User login
Pediatric Hospitalist Workload and Sustainability in University-Based Programs: Results from a National Interview-Based Survey
Pediatric hospital medicine (PHM) has grown tremendously since Wachter first described the specialty in 1996.1 Evidence of this growth is seen most markedly at the annual Pediatric Hospitalist Meeting, which has experienced an increase in attendance from 700 in 2013 to over 1,200 in 20172. Although the exact number of pediatric hospitalists in the United States is unknown, the American Academy of Pediatrics Section on Hospital Medicine (AAP SOHM) estimates that approximately 3,000-5,000 pediatric hospitalists currently practice in the country (personal communication).
As PHM programs have grown, variability has been reported in the roles, responsibilities, and workload among practitioners. Gosdin et al.3 reported large ranges and standard deviations in workload among full-time equivalents (FTEs) in academic PHM programs. However, this study’s ability to account for important nuances in program description was limited given that its data were obtained from an online survey.
Program variability, particularly regarding clinical hours and overall clinical burden (eg, in-house hours, census caps, and weekend coverage), is concerning given the well-reported increase in physician burn-out.4,5 Benchmarking data regarding the overall workload of pediatric hospitalists can offer nationally recognized guidance to assist program leaders in building successful programs. With this goal in mind, we sought to obtain data on university-based PHM programs to describe the current average workload for a 1.0 clinical FTE pediatric hospitalist and to assess the perceptions of program directors regarding the sustainability of the current workload.
METHODS
Study Design and Population
To obtain data with sufficient detail to compare programs, the authors, all of whom are practicing pediatric hospitalists at university-based programs, conducted structured interviews of PHM leaders in the United States. Given the absence of a single database for all PHM programs in the United States, the clinical division/program leaders of university-based programs were invited to participate through a post (with 2 reminders) to the AAP SOHM Listserv for PHM Division Leaders in May of 2017. To encourage participation, respondents were promised a summary of aggregate data. The study was exempted by the IRB of the University of Chicago.
Interview Content and Administration
The authors designed an 18-question structured interview regarding the current state of staffing in university-based PHM programs, with a focus on current descriptions of FTE, patient volume, and workload. Utilizing prior surveys3 as a basis, the authors iteratively determined the questions essential to understanding the programs’ current staffing models and ideal models. Considering the diversity of program models, interviews allowed for the clarification of questions and answers. A question regarding employment models was included to determine whether hospitalists were university-employed, hospital-employed, or a hybrid of the 2 modes of employment. The interview was also designed to establish a common language for work metrics (hours per year) for comparative purposes and to assess the perceived sustainability of the workload. Questions were provided in advance to provide respondents with sufficient time to collect data, thus increasing the accuracy of estimates. Respondents were asked, “Do you or your hospitalists have concerns about the sustainability of the model?” Sustainability was intentionally undefined to prevent limiting respondent perspective. For clarification, however, a follow-up comment that included examples was provided: “Faculty departures, reduction in total effort, and/or significant burn out.” The authors piloted the interview protocol by interviewing the division leaders of their own programs, and revisions were made based on feedback on feasibility and clarity. Finally, the AAP SOHM Subcommittee on Division Leaders provided feedback, which was incorporated.
Each author then interviewed 10-12 leaders (or designee) during May and June of 2017. Answers were recorded in REDCAP, an online survey and database tool that contains largely numeric data fields and has 1 field for narrative comments.
Data Analysis
Descriptive statistics were used to summarize interview responses, including median values with interquartile range. Data were compared between programs with models that were self-identified as either sustainable or unsustainable, with P-values in categorical variables from χ2-test or Fischer’s exact test and in continuous variables from Wilcoxon rank-sum test.
Spearman correlation coefficient was used to evaluate the association between average protected time (defined as the percent of funded time for nonclinical roles) and percentage working full-time clinical effort. It was also used to evaluate hours per year per 1.0 FTE and total weekends per year per 1.0 FTE and perceived sustainability. Linear regression was used to determine whether associations differed between groups identifying as sustainable versus unsustainable.
RESULTS
Participation and Program Characteristics
Administration
A wide variation was reported in the clinical time expected of a 1.0 FTE hospitalist. Clinical time for 1.0 FTE was defined as the amount of clinical service a full-time hospitalist is expected to complete in 12 months (Table 1). The median hours worked per year were 1800 (Interquartile range [IQR] 1620,1975; mean 1796). The median number of weekends worked per year was 15.0 (IQR 12.5, 21; mean 16.8). Only 30% of pediatric hospitalists were full-time clinicians, whereas the rest had protected time for nonclinical duties. The average amount of protected time was 20% per full-time hospitalist.
Sustainability and Ideal FTE
Half of the division leaders reported that they or their hospitalists have concerns about the sustainability of the current workload. Programs perceived as sustainable required significantly fewer weekends per year (13 vs. 16, P < .02; Table 2) than those perceived as unsustainable. University-employed programs were more likely to be perceived as unsustainable (64% unsustainable vs. 32% unsustainable, P < .048), whereas programs with other employment models were more likely to be perceived as sustainable (Table 2). 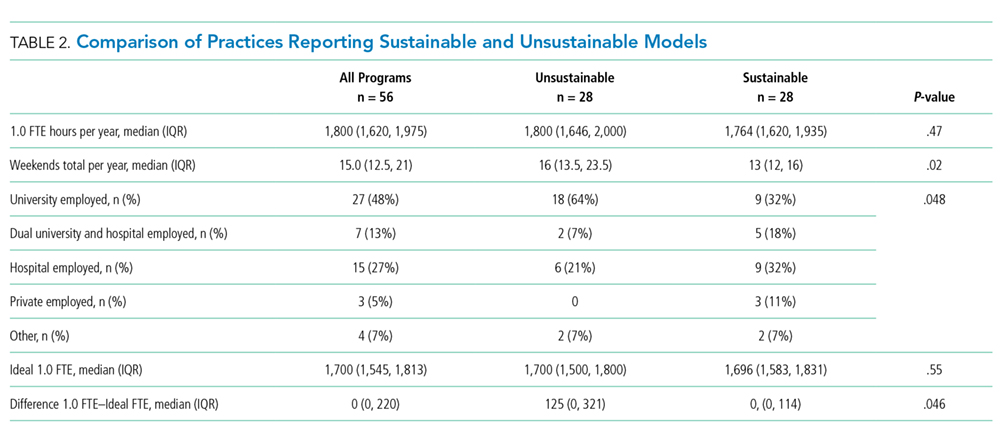
DISCUSSION
This study updates what has been previously reported about the structure and characteristics of university-based pediatric hospitalist programs.3 It also deepens our understanding of a relatively new field and the evolution of clinical coverage models. This evolution has been impacted by decreased resident work hours, increased patient complexity and acuity,6 and a broadened focus on care coordination and communication,7 while attempting to build and sustain a high-quality workforce.
This study is the first to use an interview-based method to determine the current PHM workload and to focus exclusively on university-based programs. Compared with the study by Gosdin et al,3 our study, which utilized interviews instead of surveys, was able to clarify questions and obtain workload data with a common language of hours per year. This approach allowed interviewees to incorporate subtleties, such as clinical vs. total FTE, in their responses. Our study found a slightly narrower range of clinical hours per year and extended the understanding of nonclinical duties by finding that university-based hospitalists have an average of 20% protected time from clinical duties.
In this study, we also explored the perceived sustainability of current clinical models and the ideal clinical model in hours per year. Half of respondents felt their current model was unsustainable. This result suggested that the field must continue to mitigate attrition and burnout.
Interestingly, the total number of clinical hours did not significantly differ in programs perceived to be unsustainable. Instead, a higher number of weekends worked and university employment were associated with lack of sustainability. We hypothesize that weekends have a disproportionate impact on work-life balance as compared with total hours, and that employment by a university may be a proxy for the increased academic and teaching demands of hospitalists without protected time. Future studies may better elucidate these findings and inform programmatic efforts to address sustainability.
Given that PHM is a relatively young field, considering the evolution of our clinical work model within the context of pediatric emergency medicine (PEM), a field that faces similar challenges in overnight and weekend staffing requirements, may be helpful. Gorelick et al.8 reported that total clinical work hours in PEM (combined academic and nonacademic programs) has decreased from 35.3 hours per week in 1998 to 26.7 in 2013. Extrapolating these numbers to an annual position with 5 weeks PTO/CME, the average PEM attending physician works 1254 clinical hours. These numbers demonstrate a marked difference compared with the average 1800 clinical work hours for PHM found in our study.
Although total hours trend lower in PEM, the authors noted continued challenges in sustainability with an estimated half of all PEM respondents indicating a plan to reduce hours or leave the field in the next 5 years and endorsing symptoms of burnout.6 These findings from PEM may motivate PHM leaders to be more aggressive in adjusting work models toward sustainability in the future.
Our study has several limitations. We utilized a convenience sampling approach that requires the voluntary participation of division directors. Although we had robust interest from respondents representing all major geographic areas, the respondent pool might conceivably over-represent those most interested in understanding and/or changing PHM clinical models. Overall, our sample size was smaller than that achieved by a survey approach. Nevertheless, this limitation was offset by controlling respondent type and clarifying questions, thus improving the quality of our obtained data.
CONCLUSION
This interview-based study of PHM directors describes the current state of clinical work models for university-based hospitalists. University-based PHM programs have similar mean and median total clinical hours per year. However, these hours are higher than those considered ideal by PHM directors, and many are concerned about the sustainability of current work models. Notably, programs that are university-employed or have higher weekends worked per year are more likely to be perceived as unsustainable. Future studies should explore differences between programs with sustainable work models and those with high levels of attrition and burnout.
Disclosures
The authors have no other conflicts to report.
Funding
A grant from the American Academy of Pediatrics Section on Hospital Medicine funded this study through the Subcommittee on Division and Program Leaders.
1. Wachter RM, Goldman L. The emerging role of “hospitalists” in the American health care system. N Engl J Med. 1996;335(7):514-517. DOI: 10.1056/NEJM199608153350713 PubMed
2. Chang W. Record Attendance, Key Issues Highlight Pediatric Hospital Medicine’s 10th Anniversary.
3. Gosdin C, Simmons J, Yau C, Sucharew H, Carlson D, Paciorkowski N. Survey of academic pediatric hospitalist programs in the US: organizational, administrative, and financial factors. J Hosp Med. 2013;8(6):285-291. DOI: 10.1002/jhm.2020. PubMed
4. Hinami K, Whelan CT, Wolosin RJ, Miller JA, Wetterneck TB. Worklife and satisfaction of hospitalists: toward flourishing careers. J Gen Intern Med. 2011;27(1):28-36. DOI: 10.1007/s11606-011-1780-z. PubMed
5. Hinami K, Whelan CT, Miller JA, Wolosin RJ, Wetterneck TB. Job characteristics, satisfaction, and burnout across hospitalist practice models. J Hosp Med. 2012;7(5):402-410. DOI: 10.1002/jhm.1907. PubMed
6. Barrett DJ, McGuinness GA, Cunha CA, et al. Pediatric hospital medicine: a proposed new subspecialty. Pediatrics. 2017;139(3):1-9. DOI: 10.1542/peds.2016-1823. PubMed
7. Cawley P, Deitelzweig S, Flores L, et al. The key principles and characteristics of an effective hospital medicine group: an assessment guide for hospitals and hospitalists. J Hosp Med. 2014;9(2):123-128. DOI: 10.1002/jhm.2119. PubMed
8. Gorelick MH, Schremmer R, Ruch-Ross H, Radabaugh C, Selbst S. Current workforce characteristics and burnout in pediatric emergency medicine. Acad Emerg Med. 2016;23(1):48-54. DOI: 10.1111/acem.12845. PubMed
Pediatric hospital medicine (PHM) has grown tremendously since Wachter first described the specialty in 1996.1 Evidence of this growth is seen most markedly at the annual Pediatric Hospitalist Meeting, which has experienced an increase in attendance from 700 in 2013 to over 1,200 in 20172. Although the exact number of pediatric hospitalists in the United States is unknown, the American Academy of Pediatrics Section on Hospital Medicine (AAP SOHM) estimates that approximately 3,000-5,000 pediatric hospitalists currently practice in the country (personal communication).
As PHM programs have grown, variability has been reported in the roles, responsibilities, and workload among practitioners. Gosdin et al.3 reported large ranges and standard deviations in workload among full-time equivalents (FTEs) in academic PHM programs. However, this study’s ability to account for important nuances in program description was limited given that its data were obtained from an online survey.
Program variability, particularly regarding clinical hours and overall clinical burden (eg, in-house hours, census caps, and weekend coverage), is concerning given the well-reported increase in physician burn-out.4,5 Benchmarking data regarding the overall workload of pediatric hospitalists can offer nationally recognized guidance to assist program leaders in building successful programs. With this goal in mind, we sought to obtain data on university-based PHM programs to describe the current average workload for a 1.0 clinical FTE pediatric hospitalist and to assess the perceptions of program directors regarding the sustainability of the current workload.
METHODS
Study Design and Population
To obtain data with sufficient detail to compare programs, the authors, all of whom are practicing pediatric hospitalists at university-based programs, conducted structured interviews of PHM leaders in the United States. Given the absence of a single database for all PHM programs in the United States, the clinical division/program leaders of university-based programs were invited to participate through a post (with 2 reminders) to the AAP SOHM Listserv for PHM Division Leaders in May of 2017. To encourage participation, respondents were promised a summary of aggregate data. The study was exempted by the IRB of the University of Chicago.
Interview Content and Administration
The authors designed an 18-question structured interview regarding the current state of staffing in university-based PHM programs, with a focus on current descriptions of FTE, patient volume, and workload. Utilizing prior surveys3 as a basis, the authors iteratively determined the questions essential to understanding the programs’ current staffing models and ideal models. Considering the diversity of program models, interviews allowed for the clarification of questions and answers. A question regarding employment models was included to determine whether hospitalists were university-employed, hospital-employed, or a hybrid of the 2 modes of employment. The interview was also designed to establish a common language for work metrics (hours per year) for comparative purposes and to assess the perceived sustainability of the workload. Questions were provided in advance to provide respondents with sufficient time to collect data, thus increasing the accuracy of estimates. Respondents were asked, “Do you or your hospitalists have concerns about the sustainability of the model?” Sustainability was intentionally undefined to prevent limiting respondent perspective. For clarification, however, a follow-up comment that included examples was provided: “Faculty departures, reduction in total effort, and/or significant burn out.” The authors piloted the interview protocol by interviewing the division leaders of their own programs, and revisions were made based on feedback on feasibility and clarity. Finally, the AAP SOHM Subcommittee on Division Leaders provided feedback, which was incorporated.
Each author then interviewed 10-12 leaders (or designee) during May and June of 2017. Answers were recorded in REDCAP, an online survey and database tool that contains largely numeric data fields and has 1 field for narrative comments.
Data Analysis
Descriptive statistics were used to summarize interview responses, including median values with interquartile range. Data were compared between programs with models that were self-identified as either sustainable or unsustainable, with P-values in categorical variables from χ2-test or Fischer’s exact test and in continuous variables from Wilcoxon rank-sum test.
Spearman correlation coefficient was used to evaluate the association between average protected time (defined as the percent of funded time for nonclinical roles) and percentage working full-time clinical effort. It was also used to evaluate hours per year per 1.0 FTE and total weekends per year per 1.0 FTE and perceived sustainability. Linear regression was used to determine whether associations differed between groups identifying as sustainable versus unsustainable.
RESULTS
Participation and Program Characteristics
Administration
A wide variation was reported in the clinical time expected of a 1.0 FTE hospitalist. Clinical time for 1.0 FTE was defined as the amount of clinical service a full-time hospitalist is expected to complete in 12 months (Table 1). The median hours worked per year were 1800 (Interquartile range [IQR] 1620,1975; mean 1796). The median number of weekends worked per year was 15.0 (IQR 12.5, 21; mean 16.8). Only 30% of pediatric hospitalists were full-time clinicians, whereas the rest had protected time for nonclinical duties. The average amount of protected time was 20% per full-time hospitalist.
Sustainability and Ideal FTE
Half of the division leaders reported that they or their hospitalists have concerns about the sustainability of the current workload. Programs perceived as sustainable required significantly fewer weekends per year (13 vs. 16, P < .02; Table 2) than those perceived as unsustainable. University-employed programs were more likely to be perceived as unsustainable (64% unsustainable vs. 32% unsustainable, P < .048), whereas programs with other employment models were more likely to be perceived as sustainable (Table 2).
DISCUSSION
This study updates what has been previously reported about the structure and characteristics of university-based pediatric hospitalist programs.3 It also deepens our understanding of a relatively new field and the evolution of clinical coverage models. This evolution has been impacted by decreased resident work hours, increased patient complexity and acuity,6 and a broadened focus on care coordination and communication,7 while attempting to build and sustain a high-quality workforce.
This study is the first to use an interview-based method to determine the current PHM workload and to focus exclusively on university-based programs. Compared with the study by Gosdin et al,3 our study, which utilized interviews instead of surveys, was able to clarify questions and obtain workload data with a common language of hours per year. This approach allowed interviewees to incorporate subtleties, such as clinical vs. total FTE, in their responses. Our study found a slightly narrower range of clinical hours per year and extended the understanding of nonclinical duties by finding that university-based hospitalists have an average of 20% protected time from clinical duties.
In this study, we also explored the perceived sustainability of current clinical models and the ideal clinical model in hours per year. Half of respondents felt their current model was unsustainable. This result suggested that the field must continue to mitigate attrition and burnout.
Interestingly, the total number of clinical hours did not significantly differ in programs perceived to be unsustainable. Instead, a higher number of weekends worked and university employment were associated with lack of sustainability. We hypothesize that weekends have a disproportionate impact on work-life balance as compared with total hours, and that employment by a university may be a proxy for the increased academic and teaching demands of hospitalists without protected time. Future studies may better elucidate these findings and inform programmatic efforts to address sustainability.
Given that PHM is a relatively young field, considering the evolution of our clinical work model within the context of pediatric emergency medicine (PEM), a field that faces similar challenges in overnight and weekend staffing requirements, may be helpful. Gorelick et al.8 reported that total clinical work hours in PEM (combined academic and nonacademic programs) has decreased from 35.3 hours per week in 1998 to 26.7 in 2013. Extrapolating these numbers to an annual position with 5 weeks PTO/CME, the average PEM attending physician works 1254 clinical hours. These numbers demonstrate a marked difference compared with the average 1800 clinical work hours for PHM found in our study.
Although total hours trend lower in PEM, the authors noted continued challenges in sustainability with an estimated half of all PEM respondents indicating a plan to reduce hours or leave the field in the next 5 years and endorsing symptoms of burnout.6 These findings from PEM may motivate PHM leaders to be more aggressive in adjusting work models toward sustainability in the future.
Our study has several limitations. We utilized a convenience sampling approach that requires the voluntary participation of division directors. Although we had robust interest from respondents representing all major geographic areas, the respondent pool might conceivably over-represent those most interested in understanding and/or changing PHM clinical models. Overall, our sample size was smaller than that achieved by a survey approach. Nevertheless, this limitation was offset by controlling respondent type and clarifying questions, thus improving the quality of our obtained data.
CONCLUSION
This interview-based study of PHM directors describes the current state of clinical work models for university-based hospitalists. University-based PHM programs have similar mean and median total clinical hours per year. However, these hours are higher than those considered ideal by PHM directors, and many are concerned about the sustainability of current work models. Notably, programs that are university-employed or have higher weekends worked per year are more likely to be perceived as unsustainable. Future studies should explore differences between programs with sustainable work models and those with high levels of attrition and burnout.
Disclosures
The authors have no other conflicts to report.
Funding
A grant from the American Academy of Pediatrics Section on Hospital Medicine funded this study through the Subcommittee on Division and Program Leaders.
Pediatric hospital medicine (PHM) has grown tremendously since Wachter first described the specialty in 1996.1 Evidence of this growth is seen most markedly at the annual Pediatric Hospitalist Meeting, which has experienced an increase in attendance from 700 in 2013 to over 1,200 in 20172. Although the exact number of pediatric hospitalists in the United States is unknown, the American Academy of Pediatrics Section on Hospital Medicine (AAP SOHM) estimates that approximately 3,000-5,000 pediatric hospitalists currently practice in the country (personal communication).
As PHM programs have grown, variability has been reported in the roles, responsibilities, and workload among practitioners. Gosdin et al.3 reported large ranges and standard deviations in workload among full-time equivalents (FTEs) in academic PHM programs. However, this study’s ability to account for important nuances in program description was limited given that its data were obtained from an online survey.
Program variability, particularly regarding clinical hours and overall clinical burden (eg, in-house hours, census caps, and weekend coverage), is concerning given the well-reported increase in physician burn-out.4,5 Benchmarking data regarding the overall workload of pediatric hospitalists can offer nationally recognized guidance to assist program leaders in building successful programs. With this goal in mind, we sought to obtain data on university-based PHM programs to describe the current average workload for a 1.0 clinical FTE pediatric hospitalist and to assess the perceptions of program directors regarding the sustainability of the current workload.
METHODS
Study Design and Population
To obtain data with sufficient detail to compare programs, the authors, all of whom are practicing pediatric hospitalists at university-based programs, conducted structured interviews of PHM leaders in the United States. Given the absence of a single database for all PHM programs in the United States, the clinical division/program leaders of university-based programs were invited to participate through a post (with 2 reminders) to the AAP SOHM Listserv for PHM Division Leaders in May of 2017. To encourage participation, respondents were promised a summary of aggregate data. The study was exempted by the IRB of the University of Chicago.
Interview Content and Administration
The authors designed an 18-question structured interview regarding the current state of staffing in university-based PHM programs, with a focus on current descriptions of FTE, patient volume, and workload. Utilizing prior surveys3 as a basis, the authors iteratively determined the questions essential to understanding the programs’ current staffing models and ideal models. Considering the diversity of program models, interviews allowed for the clarification of questions and answers. A question regarding employment models was included to determine whether hospitalists were university-employed, hospital-employed, or a hybrid of the 2 modes of employment. The interview was also designed to establish a common language for work metrics (hours per year) for comparative purposes and to assess the perceived sustainability of the workload. Questions were provided in advance to provide respondents with sufficient time to collect data, thus increasing the accuracy of estimates. Respondents were asked, “Do you or your hospitalists have concerns about the sustainability of the model?” Sustainability was intentionally undefined to prevent limiting respondent perspective. For clarification, however, a follow-up comment that included examples was provided: “Faculty departures, reduction in total effort, and/or significant burn out.” The authors piloted the interview protocol by interviewing the division leaders of their own programs, and revisions were made based on feedback on feasibility and clarity. Finally, the AAP SOHM Subcommittee on Division Leaders provided feedback, which was incorporated.
Each author then interviewed 10-12 leaders (or designee) during May and June of 2017. Answers were recorded in REDCAP, an online survey and database tool that contains largely numeric data fields and has 1 field for narrative comments.
Data Analysis
Descriptive statistics were used to summarize interview responses, including median values with interquartile range. Data were compared between programs with models that were self-identified as either sustainable or unsustainable, with P-values in categorical variables from χ2-test or Fischer’s exact test and in continuous variables from Wilcoxon rank-sum test.
Spearman correlation coefficient was used to evaluate the association between average protected time (defined as the percent of funded time for nonclinical roles) and percentage working full-time clinical effort. It was also used to evaluate hours per year per 1.0 FTE and total weekends per year per 1.0 FTE and perceived sustainability. Linear regression was used to determine whether associations differed between groups identifying as sustainable versus unsustainable.
RESULTS
Participation and Program Characteristics
Administration
A wide variation was reported in the clinical time expected of a 1.0 FTE hospitalist. Clinical time for 1.0 FTE was defined as the amount of clinical service a full-time hospitalist is expected to complete in 12 months (Table 1). The median hours worked per year were 1800 (Interquartile range [IQR] 1620,1975; mean 1796). The median number of weekends worked per year was 15.0 (IQR 12.5, 21; mean 16.8). Only 30% of pediatric hospitalists were full-time clinicians, whereas the rest had protected time for nonclinical duties. The average amount of protected time was 20% per full-time hospitalist.
Sustainability and Ideal FTE
Half of the division leaders reported that they or their hospitalists have concerns about the sustainability of the current workload. Programs perceived as sustainable required significantly fewer weekends per year (13 vs. 16, P < .02; Table 2) than those perceived as unsustainable. University-employed programs were more likely to be perceived as unsustainable (64% unsustainable vs. 32% unsustainable, P < .048), whereas programs with other employment models were more likely to be perceived as sustainable (Table 2).
DISCUSSION
This study updates what has been previously reported about the structure and characteristics of university-based pediatric hospitalist programs.3 It also deepens our understanding of a relatively new field and the evolution of clinical coverage models. This evolution has been impacted by decreased resident work hours, increased patient complexity and acuity,6 and a broadened focus on care coordination and communication,7 while attempting to build and sustain a high-quality workforce.
This study is the first to use an interview-based method to determine the current PHM workload and to focus exclusively on university-based programs. Compared with the study by Gosdin et al,3 our study, which utilized interviews instead of surveys, was able to clarify questions and obtain workload data with a common language of hours per year. This approach allowed interviewees to incorporate subtleties, such as clinical vs. total FTE, in their responses. Our study found a slightly narrower range of clinical hours per year and extended the understanding of nonclinical duties by finding that university-based hospitalists have an average of 20% protected time from clinical duties.
In this study, we also explored the perceived sustainability of current clinical models and the ideal clinical model in hours per year. Half of respondents felt their current model was unsustainable. This result suggested that the field must continue to mitigate attrition and burnout.
Interestingly, the total number of clinical hours did not significantly differ in programs perceived to be unsustainable. Instead, a higher number of weekends worked and university employment were associated with lack of sustainability. We hypothesize that weekends have a disproportionate impact on work-life balance as compared with total hours, and that employment by a university may be a proxy for the increased academic and teaching demands of hospitalists without protected time. Future studies may better elucidate these findings and inform programmatic efforts to address sustainability.
Given that PHM is a relatively young field, considering the evolution of our clinical work model within the context of pediatric emergency medicine (PEM), a field that faces similar challenges in overnight and weekend staffing requirements, may be helpful. Gorelick et al.8 reported that total clinical work hours in PEM (combined academic and nonacademic programs) has decreased from 35.3 hours per week in 1998 to 26.7 in 2013. Extrapolating these numbers to an annual position with 5 weeks PTO/CME, the average PEM attending physician works 1254 clinical hours. These numbers demonstrate a marked difference compared with the average 1800 clinical work hours for PHM found in our study.
Although total hours trend lower in PEM, the authors noted continued challenges in sustainability with an estimated half of all PEM respondents indicating a plan to reduce hours or leave the field in the next 5 years and endorsing symptoms of burnout.6 These findings from PEM may motivate PHM leaders to be more aggressive in adjusting work models toward sustainability in the future.
Our study has several limitations. We utilized a convenience sampling approach that requires the voluntary participation of division directors. Although we had robust interest from respondents representing all major geographic areas, the respondent pool might conceivably over-represent those most interested in understanding and/or changing PHM clinical models. Overall, our sample size was smaller than that achieved by a survey approach. Nevertheless, this limitation was offset by controlling respondent type and clarifying questions, thus improving the quality of our obtained data.
CONCLUSION
This interview-based study of PHM directors describes the current state of clinical work models for university-based hospitalists. University-based PHM programs have similar mean and median total clinical hours per year. However, these hours are higher than those considered ideal by PHM directors, and many are concerned about the sustainability of current work models. Notably, programs that are university-employed or have higher weekends worked per year are more likely to be perceived as unsustainable. Future studies should explore differences between programs with sustainable work models and those with high levels of attrition and burnout.
Disclosures
The authors have no other conflicts to report.
Funding
A grant from the American Academy of Pediatrics Section on Hospital Medicine funded this study through the Subcommittee on Division and Program Leaders.
1. Wachter RM, Goldman L. The emerging role of “hospitalists” in the American health care system. N Engl J Med. 1996;335(7):514-517. DOI: 10.1056/NEJM199608153350713 PubMed
2. Chang W. Record Attendance, Key Issues Highlight Pediatric Hospital Medicine’s 10th Anniversary.
3. Gosdin C, Simmons J, Yau C, Sucharew H, Carlson D, Paciorkowski N. Survey of academic pediatric hospitalist programs in the US: organizational, administrative, and financial factors. J Hosp Med. 2013;8(6):285-291. DOI: 10.1002/jhm.2020. PubMed
4. Hinami K, Whelan CT, Wolosin RJ, Miller JA, Wetterneck TB. Worklife and satisfaction of hospitalists: toward flourishing careers. J Gen Intern Med. 2011;27(1):28-36. DOI: 10.1007/s11606-011-1780-z. PubMed
5. Hinami K, Whelan CT, Miller JA, Wolosin RJ, Wetterneck TB. Job characteristics, satisfaction, and burnout across hospitalist practice models. J Hosp Med. 2012;7(5):402-410. DOI: 10.1002/jhm.1907. PubMed
6. Barrett DJ, McGuinness GA, Cunha CA, et al. Pediatric hospital medicine: a proposed new subspecialty. Pediatrics. 2017;139(3):1-9. DOI: 10.1542/peds.2016-1823. PubMed
7. Cawley P, Deitelzweig S, Flores L, et al. The key principles and characteristics of an effective hospital medicine group: an assessment guide for hospitals and hospitalists. J Hosp Med. 2014;9(2):123-128. DOI: 10.1002/jhm.2119. PubMed
8. Gorelick MH, Schremmer R, Ruch-Ross H, Radabaugh C, Selbst S. Current workforce characteristics and burnout in pediatric emergency medicine. Acad Emerg Med. 2016;23(1):48-54. DOI: 10.1111/acem.12845. PubMed
1. Wachter RM, Goldman L. The emerging role of “hospitalists” in the American health care system. N Engl J Med. 1996;335(7):514-517. DOI: 10.1056/NEJM199608153350713 PubMed
2. Chang W. Record Attendance, Key Issues Highlight Pediatric Hospital Medicine’s 10th Anniversary.
3. Gosdin C, Simmons J, Yau C, Sucharew H, Carlson D, Paciorkowski N. Survey of academic pediatric hospitalist programs in the US: organizational, administrative, and financial factors. J Hosp Med. 2013;8(6):285-291. DOI: 10.1002/jhm.2020. PubMed
4. Hinami K, Whelan CT, Wolosin RJ, Miller JA, Wetterneck TB. Worklife and satisfaction of hospitalists: toward flourishing careers. J Gen Intern Med. 2011;27(1):28-36. DOI: 10.1007/s11606-011-1780-z. PubMed
5. Hinami K, Whelan CT, Miller JA, Wolosin RJ, Wetterneck TB. Job characteristics, satisfaction, and burnout across hospitalist practice models. J Hosp Med. 2012;7(5):402-410. DOI: 10.1002/jhm.1907. PubMed
6. Barrett DJ, McGuinness GA, Cunha CA, et al. Pediatric hospital medicine: a proposed new subspecialty. Pediatrics. 2017;139(3):1-9. DOI: 10.1542/peds.2016-1823. PubMed
7. Cawley P, Deitelzweig S, Flores L, et al. The key principles and characteristics of an effective hospital medicine group: an assessment guide for hospitals and hospitalists. J Hosp Med. 2014;9(2):123-128. DOI: 10.1002/jhm.2119. PubMed
8. Gorelick MH, Schremmer R, Ruch-Ross H, Radabaugh C, Selbst S. Current workforce characteristics and burnout in pediatric emergency medicine. Acad Emerg Med. 2016;23(1):48-54. DOI: 10.1111/acem.12845. PubMed
© 2018 Society of Hospital Medicine
Cardiac Troponins in Low-Risk Pulmonary Embolism Patients: A Systematic Review and Meta-Analysis
Hospital stays for pulmonary embolism (PE) represent a significant cost burden to the United States healthcare system.1 The mean total hospitalization costs for treating a patient with PE ranges widely from $8,764 to $37,006, with an average reported length of stay between 4 and 5 days.2,3 This cost range is attributed to many factors, including type of PE, therapy-induced bleeding risk requiring close monitoring, comorbidities, and social determinants of health. Given that patients with low-risk PE represent the majority of the cases, changes in approaches to care for this population can significantly impact the overall healthcare costs for PE. The European Society of Cardiology (ESC) guidelines incorporate well-validated risk scores, known as the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI) score, and diagnostic test recommendations, including troponin test, echocardiography, and computed tomography, to evaluate patients with PE at varying risk for mortality.4 In these guidelines, the risk stratification algorithm for patients with a low PESI score or a sPESI score of zero does not include checking for the presence of troponin. In reality, practicing hospitalists frequently find that patients receiving a workup in the emergency department for suspected PE undergo troponin test. The ESC guidelines categorize patients with a low-risk score on PESI/sPESI, who subsequently have a positive troponin status, as intermediate low-risk and suggest consideration of hospitalization. The guidelines recommend patients with positive cardiac biomarkers to undergo assessment of right ventricular function through echocardiogram or computed tomography analysis. Moreover, the guidelines support early discharge or ambulatory treatment for low-risk patients who have a negative troponin status.4
The American College of Chest Physicians (ACCP) guidelines on venous thromboembolism (VTE) recommend that cardiac biomarkers should not be measured routinely in all patients with PE and that positive troponin status should discourage physicians from pursuing ambulatory treatment.5 Therefore, ambiguity lies within both guidelines with regard to how hospitalists should interpret a positive troponin status in patients with low risk, which in turn may lead to unnecessary hospitalizations and further imaging. This systematic review and meta-analysis aims to provide clarity, both about gaps in literature and about how practicing hospitalists should interpret troponins in patients with low-risk PE.
METHODS
Data Sources and Searches
This systematic review and meta-analysis was performed in accordance with the established methods and Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. We searched MEDLINE, SCOPUS, and Cochrane Controlled Trial Registry databases for studies published from inception to December 2016 by using the following key words: pulmonary embolism AND PESI OR “pulmonary embolism severity index.” Only articles written in English language were included. The full articles of potentially eligible studies were reviewed, and articles published only in abstract form were excluded.
Study Selection
Two investigators independently assessed the abstract of each article, and the full article was assessed if it fulfilled the following criteria: (1) the publication must be original; (2) inclusion of objectively diagnosed, hemodynamically stable patients (normotensive patients) with acute PE in the inpatient or outpatient setting; (3) inclusion of patients>19 years old; (4) use of the PESI or sPESI model to stratify patients into a low-risk group irrespective of any evidence of right ventricular dysfunction; and (5) testing of cardiac troponin levels (TnI-troponin I, TnT-troponin T, or hs-TnI/TnT-high sensitivity troponin I/T) in patients. Study design, sample size, duration of follow-up, type of troponin used, definition of hemodynamic stability, and specific type of outcome measured (endpoint) did not affect the study eligibility.
Data Extraction and Risk of Bias Assessment
Statistical Analysis
Data were summarized by using 30-day all-cause mortality only because it is the most consistent endpoint reported by all of the included studies. For each study, 30-day all-cause mortality was analyzed across the 2 troponin groups, and the results were summarized in terms of positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR), and odds ratio (OR). To quantify the uncertainty in the LRs and ORs, we calculated 95% confidence intervals (CI).
Overall measures of PPV, NPV, PLR, and NLR were calculated on the pooled collection of data from the studies. LRs are one of the best measures of diagnostic accuracy; therefore, we defined the degree of probability of disease based on simple estimations that were reported by McGee.6 These estimations are independent of pretest probability and include the following: PLR 5.0 increases the probability of the outcome by about 30%, whereas NLR 0.20 decreases the probability of the outcome by 30%. To identify reasonable performance, we defined a PLR > 5 as an increase in moderate to high probability and a NLR < 0.20 as a decrease in moderate to high probability.6
The overall association between 30-day all-cause mortality and troponin classification among patients with low-risk PE was assessed using a mixed effects logistic regression model. The model included a random intercept to account for the correlation among the measurements for patients within a study. The exponentiated regression coefficient for troponin classification is the OR for 30-day all-cause mortality, comparing troponin-positive patients to troponin-negative patients. OR is reported with a 95% CI and a P value. A continuity correction (correction = 0.5) was applied to zero cells. Heterogeneity was measured using Cochran Q statistic and Higgins I2 statistic.
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Study Characteristics
The abstracts of 117 articles were initially identified using the search strategy described above. Of these, 18 articles were deemed appropriate for review based on the criteria outlined in “Study Selection.” The full-text articles of the selected studies were obtained. Upon further evaluation, we identified 16 articles (Figure 1) eligible for the systematic review. Two studies were excluded because they did not provide the number of study participants that met the primary endpoints. The included studies were published from 2009–2016 (Table 1). For patients with low-risk PE, the number of patients with right ventricle dysfunction was either difficult to determine or not reported in all the studies.
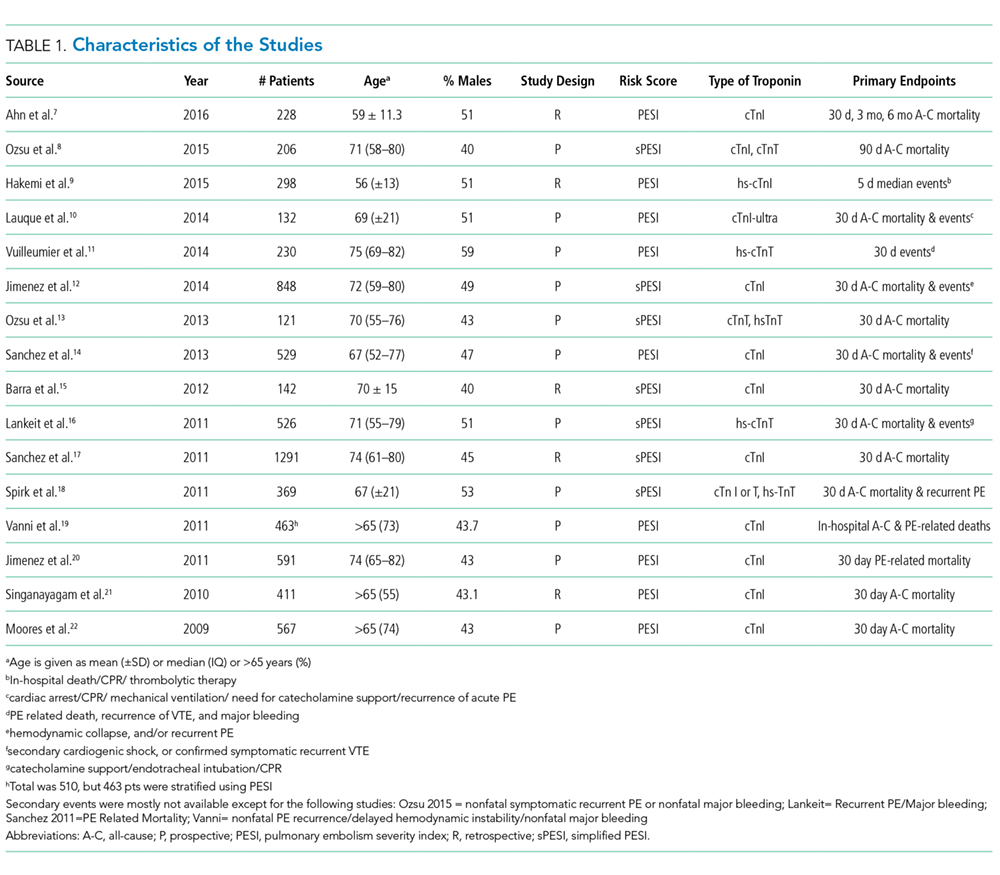
Regarding study design, 11 studies were described as prospective cohorts and the remaining 5 studies were identified as retrospective (Table 1). Seven studies stratified participants’ risk of mortality by using sPESI, and 8 studies employed the PESI score. A total of 6952 participants diagnosed with PE were obtained, and 2662 (38%) were recognized as being low-risk based on either the PESI or sPESI. The sample sizes of the individual studies ranged from 121 to 1,291. The studies used either hs-cTnT, hs-cTnI, cTnT, cTnI, or a combination of hs-cTnT and cTnI or cTnT for troponin assay. Most studies used a pre-defined cut-off value to determine positive or negative troponin status.
Thirteen studies reported 30-day event rate as one of the primary endpoints. The 3 other studies included 90-day all-cause mortality, and 2 of them included in-hospital events. Secondary event rates were only reported in 4 studies and consisted of nonfatal PE, nonfatal major bleeding, and PE-related mortality.
Our systematic review revealed that 5 of the 16 studies used either hemodynamic decompensation, cardiopulmonary resuscitation, mechanical ventilation, or a combination of any of these parameters as part of their primary or secondary endpoint. However, none of the studies specified the number of patients that reached any of these endpoints. Furthermore, 10 of the 16 studies did not specify 30-day PE-related mortality outcomes. The most common endpoint was 30-day all-cause mortality, and only 7 studies reported outcomes with positive or negative troponin status.
Outcome Data of All Studies
A total of 2662 participants were categorized as being low risk based on the PESI or sPESI risk score. The pooled rate of PE-related mortality (specified and inferred) was 5 (0.46%) from 6 studies (1,093 patients), in which only 2 studies specified PE-related mortality as the primary endpoint (Vanni [2011]19 and Jimenez [2011]20). The pooled rate of 30-day all-cause mortality was 24 (1.3%) from 12 studies (1882 patients). In 14 studies (2163 patients), the rates of recurrence of PE and major bleeding were 3 (0.14%) and 6 (0.28%), respectively.
Outcomes of Studies with Corresponding Troponin+ and Troponin –
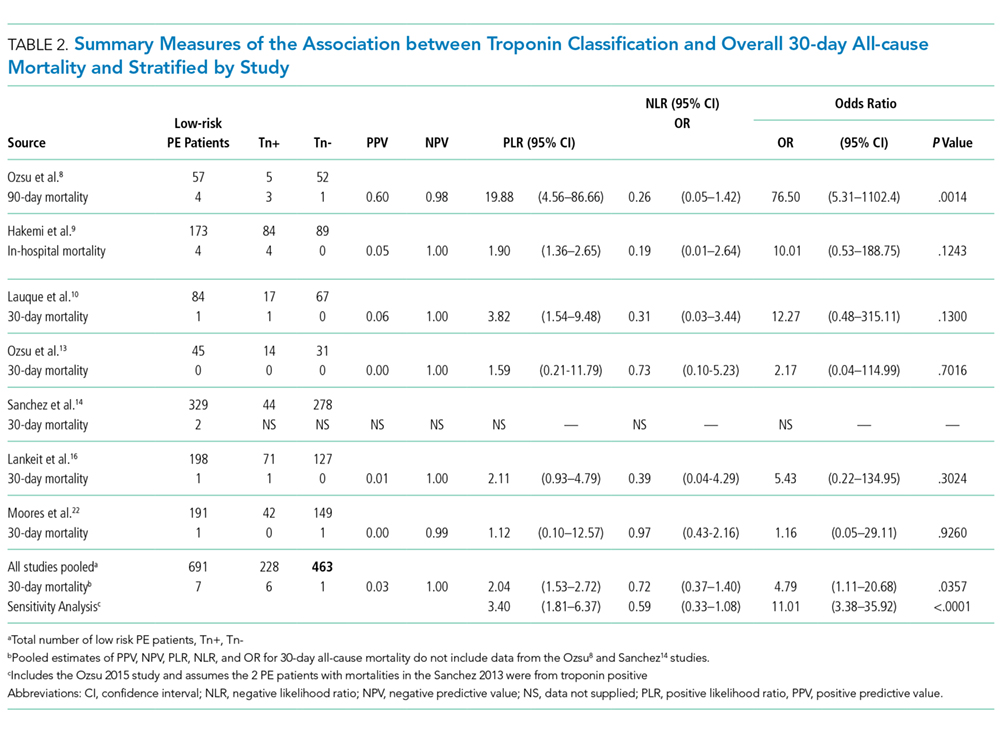
Seven studies used positive or negative troponin status as endpoint to assess low-risk participants (Table 2). However, only 5 studies were included in the final meta-analysis because some data were missing in the Sanchez14 study and the Oszu8 study’s mortality endpoint was more than 30 days. The risk of bias within the studies was evaluated, and for most studies, the quality was of moderate degree (Supplementary Table 1). Table 2 shows the results for the overall pooled data stratified by study. In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moores (2009)22 who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi(2015)9, Lauque (2014)10, and Lankeit(2011)16 studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 (95% CI, 1.53 to 2.72) and negative LR 0.72 (95% CI, 0.37 to 1.40). The OR for all-cause mortality was 4.79 (95% CI 1.11 to 20.68, P = .0357).
