User login
Treatment of Inpatient Asymptomatic Hypertension: Not a Call to Act but to Think
Your pager beeps. Your patient, Mrs. Jones, who was admitted with cellulitis and is improving, now has a blood pressure of 188/103 on routine vitals. Her nurse reports that she is comfortable and asymptomatic, but she met the “call parameters.” You review her chart and find that since admission her systolic blood pressure (SBP) has ranged from 149 to 157 mm Hg and her diastolic blood pressure (DBP) from 84 to 96 mm Hg. Her nurse asks how you would like to treat her.
While over half of inpatients have at least one hypertensive episode during their stay, evidence suggests that nearly all such episodes—estimates are between 98% and 99%1,2—should be treated over several days with oral antihypertensives, not acutely with intravenous medications.3-6 Current guidelines recommend that intravenous medications should be reserved for severe hypertensive episodes (SBP > 180, DBP > 120) with acute end-organ damage,7,8 but such “hypertensive emergencies” are rare on the general medicine wards. Still, hospitalists regularly face the dilemma posed by Mrs. Jones, and evidence shows they often prescribe intravenous antihypertensives.1,4,5 This unnecessary use can lead to unreliable drops in blood pressure and exposes our patients to potential harm.5,6
In this issue of the Journal of Hospital Medicine, two papers describe the frequency of inappropriate intravenous antihypertensive use in their hospitals and the subsequent quality improvement efforts implemented to reduce this practice. The first, by Jacobs et al., found that over a 10-month period, 11% of patients who experienced “asymptomatic hypertension” on an urban academic hospital medicine service were treated inappropriately with intravenous antihypertensives,9 with 14% of those experiencing an adverse event. The second paper, by Pasik et al., found that in their urban academic medical center there were 8.3 inappropriate intravenous antihypertensive orders placed per 1,000 patient days,10 with nearly half of those treated experiencing an adverse event. Based on these findings, each group then led interventions to reduce the use of intravenous antihypertensives.
While both groups engaged physicians and nurses as primary stakeholders, Pasik et al.10 worked to further expand nursing staff roles by empowering them to assess for underlying causes of hypertension, such as pain or anxiety, as well as end-organ damage via specific guided algorithms prior to contacting physicians. In doing so, they reduced intravenous antihypertensive use by 60% during the postintervention period, with a proportional reduction in adverse events. In addition to their educational initiative, Jacobs et al. aimed to limit calls by liberalizing the “ceiling” on standard nursing call parameters for blood pressure from 160/80 to 180/90. Following their intervention, intravenous antihypertensive orders were reduced by 40%, with the mean orders per patient with asymptomatic hypertension decreasing from 11% to 7% .
While these results are admirable, some caution in their interpretation is needed. For example, Jacobs et al. used electronic health record data to retrospectively identify hypertension as “symptomatic” or “asymptomatic” using laboratory, electrocardiogram, and imaging diagnostics as surrogate markers for “provider concern for end-organ damage.” Although it appropriately focused on concern for end-organ damage as justification for intravenous antihypertensives, this approach potentially underappreciated true hypertensive emergencies, thereby overestimating the amount of inappropriate use of intravenous antihypertensives. Pasik et al. utilized chart review of patients prescribed intravenous antihypertensives and therefore did not explore how often symptomatic hypertension occurred in patients who did not receive intravenous antihypertensives. Subsequently, this limited their ability to evaluate unintended harms of their initiative. To address this limitation, the authors followed a group of 111 patients who had elevated hypertension but did not receive intravenous antihypertensives and found no adverse outcomes.10 Because both studies were retrospective in nature, they were subject to biases from providers choosing intravenous antihypertensives for reasons that were neither captured by their datasets nor adjusted for. Additionally, neither study reported downstream impacts such as an increase in symptomatic hypertensive episodes or more rare events such as kidney injury, stroke, or myocardial infarction.
Given that guidelines discourage using intravenous antihypertensives, why were the efforts of Jacobs et al.9 and Pasik et al.10 needed in the first place? In a recent installment of Choosing Wisely: Things We Do For No Reason, Breu et al.11 cite two primary reasons: first, providers have unfounded fears that asymptomatic hypertension will quickly progress to cause organ damage; second, providers lack understanding of the potential harms from overtreatment. It is fitting, therefore, that both groups of authors focused on these topics in their education initiatives for physicians and nurses. Yet, as good quality improvement requires steps beyond education, it was promising to see that both authors additionally focused on intervening to change the systems and culture that existed around physician and nursing communication.
In the age of electronic health records, there has been a sustained focus on creating standardized order sets. While the value of these order sets has been widely demonstrated, there are downsides. For example, nursing call parameters in admission order sets are rarely patient-specific but account for a significant portion of nursing and physician communication. These one-size-fits-all orders limit nurses from using their clinical training and create unnecessary tensions as nurses are obligated to call covering hospitalists to address “abnormal” but clinically insignificant findings. Regular monitoring of vital signs is an integral part of caring for acutely ill inpatients but for most inpatients, the importance of vitals is to detect clinically meaningful changes, not to treat risk factors like hypertension that should be treated safely over the long term.
When inpatients become febrile, tachycardic, or hypoxic, hospitalists use critical thinking to diagnose the underlying causes. Unfortunately, high blood pressure is a vital sign that is treated differently. Many hospitalists see it as a number to fix, not a potential sign of a new underlying problem such as uncontrolled pain, anxiety, or medication side effects.8 Both groups of authors took the important first step of educating physicians to think critically when called about high blood pressure. Even more importantly, they took steps to change the system and culture in which providers make these decisions in the first place. Future work in this area would be wise to follow in these footsteps, by encouraging collaboration between hospitalist and nurses to create more logical and patient-specific call parameters that could potentially improve nursing-physician communication, and subsequently, patient care.
Changing the culture to limit the use of intravenous antihypertensives will not be easy, but it is necessary. We encourage readers to investigate intravenous antihypertensives in their own hospitals and consider how better communication between nurses and physicians could change their practice. Recalling Mrs. Jones above, the provider should engage her nurse to help confirm that her hypertension is “asymptomatic” and then consider underlying causes such as pain, anxiety, or withholding her home medications as reasons for her elevated blood pressure. After all, if nothing else, it seems clear that a call about inpatient hypertension is not a call to act, but to think.
Disclosures
The authors declare that they have no competing interests.
Funding
Dr. Lucas is supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development and Dartmouth SYNERGY, National Institutes of Health, National Center for Translational Science (UL1TR001086).
1. Axon RN, Cousineau L, Egan BM. Prevalence and management of hypertension in the inpatient setting: A systematic review. J Hosp Med. 2011;6(7):417- 422. doi: 10.1002/jhm.804. PubMed
2. Global status report on noncommunicable diseases 2010. Geneva, Switzerland: World Health Organization;2011. 3.
3. Herzog E, Frankenberger O, Aziz E, et al. A novel pathway for the management of hypertension for hospitalized patients. Crit Pathw Cardiol. 2007;6(4):150-160. doi: 10.1097/HPC.0b013e318160c3a7. PubMed
4. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: use of intravenous labetalol and hydralazine. J Clin Hypertens (Greenwich). 2010;12(1):29-33. doi: 10.1111/j.1751-7176.2009.00196.x. PubMed
5. Campbell P, Baker WL, Bendel SD, White WB. Intravenous hydralazine for blood pressure management in the hospitalized patient: its use is often unjustified. J Am Soc Hypertens. 2011;5(6):473-477. doi: 10.1016/j. jash.2011.07.002. PubMed
6. Gaynor MF, Wright GC, Vondracek S. Retrospective review of the use of as-needed hydralazine and labetalol for the treatment of acute hypertension in hospitalized medicine patients. Ther Adv Cardiovasc Dis. 2017;12(1):7-15. doi: 10.1177/1753944717746613. PubMed
7. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. doi: 10.1001/jama.2013.284427. PubMed
8. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):1269-1324. doi: 10.1161/HYP.0000000000000066. PubMed
9. Reducing Unnecessary Treatment of Asymptomatic Elevated Blood Pressure with Intravenous Medications on the General Internal Medicine Wards: A Quality Improvement Initiative. Jacobs ZG, Najafi N, Fang MC, et al. J Hosp Med. 2019;14:XXX-XXX. doi: 10.12788/jhm.3087. PubMed
10. Assess Before Rx: Reducing the Overtreatment of Asymptomatic Blood Pressure Elevation in the Inpatient Setting. Pasik SD, Chiu S, Yang J, et al. J Hosp Med. 2019;14:XXX-XXX. doi: 10.12788/jhm.3125. PubMed
11. Breu AC, Axon RN. Acute treatment of hypertensive urgency. J Hosp Med. 2018;13(12):860-862. doi: 10.12788/jhm.3086. PubMed
Your pager beeps. Your patient, Mrs. Jones, who was admitted with cellulitis and is improving, now has a blood pressure of 188/103 on routine vitals. Her nurse reports that she is comfortable and asymptomatic, but she met the “call parameters.” You review her chart and find that since admission her systolic blood pressure (SBP) has ranged from 149 to 157 mm Hg and her diastolic blood pressure (DBP) from 84 to 96 mm Hg. Her nurse asks how you would like to treat her.
While over half of inpatients have at least one hypertensive episode during their stay, evidence suggests that nearly all such episodes—estimates are between 98% and 99%1,2—should be treated over several days with oral antihypertensives, not acutely with intravenous medications.3-6 Current guidelines recommend that intravenous medications should be reserved for severe hypertensive episodes (SBP > 180, DBP > 120) with acute end-organ damage,7,8 but such “hypertensive emergencies” are rare on the general medicine wards. Still, hospitalists regularly face the dilemma posed by Mrs. Jones, and evidence shows they often prescribe intravenous antihypertensives.1,4,5 This unnecessary use can lead to unreliable drops in blood pressure and exposes our patients to potential harm.5,6
In this issue of the Journal of Hospital Medicine, two papers describe the frequency of inappropriate intravenous antihypertensive use in their hospitals and the subsequent quality improvement efforts implemented to reduce this practice. The first, by Jacobs et al., found that over a 10-month period, 11% of patients who experienced “asymptomatic hypertension” on an urban academic hospital medicine service were treated inappropriately with intravenous antihypertensives,9 with 14% of those experiencing an adverse event. The second paper, by Pasik et al., found that in their urban academic medical center there were 8.3 inappropriate intravenous antihypertensive orders placed per 1,000 patient days,10 with nearly half of those treated experiencing an adverse event. Based on these findings, each group then led interventions to reduce the use of intravenous antihypertensives.
While both groups engaged physicians and nurses as primary stakeholders, Pasik et al.10 worked to further expand nursing staff roles by empowering them to assess for underlying causes of hypertension, such as pain or anxiety, as well as end-organ damage via specific guided algorithms prior to contacting physicians. In doing so, they reduced intravenous antihypertensive use by 60% during the postintervention period, with a proportional reduction in adverse events. In addition to their educational initiative, Jacobs et al. aimed to limit calls by liberalizing the “ceiling” on standard nursing call parameters for blood pressure from 160/80 to 180/90. Following their intervention, intravenous antihypertensive orders were reduced by 40%, with the mean orders per patient with asymptomatic hypertension decreasing from 11% to 7% .
While these results are admirable, some caution in their interpretation is needed. For example, Jacobs et al. used electronic health record data to retrospectively identify hypertension as “symptomatic” or “asymptomatic” using laboratory, electrocardiogram, and imaging diagnostics as surrogate markers for “provider concern for end-organ damage.” Although it appropriately focused on concern for end-organ damage as justification for intravenous antihypertensives, this approach potentially underappreciated true hypertensive emergencies, thereby overestimating the amount of inappropriate use of intravenous antihypertensives. Pasik et al. utilized chart review of patients prescribed intravenous antihypertensives and therefore did not explore how often symptomatic hypertension occurred in patients who did not receive intravenous antihypertensives. Subsequently, this limited their ability to evaluate unintended harms of their initiative. To address this limitation, the authors followed a group of 111 patients who had elevated hypertension but did not receive intravenous antihypertensives and found no adverse outcomes.10 Because both studies were retrospective in nature, they were subject to biases from providers choosing intravenous antihypertensives for reasons that were neither captured by their datasets nor adjusted for. Additionally, neither study reported downstream impacts such as an increase in symptomatic hypertensive episodes or more rare events such as kidney injury, stroke, or myocardial infarction.
Given that guidelines discourage using intravenous antihypertensives, why were the efforts of Jacobs et al.9 and Pasik et al.10 needed in the first place? In a recent installment of Choosing Wisely: Things We Do For No Reason, Breu et al.11 cite two primary reasons: first, providers have unfounded fears that asymptomatic hypertension will quickly progress to cause organ damage; second, providers lack understanding of the potential harms from overtreatment. It is fitting, therefore, that both groups of authors focused on these topics in their education initiatives for physicians and nurses. Yet, as good quality improvement requires steps beyond education, it was promising to see that both authors additionally focused on intervening to change the systems and culture that existed around physician and nursing communication.
In the age of electronic health records, there has been a sustained focus on creating standardized order sets. While the value of these order sets has been widely demonstrated, there are downsides. For example, nursing call parameters in admission order sets are rarely patient-specific but account for a significant portion of nursing and physician communication. These one-size-fits-all orders limit nurses from using their clinical training and create unnecessary tensions as nurses are obligated to call covering hospitalists to address “abnormal” but clinically insignificant findings. Regular monitoring of vital signs is an integral part of caring for acutely ill inpatients but for most inpatients, the importance of vitals is to detect clinically meaningful changes, not to treat risk factors like hypertension that should be treated safely over the long term.
When inpatients become febrile, tachycardic, or hypoxic, hospitalists use critical thinking to diagnose the underlying causes. Unfortunately, high blood pressure is a vital sign that is treated differently. Many hospitalists see it as a number to fix, not a potential sign of a new underlying problem such as uncontrolled pain, anxiety, or medication side effects.8 Both groups of authors took the important first step of educating physicians to think critically when called about high blood pressure. Even more importantly, they took steps to change the system and culture in which providers make these decisions in the first place. Future work in this area would be wise to follow in these footsteps, by encouraging collaboration between hospitalist and nurses to create more logical and patient-specific call parameters that could potentially improve nursing-physician communication, and subsequently, patient care.
Changing the culture to limit the use of intravenous antihypertensives will not be easy, but it is necessary. We encourage readers to investigate intravenous antihypertensives in their own hospitals and consider how better communication between nurses and physicians could change their practice. Recalling Mrs. Jones above, the provider should engage her nurse to help confirm that her hypertension is “asymptomatic” and then consider underlying causes such as pain, anxiety, or withholding her home medications as reasons for her elevated blood pressure. After all, if nothing else, it seems clear that a call about inpatient hypertension is not a call to act, but to think.
Disclosures
The authors declare that they have no competing interests.
Funding
Dr. Lucas is supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development and Dartmouth SYNERGY, National Institutes of Health, National Center for Translational Science (UL1TR001086).
Your pager beeps. Your patient, Mrs. Jones, who was admitted with cellulitis and is improving, now has a blood pressure of 188/103 on routine vitals. Her nurse reports that she is comfortable and asymptomatic, but she met the “call parameters.” You review her chart and find that since admission her systolic blood pressure (SBP) has ranged from 149 to 157 mm Hg and her diastolic blood pressure (DBP) from 84 to 96 mm Hg. Her nurse asks how you would like to treat her.
While over half of inpatients have at least one hypertensive episode during their stay, evidence suggests that nearly all such episodes—estimates are between 98% and 99%1,2—should be treated over several days with oral antihypertensives, not acutely with intravenous medications.3-6 Current guidelines recommend that intravenous medications should be reserved for severe hypertensive episodes (SBP > 180, DBP > 120) with acute end-organ damage,7,8 but such “hypertensive emergencies” are rare on the general medicine wards. Still, hospitalists regularly face the dilemma posed by Mrs. Jones, and evidence shows they often prescribe intravenous antihypertensives.1,4,5 This unnecessary use can lead to unreliable drops in blood pressure and exposes our patients to potential harm.5,6
In this issue of the Journal of Hospital Medicine, two papers describe the frequency of inappropriate intravenous antihypertensive use in their hospitals and the subsequent quality improvement efforts implemented to reduce this practice. The first, by Jacobs et al., found that over a 10-month period, 11% of patients who experienced “asymptomatic hypertension” on an urban academic hospital medicine service were treated inappropriately with intravenous antihypertensives,9 with 14% of those experiencing an adverse event. The second paper, by Pasik et al., found that in their urban academic medical center there were 8.3 inappropriate intravenous antihypertensive orders placed per 1,000 patient days,10 with nearly half of those treated experiencing an adverse event. Based on these findings, each group then led interventions to reduce the use of intravenous antihypertensives.
While both groups engaged physicians and nurses as primary stakeholders, Pasik et al.10 worked to further expand nursing staff roles by empowering them to assess for underlying causes of hypertension, such as pain or anxiety, as well as end-organ damage via specific guided algorithms prior to contacting physicians. In doing so, they reduced intravenous antihypertensive use by 60% during the postintervention period, with a proportional reduction in adverse events. In addition to their educational initiative, Jacobs et al. aimed to limit calls by liberalizing the “ceiling” on standard nursing call parameters for blood pressure from 160/80 to 180/90. Following their intervention, intravenous antihypertensive orders were reduced by 40%, with the mean orders per patient with asymptomatic hypertension decreasing from 11% to 7% .
While these results are admirable, some caution in their interpretation is needed. For example, Jacobs et al. used electronic health record data to retrospectively identify hypertension as “symptomatic” or “asymptomatic” using laboratory, electrocardiogram, and imaging diagnostics as surrogate markers for “provider concern for end-organ damage.” Although it appropriately focused on concern for end-organ damage as justification for intravenous antihypertensives, this approach potentially underappreciated true hypertensive emergencies, thereby overestimating the amount of inappropriate use of intravenous antihypertensives. Pasik et al. utilized chart review of patients prescribed intravenous antihypertensives and therefore did not explore how often symptomatic hypertension occurred in patients who did not receive intravenous antihypertensives. Subsequently, this limited their ability to evaluate unintended harms of their initiative. To address this limitation, the authors followed a group of 111 patients who had elevated hypertension but did not receive intravenous antihypertensives and found no adverse outcomes.10 Because both studies were retrospective in nature, they were subject to biases from providers choosing intravenous antihypertensives for reasons that were neither captured by their datasets nor adjusted for. Additionally, neither study reported downstream impacts such as an increase in symptomatic hypertensive episodes or more rare events such as kidney injury, stroke, or myocardial infarction.
Given that guidelines discourage using intravenous antihypertensives, why were the efforts of Jacobs et al.9 and Pasik et al.10 needed in the first place? In a recent installment of Choosing Wisely: Things We Do For No Reason, Breu et al.11 cite two primary reasons: first, providers have unfounded fears that asymptomatic hypertension will quickly progress to cause organ damage; second, providers lack understanding of the potential harms from overtreatment. It is fitting, therefore, that both groups of authors focused on these topics in their education initiatives for physicians and nurses. Yet, as good quality improvement requires steps beyond education, it was promising to see that both authors additionally focused on intervening to change the systems and culture that existed around physician and nursing communication.
In the age of electronic health records, there has been a sustained focus on creating standardized order sets. While the value of these order sets has been widely demonstrated, there are downsides. For example, nursing call parameters in admission order sets are rarely patient-specific but account for a significant portion of nursing and physician communication. These one-size-fits-all orders limit nurses from using their clinical training and create unnecessary tensions as nurses are obligated to call covering hospitalists to address “abnormal” but clinically insignificant findings. Regular monitoring of vital signs is an integral part of caring for acutely ill inpatients but for most inpatients, the importance of vitals is to detect clinically meaningful changes, not to treat risk factors like hypertension that should be treated safely over the long term.
When inpatients become febrile, tachycardic, or hypoxic, hospitalists use critical thinking to diagnose the underlying causes. Unfortunately, high blood pressure is a vital sign that is treated differently. Many hospitalists see it as a number to fix, not a potential sign of a new underlying problem such as uncontrolled pain, anxiety, or medication side effects.8 Both groups of authors took the important first step of educating physicians to think critically when called about high blood pressure. Even more importantly, they took steps to change the system and culture in which providers make these decisions in the first place. Future work in this area would be wise to follow in these footsteps, by encouraging collaboration between hospitalist and nurses to create more logical and patient-specific call parameters that could potentially improve nursing-physician communication, and subsequently, patient care.
Changing the culture to limit the use of intravenous antihypertensives will not be easy, but it is necessary. We encourage readers to investigate intravenous antihypertensives in their own hospitals and consider how better communication between nurses and physicians could change their practice. Recalling Mrs. Jones above, the provider should engage her nurse to help confirm that her hypertension is “asymptomatic” and then consider underlying causes such as pain, anxiety, or withholding her home medications as reasons for her elevated blood pressure. After all, if nothing else, it seems clear that a call about inpatient hypertension is not a call to act, but to think.
Disclosures
The authors declare that they have no competing interests.
Funding
Dr. Lucas is supported by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development and Dartmouth SYNERGY, National Institutes of Health, National Center for Translational Science (UL1TR001086).
1. Axon RN, Cousineau L, Egan BM. Prevalence and management of hypertension in the inpatient setting: A systematic review. J Hosp Med. 2011;6(7):417- 422. doi: 10.1002/jhm.804. PubMed
2. Global status report on noncommunicable diseases 2010. Geneva, Switzerland: World Health Organization;2011. 3.
3. Herzog E, Frankenberger O, Aziz E, et al. A novel pathway for the management of hypertension for hospitalized patients. Crit Pathw Cardiol. 2007;6(4):150-160. doi: 10.1097/HPC.0b013e318160c3a7. PubMed
4. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: use of intravenous labetalol and hydralazine. J Clin Hypertens (Greenwich). 2010;12(1):29-33. doi: 10.1111/j.1751-7176.2009.00196.x. PubMed
5. Campbell P, Baker WL, Bendel SD, White WB. Intravenous hydralazine for blood pressure management in the hospitalized patient: its use is often unjustified. J Am Soc Hypertens. 2011;5(6):473-477. doi: 10.1016/j. jash.2011.07.002. PubMed
6. Gaynor MF, Wright GC, Vondracek S. Retrospective review of the use of as-needed hydralazine and labetalol for the treatment of acute hypertension in hospitalized medicine patients. Ther Adv Cardiovasc Dis. 2017;12(1):7-15. doi: 10.1177/1753944717746613. PubMed
7. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. doi: 10.1001/jama.2013.284427. PubMed
8. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):1269-1324. doi: 10.1161/HYP.0000000000000066. PubMed
9. Reducing Unnecessary Treatment of Asymptomatic Elevated Blood Pressure with Intravenous Medications on the General Internal Medicine Wards: A Quality Improvement Initiative. Jacobs ZG, Najafi N, Fang MC, et al. J Hosp Med. 2019;14:XXX-XXX. doi: 10.12788/jhm.3087. PubMed
10. Assess Before Rx: Reducing the Overtreatment of Asymptomatic Blood Pressure Elevation in the Inpatient Setting. Pasik SD, Chiu S, Yang J, et al. J Hosp Med. 2019;14:XXX-XXX. doi: 10.12788/jhm.3125. PubMed
11. Breu AC, Axon RN. Acute treatment of hypertensive urgency. J Hosp Med. 2018;13(12):860-862. doi: 10.12788/jhm.3086. PubMed
1. Axon RN, Cousineau L, Egan BM. Prevalence and management of hypertension in the inpatient setting: A systematic review. J Hosp Med. 2011;6(7):417- 422. doi: 10.1002/jhm.804. PubMed
2. Global status report on noncommunicable diseases 2010. Geneva, Switzerland: World Health Organization;2011. 3.
3. Herzog E, Frankenberger O, Aziz E, et al. A novel pathway for the management of hypertension for hospitalized patients. Crit Pathw Cardiol. 2007;6(4):150-160. doi: 10.1097/HPC.0b013e318160c3a7. PubMed
4. Weder AB, Erickson S. Treatment of hypertension in the inpatient setting: use of intravenous labetalol and hydralazine. J Clin Hypertens (Greenwich). 2010;12(1):29-33. doi: 10.1111/j.1751-7176.2009.00196.x. PubMed
5. Campbell P, Baker WL, Bendel SD, White WB. Intravenous hydralazine for blood pressure management in the hospitalized patient: its use is often unjustified. J Am Soc Hypertens. 2011;5(6):473-477. doi: 10.1016/j. jash.2011.07.002. PubMed
6. Gaynor MF, Wright GC, Vondracek S. Retrospective review of the use of as-needed hydralazine and labetalol for the treatment of acute hypertension in hospitalized medicine patients. Ther Adv Cardiovasc Dis. 2017;12(1):7-15. doi: 10.1177/1753944717746613. PubMed
7. James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507-520. doi: 10.1001/jama.2013.284427. PubMed
8. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):1269-1324. doi: 10.1161/HYP.0000000000000066. PubMed
9. Reducing Unnecessary Treatment of Asymptomatic Elevated Blood Pressure with Intravenous Medications on the General Internal Medicine Wards: A Quality Improvement Initiative. Jacobs ZG, Najafi N, Fang MC, et al. J Hosp Med. 2019;14:XXX-XXX. doi: 10.12788/jhm.3087. PubMed
10. Assess Before Rx: Reducing the Overtreatment of Asymptomatic Blood Pressure Elevation in the Inpatient Setting. Pasik SD, Chiu S, Yang J, et al. J Hosp Med. 2019;14:XXX-XXX. doi: 10.12788/jhm.3125. PubMed
11. Breu AC, Axon RN. Acute treatment of hypertensive urgency. J Hosp Med. 2018;13(12):860-862. doi: 10.12788/jhm.3086. PubMed
© 2019 Society of Hospital Medicine
Adherence to Recommended Inpatient Hepatic Encephalopathy Workup
Clinical guidelines are periodically released by medical societies with the overarching goal of improving deliverable medical care by standardizing disease management according to best available published literature and by reducing healthcare expenditure associated with unnecessary and superfluous testing.1 Unfortunately, nonadherence to guidelines is common in clinical practice2 and contributes to the rising cost of healthcare.3 Health resource utilization is particularly relevant in management of cirrhosis, a condition with an annual healthcare expenditure of $13 billion.4 Hepatic encephalopathy (HE), the most common complication of cirrhosis, is characterized by altered sensorium and is the leading indication for hospitalization among cirrhotics. The joint guidelines of the European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD) for diagnostic workup for HE recommend identification and treatment of potential precipitants.5 The guidelines also recommend against checking serum ammonia levels, which have not been shown to correlate with diagnosis or severity of HE.6-8 Currently, limited data are available on practice patterns regarding guideline adherence and unnecessary serum ammonia testing for initial evaluation of HE in hospitals. To overcome this gap in knowledge, we conducted the present study to provide granular details regarding the diagnostic workup for hospitalized patients with HE.
METHODS
This study adopted a retrospective design and recruited patients admitted to the Virginia Commonwealth University Medical Center between July 1, 2016 and July 1, 2017. The institutional review board approved the study, and the manuscript was reviewed and approved by all authors prior to submission. All chart reviews were performed by hepatologists with access to patients’ electronic medical record (EMR).
Patient Population
Patients were identified from the EMR system by using ICD-9 and ICD-10 codes for cirrhosis, hepatic encephalopathy, and altered mental status. All consecutive admissions with these diagnosis codes were considered for inclusion. Adult patients with cirrhosis resulting from any etiology of chronic liver diseases with primary reason for admission of HE were included. If patients were readmitted for HE during the study period, then only the data from index HE admission was included in the analysis and data from subsequent admissions were excluded. The other exclusion criteria included non-HE causes of confusion, acute liver failure, and those admitted with a preformulated plan (eg, direct hepatology clinic admission or outside hospital transfer). Patients who developed HE during their hospitalization where HE was not the indication for admission were also excluded. Finally, all patients admitted under the direct care of hepatology were excluded.
