User login
Cardiology News is an independent news source that provides cardiologists with timely and relevant news and commentary about clinical developments and the impact of health care policy on cardiology and the cardiologist's practice. Cardiology News Digital Network is the online destination and multimedia properties of Cardiology News, the independent news publication for cardiologists. Cardiology news is the leading source of news and commentary about clinical developments in cardiology as well as health care policy and regulations that affect the cardiologist's practice. Cardiology News Digital Network is owned by Frontline Medical Communications.
Proposed HIPAA overhaul to ease access to patient health info
The Department of Health & Human Services is proposing an overhaul of HIPAA that will make it easier to access patients’ personal health information, including the health records of patients with mental illness. The proposal would also do away with the requirement that all patients sign a notice of privacy practices.
The changes are contained in a 357-page proposed rule, which was unveiled by federal officials Dec. 10. Roger Severino, director of HHS’ Office for Civil Rights, said in a briefing that the sweeping proposal would empower patients, reduce the administrative burden for health care providers, and pave the way to better-coordinated care.
HHS estimated that the rule could save $3.2 billion over 5 years, but it’s not clear how much of that would accrue to clinical practices.
The most obvious cost-saving aspect for medical and dental practices is the proposal that practitioners would no longer have to provide and collect signed notifications of privacy practices.
“This has been a tremendous waste of time and effort and has caused massive confusion,” said Mr. Severino. He said some patients thought they were waiving privacy rights and that, in some cases, physicians refused to administer care unless patients signed the notices. “That was never the intent.”
Requiring that patients sign the form and that practices keep copies for 6 years is an “unnecessary burden,” said Mr. Severino. “We’ve lost whole forests from this regulation.”
Under the new proposal, health care providers would merely have to let patients know where to find their privacy policies.
Sharing mental health info
The rule would also ease the standard for sharing information about a patient who is in a mental health crisis, such as an exacerbation of a serious mental illness or a crisis related to a substance use disorder, including an overdose.
Currently, clinicians can choose to disclose protected health information – to a family member, a caregiver, a law enforcement official, a doctor, or an insurer – if they believe that doing so is advisable in their “professional judgment.” The rule proposes to ease that to a “good faith” belief that a disclosure would be in the best interest of the patient. In both instances, the patient can still object and block the disclosure.
As an example, HHS said that, in the case of a young adult who had experienced an overdose of opioids, a licensed health care professional could make the determination to “disclose relevant information to a parent who is involved in the patient’s treatment and who the young adult would expect, based on their relationship, to participate in or be involved with the patient’s recovery from the overdose.”
HHS is also proposing to let clinicians disclose information in cases in which an individual might be a threat to himself or others, provided the harm is “serious and reasonably foreseeable.”
Currently, information can only be disclosed if it appears there is a “serious and imminent” threat to health or safety. If an individual experienced suicidal ideation, for instance, a health care professional could notify family that the individual is at risk.
Fast, no-cost access
The rule also aims to make it easier for patients to get access to their own health care information quickly – within 15 days of a request – instead of the 30 days currently allowed, and sometimes at no cost.
The 30-day time frame is “a relic of a pre-Internet age that should be dispensed with,” said Mr. Severino.
Patients can also request that a treating physician get his or her records from a clinician who had previously treated the individual. The request would be fulfilled within 15 days, although extensions might be possible.
“That takes away the burden of coordination from the patient and puts it on those parties that are responsible for the actual provision of care and that are better positioned to do that coordination,” Mr. Severino said.
Health care professionals will also have to share with patients a fee schedule for records requests. However, if records are shared through a patient portal with view, download, and transmit capabilities, the provider can’t charge the patient for the time it took to upload the information into the system.
“We do not believe a patient’s personal medical record should be profit centers for providers,” Mr. Severino said.
Patients will be allowed to take photos with a smartphone of personal health information – such as an x-ray or sonogram – while receiving care.
The rule is open for public comment until mid-February. After that, it will become final in 180 days. The agency said it would not begin enforcement until 240 days after the final rule was published.
A version of this article originally appeared on Medscape.com.
The Department of Health & Human Services is proposing an overhaul of HIPAA that will make it easier to access patients’ personal health information, including the health records of patients with mental illness. The proposal would also do away with the requirement that all patients sign a notice of privacy practices.
The changes are contained in a 357-page proposed rule, which was unveiled by federal officials Dec. 10. Roger Severino, director of HHS’ Office for Civil Rights, said in a briefing that the sweeping proposal would empower patients, reduce the administrative burden for health care providers, and pave the way to better-coordinated care.
HHS estimated that the rule could save $3.2 billion over 5 years, but it’s not clear how much of that would accrue to clinical practices.
The most obvious cost-saving aspect for medical and dental practices is the proposal that practitioners would no longer have to provide and collect signed notifications of privacy practices.
“This has been a tremendous waste of time and effort and has caused massive confusion,” said Mr. Severino. He said some patients thought they were waiving privacy rights and that, in some cases, physicians refused to administer care unless patients signed the notices. “That was never the intent.”
Requiring that patients sign the form and that practices keep copies for 6 years is an “unnecessary burden,” said Mr. Severino. “We’ve lost whole forests from this regulation.”
Under the new proposal, health care providers would merely have to let patients know where to find their privacy policies.
Sharing mental health info
The rule would also ease the standard for sharing information about a patient who is in a mental health crisis, such as an exacerbation of a serious mental illness or a crisis related to a substance use disorder, including an overdose.
Currently, clinicians can choose to disclose protected health information – to a family member, a caregiver, a law enforcement official, a doctor, or an insurer – if they believe that doing so is advisable in their “professional judgment.” The rule proposes to ease that to a “good faith” belief that a disclosure would be in the best interest of the patient. In both instances, the patient can still object and block the disclosure.
As an example, HHS said that, in the case of a young adult who had experienced an overdose of opioids, a licensed health care professional could make the determination to “disclose relevant information to a parent who is involved in the patient’s treatment and who the young adult would expect, based on their relationship, to participate in or be involved with the patient’s recovery from the overdose.”
HHS is also proposing to let clinicians disclose information in cases in which an individual might be a threat to himself or others, provided the harm is “serious and reasonably foreseeable.”
Currently, information can only be disclosed if it appears there is a “serious and imminent” threat to health or safety. If an individual experienced suicidal ideation, for instance, a health care professional could notify family that the individual is at risk.
