User login
Clinicians ask FDA for continued ‘discretion’ to do fecal transplants
Attendees at a public meeting on Nov. 4 gave the US Food and Drug Administration conflicting views on whether the agency should continue to allow a relatively loose regulatory environment for fecal microbiota transplants (FMT) – debating the limits of “enforcement discretion” the FDA now has in place.
The question is especially relevant as use of the procedure is growing, while safety data are not being rigorously collected in all cases. The death of an immunocompromised FMT patient earlier in 2018 from an invasive bacterial infection caused by drug-resistant Escherichia coli, as reported by Medscape Medical News, is seen by some as an example of the consequences of a loose policy.
Still, the American Gastroenterological Association (AGA) presented new, unpublished follow-up data at the meeting that showed that the majority of FMT patients in a national registry had no adverse events.
Some companies developing FMT-based products argued at the meeting that the agency should impose stricter requirements, while stool banks and clinicians offering the therapy outside of clinical trials said that the current policy – in place since 2013 – in which the FDA has exercised “enforcement discretion,” should be allowed to continue.
“Enforcement discretion has been successful in enabling and overcoming key barriers to access to treatment,” said Majdi Osman, MD, clinical program director at OpenBiome, a nonprofit stool bank based in Cambridge, Mass. Dr. Osman said that 98% of the U.S. population now lives within a 2-hour drive of an FMT provider.
Amanda Kabage, a researcher and donor program coordinator for the Microbiota Therapeutics program at the University of Minnesota in Minneapolis, and herself a former recipient of FMT, said she was in favor of continuing the FDA policy.
“If enforcement discretion were to go away, patients far sicker than I was will not have access. They’ll get sicker and they will die,” Ms. Kabage said.
But, she added, the FDA had missed an opportunity by not insisting on collecting outcomes and safety data. Minnesota has established a patient registry to do just that, and physicians cannot administer FMT unless they agree to participate, she said. In response, FDA panelists noted that the agency cannot mandate data collection under an enforcement policy.
Lee Jones, founder and chief executive officer of Rebiotix/Ferring, a biotech company focused on the development of microbiome-based therapeutics, argued for tighter restrictions, however, claiming that increased access – and the FDA policy – had led to a fourfold decrease in enrollment since the company began study of its lead FMT product, RBX2660, in 2013.
“We’re dealing with an orphan indication and the patients were hard to come by to begin with,” she said at the meeting. “Enforcement discretion has slowed our clinical development and delayed patient access to FDA-approved therapies by over 2 years.”
An investigator at the University of Texas Health Science Center at Houston, Herbert DuPont, MD, who has administered FMT and is conducting a trial for Rebiotix, said his center wanted the FDA policy to continue “allowing multiple groups to perform FMT for recurrent [Clostridium difficile], because of the incredible public health need.”
But, he added, “We’re very concerned about industry and ability to do clinical trials.”
Those trials are important, Dr. DuPont said. “I think we have to address very actively how industry can move these products through,” he said, “because all of us want to remove the F from FMT,” by isolating the necessary elements of the process while not having the risk sometimes associated with human stool.
Policy slow to evolve
“I’m frustrated that it’s taken over 6 years and three draft guidances to get us this far,” Christian John Lillis, executive director of the Peggy Lillis Foundation – a group dedicated to creating awareness about the dangers of C. difficile – said at the meeting.
Mr. Lillis said that probably several thousand deaths had been prevented through increased FMT access, but that it was time to create a concrete policy that advanced the therapy.
The FDA guidance issued in 2013 allowed physicians to provide FMT for recurrent or refractory C. difficile infection without filing an investigational new drug (IND) application.
Clinicians must obtain informed consent that includes a discussion of the risks, and a statement that FMT is investigational. In March 2016, the agency issued revised draft guidance that it was aiming to require stool banks to apply for INDs, as reported by Medscape Medical News.
OpenBiome has flourished under the current policy. It has provided more than 50,000 treatments to 1,200 hospitals and clinics, and has provided FMT for 49 clinical trials and for 16 single patients who received INDs, Dr. Osman said.
But requiring INDs for all centers is a bad idea, he said. “IND requirements are insurmountable for most health centers,” Dr. Osman said, noting that most of the FMT material OpenBiome produces is sent to community-based physicians.
“These requirements would likely mean restrictions in access for stool bank–provided FMT and potentially pushing patients to physician-directed FMT or discouraging physicians from using FMT at all,” he said.
Stacy Kahn, MD, FMT director at Boston Children’s Hospital in Massachusetts, said that having ready access from a stool bank was crucial.
“Universal donor FMT is much easier, much faster and much more cost effective than what we can do as clinicians,” she said.
New safety and efficacy data
One unpublished study showed that 75% of patients treated since 2011 had a sustained cure, noted Colleen Kelly, MD, a Brown University professor of medicine and principal investigator for the National Institutes of Health–funded national FMT registry (although the data in this study were not from the FMT registry).
The study, which was a collaboration between the Alpert Medical School of Brown University, Brigham and Women’s Hospital, and Indiana University School of Medicine, attempted follow-up on 533 patients; 208 were successfully contacted, and an additional 55 had died, none due to FMT.
Dr. Kelly also presented data from the FMT National Registry showing that at 1 month posttransplant, two (1%) of 253 patients had an infection possibly related to FMT; one with Bacteroides fragilis and one with enteropathogenic E. coli. Seven hospitalizations were deemed related or possibly related to FMT, including two recurrences of C. difficile.
At 6 months posttransplant, 8 (5%) of 152 patients had a serious infection, and 23 patients reported a diagnosis of a new condition, primarily diarrhea-predominant irritable bowel syndrome, which is common post FMT, said Dr. Kelly, who presented the data on behalf of AGA, which administers the registry.
The AGA supports a continuation of the enforcement discretion as a means to maintain patient access where the evidence supports the use of FMT, but the group does not back use of FMT outside medical supervision, Dr. Kelly said.
This article originally appeared on Medscape. For more news, follow Medscape on Facebook, Twitter, Instagram, and YouTube.
Attendees at a public meeting on Nov. 4 gave the US Food and Drug Administration conflicting views on whether the agency should continue to allow a relatively loose regulatory environment for fecal microbiota transplants (FMT) – debating the limits of “enforcement discretion” the FDA now has in place.
The question is especially relevant as use of the procedure is growing, while safety data are not being rigorously collected in all cases. The death of an immunocompromised FMT patient earlier in 2018 from an invasive bacterial infection caused by drug-resistant Escherichia coli, as reported by Medscape Medical News, is seen by some as an example of the consequences of a loose policy.
Still, the American Gastroenterological Association (AGA) presented new, unpublished follow-up data at the meeting that showed that the majority of FMT patients in a national registry had no adverse events.
Some companies developing FMT-based products argued at the meeting that the agency should impose stricter requirements, while stool banks and clinicians offering the therapy outside of clinical trials said that the current policy – in place since 2013 – in which the FDA has exercised “enforcement discretion,” should be allowed to continue.
“Enforcement discretion has been successful in enabling and overcoming key barriers to access to treatment,” said Majdi Osman, MD, clinical program director at OpenBiome, a nonprofit stool bank based in Cambridge, Mass. Dr. Osman said that 98% of the U.S. population now lives within a 2-hour drive of an FMT provider.
Amanda Kabage, a researcher and donor program coordinator for the Microbiota Therapeutics program at the University of Minnesota in Minneapolis, and herself a former recipient of FMT, said she was in favor of continuing the FDA policy.
“If enforcement discretion were to go away, patients far sicker than I was will not have access. They’ll get sicker and they will die,” Ms. Kabage said.
But, she added, the FDA had missed an opportunity by not insisting on collecting outcomes and safety data. Minnesota has established a patient registry to do just that, and physicians cannot administer FMT unless they agree to participate, she said. In response, FDA panelists noted that the agency cannot mandate data collection under an enforcement policy.
Lee Jones, founder and chief executive officer of Rebiotix/Ferring, a biotech company focused on the development of microbiome-based therapeutics, argued for tighter restrictions, however, claiming that increased access – and the FDA policy – had led to a fourfold decrease in enrollment since the company began study of its lead FMT product, RBX2660, in 2013.
“We’re dealing with an orphan indication and the patients were hard to come by to begin with,” she said at the meeting. “Enforcement discretion has slowed our clinical development and delayed patient access to FDA-approved therapies by over 2 years.”
An investigator at the University of Texas Health Science Center at Houston, Herbert DuPont, MD, who has administered FMT and is conducting a trial for Rebiotix, said his center wanted the FDA policy to continue “allowing multiple groups to perform FMT for recurrent [Clostridium difficile], because of the incredible public health need.”
But, he added, “We’re very concerned about industry and ability to do clinical trials.”
Those trials are important, Dr. DuPont said. “I think we have to address very actively how industry can move these products through,” he said, “because all of us want to remove the F from FMT,” by isolating the necessary elements of the process while not having the risk sometimes associated with human stool.
Policy slow to evolve
“I’m frustrated that it’s taken over 6 years and three draft guidances to get us this far,” Christian John Lillis, executive director of the Peggy Lillis Foundation – a group dedicated to creating awareness about the dangers of C. difficile – said at the meeting.
Mr. Lillis said that probably several thousand deaths had been prevented through increased FMT access, but that it was time to create a concrete policy that advanced the therapy.
The FDA guidance issued in 2013 allowed physicians to provide FMT for recurrent or refractory C. difficile infection without filing an investigational new drug (IND) application.
Clinicians must obtain informed consent that includes a discussion of the risks, and a statement that FMT is investigational. In March 2016, the agency issued revised draft guidance that it was aiming to require stool banks to apply for INDs, as reported by Medscape Medical News.
OpenBiome has flourished under the current policy. It has provided more than 50,000 treatments to 1,200 hospitals and clinics, and has provided FMT for 49 clinical trials and for 16 single patients who received INDs, Dr. Osman said.
But requiring INDs for all centers is a bad idea, he said. “IND requirements are insurmountable for most health centers,” Dr. Osman said, noting that most of the FMT material OpenBiome produces is sent to community-based physicians.
“These requirements would likely mean restrictions in access for stool bank–provided FMT and potentially pushing patients to physician-directed FMT or discouraging physicians from using FMT at all,” he said.
Stacy Kahn, MD, FMT director at Boston Children’s Hospital in Massachusetts, said that having ready access from a stool bank was crucial.
“Universal donor FMT is much easier, much faster and much more cost effective than what we can do as clinicians,” she said.
New safety and efficacy data
One unpublished study showed that 75% of patients treated since 2011 had a sustained cure, noted Colleen Kelly, MD, a Brown University professor of medicine and principal investigator for the National Institutes of Health–funded national FMT registry (although the data in this study were not from the FMT registry).
The study, which was a collaboration between the Alpert Medical School of Brown University, Brigham and Women’s Hospital, and Indiana University School of Medicine, attempted follow-up on 533 patients; 208 were successfully contacted, and an additional 55 had died, none due to FMT.
Dr. Kelly also presented data from the FMT National Registry showing that at 1 month posttransplant, two (1%) of 253 patients had an infection possibly related to FMT; one with Bacteroides fragilis and one with enteropathogenic E. coli. Seven hospitalizations were deemed related or possibly related to FMT, including two recurrences of C. difficile.
At 6 months posttransplant, 8 (5%) of 152 patients had a serious infection, and 23 patients reported a diagnosis of a new condition, primarily diarrhea-predominant irritable bowel syndrome, which is common post FMT, said Dr. Kelly, who presented the data on behalf of AGA, which administers the registry.
The AGA supports a continuation of the enforcement discretion as a means to maintain patient access where the evidence supports the use of FMT, but the group does not back use of FMT outside medical supervision, Dr. Kelly said.
This article originally appeared on Medscape. For more news, follow Medscape on Facebook, Twitter, Instagram, and YouTube.
Attendees at a public meeting on Nov. 4 gave the US Food and Drug Administration conflicting views on whether the agency should continue to allow a relatively loose regulatory environment for fecal microbiota transplants (FMT) – debating the limits of “enforcement discretion” the FDA now has in place.
The question is especially relevant as use of the procedure is growing, while safety data are not being rigorously collected in all cases. The death of an immunocompromised FMT patient earlier in 2018 from an invasive bacterial infection caused by drug-resistant Escherichia coli, as reported by Medscape Medical News, is seen by some as an example of the consequences of a loose policy.
Still, the American Gastroenterological Association (AGA) presented new, unpublished follow-up data at the meeting that showed that the majority of FMT patients in a national registry had no adverse events.
Some companies developing FMT-based products argued at the meeting that the agency should impose stricter requirements, while stool banks and clinicians offering the therapy outside of clinical trials said that the current policy – in place since 2013 – in which the FDA has exercised “enforcement discretion,” should be allowed to continue.
“Enforcement discretion has been successful in enabling and overcoming key barriers to access to treatment,” said Majdi Osman, MD, clinical program director at OpenBiome, a nonprofit stool bank based in Cambridge, Mass. Dr. Osman said that 98% of the U.S. population now lives within a 2-hour drive of an FMT provider.
Amanda Kabage, a researcher and donor program coordinator for the Microbiota Therapeutics program at the University of Minnesota in Minneapolis, and herself a former recipient of FMT, said she was in favor of continuing the FDA policy.
“If enforcement discretion were to go away, patients far sicker than I was will not have access. They’ll get sicker and they will die,” Ms. Kabage said.
But, she added, the FDA had missed an opportunity by not insisting on collecting outcomes and safety data. Minnesota has established a patient registry to do just that, and physicians cannot administer FMT unless they agree to participate, she said. In response, FDA panelists noted that the agency cannot mandate data collection under an enforcement policy.
Lee Jones, founder and chief executive officer of Rebiotix/Ferring, a biotech company focused on the development of microbiome-based therapeutics, argued for tighter restrictions, however, claiming that increased access – and the FDA policy – had led to a fourfold decrease in enrollment since the company began study of its lead FMT product, RBX2660, in 2013.
“We’re dealing with an orphan indication and the patients were hard to come by to begin with,” she said at the meeting. “Enforcement discretion has slowed our clinical development and delayed patient access to FDA-approved therapies by over 2 years.”
An investigator at the University of Texas Health Science Center at Houston, Herbert DuPont, MD, who has administered FMT and is conducting a trial for Rebiotix, said his center wanted the FDA policy to continue “allowing multiple groups to perform FMT for recurrent [Clostridium difficile], because of the incredible public health need.”
But, he added, “We’re very concerned about industry and ability to do clinical trials.”
Those trials are important, Dr. DuPont said. “I think we have to address very actively how industry can move these products through,” he said, “because all of us want to remove the F from FMT,” by isolating the necessary elements of the process while not having the risk sometimes associated with human stool.
Policy slow to evolve
“I’m frustrated that it’s taken over 6 years and three draft guidances to get us this far,” Christian John Lillis, executive director of the Peggy Lillis Foundation – a group dedicated to creating awareness about the dangers of C. difficile – said at the meeting.
Mr. Lillis said that probably several thousand deaths had been prevented through increased FMT access, but that it was time to create a concrete policy that advanced the therapy.
The FDA guidance issued in 2013 allowed physicians to provide FMT for recurrent or refractory C. difficile infection without filing an investigational new drug (IND) application.
Clinicians must obtain informed consent that includes a discussion of the risks, and a statement that FMT is investigational. In March 2016, the agency issued revised draft guidance that it was aiming to require stool banks to apply for INDs, as reported by Medscape Medical News.
OpenBiome has flourished under the current policy. It has provided more than 50,000 treatments to 1,200 hospitals and clinics, and has provided FMT for 49 clinical trials and for 16 single patients who received INDs, Dr. Osman said.
But requiring INDs for all centers is a bad idea, he said. “IND requirements are insurmountable for most health centers,” Dr. Osman said, noting that most of the FMT material OpenBiome produces is sent to community-based physicians.
“These requirements would likely mean restrictions in access for stool bank–provided FMT and potentially pushing patients to physician-directed FMT or discouraging physicians from using FMT at all,” he said.
Stacy Kahn, MD, FMT director at Boston Children’s Hospital in Massachusetts, said that having ready access from a stool bank was crucial.
“Universal donor FMT is much easier, much faster and much more cost effective than what we can do as clinicians,” she said.
New safety and efficacy data
One unpublished study showed that 75% of patients treated since 2011 had a sustained cure, noted Colleen Kelly, MD, a Brown University professor of medicine and principal investigator for the National Institutes of Health–funded national FMT registry (although the data in this study were not from the FMT registry).
The study, which was a collaboration between the Alpert Medical School of Brown University, Brigham and Women’s Hospital, and Indiana University School of Medicine, attempted follow-up on 533 patients; 208 were successfully contacted, and an additional 55 had died, none due to FMT.
Dr. Kelly also presented data from the FMT National Registry showing that at 1 month posttransplant, two (1%) of 253 patients had an infection possibly related to FMT; one with Bacteroides fragilis and one with enteropathogenic E. coli. Seven hospitalizations were deemed related or possibly related to FMT, including two recurrences of C. difficile.
At 6 months posttransplant, 8 (5%) of 152 patients had a serious infection, and 23 patients reported a diagnosis of a new condition, primarily diarrhea-predominant irritable bowel syndrome, which is common post FMT, said Dr. Kelly, who presented the data on behalf of AGA, which administers the registry.
The AGA supports a continuation of the enforcement discretion as a means to maintain patient access where the evidence supports the use of FMT, but the group does not back use of FMT outside medical supervision, Dr. Kelly said.
This article originally appeared on Medscape. For more news, follow Medscape on Facebook, Twitter, Instagram, and YouTube.

Open Clinical Trials for Native Americans With Diabetes Mellitus(FULL)
Providing access to clinical trials for patients with diabetes mellitus can be a challenge, but a significant number of trials are now recruiting patients. The clinical trials listed below are all open as of October 31, 2019; and are focused on diabetes mellitus-related treatments for American Indians. For additional information and full inclusion/exclusion criteria, please consult clinicaltrials.gov.
Cross-Sectional and Longitudinal Studies of “Pre-Diabetes” in the Pima Indians
The Pima Indians of Arizona have the highest prevalence and incidence of type 2 diabetes of any population in the world. Prospective analyses in this population have identified insulin resistance and a defect in early insulin secretion as risk factors for the development of the disease. To identify the genetic and environmental determinants of diabetes we plan to study Pima Indian families to determine: (1) if there are genes that segregate with metabolic risk factors for diabetes which might therefore be genetic markers for type 2 diabetes; and (2) the mechanisms mediating genetic and environmental determinants of insulin resistance and impaired insulin secretion.
ID: NCT00340132
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: Clifton Bogardus, MD, [email protected]
Location: NIDDK, Phoenix, AZ
Empaglifozin in Early Diabetic Kidney Disease
Diabetes is common among American Indian people and diabetic kidney disease is a common complication. Kidney disease caused by diabetes can lead to the need for kidney replacement, by dialysis or kidney transplant, and is also associated with higher risk of early death. A new diabetes medicine called empagliflozin may slow kidney disease from type 2 diabetes. Researchers want to learn if it protects the kidneys when used in very early stages of diabetic kidney disease.
ID: NCT03173963
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: Helen C Looker, [email protected]
Location: NIDDK, Phoenix, AZ
Family Investigation of Nephropathy and Diabetes
The Family Investigation of Nephropathy and Diabetes (FIND) is a multicenter study designed to identify genetic determinants of diabetic kidney disease. FIND will be conducted in 11 centers and in many ethnic groups throughout the United States. Two different strategies will be used to localize genes predisposing to kidney disease: a family-based genetic linkage study and a case-control study that utilizes admixture linkage disequilibrium. The center will conduct family-based linkage studies among American Indian populations in the southwestern United States.
ID: NCT00342927
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: William C Knowler, MD, [email protected]
Location: NIDDK, Phoenix, AZ
Look AHEAD: Action for Health in Diabetes
The Look AHEAD study is a multi-center, randomized clinical trial to examine the long-term effects of a lifestyle intervention designed to achieve and maintain weight loss. The study will investigate the effects of the intervention on heart attacks, stroke and cardiovascular-related death in individuals with type 2 diabetes who are also overweight or obese.
ID: NCT00017953
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Location: Southwestern American Indian Center, Phoenix, AZ
Vitamin D and Type 2 Diabetes Study
The goal of the Vitamin D and type 2 diabetes (D2d) study is to determine if vitamin D supplementation works to delay the onset of type 2 diabetes in people at risk for the disease and to gain a better understand how vitamin D affects glucose (sugar) metabolism.
ID: NCT01942694
Sponsor: Tufts Medical Center
Locations: Southwest American Indian Center; Phoenix, AZ; Orlando VA Medical Center, FL; Atlanta VA Medical Center, Decatur, GA; Omaha VA Medical Center, NE
Reducing Diabetes Risk Factors in American Indian Children: Tribal Turning Point (TTP)
This study will evaluate a behavioral intervention designed to reduce risk factors for type 2 diabetes in American Indian youth aged 7-10 years.
ID: NCT03573856
Sponsor: University of Colorado, Denver
Contact: Katherine Sauder, PhD, [email protected]; Dana Dabelea, MD, PhD, [email protected]
Location: Childrens Hospital Colorado, Aurora
Strong Men, Strong Communities Diabetes Risk Reduction in American Indian Men (SMSC)
SMSC will inform the design and implementation of culturally informed, community-based lifestyle interventions for diabetes prevention in AI men in our partner communities and elsewhere, as well as in men of other minority groups who experience a heavy burden of diabetes.
ID: NCT02953977
Sponsor: Washington State University
Contact: Kaimi Sinclair, PhD, MPH, [email protected] Location: IREACH, Seattle, WA
Growing Resilience in Wind River Indian Reservation (GR)
The Growing Resilience research leverages reservation-based assets of land, family, culture, and front-line tribal health organizations to develop and evaluate home food gardens as a family-based health promotion intervention to reduce disparities suffered by Native Americans in nearly every measure of health. Home gardening interventions show great promise for enabling families to improve their health, and this study aims to fulfill that promise with university and Wind River Indian Reservation partners. The investigators will develop an empowering, scalable, and sustainable family-based health promotion intervention with, by, and for Native American families and conduct the first randomized controlled trial to assess the health impacts of home gardens.
ID: NCT02672748
Sponsor: University of Wyoming
Location: University of Wyoming, Laramie
A Comparative Effectiveness Study of Major Glycemia-lowering Medications for Treatment of Type 2 Diabetes (GRADE)
The GRADE Study is a pragmatic, unmasked clinical trial that will compare commonly used diabetes medications, when combined with metformin, on glycemia-lowering effectiveness and patient-centered outcomes.
ID: NCT01794143
Sponsor: GRADE Study Group
Location: Southwestern American Indian Center, Phoenix, AZ
Home-Based Kidney Care in Native Americans of New Mexico (HBKC)
New Mexico American Indians are experiencing an epidemic of chronic kidney disease due primarily to the high rates of obesity and diabetes. The present study entitled Home-Based Kidney Care is designed to delay / reduce rates of end stage renal disease by early interventions in chronic kidney disease (CKD). Investigators propose to assess the safety and efficacy of conducting a full-scale study to determine if home based care delivered by a collaborative team composed of community health workers, the Albuquerque Area Indian Health Board and University of New Mexico faculty will decrease the risk for the development and the progression of CKD.
ID: NCT03179085
Sponsor: University of New Mexico
Contact: Vallabh Shah, PhD, [email protected]; Kevin English, PhD, [email protected]
Location: University of New Mexico, Albuquerque
Home-based Prediabetes Care in Acoma Pueblo - Study 1
Our major goal of implementing educational interventions to slow the current rate of increase in diabetes in Native communities is aligned with the National Institute of Health (NIGMS) and New Mexico INBRE’s vision in reducing health disparity using innovative interventions. The investigators propose following aims: (1) Recruit and Screen 300 community members in Acoma Pueblo, New Mexico to identify incident cases of pre-diabetes for the proposed study of Home Based Diabetes Care (HBDC); (2) Enroll 150 Acoma Natives aged 21-70 years, at risk for type 2 diabetes mellitus and conduct HBDC for a 16-week lifestyle intervention in a longitudinal cohort study.
ID: NCT04029298
Sponsor: University of New Mexico
Contact: Matthew Bouchonville, MD, [email protected]; Vallabh Shah, PhD, [email protected]
Providing access to clinical trials for patients with diabetes mellitus can be a challenge, but a significant number of trials are now recruiting patients. The clinical trials listed below are all open as of October 31, 2019; and are focused on diabetes mellitus-related treatments for American Indians. For additional information and full inclusion/exclusion criteria, please consult clinicaltrials.gov.
Cross-Sectional and Longitudinal Studies of “Pre-Diabetes” in the Pima Indians
The Pima Indians of Arizona have the highest prevalence and incidence of type 2 diabetes of any population in the world. Prospective analyses in this population have identified insulin resistance and a defect in early insulin secretion as risk factors for the development of the disease. To identify the genetic and environmental determinants of diabetes we plan to study Pima Indian families to determine: (1) if there are genes that segregate with metabolic risk factors for diabetes which might therefore be genetic markers for type 2 diabetes; and (2) the mechanisms mediating genetic and environmental determinants of insulin resistance and impaired insulin secretion.
ID: NCT00340132
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: Clifton Bogardus, MD, [email protected]
Location: NIDDK, Phoenix, AZ
Empaglifozin in Early Diabetic Kidney Disease
Diabetes is common among American Indian people and diabetic kidney disease is a common complication. Kidney disease caused by diabetes can lead to the need for kidney replacement, by dialysis or kidney transplant, and is also associated with higher risk of early death. A new diabetes medicine called empagliflozin may slow kidney disease from type 2 diabetes. Researchers want to learn if it protects the kidneys when used in very early stages of diabetic kidney disease.
ID: NCT03173963
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: Helen C Looker, [email protected]
Location: NIDDK, Phoenix, AZ
Family Investigation of Nephropathy and Diabetes
The Family Investigation of Nephropathy and Diabetes (FIND) is a multicenter study designed to identify genetic determinants of diabetic kidney disease. FIND will be conducted in 11 centers and in many ethnic groups throughout the United States. Two different strategies will be used to localize genes predisposing to kidney disease: a family-based genetic linkage study and a case-control study that utilizes admixture linkage disequilibrium. The center will conduct family-based linkage studies among American Indian populations in the southwestern United States.
ID: NCT00342927
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: William C Knowler, MD, [email protected]
Location: NIDDK, Phoenix, AZ
Look AHEAD: Action for Health in Diabetes
The Look AHEAD study is a multi-center, randomized clinical trial to examine the long-term effects of a lifestyle intervention designed to achieve and maintain weight loss. The study will investigate the effects of the intervention on heart attacks, stroke and cardiovascular-related death in individuals with type 2 diabetes who are also overweight or obese.
ID: NCT00017953
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Location: Southwestern American Indian Center, Phoenix, AZ
Vitamin D and Type 2 Diabetes Study
The goal of the Vitamin D and type 2 diabetes (D2d) study is to determine if vitamin D supplementation works to delay the onset of type 2 diabetes in people at risk for the disease and to gain a better understand how vitamin D affects glucose (sugar) metabolism.
ID: NCT01942694
Sponsor: Tufts Medical Center
Locations: Southwest American Indian Center; Phoenix, AZ; Orlando VA Medical Center, FL; Atlanta VA Medical Center, Decatur, GA; Omaha VA Medical Center, NE
Reducing Diabetes Risk Factors in American Indian Children: Tribal Turning Point (TTP)
This study will evaluate a behavioral intervention designed to reduce risk factors for type 2 diabetes in American Indian youth aged 7-10 years.
ID: NCT03573856
Sponsor: University of Colorado, Denver
Contact: Katherine Sauder, PhD, [email protected]; Dana Dabelea, MD, PhD, [email protected]
Location: Childrens Hospital Colorado, Aurora
Strong Men, Strong Communities Diabetes Risk Reduction in American Indian Men (SMSC)
SMSC will inform the design and implementation of culturally informed, community-based lifestyle interventions for diabetes prevention in AI men in our partner communities and elsewhere, as well as in men of other minority groups who experience a heavy burden of diabetes.
ID: NCT02953977
Sponsor: Washington State University
Contact: Kaimi Sinclair, PhD, MPH, [email protected] Location: IREACH, Seattle, WA
Growing Resilience in Wind River Indian Reservation (GR)
The Growing Resilience research leverages reservation-based assets of land, family, culture, and front-line tribal health organizations to develop and evaluate home food gardens as a family-based health promotion intervention to reduce disparities suffered by Native Americans in nearly every measure of health. Home gardening interventions show great promise for enabling families to improve their health, and this study aims to fulfill that promise with university and Wind River Indian Reservation partners. The investigators will develop an empowering, scalable, and sustainable family-based health promotion intervention with, by, and for Native American families and conduct the first randomized controlled trial to assess the health impacts of home gardens.
