User login
Hernia recurrence has improved only slightly
, according to a new research letter published March 1 in JAMA. Patients who underwent minimally invasive hernia repair had a higher incidence of reoperation than those who underwent open repairs.
In the United States, surgeons perform more than 1 million hernia repairs each year, according to the U.S. Food and Drug Administration. Despite hernias being such a common condition, it is “not at the forefront of many research agendas,” senior author Dana Telem, MD, an associate professor and section chief of general surgery at University of Michigan Health in Ann Arbor, said in an interview
While many surgical outcomes are measured within 30 days of operation, recurrences generally happen within 2 to 5 years after repair, she said. The last study that looked at reoperations for hernia repair at 10 years was published in 2003 and found that about 20% of patients needed surgery for reoccurrence over a decade. “We don’t really have a good understanding of what happened after these operations,” she explained. “Without knowing that piece, it is hard to go back retrospectively and understand what is the right operation for the right person at the right time.”
To understand rates of reoperation for hernia reoccurrence in today’s U.S. population of older adults, Dr. Telem and colleagues sorted through Medicare claims data to find adult patients who had undergone ventral or incisional and umbilical hernia repair from January 1, 2007 through December 31, 2018. They identified a total of 175,735 patients, 162,292 that underwent ventral or incisional hernia repair and 13,443 that underwent umbilical hernia repair. The average age of patients was 68.9 years and 39.2% were men. Most patients were White (87.2%), 8.1% were Black, 1.9% were Hispanic, and 0.5% were Asian. Median follow-up was 5.3 years.
Over the 10-year study period, 25,061 patients required reoperation for hernia recurrence with an adjusted cumulative incidence of 16.1% (95% CI, 16.1% - 16.2%). Patients who underwent open repair had a lower incidence of recurrence over 10 years than those who underwent minimally invasive repair for all hernia types (Table 1).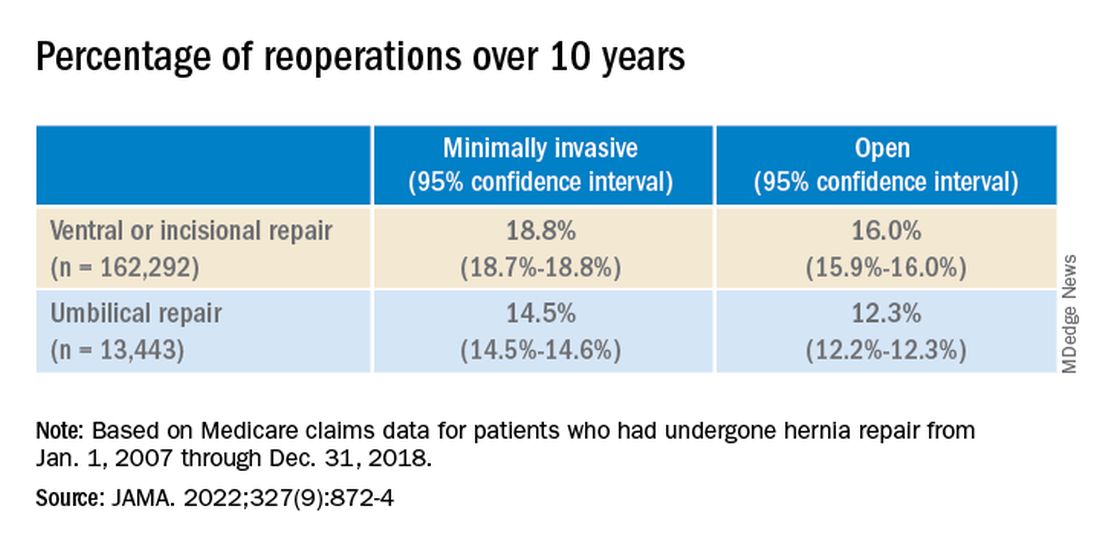
While it appears that hernia recurrence and reoperation have only marginally improved from 2003 to today, Vedra Augenstein, MD, an associate professor of surgery at the Atrium Health General & Complex Abdominal Surgery facility in Charlotte, N.C., suspects there is more to the story. “I think the reason it hasn’t gotten a whole lot better is just because we are operating on much tougher cases than we used to,” she said in an interview. “The way we are fixing hernias has changed and patients are being optimized differently.” Dr. Augenstein was not involved with the research.
To better understand how recurrence has changed over time, there needs to be more data about the comorbidities of patients, the techniques employed, and the meshes used in these surgeries, she said. Those numbers are not available in the published JAMA research letter, but Dr. Telem and colleagues will be submitting an article about this work with greater details.
Dr. Augenstein was also surprised that minimally invasive surgeries had higher incidences of reoperation for recurrence compared to open hernia surgeries. “I would think that patients who had minimally invasive repairs would actually have a lower chance of having postoperative complications because of wound issues,” she said. “Literature has shown that the recurrence rate is lower [in minimally invasive surgeries] because of fewer surgical site infections.”
While Dr. Telem also considers this research letter to be the first step in understanding modern hernia surgery outcomes, it is also a reminder that there is room for improvement in hernia repair surgeries. This includes advising patients on risk factors that may make them more likely to have a hernia recurrence, such as obesity, smoking, and diabetes, she added. “If we know it’s not a perfect science, then we have to do everything that we can upfront to help those numbers.”
Dr. Telem has reported receiving grants from the Agency for Healthcare Research and Quality and consulting fees from Medtronic. Dr. Augenstein has reported consulting for Intuitive Surgical, Medtronic, Allergan, Acelity, Vicarious Surgical, and Bard Pharmaceuticals and has received honoraria for speaking from Medtronic, Allergan, Intuitive Surgical, Acelity, and WL Gore.
A version of this article first appeared on Medscape.com.
, according to a new research letter published March 1 in JAMA. Patients who underwent minimally invasive hernia repair had a higher incidence of reoperation than those who underwent open repairs.
In the United States, surgeons perform more than 1 million hernia repairs each year, according to the U.S. Food and Drug Administration. Despite hernias being such a common condition, it is “not at the forefront of many research agendas,” senior author Dana Telem, MD, an associate professor and section chief of general surgery at University of Michigan Health in Ann Arbor, said in an interview
While many surgical outcomes are measured within 30 days of operation, recurrences generally happen within 2 to 5 years after repair, she said. The last study that looked at reoperations for hernia repair at 10 years was published in 2003 and found that about 20% of patients needed surgery for reoccurrence over a decade. “We don’t really have a good understanding of what happened after these operations,” she explained. “Without knowing that piece, it is hard to go back retrospectively and understand what is the right operation for the right person at the right time.”
To understand rates of reoperation for hernia reoccurrence in today’s U.S. population of older adults, Dr. Telem and colleagues sorted through Medicare claims data to find adult patients who had undergone ventral or incisional and umbilical hernia repair from January 1, 2007 through December 31, 2018. They identified a total of 175,735 patients, 162,292 that underwent ventral or incisional hernia repair and 13,443 that underwent umbilical hernia repair. The average age of patients was 68.9 years and 39.2% were men. Most patients were White (87.2%), 8.1% were Black, 1.9% were Hispanic, and 0.5% were Asian. Median follow-up was 5.3 years.
Over the 10-year study period, 25,061 patients required reoperation for hernia recurrence with an adjusted cumulative incidence of 16.1% (95% CI, 16.1% - 16.2%). Patients who underwent open repair had a lower incidence of recurrence over 10 years than those who underwent minimally invasive repair for all hernia types (Table 1).
While it appears that hernia recurrence and reoperation have only marginally improved from 2003 to today, Vedra Augenstein, MD, an associate professor of surgery at the Atrium Health General & Complex Abdominal Surgery facility in Charlotte, N.C., suspects there is more to the story. “I think the reason it hasn’t gotten a whole lot better is just because we are operating on much tougher cases than we used to,” she said in an interview. “The way we are fixing hernias has changed and patients are being optimized differently.” Dr. Augenstein was not involved with the research.
To better understand how recurrence has changed over time, there needs to be more data about the comorbidities of patients, the techniques employed, and the meshes used in these surgeries, she said. Those numbers are not available in the published JAMA research letter, but Dr. Telem and colleagues will be submitting an article about this work with greater details.
Dr. Augenstein was also surprised that minimally invasive surgeries had higher incidences of reoperation for recurrence compared to open hernia surgeries. “I would think that patients who had minimally invasive repairs would actually have a lower chance of having postoperative complications because of wound issues,” she said. “Literature has shown that the recurrence rate is lower [in minimally invasive surgeries] because of fewer surgical site infections.”
While Dr. Telem also considers this research letter to be the first step in understanding modern hernia surgery outcomes, it is also a reminder that there is room for improvement in hernia repair surgeries. This includes advising patients on risk factors that may make them more likely to have a hernia recurrence, such as obesity, smoking, and diabetes, she added. “If we know it’s not a perfect science, then we have to do everything that we can upfront to help those numbers.”
Dr. Telem has reported receiving grants from the Agency for Healthcare Research and Quality and consulting fees from Medtronic. Dr. Augenstein has reported consulting for Intuitive Surgical, Medtronic, Allergan, Acelity, Vicarious Surgical, and Bard Pharmaceuticals and has received honoraria for speaking from Medtronic, Allergan, Intuitive Surgical, Acelity, and WL Gore.
A version of this article first appeared on Medscape.com.
, according to a new research letter published March 1 in JAMA. Patients who underwent minimally invasive hernia repair had a higher incidence of reoperation than those who underwent open repairs.
In the United States, surgeons perform more than 1 million hernia repairs each year, according to the U.S. Food and Drug Administration. Despite hernias being such a common condition, it is “not at the forefront of many research agendas,” senior author Dana Telem, MD, an associate professor and section chief of general surgery at University of Michigan Health in Ann Arbor, said in an interview
While many surgical outcomes are measured within 30 days of operation, recurrences generally happen within 2 to 5 years after repair, she said. The last study that looked at reoperations for hernia repair at 10 years was published in 2003 and found that about 20% of patients needed surgery for reoccurrence over a decade. “We don’t really have a good understanding of what happened after these operations,” she explained. “Without knowing that piece, it is hard to go back retrospectively and understand what is the right operation for the right person at the right time.”
To understand rates of reoperation for hernia reoccurrence in today’s U.S. population of older adults, Dr. Telem and colleagues sorted through Medicare claims data to find adult patients who had undergone ventral or incisional and umbilical hernia repair from January 1, 2007 through December 31, 2018. They identified a total of 175,735 patients, 162,292 that underwent ventral or incisional hernia repair and 13,443 that underwent umbilical hernia repair. The average age of patients was 68.9 years and 39.2% were men. Most patients were White (87.2%), 8.1% were Black, 1.9% were Hispanic, and 0.5% were Asian. Median follow-up was 5.3 years.
Over the 10-year study period, 25,061 patients required reoperation for hernia recurrence with an adjusted cumulative incidence of 16.1% (95% CI, 16.1% - 16.2%). Patients who underwent open repair had a lower incidence of recurrence over 10 years than those who underwent minimally invasive repair for all hernia types (Table 1).
While it appears that hernia recurrence and reoperation have only marginally improved from 2003 to today, Vedra Augenstein, MD, an associate professor of surgery at the Atrium Health General & Complex Abdominal Surgery facility in Charlotte, N.C., suspects there is more to the story. “I think the reason it hasn’t gotten a whole lot better is just because we are operating on much tougher cases than we used to,” she said in an interview. “The way we are fixing hernias has changed and patients are being optimized differently.” Dr. Augenstein was not involved with the research.
To better understand how recurrence has changed over time, there needs to be more data about the comorbidities of patients, the techniques employed, and the meshes used in these surgeries, she said. Those numbers are not available in the published JAMA research letter, but Dr. Telem and colleagues will be submitting an article about this work with greater details.
Dr. Augenstein was also surprised that minimally invasive surgeries had higher incidences of reoperation for recurrence compared to open hernia surgeries. “I would think that patients who had minimally invasive repairs would actually have a lower chance of having postoperative complications because of wound issues,” she said. “Literature has shown that the recurrence rate is lower [in minimally invasive surgeries] because of fewer surgical site infections.”
While Dr. Telem also considers this research letter to be the first step in understanding modern hernia surgery outcomes, it is also a reminder that there is room for improvement in hernia repair surgeries. This includes advising patients on risk factors that may make them more likely to have a hernia recurrence, such as obesity, smoking, and diabetes, she added. “If we know it’s not a perfect science, then we have to do everything that we can upfront to help those numbers.”
Dr. Telem has reported receiving grants from the Agency for Healthcare Research and Quality and consulting fees from Medtronic. Dr. Augenstein has reported consulting for Intuitive Surgical, Medtronic, Allergan, Acelity, Vicarious Surgical, and Bard Pharmaceuticals and has received honoraria for speaking from Medtronic, Allergan, Intuitive Surgical, Acelity, and WL Gore.
A version of this article first appeared on Medscape.com.
FROM JAMA
Artificial intelligence aids assessment of UC activity, remission
Not only are artificial intelligence (AI) systems potentially highly accurate for assessment of disease activity and remission of ulcerative colitis (UC), but they can mitigate some limits of human assessment, according to presentations at the 17th congress of the European Crohn’s and Colitis Organisation.
Importantly, AI systems have the potential to supplement the services of expert histopathologists and endoscopists rather than replace them, several experts asserted at the meeting.
“We will always need pathologists,” reassured inflammatory bowel disease (IBD) specialist Laurent Peyrin-Biroulet, MD, PhD, of Nancy (France) University Hospital, who presented about the use of an AI-driven scoring system to measure histological disease activity in UC.
Dr. Peyrin-Biroulet, who is the president of ECCO and acts as the scientific secretary of the International Organization for the Study of IBD, added that the use of AI systems could mean that pathologists have more time to do other tasks. Not only that, but it’s also not always possible to have IBD pathologist in every center, everywhere in the world.
“If we can get something that will automatically evaluate the disease activity, I think it will be something fantastic,” Dr. Peyrin-Biroulet said, “and it’s the reason why we were thinking that there is a need for an automated method to measure histological activity in UC.”
Old concept enhancing current practice
The idea of using AI systems to aid diagnostics is not new but now makes even more sense in the post–COVID-19 era, suggested Aaron F. Pollett, MD, MSc, FRCPC, codirector of the division of diagnostic medical genetics at Mount Sinai Hospital in Toronto and a pathologist with a specialty interest in gastrointestinal pathology.
“When we talk about artificial intelligence and histology, there’s actually a very long history, it goes back over 30 years,” Dr. Pollett said, from assessing cervical samples to its use in breast screening.
What seems to be sudden flurry of activity in the world of AI and pathology in recent years comes down to having a higher capacity for looking at large images, having access to large data sets, and having a high amount of computing power, Dr. Pollet inferred. Moreover, “the capacity and the need for whole slide imaging has really grown especially in the last few years as the pandemic has forced centers to adopt.” The need to work remotely and flexibly across centers and the number of available pathologists have also played a role.
AI systems that use image-based retrieval systems are making good headway in IBD, particularly in the diagnosis of UC where “some of the initial research is showing it can be quite good,” said Dr. Pollett. The “patchiness that Crohn’s can have in comparison to UC” means that it’s still an emerging area, but can perhaps be useful for more questionable cases in which “having that degree of certainty can certainly help because there is a discrepancy between specialist and nonspecialist pathologists in the likelihood that what they predict on the biopsy will be the underlying disease.”
AI systems in IBD – do they work?
Histopathology is becoming increasingly integrated into IBD clinical trial design at the behest of the Food and Drug Administration and European associations such as ECCO. This can be a tedious procedure that can be prone to error and disagreement between scorers.
The AI-driven scoring system that Dr. Peyrin-Biroulet and associates have been working on aims to fix all that by using machine learning and image processing to set up a reproducible system. Their system, which is based on the Nancy histological index for UC, shows high correlation (87%) with histopathologists’ assessment and was 100% accurate in identifying images with high (grade 4) or no (grade 0) inflammatory activity. The accuracy decreased, however, when trying to distinguish between more moderate activity, with a 75% accuracy for identifying grade 3 and 82% accuracy for grades 1 or 2.
“I’m actually very fascinated to see how we can be supported by the AI work in our practice,” observed Francesca Rosini, a histopathologist working at S. Orsola–Malpighi University Hospital in Bologna, Italy.
Dr. Rosini, who chaired the digital oral presentation session in which Dr. Peyrin-Biroulet had presented also noted that “obviously for us as well [as AI systems] no activity or severe activity is the easiest part but when it’s in between that’s where the problems come.”
Simplifying histological scoring
Simplifying scoring for use in AI systems could be the key to their future success, as Tommaso Lorenzo Parigi, MD, from Humanitas University in Milan, and a research fellow at the University of Birmingham (England), suggested.
“Histology is particularly important to distinguish between mild activity and remission,” Dr. Parigi said. “More than 30 histological scores that have been proposed, but their adoption in clinical practice remains limited.”
Dr. Parigi has been part of an international team that has developed a simplified histological score based on “the presence of absence of neutrophils, regardless of their number,” since these are “key determinants of disease activity”.
The score, known as the Paddington International Virtual Chromoendoscopy Scre (PICaSSO) Histologic Remission Index (PHRI), has been shown to correlate well with endoscopic outcomes and thus a good measure to include in AI systems. The results of this work were published online in Gut to coincide with the ECCO congress.
“We are getting close to a world where we could screen biopsies with this kind of systems and consider skipping the pathologists result if AI detected activity,” Dr. Parigi provocatively suggested. “Of course, we need to increase and improve our sensitivity, and we are currently working on that to reduce false negatives, as well as training our model to use and apply other histological scores.”