Fast, no-cost access
The rule also aims to make it easier for patients to get access to their own health care information quickly – within 15 days of a request – instead of the 30 days currently allowed, and sometimes at no cost.
The 30-day time frame is “a relic of a pre-Internet age that should be dispensed with,” said Mr. Severino.
Patients can also request that a treating physician get his or her records from a clinician who had previously treated the individual. The request would be fulfilled within 15 days, although extensions might be possible.
“That takes away the burden of coordination from the patient and puts it on those parties that are responsible for the actual provision of care and that are better positioned to do that coordination,” Mr. Severino said.
Health care professionals will also have to share with patients a fee schedule for records requests. However, if records are shared through a patient portal with view, download, and transmit capabilities, the provider can’t charge the patient for the time it took to upload the information into the system.
“We do not believe a patient’s personal medical record should be profit centers for providers,” Mr. Severino said.
Patients will be allowed to take photos with a smartphone of personal health information – such as an x-ray or sonogram – while receiving care.
The rule is open for public comment until mid-February. After that, it will become final in 180 days. The agency said it would not begin enforcement until 240 days after the final rule was published.
A version of this article originally appeared on Medscape.com.
The Department of Health & Human Services is proposing an overhaul of HIPAA that will make it easier to access patients’ personal health information, including the health records of patients with mental illness. The proposal would also do away with the requirement that all patients sign a notice of privacy practices.
The changes are contained in a 357-page proposed rule, which was unveiled by federal officials Dec. 10. Roger Severino, director of HHS’ Office for Civil Rights, said in a briefing that the sweeping proposal would empower patients, reduce the administrative burden for health care providers, and pave the way to better-coordinated care.
HHS estimated that the rule could save $3.2 billion over 5 years, but it’s not clear how much of that would accrue to clinical practices.
The most obvious cost-saving aspect for medical and dental practices is the proposal that practitioners would no longer have to provide and collect signed notifications of privacy practices.
“This has been a tremendous waste of time and effort and has caused massive confusion,” said Mr. Severino. He said some patients thought they were waiving privacy rights and that, in some cases, physicians refused to administer care unless patients signed the notices. “That was never the intent.”
Requiring that patients sign the form and that practices keep copies for 6 years is an “unnecessary burden,” said Mr. Severino. “We’ve lost whole forests from this regulation.”
Under the new proposal, health care providers would merely have to let patients know where to find their privacy policies.
Sharing mental health info
The rule would also ease the standard for sharing information about a patient who is in a mental health crisis, such as an exacerbation of a serious mental illness or a crisis related to a substance use disorder, including an overdose.
Currently, clinicians can choose to disclose protected health information – to a family member, a caregiver, a law enforcement official, a doctor, or an insurer – if they believe that doing so is advisable in their “professional judgment.” The rule proposes to ease that to a “good faith” belief that a disclosure would be in the best interest of the patient. In both instances, the patient can still object and block the disclosure.
As an example, HHS said that, in the case of a young adult who had experienced an overdose of opioids, a licensed health care professional could make the determination to “disclose relevant information to a parent who is involved in the patient’s treatment and who the young adult would expect, based on their relationship, to participate in or be involved with the patient’s recovery from the overdose.”
HHS is also proposing to let clinicians disclose information in cases in which an individual might be a threat to himself or others, provided the harm is “serious and reasonably foreseeable.”
Currently, information can only be disclosed if it appears there is a “serious and imminent” threat to health or safety. If an individual experienced suicidal ideation, for instance, a health care professional could notify family that the individual is at risk.
Fast, no-cost access
The rule also aims to make it easier for patients to get access to their own health care information quickly – within 15 days of a request – instead of the 30 days currently allowed, and sometimes at no cost.
The 30-day time frame is “a relic of a pre-Internet age that should be dispensed with,” said Mr. Severino.
Patients can also request that a treating physician get his or her records from a clinician who had previously treated the individual. The request would be fulfilled within 15 days, although extensions might be possible.
“That takes away the burden of coordination from the patient and puts it on those parties that are responsible for the actual provision of care and that are better positioned to do that coordination,” Mr. Severino said.
Health care professionals will also have to share with patients a fee schedule for records requests. However, if records are shared through a patient portal with view, download, and transmit capabilities, the provider can’t charge the patient for the time it took to upload the information into the system.
“We do not believe a patient’s personal medical record should be profit centers for providers,” Mr. Severino said.
Patients will be allowed to take photos with a smartphone of personal health information – such as an x-ray or sonogram – while receiving care.
The rule is open for public comment until mid-February. After that, it will become final in 180 days. The agency said it would not begin enforcement until 240 days after the final rule was published.
A version of this article originally appeared on Medscape.com.
ADA 2021 standards address financial hardship in diabetes
For 2021, the American Diabetes Association offers new guidance on assessing patients’ financial and social barriers to care, especially given the COVID-19 pandemic, individualizing treatment of patients with type 2 diabetes, and use of diabetes technology.
As it does every year, the annual update incorporates new clinical information that has become available since the last guideline, with occasional revisions during the year as needed. “Standards of Medical Care in Diabetes – 2021,” was published online as a supplement to Diabetes Care.
The new standards advise that patients be assessed for food and housing insecurity, social support, and “cost-related medication nonadherence,” and those found to have difficulty referred to appropriate community resources.
“Clinicians need to be sensitive to the fact that patients may have very good reasons for not taking their medication, [as in] if they can’t afford it,” ADA chief science & medical officer Robert A. Gabbay, MD, PhD, said in an interview.
Dr. Gabbay noted that “a heightened awareness” of social determinants of health is weaved throughout the 2021 standards because of the pandemic, with information on the topic derived from a July 2020 joint consensus statement in Diabetes Care, endorsed by a number of other societies, as well as a November publication also in Diabetes Care.
“We made several recommendations that speak to social determinants of health, placing an emphasis on engaging in conversations around this subject and screening for related issues such as food insecurity that weren’t there previously,” he said.
“Screening tools are suggested. It helped us to have an in-depth scientific review of the literature to know the prevalence of this in people with diabetes. ... Having the science to put it in was a key step,” Dr. Gabbay noted.
Consider kidney, heart disease in type 2 treatment individualization
Recent data from trials such as CREDENCE and DAPA-HF, among others, have been added to inform the choice of pharmacologic treatment in patients with type 2 diabetes with comorbid diabetic kidney disease and chronic heart failure.