Diagnostic Workup
The recommendations of the AASLD and the EASL for workup for HE include obtaining detailed history and physical examination supplemented by diagnostic evaluation for potential HE precipitants including infections, electrolyte disturbances, dehydration, renal failure, glycemic disturbances, and toxin ingestion (eg, alcohol, illicit drugs).5 Based on the guideline recommendation, this study defined a “complete workup” as including all of the following elements: infection evaluation (blood culture, urinalysis/urine culture, chest radiograph, diagnostic paracentesis in the presence of ascites), electrolyte/renal evaluation (serum sodium, potassium, creatinine, and glucose), and toxin evaluation (urine drug screening). Any HE admission that was missing elements from the aforementioned battery of tests was defined as “incomplete workup.” In patients admitted with decompensated cirrhosis, serum ammonia testing was considered inappropriate unless there was a nuanced explanation supporting its use documented within the EMR. The frequency and specialty of the physician ordering serum ammonia level tests were determined. The financial burden of unnecessary ammonia testing was estimated by assigning a laboratory charge ($258) for each patient.
Statistical Analysis
Continuous and categorical variables are reported as means (± standard deviation), median (interquartile range or IQR), or proportion (%) as appropriate. Across-group differences were compared using Student t-test for normally distributed continuous variables and Mann-Whitney U test for skewed data. Fisher’s exact test was used to compare proportion. HE evaluations were quantified by the number of patients with complete workup and by the number of patients with missing components of the workup. A nominal P value of less than .05 was considered statistically significant. All statistical analyses were performed using SPSS Statistics version 24.0 (IBM Corporation, Armonk, New York).
RESULTS
Cohort Characteristics
The baseline cohort demographics are listed in the Table. Of the 145 patients identified using diagnostic codes for cirrhosis, 78 subjects met the study criteria. The most common exclusion criteria included non-HE etiology of altered mental status (n = 37) and patients with readmissions for HE during the study period (n = 30). The mean age of the study cohort was 59.3 years, and the most common etiology of cirrhosis was hepatitis C (n = 41), alcohol induced (n = 14), and nonalcoholic steatohepatitis (n = 13).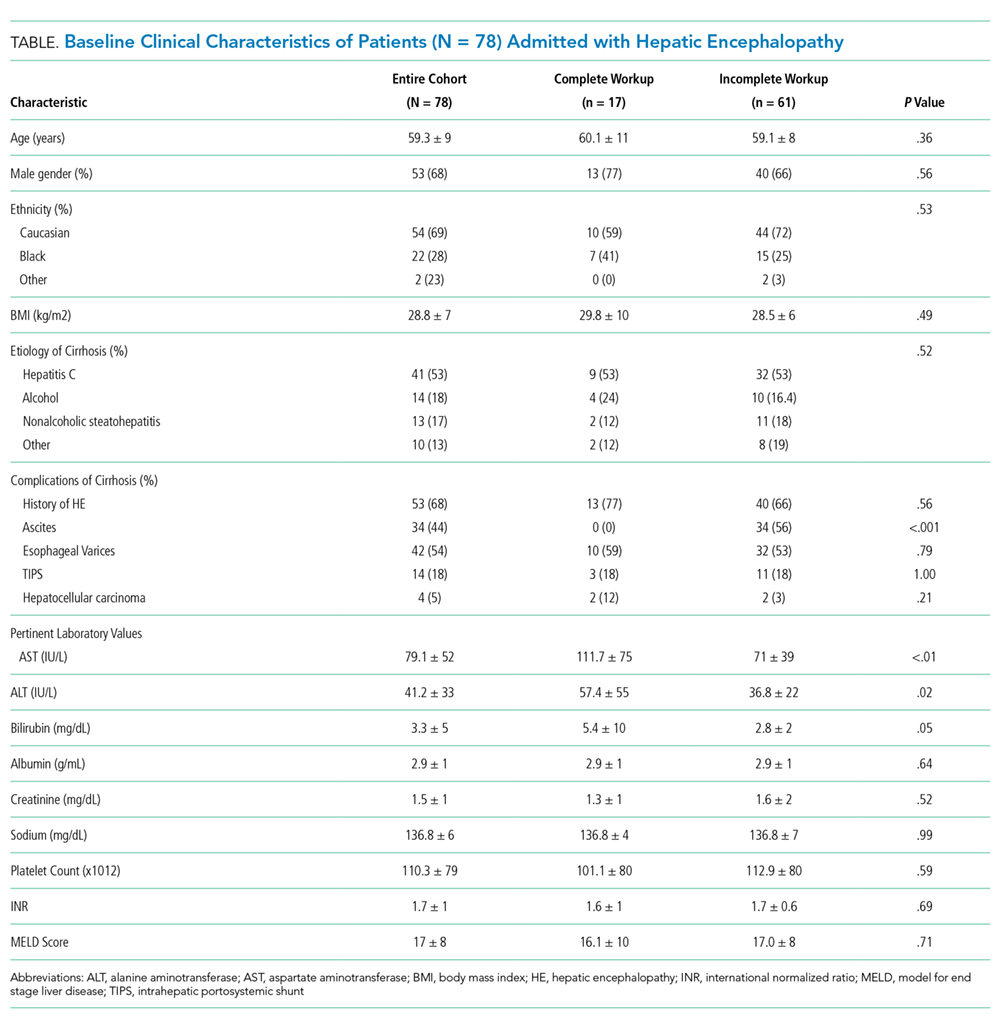
Initial Diagnostic Evaluation
The major precipitants of HE in the study cohort were ineffective lactulose dosing (n = 43), infections (n = 25), and electrolyte disturbances/renal injury (n = 6). At the time of admission, 53 patients were on therapy for HE. Only 17 (22%) patients had complete diagnostic workup within 24 hours of hospital admission. The individual components of the complete workup are shown in the Figure. Notably, 23 (30%) patients were missing blood cultures, 16 (21%) were missing urinalysis, 15 (20%) were missing chest radiograph, and 34 (44%) were missing urine drug screening. Of the 34 patients with ascites on admission, only eight (23%) had diagnostic paracentesis performed on admission to rule out spontaneous bacterial peritonitis.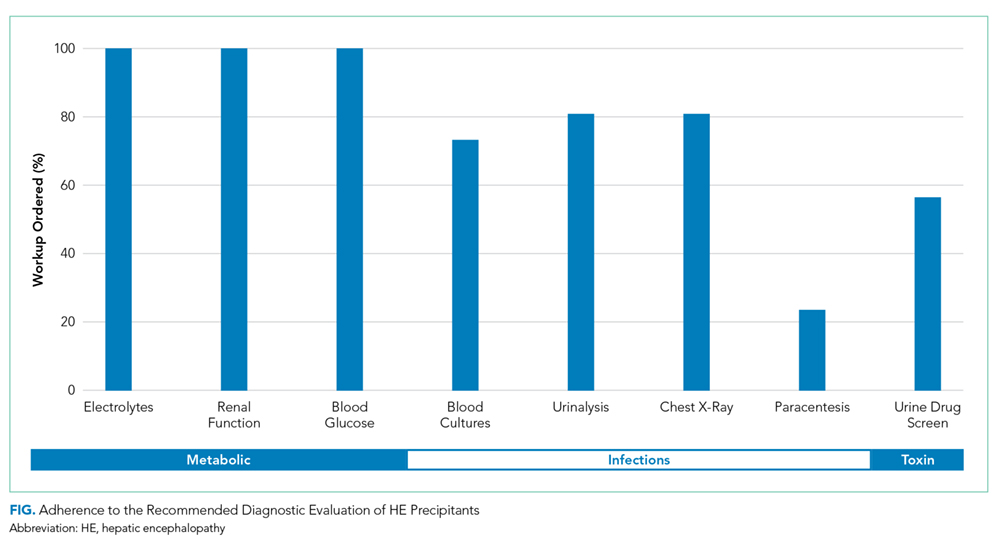
Serum Ammonia Testing
Serum ammonia testing was performed on 74 patients (94.9%), and no patient met the criteria for appropriate testing. Forty patients already had a known diagnosis of HE prior to index admission. Furthermore, 10 (14%) patients had serum ammonia testing repeated after admission without documentation in the EMR to justify repeat testing. Emergency Department (ED) physicians ordered ammonia testing in 57 cases (77%), internists ordered the testing in 11 cases (15%), and intensivists ordered the testing in two cases (3%). The patient’s charges for serum ammonia testing at the time of admission and for repeat testing were $19,092 and $2,580, respectively.
DISCUSSION
This study utilized HE in patients with decompensated cirrhosis as a framework to analyze adherence to societal guidelines. The adherence rate to AALSD/EASL recommended inpatient evaluation of HE is surprisingly low, and most patients are missing key essential elements of the diagnostic work up. While the diagnostic tests that are ordered as part of a panel are completed universally (renal function, electrolytes, and glucose testing), individual testing is less inclined to be ordered (blood cultures, urine culture/urinalysis, CXR, UDS) and procedural testing, such as diagnostic paracentesis, is often missed. This last finding is in line with published literature showing that 40% of patients admitted with ascites or HE did not have diagnostic paracentesis during hospital admission despite 24% reduction of inhospital mortality among patients undergoing the procedure.9
Although serum ammonia testing is not endorsed by the AASLD/EASL guidelines for HE,5 it is ordered nearly universally. The cost of an individual test is relatively low, but the cumulative cost of serum ammonia testing can be substantial because HE is the most common indication for hospitalization among patients with cirrhosis.4 Initiatives, such as the Choosing Wisely® campaign, encourage high-value and evidence-based care by limiting excessive and unnecessary diagnostic testing.10 The Canadian Choosing Wisely campaign specifically includes avoidance of serum ammonia testing for diagnosis of HE to provide high-value care in hepatology.11
Although the exact reasons for nonadherence to recommended HE evaluations are unclear, a potential method to mitigate excessive testing is to utilize the EMR and ordering system.3 EMR-based strategies can curb unnecessary testing in inpatient settings.12 The use of HE order sets, the inclusion of clinical decision support systems, and the restriction of access to specialized testing can be readily incorporated into the EMR to encourage adherence to guideline-based care while limiting unnecessary testing.
This study should be interpreted in the context of study limitations. Given the retrospective design of the study, salient factors in decisions behind diagnostic testing cannot be assessed. Future studies should utilize mixed-model methodology to elucidate reasons behind these decisions. The present study used a strict definition of complete workup including all the mentioned elements of the diagnostic workup for HE; however, in clinical practice, providers could be justified in not ordering certain tests if the specific clinical scenario does not lead to its use (eg, chest X-ray deferred in a patient with clear lung exam, no symptoms, or hypoxia). Similarly, UDS was included as a required element for a complete workup. While it may be ordered in a case-by-case basis to screen for illicit drug abuse, UDS is also a critical element of the workup to screen for opioid use as a precipitant of HE. Finally, considering the strict study entry criteria, we excluded repeated admissions for HE during the study period and therefore likely underestimate the cost burden of serum ammonia testing.
In conclusion, valuable guideline-based diagnostic testing is often missing in patients admitted for HE while serum ammonia testing is nearly universally ordered. These findings underscore the importance of implementing educational strategies, such as the Choosing Wisely® campaign, and EMR-based clinical decision support systems to improve health resource utilization in patients with cirrhosis and HE.
Disclosures
The authors have nothing to disclose.
1. Andrews EJ, Redmond HP. A review of clinical guidelines. Br J Surg. 2004;91:956-964. doi: 10.1002/bjs.4630 PubMed
2. Arts DL, Voncken AG, Medlock S, Abu-Hanna A, van Weert HC. Reasons for intentional guideline non-adherence: a systematic review. Int J Med Inform. 2016;89:55-62. doi: 10.1016/j.ijmedinf.2016.02.009. PubMed
3. Eaton KP, Levy K, Soong C, et al. Evidence-based guidelines to eliminate repetitive laboratory testing. JAMA Intern Med. 2017;177(12):1833-1839. doi: 10.1001/jamainternmed.2017.5152. PubMed
4. Everhart J. The burden of digestive diseases in the United States. Washington D.C.: US Department of Health and Human Services, Public Health Service, National Institutes of Health. U.S. Government Printing Office; 2008:111-114.
5. Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of Liver Diseases. Hepatology . 2014;60:715-735. doi: 10.1002/hep.27210 PubMed
6. Stahl J. Studies of the blood ammonia in liver disease: Its diagnostic, prognostic, and therapeutic significance. Ann Intern Med . 1963;58:1-24. PubMed
7. Ong JP, Aggarwal A, Kreiger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med . 2003;114:188-193. doi: 10.1016/S0002-9343(02)01477-8 PubMed
8. Nicalao F, Efrati C, Masini A, Merli M, Attili AF, Riggio O. Role of determination of partial pressure of ammonia in cirrhotic patients with and without hepatic encephalopathy. J Hepatol. 2003;38:441-446. doi: 10.1016/S0168-8278(02)00436-1 PubMed
9. Orman ES, Hayashi PH, Bataller R, Barritt AS 4th. Paracentesis is associated with reduced mortality in patients hospitalized with cirrhosis and ascites. Clin Gastroenterol Hepatol. 2014;12:496-503. doi: 10.1016/j.cgh.2013.08.025. PubMed
10. Cassek CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA. 2012;307:1801-1802. doi: 10.1001/jama.2012.476. PubMed
11. Choosing Wisely Canada. 2018. Five things patients and physicians should question. Available at: https://choosingwiselycanada.org/hepatology/ . Accessed November 18, 2018.
12. Iturrate E, Jubelt L, Volpicelli F, Hochman K. Optimize your electronic medical record to increase value: reducing laboratory overutilization. Am J Med . 2016;129:215-220. doi: 10.1016/j.amjmed.2015.09.009. PubMed
Clinical guidelines are periodically released by medical societies with the overarching goal of improving deliverable medical care by standardizing disease management according to best available published literature and by reducing healthcare expenditure associated with unnecessary and superfluous testing.1 Unfortunately, nonadherence to guidelines is common in clinical practice2 and contributes to the rising cost of healthcare.3 Health resource utilization is particularly relevant in management of cirrhosis, a condition with an annual healthcare expenditure of $13 billion.4 Hepatic encephalopathy (HE), the most common complication of cirrhosis, is characterized by altered sensorium and is the leading indication for hospitalization among cirrhotics. The joint guidelines of the European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD) for diagnostic workup for HE recommend identification and treatment of potential precipitants.5 The guidelines also recommend against checking serum ammonia levels, which have not been shown to correlate with diagnosis or severity of HE.6-8 Currently, limited data are available on practice patterns regarding guideline adherence and unnecessary serum ammonia testing for initial evaluation of HE in hospitals. To overcome this gap in knowledge, we conducted the present study to provide granular details regarding the diagnostic workup for hospitalized patients with HE.
METHODS
This study adopted a retrospective design and recruited patients admitted to the Virginia Commonwealth University Medical Center between July 1, 2016 and July 1, 2017. The institutional review board approved the study, and the manuscript was reviewed and approved by all authors prior to submission. All chart reviews were performed by hepatologists with access to patients’ electronic medical record (EMR).
Patient Population
Patients were identified from the EMR system by using ICD-9 and ICD-10 codes for cirrhosis, hepatic encephalopathy, and altered mental status. All consecutive admissions with these diagnosis codes were considered for inclusion. Adult patients with cirrhosis resulting from any etiology of chronic liver diseases with primary reason for admission of HE were included. If patients were readmitted for HE during the study period, then only the data from index HE admission was included in the analysis and data from subsequent admissions were excluded. The other exclusion criteria included non-HE causes of confusion, acute liver failure, and those admitted with a preformulated plan (eg, direct hepatology clinic admission or outside hospital transfer). Patients who developed HE during their hospitalization where HE was not the indication for admission were also excluded. Finally, all patients admitted under the direct care of hepatology were excluded.
Diagnostic Workup
The recommendations of the AASLD and the EASL for workup for HE include obtaining detailed history and physical examination supplemented by diagnostic evaluation for potential HE precipitants including infections, electrolyte disturbances, dehydration, renal failure, glycemic disturbances, and toxin ingestion (eg, alcohol, illicit drugs).5 Based on the guideline recommendation, this study defined a “complete workup” as including all of the following elements: infection evaluation (blood culture, urinalysis/urine culture, chest radiograph, diagnostic paracentesis in the presence of ascites), electrolyte/renal evaluation (serum sodium, potassium, creatinine, and glucose), and toxin evaluation (urine drug screening). Any HE admission that was missing elements from the aforementioned battery of tests was defined as “incomplete workup.” In patients admitted with decompensated cirrhosis, serum ammonia testing was considered inappropriate unless there was a nuanced explanation supporting its use documented within the EMR. The frequency and specialty of the physician ordering serum ammonia level tests were determined. The financial burden of unnecessary ammonia testing was estimated by assigning a laboratory charge ($258) for each patient.
Statistical Analysis
Continuous and categorical variables are reported as means (± standard deviation), median (interquartile range or IQR), or proportion (%) as appropriate. Across-group differences were compared using Student t-test for normally distributed continuous variables and Mann-Whitney U test for skewed data. Fisher’s exact test was used to compare proportion. HE evaluations were quantified by the number of patients with complete workup and by the number of patients with missing components of the workup. A nominal P value of less than .05 was considered statistically significant. All statistical analyses were performed using SPSS Statistics version 24.0 (IBM Corporation, Armonk, New York).
RESULTS
Cohort Characteristics
The baseline cohort demographics are listed in the Table. Of the 145 patients identified using diagnostic codes for cirrhosis, 78 subjects met the study criteria. The most common exclusion criteria included non-HE etiology of altered mental status (n = 37) and patients with readmissions for HE during the study period (n = 30). The mean age of the study cohort was 59.3 years, and the most common etiology of cirrhosis was hepatitis C (n = 41), alcohol induced (n = 14), and nonalcoholic steatohepatitis (n = 13).
Initial Diagnostic Evaluation
The major precipitants of HE in the study cohort were ineffective lactulose dosing (n = 43), infections (n = 25), and electrolyte disturbances/renal injury (n = 6). At the time of admission, 53 patients were on therapy for HE. Only 17 (22%) patients had complete diagnostic workup within 24 hours of hospital admission. The individual components of the complete workup are shown in the Figure. Notably, 23 (30%) patients were missing blood cultures, 16 (21%) were missing urinalysis, 15 (20%) were missing chest radiograph, and 34 (44%) were missing urine drug screening. Of the 34 patients with ascites on admission, only eight (23%) had diagnostic paracentesis performed on admission to rule out spontaneous bacterial peritonitis.
Serum Ammonia Testing
Serum ammonia testing was performed on 74 patients (94.9%), and no patient met the criteria for appropriate testing. Forty patients already had a known diagnosis of HE prior to index admission. Furthermore, 10 (14%) patients had serum ammonia testing repeated after admission without documentation in the EMR to justify repeat testing. Emergency Department (ED) physicians ordered ammonia testing in 57 cases (77%), internists ordered the testing in 11 cases (15%), and intensivists ordered the testing in two cases (3%). The patient’s charges for serum ammonia testing at the time of admission and for repeat testing were $19,092 and $2,580, respectively.
DISCUSSION
This study utilized HE in patients with decompensated cirrhosis as a framework to analyze adherence to societal guidelines. The adherence rate to AALSD/EASL recommended inpatient evaluation of HE is surprisingly low, and most patients are missing key essential elements of the diagnostic work up. While the diagnostic tests that are ordered as part of a panel are completed universally (renal function, electrolytes, and glucose testing), individual testing is less inclined to be ordered (blood cultures, urine culture/urinalysis, CXR, UDS) and procedural testing, such as diagnostic paracentesis, is often missed. This last finding is in line with published literature showing that 40% of patients admitted with ascites or HE did not have diagnostic paracentesis during hospital admission despite 24% reduction of inhospital mortality among patients undergoing the procedure.9
Although serum ammonia testing is not endorsed by the AASLD/EASL guidelines for HE,5 it is ordered nearly universally. The cost of an individual test is relatively low, but the cumulative cost of serum ammonia testing can be substantial because HE is the most common indication for hospitalization among patients with cirrhosis.4 Initiatives, such as the Choosing Wisely® campaign, encourage high-value and evidence-based care by limiting excessive and unnecessary diagnostic testing.10 The Canadian Choosing Wisely campaign specifically includes avoidance of serum ammonia testing for diagnosis of HE to provide high-value care in hepatology.11
Although the exact reasons for nonadherence to recommended HE evaluations are unclear, a potential method to mitigate excessive testing is to utilize the EMR and ordering system.3 EMR-based strategies can curb unnecessary testing in inpatient settings.12 The use of HE order sets, the inclusion of clinical decision support systems, and the restriction of access to specialized testing can be readily incorporated into the EMR to encourage adherence to guideline-based care while limiting unnecessary testing.
This study should be interpreted in the context of study limitations. Given the retrospective design of the study, salient factors in decisions behind diagnostic testing cannot be assessed. Future studies should utilize mixed-model methodology to elucidate reasons behind these decisions. The present study used a strict definition of complete workup including all the mentioned elements of the diagnostic workup for HE; however, in clinical practice, providers could be justified in not ordering certain tests if the specific clinical scenario does not lead to its use (eg, chest X-ray deferred in a patient with clear lung exam, no symptoms, or hypoxia). Similarly, UDS was included as a required element for a complete workup. While it may be ordered in a case-by-case basis to screen for illicit drug abuse, UDS is also a critical element of the workup to screen for opioid use as a precipitant of HE. Finally, considering the strict study entry criteria, we excluded repeated admissions for HE during the study period and therefore likely underestimate the cost burden of serum ammonia testing.
In conclusion, valuable guideline-based diagnostic testing is often missing in patients admitted for HE while serum ammonia testing is nearly universally ordered. These findings underscore the importance of implementing educational strategies, such as the Choosing Wisely® campaign, and EMR-based clinical decision support systems to improve health resource utilization in patients with cirrhosis and HE.
Disclosures
The authors have nothing to disclose.
Clinical guidelines are periodically released by medical societies with the overarching goal of improving deliverable medical care by standardizing disease management according to best available published literature and by reducing healthcare expenditure associated with unnecessary and superfluous testing.1 Unfortunately, nonadherence to guidelines is common in clinical practice2 and contributes to the rising cost of healthcare.3 Health resource utilization is particularly relevant in management of cirrhosis, a condition with an annual healthcare expenditure of $13 billion.4 Hepatic encephalopathy (HE), the most common complication of cirrhosis, is characterized by altered sensorium and is the leading indication for hospitalization among cirrhotics. The joint guidelines of the European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD) for diagnostic workup for HE recommend identification and treatment of potential precipitants.5 The guidelines also recommend against checking serum ammonia levels, which have not been shown to correlate with diagnosis or severity of HE.6-8 Currently, limited data are available on practice patterns regarding guideline adherence and unnecessary serum ammonia testing for initial evaluation of HE in hospitals. To overcome this gap in knowledge, we conducted the present study to provide granular details regarding the diagnostic workup for hospitalized patients with HE.
METHODS
This study adopted a retrospective design and recruited patients admitted to the Virginia Commonwealth University Medical Center between July 1, 2016 and July 1, 2017. The institutional review board approved the study, and the manuscript was reviewed and approved by all authors prior to submission. All chart reviews were performed by hepatologists with access to patients’ electronic medical record (EMR).
Patient Population
Patients were identified from the EMR system by using ICD-9 and ICD-10 codes for cirrhosis, hepatic encephalopathy, and altered mental status. All consecutive admissions with these diagnosis codes were considered for inclusion. Adult patients with cirrhosis resulting from any etiology of chronic liver diseases with primary reason for admission of HE were included. If patients were readmitted for HE during the study period, then only the data from index HE admission was included in the analysis and data from subsequent admissions were excluded. The other exclusion criteria included non-HE causes of confusion, acute liver failure, and those admitted with a preformulated plan (eg, direct hepatology clinic admission or outside hospital transfer). Patients who developed HE during their hospitalization where HE was not the indication for admission were also excluded. Finally, all patients admitted under the direct care of hepatology were excluded.
Diagnostic Workup
The recommendations of the AASLD and the EASL for workup for HE include obtaining detailed history and physical examination supplemented by diagnostic evaluation for potential HE precipitants including infections, electrolyte disturbances, dehydration, renal failure, glycemic disturbances, and toxin ingestion (eg, alcohol, illicit drugs).5 Based on the guideline recommendation, this study defined a “complete workup” as including all of the following elements: infection evaluation (blood culture, urinalysis/urine culture, chest radiograph, diagnostic paracentesis in the presence of ascites), electrolyte/renal evaluation (serum sodium, potassium, creatinine, and glucose), and toxin evaluation (urine drug screening). Any HE admission that was missing elements from the aforementioned battery of tests was defined as “incomplete workup.” In patients admitted with decompensated cirrhosis, serum ammonia testing was considered inappropriate unless there was a nuanced explanation supporting its use documented within the EMR. The frequency and specialty of the physician ordering serum ammonia level tests were determined. The financial burden of unnecessary ammonia testing was estimated by assigning a laboratory charge ($258) for each patient.
Statistical Analysis
Continuous and categorical variables are reported as means (± standard deviation), median (interquartile range or IQR), or proportion (%) as appropriate. Across-group differences were compared using Student t-test for normally distributed continuous variables and Mann-Whitney U test for skewed data. Fisher’s exact test was used to compare proportion. HE evaluations were quantified by the number of patients with complete workup and by the number of patients with missing components of the workup. A nominal P value of less than .05 was considered statistically significant. All statistical analyses were performed using SPSS Statistics version 24.0 (IBM Corporation, Armonk, New York).
RESULTS
Cohort Characteristics
The baseline cohort demographics are listed in the Table. Of the 145 patients identified using diagnostic codes for cirrhosis, 78 subjects met the study criteria. The most common exclusion criteria included non-HE etiology of altered mental status (n = 37) and patients with readmissions for HE during the study period (n = 30). The mean age of the study cohort was 59.3 years, and the most common etiology of cirrhosis was hepatitis C (n = 41), alcohol induced (n = 14), and nonalcoholic steatohepatitis (n = 13).
Initial Diagnostic Evaluation
The major precipitants of HE in the study cohort were ineffective lactulose dosing (n = 43), infections (n = 25), and electrolyte disturbances/renal injury (n = 6). At the time of admission, 53 patients were on therapy for HE. Only 17 (22%) patients had complete diagnostic workup within 24 hours of hospital admission. The individual components of the complete workup are shown in the Figure. Notably, 23 (30%) patients were missing blood cultures, 16 (21%) were missing urinalysis, 15 (20%) were missing chest radiograph, and 34 (44%) were missing urine drug screening. Of the 34 patients with ascites on admission, only eight (23%) had diagnostic paracentesis performed on admission to rule out spontaneous bacterial peritonitis.
Serum Ammonia Testing
Serum ammonia testing was performed on 74 patients (94.9%), and no patient met the criteria for appropriate testing. Forty patients already had a known diagnosis of HE prior to index admission. Furthermore, 10 (14%) patients had serum ammonia testing repeated after admission without documentation in the EMR to justify repeat testing. Emergency Department (ED) physicians ordered ammonia testing in 57 cases (77%), internists ordered the testing in 11 cases (15%), and intensivists ordered the testing in two cases (3%). The patient’s charges for serum ammonia testing at the time of admission and for repeat testing were $19,092 and $2,580, respectively.
DISCUSSION
This study utilized HE in patients with decompensated cirrhosis as a framework to analyze adherence to societal guidelines. The adherence rate to AALSD/EASL recommended inpatient evaluation of HE is surprisingly low, and most patients are missing key essential elements of the diagnostic work up. While the diagnostic tests that are ordered as part of a panel are completed universally (renal function, electrolytes, and glucose testing), individual testing is less inclined to be ordered (blood cultures, urine culture/urinalysis, CXR, UDS) and procedural testing, such as diagnostic paracentesis, is often missed. This last finding is in line with published literature showing that 40% of patients admitted with ascites or HE did not have diagnostic paracentesis during hospital admission despite 24% reduction of inhospital mortality among patients undergoing the procedure.9
Although serum ammonia testing is not endorsed by the AASLD/EASL guidelines for HE,5 it is ordered nearly universally. The cost of an individual test is relatively low, but the cumulative cost of serum ammonia testing can be substantial because HE is the most common indication for hospitalization among patients with cirrhosis.4 Initiatives, such as the Choosing Wisely® campaign, encourage high-value and evidence-based care by limiting excessive and unnecessary diagnostic testing.10 The Canadian Choosing Wisely campaign specifically includes avoidance of serum ammonia testing for diagnosis of HE to provide high-value care in hepatology.11
Although the exact reasons for nonadherence to recommended HE evaluations are unclear, a potential method to mitigate excessive testing is to utilize the EMR and ordering system.3 EMR-based strategies can curb unnecessary testing in inpatient settings.12 The use of HE order sets, the inclusion of clinical decision support systems, and the restriction of access to specialized testing can be readily incorporated into the EMR to encourage adherence to guideline-based care while limiting unnecessary testing.