ID: NCT02672748
Sponsor: University of Wyoming
Location: University of Wyoming, Laramie
A Comparative Effectiveness Study of Major Glycemia-lowering Medications for Treatment of Type 2 Diabetes (GRADE)
The GRADE Study is a pragmatic, unmasked clinical trial that will compare commonly used diabetes medications, when combined with metformin, on glycemia-lowering effectiveness and patient-centered outcomes.
ID: NCT01794143
Sponsor: GRADE Study Group
Location: Southwestern American Indian Center, Phoenix, AZ
Home-Based Kidney Care in Native Americans of New Mexico (HBKC)
New Mexico American Indians are experiencing an epidemic of chronic kidney disease due primarily to the high rates of obesity and diabetes. The present study entitled Home-Based Kidney Care is designed to delay / reduce rates of end stage renal disease by early interventions in chronic kidney disease (CKD). Investigators propose to assess the safety and efficacy of conducting a full-scale study to determine if home based care delivered by a collaborative team composed of community health workers, the Albuquerque Area Indian Health Board and University of New Mexico faculty will decrease the risk for the development and the progression of CKD.
ID: NCT03179085
Sponsor: University of New Mexico
Contact: Vallabh Shah, PhD, [email protected]; Kevin English, PhD, [email protected]
Location: University of New Mexico, Albuquerque
Home-based Prediabetes Care in Acoma Pueblo - Study 1
Our major goal of implementing educational interventions to slow the current rate of increase in diabetes in Native communities is aligned with the National Institute of Health (NIGMS) and New Mexico INBRE’s vision in reducing health disparity using innovative interventions. The investigators propose following aims: (1) Recruit and Screen 300 community members in Acoma Pueblo, New Mexico to identify incident cases of pre-diabetes for the proposed study of Home Based Diabetes Care (HBDC); (2) Enroll 150 Acoma Natives aged 21-70 years, at risk for type 2 diabetes mellitus and conduct HBDC for a 16-week lifestyle intervention in a longitudinal cohort study.
ID: NCT04029298
Sponsor: University of New Mexico
Contact: Matthew Bouchonville, MD, [email protected]; Vallabh Shah, PhD, [email protected]
Providing access to clinical trials for patients with diabetes mellitus can be a challenge, but a significant number of trials are now recruiting patients. The clinical trials listed below are all open as of October 31, 2019; and are focused on diabetes mellitus-related treatments for American Indians. For additional information and full inclusion/exclusion criteria, please consult clinicaltrials.gov.
Cross-Sectional and Longitudinal Studies of “Pre-Diabetes” in the Pima Indians
The Pima Indians of Arizona have the highest prevalence and incidence of type 2 diabetes of any population in the world. Prospective analyses in this population have identified insulin resistance and a defect in early insulin secretion as risk factors for the development of the disease. To identify the genetic and environmental determinants of diabetes we plan to study Pima Indian families to determine: (1) if there are genes that segregate with metabolic risk factors for diabetes which might therefore be genetic markers for type 2 diabetes; and (2) the mechanisms mediating genetic and environmental determinants of insulin resistance and impaired insulin secretion.
ID: NCT00340132
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: Clifton Bogardus, MD, [email protected]
Location: NIDDK, Phoenix, AZ
Empaglifozin in Early Diabetic Kidney Disease
Diabetes is common among American Indian people and diabetic kidney disease is a common complication. Kidney disease caused by diabetes can lead to the need for kidney replacement, by dialysis or kidney transplant, and is also associated with higher risk of early death. A new diabetes medicine called empagliflozin may slow kidney disease from type 2 diabetes. Researchers want to learn if it protects the kidneys when used in very early stages of diabetic kidney disease.
ID: NCT03173963
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: Helen C Looker, [email protected]
Location: NIDDK, Phoenix, AZ
Family Investigation of Nephropathy and Diabetes
The Family Investigation of Nephropathy and Diabetes (FIND) is a multicenter study designed to identify genetic determinants of diabetic kidney disease. FIND will be conducted in 11 centers and in many ethnic groups throughout the United States. Two different strategies will be used to localize genes predisposing to kidney disease: a family-based genetic linkage study and a case-control study that utilizes admixture linkage disequilibrium. The center will conduct family-based linkage studies among American Indian populations in the southwestern United States.
ID: NCT00342927
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Contact: William C Knowler, MD, [email protected]
Location: NIDDK, Phoenix, AZ
Look AHEAD: Action for Health in Diabetes
The Look AHEAD study is a multi-center, randomized clinical trial to examine the long-term effects of a lifestyle intervention designed to achieve and maintain weight loss. The study will investigate the effects of the intervention on heart attacks, stroke and cardiovascular-related death in individuals with type 2 diabetes who are also overweight or obese.
ID: NCT00017953
Sponsor: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)
Location: Southwestern American Indian Center, Phoenix, AZ
Vitamin D and Type 2 Diabetes Study
The goal of the Vitamin D and type 2 diabetes (D2d) study is to determine if vitamin D supplementation works to delay the onset of type 2 diabetes in people at risk for the disease and to gain a better understand how vitamin D affects glucose (sugar) metabolism.
ID: NCT01942694
Sponsor: Tufts Medical Center
Locations: Southwest American Indian Center; Phoenix, AZ; Orlando VA Medical Center, FL; Atlanta VA Medical Center, Decatur, GA; Omaha VA Medical Center, NE
Reducing Diabetes Risk Factors in American Indian Children: Tribal Turning Point (TTP)
This study will evaluate a behavioral intervention designed to reduce risk factors for type 2 diabetes in American Indian youth aged 7-10 years.
ID: NCT03573856
Sponsor: University of Colorado, Denver
Contact: Katherine Sauder, PhD, [email protected]; Dana Dabelea, MD, PhD, [email protected]
Location: Childrens Hospital Colorado, Aurora
Strong Men, Strong Communities Diabetes Risk Reduction in American Indian Men (SMSC)
SMSC will inform the design and implementation of culturally informed, community-based lifestyle interventions for diabetes prevention in AI men in our partner communities and elsewhere, as well as in men of other minority groups who experience a heavy burden of diabetes.
ID: NCT02953977
Sponsor: Washington State University
Contact: Kaimi Sinclair, PhD, MPH, [email protected] Location: IREACH, Seattle, WA
Growing Resilience in Wind River Indian Reservation (GR)
The Growing Resilience research leverages reservation-based assets of land, family, culture, and front-line tribal health organizations to develop and evaluate home food gardens as a family-based health promotion intervention to reduce disparities suffered by Native Americans in nearly every measure of health. Home gardening interventions show great promise for enabling families to improve their health, and this study aims to fulfill that promise with university and Wind River Indian Reservation partners. The investigators will develop an empowering, scalable, and sustainable family-based health promotion intervention with, by, and for Native American families and conduct the first randomized controlled trial to assess the health impacts of home gardens.
ID: NCT02672748
Sponsor: University of Wyoming
Location: University of Wyoming, Laramie
A Comparative Effectiveness Study of Major Glycemia-lowering Medications for Treatment of Type 2 Diabetes (GRADE)
The GRADE Study is a pragmatic, unmasked clinical trial that will compare commonly used diabetes medications, when combined with metformin, on glycemia-lowering effectiveness and patient-centered outcomes.
ID: NCT01794143
Sponsor: GRADE Study Group
Location: Southwestern American Indian Center, Phoenix, AZ
Home-Based Kidney Care in Native Americans of New Mexico (HBKC)
New Mexico American Indians are experiencing an epidemic of chronic kidney disease due primarily to the high rates of obesity and diabetes. The present study entitled Home-Based Kidney Care is designed to delay / reduce rates of end stage renal disease by early interventions in chronic kidney disease (CKD). Investigators propose to assess the safety and efficacy of conducting a full-scale study to determine if home based care delivered by a collaborative team composed of community health workers, the Albuquerque Area Indian Health Board and University of New Mexico faculty will decrease the risk for the development and the progression of CKD.
ID: NCT03179085
Sponsor: University of New Mexico
Contact: Vallabh Shah, PhD, [email protected]; Kevin English, PhD, [email protected]
Location: University of New Mexico, Albuquerque
Home-based Prediabetes Care in Acoma Pueblo - Study 1
Our major goal of implementing educational interventions to slow the current rate of increase in diabetes in Native communities is aligned with the National Institute of Health (NIGMS) and New Mexico INBRE’s vision in reducing health disparity using innovative interventions. The investigators propose following aims: (1) Recruit and Screen 300 community members in Acoma Pueblo, New Mexico to identify incident cases of pre-diabetes for the proposed study of Home Based Diabetes Care (HBDC); (2) Enroll 150 Acoma Natives aged 21-70 years, at risk for type 2 diabetes mellitus and conduct HBDC for a 16-week lifestyle intervention in a longitudinal cohort study.
ID: NCT04029298
Sponsor: University of New Mexico
Contact: Matthew Bouchonville, MD, [email protected]; Vallabh Shah, PhD, [email protected]
Evaluating a Program Process Change to Improve Completion of Foot Exams and Amputation Risk Assessments for Veterans with Diabetes (FULL)
Individuals with diabetes mellitus (DM), peripheral vascular disease, or end-stage renal disease are at risk for a nontraumatic lower limb amputation.1 Veterans have a high number of risk factors and are especially vulnerable. More than 70% of veterans enrolled in US Department of Veterans Affairs (VA) healthcare are at increased risk for developing DM due to excess weight, poor eating habits, and physical inactivity.2 One in 4 veterans has DM, compared with 1 in 6 in the general population.2
DM can lead to long-term complications including limb amputations. Annually in the US about 73,000 nontraumatic lower limb amputations are performed and > 60% occur among persons with DM.3 Complications from diabetic wounds are the cause of 90% of lower limb amputations, and foot ulcers are the most prevalent complication.4 Diabetic ulcers are slow to heal due to vascular impairments and nerve damage.5 Peripheral vascular disease, a common comorbid condition, contributes to restricted blood flow and can lead to tissue death or gangrene requiring amputation.6
Between 2010 and 2014, VA Portland Healthcare System (VAPORHCS) had one of the highest national amputation rates in VA.7 A clinical chart review found that annual foot examinations and amputation risk assessments (ARAs) were not completed with all at-risk veterans. In 2013, a VA Office of Inspector General (OIG) national report found that more than one-third of veterans enrolled in VA with DM had no documentation of required annual foot exams.8 In 2017, VA released Directive 1410, which outlined the scope of care required to prevent and treat lower limb complications and amputations for veterans at risk for primary or secondary limb loss.1 This national initiative is a comprehensive approach that engages multiprofessional teams to perform routine foot examinations and amputation risk assessments; identify and promptly treat foot ulcers; track, monitor and educate at-risk veterans; and participate in clinical education to enhance staff skills.
To decrease the amputation rate, VAPORHCS redesigned its foot-care program to comply with the national initiative. As is typical in VA, VAPORHCS uses a team-based approach in primary care. The basic 4-member team patient-aligned care team (PACT) consists of a physician or nurse practitioner (NP) primary care provider (PCP), a registered nurse (RN) care manager, a licensed practical nurse (LPN), and a medical staff assistant (MSA) for administrative support. Each PACT cares for about 1,800 veterans. Formerly, LPNs completed the annual diabetic foot exams, and PCPs verified the exams and completed the ARA based on the LPNs’ findings. If patients were moderate risk or high risk, they were referred to podiatry. The VAPORHCS audit found that not all at-risk veterans had both the foot exam and ARA completed, or were referred to podiatry when indicated. There was a need for a process improvement project to develop a seamless program consisting of all recommended foot care components crucial for timely care.
This quality improvement project sought to evaluate the effectiveness of the process changes by examining PCPs’ adoption of, and consistency in completing annual diabetic foot exams and ARAs with veterans. The goals of the project were to evaluate changes in the: (1) Number of accurate diabetic foot exams and amputation risk assessments completed with veterans with DM; (2) Number and timeliness of appropriate referrals to podiatry for an in-depth assessment and treatment of veterans found to be at moderate-to-high risk for lower limb amputations; and (3) Number of administrative text orders entered by PCPs for nurse care managers to offer foot care education and the completion of the education with veterans found to be at normal-to-low risk for lower limb amputations. The institutional review boards of VAPORHCS and Gonzaga University approved the study.
Methods
Established by the American Diabetes Association and endorsed by the American Association of Clinical Endocrinologists, the comprehensive foot exam includes a visual exam, pedal pulse checks, and a sensory exam.9,10 The templated Computerized Patient Record System (CPRS) electronic health record note specifies normal and abnormal parameters of each section. On the same template, the provider assigns an ARA score based on the results of the completed foot exam. Risk scores range from 0 to 3 (0, normal or no risk; 1, low risk, 2; moderate risk; 3, high risk) If the veteran has normal or low risk, the PCP can encourage the veteran to remain at low risk by entering an administrative CPRS text order for the nurse care manager to offer education about daily foot care at the same visit or at a scheduled follow-up visit. This process facilitates nurse care managers to include routine foot care as integral to their usual duties coaching veterans to engage in self-care to manage chronic conditions. If the risk is assessed as moderate or high risk, PCPs are prompted to send a referral to podiatry to repeat the foot exam, verify the ARA score, and provide appropriate foot care treatment and follow-up.
On October 31, 2017, following training on the updated foot exam and ARA template with staff at the 13 VAPORHCS outpatient clinic sites, 2 sites piloted all components of the Comprehensive Foot Care program. An in-person training was completed with PCPs to review the changes of the foot care template in CPRS and to answer their questions about it. PCPs were required to complete both the 3-part foot exam and ARA at least once annually with veterans with DM.
An electronic clinical reminder was built to alert PCPs and PACTs that a veteran was either due or overdue for an exam and risk assessment. VA podiatrists agreed to complete the reminder with veterans under their care. One of the 2 sites was randomly selected for this study. Data were collected from August 1, 2017 to July 31, 2018. Patients were identified from the Diabetes Registry, a database established at VAPORHCS in 2008 to track veterans with DM to ensure quality care.11 Veterans’ personal health identifiers from the registry were used to access their health records to complete chart reviews and assess the completion, accuracy and timeliness of all foot care components.
The Diabetes Registry lists a veterans’ upcoming appointments and tracks their most recent clinic visits; laboratory tests; physical exams; and screening exams for foot, eye, and renal care. Newly diagnosed veterans are uploaded automatically into this registry by tracking all DM-related International Classification of Diseases (ICD-10) codes, hemoglobin A1c (HbA1c) levels ≥ 6.5%, or outpatient prescriptions for insulin or oral hypoglycemic agents.11
Study Design
This quality improvement project evaluated PCPs’ actions in a program process change intended to improve foot care provided with veterans at-risk for nontraumatic lower limb amputations. Audits of CPRS records and the Diabetes Registry determined the results of the practice change. Data on the total number of foot exams, amputation risk scores, appropriate podiatry referrals, administrative orders for nurse coaching, and completed foot care education were collected during the study period. Data collected for the 3-month period preceding the process change established preimplementation comparison vs the postimplementation data. Data were collected at 3, 6, and 9 months after implementation. The foot exams and ARAs were reviewed to determine whether exams and assessments were completed accurately during the pre- and post-implementation timeframes. Incomplete or clearly incorrectly completed documentation were considered inaccurate. For example, it was considered inaccurate if only the foot exam portion was completed in the assessment and the ARA was not.
Data Analysis
Data on the total number of accurately completed foot examinations and ARAs, total number of podiatry referrals, and total number of administrative text orders placed by PCPs, and education completed by nurse care managers were assessed. Statistical significance was evaluated using χ2 and Fisher exact test as appropriate. A Pearson correlation coefficient was used to determine whether there was a statistically significant increase in accurate foot examinations and ARAs as well as total number of podiatry referrals during the study period. Statistical analyses were performed using Stata 14.1 statistical software (College Station, TX).
Results
A total of 1,242 completed diabetic foot examinations were identified from August 1, 2017 to July 31, 2018 using the Diabetes Registry (Table). For the 3 months prior to the change, there were 191 appropriately completed foot examinations and ARAs. This number increased progressively over three 3-month periods (Figure 1). Within the 1-year study period, there was a statistically significant increase in the number of appropriate foot examinations (r = 0.495). PCPs placed 34 podiatry referrals during the prechange period. After the change, the number of appropriate referrals increased statistically significantly in the 3 following 3-month-periods (r = 0.222) (Figure 2).
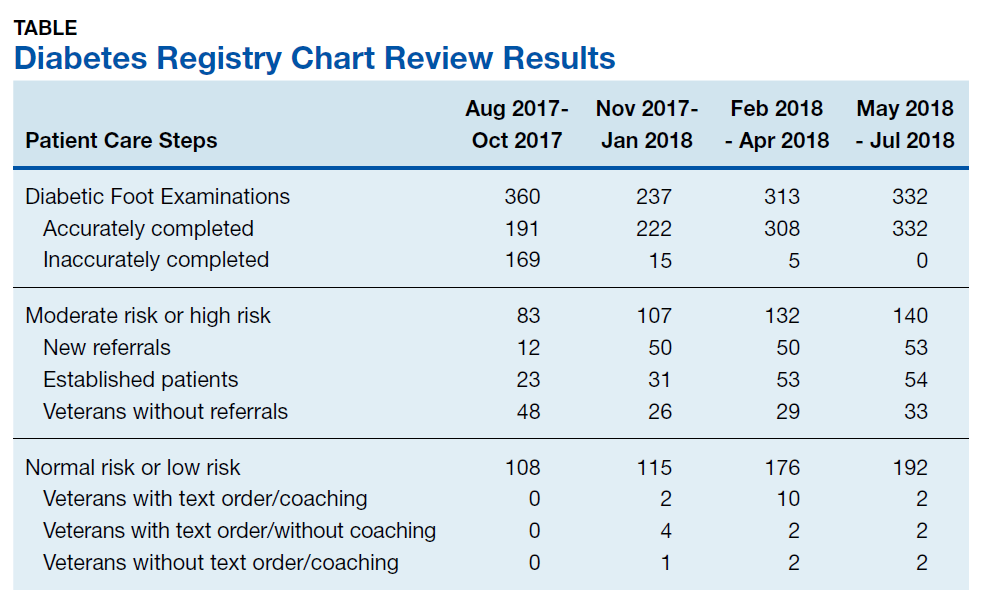
To determine the accuracy of documentation and ratio of appropriate referrals, the 3-month prech
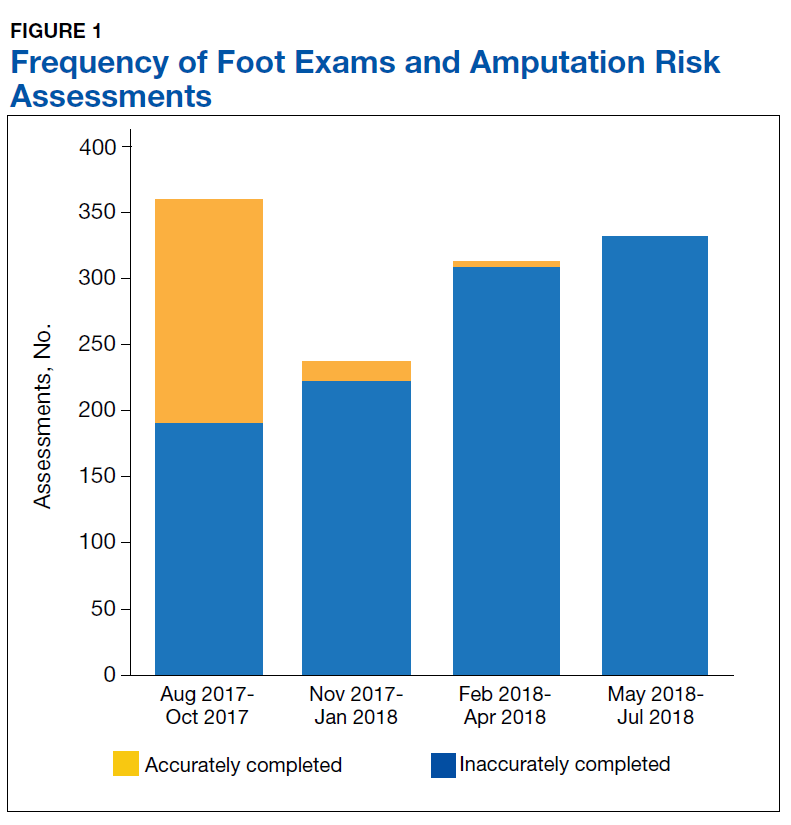
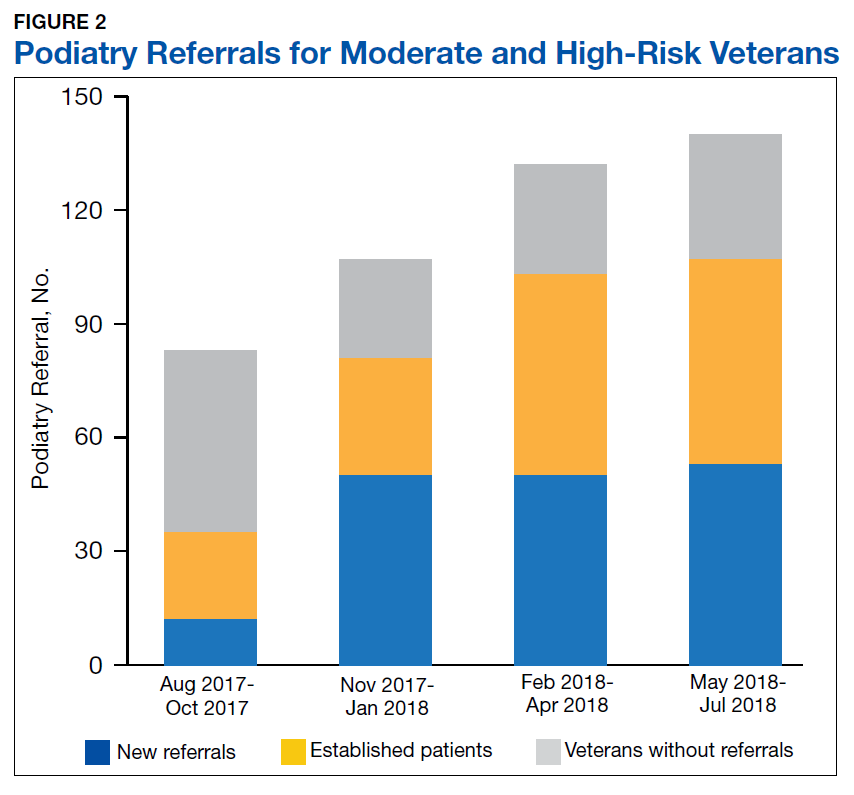
Notably, at the end of the first year of this evaluation, 119 veterans at the clinic did not show a recorded comprehensive foot examination since receiving a DM diagnosis and 299 veterans were due for an annual examination—a 25.2% gap of veterans without the recommended progression of foot care services. Of those that previously had a recorded foot examination, 51 (17.0%) veterans were found to be ≥ 2 years overdue.
Discussion
DM management requires a comprehensive team-based approach to help monitor for associated complications. At the VA, PACTs are veterans’ initial and primary point of contact for chronic condition management. PACTs have regular opportunities to engage veterans in initial and follow-up care and appropriate self-care. PCPs are critical in placing referrals for specialized care promptly to prevent and minimize complications such as foot ulcers, and ultimately, lower limb amputations.9,10,12
When PCPs assume responsibility for the entire foot examination, they are able to identify problems early.1 Left untreated, foot wounds and ulcers have the potential to grow into serious infections.9 Early risk identification and management can lead to increased patient satisfaction, improved life expectancy, quality of life, and ultimately, lower healthcare costs.12
Multiple studies have shown the clinical importance of foot examinations in preventative care. In one study, researchers found that completing foot examinations, among other early interventions, increased life expectancy and reduced foot complications.13 Diabetic foot management programs involving screening and categorizing patients into low- and high-risk groups had a 47.4% decrease in the incidence of amputations and 37.8% decrease in hospital admissions.14 Amputations were found to be inversely correlated with multidisciplinary foot care programs with reduction of lower limb amputations at 2 years.15 The Centers for Disease Control and Prevention found that comprehensive foot care programs that include a foot examination, ARA, appropriate referrals to specialists, and foot-care education and preventative services can reduce lower limb amputation rates by 45% to 85%.16
With one of the highest amputation rates in VA, VAPORHCS needed an integrated approach to ensure that appropriate foot care occurred regularly with veterans with DM. Prior to the process change, LPNs completed foot examinations and PCPs completed the ARA. Separating these clinical services resulted in few veterans receiving an amputation risk score. Of those with scores, the lack of a standardized program protocol resulted in discrepancies between ARAs from patient to patient as health care providers did not have clear or enough information to select the correct score and make the appropriate referrals. Thus, veterans previously identified as at moderate or high risk also lacked podiatric follow-up care.
The new quality-driven process change corrected the documentation process to nationally accepted standards. The goal was to create a consistent template in the electronic health record for all health care providers. The new template simplifies the documentation process and clarifies the amputation risk score assignment, which allows for proper foot care management. The PCP completes the process from assessment through referral, removing gaps in care and improving efficiency. Although this change was initially met with resistance from PCPs, it led to a significant increase in the number of patients with accurately documented examinations. Similarly, the number of appropriate referrals significantly rose during the study period. The standardized documentation process resulted in improved accurate examinations and ARAs over the past year. The new program also resulted in an increased number of appropriate podiatry referrals for those identified to be at moderate or high risk. This elevation of care is crucial for veterans to receive frequent follow-up visits for preventative care and/or treatment, including surgical modalities to promote limb salvage.
Barriers
This project identified several barriers to the Comprehensive Foot Care program. One major barrier was health care provider resistance to using the new process. For example, VAPORHCS podiatrists are not using the new template with established patients, which requires PCPs to complete the clinical reminder template for quality performance, an additional burden unrelated to clinical care. PCPs that do complete the foot examination/ARA templated note use the podiatrist’s visit note without personally assessing the patient.
PCPs also have been resistant to entering administrative text orders for preventative foot care in normal- or low-risk veterans (4.6% overall), which has resulted in decreased patient education (3.9% overall). Education for normal-risk and low-risk patients is designed to engage veterans in self-care and prevent risk progression, critical to prevention.
It was found that PCPs often did not ask nurses to coach normal- or low-risk veterans on preventative foot care, as suggested by the low rates at which patients were offered education. This is an area we will target with future quality improvement efforts. All patients with DM should have general education about risk factors and appropriate management of them to decrease their risk for complications.9 Preventative foot care education is a critical resource to share with patients during health coaching opportunities to clarify misunderstandings and support change talk when patients are ambivalent or resistant to change. Individual or group-based nurse visits can facilitate better coaching for patients.
At the VA, coaching begins with a conversation about what matters most to the veteran, facilitating the development of a personalized plan based on patients’ values, needs, preferences and goals.9,10,12,17 Coaching allows nurses to assess veterans’ knowledge and willingness to engage in healthy habits; and identify additional resources to help them achieve their goals.
Limitations
There are many limitations to this short quality improvement analysis. For example, only 1 of 2 clinics that piloted the program change was evaluated. In addition, there are 11 other clinics that need additional in-depth education on the program change. Although this analysis was overwhelmingly positive, it may not be as successful at other clinic sites and may be subject to the Hawthorne effect—since the 2 piloted locations knew they were being observed for the quality improvement program and may have made an extra effort to be compliant.18 Additionally, we were unable to track the records of veterans receiving care through the VA Choice Program for this analysis resulting in a lack of documentation of completed diabetic foot examinations and a lack of internal referrals to VA podiatry.
Another major limitation of this project involved calculating the number of referrals placed to podiatry. On January 1, 2018, about halfway through the program evaluation, a national VA decision enabled veterans to self-refer to podiatry, which may have limited the number of podiatry referrals placed by PCPs. Finally, patients could refuse podiatry referrals. In the 9-month postimplementation period, 57 (64.8%) veterans declined podiatry referrals, according to their CPRS records.
