User login
Psoriasis Journal Scan: June 2019
Management of psoriasis as a systemic disease: What is the evidence?
Korman NJ. Br J Dermatol. 2019 Jun 21.
This narrative review explores the pathophysiological relationship between psoriasis and its common comorbidities and discusses the need for new treatment paradigms that include strategies to reduce systemic inflammation in patients with moderate-to-severe psoriasis.
Managing Psoriasis in Patients with HBV or HCV Infection: Practical Considerations.
Piaserico S, Messina F, Russo FP. Am J Clin Dermatol. 2019 Jun 20.
It has been estimated that two billion individuals are infected with HBV worldwide and approximately 240 million have chronic HBV infection. Moreover, there are approximately 71 million individuals with chronic HCV infection worldwide, with a high percentage of them unaware of being infected. As patients with HBV and HCV infections are excluded from controlled clinical trials investigating new drugs, data regarding their safety in patients with psoriasis are based almost exclusively on case reports and small retrospective cohort studies and need to be constantly updated.
Effects of Online Care on Functional and Psychological Outcomes in Patients with Psoriasis: A Randomized Controlled Trial.
Young PM, Chen AY, Ford AR, Cheng MY, Lane CJ, Armstrong AW. J Am Acad Dermatol. 2019 Jun 5.
The impact of online care on patients' functional and psychological outcomes is critical to determine yet still unknown. This 12-month randomized controlled equivalency trial evaluated how a novel online health model that facilitates physician-patient collaboration compares with in-person care for improving psoriasis patients' functional status and mental health.
Feasibility and Utility of the Psoriasis Symptom Inventory (PSI) in Clinical Care Settings: A Study from the International Psoriasis Council.
Strober B, van de Kerkhof PCM, Callis Duffin K, et al. Am J Clin Dermatol. 2019 Jun 21.
The Psoriasis Symptom Inventory (PSI) is a patient-reported outcome measure designed to assess psoriasis signs and symptoms. The aim of the study was to assess the usefulness of the PSI in enhancing patient care in the clinical setting. Eight dermatology clinics in six countries enrolled adults representing the full spectrum of psoriasis severity who regularly received care at the clinic. Key benefits of PSI discussions included the following: new information regarding symptom location and severity for physicians; prompting of quality-of-life discussions; better understanding of patient treatment priorities; change in treatment regimens to target specific symptoms or areas; and improvement of patient-physician relationship.
Socioeconomic Costs and Health Inequalities from Psoriasis: A Cohort Study.
Thomsen SF, Skov L, Dodge R, Hedegaard MS, Kjellberg J. Dermatology. 2019 Jun 25:1-8.
Incentives for health care management based on patient-related outcomes and value (IMPROVE) in psoriasis and psoriatic arthritis is a project aimed at assisting movement from activity-based to outcome-based health care management. One of the key objectives in IMPROVE is to describe the disease-associated socioeconomic burden of psoriasis. The IMPROVE study was a retrospective analysis of patients with a hospital diagnosis of psoriasis identified from the Danish National Patient Registry.
Management of psoriasis as a systemic disease: What is the evidence?
Korman NJ. Br J Dermatol. 2019 Jun 21.
This narrative review explores the pathophysiological relationship between psoriasis and its common comorbidities and discusses the need for new treatment paradigms that include strategies to reduce systemic inflammation in patients with moderate-to-severe psoriasis.
Managing Psoriasis in Patients with HBV or HCV Infection: Practical Considerations.
Piaserico S, Messina F, Russo FP. Am J Clin Dermatol. 2019 Jun 20.
It has been estimated that two billion individuals are infected with HBV worldwide and approximately 240 million have chronic HBV infection. Moreover, there are approximately 71 million individuals with chronic HCV infection worldwide, with a high percentage of them unaware of being infected. As patients with HBV and HCV infections are excluded from controlled clinical trials investigating new drugs, data regarding their safety in patients with psoriasis are based almost exclusively on case reports and small retrospective cohort studies and need to be constantly updated.
Effects of Online Care on Functional and Psychological Outcomes in Patients with Psoriasis: A Randomized Controlled Trial.
Young PM, Chen AY, Ford AR, Cheng MY, Lane CJ, Armstrong AW. J Am Acad Dermatol. 2019 Jun 5.
The impact of online care on patients' functional and psychological outcomes is critical to determine yet still unknown. This 12-month randomized controlled equivalency trial evaluated how a novel online health model that facilitates physician-patient collaboration compares with in-person care for improving psoriasis patients' functional status and mental health.
Feasibility and Utility of the Psoriasis Symptom Inventory (PSI) in Clinical Care Settings: A Study from the International Psoriasis Council.
Strober B, van de Kerkhof PCM, Callis Duffin K, et al. Am J Clin Dermatol. 2019 Jun 21.
The Psoriasis Symptom Inventory (PSI) is a patient-reported outcome measure designed to assess psoriasis signs and symptoms. The aim of the study was to assess the usefulness of the PSI in enhancing patient care in the clinical setting. Eight dermatology clinics in six countries enrolled adults representing the full spectrum of psoriasis severity who regularly received care at the clinic. Key benefits of PSI discussions included the following: new information regarding symptom location and severity for physicians; prompting of quality-of-life discussions; better understanding of patient treatment priorities; change in treatment regimens to target specific symptoms or areas; and improvement of patient-physician relationship.
Socioeconomic Costs and Health Inequalities from Psoriasis: A Cohort Study.
Thomsen SF, Skov L, Dodge R, Hedegaard MS, Kjellberg J. Dermatology. 2019 Jun 25:1-8.
Incentives for health care management based on patient-related outcomes and value (IMPROVE) in psoriasis and psoriatic arthritis is a project aimed at assisting movement from activity-based to outcome-based health care management. One of the key objectives in IMPROVE is to describe the disease-associated socioeconomic burden of psoriasis. The IMPROVE study was a retrospective analysis of patients with a hospital diagnosis of psoriasis identified from the Danish National Patient Registry.
Management of psoriasis as a systemic disease: What is the evidence?
Korman NJ. Br J Dermatol. 2019 Jun 21.
This narrative review explores the pathophysiological relationship between psoriasis and its common comorbidities and discusses the need for new treatment paradigms that include strategies to reduce systemic inflammation in patients with moderate-to-severe psoriasis.
Managing Psoriasis in Patients with HBV or HCV Infection: Practical Considerations.
Piaserico S, Messina F, Russo FP. Am J Clin Dermatol. 2019 Jun 20.
It has been estimated that two billion individuals are infected with HBV worldwide and approximately 240 million have chronic HBV infection. Moreover, there are approximately 71 million individuals with chronic HCV infection worldwide, with a high percentage of them unaware of being infected. As patients with HBV and HCV infections are excluded from controlled clinical trials investigating new drugs, data regarding their safety in patients with psoriasis are based almost exclusively on case reports and small retrospective cohort studies and need to be constantly updated.
Effects of Online Care on Functional and Psychological Outcomes in Patients with Psoriasis: A Randomized Controlled Trial.
Young PM, Chen AY, Ford AR, Cheng MY, Lane CJ, Armstrong AW. J Am Acad Dermatol. 2019 Jun 5.
The impact of online care on patients' functional and psychological outcomes is critical to determine yet still unknown. This 12-month randomized controlled equivalency trial evaluated how a novel online health model that facilitates physician-patient collaboration compares with in-person care for improving psoriasis patients' functional status and mental health.
Feasibility and Utility of the Psoriasis Symptom Inventory (PSI) in Clinical Care Settings: A Study from the International Psoriasis Council.
Strober B, van de Kerkhof PCM, Callis Duffin K, et al. Am J Clin Dermatol. 2019 Jun 21.