This study should be interpreted in the context of study limitations. Given the retrospective design of the study, salient factors in decisions behind diagnostic testing cannot be assessed. Future studies should utilize mixed-model methodology to elucidate reasons behind these decisions. The present study used a strict definition of complete workup including all the mentioned elements of the diagnostic workup for HE; however, in clinical practice, providers could be justified in not ordering certain tests if the specific clinical scenario does not lead to its use (eg, chest X-ray deferred in a patient with clear lung exam, no symptoms, or hypoxia). Similarly, UDS was included as a required element for a complete workup. While it may be ordered in a case-by-case basis to screen for illicit drug abuse, UDS is also a critical element of the workup to screen for opioid use as a precipitant of HE. Finally, considering the strict study entry criteria, we excluded repeated admissions for HE during the study period and therefore likely underestimate the cost burden of serum ammonia testing.
In conclusion, valuable guideline-based diagnostic testing is often missing in patients admitted for HE while serum ammonia testing is nearly universally ordered. These findings underscore the importance of implementing educational strategies, such as the Choosing Wisely® campaign, and EMR-based clinical decision support systems to improve health resource utilization in patients with cirrhosis and HE.
Disclosures
The authors have nothing to disclose.
1. Andrews EJ, Redmond HP. A review of clinical guidelines. Br J Surg. 2004;91:956-964. doi: 10.1002/bjs.4630 PubMed
2. Arts DL, Voncken AG, Medlock S, Abu-Hanna A, van Weert HC. Reasons for intentional guideline non-adherence: a systematic review. Int J Med Inform. 2016;89:55-62. doi: 10.1016/j.ijmedinf.2016.02.009. PubMed
3. Eaton KP, Levy K, Soong C, et al. Evidence-based guidelines to eliminate repetitive laboratory testing. JAMA Intern Med. 2017;177(12):1833-1839. doi: 10.1001/jamainternmed.2017.5152. PubMed
4. Everhart J. The burden of digestive diseases in the United States. Washington D.C.: US Department of Health and Human Services, Public Health Service, National Institutes of Health. U.S. Government Printing Office; 2008:111-114.
5. Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of Liver Diseases. Hepatology . 2014;60:715-735. doi: 10.1002/hep.27210 PubMed
6. Stahl J. Studies of the blood ammonia in liver disease: Its diagnostic, prognostic, and therapeutic significance. Ann Intern Med . 1963;58:1-24. PubMed
7. Ong JP, Aggarwal A, Kreiger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med . 2003;114:188-193. doi: 10.1016/S0002-9343(02)01477-8 PubMed
8. Nicalao F, Efrati C, Masini A, Merli M, Attili AF, Riggio O. Role of determination of partial pressure of ammonia in cirrhotic patients with and without hepatic encephalopathy. J Hepatol. 2003;38:441-446. doi: 10.1016/S0168-8278(02)00436-1 PubMed
9. Orman ES, Hayashi PH, Bataller R, Barritt AS 4th. Paracentesis is associated with reduced mortality in patients hospitalized with cirrhosis and ascites. Clin Gastroenterol Hepatol. 2014;12:496-503. doi: 10.1016/j.cgh.2013.08.025. PubMed
10. Cassek CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA. 2012;307:1801-1802. doi: 10.1001/jama.2012.476. PubMed
11. Choosing Wisely Canada. 2018. Five things patients and physicians should question. Available at: https://choosingwiselycanada.org/hepatology/ . Accessed November 18, 2018.
12. Iturrate E, Jubelt L, Volpicelli F, Hochman K. Optimize your electronic medical record to increase value: reducing laboratory overutilization. Am J Med . 2016;129:215-220. doi: 10.1016/j.amjmed.2015.09.009. PubMed
1. Andrews EJ, Redmond HP. A review of clinical guidelines. Br J Surg. 2004;91:956-964. doi: 10.1002/bjs.4630 PubMed
2. Arts DL, Voncken AG, Medlock S, Abu-Hanna A, van Weert HC. Reasons for intentional guideline non-adherence: a systematic review. Int J Med Inform. 2016;89:55-62. doi: 10.1016/j.ijmedinf.2016.02.009. PubMed
3. Eaton KP, Levy K, Soong C, et al. Evidence-based guidelines to eliminate repetitive laboratory testing. JAMA Intern Med. 2017;177(12):1833-1839. doi: 10.1001/jamainternmed.2017.5152. PubMed
4. Everhart J. The burden of digestive diseases in the United States. Washington D.C.: US Department of Health and Human Services, Public Health Service, National Institutes of Health. U.S. Government Printing Office; 2008:111-114.
5. Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of Liver Diseases. Hepatology . 2014;60:715-735. doi: 10.1002/hep.27210 PubMed
6. Stahl J. Studies of the blood ammonia in liver disease: Its diagnostic, prognostic, and therapeutic significance. Ann Intern Med . 1963;58:1-24. PubMed
7. Ong JP, Aggarwal A, Kreiger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med . 2003;114:188-193. doi: 10.1016/S0002-9343(02)01477-8 PubMed
8. Nicalao F, Efrati C, Masini A, Merli M, Attili AF, Riggio O. Role of determination of partial pressure of ammonia in cirrhotic patients with and without hepatic encephalopathy. J Hepatol. 2003;38:441-446. doi: 10.1016/S0168-8278(02)00436-1 PubMed
9. Orman ES, Hayashi PH, Bataller R, Barritt AS 4th. Paracentesis is associated with reduced mortality in patients hospitalized with cirrhosis and ascites. Clin Gastroenterol Hepatol. 2014;12:496-503. doi: 10.1016/j.cgh.2013.08.025. PubMed
10. Cassek CK, Guest JA. Choosing wisely: helping physicians and patients make smart decisions about their care. JAMA. 2012;307:1801-1802. doi: 10.1001/jama.2012.476. PubMed
11. Choosing Wisely Canada. 2018. Five things patients and physicians should question. Available at: https://choosingwiselycanada.org/hepatology/ . Accessed November 18, 2018.
12. Iturrate E, Jubelt L, Volpicelli F, Hochman K. Optimize your electronic medical record to increase value: reducing laboratory overutilization. Am J Med . 2016;129:215-220. doi: 10.1016/j.amjmed.2015.09.009. PubMed
© 2019 Society of Hospital Medicine
Reviews Reenvisioned: Supporting Enhanced Practice Improvement for Hospitalists
As part of the Journal of Hospital Medicine’s® commitment to our readership, we are excited to announce innovative new review formats, designed for busy hospitalists. The state of knowledge in our field is changing rapidly, and the 21st century poses a conundrum to clinicians in the form of increasingly complex studies and guidelines amidst ever-decreasing time to digest them. As a result, it can be challenging for hospitalists to access and interpret recently published research to inform their clinical practice. Because we are committed to practical innovation for hospitalists, starting in 2019, JHM will offer focused yet informative content that places important advances into relevant clinical or methodological context and provides our readers with information that is accessible, meaningful, and actionable—all in a more concise format.
Our new Clinical Guideline Highlights for the Hospitalist is a brief, targeted review of recently published clinical guidelines, distilling the major recommendations relevant to hospital medicine and placing them in context of the available evidence. This review format also offers a critique of gaps in the literature and notes areas ripe for future study. In this issue, we debut two articles using this new approach—one aimed at adult hospitalists and the other at pediatric hospitalists—regarding recently published studies and guidelines about maintenance intravenous fluids.1-5
In 2019, we will also introduce a second new format, called Progress Notes. These reviews will be shorter than JHM’s traditional review format, and will accept two types of articles: clinical and methodological. The clinical Progress Notes will provide an update on the last several years of evidence related to diagnosis, treatment, risk stratification, and/or prevention of a clinical problem highly pertinent to hospitalists. The methodological Progress Notes will provide our readers with insight into the application of quantitative, qualitative, and quality improvement methods commonly used in work published in this journal. Our aim is to use Progress Notes as a way to enhance both clinical practice and scholarship efforts by our readers.
Finally, we will introduce “Hospital Medicine: The Year in Review,” an annual feature that concisely compiles and critiques the top articles in both adult and pediatric hospital medicine over the past year. The “Year in Review” will serve as a written corollary to the popular “Updates in Hospital Medicine” presentation at the Society of Hospital Medicine annual meeting, and will highlight important research that advanced our field or provided us a fresh perspective on hospitalist practice.
As we introduce these new review formats, it is important to note that JHM will continue to accept traditional, long-form reviews on any topic relevant to hospitalists, with a preference for rigorous systematic reviews or meta-analyses. Equally important is that JHM’s overarching commitment remains unchanged: support clinicians, leaders, and scholars in our field in their pursuit of delivering evidence-based, high-value clinical care. We hope you enjoy these new article formats and we look forward to your feedback.
Disclosures
The authors declare they have no conflicts of interest/competing interests.
1. National Clinical Guideline Centre. Intravenous Fluid Therapy: Intravenous Fluid Therapy in Adults in Hospital. London: Royal College of Physicians (UK); 2013 Dec. PubMed
2. Selmer MW, Self WH, Wanderer JP, et al. Balanced Crystalloids versus Saline in Critically Ill Adults, N Engl J Med. 2018 Mar 1;378(9):829-839. doi: 10.1056/NEJMoa1711584. PubMed
3. Fled LG, et. al. “Clinical Practice Guideline: Maintenance Intravenous Fluids in Children,” Pediatrics. 2018 Dec;142(6). doi: 10.1542/peds.2018-3083. PubMed
4. Gottenborg E, Pierce R. Clinical Guideline Highlights for the Hospitalist: The Use of Intravenous Fluids in the Hospitalized Adult. J Hosp Med. 2019;14(3):172-173. doi: 10.12788/jhm.3178. PubMed
5. Girdwood ST, Parker MW, Shaughnessy EE. Clinical Guideline Highlights for the Hospitalist: Maintenance Intravenous Fluids in Infants and Children. J Hosp Med. 2019;14(3):170-171. doi: 10.12788/jhm.3177. PubMed
As part of the Journal of Hospital Medicine’s® commitment to our readership, we are excited to announce innovative new review formats, designed for busy hospitalists. The state of knowledge in our field is changing rapidly, and the 21st century poses a conundrum to clinicians in the form of increasingly complex studies and guidelines amidst ever-decreasing time to digest them. As a result, it can be challenging for hospitalists to access and interpret recently published research to inform their clinical practice. Because we are committed to practical innovation for hospitalists, starting in 2019, JHM will offer focused yet informative content that places important advances into relevant clinical or methodological context and provides our readers with information that is accessible, meaningful, and actionable—all in a more concise format.
Our new Clinical Guideline Highlights for the Hospitalist is a brief, targeted review of recently published clinical guidelines, distilling the major recommendations relevant to hospital medicine and placing them in context of the available evidence. This review format also offers a critique of gaps in the literature and notes areas ripe for future study. In this issue, we debut two articles using this new approach—one aimed at adult hospitalists and the other at pediatric hospitalists—regarding recently published studies and guidelines about maintenance intravenous fluids.1-5
In 2019, we will also introduce a second new format, called Progress Notes. These reviews will be shorter than JHM’s traditional review format, and will accept two types of articles: clinical and methodological. The clinical Progress Notes will provide an update on the last several years of evidence related to diagnosis, treatment, risk stratification, and/or prevention of a clinical problem highly pertinent to hospitalists. The methodological Progress Notes will provide our readers with insight into the application of quantitative, qualitative, and quality improvement methods commonly used in work published in this journal. Our aim is to use Progress Notes as a way to enhance both clinical practice and scholarship efforts by our readers.
Finally, we will introduce “Hospital Medicine: The Year in Review,” an annual feature that concisely compiles and critiques the top articles in both adult and pediatric hospital medicine over the past year. The “Year in Review” will serve as a written corollary to the popular “Updates in Hospital Medicine” presentation at the Society of Hospital Medicine annual meeting, and will highlight important research that advanced our field or provided us a fresh perspective on hospitalist practice.
As we introduce these new review formats, it is important to note that JHM will continue to accept traditional, long-form reviews on any topic relevant to hospitalists, with a preference for rigorous systematic reviews or meta-analyses. Equally important is that JHM’s overarching commitment remains unchanged: support clinicians, leaders, and scholars in our field in their pursuit of delivering evidence-based, high-value clinical care. We hope you enjoy these new article formats and we look forward to your feedback.
Disclosures
The authors declare they have no conflicts of interest/competing interests.
As part of the Journal of Hospital Medicine’s® commitment to our readership, we are excited to announce innovative new review formats, designed for busy hospitalists. The state of knowledge in our field is changing rapidly, and the 21st century poses a conundrum to clinicians in the form of increasingly complex studies and guidelines amidst ever-decreasing time to digest them. As a result, it can be challenging for hospitalists to access and interpret recently published research to inform their clinical practice. Because we are committed to practical innovation for hospitalists, starting in 2019, JHM will offer focused yet informative content that places important advances into relevant clinical or methodological context and provides our readers with information that is accessible, meaningful, and actionable—all in a more concise format.
Our new Clinical Guideline Highlights for the Hospitalist is a brief, targeted review of recently published clinical guidelines, distilling the major recommendations relevant to hospital medicine and placing them in context of the available evidence. This review format also offers a critique of gaps in the literature and notes areas ripe for future study. In this issue, we debut two articles using this new approach—one aimed at adult hospitalists and the other at pediatric hospitalists—regarding recently published studies and guidelines about maintenance intravenous fluids.1-5
In 2019, we will also introduce a second new format, called Progress Notes. These reviews will be shorter than JHM’s traditional review format, and will accept two types of articles: clinical and methodological. The clinical Progress Notes will provide an update on the last several years of evidence related to diagnosis, treatment, risk stratification, and/or prevention of a clinical problem highly pertinent to hospitalists. The methodological Progress Notes will provide our readers with insight into the application of quantitative, qualitative, and quality improvement methods commonly used in work published in this journal. Our aim is to use Progress Notes as a way to enhance both clinical practice and scholarship efforts by our readers.
Finally, we will introduce “Hospital Medicine: The Year in Review,” an annual feature that concisely compiles and critiques the top articles in both adult and pediatric hospital medicine over the past year. The “Year in Review” will serve as a written corollary to the popular “Updates in Hospital Medicine” presentation at the Society of Hospital Medicine annual meeting, and will highlight important research that advanced our field or provided us a fresh perspective on hospitalist practice.
As we introduce these new review formats, it is important to note that JHM will continue to accept traditional, long-form reviews on any topic relevant to hospitalists, with a preference for rigorous systematic reviews or meta-analyses. Equally important is that JHM’s overarching commitment remains unchanged: support clinicians, leaders, and scholars in our field in their pursuit of delivering evidence-based, high-value clinical care. We hope you enjoy these new article formats and we look forward to your feedback.
Disclosures
The authors declare they have no conflicts of interest/competing interests.
1. National Clinical Guideline Centre. Intravenous Fluid Therapy: Intravenous Fluid Therapy in Adults in Hospital. London: Royal College of Physicians (UK); 2013 Dec. PubMed
2. Selmer MW, Self WH, Wanderer JP, et al. Balanced Crystalloids versus Saline in Critically Ill Adults, N Engl J Med. 2018 Mar 1;378(9):829-839. doi: 10.1056/NEJMoa1711584. PubMed
3. Fled LG, et. al. “Clinical Practice Guideline: Maintenance Intravenous Fluids in Children,” Pediatrics. 2018 Dec;142(6). doi: 10.1542/peds.2018-3083. PubMed
4. Gottenborg E, Pierce R. Clinical Guideline Highlights for the Hospitalist: The Use of Intravenous Fluids in the Hospitalized Adult. J Hosp Med. 2019;14(3):172-173. doi: 10.12788/jhm.3178. PubMed
5. Girdwood ST, Parker MW, Shaughnessy EE. Clinical Guideline Highlights for the Hospitalist: Maintenance Intravenous Fluids in Infants and Children. J Hosp Med. 2019;14(3):170-171. doi: 10.12788/jhm.3177. PubMed
1. National Clinical Guideline Centre. Intravenous Fluid Therapy: Intravenous Fluid Therapy in Adults in Hospital. London: Royal College of Physicians (UK); 2013 Dec. PubMed
2. Selmer MW, Self WH, Wanderer JP, et al. Balanced Crystalloids versus Saline in Critically Ill Adults, N Engl J Med. 2018 Mar 1;378(9):829-839. doi: 10.1056/NEJMoa1711584. PubMed
3. Fled LG, et. al. “Clinical Practice Guideline: Maintenance Intravenous Fluids in Children,” Pediatrics. 2018 Dec;142(6). doi: 10.1542/peds.2018-3083. PubMed
4. Gottenborg E, Pierce R. Clinical Guideline Highlights for the Hospitalist: The Use of Intravenous Fluids in the Hospitalized Adult. J Hosp Med. 2019;14(3):172-173. doi: 10.12788/jhm.3178. PubMed
5. Girdwood ST, Parker MW, Shaughnessy EE. Clinical Guideline Highlights for the Hospitalist: Maintenance Intravenous Fluids in Infants and Children. J Hosp Med. 2019;14(3):170-171. doi: 10.12788/jhm.3177. PubMed
© 2019 Society of Hospital Medicine
Addressing substance use in patients with intellectual disability: 5 Steps
Approximately 5% of patients with intellectual disability (ID) have a comorbid substance use disorder (SUD).1 These patients frequently abuse alcohol, tobacco, and cannabis, but are largely underdiagnosed and undertreated for SUDs. Treatment for SUDs in these patients is critical because substance abuse among patients with ID is associated with developing mood disorders, long-term health consequences, incarceration, and interpersonal instability.1 To ensure that these often-marginalized patients are adequately assessed and treated for SUDs, consider the following 5 steps.
1. Perform screening tests. Unfortunately, no substance use screening tests are validated specifically for patients with ID. When presented with mainstream screening tools, patients with ID could produce false positives or false negatives for 2 reasons:
- Patients with ID are more likely to respond in the affirmative to screening questions that they do not understand.
- Many screening questionnaires assume that patients possess an amount of knowledge and cognitive ability to abstract information that patients with ID may lack.
Clinicians should therefore adapt screening questions to better match the cognitive and communicative abilities of their patients with ID by simplifying sentences, using graphics, and avoiding negative phrases and confrontation. For example, while all-encompassing, the term “alcohol” may be confusing for some patients. Instead of broadly asking a patient, “Do you drink alcoholic beverages?” it may be necessary to specifically ask, “Do you drink wine?” or “Do you drink beer?” Similarly, it may be insufficient to ask a patient, “Do you smoke marijuana?” Instead, use colloquial terms (ie, weed, reefer) to ensure that the patient knows which substance you mean. Screening questions can be complemented by ordering urine drug testing and obtaining collateral information from caregivers.2
2. Use approved medications to treat SUDs. Medication-assisted treatment (MAT) is underprescribed for patients with ID. Medication compliance in patients with ID may be a concern; however, many of these patients are compliant with treatment because they often live with family members, in group homes, or in other settings where their medications are administered to them.
Also, be mindful of whether your patient has epilepsy. This condition is common among patients with ID,3 and some MAT can lower the seizure threshold. When starting and titrating MAT, always monitor patients carefully for benefits and adverse effects.4
3. Make a thorough assessment before recommending Alcoholics Anonymous or Narcotics Anonymous meetings. While the 12-step recovery model has proven benefits, the typical structure of 12-step meetings is not conducive to all patients with ID. Only recommend such meetings to patients who have 60- to 90-minute attention spans and demonstrate the cognitive, communicative, literacy, and social skills to fully engage during the meetings.5
4. Employ motivational interviewing. Many patients with ID have cursory knowledge of the health risks associated with substance abuse, particularly those with mild ID. Motivational interviewing techniques that include health education may help produce favorable outcomes in these patients.6
Continue to: Provide ongoing support
5. Provide ongoing support. Remember that addiction is a chronic disease with a risk of relapse. Provide continuous support for patients with ID and comorbid SUDs throughout all phases of their recovery, and refer them to addiction specialists, pain specialists, or psychotherapists as appropriate.
1. Chapman SL, Wu L. Substance abuse among individuals with intellectual disabilities. Res Dev Disabil. 2012;33(4):1147-1156.
2. Kiewik M, Vandernagel J, Engles, R, et al. Intellectually disabled and addicted: a call for evidence based tailor-made interventions. Addiction. 2017;112(45):20 67-2068.
3. Mcgrother C, Bhaumik S, Thorp C, et al. Epilepsy in adults with intellectual disabilities: prevalence, associations and service implications. Seizure. 2006;15(6):376-386.
4. Connery H. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
5. Slayter E. Disparities in access to substance abuse treatment among people with intellectual disabilities and serious mental illness. Health Soc Work. 2010;35(1):49-59.
6. Frielink N, Schuengel C, Kroon A, et al. Pretreatment for substance-abusing people with intellectual disabilities: intervening on autonomous motivation for treatment entry. J Intellect Disabil Res. 2015;59(12):1168-1182.
Approximately 5% of patients with intellectual disability (ID) have a comorbid substance use disorder (SUD).1 These patients frequently abuse alcohol, tobacco, and cannabis, but are largely underdiagnosed and undertreated for SUDs. Treatment for SUDs in these patients is critical because substance abuse among patients with ID is associated with developing mood disorders, long-term health consequences, incarceration, and interpersonal instability.1 To ensure that these often-marginalized patients are adequately assessed and treated for SUDs, consider the following 5 steps.
1. Perform screening tests. Unfortunately, no substance use screening tests are validated specifically for patients with ID. When presented with mainstream screening tools, patients with ID could produce false positives or false negatives for 2 reasons:
- Patients with ID are more likely to respond in the affirmative to screening questions that they do not understand.
- Many screening questionnaires assume that patients possess an amount of knowledge and cognitive ability to abstract information that patients with ID may lack.
Clinicians should therefore adapt screening questions to better match the cognitive and communicative abilities of their patients with ID by simplifying sentences, using graphics, and avoiding negative phrases and confrontation. For example, while all-encompassing, the term “alcohol” may be confusing for some patients. Instead of broadly asking a patient, “Do you drink alcoholic beverages?” it may be necessary to specifically ask, “Do you drink wine?” or “Do you drink beer?” Similarly, it may be insufficient to ask a patient, “Do you smoke marijuana?” Instead, use colloquial terms (ie, weed, reefer) to ensure that the patient knows which substance you mean. Screening questions can be complemented by ordering urine drug testing and obtaining collateral information from caregivers.2
2. Use approved medications to treat SUDs. Medication-assisted treatment (MAT) is underprescribed for patients with ID. Medication compliance in patients with ID may be a concern; however, many of these patients are compliant with treatment because they often live with family members, in group homes, or in other settings where their medications are administered to them.
Also, be mindful of whether your patient has epilepsy. This condition is common among patients with ID,3 and some MAT can lower the seizure threshold. When starting and titrating MAT, always monitor patients carefully for benefits and adverse effects.4
3. Make a thorough assessment before recommending Alcoholics Anonymous or Narcotics Anonymous meetings. While the 12-step recovery model has proven benefits, the typical structure of 12-step meetings is not conducive to all patients with ID. Only recommend such meetings to patients who have 60- to 90-minute attention spans and demonstrate the cognitive, communicative, literacy, and social skills to fully engage during the meetings.5
4. Employ motivational interviewing. Many patients with ID have cursory knowledge of the health risks associated with substance abuse, particularly those with mild ID. Motivational interviewing techniques that include health education may help produce favorable outcomes in these patients.6
Continue to: Provide ongoing support
5. Provide ongoing support. Remember that addiction is a chronic disease with a risk of relapse. Provide continuous support for patients with ID and comorbid SUDs throughout all phases of their recovery, and refer them to addiction specialists, pain specialists, or psychotherapists as appropriate.
Approximately 5% of patients with intellectual disability (ID) have a comorbid substance use disorder (SUD).1 These patients frequently abuse alcohol, tobacco, and cannabis, but are largely underdiagnosed and undertreated for SUDs. Treatment for SUDs in these patients is critical because substance abuse among patients with ID is associated with developing mood disorders, long-term health consequences, incarceration, and interpersonal instability.1 To ensure that these often-marginalized patients are adequately assessed and treated for SUDs, consider the following 5 steps.
1. Perform screening tests. Unfortunately, no substance use screening tests are validated specifically for patients with ID. When presented with mainstream screening tools, patients with ID could produce false positives or false negatives for 2 reasons:
- Patients with ID are more likely to respond in the affirmative to screening questions that they do not understand.
- Many screening questionnaires assume that patients possess an amount of knowledge and cognitive ability to abstract information that patients with ID may lack.
Clinicians should therefore adapt screening questions to better match the cognitive and communicative abilities of their patients with ID by simplifying sentences, using graphics, and avoiding negative phrases and confrontation. For example, while all-encompassing, the term “alcohol” may be confusing for some patients. Instead of broadly asking a patient, “Do you drink alcoholic beverages?” it may be necessary to specifically ask, “Do you drink wine?” or “Do you drink beer?” Similarly, it may be insufficient to ask a patient, “Do you smoke marijuana?” Instead, use colloquial terms (ie, weed, reefer) to ensure that the patient knows which substance you mean. Screening questions can be complemented by ordering urine drug testing and obtaining collateral information from caregivers.2
2. Use approved medications to treat SUDs. Medication-assisted treatment (MAT) is underprescribed for patients with ID. Medication compliance in patients with ID may be a concern; however, many of these patients are compliant with treatment because they often live with family members, in group homes, or in other settings where their medications are administered to them.
Also, be mindful of whether your patient has epilepsy. This condition is common among patients with ID,3 and some MAT can lower the seizure threshold. When starting and titrating MAT, always monitor patients carefully for benefits and adverse effects.4
3. Make a thorough assessment before recommending Alcoholics Anonymous or Narcotics Anonymous meetings. While the 12-step recovery model has proven benefits, the typical structure of 12-step meetings is not conducive to all patients with ID. Only recommend such meetings to patients who have 60- to 90-minute attention spans and demonstrate the cognitive, communicative, literacy, and social skills to fully engage during the meetings.5
4. Employ motivational interviewing. Many patients with ID have cursory knowledge of the health risks associated with substance abuse, particularly those with mild ID. Motivational interviewing techniques that include health education may help produce favorable outcomes in these patients.6
Continue to: Provide ongoing support
5. Provide ongoing support. Remember that addiction is a chronic disease with a risk of relapse. Provide continuous support for patients with ID and comorbid SUDs throughout all phases of their recovery, and refer them to addiction specialists, pain specialists, or psychotherapists as appropriate.
1. Chapman SL, Wu L. Substance abuse among individuals with intellectual disabilities. Res Dev Disabil. 2012;33(4):1147-1156.
2. Kiewik M, Vandernagel J, Engles, R, et al. Intellectually disabled and addicted: a call for evidence based tailor-made interventions. Addiction. 2017;112(45):20 67-2068.
3. Mcgrother C, Bhaumik S, Thorp C, et al. Epilepsy in adults with intellectual disabilities: prevalence, associations and service implications. Seizure. 2006;15(6):376-386.
4. Connery H. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
5. Slayter E. Disparities in access to substance abuse treatment among people with intellectual disabilities and serious mental illness. Health Soc Work. 2010;35(1):49-59.
6. Frielink N, Schuengel C, Kroon A, et al. Pretreatment for substance-abusing people with intellectual disabilities: intervening on autonomous motivation for treatment entry. J Intellect Disabil Res. 2015;59(12):1168-1182.
1. Chapman SL, Wu L. Substance abuse among individuals with intellectual disabilities. Res Dev Disabil. 2012;33(4):1147-1156.
2. Kiewik M, Vandernagel J, Engles, R, et al. Intellectually disabled and addicted: a call for evidence based tailor-made interventions. Addiction. 2017;112(45):20 67-2068.
3. Mcgrother C, Bhaumik S, Thorp C, et al. Epilepsy in adults with intellectual disabilities: prevalence, associations and service implications. Seizure. 2006;15(6):376-386.
4. Connery H. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
5. Slayter E. Disparities in access to substance abuse treatment among people with intellectual disabilities and serious mental illness. Health Soc Work. 2010;35(1):49-59.