A forest plot was created to visualize the PLR from each study included in the main analysis (Figure 2).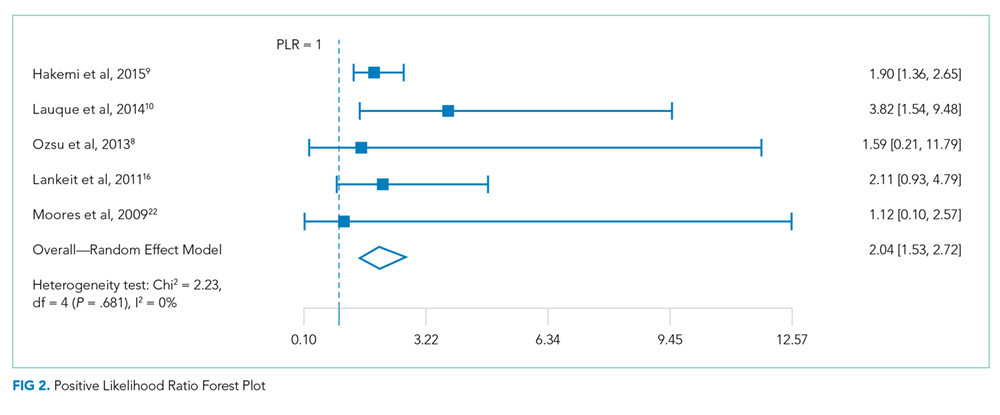
A sensitivity analysis among troponin-positive patients was conducted using 90-day all-cause mortality outcome from the study of Ozsu8 (2015) and the 2 all-cause mortality outcomes from the study of Sanchez14 (2013). The pooled estimates from the 30-day all-cause mortality differed slightly from those previously reported. The PLR increased to 3.40 (95% CI 1.81 to 6.37), and the NLR decreased to 0.59 (95% CI 0.33 to 1.08).
DISCUSSION
In this meta-analysis of 5 studies, which included 691 patients with low-risk PESI or sPESI scores, those tested positive for troponin had nearly a fivefold increased risk of 30-day all-cause mortality compared with patients who tested negative. However, the clinical significance of this association is unclear given that the CI is quite wide and mortality could be associated with PE versus other causes. Similar results were reported by other meta-analyses that consisted of patients with normotensive PE.23-25 To our knowledge, the present meta-analysis is the first to report outcomes in patients with low-risk PE stratified by the presence of cardiac troponin.
A published paper on simplifying the clinical interpretation of LRs state that a positive LR of greater than 5 and a negative LR of less than 0.20 provide dependable evidence regarding reasonable prognostic performance.6 In our analysis, the positive LR was less than 5 and the negative LR’s CI included one. These results suggest a small statistical probability that a patient with a low PESI/sPESI score and a positive troponin status would benefit from inpatient monitoring; simultaneously, a negative troponin does not necessarily translate to safe outpatient therapy, based on our statistical analysis. Previous studies also reported nonextreme positive LRs.23,24 We therefore conclude that low-risk PE patients with positive troponins may be eligible for safe ambulatory treatment or early discharge. However, the number of outcomes of interest (mortality) occurred in only 6 patients among the 228 patients who had positive troponin status. The majority of deaths were reported by Hakemi et al.9 in their retrospective cohort study; as such, drawing conclusions is difficult. Furthermore, the low 30-day all-cause mortality rate of 2.6% in the positive troponin group may have been affected by close monitoring of the patients, who commonly received hemodynamic and oxygen support. Based on these factors, our conclusion is relatively weak, and we cannot recommend a change in practice compared to existing guidelines. In general, additional prospective research is needed to determine whether patients with low-risk PE tested positive for troponin can receive care safely outside the hospital or, rather, require hospitalization similar to patients with intermediate-high risk PE.
We identified a number of other limitations in our analysis. First, aside from the relatively small number of pertinent studies in the literature, most of the studies are of low-moderate quality. Second, the troponin classification in various studies was not conducted using the same assay, and the cut-off value determining positive versus negative results in each case may have differed. These differences may have created some ambiguity or misclassification when the data were pooled together. Third, although the mixed effects logistic regression model controls for some of the variations among patients enrolled in different studies, significant differences exist in terms of patient characteristics or the protocol for follow-up care. This aspect was unaccounted for in this analysis. Lastly, pooled outcome events could not be retrieved from all of the included studies, which would have resulted in a misrepresentation of the true outcomes.
The ESC guidelines suggest avoiding cardiac biomarker testing in patients with low-risk PE because this practice does not have therapeutic implications. Moreover, ESC and ACCP guidelines both state that a positive cardiac biomarker should discourage treatment out of the hospital. The ACCP guidelines further encourage testing of cardiac biomarkers and/or evaluating right ventricular function via echocardiography when uncertainty exists regarding whether patients may require close in-hospital monitoring or not. Although no resounding evidence suggests that troponins have therapeutic implications in patients with low-risk PE, the current guidelines and our meta-analysis cannot offer an overwhelmingly convincing recommendation about whether or not patients with low-risk PE and positive cardiac biomarkers are best treated in the ambulatory or inpatient setting. Such patients may benefit from monitoring in an observation unit (eg, less than 24 or 48 hours), rather than requiring a full admission to the hospital. Nevertheless, our analysis shows that making this determination will require prospective studies that will utilize cardiac troponin status in predicting PE-related events, such as arrhythmia, acute respiratory failure, and hemodynamic decompensation, rather than all-cause mortality.
Until further studies, hospitalists should integrate the use of cardiac troponin and other clinical data, including those available from patient history, physical exam, and other laboratory testing, in determining whether or not to admit, observe, or discharge patients with low-risk PE. As the current guidelines recommend, we support consideration of right ventricular function assessment, via echocardiogram or computed tomography, in patients with positive cardiac troponins even when their PESI/sPESI score is low.
ACKNOWLEDGMENTS
The authors would like to thank Megan Therese Smith, PhD and Lishi Zhang, MS for their contribution in providing a comprehensive statistical analysis of this meta-analysis.
Disclosures
The authors declare no conflicts of interest in the work under consideration for publication. Abdullah Mahayni and Mukti Patel, MD also declared no conflicts of interest with regard to the relevant financial activities outside the submitted work. Omar Darwish, DO and Alpesh Amin, MD also declared no relevant financial activities outside the submitted work; they are speakers for Bristol Myer Squibb and Pfizer regarding the anticoagulant, Apixaban, for treatment of venous thromboembolism and atrial fibrillation.
1. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3-10 PubMed
2. Fanikos J, Rao A, Seger AC, Carter D, Piazza G, Goldhaber SZ. Hospital Costs of Acute Pulmonary Embolism. Am J Med. 2013;126(2):127-132. PubMed
3. LaMori JC, Shoheiber O, Mody SH, Bookart BK. Inpatient Resource Use and Cost Burden of Deep Vein Thrombosis and Pulmonary Embolism in the United States. Clin Ther. 2015;37(1):62-70. PubMed
4. Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galié N, et al. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(43):3033-3080. PubMed
5. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149(2):315-352. PubMed
6. McGee S. Simplifying Likelihood Ratios. J Gen Intern Med. 2002;17(8):647-650. PubMed
7. Ahn S, Lee Y, Kim WY, Lim KS, Lee J. Prognostic Value of Treatment Setting in Patients With Cancer Having Pulmonary Embolism: Comparison With the Pulmonary Embolism Severity Index. Clin Appl Thromb Hemost. 2016;23(6):615-621. PubMed
8. Ozsu S, Bektas H, Abul Y, Ozlu T, Örem A. Value of Cardiac Troponin and sPESI in Treatment of Pulmonary Thromboembolism at Outpatient Setting. Lung. 2015;193(4):559-565. PubMed
9. Hakemi EU, Alyousef T, Dang G, Hakmei J, Doukky R. The prognostic value of undetectable highly sensitive cardiac troponin I in patients with acute pulmonary embolism. Chest. 2015;147(3):685-694. PubMed
10. Lauque D, Maupas-Schwalm F, Bounes V, et al. Predictive Value of the Heart‐type Fatty Acid–binding Protein and the Pulmonary Embolism Severity Index in Patients With Acute Pulmonary Embolism in the Emergency Department. Acad Emerg Med. 2014;21(10):1143-1150. PubMed
11. Vuilleumier N, Limacher A, Méan M, Choffat J, Lescuyer P, Bounameaux H, et al. Cardiac biomarkers and clinical scores for risk stratification in elderly patients with non‐high‐risk pulmonary embolism. J Intern Med. 2014;277(6):707-716. PubMed
12. Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med. 2014;189(6):718-726. PubMed
13. Ozsu S, Abul Y, Orem A, et al. Predictive value of troponins and simplified pulmonary embolism severity index in patients with normotensive pulmonary embolism. Multidiscip Respir Med. 2013;8(1):34. PubMed
14. Sanchez O, Trinquart L, Planquette B, et al. Echocardiography and pulmonary embolism severity index have independent prognostic roles in pulmonary embolism. Eur Respir J. 2013;42(3):681-688. PubMed
15. Barra SN, Paiva L, Providéncia R, Fernandes A, Nascimento J, Marques AL. LR–PED Rule: Low Risk Pulmonary Embolism Decision Rule–A new decision score for low risk Pulmonary Embolism. Thromb Res. 2012;130(3):327-333. PubMed
16. Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive Value of the High-Sensitivity Troponin T Assay and the Simplified Pulmonary Embolism Severity Index in Hemodynamically Stable Patients With Acute Pulmonary Embolism A Prospective Validation Study. Circulation. 2011;124(24):2716-2724. PubMed
17. Sánchez D, De Miguel J, Sam A, et al. The effects of cause of death classification on prognostic assessment of patients with pulmonary embolism. J Thromb Haemost. 2011;9(11):2201-2207. PubMed
18. Spirk D, Aujesky D, Husmann M, et al. Cardiac troponin testing and the simplified Pulmonary Embolism Severity Index. J Thromb Haemost. 2011;105(05):978-984. PubMed
19. Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction‐guided approach. J Thromb Haemos. 2011;9(10):1916-1923. PubMed
20. Jiménez D, Aujesky D, Moores L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66(1):75-81. PubMed
21. Singanayagam A, Scally C, Al-Khairalla MZ, et al. Are biomarkers additive to pulmonary embolism severity index for severity assessment in normotensive patients with acute pulmonary embolism? QJM. 2010;104(2):125-131. PubMed
22. Moores L, Aujesky D, Jimenez D, et al. Pulmonary Embolism Severity Index and troponin testing for the selection of low‐risk patients with acute symptomatic pulmonary embolism. J Thromb Haemost. 2009;8(3):517-522. PubMed
23. Bajaj A, Rathor P, Sehgal V, et al. Prognostic Value of Biomarkers in Acute Non-massive Pulmonary Embolism; A Sysemative Review and Meta-Analysis. Lung. 2015;193(5):639-651. PubMed
24. Jiménez D Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism; a systematic review and metaanalysis. Chest. 2009;136(4):974-982. PubMed
25. Becattini C, Vedovati MC, Agnelli G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation. 2007;116(4):427-433. PubMed
Hospital stays for pulmonary embolism (PE) represent a significant cost burden to the United States healthcare system.1 The mean total hospitalization costs for treating a patient with PE ranges widely from $8,764 to $37,006, with an average reported length of stay between 4 and 5 days.2,3 This cost range is attributed to many factors, including type of PE, therapy-induced bleeding risk requiring close monitoring, comorbidities, and social determinants of health. Given that patients with low-risk PE represent the majority of the cases, changes in approaches to care for this population can significantly impact the overall healthcare costs for PE. The European Society of Cardiology (ESC) guidelines incorporate well-validated risk scores, known as the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI) score, and diagnostic test recommendations, including troponin test, echocardiography, and computed tomography, to evaluate patients with PE at varying risk for mortality.4 In these guidelines, the risk stratification algorithm for patients with a low PESI score or a sPESI score of zero does not include checking for the presence of troponin. In reality, practicing hospitalists frequently find that patients receiving a workup in the emergency department for suspected PE undergo troponin test. The ESC guidelines categorize patients with a low-risk score on PESI/sPESI, who subsequently have a positive troponin status, as intermediate low-risk and suggest consideration of hospitalization. The guidelines recommend patients with positive cardiac biomarkers to undergo assessment of right ventricular function through echocardiogram or computed tomography analysis. Moreover, the guidelines support early discharge or ambulatory treatment for low-risk patients who have a negative troponin status.4
The American College of Chest Physicians (ACCP) guidelines on venous thromboembolism (VTE) recommend that cardiac biomarkers should not be measured routinely in all patients with PE and that positive troponin status should discourage physicians from pursuing ambulatory treatment.5 Therefore, ambiguity lies within both guidelines with regard to how hospitalists should interpret a positive troponin status in patients with low risk, which in turn may lead to unnecessary hospitalizations and further imaging. This systematic review and meta-analysis aims to provide clarity, both about gaps in literature and about how practicing hospitalists should interpret troponins in patients with low-risk PE.
METHODS
Data Sources and Searches
This systematic review and meta-analysis was performed in accordance with the established methods and Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. We searched MEDLINE, SCOPUS, and Cochrane Controlled Trial Registry databases for studies published from inception to December 2016 by using the following key words: pulmonary embolism AND PESI OR “pulmonary embolism severity index.” Only articles written in English language were included. The full articles of potentially eligible studies were reviewed, and articles published only in abstract form were excluded.
Study Selection
Two investigators independently assessed the abstract of each article, and the full article was assessed if it fulfilled the following criteria: (1) the publication must be original; (2) inclusion of objectively diagnosed, hemodynamically stable patients (normotensive patients) with acute PE in the inpatient or outpatient setting; (3) inclusion of patients>19 years old; (4) use of the PESI or sPESI model to stratify patients into a low-risk group irrespective of any evidence of right ventricular dysfunction; and (5) testing of cardiac troponin levels (TnI-troponin I, TnT-troponin T, or hs-TnI/TnT-high sensitivity troponin I/T) in patients. Study design, sample size, duration of follow-up, type of troponin used, definition of hemodynamic stability, and specific type of outcome measured (endpoint) did not affect the study eligibility.
Data Extraction and Risk of Bias Assessment
Statistical Analysis
Data were summarized by using 30-day all-cause mortality only because it is the most consistent endpoint reported by all of the included studies. For each study, 30-day all-cause mortality was analyzed across the 2 troponin groups, and the results were summarized in terms of positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR), and odds ratio (OR). To quantify the uncertainty in the LRs and ORs, we calculated 95% confidence intervals (CI).
Overall measures of PPV, NPV, PLR, and NLR were calculated on the pooled collection of data from the studies. LRs are one of the best measures of diagnostic accuracy; therefore, we defined the degree of probability of disease based on simple estimations that were reported by McGee.6 These estimations are independent of pretest probability and include the following: PLR 5.0 increases the probability of the outcome by about 30%, whereas NLR 0.20 decreases the probability of the outcome by 30%. To identify reasonable performance, we defined a PLR > 5 as an increase in moderate to high probability and a NLR < 0.20 as a decrease in moderate to high probability.6
The overall association between 30-day all-cause mortality and troponin classification among patients with low-risk PE was assessed using a mixed effects logistic regression model. The model included a random intercept to account for the correlation among the measurements for patients within a study. The exponentiated regression coefficient for troponin classification is the OR for 30-day all-cause mortality, comparing troponin-positive patients to troponin-negative patients. OR is reported with a 95% CI and a P value. A continuity correction (correction = 0.5) was applied to zero cells. Heterogeneity was measured using Cochran Q statistic and Higgins I2 statistic.
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Study Characteristics
The abstracts of 117 articles were initially identified using the search strategy described above. Of these, 18 articles were deemed appropriate for review based on the criteria outlined in “Study Selection.” The full-text articles of the selected studies were obtained. Upon further evaluation, we identified 16 articles (Figure 1) eligible for the systematic review. Two studies were excluded because they did not provide the number of study participants that met the primary endpoints. The included studies were published from 2009–2016 (Table 1). For patients with low-risk PE, the number of patients with right ventricle dysfunction was either difficult to determine or not reported in all the studies.
Regarding study design, 11 studies were described as prospective cohorts and the remaining 5 studies were identified as retrospective (Table 1). Seven studies stratified participants’ risk of mortality by using sPESI, and 8 studies employed the PESI score. A total of 6952 participants diagnosed with PE were obtained, and 2662 (38%) were recognized as being low-risk based on either the PESI or sPESI. The sample sizes of the individual studies ranged from 121 to 1,291. The studies used either hs-cTnT, hs-cTnI, cTnT, cTnI, or a combination of hs-cTnT and cTnI or cTnT for troponin assay. Most studies used a pre-defined cut-off value to determine positive or negative troponin status.
Thirteen studies reported 30-day event rate as one of the primary endpoints. The 3 other studies included 90-day all-cause mortality, and 2 of them included in-hospital events. Secondary event rates were only reported in 4 studies and consisted of nonfatal PE, nonfatal major bleeding, and PE-related mortality.
Our systematic review revealed that 5 of the 16 studies used either hemodynamic decompensation, cardiopulmonary resuscitation, mechanical ventilation, or a combination of any of these parameters as part of their primary or secondary endpoint. However, none of the studies specified the number of patients that reached any of these endpoints. Furthermore, 10 of the 16 studies did not specify 30-day PE-related mortality outcomes. The most common endpoint was 30-day all-cause mortality, and only 7 studies reported outcomes with positive or negative troponin status.
Outcome Data of All Studies
A total of 2662 participants were categorized as being low risk based on the PESI or sPESI risk score. The pooled rate of PE-related mortality (specified and inferred) was 5 (0.46%) from 6 studies (1,093 patients), in which only 2 studies specified PE-related mortality as the primary endpoint (Vanni [2011]19 and Jimenez [2011]20). The pooled rate of 30-day all-cause mortality was 24 (1.3%) from 12 studies (1882 patients). In 14 studies (2163 patients), the rates of recurrence of PE and major bleeding were 3 (0.14%) and 6 (0.28%), respectively.
Outcomes of Studies with Corresponding Troponin+ and Troponin –
Seven studies used positive or negative troponin status as endpoint to assess low-risk participants (Table 2). However, only 5 studies were included in the final meta-analysis because some data were missing in the Sanchez14 study and the Oszu8 study’s mortality endpoint was more than 30 days. The risk of bias within the studies was evaluated, and for most studies, the quality was of moderate degree (Supplementary Table 1). Table 2 shows the results for the overall pooled data stratified by study. In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moores (2009)22 who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi(2015)9, Lauque (2014)10, and Lankeit(2011)16 studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 (95% CI, 1.53 to 2.72) and negative LR 0.72 (95% CI, 0.37 to 1.40). The OR for all-cause mortality was 4.79 (95% CI 1.11 to 20.68, P = .0357).
A forest plot was created to visualize the PLR from each study included in the main analysis (Figure 2).
A sensitivity analysis among troponin-positive patients was conducted using 90-day all-cause mortality outcome from the study of Ozsu8 (2015) and the 2 all-cause mortality outcomes from the study of Sanchez14 (2013). The pooled estimates from the 30-day all-cause mortality differed slightly from those previously reported. The PLR increased to 3.40 (95% CI 1.81 to 6.37), and the NLR decreased to 0.59 (95% CI 0.33 to 1.08).
DISCUSSION
In this meta-analysis of 5 studies, which included 691 patients with low-risk PESI or sPESI scores, those tested positive for troponin had nearly a fivefold increased risk of 30-day all-cause mortality compared with patients who tested negative. However, the clinical significance of this association is unclear given that the CI is quite wide and mortality could be associated with PE versus other causes. Similar results were reported by other meta-analyses that consisted of patients with normotensive PE.23-25 To our knowledge, the present meta-analysis is the first to report outcomes in patients with low-risk PE stratified by the presence of cardiac troponin.
A published paper on simplifying the clinical interpretation of LRs state that a positive LR of greater than 5 and a negative LR of less than 0.20 provide dependable evidence regarding reasonable prognostic performance.6 In our analysis, the positive LR was less than 5 and the negative LR’s CI included one. These results suggest a small statistical probability that a patient with a low PESI/sPESI score and a positive troponin status would benefit from inpatient monitoring; simultaneously, a negative troponin does not necessarily translate to safe outpatient therapy, based on our statistical analysis. Previous studies also reported nonextreme positive LRs.23,24 We therefore conclude that low-risk PE patients with positive troponins may be eligible for safe ambulatory treatment or early discharge. However, the number of outcomes of interest (mortality) occurred in only 6 patients among the 228 patients who had positive troponin status. The majority of deaths were reported by Hakemi et al.9 in their retrospective cohort study; as such, drawing conclusions is difficult. Furthermore, the low 30-day all-cause mortality rate of 2.6% in the positive troponin group may have been affected by close monitoring of the patients, who commonly received hemodynamic and oxygen support. Based on these factors, our conclusion is relatively weak, and we cannot recommend a change in practice compared to existing guidelines. In general, additional prospective research is needed to determine whether patients with low-risk PE tested positive for troponin can receive care safely outside the hospital or, rather, require hospitalization similar to patients with intermediate-high risk PE.
We identified a number of other limitations in our analysis. First, aside from the relatively small number of pertinent studies in the literature, most of the studies are of low-moderate quality. Second, the troponin classification in various studies was not conducted using the same assay, and the cut-off value determining positive versus negative results in each case may have differed. These differences may have created some ambiguity or misclassification when the data were pooled together. Third, although the mixed effects logistic regression model controls for some of the variations among patients enrolled in different studies, significant differences exist in terms of patient characteristics or the protocol for follow-up care. This aspect was unaccounted for in this analysis. Lastly, pooled outcome events could not be retrieved from all of the included studies, which would have resulted in a misrepresentation of the true outcomes.
The ESC guidelines suggest avoiding cardiac biomarker testing in patients with low-risk PE because this practice does not have therapeutic implications. Moreover, ESC and ACCP guidelines both state that a positive cardiac biomarker should discourage treatment out of the hospital. The ACCP guidelines further encourage testing of cardiac biomarkers and/or evaluating right ventricular function via echocardiography when uncertainty exists regarding whether patients may require close in-hospital monitoring or not. Although no resounding evidence suggests that troponins have therapeutic implications in patients with low-risk PE, the current guidelines and our meta-analysis cannot offer an overwhelmingly convincing recommendation about whether or not patients with low-risk PE and positive cardiac biomarkers are best treated in the ambulatory or inpatient setting. Such patients may benefit from monitoring in an observation unit (eg, less than 24 or 48 hours), rather than requiring a full admission to the hospital. Nevertheless, our analysis shows that making this determination will require prospective studies that will utilize cardiac troponin status in predicting PE-related events, such as arrhythmia, acute respiratory failure, and hemodynamic decompensation, rather than all-cause mortality.
Until further studies, hospitalists should integrate the use of cardiac troponin and other clinical data, including those available from patient history, physical exam, and other laboratory testing, in determining whether or not to admit, observe, or discharge patients with low-risk PE. As the current guidelines recommend, we support consideration of right ventricular function assessment, via echocardiogram or computed tomography, in patients with positive cardiac troponins even when their PESI/sPESI score is low.
ACKNOWLEDGMENTS
The authors would like to thank Megan Therese Smith, PhD and Lishi Zhang, MS for their contribution in providing a comprehensive statistical analysis of this meta-analysis.
Disclosures
The authors declare no conflicts of interest in the work under consideration for publication. Abdullah Mahayni and Mukti Patel, MD also declared no conflicts of interest with regard to the relevant financial activities outside the submitted work. Omar Darwish, DO and Alpesh Amin, MD also declared no relevant financial activities outside the submitted work; they are speakers for Bristol Myer Squibb and Pfizer regarding the anticoagulant, Apixaban, for treatment of venous thromboembolism and atrial fibrillation.
Hospital stays for pulmonary embolism (PE) represent a significant cost burden to the United States healthcare system.1 The mean total hospitalization costs for treating a patient with PE ranges widely from $8,764 to $37,006, with an average reported length of stay between 4 and 5 days.2,3 This cost range is attributed to many factors, including type of PE, therapy-induced bleeding risk requiring close monitoring, comorbidities, and social determinants of health. Given that patients with low-risk PE represent the majority of the cases, changes in approaches to care for this population can significantly impact the overall healthcare costs for PE. The European Society of Cardiology (ESC) guidelines incorporate well-validated risk scores, known as the pulmonary embolism severity index (PESI) and the simplified PESI (sPESI) score, and diagnostic test recommendations, including troponin test, echocardiography, and computed tomography, to evaluate patients with PE at varying risk for mortality.4 In these guidelines, the risk stratification algorithm for patients with a low PESI score or a sPESI score of zero does not include checking for the presence of troponin. In reality, practicing hospitalists frequently find that patients receiving a workup in the emergency department for suspected PE undergo troponin test. The ESC guidelines categorize patients with a low-risk score on PESI/sPESI, who subsequently have a positive troponin status, as intermediate low-risk and suggest consideration of hospitalization. The guidelines recommend patients with positive cardiac biomarkers to undergo assessment of right ventricular function through echocardiogram or computed tomography analysis. Moreover, the guidelines support early discharge or ambulatory treatment for low-risk patients who have a negative troponin status.4
The American College of Chest Physicians (ACCP) guidelines on venous thromboembolism (VTE) recommend that cardiac biomarkers should not be measured routinely in all patients with PE and that positive troponin status should discourage physicians from pursuing ambulatory treatment.5 Therefore, ambiguity lies within both guidelines with regard to how hospitalists should interpret a positive troponin status in patients with low risk, which in turn may lead to unnecessary hospitalizations and further imaging. This systematic review and meta-analysis aims to provide clarity, both about gaps in literature and about how practicing hospitalists should interpret troponins in patients with low-risk PE.
METHODS
Data Sources and Searches
This systematic review and meta-analysis was performed in accordance with the established methods and Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines. We searched MEDLINE, SCOPUS, and Cochrane Controlled Trial Registry databases for studies published from inception to December 2016 by using the following key words: pulmonary embolism AND PESI OR “pulmonary embolism severity index.” Only articles written in English language were included. The full articles of potentially eligible studies were reviewed, and articles published only in abstract form were excluded.
Study Selection
Two investigators independently assessed the abstract of each article, and the full article was assessed if it fulfilled the following criteria: (1) the publication must be original; (2) inclusion of objectively diagnosed, hemodynamically stable patients (normotensive patients) with acute PE in the inpatient or outpatient setting; (3) inclusion of patients>19 years old; (4) use of the PESI or sPESI model to stratify patients into a low-risk group irrespective of any evidence of right ventricular dysfunction; and (5) testing of cardiac troponin levels (TnI-troponin I, TnT-troponin T, or hs-TnI/TnT-high sensitivity troponin I/T) in patients. Study design, sample size, duration of follow-up, type of troponin used, definition of hemodynamic stability, and specific type of outcome measured (endpoint) did not affect the study eligibility.
Data Extraction and Risk of Bias Assessment
Statistical Analysis
Data were summarized by using 30-day all-cause mortality only because it is the most consistent endpoint reported by all of the included studies. For each study, 30-day all-cause mortality was analyzed across the 2 troponin groups, and the results were summarized in terms of positive predictive value (PPV), negative predictive value (NPV), positive likelihood ratio (PLR), negative likelihood ratio (NLR), and odds ratio (OR). To quantify the uncertainty in the LRs and ORs, we calculated 95% confidence intervals (CI).
Overall measures of PPV, NPV, PLR, and NLR were calculated on the pooled collection of data from the studies. LRs are one of the best measures of diagnostic accuracy; therefore, we defined the degree of probability of disease based on simple estimations that were reported by McGee.6 These estimations are independent of pretest probability and include the following: PLR 5.0 increases the probability of the outcome by about 30%, whereas NLR 0.20 decreases the probability of the outcome by 30%. To identify reasonable performance, we defined a PLR > 5 as an increase in moderate to high probability and a NLR < 0.20 as a decrease in moderate to high probability.6
The overall association between 30-day all-cause mortality and troponin classification among patients with low-risk PE was assessed using a mixed effects logistic regression model. The model included a random intercept to account for the correlation among the measurements for patients within a study. The exponentiated regression coefficient for troponin classification is the OR for 30-day all-cause mortality, comparing troponin-positive patients to troponin-negative patients. OR is reported with a 95% CI and a P value. A continuity correction (correction = 0.5) was applied to zero cells. Heterogeneity was measured using Cochran Q statistic and Higgins I2 statistic.
RESULTS
Search Results
Figure 1 represents the PRISMA flow diagram for literature search and selection process to identify eligible studies for inclusion.
Study Characteristics
The abstracts of 117 articles were initially identified using the search strategy described above. Of these, 18 articles were deemed appropriate for review based on the criteria outlined in “Study Selection.” The full-text articles of the selected studies were obtained. Upon further evaluation, we identified 16 articles (Figure 1) eligible for the systematic review. Two studies were excluded because they did not provide the number of study participants that met the primary endpoints. The included studies were published from 2009–2016 (Table 1). For patients with low-risk PE, the number of patients with right ventricle dysfunction was either difficult to determine or not reported in all the studies.
Regarding study design, 11 studies were described as prospective cohorts and the remaining 5 studies were identified as retrospective (Table 1). Seven studies stratified participants’ risk of mortality by using sPESI, and 8 studies employed the PESI score. A total of 6952 participants diagnosed with PE were obtained, and 2662 (38%) were recognized as being low-risk based on either the PESI or sPESI. The sample sizes of the individual studies ranged from 121 to 1,291. The studies used either hs-cTnT, hs-cTnI, cTnT, cTnI, or a combination of hs-cTnT and cTnI or cTnT for troponin assay. Most studies used a pre-defined cut-off value to determine positive or negative troponin status.
Thirteen studies reported 30-day event rate as one of the primary endpoints. The 3 other studies included 90-day all-cause mortality, and 2 of them included in-hospital events. Secondary event rates were only reported in 4 studies and consisted of nonfatal PE, nonfatal major bleeding, and PE-related mortality.
Our systematic review revealed that 5 of the 16 studies used either hemodynamic decompensation, cardiopulmonary resuscitation, mechanical ventilation, or a combination of any of these parameters as part of their primary or secondary endpoint. However, none of the studies specified the number of patients that reached any of these endpoints. Furthermore, 10 of the 16 studies did not specify 30-day PE-related mortality outcomes. The most common endpoint was 30-day all-cause mortality, and only 7 studies reported outcomes with positive or negative troponin status.
Outcome Data of All Studies
A total of 2662 participants were categorized as being low risk based on the PESI or sPESI risk score. The pooled rate of PE-related mortality (specified and inferred) was 5 (0.46%) from 6 studies (1,093 patients), in which only 2 studies specified PE-related mortality as the primary endpoint (Vanni [2011]19 and Jimenez [2011]20). The pooled rate of 30-day all-cause mortality was 24 (1.3%) from 12 studies (1882 patients). In 14 studies (2163 patients), the rates of recurrence of PE and major bleeding were 3 (0.14%) and 6 (0.28%), respectively.
Outcomes of Studies with Corresponding Troponin+ and Troponin –
Seven studies used positive or negative troponin status as endpoint to assess low-risk participants (Table 2). However, only 5 studies were included in the final meta-analysis because some data were missing in the Sanchez14 study and the Oszu8 study’s mortality endpoint was more than 30 days. The risk of bias within the studies was evaluated, and for most studies, the quality was of moderate degree (Supplementary Table 1). Table 2 shows the results for the overall pooled data stratified by study. In the pooled data, 463 (67%) patients tested negative for troponin and 228 (33%) tested positive. The overall mortality (from sensitivity analysis) including in-hospital, 30-day, and 90-day mortalities was 1.2%. The NPVs for all individual studies and the overall NPV are 1 or approximately 1. The overall PPVs and by study were low, ranging from 0 to 0.60. The PLRs and NLRs were not estimated for an outcome within an individual study if none of the patients experienced the outcome. When outcomes were only observed among troponin-negative patients, such as in the study of Moores (2009)22 who used 30-day all-cause mortality, the PLR had a value of zero. When outcomes were only observed among troponin-positive patients, as for 30-day all-cause mortality in the Hakemi(2015)9, Lauque (2014)10, and Lankeit(2011)16 studies, the NLR had a value of zero. For zero cells, a continuity correction of 0.5 was applied. The pooled likelihood ratios (LRs) for all-cause mortality were positive LR 2.04 (95% CI, 1.53 to 2.72) and negative LR 0.72 (95% CI, 0.37 to 1.40). The OR for all-cause mortality was 4.79 (95% CI 1.11 to 20.68, P = .0357).
A forest plot was created to visualize the PLR from each study included in the main analysis (Figure 2).
A sensitivity analysis among troponin-positive patients was conducted using 90-day all-cause mortality outcome from the study of Ozsu8 (2015) and the 2 all-cause mortality outcomes from the study of Sanchez14 (2013). The pooled estimates from the 30-day all-cause mortality differed slightly from those previously reported. The PLR increased to 3.40 (95% CI 1.81 to 6.37), and the NLR decreased to 0.59 (95% CI 0.33 to 1.08).
DISCUSSION
In this meta-analysis of 5 studies, which included 691 patients with low-risk PESI or sPESI scores, those tested positive for troponin had nearly a fivefold increased risk of 30-day all-cause mortality compared with patients who tested negative. However, the clinical significance of this association is unclear given that the CI is quite wide and mortality could be associated with PE versus other causes. Similar results were reported by other meta-analyses that consisted of patients with normotensive PE.23-25 To our knowledge, the present meta-analysis is the first to report outcomes in patients with low-risk PE stratified by the presence of cardiac troponin.
A published paper on simplifying the clinical interpretation of LRs state that a positive LR of greater than 5 and a negative LR of less than 0.20 provide dependable evidence regarding reasonable prognostic performance.6 In our analysis, the positive LR was less than 5 and the negative LR’s CI included one. These results suggest a small statistical probability that a patient with a low PESI/sPESI score and a positive troponin status would benefit from inpatient monitoring; simultaneously, a negative troponin does not necessarily translate to safe outpatient therapy, based on our statistical analysis. Previous studies also reported nonextreme positive LRs.23,24 We therefore conclude that low-risk PE patients with positive troponins may be eligible for safe ambulatory treatment or early discharge. However, the number of outcomes of interest (mortality) occurred in only 6 patients among the 228 patients who had positive troponin status. The majority of deaths were reported by Hakemi et al.9 in their retrospective cohort study; as such, drawing conclusions is difficult. Furthermore, the low 30-day all-cause mortality rate of 2.6% in the positive troponin group may have been affected by close monitoring of the patients, who commonly received hemodynamic and oxygen support. Based on these factors, our conclusion is relatively weak, and we cannot recommend a change in practice compared to existing guidelines. In general, additional prospective research is needed to determine whether patients with low-risk PE tested positive for troponin can receive care safely outside the hospital or, rather, require hospitalization similar to patients with intermediate-high risk PE.
We identified a number of other limitations in our analysis. First, aside from the relatively small number of pertinent studies in the literature, most of the studies are of low-moderate quality. Second, the troponin classification in various studies was not conducted using the same assay, and the cut-off value determining positive versus negative results in each case may have differed. These differences may have created some ambiguity or misclassification when the data were pooled together. Third, although the mixed effects logistic regression model controls for some of the variations among patients enrolled in different studies, significant differences exist in terms of patient characteristics or the protocol for follow-up care. This aspect was unaccounted for in this analysis. Lastly, pooled outcome events could not be retrieved from all of the included studies, which would have resulted in a misrepresentation of the true outcomes.
The ESC guidelines suggest avoiding cardiac biomarker testing in patients with low-risk PE because this practice does not have therapeutic implications. Moreover, ESC and ACCP guidelines both state that a positive cardiac biomarker should discourage treatment out of the hospital. The ACCP guidelines further encourage testing of cardiac biomarkers and/or evaluating right ventricular function via echocardiography when uncertainty exists regarding whether patients may require close in-hospital monitoring or not. Although no resounding evidence suggests that troponins have therapeutic implications in patients with low-risk PE, the current guidelines and our meta-analysis cannot offer an overwhelmingly convincing recommendation about whether or not patients with low-risk PE and positive cardiac biomarkers are best treated in the ambulatory or inpatient setting. Such patients may benefit from monitoring in an observation unit (eg, less than 24 or 48 hours), rather than requiring a full admission to the hospital. Nevertheless, our analysis shows that making this determination will require prospective studies that will utilize cardiac troponin status in predicting PE-related events, such as arrhythmia, acute respiratory failure, and hemodynamic decompensation, rather than all-cause mortality.
Until further studies, hospitalists should integrate the use of cardiac troponin and other clinical data, including those available from patient history, physical exam, and other laboratory testing, in determining whether or not to admit, observe, or discharge patients with low-risk PE. As the current guidelines recommend, we support consideration of right ventricular function assessment, via echocardiogram or computed tomography, in patients with positive cardiac troponins even when their PESI/sPESI score is low.
ACKNOWLEDGMENTS
The authors would like to thank Megan Therese Smith, PhD and Lishi Zhang, MS for their contribution in providing a comprehensive statistical analysis of this meta-analysis.
Disclosures
The authors declare no conflicts of interest in the work under consideration for publication. Abdullah Mahayni and Mukti Patel, MD also declared no conflicts of interest with regard to the relevant financial activities outside the submitted work. Omar Darwish, DO and Alpesh Amin, MD also declared no relevant financial activities outside the submitted work; they are speakers for Bristol Myer Squibb and Pfizer regarding the anticoagulant, Apixaban, for treatment of venous thromboembolism and atrial fibrillation.
1. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3-10 PubMed
2. Fanikos J, Rao A, Seger AC, Carter D, Piazza G, Goldhaber SZ. Hospital Costs of Acute Pulmonary Embolism. Am J Med. 2013;126(2):127-132. PubMed
3. LaMori JC, Shoheiber O, Mody SH, Bookart BK. Inpatient Resource Use and Cost Burden of Deep Vein Thrombosis and Pulmonary Embolism in the United States. Clin Ther. 2015;37(1):62-70. PubMed
4. Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galié N, et al. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(43):3033-3080. PubMed
5. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149(2):315-352. PubMed
6. McGee S. Simplifying Likelihood Ratios. J Gen Intern Med. 2002;17(8):647-650. PubMed
7. Ahn S, Lee Y, Kim WY, Lim KS, Lee J. Prognostic Value of Treatment Setting in Patients With Cancer Having Pulmonary Embolism: Comparison With the Pulmonary Embolism Severity Index. Clin Appl Thromb Hemost. 2016;23(6):615-621. PubMed
8. Ozsu S, Bektas H, Abul Y, Ozlu T, Örem A. Value of Cardiac Troponin and sPESI in Treatment of Pulmonary Thromboembolism at Outpatient Setting. Lung. 2015;193(4):559-565. PubMed
9. Hakemi EU, Alyousef T, Dang G, Hakmei J, Doukky R. The prognostic value of undetectable highly sensitive cardiac troponin I in patients with acute pulmonary embolism. Chest. 2015;147(3):685-694. PubMed
10. Lauque D, Maupas-Schwalm F, Bounes V, et al. Predictive Value of the Heart‐type Fatty Acid–binding Protein and the Pulmonary Embolism Severity Index in Patients With Acute Pulmonary Embolism in the Emergency Department. Acad Emerg Med. 2014;21(10):1143-1150. PubMed
11. Vuilleumier N, Limacher A, Méan M, Choffat J, Lescuyer P, Bounameaux H, et al. Cardiac biomarkers and clinical scores for risk stratification in elderly patients with non‐high‐risk pulmonary embolism. J Intern Med. 2014;277(6):707-716. PubMed
12. Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med. 2014;189(6):718-726. PubMed
13. Ozsu S, Abul Y, Orem A, et al. Predictive value of troponins and simplified pulmonary embolism severity index in patients with normotensive pulmonary embolism. Multidiscip Respir Med. 2013;8(1):34. PubMed
14. Sanchez O, Trinquart L, Planquette B, et al. Echocardiography and pulmonary embolism severity index have independent prognostic roles in pulmonary embolism. Eur Respir J. 2013;42(3):681-688. PubMed
15. Barra SN, Paiva L, Providéncia R, Fernandes A, Nascimento J, Marques AL. LR–PED Rule: Low Risk Pulmonary Embolism Decision Rule–A new decision score for low risk Pulmonary Embolism. Thromb Res. 2012;130(3):327-333. PubMed
16. Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive Value of the High-Sensitivity Troponin T Assay and the Simplified Pulmonary Embolism Severity Index in Hemodynamically Stable Patients With Acute Pulmonary Embolism A Prospective Validation Study. Circulation. 2011;124(24):2716-2724. PubMed
17. Sánchez D, De Miguel J, Sam A, et al. The effects of cause of death classification on prognostic assessment of patients with pulmonary embolism. J Thromb Haemost. 2011;9(11):2201-2207. PubMed
18. Spirk D, Aujesky D, Husmann M, et al. Cardiac troponin testing and the simplified Pulmonary Embolism Severity Index. J Thromb Haemost. 2011;105(05):978-984. PubMed
19. Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction‐guided approach. J Thromb Haemos. 2011;9(10):1916-1923. PubMed
20. Jiménez D, Aujesky D, Moores L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66(1):75-81. PubMed
21. Singanayagam A, Scally C, Al-Khairalla MZ, et al. Are biomarkers additive to pulmonary embolism severity index for severity assessment in normotensive patients with acute pulmonary embolism? QJM. 2010;104(2):125-131. PubMed
22. Moores L, Aujesky D, Jimenez D, et al. Pulmonary Embolism Severity Index and troponin testing for the selection of low‐risk patients with acute symptomatic pulmonary embolism. J Thromb Haemost. 2009;8(3):517-522. PubMed
23. Bajaj A, Rathor P, Sehgal V, et al. Prognostic Value of Biomarkers in Acute Non-massive Pulmonary Embolism; A Sysemative Review and Meta-Analysis. Lung. 2015;193(5):639-651. PubMed
24. Jiménez D Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism; a systematic review and metaanalysis. Chest. 2009;136(4):974-982. PubMed
25. Becattini C, Vedovati MC, Agnelli G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation. 2007;116(4):427-433. PubMed
1. Grosse SD, Nelson RE, Nyarko KA, Richardson LC, Raskob GE. The economic burden of incident venous thromboembolism in the United States: A review of estimated attributable healthcare costs. Thromb Res. 2016;137:3-10 PubMed
2. Fanikos J, Rao A, Seger AC, Carter D, Piazza G, Goldhaber SZ. Hospital Costs of Acute Pulmonary Embolism. Am J Med. 2013;126(2):127-132. PubMed
3. LaMori JC, Shoheiber O, Mody SH, Bookart BK. Inpatient Resource Use and Cost Burden of Deep Vein Thrombosis and Pulmonary Embolism in the United States. Clin Ther. 2015;37(1):62-70. PubMed
4. Konstantinides S, Torbicki A, Agnelli G, Danchin N, Fitzmaurice D, Galié N, et al. 2014 ESC Guidelines on the diagnosis and management of acute pulmonary embolism. The Task Force for the Diagnosis and Management of Acute Pulmonary Embolism of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(43):3033-3080. PubMed
5. Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic Therapy for VTE Disease: CHEST Guideline and Expert Panel Report. Chest. 2016;149(2):315-352. PubMed
6. McGee S. Simplifying Likelihood Ratios. J Gen Intern Med. 2002;17(8):647-650. PubMed
7. Ahn S, Lee Y, Kim WY, Lim KS, Lee J. Prognostic Value of Treatment Setting in Patients With Cancer Having Pulmonary Embolism: Comparison With the Pulmonary Embolism Severity Index. Clin Appl Thromb Hemost. 2016;23(6):615-621. PubMed
8. Ozsu S, Bektas H, Abul Y, Ozlu T, Örem A. Value of Cardiac Troponin and sPESI in Treatment of Pulmonary Thromboembolism at Outpatient Setting. Lung. 2015;193(4):559-565. PubMed
9. Hakemi EU, Alyousef T, Dang G, Hakmei J, Doukky R. The prognostic value of undetectable highly sensitive cardiac troponin I in patients with acute pulmonary embolism. Chest. 2015;147(3):685-694. PubMed
10. Lauque D, Maupas-Schwalm F, Bounes V, et al. Predictive Value of the Heart‐type Fatty Acid–binding Protein and the Pulmonary Embolism Severity Index in Patients With Acute Pulmonary Embolism in the Emergency Department. Acad Emerg Med. 2014;21(10):1143-1150. PubMed
11. Vuilleumier N, Limacher A, Méan M, Choffat J, Lescuyer P, Bounameaux H, et al. Cardiac biomarkers and clinical scores for risk stratification in elderly patients with non‐high‐risk pulmonary embolism. J Intern Med. 2014;277(6):707-716. PubMed
12. Jiménez D, Kopecna D, Tapson V, et al. Derivation and validation of multimarker prognostication for normotensive patients with acute symptomatic pulmonary embolism. Am J Respir Crit Care Med. 2014;189(6):718-726. PubMed
13. Ozsu S, Abul Y, Orem A, et al. Predictive value of troponins and simplified pulmonary embolism severity index in patients with normotensive pulmonary embolism. Multidiscip Respir Med. 2013;8(1):34. PubMed
14. Sanchez O, Trinquart L, Planquette B, et al. Echocardiography and pulmonary embolism severity index have independent prognostic roles in pulmonary embolism. Eur Respir J. 2013;42(3):681-688. PubMed
15. Barra SN, Paiva L, Providéncia R, Fernandes A, Nascimento J, Marques AL. LR–PED Rule: Low Risk Pulmonary Embolism Decision Rule–A new decision score for low risk Pulmonary Embolism. Thromb Res. 2012;130(3):327-333. PubMed
16. Lankeit M, Jiménez D, Kostrubiec M, et al. Predictive Value of the High-Sensitivity Troponin T Assay and the Simplified Pulmonary Embolism Severity Index in Hemodynamically Stable Patients With Acute Pulmonary Embolism A Prospective Validation Study. Circulation. 2011;124(24):2716-2724. PubMed
17. Sánchez D, De Miguel J, Sam A, et al. The effects of cause of death classification on prognostic assessment of patients with pulmonary embolism. J Thromb Haemost. 2011;9(11):2201-2207. PubMed
18. Spirk D, Aujesky D, Husmann M, et al. Cardiac troponin testing and the simplified Pulmonary Embolism Severity Index. J Thromb Haemost. 2011;105(05):978-984. PubMed
19. Vanni S, Nazerian P, Pepe G, et al. Comparison of two prognostic models for acute pulmonary embolism: clinical vs. right ventricular dysfunction‐guided approach. J Thromb Haemos. 2011;9(10):1916-1923. PubMed
20. Jiménez D, Aujesky D, Moores L, et al. Combinations of prognostic tools for identification of high-risk normotensive patients with acute symptomatic pulmonary embolism. Thorax. 2011;66(1):75-81. PubMed
21. Singanayagam A, Scally C, Al-Khairalla MZ, et al. Are biomarkers additive to pulmonary embolism severity index for severity assessment in normotensive patients with acute pulmonary embolism? QJM. 2010;104(2):125-131. PubMed
22. Moores L, Aujesky D, Jimenez D, et al. Pulmonary Embolism Severity Index and troponin testing for the selection of low‐risk patients with acute symptomatic pulmonary embolism. J Thromb Haemost. 2009;8(3):517-522. PubMed
23. Bajaj A, Rathor P, Sehgal V, et al. Prognostic Value of Biomarkers in Acute Non-massive Pulmonary Embolism; A Sysemative Review and Meta-Analysis. Lung. 2015;193(5):639-651. PubMed
24. Jiménez D Uresandi F, Otero R, et al. Troponin-based risk stratification of patients with acute nonmassive pulmonary embolism; a systematic review and metaanalysis. Chest. 2009;136(4):974-982. PubMed
25. Becattini C, Vedovati MC, Agnelli G. Prognostic Value of Troponins in Acute Pulmonary Embolism: A Meta-Analysis. Circulation. 2007;116(4):427-433. PubMed
© 2018 Society of Hospital Medicine
Characterizing Hospitalizations for Pediatric Concussion and Trends in Care
Approximately 14% of children who sustain a concussion are admitted to the hospital,1 although admission rates reportedly vary substantially among pediatric hospitals.2 Children hospitalized for concussion may be at a higher risk for persistent postconcussive symptoms,3,4 yet little is known about this subset of children and how they are managed while in the hospital. Characterizing children hospitalized for concussion and describing the inpatient care they received will promote hypothesis generation for further inquiry into indications for admission, as well as the relationship between inpatient management and concussion recovery.
