User login
On the receiving end of care
It’s tough being on the receiving end of care. I’ve tried to avoid it as much as possible, being ever mindful of the law from Samuel Shem’s The House of God: “They can always hurt you more.”
Fortunately, each procedure went more smoothly than the prior one.
The first was not so elective. I had some uncomfortable symptoms while exercising and, not wanting to totally be in denial, contacted my doctor to ensure that it was not cardiac in origin since symptoms are often atypical in women.
My physician promptly saw me, then scheduled a nuclear stress test. There was a series of needless glitches. Registration at the diagnostic center had me on their schedule but did not have an order. They would have canceled the procedure had I not been able to get hold of the doctor’s office. Why isn’t an order automatically entered when the physician schedules the test?
While I was given the euphemistic “Patient Rights” brochure, asking to have reports sent to a physician outside of the University of Pittsburgh Medical Center empire is apparently not included.
The staff canceled the stress test because I was not fasting. I had received no instructions from diagnostic cardiology. They suggested it was my internist’s responsibility.
I deliberately ate (2 hours earlier) because my trainer always wants me to eat a light meal so I don’t get hypoglycemic during our workouts, and an exercise stress test, is, of course, a workout. The nurse practitioner said that they were concerned I would vomit. I offered to sign a waiver. She parried, saying they would not be able to get adequate images, so I was out of luck.
When I expressed concern about getting hypoglycemic and having difficulty with the test if fasting, the tech said I should bring a soda and snack. Who tells a “borderline” diabetic to bring a soda?
The tech also said she had called our home to give instructions but encountered a busy signal and had not had time to call back. I had not left the house during the prior week (or most of the past 2 years), so this was a pretty lame excuse.
I suggested to the administration that the hospital offer to email the patient instructions well ahead of time (and perhaps ask for confirmation of receipt). If calling, they should try more than once. They should also have patient instruction sheets at the physician’s office and perhaps have them on their website.
It turns out that the hospital mailed me instructions, not on the date it was ordered, but with the postmark being the day of the procedure itself. With Trump donor Louis DeJoy in charge of the U.S. Postal Service, mail across town now has to travel to Baltimore, 3 hours away, be sorted, and returned.
I did finally have the stress test, which was reassuringly normal. I was not surprised, given that the fury I felt on the first attempt had not precipitated symptoms. The hospital sent a patient ombudsman to meet me there to discuss my previous complaints. I have no idea if they implemented any of the changes I had suggested. In 2021, when I urgently had to take my husband to the ED, I couldn’t see the sign pointing toward the ED and had to ask for directions at the main entrance. They said they would fix that promptly but still have not improved the signage. How I miss the friendly community hospital we had before!
Next was trigger-finger surgery. I had developed that in 1978 from using crutches after a fall. I figured that the relative lull in COVID and my activities made it as good a time as any to finally have it fixed. The surgicenter was great; the surgeon was someone I had worked with and respected for decades. The only glitch was not really knowing how long I was going to be out of commission.
The third encounter (at yet another institution) went really well, despite some early administrative glitches. My major complaint was with the lack of communication between preoperative anesthesia and the operating room and the lack of personalization of preoperative instructions. Despite EPIC, medicines were not correctly reconciled between the different encounters, even on the same day!
After about 15 years of diplopia, which has been gradually worsening, my eye doc had suggested that I consider strabismus surgery as a sort of last-ditch effort to improve my quality of life.
Anesthesiology has stock instructions, which they made no effort to individualize. For example, there is no reason to stop NSAIDs a week before such minor surgery. That’s a problem if you depend on NSAIDs for pain control. Similarly, nothing by mouth after midnight is passé and could be tailored for the patient. I felt particularly inconvenienced that I had to go out of town for the preoperative visit and then have a redundant preoperative clearance by my physician.
The nurses in the preoperative area made me feel quite comfortable and as relaxed as I could be under the circumstances. They had a good sense of humor, which helped too. And from the time I met him a few weeks earlier, I instantly liked my surgeon and felt very comfortable with him and had complete trust.
I was pleased that the chief anesthesiologist responded promptly and undefensively to my letter expressing concerns. I do believe that he will try to improve the systemic problems.
The best part: The surgery appears to have been successful and I should have a significantly improved quality of life.
Hospitals could do so much better by improving communications with patients and by viewing them as customers whose loyalty they must earn and will value. With monopolies growing, memories of such care are quickly fading, soon to be as extinct as the family doc who made house calls.
Dr. Stone is an infectious disease specialist and author of Resilience: One Family’s Story of Hope and Triumph over Evil and Conducting Clinical Research: A Practical Guide. She disclosed no relevant financial relationships. A version of this article first appeared on Medscape.com.
It’s tough being on the receiving end of care. I’ve tried to avoid it as much as possible, being ever mindful of the law from Samuel Shem’s The House of God: “They can always hurt you more.”
Fortunately, each procedure went more smoothly than the prior one.
The first was not so elective. I had some uncomfortable symptoms while exercising and, not wanting to totally be in denial, contacted my doctor to ensure that it was not cardiac in origin since symptoms are often atypical in women.
My physician promptly saw me, then scheduled a nuclear stress test. There was a series of needless glitches. Registration at the diagnostic center had me on their schedule but did not have an order. They would have canceled the procedure had I not been able to get hold of the doctor’s office. Why isn’t an order automatically entered when the physician schedules the test?
While I was given the euphemistic “Patient Rights” brochure, asking to have reports sent to a physician outside of the University of Pittsburgh Medical Center empire is apparently not included.
The staff canceled the stress test because I was not fasting. I had received no instructions from diagnostic cardiology. They suggested it was my internist’s responsibility.
I deliberately ate (2 hours earlier) because my trainer always wants me to eat a light meal so I don’t get hypoglycemic during our workouts, and an exercise stress test, is, of course, a workout. The nurse practitioner said that they were concerned I would vomit. I offered to sign a waiver. She parried, saying they would not be able to get adequate images, so I was out of luck.
When I expressed concern about getting hypoglycemic and having difficulty with the test if fasting, the tech said I should bring a soda and snack. Who tells a “borderline” diabetic to bring a soda?
The tech also said she had called our home to give instructions but encountered a busy signal and had not had time to call back. I had not left the house during the prior week (or most of the past 2 years), so this was a pretty lame excuse.
I suggested to the administration that the hospital offer to email the patient instructions well ahead of time (and perhaps ask for confirmation of receipt). If calling, they should try more than once. They should also have patient instruction sheets at the physician’s office and perhaps have them on their website.
It turns out that the hospital mailed me instructions, not on the date it was ordered, but with the postmark being the day of the procedure itself. With Trump donor Louis DeJoy in charge of the U.S. Postal Service, mail across town now has to travel to Baltimore, 3 hours away, be sorted, and returned.
I did finally have the stress test, which was reassuringly normal. I was not surprised, given that the fury I felt on the first attempt had not precipitated symptoms. The hospital sent a patient ombudsman to meet me there to discuss my previous complaints. I have no idea if they implemented any of the changes I had suggested. In 2021, when I urgently had to take my husband to the ED, I couldn’t see the sign pointing toward the ED and had to ask for directions at the main entrance. They said they would fix that promptly but still have not improved the signage. How I miss the friendly community hospital we had before!
Next was trigger-finger surgery. I had developed that in 1978 from using crutches after a fall. I figured that the relative lull in COVID and my activities made it as good a time as any to finally have it fixed. The surgicenter was great; the surgeon was someone I had worked with and respected for decades. The only glitch was not really knowing how long I was going to be out of commission.
The third encounter (at yet another institution) went really well, despite some early administrative glitches. My major complaint was with the lack of communication between preoperative anesthesia and the operating room and the lack of personalization of preoperative instructions. Despite EPIC, medicines were not correctly reconciled between the different encounters, even on the same day!
After about 15 years of diplopia, which has been gradually worsening, my eye doc had suggested that I consider strabismus surgery as a sort of last-ditch effort to improve my quality of life.
Anesthesiology has stock instructions, which they made no effort to individualize. For example, there is no reason to stop NSAIDs a week before such minor surgery. That’s a problem if you depend on NSAIDs for pain control. Similarly, nothing by mouth after midnight is passé and could be tailored for the patient. I felt particularly inconvenienced that I had to go out of town for the preoperative visit and then have a redundant preoperative clearance by my physician.
The nurses in the preoperative area made me feel quite comfortable and as relaxed as I could be under the circumstances. They had a good sense of humor, which helped too. And from the time I met him a few weeks earlier, I instantly liked my surgeon and felt very comfortable with him and had complete trust.
I was pleased that the chief anesthesiologist responded promptly and undefensively to my letter expressing concerns. I do believe that he will try to improve the systemic problems.
The best part: The surgery appears to have been successful and I should have a significantly improved quality of life.
Hospitals could do so much better by improving communications with patients and by viewing them as customers whose loyalty they must earn and will value. With monopolies growing, memories of such care are quickly fading, soon to be as extinct as the family doc who made house calls.
Dr. Stone is an infectious disease specialist and author of Resilience: One Family’s Story of Hope and Triumph over Evil and Conducting Clinical Research: A Practical Guide. She disclosed no relevant financial relationships. A version of this article first appeared on Medscape.com.
It’s tough being on the receiving end of care. I’ve tried to avoid it as much as possible, being ever mindful of the law from Samuel Shem’s The House of God: “They can always hurt you more.”
Fortunately, each procedure went more smoothly than the prior one.
The first was not so elective. I had some uncomfortable symptoms while exercising and, not wanting to totally be in denial, contacted my doctor to ensure that it was not cardiac in origin since symptoms are often atypical in women.
My physician promptly saw me, then scheduled a nuclear stress test. There was a series of needless glitches. Registration at the diagnostic center had me on their schedule but did not have an order. They would have canceled the procedure had I not been able to get hold of the doctor’s office. Why isn’t an order automatically entered when the physician schedules the test?
While I was given the euphemistic “Patient Rights” brochure, asking to have reports sent to a physician outside of the University of Pittsburgh Medical Center empire is apparently not included.
The staff canceled the stress test because I was not fasting. I had received no instructions from diagnostic cardiology. They suggested it was my internist’s responsibility.
I deliberately ate (2 hours earlier) because my trainer always wants me to eat a light meal so I don’t get hypoglycemic during our workouts, and an exercise stress test, is, of course, a workout. The nurse practitioner said that they were concerned I would vomit. I offered to sign a waiver. She parried, saying they would not be able to get adequate images, so I was out of luck.
When I expressed concern about getting hypoglycemic and having difficulty with the test if fasting, the tech said I should bring a soda and snack. Who tells a “borderline” diabetic to bring a soda?
The tech also said she had called our home to give instructions but encountered a busy signal and had not had time to call back. I had not left the house during the prior week (or most of the past 2 years), so this was a pretty lame excuse.
I suggested to the administration that the hospital offer to email the patient instructions well ahead of time (and perhaps ask for confirmation of receipt). If calling, they should try more than once. They should also have patient instruction sheets at the physician’s office and perhaps have them on their website.
It turns out that the hospital mailed me instructions, not on the date it was ordered, but with the postmark being the day of the procedure itself. With Trump donor Louis DeJoy in charge of the U.S. Postal Service, mail across town now has to travel to Baltimore, 3 hours away, be sorted, and returned.
I did finally have the stress test, which was reassuringly normal. I was not surprised, given that the fury I felt on the first attempt had not precipitated symptoms. The hospital sent a patient ombudsman to meet me there to discuss my previous complaints. I have no idea if they implemented any of the changes I had suggested. In 2021, when I urgently had to take my husband to the ED, I couldn’t see the sign pointing toward the ED and had to ask for directions at the main entrance. They said they would fix that promptly but still have not improved the signage. How I miss the friendly community hospital we had before!
Next was trigger-finger surgery. I had developed that in 1978 from using crutches after a fall. I figured that the relative lull in COVID and my activities made it as good a time as any to finally have it fixed. The surgicenter was great; the surgeon was someone I had worked with and respected for decades. The only glitch was not really knowing how long I was going to be out of commission.
The third encounter (at yet another institution) went really well, despite some early administrative glitches. My major complaint was with the lack of communication between preoperative anesthesia and the operating room and the lack of personalization of preoperative instructions. Despite EPIC, medicines were not correctly reconciled between the different encounters, even on the same day!
After about 15 years of diplopia, which has been gradually worsening, my eye doc had suggested that I consider strabismus surgery as a sort of last-ditch effort to improve my quality of life.
Anesthesiology has stock instructions, which they made no effort to individualize. For example, there is no reason to stop NSAIDs a week before such minor surgery. That’s a problem if you depend on NSAIDs for pain control. Similarly, nothing by mouth after midnight is passé and could be tailored for the patient. I felt particularly inconvenienced that I had to go out of town for the preoperative visit and then have a redundant preoperative clearance by my physician.
The nurses in the preoperative area made me feel quite comfortable and as relaxed as I could be under the circumstances. They had a good sense of humor, which helped too. And from the time I met him a few weeks earlier, I instantly liked my surgeon and felt very comfortable with him and had complete trust.
I was pleased that the chief anesthesiologist responded promptly and undefensively to my letter expressing concerns. I do believe that he will try to improve the systemic problems.
The best part: The surgery appears to have been successful and I should have a significantly improved quality of life.
Hospitals could do so much better by improving communications with patients and by viewing them as customers whose loyalty they must earn and will value. With monopolies growing, memories of such care are quickly fading, soon to be as extinct as the family doc who made house calls.
Dr. Stone is an infectious disease specialist and author of Resilience: One Family’s Story of Hope and Triumph over Evil and Conducting Clinical Research: A Practical Guide. She disclosed no relevant financial relationships. A version of this article first appeared on Medscape.com.
Global melanoma incidence high and on the rise
Even by cautious calculations,
An estimated 325,000 people worldwide received a new diagnosis of cutaneous melanoma in 2020, and if present trends continue, the incidence of new cases is predicted to increase by about 50% in 2040, with melanoma deaths expected to rise by almost 70%, Melina Arnold, PhD, from the Cancer Surveillance Branch of the International Agency for Research on Cancer in Lyon, France, and colleagues reported.
“Melanoma is the most lethal form of skin cancer; this epidemiological assessment found a heavy public health and economic burden, and our projections suggest that it will remain so in the coming decades,” they wrote in a study published online in JAMA Dermatology.
In an accompanying editorial, Mavis Obeng-Kusi, MPharm and Ivo Abraham, PhD from the Center for Health Outcomes and PharmacoEconomic Research at the University of Arizona, Tucson, commented that the findings are “sobering,” but may substantially underestimate the gravity of the problem in low- and middle-income countries (LMIC).
“The study by Arnold et al. brings to the fore a public health concern that requires global attention and initiates conversations particularly related to LMIC settings, where the incidence and mortality of melanoma is thought to be minimal and for which preventive measures may be insufficient,” they wrote.
Down Under nations lead
Dr. Arnold and colleagues looked at data on age-standardized melanoma incidence and mortality rates per 100,000 person-years (PY) by country, each of 20 world regions as defined by the United Nations, and according to the UN’s four-tier Human Development Index, which stratifies countries into low-, medium-, high-, and very high–income categories.
As noted previously, the researchers estimated that there were 325,000 new melanoma cases worldwide in 2020 (174,000 cases in males and 151,000 in females). There were 57,000 estimated melanoma deaths the same year (32,000 in males and 25,000 in females.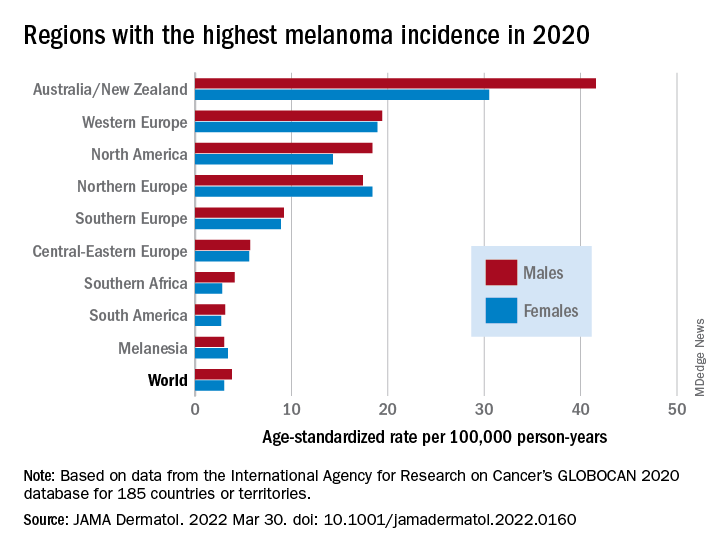
The highest incidence rates were seen in Australia and New Zealand, at 42 per 100,000 PY among males and 31 per 100,000 PY in females, followed by Western Europe with 19 per 100,000 PY in both males and females, North America with 18 and 14 cases per 100,000 PY in males and females respectively, and Northern Europe, with 17 per 100,000 PY in males, and 18 per 100,000 PY in females.
In contrast, in most African and Asian countries melanoma was rare, with rates commonly less than 1 per 100,000 PY, the investigators noted.
The melanoma mortality rate was highest in New Zealand, at 5 per 100,000 PY. Mortality rates worldwide varied less widely than incidence rates. In most other regions of the world, mortality rates were “much lower,” ranging between 0.2-1.0 per 100,000 PY, they wrote.
The authors estimated that, if 2020 rates remain stable, the global burden from melanoma in 2040 will increase to approximately 510,000 new cases and 96,000 deaths.
Public health efforts needed
In their editorial, Ms. Obeng-Kusi and Dr. Abraham pointed out that the study was hampered by the limited availability of cancer data from LMICs, leading the authors to estimate incidence and mortality rates based on proxy data, such as statistical modeling or averaged rates from neighboring countries.
They emphasized the need for going beyond the statistics: “Specific to cutaneous melanoma data, what is most important globally, knowing the exact numbers of cases and deaths or understanding the order of magnitude of the present and future epidemiology? No doubt the latter. Melanoma can be treated more easily if caught at earlier stages.”
Projections such as those provided by Dr. Arnold and colleagues could help to raise awareness of the importance of decreasing exposure to UV radiation, which accounts for three-fourths of all incident melanomas, the editorialists said.
The study was funded in part by a grant to coauthor Anna E. Cust, PhD, MPH. Dr. Cust reported receiving a fellowship from the Australian National Health and Medical Research Council outside the submitted work. Dr. Arnold had no conflicts of interested to disclose. Dr. Abraham reported financial relationships with various entities. Ms. Obeng-Kusi had no disclosures.
Even by cautious calculations,
An estimated 325,000 people worldwide received a new diagnosis of cutaneous melanoma in 2020, and if present trends continue, the incidence of new cases is predicted to increase by about 50% in 2040, with melanoma deaths expected to rise by almost 70%, Melina Arnold, PhD, from the Cancer Surveillance Branch of the International Agency for Research on Cancer in Lyon, France, and colleagues reported.
“Melanoma is the most lethal form of skin cancer; this epidemiological assessment found a heavy public health and economic burden, and our projections suggest that it will remain so in the coming decades,” they wrote in a study published online in JAMA Dermatology.
In an accompanying editorial, Mavis Obeng-Kusi, MPharm and Ivo Abraham, PhD from the Center for Health Outcomes and PharmacoEconomic Research at the University of Arizona, Tucson, commented that the findings are “sobering,” but may substantially underestimate the gravity of the problem in low- and middle-income countries (LMIC).
“The study by Arnold et al. brings to the fore a public health concern that requires global attention and initiates conversations particularly related to LMIC settings, where the incidence and mortality of melanoma is thought to be minimal and for which preventive measures may be insufficient,” they wrote.
Down Under nations lead
Dr. Arnold and colleagues looked at data on age-standardized melanoma incidence and mortality rates per 100,000 person-years (PY) by country, each of 20 world regions as defined by the United Nations, and according to the UN’s four-tier Human Development Index, which stratifies countries into low-, medium-, high-, and very high–income categories.
As noted previously, the researchers estimated that there were 325,000 new melanoma cases worldwide in 2020 (174,000 cases in males and 151,000 in females). There were 57,000 estimated melanoma deaths the same year (32,000 in males and 25,000 in females.
