User login
Skin imaging working group releases first guidelines for AI algorithms used in dermatology
The

The guidelines, published in JAMA Dermatology on Dec. 1, 2021, contain a broad range of recommendations stakeholders should consider when developing and assessing image-based AI algorithms in dermatology. The recommendations are divided into categories of data, technique, technical assessment, and application. ISIC is “an academia and industry partnership designed to facilitate the application of digital skin imaging to help reduce melanoma mortality,” and is organized into different working groups, including the AI working group, according to its website.
“Our goal with these guidelines was to create higher-quality reporting of dataset and algorithm characteristics for dermatology AI,” first author Roxana Daneshjou, MD, PhD, clinical scholar in dermatology, in the department of dermatology at Stanford (Calif.) University, said in an interview. “We hope these guidelines also aid regulatory bodies around the world when they are assessing algorithms to be used in dermatology.”
Recommendations for data
The authors recommended that datasets used by AI algorithms have image descriptions and details on image artifacts. “For photography, these include the type of camera used; whether images were taken under standardized or varying conditions; whether they were taken by professional photographers, laymen, or health care professionals; and image quality,” they wrote. They also recommended that developers include in an image description the type of lighting used and whether the photo contains pen markings, hair, tattoos, injuries, surgical effects, or other “physical perturbations.”
Exchangeable image file format data obtained from the camera, and preprocessing procedures like color normalization and “postprocessing” of images, such as filtering, should also be disclosed. In addition, developers should disclose and justify inclusion of images that have been created by an algorithm within a dataset. Any public images used in the datasets should have references, and privately used images should be made public where possible, the authors said.
The ISIC working group guidelines also provided recommendations for patient-level metadata. Each image should include a patient’s geographical location and medical center they visited as well as their age, sex and gender, ethnicity and/or race, and skin tone. Dr. Daneshjou said this was one area where she and her colleagues found a lack of transparency in AI datasets in algorithms in a recent review. “We found that many AI papers provided sparse details about the images used to train and test their algorithms,” Dr. Daneshjou explained. “For example, only 7 out of 70 papers had any information about the skin tones in the images used for developing and/or testing AI algorithms. Understanding the diversity of images used to train and test algorithms is important because algorithms that are developed on images of predominantly white skin likely won’t work as well on Black and brown skin.”
The guideline authors also asked algorithm developers to describe the limitations of not including patient-level metadata information when it is incomplete or unavailable. In addition, “we ask that algorithm developers comment on potential biases of their algorithms,” Dr. Daneshjou said. “For example, an algorithm based only on telemedicine images may not capture the full range of diseases seen within an in-person clinic.”
When describing their AI algorithm, developers should detail their reasoning for the dataset size and partitions, inclusion and exclusion criteria for images, and use of any external samples for test sets. “Authors should consider any differences between the image characteristics used for algorithm development and those that might be encountered in the real world,” the guidelines stated.
Recommendations for technique
How the images in a dataset are labeled is a unique challenge in developing AI algorithms for dermatology, the authors noted. Developers should use histopathological diagnosis in their labeling, but this can sometimes result in label noise.
“Many of the AI algorithms in dermatology use supervised learning, which requires labeled examples to help the algorithm ‘learn’ features for discriminating between lesions. We found that some papers use consensus labeling – dermatologists providing a label – to label skin cancers; however, the standard for diagnosing skin cancer is using histopathology from a biopsy,” she said. “Dermatologists can biopsy seven to eight suspected melanomas before discovering a true melanoma, so dermatologist labeling of skin cancers is prone to label noise.”
ISIC’s guidelines stated a gold standard of labeling for dermatologic images is one area that still needs future research, but currently, “diagnoses, labels and diagnostic groups used in data repositories as well as public ontologies” such as ICD-11, AnatomyMapper, and SNOMED-CT should be included in dermatologic image datasets.
AI developers should also provide a detailed description of their algorithm, which includes methods, work flows, mathematical formulas as well as the generalizability of the algorithm across more than one dataset.
Recommendations for technical assessment
“Another important recommendation is that algorithm developers should provide a way for algorithms to be publicly evaluable by researchers,” Dr. Daneshjou said. “Many dermatology AI algorithms do not share either their data or their algorithm. Algorithm sharing is important for assessing reproducibility and robustness.”
Google’s recently announced AI-powered dermatology assistant tool, for example, “has made claims about its accuracy and ability to diagnose skin disease at a dermatologist level, but there is no way for researchers to independently test these claims,” she said. Other options like Model Dermatology, developed by Seung Seog Han, MD, PhD, of the Dermatology Clinic in Seoul, South Korea, and colleagues, offer an application programming interface “that allows researchers to test the algorithm,” Dr. Daneshjou said. “This kind of openness is key for assessing algorithm robustness.”
Developers should also note in their algorithm explanations how performance markers and benchmarks would translate to proposed clinical application. “In this context,” the use case – the context in which the AI application is being used – “should be clearly described – who are the intended users and under what clinical scenario are they using the algorithm,” the authors wrote.
Recommendations for application
The guidelines note that use case for the model should also be described by the AI developers. “Our checklist includes delineating use cases for algorithms and describing what use cases may be within the scope of the algorithm versus which use cases are out of scope,” Dr. Daneshjou said. “For example, an algorithm developed to provide decision support to dermatologists, with a human in the loop, may not be accurate enough to release directly to consumers.”
As the goal of AI algorithms in dermatology is eventual implementation for clinicians and patients, the authors asked developers to consider shortcomings and potential harms of the algorithm during implementation. “Ethical considerations and impact on vulnerable populations should also be considered and discussed,” they wrote. An algorithm “suggesting aesthetic medical treatments may have negative effects given the biased nature of beauty standards,” and “an algorithm that diagnoses basal cell carcinomas but lacks any pigmented basal cell carcinomas, which are more often seen in skin of color, will not perform equitably across populations.”
Prior to implementing an AI algorithm, the ISIC working group recommended developers perform prospective clinical trials for validation. Checklists and guidelines like SPIRIT-AI and CONSORT-AI “provide guidance on how to design clinical trials to test AI algorithms,” Dr. Daneshjou said.
After implementation, “I believe we need additional research in how we monitor algorithms after they are deployed clinically, Dr. Daneshjou said. “Currently there are no [Food and Drug Administration]–approved AI algorithms in dermatology; however, there are several applications that have CE mark in Europe, and there are no mechanisms for postmarket surveillance there.
'Timely' recommendations
Commenting on the ISIC working group guidelines, Justin M. Ko, MD, MBA, director and chief of medical dermatology for Stanford Health Care, who was not involved with the work, said that the recommendations are timely and provide “a framework for a ‘common language’ around AI datasets specifically tailored to dermatology.” Dr. Ko, chair of the American Academy of Dermatology’s Ad Hoc Task Force on Augmented Intelligence, noted the work by Dr. Daneshjou and colleagues “is consistent with and builds further details” on the position statement released by the AAD AI task force in 2019.

“As machine-learning capabilities and commercial efforts continue to mature, it becomes increasingly important that we are able to ‘look under the hood,’ and evaluate all the critical factors that influence development of these capabilities,” he said in an interview. “A standard set of reporting guidelines not only allows for transparency in evaluating data and performance of models and algorithms, but also forces the consideration of issues of equity, fairness, mitigation of bias, and clinically meaningful outcomes.”
One concern is the impact of AI algorithms on societal or health systems, he noted, which is brought up in the guidelines. “The last thing we would want is the development of robust AI systems that exacerbate access challenges, or generate patient anxiety/worry, or drive low-value utilization, or adds to care team burden, or create a technological barrier to care, or increases inequity in dermatologic care,” he said.
In developing AI algorithms for dermatology, a “major practical issue” is how performance on paper will translate to real-world use, Dr. Ko explained, and the ISIC guidelines “provide a critical step in empowering clinicians, practices, and our field to shape the advent of the AI and augmented intelligence tools and systems to promote and enhance meaningful clinical outcomes, and augment the core patient-clinician relationship and ensure they are grounded in principles of fairness, equity and transparency.”
This research was funded by awards and grants to individual authors from the Charina Fund, a Google Research Award, Melanoma Research Alliance, National Health and Medical Research Council, National Institutes of Health/National Cancer Institute, National Science Foundation, and the Department of Veterans Affairs. The authors disclosed relationships with governmental entities, pharmaceutical companies, technology startups, medical publishers, charitable trusts, consulting firms, dermatology training companies, providers of medical devices, manufacturers of dermatologic products, and other organizations related to the paper in the form of supplied equipment, having founded a company; receiving grants, patents, or personal fees; holding shares; and medical reporting. Dr. Ko reported that he serves as a clinical advisor for Skin Analytics, and has an ongoing research collaboration with Google.
The
The guidelines, published in JAMA Dermatology on Dec. 1, 2021, contain a broad range of recommendations stakeholders should consider when developing and assessing image-based AI algorithms in dermatology. The recommendations are divided into categories of data, technique, technical assessment, and application. ISIC is “an academia and industry partnership designed to facilitate the application of digital skin imaging to help reduce melanoma mortality,” and is organized into different working groups, including the AI working group, according to its website.
“Our goal with these guidelines was to create higher-quality reporting of dataset and algorithm characteristics for dermatology AI,” first author Roxana Daneshjou, MD, PhD, clinical scholar in dermatology, in the department of dermatology at Stanford (Calif.) University, said in an interview. “We hope these guidelines also aid regulatory bodies around the world when they are assessing algorithms to be used in dermatology.”
Recommendations for data
The authors recommended that datasets used by AI algorithms have image descriptions and details on image artifacts. “For photography, these include the type of camera used; whether images were taken under standardized or varying conditions; whether they were taken by professional photographers, laymen, or health care professionals; and image quality,” they wrote. They also recommended that developers include in an image description the type of lighting used and whether the photo contains pen markings, hair, tattoos, injuries, surgical effects, or other “physical perturbations.”
Exchangeable image file format data obtained from the camera, and preprocessing procedures like color normalization and “postprocessing” of images, such as filtering, should also be disclosed. In addition, developers should disclose and justify inclusion of images that have been created by an algorithm within a dataset. Any public images used in the datasets should have references, and privately used images should be made public where possible, the authors said.
The ISIC working group guidelines also provided recommendations for patient-level metadata. Each image should include a patient’s geographical location and medical center they visited as well as their age, sex and gender, ethnicity and/or race, and skin tone. Dr. Daneshjou said this was one area where she and her colleagues found a lack of transparency in AI datasets in algorithms in a recent review. “We found that many AI papers provided sparse details about the images used to train and test their algorithms,” Dr. Daneshjou explained. “For example, only 7 out of 70 papers had any information about the skin tones in the images used for developing and/or testing AI algorithms. Understanding the diversity of images used to train and test algorithms is important because algorithms that are developed on images of predominantly white skin likely won’t work as well on Black and brown skin.”
The guideline authors also asked algorithm developers to describe the limitations of not including patient-level metadata information when it is incomplete or unavailable. In addition, “we ask that algorithm developers comment on potential biases of their algorithms,” Dr. Daneshjou said. “For example, an algorithm based only on telemedicine images may not capture the full range of diseases seen within an in-person clinic.”
When describing their AI algorithm, developers should detail their reasoning for the dataset size and partitions, inclusion and exclusion criteria for images, and use of any external samples for test sets. “Authors should consider any differences between the image characteristics used for algorithm development and those that might be encountered in the real world,” the guidelines stated.
Recommendations for technique
How the images in a dataset are labeled is a unique challenge in developing AI algorithms for dermatology, the authors noted. Developers should use histopathological diagnosis in their labeling, but this can sometimes result in label noise.
“Many of the AI algorithms in dermatology use supervised learning, which requires labeled examples to help the algorithm ‘learn’ features for discriminating between lesions. We found that some papers use consensus labeling – dermatologists providing a label – to label skin cancers; however, the standard for diagnosing skin cancer is using histopathology from a biopsy,” she said. “Dermatologists can biopsy seven to eight suspected melanomas before discovering a true melanoma, so dermatologist labeling of skin cancers is prone to label noise.”
ISIC’s guidelines stated a gold standard of labeling for dermatologic images is one area that still needs future research, but currently, “diagnoses, labels and diagnostic groups used in data repositories as well as public ontologies” such as ICD-11, AnatomyMapper, and SNOMED-CT should be included in dermatologic image datasets.
AI developers should also provide a detailed description of their algorithm, which includes methods, work flows, mathematical formulas as well as the generalizability of the algorithm across more than one dataset.
Recommendations for technical assessment
“Another important recommendation is that algorithm developers should provide a way for algorithms to be publicly evaluable by researchers,” Dr. Daneshjou said. “Many dermatology AI algorithms do not share either their data or their algorithm. Algorithm sharing is important for assessing reproducibility and robustness.”
Google’s recently announced AI-powered dermatology assistant tool, for example, “has made claims about its accuracy and ability to diagnose skin disease at a dermatologist level, but there is no way for researchers to independently test these claims,” she said. Other options like Model Dermatology, developed by Seung Seog Han, MD, PhD, of the Dermatology Clinic in Seoul, South Korea, and colleagues, offer an application programming interface “that allows researchers to test the algorithm,” Dr. Daneshjou said. “This kind of openness is key for assessing algorithm robustness.”
Developers should also note in their algorithm explanations how performance markers and benchmarks would translate to proposed clinical application. “In this context,” the use case – the context in which the AI application is being used – “should be clearly described – who are the intended users and under what clinical scenario are they using the algorithm,” the authors wrote.
Recommendations for application
The guidelines note that use case for the model should also be described by the AI developers. “Our checklist includes delineating use cases for algorithms and describing what use cases may be within the scope of the algorithm versus which use cases are out of scope,” Dr. Daneshjou said. “For example, an algorithm developed to provide decision support to dermatologists, with a human in the loop, may not be accurate enough to release directly to consumers.”
As the goal of AI algorithms in dermatology is eventual implementation for clinicians and patients, the authors asked developers to consider shortcomings and potential harms of the algorithm during implementation. “Ethical considerations and impact on vulnerable populations should also be considered and discussed,” they wrote. An algorithm “suggesting aesthetic medical treatments may have negative effects given the biased nature of beauty standards,” and “an algorithm that diagnoses basal cell carcinomas but lacks any pigmented basal cell carcinomas, which are more often seen in skin of color, will not perform equitably across populations.”
Prior to implementing an AI algorithm, the ISIC working group recommended developers perform prospective clinical trials for validation. Checklists and guidelines like SPIRIT-AI and CONSORT-AI “provide guidance on how to design clinical trials to test AI algorithms,” Dr. Daneshjou said.