Although, there was an improvement in the accuracy of diabetic foot examinations, ARAs, and appropriate podiatry referrals, the ultimate goal of reducing diabetic foot ulcers and lower limb amputations was not tracked due to the limited timeframe of this analysis. Tracking these endpoints with continuous plan-do-study-act cycles are needed for this ongoing quality improvement project.
Conclusion
The goal of the VAPORHCS Comprehensive Foot Care program is to provide veterans with a program that is predictable, easy and consistent to prevent and treat foot ulcers to reduce the rate of lower limb amputations. It requires multidisciplinary team collaboration for success. Implementation of this new comprehensive program has increased the number of accurate annual foot exams, ARAs and podiatry referrals. Despite these improvements, areas of future improvement include emphasizing patient education and ongoing provider compliance with annual assessments.
Author contributions
MHG proposed the program evaluation project idea. TVQ collected and analyzed the data and wrote the manuscript. MHG oversaw the project and edited the manuscript. TVQ is the guarantor of this project and takes responsibility for the contents of this journal article.
Acknowledgments
The authors thank Tyra Haebe, VAPORHCS Prevention of Amputation in Veterans Everywhere (PAVE) Manager, and the entire VAPORHCS PAVE committee for their support in this program evaluation project.
1. US Department of Veterans Affairs, Veterans Health Administration. VHA directive 1410, prevention of amputation in veterans everywhere (PAVE) program. http://vaww.medical surgical.va.gov/podiatry/docs/VHADirective_1410_PAVE.pdf. Published March 31, 2017. Accessed October 11, 2019.
2. US Department of Veterans Affairs. Close to 25 percent of VA patients have diabetes http://www.va.gov/health/NewsFeatures/20111115a.asp. Accessed 14 October 2017
3. Centers for Disease Control and Prevention. National diabetes statistics report, 2017: Estimates of Diabetes and Its Burden in the United States. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf. Accessed October 11, 2019.
4. Gibson LW, Abbas A: Limb salvage for veterans with diabetes: to care for him who has borne the battle. Crit Care Nurs Clin North Am. 2012;25(1):131-134
5. Boyko EJ, Monteiro-Soares M, Wheeler SGB. “Peripheral arterial disease, foot ulcers, lower extremity amputations, and diabetes.” In: Cowie CC, Casagrande SS, Menke A, et al, eds. Diabetes in America. 3rd ed. Bethesda, MD: National Institutes of Health Publication; 2017:20-21,20-34.
6. National Institute of Health, National Institute of Neurological Disorders and Stroke. Peripheral neuropathy fact sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Peripheral-Neuropathy-Fact-Sheet. Updated August 13, 2019. Accessed October 11, 2019.
7. US Department of Veterans Affairs, Veterans Health Administration, Support Services Center. Amputation cube, lower amputations 2015. http://vssc.med.va.gov/AlphaIndex. [Nonpublic source, not verified]
8. US Department of Veterans Affairs, Office of Inspector General. Healthcare inspection: Foot care for patients with diabetes and additional risk factors for amputation. https://www.va.gov/oig/pubs/VAOIG-11-00711-74.pdf. Published January 17, 2013. Accessed October 11, 2019.
9. American Diabetes Association. Standards of medical care in diabetes - 2017. Diabetes Care. 2017;40(suppl 1):1-142.
10. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31(8):1679-1685.
11. Yang J, McConnachie J, Renfro R, Schreiner S, Tallett S, Winterbottom L. The diabetes registry and future panel management tool https://docplayer.net/19062632-The-diabetes-registry-and.html. Accessed October 11, 2019.
12. National Institute of Health, Centers for Disease Control and Prevention, the National Diabetes Education Program. Working together to manage diabetes: a guide for pharmcy, podiatry, optometry, and dentistry. https://www.cdc.gov/diabetes/ndep/pdfs/ppod-guide.pdf. Accessed October 11, 2019.
13. Ortegon MM, Redekop WK, Niessen LW. Cost-effectiveness of prevention and treatment of the diabetic foot: a Markov analysis. Diabetes Care. 2004;27(4):901-907.
14. Lavery LA, Wunderlich RP, Tredwell JL. Disease management for the diabetic foot: effectiveness of a diabetic foot prevention program to reduce amputations and hospitalizations. Diabetes Res Clin Pract. 2005;70(1):31-37.
15. Paisey RB, Abbott A, Levenson R, et al; South-West Cardiovascular Strategic Clinical Network peer diabetic foot service review team. Diabetes-related major lower limb amputation incidence is strongly related to diabetic foot service provision and improves with enhancement of services: peer review of the south-west of England. Diabet Med. 2017;35(1):53-62.
16. Centers for Disease Control and Prevention. National diabetes fact sheet: National estimates and general information on diabetes and prediabetes in the United States, 2011. https://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Published 2011. Accessed October 11, 2019.
17. US Department of Veterans Affairs. Whole health for life. https://www.va.gov/patientcenteredcare/explore/about-whole-health.asp. Updated July 20, 2017. Accessed October 11, 2019.
18. Parsons HM. What happened at Hawthorne? New evidence suggests the Hawthorne effect resulted from operant reinforcement contingencies. Science. 1974;183(4128):922–9322.
Individuals with diabetes mellitus (DM), peripheral vascular disease, or end-stage renal disease are at risk for a nontraumatic lower limb amputation.1 Veterans have a high number of risk factors and are especially vulnerable. More than 70% of veterans enrolled in US Department of Veterans Affairs (VA) healthcare are at increased risk for developing DM due to excess weight, poor eating habits, and physical inactivity.2 One in 4 veterans has DM, compared with 1 in 6 in the general population.2
DM can lead to long-term complications including limb amputations. Annually in the US about 73,000 nontraumatic lower limb amputations are performed and > 60% occur among persons with DM.3 Complications from diabetic wounds are the cause of 90% of lower limb amputations, and foot ulcers are the most prevalent complication.4 Diabetic ulcers are slow to heal due to vascular impairments and nerve damage.5 Peripheral vascular disease, a common comorbid condition, contributes to restricted blood flow and can lead to tissue death or gangrene requiring amputation.6
Between 2010 and 2014, VA Portland Healthcare System (VAPORHCS) had one of the highest national amputation rates in VA.7 A clinical chart review found that annual foot examinations and amputation risk assessments (ARAs) were not completed with all at-risk veterans. In 2013, a VA Office of Inspector General (OIG) national report found that more than one-third of veterans enrolled in VA with DM had no documentation of required annual foot exams.8 In 2017, VA released Directive 1410, which outlined the scope of care required to prevent and treat lower limb complications and amputations for veterans at risk for primary or secondary limb loss.1 This national initiative is a comprehensive approach that engages multiprofessional teams to perform routine foot examinations and amputation risk assessments; identify and promptly treat foot ulcers; track, monitor and educate at-risk veterans; and participate in clinical education to enhance staff skills.
To decrease the amputation rate, VAPORHCS redesigned its foot-care program to comply with the national initiative. As is typical in VA, VAPORHCS uses a team-based approach in primary care. The basic 4-member team patient-aligned care team (PACT) consists of a physician or nurse practitioner (NP) primary care provider (PCP), a registered nurse (RN) care manager, a licensed practical nurse (LPN), and a medical staff assistant (MSA) for administrative support. Each PACT cares for about 1,800 veterans. Formerly, LPNs completed the annual diabetic foot exams, and PCPs verified the exams and completed the ARA based on the LPNs’ findings. If patients were moderate risk or high risk, they were referred to podiatry. The VAPORHCS audit found that not all at-risk veterans had both the foot exam and ARA completed, or were referred to podiatry when indicated. There was a need for a process improvement project to develop a seamless program consisting of all recommended foot care components crucial for timely care.
This quality improvement project sought to evaluate the effectiveness of the process changes by examining PCPs’ adoption of, and consistency in completing annual diabetic foot exams and ARAs with veterans. The goals of the project were to evaluate changes in the: (1) Number of accurate diabetic foot exams and amputation risk assessments completed with veterans with DM; (2) Number and timeliness of appropriate referrals to podiatry for an in-depth assessment and treatment of veterans found to be at moderate-to-high risk for lower limb amputations; and (3) Number of administrative text orders entered by PCPs for nurse care managers to offer foot care education and the completion of the education with veterans found to be at normal-to-low risk for lower limb amputations. The institutional review boards of VAPORHCS and Gonzaga University approved the study.
Methods
Established by the American Diabetes Association and endorsed by the American Association of Clinical Endocrinologists, the comprehensive foot exam includes a visual exam, pedal pulse checks, and a sensory exam.9,10 The templated Computerized Patient Record System (CPRS) electronic health record note specifies normal and abnormal parameters of each section. On the same template, the provider assigns an ARA score based on the results of the completed foot exam. Risk scores range from 0 to 3 (0, normal or no risk; 1, low risk, 2; moderate risk; 3, high risk) If the veteran has normal or low risk, the PCP can encourage the veteran to remain at low risk by entering an administrative CPRS text order for the nurse care manager to offer education about daily foot care at the same visit or at a scheduled follow-up visit. This process facilitates nurse care managers to include routine foot care as integral to their usual duties coaching veterans to engage in self-care to manage chronic conditions. If the risk is assessed as moderate or high risk, PCPs are prompted to send a referral to podiatry to repeat the foot exam, verify the ARA score, and provide appropriate foot care treatment and follow-up.
On October 31, 2017, following training on the updated foot exam and ARA template with staff at the 13 VAPORHCS outpatient clinic sites, 2 sites piloted all components of the Comprehensive Foot Care program. An in-person training was completed with PCPs to review the changes of the foot care template in CPRS and to answer their questions about it. PCPs were required to complete both the 3-part foot exam and ARA at least once annually with veterans with DM.
An electronic clinical reminder was built to alert PCPs and PACTs that a veteran was either due or overdue for an exam and risk assessment. VA podiatrists agreed to complete the reminder with veterans under their care. One of the 2 sites was randomly selected for this study. Data were collected from August 1, 2017 to July 31, 2018. Patients were identified from the Diabetes Registry, a database established at VAPORHCS in 2008 to track veterans with DM to ensure quality care.11 Veterans’ personal health identifiers from the registry were used to access their health records to complete chart reviews and assess the completion, accuracy and timeliness of all foot care components.
The Diabetes Registry lists a veterans’ upcoming appointments and tracks their most recent clinic visits; laboratory tests; physical exams; and screening exams for foot, eye, and renal care. Newly diagnosed veterans are uploaded automatically into this registry by tracking all DM-related International Classification of Diseases (ICD-10) codes, hemoglobin A1c (HbA1c) levels ≥ 6.5%, or outpatient prescriptions for insulin or oral hypoglycemic agents.11
Study Design
This quality improvement project evaluated PCPs’ actions in a program process change intended to improve foot care provided with veterans at-risk for nontraumatic lower limb amputations. Audits of CPRS records and the Diabetes Registry determined the results of the practice change. Data on the total number of foot exams, amputation risk scores, appropriate podiatry referrals, administrative orders for nurse coaching, and completed foot care education were collected during the study period. Data collected for the 3-month period preceding the process change established preimplementation comparison vs the postimplementation data. Data were collected at 3, 6, and 9 months after implementation. The foot exams and ARAs were reviewed to determine whether exams and assessments were completed accurately during the pre- and post-implementation timeframes. Incomplete or clearly incorrectly completed documentation were considered inaccurate. For example, it was considered inaccurate if only the foot exam portion was completed in the assessment and the ARA was not.
Data Analysis
Data on the total number of accurately completed foot examinations and ARAs, total number of podiatry referrals, and total number of administrative text orders placed by PCPs, and education completed by nurse care managers were assessed. Statistical significance was evaluated using χ2 and Fisher exact test as appropriate. A Pearson correlation coefficient was used to determine whether there was a statistically significant increase in accurate foot examinations and ARAs as well as total number of podiatry referrals during the study period. Statistical analyses were performed using Stata 14.1 statistical software (College Station, TX).
Results
A total of 1,242 completed diabetic foot examinations were identified from August 1, 2017 to July 31, 2018 using the Diabetes Registry (Table). For the 3 months prior to the change, there were 191 appropriately completed foot examinations and ARAs. This number increased progressively over three 3-month periods (Figure 1). Within the 1-year study period, there was a statistically significant increase in the number of appropriate foot examinations (r = 0.495). PCPs placed 34 podiatry referrals during the prechange period. After the change, the number of appropriate referrals increased statistically significantly in the 3 following 3-month-periods (r = 0.222) (Figure 2).
To determine the accuracy of documentation and ratio of appropriate referrals, the 3-month prech
Notably, at the end of the first year of this evaluation, 119 veterans at the clinic did not show a recorded comprehensive foot examination since receiving a DM diagnosis and 299 veterans were due for an annual examination—a 25.2% gap of veterans without the recommended progression of foot care services. Of those that previously had a recorded foot examination, 51 (17.0%) veterans were found to be ≥ 2 years overdue.
Discussion
DM management requires a comprehensive team-based approach to help monitor for associated complications. At the VA, PACTs are veterans’ initial and primary point of contact for chronic condition management. PACTs have regular opportunities to engage veterans in initial and follow-up care and appropriate self-care. PCPs are critical in placing referrals for specialized care promptly to prevent and minimize complications such as foot ulcers, and ultimately, lower limb amputations.9,10,12
When PCPs assume responsibility for the entire foot examination, they are able to identify problems early.1 Left untreated, foot wounds and ulcers have the potential to grow into serious infections.9 Early risk identification and management can lead to increased patient satisfaction, improved life expectancy, quality of life, and ultimately, lower healthcare costs.12
Multiple studies have shown the clinical importance of foot examinations in preventative care. In one study, researchers found that completing foot examinations, among other early interventions, increased life expectancy and reduced foot complications.13 Diabetic foot management programs involving screening and categorizing patients into low- and high-risk groups had a 47.4% decrease in the incidence of amputations and 37.8% decrease in hospital admissions.14 Amputations were found to be inversely correlated with multidisciplinary foot care programs with reduction of lower limb amputations at 2 years.15 The Centers for Disease Control and Prevention found that comprehensive foot care programs that include a foot examination, ARA, appropriate referrals to specialists, and foot-care education and preventative services can reduce lower limb amputation rates by 45% to 85%.16
With one of the highest amputation rates in VA, VAPORHCS needed an integrated approach to ensure that appropriate foot care occurred regularly with veterans with DM. Prior to the process change, LPNs completed foot examinations and PCPs completed the ARA. Separating these clinical services resulted in few veterans receiving an amputation risk score. Of those with scores, the lack of a standardized program protocol resulted in discrepancies between ARAs from patient to patient as health care providers did not have clear or enough information to select the correct score and make the appropriate referrals. Thus, veterans previously identified as at moderate or high risk also lacked podiatric follow-up care.
The new quality-driven process change corrected the documentation process to nationally accepted standards. The goal was to create a consistent template in the electronic health record for all health care providers. The new template simplifies the documentation process and clarifies the amputation risk score assignment, which allows for proper foot care management. The PCP completes the process from assessment through referral, removing gaps in care and improving efficiency. Although this change was initially met with resistance from PCPs, it led to a significant increase in the number of patients with accurately documented examinations. Similarly, the number of appropriate referrals significantly rose during the study period. The standardized documentation process resulted in improved accurate examinations and ARAs over the past year. The new program also resulted in an increased number of appropriate podiatry referrals for those identified to be at moderate or high risk. This elevation of care is crucial for veterans to receive frequent follow-up visits for preventative care and/or treatment, including surgical modalities to promote limb salvage.
Barriers
This project identified several barriers to the Comprehensive Foot Care program. One major barrier was health care provider resistance to using the new process. For example, VAPORHCS podiatrists are not using the new template with established patients, which requires PCPs to complete the clinical reminder template for quality performance, an additional burden unrelated to clinical care. PCPs that do complete the foot examination/ARA templated note use the podiatrist’s visit note without personally assessing the patient.
PCPs also have been resistant to entering administrative text orders for preventative foot care in normal- or low-risk veterans (4.6% overall), which has resulted in decreased patient education (3.9% overall). Education for normal-risk and low-risk patients is designed to engage veterans in self-care and prevent risk progression, critical to prevention.
It was found that PCPs often did not ask nurses to coach normal- or low-risk veterans on preventative foot care, as suggested by the low rates at which patients were offered education. This is an area we will target with future quality improvement efforts. All patients with DM should have general education about risk factors and appropriate management of them to decrease their risk for complications.9 Preventative foot care education is a critical resource to share with patients during health coaching opportunities to clarify misunderstandings and support change talk when patients are ambivalent or resistant to change. Individual or group-based nurse visits can facilitate better coaching for patients.
At the VA, coaching begins with a conversation about what matters most to the veteran, facilitating the development of a personalized plan based on patients’ values, needs, preferences and goals.9,10,12,17 Coaching allows nurses to assess veterans’ knowledge and willingness to engage in healthy habits; and identify additional resources to help them achieve their goals.
Limitations
There are many limitations to this short quality improvement analysis. For example, only 1 of 2 clinics that piloted the program change was evaluated. In addition, there are 11 other clinics that need additional in-depth education on the program change. Although this analysis was overwhelmingly positive, it may not be as successful at other clinic sites and may be subject to the Hawthorne effect—since the 2 piloted locations knew they were being observed for the quality improvement program and may have made an extra effort to be compliant.18 Additionally, we were unable to track the records of veterans receiving care through the VA Choice Program for this analysis resulting in a lack of documentation of completed diabetic foot examinations and a lack of internal referrals to VA podiatry.
Another major limitation of this project involved calculating the number of referrals placed to podiatry. On January 1, 2018, about halfway through the program evaluation, a national VA decision enabled veterans to self-refer to podiatry, which may have limited the number of podiatry referrals placed by PCPs. Finally, patients could refuse podiatry referrals. In the 9-month postimplementation period, 57 (64.8%) veterans declined podiatry referrals, according to their CPRS records.
Although, there was an improvement in the accuracy of diabetic foot examinations, ARAs, and appropriate podiatry referrals, the ultimate goal of reducing diabetic foot ulcers and lower limb amputations was not tracked due to the limited timeframe of this analysis. Tracking these endpoints with continuous plan-do-study-act cycles are needed for this ongoing quality improvement project.
Conclusion
The goal of the VAPORHCS Comprehensive Foot Care program is to provide veterans with a program that is predictable, easy and consistent to prevent and treat foot ulcers to reduce the rate of lower limb amputations. It requires multidisciplinary team collaboration for success. Implementation of this new comprehensive program has increased the number of accurate annual foot exams, ARAs and podiatry referrals. Despite these improvements, areas of future improvement include emphasizing patient education and ongoing provider compliance with annual assessments.
Author contributions
MHG proposed the program evaluation project idea. TVQ collected and analyzed the data and wrote the manuscript. MHG oversaw the project and edited the manuscript. TVQ is the guarantor of this project and takes responsibility for the contents of this journal article.
Acknowledgments
The authors thank Tyra Haebe, VAPORHCS Prevention of Amputation in Veterans Everywhere (PAVE) Manager, and the entire VAPORHCS PAVE committee for their support in this program evaluation project.
Individuals with diabetes mellitus (DM), peripheral vascular disease, or end-stage renal disease are at risk for a nontraumatic lower limb amputation.1 Veterans have a high number of risk factors and are especially vulnerable. More than 70% of veterans enrolled in US Department of Veterans Affairs (VA) healthcare are at increased risk for developing DM due to excess weight, poor eating habits, and physical inactivity.2 One in 4 veterans has DM, compared with 1 in 6 in the general population.2
DM can lead to long-term complications including limb amputations. Annually in the US about 73,000 nontraumatic lower limb amputations are performed and > 60% occur among persons with DM.3 Complications from diabetic wounds are the cause of 90% of lower limb amputations, and foot ulcers are the most prevalent complication.4 Diabetic ulcers are slow to heal due to vascular impairments and nerve damage.5 Peripheral vascular disease, a common comorbid condition, contributes to restricted blood flow and can lead to tissue death or gangrene requiring amputation.6
Between 2010 and 2014, VA Portland Healthcare System (VAPORHCS) had one of the highest national amputation rates in VA.7 A clinical chart review found that annual foot examinations and amputation risk assessments (ARAs) were not completed with all at-risk veterans. In 2013, a VA Office of Inspector General (OIG) national report found that more than one-third of veterans enrolled in VA with DM had no documentation of required annual foot exams.8 In 2017, VA released Directive 1410, which outlined the scope of care required to prevent and treat lower limb complications and amputations for veterans at risk for primary or secondary limb loss.1 This national initiative is a comprehensive approach that engages multiprofessional teams to perform routine foot examinations and amputation risk assessments; identify and promptly treat foot ulcers; track, monitor and educate at-risk veterans; and participate in clinical education to enhance staff skills.
To decrease the amputation rate, VAPORHCS redesigned its foot-care program to comply with the national initiative. As is typical in VA, VAPORHCS uses a team-based approach in primary care. The basic 4-member team patient-aligned care team (PACT) consists of a physician or nurse practitioner (NP) primary care provider (PCP), a registered nurse (RN) care manager, a licensed practical nurse (LPN), and a medical staff assistant (MSA) for administrative support. Each PACT cares for about 1,800 veterans. Formerly, LPNs completed the annual diabetic foot exams, and PCPs verified the exams and completed the ARA based on the LPNs’ findings. If patients were moderate risk or high risk, they were referred to podiatry. The VAPORHCS audit found that not all at-risk veterans had both the foot exam and ARA completed, or were referred to podiatry when indicated. There was a need for a process improvement project to develop a seamless program consisting of all recommended foot care components crucial for timely care.
This quality improvement project sought to evaluate the effectiveness of the process changes by examining PCPs’ adoption of, and consistency in completing annual diabetic foot exams and ARAs with veterans. The goals of the project were to evaluate changes in the: (1) Number of accurate diabetic foot exams and amputation risk assessments completed with veterans with DM; (2) Number and timeliness of appropriate referrals to podiatry for an in-depth assessment and treatment of veterans found to be at moderate-to-high risk for lower limb amputations; and (3) Number of administrative text orders entered by PCPs for nurse care managers to offer foot care education and the completion of the education with veterans found to be at normal-to-low risk for lower limb amputations. The institutional review boards of VAPORHCS and Gonzaga University approved the study.
Methods
Established by the American Diabetes Association and endorsed by the American Association of Clinical Endocrinologists, the comprehensive foot exam includes a visual exam, pedal pulse checks, and a sensory exam.9,10 The templated Computerized Patient Record System (CPRS) electronic health record note specifies normal and abnormal parameters of each section. On the same template, the provider assigns an ARA score based on the results of the completed foot exam. Risk scores range from 0 to 3 (0, normal or no risk; 1, low risk, 2; moderate risk; 3, high risk) If the veteran has normal or low risk, the PCP can encourage the veteran to remain at low risk by entering an administrative CPRS text order for the nurse care manager to offer education about daily foot care at the same visit or at a scheduled follow-up visit. This process facilitates nurse care managers to include routine foot care as integral to their usual duties coaching veterans to engage in self-care to manage chronic conditions. If the risk is assessed as moderate or high risk, PCPs are prompted to send a referral to podiatry to repeat the foot exam, verify the ARA score, and provide appropriate foot care treatment and follow-up.
On October 31, 2017, following training on the updated foot exam and ARA template with staff at the 13 VAPORHCS outpatient clinic sites, 2 sites piloted all components of the Comprehensive Foot Care program. An in-person training was completed with PCPs to review the changes of the foot care template in CPRS and to answer their questions about it. PCPs were required to complete both the 3-part foot exam and ARA at least once annually with veterans with DM.
An electronic clinical reminder was built to alert PCPs and PACTs that a veteran was either due or overdue for an exam and risk assessment. VA podiatrists agreed to complete the reminder with veterans under their care. One of the 2 sites was randomly selected for this study. Data were collected from August 1, 2017 to July 31, 2018. Patients were identified from the Diabetes Registry, a database established at VAPORHCS in 2008 to track veterans with DM to ensure quality care.11 Veterans’ personal health identifiers from the registry were used to access their health records to complete chart reviews and assess the completion, accuracy and timeliness of all foot care components.
The Diabetes Registry lists a veterans’ upcoming appointments and tracks their most recent clinic visits; laboratory tests; physical exams; and screening exams for foot, eye, and renal care. Newly diagnosed veterans are uploaded automatically into this registry by tracking all DM-related International Classification of Diseases (ICD-10) codes, hemoglobin A1c (HbA1c) levels ≥ 6.5%, or outpatient prescriptions for insulin or oral hypoglycemic agents.11
Study Design
This quality improvement project evaluated PCPs’ actions in a program process change intended to improve foot care provided with veterans at-risk for nontraumatic lower limb amputations. Audits of CPRS records and the Diabetes Registry determined the results of the practice change. Data on the total number of foot exams, amputation risk scores, appropriate podiatry referrals, administrative orders for nurse coaching, and completed foot care education were collected during the study period. Data collected for the 3-month period preceding the process change established preimplementation comparison vs the postimplementation data. Data were collected at 3, 6, and 9 months after implementation. The foot exams and ARAs were reviewed to determine whether exams and assessments were completed accurately during the pre- and post-implementation timeframes. Incomplete or clearly incorrectly completed documentation were considered inaccurate. For example, it was considered inaccurate if only the foot exam portion was completed in the assessment and the ARA was not.
Data Analysis
Data on the total number of accurately completed foot examinations and ARAs, total number of podiatry referrals, and total number of administrative text orders placed by PCPs, and education completed by nurse care managers were assessed. Statistical significance was evaluated using χ2 and Fisher exact test as appropriate. A Pearson correlation coefficient was used to determine whether there was a statistically significant increase in accurate foot examinations and ARAs as well as total number of podiatry referrals during the study period. Statistical analyses were performed using Stata 14.1 statistical software (College Station, TX).
Results
A total of 1,242 completed diabetic foot examinations were identified from August 1, 2017 to July 31, 2018 using the Diabetes Registry (Table). For the 3 months prior to the change, there were 191 appropriately completed foot examinations and ARAs. This number increased progressively over three 3-month periods (Figure 1). Within the 1-year study period, there was a statistically significant increase in the number of appropriate foot examinations (r = 0.495). PCPs placed 34 podiatry referrals during the prechange period. After the change, the number of appropriate referrals increased statistically significantly in the 3 following 3-month-periods (r = 0.222) (Figure 2).
To determine the accuracy of documentation and ratio of appropriate referrals, the 3-month prech
Notably, at the end of the first year of this evaluation, 119 veterans at the clinic did not show a recorded comprehensive foot examination since receiving a DM diagnosis and 299 veterans were due for an annual examination—a 25.2% gap of veterans without the recommended progression of foot care services. Of those that previously had a recorded foot examination, 51 (17.0%) veterans were found to be ≥ 2 years overdue.
Discussion
DM management requires a comprehensive team-based approach to help monitor for associated complications. At the VA, PACTs are veterans’ initial and primary point of contact for chronic condition management. PACTs have regular opportunities to engage veterans in initial and follow-up care and appropriate self-care. PCPs are critical in placing referrals for specialized care promptly to prevent and minimize complications such as foot ulcers, and ultimately, lower limb amputations.9,10,12
When PCPs assume responsibility for the entire foot examination, they are able to identify problems early.1 Left untreated, foot wounds and ulcers have the potential to grow into serious infections.9 Early risk identification and management can lead to increased patient satisfaction, improved life expectancy, quality of life, and ultimately, lower healthcare costs.12
Multiple studies have shown the clinical importance of foot examinations in preventative care. In one study, researchers found that completing foot examinations, among other early interventions, increased life expectancy and reduced foot complications.13 Diabetic foot management programs involving screening and categorizing patients into low- and high-risk groups had a 47.4% decrease in the incidence of amputations and 37.8% decrease in hospital admissions.14 Amputations were found to be inversely correlated with multidisciplinary foot care programs with reduction of lower limb amputations at 2 years.15 The Centers for Disease Control and Prevention found that comprehensive foot care programs that include a foot examination, ARA, appropriate referrals to specialists, and foot-care education and preventative services can reduce lower limb amputation rates by 45% to 85%.16
With one of the highest amputation rates in VA, VAPORHCS needed an integrated approach to ensure that appropriate foot care occurred regularly with veterans with DM. Prior to the process change, LPNs completed foot examinations and PCPs completed the ARA. Separating these clinical services resulted in few veterans receiving an amputation risk score. Of those with scores, the lack of a standardized program protocol resulted in discrepancies between ARAs from patient to patient as health care providers did not have clear or enough information to select the correct score and make the appropriate referrals. Thus, veterans previously identified as at moderate or high risk also lacked podiatric follow-up care.