“ADA has been advocating individualization of treatment based on comorbidities for a while, but we’ve taken more steps in that direction. Beyond lifestyle for all individuals with type 2 diabetes, clinicians want to think early on about which comorbidities patients have and then think about the appropriate treatment based on that,” Dr. Gabbay said.
And for the third year in a row, the section on cardiovascular disease and risk management has been endorsed by the American College of Cardiology.
“All the things in that section are very much aligned with ACC and that’s been a great partnership,” Dr. Gabbay said.
Now, ADA is in discussions with other professional societies representing relevant specialties to create further such unified messages.
“What we all want to avoid is having multiple different guidelines. We want to speak with one voice and find common ground as much as possible. … It makes it much easier for clinicians to know what to do. That’s the goal of all this,” he noted.
Diabetes technology: The rise of CGM during pandemic and beyond
New information about continuous glucose monitoring (CGM) has been added to the diabetes technology section. Use of CGM is now recommended for anyone with diabetes who takes multiple daily injections or uses an insulin pump, regardless of age or diabetes type. The document provides expanded advice on use of time in range data for glycemic monitoring, particularly during the COVID-19 pandemic when remote monitoring is preferable.
Insurers are increasingly covering CGM for patients on insulin, but it’s far from universal. While the ultimate goal is to ensure access to CGM for everyone with diabetes, those treated with multiple daily insulin doses are the priority for now.
“Our hope is that as there’s greater evidence there will be more movement towards coverage. There are still so many people for whom it’s quite clear they would benefit because they’re on insulin but don’t have access to it. That’s an important area that ADA is advocating for, and it’s reflected in the standards of care,” Dr. Gabbay said.
In another technology-related revision, the term “blinded” CGM has been replaced with “professional CGM,” because clinic-based use of the devices can be “blinded” to the patient or monitored in real-time by both the patient and clinician. Also, a new recommendation has been added to address skin reactions associated with diabetes technology use.
Information about use of CGM in hospital settings during the COVID-19 pandemic has also been added in the technology section.
The COVID-19 pandemic comes up again in the section on vaccines.
“We mention that people with diabetes should be considered high priority [for COVID-19 vaccines], and that’s something that ADA is strongly advocating for because 40% of COVID-19 deaths have been in people with diabetes,” Dr. Gabbay said.
Dr. Gabbay reported being on the advisory boards of Onduo, Health Reveal, Vida Health, Lark, and Form Health.
A version of this article originally appeared on Medscape.com.
For 2021, the American Diabetes Association offers new guidance on assessing patients’ financial and social barriers to care, especially given the COVID-19 pandemic, individualizing treatment of patients with type 2 diabetes, and use of diabetes technology.
As it does every year, the annual update incorporates new clinical information that has become available since the last guideline, with occasional revisions during the year as needed. “Standards of Medical Care in Diabetes – 2021,” was published online as a supplement to Diabetes Care.
The new standards advise that patients be assessed for food and housing insecurity, social support, and “cost-related medication nonadherence,” and those found to have difficulty referred to appropriate community resources.
“Clinicians need to be sensitive to the fact that patients may have very good reasons for not taking their medication, [as in] if they can’t afford it,” ADA chief science & medical officer Robert A. Gabbay, MD, PhD, said in an interview.
Dr. Gabbay noted that “a heightened awareness” of social determinants of health is weaved throughout the 2021 standards because of the pandemic, with information on the topic derived from a July 2020 joint consensus statement in Diabetes Care, endorsed by a number of other societies, as well as a November publication also in Diabetes Care.
“We made several recommendations that speak to social determinants of health, placing an emphasis on engaging in conversations around this subject and screening for related issues such as food insecurity that weren’t there previously,” he said.
“Screening tools are suggested. It helped us to have an in-depth scientific review of the literature to know the prevalence of this in people with diabetes. ... Having the science to put it in was a key step,” Dr. Gabbay noted.
Consider kidney, heart disease in type 2 treatment individualization
Recent data from trials such as CREDENCE and DAPA-HF, among others, have been added to inform the choice of pharmacologic treatment in patients with type 2 diabetes with comorbid diabetic kidney disease and chronic heart failure.
“ADA has been advocating individualization of treatment based on comorbidities for a while, but we’ve taken more steps in that direction. Beyond lifestyle for all individuals with type 2 diabetes, clinicians want to think early on about which comorbidities patients have and then think about the appropriate treatment based on that,” Dr. Gabbay said.
And for the third year in a row, the section on cardiovascular disease and risk management has been endorsed by the American College of Cardiology.
“All the things in that section are very much aligned with ACC and that’s been a great partnership,” Dr. Gabbay said.
Now, ADA is in discussions with other professional societies representing relevant specialties to create further such unified messages.
“What we all want to avoid is having multiple different guidelines. We want to speak with one voice and find common ground as much as possible. … It makes it much easier for clinicians to know what to do. That’s the goal of all this,” he noted.
Diabetes technology: The rise of CGM during pandemic and beyond
New information about continuous glucose monitoring (CGM) has been added to the diabetes technology section. Use of CGM is now recommended for anyone with diabetes who takes multiple daily injections or uses an insulin pump, regardless of age or diabetes type. The document provides expanded advice on use of time in range data for glycemic monitoring, particularly during the COVID-19 pandemic when remote monitoring is preferable.
Insurers are increasingly covering CGM for patients on insulin, but it’s far from universal. While the ultimate goal is to ensure access to CGM for everyone with diabetes, those treated with multiple daily insulin doses are the priority for now.
“Our hope is that as there’s greater evidence there will be more movement towards coverage. There are still so many people for whom it’s quite clear they would benefit because they’re on insulin but don’t have access to it. That’s an important area that ADA is advocating for, and it’s reflected in the standards of care,” Dr. Gabbay said.
In another technology-related revision, the term “blinded” CGM has been replaced with “professional CGM,” because clinic-based use of the devices can be “blinded” to the patient or monitored in real-time by both the patient and clinician. Also, a new recommendation has been added to address skin reactions associated with diabetes technology use.
Information about use of CGM in hospital settings during the COVID-19 pandemic has also been added in the technology section.
The COVID-19 pandemic comes up again in the section on vaccines.