Assessing the gut in real time
Perhaps one of the most exciting developments it to be able to use these AI technologies to examine the gut in real time.
“Virtual chromoendoscopy will give you the opportunity to distinguish very carefully all the details of mucosal vascular pattern,” said Marietta Iacucci MD, PhD, FASGE, AGAF, an associate professor and gastroenterology consultant at the Birmingham (England) University Hospitals.
“So AI can give you, in real time, the score but at the same time it can help to target, to do biopsies for healing,” Dr. Iacucci added when reporting the results of a study evaluating the performance of the first virtual chromoendoscopy AI system to detect endoscopic and histologic remission in UC.
The system was proven to predict endoscopic remission very accurately (94% using PICaSSO and 87% using the UC endoscopic index of severity) when compared with a human endoscopist. Rates of predicting histological remission were also high, at around 83%-85%, depending on the score used.
“For the future, this AI tool can expediate, support, and standardize the endoscopic evaluation of UC mucosal healing in clinical practice and in clinical trials,” Dr. Iacucci said.
The next steps are to combine virtual chromoendoscopy with the PHRI and to validate the tool in a multicenter, international PICaSSO-AI study.
The AI-driven scoring system presented by Dr. Peyrin-Biroulet was supported by Takeda. Dr. Peryin-Biroulet acknowledged the receipt of personal fees and grants from Takeda along with multiple other Pharma companies and owning stock options from CTMA. Dr. Iacucci has received research grants from Pentax, AbbVie, Olympus, and Fujifilm and personal fees from Pentax, AbbVie and Janssen. Dr. Pollett, Dr. Rosini, and Dr. Parigi had no financial conflicts of interest to disclose.
Not only are artificial intelligence (AI) systems potentially highly accurate for assessment of disease activity and remission of ulcerative colitis (UC), but they can mitigate some limits of human assessment, according to presentations at the 17th congress of the European Crohn’s and Colitis Organisation.
Importantly, AI systems have the potential to supplement the services of expert histopathologists and endoscopists rather than replace them, several experts asserted at the meeting.
“We will always need pathologists,” reassured inflammatory bowel disease (IBD) specialist Laurent Peyrin-Biroulet, MD, PhD, of Nancy (France) University Hospital, who presented about the use of an AI-driven scoring system to measure histological disease activity in UC.
Dr. Peyrin-Biroulet, who is the president of ECCO and acts as the scientific secretary of the International Organization for the Study of IBD, added that the use of AI systems could mean that pathologists have more time to do other tasks. Not only that, but it’s also not always possible to have IBD pathologist in every center, everywhere in the world.
“If we can get something that will automatically evaluate the disease activity, I think it will be something fantastic,” Dr. Peyrin-Biroulet said, “and it’s the reason why we were thinking that there is a need for an automated method to measure histological activity in UC.”
Old concept enhancing current practice
The idea of using AI systems to aid diagnostics is not new but now makes even more sense in the post–COVID-19 era, suggested Aaron F. Pollett, MD, MSc, FRCPC, codirector of the division of diagnostic medical genetics at Mount Sinai Hospital in Toronto and a pathologist with a specialty interest in gastrointestinal pathology.
“When we talk about artificial intelligence and histology, there’s actually a very long history, it goes back over 30 years,” Dr. Pollett said, from assessing cervical samples to its use in breast screening.
What seems to be sudden flurry of activity in the world of AI and pathology in recent years comes down to having a higher capacity for looking at large images, having access to large data sets, and having a high amount of computing power, Dr. Pollet inferred. Moreover, “the capacity and the need for whole slide imaging has really grown especially in the last few years as the pandemic has forced centers to adopt.” The need to work remotely and flexibly across centers and the number of available pathologists have also played a role.
AI systems that use image-based retrieval systems are making good headway in IBD, particularly in the diagnosis of UC where “some of the initial research is showing it can be quite good,” said Dr. Pollett. The “patchiness that Crohn’s can have in comparison to UC” means that it’s still an emerging area, but can perhaps be useful for more questionable cases in which “having that degree of certainty can certainly help because there is a discrepancy between specialist and nonspecialist pathologists in the likelihood that what they predict on the biopsy will be the underlying disease.”
AI systems in IBD – do they work?
Histopathology is becoming increasingly integrated into IBD clinical trial design at the behest of the Food and Drug Administration and European associations such as ECCO. This can be a tedious procedure that can be prone to error and disagreement between scorers.
The AI-driven scoring system that Dr. Peyrin-Biroulet and associates have been working on aims to fix all that by using machine learning and image processing to set up a reproducible system. Their system, which is based on the Nancy histological index for UC, shows high correlation (87%) with histopathologists’ assessment and was 100% accurate in identifying images with high (grade 4) or no (grade 0) inflammatory activity. The accuracy decreased, however, when trying to distinguish between more moderate activity, with a 75% accuracy for identifying grade 3 and 82% accuracy for grades 1 or 2.
“I’m actually very fascinated to see how we can be supported by the AI work in our practice,” observed Francesca Rosini, a histopathologist working at S. Orsola–Malpighi University Hospital in Bologna, Italy.
Dr. Rosini, who chaired the digital oral presentation session in which Dr. Peyrin-Biroulet had presented also noted that “obviously for us as well [as AI systems] no activity or severe activity is the easiest part but when it’s in between that’s where the problems come.”
Simplifying histological scoring
Simplifying scoring for use in AI systems could be the key to their future success, as Tommaso Lorenzo Parigi, MD, from Humanitas University in Milan, and a research fellow at the University of Birmingham (England), suggested.
“Histology is particularly important to distinguish between mild activity and remission,” Dr. Parigi said. “More than 30 histological scores that have been proposed, but their adoption in clinical practice remains limited.”
Dr. Parigi has been part of an international team that has developed a simplified histological score based on “the presence of absence of neutrophils, regardless of their number,” since these are “key determinants of disease activity”.
The score, known as the Paddington International Virtual Chromoendoscopy Scre (PICaSSO) Histologic Remission Index (PHRI), has been shown to correlate well with endoscopic outcomes and thus a good measure to include in AI systems. The results of this work were published online in Gut to coincide with the ECCO congress.
“We are getting close to a world where we could screen biopsies with this kind of systems and consider skipping the pathologists result if AI detected activity,” Dr. Parigi provocatively suggested. “Of course, we need to increase and improve our sensitivity, and we are currently working on that to reduce false negatives, as well as training our model to use and apply other histological scores.”
Assessing the gut in real time
Perhaps one of the most exciting developments it to be able to use these AI technologies to examine the gut in real time.
“Virtual chromoendoscopy will give you the opportunity to distinguish very carefully all the details of mucosal vascular pattern,” said Marietta Iacucci MD, PhD, FASGE, AGAF, an associate professor and gastroenterology consultant at the Birmingham (England) University Hospitals.
“So AI can give you, in real time, the score but at the same time it can help to target, to do biopsies for healing,” Dr. Iacucci added when reporting the results of a study evaluating the performance of the first virtual chromoendoscopy AI system to detect endoscopic and histologic remission in UC.
The system was proven to predict endoscopic remission very accurately (94% using PICaSSO and 87% using the UC endoscopic index of severity) when compared with a human endoscopist. Rates of predicting histological remission were also high, at around 83%-85%, depending on the score used.
“For the future, this AI tool can expediate, support, and standardize the endoscopic evaluation of UC mucosal healing in clinical practice and in clinical trials,” Dr. Iacucci said.
The next steps are to combine virtual chromoendoscopy with the PHRI and to validate the tool in a multicenter, international PICaSSO-AI study.
The AI-driven scoring system presented by Dr. Peyrin-Biroulet was supported by Takeda. Dr. Peryin-Biroulet acknowledged the receipt of personal fees and grants from Takeda along with multiple other Pharma companies and owning stock options from CTMA. Dr. Iacucci has received research grants from Pentax, AbbVie, Olympus, and Fujifilm and personal fees from Pentax, AbbVie and Janssen. Dr. Pollett, Dr. Rosini, and Dr. Parigi had no financial conflicts of interest to disclose.
Not only are artificial intelligence (AI) systems potentially highly accurate for assessment of disease activity and remission of ulcerative colitis (UC), but they can mitigate some limits of human assessment, according to presentations at the 17th congress of the European Crohn’s and Colitis Organisation.
Importantly, AI systems have the potential to supplement the services of expert histopathologists and endoscopists rather than replace them, several experts asserted at the meeting.
“We will always need pathologists,” reassured inflammatory bowel disease (IBD) specialist Laurent Peyrin-Biroulet, MD, PhD, of Nancy (France) University Hospital, who presented about the use of an AI-driven scoring system to measure histological disease activity in UC.
Dr. Peyrin-Biroulet, who is the president of ECCO and acts as the scientific secretary of the International Organization for the Study of IBD, added that the use of AI systems could mean that pathologists have more time to do other tasks. Not only that, but it’s also not always possible to have IBD pathologist in every center, everywhere in the world.
“If we can get something that will automatically evaluate the disease activity, I think it will be something fantastic,” Dr. Peyrin-Biroulet said, “and it’s the reason why we were thinking that there is a need for an automated method to measure histological activity in UC.”
Old concept enhancing current practice
The idea of using AI systems to aid diagnostics is not new but now makes even more sense in the post–COVID-19 era, suggested Aaron F. Pollett, MD, MSc, FRCPC, codirector of the division of diagnostic medical genetics at Mount Sinai Hospital in Toronto and a pathologist with a specialty interest in gastrointestinal pathology.
“When we talk about artificial intelligence and histology, there’s actually a very long history, it goes back over 30 years,” Dr. Pollett said, from assessing cervical samples to its use in breast screening.
What seems to be sudden flurry of activity in the world of AI and pathology in recent years comes down to having a higher capacity for looking at large images, having access to large data sets, and having a high amount of computing power, Dr. Pollet inferred. Moreover, “the capacity and the need for whole slide imaging has really grown especially in the last few years as the pandemic has forced centers to adopt.” The need to work remotely and flexibly across centers and the number of available pathologists have also played a role.
AI systems that use image-based retrieval systems are making good headway in IBD, particularly in the diagnosis of UC where “some of the initial research is showing it can be quite good,” said Dr. Pollett. The “patchiness that Crohn’s can have in comparison to UC” means that it’s still an emerging area, but can perhaps be useful for more questionable cases in which “having that degree of certainty can certainly help because there is a discrepancy between specialist and nonspecialist pathologists in the likelihood that what they predict on the biopsy will be the underlying disease.”
AI systems in IBD – do they work?
Histopathology is becoming increasingly integrated into IBD clinical trial design at the behest of the Food and Drug Administration and European associations such as ECCO. This can be a tedious procedure that can be prone to error and disagreement between scorers.
The AI-driven scoring system that Dr. Peyrin-Biroulet and associates have been working on aims to fix all that by using machine learning and image processing to set up a reproducible system. Their system, which is based on the Nancy histological index for UC, shows high correlation (87%) with histopathologists’ assessment and was 100% accurate in identifying images with high (grade 4) or no (grade 0) inflammatory activity. The accuracy decreased, however, when trying to distinguish between more moderate activity, with a 75% accuracy for identifying grade 3 and 82% accuracy for grades 1 or 2.
“I’m actually very fascinated to see how we can be supported by the AI work in our practice,” observed Francesca Rosini, a histopathologist working at S. Orsola–Malpighi University Hospital in Bologna, Italy.
Dr. Rosini, who chaired the digital oral presentation session in which Dr. Peyrin-Biroulet had presented also noted that “obviously for us as well [as AI systems] no activity or severe activity is the easiest part but when it’s in between that’s where the problems come.”
Simplifying histological scoring
Simplifying scoring for use in AI systems could be the key to their future success, as Tommaso Lorenzo Parigi, MD, from Humanitas University in Milan, and a research fellow at the University of Birmingham (England), suggested.
“Histology is particularly important to distinguish between mild activity and remission,” Dr. Parigi said. “More than 30 histological scores that have been proposed, but their adoption in clinical practice remains limited.”
Dr. Parigi has been part of an international team that has developed a simplified histological score based on “the presence of absence of neutrophils, regardless of their number,” since these are “key determinants of disease activity”.
The score, known as the Paddington International Virtual Chromoendoscopy Scre (PICaSSO) Histologic Remission Index (PHRI), has been shown to correlate well with endoscopic outcomes and thus a good measure to include in AI systems. The results of this work were published online in Gut to coincide with the ECCO congress.
“We are getting close to a world where we could screen biopsies with this kind of systems and consider skipping the pathologists result if AI detected activity,” Dr. Parigi provocatively suggested. “Of course, we need to increase and improve our sensitivity, and we are currently working on that to reduce false negatives, as well as training our model to use and apply other histological scores.”
Assessing the gut in real time
Perhaps one of the most exciting developments it to be able to use these AI technologies to examine the gut in real time.
“Virtual chromoendoscopy will give you the opportunity to distinguish very carefully all the details of mucosal vascular pattern,” said Marietta Iacucci MD, PhD, FASGE, AGAF, an associate professor and gastroenterology consultant at the Birmingham (England) University Hospitals.
“So AI can give you, in real time, the score but at the same time it can help to target, to do biopsies for healing,” Dr. Iacucci added when reporting the results of a study evaluating the performance of the first virtual chromoendoscopy AI system to detect endoscopic and histologic remission in UC.
The system was proven to predict endoscopic remission very accurately (94% using PICaSSO and 87% using the UC endoscopic index of severity) when compared with a human endoscopist. Rates of predicting histological remission were also high, at around 83%-85%, depending on the score used.
“For the future, this AI tool can expediate, support, and standardize the endoscopic evaluation of UC mucosal healing in clinical practice and in clinical trials,” Dr. Iacucci said.
The next steps are to combine virtual chromoendoscopy with the PHRI and to validate the tool in a multicenter, international PICaSSO-AI study.
The AI-driven scoring system presented by Dr. Peyrin-Biroulet was supported by Takeda. Dr. Peryin-Biroulet acknowledged the receipt of personal fees and grants from Takeda along with multiple other Pharma companies and owning stock options from CTMA. Dr. Iacucci has received research grants from Pentax, AbbVie, Olympus, and Fujifilm and personal fees from Pentax, AbbVie and Janssen. Dr. Pollett, Dr. Rosini, and Dr. Parigi had no financial conflicts of interest to disclose.
FROM ECCO 2022
Endoscopic healing of Crohn’s disease could differ by biologic
Greater endoscopic healing at 1 year might be achieved in people with Crohn’s disease if they are treated with anti–tumor necrosis factor (TNF) drugs than if they are treated with certain other biologics it appears.
In a pooled analysis of data from four different clinical trial programs, which altogether included 344 patients with Crohn’s disease, both an infliximab biosimilar and adalimumab were associated with better endoscopic healing rates of both the ileum and colon than were either vedolizumab or ustekinumab.
The difference disappeared for ileal not colonic involvement, however, if patients had been biologic naive before receiving any of the four drugs that were compared.
“Recent studies have suggested that the ileum and colon differ with regards to their ability to achieve healing in Crohn’s disease,” Neeraj Narula, MD, said in reporting the analysis at the 17th congress of the European Crohn’s and Colitis Organisation.
“Our group has shown that larger ulcers in the ileum and rectum in particular do not heal as well as other areas of the colon when using infliximab therapies,” added Dr. Narula, who is an assistant professor of medicine at McMaster University, Hamilton, Ont., and the director of the IBD clinic and staff gastroenterologist at Hamilton Health Sciences.
Whether there are differences in how the ileal and colonic regions heal in response to biologic therapy is not known, which is why Dr. Narula and colleagues carried out their analysis.
Pooling pivotal trial program data
“Our primary aim was to evaluate the efficacy of four approved biologic therapies for Crohn’s disease with regards to their ability to achieve endoscopic healing after continuous use for 1 year,” he noted.
For their analysis, original data from the EXTEND, UNITI, VERSIFY, and infliximab biosimilar CT-P13 clinical trial programs were obtained and pooled.
The extent of mucosal inflammation and thereby healing were determined using a modified version of the Simple Endoscopic Score for Crohn’s disease (SES-CD), which is a measure often used in clinical trials.
At inclusion, patients had to have had an SES-CD score of 3 or more in at least one segment of the ileum or colon and confirmed ulceration. The primary endpoint was endoscopic healing defined as an SES-CD score of 0 after 1 year’s continuous treatment.
Multivariate logistic regression was used, and adjustments were made for potential confounding factors, such as how long people had had Crohn’s disease, the use of steroids, and if there had been prior anti-TNF failure.
Main results
Overall, 299 patients were in the final analysis; most (n = 141) had been treated with the infliximab biosimilar, with 61 treated with adalimumab, 56 vedolizumab, and 41 ustekinumab.
The highest rate of endoscopic healing at 1 year for ileal involvement was seen with the infliximab biosimilar (36.7% of patients) and the lowest rate with vedolizumab (18.6%), with rates of 30% and 22.7% for adalimumab and ustekinumab, respectively. Only the comparison between the infliximab biosimilar and vedolizumab was statistically significant (P = .038).
As for ileal ulcers, there were fewer seen with both anti-TNF treatments than with either ustekinumab or vedolizumab, at 40.8% for the infliximab biosimilar, 30% for adalimumab, 17.7% for ustekinumab, and 8.7% for vedolizumab. Rates of ileal ulcer absence in biologic-naive patients were a respective 36.7%, 37.5%, 40%, and 21.9%.
In terms of colonic involvement, the lowest rate of endoscopic healing occurred in patients treated with ustekinumab, at 29%, and the highest for adalimumab (62.5%), followed by the infliximab biosimilar (52.4%) and then vedolizumab (31.3%).