We described a cohort of children admitted to 40 pediatric hospitals primarily for concussion and detailed care delivered during hospitalization. We explored individual-level factors and their association with prolonged length of stay (LOS) and emergency department (ED) readmission. Finally, we evaluated if there had been changes in inpatient care over the 8-year study period.
PATIENTS AND METHODS
Study Design
The Institutional Review Board determined that this retrospective cohort study was exempt from review.
Data Source
The Children’s Hospital Association’s Pediatric Health Information System (PHIS) is an administrative database from pediatric hospitals located within 17 major metropolitan areas in the United States. Data include: service dates, patient demographics, payer type, diagnosis codes, resource utilization information (eg, medications), and hospital characteristics.1,5 De-identified data undergo reliability and validity checks prior to inclusion.1,5 We analyzed data from 40 of 43 hospitals that contributed inpatient data during our study period. 2 hospitals were excluded due to inconsistent data submission, and 1 removed their data.
Study Population
Data were extracted for children 0 to 17 years old who were admitted to an inpatient or observational unit between January 1, 2007 and December 31, 2014 for traumatic brain injury (TBI). Children were identified using International Classification of Diseases, Clinical Modification, Ninth Revision (ICD-9-CM) diagnosis codes that denote TBI per the Centers for Disease Control (CDC): 800.0–801.9, 803.0–804.9, 850–854.1, and 959.01.6–8 To examine inpatient care for concussion, we only retained children with a primary (ie, first) concussion-related diagnosis code (850.0–850.99) for analyses. For patients with multiple visits during our study period, only the index admission was analyzed. We refined our cohort using 2 injury scores calculated from ICD-9-CM diagnosis codes using validated ICDMAP-90 injury coding software.6,10–12 The Abbreviated Injury Scale (AIS) ranges from 1 (minor injury) to 6 (not survivable). The total Injury Severity Score (ISS) is based on 6 body regions (head/neck, face, chest, abdomen, extremity, and external) and calculated by summing the squares of the 3 worst AIS scores.13 A concussion receives a head AIS score of 2 if there is an associated loss of consciousness or a score of 1 if there is not; therefore, children were excluded if the head AIS score was >2. We also excluded children with the following features, as they may be indicative of more severe injuries that were likely the cause of admission: ISS > 6, secondary diagnosis code of skull fracture or intracranial injury, intensive care unit (ICU) or operating room (OR) charges, or a LOS > 7 days. Because some children are hospitalized for potentially abusive minor head trauma pending a safe discharge plan, we excluded children 0 to 4 years of age with child abuse, which was determined using a specific set of diagnosis codes (E960-E96820, 995.54, and 995.55) similar to previous research.14
Data Elements and Outcomes
Outcomes
Based on previous reports,1,15 a LOS ≥ 2 days distinguished a typical hospitalization from a prolonged one. ED revisit was identified when a child had a visit with a TBI-related primary diagnosis code at a PHIS hospital within 30 days of initial admission and was discharged home. We limited analyses to children discharged, as children readmitted may have had an initially missed intracranial injury.
Patient Characteristics
We examined the following patient variables: age, race, sex, presence of chronic medical condition, payer type, household income, area of residence (eg, rural versus urban), and mechanism of injury. Age was categorized to represent early childhood (0 to 4 years), school age (5 to 12 years), and adolescence (12 to 17 years). Race was grouped as white, black, or other (Asian, Pacific Islander, American Indian, and “other” per PHIS). Ethnicity was described as Hispanic/Latino or not Hispanic/Latino. Children with medical conditions lasting at least 12 months and comorbidities that may impact TBI recovery were identified using a subgrouping of ICD-9-CM codes for children with “complex chronic conditions”.16 Payer type was categorized as government, private, and self-pay. We extracted a PHIS variable representing the 2010 median household income for the child’s home zip code and categorized it into quartiles based on the Federal Poverty Level for a family of 4.17,18 Area of residence was defined using a Rural–Urban Commuting Area (RUCA) classification system19 and grouped into large urban core, suburban area, large rural town, or small rural town/isolated rural area.17 Mechanism of injury was determined using E-codes and categorized using the CDC injury framework,20 with sports-related injuries identified using a previously described set of E-codes.1 Mechanisms of injury included fall, motor vehicle collision, other motorized transport (eg, all-terrain vehicles), sports-related, struck by or against (ie, objects), and all others (eg, cyclists).
Hospital Characteristics
Hospitals were characterized by region (Northeast, Central, South, and West) and size (small <200, medium 200–400, and large >400 beds). The trauma-level accreditation was identified with Level 1 reflecting the highest possible trauma resources.
Medical Care Variables
Care variables included medications, neuroimaging, and cost of stay. Medication classes included oral non-narcotic analgesics [acetaminophen, ibuprofen, and others (aspirin, tramadol, and naproxen)], oral narcotics (codeine, oxycodone, and narcotic–non-narcotic combinations), intravenous (IV) non-narcotics (ketorolac), IV narcotics (morphine, fentanyl, and hydromorphone), antiemetics [ondansetron, metoclopramide, and phenothiazines (prochlorperazine, chlorpromazine, and promethazine)], maintenance IV fluids (dextrose with electrolytes or 0.45% sodium chloride), and resuscitation IV fluids (0.9% sodium chloride or lactated Ringer’s solution). Receipt of neuroimaging was determined if head computed tomography (CT) had been conducted at the admitting hospital. Adjusted cost of stay was calculated using a hospital-specific cost-to-charge ratio with additional adjustments using the Center for Medicare & Medicaid’s Wage Index.
Statistical Analyses
Descriptive statistics were calculated for individual, injury, and hospital, and care data elements, LOS, and ED readmissions. The number of children admitted with TBI was used as the denominator to assess the proportion of pediatric TBI admissions that were due to concussions. To identify factors associated with prolonged LOS (ie, ≥2 days) and ED readmission, we employed a mixed models approach that accounted for clustering of observations within hospitals. Independent variables included age, sex, race, ethnicity, payer type, household income, RUCA code, chronic medical condition, and injury mechanism. Models were adjusted for hospital location, size, and trauma-level accreditation. The binary distribution was specified along with a logit link function. A 2-phase process determined factors associated with each outcome. First, bivariable models were developed, followed by multivariable models that included independent variables with P values < .25 in the bivariable analysis. Backward step-wise elimination was performed, deleting variables with the highest P value one at a time. After each deletion, the percentage change in odds ratios was examined; if variable removal resulted in >10% change, the variable was retained as a potential confounder. This process was repeated until all remaining variables were significant (P < .05) with the exception of potential confounders. Finally, we examined the proportion of children receiving selected care practices annually. Descriptive and trend analyses were used to analyze adjusted median cost of stay. Analyses were performed using SAS software (Version 9.3, SAS Institute Inc., Cary, North Carolina).
RESULTS
Over 8 years, 88,526 children were admitted to 40 PHIS hospitals with a TBI-related diagnosis, among whom 13,708 had a primary diagnosis of concussion. We excluded 2,973 children with 1 or more of the following characteristics: a secondary diagnosis of intracranial injury (n = 58), head AIS score > 2 (n = 218), LOS > 7 days (n = 50), OR charges (n = 132), ICU charges (n = 1947), and ISS > 6 (n = 568). Six additional children aging 0 to 4 years were excluded due to child abuse. The remaining 10,729 children, averaging 1300 hospitalizations annually, were identified as being hospitalized primarily for concussion.
Table 1 summarizes the individual characteristics for this cohort. The average (standard deviation) age was 9.5 (5.1) years. Ethnicity was missing for 25.3% and therefore excluded from the multivariable models. Almost all children had a head AIS score of 2 (99.2%), and the majority had a total ISS ≤ 4 (73.4%). The majority of admissions were admitted to Level 1 trauma-accredited hospitals (78.7%) and medium-sized hospitals (63.9%).
The most commonly delivered medication classes were non-narcotic oral analgesics (53.7%), dextrose-containing IV fluids (45.0%), and antiemetic medications (34.1%). IV and oral narcotic use occurred in 19.7% and 10.2% of the children, respectively. Among our cohort, 16.7% received none of these medication classes. Of the 8,940 receiving medication, 32.6% received a single medication class, 29.5% received 2 classes, 20.5% 3 classes, 11.9% 4 classes, and 5.5% received 5 or more medication classes. Approximately 15% (n = 1597) received only oral medications, among whom 91.2% (n = 1457) received only non-narcotic analgesics and 3.9% (n = 63) received only oral narcotic analgesics. The majority (69.5%) received a head CT.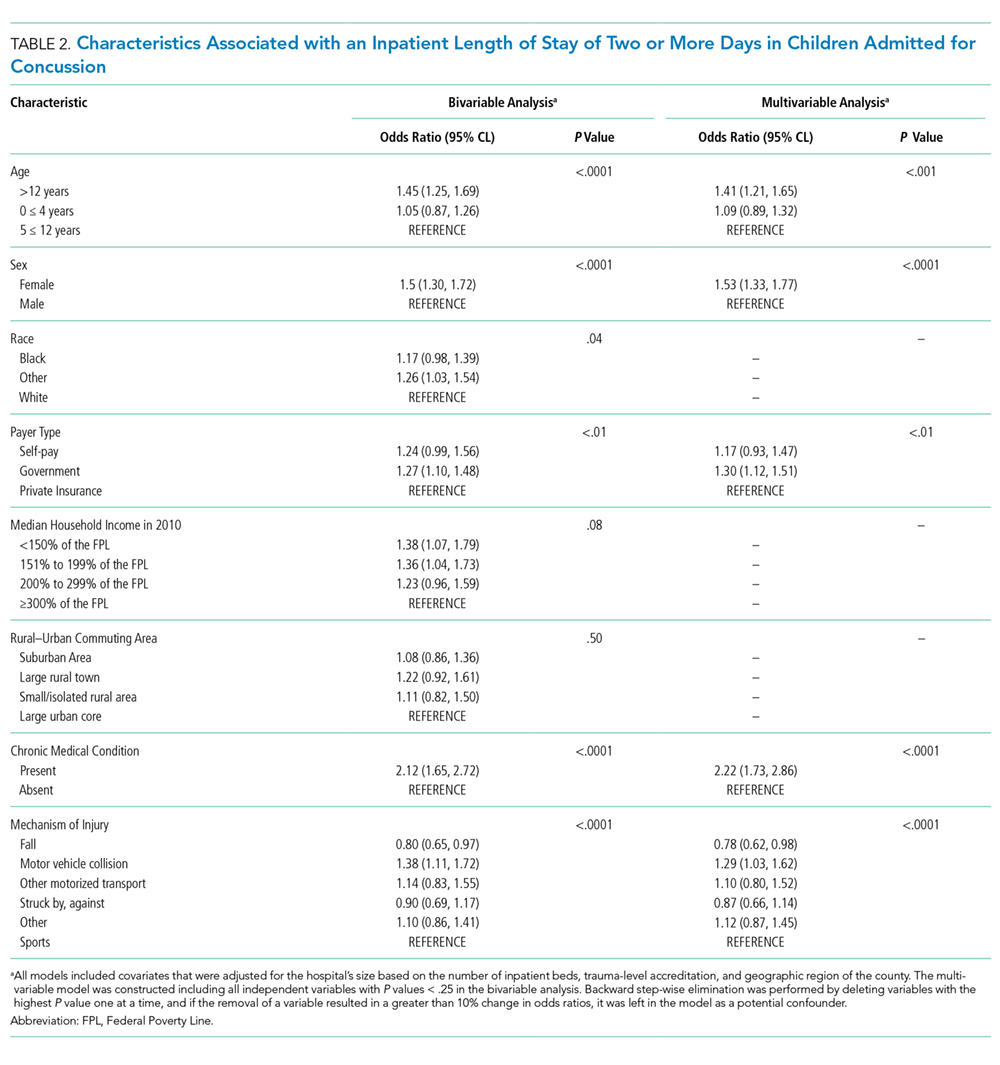
Table 4 summarizes medication administration trends over time. Oral non-narcotic administration increased significantly (slope = 0.99, P < .01) with the most pronounced change occurring in ibuprofen use (slope = 1.11, P < .001). Use of the IV non-narcotic ketorolac (slope = 0.61, P < .001) also increased significantly, as did the proportion of children receiving antiemetics (slope = 1.59, P = .001), with a substantial increase in ondansetron use (slope = 1.56, P = .001). The proportion of children receiving head CTs decreased linearly over time (slope= −1.75, P < .001), from 76.1% in 2007 to 63.7% in 2014. Median cost, adjusted for inflation, increased during our study period (P < .001) by approximately $353 each year, reaching $11,249 by 2014.
DISCUSSION
From 2007 to 2014, approximately 15% of children admitted to PHIS hospitals for TBI were admitted primarily for concussion. Since almost all children had a head AIS score of 2 and an ISS ≤ 4, our data suggest that most children had an associated loss of consciousness and that concussion was the only injury sustained, respectively. This study identified important subgroups that necessitated inpatient care but are rarely the focus of concussion research (eg, toddlers and those injured due to a motor vehicle collision). Most children (83.3%) received medications to treat common postconcussive symptoms (eg, pain and nausea), with almost half receiving 3 or more medication classes. Factors associated with the development of postconcussive syndrome (eg, female sex and adolescent age)4,21 were significantly associated with hospitalization of 2 or more days and ED revisit within 30 days of admission. In the absence of evidenced-based guidelines for inpatient concussion management, we identified significant trends in care, including increased use of specific pain [ie, oral and IV nonsteroidal anti-inflammatory drugs (NSAIDs)] and antiemetic (ie, ondansetron) medications and decreased use of head CT. Given the number of children admitted and receiving treatment for concussion symptomatology, influences on the decision to deliver specific care practices, as well as the impact and benefit of hospitalization, require closer examination.
Our study extends previous reports from the PHIS database by characterizing children admitted for concussion.1 We found that children admitted for concussion had similar characteristics to the broader population of children who sustain concussion (eg, school-aged children, male, and injured due to a fall or during sports).1,3,22 However, approximately 20% of the cohort were less than 5 years old, and less is known regarding appropriate treatment and outcomes of concussion in this age group.23 Uncertainty regarding optimal management and a young child’s inability to articulate symptoms may contribute to a physician’s decision to admit for close observation. Similar to Blinman et al., we found that a substantial proportion of children admitted with concussion were injured due to a motor vehicle collision,3 suggesting that although sports-related injuries are responsible for a significant proportion of pediatric concussions, children injured by other preventable mechanisms may also be incurring significant concussive injuries. Finally, the majority of our cohort was from an urban core, relative to a rural area, which is likely a reflection of the regionalization of trauma care, as well as variations in access to health care.
Although most children recover fully from concussion without specific interventions, 20%-30% may remain symptomatic at 1 month,3,4,21,24 and children who are hospitalized with concussion may be at higher risk for protracted symptoms. While specific individual or injury-related factors (eg, female sex, adolescent age, and injury due to motor vehicle collision) may contribute to more significant postconcussive symptoms, it is unclear how inpatient management affects recovery trajectory. Frequent sleep disruptions associated with inpatient care25 contradict current acute concussion management recommendations for physical and cognitive rest26 and could potentially impair symptom recovery. Additionally, we found widespread use of NSAIDs, although there is evidence suggesting that NSAIDs may potentially worsen concussive symptoms.26 We identified an increase in medication usage over time despite limited evidence of their effectiveness for pediatric concussion.27–29 This change may reflect improved symptom screening4,30 and/or increased awareness of specific medication safety profiles in pediatric trauma patients, especially for NSAIDs and ondansetron. Although we saw an increase in NSAID use, we did not see a proportional decrease in narcotic use. Similarly, while two-thirds of our cohort received IV medications, there is controversy about the need for IV fluids and medications for other pediatric illnesses, with research demonstrating that IV treatment may not reduce recovery time and may contribute to prolonged hospitalization and phlebitis.31,32 Thus, there is a need to understand the therapeutic effectiveness and benefits of medications and fluids on postconcussion recovery.
Neuroimaging rates for children receiving ED evaluation for concussion have been reported to be up to 60%-70%,1,22 although a more recent study spanning 2006 to 2011 found a 35-%–40% head CT rate in pediatric patients by hospital-based EDs in the United States.33 Our results appear to support decreasing head CT use over time in pediatric hospitals. Hospitalization for observation is costly1 but could decrease a child’s risk of malignancy from radiation exposure. Further work on balancing cost, risk, and shared decision-making with parents could guide decisions regarding emergent neuroimaging versus admission.
This study has limitations inherent to the use of an administrative dataset, including lack of information regarding why the child was admitted. Since the focus was to describe inpatient care of children with concussion, those discharged home from the ED were not included in this dataset. Consequently, we could not contrast the ED care of those discharged home with those who were admitted or assess trends in admission rates for concussion. Although the overall number of concussion admissions has continued to remain stable over time,1 due to a lack of prospectively collected clinical information, we are unable to determine whether observed trends in care are secondary to changes in practice or changes in concussion severity. However, there has been no research to date supporting the latter. Ethnicity was excluded due to high levels of missing data. Cost of stay was not extensively analyzed given hospital variation in designation of observational or inpatient status, which subsequently affects billing.34 Rates of neuroimaging and ED revisit may have been underestimated since children could have received care at a non-PHIS hospital. Similarly, the decrease in the proportion of children receiving neuroimaging over time may have been associated with an increase in children being transferred from a non-PHIS hospital for admission, although with increased regionalization in trauma care, we would not expect transfers of children with only concussion to have significantly increased. Finally, data were limited to the pediatric tertiary care centers participating in PHIS, thereby reducing generalizability and introducing selection bias by only including children who were able to access care at PHIS hospitals. Although the care practices we evaluated (eg, NSAIDs and head CT) are available at all hospitals, our analyses only reflect care delivered within the PHIS.
Concussion accounted for 15% of all pediatric TBI admissions during our study period. Further investigation of potential factors associated with admission and protracted recovery (eg, adolescent females needing treatment for severe symptomatology) could facilitate better understanding of how hospitalization affects recovery. Additionally, research on acute pharmacotherapies (eg, IV therapies and/or inpatient treatment until symptoms resolve) is needed to fully elucidate the acute and long-term benefits of interventions delivered to children.
ACKNOWLEDGMENTS
Colleen Mangeot: Biostatistician with extensive PHIS knowledge who contributed to database creation and statistical analysis. Yanhong (Amy) Liu: Research database programmer who developed the database, ran quality assurance measures, and cleaned all study data.
Disclosures
The authors have nothing to disclose.
Funding
This study was supported by grant R40 MC 268060102 from the Maternal and Child Health Research Program, Maternal and Child Health Bureau (Title V, Social Security Act), Health Resources and Services Administration, Department of Health and Human Services. The funding source was not involved in development of the study design; in the collection, analysis and interpretation of data; or in the writing of this report.
1. Colvin JD, Thurm C, Pate BM, Newland JG, Hall M, Meehan WP. Diagnosis and acute management of patients with concussion at children’s hospitals. Arch Dis Child. 2013;98(12):934-938. PubMed
2. Bourgeois FT, Monuteaux MC, Stack AM, Neuman MI. Variation in emergency department admission rates in US children’s hospitals. Pediatrics. 2014;134(3):539-545. PubMed
3. Blinman TA, Houseknecht E, Snyder C, Wiebe DJ, Nance ML. Postconcussive symptoms in hospitalized pediatric patients after mild traumatic brain injury. J Pediatr Surg. 2009;44(6):1223-1228. PubMed
4. Babcock L, Byczkowski T, Wade SL, Ho M, Mookerjee S, Bazarian JJ. Predicting postconcussion syndrome after mild traumatic brain injury in children and adolescents who present to the emergency department. JAMA pediatrics. 2013;167(2):156-161. PubMed
5. Conway PH, Keren R. Factors associated with variability in outcomes for children hospitalized with urinary tract infection. The Journal of pediatrics. 2009;154(6):789-796. PubMed
6. Services UDoHaH. International classification of diseases, 9th Revision, Clinical modification (ICD-9CM). Washington, DC: US Department of Health and Human Services. Public Health Service, Health Care Financing Administration 1989.
7. Marr AL, Coronado VG. Annual data submission standards. Central nervous system injury surveillance. In: US Department of Health and Human Services PHS, CDC, ed. Atlanta, GA 2001.
8. Organization WH. International classification of diseases: manual on the international statistical classification of diseases, injuries, and cause of death. In: Organization WH, ed. 9th rev. ed. Geneva, Switerland 1977.
9. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Report to Congress on mild traumatic brain injury in the United States: steps to prevent a serious public health problem. Atlanta, GA: Centers for Disease Control and Prevention; 2003.
10. Mackenzie E, Sacco WJ. ICDMAP-90 software: user’s guide. Baltimore, Maryland: Johns Hopkins University and Tri-Analytics. 1997:1-25.
11. MacKenzie EJ, Steinwachs DM, Shankar B. Classifying trauma severity based on hospital discharge diagnoses. Validation of an ICD-9CM to AIS-85 conversion table. Med Care. 1989;27(4):412-422. PubMed
12. Fleischman RJ, Mann NC, Dai M, et al. Validating the use of ICD-9 code mapping to generate injury severity scores. J Trauma Nurs. 2017;24(1):4-14. PubMed
13. Baker SP, O’Neill B, Haddon W, Jr., Long WB. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. The Journal of trauma. 1974;14(3):187-196. PubMed
14. Wood JN, Feudtner C, Medina SP, Luan X, Localio R, Rubin DM. Variation in occult injury screening for children with suspected abuse in selected US children’s hospitals. Pediatrics
15. Yang J, Phillips G, Xiang H, Allareddy V, Heiden E, Peek-Asa C. Hospitalisations for sport-related concussions in US children aged 5 to 18 years during 2000-2004. Br J Sports Med. 2008;42(8):664-669. PubMed
16. Feudtner C, Christakis DA, Connell FA. Pediatric deaths attributable to complex chronic conditions: a population-based study of Washington State, 1980-1997. Pediatrics. 2000;106(1):205-209. PubMed
17. Peltz A, Wu CL, White ML, et al. Characteristics of rural children admitted to pediatric hospitals. Pediatrics. 2016;137(5): e20153156. PubMed
18. Services UDoHaH. Annual update of the HHS Poverty Guidelines. Federal Register; 2016-03-14 2011.
19. Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95(7):1149-1155. PubMed
20. Proposed Matrix of E-code Groupings| WISQARS | Injury Center | CDC. 2016; http://www.cdc.gov/injury/wisqars/ecode_matrix.html.
21. Zemek RL, Farion KJ, Sampson M, McGahern C. Prognosticators of persistent symptoms following pediatric concussion: A systematic review. JAMA Pediatr. 2013;167(3):259-265. PubMed
22. Meehan WP, Mannix R. Pediatric concussions in United States emergency departments in the years 2002 to 2006. J Pediatr. 2010;157(6):889-893. PubMed
23. Davis GA, Purcell LK. The evaluation and management of acute concussion differs in young children. Br J Sports Med. 2014;48(2):98-101. PubMed
24. Zemek R, Barrowman N, Freedman SB, et al. Clinical risk score for persistent postconcussion symptoms among children with acute concussion in the ED. JAMA. 2016;315(10):1014-1025. PubMed
25. Hinds PS, Hockenberry M, Rai SN, et al. Nocturnal awakenings, sleep environment interruptions, and fatigue in hospitalized children with cancer. Oncol Nurs Forum. 2007;34(2):393-402. PubMed
26. Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. 2012;6:58. PubMed
27. Meehan WP. Medical therapies for concussion. Clin Sports Med. 2011;30(1):115-124, ix. PubMed
28. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533. PubMed
29. Giza CC, Kutcher JS, Ashwal S, et al. Summary of evidence-based guideline update: evaluation and management of concussion in sports: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;80(24):2250-2257. PubMed
30. Barlow KM, Crawford S, Stevenson A, Sandhu SS, Belanger F, Dewey D. Epidemiology of postconcussion syndrome in pediatric mild traumatic brain injury. Pediatrics. 2010;126(2):e374-e381. PubMed
31. Keren R, Shah SS, Srivastava R, et al. Comparative effectiveness of intravenous vs oral antibiotics for postdischarge treatment of acute osteomyelitis in children. JAMA Pediatr. 2015;169(2):120-128. PubMed
32. Hartling L, Bellemare S, Wiebe N, Russell K, Klassen TP, Craig W. Oral versus intravenous rehydration for treating dehydration due to gastroenteritis in children. Cochrane Database Syst Rev. 2006(3):CD004390. PubMed
34. Fieldston ES, Shah SS, Hall M, et al. Resource utilization for observation-status stays at children’s hospitals. Pediatrics. 2013;131(6):1050-1058. PubMed
33. Zonfrillo MR, Kim KH, Arbogast KB. Emergency Department Visits and Head Computed Tomography Utilization for Concussion Patients From 2006 to 2011. Acad Emerg Med. 2015;22(7):872-877. PubMed
Approximately 14% of children who sustain a concussion are admitted to the hospital,1 although admission rates reportedly vary substantially among pediatric hospitals.2 Children hospitalized for concussion may be at a higher risk for persistent postconcussive symptoms,3,4 yet little is known about this subset of children and how they are managed while in the hospital. Characterizing children hospitalized for concussion and describing the inpatient care they received will promote hypothesis generation for further inquiry into indications for admission, as well as the relationship between inpatient management and concussion recovery.
We described a cohort of children admitted to 40 pediatric hospitals primarily for concussion and detailed care delivered during hospitalization. We explored individual-level factors and their association with prolonged length of stay (LOS) and emergency department (ED) readmission. Finally, we evaluated if there had been changes in inpatient care over the 8-year study period.
PATIENTS AND METHODS
Study Design
The Institutional Review Board determined that this retrospective cohort study was exempt from review.
Data Source
The Children’s Hospital Association’s Pediatric Health Information System (PHIS) is an administrative database from pediatric hospitals located within 17 major metropolitan areas in the United States. Data include: service dates, patient demographics, payer type, diagnosis codes, resource utilization information (eg, medications), and hospital characteristics.1,5 De-identified data undergo reliability and validity checks prior to inclusion.1,5 We analyzed data from 40 of 43 hospitals that contributed inpatient data during our study period. 2 hospitals were excluded due to inconsistent data submission, and 1 removed their data.
Study Population
Data were extracted for children 0 to 17 years old who were admitted to an inpatient or observational unit between January 1, 2007 and December 31, 2014 for traumatic brain injury (TBI). Children were identified using International Classification of Diseases, Clinical Modification, Ninth Revision (ICD-9-CM) diagnosis codes that denote TBI per the Centers for Disease Control (CDC): 800.0–801.9, 803.0–804.9, 850–854.1, and 959.01.6–8 To examine inpatient care for concussion, we only retained children with a primary (ie, first) concussion-related diagnosis code (850.0–850.99) for analyses. For patients with multiple visits during our study period, only the index admission was analyzed. We refined our cohort using 2 injury scores calculated from ICD-9-CM diagnosis codes using validated ICDMAP-90 injury coding software.6,10–12 The Abbreviated Injury Scale (AIS) ranges from 1 (minor injury) to 6 (not survivable). The total Injury Severity Score (ISS) is based on 6 body regions (head/neck, face, chest, abdomen, extremity, and external) and calculated by summing the squares of the 3 worst AIS scores.13 A concussion receives a head AIS score of 2 if there is an associated loss of consciousness or a score of 1 if there is not; therefore, children were excluded if the head AIS score was >2. We also excluded children with the following features, as they may be indicative of more severe injuries that were likely the cause of admission: ISS > 6, secondary diagnosis code of skull fracture or intracranial injury, intensive care unit (ICU) or operating room (OR) charges, or a LOS > 7 days. Because some children are hospitalized for potentially abusive minor head trauma pending a safe discharge plan, we excluded children 0 to 4 years of age with child abuse, which was determined using a specific set of diagnosis codes (E960-E96820, 995.54, and 995.55) similar to previous research.14
Data Elements and Outcomes
Outcomes
Based on previous reports,1,15 a LOS ≥ 2 days distinguished a typical hospitalization from a prolonged one. ED revisit was identified when a child had a visit with a TBI-related primary diagnosis code at a PHIS hospital within 30 days of initial admission and was discharged home. We limited analyses to children discharged, as children readmitted may have had an initially missed intracranial injury.
Patient Characteristics
We examined the following patient variables: age, race, sex, presence of chronic medical condition, payer type, household income, area of residence (eg, rural versus urban), and mechanism of injury. Age was categorized to represent early childhood (0 to 4 years), school age (5 to 12 years), and adolescence (12 to 17 years). Race was grouped as white, black, or other (Asian, Pacific Islander, American Indian, and “other” per PHIS). Ethnicity was described as Hispanic/Latino or not Hispanic/Latino. Children with medical conditions lasting at least 12 months and comorbidities that may impact TBI recovery were identified using a subgrouping of ICD-9-CM codes for children with “complex chronic conditions”.16 Payer type was categorized as government, private, and self-pay. We extracted a PHIS variable representing the 2010 median household income for the child’s home zip code and categorized it into quartiles based on the Federal Poverty Level for a family of 4.17,18 Area of residence was defined using a Rural–Urban Commuting Area (RUCA) classification system19 and grouped into large urban core, suburban area, large rural town, or small rural town/isolated rural area.17 Mechanism of injury was determined using E-codes and categorized using the CDC injury framework,20 with sports-related injuries identified using a previously described set of E-codes.1 Mechanisms of injury included fall, motor vehicle collision, other motorized transport (eg, all-terrain vehicles), sports-related, struck by or against (ie, objects), and all others (eg, cyclists).
Hospital Characteristics
Hospitals were characterized by region (Northeast, Central, South, and West) and size (small <200, medium 200–400, and large >400 beds). The trauma-level accreditation was identified with Level 1 reflecting the highest possible trauma resources.
Medical Care Variables
Care variables included medications, neuroimaging, and cost of stay. Medication classes included oral non-narcotic analgesics [acetaminophen, ibuprofen, and others (aspirin, tramadol, and naproxen)], oral narcotics (codeine, oxycodone, and narcotic–non-narcotic combinations), intravenous (IV) non-narcotics (ketorolac), IV narcotics (morphine, fentanyl, and hydromorphone), antiemetics [ondansetron, metoclopramide, and phenothiazines (prochlorperazine, chlorpromazine, and promethazine)], maintenance IV fluids (dextrose with electrolytes or 0.45% sodium chloride), and resuscitation IV fluids (0.9% sodium chloride or lactated Ringer’s solution). Receipt of neuroimaging was determined if head computed tomography (CT) had been conducted at the admitting hospital. Adjusted cost of stay was calculated using a hospital-specific cost-to-charge ratio with additional adjustments using the Center for Medicare & Medicaid’s Wage Index.
Statistical Analyses
Descriptive statistics were calculated for individual, injury, and hospital, and care data elements, LOS, and ED readmissions. The number of children admitted with TBI was used as the denominator to assess the proportion of pediatric TBI admissions that were due to concussions. To identify factors associated with prolonged LOS (ie, ≥2 days) and ED readmission, we employed a mixed models approach that accounted for clustering of observations within hospitals. Independent variables included age, sex, race, ethnicity, payer type, household income, RUCA code, chronic medical condition, and injury mechanism. Models were adjusted for hospital location, size, and trauma-level accreditation. The binary distribution was specified along with a logit link function. A 2-phase process determined factors associated with each outcome. First, bivariable models were developed, followed by multivariable models that included independent variables with P values < .25 in the bivariable analysis. Backward step-wise elimination was performed, deleting variables with the highest P value one at a time. After each deletion, the percentage change in odds ratios was examined; if variable removal resulted in >10% change, the variable was retained as a potential confounder. This process was repeated until all remaining variables were significant (P < .05) with the exception of potential confounders. Finally, we examined the proportion of children receiving selected care practices annually. Descriptive and trend analyses were used to analyze adjusted median cost of stay. Analyses were performed using SAS software (Version 9.3, SAS Institute Inc., Cary, North Carolina).
RESULTS
Over 8 years, 88,526 children were admitted to 40 PHIS hospitals with a TBI-related diagnosis, among whom 13,708 had a primary diagnosis of concussion. We excluded 2,973 children with 1 or more of the following characteristics: a secondary diagnosis of intracranial injury (n = 58), head AIS score > 2 (n = 218), LOS > 7 days (n = 50), OR charges (n = 132), ICU charges (n = 1947), and ISS > 6 (n = 568). Six additional children aging 0 to 4 years were excluded due to child abuse. The remaining 10,729 children, averaging 1300 hospitalizations annually, were identified as being hospitalized primarily for concussion.
Table 1 summarizes the individual characteristics for this cohort. The average (standard deviation) age was 9.5 (5.1) years. Ethnicity was missing for 25.3% and therefore excluded from the multivariable models. Almost all children had a head AIS score of 2 (99.2%), and the majority had a total ISS ≤ 4 (73.4%). The majority of admissions were admitted to Level 1 trauma-accredited hospitals (78.7%) and medium-sized hospitals (63.9%).
The most commonly delivered medication classes were non-narcotic oral analgesics (53.7%), dextrose-containing IV fluids (45.0%), and antiemetic medications (34.1%). IV and oral narcotic use occurred in 19.7% and 10.2% of the children, respectively. Among our cohort, 16.7% received none of these medication classes. Of the 8,940 receiving medication, 32.6% received a single medication class, 29.5% received 2 classes, 20.5% 3 classes, 11.9% 4 classes, and 5.5% received 5 or more medication classes. Approximately 15% (n = 1597) received only oral medications, among whom 91.2% (n = 1457) received only non-narcotic analgesics and 3.9% (n = 63) received only oral narcotic analgesics. The majority (69.5%) received a head CT.
Table 4 summarizes medication administration trends over time. Oral non-narcotic administration increased significantly (slope = 0.99, P < .01) with the most pronounced change occurring in ibuprofen use (slope = 1.11, P < .001). Use of the IV non-narcotic ketorolac (slope = 0.61, P < .001) also increased significantly, as did the proportion of children receiving antiemetics (slope = 1.59, P = .001), with a substantial increase in ondansetron use (slope = 1.56, P = .001). The proportion of children receiving head CTs decreased linearly over time (slope= −1.75, P < .001), from 76.1% in 2007 to 63.7% in 2014. Median cost, adjusted for inflation, increased during our study period (P < .001) by approximately $353 each year, reaching $11,249 by 2014.
DISCUSSION
From 2007 to 2014, approximately 15% of children admitted to PHIS hospitals for TBI were admitted primarily for concussion. Since almost all children had a head AIS score of 2 and an ISS ≤ 4, our data suggest that most children had an associated loss of consciousness and that concussion was the only injury sustained, respectively. This study identified important subgroups that necessitated inpatient care but are rarely the focus of concussion research (eg, toddlers and those injured due to a motor vehicle collision). Most children (83.3%) received medications to treat common postconcussive symptoms (eg, pain and nausea), with almost half receiving 3 or more medication classes. Factors associated with the development of postconcussive syndrome (eg, female sex and adolescent age)4,21 were significantly associated with hospitalization of 2 or more days and ED revisit within 30 days of admission. In the absence of evidenced-based guidelines for inpatient concussion management, we identified significant trends in care, including increased use of specific pain [ie, oral and IV nonsteroidal anti-inflammatory drugs (NSAIDs)] and antiemetic (ie, ondansetron) medications and decreased use of head CT. Given the number of children admitted and receiving treatment for concussion symptomatology, influences on the decision to deliver specific care practices, as well as the impact and benefit of hospitalization, require closer examination.
Our study extends previous reports from the PHIS database by characterizing children admitted for concussion.1 We found that children admitted for concussion had similar characteristics to the broader population of children who sustain concussion (eg, school-aged children, male, and injured due to a fall or during sports).1,3,22 However, approximately 20% of the cohort were less than 5 years old, and less is known regarding appropriate treatment and outcomes of concussion in this age group.23 Uncertainty regarding optimal management and a young child’s inability to articulate symptoms may contribute to a physician’s decision to admit for close observation. Similar to Blinman et al., we found that a substantial proportion of children admitted with concussion were injured due to a motor vehicle collision,3 suggesting that although sports-related injuries are responsible for a significant proportion of pediatric concussions, children injured by other preventable mechanisms may also be incurring significant concussive injuries. Finally, the majority of our cohort was from an urban core, relative to a rural area, which is likely a reflection of the regionalization of trauma care, as well as variations in access to health care.
Although most children recover fully from concussion without specific interventions, 20%-30% may remain symptomatic at 1 month,3,4,21,24 and children who are hospitalized with concussion may be at higher risk for protracted symptoms. While specific individual or injury-related factors (eg, female sex, adolescent age, and injury due to motor vehicle collision) may contribute to more significant postconcussive symptoms, it is unclear how inpatient management affects recovery trajectory. Frequent sleep disruptions associated with inpatient care25 contradict current acute concussion management recommendations for physical and cognitive rest26 and could potentially impair symptom recovery. Additionally, we found widespread use of NSAIDs, although there is evidence suggesting that NSAIDs may potentially worsen concussive symptoms.26 We identified an increase in medication usage over time despite limited evidence of their effectiveness for pediatric concussion.27–29 This change may reflect improved symptom screening4,30 and/or increased awareness of specific medication safety profiles in pediatric trauma patients, especially for NSAIDs and ondansetron. Although we saw an increase in NSAID use, we did not see a proportional decrease in narcotic use. Similarly, while two-thirds of our cohort received IV medications, there is controversy about the need for IV fluids and medications for other pediatric illnesses, with research demonstrating that IV treatment may not reduce recovery time and may contribute to prolonged hospitalization and phlebitis.31,32 Thus, there is a need to understand the therapeutic effectiveness and benefits of medications and fluids on postconcussion recovery.
Neuroimaging rates for children receiving ED evaluation for concussion have been reported to be up to 60%-70%,1,22 although a more recent study spanning 2006 to 2011 found a 35-%–40% head CT rate in pediatric patients by hospital-based EDs in the United States.33 Our results appear to support decreasing head CT use over time in pediatric hospitals. Hospitalization for observation is costly1 but could decrease a child’s risk of malignancy from radiation exposure. Further work on balancing cost, risk, and shared decision-making with parents could guide decisions regarding emergent neuroimaging versus admission.
This study has limitations inherent to the use of an administrative dataset, including lack of information regarding why the child was admitted. Since the focus was to describe inpatient care of children with concussion, those discharged home from the ED were not included in this dataset. Consequently, we could not contrast the ED care of those discharged home with those who were admitted or assess trends in admission rates for concussion. Although the overall number of concussion admissions has continued to remain stable over time,1 due to a lack of prospectively collected clinical information, we are unable to determine whether observed trends in care are secondary to changes in practice or changes in concussion severity. However, there has been no research to date supporting the latter. Ethnicity was excluded due to high levels of missing data. Cost of stay was not extensively analyzed given hospital variation in designation of observational or inpatient status, which subsequently affects billing.34 Rates of neuroimaging and ED revisit may have been underestimated since children could have received care at a non-PHIS hospital. Similarly, the decrease in the proportion of children receiving neuroimaging over time may have been associated with an increase in children being transferred from a non-PHIS hospital for admission, although with increased regionalization in trauma care, we would not expect transfers of children with only concussion to have significantly increased. Finally, data were limited to the pediatric tertiary care centers participating in PHIS, thereby reducing generalizability and introducing selection bias by only including children who were able to access care at PHIS hospitals. Although the care practices we evaluated (eg, NSAIDs and head CT) are available at all hospitals, our analyses only reflect care delivered within the PHIS.