“We mention that people with diabetes should be considered high priority [for COVID-19 vaccines], and that’s something that ADA is strongly advocating for because 40% of COVID-19 deaths have been in people with diabetes,” Dr. Gabbay said.
Dr. Gabbay reported being on the advisory boards of Onduo, Health Reveal, Vida Health, Lark, and Form Health.
A version of this article originally appeared on Medscape.com.
For 2021, the American Diabetes Association offers new guidance on assessing patients’ financial and social barriers to care, especially given the COVID-19 pandemic, individualizing treatment of patients with type 2 diabetes, and use of diabetes technology.
As it does every year, the annual update incorporates new clinical information that has become available since the last guideline, with occasional revisions during the year as needed. “Standards of Medical Care in Diabetes – 2021,” was published online as a supplement to Diabetes Care.
The new standards advise that patients be assessed for food and housing insecurity, social support, and “cost-related medication nonadherence,” and those found to have difficulty referred to appropriate community resources.
“Clinicians need to be sensitive to the fact that patients may have very good reasons for not taking their medication, [as in] if they can’t afford it,” ADA chief science & medical officer Robert A. Gabbay, MD, PhD, said in an interview.
Dr. Gabbay noted that “a heightened awareness” of social determinants of health is weaved throughout the 2021 standards because of the pandemic, with information on the topic derived from a July 2020 joint consensus statement in Diabetes Care, endorsed by a number of other societies, as well as a November publication also in Diabetes Care.
“We made several recommendations that speak to social determinants of health, placing an emphasis on engaging in conversations around this subject and screening for related issues such as food insecurity that weren’t there previously,” he said.
“Screening tools are suggested. It helped us to have an in-depth scientific review of the literature to know the prevalence of this in people with diabetes. ... Having the science to put it in was a key step,” Dr. Gabbay noted.
Consider kidney, heart disease in type 2 treatment individualization
Recent data from trials such as CREDENCE and DAPA-HF, among others, have been added to inform the choice of pharmacologic treatment in patients with type 2 diabetes with comorbid diabetic kidney disease and chronic heart failure.
“ADA has been advocating individualization of treatment based on comorbidities for a while, but we’ve taken more steps in that direction. Beyond lifestyle for all individuals with type 2 diabetes, clinicians want to think early on about which comorbidities patients have and then think about the appropriate treatment based on that,” Dr. Gabbay said.
And for the third year in a row, the section on cardiovascular disease and risk management has been endorsed by the American College of Cardiology.
“All the things in that section are very much aligned with ACC and that’s been a great partnership,” Dr. Gabbay said.
Now, ADA is in discussions with other professional societies representing relevant specialties to create further such unified messages.
“What we all want to avoid is having multiple different guidelines. We want to speak with one voice and find common ground as much as possible. … It makes it much easier for clinicians to know what to do. That’s the goal of all this,” he noted.
Diabetes technology: The rise of CGM during pandemic and beyond
New information about continuous glucose monitoring (CGM) has been added to the diabetes technology section. Use of CGM is now recommended for anyone with diabetes who takes multiple daily injections or uses an insulin pump, regardless of age or diabetes type. The document provides expanded advice on use of time in range data for glycemic monitoring, particularly during the COVID-19 pandemic when remote monitoring is preferable.
Insurers are increasingly covering CGM for patients on insulin, but it’s far from universal. While the ultimate goal is to ensure access to CGM for everyone with diabetes, those treated with multiple daily insulin doses are the priority for now.
“Our hope is that as there’s greater evidence there will be more movement towards coverage. There are still so many people for whom it’s quite clear they would benefit because they’re on insulin but don’t have access to it. That’s an important area that ADA is advocating for, and it’s reflected in the standards of care,” Dr. Gabbay said.
In another technology-related revision, the term “blinded” CGM has been replaced with “professional CGM,” because clinic-based use of the devices can be “blinded” to the patient or monitored in real-time by both the patient and clinician. Also, a new recommendation has been added to address skin reactions associated with diabetes technology use.
Information about use of CGM in hospital settings during the COVID-19 pandemic has also been added in the technology section.
The COVID-19 pandemic comes up again in the section on vaccines.
“We mention that people with diabetes should be considered high priority [for COVID-19 vaccines], and that’s something that ADA is strongly advocating for because 40% of COVID-19 deaths have been in people with diabetes,” Dr. Gabbay said.
Dr. Gabbay reported being on the advisory boards of Onduo, Health Reveal, Vida Health, Lark, and Form Health.
A version of this article originally appeared on Medscape.com.
Medicare payments could get tougher for docs
More than 40 value-based payment models – from direct contracting to bundled payments – have been introduced into the Medicare program in the past 10 years, with the goal of improving care while lowering costs. Hopes were high that they would be successful.
Physicians could suffer a huge blow to their income.
Many of the value-based care models simply did not work as expected, said Seema Verma, head of the Centers for Medicare & Medicaid Services, at a recent HLTH Conference. “They are not producing the types of savings the taxpayers deserve,” Ms. Verma said.
The Medicare Payment Advisory Commission (MedPac) concluded that, while dozens of payment models were tested, most failed to generate net savings for Medicare. Even the most successful of the models produced only modest savings. MedPac elaborated: “The track record raises the question of whether changes to particular models or CMMI’s [Center for Medicare & Medicaid Innovation’s] broader strategies might be warranted.”
What will happen now, as government officials admit that their value-based programs haven’t worked? The value-based programs could become more stringent. Here’s what physicians will have to contend with.
More risk. Experts agree that risk – financial risk – will be a component of future programs. Two-sided risk is likely to be the norm. This means that both parties – the provider and the insurer – are at financial risk for the patients covered by the program.
For example, a plan with 50,000 beneficiary patients would estimate the cost of caring for those patients on the basis of multiple variables. If the actual cost is lower than anticipated, both parties share in the savings. However, both share in the loss if the cost of caring for their patient population exceeds expectations.
This may compel physicians to enhance efficiency and potentially limit the services provided to patients. Typically, however, the strategy is to make efforts to prevent services like ED visits and admissions by focusing on health maintenance.
In contrast to most current value-based models, which feature little to no downside risk for physicians, double-sided risk means physicians could lose money. The loss may incorporate a cap – 5%, for example – but programs may differ. Experts concur that double-sided risk will be a hallmark of future programs.