6. Frielink N, Schuengel C, Kroon A, et al. Pretreatment for substance-abusing people with intellectual disabilities: intervening on autonomous motivation for treatment entry. J Intellect Disabil Res. 2015;59(12):1168-1182.
Helping patients through a benzodiazepine taper
Benzodiazepines are one of the most commonly prescribed medication classes worldwide.1 Patients prescribed benzodiazepines who have no history of abuse or misuse may want to reduce or discontinue using these agents for various reasons, including adverse effects or wanting to reduce the number of medications they take. In this article, we offer strategies for creating an individualized taper plan, and describe additional nonpharmacologic interventions to help ensure that the taper is successful.
Formulating a taper plan
There is no gold-standard algorithm for tapering benzodiazepines.1,2 Even with a carefully designed plan, tapering can be challenging because approximately one-third of patients will experience difficulties such as withdrawal symptoms.1 Prior to creating a plan, carefully assess the patient’s history, including the type of benzodiazepine prescribed (short- or long-acting); the dose, dosing frequency, and duration of use; comorbid medical and psychiatric conditions; any previous experience with withdrawal symptoms; and psychosocial factors (eg, lifestyle and personality). Consider whether the patient can be safely tapered in an outpatient setting or will require hospitalization. Tapering designed to take place over several weeks or months tends to be more successful; however, patient-specific circumstances play a role in determining the duration of the taper.1,2
For the greatest chance of success, a benzodiazepine should not be reduced faster than 25% of the total daily dose per week.1 Consider which of the following pharmacologic approaches to benzodiazepine tapering might work best for your patient:
- Reduce the daily dose by one-eighth to one-tenth every 1 to 2 weeks over a 2- to 12-month period for patients with a physiological dependence.1
- Reduce the benzodiazepine dose by 10% to 25% every 2 weeks over a 4- to 8-week period.2
- Some guidelines have suggested converting the prescribed benzodiazepine to an equivalent dose of diazepam because of its long half-life, and then reducing the diazepam dose by one-eighth every 2 weeks.3
There is uncertainty in the medical literature about using a long-acting benzodiazepine to taper off a short-acting benzodiazepine, although this practice is generally clinically accepted.1,2 Similarly, there is no definitive evidence that supports using adjuvant medications to facilitate tapering.1,2
Nonpharmacologic interventions
Patients are more likely to have a successful taper if nonpharmacologic interventions are part of a comprehensive treatment plan.1
To help your patients through the challenges of a benzodiazepine taper:
- Validate their concerns, reassure them that you will support them throughout the taper, and provide information on additional resources for support.
- Provide education about the process of tapering and symptoms of withdrawal.
- Recommend therapies, such as cognitive-behavioral therapy or motivational interventions, that develop or enhance coping skills.
- Enlist the help of the patient’s family and friends for support and encouragement.
Despite some clinicians’ trepidation, 70% to 90% of patients can be successfully tapered off benzodiazepines by using an individualized approach that includes tailored tapering and nonpharmacologic interventions that provide benefits that persist after the patient completes the taper.1
1. Guina J, Merrill B. Benzodiazepines II: waking up on sedatives: providing optimal care when inheriting benzodiazepine prescriptions in transfer patients. J Clin Med. 2018;7(2):pii: E20. doi: 10.3390/jcm7020020.
2. Soyka M. Treatment of benzodiazepine dependence. N Engl J Med. 2017;376(12):1147-1157.
3. Diaper AM, Law FD, Melichar JK. Pharmacological strategies for detoxification. Br J Clin Pharmacol. 2014;77(2):302-314.
Benzodiazepines are one of the most commonly prescribed medication classes worldwide.1 Patients prescribed benzodiazepines who have no history of abuse or misuse may want to reduce or discontinue using these agents for various reasons, including adverse effects or wanting to reduce the number of medications they take. In this article, we offer strategies for creating an individualized taper plan, and describe additional nonpharmacologic interventions to help ensure that the taper is successful.
Formulating a taper plan
There is no gold-standard algorithm for tapering benzodiazepines.1,2 Even with a carefully designed plan, tapering can be challenging because approximately one-third of patients will experience difficulties such as withdrawal symptoms.1 Prior to creating a plan, carefully assess the patient’s history, including the type of benzodiazepine prescribed (short- or long-acting); the dose, dosing frequency, and duration of use; comorbid medical and psychiatric conditions; any previous experience with withdrawal symptoms; and psychosocial factors (eg, lifestyle and personality). Consider whether the patient can be safely tapered in an outpatient setting or will require hospitalization. Tapering designed to take place over several weeks or months tends to be more successful; however, patient-specific circumstances play a role in determining the duration of the taper.1,2
For the greatest chance of success, a benzodiazepine should not be reduced faster than 25% of the total daily dose per week.1 Consider which of the following pharmacologic approaches to benzodiazepine tapering might work best for your patient:
- Reduce the daily dose by one-eighth to one-tenth every 1 to 2 weeks over a 2- to 12-month period for patients with a physiological dependence.1
- Reduce the benzodiazepine dose by 10% to 25% every 2 weeks over a 4- to 8-week period.2
- Some guidelines have suggested converting the prescribed benzodiazepine to an equivalent dose of diazepam because of its long half-life, and then reducing the diazepam dose by one-eighth every 2 weeks.3
There is uncertainty in the medical literature about using a long-acting benzodiazepine to taper off a short-acting benzodiazepine, although this practice is generally clinically accepted.1,2 Similarly, there is no definitive evidence that supports using adjuvant medications to facilitate tapering.1,2
Nonpharmacologic interventions
Patients are more likely to have a successful taper if nonpharmacologic interventions are part of a comprehensive treatment plan.1
To help your patients through the challenges of a benzodiazepine taper:
- Validate their concerns, reassure them that you will support them throughout the taper, and provide information on additional resources for support.
- Provide education about the process of tapering and symptoms of withdrawal.
- Recommend therapies, such as cognitive-behavioral therapy or motivational interventions, that develop or enhance coping skills.
- Enlist the help of the patient’s family and friends for support and encouragement.
Despite some clinicians’ trepidation, 70% to 90% of patients can be successfully tapered off benzodiazepines by using an individualized approach that includes tailored tapering and nonpharmacologic interventions that provide benefits that persist after the patient completes the taper.1
Benzodiazepines are one of the most commonly prescribed medication classes worldwide.1 Patients prescribed benzodiazepines who have no history of abuse or misuse may want to reduce or discontinue using these agents for various reasons, including adverse effects or wanting to reduce the number of medications they take. In this article, we offer strategies for creating an individualized taper plan, and describe additional nonpharmacologic interventions to help ensure that the taper is successful.
Formulating a taper plan
There is no gold-standard algorithm for tapering benzodiazepines.1,2 Even with a carefully designed plan, tapering can be challenging because approximately one-third of patients will experience difficulties such as withdrawal symptoms.1 Prior to creating a plan, carefully assess the patient’s history, including the type of benzodiazepine prescribed (short- or long-acting); the dose, dosing frequency, and duration of use; comorbid medical and psychiatric conditions; any previous experience with withdrawal symptoms; and psychosocial factors (eg, lifestyle and personality). Consider whether the patient can be safely tapered in an outpatient setting or will require hospitalization. Tapering designed to take place over several weeks or months tends to be more successful; however, patient-specific circumstances play a role in determining the duration of the taper.1,2
For the greatest chance of success, a benzodiazepine should not be reduced faster than 25% of the total daily dose per week.1 Consider which of the following pharmacologic approaches to benzodiazepine tapering might work best for your patient:
- Reduce the daily dose by one-eighth to one-tenth every 1 to 2 weeks over a 2- to 12-month period for patients with a physiological dependence.1
- Reduce the benzodiazepine dose by 10% to 25% every 2 weeks over a 4- to 8-week period.2
- Some guidelines have suggested converting the prescribed benzodiazepine to an equivalent dose of diazepam because of its long half-life, and then reducing the diazepam dose by one-eighth every 2 weeks.3
There is uncertainty in the medical literature about using a long-acting benzodiazepine to taper off a short-acting benzodiazepine, although this practice is generally clinically accepted.1,2 Similarly, there is no definitive evidence that supports using adjuvant medications to facilitate tapering.1,2
Nonpharmacologic interventions
Patients are more likely to have a successful taper if nonpharmacologic interventions are part of a comprehensive treatment plan.1
To help your patients through the challenges of a benzodiazepine taper:
- Validate their concerns, reassure them that you will support them throughout the taper, and provide information on additional resources for support.
- Provide education about the process of tapering and symptoms of withdrawal.
- Recommend therapies, such as cognitive-behavioral therapy or motivational interventions, that develop or enhance coping skills.
- Enlist the help of the patient’s family and friends for support and encouragement.
Despite some clinicians’ trepidation, 70% to 90% of patients can be successfully tapered off benzodiazepines by using an individualized approach that includes tailored tapering and nonpharmacologic interventions that provide benefits that persist after the patient completes the taper.1
1. Guina J, Merrill B. Benzodiazepines II: waking up on sedatives: providing optimal care when inheriting benzodiazepine prescriptions in transfer patients. J Clin Med. 2018;7(2):pii: E20. doi: 10.3390/jcm7020020.
2. Soyka M. Treatment of benzodiazepine dependence. N Engl J Med. 2017;376(12):1147-1157.
3. Diaper AM, Law FD, Melichar JK. Pharmacological strategies for detoxification. Br J Clin Pharmacol. 2014;77(2):302-314.
1. Guina J, Merrill B. Benzodiazepines II: waking up on sedatives: providing optimal care when inheriting benzodiazepine prescriptions in transfer patients. J Clin Med. 2018;7(2):pii: E20. doi: 10.3390/jcm7020020.
2. Soyka M. Treatment of benzodiazepine dependence. N Engl J Med. 2017;376(12):1147-1157.
3. Diaper AM, Law FD, Melichar JK. Pharmacological strategies for detoxification. Br J Clin Pharmacol. 2014;77(2):302-314.
A suicide attempt, or something else?
CASE Unexplained hypoglycemia
Ms. A, age 12, is brought to the emergency department (ED) via ambulance with altered mentation and life-threatening hypoglycemia for management of a hypoglycemic seizure. Earlier that day, Ms. A’s parents had found her unresponsive and incontinent of urine. In the ED, Ms. A is minimally responsive. Her blood glucose level measurements are in the range of 30 to 39 mg/dL (reference range: 70 to 99 mg/dL), despite having received IV dextrose first from paramedics, and then in the ED. Ms. A has no history of hypoglycemia or diabetes. Her parents say that the night before coming to the ED, Ms. A had experienced flu-like symptoms, including nausea, vomiting, and diarrhea, that continued overnight and resulted in minimal food intake for 24 hours (Table 1).

A physical exam demonstrates left-sided weakness of face, arm, and leg, rightward gaze, and left-sided neglect. However, the results of CT angiography and an MRI of the brain rule out a stroke. An EEG shows right hemispheric slowing consistent with postictal paralysis, but no ongoing seizure activity. Ms. A is transferred to the pediatric intensive care unit (PICU).
Although Ms. A has no psychiatric diagnoses, she has a history of depressive symptoms, self-harm by cutting, and a suicide attempt by ingestion of an over-the-counter (OTC) medication 1 year ago. She had reported the suicide attempt to her parents several months after the fact, and asked them to find her a therapist, which her parents arranged. She also has a history of asthma, which is well-controlled with montelukast, 5 mg/d.
EVALUATION Elevated insulin levels
Subsequent investigations for organic causes of hypoglycemia are negative for adrenal insufficiency, fatty acid oxidation defect, and sepsis. Blood results demonstrate significantly elevated insulin levels of 92.4 mcIU/mL (reference range: 2.6 to 24.9 mcIU/mL) and a C-peptide level of 9.5 ng/mL (reference range: 1.1 to 4.4 ng/mL).
On Day 1 of admission to the PICU, Ms. A’s blood glucose level normalizes, and her mentation improves. Her parents report that one of them has diabetes and takes oral hypoglycemic agents at home, including glipizide immediate release (IR) tablets, 10 mg, and long-acting insulin glargine. The treatment team suspects that Ms. A may have ingested one or both of these agents, and orders a toxicologic screening for oral hypoglycemic agents.
On Day 2, the toxicology results are returned and are positive for glipizide, which Ms. A had not been prescribed. Ms. A states that she had taken only her montelukast tablet on the day of admission and adamantly denies deliberately ingesting her parent’s diabetes medications. Her parents check the home medications and state there are no missing glipizide IR tablets or insulin vials. They also report that Ms. A had no access to extended-release glipizide.
The treatment team discuss Ms. A’s clinical condition and toxicology results with the pediatric endocrinology team. The endocrinology team states that with no history of hypoglycemic episodes, it is unlikely that Ms. A had an endogenous etiology that would present so catastrophically. In their experience, inexplicable hypoglycemia in a healthy individual who lives in a household with someone who has diabetes is due to ingestion of a hypoglycemic agent until proven otherwise.
[polldaddy:10252689]
Continue to: The authors' observations
The authors’ observations
In the context of Ms. A’s prior suicide attempt and history of self-harm, the pediatric team was concerned that her presentation was consistent with a suicide attempt and consulted the psychiatry service.
Glipizide is a second-generation sulfonylurea used to treat type 2 diabetes. It lowers blood glucose by stimulating pancreatic insulin secretion. It is a rare drug of overdose.1 Although pediatric glipizide overdoses have been documented, there are currently no pediatric or adolescent glipizide pharmacokinetic studies in the literature.1-4 In adults, the immediate-release formulation has 100% oral bioavailability, with a maximum plasma concentration (Tmax) of approximately 2 hours.5 The half-life typically ranges from 4 to 6 hours in adults.6 Patients who do not have diabetes are much more susceptible to the hypoglycemic effects of glipizide because the medication simulates their fully functional pancreas to produce a vigorous insulin response.
Ms. A’s significantly elevated insulin was consistent with normal glipizide effects in a healthy child, while the elevated C-peptide was consistent with insulin being endogenously produced, which ruled out ingestion of her parent’s insulin. Importantly, the pediatric endocrinology team noted that, in their experience, a single 5- to 10-mg dose of glipizide IR was sufficient to lower blood glucose levels to the low 30s mg/dL in the context of a functional pancreas, which suggested that Ms. A might have accidentally ingested a single glipizide IR tablet, and might be telling the truth when she denies deliberately ingesting it to hurt herself.
The clinical value of pharmacokinetics
The screen of Ms. A’s toxicology sample detected glipizide. The laboratory used a semi-quantitative serum screen of several hypoglycemic agents. A positive result for each agent is based on a quantitative cut-off value, which is 3 ng/mL for glipizide. The clinical chemist on call was asked to assist in interpreting the results. The serum specimen collected on Day 1 had a significantly positive glipizide result of 86 to 130 ng/mL. The maximum effective glipizide concentration for adult patients with diabetes is 100 ng/mL.7 Thus, the glipizide level of 86 to 130 ng/mL (20.5 hours after initial symptoms) is consistent with the clinical presentation of persistent hypoglycemia requiring ongoing glucose replacement therapy.
Due to the lack of pediatric pharmacokinetic data for glipizide and only a single serum measurement, it is not possible to estimate the glipizide concentration at the time of maximal symptoms (loss of consciousness at 2:30
Continue to: Clinicians need to be aware that...
Clinicians need to be aware that although hypoglycemia usually presents rapidly, in children glipizide IR can rarely cause delayed hypoglycemia up to 16 hours after ingestion,2 and a delay of 45 hours was reported in a case of ingestion of extended-release glipizide.8 Hypoglycemia can last up to nearly 24 hours and is exacerbated if the patient has not eaten.1,2 Importantly, Ms. A’s parents reported that she had no access to extended-release glipizide. When detailed pharmacokinetic data are not available, the information provided by the patient and parents becomes extremely important, especially in distinguishing between single and multiple overdoses prior to presentation, or co-ingestions, or decreased food intake that could exacerbate hypoglycemia.
EVALUATION Safety assessment
On Day 2, Ms. A and her parents are interviewed separately, and they all are consistent in their recollection that Ms. A had been feverish with flu-like symptoms throughout the night before coming to the ED, and had still seemed mildly confused on the morning of admission.
During the interview, her parents wonder when Ms. A took her daily dose of a single montelukast tablet for asthma, and whether she had accidentally confused it with their glipizide. They report that on the morning of admission, both the glipizide and montelukast medication vials were in the same room. The vials are the same color, the same size, and labeled from the same pharmacy, and contain white, scored, round tablets that look very similar.
During the interview, Ms. A consistently denies having thoughts of hurting or killing herself on the day of admission or before that.
[polldaddy:10252690]
Continue to: The authors' observations
The authors’ observations
This case was ultimately an accidental ingestion of glipizide, rather than a suicide attempt. The initial suspicion for a suicide attempt had been reasonable in the context of Ms. A’s depressive symptoms, remote history of a prior suicide attempt by ingesting an OTC medication, and toxicologic evidence of ingesting a drug not prescribed to her. Additionally, because of the life-threatening presentation, it was easy to make the erroneous assumption that the ingestion of glipizide must have involved many tablets, and thus must have been deliberate. However, through multidisciplinary teamwork, we were able to demonstrate that this was likely an accidental ingestion by a patient who had an acute febrile illness. Her illness had caused confusion, which contributed to the accidental ingestion, and also caused reduced food intake, which enhanced the hypoglycemic effects of glipizide. Additionally, a lack of awareness of medication safety in the home had facilitated the confusion between the two medication vials.

A single tablet of glipizide IR is sufficient to produce profound clinical effects that could be mistaken by medical and psychiatric teams for a much larger and/or deliberate overdose, especially in patients with a psychiatric history. The inappropriate psychiatric hospitalization of a patient, especially a child, who has been mistakenly diagnosed as having attempted suicide, can have negative therapeutic consequences (Table 2). A psychiatric admission would have been misguided if it attempted to address safety and reduce suicidality when no such concerns were present. Additionally, it could have damaged relationships with the patient and the family, especially in a child who had historically not sought psychiatric care despite depressive symptoms and a previous suicide attempt. When assessing for suicidality, consider accidental ingestion in the differential and use specialty expertise and confirmatory testing in the evaluation, taking the pharmacokinetics of the suspected agent into account.
OUTCOME Outpatient treatment
Ms. A’s neurologic symptoms resolve within 24 hours of admission. She is offered psychiatric inpatient hospitalization to address her depressive symptoms; however, her parents prefer that she receive outpatient care. Ms. A’s parents also state that after Ms. A’s admission, they locked up all household medications and will be more mindful with medication in the home. Because her parents are arranging appropriate outpatient treatment for Ms. A’s depression and maintenance of her safety, an involuntary hospitalization is not deemed necessary.
On Day 2, Ms. A is eating normally, her blood glucose levels remain stable, and she is discharged home.
Bottom Line
Oral hypoglycemic agents can cause life-threatening syndromes in healthy patients and can clinically mimic large, intentional overdoses. Clinicians must be aware of the differential of accidental ingestion when assessing for suicidality, and can use toxicology results in their assessment.
Related Resources
- Kidemergencies.com. Emergencies: One pill can kill. http://kidemergencies.com/onepill1.html.
- Safe Kids Worldwide. Medication safety. https://www.safekids.org/medicinesafety.
- American Association of Poison Control Centers. http://www.aapcc.org/.
Drug Brand Names
Glipizide • Glucotrol
Insulin glargine • Lantus
Montelukast • Singulair
1. Spiller HA, Villalobos D, Krenzelok EP, et al. Prospective multicenter study of sulfonylurea ingestion in children. J Pediatr. 1997;131(1):141-146.
2. Quadrani DA, Spiller HA, Widder P. Five year retrospective evaluation of sulfonylurea ingestion in children. J Toxicol Clin Toxicol. 1996;34(3):267-270.
3. Borowski H, Caraccio T, Mofenson H. Sulfonylurea ingestion in children: is an 8-hour observation period sufficient? J Pediatr. 1998;133(4):584-585.
4. Little GL, Boniface KS. Are one or two dangerous? Sulfony-lurea exposure in toddlers. J Emerg Med. 2005;28(3):305-310.
5. Huupponen R, Seppala P, Iisalo E. Glipizide pharma-cokinetics and response in diabetics. Int J Clin Pharmacol Ther Toxicol. 1982;20(9):417-422.
6. Baselt RC. Disposition of toxic drugs and chemicals in man. 10th ed. Seal Beach, California: Biomedical Publications; 2014.
7. Simonson DC, Kourides IA, Feinglos M, et al; the Glipizide Gastrointestinal Therapeutic System Study Group. Efficacy, safety, and dose-response characteristics of glipizide gastrointestinal therapeutic system on glycemic control and insulin secretion in NIDDM. Results of two multicenter, randomized, placebo-controlled clinical trials. Diabetes Care. 1997;20(4):597-606.
8. Pelavin PI, Abramson E, Pon S, et al. Extended-release glipizide overdose presenting with delayed hypoglycemia and treated with subcutaneous octreotide. J Pediatr Endocrinol Metab. 2009;22(2):171-175.
CASE Unexplained hypoglycemia
Ms. A, age 12, is brought to the emergency department (ED) via ambulance with altered mentation and life-threatening hypoglycemia for management of a hypoglycemic seizure. Earlier that day, Ms. A’s parents had found her unresponsive and incontinent of urine. In the ED, Ms. A is minimally responsive. Her blood glucose level measurements are in the range of 30 to 39 mg/dL (reference range: 70 to 99 mg/dL), despite having received IV dextrose first from paramedics, and then in the ED. Ms. A has no history of hypoglycemia or diabetes. Her parents say that the night before coming to the ED, Ms. A had experienced flu-like symptoms, including nausea, vomiting, and diarrhea, that continued overnight and resulted in minimal food intake for 24 hours (Table 1).
A physical exam demonstrates left-sided weakness of face, arm, and leg, rightward gaze, and left-sided neglect. However, the results of CT angiography and an MRI of the brain rule out a stroke. An EEG shows right hemispheric slowing consistent with postictal paralysis, but no ongoing seizure activity. Ms. A is transferred to the pediatric intensive care unit (PICU).
Although Ms. A has no psychiatric diagnoses, she has a history of depressive symptoms, self-harm by cutting, and a suicide attempt by ingestion of an over-the-counter (OTC) medication 1 year ago. She had reported the suicide attempt to her parents several months after the fact, and asked them to find her a therapist, which her parents arranged. She also has a history of asthma, which is well-controlled with montelukast, 5 mg/d.
EVALUATION Elevated insulin levels
Subsequent investigations for organic causes of hypoglycemia are negative for adrenal insufficiency, fatty acid oxidation defect, and sepsis. Blood results demonstrate significantly elevated insulin levels of 92.4 mcIU/mL (reference range: 2.6 to 24.9 mcIU/mL) and a C-peptide level of 9.5 ng/mL (reference range: 1.1 to 4.4 ng/mL).
On Day 1 of admission to the PICU, Ms. A’s blood glucose level normalizes, and her mentation improves. Her parents report that one of them has diabetes and takes oral hypoglycemic agents at home, including glipizide immediate release (IR) tablets, 10 mg, and long-acting insulin glargine. The treatment team suspects that Ms. A may have ingested one or both of these agents, and orders a toxicologic screening for oral hypoglycemic agents.
On Day 2, the toxicology results are returned and are positive for glipizide, which Ms. A had not been prescribed. Ms. A states that she had taken only her montelukast tablet on the day of admission and adamantly denies deliberately ingesting her parent’s diabetes medications. Her parents check the home medications and state there are no missing glipizide IR tablets or insulin vials. They also report that Ms. A had no access to extended-release glipizide.
The treatment team discuss Ms. A’s clinical condition and toxicology results with the pediatric endocrinology team. The endocrinology team states that with no history of hypoglycemic episodes, it is unlikely that Ms. A had an endogenous etiology that would present so catastrophically. In their experience, inexplicable hypoglycemia in a healthy individual who lives in a household with someone who has diabetes is due to ingestion of a hypoglycemic agent until proven otherwise.
[polldaddy:10252689]
Continue to: The authors' observations
The authors’ observations
In the context of Ms. A’s prior suicide attempt and history of self-harm, the pediatric team was concerned that her presentation was consistent with a suicide attempt and consulted the psychiatry service.
Glipizide is a second-generation sulfonylurea used to treat type 2 diabetes. It lowers blood glucose by stimulating pancreatic insulin secretion. It is a rare drug of overdose.1 Although pediatric glipizide overdoses have been documented, there are currently no pediatric or adolescent glipizide pharmacokinetic studies in the literature.1-4 In adults, the immediate-release formulation has 100% oral bioavailability, with a maximum plasma concentration (Tmax) of approximately 2 hours.5 The half-life typically ranges from 4 to 6 hours in adults.6 Patients who do not have diabetes are much more susceptible to the hypoglycemic effects of glipizide because the medication simulates their fully functional pancreas to produce a vigorous insulin response.
Ms. A’s significantly elevated insulin was consistent with normal glipizide effects in a healthy child, while the elevated C-peptide was consistent with insulin being endogenously produced, which ruled out ingestion of her parent’s insulin. Importantly, the pediatric endocrinology team noted that, in their experience, a single 5- to 10-mg dose of glipizide IR was sufficient to lower blood glucose levels to the low 30s mg/dL in the context of a functional pancreas, which suggested that Ms. A might have accidentally ingested a single glipizide IR tablet, and might be telling the truth when she denies deliberately ingesting it to hurt herself.
The clinical value of pharmacokinetics
The screen of Ms. A’s toxicology sample detected glipizide. The laboratory used a semi-quantitative serum screen of several hypoglycemic agents. A positive result for each agent is based on a quantitative cut-off value, which is 3 ng/mL for glipizide. The clinical chemist on call was asked to assist in interpreting the results. The serum specimen collected on Day 1 had a significantly positive glipizide result of 86 to 130 ng/mL. The maximum effective glipizide concentration for adult patients with diabetes is 100 ng/mL.7 Thus, the glipizide level of 86 to 130 ng/mL (20.5 hours after initial symptoms) is consistent with the clinical presentation of persistent hypoglycemia requiring ongoing glucose replacement therapy.
Due to the lack of pediatric pharmacokinetic data for glipizide and only a single serum measurement, it is not possible to estimate the glipizide concentration at the time of maximal symptoms (loss of consciousness at 2:30
Continue to: Clinicians need to be aware that...
Clinicians need to be aware that although hypoglycemia usually presents rapidly, in children glipizide IR can rarely cause delayed hypoglycemia up to 16 hours after ingestion,2 and a delay of 45 hours was reported in a case of ingestion of extended-release glipizide.8 Hypoglycemia can last up to nearly 24 hours and is exacerbated if the patient has not eaten.1,2 Importantly, Ms. A’s parents reported that she had no access to extended-release glipizide. When detailed pharmacokinetic data are not available, the information provided by the patient and parents becomes extremely important, especially in distinguishing between single and multiple overdoses prior to presentation, or co-ingestions, or decreased food intake that could exacerbate hypoglycemia.
EVALUATION Safety assessment
On Day 2, Ms. A and her parents are interviewed separately, and they all are consistent in their recollection that Ms. A had been feverish with flu-like symptoms throughout the night before coming to the ED, and had still seemed mildly confused on the morning of admission.
During the interview, her parents wonder when Ms. A took her daily dose of a single montelukast tablet for asthma, and whether she had accidentally confused it with their glipizide. They report that on the morning of admission, both the glipizide and montelukast medication vials were in the same room. The vials are the same color, the same size, and labeled from the same pharmacy, and contain white, scored, round tablets that look very similar.
During the interview, Ms. A consistently denies having thoughts of hurting or killing herself on the day of admission or before that.
[polldaddy:10252690]
Continue to: The authors' observations
The authors’ observations
This case was ultimately an accidental ingestion of glipizide, rather than a suicide attempt. The initial suspicion for a suicide attempt had been reasonable in the context of Ms. A’s depressive symptoms, remote history of a prior suicide attempt by ingesting an OTC medication, and toxicologic evidence of ingesting a drug not prescribed to her. Additionally, because of the life-threatening presentation, it was easy to make the erroneous assumption that the ingestion of glipizide must have involved many tablets, and thus must have been deliberate. However, through multidisciplinary teamwork, we were able to demonstrate that this was likely an accidental ingestion by a patient who had an acute febrile illness. Her illness had caused confusion, which contributed to the accidental ingestion, and also caused reduced food intake, which enhanced the hypoglycemic effects of glipizide. Additionally, a lack of awareness of medication safety in the home had facilitated the confusion between the two medication vials.
