User login
Swings in four metabolic measures predicted death in healthy people
, based on data from a 5.5-year population-based study in Korea.

In a model adjusted for age, sex, smoking, alcohol consumption, regular exercise, and income status the group with high variability for all four parameters had a significantly higher risk for all-cause mortality (hazard ratio, 2.27; 95% confidence interval, 2.13-2.42), for MI (HR, 1.43; 95% CI, 1.25-1.64), and for stroke (HR, 1.41; 95% CI, 1.25-1.60), compared with the group with low variability for all four parameters. The association with risk was graded and persisted after multivariable adjustment.
“Variability in metabolic parameters may be prognostic surrogate markers for predicting mortality and cardiovascular outcomes,” wrote senior author Seung-Hwan Lee, MD, PhD, and professor of endocrinology at the College of Medicine of the Catholic University of Korea in Seoul, South Korea, and colleagues. “High variability in metabolic parameters (may be) associated with adverse health outcomes not only in a diseased population, but also in the relatively healthy population although the mechanism could be somewhat different.”
Korea has a single-payer system, the Korean National Health Insurance system, that includes health information on its entire population. The researchers selected data from 6,748,773 people who were free of diabetes mellitus, hypertension, and dyslipidemia, and who underwent three or more health examinations during 2005-2012 that documented body mass index (BMI), fasting blood glucose, systolic blood pressure, and total cholesterol. Participants were followed to the end of 2015, for a median follow-up of 5.5 years. There were 54,785 deaths (0.8%), 22,498 cases of stroke (0.3%), and 21,452 MIs (0.3%).
The research team defined high variability as the highest quartile, classifying participants according to the number of high-variability parameters. A score of 4 indicated high variability in all four metabolic parameters – body weight, systolic blood pressure, total cholesterol, and fasting blood glucose.
In the highest quartile in fasting blood glucose variability, compared with the lowest quartile, the risk of all-cause mortality increased by 20% (HR, 1.20; 95% CI, 1.18-1.23), MI by 16% (HR, 1.16; 95% CI, 1.12-1.21), and stroke by 13% (HR, 1.13; 95% CI, 1.09-1.17).
For the highest quartile in total cholesterol variability, compared with the lowest quartile, the risk of all-cause mortality increased by 31% (HR, 1.31; 95% CI, 1.28-1.34), MI by 10% (HR, 1.10; 95% CI, 1.06-1.14), and stroke by 6% (HR, 1.06; 95% CI, 1.03-1.10).
For the highest quartile in systolic BP variability, compared with the lowest quartile, the risk of all-cause mortality increased by 19% (HR, 1.19; 95% CI, 1.16-1.22), MI by 7% (HR, 1.07; 95% CI, 1.03-1.11), and stroke by 14% (HR, 1.14; 95% CI, 1.10-1.18).
For the highest quartile in BMI variability, compared with the lowest quartile, the risk of all-cause mortality increased by 53% (HR, 1.53; 95% CI, 1.50-1.57), MI by 14% (HR, 1.14; 95% CI, 1.09-1.18), and stroke by 14% (HR, 1.14; 95% CI, 1.10-1.18).
“It is not certain whether these results from Korea would apply to the United States. However, several previous studies on variability were performed in other populations, suggesting that it is likely to be a common phenomenon,” the authors wrote.
The study was supported in part by the National Research Foundation of Korea Grant funded by the Korean Government. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
SOURCE: Lee S-H et al. Circulation. 2018 Oct.
, based on data from a 5.5-year population-based study in Korea.
In a model adjusted for age, sex, smoking, alcohol consumption, regular exercise, and income status the group with high variability for all four parameters had a significantly higher risk for all-cause mortality (hazard ratio, 2.27; 95% confidence interval, 2.13-2.42), for MI (HR, 1.43; 95% CI, 1.25-1.64), and for stroke (HR, 1.41; 95% CI, 1.25-1.60), compared with the group with low variability for all four parameters. The association with risk was graded and persisted after multivariable adjustment.
“Variability in metabolic parameters may be prognostic surrogate markers for predicting mortality and cardiovascular outcomes,” wrote senior author Seung-Hwan Lee, MD, PhD, and professor of endocrinology at the College of Medicine of the Catholic University of Korea in Seoul, South Korea, and colleagues. “High variability in metabolic parameters (may be) associated with adverse health outcomes not only in a diseased population, but also in the relatively healthy population although the mechanism could be somewhat different.”
Korea has a single-payer system, the Korean National Health Insurance system, that includes health information on its entire population. The researchers selected data from 6,748,773 people who were free of diabetes mellitus, hypertension, and dyslipidemia, and who underwent three or more health examinations during 2005-2012 that documented body mass index (BMI), fasting blood glucose, systolic blood pressure, and total cholesterol. Participants were followed to the end of 2015, for a median follow-up of 5.5 years. There were 54,785 deaths (0.8%), 22,498 cases of stroke (0.3%), and 21,452 MIs (0.3%).
The research team defined high variability as the highest quartile, classifying participants according to the number of high-variability parameters. A score of 4 indicated high variability in all four metabolic parameters – body weight, systolic blood pressure, total cholesterol, and fasting blood glucose.
In the highest quartile in fasting blood glucose variability, compared with the lowest quartile, the risk of all-cause mortality increased by 20% (HR, 1.20; 95% CI, 1.18-1.23), MI by 16% (HR, 1.16; 95% CI, 1.12-1.21), and stroke by 13% (HR, 1.13; 95% CI, 1.09-1.17).
For the highest quartile in total cholesterol variability, compared with the lowest quartile, the risk of all-cause mortality increased by 31% (HR, 1.31; 95% CI, 1.28-1.34), MI by 10% (HR, 1.10; 95% CI, 1.06-1.14), and stroke by 6% (HR, 1.06; 95% CI, 1.03-1.10).
For the highest quartile in systolic BP variability, compared with the lowest quartile, the risk of all-cause mortality increased by 19% (HR, 1.19; 95% CI, 1.16-1.22), MI by 7% (HR, 1.07; 95% CI, 1.03-1.11), and stroke by 14% (HR, 1.14; 95% CI, 1.10-1.18).
For the highest quartile in BMI variability, compared with the lowest quartile, the risk of all-cause mortality increased by 53% (HR, 1.53; 95% CI, 1.50-1.57), MI by 14% (HR, 1.14; 95% CI, 1.09-1.18), and stroke by 14% (HR, 1.14; 95% CI, 1.10-1.18).
“It is not certain whether these results from Korea would apply to the United States. However, several previous studies on variability were performed in other populations, suggesting that it is likely to be a common phenomenon,” the authors wrote.
The study was supported in part by the National Research Foundation of Korea Grant funded by the Korean Government. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
SOURCE: Lee S-H et al. Circulation. 2018 Oct.
, based on data from a 5.5-year population-based study in Korea.
In a model adjusted for age, sex, smoking, alcohol consumption, regular exercise, and income status the group with high variability for all four parameters had a significantly higher risk for all-cause mortality (hazard ratio, 2.27; 95% confidence interval, 2.13-2.42), for MI (HR, 1.43; 95% CI, 1.25-1.64), and for stroke (HR, 1.41; 95% CI, 1.25-1.60), compared with the group with low variability for all four parameters. The association with risk was graded and persisted after multivariable adjustment.
“Variability in metabolic parameters may be prognostic surrogate markers for predicting mortality and cardiovascular outcomes,” wrote senior author Seung-Hwan Lee, MD, PhD, and professor of endocrinology at the College of Medicine of the Catholic University of Korea in Seoul, South Korea, and colleagues. “High variability in metabolic parameters (may be) associated with adverse health outcomes not only in a diseased population, but also in the relatively healthy population although the mechanism could be somewhat different.”
Korea has a single-payer system, the Korean National Health Insurance system, that includes health information on its entire population. The researchers selected data from 6,748,773 people who were free of diabetes mellitus, hypertension, and dyslipidemia, and who underwent three or more health examinations during 2005-2012 that documented body mass index (BMI), fasting blood glucose, systolic blood pressure, and total cholesterol. Participants were followed to the end of 2015, for a median follow-up of 5.5 years. There were 54,785 deaths (0.8%), 22,498 cases of stroke (0.3%), and 21,452 MIs (0.3%).
The research team defined high variability as the highest quartile, classifying participants according to the number of high-variability parameters. A score of 4 indicated high variability in all four metabolic parameters – body weight, systolic blood pressure, total cholesterol, and fasting blood glucose.
In the highest quartile in fasting blood glucose variability, compared with the lowest quartile, the risk of all-cause mortality increased by 20% (HR, 1.20; 95% CI, 1.18-1.23), MI by 16% (HR, 1.16; 95% CI, 1.12-1.21), and stroke by 13% (HR, 1.13; 95% CI, 1.09-1.17).
For the highest quartile in total cholesterol variability, compared with the lowest quartile, the risk of all-cause mortality increased by 31% (HR, 1.31; 95% CI, 1.28-1.34), MI by 10% (HR, 1.10; 95% CI, 1.06-1.14), and stroke by 6% (HR, 1.06; 95% CI, 1.03-1.10).
For the highest quartile in systolic BP variability, compared with the lowest quartile, the risk of all-cause mortality increased by 19% (HR, 1.19; 95% CI, 1.16-1.22), MI by 7% (HR, 1.07; 95% CI, 1.03-1.11), and stroke by 14% (HR, 1.14; 95% CI, 1.10-1.18).
For the highest quartile in BMI variability, compared with the lowest quartile, the risk of all-cause mortality increased by 53% (HR, 1.53; 95% CI, 1.50-1.57), MI by 14% (HR, 1.14; 95% CI, 1.09-1.18), and stroke by 14% (HR, 1.14; 95% CI, 1.10-1.18).
“It is not certain whether these results from Korea would apply to the United States. However, several previous studies on variability were performed in other populations, suggesting that it is likely to be a common phenomenon,” the authors wrote.
The study was supported in part by the National Research Foundation of Korea Grant funded by the Korean Government. The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
SOURCE: Lee S-H et al. Circulation. 2018 Oct.
FROM CIRCULATION
Key clinical point: Fluctuations in fasting glucose and cholesterol levels, systolic blood pressure, and body mass index are associated with a higher risk for all-cause mortality, myocardial infarction, and stroke in otherwise healthy people.
Major finding: The hazard ratios were 2.27 (95% CI, 2.13-2.42) for all-cause mortality, 1.43 (95% CI, 1.25-1.64) for MI, and 1.41 (95% CI, 1.25-1.60) for stroke.
Study details: An observational population-based study involving more than 6.7 million Koreans age 20 and older.
Disclosures: The study was funded by the National Research Foundation of Korea. The authors had no relevant conflicts of interest to declare.
Source: Lee S-H et al. Circulation. 2018 Oct.
Reply to “In Reference to 'Improving the Safety of Opioid Use for Acute Noncancer Pain in Hospitalized Adults: A Consensus Statement from the Society of Hospital Medicine'”
Hall et al. draw attention to the important question of whether some patients may benefit from a naloxone prescription when discharged from the hospital with a short-term opioid prescription for acute pain. Although all members of the working group agreed that naloxone is appropriate in some cases, we were hesitant to recommend this as a standard practice for several reasons.
First, the intent of our Consensus Statement1 was to synthesize and summarize the areas of consensus in existing guidelines; none of the existing guidelines included in our systematic review make a recommendation for naloxone prescription in the setting of short-term opioid use for acute pain.2 We believe that this may relate to the fact that the risk factors for overdose and the threshold of risk above which naloxone would be beneficial have yet to be defined for this population and are likely to differ from those defined in patients using opioids chronically.
Additionally, if practitioners follow the recommendations to limit prescribing for acute pain to the minimum dose and duration of an opioid that was presumably administered in the hospital with an observed response, then the risk of overdose and the potential benefit of naloxone will decrease. Furthermore, emerging data from randomized controlled trials demonstrating noninferiority of nonopioid analgesics in the management of acute pain suggest that we should not so readily presume opioids to be the necessary or the best option.3-5 Data questioning the benefits of opioids over other safer therapies have particularly important implications for patients in whom the risks are felt to be high enough to warrant consideration of naloxone.
Disclosures
Dr. Herzig reports receiving compensation from the Society of Hospital Medicine for her editorial role in the Journal of Hospital Medicine (unrelated to the present work). None of the other authors have any conflicts of interest to disclose.
Funding
Dr. Herzig is funded by a grant number K23AG042459 from the National Institute on Aging. Dr. Mosher is supported in part by the Department of Veterans Affairs Office of Academic Affiliations and the Office of Research and Development and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412). The views expressed in this manuscript do not necessarily represent the views of the funding agencies.
1. Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK. Improving the safety of opioid use for acute noncancer pain in hospitalized adults: a consensus statement from the Society of Hospital Medicine. J Hosp Med. 2018;13(4):263-271. doi: 10.12788/jhm.2980. PubMed
2. Herzig SJ, Calcaterra SL, Mosher HJ, et al. Safe opioid prescribing for acute noncancer pain in hospitalized adults: a systematic review of existing guidelines.. J Hosp Med. 2018;13(4):256-262. doi: 10.12788/jhm.2979. PubMed
3. Chang AK, Bijur PE, Esses D, Barnaby DP, Baer J. Effect of a single dose of oral opioid and nonopioid analgesics on acute extremity pain in the emergency department: a randomized clinical trial. JAMA. 2017;318(17):1661-1667. doi: 10.1001/jama.2017.16190. PubMed
4. Graudins A, Meek R, Parkinson J, Egerton-Warburton D, Meyer A. A randomised controlled trial of paracetamol and ibuprofen with or without codeine or oxycodone as initial analgesia for adults with moderate pain from limb injury. Emerg Med Australas. 2016;28(6):666-672. doi: 10.1111/1742-6723.12672 PubMed
5. Holdgate A, Pollock T. Nonsteroidal anti-inflammatory drugs (NSAIDs) versus opioids for acute renal colic. Cochrane Database Syst Rev. 2005:CD004137. doi: 10.1002/14651858.CD004137.pub3 PubMed
Hall et al. draw attention to the important question of whether some patients may benefit from a naloxone prescription when discharged from the hospital with a short-term opioid prescription for acute pain. Although all members of the working group agreed that naloxone is appropriate in some cases, we were hesitant to recommend this as a standard practice for several reasons.
First, the intent of our Consensus Statement1 was to synthesize and summarize the areas of consensus in existing guidelines; none of the existing guidelines included in our systematic review make a recommendation for naloxone prescription in the setting of short-term opioid use for acute pain.2 We believe that this may relate to the fact that the risk factors for overdose and the threshold of risk above which naloxone would be beneficial have yet to be defined for this population and are likely to differ from those defined in patients using opioids chronically.
Additionally, if practitioners follow the recommendations to limit prescribing for acute pain to the minimum dose and duration of an opioid that was presumably administered in the hospital with an observed response, then the risk of overdose and the potential benefit of naloxone will decrease. Furthermore, emerging data from randomized controlled trials demonstrating noninferiority of nonopioid analgesics in the management of acute pain suggest that we should not so readily presume opioids to be the necessary or the best option.3-5 Data questioning the benefits of opioids over other safer therapies have particularly important implications for patients in whom the risks are felt to be high enough to warrant consideration of naloxone.
Disclosures
Dr. Herzig reports receiving compensation from the Society of Hospital Medicine for her editorial role in the Journal of Hospital Medicine (unrelated to the present work). None of the other authors have any conflicts of interest to disclose.
Funding
Dr. Herzig is funded by a grant number K23AG042459 from the National Institute on Aging. Dr. Mosher is supported in part by the Department of Veterans Affairs Office of Academic Affiliations and the Office of Research and Development and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412). The views expressed in this manuscript do not necessarily represent the views of the funding agencies.
Hall et al. draw attention to the important question of whether some patients may benefit from a naloxone prescription when discharged from the hospital with a short-term opioid prescription for acute pain. Although all members of the working group agreed that naloxone is appropriate in some cases, we were hesitant to recommend this as a standard practice for several reasons.
First, the intent of our Consensus Statement1 was to synthesize and summarize the areas of consensus in existing guidelines; none of the existing guidelines included in our systematic review make a recommendation for naloxone prescription in the setting of short-term opioid use for acute pain.2 We believe that this may relate to the fact that the risk factors for overdose and the threshold of risk above which naloxone would be beneficial have yet to be defined for this population and are likely to differ from those defined in patients using opioids chronically.
Additionally, if practitioners follow the recommendations to limit prescribing for acute pain to the minimum dose and duration of an opioid that was presumably administered in the hospital with an observed response, then the risk of overdose and the potential benefit of naloxone will decrease. Furthermore, emerging data from randomized controlled trials demonstrating noninferiority of nonopioid analgesics in the management of acute pain suggest that we should not so readily presume opioids to be the necessary or the best option.3-5 Data questioning the benefits of opioids over other safer therapies have particularly important implications for patients in whom the risks are felt to be high enough to warrant consideration of naloxone.
Disclosures
Dr. Herzig reports receiving compensation from the Society of Hospital Medicine for her editorial role in the Journal of Hospital Medicine (unrelated to the present work). None of the other authors have any conflicts of interest to disclose.
Funding
Dr. Herzig is funded by a grant number K23AG042459 from the National Institute on Aging. Dr. Mosher is supported in part by the Department of Veterans Affairs Office of Academic Affiliations and the Office of Research and Development and Health Services Research and Development Service (HSR&D) through the Comprehensive Access and Delivery Research and Evaluation Center (CIN 13-412). The views expressed in this manuscript do not necessarily represent the views of the funding agencies.
1. Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK. Improving the safety of opioid use for acute noncancer pain in hospitalized adults: a consensus statement from the Society of Hospital Medicine. J Hosp Med. 2018;13(4):263-271. doi: 10.12788/jhm.2980. PubMed
2. Herzig SJ, Calcaterra SL, Mosher HJ, et al. Safe opioid prescribing for acute noncancer pain in hospitalized adults: a systematic review of existing guidelines.. J Hosp Med. 2018;13(4):256-262. doi: 10.12788/jhm.2979. PubMed
3. Chang AK, Bijur PE, Esses D, Barnaby DP, Baer J. Effect of a single dose of oral opioid and nonopioid analgesics on acute extremity pain in the emergency department: a randomized clinical trial. JAMA. 2017;318(17):1661-1667. doi: 10.1001/jama.2017.16190. PubMed
4. Graudins A, Meek R, Parkinson J, Egerton-Warburton D, Meyer A. A randomised controlled trial of paracetamol and ibuprofen with or without codeine or oxycodone as initial analgesia for adults with moderate pain from limb injury. Emerg Med Australas. 2016;28(6):666-672. doi: 10.1111/1742-6723.12672 PubMed
5. Holdgate A, Pollock T. Nonsteroidal anti-inflammatory drugs (NSAIDs) versus opioids for acute renal colic. Cochrane Database Syst Rev. 2005:CD004137. doi: 10.1002/14651858.CD004137.pub3 PubMed
1. Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK. Improving the safety of opioid use for acute noncancer pain in hospitalized adults: a consensus statement from the Society of Hospital Medicine. J Hosp Med. 2018;13(4):263-271. doi: 10.12788/jhm.2980. PubMed
2. Herzig SJ, Calcaterra SL, Mosher HJ, et al. Safe opioid prescribing for acute noncancer pain in hospitalized adults: a systematic review of existing guidelines.. J Hosp Med. 2018;13(4):256-262. doi: 10.12788/jhm.2979. PubMed
3. Chang AK, Bijur PE, Esses D, Barnaby DP, Baer J. Effect of a single dose of oral opioid and nonopioid analgesics on acute extremity pain in the emergency department: a randomized clinical trial. JAMA. 2017;318(17):1661-1667. doi: 10.1001/jama.2017.16190. PubMed
4. Graudins A, Meek R, Parkinson J, Egerton-Warburton D, Meyer A. A randomised controlled trial of paracetamol and ibuprofen with or without codeine or oxycodone as initial analgesia for adults with moderate pain from limb injury. Emerg Med Australas. 2016;28(6):666-672. doi: 10.1111/1742-6723.12672 PubMed
5. Holdgate A, Pollock T. Nonsteroidal anti-inflammatory drugs (NSAIDs) versus opioids for acute renal colic. Cochrane Database Syst Rev. 2005:CD004137. doi: 10.1002/14651858.CD004137.pub3 PubMed
In Reference to “Improving the Safety of Opioid Use for Acute Noncancer Pain in Hospitalized Adults: A Consensus Statement from the Society of Hospital Medicine”
We read with great interest the consensus statement on improving the safety of opioid use for acute noncancer pain by Herzig et al.1 We strongly support the recommendations outlined in the document.
However, we would like to advocate for an additional recommendation that was considered but not included by the authors. Given the proven benefit—with minimal risk—in providing naloxone to patients and family members, we encourage naloxone prescriptions at discharge for all patients at risk for opioid overdose independent of therapy duration.2 Even opioid-naive patients who are prescribed opioids at hospital discharge have a significantly higher risk for chronic opioid use.3
We support extrapolating recommendations from the Centers for Disease Control and Prevention and Substance Abuse and Mental Health Services Administration to prescribe naloxone to all patients at discharge who are at risk for an opioid overdose, including those with a history of overdose or substance use disorder as well as those receiving a prescription of ≥50 mg morphine equivalents per day or who use opioids and benzodiazepines.4,5
Given the current barriers to healthcare access, prescribing naloxone at discharge may be a rare opportunity to provide a potential life-saving intervention to prevent a fatal opioid overdose.
Disclosures
We have no relevant conflicts of interest to report. No payment or services from a third party were received for any aspect of this submitted work. We have no financial relationships with entities in the biomedical arena that could be perceived to influence, or that give the appearance of potentially influencing, what was written in this submitted work.
1. Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK. Improving the safety of opioid use for acute noncancer pain in hospitalized adults: a consensus statement from the society of hospital medicine. J Hosp Med. 2018;13(4);263-271. doi: 10.12788/jhm.2980. PubMed
2. McDonald R, Strang J. Are take-home naloxone programmes effective? Systematic review utilizing application of the Bradford Hill criteria. Addiction 2016;111(7):1177-1187. doi: 10.1111/add.13326. PubMed
3. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med. 2016;31(5):478-485. doi: 10.1007/s11606-015-3539-4. PubMed
4. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain--United States, 2016. JAMA. 2016;315(15):1624-1645. doi: 10.1001/jama.2016.1464. PubMed
5. Substance Abuse and Mental Health Services Administration. Medications for Opioid Use Disorder. Treatment Improvement Protocol (TIP) Series 63, Full Document. HHS Publication No. (SMA) 18- 5063FULLDOC. Rockville, MD: Substance Abuse and Mental Health Services Administration, 2018. Available at: https://store.samhsa.gov/shin/content//SMA18-5063FULLDOC/SMA18-5063FULLDOC.pdf. Accessed April 12, 2018.
We read with great interest the consensus statement on improving the safety of opioid use for acute noncancer pain by Herzig et al.1 We strongly support the recommendations outlined in the document.
However, we would like to advocate for an additional recommendation that was considered but not included by the authors. Given the proven benefit—with minimal risk—in providing naloxone to patients and family members, we encourage naloxone prescriptions at discharge for all patients at risk for opioid overdose independent of therapy duration.2 Even opioid-naive patients who are prescribed opioids at hospital discharge have a significantly higher risk for chronic opioid use.3
We support extrapolating recommendations from the Centers for Disease Control and Prevention and Substance Abuse and Mental Health Services Administration to prescribe naloxone to all patients at discharge who are at risk for an opioid overdose, including those with a history of overdose or substance use disorder as well as those receiving a prescription of ≥50 mg morphine equivalents per day or who use opioids and benzodiazepines.4,5
Given the current barriers to healthcare access, prescribing naloxone at discharge may be a rare opportunity to provide a potential life-saving intervention to prevent a fatal opioid overdose.
Disclosures
We have no relevant conflicts of interest to report. No payment or services from a third party were received for any aspect of this submitted work. We have no financial relationships with entities in the biomedical arena that could be perceived to influence, or that give the appearance of potentially influencing, what was written in this submitted work.
We read with great interest the consensus statement on improving the safety of opioid use for acute noncancer pain by Herzig et al.1 We strongly support the recommendations outlined in the document.
However, we would like to advocate for an additional recommendation that was considered but not included by the authors. Given the proven benefit—with minimal risk—in providing naloxone to patients and family members, we encourage naloxone prescriptions at discharge for all patients at risk for opioid overdose independent of therapy duration.2 Even opioid-naive patients who are prescribed opioids at hospital discharge have a significantly higher risk for chronic opioid use.3
We support extrapolating recommendations from the Centers for Disease Control and Prevention and Substance Abuse and Mental Health Services Administration to prescribe naloxone to all patients at discharge who are at risk for an opioid overdose, including those with a history of overdose or substance use disorder as well as those receiving a prescription of ≥50 mg morphine equivalents per day or who use opioids and benzodiazepines.4,5
Given the current barriers to healthcare access, prescribing naloxone at discharge may be a rare opportunity to provide a potential life-saving intervention to prevent a fatal opioid overdose.
Disclosures
We have no relevant conflicts of interest to report. No payment or services from a third party were received for any aspect of this submitted work. We have no financial relationships with entities in the biomedical arena that could be perceived to influence, or that give the appearance of potentially influencing, what was written in this submitted work.
1. Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK. Improving the safety of opioid use for acute noncancer pain in hospitalized adults: a consensus statement from the society of hospital medicine. J Hosp Med. 2018;13(4);263-271. doi: 10.12788/jhm.2980. PubMed
2. McDonald R, Strang J. Are take-home naloxone programmes effective? Systematic review utilizing application of the Bradford Hill criteria. Addiction 2016;111(7):1177-1187. doi: 10.1111/add.13326. PubMed
3. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med. 2016;31(5):478-485. doi: 10.1007/s11606-015-3539-4. PubMed
4. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain--United States, 2016. JAMA. 2016;315(15):1624-1645. doi: 10.1001/jama.2016.1464. PubMed
5. Substance Abuse and Mental Health Services Administration. Medications for Opioid Use Disorder. Treatment Improvement Protocol (TIP) Series 63, Full Document. HHS Publication No. (SMA) 18- 5063FULLDOC. Rockville, MD: Substance Abuse and Mental Health Services Administration, 2018. Available at: https://store.samhsa.gov/shin/content//SMA18-5063FULLDOC/SMA18-5063FULLDOC.pdf. Accessed April 12, 2018.
1. Herzig SJ, Mosher HJ, Calcaterra SL, Jena AB, Nuckols TK. Improving the safety of opioid use for acute noncancer pain in hospitalized adults: a consensus statement from the society of hospital medicine. J Hosp Med. 2018;13(4);263-271. doi: 10.12788/jhm.2980. PubMed
2. McDonald R, Strang J. Are take-home naloxone programmes effective? Systematic review utilizing application of the Bradford Hill criteria. Addiction 2016;111(7):1177-1187. doi: 10.1111/add.13326. PubMed
3. Calcaterra SL, Yamashita TE, Min SJ, Keniston A, Frank JW, Binswanger IA. Opioid prescribing at hospital discharge contributes to chronic opioid use. J Gen Intern Med. 2016;31(5):478-485. doi: 10.1007/s11606-015-3539-4. PubMed
4. Dowell D, Haegerich TM, Chou R. CDC Guideline for Prescribing Opioids for Chronic Pain--United States, 2016. JAMA. 2016;315(15):1624-1645. doi: 10.1001/jama.2016.1464. PubMed
5. Substance Abuse and Mental Health Services Administration. Medications for Opioid Use Disorder. Treatment Improvement Protocol (TIP) Series 63, Full Document. HHS Publication No. (SMA) 18- 5063FULLDOC. Rockville, MD: Substance Abuse and Mental Health Services Administration, 2018. Available at: https://store.samhsa.gov/shin/content//SMA18-5063FULLDOC/SMA18-5063FULLDOC.pdf. Accessed April 12, 2018.
© 2018 Society of Hospital Medicine
A Shooting in the Hospital: When Domestic Violence Occurs in the Hospital, Reflection, and Response
On September 12, 2017, a son walked into his mother’s room in the surgical intensive care unit (ICU) of Dartmouth-Hitchcock Medical Center (DHMC) in Lebanon, New Hampshire, and shot her with a handgun. As an actively practicing hospitalist and the Chief Clinical Officer for DHMC, I immediately became involved with our hospitals’ response to domestic violence, a homicide, and an issue that to this point we felt lived outside our walls.
Several hospital systems are struggling with violence entering their institutions, particularly in their psychiatry and emergency service areas, fueled in part by untreated mental health and the rising opioid epidemic. Although gun violence in hospitals is indeed rare, inside the hospital, it occurs often in the emergency department.1 In New Hampshire, we suffer from a woefully underfunded state mental health infrastructure and one of the highest opioid death rates in the United States.2
DHMC is a 400-bed academic medical center, level 1 trauma center, and a National Cancer Institute (NCI)-designated cancer center that serves New Hampshire and eastern Vermont with its community and critical access hospitals and community group practices across the two states. With a wide geographic catchment area, our academic hospital at DHMC has one of the highest case-mix indices in the northeastern United States and is in the top 30 among hospitals of >300 beds in the United States.
After the shooting, the patient’s son left the ICU without targeting anyone else, and despite video surveillance systems, he was not seen leaving the hospital. At the same time, a Code Blue was called to address the victim and her needs. The Critical Care staff struggled to attend to and resuscitate the victim, and my Medicine team, on call that day, was paged and rushed to the ICU to assist. In a unit trained to manage the sequelae of trauma, this event was painfully surreal. Ultimately, the surgical critical-care physician, attending to the patient, ended the resuscitation efforts when it was clear that the patient, now a homicide victim, could not be saved.
With the shooter’s whereabouts unknown, a Code Silver (Active Shooter alert) was called. Then, following our “Run-Hide-Fight” training protocol, staff, patients, and visitors exited the building in large numbers and those that could not, sheltered in place. The operating room and the emergency department were secured and continued to function.
More than 160 law enforcement officers, including trained tactical and SWAT teams, from 13 different agencies arrived on scene. Ninety minutes after the shooting, the son was apprehended at a police traffic checkpoint, attempting to leave the hospital campus.
Our involvement in this event did not end at this point. Concerned about the possibility of other suspects or devices left in the hospital, the law enforcement officers swept our hospital. With a 1.2 million square foot campus, this would take another two hours, during which we still provided care to our patients and asked the staff and families to continue to seek safe shelter.
The shock of this terrible day was immediate and profound, leading to a thorough debrief and systematic analysis of how we might improve our processes and in turn help other organizations that might unfortunately face similar situations.
We reflected on how to better secure our hospital and to strengthen our coordination and collaboration with law enforcement. We increased our security presence not only in the ICU but also in our emergency department and developed individual unit-based security measures. We fast-tracked a unit-based shutdown plan that was already in process and increased our commitments to plan and drill for larger scenarios in conjunction with law enforcement agencies.
The physical location of our hospital was important in how our response unfolded. DHMC’s unique rural location in northern New England added challenges specific to our location, which may provide an opportunity for other hospitals to consider. Although we were able to provide care, water, and transport during this tragedy on a warm day in September, caring for thousands of people outside a hospital during a typical subzero February would be a different story.
Communication during the event and how specifically to ask people to act were identified as a key area of improvement. We realized that our language and training around the various codes lacked clarity and specificity. As is familiar to many, in our hospital with Red, Blue, Black, Purple, and White codes, some staff (and certainly families and visitors) were not sure what to do in a “Code Silver.” We worked to better define our language so that in a future event or in a drill, we would state in plain language that we have “an active shooter” or a “violence with weapons” event in progress with clear instructions on next steps. Our term “Run-Hide-Fight” was changed to “Avoid-Hide-Fight” to better reflect updated training and best practice for a future event. We revised our teaching and training materials and protocols, so that in the event of a similar situation, we could provide information in plain language, across numerous formats, and with some frequency to keep people apprised, even if the situation is not changing.
Our methods of ongoing communications were also reassessed. In our reviews, it became clear that the notification systems and the computer-based alerts seen on the computers of hospital staff were different from those at the medical school. Communication protocols on pagers and mobile phones and across social media such as Facebook and Twitter were redesigned. Though our institution has long had the ability to provide cell phone notifications during emergencies, not all employees and staff had elected to activate this feature. We also improved our speaker systems so that overhead paging and alerts could be heard outside the building.
Having improved personal reference materials on hand is important. We updated the cards attached to our ID badges with clear instructions about “active shooter” or “violence with weapon” situations. We also developed different response scenarios dependent on the campus location. An event in the ICU, for example, might require leaving the scene, although sheltering-in-place might be more appropriate for an offsite administrative building.
A significant challenge to our active-shooter situation was making sure that our staff, patients, visitors, and their families were adequately supported following the event. Learning from the experiences of other hospitals and communities, we undertook a deliberate process of preparedness and healing.3 From our surgical ICU to our distant community group practices, we provided communication and avenues for personal support. Our Employee Assistance Program provided 24/7 support in a conference room in the surgical ICU and in other areas, on and off site, for all staff at Dartmouth-Hitchcock. The shooting affected those in the vicinity, as well as far away. Staff who had experienced domestic and other violence in their past were impacted in ways that required special care and attention. Some who were in adjacent rooms during the event were able to return to work immediately, whereas other staff, in separate units and more distant clinics, struggled and required leaves of absence. Through this event, we witnessed the personal and deep psychological impact of such violence. We held town halls, updated daily communications from our Incident Command Team, and maintained an open dialog across the organization.
In reflection, it is challenging to face this experience without the greater context of what we unfortunately experience all too often in America today. We have seen the spectrum from the shootings at Marjory Stoneman Douglas High School in Parkland, Florida, to the isolated events that rarely reach our national news and collective consciousness. It seems that we have already experienced a shooting at a school every week in the US.
There is even an overlap seen in domestic and mass shootings as we saw in the Sandy Hook Elementary School shootings in 2012, in which the tragic event was preceded by the shooter murdering his mother in her home.4 Today, in the US, women are disproportionally the subject of domestic violence, and more than half of all killed are done so by a male family member. The presence of a gun in domestic violence situations increases the risk for homicide for women by 500%.5- 7 Our experience indeed mirrored this reality.
Many readers of this piece will recognize how similar their situation is to that of our hospital, that this happens elsewhere, not here. Although my institution has faced this as a tragedy that has tested our organization, one cannot also be deeply troubled by the greater impact of domestic and gun violence on healthcare and the American society today. Our staff and physicians have been witness and at times subject to such violence, and this experience has now made it even more poignant. Ultimately, and sadly, we feel that we are more prepared.
Disclosures
The author has nothing to disclose.
1. Kelen GD, Catlett CL, Kunitz JG, Hsieh YH. Hospital-based shootings in the United States: 2000 to 2011. Ann Emerg Med. 2012;60(6):790-798. doi: 10.1016/j.annemergmed.2012.08.012. PubMed
2. Center for Disease Control and Preventions (CDC) Drug Overdose Death Data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Accessed April 10, 2018
3. Van Den Bos J, Creten N, Davenport S, Roberts, M. Cost of community violence to hospitals and health systems. Report for the American Hospital Association. July 26, 2017
4. Krouse WJ, Richardson DJ. Mass murder with firearms: incidents and victims, 1999-2013. Congressional Research Service. https://fas.org/sgp/crs/misc/R44126.pdf. Accessed April 10, 2018
5. Campbell JC, Webster D, Koziol-McLain J, et al. Risk factors for femicide within physically abusive intimate relationships. Am J Public Health. 2003;93(7):1089-1097. https:/doi.org/10.2105/AJPH.93.7.1089.
6. Fox JA, Zawitz MW. Homicide trends in the United States: Bureau of Justice Statistics; 2009.
7. Federal Bureau of Investigation. 2014 Crime in their United States. https://ucr.fbi.gov/crime-in-the-u.s/2014/crime-in-the-u.s.-2014/cius-home. Accessed April 10, 2018
On September 12, 2017, a son walked into his mother’s room in the surgical intensive care unit (ICU) of Dartmouth-Hitchcock Medical Center (DHMC) in Lebanon, New Hampshire, and shot her with a handgun. As an actively practicing hospitalist and the Chief Clinical Officer for DHMC, I immediately became involved with our hospitals’ response to domestic violence, a homicide, and an issue that to this point we felt lived outside our walls.
Several hospital systems are struggling with violence entering their institutions, particularly in their psychiatry and emergency service areas, fueled in part by untreated mental health and the rising opioid epidemic. Although gun violence in hospitals is indeed rare, inside the hospital, it occurs often in the emergency department.1 In New Hampshire, we suffer from a woefully underfunded state mental health infrastructure and one of the highest opioid death rates in the United States.2
DHMC is a 400-bed academic medical center, level 1 trauma center, and a National Cancer Institute (NCI)-designated cancer center that serves New Hampshire and eastern Vermont with its community and critical access hospitals and community group practices across the two states. With a wide geographic catchment area, our academic hospital at DHMC has one of the highest case-mix indices in the northeastern United States and is in the top 30 among hospitals of >300 beds in the United States.
After the shooting, the patient’s son left the ICU without targeting anyone else, and despite video surveillance systems, he was not seen leaving the hospital. At the same time, a Code Blue was called to address the victim and her needs. The Critical Care staff struggled to attend to and resuscitate the victim, and my Medicine team, on call that day, was paged and rushed to the ICU to assist. In a unit trained to manage the sequelae of trauma, this event was painfully surreal. Ultimately, the surgical critical-care physician, attending to the patient, ended the resuscitation efforts when it was clear that the patient, now a homicide victim, could not be saved.
With the shooter’s whereabouts unknown, a Code Silver (Active Shooter alert) was called. Then, following our “Run-Hide-Fight” training protocol, staff, patients, and visitors exited the building in large numbers and those that could not, sheltered in place. The operating room and the emergency department were secured and continued to function.
More than 160 law enforcement officers, including trained tactical and SWAT teams, from 13 different agencies arrived on scene. Ninety minutes after the shooting, the son was apprehended at a police traffic checkpoint, attempting to leave the hospital campus.
Our involvement in this event did not end at this point. Concerned about the possibility of other suspects or devices left in the hospital, the law enforcement officers swept our hospital. With a 1.2 million square foot campus, this would take another two hours, during which we still provided care to our patients and asked the staff and families to continue to seek safe shelter.
The shock of this terrible day was immediate and profound, leading to a thorough debrief and systematic analysis of how we might improve our processes and in turn help other organizations that might unfortunately face similar situations.
We reflected on how to better secure our hospital and to strengthen our coordination and collaboration with law enforcement. We increased our security presence not only in the ICU but also in our emergency department and developed individual unit-based security measures. We fast-tracked a unit-based shutdown plan that was already in process and increased our commitments to plan and drill for larger scenarios in conjunction with law enforcement agencies.
The physical location of our hospital was important in how our response unfolded. DHMC’s unique rural location in northern New England added challenges specific to our location, which may provide an opportunity for other hospitals to consider. Although we were able to provide care, water, and transport during this tragedy on a warm day in September, caring for thousands of people outside a hospital during a typical subzero February would be a different story.
Communication during the event and how specifically to ask people to act were identified as a key area of improvement. We realized that our language and training around the various codes lacked clarity and specificity. As is familiar to many, in our hospital with Red, Blue, Black, Purple, and White codes, some staff (and certainly families and visitors) were not sure what to do in a “Code Silver.” We worked to better define our language so that in a future event or in a drill, we would state in plain language that we have “an active shooter” or a “violence with weapons” event in progress with clear instructions on next steps. Our term “Run-Hide-Fight” was changed to “Avoid-Hide-Fight” to better reflect updated training and best practice for a future event. We revised our teaching and training materials and protocols, so that in the event of a similar situation, we could provide information in plain language, across numerous formats, and with some frequency to keep people apprised, even if the situation is not changing.
Our methods of ongoing communications were also reassessed. In our reviews, it became clear that the notification systems and the computer-based alerts seen on the computers of hospital staff were different from those at the medical school. Communication protocols on pagers and mobile phones and across social media such as Facebook and Twitter were redesigned. Though our institution has long had the ability to provide cell phone notifications during emergencies, not all employees and staff had elected to activate this feature. We also improved our speaker systems so that overhead paging and alerts could be heard outside the building.
Having improved personal reference materials on hand is important. We updated the cards attached to our ID badges with clear instructions about “active shooter” or “violence with weapon” situations. We also developed different response scenarios dependent on the campus location. An event in the ICU, for example, might require leaving the scene, although sheltering-in-place might be more appropriate for an offsite administrative building.
A significant challenge to our active-shooter situation was making sure that our staff, patients, visitors, and their families were adequately supported following the event. Learning from the experiences of other hospitals and communities, we undertook a deliberate process of preparedness and healing.3 From our surgical ICU to our distant community group practices, we provided communication and avenues for personal support. Our Employee Assistance Program provided 24/7 support in a conference room in the surgical ICU and in other areas, on and off site, for all staff at Dartmouth-Hitchcock. The shooting affected those in the vicinity, as well as far away. Staff who had experienced domestic and other violence in their past were impacted in ways that required special care and attention. Some who were in adjacent rooms during the event were able to return to work immediately, whereas other staff, in separate units and more distant clinics, struggled and required leaves of absence. Through this event, we witnessed the personal and deep psychological impact of such violence. We held town halls, updated daily communications from our Incident Command Team, and maintained an open dialog across the organization.
In reflection, it is challenging to face this experience without the greater context of what we unfortunately experience all too often in America today. We have seen the spectrum from the shootings at Marjory Stoneman Douglas High School in Parkland, Florida, to the isolated events that rarely reach our national news and collective consciousness. It seems that we have already experienced a shooting at a school every week in the US.
There is even an overlap seen in domestic and mass shootings as we saw in the Sandy Hook Elementary School shootings in 2012, in which the tragic event was preceded by the shooter murdering his mother in her home.4 Today, in the US, women are disproportionally the subject of domestic violence, and more than half of all killed are done so by a male family member. The presence of a gun in domestic violence situations increases the risk for homicide for women by 500%.5- 7 Our experience indeed mirrored this reality.
Many readers of this piece will recognize how similar their situation is to that of our hospital, that this happens elsewhere, not here. Although my institution has faced this as a tragedy that has tested our organization, one cannot also be deeply troubled by the greater impact of domestic and gun violence on healthcare and the American society today. Our staff and physicians have been witness and at times subject to such violence, and this experience has now made it even more poignant. Ultimately, and sadly, we feel that we are more prepared.
Disclosures
The author has nothing to disclose.
On September 12, 2017, a son walked into his mother’s room in the surgical intensive care unit (ICU) of Dartmouth-Hitchcock Medical Center (DHMC) in Lebanon, New Hampshire, and shot her with a handgun. As an actively practicing hospitalist and the Chief Clinical Officer for DHMC, I immediately became involved with our hospitals’ response to domestic violence, a homicide, and an issue that to this point we felt lived outside our walls.