Better data. The majority of health care services are rendered via fee-for-service: Patients receive services and physicians are paid, yet little or no information about outcomes is exchanged between insurers and physicians.
Penny Noyes, president of Health Business Navigators and contract negotiator for physicians, is not a fan of the current crop of value-based programs and feels that data transparency is positive. Sound metrics can lead to improvement, she said, adding: “It’s not money that drives physicians to make decisions; it’s what’s in the best interest of their patients and their patients’ long-term care.”
Value-based programs can work but only if applicable data are developed and given to physicians so that they can better understand their current performance and how to improve.
Mandated participation. Participation in value-based programs has been voluntary, but that may have skewed the results, which were better than what typical practice would have shown. Acknowledging this may lead CMS to call for mandated participation as a component of future programs. Physicians may be brought into programs, if only to determine whether the models really work. To date, participation in the programs has been voluntary, but that may change in the future.
Innovation. The private insurance market may end up as a key player. Over the past 6 months, health insurers have either consolidated partnerships with telemedicine companies to provide no-cost care to beneficiaries or have launched their own initiatives.
Others are focused on bringing together patients and providers operating outside of the traditional health care system, such as Aetna’s merger with CVS which now offers retail-based acute care (MinuteClinic) and chronic care (HealthHUB). Still other payers are gambling with physician practice ownership, as in the case of United Healthcare’s OptumHealth, which now boasts around 50,000 physicians throughout the country.
New practice models are emerging in private practices as well. Physicians are embracing remote care, proactively managing care transitions, and seeking out more methods to keep patients healthy and at home.
Not much was expected from value-based plans
Many are not surprised that the value-based models did not produce impressive results. Ms. Noyes doubted that positive outcomes will be achieved for physicians in comparison with what could have been attained under fee-for-service arrangements with lower administrative costs.
While the Affordable Care Act attempted to encourage alternative reimbursement, it limits the maximum medical loss ratio (MLR) a payer could achieve. For many plans, that maximum was 85%. Simply put, at least $0.85 of each premium collected had to be paid in claims; the remaining $0.15 went to margin, claims, and other administrative costs. A payer with an 82% MLR then would have to rebate the 3% difference to enrollees.
But that’s not what occurred, according to Ms. Noyes. Because value-based payments to providers are considered a claims expense, an MLR ratio of 82% allowed the payer to distribute the 3% difference to providers as value-based payments. Ms. Noyes said: “That may sound good for the provider, but the result was essentially a freeze on the provider’s fee-for-service reimbursement with the prospect of getting value-based payments like ‘shared savings.’
“When the providers tried to increase their base fee-for-service rates just to match inflation, payers often advised that any future raises had to be earned through value-based programs,” Ms. Noyes added. The value-based formulas confuse providers because payments are often made for periods as far back as 18 months, and providers do not have data systems to reconcile their payer report cards retrospectively. The result is that providers tended to accept whatever amount the payer distributed.
Executives at Lumeris, a company that helps health systems participate successfully in value-based care, see potential in a newer approach to alternative payments, such as CMS’ Direct Contracting initiative. This voluntary payment model offers options tailored to several types of organizations that aim to reduce costs while preserving or enhancing the quality of care for Medicare fee-for-service beneficiaries.
Jeff Smith, chief commercial officer for population health at Lumeris, explained that the Direct Contracting initiative can provide physicians with a more attractive option than prior value-based models because it adjusts for the complexity and fragility of patients with complex and chronic conditions. By allowing providers to participate in the savings generated, the initiative stands in stark contrast to what Mr. Smith described as the “shared savings to nothingness” experienced by providers in earlier-stage alternative payment models.
Physicians engaged with value-based programs like Direct Contracting are investing in nurses to aid with initiatives regarding health promotion and transitions of care. When a patient is discharged, for example, the nurse contacts the patient to discuss medications, schedule follow-up appointments, and so forth – tasks typically left to the patient (or caregiver) to navigate in the traditional system.
The initiative recognizes the importance of managing high-risk patients, those whom physicians identify as having an extraordinary number of ED visits and admissions. These patients, as well as so-called “rising-risk” patients, are targeted by nurses who proactively communicate with patients (and caregivers) to address patient’s needs, including social determinants of health.
Physicians who have a large load of patients in value-based programs are hiring social workers, pharmacists, and behavioral health experts to help. Of course, these personnel are costly, but that’s what the value-based programs aim to reimburse.
Still, the road ahead to value based is rocky and may not gain momentum for some time. Johns Hopkins University’s Doug Hough, PhD, an economist, recounts a government research study that sought to assess the university’s health system participation in a value-based payment program. While there were positive impacts on the program’s target population, Hough and his team discovered that the returns achieved by the optional model didn’t justify the health system’s financial support for it. The increasingly indebted health system ultimately decided to drop the optional program.
Dr. Hough indicated that the health system – Johns Hopkins Medicine – likely would have continued its support for the program had the government at least allowed it to break even. Although the payment program under study was a 3-year project, the bigger challenge, declared Dr. Hough, is that “we can’t turn an aircraft carrier that quickly.”
“Three years won’t show whether value-based care is really working,” Dr. Hough said.
Robert Zipper, MD, a hospitalist and senior policy advisor for Sound Physicians, a company that works to improve outcomes in acute care, agreed with Dr. Hough that performance tends to improve with time. Yet, Dr. Zipper doesn’t see much change in the near term, because “after all, there is nothing to replace them [the programs].”
The problem gets even stickier for private payers because patients may be on an insurance panel for as little as a year or 2. Thanks to this rapid churn of beneficiaries, even the best-designed value-based program will have little time to prove its worth.
Dr. Zipper is among the many who don’t expect significant changes in the near term, asserting that “President Biden will want to get a few policy wins first, and health care is not the easiest place to start.”
But it’s likely that payers and others will want to see more emphasis on value-based programs despite these programs’ possible value to patients, physicians, and health systems alike.
A version of this article originally appeared on Medscape.com.
More than 40 value-based payment models – from direct contracting to bundled payments – have been introduced into the Medicare program in the past 10 years, with the goal of improving care while lowering costs. Hopes were high that they would be successful.
Physicians could suffer a huge blow to their income.
Many of the value-based care models simply did not work as expected, said Seema Verma, head of the Centers for Medicare & Medicaid Services, at a recent HLTH Conference. “They are not producing the types of savings the taxpayers deserve,” Ms. Verma said.