After implementation, “I believe we need additional research in how we monitor algorithms after they are deployed clinically, Dr. Daneshjou said. “Currently there are no [Food and Drug Administration]–approved AI algorithms in dermatology; however, there are several applications that have CE mark in Europe, and there are no mechanisms for postmarket surveillance there.
'Timely' recommendations
Commenting on the ISIC working group guidelines, Justin M. Ko, MD, MBA, director and chief of medical dermatology for Stanford Health Care, who was not involved with the work, said that the recommendations are timely and provide “a framework for a ‘common language’ around AI datasets specifically tailored to dermatology.” Dr. Ko, chair of the American Academy of Dermatology’s Ad Hoc Task Force on Augmented Intelligence, noted the work by Dr. Daneshjou and colleagues “is consistent with and builds further details” on the position statement released by the AAD AI task force in 2019.
“As machine-learning capabilities and commercial efforts continue to mature, it becomes increasingly important that we are able to ‘look under the hood,’ and evaluate all the critical factors that influence development of these capabilities,” he said in an interview. “A standard set of reporting guidelines not only allows for transparency in evaluating data and performance of models and algorithms, but also forces the consideration of issues of equity, fairness, mitigation of bias, and clinically meaningful outcomes.”
One concern is the impact of AI algorithms on societal or health systems, he noted, which is brought up in the guidelines. “The last thing we would want is the development of robust AI systems that exacerbate access challenges, or generate patient anxiety/worry, or drive low-value utilization, or adds to care team burden, or create a technological barrier to care, or increases inequity in dermatologic care,” he said.
In developing AI algorithms for dermatology, a “major practical issue” is how performance on paper will translate to real-world use, Dr. Ko explained, and the ISIC guidelines “provide a critical step in empowering clinicians, practices, and our field to shape the advent of the AI and augmented intelligence tools and systems to promote and enhance meaningful clinical outcomes, and augment the core patient-clinician relationship and ensure they are grounded in principles of fairness, equity and transparency.”
This research was funded by awards and grants to individual authors from the Charina Fund, a Google Research Award, Melanoma Research Alliance, National Health and Medical Research Council, National Institutes of Health/National Cancer Institute, National Science Foundation, and the Department of Veterans Affairs. The authors disclosed relationships with governmental entities, pharmaceutical companies, technology startups, medical publishers, charitable trusts, consulting firms, dermatology training companies, providers of medical devices, manufacturers of dermatologic products, and other organizations related to the paper in the form of supplied equipment, having founded a company; receiving grants, patents, or personal fees; holding shares; and medical reporting. Dr. Ko reported that he serves as a clinical advisor for Skin Analytics, and has an ongoing research collaboration with Google.
The
The guidelines, published in JAMA Dermatology on Dec. 1, 2021, contain a broad range of recommendations stakeholders should consider when developing and assessing image-based AI algorithms in dermatology. The recommendations are divided into categories of data, technique, technical assessment, and application. ISIC is “an academia and industry partnership designed to facilitate the application of digital skin imaging to help reduce melanoma mortality,” and is organized into different working groups, including the AI working group, according to its website.
“Our goal with these guidelines was to create higher-quality reporting of dataset and algorithm characteristics for dermatology AI,” first author Roxana Daneshjou, MD, PhD, clinical scholar in dermatology, in the department of dermatology at Stanford (Calif.) University, said in an interview. “We hope these guidelines also aid regulatory bodies around the world when they are assessing algorithms to be used in dermatology.”
Recommendations for data
The authors recommended that datasets used by AI algorithms have image descriptions and details on image artifacts. “For photography, these include the type of camera used; whether images were taken under standardized or varying conditions; whether they were taken by professional photographers, laymen, or health care professionals; and image quality,” they wrote. They also recommended that developers include in an image description the type of lighting used and whether the photo contains pen markings, hair, tattoos, injuries, surgical effects, or other “physical perturbations.”
Exchangeable image file format data obtained from the camera, and preprocessing procedures like color normalization and “postprocessing” of images, such as filtering, should also be disclosed. In addition, developers should disclose and justify inclusion of images that have been created by an algorithm within a dataset. Any public images used in the datasets should have references, and privately used images should be made public where possible, the authors said.
The ISIC working group guidelines also provided recommendations for patient-level metadata. Each image should include a patient’s geographical location and medical center they visited as well as their age, sex and gender, ethnicity and/or race, and skin tone. Dr. Daneshjou said this was one area where she and her colleagues found a lack of transparency in AI datasets in algorithms in a recent review. “We found that many AI papers provided sparse details about the images used to train and test their algorithms,” Dr. Daneshjou explained. “For example, only 7 out of 70 papers had any information about the skin tones in the images used for developing and/or testing AI algorithms. Understanding the diversity of images used to train and test algorithms is important because algorithms that are developed on images of predominantly white skin likely won’t work as well on Black and brown skin.”
The guideline authors also asked algorithm developers to describe the limitations of not including patient-level metadata information when it is incomplete or unavailable. In addition, “we ask that algorithm developers comment on potential biases of their algorithms,” Dr. Daneshjou said. “For example, an algorithm based only on telemedicine images may not capture the full range of diseases seen within an in-person clinic.”
When describing their AI algorithm, developers should detail their reasoning for the dataset size and partitions, inclusion and exclusion criteria for images, and use of any external samples for test sets. “Authors should consider any differences between the image characteristics used for algorithm development and those that might be encountered in the real world,” the guidelines stated.
Recommendations for technique
How the images in a dataset are labeled is a unique challenge in developing AI algorithms for dermatology, the authors noted. Developers should use histopathological diagnosis in their labeling, but this can sometimes result in label noise.
“Many of the AI algorithms in dermatology use supervised learning, which requires labeled examples to help the algorithm ‘learn’ features for discriminating between lesions. We found that some papers use consensus labeling – dermatologists providing a label – to label skin cancers; however, the standard for diagnosing skin cancer is using histopathology from a biopsy,” she said. “Dermatologists can biopsy seven to eight suspected melanomas before discovering a true melanoma, so dermatologist labeling of skin cancers is prone to label noise.”
ISIC’s guidelines stated a gold standard of labeling for dermatologic images is one area that still needs future research, but currently, “diagnoses, labels and diagnostic groups used in data repositories as well as public ontologies” such as ICD-11, AnatomyMapper, and SNOMED-CT should be included in dermatologic image datasets.
AI developers should also provide a detailed description of their algorithm, which includes methods, work flows, mathematical formulas as well as the generalizability of the algorithm across more than one dataset.
Recommendations for technical assessment
“Another important recommendation is that algorithm developers should provide a way for algorithms to be publicly evaluable by researchers,” Dr. Daneshjou said. “Many dermatology AI algorithms do not share either their data or their algorithm. Algorithm sharing is important for assessing reproducibility and robustness.”
Google’s recently announced AI-powered dermatology assistant tool, for example, “has made claims about its accuracy and ability to diagnose skin disease at a dermatologist level, but there is no way for researchers to independently test these claims,” she said. Other options like Model Dermatology, developed by Seung Seog Han, MD, PhD, of the Dermatology Clinic in Seoul, South Korea, and colleagues, offer an application programming interface “that allows researchers to test the algorithm,” Dr. Daneshjou said. “This kind of openness is key for assessing algorithm robustness.”
Developers should also note in their algorithm explanations how performance markers and benchmarks would translate to proposed clinical application. “In this context,” the use case – the context in which the AI application is being used – “should be clearly described – who are the intended users and under what clinical scenario are they using the algorithm,” the authors wrote.
Recommendations for application
The guidelines note that use case for the model should also be described by the AI developers. “Our checklist includes delineating use cases for algorithms and describing what use cases may be within the scope of the algorithm versus which use cases are out of scope,” Dr. Daneshjou said. “For example, an algorithm developed to provide decision support to dermatologists, with a human in the loop, may not be accurate enough to release directly to consumers.”
As the goal of AI algorithms in dermatology is eventual implementation for clinicians and patients, the authors asked developers to consider shortcomings and potential harms of the algorithm during implementation. “Ethical considerations and impact on vulnerable populations should also be considered and discussed,” they wrote. An algorithm “suggesting aesthetic medical treatments may have negative effects given the biased nature of beauty standards,” and “an algorithm that diagnoses basal cell carcinomas but lacks any pigmented basal cell carcinomas, which are more often seen in skin of color, will not perform equitably across populations.”
Prior to implementing an AI algorithm, the ISIC working group recommended developers perform prospective clinical trials for validation. Checklists and guidelines like SPIRIT-AI and CONSORT-AI “provide guidance on how to design clinical trials to test AI algorithms,” Dr. Daneshjou said.
After implementation, “I believe we need additional research in how we monitor algorithms after they are deployed clinically, Dr. Daneshjou said. “Currently there are no [Food and Drug Administration]–approved AI algorithms in dermatology; however, there are several applications that have CE mark in Europe, and there are no mechanisms for postmarket surveillance there.
'Timely' recommendations
Commenting on the ISIC working group guidelines, Justin M. Ko, MD, MBA, director and chief of medical dermatology for Stanford Health Care, who was not involved with the work, said that the recommendations are timely and provide “a framework for a ‘common language’ around AI datasets specifically tailored to dermatology.” Dr. Ko, chair of the American Academy of Dermatology’s Ad Hoc Task Force on Augmented Intelligence, noted the work by Dr. Daneshjou and colleagues “is consistent with and builds further details” on the position statement released by the AAD AI task force in 2019.
“As machine-learning capabilities and commercial efforts continue to mature, it becomes increasingly important that we are able to ‘look under the hood,’ and evaluate all the critical factors that influence development of these capabilities,” he said in an interview. “A standard set of reporting guidelines not only allows for transparency in evaluating data and performance of models and algorithms, but also forces the consideration of issues of equity, fairness, mitigation of bias, and clinically meaningful outcomes.”
One concern is the impact of AI algorithms on societal or health systems, he noted, which is brought up in the guidelines. “The last thing we would want is the development of robust AI systems that exacerbate access challenges, or generate patient anxiety/worry, or drive low-value utilization, or adds to care team burden, or create a technological barrier to care, or increases inequity in dermatologic care,” he said.
In developing AI algorithms for dermatology, a “major practical issue” is how performance on paper will translate to real-world use, Dr. Ko explained, and the ISIC guidelines “provide a critical step in empowering clinicians, practices, and our field to shape the advent of the AI and augmented intelligence tools and systems to promote and enhance meaningful clinical outcomes, and augment the core patient-clinician relationship and ensure they are grounded in principles of fairness, equity and transparency.”
This research was funded by awards and grants to individual authors from the Charina Fund, a Google Research Award, Melanoma Research Alliance, National Health and Medical Research Council, National Institutes of Health/National Cancer Institute, National Science Foundation, and the Department of Veterans Affairs. The authors disclosed relationships with governmental entities, pharmaceutical companies, technology startups, medical publishers, charitable trusts, consulting firms, dermatology training companies, providers of medical devices, manufacturers of dermatologic products, and other organizations related to the paper in the form of supplied equipment, having founded a company; receiving grants, patents, or personal fees; holding shares; and medical reporting. Dr. Ko reported that he serves as a clinical advisor for Skin Analytics, and has an ongoing research collaboration with Google.
FROM JAMA DERMATOLOGY
Bamlanivimab’s effects in COVID-19 depend on antibodies
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
In the randomized controlled trial, in both the group who received bamlanivimab and the group who received placebo, higher antigen and viral RNA levels were associated with a lower proportion of patients achieving recovery.
Other studies have shown that the use of monoclonal antibodies reduces hospitalization risk in outpatients with early COVID-19, and appears to promote viral load decline in the nasopharynx, wrote Jens D. Lundgren, MD, of the University of Copenhagen and colleagues in their article published in the Annals of Internal Medicine. What had been missing prior to this new research was final results from hospitalized patients, the authors said.
In the new study, the researchers randomized 314 adults hospitalized with COVID-19 but without end-organ failure to receive 7,000 mg bamlanivimab (163 patients) or a placebo (151 patients). All patients received study-supplied remdesivir unless contraindicated. The researchers compared the efficacy of bamlanivimab versus placebo, but considered remdesivir the standard of care in this study.
At baseline, 50% of patients overall had antispike endogenous neutralizing antibodies (nAbs), and 50% had SARS-CoV-2 nucleocapsid plasma antigen levels of at least 1,000 ng/L.
The median time to sustained recovery, 19 days, was not significantly different between the bamlanivimab and placebo groups (subhazard ratio, 0.99).
“As hypothesized, among those who were negative for nAb, the difference between bamlanivimab and placebo was more evident if levels of plasma antigen or nasal-swab viral RNA were above the median entry levels,” with subhazard ratios of 1.48 and 1.89, respectively, the researchers explained.
However, the hazard ratio for death for bamlanivimab vs. placebo was 0.45 for patients negative for nAb vs. 3.53 for those positive for nAb. These differences with respect to nAb status were similar across all 90 elements of a composite safety outcome, the researchers said.
Potential benefits remain unclear
The use of neutralizing monoclonal antibodies has been extensively documented as an effective treatment for COVID-19 among ambulatory patients, corresponding author Dr. Lundgren said in an interview.
“Conversely, among admitted patients with COVID-19 pneumonia, the benefit has been questionable,” he said.
The researchers examined a hypothesis that the null finding in hospitalized patients may stem from differences in underlying mechanisms, “either from uncontrolled viral replication – which would be predicted to occur in particular among those not yet been able to mount an endogenous immune response – or from hyperinflammation among those that have mounted such a response,” Dr. Lundgren said.
The study findings supported the stated hypothesis, said Dr. Lundgren. “However, it was surprising that not only was the neutralizing antibody without any benefit among those that had mounted an endogenous immune response, but it actually may have been harmful,” he said.
Bamlanivimab was effective against the viral strain that circulated at the time of enrollment in the study, but subsequent viral strains have appeared to be unaffected by the neutralizing activity of the antibody, said Dr. Lundgren.
From a practical standpoint, “the findings would suggest that use of neutralizing monoclonal antibodies for patients admitted to a hospital with COVID pneumonia should be restricted to those that have not yet mounted an endogenous immune response, as determined by lack of detectable neutralizing antibodies at the time of admission,” Dr. Lundgren said.
Looking ahead, studies are currently underway to examine how the findings translate to vaccinated patients, he added. Other questions to be addressed include whether the benefits and harms apply to some or all neutralizing antibody products, he said.
In addition, “our research consortium is currently doing field testing of several point-of-care test candidates to examine their reliability and functionality,” for how quickly they might identify an endogenous neutralizing antibody response in an admitted COVID pneumonia patient,” Dr. Lundgren noted.
Findings show bamlanivimab’s limits
“Based on the findings of the current study, no clear subgroup of patients could be identified who would benefit from bamlanivimab when hospitalized with COVID-19,” said Suman Pal, MD, of the University of New Mexico, Albuquerque, in an interview.