The highest incidence rates were seen in Australia and New Zealand, at 42 per 100,000 PY among males and 31 per 100,000 PY in females, followed by Western Europe with 19 per 100,000 PY in both males and females, North America with 18 and 14 cases per 100,000 PY in males and females respectively, and Northern Europe, with 17 per 100,000 PY in males, and 18 per 100,000 PY in females.
In contrast, in most African and Asian countries melanoma was rare, with rates commonly less than 1 per 100,000 PY, the investigators noted.
The melanoma mortality rate was highest in New Zealand, at 5 per 100,000 PY. Mortality rates worldwide varied less widely than incidence rates. In most other regions of the world, mortality rates were “much lower,” ranging between 0.2-1.0 per 100,000 PY, they wrote.
The authors estimated that, if 2020 rates remain stable, the global burden from melanoma in 2040 will increase to approximately 510,000 new cases and 96,000 deaths.
Public health efforts needed
In their editorial, Ms. Obeng-Kusi and Dr. Abraham pointed out that the study was hampered by the limited availability of cancer data from LMICs, leading the authors to estimate incidence and mortality rates based on proxy data, such as statistical modeling or averaged rates from neighboring countries.
They emphasized the need for going beyond the statistics: “Specific to cutaneous melanoma data, what is most important globally, knowing the exact numbers of cases and deaths or understanding the order of magnitude of the present and future epidemiology? No doubt the latter. Melanoma can be treated more easily if caught at earlier stages.”
Projections such as those provided by Dr. Arnold and colleagues could help to raise awareness of the importance of decreasing exposure to UV radiation, which accounts for three-fourths of all incident melanomas, the editorialists said.
The study was funded in part by a grant to coauthor Anna E. Cust, PhD, MPH. Dr. Cust reported receiving a fellowship from the Australian National Health and Medical Research Council outside the submitted work. Dr. Arnold had no conflicts of interested to disclose. Dr. Abraham reported financial relationships with various entities. Ms. Obeng-Kusi had no disclosures.
Even by cautious calculations,
An estimated 325,000 people worldwide received a new diagnosis of cutaneous melanoma in 2020, and if present trends continue, the incidence of new cases is predicted to increase by about 50% in 2040, with melanoma deaths expected to rise by almost 70%, Melina Arnold, PhD, from the Cancer Surveillance Branch of the International Agency for Research on Cancer in Lyon, France, and colleagues reported.
“Melanoma is the most lethal form of skin cancer; this epidemiological assessment found a heavy public health and economic burden, and our projections suggest that it will remain so in the coming decades,” they wrote in a study published online in JAMA Dermatology.
In an accompanying editorial, Mavis Obeng-Kusi, MPharm and Ivo Abraham, PhD from the Center for Health Outcomes and PharmacoEconomic Research at the University of Arizona, Tucson, commented that the findings are “sobering,” but may substantially underestimate the gravity of the problem in low- and middle-income countries (LMIC).
“The study by Arnold et al. brings to the fore a public health concern that requires global attention and initiates conversations particularly related to LMIC settings, where the incidence and mortality of melanoma is thought to be minimal and for which preventive measures may be insufficient,” they wrote.
Down Under nations lead
Dr. Arnold and colleagues looked at data on age-standardized melanoma incidence and mortality rates per 100,000 person-years (PY) by country, each of 20 world regions as defined by the United Nations, and according to the UN’s four-tier Human Development Index, which stratifies countries into low-, medium-, high-, and very high–income categories.
As noted previously, the researchers estimated that there were 325,000 new melanoma cases worldwide in 2020 (174,000 cases in males and 151,000 in females). There were 57,000 estimated melanoma deaths the same year (32,000 in males and 25,000 in females.
The highest incidence rates were seen in Australia and New Zealand, at 42 per 100,000 PY among males and 31 per 100,000 PY in females, followed by Western Europe with 19 per 100,000 PY in both males and females, North America with 18 and 14 cases per 100,000 PY in males and females respectively, and Northern Europe, with 17 per 100,000 PY in males, and 18 per 100,000 PY in females.
In contrast, in most African and Asian countries melanoma was rare, with rates commonly less than 1 per 100,000 PY, the investigators noted.
The melanoma mortality rate was highest in New Zealand, at 5 per 100,000 PY. Mortality rates worldwide varied less widely than incidence rates. In most other regions of the world, mortality rates were “much lower,” ranging between 0.2-1.0 per 100,000 PY, they wrote.
The authors estimated that, if 2020 rates remain stable, the global burden from melanoma in 2040 will increase to approximately 510,000 new cases and 96,000 deaths.
Public health efforts needed
In their editorial, Ms. Obeng-Kusi and Dr. Abraham pointed out that the study was hampered by the limited availability of cancer data from LMICs, leading the authors to estimate incidence and mortality rates based on proxy data, such as statistical modeling or averaged rates from neighboring countries.
They emphasized the need for going beyond the statistics: “Specific to cutaneous melanoma data, what is most important globally, knowing the exact numbers of cases and deaths or understanding the order of magnitude of the present and future epidemiology? No doubt the latter. Melanoma can be treated more easily if caught at earlier stages.”
Projections such as those provided by Dr. Arnold and colleagues could help to raise awareness of the importance of decreasing exposure to UV radiation, which accounts for three-fourths of all incident melanomas, the editorialists said.
The study was funded in part by a grant to coauthor Anna E. Cust, PhD, MPH. Dr. Cust reported receiving a fellowship from the Australian National Health and Medical Research Council outside the submitted work. Dr. Arnold had no conflicts of interested to disclose. Dr. Abraham reported financial relationships with various entities. Ms. Obeng-Kusi had no disclosures.
FROM JAMA DERMATOLOGY
Some reproductive factors linked with risk of dementia
Certain reproductive factors are associated with greater or lower risk of dementia, according to researchers who conducted a large population-based study with UK Biobank data.
Jessica Gong, a PhD candidate at the George Institute for Global Health at University of New South Wales in Australia, and coauthors found a greater dementia risk in women with early and late menarche, women who were younger when they first gave birth, and those who had had a hysterectomy, especially those who had a hysterectomy without concomitant oophorectomy or with a previous oophorectomy.
After controlling for key confounders, the researchers found lower risk of all-cause dementia if women had ever been pregnant, ever had an abortion, had a longer reproductive span, or had later menopause.
Use of oral contraceptive pills was associated with a lower dementia risk, they found.
In this study, there was no evidence that hormone therapy (HT) was associated with dementia risk (hazard ratio, 0.99, 95% confidence interval [0.90-1.09], P =.0828).
The analysis, published online April 5 in PLOS Medicine, comprised 273,240 women and 228,957 men without prevalent dementia.
The authors noted that dementia rates are increasing. Globally, 50 million people live with dementia, and the number is expected to triple by 2050, according to Alzheimer’s Disease International.
“Our study identified certain reproductive factors related to shorter exposure to endogenous estrogen were associated with increased risk of dementia, highlighting the susceptibility in dementia risk pertaining to women,” Ms. Gong told this publication.
Risk comparison of men and women
Men were included in this study to compare the association between number of children fathered and the risk of all-cause dementia, with the association in their female counterparts.
The U-shaped associations between the number of children and dementia risk were similar for both sexes, suggesting that the risk difference in women may not be associated with factors associated with childbearing
“It may be more related to social and behavioral factors in parenthood, rather than biological factors involved in childbearing,” Ms. Gong said.
Compared with those with two children, for those without children, the multiple adjusted HR (95% CI) was 1.18 (1.04, 1.33) (P = .027) for women and 1.10 (0.98-1.23) P = .164) for men.
For those with four or more children, the HR was 1.14 (0.98, 1.33) (P = .132) for women and 1.26 (1.10-1.45) (P = .003) for men.
Rachel Buckley, PhD, assistant professor of neurology with a dual appointment at Brigham and Women’s and Massachusetts General hospitals in Boston, told this publication she found the comparison of dementia risk with number of children in men and women “fascinating.”
She said the argument usually is that if women have had more births, then they have had more estrogen through their body because women get a huge injection of hormones in pregnancy.

“The idea is that the more pregnancies you have the more protected you are. But this study put that on its head, because if men and women are showing increased [dementia] risk in the number of children they have, it suggests there must be something about having the children – not necessarily the circulating hormones – that might be having an impact,” Dr. Buckley said.
“I had never thought to compare the number of children in men. I do find that very interesting,” she said.
As for the lack of a link between HT and dementia risk, in this study she said, she wouldn’t shut the door on that discussion just yet.
She noted the long history of controversy in the field about whether there is a protective factor against dementia for estrogen or whether exposure to estrogen leads to increased risk.
Before the landmark Women’s Health Initiative (WHI) study in the 1990s, she pointed out, there was evidence in many observational studies that women who had longer exposure to estrogen – whether that was earlier age at first period and later age at menopause combined or women had taken hormone therapy at some point, had less risk for dementia.
Dr. Buckley said that in a secondary outcome of WHI, however, “there was increased risk for progression to dementia in women who were taking hormone therapy which essentially flipped the field on its ahead because until that point everybody thought that estrogen was a protective factor.”
She said although this study found no association with dementia, she still thinks HT has a role to play and that it may just need to be better tailored to individuals.
“If you think about it, we have our tailored cocktail of hormones in our body and who’s to say that my hormones are going to be the same as yours? Why should you and I be put on the same hormone therapy and assume that will give us the same outcome? I think we could do a lot better with customization and calibration of hormones to aid in women’s health.”
Lifetime approach to dementia
Ms. Gong says future dementia risk-reduction strategies should consider sex-specific risk, and consider the reproductive events that took place in women’s lifespans as well as their entire hormone history when assessing dementia risk, to ensure that the strategies are sex sensitive.
Dr. Buckley agrees: “I don’t think we should ever think about dementia in terms of 65 onwards. We know this disease is insidious and it starts very, very early.”
Regarding limitations, the authors noted that it was a retrospective study that included self-reported measures of reproductive factors, which may be inherently subject to recall bias.
A coauthor does consultant work for Amgen, Freeline, and Kirin outside the submitted work. There were no other relevant financial disclosures.
Certain reproductive factors are associated with greater or lower risk of dementia, according to researchers who conducted a large population-based study with UK Biobank data.
Jessica Gong, a PhD candidate at the George Institute for Global Health at University of New South Wales in Australia, and coauthors found a greater dementia risk in women with early and late menarche, women who were younger when they first gave birth, and those who had had a hysterectomy, especially those who had a hysterectomy without concomitant oophorectomy or with a previous oophorectomy.
After controlling for key confounders, the researchers found lower risk of all-cause dementia if women had ever been pregnant, ever had an abortion, had a longer reproductive span, or had later menopause.
Use of oral contraceptive pills was associated with a lower dementia risk, they found.
In this study, there was no evidence that hormone therapy (HT) was associated with dementia risk (hazard ratio, 0.99, 95% confidence interval [0.90-1.09], P =.0828).
The analysis, published online April 5 in PLOS Medicine, comprised 273,240 women and 228,957 men without prevalent dementia.
The authors noted that dementia rates are increasing. Globally, 50 million people live with dementia, and the number is expected to triple by 2050, according to Alzheimer’s Disease International.
“Our study identified certain reproductive factors related to shorter exposure to endogenous estrogen were associated with increased risk of dementia, highlighting the susceptibility in dementia risk pertaining to women,” Ms. Gong told this publication.
Risk comparison of men and women
Men were included in this study to compare the association between number of children fathered and the risk of all-cause dementia, with the association in their female counterparts.
The U-shaped associations between the number of children and dementia risk were similar for both sexes, suggesting that the risk difference in women may not be associated with factors associated with childbearing
“It may be more related to social and behavioral factors in parenthood, rather than biological factors involved in childbearing,” Ms. Gong said.
Compared with those with two children, for those without children, the multiple adjusted HR (95% CI) was 1.18 (1.04, 1.33) (P = .027) for women and 1.10 (0.98-1.23) P = .164) for men.
For those with four or more children, the HR was 1.14 (0.98, 1.33) (P = .132) for women and 1.26 (1.10-1.45) (P = .003) for men.
Rachel Buckley, PhD, assistant professor of neurology with a dual appointment at Brigham and Women’s and Massachusetts General hospitals in Boston, told this publication she found the comparison of dementia risk with number of children in men and women “fascinating.”
She said the argument usually is that if women have had more births, then they have had more estrogen through their body because women get a huge injection of hormones in pregnancy.
“The idea is that the more pregnancies you have the more protected you are. But this study put that on its head, because if men and women are showing increased [dementia] risk in the number of children they have, it suggests there must be something about having the children – not necessarily the circulating hormones – that might be having an impact,” Dr. Buckley said.
“I had never thought to compare the number of children in men. I do find that very interesting,” she said.
As for the lack of a link between HT and dementia risk, in this study she said, she wouldn’t shut the door on that discussion just yet.
She noted the long history of controversy in the field about whether there is a protective factor against dementia for estrogen or whether exposure to estrogen leads to increased risk.
Before the landmark Women’s Health Initiative (WHI) study in the 1990s, she pointed out, there was evidence in many observational studies that women who had longer exposure to estrogen – whether that was earlier age at first period and later age at menopause combined or women had taken hormone therapy at some point, had less risk for dementia.
Dr. Buckley said that in a secondary outcome of WHI, however, “there was increased risk for progression to dementia in women who were taking hormone therapy which essentially flipped the field on its ahead because until that point everybody thought that estrogen was a protective factor.”
She said although this study found no association with dementia, she still thinks HT has a role to play and that it may just need to be better tailored to individuals.
“If you think about it, we have our tailored cocktail of hormones in our body and who’s to say that my hormones are going to be the same as yours? Why should you and I be put on the same hormone therapy and assume that will give us the same outcome? I think we could do a lot better with customization and calibration of hormones to aid in women’s health.”
Lifetime approach to dementia
Ms. Gong says future dementia risk-reduction strategies should consider sex-specific risk, and consider the reproductive events that took place in women’s lifespans as well as their entire hormone history when assessing dementia risk, to ensure that the strategies are sex sensitive.
Dr. Buckley agrees: “I don’t think we should ever think about dementia in terms of 65 onwards. We know this disease is insidious and it starts very, very early.”
Regarding limitations, the authors noted that it was a retrospective study that included self-reported measures of reproductive factors, which may be inherently subject to recall bias.
A coauthor does consultant work for Amgen, Freeline, and Kirin outside the submitted work. There were no other relevant financial disclosures.
Certain reproductive factors are associated with greater or lower risk of dementia, according to researchers who conducted a large population-based study with UK Biobank data.
Jessica Gong, a PhD candidate at the George Institute for Global Health at University of New South Wales in Australia, and coauthors found a greater dementia risk in women with early and late menarche, women who were younger when they first gave birth, and those who had had a hysterectomy, especially those who had a hysterectomy without concomitant oophorectomy or with a previous oophorectomy.
After controlling for key confounders, the researchers found lower risk of all-cause dementia if women had ever been pregnant, ever had an abortion, had a longer reproductive span, or had later menopause.
Use of oral contraceptive pills was associated with a lower dementia risk, they found.
In this study, there was no evidence that hormone therapy (HT) was associated with dementia risk (hazard ratio, 0.99, 95% confidence interval [0.90-1.09], P =.0828).
The analysis, published online April 5 in PLOS Medicine, comprised 273,240 women and 228,957 men without prevalent dementia.
The authors noted that dementia rates are increasing. Globally, 50 million people live with dementia, and the number is expected to triple by 2050, according to Alzheimer’s Disease International.
“Our study identified certain reproductive factors related to shorter exposure to endogenous estrogen were associated with increased risk of dementia, highlighting the susceptibility in dementia risk pertaining to women,” Ms. Gong told this publication.
Risk comparison of men and women
Men were included in this study to compare the association between number of children fathered and the risk of all-cause dementia, with the association in their female counterparts.
The U-shaped associations between the number of children and dementia risk were similar for both sexes, suggesting that the risk difference in women may not be associated with factors associated with childbearing
“It may be more related to social and behavioral factors in parenthood, rather than biological factors involved in childbearing,” Ms. Gong said.
Compared with those with two children, for those without children, the multiple adjusted HR (95% CI) was 1.18 (1.04, 1.33) (P = .027) for women and 1.10 (0.98-1.23) P = .164) for men.
For those with four or more children, the HR was 1.14 (0.98, 1.33) (P = .132) for women and 1.26 (1.10-1.45) (P = .003) for men.
Rachel Buckley, PhD, assistant professor of neurology with a dual appointment at Brigham and Women’s and Massachusetts General hospitals in Boston, told this publication she found the comparison of dementia risk with number of children in men and women “fascinating.”
She said the argument usually is that if women have had more births, then they have had more estrogen through their body because women get a huge injection of hormones in pregnancy.
“The idea is that the more pregnancies you have the more protected you are. But this study put that on its head, because if men and women are showing increased [dementia] risk in the number of children they have, it suggests there must be something about having the children – not necessarily the circulating hormones – that might be having an impact,” Dr. Buckley said.
“I had never thought to compare the number of children in men. I do find that very interesting,” she said.
As for the lack of a link between HT and dementia risk, in this study she said, she wouldn’t shut the door on that discussion just yet.
She noted the long history of controversy in the field about whether there is a protective factor against dementia for estrogen or whether exposure to estrogen leads to increased risk.
Before the landmark Women’s Health Initiative (WHI) study in the 1990s, she pointed out, there was evidence in many observational studies that women who had longer exposure to estrogen – whether that was earlier age at first period and later age at menopause combined or women had taken hormone therapy at some point, had less risk for dementia.
Dr. Buckley said that in a secondary outcome of WHI, however, “there was increased risk for progression to dementia in women who were taking hormone therapy which essentially flipped the field on its ahead because until that point everybody thought that estrogen was a protective factor.”
She said although this study found no association with dementia, she still thinks HT has a role to play and that it may just need to be better tailored to individuals.
“If you think about it, we have our tailored cocktail of hormones in our body and who’s to say that my hormones are going to be the same as yours? Why should you and I be put on the same hormone therapy and assume that will give us the same outcome? I think we could do a lot better with customization and calibration of hormones to aid in women’s health.”
Lifetime approach to dementia
Ms. Gong says future dementia risk-reduction strategies should consider sex-specific risk, and consider the reproductive events that took place in women’s lifespans as well as their entire hormone history when assessing dementia risk, to ensure that the strategies are sex sensitive.
Dr. Buckley agrees: “I don’t think we should ever think about dementia in terms of 65 onwards. We know this disease is insidious and it starts very, very early.”
Regarding limitations, the authors noted that it was a retrospective study that included self-reported measures of reproductive factors, which may be inherently subject to recall bias.
A coauthor does consultant work for Amgen, Freeline, and Kirin outside the submitted work. There were no other relevant financial disclosures.
FROM PLOS MEDICINE
‘Eye-opening’ experience on the other side of the hospital bed
The 5 days that she spent at her mother’s bedside were eye-opening for an oncologist used to being on the other side of the clinician–patient relationship.
“As a physician, I thought I had a unique perspective of things that were done well – and things that were not,” commented Pamela Kunz, MD.
Dr. Kunz, who was named the 2021 Woman Oncologist of the Year, is director of the Center for Gastrointestinal Cancers at Smilow Cancer Hospital and of the Yale Cancer Center, New Haven, Conn.
But she was propelled into quite a different role when her mother was admitted to the hospital.
Her mom, who has trouble hearing, was easily confused by jargon and by “all of the people coming in and out with no introductions,” she explained.
“She needed someone to translate what was going on because she didn’t feel well,” she added.
Seeing inpatient care through her mother’s eyes was enlightening, and at times it was “shocking to be on the other side.”
Physicians get used to “checking boxes, getting through the day,” she said. “It’s easy to forget the human side.”
“Seeing a loved one sick, [struggling] through this – I just wished I had seen things done differently,” added Dr. Kunz.
Her thread has since garnered thousands of “likes” and scores of comments and retweets.
She began the Twitter thread explaining what prompted her comments:
“I spent many hours last week observing the practice of medicine while sitting at my mom’s hospital bedside and was reminded of some important communication pearls. Some musings ...”
“1. Introduce yourself by full name, role, and team and have ID badges visible. It can get very confusing for [patients] and family members with the number of people in and out of rooms. E.g. ‘My name is Dr. X. I’m the intern on the primary internal medicine team.’