Several hospital systems are struggling with violence entering their institutions, particularly in their psychiatry and emergency service areas, fueled in part by untreated mental health and the rising opioid epidemic. Although gun violence in hospitals is indeed rare, inside the hospital, it occurs often in the emergency department.1 In New Hampshire, we suffer from a woefully underfunded state mental health infrastructure and one of the highest opioid death rates in the United States.2
DHMC is a 400-bed academic medical center, level 1 trauma center, and a National Cancer Institute (NCI)-designated cancer center that serves New Hampshire and eastern Vermont with its community and critical access hospitals and community group practices across the two states. With a wide geographic catchment area, our academic hospital at DHMC has one of the highest case-mix indices in the northeastern United States and is in the top 30 among hospitals of >300 beds in the United States.
After the shooting, the patient’s son left the ICU without targeting anyone else, and despite video surveillance systems, he was not seen leaving the hospital. At the same time, a Code Blue was called to address the victim and her needs. The Critical Care staff struggled to attend to and resuscitate the victim, and my Medicine team, on call that day, was paged and rushed to the ICU to assist. In a unit trained to manage the sequelae of trauma, this event was painfully surreal. Ultimately, the surgical critical-care physician, attending to the patient, ended the resuscitation efforts when it was clear that the patient, now a homicide victim, could not be saved.
With the shooter’s whereabouts unknown, a Code Silver (Active Shooter alert) was called. Then, following our “Run-Hide-Fight” training protocol, staff, patients, and visitors exited the building in large numbers and those that could not, sheltered in place. The operating room and the emergency department were secured and continued to function.
More than 160 law enforcement officers, including trained tactical and SWAT teams, from 13 different agencies arrived on scene. Ninety minutes after the shooting, the son was apprehended at a police traffic checkpoint, attempting to leave the hospital campus.
Our involvement in this event did not end at this point. Concerned about the possibility of other suspects or devices left in the hospital, the law enforcement officers swept our hospital. With a 1.2 million square foot campus, this would take another two hours, during which we still provided care to our patients and asked the staff and families to continue to seek safe shelter.
The shock of this terrible day was immediate and profound, leading to a thorough debrief and systematic analysis of how we might improve our processes and in turn help other organizations that might unfortunately face similar situations.
We reflected on how to better secure our hospital and to strengthen our coordination and collaboration with law enforcement. We increased our security presence not only in the ICU but also in our emergency department and developed individual unit-based security measures. We fast-tracked a unit-based shutdown plan that was already in process and increased our commitments to plan and drill for larger scenarios in conjunction with law enforcement agencies.
The physical location of our hospital was important in how our response unfolded. DHMC’s unique rural location in northern New England added challenges specific to our location, which may provide an opportunity for other hospitals to consider. Although we were able to provide care, water, and transport during this tragedy on a warm day in September, caring for thousands of people outside a hospital during a typical subzero February would be a different story.
Communication during the event and how specifically to ask people to act were identified as a key area of improvement. We realized that our language and training around the various codes lacked clarity and specificity. As is familiar to many, in our hospital with Red, Blue, Black, Purple, and White codes, some staff (and certainly families and visitors) were not sure what to do in a “Code Silver.” We worked to better define our language so that in a future event or in a drill, we would state in plain language that we have “an active shooter” or a “violence with weapons” event in progress with clear instructions on next steps. Our term “Run-Hide-Fight” was changed to “Avoid-Hide-Fight” to better reflect updated training and best practice for a future event. We revised our teaching and training materials and protocols, so that in the event of a similar situation, we could provide information in plain language, across numerous formats, and with some frequency to keep people apprised, even if the situation is not changing.
Our methods of ongoing communications were also reassessed. In our reviews, it became clear that the notification systems and the computer-based alerts seen on the computers of hospital staff were different from those at the medical school. Communication protocols on pagers and mobile phones and across social media such as Facebook and Twitter were redesigned. Though our institution has long had the ability to provide cell phone notifications during emergencies, not all employees and staff had elected to activate this feature. We also improved our speaker systems so that overhead paging and alerts could be heard outside the building.
Having improved personal reference materials on hand is important. We updated the cards attached to our ID badges with clear instructions about “active shooter” or “violence with weapon” situations. We also developed different response scenarios dependent on the campus location. An event in the ICU, for example, might require leaving the scene, although sheltering-in-place might be more appropriate for an offsite administrative building.
A significant challenge to our active-shooter situation was making sure that our staff, patients, visitors, and their families were adequately supported following the event. Learning from the experiences of other hospitals and communities, we undertook a deliberate process of preparedness and healing.3 From our surgical ICU to our distant community group practices, we provided communication and avenues for personal support. Our Employee Assistance Program provided 24/7 support in a conference room in the surgical ICU and in other areas, on and off site, for all staff at Dartmouth-Hitchcock. The shooting affected those in the vicinity, as well as far away. Staff who had experienced domestic and other violence in their past were impacted in ways that required special care and attention. Some who were in adjacent rooms during the event were able to return to work immediately, whereas other staff, in separate units and more distant clinics, struggled and required leaves of absence. Through this event, we witnessed the personal and deep psychological impact of such violence. We held town halls, updated daily communications from our Incident Command Team, and maintained an open dialog across the organization.
In reflection, it is challenging to face this experience without the greater context of what we unfortunately experience all too often in America today. We have seen the spectrum from the shootings at Marjory Stoneman Douglas High School in Parkland, Florida, to the isolated events that rarely reach our national news and collective consciousness. It seems that we have already experienced a shooting at a school every week in the US.
There is even an overlap seen in domestic and mass shootings as we saw in the Sandy Hook Elementary School shootings in 2012, in which the tragic event was preceded by the shooter murdering his mother in her home.4 Today, in the US, women are disproportionally the subject of domestic violence, and more than half of all killed are done so by a male family member. The presence of a gun in domestic violence situations increases the risk for homicide for women by 500%.5- 7 Our experience indeed mirrored this reality.
Many readers of this piece will recognize how similar their situation is to that of our hospital, that this happens elsewhere, not here. Although my institution has faced this as a tragedy that has tested our organization, one cannot also be deeply troubled by the greater impact of domestic and gun violence on healthcare and the American society today. Our staff and physicians have been witness and at times subject to such violence, and this experience has now made it even more poignant. Ultimately, and sadly, we feel that we are more prepared.
Disclosures
The author has nothing to disclose.
1. Kelen GD, Catlett CL, Kunitz JG, Hsieh YH. Hospital-based shootings in the United States: 2000 to 2011. Ann Emerg Med. 2012;60(6):790-798. doi: 10.1016/j.annemergmed.2012.08.012. PubMed
2. Center for Disease Control and Preventions (CDC) Drug Overdose Death Data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Accessed April 10, 2018
3. Van Den Bos J, Creten N, Davenport S, Roberts, M. Cost of community violence to hospitals and health systems. Report for the American Hospital Association. July 26, 2017
4. Krouse WJ, Richardson DJ. Mass murder with firearms: incidents and victims, 1999-2013. Congressional Research Service. https://fas.org/sgp/crs/misc/R44126.pdf. Accessed April 10, 2018
5. Campbell JC, Webster D, Koziol-McLain J, et al. Risk factors for femicide within physically abusive intimate relationships. Am J Public Health. 2003;93(7):1089-1097. https:/doi.org/10.2105/AJPH.93.7.1089.
6. Fox JA, Zawitz MW. Homicide trends in the United States: Bureau of Justice Statistics; 2009.
7. Federal Bureau of Investigation. 2014 Crime in their United States. https://ucr.fbi.gov/crime-in-the-u.s/2014/crime-in-the-u.s.-2014/cius-home. Accessed April 10, 2018
1. Kelen GD, Catlett CL, Kunitz JG, Hsieh YH. Hospital-based shootings in the United States: 2000 to 2011. Ann Emerg Med. 2012;60(6):790-798. doi: 10.1016/j.annemergmed.2012.08.012. PubMed
2. Center for Disease Control and Preventions (CDC) Drug Overdose Death Data. https://www.cdc.gov/drugoverdose/data/statedeaths.html. Accessed April 10, 2018
3. Van Den Bos J, Creten N, Davenport S, Roberts, M. Cost of community violence to hospitals and health systems. Report for the American Hospital Association. July 26, 2017
4. Krouse WJ, Richardson DJ. Mass murder with firearms: incidents and victims, 1999-2013. Congressional Research Service. https://fas.org/sgp/crs/misc/R44126.pdf. Accessed April 10, 2018
5. Campbell JC, Webster D, Koziol-McLain J, et al. Risk factors for femicide within physically abusive intimate relationships. Am J Public Health. 2003;93(7):1089-1097. https:/doi.org/10.2105/AJPH.93.7.1089.
6. Fox JA, Zawitz MW. Homicide trends in the United States: Bureau of Justice Statistics; 2009.
7. Federal Bureau of Investigation. 2014 Crime in their United States. https://ucr.fbi.gov/crime-in-the-u.s/2014/crime-in-the-u.s.-2014/cius-home. Accessed April 10, 2018
© 2018 Society of Hospital Medicine
Inferior Vena Cava Filter Placement in Patients with Venous Thromboembolism without Contraindication to Anticoagulation
The “Things We Do for No Reason” (TWDFNR) series reviews practices that have become common parts of hospital care but may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent “black and white” conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/

A nticoagulation is the cornerstone of acute venous thromboembolism (VTE) management. Nonetheless, the use of inferior vena cava (IVC) filters in addition to anticoagulation is increasing, with wide variation in practice patterns and a growing recognition of filter-related complications. Rigorous randomized controlled data demonstrating that IVC filters, particularly the increasingly commonly placed retrievable filters, provide a mortality benefit are sparse. Given our review of IVC filter use and the lack of evidence demonstrating that IVC filters provide a mortality benefit, we recommend using anticoagulation alone for stable medical service patients admitted with acute VTE. In nuanced cases, hospitalists should engage in multidisciplinary care to develop individualized treatment options.
CASE PRESENTATION
A 65-year-old woman with a history of diabetes mellitus, metastatic breast cancer, and peptic ulcer disease presents to the Emergency Department for the evaluation of right thigh swelling, chest pain, and dyspnea after a transcontinental flight. Physical examination is notable for a pulse of 114 beats per minute, blood pressure of 136/93 mm Hg, respiratory rate of 14 breaths per minute, oxygen saturation of 95% on room air, and swelling of the right thigh. Computerized tomography imaging demonstrates multiple bilateral pulmonary emboli. Emergency department physicians begin anticoagulation and inform you that they have ordered the placement of a retrievable inferior vena cava (IVC) filter.
BACKGROUND
Acute venous thromboembolism (VTE) accounts for more than 500,000 hospitalizations in the United States each year.1 Although the management of VTE centers around anticoagulation, the concurrent use of IVC filters has increased over the past several decades.2 Several observational studies have attempted to quantify IVC filter usage and have shown that overall filter placement has increased at an impressive rate. Within two decades, the number of patients undergoing IVC filter placement has increased nearly 25 times from 2,000 in 1979 to 49,000 in 1999.2 Recent Medicare data show that claims for IVC filter placement procedures have increased from 30,756 in 1999 to 65,041 in 2008.3 IVC filter placement rates are higher in the US than in other developed countries; one review projected that in 2012, the IVC filter placement rate in a given population in the US is 25 times higher than that in a similar population in Europe.4
The guidelines for IVC filter usage are largely based on expert opinion, and solid data regarding this intervention are lacking. This combination is problematic, especially because the practice is becoming commonplace, and filter-related complications are increasingly recognized. Additionally, the appropriateness of filter use varies among providers, as evidenced by a retrospective study in which three VTE experts reviewed medical records to determine the appropriateness of filter placement. They unanimously agreed that filter use was appropriate in 51% of the cases, unanimously agreed that filter use was inappropriate in 26% of the cases, and lacked consensus on the appropriateness of filter use in 23% of the cases.5 The striking lack of consensus among experts underscores the wide range of opinion regarding the appropriateness of IVC filter placement on a case-by-case basis. Moreover, evidence suggests that physician adherence to guidelines for appropriate IVC filter use is suboptimal. One single-center study showed that only 43.5% of filters placed by interventional radiology practitioners met the guidelines established by the American College of Chest Physicians (ACCP), with a slightly increased percentage of filter placement meeting guidelines if the requesting provider is an IM-trained physician.6
WHY YOU MIGHT THINK IVC FILTER PLACEMENT IS HELPFUL IN PATIENTS WITH VTE WITHOUT CONTRAINDICATION TO ANTICOAGULATION
In theory, the concept of IVC filters makes intuitive sense—filters block the ascent of any thrombus from the lower extremities to prevent the feared complication of a pulmonary embolism (PE). Unfortunately, rigorous data are limited, and consensus guidelines vary between different specialty organizations, further obfuscating the role of IVC filter placement in the management of VTE. For example, the ACCP recommends against the use of IVC filters in most patients with VTE receiving anticoagulation and does not list any prophylactic indications.7,8 Meanwhile, the Society of Interventional Radiology lists prophylactic indications for IVC filter placement in certain patient populations, such patients with a risk of VTE and a high risk of bleeding, and notes numerous relative indications for IVC filter placement.8 Notably, these differences in expert opinion likely influence practice patterns, as evidenced by the increase in IVC filter placement for relative indications.9,10
WHY IVC FILTERS PLACEMENT IN PATIENTS WITH VTE WHO CAN BE ANTICOAGULATED IS NOT HELPFUL
The Prevention du Risque d’Embolie Pulmonaire par Interruption Cave (PRECIP) trial is the most robust study supporting the 2016 ACCP recommendation against IVC filter use in patients that can receive anticoagulation.7,11 This study randomized 400 patients with deep vein thrombosis (DVT) at high risk for PE to anticoagulation with or without permanent filter placement to address VTE and mortality rates associated with IVC filter placement. The trial showed that the VTE burden shifts in the presence of IVC filters. At 2-year follow-up, the group with IVC filters had nonsignificantly fewer PEs than the control group and an increased incidence of DVT. Mortality rates did not differ between groups.11 At eight-year follow-up this shift in VTE burden is again seen given that the number of PEs in patients who received IVC filters decreased and the incidence of DVTs increased. Again, mortality did not differ between groups.12 A subsequent study randomized 399 patients with DVT and acute symptomatic PE with at least one additional marker of severity to anticoagulation with or without retrievable IVC filter placement and showed no difference in recurrent PE or mortality at 3 or 6 months.13 These results argue against placing retrievable filters in patients receiving anticoagulation.
The identification of associated adverse events further favor the judicious use of IVC filters. A retrospective review of the long-term complications of IVC filters based on imaging data showed a 14% fracture rate, 13% IVC thrombosis rate, and a 48% perforation rate.14 Multiple studies have shown that the associated complication rates of retrievable filters are higher than those of permanent filters; such an association is concerning given that retrievable filter usage exceeds permanent filter usage.14,15 The increase in retrievable filter usage is likely attributable to their attractive risk-benefit calculation. In theory, retrievable IVC filters should be perfect for patients who have conditions that increase VTE risk but create temporary contraindications, such as trauma or major surgery, to anticoagulation. However, anticoagulation is preferred over IVC filters in the long term because the complication rates of IVC filters increase with dwell time.16 Given the reports of adverse events and concern that IVC filters are not appropriately removed, the Food and Drug Administration recommends removing retrievable IVC filters once the risk of filters outweighs the benefits, which appears to be 29-54 days after implantation.17 However, successful retrieval rates are low, both because of the low rates of removal attempts and because of the interference of complications, such as embedded or thrombosed filters, with removal.10,18 As an example, in a retrospective review of all patients who received an IVC filter at an academic medical center over the period of 2003-2011, nearly 25% of patients were discharged on anticoagulation after IVC filter placement.10 This suggests that their contraindication to anticoagulation and need for IVC placement have passed by the time of discharge. Nevertheless, clinicians attempted filter retrieval in only 9.6% of these patients, representing a significant missed opportunity of treatment with anticoagulation rather than IVC filters.10
Factors such as filter plan documentation, hematology involvement, patient age ≤70 years, and establishment of dedicated IVC filter clinics are correlated with improved rates of filter removal; these correlations emphasize the importance of a clear follow-up plan in the timely removal of these devices.18,19
WHEN MIGHT IT BE HELPFUL TO PLACE IVC FILTERS IN PATIENTS WITH NO CONTRAINDICATION TO ANTICOAGULATION?
IVC filter placement is inappropriate in the vast majority of patients with VTE who can be anticoagulated. However the ACCP does acknowledge that a small subset of patients – specifically, those with severe or massive PE – may fall outside this guideline.7 Clinicians fear that these patients have low cardiopulmonary reserve and may experience hemodynamic collapse and death with another “hit” from a recurrent PE. This recommendation is consistent with the evidence that in unstable patients with PE, IVC filter placement is associated with decreased in-hospital mortality.20 Data remain limited for this situation, and the decision to place an IVC filter in anticoagulated but unstable patients is an individualized one.
WHAT YOU SHOULD DO INSTEAD: REFRAIN FROM IVC FILTER PLACEMENT AND TREAT WITH SYSTEMIC ANTICOAGULATION
In stable patients admitted to the medical service with VTE and who can be anticoagulated, there is little evidence that placement of an IVC filter will improve short- or long-term mortality. Hospitalists should anticoagulate these patients with a vitamin-K antagonist, heparin product, or novel oral anticoagulants.
RECOMMENDATIONS
- Anticoagulate hemodynamically stable patients who are admitted to the medical service with VTE and who do not have a contraindication to anticoagulation. Do not place a permanent or retrievable IVC filter.
- IVC filter placement may benefit unstable patients who may experience hemodynamic collapse with an increased PE burden. IVC filter placement should be discussed with a multidisciplinary team.
- When discharging a patient with an IVC filter, hospitalists should improve retrieval rates by scheduling subsequent removal. The discharge summary should contain information about the IVC filter, as well as clear instructions regarding the plan for removal. The instructions should include radiology follow-up information and the designation of responsible physicians in case of questions.
CONCLUSION
Although IVC filter use is increasing, the evidence does not support their use in hemodynamically stable patients who can be anticoagulated. The patient described in the initial case has no contraindication to systemic anticoagulation. Therefore, she should be started on anticoagulation, and an IVC filter should not be placed.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason?” Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason” topics by emailing[email protected].
Disclosures
The authors do not have any conflicts of interest to disclose
1. Centers for Disease Control and Prevention. Venous thromboembolism in adult hospitalizations – United States, 2007-2009. MMWR. 2012;61:401-404. PubMed
2. Stein PD, Kayali F, Olson RE. Twenty-one-year trends in the use of inferior vena cava filters. Arch Intern Med. 2004;164(14):1541-1545. doi: 10.1001/archinte.164.14.1541 PubMed
3. Duszak R Jr, Parker L, Levin DC, Rao VM. Placement and removal of inferior vena cava filters: national trends in the Medicare population. J Am Coll Radiol. 2011;8(7):483-489. doi: 10.1016/j.jacr.2010.12.021. PubMed
4. Wang SL, Llyod AJ. Clinical review: inferior vena cava filters in the age of patient-centered outcomes. Ann Med. 2013;45(7):474-481. doi: 10.3109/07853890.2013.832951. PubMed
5. Spencer FA, Bates SM, Goldberg RJ, et al. A population-based study of inferior vena cava filters in patients with acute venous thromboembolism. Arch Intern Med.2010;170(16):1456-1462. doi: 10.1001/archinternmed.2010.272. PubMed
6. Baadh AS, Zikria JF, Rivioli S, et al. Indications for inferior vena cava filter placement: do physicians comply with guidelines? J Vasc Interv Radiol. 2012;23(8):989-995. doi: 10.1016/j.jvir.2012.04.017. PubMed
7. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149(2):315-352. doi: 10.1016/j.chest.2015.11.026. PubMed
8. Kaufman JA, Kinney TB, Streiff MB, et al. Guidelines for the use of retrievable and convertible vena cava filters: report from the Society of Interventional Radiology multidisciplinary consensus conference. J Vasc Interv Radiol. 2006;17(3):449-459. doi: 10.1097/01.rvi.0000203418.39769.0d. PubMed
9. Tao MJ, Montbriand JM, Eisenberg N, Sniderman KW, Roche-Nagle G. Temporary inferior vena cava filter indications, retrieval rates, and follow-up management at a multicenter tertiary care institution. J Vasc Surg. 2016;64(2):430-437. doi: 10.1016/j.jvs.2016.02.034. PubMed
10. Sarosiek S, Crowther M, Sloan JM. Indications, complications, and management of inferior vena cava filters. JAMA Intern Med.2013;173(7):513-517. doi: 10.1001/jamainternmed.2013.343. PubMed
11. Decousus H, Leizorovicz A, Parent F, et al. A clinical trial of vena cava filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med. 1998;338(7):409-415. doi: 10.1056/NEJM199802123380701. PubMed
12. PRECIP Study Group. Eight-year follow up of patients with permanent vena cava filters in the prevention of pulmonary embolism. Circulation. 2005;112(3):416-422. doi: 10.1161/CIRCULATIONAHA.104.512834. PubMed
13. Mismetti P, Laporte S, Pellerin O, et al. Effect of a retrievable inferior vena cava filter plus anticoagulation vs anticoagulation alone on risk of recurrent pulmonary embolism. JAMA. 2015;313(16):1627-1635. doi: 10.1001/jama.2015.3780. PubMed
14. Wang SL, Siddiqui A, Rosenthal E. Long-term complications of inferior vena cava filters. J Vasc Surg Venous Lymphat Disord. 2017;5(1):33-41. doi: 10.1016/j.jvsv.2016.07.002. PubMed
15. Andreoli JM, Lewandowski RJ, Vogelzang RL, Ryu RK. Comparison of complication rates associated with permanent and retrievable inferior vena cava filters: a review of the MAUDE database. J Vasc Interv Radiol. 2014;25(8):1181-1185. doi: 10.1016/j.jvir.2014.04.016. PubMed
16. Vijay K, Hughes JA, Burdette AS, et al. Fractured bard Recovery, G2, and G2 Express inferior vena cava filters: incidence, clinical consequences, and outcomes of removal attempts. J Vasc Interv Radiol. 2012;23(2):188-194. doi: 10.1016/j.jvir.2011.10.005. PubMed
17. Removing Retrievable Inferior Vena Cava Filters: FDA Safety Communication. FDA.gov. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm396377.htm. Published May 6, 2014. Accessed April 10, 2017.
18. Peterson EA, Yenson PR, Liu D, Lee AYY. Predictors of attempted inferior vena cava filters retrieval in a tertiary care centre. Thromb Res. 2014;134(2):300-304. doi: 10.1016/j.thromres.2014.05.029. PubMed
19. Minocha J, Idakoji I, Riaz A, et al. Improving inferior vena cava filter retrieval rates: impact of a dedicated inferior vena cava filter clinic. J Vasc Interv Radiol. 2010;21(12):1847-1851. doi: 10.1016/j.jvir.2010.09.003. PubMed
20. Stein PD, Matta F, Keyes DC, Willyerd GL. Impact of vena cava filters on in-hospital case fatality rate from pulmonary embolism. Am J Med. 2012;125(5):478-484. doi: 10.1016/j.amjmed.2011.05.025. PubMed
The “Things We Do for No Reason” (TWDFNR) series reviews practices that have become common parts of hospital care but may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent “black and white” conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
A nticoagulation is the cornerstone of acute venous thromboembolism (VTE) management. Nonetheless, the use of inferior vena cava (IVC) filters in addition to anticoagulation is increasing, with wide variation in practice patterns and a growing recognition of filter-related complications. Rigorous randomized controlled data demonstrating that IVC filters, particularly the increasingly commonly placed retrievable filters, provide a mortality benefit are sparse. Given our review of IVC filter use and the lack of evidence demonstrating that IVC filters provide a mortality benefit, we recommend using anticoagulation alone for stable medical service patients admitted with acute VTE. In nuanced cases, hospitalists should engage in multidisciplinary care to develop individualized treatment options.
CASE PRESENTATION
A 65-year-old woman with a history of diabetes mellitus, metastatic breast cancer, and peptic ulcer disease presents to the Emergency Department for the evaluation of right thigh swelling, chest pain, and dyspnea after a transcontinental flight. Physical examination is notable for a pulse of 114 beats per minute, blood pressure of 136/93 mm Hg, respiratory rate of 14 breaths per minute, oxygen saturation of 95% on room air, and swelling of the right thigh. Computerized tomography imaging demonstrates multiple bilateral pulmonary emboli. Emergency department physicians begin anticoagulation and inform you that they have ordered the placement of a retrievable inferior vena cava (IVC) filter.
BACKGROUND
Acute venous thromboembolism (VTE) accounts for more than 500,000 hospitalizations in the United States each year.1 Although the management of VTE centers around anticoagulation, the concurrent use of IVC filters has increased over the past several decades.2 Several observational studies have attempted to quantify IVC filter usage and have shown that overall filter placement has increased at an impressive rate. Within two decades, the number of patients undergoing IVC filter placement has increased nearly 25 times from 2,000 in 1979 to 49,000 in 1999.2 Recent Medicare data show that claims for IVC filter placement procedures have increased from 30,756 in 1999 to 65,041 in 2008.3 IVC filter placement rates are higher in the US than in other developed countries; one review projected that in 2012, the IVC filter placement rate in a given population in the US is 25 times higher than that in a similar population in Europe.4
The guidelines for IVC filter usage are largely based on expert opinion, and solid data regarding this intervention are lacking. This combination is problematic, especially because the practice is becoming commonplace, and filter-related complications are increasingly recognized. Additionally, the appropriateness of filter use varies among providers, as evidenced by a retrospective study in which three VTE experts reviewed medical records to determine the appropriateness of filter placement. They unanimously agreed that filter use was appropriate in 51% of the cases, unanimously agreed that filter use was inappropriate in 26% of the cases, and lacked consensus on the appropriateness of filter use in 23% of the cases.5 The striking lack of consensus among experts underscores the wide range of opinion regarding the appropriateness of IVC filter placement on a case-by-case basis. Moreover, evidence suggests that physician adherence to guidelines for appropriate IVC filter use is suboptimal. One single-center study showed that only 43.5% of filters placed by interventional radiology practitioners met the guidelines established by the American College of Chest Physicians (ACCP), with a slightly increased percentage of filter placement meeting guidelines if the requesting provider is an IM-trained physician.6
WHY YOU MIGHT THINK IVC FILTER PLACEMENT IS HELPFUL IN PATIENTS WITH VTE WITHOUT CONTRAINDICATION TO ANTICOAGULATION
In theory, the concept of IVC filters makes intuitive sense—filters block the ascent of any thrombus from the lower extremities to prevent the feared complication of a pulmonary embolism (PE). Unfortunately, rigorous data are limited, and consensus guidelines vary between different specialty organizations, further obfuscating the role of IVC filter placement in the management of VTE. For example, the ACCP recommends against the use of IVC filters in most patients with VTE receiving anticoagulation and does not list any prophylactic indications.7,8 Meanwhile, the Society of Interventional Radiology lists prophylactic indications for IVC filter placement in certain patient populations, such patients with a risk of VTE and a high risk of bleeding, and notes numerous relative indications for IVC filter placement.8 Notably, these differences in expert opinion likely influence practice patterns, as evidenced by the increase in IVC filter placement for relative indications.9,10
WHY IVC FILTERS PLACEMENT IN PATIENTS WITH VTE WHO CAN BE ANTICOAGULATED IS NOT HELPFUL
The Prevention du Risque d’Embolie Pulmonaire par Interruption Cave (PRECIP) trial is the most robust study supporting the 2016 ACCP recommendation against IVC filter use in patients that can receive anticoagulation.7,11 This study randomized 400 patients with deep vein thrombosis (DVT) at high risk for PE to anticoagulation with or without permanent filter placement to address VTE and mortality rates associated with IVC filter placement. The trial showed that the VTE burden shifts in the presence of IVC filters. At 2-year follow-up, the group with IVC filters had nonsignificantly fewer PEs than the control group and an increased incidence of DVT. Mortality rates did not differ between groups.11 At eight-year follow-up this shift in VTE burden is again seen given that the number of PEs in patients who received IVC filters decreased and the incidence of DVTs increased. Again, mortality did not differ between groups.12 A subsequent study randomized 399 patients with DVT and acute symptomatic PE with at least one additional marker of severity to anticoagulation with or without retrievable IVC filter placement and showed no difference in recurrent PE or mortality at 3 or 6 months.13 These results argue against placing retrievable filters in patients receiving anticoagulation.
The identification of associated adverse events further favor the judicious use of IVC filters. A retrospective review of the long-term complications of IVC filters based on imaging data showed a 14% fracture rate, 13% IVC thrombosis rate, and a 48% perforation rate.14 Multiple studies have shown that the associated complication rates of retrievable filters are higher than those of permanent filters; such an association is concerning given that retrievable filter usage exceeds permanent filter usage.14,15 The increase in retrievable filter usage is likely attributable to their attractive risk-benefit calculation. In theory, retrievable IVC filters should be perfect for patients who have conditions that increase VTE risk but create temporary contraindications, such as trauma or major surgery, to anticoagulation. However, anticoagulation is preferred over IVC filters in the long term because the complication rates of IVC filters increase with dwell time.16 Given the reports of adverse events and concern that IVC filters are not appropriately removed, the Food and Drug Administration recommends removing retrievable IVC filters once the risk of filters outweighs the benefits, which appears to be 29-54 days after implantation.17 However, successful retrieval rates are low, both because of the low rates of removal attempts and because of the interference of complications, such as embedded or thrombosed filters, with removal.10,18 As an example, in a retrospective review of all patients who received an IVC filter at an academic medical center over the period of 2003-2011, nearly 25% of patients were discharged on anticoagulation after IVC filter placement.10 This suggests that their contraindication to anticoagulation and need for IVC placement have passed by the time of discharge. Nevertheless, clinicians attempted filter retrieval in only 9.6% of these patients, representing a significant missed opportunity of treatment with anticoagulation rather than IVC filters.10
Factors such as filter plan documentation, hematology involvement, patient age ≤70 years, and establishment of dedicated IVC filter clinics are correlated with improved rates of filter removal; these correlations emphasize the importance of a clear follow-up plan in the timely removal of these devices.18,19
WHEN MIGHT IT BE HELPFUL TO PLACE IVC FILTERS IN PATIENTS WITH NO CONTRAINDICATION TO ANTICOAGULATION?
IVC filter placement is inappropriate in the vast majority of patients with VTE who can be anticoagulated. However the ACCP does acknowledge that a small subset of patients – specifically, those with severe or massive PE – may fall outside this guideline.7 Clinicians fear that these patients have low cardiopulmonary reserve and may experience hemodynamic collapse and death with another “hit” from a recurrent PE. This recommendation is consistent with the evidence that in unstable patients with PE, IVC filter placement is associated with decreased in-hospital mortality.20 Data remain limited for this situation, and the decision to place an IVC filter in anticoagulated but unstable patients is an individualized one.
WHAT YOU SHOULD DO INSTEAD: REFRAIN FROM IVC FILTER PLACEMENT AND TREAT WITH SYSTEMIC ANTICOAGULATION
In stable patients admitted to the medical service with VTE and who can be anticoagulated, there is little evidence that placement of an IVC filter will improve short- or long-term mortality. Hospitalists should anticoagulate these patients with a vitamin-K antagonist, heparin product, or novel oral anticoagulants.
RECOMMENDATIONS
- Anticoagulate hemodynamically stable patients who are admitted to the medical service with VTE and who do not have a contraindication to anticoagulation. Do not place a permanent or retrievable IVC filter.
- IVC filter placement may benefit unstable patients who may experience hemodynamic collapse with an increased PE burden. IVC filter placement should be discussed with a multidisciplinary team.
- When discharging a patient with an IVC filter, hospitalists should improve retrieval rates by scheduling subsequent removal. The discharge summary should contain information about the IVC filter, as well as clear instructions regarding the plan for removal. The instructions should include radiology follow-up information and the designation of responsible physicians in case of questions.
CONCLUSION
Although IVC filter use is increasing, the evidence does not support their use in hemodynamically stable patients who can be anticoagulated. The patient described in the initial case has no contraindication to systemic anticoagulation. Therefore, she should be started on anticoagulation, and an IVC filter should not be placed.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason?” Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason” topics by emailing[email protected].
Disclosures
The authors do not have any conflicts of interest to disclose
The “Things We Do for No Reason” (TWDFNR) series reviews practices that have become common parts of hospital care but may provide little value to our patients. Practices reviewed in the TWDFNR series do not represent “black and white” conclusions or clinical practice standards but are meant as a starting place for research and active discussions among hospitalists and patients. We invite you to be part of that discussion. https://www.choosingwisely.org/
A nticoagulation is the cornerstone of acute venous thromboembolism (VTE) management. Nonetheless, the use of inferior vena cava (IVC) filters in addition to anticoagulation is increasing, with wide variation in practice patterns and a growing recognition of filter-related complications. Rigorous randomized controlled data demonstrating that IVC filters, particularly the increasingly commonly placed retrievable filters, provide a mortality benefit are sparse. Given our review of IVC filter use and the lack of evidence demonstrating that IVC filters provide a mortality benefit, we recommend using anticoagulation alone for stable medical service patients admitted with acute VTE. In nuanced cases, hospitalists should engage in multidisciplinary care to develop individualized treatment options.
CASE PRESENTATION
A 65-year-old woman with a history of diabetes mellitus, metastatic breast cancer, and peptic ulcer disease presents to the Emergency Department for the evaluation of right thigh swelling, chest pain, and dyspnea after a transcontinental flight. Physical examination is notable for a pulse of 114 beats per minute, blood pressure of 136/93 mm Hg, respiratory rate of 14 breaths per minute, oxygen saturation of 95% on room air, and swelling of the right thigh. Computerized tomography imaging demonstrates multiple bilateral pulmonary emboli. Emergency department physicians begin anticoagulation and inform you that they have ordered the placement of a retrievable inferior vena cava (IVC) filter.
BACKGROUND
Acute venous thromboembolism (VTE) accounts for more than 500,000 hospitalizations in the United States each year.1 Although the management of VTE centers around anticoagulation, the concurrent use of IVC filters has increased over the past several decades.2 Several observational studies have attempted to quantify IVC filter usage and have shown that overall filter placement has increased at an impressive rate. Within two decades, the number of patients undergoing IVC filter placement has increased nearly 25 times from 2,000 in 1979 to 49,000 in 1999.2 Recent Medicare data show that claims for IVC filter placement procedures have increased from 30,756 in 1999 to 65,041 in 2008.3 IVC filter placement rates are higher in the US than in other developed countries; one review projected that in 2012, the IVC filter placement rate in a given population in the US is 25 times higher than that in a similar population in Europe.4
The guidelines for IVC filter usage are largely based on expert opinion, and solid data regarding this intervention are lacking. This combination is problematic, especially because the practice is becoming commonplace, and filter-related complications are increasingly recognized. Additionally, the appropriateness of filter use varies among providers, as evidenced by a retrospective study in which three VTE experts reviewed medical records to determine the appropriateness of filter placement. They unanimously agreed that filter use was appropriate in 51% of the cases, unanimously agreed that filter use was inappropriate in 26% of the cases, and lacked consensus on the appropriateness of filter use in 23% of the cases.5 The striking lack of consensus among experts underscores the wide range of opinion regarding the appropriateness of IVC filter placement on a case-by-case basis. Moreover, evidence suggests that physician adherence to guidelines for appropriate IVC filter use is suboptimal. One single-center study showed that only 43.5% of filters placed by interventional radiology practitioners met the guidelines established by the American College of Chest Physicians (ACCP), with a slightly increased percentage of filter placement meeting guidelines if the requesting provider is an IM-trained physician.6
WHY YOU MIGHT THINK IVC FILTER PLACEMENT IS HELPFUL IN PATIENTS WITH VTE WITHOUT CONTRAINDICATION TO ANTICOAGULATION
In theory, the concept of IVC filters makes intuitive sense—filters block the ascent of any thrombus from the lower extremities to prevent the feared complication of a pulmonary embolism (PE). Unfortunately, rigorous data are limited, and consensus guidelines vary between different specialty organizations, further obfuscating the role of IVC filter placement in the management of VTE. For example, the ACCP recommends against the use of IVC filters in most patients with VTE receiving anticoagulation and does not list any prophylactic indications.7,8 Meanwhile, the Society of Interventional Radiology lists prophylactic indications for IVC filter placement in certain patient populations, such patients with a risk of VTE and a high risk of bleeding, and notes numerous relative indications for IVC filter placement.8 Notably, these differences in expert opinion likely influence practice patterns, as evidenced by the increase in IVC filter placement for relative indications.9,10
WHY IVC FILTERS PLACEMENT IN PATIENTS WITH VTE WHO CAN BE ANTICOAGULATED IS NOT HELPFUL
The Prevention du Risque d’Embolie Pulmonaire par Interruption Cave (PRECIP) trial is the most robust study supporting the 2016 ACCP recommendation against IVC filter use in patients that can receive anticoagulation.7,11 This study randomized 400 patients with deep vein thrombosis (DVT) at high risk for PE to anticoagulation with or without permanent filter placement to address VTE and mortality rates associated with IVC filter placement. The trial showed that the VTE burden shifts in the presence of IVC filters. At 2-year follow-up, the group with IVC filters had nonsignificantly fewer PEs than the control group and an increased incidence of DVT. Mortality rates did not differ between groups.11 At eight-year follow-up this shift in VTE burden is again seen given that the number of PEs in patients who received IVC filters decreased and the incidence of DVTs increased. Again, mortality did not differ between groups.12 A subsequent study randomized 399 patients with DVT and acute symptomatic PE with at least one additional marker of severity to anticoagulation with or without retrievable IVC filter placement and showed no difference in recurrent PE or mortality at 3 or 6 months.13 These results argue against placing retrievable filters in patients receiving anticoagulation.
The identification of associated adverse events further favor the judicious use of IVC filters. A retrospective review of the long-term complications of IVC filters based on imaging data showed a 14% fracture rate, 13% IVC thrombosis rate, and a 48% perforation rate.14 Multiple studies have shown that the associated complication rates of retrievable filters are higher than those of permanent filters; such an association is concerning given that retrievable filter usage exceeds permanent filter usage.14,15 The increase in retrievable filter usage is likely attributable to their attractive risk-benefit calculation. In theory, retrievable IVC filters should be perfect for patients who have conditions that increase VTE risk but create temporary contraindications, such as trauma or major surgery, to anticoagulation. However, anticoagulation is preferred over IVC filters in the long term because the complication rates of IVC filters increase with dwell time.16 Given the reports of adverse events and concern that IVC filters are not appropriately removed, the Food and Drug Administration recommends removing retrievable IVC filters once the risk of filters outweighs the benefits, which appears to be 29-54 days after implantation.17 However, successful retrieval rates are low, both because of the low rates of removal attempts and because of the interference of complications, such as embedded or thrombosed filters, with removal.10,18 As an example, in a retrospective review of all patients who received an IVC filter at an academic medical center over the period of 2003-2011, nearly 25% of patients were discharged on anticoagulation after IVC filter placement.10 This suggests that their contraindication to anticoagulation and need for IVC placement have passed by the time of discharge. Nevertheless, clinicians attempted filter retrieval in only 9.6% of these patients, representing a significant missed opportunity of treatment with anticoagulation rather than IVC filters.10
Factors such as filter plan documentation, hematology involvement, patient age ≤70 years, and establishment of dedicated IVC filter clinics are correlated with improved rates of filter removal; these correlations emphasize the importance of a clear follow-up plan in the timely removal of these devices.18,19
WHEN MIGHT IT BE HELPFUL TO PLACE IVC FILTERS IN PATIENTS WITH NO CONTRAINDICATION TO ANTICOAGULATION?
IVC filter placement is inappropriate in the vast majority of patients with VTE who can be anticoagulated. However the ACCP does acknowledge that a small subset of patients – specifically, those with severe or massive PE – may fall outside this guideline.7 Clinicians fear that these patients have low cardiopulmonary reserve and may experience hemodynamic collapse and death with another “hit” from a recurrent PE. This recommendation is consistent with the evidence that in unstable patients with PE, IVC filter placement is associated with decreased in-hospital mortality.20 Data remain limited for this situation, and the decision to place an IVC filter in anticoagulated but unstable patients is an individualized one.
WHAT YOU SHOULD DO INSTEAD: REFRAIN FROM IVC FILTER PLACEMENT AND TREAT WITH SYSTEMIC ANTICOAGULATION
In stable patients admitted to the medical service with VTE and who can be anticoagulated, there is little evidence that placement of an IVC filter will improve short- or long-term mortality. Hospitalists should anticoagulate these patients with a vitamin-K antagonist, heparin product, or novel oral anticoagulants.
RECOMMENDATIONS
- Anticoagulate hemodynamically stable patients who are admitted to the medical service with VTE and who do not have a contraindication to anticoagulation. Do not place a permanent or retrievable IVC filter.
- IVC filter placement may benefit unstable patients who may experience hemodynamic collapse with an increased PE burden. IVC filter placement should be discussed with a multidisciplinary team.
- When discharging a patient with an IVC filter, hospitalists should improve retrieval rates by scheduling subsequent removal. The discharge summary should contain information about the IVC filter, as well as clear instructions regarding the plan for removal. The instructions should include radiology follow-up information and the designation of responsible physicians in case of questions.
CONCLUSION
Although IVC filter use is increasing, the evidence does not support their use in hemodynamically stable patients who can be anticoagulated. The patient described in the initial case has no contraindication to systemic anticoagulation. Therefore, she should be started on anticoagulation, and an IVC filter should not be placed.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason?” Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason” topics by emailing[email protected].