The new quality-driven process change corrected the documentation process to nationally accepted standards. The goal was to create a consistent template in the electronic health record for all health care providers. The new template simplifies the documentation process and clarifies the amputation risk score assignment, which allows for proper foot care management. The PCP completes the process from assessment through referral, removing gaps in care and improving efficiency. Although this change was initially met with resistance from PCPs, it led to a significant increase in the number of patients with accurately documented examinations. Similarly, the number of appropriate referrals significantly rose during the study period. The standardized documentation process resulted in improved accurate examinations and ARAs over the past year. The new program also resulted in an increased number of appropriate podiatry referrals for those identified to be at moderate or high risk. This elevation of care is crucial for veterans to receive frequent follow-up visits for preventative care and/or treatment, including surgical modalities to promote limb salvage.
Barriers
This project identified several barriers to the Comprehensive Foot Care program. One major barrier was health care provider resistance to using the new process. For example, VAPORHCS podiatrists are not using the new template with established patients, which requires PCPs to complete the clinical reminder template for quality performance, an additional burden unrelated to clinical care. PCPs that do complete the foot examination/ARA templated note use the podiatrist’s visit note without personally assessing the patient.
PCPs also have been resistant to entering administrative text orders for preventative foot care in normal- or low-risk veterans (4.6% overall), which has resulted in decreased patient education (3.9% overall). Education for normal-risk and low-risk patients is designed to engage veterans in self-care and prevent risk progression, critical to prevention.
It was found that PCPs often did not ask nurses to coach normal- or low-risk veterans on preventative foot care, as suggested by the low rates at which patients were offered education. This is an area we will target with future quality improvement efforts. All patients with DM should have general education about risk factors and appropriate management of them to decrease their risk for complications.9 Preventative foot care education is a critical resource to share with patients during health coaching opportunities to clarify misunderstandings and support change talk when patients are ambivalent or resistant to change. Individual or group-based nurse visits can facilitate better coaching for patients.
At the VA, coaching begins with a conversation about what matters most to the veteran, facilitating the development of a personalized plan based on patients’ values, needs, preferences and goals.9,10,12,17 Coaching allows nurses to assess veterans’ knowledge and willingness to engage in healthy habits; and identify additional resources to help them achieve their goals.
Limitations
There are many limitations to this short quality improvement analysis. For example, only 1 of 2 clinics that piloted the program change was evaluated. In addition, there are 11 other clinics that need additional in-depth education on the program change. Although this analysis was overwhelmingly positive, it may not be as successful at other clinic sites and may be subject to the Hawthorne effect—since the 2 piloted locations knew they were being observed for the quality improvement program and may have made an extra effort to be compliant.18 Additionally, we were unable to track the records of veterans receiving care through the VA Choice Program for this analysis resulting in a lack of documentation of completed diabetic foot examinations and a lack of internal referrals to VA podiatry.
Another major limitation of this project involved calculating the number of referrals placed to podiatry. On January 1, 2018, about halfway through the program evaluation, a national VA decision enabled veterans to self-refer to podiatry, which may have limited the number of podiatry referrals placed by PCPs. Finally, patients could refuse podiatry referrals. In the 9-month postimplementation period, 57 (64.8%) veterans declined podiatry referrals, according to their CPRS records.
Although, there was an improvement in the accuracy of diabetic foot examinations, ARAs, and appropriate podiatry referrals, the ultimate goal of reducing diabetic foot ulcers and lower limb amputations was not tracked due to the limited timeframe of this analysis. Tracking these endpoints with continuous plan-do-study-act cycles are needed for this ongoing quality improvement project.
Conclusion
The goal of the VAPORHCS Comprehensive Foot Care program is to provide veterans with a program that is predictable, easy and consistent to prevent and treat foot ulcers to reduce the rate of lower limb amputations. It requires multidisciplinary team collaboration for success. Implementation of this new comprehensive program has increased the number of accurate annual foot exams, ARAs and podiatry referrals. Despite these improvements, areas of future improvement include emphasizing patient education and ongoing provider compliance with annual assessments.
Author contributions
MHG proposed the program evaluation project idea. TVQ collected and analyzed the data and wrote the manuscript. MHG oversaw the project and edited the manuscript. TVQ is the guarantor of this project and takes responsibility for the contents of this journal article.
Acknowledgments
The authors thank Tyra Haebe, VAPORHCS Prevention of Amputation in Veterans Everywhere (PAVE) Manager, and the entire VAPORHCS PAVE committee for their support in this program evaluation project.
1. US Department of Veterans Affairs, Veterans Health Administration. VHA directive 1410, prevention of amputation in veterans everywhere (PAVE) program. http://vaww.medical surgical.va.gov/podiatry/docs/VHADirective_1410_PAVE.pdf. Published March 31, 2017. Accessed October 11, 2019.
2. US Department of Veterans Affairs. Close to 25 percent of VA patients have diabetes http://www.va.gov/health/NewsFeatures/20111115a.asp. Accessed 14 October 2017
3. Centers for Disease Control and Prevention. National diabetes statistics report, 2017: Estimates of Diabetes and Its Burden in the United States. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf. Accessed October 11, 2019.
4. Gibson LW, Abbas A: Limb salvage for veterans with diabetes: to care for him who has borne the battle. Crit Care Nurs Clin North Am. 2012;25(1):131-134
5. Boyko EJ, Monteiro-Soares M, Wheeler SGB. “Peripheral arterial disease, foot ulcers, lower extremity amputations, and diabetes.” In: Cowie CC, Casagrande SS, Menke A, et al, eds. Diabetes in America. 3rd ed. Bethesda, MD: National Institutes of Health Publication; 2017:20-21,20-34.
6. National Institute of Health, National Institute of Neurological Disorders and Stroke. Peripheral neuropathy fact sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Peripheral-Neuropathy-Fact-Sheet. Updated August 13, 2019. Accessed October 11, 2019.
7. US Department of Veterans Affairs, Veterans Health Administration, Support Services Center. Amputation cube, lower amputations 2015. http://vssc.med.va.gov/AlphaIndex. [Nonpublic source, not verified]
8. US Department of Veterans Affairs, Office of Inspector General. Healthcare inspection: Foot care for patients with diabetes and additional risk factors for amputation. https://www.va.gov/oig/pubs/VAOIG-11-00711-74.pdf. Published January 17, 2013. Accessed October 11, 2019.
9. American Diabetes Association. Standards of medical care in diabetes - 2017. Diabetes Care. 2017;40(suppl 1):1-142.
10. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31(8):1679-1685.
11. Yang J, McConnachie J, Renfro R, Schreiner S, Tallett S, Winterbottom L. The diabetes registry and future panel management tool https://docplayer.net/19062632-The-diabetes-registry-and.html. Accessed October 11, 2019.
12. National Institute of Health, Centers for Disease Control and Prevention, the National Diabetes Education Program. Working together to manage diabetes: a guide for pharmcy, podiatry, optometry, and dentistry. https://www.cdc.gov/diabetes/ndep/pdfs/ppod-guide.pdf. Accessed October 11, 2019.
13. Ortegon MM, Redekop WK, Niessen LW. Cost-effectiveness of prevention and treatment of the diabetic foot: a Markov analysis. Diabetes Care. 2004;27(4):901-907.
14. Lavery LA, Wunderlich RP, Tredwell JL. Disease management for the diabetic foot: effectiveness of a diabetic foot prevention program to reduce amputations and hospitalizations. Diabetes Res Clin Pract. 2005;70(1):31-37.
15. Paisey RB, Abbott A, Levenson R, et al; South-West Cardiovascular Strategic Clinical Network peer diabetic foot service review team. Diabetes-related major lower limb amputation incidence is strongly related to diabetic foot service provision and improves with enhancement of services: peer review of the south-west of England. Diabet Med. 2017;35(1):53-62.
16. Centers for Disease Control and Prevention. National diabetes fact sheet: National estimates and general information on diabetes and prediabetes in the United States, 2011. https://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Published 2011. Accessed October 11, 2019.
17. US Department of Veterans Affairs. Whole health for life. https://www.va.gov/patientcenteredcare/explore/about-whole-health.asp. Updated July 20, 2017. Accessed October 11, 2019.
18. Parsons HM. What happened at Hawthorne? New evidence suggests the Hawthorne effect resulted from operant reinforcement contingencies. Science. 1974;183(4128):922–9322.
1. US Department of Veterans Affairs, Veterans Health Administration. VHA directive 1410, prevention of amputation in veterans everywhere (PAVE) program. http://vaww.medical surgical.va.gov/podiatry/docs/VHADirective_1410_PAVE.pdf. Published March 31, 2017. Accessed October 11, 2019.
2. US Department of Veterans Affairs. Close to 25 percent of VA patients have diabetes http://www.va.gov/health/NewsFeatures/20111115a.asp. Accessed 14 October 2017
3. Centers for Disease Control and Prevention. National diabetes statistics report, 2017: Estimates of Diabetes and Its Burden in the United States. https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf. Accessed October 11, 2019.
4. Gibson LW, Abbas A: Limb salvage for veterans with diabetes: to care for him who has borne the battle. Crit Care Nurs Clin North Am. 2012;25(1):131-134
5. Boyko EJ, Monteiro-Soares M, Wheeler SGB. “Peripheral arterial disease, foot ulcers, lower extremity amputations, and diabetes.” In: Cowie CC, Casagrande SS, Menke A, et al, eds. Diabetes in America. 3rd ed. Bethesda, MD: National Institutes of Health Publication; 2017:20-21,20-34.
6. National Institute of Health, National Institute of Neurological Disorders and Stroke. Peripheral neuropathy fact sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Peripheral-Neuropathy-Fact-Sheet. Updated August 13, 2019. Accessed October 11, 2019.
7. US Department of Veterans Affairs, Veterans Health Administration, Support Services Center. Amputation cube, lower amputations 2015. http://vssc.med.va.gov/AlphaIndex. [Nonpublic source, not verified]
8. US Department of Veterans Affairs, Office of Inspector General. Healthcare inspection: Foot care for patients with diabetes and additional risk factors for amputation. https://www.va.gov/oig/pubs/VAOIG-11-00711-74.pdf. Published January 17, 2013. Accessed October 11, 2019.
9. American Diabetes Association. Standards of medical care in diabetes - 2017. Diabetes Care. 2017;40(suppl 1):1-142.
10. Boulton AJM, Armstrong DG, Albert SF, et al. Comprehensive foot examination and risk assessment: a report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31(8):1679-1685.
11. Yang J, McConnachie J, Renfro R, Schreiner S, Tallett S, Winterbottom L. The diabetes registry and future panel management tool https://docplayer.net/19062632-The-diabetes-registry-and.html. Accessed October 11, 2019.
12. National Institute of Health, Centers for Disease Control and Prevention, the National Diabetes Education Program. Working together to manage diabetes: a guide for pharmcy, podiatry, optometry, and dentistry. https://www.cdc.gov/diabetes/ndep/pdfs/ppod-guide.pdf. Accessed October 11, 2019.
13. Ortegon MM, Redekop WK, Niessen LW. Cost-effectiveness of prevention and treatment of the diabetic foot: a Markov analysis. Diabetes Care. 2004;27(4):901-907.
14. Lavery LA, Wunderlich RP, Tredwell JL. Disease management for the diabetic foot: effectiveness of a diabetic foot prevention program to reduce amputations and hospitalizations. Diabetes Res Clin Pract. 2005;70(1):31-37.
15. Paisey RB, Abbott A, Levenson R, et al; South-West Cardiovascular Strategic Clinical Network peer diabetic foot service review team. Diabetes-related major lower limb amputation incidence is strongly related to diabetic foot service provision and improves with enhancement of services: peer review of the south-west of England. Diabet Med. 2017;35(1):53-62.
16. Centers for Disease Control and Prevention. National diabetes fact sheet: National estimates and general information on diabetes and prediabetes in the United States, 2011. https://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Published 2011. Accessed October 11, 2019.
17. US Department of Veterans Affairs. Whole health for life. https://www.va.gov/patientcenteredcare/explore/about-whole-health.asp. Updated July 20, 2017. Accessed October 11, 2019.
18. Parsons HM. What happened at Hawthorne? New evidence suggests the Hawthorne effect resulted from operant reinforcement contingencies. Science. 1974;183(4128):922–9322.
A National Survey of Veterans Affairs Medical Centers’ Cardiology Services (FULL)
The US Department of Veterans Affairs (VA) remains the largest integrated health care system in the US serving 9 million veterans. Two recent studies that compared 30-day mortality and readmission rates between VA and non-VA hospitals among older men with acute myocardial infarction (AMI), and heart failure (HF). The studies found that hospitalization at VA hospitals was associated with lower risk-standardized 30-day all-cause mortality rates for MI and HF when compared with hospitalization at non-VA hospitals.1,2
However, it is unknown whether the delivery of cardiovascular care is optimized in the VA system. For example, in comparisons between generalist-led hospitalized care for MI and HF, several studies have demonstrated that cardiology-led care has been associated with lower rates of mortality.3-5 Although data on the types of cardiac technology and use of cardiac procedures were described previously, we have not found detailed information on the types of inpatient cardiology services provided at VA medical centers nationwide.1,6,7 To develop further improvements in delivery of cardiovascular care within the VA, a better understanding of the types of resources that are currently available within the VA system must be made available. In this article, we present results of a national survey of cardiology services at VA facilities.
Methods
From February to March of 2017, we conducted a comprehensive nation-wide survey of all VA facilities to quantify the availability of cardiology services, excluding cardiothoracic surgical services. The survey questions are listed in the Appendix. The chief of medicine and the chief of cardiology were each e-mailed 3 times at every facility. If no response was received from a facility, we e-mailed the chief of staff 3 times. If there still was no response, the remaining facilities were contacted by phone and study authors (PE and WB) spoke to individuals directly regarding the structure of cardiology services at a facility. Responses were categorized by facility level of complexity. Complexity designation was determined by the VA Central Office (VACO)—level 1 facilities represent the most complex and level 3 facilities are the least complex. VACO also divides facility complexity into sublevels, for example level 1A facilities generally are associated with academic medical centers and provide the highest levels (tertiary or quaternary) of care.8
Results were coded according to a predetermined rubric for how cardiology services are structured (admitting service, consult service, inpatient, outpatient, other) and for how they were staffed (attending only, house staff, or advanced practice providers (APPs). After the first wave of surveys, 2 additional questions were added to the survey tool; these asked about employed vs contracted cardiologist and use of APPs. The results were tabulated and simple percentages calculated to express the prevalence of each structure and staffing model.
The study was reviewed and approved by the University of Utah/Salt Lake City VA Medical Center joint institutional review board and all authors completed human subjects research training.
Results
Study authors initially identified all 168 VA medical center facilities operating in 2017. Initial polling revealed that multiple facilities either were substations or had agreements for cardiology services from larger facilities, with 1 facility having 2 campuses with different levels of service at each. After adjusting for these nuances, the total number of potential respondents was 139. We obtained a response from 122 of the 139 facilities for an overall survey completion rate of 88%. Response rates varied by complexity level (Table 1). The survey received responses from all Level 1A and 1B facilities, 96% from Level 1C facilities; 83% (20/24) from level 2 facilities, and 62% (18/30) from level 3 facilities. (Please note that in the reference document providing detailed descriptions of the VA level of complexity has different numbers for each facility type given that there has been reassignments of the levels since our survey was completed.)8
We were specifically interested in inpatient cardiology services and whether facilities provided only consult services or inpatient services led by a cardiology attending. Having inpatient services does not exclude the availability of consult-liaison services (Table 2).
Higher complexity facilities (1A and 1B) were more likely to have dedicated, cardiology-led inpatient services, while lower complexity facilities relied on a cardiology consult service. Two-thirds of Level 3 facilities did not have inpatient cardiology services available.
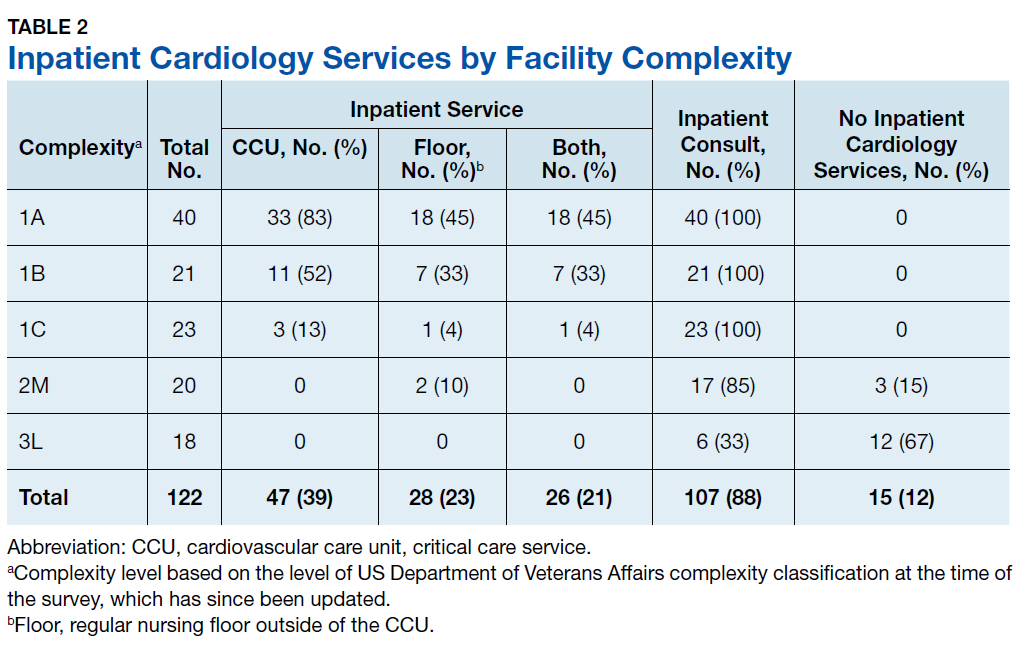
Dedicated cardiovascular care unit (CCU) teams were the most common inpatient service provided, present in more than half of all Level 1 facilities and 83% of Level 1A facilities (Table 3). Cardiology-led floor teams were available in 45% and 33% of level 1A and 1B facilities, respectively, but were much less common in Level 1C and Levels 2 and 3 facilities (4%, 10%, 0%, respectively). Only 31% of Level 1 facilities had both a CCU team and a cardiology-led inpatient floor team. Inpatient consulting cardiologists were commonly available at Levels 1 and 2 facilities; however, only 33% of Level 3 facilities had inpatient consulting cardiologists.
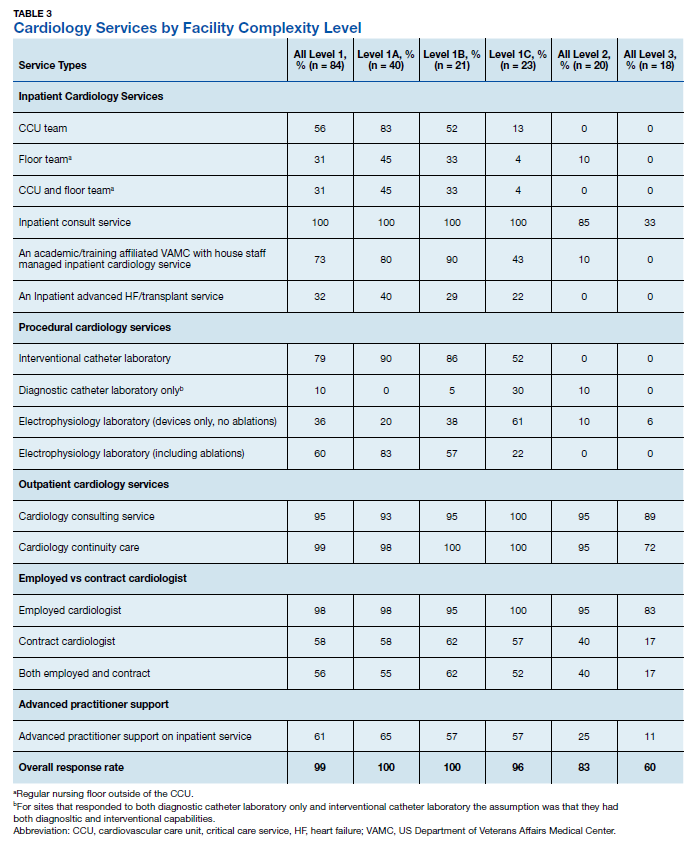
Housestaff-managed inpatient services, teams consisting of, but not limited to, medical residents in training, led by a cardiology attending were present in 73% of Level 1 facilities. Interestingly, Level 1B facilities were more likely to have housestaff-led services than were Level 1A facilities (90% and 80% respectively). Inpatient advanced heart failure services were less common and available only in Level 1 facilities. We did not survey the specific details of the other (eg, led by a noncardiology attending physician) models of inpatient cardiology care provided.
Cardiac catheterization (including interventional cardiology and electrophysiology [EP]) services, varied considerably. Ninety percent of Level 1A facilities offered interventional services, compared with only 52% of Level 1C facilities offered interventions. EP services were divided into simple (device only) and complex (ablations). As noted, complex EP services were more common in more complex facilities; for example, 10% of Level 2 facilities offered device placement but none had advanced EP services.
Outpatient services were widely available. Most facilities offered outpatient consultative cardiology services, ranging from 95% (Level 1) to 89% (Level 3) and outpatient cardiology continuity clinics 99% (Level 1) to 72% (Level 3).
Regardless of level of complexity, > 80% of facilities employed cardiologists. Many also used contract cardiologists. No facility utilized only contracted cardiologists. Use of nurse practitioners (NPs) and physician assistants (PAs) to assist with managing inpatient services was relatively common, with 61% of Level 1 facilities using such services.
Discussion
Studies of patient outcomes for various conditions, including cardiac conditions, in the 1990s found that when compared with non-VA health-care systems, patient outcomes in the VA were less favorable.9 During the late 1990s, the VA embraced quality and safety initiatives that have continued to the present time.9,10 Recent studies have found that, in most (but not all) cases, VA patient outcomes are as good as, and in many cases better, than are non-VA patient outcomes.1,10,11 The exact changes that have improved care are not clear, though studies of other health care systems have considered variation in services and costs in relationship to morbidity and mortality outcomes.12-14 In the context of better patient outcomes in VA hospitals, the present study provides insight into the cardiology services available at VA facilities throughout the nation.
Limitations
While this study provides background information that may be useful in comparing cardiology services between VA and non-VA systems, drawing causal relationships may not be warranted. For example, while the literature generally supports the concept of inpatient cardiology services led by an attending cardiologist, a substantial numbers of VA inpatient facilities have not yet adopted this model.4-6 Even among more complex, level 1 facilities, we found that only 31% offered both an inpatient CCU and floor team service led by an attending cardiologist physician. Thus, 69% of Level 1 facilities reported caring for patients with a primary cardiology problem through a noncardiology admitting services (with access to a cardiology consultation service). Additional studies should be conducted that would evaluate patient outcomes in relationship to the types of services available at a given VA medical center. Patient outcomes in relationship to service provision between the VA and non-VA health care systems also are warranted.
This study is limited by its reliance on self-reporting. Although we believe that we reached respondents who were qualified to complete the survey, the accuracy of reporting was not independently validated. Further, we asked questions about the most frequent models of cardiology care but may not have captured more novel methods. In trying to keep the survey time to < 2 minutes, we did not explore other details of cardiology services, such as the availability of a dedicated pharmacist, nor more advanced procedures such as transcatheter aortic valve replacement. Additionally, the present study is a snapshot of cardiology services for a given period, and, as noted above, did not look at patient outcomes. Further research is needed to determine which service provided is most beneficial or feasible in improving patient outcomes, which includes examining the various models of inpatient cardiology-led services for optimal care delivery.
Conclusion
Cardiology services were widely available throughout the VA system. However, the types of services available varied considerably. Predictably, facilities that were more complex generally had more advanced services available. Providing a general overview of how cardiovascular care is being delivered currently across VA systems helps to identify areas for optimization within VA facilities of various complexities with initiatives such as implementation of cardiology-led inpatient services, which may be beneficial in improving patient care outcomes as demonstrated previously in other large healthcare systems.

Acknowledgments
This material is the result of work supported with resources and use of the facilities at the George E. Wahlen Salt Lake City VA Medical Center. We are grateful to all of those who responded to our survey, and the support of the facility leadership. We are thankful for Tasia M. Nash and Tammy Jackson who helped to organize the data, and to Leigh Eleazer for her help in the manuscript preparation and formatting.
1. Nuti SV, Qin L, Rumsfeld JS, et al. Association of admission to Veterans Affairs hospitals vs non-veterans affairs hospitals with mortality and readmission rates among older men hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2016;315(6):582-592.
2. Blay E Jr, DeLancey JO, Hewitt DB, Chung JW, Bilimoria KY. Initial public reporting of quality at Veterans Affairs vs non-Veterans Affairs hospitals. JAMA Intern Med. 2017;177(6):882-885.
3. Hartz A, James PA. A systematic review of studies comparing myocardial infarction mortality for generalists and specialists: lessons for research and health policy. J Am Board Fam Med. 2006;19(3):291-302.
4. Driscoll A, Meagher S, Kennedy R, et al. What is the impact of systems of care for heart failure on patients diagnosed with heart failure: a systematic review. BMC Cardiovasc Disord. 2016;16(1):195.
5. Mitchell P, Marle D, Donkor A, et al; National Heart Failure Audit Steering Group. National heart failure audit: April 2013-March 2014. https://www.nicor.org.uk/wp-content/uploads/2019/02/hfannual13-14-updated.pdf. Published 2014. Accessed October 8, 2019.6. Mirvis DM, Graney MJ. Variations in the use of cardiac procedures in the Veterans Health Administration. Am Heart J. 1999;137(4 pt 1):706-713.
7. Wright SM, Petersen LA, Daley J. Availability of cardiac technology: trends in procedure use and outcomes for patients with acute myocardial infarction. Med Care Res Rev. 1998;55(2):239-254.
8. US Department of Veterans Affairs. Summary of VHA Facility Complexity Model. https://www.vendorportal.ecms.va.gov. [Nonpublic source, not verified]
9. Jha AK, Perlin JB, Kizer KW, Dudley RA. Effect of the transformation of the Veterans Affairs Health Care System on the quality of care. N Engl J Med. 2003;348(22):2218-2227.
10. Atkins D, Clancy C. Advancing high performance in Veterans Affairs health care. JAMA. 2017;318(19):1927-1928.
11. O’Hanlon C, Huang C, Sloss E, et al. Comparing VA and non-VA quality of care: a systematic review. J Gen Intern Med. 2017;32(1):105-121.
12. Stukel TA; Lucas FL, Wennberg DE. Long-term outcomes of regional variations in intensity of invasive vs medical management of medicare patients with acute myocardial infarction. JAMA. 2005;293(11):1329-1337.
13. Krumholz HM, Chen J, Rathore SS, Wang Y, Radford MJ. Regional variation in the treatment and outcomes of myocardial infarction: investigating New England’s advantage. Am Heart J. 2003;146(2):242-249.
14. Petersen LA, Normand SL, Leape LL, McNeil BJ. Regionalization and the underuse of angiography in the Veterans Affairs Health Care System as compared with a fee-for-service system. N Engl J Med. 2003;348(22):2209-2217.
The US Department of Veterans Affairs (VA) remains the largest integrated health care system in the US serving 9 million veterans. Two recent studies that compared 30-day mortality and readmission rates between VA and non-VA hospitals among older men with acute myocardial infarction (AMI), and heart failure (HF). The studies found that hospitalization at VA hospitals was associated with lower risk-standardized 30-day all-cause mortality rates for MI and HF when compared with hospitalization at non-VA hospitals.1,2
However, it is unknown whether the delivery of cardiovascular care is optimized in the VA system. For example, in comparisons between generalist-led hospitalized care for MI and HF, several studies have demonstrated that cardiology-led care has been associated with lower rates of mortality.3-5 Although data on the types of cardiac technology and use of cardiac procedures were described previously, we have not found detailed information on the types of inpatient cardiology services provided at VA medical centers nationwide.1,6,7 To develop further improvements in delivery of cardiovascular care within the VA, a better understanding of the types of resources that are currently available within the VA system must be made available. In this article, we present results of a national survey of cardiology services at VA facilities.
Methods
From February to March of 2017, we conducted a comprehensive nation-wide survey of all VA facilities to quantify the availability of cardiology services, excluding cardiothoracic surgical services. The survey questions are listed in the Appendix. The chief of medicine and the chief of cardiology were each e-mailed 3 times at every facility. If no response was received from a facility, we e-mailed the chief of staff 3 times. If there still was no response, the remaining facilities were contacted by phone and study authors (PE and WB) spoke to individuals directly regarding the structure of cardiology services at a facility. Responses were categorized by facility level of complexity. Complexity designation was determined by the VA Central Office (VACO)—level 1 facilities represent the most complex and level 3 facilities are the least complex. VACO also divides facility complexity into sublevels, for example level 1A facilities generally are associated with academic medical centers and provide the highest levels (tertiary or quaternary) of care.8
Results were coded according to a predetermined rubric for how cardiology services are structured (admitting service, consult service, inpatient, outpatient, other) and for how they were staffed (attending only, house staff, or advanced practice providers (APPs). After the first wave of surveys, 2 additional questions were added to the survey tool; these asked about employed vs contracted cardiologist and use of APPs. The results were tabulated and simple percentages calculated to express the prevalence of each structure and staffing model.
The study was reviewed and approved by the University of Utah/Salt Lake City VA Medical Center joint institutional review board and all authors completed human subjects research training.
Results
Study authors initially identified all 168 VA medical center facilities operating in 2017. Initial polling revealed that multiple facilities either were substations or had agreements for cardiology services from larger facilities, with 1 facility having 2 campuses with different levels of service at each. After adjusting for these nuances, the total number of potential respondents was 139. We obtained a response from 122 of the 139 facilities for an overall survey completion rate of 88%. Response rates varied by complexity level (Table 1). The survey received responses from all Level 1A and 1B facilities, 96% from Level 1C facilities; 83% (20/24) from level 2 facilities, and 62% (18/30) from level 3 facilities. (Please note that in the reference document providing detailed descriptions of the VA level of complexity has different numbers for each facility type given that there has been reassignments of the levels since our survey was completed.)8
We were specifically interested in inpatient cardiology services and whether facilities provided only consult services or inpatient services led by a cardiology attending. Having inpatient services does not exclude the availability of consult-liaison services (Table 2).
Higher complexity facilities (1A and 1B) were more likely to have dedicated, cardiology-led inpatient services, while lower complexity facilities relied on a cardiology consult service. Two-thirds of Level 3 facilities did not have inpatient cardiology services available.
Dedicated cardiovascular care unit (CCU) teams were the most common inpatient service provided, present in more than half of all Level 1 facilities and 83% of Level 1A facilities (Table 3). Cardiology-led floor teams were available in 45% and 33% of level 1A and 1B facilities, respectively, but were much less common in Level 1C and Levels 2 and 3 facilities (4%, 10%, 0%, respectively). Only 31% of Level 1 facilities had both a CCU team and a cardiology-led inpatient floor team. Inpatient consulting cardiologists were commonly available at Levels 1 and 2 facilities; however, only 33% of Level 3 facilities had inpatient consulting cardiologists.
Housestaff-managed inpatient services, teams consisting of, but not limited to, medical residents in training, led by a cardiology attending were present in 73% of Level 1 facilities. Interestingly, Level 1B facilities were more likely to have housestaff-led services than were Level 1A facilities (90% and 80% respectively). Inpatient advanced heart failure services were less common and available only in Level 1 facilities. We did not survey the specific details of the other (eg, led by a noncardiology attending physician) models of inpatient cardiology care provided.
Cardiac catheterization (including interventional cardiology and electrophysiology [EP]) services, varied considerably. Ninety percent of Level 1A facilities offered interventional services, compared with only 52% of Level 1C facilities offered interventions. EP services were divided into simple (device only) and complex (ablations). As noted, complex EP services were more common in more complex facilities; for example, 10% of Level 2 facilities offered device placement but none had advanced EP services.
Outpatient services were widely available. Most facilities offered outpatient consultative cardiology services, ranging from 95% (Level 1) to 89% (Level 3) and outpatient cardiology continuity clinics 99% (Level 1) to 72% (Level 3).
Regardless of level of complexity, > 80% of facilities employed cardiologists. Many also used contract cardiologists. No facility utilized only contracted cardiologists. Use of nurse practitioners (NPs) and physician assistants (PAs) to assist with managing inpatient services was relatively common, with 61% of Level 1 facilities using such services.
Discussion
Studies of patient outcomes for various conditions, including cardiac conditions, in the 1990s found that when compared with non-VA health-care systems, patient outcomes in the VA were less favorable.9 During the late 1990s, the VA embraced quality and safety initiatives that have continued to the present time.9,10 Recent studies have found that, in most (but not all) cases, VA patient outcomes are as good as, and in many cases better, than are non-VA patient outcomes.1,10,11 The exact changes that have improved care are not clear, though studies of other health care systems have considered variation in services and costs in relationship to morbidity and mortality outcomes.12-14 In the context of better patient outcomes in VA hospitals, the present study provides insight into the cardiology services available at VA facilities throughout the nation.
Limitations
While this study provides background information that may be useful in comparing cardiology services between VA and non-VA systems, drawing causal relationships may not be warranted. For example, while the literature generally supports the concept of inpatient cardiology services led by an attending cardiologist, a substantial numbers of VA inpatient facilities have not yet adopted this model.4-6 Even among more complex, level 1 facilities, we found that only 31% offered both an inpatient CCU and floor team service led by an attending cardiologist physician. Thus, 69% of Level 1 facilities reported caring for patients with a primary cardiology problem through a noncardiology admitting services (with access to a cardiology consultation service). Additional studies should be conducted that would evaluate patient outcomes in relationship to the types of services available at a given VA medical center. Patient outcomes in relationship to service provision between the VA and non-VA health care systems also are warranted.
This study is limited by its reliance on self-reporting. Although we believe that we reached respondents who were qualified to complete the survey, the accuracy of reporting was not independently validated. Further, we asked questions about the most frequent models of cardiology care but may not have captured more novel methods. In trying to keep the survey time to < 2 minutes, we did not explore other details of cardiology services, such as the availability of a dedicated pharmacist, nor more advanced procedures such as transcatheter aortic valve replacement. Additionally, the present study is a snapshot of cardiology services for a given period, and, as noted above, did not look at patient outcomes. Further research is needed to determine which service provided is most beneficial or feasible in improving patient outcomes, which includes examining the various models of inpatient cardiology-led services for optimal care delivery.
Conclusion
Cardiology services were widely available throughout the VA system. However, the types of services available varied considerably. Predictably, facilities that were more complex generally had more advanced services available. Providing a general overview of how cardiovascular care is being delivered currently across VA systems helps to identify areas for optimization within VA facilities of various complexities with initiatives such as implementation of cardiology-led inpatient services, which may be beneficial in improving patient care outcomes as demonstrated previously in other large healthcare systems.
Acknowledgments
This material is the result of work supported with resources and use of the facilities at the George E. Wahlen Salt Lake City VA Medical Center. We are grateful to all of those who responded to our survey, and the support of the facility leadership. We are thankful for Tasia M. Nash and Tammy Jackson who helped to organize the data, and to Leigh Eleazer for her help in the manuscript preparation and formatting.
The US Department of Veterans Affairs (VA) remains the largest integrated health care system in the US serving 9 million veterans. Two recent studies that compared 30-day mortality and readmission rates between VA and non-VA hospitals among older men with acute myocardial infarction (AMI), and heart failure (HF). The studies found that hospitalization at VA hospitals was associated with lower risk-standardized 30-day all-cause mortality rates for MI and HF when compared with hospitalization at non-VA hospitals.1,2
However, it is unknown whether the delivery of cardiovascular care is optimized in the VA system. For example, in comparisons between generalist-led hospitalized care for MI and HF, several studies have demonstrated that cardiology-led care has been associated with lower rates of mortality.3-5 Although data on the types of cardiac technology and use of cardiac procedures were described previously, we have not found detailed information on the types of inpatient cardiology services provided at VA medical centers nationwide.1,6,7 To develop further improvements in delivery of cardiovascular care within the VA, a better understanding of the types of resources that are currently available within the VA system must be made available. In this article, we present results of a national survey of cardiology services at VA facilities.
Methods
From February to March of 2017, we conducted a comprehensive nation-wide survey of all VA facilities to quantify the availability of cardiology services, excluding cardiothoracic surgical services. The survey questions are listed in the Appendix. The chief of medicine and the chief of cardiology were each e-mailed 3 times at every facility. If no response was received from a facility, we e-mailed the chief of staff 3 times. If there still was no response, the remaining facilities were contacted by phone and study authors (PE and WB) spoke to individuals directly regarding the structure of cardiology services at a facility. Responses were categorized by facility level of complexity. Complexity designation was determined by the VA Central Office (VACO)—level 1 facilities represent the most complex and level 3 facilities are the least complex. VACO also divides facility complexity into sublevels, for example level 1A facilities generally are associated with academic medical centers and provide the highest levels (tertiary or quaternary) of care.8
Results were coded according to a predetermined rubric for how cardiology services are structured (admitting service, consult service, inpatient, outpatient, other) and for how they were staffed (attending only, house staff, or advanced practice providers (APPs). After the first wave of surveys, 2 additional questions were added to the survey tool; these asked about employed vs contracted cardiologist and use of APPs. The results were tabulated and simple percentages calculated to express the prevalence of each structure and staffing model.
The study was reviewed and approved by the University of Utah/Salt Lake City VA Medical Center joint institutional review board and all authors completed human subjects research training.
Results
Study authors initially identified all 168 VA medical center facilities operating in 2017. Initial polling revealed that multiple facilities either were substations or had agreements for cardiology services from larger facilities, with 1 facility having 2 campuses with different levels of service at each. After adjusting for these nuances, the total number of potential respondents was 139. We obtained a response from 122 of the 139 facilities for an overall survey completion rate of 88%. Response rates varied by complexity level (Table 1). The survey received responses from all Level 1A and 1B facilities, 96% from Level 1C facilities; 83% (20/24) from level 2 facilities, and 62% (18/30) from level 3 facilities. (Please note that in the reference document providing detailed descriptions of the VA level of complexity has different numbers for each facility type given that there has been reassignments of the levels since our survey was completed.)8
We were specifically interested in inpatient cardiology services and whether facilities provided only consult services or inpatient services led by a cardiology attending. Having inpatient services does not exclude the availability of consult-liaison services (Table 2).
Higher complexity facilities (1A and 1B) were more likely to have dedicated, cardiology-led inpatient services, while lower complexity facilities relied on a cardiology consult service. Two-thirds of Level 3 facilities did not have inpatient cardiology services available.
Dedicated cardiovascular care unit (CCU) teams were the most common inpatient service provided, present in more than half of all Level 1 facilities and 83% of Level 1A facilities (Table 3). Cardiology-led floor teams were available in 45% and 33% of level 1A and 1B facilities, respectively, but were much less common in Level 1C and Levels 2 and 3 facilities (4%, 10%, 0%, respectively). Only 31% of Level 1 facilities had both a CCU team and a cardiology-led inpatient floor team. Inpatient consulting cardiologists were commonly available at Levels 1 and 2 facilities; however, only 33% of Level 3 facilities had inpatient consulting cardiologists.
Housestaff-managed inpatient services, teams consisting of, but not limited to, medical residents in training, led by a cardiology attending were present in 73% of Level 1 facilities. Interestingly, Level 1B facilities were more likely to have housestaff-led services than were Level 1A facilities (90% and 80% respectively). Inpatient advanced heart failure services were less common and available only in Level 1 facilities. We did not survey the specific details of the other (eg, led by a noncardiology attending physician) models of inpatient cardiology care provided.
Cardiac catheterization (including interventional cardiology and electrophysiology [EP]) services, varied considerably. Ninety percent of Level 1A facilities offered interventional services, compared with only 52% of Level 1C facilities offered interventions. EP services were divided into simple (device only) and complex (ablations). As noted, complex EP services were more common in more complex facilities; for example, 10% of Level 2 facilities offered device placement but none had advanced EP services.
Outpatient services were widely available. Most facilities offered outpatient consultative cardiology services, ranging from 95% (Level 1) to 89% (Level 3) and outpatient cardiology continuity clinics 99% (Level 1) to 72% (Level 3).
Regardless of level of complexity, > 80% of facilities employed cardiologists. Many also used contract cardiologists. No facility utilized only contracted cardiologists. Use of nurse practitioners (NPs) and physician assistants (PAs) to assist with managing inpatient services was relatively common, with 61% of Level 1 facilities using such services.
Discussion
Studies of patient outcomes for various conditions, including cardiac conditions, in the 1990s found that when compared with non-VA health-care systems, patient outcomes in the VA were less favorable.9 During the late 1990s, the VA embraced quality and safety initiatives that have continued to the present time.9,10 Recent studies have found that, in most (but not all) cases, VA patient outcomes are as good as, and in many cases better, than are non-VA patient outcomes.1,10,11 The exact changes that have improved care are not clear, though studies of other health care systems have considered variation in services and costs in relationship to morbidity and mortality outcomes.12-14 In the context of better patient outcomes in VA hospitals, the present study provides insight into the cardiology services available at VA facilities throughout the nation.
Limitations
While this study provides background information that may be useful in comparing cardiology services between VA and non-VA systems, drawing causal relationships may not be warranted. For example, while the literature generally supports the concept of inpatient cardiology services led by an attending cardiologist, a substantial numbers of VA inpatient facilities have not yet adopted this model.4-6 Even among more complex, level 1 facilities, we found that only 31% offered both an inpatient CCU and floor team service led by an attending cardiologist physician. Thus, 69% of Level 1 facilities reported caring for patients with a primary cardiology problem through a noncardiology admitting services (with access to a cardiology consultation service). Additional studies should be conducted that would evaluate patient outcomes in relationship to the types of services available at a given VA medical center. Patient outcomes in relationship to service provision between the VA and non-VA health care systems also are warranted.
This study is limited by its reliance on self-reporting. Although we believe that we reached respondents who were qualified to complete the survey, the accuracy of reporting was not independently validated. Further, we asked questions about the most frequent models of cardiology care but may not have captured more novel methods. In trying to keep the survey time to < 2 minutes, we did not explore other details of cardiology services, such as the availability of a dedicated pharmacist, nor more advanced procedures such as transcatheter aortic valve replacement. Additionally, the present study is a snapshot of cardiology services for a given period, and, as noted above, did not look at patient outcomes. Further research is needed to determine which service provided is most beneficial or feasible in improving patient outcomes, which includes examining the various models of inpatient cardiology-led services for optimal care delivery.
Conclusion
Cardiology services were widely available throughout the VA system. However, the types of services available varied considerably. Predictably, facilities that were more complex generally had more advanced services available. Providing a general overview of how cardiovascular care is being delivered currently across VA systems helps to identify areas for optimization within VA facilities of various complexities with initiatives such as implementation of cardiology-led inpatient services, which may be beneficial in improving patient care outcomes as demonstrated previously in other large healthcare systems.
Acknowledgments
This material is the result of work supported with resources and use of the facilities at the George E. Wahlen Salt Lake City VA Medical Center. We are grateful to all of those who responded to our survey, and the support of the facility leadership. We are thankful for Tasia M. Nash and Tammy Jackson who helped to organize the data, and to Leigh Eleazer for her help in the manuscript preparation and formatting.
1. Nuti SV, Qin L, Rumsfeld JS, et al. Association of admission to Veterans Affairs hospitals vs non-veterans affairs hospitals with mortality and readmission rates among older men hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2016;315(6):582-592.
2. Blay E Jr, DeLancey JO, Hewitt DB, Chung JW, Bilimoria KY. Initial public reporting of quality at Veterans Affairs vs non-Veterans Affairs hospitals. JAMA Intern Med. 2017;177(6):882-885.
3. Hartz A, James PA. A systematic review of studies comparing myocardial infarction mortality for generalists and specialists: lessons for research and health policy. J Am Board Fam Med. 2006;19(3):291-302.
4. Driscoll A, Meagher S, Kennedy R, et al. What is the impact of systems of care for heart failure on patients diagnosed with heart failure: a systematic review. BMC Cardiovasc Disord. 2016;16(1):195.
5. Mitchell P, Marle D, Donkor A, et al; National Heart Failure Audit Steering Group. National heart failure audit: April 2013-March 2014. https://www.nicor.org.uk/wp-content/uploads/2019/02/hfannual13-14-updated.pdf. Published 2014. Accessed October 8, 2019.6. Mirvis DM, Graney MJ. Variations in the use of cardiac procedures in the Veterans Health Administration. Am Heart J. 1999;137(4 pt 1):706-713.
7. Wright SM, Petersen LA, Daley J. Availability of cardiac technology: trends in procedure use and outcomes for patients with acute myocardial infarction. Med Care Res Rev. 1998;55(2):239-254.
8. US Department of Veterans Affairs. Summary of VHA Facility Complexity Model. https://www.vendorportal.ecms.va.gov. [Nonpublic source, not verified]
9. Jha AK, Perlin JB, Kizer KW, Dudley RA. Effect of the transformation of the Veterans Affairs Health Care System on the quality of care. N Engl J Med. 2003;348(22):2218-2227.
10. Atkins D, Clancy C. Advancing high performance in Veterans Affairs health care. JAMA. 2017;318(19):1927-1928.
11. O’Hanlon C, Huang C, Sloss E, et al. Comparing VA and non-VA quality of care: a systematic review. J Gen Intern Med. 2017;32(1):105-121.
12. Stukel TA; Lucas FL, Wennberg DE. Long-term outcomes of regional variations in intensity of invasive vs medical management of medicare patients with acute myocardial infarction. JAMA. 2005;293(11):1329-1337.
13. Krumholz HM, Chen J, Rathore SS, Wang Y, Radford MJ. Regional variation in the treatment and outcomes of myocardial infarction: investigating New England’s advantage. Am Heart J. 2003;146(2):242-249.
14. Petersen LA, Normand SL, Leape LL, McNeil BJ. Regionalization and the underuse of angiography in the Veterans Affairs Health Care System as compared with a fee-for-service system. N Engl J Med. 2003;348(22):2209-2217.
1. Nuti SV, Qin L, Rumsfeld JS, et al. Association of admission to Veterans Affairs hospitals vs non-veterans affairs hospitals with mortality and readmission rates among older men hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2016;315(6):582-592.
2. Blay E Jr, DeLancey JO, Hewitt DB, Chung JW, Bilimoria KY. Initial public reporting of quality at Veterans Affairs vs non-Veterans Affairs hospitals. JAMA Intern Med. 2017;177(6):882-885.
3. Hartz A, James PA. A systematic review of studies comparing myocardial infarction mortality for generalists and specialists: lessons for research and health policy. J Am Board Fam Med. 2006;19(3):291-302.
4. Driscoll A, Meagher S, Kennedy R, et al. What is the impact of systems of care for heart failure on patients diagnosed with heart failure: a systematic review. BMC Cardiovasc Disord. 2016;16(1):195.
5. Mitchell P, Marle D, Donkor A, et al; National Heart Failure Audit Steering Group. National heart failure audit: April 2013-March 2014. https://www.nicor.org.uk/wp-content/uploads/2019/02/hfannual13-14-updated.pdf. Published 2014. Accessed October 8, 2019.6. Mirvis DM, Graney MJ. Variations in the use of cardiac procedures in the Veterans Health Administration. Am Heart J. 1999;137(4 pt 1):706-713.
7. Wright SM, Petersen LA, Daley J. Availability of cardiac technology: trends in procedure use and outcomes for patients with acute myocardial infarction. Med Care Res Rev. 1998;55(2):239-254.
8. US Department of Veterans Affairs. Summary of VHA Facility Complexity Model. https://www.vendorportal.ecms.va.gov. [Nonpublic source, not verified]
9. Jha AK, Perlin JB, Kizer KW, Dudley RA. Effect of the transformation of the Veterans Affairs Health Care System on the quality of care. N Engl J Med. 2003;348(22):2218-2227.
10. Atkins D, Clancy C. Advancing high performance in Veterans Affairs health care. JAMA. 2017;318(19):1927-1928.
11. O’Hanlon C, Huang C, Sloss E, et al. Comparing VA and non-VA quality of care: a systematic review. J Gen Intern Med. 2017;32(1):105-121.
12. Stukel TA; Lucas FL, Wennberg DE. Long-term outcomes of regional variations in intensity of invasive vs medical management of medicare patients with acute myocardial infarction. JAMA. 2005;293(11):1329-1337.
13. Krumholz HM, Chen J, Rathore SS, Wang Y, Radford MJ. Regional variation in the treatment and outcomes of myocardial infarction: investigating New England’s advantage. Am Heart J. 2003;146(2):242-249.
14. Petersen LA, Normand SL, Leape LL, McNeil BJ. Regionalization and the underuse of angiography in the Veterans Affairs Health Care System as compared with a fee-for-service system. N Engl J Med. 2003;348(22):2209-2217.
A Health Care Provider Intervention to Address Obesity in Patients with Diabetes (FULL)
Obesity is associated with a significant increase in mortality. It increases the risk of type 2 diabetes mellitus (T2DM), hypertension, hyperlipidemia, and coronary artery disease.1 T2DM is strongly associated with obesity in all ethnic groups.
Medical nutrition therapy and weight loss are very important for DM management.2 This includes providing education about diet modification, increased physical activity, daily calorie intake evaluation, and consistent carbohydrate intake. For patients with T2DM, health care providers (HCPs) should emphasize lowering caloric intake and inducing weight loss for those who are overweight (body mass index [BMI] between 25 and 29.9) and obese (BMI ≥ 30). This can improve glycemic control by decreasing insulin resistance. Initial recommendations for weight loss and physical activity are to lose between 5% and 10% of initial body weight and to accumulate at least 30 minutes of moderate physical activity over the course of most days of the week.3,4
Several formulas are available to estimate baseline caloric intake for weight maintenance. For weight loss of 1 to 2 pounds per week, lowering 500 to 1,000 calories from daily weight maintenance calories serves the goal. The American Diabetes Association (ADA) also suggests that HCPs recommend diet, physical activity, and behavioral therapy designed to achieve > 5% weight loss to overweight and obese patients with T2DM.5
Recognizing the clinical benefits of achieving weight loss in overweight or obese patients with T2DM, we aimed to increase the number of visits in the Endocrine Clinic at Central Arkansas Veterans Healthcare System (CAVHS) in Little Rock that addressed obesity, documented calorie goal for patients who are overweight or obese, and performed an intervention with further education for the patient.
Methods
The study population included veterans with either type 1 DM (T1DM) or T2DM with BMI > 25 on any DM control regimen. We performed a health record review of the eligible patients seen in the CAVHS Endocrine Clinic from June 1, 2016 to July 31, 2016 to determine the baseline percentage of visits that addressed obesity and provided weight loss advice to patients. We obtained a list of patients seen in the clinic during the study period from Strategic Management Service Services at CAVHS. We also obtained information that age, gender, medications, BMI, and last Endocrine clinic HCP assessment from the electronic health record. We reviewed the HCPs notes, including fellows and faculty who were involved in the patients’ treatment, to determine whether their notes documented a BMI > 25 and whether they discussed an intervention for overweight or obesity with the patient. The CAVHS Institutional Review Board reviewed and approved the initiative as a quality improvement study.

Intervention
Our clinic has a defined group of HCPs that we targeted for the intervention. After getting baseline information, during August 2017 we educated these HCPs on the tools available to calculate calorie goal for the patients. We advised the HCPs to use the Mifflin St Jyor equation for estimating energy expenditure and set a goal of initial weight loss between 5% and 7% of body weight. We gave specific instructions and advice to the providers (Table 1). HCPs also received educational material to distribute to patients that provided information on the healthy plate method, discussed how to count calories, and advised them on ADA goals with carbohydrate limitation. We encouraged HCPs to recommend that patients cut between 500 and 1,000 calories daily from their current diet. HCPs also received advice to seek help from clinical dieticians and the VA MOVE! Weight Management Program when appropriate.
Study of Effect of the Intervention
To study the effect of this intervention, we reviewed documentation by HCPs and assessed patient satisfaction. We obtained a list of patients and reviewed HCP notes on patients with BMI > 25 to assess whether providers addressed obesity in November and December 2017. We also evaluated whether HCPs offered a specific intervention to address the problem, such as providing education material to the patient or an estimate of daily calorie goal, or referring them to clinical dietician and/or the MOVE program. Patients received a 5-question survey that assessed their understanding and satisfaction at the end of the visit (Table 2).
Results
Of the 100 charts reviewed prior to intervention, HCPs discussed obesity management with only 6% of patients. After the intervention, we collected data again through chart review of the patients who were overweight or obese and seen for DM in the same clinic during a 2-month period. Of the 100 charts reviewed, we noticed that recognition and management of obesity improved to 60%.
To evaluate the impact of this intervention, patients received a questionnaire at the end of the visit. Nearly all (97%) patients mentioned that the provider discussed weight management during that visit. Most (83%) patients mentioned that weight management was discussed with them during prior visits, while 70% of patients felt their knowledge on working on weight loss had improved. Almost half (46%) were interested in further referral to a dietician or the MOVE program if they did not achieve desired results, but 78% were confident that they could implement the discussed weight management measures.
Discussion
Increased body weight is associated with worsening of DM and can result in poor glycemic control. Achieving weight loss in overweight or obese patients with DM can lead to clinical benefits; however, this is a challenge. In one study, a DM prevention program with lifestyle intervention leading to weight loss significantly reduced the rate of progression from impaired glucose tolerance to DM over a 3-year period and improved cardiovascular risk factors like elevated blood pressure and dyslipidemia.6 A randomized trial of an intensive lifestyle intervention to increase physical activity and decrease caloric intake vs standard DM education in people with T2DM showed a modest weight loss of 8.6% of initial weight at 1 year.7 This weight loss was associated with significant improvement in blood pressure, glycemic control, fasting blood glucose, high-density lipoprotein (HDL) cholesterol, and triglyceride levels and significant reductions in the use of DM, hypertension, and lipid-lowering medications.7 Obesity attributes to dyslipidemia with increased levels of cholesterol, low-density lipoprotein, very low-density lipoprotein, triglycerides, and decreased levels of HDL by about 5%.8 Obesity also is associated with hypertension, coronary heart disease, heart failure, and cardiovascular and all-cause mortality.9
Limitations
Limitations of this study include the small sample size and that multiple HCPs were involved. The nature of intervention might have differed with different HCPs or in a different setting than a VA clinic. In addition, we did not evaluate the effect on weight loss in specific patients as we only reviewed charts to check whether HCPs addressed weight loss. Nevertheless, our intervention was effective because it improved patient and provider awareness. It also gave us the opportunity to create framework for further collaborations and community building. The Endocrinology department at CAVHS is currently collaborating with the MOVE program, which is a part of the nutrition and food services. We hope to have an endocrinologist involved to provide guidance on medication management for obesity.
Conclusion
At CAVHS a simple intervention was instituted to evaluate whether HCPs were discussing weight loss in patients with DM, providing them with information to assess patients’ daily calorie goal, and prompting them for intervention to achieve weight loss. The intervention led to better management of patients with DM and obesity and greater engagement in weight loss from patients.
This project was a team effort. The clinic nurse documented patient’s BMI on the check in slip. HCPs discussed the problem and specific intervention. The clinical dieticians provided focused education for patients. The clerks collected the patient responses to questionnaire. This project also improved communication within the Endocrine Clinic team. Documentation of HCPs pertaining to addressing obesity improved by 54%. Improved patient satisfaction and insight was evident on patient responses to the questionnaire.
We believe that HCP apathy is a major contributor to the problem of obesity. Small steps like these go a long way for further management of obesity. Most VA hospitals have MOVE programs that provide dietary advice and encourage behavioral changes. However, getting patients to commit to these programs is a challenge. Primary care and endocrine clinics are important services that may help with patient awareness.
This project helped us better recognize patients with obesity and provide them with initial counseling and dietary advice. We received help from clinical dieticians and gave patients the option to join MOVE in situations where initial advice did not yield results and for more consistent follow up.
We tried to improve the care for patients with DM who were overweight or obese at CAVHS by prompting HCPs to focus on obesity as a problem and perform interventions to address this problem. The activities carried out and the data collected were used for internal quality improvement and for encouraging further interventions in the care of these patients.
1. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
2. Evert AB, Boucher JL, Cypress M, et al; American Diabetes Association. Nutrition therapy recommendations for the management of adults with diabetes. Diabetes Care. 2013;36(11):3821-3842.
3. NHLBI Obesity Education Initiative Expert Panel on the Identification, Evaluation, and Treatment of Obesity in Adults (US). Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults: The Evidence Report. Bethesda, MD: National Heart, Lung, and Blood Institute; 1998.
4. US Department of Health and Human Services. Physical Activity and Health: A Report of the Surgeon General. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion; 1996.
5. American Diabetes Association. 7. Obesity management for the treatment of type 2 diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41(Suppl 1):S65-S72.
6. Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
7. Look AHEAD Research Group; Pi-Sunyer X, Blackburn G, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care. 2007;30(6):1374-1383.
8. Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26(5):968-976.
9. Aune D, Sen A, Norat T, et al. Body mass index, abdominal fatness, and heart failure incidence and mortality: a systematic review and dose-response meta-analysis of prospective studies. Circulation. 2016;133(7):639-649.
Obesity is associated with a significant increase in mortality. It increases the risk of type 2 diabetes mellitus (T2DM), hypertension, hyperlipidemia, and coronary artery disease.1 T2DM is strongly associated with obesity in all ethnic groups.
Medical nutrition therapy and weight loss are very important for DM management.2 This includes providing education about diet modification, increased physical activity, daily calorie intake evaluation, and consistent carbohydrate intake. For patients with T2DM, health care providers (HCPs) should emphasize lowering caloric intake and inducing weight loss for those who are overweight (body mass index [BMI] between 25 and 29.9) and obese (BMI ≥ 30). This can improve glycemic control by decreasing insulin resistance. Initial recommendations for weight loss and physical activity are to lose between 5% and 10% of initial body weight and to accumulate at least 30 minutes of moderate physical activity over the course of most days of the week.3,4
Several formulas are available to estimate baseline caloric intake for weight maintenance. For weight loss of 1 to 2 pounds per week, lowering 500 to 1,000 calories from daily weight maintenance calories serves the goal. The American Diabetes Association (ADA) also suggests that HCPs recommend diet, physical activity, and behavioral therapy designed to achieve > 5% weight loss to overweight and obese patients with T2DM.5
Recognizing the clinical benefits of achieving weight loss in overweight or obese patients with T2DM, we aimed to increase the number of visits in the Endocrine Clinic at Central Arkansas Veterans Healthcare System (CAVHS) in Little Rock that addressed obesity, documented calorie goal for patients who are overweight or obese, and performed an intervention with further education for the patient.
Methods
The study population included veterans with either type 1 DM (T1DM) or T2DM with BMI > 25 on any DM control regimen. We performed a health record review of the eligible patients seen in the CAVHS Endocrine Clinic from June 1, 2016 to July 31, 2016 to determine the baseline percentage of visits that addressed obesity and provided weight loss advice to patients. We obtained a list of patients seen in the clinic during the study period from Strategic Management Service Services at CAVHS. We also obtained information that age, gender, medications, BMI, and last Endocrine clinic HCP assessment from the electronic health record. We reviewed the HCPs notes, including fellows and faculty who were involved in the patients’ treatment, to determine whether their notes documented a BMI > 25 and whether they discussed an intervention for overweight or obesity with the patient. The CAVHS Institutional Review Board reviewed and approved the initiative as a quality improvement study.
Intervention
Our clinic has a defined group of HCPs that we targeted for the intervention. After getting baseline information, during August 2017 we educated these HCPs on the tools available to calculate calorie goal for the patients. We advised the HCPs to use the Mifflin St Jyor equation for estimating energy expenditure and set a goal of initial weight loss between 5% and 7% of body weight. We gave specific instructions and advice to the providers (Table 1). HCPs also received educational material to distribute to patients that provided information on the healthy plate method, discussed how to count calories, and advised them on ADA goals with carbohydrate limitation. We encouraged HCPs to recommend that patients cut between 500 and 1,000 calories daily from their current diet. HCPs also received advice to seek help from clinical dieticians and the VA MOVE! Weight Management Program when appropriate.
Study of Effect of the Intervention
To study the effect of this intervention, we reviewed documentation by HCPs and assessed patient satisfaction. We obtained a list of patients and reviewed HCP notes on patients with BMI > 25 to assess whether providers addressed obesity in November and December 2017. We also evaluated whether HCPs offered a specific intervention to address the problem, such as providing education material to the patient or an estimate of daily calorie goal, or referring them to clinical dietician and/or the MOVE program. Patients received a 5-question survey that assessed their understanding and satisfaction at the end of the visit (Table 2).
Results
Of the 100 charts reviewed prior to intervention, HCPs discussed obesity management with only 6% of patients. After the intervention, we collected data again through chart review of the patients who were overweight or obese and seen for DM in the same clinic during a 2-month period. Of the 100 charts reviewed, we noticed that recognition and management of obesity improved to 60%.
To evaluate the impact of this intervention, patients received a questionnaire at the end of the visit. Nearly all (97%) patients mentioned that the provider discussed weight management during that visit. Most (83%) patients mentioned that weight management was discussed with them during prior visits, while 70% of patients felt their knowledge on working on weight loss had improved. Almost half (46%) were interested in further referral to a dietician or the MOVE program if they did not achieve desired results, but 78% were confident that they could implement the discussed weight management measures.
Discussion
Increased body weight is associated with worsening of DM and can result in poor glycemic control. Achieving weight loss in overweight or obese patients with DM can lead to clinical benefits; however, this is a challenge. In one study, a DM prevention program with lifestyle intervention leading to weight loss significantly reduced the rate of progression from impaired glucose tolerance to DM over a 3-year period and improved cardiovascular risk factors like elevated blood pressure and dyslipidemia.6 A randomized trial of an intensive lifestyle intervention to increase physical activity and decrease caloric intake vs standard DM education in people with T2DM showed a modest weight loss of 8.6% of initial weight at 1 year.7 This weight loss was associated with significant improvement in blood pressure, glycemic control, fasting blood glucose, high-density lipoprotein (HDL) cholesterol, and triglyceride levels and significant reductions in the use of DM, hypertension, and lipid-lowering medications.7 Obesity attributes to dyslipidemia with increased levels of cholesterol, low-density lipoprotein, very low-density lipoprotein, triglycerides, and decreased levels of HDL by about 5%.8 Obesity also is associated with hypertension, coronary heart disease, heart failure, and cardiovascular and all-cause mortality.9
Limitations
Limitations of this study include the small sample size and that multiple HCPs were involved. The nature of intervention might have differed with different HCPs or in a different setting than a VA clinic. In addition, we did not evaluate the effect on weight loss in specific patients as we only reviewed charts to check whether HCPs addressed weight loss. Nevertheless, our intervention was effective because it improved patient and provider awareness. It also gave us the opportunity to create framework for further collaborations and community building. The Endocrinology department at CAVHS is currently collaborating with the MOVE program, which is a part of the nutrition and food services. We hope to have an endocrinologist involved to provide guidance on medication management for obesity.
Conclusion
At CAVHS a simple intervention was instituted to evaluate whether HCPs were discussing weight loss in patients with DM, providing them with information to assess patients’ daily calorie goal, and prompting them for intervention to achieve weight loss. The intervention led to better management of patients with DM and obesity and greater engagement in weight loss from patients.
This project was a team effort. The clinic nurse documented patient’s BMI on the check in slip. HCPs discussed the problem and specific intervention. The clinical dieticians provided focused education for patients. The clerks collected the patient responses to questionnaire. This project also improved communication within the Endocrine Clinic team. Documentation of HCPs pertaining to addressing obesity improved by 54%. Improved patient satisfaction and insight was evident on patient responses to the questionnaire.
We believe that HCP apathy is a major contributor to the problem of obesity. Small steps like these go a long way for further management of obesity. Most VA hospitals have MOVE programs that provide dietary advice and encourage behavioral changes. However, getting patients to commit to these programs is a challenge. Primary care and endocrine clinics are important services that may help with patient awareness.
This project helped us better recognize patients with obesity and provide them with initial counseling and dietary advice. We received help from clinical dieticians and gave patients the option to join MOVE in situations where initial advice did not yield results and for more consistent follow up.
We tried to improve the care for patients with DM who were overweight or obese at CAVHS by prompting HCPs to focus on obesity as a problem and perform interventions to address this problem. The activities carried out and the data collected were used for internal quality improvement and for encouraging further interventions in the care of these patients.
Obesity is associated with a significant increase in mortality. It increases the risk of type 2 diabetes mellitus (T2DM), hypertension, hyperlipidemia, and coronary artery disease.1 T2DM is strongly associated with obesity in all ethnic groups.
Medical nutrition therapy and weight loss are very important for DM management.2 This includes providing education about diet modification, increased physical activity, daily calorie intake evaluation, and consistent carbohydrate intake. For patients with T2DM, health care providers (HCPs) should emphasize lowering caloric intake and inducing weight loss for those who are overweight (body mass index [BMI] between 25 and 29.9) and obese (BMI ≥ 30). This can improve glycemic control by decreasing insulin resistance. Initial recommendations for weight loss and physical activity are to lose between 5% and 10% of initial body weight and to accumulate at least 30 minutes of moderate physical activity over the course of most days of the week.3,4
Several formulas are available to estimate baseline caloric intake for weight maintenance. For weight loss of 1 to 2 pounds per week, lowering 500 to 1,000 calories from daily weight maintenance calories serves the goal. The American Diabetes Association (ADA) also suggests that HCPs recommend diet, physical activity, and behavioral therapy designed to achieve > 5% weight loss to overweight and obese patients with T2DM.5
Recognizing the clinical benefits of achieving weight loss in overweight or obese patients with T2DM, we aimed to increase the number of visits in the Endocrine Clinic at Central Arkansas Veterans Healthcare System (CAVHS) in Little Rock that addressed obesity, documented calorie goal for patients who are overweight or obese, and performed an intervention with further education for the patient.
Methods
The study population included veterans with either type 1 DM (T1DM) or T2DM with BMI > 25 on any DM control regimen. We performed a health record review of the eligible patients seen in the CAVHS Endocrine Clinic from June 1, 2016 to July 31, 2016 to determine the baseline percentage of visits that addressed obesity and provided weight loss advice to patients. We obtained a list of patients seen in the clinic during the study period from Strategic Management Service Services at CAVHS. We also obtained information that age, gender, medications, BMI, and last Endocrine clinic HCP assessment from the electronic health record. We reviewed the HCPs notes, including fellows and faculty who were involved in the patients’ treatment, to determine whether their notes documented a BMI > 25 and whether they discussed an intervention for overweight or obesity with the patient. The CAVHS Institutional Review Board reviewed and approved the initiative as a quality improvement study.
Intervention
Our clinic has a defined group of HCPs that we targeted for the intervention. After getting baseline information, during August 2017 we educated these HCPs on the tools available to calculate calorie goal for the patients. We advised the HCPs to use the Mifflin St Jyor equation for estimating energy expenditure and set a goal of initial weight loss between 5% and 7% of body weight. We gave specific instructions and advice to the providers (Table 1). HCPs also received educational material to distribute to patients that provided information on the healthy plate method, discussed how to count calories, and advised them on ADA goals with carbohydrate limitation. We encouraged HCPs to recommend that patients cut between 500 and 1,000 calories daily from their current diet. HCPs also received advice to seek help from clinical dieticians and the VA MOVE! Weight Management Program when appropriate.
Study of Effect of the Intervention
To study the effect of this intervention, we reviewed documentation by HCPs and assessed patient satisfaction. We obtained a list of patients and reviewed HCP notes on patients with BMI > 25 to assess whether providers addressed obesity in November and December 2017. We also evaluated whether HCPs offered a specific intervention to address the problem, such as providing education material to the patient or an estimate of daily calorie goal, or referring them to clinical dietician and/or the MOVE program. Patients received a 5-question survey that assessed their understanding and satisfaction at the end of the visit (Table 2).
Results
Of the 100 charts reviewed prior to intervention, HCPs discussed obesity management with only 6% of patients. After the intervention, we collected data again through chart review of the patients who were overweight or obese and seen for DM in the same clinic during a 2-month period. Of the 100 charts reviewed, we noticed that recognition and management of obesity improved to 60%.
To evaluate the impact of this intervention, patients received a questionnaire at the end of the visit. Nearly all (97%) patients mentioned that the provider discussed weight management during that visit. Most (83%) patients mentioned that weight management was discussed with them during prior visits, while 70% of patients felt their knowledge on working on weight loss had improved. Almost half (46%) were interested in further referral to a dietician or the MOVE program if they did not achieve desired results, but 78% were confident that they could implement the discussed weight management measures.
Discussion
Increased body weight is associated with worsening of DM and can result in poor glycemic control. Achieving weight loss in overweight or obese patients with DM can lead to clinical benefits; however, this is a challenge. In one study, a DM prevention program with lifestyle intervention leading to weight loss significantly reduced the rate of progression from impaired glucose tolerance to DM over a 3-year period and improved cardiovascular risk factors like elevated blood pressure and dyslipidemia.6 A randomized trial of an intensive lifestyle intervention to increase physical activity and decrease caloric intake vs standard DM education in people with T2DM showed a modest weight loss of 8.6% of initial weight at 1 year.7 This weight loss was associated with significant improvement in blood pressure, glycemic control, fasting blood glucose, high-density lipoprotein (HDL) cholesterol, and triglyceride levels and significant reductions in the use of DM, hypertension, and lipid-lowering medications.7 Obesity attributes to dyslipidemia with increased levels of cholesterol, low-density lipoprotein, very low-density lipoprotein, triglycerides, and decreased levels of HDL by about 5%.8 Obesity also is associated with hypertension, coronary heart disease, heart failure, and cardiovascular and all-cause mortality.9
Limitations
Limitations of this study include the small sample size and that multiple HCPs were involved. The nature of intervention might have differed with different HCPs or in a different setting than a VA clinic. In addition, we did not evaluate the effect on weight loss in specific patients as we only reviewed charts to check whether HCPs addressed weight loss. Nevertheless, our intervention was effective because it improved patient and provider awareness. It also gave us the opportunity to create framework for further collaborations and community building. The Endocrinology department at CAVHS is currently collaborating with the MOVE program, which is a part of the nutrition and food services. We hope to have an endocrinologist involved to provide guidance on medication management for obesity.
Conclusion
At CAVHS a simple intervention was instituted to evaluate whether HCPs were discussing weight loss in patients with DM, providing them with information to assess patients’ daily calorie goal, and prompting them for intervention to achieve weight loss. The intervention led to better management of patients with DM and obesity and greater engagement in weight loss from patients.
This project was a team effort. The clinic nurse documented patient’s BMI on the check in slip. HCPs discussed the problem and specific intervention. The clinical dieticians provided focused education for patients. The clerks collected the patient responses to questionnaire. This project also improved communication within the Endocrine Clinic team. Documentation of HCPs pertaining to addressing obesity improved by 54%. Improved patient satisfaction and insight was evident on patient responses to the questionnaire.
We believe that HCP apathy is a major contributor to the problem of obesity. Small steps like these go a long way for further management of obesity. Most VA hospitals have MOVE programs that provide dietary advice and encourage behavioral changes. However, getting patients to commit to these programs is a challenge. Primary care and endocrine clinics are important services that may help with patient awareness.
This project helped us better recognize patients with obesity and provide them with initial counseling and dietary advice. We received help from clinical dieticians and gave patients the option to join MOVE in situations where initial advice did not yield results and for more consistent follow up.
We tried to improve the care for patients with DM who were overweight or obese at CAVHS by prompting HCPs to focus on obesity as a problem and perform interventions to address this problem. The activities carried out and the data collected were used for internal quality improvement and for encouraging further interventions in the care of these patients.
1. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
2. Evert AB, Boucher JL, Cypress M, et al; American Diabetes Association. Nutrition therapy recommendations for the management of adults with diabetes. Diabetes Care. 2013;36(11):3821-3842.
3. NHLBI Obesity Education Initiative Expert Panel on the Identification, Evaluation, and Treatment of Obesity in Adults (US). Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults: The Evidence Report. Bethesda, MD: National Heart, Lung, and Blood Institute; 1998.
4. US Department of Health and Human Services. Physical Activity and Health: A Report of the Surgeon General. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion; 1996.
5. American Diabetes Association. 7. Obesity management for the treatment of type 2 diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41(Suppl 1):S65-S72.
6. Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
7. Look AHEAD Research Group; Pi-Sunyer X, Blackburn G, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care. 2007;30(6):1374-1383.
8. Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26(5):968-976.
9. Aune D, Sen A, Norat T, et al. Body mass index, abdominal fatness, and heart failure incidence and mortality: a systematic review and dose-response meta-analysis of prospective studies. Circulation. 2016;133(7):639-649.
1. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
2. Evert AB, Boucher JL, Cypress M, et al; American Diabetes Association. Nutrition therapy recommendations for the management of adults with diabetes. Diabetes Care. 2013;36(11):3821-3842.
3. NHLBI Obesity Education Initiative Expert Panel on the Identification, Evaluation, and Treatment of Obesity in Adults (US). Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults: The Evidence Report. Bethesda, MD: National Heart, Lung, and Blood Institute; 1998.
4. US Department of Health and Human Services. Physical Activity and Health: A Report of the Surgeon General. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion; 1996.
5. American Diabetes Association. 7. Obesity management for the treatment of type 2 diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care. 2018;41(Suppl 1):S65-S72.
6. Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393-403.
7. Look AHEAD Research Group; Pi-Sunyer X, Blackburn G, et al. Reduction in weight and cardiovascular disease risk factors in individuals with type 2 diabetes: one-year results of the look AHEAD trial. Diabetes Care. 2007;30(6):1374-1383.
8. Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26(5):968-976.
9. Aune D, Sen A, Norat T, et al. Body mass index, abdominal fatness, and heart failure incidence and mortality: a systematic review and dose-response meta-analysis of prospective studies. Circulation. 2016;133(7):639-649.
Emotional processing of scenes in bipolar I appears intact
New findings contradict previous studies on processing of faces in bipolar
Differences in self-reported and EEG-measured responses to emotional scenes between patients with bipolar I disorder with and without a history of psychosis, and healthy controls are negligible, results of a cross-sectional study suggest.
“While prior research supports abnormalities in the emotional face response, this study suggests these neural and behavior differences do not fully generalize to scenes, indicating that nonsocial emotional responding may be intact in these patients,” reported Rebekah L. Trotti and colleagues.
The investigators conducted a multisite study among 130 participants with bipolar and a history of psychosis, 75 with bipolar and no history of psychosis, and 181 healthy controls. Although the investigators had hypothesized that, in keeping with findings from face-processing studies, emotional responses would be reduced in patients with bipolar I disorder, The groups were presented with the same 60 scenes that were unpleasant, neutral, or pleasant. The study was published in the Journal of Psychiatric Research.
Participants rated each scene according to the Self-Assessment Manikin after the respective EEG readings were taken. No significant statistical differences were seen on these ratings between groups, reported Ms. Trotti, a graduate student in the behavioral and brain sciences program at the University of Georgia, Athens, and colleagues.
The investigators also assessed whether participants had psychosis and looked at medications they were taking. However, those analyses also showed no statistically significant differences between participants with bipolar I and a history of psychosis, those with bipolar and no history of psychosis, and healthy controls in the processing of emotional scenes. Ms. Trotti and colleagues noted that other ways of differentiating subtypes in this heterogeneous disorder, such as those based on biomarkers and brain structure rather than those laid out by the DSM, might yield the differences in neural activity they had expected.
“Future research on this topic should focus on neurocognitive subtypes of mood and psychotic disorders, as well as other domains of emotional responding and behavior,” Ms. Trotti and colleagues wrote.
SOURCE: Trotti RL et al. J Psychiatr Res. 2019. doi: 10.1016/j.jpsychires.2019.10.005.
New findings contradict previous studies on processing of faces in bipolar
New findings contradict previous studies on processing of faces in bipolar
Differences in self-reported and EEG-measured responses to emotional scenes between patients with bipolar I disorder with and without a history of psychosis, and healthy controls are negligible, results of a cross-sectional study suggest.
“While prior research supports abnormalities in the emotional face response, this study suggests these neural and behavior differences do not fully generalize to scenes, indicating that nonsocial emotional responding may be intact in these patients,” reported Rebekah L. Trotti and colleagues.
The investigators conducted a multisite study among 130 participants with bipolar and a history of psychosis, 75 with bipolar and no history of psychosis, and 181 healthy controls. Although the investigators had hypothesized that, in keeping with findings from face-processing studies, emotional responses would be reduced in patients with bipolar I disorder, The groups were presented with the same 60 scenes that were unpleasant, neutral, or pleasant. The study was published in the Journal of Psychiatric Research.
Participants rated each scene according to the Self-Assessment Manikin after the respective EEG readings were taken. No significant statistical differences were seen on these ratings between groups, reported Ms. Trotti, a graduate student in the behavioral and brain sciences program at the University of Georgia, Athens, and colleagues.
The investigators also assessed whether participants had psychosis and looked at medications they were taking. However, those analyses also showed no statistically significant differences between participants with bipolar I and a history of psychosis, those with bipolar and no history of psychosis, and healthy controls in the processing of emotional scenes. Ms. Trotti and colleagues noted that other ways of differentiating subtypes in this heterogeneous disorder, such as those based on biomarkers and brain structure rather than those laid out by the DSM, might yield the differences in neural activity they had expected.
“Future research on this topic should focus on neurocognitive subtypes of mood and psychotic disorders, as well as other domains of emotional responding and behavior,” Ms. Trotti and colleagues wrote.
SOURCE: Trotti RL et al. J Psychiatr Res. 2019. doi: 10.1016/j.jpsychires.2019.10.005.
Differences in self-reported and EEG-measured responses to emotional scenes between patients with bipolar I disorder with and without a history of psychosis, and healthy controls are negligible, results of a cross-sectional study suggest.
“While prior research supports abnormalities in the emotional face response, this study suggests these neural and behavior differences do not fully generalize to scenes, indicating that nonsocial emotional responding may be intact in these patients,” reported Rebekah L. Trotti and colleagues.
The investigators conducted a multisite study among 130 participants with bipolar and a history of psychosis, 75 with bipolar and no history of psychosis, and 181 healthy controls. Although the investigators had hypothesized that, in keeping with findings from face-processing studies, emotional responses would be reduced in patients with bipolar I disorder, The groups were presented with the same 60 scenes that were unpleasant, neutral, or pleasant. The study was published in the Journal of Psychiatric Research.
Participants rated each scene according to the Self-Assessment Manikin after the respective EEG readings were taken. No significant statistical differences were seen on these ratings between groups, reported Ms. Trotti, a graduate student in the behavioral and brain sciences program at the University of Georgia, Athens, and colleagues.
The investigators also assessed whether participants had psychosis and looked at medications they were taking. However, those analyses also showed no statistically significant differences between participants with bipolar I and a history of psychosis, those with bipolar and no history of psychosis, and healthy controls in the processing of emotional scenes. Ms. Trotti and colleagues noted that other ways of differentiating subtypes in this heterogeneous disorder, such as those based on biomarkers and brain structure rather than those laid out by the DSM, might yield the differences in neural activity they had expected.
“Future research on this topic should focus on neurocognitive subtypes of mood and psychotic disorders, as well as other domains of emotional responding and behavior,” Ms. Trotti and colleagues wrote.
SOURCE: Trotti RL et al. J Psychiatr Res. 2019. doi: 10.1016/j.jpsychires.2019.10.005.
FROM THE JOURNAL OF PSYCHIATRIC RESEARCH
Limited Use of Outpatient Stress Testing in Young Patients With Atypical Chest Pain (FULL)
Low prevalence of coronary artery disease within this population suggests that younger patients may not require stress testing for chest pain evaluations as long as pretest likelihood is low.
The decision to perform stress testing in the evaluation of chest pain is often based on the pretest likelihood of coronary artery disease (CAD).1-7 Cardiac risk scores, which incorporate smoking status, blood pressure, diabetes mellitus, and cholesterol levels, also may provide further risk stratification.8-11 Assuming that the prevalence of CAD increases with age, young adults could be deemed low risk, not warranting cardiac screening.12
Professional society guidelines from the American College of Cardiology/American Heart Association and American College of Physicians4,5 recommend stress testing as the initial diagnostic test for CAD in symptomatic patients; additionally, the guidelines also suggest that screening stress tests may confer primary prevention benefit in intermediate-risk asymptomatic patients.9,13 Exercise treadmill testing is considered the initial modality of choice, given its technical ease and lower cost, compared with stress echocardiography.14
Previously published reports have shown the limited use of stress testing to screen young asymptomatic adults.15-17 Because this patient demographic typically has a low pretest likelihood of CAD, positive stress tests are often false-positive results.7,18 The consequence of false-positive testing may be unnecessary additional cardiac testing, potentially leading to more patient harm than benefit.18,19 For active-duty service members, false-positive testing also has the potential to affect worldwide deployability and/or sea duty status while further risk stratification is performed; as a result, mission readiness may be impacted.Although the number of clinic visits for chest pain has declined, there has been a discordant increase in the rates of stress testing in the US.20-22 Additionally, the rate of stress testing among young adults, specifically in the 25- to 34-year age group, has increased in recent years. Given the rising use of stress tests in the young patient population, the clinical use of stress testing needs to be reassessed.
Although much of the literature has already demonstrated the low value of stress testing in young asymptomatic adults, no data currently exist regarding its outpatient use in evaluating young symptomatic patients. The military represents a predominantly young cross-section of the general population suitable for exploring this topic. Using a cohort of active-duty service members, we aimed to determine the use of outpatient stress testing in evaluating young patients with atypical chest pain.
Methods
The US Department of Defense (DoD) Military Health System Database Repository (MDR) and Comprehensive Ambulatory Professional Encounter Record (CAPER) were the data sources for this study. The MDR contains continually updated, longitudinal electronic medical records (EMRs) for nearly 1.4 million active-duty service members and is composed of administrative, medical, pharmacy, and clinical data. The Naval Medical Center Portsmouth (NMCP) Institutional Review Board approved this study.
Study Cohort
We performed chart reviews of service members aged 18 to 35 years who received cardiac stress testing at NMCP, an academic tertiary care center, within 30 days after an office visit for atypical chest pain between October 1, 2010, and September 30, 2015. Atypical chest pain was defined as any outpatient claim with ICD-9 code, 786.5x, in the primary diagnosis field (Table 1).4 
Demographics and cardiac risk factors (ie, hypertension, hyperlipidemia, diabetes mellitus, and smoking status) were assessed prior to index chest pain evaluations and defined via ICD-9 codes within outpatient records.
Cardiac Testing Outcomes
Patients were initially categorized by the results of baseline electrocardiograms (ECG) and index stress tests (ie, exercise treadmill or stress echocardiography, exercise or Lexiscan myocardial perfusion imaging, dobutamine stress echocardiography). Positive tests were defined as those having electrical or structural ischemic changes. Chronotropic changes were infrequent and nonpathologic and were not counted. Patient endpoints were either additional cardiac testing or negative index stress test without additional testing.
Statistical Analysis
The agreement between both baseline ECG and index stress test as well as index stress test and additional cardiac testing were analyzed using McNemar test and matched-pair odds ratios (ORs) with corresponding 95% CIs. Analyses were stratified by demographics and cardiac risk factors to assess for potential confounding. Analyses were performed using SAS version 9.4 (Cary, NC).
Results
A total of 1,036 patients were evaluated for atypical chest pain and had index stress testing between October 1, 2010 and September 30, 2015. The study cohort was 69% male with a mean (SD) age of 27.3 (4.7) years. More than 60% of the cohort was older than aged > 25 years. 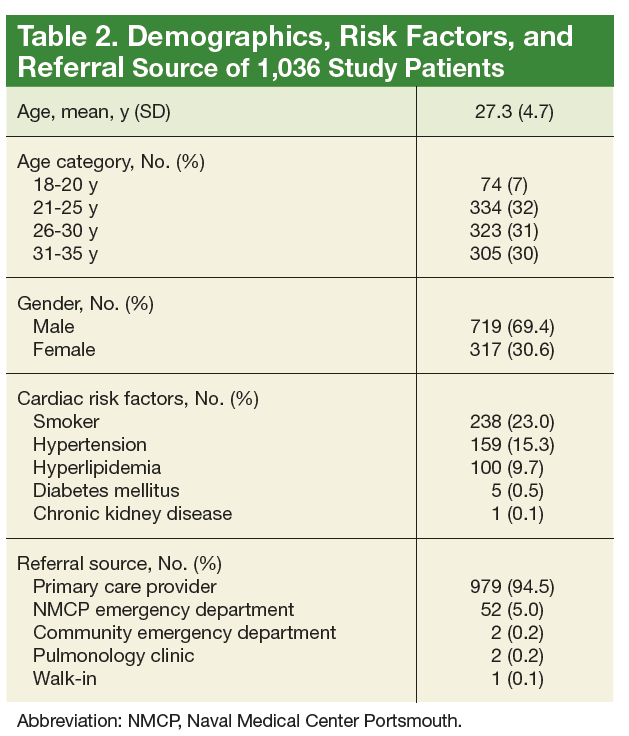
In the initial testing cohort, exercise treadmill test (59.3%) and exercise echocardiogram (37.1%) were the most common stress testing modalities. The mean (SD) metabolic equivalents (METS) achieved among individuals who performed exercise stress testing was 13.9 (2.8). There were 65 patients who had a positive baseline ECG/negative index stress test, 958 patients had a negative ECG/negative index test, and 8 patients had a negative ECG/positive index test. 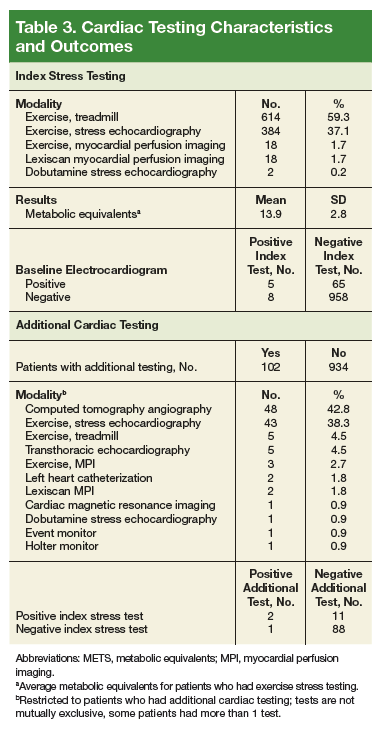
There were 102 patients (10%) who performed additional cardiac testing. Among this subgroup, 13 patients (1.3%) had additional testing for further evaluation of a positive index stress test (Table 4) and 89 patients (8.6%) had testing for continuing atypical chest pain despite a negative index stress test. 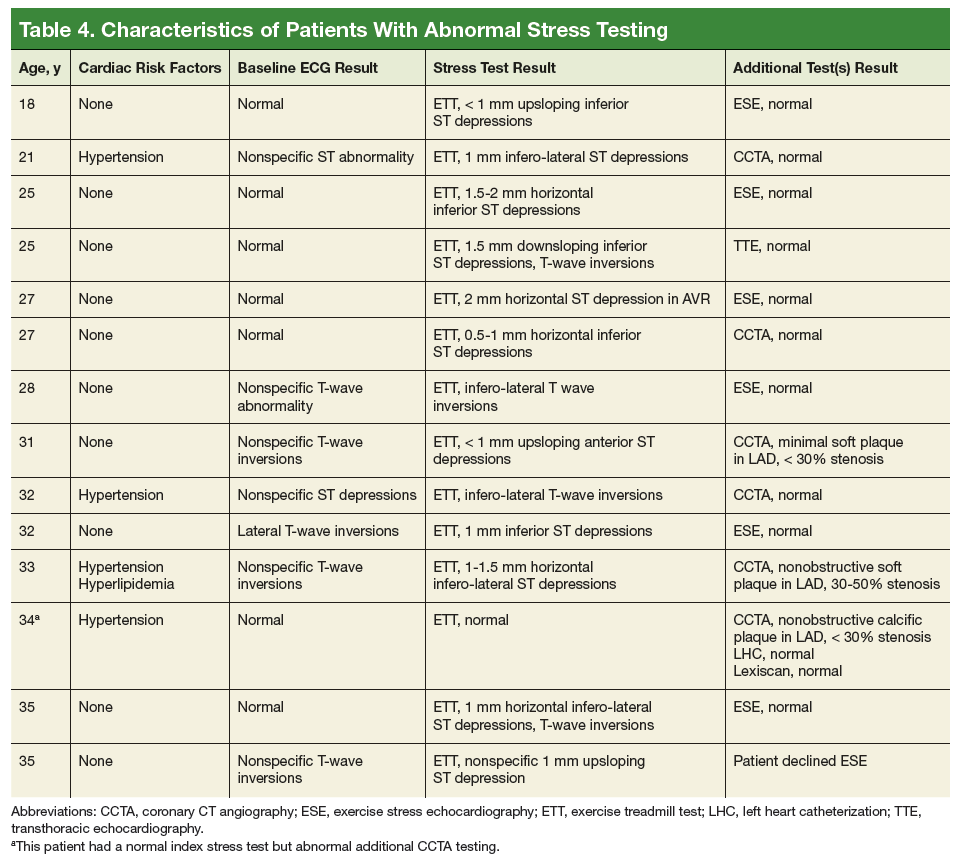
Coronary computed tomography angiography (CCTA) demonstrated nonobstructive CAD in 3 patients (0.3%) within the study cohort. There was no obstructive CAD identified in our cohort. Two patients had negative left heart catheterizations (LHC). One of these patients had a negative LHC and a negative Lexiscan after a CCTA showed CAD; all 3 of these additional tests were performed for evaluation of continued chest pain despite negative index stress testing. The positive predictive value of cardiac stress testing for nonobstructive CAD in this low-risk population was 15.4% (2 of 13). Stratification by demographics, CAD risk factors, and cardiac test results revealed no presence of confounding factors during analyses.
Discussion
In this retrospective, observational study of 1,036 young patients with atypical chest pain who had stress testing, there was relatively strong agreement between baseline ECG and index stress test results. Individuals also were 8 times more likely to have positive baseline ECGs and negative stress testing than having the opposite finding. Additional cardiac testing similarly demonstrated congruency with index stress testing and showed the propensity for false-positive stress tests. Further testing with CCTA demonstrated minimal nonobstructive CAD in < 1% of the study cohort and 2 LHC were negative. Despite the low prevalence of CAD and apparent low diagnostic use of stress testing in our young cohort, symptomatic service members still require stress testing to determine deployment suitability.
The low yield of outpatient stress testing in our young population is rooted in Bayes’ theorem, which highlights the importance of pretest likelihood in the diagnosis of CAD.7,23 Because our cohort had a low prevalence and low pretest likelihood of CAD, positive index stress tests were often false-positive results and consequently did not increase the posttest likelihood of CAD, resulting in low positive predictive value. Additional cardiac testing had limited clinical value in our cohort. The 3 cases of nonobstructive CAD were unlikely to be pathologic given the minimal degree of observed stenosis and the 2 LHC did not require revascularization. These results are similar to those shown by Christman and colleagues and Mudrick and colleagues, which highlighted the low yield of additional cardiac studies and low rate of revascularization among symptomatic patients without known cardiac disease, respectively.18,19
This is the first study, to our knowledge, to quantitatively demonstrate the low use of outpatient stress testing for young adults with atypical chest pain. Previous studies that assessed stress testing for young patients with chest pain in acute settings such as emergency departments and chest pain observation units, similarly demonstrated minimal yield of routine diagnostic testing.23,24 This further highlights the premise that outpatient and even emergent-setting stress testing in low cardiac risk individuals may be of limited value and not always necessary.
Limitations
There were several study limitations. As a single-center, cross-sectional review, we may not be able to extrapolate our findings to the general population. However, given the low prevalence of CAD in young adults, stress testing would likely have limited value regardless of the sample distribution; so it may be possible to extend our findings beyond our cohort. Also, neither baseline ECG nor index stress test (irrespective of modality) could be given a diagnostic value in predicting ischemia alone; doing so would require comparison with the gold standard—heart catheterization. Although referral bias has been associated with diagnostic performance of stress testing, we did not adjust for this phenomenon.25 Given the higher average metabolic equivalents achieved in our cohort, this potential bias likely did not affect diagnostic performance.
Conclusion
There was low diagnostic use of outpatient stress testing and additional cardiac testing for CAD among young patients with atypical chest pain. The limited value of cardiac stress testing is likely a function of the low CAD prevalence within this population, suggesting that younger patients may not necessarily require stress testing for chest pain evaluations as long as pretest likelihood is low. Despite our results, we maintain that the decision to perform stress testing should still be guided by clinical judgment, but perhaps our findings may alleviate physicians’ concerns over the urgency of when to refer low-risk patients for testing. Although we are cautious in inferring our findings to the general population, the similarity it shares with those from other published reports may suggest its applicability beyond our study cohort.
1. Fowler-Brown A, Pignone M, Pletcher M, et al. Exercise tolerance testing to screen for coronary heart disease: a systematic review for the technical support for the U.S. Preventive Services Task Force. Ann Intern Med. 2004;140(7):W9-W24.
2. Gibbons RJ, Balady GJ, Bricker JT, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Committee to Update the 1997 Exercise Testing Guidelines. ACC/AHA 2002 guideline update for exercise testing: summary article. A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (Committee to Update the 1997 Exercise Testing Guidelines). J Am Coll Cardiol. 2002;40(8):1531-1540.
3. Chou R, Arora B, Dana T, Fu R, Miranda Walker M, Humphrey L. Screening Asymptomatic Adults for Coronary Heart Disease With Resting or Exercise Electrocardiography: Systematic Review to Update the 2004 U.S. Preventive Services Task Force recommendation. Evidence Synthesis No. 88. AHRQ Publication No. 11-05158-EF-1. Rockville, MD: Agency for Healthcare Research and Quality; September 2011.
4. Fihn S, Gardin J, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiology. 2012;60(24):e44-e164.
5. Gibbons RJ, Balady GJ, Beasley JW, et al. ACC/AHA guidelines for exercise testing: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Exercise Testing). Circulation. 1997;96(1):345-354.
6. Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. A report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2010;56(25):e50-e103.
7. Diamond G, Forrester J. Analysis of probability as an aid in the clinical diagnosis of coronary artery disease. N Engl J Med. 1979;300(24):1350-1358.
8. Goff D, Lloyd-Jones D, Bennett G, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25)(suppl 2):S49-S73.
9. Greenland P, Gaziano J. Selecting asymptomatic patients for coronary computed tomography or electrocardiographic exercise testing. N Engl J Med. 2003;349(5):465-473.
10. Shah N, Soon K, Wong C, Kellu AM. Screening for asymptomatic coronary heart disease in the young ‘at risk’ population: who and how? Int J Cardiol Heart Vasc. 2014;6:60-65.
11. Morise A, Evans M, Jalisi F, Shetty R, Stauffer M. A pretest prognostic score to assess patients undergoing exercise or pharmacological stress testing. Heart. 2007;93(2):200-204.
12. Amsterdam EA, Kirk JD, Bluemke DA, et al; American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee of the Council on Clinical Cardiology, Council on Cardiovascular Nursing, and Interdisciplinary Council on Quality of Care and Outcomes Research. Testing of low-risk patients presenting to the emergency department with chest pain: a scientific statement from the American Heart Association. Circulation. 2010;122(17):1756-1776.
13. Livschitz S, Sharabi Y, Yushin J, et al. Limited clinical value of exercise stress test for the screening of coronary artery disease in young, asymptomatic adult men. Am J Cardiol. 2000;86(4):462-464.
14. Miller T. Stress testing: the case for the standard treadmill test. Curr Opin Cardiol. 2011;26(5):363-369.
15. La Gerche A, Baggish A, Knuuti J, et al. Cardiac imaging and stress testing asymptomatic athletes to identify those at risk of sudden cardiac death. JACC Cardiovasc Imaging. 2013;6(9):993-1007.
16. Lauer M, Froelicher ES, Williams M, Kligfield P; American Heart Association Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Exercise testing in asymptomatic adults: a statement for professionals from the American Heart Association Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Circulation. 2005;112(5):771-776.
17. Sammito S, Gundlach N, Bockelmann I. Prevalence of cardiac arrhythmia under stress conditions in occupational health assessments of young military servicemen and servicewomen. Mil Med. 2016;181(4):369-372.
18. Mudrick DW, Cowper PA, Shah BR, et al. Downstream procedures and outcomes after stress testing for chest pain without known coronary artery disease in the United States. Am Heart J. 2012;163(3):454-461.
19. Christman MP, Bittencourt MS, Hulten E, et al. Yield of downstream tests after exercise treadmill testing. J Am Coll Cardiol. 2014;63(13):1264-1274.
20. Will J, Loustalot F, Hong Y. National trends in visits to physician offices and outpatient clinics for angina 1995 to 2010. Circ Cardiovasc Qual Outcomes. 2014;7(1):110-117.
21. Kini V, McCarthy F, Dayoub E, et al. Cardiac stress test trends among US patients younger than 65 years, 2005-2012. JAMA Cardiol. 2016;1(9):1038-1042.
22. Ladapo JA, Blecker S, Douglas PS. Physician decision making and trends in the use of cardiac stress testing in the United States: an analysis of repeated cross-sectional data. Ann Intern Med. 2014;161(7):482-490.
23. Winchester DE, Brandt J, Schmidt C, Schmidt C, Allen B, Payton T, Amsterdam EA. Diagnostic yield of routine noninvasive cardiovascular testing in low-risk acute chest pain patients. Am J Cardiol. 2015;116(2):204-207.
24. Hermann L, Weingart SD, Duvall W, Henzlova MJ. The limited utility of routine cardiac stress testing in emergency department chest pain patients younger than 40 years. Ann Emerg Med. 2009;54(1):12-16.
25. Ladapo JA, Blecker S, Elashoff MR, et al. Clinical implications of referral bias in the diagnostic performance of exercise testing for coronary artery disease. J Am Heart Assoc. 2013;2(6):e000505.
Low prevalence of coronary artery disease within this population suggests that younger patients may not require stress testing for chest pain evaluations as long as pretest likelihood is low.
Low prevalence of coronary artery disease within this population suggests that younger patients may not require stress testing for chest pain evaluations as long as pretest likelihood is low.
The decision to perform stress testing in the evaluation of chest pain is often based on the pretest likelihood of coronary artery disease (CAD).1-7 Cardiac risk scores, which incorporate smoking status, blood pressure, diabetes mellitus, and cholesterol levels, also may provide further risk stratification.8-11 Assuming that the prevalence of CAD increases with age, young adults could be deemed low risk, not warranting cardiac screening.12
Professional society guidelines from the American College of Cardiology/American Heart Association and American College of Physicians4,5 recommend stress testing as the initial diagnostic test for CAD in symptomatic patients; additionally, the guidelines also suggest that screening stress tests may confer primary prevention benefit in intermediate-risk asymptomatic patients.9,13 Exercise treadmill testing is considered the initial modality of choice, given its technical ease and lower cost, compared with stress echocardiography.14
Previously published reports have shown the limited use of stress testing to screen young asymptomatic adults.15-17 Because this patient demographic typically has a low pretest likelihood of CAD, positive stress tests are often false-positive results.7,18 The consequence of false-positive testing may be unnecessary additional cardiac testing, potentially leading to more patient harm than benefit.18,19 For active-duty service members, false-positive testing also has the potential to affect worldwide deployability and/or sea duty status while further risk stratification is performed; as a result, mission readiness may be impacted.Although the number of clinic visits for chest pain has declined, there has been a discordant increase in the rates of stress testing in the US.20-22 Additionally, the rate of stress testing among young adults, specifically in the 25- to 34-year age group, has increased in recent years. Given the rising use of stress tests in the young patient population, the clinical use of stress testing needs to be reassessed.
Although much of the literature has already demonstrated the low value of stress testing in young asymptomatic adults, no data currently exist regarding its outpatient use in evaluating young symptomatic patients. The military represents a predominantly young cross-section of the general population suitable for exploring this topic. Using a cohort of active-duty service members, we aimed to determine the use of outpatient stress testing in evaluating young patients with atypical chest pain.
Methods
The US Department of Defense (DoD) Military Health System Database Repository (MDR) and Comprehensive Ambulatory Professional Encounter Record (CAPER) were the data sources for this study. The MDR contains continually updated, longitudinal electronic medical records (EMRs) for nearly 1.4 million active-duty service members and is composed of administrative, medical, pharmacy, and clinical data. The Naval Medical Center Portsmouth (NMCP) Institutional Review Board approved this study.
Study Cohort
We performed chart reviews of service members aged 18 to 35 years who received cardiac stress testing at NMCP, an academic tertiary care center, within 30 days after an office visit for atypical chest pain between October 1, 2010, and September 30, 2015. Atypical chest pain was defined as any outpatient claim with ICD-9 code, 786.5x, in the primary diagnosis field (Table 1).4
Demographics and cardiac risk factors (ie, hypertension, hyperlipidemia, diabetes mellitus, and smoking status) were assessed prior to index chest pain evaluations and defined via ICD-9 codes within outpatient records.
Cardiac Testing Outcomes
Patients were initially categorized by the results of baseline electrocardiograms (ECG) and index stress tests (ie, exercise treadmill or stress echocardiography, exercise or Lexiscan myocardial perfusion imaging, dobutamine stress echocardiography). Positive tests were defined as those having electrical or structural ischemic changes. Chronotropic changes were infrequent and nonpathologic and were not counted. Patient endpoints were either additional cardiac testing or negative index stress test without additional testing.
Statistical Analysis
The agreement between both baseline ECG and index stress test as well as index stress test and additional cardiac testing were analyzed using McNemar test and matched-pair odds ratios (ORs) with corresponding 95% CIs. Analyses were stratified by demographics and cardiac risk factors to assess for potential confounding. Analyses were performed using SAS version 9.4 (Cary, NC).
Results
A total of 1,036 patients were evaluated for atypical chest pain and had index stress testing between October 1, 2010 and September 30, 2015. The study cohort was 69% male with a mean (SD) age of 27.3 (4.7) years. More than 60% of the cohort was older than aged > 25 years.
In the initial testing cohort, exercise treadmill test (59.3%) and exercise echocardiogram (37.1%) were the most common stress testing modalities. The mean (SD) metabolic equivalents (METS) achieved among individuals who performed exercise stress testing was 13.9 (2.8). There were 65 patients who had a positive baseline ECG/negative index stress test, 958 patients had a negative ECG/negative index test, and 8 patients had a negative ECG/positive index test.
There were 102 patients (10%) who performed additional cardiac testing. Among this subgroup, 13 patients (1.3%) had additional testing for further evaluation of a positive index stress test (Table 4) and 89 patients (8.6%) had testing for continuing atypical chest pain despite a negative index stress test.
Coronary computed tomography angiography (CCTA) demonstrated nonobstructive CAD in 3 patients (0.3%) within the study cohort. There was no obstructive CAD identified in our cohort. Two patients had negative left heart catheterizations (LHC). One of these patients had a negative LHC and a negative Lexiscan after a CCTA showed CAD; all 3 of these additional tests were performed for evaluation of continued chest pain despite negative index stress testing. The positive predictive value of cardiac stress testing for nonobstructive CAD in this low-risk population was 15.4% (2 of 13). Stratification by demographics, CAD risk factors, and cardiac test results revealed no presence of confounding factors during analyses.
Discussion
In this retrospective, observational study of 1,036 young patients with atypical chest pain who had stress testing, there was relatively strong agreement between baseline ECG and index stress test results. Individuals also were 8 times more likely to have positive baseline ECGs and negative stress testing than having the opposite finding. Additional cardiac testing similarly demonstrated congruency with index stress testing and showed the propensity for false-positive stress tests. Further testing with CCTA demonstrated minimal nonobstructive CAD in < 1% of the study cohort and 2 LHC were negative. Despite the low prevalence of CAD and apparent low diagnostic use of stress testing in our young cohort, symptomatic service members still require stress testing to determine deployment suitability.
The low yield of outpatient stress testing in our young population is rooted in Bayes’ theorem, which highlights the importance of pretest likelihood in the diagnosis of CAD.7,23 Because our cohort had a low prevalence and low pretest likelihood of CAD, positive index stress tests were often false-positive results and consequently did not increase the posttest likelihood of CAD, resulting in low positive predictive value. Additional cardiac testing had limited clinical value in our cohort. The 3 cases of nonobstructive CAD were unlikely to be pathologic given the minimal degree of observed stenosis and the 2 LHC did not require revascularization. These results are similar to those shown by Christman and colleagues and Mudrick and colleagues, which highlighted the low yield of additional cardiac studies and low rate of revascularization among symptomatic patients without known cardiac disease, respectively.18,19
This is the first study, to our knowledge, to quantitatively demonstrate the low use of outpatient stress testing for young adults with atypical chest pain. Previous studies that assessed stress testing for young patients with chest pain in acute settings such as emergency departments and chest pain observation units, similarly demonstrated minimal yield of routine diagnostic testing.23,24 This further highlights the premise that outpatient and even emergent-setting stress testing in low cardiac risk individuals may be of limited value and not always necessary.
Limitations
There were several study limitations. As a single-center, cross-sectional review, we may not be able to extrapolate our findings to the general population. However, given the low prevalence of CAD in young adults, stress testing would likely have limited value regardless of the sample distribution; so it may be possible to extend our findings beyond our cohort. Also, neither baseline ECG nor index stress test (irrespective of modality) could be given a diagnostic value in predicting ischemia alone; doing so would require comparison with the gold standard—heart catheterization. Although referral bias has been associated with diagnostic performance of stress testing, we did not adjust for this phenomenon.25 Given the higher average metabolic equivalents achieved in our cohort, this potential bias likely did not affect diagnostic performance.
Conclusion
There was low diagnostic use of outpatient stress testing and additional cardiac testing for CAD among young patients with atypical chest pain. The limited value of cardiac stress testing is likely a function of the low CAD prevalence within this population, suggesting that younger patients may not necessarily require stress testing for chest pain evaluations as long as pretest likelihood is low. Despite our results, we maintain that the decision to perform stress testing should still be guided by clinical judgment, but perhaps our findings may alleviate physicians’ concerns over the urgency of when to refer low-risk patients for testing. Although we are cautious in inferring our findings to the general population, the similarity it shares with those from other published reports may suggest its applicability beyond our study cohort.
The decision to perform stress testing in the evaluation of chest pain is often based on the pretest likelihood of coronary artery disease (CAD).1-7 Cardiac risk scores, which incorporate smoking status, blood pressure, diabetes mellitus, and cholesterol levels, also may provide further risk stratification.8-11 Assuming that the prevalence of CAD increases with age, young adults could be deemed low risk, not warranting cardiac screening.12
Professional society guidelines from the American College of Cardiology/American Heart Association and American College of Physicians4,5 recommend stress testing as the initial diagnostic test for CAD in symptomatic patients; additionally, the guidelines also suggest that screening stress tests may confer primary prevention benefit in intermediate-risk asymptomatic patients.9,13 Exercise treadmill testing is considered the initial modality of choice, given its technical ease and lower cost, compared with stress echocardiography.14
Previously published reports have shown the limited use of stress testing to screen young asymptomatic adults.15-17 Because this patient demographic typically has a low pretest likelihood of CAD, positive stress tests are often false-positive results.7,18 The consequence of false-positive testing may be unnecessary additional cardiac testing, potentially leading to more patient harm than benefit.18,19 For active-duty service members, false-positive testing also has the potential to affect worldwide deployability and/or sea duty status while further risk stratification is performed; as a result, mission readiness may be impacted.Although the number of clinic visits for chest pain has declined, there has been a discordant increase in the rates of stress testing in the US.20-22 Additionally, the rate of stress testing among young adults, specifically in the 25- to 34-year age group, has increased in recent years. Given the rising use of stress tests in the young patient population, the clinical use of stress testing needs to be reassessed.
Although much of the literature has already demonstrated the low value of stress testing in young asymptomatic adults, no data currently exist regarding its outpatient use in evaluating young symptomatic patients. The military represents a predominantly young cross-section of the general population suitable for exploring this topic. Using a cohort of active-duty service members, we aimed to determine the use of outpatient stress testing in evaluating young patients with atypical chest pain.
Methods
The US Department of Defense (DoD) Military Health System Database Repository (MDR) and Comprehensive Ambulatory Professional Encounter Record (CAPER) were the data sources for this study. The MDR contains continually updated, longitudinal electronic medical records (EMRs) for nearly 1.4 million active-duty service members and is composed of administrative, medical, pharmacy, and clinical data. The Naval Medical Center Portsmouth (NMCP) Institutional Review Board approved this study.
Study Cohort
We performed chart reviews of service members aged 18 to 35 years who received cardiac stress testing at NMCP, an academic tertiary care center, within 30 days after an office visit for atypical chest pain between October 1, 2010, and September 30, 2015. Atypical chest pain was defined as any outpatient claim with ICD-9 code, 786.5x, in the primary diagnosis field (Table 1).4
Demographics and cardiac risk factors (ie, hypertension, hyperlipidemia, diabetes mellitus, and smoking status) were assessed prior to index chest pain evaluations and defined via ICD-9 codes within outpatient records.
Cardiac Testing Outcomes
Patients were initially categorized by the results of baseline electrocardiograms (ECG) and index stress tests (ie, exercise treadmill or stress echocardiography, exercise or Lexiscan myocardial perfusion imaging, dobutamine stress echocardiography). Positive tests were defined as those having electrical or structural ischemic changes. Chronotropic changes were infrequent and nonpathologic and were not counted. Patient endpoints were either additional cardiac testing or negative index stress test without additional testing.
Statistical Analysis
The agreement between both baseline ECG and index stress test as well as index stress test and additional cardiac testing were analyzed using McNemar test and matched-pair odds ratios (ORs) with corresponding 95% CIs. Analyses were stratified by demographics and cardiac risk factors to assess for potential confounding. Analyses were performed using SAS version 9.4 (Cary, NC).
Results
A total of 1,036 patients were evaluated for atypical chest pain and had index stress testing between October 1, 2010 and September 30, 2015. The study cohort was 69% male with a mean (SD) age of 27.3 (4.7) years. More than 60% of the cohort was older than aged > 25 years.
In the initial testing cohort, exercise treadmill test (59.3%) and exercise echocardiogram (37.1%) were the most common stress testing modalities. The mean (SD) metabolic equivalents (METS) achieved among individuals who performed exercise stress testing was 13.9 (2.8). There were 65 patients who had a positive baseline ECG/negative index stress test, 958 patients had a negative ECG/negative index test, and 8 patients had a negative ECG/positive index test.
There were 102 patients (10%) who performed additional cardiac testing. Among this subgroup, 13 patients (1.3%) had additional testing for further evaluation of a positive index stress test (Table 4) and 89 patients (8.6%) had testing for continuing atypical chest pain despite a negative index stress test.
Coronary computed tomography angiography (CCTA) demonstrated nonobstructive CAD in 3 patients (0.3%) within the study cohort. There was no obstructive CAD identified in our cohort. Two patients had negative left heart catheterizations (LHC). One of these patients had a negative LHC and a negative Lexiscan after a CCTA showed CAD; all 3 of these additional tests were performed for evaluation of continued chest pain despite negative index stress testing. The positive predictive value of cardiac stress testing for nonobstructive CAD in this low-risk population was 15.4% (2 of 13). Stratification by demographics, CAD risk factors, and cardiac test results revealed no presence of confounding factors during analyses.
Discussion
In this retrospective, observational study of 1,036 young patients with atypical chest pain who had stress testing, there was relatively strong agreement between baseline ECG and index stress test results. Individuals also were 8 times more likely to have positive baseline ECGs and negative stress testing than having the opposite finding. Additional cardiac testing similarly demonstrated congruency with index stress testing and showed the propensity for false-positive stress tests. Further testing with CCTA demonstrated minimal nonobstructive CAD in < 1% of the study cohort and 2 LHC were negative. Despite the low prevalence of CAD and apparent low diagnostic use of stress testing in our young cohort, symptomatic service members still require stress testing to determine deployment suitability.
The low yield of outpatient stress testing in our young population is rooted in Bayes’ theorem, which highlights the importance of pretest likelihood in the diagnosis of CAD.7,23 Because our cohort had a low prevalence and low pretest likelihood of CAD, positive index stress tests were often false-positive results and consequently did not increase the posttest likelihood of CAD, resulting in low positive predictive value. Additional cardiac testing had limited clinical value in our cohort. The 3 cases of nonobstructive CAD were unlikely to be pathologic given the minimal degree of observed stenosis and the 2 LHC did not require revascularization. These results are similar to those shown by Christman and colleagues and Mudrick and colleagues, which highlighted the low yield of additional cardiac studies and low rate of revascularization among symptomatic patients without known cardiac disease, respectively.18,19
This is the first study, to our knowledge, to quantitatively demonstrate the low use of outpatient stress testing for young adults with atypical chest pain. Previous studies that assessed stress testing for young patients with chest pain in acute settings such as emergency departments and chest pain observation units, similarly demonstrated minimal yield of routine diagnostic testing.23,24 This further highlights the premise that outpatient and even emergent-setting stress testing in low cardiac risk individuals may be of limited value and not always necessary.