2. End your patient visit with a summary of the plan for the day.
3. Avoid medical jargon & speak slowly, clearly, and logically. Remember you are a teacher for your [patients] and their family.
4. Masks make it harder to hear, especially for [patients] with hearing loss (and they no longer have the aid of lip reading).
5. Many older [patients] get confused in the hospital. Repetition is a good thing.
6. Speak to a family member at least once per day to relay the plan.
7. Try to avoid last minute or surprise discharges – they make [patients] and family members anxious. Talk about discharge planning from day 1 and what milestones must occur prior to a safe discharge. ‘In order for you to leave the hospital, X, Y, X must happen.’
8. Talk with your [patients] about something other than what brought them to the hospital (a tip I once learned from a wise mentor).
9. When possible, sit at eye level with your patient (I love these stools from @YNHH).
10. Take time to listen.”
Dr. Kunz closed with her golden rule: “Lastly, treat your patients how you would want your own family member treated.”
Twitter user @BrunaPellini replied: “I love this, especially ‘Treat your patients how you would want your own family member treated.’ My mom and grandma always said that to me since I was a med student, and this is definitely one of my core values.”
Other clinicians shared similar experiences, and some added to Dr. Kunz’s list.
“Agree entirely, love the list – and while none of us can always practice perfectly, my experiences with my own mother’s illness taught me an enormous amount about communication,” @hoperugo responded.
Twitter user @mariejacork added: “Everyone in health care please read ... if you are lucky enough to not have had a loved one unwell in hospital, these may get forgotten. Having sat with my dad for a few days before he died a few years ago, I felt a lot of these, and it changed my practice forever.”
@bjcohenmd provided additional advice: “And use the dry erase board that should be in every room. Never start a medication without explaining it. Many docs will see the patient and then go to the computer, decide to order a med, but never go back to explain it.”
Patients also shared experiences and offered suggestions.
“As a chronic pain patient I’d add – we know it’s frustrating you can’t cure us but PLEASE do not SIGH if we say something didn’t work or [tell] us to be more positive. Just say ‘I know this is very hard, I’m here to listen.’ We don’t expect a cure, we do expect to be believed,” said @ppenguinsmt. “It makes me feel like I’m causing distress to you if I say the pain has been unrelenting. I leave feeling worse. ...You may have heard 10 [people] in pain before me but this is MY only [appointment].”
Twitter user @KatieCahoots added: “These are perfect. I wish doctors would do this not only in the hospital but in the doctor’s office, as well. I would add one caveat: When you try not to use medical jargon, don’t dumb it down as though I don’t know anything about science or haven’t done any of my own research.”
Dr. Kunz said she was taken aback but pleased by the response to her Tweet.
“It’s an example of the human side of medicine, so it resonates with physicians and with patients,” she commented. Seeing through her mom’s eyes how care was provided made her realize that medical training should include more emphasis on communication, including “real-time feedback to interns, residents, fellows, and students.”
Yes, it takes time, and “we don’t all have a lot of extra time,” she acknowledged.
“But some of these elements don’t take that much more time to do. They can help build trust and can, in the long run, actually save time if patients understand and family members feel engaged and like they are participants,” she said. “I think a little time investment will go a long way.”
In her case, she very much appreciated the one trainee who tried to call her and update her about her mother’s care each afternoon. “I really valued that,” she said.
A version of this article first appeared on Medscape.com.
The 5 days that she spent at her mother’s bedside were eye-opening for an oncologist used to being on the other side of the clinician–patient relationship.
“As a physician, I thought I had a unique perspective of things that were done well – and things that were not,” commented Pamela Kunz, MD.
Dr. Kunz, who was named the 2021 Woman Oncologist of the Year, is director of the Center for Gastrointestinal Cancers at Smilow Cancer Hospital and of the Yale Cancer Center, New Haven, Conn.
But she was propelled into quite a different role when her mother was admitted to the hospital.
Her mom, who has trouble hearing, was easily confused by jargon and by “all of the people coming in and out with no introductions,” she explained.
“She needed someone to translate what was going on because she didn’t feel well,” she added.
Seeing inpatient care through her mother’s eyes was enlightening, and at times it was “shocking to be on the other side.”
Physicians get used to “checking boxes, getting through the day,” she said. “It’s easy to forget the human side.”
“Seeing a loved one sick, [struggling] through this – I just wished I had seen things done differently,” added Dr. Kunz.
Her thread has since garnered thousands of “likes” and scores of comments and retweets.
She began the Twitter thread explaining what prompted her comments:
“I spent many hours last week observing the practice of medicine while sitting at my mom’s hospital bedside and was reminded of some important communication pearls. Some musings ...”
“1. Introduce yourself by full name, role, and team and have ID badges visible. It can get very confusing for [patients] and family members with the number of people in and out of rooms. E.g. ‘My name is Dr. X. I’m the intern on the primary internal medicine team.’
2. End your patient visit with a summary of the plan for the day.
3. Avoid medical jargon & speak slowly, clearly, and logically. Remember you are a teacher for your [patients] and their family.
4. Masks make it harder to hear, especially for [patients] with hearing loss (and they no longer have the aid of lip reading).
5. Many older [patients] get confused in the hospital. Repetition is a good thing.
6. Speak to a family member at least once per day to relay the plan.
7. Try to avoid last minute or surprise discharges – they make [patients] and family members anxious. Talk about discharge planning from day 1 and what milestones must occur prior to a safe discharge. ‘In order for you to leave the hospital, X, Y, X must happen.’
8. Talk with your [patients] about something other than what brought them to the hospital (a tip I once learned from a wise mentor).
9. When possible, sit at eye level with your patient (I love these stools from @YNHH).
10. Take time to listen.”
Dr. Kunz closed with her golden rule: “Lastly, treat your patients how you would want your own family member treated.”
Twitter user @BrunaPellini replied: “I love this, especially ‘Treat your patients how you would want your own family member treated.’ My mom and grandma always said that to me since I was a med student, and this is definitely one of my core values.”
Other clinicians shared similar experiences, and some added to Dr. Kunz’s list.
“Agree entirely, love the list – and while none of us can always practice perfectly, my experiences with my own mother’s illness taught me an enormous amount about communication,” @hoperugo responded.
Twitter user @mariejacork added: “Everyone in health care please read ... if you are lucky enough to not have had a loved one unwell in hospital, these may get forgotten. Having sat with my dad for a few days before he died a few years ago, I felt a lot of these, and it changed my practice forever.”
@bjcohenmd provided additional advice: “And use the dry erase board that should be in every room. Never start a medication without explaining it. Many docs will see the patient and then go to the computer, decide to order a med, but never go back to explain it.”
Patients also shared experiences and offered suggestions.
“As a chronic pain patient I’d add – we know it’s frustrating you can’t cure us but PLEASE do not SIGH if we say something didn’t work or [tell] us to be more positive. Just say ‘I know this is very hard, I’m here to listen.’ We don’t expect a cure, we do expect to be believed,” said @ppenguinsmt. “It makes me feel like I’m causing distress to you if I say the pain has been unrelenting. I leave feeling worse. ...You may have heard 10 [people] in pain before me but this is MY only [appointment].”
Twitter user @KatieCahoots added: “These are perfect. I wish doctors would do this not only in the hospital but in the doctor’s office, as well. I would add one caveat: When you try not to use medical jargon, don’t dumb it down as though I don’t know anything about science or haven’t done any of my own research.”
Dr. Kunz said she was taken aback but pleased by the response to her Tweet.
“It’s an example of the human side of medicine, so it resonates with physicians and with patients,” she commented. Seeing through her mom’s eyes how care was provided made her realize that medical training should include more emphasis on communication, including “real-time feedback to interns, residents, fellows, and students.”
Yes, it takes time, and “we don’t all have a lot of extra time,” she acknowledged.
“But some of these elements don’t take that much more time to do. They can help build trust and can, in the long run, actually save time if patients understand and family members feel engaged and like they are participants,” she said. “I think a little time investment will go a long way.”
In her case, she very much appreciated the one trainee who tried to call her and update her about her mother’s care each afternoon. “I really valued that,” she said.
A version of this article first appeared on Medscape.com.
The 5 days that she spent at her mother’s bedside were eye-opening for an oncologist used to being on the other side of the clinician–patient relationship.
“As a physician, I thought I had a unique perspective of things that were done well – and things that were not,” commented Pamela Kunz, MD.
Dr. Kunz, who was named the 2021 Woman Oncologist of the Year, is director of the Center for Gastrointestinal Cancers at Smilow Cancer Hospital and of the Yale Cancer Center, New Haven, Conn.
But she was propelled into quite a different role when her mother was admitted to the hospital.
Her mom, who has trouble hearing, was easily confused by jargon and by “all of the people coming in and out with no introductions,” she explained.
“She needed someone to translate what was going on because she didn’t feel well,” she added.
Seeing inpatient care through her mother’s eyes was enlightening, and at times it was “shocking to be on the other side.”
Physicians get used to “checking boxes, getting through the day,” she said. “It’s easy to forget the human side.”
“Seeing a loved one sick, [struggling] through this – I just wished I had seen things done differently,” added Dr. Kunz.
Her thread has since garnered thousands of “likes” and scores of comments and retweets.
She began the Twitter thread explaining what prompted her comments:
“I spent many hours last week observing the practice of medicine while sitting at my mom’s hospital bedside and was reminded of some important communication pearls. Some musings ...”
“1. Introduce yourself by full name, role, and team and have ID badges visible. It can get very confusing for [patients] and family members with the number of people in and out of rooms. E.g. ‘My name is Dr. X. I’m the intern on the primary internal medicine team.’
2. End your patient visit with a summary of the plan for the day.
3. Avoid medical jargon & speak slowly, clearly, and logically. Remember you are a teacher for your [patients] and their family.
4. Masks make it harder to hear, especially for [patients] with hearing loss (and they no longer have the aid of lip reading).
5. Many older [patients] get confused in the hospital. Repetition is a good thing.
6. Speak to a family member at least once per day to relay the plan.
7. Try to avoid last minute or surprise discharges – they make [patients] and family members anxious. Talk about discharge planning from day 1 and what milestones must occur prior to a safe discharge. ‘In order for you to leave the hospital, X, Y, X must happen.’
8. Talk with your [patients] about something other than what brought them to the hospital (a tip I once learned from a wise mentor).
9. When possible, sit at eye level with your patient (I love these stools from @YNHH).
10. Take time to listen.”
Dr. Kunz closed with her golden rule: “Lastly, treat your patients how you would want your own family member treated.”
Twitter user @BrunaPellini replied: “I love this, especially ‘Treat your patients how you would want your own family member treated.’ My mom and grandma always said that to me since I was a med student, and this is definitely one of my core values.”
Other clinicians shared similar experiences, and some added to Dr. Kunz’s list.
“Agree entirely, love the list – and while none of us can always practice perfectly, my experiences with my own mother’s illness taught me an enormous amount about communication,” @hoperugo responded.
Twitter user @mariejacork added: “Everyone in health care please read ... if you are lucky enough to not have had a loved one unwell in hospital, these may get forgotten. Having sat with my dad for a few days before he died a few years ago, I felt a lot of these, and it changed my practice forever.”
@bjcohenmd provided additional advice: “And use the dry erase board that should be in every room. Never start a medication without explaining it. Many docs will see the patient and then go to the computer, decide to order a med, but never go back to explain it.”
Patients also shared experiences and offered suggestions.
“As a chronic pain patient I’d add – we know it’s frustrating you can’t cure us but PLEASE do not SIGH if we say something didn’t work or [tell] us to be more positive. Just say ‘I know this is very hard, I’m here to listen.’ We don’t expect a cure, we do expect to be believed,” said @ppenguinsmt. “It makes me feel like I’m causing distress to you if I say the pain has been unrelenting. I leave feeling worse. ...You may have heard 10 [people] in pain before me but this is MY only [appointment].”
Twitter user @KatieCahoots added: “These are perfect. I wish doctors would do this not only in the hospital but in the doctor’s office, as well. I would add one caveat: When you try not to use medical jargon, don’t dumb it down as though I don’t know anything about science or haven’t done any of my own research.”
Dr. Kunz said she was taken aback but pleased by the response to her Tweet.
“It’s an example of the human side of medicine, so it resonates with physicians and with patients,” she commented. Seeing through her mom’s eyes how care was provided made her realize that medical training should include more emphasis on communication, including “real-time feedback to interns, residents, fellows, and students.”
Yes, it takes time, and “we don’t all have a lot of extra time,” she acknowledged.
“But some of these elements don’t take that much more time to do. They can help build trust and can, in the long run, actually save time if patients understand and family members feel engaged and like they are participants,” she said. “I think a little time investment will go a long way.”
In her case, she very much appreciated the one trainee who tried to call her and update her about her mother’s care each afternoon. “I really valued that,” she said.
A version of this article first appeared on Medscape.com.
We all struggle with the unwritten rules of medical culture
There is a two-lane bridge in my town. It is quaint and picturesque, and when we first moved here, I would gaze out at the water as I drove, letting my mind wander along with the seagulls drifting alongside the car. Until one day, crossing back over, I passed a school bus stopped in the other lane, and instead of waving back, the driver gave me such a fierce look of disapproval I felt like I’d been to the principal’s office. What had I done?
I started paying more attention to the pattern of the other cars on the bridge. Although it appeared to be a standard two-lane width, the lanes weren’t quite wide enough if a school bus or large truck needed to cross at the same time as a car coming from the opposite direction. They had to wait until the other lane was clear. It was an unwritten rule of the town that if you saw a school bus on the other side, you stopped your car and yielded the bridge to the bus. It took me weeks to figure this out. When I did, I felt like I finally belonged in the community. Before, I’d been an outsider.
This got me thinking about culture. Every place has its unwritten rules, whether a community or a workplace. But how do we know the culture of a place? It’s pretty much impossible until we experience it for ourselves.
When I did figure out the bridge, I had a little bit of anger, to be honest. How was I supposed to know about the lanes? There weren’t any signs. Geez.
Now, when I approach the bridge, I don’t even think about it. I know what to do if I see a bus coming.
But sometimes I remember that time of confusion before I deciphered the unwritten rule. I still have a twinge of guilt for having done something wrong, even though it hadn’t been my fault.
It reminded me of a memory from medical training. I was an MS4, and my ER rotation was in a busy county hospital with a level I trauma center. To say that the place was chaotic would be an understatement.
On the first morning, I was shown the chart rack (yes, this was back in the day of paper charts). Charts were placed in the order that patients arrived. Med students and residents were to take a chart in chronological order, go triage and assess the patient, and then find an attending. Once finished, you put the chart back on the rack and picked up the next one. This was the extent of my orientation to the ER.
The days and weeks of the rotation flew by. It was a busy and exciting time. By the end of the month, I’d come to feel a part of the team.
Until one day, after finishing discharging a patient, an attending asked me, “Where’s the billing sheet?”
I had no idea what she was talking about. No one had ever shown me a billing sheet. But by this point, as an MS4, I knew well that if an attending asked you something you didn’t know the answer to, you shouldn’t just say that you didn’t know. You should try to figure out if you could at least approximate an answer first.
As I scrambled in my mind to figure out what she was asking me, she took one look at the apprehension in my eyes and asked again, raising her voice, “You haven’t been doing the billing sheets?”
I thought back to the first day of the rotation. The cursory 30-second orientation. Chart rack. Take one. See the patient. Put it back. See the next patient. Nothing about billing sheets.
“No,” I said. “No one ever told me about – ”
But the attending didn’t care that I hadn’t been instructed on the billing sheets. She ripped into me, yelling about how she couldn’t believe I’d been working there the entire month and was not doing the billing sheets. She showed me what they were and where they were supposed to be going and, in front of the whole staff, treated me like not only the biggest idiot she’d ever worked with but that the hospital had ever seen.
As she berated me, I thought about all the patients I’d seen that month. All the billing sheets I hadn’t placed in the pile. All the attendings who hadn’t gotten credit for the patients they’d staffed with me.
But how could I have known? I wanted to ask. How could I have known if nobody showed me or told me?
It was like the bridge. I was in a new environment and somehow expected to know the rules without anyone telling me; and when I didn’t know, people treated me like I’d done it the wrong way on purpose.
I didn’t end up saying anything more to that attending. What could I have said? She had already unleashed a mountain of her pent-up anger at me.
What I did decide in that moment was that I would never be an attending like that.
Like the bridge, this memory years later can still make me feel guilt and shame for doing something wrong. Even though it wasn’t my fault.
I was thinking about this recently with the Match. Thousands of freshly graduated medical students embarking on their new positions as interns in teaching hospitals across the country.
If someone treats you poorly for not knowing something, you are not an idiot. You’ve worked incredibly hard to get where you are, and you deserve to be there.
For attendings and more senior trainees, remember what it was like to be starting in a new place. We all make mistakes, and often it’s simply because of a lack of information.
Trainees shouldn’t have to suffer and be made to feel like outsiders until they figure out the unwritten rules of the place. They belong.
Dr. Lycette is medical director of Providence Oncology and Hematology Care Clinic, Seaside, Ore. She disclosed no relevant conflicts of interest. A version of this article first appeared on Medscape.com.
There is a two-lane bridge in my town. It is quaint and picturesque, and when we first moved here, I would gaze out at the water as I drove, letting my mind wander along with the seagulls drifting alongside the car. Until one day, crossing back over, I passed a school bus stopped in the other lane, and instead of waving back, the driver gave me such a fierce look of disapproval I felt like I’d been to the principal’s office. What had I done?
I started paying more attention to the pattern of the other cars on the bridge. Although it appeared to be a standard two-lane width, the lanes weren’t quite wide enough if a school bus or large truck needed to cross at the same time as a car coming from the opposite direction. They had to wait until the other lane was clear. It was an unwritten rule of the town that if you saw a school bus on the other side, you stopped your car and yielded the bridge to the bus. It took me weeks to figure this out. When I did, I felt like I finally belonged in the community. Before, I’d been an outsider.
This got me thinking about culture. Every place has its unwritten rules, whether a community or a workplace. But how do we know the culture of a place? It’s pretty much impossible until we experience it for ourselves.
When I did figure out the bridge, I had a little bit of anger, to be honest. How was I supposed to know about the lanes? There weren’t any signs. Geez.
Now, when I approach the bridge, I don’t even think about it. I know what to do if I see a bus coming.
But sometimes I remember that time of confusion before I deciphered the unwritten rule. I still have a twinge of guilt for having done something wrong, even though it hadn’t been my fault.
It reminded me of a memory from medical training. I was an MS4, and my ER rotation was in a busy county hospital with a level I trauma center. To say that the place was chaotic would be an understatement.
On the first morning, I was shown the chart rack (yes, this was back in the day of paper charts). Charts were placed in the order that patients arrived. Med students and residents were to take a chart in chronological order, go triage and assess the patient, and then find an attending. Once finished, you put the chart back on the rack and picked up the next one. This was the extent of my orientation to the ER.
The days and weeks of the rotation flew by. It was a busy and exciting time. By the end of the month, I’d come to feel a part of the team.
Until one day, after finishing discharging a patient, an attending asked me, “Where’s the billing sheet?”
I had no idea what she was talking about. No one had ever shown me a billing sheet. But by this point, as an MS4, I knew well that if an attending asked you something you didn’t know the answer to, you shouldn’t just say that you didn’t know. You should try to figure out if you could at least approximate an answer first.
As I scrambled in my mind to figure out what she was asking me, she took one look at the apprehension in my eyes and asked again, raising her voice, “You haven’t been doing the billing sheets?”
I thought back to the first day of the rotation. The cursory 30-second orientation. Chart rack. Take one. See the patient. Put it back. See the next patient. Nothing about billing sheets.
“No,” I said. “No one ever told me about – ”
But the attending didn’t care that I hadn’t been instructed on the billing sheets. She ripped into me, yelling about how she couldn’t believe I’d been working there the entire month and was not doing the billing sheets. She showed me what they were and where they were supposed to be going and, in front of the whole staff, treated me like not only the biggest idiot she’d ever worked with but that the hospital had ever seen.
As she berated me, I thought about all the patients I’d seen that month. All the billing sheets I hadn’t placed in the pile. All the attendings who hadn’t gotten credit for the patients they’d staffed with me.
But how could I have known? I wanted to ask. How could I have known if nobody showed me or told me?