Disclosures
The authors do not have any conflicts of interest to disclose
1. Centers for Disease Control and Prevention. Venous thromboembolism in adult hospitalizations – United States, 2007-2009. MMWR. 2012;61:401-404. PubMed
2. Stein PD, Kayali F, Olson RE. Twenty-one-year trends in the use of inferior vena cava filters. Arch Intern Med. 2004;164(14):1541-1545. doi: 10.1001/archinte.164.14.1541 PubMed
3. Duszak R Jr, Parker L, Levin DC, Rao VM. Placement and removal of inferior vena cava filters: national trends in the Medicare population. J Am Coll Radiol. 2011;8(7):483-489. doi: 10.1016/j.jacr.2010.12.021. PubMed
4. Wang SL, Llyod AJ. Clinical review: inferior vena cava filters in the age of patient-centered outcomes. Ann Med. 2013;45(7):474-481. doi: 10.3109/07853890.2013.832951. PubMed
5. Spencer FA, Bates SM, Goldberg RJ, et al. A population-based study of inferior vena cava filters in patients with acute venous thromboembolism. Arch Intern Med.2010;170(16):1456-1462. doi: 10.1001/archinternmed.2010.272. PubMed
6. Baadh AS, Zikria JF, Rivioli S, et al. Indications for inferior vena cava filter placement: do physicians comply with guidelines? J Vasc Interv Radiol. 2012;23(8):989-995. doi: 10.1016/j.jvir.2012.04.017. PubMed
7. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149(2):315-352. doi: 10.1016/j.chest.2015.11.026. PubMed
8. Kaufman JA, Kinney TB, Streiff MB, et al. Guidelines for the use of retrievable and convertible vena cava filters: report from the Society of Interventional Radiology multidisciplinary consensus conference. J Vasc Interv Radiol. 2006;17(3):449-459. doi: 10.1097/01.rvi.0000203418.39769.0d. PubMed
9. Tao MJ, Montbriand JM, Eisenberg N, Sniderman KW, Roche-Nagle G. Temporary inferior vena cava filter indications, retrieval rates, and follow-up management at a multicenter tertiary care institution. J Vasc Surg. 2016;64(2):430-437. doi: 10.1016/j.jvs.2016.02.034. PubMed
10. Sarosiek S, Crowther M, Sloan JM. Indications, complications, and management of inferior vena cava filters. JAMA Intern Med.2013;173(7):513-517. doi: 10.1001/jamainternmed.2013.343. PubMed
11. Decousus H, Leizorovicz A, Parent F, et al. A clinical trial of vena cava filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med. 1998;338(7):409-415. doi: 10.1056/NEJM199802123380701. PubMed
12. PRECIP Study Group. Eight-year follow up of patients with permanent vena cava filters in the prevention of pulmonary embolism. Circulation. 2005;112(3):416-422. doi: 10.1161/CIRCULATIONAHA.104.512834. PubMed
13. Mismetti P, Laporte S, Pellerin O, et al. Effect of a retrievable inferior vena cava filter plus anticoagulation vs anticoagulation alone on risk of recurrent pulmonary embolism. JAMA. 2015;313(16):1627-1635. doi: 10.1001/jama.2015.3780. PubMed
14. Wang SL, Siddiqui A, Rosenthal E. Long-term complications of inferior vena cava filters. J Vasc Surg Venous Lymphat Disord. 2017;5(1):33-41. doi: 10.1016/j.jvsv.2016.07.002. PubMed
15. Andreoli JM, Lewandowski RJ, Vogelzang RL, Ryu RK. Comparison of complication rates associated with permanent and retrievable inferior vena cava filters: a review of the MAUDE database. J Vasc Interv Radiol. 2014;25(8):1181-1185. doi: 10.1016/j.jvir.2014.04.016. PubMed
16. Vijay K, Hughes JA, Burdette AS, et al. Fractured bard Recovery, G2, and G2 Express inferior vena cava filters: incidence, clinical consequences, and outcomes of removal attempts. J Vasc Interv Radiol. 2012;23(2):188-194. doi: 10.1016/j.jvir.2011.10.005. PubMed
17. Removing Retrievable Inferior Vena Cava Filters: FDA Safety Communication. FDA.gov. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm396377.htm. Published May 6, 2014. Accessed April 10, 2017.
18. Peterson EA, Yenson PR, Liu D, Lee AYY. Predictors of attempted inferior vena cava filters retrieval in a tertiary care centre. Thromb Res. 2014;134(2):300-304. doi: 10.1016/j.thromres.2014.05.029. PubMed
19. Minocha J, Idakoji I, Riaz A, et al. Improving inferior vena cava filter retrieval rates: impact of a dedicated inferior vena cava filter clinic. J Vasc Interv Radiol. 2010;21(12):1847-1851. doi: 10.1016/j.jvir.2010.09.003. PubMed
20. Stein PD, Matta F, Keyes DC, Willyerd GL. Impact of vena cava filters on in-hospital case fatality rate from pulmonary embolism. Am J Med. 2012;125(5):478-484. doi: 10.1016/j.amjmed.2011.05.025. PubMed
1. Centers for Disease Control and Prevention. Venous thromboembolism in adult hospitalizations – United States, 2007-2009. MMWR. 2012;61:401-404. PubMed
2. Stein PD, Kayali F, Olson RE. Twenty-one-year trends in the use of inferior vena cava filters. Arch Intern Med. 2004;164(14):1541-1545. doi: 10.1001/archinte.164.14.1541 PubMed
3. Duszak R Jr, Parker L, Levin DC, Rao VM. Placement and removal of inferior vena cava filters: national trends in the Medicare population. J Am Coll Radiol. 2011;8(7):483-489. doi: 10.1016/j.jacr.2010.12.021. PubMed
4. Wang SL, Llyod AJ. Clinical review: inferior vena cava filters in the age of patient-centered outcomes. Ann Med. 2013;45(7):474-481. doi: 10.3109/07853890.2013.832951. PubMed
5. Spencer FA, Bates SM, Goldberg RJ, et al. A population-based study of inferior vena cava filters in patients with acute venous thromboembolism. Arch Intern Med.2010;170(16):1456-1462. doi: 10.1001/archinternmed.2010.272. PubMed
6. Baadh AS, Zikria JF, Rivioli S, et al. Indications for inferior vena cava filter placement: do physicians comply with guidelines? J Vasc Interv Radiol. 2012;23(8):989-995. doi: 10.1016/j.jvir.2012.04.017. PubMed
7. Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149(2):315-352. doi: 10.1016/j.chest.2015.11.026. PubMed
8. Kaufman JA, Kinney TB, Streiff MB, et al. Guidelines for the use of retrievable and convertible vena cava filters: report from the Society of Interventional Radiology multidisciplinary consensus conference. J Vasc Interv Radiol. 2006;17(3):449-459. doi: 10.1097/01.rvi.0000203418.39769.0d. PubMed
9. Tao MJ, Montbriand JM, Eisenberg N, Sniderman KW, Roche-Nagle G. Temporary inferior vena cava filter indications, retrieval rates, and follow-up management at a multicenter tertiary care institution. J Vasc Surg. 2016;64(2):430-437. doi: 10.1016/j.jvs.2016.02.034. PubMed
10. Sarosiek S, Crowther M, Sloan JM. Indications, complications, and management of inferior vena cava filters. JAMA Intern Med.2013;173(7):513-517. doi: 10.1001/jamainternmed.2013.343. PubMed
11. Decousus H, Leizorovicz A, Parent F, et al. A clinical trial of vena cava filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med. 1998;338(7):409-415. doi: 10.1056/NEJM199802123380701. PubMed
12. PRECIP Study Group. Eight-year follow up of patients with permanent vena cava filters in the prevention of pulmonary embolism. Circulation. 2005;112(3):416-422. doi: 10.1161/CIRCULATIONAHA.104.512834. PubMed
13. Mismetti P, Laporte S, Pellerin O, et al. Effect of a retrievable inferior vena cava filter plus anticoagulation vs anticoagulation alone on risk of recurrent pulmonary embolism. JAMA. 2015;313(16):1627-1635. doi: 10.1001/jama.2015.3780. PubMed
14. Wang SL, Siddiqui A, Rosenthal E. Long-term complications of inferior vena cava filters. J Vasc Surg Venous Lymphat Disord. 2017;5(1):33-41. doi: 10.1016/j.jvsv.2016.07.002. PubMed
15. Andreoli JM, Lewandowski RJ, Vogelzang RL, Ryu RK. Comparison of complication rates associated with permanent and retrievable inferior vena cava filters: a review of the MAUDE database. J Vasc Interv Radiol. 2014;25(8):1181-1185. doi: 10.1016/j.jvir.2014.04.016. PubMed
16. Vijay K, Hughes JA, Burdette AS, et al. Fractured bard Recovery, G2, and G2 Express inferior vena cava filters: incidence, clinical consequences, and outcomes of removal attempts. J Vasc Interv Radiol. 2012;23(2):188-194. doi: 10.1016/j.jvir.2011.10.005. PubMed
17. Removing Retrievable Inferior Vena Cava Filters: FDA Safety Communication. FDA.gov. https://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/ucm396377.htm. Published May 6, 2014. Accessed April 10, 2017.
18. Peterson EA, Yenson PR, Liu D, Lee AYY. Predictors of attempted inferior vena cava filters retrieval in a tertiary care centre. Thromb Res. 2014;134(2):300-304. doi: 10.1016/j.thromres.2014.05.029. PubMed
19. Minocha J, Idakoji I, Riaz A, et al. Improving inferior vena cava filter retrieval rates: impact of a dedicated inferior vena cava filter clinic. J Vasc Interv Radiol. 2010;21(12):1847-1851. doi: 10.1016/j.jvir.2010.09.003. PubMed
20. Stein PD, Matta F, Keyes DC, Willyerd GL. Impact of vena cava filters on in-hospital case fatality rate from pulmonary embolism. Am J Med. 2012;125(5):478-484. doi: 10.1016/j.amjmed.2011.05.025. PubMed
© 2018 Society of Hospital Medicine
The Role of Hospital Medicine in Emergency Preparedness: A Framework for Hospitalist Leadership in Disaster Preparedness, Response, and Recovery
Recent events, domestically and globally, have highlighted the numerous complex challenges that disasters and mass casualty incidents (MCIs) impose on hospitals. Mass casualty events result from natural phenomena (eg, hurricanes, tornadoes, and wildfires), accidents (eg, plane crashes, building collapses, and toxic waste spills), or man-made crises (eg, terrorism).1-4 These events feature the potential to cause an acute surge of patients, which can overwhelm available hospital resources and personnel. Mass effect incidents are sustained crises, which often occur due to loss of infrastructure, epidemic infectious diseases, or need for hospital evacuations, and can completely overtax local and regional resources, thus requiring federal and state coordination.5
Hospital disaster response plans have traditionally centered on responses by the emergency department (ED) and associated surgical services to mass trauma-type events, without commensurate involvement of other hospital departments in either incident management operations or the planning process for mass effect incidents.6,7 In particular, the role of hospitalists in the leadership structure of various hospital disaster command structures remains undefined.8 However, recent disasters suggest that hospitalist involvement will highly benefit hospital emergency preparedness.9 Hospitalists possess specialized expertise in patient triage and disposition; medical comanagement with surgical services; coordination of complex medical care (usually with continuous 24/7 in-house coverage); integration with the full spectrum of affiliated services, such as case management or patient rehabilitation; and quality improvement research.10-12 At our institution, hospitalists are involved in the direct care of over 60% of the patients admitted across all medical and surgical services. Thus, we believe that hospitalists are uniquely qualified to offer leadership in disaster preparation, response, and recovery if integrated into hospitals’ incident command architectures. For example, although numerous acute patient surges are due to trauma MCIs, hospitalists may nevertheless act as the primary care providers in disasters that are medical in nature or that require rapid hospital evacuation and patient transfer (Table 1).
Although truly large-scale disasters are uncommon, several recent incidents exemplify scenarios with remarkably extreme acute patient surges (defined as >20% of normal patient volumes), which completely overwhelm a hospital’s capacity to maintain normal operations and require response from all available medical personnel, ideally in a preplanned and organized manner.13 The Las Vegas shooting on October 1, 2017, for example, resulted in 546 trauma victims, inundating two local hospitals and one regional facility.14,15 In another case, the deadliest tornado in modern US history struck Joplin, Missouri on May 22, 2011, destroying one of the two hospitals in the city and leaving an estimated 1,371 people injured, many of whom were presented to the one remaining area hospital.16 One of our team members (J.P.), a storm chaser from out-of-town, reported to the remaining functioning hospital and oversaw an impromptu hospital unit that received patients from the damaged hospital, ultimately caring for approximately 40 patients with a combination of medical and surgical issues from presentation through eventual disposition or transfer to outlying hospitals.17 Such incidents illustrate that during trauma events, hospitalists play critical roles for continuity of care for hospitalized disaster victims.
Therefore, we propose a means for incorporating hospitalists into the coordinated hospital disaster response effort, first by providing hospitalists with an overview of disaster management principles to allow their engagement with hospitals’ disaster management system with working fluency and second, by proposing a framework for hospitalist involvement in hospital emergency response. These recommendations stem from our experience and from similar recommendations from a number of evidence-based articles on intensive care medicine, disaster preparedness, and emergency medicine literature cited in this article. To our knowledge, no evidence-based literature discusses hospital medicine or internal medicine specific to emergency preparedness. We aim to change such condition with this article.
KEY PRINCIPLES OF INCIDENT MANAGEMENT AND PREPAREDNESS: A PRIMER FOR HOSPITALISTS
Effective participation in disaster response and planning requires a basic understanding of the organizational structures for incident management.18,19 Overall disaster response within the United States is guided by the National Response Framework, a national-level strategy that directs coordination between military and civilian response efforts, the latter of which are structured by the National Incident Management System (NIMS).20 NIMS organizes emergency management across all government levels (federal, state, and local) and the private sector under a common operational language and command structure. Both systems grew out of analyses of the September 11, 2001 attacks and Hurricane Katrina, indicating the need for a wider systemic organization to response efforts.1 State-level efforts are designed to mobilize resources to assist in community-level operations. Incident management always falls to the local authorities. At this local level, discrete hospitals often take part in healthcare coalitions that act in conjunction with other health entities, local public health departments, and emergency medical services, forming a multiagency coordination system.5 This healthcare coalition (emergency support function #8 health and medical), in support of emergency managers of city and county governments, forms the core of the medical response. One commonality to all emergency management is the concept of an “all-hazards” approach, which aims to develop a broad and flexible strategy for efficient management of nearly any type of incident. This “all-hazards” approach allows effective management through each of the four phases of incident management: preparation, response, recovery, and ongoing mitigation.
Direct supervision over incident management is achieved through an Incident Command System (ICS), a hierarchical organization of positions involved in response. The top supervisory structure of ICS (Incident Command and General Staff) is the same regardless of the locale in which it operates, allowing coherent interoperability with other agencies. Incidents of any size are managed with a scalable approach; subordinate ICS positions, which are tailored according to specific needs, can be activated as needed. Healthcare implementation of the ICS structure led to the development of the Hospital Incident Command System (HICS), which now serves as the national standard for hospital-based incident management and facilitates the capacity of individual hospitals to coordinate with other resources regionally and is a part of NIMS for emergency response (Figure 1).21 The success of HICS-led regulatory agencies (namely the Centers for Medicare and Medicaid Services and the Joint Commission) to require ICS/HICS in-hospital incident response plans.22 The most recent HICS (Version V) excludes physician involvement in the overall management chart. However, we demonstrate how the inherent flexibility in ICS can adapt to involve hospitalists. Although HICS serves as a backbone that requires institutionally specific modifications, other institutions, such as ours, commonly have entire branches or positions renamed, reapportioned, or created to fill their specific needs. Specialized training in ICS, NIMS, and other aspects of hospital emergency response is beyond the scope of this article but is available for free through the Department of Homeland Security and FEMA.23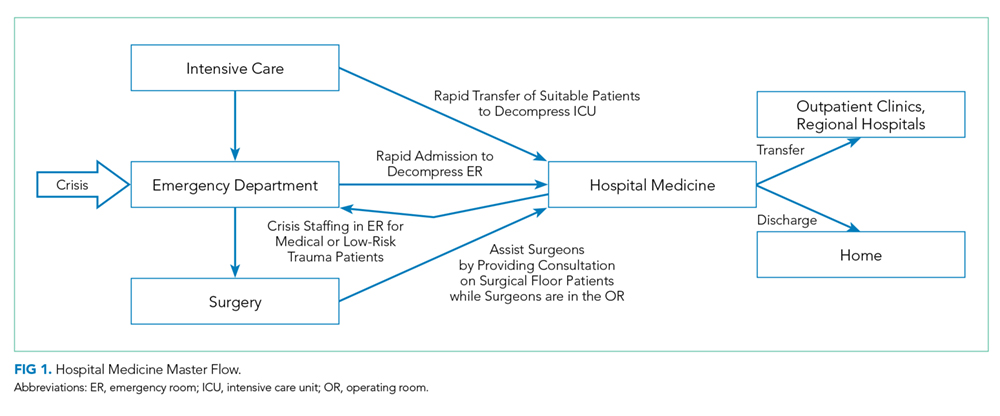
Perhaps, the defining feature of ICS/HICS is its expandability, allowing the response efforts to be scaled and tailored in size, scope, and complexity of that of the incident.24 At the same time, the principles of span of control and unity of command promote efficient command structure by mandating each participant within the disaster response process to report to only one superior, whereas these superiors are limited to a manageable number of subordinates. For example, in Figure 2, all Strike Team Leaders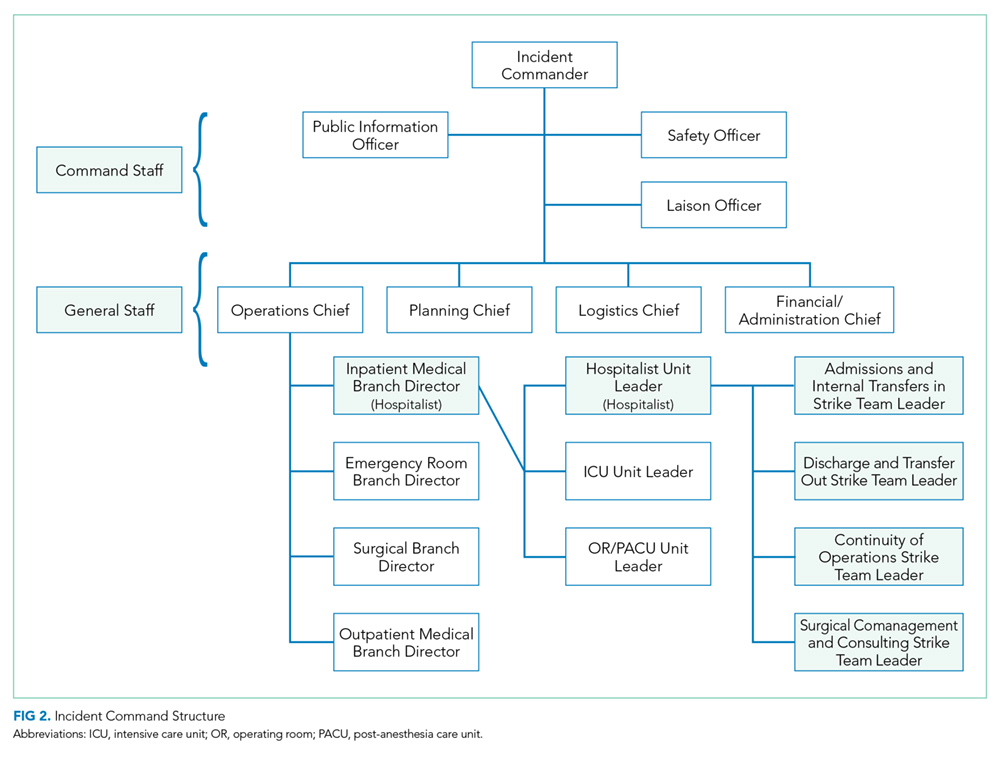
PROPOSED FRAMEWORK FOR HOSPITALIST INVOLVEMENT
Although incidents vary in terms of their severity, acuity of onset, duration, and composition of patients, a defining feature of MCIs is the rapid surge of patients with acute needs. Many MCIs are easily absorbed by local facilities. However, smaller hospitals or hospitals receiving patients from larger-scale incidents may become overwhelmed, in which larger incidents may result in an acute surge of over 20% of hospital capacity.13 Moreover, hospital surge capabilities have markedly diminished over the past decade due to overcrowding of emergency rooms, in part by admitted patients occupying the room space within the ED (“boarding”), further decreasing the hospitals’ capacities to accept new patients.25
Our proposed framework for hospitalist involvement in MCI disaster response focuses on such a situation, with emphasis on augmentation of hospital surge capacity and facilitation of patient throughput and discharge. Notably, these goals are modified from the standard HICS architecture (Figures 1-2 and Table 2). In this framework, hospitalists can play a critical role in decompressing the emergency room through admitting medical patients as rapidly as possible (even if preliminary workup is still pending), facilitating rapid discharge of patients to allow newer admissions to reach the floor, and prioritizing patients that could be transferred to other facilities or services and thus opening additional beds for admission (eg, accepting patients from the ICU or surgical floors to increase capacities on those services). Additionally, hospitalists can comanage surgical patients while surgeons are operating, assist intensivists with medical issues, and facilitate care of patients with minor injuries.
Using the HICS framework, each of those domains would be handled by a Strike Team led by one Team Leader whose goal is to operationalize various assets into a cohesive team specializing in those goals. Table 2 summarizes these goals, as presented in the context of patient examples.
To keep up with the ICS fundamentals, Hospitalist Unit Leaders may address a large MCI with all four strike teams or may only activate the strike teams needed for a less intensive MCI. For example, a bombing may result in a patient surge of 30% more than normal operations and thus demand a full response that includes all the strike teams noted above. By contrast, a bus accident with 20 injured patients may only require a Hospitalist Unit Leader to activate the “Admissions and Internal Transfers In” Strike Team to help offload a busy emergency room.
HOSPITALIST LEADERSHIP IN HOSPITAL EMERGENCY OPERATION PLAN DEVELOPMENT
Emergency management is comprised of four phases: preparation, response, recovery, and mitigation. The latter two phases are beyond the scope of this paper. Although most of our review has focused on modeling disaster response, hospitalist leadership remains critical in preparing for disasters. A disaster often psychologically overwhelms care providers, who feel compelled to help but are uncertain where to begin. To aid the members of a disaster response team, a state-of-the-art hospitalist group creates Job Action Sheets (JASs) for each position in their HICS organizational chart; these sheets codify how to respond and what roles are needed. These formal, protocolized sheets provide individuals assigned to these positions a description of their roles and responsibilities, including to whom they report and over whom they supervise, and include detailed checklists to aid in reaching critical milestones during the response phase. For example, the “Surgical Comanagement and Consulting” Strike Team Leader JAS would likely include the expectations of surgeons for assisting in patient management (ie, auto-consulting on all postoperative patients) and whether nursing phone calls on surgical patients would be temporarily routed to the Strike Team during periods of OR surge.
Hospitalists are well suited as leaders in disaster preparation given their ability to coordinate care among a large spectrum of stakeholders. For example, case managers and social workers are essential members of a well-structured Discharge Strike Team. Their input is critical to ensure that disaster tactics – such as care coordination contracts with local skilled nursing facilities willing to expedite discharge in emergencies to their facilities – are in-place before a real MCI. During Hurricane Sandy, mass evacuation of affected hospitals was effective through the Healthcare Facility Evacuation Center (a healthcare coalition of the New York Hospital Association) but nevertheless plagued with issues regarding situational awareness, poor communication between facilities, and difficulty bundling patients with medical records to receiving facilities – items which can be identified, anticipated, and thoroughly vetted by hospitalists well in advance of a real-world evacuation.26, 27
As the Joint Commission mandates regular exercises of the emergency plan, protocols must be drilled regularly to uncover deficiencies and areas for improvement.18 The most common failure patterns in Emergency Operation Plans (EOPs) include unrealistic and ineffective expectations and poor communication between different personnel and groups, resulting in confusion and obfuscation.28-30 Therefore, EOPs need to be both comprehensive and realistic – characteristics that can only be tested through repeated drills. These characteristics can be tested during tabletop exercises, where hospitalists assume the role of a part of the ICS structure and with JAS in hand, attempt to reason how to respond to a given scenario.31 Our experience is that small-scale drills conducted more frequently than the bare minimum mandated by the Joint Commission are far more effective for success in real-life situations.
Although no hospital EOP can anticipate every contingency, hospitalists can proactively practice contingency planning for sustained system-wide mass effect incidents, in which hospitals are unable to maintain normal operations and shift from standard to crisis conventions of care. For example, mass effect incidents (ie, hospital damage from an earthquake or a massive and persistent regional power failure), require planning for how a hospital-wide mass evacuation would unfold and how efforts from multiple ancillary hospital services (engineering, nursing, security, and patient transport) would be integrated. As of 2015, over 90% of hospitals have adopted an electronic health record, but only two-thirds of hospitals feature EOPs for information technology failures.32,33 Given the large footprint of hospitalists in clinical practice, HICS principles appear ripe for application in IT outages and through development of ICS positions structured specifically to this type of contingency.34
CONCLUSION
Disasters unfold rapidly with marked patient surges and the potential to strain healthcare systems over an extended period. However, in both instances, hospitalists are possibly some of the most qualified clinicians to prepare for and respond to such events. Hospitalists need to assume a leadership role in emergency preparedness to integrate seamlessly into hospital incident command structures and to shape the interdepartmental relationships vital to success – skills at which hospitalists excel. Although no plan can address all possible disasters, familiarity with HICS and well-prepared and well-written JASs should help groups respond and succeed in almost all hazards.
Disclosures
None of the authors have any conflicts of interest to report.
1. Homeland Security Presidential Directive-5. 2003.
2. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: I. General principles of response and management. J Am Acad Orthop Surg. 2007;15(7):388-396. doi: 10.5435/00124635-200707000-00004. PubMed
3. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: II. explosive, biologic, chemical, and nuclear agents. J Am Acad Orthop Surg. 2007;15(8):461-473. PubMed
4. Christian MD, Hawryluck L, Wax RS, et al., Development of a triage protocol for critical care during an influenza pandemic. CMAJ. 2006;175(11):1377-1381. doi: 10.1503/cmaj.060911. PubMed
5. Barbera JA, Macintyre AG. Medical Surge Capacity and Capability: The Healthcare Coalition in Emergency Response and Recovery. In: Knebel A, Trabert E, eds. Department of Health and Human Services. 2007.
6. Roccaforte JD, Cushman JG. Disaster preparation and management for the intensive care unit. Curr Opin Crit Care. 2002;8(6):607-615. PubMed
7. Roccaforte JD, Cushman JG. Disaster preparedness, triage, and surge capacity for hospital definitive care areas: optimizing outcomes when demands exceed resources. Anesthesiol Clin. 2007;25(1):161-177, xi. doi: 10.1016/j.anclin.2007.01.002. PubMed
8. Emergency Medical Services of California. Hospital Incident Command System V. 2014 [cited 2018 February 14th]. https://emsa.ca.gov/wp-content/uploads/sites/47/2017/09/HICS_Guidebook_2014_11.pdf. Accessed June 1, 2018.
9. Sprung CL, Zimmerman JL, Christian MD, et al. Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: Summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med. 2010;36(3):428-443. doi: 10.1007/s00134-010-1759-y. PubMed
10. Inpatient specialists help cut costs, reduce LOS. Hospitalists partner with case managers. Hosp Case Manag. 1997;5(5):79-81. PubMed
11. Thompson RE, Pfeifer K, Grant PJ, et al. Hospital medicine and perioperative care: A framework for high-quality, high-value collaborative care. J Hosp Med. 2017;12(4):277-282. doi: 10.12788/jhm.2717. PubMed
12. Gupta R, Moriates C, Harrison JD, et al. Development of a high-value care culture survey: A modified Delphi process and psychometric evaluation. BMJ Qual Saf. 2017;26(6):475-483. doi: 10.1136/bmjqs-2016-005612. PubMed
13. Tadmor B, McManus J, Koenig KL. The art and science of surge: Experience from Israel and the U.S. military. Acad Emerg Med. 2006;13(11):1130-1134. doi: 10.1197/j.aem.2006.06.043. PubMed
14. Myers AL. Vegas Hospitals Swamped With Victims After High-Rise Attack. Associated Press; 2017. https://www.msn.com/en-us/news/breakingnews/vegas-hospitals-swamped-with-victims-after-high-rise-attack/ar-AAsQyZ8?ocid=HPCDHP. Las Vegas. Accessed June 1, 2018.
15. Craig T. As the Wounded Kept Coming, Las Vegas Hospitals Dealt With Injuries Rarely Seen in the US. In: Mello F, Sun L, eds. Washington Post: Washington Post; Oct 3, 2017.
16. Porth L. Preparedness and Partnerships: Lessons learned from the Missouri disasters of 2011. A Focus on Joplin. 2012, Missouri Hospital Association. PubMed
17. Persoff J. First Response Mode: May 22, 2011, Joplin Tornado. June 5, 2011; Available from: http://stormdoctor.blogspot.com/2011/06/first-response-mode-may-22-2011-joplin.html. Accessed June 1, 2018.
18. Dichter JR, Kanter RK, Dries D, et al. System-level planning, coordination, and communication: care of the critically ill and injured during pandemics and disasters: CHEST consensus statement. Chest. 2014;146(4 Suppl):e87S-e102S. doi: 10.1378/chest.14-0738. PubMed
19. Thomas TL, Hsu EB, Kim HK, Colli S, Arana G, Green GB. The incident command system in disasters: Evaluation methods for a hospital-based exercise. Prehosp Disaster Med. 2005;20(1):14-23. doi: 10.1017/S1049023X00002090. PubMed
20. FEMA. The Historical Contex of Emergency Management. [cited 2018 February 14th]; Available from: https://training.fema.gov/emi.aspx. Accessed June 1, 2018.
21. Backer H. Hospital Incident Command System Guidebook 5th Edition. In: Smiley D, Schoenthal L, eds. California Emergency Medical Services Authority, 2014. Accessed June 1, 2018.
22. Emergency Management Resources. Available from: https://www.jointcommission.org/emergency_management.aspx. Accessed June 1, 2018.
23. Incident Command System Training Program. Available from: https://training.fema.gov/emiweb/is/icsresource/trainingmaterials.htm.
24. Agency, F.E.M. NIMS and the Incident Command System. Nov 23, 2004; Available from: https://www.fema.gov/txt/nims/nims_ics_position_paper.txt. Accessed June 1, 2018.
25. Peleg K, Kellermann AL. Enhancing hospital surge capacity for mass casualty events. JAMA. 2009;302(5):565-567. doi: 10.1001/jama.2009.1119. PubMed
26. Adalja AA, Watson M, Bouri N, et al. Absorbing citywide patient surge during Hurricane Sandy: a case study in accommodating multiple hospital evacuations. Ann Emerg Med. 2014;64(1):66-73.e1. doi: 10.1016/j.annemergmed.2013.12.010. PubMed
27. Adalja AA, Watson M, Wollner S, Rambhia KJ, Toner ES. Response to the sudden closure of St. Vincent’s Hospital: learning from a real, no-notice, prolonged surge event. Biosecur Bioterror. 2011;9(2):153-161. doi: 10.1089/bsp.2011.0002. PubMed
28. Klein JS, Weigelt JA. Disaster management. Lessons learned. Surg Clin North Am. 1991;71(2):257-266. PubMed
29. Frykberg ER. Medical management of disasters and mass casualties from terrorist bombings: How can we cope? J Trauma. 2002;53(2):201-212. doi: 10.1097/00005373-200208000-00001. PubMed
30. Lynn M, Gurr D, Memon A, Kaliff J. Management of conventional mass casualty incidents: Ten commandments for hospital planning. J Burn Care Res. 2006;27(5):649-658. doi: 10.1097/01.BCR.0000238119.29269.2B. PubMed
31. Williams J, Nocera M, Casteel C. The effectiveness of disaster training for health care workers: A systematic review. Ann Emerg Med. 2008;52(3):211-22, 222.e1-2. doi: 10.1016/j.annemergmed.2007.09.030. PubMed
32. Percent of Hospitals, By Type, that Possess Certified Health IT. 2015, US Department of Health and Human Services: Office of the National Coordinator for Health Information Technology.
33. Lee C, Robinson KM, Wendt K, Williamson D, et al. The preparedness of hospital Health Information Services for system failures due to internal disasters. Health Inf Manag. 2009;38(2):18-25. doi: 10.1177/183335830903800203. PubMed
34. Situations, C.o.G.f.E.C.S.o.C.f.U.i.D. and I.o. Medicine, Crisis Standards of Care: A Systems Framework for Catastrophic Disaster Response. Mar 21, 2012, Washington (DC): National Academies Press (US). PubMed
Recent events, domestically and globally, have highlighted the numerous complex challenges that disasters and mass casualty incidents (MCIs) impose on hospitals. Mass casualty events result from natural phenomena (eg, hurricanes, tornadoes, and wildfires), accidents (eg, plane crashes, building collapses, and toxic waste spills), or man-made crises (eg, terrorism).1-4 These events feature the potential to cause an acute surge of patients, which can overwhelm available hospital resources and personnel. Mass effect incidents are sustained crises, which often occur due to loss of infrastructure, epidemic infectious diseases, or need for hospital evacuations, and can completely overtax local and regional resources, thus requiring federal and state coordination.5
Hospital disaster response plans have traditionally centered on responses by the emergency department (ED) and associated surgical services to mass trauma-type events, without commensurate involvement of other hospital departments in either incident management operations or the planning process for mass effect incidents.6,7 In particular, the role of hospitalists in the leadership structure of various hospital disaster command structures remains undefined.8 However, recent disasters suggest that hospitalist involvement will highly benefit hospital emergency preparedness.9 Hospitalists possess specialized expertise in patient triage and disposition; medical comanagement with surgical services; coordination of complex medical care (usually with continuous 24/7 in-house coverage); integration with the full spectrum of affiliated services, such as case management or patient rehabilitation; and quality improvement research.10-12 At our institution, hospitalists are involved in the direct care of over 60% of the patients admitted across all medical and surgical services. Thus, we believe that hospitalists are uniquely qualified to offer leadership in disaster preparation, response, and recovery if integrated into hospitals’ incident command architectures. For example, although numerous acute patient surges are due to trauma MCIs, hospitalists may nevertheless act as the primary care providers in disasters that are medical in nature or that require rapid hospital evacuation and patient transfer (Table 1).
Although truly large-scale disasters are uncommon, several recent incidents exemplify scenarios with remarkably extreme acute patient surges (defined as >20% of normal patient volumes), which completely overwhelm a hospital’s capacity to maintain normal operations and require response from all available medical personnel, ideally in a preplanned and organized manner.13 The Las Vegas shooting on October 1, 2017, for example, resulted in 546 trauma victims, inundating two local hospitals and one regional facility.14,15 In another case, the deadliest tornado in modern US history struck Joplin, Missouri on May 22, 2011, destroying one of the two hospitals in the city and leaving an estimated 1,371 people injured, many of whom were presented to the one remaining area hospital.16 One of our team members (J.P.), a storm chaser from out-of-town, reported to the remaining functioning hospital and oversaw an impromptu hospital unit that received patients from the damaged hospital, ultimately caring for approximately 40 patients with a combination of medical and surgical issues from presentation through eventual disposition or transfer to outlying hospitals.17 Such incidents illustrate that during trauma events, hospitalists play critical roles for continuity of care for hospitalized disaster victims.
Therefore, we propose a means for incorporating hospitalists into the coordinated hospital disaster response effort, first by providing hospitalists with an overview of disaster management principles to allow their engagement with hospitals’ disaster management system with working fluency and second, by proposing a framework for hospitalist involvement in hospital emergency response. These recommendations stem from our experience and from similar recommendations from a number of evidence-based articles on intensive care medicine, disaster preparedness, and emergency medicine literature cited in this article. To our knowledge, no evidence-based literature discusses hospital medicine or internal medicine specific to emergency preparedness. We aim to change such condition with this article.
KEY PRINCIPLES OF INCIDENT MANAGEMENT AND PREPAREDNESS: A PRIMER FOR HOSPITALISTS
Effective participation in disaster response and planning requires a basic understanding of the organizational structures for incident management.18,19 Overall disaster response within the United States is guided by the National Response Framework, a national-level strategy that directs coordination between military and civilian response efforts, the latter of which are structured by the National Incident Management System (NIMS).20 NIMS organizes emergency management across all government levels (federal, state, and local) and the private sector under a common operational language and command structure. Both systems grew out of analyses of the September 11, 2001 attacks and Hurricane Katrina, indicating the need for a wider systemic organization to response efforts.1 State-level efforts are designed to mobilize resources to assist in community-level operations. Incident management always falls to the local authorities. At this local level, discrete hospitals often take part in healthcare coalitions that act in conjunction with other health entities, local public health departments, and emergency medical services, forming a multiagency coordination system.5 This healthcare coalition (emergency support function #8 health and medical), in support of emergency managers of city and county governments, forms the core of the medical response. One commonality to all emergency management is the concept of an “all-hazards” approach, which aims to develop a broad and flexible strategy for efficient management of nearly any type of incident. This “all-hazards” approach allows effective management through each of the four phases of incident management: preparation, response, recovery, and ongoing mitigation.
Direct supervision over incident management is achieved through an Incident Command System (ICS), a hierarchical organization of positions involved in response. The top supervisory structure of ICS (Incident Command and General Staff) is the same regardless of the locale in which it operates, allowing coherent interoperability with other agencies. Incidents of any size are managed with a scalable approach; subordinate ICS positions, which are tailored according to specific needs, can be activated as needed. Healthcare implementation of the ICS structure led to the development of the Hospital Incident Command System (HICS), which now serves as the national standard for hospital-based incident management and facilitates the capacity of individual hospitals to coordinate with other resources regionally and is a part of NIMS for emergency response (Figure 1).21 The success of HICS-led regulatory agencies (namely the Centers for Medicare and Medicaid Services and the Joint Commission) to require ICS/HICS in-hospital incident response plans.22 The most recent HICS (Version V) excludes physician involvement in the overall management chart. However, we demonstrate how the inherent flexibility in ICS can adapt to involve hospitalists. Although HICS serves as a backbone that requires institutionally specific modifications, other institutions, such as ours, commonly have entire branches or positions renamed, reapportioned, or created to fill their specific needs. Specialized training in ICS, NIMS, and other aspects of hospital emergency response is beyond the scope of this article but is available for free through the Department of Homeland Security and FEMA.23
Perhaps, the defining feature of ICS/HICS is its expandability, allowing the response efforts to be scaled and tailored in size, scope, and complexity of that of the incident.24 At the same time, the principles of span of control and unity of command promote efficient command structure by mandating each participant within the disaster response process to report to only one superior, whereas these superiors are limited to a manageable number of subordinates. For example, in Figure 2, all Strike Team Leaders
PROPOSED FRAMEWORK FOR HOSPITALIST INVOLVEMENT
Although incidents vary in terms of their severity, acuity of onset, duration, and composition of patients, a defining feature of MCIs is the rapid surge of patients with acute needs. Many MCIs are easily absorbed by local facilities. However, smaller hospitals or hospitals receiving patients from larger-scale incidents may become overwhelmed, in which larger incidents may result in an acute surge of over 20% of hospital capacity.13 Moreover, hospital surge capabilities have markedly diminished over the past decade due to overcrowding of emergency rooms, in part by admitted patients occupying the room space within the ED (“boarding”), further decreasing the hospitals’ capacities to accept new patients.25
Our proposed framework for hospitalist involvement in MCI disaster response focuses on such a situation, with emphasis on augmentation of hospital surge capacity and facilitation of patient throughput and discharge. Notably, these goals are modified from the standard HICS architecture (Figures 1-2 and Table 2). In this framework, hospitalists can play a critical role in decompressing the emergency room through admitting medical patients as rapidly as possible (even if preliminary workup is still pending), facilitating rapid discharge of patients to allow newer admissions to reach the floor, and prioritizing patients that could be transferred to other facilities or services and thus opening additional beds for admission (eg, accepting patients from the ICU or surgical floors to increase capacities on those services). Additionally, hospitalists can comanage surgical patients while surgeons are operating, assist intensivists with medical issues, and facilitate care of patients with minor injuries.
Using the HICS framework, each of those domains would be handled by a Strike Team led by one Team Leader whose goal is to operationalize various assets into a cohesive team specializing in those goals. Table 2 summarizes these goals, as presented in the context of patient examples.
To keep up with the ICS fundamentals, Hospitalist Unit Leaders may address a large MCI with all four strike teams or may only activate the strike teams needed for a less intensive MCI. For example, a bombing may result in a patient surge of 30% more than normal operations and thus demand a full response that includes all the strike teams noted above. By contrast, a bus accident with 20 injured patients may only require a Hospitalist Unit Leader to activate the “Admissions and Internal Transfers In” Strike Team to help offload a busy emergency room.
HOSPITALIST LEADERSHIP IN HOSPITAL EMERGENCY OPERATION PLAN DEVELOPMENT
Emergency management is comprised of four phases: preparation, response, recovery, and mitigation. The latter two phases are beyond the scope of this paper. Although most of our review has focused on modeling disaster response, hospitalist leadership remains critical in preparing for disasters. A disaster often psychologically overwhelms care providers, who feel compelled to help but are uncertain where to begin. To aid the members of a disaster response team, a state-of-the-art hospitalist group creates Job Action Sheets (JASs) for each position in their HICS organizational chart; these sheets codify how to respond and what roles are needed. These formal, protocolized sheets provide individuals assigned to these positions a description of their roles and responsibilities, including to whom they report and over whom they supervise, and include detailed checklists to aid in reaching critical milestones during the response phase. For example, the “Surgical Comanagement and Consulting” Strike Team Leader JAS would likely include the expectations of surgeons for assisting in patient management (ie, auto-consulting on all postoperative patients) and whether nursing phone calls on surgical patients would be temporarily routed to the Strike Team during periods of OR surge.
Hospitalists are well suited as leaders in disaster preparation given their ability to coordinate care among a large spectrum of stakeholders. For example, case managers and social workers are essential members of a well-structured Discharge Strike Team. Their input is critical to ensure that disaster tactics – such as care coordination contracts with local skilled nursing facilities willing to expedite discharge in emergencies to their facilities – are in-place before a real MCI. During Hurricane Sandy, mass evacuation of affected hospitals was effective through the Healthcare Facility Evacuation Center (a healthcare coalition of the New York Hospital Association) but nevertheless plagued with issues regarding situational awareness, poor communication between facilities, and difficulty bundling patients with medical records to receiving facilities – items which can be identified, anticipated, and thoroughly vetted by hospitalists well in advance of a real-world evacuation.26, 27
As the Joint Commission mandates regular exercises of the emergency plan, protocols must be drilled regularly to uncover deficiencies and areas for improvement.18 The most common failure patterns in Emergency Operation Plans (EOPs) include unrealistic and ineffective expectations and poor communication between different personnel and groups, resulting in confusion and obfuscation.28-30 Therefore, EOPs need to be both comprehensive and realistic – characteristics that can only be tested through repeated drills. These characteristics can be tested during tabletop exercises, where hospitalists assume the role of a part of the ICS structure and with JAS in hand, attempt to reason how to respond to a given scenario.31 Our experience is that small-scale drills conducted more frequently than the bare minimum mandated by the Joint Commission are far more effective for success in real-life situations.
Although no hospital EOP can anticipate every contingency, hospitalists can proactively practice contingency planning for sustained system-wide mass effect incidents, in which hospitals are unable to maintain normal operations and shift from standard to crisis conventions of care. For example, mass effect incidents (ie, hospital damage from an earthquake or a massive and persistent regional power failure), require planning for how a hospital-wide mass evacuation would unfold and how efforts from multiple ancillary hospital services (engineering, nursing, security, and patient transport) would be integrated. As of 2015, over 90% of hospitals have adopted an electronic health record, but only two-thirds of hospitals feature EOPs for information technology failures.32,33 Given the large footprint of hospitalists in clinical practice, HICS principles appear ripe for application in IT outages and through development of ICS positions structured specifically to this type of contingency.34
CONCLUSION
Disasters unfold rapidly with marked patient surges and the potential to strain healthcare systems over an extended period. However, in both instances, hospitalists are possibly some of the most qualified clinicians to prepare for and respond to such events. Hospitalists need to assume a leadership role in emergency preparedness to integrate seamlessly into hospital incident command structures and to shape the interdepartmental relationships vital to success – skills at which hospitalists excel. Although no plan can address all possible disasters, familiarity with HICS and well-prepared and well-written JASs should help groups respond and succeed in almost all hazards.