Absence of colonic ulcers was similarly low for ustekinumab (29.6%) and higher for the other three groups (64.9%, 70.5%, and 41.2%, respectively). When considering biologic-naive patients, there was a significant difference in the absence of colonic ulcers comparing adalimumab (66.7; P = .004) and the infliximab biosimilar (52.4%; P = .022), but not vedolizumab (37.1%) versus ustekinumab (29.4%).
Lots of questions and limitations
Dr. Narula’s presentation garnered a lot of questions, with viewers noting that the number of patients was too small or methodologically too flawed to be able to draw any sound conclusions.
“We acknowledge that our study cannot substitute for head-to-head trials of biologics in Crohn’s disease since we cannot account for all confounding variables,” said Dr. Narula.
“We did try to account for this limitation by performing some subgroup analyses to account for biologic-naive patients only,” he added, alongside the multivariate analyses.
Also, there might be a difference in the duration of treatment needed before endoscopic healing is seen, as the biologics studied all have a different duration of onset. The dosages used may also be important, and Dr. Narula conceded that their analyses were done assuming standard doses, which may not have been optimized.
There were several demographic differences between the infliximab arm and the other treatments. Of note, 76% of patients had been given immunomodulators at the same time, which is known to enhance the effects of infliximab.
Dr. Narula pointed out, however, that baseline characteristic were pretty similar in the other three study arms, and adalimumab still showed superiority in the analyses that were performed.
So are anti-TNFs the best choice?
“Ultimately, we always factor in the therapeutic index of therapy, trying to weigh benefit versus risk,” Dr. Narula said in answering a question from the chair of the session on the risks associated with anti-TNFs.
“We didn’t compare risk within this clinical trial, but certainly risk can be compared, and there’s things like number needed to treat versus number needed to harm to ultimately come at a best answer for the patient,” he added.
Dr. Narula disclosed receiving grants from Takeda and Pfizer; personal fees from AbbVie, Janssen, Takeda, Pfizer, Merck, Amgen, and Sandoz; and nonfinancial support from AbbVie, Janssen, Takeda, Pfizer, Ferring, and Lupin. The data used in the analysis were obtained through YODA Project #2021-4778 which has an agreement with Janssen Research & Developmen and via Vivli, which has access to data from AbbVie and Takeda. Data were also obtained with permission from Celltrion.
Greater endoscopic healing at 1 year might be achieved in people with Crohn’s disease if they are treated with anti–tumor necrosis factor (TNF) drugs than if they are treated with certain other biologics it appears.
In a pooled analysis of data from four different clinical trial programs, which altogether included 344 patients with Crohn’s disease, both an infliximab biosimilar and adalimumab were associated with better endoscopic healing rates of both the ileum and colon than were either vedolizumab or ustekinumab.
The difference disappeared for ileal not colonic involvement, however, if patients had been biologic naive before receiving any of the four drugs that were compared.
“Recent studies have suggested that the ileum and colon differ with regards to their ability to achieve healing in Crohn’s disease,” Neeraj Narula, MD, said in reporting the analysis at the 17th congress of the European Crohn’s and Colitis Organisation.
“Our group has shown that larger ulcers in the ileum and rectum in particular do not heal as well as other areas of the colon when using infliximab therapies,” added Dr. Narula, who is an assistant professor of medicine at McMaster University, Hamilton, Ont., and the director of the IBD clinic and staff gastroenterologist at Hamilton Health Sciences.
Whether there are differences in how the ileal and colonic regions heal in response to biologic therapy is not known, which is why Dr. Narula and colleagues carried out their analysis.
Pooling pivotal trial program data
“Our primary aim was to evaluate the efficacy of four approved biologic therapies for Crohn’s disease with regards to their ability to achieve endoscopic healing after continuous use for 1 year,” he noted.
For their analysis, original data from the EXTEND, UNITI, VERSIFY, and infliximab biosimilar CT-P13 clinical trial programs were obtained and pooled.
The extent of mucosal inflammation and thereby healing were determined using a modified version of the Simple Endoscopic Score for Crohn’s disease (SES-CD), which is a measure often used in clinical trials.
At inclusion, patients had to have had an SES-CD score of 3 or more in at least one segment of the ileum or colon and confirmed ulceration. The primary endpoint was endoscopic healing defined as an SES-CD score of 0 after 1 year’s continuous treatment.
Multivariate logistic regression was used, and adjustments were made for potential confounding factors, such as how long people had had Crohn’s disease, the use of steroids, and if there had been prior anti-TNF failure.
Main results
Overall, 299 patients were in the final analysis; most (n = 141) had been treated with the infliximab biosimilar, with 61 treated with adalimumab, 56 vedolizumab, and 41 ustekinumab.
The highest rate of endoscopic healing at 1 year for ileal involvement was seen with the infliximab biosimilar (36.7% of patients) and the lowest rate with vedolizumab (18.6%), with rates of 30% and 22.7% for adalimumab and ustekinumab, respectively. Only the comparison between the infliximab biosimilar and vedolizumab was statistically significant (P = .038).
As for ileal ulcers, there were fewer seen with both anti-TNF treatments than with either ustekinumab or vedolizumab, at 40.8% for the infliximab biosimilar, 30% for adalimumab, 17.7% for ustekinumab, and 8.7% for vedolizumab. Rates of ileal ulcer absence in biologic-naive patients were a respective 36.7%, 37.5%, 40%, and 21.9%.
In terms of colonic involvement, the lowest rate of endoscopic healing occurred in patients treated with ustekinumab, at 29%, and the highest for adalimumab (62.5%), followed by the infliximab biosimilar (52.4%) and then vedolizumab (31.3%).
Absence of colonic ulcers was similarly low for ustekinumab (29.6%) and higher for the other three groups (64.9%, 70.5%, and 41.2%, respectively). When considering biologic-naive patients, there was a significant difference in the absence of colonic ulcers comparing adalimumab (66.7; P = .004) and the infliximab biosimilar (52.4%; P = .022), but not vedolizumab (37.1%) versus ustekinumab (29.4%).
Lots of questions and limitations
Dr. Narula’s presentation garnered a lot of questions, with viewers noting that the number of patients was too small or methodologically too flawed to be able to draw any sound conclusions.
“We acknowledge that our study cannot substitute for head-to-head trials of biologics in Crohn’s disease since we cannot account for all confounding variables,” said Dr. Narula.
“We did try to account for this limitation by performing some subgroup analyses to account for biologic-naive patients only,” he added, alongside the multivariate analyses.
Also, there might be a difference in the duration of treatment needed before endoscopic healing is seen, as the biologics studied all have a different duration of onset. The dosages used may also be important, and Dr. Narula conceded that their analyses were done assuming standard doses, which may not have been optimized.
There were several demographic differences between the infliximab arm and the other treatments. Of note, 76% of patients had been given immunomodulators at the same time, which is known to enhance the effects of infliximab.
Dr. Narula pointed out, however, that baseline characteristic were pretty similar in the other three study arms, and adalimumab still showed superiority in the analyses that were performed.
So are anti-TNFs the best choice?
“Ultimately, we always factor in the therapeutic index of therapy, trying to weigh benefit versus risk,” Dr. Narula said in answering a question from the chair of the session on the risks associated with anti-TNFs.
“We didn’t compare risk within this clinical trial, but certainly risk can be compared, and there’s things like number needed to treat versus number needed to harm to ultimately come at a best answer for the patient,” he added.
Dr. Narula disclosed receiving grants from Takeda and Pfizer; personal fees from AbbVie, Janssen, Takeda, Pfizer, Merck, Amgen, and Sandoz; and nonfinancial support from AbbVie, Janssen, Takeda, Pfizer, Ferring, and Lupin. The data used in the analysis were obtained through YODA Project #2021-4778 which has an agreement with Janssen Research & Developmen and via Vivli, which has access to data from AbbVie and Takeda. Data were also obtained with permission from Celltrion.
Greater endoscopic healing at 1 year might be achieved in people with Crohn’s disease if they are treated with anti–tumor necrosis factor (TNF) drugs than if they are treated with certain other biologics it appears.
In a pooled analysis of data from four different clinical trial programs, which altogether included 344 patients with Crohn’s disease, both an infliximab biosimilar and adalimumab were associated with better endoscopic healing rates of both the ileum and colon than were either vedolizumab or ustekinumab.
The difference disappeared for ileal not colonic involvement, however, if patients had been biologic naive before receiving any of the four drugs that were compared.
“Recent studies have suggested that the ileum and colon differ with regards to their ability to achieve healing in Crohn’s disease,” Neeraj Narula, MD, said in reporting the analysis at the 17th congress of the European Crohn’s and Colitis Organisation.
“Our group has shown that larger ulcers in the ileum and rectum in particular do not heal as well as other areas of the colon when using infliximab therapies,” added Dr. Narula, who is an assistant professor of medicine at McMaster University, Hamilton, Ont., and the director of the IBD clinic and staff gastroenterologist at Hamilton Health Sciences.
Whether there are differences in how the ileal and colonic regions heal in response to biologic therapy is not known, which is why Dr. Narula and colleagues carried out their analysis.
Pooling pivotal trial program data
“Our primary aim was to evaluate the efficacy of four approved biologic therapies for Crohn’s disease with regards to their ability to achieve endoscopic healing after continuous use for 1 year,” he noted.
For their analysis, original data from the EXTEND, UNITI, VERSIFY, and infliximab biosimilar CT-P13 clinical trial programs were obtained and pooled.
The extent of mucosal inflammation and thereby healing were determined using a modified version of the Simple Endoscopic Score for Crohn’s disease (SES-CD), which is a measure often used in clinical trials.
At inclusion, patients had to have had an SES-CD score of 3 or more in at least one segment of the ileum or colon and confirmed ulceration. The primary endpoint was endoscopic healing defined as an SES-CD score of 0 after 1 year’s continuous treatment.
Multivariate logistic regression was used, and adjustments were made for potential confounding factors, such as how long people had had Crohn’s disease, the use of steroids, and if there had been prior anti-TNF failure.
Main results
Overall, 299 patients were in the final analysis; most (n = 141) had been treated with the infliximab biosimilar, with 61 treated with adalimumab, 56 vedolizumab, and 41 ustekinumab.
The highest rate of endoscopic healing at 1 year for ileal involvement was seen with the infliximab biosimilar (36.7% of patients) and the lowest rate with vedolizumab (18.6%), with rates of 30% and 22.7% for adalimumab and ustekinumab, respectively. Only the comparison between the infliximab biosimilar and vedolizumab was statistically significant (P = .038).
As for ileal ulcers, there were fewer seen with both anti-TNF treatments than with either ustekinumab or vedolizumab, at 40.8% for the infliximab biosimilar, 30% for adalimumab, 17.7% for ustekinumab, and 8.7% for vedolizumab. Rates of ileal ulcer absence in biologic-naive patients were a respective 36.7%, 37.5%, 40%, and 21.9%.
In terms of colonic involvement, the lowest rate of endoscopic healing occurred in patients treated with ustekinumab, at 29%, and the highest for adalimumab (62.5%), followed by the infliximab biosimilar (52.4%) and then vedolizumab (31.3%).
Absence of colonic ulcers was similarly low for ustekinumab (29.6%) and higher for the other three groups (64.9%, 70.5%, and 41.2%, respectively). When considering biologic-naive patients, there was a significant difference in the absence of colonic ulcers comparing adalimumab (66.7; P = .004) and the infliximab biosimilar (52.4%; P = .022), but not vedolizumab (37.1%) versus ustekinumab (29.4%).
Lots of questions and limitations
Dr. Narula’s presentation garnered a lot of questions, with viewers noting that the number of patients was too small or methodologically too flawed to be able to draw any sound conclusions.
“We acknowledge that our study cannot substitute for head-to-head trials of biologics in Crohn’s disease since we cannot account for all confounding variables,” said Dr. Narula.
“We did try to account for this limitation by performing some subgroup analyses to account for biologic-naive patients only,” he added, alongside the multivariate analyses.
Also, there might be a difference in the duration of treatment needed before endoscopic healing is seen, as the biologics studied all have a different duration of onset. The dosages used may also be important, and Dr. Narula conceded that their analyses were done assuming standard doses, which may not have been optimized.
There were several demographic differences between the infliximab arm and the other treatments. Of note, 76% of patients had been given immunomodulators at the same time, which is known to enhance the effects of infliximab.
Dr. Narula pointed out, however, that baseline characteristic were pretty similar in the other three study arms, and adalimumab still showed superiority in the analyses that were performed.
So are anti-TNFs the best choice?
“Ultimately, we always factor in the therapeutic index of therapy, trying to weigh benefit versus risk,” Dr. Narula said in answering a question from the chair of the session on the risks associated with anti-TNFs.
“We didn’t compare risk within this clinical trial, but certainly risk can be compared, and there’s things like number needed to treat versus number needed to harm to ultimately come at a best answer for the patient,” he added.
Dr. Narula disclosed receiving grants from Takeda and Pfizer; personal fees from AbbVie, Janssen, Takeda, Pfizer, Merck, Amgen, and Sandoz; and nonfinancial support from AbbVie, Janssen, Takeda, Pfizer, Ferring, and Lupin. The data used in the analysis were obtained through YODA Project #2021-4778 which has an agreement with Janssen Research & Developmen and via Vivli, which has access to data from AbbVie and Takeda. Data were also obtained with permission from Celltrion.
FROM ECCO 2022
Azithromycin doesn’t prevent recurrent wheezing after acute infant RSV
Azithromycin administered for severe early-life respiratory syncytial virus (RSV) bronchiolitis did not prevent recurrent wheezing in affected children over the next 2-4 years, a randomized, single-center study found.
Antibiotics are frequently given to patients with RSV bronchiolitis, although this practice is not supported by American Academy of Pediatrics clinical guidelines. Many doctors will prescribe them anyway if they see redness in the ears or other signs of infection, lead author Avraham Beigelman, MD, a pediatric allergist and immunologist at Washington University in St. Louis, said in an interview.

The double-blind, placebo-controlled trial, presented at the 2022 meeting of the American Academy of Allergy, Asthma & Immunology in Phoenix, was simultaneously published online Feb. 27, 2022, in the New England Journal of Medicine–Evidence.
Since azithromycin has shown anti-inflammatory benefit in chronic lung diseases and is a mainstay of care in cystic fibrosis and had shown previous effects in RSV patients, this trial examined its potential for preventing future recurrent wheezing in infants hospitalized with RSV who are at risk for developing asthma later. About half of children admitted to the hospital for RSV will develop asthma by age 7, Dr. Beigelman said.
“We were very surprised that azithromycin didn’t help in this trial given our previous findings,” Dr. Beigelman said.
And while those given azithromycin versus those given a placebo showed no significant decrease in recurrent wheezing, there was a slight suggestion that treatment with antibiotics of any kind may increase the risk of later wheezing in infants hospitalized with the virus.
“The study was not designed to tease at the effects of different antibiotics or combinations of antibiotics, so we have to be very cautious about this trend,” Dr. Beigelman said. “There may be short-term effects and long-term effects. Certain antibiotics may affect the infant microbiome in other parts of the body, such as the gut, [in] a way that may predispose to asthma. But all these associations suggest that early-life antibiotics for viral infections are not good for you.”
He pointed to the longstanding question among clinicians whether it is the antibiotic that’s increasing the risk of the harm or the condition for which the antibiotic is prescribed. These exploratory data, however, suggest that antibiotics for RSV may be causing harm.
In pursuit of that hypothesis, his group has collected airway microbiome samples from these infants and plan to investigate whether bacteria colonizing the airway may interact with the antibiotics to increase wheezing. The researchers will analyze stool samples from the babies to see whether the gut microbiome may also play a role in wheezing and the subsequent risk of developing childhood asthma.
Study details
The trial prospectively enrolled 200 otherwise healthy babies aged 1-18 months who were hospitalized at St. Louis Children’s Hospital for acute RSV bronchiolitis. Although RSV is a very common pediatric virus, only bout 3% of babies will require hospitalization in order to receive oxygen, Dr. Beigelman said.
Babies were randomly assigned to receive placebo or oral azithromycin at 10 mg/kg daily for 7 days, followed by 5 mg/kg daily for 7 days. Randomization was stratified by recent open-label antibiotic use. The primary outcome was recurrent wheeze, defined as a third episode of post-RSV wheeze over the following 2-4 years.
The biologic activity of azithromycin was clear since nasal-wash interleukin at day 14 after randomization was lower in azithromycin-treated infants. But despite evidence of activity, the risk of post-RSV recurrent wheeze was similar in both arms: 47% in the azithromycin group versus 36% in the placebo group, for an adjusted hazard ratio of 1.45 (95% confidence interval, 0.92-2.29; P = .11).
Nor did azithromycin lower the risk of recurrent wheeze in babies already receiving other antibiotics at the time of enrollment (HR, 0.94; 95% CI, 0.43-2.07). As for antibiotic-naive participants receiving azithromycin, there was a slight signal of potential increased risk of developing recurrent wheezing (HR, 1.79; 95% CI, 1.03-3.1).
The bottom line? The findings support current clinical guidelines recommending against the use of antibiotics for RSV. “At the very least, azithromycin and antibiotics in general have no benefit in preventing recurrent wheeze, and there is a possibility they may be harmful,” Dr. Beigelman said.
This trial is funded by the National Heart, Lung, and Blood Institute. Dr. Beigelman reported relationships with AstraZeneca, Novartis, and Sanofi. Two study coauthors disclosed various ties to industry.
Azithromycin administered for severe early-life respiratory syncytial virus (RSV) bronchiolitis did not prevent recurrent wheezing in affected children over the next 2-4 years, a randomized, single-center study found.