It was like the bridge. I was in a new environment and somehow expected to know the rules without anyone telling me; and when I didn’t know, people treated me like I’d done it the wrong way on purpose.
I didn’t end up saying anything more to that attending. What could I have said? She had already unleashed a mountain of her pent-up anger at me.
What I did decide in that moment was that I would never be an attending like that.
Like the bridge, this memory years later can still make me feel guilt and shame for doing something wrong. Even though it wasn’t my fault.
I was thinking about this recently with the Match. Thousands of freshly graduated medical students embarking on their new positions as interns in teaching hospitals across the country.
If someone treats you poorly for not knowing something, you are not an idiot. You’ve worked incredibly hard to get where you are, and you deserve to be there.
For attendings and more senior trainees, remember what it was like to be starting in a new place. We all make mistakes, and often it’s simply because of a lack of information.
Trainees shouldn’t have to suffer and be made to feel like outsiders until they figure out the unwritten rules of the place. They belong.
Dr. Lycette is medical director of Providence Oncology and Hematology Care Clinic, Seaside, Ore. She disclosed no relevant conflicts of interest. A version of this article first appeared on Medscape.com.
There is a two-lane bridge in my town. It is quaint and picturesque, and when we first moved here, I would gaze out at the water as I drove, letting my mind wander along with the seagulls drifting alongside the car. Until one day, crossing back over, I passed a school bus stopped in the other lane, and instead of waving back, the driver gave me such a fierce look of disapproval I felt like I’d been to the principal’s office. What had I done?
I started paying more attention to the pattern of the other cars on the bridge. Although it appeared to be a standard two-lane width, the lanes weren’t quite wide enough if a school bus or large truck needed to cross at the same time as a car coming from the opposite direction. They had to wait until the other lane was clear. It was an unwritten rule of the town that if you saw a school bus on the other side, you stopped your car and yielded the bridge to the bus. It took me weeks to figure this out. When I did, I felt like I finally belonged in the community. Before, I’d been an outsider.
This got me thinking about culture. Every place has its unwritten rules, whether a community or a workplace. But how do we know the culture of a place? It’s pretty much impossible until we experience it for ourselves.
When I did figure out the bridge, I had a little bit of anger, to be honest. How was I supposed to know about the lanes? There weren’t any signs. Geez.
Now, when I approach the bridge, I don’t even think about it. I know what to do if I see a bus coming.
But sometimes I remember that time of confusion before I deciphered the unwritten rule. I still have a twinge of guilt for having done something wrong, even though it hadn’t been my fault.
It reminded me of a memory from medical training. I was an MS4, and my ER rotation was in a busy county hospital with a level I trauma center. To say that the place was chaotic would be an understatement.
On the first morning, I was shown the chart rack (yes, this was back in the day of paper charts). Charts were placed in the order that patients arrived. Med students and residents were to take a chart in chronological order, go triage and assess the patient, and then find an attending. Once finished, you put the chart back on the rack and picked up the next one. This was the extent of my orientation to the ER.
The days and weeks of the rotation flew by. It was a busy and exciting time. By the end of the month, I’d come to feel a part of the team.
Until one day, after finishing discharging a patient, an attending asked me, “Where’s the billing sheet?”
I had no idea what she was talking about. No one had ever shown me a billing sheet. But by this point, as an MS4, I knew well that if an attending asked you something you didn’t know the answer to, you shouldn’t just say that you didn’t know. You should try to figure out if you could at least approximate an answer first.
As I scrambled in my mind to figure out what she was asking me, she took one look at the apprehension in my eyes and asked again, raising her voice, “You haven’t been doing the billing sheets?”
I thought back to the first day of the rotation. The cursory 30-second orientation. Chart rack. Take one. See the patient. Put it back. See the next patient. Nothing about billing sheets.
“No,” I said. “No one ever told me about – ”
But the attending didn’t care that I hadn’t been instructed on the billing sheets. She ripped into me, yelling about how she couldn’t believe I’d been working there the entire month and was not doing the billing sheets. She showed me what they were and where they were supposed to be going and, in front of the whole staff, treated me like not only the biggest idiot she’d ever worked with but that the hospital had ever seen.
As she berated me, I thought about all the patients I’d seen that month. All the billing sheets I hadn’t placed in the pile. All the attendings who hadn’t gotten credit for the patients they’d staffed with me.
But how could I have known? I wanted to ask. How could I have known if nobody showed me or told me?
It was like the bridge. I was in a new environment and somehow expected to know the rules without anyone telling me; and when I didn’t know, people treated me like I’d done it the wrong way on purpose.
I didn’t end up saying anything more to that attending. What could I have said? She had already unleashed a mountain of her pent-up anger at me.
What I did decide in that moment was that I would never be an attending like that.
Like the bridge, this memory years later can still make me feel guilt and shame for doing something wrong. Even though it wasn’t my fault.
I was thinking about this recently with the Match. Thousands of freshly graduated medical students embarking on their new positions as interns in teaching hospitals across the country.
If someone treats you poorly for not knowing something, you are not an idiot. You’ve worked incredibly hard to get where you are, and you deserve to be there.
For attendings and more senior trainees, remember what it was like to be starting in a new place. We all make mistakes, and often it’s simply because of a lack of information.
Trainees shouldn’t have to suffer and be made to feel like outsiders until they figure out the unwritten rules of the place. They belong.
Dr. Lycette is medical director of Providence Oncology and Hematology Care Clinic, Seaside, Ore. She disclosed no relevant conflicts of interest. A version of this article first appeared on Medscape.com.
Flu vaccines cut seasonal death in heart failure patients
WASHINGTON – Patients with heart failure who received an annual influenza vaccine for 3 years running had significantly fewer all-cause hospitalizations and significantly fewer cases of pneumonia during that time, compared with placebo-treated patients with heart failure, in a prospective, randomized, global trial with 5,129 participants.
Although the results failed to show a significant reduction in all-cause deaths linked to influenza vaccination, compared with controls during the entire 3 years of the study, the results did show a significant 21% relative mortality-risk reduction by vaccination during periods of peak influenza circulation, and a significant 23% reduction in cardiovascular deaths, compared with controls during peak seasons.

“This is the first randomized, controlled trial of influenza vaccine in patients with heart failure, and we showed that vaccination reduces deaths” during peak influenza seasons, Mark Loeb, MD, said during a press briefing at the annual scientific sessions of the American College of Cardiology. The results send “an important global message that patients with heart failure should receive the influenza vaccine,” said Dr. Loeb, a professor at McMaster University, Hamilton, Ont., who specializes in clinical epidemiology and infectious diseases.
Dr. Loeb admitted that he and his associates erred when they picked the time window to assess the two primary endpoints for the trial: the combined rate of cardiovascular death, nonfatal MI, and nonfatal stroke, and this combined endpoint plus hospitalizations for heart failure.
The time window they selected was the entirety of all 3 years following three annual immunizations. That was a mistake.
No flu vaccine benefit outside flu season
“We know that the influenza vaccine will not have any effect outside of when influenza is circulating. In retrospect, we should have done that,” Dr. Loeb bemoaned during his talk. He chalked up the bad choice to concern over collecting enough endpoints to see a significant between-group difference when the researchers designed the study.
For the entire 3 years of follow-up, influenza vaccination was tied to a nonsignificant 7% relative risk reduction for the first primary endpoint, and a nonsignificant 9% relative risk reduction for the second primary endpoint, he reported.
But Dr. Loeb lobbied for the relevance of several significant secondary endpoints that collectively showed a compelling pattern of benefit during his talk. These included, for the full 3-years of follow-up, important, significant reductions relative to placebo of 16% for first all-cause hospitalizations (P = .01), and a 42% relative risk reduction in first cases of pneumonia (P = .0006).
Then there were the benefits that appeared during influenza season. In that analysis, first events for the first primary endpoint fell after vaccination by a significant 18% relative to placebo. The in-season analysis also showed the significant cuts in both all-cause and cardiovascular deaths.
Despite the neutral primary endpoints, “if you look at these data as a whole I think they speak to the importance of vaccinating patients with heart failure against influenza,” Dr. Loeb maintained.
‘Totality of evidence supports vaccination’
“I agree that the totality of evidence supports influenza vaccination,” commented Mark H. Drazner, MD, professor and clinical chief of cardiology at the University of Texas Southwestern Medical Center, Dallas, who was designated discussant for the report.

“The message should be to offer influenza vaccine to patients with heart failure,” Dr. Drazner said in an interview. “Previous data on influenza vaccine in patients with heart failure were largely observational. This was a randomized, prospective, placebo-controlled trial. That’s a step forward. Proving efficacy in a randomized trial is important.”
Dr Drazner added that his institution already promotes a “strong mandate” to vaccinate patients with heart failure against influenza.
“The influenza vaccine is a very effective and cost-efficient public health measure. Preventing hospitalizations of patients with heart failure has so many benefits,” commented Craig Beavers, PharmD, vice president of professional services at Baptist Health in Paducah, Ky., and a discussant during the press briefing.

The Influenza Vaccine To Prevent Adverse Vascular Events (IVVE) trial enrolled people with heart failure in New York Heart Association functional class II, III, or IV from any of 10 low- and middle-income countries including China, India, the Philippines, and multiple countries from Africa and the Middle East. They averaged 57 years of age, and slightly more than half were women.
IVVE was sponsored by McMaster University; the only commercial support that IVVE received was a free supply of influenza vaccine from Sanofi Pasteur. Dr. Loeb, Dr. Drazner, and Dr. Beavers had no disclosures.
WASHINGTON – Patients with heart failure who received an annual influenza vaccine for 3 years running had significantly fewer all-cause hospitalizations and significantly fewer cases of pneumonia during that time, compared with placebo-treated patients with heart failure, in a prospective, randomized, global trial with 5,129 participants.
Although the results failed to show a significant reduction in all-cause deaths linked to influenza vaccination, compared with controls during the entire 3 years of the study, the results did show a significant 21% relative mortality-risk reduction by vaccination during periods of peak influenza circulation, and a significant 23% reduction in cardiovascular deaths, compared with controls during peak seasons.
“This is the first randomized, controlled trial of influenza vaccine in patients with heart failure, and we showed that vaccination reduces deaths” during peak influenza seasons, Mark Loeb, MD, said during a press briefing at the annual scientific sessions of the American College of Cardiology. The results send “an important global message that patients with heart failure should receive the influenza vaccine,” said Dr. Loeb, a professor at McMaster University, Hamilton, Ont., who specializes in clinical epidemiology and infectious diseases.
Dr. Loeb admitted that he and his associates erred when they picked the time window to assess the two primary endpoints for the trial: the combined rate of cardiovascular death, nonfatal MI, and nonfatal stroke, and this combined endpoint plus hospitalizations for heart failure.
The time window they selected was the entirety of all 3 years following three annual immunizations. That was a mistake.
No flu vaccine benefit outside flu season
“We know that the influenza vaccine will not have any effect outside of when influenza is circulating. In retrospect, we should have done that,” Dr. Loeb bemoaned during his talk. He chalked up the bad choice to concern over collecting enough endpoints to see a significant between-group difference when the researchers designed the study.
For the entire 3 years of follow-up, influenza vaccination was tied to a nonsignificant 7% relative risk reduction for the first primary endpoint, and a nonsignificant 9% relative risk reduction for the second primary endpoint, he reported.
But Dr. Loeb lobbied for the relevance of several significant secondary endpoints that collectively showed a compelling pattern of benefit during his talk. These included, for the full 3-years of follow-up, important, significant reductions relative to placebo of 16% for first all-cause hospitalizations (P = .01), and a 42% relative risk reduction in first cases of pneumonia (P = .0006).
Then there were the benefits that appeared during influenza season. In that analysis, first events for the first primary endpoint fell after vaccination by a significant 18% relative to placebo. The in-season analysis also showed the significant cuts in both all-cause and cardiovascular deaths.
Despite the neutral primary endpoints, “if you look at these data as a whole I think they speak to the importance of vaccinating patients with heart failure against influenza,” Dr. Loeb maintained.
‘Totality of evidence supports vaccination’
“I agree that the totality of evidence supports influenza vaccination,” commented Mark H. Drazner, MD, professor and clinical chief of cardiology at the University of Texas Southwestern Medical Center, Dallas, who was designated discussant for the report.
“The message should be to offer influenza vaccine to patients with heart failure,” Dr. Drazner said in an interview. “Previous data on influenza vaccine in patients with heart failure were largely observational. This was a randomized, prospective, placebo-controlled trial. That’s a step forward. Proving efficacy in a randomized trial is important.”
Dr Drazner added that his institution already promotes a “strong mandate” to vaccinate patients with heart failure against influenza.
“The influenza vaccine is a very effective and cost-efficient public health measure. Preventing hospitalizations of patients with heart failure has so many benefits,” commented Craig Beavers, PharmD, vice president of professional services at Baptist Health in Paducah, Ky., and a discussant during the press briefing.
The Influenza Vaccine To Prevent Adverse Vascular Events (IVVE) trial enrolled people with heart failure in New York Heart Association functional class II, III, or IV from any of 10 low- and middle-income countries including China, India, the Philippines, and multiple countries from Africa and the Middle East. They averaged 57 years of age, and slightly more than half were women.
IVVE was sponsored by McMaster University; the only commercial support that IVVE received was a free supply of influenza vaccine from Sanofi Pasteur. Dr. Loeb, Dr. Drazner, and Dr. Beavers had no disclosures.
WASHINGTON – Patients with heart failure who received an annual influenza vaccine for 3 years running had significantly fewer all-cause hospitalizations and significantly fewer cases of pneumonia during that time, compared with placebo-treated patients with heart failure, in a prospective, randomized, global trial with 5,129 participants.
Although the results failed to show a significant reduction in all-cause deaths linked to influenza vaccination, compared with controls during the entire 3 years of the study, the results did show a significant 21% relative mortality-risk reduction by vaccination during periods of peak influenza circulation, and a significant 23% reduction in cardiovascular deaths, compared with controls during peak seasons.
“This is the first randomized, controlled trial of influenza vaccine in patients with heart failure, and we showed that vaccination reduces deaths” during peak influenza seasons, Mark Loeb, MD, said during a press briefing at the annual scientific sessions of the American College of Cardiology. The results send “an important global message that patients with heart failure should receive the influenza vaccine,” said Dr. Loeb, a professor at McMaster University, Hamilton, Ont., who specializes in clinical epidemiology and infectious diseases.
Dr. Loeb admitted that he and his associates erred when they picked the time window to assess the two primary endpoints for the trial: the combined rate of cardiovascular death, nonfatal MI, and nonfatal stroke, and this combined endpoint plus hospitalizations for heart failure.
The time window they selected was the entirety of all 3 years following three annual immunizations. That was a mistake.
No flu vaccine benefit outside flu season
“We know that the influenza vaccine will not have any effect outside of when influenza is circulating. In retrospect, we should have done that,” Dr. Loeb bemoaned during his talk. He chalked up the bad choice to concern over collecting enough endpoints to see a significant between-group difference when the researchers designed the study.
For the entire 3 years of follow-up, influenza vaccination was tied to a nonsignificant 7% relative risk reduction for the first primary endpoint, and a nonsignificant 9% relative risk reduction for the second primary endpoint, he reported.
But Dr. Loeb lobbied for the relevance of several significant secondary endpoints that collectively showed a compelling pattern of benefit during his talk. These included, for the full 3-years of follow-up, important, significant reductions relative to placebo of 16% for first all-cause hospitalizations (P = .01), and a 42% relative risk reduction in first cases of pneumonia (P = .0006).
Then there were the benefits that appeared during influenza season. In that analysis, first events for the first primary endpoint fell after vaccination by a significant 18% relative to placebo. The in-season analysis also showed the significant cuts in both all-cause and cardiovascular deaths.
Despite the neutral primary endpoints, “if you look at these data as a whole I think they speak to the importance of vaccinating patients with heart failure against influenza,” Dr. Loeb maintained.
‘Totality of evidence supports vaccination’
“I agree that the totality of evidence supports influenza vaccination,” commented Mark H. Drazner, MD, professor and clinical chief of cardiology at the University of Texas Southwestern Medical Center, Dallas, who was designated discussant for the report.
“The message should be to offer influenza vaccine to patients with heart failure,” Dr. Drazner said in an interview. “Previous data on influenza vaccine in patients with heart failure were largely observational. This was a randomized, prospective, placebo-controlled trial. That’s a step forward. Proving efficacy in a randomized trial is important.”
Dr Drazner added that his institution already promotes a “strong mandate” to vaccinate patients with heart failure against influenza.
“The influenza vaccine is a very effective and cost-efficient public health measure. Preventing hospitalizations of patients with heart failure has so many benefits,” commented Craig Beavers, PharmD, vice president of professional services at Baptist Health in Paducah, Ky., and a discussant during the press briefing.
The Influenza Vaccine To Prevent Adverse Vascular Events (IVVE) trial enrolled people with heart failure in New York Heart Association functional class II, III, or IV from any of 10 low- and middle-income countries including China, India, the Philippines, and multiple countries from Africa and the Middle East. They averaged 57 years of age, and slightly more than half were women.
IVVE was sponsored by McMaster University; the only commercial support that IVVE received was a free supply of influenza vaccine from Sanofi Pasteur. Dr. Loeb, Dr. Drazner, and Dr. Beavers had no disclosures.
AT ACC 2022
Ukraine war likely to cause infection outbreaks that will spread beyond borders
Every day we see stark images of the war in Ukraine – bombed-out buildings, explosions, and bodies lying in the streets. But there’s another, less visible war against the bacteria and viruses that are gathering their forces together. They, too, will infect parts of the population and may spread throughout Europe. Here’s what Ukrainians, and their neighbors, are facing on the infectious disease front.
Andrey Zinchuk, MD, MHS, a pulmonary/critical care physician at Yale and a native of Ukraine who immigrated to the U.S. at the age of 14 with his family, set the background for understanding this crisis. He said that TB and HIV rates in Ukraine have long been especially high, even before the current conflict: “Part of the challenge of the health care system in Ukraine is that it’s difficult to maintain a steady policy because of political instability,” he said. “We’ve had three revolutions in the last 20 years,” not counting the current Russian invasion.
The first was the breakup of the Soviet Union, which led to “an epidemic of people with HIV, hepatitis, and opioid use.” Next was the Orange Revolution in 2004 over fraud during a presidential election. In 2014 came the Maiden Revolution, after the government chose closer ties to Russia rather than Europe. Then-president Viktor Yanukovych fled to Russia.
“That’s when Russia annexed Crimea. There was essentially infiltration in Russian propaganda in the east of the country,” Dr. Zinchuk said. “This helped the Russians manufacture uprisings there to create a separatist state (the Luhansk and Donetsk People’s Republics) which were mostly Russian-speaking parts of the country,” an area known as the Donbas. This resulted in a war in eastern Ukraine that began 2014, with more than 10,000 deaths.
After the 2014 revolution, Dr. Zinchuk said, “There was a tremendous change in the way ... medical care was provided, and tremendous growth and stability in the medical supply for those chronic medical conditions.”
Nevertheless, health care expenditures in Ukraine have been quite low. Even before the current conflict, Dr. Zinchuk noted, annual health care expenditures in Ukraine were about $600 per capita. In comparison, it’s about $4,500 per person in Germany and $12,530 in the United States.
Despite those low per-capita expenditures in Ukraine, access to medicines – such as insulin for diabetes and antibiotics for tuberculosis – was stable before the war. But now, Dr. Zinchuk said, his aunt and uncle have had to flee Kyiv for the countryside and, while safe, they have “no plumbing and have to heat the house by burning firewood.” More significantly, their supply of medicine is unstable.
Asked what infections are of most immediate concern, Sten Vermund, MD, PhD, Dean of the Yale School of Public Health, told this news organization that it was “diarrheal diseases, especially in kids ... The water supply [of Mariupol] is no longer potable, but people are drinking it anyway. And sewage systems are destroyed, and raw sewage is just released into the rivers and streams. So the whole family of diarrheal diseases and war are bedfellows. So are respiratory diseases, whenever we have mass migrations and mixing of ... homeless people and transients.”