The Psoriasis Symptom Inventory (PSI) is a patient-reported outcome measure designed to assess psoriasis signs and symptoms. The aim of the study was to assess the usefulness of the PSI in enhancing patient care in the clinical setting. Eight dermatology clinics in six countries enrolled adults representing the full spectrum of psoriasis severity who regularly received care at the clinic. Key benefits of PSI discussions included the following: new information regarding symptom location and severity for physicians; prompting of quality-of-life discussions; better understanding of patient treatment priorities; change in treatment regimens to target specific symptoms or areas; and improvement of patient-physician relationship.
Socioeconomic Costs and Health Inequalities from Psoriasis: A Cohort Study.
Thomsen SF, Skov L, Dodge R, Hedegaard MS, Kjellberg J. Dermatology. 2019 Jun 25:1-8.
Incentives for health care management based on patient-related outcomes and value (IMPROVE) in psoriasis and psoriatic arthritis is a project aimed at assisting movement from activity-based to outcome-based health care management. One of the key objectives in IMPROVE is to describe the disease-associated socioeconomic burden of psoriasis. The IMPROVE study was a retrospective analysis of patients with a hospital diagnosis of psoriasis identified from the Danish National Patient Registry.
Psoriatic Arthritis Journal Scan: June 2019
Treating Psoriatic Arthritis to Target: Defining Psoriatic Arthritis Disease Activity Score (PASDAS) That Reflects State Of Minimal Disease Activity (MDA).
Perruccio AV, Got M, Li S, Ye Y, Gladman DD, Chandran V. J Rheumatol. 2019 Jun 15.
PsA Disease Activity Score (PASDAS) is a composite disease activity measure (range 0-10) for psoriatic arthritis. The study aimed to validate a cutoff value of PASDAS that defines minimal disease activity state, as well as validate previously defined PASDAS cutoffs for low and high disease activity.
Evaluating current definitions of low disease activity in psoriatic arthritis using ultrasound.
Bosch P, Husic R, Ficjan A, et al. Rheumatology (Oxford). 2019 Jun 14.
The aim of the study was to evaluate low disease activity (LDA) cut-offs in psoriatic arthritis (PsA) using ultrasound. Eighty-three PsA patients underwent clinical and ultrasound examinations at two visits. Pain and pain-related items are the main reason why PsA patients without signs of ultrasound inflammation are classified with higher disease activity.
A Threshold of Meaning for Work Disability Improvement in Psoriatic Arthritis Measured by the Work Productivity and Activity Impairment Questionnaire.
Tillett W, Lin CY, Zbrozek A, Sprabery AT, Birt J. Rheumatol Ther. 2019 Jun 1.
The Work Productivity and Activity Impairment Specific Health Problem Questionnaire (WPAI:SHP) is used to assess the impact of an intervention on work productivity in patients with psoriatic arthritis (PsA). Unfortunately, studies reporting changes or improvements in domains of WPAI:SHP by patients with PsA have a limited threshold of meaning due to the absence of published minimal clinically important differences (MCIDs). The objective of the study was to determine the MCIDs for improvement in WPAI:SHP in patients with active PsA.
Measuring Psoriatic Arthritis Symptoms, A Core Domain in Psoriasis Clinical Trials.
Perez-Chada LM, Gottlieb AB, Cohen J, et al. J Am Acad Dermatol. 2019 Jun 1.
The International Dermatology Outcome Measures (IDEOM) established a set of core domains to be measured in all psoriasis trials. This set indicates that symptoms of psoriatic arthritis (PsA) should be measured in all psoriasis studies. The objective of the study was to identify the approach to PsA screening, and the most appropriate outcome measure for capturing PsA symptoms. The overwhelming majority of expert stakeholders agreed that all psoriasis trial subjects should be screened for PsA with subsequent measurement of PsA symptoms with use of the PsAID9 (with the RAPID3 as an acceptable alternative measure).
The Genetics of Psoriasis and Psoriatic Arthritis.
O'Rielly DD, Jani M, Rahman P, Elder JT. J Rheumatol Suppl. 2019 Jun;95:46-50.
Psoriatic arthritis (PsA) is an inflammatory arthritis that manifests in 20-30% of patients diagnosed with psoriasis. Epidemiologic studies suggest a substantial genetic contribution to PsA. There is a strong need for genome-wide association studies on patients with PsA, including PsA-weighted or specific variants. Genomics and serological factors may also predict treatment response in tumor necrosis factor inhibitors (TNFi) in PsA, and genetics may play a role in treatment response to TNFi. Collaborations through the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) are essential to increase study population size, which will enhance the ability to detect the genetic variants that create a predisposition to psoriatic disease and to predict response to biological therapy.
Treating Psoriatic Arthritis to Target: Defining Psoriatic Arthritis Disease Activity Score (PASDAS) That Reflects State Of Minimal Disease Activity (MDA).
Perruccio AV, Got M, Li S, Ye Y, Gladman DD, Chandran V. J Rheumatol. 2019 Jun 15.
PsA Disease Activity Score (PASDAS) is a composite disease activity measure (range 0-10) for psoriatic arthritis. The study aimed to validate a cutoff value of PASDAS that defines minimal disease activity state, as well as validate previously defined PASDAS cutoffs for low and high disease activity.
Evaluating current definitions of low disease activity in psoriatic arthritis using ultrasound.
Bosch P, Husic R, Ficjan A, et al. Rheumatology (Oxford). 2019 Jun 14.
The aim of the study was to evaluate low disease activity (LDA) cut-offs in psoriatic arthritis (PsA) using ultrasound. Eighty-three PsA patients underwent clinical and ultrasound examinations at two visits. Pain and pain-related items are the main reason why PsA patients without signs of ultrasound inflammation are classified with higher disease activity.
A Threshold of Meaning for Work Disability Improvement in Psoriatic Arthritis Measured by the Work Productivity and Activity Impairment Questionnaire.
Tillett W, Lin CY, Zbrozek A, Sprabery AT, Birt J. Rheumatol Ther. 2019 Jun 1.
The Work Productivity and Activity Impairment Specific Health Problem Questionnaire (WPAI:SHP) is used to assess the impact of an intervention on work productivity in patients with psoriatic arthritis (PsA). Unfortunately, studies reporting changes or improvements in domains of WPAI:SHP by patients with PsA have a limited threshold of meaning due to the absence of published minimal clinically important differences (MCIDs). The objective of the study was to determine the MCIDs for improvement in WPAI:SHP in patients with active PsA.
Measuring Psoriatic Arthritis Symptoms, A Core Domain in Psoriasis Clinical Trials.
Perez-Chada LM, Gottlieb AB, Cohen J, et al. J Am Acad Dermatol. 2019 Jun 1.
The International Dermatology Outcome Measures (IDEOM) established a set of core domains to be measured in all psoriasis trials. This set indicates that symptoms of psoriatic arthritis (PsA) should be measured in all psoriasis studies. The objective of the study was to identify the approach to PsA screening, and the most appropriate outcome measure for capturing PsA symptoms. The overwhelming majority of expert stakeholders agreed that all psoriasis trial subjects should be screened for PsA with subsequent measurement of PsA symptoms with use of the PsAID9 (with the RAPID3 as an acceptable alternative measure).
The Genetics of Psoriasis and Psoriatic Arthritis.
O'Rielly DD, Jani M, Rahman P, Elder JT. J Rheumatol Suppl. 2019 Jun;95:46-50.
Psoriatic arthritis (PsA) is an inflammatory arthritis that manifests in 20-30% of patients diagnosed with psoriasis. Epidemiologic studies suggest a substantial genetic contribution to PsA. There is a strong need for genome-wide association studies on patients with PsA, including PsA-weighted or specific variants. Genomics and serological factors may also predict treatment response in tumor necrosis factor inhibitors (TNFi) in PsA, and genetics may play a role in treatment response to TNFi. Collaborations through the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) are essential to increase study population size, which will enhance the ability to detect the genetic variants that create a predisposition to psoriatic disease and to predict response to biological therapy.
