. In a retrospective study of patients receiving femoral nerve block in addition to general anesthesia, Dold and colleagues16 noted postoperative pain at 0, 15, 30, 45, and 60 minutes following arrival in the PACU, and discovered a statistically significantly lower level of pain at 60 minutes compared with inpatients receiving general anesthesia alone. YaDeau and colleagues18 found a significantly lower level of pain at rest in the PACU for those receiving CSE and lumbar plexus blockade compared with those receiving CSE alone. This significant difference did not persist at 24 hours or 6 months after the procedure, nor did it exist for pain with movement at any time point. Similarly, Schroeder and colleagues17 examined patients receiving general anesthesia and lumbar plexus block and found a significant reduction in pain immediately postoperatively in the PACU, though these effects disappeared the day following the procedure. Krych and colleagues27 also reported on postoperative pain in patients undergoing fascia iliaca blockade, although they did not include a comparator group. Outcome comparison between patients who received PNB and controls in the PACU and 1 day following the procedure are presented in Table 4.
ANALGESIC USE
Four studies reported on analgesic use after PNB, and these data are presented in Table 3. Dold and colleagues16 noted analgesic use intraoperatively, in the PACU, and in the surgical day care unit (SDCU). These authors found a significant reduction in morphine equivalent dose given in the operating room and in the PACU in the group receiving PNB, with a nonsignificant trend toward lower use of oxycodone in the SDCU. Schroeder and colleagues17 similarly reported significant reductions in morphine equivalent dose intraoperatively and in Phase I recovery for patients receiving PNB, and these differences disappeared in Phase II recovery as well as intraoperatively if the block dose was considered. In addition, these authors found a significant reduction in the use of fentanyl and hydromorphone in the operating room in the PNB group, as well as a significant reduction in the proportion of patients receiving ketorolac in the operating room or PACU. Finally, YaDeau and colleagues18 reported total analgesic usage in the PACU among PNB patients compared with those receiving CSE alone and showed a strong trend toward reduced use in the PNB group, although this difference was not significant (P = .051). PACU analgesic use is presented in Table 4.
Continue to: Postoperative nausea...
POSTOPERATIVE NAUSEA/VOMITING AND ANTIEMETIC USE
Five studies presented data on nausea, vomiting, or antiemetic use following PNB and are shown in Table 3. YaDeau and colleagues18 reported nausea among 34% of patients in the PNB group, compared with 20% in the control group, vomiting in 2% and 7%, respectively, and antiemetic use in 12% of both groups. Dold and colleagues16 identified a similar trend, with 41.1% of patients in the PNB group and 32.5% of patients in the control group experiencing postoperative nausea or vomiting, while Krych and colleagues27 noted only 10% of PNB patients with mild nausea and none requiring antiemetic use. In their study of patients receiving PNB, Schroeder and colleagues17 found a significant reduction in antiemetic use among PNB patients compared with those receiving general anesthesia alone. Similarly, Ward and colleagues29 noted a significant difference in postoperative nausea, with 10% of patients in the PNB group experiencing postoperative nausea compared with 75% of those in the comparator group who received intravenous morphine. The mean percentage of patients experiencing postoperative nausea and/or vomiting is shown in Table 4.
DISCHARGE TIME
Four studies presented data on discharge time from the PACU and are summarized in Table 3. Three of these studies included a comparator group. Both Dold and colleagues16 and YaDeau and colleagues18 reported an increase in the time to discharge for patients receiving PNB, although these differences were not significant. The study by Ward and colleagues,29 on the other hand, noted a significant reduction in the time to discharge for the PNB group. In addition to these studies, Krych and colleagues27 examined the time from skin closure to discharge for patients receiving PNB, noting a mean 199 minutes for the patients in their study. Mean times to discharge for the PNB and control groups are presented in Table 4.
INPATIENT ADMISSION
Four studies presented data on the proportion of study participants who were admitted as inpatients, and these data are shown in Table 3. Dold and colleagues16 reported no inpatient admissions in their PNB group compared with 5.0% for the control group (both cases of pain control), while YaDeau and colleagues18 found that 3 admissions occurred, with 2 in the control group (1 for oxygen desaturation and the other for intractable pain and nausea) and 1 from the PNB group (epidural spread and urinary retention). Two additional studies reported data on PNB groups alone. Krych and colleagues27 observed no overnight admissions in their study, while Nye and colleagues28 reported 1 readmission for bilateral leg numbness and weakness due to epidural spread, which resolved following discontinuation of the block. The mean proportion of inpatient admissions is presented in Table 4.
SATISFACTION
A total of 3 studies examined patient satisfaction, and these data are presented in Table 3. In their study, Ward and colleagues29 reported a significantly greater rate of satisfaction at 1 day postoperatively among the patients in the PNB group (90%) than among patients who received intravenous morphine (25%) (P < .0001). Similarly, YaDeau and colleagues18 noted greater satisfaction among the PNB group than among the control group, with PNB patients rating their satisfaction at a mean of 8.6 and control patients at a mean of 7.9 on a 10-point scale (0-10) 24 hours postoperatively, although this difference was not significant. Finally, Krych and colleagues27 found that 67% of patients were “very satisfied” and 33% were “satisfied”, based on a Likert scale.
COMPLICATIONS
Four studies presented data on complications, and these findings are summarized in Table 3. In their work, Nye and colleagues28 reported most extensively on complications associated with PNB. Overall, the authors found a rate of significant complications of 3.8%. In terms of specific complications, they noted local anesthetic systemic toxicity (0.9%), epidural spread (0.5%), sensory or motor deficits (9.4%), falls (0.5%), and catheter issues. In their study of patients receiving PNB and CSE, YaDeau and colleagues18 identified 1 patient in the PNB group with epidural spread and urinary retention, while they noted 1 case of oxygen desaturation and another case of intractable pain and nausea in the group receiving CSE alone, all 3 of which required inpatient admission. They found no permanent adverse events attributable to the PNB. In another study, Dold and colleagues16 observed no complications in patients receiving PNB compared with those in 2 admissions in the control group for inadequate pain control. Similarly, Krych and colleagues27 identified no complications in patients who received PNB in their study.
DISCUSSION
Hip arthroscopy has experienced a substantial gain in popularity in recent years, emerging as a beneficial technique for both the diagnosis and treatment of diverse hip pathologies in patients spanning a variety of demographics. Nevertheless, postoperative pain control, as well as medication side effects and unwanted patient admissions, present major challenges to the treating surgeon. As an adjuvant measure, peripheral nerve block represents one option to improve postoperative pain management, while at the same time addressing the adverse effects of considerable opioid use, which is commonly seen in these patients. Early experience with this method in hip arthroscopy was reported in a case series by Lee and colleagues.12 In an attempt to reduce postoperative pain, as well as limit the adverse effects and delay in discharge associated with considerable opioid use in the PACU, the authors used preoperative paravertebral blocks of L1 and L2 in 2 patients requiring hip arthroscopy with encouraging results. Since then, a number of studies have attempted the use of PNB in hip arthroscopy.16-18,27-29 However, we were unable to identify any prior reviews reporting on peripheral nerve blockade in hip arthroscopy, and thus this study is unique in providing a greater understanding of the outcomes associated with PNB use.
In general, we found that PNB was associated with improved outcomes. Based on the studies included in this review, there was a statistically significantly lower level of pain in the PACU for femoral nerve block (compared with general anesthesia alone)16 and lumbar plexus blockade (compared with general anesthesia17 and CSE18 alone). Nevertheless, these effects are likely short-lived, with differences disappearing the day following the procedure. In terms of analgesic use, 2 studies report significant reductions in analgesic use intraoperatively and in the PACU/Phase I recovery,16,17 with a third reporting a strong trend toward reduced analgesic use in the PACU (P = .051).18 Finally, we report fewer admissions for the PNB group, as well as high rates of satisfaction and few complications across these studies.
Continue to: Unlike these measures...
Unlike these measures, postoperative nausea, vomiting, and antiemetic use, as well as time to discharge, showed more mixed results. With regard to nausea/vomiting, 2 studies16,18 reported nonsignificantly increased rates in the PNB group, whereas others reported significant reductions in nausea/vomiting29 and in the proportion of patients receiving antiemetics.17 Similarly, mixed results were seen in terms of patient discharge time from the PACU. Two studies16,18 reported a nonsignificant increase in time to discharge for the PNB group, while another29 noted a significant reduction for the PNB group compared with those receiving intravenous morphine. These mixed results were surprising, as we expected reductions in opioid use to result in fewer instances of nausea/vomiting and a quicker time to discharge. The reasons underlying these findings are not clear, although it has been suggested that current discharge guidelines and clinical pathways limit the ability to take advantage of the accelerated timeline offered by regional anesthesia.16,30 As experience with PNB grows, our guidelines and pathways are likely to adapt to capitalize on these advantages, and future studies may show more reliable improvements in these measures.
While rare, the risk of bleeding requiring blood transfusion following hip arthroscopy is one of the most common complications of this procedure. Though the studies included in this review did not report on the need for transfusion, a recent study by Cvetanovich and colleagues10 used a national database and found that, of patients undergoing hip arthroscopy (n = 1338), 0.4% (n = 5) had bleeding requiring a transfusion, with 0.3% (n = 4) requiring return to the operating room, similar to an earlier study by Clarke and colleagues,31 who noted bleeding from the portal site in 0.4% of hip arthroscopy patients. In terms of risk factors, Cvetanovich and colleagues10 found that ASA class, older age, and prior cardiac surgery were significantly associated with minor and overall complications, whereas both regional anesthesia/monitored anesthesia care and alcohol consumption of >2 drinks a day were significantly associated with minor complications, including bleeding requiring transfusions. They noted, however, that these risk factors accounted for only 5% of the variance in complication rates, indicating that other unidentified variables better explained the variance in complication rates. These authors concluded that complications associated with hip arthroscopy are so rare that we may not be able to predict which risk factors or anesthesia types are more likely to cause them. Further characterization of bleeding following hip arthroscopy and its associated risk factors is a valuable area for future research.
LIMITATIONS
Our study contains a number of limitations. This review included studies whose level of evidence varied from I to IV; therefore, our study is limited by any bias or heterogeneity introduced in patient recruitment, selection, variability of technique, data collection, and analysis used in these studies. This heterogeneity is most apparent in the block types and comparator groups. Furthermore, several different outcome measures were reported across the 6 studies used in this review, which decreased the relevance of any one of these individual outcomes. Finally, given the limited data that currently exist for the use of PNB in hip arthroscopy, we are unable to note meaningful differences between various types of PNBs, such as differences in postoperative pain or other measures such as quadriceps weakness, which can accompany femoral nerve block.12 While it is important to read our work with these limitations in mind, this systematic review is, to our knowledge, the only comprehensive review to date of studies reporting on PNB in hip arthroscopy, providing clinicians and patients with a greater understanding of the associated outcomes across these studies.
CONCLUSION
This systematic review shows improved outcomes and few complications with PNB use in hip arthroscopy, with reductions in postoperative pain, analgesic use, and the rate of inpatient admissions. Although opioid use was reduced in these studies, we found similar rates of postoperative nausea/vomiting as well as similar time to discharge from the PACU, which may reflect our continued reliance on outdated discharge guidelines and clinical pathways. Current attempts to provide peripheral nerve blockade are quite varied, with studies targeting femoral nerve, fascia iliaca, L1/L2 paravertebral, and lumbar plexus blockade. Future research efforts with a large prospective trial investigating these techniques should focus on which of these PNBs presents the optimal risk-benefit profile for hip arthroscopy patients and thus appropriately address the clinical questions at hand.
This paper will be judged for the Resident Writer’s Award.
- Baber YF, Robinson AH, Villar RN. Is diagnostic arthroscopy of the hip worthwhile? A prospective review of 328 adults investigated for hip pain. J Bone Joint Surg Br. 1999;81:600-603.
- Byrd JW, Jones KS. Arthroscopic management of femoroacetabular impingement: minimum 2-year follow-up. Arthroscopy. 2011;27:1379-1388.
- Larson CM, Giveans MR. Arthroscopic management of femoroacetabular impingement: early outcomes measures. Arthroscopy. 2008;24:540-546.
- O'Leary JA, Berend K, Vail TP. The relationship between diagnosis and outcome in arthroscopy of the hip. Arthroscopy. 2001;17:181-188.
- Philippon M, Schenker M, Briggs K, Kuppersmith D. Femoroacetabular impingement in 45 professional athletes: associated pathologies and return to sport following arthroscopic decompression. Knee Surg Sports Traumatol Arthrosc. 2007;15:908-914.
- Potter BK, Freedman BA, Andersen RC, Bojescul JA, Kuklo TR, Murphy KP. Correlation of Short Form-36 and disability status with outcomes of arthroscopic acetabular labral debridement. Am J Sports Med. 2005;33:864-870.