There is one notable piece of good news that may reduce the spread of infectious diseases. Unlike the aftermath of World War II or the ongoing conflicts in the Middle East, Africa, and South Asia, refugees from the war in Ukraine are being taken into individual households throughout Poland, Germany, and other countries and are not being held in large displaced-persons camps. Dr. Vermund added, “The Syrian refugee camps in Lebanon are just tent camps with a million, 2 million people in them ... In theory, what the Poles are doing is a good thing from the point of view of preventing the spread of infection.”
One way of examining infections in war zones is by considering them based on how they are spread.
Respiratory infections
Although not as high on the list of concerns as TB or HIV, COVID-19 remains a big problem for infectious disease experts. Last fall, Ukraine ranked just behind the U.S. and Russia in deaths from COVID and in the top 10 in infections. Despite these dismal numbers, only 35% of people had completed the initial vaccination series.
The same conditions that fuel TB and COVID – crowding, especially in poorly ventilated settings – could lead to another measles outbreak. One occurred in Ukraine from 2017-2020, resulting in more than 115,000 cases. Even though the immunization rate for measles has now reached about 80%, the CDC considers Ukraine at high risk for another large outbreak since measles is so highly contagious.
According to the European Centre for Disease Prevention and Control (ECDC), Ukraine reported the second-highest number of TB cases in Europe (28,539). It is also one of the top 10 countries globally with the highest burden of multidrug-resistant tuberculosis (MDR-TB) – 27%. Equally disturbing is its ranking as having the second-highest rate of HIV/TB co-infection (26%) even before the war. Experts say war is a perfect breeding ground for TB, since starvation and overcrowding in poorly ventilated spaces encourages its spread.
Before the war, COVID had already caused severe disruptions in TB diagnosis and treatment access in Ukraine, and the World Health Organization suggested that the pandemic has set back efforts to end TB by more than a decade.
Drug-resistant TB has been one of the biggest worries. In their report on TB in Ukraine, British tuberculosis experts Tom Wingfield, MBChB, PhD, from the Liverpool School of Tropical Medicine, and Jessica Potter MBBCh, PhD, from Queen Mary University of London, pointed out that “drug resistance thrives on fractured health systems and sporadic medicine supply.”
Frederick Altice, MD, a Yale epidemiologist and addiction specialist, noted, “[if] medication for tuberculosis is discontinued, that not only causes potential recurrence of disease but multidrug-resistant TB disease,” and patients could become infectious again.
Dr. Wingfield expressed concern that people will not seek care because they see it as unaffordable, although he told this news organization that he’s impressed at the Polish government’s efforts to ensure care. Especially with the triad of HIV, TB, and opioid use, Dr. Wingfield and Dr. Potter emphasized that these problems reflect the social determinants of health – “the experiences and conditions in which people live.” These medical conditions are all quite treatable with support, and once treated they pose no risk to others.
HIV and opioid use
Before the war, an estimated 260,000 people were living with HIV in Ukraine. Their rate of new HIV diagnoses in 2017 was second highest in the world – 37 out of every 100,000, exceeded only by Russia, with 71 out of 100,000.
Dr. Vermund told this news organization that “when Crimea was seized by the Russians in 2014, there was an immediate crisis among injection drug users who were in drug treatment programs, because it’s illegal in Russia to use buprenorphine or methadone ... So immediately, those programs were shut down, and all the drug users who were holding jobs, supporting their families, were withdrawing from their addictions and searching for a replacement, which was illegal heroin.”
Dr. Altice added that of 800 patients in the region who had to go cold turkey, “ten percent were dead within 6 months. Dependent on unreliable street drugs, some overdosed or committed suicide because they could not get treatment. They went through terrible withdrawal and stress.”
And as they relapsed, the HIV rate soared. “Fifty percent of the methadone patients have got HIV,” Dr. Altice said, “and if they stop taking the methadone, they’re going to stop taking their HIV medications as well. Their lives will become chaotic and very destabilized.”
This experience may soon repeat itself. There were two methadone factories in Ukraine – in Odessa and Kharkiv – that are now shut down by the war. Although there are efforts to import methadone and many other drugs, supply chain issues are “devastating,” Dr. Altice said. “If their medication for tuberculosis is discontinued, that not only causes potential recurrence of disease but multidrug-resistant TB disease,” and they could become infectious again. “[With a] lack of medication, lack of sterile syringes, people will be sharing syringes; they’ll be desperate. So as the desperation level goes up, the risk environment goes up, so that people have decreased opportunities to protect themselves,” and there will be an explosion in HIV.
Dr. Altice observed that with the immigration to Poland and the west, many Ukrainian refugees “are relying on the kindness of strangers.” They are likely to be “fearful to disclose either their HIV or their TB treatment status,” being afraid of being regarded as modern-day lepers, even though they are likely not infectious. Both Dr. Altice and Dr. Potter emphasized the need for the governments of Poland and other receiving countries to provide the refugees with “reassurance that their health information will not be shared with others.” Dr. Altice emphasized that “this is one of the things that I would say that these other countries have to get right.”
Dr. Potter echoed that, noting that extraordinary care needs to be taken so that shared information is not used for deportation.
When refugees are housed with rural hosts, transportation problems sometimes arise, creating major barriers to accessing care and treatment. In particular, refugees with TB, HIV, and addiction who are placed in small, remote locations may have difficulty securing transportation to sites where treatments for their complex illnesses are available, including specialists and medications.
Ukrainian-born microbiologist Olena Rzhepishevska, PhD, of Umeå University in Sweden, said in an interview that a network of European TB researchers have developed a database on TBNet where patients with TB can be specifically placed with understanding and helpful hosts outside of Ukraine. They can receive housing and medication through this network.
So far, 4 million Ukrainians have fled the country and millions more have been displaced internally. Dr. Altice noted that there is an “increased vulnerability beyond the vulnerability that they already [have] just by being a refugee” that we generally don’t recognize. Additionally, Poland and Hungary are not very progressive about methadone therapy nor are those nations well-equipped to provide it.
Dr. Altice explained that even within Ukraine, those who want to move to better their chance of getting their methadone are then at risk of being conscripted. He spoke of the grave calculations men must make, choosing to become internally displaced and risk conscription or losing life-saving methadone or medicines for HIV or TB.
One other unfortunate consequence of war might be a spike in rape, sexual abuse, prostitution, unwanted pregnancies, HIV, and sexually transmitted infections.
There were an estimated 80,100 female sex workers in Ukraine in 2016, with 5.2% HIV positive. In times of war, with no home or income, some women turn to prostitution to survive. Others are victims of sex trafficking, both within Ukraine and as refugees. The Russian invasion increased the risks of a surge in HIV infections, unwanted pregnancies, and abortions. Women who find themselves pregnant due to rape (a common tool of war) or sex trafficking may also struggle to access safe abortions. Poland, for example, has severe restrictions on abortion, and Ukrainian women may turn to unsafe, back-alley abortions, with their resulting high risk of infection.
Waterborne infections
Another concern involves waterborne infections. In addition to the common diarrheal diseases such as E coli, which can be expected from poor sanitation, polio is a significant concern. In the fall of 2021, Ukraine had an outbreak of vaccine-derived polio, with two cases of paralysis and 20 additional cases. As polio only paralyzes 1 person in 200 of those infected, many other cases were likely undetected. A vaccination campaign was just beginning when the war began.
Wound infections and antimicrobial resistance
The ECDC also reports high rates of antimicrobial resistance (AMR) in Ukraine, particularly involving common gram-negative bacteria, including Escherichia coli (53% resistance to third-generation cephalosporins), Klebsiella pneumoniae (54% resistance to carbapenems), and Acinetobacter spp. (77% resistance to carbapenems). Because of this, they recommend refugees requiring hospital admission be isolated on admission and screened for AMR. These AMR often complicate traumatic injuries of war.
Prevention
Many of these potential problems stemming from the war in Ukraine and the displacement of millions of its citizens can be avoided.
Attempts are being made to immunize refugees. WHO has made working with countries receiving refugees a priority, particularly by vaccinating children against measles, rubella, and COVID. The European Union has also purchased vaccines for polio and tuberculosis.
But Russia has waged an active anti-vaccine campaign against COVID in Ukraine, while at the same time advocating for vaccines in Russia. According to UNICEF, other countries with relatively low vaccination rates and high vaccine skepticism – Moldova, Romania, and Bulgaria – are at higher risk of polio and measles than those with high vaccination levels.
The continuing war in Ukraine has exacerbated the medical challenges the citizens of Ukraine face at home and as refugees fleeing to neighboring countries. Improving communication among agencies and governments and building trust with the refugees could go a long way toward limiting the spread of preventable infectious diseases as a result of the war.
Continuing to try to keep supply chains open within Ukraine and ensuring adequate supplies of medications and vaccines to refugees will also be essential. But, of course, the better solution is to end the war.
Dr. Altice, Dr. Potter, Dr. Wingfield, Dr. Vermund, and Dr. Zinchuk all report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Every day we see stark images of the war in Ukraine – bombed-out buildings, explosions, and bodies lying in the streets. But there’s another, less visible war against the bacteria and viruses that are gathering their forces together. They, too, will infect parts of the population and may spread throughout Europe. Here’s what Ukrainians, and their neighbors, are facing on the infectious disease front.
Andrey Zinchuk, MD, MHS, a pulmonary/critical care physician at Yale and a native of Ukraine who immigrated to the U.S. at the age of 14 with his family, set the background for understanding this crisis. He said that TB and HIV rates in Ukraine have long been especially high, even before the current conflict: “Part of the challenge of the health care system in Ukraine is that it’s difficult to maintain a steady policy because of political instability,” he said. “We’ve had three revolutions in the last 20 years,” not counting the current Russian invasion.
The first was the breakup of the Soviet Union, which led to “an epidemic of people with HIV, hepatitis, and opioid use.” Next was the Orange Revolution in 2004 over fraud during a presidential election. In 2014 came the Maiden Revolution, after the government chose closer ties to Russia rather than Europe. Then-president Viktor Yanukovych fled to Russia.
“That’s when Russia annexed Crimea. There was essentially infiltration in Russian propaganda in the east of the country,” Dr. Zinchuk said. “This helped the Russians manufacture uprisings there to create a separatist state (the Luhansk and Donetsk People’s Republics) which were mostly Russian-speaking parts of the country,” an area known as the Donbas. This resulted in a war in eastern Ukraine that began 2014, with more than 10,000 deaths.
After the 2014 revolution, Dr. Zinchuk said, “There was a tremendous change in the way ... medical care was provided, and tremendous growth and stability in the medical supply for those chronic medical conditions.”
Nevertheless, health care expenditures in Ukraine have been quite low. Even before the current conflict, Dr. Zinchuk noted, annual health care expenditures in Ukraine were about $600 per capita. In comparison, it’s about $4,500 per person in Germany and $12,530 in the United States.
Despite those low per-capita expenditures in Ukraine, access to medicines – such as insulin for diabetes and antibiotics for tuberculosis – was stable before the war. But now, Dr. Zinchuk said, his aunt and uncle have had to flee Kyiv for the countryside and, while safe, they have “no plumbing and have to heat the house by burning firewood.” More significantly, their supply of medicine is unstable.
Asked what infections are of most immediate concern, Sten Vermund, MD, PhD, Dean of the Yale School of Public Health, told this news organization that it was “diarrheal diseases, especially in kids ... The water supply [of Mariupol] is no longer potable, but people are drinking it anyway. And sewage systems are destroyed, and raw sewage is just released into the rivers and streams. So the whole family of diarrheal diseases and war are bedfellows. So are respiratory diseases, whenever we have mass migrations and mixing of ... homeless people and transients.”
There is one notable piece of good news that may reduce the spread of infectious diseases. Unlike the aftermath of World War II or the ongoing conflicts in the Middle East, Africa, and South Asia, refugees from the war in Ukraine are being taken into individual households throughout Poland, Germany, and other countries and are not being held in large displaced-persons camps. Dr. Vermund added, “The Syrian refugee camps in Lebanon are just tent camps with a million, 2 million people in them ... In theory, what the Poles are doing is a good thing from the point of view of preventing the spread of infection.”
One way of examining infections in war zones is by considering them based on how they are spread.
Respiratory infections
Although not as high on the list of concerns as TB or HIV, COVID-19 remains a big problem for infectious disease experts. Last fall, Ukraine ranked just behind the U.S. and Russia in deaths from COVID and in the top 10 in infections. Despite these dismal numbers, only 35% of people had completed the initial vaccination series.
The same conditions that fuel TB and COVID – crowding, especially in poorly ventilated settings – could lead to another measles outbreak. One occurred in Ukraine from 2017-2020, resulting in more than 115,000 cases. Even though the immunization rate for measles has now reached about 80%, the CDC considers Ukraine at high risk for another large outbreak since measles is so highly contagious.
According to the European Centre for Disease Prevention and Control (ECDC), Ukraine reported the second-highest number of TB cases in Europe (28,539). It is also one of the top 10 countries globally with the highest burden of multidrug-resistant tuberculosis (MDR-TB) – 27%. Equally disturbing is its ranking as having the second-highest rate of HIV/TB co-infection (26%) even before the war. Experts say war is a perfect breeding ground for TB, since starvation and overcrowding in poorly ventilated spaces encourages its spread.
Before the war, COVID had already caused severe disruptions in TB diagnosis and treatment access in Ukraine, and the World Health Organization suggested that the pandemic has set back efforts to end TB by more than a decade.
Drug-resistant TB has been one of the biggest worries. In their report on TB in Ukraine, British tuberculosis experts Tom Wingfield, MBChB, PhD, from the Liverpool School of Tropical Medicine, and Jessica Potter MBBCh, PhD, from Queen Mary University of London, pointed out that “drug resistance thrives on fractured health systems and sporadic medicine supply.”
Frederick Altice, MD, a Yale epidemiologist and addiction specialist, noted, “[if] medication for tuberculosis is discontinued, that not only causes potential recurrence of disease but multidrug-resistant TB disease,” and patients could become infectious again.
Dr. Wingfield expressed concern that people will not seek care because they see it as unaffordable, although he told this news organization that he’s impressed at the Polish government’s efforts to ensure care. Especially with the triad of HIV, TB, and opioid use, Dr. Wingfield and Dr. Potter emphasized that these problems reflect the social determinants of health – “the experiences and conditions in which people live.” These medical conditions are all quite treatable with support, and once treated they pose no risk to others.
HIV and opioid use
Before the war, an estimated 260,000 people were living with HIV in Ukraine. Their rate of new HIV diagnoses in 2017 was second highest in the world – 37 out of every 100,000, exceeded only by Russia, with 71 out of 100,000.
Dr. Vermund told this news organization that “when Crimea was seized by the Russians in 2014, there was an immediate crisis among injection drug users who were in drug treatment programs, because it’s illegal in Russia to use buprenorphine or methadone ... So immediately, those programs were shut down, and all the drug users who were holding jobs, supporting their families, were withdrawing from their addictions and searching for a replacement, which was illegal heroin.”
Dr. Altice added that of 800 patients in the region who had to go cold turkey, “ten percent were dead within 6 months. Dependent on unreliable street drugs, some overdosed or committed suicide because they could not get treatment. They went through terrible withdrawal and stress.”
And as they relapsed, the HIV rate soared. “Fifty percent of the methadone patients have got HIV,” Dr. Altice said, “and if they stop taking the methadone, they’re going to stop taking their HIV medications as well. Their lives will become chaotic and very destabilized.”
This experience may soon repeat itself. There were two methadone factories in Ukraine – in Odessa and Kharkiv – that are now shut down by the war. Although there are efforts to import methadone and many other drugs, supply chain issues are “devastating,” Dr. Altice said. “If their medication for tuberculosis is discontinued, that not only causes potential recurrence of disease but multidrug-resistant TB disease,” and they could become infectious again. “[With a] lack of medication, lack of sterile syringes, people will be sharing syringes; they’ll be desperate. So as the desperation level goes up, the risk environment goes up, so that people have decreased opportunities to protect themselves,” and there will be an explosion in HIV.
Dr. Altice observed that with the immigration to Poland and the west, many Ukrainian refugees “are relying on the kindness of strangers.” They are likely to be “fearful to disclose either their HIV or their TB treatment status,” being afraid of being regarded as modern-day lepers, even though they are likely not infectious. Both Dr. Altice and Dr. Potter emphasized the need for the governments of Poland and other receiving countries to provide the refugees with “reassurance that their health information will not be shared with others.” Dr. Altice emphasized that “this is one of the things that I would say that these other countries have to get right.”
Dr. Potter echoed that, noting that extraordinary care needs to be taken so that shared information is not used for deportation.
When refugees are housed with rural hosts, transportation problems sometimes arise, creating major barriers to accessing care and treatment. In particular, refugees with TB, HIV, and addiction who are placed in small, remote locations may have difficulty securing transportation to sites where treatments for their complex illnesses are available, including specialists and medications.
Ukrainian-born microbiologist Olena Rzhepishevska, PhD, of Umeå University in Sweden, said in an interview that a network of European TB researchers have developed a database on TBNet where patients with TB can be specifically placed with understanding and helpful hosts outside of Ukraine. They can receive housing and medication through this network.
So far, 4 million Ukrainians have fled the country and millions more have been displaced internally. Dr. Altice noted that there is an “increased vulnerability beyond the vulnerability that they already [have] just by being a refugee” that we generally don’t recognize. Additionally, Poland and Hungary are not very progressive about methadone therapy nor are those nations well-equipped to provide it.
Dr. Altice explained that even within Ukraine, those who want to move to better their chance of getting their methadone are then at risk of being conscripted. He spoke of the grave calculations men must make, choosing to become internally displaced and risk conscription or losing life-saving methadone or medicines for HIV or TB.
One other unfortunate consequence of war might be a spike in rape, sexual abuse, prostitution, unwanted pregnancies, HIV, and sexually transmitted infections.
There were an estimated 80,100 female sex workers in Ukraine in 2016, with 5.2% HIV positive. In times of war, with no home or income, some women turn to prostitution to survive. Others are victims of sex trafficking, both within Ukraine and as refugees. The Russian invasion increased the risks of a surge in HIV infections, unwanted pregnancies, and abortions. Women who find themselves pregnant due to rape (a common tool of war) or sex trafficking may also struggle to access safe abortions. Poland, for example, has severe restrictions on abortion, and Ukrainian women may turn to unsafe, back-alley abortions, with their resulting high risk of infection.
Waterborne infections
Another concern involves waterborne infections. In addition to the common diarrheal diseases such as E coli, which can be expected from poor sanitation, polio is a significant concern. In the fall of 2021, Ukraine had an outbreak of vaccine-derived polio, with two cases of paralysis and 20 additional cases. As polio only paralyzes 1 person in 200 of those infected, many other cases were likely undetected. A vaccination campaign was just beginning when the war began.
Wound infections and antimicrobial resistance
The ECDC also reports high rates of antimicrobial resistance (AMR) in Ukraine, particularly involving common gram-negative bacteria, including Escherichia coli (53% resistance to third-generation cephalosporins), Klebsiella pneumoniae (54% resistance to carbapenems), and Acinetobacter spp. (77% resistance to carbapenems). Because of this, they recommend refugees requiring hospital admission be isolated on admission and screened for AMR. These AMR often complicate traumatic injuries of war.
Prevention
Many of these potential problems stemming from the war in Ukraine and the displacement of millions of its citizens can be avoided.
Attempts are being made to immunize refugees. WHO has made working with countries receiving refugees a priority, particularly by vaccinating children against measles, rubella, and COVID. The European Union has also purchased vaccines for polio and tuberculosis.
But Russia has waged an active anti-vaccine campaign against COVID in Ukraine, while at the same time advocating for vaccines in Russia. According to UNICEF, other countries with relatively low vaccination rates and high vaccine skepticism – Moldova, Romania, and Bulgaria – are at higher risk of polio and measles than those with high vaccination levels.
The continuing war in Ukraine has exacerbated the medical challenges the citizens of Ukraine face at home and as refugees fleeing to neighboring countries. Improving communication among agencies and governments and building trust with the refugees could go a long way toward limiting the spread of preventable infectious diseases as a result of the war.
Continuing to try to keep supply chains open within Ukraine and ensuring adequate supplies of medications and vaccines to refugees will also be essential. But, of course, the better solution is to end the war.