Concussion accounted for 15% of all pediatric TBI admissions during our study period. Further investigation of potential factors associated with admission and protracted recovery (eg, adolescent females needing treatment for severe symptomatology) could facilitate better understanding of how hospitalization affects recovery. Additionally, research on acute pharmacotherapies (eg, IV therapies and/or inpatient treatment until symptoms resolve) is needed to fully elucidate the acute and long-term benefits of interventions delivered to children.
ACKNOWLEDGMENTS
Colleen Mangeot: Biostatistician with extensive PHIS knowledge who contributed to database creation and statistical analysis. Yanhong (Amy) Liu: Research database programmer who developed the database, ran quality assurance measures, and cleaned all study data.
Disclosures
The authors have nothing to disclose.
Funding
This study was supported by grant R40 MC 268060102 from the Maternal and Child Health Research Program, Maternal and Child Health Bureau (Title V, Social Security Act), Health Resources and Services Administration, Department of Health and Human Services. The funding source was not involved in development of the study design; in the collection, analysis and interpretation of data; or in the writing of this report.
Approximately 14% of children who sustain a concussion are admitted to the hospital,1 although admission rates reportedly vary substantially among pediatric hospitals.2 Children hospitalized for concussion may be at a higher risk for persistent postconcussive symptoms,3,4 yet little is known about this subset of children and how they are managed while in the hospital. Characterizing children hospitalized for concussion and describing the inpatient care they received will promote hypothesis generation for further inquiry into indications for admission, as well as the relationship between inpatient management and concussion recovery.
We described a cohort of children admitted to 40 pediatric hospitals primarily for concussion and detailed care delivered during hospitalization. We explored individual-level factors and their association with prolonged length of stay (LOS) and emergency department (ED) readmission. Finally, we evaluated if there had been changes in inpatient care over the 8-year study period.
PATIENTS AND METHODS
Study Design
The Institutional Review Board determined that this retrospective cohort study was exempt from review.
Data Source
The Children’s Hospital Association’s Pediatric Health Information System (PHIS) is an administrative database from pediatric hospitals located within 17 major metropolitan areas in the United States. Data include: service dates, patient demographics, payer type, diagnosis codes, resource utilization information (eg, medications), and hospital characteristics.1,5 De-identified data undergo reliability and validity checks prior to inclusion.1,5 We analyzed data from 40 of 43 hospitals that contributed inpatient data during our study period. 2 hospitals were excluded due to inconsistent data submission, and 1 removed their data.
Study Population
Data were extracted for children 0 to 17 years old who were admitted to an inpatient or observational unit between January 1, 2007 and December 31, 2014 for traumatic brain injury (TBI). Children were identified using International Classification of Diseases, Clinical Modification, Ninth Revision (ICD-9-CM) diagnosis codes that denote TBI per the Centers for Disease Control (CDC): 800.0–801.9, 803.0–804.9, 850–854.1, and 959.01.6–8 To examine inpatient care for concussion, we only retained children with a primary (ie, first) concussion-related diagnosis code (850.0–850.99) for analyses. For patients with multiple visits during our study period, only the index admission was analyzed. We refined our cohort using 2 injury scores calculated from ICD-9-CM diagnosis codes using validated ICDMAP-90 injury coding software.6,10–12 The Abbreviated Injury Scale (AIS) ranges from 1 (minor injury) to 6 (not survivable). The total Injury Severity Score (ISS) is based on 6 body regions (head/neck, face, chest, abdomen, extremity, and external) and calculated by summing the squares of the 3 worst AIS scores.13 A concussion receives a head AIS score of 2 if there is an associated loss of consciousness or a score of 1 if there is not; therefore, children were excluded if the head AIS score was >2. We also excluded children with the following features, as they may be indicative of more severe injuries that were likely the cause of admission: ISS > 6, secondary diagnosis code of skull fracture or intracranial injury, intensive care unit (ICU) or operating room (OR) charges, or a LOS > 7 days. Because some children are hospitalized for potentially abusive minor head trauma pending a safe discharge plan, we excluded children 0 to 4 years of age with child abuse, which was determined using a specific set of diagnosis codes (E960-E96820, 995.54, and 995.55) similar to previous research.14
Data Elements and Outcomes
Outcomes
Based on previous reports,1,15 a LOS ≥ 2 days distinguished a typical hospitalization from a prolonged one. ED revisit was identified when a child had a visit with a TBI-related primary diagnosis code at a PHIS hospital within 30 days of initial admission and was discharged home. We limited analyses to children discharged, as children readmitted may have had an initially missed intracranial injury.
Patient Characteristics
We examined the following patient variables: age, race, sex, presence of chronic medical condition, payer type, household income, area of residence (eg, rural versus urban), and mechanism of injury. Age was categorized to represent early childhood (0 to 4 years), school age (5 to 12 years), and adolescence (12 to 17 years). Race was grouped as white, black, or other (Asian, Pacific Islander, American Indian, and “other” per PHIS). Ethnicity was described as Hispanic/Latino or not Hispanic/Latino. Children with medical conditions lasting at least 12 months and comorbidities that may impact TBI recovery were identified using a subgrouping of ICD-9-CM codes for children with “complex chronic conditions”.16 Payer type was categorized as government, private, and self-pay. We extracted a PHIS variable representing the 2010 median household income for the child’s home zip code and categorized it into quartiles based on the Federal Poverty Level for a family of 4.17,18 Area of residence was defined using a Rural–Urban Commuting Area (RUCA) classification system19 and grouped into large urban core, suburban area, large rural town, or small rural town/isolated rural area.17 Mechanism of injury was determined using E-codes and categorized using the CDC injury framework,20 with sports-related injuries identified using a previously described set of E-codes.1 Mechanisms of injury included fall, motor vehicle collision, other motorized transport (eg, all-terrain vehicles), sports-related, struck by or against (ie, objects), and all others (eg, cyclists).
Hospital Characteristics
Hospitals were characterized by region (Northeast, Central, South, and West) and size (small <200, medium 200–400, and large >400 beds). The trauma-level accreditation was identified with Level 1 reflecting the highest possible trauma resources.
Medical Care Variables
Care variables included medications, neuroimaging, and cost of stay. Medication classes included oral non-narcotic analgesics [acetaminophen, ibuprofen, and others (aspirin, tramadol, and naproxen)], oral narcotics (codeine, oxycodone, and narcotic–non-narcotic combinations), intravenous (IV) non-narcotics (ketorolac), IV narcotics (morphine, fentanyl, and hydromorphone), antiemetics [ondansetron, metoclopramide, and phenothiazines (prochlorperazine, chlorpromazine, and promethazine)], maintenance IV fluids (dextrose with electrolytes or 0.45% sodium chloride), and resuscitation IV fluids (0.9% sodium chloride or lactated Ringer’s solution). Receipt of neuroimaging was determined if head computed tomography (CT) had been conducted at the admitting hospital. Adjusted cost of stay was calculated using a hospital-specific cost-to-charge ratio with additional adjustments using the Center for Medicare & Medicaid’s Wage Index.
Statistical Analyses
Descriptive statistics were calculated for individual, injury, and hospital, and care data elements, LOS, and ED readmissions. The number of children admitted with TBI was used as the denominator to assess the proportion of pediatric TBI admissions that were due to concussions. To identify factors associated with prolonged LOS (ie, ≥2 days) and ED readmission, we employed a mixed models approach that accounted for clustering of observations within hospitals. Independent variables included age, sex, race, ethnicity, payer type, household income, RUCA code, chronic medical condition, and injury mechanism. Models were adjusted for hospital location, size, and trauma-level accreditation. The binary distribution was specified along with a logit link function. A 2-phase process determined factors associated with each outcome. First, bivariable models were developed, followed by multivariable models that included independent variables with P values < .25 in the bivariable analysis. Backward step-wise elimination was performed, deleting variables with the highest P value one at a time. After each deletion, the percentage change in odds ratios was examined; if variable removal resulted in >10% change, the variable was retained as a potential confounder. This process was repeated until all remaining variables were significant (P < .05) with the exception of potential confounders. Finally, we examined the proportion of children receiving selected care practices annually. Descriptive and trend analyses were used to analyze adjusted median cost of stay. Analyses were performed using SAS software (Version 9.3, SAS Institute Inc., Cary, North Carolina).
RESULTS
Over 8 years, 88,526 children were admitted to 40 PHIS hospitals with a TBI-related diagnosis, among whom 13,708 had a primary diagnosis of concussion. We excluded 2,973 children with 1 or more of the following characteristics: a secondary diagnosis of intracranial injury (n = 58), head AIS score > 2 (n = 218), LOS > 7 days (n = 50), OR charges (n = 132), ICU charges (n = 1947), and ISS > 6 (n = 568). Six additional children aging 0 to 4 years were excluded due to child abuse. The remaining 10,729 children, averaging 1300 hospitalizations annually, were identified as being hospitalized primarily for concussion.
Table 1 summarizes the individual characteristics for this cohort. The average (standard deviation) age was 9.5 (5.1) years. Ethnicity was missing for 25.3% and therefore excluded from the multivariable models. Almost all children had a head AIS score of 2 (99.2%), and the majority had a total ISS ≤ 4 (73.4%). The majority of admissions were admitted to Level 1 trauma-accredited hospitals (78.7%) and medium-sized hospitals (63.9%).
The most commonly delivered medication classes were non-narcotic oral analgesics (53.7%), dextrose-containing IV fluids (45.0%), and antiemetic medications (34.1%). IV and oral narcotic use occurred in 19.7% and 10.2% of the children, respectively. Among our cohort, 16.7% received none of these medication classes. Of the 8,940 receiving medication, 32.6% received a single medication class, 29.5% received 2 classes, 20.5% 3 classes, 11.9% 4 classes, and 5.5% received 5 or more medication classes. Approximately 15% (n = 1597) received only oral medications, among whom 91.2% (n = 1457) received only non-narcotic analgesics and 3.9% (n = 63) received only oral narcotic analgesics. The majority (69.5%) received a head CT.
Table 4 summarizes medication administration trends over time. Oral non-narcotic administration increased significantly (slope = 0.99, P < .01) with the most pronounced change occurring in ibuprofen use (slope = 1.11, P < .001). Use of the IV non-narcotic ketorolac (slope = 0.61, P < .001) also increased significantly, as did the proportion of children receiving antiemetics (slope = 1.59, P = .001), with a substantial increase in ondansetron use (slope = 1.56, P = .001). The proportion of children receiving head CTs decreased linearly over time (slope= −1.75, P < .001), from 76.1% in 2007 to 63.7% in 2014. Median cost, adjusted for inflation, increased during our study period (P < .001) by approximately $353 each year, reaching $11,249 by 2014.
DISCUSSION
From 2007 to 2014, approximately 15% of children admitted to PHIS hospitals for TBI were admitted primarily for concussion. Since almost all children had a head AIS score of 2 and an ISS ≤ 4, our data suggest that most children had an associated loss of consciousness and that concussion was the only injury sustained, respectively. This study identified important subgroups that necessitated inpatient care but are rarely the focus of concussion research (eg, toddlers and those injured due to a motor vehicle collision). Most children (83.3%) received medications to treat common postconcussive symptoms (eg, pain and nausea), with almost half receiving 3 or more medication classes. Factors associated with the development of postconcussive syndrome (eg, female sex and adolescent age)4,21 were significantly associated with hospitalization of 2 or more days and ED revisit within 30 days of admission. In the absence of evidenced-based guidelines for inpatient concussion management, we identified significant trends in care, including increased use of specific pain [ie, oral and IV nonsteroidal anti-inflammatory drugs (NSAIDs)] and antiemetic (ie, ondansetron) medications and decreased use of head CT. Given the number of children admitted and receiving treatment for concussion symptomatology, influences on the decision to deliver specific care practices, as well as the impact and benefit of hospitalization, require closer examination.
Our study extends previous reports from the PHIS database by characterizing children admitted for concussion.1 We found that children admitted for concussion had similar characteristics to the broader population of children who sustain concussion (eg, school-aged children, male, and injured due to a fall or during sports).1,3,22 However, approximately 20% of the cohort were less than 5 years old, and less is known regarding appropriate treatment and outcomes of concussion in this age group.23 Uncertainty regarding optimal management and a young child’s inability to articulate symptoms may contribute to a physician’s decision to admit for close observation. Similar to Blinman et al., we found that a substantial proportion of children admitted with concussion were injured due to a motor vehicle collision,3 suggesting that although sports-related injuries are responsible for a significant proportion of pediatric concussions, children injured by other preventable mechanisms may also be incurring significant concussive injuries. Finally, the majority of our cohort was from an urban core, relative to a rural area, which is likely a reflection of the regionalization of trauma care, as well as variations in access to health care.
Although most children recover fully from concussion without specific interventions, 20%-30% may remain symptomatic at 1 month,3,4,21,24 and children who are hospitalized with concussion may be at higher risk for protracted symptoms. While specific individual or injury-related factors (eg, female sex, adolescent age, and injury due to motor vehicle collision) may contribute to more significant postconcussive symptoms, it is unclear how inpatient management affects recovery trajectory. Frequent sleep disruptions associated with inpatient care25 contradict current acute concussion management recommendations for physical and cognitive rest26 and could potentially impair symptom recovery. Additionally, we found widespread use of NSAIDs, although there is evidence suggesting that NSAIDs may potentially worsen concussive symptoms.26 We identified an increase in medication usage over time despite limited evidence of their effectiveness for pediatric concussion.27–29 This change may reflect improved symptom screening4,30 and/or increased awareness of specific medication safety profiles in pediatric trauma patients, especially for NSAIDs and ondansetron. Although we saw an increase in NSAID use, we did not see a proportional decrease in narcotic use. Similarly, while two-thirds of our cohort received IV medications, there is controversy about the need for IV fluids and medications for other pediatric illnesses, with research demonstrating that IV treatment may not reduce recovery time and may contribute to prolonged hospitalization and phlebitis.31,32 Thus, there is a need to understand the therapeutic effectiveness and benefits of medications and fluids on postconcussion recovery.
Neuroimaging rates for children receiving ED evaluation for concussion have been reported to be up to 60%-70%,1,22 although a more recent study spanning 2006 to 2011 found a 35-%–40% head CT rate in pediatric patients by hospital-based EDs in the United States.33 Our results appear to support decreasing head CT use over time in pediatric hospitals. Hospitalization for observation is costly1 but could decrease a child’s risk of malignancy from radiation exposure. Further work on balancing cost, risk, and shared decision-making with parents could guide decisions regarding emergent neuroimaging versus admission.
This study has limitations inherent to the use of an administrative dataset, including lack of information regarding why the child was admitted. Since the focus was to describe inpatient care of children with concussion, those discharged home from the ED were not included in this dataset. Consequently, we could not contrast the ED care of those discharged home with those who were admitted or assess trends in admission rates for concussion. Although the overall number of concussion admissions has continued to remain stable over time,1 due to a lack of prospectively collected clinical information, we are unable to determine whether observed trends in care are secondary to changes in practice or changes in concussion severity. However, there has been no research to date supporting the latter. Ethnicity was excluded due to high levels of missing data. Cost of stay was not extensively analyzed given hospital variation in designation of observational or inpatient status, which subsequently affects billing.34 Rates of neuroimaging and ED revisit may have been underestimated since children could have received care at a non-PHIS hospital. Similarly, the decrease in the proportion of children receiving neuroimaging over time may have been associated with an increase in children being transferred from a non-PHIS hospital for admission, although with increased regionalization in trauma care, we would not expect transfers of children with only concussion to have significantly increased. Finally, data were limited to the pediatric tertiary care centers participating in PHIS, thereby reducing generalizability and introducing selection bias by only including children who were able to access care at PHIS hospitals. Although the care practices we evaluated (eg, NSAIDs and head CT) are available at all hospitals, our analyses only reflect care delivered within the PHIS.
Concussion accounted for 15% of all pediatric TBI admissions during our study period. Further investigation of potential factors associated with admission and protracted recovery (eg, adolescent females needing treatment for severe symptomatology) could facilitate better understanding of how hospitalization affects recovery. Additionally, research on acute pharmacotherapies (eg, IV therapies and/or inpatient treatment until symptoms resolve) is needed to fully elucidate the acute and long-term benefits of interventions delivered to children.
ACKNOWLEDGMENTS
Colleen Mangeot: Biostatistician with extensive PHIS knowledge who contributed to database creation and statistical analysis. Yanhong (Amy) Liu: Research database programmer who developed the database, ran quality assurance measures, and cleaned all study data.
Disclosures
The authors have nothing to disclose.
Funding
This study was supported by grant R40 MC 268060102 from the Maternal and Child Health Research Program, Maternal and Child Health Bureau (Title V, Social Security Act), Health Resources and Services Administration, Department of Health and Human Services. The funding source was not involved in development of the study design; in the collection, analysis and interpretation of data; or in the writing of this report.
1. Colvin JD, Thurm C, Pate BM, Newland JG, Hall M, Meehan WP. Diagnosis and acute management of patients with concussion at children’s hospitals. Arch Dis Child. 2013;98(12):934-938. PubMed
2. Bourgeois FT, Monuteaux MC, Stack AM, Neuman MI. Variation in emergency department admission rates in US children’s hospitals. Pediatrics. 2014;134(3):539-545. PubMed
3. Blinman TA, Houseknecht E, Snyder C, Wiebe DJ, Nance ML. Postconcussive symptoms in hospitalized pediatric patients after mild traumatic brain injury. J Pediatr Surg. 2009;44(6):1223-1228. PubMed
4. Babcock L, Byczkowski T, Wade SL, Ho M, Mookerjee S, Bazarian JJ. Predicting postconcussion syndrome after mild traumatic brain injury in children and adolescents who present to the emergency department. JAMA pediatrics. 2013;167(2):156-161. PubMed
5. Conway PH, Keren R. Factors associated with variability in outcomes for children hospitalized with urinary tract infection. The Journal of pediatrics. 2009;154(6):789-796. PubMed
6. Services UDoHaH. International classification of diseases, 9th Revision, Clinical modification (ICD-9CM). Washington, DC: US Department of Health and Human Services. Public Health Service, Health Care Financing Administration 1989.
7. Marr AL, Coronado VG. Annual data submission standards. Central nervous system injury surveillance. In: US Department of Health and Human Services PHS, CDC, ed. Atlanta, GA 2001.
8. Organization WH. International classification of diseases: manual on the international statistical classification of diseases, injuries, and cause of death. In: Organization WH, ed. 9th rev. ed. Geneva, Switerland 1977.
9. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Report to Congress on mild traumatic brain injury in the United States: steps to prevent a serious public health problem. Atlanta, GA: Centers for Disease Control and Prevention; 2003.
10. Mackenzie E, Sacco WJ. ICDMAP-90 software: user’s guide. Baltimore, Maryland: Johns Hopkins University and Tri-Analytics. 1997:1-25.
11. MacKenzie EJ, Steinwachs DM, Shankar B. Classifying trauma severity based on hospital discharge diagnoses. Validation of an ICD-9CM to AIS-85 conversion table. Med Care. 1989;27(4):412-422. PubMed
12. Fleischman RJ, Mann NC, Dai M, et al. Validating the use of ICD-9 code mapping to generate injury severity scores. J Trauma Nurs. 2017;24(1):4-14. PubMed
13. Baker SP, O’Neill B, Haddon W, Jr., Long WB. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. The Journal of trauma. 1974;14(3):187-196. PubMed
14. Wood JN, Feudtner C, Medina SP, Luan X, Localio R, Rubin DM. Variation in occult injury screening for children with suspected abuse in selected US children’s hospitals. Pediatrics
15. Yang J, Phillips G, Xiang H, Allareddy V, Heiden E, Peek-Asa C. Hospitalisations for sport-related concussions in US children aged 5 to 18 years during 2000-2004. Br J Sports Med. 2008;42(8):664-669. PubMed
16. Feudtner C, Christakis DA, Connell FA. Pediatric deaths attributable to complex chronic conditions: a population-based study of Washington State, 1980-1997. Pediatrics. 2000;106(1):205-209. PubMed
17. Peltz A, Wu CL, White ML, et al. Characteristics of rural children admitted to pediatric hospitals. Pediatrics. 2016;137(5): e20153156. PubMed
18. Services UDoHaH. Annual update of the HHS Poverty Guidelines. Federal Register; 2016-03-14 2011.
19. Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95(7):1149-1155. PubMed
20. Proposed Matrix of E-code Groupings| WISQARS | Injury Center | CDC. 2016; http://www.cdc.gov/injury/wisqars/ecode_matrix.html.
21. Zemek RL, Farion KJ, Sampson M, McGahern C. Prognosticators of persistent symptoms following pediatric concussion: A systematic review. JAMA Pediatr. 2013;167(3):259-265. PubMed
22. Meehan WP, Mannix R. Pediatric concussions in United States emergency departments in the years 2002 to 2006. J Pediatr. 2010;157(6):889-893. PubMed
23. Davis GA, Purcell LK. The evaluation and management of acute concussion differs in young children. Br J Sports Med. 2014;48(2):98-101. PubMed
24. Zemek R, Barrowman N, Freedman SB, et al. Clinical risk score for persistent postconcussion symptoms among children with acute concussion in the ED. JAMA. 2016;315(10):1014-1025. PubMed
25. Hinds PS, Hockenberry M, Rai SN, et al. Nocturnal awakenings, sleep environment interruptions, and fatigue in hospitalized children with cancer. Oncol Nurs Forum. 2007;34(2):393-402. PubMed
26. Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. 2012;6:58. PubMed
27. Meehan WP. Medical therapies for concussion. Clin Sports Med. 2011;30(1):115-124, ix. PubMed
28. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533. PubMed
29. Giza CC, Kutcher JS, Ashwal S, et al. Summary of evidence-based guideline update: evaluation and management of concussion in sports: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;80(24):2250-2257. PubMed
30. Barlow KM, Crawford S, Stevenson A, Sandhu SS, Belanger F, Dewey D. Epidemiology of postconcussion syndrome in pediatric mild traumatic brain injury. Pediatrics. 2010;126(2):e374-e381. PubMed
31. Keren R, Shah SS, Srivastava R, et al. Comparative effectiveness of intravenous vs oral antibiotics for postdischarge treatment of acute osteomyelitis in children. JAMA Pediatr. 2015;169(2):120-128. PubMed
32. Hartling L, Bellemare S, Wiebe N, Russell K, Klassen TP, Craig W. Oral versus intravenous rehydration for treating dehydration due to gastroenteritis in children. Cochrane Database Syst Rev. 2006(3):CD004390. PubMed
34. Fieldston ES, Shah SS, Hall M, et al. Resource utilization for observation-status stays at children’s hospitals. Pediatrics. 2013;131(6):1050-1058. PubMed
33. Zonfrillo MR, Kim KH, Arbogast KB. Emergency Department Visits and Head Computed Tomography Utilization for Concussion Patients From 2006 to 2011. Acad Emerg Med. 2015;22(7):872-877. PubMed
1. Colvin JD, Thurm C, Pate BM, Newland JG, Hall M, Meehan WP. Diagnosis and acute management of patients with concussion at children’s hospitals. Arch Dis Child. 2013;98(12):934-938. PubMed
2. Bourgeois FT, Monuteaux MC, Stack AM, Neuman MI. Variation in emergency department admission rates in US children’s hospitals. Pediatrics. 2014;134(3):539-545. PubMed
3. Blinman TA, Houseknecht E, Snyder C, Wiebe DJ, Nance ML. Postconcussive symptoms in hospitalized pediatric patients after mild traumatic brain injury. J Pediatr Surg. 2009;44(6):1223-1228. PubMed
4. Babcock L, Byczkowski T, Wade SL, Ho M, Mookerjee S, Bazarian JJ. Predicting postconcussion syndrome after mild traumatic brain injury in children and adolescents who present to the emergency department. JAMA pediatrics. 2013;167(2):156-161. PubMed
5. Conway PH, Keren R. Factors associated with variability in outcomes for children hospitalized with urinary tract infection. The Journal of pediatrics. 2009;154(6):789-796. PubMed
6. Services UDoHaH. International classification of diseases, 9th Revision, Clinical modification (ICD-9CM). Washington, DC: US Department of Health and Human Services. Public Health Service, Health Care Financing Administration 1989.
7. Marr AL, Coronado VG. Annual data submission standards. Central nervous system injury surveillance. In: US Department of Health and Human Services PHS, CDC, ed. Atlanta, GA 2001.
8. Organization WH. International classification of diseases: manual on the international statistical classification of diseases, injuries, and cause of death. In: Organization WH, ed. 9th rev. ed. Geneva, Switerland 1977.
9. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Report to Congress on mild traumatic brain injury in the United States: steps to prevent a serious public health problem. Atlanta, GA: Centers for Disease Control and Prevention; 2003.
10. Mackenzie E, Sacco WJ. ICDMAP-90 software: user’s guide. Baltimore, Maryland: Johns Hopkins University and Tri-Analytics. 1997:1-25.
11. MacKenzie EJ, Steinwachs DM, Shankar B. Classifying trauma severity based on hospital discharge diagnoses. Validation of an ICD-9CM to AIS-85 conversion table. Med Care. 1989;27(4):412-422. PubMed
12. Fleischman RJ, Mann NC, Dai M, et al. Validating the use of ICD-9 code mapping to generate injury severity scores. J Trauma Nurs. 2017;24(1):4-14. PubMed
13. Baker SP, O’Neill B, Haddon W, Jr., Long WB. The injury severity score: a method for describing patients with multiple injuries and evaluating emergency care. The Journal of trauma. 1974;14(3):187-196. PubMed
14. Wood JN, Feudtner C, Medina SP, Luan X, Localio R, Rubin DM. Variation in occult injury screening for children with suspected abuse in selected US children’s hospitals. Pediatrics
15. Yang J, Phillips G, Xiang H, Allareddy V, Heiden E, Peek-Asa C. Hospitalisations for sport-related concussions in US children aged 5 to 18 years during 2000-2004. Br J Sports Med. 2008;42(8):664-669. PubMed
16. Feudtner C, Christakis DA, Connell FA. Pediatric deaths attributable to complex chronic conditions: a population-based study of Washington State, 1980-1997. Pediatrics. 2000;106(1):205-209. PubMed
17. Peltz A, Wu CL, White ML, et al. Characteristics of rural children admitted to pediatric hospitals. Pediatrics. 2016;137(5): e20153156. PubMed
18. Services UDoHaH. Annual update of the HHS Poverty Guidelines. Federal Register; 2016-03-14 2011.
19. Hart LG, Larson EH, Lishner DM. Rural definitions for health policy and research. Am J Public Health. 2005;95(7):1149-1155. PubMed
20. Proposed Matrix of E-code Groupings| WISQARS | Injury Center | CDC. 2016; http://www.cdc.gov/injury/wisqars/ecode_matrix.html.
21. Zemek RL, Farion KJ, Sampson M, McGahern C. Prognosticators of persistent symptoms following pediatric concussion: A systematic review. JAMA Pediatr. 2013;167(3):259-265. PubMed
22. Meehan WP, Mannix R. Pediatric concussions in United States emergency departments in the years 2002 to 2006. J Pediatr. 2010;157(6):889-893. PubMed
23. Davis GA, Purcell LK. The evaluation and management of acute concussion differs in young children. Br J Sports Med. 2014;48(2):98-101. PubMed
24. Zemek R, Barrowman N, Freedman SB, et al. Clinical risk score for persistent postconcussion symptoms among children with acute concussion in the ED. JAMA. 2016;315(10):1014-1025. PubMed
25. Hinds PS, Hockenberry M, Rai SN, et al. Nocturnal awakenings, sleep environment interruptions, and fatigue in hospitalized children with cancer. Oncol Nurs Forum. 2007;34(2):393-402. PubMed
26. Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. 2012;6:58. PubMed
27. Meehan WP. Medical therapies for concussion. Clin Sports Med. 2011;30(1):115-124, ix. PubMed
28. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533. PubMed
29. Giza CC, Kutcher JS, Ashwal S, et al. Summary of evidence-based guideline update: evaluation and management of concussion in sports: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;80(24):2250-2257. PubMed
30. Barlow KM, Crawford S, Stevenson A, Sandhu SS, Belanger F, Dewey D. Epidemiology of postconcussion syndrome in pediatric mild traumatic brain injury. Pediatrics. 2010;126(2):e374-e381. PubMed
31. Keren R, Shah SS, Srivastava R, et al. Comparative effectiveness of intravenous vs oral antibiotics for postdischarge treatment of acute osteomyelitis in children. JAMA Pediatr. 2015;169(2):120-128. PubMed
32. Hartling L, Bellemare S, Wiebe N, Russell K, Klassen TP, Craig W. Oral versus intravenous rehydration for treating dehydration due to gastroenteritis in children. Cochrane Database Syst Rev. 2006(3):CD004390. PubMed
34. Fieldston ES, Shah SS, Hall M, et al. Resource utilization for observation-status stays at children’s hospitals. Pediatrics. 2013;131(6):1050-1058. PubMed
33. Zonfrillo MR, Kim KH, Arbogast KB. Emergency Department Visits and Head Computed Tomography Utilization for Concussion Patients From 2006 to 2011. Acad Emerg Med. 2015;22(7):872-877. PubMed
© 2018 Society of Hospital Medicine
Focused Ethnography of Diagnosis in Academic Medical Centers
Diagnostic error—defined as a failure to establish an accurate and timely explanation of the patient’s health problem—is an important source of patient harm.1 Data suggest that all patients will experience at least 1 diagnostic error in their lifetime.2-4 Not surprisingly, diagnostic errors are among the leading categories of paid malpractice claims in the United States.5
Despite diagnostic errors being morbid and sometimes deadly in the hospital,6,7 little is known about how residents and learners approach diagnostic decision making. Errors in diagnosis are believed to stem from cognitive or system failures,8 with errors in cognition believed to occur due to rapid, reflexive thinking operating in the absence of a more analytical, deliberate process. System-based problems (eg, lack of expert availability, technology barriers, and access to data) have also been cited as contributors.9 However, whether and how these apply to trainees is not known.
Therefore, we conducted a focused ethnography of inpatient medicine teams (ie, attendings, residents, interns, and medical students) in 2 affiliated teaching hospitals, aiming to (a) observe the process of diagnosis by trainees and (b) identify methods to improve the diagnostic process and prevent errors.
METHODS
We designed a multimethod, focused ethnographic study to examine diagnostic decision making in hospital settings.10,11 In contrast to anthropologic ethnographies that study entire fields using open-ended questions, our study was designed to examine the process of diagnosis from the perspective of clinicians engaged in this activity.11 This approach allowed us to capture diagnostic decisions and cognitive and system-based factors in a manner currently lacking in the literature.12
Setting and Participants
Between January 2016 and May 2016, we observed the members of four inpatient internal medicine teaching teams at 2 affiliated teaching hospitals. We purposefully selected teaching teams for observation because they are the primary model of care in academic settings and we have expertise in carrying out similar studies.13,14 Teaching teams typically consisted of a medical attending (senior-level physician), 1 senior resident (a second- or third-year postgraduate trainee), two interns (a trainee in their first postgraduate year), and two to four medical students. Teams were selected at random using existing schedules and followed Monday to Friday so as to permit observation of work on call and noncall days. Owing to manpower limitations, weekend and night shifts were not observed. However, overnight events were captured during morning rounds.
Most of the teams began rounds at 8:30 AM. Typically, rounds lasted for 90–120 min and concluded with a recap (ie, “running the list”) with a review of explicit plans for patients after they had been evaluated by the attending. This discussion often occurred in the team rooms, with the attending leading the discussion with the trainees.
Data Collection
A multidisciplinary team, including clinicians (eg, physicians, nurses), nonclinicians (eg, qualitative researchers, social scientists), and healthcare engineers, conducted the observations. We observed preround activities of interns and residents before arrival of the attending (7:00 AM - 8:30 AM), followed by morning rounds with the entire team, and afternoon work that included senior residents, interns, and students.
To capture multiple aspects of the diagnostic process, we collected data using field notes modeled on components of the National Academy of Science model for diagnosis (Appendix).1,15 This model encompasses phases of the diagnostic process (eg, data gathering, integration, formulation of a working diagnosis, treatment delivery, and outcomes) and the work system (team members, organization, technology and tools, physical environment, tasks).
Focus Groups and Interviews
At the end of weekly observations, we conducted focus groups with the residents and one-on- one interviews with the attendings. Focus groups with the residents were conducted to encourage a group discussion about the diagnostic process. Separate interviews with the attendings were performed to ensure that power differentials did not influence discussions. During focus groups, we specifically asked about challenges and possible solutions to improve diagnosis. Experienced qualitative methodologists (J.F., M.H., M.Q.) used semistructured interview guides for discussions (Appendix).
Data Analysis
After aggregating and reading the data, three reviewers (V.C., S.K., S.S.) began inductive analysis by handwriting notes and initial reflective thoughts to create preliminary codes. Multiple team members then reread the original field notes and the focus group/interview data to refine the preliminary codes and develop additional codes. Next, relationships between codes were identified and used to develop key themes. Triangulation of data collected from observations and interview/focus group sessions was carried out to compare data that we surmised with data that were verbalized by the team. The developed themes were discussed as a group to ensure consistency of major findings.
Ethical and Regulatory Oversight
This study was reviewed and approved by the Institutional Review Boards at the University of Michigan Health System (HUM-00106657) and the VA Ann Arbor Healthcare System (1-2016-010040).
RESULTS
Four teaching teams (4 attendings, 4 senior residents, 9 interns, and 14 medical students) were observed over 33 distinct shifts and 168 hours. Observations included morning rounds (96 h), postround call days (52 h), and postround non-call days (20 h). Morning rounds lasted an average of 127 min (range: 48-232 min) and included an average of 9 patients (range: 4-16 patients).
Themes Regarding the Diagnostic Process
We identified the following 4 primary themes related to the diagnostic process in teaching hospitals: (1) diagnosis is a social phenomenon; (2) data necessary to make diagnoses are fragmented; (3) distractions undermine the diagnostic process; and (4) time pressures interfere with diagnostic decision making (Appendix Table 1).
(1) Diagnosis is a Social Phenomenon.
Team members viewed the process of diagnosis as a social exchange of facts, findings, and strategies within a defined structure. The opportunity to discuss impressions with others was valued as a means to share, test, and process assumptions.
“Rounds are the most important part of the process. That is where we make most decisions in a collective, collaborative way with the attending present. We bounce ideas off each other.” (Intern)
Typical of social processes, variations based on time of day and schedule were observed. For instance, during call days, learners gathered data and formed working diagnosis and treatment plans with minimal attending interaction. This separation of roles and responsibilities introduced a hierarchy within diagnosis as follows:
“The interns would not call me first; they would talk to the senior resident and then if the senior thought he should chat with me, then they would call. But for the most part, they gather information and come up with the plan.” (Attending).
The work system was suited to facilitate social interactions. For instance, designated rooms (with team members informally assigned to a computer) provided physical proximity of the resident to interns and medical students. In this space, numerous informal discussions between team members (eg, “What do you think about this test?” “I’m not sure what to do about this finding.” “Should I call a [consult] on this patient?”) were observed. Although proximity to each other was viewed as beneficial, dangers to the social nature of diagnosis in the form of anchoring (ie, a cognitive bias where emphasis is placed on the first piece of data)16 were also mentioned. Similarly, the paradox associated with social proof (ie, the pressure to assume conformity within a group) was also observed as disagreement between team members and attendings rarely occurred during observations.
“I mean, they’re the attending, right? It’s hard to argue with them when they want a test or something done. When I do push back, it’s rare that others will support me–so it’s usually me and the attending.” (Resident)
“I would push back if I think it’s really bad for the patient or could cause harm–but the truth is, it doesn’t happen much.” (Intern)
(2) Data Necessary to Make Diagnoses are Fragmented
Team members universally cited fragmentation in data delivery, retrieval, and processing as a barrier to diagnosis. Team members indicated that test results might not be looked at or acted upon in a timely manner, and participants pointed to the electronic medical record as a source of this challenge.
“Before I knew about [the app for Epic], I would literally sit on the computer to get all the information we would need on rounds. Its key to making decisions. We often say we will do something, only to find the test result doesn’t support it–and then we’re back to square 1.” (Intern)
Information used by teams came from myriad sources (eg, patients, family members, electronic records) and from various settings (eg, emergency department, patient rooms, discussions with consultants). Additionally, test results often appeared without warning. Thus, availability of information was poorly aligned with clinical duties.
“They (the lab) will call us when a blood culture is positive or something is off. That is very helpful but it often comes later in the day, when we’re done with rounds.” (Resident)
The work system was highlighted as a key contributor to data fragmentation. Peculiarities of our electronic medical record (EMR) and how data were collected, stored, or presented were described as “frustrating,” and “unsafe,” by team members. Correspondingly, we frequently observed interns asking for assistance for tasks such as ordering tests or finding information despite being “trained” to use the EMR.
“People have to learn how to filter, how to recognize the most important points and link data streams together in terms of causality. But we assume they know where to find that information. It’s actually a very hard thing to do, for both the house staff and me.” (Attending)
(3) Distractions Undermine the Diagnostic Process
Distractions often created cognitive difficulties. For example, ambient noise and interruptions from neighbors working on other teams were cited as barriers to diagnosis. In addition, we observed several team members using headphones to drown out ambient noise while working on the computer.
“I know I shouldn’t do it (wear headphones), but I have no other way of turning down the noise so I can concentrate.” (Intern)
Similarly, the unpredictable nature and the volume of pages often interrupted thinking about diagnosis.
“Sometimes the pager just goes off all the time and (after making sure its not an urgent issue), I will just ignore it for a bit, especially if I am in the middle of something. It would be great if I could finish my thought process knowing I would not be interrupted.” (Resident)
To mitigate this problem, 1 attending described how he would proactively seek out nurses caring for his patients to “head off” questions (eg, “I will renew the restraints and medications this morning,” and “Is there anything you need in terms of orders for this patient that I can take care of now?”) that might lead to pages. Another resident described his approach as follows:
“I make it a point to tell the nurses where I will be hanging out and where they can find me if they have any questions. I tell them to come talk to me rather than page me since that will be less distracting.” (Resident).
Most of the interns described documentation work such as writing admission and progress notes in negative terms (“an academic exercise,” “part of the billing activity”). However, in the context of interruptions, some described this as helpful.
“The most valuable part of the thinking process was writing the assessment and plan because that’s actually my schema for all problems. It literally is the only time where I can sit and collect my thoughts to formulate a diagnosis and plan.” (Intern)
(4) Time Pressures Interfere With Diagnostic Decision Making
All team members spoke about the challenge of finding time for diagnosis during the workday. Often, they had to skip learning sessions for this purpose.
“They tell us we should go to morning report or noon conference but when I’m running around trying to get things done. I hate having to choose between my education and doing what’s best for the patient–but that’s often what it comes down to.” (Intern)
When specifically asked whether setting aside dedicated time to specifically review and formulate diagnoses would be valuable, respondents were uniformly enthusiastic. Team members described attentional conflicts as being the worst when “cross covering” other teams on call days, as their patient load effectively doubled during this time. Of note, cross-covering occurred when teams were also on call—and thus took them away from important diagnostic activities such as data gathering or synthesis for patients they were admitting.
“If you were to ever design a system where errors were likely–this is how you would design it: take a team with little supervision, double their patient load, keep them busy with new challenging cases and then ask questions about patients they know little about.” (Resident)
DISCUSSION
Although diagnostic errors have been called “the next frontier for patient safety,”17 little is known about the process, barriers, and facilitators to diagnosis in teaching hospitals. In this focused ethnography conducted at 2 academic medical centers, we identified multiple cognitive and system-level challenges and potential strategies to improve diagnosis from trainees engaged in this activity. Key themes identified by those we observed included the social nature of diagnosis, fragmented information delivery, constant distractions and interruptions, and time pressures. In turn, these insights allow us to generate strategies that can be applied to improve the diagnostic process in teaching hospitals.
Our study underscores the importance of social interactions in diagnosis. In contrast, most of the interventions to prevent diagnostic errors target individual providers through practices such as metacognition and “thinking about thinking.”18-20 These interventions are based on Daniel Kahnemann’s work on dual thought process. Type 1 thought processes are fast, subconscious, reflexive, largely intuitive, and more vulnerable to error. In contrast, Type 2 processes are slower, deliberate, analytic, and less prone to error.21 Although an individual’s Type 2 thought capacity is limited, a major goal of cognitive interventions is to encourage Type 2 over Type 1 thinking, an approach termed “de-biasing.”22-24 Unfortunately, cognitive interventions testing such approaches have suffered mixed results–perhaps because of lack of focus on collective wisdom or group thinking, which may be key to diagnosis from our findings.9,25 In this sense, morning rounds were a social gathering used to strategize and develop care plans, but with limited time to think about diagnosis.26 Introduction of defined periods for individuals to engage in diagnostic activities such as de-biasing (ie, asking “what else could this be)27 before or after rounds may provide an opportunity for reflection and improving diagnosis. In addition, embedding tools such as diagnosis expanders and checklists within these defined time slots28,29 may prove to be useful in reflecting on diagnosis and preventing diagnostic errors.
An unexpected yet important finding from this study were the challenges posed by distractions and the physical environment. Potentially maladaptive workarounds to these interruptions included use of headphones; more productive strategies included updating nurses with plans to avert pages and creating a list of activities to ensure that key tasks were not forgotten.30,31 Applying lessons from aviation, a focused effort to limit distractions during key portions of the day, might be worth considering for diagnostic safety.32 Similarly, improving the environment in which diagnosis occurs—including creating spaces that are quiet, orderly, and optimized for thinking—may be valuable.33Our study has limitations. First, our findings are limited to direct observations; we are thus unable to comment on how unobserved aspects of care (eg, cognitive processes) might have influenced our findings. Our observations of clinical care might also have introduced a Hawthorne effect. However, because we were closely integrated with teams and conducted focus groups to corroborate our assessments, we believe that this was not the case. Second, we did not identify diagnostic errors or link processes we observed to errors. Third, our approach is limited to 2 teaching centers, thereby limiting the generalizability of findings. Relatedly, we were only able to conduct observations during weekdays; differences in weekend and night resources might affect our insights.
The cognitive and system-based barriers faced by clinicians in teaching hospitals suggest that new methods to improve diagnosis are needed. Future interventions such as defined “time-outs” for diagnosis, strategies focused on limiting distractions, and methods to improve communication between team members are novel and have parallels in other industries. As challenges to quantify diagnostic errors abound,34 improving cognitive- and system-based factors via reflection through communication, concentration, and organization is necessary to improve medical decision making in academic medical centers.
Disclosures
None declared for all coauthors.
Funding
This project was supported by grant number P30HS024385 from the Agency for Healthcare Research and Quality. The funding source played no role in study design, data acquisition, analysis or decision to report these data. Dr. Chopra is supported by a career development award from the Agency of Healthcare Research and Quality (1-K08-HS022835-01). Dr. Krein is supported by a VA Health Services Research and Development Research Career Scientist Award (RCS 11-222). Dr. Singh is partially supported by Houston VA HSR&D Center for Innovations in Quality, Effectiveness and Safety (CIN 13-413). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Agency for Healthcare Research and Quality or the Department of Veterans Affairs.
1. National Academies of Sciences, Engineering, and Medicine. 2015. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press. http://www.nap.edu/21794. Accessed November 1; 2016:2015. https://doi.org/10.17226/21794.