The Medicare Payment Advisory Commission (MedPac) concluded that, while dozens of payment models were tested, most failed to generate net savings for Medicare. Even the most successful of the models produced only modest savings. MedPac elaborated: “The track record raises the question of whether changes to particular models or CMMI’s [Center for Medicare & Medicaid Innovation’s] broader strategies might be warranted.”
What will happen now, as government officials admit that their value-based programs haven’t worked? The value-based programs could become more stringent. Here’s what physicians will have to contend with.
More risk. Experts agree that risk – financial risk – will be a component of future programs. Two-sided risk is likely to be the norm. This means that both parties – the provider and the insurer – are at financial risk for the patients covered by the program.
For example, a plan with 50,000 beneficiary patients would estimate the cost of caring for those patients on the basis of multiple variables. If the actual cost is lower than anticipated, both parties share in the savings. However, both share in the loss if the cost of caring for their patient population exceeds expectations.
This may compel physicians to enhance efficiency and potentially limit the services provided to patients. Typically, however, the strategy is to make efforts to prevent services like ED visits and admissions by focusing on health maintenance.
In contrast to most current value-based models, which feature little to no downside risk for physicians, double-sided risk means physicians could lose money. The loss may incorporate a cap – 5%, for example – but programs may differ. Experts concur that double-sided risk will be a hallmark of future programs.
Better data. The majority of health care services are rendered via fee-for-service: Patients receive services and physicians are paid, yet little or no information about outcomes is exchanged between insurers and physicians.
Penny Noyes, president of Health Business Navigators and contract negotiator for physicians, is not a fan of the current crop of value-based programs and feels that data transparency is positive. Sound metrics can lead to improvement, she said, adding: “It’s not money that drives physicians to make decisions; it’s what’s in the best interest of their patients and their patients’ long-term care.”
Value-based programs can work but only if applicable data are developed and given to physicians so that they can better understand their current performance and how to improve.
Mandated participation. Participation in value-based programs has been voluntary, but that may have skewed the results, which were better than what typical practice would have shown. Acknowledging this may lead CMS to call for mandated participation as a component of future programs. Physicians may be brought into programs, if only to determine whether the models really work. To date, participation in the programs has been voluntary, but that may change in the future.
Innovation. The private insurance market may end up as a key player. Over the past 6 months, health insurers have either consolidated partnerships with telemedicine companies to provide no-cost care to beneficiaries or have launched their own initiatives.
Others are focused on bringing together patients and providers operating outside of the traditional health care system, such as Aetna’s merger with CVS which now offers retail-based acute care (MinuteClinic) and chronic care (HealthHUB). Still other payers are gambling with physician practice ownership, as in the case of United Healthcare’s OptumHealth, which now boasts around 50,000 physicians throughout the country.
New practice models are emerging in private practices as well. Physicians are embracing remote care, proactively managing care transitions, and seeking out more methods to keep patients healthy and at home.
Not much was expected from value-based plans
Many are not surprised that the value-based models did not produce impressive results. Ms. Noyes doubted that positive outcomes will be achieved for physicians in comparison with what could have been attained under fee-for-service arrangements with lower administrative costs.
While the Affordable Care Act attempted to encourage alternative reimbursement, it limits the maximum medical loss ratio (MLR) a payer could achieve. For many plans, that maximum was 85%. Simply put, at least $0.85 of each premium collected had to be paid in claims; the remaining $0.15 went to margin, claims, and other administrative costs. A payer with an 82% MLR then would have to rebate the 3% difference to enrollees.
But that’s not what occurred, according to Ms. Noyes. Because value-based payments to providers are considered a claims expense, an MLR ratio of 82% allowed the payer to distribute the 3% difference to providers as value-based payments. Ms. Noyes said: “That may sound good for the provider, but the result was essentially a freeze on the provider’s fee-for-service reimbursement with the prospect of getting value-based payments like ‘shared savings.’
“When the providers tried to increase their base fee-for-service rates just to match inflation, payers often advised that any future raises had to be earned through value-based programs,” Ms. Noyes added. The value-based formulas confuse providers because payments are often made for periods as far back as 18 months, and providers do not have data systems to reconcile their payer report cards retrospectively. The result is that providers tended to accept whatever amount the payer distributed.
Executives at Lumeris, a company that helps health systems participate successfully in value-based care, see potential in a newer approach to alternative payments, such as CMS’ Direct Contracting initiative. This voluntary payment model offers options tailored to several types of organizations that aim to reduce costs while preserving or enhancing the quality of care for Medicare fee-for-service beneficiaries.
Jeff Smith, chief commercial officer for population health at Lumeris, explained that the Direct Contracting initiative can provide physicians with a more attractive option than prior value-based models because it adjusts for the complexity and fragility of patients with complex and chronic conditions. By allowing providers to participate in the savings generated, the initiative stands in stark contrast to what Mr. Smith described as the “shared savings to nothingness” experienced by providers in earlier-stage alternative payment models.
Physicians engaged with value-based programs like Direct Contracting are investing in nurses to aid with initiatives regarding health promotion and transitions of care. When a patient is discharged, for example, the nurse contacts the patient to discuss medications, schedule follow-up appointments, and so forth – tasks typically left to the patient (or caregiver) to navigate in the traditional system.
The initiative recognizes the importance of managing high-risk patients, those whom physicians identify as having an extraordinary number of ED visits and admissions. These patients, as well as so-called “rising-risk” patients, are targeted by nurses who proactively communicate with patients (and caregivers) to address patient’s needs, including social determinants of health.
Physicians who have a large load of patients in value-based programs are hiring social workers, pharmacists, and behavioral health experts to help. Of course, these personnel are costly, but that’s what the value-based programs aim to reimburse.
Still, the road ahead to value based is rocky and may not gain momentum for some time. Johns Hopkins University’s Doug Hough, PhD, an economist, recounts a government research study that sought to assess the university’s health system participation in a value-based payment program. While there were positive impacts on the program’s target population, Hough and his team discovered that the returns achieved by the optional model didn’t justify the health system’s financial support for it. The increasingly indebted health system ultimately decided to drop the optional program.
Dr. Hough indicated that the health system – Johns Hopkins Medicine – likely would have continued its support for the program had the government at least allowed it to break even. Although the payment program under study was a 3-year project, the bigger challenge, declared Dr. Hough, is that “we can’t turn an aircraft carrier that quickly.”
“Three years won’t show whether value-based care is really working,” Dr. Hough said.
Robert Zipper, MD, a hospitalist and senior policy advisor for Sound Physicians, a company that works to improve outcomes in acute care, agreed with Dr. Hough that performance tends to improve with time. Yet, Dr. Zipper doesn’t see much change in the near term, because “after all, there is nothing to replace them [the programs].”
The problem gets even stickier for private payers because patients may be on an insurance panel for as little as a year or 2. Thanks to this rapid churn of beneficiaries, even the best-designed value-based program will have little time to prove its worth.
Dr. Zipper is among the many who don’t expect significant changes in the near term, asserting that “President Biden will want to get a few policy wins first, and health care is not the easiest place to start.”
But it’s likely that payers and others will want to see more emphasis on value-based programs despite these programs’ possible value to patients, physicians, and health systems alike.
A version of this article originally appeared on Medscape.com.
More than 40 value-based payment models – from direct contracting to bundled payments – have been introduced into the Medicare program in the past 10 years, with the goal of improving care while lowering costs. Hopes were high that they would be successful.
Physicians could suffer a huge blow to their income.
Many of the value-based care models simply did not work as expected, said Seema Verma, head of the Centers for Medicare & Medicaid Services, at a recent HLTH Conference. “They are not producing the types of savings the taxpayers deserve,” Ms. Verma said.
The Medicare Payment Advisory Commission (MedPac) concluded that, while dozens of payment models were tested, most failed to generate net savings for Medicare. Even the most successful of the models produced only modest savings. MedPac elaborated: “The track record raises the question of whether changes to particular models or CMMI’s [Center for Medicare & Medicaid Innovation’s] broader strategies might be warranted.”
What will happen now, as government officials admit that their value-based programs haven’t worked? The value-based programs could become more stringent. Here’s what physicians will have to contend with.
More risk. Experts agree that risk – financial risk – will be a component of future programs. Two-sided risk is likely to be the norm. This means that both parties – the provider and the insurer – are at financial risk for the patients covered by the program.
For example, a plan with 50,000 beneficiary patients would estimate the cost of caring for those patients on the basis of multiple variables. If the actual cost is lower than anticipated, both parties share in the savings. However, both share in the loss if the cost of caring for their patient population exceeds expectations.
This may compel physicians to enhance efficiency and potentially limit the services provided to patients. Typically, however, the strategy is to make efforts to prevent services like ED visits and admissions by focusing on health maintenance.
In contrast to most current value-based models, which feature little to no downside risk for physicians, double-sided risk means physicians could lose money. The loss may incorporate a cap – 5%, for example – but programs may differ. Experts concur that double-sided risk will be a hallmark of future programs.
Better data. The majority of health care services are rendered via fee-for-service: Patients receive services and physicians are paid, yet little or no information about outcomes is exchanged between insurers and physicians.
Penny Noyes, president of Health Business Navigators and contract negotiator for physicians, is not a fan of the current crop of value-based programs and feels that data transparency is positive. Sound metrics can lead to improvement, she said, adding: “It’s not money that drives physicians to make decisions; it’s what’s in the best interest of their patients and their patients’ long-term care.”
Value-based programs can work but only if applicable data are developed and given to physicians so that they can better understand their current performance and how to improve.
Mandated participation. Participation in value-based programs has been voluntary, but that may have skewed the results, which were better than what typical practice would have shown. Acknowledging this may lead CMS to call for mandated participation as a component of future programs. Physicians may be brought into programs, if only to determine whether the models really work. To date, participation in the programs has been voluntary, but that may change in the future.
Innovation. The private insurance market may end up as a key player. Over the past 6 months, health insurers have either consolidated partnerships with telemedicine companies to provide no-cost care to beneficiaries or have launched their own initiatives.
Others are focused on bringing together patients and providers operating outside of the traditional health care system, such as Aetna’s merger with CVS which now offers retail-based acute care (MinuteClinic) and chronic care (HealthHUB). Still other payers are gambling with physician practice ownership, as in the case of United Healthcare’s OptumHealth, which now boasts around 50,000 physicians throughout the country.
New practice models are emerging in private practices as well. Physicians are embracing remote care, proactively managing care transitions, and seeking out more methods to keep patients healthy and at home.
Not much was expected from value-based plans
Many are not surprised that the value-based models did not produce impressive results. Ms. Noyes doubted that positive outcomes will be achieved for physicians in comparison with what could have been attained under fee-for-service arrangements with lower administrative costs.
While the Affordable Care Act attempted to encourage alternative reimbursement, it limits the maximum medical loss ratio (MLR) a payer could achieve. For many plans, that maximum was 85%. Simply put, at least $0.85 of each premium collected had to be paid in claims; the remaining $0.15 went to margin, claims, and other administrative costs. A payer with an 82% MLR then would have to rebate the 3% difference to enrollees.
But that’s not what occurred, according to Ms. Noyes. Because value-based payments to providers are considered a claims expense, an MLR ratio of 82% allowed the payer to distribute the 3% difference to providers as value-based payments. Ms. Noyes said: “That may sound good for the provider, but the result was essentially a freeze on the provider’s fee-for-service reimbursement with the prospect of getting value-based payments like ‘shared savings.’
“When the providers tried to increase their base fee-for-service rates just to match inflation, payers often advised that any future raises had to be earned through value-based programs,” Ms. Noyes added. The value-based formulas confuse providers because payments are often made for periods as far back as 18 months, and providers do not have data systems to reconcile their payer report cards retrospectively. The result is that providers tended to accept whatever amount the payer distributed.
Executives at Lumeris, a company that helps health systems participate successfully in value-based care, see potential in a newer approach to alternative payments, such as CMS’ Direct Contracting initiative. This voluntary payment model offers options tailored to several types of organizations that aim to reduce costs while preserving or enhancing the quality of care for Medicare fee-for-service beneficiaries.
Jeff Smith, chief commercial officer for population health at Lumeris, explained that the Direct Contracting initiative can provide physicians with a more attractive option than prior value-based models because it adjusts for the complexity and fragility of patients with complex and chronic conditions. By allowing providers to participate in the savings generated, the initiative stands in stark contrast to what Mr. Smith described as the “shared savings to nothingness” experienced by providers in earlier-stage alternative payment models.