Dr. Altice, Dr. Potter, Dr. Wingfield, Dr. Vermund, and Dr. Zinchuk all report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Every day we see stark images of the war in Ukraine – bombed-out buildings, explosions, and bodies lying in the streets. But there’s another, less visible war against the bacteria and viruses that are gathering their forces together. They, too, will infect parts of the population and may spread throughout Europe. Here’s what Ukrainians, and their neighbors, are facing on the infectious disease front.
Andrey Zinchuk, MD, MHS, a pulmonary/critical care physician at Yale and a native of Ukraine who immigrated to the U.S. at the age of 14 with his family, set the background for understanding this crisis. He said that TB and HIV rates in Ukraine have long been especially high, even before the current conflict: “Part of the challenge of the health care system in Ukraine is that it’s difficult to maintain a steady policy because of political instability,” he said. “We’ve had three revolutions in the last 20 years,” not counting the current Russian invasion.
The first was the breakup of the Soviet Union, which led to “an epidemic of people with HIV, hepatitis, and opioid use.” Next was the Orange Revolution in 2004 over fraud during a presidential election. In 2014 came the Maiden Revolution, after the government chose closer ties to Russia rather than Europe. Then-president Viktor Yanukovych fled to Russia.
“That’s when Russia annexed Crimea. There was essentially infiltration in Russian propaganda in the east of the country,” Dr. Zinchuk said. “This helped the Russians manufacture uprisings there to create a separatist state (the Luhansk and Donetsk People’s Republics) which were mostly Russian-speaking parts of the country,” an area known as the Donbas. This resulted in a war in eastern Ukraine that began 2014, with more than 10,000 deaths.
After the 2014 revolution, Dr. Zinchuk said, “There was a tremendous change in the way ... medical care was provided, and tremendous growth and stability in the medical supply for those chronic medical conditions.”
Nevertheless, health care expenditures in Ukraine have been quite low. Even before the current conflict, Dr. Zinchuk noted, annual health care expenditures in Ukraine were about $600 per capita. In comparison, it’s about $4,500 per person in Germany and $12,530 in the United States.
Despite those low per-capita expenditures in Ukraine, access to medicines – such as insulin for diabetes and antibiotics for tuberculosis – was stable before the war. But now, Dr. Zinchuk said, his aunt and uncle have had to flee Kyiv for the countryside and, while safe, they have “no plumbing and have to heat the house by burning firewood.” More significantly, their supply of medicine is unstable.
Asked what infections are of most immediate concern, Sten Vermund, MD, PhD, Dean of the Yale School of Public Health, told this news organization that it was “diarrheal diseases, especially in kids ... The water supply [of Mariupol] is no longer potable, but people are drinking it anyway. And sewage systems are destroyed, and raw sewage is just released into the rivers and streams. So the whole family of diarrheal diseases and war are bedfellows. So are respiratory diseases, whenever we have mass migrations and mixing of ... homeless people and transients.”
There is one notable piece of good news that may reduce the spread of infectious diseases. Unlike the aftermath of World War II or the ongoing conflicts in the Middle East, Africa, and South Asia, refugees from the war in Ukraine are being taken into individual households throughout Poland, Germany, and other countries and are not being held in large displaced-persons camps. Dr. Vermund added, “The Syrian refugee camps in Lebanon are just tent camps with a million, 2 million people in them ... In theory, what the Poles are doing is a good thing from the point of view of preventing the spread of infection.”
One way of examining infections in war zones is by considering them based on how they are spread.
Respiratory infections
Although not as high on the list of concerns as TB or HIV, COVID-19 remains a big problem for infectious disease experts. Last fall, Ukraine ranked just behind the U.S. and Russia in deaths from COVID and in the top 10 in infections. Despite these dismal numbers, only 35% of people had completed the initial vaccination series.
The same conditions that fuel TB and COVID – crowding, especially in poorly ventilated settings – could lead to another measles outbreak. One occurred in Ukraine from 2017-2020, resulting in more than 115,000 cases. Even though the immunization rate for measles has now reached about 80%, the CDC considers Ukraine at high risk for another large outbreak since measles is so highly contagious.
According to the European Centre for Disease Prevention and Control (ECDC), Ukraine reported the second-highest number of TB cases in Europe (28,539). It is also one of the top 10 countries globally with the highest burden of multidrug-resistant tuberculosis (MDR-TB) – 27%. Equally disturbing is its ranking as having the second-highest rate of HIV/TB co-infection (26%) even before the war. Experts say war is a perfect breeding ground for TB, since starvation and overcrowding in poorly ventilated spaces encourages its spread.
Before the war, COVID had already caused severe disruptions in TB diagnosis and treatment access in Ukraine, and the World Health Organization suggested that the pandemic has set back efforts to end TB by more than a decade.
Drug-resistant TB has been one of the biggest worries. In their report on TB in Ukraine, British tuberculosis experts Tom Wingfield, MBChB, PhD, from the Liverpool School of Tropical Medicine, and Jessica Potter MBBCh, PhD, from Queen Mary University of London, pointed out that “drug resistance thrives on fractured health systems and sporadic medicine supply.”
Frederick Altice, MD, a Yale epidemiologist and addiction specialist, noted, “[if] medication for tuberculosis is discontinued, that not only causes potential recurrence of disease but multidrug-resistant TB disease,” and patients could become infectious again.
Dr. Wingfield expressed concern that people will not seek care because they see it as unaffordable, although he told this news organization that he’s impressed at the Polish government’s efforts to ensure care. Especially with the triad of HIV, TB, and opioid use, Dr. Wingfield and Dr. Potter emphasized that these problems reflect the social determinants of health – “the experiences and conditions in which people live.” These medical conditions are all quite treatable with support, and once treated they pose no risk to others.
HIV and opioid use
Before the war, an estimated 260,000 people were living with HIV in Ukraine. Their rate of new HIV diagnoses in 2017 was second highest in the world – 37 out of every 100,000, exceeded only by Russia, with 71 out of 100,000.
Dr. Vermund told this news organization that “when Crimea was seized by the Russians in 2014, there was an immediate crisis among injection drug users who were in drug treatment programs, because it’s illegal in Russia to use buprenorphine or methadone ... So immediately, those programs were shut down, and all the drug users who were holding jobs, supporting their families, were withdrawing from their addictions and searching for a replacement, which was illegal heroin.”
Dr. Altice added that of 800 patients in the region who had to go cold turkey, “ten percent were dead within 6 months. Dependent on unreliable street drugs, some overdosed or committed suicide because they could not get treatment. They went through terrible withdrawal and stress.”
And as they relapsed, the HIV rate soared. “Fifty percent of the methadone patients have got HIV,” Dr. Altice said, “and if they stop taking the methadone, they’re going to stop taking their HIV medications as well. Their lives will become chaotic and very destabilized.”
This experience may soon repeat itself. There were two methadone factories in Ukraine – in Odessa and Kharkiv – that are now shut down by the war. Although there are efforts to import methadone and many other drugs, supply chain issues are “devastating,” Dr. Altice said. “If their medication for tuberculosis is discontinued, that not only causes potential recurrence of disease but multidrug-resistant TB disease,” and they could become infectious again. “[With a] lack of medication, lack of sterile syringes, people will be sharing syringes; they’ll be desperate. So as the desperation level goes up, the risk environment goes up, so that people have decreased opportunities to protect themselves,” and there will be an explosion in HIV.
Dr. Altice observed that with the immigration to Poland and the west, many Ukrainian refugees “are relying on the kindness of strangers.” They are likely to be “fearful to disclose either their HIV or their TB treatment status,” being afraid of being regarded as modern-day lepers, even though they are likely not infectious. Both Dr. Altice and Dr. Potter emphasized the need for the governments of Poland and other receiving countries to provide the refugees with “reassurance that their health information will not be shared with others.” Dr. Altice emphasized that “this is one of the things that I would say that these other countries have to get right.”
Dr. Potter echoed that, noting that extraordinary care needs to be taken so that shared information is not used for deportation.
When refugees are housed with rural hosts, transportation problems sometimes arise, creating major barriers to accessing care and treatment. In particular, refugees with TB, HIV, and addiction who are placed in small, remote locations may have difficulty securing transportation to sites where treatments for their complex illnesses are available, including specialists and medications.
Ukrainian-born microbiologist Olena Rzhepishevska, PhD, of Umeå University in Sweden, said in an interview that a network of European TB researchers have developed a database on TBNet where patients with TB can be specifically placed with understanding and helpful hosts outside of Ukraine. They can receive housing and medication through this network.
So far, 4 million Ukrainians have fled the country and millions more have been displaced internally. Dr. Altice noted that there is an “increased vulnerability beyond the vulnerability that they already [have] just by being a refugee” that we generally don’t recognize. Additionally, Poland and Hungary are not very progressive about methadone therapy nor are those nations well-equipped to provide it.
Dr. Altice explained that even within Ukraine, those who want to move to better their chance of getting their methadone are then at risk of being conscripted. He spoke of the grave calculations men must make, choosing to become internally displaced and risk conscription or losing life-saving methadone or medicines for HIV or TB.
One other unfortunate consequence of war might be a spike in rape, sexual abuse, prostitution, unwanted pregnancies, HIV, and sexually transmitted infections.
There were an estimated 80,100 female sex workers in Ukraine in 2016, with 5.2% HIV positive. In times of war, with no home or income, some women turn to prostitution to survive. Others are victims of sex trafficking, both within Ukraine and as refugees. The Russian invasion increased the risks of a surge in HIV infections, unwanted pregnancies, and abortions. Women who find themselves pregnant due to rape (a common tool of war) or sex trafficking may also struggle to access safe abortions. Poland, for example, has severe restrictions on abortion, and Ukrainian women may turn to unsafe, back-alley abortions, with their resulting high risk of infection.
Waterborne infections
Another concern involves waterborne infections. In addition to the common diarrheal diseases such as E coli, which can be expected from poor sanitation, polio is a significant concern. In the fall of 2021, Ukraine had an outbreak of vaccine-derived polio, with two cases of paralysis and 20 additional cases. As polio only paralyzes 1 person in 200 of those infected, many other cases were likely undetected. A vaccination campaign was just beginning when the war began.
Wound infections and antimicrobial resistance
The ECDC also reports high rates of antimicrobial resistance (AMR) in Ukraine, particularly involving common gram-negative bacteria, including Escherichia coli (53% resistance to third-generation cephalosporins), Klebsiella pneumoniae (54% resistance to carbapenems), and Acinetobacter spp. (77% resistance to carbapenems). Because of this, they recommend refugees requiring hospital admission be isolated on admission and screened for AMR. These AMR often complicate traumatic injuries of war.
Prevention
Many of these potential problems stemming from the war in Ukraine and the displacement of millions of its citizens can be avoided.
Attempts are being made to immunize refugees. WHO has made working with countries receiving refugees a priority, particularly by vaccinating children against measles, rubella, and COVID. The European Union has also purchased vaccines for polio and tuberculosis.
But Russia has waged an active anti-vaccine campaign against COVID in Ukraine, while at the same time advocating for vaccines in Russia. According to UNICEF, other countries with relatively low vaccination rates and high vaccine skepticism – Moldova, Romania, and Bulgaria – are at higher risk of polio and measles than those with high vaccination levels.
The continuing war in Ukraine has exacerbated the medical challenges the citizens of Ukraine face at home and as refugees fleeing to neighboring countries. Improving communication among agencies and governments and building trust with the refugees could go a long way toward limiting the spread of preventable infectious diseases as a result of the war.
Continuing to try to keep supply chains open within Ukraine and ensuring adequate supplies of medications and vaccines to refugees will also be essential. But, of course, the better solution is to end the war.
Dr. Altice, Dr. Potter, Dr. Wingfield, Dr. Vermund, and Dr. Zinchuk all report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The importance of treating insomnia in psychiatric illness
Data suggests this symptom, defined as chronic sleep onset and/or sleep continuity problems associated with impaired daytime functioning, is common in psychiatric illnesses, and can worsen their course.2
The incidence of psychiatric illness in patients with insomnia is estimated at near 50%, with the highest rates found in mood disorders such as depression and bipolar disorder, as well as anxiety disorders.3 In patients with diagnosed major depressive disorder, insomnia rates can approach 90%.4-6

Insomnia has been identified as a risk factor for development of mental illness, including doubling the risk of major depressive disorder and tripling the risk of any depressive or anxiety disorder.7,8 It can also significantly increase the risk of alcohol abuse and psychosis.8
Sleep disturbances can worsen symptoms of diagnosed mental illness, including substance abuse, mood and psychotic disorders.9-10 In one study, nearly 75% of patients with a diagnosis of schizophrenia or bipolar spectrum disorder had at least one type of sleep disturbance (insomnia, hypersomnia, or delayed sleep phase).10 This was almost twice the rate in healthy controls. Importantly, compared with well-rested subjects with mental illness in this study, sleep-disordered participants had higher rates of negative and depressive symptoms on the Positive and Negative Syndrome Scale, as well as significantly lower function via the global assessment of functioning.11,12
Additional data suggests simply being awake during the night (00:00-05:59) elevates risk of suicide. The mean incident rate of completed suicide in one study was a striking four times the rate noted during daytime hours (06:00-23:59 ) (P < .001).13
Although insomnia symptoms can resolve after relief from a particular life stressor, as many as half of patients with more severe symptoms develop a chronic course.14 This then leads to an extended use of many types of sedative-hypnotics designed and studied primarily for short-term use.15 In a survey reviewing national use of prescription drugs for insomnia, as many as 20% of individuals use a medication to target insomnia in a given month.16
Fortunately, despite the many challenges posed by COVID-19, particularly for those with psychiatric illness and limited access to care, telehealth has become more readily available. Additionally, digital versions of evidence-based treatments specifically for sleep problems, such as cognitive-behavioral therapy for insomnia (CBT-I), are regularly being developed.
The benefits of CBT-I have been demonstrated repeatedly and it is recommended as the first line treatment for insomnia by the Clinical Guidelines of the American Academy of Sleep Medicine, the Centers for Disease Control and Prevention, and the National Institutes of Health.17-21 Studies suggest benefits persist long-term, even after completing the therapy sessions, which differ in durability from medication choices.18
One group that may be particularly suited for treatment with CBT-I is women with insomnia during pregnancy or the postpartum period. In these women, options for treatment may be limited by risk of medication during breastfeeding, as well as difficulty traveling to a physician’s or therapist’s office to receive psychotherapy. However, two recent studies evaluated the use of digital CBT-I to treat insomnia during pregnancy and in the postpartum period, respectively.22-23
In both studies,the same group of women with insomnia diagnosed during pregnancy were given six weekly 20-minute sessions of digital CBT-I or standard treatment for insomnia, including medication and psychotherapy per their usual provider.
By study end, the pregnant women receiving the CBT-I intervention not only had significantly improved severity of insomnia, they also experienced improved depression and anxiety symptoms, and a decrease in the use of prescription or over-the-counter sleep aides, compared with the standard treatment group, lowering the fetal exposure to medication during pregnancy.22
In the more recent study, the same group was followed for 6 months post partum.23 Results were again notable, with the women who received CBT-I reporting significantly less insomnia, as well as significantly lower rates of probable major depression at 3 and 6 months (18% vs. 4%, 10% vs. 0%, respectively.) They also exhibited lower rates of moderate to severe anxiety (17% vs. 4%) at 3 months, compared with those receiving standard care. With as many as one in seven women suffering from postpartum depression, these findings represent a substantial public health benefit.
In summary, insomnia is a critical area of focus for any provider diagnosing and treating psychiatric illness. Attempts to optimize sleep, whether through CBT-I or other psychotherapy approaches, or evidence-based medications dosed for appropriate lengths and at safe doses, should be a part of most, if not all, clinical encounters.
Dr. Reid is a board-certified psychiatrist and award-winning medical educator with a private practice in Philadelphia, as well as a clinical faculty role at the University of Pennsylvania, also in Philadelphia. She attended medical school at Columbia University, New York, and completed her psychiatry residency at the University of California, Los Angeles. Dr. Reid is a regular contributor to Psychology Today with her blog, “Think Like a Shrink,” and writes and podcasts as The Reflective Doc.
References
1. Voitsidis P et al. Psychiatry Res. 2020 Jul;289:113076. doi: 10.1016/j.psychres.2020.113076.
2. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, Va.: American Psychiatric Publishing, 2013.
3. Ford DE and Kamerow DB. JAMA. 1989;262(11):1479-84. doi: 10.1001/jama.1989.03430110069030.
4. Ohayon MM and Roth T. J Psychiatr Res. Jan-Feb 2003;37(1):9-15. doi: 10.1016/s0022-3956(02)00052-3.
5. Seow LSE et al. J Ment Health. 2016 Dec;25(6):492-9. doi: 10.3109/09638237.2015.1124390.
6. Thase ME. J Clin Psychiatry. 1999;60 Suppl 17:28-31; discussion 46-8.
7. Baglioni C et al. J Affect Disord. 2011 Dec;135(1-3):10-9. doi: 10.1016/j.jad.2011.01.011.
8. Hertenstein E et al. Sleep Med Rev. 2019 Feb;43:96-105. doi: 10.1016/j.smrv.2018.10.006.
9. Brower KJ et al. Medical Hypotheses. 2010;74(5):928-33. doi: 10.1016/j.mehy.2009.10.020.
10. Laskemoen JF et al. Compr Psychiatry. 2019 May;91:6-12. doi: 10.1016/j.comppsych.2019.02.006.
11. Kay SR et al. Schizophr Bull. 1987;13(2):261-76. doi: 10.1093/schbul/13.2.261.
12. Hall R. Psychosomatics. May-Jun 1995;36(3):267-75. doi: 10.1016/S0033-3182(95)71666-8.
13. Perlis ML et al. J Clin Psychiatry. 2016 Jun;77(6):e726-33. doi: 10.4088/JCP.15m10131.
14. Morin CM et al. Arch Intern Med. 2009 Mar 9. doi: 10.1001/archinternmed.2008.610.
15. Cheung J et al. Sleep Med Clin. 2019 Jun;14(2):253-65. doi: 10.1016/j.jsmc.2019.01.006.
16. Bertisch SM et al. Sleep. 2014 Feb 1. doi: 10.5665/sleep.3410.
17. Okajima I et al. Sleep Biol Rhythms. 2010 Nov 28. doi: 10.1111/j.1479-8425.2010.00481.x.
18. Trauer JM et al. Ann Intern Med. 2015 Aug 4. doi: 10.7326/M14-2841.
19. Edinger J et al. J Clin Sleep Med. 2021 Feb 1. doi: 10.5664/jcsm.8986.
20. U.S. Centers for Disease Control and Prevention. https://www.cdc.gov/sleep/for-clinicians.html.
21. National Institutes of Health. Sleep Health. https://www.nhlbi.nih.gov/health-topics/education-and-awareness/sleep-health.
22. Felder JN et al. JAMA Psychiatry. 2020;77(5):484-92. doi:10.1001/jamapsychiatry.2019.4491.
23. Felder JN et al. Sleep. 2022 Feb 14. doi: 10.1093/sleep/zsab280.
Data suggests this symptom, defined as chronic sleep onset and/or sleep continuity problems associated with impaired daytime functioning, is common in psychiatric illnesses, and can worsen their course.2
The incidence of psychiatric illness in patients with insomnia is estimated at near 50%, with the highest rates found in mood disorders such as depression and bipolar disorder, as well as anxiety disorders.3 In patients with diagnosed major depressive disorder, insomnia rates can approach 90%.4-6
Insomnia has been identified as a risk factor for development of mental illness, including doubling the risk of major depressive disorder and tripling the risk of any depressive or anxiety disorder.7,8 It can also significantly increase the risk of alcohol abuse and psychosis.8
Sleep disturbances can worsen symptoms of diagnosed mental illness, including substance abuse, mood and psychotic disorders.9-10 In one study, nearly 75% of patients with a diagnosis of schizophrenia or bipolar spectrum disorder had at least one type of sleep disturbance (insomnia, hypersomnia, or delayed sleep phase).10 This was almost twice the rate in healthy controls. Importantly, compared with well-rested subjects with mental illness in this study, sleep-disordered participants had higher rates of negative and depressive symptoms on the Positive and Negative Syndrome Scale, as well as significantly lower function via the global assessment of functioning.11,12
Additional data suggests simply being awake during the night (00:00-05:59) elevates risk of suicide. The mean incident rate of completed suicide in one study was a striking four times the rate noted during daytime hours (06:00-23:59 ) (P < .001).13
Although insomnia symptoms can resolve after relief from a particular life stressor, as many as half of patients with more severe symptoms develop a chronic course.14 This then leads to an extended use of many types of sedative-hypnotics designed and studied primarily for short-term use.15 In a survey reviewing national use of prescription drugs for insomnia, as many as 20% of individuals use a medication to target insomnia in a given month.16
Fortunately, despite the many challenges posed by COVID-19, particularly for those with psychiatric illness and limited access to care, telehealth has become more readily available. Additionally, digital versions of evidence-based treatments specifically for sleep problems, such as cognitive-behavioral therapy for insomnia (CBT-I), are regularly being developed.