Antibiotics are frequently given to patients with RSV bronchiolitis, although this practice is not supported by American Academy of Pediatrics clinical guidelines. Many doctors will prescribe them anyway if they see redness in the ears or other signs of infection, lead author Avraham Beigelman, MD, a pediatric allergist and immunologist at Washington University in St. Louis, said in an interview.
The double-blind, placebo-controlled trial, presented at the 2022 meeting of the American Academy of Allergy, Asthma & Immunology in Phoenix, was simultaneously published online Feb. 27, 2022, in the New England Journal of Medicine–Evidence.
Since azithromycin has shown anti-inflammatory benefit in chronic lung diseases and is a mainstay of care in cystic fibrosis and had shown previous effects in RSV patients, this trial examined its potential for preventing future recurrent wheezing in infants hospitalized with RSV who are at risk for developing asthma later. About half of children admitted to the hospital for RSV will develop asthma by age 7, Dr. Beigelman said.
“We were very surprised that azithromycin didn’t help in this trial given our previous findings,” Dr. Beigelman said.
And while those given azithromycin versus those given a placebo showed no significant decrease in recurrent wheezing, there was a slight suggestion that treatment with antibiotics of any kind may increase the risk of later wheezing in infants hospitalized with the virus.
“The study was not designed to tease at the effects of different antibiotics or combinations of antibiotics, so we have to be very cautious about this trend,” Dr. Beigelman said. “There may be short-term effects and long-term effects. Certain antibiotics may affect the infant microbiome in other parts of the body, such as the gut, [in] a way that may predispose to asthma. But all these associations suggest that early-life antibiotics for viral infections are not good for you.”
He pointed to the longstanding question among clinicians whether it is the antibiotic that’s increasing the risk of the harm or the condition for which the antibiotic is prescribed. These exploratory data, however, suggest that antibiotics for RSV may be causing harm.
In pursuit of that hypothesis, his group has collected airway microbiome samples from these infants and plan to investigate whether bacteria colonizing the airway may interact with the antibiotics to increase wheezing. The researchers will analyze stool samples from the babies to see whether the gut microbiome may also play a role in wheezing and the subsequent risk of developing childhood asthma.
Study details
The trial prospectively enrolled 200 otherwise healthy babies aged 1-18 months who were hospitalized at St. Louis Children’s Hospital for acute RSV bronchiolitis. Although RSV is a very common pediatric virus, only bout 3% of babies will require hospitalization in order to receive oxygen, Dr. Beigelman said.
Babies were randomly assigned to receive placebo or oral azithromycin at 10 mg/kg daily for 7 days, followed by 5 mg/kg daily for 7 days. Randomization was stratified by recent open-label antibiotic use. The primary outcome was recurrent wheeze, defined as a third episode of post-RSV wheeze over the following 2-4 years.
The biologic activity of azithromycin was clear since nasal-wash interleukin at day 14 after randomization was lower in azithromycin-treated infants. But despite evidence of activity, the risk of post-RSV recurrent wheeze was similar in both arms: 47% in the azithromycin group versus 36% in the placebo group, for an adjusted hazard ratio of 1.45 (95% confidence interval, 0.92-2.29; P = .11).
Nor did azithromycin lower the risk of recurrent wheeze in babies already receiving other antibiotics at the time of enrollment (HR, 0.94; 95% CI, 0.43-2.07). As for antibiotic-naive participants receiving azithromycin, there was a slight signal of potential increased risk of developing recurrent wheezing (HR, 1.79; 95% CI, 1.03-3.1).
The bottom line? The findings support current clinical guidelines recommending against the use of antibiotics for RSV. “At the very least, azithromycin and antibiotics in general have no benefit in preventing recurrent wheeze, and there is a possibility they may be harmful,” Dr. Beigelman said.
This trial is funded by the National Heart, Lung, and Blood Institute. Dr. Beigelman reported relationships with AstraZeneca, Novartis, and Sanofi. Two study coauthors disclosed various ties to industry.
Azithromycin administered for severe early-life respiratory syncytial virus (RSV) bronchiolitis did not prevent recurrent wheezing in affected children over the next 2-4 years, a randomized, single-center study found.
Antibiotics are frequently given to patients with RSV bronchiolitis, although this practice is not supported by American Academy of Pediatrics clinical guidelines. Many doctors will prescribe them anyway if they see redness in the ears or other signs of infection, lead author Avraham Beigelman, MD, a pediatric allergist and immunologist at Washington University in St. Louis, said in an interview.
The double-blind, placebo-controlled trial, presented at the 2022 meeting of the American Academy of Allergy, Asthma & Immunology in Phoenix, was simultaneously published online Feb. 27, 2022, in the New England Journal of Medicine–Evidence.
Since azithromycin has shown anti-inflammatory benefit in chronic lung diseases and is a mainstay of care in cystic fibrosis and had shown previous effects in RSV patients, this trial examined its potential for preventing future recurrent wheezing in infants hospitalized with RSV who are at risk for developing asthma later. About half of children admitted to the hospital for RSV will develop asthma by age 7, Dr. Beigelman said.
“We were very surprised that azithromycin didn’t help in this trial given our previous findings,” Dr. Beigelman said.
And while those given azithromycin versus those given a placebo showed no significant decrease in recurrent wheezing, there was a slight suggestion that treatment with antibiotics of any kind may increase the risk of later wheezing in infants hospitalized with the virus.
“The study was not designed to tease at the effects of different antibiotics or combinations of antibiotics, so we have to be very cautious about this trend,” Dr. Beigelman said. “There may be short-term effects and long-term effects. Certain antibiotics may affect the infant microbiome in other parts of the body, such as the gut, [in] a way that may predispose to asthma. But all these associations suggest that early-life antibiotics for viral infections are not good for you.”
He pointed to the longstanding question among clinicians whether it is the antibiotic that’s increasing the risk of the harm or the condition for which the antibiotic is prescribed. These exploratory data, however, suggest that antibiotics for RSV may be causing harm.
In pursuit of that hypothesis, his group has collected airway microbiome samples from these infants and plan to investigate whether bacteria colonizing the airway may interact with the antibiotics to increase wheezing. The researchers will analyze stool samples from the babies to see whether the gut microbiome may also play a role in wheezing and the subsequent risk of developing childhood asthma.
Study details
The trial prospectively enrolled 200 otherwise healthy babies aged 1-18 months who were hospitalized at St. Louis Children’s Hospital for acute RSV bronchiolitis. Although RSV is a very common pediatric virus, only bout 3% of babies will require hospitalization in order to receive oxygen, Dr. Beigelman said.
Babies were randomly assigned to receive placebo or oral azithromycin at 10 mg/kg daily for 7 days, followed by 5 mg/kg daily for 7 days. Randomization was stratified by recent open-label antibiotic use. The primary outcome was recurrent wheeze, defined as a third episode of post-RSV wheeze over the following 2-4 years.
The biologic activity of azithromycin was clear since nasal-wash interleukin at day 14 after randomization was lower in azithromycin-treated infants. But despite evidence of activity, the risk of post-RSV recurrent wheeze was similar in both arms: 47% in the azithromycin group versus 36% in the placebo group, for an adjusted hazard ratio of 1.45 (95% confidence interval, 0.92-2.29; P = .11).
Nor did azithromycin lower the risk of recurrent wheeze in babies already receiving other antibiotics at the time of enrollment (HR, 0.94; 95% CI, 0.43-2.07). As for antibiotic-naive participants receiving azithromycin, there was a slight signal of potential increased risk of developing recurrent wheezing (HR, 1.79; 95% CI, 1.03-3.1).
The bottom line? The findings support current clinical guidelines recommending against the use of antibiotics for RSV. “At the very least, azithromycin and antibiotics in general have no benefit in preventing recurrent wheeze, and there is a possibility they may be harmful,” Dr. Beigelman said.
This trial is funded by the National Heart, Lung, and Blood Institute. Dr. Beigelman reported relationships with AstraZeneca, Novartis, and Sanofi. Two study coauthors disclosed various ties to industry.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE–EVIDENCE
Oncology care model reduces cost of supportive care meds
The Oncology Care Model (OCM), launched by the Centers for Medicare & Medicaid Services (CMS) with the goal of reducing spending for Medicare beneficiaries, was “associated with meaningful changes in the use of supportive care medications during chemotherapy treatment episodes,” according to new findings.
The OCM led to a statistically significant reduction in the use of denosumab – a pricier bone-modifying drug – by patients with bone metastases without changing the overall use of bone-modifying medications. The OCM also prompted more rapid adoption of a less expensive white blood cell growth factor agent – the biosimilar filgrastim – and more selective use of costly antiemetics as primary prophylaxis for chemotherapy-induced nausea.
study author Gabriel A. Brooks, MD, MPH, of the Dartmouth Institute for Health Policy and Clinical Practice, Geisel School of Medicine, Lebanon, N.H., and colleagues write.
The study was published online Feb. 25 in the Journal of Clinical Oncology.
Since the OCM was launched in 2016, several studies have evaluated whether the alternative payment model reached its goal of reducing spending while improving or maintaining the quality of cancer care.
The results have been decidedly mixed.
As previously reported by this news organization, one study found that after 4 years, the OCM led to a $155 million net loss to Medicare. During that time, physician participation in the program also declined, with the number of practices dropping almost 30% between 2016 and 2020.
Other studies, however, have highlighted more positive results.
One large community practice reported saving Medicare $3 million over the course of 1 year. Another analysis found that among community practices that adopted the OCM, in the first year of the program, there was less physician-administered drug use by patients with prostate cancer, lower drug costs by patients with lung and prostate cancer, fewer visits by patients with breast or colon cancer, and lower office-based costs in all cancers analyzed. However, these savings were largely offset by the costs of these programs.
In the current study, DR. Brooks and colleagues compared the use of supportive care medications – bone-modifying drugs as well as prophylactic white blood cell (WBC) growth factors and antiemetics – in practices that adopted the OCM and those that didn’t.
More specifically, the authors zeroed in on the bone-modifying agent denosumab for patients with breast, lung, or prostate cancer and the WBC growth factor biosimilar filgrastim for those receiving chemotherapy for breast, lung, or colorectal cancer. Prophylactic use of higher-cost neurokinin-1 (NK1) antagonists and long-acting serotonin antagonists for patients receiving chemotherapy for any type of cancer was also evaluated.
The authors evaluated chemotherapy episodes assigned to OCM (n = 201) and comparison practices (n = 534) using Medicare claims from 2013-2019.
There was a total of 255,638 treatment episodes for bone metastases. The authors found that the OCM led to relative reductions in the use of denosumab but not in the overall use of bone-modifying medications, which included the less costly options zoledronic acid and pamidronate. The use of denosumab was similar for OCM and comparison practices during the baseline period, but during the intervention period, there were statistically significant relative reductions in the use of denosumab at OCM practices for breast (-5.0%), prostate (-4.0%), and lung cancer (-4.1%).
For WBC growth factors, 164,310 episodes were included in analyses. The OCM did not affect the use of prophylactic WBC growth factors during breast cancer chemotherapy for those at high risk of febrile neutropenia but did lead to a relative decrease during intermediate-risk chemotherapy (-7.6%). The authors observed no OCM impact on the use of prophylactic WBC growth factors among intermediate-risk lung or colorectal cancer patients. But, during the intervention period, OCM practices did demonstrate an increased use of originator or biosimilar filgrastim (57.3%) compared to other practices (47.6%), and the quarterly rate of increase in the use of the biosimilar grew 2.6 percentage points faster in OCM practices.
The authors report that there were 414,792 treatment episodes involving the use of prophylactic antiemetics. Overall, among patients receiving chemotherapy with high or moderate emetic risk, the OCM led to reductions in the prophylactic use of NK1 antagonists and long-acting serotonin antagonists. The authors report a 6.0 percentage point reduction in the use of NK1 antagonists during high-emetic-risk chemotherapy.
“We found that OCM was associated with meaningful changes in the use of supportive care medications during chemotherapy treatment episodes consistent with value-based care redesign,” the authors conclude. “These impacts on supportive care medication use align with previously reported spending reductions attributable to OCM and suggest that alternative payment models have potential to drive value-based changes in supportive care during cancer treatment.”
The study was supported by CMS. Several of the coauthors have reported relationships with industry, as noted in the article.
A version of this article first appeared on Medscape.com.
The Oncology Care Model (OCM), launched by the Centers for Medicare & Medicaid Services (CMS) with the goal of reducing spending for Medicare beneficiaries, was “associated with meaningful changes in the use of supportive care medications during chemotherapy treatment episodes,” according to new findings.
The OCM led to a statistically significant reduction in the use of denosumab – a pricier bone-modifying drug – by patients with bone metastases without changing the overall use of bone-modifying medications. The OCM also prompted more rapid adoption of a less expensive white blood cell growth factor agent – the biosimilar filgrastim – and more selective use of costly antiemetics as primary prophylaxis for chemotherapy-induced nausea.
study author Gabriel A. Brooks, MD, MPH, of the Dartmouth Institute for Health Policy and Clinical Practice, Geisel School of Medicine, Lebanon, N.H., and colleagues write.
The study was published online Feb. 25 in the Journal of Clinical Oncology.
Since the OCM was launched in 2016, several studies have evaluated whether the alternative payment model reached its goal of reducing spending while improving or maintaining the quality of cancer care.
The results have been decidedly mixed.
As previously reported by this news organization, one study found that after 4 years, the OCM led to a $155 million net loss to Medicare. During that time, physician participation in the program also declined, with the number of practices dropping almost 30% between 2016 and 2020.
Other studies, however, have highlighted more positive results.
One large community practice reported saving Medicare $3 million over the course of 1 year. Another analysis found that among community practices that adopted the OCM, in the first year of the program, there was less physician-administered drug use by patients with prostate cancer, lower drug costs by patients with lung and prostate cancer, fewer visits by patients with breast or colon cancer, and lower office-based costs in all cancers analyzed. However, these savings were largely offset by the costs of these programs.
In the current study, DR. Brooks and colleagues compared the use of supportive care medications – bone-modifying drugs as well as prophylactic white blood cell (WBC) growth factors and antiemetics – in practices that adopted the OCM and those that didn’t.
More specifically, the authors zeroed in on the bone-modifying agent denosumab for patients with breast, lung, or prostate cancer and the WBC growth factor biosimilar filgrastim for those receiving chemotherapy for breast, lung, or colorectal cancer. Prophylactic use of higher-cost neurokinin-1 (NK1) antagonists and long-acting serotonin antagonists for patients receiving chemotherapy for any type of cancer was also evaluated.
The authors evaluated chemotherapy episodes assigned to OCM (n = 201) and comparison practices (n = 534) using Medicare claims from 2013-2019.
There was a total of 255,638 treatment episodes for bone metastases. The authors found that the OCM led to relative reductions in the use of denosumab but not in the overall use of bone-modifying medications, which included the less costly options zoledronic acid and pamidronate. The use of denosumab was similar for OCM and comparison practices during the baseline period, but during the intervention period, there were statistically significant relative reductions in the use of denosumab at OCM practices for breast (-5.0%), prostate (-4.0%), and lung cancer (-4.1%).
For WBC growth factors, 164,310 episodes were included in analyses. The OCM did not affect the use of prophylactic WBC growth factors during breast cancer chemotherapy for those at high risk of febrile neutropenia but did lead to a relative decrease during intermediate-risk chemotherapy (-7.6%). The authors observed no OCM impact on the use of prophylactic WBC growth factors among intermediate-risk lung or colorectal cancer patients. But, during the intervention period, OCM practices did demonstrate an increased use of originator or biosimilar filgrastim (57.3%) compared to other practices (47.6%), and the quarterly rate of increase in the use of the biosimilar grew 2.6 percentage points faster in OCM practices.
The authors report that there were 414,792 treatment episodes involving the use of prophylactic antiemetics. Overall, among patients receiving chemotherapy with high or moderate emetic risk, the OCM led to reductions in the prophylactic use of NK1 antagonists and long-acting serotonin antagonists. The authors report a 6.0 percentage point reduction in the use of NK1 antagonists during high-emetic-risk chemotherapy.
“We found that OCM was associated with meaningful changes in the use of supportive care medications during chemotherapy treatment episodes consistent with value-based care redesign,” the authors conclude. “These impacts on supportive care medication use align with previously reported spending reductions attributable to OCM and suggest that alternative payment models have potential to drive value-based changes in supportive care during cancer treatment.”
The study was supported by CMS. Several of the coauthors have reported relationships with industry, as noted in the article.
A version of this article first appeared on Medscape.com.
The Oncology Care Model (OCM), launched by the Centers for Medicare & Medicaid Services (CMS) with the goal of reducing spending for Medicare beneficiaries, was “associated with meaningful changes in the use of supportive care medications during chemotherapy treatment episodes,” according to new findings.
The OCM led to a statistically significant reduction in the use of denosumab – a pricier bone-modifying drug – by patients with bone metastases without changing the overall use of bone-modifying medications. The OCM also prompted more rapid adoption of a less expensive white blood cell growth factor agent – the biosimilar filgrastim – and more selective use of costly antiemetics as primary prophylaxis for chemotherapy-induced nausea.
study author Gabriel A. Brooks, MD, MPH, of the Dartmouth Institute for Health Policy and Clinical Practice, Geisel School of Medicine, Lebanon, N.H., and colleagues write.
The study was published online Feb. 25 in the Journal of Clinical Oncology.
Since the OCM was launched in 2016, several studies have evaluated whether the alternative payment model reached its goal of reducing spending while improving or maintaining the quality of cancer care.
The results have been decidedly mixed.
As previously reported by this news organization, one study found that after 4 years, the OCM led to a $155 million net loss to Medicare. During that time, physician participation in the program also declined, with the number of practices dropping almost 30% between 2016 and 2020.