A single tablet of glipizide IR is sufficient to produce profound clinical effects that could be mistaken by medical and psychiatric teams for a much larger and/or deliberate overdose, especially in patients with a psychiatric history. The inappropriate psychiatric hospitalization of a patient, especially a child, who has been mistakenly diagnosed as having attempted suicide, can have negative therapeutic consequences (Table 2). A psychiatric admission would have been misguided if it attempted to address safety and reduce suicidality when no such concerns were present. Additionally, it could have damaged relationships with the patient and the family, especially in a child who had historically not sought psychiatric care despite depressive symptoms and a previous suicide attempt. When assessing for suicidality, consider accidental ingestion in the differential and use specialty expertise and confirmatory testing in the evaluation, taking the pharmacokinetics of the suspected agent into account.
OUTCOME Outpatient treatment
Ms. A’s neurologic symptoms resolve within 24 hours of admission. She is offered psychiatric inpatient hospitalization to address her depressive symptoms; however, her parents prefer that she receive outpatient care. Ms. A’s parents also state that after Ms. A’s admission, they locked up all household medications and will be more mindful with medication in the home. Because her parents are arranging appropriate outpatient treatment for Ms. A’s depression and maintenance of her safety, an involuntary hospitalization is not deemed necessary.
On Day 2, Ms. A is eating normally, her blood glucose levels remain stable, and she is discharged home.
Bottom Line
Oral hypoglycemic agents can cause life-threatening syndromes in healthy patients and can clinically mimic large, intentional overdoses. Clinicians must be aware of the differential of accidental ingestion when assessing for suicidality, and can use toxicology results in their assessment.
Related Resources
- Kidemergencies.com. Emergencies: One pill can kill. http://kidemergencies.com/onepill1.html.
- Safe Kids Worldwide. Medication safety. https://www.safekids.org/medicinesafety.
- American Association of Poison Control Centers. http://www.aapcc.org/.
Drug Brand Names
Glipizide • Glucotrol
Insulin glargine • Lantus
Montelukast • Singulair
CASE Unexplained hypoglycemia
Ms. A, age 12, is brought to the emergency department (ED) via ambulance with altered mentation and life-threatening hypoglycemia for management of a hypoglycemic seizure. Earlier that day, Ms. A’s parents had found her unresponsive and incontinent of urine. In the ED, Ms. A is minimally responsive. Her blood glucose level measurements are in the range of 30 to 39 mg/dL (reference range: 70 to 99 mg/dL), despite having received IV dextrose first from paramedics, and then in the ED. Ms. A has no history of hypoglycemia or diabetes. Her parents say that the night before coming to the ED, Ms. A had experienced flu-like symptoms, including nausea, vomiting, and diarrhea, that continued overnight and resulted in minimal food intake for 24 hours (Table 1).
A physical exam demonstrates left-sided weakness of face, arm, and leg, rightward gaze, and left-sided neglect. However, the results of CT angiography and an MRI of the brain rule out a stroke. An EEG shows right hemispheric slowing consistent with postictal paralysis, but no ongoing seizure activity. Ms. A is transferred to the pediatric intensive care unit (PICU).
Although Ms. A has no psychiatric diagnoses, she has a history of depressive symptoms, self-harm by cutting, and a suicide attempt by ingestion of an over-the-counter (OTC) medication 1 year ago. She had reported the suicide attempt to her parents several months after the fact, and asked them to find her a therapist, which her parents arranged. She also has a history of asthma, which is well-controlled with montelukast, 5 mg/d.
EVALUATION Elevated insulin levels
Subsequent investigations for organic causes of hypoglycemia are negative for adrenal insufficiency, fatty acid oxidation defect, and sepsis. Blood results demonstrate significantly elevated insulin levels of 92.4 mcIU/mL (reference range: 2.6 to 24.9 mcIU/mL) and a C-peptide level of 9.5 ng/mL (reference range: 1.1 to 4.4 ng/mL).
On Day 1 of admission to the PICU, Ms. A’s blood glucose level normalizes, and her mentation improves. Her parents report that one of them has diabetes and takes oral hypoglycemic agents at home, including glipizide immediate release (IR) tablets, 10 mg, and long-acting insulin glargine. The treatment team suspects that Ms. A may have ingested one or both of these agents, and orders a toxicologic screening for oral hypoglycemic agents.
On Day 2, the toxicology results are returned and are positive for glipizide, which Ms. A had not been prescribed. Ms. A states that she had taken only her montelukast tablet on the day of admission and adamantly denies deliberately ingesting her parent’s diabetes medications. Her parents check the home medications and state there are no missing glipizide IR tablets or insulin vials. They also report that Ms. A had no access to extended-release glipizide.
The treatment team discuss Ms. A’s clinical condition and toxicology results with the pediatric endocrinology team. The endocrinology team states that with no history of hypoglycemic episodes, it is unlikely that Ms. A had an endogenous etiology that would present so catastrophically. In their experience, inexplicable hypoglycemia in a healthy individual who lives in a household with someone who has diabetes is due to ingestion of a hypoglycemic agent until proven otherwise.
[polldaddy:10252689]
Continue to: The authors' observations
The authors’ observations
In the context of Ms. A’s prior suicide attempt and history of self-harm, the pediatric team was concerned that her presentation was consistent with a suicide attempt and consulted the psychiatry service.
Glipizide is a second-generation sulfonylurea used to treat type 2 diabetes. It lowers blood glucose by stimulating pancreatic insulin secretion. It is a rare drug of overdose.1 Although pediatric glipizide overdoses have been documented, there are currently no pediatric or adolescent glipizide pharmacokinetic studies in the literature.1-4 In adults, the immediate-release formulation has 100% oral bioavailability, with a maximum plasma concentration (Tmax) of approximately 2 hours.5 The half-life typically ranges from 4 to 6 hours in adults.6 Patients who do not have diabetes are much more susceptible to the hypoglycemic effects of glipizide because the medication simulates their fully functional pancreas to produce a vigorous insulin response.
Ms. A’s significantly elevated insulin was consistent with normal glipizide effects in a healthy child, while the elevated C-peptide was consistent with insulin being endogenously produced, which ruled out ingestion of her parent’s insulin. Importantly, the pediatric endocrinology team noted that, in their experience, a single 5- to 10-mg dose of glipizide IR was sufficient to lower blood glucose levels to the low 30s mg/dL in the context of a functional pancreas, which suggested that Ms. A might have accidentally ingested a single glipizide IR tablet, and might be telling the truth when she denies deliberately ingesting it to hurt herself.
The clinical value of pharmacokinetics
The screen of Ms. A’s toxicology sample detected glipizide. The laboratory used a semi-quantitative serum screen of several hypoglycemic agents. A positive result for each agent is based on a quantitative cut-off value, which is 3 ng/mL for glipizide. The clinical chemist on call was asked to assist in interpreting the results. The serum specimen collected on Day 1 had a significantly positive glipizide result of 86 to 130 ng/mL. The maximum effective glipizide concentration for adult patients with diabetes is 100 ng/mL.7 Thus, the glipizide level of 86 to 130 ng/mL (20.5 hours after initial symptoms) is consistent with the clinical presentation of persistent hypoglycemia requiring ongoing glucose replacement therapy.
Due to the lack of pediatric pharmacokinetic data for glipizide and only a single serum measurement, it is not possible to estimate the glipizide concentration at the time of maximal symptoms (loss of consciousness at 2:30
Continue to: Clinicians need to be aware that...
Clinicians need to be aware that although hypoglycemia usually presents rapidly, in children glipizide IR can rarely cause delayed hypoglycemia up to 16 hours after ingestion,2 and a delay of 45 hours was reported in a case of ingestion of extended-release glipizide.8 Hypoglycemia can last up to nearly 24 hours and is exacerbated if the patient has not eaten.1,2 Importantly, Ms. A’s parents reported that she had no access to extended-release glipizide. When detailed pharmacokinetic data are not available, the information provided by the patient and parents becomes extremely important, especially in distinguishing between single and multiple overdoses prior to presentation, or co-ingestions, or decreased food intake that could exacerbate hypoglycemia.
EVALUATION Safety assessment
On Day 2, Ms. A and her parents are interviewed separately, and they all are consistent in their recollection that Ms. A had been feverish with flu-like symptoms throughout the night before coming to the ED, and had still seemed mildly confused on the morning of admission.
During the interview, her parents wonder when Ms. A took her daily dose of a single montelukast tablet for asthma, and whether she had accidentally confused it with their glipizide. They report that on the morning of admission, both the glipizide and montelukast medication vials were in the same room. The vials are the same color, the same size, and labeled from the same pharmacy, and contain white, scored, round tablets that look very similar.
During the interview, Ms. A consistently denies having thoughts of hurting or killing herself on the day of admission or before that.
[polldaddy:10252690]
Continue to: The authors' observations
The authors’ observations
This case was ultimately an accidental ingestion of glipizide, rather than a suicide attempt. The initial suspicion for a suicide attempt had been reasonable in the context of Ms. A’s depressive symptoms, remote history of a prior suicide attempt by ingesting an OTC medication, and toxicologic evidence of ingesting a drug not prescribed to her. Additionally, because of the life-threatening presentation, it was easy to make the erroneous assumption that the ingestion of glipizide must have involved many tablets, and thus must have been deliberate. However, through multidisciplinary teamwork, we were able to demonstrate that this was likely an accidental ingestion by a patient who had an acute febrile illness. Her illness had caused confusion, which contributed to the accidental ingestion, and also caused reduced food intake, which enhanced the hypoglycemic effects of glipizide. Additionally, a lack of awareness of medication safety in the home had facilitated the confusion between the two medication vials.
A single tablet of glipizide IR is sufficient to produce profound clinical effects that could be mistaken by medical and psychiatric teams for a much larger and/or deliberate overdose, especially in patients with a psychiatric history. The inappropriate psychiatric hospitalization of a patient, especially a child, who has been mistakenly diagnosed as having attempted suicide, can have negative therapeutic consequences (Table 2). A psychiatric admission would have been misguided if it attempted to address safety and reduce suicidality when no such concerns were present. Additionally, it could have damaged relationships with the patient and the family, especially in a child who had historically not sought psychiatric care despite depressive symptoms and a previous suicide attempt. When assessing for suicidality, consider accidental ingestion in the differential and use specialty expertise and confirmatory testing in the evaluation, taking the pharmacokinetics of the suspected agent into account.
OUTCOME Outpatient treatment
Ms. A’s neurologic symptoms resolve within 24 hours of admission. She is offered psychiatric inpatient hospitalization to address her depressive symptoms; however, her parents prefer that she receive outpatient care. Ms. A’s parents also state that after Ms. A’s admission, they locked up all household medications and will be more mindful with medication in the home. Because her parents are arranging appropriate outpatient treatment for Ms. A’s depression and maintenance of her safety, an involuntary hospitalization is not deemed necessary.
On Day 2, Ms. A is eating normally, her blood glucose levels remain stable, and she is discharged home.
Bottom Line
Oral hypoglycemic agents can cause life-threatening syndromes in healthy patients and can clinically mimic large, intentional overdoses. Clinicians must be aware of the differential of accidental ingestion when assessing for suicidality, and can use toxicology results in their assessment.
Related Resources
- Kidemergencies.com. Emergencies: One pill can kill. http://kidemergencies.com/onepill1.html.
- Safe Kids Worldwide. Medication safety. https://www.safekids.org/medicinesafety.
- American Association of Poison Control Centers. http://www.aapcc.org/.
Drug Brand Names
Glipizide • Glucotrol
Insulin glargine • Lantus
Montelukast • Singulair
1. Spiller HA, Villalobos D, Krenzelok EP, et al. Prospective multicenter study of sulfonylurea ingestion in children. J Pediatr. 1997;131(1):141-146.
2. Quadrani DA, Spiller HA, Widder P. Five year retrospective evaluation of sulfonylurea ingestion in children. J Toxicol Clin Toxicol. 1996;34(3):267-270.
3. Borowski H, Caraccio T, Mofenson H. Sulfonylurea ingestion in children: is an 8-hour observation period sufficient? J Pediatr. 1998;133(4):584-585.
4. Little GL, Boniface KS. Are one or two dangerous? Sulfony-lurea exposure in toddlers. J Emerg Med. 2005;28(3):305-310.
5. Huupponen R, Seppala P, Iisalo E. Glipizide pharma-cokinetics and response in diabetics. Int J Clin Pharmacol Ther Toxicol. 1982;20(9):417-422.
6. Baselt RC. Disposition of toxic drugs and chemicals in man. 10th ed. Seal Beach, California: Biomedical Publications; 2014.
7. Simonson DC, Kourides IA, Feinglos M, et al; the Glipizide Gastrointestinal Therapeutic System Study Group. Efficacy, safety, and dose-response characteristics of glipizide gastrointestinal therapeutic system on glycemic control and insulin secretion in NIDDM. Results of two multicenter, randomized, placebo-controlled clinical trials. Diabetes Care. 1997;20(4):597-606.
8. Pelavin PI, Abramson E, Pon S, et al. Extended-release glipizide overdose presenting with delayed hypoglycemia and treated with subcutaneous octreotide. J Pediatr Endocrinol Metab. 2009;22(2):171-175.
1. Spiller HA, Villalobos D, Krenzelok EP, et al. Prospective multicenter study of sulfonylurea ingestion in children. J Pediatr. 1997;131(1):141-146.
2. Quadrani DA, Spiller HA, Widder P. Five year retrospective evaluation of sulfonylurea ingestion in children. J Toxicol Clin Toxicol. 1996;34(3):267-270.
3. Borowski H, Caraccio T, Mofenson H. Sulfonylurea ingestion in children: is an 8-hour observation period sufficient? J Pediatr. 1998;133(4):584-585.
4. Little GL, Boniface KS. Are one or two dangerous? Sulfony-lurea exposure in toddlers. J Emerg Med. 2005;28(3):305-310.
5. Huupponen R, Seppala P, Iisalo E. Glipizide pharma-cokinetics and response in diabetics. Int J Clin Pharmacol Ther Toxicol. 1982;20(9):417-422.
6. Baselt RC. Disposition of toxic drugs and chemicals in man. 10th ed. Seal Beach, California: Biomedical Publications; 2014.
7. Simonson DC, Kourides IA, Feinglos M, et al; the Glipizide Gastrointestinal Therapeutic System Study Group. Efficacy, safety, and dose-response characteristics of glipizide gastrointestinal therapeutic system on glycemic control and insulin secretion in NIDDM. Results of two multicenter, randomized, placebo-controlled clinical trials. Diabetes Care. 1997;20(4):597-606.
8. Pelavin PI, Abramson E, Pon S, et al. Extended-release glipizide overdose presenting with delayed hypoglycemia and treated with subcutaneous octreotide. J Pediatr Endocrinol Metab. 2009;22(2):171-175.
Motherhood and the working psychiatrist
Raising a child is difficult. For working professional women, including doctors, that difficulty extends beyond bottles, bath time, and burping; it impacts day-to-day physiological function, time management, and emotional well-being.
The 1950s upheld a family model with traditional gender roles. By 1960, the family portrait of a breadwinner father and a stay-at-home mother with one or more children comprised 62% of American households.1 Precipitous changes occurred over the next decades as the housing market soared, education costs increased, and divorce rates rose. The 1980s ushered the arrival of women’s power suits and the notion of women “having it all.”1
Fast-forward to modern times. Medicine is changing, too. Women are slowly but surely starting to rise in this once male-led field. In 2017, for the first time more women than men enrolled in medical schools in the United States.2 In a 2015 report, the Association of American Medical Colleges found that 57% of residents who were pursuing psychiatry were women.3 And the median age of women applying to medical school who enrolled in 2017 or 2018 was 23 years.4
Choosing to parent as a physician poses challenges for women and men alike. As the rates of women in medicine and psychiatry are increasing, this article focuses on unique obstacles faced by mothers and aims to:
- explore the dueling duties of mothers who practice medicine
- consider the dilemma women face when returning to the workforce during the postpartum period
- discuss options for enhanced recognition and care of maternal and child well-being.
Duty: Being both parent and physician
The working psychiatrist mother has a duty to her patients and profession—not to mention a duty to her child. The demands are endless on both sides. No matter what stage of her professional career (medical school, residency, fellowship, or beyond) she chooses to begin motherhood, the responsibilities and expectations can be overwhelming. Doctor appointments, nausea and vomiting, fatigue, discomfort, and stress do not fit well within a schedule of intensive studying, working 24-hour shifts, navigating complex schedules, treating patients, and sorting out the financial heft of loan repayment, home ownership, contract negotiation, or relocation.5
Psychiatry carries a notable dichotomy of lecturing at length on the importance of maternal-infant attachment. John Bowlby argued that a child’s attachment to the mother is instinctual and primary, noting that early loss creates true mourning due to the primal ties of child to mother.6 Bowlby also asserted that personality development and psychopathology are rooted in the concept of attachment and the emotional security built through early childhood experiences.6
Continue to: Dr. Donald Winnnicott introduced the concept of...
Dr. Donald Winnicott introduced the concept of a “good-enough” mother in 1953.7 Today, although Winnicott’s teachings are explored in psychiatry training programs and practice, his concept does not resonate with many working mothers. Most physicians strive for perfection while struggling to balance their personal and professional lives.7
It’s no wonder that tales abound of female physicians being praised for their ability to take on grueling shifts up to their due date, forego lunch to pump breast milk, or cover shifts beyond child daycare closing times. This raises an interesting dilemma: Is the primary goal the efficiency of promoting commerce, patient numbers, and the workings of the health care system? Or is it the wellness of expecting mothers and the development and attachment of an infant to the parent? Is the goal to slowly and carefully craft our next generation of young humans? Or is there a way to “have it all”?
Dilemma: Misperceptions after returning to work
As they regain control of their bodies, sleep, and overall health, women who return to work during the postpartum period battle a myriad of misperceptions along with the logistical hurdles of breast-feeding. In a study of surgical residencies, 61% of program directors reported that female trainees’ work was negatively affected by becoming parents.8 But other evidence suggests there is a disparity between perception and reality.
As a result, misperceptions can negatively affect maternity leave or lactation time. Women often rightfully fear they may be viewed as taking leisure time or making convenient excuses to shirk responsibility, rather than focusing on the necessities for recovery, care, and bonding. Such pressures can lead to burnout and resentment. The struggle with breast-feeding is pervasive across all medical specialties. In a 2018 survey of 347 women who had children during surgical residency, 39% of respondents strongly considered leaving their training, 95.6% indicated that breast-feeding was important to them, and 58.1% stopped breast-feeding earlier than desired due to challenges faced in the workplace, such as poor access to lactation facilities and difficulty leaving the operating room to express milk.11
The American Academy of Pediatrics (AAP) recommends exclusive breast-feeding through 6 months of the postpartum period, and continued breast-feeding until the infant is at least 12 months old. Breast-feeding confers benefits to both the infant and mother, including positive impacts on the child’s cognitive development and health into adulthood, as well as higher productivity and lower absenteeism for breast-feeding mothers.12 By 2009, only 23 states had adopted laws to encourage breast-feeding in the workplace. In 2010, the United States government enacted the “reasonable break time” provision in Section 4207 of the Patient Protection and Affordable Care Act (ACA), which requires all employers to provide a period of time and private space other than a bathroom in which female employees can express milk for a child up to age 1.12
Continue to: In 2016...
In 2016, a follow-up national survey of employed women explored workplace changes after the ACA, and noted that only 40% of women had access to both break time and a private space for lactation.13 If the goal is to give working women a true choice of whether to continue breast-feeding after returning to work, these mothers need to be provided with the proper social and structural supports in order to allow for that personal decision.14
Discussion: Barriers to change
Breast-feeding, it has been argued, is the most enduring investment in women’s physical, cognitive, and social capacities, and provides protection for children against death, disease, and poverty.15 Research has shown that breast-feeding every child until age 1 would yield medical benefits, including fewer infections, increased intelligence in children, protection against breast cancer in mothers, and economic savings of $300 billion for the United States.15
We are no longer in the 1950s, but modern times still present challenges for mothers who are working as physicians. Although the AAP recommends that new parents receive 12 weeks leave from work, policies for faculty at the 12 top medical schools in the United States offer new mothers only approximately 2 months of paid leave.16 There also are problems of inconsistency among approaches to parenthood in graduate medical education (GME) training, different specialty clinical requirements, and different residency training programs. These factors all contribute to negative attitudes towards parenthood.17
We know the barriers for women.18 With more women entering the medical profession, we need to continue finding creative and workable solutions as these problems become more pressing.19 In a 2018 Time article, Lily Rothman wrote, “you can’t talk about breastfeeding in the United States without pointing out that every other wealthy country has found a way to accommodate breastfeeding mothers, and usually in the form of lengthy paid maternity leave.”20 However, maternity leave in the United States today dictates that mothers return to work while their children would still benefit from nursing.21
When it comes to GME and medical institutions, programs could look at barriers such as lack of accommodations for trainees who are pregnant or have young children. Addressing these barriers could include making private lactation rooms available and instituting flexible scheduling. It would be best if scheduling accommodations and policies were established by an institution’s administration, rather than leaving coverage up to the students or residents. Going further, institutions could consider offering flexible maternity leave and work schedules, allowing breaks for those who are breast-feeding, and creating lactation facilities.22 This could take the form of a breast-feeding support program that fits available budget resources.23
Continue to: Psychiatrists frequently discuss...
Psychiatrists frequently discuss Winnicott’s “good-enough mother” concept, with the mother transitioning from focusing on her baby’s needs to her own sense of personhood that is unable to respond to her baby’s every wish.6 This concept was established well before the shifting demographics of the nuclear family, the short maternity leaves and early returns to work, early separation of one’s infants to childcare settings, and experiences with pumped lactation milk that working mothers experience today. Is it any wonder childbearing female psychiatrists face a special kind of working-mother guilt?
1. Collins G. When everything changed: the amazing journey of American women from 1960 to the present. New York, NY: Little, Brown and Company; 2009;271, 301.
2. AAMCNews. More women than men enrolled in U.S. medical schools in 2017. Association of American Medical Colleges. https://news.aamc.org/press-releases/article/applicant-enrollment-2017/. Published December 18, 2017. Accessed November 21, 2018.
3. Vassar L. How medical specialties vary by gender. American Medical Association. https://wire.ama-assn.org/education/how-medical-specialties-vary-gender. Published February 18, 2015. Accessed November 21, 2018.
4. Association of American Medical Colleges. Table A-6: age of applicants to U.S. medical schools at anticipated matriculation by sex and race/ethnicity, 2014-2015 through 2017-2018. https://www.aamc.org/download/321468/data/factstablea6.pdf. Published November 30, 2017. Accessed February 7, 2019.
5. Jones V. Best time to have a baby as a physician? It depends. Doximity. https://opmed.doximity.com/articles/the-best-time-to-have-a-baby-as-a-physician-it-depends-c8064a92156c. Published September 11, 2017. Accessed November 21, 2018.
6. Mitchell SA, Black MJ. The British object relations school: W.R.D. Fairbairn and D.W. Winnicott. In: Freud and beyond: a history of modern psychoanalytic thought. New York, NY: Basic Books; 1995:125-126, 137.
7. Ratnapalan S, Batty H. To be good enough. Can Fam Physician. 2009;55(3):239-242.
8. Sandler BJ, Tackett JJ, Longo WE, et al. Pregnancy and parenthood among surgery residents: results of the first nationwide survey of general surgery residency program Directors. J Am Coll Surg. 2016;222(6):1090-1096.
9. Kmec JA. Are motherhood penalties and fatherhood bonuses warranted? Comparing pro-work behaviors and conditions of mothers, fathers, and non-parents. Social Science Research. 2011;40(2):444-459.
10. Hampton R. Working moms don’t deserve the blame for unfair work expectations. Slate. https://slate.com/human-interest/2018/05/working-moms-dont-deserve-blame-for-unfair-work-expectations.html. Published May 18, 2018. Accessed November 25, 2018.
11. Rangel EL, Smink DS, Castillo-Angeles M, et al. Pregnancy and motherhood during surgical training. JAMA Surgery. 2018;153(7):644-652.
12. Murtagh L, Moulton AD. Working mothers, breastfeeding, and the law. Am J Public Health. 2011;101(2):217-223.
13. Kozhimannil KB, Jou J, Gjerdingen DK, et al. Access to workplace accommodations to support breastfeeding after passage of the Affordable Care Act. Womens Health Issues. 2016;26(1):6-13.
14. Dinour LM, Bai YK. Breastfeeding: the illusion of choice. Womens Health Issues. 2016;26(5):479-482.
15. Hansen K. Breastfeeding: a smart investment in people and in economies. Lancet. 2016;387(10017):416.
16. Greenfield R. Even America’s top doctors aren’t getting the parental leave doctors recommend. Bloomberg. https://www.bloomberg.com/news/articles/2018-02-13/even-america-s-top-doctors-aren-t-getting-the-parental-leave-doctors-recommend. Published February 13, 2018. Accessed November 21, 2018.
17. Humphries LS, Lyon S, Garza R, et al. Parental leave policies in graduate medical education: a systematic review. American J Surg. 2017;214(4):634-639.
18. Raju TNK. Continued barriers for breast-feeding in public and the workplace. J Pediatr. 2006;148(5):677-679.
19. Stewart DE, Robinson GE. Combining motherhood with psychiatric training and practice. Can J Psychiatry. 1985;30(1):28-34.
20. Rothman L. D esperate women, desperate doctors and the surprising history behind the breastfeeding debate. Time. http://time.com/5353068/breastfeeding-debate-history/. Published July 31, 2018. Accessed November 21, 2018.
21. Livingston G. Among 41 nations, U.S. is the outlier when it comes to paid parental leave. Pew Research Center. http://www.pewresearch.org/fact-tank/2016/09/26/u-s-lacks-mandated-paid-parental-leave/. Published September 26, 2016. Accessed November 21, 2018.
22. McCluskey PD. Long hours, short leaves force moms to reconsider jobs as surgeons. Boston Globe. https://www.bostonglobe.com/metro/2018/03/21/new-survey-says-female-surgical-residents-struggle-balance-training-motherhood/2ENQU1aPZmIJYy20iaRlLL/story.html. Published March 21, 2018. Accessed November 21, 2018.
23. Dinour LM, Szaro JM. Employer-based programs to support breastfeeding among working mothers: a systematic review. Breastfeeding Med. 2017;12:131-141.
Raising a child is difficult. For working professional women, including doctors, that difficulty extends beyond bottles, bath time, and burping; it impacts day-to-day physiological function, time management, and emotional well-being.
The 1950s upheld a family model with traditional gender roles. By 1960, the family portrait of a breadwinner father and a stay-at-home mother with one or more children comprised 62% of American households.1 Precipitous changes occurred over the next decades as the housing market soared, education costs increased, and divorce rates rose. The 1980s ushered the arrival of women’s power suits and the notion of women “having it all.”1
Fast-forward to modern times. Medicine is changing, too. Women are slowly but surely starting to rise in this once male-led field. In 2017, for the first time more women than men enrolled in medical schools in the United States.2 In a 2015 report, the Association of American Medical Colleges found that 57% of residents who were pursuing psychiatry were women.3 And the median age of women applying to medical school who enrolled in 2017 or 2018 was 23 years.4
Choosing to parent as a physician poses challenges for women and men alike. As the rates of women in medicine and psychiatry are increasing, this article focuses on unique obstacles faced by mothers and aims to:
- explore the dueling duties of mothers who practice medicine
- consider the dilemma women face when returning to the workforce during the postpartum period
- discuss options for enhanced recognition and care of maternal and child well-being.
Duty: Being both parent and physician
The working psychiatrist mother has a duty to her patients and profession—not to mention a duty to her child. The demands are endless on both sides. No matter what stage of her professional career (medical school, residency, fellowship, or beyond) she chooses to begin motherhood, the responsibilities and expectations can be overwhelming. Doctor appointments, nausea and vomiting, fatigue, discomfort, and stress do not fit well within a schedule of intensive studying, working 24-hour shifts, navigating complex schedules, treating patients, and sorting out the financial heft of loan repayment, home ownership, contract negotiation, or relocation.5
Psychiatry carries a notable dichotomy of lecturing at length on the importance of maternal-infant attachment. John Bowlby argued that a child’s attachment to the mother is instinctual and primary, noting that early loss creates true mourning due to the primal ties of child to mother.6 Bowlby also asserted that personality development and psychopathology are rooted in the concept of attachment and the emotional security built through early childhood experiences.6
Continue to: Dr. Donald Winnnicott introduced the concept of...