Treating Psoriatic Arthritis to Target: Defining Psoriatic Arthritis Disease Activity Score (PASDAS) That Reflects State Of Minimal Disease Activity (MDA).
Perruccio AV, Got M, Li S, Ye Y, Gladman DD, Chandran V. J Rheumatol. 2019 Jun 15.
PsA Disease Activity Score (PASDAS) is a composite disease activity measure (range 0-10) for psoriatic arthritis. The study aimed to validate a cutoff value of PASDAS that defines minimal disease activity state, as well as validate previously defined PASDAS cutoffs for low and high disease activity.
Evaluating current definitions of low disease activity in psoriatic arthritis using ultrasound.
Bosch P, Husic R, Ficjan A, et al. Rheumatology (Oxford). 2019 Jun 14.
The aim of the study was to evaluate low disease activity (LDA) cut-offs in psoriatic arthritis (PsA) using ultrasound. Eighty-three PsA patients underwent clinical and ultrasound examinations at two visits. Pain and pain-related items are the main reason why PsA patients without signs of ultrasound inflammation are classified with higher disease activity.
A Threshold of Meaning for Work Disability Improvement in Psoriatic Arthritis Measured by the Work Productivity and Activity Impairment Questionnaire.
Tillett W, Lin CY, Zbrozek A, Sprabery AT, Birt J. Rheumatol Ther. 2019 Jun 1.
The Work Productivity and Activity Impairment Specific Health Problem Questionnaire (WPAI:SHP) is used to assess the impact of an intervention on work productivity in patients with psoriatic arthritis (PsA). Unfortunately, studies reporting changes or improvements in domains of WPAI:SHP by patients with PsA have a limited threshold of meaning due to the absence of published minimal clinically important differences (MCIDs). The objective of the study was to determine the MCIDs for improvement in WPAI:SHP in patients with active PsA.
Measuring Psoriatic Arthritis Symptoms, A Core Domain in Psoriasis Clinical Trials.
Perez-Chada LM, Gottlieb AB, Cohen J, et al. J Am Acad Dermatol. 2019 Jun 1.
The International Dermatology Outcome Measures (IDEOM) established a set of core domains to be measured in all psoriasis trials. This set indicates that symptoms of psoriatic arthritis (PsA) should be measured in all psoriasis studies. The objective of the study was to identify the approach to PsA screening, and the most appropriate outcome measure for capturing PsA symptoms. The overwhelming majority of expert stakeholders agreed that all psoriasis trial subjects should be screened for PsA with subsequent measurement of PsA symptoms with use of the PsAID9 (with the RAPID3 as an acceptable alternative measure).
The Genetics of Psoriasis and Psoriatic Arthritis.
O'Rielly DD, Jani M, Rahman P, Elder JT. J Rheumatol Suppl. 2019 Jun;95:46-50.
Psoriatic arthritis (PsA) is an inflammatory arthritis that manifests in 20-30% of patients diagnosed with psoriasis. Epidemiologic studies suggest a substantial genetic contribution to PsA. There is a strong need for genome-wide association studies on patients with PsA, including PsA-weighted or specific variants. Genomics and serological factors may also predict treatment response in tumor necrosis factor inhibitors (TNFi) in PsA, and genetics may play a role in treatment response to TNFi. Collaborations through the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) are essential to increase study population size, which will enhance the ability to detect the genetic variants that create a predisposition to psoriatic disease and to predict response to biological therapy.
Infections linked with transition to psoriatic arthritis
MADRID – Several novel risk associations with psoriasis progression were found to differ by sex, and collectively appeared to implicate infections and the “stress response” as a trigger of psoriatic arthritis.

The findings come from a risk factor analysis of a U.S. claims database of more than 200,000 adults with psoriasis including more than 4,000 patients who progressed to psoriatic arthritis during nearly 6 years of follow-up.
The new analysis confirmed several previously described risk associations linked with progression to psoriatic arthritis (PsA) that have roughly equal impact on both women and men: fatigue, obesity, and depression, Alexis Ogdie, MD, said at the European Congress of Rheumatology. The new findings also showed several novel, sex-specific associations. In women, these associations included salmonella infection, sepsis, and uveitis; in men, they included gangrene, encephalitis, and hidradenitis suppurativa.
The links with various infections were generally rare; they showed strong nominal associations in multivariate analyses but with wide confidence limits. The findings suggest that events that induce major stress responses, such as infections, often preceded the progression of psoriasis to a diagnosis of PsA, said Dr. Ogdie, director of the psoriatic arthritis clinic at the University of Pennsylvania in Philadelphia. Other, noninfectious clinical features that significantly linked with PsA development but at a lower magnitude included anemia and diabetes in women, and irritable bowel syndrome and venous thromboembolism in men.
Dr. Ogdie cautioned that the findings were preliminary and need confirmation in different data sets, as well as in additional subgroup analyses of the data used in the current analysis, taken from the electronic medical records of 215,386 U.S. residents diagnosed with psoriasis in the Optum medical-claims database for 2006-2017.
The analysis focused on patients who received a second diagnostic code in their EMR for psoriasis during the 12 months after the index psoriasis entry. The identified group averaged 50 years old; 55% of the psoriasis patients were women, and 86% were white.
During the year after their first diagnostic-code entry for psoriasis, 4.6% of the patients received a biological drug and 4.2% received an oral drug for their psoriasis. During 5.6 years of follow-up, 4,288 patients (2%) developed PsA, a rate of 3.5 cases/1,000 patient-years. Dr. Ogdie noted that prior studies have documented the challenge of diagnosing PsA in patients with psoriasis, so this may be a conservative estimate of the progression rate.
The researchers assessed possible linkage with PsA progression for more than 250 different entries in the EMR, but the analysis was limited by the absence of measures of rheumatoid susceptibility, such as immunologic markers, which were not included in the EMR. In multivariate analysis of the full cohort, fatigue at baseline was linked with a 77% higher rate of progression to PsA, obesity was linked with a 48% higher rate, and depression with a 29% higher rate of progression when compared with psoriasis patients without each of these factors. All three differences were statistically significant. Dr. Ogdie cited an article she recently coauthored that detailed the background to this approach in studying the etiology of PsA (Nat Rev Rheumatol. 2019 March;15:153-66).
This is the first study to report sex-linked differences in clinical measures that link with progression to PsA, Dr. Ogdie noted. In women, salmonella infection linked with a 9-fold higher rate of PsA development compared with women with psoriasis without salmonella infection, women with uveitis had a 2.9-fold higher rate of PsA development, and those with sepsis had a 2.4-fold increased rate of PsA. Among men, those with gangrene, encephalitis, or hidradenitis suppurativa each had a greater than 4-fold higher rate of developing PsA, and men with osteomyelitis had a 2.7-fold increase.
All these between-group differences were statistically significant. But because each of these was a relatively rare event, the confidence intervals around these point estimates were wide. For example, in women with salmonella infection from a statistical standpoint the possible range of increased risk could be anywhere from 1.3 to 66. The analysis identified among women and men several additional sex-specific risk associations that were statistically significant but with smaller point estimates.
SOURCE: Ogdie A et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):131-2. Abstract OP0115. doi: 10.1136/annrheumdis-2019-eular.4390.
MADRID – Several novel risk associations with psoriasis progression were found to differ by sex, and collectively appeared to implicate infections and the “stress response” as a trigger of psoriatic arthritis.
The findings come from a risk factor analysis of a U.S. claims database of more than 200,000 adults with psoriasis including more than 4,000 patients who progressed to psoriatic arthritis during nearly 6 years of follow-up.