Other studies, however, have highlighted more positive results.
One large community practice reported saving Medicare $3 million over the course of 1 year. Another analysis found that among community practices that adopted the OCM, in the first year of the program, there was less physician-administered drug use by patients with prostate cancer, lower drug costs by patients with lung and prostate cancer, fewer visits by patients with breast or colon cancer, and lower office-based costs in all cancers analyzed. However, these savings were largely offset by the costs of these programs.
In the current study, DR. Brooks and colleagues compared the use of supportive care medications – bone-modifying drugs as well as prophylactic white blood cell (WBC) growth factors and antiemetics – in practices that adopted the OCM and those that didn’t.
More specifically, the authors zeroed in on the bone-modifying agent denosumab for patients with breast, lung, or prostate cancer and the WBC growth factor biosimilar filgrastim for those receiving chemotherapy for breast, lung, or colorectal cancer. Prophylactic use of higher-cost neurokinin-1 (NK1) antagonists and long-acting serotonin antagonists for patients receiving chemotherapy for any type of cancer was also evaluated.
The authors evaluated chemotherapy episodes assigned to OCM (n = 201) and comparison practices (n = 534) using Medicare claims from 2013-2019.
There was a total of 255,638 treatment episodes for bone metastases. The authors found that the OCM led to relative reductions in the use of denosumab but not in the overall use of bone-modifying medications, which included the less costly options zoledronic acid and pamidronate. The use of denosumab was similar for OCM and comparison practices during the baseline period, but during the intervention period, there were statistically significant relative reductions in the use of denosumab at OCM practices for breast (-5.0%), prostate (-4.0%), and lung cancer (-4.1%).
For WBC growth factors, 164,310 episodes were included in analyses. The OCM did not affect the use of prophylactic WBC growth factors during breast cancer chemotherapy for those at high risk of febrile neutropenia but did lead to a relative decrease during intermediate-risk chemotherapy (-7.6%). The authors observed no OCM impact on the use of prophylactic WBC growth factors among intermediate-risk lung or colorectal cancer patients. But, during the intervention period, OCM practices did demonstrate an increased use of originator or biosimilar filgrastim (57.3%) compared to other practices (47.6%), and the quarterly rate of increase in the use of the biosimilar grew 2.6 percentage points faster in OCM practices.
The authors report that there were 414,792 treatment episodes involving the use of prophylactic antiemetics. Overall, among patients receiving chemotherapy with high or moderate emetic risk, the OCM led to reductions in the prophylactic use of NK1 antagonists and long-acting serotonin antagonists. The authors report a 6.0 percentage point reduction in the use of NK1 antagonists during high-emetic-risk chemotherapy.
“We found that OCM was associated with meaningful changes in the use of supportive care medications during chemotherapy treatment episodes consistent with value-based care redesign,” the authors conclude. “These impacts on supportive care medication use align with previously reported spending reductions attributable to OCM and suggest that alternative payment models have potential to drive value-based changes in supportive care during cancer treatment.”
The study was supported by CMS. Several of the coauthors have reported relationships with industry, as noted in the article.
A version of this article first appeared on Medscape.com.
FROM JOURNAL OF CLINICAL ONCOLOGY
Practicing across state lines: A challenge for telemental health
I was taught to think clinically first and legally second. There are moments when following every regulation is clearly detrimental to the well-being of both the patient and the medical community at large, and these challenges have been highlighted by issues with telemental health during the pandemic.
A friend emailed me with a problem: He has a son who is a traveling nurse and is currently in psychotherapy. The therapist has, in accordance with licensing requirements, told his son that she can not see him when assignments take him to any state where she is not licensed. The patient needs to physically be in the same state where the clinician holds a license, technically for every appointment. The nursing assignments last for 3 months and he will be going to a variety of states. Does he really need to get a new therapist every 90 days?

The logistics seem mind-boggling in a time when there is a shortage of mental health professionals, and there are often long wait lists to get care. And even if it was all easy, I’ll point out that working with a therapist is a bit different then going to an urgent care center to have sutures removed or to obtain antibiotics for strep throat: The relationship is not easily interchangeable, and I know of no one who would think it clinically optimal for anyone to change psychotherapists every 3 months. The traveling nurse does not just need to find a “provider” in each state, he needs to find one he is comfortable with and he will have to spend several sessions relaying his history and forming a new therapeutic alliance. And given the ambiguities of psychotherapy, he would optimally see therapists who do not make conflicting interpretations or recommendations. Mind-boggling. And while none of us are irreplaceable, it feels heartless to tell someone who is traveling to provide medical care to others during a pandemic that they can’t have mental health care when our technology would allow for it.
In the “old days” it was simpler: Patients came to the office and both the patient and the clinician were physically located in the same state, even if the patient resided in another state and commuted hours to the appointment. Telemental health was done in select rural areas or in military settings, and most physicians did not consider the option for video visits, much less full video treatment. For the average practitioner, issues of location were not relevant. The exception was for college students who might reside in one state and see a psychiatrist or therapist in another, but typically everyone was comfortable taking a break from therapy when the patient could not meet with the therapist in person. If psychiatrists were having phone or video sessions with out-of-state patients on an occasional basis, it may have been because there was less scrutiny and it was less obvious that this was not permitted.
When the pandemic forced treatment to go online, the issues changed. At the beginning, issues related to state licensing were waived. Now each state has a different requirement with regard to out-of-state physicians; some allow their residents to be seen, while others require the physician to get licensed in their state and the process may or may not be costly or arduous for the provider. The regulations change frequently, and can be quite confusing to follow. Since psychiatry is a shortage field, many psychiatrists are not looking to have more patients from other states and are not motivated to apply for extra licenses.
Life as a practicing psychiatrist has been a moving target: I reopened my practice for some in-person visits for vaccinated patients in June 2021, then closed it when the Omicron surge seemed too risky, and I’ll be reopening soon. Patients, too, have had unpredictable lives.
For the practitioner who is following the rules precisely, the issues can be sticky. It may be fine to have Zoom visits with a patient who lives across the street, but not with the elderly patient who has to drive 90 minutes across a state line, and it’s always fine to have a video session with a patient in Guam. If a patient signs on for a video visit with a doctor licensed in Maine and announces there will be a visit to a brother in Michigan, does the clinician abruptly end the session? Does he charge for the then missed appointment, and don’t we feel this is a waste of the psychiatrist’s time when appointments are limited?
If college students started with therapists in their home states when universities shut down in the spring of 2020, must they now try to get treatment in the states where their college campuses are located? What if the university has a long wait for services, there are no local psychiatrists taking on new patients, or the student feels he is making good progress with the doctor he is working with? And how do we even know for sure where our patients are located? Are we obligated to ask for a precise location at the beginning of each session? What if patients do not offer their locations, or lie about where they are?
Oddly, the issue is with the location of the patient; the doctor can be anywhere as long as the patient’s body is in a state where he or she is licensed. And it has never been a problem to send prescriptions to pharmacies in other states, though this seems to me the essence of practicing across state lines.
In the State of the Union Address on March 1, President Biden had a hefty agenda: The Russian invasion of Ukraine, a global pandemic, spiraling inflation, and for the first time in a SOTU address, our president discussed a strategy to address our National Mental Health Crisis. The fact sheet released by the White House details many long-awaited changes to increase the mental health workforce to address shortages, instituting a “988” crisis line to initiate “someone to call, someone to respond, and somewhere for every American in crisis to go.” The proposals call for a sweeping reform in providing access to services, strengthening parity, and improving community, veterans, and university services – and the Biden administration specifically addresses telemental health. “To maintain continuity of access, the Administration will work with Congress to ensure coverage of tele-behavioral health across health plans, and support appropriate delivery of telemedicine across state lines.”
This is good news, as it’s time we concentrated on allowing for access to care in a consumer-oriented way. It may let us focus on offering good clinical care and not focus on following outdated regulations. Hopefully, those who want help will be able to access it, and perhaps soon a traveling nurse will be permitted to get mental health care with continuity of treatment.
Dr. Miller is a coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016). She has a private practice and is assistant professor of psychiatry and behavioral sciences at Johns Hopkins in Baltimore. Dr. Miller has no conflicts of interest.
I was taught to think clinically first and legally second. There are moments when following every regulation is clearly detrimental to the well-being of both the patient and the medical community at large, and these challenges have been highlighted by issues with telemental health during the pandemic.
A friend emailed me with a problem: He has a son who is a traveling nurse and is currently in psychotherapy. The therapist has, in accordance with licensing requirements, told his son that she can not see him when assignments take him to any state where she is not licensed. The patient needs to physically be in the same state where the clinician holds a license, technically for every appointment. The nursing assignments last for 3 months and he will be going to a variety of states. Does he really need to get a new therapist every 90 days?
The logistics seem mind-boggling in a time when there is a shortage of mental health professionals, and there are often long wait lists to get care. And even if it was all easy, I’ll point out that working with a therapist is a bit different then going to an urgent care center to have sutures removed or to obtain antibiotics for strep throat: The relationship is not easily interchangeable, and I know of no one who would think it clinically optimal for anyone to change psychotherapists every 3 months. The traveling nurse does not just need to find a “provider” in each state, he needs to find one he is comfortable with and he will have to spend several sessions relaying his history and forming a new therapeutic alliance. And given the ambiguities of psychotherapy, he would optimally see therapists who do not make conflicting interpretations or recommendations. Mind-boggling. And while none of us are irreplaceable, it feels heartless to tell someone who is traveling to provide medical care to others during a pandemic that they can’t have mental health care when our technology would allow for it.
In the “old days” it was simpler: Patients came to the office and both the patient and the clinician were physically located in the same state, even if the patient resided in another state and commuted hours to the appointment. Telemental health was done in select rural areas or in military settings, and most physicians did not consider the option for video visits, much less full video treatment. For the average practitioner, issues of location were not relevant. The exception was for college students who might reside in one state and see a psychiatrist or therapist in another, but typically everyone was comfortable taking a break from therapy when the patient could not meet with the therapist in person. If psychiatrists were having phone or video sessions with out-of-state patients on an occasional basis, it may have been because there was less scrutiny and it was less obvious that this was not permitted.
When the pandemic forced treatment to go online, the issues changed. At the beginning, issues related to state licensing were waived. Now each state has a different requirement with regard to out-of-state physicians; some allow their residents to be seen, while others require the physician to get licensed in their state and the process may or may not be costly or arduous for the provider. The regulations change frequently, and can be quite confusing to follow. Since psychiatry is a shortage field, many psychiatrists are not looking to have more patients from other states and are not motivated to apply for extra licenses.
Life as a practicing psychiatrist has been a moving target: I reopened my practice for some in-person visits for vaccinated patients in June 2021, then closed it when the Omicron surge seemed too risky, and I’ll be reopening soon. Patients, too, have had unpredictable lives.
For the practitioner who is following the rules precisely, the issues can be sticky. It may be fine to have Zoom visits with a patient who lives across the street, but not with the elderly patient who has to drive 90 minutes across a state line, and it’s always fine to have a video session with a patient in Guam. If a patient signs on for a video visit with a doctor licensed in Maine and announces there will be a visit to a brother in Michigan, does the clinician abruptly end the session? Does he charge for the then missed appointment, and don’t we feel this is a waste of the psychiatrist’s time when appointments are limited?
If college students started with therapists in their home states when universities shut down in the spring of 2020, must they now try to get treatment in the states where their college campuses are located? What if the university has a long wait for services, there are no local psychiatrists taking on new patients, or the student feels he is making good progress with the doctor he is working with? And how do we even know for sure where our patients are located? Are we obligated to ask for a precise location at the beginning of each session? What if patients do not offer their locations, or lie about where they are?
Oddly, the issue is with the location of the patient; the doctor can be anywhere as long as the patient’s body is in a state where he or she is licensed. And it has never been a problem to send prescriptions to pharmacies in other states, though this seems to me the essence of practicing across state lines.
In the State of the Union Address on March 1, President Biden had a hefty agenda: The Russian invasion of Ukraine, a global pandemic, spiraling inflation, and for the first time in a SOTU address, our president discussed a strategy to address our National Mental Health Crisis. The fact sheet released by the White House details many long-awaited changes to increase the mental health workforce to address shortages, instituting a “988” crisis line to initiate “someone to call, someone to respond, and somewhere for every American in crisis to go.” The proposals call for a sweeping reform in providing access to services, strengthening parity, and improving community, veterans, and university services – and the Biden administration specifically addresses telemental health. “To maintain continuity of access, the Administration will work with Congress to ensure coverage of tele-behavioral health across health plans, and support appropriate delivery of telemedicine across state lines.”
This is good news, as it’s time we concentrated on allowing for access to care in a consumer-oriented way. It may let us focus on offering good clinical care and not focus on following outdated regulations. Hopefully, those who want help will be able to access it, and perhaps soon a traveling nurse will be permitted to get mental health care with continuity of treatment.
Dr. Miller is a coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016). She has a private practice and is assistant professor of psychiatry and behavioral sciences at Johns Hopkins in Baltimore. Dr. Miller has no conflicts of interest.
I was taught to think clinically first and legally second. There are moments when following every regulation is clearly detrimental to the well-being of both the patient and the medical community at large, and these challenges have been highlighted by issues with telemental health during the pandemic.
A friend emailed me with a problem: He has a son who is a traveling nurse and is currently in psychotherapy. The therapist has, in accordance with licensing requirements, told his son that she can not see him when assignments take him to any state where she is not licensed. The patient needs to physically be in the same state where the clinician holds a license, technically for every appointment. The nursing assignments last for 3 months and he will be going to a variety of states. Does he really need to get a new therapist every 90 days?
The logistics seem mind-boggling in a time when there is a shortage of mental health professionals, and there are often long wait lists to get care. And even if it was all easy, I’ll point out that working with a therapist is a bit different then going to an urgent care center to have sutures removed or to obtain antibiotics for strep throat: The relationship is not easily interchangeable, and I know of no one who would think it clinically optimal for anyone to change psychotherapists every 3 months. The traveling nurse does not just need to find a “provider” in each state, he needs to find one he is comfortable with and he will have to spend several sessions relaying his history and forming a new therapeutic alliance. And given the ambiguities of psychotherapy, he would optimally see therapists who do not make conflicting interpretations or recommendations. Mind-boggling. And while none of us are irreplaceable, it feels heartless to tell someone who is traveling to provide medical care to others during a pandemic that they can’t have mental health care when our technology would allow for it.
In the “old days” it was simpler: Patients came to the office and both the patient and the clinician were physically located in the same state, even if the patient resided in another state and commuted hours to the appointment. Telemental health was done in select rural areas or in military settings, and most physicians did not consider the option for video visits, much less full video treatment. For the average practitioner, issues of location were not relevant. The exception was for college students who might reside in one state and see a psychiatrist or therapist in another, but typically everyone was comfortable taking a break from therapy when the patient could not meet with the therapist in person. If psychiatrists were having phone or video sessions with out-of-state patients on an occasional basis, it may have been because there was less scrutiny and it was less obvious that this was not permitted.
When the pandemic forced treatment to go online, the issues changed. At the beginning, issues related to state licensing were waived. Now each state has a different requirement with regard to out-of-state physicians; some allow their residents to be seen, while others require the physician to get licensed in their state and the process may or may not be costly or arduous for the provider. The regulations change frequently, and can be quite confusing to follow. Since psychiatry is a shortage field, many psychiatrists are not looking to have more patients from other states and are not motivated to apply for extra licenses.
Life as a practicing psychiatrist has been a moving target: I reopened my practice for some in-person visits for vaccinated patients in June 2021, then closed it when the Omicron surge seemed too risky, and I’ll be reopening soon. Patients, too, have had unpredictable lives.
For the practitioner who is following the rules precisely, the issues can be sticky. It may be fine to have Zoom visits with a patient who lives across the street, but not with the elderly patient who has to drive 90 minutes across a state line, and it’s always fine to have a video session with a patient in Guam. If a patient signs on for a video visit with a doctor licensed in Maine and announces there will be a visit to a brother in Michigan, does the clinician abruptly end the session? Does he charge for the then missed appointment, and don’t we feel this is a waste of the psychiatrist’s time when appointments are limited?
If college students started with therapists in their home states when universities shut down in the spring of 2020, must they now try to get treatment in the states where their college campuses are located? What if the university has a long wait for services, there are no local psychiatrists taking on new patients, or the student feels he is making good progress with the doctor he is working with? And how do we even know for sure where our patients are located? Are we obligated to ask for a precise location at the beginning of each session? What if patients do not offer their locations, or lie about where they are?
Oddly, the issue is with the location of the patient; the doctor can be anywhere as long as the patient’s body is in a state where he or she is licensed. And it has never been a problem to send prescriptions to pharmacies in other states, though this seems to me the essence of practicing across state lines.
In the State of the Union Address on March 1, President Biden had a hefty agenda: The Russian invasion of Ukraine, a global pandemic, spiraling inflation, and for the first time in a SOTU address, our president discussed a strategy to address our National Mental Health Crisis. The fact sheet released by the White House details many long-awaited changes to increase the mental health workforce to address shortages, instituting a “988” crisis line to initiate “someone to call, someone to respond, and somewhere for every American in crisis to go.” The proposals call for a sweeping reform in providing access to services, strengthening parity, and improving community, veterans, and university services – and the Biden administration specifically addresses telemental health. “To maintain continuity of access, the Administration will work with Congress to ensure coverage of tele-behavioral health across health plans, and support appropriate delivery of telemedicine across state lines.”
This is good news, as it’s time we concentrated on allowing for access to care in a consumer-oriented way. It may let us focus on offering good clinical care and not focus on following outdated regulations. Hopefully, those who want help will be able to access it, and perhaps soon a traveling nurse will be permitted to get mental health care with continuity of treatment.
