User login
Most nonemergent diagnoses can’t be predicted at ED presentation
based on an analysis of a national health care database presented at the annual meeting of the American College of Emergency Physicians and later published in JAMA Network Open.
The findings have important policy implications, as a large health care insurer recently rolled out a program to deny coverage for ED visits that conclude with a nonemergent diagnosis.
“Nonemergent diagnoses correlate poorly with visit severity and the need for multiple diagnostic tests and hospital care,” Shih-Chuan Chou, MD, of Brigham and Women’s Hospital in Boston, said at the meeting. “Nearly 9 out of 10 ED patients will present with some sort of symptoms that may potentially lead to a nonemergent diagnosis.”
Anthem initiated the decision to deny coverage for nonemergent conditions in 2017. Anthem’s policy is active in six states: Georgia, Indiana, Kentucky, Missouri, New Hampshire, and Ohio. ACEP and the Medical Association of Georgia have filed a federal lawsuit asserting that Anthem Blue Cross/Blue Shield of Georgia is violating the prudent layperson standard, which is a federal law requiring insurance companies to cover the costs of emergency care based on a patient’s symptoms – not their final diagnosis.
In the study reported by Dr. Chou, the impact of the change in reimbursement was applied to ED visit data from the National Hospital Ambulatory Medical Care Survey (NHAMCS-ED).
Of the 29.6 million adult ED visits by commercially insured patients in the NHAMCS-ED database over the study period, 15.7%, or approximately 4.6 million visits, would have been denied reimbursement based on the new Anthem policy. Of these, 24.5% of the visits were initially triaged by the ED staff as urgent or emergent. Another 26% of the visits resulted in two or more diagnostic tests, suggesting that ED staff were concerned that the underlying disease was potentially serious.
From another perspective, 87.9% of patients with a diagnosis that met criteria for reimbursement had symptoms similar to those of patients who would have been denied reimbursement. In other words, according to Dr. Chou, neither patients nor ED staff would likely be able to distinguish on the basis of symptoms alone which patients would ultimately be diagnosed with a disease that was or was not eligible for reimbursement.
“If commercial insurers begin adopting similar policies and retrospectively deny coverage for ED visits using discharge diagnoses, patients will be forced to weigh the odds of foregoing potentially necessary care against the risk of facing a significant financial burden if they guessed wrong,” Dr. Chou said.
Such policies are “likely to disproportionally impact low income populations,” he added, noting that many patient advocacy groups, as well as the American Medical Association, have expressed opposition to Anthem’s approach.
New strategies are needed to reduce reliance on ED visits for acute but nonemergent diseases, Dr. Chou said, but the data argue against denial of reimbursement as a method consistent with delivery of good health care.
Dr. Chou reported no financial relationships relevant to this study.
SOURCE: Chou S-C et al. JAMA Netw Open. 2018 Oct 19. doi: 10.1001/jamanetworkopen.2018.3731.
based on an analysis of a national health care database presented at the annual meeting of the American College of Emergency Physicians and later published in JAMA Network Open.
The findings have important policy implications, as a large health care insurer recently rolled out a program to deny coverage for ED visits that conclude with a nonemergent diagnosis.
“Nonemergent diagnoses correlate poorly with visit severity and the need for multiple diagnostic tests and hospital care,” Shih-Chuan Chou, MD, of Brigham and Women’s Hospital in Boston, said at the meeting. “Nearly 9 out of 10 ED patients will present with some sort of symptoms that may potentially lead to a nonemergent diagnosis.”
Anthem initiated the decision to deny coverage for nonemergent conditions in 2017. Anthem’s policy is active in six states: Georgia, Indiana, Kentucky, Missouri, New Hampshire, and Ohio. ACEP and the Medical Association of Georgia have filed a federal lawsuit asserting that Anthem Blue Cross/Blue Shield of Georgia is violating the prudent layperson standard, which is a federal law requiring insurance companies to cover the costs of emergency care based on a patient’s symptoms – not their final diagnosis.
In the study reported by Dr. Chou, the impact of the change in reimbursement was applied to ED visit data from the National Hospital Ambulatory Medical Care Survey (NHAMCS-ED).
Of the 29.6 million adult ED visits by commercially insured patients in the NHAMCS-ED database over the study period, 15.7%, or approximately 4.6 million visits, would have been denied reimbursement based on the new Anthem policy. Of these, 24.5% of the visits were initially triaged by the ED staff as urgent or emergent. Another 26% of the visits resulted in two or more diagnostic tests, suggesting that ED staff were concerned that the underlying disease was potentially serious.
From another perspective, 87.9% of patients with a diagnosis that met criteria for reimbursement had symptoms similar to those of patients who would have been denied reimbursement. In other words, according to Dr. Chou, neither patients nor ED staff would likely be able to distinguish on the basis of symptoms alone which patients would ultimately be diagnosed with a disease that was or was not eligible for reimbursement.
“If commercial insurers begin adopting similar policies and retrospectively deny coverage for ED visits using discharge diagnoses, patients will be forced to weigh the odds of foregoing potentially necessary care against the risk of facing a significant financial burden if they guessed wrong,” Dr. Chou said.
Such policies are “likely to disproportionally impact low income populations,” he added, noting that many patient advocacy groups, as well as the American Medical Association, have expressed opposition to Anthem’s approach.
New strategies are needed to reduce reliance on ED visits for acute but nonemergent diseases, Dr. Chou said, but the data argue against denial of reimbursement as a method consistent with delivery of good health care.
Dr. Chou reported no financial relationships relevant to this study.
SOURCE: Chou S-C et al. JAMA Netw Open. 2018 Oct 19. doi: 10.1001/jamanetworkopen.2018.3731.
based on an analysis of a national health care database presented at the annual meeting of the American College of Emergency Physicians and later published in JAMA Network Open.
The findings have important policy implications, as a large health care insurer recently rolled out a program to deny coverage for ED visits that conclude with a nonemergent diagnosis.
“Nonemergent diagnoses correlate poorly with visit severity and the need for multiple diagnostic tests and hospital care,” Shih-Chuan Chou, MD, of Brigham and Women’s Hospital in Boston, said at the meeting. “Nearly 9 out of 10 ED patients will present with some sort of symptoms that may potentially lead to a nonemergent diagnosis.”
Anthem initiated the decision to deny coverage for nonemergent conditions in 2017. Anthem’s policy is active in six states: Georgia, Indiana, Kentucky, Missouri, New Hampshire, and Ohio. ACEP and the Medical Association of Georgia have filed a federal lawsuit asserting that Anthem Blue Cross/Blue Shield of Georgia is violating the prudent layperson standard, which is a federal law requiring insurance companies to cover the costs of emergency care based on a patient’s symptoms – not their final diagnosis.
In the study reported by Dr. Chou, the impact of the change in reimbursement was applied to ED visit data from the National Hospital Ambulatory Medical Care Survey (NHAMCS-ED).
Of the 29.6 million adult ED visits by commercially insured patients in the NHAMCS-ED database over the study period, 15.7%, or approximately 4.6 million visits, would have been denied reimbursement based on the new Anthem policy. Of these, 24.5% of the visits were initially triaged by the ED staff as urgent or emergent. Another 26% of the visits resulted in two or more diagnostic tests, suggesting that ED staff were concerned that the underlying disease was potentially serious.
From another perspective, 87.9% of patients with a diagnosis that met criteria for reimbursement had symptoms similar to those of patients who would have been denied reimbursement. In other words, according to Dr. Chou, neither patients nor ED staff would likely be able to distinguish on the basis of symptoms alone which patients would ultimately be diagnosed with a disease that was or was not eligible for reimbursement.
“If commercial insurers begin adopting similar policies and retrospectively deny coverage for ED visits using discharge diagnoses, patients will be forced to weigh the odds of foregoing potentially necessary care against the risk of facing a significant financial burden if they guessed wrong,” Dr. Chou said.
Such policies are “likely to disproportionally impact low income populations,” he added, noting that many patient advocacy groups, as well as the American Medical Association, have expressed opposition to Anthem’s approach.
New strategies are needed to reduce reliance on ED visits for acute but nonemergent diseases, Dr. Chou said, but the data argue against denial of reimbursement as a method consistent with delivery of good health care.
Dr. Chou reported no financial relationships relevant to this study.
SOURCE: Chou S-C et al. JAMA Netw Open. 2018 Oct 19. doi: 10.1001/jamanetworkopen.2018.3731.
REPORTING FROM ACEP18
Key clinical point: A program to deny coverage for nonemergent visits to the ED is likely to have an adverse impact on patient care.
Major finding: Of patients with serious diseases eligible for reimbursement, 87.9% also have symptoms associated with nonurgent diseases.
Study details: An analysis of 29.6 million adult ED visits by commercially insured patients in the National Hospital Ambulatory Medical Care Survey.
Disclosures: Dr. Chou reported no financial relationships relevant to this study.
Source: Chou S-C et al. JAMA Netw Open. 2018 Oct 19. doi: 10.1001/jamanetworkopen.2018.3731.
Nipple-sparing mastectomy safe in older patients
BOSTON – For women undergoing results of recent studies suggest.

The procedure was “surgically safe” in older patients, with complication rates comparable to those seen in younger patients, Solange E. Cox, MD, of MedStar Georgetown University Hospital, Washington, said in a presentation of one those two retrospective analyses at the annual clinical congress of the American College of Surgeons.
“From this, we think that eligible older patients should be offered a nipple-sparing mastectomy as a surgical option for breast cancer, and age alone should not be used as criteria to exclude these patients from the option,” she said.
The second retrospective study showed that patients undergoing neoadjuvant chemotherapy had a rate of surgical complications and unintended reoperations comparable to what was seen in women undergoing primary surgery.
“Our big-picture takeaway from this study is that receipt of neoadjuvant chemotherapy is not a contraindication for nipple-sparing mastectomy,” said investigator Alex J. Bartholomew, MS, also of Medstar Georgetown University Hospital.
Mr. Bartholomew’s conclusion was based on an analysis of the nipple-sparing mastectomy registry of the American Society of Breast Surgeons that included a total of 3,125 breasts. Neoadjuvant chemotherapy was used in 528, or 16.9%, while primary surgery was performed in 2,597, or 83.1%.
The overall rate of complications was 11%, with nonsignificant differences between the neoadjuvant chemotherapy and primary surgery groups at 12.7% and 10.7%, respectively.

The rate of unintended reoperation, at 4.9%, was not significantly different in the neoadjuvant chemotherapy and primary surgery groups, at 5.2% and 4.8%, Mr. Bartholomew said. Similarly, he found that the rate of nipple areolar complex loss of 1% overall was not different between groups.
Advanced age was likewise not associated with increased complications in the study presented by Dr. Cox, which was a retrospective review of data for patients undergoing nipple-sparing mastectomy from 1998 to 2015 at a single institution. That cohort included 38 patients age 60 years or older, and 358 younger patients.
The rate of complications was 15.5% for patients over age 60 years, and similarly, 13.0% for their younger counterparts (P = .590), Dr. Cox reported. Likewise, the rate of unintended operations was 13.3% and 15.3% for older and younger patients, respectively (P = .274).
These findings are important because advancing age has been associated with a decrease in the likelihood of nipple-sparing mastectomy, according to Dr. Cox.
For mastectomies in general, advanced age has been implicated as a potential risk factor for necrosis, technical complications, and poor outcomes with mastectomies. However, no prior studies had been done specifically to evaluate nipple-sparing mastectomies in older breast cancer patients, Dr. Cox said.
Nipple-sparing mastectomy provides both cosmetic and psychosocial benefits to patients, according to the researchers, because the procedure spares the nipple-areolar complex.
The researchers who had no relevant disclosures.
SOURCES: Cox S et al. SF310 abstract; Bartholomew AJ et al. SF310 abstract ACS Clinical Congress 2018
BOSTON – For women undergoing results of recent studies suggest.
The procedure was “surgically safe” in older patients, with complication rates comparable to those seen in younger patients, Solange E. Cox, MD, of MedStar Georgetown University Hospital, Washington, said in a presentation of one those two retrospective analyses at the annual clinical congress of the American College of Surgeons.
“From this, we think that eligible older patients should be offered a nipple-sparing mastectomy as a surgical option for breast cancer, and age alone should not be used as criteria to exclude these patients from the option,” she said.
The second retrospective study showed that patients undergoing neoadjuvant chemotherapy had a rate of surgical complications and unintended reoperations comparable to what was seen in women undergoing primary surgery.
“Our big-picture takeaway from this study is that receipt of neoadjuvant chemotherapy is not a contraindication for nipple-sparing mastectomy,” said investigator Alex J. Bartholomew, MS, also of Medstar Georgetown University Hospital.
Mr. Bartholomew’s conclusion was based on an analysis of the nipple-sparing mastectomy registry of the American Society of Breast Surgeons that included a total of 3,125 breasts. Neoadjuvant chemotherapy was used in 528, or 16.9%, while primary surgery was performed in 2,597, or 83.1%.
The overall rate of complications was 11%, with nonsignificant differences between the neoadjuvant chemotherapy and primary surgery groups at 12.7% and 10.7%, respectively.
The rate of unintended reoperation, at 4.9%, was not significantly different in the neoadjuvant chemotherapy and primary surgery groups, at 5.2% and 4.8%, Mr. Bartholomew said. Similarly, he found that the rate of nipple areolar complex loss of 1% overall was not different between groups.
Advanced age was likewise not associated with increased complications in the study presented by Dr. Cox, which was a retrospective review of data for patients undergoing nipple-sparing mastectomy from 1998 to 2015 at a single institution. That cohort included 38 patients age 60 years or older, and 358 younger patients.
The rate of complications was 15.5% for patients over age 60 years, and similarly, 13.0% for their younger counterparts (P = .590), Dr. Cox reported. Likewise, the rate of unintended operations was 13.3% and 15.3% for older and younger patients, respectively (P = .274).
These findings are important because advancing age has been associated with a decrease in the likelihood of nipple-sparing mastectomy, according to Dr. Cox.
For mastectomies in general, advanced age has been implicated as a potential risk factor for necrosis, technical complications, and poor outcomes with mastectomies. However, no prior studies had been done specifically to evaluate nipple-sparing mastectomies in older breast cancer patients, Dr. Cox said.
Nipple-sparing mastectomy provides both cosmetic and psychosocial benefits to patients, according to the researchers, because the procedure spares the nipple-areolar complex.
The researchers who had no relevant disclosures.
SOURCES: Cox S et al. SF310 abstract; Bartholomew AJ et al. SF310 abstract ACS Clinical Congress 2018
BOSTON – For women undergoing results of recent studies suggest.
The procedure was “surgically safe” in older patients, with complication rates comparable to those seen in younger patients, Solange E. Cox, MD, of MedStar Georgetown University Hospital, Washington, said in a presentation of one those two retrospective analyses at the annual clinical congress of the American College of Surgeons.
“From this, we think that eligible older patients should be offered a nipple-sparing mastectomy as a surgical option for breast cancer, and age alone should not be used as criteria to exclude these patients from the option,” she said.
The second retrospective study showed that patients undergoing neoadjuvant chemotherapy had a rate of surgical complications and unintended reoperations comparable to what was seen in women undergoing primary surgery.
“Our big-picture takeaway from this study is that receipt of neoadjuvant chemotherapy is not a contraindication for nipple-sparing mastectomy,” said investigator Alex J. Bartholomew, MS, also of Medstar Georgetown University Hospital.
Mr. Bartholomew’s conclusion was based on an analysis of the nipple-sparing mastectomy registry of the American Society of Breast Surgeons that included a total of 3,125 breasts. Neoadjuvant chemotherapy was used in 528, or 16.9%, while primary surgery was performed in 2,597, or 83.1%.
The overall rate of complications was 11%, with nonsignificant differences between the neoadjuvant chemotherapy and primary surgery groups at 12.7% and 10.7%, respectively.
The rate of unintended reoperation, at 4.9%, was not significantly different in the neoadjuvant chemotherapy and primary surgery groups, at 5.2% and 4.8%, Mr. Bartholomew said. Similarly, he found that the rate of nipple areolar complex loss of 1% overall was not different between groups.
Advanced age was likewise not associated with increased complications in the study presented by Dr. Cox, which was a retrospective review of data for patients undergoing nipple-sparing mastectomy from 1998 to 2015 at a single institution. That cohort included 38 patients age 60 years or older, and 358 younger patients.
The rate of complications was 15.5% for patients over age 60 years, and similarly, 13.0% for their younger counterparts (P = .590), Dr. Cox reported. Likewise, the rate of unintended operations was 13.3% and 15.3% for older and younger patients, respectively (P = .274).
These findings are important because advancing age has been associated with a decrease in the likelihood of nipple-sparing mastectomy, according to Dr. Cox.
For mastectomies in general, advanced age has been implicated as a potential risk factor for necrosis, technical complications, and poor outcomes with mastectomies. However, no prior studies had been done specifically to evaluate nipple-sparing mastectomies in older breast cancer patients, Dr. Cox said.
Nipple-sparing mastectomy provides both cosmetic and psychosocial benefits to patients, according to the researchers, because the procedure spares the nipple-areolar complex.
The researchers who had no relevant disclosures.
SOURCES: Cox S et al. SF310 abstract; Bartholomew AJ et al. SF310 abstract ACS Clinical Congress 2018
REPORTING FROM THE ACS CLINICAL CONGRESS
Key clinical point: Neoadjuvant chemotherapy and advancing age were not associated with increased rates of complications in women undergoing nipple-sparing mastectomy in two recent studies.
Major finding: The overall rate of complications was not significantly different for neoadjuvant chemotherapy vs. primary surgery (12.7% vs. 10.7%) or for age over 60 years vs. younger age (15.5% vs. 13.0%).
Study details: Retrospective studies of a nipple-sparing mastectomy registry including more than 3,000 breasts (neoadjuvant chemotherapy vs. primary surgery study) and a single-institution study of nearly 400 patients (older vs. younger study).
Disclosures: The authors reported no conflicts of interest.
Source: Cox S et al. and Bartholomew AJ et al. Session SF310 ACS Clinical Congress 2018.
Striking racial/ethnic differences seen in RCC features
In a southwestern U.S. population having renal cell carcinoma (RCC), patient and disease characteristics differ by race/ethnicity in ways that may have implications for prevention, diagnosis, prognosis, and treatment, finds a single-center cohort study.
Investigators led by Ken Batai, PhD, of University of Arizona, Tucson, retrospectively reviewed the medical records of 294 patients with RCC as their first cancer who underwent partial or radical nephrectomy: 151 European Americans, 95 Hispanic Americans, 22 Native Americans, 9 African Americans, and 17 other race/ethnicity. About 12% overall had metastases at presentation.
On average, compared with European Americans, Hispanic Americans were about 5 years younger at diagnosis (55.8 vs. 60.5) and had higher odds of diagnosis before the age of 50 (odds ratio, 2.77), according to results published in Clinical Genitourinary Cancer.
Native Americans were even younger (49.7) and had dramatically elevated odds of diagnosis before that age (odds ratio, 6.23).
Relative to their European American counterparts, Hispanic Americans less commonly smoked (30.5% vs 48.6%) and African Americans more commonly had chronic kidney disease (37.5% vs. 5.8%). Both groups had higher prevalence of diabetes (45.6% and 54.5% vs. 21.7%). In addition, Native Americans had higher body mass index (35.2 vs. 30.7).
Clear cell histology was seen in 78.8% of European Americans, but in 92.6% of Hispanic Americans (odds ratio, 2.79) and 86.4% of Native Americans. African Americans more commonly had stage III or IV disease at diagnosis (77.8% vs. 35.3%; odds ratio, 6.51), but the racial/ethnic groups did not differ significantly on grade, tumor size, or presence of necrosis.
Among the Hispanic American patients undergoing radical nephrectomy, disease was more commonly of stage III or IV at diagnosis in those who were aged 65 or older (odds ratio, 10.48) and those who spoke Spanish as their primary language (odds ratios, 4.61).
The reasons for the observed racial/ethnic disparities remain unclear, according to Dr. Batai and his coinvestigators. Nonetheless, “it is necessary to better understand the clinical characteristics of these underserved Hispanic American and Native American populations with high kidney cancer burden,” they wrote.
“Our findings can direct future research toward elucidating the difference in tumor behavior among the different ethnic groups and health care issues causing poor outcomes,” they concluded. The findings also “bring ... awareness to practitioners treating patients from these racial/ethnic minority groups regarding the clinical characteristics and underlying issues in these patient populations.”
The study was supported by the American Cancer Society and the Partnership for Native American Cancer Prevention.
SOURCE: Batai K et al. Clin Genitourin Cancer. 2018 Oct 26. doi: 10.1016/j.clgc.2018.10.012.
In a southwestern U.S. population having renal cell carcinoma (RCC), patient and disease characteristics differ by race/ethnicity in ways that may have implications for prevention, diagnosis, prognosis, and treatment, finds a single-center cohort study.
Investigators led by Ken Batai, PhD, of University of Arizona, Tucson, retrospectively reviewed the medical records of 294 patients with RCC as their first cancer who underwent partial or radical nephrectomy: 151 European Americans, 95 Hispanic Americans, 22 Native Americans, 9 African Americans, and 17 other race/ethnicity. About 12% overall had metastases at presentation.
On average, compared with European Americans, Hispanic Americans were about 5 years younger at diagnosis (55.8 vs. 60.5) and had higher odds of diagnosis before the age of 50 (odds ratio, 2.77), according to results published in Clinical Genitourinary Cancer.
Native Americans were even younger (49.7) and had dramatically elevated odds of diagnosis before that age (odds ratio, 6.23).
Relative to their European American counterparts, Hispanic Americans less commonly smoked (30.5% vs 48.6%) and African Americans more commonly had chronic kidney disease (37.5% vs. 5.8%). Both groups had higher prevalence of diabetes (45.6% and 54.5% vs. 21.7%). In addition, Native Americans had higher body mass index (35.2 vs. 30.7).
Clear cell histology was seen in 78.8% of European Americans, but in 92.6% of Hispanic Americans (odds ratio, 2.79) and 86.4% of Native Americans. African Americans more commonly had stage III or IV disease at diagnosis (77.8% vs. 35.3%; odds ratio, 6.51), but the racial/ethnic groups did not differ significantly on grade, tumor size, or presence of necrosis.
Among the Hispanic American patients undergoing radical nephrectomy, disease was more commonly of stage III or IV at diagnosis in those who were aged 65 or older (odds ratio, 10.48) and those who spoke Spanish as their primary language (odds ratios, 4.61).
The reasons for the observed racial/ethnic disparities remain unclear, according to Dr. Batai and his coinvestigators. Nonetheless, “it is necessary to better understand the clinical characteristics of these underserved Hispanic American and Native American populations with high kidney cancer burden,” they wrote.
“Our findings can direct future research toward elucidating the difference in tumor behavior among the different ethnic groups and health care issues causing poor outcomes,” they concluded. The findings also “bring ... awareness to practitioners treating patients from these racial/ethnic minority groups regarding the clinical characteristics and underlying issues in these patient populations.”