Disclosures
None of the authors have any conflicts of interest to report.
Recent events, domestically and globally, have highlighted the numerous complex challenges that disasters and mass casualty incidents (MCIs) impose on hospitals. Mass casualty events result from natural phenomena (eg, hurricanes, tornadoes, and wildfires), accidents (eg, plane crashes, building collapses, and toxic waste spills), or man-made crises (eg, terrorism).1-4 These events feature the potential to cause an acute surge of patients, which can overwhelm available hospital resources and personnel. Mass effect incidents are sustained crises, which often occur due to loss of infrastructure, epidemic infectious diseases, or need for hospital evacuations, and can completely overtax local and regional resources, thus requiring federal and state coordination.5
Hospital disaster response plans have traditionally centered on responses by the emergency department (ED) and associated surgical services to mass trauma-type events, without commensurate involvement of other hospital departments in either incident management operations or the planning process for mass effect incidents.6,7 In particular, the role of hospitalists in the leadership structure of various hospital disaster command structures remains undefined.8 However, recent disasters suggest that hospitalist involvement will highly benefit hospital emergency preparedness.9 Hospitalists possess specialized expertise in patient triage and disposition; medical comanagement with surgical services; coordination of complex medical care (usually with continuous 24/7 in-house coverage); integration with the full spectrum of affiliated services, such as case management or patient rehabilitation; and quality improvement research.10-12 At our institution, hospitalists are involved in the direct care of over 60% of the patients admitted across all medical and surgical services. Thus, we believe that hospitalists are uniquely qualified to offer leadership in disaster preparation, response, and recovery if integrated into hospitals’ incident command architectures. For example, although numerous acute patient surges are due to trauma MCIs, hospitalists may nevertheless act as the primary care providers in disasters that are medical in nature or that require rapid hospital evacuation and patient transfer (Table 1).
Although truly large-scale disasters are uncommon, several recent incidents exemplify scenarios with remarkably extreme acute patient surges (defined as >20% of normal patient volumes), which completely overwhelm a hospital’s capacity to maintain normal operations and require response from all available medical personnel, ideally in a preplanned and organized manner.13 The Las Vegas shooting on October 1, 2017, for example, resulted in 546 trauma victims, inundating two local hospitals and one regional facility.14,15 In another case, the deadliest tornado in modern US history struck Joplin, Missouri on May 22, 2011, destroying one of the two hospitals in the city and leaving an estimated 1,371 people injured, many of whom were presented to the one remaining area hospital.16 One of our team members (J.P.), a storm chaser from out-of-town, reported to the remaining functioning hospital and oversaw an impromptu hospital unit that received patients from the damaged hospital, ultimately caring for approximately 40 patients with a combination of medical and surgical issues from presentation through eventual disposition or transfer to outlying hospitals.17 Such incidents illustrate that during trauma events, hospitalists play critical roles for continuity of care for hospitalized disaster victims.
Therefore, we propose a means for incorporating hospitalists into the coordinated hospital disaster response effort, first by providing hospitalists with an overview of disaster management principles to allow their engagement with hospitals’ disaster management system with working fluency and second, by proposing a framework for hospitalist involvement in hospital emergency response. These recommendations stem from our experience and from similar recommendations from a number of evidence-based articles on intensive care medicine, disaster preparedness, and emergency medicine literature cited in this article. To our knowledge, no evidence-based literature discusses hospital medicine or internal medicine specific to emergency preparedness. We aim to change such condition with this article.
KEY PRINCIPLES OF INCIDENT MANAGEMENT AND PREPAREDNESS: A PRIMER FOR HOSPITALISTS
Effective participation in disaster response and planning requires a basic understanding of the organizational structures for incident management.18,19 Overall disaster response within the United States is guided by the National Response Framework, a national-level strategy that directs coordination between military and civilian response efforts, the latter of which are structured by the National Incident Management System (NIMS).20 NIMS organizes emergency management across all government levels (federal, state, and local) and the private sector under a common operational language and command structure. Both systems grew out of analyses of the September 11, 2001 attacks and Hurricane Katrina, indicating the need for a wider systemic organization to response efforts.1 State-level efforts are designed to mobilize resources to assist in community-level operations. Incident management always falls to the local authorities. At this local level, discrete hospitals often take part in healthcare coalitions that act in conjunction with other health entities, local public health departments, and emergency medical services, forming a multiagency coordination system.5 This healthcare coalition (emergency support function #8 health and medical), in support of emergency managers of city and county governments, forms the core of the medical response. One commonality to all emergency management is the concept of an “all-hazards” approach, which aims to develop a broad and flexible strategy for efficient management of nearly any type of incident. This “all-hazards” approach allows effective management through each of the four phases of incident management: preparation, response, recovery, and ongoing mitigation.
Direct supervision over incident management is achieved through an Incident Command System (ICS), a hierarchical organization of positions involved in response. The top supervisory structure of ICS (Incident Command and General Staff) is the same regardless of the locale in which it operates, allowing coherent interoperability with other agencies. Incidents of any size are managed with a scalable approach; subordinate ICS positions, which are tailored according to specific needs, can be activated as needed. Healthcare implementation of the ICS structure led to the development of the Hospital Incident Command System (HICS), which now serves as the national standard for hospital-based incident management and facilitates the capacity of individual hospitals to coordinate with other resources regionally and is a part of NIMS for emergency response (Figure 1).21 The success of HICS-led regulatory agencies (namely the Centers for Medicare and Medicaid Services and the Joint Commission) to require ICS/HICS in-hospital incident response plans.22 The most recent HICS (Version V) excludes physician involvement in the overall management chart. However, we demonstrate how the inherent flexibility in ICS can adapt to involve hospitalists. Although HICS serves as a backbone that requires institutionally specific modifications, other institutions, such as ours, commonly have entire branches or positions renamed, reapportioned, or created to fill their specific needs. Specialized training in ICS, NIMS, and other aspects of hospital emergency response is beyond the scope of this article but is available for free through the Department of Homeland Security and FEMA.23
Perhaps, the defining feature of ICS/HICS is its expandability, allowing the response efforts to be scaled and tailored in size, scope, and complexity of that of the incident.24 At the same time, the principles of span of control and unity of command promote efficient command structure by mandating each participant within the disaster response process to report to only one superior, whereas these superiors are limited to a manageable number of subordinates. For example, in Figure 2, all Strike Team Leaders
PROPOSED FRAMEWORK FOR HOSPITALIST INVOLVEMENT
Although incidents vary in terms of their severity, acuity of onset, duration, and composition of patients, a defining feature of MCIs is the rapid surge of patients with acute needs. Many MCIs are easily absorbed by local facilities. However, smaller hospitals or hospitals receiving patients from larger-scale incidents may become overwhelmed, in which larger incidents may result in an acute surge of over 20% of hospital capacity.13 Moreover, hospital surge capabilities have markedly diminished over the past decade due to overcrowding of emergency rooms, in part by admitted patients occupying the room space within the ED (“boarding”), further decreasing the hospitals’ capacities to accept new patients.25
Our proposed framework for hospitalist involvement in MCI disaster response focuses on such a situation, with emphasis on augmentation of hospital surge capacity and facilitation of patient throughput and discharge. Notably, these goals are modified from the standard HICS architecture (Figures 1-2 and Table 2). In this framework, hospitalists can play a critical role in decompressing the emergency room through admitting medical patients as rapidly as possible (even if preliminary workup is still pending), facilitating rapid discharge of patients to allow newer admissions to reach the floor, and prioritizing patients that could be transferred to other facilities or services and thus opening additional beds for admission (eg, accepting patients from the ICU or surgical floors to increase capacities on those services). Additionally, hospitalists can comanage surgical patients while surgeons are operating, assist intensivists with medical issues, and facilitate care of patients with minor injuries.
Using the HICS framework, each of those domains would be handled by a Strike Team led by one Team Leader whose goal is to operationalize various assets into a cohesive team specializing in those goals. Table 2 summarizes these goals, as presented in the context of patient examples.
To keep up with the ICS fundamentals, Hospitalist Unit Leaders may address a large MCI with all four strike teams or may only activate the strike teams needed for a less intensive MCI. For example, a bombing may result in a patient surge of 30% more than normal operations and thus demand a full response that includes all the strike teams noted above. By contrast, a bus accident with 20 injured patients may only require a Hospitalist Unit Leader to activate the “Admissions and Internal Transfers In” Strike Team to help offload a busy emergency room.
HOSPITALIST LEADERSHIP IN HOSPITAL EMERGENCY OPERATION PLAN DEVELOPMENT
Emergency management is comprised of four phases: preparation, response, recovery, and mitigation. The latter two phases are beyond the scope of this paper. Although most of our review has focused on modeling disaster response, hospitalist leadership remains critical in preparing for disasters. A disaster often psychologically overwhelms care providers, who feel compelled to help but are uncertain where to begin. To aid the members of a disaster response team, a state-of-the-art hospitalist group creates Job Action Sheets (JASs) for each position in their HICS organizational chart; these sheets codify how to respond and what roles are needed. These formal, protocolized sheets provide individuals assigned to these positions a description of their roles and responsibilities, including to whom they report and over whom they supervise, and include detailed checklists to aid in reaching critical milestones during the response phase. For example, the “Surgical Comanagement and Consulting” Strike Team Leader JAS would likely include the expectations of surgeons for assisting in patient management (ie, auto-consulting on all postoperative patients) and whether nursing phone calls on surgical patients would be temporarily routed to the Strike Team during periods of OR surge.
Hospitalists are well suited as leaders in disaster preparation given their ability to coordinate care among a large spectrum of stakeholders. For example, case managers and social workers are essential members of a well-structured Discharge Strike Team. Their input is critical to ensure that disaster tactics – such as care coordination contracts with local skilled nursing facilities willing to expedite discharge in emergencies to their facilities – are in-place before a real MCI. During Hurricane Sandy, mass evacuation of affected hospitals was effective through the Healthcare Facility Evacuation Center (a healthcare coalition of the New York Hospital Association) but nevertheless plagued with issues regarding situational awareness, poor communication between facilities, and difficulty bundling patients with medical records to receiving facilities – items which can be identified, anticipated, and thoroughly vetted by hospitalists well in advance of a real-world evacuation.26, 27
As the Joint Commission mandates regular exercises of the emergency plan, protocols must be drilled regularly to uncover deficiencies and areas for improvement.18 The most common failure patterns in Emergency Operation Plans (EOPs) include unrealistic and ineffective expectations and poor communication between different personnel and groups, resulting in confusion and obfuscation.28-30 Therefore, EOPs need to be both comprehensive and realistic – characteristics that can only be tested through repeated drills. These characteristics can be tested during tabletop exercises, where hospitalists assume the role of a part of the ICS structure and with JAS in hand, attempt to reason how to respond to a given scenario.31 Our experience is that small-scale drills conducted more frequently than the bare minimum mandated by the Joint Commission are far more effective for success in real-life situations.
Although no hospital EOP can anticipate every contingency, hospitalists can proactively practice contingency planning for sustained system-wide mass effect incidents, in which hospitals are unable to maintain normal operations and shift from standard to crisis conventions of care. For example, mass effect incidents (ie, hospital damage from an earthquake or a massive and persistent regional power failure), require planning for how a hospital-wide mass evacuation would unfold and how efforts from multiple ancillary hospital services (engineering, nursing, security, and patient transport) would be integrated. As of 2015, over 90% of hospitals have adopted an electronic health record, but only two-thirds of hospitals feature EOPs for information technology failures.32,33 Given the large footprint of hospitalists in clinical practice, HICS principles appear ripe for application in IT outages and through development of ICS positions structured specifically to this type of contingency.34
CONCLUSION
Disasters unfold rapidly with marked patient surges and the potential to strain healthcare systems over an extended period. However, in both instances, hospitalists are possibly some of the most qualified clinicians to prepare for and respond to such events. Hospitalists need to assume a leadership role in emergency preparedness to integrate seamlessly into hospital incident command structures and to shape the interdepartmental relationships vital to success – skills at which hospitalists excel. Although no plan can address all possible disasters, familiarity with HICS and well-prepared and well-written JASs should help groups respond and succeed in almost all hazards.
Disclosures
None of the authors have any conflicts of interest to report.
1. Homeland Security Presidential Directive-5. 2003.
2. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: I. General principles of response and management. J Am Acad Orthop Surg. 2007;15(7):388-396. doi: 10.5435/00124635-200707000-00004. PubMed
3. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: II. explosive, biologic, chemical, and nuclear agents. J Am Acad Orthop Surg. 2007;15(8):461-473. PubMed
4. Christian MD, Hawryluck L, Wax RS, et al., Development of a triage protocol for critical care during an influenza pandemic. CMAJ. 2006;175(11):1377-1381. doi: 10.1503/cmaj.060911. PubMed
5. Barbera JA, Macintyre AG. Medical Surge Capacity and Capability: The Healthcare Coalition in Emergency Response and Recovery. In: Knebel A, Trabert E, eds. Department of Health and Human Services. 2007.
6. Roccaforte JD, Cushman JG. Disaster preparation and management for the intensive care unit. Curr Opin Crit Care. 2002;8(6):607-615. PubMed
7. Roccaforte JD, Cushman JG. Disaster preparedness, triage, and surge capacity for hospital definitive care areas: optimizing outcomes when demands exceed resources. Anesthesiol Clin. 2007;25(1):161-177, xi. doi: 10.1016/j.anclin.2007.01.002. PubMed
8. Emergency Medical Services of California. Hospital Incident Command System V. 2014 [cited 2018 February 14th]. https://emsa.ca.gov/wp-content/uploads/sites/47/2017/09/HICS_Guidebook_2014_11.pdf. Accessed June 1, 2018.
9. Sprung CL, Zimmerman JL, Christian MD, et al. Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: Summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med. 2010;36(3):428-443. doi: 10.1007/s00134-010-1759-y. PubMed
10. Inpatient specialists help cut costs, reduce LOS. Hospitalists partner with case managers. Hosp Case Manag. 1997;5(5):79-81. PubMed
11. Thompson RE, Pfeifer K, Grant PJ, et al. Hospital medicine and perioperative care: A framework for high-quality, high-value collaborative care. J Hosp Med. 2017;12(4):277-282. doi: 10.12788/jhm.2717. PubMed
12. Gupta R, Moriates C, Harrison JD, et al. Development of a high-value care culture survey: A modified Delphi process and psychometric evaluation. BMJ Qual Saf. 2017;26(6):475-483. doi: 10.1136/bmjqs-2016-005612. PubMed
13. Tadmor B, McManus J, Koenig KL. The art and science of surge: Experience from Israel and the U.S. military. Acad Emerg Med. 2006;13(11):1130-1134. doi: 10.1197/j.aem.2006.06.043. PubMed
14. Myers AL. Vegas Hospitals Swamped With Victims After High-Rise Attack. Associated Press; 2017. https://www.msn.com/en-us/news/breakingnews/vegas-hospitals-swamped-with-victims-after-high-rise-attack/ar-AAsQyZ8?ocid=HPCDHP. Las Vegas. Accessed June 1, 2018.
15. Craig T. As the Wounded Kept Coming, Las Vegas Hospitals Dealt With Injuries Rarely Seen in the US. In: Mello F, Sun L, eds. Washington Post: Washington Post; Oct 3, 2017.
16. Porth L. Preparedness and Partnerships: Lessons learned from the Missouri disasters of 2011. A Focus on Joplin. 2012, Missouri Hospital Association. PubMed
17. Persoff J. First Response Mode: May 22, 2011, Joplin Tornado. June 5, 2011; Available from: http://stormdoctor.blogspot.com/2011/06/first-response-mode-may-22-2011-joplin.html. Accessed June 1, 2018.
18. Dichter JR, Kanter RK, Dries D, et al. System-level planning, coordination, and communication: care of the critically ill and injured during pandemics and disasters: CHEST consensus statement. Chest. 2014;146(4 Suppl):e87S-e102S. doi: 10.1378/chest.14-0738. PubMed
19. Thomas TL, Hsu EB, Kim HK, Colli S, Arana G, Green GB. The incident command system in disasters: Evaluation methods for a hospital-based exercise. Prehosp Disaster Med. 2005;20(1):14-23. doi: 10.1017/S1049023X00002090. PubMed
20. FEMA. The Historical Contex of Emergency Management. [cited 2018 February 14th]; Available from: https://training.fema.gov/emi.aspx. Accessed June 1, 2018.
21. Backer H. Hospital Incident Command System Guidebook 5th Edition. In: Smiley D, Schoenthal L, eds. California Emergency Medical Services Authority, 2014. Accessed June 1, 2018.
22. Emergency Management Resources. Available from: https://www.jointcommission.org/emergency_management.aspx. Accessed June 1, 2018.
23. Incident Command System Training Program. Available from: https://training.fema.gov/emiweb/is/icsresource/trainingmaterials.htm.
24. Agency, F.E.M. NIMS and the Incident Command System. Nov 23, 2004; Available from: https://www.fema.gov/txt/nims/nims_ics_position_paper.txt. Accessed June 1, 2018.
25. Peleg K, Kellermann AL. Enhancing hospital surge capacity for mass casualty events. JAMA. 2009;302(5):565-567. doi: 10.1001/jama.2009.1119. PubMed
26. Adalja AA, Watson M, Bouri N, et al. Absorbing citywide patient surge during Hurricane Sandy: a case study in accommodating multiple hospital evacuations. Ann Emerg Med. 2014;64(1):66-73.e1. doi: 10.1016/j.annemergmed.2013.12.010. PubMed
27. Adalja AA, Watson M, Wollner S, Rambhia KJ, Toner ES. Response to the sudden closure of St. Vincent’s Hospital: learning from a real, no-notice, prolonged surge event. Biosecur Bioterror. 2011;9(2):153-161. doi: 10.1089/bsp.2011.0002. PubMed
28. Klein JS, Weigelt JA. Disaster management. Lessons learned. Surg Clin North Am. 1991;71(2):257-266. PubMed
29. Frykberg ER. Medical management of disasters and mass casualties from terrorist bombings: How can we cope? J Trauma. 2002;53(2):201-212. doi: 10.1097/00005373-200208000-00001. PubMed
30. Lynn M, Gurr D, Memon A, Kaliff J. Management of conventional mass casualty incidents: Ten commandments for hospital planning. J Burn Care Res. 2006;27(5):649-658. doi: 10.1097/01.BCR.0000238119.29269.2B. PubMed
31. Williams J, Nocera M, Casteel C. The effectiveness of disaster training for health care workers: A systematic review. Ann Emerg Med. 2008;52(3):211-22, 222.e1-2. doi: 10.1016/j.annemergmed.2007.09.030. PubMed
32. Percent of Hospitals, By Type, that Possess Certified Health IT. 2015, US Department of Health and Human Services: Office of the National Coordinator for Health Information Technology.
33. Lee C, Robinson KM, Wendt K, Williamson D, et al. The preparedness of hospital Health Information Services for system failures due to internal disasters. Health Inf Manag. 2009;38(2):18-25. doi: 10.1177/183335830903800203. PubMed
34. Situations, C.o.G.f.E.C.S.o.C.f.U.i.D. and I.o. Medicine, Crisis Standards of Care: A Systems Framework for Catastrophic Disaster Response. Mar 21, 2012, Washington (DC): National Academies Press (US). PubMed
1. Homeland Security Presidential Directive-5. 2003.
2. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: I. General principles of response and management. J Am Acad Orthop Surg. 2007;15(7):388-396. doi: 10.5435/00124635-200707000-00004. PubMed
3. Born CT, Briggs SM, Ciraulo DL, et al. Disasters and mass casualties: II. explosive, biologic, chemical, and nuclear agents. J Am Acad Orthop Surg. 2007;15(8):461-473. PubMed
4. Christian MD, Hawryluck L, Wax RS, et al., Development of a triage protocol for critical care during an influenza pandemic. CMAJ. 2006;175(11):1377-1381. doi: 10.1503/cmaj.060911. PubMed
5. Barbera JA, Macintyre AG. Medical Surge Capacity and Capability: The Healthcare Coalition in Emergency Response and Recovery. In: Knebel A, Trabert E, eds. Department of Health and Human Services. 2007.
6. Roccaforte JD, Cushman JG. Disaster preparation and management for the intensive care unit. Curr Opin Crit Care. 2002;8(6):607-615. PubMed
7. Roccaforte JD, Cushman JG. Disaster preparedness, triage, and surge capacity for hospital definitive care areas: optimizing outcomes when demands exceed resources. Anesthesiol Clin. 2007;25(1):161-177, xi. doi: 10.1016/j.anclin.2007.01.002. PubMed
8. Emergency Medical Services of California. Hospital Incident Command System V. 2014 [cited 2018 February 14th]. https://emsa.ca.gov/wp-content/uploads/sites/47/2017/09/HICS_Guidebook_2014_11.pdf. Accessed June 1, 2018.
9. Sprung CL, Zimmerman JL, Christian MD, et al. Recommendations for intensive care unit and hospital preparations for an influenza epidemic or mass disaster: Summary report of the European Society of Intensive Care Medicine’s Task Force for intensive care unit triage during an influenza epidemic or mass disaster. Intensive Care Med. 2010;36(3):428-443. doi: 10.1007/s00134-010-1759-y. PubMed
10. Inpatient specialists help cut costs, reduce LOS. Hospitalists partner with case managers. Hosp Case Manag. 1997;5(5):79-81. PubMed
11. Thompson RE, Pfeifer K, Grant PJ, et al. Hospital medicine and perioperative care: A framework for high-quality, high-value collaborative care. J Hosp Med. 2017;12(4):277-282. doi: 10.12788/jhm.2717. PubMed
12. Gupta R, Moriates C, Harrison JD, et al. Development of a high-value care culture survey: A modified Delphi process and psychometric evaluation. BMJ Qual Saf. 2017;26(6):475-483. doi: 10.1136/bmjqs-2016-005612. PubMed
13. Tadmor B, McManus J, Koenig KL. The art and science of surge: Experience from Israel and the U.S. military. Acad Emerg Med. 2006;13(11):1130-1134. doi: 10.1197/j.aem.2006.06.043. PubMed
14. Myers AL. Vegas Hospitals Swamped With Victims After High-Rise Attack. Associated Press; 2017. https://www.msn.com/en-us/news/breakingnews/vegas-hospitals-swamped-with-victims-after-high-rise-attack/ar-AAsQyZ8?ocid=HPCDHP. Las Vegas. Accessed June 1, 2018.
15. Craig T. As the Wounded Kept Coming, Las Vegas Hospitals Dealt With Injuries Rarely Seen in the US. In: Mello F, Sun L, eds. Washington Post: Washington Post; Oct 3, 2017.
16. Porth L. Preparedness and Partnerships: Lessons learned from the Missouri disasters of 2011. A Focus on Joplin. 2012, Missouri Hospital Association. PubMed
17. Persoff J. First Response Mode: May 22, 2011, Joplin Tornado. June 5, 2011; Available from: http://stormdoctor.blogspot.com/2011/06/first-response-mode-may-22-2011-joplin.html. Accessed June 1, 2018.
18. Dichter JR, Kanter RK, Dries D, et al. System-level planning, coordination, and communication: care of the critically ill and injured during pandemics and disasters: CHEST consensus statement. Chest. 2014;146(4 Suppl):e87S-e102S. doi: 10.1378/chest.14-0738. PubMed
19. Thomas TL, Hsu EB, Kim HK, Colli S, Arana G, Green GB. The incident command system in disasters: Evaluation methods for a hospital-based exercise. Prehosp Disaster Med. 2005;20(1):14-23. doi: 10.1017/S1049023X00002090. PubMed
20. FEMA. The Historical Contex of Emergency Management. [cited 2018 February 14th]; Available from: https://training.fema.gov/emi.aspx. Accessed June 1, 2018.
21. Backer H. Hospital Incident Command System Guidebook 5th Edition. In: Smiley D, Schoenthal L, eds. California Emergency Medical Services Authority, 2014. Accessed June 1, 2018.
22. Emergency Management Resources. Available from: https://www.jointcommission.org/emergency_management.aspx. Accessed June 1, 2018.
23. Incident Command System Training Program. Available from: https://training.fema.gov/emiweb/is/icsresource/trainingmaterials.htm.
24. Agency, F.E.M. NIMS and the Incident Command System. Nov 23, 2004; Available from: https://www.fema.gov/txt/nims/nims_ics_position_paper.txt. Accessed June 1, 2018.
25. Peleg K, Kellermann AL. Enhancing hospital surge capacity for mass casualty events. JAMA. 2009;302(5):565-567. doi: 10.1001/jama.2009.1119. PubMed
26. Adalja AA, Watson M, Bouri N, et al. Absorbing citywide patient surge during Hurricane Sandy: a case study in accommodating multiple hospital evacuations. Ann Emerg Med. 2014;64(1):66-73.e1. doi: 10.1016/j.annemergmed.2013.12.010. PubMed
27. Adalja AA, Watson M, Wollner S, Rambhia KJ, Toner ES. Response to the sudden closure of St. Vincent’s Hospital: learning from a real, no-notice, prolonged surge event. Biosecur Bioterror. 2011;9(2):153-161. doi: 10.1089/bsp.2011.0002. PubMed
28. Klein JS, Weigelt JA. Disaster management. Lessons learned. Surg Clin North Am. 1991;71(2):257-266. PubMed
29. Frykberg ER. Medical management of disasters and mass casualties from terrorist bombings: How can we cope? J Trauma. 2002;53(2):201-212. doi: 10.1097/00005373-200208000-00001. PubMed
30. Lynn M, Gurr D, Memon A, Kaliff J. Management of conventional mass casualty incidents: Ten commandments for hospital planning. J Burn Care Res. 2006;27(5):649-658. doi: 10.1097/01.BCR.0000238119.29269.2B. PubMed
31. Williams J, Nocera M, Casteel C. The effectiveness of disaster training for health care workers: A systematic review. Ann Emerg Med. 2008;52(3):211-22, 222.e1-2. doi: 10.1016/j.annemergmed.2007.09.030. PubMed
32. Percent of Hospitals, By Type, that Possess Certified Health IT. 2015, US Department of Health and Human Services: Office of the National Coordinator for Health Information Technology.
33. Lee C, Robinson KM, Wendt K, Williamson D, et al. The preparedness of hospital Health Information Services for system failures due to internal disasters. Health Inf Manag. 2009;38(2):18-25. doi: 10.1177/183335830903800203. PubMed
34. Situations, C.o.G.f.E.C.S.o.C.f.U.i.D. and I.o. Medicine, Crisis Standards of Care: A Systems Framework for Catastrophic Disaster Response. Mar 21, 2012, Washington (DC): National Academies Press (US). PubMed
© 2018 Society of Hospital Medicine
Perioperative Management of ACE Inhibitor Therapy: Challenges of Clinical Decision-Making Based on Surrogate Endpoints
Renin-angiotensin inhibitors, which include angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs), have demonstrated benefits in the treatment of several common cardiovascular and renal conditions. For example, they are prescribed to individuals with hypertension, heart failure with reduced ejection fraction (HFrEF), prior myocardial infarction, and chronic kidney disease with proteinuria. Perhaps unsurprisingly, many individuals presenting for surgery are already on long-term ACE inhibitor or ARB therapy. For example, such individuals comprised approximately one-third of the sample in the Vascular Events In Noncardiac Surgery Patients Cohort Evaluation (VISION) multicenter prospective cohort study of major inpatient noncardiac surgery.1
There is considerable controversy regarding how best to manage these cardiovascular medications during the perioperative period. The critical question pertains to whether renin-angiotensin inhibitors should be temporarily withdrawn 24 hours before surgery or continued uninterrupted up to the day of surgery. The main argument for withdrawing these medications is concern that they cause perioperative hypotension. For example, a recent systematic review of randomized controlled trials (RCTs) and cohort studies found that preoperative continuation of renin-angiotensin inhibitor therapy led to a significantly increased risk of intraoperative hypotension, albeit without associated effects on rates of death, major adverse cardiac events, or postoperative hypotension.2 Notably, randomized trial evidence in this meta-analysis was limited to only five trials with a total of 774 participants. Conversely, preoperative interruption of renin-angiotensin inhibitor therapy also has risks. For example, there is a potential for unintended permanent discontinuation of medications with long-term benefits.3 Furthermore, some prior cohort studies have demonstrated that the failure to resume renin-angiotensin inhibitor therapy promptly after surgery is associated with an elevated risk of postoperative mortality.4,5 While these studies have methodological limitations related to survivorship bias and unmeasured confounders, they still raise concerns that the abrupt withdrawal of long-term cardiovascular therapy before major surgery can have adverse effects. While ACE inhibitor withdrawal has not shown adverse physiological effects in the perioperative setting, it has led to rebound myocardial ischemia in patients with prior myocardial infarction.6
Given this controversy, there is variation across hospitals1 and practice guidelines with respect to perioperative management of renin-angiotensin inhibitors. For example, the 2017 Canadian Cardiovascular Society guidelines recommend that renin-angiotensin inhibitors be stopped temporarily 24 hours before major inpatient surgery,7 and the 2014 European guidelines recommend continuing therapy in patients with HFrEF but temporarily interrupting therapy in patients with hypertension.8 The 2014 American Heart Association and American College of Cardiology guidelines suggest that either continuation or interruption are reasonable options, but any interrupted therapy should be restarted postoperatively as soon as clinically feasible.9
In this issue of the Journal of Hospital Medicine, Shiffermiller and colleagues present a single-center RCT that provides additional high-quality data to improve our understanding of this important clinical issue.10 In a sample of 275 patients undergoing nonvascular inpatient noncardiac surgery, omission of the final dose of preoperative ACE inhibitor therapy reduced the risk of intraoperative hypotension across multiple definitions, including any episode of systolic blood pressure less than 80 mmHg (number needed to treat: 8), any episode of a systolic blood pressure less than 80 mmHg necessitating vasopressor therapy (number needed to treat: 6), and total cumulative duration of intraoperative systolic blood pressure less than 80 mmHg. In addition, the investigators found that preoperative interruption of ACE inhibitor therapy reduced the risk of postoperative hypotension (number needed to treat: 9), increased the risk of severe postoperative hypertension (number needed to harm: 9), and had no effect on clinical outcomes (eg, acute kidney injury, major adverse cardiac events). In conjunction with a recent systematic review,2 these new data demonstrate that temporary preoperative discontinuation of renin-angiotensin inhibitors leads to reduced risks of intraoperative and postoperative hypotension, with the only major identified risk being episodes of postoperative hypertension.
This current evidence base suggests that, in most cases, perioperative physicians should temporarily interrupt renin-angiotensin inhibitor therapy before inpatient noncardiac surgery, provided that protocols are in place to resume treatment postoperatively as soon as clinically feasible. Nonetheless, clinicians must also be cognizant of the key limitations to current data, namely that hypotension, be it intraoperative or postoperative, remains essentially a surrogate endpoint.11,12 Stated otherwise, the clinical importance of perioperative hypotension is largely predicated on its close association with clinically important or patient-relevant outcomes such as cardiovascular complications, acute kidney injury, and death.13–16 There is an implicit assumption that a reduction in the risk of hypotension will necessarily lead to reduced rates of clinical adverse events. This assumption is unlikely to be true, especially since many different underlying mechanisms lead to hypotension in the dynamic perioperative environment, including decreased cardiac contractility, decreased heart rate, decreased intravascular volume status, and vasodilation. Consistent with this possibility, different perioperative interventions with similar effects on hypotension have shown quite different effects on clinical outcomes. For example, epidural analgesia invariably reduces perioperative blood pressure, yet it does not appear to increase the risk of postoperative complications.17 Similarly, both beta-blockers and clonidine increase the risk of significant perioperative hypotension and bradycardia, yet only beta-blockers appear to lead to increased rates of mortality after noncardiac surgery.18,19 Thus, the relationship between perioperative hypotension and outcomes is clearly complex. Unless a RCT demonstrates that a hypotension-reduction strategy leads to an improvement in clinical outcomes,20 perioperative physicians should not assume that prevention of hypotension will always lead to improvements in patient-relevant clinical outcomes. Similar assumptions about other surrogate endpoints in cardiovascular medicine have sometimes been spectacularly incorrect.12,21 To more definitively address this important clinical issue, RCTs must be specifically designed to compare the effects of renin-angiotensin inhibitor therapy withdrawal versus continuation on patient-relevant and clinically important outcomes, such as death, myocardial infarction, and stroke. Fortunately, some ongoing trials will address this question, either directly (ClinicalTrials.gov NCT03374449) or as a component of a hypotension-avoidance strategy (ClinicalTrials.gov NCT03505723).
Overall, perioperative physicians should now adopt the standard approach of temporarily withdrawing renin-angiotensin inhibitor therapy 24 hours before major inpatient noncardiac surgery. Nonetheless, they should do so cautiously, recognizing that the data underpinning this strategy remain weak. As with many aspects of perioperative medicine, more research remains needed.
Disclosures
The authors have nothing to report.Funding: DNW is supported in part by a New Investigator Award from the Canadian Institutes of Health Research, and a Merit Award from the Department of Anesthesia at the University of Toronto.
1. Roshanov PS, Rochwerg B, Patel A, et al. Withholding versus continuing angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers before noncardiac surgery: An Analysis of the Vascular events In noncardiac Surgery patients cohort evaluation prospective cohort. Anesthesiology. 2017;126(1):16-27. doi: 10.1097/ALN.0000000000001404. PubMed
2. Hollmann C, Fernandes NL, Biccard BM. A systematic review of outcomes associated with withholding or continuing angiotensin-converting enzyme inhibitors and angiotensin receptor blockers before noncardiac surgery [published online ahead of print January 29, 2018]. Anesth Analg. doi: 10.1213/ANE.0000000000002837. PubMed
3. Bell CM, Bajcar J, Bierman AS, Li P, Mamdani MM, Urbach DR. Potentially unintended discontinuation of long-term medication use after elective surgical procedures. Arch Intern Med. 2006;166(22):2525-2531. doi: 10.1001/archinte.166.22.2525. PubMed
4. Mudumbai SC, Takemoto S, Cason BA, Au S, Upadhyay A, Wallace AW. Thirty-day mortality risk associated with the postoperative nonresumption of angiotensin-converting enzyme inhibitors: a retrospective study of the Veterans Affairs Healthcare System. J Hosp Med. 2014;9(5):289-296. doi: 10.1002/jhm.2182. PubMed
5. Lee SM, Takemoto S, Wallace AW. Association between withholding angiotensin receptor blockers in the early postoperative period and 30-day mortality: a cohort study of the Veterans Affairs Healthcare System. Anesthesiology. 2015;123(2):288-306. doi: 10.1097/ALN.0000000000000739. PubMed
6. van den Heuvel AF, van Gilst WH, van Veldhuisen DJ, de Vries RJ, Dunselman PH, Kingma JH. Long-term anti-ischemic effects of angiotensin-converting enzyme inhibition in patients after myocardial infarction. J Am Coll Cardiol. 1997;30(2):400-405. doi: 10.1016/S0735-1097(97)00183-6 PubMed
7. Duceppe E, Parlow J, MacDonald P, et al. Canadian Cardiovascular Society guidelines on perioperative cardiac risk assessment and management for patients who undergo noncardiac surgery. Can J Cardiol. 2017;33(1):17-32. doi: 10.1016/j.cjca.2016.09.008. PubMed
8. Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management./ The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J. 2014;35(35):2383-2431. doi: 10.1093/eurheartj/ehu282. PubMed
9. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130(24):e278-e333. doi: 10.1161/CIR.0000000000000105. PubMed
10. Shiffermiller JF, Monson BJ, Vokoun CW, et al. Prospective randomized evaluation of preoperative angiotensin converting enzyme inhibition (PREOP-ACEI) [published online ahead of print July 25, 2018]. J Hosp Med. doi: 10.12788/jhm.3036. PubMed
11. Psaty BM, Weiss NS, Furberg CD, et al. Surrogate end points, health outcomes, and the drug-approval process for the treatment of risk factors for cardiovascular disease. JAMA. 1999;282(8):786-790. doi: 10.1001/jama.282.8.786. PubMed
12. Vanderweele TJ. Surrogate measures and consistent surrogates. Biometrics. 2013;69(3):561-569. doi: 10.1111/biom.12071. PubMed
13. Sun LY, Wijeysundera DN, Tait GA, Beattie WS. Association of intraoperative hypotension with acute kidney injury after elective noncardiac surgery. Anesthesiology. 2015;123(3):515-523. doi: 10.1097/ALN.0000000000000765. PubMed
14. van Waes JA, van Klei WA, Wijeysundera DN, van Wolfswinkel L, Lindsay TF, Beattie WS. Association between intraoperative hypotension and myocardial injury after vascular surgery. Anesthesiology. 2016;124(1):35-44. doi: 10.1097/ALN.0000000000000922. PubMed
15. Salmasi V, Maheshwari K, Yang D, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126(1):47-65. doi: 10.1097/ALN.0000000000001432. PubMed
16. Monk TG, Bronsert MR, Henderson WG, et al. Association between intraoperative hypotension and hypertension and 30-day postoperative mortality in noncardiac surgery. Anesthesiology. 2015;123(2):307-319.doi: 10.1097/ALN.0000000000000756. PubMed
17. Rigg JR, Jamrozik K, Myles PS, et al. Epidural anaesthesia and analgesia and outcome of major surgery: a randomised trial. Lancet. 2002;359(9314):1276-1282. doi: 10.1016/S0140-6736(02)08266-1. PubMed
18. POISE Study Group. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371(9627):1839-1847. doi: 10.1016/S0140-6736(08)60601-7. PubMed
19. Devereaux PJ, Sessler DI, Leslie K, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med. 2014;370(16):1504-1513. doi: 10.1056/NEJMoa1401106. PubMed
20. Futier E, Lefrant JY, Guinot PG, et al. Effect of individualized vs standard blood pressure management strategies on postoperative organ dysfunction among high-risk patients undergoing major surgery: a randomized clinical trial. JAMA. 2017;318(14):1346-1357. doi: 10.1001/jama.2017.14172. PubMed
21. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629 PubMed
Renin-angiotensin inhibitors, which include angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs), have demonstrated benefits in the treatment of several common cardiovascular and renal conditions. For example, they are prescribed to individuals with hypertension, heart failure with reduced ejection fraction (HFrEF), prior myocardial infarction, and chronic kidney disease with proteinuria. Perhaps unsurprisingly, many individuals presenting for surgery are already on long-term ACE inhibitor or ARB therapy. For example, such individuals comprised approximately one-third of the sample in the Vascular Events In Noncardiac Surgery Patients Cohort Evaluation (VISION) multicenter prospective cohort study of major inpatient noncardiac surgery.1
There is considerable controversy regarding how best to manage these cardiovascular medications during the perioperative period. The critical question pertains to whether renin-angiotensin inhibitors should be temporarily withdrawn 24 hours before surgery or continued uninterrupted up to the day of surgery. The main argument for withdrawing these medications is concern that they cause perioperative hypotension. For example, a recent systematic review of randomized controlled trials (RCTs) and cohort studies found that preoperative continuation of renin-angiotensin inhibitor therapy led to a significantly increased risk of intraoperative hypotension, albeit without associated effects on rates of death, major adverse cardiac events, or postoperative hypotension.2 Notably, randomized trial evidence in this meta-analysis was limited to only five trials with a total of 774 participants. Conversely, preoperative interruption of renin-angiotensin inhibitor therapy also has risks. For example, there is a potential for unintended permanent discontinuation of medications with long-term benefits.3 Furthermore, some prior cohort studies have demonstrated that the failure to resume renin-angiotensin inhibitor therapy promptly after surgery is associated with an elevated risk of postoperative mortality.4,5 While these studies have methodological limitations related to survivorship bias and unmeasured confounders, they still raise concerns that the abrupt withdrawal of long-term cardiovascular therapy before major surgery can have adverse effects. While ACE inhibitor withdrawal has not shown adverse physiological effects in the perioperative setting, it has led to rebound myocardial ischemia in patients with prior myocardial infarction.6
Given this controversy, there is variation across hospitals1 and practice guidelines with respect to perioperative management of renin-angiotensin inhibitors. For example, the 2017 Canadian Cardiovascular Society guidelines recommend that renin-angiotensin inhibitors be stopped temporarily 24 hours before major inpatient surgery,7 and the 2014 European guidelines recommend continuing therapy in patients with HFrEF but temporarily interrupting therapy in patients with hypertension.8 The 2014 American Heart Association and American College of Cardiology guidelines suggest that either continuation or interruption are reasonable options, but any interrupted therapy should be restarted postoperatively as soon as clinically feasible.9
In this issue of the Journal of Hospital Medicine, Shiffermiller and colleagues present a single-center RCT that provides additional high-quality data to improve our understanding of this important clinical issue.10 In a sample of 275 patients undergoing nonvascular inpatient noncardiac surgery, omission of the final dose of preoperative ACE inhibitor therapy reduced the risk of intraoperative hypotension across multiple definitions, including any episode of systolic blood pressure less than 80 mmHg (number needed to treat: 8), any episode of a systolic blood pressure less than 80 mmHg necessitating vasopressor therapy (number needed to treat: 6), and total cumulative duration of intraoperative systolic blood pressure less than 80 mmHg. In addition, the investigators found that preoperative interruption of ACE inhibitor therapy reduced the risk of postoperative hypotension (number needed to treat: 9), increased the risk of severe postoperative hypertension (number needed to harm: 9), and had no effect on clinical outcomes (eg, acute kidney injury, major adverse cardiac events). In conjunction with a recent systematic review,2 these new data demonstrate that temporary preoperative discontinuation of renin-angiotensin inhibitors leads to reduced risks of intraoperative and postoperative hypotension, with the only major identified risk being episodes of postoperative hypertension.
This current evidence base suggests that, in most cases, perioperative physicians should temporarily interrupt renin-angiotensin inhibitor therapy before inpatient noncardiac surgery, provided that protocols are in place to resume treatment postoperatively as soon as clinically feasible. Nonetheless, clinicians must also be cognizant of the key limitations to current data, namely that hypotension, be it intraoperative or postoperative, remains essentially a surrogate endpoint.11,12 Stated otherwise, the clinical importance of perioperative hypotension is largely predicated on its close association with clinically important or patient-relevant outcomes such as cardiovascular complications, acute kidney injury, and death.13–16 There is an implicit assumption that a reduction in the risk of hypotension will necessarily lead to reduced rates of clinical adverse events. This assumption is unlikely to be true, especially since many different underlying mechanisms lead to hypotension in the dynamic perioperative environment, including decreased cardiac contractility, decreased heart rate, decreased intravascular volume status, and vasodilation. Consistent with this possibility, different perioperative interventions with similar effects on hypotension have shown quite different effects on clinical outcomes. For example, epidural analgesia invariably reduces perioperative blood pressure, yet it does not appear to increase the risk of postoperative complications.17 Similarly, both beta-blockers and clonidine increase the risk of significant perioperative hypotension and bradycardia, yet only beta-blockers appear to lead to increased rates of mortality after noncardiac surgery.18,19 Thus, the relationship between perioperative hypotension and outcomes is clearly complex. Unless a RCT demonstrates that a hypotension-reduction strategy leads to an improvement in clinical outcomes,20 perioperative physicians should not assume that prevention of hypotension will always lead to improvements in patient-relevant clinical outcomes. Similar assumptions about other surrogate endpoints in cardiovascular medicine have sometimes been spectacularly incorrect.12,21 To more definitively address this important clinical issue, RCTs must be specifically designed to compare the effects of renin-angiotensin inhibitor therapy withdrawal versus continuation on patient-relevant and clinically important outcomes, such as death, myocardial infarction, and stroke. Fortunately, some ongoing trials will address this question, either directly (ClinicalTrials.gov NCT03374449) or as a component of a hypotension-avoidance strategy (ClinicalTrials.gov NCT03505723).