The benefits of CBT-I have been demonstrated repeatedly and it is recommended as the first line treatment for insomnia by the Clinical Guidelines of the American Academy of Sleep Medicine, the Centers for Disease Control and Prevention, and the National Institutes of Health.17-21 Studies suggest benefits persist long-term, even after completing the therapy sessions, which differ in durability from medication choices.18
One group that may be particularly suited for treatment with CBT-I is women with insomnia during pregnancy or the postpartum period. In these women, options for treatment may be limited by risk of medication during breastfeeding, as well as difficulty traveling to a physician’s or therapist’s office to receive psychotherapy. However, two recent studies evaluated the use of digital CBT-I to treat insomnia during pregnancy and in the postpartum period, respectively.22-23
In both studies,the same group of women with insomnia diagnosed during pregnancy were given six weekly 20-minute sessions of digital CBT-I or standard treatment for insomnia, including medication and psychotherapy per their usual provider.
By study end, the pregnant women receiving the CBT-I intervention not only had significantly improved severity of insomnia, they also experienced improved depression and anxiety symptoms, and a decrease in the use of prescription or over-the-counter sleep aides, compared with the standard treatment group, lowering the fetal exposure to medication during pregnancy.22
In the more recent study, the same group was followed for 6 months post partum.23 Results were again notable, with the women who received CBT-I reporting significantly less insomnia, as well as significantly lower rates of probable major depression at 3 and 6 months (18% vs. 4%, 10% vs. 0%, respectively.) They also exhibited lower rates of moderate to severe anxiety (17% vs. 4%) at 3 months, compared with those receiving standard care. With as many as one in seven women suffering from postpartum depression, these findings represent a substantial public health benefit.
In summary, insomnia is a critical area of focus for any provider diagnosing and treating psychiatric illness. Attempts to optimize sleep, whether through CBT-I or other psychotherapy approaches, or evidence-based medications dosed for appropriate lengths and at safe doses, should be a part of most, if not all, clinical encounters.
Dr. Reid is a board-certified psychiatrist and award-winning medical educator with a private practice in Philadelphia, as well as a clinical faculty role at the University of Pennsylvania, also in Philadelphia. She attended medical school at Columbia University, New York, and completed her psychiatry residency at the University of California, Los Angeles. Dr. Reid is a regular contributor to Psychology Today with her blog, “Think Like a Shrink,” and writes and podcasts as The Reflective Doc.
References
1. Voitsidis P et al. Psychiatry Res. 2020 Jul;289:113076. doi: 10.1016/j.psychres.2020.113076.
2. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, Va.: American Psychiatric Publishing, 2013.
3. Ford DE and Kamerow DB. JAMA. 1989;262(11):1479-84. doi: 10.1001/jama.1989.03430110069030.
4. Ohayon MM and Roth T. J Psychiatr Res. Jan-Feb 2003;37(1):9-15. doi: 10.1016/s0022-3956(02)00052-3.
5. Seow LSE et al. J Ment Health. 2016 Dec;25(6):492-9. doi: 10.3109/09638237.2015.1124390.
6. Thase ME. J Clin Psychiatry. 1999;60 Suppl 17:28-31; discussion 46-8.
7. Baglioni C et al. J Affect Disord. 2011 Dec;135(1-3):10-9. doi: 10.1016/j.jad.2011.01.011.
8. Hertenstein E et al. Sleep Med Rev. 2019 Feb;43:96-105. doi: 10.1016/j.smrv.2018.10.006.
9. Brower KJ et al. Medical Hypotheses. 2010;74(5):928-33. doi: 10.1016/j.mehy.2009.10.020.
10. Laskemoen JF et al. Compr Psychiatry. 2019 May;91:6-12. doi: 10.1016/j.comppsych.2019.02.006.
11. Kay SR et al. Schizophr Bull. 1987;13(2):261-76. doi: 10.1093/schbul/13.2.261.
12. Hall R. Psychosomatics. May-Jun 1995;36(3):267-75. doi: 10.1016/S0033-3182(95)71666-8.
13. Perlis ML et al. J Clin Psychiatry. 2016 Jun;77(6):e726-33. doi: 10.4088/JCP.15m10131.
14. Morin CM et al. Arch Intern Med. 2009 Mar 9. doi: 10.1001/archinternmed.2008.610.
15. Cheung J et al. Sleep Med Clin. 2019 Jun;14(2):253-65. doi: 10.1016/j.jsmc.2019.01.006.
16. Bertisch SM et al. Sleep. 2014 Feb 1. doi: 10.5665/sleep.3410.
17. Okajima I et al. Sleep Biol Rhythms. 2010 Nov 28. doi: 10.1111/j.1479-8425.2010.00481.x.
18. Trauer JM et al. Ann Intern Med. 2015 Aug 4. doi: 10.7326/M14-2841.
19. Edinger J et al. J Clin Sleep Med. 2021 Feb 1. doi: 10.5664/jcsm.8986.
20. U.S. Centers for Disease Control and Prevention. https://www.cdc.gov/sleep/for-clinicians.html.
21. National Institutes of Health. Sleep Health. https://www.nhlbi.nih.gov/health-topics/education-and-awareness/sleep-health.
22. Felder JN et al. JAMA Psychiatry. 2020;77(5):484-92. doi:10.1001/jamapsychiatry.2019.4491.
23. Felder JN et al. Sleep. 2022 Feb 14. doi: 10.1093/sleep/zsab280.
Data suggests this symptom, defined as chronic sleep onset and/or sleep continuity problems associated with impaired daytime functioning, is common in psychiatric illnesses, and can worsen their course.2
The incidence of psychiatric illness in patients with insomnia is estimated at near 50%, with the highest rates found in mood disorders such as depression and bipolar disorder, as well as anxiety disorders.3 In patients with diagnosed major depressive disorder, insomnia rates can approach 90%.4-6
Insomnia has been identified as a risk factor for development of mental illness, including doubling the risk of major depressive disorder and tripling the risk of any depressive or anxiety disorder.7,8 It can also significantly increase the risk of alcohol abuse and psychosis.8
Sleep disturbances can worsen symptoms of diagnosed mental illness, including substance abuse, mood and psychotic disorders.9-10 In one study, nearly 75% of patients with a diagnosis of schizophrenia or bipolar spectrum disorder had at least one type of sleep disturbance (insomnia, hypersomnia, or delayed sleep phase).10 This was almost twice the rate in healthy controls. Importantly, compared with well-rested subjects with mental illness in this study, sleep-disordered participants had higher rates of negative and depressive symptoms on the Positive and Negative Syndrome Scale, as well as significantly lower function via the global assessment of functioning.11,12
Additional data suggests simply being awake during the night (00:00-05:59) elevates risk of suicide. The mean incident rate of completed suicide in one study was a striking four times the rate noted during daytime hours (06:00-23:59 ) (P < .001).13
Although insomnia symptoms can resolve after relief from a particular life stressor, as many as half of patients with more severe symptoms develop a chronic course.14 This then leads to an extended use of many types of sedative-hypnotics designed and studied primarily for short-term use.15 In a survey reviewing national use of prescription drugs for insomnia, as many as 20% of individuals use a medication to target insomnia in a given month.16
Fortunately, despite the many challenges posed by COVID-19, particularly for those with psychiatric illness and limited access to care, telehealth has become more readily available. Additionally, digital versions of evidence-based treatments specifically for sleep problems, such as cognitive-behavioral therapy for insomnia (CBT-I), are regularly being developed.
The benefits of CBT-I have been demonstrated repeatedly and it is recommended as the first line treatment for insomnia by the Clinical Guidelines of the American Academy of Sleep Medicine, the Centers for Disease Control and Prevention, and the National Institutes of Health.17-21 Studies suggest benefits persist long-term, even after completing the therapy sessions, which differ in durability from medication choices.18
One group that may be particularly suited for treatment with CBT-I is women with insomnia during pregnancy or the postpartum period. In these women, options for treatment may be limited by risk of medication during breastfeeding, as well as difficulty traveling to a physician’s or therapist’s office to receive psychotherapy. However, two recent studies evaluated the use of digital CBT-I to treat insomnia during pregnancy and in the postpartum period, respectively.22-23
In both studies,the same group of women with insomnia diagnosed during pregnancy were given six weekly 20-minute sessions of digital CBT-I or standard treatment for insomnia, including medication and psychotherapy per their usual provider.
By study end, the pregnant women receiving the CBT-I intervention not only had significantly improved severity of insomnia, they also experienced improved depression and anxiety symptoms, and a decrease in the use of prescription or over-the-counter sleep aides, compared with the standard treatment group, lowering the fetal exposure to medication during pregnancy.22
In the more recent study, the same group was followed for 6 months post partum.23 Results were again notable, with the women who received CBT-I reporting significantly less insomnia, as well as significantly lower rates of probable major depression at 3 and 6 months (18% vs. 4%, 10% vs. 0%, respectively.) They also exhibited lower rates of moderate to severe anxiety (17% vs. 4%) at 3 months, compared with those receiving standard care. With as many as one in seven women suffering from postpartum depression, these findings represent a substantial public health benefit.
In summary, insomnia is a critical area of focus for any provider diagnosing and treating psychiatric illness. Attempts to optimize sleep, whether through CBT-I or other psychotherapy approaches, or evidence-based medications dosed for appropriate lengths and at safe doses, should be a part of most, if not all, clinical encounters.
Dr. Reid is a board-certified psychiatrist and award-winning medical educator with a private practice in Philadelphia, as well as a clinical faculty role at the University of Pennsylvania, also in Philadelphia. She attended medical school at Columbia University, New York, and completed her psychiatry residency at the University of California, Los Angeles. Dr. Reid is a regular contributor to Psychology Today with her blog, “Think Like a Shrink,” and writes and podcasts as The Reflective Doc.
References
1. Voitsidis P et al. Psychiatry Res. 2020 Jul;289:113076. doi: 10.1016/j.psychres.2020.113076.
2. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington, Va.: American Psychiatric Publishing, 2013.
3. Ford DE and Kamerow DB. JAMA. 1989;262(11):1479-84. doi: 10.1001/jama.1989.03430110069030.
4. Ohayon MM and Roth T. J Psychiatr Res. Jan-Feb 2003;37(1):9-15. doi: 10.1016/s0022-3956(02)00052-3.
5. Seow LSE et al. J Ment Health. 2016 Dec;25(6):492-9. doi: 10.3109/09638237.2015.1124390.
6. Thase ME. J Clin Psychiatry. 1999;60 Suppl 17:28-31; discussion 46-8.
7. Baglioni C et al. J Affect Disord. 2011 Dec;135(1-3):10-9. doi: 10.1016/j.jad.2011.01.011.
8. Hertenstein E et al. Sleep Med Rev. 2019 Feb;43:96-105. doi: 10.1016/j.smrv.2018.10.006.
9. Brower KJ et al. Medical Hypotheses. 2010;74(5):928-33. doi: 10.1016/j.mehy.2009.10.020.
10. Laskemoen JF et al. Compr Psychiatry. 2019 May;91:6-12. doi: 10.1016/j.comppsych.2019.02.006.
11. Kay SR et al. Schizophr Bull. 1987;13(2):261-76. doi: 10.1093/schbul/13.2.261.
12. Hall R. Psychosomatics. May-Jun 1995;36(3):267-75. doi: 10.1016/S0033-3182(95)71666-8.
13. Perlis ML et al. J Clin Psychiatry. 2016 Jun;77(6):e726-33. doi: 10.4088/JCP.15m10131.
14. Morin CM et al. Arch Intern Med. 2009 Mar 9. doi: 10.1001/archinternmed.2008.610.
15. Cheung J et al. Sleep Med Clin. 2019 Jun;14(2):253-65. doi: 10.1016/j.jsmc.2019.01.006.
16. Bertisch SM et al. Sleep. 2014 Feb 1. doi: 10.5665/sleep.3410.
17. Okajima I et al. Sleep Biol Rhythms. 2010 Nov 28. doi: 10.1111/j.1479-8425.2010.00481.x.
18. Trauer JM et al. Ann Intern Med. 2015 Aug 4. doi: 10.7326/M14-2841.
19. Edinger J et al. J Clin Sleep Med. 2021 Feb 1. doi: 10.5664/jcsm.8986.
20. U.S. Centers for Disease Control and Prevention. https://www.cdc.gov/sleep/for-clinicians.html.
21. National Institutes of Health. Sleep Health. https://www.nhlbi.nih.gov/health-topics/education-and-awareness/sleep-health.
22. Felder JN et al. JAMA Psychiatry. 2020;77(5):484-92. doi:10.1001/jamapsychiatry.2019.4491.
23. Felder JN et al. Sleep. 2022 Feb 14. doi: 10.1093/sleep/zsab280.
IBD: Patients struggle with presenteeism, mental health
Anxiety and depression are common among individuals with inflammatory bowel disease (IBD) who are in remission, and the conditions are linked to unemployment and presenteeism, according to a prospective study.
Previous studies have found a heightened risk of anxiety and depression in IBD, as well as an association with poor treatment compliance and greater morbidity. Presenteeism, defined as reduced productivity due to a physical or mental condition, is increasingly recognized as an indirect economic cost of chronic conditions that may exact a higher cost than absenteeism.

More than one-third of patients with IBD experienced presenteeism in one study. Other studies have examined exercise in chronic diseases, and most find an association between more exercise and lower rates of anxiety and depression. To date, few studies have examined a combination of physical activity, mental health, and presenteeism in the context of IBD.
The new study, published in the Journal of Crohn’s and Colitis, “is very important, as presenteeism is not commonly discussed in any formal, measurable way in the IBD space,” said Laurie Keefer, PhD, professor of medicine and a gastropsychologist at Icahn School of Medicine, who was asked to comment on the research. “While conducted in Europe and Israel, the study also has relevance in the U.S. since, here, employers typically pay for health care – so absenteeism, presenteeism, and health care cost are all intertwined.”

“The study confirmed high rates of depression and anxiety after a diagnosis of IBD. One high-quality aspect of this study was that the diagnosis of depression and anxiety was confirmed by a medical practitioner,” said Dr. Keefer.
The results suggest that current efforts to screen IBD patients for depression and anxiety may not be enough, according to Stephen Lupe, PsyD, who was also asked to comment on the study. He noted that almost half of the patients had symptoms of anxiety or depression as measured by the Hospital Anxiety and Depression Score (HADS). “We’re missing all kinds of people,” said Dr. Lupe, who is director of behavioral medicine for the department of gastroenterology, hepatology, and nutrition at the Cleveland Clinic. He cited other data that show that, if someone develops depression, they are more likely to have a surgical outcome, and they are more likely to have problems with medication adherence. They likely will have more flares and less time in remission. “We need to be screening for it as part of care,” he said.
The researchers prospectively studied 585 IBD patients who were in clinical remission at 8 centers in Europe and Israel between September 2020 and March 2021. Participants filled out the HADS and The Stanford Presenteeism scale (SPS-6). A total of 62.2% of the participants had CD; 53.0% were male. Participants’ mean age was 39 years. Among the group, 10.8% had a pre existing diagnosis of anxiety or depression at the time of IBD diagnosis, and an additional 14.2% were later diagnosed with anxiety or depression.
Less than half, 46.1%, of IBD patients had a score 8 or higher in the HADS-anxiety (HADS-A) or HADS-depression (HADS-D) subscale, a cutoff that suggests evidence of clinical depression or anxiety. A total of 27.4% had a score of 11 or higher, indicating a mood disorder. High HADS-A score was associated with female gender (odds ratio, 1.91; P < 0.05), long duration of disease (OR, 1.04; P < 0.01), and perianal disease (P < 0.023). The authors speculate that the latter result may be due to a higher burden of symptoms. Three-quarters, 74.5%, of patients were employed; 34.0% experienced presenteeism as defined by SPS-6 score less than or equal to 18.
The researchers found that 23.0% of the patients were sedentary, and this was more common among individuals with HADS-A or HADS-D scores greater than or equal to 8. Among those experiencing presenteeism, 50% were sedentary, 29.4% were active, and 20.6% were moderately active (P < 0.01). Individuals with higher HADS-A or HADS-D scores had a greater likelihood of being sedentary (P < 0.05).
One limitation of the study was that the questionnaires were translated into the respective languages and scores were taken at only one time point.
“Rather than relying on the patient to come forward and seek help,” physicians should familiarize themselves with these validated screening tools “in order to increase the diagnostic rate of such pathologies and enable a better holistic care for the IBD patients,” the authors concluded. “Active involvement of a psychologist and/or a psychiatrist, as part of the IBD team, should be pursued to further improve the patients’ quality of life, which has emerged as one of the top priority outcomes in IBD.”
Dr. Lupe said that the findings regarding presenteeism are consistent with his experience. He pointed out that IBD patients must be more aware of their body and vigilant in managing symptoms, and he speculated that that could detract from concentration at work. He said that the study shows the need for a holistic approach to treatment. “When someone is coping with a chronic disease, like ulcerative colitis or Crohn’s disease, it affects the whole person,” including psychologically, professionally, and personally. “These are bidirectional relationships, so that if someone’s social life starts falling down, it’s more contributory to the development of something like depression and anxiety, and maybe that’s contributory to complications that come up in a disease state like Crohn’s disease or ulcerative colitis.”
The study did not receive funding, but two authors disclosed relations with AbbVie, Janssen, Pfizer, and other companies. Dr. Keefer is a cofounder and has equity ownership In Trellus Health. Dr. Lupe has no relevant financial disclosures.
Anxiety and depression are common among individuals with inflammatory bowel disease (IBD) who are in remission, and the conditions are linked to unemployment and presenteeism, according to a prospective study.
Previous studies have found a heightened risk of anxiety and depression in IBD, as well as an association with poor treatment compliance and greater morbidity. Presenteeism, defined as reduced productivity due to a physical or mental condition, is increasingly recognized as an indirect economic cost of chronic conditions that may exact a higher cost than absenteeism.
More than one-third of patients with IBD experienced presenteeism in one study. Other studies have examined exercise in chronic diseases, and most find an association between more exercise and lower rates of anxiety and depression. To date, few studies have examined a combination of physical activity, mental health, and presenteeism in the context of IBD.
The new study, published in the Journal of Crohn’s and Colitis, “is very important, as presenteeism is not commonly discussed in any formal, measurable way in the IBD space,” said Laurie Keefer, PhD, professor of medicine and a gastropsychologist at Icahn School of Medicine, who was asked to comment on the research. “While conducted in Europe and Israel, the study also has relevance in the U.S. since, here, employers typically pay for health care – so absenteeism, presenteeism, and health care cost are all intertwined.”
“The study confirmed high rates of depression and anxiety after a diagnosis of IBD. One high-quality aspect of this study was that the diagnosis of depression and anxiety was confirmed by a medical practitioner,” said Dr. Keefer.
The results suggest that current efforts to screen IBD patients for depression and anxiety may not be enough, according to Stephen Lupe, PsyD, who was also asked to comment on the study. He noted that almost half of the patients had symptoms of anxiety or depression as measured by the Hospital Anxiety and Depression Score (HADS). “We’re missing all kinds of people,” said Dr. Lupe, who is director of behavioral medicine for the department of gastroenterology, hepatology, and nutrition at the Cleveland Clinic. He cited other data that show that, if someone develops depression, they are more likely to have a surgical outcome, and they are more likely to have problems with medication adherence. They likely will have more flares and less time in remission. “We need to be screening for it as part of care,” he said.