Dr. Miller is a coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016). She has a private practice and is assistant professor of psychiatry and behavioral sciences at Johns Hopkins in Baltimore. Dr. Miller has no conflicts of interest.
Mental illness tied to increased dementia risk
Results of a large, longitudinal, population-based study show that individuals hospitalized for a mental health disorder had a fourfold increased relative risk (RR) for developing dementia, compared with those who were not hospitalized with a mental illness.
In addition, those with dementia plus a mental disorder developed dementia almost 6 years earlier than those without a mental illness.
The findings were consistent among men and women, in patients with early- and late-onset dementia, in those with Alzheimer’s and non-Alzheimer’s dementia, and across all mental health disorders – and remained so after accounting for pre-existing physical illness and socioeconomic factors.
“Dementia is not typically treated until later in life, but our study suggests that we need to be thinking about dementia prevention much earlier in the life course,” study investigator Leah Richmond-Rakerd, PhD, assistant professor, department of psychology, University of Michigan, said in an interview.
“Supporting young people’s mental health could be a window of opportunity to help reduce the burden of dementia in older adults,” she said.
The findings were published online Feb. 16.
Underappreciated risk factor
“Recognition of the outsized influence of dementia on later-life functioning has fueled research into modifiable risk factors and prevention targets,” the investigators write.
Previous research suggests mental disorders may “comprise an underappreciated category of modifiable risk factors.” However, those studies focused primarily on midlife and older individuals, not on capturing mental disorders during young adulthood, which is the time of “peak prevalence,” they add. In addition, most studies have not explored the full range of mental disorders.
Dr. Richmond-Rakerd noted that it is well known that mental health disorders peak in adolescence and young adulthood – and are treatable.
“If the same people who have mental disorders when they are young tend to develop dementia when they are older, that would mean that preventing mental health problems in younger people might reduce or delay the burden of dementia in older people,” she said.
The investigators assessed records from the New Zealand Integrated Data Infrastructure, which is a de-identified register that includes the entire New Zealand population. They also examined information about hospitalizations and diagnoses from records kept by the New Zealand Ministry of Health.
The researchers followed 1,711,386 individuals born between 1928 and 1967 (50.6% men, aged 21 to 60 years at baseline) for 30 years. The population was subdivided into age groups based on birth years: 1928-1937 (14.8%), 1938-1947 (20.85%), 1948-1957 (29.35%), and 1958-1967 (35.1%).
Earlier onset
During the study period, 3.8% of individuals were identified as having a mental disorder, and 2% were identified as having dementia. Similar percentages of men and women had a mental disorder, and similar percentages had dementia.
Dementia was “over-represented” among participants with versus without a mental disorder (6.1% vs. 1.8%). This finding held across all age groups.
Those diagnosed with a mental disorder were also more likely to develop dementia, compared with their peers without a mental disorder (RR, 3.51; 95% confidence interval, 3.39-3.64), which is a larger association than that between physical diseases and dementia (RR, 1.19; 95% CI, 1.16-1.21).
These associations were present in both sexes and in all age groups, although the associations were stronger in more recently born cohorts.
A sixfold higher risk for dementia remained even after adjusting for pre-existing physical illnesses (HR, 6.49; 95% CI, 6.25-6.73); and the elevated risk was evident across different lengths of follow-up from the index mental disorder.
When the researchers focused specifically on individuals diagnosed with dementia, they found that those diagnosed with a mental disorder developed dementia a mean of 5.60 years earlier than those without a mental disorder diagnosis – an association observed across both sexes and all age groups.
“Individuals diagnosed with psychotic, substance use, mood, neurotic, and all other mental disorders and who engaged in self-harm were all more likely than those without a mental disorder to be diagnosed with subsequent dementia, even after accounting for their physical disease histories,” the investigators write.
Although there was a link between mental disorders in both Alzheimer’s and non-Alzheimer’s dementias, the association was larger in non-Alzheimer’s.
The researchers note that the study has several limitations, including the fact that it was conducted in New Zealand and therefore the results may not be generalizable to other regions. In addition, inpatient hospital records do not capture less severe mental disorder cases treated in the outpatient setting.
Dr. Richmond-Rakerd suggested several potential mechanisms that could account for the link between mental illness and dementia, including poor lifestyle choices and metabolic side effects associated with some psychiatric medications.
“There could also be shared risk factors for both mental disorders and dementia, such as shared genetics, or individuals may experience a lifelong brain vulnerability that shows up as mental health problems earlier in life and shows up as dementia later in life,” she said.
An important risk factor
Commenting for this article, Ken Duckworth, MD, chief medical officer of the National Alliance on Mental Illness, said a major strength of the study was its longitudinal scope and large population size.
He described the study as allowing clinicians to “watch the movie,” as opposed to looking at a “snapshot” of data.
“Although you can learn things from snapshots, a large, comprehensive public health system looking at 30 years of claims – something not possible in the U.S. because of our more fragmented health care system – offers more insight,” said Dr. Duckworth, who was not involved with the research.
The investigators are “painting a picture of a correlation of risk, and to me, that’s the beginning of further inquiry,” he added. “Would preventive efforts targeting dementia, such as exercise and socialization, be helpful? It’s a great study that raises these interesting questions.”
Also commenting in an interview, Claire Sexton, DPhil, director of scientific programs and outreach at the Alzheimer’s Association, said the study “adds a wealth of data to our understanding” of mental disorders as a dementia risk factor.
However, the study was observational, so “the findings cannot imply causation, [and just] because someone has depression, that does not mean they will go on to develop Alzheimer’s,” said Dr. Sexton, who also was not involved with the research.
Still, “these data support the idea that taking care of one’s mental health is incredibly important for overall wellbeing. For providers, it’s important to have mental health evaluation be a part of your patient’s regular checkups,” she added.
Dr. Richmond-Rakerd noted that even if mental health conditions are not a causal risk factor for dementia, “the presence of a mental health problem is still an important indicator of risk. Mental health providers may wish to target other risk factors for dementia that are more common in individuals with mental health conditions, such as social disconnection.”
The study was funded by grants from the National Institute on Aging, the U.K. Medical Research Council, the National Institute of Child Health and Development through the Duke Population Research Center, and the National Institute on Aging through the Center for Advancing Sociodemographic and Economic Study of Alzheimer’s Disease and Related Dementias. Dr. Richmond-Rakerd reports no relevant financial relationships. The other investigators’ disclosures are listed in the original article. Dr. Sexton and Dr. Duckworth report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Results of a large, longitudinal, population-based study show that individuals hospitalized for a mental health disorder had a fourfold increased relative risk (RR) for developing dementia, compared with those who were not hospitalized with a mental illness.
In addition, those with dementia plus a mental disorder developed dementia almost 6 years earlier than those without a mental illness.
The findings were consistent among men and women, in patients with early- and late-onset dementia, in those with Alzheimer’s and non-Alzheimer’s dementia, and across all mental health disorders – and remained so after accounting for pre-existing physical illness and socioeconomic factors.
“Dementia is not typically treated until later in life, but our study suggests that we need to be thinking about dementia prevention much earlier in the life course,” study investigator Leah Richmond-Rakerd, PhD, assistant professor, department of psychology, University of Michigan, said in an interview.
“Supporting young people’s mental health could be a window of opportunity to help reduce the burden of dementia in older adults,” she said.
The findings were published online Feb. 16.
Underappreciated risk factor
“Recognition of the outsized influence of dementia on later-life functioning has fueled research into modifiable risk factors and prevention targets,” the investigators write.
Previous research suggests mental disorders may “comprise an underappreciated category of modifiable risk factors.” However, those studies focused primarily on midlife and older individuals, not on capturing mental disorders during young adulthood, which is the time of “peak prevalence,” they add. In addition, most studies have not explored the full range of mental disorders.
Dr. Richmond-Rakerd noted that it is well known that mental health disorders peak in adolescence and young adulthood – and are treatable.
“If the same people who have mental disorders when they are young tend to develop dementia when they are older, that would mean that preventing mental health problems in younger people might reduce or delay the burden of dementia in older people,” she said.
The investigators assessed records from the New Zealand Integrated Data Infrastructure, which is a de-identified register that includes the entire New Zealand population. They also examined information about hospitalizations and diagnoses from records kept by the New Zealand Ministry of Health.
The researchers followed 1,711,386 individuals born between 1928 and 1967 (50.6% men, aged 21 to 60 years at baseline) for 30 years. The population was subdivided into age groups based on birth years: 1928-1937 (14.8%), 1938-1947 (20.85%), 1948-1957 (29.35%), and 1958-1967 (35.1%).
Earlier onset
During the study period, 3.8% of individuals were identified as having a mental disorder, and 2% were identified as having dementia. Similar percentages of men and women had a mental disorder, and similar percentages had dementia.
Dementia was “over-represented” among participants with versus without a mental disorder (6.1% vs. 1.8%). This finding held across all age groups.
Those diagnosed with a mental disorder were also more likely to develop dementia, compared with their peers without a mental disorder (RR, 3.51; 95% confidence interval, 3.39-3.64), which is a larger association than that between physical diseases and dementia (RR, 1.19; 95% CI, 1.16-1.21).
These associations were present in both sexes and in all age groups, although the associations were stronger in more recently born cohorts.
A sixfold higher risk for dementia remained even after adjusting for pre-existing physical illnesses (HR, 6.49; 95% CI, 6.25-6.73); and the elevated risk was evident across different lengths of follow-up from the index mental disorder.
When the researchers focused specifically on individuals diagnosed with dementia, they found that those diagnosed with a mental disorder developed dementia a mean of 5.60 years earlier than those without a mental disorder diagnosis – an association observed across both sexes and all age groups.
“Individuals diagnosed with psychotic, substance use, mood, neurotic, and all other mental disorders and who engaged in self-harm were all more likely than those without a mental disorder to be diagnosed with subsequent dementia, even after accounting for their physical disease histories,” the investigators write.
Although there was a link between mental disorders in both Alzheimer’s and non-Alzheimer’s dementias, the association was larger in non-Alzheimer’s.
The researchers note that the study has several limitations, including the fact that it was conducted in New Zealand and therefore the results may not be generalizable to other regions. In addition, inpatient hospital records do not capture less severe mental disorder cases treated in the outpatient setting.
Dr. Richmond-Rakerd suggested several potential mechanisms that could account for the link between mental illness and dementia, including poor lifestyle choices and metabolic side effects associated with some psychiatric medications.
“There could also be shared risk factors for both mental disorders and dementia, such as shared genetics, or individuals may experience a lifelong brain vulnerability that shows up as mental health problems earlier in life and shows up as dementia later in life,” she said.
An important risk factor
Commenting for this article, Ken Duckworth, MD, chief medical officer of the National Alliance on Mental Illness, said a major strength of the study was its longitudinal scope and large population size.
He described the study as allowing clinicians to “watch the movie,” as opposed to looking at a “snapshot” of data.
“Although you can learn things from snapshots, a large, comprehensive public health system looking at 30 years of claims – something not possible in the U.S. because of our more fragmented health care system – offers more insight,” said Dr. Duckworth, who was not involved with the research.
The investigators are “painting a picture of a correlation of risk, and to me, that’s the beginning of further inquiry,” he added. “Would preventive efforts targeting dementia, such as exercise and socialization, be helpful? It’s a great study that raises these interesting questions.”
Also commenting in an interview, Claire Sexton, DPhil, director of scientific programs and outreach at the Alzheimer’s Association, said the study “adds a wealth of data to our understanding” of mental disorders as a dementia risk factor.
However, the study was observational, so “the findings cannot imply causation, [and just] because someone has depression, that does not mean they will go on to develop Alzheimer’s,” said Dr. Sexton, who also was not involved with the research.
Still, “these data support the idea that taking care of one’s mental health is incredibly important for overall wellbeing. For providers, it’s important to have mental health evaluation be a part of your patient’s regular checkups,” she added.
Dr. Richmond-Rakerd noted that even if mental health conditions are not a causal risk factor for dementia, “the presence of a mental health problem is still an important indicator of risk. Mental health providers may wish to target other risk factors for dementia that are more common in individuals with mental health conditions, such as social disconnection.”
The study was funded by grants from the National Institute on Aging, the U.K. Medical Research Council, the National Institute of Child Health and Development through the Duke Population Research Center, and the National Institute on Aging through the Center for Advancing Sociodemographic and Economic Study of Alzheimer’s Disease and Related Dementias. Dr. Richmond-Rakerd reports no relevant financial relationships. The other investigators’ disclosures are listed in the original article. Dr. Sexton and Dr. Duckworth report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Results of a large, longitudinal, population-based study show that individuals hospitalized for a mental health disorder had a fourfold increased relative risk (RR) for developing dementia, compared with those who were not hospitalized with a mental illness.
In addition, those with dementia plus a mental disorder developed dementia almost 6 years earlier than those without a mental illness.
The findings were consistent among men and women, in patients with early- and late-onset dementia, in those with Alzheimer’s and non-Alzheimer’s dementia, and across all mental health disorders – and remained so after accounting for pre-existing physical illness and socioeconomic factors.
“Dementia is not typically treated until later in life, but our study suggests that we need to be thinking about dementia prevention much earlier in the life course,” study investigator Leah Richmond-Rakerd, PhD, assistant professor, department of psychology, University of Michigan, said in an interview.
“Supporting young people’s mental health could be a window of opportunity to help reduce the burden of dementia in older adults,” she said.
The findings were published online Feb. 16.
Underappreciated risk factor
“Recognition of the outsized influence of dementia on later-life functioning has fueled research into modifiable risk factors and prevention targets,” the investigators write.
Previous research suggests mental disorders may “comprise an underappreciated category of modifiable risk factors.” However, those studies focused primarily on midlife and older individuals, not on capturing mental disorders during young adulthood, which is the time of “peak prevalence,” they add. In addition, most studies have not explored the full range of mental disorders.
Dr. Richmond-Rakerd noted that it is well known that mental health disorders peak in adolescence and young adulthood – and are treatable.
“If the same people who have mental disorders when they are young tend to develop dementia when they are older, that would mean that preventing mental health problems in younger people might reduce or delay the burden of dementia in older people,” she said.
The investigators assessed records from the New Zealand Integrated Data Infrastructure, which is a de-identified register that includes the entire New Zealand population. They also examined information about hospitalizations and diagnoses from records kept by the New Zealand Ministry of Health.
The researchers followed 1,711,386 individuals born between 1928 and 1967 (50.6% men, aged 21 to 60 years at baseline) for 30 years. The population was subdivided into age groups based on birth years: 1928-1937 (14.8%), 1938-1947 (20.85%), 1948-1957 (29.35%), and 1958-1967 (35.1%).
Earlier onset
During the study period, 3.8% of individuals were identified as having a mental disorder, and 2% were identified as having dementia. Similar percentages of men and women had a mental disorder, and similar percentages had dementia.
Dementia was “over-represented” among participants with versus without a mental disorder (6.1% vs. 1.8%). This finding held across all age groups.
Those diagnosed with a mental disorder were also more likely to develop dementia, compared with their peers without a mental disorder (RR, 3.51; 95% confidence interval, 3.39-3.64), which is a larger association than that between physical diseases and dementia (RR, 1.19; 95% CI, 1.16-1.21).
These associations were present in both sexes and in all age groups, although the associations were stronger in more recently born cohorts.
A sixfold higher risk for dementia remained even after adjusting for pre-existing physical illnesses (HR, 6.49; 95% CI, 6.25-6.73); and the elevated risk was evident across different lengths of follow-up from the index mental disorder.
When the researchers focused specifically on individuals diagnosed with dementia, they found that those diagnosed with a mental disorder developed dementia a mean of 5.60 years earlier than those without a mental disorder diagnosis – an association observed across both sexes and all age groups.
“Individuals diagnosed with psychotic, substance use, mood, neurotic, and all other mental disorders and who engaged in self-harm were all more likely than those without a mental disorder to be diagnosed with subsequent dementia, even after accounting for their physical disease histories,” the investigators write.
Although there was a link between mental disorders in both Alzheimer’s and non-Alzheimer’s dementias, the association was larger in non-Alzheimer’s.
The researchers note that the study has several limitations, including the fact that it was conducted in New Zealand and therefore the results may not be generalizable to other regions. In addition, inpatient hospital records do not capture less severe mental disorder cases treated in the outpatient setting.
Dr. Richmond-Rakerd suggested several potential mechanisms that could account for the link between mental illness and dementia, including poor lifestyle choices and metabolic side effects associated with some psychiatric medications.
“There could also be shared risk factors for both mental disorders and dementia, such as shared genetics, or individuals may experience a lifelong brain vulnerability that shows up as mental health problems earlier in life and shows up as dementia later in life,” she said.
An important risk factor
Commenting for this article, Ken Duckworth, MD, chief medical officer of the National Alliance on Mental Illness, said a major strength of the study was its longitudinal scope and large population size.
He described the study as allowing clinicians to “watch the movie,” as opposed to looking at a “snapshot” of data.
“Although you can learn things from snapshots, a large, comprehensive public health system looking at 30 years of claims – something not possible in the U.S. because of our more fragmented health care system – offers more insight,” said Dr. Duckworth, who was not involved with the research.
The investigators are “painting a picture of a correlation of risk, and to me, that’s the beginning of further inquiry,” he added. “Would preventive efforts targeting dementia, such as exercise and socialization, be helpful? It’s a great study that raises these interesting questions.”
Also commenting in an interview, Claire Sexton, DPhil, director of scientific programs and outreach at the Alzheimer’s Association, said the study “adds a wealth of data to our understanding” of mental disorders as a dementia risk factor.