Overall, perioperative physicians should now adopt the standard approach of temporarily withdrawing renin-angiotensin inhibitor therapy 24 hours before major inpatient noncardiac surgery. Nonetheless, they should do so cautiously, recognizing that the data underpinning this strategy remain weak. As with many aspects of perioperative medicine, more research remains needed.
Disclosures
The authors have nothing to report.Funding: DNW is supported in part by a New Investigator Award from the Canadian Institutes of Health Research, and a Merit Award from the Department of Anesthesia at the University of Toronto.
Renin-angiotensin inhibitors, which include angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs), have demonstrated benefits in the treatment of several common cardiovascular and renal conditions. For example, they are prescribed to individuals with hypertension, heart failure with reduced ejection fraction (HFrEF), prior myocardial infarction, and chronic kidney disease with proteinuria. Perhaps unsurprisingly, many individuals presenting for surgery are already on long-term ACE inhibitor or ARB therapy. For example, such individuals comprised approximately one-third of the sample in the Vascular Events In Noncardiac Surgery Patients Cohort Evaluation (VISION) multicenter prospective cohort study of major inpatient noncardiac surgery.1
There is considerable controversy regarding how best to manage these cardiovascular medications during the perioperative period. The critical question pertains to whether renin-angiotensin inhibitors should be temporarily withdrawn 24 hours before surgery or continued uninterrupted up to the day of surgery. The main argument for withdrawing these medications is concern that they cause perioperative hypotension. For example, a recent systematic review of randomized controlled trials (RCTs) and cohort studies found that preoperative continuation of renin-angiotensin inhibitor therapy led to a significantly increased risk of intraoperative hypotension, albeit without associated effects on rates of death, major adverse cardiac events, or postoperative hypotension.2 Notably, randomized trial evidence in this meta-analysis was limited to only five trials with a total of 774 participants. Conversely, preoperative interruption of renin-angiotensin inhibitor therapy also has risks. For example, there is a potential for unintended permanent discontinuation of medications with long-term benefits.3 Furthermore, some prior cohort studies have demonstrated that the failure to resume renin-angiotensin inhibitor therapy promptly after surgery is associated with an elevated risk of postoperative mortality.4,5 While these studies have methodological limitations related to survivorship bias and unmeasured confounders, they still raise concerns that the abrupt withdrawal of long-term cardiovascular therapy before major surgery can have adverse effects. While ACE inhibitor withdrawal has not shown adverse physiological effects in the perioperative setting, it has led to rebound myocardial ischemia in patients with prior myocardial infarction.6
Given this controversy, there is variation across hospitals1 and practice guidelines with respect to perioperative management of renin-angiotensin inhibitors. For example, the 2017 Canadian Cardiovascular Society guidelines recommend that renin-angiotensin inhibitors be stopped temporarily 24 hours before major inpatient surgery,7 and the 2014 European guidelines recommend continuing therapy in patients with HFrEF but temporarily interrupting therapy in patients with hypertension.8 The 2014 American Heart Association and American College of Cardiology guidelines suggest that either continuation or interruption are reasonable options, but any interrupted therapy should be restarted postoperatively as soon as clinically feasible.9
In this issue of the Journal of Hospital Medicine, Shiffermiller and colleagues present a single-center RCT that provides additional high-quality data to improve our understanding of this important clinical issue.10 In a sample of 275 patients undergoing nonvascular inpatient noncardiac surgery, omission of the final dose of preoperative ACE inhibitor therapy reduced the risk of intraoperative hypotension across multiple definitions, including any episode of systolic blood pressure less than 80 mmHg (number needed to treat: 8), any episode of a systolic blood pressure less than 80 mmHg necessitating vasopressor therapy (number needed to treat: 6), and total cumulative duration of intraoperative systolic blood pressure less than 80 mmHg. In addition, the investigators found that preoperative interruption of ACE inhibitor therapy reduced the risk of postoperative hypotension (number needed to treat: 9), increased the risk of severe postoperative hypertension (number needed to harm: 9), and had no effect on clinical outcomes (eg, acute kidney injury, major adverse cardiac events). In conjunction with a recent systematic review,2 these new data demonstrate that temporary preoperative discontinuation of renin-angiotensin inhibitors leads to reduced risks of intraoperative and postoperative hypotension, with the only major identified risk being episodes of postoperative hypertension.
This current evidence base suggests that, in most cases, perioperative physicians should temporarily interrupt renin-angiotensin inhibitor therapy before inpatient noncardiac surgery, provided that protocols are in place to resume treatment postoperatively as soon as clinically feasible. Nonetheless, clinicians must also be cognizant of the key limitations to current data, namely that hypotension, be it intraoperative or postoperative, remains essentially a surrogate endpoint.11,12 Stated otherwise, the clinical importance of perioperative hypotension is largely predicated on its close association with clinically important or patient-relevant outcomes such as cardiovascular complications, acute kidney injury, and death.13–16 There is an implicit assumption that a reduction in the risk of hypotension will necessarily lead to reduced rates of clinical adverse events. This assumption is unlikely to be true, especially since many different underlying mechanisms lead to hypotension in the dynamic perioperative environment, including decreased cardiac contractility, decreased heart rate, decreased intravascular volume status, and vasodilation. Consistent with this possibility, different perioperative interventions with similar effects on hypotension have shown quite different effects on clinical outcomes. For example, epidural analgesia invariably reduces perioperative blood pressure, yet it does not appear to increase the risk of postoperative complications.17 Similarly, both beta-blockers and clonidine increase the risk of significant perioperative hypotension and bradycardia, yet only beta-blockers appear to lead to increased rates of mortality after noncardiac surgery.18,19 Thus, the relationship between perioperative hypotension and outcomes is clearly complex. Unless a RCT demonstrates that a hypotension-reduction strategy leads to an improvement in clinical outcomes,20 perioperative physicians should not assume that prevention of hypotension will always lead to improvements in patient-relevant clinical outcomes. Similar assumptions about other surrogate endpoints in cardiovascular medicine have sometimes been spectacularly incorrect.12,21 To more definitively address this important clinical issue, RCTs must be specifically designed to compare the effects of renin-angiotensin inhibitor therapy withdrawal versus continuation on patient-relevant and clinically important outcomes, such as death, myocardial infarction, and stroke. Fortunately, some ongoing trials will address this question, either directly (ClinicalTrials.gov NCT03374449) or as a component of a hypotension-avoidance strategy (ClinicalTrials.gov NCT03505723).
Overall, perioperative physicians should now adopt the standard approach of temporarily withdrawing renin-angiotensin inhibitor therapy 24 hours before major inpatient noncardiac surgery. Nonetheless, they should do so cautiously, recognizing that the data underpinning this strategy remain weak. As with many aspects of perioperative medicine, more research remains needed.
Disclosures
The authors have nothing to report.Funding: DNW is supported in part by a New Investigator Award from the Canadian Institutes of Health Research, and a Merit Award from the Department of Anesthesia at the University of Toronto.
1. Roshanov PS, Rochwerg B, Patel A, et al. Withholding versus continuing angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers before noncardiac surgery: An Analysis of the Vascular events In noncardiac Surgery patients cohort evaluation prospective cohort. Anesthesiology. 2017;126(1):16-27. doi: 10.1097/ALN.0000000000001404. PubMed
2. Hollmann C, Fernandes NL, Biccard BM. A systematic review of outcomes associated with withholding or continuing angiotensin-converting enzyme inhibitors and angiotensin receptor blockers before noncardiac surgery [published online ahead of print January 29, 2018]. Anesth Analg. doi: 10.1213/ANE.0000000000002837. PubMed
3. Bell CM, Bajcar J, Bierman AS, Li P, Mamdani MM, Urbach DR. Potentially unintended discontinuation of long-term medication use after elective surgical procedures. Arch Intern Med. 2006;166(22):2525-2531. doi: 10.1001/archinte.166.22.2525. PubMed
4. Mudumbai SC, Takemoto S, Cason BA, Au S, Upadhyay A, Wallace AW. Thirty-day mortality risk associated with the postoperative nonresumption of angiotensin-converting enzyme inhibitors: a retrospective study of the Veterans Affairs Healthcare System. J Hosp Med. 2014;9(5):289-296. doi: 10.1002/jhm.2182. PubMed
5. Lee SM, Takemoto S, Wallace AW. Association between withholding angiotensin receptor blockers in the early postoperative period and 30-day mortality: a cohort study of the Veterans Affairs Healthcare System. Anesthesiology. 2015;123(2):288-306. doi: 10.1097/ALN.0000000000000739. PubMed
6. van den Heuvel AF, van Gilst WH, van Veldhuisen DJ, de Vries RJ, Dunselman PH, Kingma JH. Long-term anti-ischemic effects of angiotensin-converting enzyme inhibition in patients after myocardial infarction. J Am Coll Cardiol. 1997;30(2):400-405. doi: 10.1016/S0735-1097(97)00183-6 PubMed
7. Duceppe E, Parlow J, MacDonald P, et al. Canadian Cardiovascular Society guidelines on perioperative cardiac risk assessment and management for patients who undergo noncardiac surgery. Can J Cardiol. 2017;33(1):17-32. doi: 10.1016/j.cjca.2016.09.008. PubMed
8. Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management./ The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J. 2014;35(35):2383-2431. doi: 10.1093/eurheartj/ehu282. PubMed
9. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130(24):e278-e333. doi: 10.1161/CIR.0000000000000105. PubMed
10. Shiffermiller JF, Monson BJ, Vokoun CW, et al. Prospective randomized evaluation of preoperative angiotensin converting enzyme inhibition (PREOP-ACEI) [published online ahead of print July 25, 2018]. J Hosp Med. doi: 10.12788/jhm.3036. PubMed
11. Psaty BM, Weiss NS, Furberg CD, et al. Surrogate end points, health outcomes, and the drug-approval process for the treatment of risk factors for cardiovascular disease. JAMA. 1999;282(8):786-790. doi: 10.1001/jama.282.8.786. PubMed
12. Vanderweele TJ. Surrogate measures and consistent surrogates. Biometrics. 2013;69(3):561-569. doi: 10.1111/biom.12071. PubMed
13. Sun LY, Wijeysundera DN, Tait GA, Beattie WS. Association of intraoperative hypotension with acute kidney injury after elective noncardiac surgery. Anesthesiology. 2015;123(3):515-523. doi: 10.1097/ALN.0000000000000765. PubMed
14. van Waes JA, van Klei WA, Wijeysundera DN, van Wolfswinkel L, Lindsay TF, Beattie WS. Association between intraoperative hypotension and myocardial injury after vascular surgery. Anesthesiology. 2016;124(1):35-44. doi: 10.1097/ALN.0000000000000922. PubMed
15. Salmasi V, Maheshwari K, Yang D, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126(1):47-65. doi: 10.1097/ALN.0000000000001432. PubMed
16. Monk TG, Bronsert MR, Henderson WG, et al. Association between intraoperative hypotension and hypertension and 30-day postoperative mortality in noncardiac surgery. Anesthesiology. 2015;123(2):307-319.doi: 10.1097/ALN.0000000000000756. PubMed
17. Rigg JR, Jamrozik K, Myles PS, et al. Epidural anaesthesia and analgesia and outcome of major surgery: a randomised trial. Lancet. 2002;359(9314):1276-1282. doi: 10.1016/S0140-6736(02)08266-1. PubMed
18. POISE Study Group. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371(9627):1839-1847. doi: 10.1016/S0140-6736(08)60601-7. PubMed
19. Devereaux PJ, Sessler DI, Leslie K, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med. 2014;370(16):1504-1513. doi: 10.1056/NEJMoa1401106. PubMed
20. Futier E, Lefrant JY, Guinot PG, et al. Effect of individualized vs standard blood pressure management strategies on postoperative organ dysfunction among high-risk patients undergoing major surgery: a randomized clinical trial. JAMA. 2017;318(14):1346-1357. doi: 10.1001/jama.2017.14172. PubMed
21. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629 PubMed
1. Roshanov PS, Rochwerg B, Patel A, et al. Withholding versus continuing angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers before noncardiac surgery: An Analysis of the Vascular events In noncardiac Surgery patients cohort evaluation prospective cohort. Anesthesiology. 2017;126(1):16-27. doi: 10.1097/ALN.0000000000001404. PubMed
2. Hollmann C, Fernandes NL, Biccard BM. A systematic review of outcomes associated with withholding or continuing angiotensin-converting enzyme inhibitors and angiotensin receptor blockers before noncardiac surgery [published online ahead of print January 29, 2018]. Anesth Analg. doi: 10.1213/ANE.0000000000002837. PubMed
3. Bell CM, Bajcar J, Bierman AS, Li P, Mamdani MM, Urbach DR. Potentially unintended discontinuation of long-term medication use after elective surgical procedures. Arch Intern Med. 2006;166(22):2525-2531. doi: 10.1001/archinte.166.22.2525. PubMed
4. Mudumbai SC, Takemoto S, Cason BA, Au S, Upadhyay A, Wallace AW. Thirty-day mortality risk associated with the postoperative nonresumption of angiotensin-converting enzyme inhibitors: a retrospective study of the Veterans Affairs Healthcare System. J Hosp Med. 2014;9(5):289-296. doi: 10.1002/jhm.2182. PubMed
5. Lee SM, Takemoto S, Wallace AW. Association between withholding angiotensin receptor blockers in the early postoperative period and 30-day mortality: a cohort study of the Veterans Affairs Healthcare System. Anesthesiology. 2015;123(2):288-306. doi: 10.1097/ALN.0000000000000739. PubMed
6. van den Heuvel AF, van Gilst WH, van Veldhuisen DJ, de Vries RJ, Dunselman PH, Kingma JH. Long-term anti-ischemic effects of angiotensin-converting enzyme inhibition in patients after myocardial infarction. J Am Coll Cardiol. 1997;30(2):400-405. doi: 10.1016/S0735-1097(97)00183-6 PubMed
7. Duceppe E, Parlow J, MacDonald P, et al. Canadian Cardiovascular Society guidelines on perioperative cardiac risk assessment and management for patients who undergo noncardiac surgery. Can J Cardiol. 2017;33(1):17-32. doi: 10.1016/j.cjca.2016.09.008. PubMed
8. Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management./ The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J. 2014;35(35):2383-2431. doi: 10.1093/eurheartj/ehu282. PubMed
9. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130(24):e278-e333. doi: 10.1161/CIR.0000000000000105. PubMed
10. Shiffermiller JF, Monson BJ, Vokoun CW, et al. Prospective randomized evaluation of preoperative angiotensin converting enzyme inhibition (PREOP-ACEI) [published online ahead of print July 25, 2018]. J Hosp Med. doi: 10.12788/jhm.3036. PubMed
11. Psaty BM, Weiss NS, Furberg CD, et al. Surrogate end points, health outcomes, and the drug-approval process for the treatment of risk factors for cardiovascular disease. JAMA. 1999;282(8):786-790. doi: 10.1001/jama.282.8.786. PubMed
12. Vanderweele TJ. Surrogate measures and consistent surrogates. Biometrics. 2013;69(3):561-569. doi: 10.1111/biom.12071. PubMed
13. Sun LY, Wijeysundera DN, Tait GA, Beattie WS. Association of intraoperative hypotension with acute kidney injury after elective noncardiac surgery. Anesthesiology. 2015;123(3):515-523. doi: 10.1097/ALN.0000000000000765. PubMed
14. van Waes JA, van Klei WA, Wijeysundera DN, van Wolfswinkel L, Lindsay TF, Beattie WS. Association between intraoperative hypotension and myocardial injury after vascular surgery. Anesthesiology. 2016;124(1):35-44. doi: 10.1097/ALN.0000000000000922. PubMed
15. Salmasi V, Maheshwari K, Yang D, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126(1):47-65. doi: 10.1097/ALN.0000000000001432. PubMed
16. Monk TG, Bronsert MR, Henderson WG, et al. Association between intraoperative hypotension and hypertension and 30-day postoperative mortality in noncardiac surgery. Anesthesiology. 2015;123(2):307-319.doi: 10.1097/ALN.0000000000000756. PubMed
17. Rigg JR, Jamrozik K, Myles PS, et al. Epidural anaesthesia and analgesia and outcome of major surgery: a randomised trial. Lancet. 2002;359(9314):1276-1282. doi: 10.1016/S0140-6736(02)08266-1. PubMed
18. POISE Study Group. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008;371(9627):1839-1847. doi: 10.1016/S0140-6736(08)60601-7. PubMed
19. Devereaux PJ, Sessler DI, Leslie K, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med. 2014;370(16):1504-1513. doi: 10.1056/NEJMoa1401106. PubMed
20. Futier E, Lefrant JY, Guinot PG, et al. Effect of individualized vs standard blood pressure management strategies on postoperative organ dysfunction among high-risk patients undergoing major surgery: a randomized clinical trial. JAMA. 2017;318(14):1346-1357. doi: 10.1001/jama.2017.14172. PubMed
21. Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321(6):406-412. doi:10.1056/NEJM198908103210629 PubMed
© 2018 Society of Hospital Medicine
Does Patient Experience Predict 30-Day Readmission? A Patient-Level Analysis of HCAHPS Data
Patient experience and 30-day readmission are important measures of quality of care for hospitalized patients. Performance on both of these measures impact hospitals financially. Performance on the Hospital Consumer Assessment of Healthcare Systems and Providers (HCAHPS) survey is linked to 25% of the incentive payment under Value Based Purchasing (VBP) Program.1 Starting in 2012, the Centers for Medicare and Medicaid Services (CMS) introduced the Readmission Reduction Program, penalizing hospitals financially for excessive readmissions.2
A relationship between patient experience and readmissions has been explored at the hospital level. Studies have mostly found that higher patient experience scores are associated with lower 30-day readmission rates. In a study of the relationship between 30-day risk-standardized readmission rates for three medical conditions (acute myocardial infarction, heart failure, and pneumonia) and patient experience, the authors noted that higher experience scores for overall care and discharge planning were associated with lower readmission rates for these conditions. They also concluded that patient experience scores were more predictive of 30-day readmission than clinical performance measures. Additionally, the authors predicted that if a hospital increased its total experience scores from the 25th percentile to the 75th percentile, there would be an associated decrease in readmissions by at least 2.3% for each of these conditions.3 Practice management companies and the media have cited this finding to conclude that higher patient experience drives clinical outcomes such as 30-day readmission and that patients are often the best judges of the quality of care delivered.4,5
Other hospital-level studies have found that high 30-day readmission rates are associated with lower overall experience scores in a mixed surgical patient population; worse reports of pain control and overall care in the colorectal surgery population; lower experience scores with discharge preparedness in vascular surgery patients; and lower experience scores with physician communication, nurse communication, and discharge preparedness.6-9 A patient-level study noted higher readmissions are associated with worse experience with physician and nursing communication along with a paradoxically better experience with discharge information.10
Because these studies used an observational design, they demonstrated associations rather than causality. An alternative hypothesis is that readmitted patients complete their patient experience survey after readmission and the low experience is the result, rather than the cause, of their readmission. For patients who are readmitted, it is unclear whether there is an opportunity to complete the survey prior to readmission and whether being readmitted may impact patient perception of quality of care. Using patient-level data, we sought to assess HCAHPS patient-experience responses linked to the index admission of the patients who were readmitted in 30 days and compare it with those patients who were not readmitted during this time period. We paid particular attention to when the surveys were returned.
METHODS
Study Design
We conducted a retrospective analysis of prospectively collected 10-year HCAHPS and Press Ganey patient survey data for a single tertiary care academic hospital.
Participants
All adult patients discharged from the hospital and who responded to the routinely sent patient-experience survey were included. Surveys were sent to a random sample of 50% of the discharged patients.
The exposure group was comprised of patients who responded to the survey and were readmitted within 30 days of discharge. After subtracting 5 days from the survey receipt date for expected delays related to mail delivery time and processing time, survey response date was calculated. The exposure group was further divided into patients who responded to the survey prior to their 30-day readmission (“Pre-readmission responders”) and those that responded to the survey after their readmission (“Postreadmission responders”). A sensitivity analysis was performed by changing the number of days subtracted from the survey receipt date by 2 days in either direction. This approach did not result in any significant changes in the results.
The control group comprised patients who were not readmitted to the hospital within 30 days of discharge and who did not have an admission in the previous 30 days as well (“Not readmitted” group). An additional comparison group for exploratory analysis included patients who had experienced an admission in the prior 30 days but were not readmitted after the admission linked to the survey. These patients responded to the patient-experience surveys that were linked to their second admission in 30 days (“2nd-admission responders” group; Figure).
Time Periods
All survey responders from the third quarter of 2006 to the first quarter of 2016 were included in the study. Additionally, administrative data on non-responders were available from 7/2006 to 8/2012. These data were used to estimate response rates. Patient level experience and administrative data were obtained in a linked fashion for these time periods.
Instruments
Press Ganey and HCAHPS surveys were sent via mail in the same envelope. Fifty percent of the discharged patients were randomized to receive the surveys. The Press Ganey survey contained 33 items encompassing several subdomains, including room, meal, nursing, physician, ancillary staff, visitor, discharge, and overall experience.
The HCAHPS survey contained 29 CMS-mandated items, of which 21 are related to patient experience. The development, testing, and methods for administration and reporting of the HCAHPS survey have been previously described and studies using this instrument have been reported in the literature.11 Press Ganey patient satisfaction survey results have also been reported in the literature.12
Outcome Variables and Covariates
HCAHPS and Press Ganey experience survey individual item responses were the primary outcome variables of this study. Age, self-reported health status, education, primary language spoken, service line, and time taken to respond to the surveys served as the covariates. These variables are used by CMS for patient-mix adjustment and are collected on the HCAHPS survey. Additionally, the number of days to respond to the survey were included in all regression analysis to adjust for early responder effect.13-15
Statistical Analysis
“Percent top-box” scores were calculated for each survey item for patients in each group. The percent top-box scores were calculated as the percent of patients who responded “very good” for a given item on Press Ganey survey items and “always” or “definitely yes” or “yes” or “9” or “10” on HCAHPS survey items. CMS utilizes “percent top-box scores” to calculate payments under the VBP program and to report the results publicly. Numerous studies have also reported percent top-box scores for HCAHPS survey results.12
We hypothesized that whether patients complete the HCAHPS survey before or after the readmission influences their reporting of experience. To test this hypothesis, HCAHPS and Press Ganey item top-box scores of “Pre-readmission responders” and “Postreadmission responders” were compared with those of the control group using multivariate logistic regression. “Pre-readmission responders” were also compared with “Postreadmission responders”.
“2nd-admission responders” were similarly compared with the control group for an exploratory analysis. Finally, “Postreadmission responders” and “2nd-admission responders” were compared in another exploratory analysis since both these groups responded to the survey after being exposed to the readmission, even though the “Postreadmission responders” group is administratively linked to the index admission.
The Johns Hopkins Institutional Review Board approved this study.
RESULTS
There were 43,737 survey responders, among whom 4,707 were subsequently readmitted within 30 days of discharge. Among the readmitted patients who responded to the surveys linked to their index admission, only 15.8% returned the survey before readmission (pre-readmission responders’) and 84.2% returned the survey after readmission (postreadmission responders). Additionally, 1,663 patients responded to experience surveys linked to their readmission. There were 37,365 patients in the control arm (ie, patients who responded to the survey and were not readmitted within 30 days of discharge or in the prior 30 days; Figure 1). The readmission rate among survey responders was 10.6%. Among the readmitted patients, the median number of days to readmission was 10 days while the median number of days to respond to the survey for this group was 33 days. Among the nonreadmitted patients, the median number of days to return the survey was 29 days.

We also conducted an exploratory analysis of the postreadmission responders, comparing them with patients who received patient-experience surveys linked to their second admission in 30 days. Both of these groups were exposed to a readmission before they completed the surveys. There were no significant differences between these two groups on patient experience scores. Additionally, the patients who received the survey linked to their readmission had a broad dissatisfaction pattern on HCAHPS survey items that appeared similar to that of the postreadmission group when compared to the non-readmitted group (Table 3).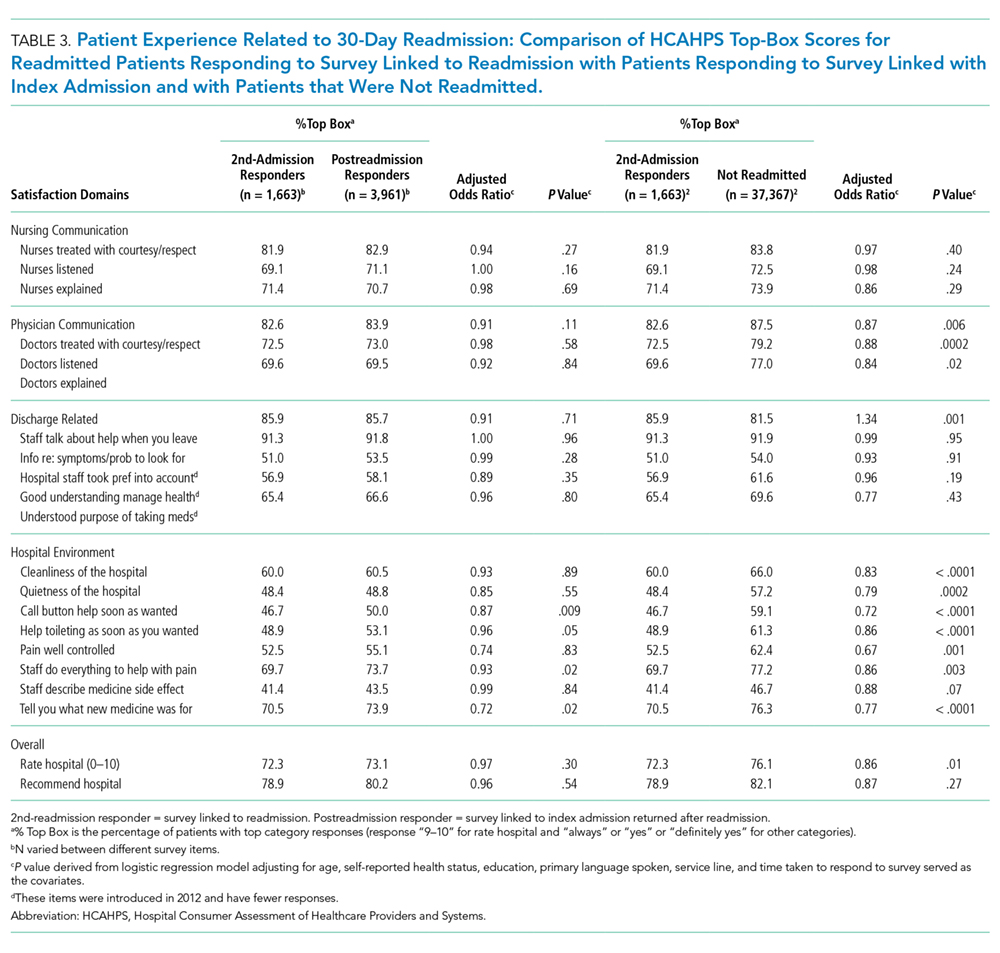
DISCUSSION
In this retrospective analysis of prospectively collected Press Ganey and HCAHPS patient-experience survey data, we found that the overwhelming majority of patients readmitted within 30 days of discharge respond to HCAHPS surveys after readmission even though the survey is sent linked to the first admission. This is not unexpected since the median time to survey response is 33 days for this group, while median time to readmission is 10 days. The dissatisfaction pattern of Postreadmission responders was similar to those who responded to the survey linked to the readmission. When a patient is readmitted prior to completing the survey, their responses appear to reflect the cumulative experience of the index admission and the readmission. The lower scores of those who respond to the survey after their readmission appear to be a driver for lower patient-experience scores related to readmissions. Overall, readmission was associated with lower scores on items in five of the nine domains used to calculate patient experience related payments under VBP.16
These findings have important implications in inferring the direction of potential causal relationship between readmissions and patient experience at the hospital level. Additionally, these patients show broad dissatisfaction with areas beyond physician communication and discharge planning. These include staff responsiveness, phlebotomy, meals, hospital cleanliness, and noise level. This pattern of dissatisfaction may represent impatience and frustration with spending additional time in the hospital environment.
Our results are consistent with findings of many of the earlier studies, but our study goes a step further by using patient-level data and incorporating survey response time in our analysis.3,7,9,10 By separating out the readmitted patients who responded to the survey prior to admission, we attempted to address the ability of patients’ perception of care to predict future readmissions. Our results do not support this idea, since pre-readmission responders had similar experience scores to non-readmitted patients. However, because of the low numbers of pre-readmission responders, the comparison lacks precision. Current HCAHPS and Press Ganey questions may lack the ability to predict future readmissions because of the timing of the survey (postdischarge) or the questions themselves.
Overall, postreadmission responders are dissatisfied with multiple domains of hospital care. Many of these survey responses may simply be related to general frustration. Alternatively, they may represent a patient population with a high degree of needs that are not as easily met by a hospital’s routine processes of care. Even though the readmission rates were 10.6% among survey responders, 14.6% of the survey responses were associated with readmissions after accounting for those who respond to surveys linked to readmission. These patients could have significant impact on cumulative experience scores.
Our study has a few limitations. First, it involves a single tertiary care academic center study, and our results may not be generalizable. Second, we did not adjust for some of the patient characteristics associated with readmissions. Patients who were admitted within 30 days are different than those not readmitted based on payor, race, length of stay, and severity of illness, and we did not adjust for these factors in our analysis. This was intentional, however. Our goal was to better understand the relationship between 30-day readmission and patient experience scores as they are used for hospital-level studies, VBP, and public reporting. For these purposes, the scores are not adjusted for factors, such as payor and length of stay. We did adjust for patient-mix adjustment factors used by CMS. Third, the response rates to the HCAHPS were low and may have biased the scores. However, HCAHPS is widely used for comparisons between hospitals has been validated, and our study results have implications with regard to comparing hospital-level performance. HCAHPS results are relevant to policy and have financial consequences.17 Fourth, our study did not directly compare whether the relationship between patient experience for the postreadmission group and nonreadmitted group was different from the relationship between the pre-readmission group and postreadmission group. It is possible that there is no difference in relationship between the groups. However, despite the small number of pre-readmission responders, these patients tended to have more favorable experience responses than those who responded after being readmitted, even after adjusting for response time. Although the P values are nonsignificant for many comparisons, the directionality of the effect is relatively consistent. Also, the vast majority of the patients fall in the postreadmission group, and these patients appear to drive the overall experience related to readmissions. Finally, since relatively few patients turned in surveys prior to readmission, we had limited power to detect a significant difference between these pre-readmission responders and nonreadmitted patients.
Our study has implications for policy makers, researchers, and providers. The HCAHPS scores of patients who are readmitted and completed the survey after being readmitted reflects their experience of both the index admission and the readmission. We did not find evidence to support that HCAHPS survey responses predict future readmissions at the patient level. Our findings do support the concept that lower readmissions rates (whether due to the patient population or processes of care that decrease readmission rates) may improve HCAHPS scores. We suggest caution in assuming that improving patient experience is likely to reduce readmission rates.
Disclosures
The authors declare no conflicts of interest.
1. Hospital value-based purchasing. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/downloads/Hospital_VBPurchasing_Fact_Sheet_ICN907664.pdf. Accessed June 25, 2016.
2. Readmissions reduction program (HRRP). Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Accessed June 25, 2016.
3. Boulding W, Glickman SW, Manary MP, Schulman KA, Staelin R. Relationship between patient satisfaction with inpatient care and hospital readmission within 30 days. Am J Manag Care. 2011;17(1):41-48. PubMed
4. Buum HA, Duran-Nelson AM, Menk J, Nixon LJ. Duty-hours monitoring revisited: self-report may not be adequate. Am J Med. 2013;126(4):362-365. doi: 10.1016/j.amjmed.2012.12.003 PubMed
5. Choma NN, Vasilevskis EE, Sponsler KC, Hathaway J, Kripalani S. Effect of the ACGME 16-hour rule on efficiency and quality of care: duty hours 2.0. JAMA Int Med. 2013;173(9):819-821. doi: 10.1001/jamainternmed.2013.3014 PubMed
6. Brooke BS, Samourjian E, Sarfati MR, Nguyen TT, Greer D, Kraiss LW. RR3. Patient-reported readiness at time of discharge predicts readmission following vascular surgery. J Vasc Surg. 2015;61(6):188S. doi: 10.1016/j.jvs.2015.04.356
7. Duraes LC, Merlino J, Stocchi L, et al. 756 readmission decreases patient satisfaction in colorectal surgery. Gastroenterology. 2014;146(5):S-1029. doi: 10.1016/S0016-5085(14)63751-3
8. Mitchell JP. Association of provider communication and discharge instructions on lower readmissions. J Healthc Qual. 2015;37(1):33-40. doi: 10.1097/01.JHQ.0000460126.88382.13 PubMed
9. Tsai TC, Orav EJ, Jha AK. Patient satisfaction and quality of surgical care in US hospitals. Ann Surg. 2015;261(1):2-8. doi: 10.1097/SLA.0000000000000765 PubMed
10. Hachem F, Canar J, Fullam M, Andrew S, Hohmann S, Johnson C. The relationships between HCAHPS communication and discharge satisfaction items and hospital readmissions. Patient Exp J. 2014;1(2):71-77.
11. Irby DM, Cooke M, Lowenstein D, Richards B. The academy movement: a structural approach to reinvigorating the educational mission. Acad Med. 2004;79(8):729-736. doi: 10.1097/00001888-200408000-00003 PubMed
12. Siddiqui ZK, Zuccarelli R, Durkin N, Wu AW, Brotman DJ. Changes in patient satisfaction related to hospital renovation: experience with a new clinical building. J Hosp Med. 2015;10(3):165-171. doi: 10.1002/jhm.2297 PubMed
13. Nair BR, Coughlan JL, Hensley MJ. Student and patient perspectives on bedside teaching. Med Educ. 1997;31(5):341-346. doi: 10.1046/j.1365-2923.1997.00673.x PubMed
14. Elliott MN, Zaslavsky AM, Goldstein E, et al. Effects of survey mode, patient mix, and nonresponse on CAHPS® hospital survey scores. BMC Health Serv Res. 2009;44(2p1):501-518. doi: 10.1111/j.1475-6773.2008.00914.x PubMed
15. Saunders CL, Elliott MN, Lyratzopoulos G, Abel GA. Do differential response rates to patient surveys between organizations lead to unfair performance comparisons?: evidence from the English Cancer Patient Experience Survey. Medical care. 2016;54(1):45. doi: 10.1097/MLR.0000000000000457 PubMed
16. Sabel E, Archer J. “Medical education is the ugly duckling of the medical world” and other challenges to medical educators’ identity construction: a qualitative study. Acad Med. 2014;89(11):1474-1480. doi: 10.1097/ACM.0000000000000420 PubMed
17. O’Malley AJ, Zaslavsky AM, Elliott MN, Zaborski L, Cleary PD. Case‐Mix adjustment of the CAHPS® Hospital Survey. BMC Health Serv Res. 2005;40(6p2):2162-2181. doi: 10.1111/j.1475-6773.2005.00470.x
Patient experience and 30-day readmission are important measures of quality of care for hospitalized patients. Performance on both of these measures impact hospitals financially. Performance on the Hospital Consumer Assessment of Healthcare Systems and Providers (HCAHPS) survey is linked to 25% of the incentive payment under Value Based Purchasing (VBP) Program.1 Starting in 2012, the Centers for Medicare and Medicaid Services (CMS) introduced the Readmission Reduction Program, penalizing hospitals financially for excessive readmissions.2
A relationship between patient experience and readmissions has been explored at the hospital level. Studies have mostly found that higher patient experience scores are associated with lower 30-day readmission rates. In a study of the relationship between 30-day risk-standardized readmission rates for three medical conditions (acute myocardial infarction, heart failure, and pneumonia) and patient experience, the authors noted that higher experience scores for overall care and discharge planning were associated with lower readmission rates for these conditions. They also concluded that patient experience scores were more predictive of 30-day readmission than clinical performance measures. Additionally, the authors predicted that if a hospital increased its total experience scores from the 25th percentile to the 75th percentile, there would be an associated decrease in readmissions by at least 2.3% for each of these conditions.3 Practice management companies and the media have cited this finding to conclude that higher patient experience drives clinical outcomes such as 30-day readmission and that patients are often the best judges of the quality of care delivered.4,5
Other hospital-level studies have found that high 30-day readmission rates are associated with lower overall experience scores in a mixed surgical patient population; worse reports of pain control and overall care in the colorectal surgery population; lower experience scores with discharge preparedness in vascular surgery patients; and lower experience scores with physician communication, nurse communication, and discharge preparedness.6-9 A patient-level study noted higher readmissions are associated with worse experience with physician and nursing communication along with a paradoxically better experience with discharge information.10
Because these studies used an observational design, they demonstrated associations rather than causality. An alternative hypothesis is that readmitted patients complete their patient experience survey after readmission and the low experience is the result, rather than the cause, of their readmission. For patients who are readmitted, it is unclear whether there is an opportunity to complete the survey prior to readmission and whether being readmitted may impact patient perception of quality of care. Using patient-level data, we sought to assess HCAHPS patient-experience responses linked to the index admission of the patients who were readmitted in 30 days and compare it with those patients who were not readmitted during this time period. We paid particular attention to when the surveys were returned.
METHODS
Study Design
We conducted a retrospective analysis of prospectively collected 10-year HCAHPS and Press Ganey patient survey data for a single tertiary care academic hospital.
Participants
All adult patients discharged from the hospital and who responded to the routinely sent patient-experience survey were included. Surveys were sent to a random sample of 50% of the discharged patients.
The exposure group was comprised of patients who responded to the survey and were readmitted within 30 days of discharge. After subtracting 5 days from the survey receipt date for expected delays related to mail delivery time and processing time, survey response date was calculated. The exposure group was further divided into patients who responded to the survey prior to their 30-day readmission (“Pre-readmission responders”) and those that responded to the survey after their readmission (“Postreadmission responders”). A sensitivity analysis was performed by changing the number of days subtracted from the survey receipt date by 2 days in either direction. This approach did not result in any significant changes in the results.
The control group comprised patients who were not readmitted to the hospital within 30 days of discharge and who did not have an admission in the previous 30 days as well (“Not readmitted” group). An additional comparison group for exploratory analysis included patients who had experienced an admission in the prior 30 days but were not readmitted after the admission linked to the survey. These patients responded to the patient-experience surveys that were linked to their second admission in 30 days (“2nd-admission responders” group; Figure).
Time Periods
All survey responders from the third quarter of 2006 to the first quarter of 2016 were included in the study. Additionally, administrative data on non-responders were available from 7/2006 to 8/2012. These data were used to estimate response rates. Patient level experience and administrative data were obtained in a linked fashion for these time periods.
Instruments
Press Ganey and HCAHPS surveys were sent via mail in the same envelope. Fifty percent of the discharged patients were randomized to receive the surveys. The Press Ganey survey contained 33 items encompassing several subdomains, including room, meal, nursing, physician, ancillary staff, visitor, discharge, and overall experience.
The HCAHPS survey contained 29 CMS-mandated items, of which 21 are related to patient experience. The development, testing, and methods for administration and reporting of the HCAHPS survey have been previously described and studies using this instrument have been reported in the literature.11 Press Ganey patient satisfaction survey results have also been reported in the literature.12
Outcome Variables and Covariates
HCAHPS and Press Ganey experience survey individual item responses were the primary outcome variables of this study. Age, self-reported health status, education, primary language spoken, service line, and time taken to respond to the surveys served as the covariates. These variables are used by CMS for patient-mix adjustment and are collected on the HCAHPS survey. Additionally, the number of days to respond to the survey were included in all regression analysis to adjust for early responder effect.13-15
Statistical Analysis
“Percent top-box” scores were calculated for each survey item for patients in each group. The percent top-box scores were calculated as the percent of patients who responded “very good” for a given item on Press Ganey survey items and “always” or “definitely yes” or “yes” or “9” or “10” on HCAHPS survey items. CMS utilizes “percent top-box scores” to calculate payments under the VBP program and to report the results publicly. Numerous studies have also reported percent top-box scores for HCAHPS survey results.12
We hypothesized that whether patients complete the HCAHPS survey before or after the readmission influences their reporting of experience. To test this hypothesis, HCAHPS and Press Ganey item top-box scores of “Pre-readmission responders” and “Postreadmission responders” were compared with those of the control group using multivariate logistic regression. “Pre-readmission responders” were also compared with “Postreadmission responders”.
“2nd-admission responders” were similarly compared with the control group for an exploratory analysis. Finally, “Postreadmission responders” and “2nd-admission responders” were compared in another exploratory analysis since both these groups responded to the survey after being exposed to the readmission, even though the “Postreadmission responders” group is administratively linked to the index admission.
The Johns Hopkins Institutional Review Board approved this study.
RESULTS
There were 43,737 survey responders, among whom 4,707 were subsequently readmitted within 30 days of discharge. Among the readmitted patients who responded to the surveys linked to their index admission, only 15.8% returned the survey before readmission (pre-readmission responders’) and 84.2% returned the survey after readmission (postreadmission responders). Additionally, 1,663 patients responded to experience surveys linked to their readmission. There were 37,365 patients in the control arm (ie, patients who responded to the survey and were not readmitted within 30 days of discharge or in the prior 30 days; Figure 1). The readmission rate among survey responders was 10.6%. Among the readmitted patients, the median number of days to readmission was 10 days while the median number of days to respond to the survey for this group was 33 days. Among the nonreadmitted patients, the median number of days to return the survey was 29 days.
We also conducted an exploratory analysis of the postreadmission responders, comparing them with patients who received patient-experience surveys linked to their second admission in 30 days. Both of these groups were exposed to a readmission before they completed the surveys. There were no significant differences between these two groups on patient experience scores. Additionally, the patients who received the survey linked to their readmission had a broad dissatisfaction pattern on HCAHPS survey items that appeared similar to that of the postreadmission group when compared to the non-readmitted group (Table 3).