The study was supported by the American Cancer Society and the Partnership for Native American Cancer Prevention.
SOURCE: Batai K et al. Clin Genitourin Cancer. 2018 Oct 26. doi: 10.1016/j.clgc.2018.10.012.
In a southwestern U.S. population having renal cell carcinoma (RCC), patient and disease characteristics differ by race/ethnicity in ways that may have implications for prevention, diagnosis, prognosis, and treatment, finds a single-center cohort study.
Investigators led by Ken Batai, PhD, of University of Arizona, Tucson, retrospectively reviewed the medical records of 294 patients with RCC as their first cancer who underwent partial or radical nephrectomy: 151 European Americans, 95 Hispanic Americans, 22 Native Americans, 9 African Americans, and 17 other race/ethnicity. About 12% overall had metastases at presentation.
On average, compared with European Americans, Hispanic Americans were about 5 years younger at diagnosis (55.8 vs. 60.5) and had higher odds of diagnosis before the age of 50 (odds ratio, 2.77), according to results published in Clinical Genitourinary Cancer.
Native Americans were even younger (49.7) and had dramatically elevated odds of diagnosis before that age (odds ratio, 6.23).
Relative to their European American counterparts, Hispanic Americans less commonly smoked (30.5% vs 48.6%) and African Americans more commonly had chronic kidney disease (37.5% vs. 5.8%). Both groups had higher prevalence of diabetes (45.6% and 54.5% vs. 21.7%). In addition, Native Americans had higher body mass index (35.2 vs. 30.7).
Clear cell histology was seen in 78.8% of European Americans, but in 92.6% of Hispanic Americans (odds ratio, 2.79) and 86.4% of Native Americans. African Americans more commonly had stage III or IV disease at diagnosis (77.8% vs. 35.3%; odds ratio, 6.51), but the racial/ethnic groups did not differ significantly on grade, tumor size, or presence of necrosis.
Among the Hispanic American patients undergoing radical nephrectomy, disease was more commonly of stage III or IV at diagnosis in those who were aged 65 or older (odds ratio, 10.48) and those who spoke Spanish as their primary language (odds ratios, 4.61).
The reasons for the observed racial/ethnic disparities remain unclear, according to Dr. Batai and his coinvestigators. Nonetheless, “it is necessary to better understand the clinical characteristics of these underserved Hispanic American and Native American populations with high kidney cancer burden,” they wrote.
“Our findings can direct future research toward elucidating the difference in tumor behavior among the different ethnic groups and health care issues causing poor outcomes,” they concluded. The findings also “bring ... awareness to practitioners treating patients from these racial/ethnic minority groups regarding the clinical characteristics and underlying issues in these patient populations.”
The study was supported by the American Cancer Society and the Partnership for Native American Cancer Prevention.
SOURCE: Batai K et al. Clin Genitourin Cancer. 2018 Oct 26. doi: 10.1016/j.clgc.2018.10.012.
FROM CLINICAL GENITOURINARY CANCER
Key clinical point: .
Major finding: Compared with European Americans, Hispanic Americans and Native Americans were younger at diagnosis (55.8 and 49.7 vs. 60.5 years) and more often had clear cell histology (92.6% and 86.4% vs. 78.8%), and African Americans more often had stage III/IV disease (77.8% vs. 35.3%).
Study details: U.S. single-center retrospective cohort study of 294 patients with RCC who underwent partial or radical nephrectomy.
Disclosures: The authors declared that they did not have any conflicts of interest. The study was supported by the American Cancer Society and the Partnership for Native American Cancer Prevention.
Source: Batai K et al. Clin Genitourin Cancer. 2018 Oct 26. doi: 10.1016/j.clgc.2018.10.012.
Propolis plus L-dopa buffers PD symptoms in rat model
NEW YORK – A bee-derived supplement known to have anti-inflammatory properties showed promise in potentiating the effect of levodopa in a rodent model of Parkinson’s disease, with biochemical and behavioral improvements seen that exceeded high-dose levodopa alone.
Levodopa (L-dopa) has long been a keystone in treatment for individuals with Parkinson’s disease (PD). However, its effectiveness wanes with time and long-term use is associated with significant undesirable side effects, including dyskinesias.
A dopamine precursor, L-dopa, once converted to dopamine, replaces the dopamine no longer made by the substantia nigra. Increasingly, neuroinflammation and oxidative stress are felt to play a role in the natural history of PD, said Azza Ali, PhD, who presented the findings at a poster session of the International Conference on Parkinson’s Disease and Movement Disorders. Whether mitigating these effects can alter the disease course for individuals with PD has not been well investigated, she added.
Dr. Ali and her research group at Al-Azhar University, Cairo, Egypt, where she heads the department of pharmacology and toxicology, are investigating several nutritional and supplement strategies to dampen inflammation. Among the substances she and her colleagues are investigating is propolis, a plant-derived product produced by bees and distinct from honey or beeswax.
Propolis has been shown to have antioxidant properties in addition to other reported health benefits.
Dr. Ali and her colleagues used a rodent model for PD: Rats were dosed with rotenone, which is known to induce a histologically verifiable parkinsonian syndrome. The investigators used six groups of rats; one group was the normal control, while the others all had rotenone-induced parkinsonism.
Of the remaining groups, one rotenone-dosed group was given neither L-dopa nor propolis. Two groups were treated with oral L-dopa alone, at 10 or 25 mg/kg per day. Another group was given an oral dose of 300 mg/kg per day of propolis, and the last group received propolis at that dose while also receiving the lower dose of L-dopa.
All groups were subject to behavioral assessments including a swim test, an open field test, a maze test, and other tasks designed to assess motor function and other aspects of behavior. Additionally, Dr. Ali and her collaborators assessed a variety of biochemical parameters that looked at oxidative stress and neuroinflammation in all rodent groups.
For most parameters, the rotenone-dosed rats that showed results most like normal controls were those who received both low-dose L-dopa and propolis. In particular, the rats receiving both L-dopa and propolis outperformed the other rotenone groups in the behavioral open field test and a grid test, where their performance neared the control group.
Levels of interleukin-1 beta fell for all treated rodents, but fell the most for those treated with the combination. Tissue dopamine levels were also lower for the rats who received combination treatment, and acetylcholinesterase and malondialdehyde levels also fell to near normal for rats receiving the combination treatment, but not for the other groups.
“Propolis is efficient in protection from PD development and represents a suitable adjuvant therapy, which can be translated to serious reduction of the long-term therapy side effects of the mainstay drug L-dopa,” wrote Dr. Ali and her coauthors. “Consequently, propolis could be recommended as a disease-modifying therapy of PD as well as a promising adjunct therapy with L-dopa especially when given early in the treatment course.”
NEW YORK – A bee-derived supplement known to have anti-inflammatory properties showed promise in potentiating the effect of levodopa in a rodent model of Parkinson’s disease, with biochemical and behavioral improvements seen that exceeded high-dose levodopa alone.
Levodopa (L-dopa) has long been a keystone in treatment for individuals with Parkinson’s disease (PD). However, its effectiveness wanes with time and long-term use is associated with significant undesirable side effects, including dyskinesias.
A dopamine precursor, L-dopa, once converted to dopamine, replaces the dopamine no longer made by the substantia nigra. Increasingly, neuroinflammation and oxidative stress are felt to play a role in the natural history of PD, said Azza Ali, PhD, who presented the findings at a poster session of the International Conference on Parkinson’s Disease and Movement Disorders. Whether mitigating these effects can alter the disease course for individuals with PD has not been well investigated, she added.
Dr. Ali and her research group at Al-Azhar University, Cairo, Egypt, where she heads the department of pharmacology and toxicology, are investigating several nutritional and supplement strategies to dampen inflammation. Among the substances she and her colleagues are investigating is propolis, a plant-derived product produced by bees and distinct from honey or beeswax.
Propolis has been shown to have antioxidant properties in addition to other reported health benefits.
Dr. Ali and her colleagues used a rodent model for PD: Rats were dosed with rotenone, which is known to induce a histologically verifiable parkinsonian syndrome. The investigators used six groups of rats; one group was the normal control, while the others all had rotenone-induced parkinsonism.
Of the remaining groups, one rotenone-dosed group was given neither L-dopa nor propolis. Two groups were treated with oral L-dopa alone, at 10 or 25 mg/kg per day. Another group was given an oral dose of 300 mg/kg per day of propolis, and the last group received propolis at that dose while also receiving the lower dose of L-dopa.
All groups were subject to behavioral assessments including a swim test, an open field test, a maze test, and other tasks designed to assess motor function and other aspects of behavior. Additionally, Dr. Ali and her collaborators assessed a variety of biochemical parameters that looked at oxidative stress and neuroinflammation in all rodent groups.
For most parameters, the rotenone-dosed rats that showed results most like normal controls were those who received both low-dose L-dopa and propolis. In particular, the rats receiving both L-dopa and propolis outperformed the other rotenone groups in the behavioral open field test and a grid test, where their performance neared the control group.
Levels of interleukin-1 beta fell for all treated rodents, but fell the most for those treated with the combination. Tissue dopamine levels were also lower for the rats who received combination treatment, and acetylcholinesterase and malondialdehyde levels also fell to near normal for rats receiving the combination treatment, but not for the other groups.
“Propolis is efficient in protection from PD development and represents a suitable adjuvant therapy, which can be translated to serious reduction of the long-term therapy side effects of the mainstay drug L-dopa,” wrote Dr. Ali and her coauthors. “Consequently, propolis could be recommended as a disease-modifying therapy of PD as well as a promising adjunct therapy with L-dopa especially when given early in the treatment course.”
NEW YORK – A bee-derived supplement known to have anti-inflammatory properties showed promise in potentiating the effect of levodopa in a rodent model of Parkinson’s disease, with biochemical and behavioral improvements seen that exceeded high-dose levodopa alone.
Levodopa (L-dopa) has long been a keystone in treatment for individuals with Parkinson’s disease (PD). However, its effectiveness wanes with time and long-term use is associated with significant undesirable side effects, including dyskinesias.
A dopamine precursor, L-dopa, once converted to dopamine, replaces the dopamine no longer made by the substantia nigra. Increasingly, neuroinflammation and oxidative stress are felt to play a role in the natural history of PD, said Azza Ali, PhD, who presented the findings at a poster session of the International Conference on Parkinson’s Disease and Movement Disorders. Whether mitigating these effects can alter the disease course for individuals with PD has not been well investigated, she added.
Dr. Ali and her research group at Al-Azhar University, Cairo, Egypt, where she heads the department of pharmacology and toxicology, are investigating several nutritional and supplement strategies to dampen inflammation. Among the substances she and her colleagues are investigating is propolis, a plant-derived product produced by bees and distinct from honey or beeswax.
Propolis has been shown to have antioxidant properties in addition to other reported health benefits.
Dr. Ali and her colleagues used a rodent model for PD: Rats were dosed with rotenone, which is known to induce a histologically verifiable parkinsonian syndrome. The investigators used six groups of rats; one group was the normal control, while the others all had rotenone-induced parkinsonism.
Of the remaining groups, one rotenone-dosed group was given neither L-dopa nor propolis. Two groups were treated with oral L-dopa alone, at 10 or 25 mg/kg per day. Another group was given an oral dose of 300 mg/kg per day of propolis, and the last group received propolis at that dose while also receiving the lower dose of L-dopa.
All groups were subject to behavioral assessments including a swim test, an open field test, a maze test, and other tasks designed to assess motor function and other aspects of behavior. Additionally, Dr. Ali and her collaborators assessed a variety of biochemical parameters that looked at oxidative stress and neuroinflammation in all rodent groups.
For most parameters, the rotenone-dosed rats that showed results most like normal controls were those who received both low-dose L-dopa and propolis. In particular, the rats receiving both L-dopa and propolis outperformed the other rotenone groups in the behavioral open field test and a grid test, where their performance neared the control group.
Levels of interleukin-1 beta fell for all treated rodents, but fell the most for those treated with the combination. Tissue dopamine levels were also lower for the rats who received combination treatment, and acetylcholinesterase and malondialdehyde levels also fell to near normal for rats receiving the combination treatment, but not for the other groups.
“Propolis is efficient in protection from PD development and represents a suitable adjuvant therapy, which can be translated to serious reduction of the long-term therapy side effects of the mainstay drug L-dopa,” wrote Dr. Ali and her coauthors. “Consequently, propolis could be recommended as a disease-modifying therapy of PD as well as a promising adjunct therapy with L-dopa especially when given early in the treatment course.”
REPORTING FROM ICPDMD 2018
November is Diabetes Awareness Month
A new flier on diabetes and vascular disease is now available in English and Spanish as an instant download. This is the second new flier produced by the SVS Foundation as part of its awareness and prevention mission. Please download and share this important information. It’s a good opportunity to remind your patients of the effects of diabetes on their vascular system. We are offering two versions of PDFs on which you can easily type your office contact information. As you share with other physician referrers and your patients, also send them to our Diabetes Information page, where physicians can find the latest vascular and diabetes research, and patients can find useful health information.
A new flier on diabetes and vascular disease is now available in English and Spanish as an instant download. This is the second new flier produced by the SVS Foundation as part of its awareness and prevention mission. Please download and share this important information. It’s a good opportunity to remind your patients of the effects of diabetes on their vascular system. We are offering two versions of PDFs on which you can easily type your office contact information. As you share with other physician referrers and your patients, also send them to our Diabetes Information page, where physicians can find the latest vascular and diabetes research, and patients can find useful health information.
A new flier on diabetes and vascular disease is now available in English and Spanish as an instant download. This is the second new flier produced by the SVS Foundation as part of its awareness and prevention mission. Please download and share this important information. It’s a good opportunity to remind your patients of the effects of diabetes on their vascular system. We are offering two versions of PDFs on which you can easily type your office contact information. As you share with other physician referrers and your patients, also send them to our Diabetes Information page, where physicians can find the latest vascular and diabetes research, and patients can find useful health information.
Combination supplements show neuroprotective potential in Parkinson’s
NEW YORK – Preclinical studies have shown the natural supplements vinpocetine and pomegranate, along with vitamin B complex and vitamin E, may have some effect individually in providing neuroprotection in Parkinson’s disease, but may have a more profound effect when used in combination, a researcher from Egypt reported at the International Conference on Parkinson’s Disease and Movement Disorders.

“We need to carry out a clinical trial to ensure that this multiple direct strategy can provide protection in different stages of neurodegenerative disease – during induction and even during the progression of the disease,” said Azza Ali, PhD, of Al-Azhar University, Cairo. She presented a poster of her research in an unspecified number of rats with manganese-induced Parkinsonian symptoms. The goal of the study was to compare the oral supplements to each other and to evaluate their impact in combinations. Excessive levels of manganese have been associated with movement disorders similar to Parkinson’s disease.
The research involved histologic studies to evaluate the impact of the supplements in the brains, and evaluated biochemical, neuroinflammatory, apoptotic, and oxidative markers. Behavioral tests evaluated cognition, memory, and motor skills.
Histological studies of manganese-induced brains exhibited nuclear pyknosis – clumping of chromosomes, excessive chromatic aberrations, and shrinkage of the nucleus – in the neurons of the cerebral cortex as well as in some areas of the hippocampus, although no alteration was seen in the subiculum, Dr. Ali reported. The stria showed multiple plaque formations with nuclear pyknosis and degeneration in some neurons.
All the studied treatments improved motor, memory, and cognitive decline induced by manganese, with pomegranate and vinpocetine yielding the best results, Dr. Ali said. However, a combination of treatments showed more pronounced improvements in some biochemical markers, as well as the neuroinflammatory, apoptotic, and oxidative markers. “They have a high antioxidant and antiapoptotic effect,” Dr. Ali said. Histopathologic studies confirmed those results, she noted.
Pomegranate (150 mg/kg) had a somewhat positive effect in the subiculum and fascia dentate areas of the hippocampus, although the stria appeared similar to manganese-induced brains. With vinpocetine (20 mg/kg), neurons in the cerebral cortex showed intact histological structure with some degeneration and nuclear pyknosis in the subiculum. There was no alteration in the neurons of the fascia dentate, hilus, and stria of the hippocampus.
Histologic studies of induced brains after treatment with vitamin-B complex (8.5 mg/kg) showed nuclear pyknosis and degeneration in the neurons of the cerebral cortex and hippocampus, including the subiculum. The stria also showed multiple focal eosinophilic plaques with nuclear pyknosis and degeneration in some neurons. Vitamin E (100 mg/kg) resulted in intact neurons in the cerebral cortex but not in the hippocampus.
Histopathologic studies of brains that received combination treatment showed no alteration in the neurons of the cerebral cortex or in the subiculum and fascia dentate of the hippocampus, although a few neurons in the stria showed nuclear pyknosis and degeneration, Dr. Ali said.
She had no financial relationships to disclose.
NEW YORK – Preclinical studies have shown the natural supplements vinpocetine and pomegranate, along with vitamin B complex and vitamin E, may have some effect individually in providing neuroprotection in Parkinson’s disease, but may have a more profound effect when used in combination, a researcher from Egypt reported at the International Conference on Parkinson’s Disease and Movement Disorders.
“We need to carry out a clinical trial to ensure that this multiple direct strategy can provide protection in different stages of neurodegenerative disease – during induction and even during the progression of the disease,” said Azza Ali, PhD, of Al-Azhar University, Cairo. She presented a poster of her research in an unspecified number of rats with manganese-induced Parkinsonian symptoms. The goal of the study was to compare the oral supplements to each other and to evaluate their impact in combinations. Excessive levels of manganese have been associated with movement disorders similar to Parkinson’s disease.
The research involved histologic studies to evaluate the impact of the supplements in the brains, and evaluated biochemical, neuroinflammatory, apoptotic, and oxidative markers. Behavioral tests evaluated cognition, memory, and motor skills.
Histological studies of manganese-induced brains exhibited nuclear pyknosis – clumping of chromosomes, excessive chromatic aberrations, and shrinkage of the nucleus – in the neurons of the cerebral cortex as well as in some areas of the hippocampus, although no alteration was seen in the subiculum, Dr. Ali reported. The stria showed multiple plaque formations with nuclear pyknosis and degeneration in some neurons.
All the studied treatments improved motor, memory, and cognitive decline induced by manganese, with pomegranate and vinpocetine yielding the best results, Dr. Ali said. However, a combination of treatments showed more pronounced improvements in some biochemical markers, as well as the neuroinflammatory, apoptotic, and oxidative markers. “They have a high antioxidant and antiapoptotic effect,” Dr. Ali said. Histopathologic studies confirmed those results, she noted.
Pomegranate (150 mg/kg) had a somewhat positive effect in the subiculum and fascia dentate areas of the hippocampus, although the stria appeared similar to manganese-induced brains. With vinpocetine (20 mg/kg), neurons in the cerebral cortex showed intact histological structure with some degeneration and nuclear pyknosis in the subiculum. There was no alteration in the neurons of the fascia dentate, hilus, and stria of the hippocampus.
Histologic studies of induced brains after treatment with vitamin-B complex (8.5 mg/kg) showed nuclear pyknosis and degeneration in the neurons of the cerebral cortex and hippocampus, including the subiculum. The stria also showed multiple focal eosinophilic plaques with nuclear pyknosis and degeneration in some neurons. Vitamin E (100 mg/kg) resulted in intact neurons in the cerebral cortex but not in the hippocampus.
Histopathologic studies of brains that received combination treatment showed no alteration in the neurons of the cerebral cortex or in the subiculum and fascia dentate of the hippocampus, although a few neurons in the stria showed nuclear pyknosis and degeneration, Dr. Ali said.
She had no financial relationships to disclose.
NEW YORK – Preclinical studies have shown the natural supplements vinpocetine and pomegranate, along with vitamin B complex and vitamin E, may have some effect individually in providing neuroprotection in Parkinson’s disease, but may have a more profound effect when used in combination, a researcher from Egypt reported at the International Conference on Parkinson’s Disease and Movement Disorders.
“We need to carry out a clinical trial to ensure that this multiple direct strategy can provide protection in different stages of neurodegenerative disease – during induction and even during the progression of the disease,” said Azza Ali, PhD, of Al-Azhar University, Cairo. She presented a poster of her research in an unspecified number of rats with manganese-induced Parkinsonian symptoms. The goal of the study was to compare the oral supplements to each other and to evaluate their impact in combinations. Excessive levels of manganese have been associated with movement disorders similar to Parkinson’s disease.
The research involved histologic studies to evaluate the impact of the supplements in the brains, and evaluated biochemical, neuroinflammatory, apoptotic, and oxidative markers. Behavioral tests evaluated cognition, memory, and motor skills.
Histological studies of manganese-induced brains exhibited nuclear pyknosis – clumping of chromosomes, excessive chromatic aberrations, and shrinkage of the nucleus – in the neurons of the cerebral cortex as well as in some areas of the hippocampus, although no alteration was seen in the subiculum, Dr. Ali reported. The stria showed multiple plaque formations with nuclear pyknosis and degeneration in some neurons.
All the studied treatments improved motor, memory, and cognitive decline induced by manganese, with pomegranate and vinpocetine yielding the best results, Dr. Ali said. However, a combination of treatments showed more pronounced improvements in some biochemical markers, as well as the neuroinflammatory, apoptotic, and oxidative markers. “They have a high antioxidant and antiapoptotic effect,” Dr. Ali said. Histopathologic studies confirmed those results, she noted.
Pomegranate (150 mg/kg) had a somewhat positive effect in the subiculum and fascia dentate areas of the hippocampus, although the stria appeared similar to manganese-induced brains. With vinpocetine (20 mg/kg), neurons in the cerebral cortex showed intact histological structure with some degeneration and nuclear pyknosis in the subiculum. There was no alteration in the neurons of the fascia dentate, hilus, and stria of the hippocampus.
Histologic studies of induced brains after treatment with vitamin-B complex (8.5 mg/kg) showed nuclear pyknosis and degeneration in the neurons of the cerebral cortex and hippocampus, including the subiculum. The stria also showed multiple focal eosinophilic plaques with nuclear pyknosis and degeneration in some neurons. Vitamin E (100 mg/kg) resulted in intact neurons in the cerebral cortex but not in the hippocampus.
Histopathologic studies of brains that received combination treatment showed no alteration in the neurons of the cerebral cortex or in the subiculum and fascia dentate of the hippocampus, although a few neurons in the stria showed nuclear pyknosis and degeneration, Dr. Ali said.
She had no financial relationships to disclose.
REPORTING FROM ICPDMD 2018
Playing harmonica improves COPD
SAN ANTONIO – Playing while also boosting their quality of life, suggest the findings from a small pilot study.

Three months of playing the harmonica about a half hour a day most days of the week led to several improved pulmonary outcome measures in participants, Mary Hart, RRT, MS, of Baylor Scott & White Health in Dallas, reported at the annual meeting of the American College of Chest Physicians.
Ms. Hart played a bit of harmonica during her presentation to demonstrate how playing can help with breathing.
“The harder I push with my diaphragm, the louder I was blowing,” she told attendees. “There’s actually a different amount of effort that you have to use to create sounds with using the harmonica notes.”
Hart said her team found a news article from 1999 about the benefits of playing harmonica, and they became interested in exploring whether it might be a helpful adjunct to respiratory therapy.
Though some previous research has explored potential benefits of harmonica playing in patients with lung disease, one study was too short to demonstrate significant improvement and the other looked at multiple different pulmonary conditions, Ms. Hart said.
The cohort study began with 14 former smokers, average age 72 years, who had completed pulmonary rehabilitation at least 6 months prior to joining the “Harmaniacs,” as the group eventually called themselves.
All participants received a harmonica, an instruction booklet with audio and video supplements, and sheet music for a harmonica in the key of C.
They attended a 2-hour group session once a week with a respiratory therapist and music therapist. The classes focused initially on breathing and relaxation techniques, pacing, and basic harmonica instruction, but the amount of actual playing time increased as the 12-week course went on. Participants were expected to practice their playing for at least a half hour 5 days a week at home.
The group began with the songs “Taps” and “Happy Birthday” because these songs were easy to play. Then they added a song each week, such as “America the Beautiful” and “You Are My Sunshine,” then seasonal favorites such as “We Wish You a Merry Christmas” and “Silent Night,” and easy pop tunes.
The researchers measured both respiratory and quality of life outcomes. Assessments included spirometry, the Six Minute Walk Test, maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP), the COPD Assessment Test, the modified Medical Research Council Dyspnea Scale, the Patient Health Questionnaire for depression, the St. George’s Respiratory Questionnaire for quality of life, perceived exertion using the Borg scale and assessments by the respiratory therapist and music therapist.
The music therapist listened to and documented participants’ “stories about how they felt about life living with COPD,” and Ms. Hart and her colleagues conducted a respiratory assessment that included data on medication management, adherence to medication, previous hospitalizations and length of stay, perceived shortness of breath, and daily living activities.
In addition to those assessments, the researchers collected data on the length of practice sessions, Borg scores before and after playing, the percentage of time taken for participation in class, the participants’ ability to make a sound, their challenges and triumphs, their tiredness and/or soreness after playing, and the number of people who continued playing after training.
Among the 11 participants who completed the training and all evaluations, the MIP increased by an average 15.36 cmH20 (P = .0017), and their MEP increased by an average 14.36 cmH20 (P = .0061).
Participants increased their distance in the Six Minute Walk Test by an average 60.55 meters (P = .0280), and Ms. Hart reported an improvement in quality of life scores.
In addition to home practice, participants were expected to keep a daily log of how it felt to play and what their biggest challenges and rewards were. The comments they wrote revealed benefits that sometimes surprised even the researchers:
“I can do laundry now.”
“I am more confident.”
“It is relaxing.”
“I want to keep playing forever.”
“It helps me cough up phlegm.”
“I lose track of time and enjoy my playing.”
“I played Happy Birthday at a party for my friend.”
Others express their difficulties as well, such as one person who wrote of being “really frustrated” and another who claimed to “have a hard time playing just one note.”
But the players learned to play as a group as well, even ordering T-shirts for themselves to give concerts. The group now has about 30 songs in its repertoire, Ms. Hart said, and they recently gave a 2-hour concert during which they played all 30 songs twice.
One consistent theme that emerged, Ms. Hart said, was improved control of breathing since playing the harmonica required participants to purse their lips (similar to the way needed for expiratory maneuvers), breathe from their diaphragms, and pace themselves. Playing exercised “the muscles that help pull air in and push air out of the lungs,” Ms. Hart said, and strengthened participants’ abdominal muscles, allowing more effective coughing.
Playing harmonica also increased self-confidence. It provided stress relief for some, and others simply found it fun or enjoyed the socializing opportunities.
The study’s small size and lack of a control group limit the generalizability of its findings.
Baylor Scott & White Central Texas Foundation funded the research. Ms. Hart reported no conflicts of interest.
SOURCE: Hart M et al. CHEST 2018. doi: 10.1016/j.chest.2018.08.669.
SAN ANTONIO – Playing while also boosting their quality of life, suggest the findings from a small pilot study.
Three months of playing the harmonica about a half hour a day most days of the week led to several improved pulmonary outcome measures in participants, Mary Hart, RRT, MS, of Baylor Scott & White Health in Dallas, reported at the annual meeting of the American College of Chest Physicians.
Ms. Hart played a bit of harmonica during her presentation to demonstrate how playing can help with breathing.
“The harder I push with my diaphragm, the louder I was blowing,” she told attendees. “There’s actually a different amount of effort that you have to use to create sounds with using the harmonica notes.”
Hart said her team found a news article from 1999 about the benefits of playing harmonica, and they became interested in exploring whether it might be a helpful adjunct to respiratory therapy.
Though some previous research has explored potential benefits of harmonica playing in patients with lung disease, one study was too short to demonstrate significant improvement and the other looked at multiple different pulmonary conditions, Ms. Hart said.
The cohort study began with 14 former smokers, average age 72 years, who had completed pulmonary rehabilitation at least 6 months prior to joining the “Harmaniacs,” as the group eventually called themselves.
All participants received a harmonica, an instruction booklet with audio and video supplements, and sheet music for a harmonica in the key of C.
They attended a 2-hour group session once a week with a respiratory therapist and music therapist. The classes focused initially on breathing and relaxation techniques, pacing, and basic harmonica instruction, but the amount of actual playing time increased as the 12-week course went on. Participants were expected to practice their playing for at least a half hour 5 days a week at home.
The group began with the songs “Taps” and “Happy Birthday” because these songs were easy to play. Then they added a song each week, such as “America the Beautiful” and “You Are My Sunshine,” then seasonal favorites such as “We Wish You a Merry Christmas” and “Silent Night,” and easy pop tunes.
The researchers measured both respiratory and quality of life outcomes. Assessments included spirometry, the Six Minute Walk Test, maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP), the COPD Assessment Test, the modified Medical Research Council Dyspnea Scale, the Patient Health Questionnaire for depression, the St. George’s Respiratory Questionnaire for quality of life, perceived exertion using the Borg scale and assessments by the respiratory therapist and music therapist.
The music therapist listened to and documented participants’ “stories about how they felt about life living with COPD,” and Ms. Hart and her colleagues conducted a respiratory assessment that included data on medication management, adherence to medication, previous hospitalizations and length of stay, perceived shortness of breath, and daily living activities.
In addition to those assessments, the researchers collected data on the length of practice sessions, Borg scores before and after playing, the percentage of time taken for participation in class, the participants’ ability to make a sound, their challenges and triumphs, their tiredness and/or soreness after playing, and the number of people who continued playing after training.
Among the 11 participants who completed the training and all evaluations, the MIP increased by an average 15.36 cmH20 (P = .0017), and their MEP increased by an average 14.36 cmH20 (P = .0061).
Participants increased their distance in the Six Minute Walk Test by an average 60.55 meters (P = .0280), and Ms. Hart reported an improvement in quality of life scores.
In addition to home practice, participants were expected to keep a daily log of how it felt to play and what their biggest challenges and rewards were. The comments they wrote revealed benefits that sometimes surprised even the researchers:
“I can do laundry now.”
“I am more confident.”
“It is relaxing.”
“I want to keep playing forever.”
“It helps me cough up phlegm.”
“I lose track of time and enjoy my playing.”
“I played Happy Birthday at a party for my friend.”
Others express their difficulties as well, such as one person who wrote of being “really frustrated” and another who claimed to “have a hard time playing just one note.”
But the players learned to play as a group as well, even ordering T-shirts for themselves to give concerts. The group now has about 30 songs in its repertoire, Ms. Hart said, and they recently gave a 2-hour concert during which they played all 30 songs twice.
One consistent theme that emerged, Ms. Hart said, was improved control of breathing since playing the harmonica required participants to purse their lips (similar to the way needed for expiratory maneuvers), breathe from their diaphragms, and pace themselves. Playing exercised “the muscles that help pull air in and push air out of the lungs,” Ms. Hart said, and strengthened participants’ abdominal muscles, allowing more effective coughing.
Playing harmonica also increased self-confidence. It provided stress relief for some, and others simply found it fun or enjoyed the socializing opportunities.
The study’s small size and lack of a control group limit the generalizability of its findings.
Baylor Scott & White Central Texas Foundation funded the research. Ms. Hart reported no conflicts of interest.
SOURCE: Hart M et al. CHEST 2018. doi: 10.1016/j.chest.2018.08.669.
SAN ANTONIO – Playing while also boosting their quality of life, suggest the findings from a small pilot study.
Three months of playing the harmonica about a half hour a day most days of the week led to several improved pulmonary outcome measures in participants, Mary Hart, RRT, MS, of Baylor Scott & White Health in Dallas, reported at the annual meeting of the American College of Chest Physicians.
Ms. Hart played a bit of harmonica during her presentation to demonstrate how playing can help with breathing.
“The harder I push with my diaphragm, the louder I was blowing,” she told attendees. “There’s actually a different amount of effort that you have to use to create sounds with using the harmonica notes.”
Hart said her team found a news article from 1999 about the benefits of playing harmonica, and they became interested in exploring whether it might be a helpful adjunct to respiratory therapy.
Though some previous research has explored potential benefits of harmonica playing in patients with lung disease, one study was too short to demonstrate significant improvement and the other looked at multiple different pulmonary conditions, Ms. Hart said.
The cohort study began with 14 former smokers, average age 72 years, who had completed pulmonary rehabilitation at least 6 months prior to joining the “Harmaniacs,” as the group eventually called themselves.
All participants received a harmonica, an instruction booklet with audio and video supplements, and sheet music for a harmonica in the key of C.
They attended a 2-hour group session once a week with a respiratory therapist and music therapist. The classes focused initially on breathing and relaxation techniques, pacing, and basic harmonica instruction, but the amount of actual playing time increased as the 12-week course went on. Participants were expected to practice their playing for at least a half hour 5 days a week at home.
The group began with the songs “Taps” and “Happy Birthday” because these songs were easy to play. Then they added a song each week, such as “America the Beautiful” and “You Are My Sunshine,” then seasonal favorites such as “We Wish You a Merry Christmas” and “Silent Night,” and easy pop tunes.
The researchers measured both respiratory and quality of life outcomes. Assessments included spirometry, the Six Minute Walk Test, maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP), the COPD Assessment Test, the modified Medical Research Council Dyspnea Scale, the Patient Health Questionnaire for depression, the St. George’s Respiratory Questionnaire for quality of life, perceived exertion using the Borg scale and assessments by the respiratory therapist and music therapist.
The music therapist listened to and documented participants’ “stories about how they felt about life living with COPD,” and Ms. Hart and her colleagues conducted a respiratory assessment that included data on medication management, adherence to medication, previous hospitalizations and length of stay, perceived shortness of breath, and daily living activities.
In addition to those assessments, the researchers collected data on the length of practice sessions, Borg scores before and after playing, the percentage of time taken for participation in class, the participants’ ability to make a sound, their challenges and triumphs, their tiredness and/or soreness after playing, and the number of people who continued playing after training.
Among the 11 participants who completed the training and all evaluations, the MIP increased by an average 15.36 cmH20 (P = .0017), and their MEP increased by an average 14.36 cmH20 (P = .0061).
Participants increased their distance in the Six Minute Walk Test by an average 60.55 meters (P = .0280), and Ms. Hart reported an improvement in quality of life scores.
In addition to home practice, participants were expected to keep a daily log of how it felt to play and what their biggest challenges and rewards were. The comments they wrote revealed benefits that sometimes surprised even the researchers:
“I can do laundry now.”
“I am more confident.”
“It is relaxing.”
“I want to keep playing forever.”
“It helps me cough up phlegm.”
“I lose track of time and enjoy my playing.”
“I played Happy Birthday at a party for my friend.”
Others express their difficulties as well, such as one person who wrote of being “really frustrated” and another who claimed to “have a hard time playing just one note.”
But the players learned to play as a group as well, even ordering T-shirts for themselves to give concerts. The group now has about 30 songs in its repertoire, Ms. Hart said, and they recently gave a 2-hour concert during which they played all 30 songs twice.
One consistent theme that emerged, Ms. Hart said, was improved control of breathing since playing the harmonica required participants to purse their lips (similar to the way needed for expiratory maneuvers), breathe from their diaphragms, and pace themselves. Playing exercised “the muscles that help pull air in and push air out of the lungs,” Ms. Hart said, and strengthened participants’ abdominal muscles, allowing more effective coughing.
Playing harmonica also increased self-confidence. It provided stress relief for some, and others simply found it fun or enjoyed the socializing opportunities.
The study’s small size and lack of a control group limit the generalizability of its findings.
Baylor Scott & White Central Texas Foundation funded the research. Ms. Hart reported no conflicts of interest.
SOURCE: Hart M et al. CHEST 2018. doi: 10.1016/j.chest.2018.08.669.
REPORTING FROM CHEST 2018
Key clinical point: Playing harmonica improved pulmonary and quality of life outcomes in patients with COPD.
Major finding: Maximal inspiratory pressure increased by an average 15.36 cmH20, maximal expiratory pressure increased by an average 14.36 cmH20, and Six Minute Walk Test distance increased by an average 60.55 meters.
Data source: Cohort study completed by 11 participants with COPD, at least 45 years old, who completed a 12-week harmonica training course.
Disclosures: Baylor Scott & White Central Texas Foundation funded the research. Ms. Hart reported no conflicts of interest.
Source: Hart M et al. CHEST 2018. 10.1016/j.chest.2018.08.669.
Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) Polypharmacy Clinic
An interprofessional polypharmacy clinic for intensive management of medication regimens helps high-risk patients manage their medications.
In 2011, 5 VA medical centers (VAMCs) were selected by the Office of Academic Affiliations (OAA) to establish CoEPCE. Part of the VA New Models of Care initiative, the 5 Centers of Excellence (CoE) in Boise, Idaho; Cleveland, Ohio; San Francisco, California; Seattle, Washington; and West Haven, Connecticut, are utilizing VA primary care settings to develop and test innovative approaches to prepare physician residents and students, advanced practice nurse residents and undergraduate nursing students, and other professions of health trainees (eg, pharmacy, social work, psychology, physician assistants [PAs], physical therapists) for primary care practice in the 21st century. The CoEs are developing, implementing, and evaluating curricula designed to prepare learners from relevant professions to practice in patient-centered, interprofessional team-based primary care settings. The curricula at all CoEs must address 4 core domains (Table).
Health care professional education programs do not have many opportunities for workplace learning where trainees from different professions can learn and work together to provide care to patients in real time. 
The VA Connecticut Healthcare System CoEPCE developed and implemented an education and practice-based immersion learning model with physician residents, nurse practitioner (NP) residents and NP students, pharmacy residents, postdoctorate psychology learners, and PA and physical therapy learners and faculty. This interprofessional, collaborative team model breaks from the traditional independent model of siloed primary care providers (PCPs) caring for a panel of patients.
Methods
In 2015, OAA evaluators reviewed background documents and conducted open-ended interviews with 12 West Haven CoEPCE staff, participating trainees, VA faculty, VA facility leadership, and affiliate faculty. Informants described their involvement, challenges encountered, and benefits of the Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) program to trainees, veterans, and the VA.
Lack of Clinical Approaches to Interprofessional Education and Care
Polypharmacy is a common problem among older adults with multiple chronic conditions, which places patients at higher risk for multiple negative health outcomes.2,3 The typical primary care visit rarely allows for a thorough review of a patient’s medications, much less the identification of strategies to reduce polypharmacy and improve medication management. Rather, the complexity inherent to polypharmacy makes it an ideal challenge for a team-based approach.
Team Approach to Medication Needs
A key CoEPCE program aim is to expand workplace learning instruction strategies and to create more clinical opportunities for CoEPCE trainees to work together as a team to anticipate and address the health care needs of veterans. To address this training need, the West Haven CoEPCE developed IMPROVE to focus on high-need patients and provides a venue in which trainees and supervisors from different professions can collaborate on a specific patient case, using a patient-centered framework. IMPROVE can be easily applied to a range of medication-related aims, such as reducing medications, managing medications and adherence, and addressing adverse effects (AEs). These goals are 2-fold: (1) implement a trainee-led performance improvement project that reduces polypharmacy in elderly veterans; and (2) develop a hands-on, experiential geriatrics training program that enhances trainee skills and knowledge related to safe prescribing.
Planning and Implementation
IMPROVE has its origins in a scholarly project developed by a West Haven CoE physician resident trainee. Development of the IMPROVE program involved VA health psychology, internal medicine faculty, geriatric medicine faculty, NP faculty, and geriatric pharmacy residents and faculty. Planning started in 2013 with a series of pilot clinics and became an official project of the West Haven CoE in September 2014. The intervention required no change in West Haven VAMC policy. However, the initiative required buy-in from West Haven CoE leadership and the director of the West Haven primary care clinic.
Curriculum
IMPROVE is an educational, workplace learning, and clinical activity that combines a 1-hour trainee teaching session, a 45-minute group visit, and a 60-minute individual clinic visit to address the complex problem of polypharmacy. It emphasizes the sharing of trainee and faculty backgrounds by serving as a venue for interprofessional trainees and providers to discuss pharmacologic and nonpharmacologic treatment in the elderly and brainstorm strategies to optimize treatment regimens, minimize risk, and execute medication plans with patients.
All CoEPCE trainees in West Haven are required to participate in IMPROVE and on average, each trainee presents and sees one of their patients at least 3 times per year in the program. Up to 5 trainees participate in each IMPROVE session. Trainees are responsible for reviewing their panels to identify patients who might benefit from participation, followed by inviting the patient to participate. Patients are instructed to bring their pill bottles to the visit. To prepare for the polypharmacy clinic, the trainees, the geriatrician, and the geriatric pharmacist perform an extensive medication chart review, using the medication review worksheet developed by West Haven VAMC providers.4 They also work with a protocol for medication discontinuation, which was compiled by West Haven VAMC clinicians. The teams use a variety of tools that guide appropriate prescribing in older adult populations.5,6 During a preclinic conference, trainees present their patients to the interprofessional team for discussion and participate in a short discussion led by a pharmacist, geriatrician, or health psychologist on a topic related to prescribing safety in older adults or nonpharmacologic treatments.
IMPROVE emphasizes a patient-centered approach to develop, execute, and monitor medication plans. Patients and their family members are invited by their trainee clinician to participate in a group visit. Typically, trainees invite patients aged ≥ 65 years who have ≥ 10 medications and are considered appropriate for a group visit. 
The group visit process is based on health psychology strategies, which often incorporate group-based engagement with patients. The health psychologist can give advice to facilitate the visit and optimize participant involvement. There is a discussion facilitator guide that lists the education points to be covered by a designated trainee facilitator and sample questions to guide the discussion.7 A health psychology resident and other rotating trainees cofacilitate the group visit with a goal to reach out to each group member, including family members, and have them discuss perceptions and share concerns and treatment goals. There is shared responsibility among the trainees to address the educational material as well as involve their respective patients during the sessions.
Immediately following the group visit, trainees conduct a 1-hour clinic session that includes medication reconciliation, a review of an IMPROVE questionnaire, orthostatic vital signs, and the St. Louis University Mental Status (SLUMS) exam to assess changes in cognition.7,8 Discussion involved the patient’s medication list as well as possible changes that could be made to the list. Using shared decision-making techniques, this conversation considers the patients’ treatment goals, feelings about the medications, which medications they would like to stop, and AEs they may be experiencing. After the individual visit is completed, the trainee participates in a 10-minute interprofessional precepting session, which may include a geriatrician, a pharmacist, and a health psychologist. In the session they may discuss adjustments to medications and a safe follow-up plan, including appropriate referrals. Trainees discuss the plan with the patient and send a letter describing the plan shortly after the visit.
IMPROVE combines didactic teaching with experiential education. It embodies the 4 core domains that shape the CoEPCE curriculum. First, trainees learn interprofessional collaboration concepts, including highlighting the roles of each profession and working with an interprofessional team to solve problems. Second, CoEPCE trainees learn performance improvement under the supervision of faculty. Third, IMPROVE allows trainees to develop sustained relationships with other team members while improving the quality of the clinic experience as well as with patients through increased continuity of care. Trainees see patients on their panel and are responsible for outreach before and after the visit. Finally, with a focus on personalized patient goals, trainees have the opportunity to further develop skills in shared decision making (SDM).
Related: Reducing Benzodiazepine Prescribing in Older Veterans: A Direct-to-Consumer Educational Brochure
The IMPROVE model continues to evolve. The original curriculum involved an hour-long preclinic preparation session before the group visit in which trainees and faculty discussed the medication review for each patient scheduled that day. This preparation session was later shortened to 40 minutes, and a 20-minute didactic component was added to create the current preclinic session. The didactic component focused on a specific topic in appropriate prescribing for older patients. For example, one didactic lesson is on a particular class of medications, its common AEs, and practical prescribing and “deprescribing” strategies for that class. Initially, the oldest patients or patients who could be grouped thematically, such as those taking both narcotics and benzodiazepines, were invited to participate, but that limited the number of appropriate patients within the CoEPCE. Currently, trainees identify patients from their panels who might benefit, based on age, number of medications, or potential medication-related concerns, such as falls, cognitive impairment, or other concerns for adverse drug effects. These trainees have the unique opportunity to apply learned strategies to their patients to continue to optimize the medication regimen even after the IMPROVE visit. Another significant change was the inclusion of veterans who are comanaged with PCPs outside the VA, because we found that patients with multiple providers could benefit from improved coordination of care.
Faculty Role
CoE faculty and non-CoE VA faculty participate in supervisory, consulting, teaching and precepting roles. Some faculty members such as the health psychologists are already located in or near the VA primary care clinic, so they can assist in curriculum development and execution during their regular clinic duties. The geriatrician reviews the patients’ health records before the patients come into the clinic, participates in the group visit, and coprecepts during the 1:1 patient visits. Collaboration is inherent in IMPROVE. For example, the geriatrician works with the geriatric pharmacist to identify and teach an educational topic. IMPROVE is characterized by a strong faculty/trainee partnership, with trainees playing roles as both teacher and facilitator in addition to learning how to take a team approach to polypharmacy.
Resources
IMPROVE requires administrative and academic support, especially faculty and trainee preparation of education sessions. The CoEPCE internal medicine resident and the internal medicine chief resident work with the health technicians for each patient aligned care team (PACT) to enter the information into the VA medical scheduling system. Trainee clinic time is blocked for their group visits in advance. Patients are scheduled 1 to 3 weeks in advance. Trainees and faculty are expected to review the medication review worksheet and resources prior to the visit. One CoEPCE faculty member reviews patients prior to the preclinic session (about an hour of preparation per session). Sufficient space also is required: a room large enough to accommodate up to 10 people for both didactic lessons and preclinic sessions, a facility patient education conference room for the group visit, and up to 5 clinic exam rooms. CoEPCE staff developed a templated note in the VA Computerized Patient Record System (CPRS), the VA electronic health record system to guide trainees step-by-step through the clinic visit and allow them to directly enter information into the system.7
Monitoring and Assessment
CoEPCE staff are evaluating IMPROVE by building a database for patient-level and trainee-level outcomes, including changes in trainee knowledge and attitudes over time. The CoEPCE also validated the polypharmacy knowledge assessment tool for medicine and NP trainees.
Partnerships
IMPROVE has greatly benefited from partnerships with facility department leadership, particularly involvement of pharmacy staff. In addition, we have partnered with both the health psychology and pharmacy faculty and trainees to participate in the program. Geriatrics faculty and trainees also have contributed extensively to IMPROVE. Future goals include offering the program to non-COEPCE patients throughout primary care.
The Yale Primary Care Internal Medicine Residency program and the Yale Categorical Internal Medicine Residency Program are integral partners to the CoEPCE. IMPROVE supports their mandate to encourage interprofessional teamwork in primary care, meet the Accreditation Council for Graduate Medical Education interprofessional milestones, and promote individual trainee scholarship and performance improvement in areas of broad applicability. IMPROVE also is an opportunity to share ideas across institutions and stimulate new collaborations and dissemination of the model to other primary care settings outside the VA.
Challenges and Solutions
The demand for increased direct patient care pressures programs like IMPROVE, which is a time-intensive process with high impact on a few complex patients. The assumption is that managing medications will save money in the long run, but in the short-term, a strong case has to be made for securing resources, particularly blocking provider time and securing an education room for group visits and clinic exam rooms for individual visits. First, decision makers need to be convinced that polypharmacy is important and should be a training priority. The CoEPCE has tried different configurations to increase the number of patients being seen, such as having ≥ 1 IMPROVE session in an afternoon, but trainees found this to be labor intensive and stressful.
Second, patients with medications prescribed by providers outside the VA require additional communication and coordination to reduce medications. The CoEPCE initially excluded these patients, but after realizing that some of these patients needed the most help, it developed a process for reaching out to non-VA providers and coordinating care. Additionally, there is significant diversity in patient polypharmacy needs. These can range from adherence problems to the challenge of complex psychosocial needs that are more easily (but less effectively) addressed with medications. The issue of polypharmacy is further complicated by evolving understanding of medications’ relative risks and benefits in older adults with multiple chronic conditions. IMPROVE is an effective vehicle for synthesizing current science in medications and their management, especially in complex older patients with multiple chronic conditions.
Other challenges include developing a templated CPRS electronic note that interfaces with the VA information technology system. The process of creating a template, obtaining approval from the forms committee, and working with information technology personnel to implement the template was more time intensive than anticipated and required multiple iterations of proofreading and editing.
Factors for Success
The commitment to support new models of trainee education by West Haven CoEPCE faculty and leadership, and West Haven VAMC and primary care clinic leadership facilitated the implementation of IMPROVE. Additionally, there is strong CoEPCE collaboration at all levels—codirectors, faculty, and trainees—for the program. High interprofessional trainee interest, organizational insight, and an academic orientation were critical for developing and launching IMPROVE.
Additionally, there is synergy with other team-based professions. Geriatrics has a tradition of working in multidisciplinary teams as well as working with SDM concepts as part of care discussions. High interest and collaboration by a geriatrician and an experienced geriatric pharmacist has been key. The 2 specialties complement each other and address the complex health needs of participating veterans. Health psychologists transition patients to nonpharmacologic treatments, such as sleep hygiene education and cognitive behavioral therapy, in addition to exploring barriers to behavior change.
Another factor for success has been the CoEPCE framework and expertise in interprofessional education. While refining the model, program planners tapped into existing expertise in polypharmacy within the VA from the geriatrics, pharmacy, and clinical health psychology departments. The success of the individual components—the preparation session, the group visit, and the 1:1 patient visit—is in large part the result of a collective effort by CoEPCE staff and the integration of CoEPCE staff through coordination, communications, logistics, quality improvement, and faculty involvement from multiple professions.
The IMPROVE model is flexible and can accommodate diverse patient interests and issues. Model components are based on sound practices that have demonstrated success in other arenas, such as diabetes mellitus group visits. The model can also accommodate diverse trainee levels. Senior trainees can be more independent in developing their care plans, teaching the didactic topic, or precepting during the 1:1 patient exam.
Accomplishments and Benefits
Trainees are using team skills to provide patient-centered care. They are strengthening their clinical skills through exposure to patients in a group visit and 1:1 clinic visit. There have been significant improvements in the trainees’ provision of individual patient care. Key IMPROVE outcomes are outlined below.
Interprofessional Education
Unlike a traditional didactic, IMPROVE is an opportunity for health care professionals to work together to provide care in a clinic setting. It also expands CoEPCE interprofessional education capacity through colocation of different trainee and faculty professions during the conference session. This combination trains participants to work as a team and reflect on patients together, which has strengthened communications among professions. The model provides sufficient time and expertise to discuss the medications in detail and as a team, something that would not normally happen during a regular primary care visit.
CoEPCE trainees learn about medication management, its importance in preventing complications and improving patient health outcomes. Trainees of all professions learn to translate the skills they learn in IMPROVE to other patients, such as how to perform a complete medication reconciliation or lead a discussion using SDM. IMPROVE also provides techniques useful in other contexts, such as group visits and consideration of different medication options for patients who have been cared for by other (VA and non-VA) providers.
Interprofessional Collaboration
Understanding and leveraging the expertise of trainees and faculty from different professions is a primary goal of IMPROVE. Education sessions, the group visit, and precepting model are intentionally designed to break down silos and foster a team approach to care, which supports the PACT team model. Trainees and faculty all have their unique strengths and look at the issue from a different perspective, which increases the likelihood that the patient will hear a cohesive solution or strategy. The result is that trainees are more well rounded and become better practitioners who seek advice from other professions and work well in teams.
Trainees are expected to learn about other professions and their skill sets. For example, trainees learn early about the roles and scopes of practice of pharmacists and health psychologists for more effective referrals. Discussions during the session before the group visit may bring conditions like depression or dementia to the trainees’ attention. This is significant because issues like patient motivation may be better handled from a behavioral perspective.
Expanded Clinical Performance
IMPROVE is an opportunity for CoEPCE trainees to expand their clinical expertise. It provides exposure to a variety of patients and patient care needs and is an opportunity to present a high-risk patient to colleagues of various professions. As of December 2015, about 30 internal medicine residents and 6 NP residents have seen patients in the polypharmacy clinic. Each year, 4 NP residents, 2 health psychology residents, 4 clinical pharmacy residents, and 1 geriatric pharmacy resident participate in the IMPROVE clinic during their yearlong training program. During their 3-year training program, 17 to 19 internal medicine residents participate in IMPROVE.
A structured forum for discussing patients and their care options supports professionals’ utilization of the full scope of their practice. Trainees learn and apply team skills, such as communication and the warm handoff, which can be used in other clinic settings. A warm handoff is often described as an intervention in which “a clinician directly introduces a patient to another clinician at the time of the patient’s visit and often a brief encounter between the patient and the health care professional occurs.”9 An interprofessional care plan supports trainee clinical performance, providing a more robust approach to patient care than individual providers might on their own.
Patient Outcomes
IMPROVE is an enriched care plan informed by multiple professions with the potential to improve medication use and provide better care. Veterans also are receiving better medication education as well as access to a health psychologist who can help them with goal setting and effective behavioral interventions. On average, 5 patients participate each month. As of December 2015, 68 patients have participated in IMPROVE.
The group visit and the 1:1 patient visits focus exclusively on medication issues and solutions, which would be less common in a typical primary care visit with a complex patient who brings a list of agenda items. In addition to taking a thorough look at their medications and related problems, it also educates patients on related issues such as sleep hygiene. Participating veterans also are encouraged to share their concerns, experiences, and solutions with the group, which may increase the saliency of the message beyond what is offered in counseling from a provider.
To date, preliminary data suggest that in some patients, cognition (as measured with SLUMS after 6 months) has modestly improved after decreasing their medications. Other outcomes being monitored in follow-up are utilization of care, reported history of falls, number of medications, and vital signs at initial and follow-up visits.
Patients experience increased continuity of care because the patient now has a team focusing on his or her care. Team members have a shared understanding of the patient’s situation and are better able to establish therapeutic rapport with patients during the group visit. Moreover, CoEPCE trainees and faculty try to ensure that everyone knows about and concurs with medication changes, including outside providers and family members.
Satisfaction Questionnaire
Patients that are presented at IMPROVE can be particularly challenging, and there may be a psychological benefit to working with a team to develop a new care plan. Providers are able to get input and look at the patient in a new light.
Results of postvisit patient satisfaction questionnaires are encouraging and result in a high level of patient satisfaction and perception of clinical benefit. Patients identify an improvement in the understanding of their medications, feel they are able to safely decrease their medications, and are interested in participating again.
CoEPCE Benefits
IMPROVE expands the prevention and treatment options for populations at risk of hospitalization and adverse outcomes from medication complications, such as AEs and drug-drug or drug-disease interactions. Embedding the polypharmacy clinic within the primary care setting rather than in a separate specialty clinic results in an increased likelihood of implementation of pharmacist and geriatrician recommendations for polypharmacy and allows for direct interprofessional education and collaboration.
IMPROVE also combines key components of interprofessional education—an enriched clinical training model and knowledge of medications in an elderly population—into a training activity that complements other CoEPCE activities. The model not only has strengthened CoEPCE partnerships with other VA departments and specialties, but also revealed opportunities for collaboration with academic affiliates as a means to break down traditional silos among medicine, nursing, pharmacy, geriatrics, and psychology.
IMPROVE combines key components of interprofessional education, including all 4 CoEPCE core domains, to provide hands-on experience with knowledge learned in other aspects of the CoEPCE training program (eg, shared decision-making strategies for eliciting patient goals, weighing risks and benefits in complex clinical situations). Physician and NP trainees work together with trainees in pharmacy and health psychology in the complex approach to polypharmacy. IMPROVE provides the framework for an interprofessional clinic that could be used in the treatment of other complex or high-risk chronic conditions.
The Future
An opportunity for improvement and expansion includes increased patient involvement (as patients continue to learn they have a team working on their behalf). Opportunities exist to connect with patients who have several clinicians prescribing medications outside the CoEPCE to provide comprehensive care and decrease medication complexity.
The CoEPCE has been proactive in increasing the visibility of IMPROVE through multiple presentations at local and national meetings, facilitating collaborations and greater adoption in primary care. Individual and collective IMPROVE components can be adapted to other contexts. For example, the 20-minute geriatrics education session and the forms completed prior and during the patient visit can be readily applied to other complex patients that trainees meet in clinic. Under stage 2 of the CoEPCE program, the CoEPCE is developing an implementation kit that describes the training process and includes the medication worksheet, assessment tools, and directions for conducting the group visit.
It is hoped that working collaboratively with the West Haven COEPCE polypharmacy faculty, a similar model of education and training will be implemented at other health professional training sites at Yale University in New Haven, Connecticut. Additionally, the West Haven CoEPCE is planning to partner with the other original CoEPCE program sites to implement similar interprofessional polypharmacy clinics.
1. US Department of Health and Human Services, Agency for Health Research and Quality. Transforming the organization and delivery of primary care. http://www.pcmh.ahrq .gov/. Accessed August 14, 2018.
2. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831.
3. Fried TR, O’Leary J, Towle V, Goldstein MK, Trentalange M, Martin DK. Health outcomes associated with polypharmacy in community-dwelling older adults: a systematic review. J Am Geriatr Soc. 2014;62(12):2261-2272.
4. Mecca M, Niehoff K, Grammas M. Medication review worksheet 2015. http://pogoe.org/productid/21872. Accessed August 14, 2018.
5. American Geriatrics Society 2015 Beers criteria update expert panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
6. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213-218.
7. Yale University. IMPROVE Polypharmacy Project. http://improvepolypharmacy.yale.edu. Accessed August 14, 2018.
8. Tariq SH, Tumosa N, Chibnall JT, Perry MH III, Morley JE. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
9. Cohen DJ, Balasubramanian BA, Davis M, et al. Understanding care integration from the ground up: Five organizing constructs that shape integrated practices. J Am Board Fam Med. 2015;28(suppl):S7-S20.
An interprofessional polypharmacy clinic for intensive management of medication regimens helps high-risk patients manage their medications.
An interprofessional polypharmacy clinic for intensive management of medication regimens helps high-risk patients manage their medications.
In 2011, 5 VA medical centers (VAMCs) were selected by the Office of Academic Affiliations (OAA) to establish CoEPCE. Part of the VA New Models of Care initiative, the 5 Centers of Excellence (CoE) in Boise, Idaho; Cleveland, Ohio; San Francisco, California; Seattle, Washington; and West Haven, Connecticut, are utilizing VA primary care settings to develop and test innovative approaches to prepare physician residents and students, advanced practice nurse residents and undergraduate nursing students, and other professions of health trainees (eg, pharmacy, social work, psychology, physician assistants [PAs], physical therapists) for primary care practice in the 21st century. The CoEs are developing, implementing, and evaluating curricula designed to prepare learners from relevant professions to practice in patient-centered, interprofessional team-based primary care settings. The curricula at all CoEs must address 4 core domains (Table).
Health care professional education programs do not have many opportunities for workplace learning where trainees from different professions can learn and work together to provide care to patients in real time.
The VA Connecticut Healthcare System CoEPCE developed and implemented an education and practice-based immersion learning model with physician residents, nurse practitioner (NP) residents and NP students, pharmacy residents, postdoctorate psychology learners, and PA and physical therapy learners and faculty. This interprofessional, collaborative team model breaks from the traditional independent model of siloed primary care providers (PCPs) caring for a panel of patients.
Methods
In 2015, OAA evaluators reviewed background documents and conducted open-ended interviews with 12 West Haven CoEPCE staff, participating trainees, VA faculty, VA facility leadership, and affiliate faculty. Informants described their involvement, challenges encountered, and benefits of the Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) program to trainees, veterans, and the VA.
Lack of Clinical Approaches to Interprofessional Education and Care
Polypharmacy is a common problem among older adults with multiple chronic conditions, which places patients at higher risk for multiple negative health outcomes.2,3 The typical primary care visit rarely allows for a thorough review of a patient’s medications, much less the identification of strategies to reduce polypharmacy and improve medication management. Rather, the complexity inherent to polypharmacy makes it an ideal challenge for a team-based approach.
Team Approach to Medication Needs
A key CoEPCE program aim is to expand workplace learning instruction strategies and to create more clinical opportunities for CoEPCE trainees to work together as a team to anticipate and address the health care needs of veterans. To address this training need, the West Haven CoEPCE developed IMPROVE to focus on high-need patients and provides a venue in which trainees and supervisors from different professions can collaborate on a specific patient case, using a patient-centered framework. IMPROVE can be easily applied to a range of medication-related aims, such as reducing medications, managing medications and adherence, and addressing adverse effects (AEs). These goals are 2-fold: (1) implement a trainee-led performance improvement project that reduces polypharmacy in elderly veterans; and (2) develop a hands-on, experiential geriatrics training program that enhances trainee skills and knowledge related to safe prescribing.
Planning and Implementation
IMPROVE has its origins in a scholarly project developed by a West Haven CoE physician resident trainee. Development of the IMPROVE program involved VA health psychology, internal medicine faculty, geriatric medicine faculty, NP faculty, and geriatric pharmacy residents and faculty. Planning started in 2013 with a series of pilot clinics and became an official project of the West Haven CoE in September 2014. The intervention required no change in West Haven VAMC policy. However, the initiative required buy-in from West Haven CoE leadership and the director of the West Haven primary care clinic.
Curriculum
IMPROVE is an educational, workplace learning, and clinical activity that combines a 1-hour trainee teaching session, a 45-minute group visit, and a 60-minute individual clinic visit to address the complex problem of polypharmacy. It emphasizes the sharing of trainee and faculty backgrounds by serving as a venue for interprofessional trainees and providers to discuss pharmacologic and nonpharmacologic treatment in the elderly and brainstorm strategies to optimize treatment regimens, minimize risk, and execute medication plans with patients.
All CoEPCE trainees in West Haven are required to participate in IMPROVE and on average, each trainee presents and sees one of their patients at least 3 times per year in the program. Up to 5 trainees participate in each IMPROVE session. Trainees are responsible for reviewing their panels to identify patients who might benefit from participation, followed by inviting the patient to participate. Patients are instructed to bring their pill bottles to the visit. To prepare for the polypharmacy clinic, the trainees, the geriatrician, and the geriatric pharmacist perform an extensive medication chart review, using the medication review worksheet developed by West Haven VAMC providers.4 They also work with a protocol for medication discontinuation, which was compiled by West Haven VAMC clinicians. The teams use a variety of tools that guide appropriate prescribing in older adult populations.5,6 During a preclinic conference, trainees present their patients to the interprofessional team for discussion and participate in a short discussion led by a pharmacist, geriatrician, or health psychologist on a topic related to prescribing safety in older adults or nonpharmacologic treatments.
IMPROVE emphasizes a patient-centered approach to develop, execute, and monitor medication plans. Patients and their family members are invited by their trainee clinician to participate in a group visit. Typically, trainees invite patients aged ≥ 65 years who have ≥ 10 medications and are considered appropriate for a group visit.
The group visit process is based on health psychology strategies, which often incorporate group-based engagement with patients. The health psychologist can give advice to facilitate the visit and optimize participant involvement. There is a discussion facilitator guide that lists the education points to be covered by a designated trainee facilitator and sample questions to guide the discussion.7 A health psychology resident and other rotating trainees cofacilitate the group visit with a goal to reach out to each group member, including family members, and have them discuss perceptions and share concerns and treatment goals. There is shared responsibility among the trainees to address the educational material as well as involve their respective patients during the sessions.
Immediately following the group visit, trainees conduct a 1-hour clinic session that includes medication reconciliation, a review of an IMPROVE questionnaire, orthostatic vital signs, and the St. Louis University Mental Status (SLUMS) exam to assess changes in cognition.7,8 Discussion involved the patient’s medication list as well as possible changes that could be made to the list. Using shared decision-making techniques, this conversation considers the patients’ treatment goals, feelings about the medications, which medications they would like to stop, and AEs they may be experiencing. After the individual visit is completed, the trainee participates in a 10-minute interprofessional precepting session, which may include a geriatrician, a pharmacist, and a health psychologist. In the session they may discuss adjustments to medications and a safe follow-up plan, including appropriate referrals. Trainees discuss the plan with the patient and send a letter describing the plan shortly after the visit.
IMPROVE combines didactic teaching with experiential education. It embodies the 4 core domains that shape the CoEPCE curriculum. First, trainees learn interprofessional collaboration concepts, including highlighting the roles of each profession and working with an interprofessional team to solve problems. Second, CoEPCE trainees learn performance improvement under the supervision of faculty. Third, IMPROVE allows trainees to develop sustained relationships with other team members while improving the quality of the clinic experience as well as with patients through increased continuity of care. Trainees see patients on their panel and are responsible for outreach before and after the visit. Finally, with a focus on personalized patient goals, trainees have the opportunity to further develop skills in shared decision making (SDM).
Related: Reducing Benzodiazepine Prescribing in Older Veterans: A Direct-to-Consumer Educational Brochure
The IMPROVE model continues to evolve. The original curriculum involved an hour-long preclinic preparation session before the group visit in which trainees and faculty discussed the medication review for each patient scheduled that day. This preparation session was later shortened to 40 minutes, and a 20-minute didactic component was added to create the current preclinic session. The didactic component focused on a specific topic in appropriate prescribing for older patients. For example, one didactic lesson is on a particular class of medications, its common AEs, and practical prescribing and “deprescribing” strategies for that class. Initially, the oldest patients or patients who could be grouped thematically, such as those taking both narcotics and benzodiazepines, were invited to participate, but that limited the number of appropriate patients within the CoEPCE. Currently, trainees identify patients from their panels who might benefit, based on age, number of medications, or potential medication-related concerns, such as falls, cognitive impairment, or other concerns for adverse drug effects. These trainees have the unique opportunity to apply learned strategies to their patients to continue to optimize the medication regimen even after the IMPROVE visit. Another significant change was the inclusion of veterans who are comanaged with PCPs outside the VA, because we found that patients with multiple providers could benefit from improved coordination of care.
Faculty Role
CoE faculty and non-CoE VA faculty participate in supervisory, consulting, teaching and precepting roles. Some faculty members such as the health psychologists are already located in or near the VA primary care clinic, so they can assist in curriculum development and execution during their regular clinic duties. The geriatrician reviews the patients’ health records before the patients come into the clinic, participates in the group visit, and coprecepts during the 1:1 patient visits. Collaboration is inherent in IMPROVE. For example, the geriatrician works with the geriatric pharmacist to identify and teach an educational topic. IMPROVE is characterized by a strong faculty/trainee partnership, with trainees playing roles as both teacher and facilitator in addition to learning how to take a team approach to polypharmacy.
Resources
IMPROVE requires administrative and academic support, especially faculty and trainee preparation of education sessions. The CoEPCE internal medicine resident and the internal medicine chief resident work with the health technicians for each patient aligned care team (PACT) to enter the information into the VA medical scheduling system. Trainee clinic time is blocked for their group visits in advance. Patients are scheduled 1 to 3 weeks in advance. Trainees and faculty are expected to review the medication review worksheet and resources prior to the visit. One CoEPCE faculty member reviews patients prior to the preclinic session (about an hour of preparation per session). Sufficient space also is required: a room large enough to accommodate up to 10 people for both didactic lessons and preclinic sessions, a facility patient education conference room for the group visit, and up to 5 clinic exam rooms. CoEPCE staff developed a templated note in the VA Computerized Patient Record System (CPRS), the VA electronic health record system to guide trainees step-by-step through the clinic visit and allow them to directly enter information into the system.7
Monitoring and Assessment
CoEPCE staff are evaluating IMPROVE by building a database for patient-level and trainee-level outcomes, including changes in trainee knowledge and attitudes over time. The CoEPCE also validated the polypharmacy knowledge assessment tool for medicine and NP trainees.
Partnerships
IMPROVE has greatly benefited from partnerships with facility department leadership, particularly involvement of pharmacy staff. In addition, we have partnered with both the health psychology and pharmacy faculty and trainees to participate in the program. Geriatrics faculty and trainees also have contributed extensively to IMPROVE. Future goals include offering the program to non-COEPCE patients throughout primary care.
The Yale Primary Care Internal Medicine Residency program and the Yale Categorical Internal Medicine Residency Program are integral partners to the CoEPCE. IMPROVE supports their mandate to encourage interprofessional teamwork in primary care, meet the Accreditation Council for Graduate Medical Education interprofessional milestones, and promote individual trainee scholarship and performance improvement in areas of broad applicability. IMPROVE also is an opportunity to share ideas across institutions and stimulate new collaborations and dissemination of the model to other primary care settings outside the VA.
Challenges and Solutions
The demand for increased direct patient care pressures programs like IMPROVE, which is a time-intensive process with high impact on a few complex patients. The assumption is that managing medications will save money in the long run, but in the short-term, a strong case has to be made for securing resources, particularly blocking provider time and securing an education room for group visits and clinic exam rooms for individual visits. First, decision makers need to be convinced that polypharmacy is important and should be a training priority. The CoEPCE has tried different configurations to increase the number of patients being seen, such as having ≥ 1 IMPROVE session in an afternoon, but trainees found this to be labor intensive and stressful.
Second, patients with medications prescribed by providers outside the VA require additional communication and coordination to reduce medications. The CoEPCE initially excluded these patients, but after realizing that some of these patients needed the most help, it developed a process for reaching out to non-VA providers and coordinating care. Additionally, there is significant diversity in patient polypharmacy needs. These can range from adherence problems to the challenge of complex psychosocial needs that are more easily (but less effectively) addressed with medications. The issue of polypharmacy is further complicated by evolving understanding of medications’ relative risks and benefits in older adults with multiple chronic conditions. IMPROVE is an effective vehicle for synthesizing current science in medications and their management, especially in complex older patients with multiple chronic conditions.
Other challenges include developing a templated CPRS electronic note that interfaces with the VA information technology system. The process of creating a template, obtaining approval from the forms committee, and working with information technology personnel to implement the template was more time intensive than anticipated and required multiple iterations of proofreading and editing.
Factors for Success
The commitment to support new models of trainee education by West Haven CoEPCE faculty and leadership, and West Haven VAMC and primary care clinic leadership facilitated the implementation of IMPROVE. Additionally, there is strong CoEPCE collaboration at all levels—codirectors, faculty, and trainees—for the program. High interprofessional trainee interest, organizational insight, and an academic orientation were critical for developing and launching IMPROVE.
Additionally, there is synergy with other team-based professions. Geriatrics has a tradition of working in multidisciplinary teams as well as working with SDM concepts as part of care discussions. High interest and collaboration by a geriatrician and an experienced geriatric pharmacist has been key. The 2 specialties complement each other and address the complex health needs of participating veterans. Health psychologists transition patients to nonpharmacologic treatments, such as sleep hygiene education and cognitive behavioral therapy, in addition to exploring barriers to behavior change.
Another factor for success has been the CoEPCE framework and expertise in interprofessional education. While refining the model, program planners tapped into existing expertise in polypharmacy within the VA from the geriatrics, pharmacy, and clinical health psychology departments. The success of the individual components—the preparation session, the group visit, and the 1:1 patient visit—is in large part the result of a collective effort by CoEPCE staff and the integration of CoEPCE staff through coordination, communications, logistics, quality improvement, and faculty involvement from multiple professions.
The IMPROVE model is flexible and can accommodate diverse patient interests and issues. Model components are based on sound practices that have demonstrated success in other arenas, such as diabetes mellitus group visits. The model can also accommodate diverse trainee levels. Senior trainees can be more independent in developing their care plans, teaching the didactic topic, or precepting during the 1:1 patient exam.
Accomplishments and Benefits
Trainees are using team skills to provide patient-centered care. They are strengthening their clinical skills through exposure to patients in a group visit and 1:1 clinic visit. There have been significant improvements in the trainees’ provision of individual patient care. Key IMPROVE outcomes are outlined below.
Interprofessional Education
Unlike a traditional didactic, IMPROVE is an opportunity for health care professionals to work together to provide care in a clinic setting. It also expands CoEPCE interprofessional education capacity through colocation of different trainee and faculty professions during the conference session. This combination trains participants to work as a team and reflect on patients together, which has strengthened communications among professions. The model provides sufficient time and expertise to discuss the medications in detail and as a team, something that would not normally happen during a regular primary care visit.
CoEPCE trainees learn about medication management, its importance in preventing complications and improving patient health outcomes. Trainees of all professions learn to translate the skills they learn in IMPROVE to other patients, such as how to perform a complete medication reconciliation or lead a discussion using SDM. IMPROVE also provides techniques useful in other contexts, such as group visits and consideration of different medication options for patients who have been cared for by other (VA and non-VA) providers.
Interprofessional Collaboration
Understanding and leveraging the expertise of trainees and faculty from different professions is a primary goal of IMPROVE. Education sessions, the group visit, and precepting model are intentionally designed to break down silos and foster a team approach to care, which supports the PACT team model. Trainees and faculty all have their unique strengths and look at the issue from a different perspective, which increases the likelihood that the patient will hear a cohesive solution or strategy. The result is that trainees are more well rounded and become better practitioners who seek advice from other professions and work well in teams.
Trainees are expected to learn about other professions and their skill sets. For example, trainees learn early about the roles and scopes of practice of pharmacists and health psychologists for more effective referrals. Discussions during the session before the group visit may bring conditions like depression or dementia to the trainees’ attention. This is significant because issues like patient motivation may be better handled from a behavioral perspective.
Expanded Clinical Performance
IMPROVE is an opportunity for CoEPCE trainees to expand their clinical expertise. It provides exposure to a variety of patients and patient care needs and is an opportunity to present a high-risk patient to colleagues of various professions. As of December 2015, about 30 internal medicine residents and 6 NP residents have seen patients in the polypharmacy clinic. Each year, 4 NP residents, 2 health psychology residents, 4 clinical pharmacy residents, and 1 geriatric pharmacy resident participate in the IMPROVE clinic during their yearlong training program. During their 3-year training program, 17 to 19 internal medicine residents participate in IMPROVE.
A structured forum for discussing patients and their care options supports professionals’ utilization of the full scope of their practice. Trainees learn and apply team skills, such as communication and the warm handoff, which can be used in other clinic settings. A warm handoff is often described as an intervention in which “a clinician directly introduces a patient to another clinician at the time of the patient’s visit and often a brief encounter between the patient and the health care professional occurs.”9 An interprofessional care plan supports trainee clinical performance, providing a more robust approach to patient care than individual providers might on their own.
Patient Outcomes
IMPROVE is an enriched care plan informed by multiple professions with the potential to improve medication use and provide better care. Veterans also are receiving better medication education as well as access to a health psychologist who can help them with goal setting and effective behavioral interventions. On average, 5 patients participate each month. As of December 2015, 68 patients have participated in IMPROVE.
The group visit and the 1:1 patient visits focus exclusively on medication issues and solutions, which would be less common in a typical primary care visit with a complex patient who brings a list of agenda items. In addition to taking a thorough look at their medications and related problems, it also educates patients on related issues such as sleep hygiene. Participating veterans also are encouraged to share their concerns, experiences, and solutions with the group, which may increase the saliency of the message beyond what is offered in counseling from a provider.
To date, preliminary data suggest that in some patients, cognition (as measured with SLUMS after 6 months) has modestly improved after decreasing their medications. Other outcomes being monitored in follow-up are utilization of care, reported history of falls, number of medications, and vital signs at initial and follow-up visits.
Patients experience increased continuity of care because the patient now has a team focusing on his or her care. Team members have a shared understanding of the patient’s situation and are better able to establish therapeutic rapport with patients during the group visit. Moreover, CoEPCE trainees and faculty try to ensure that everyone knows about and concurs with medication changes, including outside providers and family members.
Satisfaction Questionnaire
Patients that are presented at IMPROVE can be particularly challenging, and there may be a psychological benefit to working with a team to develop a new care plan. Providers are able to get input and look at the patient in a new light.
Results of postvisit patient satisfaction questionnaires are encouraging and result in a high level of patient satisfaction and perception of clinical benefit. Patients identify an improvement in the understanding of their medications, feel they are able to safely decrease their medications, and are interested in participating again.
CoEPCE Benefits
IMPROVE expands the prevention and treatment options for populations at risk of hospitalization and adverse outcomes from medication complications, such as AEs and drug-drug or drug-disease interactions. Embedding the polypharmacy clinic within the primary care setting rather than in a separate specialty clinic results in an increased likelihood of implementation of pharmacist and geriatrician recommendations for polypharmacy and allows for direct interprofessional education and collaboration.
IMPROVE also combines key components of interprofessional education—an enriched clinical training model and knowledge of medications in an elderly population—into a training activity that complements other CoEPCE activities. The model not only has strengthened CoEPCE partnerships with other VA departments and specialties, but also revealed opportunities for collaboration with academic affiliates as a means to break down traditional silos among medicine, nursing, pharmacy, geriatrics, and psychology.
IMPROVE combines key components of interprofessional education, including all 4 CoEPCE core domains, to provide hands-on experience with knowledge learned in other aspects of the CoEPCE training program (eg, shared decision-making strategies for eliciting patient goals, weighing risks and benefits in complex clinical situations). Physician and NP trainees work together with trainees in pharmacy and health psychology in the complex approach to polypharmacy. IMPROVE provides the framework for an interprofessional clinic that could be used in the treatment of other complex or high-risk chronic conditions.
The Future
An opportunity for improvement and expansion includes increased patient involvement (as patients continue to learn they have a team working on their behalf). Opportunities exist to connect with patients who have several clinicians prescribing medications outside the CoEPCE to provide comprehensive care and decrease medication complexity.
The CoEPCE has been proactive in increasing the visibility of IMPROVE through multiple presentations at local and national meetings, facilitating collaborations and greater adoption in primary care. Individual and collective IMPROVE components can be adapted to other contexts. For example, the 20-minute geriatrics education session and the forms completed prior and during the patient visit can be readily applied to other complex patients that trainees meet in clinic. Under stage 2 of the CoEPCE program, the CoEPCE is developing an implementation kit that describes the training process and includes the medication worksheet, assessment tools, and directions for conducting the group visit.
It is hoped that working collaboratively with the West Haven COEPCE polypharmacy faculty, a similar model of education and training will be implemented at other health professional training sites at Yale University in New Haven, Connecticut. Additionally, the West Haven CoEPCE is planning to partner with the other original CoEPCE program sites to implement similar interprofessional polypharmacy clinics.
In 2011, 5 VA medical centers (VAMCs) were selected by the Office of Academic Affiliations (OAA) to establish CoEPCE. Part of the VA New Models of Care initiative, the 5 Centers of Excellence (CoE) in Boise, Idaho; Cleveland, Ohio; San Francisco, California; Seattle, Washington; and West Haven, Connecticut, are utilizing VA primary care settings to develop and test innovative approaches to prepare physician residents and students, advanced practice nurse residents and undergraduate nursing students, and other professions of health trainees (eg, pharmacy, social work, psychology, physician assistants [PAs], physical therapists) for primary care practice in the 21st century. The CoEs are developing, implementing, and evaluating curricula designed to prepare learners from relevant professions to practice in patient-centered, interprofessional team-based primary care settings. The curricula at all CoEs must address 4 core domains (Table).
Health care professional education programs do not have many opportunities for workplace learning where trainees from different professions can learn and work together to provide care to patients in real time.
The VA Connecticut Healthcare System CoEPCE developed and implemented an education and practice-based immersion learning model with physician residents, nurse practitioner (NP) residents and NP students, pharmacy residents, postdoctorate psychology learners, and PA and physical therapy learners and faculty. This interprofessional, collaborative team model breaks from the traditional independent model of siloed primary care providers (PCPs) caring for a panel of patients.
Methods
In 2015, OAA evaluators reviewed background documents and conducted open-ended interviews with 12 West Haven CoEPCE staff, participating trainees, VA faculty, VA facility leadership, and affiliate faculty. Informants described their involvement, challenges encountered, and benefits of the Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) program to trainees, veterans, and the VA.
Lack of Clinical Approaches to Interprofessional Education and Care
Polypharmacy is a common problem among older adults with multiple chronic conditions, which places patients at higher risk for multiple negative health outcomes.2,3 The typical primary care visit rarely allows for a thorough review of a patient’s medications, much less the identification of strategies to reduce polypharmacy and improve medication management. Rather, the complexity inherent to polypharmacy makes it an ideal challenge for a team-based approach.
Team Approach to Medication Needs
A key CoEPCE program aim is to expand workplace learning instruction strategies and to create more clinical opportunities for CoEPCE trainees to work together as a team to anticipate and address the health care needs of veterans. To address this training need, the West Haven CoEPCE developed IMPROVE to focus on high-need patients and provides a venue in which trainees and supervisors from different professions can collaborate on a specific patient case, using a patient-centered framework. IMPROVE can be easily applied to a range of medication-related aims, such as reducing medications, managing medications and adherence, and addressing adverse effects (AEs). These goals are 2-fold: (1) implement a trainee-led performance improvement project that reduces polypharmacy in elderly veterans; and (2) develop a hands-on, experiential geriatrics training program that enhances trainee skills and knowledge related to safe prescribing.
Planning and Implementation
IMPROVE has its origins in a scholarly project developed by a West Haven CoE physician resident trainee. Development of the IMPROVE program involved VA health psychology, internal medicine faculty, geriatric medicine faculty, NP faculty, and geriatric pharmacy residents and faculty. Planning started in 2013 with a series of pilot clinics and became an official project of the West Haven CoE in September 2014. The intervention required no change in West Haven VAMC policy. However, the initiative required buy-in from West Haven CoE leadership and the director of the West Haven primary care clinic.
Curriculum
IMPROVE is an educational, workplace learning, and clinical activity that combines a 1-hour trainee teaching session, a 45-minute group visit, and a 60-minute individual clinic visit to address the complex problem of polypharmacy. It emphasizes the sharing of trainee and faculty backgrounds by serving as a venue for interprofessional trainees and providers to discuss pharmacologic and nonpharmacologic treatment in the elderly and brainstorm strategies to optimize treatment regimens, minimize risk, and execute medication plans with patients.
All CoEPCE trainees in West Haven are required to participate in IMPROVE and on average, each trainee presents and sees one of their patients at least 3 times per year in the program. Up to 5 trainees participate in each IMPROVE session. Trainees are responsible for reviewing their panels to identify patients who might benefit from participation, followed by inviting the patient to participate. Patients are instructed to bring their pill bottles to the visit. To prepare for the polypharmacy clinic, the trainees, the geriatrician, and the geriatric pharmacist perform an extensive medication chart review, using the medication review worksheet developed by West Haven VAMC providers.4 They also work with a protocol for medication discontinuation, which was compiled by West Haven VAMC clinicians. The teams use a variety of tools that guide appropriate prescribing in older adult populations.5,6 During a preclinic conference, trainees present their patients to the interprofessional team for discussion and participate in a short discussion led by a pharmacist, geriatrician, or health psychologist on a topic related to prescribing safety in older adults or nonpharmacologic treatments.
IMPROVE emphasizes a patient-centered approach to develop, execute, and monitor medication plans. Patients and their family members are invited by their trainee clinician to participate in a group visit. Typically, trainees invite patients aged ≥ 65 years who have ≥ 10 medications and are considered appropriate for a group visit.
The group visit process is based on health psychology strategies, which often incorporate group-based engagement with patients. The health psychologist can give advice to facilitate the visit and optimize participant involvement. There is a discussion facilitator guide that lists the education points to be covered by a designated trainee facilitator and sample questions to guide the discussion.7 A health psychology resident and other rotating trainees cofacilitate the group visit with a goal to reach out to each group member, including family members, and have them discuss perceptions and share concerns and treatment goals. There is shared responsibility among the trainees to address the educational material as well as involve their respective patients during the sessions.
Immediately following the group visit, trainees conduct a 1-hour clinic session that includes medication reconciliation, a review of an IMPROVE questionnaire, orthostatic vital signs, and the St. Louis University Mental Status (SLUMS) exam to assess changes in cognition.7,8 Discussion involved the patient’s medication list as well as possible changes that could be made to the list. Using shared decision-making techniques, this conversation considers the patients’ treatment goals, feelings about the medications, which medications they would like to stop, and AEs they may be experiencing. After the individual visit is completed, the trainee participates in a 10-minute interprofessional precepting session, which may include a geriatrician, a pharmacist, and a health psychologist. In the session they may discuss adjustments to medications and a safe follow-up plan, including appropriate referrals. Trainees discuss the plan with the patient and send a letter describing the plan shortly after the visit.
IMPROVE combines didactic teaching with experiential education. It embodies the 4 core domains that shape the CoEPCE curriculum. First, trainees learn interprofessional collaboration concepts, including highlighting the roles of each profession and working with an interprofessional team to solve problems. Second, CoEPCE trainees learn performance improvement under the supervision of faculty. Third, IMPROVE allows trainees to develop sustained relationships with other team members while improving the quality of the clinic experience as well as with patients through increased continuity of care. Trainees see patients on their panel and are responsible for outreach before and after the visit. Finally, with a focus on personalized patient goals, trainees have the opportunity to further develop skills in shared decision making (SDM).
Related: Reducing Benzodiazepine Prescribing in Older Veterans: A Direct-to-Consumer Educational Brochure
The IMPROVE model continues to evolve. The original curriculum involved an hour-long preclinic preparation session before the group visit in which trainees and faculty discussed the medication review for each patient scheduled that day. This preparation session was later shortened to 40 minutes, and a 20-minute didactic component was added to create the current preclinic session. The didactic component focused on a specific topic in appropriate prescribing for older patients. For example, one didactic lesson is on a particular class of medications, its common AEs, and practical prescribing and “deprescribing” strategies for that class. Initially, the oldest patients or patients who could be grouped thematically, such as those taking both narcotics and benzodiazepines, were invited to participate, but that limited the number of appropriate patients within the CoEPCE. Currently, trainees identify patients from their panels who might benefit, based on age, number of medications, or potential medication-related concerns, such as falls, cognitive impairment, or other concerns for adverse drug effects. These trainees have the unique opportunity to apply learned strategies to their patients to continue to optimize the medication regimen even after the IMPROVE visit. Another significant change was the inclusion of veterans who are comanaged with PCPs outside the VA, because we found that patients with multiple providers could benefit from improved coordination of care.
Faculty Role
CoE faculty and non-CoE VA faculty participate in supervisory, consulting, teaching and precepting roles. Some faculty members such as the health psychologists are already located in or near the VA primary care clinic, so they can assist in curriculum development and execution during their regular clinic duties. The geriatrician reviews the patients’ health records before the patients come into the clinic, participates in the group visit, and coprecepts during the 1:1 patient visits. Collaboration is inherent in IMPROVE. For example, the geriatrician works with the geriatric pharmacist to identify and teach an educational topic. IMPROVE is characterized by a strong faculty/trainee partnership, with trainees playing roles as both teacher and facilitator in addition to learning how to take a team approach to polypharmacy.
Resources
IMPROVE requires administrative and academic support, especially faculty and trainee preparation of education sessions. The CoEPCE internal medicine resident and the internal medicine chief resident work with the health technicians for each patient aligned care team (PACT) to enter the information into the VA medical scheduling system. Trainee clinic time is blocked for their group visits in advance. Patients are scheduled 1 to 3 weeks in advance. Trainees and faculty are expected to review the medication review worksheet and resources prior to the visit. One CoEPCE faculty member reviews patients prior to the preclinic session (about an hour of preparation per session). Sufficient space also is required: a room large enough to accommodate up to 10 people for both didactic lessons and preclinic sessions, a facility patient education conference room for the group visit, and up to 5 clinic exam rooms. CoEPCE staff developed a templated note in the VA Computerized Patient Record System (CPRS), the VA electronic health record system to guide trainees step-by-step through the clinic visit and allow them to directly enter information into the system.7
Monitoring and Assessment
CoEPCE staff are evaluating IMPROVE by building a database for patient-level and trainee-level outcomes, including changes in trainee knowledge and attitudes over time. The CoEPCE also validated the polypharmacy knowledge assessment tool for medicine and NP trainees.
Partnerships
IMPROVE has greatly benefited from partnerships with facility department leadership, particularly involvement of pharmacy staff. In addition, we have partnered with both the health psychology and pharmacy faculty and trainees to participate in the program. Geriatrics faculty and trainees also have contributed extensively to IMPROVE. Future goals include offering the program to non-COEPCE patients throughout primary care.
The Yale Primary Care Internal Medicine Residency program and the Yale Categorical Internal Medicine Residency Program are integral partners to the CoEPCE. IMPROVE supports their mandate to encourage interprofessional teamwork in primary care, meet the Accreditation Council for Graduate Medical Education interprofessional milestones, and promote individual trainee scholarship and performance improvement in areas of broad applicability. IMPROVE also is an opportunity to share ideas across institutions and stimulate new collaborations and dissemination of the model to other primary care settings outside the VA.
Challenges and Solutions
The demand for increased direct patient care pressures programs like IMPROVE, which is a time-intensive process with high impact on a few complex patients. The assumption is that managing medications will save money in the long run, but in the short-term, a strong case has to be made for securing resources, particularly blocking provider time and securing an education room for group visits and clinic exam rooms for individual visits. First, decision makers need to be convinced that polypharmacy is important and should be a training priority. The CoEPCE has tried different configurations to increase the number of patients being seen, such as having ≥ 1 IMPROVE session in an afternoon, but trainees found this to be labor intensive and stressful.
Second, patients with medications prescribed by providers outside the VA require additional communication and coordination to reduce medications. The CoEPCE initially excluded these patients, but after realizing that some of these patients needed the most help, it developed a process for reaching out to non-VA providers and coordinating care. Additionally, there is significant diversity in patient polypharmacy needs. These can range from adherence problems to the challenge of complex psychosocial needs that are more easily (but less effectively) addressed with medications. The issue of polypharmacy is further complicated by evolving understanding of medications’ relative risks and benefits in older adults with multiple chronic conditions. IMPROVE is an effective vehicle for synthesizing current science in medications and their management, especially in complex older patients with multiple chronic conditions.
Other challenges include developing a templated CPRS electronic note that interfaces with the VA information technology system. The process of creating a template, obtaining approval from the forms committee, and working with information technology personnel to implement the template was more time intensive than anticipated and required multiple iterations of proofreading and editing.
Factors for Success
The commitment to support new models of trainee education by West Haven CoEPCE faculty and leadership, and West Haven VAMC and primary care clinic leadership facilitated the implementation of IMPROVE. Additionally, there is strong CoEPCE collaboration at all levels—codirectors, faculty, and trainees—for the program. High interprofessional trainee interest, organizational insight, and an academic orientation were critical for developing and launching IMPROVE.
Additionally, there is synergy with other team-based professions. Geriatrics has a tradition of working in multidisciplinary teams as well as working with SDM concepts as part of care discussions. High interest and collaboration by a geriatrician and an experienced geriatric pharmacist has been key. The 2 specialties complement each other and address the complex health needs of participating veterans. Health psychologists transition patients to nonpharmacologic treatments, such as sleep hygiene education and cognitive behavioral therapy, in addition to exploring barriers to behavior change.
Another factor for success has been the CoEPCE framework and expertise in interprofessional education. While refining the model, program planners tapped into existing expertise in polypharmacy within the VA from the geriatrics, pharmacy, and clinical health psychology departments. The success of the individual components—the preparation session, the group visit, and the 1:1 patient visit—is in large part the result of a collective effort by CoEPCE staff and the integration of CoEPCE staff through coordination, communications, logistics, quality improvement, and faculty involvement from multiple professions.
The IMPROVE model is flexible and can accommodate diverse patient interests and issues. Model components are based on sound practices that have demonstrated success in other arenas, such as diabetes mellitus group visits. The model can also accommodate diverse trainee levels. Senior trainees can be more independent in developing their care plans, teaching the didactic topic, or precepting during the 1:1 patient exam.
Accomplishments and Benefits
Trainees are using team skills to provide patient-centered care. They are strengthening their clinical skills through exposure to patients in a group visit and 1:1 clinic visit. There have been significant improvements in the trainees’ provision of individual patient care. Key IMPROVE outcomes are outlined below.
Interprofessional Education
Unlike a traditional didactic, IMPROVE is an opportunity for health care professionals to work together to provide care in a clinic setting. It also expands CoEPCE interprofessional education capacity through colocation of different trainee and faculty professions during the conference session. This combination trains participants to work as a team and reflect on patients together, which has strengthened communications among professions. The model provides sufficient time and expertise to discuss the medications in detail and as a team, something that would not normally happen during a regular primary care visit.
CoEPCE trainees learn about medication management, its importance in preventing complications and improving patient health outcomes. Trainees of all professions learn to translate the skills they learn in IMPROVE to other patients, such as how to perform a complete medication reconciliation or lead a discussion using SDM. IMPROVE also provides techniques useful in other contexts, such as group visits and consideration of different medication options for patients who have been cared for by other (VA and non-VA) providers.
Interprofessional Collaboration
Understanding and leveraging the expertise of trainees and faculty from different professions is a primary goal of IMPROVE. Education sessions, the group visit, and precepting model are intentionally designed to break down silos and foster a team approach to care, which supports the PACT team model. Trainees and faculty all have their unique strengths and look at the issue from a different perspective, which increases the likelihood that the patient will hear a cohesive solution or strategy. The result is that trainees are more well rounded and become better practitioners who seek advice from other professions and work well in teams.
Trainees are expected to learn about other professions and their skill sets. For example, trainees learn early about the roles and scopes of practice of pharmacists and health psychologists for more effective referrals. Discussions during the session before the group visit may bring conditions like depression or dementia to the trainees’ attention. This is significant because issues like patient motivation may be better handled from a behavioral perspective.
Expanded Clinical Performance
IMPROVE is an opportunity for CoEPCE trainees to expand their clinical expertise. It provides exposure to a variety of patients and patient care needs and is an opportunity to present a high-risk patient to colleagues of various professions. As of December 2015, about 30 internal medicine residents and 6 NP residents have seen patients in the polypharmacy clinic. Each year, 4 NP residents, 2 health psychology residents, 4 clinical pharmacy residents, and 1 geriatric pharmacy resident participate in the IMPROVE clinic during their yearlong training program. During their 3-year training program, 17 to 19 internal medicine residents participate in IMPROVE.
A structured forum for discussing patients and their care options supports professionals’ utilization of the full scope of their practice. Trainees learn and apply team skills, such as communication and the warm handoff, which can be used in other clinic settings. A warm handoff is often described as an intervention in which “a clinician directly introduces a patient to another clinician at the time of the patient’s visit and often a brief encounter between the patient and the health care professional occurs.”9 An interprofessional care plan supports trainee clinical performance, providing a more robust approach to patient care than individual providers might on their own.
Patient Outcomes
IMPROVE is an enriched care plan informed by multiple professions with the potential to improve medication use and provide better care. Veterans also are receiving better medication education as well as access to a health psychologist who can help them with goal setting and effective behavioral interventions. On average, 5 patients participate each month. As of December 2015, 68 patients have participated in IMPROVE.
The group visit and the 1:1 patient visits focus exclusively on medication issues and solutions, which would be less common in a typical primary care visit with a complex patient who brings a list of agenda items. In addition to taking a thorough look at their medications and related problems, it also educates patients on related issues such as sleep hygiene. Participating veterans also are encouraged to share their concerns, experiences, and solutions with the group, which may increase the saliency of the message beyond what is offered in counseling from a provider.
To date, preliminary data suggest that in some patients, cognition (as measured with SLUMS after 6 months) has modestly improved after decreasing their medications. Other outcomes being monitored in follow-up are utilization of care, reported history of falls, number of medications, and vital signs at initial and follow-up visits.
Patients experience increased continuity of care because the patient now has a team focusing on his or her care. Team members have a shared understanding of the patient’s situation and are better able to establish therapeutic rapport with patients during the group visit. Moreover, CoEPCE trainees and faculty try to ensure that everyone knows about and concurs with medication changes, including outside providers and family members.
Satisfaction Questionnaire
Patients that are presented at IMPROVE can be particularly challenging, and there may be a psychological benefit to working with a team to develop a new care plan. Providers are able to get input and look at the patient in a new light.
Results of postvisit patient satisfaction questionnaires are encouraging and result in a high level of patient satisfaction and perception of clinical benefit. Patients identify an improvement in the understanding of their medications, feel they are able to safely decrease their medications, and are interested in participating again.
CoEPCE Benefits
IMPROVE expands the prevention and treatment options for populations at risk of hospitalization and adverse outcomes from medication complications, such as AEs and drug-drug or drug-disease interactions. Embedding the polypharmacy clinic within the primary care setting rather than in a separate specialty clinic results in an increased likelihood of implementation of pharmacist and geriatrician recommendations for polypharmacy and allows for direct interprofessional education and collaboration.
IMPROVE also combines key components of interprofessional education—an enriched clinical training model and knowledge of medications in an elderly population—into a training activity that complements other CoEPCE activities. The model not only has strengthened CoEPCE partnerships with other VA departments and specialties, but also revealed opportunities for collaboration with academic affiliates as a means to break down traditional silos among medicine, nursing, pharmacy, geriatrics, and psychology.
IMPROVE combines key components of interprofessional education, including all 4 CoEPCE core domains, to provide hands-on experience with knowledge learned in other aspects of the CoEPCE training program (eg, shared decision-making strategies for eliciting patient goals, weighing risks and benefits in complex clinical situations). Physician and NP trainees work together with trainees in pharmacy and health psychology in the complex approach to polypharmacy. IMPROVE provides the framework for an interprofessional clinic that could be used in the treatment of other complex or high-risk chronic conditions.
The Future
An opportunity for improvement and expansion includes increased patient involvement (as patients continue to learn they have a team working on their behalf). Opportunities exist to connect with patients who have several clinicians prescribing medications outside the CoEPCE to provide comprehensive care and decrease medication complexity.
The CoEPCE has been proactive in increasing the visibility of IMPROVE through multiple presentations at local and national meetings, facilitating collaborations and greater adoption in primary care. Individual and collective IMPROVE components can be adapted to other contexts. For example, the 20-minute geriatrics education session and the forms completed prior and during the patient visit can be readily applied to other complex patients that trainees meet in clinic. Under stage 2 of the CoEPCE program, the CoEPCE is developing an implementation kit that describes the training process and includes the medication worksheet, assessment tools, and directions for conducting the group visit.
It is hoped that working collaboratively with the West Haven COEPCE polypharmacy faculty, a similar model of education and training will be implemented at other health professional training sites at Yale University in New Haven, Connecticut. Additionally, the West Haven CoEPCE is planning to partner with the other original CoEPCE program sites to implement similar interprofessional polypharmacy clinics.
1. US Department of Health and Human Services, Agency for Health Research and Quality. Transforming the organization and delivery of primary care. http://www.pcmh.ahrq .gov/. Accessed August 14, 2018.
2. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831.
3. Fried TR, O’Leary J, Towle V, Goldstein MK, Trentalange M, Martin DK. Health outcomes associated with polypharmacy in community-dwelling older adults: a systematic review. J Am Geriatr Soc. 2014;62(12):2261-2272.
4. Mecca M, Niehoff K, Grammas M. Medication review worksheet 2015. http://pogoe.org/productid/21872. Accessed August 14, 2018.
5. American Geriatrics Society 2015 Beers criteria update expert panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
6. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213-218.
7. Yale University. IMPROVE Polypharmacy Project. http://improvepolypharmacy.yale.edu. Accessed August 14, 2018.
8. Tariq SH, Tumosa N, Chibnall JT, Perry MH III, Morley JE. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
9. Cohen DJ, Balasubramanian BA, Davis M, et al. Understanding care integration from the ground up: Five organizing constructs that shape integrated practices. J Am Board Fam Med. 2015;28(suppl):S7-S20.
1. US Department of Health and Human Services, Agency for Health Research and Quality. Transforming the organization and delivery of primary care. http://www.pcmh.ahrq .gov/. Accessed August 14, 2018.
2. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831.
3. Fried TR, O’Leary J, Towle V, Goldstein MK, Trentalange M, Martin DK. Health outcomes associated with polypharmacy in community-dwelling older adults: a systematic review. J Am Geriatr Soc. 2014;62(12):2261-2272.
4. Mecca M, Niehoff K, Grammas M. Medication review worksheet 2015. http://pogoe.org/productid/21872. Accessed August 14, 2018.
5. American Geriatrics Society 2015 Beers criteria update expert panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
6. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213-218.
7. Yale University. IMPROVE Polypharmacy Project. http://improvepolypharmacy.yale.edu. Accessed August 14, 2018.
8. Tariq SH, Tumosa N, Chibnall JT, Perry MH III, Morley JE. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
9. Cohen DJ, Balasubramanian BA, Davis M, et al. Understanding care integration from the ground up: Five organizing constructs that shape integrated practices. J Am Board Fam Med. 2015;28(suppl):S7-S20.
Skin-Colored Papules on the Chest
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
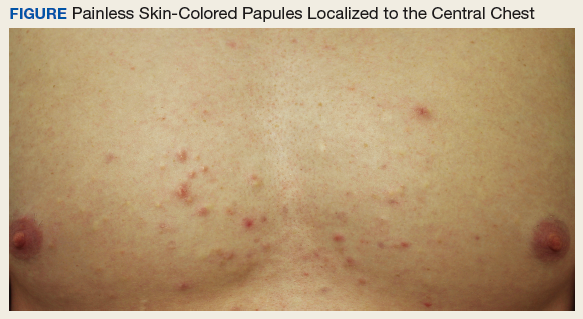
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
An otherwise healthy male presents with multiple smooth uniform painless cystic papules scattered across his central chest.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
A 25-year-old man presented with multiple sternal cysts that he first noticed when he was aged 18 years and had persisted despite treatment with topical anti-acne agents, including tretinoin. No other medications were used. The patient was unable to express purulent material from the lesions and reported no infection or additional trauma to the affected area. He had no other significant past medical history and no family history of similar skin lesions.
A physical examination revealed an otherwise healthy-appearing male with multiple uniform painless cystic papules scattered across his central chest that were smooth and flesh-colored to slightly yellow-colored, measuring 2 mm to 6 mm in diameter (Figure). A ring of erythema surrounded the lesions that had been recently manipulated by the patient. There were no overlying central puncta, and the remainder of his body was spared.
Related: Mohs Micrographic Surgery in the VHA
- What is your diagnosis?
- How would you treat this patient?
Diagnosis
The patient was diagnosed with steatocystoma multiplex based on his poor response to topical anti-acne agents, the location of his lesions, and histopathology of a biopsy specimen. Steatocystoma multiplex, sometimes termed sebocystomatosis, typically presents between puberty and the third decade of life. Lesions are usually < 2 cm in diameter and occur as multiple smooth skin-colored or yellow-colored painless papules on areas with high concentrations of hormonally sensitive sebaceous glands, especially the chest. Lesions also can be found in the axillae and on the neck.1-3 Solitary lesions can occur and are termed steatocystoma simplex.
The timing and location of presentation can easily be mistaken for acne vulgaris, but steatocystoma lesions are true sebaceous cysts, which are rare, and spontaneous resolution with increasing age does not typically occur. The diagnosis of steatocystoma often goes unreported because the disease is usually asymptomatic and mimics more common benign skin conditions, so an accurate prevalence and incidence are both unknown.
First on the differential diagnosis is acne vulgaris, which also presents at puberty and affects nearly 85% of adolescents. However, acne is less common in people of Asian or African descent and may progress along a continuum of increasingly severe and larger lesions, including the primary comedones and papules followed by pustules, nodules, and pseudocysts. Painful lesions develop from inflammation of pilosebaceous units concentrated on the face, neck, trunk, upper arms, or buttocks and are typically worse in males. Resolution often occurs spontaneously by the third decade of life, but scarring can persist.4
Related: Using Dermoscopy to Identify Melanoma and Improve Diagnostic Discrimination
Eruptive vellus hair cysts present as dozens of skin-colored small (1-4 mm) painless dome-shaped papules, sometimes with erythema and crusting. Typically these appear on the head, trunk, or flexor surfaces of infants (familial cases) or adolescents (sporadic cases) without bias for gender or ethnicity. Although benign and potential mimickers of steatocystoma and acne, these lesions can also be associated with more serious syndromes, like ectodermal dysplasias and pachyonychia congenita.2,3
Epidermoid cysts are common benign solitary skin-colored subcutaneous dome-shaped nodules that contain a central punctum through which cheeselike keratinaceous material can be expressed.4 These benign lesions arising from the dermis can enlarge to several centimeters, and adults of both genders and most ethnicities tend to develop the lesions on the trunk or face, with small cysts on the face termed milia. Ruptured cysts can incite intense inflammation, and multiple epidermoid cysts should raise concern for Gardner syndrome.2,3
About This Condition
Steatocystoma lesions are benign and thought to arise from a mutation in keratin 17. The mutation can be inherited in an autosomal dominant pattern, but sporadic nonheritable cases are more common.5 There are no distinct associations with gender or ethnicity. The dermal cysts arise from the sebaceous ducts of the pilosebaceous unit, and histopathology typically shows numerous mature sebaceous cells encased by a thin wall of stratified squamous epithelium.2 Immunohistochemical staining for the defective keratin can help diagnose biopsy specimens, and histopathology confirmed the diagnosis in this case.
Related: Recurring Bilateral Rash Concomitant With Upper Respiratory Tract Infection in a Healthy Adult Male
Treatment
Steatocystoma is usually asymptomatic, so patients mainly present to physicians for cosmetic reasons. Puncturing the cyst wall within the dermis produces translucent sebum-containing fluid, and ruptured cysts can incite inflammation, pain, and scarring.2 However, prognosis is good, and treatment consists of excision, aspiration and curettage of the cyst wall, oral isotretinoin, or laser therapy. Our patient elected to forego treatment and will consider definitive removal in the future, since the lesions will persist and potentially enlarge. Accurate diagnosis of this rare cause of chest papules improves the timeliness and efficacy of appropriate treatment, favoring good cosmesis.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
1. Zuber TJ. Minimal excision technique for epidermoid (sebaceous) cysts. Am Fam Physician. 2002;65(7):1409-1412.
2. du Vivier A. Atlas of Clinical Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012.
3. Brinster N, Liu V, Diwan AH, McKee PH. High Yield Pathology: Dermatopathology. 1st ed. Philadelphia, PA: Elsevier Saunders; 2011.
4. Wolff K, Johnson RA, Suurmond D. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 5th ed. New York: McGraw-Hill; 2005.
5. Gordon Spratt EA, Kaplan J, Patel RR, Kamino H, Ramachandran SM. Steatocystoma. Dermatol Online J. 2013;19(12):20721.
Reducing COPD Readmission Rates: Using a COPD Care Service During Care Transitions
A chronic obstructive pulmonary disease care service improves timely access to follow-up care and patient education at the time of transition from hospital to home.
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide and has an associated treatment cost of $9,800 per patient per year in the US.1-3 Within 5 years of hospital discharge for a COPD exacerbation, the rehospitalization risk is 44%, and the mortality rate is 55%.4 COPD affects more than 11 million Americans, and the disease prevalence among US veterans is 3-fold higher.5,6
Patients hospitalized for COPD have a 30-day readmission rate of 22.6%.7 Given the high patient burden, COPD was added to the Medicare Hospital Readmission Reductions Program in 2015, resulting in financial penalties for COPD readmissions within 30 days of hospital discharge.8 Ensuring timely access to follow-up care has been shown to significantly reduce risk for hospital readmissions.9 However, in a national review of Medicare claims, only 50% of patients readmitted to the hospital had a primary care provider (PCP) follow-up visit within 30 days of their hospital discharge.10 Despite the need to provide prompt patient follow-up during the transition from hospital to home, gaps within the health care system create barriers to providing timely postdischarge care.10-12 These gaps include breakdowns in practitioner and patient communication, lengthy time to follow-up, and incomplete medication reconciliation.13 To address this unmet need, clinics and hospitals require solutions that can be implemented quickly, using the resources of their current clinical models.
Pharmacists and registered nurses (RNs) within the US federal health care system are well positioned for involvement in the postdischarge care of high-risk patients with COPD. Ambulatory care practitioners within the US Department of Veterans Affairs (VA) health care system are integrated into patient aligned care teams (PACT). Each team consists of a PCP, pharmacist, RN, social worker, dietitian, licensed practical nurse, and medical scheduling support assistant.14 Each PACT team works together to provide patient education, chronic disease management, and medication optimization, and each team member contributes their unique training and expertise.
Interprofessional care is considered an integral method to improve health outcomes through effective teamwork and communication.15 Although interprofessional interventions are cited extensively in the literature highlighting medicine and nursing, a gap exists in the exploration of pharmacist contributions within interprofessional teams.16 The incorporation of clinical pharmacists in the literature is especially limited when considering transitions of care and the patient medical home.17 Given the critical and collaborative role pharmacists play within the PACT medical home, the COPD CARE (Chronic Obstructive Pulmonary Disease Coordinated Access to Reduce Exacerbations) service provides an opportunity to leverage pharmacists as prescribers with a scope of practice who coordinate transitions of care for patients with COPD.18 The service was designed to be collaborative within the PACT model and with the intent of reducing 30-day readmissions to the hospital or emergency department (ED) due to a COPD exacerbation.
This evaluation involved the identifying patients recently hospitalized for COPD; clinic follow-up, coordinated by a clinical pharmacist and nurse, within 30 days of hospital or ED discharge; the use of a COPD action plan; and timely triage of patients at high risk for COPD reexacerbation or with comorbid symptoms to PCPs. The COPD CARE service, leveraged the patient-centered medical home (PCMH) model for transitions of care after COPD exacerbations. The PCMH is a primary care model focused on the following functions: (1) comprehensive care; (2) patient-centered care; (3) coordinated care; (4) accessible service; and (5) quality and safety.19 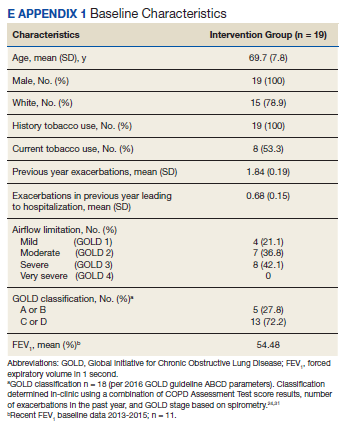
Methods
The COPD CARE service was implemented on October 1, 2015, and evaluated through March 1, 2016 (Figure 1). All veterans receiving primary care through the pilot clinic site with a hospital admission or ED visit for COPD exacerbation were offered this intervention. 
Patient Eligibility and Recruitment
Patients were excluded from the service if COPD or COPD-related diagnoses were not listed in their electronic health record (EHR) problem list. Patients who had previously received components of the intervention through consultation with specialty services were excluded. If a patient declined the service, they received the standard of care. This project was undertaken for programmatic evaluation and qualified for quality improvement (QI) exemption; as such an internal review board approval was not required.
Intervention
Participants enrolled in the COPD CARE service were scheduled for an interprofessional postdischarge follow-up visit with a pharmacist and nurse at the pilot outpatient clinic site, and this visit was termed the COPD CARE health visit. Participants ideally were seen within 30 days of discharge. The goal was to improve access to care while preventing a 30-day readmission. Within this 30-day window, the target follow-up period was 2 to 3 weeks postdischarge for the face-to-face visit. Patients who required postdischarge care for additional medical conditions received a clinic appointment with their PCP on the same day as their COPD CARE health visit. The COPD CARE health visit focused on 3 objectives: (1) COPD disease management and referrals; (2) COPD plan development; and (3) inhaler technique review and teaching.20,21
COPD Monitoring
During the 45-minute COPD CARE health visit, the pharmacist provided extensive disease management based on the GOLD guideline recommendation.22 In addition, the pharmacist administered the COPD Assessment Test (CAT) and reviewed patient COPD exacerbation history to guide prescribing.22 The patient and pharmacist also reviewed previous spirometry results if obtained within the past 2 years. COPD triggers and symptoms were assessed along with opportunities for therapeutic and lifestyle modifications.
Plan Development
Patients in the COPD CARE service also were given a COPD plan to improve health outcomes. (Figure 2). The plan included patient instructions to initiate steroid and antibiotic therapy if the patient experienced symptoms of increased cough, mucus production, and purulence, thereby reaching the high-yellow zone. 
Patient Referrals
Patient referrals also were a critical component of the COPD CARE service. Pharmacists placed referrals for tobacco treatment services, pulmonary rehabilitation, a COPD group education class, and referral to specialty care if needed.
Inhaler Technique Review
Either the pharmacist or RN review the inhaler technique, and corrections and teachback methods used to ensure patient understanding.23 Patients were encouraged to bring home inhalers into clinic for technique assessment. Demonstration inhalers also were available and used by pharmacists and nurses for inhaler teaching as needed. The pharmacist indicated through chart documentation whether the patient’s inhaler technique was correct or whether modifications were made to improve medication delivery. Medication reconciliation also was performed for inhaled devices to insure patients were using medications as prescribed.
Outcomes
The primary outcome of this evaluation was an assessment of interventions made by the interprofessional care team during the COPD CARE health visit. Secondary outcomes included assessment of 30-day readmission rates as well as patient access to the primary care team using this interprofessional care model.
Data were collected after study completion through review of the EHR at baseline and at the end of the evaluation period. Baseline demographic information was collected through a retrospective chart review. Readmission rates were calculated as a composite of ED visits and rehospitalization within 30 days of discharge due to a COPD exacerbation.
Patients’ spirometry results were used in composite with clinical symptoms and risk of exacerbations to calculate GOLD staging.24
Results
A total of 19 patients admitted to the hospital or ED received follow-up through the COPD CARE service. Patients included in this analysis were primarily older adult white males.
Referrals were placed for 53% of patients in the COPD CARE service, with 21% of patients accepting referral to tobacco treatment clinic, and 32% of patients accepting referral to pulmonary rehabilitation. COPD plans were issued to all of patients in this service. Pharmacists modified therapy 58% of the time, with a review of medications prescribed by the clinical pharmacist (eApendixes 1 and 2, available at mdedge.com/fedprac).
Patients had a 0% composite readmission rate to the ED or hospital for a COPD exacerbation within 30-days of discharge. Access to care, defined as a visit with the primary care PACT team within 30 days of discharge, was achieved in 14 of the 19 patients (73.7%). Additionally, 12 of 19 patients (63.2%) in the COPD CARE service no longer needed to see their PCP following discharge, saving their provider a visit.
The pharmacist corrected patient inhaler technique in 52.6% of the patients participating in the service.
Discussion
The intent of this QI initiative was to assess a novel clinic intervention for a high-risk patient population during COPD care transitions. The strengths of this intervention involved a rapid cycle implementation using the existing medical home model and its multiprong approach to coordinating care. This approach involved coordinating self-direction COPD plans, timely hospital follow-up, and the innovative use of the interprofessional primary care team.
The COPD CARE service improved patient access to follow-up with no COPD readmissions in the intervention group. The COPD CARE service also validated the use of a coordinated medical home consisting of clinical pharmacists and nurses who provided the initial COPD disease monitoring and plan development. This intervention also resulted in patients receiving greater access to their PACT teams within 30 days of discharge and a higher rate of referrals to tobacco cessation clinics within the COPD CARE group. In addition, use of tools that enabled patients to self-manage their care, such as the COPD plan, was greater in the COPD CARE group.
The interventions made in-clinic likely contributed to service results (eAppendix 3). 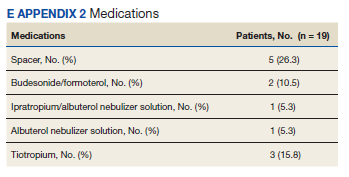
In addition, the COPD CARE service provided necessary referrals to pulmonary rehabilitation, nutrition, and tobacco treatment clinics at a higher rate than those patients in the standard of care group. The high percentage of referrals placed to tobacco treatment clinic and pulmonary rehabilitation contributes to improvements in COPD disease control long-term.25 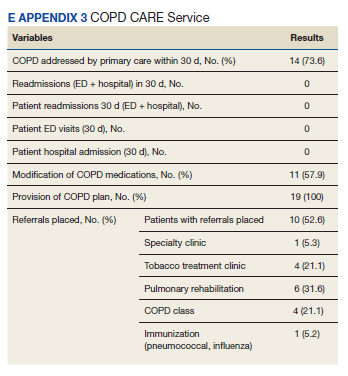
The COPD CARE service may best be described as a model for application of the interprofessional team in clinical practice, with the clinical pharmacist uniquely positioned for chronic disease management in the postacute care setting.26 Previously, literature has documented pharmacists as integral members of the team during patient care transitions. Pharmacist completion of medication reconciliation compared with usual care has shown a 28% relative risk (RR) reduction in ED visits and a 67% RR reduction in adverse drug event-related hospital revisits.27 Findings of the COPD CARE service are consistent with the literature and advance the role of pharmacists within the medical home model as prescribers for disease management.27
The interprofessional, team-based design of the COPD CARE service also is supported by recent recommendations from the COPD Foundation, as detailed in the 2nd National COPD Readmission Summit.28 Use of a proactive, team-based care model is emphasized as a central element to coordinating care transitions, with an expectation of 360 degree accountability by all team members for the patients care both during and after hospitalization. The clearly defined roles of each team member within the COPD CARE service, coupled with the expectation that each team member practices with autonomy and accountability, exemplifies the COPD Foundation vision for enhancing COPD care. In addition, the COPD CARE service uses many of the best practices detailed by the COPD Foundation, including the use of spirometry, referrals to pulmonary rehabilitation, and use of motivational interviewing for tobacco treatment clinic referral.
Limitations
This QI initiative has several limitations. By virtue of the study being designed as a practice improvement intervention with rapid implementation, the existing clinic referral structures were used to offer the service to eligible patients. This standard of care included routine telephone contact by a nurse case manager following hospital discharge. Although all patients in the COPD CARE service received the intervention, 5 patients were not seen within the 30-day window, resulting in an implementation rate of 73%. Of the 5 patients that were not seen, 4 were discharged from the ED. Timely follow-up in primary care clinic from the ED required the use of a time-intensive chart review for referral and subsequent delay in intervention delivery.A streamlined clinic referral process from the ED likely would further improve patient scheduling and result in a greater number of patients who would receive the intervention within 30 days of discharge. Despite this limitation, the COPD CARE service was able to see a large percentage of patients within the 30-day time frame postdischarge.
Future Directions
Although a major objective of this service was to reduce readmissions 30 days postdischarge, it is possible interventions made in clinic may have long-term beneficial effects.25,29 Future research should evaluate the impact of this interprofessional service on long-term disease outcomes, thereby determining whether the promising readmission results are sustained beyond 30 days postdischarge.30 In addition, incorporation of respiratory therapy and inpatient pharmacists during hospital discharge could provide a more effective and sustainable transition from hospital to home before the COPD CARE clinic visit.
Future implementations and evaluations of this COPD CARE service will in turn benefit from a key component of our intervention, which includes the collection of timely CAT scores, spirometry data, and adherence rates for COPD patients.31 Furthermore, the intervention was successfully delivered to a population recently hospitalized or seen in the ED, and therefore, at high risk for future COPD exacerbations. This initiative provides positive proof of a concept QI project using the existing PACT team model to reduce 30-day readmission rates in patients with COPD at high risk for exacerbation. Future efforts will focus on delivering this intervention to patients with mild, moderate, and severe COPD within a wide range of primary clinics.
Conslusion
The COPD CARE service involved the coordinated postdischarge care facilitated by an interprofessional team of clinical pharmacists, nurses and PCPs. The COPD CARE service leveraged an interprofessional team, centered on the PACT medical home, to make clinic interventions resulting in a 0% readmission rate and 63.2% increase in PCP access. The COPD CARE service further demonstrated the impact of coordinated efforts by interprofessional teams to optimize care for COPD management.
Acknowledgments
The authors thank Stephanie Gruber, PharmD; Lieneke Hafeman, RN; Molly Obermark, PharmD; Julia Peek, RT; Mark Regan, MD; Chris Roelke, RN; Steve Shoyer, PharmD; John Thielemann, RN; Sandy Tompkins, BS; and Wendi Wenger, RN, for their integral roles in the COPD CARE service.
1. World Health Organization. The top 10 causes of death. http://www.who.int/mediacentre/factsheets/fs310/en. Updated May 24, 2018. Accessed May 30, 2018.
2. Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged > 18 years in the United States for 2010 and projections through 2020. Chest. 2015;147(1):31-45.
3. American Lung Association. Trends in COPD (chronic bronchitis and emphysema): morbidity and mortality. http://www.lung.org/assets/documents/research/copd-trend-report.pdf. Published March 2013. Accessed May 30, 2018.
4. McGhan R, Radcliff T, Fish R, Sutherland ER, Welsh C, Make B. Predictors of rehospitalization and death after a severe exacerbation of COPD. Chest. 2007;132(6):1748-1755.
5. COPD Foundation. Patient groups back bill supporting US veterans with COPD. https://www.copdfoundation.org/About-Us/Press-Room/Press-Releases/Article/722/Patient-Groups-Back-Bill-Supporting-US-Veterans-with-COPD.aspx. Published November 9, 2010. Accessed May 30, 2018.
6. American Lung Association. Lung health and disease: how serious is COPD. http://www.lung.org/lung-health-and-diseases/lung-disease-lookup/copd/learn-about-copd/how-serious-is-copd.html. Published 2016. Accessed May 30, 2018.
7. Shah T, Press V, Huisingh-Scheetz M, White SR. COPD readmissions: addressing COPD in the era of value-based health care. Chest. 2016;150(4):916-926.
8. Mcllvennan CK, Eapen ZJ, Allen LA. Hospital readmissions reduction program. Circulation. 2015;13(20):1796-1803.
9. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122.
10. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
11. Hitch B, Parlier AB, Reed L, Galvin SL, Fagan EB, Wilson CG. Evaluation of a team-based, transition-of-care management service on 30-day readmission rates. N C Med J. 2016;77(2):87-92.
12. Stone J, Hoffman G. Medicare hospital readmissions: issues, policy options. In: Turner PM ed. Medicare: Background, Benefits and Issues. Nova Science Pub Inc; 2011:123-150.
13. Kripalani S, Jackson AT, Schnipper JL, Coleman EA. Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314-323.
14. Rosland A-M, Nelson K, Sun H, et al. The patient-centered medical home in the Veterans Health Administration. Am J Manag Care. 2013;19(7):e263-e272.
15. World Health Organization. Nursing and midwifery. http://www.who.int/hrh/nursing_midwifery/en. Accessed September 18, 2018.
16. Supper I, Catala O, Lustman M, Chemla C, Bourgueil Y, Letrilliart L. Interprofessional collaboration in primary health care: a review of facilitators and barriers perceived by involved actors. J Public Health (Oxf). 2015;37(4):716-727.
17. Melody KT, McCartney E, Sen S, Duenas G. Optimizing care transitions: the role of the community pharmacist. Integr Pharm Res Pract. 2016;5:43-51.
18. Ourth H, Groppi J, Morreale AP, Quicci-Roberts K. Clinical pharmacist prescribing activities in the Veterans Health Administration. Am J Health Syst Pharm. 2016;73(18):1406-1415.
19. US Department of Health and Human Services. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed May 29, 2018.
20. Kaplan A. The COPD action plan. Can Fam Physician. 2009;55(1):58-59.
21. Turnock AC, Walters EH, Walters JA, Wood-Baker R. Action plans for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;(4):CD005074.
22. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2017 report). http://goldcopd.org/gold-2017-global-strategy-diagnosis-management-prevention-copd. Accessed May 29, 2018.
23. Bonini M, Usmani OS. The importance of inhaler devices in the treatment of COPD. COPD Res Pract. 2015;1:9.
24. Buist AS, Anzueto A, Calverley P, DeGuia TS, Fukuch Y. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2006.
25. McCarthy B, Casey D, Devane D, Murphy K, Murphy E, Lacasse Y. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2015;(2):CD003793.
26. ASHP Research and Education Foundation. Pharmacy forecast 2016-2020: strategic planning advice. http://www.ashpfoundation.org/PharmacyForecast2016. Published December 2015. Accessed May 29, 2018.
27. Mekonnen AB, McLachlan AJ, Brien JE. Effectiveness of pharmacist-led medication reconciliation programmes on clinical outcomes at hospital transitions: a systematic review and meta-analysis. BMJ Open. 2016;6(2):e010003.
28. Willard KS, Sullivan JB, Thomashow BM, et al. The 2nd national COPD readmissions summit and beyond: from theory to implementation. Chronic Obstr Pulm Dis. 2016;3(4):778-790.
29. Scanlon PD, Connett JE, Waller LA, et al; Lung Health Study Research Group. Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease. The lung health study. Am J Respir Crit Care Med. 2000;161(2, pt 1):381-390.
30. Shah T, Press VG, Huisingh-Scheetz M, White SR. COPD readmissions: addressing COPD in the era of value-based health care. Chest. 2016;150(4):916-926.
31. GlaxoSmithKline. COPD Assessment Test (CAT). Castest Online. http://www.catestonline.org/images/UserGuides/CATHCPUser%20guideEn.pdf. Updated October 2016.
A chronic obstructive pulmonary disease care service improves timely access to follow-up care and patient education at the time of transition from hospital to home.
A chronic obstructive pulmonary disease care service improves timely access to follow-up care and patient education at the time of transition from hospital to home.
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide and has an associated treatment cost of $9,800 per patient per year in the US.1-3 Within 5 years of hospital discharge for a COPD exacerbation, the rehospitalization risk is 44%, and the mortality rate is 55%.4 COPD affects more than 11 million Americans, and the disease prevalence among US veterans is 3-fold higher.5,6
Patients hospitalized for COPD have a 30-day readmission rate of 22.6%.7 Given the high patient burden, COPD was added to the Medicare Hospital Readmission Reductions Program in 2015, resulting in financial penalties for COPD readmissions within 30 days of hospital discharge.8 Ensuring timely access to follow-up care has been shown to significantly reduce risk for hospital readmissions.9 However, in a national review of Medicare claims, only 50% of patients readmitted to the hospital had a primary care provider (PCP) follow-up visit within 30 days of their hospital discharge.10 Despite the need to provide prompt patient follow-up during the transition from hospital to home, gaps within the health care system create barriers to providing timely postdischarge care.10-12 These gaps include breakdowns in practitioner and patient communication, lengthy time to follow-up, and incomplete medication reconciliation.13 To address this unmet need, clinics and hospitals require solutions that can be implemented quickly, using the resources of their current clinical models.
Pharmacists and registered nurses (RNs) within the US federal health care system are well positioned for involvement in the postdischarge care of high-risk patients with COPD. Ambulatory care practitioners within the US Department of Veterans Affairs (VA) health care system are integrated into patient aligned care teams (PACT). Each team consists of a PCP, pharmacist, RN, social worker, dietitian, licensed practical nurse, and medical scheduling support assistant.14 Each PACT team works together to provide patient education, chronic disease management, and medication optimization, and each team member contributes their unique training and expertise.
Interprofessional care is considered an integral method to improve health outcomes through effective teamwork and communication.15 Although interprofessional interventions are cited extensively in the literature highlighting medicine and nursing, a gap exists in the exploration of pharmacist contributions within interprofessional teams.16 The incorporation of clinical pharmacists in the literature is especially limited when considering transitions of care and the patient medical home.17 Given the critical and collaborative role pharmacists play within the PACT medical home, the COPD CARE (Chronic Obstructive Pulmonary Disease Coordinated Access to Reduce Exacerbations) service provides an opportunity to leverage pharmacists as prescribers with a scope of practice who coordinate transitions of care for patients with COPD.18 The service was designed to be collaborative within the PACT model and with the intent of reducing 30-day readmissions to the hospital or emergency department (ED) due to a COPD exacerbation.
This evaluation involved the identifying patients recently hospitalized for COPD; clinic follow-up, coordinated by a clinical pharmacist and nurse, within 30 days of hospital or ED discharge; the use of a COPD action plan; and timely triage of patients at high risk for COPD reexacerbation or with comorbid symptoms to PCPs. The COPD CARE service, leveraged the patient-centered medical home (PCMH) model for transitions of care after COPD exacerbations. The PCMH is a primary care model focused on the following functions: (1) comprehensive care; (2) patient-centered care; (3) coordinated care; (4) accessible service; and (5) quality and safety.19
Methods
The COPD CARE service was implemented on October 1, 2015, and evaluated through March 1, 2016 (Figure 1). All veterans receiving primary care through the pilot clinic site with a hospital admission or ED visit for COPD exacerbation were offered this intervention.
Patient Eligibility and Recruitment
Patients were excluded from the service if COPD or COPD-related diagnoses were not listed in their electronic health record (EHR) problem list. Patients who had previously received components of the intervention through consultation with specialty services were excluded. If a patient declined the service, they received the standard of care. This project was undertaken for programmatic evaluation and qualified for quality improvement (QI) exemption; as such an internal review board approval was not required.
Intervention
Participants enrolled in the COPD CARE service were scheduled for an interprofessional postdischarge follow-up visit with a pharmacist and nurse at the pilot outpatient clinic site, and this visit was termed the COPD CARE health visit. Participants ideally were seen within 30 days of discharge. The goal was to improve access to care while preventing a 30-day readmission. Within this 30-day window, the target follow-up period was 2 to 3 weeks postdischarge for the face-to-face visit. Patients who required postdischarge care for additional medical conditions received a clinic appointment with their PCP on the same day as their COPD CARE health visit. The COPD CARE health visit focused on 3 objectives: (1) COPD disease management and referrals; (2) COPD plan development; and (3) inhaler technique review and teaching.20,21
COPD Monitoring
During the 45-minute COPD CARE health visit, the pharmacist provided extensive disease management based on the GOLD guideline recommendation.22 In addition, the pharmacist administered the COPD Assessment Test (CAT) and reviewed patient COPD exacerbation history to guide prescribing.22 The patient and pharmacist also reviewed previous spirometry results if obtained within the past 2 years. COPD triggers and symptoms were assessed along with opportunities for therapeutic and lifestyle modifications.
Plan Development
Patients in the COPD CARE service also were given a COPD plan to improve health outcomes. (Figure 2). The plan included patient instructions to initiate steroid and antibiotic therapy if the patient experienced symptoms of increased cough, mucus production, and purulence, thereby reaching the high-yellow zone.
Patient Referrals
Patient referrals also were a critical component of the COPD CARE service. Pharmacists placed referrals for tobacco treatment services, pulmonary rehabilitation, a COPD group education class, and referral to specialty care if needed.
Inhaler Technique Review
Either the pharmacist or RN review the inhaler technique, and corrections and teachback methods used to ensure patient understanding.23 Patients were encouraged to bring home inhalers into clinic for technique assessment. Demonstration inhalers also were available and used by pharmacists and nurses for inhaler teaching as needed. The pharmacist indicated through chart documentation whether the patient’s inhaler technique was correct or whether modifications were made to improve medication delivery. Medication reconciliation also was performed for inhaled devices to insure patients were using medications as prescribed.
Outcomes
The primary outcome of this evaluation was an assessment of interventions made by the interprofessional care team during the COPD CARE health visit. Secondary outcomes included assessment of 30-day readmission rates as well as patient access to the primary care team using this interprofessional care model.
Data were collected after study completion through review of the EHR at baseline and at the end of the evaluation period. Baseline demographic information was collected through a retrospective chart review. Readmission rates were calculated as a composite of ED visits and rehospitalization within 30 days of discharge due to a COPD exacerbation.
Patients’ spirometry results were used in composite with clinical symptoms and risk of exacerbations to calculate GOLD staging.24
Results
A total of 19 patients admitted to the hospital or ED received follow-up through the COPD CARE service. Patients included in this analysis were primarily older adult white males.
Referrals were placed for 53% of patients in the COPD CARE service, with 21% of patients accepting referral to tobacco treatment clinic, and 32% of patients accepting referral to pulmonary rehabilitation. COPD plans were issued to all of patients in this service. Pharmacists modified therapy 58% of the time, with a review of medications prescribed by the clinical pharmacist (eApendixes 1 and 2, available at mdedge.com/fedprac).
Patients had a 0% composite readmission rate to the ED or hospital for a COPD exacerbation within 30-days of discharge. Access to care, defined as a visit with the primary care PACT team within 30 days of discharge, was achieved in 14 of the 19 patients (73.7%). Additionally, 12 of 19 patients (63.2%) in the COPD CARE service no longer needed to see their PCP following discharge, saving their provider a visit.
The pharmacist corrected patient inhaler technique in 52.6% of the patients participating in the service.
Discussion
The intent of this QI initiative was to assess a novel clinic intervention for a high-risk patient population during COPD care transitions. The strengths of this intervention involved a rapid cycle implementation using the existing medical home model and its multiprong approach to coordinating care. This approach involved coordinating self-direction COPD plans, timely hospital follow-up, and the innovative use of the interprofessional primary care team.
The COPD CARE service improved patient access to follow-up with no COPD readmissions in the intervention group. The COPD CARE service also validated the use of a coordinated medical home consisting of clinical pharmacists and nurses who provided the initial COPD disease monitoring and plan development. This intervention also resulted in patients receiving greater access to their PACT teams within 30 days of discharge and a higher rate of referrals to tobacco cessation clinics within the COPD CARE group. In addition, use of tools that enabled patients to self-manage their care, such as the COPD plan, was greater in the COPD CARE group.
The interventions made in-clinic likely contributed to service results (eAppendix 3).
In addition, the COPD CARE service provided necessary referrals to pulmonary rehabilitation, nutrition, and tobacco treatment clinics at a higher rate than those patients in the standard of care group. The high percentage of referrals placed to tobacco treatment clinic and pulmonary rehabilitation contributes to improvements in COPD disease control long-term.25
The COPD CARE service may best be described as a model for application of the interprofessional team in clinical practice, with the clinical pharmacist uniquely positioned for chronic disease management in the postacute care setting.26 Previously, literature has documented pharmacists as integral members of the team during patient care transitions. Pharmacist completion of medication reconciliation compared with usual care has shown a 28% relative risk (RR) reduction in ED visits and a 67% RR reduction in adverse drug event-related hospital revisits.27 Findings of the COPD CARE service are consistent with the literature and advance the role of pharmacists within the medical home model as prescribers for disease management.27
The interprofessional, team-based design of the COPD CARE service also is supported by recent recommendations from the COPD Foundation, as detailed in the 2nd National COPD Readmission Summit.28 Use of a proactive, team-based care model is emphasized as a central element to coordinating care transitions, with an expectation of 360 degree accountability by all team members for the patients care both during and after hospitalization. The clearly defined roles of each team member within the COPD CARE service, coupled with the expectation that each team member practices with autonomy and accountability, exemplifies the COPD Foundation vision for enhancing COPD care. In addition, the COPD CARE service uses many of the best practices detailed by the COPD Foundation, including the use of spirometry, referrals to pulmonary rehabilitation, and use of motivational interviewing for tobacco treatment clinic referral.
Limitations
This QI initiative has several limitations. By virtue of the study being designed as a practice improvement intervention with rapid implementation, the existing clinic referral structures were used to offer the service to eligible patients. This standard of care included routine telephone contact by a nurse case manager following hospital discharge. Although all patients in the COPD CARE service received the intervention, 5 patients were not seen within the 30-day window, resulting in an implementation rate of 73%. Of the 5 patients that were not seen, 4 were discharged from the ED. Timely follow-up in primary care clinic from the ED required the use of a time-intensive chart review for referral and subsequent delay in intervention delivery.A streamlined clinic referral process from the ED likely would further improve patient scheduling and result in a greater number of patients who would receive the intervention within 30 days of discharge. Despite this limitation, the COPD CARE service was able to see a large percentage of patients within the 30-day time frame postdischarge.
Future Directions
Although a major objective of this service was to reduce readmissions 30 days postdischarge, it is possible interventions made in clinic may have long-term beneficial effects.25,29 Future research should evaluate the impact of this interprofessional service on long-term disease outcomes, thereby determining whether the promising readmission results are sustained beyond 30 days postdischarge.30 In addition, incorporation of respiratory therapy and inpatient pharmacists during hospital discharge could provide a more effective and sustainable transition from hospital to home before the COPD CARE clinic visit.
Future implementations and evaluations of this COPD CARE service will in turn benefit from a key component of our intervention, which includes the collection of timely CAT scores, spirometry data, and adherence rates for COPD patients.31 Furthermore, the intervention was successfully delivered to a population recently hospitalized or seen in the ED, and therefore, at high risk for future COPD exacerbations. This initiative provides positive proof of a concept QI project using the existing PACT team model to reduce 30-day readmission rates in patients with COPD at high risk for exacerbation. Future efforts will focus on delivering this intervention to patients with mild, moderate, and severe COPD within a wide range of primary clinics.
Conslusion
The COPD CARE service involved the coordinated postdischarge care facilitated by an interprofessional team of clinical pharmacists, nurses and PCPs. The COPD CARE service leveraged an interprofessional team, centered on the PACT medical home, to make clinic interventions resulting in a 0% readmission rate and 63.2% increase in PCP access. The COPD CARE service further demonstrated the impact of coordinated efforts by interprofessional teams to optimize care for COPD management.