The new analysis confirmed several previously described risk associations linked with progression to psoriatic arthritis (PsA) that have roughly equal impact on both women and men: fatigue, obesity, and depression, Alexis Ogdie, MD, said at the European Congress of Rheumatology. The new findings also showed several novel, sex-specific associations. In women, these associations included salmonella infection, sepsis, and uveitis; in men, they included gangrene, encephalitis, and hidradenitis suppurativa.
The links with various infections were generally rare; they showed strong nominal associations in multivariate analyses but with wide confidence limits. The findings suggest that events that induce major stress responses, such as infections, often preceded the progression of psoriasis to a diagnosis of PsA, said Dr. Ogdie, director of the psoriatic arthritis clinic at the University of Pennsylvania in Philadelphia. Other, noninfectious clinical features that significantly linked with PsA development but at a lower magnitude included anemia and diabetes in women, and irritable bowel syndrome and venous thromboembolism in men.
Dr. Ogdie cautioned that the findings were preliminary and need confirmation in different data sets, as well as in additional subgroup analyses of the data used in the current analysis, taken from the electronic medical records of 215,386 U.S. residents diagnosed with psoriasis in the Optum medical-claims database for 2006-2017.
The analysis focused on patients who received a second diagnostic code in their EMR for psoriasis during the 12 months after the index psoriasis entry. The identified group averaged 50 years old; 55% of the psoriasis patients were women, and 86% were white.
During the year after their first diagnostic-code entry for psoriasis, 4.6% of the patients received a biological drug and 4.2% received an oral drug for their psoriasis. During 5.6 years of follow-up, 4,288 patients (2%) developed PsA, a rate of 3.5 cases/1,000 patient-years. Dr. Ogdie noted that prior studies have documented the challenge of diagnosing PsA in patients with psoriasis, so this may be a conservative estimate of the progression rate.
The researchers assessed possible linkage with PsA progression for more than 250 different entries in the EMR, but the analysis was limited by the absence of measures of rheumatoid susceptibility, such as immunologic markers, which were not included in the EMR. In multivariate analysis of the full cohort, fatigue at baseline was linked with a 77% higher rate of progression to PsA, obesity was linked with a 48% higher rate, and depression with a 29% higher rate of progression when compared with psoriasis patients without each of these factors. All three differences were statistically significant. Dr. Ogdie cited an article she recently coauthored that detailed the background to this approach in studying the etiology of PsA (Nat Rev Rheumatol. 2019 March;15:153-66).
This is the first study to report sex-linked differences in clinical measures that link with progression to PsA, Dr. Ogdie noted. In women, salmonella infection linked with a 9-fold higher rate of PsA development compared with women with psoriasis without salmonella infection, women with uveitis had a 2.9-fold higher rate of PsA development, and those with sepsis had a 2.4-fold increased rate of PsA. Among men, those with gangrene, encephalitis, or hidradenitis suppurativa each had a greater than 4-fold higher rate of developing PsA, and men with osteomyelitis had a 2.7-fold increase.
All these between-group differences were statistically significant. But because each of these was a relatively rare event, the confidence intervals around these point estimates were wide. For example, in women with salmonella infection from a statistical standpoint the possible range of increased risk could be anywhere from 1.3 to 66. The analysis identified among women and men several additional sex-specific risk associations that were statistically significant but with smaller point estimates.
SOURCE: Ogdie A et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):131-2. Abstract OP0115. doi: 10.1136/annrheumdis-2019-eular.4390.
MADRID – Several novel risk associations with psoriasis progression were found to differ by sex, and collectively appeared to implicate infections and the “stress response” as a trigger of psoriatic arthritis.
The findings come from a risk factor analysis of a U.S. claims database of more than 200,000 adults with psoriasis including more than 4,000 patients who progressed to psoriatic arthritis during nearly 6 years of follow-up.
The new analysis confirmed several previously described risk associations linked with progression to psoriatic arthritis (PsA) that have roughly equal impact on both women and men: fatigue, obesity, and depression, Alexis Ogdie, MD, said at the European Congress of Rheumatology. The new findings also showed several novel, sex-specific associations. In women, these associations included salmonella infection, sepsis, and uveitis; in men, they included gangrene, encephalitis, and hidradenitis suppurativa.
The links with various infections were generally rare; they showed strong nominal associations in multivariate analyses but with wide confidence limits. The findings suggest that events that induce major stress responses, such as infections, often preceded the progression of psoriasis to a diagnosis of PsA, said Dr. Ogdie, director of the psoriatic arthritis clinic at the University of Pennsylvania in Philadelphia. Other, noninfectious clinical features that significantly linked with PsA development but at a lower magnitude included anemia and diabetes in women, and irritable bowel syndrome and venous thromboembolism in men.
Dr. Ogdie cautioned that the findings were preliminary and need confirmation in different data sets, as well as in additional subgroup analyses of the data used in the current analysis, taken from the electronic medical records of 215,386 U.S. residents diagnosed with psoriasis in the Optum medical-claims database for 2006-2017.
The analysis focused on patients who received a second diagnostic code in their EMR for psoriasis during the 12 months after the index psoriasis entry. The identified group averaged 50 years old; 55% of the psoriasis patients were women, and 86% were white.
During the year after their first diagnostic-code entry for psoriasis, 4.6% of the patients received a biological drug and 4.2% received an oral drug for their psoriasis. During 5.6 years of follow-up, 4,288 patients (2%) developed PsA, a rate of 3.5 cases/1,000 patient-years. Dr. Ogdie noted that prior studies have documented the challenge of diagnosing PsA in patients with psoriasis, so this may be a conservative estimate of the progression rate.
The researchers assessed possible linkage with PsA progression for more than 250 different entries in the EMR, but the analysis was limited by the absence of measures of rheumatoid susceptibility, such as immunologic markers, which were not included in the EMR. In multivariate analysis of the full cohort, fatigue at baseline was linked with a 77% higher rate of progression to PsA, obesity was linked with a 48% higher rate, and depression with a 29% higher rate of progression when compared with psoriasis patients without each of these factors. All three differences were statistically significant. Dr. Ogdie cited an article she recently coauthored that detailed the background to this approach in studying the etiology of PsA (Nat Rev Rheumatol. 2019 March;15:153-66).
This is the first study to report sex-linked differences in clinical measures that link with progression to PsA, Dr. Ogdie noted. In women, salmonella infection linked with a 9-fold higher rate of PsA development compared with women with psoriasis without salmonella infection, women with uveitis had a 2.9-fold higher rate of PsA development, and those with sepsis had a 2.4-fold increased rate of PsA. Among men, those with gangrene, encephalitis, or hidradenitis suppurativa each had a greater than 4-fold higher rate of developing PsA, and men with osteomyelitis had a 2.7-fold increase.
All these between-group differences were statistically significant. But because each of these was a relatively rare event, the confidence intervals around these point estimates were wide. For example, in women with salmonella infection from a statistical standpoint the possible range of increased risk could be anywhere from 1.3 to 66. The analysis identified among women and men several additional sex-specific risk associations that were statistically significant but with smaller point estimates.
SOURCE: Ogdie A et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):131-2. Abstract OP0115. doi: 10.1136/annrheumdis-2019-eular.4390.
REPORTING FROM THE EULAR 2019 CONGRESS
Ibudilast’s Efficacy Differs in Primary and Secondary Progressive MS
Key clinical point: Ibudilast’s treatment effect in a phase 2 trial for progressive multiple sclerosis (MS) primarily was driven by patients with primary progressive MS.
Major finding: The rate of brain atrophy for untreated patients with primary progressive MS was about twice as fast as that for those with secondary progressive MS.
Study details: The randomized, placebo-controlled phase 2 SPRINT-MS trial of ibudilast included 134 patients with primary progressive MS and 121 with secondary progressive MS.