DISCUSSION
In this retrospective analysis of prospectively collected Press Ganey and HCAHPS patient-experience survey data, we found that the overwhelming majority of patients readmitted within 30 days of discharge respond to HCAHPS surveys after readmission even though the survey is sent linked to the first admission. This is not unexpected since the median time to survey response is 33 days for this group, while median time to readmission is 10 days. The dissatisfaction pattern of Postreadmission responders was similar to those who responded to the survey linked to the readmission. When a patient is readmitted prior to completing the survey, their responses appear to reflect the cumulative experience of the index admission and the readmission. The lower scores of those who respond to the survey after their readmission appear to be a driver for lower patient-experience scores related to readmissions. Overall, readmission was associated with lower scores on items in five of the nine domains used to calculate patient experience related payments under VBP.16
These findings have important implications in inferring the direction of potential causal relationship between readmissions and patient experience at the hospital level. Additionally, these patients show broad dissatisfaction with areas beyond physician communication and discharge planning. These include staff responsiveness, phlebotomy, meals, hospital cleanliness, and noise level. This pattern of dissatisfaction may represent impatience and frustration with spending additional time in the hospital environment.
Our results are consistent with findings of many of the earlier studies, but our study goes a step further by using patient-level data and incorporating survey response time in our analysis.3,7,9,10 By separating out the readmitted patients who responded to the survey prior to admission, we attempted to address the ability of patients’ perception of care to predict future readmissions. Our results do not support this idea, since pre-readmission responders had similar experience scores to non-readmitted patients. However, because of the low numbers of pre-readmission responders, the comparison lacks precision. Current HCAHPS and Press Ganey questions may lack the ability to predict future readmissions because of the timing of the survey (postdischarge) or the questions themselves.
Overall, postreadmission responders are dissatisfied with multiple domains of hospital care. Many of these survey responses may simply be related to general frustration. Alternatively, they may represent a patient population with a high degree of needs that are not as easily met by a hospital’s routine processes of care. Even though the readmission rates were 10.6% among survey responders, 14.6% of the survey responses were associated with readmissions after accounting for those who respond to surveys linked to readmission. These patients could have significant impact on cumulative experience scores.
Our study has a few limitations. First, it involves a single tertiary care academic center study, and our results may not be generalizable. Second, we did not adjust for some of the patient characteristics associated with readmissions. Patients who were admitted within 30 days are different than those not readmitted based on payor, race, length of stay, and severity of illness, and we did not adjust for these factors in our analysis. This was intentional, however. Our goal was to better understand the relationship between 30-day readmission and patient experience scores as they are used for hospital-level studies, VBP, and public reporting. For these purposes, the scores are not adjusted for factors, such as payor and length of stay. We did adjust for patient-mix adjustment factors used by CMS. Third, the response rates to the HCAHPS were low and may have biased the scores. However, HCAHPS is widely used for comparisons between hospitals has been validated, and our study results have implications with regard to comparing hospital-level performance. HCAHPS results are relevant to policy and have financial consequences.17 Fourth, our study did not directly compare whether the relationship between patient experience for the postreadmission group and nonreadmitted group was different from the relationship between the pre-readmission group and postreadmission group. It is possible that there is no difference in relationship between the groups. However, despite the small number of pre-readmission responders, these patients tended to have more favorable experience responses than those who responded after being readmitted, even after adjusting for response time. Although the P values are nonsignificant for many comparisons, the directionality of the effect is relatively consistent. Also, the vast majority of the patients fall in the postreadmission group, and these patients appear to drive the overall experience related to readmissions. Finally, since relatively few patients turned in surveys prior to readmission, we had limited power to detect a significant difference between these pre-readmission responders and nonreadmitted patients.
Our study has implications for policy makers, researchers, and providers. The HCAHPS scores of patients who are readmitted and completed the survey after being readmitted reflects their experience of both the index admission and the readmission. We did not find evidence to support that HCAHPS survey responses predict future readmissions at the patient level. Our findings do support the concept that lower readmissions rates (whether due to the patient population or processes of care that decrease readmission rates) may improve HCAHPS scores. We suggest caution in assuming that improving patient experience is likely to reduce readmission rates.
Disclosures
The authors declare no conflicts of interest.
Patient experience and 30-day readmission are important measures of quality of care for hospitalized patients. Performance on both of these measures impact hospitals financially. Performance on the Hospital Consumer Assessment of Healthcare Systems and Providers (HCAHPS) survey is linked to 25% of the incentive payment under Value Based Purchasing (VBP) Program.1 Starting in 2012, the Centers for Medicare and Medicaid Services (CMS) introduced the Readmission Reduction Program, penalizing hospitals financially for excessive readmissions.2
A relationship between patient experience and readmissions has been explored at the hospital level. Studies have mostly found that higher patient experience scores are associated with lower 30-day readmission rates. In a study of the relationship between 30-day risk-standardized readmission rates for three medical conditions (acute myocardial infarction, heart failure, and pneumonia) and patient experience, the authors noted that higher experience scores for overall care and discharge planning were associated with lower readmission rates for these conditions. They also concluded that patient experience scores were more predictive of 30-day readmission than clinical performance measures. Additionally, the authors predicted that if a hospital increased its total experience scores from the 25th percentile to the 75th percentile, there would be an associated decrease in readmissions by at least 2.3% for each of these conditions.3 Practice management companies and the media have cited this finding to conclude that higher patient experience drives clinical outcomes such as 30-day readmission and that patients are often the best judges of the quality of care delivered.4,5
Other hospital-level studies have found that high 30-day readmission rates are associated with lower overall experience scores in a mixed surgical patient population; worse reports of pain control and overall care in the colorectal surgery population; lower experience scores with discharge preparedness in vascular surgery patients; and lower experience scores with physician communication, nurse communication, and discharge preparedness.6-9 A patient-level study noted higher readmissions are associated with worse experience with physician and nursing communication along with a paradoxically better experience with discharge information.10
Because these studies used an observational design, they demonstrated associations rather than causality. An alternative hypothesis is that readmitted patients complete their patient experience survey after readmission and the low experience is the result, rather than the cause, of their readmission. For patients who are readmitted, it is unclear whether there is an opportunity to complete the survey prior to readmission and whether being readmitted may impact patient perception of quality of care. Using patient-level data, we sought to assess HCAHPS patient-experience responses linked to the index admission of the patients who were readmitted in 30 days and compare it with those patients who were not readmitted during this time period. We paid particular attention to when the surveys were returned.
METHODS
Study Design
We conducted a retrospective analysis of prospectively collected 10-year HCAHPS and Press Ganey patient survey data for a single tertiary care academic hospital.
Participants
All adult patients discharged from the hospital and who responded to the routinely sent patient-experience survey were included. Surveys were sent to a random sample of 50% of the discharged patients.
The exposure group was comprised of patients who responded to the survey and were readmitted within 30 days of discharge. After subtracting 5 days from the survey receipt date for expected delays related to mail delivery time and processing time, survey response date was calculated. The exposure group was further divided into patients who responded to the survey prior to their 30-day readmission (“Pre-readmission responders”) and those that responded to the survey after their readmission (“Postreadmission responders”). A sensitivity analysis was performed by changing the number of days subtracted from the survey receipt date by 2 days in either direction. This approach did not result in any significant changes in the results.
The control group comprised patients who were not readmitted to the hospital within 30 days of discharge and who did not have an admission in the previous 30 days as well (“Not readmitted” group). An additional comparison group for exploratory analysis included patients who had experienced an admission in the prior 30 days but were not readmitted after the admission linked to the survey. These patients responded to the patient-experience surveys that were linked to their second admission in 30 days (“2nd-admission responders” group; Figure).
Time Periods
All survey responders from the third quarter of 2006 to the first quarter of 2016 were included in the study. Additionally, administrative data on non-responders were available from 7/2006 to 8/2012. These data were used to estimate response rates. Patient level experience and administrative data were obtained in a linked fashion for these time periods.
Instruments
Press Ganey and HCAHPS surveys were sent via mail in the same envelope. Fifty percent of the discharged patients were randomized to receive the surveys. The Press Ganey survey contained 33 items encompassing several subdomains, including room, meal, nursing, physician, ancillary staff, visitor, discharge, and overall experience.
The HCAHPS survey contained 29 CMS-mandated items, of which 21 are related to patient experience. The development, testing, and methods for administration and reporting of the HCAHPS survey have been previously described and studies using this instrument have been reported in the literature.11 Press Ganey patient satisfaction survey results have also been reported in the literature.12
Outcome Variables and Covariates
HCAHPS and Press Ganey experience survey individual item responses were the primary outcome variables of this study. Age, self-reported health status, education, primary language spoken, service line, and time taken to respond to the surveys served as the covariates. These variables are used by CMS for patient-mix adjustment and are collected on the HCAHPS survey. Additionally, the number of days to respond to the survey were included in all regression analysis to adjust for early responder effect.13-15
Statistical Analysis
“Percent top-box” scores were calculated for each survey item for patients in each group. The percent top-box scores were calculated as the percent of patients who responded “very good” for a given item on Press Ganey survey items and “always” or “definitely yes” or “yes” or “9” or “10” on HCAHPS survey items. CMS utilizes “percent top-box scores” to calculate payments under the VBP program and to report the results publicly. Numerous studies have also reported percent top-box scores for HCAHPS survey results.12
We hypothesized that whether patients complete the HCAHPS survey before or after the readmission influences their reporting of experience. To test this hypothesis, HCAHPS and Press Ganey item top-box scores of “Pre-readmission responders” and “Postreadmission responders” were compared with those of the control group using multivariate logistic regression. “Pre-readmission responders” were also compared with “Postreadmission responders”.
“2nd-admission responders” were similarly compared with the control group for an exploratory analysis. Finally, “Postreadmission responders” and “2nd-admission responders” were compared in another exploratory analysis since both these groups responded to the survey after being exposed to the readmission, even though the “Postreadmission responders” group is administratively linked to the index admission.
The Johns Hopkins Institutional Review Board approved this study.
RESULTS
There were 43,737 survey responders, among whom 4,707 were subsequently readmitted within 30 days of discharge. Among the readmitted patients who responded to the surveys linked to their index admission, only 15.8% returned the survey before readmission (pre-readmission responders’) and 84.2% returned the survey after readmission (postreadmission responders). Additionally, 1,663 patients responded to experience surveys linked to their readmission. There were 37,365 patients in the control arm (ie, patients who responded to the survey and were not readmitted within 30 days of discharge or in the prior 30 days; Figure 1). The readmission rate among survey responders was 10.6%. Among the readmitted patients, the median number of days to readmission was 10 days while the median number of days to respond to the survey for this group was 33 days. Among the nonreadmitted patients, the median number of days to return the survey was 29 days.
We also conducted an exploratory analysis of the postreadmission responders, comparing them with patients who received patient-experience surveys linked to their second admission in 30 days. Both of these groups were exposed to a readmission before they completed the surveys. There were no significant differences between these two groups on patient experience scores. Additionally, the patients who received the survey linked to their readmission had a broad dissatisfaction pattern on HCAHPS survey items that appeared similar to that of the postreadmission group when compared to the non-readmitted group (Table 3).
DISCUSSION
In this retrospective analysis of prospectively collected Press Ganey and HCAHPS patient-experience survey data, we found that the overwhelming majority of patients readmitted within 30 days of discharge respond to HCAHPS surveys after readmission even though the survey is sent linked to the first admission. This is not unexpected since the median time to survey response is 33 days for this group, while median time to readmission is 10 days. The dissatisfaction pattern of Postreadmission responders was similar to those who responded to the survey linked to the readmission. When a patient is readmitted prior to completing the survey, their responses appear to reflect the cumulative experience of the index admission and the readmission. The lower scores of those who respond to the survey after their readmission appear to be a driver for lower patient-experience scores related to readmissions. Overall, readmission was associated with lower scores on items in five of the nine domains used to calculate patient experience related payments under VBP.16
These findings have important implications in inferring the direction of potential causal relationship between readmissions and patient experience at the hospital level. Additionally, these patients show broad dissatisfaction with areas beyond physician communication and discharge planning. These include staff responsiveness, phlebotomy, meals, hospital cleanliness, and noise level. This pattern of dissatisfaction may represent impatience and frustration with spending additional time in the hospital environment.
Our results are consistent with findings of many of the earlier studies, but our study goes a step further by using patient-level data and incorporating survey response time in our analysis.3,7,9,10 By separating out the readmitted patients who responded to the survey prior to admission, we attempted to address the ability of patients’ perception of care to predict future readmissions. Our results do not support this idea, since pre-readmission responders had similar experience scores to non-readmitted patients. However, because of the low numbers of pre-readmission responders, the comparison lacks precision. Current HCAHPS and Press Ganey questions may lack the ability to predict future readmissions because of the timing of the survey (postdischarge) or the questions themselves.
Overall, postreadmission responders are dissatisfied with multiple domains of hospital care. Many of these survey responses may simply be related to general frustration. Alternatively, they may represent a patient population with a high degree of needs that are not as easily met by a hospital’s routine processes of care. Even though the readmission rates were 10.6% among survey responders, 14.6% of the survey responses were associated with readmissions after accounting for those who respond to surveys linked to readmission. These patients could have significant impact on cumulative experience scores.
Our study has a few limitations. First, it involves a single tertiary care academic center study, and our results may not be generalizable. Second, we did not adjust for some of the patient characteristics associated with readmissions. Patients who were admitted within 30 days are different than those not readmitted based on payor, race, length of stay, and severity of illness, and we did not adjust for these factors in our analysis. This was intentional, however. Our goal was to better understand the relationship between 30-day readmission and patient experience scores as they are used for hospital-level studies, VBP, and public reporting. For these purposes, the scores are not adjusted for factors, such as payor and length of stay. We did adjust for patient-mix adjustment factors used by CMS. Third, the response rates to the HCAHPS were low and may have biased the scores. However, HCAHPS is widely used for comparisons between hospitals has been validated, and our study results have implications with regard to comparing hospital-level performance. HCAHPS results are relevant to policy and have financial consequences.17 Fourth, our study did not directly compare whether the relationship between patient experience for the postreadmission group and nonreadmitted group was different from the relationship between the pre-readmission group and postreadmission group. It is possible that there is no difference in relationship between the groups. However, despite the small number of pre-readmission responders, these patients tended to have more favorable experience responses than those who responded after being readmitted, even after adjusting for response time. Although the P values are nonsignificant for many comparisons, the directionality of the effect is relatively consistent. Also, the vast majority of the patients fall in the postreadmission group, and these patients appear to drive the overall experience related to readmissions. Finally, since relatively few patients turned in surveys prior to readmission, we had limited power to detect a significant difference between these pre-readmission responders and nonreadmitted patients.
Our study has implications for policy makers, researchers, and providers. The HCAHPS scores of patients who are readmitted and completed the survey after being readmitted reflects their experience of both the index admission and the readmission. We did not find evidence to support that HCAHPS survey responses predict future readmissions at the patient level. Our findings do support the concept that lower readmissions rates (whether due to the patient population or processes of care that decrease readmission rates) may improve HCAHPS scores. We suggest caution in assuming that improving patient experience is likely to reduce readmission rates.
Disclosures
The authors declare no conflicts of interest.
1. Hospital value-based purchasing. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/downloads/Hospital_VBPurchasing_Fact_Sheet_ICN907664.pdf. Accessed June 25, 2016.
2. Readmissions reduction program (HRRP). Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Accessed June 25, 2016.
3. Boulding W, Glickman SW, Manary MP, Schulman KA, Staelin R. Relationship between patient satisfaction with inpatient care and hospital readmission within 30 days. Am J Manag Care. 2011;17(1):41-48. PubMed
4. Buum HA, Duran-Nelson AM, Menk J, Nixon LJ. Duty-hours monitoring revisited: self-report may not be adequate. Am J Med. 2013;126(4):362-365. doi: 10.1016/j.amjmed.2012.12.003 PubMed
5. Choma NN, Vasilevskis EE, Sponsler KC, Hathaway J, Kripalani S. Effect of the ACGME 16-hour rule on efficiency and quality of care: duty hours 2.0. JAMA Int Med. 2013;173(9):819-821. doi: 10.1001/jamainternmed.2013.3014 PubMed
6. Brooke BS, Samourjian E, Sarfati MR, Nguyen TT, Greer D, Kraiss LW. RR3. Patient-reported readiness at time of discharge predicts readmission following vascular surgery. J Vasc Surg. 2015;61(6):188S. doi: 10.1016/j.jvs.2015.04.356
7. Duraes LC, Merlino J, Stocchi L, et al. 756 readmission decreases patient satisfaction in colorectal surgery. Gastroenterology. 2014;146(5):S-1029. doi: 10.1016/S0016-5085(14)63751-3
8. Mitchell JP. Association of provider communication and discharge instructions on lower readmissions. J Healthc Qual. 2015;37(1):33-40. doi: 10.1097/01.JHQ.0000460126.88382.13 PubMed
9. Tsai TC, Orav EJ, Jha AK. Patient satisfaction and quality of surgical care in US hospitals. Ann Surg. 2015;261(1):2-8. doi: 10.1097/SLA.0000000000000765 PubMed
10. Hachem F, Canar J, Fullam M, Andrew S, Hohmann S, Johnson C. The relationships between HCAHPS communication and discharge satisfaction items and hospital readmissions. Patient Exp J. 2014;1(2):71-77.
11. Irby DM, Cooke M, Lowenstein D, Richards B. The academy movement: a structural approach to reinvigorating the educational mission. Acad Med. 2004;79(8):729-736. doi: 10.1097/00001888-200408000-00003 PubMed
12. Siddiqui ZK, Zuccarelli R, Durkin N, Wu AW, Brotman DJ. Changes in patient satisfaction related to hospital renovation: experience with a new clinical building. J Hosp Med. 2015;10(3):165-171. doi: 10.1002/jhm.2297 PubMed
13. Nair BR, Coughlan JL, Hensley MJ. Student and patient perspectives on bedside teaching. Med Educ. 1997;31(5):341-346. doi: 10.1046/j.1365-2923.1997.00673.x PubMed
14. Elliott MN, Zaslavsky AM, Goldstein E, et al. Effects of survey mode, patient mix, and nonresponse on CAHPS® hospital survey scores. BMC Health Serv Res. 2009;44(2p1):501-518. doi: 10.1111/j.1475-6773.2008.00914.x PubMed
15. Saunders CL, Elliott MN, Lyratzopoulos G, Abel GA. Do differential response rates to patient surveys between organizations lead to unfair performance comparisons?: evidence from the English Cancer Patient Experience Survey. Medical care. 2016;54(1):45. doi: 10.1097/MLR.0000000000000457 PubMed
16. Sabel E, Archer J. “Medical education is the ugly duckling of the medical world” and other challenges to medical educators’ identity construction: a qualitative study. Acad Med. 2014;89(11):1474-1480. doi: 10.1097/ACM.0000000000000420 PubMed
17. O’Malley AJ, Zaslavsky AM, Elliott MN, Zaborski L, Cleary PD. Case‐Mix adjustment of the CAHPS® Hospital Survey. BMC Health Serv Res. 2005;40(6p2):2162-2181. doi: 10.1111/j.1475-6773.2005.00470.x
1. Hospital value-based purchasing. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNProducts/downloads/Hospital_VBPurchasing_Fact_Sheet_ICN907664.pdf. Accessed June 25, 2016.
2. Readmissions reduction program (HRRP). Centers for Medicare & Medicaid Services. https://www.cms.gov/medicare/medicare-fee-for-service-payment/acuteinpatientpps/readmissions-reduction-program.html. Accessed June 25, 2016.
3. Boulding W, Glickman SW, Manary MP, Schulman KA, Staelin R. Relationship between patient satisfaction with inpatient care and hospital readmission within 30 days. Am J Manag Care. 2011;17(1):41-48. PubMed
4. Buum HA, Duran-Nelson AM, Menk J, Nixon LJ. Duty-hours monitoring revisited: self-report may not be adequate. Am J Med. 2013;126(4):362-365. doi: 10.1016/j.amjmed.2012.12.003 PubMed
5. Choma NN, Vasilevskis EE, Sponsler KC, Hathaway J, Kripalani S. Effect of the ACGME 16-hour rule on efficiency and quality of care: duty hours 2.0. JAMA Int Med. 2013;173(9):819-821. doi: 10.1001/jamainternmed.2013.3014 PubMed
6. Brooke BS, Samourjian E, Sarfati MR, Nguyen TT, Greer D, Kraiss LW. RR3. Patient-reported readiness at time of discharge predicts readmission following vascular surgery. J Vasc Surg. 2015;61(6):188S. doi: 10.1016/j.jvs.2015.04.356
7. Duraes LC, Merlino J, Stocchi L, et al. 756 readmission decreases patient satisfaction in colorectal surgery. Gastroenterology. 2014;146(5):S-1029. doi: 10.1016/S0016-5085(14)63751-3
8. Mitchell JP. Association of provider communication and discharge instructions on lower readmissions. J Healthc Qual. 2015;37(1):33-40. doi: 10.1097/01.JHQ.0000460126.88382.13 PubMed
9. Tsai TC, Orav EJ, Jha AK. Patient satisfaction and quality of surgical care in US hospitals. Ann Surg. 2015;261(1):2-8. doi: 10.1097/SLA.0000000000000765 PubMed
10. Hachem F, Canar J, Fullam M, Andrew S, Hohmann S, Johnson C. The relationships between HCAHPS communication and discharge satisfaction items and hospital readmissions. Patient Exp J. 2014;1(2):71-77.
11. Irby DM, Cooke M, Lowenstein D, Richards B. The academy movement: a structural approach to reinvigorating the educational mission. Acad Med. 2004;79(8):729-736. doi: 10.1097/00001888-200408000-00003 PubMed
12. Siddiqui ZK, Zuccarelli R, Durkin N, Wu AW, Brotman DJ. Changes in patient satisfaction related to hospital renovation: experience with a new clinical building. J Hosp Med. 2015;10(3):165-171. doi: 10.1002/jhm.2297 PubMed
13. Nair BR, Coughlan JL, Hensley MJ. Student and patient perspectives on bedside teaching. Med Educ. 1997;31(5):341-346. doi: 10.1046/j.1365-2923.1997.00673.x PubMed
14. Elliott MN, Zaslavsky AM, Goldstein E, et al. Effects of survey mode, patient mix, and nonresponse on CAHPS® hospital survey scores. BMC Health Serv Res. 2009;44(2p1):501-518. doi: 10.1111/j.1475-6773.2008.00914.x PubMed
15. Saunders CL, Elliott MN, Lyratzopoulos G, Abel GA. Do differential response rates to patient surveys between organizations lead to unfair performance comparisons?: evidence from the English Cancer Patient Experience Survey. Medical care. 2016;54(1):45. doi: 10.1097/MLR.0000000000000457 PubMed
16. Sabel E, Archer J. “Medical education is the ugly duckling of the medical world” and other challenges to medical educators’ identity construction: a qualitative study. Acad Med. 2014;89(11):1474-1480. doi: 10.1097/ACM.0000000000000420 PubMed
17. O’Malley AJ, Zaslavsky AM, Elliott MN, Zaborski L, Cleary PD. Case‐Mix adjustment of the CAHPS® Hospital Survey. BMC Health Serv Res. 2005;40(6p2):2162-2181. doi: 10.1111/j.1475-6773.2005.00470.x
© 2018 Society of Hospital Medicine
Prospective Randomized Evaluation of Preoperative Angiotensin-Converting Enzyme Inhibition (PREOP-ACEI)
Over 7 million surgeries are performed in United States hospitals each year. Among these surgeries, approximately 85% are noncardiac, nonvascular (NCNV) procedures.1,2 Although the preoperative use of an angiotensin-converting enzyme inhibitor (ACEI) can be expected in as many as 13% of these surgeries,3 the optimal preoperative ACEI management strategy for patients undergoing NCNV surgeries is poorly understood.
High-quality evidence suggests that renin–angiotensin–aldosterone system (RAAS) inhibitors are associated with intraoperative hypotension among patients undergoing cardiac or vascular surgeries.4-6 Intraoperative hypotension increases the risk of 30-day mortality,7 and the duration of intraoperative hypotension increases the risk of end organ damage.8,9 This body of evidence suggests that withholding ACEIs prior to cardiac and vascular surgeries is safer than continuing ACEIs without interruption.
The evidence concerning perioperative management of ACEIs is inconclusive for patients undergoing NCNV procedures. Some studies comparing patients taking or not taking a RAAS inhibitor preoperatively describe negligible differences in the frequency of intraoperative hypotensive episodes or complications.3,10 Others have found an increased risk of intraoperative hypotension and associated postoperative adverse events in patients continuing RAAS inhibitors preoperatively.11,12 Current guideline discrepancies reflect the uncertainty of the evidence. The guidelines set by the American College of Cardiology and American Heart Association (ACC/AHA) suggest the uninterrupted perioperative continuation of RAAS inhibitors.13 The guidelines provided by the European Society of Cardiology and European Society of Anaesthesiology also suggest the continuation of RAAS inhibitors throughout the perioperative period for patients with systolic heart failure but recommend transient discontinuation for patients with hypertension.14
This randomized study aimed to compare the effect of two practical strategies for preoperative ACEI management on the perioperative blood pressure of patients undergoing NCNV surgery. The two strategies studied were the omission of the final preoperative ACEI dose and the uninterrupted continuation of ACEI therapy. We hypothesized that patients randomized to ACEI omission would experience intraoperative hypotensive episodes less frequently than those randomized to ACEI continuation.
METHODS
Study Design and Setting
We performed a prospective randomized controlled trial (ClinicalTrials.gov: NCT01669434). The study was carried out in a preoperative evaluation clinic and its affiliated 489-bed academic medical center. Anesthesiologists and internal medicine physicians work collaboratively in the clinic to assess more than 5,000 patients annually (one-third of the institution’s elective surgeries). Patients were randomized 1:1 in block sizes of 5 and 10 and stratified by age < 65 and ≥ 65 years to the omission or continuation of the final preoperative ACEI dose (whether that dose was scheduled for the morning of surgery or the night prior). Preoperative clinicians enrolled patients and subsequently assigned them to intervention groups on the basis of a sequentially numbered list. Patients and healthcare providers were not blinded to allocation status. Intraoperative and postoperative management was provided in accordance with usual care as decided by treatment team.
Participants
Patients who presented to the preoperative evaluation clinic between May 2015 and November 2016 and who had been taking an ACEI for at least 6 weeks were eligible for inclusion. Patients taking angiotensin receptor blockers were excluded. Enrollment was limited to patients planning NCNV surgery. Patients planning intrathoracic, major vascular, organ transplant, and oncologic surgery were excluded. Patients undergoing outpatient procedures not requiring an overnight stay in the hospital were also excluded. Patients with preoperative clinic systolic blood pressure (SBP) <90 or ≥160 or diastolic blood pressure (DBP) <60 or ≥ 95 were excluded. Patients with moderate to severe or clinically decompensated heart failure (left ventricular ejection fraction < 40% or New York Heart Association class III or IV) and those with end-stage renal disease requiring dialysis were also excluded. Patients presenting more than once during the accrual period were eligible for the initial surgery only. All participating patients provided written informed consent. This project was approved by the University of Nebraska Medical Center Institutional Review Board.
Data Collection
Baseline characteristics were recorded by study personnel at the time of enrollment. We measured serum creatinine level at the preoperative visit and on postoperative day 1. An automated anesthesia information management system was used to measure intraoperative blood pressures every three minutes. Postoperative blood pressures through discharge were measured by hospital staff per usual care. During postoperative hospitalization, we queried patients about preoperative adherence to allocation. The digital abstraction of data from the electronic medical record was supplemented by chart review when necessary.
Outcomes
The primary outcome was intraoperative hypotension defined as any SBP < 80 mm Hg occurring from the administration of the first induction agent through transfer to the postanesthesia care unit (PACU). We also examined hypotension during anesthesia induction, which we defined as the 20-minute period following the administration of the first anesthesia induction agent. Episodes of SBP < 80 were defined as being associated with vasopressor administration when any vasopressor was administered during or within 10 min of the episode.
Secondary analyses included postoperative acute kidney injury (AKI), postoperative hypotensive and hypertensive episodes, cardiac events, and mortality. When comparing postoperative day 1 creatinine levels to preoperative creatinine levels, we used the Acute Kidney Injury Network definition of AKI as an increase in creatinine of 0.3 mg/dl or 50%.15 Postoperative hypotension was defined as any SBP < 90 mm Hg and postoperative hypertension as any SBP > 180 mm Hg occurring after arrival in the PACU. Major adverse cardiac events (MACE) were defined as a composite of acute coronary syndrome, acute heart failure, or new-onset arrhythmia. Discharge from the hospital served as the study endpoint for each patient.
Analysis
Fisher’s exact test was used to compare categorical outcomes between groups. The independent sample t-test or Wilcoxon rank–sum test, as appropriate, was used to compare continuous measures. We selected Fisher’s exact test over χ2-test to produce conservative estimates. Patients were maintained in their allocated group as randomized for analytical purposes regardless of adherence to allocation. We performed all analyses using SAS version 9.4 for Windows (SAS institute, Cary, North Carolina).
We estimated that a sample size of 300 patients would achieve 80% power to detect a difference of 0.17 between the group proportions of 0.33 and 0.50 at a significance level (ɑ) of 0.05 by using a two-sided z-test with continuity correction, assuming 15% loss to follow-up. This estimate allowed for 1 interim analysis using the O’Brien-Fleming spending function truncated at three standard deviations to determine the test boundaries. The monitoring boundary P values associated with the interim analysis were .003, and the threshold P value for the final analysis was .049.
RESULTS
Study Flow
A total of 453 patients were screened for eligibility. Among these patients, 162 were excluded, and the remaining 291 patients were randomized (Figure 1). Surgery was cancelled in six patients allocated to omission and in four patients allocated to continuation arms, respectively. Moreover, three patients in the omission arm were excluded from the analysis following randomization. Specifically, one was excluded because of early discharge without overnight stay, one was excluded because of withdrawal of consent, and one was excluded because of missing primary outcome data. In addition, three cases in the continuation arm were excluded following randomization because of the preoperative (permanent) discontinuation of ACEI therapy in two cases and discharge without an overnight stay in one case. Finally, 275 patients were included in the analysis: 137 in the ACEI omission group and 138 in the ACEI continuation group. Adherence to allocation was 88% and 92% in the omission and continuation groups, respectively.
Baseline Characteristics
The demographic data of patients allocated to ACEI omission and those allocated to ACEI continuation were similar (Table 1). A large majority of patients in both groups took the ACEI lisinopril. Overall, 187 of 275 (68%) patients were taking at least 1 antihypertensive agent, most commonly a diuretic, in addition to an ACEI. SBP measured during the preoperative clinic visit averaged 136.5 mm Hg and did not differ significantly between groups (P = .84).
Surgical Variables
General anesthesia was the most commonly utilized technique, although spinal and regional anesthesia were also represented (Table 1). The majority of cases in both groups were planning for orthopedic and spinal surgery. The method of anesthesia or type of surgery between patients allocated to ACEI omission and those allocated to continuation did not differ (P = .61 and P = .45 respectively).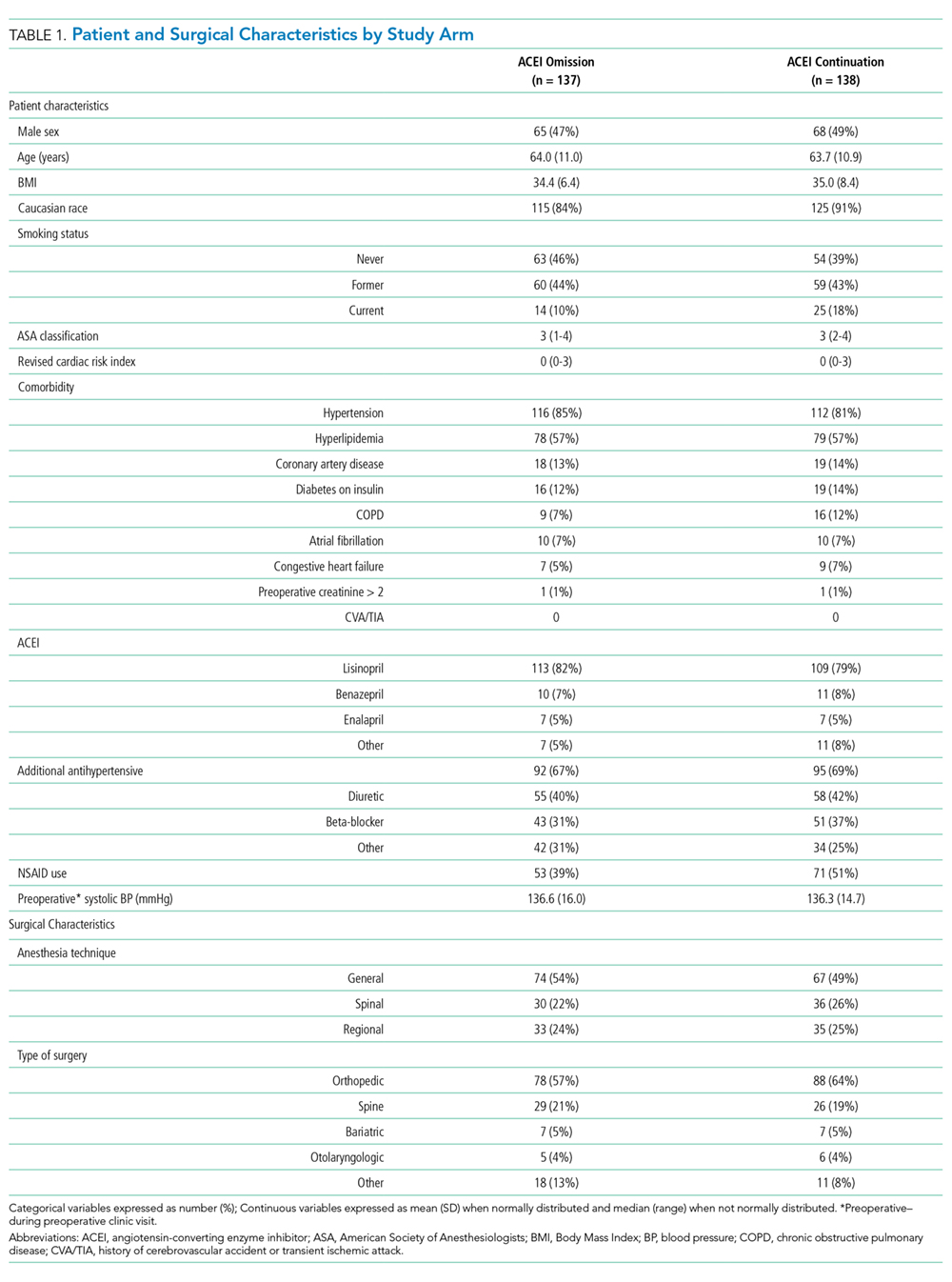
Episodes of Intraoperative Hypotension
Intraoperative SBPs are displayed in Figure 2, and hemodynamic outcomes are summarized in Table 2. Episodes of SBP < 80 mm Hg during anesthesia induction were numerically less frequent in the omission group than in the continuation group; the difference between groups, however, was not statistically significant (24 of 137 [18%] vs 38 of 138 [28%], RR: 0.64, 95% CI: 0.40 to 1.00, P = .06).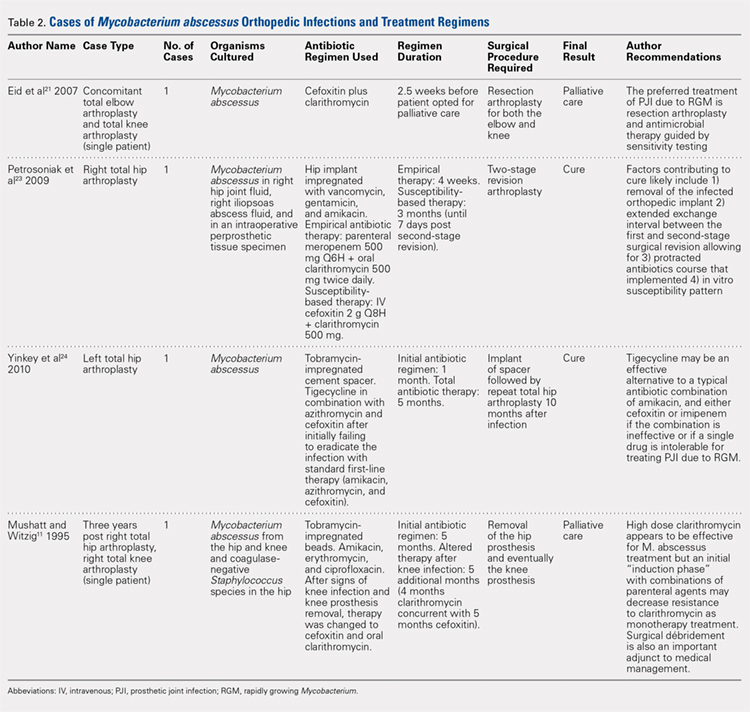
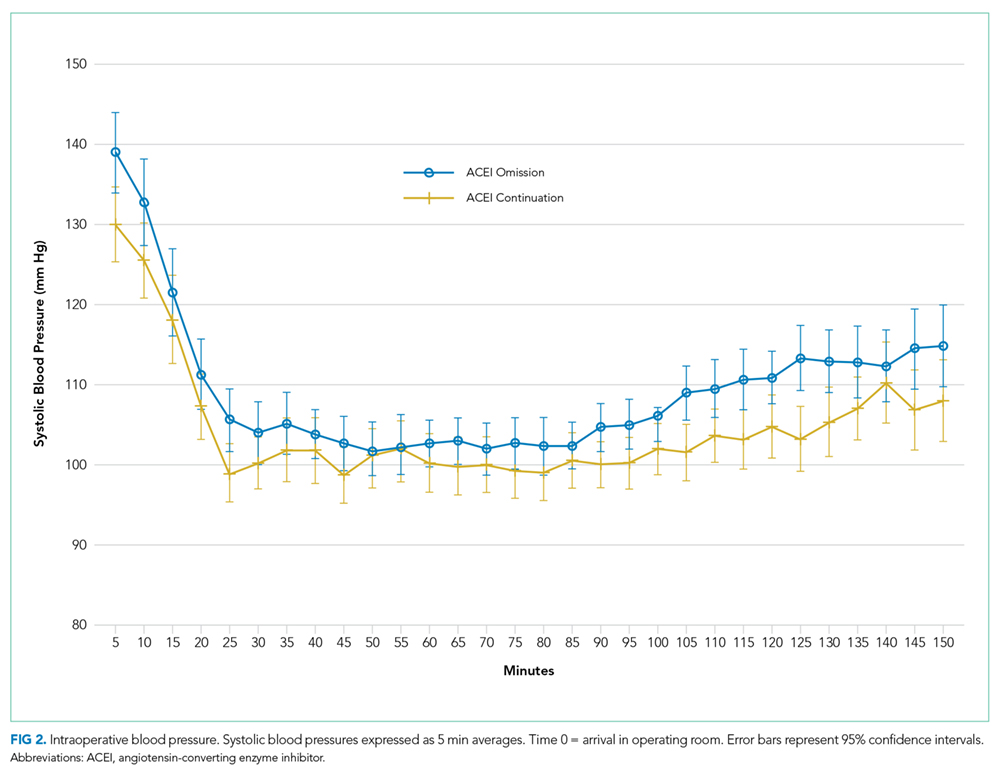
Duration of Intraoperative Hypotension
The median cumulative duration of intraoperative SBP < 80 was two minutes (range 0-41) in patients allocated to the ACEI omission group compared with seven minutes (range 0-214) in those allocated to the continuation group (P < .01). The median cumulative duration of mean arterial pressure < 55 mm Hg was also shorter in the omission group (median 0 min [range 0-39] vs 3 min [range 0-122], P < .01) than in the continuation group. The duration of surgery did not differ between groups (median 141 min [range 77-554] vs 142 min [range 57-665], P = .97).
Postoperative Outcomes
RAAS inhibitor therapy was resumed within 48 h after surgery in 122 of 137 (89%) patients allocated to the omission group and in 128 of 138 (93%) patients allocated to the continuation group (RR: 0.96, 95% CI: 0.89-1.03, P = .30).
Patients allocated to the omission group were significantly less likely to experience postoperative hypotension (15 of 137 [11%] vs 31 of 138 [22%], RR: 0.49, 95% CI: 0.28 to 0.86, P = .02) and significantly more likely to experience severe postoperative hypertension (33 of 137 [24%] vs 17 of 138 [12%], RR: 1.95, 95% CI: 1.14 to 3.34, P = .01) than those allocated to the continuation group. The occurrences of postoperative AKI (RR: 0.60, 95% CI: 0.23 to 1.60, P = .44) or MACE (RR: 4.03, 95% CI: 0.46 to 35.59, P = .21) in the omission group did not differ from the continuation group. The two groups exhibited similar PACU recovery time (mean 97.2 min) and overall hospital length of stay (mean 3.0 days) (P = .49 and P = .56 ). No episodes of inpatient mortality in either group were observed.
DISCUSSION
The omission of the final preoperative ACEI dose was associated with a significant reduction in the risk of intraoperative hypotension in patients undergoing NCNV surgery. This result confirmed our hypothesis. Coupled with the knowledge that intraoperative hypotension is associated with an increased risk of complications and mortality,7-9,16 this study favors the omission of the final preoperative ACEI dose prior to NCNV surgeries.
Our findings are in agreement with those of previous randomized studies that explored this question4,5 and help extend results from cardiac and vascular surgeries to NCNV surgeries. Previous studies on the use of RAAS inhibitors in NCNV surgeries did not employ randomization and yielded mixed results.3,10-12,17 A large single-institution study (n = 18,056) noted no difference in intraoperative blood pressure between patients taking ACEIs and a matched group of non-ACEI users.3 More recently, a subgroup analysis of the international VISION study showed that omitting RAAS inhibitors on the day of surgery reduced the risk of intraoperative hypotension.11 In that analysis, however, only a small amount of the variability in preoperative RAAS inhibitor management was explainable by modeling known factors, thus allowing for the possibility of unmeasured confounding. Our study, which minimized confounding through randomization, is the first to prospectively compare protocols for patients undergoing NCNV surgery. In contrast to previous studies, the present study was able to report the lack of difference in postoperative RAAS inhibitor administration between study groups. Postoperative RAAS inhibitor management affects complications and mortality.18,19
Our present finding that preoperative ACEI management affects postoperative hypotensive and hypertensive events conflicts with some previous findings.11,20 However, recent evidence has revealed that postoperative hypotensive episodes are associated with vascular events and mortality.11,21 In the context of that evidence, our study lends further support to the omission of the final preoperative ACEI dose. However, we did not detect any decrease in AKI, MACE, or mortality in the ACEI omission group.