- Robertson WJ, Kadrmas WR, Kelly BT. Arthroscopic management of labral tears in the hip: a systematic review of the literature. Clin Orthop Relat Res. 2007;455:88-92.
- Yusaf MA, Hame SL. Arthroscopy of the hip. Curr Sports Med Rep. 2008;7:269-274.
- Colvin AC, Harrast J, Harner C. Trends in hip arthroscopy. J Bone Joint Surg Am. 2012;94:e23.
- Cvetanovich GL, Chalmers PN, Levy DM, et al. Hip arthroscopy surgical volume trends and 30-day postoperative complications. Arthroscopy. 2016 Apr 8. [Epub before print].
- Baker JF, Byrne DP, Hunter K, Mulhall KJ. Post-operative opiate requirements after hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2011;19:1399-1402.
- Lee EM, Murphy KP, Ben-David B. Postoperative analgesia for hip arthroscopy: combined L1 and L2 paravertebral blocks. J Clin Anesth. 2008;20:462-465.
- Ganesh A, Rose JB, Wells L, et al. Continuous peripheral nerve blockade for inpatient and outpatient postoperative analgesia in children. Anesth Analg. 2007;105:1234-1242.
- Williams BA, Kentor ML, Vogt MT, et al. Femoral-sciatic nerve blocks for complex outpatient knee surgery are associated with less postoperative pain before same-day discharge: a review of 1,200 consecutive cases from the period 1996-1999. Anesthesiology. 2003;98:1206-1213.
- Zywiel MG, Stroh DA, Lee SY, Bonutti PM, Mont MA. Chronic opioid use prior to total knee arthroplasty. J Bone Joint Surg Am. 2011;93:1988-1993.
- Dold AP, Murnaghan L, Xing J, Abdallah FW, Brull R, Whelan DB. Preoperative femoral nerve block in hip arthroscopic surgery: a retrospective review of 108 consecutive cases. Am J Sports Med. 2014;42:144-149.
- Schroeder KM, Donnelly MJ, Anderson BM, Ford MP, Keene JS. The analgesic impact of preoperative lumbar plexus blocks for hip arthroscopy. A retrospective review. Hip Int. 2013;23:93-98.
- YaDeau JT, Tedore T, Goytizolo EA, et al. Lumbar plexus blockade reduces pain after hip arthroscopy: a prospective randomized controlled trial. Anesth Analg. 2012;115:968-972.
- Smart LR, Oetgen M, Noonan B, Medvecky M. Beginning hip arthroscopy: indications, positioning, portals, basic techniques, and complications. Arthroscopy. 2007;23:1348-1353.
- Stevens M, Harrison G, McGrail M. A modified fascia iliaca compartment block has significant morphine-sparing effect after total hip arthroplasty. Anaesth Intensive Care. 2007;35:949-952.
- Lehmann LJ, Loosen G, Weiss C, Schmittner MD. Interscalene plexus block versus general anaesthesia for shoulder surgery: a randomized controlled study. Eur J Orthop Surg Traumatol. 2015;25:255-261.
- Gonano C, Kettner SC, Ernstbrunner M, Schebesta K, Chiari A, Marhofer P. Comparison of economical aspects of interscalene brachial plexus blockade and general anaesthesia for arthroscopic shoulder surgery. Br J Anaesth. 2009;103:428-433.
- Hadzic A, Karaca PE, Hobeika P, et al. Peripheral nerve blocks result in superior recovery profile compared with general anesthesia in outpatient knee arthroscopy. Anesth Analg. 2005;100:976-981.
- Hsu LP, Oh S, Nuber GW, et al. Nerve block of the infrapatellar branch of the saphenous nerve in knee arthroscopy: a prospective, double-blinded, randomized, placebo-controlled trial. J Bone Joint Surg Am. 2013;95:1465-1472.
- Montes FR, Zarate E, Grueso R, et al. Comparison of spinal anesthesia with combined sciatic-femoral nerve block for outpatient knee arthroscopy. J Clin Anesth. 2008;20:415-420.
- Wulf H, Lowe J, Gnutzmann KH, Steinfeldt T. Femoral nerve block with ropivacaine or bupivacaine in day case anterior crucial ligament reconstruction. Acta Anaesthesiol Scand. 2010;54:414-420.
- Krych AJ, Baran S, Kuzma SA, Smith HM, Johnson RL, Levy BA. Utility of multimodal analgesia with fascia iliaca blockade for acute pain management following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22:843-847.
- Nye ZB, Horn JL, Crittenden W, Abrahams MS, Aziz MF. Ambulatory continuous posterior lumbar plexus blocks following hip arthroscopy: a review of 213 cases. J Clin Anesth. 2013;25:268-274.
- Ward JP, Albert DB, Altman R, Goldstein RY, Cuff G, Youm T. Are femoral nerve blocks effective for early postoperative pain management after hip arthroscopy? Arthroscopy. 2012;28:1064-1069.
- Liu SS, Strodtbeck WM, Richman JM, Wu CL. A comparison of regional versus general anesthesia for ambulatory anesthesia: a meta-analysis of randomized controlled trials. Anesth Analg. 2005;101:1634-1642.
- Clarke MT, Arora A, Villar RN. Hip arthroscopy: complications in 1054 cases. Clin Orthop Relat Res. 2003;406:84-88.
- Baber YF, Robinson AH, Villar RN. Is diagnostic arthroscopy of the hip worthwhile? A prospective review of 328 adults investigated for hip pain. J Bone Joint Surg Br. 1999;81:600-603.
- Byrd JW, Jones KS. Arthroscopic management of femoroacetabular impingement: minimum 2-year follow-up. Arthroscopy. 2011;27:1379-1388.
- Larson CM, Giveans MR. Arthroscopic management of femoroacetabular impingement: early outcomes measures. Arthroscopy. 2008;24:540-546.
- O'Leary JA, Berend K, Vail TP. The relationship between diagnosis and outcome in arthroscopy of the hip. Arthroscopy. 2001;17:181-188.
- Philippon M, Schenker M, Briggs K, Kuppersmith D. Femoroacetabular impingement in 45 professional athletes: associated pathologies and return to sport following arthroscopic decompression. Knee Surg Sports Traumatol Arthrosc. 2007;15:908-914.
- Potter BK, Freedman BA, Andersen RC, Bojescul JA, Kuklo TR, Murphy KP. Correlation of Short Form-36 and disability status with outcomes of arthroscopic acetabular labral debridement. Am J Sports Med. 2005;33:864-870.
- Robertson WJ, Kadrmas WR, Kelly BT. Arthroscopic management of labral tears in the hip: a systematic review of the literature. Clin Orthop Relat Res. 2007;455:88-92.
- Yusaf MA, Hame SL. Arthroscopy of the hip. Curr Sports Med Rep. 2008;7:269-274.
- Colvin AC, Harrast J, Harner C. Trends in hip arthroscopy. J Bone Joint Surg Am. 2012;94:e23.
- Cvetanovich GL, Chalmers PN, Levy DM, et al. Hip arthroscopy surgical volume trends and 30-day postoperative complications. Arthroscopy. 2016 Apr 8. [Epub before print].
- Baker JF, Byrne DP, Hunter K, Mulhall KJ. Post-operative opiate requirements after hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2011;19:1399-1402.
- Lee EM, Murphy KP, Ben-David B. Postoperative analgesia for hip arthroscopy: combined L1 and L2 paravertebral blocks. J Clin Anesth. 2008;20:462-465.
- Ganesh A, Rose JB, Wells L, et al. Continuous peripheral nerve blockade for inpatient and outpatient postoperative analgesia in children. Anesth Analg. 2007;105:1234-1242.
- Williams BA, Kentor ML, Vogt MT, et al. Femoral-sciatic nerve blocks for complex outpatient knee surgery are associated with less postoperative pain before same-day discharge: a review of 1,200 consecutive cases from the period 1996-1999. Anesthesiology. 2003;98:1206-1213.
- Zywiel MG, Stroh DA, Lee SY, Bonutti PM, Mont MA. Chronic opioid use prior to total knee arthroplasty. J Bone Joint Surg Am. 2011;93:1988-1993.
- Dold AP, Murnaghan L, Xing J, Abdallah FW, Brull R, Whelan DB. Preoperative femoral nerve block in hip arthroscopic surgery: a retrospective review of 108 consecutive cases. Am J Sports Med. 2014;42:144-149.
- Schroeder KM, Donnelly MJ, Anderson BM, Ford MP, Keene JS. The analgesic impact of preoperative lumbar plexus blocks for hip arthroscopy. A retrospective review. Hip Int. 2013;23:93-98.
- YaDeau JT, Tedore T, Goytizolo EA, et al. Lumbar plexus blockade reduces pain after hip arthroscopy: a prospective randomized controlled trial. Anesth Analg. 2012;115:968-972.
- Smart LR, Oetgen M, Noonan B, Medvecky M. Beginning hip arthroscopy: indications, positioning, portals, basic techniques, and complications. Arthroscopy. 2007;23:1348-1353.
- Stevens M, Harrison G, McGrail M. A modified fascia iliaca compartment block has significant morphine-sparing effect after total hip arthroplasty. Anaesth Intensive Care. 2007;35:949-952.
- Lehmann LJ, Loosen G, Weiss C, Schmittner MD. Interscalene plexus block versus general anaesthesia for shoulder surgery: a randomized controlled study. Eur J Orthop Surg Traumatol. 2015;25:255-261.
- Gonano C, Kettner SC, Ernstbrunner M, Schebesta K, Chiari A, Marhofer P. Comparison of economical aspects of interscalene brachial plexus blockade and general anaesthesia for arthroscopic shoulder surgery. Br J Anaesth. 2009;103:428-433.
- Hadzic A, Karaca PE, Hobeika P, et al. Peripheral nerve blocks result in superior recovery profile compared with general anesthesia in outpatient knee arthroscopy. Anesth Analg. 2005;100:976-981.
- Hsu LP, Oh S, Nuber GW, et al. Nerve block of the infrapatellar branch of the saphenous nerve in knee arthroscopy: a prospective, double-blinded, randomized, placebo-controlled trial. J Bone Joint Surg Am. 2013;95:1465-1472.
- Montes FR, Zarate E, Grueso R, et al. Comparison of spinal anesthesia with combined sciatic-femoral nerve block for outpatient knee arthroscopy. J Clin Anesth. 2008;20:415-420.
- Wulf H, Lowe J, Gnutzmann KH, Steinfeldt T. Femoral nerve block with ropivacaine or bupivacaine in day case anterior crucial ligament reconstruction. Acta Anaesthesiol Scand. 2010;54:414-420.
- Krych AJ, Baran S, Kuzma SA, Smith HM, Johnson RL, Levy BA. Utility of multimodal analgesia with fascia iliaca blockade for acute pain management following hip arthroscopy. Knee Surg Sports Traumatol Arthrosc. 2014;22:843-847.
- Nye ZB, Horn JL, Crittenden W, Abrahams MS, Aziz MF. Ambulatory continuous posterior lumbar plexus blocks following hip arthroscopy: a review of 213 cases. J Clin Anesth. 2013;25:268-274.
- Ward JP, Albert DB, Altman R, Goldstein RY, Cuff G, Youm T. Are femoral nerve blocks effective for early postoperative pain management after hip arthroscopy? Arthroscopy. 2012;28:1064-1069.
- Liu SS, Strodtbeck WM, Richman JM, Wu CL. A comparison of regional versus general anesthesia for ambulatory anesthesia: a meta-analysis of randomized controlled trials. Anesth Analg. 2005;101:1634-1642.
- Clarke MT, Arora A, Villar RN. Hip arthroscopy: complications in 1054 cases. Clin Orthop Relat Res. 2003;406:84-88.
TAKE-HOME POINTS
- Postoperative PACU pain was consistently reduced in the PNB group.
- Patients with PNBs had lower postoperative pain medication requirements and lower rates of inpatient admission compared with controls.
- Similar rates of nausea/vomiting and time to discharge were reported for PNB patients and controls.
- PNBs are associated with high rates of satisfaction and few complications.
- Future research should focus on comparing across PNB techniques.
Higher BMI tied to lower breast cancer risk in women before menopause
Although obesity increases the risk of breast cancer in postmenopausal women, a large multicenter analysis has confirmed the opposite effect in premenopausal women.
The association had “a greater magnitude than previously shown and across the entire distribution of body mass index,” wrote Minouk J. Schoemaker, PhD, of the Institute of Cancer Research in London, with his associates, on behalf of the Premenopausal Breast Cancer Collaborative Group. The protective effect of adiposity was strongest during young adulthood (ages 18-24 years), when it spanned breast cancer subtypes. “Understanding the biological mechanisms underlying these associations could have important preventive potential,” they wrote in JAMA Oncology.