2. Schiff GD, Hasan O, Kim S, et al. Diagnostic error in medicine: analysis of 583 physician-reported errors. Arch Intern Med. 2009;169(20):1881-1887. http://dx.doi.org/10.1001/archinternmed.2009.333. PubMed
3. Sonderegger-Iseli K, Burger S, Muntwyler J, Salomon F. Diagnostic errors in three medical eras: A necropsy study. Lancet. 2000;355(9220):2027-2031. http://dx.doi.org/10.1016/S0140-6736(00)02349-7. PubMed
4. Winters B, Custer J, Galvagno SM Jr, et al. Diagnostic errors in the intensive care unit: a systematic review of autopsy studies. BMJ Qual Saf. 2012;21(11):894-902. http://dx.doi.org/10.1136/bmjqs-2012-000803. PubMed
5. Saber Tehrani AS, Lee H, Mathews SC, et al. 25-Year summary of US malpractice claims for diagnostic errors 1986-2010: an analysis from the National Practitioner Data Bank. BMJ Qual Saf. 2013;22(8):672-680. http://dx.doi.org/10.1136/bmjqs-2012-001550. PubMed
6. Graber M, Gordon R, Franklin N. Reducing diagnostic errors in medicine: what’s the goal? Acad Med. 2002;77(10):981-992. http://dx.doi.org/10.1097/00001888-200210000-00009. PubMed
7. Gupta A, Snyder A, Kachalia A, Flanders S, Saint S, Chopra V. Malpractice claims related to diagnostic errors in the hospital. BMJ Qual Saf. 2018;27(1):53-60. 10.1136/bmjqs-2017-006774. PubMed
8. van Noord I, Eikens MP, Hamersma AM, de Bruijne MC. Application of root cause analysis on malpractice claim files related to diagnostic failures. Qual Saf Health Care. 2010;19(6):e21. http://dx.doi.org/10.1136/qshc.2008.029801. PubMed
9. Croskerry P, Petrie DA, Reilly JB, Tait G. Deciding about fast and slow decisions. Acad Med. 2014;89(2):197-200. 10.1097/ACM.0000000000000121. PubMed
10. Higginbottom GM, Pillay JJ, Boadu NY. Guidance on performing focused ethnographies with an emphasis on healthcare research. Qual Rep. 2013;18(9):1-6. https://doi.org/10.7939/R35M6287P.
11. Savage J. Participative observation: standing in the shoes of others? Qual Health Res. 2000;10(3):324-339. http://dx.doi.org/10.1177/104973200129118471. PubMed
12. Patton MQ. Qualitative Research and Evaluation Methods. 3rd ed. Thousand Oaks, CA: SAGE Publications; 2002.
13. Harrod M, Weston LE, Robinson C, Tremblay A, Greenstone CL, Forman J. “It goes beyond good camaraderie”: A qualitative study of the process of becoming an interprofessional healthcare “teamlet.” J Interprof Care. 2016;30(3):295-300. http://dx.doi.org/10.3109/13561820.2015.1130028. PubMed
14. Houchens N, Harrod M, Moody S, Fowler KE, Saint S. Techniques and behaviors associated with exemplary inpatient general medicine teaching: an exploratory qualitative study. J Hosp Med. 2017;12(7):503-509. http://dx.doi.org/10.12788/jhm.2763. PubMed
15. Mulhall A. In the field: notes on observation in qualitative research. J Adv Nurs. 2003;41(3):306-313. http://dx.doi.org/10.1046/j.1365-2648.2003.02514.x. PubMed
16. Zwaan L, Monteiro S, Sherbino J, Ilgen J, Howey B, Norman G. Is bias in the eye of the beholder? A vignette study to assess recognition of cognitive biases in clinical case workups. BMJ Qual Saf. 2017;26(2):104-110. http://dx.doi.org/10.1136/bmjqs-2015-005014. PubMed
17. Singh H, Graber ML. Improving diagnosis in health care--the next imperative for patient safety. N Engl J Med. 2015;373(26):2493-2495. http://dx.doi.org/10.1056/NEJMp1512241. PubMed
18. Croskerry P. From mindless to mindful practice--cognitive bias and clinical decision making. N Engl J Med. 2013;368(26):2445-2448. http://dx.doi.org/10.1056/NEJMp1303712. PubMed
19. van den Berge K, Mamede S. Cognitive diagnostic error in internal medicine. Eur J Intern Med. 2013;24(6):525-529. http://dx.doi.org/10.1016/j.ejim.2013.03.006. PubMed
20. Norman G, Sherbino J, Dore K, et al. The etiology of diagnostic errors: A controlled trial of system 1 versus system 2 reasoning. Acad Med. 2014;89(2):277-284. 10.1097/ACM.0000000000000105 PubMed
21. Dhaliwal G. Premature closure? Not so fast. BMJ Qual Saf. 2017;26(2):87-89. http://dx.doi.org/10.1136/bmjqs-2016-005267. PubMed
22. Croskerry P, Singhal G, Mamede S. Cognitive debiasing 1: Origins of bias and theory of debiasing. BMJ Qual Saf. 2013;22(suppl 2):ii58-iiii64. http://dx.doi.org/10.1136/bmjqs-2012-001712. PubMed
23. Croskerry P, Singhal G, Mamede S. Cognitive debiasing 2: Impediments to and strategies for change. BMJ Qual Saf. 2013;22(suppl 2):ii65-iiii72. http://dx.doi.org/10.1136/bmjqs-2012-001713. PubMed
24. Reilly JB, Ogdie AR, Von Feldt JM, Myers JS. Teaching about how doctors think: a longitudinal curriculum in cognitive bias and diagnostic error for residents. BMJ Qual Saf. 2013;22(12):1044-1050. http://dx.doi.org/10.1136/bmjqs-2013-001987. PubMed
25. Schmidt HG, Mamede S, van den Berge K, van Gog T, van Saase JL, Rikers RM. Exposure to media information about a disease can cause doctors to misdiagnose similar-looking clinical cases. Acad Med. 2014;89(2):285-291. http://dx.doi.org/10.1097/ACM.0000000000000107. PubMed
26. Hess BJ, Lipner RS, Thompson V, Holmboe ES, Graber ML. Blink or think: can further reflection improve initial diagnostic impressions? Acad Med. 2015;90(1):112-118. http://dx.doi.org/10.1097/ACM.0000000000000550. PubMed
27. Lambe KA, O’Reilly G, Kelly BD, Curristan S. Dual-process cognitive interventions to enhance diagnostic reasoning: A systematic review. BMJ Qual Saf. 2016;25(10):808-820. http://dx.doi.org/10.1136/bmjqs-2015-004417. PubMed
28. Graber ML, Kissam S, Payne VL, et al. Cognitive interventions to reduce diagnostic error: a narrative review. BMJ Qual Saf. 2012;21(7):535-557. http://dx.doi.org/10.1136/bmjqs-2011-000149. PubMed
29. McDonald KM, Matesic B, Contopoulos-Ioannidis DG, et al. Patient safety strategies targeted at diagnostic errors: a systematic review. Ann Intern Med. 2013;158(5 Pt 2):381-389. http://dx.doi.org/10.7326/0003-4819-158-5-201303051-00004. PubMed
30. Wray CM, Chaudhry S, Pincavage A, et al. Resident shift handoff strategies in US internal medicine residency programs. JAMA. 2016;316(21):2273-2275. http://dx.doi.org/10.1001/jama.2016.17786. PubMed
31. Choo KJ, Arora VM, Barach P, Johnson JK, Farnan JM. How do supervising physicians decide to entrust residents with unsupervised tasks? A qualitative analysis. J Hosp Med. 2014;9(3):169-175. http://dx.doi.org/10.1002/jhm.2150. PubMed
32. Carayon P, Wood KE. Patient safety - the role of human factors and systems engineering. Stud Health Technol Inform. 2010;153:23-46.
.http://dx.doi.org/10.1001/jama.2015.13453 PubMed
34. McGlynn EA, McDonald KM, Cassel CK. Measurement is essential for improving diagnosis and reducing diagnostic error: A report from the Institute of Medicine. JAMA. 2015;314(23):2501-2502.
.http://dx.doi.org/10.1136/bmjqs-2013-001812 PubMed
33. Carayon P, Xie A, Kianfar S. Human factors and ergonomics as a patient safety practice. BMJ Qual Saf. 2014;23(3):196-205. PubMed
Diagnostic error—defined as a failure to establish an accurate and timely explanation of the patient’s health problem—is an important source of patient harm.1 Data suggest that all patients will experience at least 1 diagnostic error in their lifetime.2-4 Not surprisingly, diagnostic errors are among the leading categories of paid malpractice claims in the United States.5
Despite diagnostic errors being morbid and sometimes deadly in the hospital,6,7 little is known about how residents and learners approach diagnostic decision making. Errors in diagnosis are believed to stem from cognitive or system failures,8 with errors in cognition believed to occur due to rapid, reflexive thinking operating in the absence of a more analytical, deliberate process. System-based problems (eg, lack of expert availability, technology barriers, and access to data) have also been cited as contributors.9 However, whether and how these apply to trainees is not known.
Therefore, we conducted a focused ethnography of inpatient medicine teams (ie, attendings, residents, interns, and medical students) in 2 affiliated teaching hospitals, aiming to (a) observe the process of diagnosis by trainees and (b) identify methods to improve the diagnostic process and prevent errors.
METHODS
We designed a multimethod, focused ethnographic study to examine diagnostic decision making in hospital settings.10,11 In contrast to anthropologic ethnographies that study entire fields using open-ended questions, our study was designed to examine the process of diagnosis from the perspective of clinicians engaged in this activity.11 This approach allowed us to capture diagnostic decisions and cognitive and system-based factors in a manner currently lacking in the literature.12
Setting and Participants
Between January 2016 and May 2016, we observed the members of four inpatient internal medicine teaching teams at 2 affiliated teaching hospitals. We purposefully selected teaching teams for observation because they are the primary model of care in academic settings and we have expertise in carrying out similar studies.13,14 Teaching teams typically consisted of a medical attending (senior-level physician), 1 senior resident (a second- or third-year postgraduate trainee), two interns (a trainee in their first postgraduate year), and two to four medical students. Teams were selected at random using existing schedules and followed Monday to Friday so as to permit observation of work on call and noncall days. Owing to manpower limitations, weekend and night shifts were not observed. However, overnight events were captured during morning rounds.
Most of the teams began rounds at 8:30 AM. Typically, rounds lasted for 90–120 min and concluded with a recap (ie, “running the list”) with a review of explicit plans for patients after they had been evaluated by the attending. This discussion often occurred in the team rooms, with the attending leading the discussion with the trainees.
Data Collection
A multidisciplinary team, including clinicians (eg, physicians, nurses), nonclinicians (eg, qualitative researchers, social scientists), and healthcare engineers, conducted the observations. We observed preround activities of interns and residents before arrival of the attending (7:00 AM - 8:30 AM), followed by morning rounds with the entire team, and afternoon work that included senior residents, interns, and students.
To capture multiple aspects of the diagnostic process, we collected data using field notes modeled on components of the National Academy of Science model for diagnosis (Appendix).1,15 This model encompasses phases of the diagnostic process (eg, data gathering, integration, formulation of a working diagnosis, treatment delivery, and outcomes) and the work system (team members, organization, technology and tools, physical environment, tasks).
Focus Groups and Interviews
At the end of weekly observations, we conducted focus groups with the residents and one-on- one interviews with the attendings. Focus groups with the residents were conducted to encourage a group discussion about the diagnostic process. Separate interviews with the attendings were performed to ensure that power differentials did not influence discussions. During focus groups, we specifically asked about challenges and possible solutions to improve diagnosis. Experienced qualitative methodologists (J.F., M.H., M.Q.) used semistructured interview guides for discussions (Appendix).
Data Analysis
After aggregating and reading the data, three reviewers (V.C., S.K., S.S.) began inductive analysis by handwriting notes and initial reflective thoughts to create preliminary codes. Multiple team members then reread the original field notes and the focus group/interview data to refine the preliminary codes and develop additional codes. Next, relationships between codes were identified and used to develop key themes. Triangulation of data collected from observations and interview/focus group sessions was carried out to compare data that we surmised with data that were verbalized by the team. The developed themes were discussed as a group to ensure consistency of major findings.
Ethical and Regulatory Oversight
This study was reviewed and approved by the Institutional Review Boards at the University of Michigan Health System (HUM-00106657) and the VA Ann Arbor Healthcare System (1-2016-010040).
RESULTS
Four teaching teams (4 attendings, 4 senior residents, 9 interns, and 14 medical students) were observed over 33 distinct shifts and 168 hours. Observations included morning rounds (96 h), postround call days (52 h), and postround non-call days (20 h). Morning rounds lasted an average of 127 min (range: 48-232 min) and included an average of 9 patients (range: 4-16 patients).
Themes Regarding the Diagnostic Process
We identified the following 4 primary themes related to the diagnostic process in teaching hospitals: (1) diagnosis is a social phenomenon; (2) data necessary to make diagnoses are fragmented; (3) distractions undermine the diagnostic process; and (4) time pressures interfere with diagnostic decision making (Appendix Table 1).
(1) Diagnosis is a Social Phenomenon.
Team members viewed the process of diagnosis as a social exchange of facts, findings, and strategies within a defined structure. The opportunity to discuss impressions with others was valued as a means to share, test, and process assumptions.
“Rounds are the most important part of the process. That is where we make most decisions in a collective, collaborative way with the attending present. We bounce ideas off each other.” (Intern)
Typical of social processes, variations based on time of day and schedule were observed. For instance, during call days, learners gathered data and formed working diagnosis and treatment plans with minimal attending interaction. This separation of roles and responsibilities introduced a hierarchy within diagnosis as follows:
“The interns would not call me first; they would talk to the senior resident and then if the senior thought he should chat with me, then they would call. But for the most part, they gather information and come up with the plan.” (Attending).
The work system was suited to facilitate social interactions. For instance, designated rooms (with team members informally assigned to a computer) provided physical proximity of the resident to interns and medical students. In this space, numerous informal discussions between team members (eg, “What do you think about this test?” “I’m not sure what to do about this finding.” “Should I call a [consult] on this patient?”) were observed. Although proximity to each other was viewed as beneficial, dangers to the social nature of diagnosis in the form of anchoring (ie, a cognitive bias where emphasis is placed on the first piece of data)16 were also mentioned. Similarly, the paradox associated with social proof (ie, the pressure to assume conformity within a group) was also observed as disagreement between team members and attendings rarely occurred during observations.
“I mean, they’re the attending, right? It’s hard to argue with them when they want a test or something done. When I do push back, it’s rare that others will support me–so it’s usually me and the attending.” (Resident)
“I would push back if I think it’s really bad for the patient or could cause harm–but the truth is, it doesn’t happen much.” (Intern)
(2) Data Necessary to Make Diagnoses are Fragmented
Team members universally cited fragmentation in data delivery, retrieval, and processing as a barrier to diagnosis. Team members indicated that test results might not be looked at or acted upon in a timely manner, and participants pointed to the electronic medical record as a source of this challenge.
“Before I knew about [the app for Epic], I would literally sit on the computer to get all the information we would need on rounds. Its key to making decisions. We often say we will do something, only to find the test result doesn’t support it–and then we’re back to square 1.” (Intern)
Information used by teams came from myriad sources (eg, patients, family members, electronic records) and from various settings (eg, emergency department, patient rooms, discussions with consultants). Additionally, test results often appeared without warning. Thus, availability of information was poorly aligned with clinical duties.
“They (the lab) will call us when a blood culture is positive or something is off. That is very helpful but it often comes later in the day, when we’re done with rounds.” (Resident)
The work system was highlighted as a key contributor to data fragmentation. Peculiarities of our electronic medical record (EMR) and how data were collected, stored, or presented were described as “frustrating,” and “unsafe,” by team members. Correspondingly, we frequently observed interns asking for assistance for tasks such as ordering tests or finding information despite being “trained” to use the EMR.
“People have to learn how to filter, how to recognize the most important points and link data streams together in terms of causality. But we assume they know where to find that information. It’s actually a very hard thing to do, for both the house staff and me.” (Attending)
(3) Distractions Undermine the Diagnostic Process
Distractions often created cognitive difficulties. For example, ambient noise and interruptions from neighbors working on other teams were cited as barriers to diagnosis. In addition, we observed several team members using headphones to drown out ambient noise while working on the computer.
“I know I shouldn’t do it (wear headphones), but I have no other way of turning down the noise so I can concentrate.” (Intern)
Similarly, the unpredictable nature and the volume of pages often interrupted thinking about diagnosis.
“Sometimes the pager just goes off all the time and (after making sure its not an urgent issue), I will just ignore it for a bit, especially if I am in the middle of something. It would be great if I could finish my thought process knowing I would not be interrupted.” (Resident)
To mitigate this problem, 1 attending described how he would proactively seek out nurses caring for his patients to “head off” questions (eg, “I will renew the restraints and medications this morning,” and “Is there anything you need in terms of orders for this patient that I can take care of now?”) that might lead to pages. Another resident described his approach as follows:
“I make it a point to tell the nurses where I will be hanging out and where they can find me if they have any questions. I tell them to come talk to me rather than page me since that will be less distracting.” (Resident).
Most of the interns described documentation work such as writing admission and progress notes in negative terms (“an academic exercise,” “part of the billing activity”). However, in the context of interruptions, some described this as helpful.
“The most valuable part of the thinking process was writing the assessment and plan because that’s actually my schema for all problems. It literally is the only time where I can sit and collect my thoughts to formulate a diagnosis and plan.” (Intern)
(4) Time Pressures Interfere With Diagnostic Decision Making
All team members spoke about the challenge of finding time for diagnosis during the workday. Often, they had to skip learning sessions for this purpose.
“They tell us we should go to morning report or noon conference but when I’m running around trying to get things done. I hate having to choose between my education and doing what’s best for the patient–but that’s often what it comes down to.” (Intern)
When specifically asked whether setting aside dedicated time to specifically review and formulate diagnoses would be valuable, respondents were uniformly enthusiastic. Team members described attentional conflicts as being the worst when “cross covering” other teams on call days, as their patient load effectively doubled during this time. Of note, cross-covering occurred when teams were also on call—and thus took them away from important diagnostic activities such as data gathering or synthesis for patients they were admitting.
“If you were to ever design a system where errors were likely–this is how you would design it: take a team with little supervision, double their patient load, keep them busy with new challenging cases and then ask questions about patients they know little about.” (Resident)
DISCUSSION
Although diagnostic errors have been called “the next frontier for patient safety,”17 little is known about the process, barriers, and facilitators to diagnosis in teaching hospitals. In this focused ethnography conducted at 2 academic medical centers, we identified multiple cognitive and system-level challenges and potential strategies to improve diagnosis from trainees engaged in this activity. Key themes identified by those we observed included the social nature of diagnosis, fragmented information delivery, constant distractions and interruptions, and time pressures. In turn, these insights allow us to generate strategies that can be applied to improve the diagnostic process in teaching hospitals.
Our study underscores the importance of social interactions in diagnosis. In contrast, most of the interventions to prevent diagnostic errors target individual providers through practices such as metacognition and “thinking about thinking.”18-20 These interventions are based on Daniel Kahnemann’s work on dual thought process. Type 1 thought processes are fast, subconscious, reflexive, largely intuitive, and more vulnerable to error. In contrast, Type 2 processes are slower, deliberate, analytic, and less prone to error.21 Although an individual’s Type 2 thought capacity is limited, a major goal of cognitive interventions is to encourage Type 2 over Type 1 thinking, an approach termed “de-biasing.”22-24 Unfortunately, cognitive interventions testing such approaches have suffered mixed results–perhaps because of lack of focus on collective wisdom or group thinking, which may be key to diagnosis from our findings.9,25 In this sense, morning rounds were a social gathering used to strategize and develop care plans, but with limited time to think about diagnosis.26 Introduction of defined periods for individuals to engage in diagnostic activities such as de-biasing (ie, asking “what else could this be)27 before or after rounds may provide an opportunity for reflection and improving diagnosis. In addition, embedding tools such as diagnosis expanders and checklists within these defined time slots28,29 may prove to be useful in reflecting on diagnosis and preventing diagnostic errors.
An unexpected yet important finding from this study were the challenges posed by distractions and the physical environment. Potentially maladaptive workarounds to these interruptions included use of headphones; more productive strategies included updating nurses with plans to avert pages and creating a list of activities to ensure that key tasks were not forgotten.30,31 Applying lessons from aviation, a focused effort to limit distractions during key portions of the day, might be worth considering for diagnostic safety.32 Similarly, improving the environment in which diagnosis occurs—including creating spaces that are quiet, orderly, and optimized for thinking—may be valuable.33Our study has limitations. First, our findings are limited to direct observations; we are thus unable to comment on how unobserved aspects of care (eg, cognitive processes) might have influenced our findings. Our observations of clinical care might also have introduced a Hawthorne effect. However, because we were closely integrated with teams and conducted focus groups to corroborate our assessments, we believe that this was not the case. Second, we did not identify diagnostic errors or link processes we observed to errors. Third, our approach is limited to 2 teaching centers, thereby limiting the generalizability of findings. Relatedly, we were only able to conduct observations during weekdays; differences in weekend and night resources might affect our insights.
The cognitive and system-based barriers faced by clinicians in teaching hospitals suggest that new methods to improve diagnosis are needed. Future interventions such as defined “time-outs” for diagnosis, strategies focused on limiting distractions, and methods to improve communication between team members are novel and have parallels in other industries. As challenges to quantify diagnostic errors abound,34 improving cognitive- and system-based factors via reflection through communication, concentration, and organization is necessary to improve medical decision making in academic medical centers.
Disclosures
None declared for all coauthors.
Funding
This project was supported by grant number P30HS024385 from the Agency for Healthcare Research and Quality. The funding source played no role in study design, data acquisition, analysis or decision to report these data. Dr. Chopra is supported by a career development award from the Agency of Healthcare Research and Quality (1-K08-HS022835-01). Dr. Krein is supported by a VA Health Services Research and Development Research Career Scientist Award (RCS 11-222). Dr. Singh is partially supported by Houston VA HSR&D Center for Innovations in Quality, Effectiveness and Safety (CIN 13-413). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Agency for Healthcare Research and Quality or the Department of Veterans Affairs.
Diagnostic error—defined as a failure to establish an accurate and timely explanation of the patient’s health problem—is an important source of patient harm.1 Data suggest that all patients will experience at least 1 diagnostic error in their lifetime.2-4 Not surprisingly, diagnostic errors are among the leading categories of paid malpractice claims in the United States.5
Despite diagnostic errors being morbid and sometimes deadly in the hospital,6,7 little is known about how residents and learners approach diagnostic decision making. Errors in diagnosis are believed to stem from cognitive or system failures,8 with errors in cognition believed to occur due to rapid, reflexive thinking operating in the absence of a more analytical, deliberate process. System-based problems (eg, lack of expert availability, technology barriers, and access to data) have also been cited as contributors.9 However, whether and how these apply to trainees is not known.
Therefore, we conducted a focused ethnography of inpatient medicine teams (ie, attendings, residents, interns, and medical students) in 2 affiliated teaching hospitals, aiming to (a) observe the process of diagnosis by trainees and (b) identify methods to improve the diagnostic process and prevent errors.
METHODS
We designed a multimethod, focused ethnographic study to examine diagnostic decision making in hospital settings.10,11 In contrast to anthropologic ethnographies that study entire fields using open-ended questions, our study was designed to examine the process of diagnosis from the perspective of clinicians engaged in this activity.11 This approach allowed us to capture diagnostic decisions and cognitive and system-based factors in a manner currently lacking in the literature.12
Setting and Participants
Between January 2016 and May 2016, we observed the members of four inpatient internal medicine teaching teams at 2 affiliated teaching hospitals. We purposefully selected teaching teams for observation because they are the primary model of care in academic settings and we have expertise in carrying out similar studies.13,14 Teaching teams typically consisted of a medical attending (senior-level physician), 1 senior resident (a second- or third-year postgraduate trainee), two interns (a trainee in their first postgraduate year), and two to four medical students. Teams were selected at random using existing schedules and followed Monday to Friday so as to permit observation of work on call and noncall days. Owing to manpower limitations, weekend and night shifts were not observed. However, overnight events were captured during morning rounds.
Most of the teams began rounds at 8:30 AM. Typically, rounds lasted for 90–120 min and concluded with a recap (ie, “running the list”) with a review of explicit plans for patients after they had been evaluated by the attending. This discussion often occurred in the team rooms, with the attending leading the discussion with the trainees.
Data Collection
A multidisciplinary team, including clinicians (eg, physicians, nurses), nonclinicians (eg, qualitative researchers, social scientists), and healthcare engineers, conducted the observations. We observed preround activities of interns and residents before arrival of the attending (7:00 AM - 8:30 AM), followed by morning rounds with the entire team, and afternoon work that included senior residents, interns, and students.
To capture multiple aspects of the diagnostic process, we collected data using field notes modeled on components of the National Academy of Science model for diagnosis (Appendix).1,15 This model encompasses phases of the diagnostic process (eg, data gathering, integration, formulation of a working diagnosis, treatment delivery, and outcomes) and the work system (team members, organization, technology and tools, physical environment, tasks).
Focus Groups and Interviews
At the end of weekly observations, we conducted focus groups with the residents and one-on- one interviews with the attendings. Focus groups with the residents were conducted to encourage a group discussion about the diagnostic process. Separate interviews with the attendings were performed to ensure that power differentials did not influence discussions. During focus groups, we specifically asked about challenges and possible solutions to improve diagnosis. Experienced qualitative methodologists (J.F., M.H., M.Q.) used semistructured interview guides for discussions (Appendix).
Data Analysis
After aggregating and reading the data, three reviewers (V.C., S.K., S.S.) began inductive analysis by handwriting notes and initial reflective thoughts to create preliminary codes. Multiple team members then reread the original field notes and the focus group/interview data to refine the preliminary codes and develop additional codes. Next, relationships between codes were identified and used to develop key themes. Triangulation of data collected from observations and interview/focus group sessions was carried out to compare data that we surmised with data that were verbalized by the team. The developed themes were discussed as a group to ensure consistency of major findings.
Ethical and Regulatory Oversight
This study was reviewed and approved by the Institutional Review Boards at the University of Michigan Health System (HUM-00106657) and the VA Ann Arbor Healthcare System (1-2016-010040).
RESULTS
Four teaching teams (4 attendings, 4 senior residents, 9 interns, and 14 medical students) were observed over 33 distinct shifts and 168 hours. Observations included morning rounds (96 h), postround call days (52 h), and postround non-call days (20 h). Morning rounds lasted an average of 127 min (range: 48-232 min) and included an average of 9 patients (range: 4-16 patients).
Themes Regarding the Diagnostic Process
We identified the following 4 primary themes related to the diagnostic process in teaching hospitals: (1) diagnosis is a social phenomenon; (2) data necessary to make diagnoses are fragmented; (3) distractions undermine the diagnostic process; and (4) time pressures interfere with diagnostic decision making (Appendix Table 1).
(1) Diagnosis is a Social Phenomenon.
Team members viewed the process of diagnosis as a social exchange of facts, findings, and strategies within a defined structure. The opportunity to discuss impressions with others was valued as a means to share, test, and process assumptions.
“Rounds are the most important part of the process. That is where we make most decisions in a collective, collaborative way with the attending present. We bounce ideas off each other.” (Intern)
Typical of social processes, variations based on time of day and schedule were observed. For instance, during call days, learners gathered data and formed working diagnosis and treatment plans with minimal attending interaction. This separation of roles and responsibilities introduced a hierarchy within diagnosis as follows:
“The interns would not call me first; they would talk to the senior resident and then if the senior thought he should chat with me, then they would call. But for the most part, they gather information and come up with the plan.” (Attending).
The work system was suited to facilitate social interactions. For instance, designated rooms (with team members informally assigned to a computer) provided physical proximity of the resident to interns and medical students. In this space, numerous informal discussions between team members (eg, “What do you think about this test?” “I’m not sure what to do about this finding.” “Should I call a [consult] on this patient?”) were observed. Although proximity to each other was viewed as beneficial, dangers to the social nature of diagnosis in the form of anchoring (ie, a cognitive bias where emphasis is placed on the first piece of data)16 were also mentioned. Similarly, the paradox associated with social proof (ie, the pressure to assume conformity within a group) was also observed as disagreement between team members and attendings rarely occurred during observations.
“I mean, they’re the attending, right? It’s hard to argue with them when they want a test or something done. When I do push back, it’s rare that others will support me–so it’s usually me and the attending.” (Resident)
“I would push back if I think it’s really bad for the patient or could cause harm–but the truth is, it doesn’t happen much.” (Intern)
(2) Data Necessary to Make Diagnoses are Fragmented
Team members universally cited fragmentation in data delivery, retrieval, and processing as a barrier to diagnosis. Team members indicated that test results might not be looked at or acted upon in a timely manner, and participants pointed to the electronic medical record as a source of this challenge.
“Before I knew about [the app for Epic], I would literally sit on the computer to get all the information we would need on rounds. Its key to making decisions. We often say we will do something, only to find the test result doesn’t support it–and then we’re back to square 1.” (Intern)
Information used by teams came from myriad sources (eg, patients, family members, electronic records) and from various settings (eg, emergency department, patient rooms, discussions with consultants). Additionally, test results often appeared without warning. Thus, availability of information was poorly aligned with clinical duties.
“They (the lab) will call us when a blood culture is positive or something is off. That is very helpful but it often comes later in the day, when we’re done with rounds.” (Resident)
The work system was highlighted as a key contributor to data fragmentation. Peculiarities of our electronic medical record (EMR) and how data were collected, stored, or presented were described as “frustrating,” and “unsafe,” by team members. Correspondingly, we frequently observed interns asking for assistance for tasks such as ordering tests or finding information despite being “trained” to use the EMR.
“People have to learn how to filter, how to recognize the most important points and link data streams together in terms of causality. But we assume they know where to find that information. It’s actually a very hard thing to do, for both the house staff and me.” (Attending)
(3) Distractions Undermine the Diagnostic Process
Distractions often created cognitive difficulties. For example, ambient noise and interruptions from neighbors working on other teams were cited as barriers to diagnosis. In addition, we observed several team members using headphones to drown out ambient noise while working on the computer.
“I know I shouldn’t do it (wear headphones), but I have no other way of turning down the noise so I can concentrate.” (Intern)
Similarly, the unpredictable nature and the volume of pages often interrupted thinking about diagnosis.
“Sometimes the pager just goes off all the time and (after making sure its not an urgent issue), I will just ignore it for a bit, especially if I am in the middle of something. It would be great if I could finish my thought process knowing I would not be interrupted.” (Resident)
To mitigate this problem, 1 attending described how he would proactively seek out nurses caring for his patients to “head off” questions (eg, “I will renew the restraints and medications this morning,” and “Is there anything you need in terms of orders for this patient that I can take care of now?”) that might lead to pages. Another resident described his approach as follows:
“I make it a point to tell the nurses where I will be hanging out and where they can find me if they have any questions. I tell them to come talk to me rather than page me since that will be less distracting.” (Resident).
Most of the interns described documentation work such as writing admission and progress notes in negative terms (“an academic exercise,” “part of the billing activity”). However, in the context of interruptions, some described this as helpful.
“The most valuable part of the thinking process was writing the assessment and plan because that’s actually my schema for all problems. It literally is the only time where I can sit and collect my thoughts to formulate a diagnosis and plan.” (Intern)
(4) Time Pressures Interfere With Diagnostic Decision Making
All team members spoke about the challenge of finding time for diagnosis during the workday. Often, they had to skip learning sessions for this purpose.
“They tell us we should go to morning report or noon conference but when I’m running around trying to get things done. I hate having to choose between my education and doing what’s best for the patient–but that’s often what it comes down to.” (Intern)
When specifically asked whether setting aside dedicated time to specifically review and formulate diagnoses would be valuable, respondents were uniformly enthusiastic. Team members described attentional conflicts as being the worst when “cross covering” other teams on call days, as their patient load effectively doubled during this time. Of note, cross-covering occurred when teams were also on call—and thus took them away from important diagnostic activities such as data gathering or synthesis for patients they were admitting.
“If you were to ever design a system where errors were likely–this is how you would design it: take a team with little supervision, double their patient load, keep them busy with new challenging cases and then ask questions about patients they know little about.” (Resident)
DISCUSSION
Although diagnostic errors have been called “the next frontier for patient safety,”17 little is known about the process, barriers, and facilitators to diagnosis in teaching hospitals. In this focused ethnography conducted at 2 academic medical centers, we identified multiple cognitive and system-level challenges and potential strategies to improve diagnosis from trainees engaged in this activity. Key themes identified by those we observed included the social nature of diagnosis, fragmented information delivery, constant distractions and interruptions, and time pressures. In turn, these insights allow us to generate strategies that can be applied to improve the diagnostic process in teaching hospitals.
Our study underscores the importance of social interactions in diagnosis. In contrast, most of the interventions to prevent diagnostic errors target individual providers through practices such as metacognition and “thinking about thinking.”18-20 These interventions are based on Daniel Kahnemann’s work on dual thought process. Type 1 thought processes are fast, subconscious, reflexive, largely intuitive, and more vulnerable to error. In contrast, Type 2 processes are slower, deliberate, analytic, and less prone to error.21 Although an individual’s Type 2 thought capacity is limited, a major goal of cognitive interventions is to encourage Type 2 over Type 1 thinking, an approach termed “de-biasing.”22-24 Unfortunately, cognitive interventions testing such approaches have suffered mixed results–perhaps because of lack of focus on collective wisdom or group thinking, which may be key to diagnosis from our findings.9,25 In this sense, morning rounds were a social gathering used to strategize and develop care plans, but with limited time to think about diagnosis.26 Introduction of defined periods for individuals to engage in diagnostic activities such as de-biasing (ie, asking “what else could this be)27 before or after rounds may provide an opportunity for reflection and improving diagnosis. In addition, embedding tools such as diagnosis expanders and checklists within these defined time slots28,29 may prove to be useful in reflecting on diagnosis and preventing diagnostic errors.
An unexpected yet important finding from this study were the challenges posed by distractions and the physical environment. Potentially maladaptive workarounds to these interruptions included use of headphones; more productive strategies included updating nurses with plans to avert pages and creating a list of activities to ensure that key tasks were not forgotten.30,31 Applying lessons from aviation, a focused effort to limit distractions during key portions of the day, might be worth considering for diagnostic safety.32 Similarly, improving the environment in which diagnosis occurs—including creating spaces that are quiet, orderly, and optimized for thinking—may be valuable.33Our study has limitations. First, our findings are limited to direct observations; we are thus unable to comment on how unobserved aspects of care (eg, cognitive processes) might have influenced our findings. Our observations of clinical care might also have introduced a Hawthorne effect. However, because we were closely integrated with teams and conducted focus groups to corroborate our assessments, we believe that this was not the case. Second, we did not identify diagnostic errors or link processes we observed to errors. Third, our approach is limited to 2 teaching centers, thereby limiting the generalizability of findings. Relatedly, we were only able to conduct observations during weekdays; differences in weekend and night resources might affect our insights.
The cognitive and system-based barriers faced by clinicians in teaching hospitals suggest that new methods to improve diagnosis are needed. Future interventions such as defined “time-outs” for diagnosis, strategies focused on limiting distractions, and methods to improve communication between team members are novel and have parallels in other industries. As challenges to quantify diagnostic errors abound,34 improving cognitive- and system-based factors via reflection through communication, concentration, and organization is necessary to improve medical decision making in academic medical centers.
Disclosures
None declared for all coauthors.
Funding
This project was supported by grant number P30HS024385 from the Agency for Healthcare Research and Quality. The funding source played no role in study design, data acquisition, analysis or decision to report these data. Dr. Chopra is supported by a career development award from the Agency of Healthcare Research and Quality (1-K08-HS022835-01). Dr. Krein is supported by a VA Health Services Research and Development Research Career Scientist Award (RCS 11-222). Dr. Singh is partially supported by Houston VA HSR&D Center for Innovations in Quality, Effectiveness and Safety (CIN 13-413). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Agency for Healthcare Research and Quality or the Department of Veterans Affairs.
1. National Academies of Sciences, Engineering, and Medicine. 2015. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press. http://www.nap.edu/21794. Accessed November 1; 2016:2015. https://doi.org/10.17226/21794.
2. Schiff GD, Hasan O, Kim S, et al. Diagnostic error in medicine: analysis of 583 physician-reported errors. Arch Intern Med. 2009;169(20):1881-1887. http://dx.doi.org/10.1001/archinternmed.2009.333. PubMed
3. Sonderegger-Iseli K, Burger S, Muntwyler J, Salomon F. Diagnostic errors in three medical eras: A necropsy study. Lancet. 2000;355(9220):2027-2031. http://dx.doi.org/10.1016/S0140-6736(00)02349-7. PubMed
4. Winters B, Custer J, Galvagno SM Jr, et al. Diagnostic errors in the intensive care unit: a systematic review of autopsy studies. BMJ Qual Saf. 2012;21(11):894-902. http://dx.doi.org/10.1136/bmjqs-2012-000803. PubMed
5. Saber Tehrani AS, Lee H, Mathews SC, et al. 25-Year summary of US malpractice claims for diagnostic errors 1986-2010: an analysis from the National Practitioner Data Bank. BMJ Qual Saf. 2013;22(8):672-680. http://dx.doi.org/10.1136/bmjqs-2012-001550. PubMed
6. Graber M, Gordon R, Franklin N. Reducing diagnostic errors in medicine: what’s the goal? Acad Med. 2002;77(10):981-992. http://dx.doi.org/10.1097/00001888-200210000-00009. PubMed
7. Gupta A, Snyder A, Kachalia A, Flanders S, Saint S, Chopra V. Malpractice claims related to diagnostic errors in the hospital. BMJ Qual Saf. 2018;27(1):53-60. 10.1136/bmjqs-2017-006774. PubMed
8. van Noord I, Eikens MP, Hamersma AM, de Bruijne MC. Application of root cause analysis on malpractice claim files related to diagnostic failures. Qual Saf Health Care. 2010;19(6):e21. http://dx.doi.org/10.1136/qshc.2008.029801. PubMed
9. Croskerry P, Petrie DA, Reilly JB, Tait G. Deciding about fast and slow decisions. Acad Med. 2014;89(2):197-200. 10.1097/ACM.0000000000000121. PubMed
10. Higginbottom GM, Pillay JJ, Boadu NY. Guidance on performing focused ethnographies with an emphasis on healthcare research. Qual Rep. 2013;18(9):1-6. https://doi.org/10.7939/R35M6287P.
11. Savage J. Participative observation: standing in the shoes of others? Qual Health Res. 2000;10(3):324-339. http://dx.doi.org/10.1177/104973200129118471. PubMed
12. Patton MQ. Qualitative Research and Evaluation Methods. 3rd ed. Thousand Oaks, CA: SAGE Publications; 2002.
13. Harrod M, Weston LE, Robinson C, Tremblay A, Greenstone CL, Forman J. “It goes beyond good camaraderie”: A qualitative study of the process of becoming an interprofessional healthcare “teamlet.” J Interprof Care. 2016;30(3):295-300. http://dx.doi.org/10.3109/13561820.2015.1130028. PubMed
14. Houchens N, Harrod M, Moody S, Fowler KE, Saint S. Techniques and behaviors associated with exemplary inpatient general medicine teaching: an exploratory qualitative study. J Hosp Med. 2017;12(7):503-509. http://dx.doi.org/10.12788/jhm.2763. PubMed
15. Mulhall A. In the field: notes on observation in qualitative research. J Adv Nurs. 2003;41(3):306-313. http://dx.doi.org/10.1046/j.1365-2648.2003.02514.x. PubMed
16. Zwaan L, Monteiro S, Sherbino J, Ilgen J, Howey B, Norman G. Is bias in the eye of the beholder? A vignette study to assess recognition of cognitive biases in clinical case workups. BMJ Qual Saf. 2017;26(2):104-110. http://dx.doi.org/10.1136/bmjqs-2015-005014. PubMed
17. Singh H, Graber ML. Improving diagnosis in health care--the next imperative for patient safety. N Engl J Med. 2015;373(26):2493-2495. http://dx.doi.org/10.1056/NEJMp1512241. PubMed
18. Croskerry P. From mindless to mindful practice--cognitive bias and clinical decision making. N Engl J Med. 2013;368(26):2445-2448. http://dx.doi.org/10.1056/NEJMp1303712. PubMed
19. van den Berge K, Mamede S. Cognitive diagnostic error in internal medicine. Eur J Intern Med. 2013;24(6):525-529. http://dx.doi.org/10.1016/j.ejim.2013.03.006. PubMed
20. Norman G, Sherbino J, Dore K, et al. The etiology of diagnostic errors: A controlled trial of system 1 versus system 2 reasoning. Acad Med. 2014;89(2):277-284. 10.1097/ACM.0000000000000105 PubMed
21. Dhaliwal G. Premature closure? Not so fast. BMJ Qual Saf. 2017;26(2):87-89. http://dx.doi.org/10.1136/bmjqs-2016-005267. PubMed
22. Croskerry P, Singhal G, Mamede S. Cognitive debiasing 1: Origins of bias and theory of debiasing. BMJ Qual Saf. 2013;22(suppl 2):ii58-iiii64. http://dx.doi.org/10.1136/bmjqs-2012-001712. PubMed
23. Croskerry P, Singhal G, Mamede S. Cognitive debiasing 2: Impediments to and strategies for change. BMJ Qual Saf. 2013;22(suppl 2):ii65-iiii72. http://dx.doi.org/10.1136/bmjqs-2012-001713. PubMed
24. Reilly JB, Ogdie AR, Von Feldt JM, Myers JS. Teaching about how doctors think: a longitudinal curriculum in cognitive bias and diagnostic error for residents. BMJ Qual Saf. 2013;22(12):1044-1050. http://dx.doi.org/10.1136/bmjqs-2013-001987. PubMed
25. Schmidt HG, Mamede S, van den Berge K, van Gog T, van Saase JL, Rikers RM. Exposure to media information about a disease can cause doctors to misdiagnose similar-looking clinical cases. Acad Med. 2014;89(2):285-291. http://dx.doi.org/10.1097/ACM.0000000000000107. PubMed
26. Hess BJ, Lipner RS, Thompson V, Holmboe ES, Graber ML. Blink or think: can further reflection improve initial diagnostic impressions? Acad Med. 2015;90(1):112-118. http://dx.doi.org/10.1097/ACM.0000000000000550. PubMed
27. Lambe KA, O’Reilly G, Kelly BD, Curristan S. Dual-process cognitive interventions to enhance diagnostic reasoning: A systematic review. BMJ Qual Saf. 2016;25(10):808-820. http://dx.doi.org/10.1136/bmjqs-2015-004417. PubMed
28. Graber ML, Kissam S, Payne VL, et al. Cognitive interventions to reduce diagnostic error: a narrative review. BMJ Qual Saf. 2012;21(7):535-557. http://dx.doi.org/10.1136/bmjqs-2011-000149. PubMed
29. McDonald KM, Matesic B, Contopoulos-Ioannidis DG, et al. Patient safety strategies targeted at diagnostic errors: a systematic review. Ann Intern Med. 2013;158(5 Pt 2):381-389. http://dx.doi.org/10.7326/0003-4819-158-5-201303051-00004. PubMed
30. Wray CM, Chaudhry S, Pincavage A, et al. Resident shift handoff strategies in US internal medicine residency programs. JAMA. 2016;316(21):2273-2275. http://dx.doi.org/10.1001/jama.2016.17786. PubMed
31. Choo KJ, Arora VM, Barach P, Johnson JK, Farnan JM. How do supervising physicians decide to entrust residents with unsupervised tasks? A qualitative analysis. J Hosp Med. 2014;9(3):169-175. http://dx.doi.org/10.1002/jhm.2150. PubMed
32. Carayon P, Wood KE. Patient safety - the role of human factors and systems engineering. Stud Health Technol Inform. 2010;153:23-46.