However, the study was observational, so “the findings cannot imply causation, [and just] because someone has depression, that does not mean they will go on to develop Alzheimer’s,” said Dr. Sexton, who also was not involved with the research.
Still, “these data support the idea that taking care of one’s mental health is incredibly important for overall wellbeing. For providers, it’s important to have mental health evaluation be a part of your patient’s regular checkups,” she added.
Dr. Richmond-Rakerd noted that even if mental health conditions are not a causal risk factor for dementia, “the presence of a mental health problem is still an important indicator of risk. Mental health providers may wish to target other risk factors for dementia that are more common in individuals with mental health conditions, such as social disconnection.”
The study was funded by grants from the National Institute on Aging, the U.K. Medical Research Council, the National Institute of Child Health and Development through the Duke Population Research Center, and the National Institute on Aging through the Center for Advancing Sociodemographic and Economic Study of Alzheimer’s Disease and Related Dementias. Dr. Richmond-Rakerd reports no relevant financial relationships. The other investigators’ disclosures are listed in the original article. Dr. Sexton and Dr. Duckworth report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA PSYCHIATRY
Robust immune response after COVID-19 boosters in those with IBD
Of the study participants, 93% had detectable antibodies after their initial vaccination series, which increased to 99.5% following an additional dose.
“Most IBD patients, including those who are immune suppressed and/or did not have detectable humoral immune responses following the initial mRNA COVID-19 vaccine series, demonstrate strong immune responses to additional doses of mRNA vaccines,” Michael D. Kappelman, MD, a pediatric gastroenterologist at the University of North Carolina at Chapel Hill, told this news organization.
“These data support an additional vaccine dose of mRNA vaccine in patients at risk for an inadequate response to the initial series,” he said.
Dr. Kappelman presented these findings on behalf of the PREVENT-COVID Study Group as an e-poster at the 17th congress of the European Crohn’s and Colitis Organisation.
A study design to measure boosters’ benefits
For people with Crohn’s disease or ulcerative colitis who are taking immunosuppressants, boosters are generally recommended, Dr. Kappelman and colleagues noted. However, “real-world data on the effectiveness and safety of additional vaccine doses are lacking.”
They studied 659 people with IBD (mean age, 45 years; 72% female), of whom 72% had Crohn’s disease and 27% had ulcerative colitis/unclassified IBD.
Of these participants, 63% received Pfizer/BioNTech vaccine and 37% received the Moderna vaccine. Five participants received the Johnson & Johnson vaccine. In 98% of cases, people who received an mRNA vaccine initially also received the same type for the additional dose.
Participants completed baseline and follow-up surveys. Their blood work was obtained and evaluated 8 weeks after completion of the initial vaccine series and 6 weeks after a booster to measure anti–receptor binding domain IgG antibody levels specific to SARS-CoV-2.
Mean increase in antibody levels was 61 µg/mL in the Pfizer vaccine group and 78 µg/mL in the Moderna vaccine group following the booster shot.
Of the 47 patients without initial antibody response, 45 (96%) had detectable antibodies following an additional dose.
Serious adverse events (AEs) associated with the booster were rare, Dr. Kappelman said. Among participants, 44% reported no AEs, 24% mild AEs, 25% moderate AEs, and 6% reported serious AEs.
“These data can be used to inform vaccine decisions in patients with a broad array of immune-medicated conditions frequently managed by immunosuppression,” the investigators note.
A ‘reassuring’ finding
“This abstract [gives us] an important understanding about how patients with inflammatory bowel disease respond to COVID-19 vaccination. There have been mixed reports in the prior studies regarding how well patients with IBD respond to vaccination,” Jason Ken Hou, MD, said when asked to comment on the research.
The main findings that 99.5% of patients had detectable antibodies after an additional dose “is reassuring, as prior studies have suggested some patients did not develop antibodies after the [initial series],” added Dr. Hou, associate professor of medicine-gastroenterology at Baylor College of Medicine in Houston.
The researchers conducted the study within a previously established, well-known Internet-based cohort of IBD patients, Dr. Hou said. Although the researchers collected information on the IBD medications that patients were taking at the time of vaccination, the analyses that were presented did not compare antibody response rates based on medication.
“Further study is still required, as there is more to vaccination response than detectable antibody alone,” he added.
A version of this article first appeared on Medscape.com.
Of the study participants, 93% had detectable antibodies after their initial vaccination series, which increased to 99.5% following an additional dose.
“Most IBD patients, including those who are immune suppressed and/or did not have detectable humoral immune responses following the initial mRNA COVID-19 vaccine series, demonstrate strong immune responses to additional doses of mRNA vaccines,” Michael D. Kappelman, MD, a pediatric gastroenterologist at the University of North Carolina at Chapel Hill, told this news organization.
“These data support an additional vaccine dose of mRNA vaccine in patients at risk for an inadequate response to the initial series,” he said.
Dr. Kappelman presented these findings on behalf of the PREVENT-COVID Study Group as an e-poster at the 17th congress of the European Crohn’s and Colitis Organisation.
A study design to measure boosters’ benefits
For people with Crohn’s disease or ulcerative colitis who are taking immunosuppressants, boosters are generally recommended, Dr. Kappelman and colleagues noted. However, “real-world data on the effectiveness and safety of additional vaccine doses are lacking.”
They studied 659 people with IBD (mean age, 45 years; 72% female), of whom 72% had Crohn’s disease and 27% had ulcerative colitis/unclassified IBD.
Of these participants, 63% received Pfizer/BioNTech vaccine and 37% received the Moderna vaccine. Five participants received the Johnson & Johnson vaccine. In 98% of cases, people who received an mRNA vaccine initially also received the same type for the additional dose.
Participants completed baseline and follow-up surveys. Their blood work was obtained and evaluated 8 weeks after completion of the initial vaccine series and 6 weeks after a booster to measure anti–receptor binding domain IgG antibody levels specific to SARS-CoV-2.
Mean increase in antibody levels was 61 µg/mL in the Pfizer vaccine group and 78 µg/mL in the Moderna vaccine group following the booster shot.
Of the 47 patients without initial antibody response, 45 (96%) had detectable antibodies following an additional dose.
Serious adverse events (AEs) associated with the booster were rare, Dr. Kappelman said. Among participants, 44% reported no AEs, 24% mild AEs, 25% moderate AEs, and 6% reported serious AEs.
“These data can be used to inform vaccine decisions in patients with a broad array of immune-medicated conditions frequently managed by immunosuppression,” the investigators note.
A ‘reassuring’ finding
“This abstract [gives us] an important understanding about how patients with inflammatory bowel disease respond to COVID-19 vaccination. There have been mixed reports in the prior studies regarding how well patients with IBD respond to vaccination,” Jason Ken Hou, MD, said when asked to comment on the research.
The main findings that 99.5% of patients had detectable antibodies after an additional dose “is reassuring, as prior studies have suggested some patients did not develop antibodies after the [initial series],” added Dr. Hou, associate professor of medicine-gastroenterology at Baylor College of Medicine in Houston.
The researchers conducted the study within a previously established, well-known Internet-based cohort of IBD patients, Dr. Hou said. Although the researchers collected information on the IBD medications that patients were taking at the time of vaccination, the analyses that were presented did not compare antibody response rates based on medication.
“Further study is still required, as there is more to vaccination response than detectable antibody alone,” he added.
A version of this article first appeared on Medscape.com.
Of the study participants, 93% had detectable antibodies after their initial vaccination series, which increased to 99.5% following an additional dose.
“Most IBD patients, including those who are immune suppressed and/or did not have detectable humoral immune responses following the initial mRNA COVID-19 vaccine series, demonstrate strong immune responses to additional doses of mRNA vaccines,” Michael D. Kappelman, MD, a pediatric gastroenterologist at the University of North Carolina at Chapel Hill, told this news organization.
“These data support an additional vaccine dose of mRNA vaccine in patients at risk for an inadequate response to the initial series,” he said.
Dr. Kappelman presented these findings on behalf of the PREVENT-COVID Study Group as an e-poster at the 17th congress of the European Crohn’s and Colitis Organisation.
A study design to measure boosters’ benefits
For people with Crohn’s disease or ulcerative colitis who are taking immunosuppressants, boosters are generally recommended, Dr. Kappelman and colleagues noted. However, “real-world data on the effectiveness and safety of additional vaccine doses are lacking.”
They studied 659 people with IBD (mean age, 45 years; 72% female), of whom 72% had Crohn’s disease and 27% had ulcerative colitis/unclassified IBD.
Of these participants, 63% received Pfizer/BioNTech vaccine and 37% received the Moderna vaccine. Five participants received the Johnson & Johnson vaccine. In 98% of cases, people who received an mRNA vaccine initially also received the same type for the additional dose.
Participants completed baseline and follow-up surveys. Their blood work was obtained and evaluated 8 weeks after completion of the initial vaccine series and 6 weeks after a booster to measure anti–receptor binding domain IgG antibody levels specific to SARS-CoV-2.
Mean increase in antibody levels was 61 µg/mL in the Pfizer vaccine group and 78 µg/mL in the Moderna vaccine group following the booster shot.
Of the 47 patients without initial antibody response, 45 (96%) had detectable antibodies following an additional dose.
Serious adverse events (AEs) associated with the booster were rare, Dr. Kappelman said. Among participants, 44% reported no AEs, 24% mild AEs, 25% moderate AEs, and 6% reported serious AEs.
“These data can be used to inform vaccine decisions in patients with a broad array of immune-medicated conditions frequently managed by immunosuppression,” the investigators note.
A ‘reassuring’ finding
“This abstract [gives us] an important understanding about how patients with inflammatory bowel disease respond to COVID-19 vaccination. There have been mixed reports in the prior studies regarding how well patients with IBD respond to vaccination,” Jason Ken Hou, MD, said when asked to comment on the research.
The main findings that 99.5% of patients had detectable antibodies after an additional dose “is reassuring, as prior studies have suggested some patients did not develop antibodies after the [initial series],” added Dr. Hou, associate professor of medicine-gastroenterology at Baylor College of Medicine in Houston.
The researchers conducted the study within a previously established, well-known Internet-based cohort of IBD patients, Dr. Hou said. Although the researchers collected information on the IBD medications that patients were taking at the time of vaccination, the analyses that were presented did not compare antibody response rates based on medication.
“Further study is still required, as there is more to vaccination response than detectable antibody alone,” he added.
A version of this article first appeared on Medscape.com.
FROM ECCO 2022
First possible case of deer-to-human COVID transmission identified
according a new preprint study that hasn’t yet been peer-reviewed.
Typically, humans spread the virus to deer, and then deer spread it to other deer. But new evidence suggests that the virus could spill over from deer into humans. The researchers identified a COVID-19 case in someone from Ontario who had recently been in contact with deer.
“This particular case, while raising a red flag, doesn’t seem to be hugely alarming,” Finlay Maguire, PhD, one of the study authors and an epidemiologist at Dalhousie University, told CBC News.
“While we haven’t seen [transmission from deer to humans] happen directly, we sampled from the human case around the same time we sampled from the deer, and we sampled from around the same location,” he said. “There is also a plausible link by which it could have happened, in that the individual involved is known to have had considerable contact with deer.”
Dr. Maguire and colleagues have been monitoring the spread of the coronavirus among animals. They analyzed nasal swabs and lymph node samples taken from hundreds of deer that were killed by hunters in fall 2021 in southwestern and eastern Ontario. Among 298 sampled deer, 17 tested positive -- all from southwestern Ontario.
During the analysis, they found a “highly divergent” coronavirus lineage, which means a cluster of samples with many mutations. Around the same time, they found a genetically similar version in a person from the same region.
The study points to the need for better surveillance of the coronavirus, Dr. Maguire told CBC News, including in humans, animals, plants, and the broader environment. Researchers aren’t quite sure how deer contract the virus from humans, but it could happen through contaminated water, direct contact, food, farming, or other animals such as mink.
The coronavirus lineage identified in the study is different from what’s circulating among humans now, and it’s not related to the Delta or Omicron variants. The closest genetic relative came from samples taken from humans and mink in Michigan in 2020, which means the divergent lineage mutated and evolved over time.
“It’s reassuring that we found no evidence of further transmission, during a time when we were doing a lot of sampling and a lot of sequencing,” Samira Mubareka, MD, one of the study authors and a virologist at Sunnybrook Health Sciences Centre, told CBC News.
“If we continue to do this surveillance, we’ll get a much better sense of what the actual risk is,” she said.
So far, the coronavirus has been found in wild white-tailed deer in the northeastern United States and central Canadian provinces.
Other known cases of transmission from animals to humans have been identified in farmed mink and potentially hamsters, the news outlet reported. But for the most part, humans transmit the virus to animals and are most likely to catch the virus from other people.
At the same time, the Public Health Agency of Canada has issued guidance for hunters, trappers, and those who handle wild deer. People should wear gloves, goggles, and a mask when they could be exposed to respiratory tissues and fluids, especially indoors.
Coronaviruses are killed by normal cooking temperatures, the agency said, and there has been no evidence that cooked venison can spread the virus.
A version of this article first appeared on WebMD.com.
according a new preprint study that hasn’t yet been peer-reviewed.
Typically, humans spread the virus to deer, and then deer spread it to other deer. But new evidence suggests that the virus could spill over from deer into humans. The researchers identified a COVID-19 case in someone from Ontario who had recently been in contact with deer.
“This particular case, while raising a red flag, doesn’t seem to be hugely alarming,” Finlay Maguire, PhD, one of the study authors and an epidemiologist at Dalhousie University, told CBC News.
“While we haven’t seen [transmission from deer to humans] happen directly, we sampled from the human case around the same time we sampled from the deer, and we sampled from around the same location,” he said. “There is also a plausible link by which it could have happened, in that the individual involved is known to have had considerable contact with deer.”
Dr. Maguire and colleagues have been monitoring the spread of the coronavirus among animals. They analyzed nasal swabs and lymph node samples taken from hundreds of deer that were killed by hunters in fall 2021 in southwestern and eastern Ontario. Among 298 sampled deer, 17 tested positive -- all from southwestern Ontario.
During the analysis, they found a “highly divergent” coronavirus lineage, which means a cluster of samples with many mutations. Around the same time, they found a genetically similar version in a person from the same region.
The study points to the need for better surveillance of the coronavirus, Dr. Maguire told CBC News, including in humans, animals, plants, and the broader environment. Researchers aren’t quite sure how deer contract the virus from humans, but it could happen through contaminated water, direct contact, food, farming, or other animals such as mink.
The coronavirus lineage identified in the study is different from what’s circulating among humans now, and it’s not related to the Delta or Omicron variants. The closest genetic relative came from samples taken from humans and mink in Michigan in 2020, which means the divergent lineage mutated and evolved over time.
“It’s reassuring that we found no evidence of further transmission, during a time when we were doing a lot of sampling and a lot of sequencing,” Samira Mubareka, MD, one of the study authors and a virologist at Sunnybrook Health Sciences Centre, told CBC News.
“If we continue to do this surveillance, we’ll get a much better sense of what the actual risk is,” she said.
So far, the coronavirus has been found in wild white-tailed deer in the northeastern United States and central Canadian provinces.
Other known cases of transmission from animals to humans have been identified in farmed mink and potentially hamsters, the news outlet reported. But for the most part, humans transmit the virus to animals and are most likely to catch the virus from other people.
At the same time, the Public Health Agency of Canada has issued guidance for hunters, trappers, and those who handle wild deer. People should wear gloves, goggles, and a mask when they could be exposed to respiratory tissues and fluids, especially indoors.
Coronaviruses are killed by normal cooking temperatures, the agency said, and there has been no evidence that cooked venison can spread the virus.
A version of this article first appeared on WebMD.com.
according a new preprint study that hasn’t yet been peer-reviewed.
Typically, humans spread the virus to deer, and then deer spread it to other deer. But new evidence suggests that the virus could spill over from deer into humans. The researchers identified a COVID-19 case in someone from Ontario who had recently been in contact with deer.
“This particular case, while raising a red flag, doesn’t seem to be hugely alarming,” Finlay Maguire, PhD, one of the study authors and an epidemiologist at Dalhousie University, told CBC News.
“While we haven’t seen [transmission from deer to humans] happen directly, we sampled from the human case around the same time we sampled from the deer, and we sampled from around the same location,” he said. “There is also a plausible link by which it could have happened, in that the individual involved is known to have had considerable contact with deer.”
Dr. Maguire and colleagues have been monitoring the spread of the coronavirus among animals. They analyzed nasal swabs and lymph node samples taken from hundreds of deer that were killed by hunters in fall 2021 in southwestern and eastern Ontario. Among 298 sampled deer, 17 tested positive -- all from southwestern Ontario.
During the analysis, they found a “highly divergent” coronavirus lineage, which means a cluster of samples with many mutations. Around the same time, they found a genetically similar version in a person from the same region.
The study points to the need for better surveillance of the coronavirus, Dr. Maguire told CBC News, including in humans, animals, plants, and the broader environment. Researchers aren’t quite sure how deer contract the virus from humans, but it could happen through contaminated water, direct contact, food, farming, or other animals such as mink.
The coronavirus lineage identified in the study is different from what’s circulating among humans now, and it’s not related to the Delta or Omicron variants. The closest genetic relative came from samples taken from humans and mink in Michigan in 2020, which means the divergent lineage mutated and evolved over time.
“It’s reassuring that we found no evidence of further transmission, during a time when we were doing a lot of sampling and a lot of sequencing,” Samira Mubareka, MD, one of the study authors and a virologist at Sunnybrook Health Sciences Centre, told CBC News.
“If we continue to do this surveillance, we’ll get a much better sense of what the actual risk is,” she said.
So far, the coronavirus has been found in wild white-tailed deer in the northeastern United States and central Canadian provinces.