Disclosures: The SPRINT-MS trial was funded by the National Institute of Neurological Disorders and Stroke. The National Multiple Sclerosis Society and MediciNova also supported the study. Dr. Goodman reported receiving research support from pharmaceutical companies, as well as personal compensation from companies for consulting, serving on a scientific advisory board, and speaking.
Citation: Goodman A et al. AAN 2019, Abstract S12.007.
Key clinical point: Ibudilast’s treatment effect in a phase 2 trial for progressive multiple sclerosis (MS) primarily was driven by patients with primary progressive MS.
Major finding: The rate of brain atrophy for untreated patients with primary progressive MS was about twice as fast as that for those with secondary progressive MS.
Study details: The randomized, placebo-controlled phase 2 SPRINT-MS trial of ibudilast included 134 patients with primary progressive MS and 121 with secondary progressive MS.
Disclosures: The SPRINT-MS trial was funded by the National Institute of Neurological Disorders and Stroke. The National Multiple Sclerosis Society and MediciNova also supported the study. Dr. Goodman reported receiving research support from pharmaceutical companies, as well as personal compensation from companies for consulting, serving on a scientific advisory board, and speaking.
Citation: Goodman A et al. AAN 2019, Abstract S12.007.
Key clinical point: Ibudilast’s treatment effect in a phase 2 trial for progressive multiple sclerosis (MS) primarily was driven by patients with primary progressive MS.
Major finding: The rate of brain atrophy for untreated patients with primary progressive MS was about twice as fast as that for those with secondary progressive MS.
Study details: The randomized, placebo-controlled phase 2 SPRINT-MS trial of ibudilast included 134 patients with primary progressive MS and 121 with secondary progressive MS.
Disclosures: The SPRINT-MS trial was funded by the National Institute of Neurological Disorders and Stroke. The National Multiple Sclerosis Society and MediciNova also supported the study. Dr. Goodman reported receiving research support from pharmaceutical companies, as well as personal compensation from companies for consulting, serving on a scientific advisory board, and speaking.
Citation: Goodman A et al. AAN 2019, Abstract S12.007.
EHR default order slashes unnecessary imaging during palliative RT
Simply adding a default order to the electronic health record that automatically opts patients out of commonly used but unnecessary radiation oncology procedures can dramatically curtail their use, suggests a stepped-wedge, cluster-randomized, controlled trial.
Daily x-ray or CT imaging is often used to better reproducibly position patients during curative radiotherapy, but guidelines consider it unnecessary during palliative radiotherapy because of limited clinical benefit, according to the investigators, led by Sonam Sharma, MD, of the Icahn School of Medicine at Mount Sinai, New York, and the Abramson Cancer Center at the University of Pennsylvania, Philadelphia. “Unnecessary imaging can increase treatment time and expense for patients in distress,” they noted.
The investigators conducted a 2-year trial among 21 radiation oncologists from five practices (one university, four community) in which they added to the EHR a default order that specified no daily imaging during palliative radiation therapy. (Radiation oncologists could select another imaging frequency if they preferred.) The default order was first rolled out in the university practice and subsequently in the community practices.
Study analyses were based on 1,019 adult patients with bone, soft tissue, or brain metastases who received 1,188 courses of palliative three-dimensional conformal radiotherapy during the trial.
Results reported in a JAMA Oncology research letter showed that the proportion of patients receiving daily imaging during their palliative radiotherapy (imaging during 80% or more of treatments) fell from 68.2% during the combined preintervention periods to 32.4% during the combined intervention periods.
After potential confounders were taken into account, implementation of the default order in the EHR was associated with a more than halving of the odds of daily imaging during palliative radiotherapy (adjusted odds ratio, 0.37; P = .003), with an adjusted percentage point reduction of –18.8.
Findings were similar in the university practice alone (aOR, 0.33; P = .01; –22.3 percentage points) and in the community practices alone (aOR, 0.45; P = .02; –27.5 percentage points).
“In a network of five radiation oncology practices, introducing a default order in the EHR reduced unnecessary daily imaging during palliative radiotherapy,” Dr. Sharma and colleagues concluded. “Our findings suggest that simple nudges, such as setting default orders, can meaningfully reduce unnecessary care.”
Dr. Sharma reported that she had no relevant conflicts of interest. The study was funded in part by the National Cancer Institute and the University of Pennsylvania Health System through the Penn Medicine Nudge Unit and the department of radiation oncology.
SOURCE: Sharma S et al. JAMA Oncol. 2019 Jun 27. doi: 10.1001/jamaoncol.2019.1432.
Simply adding a default order to the electronic health record that automatically opts patients out of commonly used but unnecessary radiation oncology procedures can dramatically curtail their use, suggests a stepped-wedge, cluster-randomized, controlled trial.
Daily x-ray or CT imaging is often used to better reproducibly position patients during curative radiotherapy, but guidelines consider it unnecessary during palliative radiotherapy because of limited clinical benefit, according to the investigators, led by Sonam Sharma, MD, of the Icahn School of Medicine at Mount Sinai, New York, and the Abramson Cancer Center at the University of Pennsylvania, Philadelphia. “Unnecessary imaging can increase treatment time and expense for patients in distress,” they noted.
The investigators conducted a 2-year trial among 21 radiation oncologists from five practices (one university, four community) in which they added to the EHR a default order that specified no daily imaging during palliative radiation therapy. (Radiation oncologists could select another imaging frequency if they preferred.) The default order was first rolled out in the university practice and subsequently in the community practices.
Study analyses were based on 1,019 adult patients with bone, soft tissue, or brain metastases who received 1,188 courses of palliative three-dimensional conformal radiotherapy during the trial.
Results reported in a JAMA Oncology research letter showed that the proportion of patients receiving daily imaging during their palliative radiotherapy (imaging during 80% or more of treatments) fell from 68.2% during the combined preintervention periods to 32.4% during the combined intervention periods.
After potential confounders were taken into account, implementation of the default order in the EHR was associated with a more than halving of the odds of daily imaging during palliative radiotherapy (adjusted odds ratio, 0.37; P = .003), with an adjusted percentage point reduction of –18.8.
Findings were similar in the university practice alone (aOR, 0.33; P = .01; –22.3 percentage points) and in the community practices alone (aOR, 0.45; P = .02; –27.5 percentage points).
“In a network of five radiation oncology practices, introducing a default order in the EHR reduced unnecessary daily imaging during palliative radiotherapy,” Dr. Sharma and colleagues concluded. “Our findings suggest that simple nudges, such as setting default orders, can meaningfully reduce unnecessary care.”
Dr. Sharma reported that she had no relevant conflicts of interest. The study was funded in part by the National Cancer Institute and the University of Pennsylvania Health System through the Penn Medicine Nudge Unit and the department of radiation oncology.
SOURCE: Sharma S et al. JAMA Oncol. 2019 Jun 27. doi: 10.1001/jamaoncol.2019.1432.
Simply adding a default order to the electronic health record that automatically opts patients out of commonly used but unnecessary radiation oncology procedures can dramatically curtail their use, suggests a stepped-wedge, cluster-randomized, controlled trial.
Daily x-ray or CT imaging is often used to better reproducibly position patients during curative radiotherapy, but guidelines consider it unnecessary during palliative radiotherapy because of limited clinical benefit, according to the investigators, led by Sonam Sharma, MD, of the Icahn School of Medicine at Mount Sinai, New York, and the Abramson Cancer Center at the University of Pennsylvania, Philadelphia. “Unnecessary imaging can increase treatment time and expense for patients in distress,” they noted.
The investigators conducted a 2-year trial among 21 radiation oncologists from five practices (one university, four community) in which they added to the EHR a default order that specified no daily imaging during palliative radiation therapy. (Radiation oncologists could select another imaging frequency if they preferred.) The default order was first rolled out in the university practice and subsequently in the community practices.
Study analyses were based on 1,019 adult patients with bone, soft tissue, or brain metastases who received 1,188 courses of palliative three-dimensional conformal radiotherapy during the trial.