Limitations
There were several study limitations. As a single-center, cross-sectional review, we may not be able to extrapolate our findings to the general population. However, given the low prevalence of CAD in young adults, stress testing would likely have limited value regardless of the sample distribution; so it may be possible to extend our findings beyond our cohort. Also, neither baseline ECG nor index stress test (irrespective of modality) could be given a diagnostic value in predicting ischemia alone; doing so would require comparison with the gold standard—heart catheterization. Although referral bias has been associated with diagnostic performance of stress testing, we did not adjust for this phenomenon.25 Given the higher average metabolic equivalents achieved in our cohort, this potential bias likely did not affect diagnostic performance.
Conclusion
There was low diagnostic use of outpatient stress testing and additional cardiac testing for CAD among young patients with atypical chest pain. The limited value of cardiac stress testing is likely a function of the low CAD prevalence within this population, suggesting that younger patients may not necessarily require stress testing for chest pain evaluations as long as pretest likelihood is low. Despite our results, we maintain that the decision to perform stress testing should still be guided by clinical judgment, but perhaps our findings may alleviate physicians’ concerns over the urgency of when to refer low-risk patients for testing. Although we are cautious in inferring our findings to the general population, the similarity it shares with those from other published reports may suggest its applicability beyond our study cohort.
1. Fowler-Brown A, Pignone M, Pletcher M, et al. Exercise tolerance testing to screen for coronary heart disease: a systematic review for the technical support for the U.S. Preventive Services Task Force. Ann Intern Med. 2004;140(7):W9-W24.
2. Gibbons RJ, Balady GJ, Bricker JT, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Committee to Update the 1997 Exercise Testing Guidelines. ACC/AHA 2002 guideline update for exercise testing: summary article. A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (Committee to Update the 1997 Exercise Testing Guidelines). J Am Coll Cardiol. 2002;40(8):1531-1540.
3. Chou R, Arora B, Dana T, Fu R, Miranda Walker M, Humphrey L. Screening Asymptomatic Adults for Coronary Heart Disease With Resting or Exercise Electrocardiography: Systematic Review to Update the 2004 U.S. Preventive Services Task Force recommendation. Evidence Synthesis No. 88. AHRQ Publication No. 11-05158-EF-1. Rockville, MD: Agency for Healthcare Research and Quality; September 2011.
4. Fihn S, Gardin J, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiology. 2012;60(24):e44-e164.
5. Gibbons RJ, Balady GJ, Beasley JW, et al. ACC/AHA guidelines for exercise testing: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Exercise Testing). Circulation. 1997;96(1):345-354.
6. Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. A report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2010;56(25):e50-e103.
7. Diamond G, Forrester J. Analysis of probability as an aid in the clinical diagnosis of coronary artery disease. N Engl J Med. 1979;300(24):1350-1358.
8. Goff D, Lloyd-Jones D, Bennett G, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25)(suppl 2):S49-S73.
9. Greenland P, Gaziano J. Selecting asymptomatic patients for coronary computed tomography or electrocardiographic exercise testing. N Engl J Med. 2003;349(5):465-473.
10. Shah N, Soon K, Wong C, Kellu AM. Screening for asymptomatic coronary heart disease in the young ‘at risk’ population: who and how? Int J Cardiol Heart Vasc. 2014;6:60-65.
11. Morise A, Evans M, Jalisi F, Shetty R, Stauffer M. A pretest prognostic score to assess patients undergoing exercise or pharmacological stress testing. Heart. 2007;93(2):200-204.
12. Amsterdam EA, Kirk JD, Bluemke DA, et al; American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee of the Council on Clinical Cardiology, Council on Cardiovascular Nursing, and Interdisciplinary Council on Quality of Care and Outcomes Research. Testing of low-risk patients presenting to the emergency department with chest pain: a scientific statement from the American Heart Association. Circulation. 2010;122(17):1756-1776.
13. Livschitz S, Sharabi Y, Yushin J, et al. Limited clinical value of exercise stress test for the screening of coronary artery disease in young, asymptomatic adult men. Am J Cardiol. 2000;86(4):462-464.
14. Miller T. Stress testing: the case for the standard treadmill test. Curr Opin Cardiol. 2011;26(5):363-369.
15. La Gerche A, Baggish A, Knuuti J, et al. Cardiac imaging and stress testing asymptomatic athletes to identify those at risk of sudden cardiac death. JACC Cardiovasc Imaging. 2013;6(9):993-1007.
16. Lauer M, Froelicher ES, Williams M, Kligfield P; American Heart Association Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Exercise testing in asymptomatic adults: a statement for professionals from the American Heart Association Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Circulation. 2005;112(5):771-776.
17. Sammito S, Gundlach N, Bockelmann I. Prevalence of cardiac arrhythmia under stress conditions in occupational health assessments of young military servicemen and servicewomen. Mil Med. 2016;181(4):369-372.
18. Mudrick DW, Cowper PA, Shah BR, et al. Downstream procedures and outcomes after stress testing for chest pain without known coronary artery disease in the United States. Am Heart J. 2012;163(3):454-461.
19. Christman MP, Bittencourt MS, Hulten E, et al. Yield of downstream tests after exercise treadmill testing. J Am Coll Cardiol. 2014;63(13):1264-1274.
20. Will J, Loustalot F, Hong Y. National trends in visits to physician offices and outpatient clinics for angina 1995 to 2010. Circ Cardiovasc Qual Outcomes. 2014;7(1):110-117.
21. Kini V, McCarthy F, Dayoub E, et al. Cardiac stress test trends among US patients younger than 65 years, 2005-2012. JAMA Cardiol. 2016;1(9):1038-1042.
22. Ladapo JA, Blecker S, Douglas PS. Physician decision making and trends in the use of cardiac stress testing in the United States: an analysis of repeated cross-sectional data. Ann Intern Med. 2014;161(7):482-490.
23. Winchester DE, Brandt J, Schmidt C, Schmidt C, Allen B, Payton T, Amsterdam EA. Diagnostic yield of routine noninvasive cardiovascular testing in low-risk acute chest pain patients. Am J Cardiol. 2015;116(2):204-207.
24. Hermann L, Weingart SD, Duvall W, Henzlova MJ. The limited utility of routine cardiac stress testing in emergency department chest pain patients younger than 40 years. Ann Emerg Med. 2009;54(1):12-16.
25. Ladapo JA, Blecker S, Elashoff MR, et al. Clinical implications of referral bias in the diagnostic performance of exercise testing for coronary artery disease. J Am Heart Assoc. 2013;2(6):e000505.
1. Fowler-Brown A, Pignone M, Pletcher M, et al. Exercise tolerance testing to screen for coronary heart disease: a systematic review for the technical support for the U.S. Preventive Services Task Force. Ann Intern Med. 2004;140(7):W9-W24.
2. Gibbons RJ, Balady GJ, Bricker JT, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Committee to Update the 1997 Exercise Testing Guidelines. ACC/AHA 2002 guideline update for exercise testing: summary article. A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines (Committee to Update the 1997 Exercise Testing Guidelines). J Am Coll Cardiol. 2002;40(8):1531-1540.
3. Chou R, Arora B, Dana T, Fu R, Miranda Walker M, Humphrey L. Screening Asymptomatic Adults for Coronary Heart Disease With Resting or Exercise Electrocardiography: Systematic Review to Update the 2004 U.S. Preventive Services Task Force recommendation. Evidence Synthesis No. 88. AHRQ Publication No. 11-05158-EF-1. Rockville, MD: Agency for Healthcare Research and Quality; September 2011.
4. Fihn S, Gardin J, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiology. 2012;60(24):e44-e164.
5. Gibbons RJ, Balady GJ, Beasley JW, et al. ACC/AHA guidelines for exercise testing: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Exercise Testing). Circulation. 1997;96(1):345-354.
6. Greenland P, Alpert JS, Beller GA, et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults. A report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2010;56(25):e50-e103.
7. Diamond G, Forrester J. Analysis of probability as an aid in the clinical diagnosis of coronary artery disease. N Engl J Med. 1979;300(24):1350-1358.
8. Goff D, Lloyd-Jones D, Bennett G, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25)(suppl 2):S49-S73.
9. Greenland P, Gaziano J. Selecting asymptomatic patients for coronary computed tomography or electrocardiographic exercise testing. N Engl J Med. 2003;349(5):465-473.
10. Shah N, Soon K, Wong C, Kellu AM. Screening for asymptomatic coronary heart disease in the young ‘at risk’ population: who and how? Int J Cardiol Heart Vasc. 2014;6:60-65.
11. Morise A, Evans M, Jalisi F, Shetty R, Stauffer M. A pretest prognostic score to assess patients undergoing exercise or pharmacological stress testing. Heart. 2007;93(2):200-204.
12. Amsterdam EA, Kirk JD, Bluemke DA, et al; American Heart Association Exercise, Cardiac Rehabilitation, and Prevention Committee of the Council on Clinical Cardiology, Council on Cardiovascular Nursing, and Interdisciplinary Council on Quality of Care and Outcomes Research. Testing of low-risk patients presenting to the emergency department with chest pain: a scientific statement from the American Heart Association. Circulation. 2010;122(17):1756-1776.
13. Livschitz S, Sharabi Y, Yushin J, et al. Limited clinical value of exercise stress test for the screening of coronary artery disease in young, asymptomatic adult men. Am J Cardiol. 2000;86(4):462-464.
14. Miller T. Stress testing: the case for the standard treadmill test. Curr Opin Cardiol. 2011;26(5):363-369.
15. La Gerche A, Baggish A, Knuuti J, et al. Cardiac imaging and stress testing asymptomatic athletes to identify those at risk of sudden cardiac death. JACC Cardiovasc Imaging. 2013;6(9):993-1007.
16. Lauer M, Froelicher ES, Williams M, Kligfield P; American Heart Association Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Exercise testing in asymptomatic adults: a statement for professionals from the American Heart Association Council on Clinical Cardiology, Subcommittee on Exercise, Cardiac Rehabilitation, and Prevention. Circulation. 2005;112(5):771-776.
17. Sammito S, Gundlach N, Bockelmann I. Prevalence of cardiac arrhythmia under stress conditions in occupational health assessments of young military servicemen and servicewomen. Mil Med. 2016;181(4):369-372.
18. Mudrick DW, Cowper PA, Shah BR, et al. Downstream procedures and outcomes after stress testing for chest pain without known coronary artery disease in the United States. Am Heart J. 2012;163(3):454-461.
19. Christman MP, Bittencourt MS, Hulten E, et al. Yield of downstream tests after exercise treadmill testing. J Am Coll Cardiol. 2014;63(13):1264-1274.
20. Will J, Loustalot F, Hong Y. National trends in visits to physician offices and outpatient clinics for angina 1995 to 2010. Circ Cardiovasc Qual Outcomes. 2014;7(1):110-117.
21. Kini V, McCarthy F, Dayoub E, et al. Cardiac stress test trends among US patients younger than 65 years, 2005-2012. JAMA Cardiol. 2016;1(9):1038-1042.
22. Ladapo JA, Blecker S, Douglas PS. Physician decision making and trends in the use of cardiac stress testing in the United States: an analysis of repeated cross-sectional data. Ann Intern Med. 2014;161(7):482-490.
23. Winchester DE, Brandt J, Schmidt C, Schmidt C, Allen B, Payton T, Amsterdam EA. Diagnostic yield of routine noninvasive cardiovascular testing in low-risk acute chest pain patients. Am J Cardiol. 2015;116(2):204-207.
24. Hermann L, Weingart SD, Duvall W, Henzlova MJ. The limited utility of routine cardiac stress testing in emergency department chest pain patients younger than 40 years. Ann Emerg Med. 2009;54(1):12-16.
25. Ladapo JA, Blecker S, Elashoff MR, et al. Clinical implications of referral bias in the diagnostic performance of exercise testing for coronary artery disease. J Am Heart Assoc. 2013;2(6):e000505.
Vaping related lung injury: Warning signs, care, & prevention
References
1. Lewis N, McCaffrey K, Sage K, et al. E-cigarette use, or vaping, practices and characteristics among persons with associated lung injury — Utah, April–October 2019. MMWR Morb Mortal Wkly Rep. 2019;68. https://www.cdc.gov/mmwr/volumes/68/wr/mm6842e1.htm?s_cid=mm6842e1_w. Published October 22, 2019. Accessed October 24, 2019.
2. Siegal DA, Jatlaoui TC, Koumans EH, et al. Update: interim guidance for health care providers evaluating and caring for patients with suspected e-cigarette, or vaping, product use associated lung injury – United States, October 2019. MMWR Morb Mortal Wkly Rep. 2019;68:919-927.
3. Centers for Disease Control and Prevention. Outbreak of lung injury associated with e-cigarette use, or vaping. https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html. Updated October 17, 2019. Accessed October 24, 2019.
References
1. Lewis N, McCaffrey K, Sage K, et al. E-cigarette use, or vaping, practices and characteristics among persons with associated lung injury — Utah, April–October 2019. MMWR Morb Mortal Wkly Rep. 2019;68. https://www.cdc.gov/mmwr/volumes/68/wr/mm6842e1.htm?s_cid=mm6842e1_w. Published October 22, 2019. Accessed October 24, 2019.
2. Siegal DA, Jatlaoui TC, Koumans EH, et al. Update: interim guidance for health care providers evaluating and caring for patients with suspected e-cigarette, or vaping, product use associated lung injury – United States, October 2019. MMWR Morb Mortal Wkly Rep. 2019;68:919-927.
3. Centers for Disease Control and Prevention. Outbreak of lung injury associated with e-cigarette use, or vaping. https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html. Updated October 17, 2019. Accessed October 24, 2019.
References
1. Lewis N, McCaffrey K, Sage K, et al. E-cigarette use, or vaping, practices and characteristics among persons with associated lung injury — Utah, April–October 2019. MMWR Morb Mortal Wkly Rep. 2019;68. https://www.cdc.gov/mmwr/volumes/68/wr/mm6842e1.htm?s_cid=mm6842e1_w. Published October 22, 2019. Accessed October 24, 2019.
2. Siegal DA, Jatlaoui TC, Koumans EH, et al. Update: interim guidance for health care providers evaluating and caring for patients with suspected e-cigarette, or vaping, product use associated lung injury – United States, October 2019. MMWR Morb Mortal Wkly Rep. 2019;68:919-927.
3. Centers for Disease Control and Prevention. Outbreak of lung injury associated with e-cigarette use, or vaping. https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html. Updated October 17, 2019. Accessed October 24, 2019.

Threshold for positivity affects FIT sensitivity for detecting CRC, advanced adenomas
Thresholds for positivity affected the sensitivity and (to a lesser extent) the specificity of quantitative fecal immunochemical tests used in the detection of colorectal cancer, which suggests that centers should consider lowering their thresholds for positivity if they have sufficient resources to handle an increase in follow-up colonoscopies, researchers wrote in Gastroenterology.
“Additional data are needed regarding the influence of sex and age on test performance,” wrote Kevin Selby, MD, of Kaiser Permanente Division of Research in Oakland, Calif., together with his associates. Additional studies also should evaluate the effect of a quantitative threshold of 10 mcg of hemoglobin per gram of feces and multiple rounds of annual testing, they added.
Fecal immunochemical tests (FITs) are recommended for colorectal cancer screening because they are diagnostically superior and are associated with higher participation rates, compared with guaiac fecal occult blood tests, the investigators noted. For screening, the optimal positivity threshold for quantitative FIT remains controversial, is likely to vary by sex and age, and also may be adjusted to reflect local health care resources. To more closely evaluate the correlates and effects of FIT cutoffs for sensitivity, the researchers searched MEDLINE, EMBASE, and the Database of Abstracts of Reviews of Effects for articles on the use of FIT for asymptomatic (screening) colorectal cancer detection in adults. This method identified 46 studies with 2.4 million participants and 6,478 detected cancers. The researchers then calculated sensitivity, specificity, numbers of detected cancers, advanced adenomas, and positive test results at positivity thresholds of up to 10 mcg, 10-20 mcg, 20-30 mcg, and more than 30 mcg of hemoglobin per gram of feces. They also examined subgroups stratified by sex and age.
The pooled sensitivity for the detection of colorectal cancer rose from 69% (95% confidence interval, 63%-75%) at a positivity threshold of more than 10 and up to 20 mcg of hemoglobin per gram of feces, to 80% at a positivity threshold of 10 mcg or less of hemoglobin per gram of feces. “At these [same] threshold values, sensitivity for detection of advanced adenomas increased from 21% (95% CI, 18%-25%) to 31% (95% CI, 27%-35%), whereas specificity decreased from 94% (95% CI, 93%-96%) to 91% (95% CI, 89%-93%),” the researchers wrote.
Only three studies stratified results by sex, and these found no statistical difference in pooled sensitivity for detecting colorectal cancer among men (77%) versus women (81%). Age, too, was stratified in only three studies and did not significantly correlate with sensitivity. “More research is needed to precisely establish FIT thresholds for each sex and age subgroup,” the researchers said.
The National Cancer Institute and the Swiss Cancer Research Foundation provided funding. The investigators reported having no conflicts of interest.
SOURCE: Selby K et al. Gastroenterology. 2019 Aug 22. doi: 10.1053/j.gastro.2019.08.023.
Quantitative fecal immunochemical tests or FITs are the most recent incarnation of screening for colorectal cancer (CRC) through the identification of occult blood in stool. Older versions of such tests were the first screening modalities shown to decrease both the incidence and mortality of CRC. FITs are much more sensitive for both CRC and advanced adenomas than are those early occult blood tests. They also are among the least costly and most easily employed CRC screening modalities. Given the quantitative nature of FITs, the question has remained as to what positivity threshold should be employed to achieve the optimal balance of sensitivity and specificity.

Future studies should examine more carefully demographic effects on FIT performance to determine if different positivity thresholds need to be employed in different demographic groups.
Reid M. Ness, MD, MPH, is an associate professor in the division of gastroenterology, hepatology and nutrition, department of medicine, Vanderbilt University Medical Center and at the Veterans Affairs Tennessee Valley Healthcare System, Nashville campus. He is also an investigator in the Vanderbilt-Ingram Cancer Center. Dr. Ness has no financial relationships to disclose.
Quantitative fecal immunochemical tests or FITs are the most recent incarnation of screening for colorectal cancer (CRC) through the identification of occult blood in stool. Older versions of such tests were the first screening modalities shown to decrease both the incidence and mortality of CRC. FITs are much more sensitive for both CRC and advanced adenomas than are those early occult blood tests. They also are among the least costly and most easily employed CRC screening modalities. Given the quantitative nature of FITs, the question has remained as to what positivity threshold should be employed to achieve the optimal balance of sensitivity and specificity.
Future studies should examine more carefully demographic effects on FIT performance to determine if different positivity thresholds need to be employed in different demographic groups.
Reid M. Ness, MD, MPH, is an associate professor in the division of gastroenterology, hepatology and nutrition, department of medicine, Vanderbilt University Medical Center and at the Veterans Affairs Tennessee Valley Healthcare System, Nashville campus. He is also an investigator in the Vanderbilt-Ingram Cancer Center. Dr. Ness has no financial relationships to disclose.
Quantitative fecal immunochemical tests or FITs are the most recent incarnation of screening for colorectal cancer (CRC) through the identification of occult blood in stool. Older versions of such tests were the first screening modalities shown to decrease both the incidence and mortality of CRC. FITs are much more sensitive for both CRC and advanced adenomas than are those early occult blood tests. They also are among the least costly and most easily employed CRC screening modalities. Given the quantitative nature of FITs, the question has remained as to what positivity threshold should be employed to achieve the optimal balance of sensitivity and specificity.
Future studies should examine more carefully demographic effects on FIT performance to determine if different positivity thresholds need to be employed in different demographic groups.
Reid M. Ness, MD, MPH, is an associate professor in the division of gastroenterology, hepatology and nutrition, department of medicine, Vanderbilt University Medical Center and at the Veterans Affairs Tennessee Valley Healthcare System, Nashville campus. He is also an investigator in the Vanderbilt-Ingram Cancer Center. Dr. Ness has no financial relationships to disclose.
Thresholds for positivity affected the sensitivity and (to a lesser extent) the specificity of quantitative fecal immunochemical tests used in the detection of colorectal cancer, which suggests that centers should consider lowering their thresholds for positivity if they have sufficient resources to handle an increase in follow-up colonoscopies, researchers wrote in Gastroenterology.
“Additional data are needed regarding the influence of sex and age on test performance,” wrote Kevin Selby, MD, of Kaiser Permanente Division of Research in Oakland, Calif., together with his associates. Additional studies also should evaluate the effect of a quantitative threshold of 10 mcg of hemoglobin per gram of feces and multiple rounds of annual testing, they added.
Fecal immunochemical tests (FITs) are recommended for colorectal cancer screening because they are diagnostically superior and are associated with higher participation rates, compared with guaiac fecal occult blood tests, the investigators noted. For screening, the optimal positivity threshold for quantitative FIT remains controversial, is likely to vary by sex and age, and also may be adjusted to reflect local health care resources. To more closely evaluate the correlates and effects of FIT cutoffs for sensitivity, the researchers searched MEDLINE, EMBASE, and the Database of Abstracts of Reviews of Effects for articles on the use of FIT for asymptomatic (screening) colorectal cancer detection in adults. This method identified 46 studies with 2.4 million participants and 6,478 detected cancers. The researchers then calculated sensitivity, specificity, numbers of detected cancers, advanced adenomas, and positive test results at positivity thresholds of up to 10 mcg, 10-20 mcg, 20-30 mcg, and more than 30 mcg of hemoglobin per gram of feces. They also examined subgroups stratified by sex and age.
The pooled sensitivity for the detection of colorectal cancer rose from 69% (95% confidence interval, 63%-75%) at a positivity threshold of more than 10 and up to 20 mcg of hemoglobin per gram of feces, to 80% at a positivity threshold of 10 mcg or less of hemoglobin per gram of feces. “At these [same] threshold values, sensitivity for detection of advanced adenomas increased from 21% (95% CI, 18%-25%) to 31% (95% CI, 27%-35%), whereas specificity decreased from 94% (95% CI, 93%-96%) to 91% (95% CI, 89%-93%),” the researchers wrote.
Only three studies stratified results by sex, and these found no statistical difference in pooled sensitivity for detecting colorectal cancer among men (77%) versus women (81%). Age, too, was stratified in only three studies and did not significantly correlate with sensitivity. “More research is needed to precisely establish FIT thresholds for each sex and age subgroup,” the researchers said.
The National Cancer Institute and the Swiss Cancer Research Foundation provided funding. The investigators reported having no conflicts of interest.
SOURCE: Selby K et al. Gastroenterology. 2019 Aug 22. doi: 10.1053/j.gastro.2019.08.023.
Thresholds for positivity affected the sensitivity and (to a lesser extent) the specificity of quantitative fecal immunochemical tests used in the detection of colorectal cancer, which suggests that centers should consider lowering their thresholds for positivity if they have sufficient resources to handle an increase in follow-up colonoscopies, researchers wrote in Gastroenterology.
“Additional data are needed regarding the influence of sex and age on test performance,” wrote Kevin Selby, MD, of Kaiser Permanente Division of Research in Oakland, Calif., together with his associates. Additional studies also should evaluate the effect of a quantitative threshold of 10 mcg of hemoglobin per gram of feces and multiple rounds of annual testing, they added.
Fecal immunochemical tests (FITs) are recommended for colorectal cancer screening because they are diagnostically superior and are associated with higher participation rates, compared with guaiac fecal occult blood tests, the investigators noted. For screening, the optimal positivity threshold for quantitative FIT remains controversial, is likely to vary by sex and age, and also may be adjusted to reflect local health care resources. To more closely evaluate the correlates and effects of FIT cutoffs for sensitivity, the researchers searched MEDLINE, EMBASE, and the Database of Abstracts of Reviews of Effects for articles on the use of FIT for asymptomatic (screening) colorectal cancer detection in adults. This method identified 46 studies with 2.4 million participants and 6,478 detected cancers. The researchers then calculated sensitivity, specificity, numbers of detected cancers, advanced adenomas, and positive test results at positivity thresholds of up to 10 mcg, 10-20 mcg, 20-30 mcg, and more than 30 mcg of hemoglobin per gram of feces. They also examined subgroups stratified by sex and age.
The pooled sensitivity for the detection of colorectal cancer rose from 69% (95% confidence interval, 63%-75%) at a positivity threshold of more than 10 and up to 20 mcg of hemoglobin per gram of feces, to 80% at a positivity threshold of 10 mcg or less of hemoglobin per gram of feces. “At these [same] threshold values, sensitivity for detection of advanced adenomas increased from 21% (95% CI, 18%-25%) to 31% (95% CI, 27%-35%), whereas specificity decreased from 94% (95% CI, 93%-96%) to 91% (95% CI, 89%-93%),” the researchers wrote.
Only three studies stratified results by sex, and these found no statistical difference in pooled sensitivity for detecting colorectal cancer among men (77%) versus women (81%). Age, too, was stratified in only three studies and did not significantly correlate with sensitivity. “More research is needed to precisely establish FIT thresholds for each sex and age subgroup,” the researchers said.
The National Cancer Institute and the Swiss Cancer Research Foundation provided funding. The investigators reported having no conflicts of interest.
SOURCE: Selby K et al. Gastroenterology. 2019 Aug 22. doi: 10.1053/j.gastro.2019.08.023.
FROM GASTROENTEROLOGY
FDA approves Ziextenzo for neutropenia-related infection reduction
The Food and Drug Administration has approved the biosimilar Ziextenzo (pegfilgrastim-bmez) to reduce the incidence of infection in patients with nonmyeloid cancer receiving suppressive anticancer drugs that are associated with febrile neutropenia.

More than 60,000 cancer patients are hospitalized in the United States each year with evidence of neutropenia, resulting in more than 4,000 deaths, according to Ziextenzo maker Sandoz, a Novartis division.
The FDA approval was based on analytical, preclinical, and clinical research, including data from a three-way pharmacokinetics and pharmacodynamics study that compared pegfilgrastim-bmez with the reference drug pegfilgrastim (Neulasta) from the United States and the European Union. Pharmacokinetic and pharmacodynamic similarity were shown between pegfilgrastim-bmez with the reference drugs, and there were no clinically significant differences in safety or immunogenicity.
The most common adverse events associated with pegfilgrastim-bmez are bone pain and pain in the extremities, according to the label.
The Food and Drug Administration has approved the biosimilar Ziextenzo (pegfilgrastim-bmez) to reduce the incidence of infection in patients with nonmyeloid cancer receiving suppressive anticancer drugs that are associated with febrile neutropenia.
More than 60,000 cancer patients are hospitalized in the United States each year with evidence of neutropenia, resulting in more than 4,000 deaths, according to Ziextenzo maker Sandoz, a Novartis division.
The FDA approval was based on analytical, preclinical, and clinical research, including data from a three-way pharmacokinetics and pharmacodynamics study that compared pegfilgrastim-bmez with the reference drug pegfilgrastim (Neulasta) from the United States and the European Union. Pharmacokinetic and pharmacodynamic similarity were shown between pegfilgrastim-bmez with the reference drugs, and there were no clinically significant differences in safety or immunogenicity.
The most common adverse events associated with pegfilgrastim-bmez are bone pain and pain in the extremities, according to the label.
The Food and Drug Administration has approved the biosimilar Ziextenzo (pegfilgrastim-bmez) to reduce the incidence of infection in patients with nonmyeloid cancer receiving suppressive anticancer drugs that are associated with febrile neutropenia.
More than 60,000 cancer patients are hospitalized in the United States each year with evidence of neutropenia, resulting in more than 4,000 deaths, according to Ziextenzo maker Sandoz, a Novartis division.
The FDA approval was based on analytical, preclinical, and clinical research, including data from a three-way pharmacokinetics and pharmacodynamics study that compared pegfilgrastim-bmez with the reference drug pegfilgrastim (Neulasta) from the United States and the European Union. Pharmacokinetic and pharmacodynamic similarity were shown between pegfilgrastim-bmez with the reference drugs, and there were no clinically significant differences in safety or immunogenicity.
The most common adverse events associated with pegfilgrastim-bmez are bone pain and pain in the extremities, according to the label.