Prior studies have linked greater body fat with reduced risk of breast cancer in younger women, but the effect has not been well characterized. For this analysis, the investigators pooled data from 19 cohort studies that included a total of 758,592 premenopausal women; median age was 40.6 years (interquartile range, 35.2-45.5 years).
For each 5-unit increase in BMI, the estimated reduction in risk of breast cancer was 23% among women aged 18-24 years (hazard ratio, 0.77; 95% confidence interval, 0.73-0.80), 15% in women aged 25-34 years, 13% in women aged 35-44 years, and 12% in women aged 45-54 years. There was no BMI threshold for risk reduction: the inverse correlation existed even when women were not overweight. Risk also did vary significantly among subgroups stratified by other risk factors for breast cancer. Adiposity was more protective against estrogen receptor-positive and progesterone-receptor positive breast cancers and less protective against hormone receptor–negative breast cancers, which “implies a hormonal mechanism,” the investigators said. “Body mass index at ages 25-54 years was not consistently associated with triple-negative or hormone receptor–negative breast cancer overall.”
Funders included Breast Cancer Now, the Institute of Cancer Research, the National Institutes of Health, and many others. The researchers reported having no relevant conflicts of interest.
SOURCE: Schoemaker MJ et al. JAMA Oncol. 2018; Jun 21. doi: 10.1001/jamaoncol.2018.1771.
Although obesity increases the risk of breast cancer in postmenopausal women, a large multicenter analysis has confirmed the opposite effect in premenopausal women.
The association had “a greater magnitude than previously shown and across the entire distribution of body mass index,” wrote Minouk J. Schoemaker, PhD, of the Institute of Cancer Research in London, with his associates, on behalf of the Premenopausal Breast Cancer Collaborative Group. The protective effect of adiposity was strongest during young adulthood (ages 18-24 years), when it spanned breast cancer subtypes. “Understanding the biological mechanisms underlying these associations could have important preventive potential,” they wrote in JAMA Oncology.
Prior studies have linked greater body fat with reduced risk of breast cancer in younger women, but the effect has not been well characterized. For this analysis, the investigators pooled data from 19 cohort studies that included a total of 758,592 premenopausal women; median age was 40.6 years (interquartile range, 35.2-45.5 years).
For each 5-unit increase in BMI, the estimated reduction in risk of breast cancer was 23% among women aged 18-24 years (hazard ratio, 0.77; 95% confidence interval, 0.73-0.80), 15% in women aged 25-34 years, 13% in women aged 35-44 years, and 12% in women aged 45-54 years. There was no BMI threshold for risk reduction: the inverse correlation existed even when women were not overweight. Risk also did vary significantly among subgroups stratified by other risk factors for breast cancer. Adiposity was more protective against estrogen receptor-positive and progesterone-receptor positive breast cancers and less protective against hormone receptor–negative breast cancers, which “implies a hormonal mechanism,” the investigators said. “Body mass index at ages 25-54 years was not consistently associated with triple-negative or hormone receptor–negative breast cancer overall.”
Funders included Breast Cancer Now, the Institute of Cancer Research, the National Institutes of Health, and many others. The researchers reported having no relevant conflicts of interest.
SOURCE: Schoemaker MJ et al. JAMA Oncol. 2018; Jun 21. doi: 10.1001/jamaoncol.2018.1771.
Although obesity increases the risk of breast cancer in postmenopausal women, a large multicenter analysis has confirmed the opposite effect in premenopausal women.
The association had “a greater magnitude than previously shown and across the entire distribution of body mass index,” wrote Minouk J. Schoemaker, PhD, of the Institute of Cancer Research in London, with his associates, on behalf of the Premenopausal Breast Cancer Collaborative Group. The protective effect of adiposity was strongest during young adulthood (ages 18-24 years), when it spanned breast cancer subtypes. “Understanding the biological mechanisms underlying these associations could have important preventive potential,” they wrote in JAMA Oncology.
Prior studies have linked greater body fat with reduced risk of breast cancer in younger women, but the effect has not been well characterized. For this analysis, the investigators pooled data from 19 cohort studies that included a total of 758,592 premenopausal women; median age was 40.6 years (interquartile range, 35.2-45.5 years).
For each 5-unit increase in BMI, the estimated reduction in risk of breast cancer was 23% among women aged 18-24 years (hazard ratio, 0.77; 95% confidence interval, 0.73-0.80), 15% in women aged 25-34 years, 13% in women aged 35-44 years, and 12% in women aged 45-54 years. There was no BMI threshold for risk reduction: the inverse correlation existed even when women were not overweight. Risk also did vary significantly among subgroups stratified by other risk factors for breast cancer. Adiposity was more protective against estrogen receptor-positive and progesterone-receptor positive breast cancers and less protective against hormone receptor–negative breast cancers, which “implies a hormonal mechanism,” the investigators said. “Body mass index at ages 25-54 years was not consistently associated with triple-negative or hormone receptor–negative breast cancer overall.”
Funders included Breast Cancer Now, the Institute of Cancer Research, the National Institutes of Health, and many others. The researchers reported having no relevant conflicts of interest.
SOURCE: Schoemaker MJ et al. JAMA Oncol. 2018; Jun 21. doi: 10.1001/jamaoncol.2018.1771.
FROM JAMA ONCOLOGY
Key clinical point: In premenopausal women, adiposity inversely correlated with risk of breast cancer, and showed a stronger protective effect than previously documented.
Major finding: For each 5-unit increase in BMI, the estimated reduction in risk of breast cancer was 23% among women aged 18-24 years (hazard ratio, 0.77; 95% confidence interval, 0.73-0.80), 15% in women aged 25-34 years, 13% in women aged 35-44 years, and 12% in women aged 45-54 years.
Study details: Multicenter analysis of 19 cohort studies.
Disclosures: Funders included Breast Cancer Now, the Institute of Cancer Research, the National Institutes of Health, and many others. The researchers reported having no relevant conflicts of interest.
Source: Schoemaker MJ et al. JAMA Oncol. 2018; Jun 21. doi: 10.1001/jamaoncol.2018.1771.
Right to Try: Mission accomplished?
On May 30, 2018, President Trump signed into law the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, guaranteeing patients with terminal illnesses the right to seek access to investigational drugs which have passed at least phase 1 testing. As he signed the bipartisan-backed bill into law, the president said he believes it will ultimately save “hundreds of thousands” of lives, scoring an apparent win-win-win for the president, for Congress, and for patients. A critical appraisal of the law – and of the supposition that patients will reap great benefit from it – requires that we ask two questions: Does the law degrade measures originally put in place to protect patients, and does it actually improve access to potentially life-saving drugs?
As a hematologist with about 2 decades of experience conducting clinical research involving patients with incurable cancer, I want as many patients as possible to have access to promising new therapies. Clinical trials, as the sole means of determining a candidate drug’s safety and efficacy, are conducted in a systematic fashion, with phase 1 trials focusing predominantly on safety. As any experienced researcher is aware, however, a phase 1 trial only provides a basic sense of a drug’s safety. It would be unimaginable (and unethical) to conduct subsequent phase 2 and 3 trials without including rigorous ongoing monitoring for adverse effects.

A later study evaluating all nonpediatric phase 1 trials sponsored by the National Cancer Institute’s Cancer Therapy Evaluation Program between 2001 and 2012, which included more than 8,300 patients, found the therapy-related death rate to be similarly low – approximately 1%.2
It is distinctly possible that the same drugs used in the analyzed trials would have been more toxic to patients who, for one reason or another, would not have met enrollment criteria. It is impossible to precisely quantify the degree to which risk may be increased under the Right to Try law, but even if it were tripled or quadrupled, compared with these historical expectations, the absolute risk of treatment-related death remains low. It should be noted that the incidence of nonfatal grade 3-4 adverse events in the same analyses was 10%-20% so a similar proportional increase in these events would impact a larger number of patients. Overall, it is difficult to know the degree to which Right to Try impacts the safety of fragile patients with terminal illnesses.
The answer to the second question – Is access to life-saving drugs improved? – is much easier to answer: No.
One reason is that these patients already have the right to seek access to investigational drugs through the FDA’s Expanded Access Program. An overview of the Expanded Access Program reported that approximately 11,000 patients over a period of 10 years submitted requests for access to investigational drugs and that 99% of the requests were granted.3
Proponents of the Right to Try law argue that the new law somehow simplifies the request process, but it is difficult to understand exactly how. Further, in a position paper on the topic of Right to Try policy, Lisa Kearns and Alison Bateman-House, PhD, of New York University, wrote, “In the more than 2½ years since the first [individual state right to try] law was signed, there have been no documented cases of anyone receiving access, because of a right to try law, to an experimental product that would not have been available via the FDA’s Expanded Access Program.”4
Finally, and most importantly, neither the existing Expanded Access Program nor the newly enacted Right to Try law require that pharmaceutical companies actually provide the requested drugs. Industry leaders, in testimony before Congress, have cited patient safety, costs, drug supply, and negative impact on existing clinical trial accrual as reasons pharmaceutical companies commonly deny requests for access to investigational products.
The bottom line is that the Right to Try law really lacks teeth. Despite the favorable optics for the president and Congress, it is virtually certain that Right to Try will never save hundreds of thousands of lives, and the suggestion that it will is either disingenuous or uninformed. Hopefully, though, Congress’s passage of Right to Try is indicative of a more general interest in bipartisan cooperation to improve access to affordable, high-quality health care.
Dr. Zonder is a professor in the department of oncology at the Barbara Ann Karmanos Cancer Institute and Wayne State University in Detroit. He is the leader of the KCI Myeloma and Amyloidosis Team. He is also a member of the Hematology News Editorial Advisory Board. He reported having no relevant financial disclosures.
References
1. Roberts TG Jr. et al. JAMA. 2004 Nov 3;292(17):2130-40.
2. Fukada YK et al. J Clin Oncol. 2014 May 20. doi: 10.1200/jco.2014.32.15_suppl.2552.
3. Jarow JP et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):177-9.
4. Kearns L et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):170-6.
On May 30, 2018, President Trump signed into law the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, guaranteeing patients with terminal illnesses the right to seek access to investigational drugs which have passed at least phase 1 testing. As he signed the bipartisan-backed bill into law, the president said he believes it will ultimately save “hundreds of thousands” of lives, scoring an apparent win-win-win for the president, for Congress, and for patients. A critical appraisal of the law – and of the supposition that patients will reap great benefit from it – requires that we ask two questions: Does the law degrade measures originally put in place to protect patients, and does it actually improve access to potentially life-saving drugs?
As a hematologist with about 2 decades of experience conducting clinical research involving patients with incurable cancer, I want as many patients as possible to have access to promising new therapies. Clinical trials, as the sole means of determining a candidate drug’s safety and efficacy, are conducted in a systematic fashion, with phase 1 trials focusing predominantly on safety. As any experienced researcher is aware, however, a phase 1 trial only provides a basic sense of a drug’s safety. It would be unimaginable (and unethical) to conduct subsequent phase 2 and 3 trials without including rigorous ongoing monitoring for adverse effects.
A later study evaluating all nonpediatric phase 1 trials sponsored by the National Cancer Institute’s Cancer Therapy Evaluation Program between 2001 and 2012, which included more than 8,300 patients, found the therapy-related death rate to be similarly low – approximately 1%.2
It is distinctly possible that the same drugs used in the analyzed trials would have been more toxic to patients who, for one reason or another, would not have met enrollment criteria. It is impossible to precisely quantify the degree to which risk may be increased under the Right to Try law, but even if it were tripled or quadrupled, compared with these historical expectations, the absolute risk of treatment-related death remains low. It should be noted that the incidence of nonfatal grade 3-4 adverse events in the same analyses was 10%-20% so a similar proportional increase in these events would impact a larger number of patients. Overall, it is difficult to know the degree to which Right to Try impacts the safety of fragile patients with terminal illnesses.
The answer to the second question – Is access to life-saving drugs improved? – is much easier to answer: No.
One reason is that these patients already have the right to seek access to investigational drugs through the FDA’s Expanded Access Program. An overview of the Expanded Access Program reported that approximately 11,000 patients over a period of 10 years submitted requests for access to investigational drugs and that 99% of the requests were granted.3
Proponents of the Right to Try law argue that the new law somehow simplifies the request process, but it is difficult to understand exactly how. Further, in a position paper on the topic of Right to Try policy, Lisa Kearns and Alison Bateman-House, PhD, of New York University, wrote, “In the more than 2½ years since the first [individual state right to try] law was signed, there have been no documented cases of anyone receiving access, because of a right to try law, to an experimental product that would not have been available via the FDA’s Expanded Access Program.”4
Finally, and most importantly, neither the existing Expanded Access Program nor the newly enacted Right to Try law require that pharmaceutical companies actually provide the requested drugs. Industry leaders, in testimony before Congress, have cited patient safety, costs, drug supply, and negative impact on existing clinical trial accrual as reasons pharmaceutical companies commonly deny requests for access to investigational products.