Dr. Donald Winnicott introduced the concept of a “good-enough” mother in 1953.7 Today, although Winnicott’s teachings are explored in psychiatry training programs and practice, his concept does not resonate with many working mothers. Most physicians strive for perfection while struggling to balance their personal and professional lives.7
It’s no wonder that tales abound of female physicians being praised for their ability to take on grueling shifts up to their due date, forego lunch to pump breast milk, or cover shifts beyond child daycare closing times. This raises an interesting dilemma: Is the primary goal the efficiency of promoting commerce, patient numbers, and the workings of the health care system? Or is it the wellness of expecting mothers and the development and attachment of an infant to the parent? Is the goal to slowly and carefully craft our next generation of young humans? Or is there a way to “have it all”?
Dilemma: Misperceptions after returning to work
As they regain control of their bodies, sleep, and overall health, women who return to work during the postpartum period battle a myriad of misperceptions along with the logistical hurdles of breast-feeding. In a study of surgical residencies, 61% of program directors reported that female trainees’ work was negatively affected by becoming parents.8 But other evidence suggests there is a disparity between perception and reality.
As a result, misperceptions can negatively affect maternity leave or lactation time. Women often rightfully fear they may be viewed as taking leisure time or making convenient excuses to shirk responsibility, rather than focusing on the necessities for recovery, care, and bonding. Such pressures can lead to burnout and resentment. The struggle with breast-feeding is pervasive across all medical specialties. In a 2018 survey of 347 women who had children during surgical residency, 39% of respondents strongly considered leaving their training, 95.6% indicated that breast-feeding was important to them, and 58.1% stopped breast-feeding earlier than desired due to challenges faced in the workplace, such as poor access to lactation facilities and difficulty leaving the operating room to express milk.11
The American Academy of Pediatrics (AAP) recommends exclusive breast-feeding through 6 months of the postpartum period, and continued breast-feeding until the infant is at least 12 months old. Breast-feeding confers benefits to both the infant and mother, including positive impacts on the child’s cognitive development and health into adulthood, as well as higher productivity and lower absenteeism for breast-feeding mothers.12 By 2009, only 23 states had adopted laws to encourage breast-feeding in the workplace. In 2010, the United States government enacted the “reasonable break time” provision in Section 4207 of the Patient Protection and Affordable Care Act (ACA), which requires all employers to provide a period of time and private space other than a bathroom in which female employees can express milk for a child up to age 1.12
Continue to: In 2016...
In 2016, a follow-up national survey of employed women explored workplace changes after the ACA, and noted that only 40% of women had access to both break time and a private space for lactation.13 If the goal is to give working women a true choice of whether to continue breast-feeding after returning to work, these mothers need to be provided with the proper social and structural supports in order to allow for that personal decision.14
Discussion: Barriers to change
Breast-feeding, it has been argued, is the most enduring investment in women’s physical, cognitive, and social capacities, and provides protection for children against death, disease, and poverty.15 Research has shown that breast-feeding every child until age 1 would yield medical benefits, including fewer infections, increased intelligence in children, protection against breast cancer in mothers, and economic savings of $300 billion for the United States.15
We are no longer in the 1950s, but modern times still present challenges for mothers who are working as physicians. Although the AAP recommends that new parents receive 12 weeks leave from work, policies for faculty at the 12 top medical schools in the United States offer new mothers only approximately 2 months of paid leave.16 There also are problems of inconsistency among approaches to parenthood in graduate medical education (GME) training, different specialty clinical requirements, and different residency training programs. These factors all contribute to negative attitudes towards parenthood.17
We know the barriers for women.18 With more women entering the medical profession, we need to continue finding creative and workable solutions as these problems become more pressing.19 In a 2018 Time article, Lily Rothman wrote, “you can’t talk about breastfeeding in the United States without pointing out that every other wealthy country has found a way to accommodate breastfeeding mothers, and usually in the form of lengthy paid maternity leave.”20 However, maternity leave in the United States today dictates that mothers return to work while their children would still benefit from nursing.21
When it comes to GME and medical institutions, programs could look at barriers such as lack of accommodations for trainees who are pregnant or have young children. Addressing these barriers could include making private lactation rooms available and instituting flexible scheduling. It would be best if scheduling accommodations and policies were established by an institution’s administration, rather than leaving coverage up to the students or residents. Going further, institutions could consider offering flexible maternity leave and work schedules, allowing breaks for those who are breast-feeding, and creating lactation facilities.22 This could take the form of a breast-feeding support program that fits available budget resources.23
Continue to: Psychiatrists frequently discuss...
Psychiatrists frequently discuss Winnicott’s “good-enough mother” concept, with the mother transitioning from focusing on her baby’s needs to her own sense of personhood that is unable to respond to her baby’s every wish.6 This concept was established well before the shifting demographics of the nuclear family, the short maternity leaves and early returns to work, early separation of one’s infants to childcare settings, and experiences with pumped lactation milk that working mothers experience today. Is it any wonder childbearing female psychiatrists face a special kind of working-mother guilt?
Raising a child is difficult. For working professional women, including doctors, that difficulty extends beyond bottles, bath time, and burping; it impacts day-to-day physiological function, time management, and emotional well-being.
The 1950s upheld a family model with traditional gender roles. By 1960, the family portrait of a breadwinner father and a stay-at-home mother with one or more children comprised 62% of American households.1 Precipitous changes occurred over the next decades as the housing market soared, education costs increased, and divorce rates rose. The 1980s ushered the arrival of women’s power suits and the notion of women “having it all.”1
Fast-forward to modern times. Medicine is changing, too. Women are slowly but surely starting to rise in this once male-led field. In 2017, for the first time more women than men enrolled in medical schools in the United States.2 In a 2015 report, the Association of American Medical Colleges found that 57% of residents who were pursuing psychiatry were women.3 And the median age of women applying to medical school who enrolled in 2017 or 2018 was 23 years.4
Choosing to parent as a physician poses challenges for women and men alike. As the rates of women in medicine and psychiatry are increasing, this article focuses on unique obstacles faced by mothers and aims to:
- explore the dueling duties of mothers who practice medicine
- consider the dilemma women face when returning to the workforce during the postpartum period
- discuss options for enhanced recognition and care of maternal and child well-being.
Duty: Being both parent and physician
The working psychiatrist mother has a duty to her patients and profession—not to mention a duty to her child. The demands are endless on both sides. No matter what stage of her professional career (medical school, residency, fellowship, or beyond) she chooses to begin motherhood, the responsibilities and expectations can be overwhelming. Doctor appointments, nausea and vomiting, fatigue, discomfort, and stress do not fit well within a schedule of intensive studying, working 24-hour shifts, navigating complex schedules, treating patients, and sorting out the financial heft of loan repayment, home ownership, contract negotiation, or relocation.5
Psychiatry carries a notable dichotomy of lecturing at length on the importance of maternal-infant attachment. John Bowlby argued that a child’s attachment to the mother is instinctual and primary, noting that early loss creates true mourning due to the primal ties of child to mother.6 Bowlby also asserted that personality development and psychopathology are rooted in the concept of attachment and the emotional security built through early childhood experiences.6
Continue to: Dr. Donald Winnnicott introduced the concept of...
Dr. Donald Winnicott introduced the concept of a “good-enough” mother in 1953.7 Today, although Winnicott’s teachings are explored in psychiatry training programs and practice, his concept does not resonate with many working mothers. Most physicians strive for perfection while struggling to balance their personal and professional lives.7
It’s no wonder that tales abound of female physicians being praised for their ability to take on grueling shifts up to their due date, forego lunch to pump breast milk, or cover shifts beyond child daycare closing times. This raises an interesting dilemma: Is the primary goal the efficiency of promoting commerce, patient numbers, and the workings of the health care system? Or is it the wellness of expecting mothers and the development and attachment of an infant to the parent? Is the goal to slowly and carefully craft our next generation of young humans? Or is there a way to “have it all”?
Dilemma: Misperceptions after returning to work
As they regain control of their bodies, sleep, and overall health, women who return to work during the postpartum period battle a myriad of misperceptions along with the logistical hurdles of breast-feeding. In a study of surgical residencies, 61% of program directors reported that female trainees’ work was negatively affected by becoming parents.8 But other evidence suggests there is a disparity between perception and reality.
As a result, misperceptions can negatively affect maternity leave or lactation time. Women often rightfully fear they may be viewed as taking leisure time or making convenient excuses to shirk responsibility, rather than focusing on the necessities for recovery, care, and bonding. Such pressures can lead to burnout and resentment. The struggle with breast-feeding is pervasive across all medical specialties. In a 2018 survey of 347 women who had children during surgical residency, 39% of respondents strongly considered leaving their training, 95.6% indicated that breast-feeding was important to them, and 58.1% stopped breast-feeding earlier than desired due to challenges faced in the workplace, such as poor access to lactation facilities and difficulty leaving the operating room to express milk.11
The American Academy of Pediatrics (AAP) recommends exclusive breast-feeding through 6 months of the postpartum period, and continued breast-feeding until the infant is at least 12 months old. Breast-feeding confers benefits to both the infant and mother, including positive impacts on the child’s cognitive development and health into adulthood, as well as higher productivity and lower absenteeism for breast-feeding mothers.12 By 2009, only 23 states had adopted laws to encourage breast-feeding in the workplace. In 2010, the United States government enacted the “reasonable break time” provision in Section 4207 of the Patient Protection and Affordable Care Act (ACA), which requires all employers to provide a period of time and private space other than a bathroom in which female employees can express milk for a child up to age 1.12
Continue to: In 2016...
In 2016, a follow-up national survey of employed women explored workplace changes after the ACA, and noted that only 40% of women had access to both break time and a private space for lactation.13 If the goal is to give working women a true choice of whether to continue breast-feeding after returning to work, these mothers need to be provided with the proper social and structural supports in order to allow for that personal decision.14
Discussion: Barriers to change
Breast-feeding, it has been argued, is the most enduring investment in women’s physical, cognitive, and social capacities, and provides protection for children against death, disease, and poverty.15 Research has shown that breast-feeding every child until age 1 would yield medical benefits, including fewer infections, increased intelligence in children, protection against breast cancer in mothers, and economic savings of $300 billion for the United States.15
We are no longer in the 1950s, but modern times still present challenges for mothers who are working as physicians. Although the AAP recommends that new parents receive 12 weeks leave from work, policies for faculty at the 12 top medical schools in the United States offer new mothers only approximately 2 months of paid leave.16 There also are problems of inconsistency among approaches to parenthood in graduate medical education (GME) training, different specialty clinical requirements, and different residency training programs. These factors all contribute to negative attitudes towards parenthood.17
We know the barriers for women.18 With more women entering the medical profession, we need to continue finding creative and workable solutions as these problems become more pressing.19 In a 2018 Time article, Lily Rothman wrote, “you can’t talk about breastfeeding in the United States without pointing out that every other wealthy country has found a way to accommodate breastfeeding mothers, and usually in the form of lengthy paid maternity leave.”20 However, maternity leave in the United States today dictates that mothers return to work while their children would still benefit from nursing.21
When it comes to GME and medical institutions, programs could look at barriers such as lack of accommodations for trainees who are pregnant or have young children. Addressing these barriers could include making private lactation rooms available and instituting flexible scheduling. It would be best if scheduling accommodations and policies were established by an institution’s administration, rather than leaving coverage up to the students or residents. Going further, institutions could consider offering flexible maternity leave and work schedules, allowing breaks for those who are breast-feeding, and creating lactation facilities.22 This could take the form of a breast-feeding support program that fits available budget resources.23
Continue to: Psychiatrists frequently discuss...
Psychiatrists frequently discuss Winnicott’s “good-enough mother” concept, with the mother transitioning from focusing on her baby’s needs to her own sense of personhood that is unable to respond to her baby’s every wish.6 This concept was established well before the shifting demographics of the nuclear family, the short maternity leaves and early returns to work, early separation of one’s infants to childcare settings, and experiences with pumped lactation milk that working mothers experience today. Is it any wonder childbearing female psychiatrists face a special kind of working-mother guilt?
1. Collins G. When everything changed: the amazing journey of American women from 1960 to the present. New York, NY: Little, Brown and Company; 2009;271, 301.
2. AAMCNews. More women than men enrolled in U.S. medical schools in 2017. Association of American Medical Colleges. https://news.aamc.org/press-releases/article/applicant-enrollment-2017/. Published December 18, 2017. Accessed November 21, 2018.
3. Vassar L. How medical specialties vary by gender. American Medical Association. https://wire.ama-assn.org/education/how-medical-specialties-vary-gender. Published February 18, 2015. Accessed November 21, 2018.
4. Association of American Medical Colleges. Table A-6: age of applicants to U.S. medical schools at anticipated matriculation by sex and race/ethnicity, 2014-2015 through 2017-2018. https://www.aamc.org/download/321468/data/factstablea6.pdf. Published November 30, 2017. Accessed February 7, 2019.
5. Jones V. Best time to have a baby as a physician? It depends. Doximity. https://opmed.doximity.com/articles/the-best-time-to-have-a-baby-as-a-physician-it-depends-c8064a92156c. Published September 11, 2017. Accessed November 21, 2018.
6. Mitchell SA, Black MJ. The British object relations school: W.R.D. Fairbairn and D.W. Winnicott. In: Freud and beyond: a history of modern psychoanalytic thought. New York, NY: Basic Books; 1995:125-126, 137.
7. Ratnapalan S, Batty H. To be good enough. Can Fam Physician. 2009;55(3):239-242.
8. Sandler BJ, Tackett JJ, Longo WE, et al. Pregnancy and parenthood among surgery residents: results of the first nationwide survey of general surgery residency program Directors. J Am Coll Surg. 2016;222(6):1090-1096.
9. Kmec JA. Are motherhood penalties and fatherhood bonuses warranted? Comparing pro-work behaviors and conditions of mothers, fathers, and non-parents. Social Science Research. 2011;40(2):444-459.
10. Hampton R. Working moms don’t deserve the blame for unfair work expectations. Slate. https://slate.com/human-interest/2018/05/working-moms-dont-deserve-blame-for-unfair-work-expectations.html. Published May 18, 2018. Accessed November 25, 2018.
11. Rangel EL, Smink DS, Castillo-Angeles M, et al. Pregnancy and motherhood during surgical training. JAMA Surgery. 2018;153(7):644-652.
12. Murtagh L, Moulton AD. Working mothers, breastfeeding, and the law. Am J Public Health. 2011;101(2):217-223.
13. Kozhimannil KB, Jou J, Gjerdingen DK, et al. Access to workplace accommodations to support breastfeeding after passage of the Affordable Care Act. Womens Health Issues. 2016;26(1):6-13.
14. Dinour LM, Bai YK. Breastfeeding: the illusion of choice. Womens Health Issues. 2016;26(5):479-482.
15. Hansen K. Breastfeeding: a smart investment in people and in economies. Lancet. 2016;387(10017):416.
16. Greenfield R. Even America’s top doctors aren’t getting the parental leave doctors recommend. Bloomberg. https://www.bloomberg.com/news/articles/2018-02-13/even-america-s-top-doctors-aren-t-getting-the-parental-leave-doctors-recommend. Published February 13, 2018. Accessed November 21, 2018.
17. Humphries LS, Lyon S, Garza R, et al. Parental leave policies in graduate medical education: a systematic review. American J Surg. 2017;214(4):634-639.
18. Raju TNK. Continued barriers for breast-feeding in public and the workplace. J Pediatr. 2006;148(5):677-679.
19. Stewart DE, Robinson GE. Combining motherhood with psychiatric training and practice. Can J Psychiatry. 1985;30(1):28-34.
20. Rothman L. D esperate women, desperate doctors and the surprising history behind the breastfeeding debate. Time. http://time.com/5353068/breastfeeding-debate-history/. Published July 31, 2018. Accessed November 21, 2018.
21. Livingston G. Among 41 nations, U.S. is the outlier when it comes to paid parental leave. Pew Research Center. http://www.pewresearch.org/fact-tank/2016/09/26/u-s-lacks-mandated-paid-parental-leave/. Published September 26, 2016. Accessed November 21, 2018.
22. McCluskey PD. Long hours, short leaves force moms to reconsider jobs as surgeons. Boston Globe. https://www.bostonglobe.com/metro/2018/03/21/new-survey-says-female-surgical-residents-struggle-balance-training-motherhood/2ENQU1aPZmIJYy20iaRlLL/story.html. Published March 21, 2018. Accessed November 21, 2018.
23. Dinour LM, Szaro JM. Employer-based programs to support breastfeeding among working mothers: a systematic review. Breastfeeding Med. 2017;12:131-141.
1. Collins G. When everything changed: the amazing journey of American women from 1960 to the present. New York, NY: Little, Brown and Company; 2009;271, 301.
2. AAMCNews. More women than men enrolled in U.S. medical schools in 2017. Association of American Medical Colleges. https://news.aamc.org/press-releases/article/applicant-enrollment-2017/. Published December 18, 2017. Accessed November 21, 2018.
3. Vassar L. How medical specialties vary by gender. American Medical Association. https://wire.ama-assn.org/education/how-medical-specialties-vary-gender. Published February 18, 2015. Accessed November 21, 2018.
4. Association of American Medical Colleges. Table A-6: age of applicants to U.S. medical schools at anticipated matriculation by sex and race/ethnicity, 2014-2015 through 2017-2018. https://www.aamc.org/download/321468/data/factstablea6.pdf. Published November 30, 2017. Accessed February 7, 2019.
5. Jones V. Best time to have a baby as a physician? It depends. Doximity. https://opmed.doximity.com/articles/the-best-time-to-have-a-baby-as-a-physician-it-depends-c8064a92156c. Published September 11, 2017. Accessed November 21, 2018.
6. Mitchell SA, Black MJ. The British object relations school: W.R.D. Fairbairn and D.W. Winnicott. In: Freud and beyond: a history of modern psychoanalytic thought. New York, NY: Basic Books; 1995:125-126, 137.
7. Ratnapalan S, Batty H. To be good enough. Can Fam Physician. 2009;55(3):239-242.
8. Sandler BJ, Tackett JJ, Longo WE, et al. Pregnancy and parenthood among surgery residents: results of the first nationwide survey of general surgery residency program Directors. J Am Coll Surg. 2016;222(6):1090-1096.
9. Kmec JA. Are motherhood penalties and fatherhood bonuses warranted? Comparing pro-work behaviors and conditions of mothers, fathers, and non-parents. Social Science Research. 2011;40(2):444-459.
10. Hampton R. Working moms don’t deserve the blame for unfair work expectations. Slate. https://slate.com/human-interest/2018/05/working-moms-dont-deserve-blame-for-unfair-work-expectations.html. Published May 18, 2018. Accessed November 25, 2018.
11. Rangel EL, Smink DS, Castillo-Angeles M, et al. Pregnancy and motherhood during surgical training. JAMA Surgery. 2018;153(7):644-652.
12. Murtagh L, Moulton AD. Working mothers, breastfeeding, and the law. Am J Public Health. 2011;101(2):217-223.
13. Kozhimannil KB, Jou J, Gjerdingen DK, et al. Access to workplace accommodations to support breastfeeding after passage of the Affordable Care Act. Womens Health Issues. 2016;26(1):6-13.
14. Dinour LM, Bai YK. Breastfeeding: the illusion of choice. Womens Health Issues. 2016;26(5):479-482.
15. Hansen K. Breastfeeding: a smart investment in people and in economies. Lancet. 2016;387(10017):416.
16. Greenfield R. Even America’s top doctors aren’t getting the parental leave doctors recommend. Bloomberg. https://www.bloomberg.com/news/articles/2018-02-13/even-america-s-top-doctors-aren-t-getting-the-parental-leave-doctors-recommend. Published February 13, 2018. Accessed November 21, 2018.
17. Humphries LS, Lyon S, Garza R, et al. Parental leave policies in graduate medical education: a systematic review. American J Surg. 2017;214(4):634-639.
18. Raju TNK. Continued barriers for breast-feeding in public and the workplace. J Pediatr. 2006;148(5):677-679.
19. Stewart DE, Robinson GE. Combining motherhood with psychiatric training and practice. Can J Psychiatry. 1985;30(1):28-34.
20. Rothman L. D esperate women, desperate doctors and the surprising history behind the breastfeeding debate. Time. http://time.com/5353068/breastfeeding-debate-history/. Published July 31, 2018. Accessed November 21, 2018.
21. Livingston G. Among 41 nations, U.S. is the outlier when it comes to paid parental leave. Pew Research Center. http://www.pewresearch.org/fact-tank/2016/09/26/u-s-lacks-mandated-paid-parental-leave/. Published September 26, 2016. Accessed November 21, 2018.
22. McCluskey PD. Long hours, short leaves force moms to reconsider jobs as surgeons. Boston Globe. https://www.bostonglobe.com/metro/2018/03/21/new-survey-says-female-surgical-residents-struggle-balance-training-motherhood/2ENQU1aPZmIJYy20iaRlLL/story.html. Published March 21, 2018. Accessed November 21, 2018.
23. Dinour LM, Szaro JM. Employer-based programs to support breastfeeding among working mothers: a systematic review. Breastfeeding Med. 2017;12:131-141.

Management of treatment-resistant depression: A review of 3 studies
An estimated 7.1% of the adults in United States had a major depressive episode in 2017, and this prevalence has been trending upward over the past few years.1 The prevalence is even higher in adults between age 18 and 25 (13.1%).1 Like other psychiatric diagnoses, major depressive disorder (MDD) has a significant impact on productivity as well as daily functioning. Only one-third of patients with MDD achieve remission on the first antidepressant medication.2 This leaves an estimated 11.47 million people in the United States in need of an alternate regimen for management of their depressive episode.
The data on evidence-based biologic treatments for treatment-resistant depression are limited (other than for electroconvulsive therapy). Pharmacologic options include switching to a different medication, combining medications, and augmentation strategies or novel approaches such as ketamine and related agents. Here we summarize the findings from 3 recent studies that investigate alternate management options for MDD.
Ketamine: Randomized controlled trial
Traditional antidepressants may reduce suicidal ideation by improving depressive symptoms, but this effect may take weeks. Ketamine, an N-methyl-
_
1. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
Grunebaum et al3 evaluated the acute effect of adjunctive subanesthetic IV ketamine on clinically significant suicidal ideation in patients with MDD, with a comparison arm that received an infusion of midazolam.
Study design
- 80 inpatients (age 18 to 65 years) with MDD who had a score ≥16 on the Hamilton Depression Rating Scale (HAM-D) and a score ≥4 on the Scale for Suicidal Ideation (SSI). Approximately one-half (54%) were taking an antidepressant
- Patients were randomly assigned to IV racemic ketamine hydrochloride, .5 mg/kg, or IV midazolam, .02 mg/kg, both administered in 100 mL normal saline over 40 minutes.
Outcomes
- Scale for Suicidal Ideation scores were assessed at screening, before infusion, 230 minutes after infusion, 24 hours after infusion, and after 1 to 6 weeks of follow-up. The average SSI score on Day 1 was 4.96 points lower in the ketamine group compared with the midazolam group. The proportion of responders (defined as patients who experienced a 50% reduction in SSI score) on Day 1 was 55% for patients in the ketamine group compared with 30% in the midazolam group.
Conclusion
- Compared with midazolam, ketamine produced a greater clinically meaningful reduction in suicidal ideation 24 hours after infusion.
Apart from the primary outcome of reduction in suicidal ideation, greater reductions were also found in overall mood disturbance, depression subscale, and fatigue subscale scores as assessed on the Profile of Mood States (POMS). Although the study noted improvement in depression scores, the proportion of responders on Day 1 in depression scales, including HAM-D and the self-rated Beck Depression Inventory, fell short of statistical significance. Overall, compared with the midazolam infusion, a single adjunctive subanesthetic ketamine infusion was associated with a greater clinically significant reduction in suicidal ideation on Day 1.
Continue to: Ketamine
Ketamine: Review and meta-analysis
Wilkinson et al4 conducted a systematic review and individual participant data meta-analysis of 11 similar comparison intervention studies examining the effects of ketamine in reducing suicidal thoughts.
2. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
Study design
- Review of 11 studies of a single dose of IV ketamine for treatment of any psychiatric disorder. Only comparison intervention trials using saline placebo or midazolam were included:
- Individual patient-level data of 298 patients were obtained from 10 of the 11 trials. Analysis was performed on 167 patients who had suicidal ideation at baseline.
- Results were assessed by clinician-administered rating scales.
Outcomes
- Ketamine reduced suicidal ideation more rapidly compared with control infusions as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) and HAM-D, with significant benefits appearing on Day 1 and extending up to Day 7. The mean MADRS score in the ketamine group decreased to 19.5 from 33.8 within 1 day of infusion, compared with a reduction to 29.2 from 32.9 in the control groups.
- The number needed to treat to be free of suicidal ideation for ketamine (compared with control) was 3.1 to 4.0 for all time points in the first week after infusion.
Conclusion
- This meta-analysis provided evidence from the largest sample to date (N = 298) that ketamine reduces suicidal ideation partially independently of mood symptoms.
While the anti-suicidal effects of ketamine appear to be robust in the above studies, the possibility of rebound suicidal ideation remains in the weeks or months following exposure. Also, these studies only prove a reduction in suicidal ideation; reduction in suicidal behavior was not studied. Nevertheless, ketamine holds considerable promise as a potential rapid-acting agent in patients at risk of suicide.
Continue to: Strategies for augmentation or switching
Strategies for augmentation or switching
Only one-third of the patients with depression achieve remission on the first antidepressant medication. The American Psychiatric Association’s current management guidelines2 for patients who do not respond to the first-choice antidepressant include multiple options. Switching strategies recommended in these guidelines include changing to an antidepressant of the same class, or to one from a different class (eg, from a selective serotonin reuptake inhibitor [SSRI] to a serotonin-norepinephrine reuptake inhibitor, or from an SSRI to a tricyclic antidepressant). Augmentation strategies include augmenting with a non-monoamine oxidase inhibitor antidepressant from a different class, lithium, thyroid hormone, or an atypical antipsychotic.
The VAST-D trial5 evaluated the relative effectiveness and safety of 3 common treatments for treatment-resistant MDD:
- switching to bupropion
- augmenting the current treatment with bupropion
- augmenting the current treatment with the second-generation antipsychotic aripiprazole.
3. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
Study design
- A multi-site, randomized, single-blind, parallel-assignment trial of 1,522 patients at 35 US Veteran Health Administration medical centers with nonpsychotic MDD with a suboptimal response to at least one antidepressant (defined as a score of ≥16 on the Quick Inventory Depressive Symptomatology-Clinician Rated questionnaire [QIDS-C16]).
- Participants were randomly assigned to 1 of 3 groups: switching to bupropion (n = 511), augmenting with bupropion (n = 506), or augmenting with aripiprazole (n = 505).
- The primary outcome was remission (defined as a QIDS-C16 score ≤5 at 2 consecutively scheduled follow-up visits). Secondary outcome was a reduction in QIDS-C16 score by ≥50%, or a Clinical Global Impression (CGI) Improvement scale score of 1 (very much improved) or 2 (much improved).
Outcomes
- The aripiprazole group showed a modest, statistically significant remission rate (28.9%) compared with the bupropion switch group (22.3%), but did not show any statistically significant difference compared with the bupropion augmentation group.
- For the secondary outcome, there was a significantly higher response rate in the aripiprazole group (74.3%) compared with the bupropion switch group (62.4%) and bupropion augmentation group (65.6%). Response measured by the CGI– Improvement scale score also favored the aripiprazole group (79%) compared with the bupropion switch group (70%) and bupropion augmentation group (74%).
Continue to: Conclusion
Conclusion
- Overall, the study found a statistically significant but modest increased likelihood of remission during 12 weeks of augmentation treatment with aripiprazole, compared with switching to bupropion monotherapy.

The studies discussed here, which are summarized in the Table,3-5 provide some potential avenues for research into interventions for patients who are acutely suicidal and those with treatment-resistant depression. Further research into long-term outcomes and adverse effects of ketamine use for suicidality in patients with depression is needed. The VAST-D trial suggests a need for further exploration into the efficacy of augmentation with second-generation antipsychotics for treatment-resistant depression.