Acknowledgments
The authors thank Stephanie Gruber, PharmD; Lieneke Hafeman, RN; Molly Obermark, PharmD; Julia Peek, RT; Mark Regan, MD; Chris Roelke, RN; Steve Shoyer, PharmD; John Thielemann, RN; Sandy Tompkins, BS; and Wendi Wenger, RN, for their integral roles in the COPD CARE service.
Chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide and has an associated treatment cost of $9,800 per patient per year in the US.1-3 Within 5 years of hospital discharge for a COPD exacerbation, the rehospitalization risk is 44%, and the mortality rate is 55%.4 COPD affects more than 11 million Americans, and the disease prevalence among US veterans is 3-fold higher.5,6
Patients hospitalized for COPD have a 30-day readmission rate of 22.6%.7 Given the high patient burden, COPD was added to the Medicare Hospital Readmission Reductions Program in 2015, resulting in financial penalties for COPD readmissions within 30 days of hospital discharge.8 Ensuring timely access to follow-up care has been shown to significantly reduce risk for hospital readmissions.9 However, in a national review of Medicare claims, only 50% of patients readmitted to the hospital had a primary care provider (PCP) follow-up visit within 30 days of their hospital discharge.10 Despite the need to provide prompt patient follow-up during the transition from hospital to home, gaps within the health care system create barriers to providing timely postdischarge care.10-12 These gaps include breakdowns in practitioner and patient communication, lengthy time to follow-up, and incomplete medication reconciliation.13 To address this unmet need, clinics and hospitals require solutions that can be implemented quickly, using the resources of their current clinical models.
Pharmacists and registered nurses (RNs) within the US federal health care system are well positioned for involvement in the postdischarge care of high-risk patients with COPD. Ambulatory care practitioners within the US Department of Veterans Affairs (VA) health care system are integrated into patient aligned care teams (PACT). Each team consists of a PCP, pharmacist, RN, social worker, dietitian, licensed practical nurse, and medical scheduling support assistant.14 Each PACT team works together to provide patient education, chronic disease management, and medication optimization, and each team member contributes their unique training and expertise.
Interprofessional care is considered an integral method to improve health outcomes through effective teamwork and communication.15 Although interprofessional interventions are cited extensively in the literature highlighting medicine and nursing, a gap exists in the exploration of pharmacist contributions within interprofessional teams.16 The incorporation of clinical pharmacists in the literature is especially limited when considering transitions of care and the patient medical home.17 Given the critical and collaborative role pharmacists play within the PACT medical home, the COPD CARE (Chronic Obstructive Pulmonary Disease Coordinated Access to Reduce Exacerbations) service provides an opportunity to leverage pharmacists as prescribers with a scope of practice who coordinate transitions of care for patients with COPD.18 The service was designed to be collaborative within the PACT model and with the intent of reducing 30-day readmissions to the hospital or emergency department (ED) due to a COPD exacerbation.
This evaluation involved the identifying patients recently hospitalized for COPD; clinic follow-up, coordinated by a clinical pharmacist and nurse, within 30 days of hospital or ED discharge; the use of a COPD action plan; and timely triage of patients at high risk for COPD reexacerbation or with comorbid symptoms to PCPs. The COPD CARE service, leveraged the patient-centered medical home (PCMH) model for transitions of care after COPD exacerbations. The PCMH is a primary care model focused on the following functions: (1) comprehensive care; (2) patient-centered care; (3) coordinated care; (4) accessible service; and (5) quality and safety.19
Methods
The COPD CARE service was implemented on October 1, 2015, and evaluated through March 1, 2016 (Figure 1). All veterans receiving primary care through the pilot clinic site with a hospital admission or ED visit for COPD exacerbation were offered this intervention.
Patient Eligibility and Recruitment
Patients were excluded from the service if COPD or COPD-related diagnoses were not listed in their electronic health record (EHR) problem list. Patients who had previously received components of the intervention through consultation with specialty services were excluded. If a patient declined the service, they received the standard of care. This project was undertaken for programmatic evaluation and qualified for quality improvement (QI) exemption; as such an internal review board approval was not required.
Intervention
Participants enrolled in the COPD CARE service were scheduled for an interprofessional postdischarge follow-up visit with a pharmacist and nurse at the pilot outpatient clinic site, and this visit was termed the COPD CARE health visit. Participants ideally were seen within 30 days of discharge. The goal was to improve access to care while preventing a 30-day readmission. Within this 30-day window, the target follow-up period was 2 to 3 weeks postdischarge for the face-to-face visit. Patients who required postdischarge care for additional medical conditions received a clinic appointment with their PCP on the same day as their COPD CARE health visit. The COPD CARE health visit focused on 3 objectives: (1) COPD disease management and referrals; (2) COPD plan development; and (3) inhaler technique review and teaching.20,21
COPD Monitoring
During the 45-minute COPD CARE health visit, the pharmacist provided extensive disease management based on the GOLD guideline recommendation.22 In addition, the pharmacist administered the COPD Assessment Test (CAT) and reviewed patient COPD exacerbation history to guide prescribing.22 The patient and pharmacist also reviewed previous spirometry results if obtained within the past 2 years. COPD triggers and symptoms were assessed along with opportunities for therapeutic and lifestyle modifications.
Plan Development
Patients in the COPD CARE service also were given a COPD plan to improve health outcomes. (Figure 2). The plan included patient instructions to initiate steroid and antibiotic therapy if the patient experienced symptoms of increased cough, mucus production, and purulence, thereby reaching the high-yellow zone.
Patient Referrals
Patient referrals also were a critical component of the COPD CARE service. Pharmacists placed referrals for tobacco treatment services, pulmonary rehabilitation, a COPD group education class, and referral to specialty care if needed.
Inhaler Technique Review
Either the pharmacist or RN review the inhaler technique, and corrections and teachback methods used to ensure patient understanding.23 Patients were encouraged to bring home inhalers into clinic for technique assessment. Demonstration inhalers also were available and used by pharmacists and nurses for inhaler teaching as needed. The pharmacist indicated through chart documentation whether the patient’s inhaler technique was correct or whether modifications were made to improve medication delivery. Medication reconciliation also was performed for inhaled devices to insure patients were using medications as prescribed.
Outcomes
The primary outcome of this evaluation was an assessment of interventions made by the interprofessional care team during the COPD CARE health visit. Secondary outcomes included assessment of 30-day readmission rates as well as patient access to the primary care team using this interprofessional care model.
Data were collected after study completion through review of the EHR at baseline and at the end of the evaluation period. Baseline demographic information was collected through a retrospective chart review. Readmission rates were calculated as a composite of ED visits and rehospitalization within 30 days of discharge due to a COPD exacerbation.
Patients’ spirometry results were used in composite with clinical symptoms and risk of exacerbations to calculate GOLD staging.24
Results
A total of 19 patients admitted to the hospital or ED received follow-up through the COPD CARE service. Patients included in this analysis were primarily older adult white males.
Referrals were placed for 53% of patients in the COPD CARE service, with 21% of patients accepting referral to tobacco treatment clinic, and 32% of patients accepting referral to pulmonary rehabilitation. COPD plans were issued to all of patients in this service. Pharmacists modified therapy 58% of the time, with a review of medications prescribed by the clinical pharmacist (eApendixes 1 and 2, available at mdedge.com/fedprac).
Patients had a 0% composite readmission rate to the ED or hospital for a COPD exacerbation within 30-days of discharge. Access to care, defined as a visit with the primary care PACT team within 30 days of discharge, was achieved in 14 of the 19 patients (73.7%). Additionally, 12 of 19 patients (63.2%) in the COPD CARE service no longer needed to see their PCP following discharge, saving their provider a visit.
The pharmacist corrected patient inhaler technique in 52.6% of the patients participating in the service.
Discussion
The intent of this QI initiative was to assess a novel clinic intervention for a high-risk patient population during COPD care transitions. The strengths of this intervention involved a rapid cycle implementation using the existing medical home model and its multiprong approach to coordinating care. This approach involved coordinating self-direction COPD plans, timely hospital follow-up, and the innovative use of the interprofessional primary care team.
The COPD CARE service improved patient access to follow-up with no COPD readmissions in the intervention group. The COPD CARE service also validated the use of a coordinated medical home consisting of clinical pharmacists and nurses who provided the initial COPD disease monitoring and plan development. This intervention also resulted in patients receiving greater access to their PACT teams within 30 days of discharge and a higher rate of referrals to tobacco cessation clinics within the COPD CARE group. In addition, use of tools that enabled patients to self-manage their care, such as the COPD plan, was greater in the COPD CARE group.
The interventions made in-clinic likely contributed to service results (eAppendix 3).
In addition, the COPD CARE service provided necessary referrals to pulmonary rehabilitation, nutrition, and tobacco treatment clinics at a higher rate than those patients in the standard of care group. The high percentage of referrals placed to tobacco treatment clinic and pulmonary rehabilitation contributes to improvements in COPD disease control long-term.25
The COPD CARE service may best be described as a model for application of the interprofessional team in clinical practice, with the clinical pharmacist uniquely positioned for chronic disease management in the postacute care setting.26 Previously, literature has documented pharmacists as integral members of the team during patient care transitions. Pharmacist completion of medication reconciliation compared with usual care has shown a 28% relative risk (RR) reduction in ED visits and a 67% RR reduction in adverse drug event-related hospital revisits.27 Findings of the COPD CARE service are consistent with the literature and advance the role of pharmacists within the medical home model as prescribers for disease management.27
The interprofessional, team-based design of the COPD CARE service also is supported by recent recommendations from the COPD Foundation, as detailed in the 2nd National COPD Readmission Summit.28 Use of a proactive, team-based care model is emphasized as a central element to coordinating care transitions, with an expectation of 360 degree accountability by all team members for the patients care both during and after hospitalization. The clearly defined roles of each team member within the COPD CARE service, coupled with the expectation that each team member practices with autonomy and accountability, exemplifies the COPD Foundation vision for enhancing COPD care. In addition, the COPD CARE service uses many of the best practices detailed by the COPD Foundation, including the use of spirometry, referrals to pulmonary rehabilitation, and use of motivational interviewing for tobacco treatment clinic referral.
Limitations
This QI initiative has several limitations. By virtue of the study being designed as a practice improvement intervention with rapid implementation, the existing clinic referral structures were used to offer the service to eligible patients. This standard of care included routine telephone contact by a nurse case manager following hospital discharge. Although all patients in the COPD CARE service received the intervention, 5 patients were not seen within the 30-day window, resulting in an implementation rate of 73%. Of the 5 patients that were not seen, 4 were discharged from the ED. Timely follow-up in primary care clinic from the ED required the use of a time-intensive chart review for referral and subsequent delay in intervention delivery.A streamlined clinic referral process from the ED likely would further improve patient scheduling and result in a greater number of patients who would receive the intervention within 30 days of discharge. Despite this limitation, the COPD CARE service was able to see a large percentage of patients within the 30-day time frame postdischarge.
Future Directions
Although a major objective of this service was to reduce readmissions 30 days postdischarge, it is possible interventions made in clinic may have long-term beneficial effects.25,29 Future research should evaluate the impact of this interprofessional service on long-term disease outcomes, thereby determining whether the promising readmission results are sustained beyond 30 days postdischarge.30 In addition, incorporation of respiratory therapy and inpatient pharmacists during hospital discharge could provide a more effective and sustainable transition from hospital to home before the COPD CARE clinic visit.
Future implementations and evaluations of this COPD CARE service will in turn benefit from a key component of our intervention, which includes the collection of timely CAT scores, spirometry data, and adherence rates for COPD patients.31 Furthermore, the intervention was successfully delivered to a population recently hospitalized or seen in the ED, and therefore, at high risk for future COPD exacerbations. This initiative provides positive proof of a concept QI project using the existing PACT team model to reduce 30-day readmission rates in patients with COPD at high risk for exacerbation. Future efforts will focus on delivering this intervention to patients with mild, moderate, and severe COPD within a wide range of primary clinics.
Conslusion
The COPD CARE service involved the coordinated postdischarge care facilitated by an interprofessional team of clinical pharmacists, nurses and PCPs. The COPD CARE service leveraged an interprofessional team, centered on the PACT medical home, to make clinic interventions resulting in a 0% readmission rate and 63.2% increase in PCP access. The COPD CARE service further demonstrated the impact of coordinated efforts by interprofessional teams to optimize care for COPD management.
Acknowledgments
The authors thank Stephanie Gruber, PharmD; Lieneke Hafeman, RN; Molly Obermark, PharmD; Julia Peek, RT; Mark Regan, MD; Chris Roelke, RN; Steve Shoyer, PharmD; John Thielemann, RN; Sandy Tompkins, BS; and Wendi Wenger, RN, for their integral roles in the COPD CARE service.
1. World Health Organization. The top 10 causes of death. http://www.who.int/mediacentre/factsheets/fs310/en. Updated May 24, 2018. Accessed May 30, 2018.
2. Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged > 18 years in the United States for 2010 and projections through 2020. Chest. 2015;147(1):31-45.
3. American Lung Association. Trends in COPD (chronic bronchitis and emphysema): morbidity and mortality. http://www.lung.org/assets/documents/research/copd-trend-report.pdf. Published March 2013. Accessed May 30, 2018.
4. McGhan R, Radcliff T, Fish R, Sutherland ER, Welsh C, Make B. Predictors of rehospitalization and death after a severe exacerbation of COPD. Chest. 2007;132(6):1748-1755.
5. COPD Foundation. Patient groups back bill supporting US veterans with COPD. https://www.copdfoundation.org/About-Us/Press-Room/Press-Releases/Article/722/Patient-Groups-Back-Bill-Supporting-US-Veterans-with-COPD.aspx. Published November 9, 2010. Accessed May 30, 2018.
6. American Lung Association. Lung health and disease: how serious is COPD. http://www.lung.org/lung-health-and-diseases/lung-disease-lookup/copd/learn-about-copd/how-serious-is-copd.html. Published 2016. Accessed May 30, 2018.
7. Shah T, Press V, Huisingh-Scheetz M, White SR. COPD readmissions: addressing COPD in the era of value-based health care. Chest. 2016;150(4):916-926.
8. Mcllvennan CK, Eapen ZJ, Allen LA. Hospital readmissions reduction program. Circulation. 2015;13(20):1796-1803.
9. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122.
10. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
11. Hitch B, Parlier AB, Reed L, Galvin SL, Fagan EB, Wilson CG. Evaluation of a team-based, transition-of-care management service on 30-day readmission rates. N C Med J. 2016;77(2):87-92.
12. Stone J, Hoffman G. Medicare hospital readmissions: issues, policy options. In: Turner PM ed. Medicare: Background, Benefits and Issues. Nova Science Pub Inc; 2011:123-150.
13. Kripalani S, Jackson AT, Schnipper JL, Coleman EA. Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314-323.
14. Rosland A-M, Nelson K, Sun H, et al. The patient-centered medical home in the Veterans Health Administration. Am J Manag Care. 2013;19(7):e263-e272.
15. World Health Organization. Nursing and midwifery. http://www.who.int/hrh/nursing_midwifery/en. Accessed September 18, 2018.
16. Supper I, Catala O, Lustman M, Chemla C, Bourgueil Y, Letrilliart L. Interprofessional collaboration in primary health care: a review of facilitators and barriers perceived by involved actors. J Public Health (Oxf). 2015;37(4):716-727.
17. Melody KT, McCartney E, Sen S, Duenas G. Optimizing care transitions: the role of the community pharmacist. Integr Pharm Res Pract. 2016;5:43-51.
18. Ourth H, Groppi J, Morreale AP, Quicci-Roberts K. Clinical pharmacist prescribing activities in the Veterans Health Administration. Am J Health Syst Pharm. 2016;73(18):1406-1415.
19. US Department of Health and Human Services. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed May 29, 2018.
20. Kaplan A. The COPD action plan. Can Fam Physician. 2009;55(1):58-59.
21. Turnock AC, Walters EH, Walters JA, Wood-Baker R. Action plans for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;(4):CD005074.
22. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2017 report). http://goldcopd.org/gold-2017-global-strategy-diagnosis-management-prevention-copd. Accessed May 29, 2018.
23. Bonini M, Usmani OS. The importance of inhaler devices in the treatment of COPD. COPD Res Pract. 2015;1:9.
24. Buist AS, Anzueto A, Calverley P, DeGuia TS, Fukuch Y. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2006.
25. McCarthy B, Casey D, Devane D, Murphy K, Murphy E, Lacasse Y. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2015;(2):CD003793.
26. ASHP Research and Education Foundation. Pharmacy forecast 2016-2020: strategic planning advice. http://www.ashpfoundation.org/PharmacyForecast2016. Published December 2015. Accessed May 29, 2018.
27. Mekonnen AB, McLachlan AJ, Brien JE. Effectiveness of pharmacist-led medication reconciliation programmes on clinical outcomes at hospital transitions: a systematic review and meta-analysis. BMJ Open. 2016;6(2):e010003.
28. Willard KS, Sullivan JB, Thomashow BM, et al. The 2nd national COPD readmissions summit and beyond: from theory to implementation. Chronic Obstr Pulm Dis. 2016;3(4):778-790.
29. Scanlon PD, Connett JE, Waller LA, et al; Lung Health Study Research Group. Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease. The lung health study. Am J Respir Crit Care Med. 2000;161(2, pt 1):381-390.
30. Shah T, Press VG, Huisingh-Scheetz M, White SR. COPD readmissions: addressing COPD in the era of value-based health care. Chest. 2016;150(4):916-926.
31. GlaxoSmithKline. COPD Assessment Test (CAT). Castest Online. http://www.catestonline.org/images/UserGuides/CATHCPUser%20guideEn.pdf. Updated October 2016.
1. World Health Organization. The top 10 causes of death. http://www.who.int/mediacentre/factsheets/fs310/en. Updated May 24, 2018. Accessed May 30, 2018.
2. Ford ES, Murphy LB, Khavjou O, Giles WH, Holt JB, Croft JB. Total and state-specific medical and absenteeism costs of COPD among adults aged > 18 years in the United States for 2010 and projections through 2020. Chest. 2015;147(1):31-45.
3. American Lung Association. Trends in COPD (chronic bronchitis and emphysema): morbidity and mortality. http://www.lung.org/assets/documents/research/copd-trend-report.pdf. Published March 2013. Accessed May 30, 2018.
4. McGhan R, Radcliff T, Fish R, Sutherland ER, Welsh C, Make B. Predictors of rehospitalization and death after a severe exacerbation of COPD. Chest. 2007;132(6):1748-1755.
5. COPD Foundation. Patient groups back bill supporting US veterans with COPD. https://www.copdfoundation.org/About-Us/Press-Room/Press-Releases/Article/722/Patient-Groups-Back-Bill-Supporting-US-Veterans-with-COPD.aspx. Published November 9, 2010. Accessed May 30, 2018.
6. American Lung Association. Lung health and disease: how serious is COPD. http://www.lung.org/lung-health-and-diseases/lung-disease-lookup/copd/learn-about-copd/how-serious-is-copd.html. Published 2016. Accessed May 30, 2018.
7. Shah T, Press V, Huisingh-Scheetz M, White SR. COPD readmissions: addressing COPD in the era of value-based health care. Chest. 2016;150(4):916-926.
8. Mcllvennan CK, Eapen ZJ, Allen LA. Hospital readmissions reduction program. Circulation. 2015;13(20):1796-1803.
9. Jackson C, Shahsahebi M, Wedlake T, DuBard CA. Timeliness of outpatient follow-up: an evidence-based approach for planning after hospital discharge. Ann Fam Med. 2015;13(2):115-122.
10. Jencks SF, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
11. Hitch B, Parlier AB, Reed L, Galvin SL, Fagan EB, Wilson CG. Evaluation of a team-based, transition-of-care management service on 30-day readmission rates. N C Med J. 2016;77(2):87-92.
12. Stone J, Hoffman G. Medicare hospital readmissions: issues, policy options. In: Turner PM ed. Medicare: Background, Benefits and Issues. Nova Science Pub Inc; 2011:123-150.
13. Kripalani S, Jackson AT, Schnipper JL, Coleman EA. Promoting effective transitions of care at hospital discharge: a review of key issues for hospitalists. J Hosp Med. 2007;2(5):314-323.
14. Rosland A-M, Nelson K, Sun H, et al. The patient-centered medical home in the Veterans Health Administration. Am J Manag Care. 2013;19(7):e263-e272.
15. World Health Organization. Nursing and midwifery. http://www.who.int/hrh/nursing_midwifery/en. Accessed September 18, 2018.
16. Supper I, Catala O, Lustman M, Chemla C, Bourgueil Y, Letrilliart L. Interprofessional collaboration in primary health care: a review of facilitators and barriers perceived by involved actors. J Public Health (Oxf). 2015;37(4):716-727.
17. Melody KT, McCartney E, Sen S, Duenas G. Optimizing care transitions: the role of the community pharmacist. Integr Pharm Res Pract. 2016;5:43-51.
18. Ourth H, Groppi J, Morreale AP, Quicci-Roberts K. Clinical pharmacist prescribing activities in the Veterans Health Administration. Am J Health Syst Pharm. 2016;73(18):1406-1415.
19. US Department of Health and Human Services. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed May 29, 2018.
20. Kaplan A. The COPD action plan. Can Fam Physician. 2009;55(1):58-59.
21. Turnock AC, Walters EH, Walters JA, Wood-Baker R. Action plans for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2005;(4):CD005074.
22. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2017 report). http://goldcopd.org/gold-2017-global-strategy-diagnosis-management-prevention-copd. Accessed May 29, 2018.
23. Bonini M, Usmani OS. The importance of inhaler devices in the treatment of COPD. COPD Res Pract. 2015;1:9.
24. Buist AS, Anzueto A, Calverley P, DeGuia TS, Fukuch Y. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2006.
25. McCarthy B, Casey D, Devane D, Murphy K, Murphy E, Lacasse Y. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2015;(2):CD003793.
26. ASHP Research and Education Foundation. Pharmacy forecast 2016-2020: strategic planning advice. http://www.ashpfoundation.org/PharmacyForecast2016. Published December 2015. Accessed May 29, 2018.
27. Mekonnen AB, McLachlan AJ, Brien JE. Effectiveness of pharmacist-led medication reconciliation programmes on clinical outcomes at hospital transitions: a systematic review and meta-analysis. BMJ Open. 2016;6(2):e010003.
28. Willard KS, Sullivan JB, Thomashow BM, et al. The 2nd national COPD readmissions summit and beyond: from theory to implementation. Chronic Obstr Pulm Dis. 2016;3(4):778-790.
29. Scanlon PD, Connett JE, Waller LA, et al; Lung Health Study Research Group. Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease. The lung health study. Am J Respir Crit Care Med. 2000;161(2, pt 1):381-390.
30. Shah T, Press VG, Huisingh-Scheetz M, White SR. COPD readmissions: addressing COPD in the era of value-based health care. Chest. 2016;150(4):916-926.
31. GlaxoSmithKline. COPD Assessment Test (CAT). Castest Online. http://www.catestonline.org/images/UserGuides/CATHCPUser%20guideEn.pdf. Updated October 2016.