“The study findings also show possible harm of using bamlanivimab in hospitalized COVID-19 patients who were seropositive for neutralizing antibodies prior to receiving therapy,” Dr. Pal emphasized. “Moreover, the study did not include participants with COVID-19 from variant strains, such as delta and omicron, which currently account for a large number of cases.” “Therefore, the results of this study do not support the use of bamlanivimab in the clinical setting until further evidence is available to guide the selection of patients who may benefit from therapy,” he explained.
“The possible benefit of bamlanivimab does not outweigh the risks in patients hospitalized with COVID-19,” he concluded.
Dr. Pal emphasized the need for larger prospective studies to establish whether bamlanivimab may have benefits in a subgroup of patients, but “well-validated point-of-care tests to identify such patients need to be readily available before this therapy can be considered by clinicians at the bedside,” he concluded.
Diligent screening required before use
Monoclonal antibody treatment has been administered to individuals with diagnosis of COVID-19 infection as outpatients as well as for hospitalized inpatients, said Noel Deep, MD, an internist in Antigo, Wisc., in an interview. “This study is important because it helps physicians and health care institutions to evaluate whether continued use of the monoclonal antibodies would be beneficial and, if so, in what patient populations,” he said.
The findings present interesting implications for the care of COVID-19 patients, said Dr. Deep. “This study indicates that bamlanivimab does not provide the benefit that was initially envisioned when the monoclonal antibody infusions were initially initiated in the treatment of COVID-19 infections. “Serological screening of the patients would help to identify that subgroup of individuals who could benefit from this monoclonal antibody rather than administering it to every COVID-19–positive individual,” he explained.
However, “it is important to note that the emergency use authorization (EUA) for single-agent bamlanivimab has been revoked,” Dr. Deep said.
“The potential benefits of bamlanivimab can be realized only if adequate attention is paid to identifying the appropriate candidates based on serological screening, and administering bamlanivimab to those who are already producing endogenous antibodies could lead to increased risk to those individuals,” he said. Dr. Deep added that he would favor administration of bamlanivimab “in those appropriately screened and eligible candidates, and it is my opinion that the benefits outweigh the risks in those individuals.”
Although the EUA for single-agent bamlanivimab has been revoked, “alternative monoclonal antibody therapies remain available under EUA, including REGEN-COV (casirivimab and imdevimab, administered together), and bamlanivimab and etesevimab administered together, for the same uses as previously authorized for bamlanivimab alone,” Dr. Deep said. “The FDA believes that these alternative monoclonal antibody therapies remain appropriate to treat patients with COVID-19, and I would like to see some data about the benefits and risks of these agents,” he noted.
Limitations, funding, and disclosures
The main limitation of the study was the small size and the fact that it was a subgroup analysis of a trial that ended early because of futility, the researchers wrote. However, the Therapeutics for Inpatients With COVID-19 (TICO) platform will proceed with clinical evaluation of additional COVID-19 treatments, they said.
The study was supported primarily by the U.S. government Operation Warp Speed and the National Institute of Allergy and Infectious Diseases. Other funding sources included the Division of Clinical Research and Leidos Biomedical Research for the INSIGHT (International Network for Strategic Initiatives in Global HIV Trials) Network, as well as an agreement between the National Heart, Lung, and Blood Institute and the Research Triangle Institute for the PETAL (Prevention & Early Treatment of Acute Lung Injury) Network and CTSN (Cardiothoracic Surgical Trials Network). Other support came from the U.S. Department of Veterans Affairs and the governments of Denmark (National Research Foundation), Australia (National Health and Medical Research Council), and the United Kingdom (Medical Research Council).
The medications used in the study were donated by Gilead Sciences and Eli Lilly.
The researchers had no financial conflicts do disclose. Dr. Deep and Dr. Pal had no relevant financial conflicts to disclose.
FROM ANNALS OF INTERNAL MEDICINE
Children and COVID: New cases up slightly, vaccinations continue to slow
New COVID-19 vaccinations in children were down by almost 24% in the last week as new cases rose by just 3.5%, based on new data.
That fairly low number suggests the latest case count from the American Academy of Pediatrics and the Children’s Hospital Association has not caught up yet to the reality of the Omicron variant, which has sent new cases climbing among all ages and now represents the majority of COVID-19 infections nationwide, the Centers for Disease Control and Prevention said.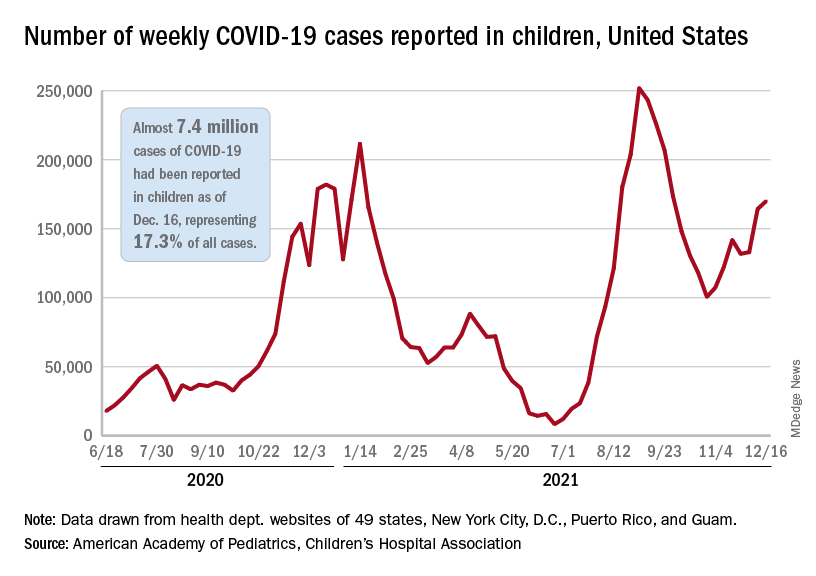
Meanwhile, in the midst of the latest surge, the United States just passed yet another sobering COVID milestone: 1,000 deaths in children aged 17 and under. The total as of Dec. 20 was 1,015, according to the CDC, with the largest share, almost 32%, occurring in children less than 5 years of age.
Regionally, the majority of that increase came in the Northeast, with a small rise in the South and decreases in the Midwest and West, the AAP and CHA said in their weekly COVID report.
At the state level, the largest percent increases in cases over the past 2 weeks were seen in Maine and New Hampshire, as well as Vermont, which has the nation’s highest vaccination rates for children aged 5-11 (51%) and 12-17 (84%), the AAP said in its vaccination trends report.
Nationally, new COVID vaccinations in children continue to trend downward. The number of children aged 5-17 years who had received at least one dose increased by about 498,000 for the week of Dec. 13-19, down from 654,000 (–23.9%) the previous week. Children aged 5-11 years still represented the largest share (22.7%) of all vaccine initiators in the last 2 weeks, but that proportion was 42.8% just before Thanksgiving, according to data from the CDC.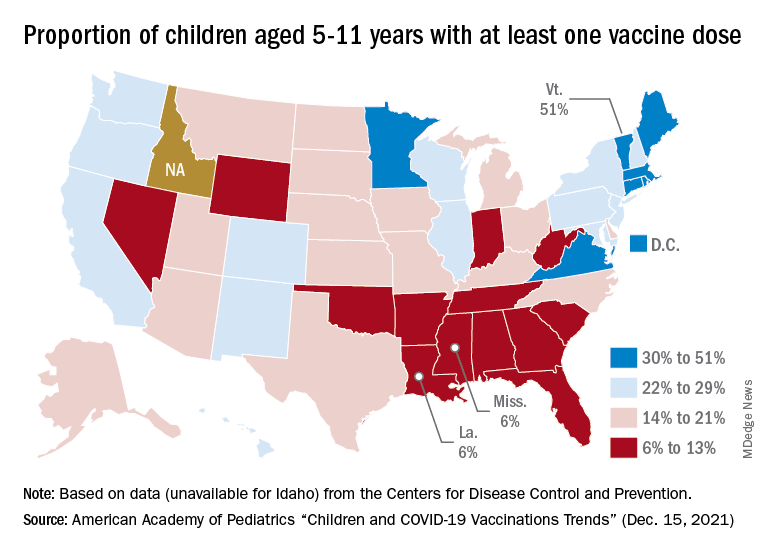
On a more positive note, children aged 5-11 made up 51% of all Americans who completed the vaccine regimen during the 2 weeks ending Dec. 20. The cumulative completion count is 3.6 million in that age group, along with almost 13.4 million children aged 12-17, and the CDC data show that 6.1 million children aged 5-11 and 15.9 million children aged 12-17 have received at least one dose.
On a less positive note, however, that means almost half (47%) of 12- to 17-year-olds still are not fully vaccinated and that over a third (37%) have received no vaccine at all, according to the COVID Data Tracker.
New COVID-19 vaccinations in children were down by almost 24% in the last week as new cases rose by just 3.5%, based on new data.
That fairly low number suggests the latest case count from the American Academy of Pediatrics and the Children’s Hospital Association has not caught up yet to the reality of the Omicron variant, which has sent new cases climbing among all ages and now represents the majority of COVID-19 infections nationwide, the Centers for Disease Control and Prevention said.
Meanwhile, in the midst of the latest surge, the United States just passed yet another sobering COVID milestone: 1,000 deaths in children aged 17 and under. The total as of Dec. 20 was 1,015, according to the CDC, with the largest share, almost 32%, occurring in children less than 5 years of age.
Regionally, the majority of that increase came in the Northeast, with a small rise in the South and decreases in the Midwest and West, the AAP and CHA said in their weekly COVID report.
At the state level, the largest percent increases in cases over the past 2 weeks were seen in Maine and New Hampshire, as well as Vermont, which has the nation’s highest vaccination rates for children aged 5-11 (51%) and 12-17 (84%), the AAP said in its vaccination trends report.
Nationally, new COVID vaccinations in children continue to trend downward. The number of children aged 5-17 years who had received at least one dose increased by about 498,000 for the week of Dec. 13-19, down from 654,000 (–23.9%) the previous week. Children aged 5-11 years still represented the largest share (22.7%) of all vaccine initiators in the last 2 weeks, but that proportion was 42.8% just before Thanksgiving, according to data from the CDC.
On a more positive note, children aged 5-11 made up 51% of all Americans who completed the vaccine regimen during the 2 weeks ending Dec. 20. The cumulative completion count is 3.6 million in that age group, along with almost 13.4 million children aged 12-17, and the CDC data show that 6.1 million children aged 5-11 and 15.9 million children aged 12-17 have received at least one dose.
On a less positive note, however, that means almost half (47%) of 12- to 17-year-olds still are not fully vaccinated and that over a third (37%) have received no vaccine at all, according to the COVID Data Tracker.
New COVID-19 vaccinations in children were down by almost 24% in the last week as new cases rose by just 3.5%, based on new data.
That fairly low number suggests the latest case count from the American Academy of Pediatrics and the Children’s Hospital Association has not caught up yet to the reality of the Omicron variant, which has sent new cases climbing among all ages and now represents the majority of COVID-19 infections nationwide, the Centers for Disease Control and Prevention said.
Meanwhile, in the midst of the latest surge, the United States just passed yet another sobering COVID milestone: 1,000 deaths in children aged 17 and under. The total as of Dec. 20 was 1,015, according to the CDC, with the largest share, almost 32%, occurring in children less than 5 years of age.
Regionally, the majority of that increase came in the Northeast, with a small rise in the South and decreases in the Midwest and West, the AAP and CHA said in their weekly COVID report.
At the state level, the largest percent increases in cases over the past 2 weeks were seen in Maine and New Hampshire, as well as Vermont, which has the nation’s highest vaccination rates for children aged 5-11 (51%) and 12-17 (84%), the AAP said in its vaccination trends report.
Nationally, new COVID vaccinations in children continue to trend downward. The number of children aged 5-17 years who had received at least one dose increased by about 498,000 for the week of Dec. 13-19, down from 654,000 (–23.9%) the previous week. Children aged 5-11 years still represented the largest share (22.7%) of all vaccine initiators in the last 2 weeks, but that proportion was 42.8% just before Thanksgiving, according to data from the CDC.
On a more positive note, children aged 5-11 made up 51% of all Americans who completed the vaccine regimen during the 2 weeks ending Dec. 20. The cumulative completion count is 3.6 million in that age group, along with almost 13.4 million children aged 12-17, and the CDC data show that 6.1 million children aged 5-11 and 15.9 million children aged 12-17 have received at least one dose.
On a less positive note, however, that means almost half (47%) of 12- to 17-year-olds still are not fully vaccinated and that over a third (37%) have received no vaccine at all, according to the COVID Data Tracker.
EMA panel backs linzagolix for uterine fibroid symptoms
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on December 17 recommended approval of linzagolix (Yselty, ObsEva), an oral gonadotropin-releasing hormone (GnRH) antagonist, for the management of moderate to severe symptoms of uterine fibroids (UF) in adult women of reproductive age.
If approved, linzagolix – which is taken once per day – would become the first GnRH receptor antagonist with a nonhormonal option to reach the market. The U.S. Food and Drug Administration in November accepted ObsEva’s new drug application for the medication, with a decision expected by September 2022.
“The positive CHMP opinion is an important milestone for millions of women in the EU living with UF to address the diverse medical needs of the women who suffer from this condition,” said Brian O’Callaghan, CEO of ObsEva, in a statement. “We will continue our productive, ongoing dialogue with [the] EMA toward potential marketing authorization in the EU and, in parallel, continue to work with the FDA to advance linzagolix through the U.S. regulatory process.”
The committee’s positive opinion was based on 52-week results from PRIMROSE 1 and PRIMROSE 2 phase 3 trials, involving more than 1,000 patients in the United States and Europe, as well as results from 76-week follow-up studies of patients in those trials. The two phase 3 trials assessed a 200-mg and 100-mg dose of linzagolix, with and without hormone add-back therapy (ABT; 1 mg estradiol and 0.5 mg norethisterone acetate).
According to ObsEVA, both trials met their primary endpoints, with all doses showing statistically significant and clinically relevant reductions in heavy menstrual bleeding (HMB) compared to placebo. The trials also achieved several secondary endpoints, including reduction in pain, rates of amenorrhea, time to reduced HMB, and amenorrhea and for the high dose without ABT, reductions in uterine and fibroid volume, the company said.
A version of this article first appeared on Medscape.com.
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on December 17 recommended approval of linzagolix (Yselty, ObsEva), an oral gonadotropin-releasing hormone (GnRH) antagonist, for the management of moderate to severe symptoms of uterine fibroids (UF) in adult women of reproductive age.