Results reported in a JAMA Oncology research letter showed that the proportion of patients receiving daily imaging during their palliative radiotherapy (imaging during 80% or more of treatments) fell from 68.2% during the combined preintervention periods to 32.4% during the combined intervention periods.
After potential confounders were taken into account, implementation of the default order in the EHR was associated with a more than halving of the odds of daily imaging during palliative radiotherapy (adjusted odds ratio, 0.37; P = .003), with an adjusted percentage point reduction of –18.8.
Findings were similar in the university practice alone (aOR, 0.33; P = .01; –22.3 percentage points) and in the community practices alone (aOR, 0.45; P = .02; –27.5 percentage points).
“In a network of five radiation oncology practices, introducing a default order in the EHR reduced unnecessary daily imaging during palliative radiotherapy,” Dr. Sharma and colleagues concluded. “Our findings suggest that simple nudges, such as setting default orders, can meaningfully reduce unnecessary care.”
Dr. Sharma reported that she had no relevant conflicts of interest. The study was funded in part by the National Cancer Institute and the University of Pennsylvania Health System through the Penn Medicine Nudge Unit and the department of radiation oncology.
SOURCE: Sharma S et al. JAMA Oncol. 2019 Jun 27. doi: 10.1001/jamaoncol.2019.1432.
FROM JAMA ONCOLOGY
Eculizumab Cuts Relapse Risk in NMO Spectrum Disorder
Key clinical point: Treatment with eculizumab substantially reduced risk of relapse versus placebo in patients with aquaporin-4 positive neuromyelitis optica spectrum disorder.
Major finding: Time to first adjudicated relapse on trial, the primary endpoint of the study, showed a significant (P less than .0001) effect in favor of monoclonal antibody treatment over placebo, with a 94.2% reduction in risk of relapse.
Study details: A phase 3, randomized, double-blind, placebo-controlled, multicenter trial (PREVENT) including 143 adult patients.
Disclosures: The study was supported by Alexion Pharmaceuticals. Dr. Pittock provided disclosures related to Alexion Pharmaceuticals, MedImmune, and Grifols, along with patents related to administration of eculizumab and cancer markers in neuromyelitis optica.
Citation: Pittock SJ et al. AAN 2019, Emerging Science Abstract 009.
Key clinical point: Treatment with eculizumab substantially reduced risk of relapse versus placebo in patients with aquaporin-4 positive neuromyelitis optica spectrum disorder.
Major finding: Time to first adjudicated relapse on trial, the primary endpoint of the study, showed a significant (P less than .0001) effect in favor of monoclonal antibody treatment over placebo, with a 94.2% reduction in risk of relapse.
Study details: A phase 3, randomized, double-blind, placebo-controlled, multicenter trial (PREVENT) including 143 adult patients.
Disclosures: The study was supported by Alexion Pharmaceuticals. Dr. Pittock provided disclosures related to Alexion Pharmaceuticals, MedImmune, and Grifols, along with patents related to administration of eculizumab and cancer markers in neuromyelitis optica.
Citation: Pittock SJ et al. AAN 2019, Emerging Science Abstract 009.
Key clinical point: Treatment with eculizumab substantially reduced risk of relapse versus placebo in patients with aquaporin-4 positive neuromyelitis optica spectrum disorder.
Major finding: Time to first adjudicated relapse on trial, the primary endpoint of the study, showed a significant (P less than .0001) effect in favor of monoclonal antibody treatment over placebo, with a 94.2% reduction in risk of relapse.
Study details: A phase 3, randomized, double-blind, placebo-controlled, multicenter trial (PREVENT) including 143 adult patients.
Disclosures: The study was supported by Alexion Pharmaceuticals. Dr. Pittock provided disclosures related to Alexion Pharmaceuticals, MedImmune, and Grifols, along with patents related to administration of eculizumab and cancer markers in neuromyelitis optica.
Citation: Pittock SJ et al. AAN 2019, Emerging Science Abstract 009.
Pediatric MS May Go Untreated in Year After Diagnosis
Key clinical point: Physicians face considerable uncertainty regarding how to treat pediatric patients with MS.
Major finding: About 65% of pediatric patients with multiple sclerosis do not receive disease-modifying therapy within 1 year of diagnosis.
Study details: Retrospective, observational study of claims data from 288 patients with pediatric MS.
Disclosures: Novartis funded the study, and Dr. Deshpande, who presented the findings, and a coauthor are employees of Novartis. Other coauthors reported consulting fees from Novartis, as well as consulting fees and grant funding from other pharmaceutical companies.
Citation: Greenberg B et al. CMSC 2019, Abstract DXM02.
Key clinical point: Physicians face considerable uncertainty regarding how to treat pediatric patients with MS.
Major finding: About 65% of pediatric patients with multiple sclerosis do not receive disease-modifying therapy within 1 year of diagnosis.
Study details: Retrospective, observational study of claims data from 288 patients with pediatric MS.
Disclosures: Novartis funded the study, and Dr. Deshpande, who presented the findings, and a coauthor are employees of Novartis. Other coauthors reported consulting fees from Novartis, as well as consulting fees and grant funding from other pharmaceutical companies.
Citation: Greenberg B et al. CMSC 2019, Abstract DXM02.
Key clinical point: Physicians face considerable uncertainty regarding how to treat pediatric patients with MS.
Major finding: About 65% of pediatric patients with multiple sclerosis do not receive disease-modifying therapy within 1 year of diagnosis.
Study details: Retrospective, observational study of claims data from 288 patients with pediatric MS.
Disclosures: Novartis funded the study, and Dr. Deshpande, who presented the findings, and a coauthor are employees of Novartis. Other coauthors reported consulting fees from Novartis, as well as consulting fees and grant funding from other pharmaceutical companies.
Citation: Greenberg B et al. CMSC 2019, Abstract DXM02.
What is your diagnosis? - July 2019
The diagnosis
von Hippel-Lindau disease
The diagnosis is von Hippel-Lindau disease (VHL). Subsequent brain and renal magnetic resonance imaging showed features suggestive of a 5-mm right cerebellar hemangioblastoma and right renal cell carcinoma (RCC), respectively. Fundoscopy showed bilateral small retinal angiomas. Plasma and 24-hour urinary metenephrine levels were normal. Genetic testing confirmed a germline VHL mutation.
VHL is a rare autosomal-dominant hereditary multicancer condition characterized by germline mutations of the VHL tumor suppressor gene, with an incidence of 1 in 36,000 live births. The commonest associated tumors are retinal and central nervous system hemangioblastomas, RCC, pheochromocytoma, pancreatic islet cell tumors, and endolymphatic sac tumors.1 Cystic lesions may also be seen in other viscera such as the liver and ovaries. Clinical diagnostic criteria require the presence of any of these tumors in a patient with a positive family history, or alternatively, at least 2 retinal or cerebellar hemangioblastomas, or 1 hemangioblastoma plus 1 visceral tumor.2
Pancreatic involvement occurs in 65%–77% of patients with VHL, and may be the sole manifestation in 7.6%. Findings include multiple true cysts (91%), microcystic serous cystadenomas (12%), solid pancreatic neuroendocrine tumors (5%–10%), or a combination (11.5%). Most lesions are asymptomatic, but may present with vague symptoms of epigastric pain, diarrhea, dyspepsia, obstructive jaundice, or endocrine and/or exocrine pancreatic insufficiency. Surgery is required for symptomatic cysts or large pancreatic neuroendocrine tumors. The main causes of death are RCC and central nervous system hemangioblastomas.3 Our patient underwent laser therapy for her retinal angiomas, and is currently undergoing close regular surveillance. Clinicians should have a high index of suspicion for diagnosing VHL in patients with multiple pancreatic cysts. Because EUS is now widely used for the evaluation of pancreatic cysts, gastroenterologists may be first in making the diagnosis, as in this patient.
References
1. Lonser R.R., Glenn G.M., Walther M. et al. von Hippel-Lindau disease. Lancet. 2003;361:2059-67.
2. Melmon K., Rosen S. Lindau’s disease. Am J Med. 1964;36:595-617
3. Hammel P.R., Vilgrain V., Terris B. et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087-95.
The diagnosis
von Hippel-Lindau disease
The diagnosis is von Hippel-Lindau disease (VHL). Subsequent brain and renal magnetic resonance imaging showed features suggestive of a 5-mm right cerebellar hemangioblastoma and right renal cell carcinoma (RCC), respectively. Fundoscopy showed bilateral small retinal angiomas. Plasma and 24-hour urinary metenephrine levels were normal. Genetic testing confirmed a germline VHL mutation.
VHL is a rare autosomal-dominant hereditary multicancer condition characterized by germline mutations of the VHL tumor suppressor gene, with an incidence of 1 in 36,000 live births. The commonest associated tumors are retinal and central nervous system hemangioblastomas, RCC, pheochromocytoma, pancreatic islet cell tumors, and endolymphatic sac tumors.1 Cystic lesions may also be seen in other viscera such as the liver and ovaries. Clinical diagnostic criteria require the presence of any of these tumors in a patient with a positive family history, or alternatively, at least 2 retinal or cerebellar hemangioblastomas, or 1 hemangioblastoma plus 1 visceral tumor.2
Pancreatic involvement occurs in 65%–77% of patients with VHL, and may be the sole manifestation in 7.6%. Findings include multiple true cysts (91%), microcystic serous cystadenomas (12%), solid pancreatic neuroendocrine tumors (5%–10%), or a combination (11.5%). Most lesions are asymptomatic, but may present with vague symptoms of epigastric pain, diarrhea, dyspepsia, obstructive jaundice, or endocrine and/or exocrine pancreatic insufficiency. Surgery is required for symptomatic cysts or large pancreatic neuroendocrine tumors. The main causes of death are RCC and central nervous system hemangioblastomas.3 Our patient underwent laser therapy for her retinal angiomas, and is currently undergoing close regular surveillance. Clinicians should have a high index of suspicion for diagnosing VHL in patients with multiple pancreatic cysts. Because EUS is now widely used for the evaluation of pancreatic cysts, gastroenterologists may be first in making the diagnosis, as in this patient.
References
1. Lonser R.R., Glenn G.M., Walther M. et al. von Hippel-Lindau disease. Lancet. 2003;361:2059-67.
2. Melmon K., Rosen S. Lindau’s disease. Am J Med. 1964;36:595-617
3. Hammel P.R., Vilgrain V., Terris B. et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087-95.
The diagnosis
von Hippel-Lindau disease
The diagnosis is von Hippel-Lindau disease (VHL). Subsequent brain and renal magnetic resonance imaging showed features suggestive of a 5-mm right cerebellar hemangioblastoma and right renal cell carcinoma (RCC), respectively. Fundoscopy showed bilateral small retinal angiomas. Plasma and 24-hour urinary metenephrine levels were normal. Genetic testing confirmed a germline VHL mutation.
VHL is a rare autosomal-dominant hereditary multicancer condition characterized by germline mutations of the VHL tumor suppressor gene, with an incidence of 1 in 36,000 live births. The commonest associated tumors are retinal and central nervous system hemangioblastomas, RCC, pheochromocytoma, pancreatic islet cell tumors, and endolymphatic sac tumors.1 Cystic lesions may also be seen in other viscera such as the liver and ovaries. Clinical diagnostic criteria require the presence of any of these tumors in a patient with a positive family history, or alternatively, at least 2 retinal or cerebellar hemangioblastomas, or 1 hemangioblastoma plus 1 visceral tumor.2
Pancreatic involvement occurs in 65%–77% of patients with VHL, and may be the sole manifestation in 7.6%. Findings include multiple true cysts (91%), microcystic serous cystadenomas (12%), solid pancreatic neuroendocrine tumors (5%–10%), or a combination (11.5%). Most lesions are asymptomatic, but may present with vague symptoms of epigastric pain, diarrhea, dyspepsia, obstructive jaundice, or endocrine and/or exocrine pancreatic insufficiency. Surgery is required for symptomatic cysts or large pancreatic neuroendocrine tumors. The main causes of death are RCC and central nervous system hemangioblastomas.3 Our patient underwent laser therapy for her retinal angiomas, and is currently undergoing close regular surveillance. Clinicians should have a high index of suspicion for diagnosing VHL in patients with multiple pancreatic cysts. Because EUS is now widely used for the evaluation of pancreatic cysts, gastroenterologists may be first in making the diagnosis, as in this patient.
References
1. Lonser R.R., Glenn G.M., Walther M. et al. von Hippel-Lindau disease. Lancet. 2003;361:2059-67.
2. Melmon K., Rosen S. Lindau’s disease. Am J Med. 1964;36:595-617
3. Hammel P.R., Vilgrain V., Terris B. et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology. 2000;119:1087-95.
A 32-year-old Filipino woman was referred for endoscopic ultrasound (EUS) imaging of the pancreas from another hospital where she had presented with a history of intermittent abdominal pain with radiation to the back precipitated by alcohol, and recurrent palpitations. During outpatient review before EUS, she gave a background history of previous laparoscopic ovarian cystectomy, as well as multiple previous admissions with supraventricular tachycardia requiring cardioversion on 1 occasion. One of her brothers had undergone brain surgery to remove a cyst, and another had died of an unspecified brain tumor at 25 years of age. Her mother had died of ovarian cancer.

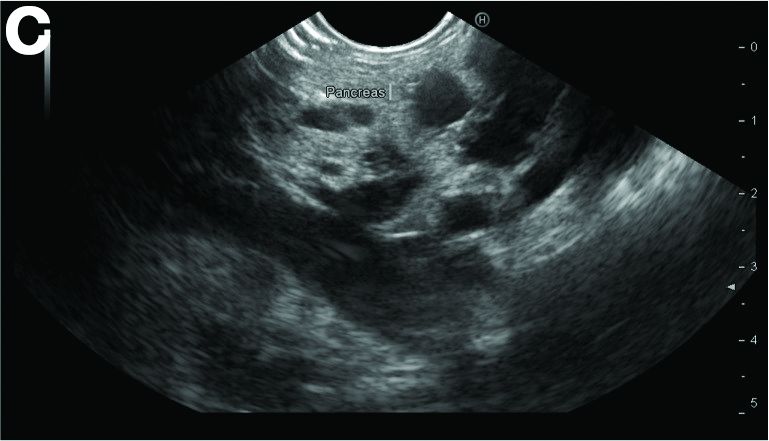
Physical examination was unremarkable, with a normal pulse rate and blood pressure and no anemia, jaundice, or lymphadenopathy. Laboratory investigations including a full blood count, urea and electrolytes, liver function tests, thyroid function tests, and serum lipase were all normal. Abdominal computed tomography and ultrasound imaging revealed multiple cysts of varying sizes throughout the pancreas (Figure A), as well as multiple small benign-looking cysts in the liver.

In addition, there was a 17-mm hyperdense solid lesion in the midpole of her right kidney visualized on computed tomography scan (Figure B). EUS revealed multiple thinly septated anechoic cysts throughout the pancreas, the largest measuring 36 mm located in the body, with no associated masses (Figure C).
What is the likely diagnosis? What other investigations would you do for confirmation?
Daratumumab wins new indication for newly diagnosed myeloma patients
Janssen’s daratumumab (Darzalex) has picked up a sixth adult multiple myeloma indication, this time in combination with lenalidomide (Revlimid) and dexamethasone in newly diagnosed patients ineligible for autologous stem cell transplants.