The researchers prospectively studied 585 IBD patients who were in clinical remission at 8 centers in Europe and Israel between September 2020 and March 2021. Participants filled out the HADS and The Stanford Presenteeism scale (SPS-6). A total of 62.2% of the participants had CD; 53.0% were male. Participants’ mean age was 39 years. Among the group, 10.8% had a pre existing diagnosis of anxiety or depression at the time of IBD diagnosis, and an additional 14.2% were later diagnosed with anxiety or depression.
Less than half, 46.1%, of IBD patients had a score 8 or higher in the HADS-anxiety (HADS-A) or HADS-depression (HADS-D) subscale, a cutoff that suggests evidence of clinical depression or anxiety. A total of 27.4% had a score of 11 or higher, indicating a mood disorder. High HADS-A score was associated with female gender (odds ratio, 1.91; P < 0.05), long duration of disease (OR, 1.04; P < 0.01), and perianal disease (P < 0.023). The authors speculate that the latter result may be due to a higher burden of symptoms. Three-quarters, 74.5%, of patients were employed; 34.0% experienced presenteeism as defined by SPS-6 score less than or equal to 18.
The researchers found that 23.0% of the patients were sedentary, and this was more common among individuals with HADS-A or HADS-D scores greater than or equal to 8. Among those experiencing presenteeism, 50% were sedentary, 29.4% were active, and 20.6% were moderately active (P < 0.01). Individuals with higher HADS-A or HADS-D scores had a greater likelihood of being sedentary (P < 0.05).
One limitation of the study was that the questionnaires were translated into the respective languages and scores were taken at only one time point.
“Rather than relying on the patient to come forward and seek help,” physicians should familiarize themselves with these validated screening tools “in order to increase the diagnostic rate of such pathologies and enable a better holistic care for the IBD patients,” the authors concluded. “Active involvement of a psychologist and/or a psychiatrist, as part of the IBD team, should be pursued to further improve the patients’ quality of life, which has emerged as one of the top priority outcomes in IBD.”
Dr. Lupe said that the findings regarding presenteeism are consistent with his experience. He pointed out that IBD patients must be more aware of their body and vigilant in managing symptoms, and he speculated that that could detract from concentration at work. He said that the study shows the need for a holistic approach to treatment. “When someone is coping with a chronic disease, like ulcerative colitis or Crohn’s disease, it affects the whole person,” including psychologically, professionally, and personally. “These are bidirectional relationships, so that if someone’s social life starts falling down, it’s more contributory to the development of something like depression and anxiety, and maybe that’s contributory to complications that come up in a disease state like Crohn’s disease or ulcerative colitis.”
The study did not receive funding, but two authors disclosed relations with AbbVie, Janssen, Pfizer, and other companies. Dr. Keefer is a cofounder and has equity ownership In Trellus Health. Dr. Lupe has no relevant financial disclosures.
Anxiety and depression are common among individuals with inflammatory bowel disease (IBD) who are in remission, and the conditions are linked to unemployment and presenteeism, according to a prospective study.
Previous studies have found a heightened risk of anxiety and depression in IBD, as well as an association with poor treatment compliance and greater morbidity. Presenteeism, defined as reduced productivity due to a physical or mental condition, is increasingly recognized as an indirect economic cost of chronic conditions that may exact a higher cost than absenteeism.
More than one-third of patients with IBD experienced presenteeism in one study. Other studies have examined exercise in chronic diseases, and most find an association between more exercise and lower rates of anxiety and depression. To date, few studies have examined a combination of physical activity, mental health, and presenteeism in the context of IBD.
The new study, published in the Journal of Crohn’s and Colitis, “is very important, as presenteeism is not commonly discussed in any formal, measurable way in the IBD space,” said Laurie Keefer, PhD, professor of medicine and a gastropsychologist at Icahn School of Medicine, who was asked to comment on the research. “While conducted in Europe and Israel, the study also has relevance in the U.S. since, here, employers typically pay for health care – so absenteeism, presenteeism, and health care cost are all intertwined.”
“The study confirmed high rates of depression and anxiety after a diagnosis of IBD. One high-quality aspect of this study was that the diagnosis of depression and anxiety was confirmed by a medical practitioner,” said Dr. Keefer.
The results suggest that current efforts to screen IBD patients for depression and anxiety may not be enough, according to Stephen Lupe, PsyD, who was also asked to comment on the study. He noted that almost half of the patients had symptoms of anxiety or depression as measured by the Hospital Anxiety and Depression Score (HADS). “We’re missing all kinds of people,” said Dr. Lupe, who is director of behavioral medicine for the department of gastroenterology, hepatology, and nutrition at the Cleveland Clinic. He cited other data that show that, if someone develops depression, they are more likely to have a surgical outcome, and they are more likely to have problems with medication adherence. They likely will have more flares and less time in remission. “We need to be screening for it as part of care,” he said.
The researchers prospectively studied 585 IBD patients who were in clinical remission at 8 centers in Europe and Israel between September 2020 and March 2021. Participants filled out the HADS and The Stanford Presenteeism scale (SPS-6). A total of 62.2% of the participants had CD; 53.0% were male. Participants’ mean age was 39 years. Among the group, 10.8% had a pre existing diagnosis of anxiety or depression at the time of IBD diagnosis, and an additional 14.2% were later diagnosed with anxiety or depression.
Less than half, 46.1%, of IBD patients had a score 8 or higher in the HADS-anxiety (HADS-A) or HADS-depression (HADS-D) subscale, a cutoff that suggests evidence of clinical depression or anxiety. A total of 27.4% had a score of 11 or higher, indicating a mood disorder. High HADS-A score was associated with female gender (odds ratio, 1.91; P < 0.05), long duration of disease (OR, 1.04; P < 0.01), and perianal disease (P < 0.023). The authors speculate that the latter result may be due to a higher burden of symptoms. Three-quarters, 74.5%, of patients were employed; 34.0% experienced presenteeism as defined by SPS-6 score less than or equal to 18.
The researchers found that 23.0% of the patients were sedentary, and this was more common among individuals with HADS-A or HADS-D scores greater than or equal to 8. Among those experiencing presenteeism, 50% were sedentary, 29.4% were active, and 20.6% were moderately active (P < 0.01). Individuals with higher HADS-A or HADS-D scores had a greater likelihood of being sedentary (P < 0.05).
One limitation of the study was that the questionnaires were translated into the respective languages and scores were taken at only one time point.
“Rather than relying on the patient to come forward and seek help,” physicians should familiarize themselves with these validated screening tools “in order to increase the diagnostic rate of such pathologies and enable a better holistic care for the IBD patients,” the authors concluded. “Active involvement of a psychologist and/or a psychiatrist, as part of the IBD team, should be pursued to further improve the patients’ quality of life, which has emerged as one of the top priority outcomes in IBD.”
Dr. Lupe said that the findings regarding presenteeism are consistent with his experience. He pointed out that IBD patients must be more aware of their body and vigilant in managing symptoms, and he speculated that that could detract from concentration at work. He said that the study shows the need for a holistic approach to treatment. “When someone is coping with a chronic disease, like ulcerative colitis or Crohn’s disease, it affects the whole person,” including psychologically, professionally, and personally. “These are bidirectional relationships, so that if someone’s social life starts falling down, it’s more contributory to the development of something like depression and anxiety, and maybe that’s contributory to complications that come up in a disease state like Crohn’s disease or ulcerative colitis.”
The study did not receive funding, but two authors disclosed relations with AbbVie, Janssen, Pfizer, and other companies. Dr. Keefer is a cofounder and has equity ownership In Trellus Health. Dr. Lupe has no relevant financial disclosures.
REPORTING FROM JOURNAL OF CROHN'S AND COLITIS
Locoregional therapy lowers waitlist dropout in HCC
The use of bridging locoregional therapy (LRT) before liver transplantation in patients with hepatocellular carcinoma (HCC) has significantly increased in the United States within the past 15 years, a recent analysis suggests. Data show that liver transplant candidates with HCC who have elevated tumor burden and patients with more compensated liver disease have received a greater number of treatments while awaiting transplant.
According to the researchers, led by Allison Kwong, MD, of Stanford (Calif.) University, liver transplant remains a curative option for individuals with unresectable HCC who meet prespecified size criteria. In the United States, a mandated waiting period of 6 months prior “to gaining exception points has been implemented” in an effort “to allow for consideration of tumor biology and reduce the disparities in waitlist dropout between HCC and non-HCC patients,” the researchers wrote.
Several forms of LRT are now available for HCC, including chemoembolization, external beam radiation, radioembolization, and radiofrequency or microwave ablation. In the liver transplant setting, these LRT options enable management of intrahepatic disease in patients who are waiting for liver transplant, Dr. Kwong and colleagues explained.
The researchers, who published their study findings online August 3 in Clinical Gastroenterology and Hepatology, sought to examine the national temporal trends and waitlist outcomes of LRT in 31,609 patients eligible for liver transplant with greater than or equal to 1 approved HCC exception application in the United States.
Patient data were obtained from the Organ Procurement and Transplantation Network database and comprised primary adult LT candidates who were listed from the years 2003 to 2018. The investigators assessed explant histology and performed multivariable competing risk analysis to examine the relationship between the type of first LRT and time to waitlist dropout.
The waitlist dropout variable was defined by list removal due to death or excessive illness. The researchers noted that list removal likely represents disease progression “beyond transplantable criteria and beyond which patients were unlikely to benefit from or be eligible for further LRT.”
In the study population, the median age was 59 years, and approximately 77% of patients were male. More than half (53.1%) of the cohort had hepatitis C as the predominant liver disease etiology. Patients had a median follow-up period of 214 days on the waitlist.
Most patients (79%) received deceased or living-donor transplants, and 18.6% of patients were removed from the waitlist. Between the 2003 and 2006 period, the median waitlist time was 123 days, but this median waitlist duration increased to 257 days for patients listed between 2015 and 2018.
A total of 34,610 LRTs were performed among 24,145 liver transplant candidates during the study period. From 2003 to 2018, the proportion of patients with greater than or equal to 1 LRT recorded in the database rose from 42.3% to 92.4%, respectively. Most patients (67.8%) who received liver-directed therapy had a single LRT, while 23.8% of patients had 2 LRTs, 6.2% had 3 LRTs, and 2.2% had greater than or equal to 4 LRTs.
The most frequent type of LRT performed was chemoembolization, followed by thermal ablation. Radioembolization increased from less than 5% in 2013 to 19% in 2018. Moreover, in 2018, chemoembolization accounted for 50% of LRTs, while thermal ablation accounted for 22% of LRTs.
The incidence rates of LRT per 100 waitlist days was above average in patients who had an initial tumor burden beyond the Milan criteria (0.188), an alpha-fetoprotein level of 21-40 (0.171) or 41-500 ng/mL (0.179), Child-Pugh class A (0.160), patients in short (0.151) and medium (0.154) wait-time regions, as well as patients who were listed following implementation of cap-and-delay in October 2015 (0.192).
In the multivariable competing-risk analysis for waitlist dropout, adjusting for initial tumor burden and AFP, Child-Pugh class, wait region, and listing era, no locoregional therapy was associated with an increased risk of waitlist dropout versus chemoembolization as the first LRT in a multivariable competing-risk analysis (subhazard ratio [sHR], 1.37; 95% CI, 1.28-1.47). The inverse probability of treatment weighting–adjusted analysis found an association between radioembolization, when compared with chemoembolization, and a reduced risk of waitlist dropout (sHR, 0.85; 95% CI, 0.81-0.89). Thermal ablation was also associated with a reduced risk of waitlist dropout, compared with chemoembolization (sHR, 0.95; 95% CI, 0.91-0.99). “Radioembolization and thermal ablation may be superior to chemoembolization and prove to be more cost-effective options, depending on the clinical context,” the researchers wrote.
The researchers noted that they were unable to distinguish patients who were removed from the waitlist between those with disease progression versus liver failure.
The researchers reported no conflicts of interest with the pharmaceutical industry. The study received no industry funding.
The use of bridging locoregional therapy (LRT) before liver transplantation in patients with hepatocellular carcinoma (HCC) has significantly increased in the United States within the past 15 years, a recent analysis suggests. Data show that liver transplant candidates with HCC who have elevated tumor burden and patients with more compensated liver disease have received a greater number of treatments while awaiting transplant.
According to the researchers, led by Allison Kwong, MD, of Stanford (Calif.) University, liver transplant remains a curative option for individuals with unresectable HCC who meet prespecified size criteria. In the United States, a mandated waiting period of 6 months prior “to gaining exception points has been implemented” in an effort “to allow for consideration of tumor biology and reduce the disparities in waitlist dropout between HCC and non-HCC patients,” the researchers wrote.
Several forms of LRT are now available for HCC, including chemoembolization, external beam radiation, radioembolization, and radiofrequency or microwave ablation. In the liver transplant setting, these LRT options enable management of intrahepatic disease in patients who are waiting for liver transplant, Dr. Kwong and colleagues explained.
The researchers, who published their study findings online August 3 in Clinical Gastroenterology and Hepatology, sought to examine the national temporal trends and waitlist outcomes of LRT in 31,609 patients eligible for liver transplant with greater than or equal to 1 approved HCC exception application in the United States.
Patient data were obtained from the Organ Procurement and Transplantation Network database and comprised primary adult LT candidates who were listed from the years 2003 to 2018. The investigators assessed explant histology and performed multivariable competing risk analysis to examine the relationship between the type of first LRT and time to waitlist dropout.
The waitlist dropout variable was defined by list removal due to death or excessive illness. The researchers noted that list removal likely represents disease progression “beyond transplantable criteria and beyond which patients were unlikely to benefit from or be eligible for further LRT.”
In the study population, the median age was 59 years, and approximately 77% of patients were male. More than half (53.1%) of the cohort had hepatitis C as the predominant liver disease etiology. Patients had a median follow-up period of 214 days on the waitlist.
Most patients (79%) received deceased or living-donor transplants, and 18.6% of patients were removed from the waitlist. Between the 2003 and 2006 period, the median waitlist time was 123 days, but this median waitlist duration increased to 257 days for patients listed between 2015 and 2018.
A total of 34,610 LRTs were performed among 24,145 liver transplant candidates during the study period. From 2003 to 2018, the proportion of patients with greater than or equal to 1 LRT recorded in the database rose from 42.3% to 92.4%, respectively. Most patients (67.8%) who received liver-directed therapy had a single LRT, while 23.8% of patients had 2 LRTs, 6.2% had 3 LRTs, and 2.2% had greater than or equal to 4 LRTs.
The most frequent type of LRT performed was chemoembolization, followed by thermal ablation. Radioembolization increased from less than 5% in 2013 to 19% in 2018. Moreover, in 2018, chemoembolization accounted for 50% of LRTs, while thermal ablation accounted for 22% of LRTs.
The incidence rates of LRT per 100 waitlist days was above average in patients who had an initial tumor burden beyond the Milan criteria (0.188), an alpha-fetoprotein level of 21-40 (0.171) or 41-500 ng/mL (0.179), Child-Pugh class A (0.160), patients in short (0.151) and medium (0.154) wait-time regions, as well as patients who were listed following implementation of cap-and-delay in October 2015 (0.192).
In the multivariable competing-risk analysis for waitlist dropout, adjusting for initial tumor burden and AFP, Child-Pugh class, wait region, and listing era, no locoregional therapy was associated with an increased risk of waitlist dropout versus chemoembolization as the first LRT in a multivariable competing-risk analysis (subhazard ratio [sHR], 1.37; 95% CI, 1.28-1.47). The inverse probability of treatment weighting–adjusted analysis found an association between radioembolization, when compared with chemoembolization, and a reduced risk of waitlist dropout (sHR, 0.85; 95% CI, 0.81-0.89). Thermal ablation was also associated with a reduced risk of waitlist dropout, compared with chemoembolization (sHR, 0.95; 95% CI, 0.91-0.99). “Radioembolization and thermal ablation may be superior to chemoembolization and prove to be more cost-effective options, depending on the clinical context,” the researchers wrote.
The researchers noted that they were unable to distinguish patients who were removed from the waitlist between those with disease progression versus liver failure.
The researchers reported no conflicts of interest with the pharmaceutical industry. The study received no industry funding.
The use of bridging locoregional therapy (LRT) before liver transplantation in patients with hepatocellular carcinoma (HCC) has significantly increased in the United States within the past 15 years, a recent analysis suggests. Data show that liver transplant candidates with HCC who have elevated tumor burden and patients with more compensated liver disease have received a greater number of treatments while awaiting transplant.
According to the researchers, led by Allison Kwong, MD, of Stanford (Calif.) University, liver transplant remains a curative option for individuals with unresectable HCC who meet prespecified size criteria. In the United States, a mandated waiting period of 6 months prior “to gaining exception points has been implemented” in an effort “to allow for consideration of tumor biology and reduce the disparities in waitlist dropout between HCC and non-HCC patients,” the researchers wrote.
Several forms of LRT are now available for HCC, including chemoembolization, external beam radiation, radioembolization, and radiofrequency or microwave ablation. In the liver transplant setting, these LRT options enable management of intrahepatic disease in patients who are waiting for liver transplant, Dr. Kwong and colleagues explained.
The researchers, who published their study findings online August 3 in Clinical Gastroenterology and Hepatology, sought to examine the national temporal trends and waitlist outcomes of LRT in 31,609 patients eligible for liver transplant with greater than or equal to 1 approved HCC exception application in the United States.
Patient data were obtained from the Organ Procurement and Transplantation Network database and comprised primary adult LT candidates who were listed from the years 2003 to 2018. The investigators assessed explant histology and performed multivariable competing risk analysis to examine the relationship between the type of first LRT and time to waitlist dropout.
The waitlist dropout variable was defined by list removal due to death or excessive illness. The researchers noted that list removal likely represents disease progression “beyond transplantable criteria and beyond which patients were unlikely to benefit from or be eligible for further LRT.”
In the study population, the median age was 59 years, and approximately 77% of patients were male. More than half (53.1%) of the cohort had hepatitis C as the predominant liver disease etiology. Patients had a median follow-up period of 214 days on the waitlist.
Most patients (79%) received deceased or living-donor transplants, and 18.6% of patients were removed from the waitlist. Between the 2003 and 2006 period, the median waitlist time was 123 days, but this median waitlist duration increased to 257 days for patients listed between 2015 and 2018.
A total of 34,610 LRTs were performed among 24,145 liver transplant candidates during the study period. From 2003 to 2018, the proportion of patients with greater than or equal to 1 LRT recorded in the database rose from 42.3% to 92.4%, respectively. Most patients (67.8%) who received liver-directed therapy had a single LRT, while 23.8% of patients had 2 LRTs, 6.2% had 3 LRTs, and 2.2% had greater than or equal to 4 LRTs.
The most frequent type of LRT performed was chemoembolization, followed by thermal ablation. Radioembolization increased from less than 5% in 2013 to 19% in 2018. Moreover, in 2018, chemoembolization accounted for 50% of LRTs, while thermal ablation accounted for 22% of LRTs.
The incidence rates of LRT per 100 waitlist days was above average in patients who had an initial tumor burden beyond the Milan criteria (0.188), an alpha-fetoprotein level of 21-40 (0.171) or 41-500 ng/mL (0.179), Child-Pugh class A (0.160), patients in short (0.151) and medium (0.154) wait-time regions, as well as patients who were listed following implementation of cap-and-delay in October 2015 (0.192).
In the multivariable competing-risk analysis for waitlist dropout, adjusting for initial tumor burden and AFP, Child-Pugh class, wait region, and listing era, no locoregional therapy was associated with an increased risk of waitlist dropout versus chemoembolization as the first LRT in a multivariable competing-risk analysis (subhazard ratio [sHR], 1.37; 95% CI, 1.28-1.47). The inverse probability of treatment weighting–adjusted analysis found an association between radioembolization, when compared with chemoembolization, and a reduced risk of waitlist dropout (sHR, 0.85; 95% CI, 0.81-0.89). Thermal ablation was also associated with a reduced risk of waitlist dropout, compared with chemoembolization (sHR, 0.95; 95% CI, 0.91-0.99). “Radioembolization and thermal ablation may be superior to chemoembolization and prove to be more cost-effective options, depending on the clinical context,” the researchers wrote.
The researchers noted that they were unable to distinguish patients who were removed from the waitlist between those with disease progression versus liver failure.
The researchers reported no conflicts of interest with the pharmaceutical industry. The study received no industry funding.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY