User login
Migraine: Erenumab demonstrates superior efficacy over topiramate
Key clinical point: Erenumab demonstrated an early onset of and superior efficacy in achieving ≥50% reduction in monthly migraine days (MMD) than topiramate in patients with migraine.
Major finding: A significantly higher proportion of patients receiving erenumab vs topiramate reported ≥50% reduction in MMD at 1 month (39.2% vs 24.0%) and during the last 3 months (60.3% vs 43.3%) of treatment (both P < .001). Reductions in MMD over last 3 months were significantly higher with erenumab vs topiramate (mean difference −1.24 days; P < .001).
Study details: This post hoc analysis of the phase 4 HER-MES trial included 776 patients with migraine who were naive to migraine prophylactics or had failed or were ill-suited for metoprolol/propranolol, amitriptyline, or flunarizine and were randomized to receive erenumab (70 or 140 mg/month) or topiramate (50-100 mg/day).
Disclosures: This study was funded by Novartis Pharma GmbH, Germany. Four authors, including the first author, declared being employees of and holding stocks in Novartis. U Reuter reported receiving grants, personal fees, and other supports from Novartis and various other sources.
Source: Ehrlich M et al. Erenumab versus topiramate: Post hoc efficacy analysis from the HER‑MES study. J Headache Pain. 2022;23:141 (Nov 15). Doi: 10.1186/s10194-022-01511-y
Key clinical point: Erenumab demonstrated an early onset of and superior efficacy in achieving ≥50% reduction in monthly migraine days (MMD) than topiramate in patients with migraine.
Major finding: A significantly higher proportion of patients receiving erenumab vs topiramate reported ≥50% reduction in MMD at 1 month (39.2% vs 24.0%) and during the last 3 months (60.3% vs 43.3%) of treatment (both P < .001). Reductions in MMD over last 3 months were significantly higher with erenumab vs topiramate (mean difference −1.24 days; P < .001).
Study details: This post hoc analysis of the phase 4 HER-MES trial included 776 patients with migraine who were naive to migraine prophylactics or had failed or were ill-suited for metoprolol/propranolol, amitriptyline, or flunarizine and were randomized to receive erenumab (70 or 140 mg/month) or topiramate (50-100 mg/day).
Disclosures: This study was funded by Novartis Pharma GmbH, Germany. Four authors, including the first author, declared being employees of and holding stocks in Novartis. U Reuter reported receiving grants, personal fees, and other supports from Novartis and various other sources.
Source: Ehrlich M et al. Erenumab versus topiramate: Post hoc efficacy analysis from the HER‑MES study. J Headache Pain. 2022;23:141 (Nov 15). Doi: 10.1186/s10194-022-01511-y
Key clinical point: Erenumab demonstrated an early onset of and superior efficacy in achieving ≥50% reduction in monthly migraine days (MMD) than topiramate in patients with migraine.
Major finding: A significantly higher proportion of patients receiving erenumab vs topiramate reported ≥50% reduction in MMD at 1 month (39.2% vs 24.0%) and during the last 3 months (60.3% vs 43.3%) of treatment (both P < .001). Reductions in MMD over last 3 months were significantly higher with erenumab vs topiramate (mean difference −1.24 days; P < .001).
Study details: This post hoc analysis of the phase 4 HER-MES trial included 776 patients with migraine who were naive to migraine prophylactics or had failed or were ill-suited for metoprolol/propranolol, amitriptyline, or flunarizine and were randomized to receive erenumab (70 or 140 mg/month) or topiramate (50-100 mg/day).
Disclosures: This study was funded by Novartis Pharma GmbH, Germany. Four authors, including the first author, declared being employees of and holding stocks in Novartis. U Reuter reported receiving grants, personal fees, and other supports from Novartis and various other sources.
Source: Ehrlich M et al. Erenumab versus topiramate: Post hoc efficacy analysis from the HER‑MES study. J Headache Pain. 2022;23:141 (Nov 15). Doi: 10.1186/s10194-022-01511-y
Some patients with resistant chronic migraine may derive long-term benefits with erenumab
Key clinical point: Monthly erenumab demonstrated promising short-term clinical effectiveness in patients with difficult-to-treat chronic migraine; however, less than one-fourth of the patients sustained efficacy over 2 years.
Major finding: The monthly migraine days (MMD) reduced significantly after 6 months of erenumab treatment (mean reduction [MR] 7.5 days; P < .001), with 48% of patients achieving ≥30% reduction in MMD. At months 12 and 24, 38% and 23% of patients remained ≥30% responders, respectively.
Study details: Findings are from a 2-year real-world prospective analysis of a clinical audit including 160 patients with difficult-to-treat chronic migraine who failed an average of 8.3 preventive treatments and received monthly erenumab.
Disclosures: This study did not receive any funding. Three authors declared receiving honoraria for speaking or participation in advisory boards or funding for travel from various sources.
Source: Andreou AP et al. Two-year effectiveness of erenumab in resistant chronic migraine: a prospective real-world analysis. J Headache Pain. 2022;23:139 (Nov 4). Doi: 10.1186/s10194-022-01507-8
Key clinical point: Monthly erenumab demonstrated promising short-term clinical effectiveness in patients with difficult-to-treat chronic migraine; however, less than one-fourth of the patients sustained efficacy over 2 years.
Major finding: The monthly migraine days (MMD) reduced significantly after 6 months of erenumab treatment (mean reduction [MR] 7.5 days; P < .001), with 48% of patients achieving ≥30% reduction in MMD. At months 12 and 24, 38% and 23% of patients remained ≥30% responders, respectively.
Study details: Findings are from a 2-year real-world prospective analysis of a clinical audit including 160 patients with difficult-to-treat chronic migraine who failed an average of 8.3 preventive treatments and received monthly erenumab.
Disclosures: This study did not receive any funding. Three authors declared receiving honoraria for speaking or participation in advisory boards or funding for travel from various sources.
Source: Andreou AP et al. Two-year effectiveness of erenumab in resistant chronic migraine: a prospective real-world analysis. J Headache Pain. 2022;23:139 (Nov 4). Doi: 10.1186/s10194-022-01507-8
Key clinical point: Monthly erenumab demonstrated promising short-term clinical effectiveness in patients with difficult-to-treat chronic migraine; however, less than one-fourth of the patients sustained efficacy over 2 years.
Major finding: The monthly migraine days (MMD) reduced significantly after 6 months of erenumab treatment (mean reduction [MR] 7.5 days; P < .001), with 48% of patients achieving ≥30% reduction in MMD. At months 12 and 24, 38% and 23% of patients remained ≥30% responders, respectively.
Study details: Findings are from a 2-year real-world prospective analysis of a clinical audit including 160 patients with difficult-to-treat chronic migraine who failed an average of 8.3 preventive treatments and received monthly erenumab.
Disclosures: This study did not receive any funding. Three authors declared receiving honoraria for speaking or participation in advisory boards or funding for travel from various sources.
Source: Andreou AP et al. Two-year effectiveness of erenumab in resistant chronic migraine: a prospective real-world analysis. J Headache Pain. 2022;23:139 (Nov 4). Doi: 10.1186/s10194-022-01507-8
Prior proton-pump inhibitor use ups migraine risk
Key clinical point: Prior use of proton pump inhibitors (PPI) increased the risk for incident migraine with or without aura irrespective of the history and duration of use.
Major finding: Compared with non-use, past and current use of PPI increased the odds of migraine by 2.56-fold (P < .001) and 4.66-fold (P < .001), respectively, with the risk being persistent for migraine with or without aura (P < .001) and higher with PPI use for ≥30 (adjusted odds ratio [aOR] 4.41; P < .001) vs <30 (aOR 2.49; P < .001) days.
Study details: This retrospective, nested case-control study included 28,159 patients with incident migraine with or without aura and 112,636 propensity score-matched control participants.
Disclosures: This study was funded by the National Research Foundation of Korea from the Korean Ministry of Science and ICT. The authors declared no conflicts of interest.
Source: Kang HS et al. Association between migraines and prior proton pump inhibitor use: A nested case-control study using a national health screening cohort. Pharmaceuticals (Basel). 2022;15(11):1385 (Nov 10). Doi: 10.3390/ph15111385
Key clinical point: Prior use of proton pump inhibitors (PPI) increased the risk for incident migraine with or without aura irrespective of the history and duration of use.
Major finding: Compared with non-use, past and current use of PPI increased the odds of migraine by 2.56-fold (P < .001) and 4.66-fold (P < .001), respectively, with the risk being persistent for migraine with or without aura (P < .001) and higher with PPI use for ≥30 (adjusted odds ratio [aOR] 4.41; P < .001) vs <30 (aOR 2.49; P < .001) days.
Study details: This retrospective, nested case-control study included 28,159 patients with incident migraine with or without aura and 112,636 propensity score-matched control participants.
Disclosures: This study was funded by the National Research Foundation of Korea from the Korean Ministry of Science and ICT. The authors declared no conflicts of interest.
Source: Kang HS et al. Association between migraines and prior proton pump inhibitor use: A nested case-control study using a national health screening cohort. Pharmaceuticals (Basel). 2022;15(11):1385 (Nov 10). Doi: 10.3390/ph15111385
Key clinical point: Prior use of proton pump inhibitors (PPI) increased the risk for incident migraine with or without aura irrespective of the history and duration of use.
Major finding: Compared with non-use, past and current use of PPI increased the odds of migraine by 2.56-fold (P < .001) and 4.66-fold (P < .001), respectively, with the risk being persistent for migraine with or without aura (P < .001) and higher with PPI use for ≥30 (adjusted odds ratio [aOR] 4.41; P < .001) vs <30 (aOR 2.49; P < .001) days.
Study details: This retrospective, nested case-control study included 28,159 patients with incident migraine with or without aura and 112,636 propensity score-matched control participants.
Disclosures: This study was funded by the National Research Foundation of Korea from the Korean Ministry of Science and ICT. The authors declared no conflicts of interest.
Source: Kang HS et al. Association between migraines and prior proton pump inhibitor use: A nested case-control study using a national health screening cohort. Pharmaceuticals (Basel). 2022;15(11):1385 (Nov 10). Doi: 10.3390/ph15111385
Alcohol as a trigger for migraine: What is the link?
Key clinical point: Alcohol intake slightly reduced the likelihood of migraine attacks 48 hours after consumption in an English-speaking cohort of patients with episodic migraine who identified themselves as mostly low-dose alcohol consumers.
Major finding: The probability of migraine attack 48 hours after consuming alcohol was 25% lower than that after no alcohol consumption (adjusted odds ratio 0.75; 95% CI 0.68-0.82); however, alcohol consumption had no significant effect on migraine probability 24 hours after consumption.
Study details: This observational prospective cohort study included 487 patients with episodic migraine who reported 5913 migraine attacks and were alcohol consumers.
Disclosures: The study was partially funded by Curelator, Inc. M Vives-Mestres and A Casanova declared receiving consulting fees and holding stock options in Curelator, Inc. N Rosen reported ties with a headache society, journals, and various other sources.
Source: Vives-Mestres M et al. Alcohol as a trigger of migraine attacks in people with migraine. Results from a large prospective cohort study in English-speaking countries. Headache. 2022;62:1329-1338. (Nov 27). Doi: 10.1111/head.14428
Key clinical point: Alcohol intake slightly reduced the likelihood of migraine attacks 48 hours after consumption in an English-speaking cohort of patients with episodic migraine who identified themselves as mostly low-dose alcohol consumers.
Major finding: The probability of migraine attack 48 hours after consuming alcohol was 25% lower than that after no alcohol consumption (adjusted odds ratio 0.75; 95% CI 0.68-0.82); however, alcohol consumption had no significant effect on migraine probability 24 hours after consumption.
Study details: This observational prospective cohort study included 487 patients with episodic migraine who reported 5913 migraine attacks and were alcohol consumers.
Disclosures: The study was partially funded by Curelator, Inc. M Vives-Mestres and A Casanova declared receiving consulting fees and holding stock options in Curelator, Inc. N Rosen reported ties with a headache society, journals, and various other sources.
Source: Vives-Mestres M et al. Alcohol as a trigger of migraine attacks in people with migraine. Results from a large prospective cohort study in English-speaking countries. Headache. 2022;62:1329-1338. (Nov 27). Doi: 10.1111/head.14428
Key clinical point: Alcohol intake slightly reduced the likelihood of migraine attacks 48 hours after consumption in an English-speaking cohort of patients with episodic migraine who identified themselves as mostly low-dose alcohol consumers.
Major finding: The probability of migraine attack 48 hours after consuming alcohol was 25% lower than that after no alcohol consumption (adjusted odds ratio 0.75; 95% CI 0.68-0.82); however, alcohol consumption had no significant effect on migraine probability 24 hours after consumption.
Study details: This observational prospective cohort study included 487 patients with episodic migraine who reported 5913 migraine attacks and were alcohol consumers.
Disclosures: The study was partially funded by Curelator, Inc. M Vives-Mestres and A Casanova declared receiving consulting fees and holding stock options in Curelator, Inc. N Rosen reported ties with a headache society, journals, and various other sources.
Source: Vives-Mestres M et al. Alcohol as a trigger of migraine attacks in people with migraine. Results from a large prospective cohort study in English-speaking countries. Headache. 2022;62:1329-1338. (Nov 27). Doi: 10.1111/head.14428
Evidence supporting atogepant as a promising treatment for migraine prevention
Key clinical point: Once-daily atogepant at doses of 30 and 60 mg significantly improved the performance of social, work-related, and daily activities compared with placebo in patients with episodic migraine.
Major finding: At week 12, atogepant (30 and 60 mg) vs placebo significantly improved Migraine-Specific Quality of Life Questionnaire version 2.1 Role Function-Restrictive (least-squares mean difference [LSMD] 10.08 and 10.8, respectively; both P < .0001). Atogepant at doses of 30 and 60 mg significantly improved monthly Activity Impairment in Migraine-Diary Performance of Daily Activities (P = .0003 and P < .0001, respectively) and Physical Impairment (P = .001 and P < .0001, respectively) scores across the 12-week treatment.
Study details: Findings are from the phase 3, ADVANCE trial including 873 patients with episodic migraine who were randomly assigned to receive once-daily atogepant (10, 30, or 60 mg) or placebo.
Disclosures: This study was sponsored by AbbVie/Allergan. Four authors declared being current or former employees of or holding stocks in AbbVie. The lead author and several other authors reported ties with various sources, including AbbVie.
Source: Lipton RB et al. Effect of atogepant for preventive migraine treatment on patient-reported outcomes in the randomized, double-blind, phase 3 ADVANCE trial. Neurology. 2022 (Nov 17). Doi: 10.1212/WNL.0000000000201568
Key clinical point: Once-daily atogepant at doses of 30 and 60 mg significantly improved the performance of social, work-related, and daily activities compared with placebo in patients with episodic migraine.
Major finding: At week 12, atogepant (30 and 60 mg) vs placebo significantly improved Migraine-Specific Quality of Life Questionnaire version 2.1 Role Function-Restrictive (least-squares mean difference [LSMD] 10.08 and 10.8, respectively; both P < .0001). Atogepant at doses of 30 and 60 mg significantly improved monthly Activity Impairment in Migraine-Diary Performance of Daily Activities (P = .0003 and P < .0001, respectively) and Physical Impairment (P = .001 and P < .0001, respectively) scores across the 12-week treatment.
Study details: Findings are from the phase 3, ADVANCE trial including 873 patients with episodic migraine who were randomly assigned to receive once-daily atogepant (10, 30, or 60 mg) or placebo.
Disclosures: This study was sponsored by AbbVie/Allergan. Four authors declared being current or former employees of or holding stocks in AbbVie. The lead author and several other authors reported ties with various sources, including AbbVie.
Source: Lipton RB et al. Effect of atogepant for preventive migraine treatment on patient-reported outcomes in the randomized, double-blind, phase 3 ADVANCE trial. Neurology. 2022 (Nov 17). Doi: 10.1212/WNL.0000000000201568
Key clinical point: Once-daily atogepant at doses of 30 and 60 mg significantly improved the performance of social, work-related, and daily activities compared with placebo in patients with episodic migraine.
Major finding: At week 12, atogepant (30 and 60 mg) vs placebo significantly improved Migraine-Specific Quality of Life Questionnaire version 2.1 Role Function-Restrictive (least-squares mean difference [LSMD] 10.08 and 10.8, respectively; both P < .0001). Atogepant at doses of 30 and 60 mg significantly improved monthly Activity Impairment in Migraine-Diary Performance of Daily Activities (P = .0003 and P < .0001, respectively) and Physical Impairment (P = .001 and P < .0001, respectively) scores across the 12-week treatment.
Study details: Findings are from the phase 3, ADVANCE trial including 873 patients with episodic migraine who were randomly assigned to receive once-daily atogepant (10, 30, or 60 mg) or placebo.
Disclosures: This study was sponsored by AbbVie/Allergan. Four authors declared being current or former employees of or holding stocks in AbbVie. The lead author and several other authors reported ties with various sources, including AbbVie.
Source: Lipton RB et al. Effect of atogepant for preventive migraine treatment on patient-reported outcomes in the randomized, double-blind, phase 3 ADVANCE trial. Neurology. 2022 (Nov 17). Doi: 10.1212/WNL.0000000000201568
Can nanotechnology help cure IBD?
Finding a cure for inflammatory bowel disease is a big goal. But the key to achieving it might be to think small.
University of Wisconsin–Madison researchers are developing nanoparticles – particles measuring between 1 and 100 nanometers (one-billionth of a meter) – designed to treat IBD, including Crohn’s disease and ulcerative colitis. (For context: A sheet of paper is about 100,000 nanometers thick.)
Described in a paper in Science Advances, these , a compatible compound commonly used in medicine.
The nanoparticles – the researchers call them “backpacks” – can be attached to probiotics, which deliver them to the gut.
“Due to the colonizing property of probiotics in colon tissues, the nanoparticles could be delivered to colon tissues by probiotics and released slowly,” says study author Quanyin Hu, PhD, a biomedical engineer and assistant professor at the University of Wisconsin–Madison School of Pharmacy.
This helps give the nanoparticles time to bring the ROS level back down to normal. But that’s only part of the IBD treatment the researchers envision.
The technology builds on a previous development from Dr. Hu and his team – a protective probiotic shell coating. The coating, which is about 330 nanometers thick, helps probiotics survive long enough to establish and multiply in the gut.
“The harsh environment of gastric acid and bile salt would kill most probiotics,” Dr. Hu says. “Moreover, antibiotics usually used in inflammatory bowel disease treatment also harm probiotic growth.”
Early results are promising, he says. Mice with IBD that received the full treatment – combining the ROS-targeting nanoparticles with the coated probiotics – had fewer IBD symptoms, like less weight loss and colon shortening, than those treated with the encapsulated probiotics alone.
By attacking the disease on multiple fronts – reducing the ROS and improving the balance of gut microbiota – a healthy gut environment could be restored, Dr. Hu says. In other words: “[It] might be possible to finally cure inflammatory bowel disease.”
Nanotechnology offers all kinds of unique advantages over traditional IBD treatments, he says. Nanoparticles can be designed to target specific tissues, like colon tissues. And, compared with small molecules, they can circulate throughout the body longer, so they have more time to build up and do their job.
The next steps will be to test the treatment in large animals and “to develop a stable formulation that can be stored for a long time and produced in a scalable and economical manner,” Dr. Hu says.
Current IBD treatments “can only relieve symptoms,” not cure the disease, he says.
“This study is our first try to fundamentally treat inflammatory bowel disease by recovering a healthy microenvironment in the intestines, and our preliminary data demonstrated that this strategy is delivering promises to pave a new treatment strategy for IBD,” Dr. Hu says.
A version of this article first appeared on WebMD.com.
Finding a cure for inflammatory bowel disease is a big goal. But the key to achieving it might be to think small.
University of Wisconsin–Madison researchers are developing nanoparticles – particles measuring between 1 and 100 nanometers (one-billionth of a meter) – designed to treat IBD, including Crohn’s disease and ulcerative colitis. (For context: A sheet of paper is about 100,000 nanometers thick.)
Described in a paper in Science Advances, these , a compatible compound commonly used in medicine.
The nanoparticles – the researchers call them “backpacks” – can be attached to probiotics, which deliver them to the gut.
“Due to the colonizing property of probiotics in colon tissues, the nanoparticles could be delivered to colon tissues by probiotics and released slowly,” says study author Quanyin Hu, PhD, a biomedical engineer and assistant professor at the University of Wisconsin–Madison School of Pharmacy.
This helps give the nanoparticles time to bring the ROS level back down to normal. But that’s only part of the IBD treatment the researchers envision.
The technology builds on a previous development from Dr. Hu and his team – a protective probiotic shell coating. The coating, which is about 330 nanometers thick, helps probiotics survive long enough to establish and multiply in the gut.
“The harsh environment of gastric acid and bile salt would kill most probiotics,” Dr. Hu says. “Moreover, antibiotics usually used in inflammatory bowel disease treatment also harm probiotic growth.”
Early results are promising, he says. Mice with IBD that received the full treatment – combining the ROS-targeting nanoparticles with the coated probiotics – had fewer IBD symptoms, like less weight loss and colon shortening, than those treated with the encapsulated probiotics alone.
By attacking the disease on multiple fronts – reducing the ROS and improving the balance of gut microbiota – a healthy gut environment could be restored, Dr. Hu says. In other words: “[It] might be possible to finally cure inflammatory bowel disease.”
Nanotechnology offers all kinds of unique advantages over traditional IBD treatments, he says. Nanoparticles can be designed to target specific tissues, like colon tissues. And, compared with small molecules, they can circulate throughout the body longer, so they have more time to build up and do their job.
The next steps will be to test the treatment in large animals and “to develop a stable formulation that can be stored for a long time and produced in a scalable and economical manner,” Dr. Hu says.
Current IBD treatments “can only relieve symptoms,” not cure the disease, he says.
“This study is our first try to fundamentally treat inflammatory bowel disease by recovering a healthy microenvironment in the intestines, and our preliminary data demonstrated that this strategy is delivering promises to pave a new treatment strategy for IBD,” Dr. Hu says.
A version of this article first appeared on WebMD.com.
Finding a cure for inflammatory bowel disease is a big goal. But the key to achieving it might be to think small.
University of Wisconsin–Madison researchers are developing nanoparticles – particles measuring between 1 and 100 nanometers (one-billionth of a meter) – designed to treat IBD, including Crohn’s disease and ulcerative colitis. (For context: A sheet of paper is about 100,000 nanometers thick.)
Described in a paper in Science Advances, these , a compatible compound commonly used in medicine.
The nanoparticles – the researchers call them “backpacks” – can be attached to probiotics, which deliver them to the gut.
“Due to the colonizing property of probiotics in colon tissues, the nanoparticles could be delivered to colon tissues by probiotics and released slowly,” says study author Quanyin Hu, PhD, a biomedical engineer and assistant professor at the University of Wisconsin–Madison School of Pharmacy.
This helps give the nanoparticles time to bring the ROS level back down to normal. But that’s only part of the IBD treatment the researchers envision.
The technology builds on a previous development from Dr. Hu and his team – a protective probiotic shell coating. The coating, which is about 330 nanometers thick, helps probiotics survive long enough to establish and multiply in the gut.
“The harsh environment of gastric acid and bile salt would kill most probiotics,” Dr. Hu says. “Moreover, antibiotics usually used in inflammatory bowel disease treatment also harm probiotic growth.”
Early results are promising, he says. Mice with IBD that received the full treatment – combining the ROS-targeting nanoparticles with the coated probiotics – had fewer IBD symptoms, like less weight loss and colon shortening, than those treated with the encapsulated probiotics alone.
By attacking the disease on multiple fronts – reducing the ROS and improving the balance of gut microbiota – a healthy gut environment could be restored, Dr. Hu says. In other words: “[It] might be possible to finally cure inflammatory bowel disease.”
Nanotechnology offers all kinds of unique advantages over traditional IBD treatments, he says. Nanoparticles can be designed to target specific tissues, like colon tissues. And, compared with small molecules, they can circulate throughout the body longer, so they have more time to build up and do their job.
The next steps will be to test the treatment in large animals and “to develop a stable formulation that can be stored for a long time and produced in a scalable and economical manner,” Dr. Hu says.
Current IBD treatments “can only relieve symptoms,” not cure the disease, he says.
“This study is our first try to fundamentally treat inflammatory bowel disease by recovering a healthy microenvironment in the intestines, and our preliminary data demonstrated that this strategy is delivering promises to pave a new treatment strategy for IBD,” Dr. Hu says.
A version of this article first appeared on WebMD.com.
FROM SCIENCE ADVANCES
HIV vaccine trial makes pivotal leap toward making ‘super antibodies’
The announcement comes from the journal Science, which published phase 1 results of a small clinical trial for a vaccine technology that aims to cause the body to create a rare kind of cell.
“At the most general level, the trial results show that one can design vaccines that induce antibodies with prespecified genetic features, and this may herald a new era of precision vaccines,” William Schief, PhD, a researcher at the Scripps Research Institute and study coauthor, told the American Association for the Advancement of Science.
The study was the first to test the approach in humans and was effective in 97% – or 35 of 36 – participants. The vaccine technology is called “germline targeting.” Trial results show that “one can design a vaccine that elicits made-to-order antibodies in humans,” Dr. Schief said in a news release.
In addition to possibly being a breakthrough for the treatment of HIV, the vaccine technology could also impact the development of treatments for flu, hepatitis C, and coronaviruses, study authors wrote.
There is no cure for HIV, but there are treatments to manage how the disease progresses. HIV attacks the body’s immune system, destroys white blood cells, and increases susceptibility to other infections, AAAS summarized. More than 1 million people in the United States and 38 million people worldwide have HIV.
Previous HIV vaccine attempts were not able to cause the production of specialized cells known as “broadly neutralizing antibodies,” CNN reported.
“Call them super antibodies, if you want,” University of Minnesota HIV researcher Timothy Schacker, MD, who was not involved in the research, told CNN. “The hope is that if you can induce this kind of immunity in people, you can protect them from some of these viruses that we’ve had a very hard time designing vaccines for that are effective. So this is an important step forward.”
Study authors said this is just the first step in the multiphase vaccine design, which so far is a theory. Further study is needed to see if the next steps also work in humans, and then if all the steps can be linked together and can be effective against HIV.
A version of this article first appeared on WebMD.com.
The announcement comes from the journal Science, which published phase 1 results of a small clinical trial for a vaccine technology that aims to cause the body to create a rare kind of cell.
“At the most general level, the trial results show that one can design vaccines that induce antibodies with prespecified genetic features, and this may herald a new era of precision vaccines,” William Schief, PhD, a researcher at the Scripps Research Institute and study coauthor, told the American Association for the Advancement of Science.
The study was the first to test the approach in humans and was effective in 97% – or 35 of 36 – participants. The vaccine technology is called “germline targeting.” Trial results show that “one can design a vaccine that elicits made-to-order antibodies in humans,” Dr. Schief said in a news release.
In addition to possibly being a breakthrough for the treatment of HIV, the vaccine technology could also impact the development of treatments for flu, hepatitis C, and coronaviruses, study authors wrote.
There is no cure for HIV, but there are treatments to manage how the disease progresses. HIV attacks the body’s immune system, destroys white blood cells, and increases susceptibility to other infections, AAAS summarized. More than 1 million people in the United States and 38 million people worldwide have HIV.
Previous HIV vaccine attempts were not able to cause the production of specialized cells known as “broadly neutralizing antibodies,” CNN reported.
“Call them super antibodies, if you want,” University of Minnesota HIV researcher Timothy Schacker, MD, who was not involved in the research, told CNN. “The hope is that if you can induce this kind of immunity in people, you can protect them from some of these viruses that we’ve had a very hard time designing vaccines for that are effective. So this is an important step forward.”
Study authors said this is just the first step in the multiphase vaccine design, which so far is a theory. Further study is needed to see if the next steps also work in humans, and then if all the steps can be linked together and can be effective against HIV.
A version of this article first appeared on WebMD.com.
The announcement comes from the journal Science, which published phase 1 results of a small clinical trial for a vaccine technology that aims to cause the body to create a rare kind of cell.
“At the most general level, the trial results show that one can design vaccines that induce antibodies with prespecified genetic features, and this may herald a new era of precision vaccines,” William Schief, PhD, a researcher at the Scripps Research Institute and study coauthor, told the American Association for the Advancement of Science.
The study was the first to test the approach in humans and was effective in 97% – or 35 of 36 – participants. The vaccine technology is called “germline targeting.” Trial results show that “one can design a vaccine that elicits made-to-order antibodies in humans,” Dr. Schief said in a news release.
In addition to possibly being a breakthrough for the treatment of HIV, the vaccine technology could also impact the development of treatments for flu, hepatitis C, and coronaviruses, study authors wrote.
There is no cure for HIV, but there are treatments to manage how the disease progresses. HIV attacks the body’s immune system, destroys white blood cells, and increases susceptibility to other infections, AAAS summarized. More than 1 million people in the United States and 38 million people worldwide have HIV.
Previous HIV vaccine attempts were not able to cause the production of specialized cells known as “broadly neutralizing antibodies,” CNN reported.
“Call them super antibodies, if you want,” University of Minnesota HIV researcher Timothy Schacker, MD, who was not involved in the research, told CNN. “The hope is that if you can induce this kind of immunity in people, you can protect them from some of these viruses that we’ve had a very hard time designing vaccines for that are effective. So this is an important step forward.”
Study authors said this is just the first step in the multiphase vaccine design, which so far is a theory. Further study is needed to see if the next steps also work in humans, and then if all the steps can be linked together and can be effective against HIV.
A version of this article first appeared on WebMD.com.
FROM SCIENCE
The new obesity breakthrough drugs
This article was originally published December 10 on Medscape editor-in-chief Eric Topol’s Substack ”Ground Truths.”
– achieving a substantial amount of weight loss without serious side effects. Many attempts to get there now fill a graveyard of failed drugs, such as fen-phen in the 1990s when a single small study of this drug combination in 121 people unleashed millions of prescriptions, some leading to serious heart valve lesions that resulted in withdrawal of the drug in 1995. The drug rimonabant, an endocannabinoid receptor blocker (think of blocking the munchies after marijuana) looked encouraging in randomized trials. However, subsequently, in a trial that I led of nearly 19,000 participants in 42 countries around the world, there was a significant excess of depression, neuropsychiatric side-effects and suicidal ideation which spelled the end of that drug’s life.
In the United States, where there had not been an antiobesity drug approved by the Food and Drug Administration since 2014, Wegovy (semaglutide), a once-weekly injection was approved in June 2021. The same drug, at a lower dose, is known as Ozempic (as in O-O-O, Ozempic, the ubiquitous commercial that you undoubtedly hear and see on TV) and had already been approved in January 2020 for improving glucose regulation in diabetes. The next drug on fast track at FDA to be imminently approved is tirzepatide (Mounjaro) following its approval for diabetes in May 2022. It is noteworthy that the discovery of these drugs for weight loss was serendipitous: they were being developed for improving glucose regulation and unexpectedly were found to achieve significant weight reduction.
Both semaglutide and tirzepatide underwent randomized, placebo-controlled trials for obesity, with marked reduction of weight as shown below. Tirzepatide at dose of 10-15 mg per week achieved greater than 20% body weight reduction. Semaglutide at a dose of 2.4 mg achieved about 17% reduction. These per cent changes in body weight are 7-9 fold more than seen with placebo (2%-3% reduction). Note: these levels of percent body-weight reduction resemble what is typically achieved with the different types of bariatric surgery, such as gastric bypass.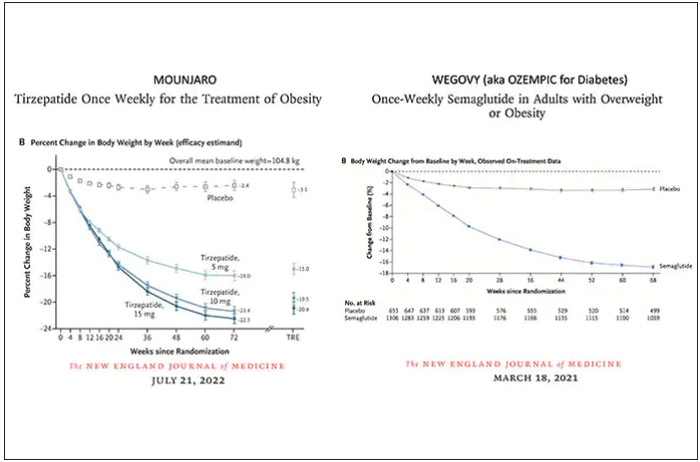
Another way to present the data for the two trials is shown here, with an edge for tirzepatide at high (10-15 mg) doses, extending to greater than 25% body-weight reduction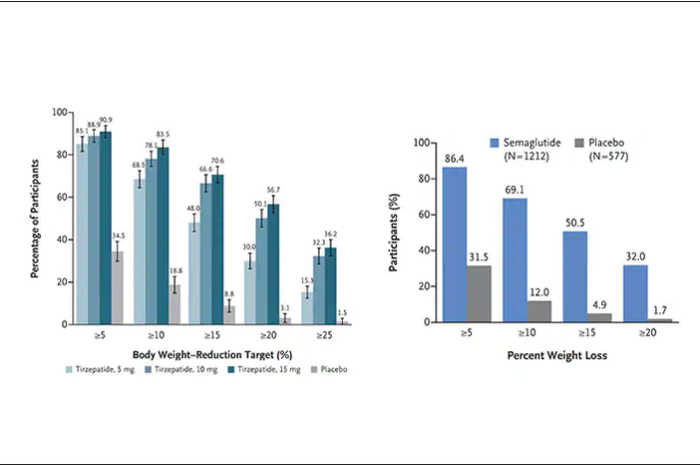
The results with semaglutide were extended to teens in a randomized trial (as shown below), and a similar trial with tirzepatide is in progress.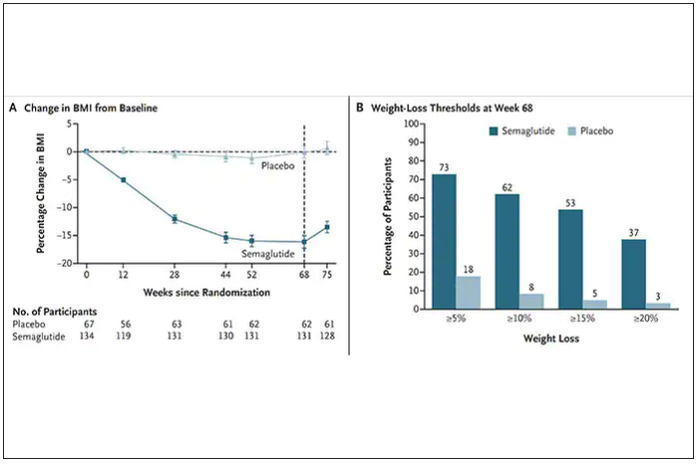
How do these drugs work?
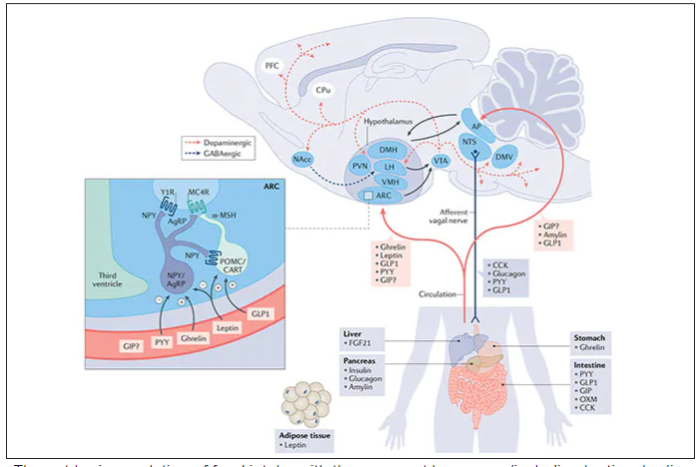
These are peptides in the class of incretins, mimicking gut hormones that are secreted after food intake which stimulate insulin secretion.
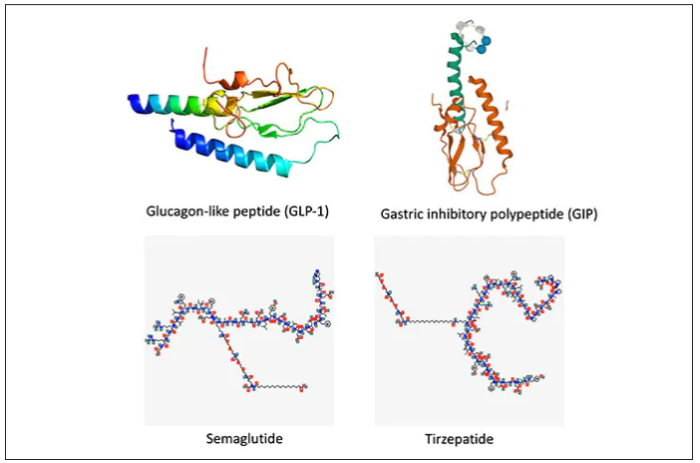
These two drugs have in common long half-lives (about 5 days), which affords once-weekly dosing, but have different mechanisms of action. Semaglutide activates (an agonist) the glucagonlike peptide–1 receptor, while tirzepatide is in a new class of dual agonists: It activates (mimics) both the GLP-1 receptor and GIP receptors (Gastric inhibit polypeptide is also known as glucose-dependent insulinotropic polypeptide.) The potency of activation for tirzepatide is fivefold more for GIPR than GLP1. As seen below, there are body wide effects that include the brain, liver, pancreas, stomach, intestine, skeletal muscle and fat tissue. While their mode of action is somewhat different, their clinical effects are overlapping, which include enhancing satiety, delaying gastric emptying, increasing insulin and its sensitivity, decreasing glucagon, and, of course, reducing high glucose levels. The overlap extends to side effects of nausea, vomiting, abdominal pain, constipation and diarrhea. Yet only 4%-6% of participants discontinued the drug in these trials, mostly owing to these GI side effects (and 1%-2% in the placebo group discontinued the study drug for the same reasons).
In randomized trials among people with type 2 diabetes, the drugs achieved hemoglobin A1c reduction of at least an absolute 2 percentage points which led to their FDA approvals (For semaglutide in January 2020, and for tirzepatide in May 2022). The edge that tirzepatide has exhibited for weight-loss reduction may be related to its dual agonist role, but the enhancement via GIP receptor activation is not fully resolved (as seen below with GIP? designation). The Amgen drug in development (AMG-133) has a marked weight loss effect but inhibits GIP rather than mimics it, clouding our precise understanding of the mechanism.
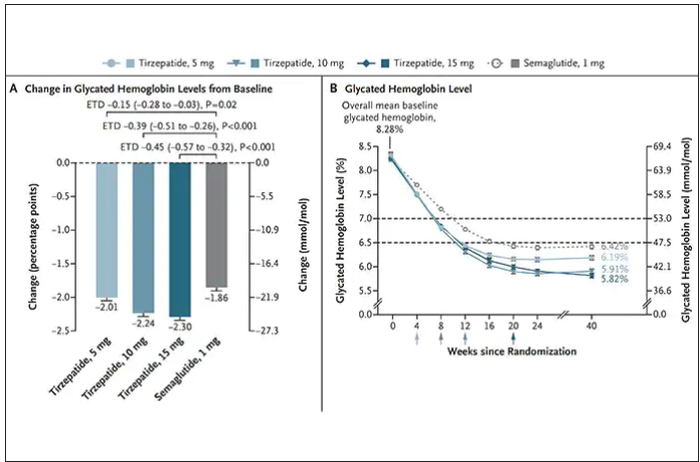
Nevertheless, when the two drugs were directly compared in a randomized trial for improving glucose regulation, tirzepatide was superior to semaglutide, as shown below. Of note, both drugs achieved very favorable effects on lipids, reducing triglycerides and LDL cholesterol and raising HDL cholesterol, along with reduction of blood pressure, an outgrowth of the indirect effect of weight reduction and direct metabolic effects of the drugs.
While there has been a concern about other side effects besides the GI ones noted above, review of all the trials to date in these classes of medication do not reinforce a risk of acute pancreatitis. Other rare side effects that have been noted with these drugs include allergic reactions, gallstones (which can occur with a large amount of weight loss), and potential of medullary thyroid cancer (so far only documented in rats, not people), which is why they are contraindicated in people with Type 2 multiple endocrine neoplasia syndrome.
How they are given and practical considerations
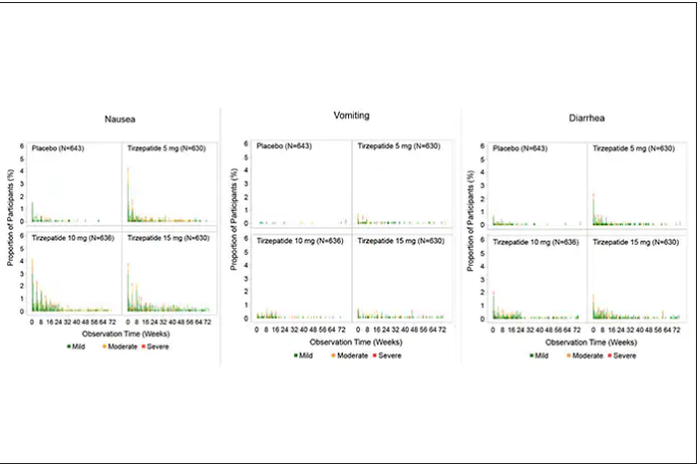
For semaglutide, which has FDA approval, the indication is a body mass index of 30 kg/m2 or greater than 27 and a weight-related medical condition (such as hypertension, hypercholesterolemia, or diabetes). To reduce the GI side effects, which mainly occur in the early dose escalation period, semaglutide is given in increasing doses by a prefilled pen by self-injection under the skin (abdomen, thigh, or arm) starting at 0.25 mg for a month and gradual increases each month reaching the maximum dose of 2.4 mg at month 5. The FDA label for dosing of tirzepatide has not been provided yet but in the weight loss trial there was a similar dose escalation from 2.5 mg up to 15 mg by month 5. The escalation is essential to reduce the frequent GI side effects, such as seen below in the tirzepatide trial.
Semaglutide is very expensive, about $1,500 per month, and not covered by Medicare. There are manufacturer starter coupons from Novo Nordisk, but that is just for the first month. These drugs have to be taken for a year to 18 months to have their full effect and without changes in lifestyle that are durable, it is likely that weight will be regained after stopping them.
What does this mean?
More than 650 million adults and 340 million children aged 5-18 are obese. The global obesity epidemic has been relentless, worsening each year, and a driver of “diabesity,” the combined dual epidemic. We now have a breakthrough class of drugs that can achieve profound weight loss equivalent to bariatric surgery, along with the side benefits of reducing cardiovascular risk factors (hypertension and hyperlipidemia), improving glucose regulation, reversing fatty liver, and the many detrimental long-term effects of obesity such as osteoarthritis and various cancers. That, in itself, is remarkable. Revolutionary.
But the downsides are also obvious. Self-injections, even though they are once a week, are not palatable for many. We have seen far more of these injectables in recent years such as the proprotein convertase subtilisin/kexin type 9 inhibitors for hypercholesterolemia or the tumor necrosis factor blockers for autoimmune conditions. That still will not make them a popular item for such an enormous population of potential users.
That brings me to Rybelsus, the oral form of semaglutide, which is approved for glucose regulation improvement but not obesity. It effects for weight loss have been modest, compared with Wegovy (5 to 8 pounds for the 7- and 14-mg dose, respectively). But the potential for the very high efficacy of an injectable to be achievable via a pill represents an important path going forward—it could help markedly reduce the cost and uptake.
The problem of discontinuation of the drugs is big, since there are limited data and the likelihood is that the weight will be regained unless there are substantial changes in lifestyle. We know how hard it is to durably achieve such changes, along with the undesirability (and uncertainty with respect to unknown side effects) of having to take injectable drugs for many years, no less the cost of doing that.
The cost of these drugs will clearly and profoundly exacerbate inequities, since they are eminently affordable by the rich, but the need is extreme among the indigent. We’ve already seen celebrities take Wegovy for weight loss who are not obese, a window into how these drugs can and will be used without supportive data. As one physician recently observed, “Other than Viagra and Botox, I’ve seen no other medication so quickly become part of modern culture’s social vernacular.” Already there are concerns that such use is preventing access to the drugs for those who qualify and need them.
There are multiple agents in the class under development which should help increase competition and reduce cost, but they will remain expensive. There is private insurance reimbursement, often with a significant copay, for people who tightly fit the inclusion criteria. Eventual coverage by Medicare will markedly expand their use, and we can expect cost-effectiveness studies to be published showing how much saving there is for the drugs compared with bariatric surgery or not achieving the weight loss. But that doesn’t change the cost at the societal level. Even as we’ve seen with generics, which will ultimately be available, the alleviation of the cost problem isn’t what we’d hoped.
This is not unlike the recent triumphs of gene therapy, as in $3.5 million for a cure of hemophilia that just got FDA approval, but instead of a rare disease we are talking about the most common medical condition in the world. We finally get across the long sought after (what many would qualify as miraculous) goal line, but the economics collide with the uptake and real benefit.
These concerns can’t be put aside in the health inequity-laden world we live in, that will unquestionably be exacerbated. However, we cannot miss that this represents one of the most important, biggest medical breakthroughs in history. This may signify the end or marked reduction in the need for bariatric surgery. These drugs will likely become some of the most prescribed of all medications in the upcoming years. While there are many drawbacks, we shouldn’t miss such an extraordinary advance in medicine – the first real, potent and safe treatment of obesity.
Thanks for reading Ground Truths. I hope you will share these posts and subscribe, to be sure you don’t miss them.
Dr. Topol is director, Scripps Translational Science Institute; executive vice president and professor of molecular medicine at The Scripps Research Institute and senior consultant, division of cardiovascular diseases, at the Scripps Clinic, both in La Jolla, Calif. He disclosed relevant financial relationships with Dexcom, Illumina, Molecular Stethoscope, Walgreens, Quest Diagnostics, MyoKardia, and National Institutes of Health. A version of this article first appeared on Medscape.com.
This article was originally published December 10 on Medscape editor-in-chief Eric Topol’s Substack ”Ground Truths.”
– achieving a substantial amount of weight loss without serious side effects. Many attempts to get there now fill a graveyard of failed drugs, such as fen-phen in the 1990s when a single small study of this drug combination in 121 people unleashed millions of prescriptions, some leading to serious heart valve lesions that resulted in withdrawal of the drug in 1995. The drug rimonabant, an endocannabinoid receptor blocker (think of blocking the munchies after marijuana) looked encouraging in randomized trials. However, subsequently, in a trial that I led of nearly 19,000 participants in 42 countries around the world, there was a significant excess of depression, neuropsychiatric side-effects and suicidal ideation which spelled the end of that drug’s life.
In the United States, where there had not been an antiobesity drug approved by the Food and Drug Administration since 2014, Wegovy (semaglutide), a once-weekly injection was approved in June 2021. The same drug, at a lower dose, is known as Ozempic (as in O-O-O, Ozempic, the ubiquitous commercial that you undoubtedly hear and see on TV) and had already been approved in January 2020 for improving glucose regulation in diabetes. The next drug on fast track at FDA to be imminently approved is tirzepatide (Mounjaro) following its approval for diabetes in May 2022. It is noteworthy that the discovery of these drugs for weight loss was serendipitous: they were being developed for improving glucose regulation and unexpectedly were found to achieve significant weight reduction.
Both semaglutide and tirzepatide underwent randomized, placebo-controlled trials for obesity, with marked reduction of weight as shown below. Tirzepatide at dose of 10-15 mg per week achieved greater than 20% body weight reduction. Semaglutide at a dose of 2.4 mg achieved about 17% reduction. These per cent changes in body weight are 7-9 fold more than seen with placebo (2%-3% reduction). Note: these levels of percent body-weight reduction resemble what is typically achieved with the different types of bariatric surgery, such as gastric bypass.
Another way to present the data for the two trials is shown here, with an edge for tirzepatide at high (10-15 mg) doses, extending to greater than 25% body-weight reduction
The results with semaglutide were extended to teens in a randomized trial (as shown below), and a similar trial with tirzepatide is in progress.
How do these drugs work?
These are peptides in the class of incretins, mimicking gut hormones that are secreted after food intake which stimulate insulin secretion.
These two drugs have in common long half-lives (about 5 days), which affords once-weekly dosing, but have different mechanisms of action. Semaglutide activates (an agonist) the glucagonlike peptide–1 receptor, while tirzepatide is in a new class of dual agonists: It activates (mimics) both the GLP-1 receptor and GIP receptors (Gastric inhibit polypeptide is also known as glucose-dependent insulinotropic polypeptide.) The potency of activation for tirzepatide is fivefold more for GIPR than GLP1. As seen below, there are body wide effects that include the brain, liver, pancreas, stomach, intestine, skeletal muscle and fat tissue. While their mode of action is somewhat different, their clinical effects are overlapping, which include enhancing satiety, delaying gastric emptying, increasing insulin and its sensitivity, decreasing glucagon, and, of course, reducing high glucose levels. The overlap extends to side effects of nausea, vomiting, abdominal pain, constipation and diarrhea. Yet only 4%-6% of participants discontinued the drug in these trials, mostly owing to these GI side effects (and 1%-2% in the placebo group discontinued the study drug for the same reasons).
In randomized trials among people with type 2 diabetes, the drugs achieved hemoglobin A1c reduction of at least an absolute 2 percentage points which led to their FDA approvals (For semaglutide in January 2020, and for tirzepatide in May 2022). The edge that tirzepatide has exhibited for weight-loss reduction may be related to its dual agonist role, but the enhancement via GIP receptor activation is not fully resolved (as seen below with GIP? designation). The Amgen drug in development (AMG-133) has a marked weight loss effect but inhibits GIP rather than mimics it, clouding our precise understanding of the mechanism.
Nevertheless, when the two drugs were directly compared in a randomized trial for improving glucose regulation, tirzepatide was superior to semaglutide, as shown below. Of note, both drugs achieved very favorable effects on lipids, reducing triglycerides and LDL cholesterol and raising HDL cholesterol, along with reduction of blood pressure, an outgrowth of the indirect effect of weight reduction and direct metabolic effects of the drugs.
While there has been a concern about other side effects besides the GI ones noted above, review of all the trials to date in these classes of medication do not reinforce a risk of acute pancreatitis. Other rare side effects that have been noted with these drugs include allergic reactions, gallstones (which can occur with a large amount of weight loss), and potential of medullary thyroid cancer (so far only documented in rats, not people), which is why they are contraindicated in people with Type 2 multiple endocrine neoplasia syndrome.
How they are given and practical considerations
For semaglutide, which has FDA approval, the indication is a body mass index of 30 kg/m2 or greater than 27 and a weight-related medical condition (such as hypertension, hypercholesterolemia, or diabetes). To reduce the GI side effects, which mainly occur in the early dose escalation period, semaglutide is given in increasing doses by a prefilled pen by self-injection under the skin (abdomen, thigh, or arm) starting at 0.25 mg for a month and gradual increases each month reaching the maximum dose of 2.4 mg at month 5. The FDA label for dosing of tirzepatide has not been provided yet but in the weight loss trial there was a similar dose escalation from 2.5 mg up to 15 mg by month 5. The escalation is essential to reduce the frequent GI side effects, such as seen below in the tirzepatide trial.
Semaglutide is very expensive, about $1,500 per month, and not covered by Medicare. There are manufacturer starter coupons from Novo Nordisk, but that is just for the first month. These drugs have to be taken for a year to 18 months to have their full effect and without changes in lifestyle that are durable, it is likely that weight will be regained after stopping them.
What does this mean?
More than 650 million adults and 340 million children aged 5-18 are obese. The global obesity epidemic has been relentless, worsening each year, and a driver of “diabesity,” the combined dual epidemic. We now have a breakthrough class of drugs that can achieve profound weight loss equivalent to bariatric surgery, along with the side benefits of reducing cardiovascular risk factors (hypertension and hyperlipidemia), improving glucose regulation, reversing fatty liver, and the many detrimental long-term effects of obesity such as osteoarthritis and various cancers. That, in itself, is remarkable. Revolutionary.
But the downsides are also obvious. Self-injections, even though they are once a week, are not palatable for many. We have seen far more of these injectables in recent years such as the proprotein convertase subtilisin/kexin type 9 inhibitors for hypercholesterolemia or the tumor necrosis factor blockers for autoimmune conditions. That still will not make them a popular item for such an enormous population of potential users.
That brings me to Rybelsus, the oral form of semaglutide, which is approved for glucose regulation improvement but not obesity. It effects for weight loss have been modest, compared with Wegovy (5 to 8 pounds for the 7- and 14-mg dose, respectively). But the potential for the very high efficacy of an injectable to be achievable via a pill represents an important path going forward—it could help markedly reduce the cost and uptake.
The problem of discontinuation of the drugs is big, since there are limited data and the likelihood is that the weight will be regained unless there are substantial changes in lifestyle. We know how hard it is to durably achieve such changes, along with the undesirability (and uncertainty with respect to unknown side effects) of having to take injectable drugs for many years, no less the cost of doing that.
The cost of these drugs will clearly and profoundly exacerbate inequities, since they are eminently affordable by the rich, but the need is extreme among the indigent. We’ve already seen celebrities take Wegovy for weight loss who are not obese, a window into how these drugs can and will be used without supportive data. As one physician recently observed, “Other than Viagra and Botox, I’ve seen no other medication so quickly become part of modern culture’s social vernacular.” Already there are concerns that such use is preventing access to the drugs for those who qualify and need them.
There are multiple agents in the class under development which should help increase competition and reduce cost, but they will remain expensive. There is private insurance reimbursement, often with a significant copay, for people who tightly fit the inclusion criteria. Eventual coverage by Medicare will markedly expand their use, and we can expect cost-effectiveness studies to be published showing how much saving there is for the drugs compared with bariatric surgery or not achieving the weight loss. But that doesn’t change the cost at the societal level. Even as we’ve seen with generics, which will ultimately be available, the alleviation of the cost problem isn’t what we’d hoped.
This is not unlike the recent triumphs of gene therapy, as in $3.5 million for a cure of hemophilia that just got FDA approval, but instead of a rare disease we are talking about the most common medical condition in the world. We finally get across the long sought after (what many would qualify as miraculous) goal line, but the economics collide with the uptake and real benefit.
These concerns can’t be put aside in the health inequity-laden world we live in, that will unquestionably be exacerbated. However, we cannot miss that this represents one of the most important, biggest medical breakthroughs in history. This may signify the end or marked reduction in the need for bariatric surgery. These drugs will likely become some of the most prescribed of all medications in the upcoming years. While there are many drawbacks, we shouldn’t miss such an extraordinary advance in medicine – the first real, potent and safe treatment of obesity.
Thanks for reading Ground Truths. I hope you will share these posts and subscribe, to be sure you don’t miss them.
Dr. Topol is director, Scripps Translational Science Institute; executive vice president and professor of molecular medicine at The Scripps Research Institute and senior consultant, division of cardiovascular diseases, at the Scripps Clinic, both in La Jolla, Calif. He disclosed relevant financial relationships with Dexcom, Illumina, Molecular Stethoscope, Walgreens, Quest Diagnostics, MyoKardia, and National Institutes of Health. A version of this article first appeared on Medscape.com.
This article was originally published December 10 on Medscape editor-in-chief Eric Topol’s Substack ”Ground Truths.”
– achieving a substantial amount of weight loss without serious side effects. Many attempts to get there now fill a graveyard of failed drugs, such as fen-phen in the 1990s when a single small study of this drug combination in 121 people unleashed millions of prescriptions, some leading to serious heart valve lesions that resulted in withdrawal of the drug in 1995. The drug rimonabant, an endocannabinoid receptor blocker (think of blocking the munchies after marijuana) looked encouraging in randomized trials. However, subsequently, in a trial that I led of nearly 19,000 participants in 42 countries around the world, there was a significant excess of depression, neuropsychiatric side-effects and suicidal ideation which spelled the end of that drug’s life.
In the United States, where there had not been an antiobesity drug approved by the Food and Drug Administration since 2014, Wegovy (semaglutide), a once-weekly injection was approved in June 2021. The same drug, at a lower dose, is known as Ozempic (as in O-O-O, Ozempic, the ubiquitous commercial that you undoubtedly hear and see on TV) and had already been approved in January 2020 for improving glucose regulation in diabetes. The next drug on fast track at FDA to be imminently approved is tirzepatide (Mounjaro) following its approval for diabetes in May 2022. It is noteworthy that the discovery of these drugs for weight loss was serendipitous: they were being developed for improving glucose regulation and unexpectedly were found to achieve significant weight reduction.
Both semaglutide and tirzepatide underwent randomized, placebo-controlled trials for obesity, with marked reduction of weight as shown below. Tirzepatide at dose of 10-15 mg per week achieved greater than 20% body weight reduction. Semaglutide at a dose of 2.4 mg achieved about 17% reduction. These per cent changes in body weight are 7-9 fold more than seen with placebo (2%-3% reduction). Note: these levels of percent body-weight reduction resemble what is typically achieved with the different types of bariatric surgery, such as gastric bypass.
Another way to present the data for the two trials is shown here, with an edge for tirzepatide at high (10-15 mg) doses, extending to greater than 25% body-weight reduction
The results with semaglutide were extended to teens in a randomized trial (as shown below), and a similar trial with tirzepatide is in progress.
How do these drugs work?
These are peptides in the class of incretins, mimicking gut hormones that are secreted after food intake which stimulate insulin secretion.
These two drugs have in common long half-lives (about 5 days), which affords once-weekly dosing, but have different mechanisms of action. Semaglutide activates (an agonist) the glucagonlike peptide–1 receptor, while tirzepatide is in a new class of dual agonists: It activates (mimics) both the GLP-1 receptor and GIP receptors (Gastric inhibit polypeptide is also known as glucose-dependent insulinotropic polypeptide.) The potency of activation for tirzepatide is fivefold more for GIPR than GLP1. As seen below, there are body wide effects that include the brain, liver, pancreas, stomach, intestine, skeletal muscle and fat tissue. While their mode of action is somewhat different, their clinical effects are overlapping, which include enhancing satiety, delaying gastric emptying, increasing insulin and its sensitivity, decreasing glucagon, and, of course, reducing high glucose levels. The overlap extends to side effects of nausea, vomiting, abdominal pain, constipation and diarrhea. Yet only 4%-6% of participants discontinued the drug in these trials, mostly owing to these GI side effects (and 1%-2% in the placebo group discontinued the study drug for the same reasons).
In randomized trials among people with type 2 diabetes, the drugs achieved hemoglobin A1c reduction of at least an absolute 2 percentage points which led to their FDA approvals (For semaglutide in January 2020, and for tirzepatide in May 2022). The edge that tirzepatide has exhibited for weight-loss reduction may be related to its dual agonist role, but the enhancement via GIP receptor activation is not fully resolved (as seen below with GIP? designation). The Amgen drug in development (AMG-133) has a marked weight loss effect but inhibits GIP rather than mimics it, clouding our precise understanding of the mechanism.
Nevertheless, when the two drugs were directly compared in a randomized trial for improving glucose regulation, tirzepatide was superior to semaglutide, as shown below. Of note, both drugs achieved very favorable effects on lipids, reducing triglycerides and LDL cholesterol and raising HDL cholesterol, along with reduction of blood pressure, an outgrowth of the indirect effect of weight reduction and direct metabolic effects of the drugs.
While there has been a concern about other side effects besides the GI ones noted above, review of all the trials to date in these classes of medication do not reinforce a risk of acute pancreatitis. Other rare side effects that have been noted with these drugs include allergic reactions, gallstones (which can occur with a large amount of weight loss), and potential of medullary thyroid cancer (so far only documented in rats, not people), which is why they are contraindicated in people with Type 2 multiple endocrine neoplasia syndrome.
How they are given and practical considerations
For semaglutide, which has FDA approval, the indication is a body mass index of 30 kg/m2 or greater than 27 and a weight-related medical condition (such as hypertension, hypercholesterolemia, or diabetes). To reduce the GI side effects, which mainly occur in the early dose escalation period, semaglutide is given in increasing doses by a prefilled pen by self-injection under the skin (abdomen, thigh, or arm) starting at 0.25 mg for a month and gradual increases each month reaching the maximum dose of 2.4 mg at month 5. The FDA label for dosing of tirzepatide has not been provided yet but in the weight loss trial there was a similar dose escalation from 2.5 mg up to 15 mg by month 5. The escalation is essential to reduce the frequent GI side effects, such as seen below in the tirzepatide trial.
Semaglutide is very expensive, about $1,500 per month, and not covered by Medicare. There are manufacturer starter coupons from Novo Nordisk, but that is just for the first month. These drugs have to be taken for a year to 18 months to have their full effect and without changes in lifestyle that are durable, it is likely that weight will be regained after stopping them.
What does this mean?
More than 650 million adults and 340 million children aged 5-18 are obese. The global obesity epidemic has been relentless, worsening each year, and a driver of “diabesity,” the combined dual epidemic. We now have a breakthrough class of drugs that can achieve profound weight loss equivalent to bariatric surgery, along with the side benefits of reducing cardiovascular risk factors (hypertension and hyperlipidemia), improving glucose regulation, reversing fatty liver, and the many detrimental long-term effects of obesity such as osteoarthritis and various cancers. That, in itself, is remarkable. Revolutionary.
But the downsides are also obvious. Self-injections, even though they are once a week, are not palatable for many. We have seen far more of these injectables in recent years such as the proprotein convertase subtilisin/kexin type 9 inhibitors for hypercholesterolemia or the tumor necrosis factor blockers for autoimmune conditions. That still will not make them a popular item for such an enormous population of potential users.
That brings me to Rybelsus, the oral form of semaglutide, which is approved for glucose regulation improvement but not obesity. It effects for weight loss have been modest, compared with Wegovy (5 to 8 pounds for the 7- and 14-mg dose, respectively). But the potential for the very high efficacy of an injectable to be achievable via a pill represents an important path going forward—it could help markedly reduce the cost and uptake.
The problem of discontinuation of the drugs is big, since there are limited data and the likelihood is that the weight will be regained unless there are substantial changes in lifestyle. We know how hard it is to durably achieve such changes, along with the undesirability (and uncertainty with respect to unknown side effects) of having to take injectable drugs for many years, no less the cost of doing that.
The cost of these drugs will clearly and profoundly exacerbate inequities, since they are eminently affordable by the rich, but the need is extreme among the indigent. We’ve already seen celebrities take Wegovy for weight loss who are not obese, a window into how these drugs can and will be used without supportive data. As one physician recently observed, “Other than Viagra and Botox, I’ve seen no other medication so quickly become part of modern culture’s social vernacular.” Already there are concerns that such use is preventing access to the drugs for those who qualify and need them.
There are multiple agents in the class under development which should help increase competition and reduce cost, but they will remain expensive. There is private insurance reimbursement, often with a significant copay, for people who tightly fit the inclusion criteria. Eventual coverage by Medicare will markedly expand their use, and we can expect cost-effectiveness studies to be published showing how much saving there is for the drugs compared with bariatric surgery or not achieving the weight loss. But that doesn’t change the cost at the societal level. Even as we’ve seen with generics, which will ultimately be available, the alleviation of the cost problem isn’t what we’d hoped.
This is not unlike the recent triumphs of gene therapy, as in $3.5 million for a cure of hemophilia that just got FDA approval, but instead of a rare disease we are talking about the most common medical condition in the world. We finally get across the long sought after (what many would qualify as miraculous) goal line, but the economics collide with the uptake and real benefit.
These concerns can’t be put aside in the health inequity-laden world we live in, that will unquestionably be exacerbated. However, we cannot miss that this represents one of the most important, biggest medical breakthroughs in history. This may signify the end or marked reduction in the need for bariatric surgery. These drugs will likely become some of the most prescribed of all medications in the upcoming years. While there are many drawbacks, we shouldn’t miss such an extraordinary advance in medicine – the first real, potent and safe treatment of obesity.
Thanks for reading Ground Truths. I hope you will share these posts and subscribe, to be sure you don’t miss them.
Dr. Topol is director, Scripps Translational Science Institute; executive vice president and professor of molecular medicine at The Scripps Research Institute and senior consultant, division of cardiovascular diseases, at the Scripps Clinic, both in La Jolla, Calif. He disclosed relevant financial relationships with Dexcom, Illumina, Molecular Stethoscope, Walgreens, Quest Diagnostics, MyoKardia, and National Institutes of Health. A version of this article first appeared on Medscape.com.
Can a Mediterranean diet ease depression in young men?
This transcript has been edited for clarity.
Drew Ramsey, MD: Welcome back, everyone. I’m Dr. Drew Ramsey. I’m on the editorial board with Medscape Psychiatry and I’m an assistant clinical professor of psychiatry at Columbia University. We have a special guest today.
I’m here with nutritionist Jessica Bayes, who’s at the University of Technology Sydney, and she’s the lead author of the AMMEND trial. [Editor’s note: Since completing her PhD, Bayes is now at Southern Cross University.]
Jessica, welcome to Medscape.
Jessica Bayes, PhD: Thank you for having me.
The AMMEND Trial
Dr. Ramsey: Thank you for coming on board and helping all of us as clinicians understand some of your research and some of what is suggested by your research – that young men can change their diet and it helped their depression. Tell us a little bit about the AMMEND trial.
Dr. Bayes: The AMMEND trial was a 12-week randomized controlled trial in young men, 18-25 years old, who had diagnosed moderate to severe clinical depression. They had a poor baseline diet and we got them to eat a healthy Mediterranean diet, which improved their symptoms of depression.
Dr. Ramsey: It was a remarkable trial. Jessica, if I recall, you helped individuals improve the Mediterranean dietary pattern score by 8 points on a 14-point scale. That led to a 20-point reduction in their Beck Depression Inventory. Tell us what that looked like on the ground.
Dr. Bayes: It’s a huge improvement. Obviously, they were feeling much better in the end in terms of their depressive symptoms, but we also measured their energy, sleep, and quality of life. Many of them at the end were at a score cutoff that suggests no depression or in remission.
Dr. Ramsey: There were 72 people in your total trial, so 36% in your intervention arm went into full remission.
Dr. Bayes: Which is just amazing.
Dr. Ramsey: It also follows up the SMILES trial, which was a little bit of a different trial. You had two nutritional counseling sessions and the SMILES trial had seven, but in the SMILES trial, 32.3% of the patients went into full remission when they adopted a Mediterranean-style diet.
Jessica, what is the secret that you and your team know? I think many clinicians, especially clinicians who are parents and have teens, are kind of shaking their heads in disbelief. They’ve been telling their kids to eat healthy. What do you guys know about how to help young men change their diet?
How to Aid Adherence to Mediterranean Diet
Dr. Bayes: Prior to starting this, when I would say this idea to people, everyone would say, “Great idea. There’s no way you’re going to get depressed young men to change their diet. Not going to happen.” We went to them and we asked them. We said, “We’re going to do this study. What do you want from us? What resources would you need? How many appointments would you like? What’s too little or too many?”
We really got their feedback on board when we designed the study, and that obviously paid off. We had a personalized approach and we met them where they were at. We gave them the skills, resources, recipes, meal ideas – all those things – so we could really set them up to succeed.
Dr. Ramsey: You were telling me earlier about a few of the dietary changes that you felt made a big difference for these young men. What were those?
Dr. Bayes: Increasing the vegetables, olive oil, and legumes are probably the big ones that most of them were really not doing beforehand. They were really able to take that on board and make significant improvements in those areas.
Dr. Ramsey: These are really some of the top food categories in nutritional psychiatry as we think about how we help our efforts to improve mental health by thinking about nutrition, nutritional quality, and nutritional density. Certainly, those food categories – nuts and legumes, plants, and olive oil – are really what help get us there.
You also gave the students a food hamper. If you were going to be in charge of mental health in Australia and America and you got to give every college freshman a little box with a note, what would be in that box?
Dr. Bayes: I’d want to put everything in that box! It would be full of brightly colored fruits and vegetables, different nuts and seeds, and legumes. It would be full of recipes and ideas of how to cook things and how to prepare really delicious things. It would be full of different herbs and spices and all of those things to get people really excited about food.
Dr. Ramsey: Did the young men pick up on your enthusiasm and excitement around food? Did they begin to adopt some of that, shifting their view of how they saw the food and how they saw that it is related to their depression?
Dr. Bayes: Hopefully. I do think energy is infectious. I’m sure that played a role somewhat, but trying to get them excited about food can be really quite daunting, thinking, I’ve got to change my entire diet and I’ve got to learn to cook and go out and buy groceries. I don’t even know what to do with a piece of salmon. Trying to get them curious, interested, and just reminding them that it’s not all-or-nothing. Make small changes, give it a go, and have fun.
Dr. Ramsey: You also have a unique aspect of your research that you’re interested in male mental health, and that’s not something that’s been widely researched. Can you tell us a little bit about what these men were like in terms of coming into your trial as depressed young men?
Dr. Bayes: In the context of the COVID-19 pandemic, mental health was at the forefront of many people’s minds. They joined the study saying, “I’ve never seen anything like this before. I’ve never seen myself represented in research. I wanted to contribute. I want to add to that conversation because I feel like we are overlooked.”
Dr. Ramsey: I love hearing this notion that maybe young men aren’t quite who we think they are. They are wanting to be seen around their mental health. They can learn to use olive oil and to cook, and they can engage in mental health interventions that work. We just need to ask, give them some food, encourage them, and it makes a big difference.
Jessica Bayes, thank you so much for joining us and sharing some of your research. Everyone, it’s the AMMEND trial. We will drop a link to the trial below so you can take a peek and tell us what you think.
Please, in the comments, let us know what you think about this notion of helping young men with depression through nutritional interventions. Take a peek at the great work that Jessica and Professor Sibbritt from the University of Technology Sydney have published and put out into the scientific literature for us all.
Thanks so much, Jessica. I look forward to seeing you soon.
Dr. Bayes: Thank you.
Dr. Ramsey is assistant clinical professor, department of psychiatry, Columbia University, New York. He has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for InterContinental Hotels Group; National Kale Day 501(c)3. Received income in an amount equal to or greater than $250 from: Sharecare. Dr. Bayes is a postdoctoral research fellow; clinical nutritionist, Southern Cross University, National Center for Naturopathic Medicine, Lismore, New South Wales, Australia. She has disclosed the following relevant financial relationships: Received research grant from Endeavour College. A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Drew Ramsey, MD: Welcome back, everyone. I’m Dr. Drew Ramsey. I’m on the editorial board with Medscape Psychiatry and I’m an assistant clinical professor of psychiatry at Columbia University. We have a special guest today.
I’m here with nutritionist Jessica Bayes, who’s at the University of Technology Sydney, and she’s the lead author of the AMMEND trial. [Editor’s note: Since completing her PhD, Bayes is now at Southern Cross University.]
Jessica, welcome to Medscape.
Jessica Bayes, PhD: Thank you for having me.
The AMMEND Trial
Dr. Ramsey: Thank you for coming on board and helping all of us as clinicians understand some of your research and some of what is suggested by your research – that young men can change their diet and it helped their depression. Tell us a little bit about the AMMEND trial.
Dr. Bayes: The AMMEND trial was a 12-week randomized controlled trial in young men, 18-25 years old, who had diagnosed moderate to severe clinical depression. They had a poor baseline diet and we got them to eat a healthy Mediterranean diet, which improved their symptoms of depression.
Dr. Ramsey: It was a remarkable trial. Jessica, if I recall, you helped individuals improve the Mediterranean dietary pattern score by 8 points on a 14-point scale. That led to a 20-point reduction in their Beck Depression Inventory. Tell us what that looked like on the ground.
Dr. Bayes: It’s a huge improvement. Obviously, they were feeling much better in the end in terms of their depressive symptoms, but we also measured their energy, sleep, and quality of life. Many of them at the end were at a score cutoff that suggests no depression or in remission.
Dr. Ramsey: There were 72 people in your total trial, so 36% in your intervention arm went into full remission.
Dr. Bayes: Which is just amazing.
Dr. Ramsey: It also follows up the SMILES trial, which was a little bit of a different trial. You had two nutritional counseling sessions and the SMILES trial had seven, but in the SMILES trial, 32.3% of the patients went into full remission when they adopted a Mediterranean-style diet.
Jessica, what is the secret that you and your team know? I think many clinicians, especially clinicians who are parents and have teens, are kind of shaking their heads in disbelief. They’ve been telling their kids to eat healthy. What do you guys know about how to help young men change their diet?
How to Aid Adherence to Mediterranean Diet
Dr. Bayes: Prior to starting this, when I would say this idea to people, everyone would say, “Great idea. There’s no way you’re going to get depressed young men to change their diet. Not going to happen.” We went to them and we asked them. We said, “We’re going to do this study. What do you want from us? What resources would you need? How many appointments would you like? What’s too little or too many?”
We really got their feedback on board when we designed the study, and that obviously paid off. We had a personalized approach and we met them where they were at. We gave them the skills, resources, recipes, meal ideas – all those things – so we could really set them up to succeed.
Dr. Ramsey: You were telling me earlier about a few of the dietary changes that you felt made a big difference for these young men. What were those?
Dr. Bayes: Increasing the vegetables, olive oil, and legumes are probably the big ones that most of them were really not doing beforehand. They were really able to take that on board and make significant improvements in those areas.
Dr. Ramsey: These are really some of the top food categories in nutritional psychiatry as we think about how we help our efforts to improve mental health by thinking about nutrition, nutritional quality, and nutritional density. Certainly, those food categories – nuts and legumes, plants, and olive oil – are really what help get us there.
You also gave the students a food hamper. If you were going to be in charge of mental health in Australia and America and you got to give every college freshman a little box with a note, what would be in that box?
Dr. Bayes: I’d want to put everything in that box! It would be full of brightly colored fruits and vegetables, different nuts and seeds, and legumes. It would be full of recipes and ideas of how to cook things and how to prepare really delicious things. It would be full of different herbs and spices and all of those things to get people really excited about food.
Dr. Ramsey: Did the young men pick up on your enthusiasm and excitement around food? Did they begin to adopt some of that, shifting their view of how they saw the food and how they saw that it is related to their depression?
Dr. Bayes: Hopefully. I do think energy is infectious. I’m sure that played a role somewhat, but trying to get them excited about food can be really quite daunting, thinking, I’ve got to change my entire diet and I’ve got to learn to cook and go out and buy groceries. I don’t even know what to do with a piece of salmon. Trying to get them curious, interested, and just reminding them that it’s not all-or-nothing. Make small changes, give it a go, and have fun.
Dr. Ramsey: You also have a unique aspect of your research that you’re interested in male mental health, and that’s not something that’s been widely researched. Can you tell us a little bit about what these men were like in terms of coming into your trial as depressed young men?
Dr. Bayes: In the context of the COVID-19 pandemic, mental health was at the forefront of many people’s minds. They joined the study saying, “I’ve never seen anything like this before. I’ve never seen myself represented in research. I wanted to contribute. I want to add to that conversation because I feel like we are overlooked.”
Dr. Ramsey: I love hearing this notion that maybe young men aren’t quite who we think they are. They are wanting to be seen around their mental health. They can learn to use olive oil and to cook, and they can engage in mental health interventions that work. We just need to ask, give them some food, encourage them, and it makes a big difference.
Jessica Bayes, thank you so much for joining us and sharing some of your research. Everyone, it’s the AMMEND trial. We will drop a link to the trial below so you can take a peek and tell us what you think.
Please, in the comments, let us know what you think about this notion of helping young men with depression through nutritional interventions. Take a peek at the great work that Jessica and Professor Sibbritt from the University of Technology Sydney have published and put out into the scientific literature for us all.
Thanks so much, Jessica. I look forward to seeing you soon.
Dr. Bayes: Thank you.
Dr. Ramsey is assistant clinical professor, department of psychiatry, Columbia University, New York. He has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for InterContinental Hotels Group; National Kale Day 501(c)3. Received income in an amount equal to or greater than $250 from: Sharecare. Dr. Bayes is a postdoctoral research fellow; clinical nutritionist, Southern Cross University, National Center for Naturopathic Medicine, Lismore, New South Wales, Australia. She has disclosed the following relevant financial relationships: Received research grant from Endeavour College. A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Drew Ramsey, MD: Welcome back, everyone. I’m Dr. Drew Ramsey. I’m on the editorial board with Medscape Psychiatry and I’m an assistant clinical professor of psychiatry at Columbia University. We have a special guest today.
I’m here with nutritionist Jessica Bayes, who’s at the University of Technology Sydney, and she’s the lead author of the AMMEND trial. [Editor’s note: Since completing her PhD, Bayes is now at Southern Cross University.]
Jessica, welcome to Medscape.
Jessica Bayes, PhD: Thank you for having me.
The AMMEND Trial
Dr. Ramsey: Thank you for coming on board and helping all of us as clinicians understand some of your research and some of what is suggested by your research – that young men can change their diet and it helped their depression. Tell us a little bit about the AMMEND trial.
Dr. Bayes: The AMMEND trial was a 12-week randomized controlled trial in young men, 18-25 years old, who had diagnosed moderate to severe clinical depression. They had a poor baseline diet and we got them to eat a healthy Mediterranean diet, which improved their symptoms of depression.
Dr. Ramsey: It was a remarkable trial. Jessica, if I recall, you helped individuals improve the Mediterranean dietary pattern score by 8 points on a 14-point scale. That led to a 20-point reduction in their Beck Depression Inventory. Tell us what that looked like on the ground.
Dr. Bayes: It’s a huge improvement. Obviously, they were feeling much better in the end in terms of their depressive symptoms, but we also measured their energy, sleep, and quality of life. Many of them at the end were at a score cutoff that suggests no depression or in remission.
Dr. Ramsey: There were 72 people in your total trial, so 36% in your intervention arm went into full remission.
Dr. Bayes: Which is just amazing.
Dr. Ramsey: It also follows up the SMILES trial, which was a little bit of a different trial. You had two nutritional counseling sessions and the SMILES trial had seven, but in the SMILES trial, 32.3% of the patients went into full remission when they adopted a Mediterranean-style diet.
Jessica, what is the secret that you and your team know? I think many clinicians, especially clinicians who are parents and have teens, are kind of shaking their heads in disbelief. They’ve been telling their kids to eat healthy. What do you guys know about how to help young men change their diet?
How to Aid Adherence to Mediterranean Diet
Dr. Bayes: Prior to starting this, when I would say this idea to people, everyone would say, “Great idea. There’s no way you’re going to get depressed young men to change their diet. Not going to happen.” We went to them and we asked them. We said, “We’re going to do this study. What do you want from us? What resources would you need? How many appointments would you like? What’s too little or too many?”
We really got their feedback on board when we designed the study, and that obviously paid off. We had a personalized approach and we met them where they were at. We gave them the skills, resources, recipes, meal ideas – all those things – so we could really set them up to succeed.
Dr. Ramsey: You were telling me earlier about a few of the dietary changes that you felt made a big difference for these young men. What were those?
Dr. Bayes: Increasing the vegetables, olive oil, and legumes are probably the big ones that most of them were really not doing beforehand. They were really able to take that on board and make significant improvements in those areas.
Dr. Ramsey: These are really some of the top food categories in nutritional psychiatry as we think about how we help our efforts to improve mental health by thinking about nutrition, nutritional quality, and nutritional density. Certainly, those food categories – nuts and legumes, plants, and olive oil – are really what help get us there.
You also gave the students a food hamper. If you were going to be in charge of mental health in Australia and America and you got to give every college freshman a little box with a note, what would be in that box?
Dr. Bayes: I’d want to put everything in that box! It would be full of brightly colored fruits and vegetables, different nuts and seeds, and legumes. It would be full of recipes and ideas of how to cook things and how to prepare really delicious things. It would be full of different herbs and spices and all of those things to get people really excited about food.
Dr. Ramsey: Did the young men pick up on your enthusiasm and excitement around food? Did they begin to adopt some of that, shifting their view of how they saw the food and how they saw that it is related to their depression?
Dr. Bayes: Hopefully. I do think energy is infectious. I’m sure that played a role somewhat, but trying to get them excited about food can be really quite daunting, thinking, I’ve got to change my entire diet and I’ve got to learn to cook and go out and buy groceries. I don’t even know what to do with a piece of salmon. Trying to get them curious, interested, and just reminding them that it’s not all-or-nothing. Make small changes, give it a go, and have fun.
Dr. Ramsey: You also have a unique aspect of your research that you’re interested in male mental health, and that’s not something that’s been widely researched. Can you tell us a little bit about what these men were like in terms of coming into your trial as depressed young men?
Dr. Bayes: In the context of the COVID-19 pandemic, mental health was at the forefront of many people’s minds. They joined the study saying, “I’ve never seen anything like this before. I’ve never seen myself represented in research. I wanted to contribute. I want to add to that conversation because I feel like we are overlooked.”
Dr. Ramsey: I love hearing this notion that maybe young men aren’t quite who we think they are. They are wanting to be seen around their mental health. They can learn to use olive oil and to cook, and they can engage in mental health interventions that work. We just need to ask, give them some food, encourage them, and it makes a big difference.
Jessica Bayes, thank you so much for joining us and sharing some of your research. Everyone, it’s the AMMEND trial. We will drop a link to the trial below so you can take a peek and tell us what you think.
Please, in the comments, let us know what you think about this notion of helping young men with depression through nutritional interventions. Take a peek at the great work that Jessica and Professor Sibbritt from the University of Technology Sydney have published and put out into the scientific literature for us all.
Thanks so much, Jessica. I look forward to seeing you soon.
Dr. Bayes: Thank you.
Dr. Ramsey is assistant clinical professor, department of psychiatry, Columbia University, New York. He has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for InterContinental Hotels Group; National Kale Day 501(c)3. Received income in an amount equal to or greater than $250 from: Sharecare. Dr. Bayes is a postdoctoral research fellow; clinical nutritionist, Southern Cross University, National Center for Naturopathic Medicine, Lismore, New South Wales, Australia. She has disclosed the following relevant financial relationships: Received research grant from Endeavour College. A version of this article first appeared on Medscape.com.
Noninvasive laser therapy tied to improved short-term memory
Investigators compared the effect of 1,064 nm of tPBM delivered over a 12-minute session to the right PFC vs. three other treatment arms: delivery of the same intervention to the left PFC, delivery of the intervention at a lower frequency, and a sham intervention.
All participants were shown a series of items prior to the intervention and asked to recall them after the intervention. Those who received tPBM 1,064 nm to the right PFC showed a superior performance of up to 25% in the memory tasks compared with the other groups.
Patients with attention-related conditions, such as attention deficit hyperactivity disorder, “could benefit from this type of treatment, which is safe, simple, and noninvasive, with no side effects,” coinvestigator Dongwei Li, a visiting PhD student at the Centre for Human Brain Health, University of Birmingham, England, said in a news release.
The findings were published online in Science Advances.
Differing wavelengths
The researchers note that “in the past decades,” noninvasive brain stimulation technology using transcranial application of direct or alternating electrical or magnetic fields “has been proven to be useful” in the improvement of working memory (WM).
When applied to the right PFC, tPBM has been shown to improve accuracy and speed of reaction time in WM tasks and improvements in “high-order cognitive functions,” such as sustained attention, emotion, and executive functions.
The investigators wanted to assess the impact of tPBM applied to different parts of the brain and at different wavelengths. They conducted four double-blind, sham-controlled experiments encompassing 90 neurotypical college students (mean age, 22 years). Each student participated in only one of the four experiments.
All completed two different tPBM sessions, separated by a week, in which sham and active tPBM were compared. Two different types of change-detection memory tasks were given: one requiring participants to remember the orientation of a series of items before and after the intervention and one other requiring them to remember the color of the items (experiments 1 and 2).
A series of follow-up experiments focused on comparing different wavelengths (1,064 nm vs. 852 nm) and different stimulation sites (right vs. left PFC; experiments 3 and 4).
EEG recordings were obtained during the intervention and the memory tasks.
Each experiment consisted of one active tPBM session and one sham tPBM session, with sessions consisting of 12 minutes of laser light (or sham) intervention. These sessions were conducted on the first and the seventh day; then, on the eighth day, participants were asked to report (or guess) which session was the active tPBM session.
Stimulating astrocytes
Results showed that, compared with sham tPBM, there was an improvement in WM capacity and scores by the 1,064 nm intervention in the orientation as well as the color task.
Participants who received the targeted treatment were able to remember between four and five test objects, whereas those with the treatment variations were only able to remember between three and four objects.
“These results support the hypothesis that 1,064 nm tPBM on the right PFC enhances WM capacity,” the investigators wrote.
They also found improvements in WM in participants receiving tPBM vs. sham regardless of whether their performance in the WM task was at a low or high level. This finding held true in both the orientation and the color tasks.
“Therefore, participants with good and poor WM capacity improved after 1,064 nm tPBM,” the researchers noted.
In addition, participants were unable to guess or report whether they had received sham or active tPBM.
EEG monitoring showed changes in brain activity that predicted the improvements in memory performance. In particular, 1,064 tPBM applied to the right PFC increased occipitoparietal contralateral delay activity (CDA), with CDA mediating the WM improvement.
This is “consistent with previous research that CDA is indicative of the number of maintained objects in visual working memory,” the investigators wrote.
Pearson correlation analyses showed that the differences in CDA set-size effects between active and sham session “correlated positively” with the behavioral differences between these sessions. For the orientation task, the r was 0.446 (P < .04); and for the color task, the r was .563 (P < .02).
No similar improvements were found with the 852 nm tPBM.
“We need further research to understand exactly why the tPBM is having this positive effect,” coinvestigator Ole Jensen, PhD, professor in translational neuroscience and codirector of the Centre for Human Brain Health, said in the release.
“It’s possible that the light is stimulating the astrocytes – the powerplants – in the nerve cells within the PFC, and this has a positive effect on the cells’ efficiency,” he noted.
Dr. Jensen added that his team “will also be investigating how long the effects might last. Clearly, if these experiments are to lead to a clinical intervention, we will need to see long-lasting benefits.”
Beneficial cognitive, emotional effects
Commenting for this news organization, Francisco Gonzalez-Lima, PhD, professor in the department of psychology, University of Texas at Austin, called the study “well done.”
Dr. Gonzalez-Lima was one of the first researchers to demonstrate that 1,064 nm transcranial infrared laser stimulation “produces beneficial cognitive and emotional effects in humans, including improving visual working memory,” he said.
The current study “reported an additional brain effect linked to the improved visual working memory that consists of an EEG-derived response, which is a new finding,” noted Dr. Gonzales-Lima, who was not involved with the new research.
He added that the same laser method “has been found by the Gonzalez-Lima lab to be effective at improving cognition in older adults and depressed and bipolar patients.”
The study was supported by the National Natural Science Foundation of China, the Ministry of Science and Technology of the People’s Republic of China, and the National Defence Basic Scientific Research Program of China. The investigators and Dr. Gonzalez-Lima report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Investigators compared the effect of 1,064 nm of tPBM delivered over a 12-minute session to the right PFC vs. three other treatment arms: delivery of the same intervention to the left PFC, delivery of the intervention at a lower frequency, and a sham intervention.
All participants were shown a series of items prior to the intervention and asked to recall them after the intervention. Those who received tPBM 1,064 nm to the right PFC showed a superior performance of up to 25% in the memory tasks compared with the other groups.
Patients with attention-related conditions, such as attention deficit hyperactivity disorder, “could benefit from this type of treatment, which is safe, simple, and noninvasive, with no side effects,” coinvestigator Dongwei Li, a visiting PhD student at the Centre for Human Brain Health, University of Birmingham, England, said in a news release.
The findings were published online in Science Advances.
Differing wavelengths
The researchers note that “in the past decades,” noninvasive brain stimulation technology using transcranial application of direct or alternating electrical or magnetic fields “has been proven to be useful” in the improvement of working memory (WM).
When applied to the right PFC, tPBM has been shown to improve accuracy and speed of reaction time in WM tasks and improvements in “high-order cognitive functions,” such as sustained attention, emotion, and executive functions.
The investigators wanted to assess the impact of tPBM applied to different parts of the brain and at different wavelengths. They conducted four double-blind, sham-controlled experiments encompassing 90 neurotypical college students (mean age, 22 years). Each student participated in only one of the four experiments.
All completed two different tPBM sessions, separated by a week, in which sham and active tPBM were compared. Two different types of change-detection memory tasks were given: one requiring participants to remember the orientation of a series of items before and after the intervention and one other requiring them to remember the color of the items (experiments 1 and 2).
A series of follow-up experiments focused on comparing different wavelengths (1,064 nm vs. 852 nm) and different stimulation sites (right vs. left PFC; experiments 3 and 4).
EEG recordings were obtained during the intervention and the memory tasks.
Each experiment consisted of one active tPBM session and one sham tPBM session, with sessions consisting of 12 minutes of laser light (or sham) intervention. These sessions were conducted on the first and the seventh day; then, on the eighth day, participants were asked to report (or guess) which session was the active tPBM session.
Stimulating astrocytes
Results showed that, compared with sham tPBM, there was an improvement in WM capacity and scores by the 1,064 nm intervention in the orientation as well as the color task.
Participants who received the targeted treatment were able to remember between four and five test objects, whereas those with the treatment variations were only able to remember between three and four objects.
“These results support the hypothesis that 1,064 nm tPBM on the right PFC enhances WM capacity,” the investigators wrote.
They also found improvements in WM in participants receiving tPBM vs. sham regardless of whether their performance in the WM task was at a low or high level. This finding held true in both the orientation and the color tasks.
“Therefore, participants with good and poor WM capacity improved after 1,064 nm tPBM,” the researchers noted.
In addition, participants were unable to guess or report whether they had received sham or active tPBM.
EEG monitoring showed changes in brain activity that predicted the improvements in memory performance. In particular, 1,064 tPBM applied to the right PFC increased occipitoparietal contralateral delay activity (CDA), with CDA mediating the WM improvement.
This is “consistent with previous research that CDA is indicative of the number of maintained objects in visual working memory,” the investigators wrote.
Pearson correlation analyses showed that the differences in CDA set-size effects between active and sham session “correlated positively” with the behavioral differences between these sessions. For the orientation task, the r was 0.446 (P < .04); and for the color task, the r was .563 (P < .02).
No similar improvements were found with the 852 nm tPBM.
“We need further research to understand exactly why the tPBM is having this positive effect,” coinvestigator Ole Jensen, PhD, professor in translational neuroscience and codirector of the Centre for Human Brain Health, said in the release.
“It’s possible that the light is stimulating the astrocytes – the powerplants – in the nerve cells within the PFC, and this has a positive effect on the cells’ efficiency,” he noted.
Dr. Jensen added that his team “will also be investigating how long the effects might last. Clearly, if these experiments are to lead to a clinical intervention, we will need to see long-lasting benefits.”
Beneficial cognitive, emotional effects
Commenting for this news organization, Francisco Gonzalez-Lima, PhD, professor in the department of psychology, University of Texas at Austin, called the study “well done.”
Dr. Gonzalez-Lima was one of the first researchers to demonstrate that 1,064 nm transcranial infrared laser stimulation “produces beneficial cognitive and emotional effects in humans, including improving visual working memory,” he said.
The current study “reported an additional brain effect linked to the improved visual working memory that consists of an EEG-derived response, which is a new finding,” noted Dr. Gonzales-Lima, who was not involved with the new research.
He added that the same laser method “has been found by the Gonzalez-Lima lab to be effective at improving cognition in older adults and depressed and bipolar patients.”
The study was supported by the National Natural Science Foundation of China, the Ministry of Science and Technology of the People’s Republic of China, and the National Defence Basic Scientific Research Program of China. The investigators and Dr. Gonzalez-Lima report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Investigators compared the effect of 1,064 nm of tPBM delivered over a 12-minute session to the right PFC vs. three other treatment arms: delivery of the same intervention to the left PFC, delivery of the intervention at a lower frequency, and a sham intervention.
All participants were shown a series of items prior to the intervention and asked to recall them after the intervention. Those who received tPBM 1,064 nm to the right PFC showed a superior performance of up to 25% in the memory tasks compared with the other groups.
Patients with attention-related conditions, such as attention deficit hyperactivity disorder, “could benefit from this type of treatment, which is safe, simple, and noninvasive, with no side effects,” coinvestigator Dongwei Li, a visiting PhD student at the Centre for Human Brain Health, University of Birmingham, England, said in a news release.
The findings were published online in Science Advances.
Differing wavelengths
The researchers note that “in the past decades,” noninvasive brain stimulation technology using transcranial application of direct or alternating electrical or magnetic fields “has been proven to be useful” in the improvement of working memory (WM).
When applied to the right PFC, tPBM has been shown to improve accuracy and speed of reaction time in WM tasks and improvements in “high-order cognitive functions,” such as sustained attention, emotion, and executive functions.
The investigators wanted to assess the impact of tPBM applied to different parts of the brain and at different wavelengths. They conducted four double-blind, sham-controlled experiments encompassing 90 neurotypical college students (mean age, 22 years). Each student participated in only one of the four experiments.
All completed two different tPBM sessions, separated by a week, in which sham and active tPBM were compared. Two different types of change-detection memory tasks were given: one requiring participants to remember the orientation of a series of items before and after the intervention and one other requiring them to remember the color of the items (experiments 1 and 2).
A series of follow-up experiments focused on comparing different wavelengths (1,064 nm vs. 852 nm) and different stimulation sites (right vs. left PFC; experiments 3 and 4).
EEG recordings were obtained during the intervention and the memory tasks.
Each experiment consisted of one active tPBM session and one sham tPBM session, with sessions consisting of 12 minutes of laser light (or sham) intervention. These sessions were conducted on the first and the seventh day; then, on the eighth day, participants were asked to report (or guess) which session was the active tPBM session.
Stimulating astrocytes
Results showed that, compared with sham tPBM, there was an improvement in WM capacity and scores by the 1,064 nm intervention in the orientation as well as the color task.
Participants who received the targeted treatment were able to remember between four and five test objects, whereas those with the treatment variations were only able to remember between three and four objects.
“These results support the hypothesis that 1,064 nm tPBM on the right PFC enhances WM capacity,” the investigators wrote.
They also found improvements in WM in participants receiving tPBM vs. sham regardless of whether their performance in the WM task was at a low or high level. This finding held true in both the orientation and the color tasks.
“Therefore, participants with good and poor WM capacity improved after 1,064 nm tPBM,” the researchers noted.
In addition, participants were unable to guess or report whether they had received sham or active tPBM.
EEG monitoring showed changes in brain activity that predicted the improvements in memory performance. In particular, 1,064 tPBM applied to the right PFC increased occipitoparietal contralateral delay activity (CDA), with CDA mediating the WM improvement.
This is “consistent with previous research that CDA is indicative of the number of maintained objects in visual working memory,” the investigators wrote.
Pearson correlation analyses showed that the differences in CDA set-size effects between active and sham session “correlated positively” with the behavioral differences between these sessions. For the orientation task, the r was 0.446 (P < .04); and for the color task, the r was .563 (P < .02).
No similar improvements were found with the 852 nm tPBM.
“We need further research to understand exactly why the tPBM is having this positive effect,” coinvestigator Ole Jensen, PhD, professor in translational neuroscience and codirector of the Centre for Human Brain Health, said in the release.
“It’s possible that the light is stimulating the astrocytes – the powerplants – in the nerve cells within the PFC, and this has a positive effect on the cells’ efficiency,” he noted.
Dr. Jensen added that his team “will also be investigating how long the effects might last. Clearly, if these experiments are to lead to a clinical intervention, we will need to see long-lasting benefits.”
Beneficial cognitive, emotional effects
Commenting for this news organization, Francisco Gonzalez-Lima, PhD, professor in the department of psychology, University of Texas at Austin, called the study “well done.”
Dr. Gonzalez-Lima was one of the first researchers to demonstrate that 1,064 nm transcranial infrared laser stimulation “produces beneficial cognitive and emotional effects in humans, including improving visual working memory,” he said.
The current study “reported an additional brain effect linked to the improved visual working memory that consists of an EEG-derived response, which is a new finding,” noted Dr. Gonzales-Lima, who was not involved with the new research.
He added that the same laser method “has been found by the Gonzalez-Lima lab to be effective at improving cognition in older adults and depressed and bipolar patients.”
The study was supported by the National Natural Science Foundation of China, the Ministry of Science and Technology of the People’s Republic of China, and the National Defence Basic Scientific Research Program of China. The investigators and Dr. Gonzalez-Lima report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM SCIENCE ADVANCES