.http://dx.doi.org/10.1001/jama.2015.13453 PubMed
34. McGlynn EA, McDonald KM, Cassel CK. Measurement is essential for improving diagnosis and reducing diagnostic error: A report from the Institute of Medicine. JAMA. 2015;314(23):2501-2502.
.http://dx.doi.org/10.1136/bmjqs-2013-001812 PubMed
33. Carayon P, Xie A, Kianfar S. Human factors and ergonomics as a patient safety practice. BMJ Qual Saf. 2014;23(3):196-205. PubMed
1. National Academies of Sciences, Engineering, and Medicine. 2015. Improving Diagnosis in Health Care. Washington, DC: The National Academies Press. http://www.nap.edu/21794. Accessed November 1; 2016:2015. https://doi.org/10.17226/21794.
2. Schiff GD, Hasan O, Kim S, et al. Diagnostic error in medicine: analysis of 583 physician-reported errors. Arch Intern Med. 2009;169(20):1881-1887. http://dx.doi.org/10.1001/archinternmed.2009.333. PubMed
3. Sonderegger-Iseli K, Burger S, Muntwyler J, Salomon F. Diagnostic errors in three medical eras: A necropsy study. Lancet. 2000;355(9220):2027-2031. http://dx.doi.org/10.1016/S0140-6736(00)02349-7. PubMed
4. Winters B, Custer J, Galvagno SM Jr, et al. Diagnostic errors in the intensive care unit: a systematic review of autopsy studies. BMJ Qual Saf. 2012;21(11):894-902. http://dx.doi.org/10.1136/bmjqs-2012-000803. PubMed
5. Saber Tehrani AS, Lee H, Mathews SC, et al. 25-Year summary of US malpractice claims for diagnostic errors 1986-2010: an analysis from the National Practitioner Data Bank. BMJ Qual Saf. 2013;22(8):672-680. http://dx.doi.org/10.1136/bmjqs-2012-001550. PubMed
6. Graber M, Gordon R, Franklin N. Reducing diagnostic errors in medicine: what’s the goal? Acad Med. 2002;77(10):981-992. http://dx.doi.org/10.1097/00001888-200210000-00009. PubMed
7. Gupta A, Snyder A, Kachalia A, Flanders S, Saint S, Chopra V. Malpractice claims related to diagnostic errors in the hospital. BMJ Qual Saf. 2018;27(1):53-60. 10.1136/bmjqs-2017-006774. PubMed
8. van Noord I, Eikens MP, Hamersma AM, de Bruijne MC. Application of root cause analysis on malpractice claim files related to diagnostic failures. Qual Saf Health Care. 2010;19(6):e21. http://dx.doi.org/10.1136/qshc.2008.029801. PubMed
9. Croskerry P, Petrie DA, Reilly JB, Tait G. Deciding about fast and slow decisions. Acad Med. 2014;89(2):197-200. 10.1097/ACM.0000000000000121. PubMed
10. Higginbottom GM, Pillay JJ, Boadu NY. Guidance on performing focused ethnographies with an emphasis on healthcare research. Qual Rep. 2013;18(9):1-6. https://doi.org/10.7939/R35M6287P.
11. Savage J. Participative observation: standing in the shoes of others? Qual Health Res. 2000;10(3):324-339. http://dx.doi.org/10.1177/104973200129118471. PubMed
12. Patton MQ. Qualitative Research and Evaluation Methods. 3rd ed. Thousand Oaks, CA: SAGE Publications; 2002.
13. Harrod M, Weston LE, Robinson C, Tremblay A, Greenstone CL, Forman J. “It goes beyond good camaraderie”: A qualitative study of the process of becoming an interprofessional healthcare “teamlet.” J Interprof Care. 2016;30(3):295-300. http://dx.doi.org/10.3109/13561820.2015.1130028. PubMed
14. Houchens N, Harrod M, Moody S, Fowler KE, Saint S. Techniques and behaviors associated with exemplary inpatient general medicine teaching: an exploratory qualitative study. J Hosp Med. 2017;12(7):503-509. http://dx.doi.org/10.12788/jhm.2763. PubMed
15. Mulhall A. In the field: notes on observation in qualitative research. J Adv Nurs. 2003;41(3):306-313. http://dx.doi.org/10.1046/j.1365-2648.2003.02514.x. PubMed
16. Zwaan L, Monteiro S, Sherbino J, Ilgen J, Howey B, Norman G. Is bias in the eye of the beholder? A vignette study to assess recognition of cognitive biases in clinical case workups. BMJ Qual Saf. 2017;26(2):104-110. http://dx.doi.org/10.1136/bmjqs-2015-005014. PubMed
17. Singh H, Graber ML. Improving diagnosis in health care--the next imperative for patient safety. N Engl J Med. 2015;373(26):2493-2495. http://dx.doi.org/10.1056/NEJMp1512241. PubMed
18. Croskerry P. From mindless to mindful practice--cognitive bias and clinical decision making. N Engl J Med. 2013;368(26):2445-2448. http://dx.doi.org/10.1056/NEJMp1303712. PubMed
19. van den Berge K, Mamede S. Cognitive diagnostic error in internal medicine. Eur J Intern Med. 2013;24(6):525-529. http://dx.doi.org/10.1016/j.ejim.2013.03.006. PubMed
20. Norman G, Sherbino J, Dore K, et al. The etiology of diagnostic errors: A controlled trial of system 1 versus system 2 reasoning. Acad Med. 2014;89(2):277-284. 10.1097/ACM.0000000000000105 PubMed
21. Dhaliwal G. Premature closure? Not so fast. BMJ Qual Saf. 2017;26(2):87-89. http://dx.doi.org/10.1136/bmjqs-2016-005267. PubMed
22. Croskerry P, Singhal G, Mamede S. Cognitive debiasing 1: Origins of bias and theory of debiasing. BMJ Qual Saf. 2013;22(suppl 2):ii58-iiii64. http://dx.doi.org/10.1136/bmjqs-2012-001712. PubMed
23. Croskerry P, Singhal G, Mamede S. Cognitive debiasing 2: Impediments to and strategies for change. BMJ Qual Saf. 2013;22(suppl 2):ii65-iiii72. http://dx.doi.org/10.1136/bmjqs-2012-001713. PubMed
24. Reilly JB, Ogdie AR, Von Feldt JM, Myers JS. Teaching about how doctors think: a longitudinal curriculum in cognitive bias and diagnostic error for residents. BMJ Qual Saf. 2013;22(12):1044-1050. http://dx.doi.org/10.1136/bmjqs-2013-001987. PubMed
25. Schmidt HG, Mamede S, van den Berge K, van Gog T, van Saase JL, Rikers RM. Exposure to media information about a disease can cause doctors to misdiagnose similar-looking clinical cases. Acad Med. 2014;89(2):285-291. http://dx.doi.org/10.1097/ACM.0000000000000107. PubMed
26. Hess BJ, Lipner RS, Thompson V, Holmboe ES, Graber ML. Blink or think: can further reflection improve initial diagnostic impressions? Acad Med. 2015;90(1):112-118. http://dx.doi.org/10.1097/ACM.0000000000000550. PubMed
27. Lambe KA, O’Reilly G, Kelly BD, Curristan S. Dual-process cognitive interventions to enhance diagnostic reasoning: A systematic review. BMJ Qual Saf. 2016;25(10):808-820. http://dx.doi.org/10.1136/bmjqs-2015-004417. PubMed
28. Graber ML, Kissam S, Payne VL, et al. Cognitive interventions to reduce diagnostic error: a narrative review. BMJ Qual Saf. 2012;21(7):535-557. http://dx.doi.org/10.1136/bmjqs-2011-000149. PubMed
29. McDonald KM, Matesic B, Contopoulos-Ioannidis DG, et al. Patient safety strategies targeted at diagnostic errors: a systematic review. Ann Intern Med. 2013;158(5 Pt 2):381-389. http://dx.doi.org/10.7326/0003-4819-158-5-201303051-00004. PubMed
30. Wray CM, Chaudhry S, Pincavage A, et al. Resident shift handoff strategies in US internal medicine residency programs. JAMA. 2016;316(21):2273-2275. http://dx.doi.org/10.1001/jama.2016.17786. PubMed
31. Choo KJ, Arora VM, Barach P, Johnson JK, Farnan JM. How do supervising physicians decide to entrust residents with unsupervised tasks? A qualitative analysis. J Hosp Med. 2014;9(3):169-175. http://dx.doi.org/10.1002/jhm.2150. PubMed
32. Carayon P, Wood KE. Patient safety - the role of human factors and systems engineering. Stud Health Technol Inform. 2010;153:23-46.
.http://dx.doi.org/10.1001/jama.2015.13453 PubMed
34. McGlynn EA, McDonald KM, Cassel CK. Measurement is essential for improving diagnosis and reducing diagnostic error: A report from the Institute of Medicine. JAMA. 2015;314(23):2501-2502.
.http://dx.doi.org/10.1136/bmjqs-2013-001812 PubMed
33. Carayon P, Xie A, Kianfar S. Human factors and ergonomics as a patient safety practice. BMJ Qual Saf. 2014;23(3):196-205. PubMed
© 2018 Society of Hospital Medicine
Appraising the Evidence Supporting Choosing Wisely® Recommendations
As healthcare costs rise, physicians and other stakeholders are now seeking innovative and effective ways to reduce the provision of low-value services.1,2 The Choosing Wisely® campaign aims to further this goal by promoting lists of specific procedures, tests, and treatments that providers should avoid in selected clinical settings.3 On February 21, 2013, the Society of Hospital Medicine (SHM) released 2 Choosing Wisely® lists consisting of adult and pediatric services that are seen as costly to consumers and to the healthcare system, but which are often nonbeneficial or even harmful.4,5 A total of 80 physician and nurse specialty societies have joined in submitting additional lists.
Despite the growing enthusiasm for this effort, questions remain regarding the Choosing Wisely® campaign’s ability to initiate the meaningful de-adoption of low-value services. Specifically, prior efforts to reduce the use of services deemed to be of questionable benefit have met several challenges.2,6 Early analyses of the Choosing Wisely® recommendations reveal similar roadblocks and variable uptakes of several recommendations.7-10 While the reasons for difficulties in achieving de-adoption are broad, one important factor in whether clinicians are willing to follow guideline recommendations from such initiatives as Choosing Wisely®is the extent to which they believe in the underlying evidence.11 The current work seeks to formally evaluate the evidence supporting the Choosing Wisely® recommendations, and to compare the quality of evidence supporting SHM lists to other published Choosing Wisely® lists.
METHODS
Data Sources
Using the online listing of published Choosing Wisely® recommendations, a dataset was generated incorporating all 320 recommendations comprising the 58 lists published through August, 2014; these include both the adult and pediatric hospital medicine lists released by the SHM.4,5,12 Although data collection ended at this point, this represents a majority of all 81 lists and 535 recommendations published through December, 2017. The reviewers (A.J.A., A.G., M.W., T.S.V., M.S., and C.R.C) extracted information about the references cited for each recommendation.
Data Analysis
The reviewers obtained each reference cited by a Choosing Wisely® recommendation and categorized it by evidence strength along the following hierarchy: clinical practice guideline (CPG), primary research, review article, expert opinion, book, or others/unknown. CPGs were used as the highest level of evidence based on standard expectations for methodological rigor.13 Primary research was further rated as follows: systematic reviews and meta-analyses, randomized controlled trials (RCTs), observational studies, and case series. Each recommendation was graded using only the strongest piece of evidence cited.
Guideline Appraisal
We further sought to evaluate the strength of referenced CPGs. To accomplish this, a 10% random sample of the Choosing Wisely® recommendations citing CPGs was selected, and the referenced CPGs were obtained. Separately, CPGs referenced by the SHM-published adult and pediatric lists were also obtained. For both groups, one CPG was randomly selected when a recommendation cited more than one CPG. These guidelines were assessed using the Appraisal of Guidelines for Research and Evaluation (AGREE) II instrument, a widely used instrument designed to assess CPG quality.14,15 AGREE II consists of 25 questions categorized into 6 domains: scope and purpose, stakeholder involvement, rigor of development, clarity of presentation, applicability, and editorial independence. Guidelines are also assigned an overall score. Two trained reviewers (A.J.A. and A.G.) assessed each of the sampled CPGs using a standardized form. Scores were then standardized using the method recommended by the instrument and reported as a percentage of available points. Although a standard interpretation of scores is not provided by the instrument, prior applications deemed scores below 50% as deficient16,17. When a recommendation item cited multiple CPGs, one was randomly selected. We also abstracted data on the year of publication, the evidence grade assigned to specific items recommended by Choosing Wisely®, and whether the CPG addressed the referring recommendation. All data management and analysis were conducted using Stata (V14.2, StataCorp, College Station, Texas).
RESULTS
A total of 320 recommendations were considered in our analysis, including 10 published across the 2 hospital medicine lists. When limited to the highest quality citation for each of the recommendations, 225 (70.3%) cited CPGs, whereas 71 (22.2%) cited primary research articles (Table 1). Specifically, 29 (9.1%) cited systematic reviews and meta-analyses, 28 (8.8%) cited observational studies, and 13 (4.1%) cited RCTs. One recommendation (0.3%) cited a case series as its highest level of evidence, 7 (2.2%) cited review articles, 7 (2.2%) cited editorials or opinion pieces, and 10 (3.1%) cited other types of documents, such as websites or books. Among hospital medicine recommendations, 9 (90%) referenced CPGs and 1 (10%) cited an observational study.
For the AGREE II assessment, we included 23 CPGs from the 225 referenced across all recommendations, after which we separately selected 6 CPGs from the hospital medicine recommendations. There was no overlap. Notably, 4 hospital medicine recommendations referenced a common CPG. Among the random sample of referenced CPGs, the median overall score obtained by using AGREE II was 54.2% (IQR 33.3%-70.8%, Table 2). This was similar to the median overall among hospital medicine guidelines (58.2%, IQR 50.0%-83.3%). Both hospital medicine and other sampled guidelines tended to score poorly in stakeholder involvement (48.6%, IQR 44.1%-61.1% and 47.2%, IQR 38.9%-61.1%, respectively). There were no significant differences between hospital medicine-referenced CPGs and the larger sample of CPGs in any AGREE II subdomains. The median age from the CPG publication to the list publication was 7 years (IQR 4–7) for hospital medicine recommendations and 3 years (IQR 2–6) for the nonhospital medicine recommendations. Substantial agreement was found between raters on the overall guideline assessment (ICC 0.80, 95% CI 0.58-0.91; Supplementary Table 1).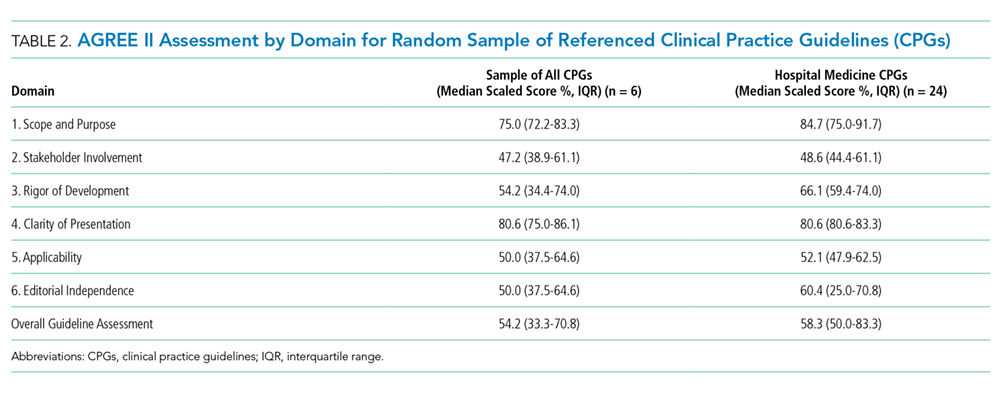
In terms of recommendation strengths and evidence grades, several recommendations were backed by Grades II–III (on a scale of I-III) evidence and level C (on a scale of A–C) recommendations in the reviewed CPG (Society of Maternal-Fetal Medicine, Recommendation 4, and Heart Rhythm Society, Recommendation 1). In one other case, the cited CPG did not directly address the Choosing Wisely® item (Society of Vascular Medicine, Recommendation 2).
DISCUSSION
Given the rising costs and the potential for iatrogenic harm, curbing ineffective practices has become an urgent concern. To achieve this, the Choosing Wisely® campaign has taken an important step by targeting certain low-value practices for de-adoption. However, the evidence supporting recommendations is variable. Specifically, 25 recommendations cited case series, review articles, or lower quality evidence as their highest level of support; moreover, among recommendations citing CPGs, quality, timeliness, and support for the recommendation item were variable. Although the hospital medicine lists tended to cite higher-quality evidence in the form of CPGs, these CPGs were often less recent than the guidelines referenced by other lists.
Our findings parallel those of other works that evaluate evidence among Choosing Wisely® recommendations and, more broadly, among CPGs.18–21 Lin and Yancey evaluated the quality of primary care-focused Choosing Wisely® recommendations using the Strength of Recommendation Taxonomy, a ranking system that evaluates evidence quality, consistency, and patient-centeredness.18 In their analysis, the authors found that many recommendations were based on lower quality evidence or relied on nonpatent-centered intermediate outcomes. Several groups, meanwhile, have evaluated the quality of evidence supporting CPG recommendations, finding them to be highly variable as well.19–21 These findings likely reflect inherent difficulties in the process, by which guideline development groups distill a broad evidence base into useful clinical recommendations, a reality that may have influenced the Choosing Wisely® list development groups seeking to make similar recommendations on low-value services.
These data should be taken in context due to several limitations. First, our sample of referenced CPGs includes only a small sample of all CPGs cited; thus, it may not be representative of all referenced guidelines. Second, the AGREE II assessment is inherently subjective, despite the availability of training materials. Third, data collection ended in April, 2014. Although this represents a majority of published lists to date, it is possible that more recent Choosing Wisely®lists include a stronger focus on evidence quality. Finally, references cited by Choosing Wisely®may not be representative of the entirety of the dataset that was considered when formulating the recommendations.
Despite these limitations, our findings suggest that Choosing Wisely®recommendations vary in terms of evidence strength. Although our results reveal that the majority of recommendations cite guidelines or high-quality original research, evidence gaps remain, with a small number citing low-quality evidence or low-quality CPGs as their highest form of support. Given the barriers to the successful de-implementation of low-value services, such campaigns as Choosing Wisely®face an uphill battle in their attempt to prompt behavior changes among providers and consumers.6-9 As a result, it is incumbent on funding agencies and medical journals to promote studies evaluating the harms and overall value of the care we deliver.
CONCLUSIONS
Although a majority of Choosing Wisely® recommendations cite high-quality evidence, some reference low-quality evidence or low-quality CPGs as their highest form of support. To overcome clinical inertia and other barriers to the successful de-implementation of low-value services, a clear rationale for the impetus to eradicate entrenched practices is critical.2,22 Choosing Wisely® has provided visionary leadership and a powerful platform to question low-value care. To expand the campaign’s efforts, the medical field must be able to generate the high-quality evidence necessary to support these efforts; further, list development groups must consider the availability of strong evidence when targeting services for de-implementation.
ACKNOWLEDGMENT
This work was supported, in part, by a grant from the Agency for Healthcare Research and Quality (No. K08HS020672, Dr. Cooke).
Disclosures
The authors have nothing to disclose.
1. Institute of Medicine Roundtable on Evidence-Based Medicine. The Healthcare Imperative: Lowering Costs and Improving Outcomes: Workshop Series Summary. Yong P, Saudners R, Olsen L, editors. Washington, D.C.: National Academies Press; 2010. PubMed
2. Weinberger SE. Providing high-value, cost-conscious care: a critical seventh general competency for physicians. Ann Intern Med. 2011;155(6):386-388. PubMed
3. Cassel CK, Guest JA. Choosing wisely: Helping physicians and patients make smart decisions about their care. JAMA. 2012;307(17):1801-1802. PubMed
4. Bulger J, Nickel W, Messler J, Goldstein J, O’Callaghan J, Auron M, et al. Choosing wisely in adult hospital medicine: Five opportunities for improved healthcare value. J Hosp Med. 2013;8(9):486-492. PubMed
5. Quinonez RA, Garber MD, Schroeder AR, Alverson BK, Nickel W, Goldstein J, et al. Choosing wisely in pediatric hospital medicine: Five opportunities for improved healthcare value. J Hosp Med. 2013;8(9):479-485. PubMed
6. Prasad V, Ioannidis JP. Evidence-based de-implementation for contradicted, unproven, and aspiring healthcare practices. Implement Sci. 2014;9:1. PubMed
7. Rosenberg A, Agiro A, Gottlieb M, Barron J, Brady P, Liu Y, et al. Early trends among seven recommendations from the Choosing Wisely campaign. JAMA Intern Med. 2015;175(12):1913-1920. PubMed
8. Zikmund-Fisher BJ, Kullgren JT, Fagerlin A, Klamerus ML, Bernstein SJ, Kerr EA. Perceived barriers to implementing individual Choosing Wisely® recommendations in two national surveys of primary care providers. J Gen Intern Med. 2017;32(2):210-217. PubMed
9. Bishop TF, Cea M, Miranda Y, Kim R, Lash-Dardia M, Lee JI, et al. Academic physicians’ views on low-value services and the choosing wisely campaign: A qualitative study. Healthc (Amsterdam, Netherlands). 2017;5(1-2):17-22. PubMed
10. Prochaska MT, Hohmann SF, Modes M, Arora VM. Trends in Troponin-only testing for AMI in academic teaching hospitals and the impact of Choosing Wisely®. J Hosp Med. 2017;12(12):957-962. PubMed
11. Cabana MD, Rand CS, Powe NR, Wu AW, Wilson MH, Abboud PA, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA. 1999;282(15):1458-1465. PubMed
12. ABIM Foundation. ChoosingWisely.org Search Recommendations. 2014.
13. Institute of Medicine (US) Committee on Standards for Developing Trustworthy Clinical Practice Guidelines. Clinical Practice Guidelines We Can Trust. Graham R, Mancher M, Miller Wolman D, Greenfield S, Steinberg E, editors. Washington, D.C.: National Academies Press; 2011. PubMed
14. Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. AGREE II: Advancing guideline development, reporting, and evaluation in health care. Prev Med (Baltim). 2010;51(5):421-424. PubMed
15. Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. Development of the AGREE II, part 2: Assessment of validity of items and tools to support application. CMAJ. 2010;182(10):E472-E478. PubMed
16. He Z, Tian H, Song A, Jin L, Zhou X, Liu X, et al. Quality appraisal of clinical practice guidelines on pancreatic cancer. Medicine (Baltimore). 2015;94(12):e635. PubMed
17. Isaac A, Saginur M, Hartling L, Robinson JL. Quality of reporting and evidence in American Academy of Pediatrics guidelines. Pediatrics. 2013;131(4):732-738. PubMed
18. Lin KW, Yancey JR. Evaluating the Evidence for Choosing WiselyTM in Primary Care Using the Strength of Recommendation Taxonomy (SORT). J Am Board Fam Med. 2016;29(4):512-515. PubMed
19. McAlister FA, van Diepen S, Padwal RS, Johnson JA, Majumdar SR. How evidence-based are the recommendations in evidence-based guidelines? PLoS Med. 2007;4(8):e250. PubMed
20. Tricoci P, Allen JM, Kramer JM, Califf RM, Smith SC. Scientific evidence underlying the ACC/AHA clinical practice guidelines. JAMA. 2009;301(8):831-841. PubMed
21. Feuerstein JD, Gifford AE, Akbari M, Goldman J, Leffler DA, Sheth SG, et al. Systematic analysis underlying the quality of the scientific evidence and conflicts of interest in gastroenterology practice guidelines. Am J Gastroenterol. 2013;108(11):1686-1693. PubMed
22. Robert G, Harlock J, Williams I. Disentangling rhetoric and reality: an international Delphi study of factors and processes that facilitate the successful implementation of decisions to decommission healthcare services. Implement Sci. 2014;9:123. PubMed
As healthcare costs rise, physicians and other stakeholders are now seeking innovative and effective ways to reduce the provision of low-value services.1,2 The Choosing Wisely® campaign aims to further this goal by promoting lists of specific procedures, tests, and treatments that providers should avoid in selected clinical settings.3 On February 21, 2013, the Society of Hospital Medicine (SHM) released 2 Choosing Wisely® lists consisting of adult and pediatric services that are seen as costly to consumers and to the healthcare system, but which are often nonbeneficial or even harmful.4,5 A total of 80 physician and nurse specialty societies have joined in submitting additional lists.
Despite the growing enthusiasm for this effort, questions remain regarding the Choosing Wisely® campaign’s ability to initiate the meaningful de-adoption of low-value services. Specifically, prior efforts to reduce the use of services deemed to be of questionable benefit have met several challenges.2,6 Early analyses of the Choosing Wisely® recommendations reveal similar roadblocks and variable uptakes of several recommendations.7-10 While the reasons for difficulties in achieving de-adoption are broad, one important factor in whether clinicians are willing to follow guideline recommendations from such initiatives as Choosing Wisely®is the extent to which they believe in the underlying evidence.11 The current work seeks to formally evaluate the evidence supporting the Choosing Wisely® recommendations, and to compare the quality of evidence supporting SHM lists to other published Choosing Wisely® lists.
METHODS
Data Sources
Using the online listing of published Choosing Wisely® recommendations, a dataset was generated incorporating all 320 recommendations comprising the 58 lists published through August, 2014; these include both the adult and pediatric hospital medicine lists released by the SHM.4,5,12 Although data collection ended at this point, this represents a majority of all 81 lists and 535 recommendations published through December, 2017. The reviewers (A.J.A., A.G., M.W., T.S.V., M.S., and C.R.C) extracted information about the references cited for each recommendation.
Data Analysis
The reviewers obtained each reference cited by a Choosing Wisely® recommendation and categorized it by evidence strength along the following hierarchy: clinical practice guideline (CPG), primary research, review article, expert opinion, book, or others/unknown. CPGs were used as the highest level of evidence based on standard expectations for methodological rigor.13 Primary research was further rated as follows: systematic reviews and meta-analyses, randomized controlled trials (RCTs), observational studies, and case series. Each recommendation was graded using only the strongest piece of evidence cited.
Guideline Appraisal
We further sought to evaluate the strength of referenced CPGs. To accomplish this, a 10% random sample of the Choosing Wisely® recommendations citing CPGs was selected, and the referenced CPGs were obtained. Separately, CPGs referenced by the SHM-published adult and pediatric lists were also obtained. For both groups, one CPG was randomly selected when a recommendation cited more than one CPG. These guidelines were assessed using the Appraisal of Guidelines for Research and Evaluation (AGREE) II instrument, a widely used instrument designed to assess CPG quality.14,15 AGREE II consists of 25 questions categorized into 6 domains: scope and purpose, stakeholder involvement, rigor of development, clarity of presentation, applicability, and editorial independence. Guidelines are also assigned an overall score. Two trained reviewers (A.J.A. and A.G.) assessed each of the sampled CPGs using a standardized form. Scores were then standardized using the method recommended by the instrument and reported as a percentage of available points. Although a standard interpretation of scores is not provided by the instrument, prior applications deemed scores below 50% as deficient16,17. When a recommendation item cited multiple CPGs, one was randomly selected. We also abstracted data on the year of publication, the evidence grade assigned to specific items recommended by Choosing Wisely®, and whether the CPG addressed the referring recommendation. All data management and analysis were conducted using Stata (V14.2, StataCorp, College Station, Texas).
RESULTS
A total of 320 recommendations were considered in our analysis, including 10 published across the 2 hospital medicine lists. When limited to the highest quality citation for each of the recommendations, 225 (70.3%) cited CPGs, whereas 71 (22.2%) cited primary research articles (Table 1). Specifically, 29 (9.1%) cited systematic reviews and meta-analyses, 28 (8.8%) cited observational studies, and 13 (4.1%) cited RCTs. One recommendation (0.3%) cited a case series as its highest level of evidence, 7 (2.2%) cited review articles, 7 (2.2%) cited editorials or opinion pieces, and 10 (3.1%) cited other types of documents, such as websites or books. Among hospital medicine recommendations, 9 (90%) referenced CPGs and 1 (10%) cited an observational study.
For the AGREE II assessment, we included 23 CPGs from the 225 referenced across all recommendations, after which we separately selected 6 CPGs from the hospital medicine recommendations. There was no overlap. Notably, 4 hospital medicine recommendations referenced a common CPG. Among the random sample of referenced CPGs, the median overall score obtained by using AGREE II was 54.2% (IQR 33.3%-70.8%, Table 2). This was similar to the median overall among hospital medicine guidelines (58.2%, IQR 50.0%-83.3%). Both hospital medicine and other sampled guidelines tended to score poorly in stakeholder involvement (48.6%, IQR 44.1%-61.1% and 47.2%, IQR 38.9%-61.1%, respectively). There were no significant differences between hospital medicine-referenced CPGs and the larger sample of CPGs in any AGREE II subdomains. The median age from the CPG publication to the list publication was 7 years (IQR 4–7) for hospital medicine recommendations and 3 years (IQR 2–6) for the nonhospital medicine recommendations. Substantial agreement was found between raters on the overall guideline assessment (ICC 0.80, 95% CI 0.58-0.91; Supplementary Table 1).
In terms of recommendation strengths and evidence grades, several recommendations were backed by Grades II–III (on a scale of I-III) evidence and level C (on a scale of A–C) recommendations in the reviewed CPG (Society of Maternal-Fetal Medicine, Recommendation 4, and Heart Rhythm Society, Recommendation 1). In one other case, the cited CPG did not directly address the Choosing Wisely® item (Society of Vascular Medicine, Recommendation 2).
DISCUSSION
Given the rising costs and the potential for iatrogenic harm, curbing ineffective practices has become an urgent concern. To achieve this, the Choosing Wisely® campaign has taken an important step by targeting certain low-value practices for de-adoption. However, the evidence supporting recommendations is variable. Specifically, 25 recommendations cited case series, review articles, or lower quality evidence as their highest level of support; moreover, among recommendations citing CPGs, quality, timeliness, and support for the recommendation item were variable. Although the hospital medicine lists tended to cite higher-quality evidence in the form of CPGs, these CPGs were often less recent than the guidelines referenced by other lists.
Our findings parallel those of other works that evaluate evidence among Choosing Wisely® recommendations and, more broadly, among CPGs.18–21 Lin and Yancey evaluated the quality of primary care-focused Choosing Wisely® recommendations using the Strength of Recommendation Taxonomy, a ranking system that evaluates evidence quality, consistency, and patient-centeredness.18 In their analysis, the authors found that many recommendations were based on lower quality evidence or relied on nonpatent-centered intermediate outcomes. Several groups, meanwhile, have evaluated the quality of evidence supporting CPG recommendations, finding them to be highly variable as well.19–21 These findings likely reflect inherent difficulties in the process, by which guideline development groups distill a broad evidence base into useful clinical recommendations, a reality that may have influenced the Choosing Wisely® list development groups seeking to make similar recommendations on low-value services.
These data should be taken in context due to several limitations. First, our sample of referenced CPGs includes only a small sample of all CPGs cited; thus, it may not be representative of all referenced guidelines. Second, the AGREE II assessment is inherently subjective, despite the availability of training materials. Third, data collection ended in April, 2014. Although this represents a majority of published lists to date, it is possible that more recent Choosing Wisely®lists include a stronger focus on evidence quality. Finally, references cited by Choosing Wisely®may not be representative of the entirety of the dataset that was considered when formulating the recommendations.
Despite these limitations, our findings suggest that Choosing Wisely®recommendations vary in terms of evidence strength. Although our results reveal that the majority of recommendations cite guidelines or high-quality original research, evidence gaps remain, with a small number citing low-quality evidence or low-quality CPGs as their highest form of support. Given the barriers to the successful de-implementation of low-value services, such campaigns as Choosing Wisely®face an uphill battle in their attempt to prompt behavior changes among providers and consumers.6-9 As a result, it is incumbent on funding agencies and medical journals to promote studies evaluating the harms and overall value of the care we deliver.
CONCLUSIONS
Although a majority of Choosing Wisely® recommendations cite high-quality evidence, some reference low-quality evidence or low-quality CPGs as their highest form of support. To overcome clinical inertia and other barriers to the successful de-implementation of low-value services, a clear rationale for the impetus to eradicate entrenched practices is critical.2,22 Choosing Wisely® has provided visionary leadership and a powerful platform to question low-value care. To expand the campaign’s efforts, the medical field must be able to generate the high-quality evidence necessary to support these efforts; further, list development groups must consider the availability of strong evidence when targeting services for de-implementation.
ACKNOWLEDGMENT
This work was supported, in part, by a grant from the Agency for Healthcare Research and Quality (No. K08HS020672, Dr. Cooke).
Disclosures
The authors have nothing to disclose.
As healthcare costs rise, physicians and other stakeholders are now seeking innovative and effective ways to reduce the provision of low-value services.1,2 The Choosing Wisely® campaign aims to further this goal by promoting lists of specific procedures, tests, and treatments that providers should avoid in selected clinical settings.3 On February 21, 2013, the Society of Hospital Medicine (SHM) released 2 Choosing Wisely® lists consisting of adult and pediatric services that are seen as costly to consumers and to the healthcare system, but which are often nonbeneficial or even harmful.4,5 A total of 80 physician and nurse specialty societies have joined in submitting additional lists.
Despite the growing enthusiasm for this effort, questions remain regarding the Choosing Wisely® campaign’s ability to initiate the meaningful de-adoption of low-value services. Specifically, prior efforts to reduce the use of services deemed to be of questionable benefit have met several challenges.2,6 Early analyses of the Choosing Wisely® recommendations reveal similar roadblocks and variable uptakes of several recommendations.7-10 While the reasons for difficulties in achieving de-adoption are broad, one important factor in whether clinicians are willing to follow guideline recommendations from such initiatives as Choosing Wisely®is the extent to which they believe in the underlying evidence.11 The current work seeks to formally evaluate the evidence supporting the Choosing Wisely® recommendations, and to compare the quality of evidence supporting SHM lists to other published Choosing Wisely® lists.
METHODS
Data Sources
Using the online listing of published Choosing Wisely® recommendations, a dataset was generated incorporating all 320 recommendations comprising the 58 lists published through August, 2014; these include both the adult and pediatric hospital medicine lists released by the SHM.4,5,12 Although data collection ended at this point, this represents a majority of all 81 lists and 535 recommendations published through December, 2017. The reviewers (A.J.A., A.G., M.W., T.S.V., M.S., and C.R.C) extracted information about the references cited for each recommendation.
Data Analysis
The reviewers obtained each reference cited by a Choosing Wisely® recommendation and categorized it by evidence strength along the following hierarchy: clinical practice guideline (CPG), primary research, review article, expert opinion, book, or others/unknown. CPGs were used as the highest level of evidence based on standard expectations for methodological rigor.13 Primary research was further rated as follows: systematic reviews and meta-analyses, randomized controlled trials (RCTs), observational studies, and case series. Each recommendation was graded using only the strongest piece of evidence cited.
Guideline Appraisal
We further sought to evaluate the strength of referenced CPGs. To accomplish this, a 10% random sample of the Choosing Wisely® recommendations citing CPGs was selected, and the referenced CPGs were obtained. Separately, CPGs referenced by the SHM-published adult and pediatric lists were also obtained. For both groups, one CPG was randomly selected when a recommendation cited more than one CPG. These guidelines were assessed using the Appraisal of Guidelines for Research and Evaluation (AGREE) II instrument, a widely used instrument designed to assess CPG quality.14,15 AGREE II consists of 25 questions categorized into 6 domains: scope and purpose, stakeholder involvement, rigor of development, clarity of presentation, applicability, and editorial independence. Guidelines are also assigned an overall score. Two trained reviewers (A.J.A. and A.G.) assessed each of the sampled CPGs using a standardized form. Scores were then standardized using the method recommended by the instrument and reported as a percentage of available points. Although a standard interpretation of scores is not provided by the instrument, prior applications deemed scores below 50% as deficient16,17. When a recommendation item cited multiple CPGs, one was randomly selected. We also abstracted data on the year of publication, the evidence grade assigned to specific items recommended by Choosing Wisely®, and whether the CPG addressed the referring recommendation. All data management and analysis were conducted using Stata (V14.2, StataCorp, College Station, Texas).
RESULTS
A total of 320 recommendations were considered in our analysis, including 10 published across the 2 hospital medicine lists. When limited to the highest quality citation for each of the recommendations, 225 (70.3%) cited CPGs, whereas 71 (22.2%) cited primary research articles (Table 1). Specifically, 29 (9.1%) cited systematic reviews and meta-analyses, 28 (8.8%) cited observational studies, and 13 (4.1%) cited RCTs. One recommendation (0.3%) cited a case series as its highest level of evidence, 7 (2.2%) cited review articles, 7 (2.2%) cited editorials or opinion pieces, and 10 (3.1%) cited other types of documents, such as websites or books. Among hospital medicine recommendations, 9 (90%) referenced CPGs and 1 (10%) cited an observational study.
For the AGREE II assessment, we included 23 CPGs from the 225 referenced across all recommendations, after which we separately selected 6 CPGs from the hospital medicine recommendations. There was no overlap. Notably, 4 hospital medicine recommendations referenced a common CPG. Among the random sample of referenced CPGs, the median overall score obtained by using AGREE II was 54.2% (IQR 33.3%-70.8%, Table 2). This was similar to the median overall among hospital medicine guidelines (58.2%, IQR 50.0%-83.3%). Both hospital medicine and other sampled guidelines tended to score poorly in stakeholder involvement (48.6%, IQR 44.1%-61.1% and 47.2%, IQR 38.9%-61.1%, respectively). There were no significant differences between hospital medicine-referenced CPGs and the larger sample of CPGs in any AGREE II subdomains. The median age from the CPG publication to the list publication was 7 years (IQR 4–7) for hospital medicine recommendations and 3 years (IQR 2–6) for the nonhospital medicine recommendations. Substantial agreement was found between raters on the overall guideline assessment (ICC 0.80, 95% CI 0.58-0.91; Supplementary Table 1).
In terms of recommendation strengths and evidence grades, several recommendations were backed by Grades II–III (on a scale of I-III) evidence and level C (on a scale of A–C) recommendations in the reviewed CPG (Society of Maternal-Fetal Medicine, Recommendation 4, and Heart Rhythm Society, Recommendation 1). In one other case, the cited CPG did not directly address the Choosing Wisely® item (Society of Vascular Medicine, Recommendation 2).
DISCUSSION
Given the rising costs and the potential for iatrogenic harm, curbing ineffective practices has become an urgent concern. To achieve this, the Choosing Wisely® campaign has taken an important step by targeting certain low-value practices for de-adoption. However, the evidence supporting recommendations is variable. Specifically, 25 recommendations cited case series, review articles, or lower quality evidence as their highest level of support; moreover, among recommendations citing CPGs, quality, timeliness, and support for the recommendation item were variable. Although the hospital medicine lists tended to cite higher-quality evidence in the form of CPGs, these CPGs were often less recent than the guidelines referenced by other lists.
Our findings parallel those of other works that evaluate evidence among Choosing Wisely® recommendations and, more broadly, among CPGs.18–21 Lin and Yancey evaluated the quality of primary care-focused Choosing Wisely® recommendations using the Strength of Recommendation Taxonomy, a ranking system that evaluates evidence quality, consistency, and patient-centeredness.18 In their analysis, the authors found that many recommendations were based on lower quality evidence or relied on nonpatent-centered intermediate outcomes. Several groups, meanwhile, have evaluated the quality of evidence supporting CPG recommendations, finding them to be highly variable as well.19–21 These findings likely reflect inherent difficulties in the process, by which guideline development groups distill a broad evidence base into useful clinical recommendations, a reality that may have influenced the Choosing Wisely® list development groups seeking to make similar recommendations on low-value services.
These data should be taken in context due to several limitations. First, our sample of referenced CPGs includes only a small sample of all CPGs cited; thus, it may not be representative of all referenced guidelines. Second, the AGREE II assessment is inherently subjective, despite the availability of training materials. Third, data collection ended in April, 2014. Although this represents a majority of published lists to date, it is possible that more recent Choosing Wisely®lists include a stronger focus on evidence quality. Finally, references cited by Choosing Wisely®may not be representative of the entirety of the dataset that was considered when formulating the recommendations.
Despite these limitations, our findings suggest that Choosing Wisely®recommendations vary in terms of evidence strength. Although our results reveal that the majority of recommendations cite guidelines or high-quality original research, evidence gaps remain, with a small number citing low-quality evidence or low-quality CPGs as their highest form of support. Given the barriers to the successful de-implementation of low-value services, such campaigns as Choosing Wisely®face an uphill battle in their attempt to prompt behavior changes among providers and consumers.6-9 As a result, it is incumbent on funding agencies and medical journals to promote studies evaluating the harms and overall value of the care we deliver.
CONCLUSIONS
Although a majority of Choosing Wisely® recommendations cite high-quality evidence, some reference low-quality evidence or low-quality CPGs as their highest form of support. To overcome clinical inertia and other barriers to the successful de-implementation of low-value services, a clear rationale for the impetus to eradicate entrenched practices is critical.2,22 Choosing Wisely® has provided visionary leadership and a powerful platform to question low-value care. To expand the campaign’s efforts, the medical field must be able to generate the high-quality evidence necessary to support these efforts; further, list development groups must consider the availability of strong evidence when targeting services for de-implementation.
ACKNOWLEDGMENT
This work was supported, in part, by a grant from the Agency for Healthcare Research and Quality (No. K08HS020672, Dr. Cooke).
Disclosures
The authors have nothing to disclose.
1. Institute of Medicine Roundtable on Evidence-Based Medicine. The Healthcare Imperative: Lowering Costs and Improving Outcomes: Workshop Series Summary. Yong P, Saudners R, Olsen L, editors. Washington, D.C.: National Academies Press; 2010. PubMed
2. Weinberger SE. Providing high-value, cost-conscious care: a critical seventh general competency for physicians. Ann Intern Med. 2011;155(6):386-388. PubMed
3. Cassel CK, Guest JA. Choosing wisely: Helping physicians and patients make smart decisions about their care. JAMA. 2012;307(17):1801-1802. PubMed
4. Bulger J, Nickel W, Messler J, Goldstein J, O’Callaghan J, Auron M, et al. Choosing wisely in adult hospital medicine: Five opportunities for improved healthcare value. J Hosp Med. 2013;8(9):486-492. PubMed
5. Quinonez RA, Garber MD, Schroeder AR, Alverson BK, Nickel W, Goldstein J, et al. Choosing wisely in pediatric hospital medicine: Five opportunities for improved healthcare value. J Hosp Med. 2013;8(9):479-485. PubMed
6. Prasad V, Ioannidis JP. Evidence-based de-implementation for contradicted, unproven, and aspiring healthcare practices. Implement Sci. 2014;9:1. PubMed
7. Rosenberg A, Agiro A, Gottlieb M, Barron J, Brady P, Liu Y, et al. Early trends among seven recommendations from the Choosing Wisely campaign. JAMA Intern Med. 2015;175(12):1913-1920. PubMed
8. Zikmund-Fisher BJ, Kullgren JT, Fagerlin A, Klamerus ML, Bernstein SJ, Kerr EA. Perceived barriers to implementing individual Choosing Wisely® recommendations in two national surveys of primary care providers. J Gen Intern Med. 2017;32(2):210-217. PubMed
9. Bishop TF, Cea M, Miranda Y, Kim R, Lash-Dardia M, Lee JI, et al. Academic physicians’ views on low-value services and the choosing wisely campaign: A qualitative study. Healthc (Amsterdam, Netherlands). 2017;5(1-2):17-22. PubMed
10. Prochaska MT, Hohmann SF, Modes M, Arora VM. Trends in Troponin-only testing for AMI in academic teaching hospitals and the impact of Choosing Wisely®. J Hosp Med. 2017;12(12):957-962. PubMed
11. Cabana MD, Rand CS, Powe NR, Wu AW, Wilson MH, Abboud PA, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA. 1999;282(15):1458-1465. PubMed
12. ABIM Foundation. ChoosingWisely.org Search Recommendations. 2014.
13. Institute of Medicine (US) Committee on Standards for Developing Trustworthy Clinical Practice Guidelines. Clinical Practice Guidelines We Can Trust. Graham R, Mancher M, Miller Wolman D, Greenfield S, Steinberg E, editors. Washington, D.C.: National Academies Press; 2011. PubMed
14. Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. AGREE II: Advancing guideline development, reporting, and evaluation in health care. Prev Med (Baltim). 2010;51(5):421-424. PubMed
15. Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. Development of the AGREE II, part 2: Assessment of validity of items and tools to support application. CMAJ. 2010;182(10):E472-E478. PubMed
16. He Z, Tian H, Song A, Jin L, Zhou X, Liu X, et al. Quality appraisal of clinical practice guidelines on pancreatic cancer. Medicine (Baltimore). 2015;94(12):e635. PubMed
17. Isaac A, Saginur M, Hartling L, Robinson JL. Quality of reporting and evidence in American Academy of Pediatrics guidelines. Pediatrics. 2013;131(4):732-738. PubMed
18. Lin KW, Yancey JR. Evaluating the Evidence for Choosing WiselyTM in Primary Care Using the Strength of Recommendation Taxonomy (SORT). J Am Board Fam Med. 2016;29(4):512-515. PubMed
19. McAlister FA, van Diepen S, Padwal RS, Johnson JA, Majumdar SR. How evidence-based are the recommendations in evidence-based guidelines? PLoS Med. 2007;4(8):e250. PubMed
20. Tricoci P, Allen JM, Kramer JM, Califf RM, Smith SC. Scientific evidence underlying the ACC/AHA clinical practice guidelines. JAMA. 2009;301(8):831-841. PubMed
21. Feuerstein JD, Gifford AE, Akbari M, Goldman J, Leffler DA, Sheth SG, et al. Systematic analysis underlying the quality of the scientific evidence and conflicts of interest in gastroenterology practice guidelines. Am J Gastroenterol. 2013;108(11):1686-1693. PubMed
22. Robert G, Harlock J, Williams I. Disentangling rhetoric and reality: an international Delphi study of factors and processes that facilitate the successful implementation of decisions to decommission healthcare services. Implement Sci. 2014;9:123. PubMed
1. Institute of Medicine Roundtable on Evidence-Based Medicine. The Healthcare Imperative: Lowering Costs and Improving Outcomes: Workshop Series Summary. Yong P, Saudners R, Olsen L, editors. Washington, D.C.: National Academies Press; 2010. PubMed
2. Weinberger SE. Providing high-value, cost-conscious care: a critical seventh general competency for physicians. Ann Intern Med. 2011;155(6):386-388. PubMed
3. Cassel CK, Guest JA. Choosing wisely: Helping physicians and patients make smart decisions about their care. JAMA. 2012;307(17):1801-1802. PubMed
4. Bulger J, Nickel W, Messler J, Goldstein J, O’Callaghan J, Auron M, et al. Choosing wisely in adult hospital medicine: Five opportunities for improved healthcare value. J Hosp Med. 2013;8(9):486-492. PubMed
5. Quinonez RA, Garber MD, Schroeder AR, Alverson BK, Nickel W, Goldstein J, et al. Choosing wisely in pediatric hospital medicine: Five opportunities for improved healthcare value. J Hosp Med. 2013;8(9):479-485. PubMed
6. Prasad V, Ioannidis JP. Evidence-based de-implementation for contradicted, unproven, and aspiring healthcare practices. Implement Sci. 2014;9:1. PubMed
7. Rosenberg A, Agiro A, Gottlieb M, Barron J, Brady P, Liu Y, et al. Early trends among seven recommendations from the Choosing Wisely campaign. JAMA Intern Med. 2015;175(12):1913-1920. PubMed
8. Zikmund-Fisher BJ, Kullgren JT, Fagerlin A, Klamerus ML, Bernstein SJ, Kerr EA. Perceived barriers to implementing individual Choosing Wisely® recommendations in two national surveys of primary care providers. J Gen Intern Med. 2017;32(2):210-217. PubMed
9. Bishop TF, Cea M, Miranda Y, Kim R, Lash-Dardia M, Lee JI, et al. Academic physicians’ views on low-value services and the choosing wisely campaign: A qualitative study. Healthc (Amsterdam, Netherlands). 2017;5(1-2):17-22. PubMed
10. Prochaska MT, Hohmann SF, Modes M, Arora VM. Trends in Troponin-only testing for AMI in academic teaching hospitals and the impact of Choosing Wisely®. J Hosp Med. 2017;12(12):957-962. PubMed
11. Cabana MD, Rand CS, Powe NR, Wu AW, Wilson MH, Abboud PA, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA. 1999;282(15):1458-1465. PubMed
12. ABIM Foundation. ChoosingWisely.org Search Recommendations. 2014.
13. Institute of Medicine (US) Committee on Standards for Developing Trustworthy Clinical Practice Guidelines. Clinical Practice Guidelines We Can Trust. Graham R, Mancher M, Miller Wolman D, Greenfield S, Steinberg E, editors. Washington, D.C.: National Academies Press; 2011. PubMed
14. Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. AGREE II: Advancing guideline development, reporting, and evaluation in health care. Prev Med (Baltim). 2010;51(5):421-424. PubMed
15. Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. Development of the AGREE II, part 2: Assessment of validity of items and tools to support application. CMAJ. 2010;182(10):E472-E478. PubMed
16. He Z, Tian H, Song A, Jin L, Zhou X, Liu X, et al. Quality appraisal of clinical practice guidelines on pancreatic cancer. Medicine (Baltimore). 2015;94(12):e635. PubMed
17. Isaac A, Saginur M, Hartling L, Robinson JL. Quality of reporting and evidence in American Academy of Pediatrics guidelines. Pediatrics. 2013;131(4):732-738. PubMed
18. Lin KW, Yancey JR. Evaluating the Evidence for Choosing WiselyTM in Primary Care Using the Strength of Recommendation Taxonomy (SORT). J Am Board Fam Med. 2016;29(4):512-515. PubMed
19. McAlister FA, van Diepen S, Padwal RS, Johnson JA, Majumdar SR. How evidence-based are the recommendations in evidence-based guidelines? PLoS Med. 2007;4(8):e250. PubMed
20. Tricoci P, Allen JM, Kramer JM, Califf RM, Smith SC. Scientific evidence underlying the ACC/AHA clinical practice guidelines. JAMA. 2009;301(8):831-841. PubMed
21. Feuerstein JD, Gifford AE, Akbari M, Goldman J, Leffler DA, Sheth SG, et al. Systematic analysis underlying the quality of the scientific evidence and conflicts of interest in gastroenterology practice guidelines. Am J Gastroenterol. 2013;108(11):1686-1693. PubMed
22. Robert G, Harlock J, Williams I. Disentangling rhetoric and reality: an international Delphi study of factors and processes that facilitate the successful implementation of decisions to decommission healthcare services. Implement Sci. 2014;9:123. PubMed
© 2018 Society of Hospital Medicine
Patient Perceptions of Readmission Risk: An Exploratory Survey
Recent years have seen a proliferation of programs designed to prevent readmissions, including patient education initiatives, financial assistance programs, postdischarge services, and clinical personnel assigned to help patients navigate their posthospitalization clinical care. Although some strategies do not require direct patient participation (such as timely and effective handoffs between inpatient and outpatient care teams), many rely upon a commitment by the patient to participate in the postdischarge care plan. At our hospital, we have found that only about 2/3 of patients who are offered transitional interventions (such as postdischarge phone calls by nurses or home nursing through a “transition guide” program) receive the intended interventions, and those who do not receive them are more likely to be readmitted.1 While limited patient uptake may relate, in part, to factors that are difficult to overcome, such as inadequate housing or phone service, we have also encountered patients whose values, beliefs, or preferences about their care do not align with those of the care team. The purposes of this exploratory study were to (1) assess patient attitudes surrounding readmission, (2) ascertain whether these attitudes are associated with actual readmission, and (3) determine whether patients can estimate their own risk of readmission.
METHODS
From January 2014 to September 2016, we circulated surveys to patients on internal medicine nursing units who were being discharged home within 24 hours. Blank surveys were distributed to nursing units by the researchers. Unit clerks and support staff were educated on the purpose of the project and asked to distribute surveys to patients who were identified by unit case managers or nurses as slated for discharge. Staff members were not asked to help with or supervise survey completion. Surveys were generally filled out by patients, but we allowed family members to assist patients if needed, and to indicate so with a checkbox. There were no exclusion criteria. Because surveys were distributed by clinical staff, the received surveys can be considered a convenience sample. Patients were asked 5 questions with 4- or 5-point Likert scale responses:
(1) “How likely is it that you will be admitted to the hospital (have to stay in the hospital overnight) again within the next 30 days after you leave the hospital this time?” [answers ranging from “Very Unlikely (<5% chance)” to “Very Likely (>50% chance)”];
(2) “How would you feel about being rehospitalized in the next month?” [answers ranging from “Very sad, frustrated, or disappointed” to “Very happy or relieved”];
(3) “How much do you think that you personally can control whether or not you will be rehospitalized (based on what you do to take care of your body, take your medicines, and follow-up with your healthcare team)?” [answers ranging from “I have no control over whether I will be rehospitalized” to “I have complete control over whether I will be rehospitalized”];
(4) “Which of the options below best describes how you plan to follow the medical instructions after you leave the hospital?” [answers ranging from “I do NOT plan to do very much of what I am being asked to do by the doctors, nurses, therapists, and other members of the care team” to “I plan to do EVERYTHING I am being asked to do by the doctors, nurses, therapists and other members of the care team”]; and
(5) “Pick the item below that best describes YOUR OWN VIEW of the care team’s recommendations:” [answers ranging from “I DO NOT AGREE AT ALL that the best way to be healthy is to do exactly what I am being asked to do by the doctors, nurses, therapists, and other members of the care team” to “I FULLY AGREE that the best way to be healthy is to do exactly what I am being asked to do by the doctors, nurses, therapists, and other members of the care team”].
Responses were linked, based on discharge date and medical record number, to administrative data, including age, sex, race, payer, and clinical data. Subsequent hospitalizations to our hospital were ascertained from administrative data. We estimated expected risk of readmission using the all payer refined diagnosis related group coupled with the associated severity-of-illness (SOI) score, as we have reported previously.2-5 We restricted our analysis to patients who answered the question related to the likelihood of readmission. Logistic regression models were constructed using actual 30-day readmission as the dependent variable to determine whether patients could predict their own readmissions and whether patient attitudes and beliefs about their care were predictive of subsequent readmission. Patient survey responses were entered as continuous independent variables (ranging from 1-4 or 1-5, as appropriate). Multivariable logistic regression was used to determine whether patients could predict their readmissions independent of demographic variables and expected readmission rate (modeled continuously); we repeated this model after dichotomizing the patient’s estimate of the likelihood of readmission as either “unlikely” or “likely.” Patients with missing survey responses were excluded from individual models without imputation. The study was approved by the Johns Hopkins institutional review board.
RESULTS
Responses were obtained from 895 patients. Their median age was 56 years [interquartile range, 43-67], 51.4% were female, and 41.7% were white. Mean SOI was 2.53 (on a 1-4 scale), and median length-of-stay was representative for our medical service at 5.2 days (range, 1-66 days). Family members reported filling out the survey in 57 cases. The primary payer was Medicare in 40.7%, Medicaid in 24.9%, and other in 34.4%. A total of 138 patients (15.4%) were readmitted within 30 days. The Table shows survey responses and associated readmission rates. None of the attitudes related to readmission were predictive of actual readmission. However, patients were able to predict their own readmissions (P = .002 for linear trend). After adjustment for expected readmission rate, race, sex, age, and payer, the trend remained significant (P = .005). Other significant predictors of readmissions in this model included expected readmission rate (P = .002), age (P = .02), and payer (P = .002). After dichotomizing the patient estimate of readmission rate as “unlikely” (N = 581) or “likely” (N = 314), the unadjusted odds ratio associating a patient-estimated risk of readmission as “likely” with actual readmission was 1.8 (95% confidence interval, 1.2-2.5). The adjusted odds ratio (including the variables above) was 1.6 (1.1-2.4).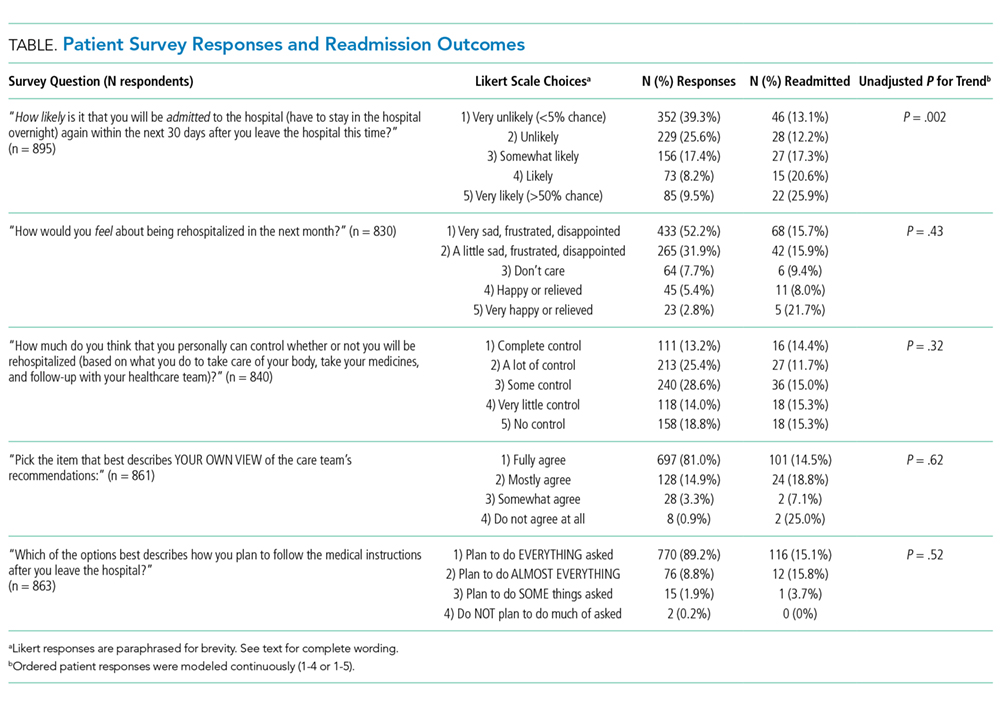
DISCUSSION
Our findings demonstrate that patients are able to quantify their own readmission risk. This was true even after adjustment for expected readmission rate, age, sex, race, and payer. However, we did not identify any patient attitudes, beliefs, or preferences related to readmission or discharge instructions that were associated with subsequent rehospitalization. Reassuringly, more than 80% of patients who responded to the survey indicated that they would be sad, frustrated, or disappointed should readmission occur. This suggests that most patients are invested in preventing rehospitalization. Also reassuring was that patients indicated that they agreed with the discharge care plan and intended to follow their discharge instructions.
The major limitation of this study is that it was a convenience sample. Surveys were distributed inconsistently by nursing unit staff, preventing us from calculating a response rate. Further, it is possible, if not likely, that those patients with higher levels of engagement were more likely to take the time to respond, enriching our sample with activated patients. Although we allowed family members to fill out surveys on behalf of patients, this was done in fewer than 10% of instances; as such, our data may have limited applicability to patients who are physically or cognitively unable to participate in the discharge process. Finally, in this study, we did not capture readmissions to other facilities.
We conclude that patients are able to predict their own readmissions, even after accounting for other potential predictors of readmission. However, we found no evidence to support the possibility that low levels of engagement, limited trust in the healthcare team, or nonchalance about being readmitted are associated with subsequent rehospitalization. Whether asking patients about their perceived risk of readmission might help target readmission prevention programs deserves further study.
Acknowledgments
Dr. Daniel J. Brotman had full access to the data in the study and takes responsibility for the integrity of the study data and the accuracy of the data analysis. The authors also thank the following individuals for their contributions: Drafting the manuscript (Brotman); revising the manuscript for important intellectual content (Brotman, Shihab, Tieu, Cheng, Bertram, Hoyer, Deutschendorf); acquiring the data (Brotman, Shihab, Tieu, Cheng, Bertram, Deutschendorf); interpreting the data (Brotman, Shihab, Tieu, Cheng, Bertram, Hoyer, Deutschendorf); and analyzing the data (Brotman). The authors thank nursing leadership and nursing unit staff for their assistance in distributing surveys.
Funding support: Johns Hopkins Hospitalist Scholars Program
Disclosures: The authors have declared no conflicts of interest.
1. Hoyer EH, Brotman DJ, Apfel A, et al. Improving outcomes after hospitalization: a prospective observational multi-center evaluation of care-coordination strategies on 30-day readmissions to Maryland hospitals. J Gen Int Med. 2017 (in press). PubMed
2. Oduyebo I, Lehmann CU, Pollack CE, et al. Association of self-reported hospital discharge handoffs with 30-day readmissions. JAMA Intern Med. 2013;173(8):624-629. PubMed
3. Hoyer EH, Needham DM, Atanelov L, Knox B, Friedman M, Brotman DJ. Association of impaired functional status at hospital discharge and subsequent rehospitalization. J Hosp Med. 2014;9(5):277-282. PubMed
4. Hoyer EH, Needham DM, Miller J, Deutschendorf A, Friedman M, Brotman DJ. Functional status impairment is associated with unplanned readmissions. Arch Phys Med Rehabil. 2013;94(10):1951-1958. PubMed
5. Hoyer EH, Odonkor CA, Bhatia SN, Leung C, Deutschendorf A, Brotman DJ. Association between days to complete inpatient discharge summaries with all-payer hospital readmissions in Maryland. J Hosp Med. 2016;11(6):393-400. PubMed
Recent years have seen a proliferation of programs designed to prevent readmissions, including patient education initiatives, financial assistance programs, postdischarge services, and clinical personnel assigned to help patients navigate their posthospitalization clinical care. Although some strategies do not require direct patient participation (such as timely and effective handoffs between inpatient and outpatient care teams), many rely upon a commitment by the patient to participate in the postdischarge care plan. At our hospital, we have found that only about 2/3 of patients who are offered transitional interventions (such as postdischarge phone calls by nurses or home nursing through a “transition guide” program) receive the intended interventions, and those who do not receive them are more likely to be readmitted.1 While limited patient uptake may relate, in part, to factors that are difficult to overcome, such as inadequate housing or phone service, we have also encountered patients whose values, beliefs, or preferences about their care do not align with those of the care team. The purposes of this exploratory study were to (1) assess patient attitudes surrounding readmission, (2) ascertain whether these attitudes are associated with actual readmission, and (3) determine whether patients can estimate their own risk of readmission.
METHODS
From January 2014 to September 2016, we circulated surveys to patients on internal medicine nursing units who were being discharged home within 24 hours. Blank surveys were distributed to nursing units by the researchers. Unit clerks and support staff were educated on the purpose of the project and asked to distribute surveys to patients who were identified by unit case managers or nurses as slated for discharge. Staff members were not asked to help with or supervise survey completion. Surveys were generally filled out by patients, but we allowed family members to assist patients if needed, and to indicate so with a checkbox. There were no exclusion criteria. Because surveys were distributed by clinical staff, the received surveys can be considered a convenience sample. Patients were asked 5 questions with 4- or 5-point Likert scale responses:
(1) “How likely is it that you will be admitted to the hospital (have to stay in the hospital overnight) again within the next 30 days after you leave the hospital this time?” [answers ranging from “Very Unlikely (<5% chance)” to “Very Likely (>50% chance)”];
(2) “How would you feel about being rehospitalized in the next month?” [answers ranging from “Very sad, frustrated, or disappointed” to “Very happy or relieved”];
(3) “How much do you think that you personally can control whether or not you will be rehospitalized (based on what you do to take care of your body, take your medicines, and follow-up with your healthcare team)?” [answers ranging from “I have no control over whether I will be rehospitalized” to “I have complete control over whether I will be rehospitalized”];
(4) “Which of the options below best describes how you plan to follow the medical instructions after you leave the hospital?” [answers ranging from “I do NOT plan to do very much of what I am being asked to do by the doctors, nurses, therapists, and other members of the care team” to “I plan to do EVERYTHING I am being asked to do by the doctors, nurses, therapists and other members of the care team”]; and
(5) “Pick the item below that best describes YOUR OWN VIEW of the care team’s recommendations:” [answers ranging from “I DO NOT AGREE AT ALL that the best way to be healthy is to do exactly what I am being asked to do by the doctors, nurses, therapists, and other members of the care team” to “I FULLY AGREE that the best way to be healthy is to do exactly what I am being asked to do by the doctors, nurses, therapists, and other members of the care team”].
Responses were linked, based on discharge date and medical record number, to administrative data, including age, sex, race, payer, and clinical data. Subsequent hospitalizations to our hospital were ascertained from administrative data. We estimated expected risk of readmission using the all payer refined diagnosis related group coupled with the associated severity-of-illness (SOI) score, as we have reported previously.2-5 We restricted our analysis to patients who answered the question related to the likelihood of readmission. Logistic regression models were constructed using actual 30-day readmission as the dependent variable to determine whether patients could predict their own readmissions and whether patient attitudes and beliefs about their care were predictive of subsequent readmission. Patient survey responses were entered as continuous independent variables (ranging from 1-4 or 1-5, as appropriate). Multivariable logistic regression was used to determine whether patients could predict their readmissions independent of demographic variables and expected readmission rate (modeled continuously); we repeated this model after dichotomizing the patient’s estimate of the likelihood of readmission as either “unlikely” or “likely.” Patients with missing survey responses were excluded from individual models without imputation. The study was approved by the Johns Hopkins institutional review board.
RESULTS
Responses were obtained from 895 patients. Their median age was 56 years [interquartile range, 43-67], 51.4% were female, and 41.7% were white. Mean SOI was 2.53 (on a 1-4 scale), and median length-of-stay was representative for our medical service at 5.2 days (range, 1-66 days). Family members reported filling out the survey in 57 cases. The primary payer was Medicare in 40.7%, Medicaid in 24.9%, and other in 34.4%. A total of 138 patients (15.4%) were readmitted within 30 days. The Table shows survey responses and associated readmission rates. None of the attitudes related to readmission were predictive of actual readmission. However, patients were able to predict their own readmissions (P = .002 for linear trend). After adjustment for expected readmission rate, race, sex, age, and payer, the trend remained significant (P = .005). Other significant predictors of readmissions in this model included expected readmission rate (P = .002), age (P = .02), and payer (P = .002). After dichotomizing the patient estimate of readmission rate as “unlikely” (N = 581) or “likely” (N = 314), the unadjusted odds ratio associating a patient-estimated risk of readmission as “likely” with actual readmission was 1.8 (95% confidence interval, 1.2-2.5). The adjusted odds ratio (including the variables above) was 1.6 (1.1-2.4).
DISCUSSION
Our findings demonstrate that patients are able to quantify their own readmission risk. This was true even after adjustment for expected readmission rate, age, sex, race, and payer. However, we did not identify any patient attitudes, beliefs, or preferences related to readmission or discharge instructions that were associated with subsequent rehospitalization. Reassuringly, more than 80% of patients who responded to the survey indicated that they would be sad, frustrated, or disappointed should readmission occur. This suggests that most patients are invested in preventing rehospitalization. Also reassuring was that patients indicated that they agreed with the discharge care plan and intended to follow their discharge instructions.
The major limitation of this study is that it was a convenience sample. Surveys were distributed inconsistently by nursing unit staff, preventing us from calculating a response rate. Further, it is possible, if not likely, that those patients with higher levels of engagement were more likely to take the time to respond, enriching our sample with activated patients. Although we allowed family members to fill out surveys on behalf of patients, this was done in fewer than 10% of instances; as such, our data may have limited applicability to patients who are physically or cognitively unable to participate in the discharge process. Finally, in this study, we did not capture readmissions to other facilities.
We conclude that patients are able to predict their own readmissions, even after accounting for other potential predictors of readmission. However, we found no evidence to support the possibility that low levels of engagement, limited trust in the healthcare team, or nonchalance about being readmitted are associated with subsequent rehospitalization. Whether asking patients about their perceived risk of readmission might help target readmission prevention programs deserves further study.
Acknowledgments
Dr. Daniel J. Brotman had full access to the data in the study and takes responsibility for the integrity of the study data and the accuracy of the data analysis. The authors also thank the following individuals for their contributions: Drafting the manuscript (Brotman); revising the manuscript for important intellectual content (Brotman, Shihab, Tieu, Cheng, Bertram, Hoyer, Deutschendorf); acquiring the data (Brotman, Shihab, Tieu, Cheng, Bertram, Deutschendorf); interpreting the data (Brotman, Shihab, Tieu, Cheng, Bertram, Hoyer, Deutschendorf); and analyzing the data (Brotman). The authors thank nursing leadership and nursing unit staff for their assistance in distributing surveys.
Funding support: Johns Hopkins Hospitalist Scholars Program
Disclosures: The authors have declared no conflicts of interest.
Recent years have seen a proliferation of programs designed to prevent readmissions, including patient education initiatives, financial assistance programs, postdischarge services, and clinical personnel assigned to help patients navigate their posthospitalization clinical care. Although some strategies do not require direct patient participation (such as timely and effective handoffs between inpatient and outpatient care teams), many rely upon a commitment by the patient to participate in the postdischarge care plan. At our hospital, we have found that only about 2/3 of patients who are offered transitional interventions (such as postdischarge phone calls by nurses or home nursing through a “transition guide” program) receive the intended interventions, and those who do not receive them are more likely to be readmitted.1 While limited patient uptake may relate, in part, to factors that are difficult to overcome, such as inadequate housing or phone service, we have also encountered patients whose values, beliefs, or preferences about their care do not align with those of the care team. The purposes of this exploratory study were to (1) assess patient attitudes surrounding readmission, (2) ascertain whether these attitudes are associated with actual readmission, and (3) determine whether patients can estimate their own risk of readmission.
METHODS
From January 2014 to September 2016, we circulated surveys to patients on internal medicine nursing units who were being discharged home within 24 hours. Blank surveys were distributed to nursing units by the researchers. Unit clerks and support staff were educated on the purpose of the project and asked to distribute surveys to patients who were identified by unit case managers or nurses as slated for discharge. Staff members were not asked to help with or supervise survey completion. Surveys were generally filled out by patients, but we allowed family members to assist patients if needed, and to indicate so with a checkbox. There were no exclusion criteria. Because surveys were distributed by clinical staff, the received surveys can be considered a convenience sample. Patients were asked 5 questions with 4- or 5-point Likert scale responses:
(1) “How likely is it that you will be admitted to the hospital (have to stay in the hospital overnight) again within the next 30 days after you leave the hospital this time?” [answers ranging from “Very Unlikely (<5% chance)” to “Very Likely (>50% chance)”];
(2) “How would you feel about being rehospitalized in the next month?” [answers ranging from “Very sad, frustrated, or disappointed” to “Very happy or relieved”];
(3) “How much do you think that you personally can control whether or not you will be rehospitalized (based on what you do to take care of your body, take your medicines, and follow-up with your healthcare team)?” [answers ranging from “I have no control over whether I will be rehospitalized” to “I have complete control over whether I will be rehospitalized”];
(4) “Which of the options below best describes how you plan to follow the medical instructions after you leave the hospital?” [answers ranging from “I do NOT plan to do very much of what I am being asked to do by the doctors, nurses, therapists, and other members of the care team” to “I plan to do EVERYTHING I am being asked to do by the doctors, nurses, therapists and other members of the care team”]; and
(5) “Pick the item below that best describes YOUR OWN VIEW of the care team’s recommendations:” [answers ranging from “I DO NOT AGREE AT ALL that the best way to be healthy is to do exactly what I am being asked to do by the doctors, nurses, therapists, and other members of the care team” to “I FULLY AGREE that the best way to be healthy is to do exactly what I am being asked to do by the doctors, nurses, therapists, and other members of the care team”].
Responses were linked, based on discharge date and medical record number, to administrative data, including age, sex, race, payer, and clinical data. Subsequent hospitalizations to our hospital were ascertained from administrative data. We estimated expected risk of readmission using the all payer refined diagnosis related group coupled with the associated severity-of-illness (SOI) score, as we have reported previously.2-5 We restricted our analysis to patients who answered the question related to the likelihood of readmission. Logistic regression models were constructed using actual 30-day readmission as the dependent variable to determine whether patients could predict their own readmissions and whether patient attitudes and beliefs about their care were predictive of subsequent readmission. Patient survey responses were entered as continuous independent variables (ranging from 1-4 or 1-5, as appropriate). Multivariable logistic regression was used to determine whether patients could predict their readmissions independent of demographic variables and expected readmission rate (modeled continuously); we repeated this model after dichotomizing the patient’s estimate of the likelihood of readmission as either “unlikely” or “likely.” Patients with missing survey responses were excluded from individual models without imputation. The study was approved by the Johns Hopkins institutional review board.
RESULTS
Responses were obtained from 895 patients. Their median age was 56 years [interquartile range, 43-67], 51.4% were female, and 41.7% were white. Mean SOI was 2.53 (on a 1-4 scale), and median length-of-stay was representative for our medical service at 5.2 days (range, 1-66 days). Family members reported filling out the survey in 57 cases. The primary payer was Medicare in 40.7%, Medicaid in 24.9%, and other in 34.4%. A total of 138 patients (15.4%) were readmitted within 30 days. The Table shows survey responses and associated readmission rates. None of the attitudes related to readmission were predictive of actual readmission. However, patients were able to predict their own readmissions (P = .002 for linear trend). After adjustment for expected readmission rate, race, sex, age, and payer, the trend remained significant (P = .005). Other significant predictors of readmissions in this model included expected readmission rate (P = .002), age (P = .02), and payer (P = .002). After dichotomizing the patient estimate of readmission rate as “unlikely” (N = 581) or “likely” (N = 314), the unadjusted odds ratio associating a patient-estimated risk of readmission as “likely” with actual readmission was 1.8 (95% confidence interval, 1.2-2.5). The adjusted odds ratio (including the variables above) was 1.6 (1.1-2.4).
DISCUSSION
Our findings demonstrate that patients are able to quantify their own readmission risk. This was true even after adjustment for expected readmission rate, age, sex, race, and payer. However, we did not identify any patient attitudes, beliefs, or preferences related to readmission or discharge instructions that were associated with subsequent rehospitalization. Reassuringly, more than 80% of patients who responded to the survey indicated that they would be sad, frustrated, or disappointed should readmission occur. This suggests that most patients are invested in preventing rehospitalization. Also reassuring was that patients indicated that they agreed with the discharge care plan and intended to follow their discharge instructions.
The major limitation of this study is that it was a convenience sample. Surveys were distributed inconsistently by nursing unit staff, preventing us from calculating a response rate. Further, it is possible, if not likely, that those patients with higher levels of engagement were more likely to take the time to respond, enriching our sample with activated patients. Although we allowed family members to fill out surveys on behalf of patients, this was done in fewer than 10% of instances; as such, our data may have limited applicability to patients who are physically or cognitively unable to participate in the discharge process. Finally, in this study, we did not capture readmissions to other facilities.
We conclude that patients are able to predict their own readmissions, even after accounting for other potential predictors of readmission. However, we found no evidence to support the possibility that low levels of engagement, limited trust in the healthcare team, or nonchalance about being readmitted are associated with subsequent rehospitalization. Whether asking patients about their perceived risk of readmission might help target readmission prevention programs deserves further study.
Acknowledgments
Dr. Daniel J. Brotman had full access to the data in the study and takes responsibility for the integrity of the study data and the accuracy of the data analysis. The authors also thank the following individuals for their contributions: Drafting the manuscript (Brotman); revising the manuscript for important intellectual content (Brotman, Shihab, Tieu, Cheng, Bertram, Hoyer, Deutschendorf); acquiring the data (Brotman, Shihab, Tieu, Cheng, Bertram, Deutschendorf); interpreting the data (Brotman, Shihab, Tieu, Cheng, Bertram, Hoyer, Deutschendorf); and analyzing the data (Brotman). The authors thank nursing leadership and nursing unit staff for their assistance in distributing surveys.
Funding support: Johns Hopkins Hospitalist Scholars Program
Disclosures: The authors have declared no conflicts of interest.
1. Hoyer EH, Brotman DJ, Apfel A, et al. Improving outcomes after hospitalization: a prospective observational multi-center evaluation of care-coordination strategies on 30-day readmissions to Maryland hospitals. J Gen Int Med. 2017 (in press). PubMed
2. Oduyebo I, Lehmann CU, Pollack CE, et al. Association of self-reported hospital discharge handoffs with 30-day readmissions. JAMA Intern Med. 2013;173(8):624-629. PubMed
3. Hoyer EH, Needham DM, Atanelov L, Knox B, Friedman M, Brotman DJ. Association of impaired functional status at hospital discharge and subsequent rehospitalization. J Hosp Med. 2014;9(5):277-282. PubMed
4. Hoyer EH, Needham DM, Miller J, Deutschendorf A, Friedman M, Brotman DJ. Functional status impairment is associated with unplanned readmissions. Arch Phys Med Rehabil. 2013;94(10):1951-1958. PubMed
5. Hoyer EH, Odonkor CA, Bhatia SN, Leung C, Deutschendorf A, Brotman DJ. Association between days to complete inpatient discharge summaries with all-payer hospital readmissions in Maryland. J Hosp Med. 2016;11(6):393-400. PubMed
1. Hoyer EH, Brotman DJ, Apfel A, et al. Improving outcomes after hospitalization: a prospective observational multi-center evaluation of care-coordination strategies on 30-day readmissions to Maryland hospitals. J Gen Int Med. 2017 (in press). PubMed
2. Oduyebo I, Lehmann CU, Pollack CE, et al. Association of self-reported hospital discharge handoffs with 30-day readmissions. JAMA Intern Med. 2013;173(8):624-629. PubMed
3. Hoyer EH, Needham DM, Atanelov L, Knox B, Friedman M, Brotman DJ. Association of impaired functional status at hospital discharge and subsequent rehospitalization. J Hosp Med. 2014;9(5):277-282. PubMed
4. Hoyer EH, Needham DM, Miller J, Deutschendorf A, Friedman M, Brotman DJ. Functional status impairment is associated with unplanned readmissions. Arch Phys Med Rehabil. 2013;94(10):1951-1958. PubMed
5. Hoyer EH, Odonkor CA, Bhatia SN, Leung C, Deutschendorf A, Brotman DJ. Association between days to complete inpatient discharge summaries with all-payer hospital readmissions in Maryland. J Hosp Med. 2016;11(6):393-400. PubMed
© 2018 Society of Hospital Medicine
The Influence of Hospitalist Continuity on the Likelihood of Patient Discharge in General Medicine Patients
In addition to treating patients, physicians frequently have other time commitments that could include administrative, teaching, research, and family duties. Inpatient medicine is particularly unforgiving to these nonclinical duties since patients have to be assessed on a daily basis. Because of this characteristic, it is not uncommon for inpatient care responsibility to be switched between physicians to create time for nonclinical duties and personal health.
In contrast to the ambulatory setting, the influence of physician continuity of care on inpatient outcomes has not been studied frequently. Studies of inpatient continuity have primarily focused on patient discharge (likely because of its objective nature) over the weekends (likely because weekend cross-coverage is common) and have reported conflicting results.1-3 However, discontinuity of care is not isolated to the weekend since hospitalist-switches can occur at any time. In addition, expressing hospitalist continuity of care as a dichotomous variable (Was there weekend cross-coverage?) could incompletely express continuity since discharge likelihood might change with the consecutive number of days that a hospitalist is on service. This study measured the influence of hospitalist continuity throughout the patient’s hospitalization (rather than just the weekend) on daily patient discharge.
METHODS
Study Setting and Databases Used for Analysis
The study was conducted at The Ottawa Hospital, Ontario, Canada, a 1000-bed teaching hospital with 2 campuses and the primary referral center in our region. The division of general internal medicine has 6 patient services (or “teams”) at two campuses led by a staff hospitalist (exclusively general internists), a senior medical resident (2nd year of training), and various numbers of interns and medical students. Staff hospitalists do not treat more than one patient service even on the weekends.
Patients are admitted to each service on a daily basis and almost exclusively from the emergency room. Assignment of patients is essentially random since all services have the same clinical expertise. At a particular campus, the number of patients assigned daily to each service is usually equivalent between teams. Patients almost never switch between teams but may be transferred to another specialty. The study was approved by our local research ethics board.
The Patient Registry Database records for each patient the date and time of admissions (defined as the moment that a patient’s admission request is entered into the database), death or discharge from hospital (defined as the time when the patient’s discharge from hospital was entered into the database), or transfer to another specialty. It also records emergency visits, patient demographics, and location during admission. The Laboratory Database records all laboratory tests and their results.
Study Cohort
The Patient Registry Database was used to identify all individuals who were admitted to the general medicine services between January 1 and December 31, 2015. This time frame was selected to ensure that data were complete and current. General medicine services were analyzed because they are collectively the largest inpatient specialty in the hospital.
Study Outcome
The primary outcome was discharge from hospital as determined from the Patient Registry Database. Patients who died or were transferred to another service were not counted as outcomes.
Covariables
The primary exposure variable was the consecutive number of days (including weekends) that a particular hospitalist rounded on patients on a particular general medicine service. This was measured using call schedules. Other covariates included tomorrow’s expected number of discharges (TEND) daily discharge probability and its components. The TEND model4 used patient factors (age, Laboratory Abnormality Physiological Score [LAPS]5 calculated at admission) and hospitalization factors (hospital campus and service, admission urgency, day of the week, ICU status) to predict the daily discharge probability. In a validation population, these daily discharge probabilities (when summed over a particular day) strongly predicted the daily number of discharges (adjusted R2 of 89.2% [P < .001], median relative difference between observed and expected number of discharges of only 1.4% [Interquartile range,IQR: −5.5% to 7.1%]). The expected annual death risk was determined using the HOMR-now! model.6 This model used routinely collected data available at patient admission regarding the patient (sex, life-table-estimated 1-year death risk, Charlson score, current living location, previous cancer clinic status, and number of emergency department visits in the previous year) and the hospitalization (urgency, service, and LAPS score). The model explained more than half of the total variability in death likelihood of death (Nagelkirke’s R2 value of 0.53),7 was highly discriminative (C-statistic 0.92), and accurately predicted death risk (calibration slope 0.98).
Analysis
Logistic generalized estimating equation (GEE) methods were used to model the adjusted daily discharge probability.8 Data in the analytical dataset were expressed in a patient-day format (each dataset row represented one day for a particular patient). This permitted the inclusion of time-dependent covariates and allowed the GEE model to cluster hospitalization days within patients.
Model construction started with the TEND daily discharge probability and the HOMR-now! expected annual death risk (both expressed as log-odds). Then, hospitalist continuity was entered as a time-dependent covariate (ie, its value changed every day). Linear, square root, and natural logarithm forms of physician continuity were examined to determine the best fit (determined using the QIC statistic9). Finally, individual components of the TEND model were also offered to the model with those which significantly improving fit kept in the model. The GEE model used an independent correlation structure since this minimized the QIC statistic in the base model. All covariates in the final daily discharge probability model were used in the hospital death model. Analyses were conducted using SAS 9.4 (Cary, NC).
RESULTS
There were 6,405 general medicine admissions involving 5208 patients and 38,967 patient-days between January 1 and December 31, 2015 (Appendix A). Patients were elderly and were evenly divided in terms of gender, with 85% of them being admitted from the community. Comorbidities were common (median coded Charlson score was 2), with 6.0% of patients known to our cancer clinic. The median length of stay was 4 days (IQR, 2–7), with 378 admissions (5.9%) ending in death and 121 admissions (1.9%) ending in a transfer to another service.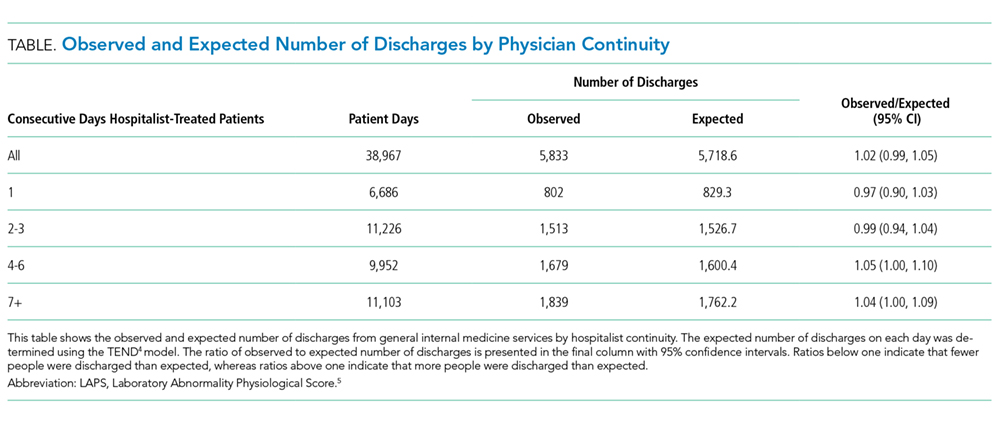
There were 41 different staff people having at least 1 day on service. The median total service by physicians was 9 weeks (IQR 1.8–10.9 weeks). Changes in hospitalist coverage were common; hospitalizations had a median of 1 (IQR 1–2) physician switches and a median of 1 (IQR 1–2) different physicians. However, patients spent a median of 100% (IQR 66.7%–100%] of their total hospitalization with their primary hospitalist. The median duration of individual physician “stints” on service was 5 days (IQR 2–7, range 1–42).
The TEND model accurately estimated daily discharge probability for the entire cohort with 5833 and 5718.6 observed and expected discharges, respectively, during 38,967 patient-days (O/E 1.02, 95% CI 0.99–1.05). Discharge probability increased as hospitalist continuity increased, but this was statistically significant only when hospitalist continuity exceeded 4 days. Other covariables also significantly influenced discharge probability (Appendix B).
After adjusting for important covariables (Appendix C), hospitalist continuity was significantly associated with daily discharge probability (Figure). Discharge probability increased linearly with increasing consecutive days that hospitalists treated patients. For each additional consecutive day with the same hospitalist, the adjusted daily odds increased by 2% (Adj-odds ratio [OR] 1.02, 95% CI 1.01–1.02, Appendix C). When the consecutive number of days that hospitalists remained on service increased from 1 to 28 days, the adjusted discharge probability for the average patient increased from 18.1% to 25.7%, respectively. Discharge was significantly influenced by other factors (Appendix C). Continuity did not influence the risk of death in hospital (Appendix D).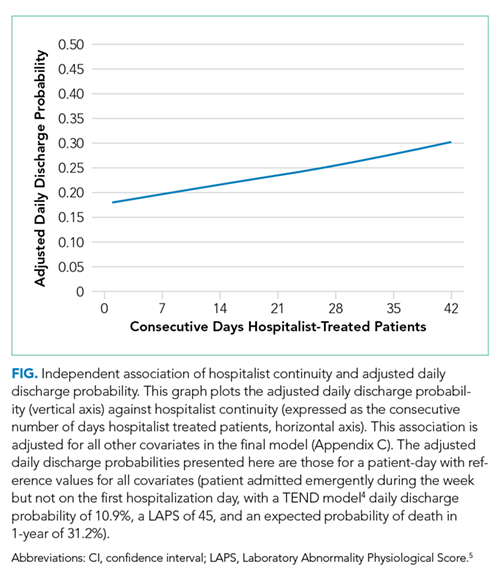
DISCUSSION
In a general medicine service at a large teaching hospital, this study found that greater hospitalist continuity was associated with a significantly increased adjusted daily discharge probability, increasing (in the average patient) from 18.1% to 25.7% when the consecutive number of hospitalist days on service increased from 1 to 28 days, respectively.
The study demonstrated some interesting findings. First, it shows that shifting patient care between physicians can significantly influence patient outcomes. This could be a function of incomplete transfer of knowledge between physicians, a phenomenon that should be expected given the extensive amount of information–both explicit and implicit–that physicians collect about particular patients during their hospitalization. Second, continuity of care could increase a physician’s and a patient’s confidence in clinical decision-making. Perhaps physicians are subconsciously more trusting of their instincts (and the decisions based on those instincts) when they have been on service for a while. It is also possible that patients more readily trust recommendations of a physician they have had throughout their stay. Finally, people wishing to decrease patient length of stay might consider minimizing the extent that hospitalists sign over patient care to colleagues.
Several issues should be noted when interpreting the results of the study. First, the study examined only patient discharge and death. These are by no means the only or the most important outcomes that might be influenced by hospitalist continuity. Second, this study was limited to a single service at a single center. Third, the analysis did not account for house-staff continuity. Since hospitalist and house-staff at the study hospital invariably switched at different times, it is unlikely that hospitalist continuity was a surrogate for house-staff continuity.
Disclosures
This study was supported by the Department of Medicine, University of Ottawa, Ottawa, Ontario, Canada. The author has nothing to disclose.
1. Ali NA, Hammersley J, Hoffmann SP et al. Continuity of care in intensive care units: a cluster-randomized trial of intensivist staffing. Am J Respir Crit Care Med. 2011;184(7):803-808. PubMed
2. Epstein K, Juarez E, Epstein A, Loya K, Singer A. The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335-338. PubMed
3. Blecker S, Shine D, Park N et al. Association of weekend continuity of care with hospital length of stay. Int J Qual Health Care. 2014;26(5):530-537. PubMed
4. van Walraven C, Forster AJ. The TEND (Tomorrow’s Expected Number of Discharges) model accurately predicted the number of patients who were discharged from the hospital in the next day. J Hosp Med. In press. PubMed
5. Escobar GJ, Greene JD, Scheirer P, Gardner MN, Draper D, Kipnis P. Risk-adjusting hospital inpatient mortality using automated inpatient, outpatient, and laboratory databases. Med Care. 2008;46(3):232-239. PubMed
6. van Walraven C, Forster AJ. HOMR-now! A modification of the HOMR score that predicts 1-year death risk for hospitalized patients using data immediately available at patient admission. Am J Med. In press. PubMed
7. Nagelkerke NJ. A note on a general definition of the coefficient of determination. Biometrika. 1991;78(3):691-692.
8. Stokes ME, Davis CS, Koch GG. Generalized estimating equations. Categorical Data Analysis Using the SAS System. 2nd ed. Cary, NC: SAS Institute Inc; 2000;469-549.
9. Pan W. Akaike’s information criterion in generalized estimating equations. Biometrics. 2001;57(1):120-125. PubMed
In addition to treating patients, physicians frequently have other time commitments that could include administrative, teaching, research, and family duties. Inpatient medicine is particularly unforgiving to these nonclinical duties since patients have to be assessed on a daily basis. Because of this characteristic, it is not uncommon for inpatient care responsibility to be switched between physicians to create time for nonclinical duties and personal health.
In contrast to the ambulatory setting, the influence of physician continuity of care on inpatient outcomes has not been studied frequently. Studies of inpatient continuity have primarily focused on patient discharge (likely because of its objective nature) over the weekends (likely because weekend cross-coverage is common) and have reported conflicting results.1-3 However, discontinuity of care is not isolated to the weekend since hospitalist-switches can occur at any time. In addition, expressing hospitalist continuity of care as a dichotomous variable (Was there weekend cross-coverage?) could incompletely express continuity since discharge likelihood might change with the consecutive number of days that a hospitalist is on service. This study measured the influence of hospitalist continuity throughout the patient’s hospitalization (rather than just the weekend) on daily patient discharge.
METHODS
Study Setting and Databases Used for Analysis
The study was conducted at The Ottawa Hospital, Ontario, Canada, a 1000-bed teaching hospital with 2 campuses and the primary referral center in our region. The division of general internal medicine has 6 patient services (or “teams”) at two campuses led by a staff hospitalist (exclusively general internists), a senior medical resident (2nd year of training), and various numbers of interns and medical students. Staff hospitalists do not treat more than one patient service even on the weekends.
Patients are admitted to each service on a daily basis and almost exclusively from the emergency room. Assignment of patients is essentially random since all services have the same clinical expertise. At a particular campus, the number of patients assigned daily to each service is usually equivalent between teams. Patients almost never switch between teams but may be transferred to another specialty. The study was approved by our local research ethics board.
The Patient Registry Database records for each patient the date and time of admissions (defined as the moment that a patient’s admission request is entered into the database), death or discharge from hospital (defined as the time when the patient’s discharge from hospital was entered into the database), or transfer to another specialty. It also records emergency visits, patient demographics, and location during admission. The Laboratory Database records all laboratory tests and their results.
Study Cohort
The Patient Registry Database was used to identify all individuals who were admitted to the general medicine services between January 1 and December 31, 2015. This time frame was selected to ensure that data were complete and current. General medicine services were analyzed because they are collectively the largest inpatient specialty in the hospital.
Study Outcome
The primary outcome was discharge from hospital as determined from the Patient Registry Database. Patients who died or were transferred to another service were not counted as outcomes.
Covariables
The primary exposure variable was the consecutive number of days (including weekends) that a particular hospitalist rounded on patients on a particular general medicine service. This was measured using call schedules. Other covariates included tomorrow’s expected number of discharges (TEND) daily discharge probability and its components. The TEND model4 used patient factors (age, Laboratory Abnormality Physiological Score [LAPS]5 calculated at admission) and hospitalization factors (hospital campus and service, admission urgency, day of the week, ICU status) to predict the daily discharge probability. In a validation population, these daily discharge probabilities (when summed over a particular day) strongly predicted the daily number of discharges (adjusted R2 of 89.2% [P < .001], median relative difference between observed and expected number of discharges of only 1.4% [Interquartile range,IQR: −5.5% to 7.1%]). The expected annual death risk was determined using the HOMR-now! model.6 This model used routinely collected data available at patient admission regarding the patient (sex, life-table-estimated 1-year death risk, Charlson score, current living location, previous cancer clinic status, and number of emergency department visits in the previous year) and the hospitalization (urgency, service, and LAPS score). The model explained more than half of the total variability in death likelihood of death (Nagelkirke’s R2 value of 0.53),7 was highly discriminative (C-statistic 0.92), and accurately predicted death risk (calibration slope 0.98).
Analysis
Logistic generalized estimating equation (GEE) methods were used to model the adjusted daily discharge probability.8 Data in the analytical dataset were expressed in a patient-day format (each dataset row represented one day for a particular patient). This permitted the inclusion of time-dependent covariates and allowed the GEE model to cluster hospitalization days within patients.
Model construction started with the TEND daily discharge probability and the HOMR-now! expected annual death risk (both expressed as log-odds). Then, hospitalist continuity was entered as a time-dependent covariate (ie, its value changed every day). Linear, square root, and natural logarithm forms of physician continuity were examined to determine the best fit (determined using the QIC statistic9). Finally, individual components of the TEND model were also offered to the model with those which significantly improving fit kept in the model. The GEE model used an independent correlation structure since this minimized the QIC statistic in the base model. All covariates in the final daily discharge probability model were used in the hospital death model. Analyses were conducted using SAS 9.4 (Cary, NC).
RESULTS
There were 6,405 general medicine admissions involving 5208 patients and 38,967 patient-days between January 1 and December 31, 2015 (Appendix A). Patients were elderly and were evenly divided in terms of gender, with 85% of them being admitted from the community. Comorbidities were common (median coded Charlson score was 2), with 6.0% of patients known to our cancer clinic. The median length of stay was 4 days (IQR, 2–7), with 378 admissions (5.9%) ending in death and 121 admissions (1.9%) ending in a transfer to another service.
There were 41 different staff people having at least 1 day on service. The median total service by physicians was 9 weeks (IQR 1.8–10.9 weeks). Changes in hospitalist coverage were common; hospitalizations had a median of 1 (IQR 1–2) physician switches and a median of 1 (IQR 1–2) different physicians. However, patients spent a median of 100% (IQR 66.7%–100%] of their total hospitalization with their primary hospitalist. The median duration of individual physician “stints” on service was 5 days (IQR 2–7, range 1–42).
The TEND model accurately estimated daily discharge probability for the entire cohort with 5833 and 5718.6 observed and expected discharges, respectively, during 38,967 patient-days (O/E 1.02, 95% CI 0.99–1.05). Discharge probability increased as hospitalist continuity increased, but this was statistically significant only when hospitalist continuity exceeded 4 days. Other covariables also significantly influenced discharge probability (Appendix B).
After adjusting for important covariables (Appendix C), hospitalist continuity was significantly associated with daily discharge probability (Figure). Discharge probability increased linearly with increasing consecutive days that hospitalists treated patients. For each additional consecutive day with the same hospitalist, the adjusted daily odds increased by 2% (Adj-odds ratio [OR] 1.02, 95% CI 1.01–1.02, Appendix C). When the consecutive number of days that hospitalists remained on service increased from 1 to 28 days, the adjusted discharge probability for the average patient increased from 18.1% to 25.7%, respectively. Discharge was significantly influenced by other factors (Appendix C). Continuity did not influence the risk of death in hospital (Appendix D).
DISCUSSION
In a general medicine service at a large teaching hospital, this study found that greater hospitalist continuity was associated with a significantly increased adjusted daily discharge probability, increasing (in the average patient) from 18.1% to 25.7% when the consecutive number of hospitalist days on service increased from 1 to 28 days, respectively.
The study demonstrated some interesting findings. First, it shows that shifting patient care between physicians can significantly influence patient outcomes. This could be a function of incomplete transfer of knowledge between physicians, a phenomenon that should be expected given the extensive amount of information–both explicit and implicit–that physicians collect about particular patients during their hospitalization. Second, continuity of care could increase a physician’s and a patient’s confidence in clinical decision-making. Perhaps physicians are subconsciously more trusting of their instincts (and the decisions based on those instincts) when they have been on service for a while. It is also possible that patients more readily trust recommendations of a physician they have had throughout their stay. Finally, people wishing to decrease patient length of stay might consider minimizing the extent that hospitalists sign over patient care to colleagues.
Several issues should be noted when interpreting the results of the study. First, the study examined only patient discharge and death. These are by no means the only or the most important outcomes that might be influenced by hospitalist continuity. Second, this study was limited to a single service at a single center. Third, the analysis did not account for house-staff continuity. Since hospitalist and house-staff at the study hospital invariably switched at different times, it is unlikely that hospitalist continuity was a surrogate for house-staff continuity.
Disclosures
This study was supported by the Department of Medicine, University of Ottawa, Ottawa, Ontario, Canada. The author has nothing to disclose.
In addition to treating patients, physicians frequently have other time commitments that could include administrative, teaching, research, and family duties. Inpatient medicine is particularly unforgiving to these nonclinical duties since patients have to be assessed on a daily basis. Because of this characteristic, it is not uncommon for inpatient care responsibility to be switched between physicians to create time for nonclinical duties and personal health.
In contrast to the ambulatory setting, the influence of physician continuity of care on inpatient outcomes has not been studied frequently. Studies of inpatient continuity have primarily focused on patient discharge (likely because of its objective nature) over the weekends (likely because weekend cross-coverage is common) and have reported conflicting results.1-3 However, discontinuity of care is not isolated to the weekend since hospitalist-switches can occur at any time. In addition, expressing hospitalist continuity of care as a dichotomous variable (Was there weekend cross-coverage?) could incompletely express continuity since discharge likelihood might change with the consecutive number of days that a hospitalist is on service. This study measured the influence of hospitalist continuity throughout the patient’s hospitalization (rather than just the weekend) on daily patient discharge.
METHODS
Study Setting and Databases Used for Analysis
The study was conducted at The Ottawa Hospital, Ontario, Canada, a 1000-bed teaching hospital with 2 campuses and the primary referral center in our region. The division of general internal medicine has 6 patient services (or “teams”) at two campuses led by a staff hospitalist (exclusively general internists), a senior medical resident (2nd year of training), and various numbers of interns and medical students. Staff hospitalists do not treat more than one patient service even on the weekends.
Patients are admitted to each service on a daily basis and almost exclusively from the emergency room. Assignment of patients is essentially random since all services have the same clinical expertise. At a particular campus, the number of patients assigned daily to each service is usually equivalent between teams. Patients almost never switch between teams but may be transferred to another specialty. The study was approved by our local research ethics board.
The Patient Registry Database records for each patient the date and time of admissions (defined as the moment that a patient’s admission request is entered into the database), death or discharge from hospital (defined as the time when the patient’s discharge from hospital was entered into the database), or transfer to another specialty. It also records emergency visits, patient demographics, and location during admission. The Laboratory Database records all laboratory tests and their results.
Study Cohort
The Patient Registry Database was used to identify all individuals who were admitted to the general medicine services between January 1 and December 31, 2015. This time frame was selected to ensure that data were complete and current. General medicine services were analyzed because they are collectively the largest inpatient specialty in the hospital.
Study Outcome
The primary outcome was discharge from hospital as determined from the Patient Registry Database. Patients who died or were transferred to another service were not counted as outcomes.
Covariables
The primary exposure variable was the consecutive number of days (including weekends) that a particular hospitalist rounded on patients on a particular general medicine service. This was measured using call schedules. Other covariates included tomorrow’s expected number of discharges (TEND) daily discharge probability and its components. The TEND model4 used patient factors (age, Laboratory Abnormality Physiological Score [LAPS]5 calculated at admission) and hospitalization factors (hospital campus and service, admission urgency, day of the week, ICU status) to predict the daily discharge probability. In a validation population, these daily discharge probabilities (when summed over a particular day) strongly predicted the daily number of discharges (adjusted R2 of 89.2% [P < .001], median relative difference between observed and expected number of discharges of only 1.4% [Interquartile range,IQR: −5.5% to 7.1%]). The expected annual death risk was determined using the HOMR-now! model.6 This model used routinely collected data available at patient admission regarding the patient (sex, life-table-estimated 1-year death risk, Charlson score, current living location, previous cancer clinic status, and number of emergency department visits in the previous year) and the hospitalization (urgency, service, and LAPS score). The model explained more than half of the total variability in death likelihood of death (Nagelkirke’s R2 value of 0.53),7 was highly discriminative (C-statistic 0.92), and accurately predicted death risk (calibration slope 0.98).
Analysis
Logistic generalized estimating equation (GEE) methods were used to model the adjusted daily discharge probability.8 Data in the analytical dataset were expressed in a patient-day format (each dataset row represented one day for a particular patient). This permitted the inclusion of time-dependent covariates and allowed the GEE model to cluster hospitalization days within patients.
Model construction started with the TEND daily discharge probability and the HOMR-now! expected annual death risk (both expressed as log-odds). Then, hospitalist continuity was entered as a time-dependent covariate (ie, its value changed every day). Linear, square root, and natural logarithm forms of physician continuity were examined to determine the best fit (determined using the QIC statistic9). Finally, individual components of the TEND model were also offered to the model with those which significantly improving fit kept in the model. The GEE model used an independent correlation structure since this minimized the QIC statistic in the base model. All covariates in the final daily discharge probability model were used in the hospital death model. Analyses were conducted using SAS 9.4 (Cary, NC).
RESULTS
There were 6,405 general medicine admissions involving 5208 patients and 38,967 patient-days between January 1 and December 31, 2015 (Appendix A). Patients were elderly and were evenly divided in terms of gender, with 85% of them being admitted from the community. Comorbidities were common (median coded Charlson score was 2), with 6.0% of patients known to our cancer clinic. The median length of stay was 4 days (IQR, 2–7), with 378 admissions (5.9%) ending in death and 121 admissions (1.9%) ending in a transfer to another service.
There were 41 different staff people having at least 1 day on service. The median total service by physicians was 9 weeks (IQR 1.8–10.9 weeks). Changes in hospitalist coverage were common; hospitalizations had a median of 1 (IQR 1–2) physician switches and a median of 1 (IQR 1–2) different physicians. However, patients spent a median of 100% (IQR 66.7%–100%] of their total hospitalization with their primary hospitalist. The median duration of individual physician “stints” on service was 5 days (IQR 2–7, range 1–42).
The TEND model accurately estimated daily discharge probability for the entire cohort with 5833 and 5718.6 observed and expected discharges, respectively, during 38,967 patient-days (O/E 1.02, 95% CI 0.99–1.05). Discharge probability increased as hospitalist continuity increased, but this was statistically significant only when hospitalist continuity exceeded 4 days. Other covariables also significantly influenced discharge probability (Appendix B).
After adjusting for important covariables (Appendix C), hospitalist continuity was significantly associated with daily discharge probability (Figure). Discharge probability increased linearly with increasing consecutive days that hospitalists treated patients. For each additional consecutive day with the same hospitalist, the adjusted daily odds increased by 2% (Adj-odds ratio [OR] 1.02, 95% CI 1.01–1.02, Appendix C). When the consecutive number of days that hospitalists remained on service increased from 1 to 28 days, the adjusted discharge probability for the average patient increased from 18.1% to 25.7%, respectively. Discharge was significantly influenced by other factors (Appendix C). Continuity did not influence the risk of death in hospital (Appendix D).
DISCUSSION
In a general medicine service at a large teaching hospital, this study found that greater hospitalist continuity was associated with a significantly increased adjusted daily discharge probability, increasing (in the average patient) from 18.1% to 25.7% when the consecutive number of hospitalist days on service increased from 1 to 28 days, respectively.
The study demonstrated some interesting findings. First, it shows that shifting patient care between physicians can significantly influence patient outcomes. This could be a function of incomplete transfer of knowledge between physicians, a phenomenon that should be expected given the extensive amount of information–both explicit and implicit–that physicians collect about particular patients during their hospitalization. Second, continuity of care could increase a physician’s and a patient’s confidence in clinical decision-making. Perhaps physicians are subconsciously more trusting of their instincts (and the decisions based on those instincts) when they have been on service for a while. It is also possible that patients more readily trust recommendations of a physician they have had throughout their stay. Finally, people wishing to decrease patient length of stay might consider minimizing the extent that hospitalists sign over patient care to colleagues.
Several issues should be noted when interpreting the results of the study. First, the study examined only patient discharge and death. These are by no means the only or the most important outcomes that might be influenced by hospitalist continuity. Second, this study was limited to a single service at a single center. Third, the analysis did not account for house-staff continuity. Since hospitalist and house-staff at the study hospital invariably switched at different times, it is unlikely that hospitalist continuity was a surrogate for house-staff continuity.
Disclosures
This study was supported by the Department of Medicine, University of Ottawa, Ottawa, Ontario, Canada. The author has nothing to disclose.
1. Ali NA, Hammersley J, Hoffmann SP et al. Continuity of care in intensive care units: a cluster-randomized trial of intensivist staffing. Am J Respir Crit Care Med. 2011;184(7):803-808. PubMed
2. Epstein K, Juarez E, Epstein A, Loya K, Singer A. The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335-338. PubMed
3. Blecker S, Shine D, Park N et al. Association of weekend continuity of care with hospital length of stay. Int J Qual Health Care. 2014;26(5):530-537. PubMed
4. van Walraven C, Forster AJ. The TEND (Tomorrow’s Expected Number of Discharges) model accurately predicted the number of patients who were discharged from the hospital in the next day. J Hosp Med. In press. PubMed
5. Escobar GJ, Greene JD, Scheirer P, Gardner MN, Draper D, Kipnis P. Risk-adjusting hospital inpatient mortality using automated inpatient, outpatient, and laboratory databases. Med Care. 2008;46(3):232-239. PubMed
6. van Walraven C, Forster AJ. HOMR-now! A modification of the HOMR score that predicts 1-year death risk for hospitalized patients using data immediately available at patient admission. Am J Med. In press. PubMed
7. Nagelkerke NJ. A note on a general definition of the coefficient of determination. Biometrika. 1991;78(3):691-692.
8. Stokes ME, Davis CS, Koch GG. Generalized estimating equations. Categorical Data Analysis Using the SAS System. 2nd ed. Cary, NC: SAS Institute Inc; 2000;469-549.
9. Pan W. Akaike’s information criterion in generalized estimating equations. Biometrics. 2001;57(1):120-125. PubMed
1. Ali NA, Hammersley J, Hoffmann SP et al. Continuity of care in intensive care units: a cluster-randomized trial of intensivist staffing. Am J Respir Crit Care Med. 2011;184(7):803-808. PubMed
2. Epstein K, Juarez E, Epstein A, Loya K, Singer A. The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335-338. PubMed
3. Blecker S, Shine D, Park N et al. Association of weekend continuity of care with hospital length of stay. Int J Qual Health Care. 2014;26(5):530-537. PubMed
4. van Walraven C, Forster AJ. The TEND (Tomorrow’s Expected Number of Discharges) model accurately predicted the number of patients who were discharged from the hospital in the next day. J Hosp Med. In press. PubMed
5. Escobar GJ, Greene JD, Scheirer P, Gardner MN, Draper D, Kipnis P. Risk-adjusting hospital inpatient mortality using automated inpatient, outpatient, and laboratory databases. Med Care. 2008;46(3):232-239. PubMed
6. van Walraven C, Forster AJ. HOMR-now! A modification of the HOMR score that predicts 1-year death risk for hospitalized patients using data immediately available at patient admission. Am J Med. In press. PubMed
7. Nagelkerke NJ. A note on a general definition of the coefficient of determination. Biometrika. 1991;78(3):691-692.
8. Stokes ME, Davis CS, Koch GG. Generalized estimating equations. Categorical Data Analysis Using the SAS System. 2nd ed. Cary, NC: SAS Institute Inc; 2000;469-549.
9. Pan W. Akaike’s information criterion in generalized estimating equations. Biometrics. 2001;57(1):120-125. PubMed
© 2018 Society of Hospital Medicine
Mental health visits, boarding continue to climb
SAN DIEGO – Between 2009 and 2015, the number of emergency department visits related to mental health increased 56% among children and 41% among adults, results from an analysis of national hospital data showed.

According to Dr. Santillanes, an emergency medicine physician at the University of Southern California, Los Angeles, data from older studies and non–nationally representative studies have demonstrated that mental health–related ED visits have been increasing. “Emergency physicians know from experience that mental health–related ED visits are increasing and that patients are boarding in EDs longer, but there had not been recent nationally representative studies analyzing these trends in visits by pediatric and adult patients,” she said.
In an effort to describe recent trends in mental health–related ED visits for patients of different ages, Dr. Santillanes and her colleagues retrospectively analyzed data from the National Hospital Ambulatory Medical Care Survey, which generates nationally representative annual estimates of ambulatory care visits to general, nonfederal, short-stay U.S. hospitals. They calculated the proportion of ED visits during 2009-2015 in which one of the first three discharge diagnoses was a mental health or substance abuse diagnosis as defined by the Healthcare Cost and Utilization Project’s Clinical Classifications Software categorization scheme. They excluded patients diagnosed with developmental disorders, as well as those with a diagnosis of delirium, dementia, amnestic and other cognitive disorders, fetal/newborn complications of alcohol and substance abuse, and chronic medical complications of alcohol abuse. The researchers also calculated ED length of stay (LOS) and the proportion of mental health ED visits resulting in inpatient care in the form of admission, observation, or transfer and used linear regression models to examined time trends of the survey data estimates.
Dr. Santillanes and her associates found that mental health–related ED visits for children jumped from 699,677 visits in 2009 to 1,095,313 visits in 2015, an increase of 56%. Adult mental health–related ED visits rose from 7.1 million in 2009 to 10 million in 2015, an increase of 41%. The researchers also found that encounters with a mental health discharge diagnosis rose from 2.1% of pediatric ED visits in 2009 to 3.4% of visits in 2015 (P = .006). For adults, the proportion of ED visits with a mental health discharge diagnosis rose from 6.9% in 2009 to 9.9% in 2015 (P less than .001). In 2015, more than 10% of visits in patients aged 15-64 years and 8.9% of visits in children aged 10-14 years resulted in a mental health discharge diagnosis visits. During this period, the proportion of ED mental health visits that resulted in inpatient care declined from 29.8% to 20.4%, or an average of –2.3% per year; P = .004). At the same time, ED LOS for patients receiving inpatient care increased from 401 to 528 minutes (a 31.7% increase, or an average of 28.6 minutes per year; P = .006), while ED LOS for discharged patients averaged 259.5 minutes and did not significantly change over the study period (P = .660).
“The mean length of stay for these visits was 8.8 hours in 2015,” Dr. Santillanes said. “That represents more than a 30% increase in length of stay from 2009. When we board patients awaiting admission for other medical conditions, we treat their medical conditions while they wait for an inpatient bed. In most EDs, when patients board awaiting a psychiatric bed, they are not receiving treatment for their psychiatric condition. Besides adding to ED crowding, those prolonged boarding times result in patients who need intensive mental health treatment spending many hours in an ED without treatment for their mental health condition.”
Knowing that patients with mental health disorders represent a significant and increasing portion of the patients treated in our EDs, Dr. Santillanes continued, emergency physicians need to determine how to best treat patients with mental health emergencies. “We have dramatically improved the care we provide to patients with medical and surgical conditions,” she said. “We need to do the same for patients with mental health emergencies. Patients with mental health emergencies represent a large proportion of the patients we treat, so we need to be prepared to provide compassionate and evidence-based care for patients with mental health conditions. At the same time, we also need to continue to advocate for improved access to mental health care for all patients so that [they] do not have to rely on emergency departments to access care.”
The researchers reported having no financial disclosures.
SOURCE: Santillanes G et al. Ann Emerg Med. 2018 Oct. doi. 10.1016/j.annemergmed.2018.08.050.
SAN DIEGO – Between 2009 and 2015, the number of emergency department visits related to mental health increased 56% among children and 41% among adults, results from an analysis of national hospital data showed.
According to Dr. Santillanes, an emergency medicine physician at the University of Southern California, Los Angeles, data from older studies and non–nationally representative studies have demonstrated that mental health–related ED visits have been increasing. “Emergency physicians know from experience that mental health–related ED visits are increasing and that patients are boarding in EDs longer, but there had not been recent nationally representative studies analyzing these trends in visits by pediatric and adult patients,” she said.
In an effort to describe recent trends in mental health–related ED visits for patients of different ages, Dr. Santillanes and her colleagues retrospectively analyzed data from the National Hospital Ambulatory Medical Care Survey, which generates nationally representative annual estimates of ambulatory care visits to general, nonfederal, short-stay U.S. hospitals. They calculated the proportion of ED visits during 2009-2015 in which one of the first three discharge diagnoses was a mental health or substance abuse diagnosis as defined by the Healthcare Cost and Utilization Project’s Clinical Classifications Software categorization scheme. They excluded patients diagnosed with developmental disorders, as well as those with a diagnosis of delirium, dementia, amnestic and other cognitive disorders, fetal/newborn complications of alcohol and substance abuse, and chronic medical complications of alcohol abuse. The researchers also calculated ED length of stay (LOS) and the proportion of mental health ED visits resulting in inpatient care in the form of admission, observation, or transfer and used linear regression models to examined time trends of the survey data estimates.
Dr. Santillanes and her associates found that mental health–related ED visits for children jumped from 699,677 visits in 2009 to 1,095,313 visits in 2015, an increase of 56%. Adult mental health–related ED visits rose from 7.1 million in 2009 to 10 million in 2015, an increase of 41%. The researchers also found that encounters with a mental health discharge diagnosis rose from 2.1% of pediatric ED visits in 2009 to 3.4% of visits in 2015 (P = .006). For adults, the proportion of ED visits with a mental health discharge diagnosis rose from 6.9% in 2009 to 9.9% in 2015 (P less than .001). In 2015, more than 10% of visits in patients aged 15-64 years and 8.9% of visits in children aged 10-14 years resulted in a mental health discharge diagnosis visits. During this period, the proportion of ED mental health visits that resulted in inpatient care declined from 29.8% to 20.4%, or an average of –2.3% per year; P = .004). At the same time, ED LOS for patients receiving inpatient care increased from 401 to 528 minutes (a 31.7% increase, or an average of 28.6 minutes per year; P = .006), while ED LOS for discharged patients averaged 259.5 minutes and did not significantly change over the study period (P = .660).
“The mean length of stay for these visits was 8.8 hours in 2015,” Dr. Santillanes said. “That represents more than a 30% increase in length of stay from 2009. When we board patients awaiting admission for other medical conditions, we treat their medical conditions while they wait for an inpatient bed. In most EDs, when patients board awaiting a psychiatric bed, they are not receiving treatment for their psychiatric condition. Besides adding to ED crowding, those prolonged boarding times result in patients who need intensive mental health treatment spending many hours in an ED without treatment for their mental health condition.”
Knowing that patients with mental health disorders represent a significant and increasing portion of the patients treated in our EDs, Dr. Santillanes continued, emergency physicians need to determine how to best treat patients with mental health emergencies. “We have dramatically improved the care we provide to patients with medical and surgical conditions,” she said. “We need to do the same for patients with mental health emergencies. Patients with mental health emergencies represent a large proportion of the patients we treat, so we need to be prepared to provide compassionate and evidence-based care for patients with mental health conditions. At the same time, we also need to continue to advocate for improved access to mental health care for all patients so that [they] do not have to rely on emergency departments to access care.”
The researchers reported having no financial disclosures.
SOURCE: Santillanes G et al. Ann Emerg Med. 2018 Oct. doi. 10.1016/j.annemergmed.2018.08.050.
SAN DIEGO – Between 2009 and 2015, the number of emergency department visits related to mental health increased 56% among children and 41% among adults, results from an analysis of national hospital data showed.
According to Dr. Santillanes, an emergency medicine physician at the University of Southern California, Los Angeles, data from older studies and non–nationally representative studies have demonstrated that mental health–related ED visits have been increasing. “Emergency physicians know from experience that mental health–related ED visits are increasing and that patients are boarding in EDs longer, but there had not been recent nationally representative studies analyzing these trends in visits by pediatric and adult patients,” she said.
In an effort to describe recent trends in mental health–related ED visits for patients of different ages, Dr. Santillanes and her colleagues retrospectively analyzed data from the National Hospital Ambulatory Medical Care Survey, which generates nationally representative annual estimates of ambulatory care visits to general, nonfederal, short-stay U.S. hospitals. They calculated the proportion of ED visits during 2009-2015 in which one of the first three discharge diagnoses was a mental health or substance abuse diagnosis as defined by the Healthcare Cost and Utilization Project’s Clinical Classifications Software categorization scheme. They excluded patients diagnosed with developmental disorders, as well as those with a diagnosis of delirium, dementia, amnestic and other cognitive disorders, fetal/newborn complications of alcohol and substance abuse, and chronic medical complications of alcohol abuse. The researchers also calculated ED length of stay (LOS) and the proportion of mental health ED visits resulting in inpatient care in the form of admission, observation, or transfer and used linear regression models to examined time trends of the survey data estimates.
Dr. Santillanes and her associates found that mental health–related ED visits for children jumped from 699,677 visits in 2009 to 1,095,313 visits in 2015, an increase of 56%. Adult mental health–related ED visits rose from 7.1 million in 2009 to 10 million in 2015, an increase of 41%. The researchers also found that encounters with a mental health discharge diagnosis rose from 2.1% of pediatric ED visits in 2009 to 3.4% of visits in 2015 (P = .006). For adults, the proportion of ED visits with a mental health discharge diagnosis rose from 6.9% in 2009 to 9.9% in 2015 (P less than .001). In 2015, more than 10% of visits in patients aged 15-64 years and 8.9% of visits in children aged 10-14 years resulted in a mental health discharge diagnosis visits. During this period, the proportion of ED mental health visits that resulted in inpatient care declined from 29.8% to 20.4%, or an average of –2.3% per year; P = .004). At the same time, ED LOS for patients receiving inpatient care increased from 401 to 528 minutes (a 31.7% increase, or an average of 28.6 minutes per year; P = .006), while ED LOS for discharged patients averaged 259.5 minutes and did not significantly change over the study period (P = .660).
“The mean length of stay for these visits was 8.8 hours in 2015,” Dr. Santillanes said. “That represents more than a 30% increase in length of stay from 2009. When we board patients awaiting admission for other medical conditions, we treat their medical conditions while they wait for an inpatient bed. In most EDs, when patients board awaiting a psychiatric bed, they are not receiving treatment for their psychiatric condition. Besides adding to ED crowding, those prolonged boarding times result in patients who need intensive mental health treatment spending many hours in an ED without treatment for their mental health condition.”
Knowing that patients with mental health disorders represent a significant and increasing portion of the patients treated in our EDs, Dr. Santillanes continued, emergency physicians need to determine how to best treat patients with mental health emergencies. “We have dramatically improved the care we provide to patients with medical and surgical conditions,” she said. “We need to do the same for patients with mental health emergencies. Patients with mental health emergencies represent a large proportion of the patients we treat, so we need to be prepared to provide compassionate and evidence-based care for patients with mental health conditions. At the same time, we also need to continue to advocate for improved access to mental health care for all patients so that [they] do not have to rely on emergency departments to access care.”
The researchers reported having no financial disclosures.
SOURCE: Santillanes G et al. Ann Emerg Med. 2018 Oct. doi. 10.1016/j.annemergmed.2018.08.050.
REPORTING FROM ACEP18
Key clinical point: Mental health ED visits are rising, in total number and as a proportion of all ED visits for children and adults.
Major finding: Between 2009 and 2015, the proportion of children and adults who made mental health–related ED visits increased 56% and 41%, respectively.
Study details: A retrospective analysis of National Hospital Ambulatory Medical Care Survey data between 2009 and 2015.
Disclosures: The researchers reported having no financial disclosures.
Source: Santillanes G et al. Ann Emerg Med. 2018 Oct. doi. 10.1016/j.annemergmed.2018.08.050.
October 2018 Digital Edition
Click here to access the October 2018 Digital Edition.
Table of Contents
- Comparison of Cardiovascular Outcomes Between Statin Monotherapy and Fish Oil and Statin Combination Therapy in a Veteran Population
- Improving Team-Based Care Coordination Delivery and Documentation in the Health Record
- An Overview of Pharmacotherapy Options for Alcohol Use Disorder
- An Imposter Twice Over: A Case of IgG4-Related Disease
- Happy Federal New Year
- FDA Update

Click here to access the October 2018 Digital Edition.
Table of Contents
- Comparison of Cardiovascular Outcomes Between Statin Monotherapy and Fish Oil and Statin Combination Therapy in a Veteran Population
- Improving Team-Based Care Coordination Delivery and Documentation in the Health Record
- An Overview of Pharmacotherapy Options for Alcohol Use Disorder
- An Imposter Twice Over: A Case of IgG4-Related Disease
- Happy Federal New Year
- FDA Update
Click here to access the October 2018 Digital Edition.
Table of Contents
- Comparison of Cardiovascular Outcomes Between Statin Monotherapy and Fish Oil and Statin Combination Therapy in a Veteran Population
- Improving Team-Based Care Coordination Delivery and Documentation in the Health Record
- An Overview of Pharmacotherapy Options for Alcohol Use Disorder
- An Imposter Twice Over: A Case of IgG4-Related Disease
- Happy Federal New Year
- FDA Update
AGA Research Foundation researcher of the month: David L. Boone, PhD
AGA Research Foundation pilot awards are an invaluable tool for investigators – they provide seed funding to explore promising new lines of research and generate preliminary data for larger grants. So, when David L. Boone, PhD, received the 2017 AGA-Pfizer Young Investigator Pilot Research Award in Inflammatory Bowel Disease from the AGA Research Foundation, he was able to double-down on a very targeted project studying innate immunity in IBD. Based on his recent accomplishments – both in and out of the lab – we’re excited for you to get to know Dr. Boone, associate professor of microbiology and immunology at Indiana University School of Medicine-South Bend, and our AGA Research Foundation researcher of the month.

Bench to bedside: working toward new treatment options in IBD
The Boone lab AGA-funded project is specifically focused on JAK inhibitors, which are becoming a more popular treatment option for patients with IBD, especially for those patients who don’t respond to anti-TNF therapy. Dr. Boone is committed to enhancing our understanding of how these JAK inhibitors work at a cellular level. If we can understand this, Dr. Boone is optimistic it will lead to new approaches for treating inflammation in IBD.
With his AGA Research Foundation grant, Dr. Boone and his lab characterized a new robust mouse model of colitis that is entirely driven by innate immune mechanisms. With this model, his team is investigating the cellular and molecular mechanisms that drive innate immune-mediated inflammation in the intestine, which will provide important insights for future IBD drug development. You can read the specifics of Dr. Boone’s research in his recently published work in Mucosal Immunology.
Pilot award provides a stepping stone
Dr. Boone’s AGA Research Foundation pilot grant has paved the way for future success. Using the data from his AGA-funded project, as well as the constructive feedback he received from the AGA awards panel, Dr. Boone went on to successfully obtain new funding in the form of a Pfizer ASPIRE research grant. This work is building the foundation for Dr. Boone’s next big grant venture: an NIH R01 grant.
Two postdocs, a graduate student, a technician, and a dog named Boone
Dr. Boone shared with us that the best outcome from his AGA grant was that the additional funding made it possible to grow his lab by a postdoctoral researcher and lab technician. One of Dr. Boone’s great passions is training the next generation of scientists, both in the lab and through his role as a microbiology and immunology professor for first-year medical students at Indiana University Medical School.

Beyond the lab – a commitment to IBD patients
Dr. Boone wanted to do more to support patients with IBD. He had heard of Camp Oasis – the Crohn’s & Colitis Foundation regional camp for patients with IBD – and knew of physicians who provided medical services at the camp. After looking into making a donation to Camp Oasis Michigan, Dr. Boone learned that what the camp really needed was male counselors. So, despite being “older than an average camp counselor,” Dr. Boone packed his bags for Michigan. Participating in Camp Oasis the last 2 years has been a great joy for Dr. Boone and provides added inspiration and motivation for his work in the lab.

The AGA Research Foundation is proud to fund researchers who are committed to improving the lives of patients – both in and out of the lab. You can help keep great researchers in GI by making a gift to the AGA Research Foundation, www.gastro.org/foundation.
AGA Research Foundation pilot awards are an invaluable tool for investigators – they provide seed funding to explore promising new lines of research and generate preliminary data for larger grants. So, when David L. Boone, PhD, received the 2017 AGA-Pfizer Young Investigator Pilot Research Award in Inflammatory Bowel Disease from the AGA Research Foundation, he was able to double-down on a very targeted project studying innate immunity in IBD. Based on his recent accomplishments – both in and out of the lab – we’re excited for you to get to know Dr. Boone, associate professor of microbiology and immunology at Indiana University School of Medicine-South Bend, and our AGA Research Foundation researcher of the month.
Bench to bedside: working toward new treatment options in IBD
The Boone lab AGA-funded project is specifically focused on JAK inhibitors, which are becoming a more popular treatment option for patients with IBD, especially for those patients who don’t respond to anti-TNF therapy. Dr. Boone is committed to enhancing our understanding of how these JAK inhibitors work at a cellular level. If we can understand this, Dr. Boone is optimistic it will lead to new approaches for treating inflammation in IBD.
With his AGA Research Foundation grant, Dr. Boone and his lab characterized a new robust mouse model of colitis that is entirely driven by innate immune mechanisms. With this model, his team is investigating the cellular and molecular mechanisms that drive innate immune-mediated inflammation in the intestine, which will provide important insights for future IBD drug development. You can read the specifics of Dr. Boone’s research in his recently published work in Mucosal Immunology.
Pilot award provides a stepping stone
Dr. Boone’s AGA Research Foundation pilot grant has paved the way for future success. Using the data from his AGA-funded project, as well as the constructive feedback he received from the AGA awards panel, Dr. Boone went on to successfully obtain new funding in the form of a Pfizer ASPIRE research grant. This work is building the foundation for Dr. Boone’s next big grant venture: an NIH R01 grant.
Two postdocs, a graduate student, a technician, and a dog named Boone
Dr. Boone shared with us that the best outcome from his AGA grant was that the additional funding made it possible to grow his lab by a postdoctoral researcher and lab technician. One of Dr. Boone’s great passions is training the next generation of scientists, both in the lab and through his role as a microbiology and immunology professor for first-year medical students at Indiana University Medical School.
Beyond the lab – a commitment to IBD patients
Dr. Boone wanted to do more to support patients with IBD. He had heard of Camp Oasis – the Crohn’s & Colitis Foundation regional camp for patients with IBD – and knew of physicians who provided medical services at the camp. After looking into making a donation to Camp Oasis Michigan, Dr. Boone learned that what the camp really needed was male counselors. So, despite being “older than an average camp counselor,” Dr. Boone packed his bags for Michigan. Participating in Camp Oasis the last 2 years has been a great joy for Dr. Boone and provides added inspiration and motivation for his work in the lab.
The AGA Research Foundation is proud to fund researchers who are committed to improving the lives of patients – both in and out of the lab. You can help keep great researchers in GI by making a gift to the AGA Research Foundation, www.gastro.org/foundation.
AGA Research Foundation pilot awards are an invaluable tool for investigators – they provide seed funding to explore promising new lines of research and generate preliminary data for larger grants. So, when David L. Boone, PhD, received the 2017 AGA-Pfizer Young Investigator Pilot Research Award in Inflammatory Bowel Disease from the AGA Research Foundation, he was able to double-down on a very targeted project studying innate immunity in IBD. Based on his recent accomplishments – both in and out of the lab – we’re excited for you to get to know Dr. Boone, associate professor of microbiology and immunology at Indiana University School of Medicine-South Bend, and our AGA Research Foundation researcher of the month.
Bench to bedside: working toward new treatment options in IBD
The Boone lab AGA-funded project is specifically focused on JAK inhibitors, which are becoming a more popular treatment option for patients with IBD, especially for those patients who don’t respond to anti-TNF therapy. Dr. Boone is committed to enhancing our understanding of how these JAK inhibitors work at a cellular level. If we can understand this, Dr. Boone is optimistic it will lead to new approaches for treating inflammation in IBD.
With his AGA Research Foundation grant, Dr. Boone and his lab characterized a new robust mouse model of colitis that is entirely driven by innate immune mechanisms. With this model, his team is investigating the cellular and molecular mechanisms that drive innate immune-mediated inflammation in the intestine, which will provide important insights for future IBD drug development. You can read the specifics of Dr. Boone’s research in his recently published work in Mucosal Immunology.
Pilot award provides a stepping stone
Dr. Boone’s AGA Research Foundation pilot grant has paved the way for future success. Using the data from his AGA-funded project, as well as the constructive feedback he received from the AGA awards panel, Dr. Boone went on to successfully obtain new funding in the form of a Pfizer ASPIRE research grant. This work is building the foundation for Dr. Boone’s next big grant venture: an NIH R01 grant.
Two postdocs, a graduate student, a technician, and a dog named Boone
Dr. Boone shared with us that the best outcome from his AGA grant was that the additional funding made it possible to grow his lab by a postdoctoral researcher and lab technician. One of Dr. Boone’s great passions is training the next generation of scientists, both in the lab and through his role as a microbiology and immunology professor for first-year medical students at Indiana University Medical School.
Beyond the lab – a commitment to IBD patients
Dr. Boone wanted to do more to support patients with IBD. He had heard of Camp Oasis – the Crohn’s & Colitis Foundation regional camp for patients with IBD – and knew of physicians who provided medical services at the camp. After looking into making a donation to Camp Oasis Michigan, Dr. Boone learned that what the camp really needed was male counselors. So, despite being “older than an average camp counselor,” Dr. Boone packed his bags for Michigan. Participating in Camp Oasis the last 2 years has been a great joy for Dr. Boone and provides added inspiration and motivation for his work in the lab.
The AGA Research Foundation is proud to fund researchers who are committed to improving the lives of patients – both in and out of the lab. You can help keep great researchers in GI by making a gift to the AGA Research Foundation, www.gastro.org/foundation.