The phase 3 MAIA trial found that 97 of 368 patients (26.4%) treated with the combination – dubbed DRd – progressed or died at a median follow-up of 28 months, versus 143 of 269 (38.8%) treated with lenalidomide and dexamethasone alone (Rd). An estimated 55.6% of patients on lenalidomide and dexamethasone, versus 70.6% with the daratumumab add-on, were alive without progression at 30 months (N Engl J Med. 2019 May 30;380[22]:2104-15).
Previously approved indications for daratumumab include relapsed or refractory disease after at least one other therapy; and combination treatment with bortezomib, melphalan, and prednisone, also in newly diagnosed patients who are ineligible for transplant.
The most common grade 3 and 4 adverse events reported in the MAIA trial were neutropenia (50.0% for the DRd group versus 35.3% for the Rd group), anemia (11.8% vs. 19.7%), lymphopenia (15.1% vs. 10.7%), and pneumonia (13.7% vs. 7.9%).
Janssen’s daratumumab (Darzalex) has picked up a sixth adult multiple myeloma indication, this time in combination with lenalidomide (Revlimid) and dexamethasone in newly diagnosed patients ineligible for autologous stem cell transplants.
The phase 3 MAIA trial found that 97 of 368 patients (26.4%) treated with the combination – dubbed DRd – progressed or died at a median follow-up of 28 months, versus 143 of 269 (38.8%) treated with lenalidomide and dexamethasone alone (Rd). An estimated 55.6% of patients on lenalidomide and dexamethasone, versus 70.6% with the daratumumab add-on, were alive without progression at 30 months (N Engl J Med. 2019 May 30;380[22]:2104-15).
Previously approved indications for daratumumab include relapsed or refractory disease after at least one other therapy; and combination treatment with bortezomib, melphalan, and prednisone, also in newly diagnosed patients who are ineligible for transplant.
The most common grade 3 and 4 adverse events reported in the MAIA trial were neutropenia (50.0% for the DRd group versus 35.3% for the Rd group), anemia (11.8% vs. 19.7%), lymphopenia (15.1% vs. 10.7%), and pneumonia (13.7% vs. 7.9%).
Janssen’s daratumumab (Darzalex) has picked up a sixth adult multiple myeloma indication, this time in combination with lenalidomide (Revlimid) and dexamethasone in newly diagnosed patients ineligible for autologous stem cell transplants.
The phase 3 MAIA trial found that 97 of 368 patients (26.4%) treated with the combination – dubbed DRd – progressed or died at a median follow-up of 28 months, versus 143 of 269 (38.8%) treated with lenalidomide and dexamethasone alone (Rd). An estimated 55.6% of patients on lenalidomide and dexamethasone, versus 70.6% with the daratumumab add-on, were alive without progression at 30 months (N Engl J Med. 2019 May 30;380[22]:2104-15).
Previously approved indications for daratumumab include relapsed or refractory disease after at least one other therapy; and combination treatment with bortezomib, melphalan, and prednisone, also in newly diagnosed patients who are ineligible for transplant.
The most common grade 3 and 4 adverse events reported in the MAIA trial were neutropenia (50.0% for the DRd group versus 35.3% for the Rd group), anemia (11.8% vs. 19.7%), lymphopenia (15.1% vs. 10.7%), and pneumonia (13.7% vs. 7.9%).
Allergic Contact Dermatitis With Sparing of Exposed Psoriasis Plaques
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
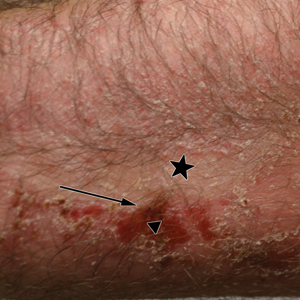
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
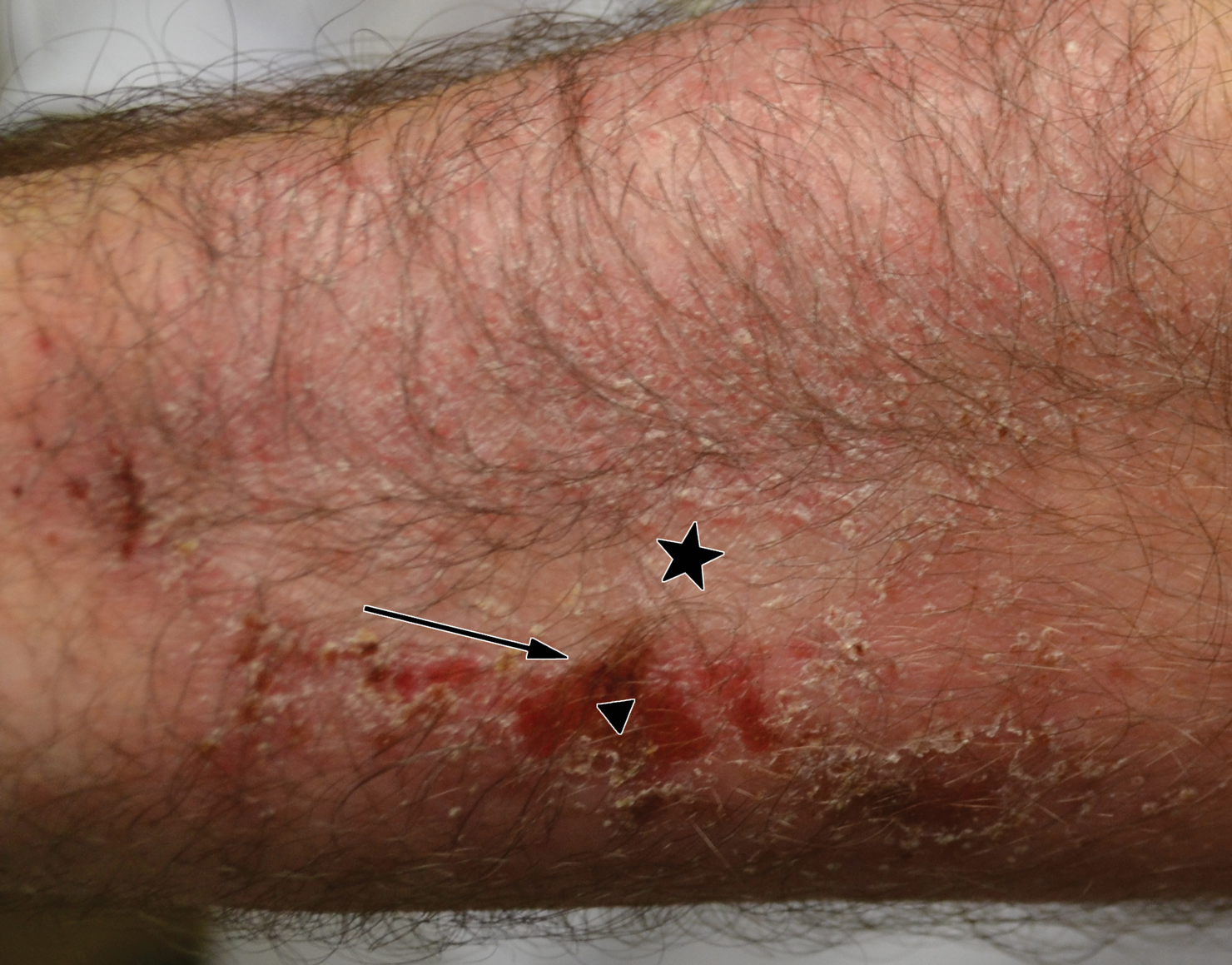
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
Practice Points
- Patients with plaque-type psoriasis who experience allergic contact dermatitis (ACD) may present with sparing of exposed psoriatic plaques.
- The divergent immunologic milieus present in ACD and psoriasis likely underly the decreased incidence of ACD in patients with psoriasis.