1. Substance Abuse and Mental Health Services Administration. Reports and detailed tables from the 2017 National Survey on Drug Use and Health (NSDUH). https://www.samhsa.gov/data/nsduh/reports-detailed-tables-2017-NSDUH. Accessed November 12, 2018.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. 3rd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published 2010. Accessed November 12, 2018.
3. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
4. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
5. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
An estimated 7.1% of the adults in United States had a major depressive episode in 2017, and this prevalence has been trending upward over the past few years.1 The prevalence is even higher in adults between age 18 and 25 (13.1%).1 Like other psychiatric diagnoses, major depressive disorder (MDD) has a significant impact on productivity as well as daily functioning. Only one-third of patients with MDD achieve remission on the first antidepressant medication.2 This leaves an estimated 11.47 million people in the United States in need of an alternate regimen for management of their depressive episode.
The data on evidence-based biologic treatments for treatment-resistant depression are limited (other than for electroconvulsive therapy). Pharmacologic options include switching to a different medication, combining medications, and augmentation strategies or novel approaches such as ketamine and related agents. Here we summarize the findings from 3 recent studies that investigate alternate management options for MDD.
Ketamine: Randomized controlled trial
Traditional antidepressants may reduce suicidal ideation by improving depressive symptoms, but this effect may take weeks. Ketamine, an N-methyl-
_
1. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
Grunebaum et al3 evaluated the acute effect of adjunctive subanesthetic IV ketamine on clinically significant suicidal ideation in patients with MDD, with a comparison arm that received an infusion of midazolam.
Study design
- 80 inpatients (age 18 to 65 years) with MDD who had a score ≥16 on the Hamilton Depression Rating Scale (HAM-D) and a score ≥4 on the Scale for Suicidal Ideation (SSI). Approximately one-half (54%) were taking an antidepressant
- Patients were randomly assigned to IV racemic ketamine hydrochloride, .5 mg/kg, or IV midazolam, .02 mg/kg, both administered in 100 mL normal saline over 40 minutes.
Outcomes
- Scale for Suicidal Ideation scores were assessed at screening, before infusion, 230 minutes after infusion, 24 hours after infusion, and after 1 to 6 weeks of follow-up. The average SSI score on Day 1 was 4.96 points lower in the ketamine group compared with the midazolam group. The proportion of responders (defined as patients who experienced a 50% reduction in SSI score) on Day 1 was 55% for patients in the ketamine group compared with 30% in the midazolam group.
Conclusion
- Compared with midazolam, ketamine produced a greater clinically meaningful reduction in suicidal ideation 24 hours after infusion.
Apart from the primary outcome of reduction in suicidal ideation, greater reductions were also found in overall mood disturbance, depression subscale, and fatigue subscale scores as assessed on the Profile of Mood States (POMS). Although the study noted improvement in depression scores, the proportion of responders on Day 1 in depression scales, including HAM-D and the self-rated Beck Depression Inventory, fell short of statistical significance. Overall, compared with the midazolam infusion, a single adjunctive subanesthetic ketamine infusion was associated with a greater clinically significant reduction in suicidal ideation on Day 1.
Continue to: Ketamine
Ketamine: Review and meta-analysis
Wilkinson et al4 conducted a systematic review and individual participant data meta-analysis of 11 similar comparison intervention studies examining the effects of ketamine in reducing suicidal thoughts.
2. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
Study design
- Review of 11 studies of a single dose of IV ketamine for treatment of any psychiatric disorder. Only comparison intervention trials using saline placebo or midazolam were included:
- Individual patient-level data of 298 patients were obtained from 10 of the 11 trials. Analysis was performed on 167 patients who had suicidal ideation at baseline.
- Results were assessed by clinician-administered rating scales.
Outcomes
- Ketamine reduced suicidal ideation more rapidly compared with control infusions as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) and HAM-D, with significant benefits appearing on Day 1 and extending up to Day 7. The mean MADRS score in the ketamine group decreased to 19.5 from 33.8 within 1 day of infusion, compared with a reduction to 29.2 from 32.9 in the control groups.
- The number needed to treat to be free of suicidal ideation for ketamine (compared with control) was 3.1 to 4.0 for all time points in the first week after infusion.
Conclusion
- This meta-analysis provided evidence from the largest sample to date (N = 298) that ketamine reduces suicidal ideation partially independently of mood symptoms.
While the anti-suicidal effects of ketamine appear to be robust in the above studies, the possibility of rebound suicidal ideation remains in the weeks or months following exposure. Also, these studies only prove a reduction in suicidal ideation; reduction in suicidal behavior was not studied. Nevertheless, ketamine holds considerable promise as a potential rapid-acting agent in patients at risk of suicide.
Continue to: Strategies for augmentation or switching
Strategies for augmentation or switching
Only one-third of the patients with depression achieve remission on the first antidepressant medication. The American Psychiatric Association’s current management guidelines2 for patients who do not respond to the first-choice antidepressant include multiple options. Switching strategies recommended in these guidelines include changing to an antidepressant of the same class, or to one from a different class (eg, from a selective serotonin reuptake inhibitor [SSRI] to a serotonin-norepinephrine reuptake inhibitor, or from an SSRI to a tricyclic antidepressant). Augmentation strategies include augmenting with a non-monoamine oxidase inhibitor antidepressant from a different class, lithium, thyroid hormone, or an atypical antipsychotic.
The VAST-D trial5 evaluated the relative effectiveness and safety of 3 common treatments for treatment-resistant MDD:
- switching to bupropion
- augmenting the current treatment with bupropion
- augmenting the current treatment with the second-generation antipsychotic aripiprazole.
3. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
Study design
- A multi-site, randomized, single-blind, parallel-assignment trial of 1,522 patients at 35 US Veteran Health Administration medical centers with nonpsychotic MDD with a suboptimal response to at least one antidepressant (defined as a score of ≥16 on the Quick Inventory Depressive Symptomatology-Clinician Rated questionnaire [QIDS-C16]).
- Participants were randomly assigned to 1 of 3 groups: switching to bupropion (n = 511), augmenting with bupropion (n = 506), or augmenting with aripiprazole (n = 505).
- The primary outcome was remission (defined as a QIDS-C16 score ≤5 at 2 consecutively scheduled follow-up visits). Secondary outcome was a reduction in QIDS-C16 score by ≥50%, or a Clinical Global Impression (CGI) Improvement scale score of 1 (very much improved) or 2 (much improved).
Outcomes
- The aripiprazole group showed a modest, statistically significant remission rate (28.9%) compared with the bupropion switch group (22.3%), but did not show any statistically significant difference compared with the bupropion augmentation group.
- For the secondary outcome, there was a significantly higher response rate in the aripiprazole group (74.3%) compared with the bupropion switch group (62.4%) and bupropion augmentation group (65.6%). Response measured by the CGI– Improvement scale score also favored the aripiprazole group (79%) compared with the bupropion switch group (70%) and bupropion augmentation group (74%).
Continue to: Conclusion
Conclusion
- Overall, the study found a statistically significant but modest increased likelihood of remission during 12 weeks of augmentation treatment with aripiprazole, compared with switching to bupropion monotherapy.
The studies discussed here, which are summarized in the Table,3-5 provide some potential avenues for research into interventions for patients who are acutely suicidal and those with treatment-resistant depression. Further research into long-term outcomes and adverse effects of ketamine use for suicidality in patients with depression is needed. The VAST-D trial suggests a need for further exploration into the efficacy of augmentation with second-generation antipsychotics for treatment-resistant depression.
An estimated 7.1% of the adults in United States had a major depressive episode in 2017, and this prevalence has been trending upward over the past few years.1 The prevalence is even higher in adults between age 18 and 25 (13.1%).1 Like other psychiatric diagnoses, major depressive disorder (MDD) has a significant impact on productivity as well as daily functioning. Only one-third of patients with MDD achieve remission on the first antidepressant medication.2 This leaves an estimated 11.47 million people in the United States in need of an alternate regimen for management of their depressive episode.
The data on evidence-based biologic treatments for treatment-resistant depression are limited (other than for electroconvulsive therapy). Pharmacologic options include switching to a different medication, combining medications, and augmentation strategies or novel approaches such as ketamine and related agents. Here we summarize the findings from 3 recent studies that investigate alternate management options for MDD.
Ketamine: Randomized controlled trial
Traditional antidepressants may reduce suicidal ideation by improving depressive symptoms, but this effect may take weeks. Ketamine, an N-methyl-
_
1. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
Grunebaum et al3 evaluated the acute effect of adjunctive subanesthetic IV ketamine on clinically significant suicidal ideation in patients with MDD, with a comparison arm that received an infusion of midazolam.
Study design
- 80 inpatients (age 18 to 65 years) with MDD who had a score ≥16 on the Hamilton Depression Rating Scale (HAM-D) and a score ≥4 on the Scale for Suicidal Ideation (SSI). Approximately one-half (54%) were taking an antidepressant
- Patients were randomly assigned to IV racemic ketamine hydrochloride, .5 mg/kg, or IV midazolam, .02 mg/kg, both administered in 100 mL normal saline over 40 minutes.
Outcomes
- Scale for Suicidal Ideation scores were assessed at screening, before infusion, 230 minutes after infusion, 24 hours after infusion, and after 1 to 6 weeks of follow-up. The average SSI score on Day 1 was 4.96 points lower in the ketamine group compared with the midazolam group. The proportion of responders (defined as patients who experienced a 50% reduction in SSI score) on Day 1 was 55% for patients in the ketamine group compared with 30% in the midazolam group.
Conclusion
- Compared with midazolam, ketamine produced a greater clinically meaningful reduction in suicidal ideation 24 hours after infusion.
Apart from the primary outcome of reduction in suicidal ideation, greater reductions were also found in overall mood disturbance, depression subscale, and fatigue subscale scores as assessed on the Profile of Mood States (POMS). Although the study noted improvement in depression scores, the proportion of responders on Day 1 in depression scales, including HAM-D and the self-rated Beck Depression Inventory, fell short of statistical significance. Overall, compared with the midazolam infusion, a single adjunctive subanesthetic ketamine infusion was associated with a greater clinically significant reduction in suicidal ideation on Day 1.
Continue to: Ketamine
Ketamine: Review and meta-analysis
Wilkinson et al4 conducted a systematic review and individual participant data meta-analysis of 11 similar comparison intervention studies examining the effects of ketamine in reducing suicidal thoughts.
2. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
Study design
- Review of 11 studies of a single dose of IV ketamine for treatment of any psychiatric disorder. Only comparison intervention trials using saline placebo or midazolam were included:
- Individual patient-level data of 298 patients were obtained from 10 of the 11 trials. Analysis was performed on 167 patients who had suicidal ideation at baseline.
- Results were assessed by clinician-administered rating scales.
Outcomes
- Ketamine reduced suicidal ideation more rapidly compared with control infusions as assessed by the Montgomery-Åsberg Depression Rating Scale (MADRS) and HAM-D, with significant benefits appearing on Day 1 and extending up to Day 7. The mean MADRS score in the ketamine group decreased to 19.5 from 33.8 within 1 day of infusion, compared with a reduction to 29.2 from 32.9 in the control groups.
- The number needed to treat to be free of suicidal ideation for ketamine (compared with control) was 3.1 to 4.0 for all time points in the first week after infusion.
Conclusion
- This meta-analysis provided evidence from the largest sample to date (N = 298) that ketamine reduces suicidal ideation partially independently of mood symptoms.
While the anti-suicidal effects of ketamine appear to be robust in the above studies, the possibility of rebound suicidal ideation remains in the weeks or months following exposure. Also, these studies only prove a reduction in suicidal ideation; reduction in suicidal behavior was not studied. Nevertheless, ketamine holds considerable promise as a potential rapid-acting agent in patients at risk of suicide.
Continue to: Strategies for augmentation or switching
Strategies for augmentation or switching
Only one-third of the patients with depression achieve remission on the first antidepressant medication. The American Psychiatric Association’s current management guidelines2 for patients who do not respond to the first-choice antidepressant include multiple options. Switching strategies recommended in these guidelines include changing to an antidepressant of the same class, or to one from a different class (eg, from a selective serotonin reuptake inhibitor [SSRI] to a serotonin-norepinephrine reuptake inhibitor, or from an SSRI to a tricyclic antidepressant). Augmentation strategies include augmenting with a non-monoamine oxidase inhibitor antidepressant from a different class, lithium, thyroid hormone, or an atypical antipsychotic.
The VAST-D trial5 evaluated the relative effectiveness and safety of 3 common treatments for treatment-resistant MDD:
- switching to bupropion
- augmenting the current treatment with bupropion
- augmenting the current treatment with the second-generation antipsychotic aripiprazole.
3. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
Study design
- A multi-site, randomized, single-blind, parallel-assignment trial of 1,522 patients at 35 US Veteran Health Administration medical centers with nonpsychotic MDD with a suboptimal response to at least one antidepressant (defined as a score of ≥16 on the Quick Inventory Depressive Symptomatology-Clinician Rated questionnaire [QIDS-C16]).
- Participants were randomly assigned to 1 of 3 groups: switching to bupropion (n = 511), augmenting with bupropion (n = 506), or augmenting with aripiprazole (n = 505).
- The primary outcome was remission (defined as a QIDS-C16 score ≤5 at 2 consecutively scheduled follow-up visits). Secondary outcome was a reduction in QIDS-C16 score by ≥50%, or a Clinical Global Impression (CGI) Improvement scale score of 1 (very much improved) or 2 (much improved).
Outcomes
- The aripiprazole group showed a modest, statistically significant remission rate (28.9%) compared with the bupropion switch group (22.3%), but did not show any statistically significant difference compared with the bupropion augmentation group.
- For the secondary outcome, there was a significantly higher response rate in the aripiprazole group (74.3%) compared with the bupropion switch group (62.4%) and bupropion augmentation group (65.6%). Response measured by the CGI– Improvement scale score also favored the aripiprazole group (79%) compared with the bupropion switch group (70%) and bupropion augmentation group (74%).
Continue to: Conclusion
Conclusion
- Overall, the study found a statistically significant but modest increased likelihood of remission during 12 weeks of augmentation treatment with aripiprazole, compared with switching to bupropion monotherapy.
The studies discussed here, which are summarized in the Table,3-5 provide some potential avenues for research into interventions for patients who are acutely suicidal and those with treatment-resistant depression. Further research into long-term outcomes and adverse effects of ketamine use for suicidality in patients with depression is needed. The VAST-D trial suggests a need for further exploration into the efficacy of augmentation with second-generation antipsychotics for treatment-resistant depression.
1. Substance Abuse and Mental Health Services Administration. Reports and detailed tables from the 2017 National Survey on Drug Use and Health (NSDUH). https://www.samhsa.gov/data/nsduh/reports-detailed-tables-2017-NSDUH. Accessed November 12, 2018.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. 3rd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published 2010. Accessed November 12, 2018.
3. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
4. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
5. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.
1. Substance Abuse and Mental Health Services Administration. Reports and detailed tables from the 2017 National Survey on Drug Use and Health (NSDUH). https://www.samhsa.gov/data/nsduh/reports-detailed-tables-2017-NSDUH. Accessed November 12, 2018.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. 3rd ed. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Published 2010. Accessed November 12, 2018.
3. Grunebaum MF, Galfalvy HC, Choo TH, et al. Ketamine for rapid reduction of suicidal thoughts in major depression: a midazolam-controlled randomized clinical trial. Am J Psychiatry. 2018;175(4):327-335.
4. Wilkinson ST, Ballard ED, Bloch MH, et al. The effect of a single dose of intravenous ketamine on suicidal ideation: a systematic review and individual participant data meta-analysis. Am J Psychiatry. 2018;175(2):150-158.
5. Mohamed S, Johnson GR, Chen P, et al. Effect of antidepressant switching vs augmentation on remission among patients with major depressive disorder unresponsive to antidepressant treatment: the VAST-D randomized clinical trial. JAMA. 2017;318(2):132-145.

Career Choices: Addiction psychiatry
Editor’s note: Career Choices features a psychiatry resident/fellow interviewing a psychiatrist about why he or she has chosen a specific career path. The goal is to inform trainees about the various psychiatric career options, and to give them a feel for the pros and cons of the various paths.
In this Career Choices, Saeed Ahmed, MD, talked with Cornel Stanciu, MD. Dr. Stanciu is an addiction psychiatrist at Dartmouth’s Geisel School of Medicine, where he is an Assistant Professor, and serves as the Director of Addiction Services at New Hampshire Hospital. He provides support to clinicians managing patients with addictive disorders in a multitude of settings, and also assists with policy making and delivery of addiction care at the state level. He is also the author of Deciphering the Addicted Brain, a guide to help families and the general public better understand addictive disorders.
Dr. Ahmed: What attracted you to pursue subspecialty training in addictive disorders?
Dr. Stanciu: In the early stages of my training, I frequently encountered individuals with medical and mental health disorders whose treatment was impacted by underlying substance use. I soon came to realize any attempts at (for example) managing hypertension in someone with cocaine use disorder, or managing schizophrenia in someone with ongoing cannabis use, were futile. Almost all of my patients receiving treatment for mental health disorders were dependent on tobacco or other substances, and most were interested in cessation. Through mentorship from addiction-trained residency faculty members, I was able to get a taste of the neurobiologic complexities of the disease, something that left me with a desire to develop a deeper understanding of the disease process. Witnessing strikingly positive outcomes with implementation of evidence-based treatment modalities further solidified my path to subspecialty training. Even during that early phase, because I expressed interest in managing these conditions, I was immediately put in a position to share and disseminate any newly acquired knowledge to other specialties as well as the public.
Dr. Ahmed: Could one manage addictive disorders with just general psychiatry training, and what are the differences between the different paths to certification that a resident could undertake?
Dr. Stanciu: Addictive disorders fall under the general umbrella of psychiatric care. Most individuals with these disorders exhibit some degree of mental illness. Medical school curriculum offers on average 2 hours of addiction-related didactics during 4 years. General psychiatry training programs vary significantly in the type of exposure to addiction—some residencies have an affiliated addiction fellowship, others have addiction-trained psychiatrists on staff, but most have none. Ultimately, there is great variability in the degree of comfort in working with individuals with addictive disorders post-residency. Being able to prescribe medications for the treatment of addictive disorders is very different from being familiar with the latest evidence-based recommendations and guidelines; the latter is
Addiction medicine is a fairly new route initially intended to allow non-psychiatric specialties access to addictive disorders training and certification. This is offered through the American Board of Preventive Medicine. There are currently 2 routes to sitting for the exam: through completion of a 1-year addiction medicine fellowship, or through the “practice pathway” still available until 2020. To be eligible for the latter, individuals must provide documentation of clinical experience post-residency, which is quantified as number of hours spent treating patients with addictions, plus any additional courses or training, and must be endorsed by a certified addictionologist.
Continue to: What was your fellowship experience link...
Dr. Ahmed: What was your fellowship experience like, and what should one consider when choosing a program?
Dr. Stanciu: I completed my fellowship training through Dartmouth’s Geisel School of Medicine, and the experience was tremendously valuable. In evaluating programs, one of the starting points is whether you have interest in a formal research track, because several programs include an optional year for that. Most programs tend to provide exposure to the Veterans Affairs system. The 1 year should provide you with broad exposure to all possible settings, all addictive disorders and patient populations, and all treatment modalities, in addition to rigorous didactic sessions. The ideal program should include rotations through methadone treatment centers, intensive outpatient programs, pain and interdisciplinary clinics, detoxification units, and centers for treatment of adolescent and young adults, as well as general medical settings and infectious disease clinics. There should also be close collaboration with psychologists who can provide training in evidence-based therapeutic modalities. During this year, it is vital to expand your knowledge of the ethical and legal regulations of treatment programs, state and federal requirements, insurance complexities, and requirements for privacy and protection of health information. The size of these programs can vary significantly, which may limit the one-on-one time devoted to your training, which is something I personally valued. My faculty was very supportive of academic endeavors, providing guidance, funding, and encouragement for attending and presenting at conferences, publishing papers, and other academic pursuits. Additionally, faculty should be current with emerging literature and willing to develop or implement new protocols and evaluate new pharmacologic therapies.
Dr. Ahmed: What are some of the career options and work settings for addiction psychiatrists?
Dr. Stanciu: Addiction psychiatrists work in numerous settings and various capacities. They can provide subspecialty care directly by seeing patients in outpatient clinics or inpatient addiction treatment centers for detoxification or rehabilitation, or they can work with dual-diagnosis populations in inpatient units. The expansion of telemedicine also holds promise for a role through virtual services. Indirectly, they can serve as a resource for expertise in the field through consultations in medical and psychiatric settings, or through policy making by working with the legislature and public health departments. Additionally, they can help create and integrate new knowledge into practice and educate future generations of physicians and the public.
Dr. Ahmed: What are some of the prevalent disorders and reasons for consultation that you encounter in your daily practice?
Continue to: Dr. Stanciu's response...
Dr. Stanciu: This can vary significantly depending on the setting, geographical region, and demographics of the population. My main non-administrative responsibilities are primarily consultative assisting clinicians at a 200-bed psychiatric hospital to address co-occurring addictive disorders. In short-term units, I am primarily asked to provide input on issues related to various toxidromes and withdrawals and the use of relapse prevention medications for alcohol use disorders as well as the use of buprenorphine or other forms of medication-assisted treatment. I work closely with licensed drug and alcohol counselors in implementing brief interventions as well as facilitating outpatient treatment referrals. Clinicians in longer term units may consult on issues related to pain management in individuals who have addictive disorders, the use of evidence-based pharmacologic agents to address cravings, or the use of relapse prevention medications for someone close to discharge. In terms of specific drugs of abuse, although opioids have recently received a tremendous amount of attention due to the visible costs through overdose deaths, the magnitude of individuals who are losing years of quality life through the use of alcohol and tobacco is significant, and hence this is a large portion of the conditions I encounter. I have also seen an abundance of marijuana use due to decreased perception of harm and increased access.
Dr. Ahmed: What are some of the challenges in working in this field?
Dr. Stanciu: Historically, funding for services has been an issue for clinicians working primarily with addictive disorders from the standpoint of reimbursement, patient access to evidence-based pharmacotherapy, and ability to collaborate with existing levels of care. In recent years, federal funding and policies have changed this, and after numerous studies have found increased cost savings, commercial insurances are providing coverage. A significant challenge also has been public stigma and dealing with a condition that is relapsing-remitting, poorly understood by other specialties and the general public, and sometimes labeled as a defect of character. Several efforts in education have lessened this; however, the impact still takes a toll on patients, who may feel ashamed of their disorder and sometimes are hesitant to take medications because they may believe that they are not “clean” if they depend on a medication for remission. Lastly, recent changes in marijuana policies make conversations about this drug quite difficult because patients often view it as harmless, and the laws governing legality and indications for therapeutic use are slightly ahead of the evidence.
Dr. Ahmed: In what direction do you believe the subspecialty is headed?
Dr. Stanciu: Currently, there are approximately 1,000 certified addiction psychiatrists for the 45 million Americans who have addictive disorders. Smoking and other forms of tobacco use pose significant threats to the 2020 Healthy People Tobacco Use objectives. There is a significant demand for addictionologists in both public and private sectors. As with mental health, demand exceeds supply, and efforts are underway to expand downstream education and increase access to specialists. Several federal laws have been put in place to remove barriers and expand access to care and have paved the way to a brighter future. One is the Affordable Care Act, which requires all insurances including Medicaid to cover the cost of treatment. Second is the Mental Health Parity and Addiction Equity Act, which ensures that the duration and dollar amount of coverage for substance use disorders is comparable to that of medical and surgical care.
Continue to: Another exciting possibility...
Another exciting possibility comes from the world of pharmaceuticals. Some medications have modest efficacy for addressing addictive disorders; however, historically these have been poorly utilized. Enhanced understanding of the neurobiology combined with increased insurance reimbursement should prompt research and new drug development. Some promising agents are already in the pipeline. Research into molecular and gene therapy as a way to better individualize care is also underway.
Going forward, I think we will also encounter a different landscape of drugs. Synthetic agents are emerging and increasing in popularity. Alarmingly, public perception of harm is decreasing. When it comes to cannabis use, I see a rise in pathologic use and the ramifications of this will have a drastic impact, particularly on patients with mental health conditions. We will need to undertake better efforts in monitoring, staying updated, and providing public education campaigns.
Dr. Ahmed: What advice do you have for trainees contemplating subspecialty training in addiction psychiatry?
Dr. Stanciu: I cannot emphasize enough the importance of mentorship. The American Academy of Addiction Psychiatry has a robust system for connecting mentees with mentors at all stages in their careers. This can be extremely helpful, especially in situations where the residency program does not have addiction-trained faculty or rotations through treatment centers. Joining such an organization also grants you access to resources that can help further your enthusiasm. Those interested should also familiarize themselves with currently available pharmacotherapeutic treatments that have evidence supporting efficacy for various addictive disorders, and begin to incorporate these medications into general mental health practice, along with attempts at motivational interviewing. For example, begin discussing naltrexone with patients who have comorbid alcohol use disorders and are interested in reducing their drinking; and varenicline with patients who smoke and are interested in quitting. The outcomes should automatically elicit an interest in pursuing further training in the field!
Editor’s note: Career Choices features a psychiatry resident/fellow interviewing a psychiatrist about why he or she has chosen a specific career path. The goal is to inform trainees about the various psychiatric career options, and to give them a feel for the pros and cons of the various paths.
In this Career Choices, Saeed Ahmed, MD, talked with Cornel Stanciu, MD. Dr. Stanciu is an addiction psychiatrist at Dartmouth’s Geisel School of Medicine, where he is an Assistant Professor, and serves as the Director of Addiction Services at New Hampshire Hospital. He provides support to clinicians managing patients with addictive disorders in a multitude of settings, and also assists with policy making and delivery of addiction care at the state level. He is also the author of Deciphering the Addicted Brain, a guide to help families and the general public better understand addictive disorders.
Dr. Ahmed: What attracted you to pursue subspecialty training in addictive disorders?
Dr. Stanciu: In the early stages of my training, I frequently encountered individuals with medical and mental health disorders whose treatment was impacted by underlying substance use. I soon came to realize any attempts at (for example) managing hypertension in someone with cocaine use disorder, or managing schizophrenia in someone with ongoing cannabis use, were futile. Almost all of my patients receiving treatment for mental health disorders were dependent on tobacco or other substances, and most were interested in cessation. Through mentorship from addiction-trained residency faculty members, I was able to get a taste of the neurobiologic complexities of the disease, something that left me with a desire to develop a deeper understanding of the disease process. Witnessing strikingly positive outcomes with implementation of evidence-based treatment modalities further solidified my path to subspecialty training. Even during that early phase, because I expressed interest in managing these conditions, I was immediately put in a position to share and disseminate any newly acquired knowledge to other specialties as well as the public.
Dr. Ahmed: Could one manage addictive disorders with just general psychiatry training, and what are the differences between the different paths to certification that a resident could undertake?
Dr. Stanciu: Addictive disorders fall under the general umbrella of psychiatric care. Most individuals with these disorders exhibit some degree of mental illness. Medical school curriculum offers on average 2 hours of addiction-related didactics during 4 years. General psychiatry training programs vary significantly in the type of exposure to addiction—some residencies have an affiliated addiction fellowship, others have addiction-trained psychiatrists on staff, but most have none. Ultimately, there is great variability in the degree of comfort in working with individuals with addictive disorders post-residency. Being able to prescribe medications for the treatment of addictive disorders is very different from being familiar with the latest evidence-based recommendations and guidelines; the latter is
Addiction medicine is a fairly new route initially intended to allow non-psychiatric specialties access to addictive disorders training and certification. This is offered through the American Board of Preventive Medicine. There are currently 2 routes to sitting for the exam: through completion of a 1-year addiction medicine fellowship, or through the “practice pathway” still available until 2020. To be eligible for the latter, individuals must provide documentation of clinical experience post-residency, which is quantified as number of hours spent treating patients with addictions, plus any additional courses or training, and must be endorsed by a certified addictionologist.
Continue to: What was your fellowship experience link...
Dr. Ahmed: What was your fellowship experience like, and what should one consider when choosing a program?