The bottom line is that the Right to Try law really lacks teeth. Despite the favorable optics for the president and Congress, it is virtually certain that Right to Try will never save hundreds of thousands of lives, and the suggestion that it will is either disingenuous or uninformed. Hopefully, though, Congress’s passage of Right to Try is indicative of a more general interest in bipartisan cooperation to improve access to affordable, high-quality health care.
Dr. Zonder is a professor in the department of oncology at the Barbara Ann Karmanos Cancer Institute and Wayne State University in Detroit. He is the leader of the KCI Myeloma and Amyloidosis Team. He is also a member of the Hematology News Editorial Advisory Board. He reported having no relevant financial disclosures.
References
1. Roberts TG Jr. et al. JAMA. 2004 Nov 3;292(17):2130-40.
2. Fukada YK et al. J Clin Oncol. 2014 May 20. doi: 10.1200/jco.2014.32.15_suppl.2552.
3. Jarow JP et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):177-9.
4. Kearns L et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):170-6.
On May 30, 2018, President Trump signed into law the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, guaranteeing patients with terminal illnesses the right to seek access to investigational drugs which have passed at least phase 1 testing. As he signed the bipartisan-backed bill into law, the president said he believes it will ultimately save “hundreds of thousands” of lives, scoring an apparent win-win-win for the president, for Congress, and for patients. A critical appraisal of the law – and of the supposition that patients will reap great benefit from it – requires that we ask two questions: Does the law degrade measures originally put in place to protect patients, and does it actually improve access to potentially life-saving drugs?
As a hematologist with about 2 decades of experience conducting clinical research involving patients with incurable cancer, I want as many patients as possible to have access to promising new therapies. Clinical trials, as the sole means of determining a candidate drug’s safety and efficacy, are conducted in a systematic fashion, with phase 1 trials focusing predominantly on safety. As any experienced researcher is aware, however, a phase 1 trial only provides a basic sense of a drug’s safety. It would be unimaginable (and unethical) to conduct subsequent phase 2 and 3 trials without including rigorous ongoing monitoring for adverse effects.
A later study evaluating all nonpediatric phase 1 trials sponsored by the National Cancer Institute’s Cancer Therapy Evaluation Program between 2001 and 2012, which included more than 8,300 patients, found the therapy-related death rate to be similarly low – approximately 1%.2
It is distinctly possible that the same drugs used in the analyzed trials would have been more toxic to patients who, for one reason or another, would not have met enrollment criteria. It is impossible to precisely quantify the degree to which risk may be increased under the Right to Try law, but even if it were tripled or quadrupled, compared with these historical expectations, the absolute risk of treatment-related death remains low. It should be noted that the incidence of nonfatal grade 3-4 adverse events in the same analyses was 10%-20% so a similar proportional increase in these events would impact a larger number of patients. Overall, it is difficult to know the degree to which Right to Try impacts the safety of fragile patients with terminal illnesses.
The answer to the second question – Is access to life-saving drugs improved? – is much easier to answer: No.
One reason is that these patients already have the right to seek access to investigational drugs through the FDA’s Expanded Access Program. An overview of the Expanded Access Program reported that approximately 11,000 patients over a period of 10 years submitted requests for access to investigational drugs and that 99% of the requests were granted.3
Proponents of the Right to Try law argue that the new law somehow simplifies the request process, but it is difficult to understand exactly how. Further, in a position paper on the topic of Right to Try policy, Lisa Kearns and Alison Bateman-House, PhD, of New York University, wrote, “In the more than 2½ years since the first [individual state right to try] law was signed, there have been no documented cases of anyone receiving access, because of a right to try law, to an experimental product that would not have been available via the FDA’s Expanded Access Program.”4
Finally, and most importantly, neither the existing Expanded Access Program nor the newly enacted Right to Try law require that pharmaceutical companies actually provide the requested drugs. Industry leaders, in testimony before Congress, have cited patient safety, costs, drug supply, and negative impact on existing clinical trial accrual as reasons pharmaceutical companies commonly deny requests for access to investigational products.
The bottom line is that the Right to Try law really lacks teeth. Despite the favorable optics for the president and Congress, it is virtually certain that Right to Try will never save hundreds of thousands of lives, and the suggestion that it will is either disingenuous or uninformed. Hopefully, though, Congress’s passage of Right to Try is indicative of a more general interest in bipartisan cooperation to improve access to affordable, high-quality health care.
Dr. Zonder is a professor in the department of oncology at the Barbara Ann Karmanos Cancer Institute and Wayne State University in Detroit. He is the leader of the KCI Myeloma and Amyloidosis Team. He is also a member of the Hematology News Editorial Advisory Board. He reported having no relevant financial disclosures.
References
1. Roberts TG Jr. et al. JAMA. 2004 Nov 3;292(17):2130-40.
2. Fukada YK et al. J Clin Oncol. 2014 May 20. doi: 10.1200/jco.2014.32.15_suppl.2552.
3. Jarow JP et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):177-9.
4. Kearns L et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):170-6.
New SLE classification criteria reset disease definition
AMSTERDAM – The new systemic lupus erythematosus classification criteria of the American College of Rheumatology and the European League Against Rheumatism are based on a point system that will produce a “paradigm shift” in how the disease gets studied going forward, said Sindhu Johnson, MD, while presenting the latest version of the newly revised classification scheme at the European Congress of Rheumatology.
Until now, classification of systemic lupus erythematosus (SLE) was a yes-or-no decision, based on whether the patient had a minimum number of characteristic signs or symptoms. The new criteria, which are on track for formal endorsement before the end of 2018 by the two medical societies that sponsored the revision, instead use a point system that gives varying weight to each of the 22 criteria. A patient needs to score at least 10 points from these criteria, and all patients classified with SLE also must have an antinuclear antibody (ANA) titer of at least 1:80 on HEp-2 cells or an equivalent positive test. This means that the criteria also can define patients who just miss classification with SLE by meeting the ANA standard and by tallying 8 or 9 points, and the criteria also identify patients who far exceed the classification threshold by having the requisite ANA plus racking up as many as, perhaps, 20 or 30 points.
“This is a real research opportunity,” to follow patients who fall just short with 8 or 9 points to assess their longer-term prognosis, as well as to study whether “higher scores mean a higher risk for developing a bad outcome,” said Dr. Johnson, a rheumatologist at the University of Toronto and director of the Toronto Scleroderma Program. Other areas for future research with the new criteria include seeing how they work in various SLE subgroups, such as patients with renal-predominant disease or skin-predominant disease, and also seeing how they work in various ethnic populations.
“Diagnosis of lupus still falls within the realm of the treating physician,” but the classification criteria “inform our concept of the disease,” Dr. Johnson said in a video interview. “The new criteria allow for a shift in the way we think of the disease.”
For example, for the first time, the new criteria includes fever as a classification criterion, which receives 2 points if an infectious or other non-SLE cause can be discounted. Fever has recently been identified as a marker of early-stage SLE in at least some patients, and its addition to the classification criteria “adds a new dimension to how we think about the disease and allows us to distinguish early disease from mimicking diseases,” she explained. At the other end of the classification spectrum, a finding of class III or IV lupus nephritis on renal biopsy receives 10 points, and hence, this one finding plus having a high enough level of ANA leads to SLE classification regardless of whether the patient has any other signs or symptoms of the disease.
That’s because “85% of our experts said that they would feel confident classifying a patient as having lupus based only on a renal biopsy” and ANA positivity, said Dr. Johnson, who served as the ACR-appointed cochair of the criteria-writing panel along with a cochair selected by EULAR, Martin Aringer, MD, PhD, of the Technical University of Dresden (Germany). She cautioned that other levels of lupus nephritis, class II or V, confer only 8 points to the classification and so by themselves are not enough to label a person as having lupus.
During her presentation, Dr. Johnson cited the high levels of sensitivity and specificity that the new classification criteria demonstrated in a validation cohort of more than 1,000 cases and controls. In the validation analysis, the new criteria had a sensitivity of 96.12% and specificity of 94.43% for classifying SLE, giving the new criteria a better result on both these measures than either the 1997 ACR criteria (Arthritis Rheum. 1997 Sept;40[9]:1725) or the 2012 Systemic Lupus International Collaborating Clinics criteria (Arthritis Rheum. 2012 Aug;64[8]:2677-86).
The 22 criteria cluster into seven separate clinical domains and three different immunologic domains. The point values assigned to each criterion range from 2 to 10 points.
Dr. Johnson had no disclosures.
AMSTERDAM – The new systemic lupus erythematosus classification criteria of the American College of Rheumatology and the European League Against Rheumatism are based on a point system that will produce a “paradigm shift” in how the disease gets studied going forward, said Sindhu Johnson, MD, while presenting the latest version of the newly revised classification scheme at the European Congress of Rheumatology.
Until now, classification of systemic lupus erythematosus (SLE) was a yes-or-no decision, based on whether the patient had a minimum number of characteristic signs or symptoms. The new criteria, which are on track for formal endorsement before the end of 2018 by the two medical societies that sponsored the revision, instead use a point system that gives varying weight to each of the 22 criteria. A patient needs to score at least 10 points from these criteria, and all patients classified with SLE also must have an antinuclear antibody (ANA) titer of at least 1:80 on HEp-2 cells or an equivalent positive test. This means that the criteria also can define patients who just miss classification with SLE by meeting the ANA standard and by tallying 8 or 9 points, and the criteria also identify patients who far exceed the classification threshold by having the requisite ANA plus racking up as many as, perhaps, 20 or 30 points.
“This is a real research opportunity,” to follow patients who fall just short with 8 or 9 points to assess their longer-term prognosis, as well as to study whether “higher scores mean a higher risk for developing a bad outcome,” said Dr. Johnson, a rheumatologist at the University of Toronto and director of the Toronto Scleroderma Program. Other areas for future research with the new criteria include seeing how they work in various SLE subgroups, such as patients with renal-predominant disease or skin-predominant disease, and also seeing how they work in various ethnic populations.
“Diagnosis of lupus still falls within the realm of the treating physician,” but the classification criteria “inform our concept of the disease,” Dr. Johnson said in a video interview. “The new criteria allow for a shift in the way we think of the disease.”
For example, for the first time, the new criteria includes fever as a classification criterion, which receives 2 points if an infectious or other non-SLE cause can be discounted. Fever has recently been identified as a marker of early-stage SLE in at least some patients, and its addition to the classification criteria “adds a new dimension to how we think about the disease and allows us to distinguish early disease from mimicking diseases,” she explained. At the other end of the classification spectrum, a finding of class III or IV lupus nephritis on renal biopsy receives 10 points, and hence, this one finding plus having a high enough level of ANA leads to SLE classification regardless of whether the patient has any other signs or symptoms of the disease.
That’s because “85% of our experts said that they would feel confident classifying a patient as having lupus based only on a renal biopsy” and ANA positivity, said Dr. Johnson, who served as the ACR-appointed cochair of the criteria-writing panel along with a cochair selected by EULAR, Martin Aringer, MD, PhD, of the Technical University of Dresden (Germany). She cautioned that other levels of lupus nephritis, class II or V, confer only 8 points to the classification and so by themselves are not enough to label a person as having lupus.
During her presentation, Dr. Johnson cited the high levels of sensitivity and specificity that the new classification criteria demonstrated in a validation cohort of more than 1,000 cases and controls. In the validation analysis, the new criteria had a sensitivity of 96.12% and specificity of 94.43% for classifying SLE, giving the new criteria a better result on both these measures than either the 1997 ACR criteria (Arthritis Rheum. 1997 Sept;40[9]:1725) or the 2012 Systemic Lupus International Collaborating Clinics criteria (Arthritis Rheum. 2012 Aug;64[8]:2677-86).
The 22 criteria cluster into seven separate clinical domains and three different immunologic domains. The point values assigned to each criterion range from 2 to 10 points.
Dr. Johnson had no disclosures.
AMSTERDAM – The new systemic lupus erythematosus classification criteria of the American College of Rheumatology and the European League Against Rheumatism are based on a point system that will produce a “paradigm shift” in how the disease gets studied going forward, said Sindhu Johnson, MD, while presenting the latest version of the newly revised classification scheme at the European Congress of Rheumatology.
Until now, classification of systemic lupus erythematosus (SLE) was a yes-or-no decision, based on whether the patient had a minimum number of characteristic signs or symptoms. The new criteria, which are on track for formal endorsement before the end of 2018 by the two medical societies that sponsored the revision, instead use a point system that gives varying weight to each of the 22 criteria. A patient needs to score at least 10 points from these criteria, and all patients classified with SLE also must have an antinuclear antibody (ANA) titer of at least 1:80 on HEp-2 cells or an equivalent positive test. This means that the criteria also can define patients who just miss classification with SLE by meeting the ANA standard and by tallying 8 or 9 points, and the criteria also identify patients who far exceed the classification threshold by having the requisite ANA plus racking up as many as, perhaps, 20 or 30 points.
“This is a real research opportunity,” to follow patients who fall just short with 8 or 9 points to assess their longer-term prognosis, as well as to study whether “higher scores mean a higher risk for developing a bad outcome,” said Dr. Johnson, a rheumatologist at the University of Toronto and director of the Toronto Scleroderma Program. Other areas for future research with the new criteria include seeing how they work in various SLE subgroups, such as patients with renal-predominant disease or skin-predominant disease, and also seeing how they work in various ethnic populations.
“Diagnosis of lupus still falls within the realm of the treating physician,” but the classification criteria “inform our concept of the disease,” Dr. Johnson said in a video interview. “The new criteria allow for a shift in the way we think of the disease.”
For example, for the first time, the new criteria includes fever as a classification criterion, which receives 2 points if an infectious or other non-SLE cause can be discounted. Fever has recently been identified as a marker of early-stage SLE in at least some patients, and its addition to the classification criteria “adds a new dimension to how we think about the disease and allows us to distinguish early disease from mimicking diseases,” she explained. At the other end of the classification spectrum, a finding of class III or IV lupus nephritis on renal biopsy receives 10 points, and hence, this one finding plus having a high enough level of ANA leads to SLE classification regardless of whether the patient has any other signs or symptoms of the disease.
That’s because “85% of our experts said that they would feel confident classifying a patient as having lupus based only on a renal biopsy” and ANA positivity, said Dr. Johnson, who served as the ACR-appointed cochair of the criteria-writing panel along with a cochair selected by EULAR, Martin Aringer, MD, PhD, of the Technical University of Dresden (Germany). She cautioned that other levels of lupus nephritis, class II or V, confer only 8 points to the classification and so by themselves are not enough to label a person as having lupus.
During her presentation, Dr. Johnson cited the high levels of sensitivity and specificity that the new classification criteria demonstrated in a validation cohort of more than 1,000 cases and controls. In the validation analysis, the new criteria had a sensitivity of 96.12% and specificity of 94.43% for classifying SLE, giving the new criteria a better result on both these measures than either the 1997 ACR criteria (Arthritis Rheum. 1997 Sept;40[9]:1725) or the 2012 Systemic Lupus International Collaborating Clinics criteria (Arthritis Rheum. 2012 Aug;64[8]:2677-86).
The 22 criteria cluster into seven separate clinical domains and three different immunologic domains. The point values assigned to each criterion range from 2 to 10 points.
Dr. Johnson had no disclosures.
REPORTING FROM THE EULAR 2018 CONGRESS
Norovirus vaccine appears promising in children
MALMO, SWEDEN – in an interim analysis of an ongoing phase 2 study, Taisei Masuda, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
The randomized, double-blind, multinational trial remains blinded because follow-up is continuing, so – to the disappointment of the ESPID audience – there are as yet no data on duration of antibody persistence or clinical efficacy.

However, an earlier phase 2 study in 420 healthy participants aged 18-64 years showed that the Takeda vaccine elicited persistent immune responses 1 year post vaccination and that higher antibody levels correlated with a reduced frequency of moderate to severe vomiting and diarrheal illness following oral challenge with norovirus (Clin Vaccine Immunol. 2015 Aug;22[8]:923-9). Follow-up will continue in order to learn how long the protective immune response lasts in adults, according to Dr. Masuda, of Takeda Pharmaceuticals in Zurich.
The bivalent Takeda vaccine is the first candidate vaccine to reach the randomized trial stage. An oral vaccine in tablet form under development by Vaxart, a San Francisco Bay Area biotech company, recently completed preliminary phase 1 studies.
Dr. Masuda explained that the Takeda vaccine contains virus-like particle antigens from norovirus strains GI.1 and GII.4c, which together account for the majority of human norovirus illness. These virus-like particles are formed on the outer surface of the virus. Of note, virus-like particle–based vaccines against hepatitis B and human papillomavirus have won regulatory approval in the United States, Europe, and elsewhere.
He presented data on 120 healthy subjects aged 1 year to less than 4 years old and another 120 aged 4 years to less than 9 years. They are part of a larger phase 2 study of 840 children as young as age 6 weeks. This was a dose-finding study, so participants received various doses of the vaccine on day 1 and either a second dose or a saline injection 28 days later. The vaccine, which contains aluminum hydroxide to enhance immunogenicity, comes in prefilled syringes.
At 57 days of follow-up in this interim analysis, protective seroresponse rates as defined by at least a fourfold increase in histo-blood group antigen–blocking titers approached 100%. In the older group, this was typically achieved with a single dose of vaccine. However, the younger group of children generally derived further benefit from a second dose, according to Dr. Masuda.
In terms of safety concerns, he said no serious adverse events occurred in the study and no one withdrew from the trial because of vaccine-related side effects. The overall safety picture was the same in the two age groups. The incidence of fever of 38° C or higher was similar after administration of vaccine and placebo. Injection site pain occurred in one-quarter of younger vaccine recipients, in 38%-63% of those aged 4 years or older, and in 17%-22% who got placebo injections. Those and other local and systemic adverse events were mostly mild and transient. Their incidence and severity weren’t related to vaccine dosage.
In sum, Dr. Masuda deemed the safety profile “clinically acceptable.”
Session chair Karina Butler, MD, of Temple Street Children’s University Hospital, Dublin, raised the question of how might this vaccine, which may require two doses in younger children, fit into an already crowded pediatric immunization schedule – will parents and physicians embrace it?
Dr. Masuda replied that noroviruses are the No. 1 cause of acute gastroenteritis worldwide and there is a clamor for development of effective vaccines to protect the groups that bear the greatest burden of disease, including children, the elderly, military personnel, cruise ship vacationers, and others who experience crowded conditions. He expressed confidence that a safe and effective vaccine will be in high demand.
“In the future, we’ll look at the possibility of a combination vaccine,” he added.
In response to audience questions, Dr. Masuda said that in adult studies higher levels of immunogenicity have been achieved after vaccination, compared with natural infection; however, there are as yet no pediatric data on that score. Also, investigators have seen evidence of cross-reactivity to the vaccine in some but not all naturally circulating nonvaccine strains.
The vaccine formulation being carried forward into advanced clinical trials in adults is 15 mcg of GI.1/50 mcg of GII.4c (J Infect Dis. 2018 Jan 30;217[4]:597-607).
The phase 2 study presented by Dr. Masuda was supported by the U.S. Army.
MALMO, SWEDEN – in an interim analysis of an ongoing phase 2 study, Taisei Masuda, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
The randomized, double-blind, multinational trial remains blinded because follow-up is continuing, so – to the disappointment of the ESPID audience – there are as yet no data on duration of antibody persistence or clinical efficacy.
However, an earlier phase 2 study in 420 healthy participants aged 18-64 years showed that the Takeda vaccine elicited persistent immune responses 1 year post vaccination and that higher antibody levels correlated with a reduced frequency of moderate to severe vomiting and diarrheal illness following oral challenge with norovirus (Clin Vaccine Immunol. 2015 Aug;22[8]:923-9). Follow-up will continue in order to learn how long the protective immune response lasts in adults, according to Dr. Masuda, of Takeda Pharmaceuticals in Zurich.
The bivalent Takeda vaccine is the first candidate vaccine to reach the randomized trial stage. An oral vaccine in tablet form under development by Vaxart, a San Francisco Bay Area biotech company, recently completed preliminary phase 1 studies.
Dr. Masuda explained that the Takeda vaccine contains virus-like particle antigens from norovirus strains GI.1 and GII.4c, which together account for the majority of human norovirus illness. These virus-like particles are formed on the outer surface of the virus. Of note, virus-like particle–based vaccines against hepatitis B and human papillomavirus have won regulatory approval in the United States, Europe, and elsewhere.
He presented data on 120 healthy subjects aged 1 year to less than 4 years old and another 120 aged 4 years to less than 9 years. They are part of a larger phase 2 study of 840 children as young as age 6 weeks. This was a dose-finding study, so participants received various doses of the vaccine on day 1 and either a second dose or a saline injection 28 days later. The vaccine, which contains aluminum hydroxide to enhance immunogenicity, comes in prefilled syringes.
At 57 days of follow-up in this interim analysis, protective seroresponse rates as defined by at least a fourfold increase in histo-blood group antigen–blocking titers approached 100%. In the older group, this was typically achieved with a single dose of vaccine. However, the younger group of children generally derived further benefit from a second dose, according to Dr. Masuda.
In terms of safety concerns, he said no serious adverse events occurred in the study and no one withdrew from the trial because of vaccine-related side effects. The overall safety picture was the same in the two age groups. The incidence of fever of 38° C or higher was similar after administration of vaccine and placebo. Injection site pain occurred in one-quarter of younger vaccine recipients, in 38%-63% of those aged 4 years or older, and in 17%-22% who got placebo injections. Those and other local and systemic adverse events were mostly mild and transient. Their incidence and severity weren’t related to vaccine dosage.
In sum, Dr. Masuda deemed the safety profile “clinically acceptable.”
Session chair Karina Butler, MD, of Temple Street Children’s University Hospital, Dublin, raised the question of how might this vaccine, which may require two doses in younger children, fit into an already crowded pediatric immunization schedule – will parents and physicians embrace it?
Dr. Masuda replied that noroviruses are the No. 1 cause of acute gastroenteritis worldwide and there is a clamor for development of effective vaccines to protect the groups that bear the greatest burden of disease, including children, the elderly, military personnel, cruise ship vacationers, and others who experience crowded conditions. He expressed confidence that a safe and effective vaccine will be in high demand.
“In the future, we’ll look at the possibility of a combination vaccine,” he added.
In response to audience questions, Dr. Masuda said that in adult studies higher levels of immunogenicity have been achieved after vaccination, compared with natural infection; however, there are as yet no pediatric data on that score. Also, investigators have seen evidence of cross-reactivity to the vaccine in some but not all naturally circulating nonvaccine strains.
The vaccine formulation being carried forward into advanced clinical trials in adults is 15 mcg of GI.1/50 mcg of GII.4c (J Infect Dis. 2018 Jan 30;217[4]:597-607).
The phase 2 study presented by Dr. Masuda was supported by the U.S. Army.
MALMO, SWEDEN – in an interim analysis of an ongoing phase 2 study, Taisei Masuda, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
The randomized, double-blind, multinational trial remains blinded because follow-up is continuing, so – to the disappointment of the ESPID audience – there are as yet no data on duration of antibody persistence or clinical efficacy.
However, an earlier phase 2 study in 420 healthy participants aged 18-64 years showed that the Takeda vaccine elicited persistent immune responses 1 year post vaccination and that higher antibody levels correlated with a reduced frequency of moderate to severe vomiting and diarrheal illness following oral challenge with norovirus (Clin Vaccine Immunol. 2015 Aug;22[8]:923-9). Follow-up will continue in order to learn how long the protective immune response lasts in adults, according to Dr. Masuda, of Takeda Pharmaceuticals in Zurich.
The bivalent Takeda vaccine is the first candidate vaccine to reach the randomized trial stage. An oral vaccine in tablet form under development by Vaxart, a San Francisco Bay Area biotech company, recently completed preliminary phase 1 studies.
Dr. Masuda explained that the Takeda vaccine contains virus-like particle antigens from norovirus strains GI.1 and GII.4c, which together account for the majority of human norovirus illness. These virus-like particles are formed on the outer surface of the virus. Of note, virus-like particle–based vaccines against hepatitis B and human papillomavirus have won regulatory approval in the United States, Europe, and elsewhere.
He presented data on 120 healthy subjects aged 1 year to less than 4 years old and another 120 aged 4 years to less than 9 years. They are part of a larger phase 2 study of 840 children as young as age 6 weeks. This was a dose-finding study, so participants received various doses of the vaccine on day 1 and either a second dose or a saline injection 28 days later. The vaccine, which contains aluminum hydroxide to enhance immunogenicity, comes in prefilled syringes.
At 57 days of follow-up in this interim analysis, protective seroresponse rates as defined by at least a fourfold increase in histo-blood group antigen–blocking titers approached 100%. In the older group, this was typically achieved with a single dose of vaccine. However, the younger group of children generally derived further benefit from a second dose, according to Dr. Masuda.
In terms of safety concerns, he said no serious adverse events occurred in the study and no one withdrew from the trial because of vaccine-related side effects. The overall safety picture was the same in the two age groups. The incidence of fever of 38° C or higher was similar after administration of vaccine and placebo. Injection site pain occurred in one-quarter of younger vaccine recipients, in 38%-63% of those aged 4 years or older, and in 17%-22% who got placebo injections. Those and other local and systemic adverse events were mostly mild and transient. Their incidence and severity weren’t related to vaccine dosage.
In sum, Dr. Masuda deemed the safety profile “clinically acceptable.”
Session chair Karina Butler, MD, of Temple Street Children’s University Hospital, Dublin, raised the question of how might this vaccine, which may require two doses in younger children, fit into an already crowded pediatric immunization schedule – will parents and physicians embrace it?
Dr. Masuda replied that noroviruses are the No. 1 cause of acute gastroenteritis worldwide and there is a clamor for development of effective vaccines to protect the groups that bear the greatest burden of disease, including children, the elderly, military personnel, cruise ship vacationers, and others who experience crowded conditions. He expressed confidence that a safe and effective vaccine will be in high demand.
“In the future, we’ll look at the possibility of a combination vaccine,” he added.
In response to audience questions, Dr. Masuda said that in adult studies higher levels of immunogenicity have been achieved after vaccination, compared with natural infection; however, there are as yet no pediatric data on that score. Also, investigators have seen evidence of cross-reactivity to the vaccine in some but not all naturally circulating nonvaccine strains.
The vaccine formulation being carried forward into advanced clinical trials in adults is 15 mcg of GI.1/50 mcg of GII.4c (J Infect Dis. 2018 Jan 30;217[4]:597-607).
The phase 2 study presented by Dr. Masuda was supported by the U.S. Army.
REPORTING FROM ESPID 2018
Key clinical point: Hope runs high that an effective norovirus vaccine is in the works.
Major finding: Protective seroresponse rates against the two chief disease-causing strains of norovirus were seen in nearly 100% of vaccinated children aged 1-8 years.
Study details: This is an ongoing prospective, multicenter, double-blind, phase 2 randomized trial including 840 children.
Disclosures: The study was supported by the U.S. Army and presented by an employee of Takeda Pharmaceuticals.
FDA: MiniMed 670G now available for younger diabetes patients
The MiniMed 670G hybrid closed loop system has been approved to help manage basal insulin levels in patients aged 7-13 years who have type 1 diabetes, according to a Food and Drug Administration announcement.
The system, manufactured by Medtronic, automatically measures insulin levels every 5 minutes using an included sensor and then delivers insulin as needed through its insulin pump and attached infusion patch.
As part of this approval for children aged 7-13 years, the FDA is requiring the product developer to perform a postmarket study to evaluate how the device performs in this age group in real-world settings.
“Caregivers and families of young patients with diabetes face unique challenges in managing this disease, in particular the round-the-clock glucose monitoring that can be disruptive to people’s lives,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The device was approved in September 2017 for use in patients aged 14 years and older.
Read more about this approval in the full FDA announcement.
The MiniMed 670G hybrid closed loop system has been approved to help manage basal insulin levels in patients aged 7-13 years who have type 1 diabetes, according to a Food and Drug Administration announcement.
The system, manufactured by Medtronic, automatically measures insulin levels every 5 minutes using an included sensor and then delivers insulin as needed through its insulin pump and attached infusion patch.
As part of this approval for children aged 7-13 years, the FDA is requiring the product developer to perform a postmarket study to evaluate how the device performs in this age group in real-world settings.
“Caregivers and families of young patients with diabetes face unique challenges in managing this disease, in particular the round-the-clock glucose monitoring that can be disruptive to people’s lives,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The device was approved in September 2017 for use in patients aged 14 years and older.
Read more about this approval in the full FDA announcement.
The MiniMed 670G hybrid closed loop system has been approved to help manage basal insulin levels in patients aged 7-13 years who have type 1 diabetes, according to a Food and Drug Administration announcement.
The system, manufactured by Medtronic, automatically measures insulin levels every 5 minutes using an included sensor and then delivers insulin as needed through its insulin pump and attached infusion patch.
As part of this approval for children aged 7-13 years, the FDA is requiring the product developer to perform a postmarket study to evaluate how the device performs in this age group in real-world settings.
“Caregivers and families of young patients with diabetes face unique challenges in managing this disease, in particular the round-the-clock glucose monitoring that can be disruptive to people’s lives,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The device was approved in September 2017 for use in patients aged 14 years and older.
Read more about this approval in the full FDA announcement.
Trifluridine/tipiracil improves survival of metastatic gastric cancer
BARCELONA – In patients with heavily pretreated metastatic gastric cancer, trifluridine/tipiracil was associated with a brief but statistically significant survival benefit, compared with placebo.
In the randomized, controlled TAGS (TAS-102 Gastric Study), median overall survival, the primary endpoint, was 5.7 months for patients assigned to receive trifluridine/tipiracil, compared with 3.6 months for patients randomized to placebo, reported Josep Tabernero, MD, PhD, of Vall d’Hebron Institute of Oncology in Barcelona.

Trifluridine/tipiracil (Lonsurf) is a combination of an oral thymidine analog (trifluridine, or FTD) and a thymidine phosphorylase inhibitor (tipiracil, or TPI).
It was approved by the Food and Drug Administration in 2015 for the treatment of patients with metastatic colorectal cancer who have been previously treated with chemotherapy and biological therapy.
In a phase 2 study conducted in Japan with 29 patients with metastatic gastric cancer that had progressed after chemotherapy with fluoropyrimidine, platinum, taxanes, or irinotecan, FTD/TPI was associated with a median overall survival of 8.7 months and an investigator-assessed disease control rate of 65.5%, Dr. Tabernero noted.
In the TAGS trial, patients with metastatic gastric cancer, including cancers of the gastroesophageal junction who had disease progression after two or more prior lines of therapy were randomized on a 2:1 basis to receive either oral FTD/TPI 35 mg/m2 twice daily on days 1-5 and 8-12 of each 28-day cycle or to receive placebo. In both arms, patients also received best supportive care.
As noted, the trial met its primary endpoint, with a 2.1 month improvement in median survival over placebo, which translated into a hazard ratio for death with FTD/TPI of 0.69 (P = .0003).
The 12-month overall survival rate for the FTD/TPI group was 21%, compared with 13% for the placebo group.
Median progression-free survival, a secondary endpoint, was also slightly but significantly better with FTD/TPI at 2.0 vs. 1.8 months (HR, 0.57; P less than .0001). Six-month progression-free survival rates were 15% and 6%, respectively.
Grade 3 or greater adverse events occurred in 80% of patients on FTD/TPI versus 58% of those on placebo. The drug combination was associated with more treatment-related adverse events (81% vs. 57%) but fewer adverse events leading to discontinuation (13% vs. 17%) and fewer treatment-related deaths (0.3 % vs. 0.6%).
FTD/TPI was also associated with more cases of grade 3 or 4 neutropenia, leukopenia, lymphocytopenia, anemia, and thrombocytopenia. Six patients (2%) treated with FTD/TPI had grade 3 or 4 febrile neutropenia, compared with none in the placebo group.
“The safety profile of trifluridine/tipiracil was predictable, manageable, and comparable to the population previously evaluated with metastatic colorectal cancer, with no new safety signals,” Dr. Tabernero said.
SOURCE: Tabernero J et al. ESMO GI 2018, Abstract LBA 002.
BARCELONA – In patients with heavily pretreated metastatic gastric cancer, trifluridine/tipiracil was associated with a brief but statistically significant survival benefit, compared with placebo.
In the randomized, controlled TAGS (TAS-102 Gastric Study), median overall survival, the primary endpoint, was 5.7 months for patients assigned to receive trifluridine/tipiracil, compared with 3.6 months for patients randomized to placebo, reported Josep Tabernero, MD, PhD, of Vall d’Hebron Institute of Oncology in Barcelona.
Trifluridine/tipiracil (Lonsurf) is a combination of an oral thymidine analog (trifluridine, or FTD) and a thymidine phosphorylase inhibitor (tipiracil, or TPI).
It was approved by the Food and Drug Administration in 2015 for the treatment of patients with metastatic colorectal cancer who have been previously treated with chemotherapy and biological therapy.
In a phase 2 study conducted in Japan with 29 patients with metastatic gastric cancer that had progressed after chemotherapy with fluoropyrimidine, platinum, taxanes, or irinotecan, FTD/TPI was associated with a median overall survival of 8.7 months and an investigator-assessed disease control rate of 65.5%, Dr. Tabernero noted.
In the TAGS trial, patients with metastatic gastric cancer, including cancers of the gastroesophageal junction who had disease progression after two or more prior lines of therapy were randomized on a 2:1 basis to receive either oral FTD/TPI 35 mg/m2 twice daily on days 1-5 and 8-12 of each 28-day cycle or to receive placebo. In both arms, patients also received best supportive care.
As noted, the trial met its primary endpoint, with a 2.1 month improvement in median survival over placebo, which translated into a hazard ratio for death with FTD/TPI of 0.69 (P = .0003).
The 12-month overall survival rate for the FTD/TPI group was 21%, compared with 13% for the placebo group.
Median progression-free survival, a secondary endpoint, was also slightly but significantly better with FTD/TPI at 2.0 vs. 1.8 months (HR, 0.57; P less than .0001). Six-month progression-free survival rates were 15% and 6%, respectively.
Grade 3 or greater adverse events occurred in 80% of patients on FTD/TPI versus 58% of those on placebo. The drug combination was associated with more treatment-related adverse events (81% vs. 57%) but fewer adverse events leading to discontinuation (13% vs. 17%) and fewer treatment-related deaths (0.3 % vs. 0.6%).
FTD/TPI was also associated with more cases of grade 3 or 4 neutropenia, leukopenia, lymphocytopenia, anemia, and thrombocytopenia. Six patients (2%) treated with FTD/TPI had grade 3 or 4 febrile neutropenia, compared with none in the placebo group.
“The safety profile of trifluridine/tipiracil was predictable, manageable, and comparable to the population previously evaluated with metastatic colorectal cancer, with no new safety signals,” Dr. Tabernero said.
SOURCE: Tabernero J et al. ESMO GI 2018, Abstract LBA 002.
BARCELONA – In patients with heavily pretreated metastatic gastric cancer, trifluridine/tipiracil was associated with a brief but statistically significant survival benefit, compared with placebo.
In the randomized, controlled TAGS (TAS-102 Gastric Study), median overall survival, the primary endpoint, was 5.7 months for patients assigned to receive trifluridine/tipiracil, compared with 3.6 months for patients randomized to placebo, reported Josep Tabernero, MD, PhD, of Vall d’Hebron Institute of Oncology in Barcelona.
Trifluridine/tipiracil (Lonsurf) is a combination of an oral thymidine analog (trifluridine, or FTD) and a thymidine phosphorylase inhibitor (tipiracil, or TPI).
It was approved by the Food and Drug Administration in 2015 for the treatment of patients with metastatic colorectal cancer who have been previously treated with chemotherapy and biological therapy.
In a phase 2 study conducted in Japan with 29 patients with metastatic gastric cancer that had progressed after chemotherapy with fluoropyrimidine, platinum, taxanes, or irinotecan, FTD/TPI was associated with a median overall survival of 8.7 months and an investigator-assessed disease control rate of 65.5%, Dr. Tabernero noted.
In the TAGS trial, patients with metastatic gastric cancer, including cancers of the gastroesophageal junction who had disease progression after two or more prior lines of therapy were randomized on a 2:1 basis to receive either oral FTD/TPI 35 mg/m2 twice daily on days 1-5 and 8-12 of each 28-day cycle or to receive placebo. In both arms, patients also received best supportive care.
As noted, the trial met its primary endpoint, with a 2.1 month improvement in median survival over placebo, which translated into a hazard ratio for death with FTD/TPI of 0.69 (P = .0003).
The 12-month overall survival rate for the FTD/TPI group was 21%, compared with 13% for the placebo group.
Median progression-free survival, a secondary endpoint, was also slightly but significantly better with FTD/TPI at 2.0 vs. 1.8 months (HR, 0.57; P less than .0001). Six-month progression-free survival rates were 15% and 6%, respectively.
Grade 3 or greater adverse events occurred in 80% of patients on FTD/TPI versus 58% of those on placebo. The drug combination was associated with more treatment-related adverse events (81% vs. 57%) but fewer adverse events leading to discontinuation (13% vs. 17%) and fewer treatment-related deaths (0.3 % vs. 0.6%).
FTD/TPI was also associated with more cases of grade 3 or 4 neutropenia, leukopenia, lymphocytopenia, anemia, and thrombocytopenia. Six patients (2%) treated with FTD/TPI had grade 3 or 4 febrile neutropenia, compared with none in the placebo group.