If approved, linzagolix – which is taken once per day – would become the first GnRH receptor antagonist with a nonhormonal option to reach the market. The U.S. Food and Drug Administration in November accepted ObsEva’s new drug application for the medication, with a decision expected by September 2022.
“The positive CHMP opinion is an important milestone for millions of women in the EU living with UF to address the diverse medical needs of the women who suffer from this condition,” said Brian O’Callaghan, CEO of ObsEva, in a statement. “We will continue our productive, ongoing dialogue with [the] EMA toward potential marketing authorization in the EU and, in parallel, continue to work with the FDA to advance linzagolix through the U.S. regulatory process.”
The committee’s positive opinion was based on 52-week results from PRIMROSE 1 and PRIMROSE 2 phase 3 trials, involving more than 1,000 patients in the United States and Europe, as well as results from 76-week follow-up studies of patients in those trials. The two phase 3 trials assessed a 200-mg and 100-mg dose of linzagolix, with and without hormone add-back therapy (ABT; 1 mg estradiol and 0.5 mg norethisterone acetate).
According to ObsEVA, both trials met their primary endpoints, with all doses showing statistically significant and clinically relevant reductions in heavy menstrual bleeding (HMB) compared to placebo. The trials also achieved several secondary endpoints, including reduction in pain, rates of amenorrhea, time to reduced HMB, and amenorrhea and for the high dose without ABT, reductions in uterine and fibroid volume, the company said.
A version of this article first appeared on Medscape.com.
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on December 17 recommended approval of linzagolix (Yselty, ObsEva), an oral gonadotropin-releasing hormone (GnRH) antagonist, for the management of moderate to severe symptoms of uterine fibroids (UF) in adult women of reproductive age.
If approved, linzagolix – which is taken once per day – would become the first GnRH receptor antagonist with a nonhormonal option to reach the market. The U.S. Food and Drug Administration in November accepted ObsEva’s new drug application for the medication, with a decision expected by September 2022.
“The positive CHMP opinion is an important milestone for millions of women in the EU living with UF to address the diverse medical needs of the women who suffer from this condition,” said Brian O’Callaghan, CEO of ObsEva, in a statement. “We will continue our productive, ongoing dialogue with [the] EMA toward potential marketing authorization in the EU and, in parallel, continue to work with the FDA to advance linzagolix through the U.S. regulatory process.”
The committee’s positive opinion was based on 52-week results from PRIMROSE 1 and PRIMROSE 2 phase 3 trials, involving more than 1,000 patients in the United States and Europe, as well as results from 76-week follow-up studies of patients in those trials. The two phase 3 trials assessed a 200-mg and 100-mg dose of linzagolix, with and without hormone add-back therapy (ABT; 1 mg estradiol and 0.5 mg norethisterone acetate).
According to ObsEVA, both trials met their primary endpoints, with all doses showing statistically significant and clinically relevant reductions in heavy menstrual bleeding (HMB) compared to placebo. The trials also achieved several secondary endpoints, including reduction in pain, rates of amenorrhea, time to reduced HMB, and amenorrhea and for the high dose without ABT, reductions in uterine and fibroid volume, the company said.
A version of this article first appeared on Medscape.com.
Fish oil: ‘No net benefit’ for depression prevention?
Fish oil supplementation does not help prevent depression or boost mood, new research suggests.
The VITAL-DEP study included more than 18,000 participants. Among adults aged 50 years or older free of clinically relevant depressive symptoms at baseline, long-term use of marine omega-3 fatty acid (omega-3) supplements did not reduce risk for depression or clinically relevant depressive symptoms — or make a difference in the quality of mood.
“While a small increase in risk of depression was inside the statistical margin of significance, there was no harmful or beneficial effect of omega-3 on the overall course of mood during the roughly 5 to 7 years of follow-up,” lead author Olivia I. Okereke, MD, Massachusetts General Hospital and Harvard Medical School, Boston, told Medscape Medical News.
“The takeaway from our study is that there is no net benefit of long-term use of daily omega-3 fish oil supplements for preventing depression or boosting mood,” Okereke said.
The findings were published online Dec. 21 in JAMA.
Assessing general population risk
For many years, experts have recommended omega-3 supplements for reduction in depression recurrence in some high-risk patients, Okereke noted.
“However, there are no guidelines related to the use of omega-3 supplements for preventing depression in the general population. Therefore, we undertook this study to provide clarity in the issue,” she said.
The VITAL-DEP study enrolled 18,353 older adults (mean age, 67.5 years; 49% women). Of these, 16,657 were at risk for incident depression, defined as having no previous history of depression; and 1696 were at risk for recurrent depression, defined as having a history of depression but not having undergone treatment for depression within the past 2 years.
Roughly half the participants were randomly assigned to receive marine omega-3 fatty acids (1 g/d of fish oil, including 465 mg of eicosapentaenoic acid [EPA] and 375 mg of docosahexaenoic acid [DHA]) and the other half to matching placebo for an average of 5.3 years.
“Because of the large sample size and long follow-up, we were able to test the effects of daily omega-3 fish oil supplements on universal prevention of depression in the adult population,” Okereke said.
No significant benefit
Results showed risk for depression or clinically relevant depressive symptoms (total of incident and recurrent cases) was not significantly different between the omega-3 group and the placebo group.
The omega-3 group had 651 depression or clinically relevant depressive symptom events (13.9 per 1000 person-years), and the placebo group had 583 depression or clinically relevant depressive symptom events (12.3 per 1000 person-years). The hazard ratio was 1.13 (95% CI, 1.01 - 1.26; P = .03).
There were also no significant between-group differences in longitudinal mood scores. The mean difference in change in 8-item Patient Health Questionnaire (PHQ-8) score was 0.03 points (95% CI, −0.01 to 0.07; P = .19).
“Patients, physicians, and other clinicians should understand that there are still many reasons for some people, under the guidance of their health care providers, to take omega-3 fish oil supplements,” Okereke noted.
“These supplements increasingly have been found to have benefits for cardiac disease prevention and treatment of inflammatory conditions, in addition to being used for management of existing depressive disorders in some high-risk patients,” she said.
“However, the results of our study indicate there is no reason for adults in the general population to be taking daily omega-3 fish oil supplements solely for the purpose of preventing depression or for maintaining a positive mood,” she added.
Okereke noted, however, that the VITAL-DEP study used 1 g/day of omega-3 fatty acids and there may be a greater benefit from taking higher doses, such as 4 g/day.
Cautionary notes
Commenting on the study for Medscape Medical News, Kuan-Pin Su, MD, PhD, chief of the Department of General Psychiatry, China Medical University, Taichung, Taiwan, highlighted some of the limitations cited by the investigators.
First, depression or depressive symptoms were defined using self-rating scales, which are “convenient to screen for depressive disorders, but a high score obtained on a self-rating scale does not necessarily indicate the presence of depressive psychopathology,” said Su, who was not involved with the research.
He also noted that use of 465 mg of EPA and 375 mg of DHA in VITAL-DEP “might be too low” to have an impact.
Finally, Su said it is “very important to also address the potential for type I error, which makes the secondary and subgroup analyses less reliable.”
VITAL-DEP was supported by a grant from the National Institute of Mental Health. Pronova BioPharma donated the fish oil and matching placebo. Okereke reported receiving royalties from Springer Publishing. Su is a founding committee member of the International Society for Nutritional Psychiatry Research, the board director of the International Society for the Study of Fatty Acids, and an associate editor of the journal Brain, Behavior, and Immunity.
A version of this article first appeared on Medscape.com.
Fish oil supplementation does not help prevent depression or boost mood, new research suggests.
The VITAL-DEP study included more than 18,000 participants. Among adults aged 50 years or older free of clinically relevant depressive symptoms at baseline, long-term use of marine omega-3 fatty acid (omega-3) supplements did not reduce risk for depression or clinically relevant depressive symptoms — or make a difference in the quality of mood.
“While a small increase in risk of depression was inside the statistical margin of significance, there was no harmful or beneficial effect of omega-3 on the overall course of mood during the roughly 5 to 7 years of follow-up,” lead author Olivia I. Okereke, MD, Massachusetts General Hospital and Harvard Medical School, Boston, told Medscape Medical News.
“The takeaway from our study is that there is no net benefit of long-term use of daily omega-3 fish oil supplements for preventing depression or boosting mood,” Okereke said.
The findings were published online Dec. 21 in JAMA.
Assessing general population risk
For many years, experts have recommended omega-3 supplements for reduction in depression recurrence in some high-risk patients, Okereke noted.
“However, there are no guidelines related to the use of omega-3 supplements for preventing depression in the general population. Therefore, we undertook this study to provide clarity in the issue,” she said.
The VITAL-DEP study enrolled 18,353 older adults (mean age, 67.5 years; 49% women). Of these, 16,657 were at risk for incident depression, defined as having no previous history of depression; and 1696 were at risk for recurrent depression, defined as having a history of depression but not having undergone treatment for depression within the past 2 years.
Roughly half the participants were randomly assigned to receive marine omega-3 fatty acids (1 g/d of fish oil, including 465 mg of eicosapentaenoic acid [EPA] and 375 mg of docosahexaenoic acid [DHA]) and the other half to matching placebo for an average of 5.3 years.
“Because of the large sample size and long follow-up, we were able to test the effects of daily omega-3 fish oil supplements on universal prevention of depression in the adult population,” Okereke said.
No significant benefit
Results showed risk for depression or clinically relevant depressive symptoms (total of incident and recurrent cases) was not significantly different between the omega-3 group and the placebo group.
The omega-3 group had 651 depression or clinically relevant depressive symptom events (13.9 per 1000 person-years), and the placebo group had 583 depression or clinically relevant depressive symptom events (12.3 per 1000 person-years). The hazard ratio was 1.13 (95% CI, 1.01 - 1.26; P = .03).
There were also no significant between-group differences in longitudinal mood scores. The mean difference in change in 8-item Patient Health Questionnaire (PHQ-8) score was 0.03 points (95% CI, −0.01 to 0.07; P = .19).
“Patients, physicians, and other clinicians should understand that there are still many reasons for some people, under the guidance of their health care providers, to take omega-3 fish oil supplements,” Okereke noted.
“These supplements increasingly have been found to have benefits for cardiac disease prevention and treatment of inflammatory conditions, in addition to being used for management of existing depressive disorders in some high-risk patients,” she said.
“However, the results of our study indicate there is no reason for adults in the general population to be taking daily omega-3 fish oil supplements solely for the purpose of preventing depression or for maintaining a positive mood,” she added.
Okereke noted, however, that the VITAL-DEP study used 1 g/day of omega-3 fatty acids and there may be a greater benefit from taking higher doses, such as 4 g/day.
Cautionary notes
Commenting on the study for Medscape Medical News, Kuan-Pin Su, MD, PhD, chief of the Department of General Psychiatry, China Medical University, Taichung, Taiwan, highlighted some of the limitations cited by the investigators.
First, depression or depressive symptoms were defined using self-rating scales, which are “convenient to screen for depressive disorders, but a high score obtained on a self-rating scale does not necessarily indicate the presence of depressive psychopathology,” said Su, who was not involved with the research.
He also noted that use of 465 mg of EPA and 375 mg of DHA in VITAL-DEP “might be too low” to have an impact.
Finally, Su said it is “very important to also address the potential for type I error, which makes the secondary and subgroup analyses less reliable.”
VITAL-DEP was supported by a grant from the National Institute of Mental Health. Pronova BioPharma donated the fish oil and matching placebo. Okereke reported receiving royalties from Springer Publishing. Su is a founding committee member of the International Society for Nutritional Psychiatry Research, the board director of the International Society for the Study of Fatty Acids, and an associate editor of the journal Brain, Behavior, and Immunity.
A version of this article first appeared on Medscape.com.
Fish oil supplementation does not help prevent depression or boost mood, new research suggests.
The VITAL-DEP study included more than 18,000 participants. Among adults aged 50 years or older free of clinically relevant depressive symptoms at baseline, long-term use of marine omega-3 fatty acid (omega-3) supplements did not reduce risk for depression or clinically relevant depressive symptoms — or make a difference in the quality of mood.
“While a small increase in risk of depression was inside the statistical margin of significance, there was no harmful or beneficial effect of omega-3 on the overall course of mood during the roughly 5 to 7 years of follow-up,” lead author Olivia I. Okereke, MD, Massachusetts General Hospital and Harvard Medical School, Boston, told Medscape Medical News.
“The takeaway from our study is that there is no net benefit of long-term use of daily omega-3 fish oil supplements for preventing depression or boosting mood,” Okereke said.
The findings were published online Dec. 21 in JAMA.
Assessing general population risk
For many years, experts have recommended omega-3 supplements for reduction in depression recurrence in some high-risk patients, Okereke noted.
“However, there are no guidelines related to the use of omega-3 supplements for preventing depression in the general population. Therefore, we undertook this study to provide clarity in the issue,” she said.
The VITAL-DEP study enrolled 18,353 older adults (mean age, 67.5 years; 49% women). Of these, 16,657 were at risk for incident depression, defined as having no previous history of depression; and 1696 were at risk for recurrent depression, defined as having a history of depression but not having undergone treatment for depression within the past 2 years.
Roughly half the participants were randomly assigned to receive marine omega-3 fatty acids (1 g/d of fish oil, including 465 mg of eicosapentaenoic acid [EPA] and 375 mg of docosahexaenoic acid [DHA]) and the other half to matching placebo for an average of 5.3 years.
“Because of the large sample size and long follow-up, we were able to test the effects of daily omega-3 fish oil supplements on universal prevention of depression in the adult population,” Okereke said.
No significant benefit
Results showed risk for depression or clinically relevant depressive symptoms (total of incident and recurrent cases) was not significantly different between the omega-3 group and the placebo group.
The omega-3 group had 651 depression or clinically relevant depressive symptom events (13.9 per 1000 person-years), and the placebo group had 583 depression or clinically relevant depressive symptom events (12.3 per 1000 person-years). The hazard ratio was 1.13 (95% CI, 1.01 - 1.26; P = .03).
There were also no significant between-group differences in longitudinal mood scores. The mean difference in change in 8-item Patient Health Questionnaire (PHQ-8) score was 0.03 points (95% CI, −0.01 to 0.07; P = .19).
“Patients, physicians, and other clinicians should understand that there are still many reasons for some people, under the guidance of their health care providers, to take omega-3 fish oil supplements,” Okereke noted.
“These supplements increasingly have been found to have benefits for cardiac disease prevention and treatment of inflammatory conditions, in addition to being used for management of existing depressive disorders in some high-risk patients,” she said.
“However, the results of our study indicate there is no reason for adults in the general population to be taking daily omega-3 fish oil supplements solely for the purpose of preventing depression or for maintaining a positive mood,” she added.
Okereke noted, however, that the VITAL-DEP study used 1 g/day of omega-3 fatty acids and there may be a greater benefit from taking higher doses, such as 4 g/day.
Cautionary notes
Commenting on the study for Medscape Medical News, Kuan-Pin Su, MD, PhD, chief of the Department of General Psychiatry, China Medical University, Taichung, Taiwan, highlighted some of the limitations cited by the investigators.
First, depression or depressive symptoms were defined using self-rating scales, which are “convenient to screen for depressive disorders, but a high score obtained on a self-rating scale does not necessarily indicate the presence of depressive psychopathology,” said Su, who was not involved with the research.
He also noted that use of 465 mg of EPA and 375 mg of DHA in VITAL-DEP “might be too low” to have an impact.
Finally, Su said it is “very important to also address the potential for type I error, which makes the secondary and subgroup analyses less reliable.”
VITAL-DEP was supported by a grant from the National Institute of Mental Health. Pronova BioPharma donated the fish oil and matching placebo. Okereke reported receiving royalties from Springer Publishing. Su is a founding committee member of the International Society for Nutritional Psychiatry Research, the board director of the International Society for the Study of Fatty Acids, and an associate editor of the journal Brain, Behavior, and Immunity.
A version of this article first appeared on Medscape.com.
GERD: Upper endoscopy may reduce GI cancer mortality
Among individuals with gastroesophageal reflux disease (GERD), a negative upper endoscopy is associated with decreased risk in incidence and mortality from gastrointestinal cancer. The benefit persisted through 5-10 years following the procedure.
The finding is similar to the survival benefit seen with colonoscopies and colorectal cancer, and may be attributable to endoscopic treatment of premalignant lesions.
“The relatively high incidence rate of upper gastrointestinal cancer in patients with GERD indicates that a one-time upper endoscopy may be beneficial,” wrote the authors, who were led by Dag Holmberg, MD, PhD, of the department of molecular medicine and surgery at the Karolinska Institutet and Karolinska University Hospital, both in Stockholm. The study was published in Gastroenterology.
GERD is the most frequent reason patients undergo an upper endoscopy, but the results are often negative. It is generally a benign condition, but can lead to Barrett’s esophagus, as well as esophageal and gastric cardia adenocarcinoma. Upper endoscopy can identify other esophageal cancers like gastric noncardia cancer and duodenal cancer, which may cause dyspepsia or GERD-like symptoms.
To determine the potential benefit of upper endoscopy, the researchers conducted a population-based, four-nation cohort study that included 1,062,740 individuals with GERD in Denmark, Finland, Norway, and Sweden. The data were gathered from national patient registries, cancer registries, and cause of death registries. The study encompassed data from 1979 through the end of 2018.
The median age was 58 years, and 52% of participants were women.
The researchers defined a negative endoscopy as no diagnosis of gastrointestinal cancer within 6 months of the procedure; 69.3% of procedures were negative.
During the follow-up period, 0.34% of participants developed and 0.27% died of upper gastrointestinal cancer. Among those with negative endoscopies, 0.23% developed and 0.22% died from upper gastrointestinal cancer.
Participants with a negative endoscopy had a lower risk of being diagnosed with upper gastrointestinal cancer during the follow-up period (adjusted hazard ratio, 0.45; 95% confidence interval, 0.43-0.48). The reduction in risk was similar across age sexes and age groups, but among procedures performed after 2008, the risk reduction was even higher (aHR, 0.34; P < .001).
The effect was strongest in the first year after the procedure, but it persisted out to 5 years before returning to baseline risk levels.
A negative endoscopy was also associated with decreased mortality risk from upper gastrointestinal cancer versus those who hadn’t had an endoscopy (aHR, 0.39; 95% CI, 0.37-0.42). The protective value continued for at least 10 years.
Esophageal adenocarcinoma developed in 0.12% of participants, and 0.10% died of the disease. Among those with a negative endoscopy, 0.09% developed adenocarcinoma, and 0.07% died (aHR vs. no upper endoscopy, 0.33; 95% CI, 0.30-0.37).
The rapid return to baseline risk was notable, and different from what occurs after negative colonoscopies. However, new tumors can readily form within one year, and the risk may reflect early malignant or premalignant lesions that were missed during the procedure.
In fact, a meta-analysis found that 11.3% of upper gastrointestinal cancers had escaped detection during an endoscopy in the previous 3 years before diagnosis, and case reviews of patients diagnosed with gastrointestinal cancer soon after an upper endoscopy usually reveal suspicious or indeterminate results that the endoscopist or pathologist missed.
Quality indicators for upper endoscopy include procedure time, rate of targeted biopsies, and computer-aided detection, but it isn’t clear what impact these measures have on outcomes. However, the greater risk reduction found with endoscopies performed more recently suggests that newer quality indicators and technological improvements may be improving outcomes.
The relatively low incidence of esophageal and gastric cancer in Western countries has discouraged widespread adoption of endoscopic screening, but the researchers point out that the risk of gastrointestinal cancer among individuals with GERD is similar to the risk of colorectal cancer in the 60-69 age group in the United States, for whom colonoscopy is recommended.
“The present study indicates that upper endoscopy may be beneficial for patients with GERD, but to make upper endoscopy screening more cost beneficial at its initiation, the target group may be limited to include patients at highest risk of cancer. Such previous cost-effectiveness studies have indicated that endoscopy is cost effective in men at aged 50 years or older with chronic GERD,” the authors wrote.
The study was funded by Swedish Research Council and Swedish Cancer Society. The authors disclosed no relevant conflicts of interest.
Among individuals with gastroesophageal reflux disease (GERD), a negative upper endoscopy is associated with decreased risk in incidence and mortality from gastrointestinal cancer. The benefit persisted through 5-10 years following the procedure.
The finding is similar to the survival benefit seen with colonoscopies and colorectal cancer, and may be attributable to endoscopic treatment of premalignant lesions.
“The relatively high incidence rate of upper gastrointestinal cancer in patients with GERD indicates that a one-time upper endoscopy may be beneficial,” wrote the authors, who were led by Dag Holmberg, MD, PhD, of the department of molecular medicine and surgery at the Karolinska Institutet and Karolinska University Hospital, both in Stockholm. The study was published in Gastroenterology.
GERD is the most frequent reason patients undergo an upper endoscopy, but the results are often negative. It is generally a benign condition, but can lead to Barrett’s esophagus, as well as esophageal and gastric cardia adenocarcinoma. Upper endoscopy can identify other esophageal cancers like gastric noncardia cancer and duodenal cancer, which may cause dyspepsia or GERD-like symptoms.
To determine the potential benefit of upper endoscopy, the researchers conducted a population-based, four-nation cohort study that included 1,062,740 individuals with GERD in Denmark, Finland, Norway, and Sweden. The data were gathered from national patient registries, cancer registries, and cause of death registries. The study encompassed data from 1979 through the end of 2018.
The median age was 58 years, and 52% of participants were women.
The researchers defined a negative endoscopy as no diagnosis of gastrointestinal cancer within 6 months of the procedure; 69.3% of procedures were negative.
During the follow-up period, 0.34% of participants developed and 0.27% died of upper gastrointestinal cancer. Among those with negative endoscopies, 0.23% developed and 0.22% died from upper gastrointestinal cancer.
Participants with a negative endoscopy had a lower risk of being diagnosed with upper gastrointestinal cancer during the follow-up period (adjusted hazard ratio, 0.45; 95% confidence interval, 0.43-0.48). The reduction in risk was similar across age sexes and age groups, but among procedures performed after 2008, the risk reduction was even higher (aHR, 0.34; P < .001).
The effect was strongest in the first year after the procedure, but it persisted out to 5 years before returning to baseline risk levels.
A negative endoscopy was also associated with decreased mortality risk from upper gastrointestinal cancer versus those who hadn’t had an endoscopy (aHR, 0.39; 95% CI, 0.37-0.42). The protective value continued for at least 10 years.
Esophageal adenocarcinoma developed in 0.12% of participants, and 0.10% died of the disease. Among those with a negative endoscopy, 0.09% developed adenocarcinoma, and 0.07% died (aHR vs. no upper endoscopy, 0.33; 95% CI, 0.30-0.37).
The rapid return to baseline risk was notable, and different from what occurs after negative colonoscopies. However, new tumors can readily form within one year, and the risk may reflect early malignant or premalignant lesions that were missed during the procedure.
In fact, a meta-analysis found that 11.3% of upper gastrointestinal cancers had escaped detection during an endoscopy in the previous 3 years before diagnosis, and case reviews of patients diagnosed with gastrointestinal cancer soon after an upper endoscopy usually reveal suspicious or indeterminate results that the endoscopist or pathologist missed.
Quality indicators for upper endoscopy include procedure time, rate of targeted biopsies, and computer-aided detection, but it isn’t clear what impact these measures have on outcomes. However, the greater risk reduction found with endoscopies performed more recently suggests that newer quality indicators and technological improvements may be improving outcomes.
The relatively low incidence of esophageal and gastric cancer in Western countries has discouraged widespread adoption of endoscopic screening, but the researchers point out that the risk of gastrointestinal cancer among individuals with GERD is similar to the risk of colorectal cancer in the 60-69 age group in the United States, for whom colonoscopy is recommended.
“The present study indicates that upper endoscopy may be beneficial for patients with GERD, but to make upper endoscopy screening more cost beneficial at its initiation, the target group may be limited to include patients at highest risk of cancer. Such previous cost-effectiveness studies have indicated that endoscopy is cost effective in men at aged 50 years or older with chronic GERD,” the authors wrote.
The study was funded by Swedish Research Council and Swedish Cancer Society. The authors disclosed no relevant conflicts of interest.
Among individuals with gastroesophageal reflux disease (GERD), a negative upper endoscopy is associated with decreased risk in incidence and mortality from gastrointestinal cancer. The benefit persisted through 5-10 years following the procedure.
The finding is similar to the survival benefit seen with colonoscopies and colorectal cancer, and may be attributable to endoscopic treatment of premalignant lesions.
“The relatively high incidence rate of upper gastrointestinal cancer in patients with GERD indicates that a one-time upper endoscopy may be beneficial,” wrote the authors, who were led by Dag Holmberg, MD, PhD, of the department of molecular medicine and surgery at the Karolinska Institutet and Karolinska University Hospital, both in Stockholm. The study was published in Gastroenterology.
GERD is the most frequent reason patients undergo an upper endoscopy, but the results are often negative. It is generally a benign condition, but can lead to Barrett’s esophagus, as well as esophageal and gastric cardia adenocarcinoma. Upper endoscopy can identify other esophageal cancers like gastric noncardia cancer and duodenal cancer, which may cause dyspepsia or GERD-like symptoms.
To determine the potential benefit of upper endoscopy, the researchers conducted a population-based, four-nation cohort study that included 1,062,740 individuals with GERD in Denmark, Finland, Norway, and Sweden. The data were gathered from national patient registries, cancer registries, and cause of death registries. The study encompassed data from 1979 through the end of 2018.
The median age was 58 years, and 52% of participants were women.
The researchers defined a negative endoscopy as no diagnosis of gastrointestinal cancer within 6 months of the procedure; 69.3% of procedures were negative.
During the follow-up period, 0.34% of participants developed and 0.27% died of upper gastrointestinal cancer. Among those with negative endoscopies, 0.23% developed and 0.22% died from upper gastrointestinal cancer.
Participants with a negative endoscopy had a lower risk of being diagnosed with upper gastrointestinal cancer during the follow-up period (adjusted hazard ratio, 0.45; 95% confidence interval, 0.43-0.48). The reduction in risk was similar across age sexes and age groups, but among procedures performed after 2008, the risk reduction was even higher (aHR, 0.34; P < .001).
The effect was strongest in the first year after the procedure, but it persisted out to 5 years before returning to baseline risk levels.
A negative endoscopy was also associated with decreased mortality risk from upper gastrointestinal cancer versus those who hadn’t had an endoscopy (aHR, 0.39; 95% CI, 0.37-0.42). The protective value continued for at least 10 years.
Esophageal adenocarcinoma developed in 0.12% of participants, and 0.10% died of the disease. Among those with a negative endoscopy, 0.09% developed adenocarcinoma, and 0.07% died (aHR vs. no upper endoscopy, 0.33; 95% CI, 0.30-0.37).
The rapid return to baseline risk was notable, and different from what occurs after negative colonoscopies. However, new tumors can readily form within one year, and the risk may reflect early malignant or premalignant lesions that were missed during the procedure.
In fact, a meta-analysis found that 11.3% of upper gastrointestinal cancers had escaped detection during an endoscopy in the previous 3 years before diagnosis, and case reviews of patients diagnosed with gastrointestinal cancer soon after an upper endoscopy usually reveal suspicious or indeterminate results that the endoscopist or pathologist missed.
Quality indicators for upper endoscopy include procedure time, rate of targeted biopsies, and computer-aided detection, but it isn’t clear what impact these measures have on outcomes. However, the greater risk reduction found with endoscopies performed more recently suggests that newer quality indicators and technological improvements may be improving outcomes.
The relatively low incidence of esophageal and gastric cancer in Western countries has discouraged widespread adoption of endoscopic screening, but the researchers point out that the risk of gastrointestinal cancer among individuals with GERD is similar to the risk of colorectal cancer in the 60-69 age group in the United States, for whom colonoscopy is recommended.
“The present study indicates that upper endoscopy may be beneficial for patients with GERD, but to make upper endoscopy screening more cost beneficial at its initiation, the target group may be limited to include patients at highest risk of cancer. Such previous cost-effectiveness studies have indicated that endoscopy is cost effective in men at aged 50 years or older with chronic GERD,” the authors wrote.
The study was funded by Swedish Research Council and Swedish Cancer Society. The authors disclosed no relevant conflicts of interest.
FROM GASTROENTEROLOGY
Single-use duodenoscope is cost effective in ERCP
The EXALT Model-D single-use duodenoscope is a cost-effective alternative to high-level disinfection (HLD) of reusable duodenoscopes, according to a new analysis.
The study compared the EXALT Model-D, HLD, culture-and-quarantine (CQ), and ethylene oxide sterilization (ETO). The results came from a simulated cohort of patients undergoing endoscopic retrograde cholangiopancreatography (ERCP) to treat choledocholithiasis.
Although EXALT was the costliest option and HLD the cheapest, EXALT produced the most quality-adjusted life years (QALYs) and allowed the hospital to decrease net costs, and sensitivity analysis showed that it was a better option than HLD over a range of willingness-to-pay values.
“When evaluating technologies based on cost-effectiveness and additionally in the context of TPT [transitional passthrough] or NTAP [new technology add-on payment], the EXALT approach meets typically used cost-effectiveness thresholds compared to all other evaluated strategies and should be considered for standard practice,” wrote the authors, who were led by Ananya Das, MD, of the Arizona Centers for Digestive Health, Gilbert. The study was published in Techniques and Innovations in Gastrointestinal Endoscopy.
Duodenoscope contamination has resulted in outbreaks of various multidrug-resistant organisms in hospital settings, which has led to the publication of various reprocessing guidelines. Although many hospitals have adopted HLD protocols, others use additional or alternative reprocessing methods such as CQ or ETO. Despite these efforts, a recent Food and Drug Administration study found that 1.9%-22% of samples taken from duodenoscopes tested positive for bacteria of concern, such as pathogens. Those and other findings have led some to suggest that it would be best to move away from HLD, and instead employ sterilizable or disposable endoscopes.
In another study, The EXALT Model-D (Boston Scientific) had been shown to be a good alternative to standard reusable duodenoscopes.
The researchers used a Markov-model to determine the cost-effectiveness of EXALT Model-D against other approaches in a simulated cohort. They found that EXALT Model-D created the most QALYs (21.9265) at the highest cost ($3,000), and HLD the fewest QALYs (21.8938) at the lowest cost ($962). Compared with HLD, the incremental cost-effectiveness ratio (ICER) of EXALT was $62,185, and $38,461 for ETO gas sterilization. CQ was dominated, indicating that it had a higher cost but was not more effective than HLD.
The researchers conducted a subanalysis of ERCP and Medicare patients to consider the recently approved TPT payment and the NTAP, in both hospital outpatient and inpatient settings. With TPT, EXALT had no cost after reimbursement, with a net saving of $962 per patient when compared with HLD, plus an increase in 0.033 QALYs (0.15%). The other procedures cost more and were less effective. With NTAP, EXALT had a net cost of $323 versus HLD, with a similar QALY benefit.
A Monte Carlo analysis of EXALT versus HLD found reductions in duodenoscope infection-related ICU admission (relative risk reduction, 0.996; 95% confidence interval, 0.936-1.0; number needed to treat, 79; 95% CI, 67-95) and death (RRR, 0.973; 95% CI, 0.552-0.998; number needed to treat, 556; 95% CI, 350-997).
In willingness-to-pay estimates from $50,000 to $100,000, EXALT was cost effective in 67.28% of trials with ICER under $100,000 per QALY.
The study did not consider medicolegal costs, which could lead to an underestimation of EXALT’s cost-effectiveness. The study also relied on available published information to determine cost per patient of hospital outbreaks in the United States and Europe since 2012, but the authors did not include costs of administrative sanctions, litigation, and poor publicity due to inconsistencies in the literature.
“While more research is needed to understand and quantify the determinants of the natural history after exposure to contaminated duodenoscopes, such as the risk of transmission and the subsequent development of serious clinical infections, this economic analysis demonstrates an approach using EXALT Model-D is cost effective in the U.S. health care system when compared to the currently utilized strategies of duodenoscope reprocessing,” the researchers concluded.
The study did not receive any funding. One of the authors is an employee and stockholder of Boston Scientific, which manufactures and markets EXALT. The other two authors have consulted for Boston Scientific.
The EXALT Model-D single-use duodenoscope is a cost-effective alternative to high-level disinfection (HLD) of reusable duodenoscopes, according to a new analysis.
The study compared the EXALT Model-D, HLD, culture-and-quarantine (CQ), and ethylene oxide sterilization (ETO). The results came from a simulated cohort of patients undergoing endoscopic retrograde cholangiopancreatography (ERCP) to treat choledocholithiasis.
Although EXALT was the costliest option and HLD the cheapest, EXALT produced the most quality-adjusted life years (QALYs) and allowed the hospital to decrease net costs, and sensitivity analysis showed that it was a better option than HLD over a range of willingness-to-pay values.
“When evaluating technologies based on cost-effectiveness and additionally in the context of TPT [transitional passthrough] or NTAP [new technology add-on payment], the EXALT approach meets typically used cost-effectiveness thresholds compared to all other evaluated strategies and should be considered for standard practice,” wrote the authors, who were led by Ananya Das, MD, of the Arizona Centers for Digestive Health, Gilbert. The study was published in Techniques and Innovations in Gastrointestinal Endoscopy.
Duodenoscope contamination has resulted in outbreaks of various multidrug-resistant organisms in hospital settings, which has led to the publication of various reprocessing guidelines. Although many hospitals have adopted HLD protocols, others use additional or alternative reprocessing methods such as CQ or ETO. Despite these efforts, a recent Food and Drug Administration study found that 1.9%-22% of samples taken from duodenoscopes tested positive for bacteria of concern, such as pathogens. Those and other findings have led some to suggest that it would be best to move away from HLD, and instead employ sterilizable or disposable endoscopes.
In another study, The EXALT Model-D (Boston Scientific) had been shown to be a good alternative to standard reusable duodenoscopes.
The researchers used a Markov-model to determine the cost-effectiveness of EXALT Model-D against other approaches in a simulated cohort. They found that EXALT Model-D created the most QALYs (21.9265) at the highest cost ($3,000), and HLD the fewest QALYs (21.8938) at the lowest cost ($962). Compared with HLD, the incremental cost-effectiveness ratio (ICER) of EXALT was $62,185, and $38,461 for ETO gas sterilization. CQ was dominated, indicating that it had a higher cost but was not more effective than HLD.
The researchers conducted a subanalysis of ERCP and Medicare patients to consider the recently approved TPT payment and the NTAP, in both hospital outpatient and inpatient settings. With TPT, EXALT had no cost after reimbursement, with a net saving of $962 per patient when compared with HLD, plus an increase in 0.033 QALYs (0.15%). The other procedures cost more and were less effective. With NTAP, EXALT had a net cost of $323 versus HLD, with a similar QALY benefit.
A Monte Carlo analysis of EXALT versus HLD found reductions in duodenoscope infection-related ICU admission (relative risk reduction, 0.996; 95% confidence interval, 0.936-1.0; number needed to treat, 79; 95% CI, 67-95) and death (RRR, 0.973; 95% CI, 0.552-0.998; number needed to treat, 556; 95% CI, 350-997).
In willingness-to-pay estimates from $50,000 to $100,000, EXALT was cost effective in 67.28% of trials with ICER under $100,000 per QALY.
The study did not consider medicolegal costs, which could lead to an underestimation of EXALT’s cost-effectiveness. The study also relied on available published information to determine cost per patient of hospital outbreaks in the United States and Europe since 2012, but the authors did not include costs of administrative sanctions, litigation, and poor publicity due to inconsistencies in the literature.
“While more research is needed to understand and quantify the determinants of the natural history after exposure to contaminated duodenoscopes, such as the risk of transmission and the subsequent development of serious clinical infections, this economic analysis demonstrates an approach using EXALT Model-D is cost effective in the U.S. health care system when compared to the currently utilized strategies of duodenoscope reprocessing,” the researchers concluded.
The study did not receive any funding. One of the authors is an employee and stockholder of Boston Scientific, which manufactures and markets EXALT. The other two authors have consulted for Boston Scientific.
The EXALT Model-D single-use duodenoscope is a cost-effective alternative to high-level disinfection (HLD) of reusable duodenoscopes, according to a new analysis.
The study compared the EXALT Model-D, HLD, culture-and-quarantine (CQ), and ethylene oxide sterilization (ETO). The results came from a simulated cohort of patients undergoing endoscopic retrograde cholangiopancreatography (ERCP) to treat choledocholithiasis.
Although EXALT was the costliest option and HLD the cheapest, EXALT produced the most quality-adjusted life years (QALYs) and allowed the hospital to decrease net costs, and sensitivity analysis showed that it was a better option than HLD over a range of willingness-to-pay values.
“When evaluating technologies based on cost-effectiveness and additionally in the context of TPT [transitional passthrough] or NTAP [new technology add-on payment], the EXALT approach meets typically used cost-effectiveness thresholds compared to all other evaluated strategies and should be considered for standard practice,” wrote the authors, who were led by Ananya Das, MD, of the Arizona Centers for Digestive Health, Gilbert. The study was published in Techniques and Innovations in Gastrointestinal Endoscopy.
Duodenoscope contamination has resulted in outbreaks of various multidrug-resistant organisms in hospital settings, which has led to the publication of various reprocessing guidelines. Although many hospitals have adopted HLD protocols, others use additional or alternative reprocessing methods such as CQ or ETO. Despite these efforts, a recent Food and Drug Administration study found that 1.9%-22% of samples taken from duodenoscopes tested positive for bacteria of concern, such as pathogens. Those and other findings have led some to suggest that it would be best to move away from HLD, and instead employ sterilizable or disposable endoscopes.
In another study, The EXALT Model-D (Boston Scientific) had been shown to be a good alternative to standard reusable duodenoscopes.
The researchers used a Markov-model to determine the cost-effectiveness of EXALT Model-D against other approaches in a simulated cohort. They found that EXALT Model-D created the most QALYs (21.9265) at the highest cost ($3,000), and HLD the fewest QALYs (21.8938) at the lowest cost ($962). Compared with HLD, the incremental cost-effectiveness ratio (ICER) of EXALT was $62,185, and $38,461 for ETO gas sterilization. CQ was dominated, indicating that it had a higher cost but was not more effective than HLD.
The researchers conducted a subanalysis of ERCP and Medicare patients to consider the recently approved TPT payment and the NTAP, in both hospital outpatient and inpatient settings. With TPT, EXALT had no cost after reimbursement, with a net saving of $962 per patient when compared with HLD, plus an increase in 0.033 QALYs (0.15%). The other procedures cost more and were less effective. With NTAP, EXALT had a net cost of $323 versus HLD, with a similar QALY benefit.
A Monte Carlo analysis of EXALT versus HLD found reductions in duodenoscope infection-related ICU admission (relative risk reduction, 0.996; 95% confidence interval, 0.936-1.0; number needed to treat, 79; 95% CI, 67-95) and death (RRR, 0.973; 95% CI, 0.552-0.998; number needed to treat, 556; 95% CI, 350-997).
In willingness-to-pay estimates from $50,000 to $100,000, EXALT was cost effective in 67.28% of trials with ICER under $100,000 per QALY.
The study did not consider medicolegal costs, which could lead to an underestimation of EXALT’s cost-effectiveness. The study also relied on available published information to determine cost per patient of hospital outbreaks in the United States and Europe since 2012, but the authors did not include costs of administrative sanctions, litigation, and poor publicity due to inconsistencies in the literature.
“While more research is needed to understand and quantify the determinants of the natural history after exposure to contaminated duodenoscopes, such as the risk of transmission and the subsequent development of serious clinical infections, this economic analysis demonstrates an approach using EXALT Model-D is cost effective in the U.S. health care system when compared to the currently utilized strategies of duodenoscope reprocessing,” the researchers concluded.
The study did not receive any funding. One of the authors is an employee and stockholder of Boston Scientific, which manufactures and markets EXALT. The other two authors have consulted for Boston Scientific.
FROM TECHNIQUES AND INNOVATIONS IN GASTROINTESTINAL ENDOSCOPY
A common problem improved but not solved
Phoenix has only a few months each year to use my hot tub, so winter is when I catch up on a lot of my reading. Recently I was reading the November Lancet, which had some interesting statistics about migraine.
- It’s the second leading cause (behind back pain) of years lived with disability.
- There are 10 million people with migraines in the United Kingdom (population roughly 70 million).
- In the last 5 years, migraine use of emergency rooms has increased 14%.
- According to the U.K. National Health Service, over 16,000 ER visits for migraine could be avoided.
These are compelling statistics, and probably (taking into account population differences) similar to numbers here in the United States or Canada.
Like all neurologists, I see my share of migraine.
Like many neurologists, I also get migraines. Not many, maybe 2-3 per month, effectively treated with a triptan. So I have a decent understanding that they aren’t pleasant.
Fortunately, migraine advances have been impressive, with seven new CGRP drugs in the last 3 years, bringing successful treatment closer for many.
But the problem is far from solved, a point that was driven home yesterday.
I awoke early yesterday morning with a migraine, and took an Imitrex. But instead of feeling better in an hour, it kept worsening until I was literally disabled by it. I took some Excedrin Migraine. The last time I had a migraine this bad was in 1998, during my fellowship, and my attending had to drive me home (thanks, Joe).
It was showing no signs of letting up. I thought about going to emergency department. After all, aren’t we trained for that when we hear “worst headache of my life?” but figured it was more likely just a migraine, and didn’t want to bog down my ED colleagues in the midst of another COVID-19 wave.
I took another Imitrex. I found a sample of Ubrelvy that I’d brought home out of curiosity, and took that, too. I think I have an old, nearly empty, bottle of Norco, somewhere, from a 2014 dental surgery, but was too photophobic to go looking for it (if I still have it at all).
I lay down in bed under the ceiling fan, and somehow fell asleep.
When I woke about 90 minutes later it was gone, like a switch had been flipped. Maybe it was all, or just one of, the meds I’d taken. I’ll never know. I could now resume my regularly scheduled program.
The migraine had cost me 7 hours. Like most small business owners, I’m trying to get all the year-end paperwork wrapped up, in addition to reviewing cases, writing up reports, and spending time with my family. So none of that happened that Saturday morning. If I’d had to see patients that morning there’s no way I could have done it.
Fortunately, as I said, that’s only the second time that’s happened to me, and it’s been 25 years since the last one.
But I’m lucky. There are those who have them far more frequently, limiting their ability to work, raise families, spend time with friends. … Have a life.
Migraine is far from a deadly disease. In neurology we treat far worse conditions. But in sheer numbers migraine affects far more people, and (indirectly) an even larger group of coworkers, parents, friends, and children who have to cover unpredictably when the other person is out with one.
For all of them,
Phoenix has only a few months each year to use my hot tub, so winter is when I catch up on a lot of my reading. Recently I was reading the November Lancet, which had some interesting statistics about migraine.
- It’s the second leading cause (behind back pain) of years lived with disability.
- There are 10 million people with migraines in the United Kingdom (population roughly 70 million).
- In the last 5 years, migraine use of emergency rooms has increased 14%.
- According to the U.K. National Health Service, over 16,000 ER visits for migraine could be avoided.
These are compelling statistics, and probably (taking into account population differences) similar to numbers here in the United States or Canada.
Like all neurologists, I see my share of migraine.
Like many neurologists, I also get migraines. Not many, maybe 2-3 per month, effectively treated with a triptan. So I have a decent understanding that they aren’t pleasant.
Fortunately, migraine advances have been impressive, with seven new CGRP drugs in the last 3 years, bringing successful treatment closer for many.
But the problem is far from solved, a point that was driven home yesterday.
I awoke early yesterday morning with a migraine, and took an Imitrex. But instead of feeling better in an hour, it kept worsening until I was literally disabled by it. I took some Excedrin Migraine. The last time I had a migraine this bad was in 1998, during my fellowship, and my attending had to drive me home (thanks, Joe).
It was showing no signs of letting up. I thought about going to emergency department. After all, aren’t we trained for that when we hear “worst headache of my life?” but figured it was more likely just a migraine, and didn’t want to bog down my ED colleagues in the midst of another COVID-19 wave.
I took another Imitrex. I found a sample of Ubrelvy that I’d brought home out of curiosity, and took that, too. I think I have an old, nearly empty, bottle of Norco, somewhere, from a 2014 dental surgery, but was too photophobic to go looking for it (if I still have it at all).
I lay down in bed under the ceiling fan, and somehow fell asleep.
When I woke about 90 minutes later it was gone, like a switch had been flipped. Maybe it was all, or just one of, the meds I’d taken. I’ll never know. I could now resume my regularly scheduled program.
The migraine had cost me 7 hours. Like most small business owners, I’m trying to get all the year-end paperwork wrapped up, in addition to reviewing cases, writing up reports, and spending time with my family. So none of that happened that Saturday morning. If I’d had to see patients that morning there’s no way I could have done it.
Fortunately, as I said, that’s only the second time that’s happened to me, and it’s been 25 years since the last one.
But I’m lucky. There are those who have them far more frequently, limiting their ability to work, raise families, spend time with friends. … Have a life.
Migraine is far from a deadly disease. In neurology we treat far worse conditions. But in sheer numbers migraine affects far more people, and (indirectly) an even larger group of coworkers, parents, friends, and children who have to cover unpredictably when the other person is out with one.
For all of them,
Phoenix has only a few months each year to use my hot tub, so winter is when I catch up on a lot of my reading. Recently I was reading the November Lancet, which had some interesting statistics about migraine.
- It’s the second leading cause (behind back pain) of years lived with disability.
- There are 10 million people with migraines in the United Kingdom (population roughly 70 million).
- In the last 5 years, migraine use of emergency rooms has increased 14%.
- According to the U.K. National Health Service, over 16,000 ER visits for migraine could be avoided.
These are compelling statistics, and probably (taking into account population differences) similar to numbers here in the United States or Canada.
Like all neurologists, I see my share of migraine.
Like many neurologists, I also get migraines. Not many, maybe 2-3 per month, effectively treated with a triptan. So I have a decent understanding that they aren’t pleasant.
Fortunately, migraine advances have been impressive, with seven new CGRP drugs in the last 3 years, bringing successful treatment closer for many.
But the problem is far from solved, a point that was driven home yesterday.
I awoke early yesterday morning with a migraine, and took an Imitrex. But instead of feeling better in an hour, it kept worsening until I was literally disabled by it. I took some Excedrin Migraine. The last time I had a migraine this bad was in 1998, during my fellowship, and my attending had to drive me home (thanks, Joe).
It was showing no signs of letting up. I thought about going to emergency department. After all, aren’t we trained for that when we hear “worst headache of my life?” but figured it was more likely just a migraine, and didn’t want to bog down my ED colleagues in the midst of another COVID-19 wave.
I took another Imitrex. I found a sample of Ubrelvy that I’d brought home out of curiosity, and took that, too. I think I have an old, nearly empty, bottle of Norco, somewhere, from a 2014 dental surgery, but was too photophobic to go looking for it (if I still have it at all).
I lay down in bed under the ceiling fan, and somehow fell asleep.
When I woke about 90 minutes later it was gone, like a switch had been flipped. Maybe it was all, or just one of, the meds I’d taken. I’ll never know. I could now resume my regularly scheduled program.
The migraine had cost me 7 hours. Like most small business owners, I’m trying to get all the year-end paperwork wrapped up, in addition to reviewing cases, writing up reports, and spending time with my family. So none of that happened that Saturday morning. If I’d had to see patients that morning there’s no way I could have done it.
Fortunately, as I said, that’s only the second time that’s happened to me, and it’s been 25 years since the last one.
But I’m lucky. There are those who have them far more frequently, limiting their ability to work, raise families, spend time with friends. … Have a life.
Migraine is far from a deadly disease. In neurology we treat far worse conditions. But in sheer numbers migraine affects far more people, and (indirectly) an even larger group of coworkers, parents, friends, and children who have to cover unpredictably when the other person is out with one.
For all of them,
Is it safe to pair low-power fractional diode lasers with cosmetic injectables in a single session?
, results from a 6-year, single-center review showed.

“These treatments can be complementary in single-session treatments and can offer increased convenience for both patients and physicians,” primary study author Jordan V. Wang, MD, MBE, MBA, said during a virtual abstract session at the annual meeting of the American Society for Dermatologic Surgery.
To date, limited studies have demonstrated the safety of pairing botulinum neurotoxin type A and soft-tissue fillers with laser and other energy-based devices during the same treatment session on the same day, said Dr. Wang, medical research director at the Laser & Skin Surgery Center of New York. “Some concerns remain, though, regarding patient safety and efficacy,” he said. “Data on single-session treatment with low-power, low-density 1,927-nm and 1,440-nm fractional diode lasers and either botulinum neurotoxin or fillers are lacking.”
In a retrospective review of electronic medical records conducted from May 2015 to April 2021, Dr. Wang, Roy G. Geronemus, MD, and Carolyn Kushner, MD evaluated patients who received a single-session facial treatment with either BoNT-A or soft-tissue fillers and the low-power, low-density 1,927-nm and 1,440-nm fractional diode lasers (Clear+Brilliant Perméa and Original, Solta, Pleasanton, Calif.). Safety was assessed by documenting adverse events related to the spread of BoNT-A and fillers or laser treatment of filled areas within 4 weeks.
Adverse events they looked for related to botulinum neurotoxin use included eyelid ptosis; neck weakness or spasms; impairments in chewing, swallowing, speech, and respiration; and prescriptions of apraclonidine eye drops. Filler-related adverse events they looked for included product migration, unexpected loss of filler volume, vascular occlusion, acute pain, necrosis, blindness, and burn. “For both, we looked at hospital or emergency room transfers or admissions and referrals to ENT or ophthalmology,” Dr. Wang said.
During the 6-year study period, 525 patients had 1,562 single-session laser treatments with a mean 46.4 units of BoNT-A, and 398 patients had 1,237 single-session treatments with a mean 1.6 soft-tissue filler syringes. Among those who received BoNT-A, most (93%) were female, their mean age was 51 years, and 99% were treated with a 1,927-nm wavelength at a medium setting in 87% of cases. The top five injection sites were glabella (82%), forehead (69%), periorbital area (64%), neck (40%), and jawline and/or masseters (13%).
The researchers noted one case (0.06%) where apraclonidine eye drops were prescribed for ptosis. The patient had undergone eight other single-session treatments without issue. There were no other documented adverse events directly related to spread of BoNT-A. According to Dr. Wang, this rate of ptosis is lower than the incidence with BoNT-A alone in two landmark trials studying its effects on glabellar lines, which was reported as 5.4% and 1.0%.
Among the 398 patients who received soft-tissue fillers, most (94%) were female, their mean age was 54 years, and 99% were treated with a 1927nm wavelength at a medium setting in 97% of cases. The top five injection sites were cheeks and/or tear troughs (89%), perioral area and/or marionette lines (77%), lips (34%), nasolabial folds (19%), and temples (11%), and the mean number of filler syringes per treatment was 1.6. Slightly more than half (51%) had 1 session, while the remainder had 2 to greater than 10 sessions. The researchers observed no documented adverse events related to spread of fillers or laser treatment of filled areas.
“This laser is a low-powered device that creates small, superficial, and transient microchannels, which likely contributes to the safety of single-session treatments with cosmetic injectables,” Dr. Wang said. However, prospective studies are needed to further validate these results, he added.

“With this very mild laser, it is not surprising that combined treatment had no effect,” said Eric F. Bernstein, MD, MSE, director of the Main Line Center for Laser Surgery in Ardmore, Pa., who was asked to comment on the study results. “There have been numerous anecdotal reports of spreading of botulinum toxin effect to areas not in the target area for treatment following a variety of lasers, including the more powerful version of the laser used in this study. In addition, spread following vascular and other lasers has been reported,” he noted
The laser used in this study, Dr. Bernstein continued, “is low powered and emits a wavelength that is very superficially absorbed, resulting in injury to the stratum corneum, superficial epidermis, or possibly the very superficial dermis, and is often used by physician extenders and not physicians – although I suspect this is not the case in the current study. One can have a reasonable degree of confidence when combining this laser with injectables, but these results cannot be extrapolated to other devices.”
The abstract received the annual ASDS Carruthers Award during the meeting. Dr. Wang reported that he is a consultant or advisor to Allergan, Alastin, AVAVA, Cynosure, Lutronic, Novoxel, Sofwave, and Solta. Dr. Bernstein reported having received research funding from Cynosure, Candela, and Acclaro. He also has received consulting fees from Cynosure and holds ownership interest in Candela, Novoxel, OnSite, Joylux, and Acclaro and has served on the advisory board for Novoxel, Cynosure, and Acclaro.
, results from a 6-year, single-center review showed.
“These treatments can be complementary in single-session treatments and can offer increased convenience for both patients and physicians,” primary study author Jordan V. Wang, MD, MBE, MBA, said during a virtual abstract session at the annual meeting of the American Society for Dermatologic Surgery.
To date, limited studies have demonstrated the safety of pairing botulinum neurotoxin type A and soft-tissue fillers with laser and other energy-based devices during the same treatment session on the same day, said Dr. Wang, medical research director at the Laser & Skin Surgery Center of New York. “Some concerns remain, though, regarding patient safety and efficacy,” he said. “Data on single-session treatment with low-power, low-density 1,927-nm and 1,440-nm fractional diode lasers and either botulinum neurotoxin or fillers are lacking.”
In a retrospective review of electronic medical records conducted from May 2015 to April 2021, Dr. Wang, Roy G. Geronemus, MD, and Carolyn Kushner, MD evaluated patients who received a single-session facial treatment with either BoNT-A or soft-tissue fillers and the low-power, low-density 1,927-nm and 1,440-nm fractional diode lasers (Clear+Brilliant Perméa and Original, Solta, Pleasanton, Calif.). Safety was assessed by documenting adverse events related to the spread of BoNT-A and fillers or laser treatment of filled areas within 4 weeks.
Adverse events they looked for related to botulinum neurotoxin use included eyelid ptosis; neck weakness or spasms; impairments in chewing, swallowing, speech, and respiration; and prescriptions of apraclonidine eye drops. Filler-related adverse events they looked for included product migration, unexpected loss of filler volume, vascular occlusion, acute pain, necrosis, blindness, and burn. “For both, we looked at hospital or emergency room transfers or admissions and referrals to ENT or ophthalmology,” Dr. Wang said.
During the 6-year study period, 525 patients had 1,562 single-session laser treatments with a mean 46.4 units of BoNT-A, and 398 patients had 1,237 single-session treatments with a mean 1.6 soft-tissue filler syringes. Among those who received BoNT-A, most (93%) were female, their mean age was 51 years, and 99% were treated with a 1,927-nm wavelength at a medium setting in 87% of cases. The top five injection sites were glabella (82%), forehead (69%), periorbital area (64%), neck (40%), and jawline and/or masseters (13%).
The researchers noted one case (0.06%) where apraclonidine eye drops were prescribed for ptosis. The patient had undergone eight other single-session treatments without issue. There were no other documented adverse events directly related to spread of BoNT-A. According to Dr. Wang, this rate of ptosis is lower than the incidence with BoNT-A alone in two landmark trials studying its effects on glabellar lines, which was reported as 5.4% and 1.0%.
Among the 398 patients who received soft-tissue fillers, most (94%) were female, their mean age was 54 years, and 99% were treated with a 1927nm wavelength at a medium setting in 97% of cases. The top five injection sites were cheeks and/or tear troughs (89%), perioral area and/or marionette lines (77%), lips (34%), nasolabial folds (19%), and temples (11%), and the mean number of filler syringes per treatment was 1.6. Slightly more than half (51%) had 1 session, while the remainder had 2 to greater than 10 sessions. The researchers observed no documented adverse events related to spread of fillers or laser treatment of filled areas.
“This laser is a low-powered device that creates small, superficial, and transient microchannels, which likely contributes to the safety of single-session treatments with cosmetic injectables,” Dr. Wang said. However, prospective studies are needed to further validate these results, he added.
“With this very mild laser, it is not surprising that combined treatment had no effect,” said Eric F. Bernstein, MD, MSE, director of the Main Line Center for Laser Surgery in Ardmore, Pa., who was asked to comment on the study results. “There have been numerous anecdotal reports of spreading of botulinum toxin effect to areas not in the target area for treatment following a variety of lasers, including the more powerful version of the laser used in this study. In addition, spread following vascular and other lasers has been reported,” he noted
The laser used in this study, Dr. Bernstein continued, “is low powered and emits a wavelength that is very superficially absorbed, resulting in injury to the stratum corneum, superficial epidermis, or possibly the very superficial dermis, and is often used by physician extenders and not physicians – although I suspect this is not the case in the current study. One can have a reasonable degree of confidence when combining this laser with injectables, but these results cannot be extrapolated to other devices.”
The abstract received the annual ASDS Carruthers Award during the meeting. Dr. Wang reported that he is a consultant or advisor to Allergan, Alastin, AVAVA, Cynosure, Lutronic, Novoxel, Sofwave, and Solta. Dr. Bernstein reported having received research funding from Cynosure, Candela, and Acclaro. He also has received consulting fees from Cynosure and holds ownership interest in Candela, Novoxel, OnSite, Joylux, and Acclaro and has served on the advisory board for Novoxel, Cynosure, and Acclaro.
, results from a 6-year, single-center review showed.
“These treatments can be complementary in single-session treatments and can offer increased convenience for both patients and physicians,” primary study author Jordan V. Wang, MD, MBE, MBA, said during a virtual abstract session at the annual meeting of the American Society for Dermatologic Surgery.
To date, limited studies have demonstrated the safety of pairing botulinum neurotoxin type A and soft-tissue fillers with laser and other energy-based devices during the same treatment session on the same day, said Dr. Wang, medical research director at the Laser & Skin Surgery Center of New York. “Some concerns remain, though, regarding patient safety and efficacy,” he said. “Data on single-session treatment with low-power, low-density 1,927-nm and 1,440-nm fractional diode lasers and either botulinum neurotoxin or fillers are lacking.”
In a retrospective review of electronic medical records conducted from May 2015 to April 2021, Dr. Wang, Roy G. Geronemus, MD, and Carolyn Kushner, MD evaluated patients who received a single-session facial treatment with either BoNT-A or soft-tissue fillers and the low-power, low-density 1,927-nm and 1,440-nm fractional diode lasers (Clear+Brilliant Perméa and Original, Solta, Pleasanton, Calif.). Safety was assessed by documenting adverse events related to the spread of BoNT-A and fillers or laser treatment of filled areas within 4 weeks.