Dr. Stanciu: I completed my fellowship training through Dartmouth’s Geisel School of Medicine, and the experience was tremendously valuable. In evaluating programs, one of the starting points is whether you have interest in a formal research track, because several programs include an optional year for that. Most programs tend to provide exposure to the Veterans Affairs system. The 1 year should provide you with broad exposure to all possible settings, all addictive disorders and patient populations, and all treatment modalities, in addition to rigorous didactic sessions. The ideal program should include rotations through methadone treatment centers, intensive outpatient programs, pain and interdisciplinary clinics, detoxification units, and centers for treatment of adolescent and young adults, as well as general medical settings and infectious disease clinics. There should also be close collaboration with psychologists who can provide training in evidence-based therapeutic modalities. During this year, it is vital to expand your knowledge of the ethical and legal regulations of treatment programs, state and federal requirements, insurance complexities, and requirements for privacy and protection of health information. The size of these programs can vary significantly, which may limit the one-on-one time devoted to your training, which is something I personally valued. My faculty was very supportive of academic endeavors, providing guidance, funding, and encouragement for attending and presenting at conferences, publishing papers, and other academic pursuits. Additionally, faculty should be current with emerging literature and willing to develop or implement new protocols and evaluate new pharmacologic therapies.
Dr. Ahmed: What are some of the career options and work settings for addiction psychiatrists?
Dr. Stanciu: Addiction psychiatrists work in numerous settings and various capacities. They can provide subspecialty care directly by seeing patients in outpatient clinics or inpatient addiction treatment centers for detoxification or rehabilitation, or they can work with dual-diagnosis populations in inpatient units. The expansion of telemedicine also holds promise for a role through virtual services. Indirectly, they can serve as a resource for expertise in the field through consultations in medical and psychiatric settings, or through policy making by working with the legislature and public health departments. Additionally, they can help create and integrate new knowledge into practice and educate future generations of physicians and the public.
Dr. Ahmed: What are some of the prevalent disorders and reasons for consultation that you encounter in your daily practice?
Continue to: Dr. Stanciu's response...
Dr. Stanciu: This can vary significantly depending on the setting, geographical region, and demographics of the population. My main non-administrative responsibilities are primarily consultative assisting clinicians at a 200-bed psychiatric hospital to address co-occurring addictive disorders. In short-term units, I am primarily asked to provide input on issues related to various toxidromes and withdrawals and the use of relapse prevention medications for alcohol use disorders as well as the use of buprenorphine or other forms of medication-assisted treatment. I work closely with licensed drug and alcohol counselors in implementing brief interventions as well as facilitating outpatient treatment referrals. Clinicians in longer term units may consult on issues related to pain management in individuals who have addictive disorders, the use of evidence-based pharmacologic agents to address cravings, or the use of relapse prevention medications for someone close to discharge. In terms of specific drugs of abuse, although opioids have recently received a tremendous amount of attention due to the visible costs through overdose deaths, the magnitude of individuals who are losing years of quality life through the use of alcohol and tobacco is significant, and hence this is a large portion of the conditions I encounter. I have also seen an abundance of marijuana use due to decreased perception of harm and increased access.
Dr. Ahmed: What are some of the challenges in working in this field?
Dr. Stanciu: Historically, funding for services has been an issue for clinicians working primarily with addictive disorders from the standpoint of reimbursement, patient access to evidence-based pharmacotherapy, and ability to collaborate with existing levels of care. In recent years, federal funding and policies have changed this, and after numerous studies have found increased cost savings, commercial insurances are providing coverage. A significant challenge also has been public stigma and dealing with a condition that is relapsing-remitting, poorly understood by other specialties and the general public, and sometimes labeled as a defect of character. Several efforts in education have lessened this; however, the impact still takes a toll on patients, who may feel ashamed of their disorder and sometimes are hesitant to take medications because they may believe that they are not “clean” if they depend on a medication for remission. Lastly, recent changes in marijuana policies make conversations about this drug quite difficult because patients often view it as harmless, and the laws governing legality and indications for therapeutic use are slightly ahead of the evidence.
Dr. Ahmed: In what direction do you believe the subspecialty is headed?
Dr. Stanciu: Currently, there are approximately 1,000 certified addiction psychiatrists for the 45 million Americans who have addictive disorders. Smoking and other forms of tobacco use pose significant threats to the 2020 Healthy People Tobacco Use objectives. There is a significant demand for addictionologists in both public and private sectors. As with mental health, demand exceeds supply, and efforts are underway to expand downstream education and increase access to specialists. Several federal laws have been put in place to remove barriers and expand access to care and have paved the way to a brighter future. One is the Affordable Care Act, which requires all insurances including Medicaid to cover the cost of treatment. Second is the Mental Health Parity and Addiction Equity Act, which ensures that the duration and dollar amount of coverage for substance use disorders is comparable to that of medical and surgical care.
Continue to: Another exciting possibility...
Another exciting possibility comes from the world of pharmaceuticals. Some medications have modest efficacy for addressing addictive disorders; however, historically these have been poorly utilized. Enhanced understanding of the neurobiology combined with increased insurance reimbursement should prompt research and new drug development. Some promising agents are already in the pipeline. Research into molecular and gene therapy as a way to better individualize care is also underway.
Going forward, I think we will also encounter a different landscape of drugs. Synthetic agents are emerging and increasing in popularity. Alarmingly, public perception of harm is decreasing. When it comes to cannabis use, I see a rise in pathologic use and the ramifications of this will have a drastic impact, particularly on patients with mental health conditions. We will need to undertake better efforts in monitoring, staying updated, and providing public education campaigns.
Dr. Ahmed: What advice do you have for trainees contemplating subspecialty training in addiction psychiatry?
Dr. Stanciu: I cannot emphasize enough the importance of mentorship. The American Academy of Addiction Psychiatry has a robust system for connecting mentees with mentors at all stages in their careers. This can be extremely helpful, especially in situations where the residency program does not have addiction-trained faculty or rotations through treatment centers. Joining such an organization also grants you access to resources that can help further your enthusiasm. Those interested should also familiarize themselves with currently available pharmacotherapeutic treatments that have evidence supporting efficacy for various addictive disorders, and begin to incorporate these medications into general mental health practice, along with attempts at motivational interviewing. For example, begin discussing naltrexone with patients who have comorbid alcohol use disorders and are interested in reducing their drinking; and varenicline with patients who smoke and are interested in quitting. The outcomes should automatically elicit an interest in pursuing further training in the field!
Editor’s note: Career Choices features a psychiatry resident/fellow interviewing a psychiatrist about why he or she has chosen a specific career path. The goal is to inform trainees about the various psychiatric career options, and to give them a feel for the pros and cons of the various paths.
In this Career Choices, Saeed Ahmed, MD, talked with Cornel Stanciu, MD. Dr. Stanciu is an addiction psychiatrist at Dartmouth’s Geisel School of Medicine, where he is an Assistant Professor, and serves as the Director of Addiction Services at New Hampshire Hospital. He provides support to clinicians managing patients with addictive disorders in a multitude of settings, and also assists with policy making and delivery of addiction care at the state level. He is also the author of Deciphering the Addicted Brain, a guide to help families and the general public better understand addictive disorders.
Dr. Ahmed: What attracted you to pursue subspecialty training in addictive disorders?
Dr. Stanciu: In the early stages of my training, I frequently encountered individuals with medical and mental health disorders whose treatment was impacted by underlying substance use. I soon came to realize any attempts at (for example) managing hypertension in someone with cocaine use disorder, or managing schizophrenia in someone with ongoing cannabis use, were futile. Almost all of my patients receiving treatment for mental health disorders were dependent on tobacco or other substances, and most were interested in cessation. Through mentorship from addiction-trained residency faculty members, I was able to get a taste of the neurobiologic complexities of the disease, something that left me with a desire to develop a deeper understanding of the disease process. Witnessing strikingly positive outcomes with implementation of evidence-based treatment modalities further solidified my path to subspecialty training. Even during that early phase, because I expressed interest in managing these conditions, I was immediately put in a position to share and disseminate any newly acquired knowledge to other specialties as well as the public.
Dr. Ahmed: Could one manage addictive disorders with just general psychiatry training, and what are the differences between the different paths to certification that a resident could undertake?
Dr. Stanciu: Addictive disorders fall under the general umbrella of psychiatric care. Most individuals with these disorders exhibit some degree of mental illness. Medical school curriculum offers on average 2 hours of addiction-related didactics during 4 years. General psychiatry training programs vary significantly in the type of exposure to addiction—some residencies have an affiliated addiction fellowship, others have addiction-trained psychiatrists on staff, but most have none. Ultimately, there is great variability in the degree of comfort in working with individuals with addictive disorders post-residency. Being able to prescribe medications for the treatment of addictive disorders is very different from being familiar with the latest evidence-based recommendations and guidelines; the latter is
Addiction medicine is a fairly new route initially intended to allow non-psychiatric specialties access to addictive disorders training and certification. This is offered through the American Board of Preventive Medicine. There are currently 2 routes to sitting for the exam: through completion of a 1-year addiction medicine fellowship, or through the “practice pathway” still available until 2020. To be eligible for the latter, individuals must provide documentation of clinical experience post-residency, which is quantified as number of hours spent treating patients with addictions, plus any additional courses or training, and must be endorsed by a certified addictionologist.
Continue to: What was your fellowship experience link...
Dr. Ahmed: What was your fellowship experience like, and what should one consider when choosing a program?
Dr. Stanciu: I completed my fellowship training through Dartmouth’s Geisel School of Medicine, and the experience was tremendously valuable. In evaluating programs, one of the starting points is whether you have interest in a formal research track, because several programs include an optional year for that. Most programs tend to provide exposure to the Veterans Affairs system. The 1 year should provide you with broad exposure to all possible settings, all addictive disorders and patient populations, and all treatment modalities, in addition to rigorous didactic sessions. The ideal program should include rotations through methadone treatment centers, intensive outpatient programs, pain and interdisciplinary clinics, detoxification units, and centers for treatment of adolescent and young adults, as well as general medical settings and infectious disease clinics. There should also be close collaboration with psychologists who can provide training in evidence-based therapeutic modalities. During this year, it is vital to expand your knowledge of the ethical and legal regulations of treatment programs, state and federal requirements, insurance complexities, and requirements for privacy and protection of health information. The size of these programs can vary significantly, which may limit the one-on-one time devoted to your training, which is something I personally valued. My faculty was very supportive of academic endeavors, providing guidance, funding, and encouragement for attending and presenting at conferences, publishing papers, and other academic pursuits. Additionally, faculty should be current with emerging literature and willing to develop or implement new protocols and evaluate new pharmacologic therapies.
Dr. Ahmed: What are some of the career options and work settings for addiction psychiatrists?
Dr. Stanciu: Addiction psychiatrists work in numerous settings and various capacities. They can provide subspecialty care directly by seeing patients in outpatient clinics or inpatient addiction treatment centers for detoxification or rehabilitation, or they can work with dual-diagnosis populations in inpatient units. The expansion of telemedicine also holds promise for a role through virtual services. Indirectly, they can serve as a resource for expertise in the field through consultations in medical and psychiatric settings, or through policy making by working with the legislature and public health departments. Additionally, they can help create and integrate new knowledge into practice and educate future generations of physicians and the public.
Dr. Ahmed: What are some of the prevalent disorders and reasons for consultation that you encounter in your daily practice?
Continue to: Dr. Stanciu's response...
Dr. Stanciu: This can vary significantly depending on the setting, geographical region, and demographics of the population. My main non-administrative responsibilities are primarily consultative assisting clinicians at a 200-bed psychiatric hospital to address co-occurring addictive disorders. In short-term units, I am primarily asked to provide input on issues related to various toxidromes and withdrawals and the use of relapse prevention medications for alcohol use disorders as well as the use of buprenorphine or other forms of medication-assisted treatment. I work closely with licensed drug and alcohol counselors in implementing brief interventions as well as facilitating outpatient treatment referrals. Clinicians in longer term units may consult on issues related to pain management in individuals who have addictive disorders, the use of evidence-based pharmacologic agents to address cravings, or the use of relapse prevention medications for someone close to discharge. In terms of specific drugs of abuse, although opioids have recently received a tremendous amount of attention due to the visible costs through overdose deaths, the magnitude of individuals who are losing years of quality life through the use of alcohol and tobacco is significant, and hence this is a large portion of the conditions I encounter. I have also seen an abundance of marijuana use due to decreased perception of harm and increased access.
Dr. Ahmed: What are some of the challenges in working in this field?
Dr. Stanciu: Historically, funding for services has been an issue for clinicians working primarily with addictive disorders from the standpoint of reimbursement, patient access to evidence-based pharmacotherapy, and ability to collaborate with existing levels of care. In recent years, federal funding and policies have changed this, and after numerous studies have found increased cost savings, commercial insurances are providing coverage. A significant challenge also has been public stigma and dealing with a condition that is relapsing-remitting, poorly understood by other specialties and the general public, and sometimes labeled as a defect of character. Several efforts in education have lessened this; however, the impact still takes a toll on patients, who may feel ashamed of their disorder and sometimes are hesitant to take medications because they may believe that they are not “clean” if they depend on a medication for remission. Lastly, recent changes in marijuana policies make conversations about this drug quite difficult because patients often view it as harmless, and the laws governing legality and indications for therapeutic use are slightly ahead of the evidence.
Dr. Ahmed: In what direction do you believe the subspecialty is headed?
Dr. Stanciu: Currently, there are approximately 1,000 certified addiction psychiatrists for the 45 million Americans who have addictive disorders. Smoking and other forms of tobacco use pose significant threats to the 2020 Healthy People Tobacco Use objectives. There is a significant demand for addictionologists in both public and private sectors. As with mental health, demand exceeds supply, and efforts are underway to expand downstream education and increase access to specialists. Several federal laws have been put in place to remove barriers and expand access to care and have paved the way to a brighter future. One is the Affordable Care Act, which requires all insurances including Medicaid to cover the cost of treatment. Second is the Mental Health Parity and Addiction Equity Act, which ensures that the duration and dollar amount of coverage for substance use disorders is comparable to that of medical and surgical care.
Continue to: Another exciting possibility...
Another exciting possibility comes from the world of pharmaceuticals. Some medications have modest efficacy for addressing addictive disorders; however, historically these have been poorly utilized. Enhanced understanding of the neurobiology combined with increased insurance reimbursement should prompt research and new drug development. Some promising agents are already in the pipeline. Research into molecular and gene therapy as a way to better individualize care is also underway.
Going forward, I think we will also encounter a different landscape of drugs. Synthetic agents are emerging and increasing in popularity. Alarmingly, public perception of harm is decreasing. When it comes to cannabis use, I see a rise in pathologic use and the ramifications of this will have a drastic impact, particularly on patients with mental health conditions. We will need to undertake better efforts in monitoring, staying updated, and providing public education campaigns.
Dr. Ahmed: What advice do you have for trainees contemplating subspecialty training in addiction psychiatry?
Dr. Stanciu: I cannot emphasize enough the importance of mentorship. The American Academy of Addiction Psychiatry has a robust system for connecting mentees with mentors at all stages in their careers. This can be extremely helpful, especially in situations where the residency program does not have addiction-trained faculty or rotations through treatment centers. Joining such an organization also grants you access to resources that can help further your enthusiasm. Those interested should also familiarize themselves with currently available pharmacotherapeutic treatments that have evidence supporting efficacy for various addictive disorders, and begin to incorporate these medications into general mental health practice, along with attempts at motivational interviewing. For example, begin discussing naltrexone with patients who have comorbid alcohol use disorders and are interested in reducing their drinking; and varenicline with patients who smoke and are interested in quitting. The outcomes should automatically elicit an interest in pursuing further training in the field!

Shining a spotlight on physician well-being, more
Shining a spotlight on physician well-being
In “Physician impairment”1 (C
Dr. Farrell’s article does not acknowledge the rule of law. I do not understand why anyone wanting to help physicians would not want them to be aware of their employment rights under the ADA or advise that their ADA rights protect them from unwarranted medical inquiries and referrals to physician health programs (PHPs) or other entities for evaluation.
Dr. Farrell also claims that burnout, poor well-being, and mental disorders cause medical errors and low quality of patient care, but there are many reasons to doubt that this is the case.3,4 Readers should be wary of medical journal articles that cover topics related to physician well-being. Articles related to PHPs, in particular, typically paint an overly rosy picture of the effectiveness of these programs and fail to note important problematic aspects.5,6
Nicholas D. Lawson, MD
Georgetown University Law Center
Washington, DC
References
1. Lawson ND. Physician impairment. Current Psychiatry. 2017;16(10):8.
2. Farrell H. Physician impairment: a need for prevention. Current Psychiatry. 2018;17(9):41-44.
3. Lawson ND. Burnout is not associated with increased medical errors. Mayo Clin Proc. 2018;93(11):1683.
4. Tyssen R. What is the level of burnout that impairs functioning? J Intern Med. 2018;283(6):594-596.
5. Lawson ND, Boyd JW. Flaws in the methods and reporting of physician health program outcome studies. Gen Hosp Psychiatry. 2018;54:65-66.
6. Lawson ND, Boyd JW. Physician health program outcome data should be viewed with caution. Judges J. 2018;57(4):36.
The author responds
I thank Dr. Lawson for his interest in my article. In this extremely challenging work that we do as psychiatrists, which can sometimes be quite isolating, there is a long continuum of experience, reward, and challenge. Dr. Lawson’s research and publication on the topic of physician’s health issues are very much respected and appreciated. In fact, I see no conflict between Dr. Lawson’s letter and my 2018 column on the prevention of impairment.
Given the extensive continuum of our work, my article on physician’s health issues sought to shine a bright spotlight solely on the topic of prevention. As colleagues, there is significant value in supporting rather than reporting one another. Awareness of and sensitivity to physician vulnerability, early detection, and prevention will hopefully continue to gain traction in the future.
By putting the focus on proactively helping colleagues, my hope is that my article will spark an ongoing conversation about how we can work collaboratively to make well-being a priority.
Dr. Lawson’s thoughtful letter is much appreciated because it continues the discussion by shining a spotlight further down the continuum. He focuses on the aftermath of impairment and aptly points out the complications in reporting, confusion about duty, and the protections provided by the ADA. Also, I support Dr. Lawson’s cautions regarding PHPs—all the more reason to join together in shifting the dialogue from management of a crisis to prevention of it.
Helen M. Farrell, MD
Lecturer
Harvard Medical School
Psychiatrist
Beth Israel Deaconess Medical Center
Boston, Massachusetts
Continue to: Neuropolitics
Neuropolitics: Psychiatrists’ responsibility
Regarding Dr. Nasrallah’s editorial “Neuropolitics in the age of extremism: Brain regions involved in hatred” (C
I agree with Dr. Nasrallah that “even the most skillful psychiatrists” cannot “repair a nation caught up in poisonous emotional turmoil”—at least not by employing clinical skills alone. But that doesn’t mean we shouldn’t try, and the American Psychiatric Association (APA) ethics code (Sections 1.2, 3, and 7) compels us to speak out when our patients or the public are being harmed by public policy.1 We are much more likely to have an impact when we speak with one voice, as is the case with professional medical organizations such as the APA. In December 2018, APA President Dr. Altha J. Stewart issued a call to action addressing “the current climate of hateful and divisive rhetoric that leads to senseless violence and tragic loss of life,” stating “… we members must speak out, use our specialized training and expertise for the public’s benefit, and apply it to not only healing, but also preventing psychological trauma and senseless tragedies.”2
James L. Fleming, MD
Psychiatric Medical Care
Lee’s Summit, Missouri
References
1. American Psychiatric Association. The principles of medical ethics with annotations especially applicable to psychiatry, 2013 edition. https://www.psychiatry.org/psychiatrists/practice/ethics. Published 2013. Accessed February 5, 2019.
2. Stewart A, Pozios, V. Forget about staying in our lane: let’s connect the dots. American Psychiatric Association Publishing. https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2018.12a20. Published December 3, 2018. Accessed February 3, 2019.
Shining a spotlight on physician well-being
In “Physician impairment”1 (C
Dr. Farrell’s article does not acknowledge the rule of law. I do not understand why anyone wanting to help physicians would not want them to be aware of their employment rights under the ADA or advise that their ADA rights protect them from unwarranted medical inquiries and referrals to physician health programs (PHPs) or other entities for evaluation.
Dr. Farrell also claims that burnout, poor well-being, and mental disorders cause medical errors and low quality of patient care, but there are many reasons to doubt that this is the case.3,4 Readers should be wary of medical journal articles that cover topics related to physician well-being. Articles related to PHPs, in particular, typically paint an overly rosy picture of the effectiveness of these programs and fail to note important problematic aspects.5,6
Nicholas D. Lawson, MD
Georgetown University Law Center
Washington, DC
References
1. Lawson ND. Physician impairment. Current Psychiatry. 2017;16(10):8.
2. Farrell H. Physician impairment: a need for prevention. Current Psychiatry. 2018;17(9):41-44.
3. Lawson ND. Burnout is not associated with increased medical errors. Mayo Clin Proc. 2018;93(11):1683.
4. Tyssen R. What is the level of burnout that impairs functioning? J Intern Med. 2018;283(6):594-596.
5. Lawson ND, Boyd JW. Flaws in the methods and reporting of physician health program outcome studies. Gen Hosp Psychiatry. 2018;54:65-66.
6. Lawson ND, Boyd JW. Physician health program outcome data should be viewed with caution. Judges J. 2018;57(4):36.
The author responds
I thank Dr. Lawson for his interest in my article. In this extremely challenging work that we do as psychiatrists, which can sometimes be quite isolating, there is a long continuum of experience, reward, and challenge. Dr. Lawson’s research and publication on the topic of physician’s health issues are very much respected and appreciated. In fact, I see no conflict between Dr. Lawson’s letter and my 2018 column on the prevention of impairment.
Given the extensive continuum of our work, my article on physician’s health issues sought to shine a bright spotlight solely on the topic of prevention. As colleagues, there is significant value in supporting rather than reporting one another. Awareness of and sensitivity to physician vulnerability, early detection, and prevention will hopefully continue to gain traction in the future.
By putting the focus on proactively helping colleagues, my hope is that my article will spark an ongoing conversation about how we can work collaboratively to make well-being a priority.
Dr. Lawson’s thoughtful letter is much appreciated because it continues the discussion by shining a spotlight further down the continuum. He focuses on the aftermath of impairment and aptly points out the complications in reporting, confusion about duty, and the protections provided by the ADA. Also, I support Dr. Lawson’s cautions regarding PHPs—all the more reason to join together in shifting the dialogue from management of a crisis to prevention of it.
Helen M. Farrell, MD
Lecturer
Harvard Medical School
Psychiatrist
Beth Israel Deaconess Medical Center
Boston, Massachusetts
Continue to: Neuropolitics
Neuropolitics: Psychiatrists’ responsibility
Regarding Dr. Nasrallah’s editorial “Neuropolitics in the age of extremism: Brain regions involved in hatred” (C
I agree with Dr. Nasrallah that “even the most skillful psychiatrists” cannot “repair a nation caught up in poisonous emotional turmoil”—at least not by employing clinical skills alone. But that doesn’t mean we shouldn’t try, and the American Psychiatric Association (APA) ethics code (Sections 1.2, 3, and 7) compels us to speak out when our patients or the public are being harmed by public policy.1 We are much more likely to have an impact when we speak with one voice, as is the case with professional medical organizations such as the APA. In December 2018, APA President Dr. Altha J. Stewart issued a call to action addressing “the current climate of hateful and divisive rhetoric that leads to senseless violence and tragic loss of life,” stating “… we members must speak out, use our specialized training and expertise for the public’s benefit, and apply it to not only healing, but also preventing psychological trauma and senseless tragedies.”2
James L. Fleming, MD
Psychiatric Medical Care
Lee’s Summit, Missouri
References
1. American Psychiatric Association. The principles of medical ethics with annotations especially applicable to psychiatry, 2013 edition. https://www.psychiatry.org/psychiatrists/practice/ethics. Published 2013. Accessed February 5, 2019.
2. Stewart A, Pozios, V. Forget about staying in our lane: let’s connect the dots. American Psychiatric Association Publishing. https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2018.12a20. Published December 3, 2018. Accessed February 3, 2019.
Shining a spotlight on physician well-being
In “Physician impairment”1 (C
Dr. Farrell’s article does not acknowledge the rule of law. I do not understand why anyone wanting to help physicians would not want them to be aware of their employment rights under the ADA or advise that their ADA rights protect them from unwarranted medical inquiries and referrals to physician health programs (PHPs) or other entities for evaluation.
Dr. Farrell also claims that burnout, poor well-being, and mental disorders cause medical errors and low quality of patient care, but there are many reasons to doubt that this is the case.3,4 Readers should be wary of medical journal articles that cover topics related to physician well-being. Articles related to PHPs, in particular, typically paint an overly rosy picture of the effectiveness of these programs and fail to note important problematic aspects.5,6
Nicholas D. Lawson, MD
Georgetown University Law Center
Washington, DC
References
1. Lawson ND. Physician impairment. Current Psychiatry. 2017;16(10):8.
2. Farrell H. Physician impairment: a need for prevention. Current Psychiatry. 2018;17(9):41-44.
3. Lawson ND. Burnout is not associated with increased medical errors. Mayo Clin Proc. 2018;93(11):1683.
4. Tyssen R. What is the level of burnout that impairs functioning? J Intern Med. 2018;283(6):594-596.
5. Lawson ND, Boyd JW. Flaws in the methods and reporting of physician health program outcome studies. Gen Hosp Psychiatry. 2018;54:65-66.
6. Lawson ND, Boyd JW. Physician health program outcome data should be viewed with caution. Judges J. 2018;57(4):36.
The author responds
I thank Dr. Lawson for his interest in my article. In this extremely challenging work that we do as psychiatrists, which can sometimes be quite isolating, there is a long continuum of experience, reward, and challenge. Dr. Lawson’s research and publication on the topic of physician’s health issues are very much respected and appreciated. In fact, I see no conflict between Dr. Lawson’s letter and my 2018 column on the prevention of impairment.
Given the extensive continuum of our work, my article on physician’s health issues sought to shine a bright spotlight solely on the topic of prevention. As colleagues, there is significant value in supporting rather than reporting one another. Awareness of and sensitivity to physician vulnerability, early detection, and prevention will hopefully continue to gain traction in the future.
By putting the focus on proactively helping colleagues, my hope is that my article will spark an ongoing conversation about how we can work collaboratively to make well-being a priority.
Dr. Lawson’s thoughtful letter is much appreciated because it continues the discussion by shining a spotlight further down the continuum. He focuses on the aftermath of impairment and aptly points out the complications in reporting, confusion about duty, and the protections provided by the ADA. Also, I support Dr. Lawson’s cautions regarding PHPs—all the more reason to join together in shifting the dialogue from management of a crisis to prevention of it.
Helen M. Farrell, MD
Lecturer
Harvard Medical School
Psychiatrist
Beth Israel Deaconess Medical Center
Boston, Massachusetts
Continue to: Neuropolitics
Neuropolitics: Psychiatrists’ responsibility
Regarding Dr. Nasrallah’s editorial “Neuropolitics in the age of extremism: Brain regions involved in hatred” (C
I agree with Dr. Nasrallah that “even the most skillful psychiatrists” cannot “repair a nation caught up in poisonous emotional turmoil”—at least not by employing clinical skills alone. But that doesn’t mean we shouldn’t try, and the American Psychiatric Association (APA) ethics code (Sections 1.2, 3, and 7) compels us to speak out when our patients or the public are being harmed by public policy.1 We are much more likely to have an impact when we speak with one voice, as is the case with professional medical organizations such as the APA. In December 2018, APA President Dr. Altha J. Stewart issued a call to action addressing “the current climate of hateful and divisive rhetoric that leads to senseless violence and tragic loss of life,” stating “… we members must speak out, use our specialized training and expertise for the public’s benefit, and apply it to not only healing, but also preventing psychological trauma and senseless tragedies.”2
James L. Fleming, MD
Psychiatric Medical Care
Lee’s Summit, Missouri
References
1. American Psychiatric Association. The principles of medical ethics with annotations especially applicable to psychiatry, 2013 edition. https://www.psychiatry.org/psychiatrists/practice/ethics. Published 2013. Accessed February 5, 2019.
2. Stewart A, Pozios, V. Forget about staying in our lane: let’s connect the dots. American Psychiatric Association Publishing. https://psychnews.psychiatryonline.org/doi/10.1176/appi.pn.2018.12a20. Published December 3, 2018. Accessed February 3, 2019.