This study should be considered in light of its limitations. The pragmatic nature of the study allowed for certain potential biases. Although adherence to allocation was high, the specific ACEI agent taken and the exact timing of the final dose in relation to surgery were not controlled. Anesthetic and postoperative management decisions were made by the treatment team and may have systematically varied given that the treatment team was not blinded to allocation. Furthermore, all outcome data were collected as part of routine care and may not have captured events with great fidelity. Generalizability is limited by the execution of the study at a single academic institution, the preponderance of orthopedic and spine surgeries, and by the negligible representation of ethnicities other than Caucasian. Additionally, recruitment from the preoperative evaluation clinic likely resulted in a patient group with greater comorbidity than the overall population of patients undergoing NCNV surgery. This study was powered for intraoperative hypotension and not postoperative outcomes. Our primary outcome, intraoperative hypotension, is an intermediate measure but one that has well-established associations with adverse outcomes, including mortality. One study showed that sustaining an intraoperative SBP below 70 mm Hg for longer than 5 min increased the risk of mortality from less than 1% to nearly 6%.16 A large study detected an increase in mortality associated with SBP sustained below 80 mm Hg for 10 min or longer.7 Intraoperative hypotension has also been associated with postoperative AKI and myocardial injury.8,9,12
Many of the limitations of the current study could be addressed by a large randomized controlled trial of ACEI management prior to NCNV surgeries that examines clinically important endpoints beyond intraoperative hypotension. Several specific aspects of perioperative RAAS inhibitor management also deserve further investigation. Our findings may not be generalizable to patients taking ARBs or to patients with congestive heart failure. The preoperative management of ARBs and the preoperative management of RAAS inhibitors in those with congestive heart failure are important areas of focus for future research. Lastly, our finding that preoperative ACEI management decisions can affect postoperative hypotensive and hypertensive events should be substantiated by future research, and any negative consequences of those events should be further explored.
Nonetheless, our study is the largest randomized study of preoperative RAAS inhibition published to date. More than twice as many patients were randomized in this study than in all previous randomized studies combined.4-6 To the best of our knowledge, this is also the first randomized study evaluating NCNV surgeries. Finally, our use of a practical ACEI omission protocol based on known pharmacokinetics allows for direct application to clinical practice.
CONCLUSION
Hypertension is among the most common chronic conditions encountered in patients planning surgery, and ACEIs are among the most frequently prescribed antihypertensive medications. This study showed that ACEI continuation is associated with an increased frequency and cumulative duration of intraoperative hypotension. These findings, while at odds with current ACC/AHA guidelines, align with the findings of a meta-analysis on this subject and with recent literature.3,11-13,22
Acknowledgments
The authors wish to thank Miranda M Fricke, MS, PA-C; Tiffany K Hillyard, APRN-FNP; and Barbara Sink, MPAS, PA-C who assisted in the design and conduct of patient enrollment and randomization procedures.
Disclosures
The authors have no relevant financial conflicts of interest to report.
Funding
This study was subsidized by a grant from the University of Nebraska Medical Center Research Support Fund. The funding source had no role in the design, conduct, analysis, or reporting of the study.
1. Steiner CA KZ, Moore BJ, Imshaug MC, Pickens G. Surgeries in hospital-based ambulatory surgery and hospital inpatient settings, 2014. Statistical Brief 2017; 1-18. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb223-Ambulatory-Inpatient-Surgeries-2014.pdf. Accessed August 30, 2017. PubMed
2. Rate of all-listed procedures for discharges from short-stay hospitals, by procedure category and age: United States, 2010. National Hospital Discharge Survey 2010; https://www.cdc.gov/nchs/nhds/nhds_tables.htm. Accessed August 30, 2017.
3. Turan A, You J, Shiba A, Kurz A, Saager L, Sessler DI. Angiotensin converting enzyme inhibitors are not associated with respiratory complications or mortality after noncardiac surgery. Anesth Analg. 2012;114(3):552-560. doi: 10.1213/ANE.0b013e318241f6af. PubMed
4. Coriat P, Richer C, Douraki T, et al. Influence of chronic angiotensin-converting enzyme inhibition on anesthetic induction. Anesthesiology. 1994;81:299-307. PubMed
5. Pigott DW, Nagle C, Allman K, S. W, D. ER. Effect of omitting regular ACE inhibitor medication before cardiac surgery on haemodynamic variables and vasoactive drug requirements. Br J Anaesth. 1999;83:715-720. doi: 10.1093/bja/83.5.715 PubMed
6. Bertrand M, Godet G, Meersschaert K, Brun L, Salcedo E, Coriat P. Should the angiotensin II antagonists be discontinued before surgery? Anesth Analg. 2001;92:26-30. PubMed
7. Mascha EJ, Yang D, Weiss S, Sessler DI. Intraoperative mean arterial pressure variability and 30-day mortality in patients having noncardiac surgery. Anesthesiology. 2015;123(1):79-91. doi: 10.1097/ALN.0000000000000686. PubMed
8. Walsh M, Devereaux PJ, Garg AX, et al. Relationship between intraoperative mean arterial pressure and clinical outcomes after noncardiac surgery: toward an empirical definition of hypotension. Anesthesiology. 2013;119(3):507-515. doi: 10.1097/ALN.0b013e3182a10e26. PubMed
9. Salmasi V, Maheshwari K, Yang D, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126(1):47-65. doi: 10.1097/ALN.0000000000001432. PubMed
10. Comfere T, Sprung J, Kumar MM, et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100(3):636-644. doi: 10.1213/01.ANE.0000146521.68059.A1. PubMed
11. Roshanov PS, Rochwerg B, Patel A, et al. Withholding versus continuing angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers before noncardiac surgery: an analysis of the vascular events in noncardiac surgery patIents cohort evaluation prospective cohort. Anesthesiology. 2017;126(1):16-27. doi: 10.1097/ALN.0000000000001404. PubMed
12. Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288. doi: 10.1002/jhm.2155. PubMed
13. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. 2014;64(22):e77-137. doi: 10.1016/j.jacc.2014.07.944. PubMed
14. Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J. 2014;35(35):2383-2431. doi: 10.1093/eurheartj/ehu282 PubMed
15. Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. doi: 10.1186/cc5713 PubMed
16. Monk TG, Bronsert MR, Henderson WG, et al. Association between intraoperative hypotension and hypertension and 30-day postoperative mortality in noncardiac surgery. Anesthesiology. 2015;123(2):307-319. doi: 10.1097/ALN.0000000000000756. PubMed
17. Kheterpal S, Khodaparast O, Shanks A, O’Reilly M, Tremper KK. Chronic angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy combined with diuretic therapy is associated with increased episodes of hypotension in noncardiac surgery. J Cardiothorac Vasc Anesth. 2008;22(2):180-186. 10.1053/j.jvca.2007.12.020. PubMed
18. Lee SM, Takemoto S, Wallace AW. Association between withholding angiotensin receptor blockers in the early postoperative period and 30-day mortality: a cohort study of the veterans affairs healthcare system. Anesthesiology. 2015;123(2):288-306. 10.1097/ALN.0000000000000739. PubMed
19. Drenger B, Fontes ML, Miao Y, et al. Patterns of use of perioperative angiotensin-converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in-hospital morbidity and mortality. Circulation. 2012;126(3):261-269. doi: 10.1161/CIRCULATIONAHA.111.059527. PubMed
20. Twersky RS, Goel V, Narayan P, Weedon J. The risk of hypertension after preoperative discontinuation of angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists in ambulatory and same-day admission patients. Anesth Analg. 2014;118(5):938-944. doi: 10.1213/ANE.0000000000000076. PubMed
21. Tan TW, Eslami MH, Kalish JA, et al. The need for treatment of hemodynamic instability following carotid endarterectomy is associated with increased perioperative and 1-year morbidity and mortality. J Vasc Surg. 2014;59(1):16-24 e11-12. https://doi.org/10.1053/j.jvca.2014.12.002 PubMed
22. Rosenman DJ, McDonald FS, Ebbert JO, Erwin PJ, LaBella M, Montori VM. Clinical consequences of withholding versus administering renin-angiotensin-aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319-325. doi: 10.1002/jhm.323. PubMed
Over 7 million surgeries are performed in United States hospitals each year. Among these surgeries, approximately 85% are noncardiac, nonvascular (NCNV) procedures.1,2 Although the preoperative use of an angiotensin-converting enzyme inhibitor (ACEI) can be expected in as many as 13% of these surgeries,3 the optimal preoperative ACEI management strategy for patients undergoing NCNV surgeries is poorly understood.
High-quality evidence suggests that renin–angiotensin–aldosterone system (RAAS) inhibitors are associated with intraoperative hypotension among patients undergoing cardiac or vascular surgeries.4-6 Intraoperative hypotension increases the risk of 30-day mortality,7 and the duration of intraoperative hypotension increases the risk of end organ damage.8,9 This body of evidence suggests that withholding ACEIs prior to cardiac and vascular surgeries is safer than continuing ACEIs without interruption.
The evidence concerning perioperative management of ACEIs is inconclusive for patients undergoing NCNV procedures. Some studies comparing patients taking or not taking a RAAS inhibitor preoperatively describe negligible differences in the frequency of intraoperative hypotensive episodes or complications.3,10 Others have found an increased risk of intraoperative hypotension and associated postoperative adverse events in patients continuing RAAS inhibitors preoperatively.11,12 Current guideline discrepancies reflect the uncertainty of the evidence. The guidelines set by the American College of Cardiology and American Heart Association (ACC/AHA) suggest the uninterrupted perioperative continuation of RAAS inhibitors.13 The guidelines provided by the European Society of Cardiology and European Society of Anaesthesiology also suggest the continuation of RAAS inhibitors throughout the perioperative period for patients with systolic heart failure but recommend transient discontinuation for patients with hypertension.14
This randomized study aimed to compare the effect of two practical strategies for preoperative ACEI management on the perioperative blood pressure of patients undergoing NCNV surgery. The two strategies studied were the omission of the final preoperative ACEI dose and the uninterrupted continuation of ACEI therapy. We hypothesized that patients randomized to ACEI omission would experience intraoperative hypotensive episodes less frequently than those randomized to ACEI continuation.
METHODS
Study Design and Setting
We performed a prospective randomized controlled trial (ClinicalTrials.gov: NCT01669434). The study was carried out in a preoperative evaluation clinic and its affiliated 489-bed academic medical center. Anesthesiologists and internal medicine physicians work collaboratively in the clinic to assess more than 5,000 patients annually (one-third of the institution’s elective surgeries). Patients were randomized 1:1 in block sizes of 5 and 10 and stratified by age < 65 and ≥ 65 years to the omission or continuation of the final preoperative ACEI dose (whether that dose was scheduled for the morning of surgery or the night prior). Preoperative clinicians enrolled patients and subsequently assigned them to intervention groups on the basis of a sequentially numbered list. Patients and healthcare providers were not blinded to allocation status. Intraoperative and postoperative management was provided in accordance with usual care as decided by treatment team.
Participants
Patients who presented to the preoperative evaluation clinic between May 2015 and November 2016 and who had been taking an ACEI for at least 6 weeks were eligible for inclusion. Patients taking angiotensin receptor blockers were excluded. Enrollment was limited to patients planning NCNV surgery. Patients planning intrathoracic, major vascular, organ transplant, and oncologic surgery were excluded. Patients undergoing outpatient procedures not requiring an overnight stay in the hospital were also excluded. Patients with preoperative clinic systolic blood pressure (SBP) <90 or ≥160 or diastolic blood pressure (DBP) <60 or ≥ 95 were excluded. Patients with moderate to severe or clinically decompensated heart failure (left ventricular ejection fraction < 40% or New York Heart Association class III or IV) and those with end-stage renal disease requiring dialysis were also excluded. Patients presenting more than once during the accrual period were eligible for the initial surgery only. All participating patients provided written informed consent. This project was approved by the University of Nebraska Medical Center Institutional Review Board.
Data Collection
Baseline characteristics were recorded by study personnel at the time of enrollment. We measured serum creatinine level at the preoperative visit and on postoperative day 1. An automated anesthesia information management system was used to measure intraoperative blood pressures every three minutes. Postoperative blood pressures through discharge were measured by hospital staff per usual care. During postoperative hospitalization, we queried patients about preoperative adherence to allocation. The digital abstraction of data from the electronic medical record was supplemented by chart review when necessary.
Outcomes
The primary outcome was intraoperative hypotension defined as any SBP < 80 mm Hg occurring from the administration of the first induction agent through transfer to the postanesthesia care unit (PACU). We also examined hypotension during anesthesia induction, which we defined as the 20-minute period following the administration of the first anesthesia induction agent. Episodes of SBP < 80 were defined as being associated with vasopressor administration when any vasopressor was administered during or within 10 min of the episode.
Secondary analyses included postoperative acute kidney injury (AKI), postoperative hypotensive and hypertensive episodes, cardiac events, and mortality. When comparing postoperative day 1 creatinine levels to preoperative creatinine levels, we used the Acute Kidney Injury Network definition of AKI as an increase in creatinine of 0.3 mg/dl or 50%.15 Postoperative hypotension was defined as any SBP < 90 mm Hg and postoperative hypertension as any SBP > 180 mm Hg occurring after arrival in the PACU. Major adverse cardiac events (MACE) were defined as a composite of acute coronary syndrome, acute heart failure, or new-onset arrhythmia. Discharge from the hospital served as the study endpoint for each patient.
Analysis
Fisher’s exact test was used to compare categorical outcomes between groups. The independent sample t-test or Wilcoxon rank–sum test, as appropriate, was used to compare continuous measures. We selected Fisher’s exact test over χ2-test to produce conservative estimates. Patients were maintained in their allocated group as randomized for analytical purposes regardless of adherence to allocation. We performed all analyses using SAS version 9.4 for Windows (SAS institute, Cary, North Carolina).
We estimated that a sample size of 300 patients would achieve 80% power to detect a difference of 0.17 between the group proportions of 0.33 and 0.50 at a significance level (ɑ) of 0.05 by using a two-sided z-test with continuity correction, assuming 15% loss to follow-up. This estimate allowed for 1 interim analysis using the O’Brien-Fleming spending function truncated at three standard deviations to determine the test boundaries. The monitoring boundary P values associated with the interim analysis were .003, and the threshold P value for the final analysis was .049.
RESULTS
Study Flow
A total of 453 patients were screened for eligibility. Among these patients, 162 were excluded, and the remaining 291 patients were randomized (Figure 1). Surgery was cancelled in six patients allocated to omission and in four patients allocated to continuation arms, respectively. Moreover, three patients in the omission arm were excluded from the analysis following randomization. Specifically, one was excluded because of early discharge without overnight stay, one was excluded because of withdrawal of consent, and one was excluded because of missing primary outcome data. In addition, three cases in the continuation arm were excluded following randomization because of the preoperative (permanent) discontinuation of ACEI therapy in two cases and discharge without an overnight stay in one case. Finally, 275 patients were included in the analysis: 137 in the ACEI omission group and 138 in the ACEI continuation group. Adherence to allocation was 88% and 92% in the omission and continuation groups, respectively.
Baseline Characteristics
The demographic data of patients allocated to ACEI omission and those allocated to ACEI continuation were similar (Table 1). A large majority of patients in both groups took the ACEI lisinopril. Overall, 187 of 275 (68%) patients were taking at least 1 antihypertensive agent, most commonly a diuretic, in addition to an ACEI. SBP measured during the preoperative clinic visit averaged 136.5 mm Hg and did not differ significantly between groups (P = .84).
Surgical Variables
General anesthesia was the most commonly utilized technique, although spinal and regional anesthesia were also represented (Table 1). The majority of cases in both groups were planning for orthopedic and spinal surgery. The method of anesthesia or type of surgery between patients allocated to ACEI omission and those allocated to continuation did not differ (P = .61 and P = .45 respectively).
Episodes of Intraoperative Hypotension
Intraoperative SBPs are displayed in Figure 2, and hemodynamic outcomes are summarized in Table 2. Episodes of SBP < 80 mm Hg during anesthesia induction were numerically less frequent in the omission group than in the continuation group; the difference between groups, however, was not statistically significant (24 of 137 [18%] vs 38 of 138 [28%], RR: 0.64, 95% CI: 0.40 to 1.00, P = .06).
Duration of Intraoperative Hypotension
The median cumulative duration of intraoperative SBP < 80 was two minutes (range 0-41) in patients allocated to the ACEI omission group compared with seven minutes (range 0-214) in those allocated to the continuation group (P < .01). The median cumulative duration of mean arterial pressure < 55 mm Hg was also shorter in the omission group (median 0 min [range 0-39] vs 3 min [range 0-122], P < .01) than in the continuation group. The duration of surgery did not differ between groups (median 141 min [range 77-554] vs 142 min [range 57-665], P = .97).
Postoperative Outcomes
RAAS inhibitor therapy was resumed within 48 h after surgery in 122 of 137 (89%) patients allocated to the omission group and in 128 of 138 (93%) patients allocated to the continuation group (RR: 0.96, 95% CI: 0.89-1.03, P = .30).
Patients allocated to the omission group were significantly less likely to experience postoperative hypotension (15 of 137 [11%] vs 31 of 138 [22%], RR: 0.49, 95% CI: 0.28 to 0.86, P = .02) and significantly more likely to experience severe postoperative hypertension (33 of 137 [24%] vs 17 of 138 [12%], RR: 1.95, 95% CI: 1.14 to 3.34, P = .01) than those allocated to the continuation group. The occurrences of postoperative AKI (RR: 0.60, 95% CI: 0.23 to 1.60, P = .44) or MACE (RR: 4.03, 95% CI: 0.46 to 35.59, P = .21) in the omission group did not differ from the continuation group. The two groups exhibited similar PACU recovery time (mean 97.2 min) and overall hospital length of stay (mean 3.0 days) (P = .49 and P = .56 ). No episodes of inpatient mortality in either group were observed.
DISCUSSION
The omission of the final preoperative ACEI dose was associated with a significant reduction in the risk of intraoperative hypotension in patients undergoing NCNV surgery. This result confirmed our hypothesis. Coupled with the knowledge that intraoperative hypotension is associated with an increased risk of complications and mortality,7-9,16 this study favors the omission of the final preoperative ACEI dose prior to NCNV surgeries.
Our findings are in agreement with those of previous randomized studies that explored this question4,5 and help extend results from cardiac and vascular surgeries to NCNV surgeries. Previous studies on the use of RAAS inhibitors in NCNV surgeries did not employ randomization and yielded mixed results.3,10-12,17 A large single-institution study (n = 18,056) noted no difference in intraoperative blood pressure between patients taking ACEIs and a matched group of non-ACEI users.3 More recently, a subgroup analysis of the international VISION study showed that omitting RAAS inhibitors on the day of surgery reduced the risk of intraoperative hypotension.11 In that analysis, however, only a small amount of the variability in preoperative RAAS inhibitor management was explainable by modeling known factors, thus allowing for the possibility of unmeasured confounding. Our study, which minimized confounding through randomization, is the first to prospectively compare protocols for patients undergoing NCNV surgery. In contrast to previous studies, the present study was able to report the lack of difference in postoperative RAAS inhibitor administration between study groups. Postoperative RAAS inhibitor management affects complications and mortality.18,19
Our present finding that preoperative ACEI management affects postoperative hypotensive and hypertensive events conflicts with some previous findings.11,20 However, recent evidence has revealed that postoperative hypotensive episodes are associated with vascular events and mortality.11,21 In the context of that evidence, our study lends further support to the omission of the final preoperative ACEI dose. However, we did not detect any decrease in AKI, MACE, or mortality in the ACEI omission group.
This study should be considered in light of its limitations. The pragmatic nature of the study allowed for certain potential biases. Although adherence to allocation was high, the specific ACEI agent taken and the exact timing of the final dose in relation to surgery were not controlled. Anesthetic and postoperative management decisions were made by the treatment team and may have systematically varied given that the treatment team was not blinded to allocation. Furthermore, all outcome data were collected as part of routine care and may not have captured events with great fidelity. Generalizability is limited by the execution of the study at a single academic institution, the preponderance of orthopedic and spine surgeries, and by the negligible representation of ethnicities other than Caucasian. Additionally, recruitment from the preoperative evaluation clinic likely resulted in a patient group with greater comorbidity than the overall population of patients undergoing NCNV surgery. This study was powered for intraoperative hypotension and not postoperative outcomes. Our primary outcome, intraoperative hypotension, is an intermediate measure but one that has well-established associations with adverse outcomes, including mortality. One study showed that sustaining an intraoperative SBP below 70 mm Hg for longer than 5 min increased the risk of mortality from less than 1% to nearly 6%.16 A large study detected an increase in mortality associated with SBP sustained below 80 mm Hg for 10 min or longer.7 Intraoperative hypotension has also been associated with postoperative AKI and myocardial injury.8,9,12
Many of the limitations of the current study could be addressed by a large randomized controlled trial of ACEI management prior to NCNV surgeries that examines clinically important endpoints beyond intraoperative hypotension. Several specific aspects of perioperative RAAS inhibitor management also deserve further investigation. Our findings may not be generalizable to patients taking ARBs or to patients with congestive heart failure. The preoperative management of ARBs and the preoperative management of RAAS inhibitors in those with congestive heart failure are important areas of focus for future research. Lastly, our finding that preoperative ACEI management decisions can affect postoperative hypotensive and hypertensive events should be substantiated by future research, and any negative consequences of those events should be further explored.
Nonetheless, our study is the largest randomized study of preoperative RAAS inhibition published to date. More than twice as many patients were randomized in this study than in all previous randomized studies combined.4-6 To the best of our knowledge, this is also the first randomized study evaluating NCNV surgeries. Finally, our use of a practical ACEI omission protocol based on known pharmacokinetics allows for direct application to clinical practice.
CONCLUSION
Hypertension is among the most common chronic conditions encountered in patients planning surgery, and ACEIs are among the most frequently prescribed antihypertensive medications. This study showed that ACEI continuation is associated with an increased frequency and cumulative duration of intraoperative hypotension. These findings, while at odds with current ACC/AHA guidelines, align with the findings of a meta-analysis on this subject and with recent literature.3,11-13,22
Acknowledgments
The authors wish to thank Miranda M Fricke, MS, PA-C; Tiffany K Hillyard, APRN-FNP; and Barbara Sink, MPAS, PA-C who assisted in the design and conduct of patient enrollment and randomization procedures.
Disclosures
The authors have no relevant financial conflicts of interest to report.
Funding
This study was subsidized by a grant from the University of Nebraska Medical Center Research Support Fund. The funding source had no role in the design, conduct, analysis, or reporting of the study.
Over 7 million surgeries are performed in United States hospitals each year. Among these surgeries, approximately 85% are noncardiac, nonvascular (NCNV) procedures.1,2 Although the preoperative use of an angiotensin-converting enzyme inhibitor (ACEI) can be expected in as many as 13% of these surgeries,3 the optimal preoperative ACEI management strategy for patients undergoing NCNV surgeries is poorly understood.
High-quality evidence suggests that renin–angiotensin–aldosterone system (RAAS) inhibitors are associated with intraoperative hypotension among patients undergoing cardiac or vascular surgeries.4-6 Intraoperative hypotension increases the risk of 30-day mortality,7 and the duration of intraoperative hypotension increases the risk of end organ damage.8,9 This body of evidence suggests that withholding ACEIs prior to cardiac and vascular surgeries is safer than continuing ACEIs without interruption.
The evidence concerning perioperative management of ACEIs is inconclusive for patients undergoing NCNV procedures. Some studies comparing patients taking or not taking a RAAS inhibitor preoperatively describe negligible differences in the frequency of intraoperative hypotensive episodes or complications.3,10 Others have found an increased risk of intraoperative hypotension and associated postoperative adverse events in patients continuing RAAS inhibitors preoperatively.11,12 Current guideline discrepancies reflect the uncertainty of the evidence. The guidelines set by the American College of Cardiology and American Heart Association (ACC/AHA) suggest the uninterrupted perioperative continuation of RAAS inhibitors.13 The guidelines provided by the European Society of Cardiology and European Society of Anaesthesiology also suggest the continuation of RAAS inhibitors throughout the perioperative period for patients with systolic heart failure but recommend transient discontinuation for patients with hypertension.14
This randomized study aimed to compare the effect of two practical strategies for preoperative ACEI management on the perioperative blood pressure of patients undergoing NCNV surgery. The two strategies studied were the omission of the final preoperative ACEI dose and the uninterrupted continuation of ACEI therapy. We hypothesized that patients randomized to ACEI omission would experience intraoperative hypotensive episodes less frequently than those randomized to ACEI continuation.
METHODS
Study Design and Setting
We performed a prospective randomized controlled trial (ClinicalTrials.gov: NCT01669434). The study was carried out in a preoperative evaluation clinic and its affiliated 489-bed academic medical center. Anesthesiologists and internal medicine physicians work collaboratively in the clinic to assess more than 5,000 patients annually (one-third of the institution’s elective surgeries). Patients were randomized 1:1 in block sizes of 5 and 10 and stratified by age < 65 and ≥ 65 years to the omission or continuation of the final preoperative ACEI dose (whether that dose was scheduled for the morning of surgery or the night prior). Preoperative clinicians enrolled patients and subsequently assigned them to intervention groups on the basis of a sequentially numbered list. Patients and healthcare providers were not blinded to allocation status. Intraoperative and postoperative management was provided in accordance with usual care as decided by treatment team.
Participants
Patients who presented to the preoperative evaluation clinic between May 2015 and November 2016 and who had been taking an ACEI for at least 6 weeks were eligible for inclusion. Patients taking angiotensin receptor blockers were excluded. Enrollment was limited to patients planning NCNV surgery. Patients planning intrathoracic, major vascular, organ transplant, and oncologic surgery were excluded. Patients undergoing outpatient procedures not requiring an overnight stay in the hospital were also excluded. Patients with preoperative clinic systolic blood pressure (SBP) <90 or ≥160 or diastolic blood pressure (DBP) <60 or ≥ 95 were excluded. Patients with moderate to severe or clinically decompensated heart failure (left ventricular ejection fraction < 40% or New York Heart Association class III or IV) and those with end-stage renal disease requiring dialysis were also excluded. Patients presenting more than once during the accrual period were eligible for the initial surgery only. All participating patients provided written informed consent. This project was approved by the University of Nebraska Medical Center Institutional Review Board.
Data Collection
Baseline characteristics were recorded by study personnel at the time of enrollment. We measured serum creatinine level at the preoperative visit and on postoperative day 1. An automated anesthesia information management system was used to measure intraoperative blood pressures every three minutes. Postoperative blood pressures through discharge were measured by hospital staff per usual care. During postoperative hospitalization, we queried patients about preoperative adherence to allocation. The digital abstraction of data from the electronic medical record was supplemented by chart review when necessary.
Outcomes
The primary outcome was intraoperative hypotension defined as any SBP < 80 mm Hg occurring from the administration of the first induction agent through transfer to the postanesthesia care unit (PACU). We also examined hypotension during anesthesia induction, which we defined as the 20-minute period following the administration of the first anesthesia induction agent. Episodes of SBP < 80 were defined as being associated with vasopressor administration when any vasopressor was administered during or within 10 min of the episode.
Secondary analyses included postoperative acute kidney injury (AKI), postoperative hypotensive and hypertensive episodes, cardiac events, and mortality. When comparing postoperative day 1 creatinine levels to preoperative creatinine levels, we used the Acute Kidney Injury Network definition of AKI as an increase in creatinine of 0.3 mg/dl or 50%.15 Postoperative hypotension was defined as any SBP < 90 mm Hg and postoperative hypertension as any SBP > 180 mm Hg occurring after arrival in the PACU. Major adverse cardiac events (MACE) were defined as a composite of acute coronary syndrome, acute heart failure, or new-onset arrhythmia. Discharge from the hospital served as the study endpoint for each patient.
Analysis
Fisher’s exact test was used to compare categorical outcomes between groups. The independent sample t-test or Wilcoxon rank–sum test, as appropriate, was used to compare continuous measures. We selected Fisher’s exact test over χ2-test to produce conservative estimates. Patients were maintained in their allocated group as randomized for analytical purposes regardless of adherence to allocation. We performed all analyses using SAS version 9.4 for Windows (SAS institute, Cary, North Carolina).
We estimated that a sample size of 300 patients would achieve 80% power to detect a difference of 0.17 between the group proportions of 0.33 and 0.50 at a significance level (ɑ) of 0.05 by using a two-sided z-test with continuity correction, assuming 15% loss to follow-up. This estimate allowed for 1 interim analysis using the O’Brien-Fleming spending function truncated at three standard deviations to determine the test boundaries. The monitoring boundary P values associated with the interim analysis were .003, and the threshold P value for the final analysis was .049.
RESULTS
Study Flow
A total of 453 patients were screened for eligibility. Among these patients, 162 were excluded, and the remaining 291 patients were randomized (Figure 1). Surgery was cancelled in six patients allocated to omission and in four patients allocated to continuation arms, respectively. Moreover, three patients in the omission arm were excluded from the analysis following randomization. Specifically, one was excluded because of early discharge without overnight stay, one was excluded because of withdrawal of consent, and one was excluded because of missing primary outcome data. In addition, three cases in the continuation arm were excluded following randomization because of the preoperative (permanent) discontinuation of ACEI therapy in two cases and discharge without an overnight stay in one case. Finally, 275 patients were included in the analysis: 137 in the ACEI omission group and 138 in the ACEI continuation group. Adherence to allocation was 88% and 92% in the omission and continuation groups, respectively.
Baseline Characteristics
The demographic data of patients allocated to ACEI omission and those allocated to ACEI continuation were similar (Table 1). A large majority of patients in both groups took the ACEI lisinopril. Overall, 187 of 275 (68%) patients were taking at least 1 antihypertensive agent, most commonly a diuretic, in addition to an ACEI. SBP measured during the preoperative clinic visit averaged 136.5 mm Hg and did not differ significantly between groups (P = .84).
Surgical Variables
General anesthesia was the most commonly utilized technique, although spinal and regional anesthesia were also represented (Table 1). The majority of cases in both groups were planning for orthopedic and spinal surgery. The method of anesthesia or type of surgery between patients allocated to ACEI omission and those allocated to continuation did not differ (P = .61 and P = .45 respectively).
Episodes of Intraoperative Hypotension
Intraoperative SBPs are displayed in Figure 2, and hemodynamic outcomes are summarized in Table 2. Episodes of SBP < 80 mm Hg during anesthesia induction were numerically less frequent in the omission group than in the continuation group; the difference between groups, however, was not statistically significant (24 of 137 [18%] vs 38 of 138 [28%], RR: 0.64, 95% CI: 0.40 to 1.00, P = .06).
Duration of Intraoperative Hypotension
The median cumulative duration of intraoperative SBP < 80 was two minutes (range 0-41) in patients allocated to the ACEI omission group compared with seven minutes (range 0-214) in those allocated to the continuation group (P < .01). The median cumulative duration of mean arterial pressure < 55 mm Hg was also shorter in the omission group (median 0 min [range 0-39] vs 3 min [range 0-122], P < .01) than in the continuation group. The duration of surgery did not differ between groups (median 141 min [range 77-554] vs 142 min [range 57-665], P = .97).
Postoperative Outcomes
RAAS inhibitor therapy was resumed within 48 h after surgery in 122 of 137 (89%) patients allocated to the omission group and in 128 of 138 (93%) patients allocated to the continuation group (RR: 0.96, 95% CI: 0.89-1.03, P = .30).
Patients allocated to the omission group were significantly less likely to experience postoperative hypotension (15 of 137 [11%] vs 31 of 138 [22%], RR: 0.49, 95% CI: 0.28 to 0.86, P = .02) and significantly more likely to experience severe postoperative hypertension (33 of 137 [24%] vs 17 of 138 [12%], RR: 1.95, 95% CI: 1.14 to 3.34, P = .01) than those allocated to the continuation group. The occurrences of postoperative AKI (RR: 0.60, 95% CI: 0.23 to 1.60, P = .44) or MACE (RR: 4.03, 95% CI: 0.46 to 35.59, P = .21) in the omission group did not differ from the continuation group. The two groups exhibited similar PACU recovery time (mean 97.2 min) and overall hospital length of stay (mean 3.0 days) (P = .49 and P = .56 ). No episodes of inpatient mortality in either group were observed.
DISCUSSION
The omission of the final preoperative ACEI dose was associated with a significant reduction in the risk of intraoperative hypotension in patients undergoing NCNV surgery. This result confirmed our hypothesis. Coupled with the knowledge that intraoperative hypotension is associated with an increased risk of complications and mortality,7-9,16 this study favors the omission of the final preoperative ACEI dose prior to NCNV surgeries.
Our findings are in agreement with those of previous randomized studies that explored this question4,5 and help extend results from cardiac and vascular surgeries to NCNV surgeries. Previous studies on the use of RAAS inhibitors in NCNV surgeries did not employ randomization and yielded mixed results.3,10-12,17 A large single-institution study (n = 18,056) noted no difference in intraoperative blood pressure between patients taking ACEIs and a matched group of non-ACEI users.3 More recently, a subgroup analysis of the international VISION study showed that omitting RAAS inhibitors on the day of surgery reduced the risk of intraoperative hypotension.11 In that analysis, however, only a small amount of the variability in preoperative RAAS inhibitor management was explainable by modeling known factors, thus allowing for the possibility of unmeasured confounding. Our study, which minimized confounding through randomization, is the first to prospectively compare protocols for patients undergoing NCNV surgery. In contrast to previous studies, the present study was able to report the lack of difference in postoperative RAAS inhibitor administration between study groups. Postoperative RAAS inhibitor management affects complications and mortality.18,19
Our present finding that preoperative ACEI management affects postoperative hypotensive and hypertensive events conflicts with some previous findings.11,20 However, recent evidence has revealed that postoperative hypotensive episodes are associated with vascular events and mortality.11,21 In the context of that evidence, our study lends further support to the omission of the final preoperative ACEI dose. However, we did not detect any decrease in AKI, MACE, or mortality in the ACEI omission group.
This study should be considered in light of its limitations. The pragmatic nature of the study allowed for certain potential biases. Although adherence to allocation was high, the specific ACEI agent taken and the exact timing of the final dose in relation to surgery were not controlled. Anesthetic and postoperative management decisions were made by the treatment team and may have systematically varied given that the treatment team was not blinded to allocation. Furthermore, all outcome data were collected as part of routine care and may not have captured events with great fidelity. Generalizability is limited by the execution of the study at a single academic institution, the preponderance of orthopedic and spine surgeries, and by the negligible representation of ethnicities other than Caucasian. Additionally, recruitment from the preoperative evaluation clinic likely resulted in a patient group with greater comorbidity than the overall population of patients undergoing NCNV surgery. This study was powered for intraoperative hypotension and not postoperative outcomes. Our primary outcome, intraoperative hypotension, is an intermediate measure but one that has well-established associations with adverse outcomes, including mortality. One study showed that sustaining an intraoperative SBP below 70 mm Hg for longer than 5 min increased the risk of mortality from less than 1% to nearly 6%.16 A large study detected an increase in mortality associated with SBP sustained below 80 mm Hg for 10 min or longer.7 Intraoperative hypotension has also been associated with postoperative AKI and myocardial injury.8,9,12
Many of the limitations of the current study could be addressed by a large randomized controlled trial of ACEI management prior to NCNV surgeries that examines clinically important endpoints beyond intraoperative hypotension. Several specific aspects of perioperative RAAS inhibitor management also deserve further investigation. Our findings may not be generalizable to patients taking ARBs or to patients with congestive heart failure. The preoperative management of ARBs and the preoperative management of RAAS inhibitors in those with congestive heart failure are important areas of focus for future research. Lastly, our finding that preoperative ACEI management decisions can affect postoperative hypotensive and hypertensive events should be substantiated by future research, and any negative consequences of those events should be further explored.
Nonetheless, our study is the largest randomized study of preoperative RAAS inhibition published to date. More than twice as many patients were randomized in this study than in all previous randomized studies combined.4-6 To the best of our knowledge, this is also the first randomized study evaluating NCNV surgeries. Finally, our use of a practical ACEI omission protocol based on known pharmacokinetics allows for direct application to clinical practice.
CONCLUSION
Hypertension is among the most common chronic conditions encountered in patients planning surgery, and ACEIs are among the most frequently prescribed antihypertensive medications. This study showed that ACEI continuation is associated with an increased frequency and cumulative duration of intraoperative hypotension. These findings, while at odds with current ACC/AHA guidelines, align with the findings of a meta-analysis on this subject and with recent literature.3,11-13,22
Acknowledgments
The authors wish to thank Miranda M Fricke, MS, PA-C; Tiffany K Hillyard, APRN-FNP; and Barbara Sink, MPAS, PA-C who assisted in the design and conduct of patient enrollment and randomization procedures.
Disclosures
The authors have no relevant financial conflicts of interest to report.
Funding
This study was subsidized by a grant from the University of Nebraska Medical Center Research Support Fund. The funding source had no role in the design, conduct, analysis, or reporting of the study.
1. Steiner CA KZ, Moore BJ, Imshaug MC, Pickens G. Surgeries in hospital-based ambulatory surgery and hospital inpatient settings, 2014. Statistical Brief 2017; 1-18. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb223-Ambulatory-Inpatient-Surgeries-2014.pdf. Accessed August 30, 2017. PubMed
2. Rate of all-listed procedures for discharges from short-stay hospitals, by procedure category and age: United States, 2010. National Hospital Discharge Survey 2010; https://www.cdc.gov/nchs/nhds/nhds_tables.htm. Accessed August 30, 2017.
3. Turan A, You J, Shiba A, Kurz A, Saager L, Sessler DI. Angiotensin converting enzyme inhibitors are not associated with respiratory complications or mortality after noncardiac surgery. Anesth Analg. 2012;114(3):552-560. doi: 10.1213/ANE.0b013e318241f6af. PubMed
4. Coriat P, Richer C, Douraki T, et al. Influence of chronic angiotensin-converting enzyme inhibition on anesthetic induction. Anesthesiology. 1994;81:299-307. PubMed
5. Pigott DW, Nagle C, Allman K, S. W, D. ER. Effect of omitting regular ACE inhibitor medication before cardiac surgery on haemodynamic variables and vasoactive drug requirements. Br J Anaesth. 1999;83:715-720. doi: 10.1093/bja/83.5.715 PubMed
6. Bertrand M, Godet G, Meersschaert K, Brun L, Salcedo E, Coriat P. Should the angiotensin II antagonists be discontinued before surgery? Anesth Analg. 2001;92:26-30. PubMed
7. Mascha EJ, Yang D, Weiss S, Sessler DI. Intraoperative mean arterial pressure variability and 30-day mortality in patients having noncardiac surgery. Anesthesiology. 2015;123(1):79-91. doi: 10.1097/ALN.0000000000000686. PubMed
8. Walsh M, Devereaux PJ, Garg AX, et al. Relationship between intraoperative mean arterial pressure and clinical outcomes after noncardiac surgery: toward an empirical definition of hypotension. Anesthesiology. 2013;119(3):507-515. doi: 10.1097/ALN.0b013e3182a10e26. PubMed
9. Salmasi V, Maheshwari K, Yang D, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126(1):47-65. doi: 10.1097/ALN.0000000000001432. PubMed
10. Comfere T, Sprung J, Kumar MM, et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100(3):636-644. doi: 10.1213/01.ANE.0000146521.68059.A1. PubMed
11. Roshanov PS, Rochwerg B, Patel A, et al. Withholding versus continuing angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers before noncardiac surgery: an analysis of the vascular events in noncardiac surgery patIents cohort evaluation prospective cohort. Anesthesiology. 2017;126(1):16-27. doi: 10.1097/ALN.0000000000001404. PubMed
12. Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288. doi: 10.1002/jhm.2155. PubMed
13. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. 2014;64(22):e77-137. doi: 10.1016/j.jacc.2014.07.944. PubMed
14. Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J. 2014;35(35):2383-2431. doi: 10.1093/eurheartj/ehu282 PubMed
15. Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. doi: 10.1186/cc5713 PubMed
16. Monk TG, Bronsert MR, Henderson WG, et al. Association between intraoperative hypotension and hypertension and 30-day postoperative mortality in noncardiac surgery. Anesthesiology. 2015;123(2):307-319. doi: 10.1097/ALN.0000000000000756. PubMed
17. Kheterpal S, Khodaparast O, Shanks A, O’Reilly M, Tremper KK. Chronic angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy combined with diuretic therapy is associated with increased episodes of hypotension in noncardiac surgery. J Cardiothorac Vasc Anesth. 2008;22(2):180-186. 10.1053/j.jvca.2007.12.020. PubMed
18. Lee SM, Takemoto S, Wallace AW. Association between withholding angiotensin receptor blockers in the early postoperative period and 30-day mortality: a cohort study of the veterans affairs healthcare system. Anesthesiology. 2015;123(2):288-306. 10.1097/ALN.0000000000000739. PubMed
19. Drenger B, Fontes ML, Miao Y, et al. Patterns of use of perioperative angiotensin-converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in-hospital morbidity and mortality. Circulation. 2012;126(3):261-269. doi: 10.1161/CIRCULATIONAHA.111.059527. PubMed
20. Twersky RS, Goel V, Narayan P, Weedon J. The risk of hypertension after preoperative discontinuation of angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists in ambulatory and same-day admission patients. Anesth Analg. 2014;118(5):938-944. doi: 10.1213/ANE.0000000000000076. PubMed
21. Tan TW, Eslami MH, Kalish JA, et al. The need for treatment of hemodynamic instability following carotid endarterectomy is associated with increased perioperative and 1-year morbidity and mortality. J Vasc Surg. 2014;59(1):16-24 e11-12. https://doi.org/10.1053/j.jvca.2014.12.002 PubMed
22. Rosenman DJ, McDonald FS, Ebbert JO, Erwin PJ, LaBella M, Montori VM. Clinical consequences of withholding versus administering renin-angiotensin-aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319-325. doi: 10.1002/jhm.323. PubMed
1. Steiner CA KZ, Moore BJ, Imshaug MC, Pickens G. Surgeries in hospital-based ambulatory surgery and hospital inpatient settings, 2014. Statistical Brief 2017; 1-18. https://www.hcup-us.ahrq.gov/reports/statbriefs/sb223-Ambulatory-Inpatient-Surgeries-2014.pdf. Accessed August 30, 2017. PubMed
2. Rate of all-listed procedures for discharges from short-stay hospitals, by procedure category and age: United States, 2010. National Hospital Discharge Survey 2010; https://www.cdc.gov/nchs/nhds/nhds_tables.htm. Accessed August 30, 2017.
3. Turan A, You J, Shiba A, Kurz A, Saager L, Sessler DI. Angiotensin converting enzyme inhibitors are not associated with respiratory complications or mortality after noncardiac surgery. Anesth Analg. 2012;114(3):552-560. doi: 10.1213/ANE.0b013e318241f6af. PubMed
4. Coriat P, Richer C, Douraki T, et al. Influence of chronic angiotensin-converting enzyme inhibition on anesthetic induction. Anesthesiology. 1994;81:299-307. PubMed
5. Pigott DW, Nagle C, Allman K, S. W, D. ER. Effect of omitting regular ACE inhibitor medication before cardiac surgery on haemodynamic variables and vasoactive drug requirements. Br J Anaesth. 1999;83:715-720. doi: 10.1093/bja/83.5.715 PubMed
6. Bertrand M, Godet G, Meersschaert K, Brun L, Salcedo E, Coriat P. Should the angiotensin II antagonists be discontinued before surgery? Anesth Analg. 2001;92:26-30. PubMed
7. Mascha EJ, Yang D, Weiss S, Sessler DI. Intraoperative mean arterial pressure variability and 30-day mortality in patients having noncardiac surgery. Anesthesiology. 2015;123(1):79-91. doi: 10.1097/ALN.0000000000000686. PubMed
8. Walsh M, Devereaux PJ, Garg AX, et al. Relationship between intraoperative mean arterial pressure and clinical outcomes after noncardiac surgery: toward an empirical definition of hypotension. Anesthesiology. 2013;119(3):507-515. doi: 10.1097/ALN.0b013e3182a10e26. PubMed
9. Salmasi V, Maheshwari K, Yang D, et al. Relationship between intraoperative hypotension, defined by either reduction from baseline or absolute thresholds, and acute kidney and myocardial injury after noncardiac surgery: a retrospective cohort analysis. Anesthesiology. 2017;126(1):47-65. doi: 10.1097/ALN.0000000000001432. PubMed
10. Comfere T, Sprung J, Kumar MM, et al. Angiotensin system inhibitors in a general surgical population. Anesth Analg. 2005;100(3):636-644. doi: 10.1213/01.ANE.0000146521.68059.A1. PubMed
11. Roshanov PS, Rochwerg B, Patel A, et al. Withholding versus continuing angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers before noncardiac surgery: an analysis of the vascular events in noncardiac surgery patIents cohort evaluation prospective cohort. Anesthesiology. 2017;126(1):16-27. doi: 10.1097/ALN.0000000000001404. PubMed
12. Nielson E, Hennrikus E, Lehman E, Mets B. Angiotensin axis blockade, hypotension, and acute kidney injury in elective major orthopedic surgery. J Hosp Med. 2014;9(5):283-288. doi: 10.1002/jhm.2155. PubMed
13. Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol. 2014;64(22):e77-137. doi: 10.1016/j.jacc.2014.07.944. PubMed
14. Kristensen SD, Knuuti J, Saraste A, et al. 2014 ESC/ESA Guidelines on non-cardiac surgery: cardiovascular assessment and management: The Joint Task Force on non-cardiac surgery: cardiovascular assessment and management of the European Society of Cardiology (ESC) and the European Society of Anaesthesiology (ESA). Eur Heart J. 2014;35(35):2383-2431. doi: 10.1093/eurheartj/ehu282 PubMed
15. Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. doi: 10.1186/cc5713 PubMed
16. Monk TG, Bronsert MR, Henderson WG, et al. Association between intraoperative hypotension and hypertension and 30-day postoperative mortality in noncardiac surgery. Anesthesiology. 2015;123(2):307-319. doi: 10.1097/ALN.0000000000000756. PubMed
17. Kheterpal S, Khodaparast O, Shanks A, O’Reilly M, Tremper KK. Chronic angiotensin-converting enzyme inhibitor or angiotensin receptor blocker therapy combined with diuretic therapy is associated with increased episodes of hypotension in noncardiac surgery. J Cardiothorac Vasc Anesth. 2008;22(2):180-186. 10.1053/j.jvca.2007.12.020. PubMed
18. Lee SM, Takemoto S, Wallace AW. Association between withholding angiotensin receptor blockers in the early postoperative period and 30-day mortality: a cohort study of the veterans affairs healthcare system. Anesthesiology. 2015;123(2):288-306. 10.1097/ALN.0000000000000739. PubMed
19. Drenger B, Fontes ML, Miao Y, et al. Patterns of use of perioperative angiotensin-converting enzyme inhibitors in coronary artery bypass graft surgery with cardiopulmonary bypass: effects on in-hospital morbidity and mortality. Circulation. 2012;126(3):261-269. doi: 10.1161/CIRCULATIONAHA.111.059527. PubMed
20. Twersky RS, Goel V, Narayan P, Weedon J. The risk of hypertension after preoperative discontinuation of angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists in ambulatory and same-day admission patients. Anesth Analg. 2014;118(5):938-944. doi: 10.1213/ANE.0000000000000076. PubMed
21. Tan TW, Eslami MH, Kalish JA, et al. The need for treatment of hemodynamic instability following carotid endarterectomy is associated with increased perioperative and 1-year morbidity and mortality. J Vasc Surg. 2014;59(1):16-24 e11-12. https://doi.org/10.1053/j.jvca.2014.12.002 PubMed
22. Rosenman DJ, McDonald FS, Ebbert JO, Erwin PJ, LaBella M, Montori VM. Clinical consequences of withholding versus administering renin-angiotensin-aldosterone system antagonists in the preoperative period. J Hosp Med. 2008;3(4):319-325. doi: 10.1002/jhm.323. PubMed
© 2018 Society of Hospital Medicine
The Association of Inpatient Occupancy with Hospital-Acquired Clostridium difficile Infection
High hospital occupancy is a fundamental challenge faced by healthcare systems in the United States.1-3 However, few studies have examined the effect of high occupancy on outcomes in the inpatient setting,4-9 and these showed mixed results. Hospital-acquired conditions (HACs), such as Clostridium difficile infection (CDI), are quality indicators for inpatient care and part of the Centers for Medicare and Medicaid Services’ Hospital-Acquired Conditions Reductions Program.10-12 However, few studies—largely conducted outside of the US—have evaluated the association between inpatient occupancy and HACs. These studies showed increasing hospital-acquired infection rates with increasing occupancy.13-15 Past studies of hospital occupancy have relied on annual average licensed bed counts, which are not a reliable measure of available and staffed beds and do not account for variations in patient volume and bed supply.16 Using a novel measure of inpatient occupancy, we tested the hypothesis that increasing inpatient occupancy is associated with a greater likelihood of CDI.
METHODS
We performed a retrospective analysis of administrative data from non-federal, acute care hospitals in California during 2008–2012 using the Office of Statewide Health Planning and Development (OSHPD) Patient Discharge Data set, a complete census of all CA licensed general acute care hospital discharge records. This study was approved by the OSHPD Committee for the Protection of Human Subjects and was deemed exempt by our institution’s Institutional Review Board.
Selection of Participants
The study population consisted of fee-for-service Medicare enrollees ≥65 years admitted through the emergency department (ED) with a hospital length of stay (HLOS) <50 days and a primary discharge diagnosis of acute myocardial infarction (MI), pneumonia (PNA), or heart failure (HF; [identified through the respective Clinical Classification Software [CCS]).
The sample was restricted to discharges with a HLOS of <50 days, because those with longer HLOS (0.01% of study sample) were likely different in ways that may bias our findings (eg, they will likely be sicker). We limited our study to admissions through the ED to reduce potential selection bias by excluding elective admissions and hospital-to-hospital transfers, which are likely dependent on occupancy. MI, HF, and PNA diagnoses were selected because they are prevalent and have high inpatient mortality, allowing us to examine the effect of occupancy on some of the sickest inpatients.17
Hospital-acquired cases of CDI were identified as discharges (using ICD-9 code 008.45 for CDI) that were not marked as present-on-admission (POA) using the method described by Zhan et al.18 To avoid small facility outlying effects, we included hospitals that had 100 or more MI, HF, and PNA discharges that met the inclusion criteria over the study years.
OSHPD inpatient data were combined with OSHPD hospital annual financial data that contain hospital-level variables including ownership (City/County, District, Investor, and Non-Profit), geography (based on health services area), teaching status, urbanicity, and size based on the number of average annual licensed beds. If characteristics were not available for a given hospital for 1 or more years, the information from the closest available year was used for that hospital (replacement required for 10,504 (1.5%) cases; 4,856 otherwise eligible cases (0.7%) were dropped because the hospital was not included in the annual financial data for any year. Approximately 0.2% of records had invalid values for disposition, payer, or admission route, and were therefore dropped. Patient residence zip code-level socioeconomic status was measured using the percentage of families living below the poverty line, median family income, and the percentage of individuals with less than a high school degree among those aged ≥ 25 years19; these measures were divided into 3 groups (bottom quartile, top quartile, and middle 50%) for analysis.
Measure of Occupancy
Calculating Daily Census and Bed Capacity
We calculated the daily census using admission date and HLOS for each observation in our dataset. We approximated the bed capacity as the maximum daily census in the 121-day window (+/- 60 days) around each census day in each hospital. The 121-day window was chosen to increase the likelihood of capturing changes in bed availability (eg, due to unit closures) and seasonal variability. Our daily census does not include patients admitted with psychiatric and obstetrics diagnoses and long-term care/rehabilitation stays (identified through CCS categories and excluded) because these patients are not likely to compete for the same hospital resources as those receiving care for MI, HF, and PNA. See Appendix Table 1 for definition of the occupancy terms.
Calculating Relative Daily Occupancy
We developed a raw hospital-specific occupancy measure by dividing the daily census by the maximum census in each 121-day window for each hospital. We converted these raw measures to percentiles within the 121-day window to create a daily relative occupancy measure. For example, median level occupancy day would correspond to an occupancy of 0.5; a minimum or maximum occupancy day would correspond to 0 or 1, respectively. We preferred a relative occupancy measure because it assumes that what constitutes “high occupancy” likely depends on the usual occupancy level of the facility.
Measuring Admission Day Occupancy and Average Occupancy over Hospitalization
Using the relative daily occupancy values, we constructed patient-level variables representing occupancy on admission day and average occupancy during hospitalization.
Data Analysis
First, we estimated descriptive statistics of the sample for occupancy, patient-level (eg, age, race, gender, and severity of illness), hospital-level (eg, size, teaching status, and urbanicity), and incident-level (day-of-the-week and season) variables. Next, we used logistic regression with cluster standard errors to estimate the adjusted and unadjusted association of occupancy with CDI. For this analysis, occupancy was broken into 4 groups: 0.00-0.25 (low occupancy); 0.26-0.50; 0.51-0.75; and 0.76-1.00 (high occupancy), with the 0.0-0.25 group treated as the reference level. We fit separate models for admission and average occupancy and re-ran the latter model including HLOS as a sensitivity analysis.
RESULTS
Study Population and Hospitals
Across 327 hospitals, 558,829 discharges (including deaths) met our inclusion criteria and there were 2045 admissions with CDI. The hospital and discharge characteristics are reported in Appendix Table 2.
Relationship of Occupancy with CDI
With regard to admission occupancy, the 0.26-0.50 group did not have a significantly higher rate of CDI than the low occupancy group. Both the 0.51-0.75 and the 0.76-1.00 occupancy groups had 15% lower odds of CDI compared to the low occupancy group (Table). The adjusted results were similar, although the comparison between the low and high occupancy groups was marginally nonsignificant.
With regard to average occupancy, intermediate levels of occupancy (ie, 0.26-0.50 and 0.51-0.75 groups) had over 3-fold increased odds of CDI relative to the low occupancy group; the high occupancy group did not have significantly different odds of CDI compared to the low occupancy group (Table 1). The adjusted results were similar with no changes in statistical significance. Including HLOS tempered the adjusted odds of CDI to 1.6 for intermediate levels of occupancy, but these remained significantly higher than high or low occupancy.
DISCUSSION
Hospital occupancy is related to CDI. However, contrary to expectation, we found that higher admission and average occupancy over hospitalization were not related to more hospital-acquired CDI. CDI rates were highest for intermediate levels of average occupancy with lower CDI rates at high and low occupancy. CDI had an inverse relationship with admission occupancy.
These findings suggest that an exploration of the processes associated with hospitals accommodating higher occupancy might elucidate measures to reduce CDI. How do staffing, implementation of policies, and routine procedures vary when hospitals are busy or quiet? What aspects of care delivery that function well during high and low occupancy periods breakdown during intermediate occupancy? Hospital policies, practices, and procedures during different phases of occupancy might inform best practices. These data suggest that hospital occupancy level should be a routinely collected data element by infection control officers and that this should be linked with protocols triggered or modified with high or low occupancy that might affect HACs.
Previous studies in Europe found increasing hospital-acquired infection rates with increasing occupancy.13-15 The authors postulated that increasing occupancy may limit available resources and increase nursing workloads, negatively impacting adherence to hand hygiene and cleaning protocols .8 However, these studies did not account for infections that were POA. In addition, our study examined hospitals in California after the 2006 implementation of the minimum nurse staffing policy, which means that staff to patient ratios could not fall below fixed thresholds that were typically higher than pre-policy ratios.19
This study had limitations pertaining to coded administrative data, including quality of coding and data validity. However, OSHPD has strict data reporting processes.20 This study focused on 1 state; however, California is large with a demographically diverse population and hospital types, characteristics that would help generalize findings. Furthermore, when using the average occupancy measure, we could not determine whether the complication was acquired during the high occupancy period of the hospitalization.
Higher admission day occupancy was associated with lower likelihood of CDI, and CDI rates were lower at high and low average occupancy. These findings should prompt exploration of how hospitals react to occupancy changes and how those care processes translate into HACs in order to inform best practices for hospital care.
Acknowledgments
The authors would like to thank Ms. Amanda Kogowski, MPH and Mr. Rekar Taymour, MS for their editorial assistance with drafting the manuscript.
Disclosures
The authors have no conflicts to disclose.
Funding
This study was funded by the National Institute on Aging.
1. Siegel B, Wilson MJ, Sickler D. Enhancing work flow to reduce crowding. Jt Comm J Qual Patient Saf. 2007;33(11):57-67. PubMed
2. Institute of Medicine Committee on the Future of Emergency Care in the U. S. Health System. The future of emergency care in the United States health system. Ann Emerg Med. 2006;48(2):115-120. DOI:10.1016/j.annemergmed.2006.06.015. PubMed
3. Weissman JS, Rothschild JM, Bendavid E, et al. Hospital workload and adverse events. Med Care. 2007;45(5):448-455. DOI: 10.1097/01.mlr.0000257231.86368.09. PubMed
4. Fieldston ES, Hall M, Shah SS, et al. Addressing inpatient crowding by smoothing occupancy at children’s hospitals. JHM. 2011;6(8):466-473. DOI: 10.1186/s12245-014-0025-4. PubMed
5. Evans WN, Kim B. Patient outcomes when hospitals experience a surge in admissions. J Health Econ. 2006;25(2):365-388. DOI: 10.1016/j.jhealeco.2005.10.003. PubMed
6. Bair AE, Song WT, Chen Y-C, Morris BA. The impact of inpatient boarding on ED efficiency: a discrete-event simulation study. J Med Syst. 2010;34(5):919-929. DOI: 10.1007/s10916-009-9307-4. PubMed
7. Schilling PL, Campbell Jr DA, Englesbe MJ, Davis MM. A comparison of in-hospital mortality risk conferred by high hospital occupancy, differences in nurse staffing levels, weekend admission, and seasonal influenza. Med Care. 2010;48(3):224-232. DOI: 10.1097/MLR.0b013e3181c162c0. PubMed
8. Schwierz C, Augurzky B, Focke A, Wasem J. Demand, selection and patient outcomes in German acute care hospitals. Health Econ. 2012;21(3):209-221. PubMed
9. Sharma R, Stano M, Gehring R. Short‐term fluctuations in hospital demand: implications for admission, discharge, and discriminatory behavior. RAND J. Econ. 2008;39(2):586-606. PubMed
10. Centers for Medicare and Medicaid Services. Hospital-Acquired Condition Reduction Program (HACRP). 2016; https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/HAC-Reduction-Program.html. Accessed October 05, 2017.
11. Cunningham JB, Kernohan G, Rush T. Bed occupancy, turnover intervals and MRSA rates in English hospitals. Br J Nurs. 2006;15(12):656-660. DOI: 10.12968/bjon.2006.15.12.21398. PubMed
12. Cunningham JB, Kernohan WG, Rush T. Bed occupancy, turnover interval and MRSA rates in Northern Ireland. Br J Nurs. 2006;15(6):324-328. DOI: 10.12968/bjon.2006.15.6.20680. PubMed
13. Kaier K, Luft D, Dettenkofer M, Kist M, Frank U. Correlations between bed occupancy rates and Clostridium difficile infections: a time-series analysis. Epidemiol Infect. 2011;139(3):482-485. DOI: 10.1017/S0950268810001214. PubMed
14. Rafferty AM, Clarke SP, Coles J, et al. Outcomes of variation in hospital nurse staffing in English hospitals: cross-sectional analysis of survey data and discharge records. Int J Nurs Stud. 2007;44(2):175-182. DOI: 10.1016/j.ijnurstu.2006.08.003. PubMed
15. Bell CM, Redelmeier DA. Mortality among patients admitted to hospitals on weekends as compared with weekdays. N Engl J Med. 2001;345(9):663-668. DOI: 10.1056/NEJMsa003376. PubMed
16. Zhan C, Elixhauser A, Richards CL Jr, et al. Identification of hospital-acquired catheter-associated urinary tract infections from Medicare claims: sensitivity and positive predictive value. Med Care. 2009;47(3):364-369. DOI: 10.1097/MLR.0b013e31818af83d. PubMed
17. U.S. American factfinder. United States Census Bureau; 2016.
18. McHugh MD, Ma C. Hospital nursing and 30-day readmissions among Medicare patients with heart failure, acute myocardial infarction, and pneumonia. Med Care. 2013;51(1):52. DOI: 10.1097/MLR.0b013e3182763284. PubMed
19. Coffman JM, Seago JA, Spetz J. Minimum nurse-to-patient ratios in acute care hospitals in California. Health Aff. 2002;21(5):53-64. DOI:10.1377/hlthaff.21.5.53 PubMed
20. State of California. Medical Information Reporting for California (MIRCal) Regulations. 2016.
High hospital occupancy is a fundamental challenge faced by healthcare systems in the United States.1-3 However, few studies have examined the effect of high occupancy on outcomes in the inpatient setting,4-9 and these showed mixed results. Hospital-acquired conditions (HACs), such as Clostridium difficile infection (CDI), are quality indicators for inpatient care and part of the Centers for Medicare and Medicaid Services’ Hospital-Acquired Conditions Reductions Program.10-12 However, few studies—largely conducted outside of the US—have evaluated the association between inpatient occupancy and HACs. These studies showed increasing hospital-acquired infection rates with increasing occupancy.13-15 Past studies of hospital occupancy have relied on annual average licensed bed counts, which are not a reliable measure of available and staffed beds and do not account for variations in patient volume and bed supply.16 Using a novel measure of inpatient occupancy, we tested the hypothesis that increasing inpatient occupancy is associated with a greater likelihood of CDI.
METHODS
We performed a retrospective analysis of administrative data from non-federal, acute care hospitals in California during 2008–2012 using the Office of Statewide Health Planning and Development (OSHPD) Patient Discharge Data set, a complete census of all CA licensed general acute care hospital discharge records. This study was approved by the OSHPD Committee for the Protection of Human Subjects and was deemed exempt by our institution’s Institutional Review Board.
Selection of Participants
The study population consisted of fee-for-service Medicare enrollees ≥65 years admitted through the emergency department (ED) with a hospital length of stay (HLOS) <50 days and a primary discharge diagnosis of acute myocardial infarction (MI), pneumonia (PNA), or heart failure (HF; [identified through the respective Clinical Classification Software [CCS]).
The sample was restricted to discharges with a HLOS of <50 days, because those with longer HLOS (0.01% of study sample) were likely different in ways that may bias our findings (eg, they will likely be sicker). We limited our study to admissions through the ED to reduce potential selection bias by excluding elective admissions and hospital-to-hospital transfers, which are likely dependent on occupancy. MI, HF, and PNA diagnoses were selected because they are prevalent and have high inpatient mortality, allowing us to examine the effect of occupancy on some of the sickest inpatients.17
Hospital-acquired cases of CDI were identified as discharges (using ICD-9 code 008.45 for CDI) that were not marked as present-on-admission (POA) using the method described by Zhan et al.18 To avoid small facility outlying effects, we included hospitals that had 100 or more MI, HF, and PNA discharges that met the inclusion criteria over the study years.
OSHPD inpatient data were combined with OSHPD hospital annual financial data that contain hospital-level variables including ownership (City/County, District, Investor, and Non-Profit), geography (based on health services area), teaching status, urbanicity, and size based on the number of average annual licensed beds. If characteristics were not available for a given hospital for 1 or more years, the information from the closest available year was used for that hospital (replacement required for 10,504 (1.5%) cases; 4,856 otherwise eligible cases (0.7%) were dropped because the hospital was not included in the annual financial data for any year. Approximately 0.2% of records had invalid values for disposition, payer, or admission route, and were therefore dropped. Patient residence zip code-level socioeconomic status was measured using the percentage of families living below the poverty line, median family income, and the percentage of individuals with less than a high school degree among those aged ≥ 25 years19; these measures were divided into 3 groups (bottom quartile, top quartile, and middle 50%) for analysis.
Measure of Occupancy
Calculating Daily Census and Bed Capacity
We calculated the daily census using admission date and HLOS for each observation in our dataset. We approximated the bed capacity as the maximum daily census in the 121-day window (+/- 60 days) around each census day in each hospital. The 121-day window was chosen to increase the likelihood of capturing changes in bed availability (eg, due to unit closures) and seasonal variability. Our daily census does not include patients admitted with psychiatric and obstetrics diagnoses and long-term care/rehabilitation stays (identified through CCS categories and excluded) because these patients are not likely to compete for the same hospital resources as those receiving care for MI, HF, and PNA. See Appendix Table 1 for definition of the occupancy terms.
Calculating Relative Daily Occupancy
We developed a raw hospital-specific occupancy measure by dividing the daily census by the maximum census in each 121-day window for each hospital. We converted these raw measures to percentiles within the 121-day window to create a daily relative occupancy measure. For example, median level occupancy day would correspond to an occupancy of 0.5; a minimum or maximum occupancy day would correspond to 0 or 1, respectively. We preferred a relative occupancy measure because it assumes that what constitutes “high occupancy” likely depends on the usual occupancy level of the facility.
Measuring Admission Day Occupancy and Average Occupancy over Hospitalization
Using the relative daily occupancy values, we constructed patient-level variables representing occupancy on admission day and average occupancy during hospitalization.
Data Analysis
First, we estimated descriptive statistics of the sample for occupancy, patient-level (eg, age, race, gender, and severity of illness), hospital-level (eg, size, teaching status, and urbanicity), and incident-level (day-of-the-week and season) variables. Next, we used logistic regression with cluster standard errors to estimate the adjusted and unadjusted association of occupancy with CDI. For this analysis, occupancy was broken into 4 groups: 0.00-0.25 (low occupancy); 0.26-0.50; 0.51-0.75; and 0.76-1.00 (high occupancy), with the 0.0-0.25 group treated as the reference level. We fit separate models for admission and average occupancy and re-ran the latter model including HLOS as a sensitivity analysis.
RESULTS
Study Population and Hospitals
Across 327 hospitals, 558,829 discharges (including deaths) met our inclusion criteria and there were 2045 admissions with CDI. The hospital and discharge characteristics are reported in Appendix Table 2.
Relationship of Occupancy with CDI
With regard to admission occupancy, the 0.26-0.50 group did not have a significantly higher rate of CDI than the low occupancy group. Both the 0.51-0.75 and the 0.76-1.00 occupancy groups had 15% lower odds of CDI compared to the low occupancy group (Table). The adjusted results were similar, although the comparison between the low and high occupancy groups was marginally nonsignificant.
With regard to average occupancy, intermediate levels of occupancy (ie, 0.26-0.50 and 0.51-0.75 groups) had over 3-fold increased odds of CDI relative to the low occupancy group; the high occupancy group did not have significantly different odds of CDI compared to the low occupancy group (Table 1). The adjusted results were similar with no changes in statistical significance. Including HLOS tempered the adjusted odds of CDI to 1.6 for intermediate levels of occupancy, but these remained significantly higher than high or low occupancy.
DISCUSSION
Hospital occupancy is related to CDI. However, contrary to expectation, we found that higher admission and average occupancy over hospitalization were not related to more hospital-acquired CDI. CDI rates were highest for intermediate levels of average occupancy with lower CDI rates at high and low occupancy. CDI had an inverse relationship with admission occupancy.
These findings suggest that an exploration of the processes associated with hospitals accommodating higher occupancy might elucidate measures to reduce CDI. How do staffing, implementation of policies, and routine procedures vary when hospitals are busy or quiet? What aspects of care delivery that function well during high and low occupancy periods breakdown during intermediate occupancy? Hospital policies, practices, and procedures during different phases of occupancy might inform best practices. These data suggest that hospital occupancy level should be a routinely collected data element by infection control officers and that this should be linked with protocols triggered or modified with high or low occupancy that might affect HACs.
Previous studies in Europe found increasing hospital-acquired infection rates with increasing occupancy.13-15 The authors postulated that increasing occupancy may limit available resources and increase nursing workloads, negatively impacting adherence to hand hygiene and cleaning protocols .8 However, these studies did not account for infections that were POA. In addition, our study examined hospitals in California after the 2006 implementation of the minimum nurse staffing policy, which means that staff to patient ratios could not fall below fixed thresholds that were typically higher than pre-policy ratios.19
This study had limitations pertaining to coded administrative data, including quality of coding and data validity. However, OSHPD has strict data reporting processes.20 This study focused on 1 state; however, California is large with a demographically diverse population and hospital types, characteristics that would help generalize findings. Furthermore, when using the average occupancy measure, we could not determine whether the complication was acquired during the high occupancy period of the hospitalization.
Higher admission day occupancy was associated with lower likelihood of CDI, and CDI rates were lower at high and low average occupancy. These findings should prompt exploration of how hospitals react to occupancy changes and how those care processes translate into HACs in order to inform best practices for hospital care.
Acknowledgments
The authors would like to thank Ms. Amanda Kogowski, MPH and Mr. Rekar Taymour, MS for their editorial assistance with drafting the manuscript.
Disclosures
The authors have no conflicts to disclose.
Funding
This study was funded by the National Institute on Aging.
High hospital occupancy is a fundamental challenge faced by healthcare systems in the United States.1-3 However, few studies have examined the effect of high occupancy on outcomes in the inpatient setting,4-9 and these showed mixed results. Hospital-acquired conditions (HACs), such as Clostridium difficile infection (CDI), are quality indicators for inpatient care and part of the Centers for Medicare and Medicaid Services’ Hospital-Acquired Conditions Reductions Program.10-12 However, few studies—largely conducted outside of the US—have evaluated the association between inpatient occupancy and HACs. These studies showed increasing hospital-acquired infection rates with increasing occupancy.13-15 Past studies of hospital occupancy have relied on annual average licensed bed counts, which are not a reliable measure of available and staffed beds and do not account for variations in patient volume and bed supply.16 Using a novel measure of inpatient occupancy, we tested the hypothesis that increasing inpatient occupancy is associated with a greater likelihood of CDI.
METHODS
We performed a retrospective analysis of administrative data from non-federal, acute care hospitals in California during 2008–2012 using the Office of Statewide Health Planning and Development (OSHPD) Patient Discharge Data set, a complete census of all CA licensed general acute care hospital discharge records. This study was approved by the OSHPD Committee for the Protection of Human Subjects and was deemed exempt by our institution’s Institutional Review Board.
Selection of Participants
The study population consisted of fee-for-service Medicare enrollees ≥65 years admitted through the emergency department (ED) with a hospital length of stay (HLOS) <50 days and a primary discharge diagnosis of acute myocardial infarction (MI), pneumonia (PNA), or heart failure (HF; [identified through the respective Clinical Classification Software [CCS]).
The sample was restricted to discharges with a HLOS of <50 days, because those with longer HLOS (0.01% of study sample) were likely different in ways that may bias our findings (eg, they will likely be sicker). We limited our study to admissions through the ED to reduce potential selection bias by excluding elective admissions and hospital-to-hospital transfers, which are likely dependent on occupancy. MI, HF, and PNA diagnoses were selected because they are prevalent and have high inpatient mortality, allowing us to examine the effect of occupancy on some of the sickest inpatients.17
Hospital-acquired cases of CDI were identified as discharges (using ICD-9 code 008.45 for CDI) that were not marked as present-on-admission (POA) using the method described by Zhan et al.18 To avoid small facility outlying effects, we included hospitals that had 100 or more MI, HF, and PNA discharges that met the inclusion criteria over the study years.
OSHPD inpatient data were combined with OSHPD hospital annual financial data that contain hospital-level variables including ownership (City/County, District, Investor, and Non-Profit), geography (based on health services area), teaching status, urbanicity, and size based on the number of average annual licensed beds. If characteristics were not available for a given hospital for 1 or more years, the information from the closest available year was used for that hospital (replacement required for 10,504 (1.5%) cases; 4,856 otherwise eligible cases (0.7%) were dropped because the hospital was not included in the annual financial data for any year. Approximately 0.2% of records had invalid values for disposition, payer, or admission route, and were therefore dropped. Patient residence zip code-level socioeconomic status was measured using the percentage of families living below the poverty line, median family income, and the percentage of individuals with less than a high school degree among those aged ≥ 25 years19; these measures were divided into 3 groups (bottom quartile, top quartile, and middle 50%) for analysis.
Measure of Occupancy
Calculating Daily Census and Bed Capacity
We calculated the daily census using admission date and HLOS for each observation in our dataset. We approximated the bed capacity as the maximum daily census in the 121-day window (+/- 60 days) around each census day in each hospital. The 121-day window was chosen to increase the likelihood of capturing changes in bed availability (eg, due to unit closures) and seasonal variability. Our daily census does not include patients admitted with psychiatric and obstetrics diagnoses and long-term care/rehabilitation stays (identified through CCS categories and excluded) because these patients are not likely to compete for the same hospital resources as those receiving care for MI, HF, and PNA. See Appendix Table 1 for definition of the occupancy terms.
Calculating Relative Daily Occupancy
We developed a raw hospital-specific occupancy measure by dividing the daily census by the maximum census in each 121-day window for each hospital. We converted these raw measures to percentiles within the 121-day window to create a daily relative occupancy measure. For example, median level occupancy day would correspond to an occupancy of 0.5; a minimum or maximum occupancy day would correspond to 0 or 1, respectively. We preferred a relative occupancy measure because it assumes that what constitutes “high occupancy” likely depends on the usual occupancy level of the facility.
Measuring Admission Day Occupancy and Average Occupancy over Hospitalization
Using the relative daily occupancy values, we constructed patient-level variables representing occupancy on admission day and average occupancy during hospitalization.
Data Analysis
First, we estimated descriptive statistics of the sample for occupancy, patient-level (eg, age, race, gender, and severity of illness), hospital-level (eg, size, teaching status, and urbanicity), and incident-level (day-of-the-week and season) variables. Next, we used logistic regression with cluster standard errors to estimate the adjusted and unadjusted association of occupancy with CDI. For this analysis, occupancy was broken into 4 groups: 0.00-0.25 (low occupancy); 0.26-0.50; 0.51-0.75; and 0.76-1.00 (high occupancy), with the 0.0-0.25 group treated as the reference level. We fit separate models for admission and average occupancy and re-ran the latter model including HLOS as a sensitivity analysis.
RESULTS
Study Population and Hospitals
Across 327 hospitals, 558,829 discharges (including deaths) met our inclusion criteria and there were 2045 admissions with CDI. The hospital and discharge characteristics are reported in Appendix Table 2.
Relationship of Occupancy with CDI
With regard to admission occupancy, the 0.26-0.50 group did not have a significantly higher rate of CDI than the low occupancy group. Both the 0.51-0.75 and the 0.76-1.00 occupancy groups had 15% lower odds of CDI compared to the low occupancy group (Table). The adjusted results were similar, although the comparison between the low and high occupancy groups was marginally nonsignificant.
With regard to average occupancy, intermediate levels of occupancy (ie, 0.26-0.50 and 0.51-0.75 groups) had over 3-fold increased odds of CDI relative to the low occupancy group; the high occupancy group did not have significantly different odds of CDI compared to the low occupancy group (Table 1). The adjusted results were similar with no changes in statistical significance. Including HLOS tempered the adjusted odds of CDI to 1.6 for intermediate levels of occupancy, but these remained significantly higher than high or low occupancy.
DISCUSSION
Hospital occupancy is related to CDI. However, contrary to expectation, we found that higher admission and average occupancy over hospitalization were not related to more hospital-acquired CDI. CDI rates were highest for intermediate levels of average occupancy with lower CDI rates at high and low occupancy. CDI had an inverse relationship with admission occupancy.
These findings suggest that an exploration of the processes associated with hospitals accommodating higher occupancy might elucidate measures to reduce CDI. How do staffing, implementation of policies, and routine procedures vary when hospitals are busy or quiet? What aspects of care delivery that function well during high and low occupancy periods breakdown during intermediate occupancy? Hospital policies, practices, and procedures during different phases of occupancy might inform best practices. These data suggest that hospital occupancy level should be a routinely collected data element by infection control officers and that this should be linked with protocols triggered or modified with high or low occupancy that might affect HACs.
Previous studies in Europe found increasing hospital-acquired infection rates with increasing occupancy.13-15 The authors postulated that increasing occupancy may limit available resources and increase nursing workloads, negatively impacting adherence to hand hygiene and cleaning protocols .8 However, these studies did not account for infections that were POA. In addition, our study examined hospitals in California after the 2006 implementation of the minimum nurse staffing policy, which means that staff to patient ratios could not fall below fixed thresholds that were typically higher than pre-policy ratios.19
This study had limitations pertaining to coded administrative data, including quality of coding and data validity. However, OSHPD has strict data reporting processes.20 This study focused on 1 state; however, California is large with a demographically diverse population and hospital types, characteristics that would help generalize findings. Furthermore, when using the average occupancy measure, we could not determine whether the complication was acquired during the high occupancy period of the hospitalization.
Higher admission day occupancy was associated with lower likelihood of CDI, and CDI rates were lower at high and low average occupancy. These findings should prompt exploration of how hospitals react to occupancy changes and how those care processes translate into HACs in order to inform best practices for hospital care.
Acknowledgments
The authors would like to thank Ms. Amanda Kogowski, MPH and Mr. Rekar Taymour, MS for their editorial assistance with drafting the manuscript.
Disclosures
The authors have no conflicts to disclose.
Funding
This study was funded by the National Institute on Aging.
1. Siegel B, Wilson MJ, Sickler D. Enhancing work flow to reduce crowding. Jt Comm J Qual Patient Saf. 2007;33(11):57-67. PubMed
2. Institute of Medicine Committee on the Future of Emergency Care in the U. S. Health System. The future of emergency care in the United States health system. Ann Emerg Med. 2006;48(2):115-120. DOI:10.1016/j.annemergmed.2006.06.015. PubMed
3. Weissman JS, Rothschild JM, Bendavid E, et al. Hospital workload and adverse events. Med Care. 2007;45(5):448-455. DOI: 10.1097/01.mlr.0000257231.86368.09. PubMed
4. Fieldston ES, Hall M, Shah SS, et al. Addressing inpatient crowding by smoothing occupancy at children’s hospitals. JHM. 2011;6(8):466-473. DOI: 10.1186/s12245-014-0025-4. PubMed
5. Evans WN, Kim B. Patient outcomes when hospitals experience a surge in admissions. J Health Econ. 2006;25(2):365-388. DOI: 10.1016/j.jhealeco.2005.10.003. PubMed
6. Bair AE, Song WT, Chen Y-C, Morris BA. The impact of inpatient boarding on ED efficiency: a discrete-event simulation study. J Med Syst. 2010;34(5):919-929. DOI: 10.1007/s10916-009-9307-4. PubMed
7. Schilling PL, Campbell Jr DA, Englesbe MJ, Davis MM. A comparison of in-hospital mortality risk conferred by high hospital occupancy, differences in nurse staffing levels, weekend admission, and seasonal influenza. Med Care. 2010;48(3):224-232. DOI: 10.1097/MLR.0b013e3181c162c0. PubMed
8. Schwierz C, Augurzky B, Focke A, Wasem J. Demand, selection and patient outcomes in German acute care hospitals. Health Econ. 2012;21(3):209-221. PubMed
9. Sharma R, Stano M, Gehring R. Short‐term fluctuations in hospital demand: implications for admission, discharge, and discriminatory behavior. RAND J. Econ. 2008;39(2):586-606. PubMed
10. Centers for Medicare and Medicaid Services. Hospital-Acquired Condition Reduction Program (HACRP). 2016; https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/HAC-Reduction-Program.html. Accessed October 05, 2017.
11. Cunningham JB, Kernohan G, Rush T. Bed occupancy, turnover intervals and MRSA rates in English hospitals. Br J Nurs. 2006;15(12):656-660. DOI: 10.12968/bjon.2006.15.12.21398. PubMed
12. Cunningham JB, Kernohan WG, Rush T. Bed occupancy, turnover interval and MRSA rates in Northern Ireland. Br J Nurs. 2006;15(6):324-328. DOI: 10.12968/bjon.2006.15.6.20680. PubMed
13. Kaier K, Luft D, Dettenkofer M, Kist M, Frank U. Correlations between bed occupancy rates and Clostridium difficile infections: a time-series analysis. Epidemiol Infect. 2011;139(3):482-485. DOI: 10.1017/S0950268810001214. PubMed
14. Rafferty AM, Clarke SP, Coles J, et al. Outcomes of variation in hospital nurse staffing in English hospitals: cross-sectional analysis of survey data and discharge records. Int J Nurs Stud. 2007;44(2):175-182. DOI: 10.1016/j.ijnurstu.2006.08.003. PubMed
15. Bell CM, Redelmeier DA. Mortality among patients admitted to hospitals on weekends as compared with weekdays. N Engl J Med. 2001;345(9):663-668. DOI: 10.1056/NEJMsa003376. PubMed
16. Zhan C, Elixhauser A, Richards CL Jr, et al. Identification of hospital-acquired catheter-associated urinary tract infections from Medicare claims: sensitivity and positive predictive value. Med Care. 2009;47(3):364-369. DOI: 10.1097/MLR.0b013e31818af83d. PubMed
17. U.S. American factfinder. United States Census Bureau; 2016.
18. McHugh MD, Ma C. Hospital nursing and 30-day readmissions among Medicare patients with heart failure, acute myocardial infarction, and pneumonia. Med Care. 2013;51(1):52. DOI: 10.1097/MLR.0b013e3182763284. PubMed
19. Coffman JM, Seago JA, Spetz J. Minimum nurse-to-patient ratios in acute care hospitals in California. Health Aff. 2002;21(5):53-64. DOI:10.1377/hlthaff.21.5.53 PubMed
20. State of California. Medical Information Reporting for California (MIRCal) Regulations. 2016.
1. Siegel B, Wilson MJ, Sickler D. Enhancing work flow to reduce crowding. Jt Comm J Qual Patient Saf. 2007;33(11):57-67. PubMed
2. Institute of Medicine Committee on the Future of Emergency Care in the U. S. Health System. The future of emergency care in the United States health system. Ann Emerg Med. 2006;48(2):115-120. DOI:10.1016/j.annemergmed.2006.06.015. PubMed
3. Weissman JS, Rothschild JM, Bendavid E, et al. Hospital workload and adverse events. Med Care. 2007;45(5):448-455. DOI: 10.1097/01.mlr.0000257231.86368.09. PubMed
4. Fieldston ES, Hall M, Shah SS, et al. Addressing inpatient crowding by smoothing occupancy at children’s hospitals. JHM. 2011;6(8):466-473. DOI: 10.1186/s12245-014-0025-4. PubMed
5. Evans WN, Kim B. Patient outcomes when hospitals experience a surge in admissions. J Health Econ. 2006;25(2):365-388. DOI: 10.1016/j.jhealeco.2005.10.003. PubMed
6. Bair AE, Song WT, Chen Y-C, Morris BA. The impact of inpatient boarding on ED efficiency: a discrete-event simulation study. J Med Syst. 2010;34(5):919-929. DOI: 10.1007/s10916-009-9307-4. PubMed
7. Schilling PL, Campbell Jr DA, Englesbe MJ, Davis MM. A comparison of in-hospital mortality risk conferred by high hospital occupancy, differences in nurse staffing levels, weekend admission, and seasonal influenza. Med Care. 2010;48(3):224-232. DOI: 10.1097/MLR.0b013e3181c162c0. PubMed
8. Schwierz C, Augurzky B, Focke A, Wasem J. Demand, selection and patient outcomes in German acute care hospitals. Health Econ. 2012;21(3):209-221. PubMed
9. Sharma R, Stano M, Gehring R. Short‐term fluctuations in hospital demand: implications for admission, discharge, and discriminatory behavior. RAND J. Econ. 2008;39(2):586-606. PubMed
10. Centers for Medicare and Medicaid Services. Hospital-Acquired Condition Reduction Program (HACRP). 2016; https://www.cms.gov/Medicare/Medicare-Fee-for-Service-Payment/AcuteInpatientPPS/HAC-Reduction-Program.html. Accessed October 05, 2017.
11. Cunningham JB, Kernohan G, Rush T. Bed occupancy, turnover intervals and MRSA rates in English hospitals. Br J Nurs. 2006;15(12):656-660. DOI: 10.12968/bjon.2006.15.12.21398. PubMed
12. Cunningham JB, Kernohan WG, Rush T. Bed occupancy, turnover interval and MRSA rates in Northern Ireland. Br J Nurs. 2006;15(6):324-328. DOI: 10.12968/bjon.2006.15.6.20680. PubMed
13. Kaier K, Luft D, Dettenkofer M, Kist M, Frank U. Correlations between bed occupancy rates and Clostridium difficile infections: a time-series analysis. Epidemiol Infect. 2011;139(3):482-485. DOI: 10.1017/S0950268810001214. PubMed
14. Rafferty AM, Clarke SP, Coles J, et al. Outcomes of variation in hospital nurse staffing in English hospitals: cross-sectional analysis of survey data and discharge records. Int J Nurs Stud. 2007;44(2):175-182. DOI: 10.1016/j.ijnurstu.2006.08.003. PubMed
15. Bell CM, Redelmeier DA. Mortality among patients admitted to hospitals on weekends as compared with weekdays. N Engl J Med. 2001;345(9):663-668. DOI: 10.1056/NEJMsa003376. PubMed
16. Zhan C, Elixhauser A, Richards CL Jr, et al. Identification of hospital-acquired catheter-associated urinary tract infections from Medicare claims: sensitivity and positive predictive value. Med Care. 2009;47(3):364-369. DOI: 10.1097/MLR.0b013e31818af83d. PubMed
17. U.S. American factfinder. United States Census Bureau; 2016.
18. McHugh MD, Ma C. Hospital nursing and 30-day readmissions among Medicare patients with heart failure, acute myocardial infarction, and pneumonia. Med Care. 2013;51(1):52. DOI: 10.1097/MLR.0b013e3182763284. PubMed
19. Coffman JM, Seago JA, Spetz J. Minimum nurse-to-patient ratios in acute care hospitals in California. Health Aff. 2002;21(5):53-64. DOI:10.1377/hlthaff.21.5.53 PubMed
20. State of California. Medical Information Reporting for California (MIRCal) Regulations. 2016.
© 2018 Society of Hospital Medicine