Adverse events they looked for related to botulinum neurotoxin use included eyelid ptosis; neck weakness or spasms; impairments in chewing, swallowing, speech, and respiration; and prescriptions of apraclonidine eye drops. Filler-related adverse events they looked for included product migration, unexpected loss of filler volume, vascular occlusion, acute pain, necrosis, blindness, and burn. “For both, we looked at hospital or emergency room transfers or admissions and referrals to ENT or ophthalmology,” Dr. Wang said.
During the 6-year study period, 525 patients had 1,562 single-session laser treatments with a mean 46.4 units of BoNT-A, and 398 patients had 1,237 single-session treatments with a mean 1.6 soft-tissue filler syringes. Among those who received BoNT-A, most (93%) were female, their mean age was 51 years, and 99% were treated with a 1,927-nm wavelength at a medium setting in 87% of cases. The top five injection sites were glabella (82%), forehead (69%), periorbital area (64%), neck (40%), and jawline and/or masseters (13%).
The researchers noted one case (0.06%) where apraclonidine eye drops were prescribed for ptosis. The patient had undergone eight other single-session treatments without issue. There were no other documented adverse events directly related to spread of BoNT-A. According to Dr. Wang, this rate of ptosis is lower than the incidence with BoNT-A alone in two landmark trials studying its effects on glabellar lines, which was reported as 5.4% and 1.0%.
Among the 398 patients who received soft-tissue fillers, most (94%) were female, their mean age was 54 years, and 99% were treated with a 1927nm wavelength at a medium setting in 97% of cases. The top five injection sites were cheeks and/or tear troughs (89%), perioral area and/or marionette lines (77%), lips (34%), nasolabial folds (19%), and temples (11%), and the mean number of filler syringes per treatment was 1.6. Slightly more than half (51%) had 1 session, while the remainder had 2 to greater than 10 sessions. The researchers observed no documented adverse events related to spread of fillers or laser treatment of filled areas.
“This laser is a low-powered device that creates small, superficial, and transient microchannels, which likely contributes to the safety of single-session treatments with cosmetic injectables,” Dr. Wang said. However, prospective studies are needed to further validate these results, he added.
“With this very mild laser, it is not surprising that combined treatment had no effect,” said Eric F. Bernstein, MD, MSE, director of the Main Line Center for Laser Surgery in Ardmore, Pa., who was asked to comment on the study results. “There have been numerous anecdotal reports of spreading of botulinum toxin effect to areas not in the target area for treatment following a variety of lasers, including the more powerful version of the laser used in this study. In addition, spread following vascular and other lasers has been reported,” he noted
The laser used in this study, Dr. Bernstein continued, “is low powered and emits a wavelength that is very superficially absorbed, resulting in injury to the stratum corneum, superficial epidermis, or possibly the very superficial dermis, and is often used by physician extenders and not physicians – although I suspect this is not the case in the current study. One can have a reasonable degree of confidence when combining this laser with injectables, but these results cannot be extrapolated to other devices.”
The abstract received the annual ASDS Carruthers Award during the meeting. Dr. Wang reported that he is a consultant or advisor to Allergan, Alastin, AVAVA, Cynosure, Lutronic, Novoxel, Sofwave, and Solta. Dr. Bernstein reported having received research funding from Cynosure, Candela, and Acclaro. He also has received consulting fees from Cynosure and holds ownership interest in Candela, Novoxel, OnSite, Joylux, and Acclaro and has served on the advisory board for Novoxel, Cynosure, and Acclaro.
FROM ASDS 2021
US Multi-Society Task Force lowers recommended CRC screening age
The U.S. Multi-Society Task Force on Colorectal Cancer (CRC) has lowered the recommended age to start CRC screening from 50 to 45 years of age for all average-risk individuals.
Although no studies have directly demonstrated the result of lowering the age of screening, lead author Swati G. Patel, MD, of University of Colorado Anschutz Medical Center, Aurora, and colleagues suggested that the increasing incidence of advanced CRC among younger individuals, coupled with the net benefit of screening, warrant a lower age threshold.
“Recent data ... show that CRC incidence rates in individuals ages 50 to 64 have increased by 1% annually between 2011 and 2016,” the authors wrote in Gastroenterology. “Similarly, CRC incidence and mortality rates in persons under age 50, termed early-age onset CRC (EAO-CRC), are also increasing.”
The task force of nine experts, representing the American Gastroenterological Association, the American College of Gastroenterology, and the American Society for Gastrointestinal Endoscopy, conducted a literature review and generated recommendations using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) criteria. In addition to recommending a lower age for initial screening, Dr. Patel and colleagues provided guidance for cessation of screening among older individuals.
Guidance for screening initiation
According to the authors, the present risk of CRC among younger individuals mirrors the historical risk for older individuals before screening was prevalent.
“The current CRC incidence rates in individuals ages 45 to 49 are similar to the incidence rates observed in 50-year-olds in 1992, before widespread CRC screening was performed,” they wrote.
Elevated rates among younger people have been disproportionately driven by rectal cancer, according to the authors. From 2006 to 2015, incidence of rectal cancer among Americans under 50 increased 1.7% per year, compared with 0.7% per year for colon cancer, based on data from the North American Association of Central Cancer Registries.
Associated mortality rates also increased, the authors noted. From 1999-2019, mortality from colon cancer among people 45-49 years increased from 6.4 to 6.6 deaths per 100,000 individuals, while deaths from rectal cancer increased from 1.3 to 1.7 per 100,000, according to the CDC. Concurrently, CRC-associated mortality rates among older individuals generally declined.
While these findings suggest a growing disease burden among the under-50-year age group, controlled data demonstrating the effects of earlier screening are lacking, Dr. Patel and colleagues noted. Still, they predicted that expanded screening would generate a net benefit.
“Although there are no CRC screening safety data for average-risk individuals [younger than] 50, there are ample data that colonoscopy for other indications (screening based on family history, symptom evaluation, etc.) is safer when comparing younger versus older individuals,” they wrote.
Supporting this claim, the authors cited three independently generated microsimulation models from the Agency for Healthcare Research and Quality that “showed a favorable balance of life-years gained compared with adverse events,” given 100% compliance.
Guidance for screening cessation
Like the situation with younger individuals, minimal data are available to determine the best time for screening cessation, according to the task force.
“There are no randomized or observational studies after 2017 that enrolled individuals over age 75 to inform the appropriate time to stop CRC screening,” the authors wrote. “In our search of 37 relevant articles, only one presented primary data for when to stop screening.”
This one available study showed that some individuals older than 74 do in fact gain benefit from screening,
“For example,” Dr. Patel and colleagues wrote, “women without a history of screening and no comorbidities benefitted from annual fecal immunochemical test (FIT) screening until age 90, whereas unscreened men with or without comorbidities benefited from annual FIT screening until age 88. Conversely, screening was not beneficial beyond age 66 in men or women with severe comorbidities.”
The task force therefore recommended personalized screening for individuals 76-85 years of age “based on the balance of benefits and harms and individual patient clinical factors and preferences.”
Screening for individuals 86 years and older, according to the task force, is unnecessary.
The authors disclosed relationships with Olympus America, Bayer Pharmaceuticals, Janssen Pharmaceuticals, and others.
This article was updated on Jan. 3, 2022.
The U.S. Multi-Society Task Force on Colorectal Cancer (CRC) has lowered the recommended age to start CRC screening from 50 to 45 years of age for all average-risk individuals.
Although no studies have directly demonstrated the result of lowering the age of screening, lead author Swati G. Patel, MD, of University of Colorado Anschutz Medical Center, Aurora, and colleagues suggested that the increasing incidence of advanced CRC among younger individuals, coupled with the net benefit of screening, warrant a lower age threshold.
“Recent data ... show that CRC incidence rates in individuals ages 50 to 64 have increased by 1% annually between 2011 and 2016,” the authors wrote in Gastroenterology. “Similarly, CRC incidence and mortality rates in persons under age 50, termed early-age onset CRC (EAO-CRC), are also increasing.”
The task force of nine experts, representing the American Gastroenterological Association, the American College of Gastroenterology, and the American Society for Gastrointestinal Endoscopy, conducted a literature review and generated recommendations using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) criteria. In addition to recommending a lower age for initial screening, Dr. Patel and colleagues provided guidance for cessation of screening among older individuals.
Guidance for screening initiation
According to the authors, the present risk of CRC among younger individuals mirrors the historical risk for older individuals before screening was prevalent.
“The current CRC incidence rates in individuals ages 45 to 49 are similar to the incidence rates observed in 50-year-olds in 1992, before widespread CRC screening was performed,” they wrote.
Elevated rates among younger people have been disproportionately driven by rectal cancer, according to the authors. From 2006 to 2015, incidence of rectal cancer among Americans under 50 increased 1.7% per year, compared with 0.7% per year for colon cancer, based on data from the North American Association of Central Cancer Registries.
Associated mortality rates also increased, the authors noted. From 1999-2019, mortality from colon cancer among people 45-49 years increased from 6.4 to 6.6 deaths per 100,000 individuals, while deaths from rectal cancer increased from 1.3 to 1.7 per 100,000, according to the CDC. Concurrently, CRC-associated mortality rates among older individuals generally declined.
While these findings suggest a growing disease burden among the under-50-year age group, controlled data demonstrating the effects of earlier screening are lacking, Dr. Patel and colleagues noted. Still, they predicted that expanded screening would generate a net benefit.
“Although there are no CRC screening safety data for average-risk individuals [younger than] 50, there are ample data that colonoscopy for other indications (screening based on family history, symptom evaluation, etc.) is safer when comparing younger versus older individuals,” they wrote.
Supporting this claim, the authors cited three independently generated microsimulation models from the Agency for Healthcare Research and Quality that “showed a favorable balance of life-years gained compared with adverse events,” given 100% compliance.
Guidance for screening cessation
Like the situation with younger individuals, minimal data are available to determine the best time for screening cessation, according to the task force.
“There are no randomized or observational studies after 2017 that enrolled individuals over age 75 to inform the appropriate time to stop CRC screening,” the authors wrote. “In our search of 37 relevant articles, only one presented primary data for when to stop screening.”
This one available study showed that some individuals older than 74 do in fact gain benefit from screening,
“For example,” Dr. Patel and colleagues wrote, “women without a history of screening and no comorbidities benefitted from annual fecal immunochemical test (FIT) screening until age 90, whereas unscreened men with or without comorbidities benefited from annual FIT screening until age 88. Conversely, screening was not beneficial beyond age 66 in men or women with severe comorbidities.”
The task force therefore recommended personalized screening for individuals 76-85 years of age “based on the balance of benefits and harms and individual patient clinical factors and preferences.”
Screening for individuals 86 years and older, according to the task force, is unnecessary.
The authors disclosed relationships with Olympus America, Bayer Pharmaceuticals, Janssen Pharmaceuticals, and others.
This article was updated on Jan. 3, 2022.
The U.S. Multi-Society Task Force on Colorectal Cancer (CRC) has lowered the recommended age to start CRC screening from 50 to 45 years of age for all average-risk individuals.
Although no studies have directly demonstrated the result of lowering the age of screening, lead author Swati G. Patel, MD, of University of Colorado Anschutz Medical Center, Aurora, and colleagues suggested that the increasing incidence of advanced CRC among younger individuals, coupled with the net benefit of screening, warrant a lower age threshold.
“Recent data ... show that CRC incidence rates in individuals ages 50 to 64 have increased by 1% annually between 2011 and 2016,” the authors wrote in Gastroenterology. “Similarly, CRC incidence and mortality rates in persons under age 50, termed early-age onset CRC (EAO-CRC), are also increasing.”
The task force of nine experts, representing the American Gastroenterological Association, the American College of Gastroenterology, and the American Society for Gastrointestinal Endoscopy, conducted a literature review and generated recommendations using the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) criteria. In addition to recommending a lower age for initial screening, Dr. Patel and colleagues provided guidance for cessation of screening among older individuals.
Guidance for screening initiation
According to the authors, the present risk of CRC among younger individuals mirrors the historical risk for older individuals before screening was prevalent.
“The current CRC incidence rates in individuals ages 45 to 49 are similar to the incidence rates observed in 50-year-olds in 1992, before widespread CRC screening was performed,” they wrote.
Elevated rates among younger people have been disproportionately driven by rectal cancer, according to the authors. From 2006 to 2015, incidence of rectal cancer among Americans under 50 increased 1.7% per year, compared with 0.7% per year for colon cancer, based on data from the North American Association of Central Cancer Registries.
Associated mortality rates also increased, the authors noted. From 1999-2019, mortality from colon cancer among people 45-49 years increased from 6.4 to 6.6 deaths per 100,000 individuals, while deaths from rectal cancer increased from 1.3 to 1.7 per 100,000, according to the CDC. Concurrently, CRC-associated mortality rates among older individuals generally declined.
While these findings suggest a growing disease burden among the under-50-year age group, controlled data demonstrating the effects of earlier screening are lacking, Dr. Patel and colleagues noted. Still, they predicted that expanded screening would generate a net benefit.
“Although there are no CRC screening safety data for average-risk individuals [younger than] 50, there are ample data that colonoscopy for other indications (screening based on family history, symptom evaluation, etc.) is safer when comparing younger versus older individuals,” they wrote.
Supporting this claim, the authors cited three independently generated microsimulation models from the Agency for Healthcare Research and Quality that “showed a favorable balance of life-years gained compared with adverse events,” given 100% compliance.
Guidance for screening cessation
Like the situation with younger individuals, minimal data are available to determine the best time for screening cessation, according to the task force.
“There are no randomized or observational studies after 2017 that enrolled individuals over age 75 to inform the appropriate time to stop CRC screening,” the authors wrote. “In our search of 37 relevant articles, only one presented primary data for when to stop screening.”
This one available study showed that some individuals older than 74 do in fact gain benefit from screening,
“For example,” Dr. Patel and colleagues wrote, “women without a history of screening and no comorbidities benefitted from annual fecal immunochemical test (FIT) screening until age 90, whereas unscreened men with or without comorbidities benefited from annual FIT screening until age 88. Conversely, screening was not beneficial beyond age 66 in men or women with severe comorbidities.”
The task force therefore recommended personalized screening for individuals 76-85 years of age “based on the balance of benefits and harms and individual patient clinical factors and preferences.”
Screening for individuals 86 years and older, according to the task force, is unnecessary.
The authors disclosed relationships with Olympus America, Bayer Pharmaceuticals, Janssen Pharmaceuticals, and others.
This article was updated on Jan. 3, 2022.
FROM GASTROENTEROLOGY