“The safety profile of trifluridine/tipiracil was predictable, manageable, and comparable to the population previously evaluated with metastatic colorectal cancer, with no new safety signals,” Dr. Tabernero said.
SOURCE: Tabernero J et al. ESMO GI 2018, Abstract LBA 002.
REPORTING FROM ESMO GI 2018
Key clinical point: Trifluridine/tipiracil (FTD/TIP) produced a small but significant survival benefit in patients with advanced gastric cancer.
Major finding: Median overall survival was 5.7 months with trifluridine/tipiracil versus 3.6 months with placebo.
Study details: Randomized, placebo-controlled trial in 506 patients with metastatic gastric cancer that progressed on at least two prior lines of therapy.
Disclosures: The study was sponsored by Taiho Oncology. Dr. Tabernero disclosed a consulting or advisory role for the company and others.
Source: Tabernero J et al. ESMO GI 2018, Abstract LBA 002.
JAK inhibitor therapy promising for refractory dermatomyositis
ORLANDO – The according to Ruth Ann Vleugels, MD, director of the autoimmune skin diseases program at Brigham and Women’s Hospital, Boston.
“We will have patients who essentially fail all of our typical therapies and are still coming to us for help. This is a huge challenge,” said Dr. Vleugels, who, several years ago, started to use tofacitinib to treat these patients. “Similar to my colleagues who use tofacitinib to treat alopecia areata, we often have to push” beyond the dose used to treat rheumatoid arthritis, to 10 mg twice a day, she said at the International Conference on Cutaneous Lupus Erythematosus. Tofacitinib helps counter the overexpression of interferon in DM.

Getting insurance coverage for this off-label indication can be tough, however, but Dr. Vleugels said she’s had success when she tells insurers that tofacitinib will likely reduce the need for IVIg.
It’s safe to keep patients on methotrexate if there are concerns about muscle involvement while their skin is brought under control with tofacitinib. In terms of side effects, “we see increased shingles,” so recommending the shingles vaccine for these patients is a good idea, she added.

It’s also important to counsel DM patients that they are at particular risk for skin reactions with antimalarials, which can be serious, so that, “if there is a drug reaction that develops, it’s noticed right away” and the drug can be stopped, she said. “If you have a patient who has very severe disease, I might skip over an antimalarial altogether,” she commented.
Methotrexate is the next option, especially if there are work ups for cancer or the patients have cancer, but Dr. Femia, director of inpatient dermatology at NYU, said she leans towards mycophenolate if there’s concern about lung involvement.
The next step, if necessary, is IVIg, which she said is “particularly helpful” for recalcitrant skin disease and can help some patients discontinue other immunosuppressives. To counter headache, a common side effect, she will space dosing out over 3 days, instead of the usual 2, and have a bag of saline administered before and after the infusion to keep patients hydrated; this counters the headache-inducing viscosity of IVIg.
Patients often see a result after the first infusion, but if there’s no benefit by the third cycle, “it’s probably time to move on,” she said. “If you have a refractory muscle disease patient and skin isn’t the main issue, rituximab is reasonable to try,” she added, noting that the benefit of tumor necrosis factor blockers, “at best, is very mixed in the DM population. They are very low down in the treatment algorithm.”
Dr. Vleugels and Dr. Femia are both Pfizer investigators.
ORLANDO – The according to Ruth Ann Vleugels, MD, director of the autoimmune skin diseases program at Brigham and Women’s Hospital, Boston.
“We will have patients who essentially fail all of our typical therapies and are still coming to us for help. This is a huge challenge,” said Dr. Vleugels, who, several years ago, started to use tofacitinib to treat these patients. “Similar to my colleagues who use tofacitinib to treat alopecia areata, we often have to push” beyond the dose used to treat rheumatoid arthritis, to 10 mg twice a day, she said at the International Conference on Cutaneous Lupus Erythematosus. Tofacitinib helps counter the overexpression of interferon in DM.
Getting insurance coverage for this off-label indication can be tough, however, but Dr. Vleugels said she’s had success when she tells insurers that tofacitinib will likely reduce the need for IVIg.
It’s safe to keep patients on methotrexate if there are concerns about muscle involvement while their skin is brought under control with tofacitinib. In terms of side effects, “we see increased shingles,” so recommending the shingles vaccine for these patients is a good idea, she added.
It’s also important to counsel DM patients that they are at particular risk for skin reactions with antimalarials, which can be serious, so that, “if there is a drug reaction that develops, it’s noticed right away” and the drug can be stopped, she said. “If you have a patient who has very severe disease, I might skip over an antimalarial altogether,” she commented.
Methotrexate is the next option, especially if there are work ups for cancer or the patients have cancer, but Dr. Femia, director of inpatient dermatology at NYU, said she leans towards mycophenolate if there’s concern about lung involvement.
The next step, if necessary, is IVIg, which she said is “particularly helpful” for recalcitrant skin disease and can help some patients discontinue other immunosuppressives. To counter headache, a common side effect, she will space dosing out over 3 days, instead of the usual 2, and have a bag of saline administered before and after the infusion to keep patients hydrated; this counters the headache-inducing viscosity of IVIg.
Patients often see a result after the first infusion, but if there’s no benefit by the third cycle, “it’s probably time to move on,” she said. “If you have a refractory muscle disease patient and skin isn’t the main issue, rituximab is reasonable to try,” she added, noting that the benefit of tumor necrosis factor blockers, “at best, is very mixed in the DM population. They are very low down in the treatment algorithm.”
Dr. Vleugels and Dr. Femia are both Pfizer investigators.
ORLANDO – The according to Ruth Ann Vleugels, MD, director of the autoimmune skin diseases program at Brigham and Women’s Hospital, Boston.
“We will have patients who essentially fail all of our typical therapies and are still coming to us for help. This is a huge challenge,” said Dr. Vleugels, who, several years ago, started to use tofacitinib to treat these patients. “Similar to my colleagues who use tofacitinib to treat alopecia areata, we often have to push” beyond the dose used to treat rheumatoid arthritis, to 10 mg twice a day, she said at the International Conference on Cutaneous Lupus Erythematosus. Tofacitinib helps counter the overexpression of interferon in DM.
Getting insurance coverage for this off-label indication can be tough, however, but Dr. Vleugels said she’s had success when she tells insurers that tofacitinib will likely reduce the need for IVIg.
It’s safe to keep patients on methotrexate if there are concerns about muscle involvement while their skin is brought under control with tofacitinib. In terms of side effects, “we see increased shingles,” so recommending the shingles vaccine for these patients is a good idea, she added.
It’s also important to counsel DM patients that they are at particular risk for skin reactions with antimalarials, which can be serious, so that, “if there is a drug reaction that develops, it’s noticed right away” and the drug can be stopped, she said. “If you have a patient who has very severe disease, I might skip over an antimalarial altogether,” she commented.
Methotrexate is the next option, especially if there are work ups for cancer or the patients have cancer, but Dr. Femia, director of inpatient dermatology at NYU, said she leans towards mycophenolate if there’s concern about lung involvement.
The next step, if necessary, is IVIg, which she said is “particularly helpful” for recalcitrant skin disease and can help some patients discontinue other immunosuppressives. To counter headache, a common side effect, she will space dosing out over 3 days, instead of the usual 2, and have a bag of saline administered before and after the infusion to keep patients hydrated; this counters the headache-inducing viscosity of IVIg.
Patients often see a result after the first infusion, but if there’s no benefit by the third cycle, “it’s probably time to move on,” she said. “If you have a refractory muscle disease patient and skin isn’t the main issue, rituximab is reasonable to try,” she added, noting that the benefit of tumor necrosis factor blockers, “at best, is very mixed in the DM population. They are very low down in the treatment algorithm.”
Dr. Vleugels and Dr. Femia are both Pfizer investigators.
EXPERT ANALYSIS FROM ICCLE 2018
SLE classification criteria perform well in validation study
AMSTERDAM – The first European League Against Rheumatism and American College of Rheumatology joint criteria for classifying systemic lupus erythematosus have a sensitivity and a specificity of more than 90%.
This is important because they improve upon the existing ACR and Systemic Lupus International Collaborating Clinics (SLICC) criteria, said Martin Aringer, MD, PhD, who cochaired the Steering Committee that produced the new classification criteria.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Most clinicians working with lupus are familiar with the 1997 ACR criteria for the classification of systemic lupus erythematosus (SLE), which “had a relatively simple structure,” Dr. Aringer said during the opening plenary abstract session at the European Congress of Rheumatology. These considered items such as the presence of malar or discoid rash, photosensitivity, oral ulcers and arthritis, among others. These had a high specificity but a lower sensitivity. The development of the SLICC criteria in 2012 improved upon the sensitivity of the ACR criteria (92%-99% vs. 77%-91%), but at a loss in specificity (74%–88% vs. 91%-96%).
The SLICC criteria introduced two novel ideas, said Dr. Aringer, professor of medicine and chief of the division of rheumatology at the Technical University of Dresden (Germany). The first was that there had to be at least one immunologic criterion met, and the second was that biopsy-proven lupus nephritis had to be present with antinuclear antibodies (ANA) and anti-DNA antibodies detected.
One of the goals in developing the joint EULAR/ACR criteria therefore was to try to maintain the respective sensitivity and specificity achieved with the SLICC and ACR criteria. One of the key things that the new criteria looked at was to see if ANA could be used as an entry criterion. Investigations involving more than 13,000 patients with SLE showed that it could, with a antibody titer threshold of 1:80, exhibit a sensitivity of 98% (Arthritis Care Res. 2018;70[3]:428-38). Another goal was to see if histology-proven nephritis was a stronger predictor of SLE than clinical factors, such as oral ulcers, and to identify items that would only be included if there was no other more likely explanation (Lupus. 2016;25[8]:805-11).
Draft SLE classification criteria were developed based on an expert Delphi process and included ANA as an entry criterion and weighted items according to the likelihood of being associated with lupus. Items considered included the presence and severity of lupus nephritis, serology and other antibody tests, skin and central nervous system involvement, and hematologic and immunologic criteria such as the presence of thrombocytopenia and low complement (C3 and/or C4).

The final, simplified draft SLE classification criteria include 22 items in addition to the presence of ANA. A cut-off score of 10 or more is required for a classification of SLE. For example, a patient with an ANA of 1:80 or higher plus class III/IV nephritis (scoring 10) would be classified as having SLE. A patient with class II/V nephritis (scoring 8) would need another factor to be classified as having lupus, such as the presence of arthritis (scoring 6).
“Performance characteristics find sensitivity similar to the SLICC criteria while maintaining the specificity of the ACR 1997 criteria,” Dr. Aringer said, adding that these criteria will now be formally submitted to and reviewed by EULAR and ACR.
The sensitivity and specificity of the new criteria were 98% and 96% in the derivation cohort and 96% and 93% in the validation cohort.
“I was really very pleased and very happy to see that the revised or the new ACR/EULAR classification criteria had sensitivity and specificity of above 90%,” Thomas Dörner, MD, PhD, said in an interview at the congress. Dr. Dörner was a codeveloper of these criteria.
Over the past 10-15 years there have been several therapies that have failed to live up to their early promise as a potential treatment for lupus, said Dr. Dörner, professor of medicine at Charité–Universitätsmedizin Berlin. He noted that the failed treatment trials had led investigators to try to determine ways in which lupus might be best treated, such as by a “treat-to-target” approach to attain remission and low-disease activity. It also led to the reevaluation of how lupus is classified to see if that might be affecting the population of patients recruited into clinical trials.
“We had the feeling, and this is now confirmed by the new classification criteria, that a number of patients studied in earlier trials may have not fulfilled what we think is the classical lupus profile, so-called lupus or SLE mimickers,” Dr. Dörner said. This could have affected the chances of a treatment approach being successful versus placebo.
The new classification criteria are similar to those in other rheumatic diseases in that they give different weight to the effects on different organ systems, Dr. Dörner said. The stipulation that there must be a positive ANA test is also an important step, “really to make sure that we are looking at an autoimmune disease and nothing else,” he observed.
For patients who do not have a positive ANA test, they can of course still be treated, Dr. Dörner reassured, but for the classification criteria and entering patients into clinical trials, it’s really important to have strict classification criteria so that the results may be compared.
Dr. Aringer and Dr. Dörner had no relevant disclosures besides their involvement in developing the new classification criteria.
SOURCE: Aringer M et al. Ann Rheum Dis. 2018;77(Suppl 2):60. Abstract OP0020.
AMSTERDAM – The first European League Against Rheumatism and American College of Rheumatology joint criteria for classifying systemic lupus erythematosus have a sensitivity and a specificity of more than 90%.