Other known cases of transmission from animals to humans have been identified in farmed mink and potentially hamsters, the news outlet reported. But for the most part, humans transmit the virus to animals and are most likely to catch the virus from other people.
At the same time, the Public Health Agency of Canada has issued guidance for hunters, trappers, and those who handle wild deer. People should wear gloves, goggles, and a mask when they could be exposed to respiratory tissues and fluids, especially indoors.
Coronaviruses are killed by normal cooking temperatures, the agency said, and there has been no evidence that cooked venison can spread the virus.
A version of this article first appeared on WebMD.com.
Direct specialty care: Concierge service without the price tag
Four years ago, I was fully employed in a “traditional” rheumatology clinic. I met Alan, a 42-year-old gentleman who was a high school math teacher in my town. He was the first patient on my panel that day. Once I entered the examining room, Alan greeted me with: “You are the third rheumatologist who I have consulted for what everybody believes is fibromyalgia. I am paying out of pocket to see you as you are not on my insurance panel. I have researched your background, and I have high expectations of you.” He was cutting to the chase.

Alan had struggled with pain for about 1½ years. He insisted that he was very healthy before his symptoms started abruptly. In the past 2 years, his personal life had been under much stress as he was caring for a disabled child and facing an imminent divorce. While his symptoms were suggestive of an inflammatory arthritis, his workup was not. Unfortunately, the allocated time with Alan was 15 minutes – too short to cover both medical and personal struggles. Meanwhile, my nurses had to room in another two patients. I felt rushed and responsible for not letting the others wait. I asked Alan to keep a diary of his symptoms and come back in 2 weeks. A few minutes after discharging Alan, my nurse followed and asked me: “Where would you like me to add this patient, as you have no openings for 4 months ?”
“Overbook him!” I said.
This was happening almost every day. Scheduled patients, overbooked patients, tens of emails, calls to patients, and fights with insurance companies to approve tests and medications. Nearly every day I was getting home, preparing dinner, feeding my family, and going back to writing notes, as I would be financially penalized if my notes were not submitted in 24-48 hours. I had no time for my family and didn’t even think about any hobbies.
In 2 weeks, Alan came back for his visit. That day, I paid someone to take my kids to school and came to my office earlier. We had 1 hour to talk about his history. At the end of the visit, Alan said: “What kind of doctor are you? You looked into my eyes while I was talking, and you didn’t touch the computer keyboard?!” His remark was not uncommon for me. Most patients complain that physicians spend more time typing than looking at them. Maybe patients do not realize, but this is the only way that physicians get paid: writing the “proper notes” and placing the correct billing code.
Alan was diagnosed and treated successfully for seronegative rheumatoid arthritis. In 1 year, paying out of pocket to see me, he ended up spending many, many thousands of dollars. As you can imagine, I was not in control of those bills.
After 4 years in the traditional system, I decided to change something for my patients and for myself as their physician, and as a mother of three kids, a wife, a daughter, and a sister.
I decided to create a clinic where I am comfortable practicing “uncomplicated” medicine, as a friend of mine said. Today, insurance companies are restricting patients to limited panels of specialists. They dictate patients’ care, giving the false impression that they will save money. Insurance companies interfere with the physician’s medical judgment. They make algorithms to approve tests and have preferred lists of medication. They decide whether a test or a medication is appropriate for you. In addition, they don’t disclose how much they pay for your consultation, tests, and medication, and they ban the contracted parties from disclosing this information. They force patients to use their testing facilities and mailing pharmacies. Although patients and employers are the payers, they do not have access to their insurance companies’ “real” prices.
I decided that it was time to take control of my time spent with patients to make my services available when patients need me, without becoming a financial burden. I created a clinic where patients do not have copayments and will never receive a “surprise bill.” All costs are transparent to patients, including laboratory and imaging tests. Patients can talk to me on the phone, send a text, or email. A clinic where patients can talk to the physician on the phone or send a text or email? This is direct specialty care.
Is direct care a new concept? No, not at all. Is direct care the same as concierge medicine? I think it is a type of concierge service, but without the price tag.
Why?
Physicians practicing the traditional concierge medicine model here in the United States still bill patients’ insurance. In addition, to make their practice profitable, they charge a retainer fee that will allow them to keep a small patient panel. In contrast, direct care specialists do not have a contract with insurance companies.
I believe that both concierge medicine and direct care specialists offer exceptional care and better access to physicians. The difference is in costs: One is more expensive than the other. Traditional concierge medicine practices usually ask for high retainer fees in addition to copayments for visits. They do not offer any access to discounted pricing for laboratory or imaging tests. Patients continue to receive surprise bills from their insurance company.
Why don’t direct specialty care practices contract with insurance companies? Contracting with insurance companies increases a practice’s overhead costs (as more money is spent on coding and billing services and more office staff). When practice overhead is lower, the cost of patient care can be significantly lower. Patients pay a monthly membership to become a direct specialty care practice member. The membership covers the cost of visits and access to the benefits of the practice. In addition, direct care specialists do not charge copayments or send surprise bills. They can contract directly with laboratory and imaging centers and offer discounted prices. Patients with insurance are welcome to use it to cover tests, imaging, and medication. The patient has the power to choose between paying a cash price versus a “covered” service.
Most young patients, like Alan, have a high-deductible plan. A few regular blood tests might cost a patient hundreds of dollars before meeting a deductible. One MRI scan costs $4,000-$6,000. Patients who join a direct specialty care practice pay $30-$40 for regular labs and $400-$500 for an MRI.
I am now 2 years into practicing medicine as a direct care specialist. It is not a dream anymore. Yes, you may call it “concierge medicine without the price tag.” I call it “direct specialty care.” My patients and I are both accountable to one another. Together, we make a plan, and we have the time to implement it.
I am not alone. Other specialists are embracing this model. That is why we created the Direct Specialty Care Alliance, a place where physicians are welcome to network and share with others what they have learned along their journeys.
After I started my company, Alan was one of the first patients to join. He embraced my practice model and became one of the ambassadors of the direct specialty care movement. He is back to a normal life of taking care of his family, getting his wife back, and teaching math to high school kids.
Dr Girnita is the CEO and founder of RheumatologistOnCall, actively seeing patients via telemedicine in 10 U.S. states. She is an advocate for digital health and telemedicine that will empower physicians and patients to take charge of their medical care. She is a cofounder of the Direct Specialty Care Alliance. She disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Four years ago, I was fully employed in a “traditional” rheumatology clinic. I met Alan, a 42-year-old gentleman who was a high school math teacher in my town. He was the first patient on my panel that day. Once I entered the examining room, Alan greeted me with: “You are the third rheumatologist who I have consulted for what everybody believes is fibromyalgia. I am paying out of pocket to see you as you are not on my insurance panel. I have researched your background, and I have high expectations of you.” He was cutting to the chase.
Alan had struggled with pain for about 1½ years. He insisted that he was very healthy before his symptoms started abruptly. In the past 2 years, his personal life had been under much stress as he was caring for a disabled child and facing an imminent divorce. While his symptoms were suggestive of an inflammatory arthritis, his workup was not. Unfortunately, the allocated time with Alan was 15 minutes – too short to cover both medical and personal struggles. Meanwhile, my nurses had to room in another two patients. I felt rushed and responsible for not letting the others wait. I asked Alan to keep a diary of his symptoms and come back in 2 weeks. A few minutes after discharging Alan, my nurse followed and asked me: “Where would you like me to add this patient, as you have no openings for 4 months ?”
“Overbook him!” I said.
This was happening almost every day. Scheduled patients, overbooked patients, tens of emails, calls to patients, and fights with insurance companies to approve tests and medications. Nearly every day I was getting home, preparing dinner, feeding my family, and going back to writing notes, as I would be financially penalized if my notes were not submitted in 24-48 hours. I had no time for my family and didn’t even think about any hobbies.
In 2 weeks, Alan came back for his visit. That day, I paid someone to take my kids to school and came to my office earlier. We had 1 hour to talk about his history. At the end of the visit, Alan said: “What kind of doctor are you? You looked into my eyes while I was talking, and you didn’t touch the computer keyboard?!” His remark was not uncommon for me. Most patients complain that physicians spend more time typing than looking at them. Maybe patients do not realize, but this is the only way that physicians get paid: writing the “proper notes” and placing the correct billing code.
Alan was diagnosed and treated successfully for seronegative rheumatoid arthritis. In 1 year, paying out of pocket to see me, he ended up spending many, many thousands of dollars. As you can imagine, I was not in control of those bills.
After 4 years in the traditional system, I decided to change something for my patients and for myself as their physician, and as a mother of three kids, a wife, a daughter, and a sister.
I decided to create a clinic where I am comfortable practicing “uncomplicated” medicine, as a friend of mine said. Today, insurance companies are restricting patients to limited panels of specialists. They dictate patients’ care, giving the false impression that they will save money. Insurance companies interfere with the physician’s medical judgment. They make algorithms to approve tests and have preferred lists of medication. They decide whether a test or a medication is appropriate for you. In addition, they don’t disclose how much they pay for your consultation, tests, and medication, and they ban the contracted parties from disclosing this information. They force patients to use their testing facilities and mailing pharmacies. Although patients and employers are the payers, they do not have access to their insurance companies’ “real” prices.
I decided that it was time to take control of my time spent with patients to make my services available when patients need me, without becoming a financial burden. I created a clinic where patients do not have copayments and will never receive a “surprise bill.” All costs are transparent to patients, including laboratory and imaging tests. Patients can talk to me on the phone, send a text, or email. A clinic where patients can talk to the physician on the phone or send a text or email? This is direct specialty care.
Is direct care a new concept? No, not at all. Is direct care the same as concierge medicine? I think it is a type of concierge service, but without the price tag.
Why?
Physicians practicing the traditional concierge medicine model here in the United States still bill patients’ insurance. In addition, to make their practice profitable, they charge a retainer fee that will allow them to keep a small patient panel. In contrast, direct care specialists do not have a contract with insurance companies.
I believe that both concierge medicine and direct care specialists offer exceptional care and better access to physicians. The difference is in costs: One is more expensive than the other. Traditional concierge medicine practices usually ask for high retainer fees in addition to copayments for visits. They do not offer any access to discounted pricing for laboratory or imaging tests. Patients continue to receive surprise bills from their insurance company.
Why don’t direct specialty care practices contract with insurance companies? Contracting with insurance companies increases a practice’s overhead costs (as more money is spent on coding and billing services and more office staff). When practice overhead is lower, the cost of patient care can be significantly lower. Patients pay a monthly membership to become a direct specialty care practice member. The membership covers the cost of visits and access to the benefits of the practice. In addition, direct care specialists do not charge copayments or send surprise bills. They can contract directly with laboratory and imaging centers and offer discounted prices. Patients with insurance are welcome to use it to cover tests, imaging, and medication. The patient has the power to choose between paying a cash price versus a “covered” service.
Most young patients, like Alan, have a high-deductible plan. A few regular blood tests might cost a patient hundreds of dollars before meeting a deductible. One MRI scan costs $4,000-$6,000. Patients who join a direct specialty care practice pay $30-$40 for regular labs and $400-$500 for an MRI.
I am now 2 years into practicing medicine as a direct care specialist. It is not a dream anymore. Yes, you may call it “concierge medicine without the price tag.” I call it “direct specialty care.” My patients and I are both accountable to one another. Together, we make a plan, and we have the time to implement it.
I am not alone. Other specialists are embracing this model. That is why we created the Direct Specialty Care Alliance, a place where physicians are welcome to network and share with others what they have learned along their journeys.
After I started my company, Alan was one of the first patients to join. He embraced my practice model and became one of the ambassadors of the direct specialty care movement. He is back to a normal life of taking care of his family, getting his wife back, and teaching math to high school kids.
Dr Girnita is the CEO and founder of RheumatologistOnCall, actively seeing patients via telemedicine in 10 U.S. states. She is an advocate for digital health and telemedicine that will empower physicians and patients to take charge of their medical care. She is a cofounder of the Direct Specialty Care Alliance. She disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Four years ago, I was fully employed in a “traditional” rheumatology clinic. I met Alan, a 42-year-old gentleman who was a high school math teacher in my town. He was the first patient on my panel that day. Once I entered the examining room, Alan greeted me with: “You are the third rheumatologist who I have consulted for what everybody believes is fibromyalgia. I am paying out of pocket to see you as you are not on my insurance panel. I have researched your background, and I have high expectations of you.” He was cutting to the chase.
Alan had struggled with pain for about 1½ years. He insisted that he was very healthy before his symptoms started abruptly. In the past 2 years, his personal life had been under much stress as he was caring for a disabled child and facing an imminent divorce. While his symptoms were suggestive of an inflammatory arthritis, his workup was not. Unfortunately, the allocated time with Alan was 15 minutes – too short to cover both medical and personal struggles. Meanwhile, my nurses had to room in another two patients. I felt rushed and responsible for not letting the others wait. I asked Alan to keep a diary of his symptoms and come back in 2 weeks. A few minutes after discharging Alan, my nurse followed and asked me: “Where would you like me to add this patient, as you have no openings for 4 months ?”
“Overbook him!” I said.
This was happening almost every day. Scheduled patients, overbooked patients, tens of emails, calls to patients, and fights with insurance companies to approve tests and medications. Nearly every day I was getting home, preparing dinner, feeding my family, and going back to writing notes, as I would be financially penalized if my notes were not submitted in 24-48 hours. I had no time for my family and didn’t even think about any hobbies.
In 2 weeks, Alan came back for his visit. That day, I paid someone to take my kids to school and came to my office earlier. We had 1 hour to talk about his history. At the end of the visit, Alan said: “What kind of doctor are you? You looked into my eyes while I was talking, and you didn’t touch the computer keyboard?!” His remark was not uncommon for me. Most patients complain that physicians spend more time typing than looking at them. Maybe patients do not realize, but this is the only way that physicians get paid: writing the “proper notes” and placing the correct billing code.
Alan was diagnosed and treated successfully for seronegative rheumatoid arthritis. In 1 year, paying out of pocket to see me, he ended up spending many, many thousands of dollars. As you can imagine, I was not in control of those bills.
After 4 years in the traditional system, I decided to change something for my patients and for myself as their physician, and as a mother of three kids, a wife, a daughter, and a sister.
I decided to create a clinic where I am comfortable practicing “uncomplicated” medicine, as a friend of mine said. Today, insurance companies are restricting patients to limited panels of specialists. They dictate patients’ care, giving the false impression that they will save money. Insurance companies interfere with the physician’s medical judgment. They make algorithms to approve tests and have preferred lists of medication. They decide whether a test or a medication is appropriate for you. In addition, they don’t disclose how much they pay for your consultation, tests, and medication, and they ban the contracted parties from disclosing this information. They force patients to use their testing facilities and mailing pharmacies. Although patients and employers are the payers, they do not have access to their insurance companies’ “real” prices.
I decided that it was time to take control of my time spent with patients to make my services available when patients need me, without becoming a financial burden. I created a clinic where patients do not have copayments and will never receive a “surprise bill.” All costs are transparent to patients, including laboratory and imaging tests. Patients can talk to me on the phone, send a text, or email. A clinic where patients can talk to the physician on the phone or send a text or email? This is direct specialty care.
Is direct care a new concept? No, not at all. Is direct care the same as concierge medicine? I think it is a type of concierge service, but without the price tag.
Why?
Physicians practicing the traditional concierge medicine model here in the United States still bill patients’ insurance. In addition, to make their practice profitable, they charge a retainer fee that will allow them to keep a small patient panel. In contrast, direct care specialists do not have a contract with insurance companies.
I believe that both concierge medicine and direct care specialists offer exceptional care and better access to physicians. The difference is in costs: One is more expensive than the other. Traditional concierge medicine practices usually ask for high retainer fees in addition to copayments for visits. They do not offer any access to discounted pricing for laboratory or imaging tests. Patients continue to receive surprise bills from their insurance company.
Why don’t direct specialty care practices contract with insurance companies? Contracting with insurance companies increases a practice’s overhead costs (as more money is spent on coding and billing services and more office staff). When practice overhead is lower, the cost of patient care can be significantly lower. Patients pay a monthly membership to become a direct specialty care practice member. The membership covers the cost of visits and access to the benefits of the practice. In addition, direct care specialists do not charge copayments or send surprise bills. They can contract directly with laboratory and imaging centers and offer discounted prices. Patients with insurance are welcome to use it to cover tests, imaging, and medication. The patient has the power to choose between paying a cash price versus a “covered” service.
Most young patients, like Alan, have a high-deductible plan. A few regular blood tests might cost a patient hundreds of dollars before meeting a deductible. One MRI scan costs $4,000-$6,000. Patients who join a direct specialty care practice pay $30-$40 for regular labs and $400-$500 for an MRI.
I am now 2 years into practicing medicine as a direct care specialist. It is not a dream anymore. Yes, you may call it “concierge medicine without the price tag.” I call it “direct specialty care.” My patients and I are both accountable to one another. Together, we make a plan, and we have the time to implement it.
I am not alone. Other specialists are embracing this model. That is why we created the Direct Specialty Care Alliance, a place where physicians are welcome to network and share with others what they have learned along their journeys.
After I started my company, Alan was one of the first patients to join. He embraced my practice model and became one of the ambassadors of the direct specialty care movement. He is back to a normal life of taking care of his family, getting his wife back, and teaching math to high school kids.
Dr Girnita is the CEO and founder of RheumatologistOnCall, actively seeing patients via telemedicine in 10 U.S. states. She is an advocate for digital health and telemedicine that will empower physicians and patients to take charge of their medical care. She is a cofounder of the Direct Specialty Care Alliance. She disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.