This is important because they improve upon the existing ACR and Systemic Lupus International Collaborating Clinics (SLICC) criteria, said Martin Aringer, MD, PhD, who cochaired the Steering Committee that produced the new classification criteria.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Most clinicians working with lupus are familiar with the 1997 ACR criteria for the classification of systemic lupus erythematosus (SLE), which “had a relatively simple structure,” Dr. Aringer said during the opening plenary abstract session at the European Congress of Rheumatology. These considered items such as the presence of malar or discoid rash, photosensitivity, oral ulcers and arthritis, among others. These had a high specificity but a lower sensitivity. The development of the SLICC criteria in 2012 improved upon the sensitivity of the ACR criteria (92%-99% vs. 77%-91%), but at a loss in specificity (74%–88% vs. 91%-96%).
The SLICC criteria introduced two novel ideas, said Dr. Aringer, professor of medicine and chief of the division of rheumatology at the Technical University of Dresden (Germany). The first was that there had to be at least one immunologic criterion met, and the second was that biopsy-proven lupus nephritis had to be present with antinuclear antibodies (ANA) and anti-DNA antibodies detected.
One of the goals in developing the joint EULAR/ACR criteria therefore was to try to maintain the respective sensitivity and specificity achieved with the SLICC and ACR criteria. One of the key things that the new criteria looked at was to see if ANA could be used as an entry criterion. Investigations involving more than 13,000 patients with SLE showed that it could, with a antibody titer threshold of 1:80, exhibit a sensitivity of 98% (Arthritis Care Res. 2018;70[3]:428-38). Another goal was to see if histology-proven nephritis was a stronger predictor of SLE than clinical factors, such as oral ulcers, and to identify items that would only be included if there was no other more likely explanation (Lupus. 2016;25[8]:805-11).
Draft SLE classification criteria were developed based on an expert Delphi process and included ANA as an entry criterion and weighted items according to the likelihood of being associated with lupus. Items considered included the presence and severity of lupus nephritis, serology and other antibody tests, skin and central nervous system involvement, and hematologic and immunologic criteria such as the presence of thrombocytopenia and low complement (C3 and/or C4).
The final, simplified draft SLE classification criteria include 22 items in addition to the presence of ANA. A cut-off score of 10 or more is required for a classification of SLE. For example, a patient with an ANA of 1:80 or higher plus class III/IV nephritis (scoring 10) would be classified as having SLE. A patient with class II/V nephritis (scoring 8) would need another factor to be classified as having lupus, such as the presence of arthritis (scoring 6).
“Performance characteristics find sensitivity similar to the SLICC criteria while maintaining the specificity of the ACR 1997 criteria,” Dr. Aringer said, adding that these criteria will now be formally submitted to and reviewed by EULAR and ACR.
The sensitivity and specificity of the new criteria were 98% and 96% in the derivation cohort and 96% and 93% in the validation cohort.
“I was really very pleased and very happy to see that the revised or the new ACR/EULAR classification criteria had sensitivity and specificity of above 90%,” Thomas Dörner, MD, PhD, said in an interview at the congress. Dr. Dörner was a codeveloper of these criteria.
Over the past 10-15 years there have been several therapies that have failed to live up to their early promise as a potential treatment for lupus, said Dr. Dörner, professor of medicine at Charité–Universitätsmedizin Berlin. He noted that the failed treatment trials had led investigators to try to determine ways in which lupus might be best treated, such as by a “treat-to-target” approach to attain remission and low-disease activity. It also led to the reevaluation of how lupus is classified to see if that might be affecting the population of patients recruited into clinical trials.
“We had the feeling, and this is now confirmed by the new classification criteria, that a number of patients studied in earlier trials may have not fulfilled what we think is the classical lupus profile, so-called lupus or SLE mimickers,” Dr. Dörner said. This could have affected the chances of a treatment approach being successful versus placebo.
The new classification criteria are similar to those in other rheumatic diseases in that they give different weight to the effects on different organ systems, Dr. Dörner said. The stipulation that there must be a positive ANA test is also an important step, “really to make sure that we are looking at an autoimmune disease and nothing else,” he observed.
For patients who do not have a positive ANA test, they can of course still be treated, Dr. Dörner reassured, but for the classification criteria and entering patients into clinical trials, it’s really important to have strict classification criteria so that the results may be compared.
Dr. Aringer and Dr. Dörner had no relevant disclosures besides their involvement in developing the new classification criteria.
SOURCE: Aringer M et al. Ann Rheum Dis. 2018;77(Suppl 2):60. Abstract OP0020.
AMSTERDAM – The first European League Against Rheumatism and American College of Rheumatology joint criteria for classifying systemic lupus erythematosus have a sensitivity and a specificity of more than 90%.
This is important because they improve upon the existing ACR and Systemic Lupus International Collaborating Clinics (SLICC) criteria, said Martin Aringer, MD, PhD, who cochaired the Steering Committee that produced the new classification criteria.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Most clinicians working with lupus are familiar with the 1997 ACR criteria for the classification of systemic lupus erythematosus (SLE), which “had a relatively simple structure,” Dr. Aringer said during the opening plenary abstract session at the European Congress of Rheumatology. These considered items such as the presence of malar or discoid rash, photosensitivity, oral ulcers and arthritis, among others. These had a high specificity but a lower sensitivity. The development of the SLICC criteria in 2012 improved upon the sensitivity of the ACR criteria (92%-99% vs. 77%-91%), but at a loss in specificity (74%–88% vs. 91%-96%).
The SLICC criteria introduced two novel ideas, said Dr. Aringer, professor of medicine and chief of the division of rheumatology at the Technical University of Dresden (Germany). The first was that there had to be at least one immunologic criterion met, and the second was that biopsy-proven lupus nephritis had to be present with antinuclear antibodies (ANA) and anti-DNA antibodies detected.
One of the goals in developing the joint EULAR/ACR criteria therefore was to try to maintain the respective sensitivity and specificity achieved with the SLICC and ACR criteria. One of the key things that the new criteria looked at was to see if ANA could be used as an entry criterion. Investigations involving more than 13,000 patients with SLE showed that it could, with a antibody titer threshold of 1:80, exhibit a sensitivity of 98% (Arthritis Care Res. 2018;70[3]:428-38). Another goal was to see if histology-proven nephritis was a stronger predictor of SLE than clinical factors, such as oral ulcers, and to identify items that would only be included if there was no other more likely explanation (Lupus. 2016;25[8]:805-11).
Draft SLE classification criteria were developed based on an expert Delphi process and included ANA as an entry criterion and weighted items according to the likelihood of being associated with lupus. Items considered included the presence and severity of lupus nephritis, serology and other antibody tests, skin and central nervous system involvement, and hematologic and immunologic criteria such as the presence of thrombocytopenia and low complement (C3 and/or C4).
The final, simplified draft SLE classification criteria include 22 items in addition to the presence of ANA. A cut-off score of 10 or more is required for a classification of SLE. For example, a patient with an ANA of 1:80 or higher plus class III/IV nephritis (scoring 10) would be classified as having SLE. A patient with class II/V nephritis (scoring 8) would need another factor to be classified as having lupus, such as the presence of arthritis (scoring 6).
“Performance characteristics find sensitivity similar to the SLICC criteria while maintaining the specificity of the ACR 1997 criteria,” Dr. Aringer said, adding that these criteria will now be formally submitted to and reviewed by EULAR and ACR.
The sensitivity and specificity of the new criteria were 98% and 96% in the derivation cohort and 96% and 93% in the validation cohort.
“I was really very pleased and very happy to see that the revised or the new ACR/EULAR classification criteria had sensitivity and specificity of above 90%,” Thomas Dörner, MD, PhD, said in an interview at the congress. Dr. Dörner was a codeveloper of these criteria.
Over the past 10-15 years there have been several therapies that have failed to live up to their early promise as a potential treatment for lupus, said Dr. Dörner, professor of medicine at Charité–Universitätsmedizin Berlin. He noted that the failed treatment trials had led investigators to try to determine ways in which lupus might be best treated, such as by a “treat-to-target” approach to attain remission and low-disease activity. It also led to the reevaluation of how lupus is classified to see if that might be affecting the population of patients recruited into clinical trials.
“We had the feeling, and this is now confirmed by the new classification criteria, that a number of patients studied in earlier trials may have not fulfilled what we think is the classical lupus profile, so-called lupus or SLE mimickers,” Dr. Dörner said. This could have affected the chances of a treatment approach being successful versus placebo.
The new classification criteria are similar to those in other rheumatic diseases in that they give different weight to the effects on different organ systems, Dr. Dörner said. The stipulation that there must be a positive ANA test is also an important step, “really to make sure that we are looking at an autoimmune disease and nothing else,” he observed.
For patients who do not have a positive ANA test, they can of course still be treated, Dr. Dörner reassured, but for the classification criteria and entering patients into clinical trials, it’s really important to have strict classification criteria so that the results may be compared.
Dr. Aringer and Dr. Dörner had no relevant disclosures besides their involvement in developing the new classification criteria.
SOURCE: Aringer M et al. Ann Rheum Dis. 2018;77(Suppl 2):60. Abstract OP0020.
REPORTING FROM THE EULAR 2018 CONGRESS
Key clinical point: New classification criteria for systemic lupus erythematosus (SLE) achieve both high sensitivity and specificity.
Major finding: The sensitivity and specificity of the new criteria were 98% and 96% in the derivation cohort and 96% and 93% in the validation cohort.
Study details: An international cohort of 1,160 SLE patients and 1,058 non-SLE patients in whom the new criteria were tested and validated.
Disclosures: Dr. Aringer and Dr. Dörner had no relevant disclosures besides their involvement in developing the new classification criteria.
Source: Aringer M et al. Ann Rheum Dis. 2018;77(Suppl 2):60. Abstract OP0020.
Cost of medical care for older adults with epilepsy steadily grows
An analysis of health care expenditures faced by elderly patients with epilepsy over a 12-year period in the United States found rising annual costs over time that approached a mean of $20,000, according to researchers from the Medical University of South Carolina, Charleston.
Alain Lekoubou, MD, and his colleagues estimated health care expenditures during 2003-2014 for patients aged 65 years and older with and without epilepsy by extrapolating data from the Medical Expenditure Panel Survey Household Component to more than 37 million elderly individuals in the United States. The investigators published their findings in Epilepsia.
The unadjusted mean direct health care expenditures for elderly people with epilepsy rose from $15,850 in 2003-2006 to $22,038 in 2007-2010 but dropped in 2011-2014 to $17,985. Figures for the same period for elderly people without epilepsy went from $10,214 to $10,358 to $9,965.
“The high prevalence of epilepsy and high health care expenditures in elderly patients should give priority to epilepsy in the elderly in the medical and public health communities’ agenda,” the authors wrote.
SOURCE: Lekoubou A et al. Epilepsia. 2018 June 19. doi: 10.1111/epi.14455.
An analysis of health care expenditures faced by elderly patients with epilepsy over a 12-year period in the United States found rising annual costs over time that approached a mean of $20,000, according to researchers from the Medical University of South Carolina, Charleston.
Alain Lekoubou, MD, and his colleagues estimated health care expenditures during 2003-2014 for patients aged 65 years and older with and without epilepsy by extrapolating data from the Medical Expenditure Panel Survey Household Component to more than 37 million elderly individuals in the United States. The investigators published their findings in Epilepsia.
The unadjusted mean direct health care expenditures for elderly people with epilepsy rose from $15,850 in 2003-2006 to $22,038 in 2007-2010 but dropped in 2011-2014 to $17,985. Figures for the same period for elderly people without epilepsy went from $10,214 to $10,358 to $9,965.
“The high prevalence of epilepsy and high health care expenditures in elderly patients should give priority to epilepsy in the elderly in the medical and public health communities’ agenda,” the authors wrote.
SOURCE: Lekoubou A et al. Epilepsia. 2018 June 19. doi: 10.1111/epi.14455.
An analysis of health care expenditures faced by elderly patients with epilepsy over a 12-year period in the United States found rising annual costs over time that approached a mean of $20,000, according to researchers from the Medical University of South Carolina, Charleston.
Alain Lekoubou, MD, and his colleagues estimated health care expenditures during 2003-2014 for patients aged 65 years and older with and without epilepsy by extrapolating data from the Medical Expenditure Panel Survey Household Component to more than 37 million elderly individuals in the United States. The investigators published their findings in Epilepsia.
The unadjusted mean direct health care expenditures for elderly people with epilepsy rose from $15,850 in 2003-2006 to $22,038 in 2007-2010 but dropped in 2011-2014 to $17,985. Figures for the same period for elderly people without epilepsy went from $10,214 to $10,358 to $9,965.
“The high prevalence of epilepsy and high health care expenditures in elderly patients should give priority to epilepsy in the elderly in the medical and public health communities’ agenda,” the authors wrote.
SOURCE: Lekoubou A et al. Epilepsia. 2018 June 19. doi: 10.1111/epi.14455.
FROM EPILEPSIA