Physicians engaged with value-based programs like Direct Contracting are investing in nurses to aid with initiatives regarding health promotion and transitions of care. When a patient is discharged, for example, the nurse contacts the patient to discuss medications, schedule follow-up appointments, and so forth – tasks typically left to the patient (or caregiver) to navigate in the traditional system.
The initiative recognizes the importance of managing high-risk patients, those whom physicians identify as having an extraordinary number of ED visits and admissions. These patients, as well as so-called “rising-risk” patients, are targeted by nurses who proactively communicate with patients (and caregivers) to address patient’s needs, including social determinants of health.
Physicians who have a large load of patients in value-based programs are hiring social workers, pharmacists, and behavioral health experts to help. Of course, these personnel are costly, but that’s what the value-based programs aim to reimburse.
Still, the road ahead to value based is rocky and may not gain momentum for some time. Johns Hopkins University’s Doug Hough, PhD, an economist, recounts a government research study that sought to assess the university’s health system participation in a value-based payment program. While there were positive impacts on the program’s target population, Hough and his team discovered that the returns achieved by the optional model didn’t justify the health system’s financial support for it. The increasingly indebted health system ultimately decided to drop the optional program.
Dr. Hough indicated that the health system – Johns Hopkins Medicine – likely would have continued its support for the program had the government at least allowed it to break even. Although the payment program under study was a 3-year project, the bigger challenge, declared Dr. Hough, is that “we can’t turn an aircraft carrier that quickly.”
“Three years won’t show whether value-based care is really working,” Dr. Hough said.
Robert Zipper, MD, a hospitalist and senior policy advisor for Sound Physicians, a company that works to improve outcomes in acute care, agreed with Dr. Hough that performance tends to improve with time. Yet, Dr. Zipper doesn’t see much change in the near term, because “after all, there is nothing to replace them [the programs].”
The problem gets even stickier for private payers because patients may be on an insurance panel for as little as a year or 2. Thanks to this rapid churn of beneficiaries, even the best-designed value-based program will have little time to prove its worth.
Dr. Zipper is among the many who don’t expect significant changes in the near term, asserting that “President Biden will want to get a few policy wins first, and health care is not the easiest place to start.”
But it’s likely that payers and others will want to see more emphasis on value-based programs despite these programs’ possible value to patients, physicians, and health systems alike.
A version of this article originally appeared on Medscape.com.
Sac/val heart failure benefit extends to diabetes patients
The beneficial effects of sacubitril/valsartan on reverse cardiac remodeling in patients with heart failure and reduced ejection fraction have been well established, but those benefits haven’t been as clearly demonstrated to carry over to HFrEF patients who also have type 2 diabetes mellitus (T2DM).

Now, a post-hoc analysis of a pivotal clinical trial reports that those benefits do extend to patients with HFrEF and T2DM.
“It’s really not about a Sophie’s choice of whether you give this or that drug in these patients,” said corresponding author Javed Butler, MD, MPH, MBA. “We really ought to be giving all of these drugs – the angiotensin receptor neprilysin inhibitors (ARNIs) and sodium-glucose transporter 2 (SGLT-2) inhibitors – to our patients for the best outcomes.”
The post-hoc analysis, published in JACC: Heart Failure, evaluated 361 patients with T2DM who were enrolled in the PROVE-HF trial of sac/val therapy for HF, published in JAMA.
PROVE-HF evaluated biomarkers, myocardial remodeling, and outcomes through a year of treatment with sac/val. The primary endpoint was the level of changes in natriuretic peptide (NT-proBNP) concentrations, left-ventricle ejection fraction (LVEF) and overall Kansas City Cardiomyopathy Questionnaire (KCCQ)-23 scores through 12 months of treatment.
The post hoc study reported that baseline NT-proBNP concentrations were higher in the DM patients (854 pg/mL vs. 706 pg/mL), but at 12 months those levels were 513 and 441 pg/mL, respectively.
LVEF changed similarly from baseline to 12 months in both groups: from 28.3% to 37% in the DM patients and from 28.1% to 38.34% in non-DM patients. Overall KCCQ-23 scores improved similarly in both groups, but longitudinal analyses found modestly higher gains in the T2DM group, 9.3 vs. 8.6 points (P = .07).
“The real reason I wanted to do this study is that I’m a huge fan of all the SGLT-2 inhibitors, and I’m very involved in those trials, and there is right now so much momentum behind SGLT-2 inhibitors that I don’t want people to forget that ARNI is still the base therapy for HF,” said Dr. Butler, chair of cardiovascular research and the department of medicine at the University of Mississippi in Jackson.
He noted that the size of the diabetes cohort in PROVE-HF “is a nonissue” for evaluating power of the post hoc analysis because it tracked key measures in the study population continuously at eight intervals over the 12 months.
The analysis further demonstrates the synergistic effects of ARNI and SGLT-2 inhibitors in patients with T2DM and HF that were also reported in the PARADIGM-HF study, Dr. Butler said.
“We have sort of moved on, saying that SGLT-2 inhibitors have a benefit on the heart, but the reverse is also true: ARNIs are still heart failure drugs, and we don’t think of them as diabetes drugs, but the PARADIGM-HF data showed that there was a substantial reduction in hemoglobin A1c in those who had diabetes,” he said.
The researchers noted that an absence of a control group may contribute to an overestimation of reverse cardiac remodeling in the T2DM patients, and that the PROVE-HF study wasn’t prospectively powered to delineate differences in how sac/val therapy affected patients with or without diabetes. “Future investigations seeking to evaluate differences by T2DM status after sacubitril/valsartan initiation may use our findings to plan prospective sample sizes,” the researchers wrote.
Dr. Butler disclosed financial relationships with Abbott, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, Eli Lilly, G3 Pharmaceutical, Impulse Dynamics, Innolife, Janssen, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana, StealthPeptide and Vifor. Lead author Muhammad Shahzeb Khan, MD, MSc, a professor at the University of Mississippi, has no relevant financial relationships to disclose.
SOURCE: Kahn MS et al. JACC: HF. 2020 Dec 9. doi: 10.1016/j.jchf.2020.09.014.
The beneficial effects of sacubitril/valsartan on reverse cardiac remodeling in patients with heart failure and reduced ejection fraction have been well established, but those benefits haven’t been as clearly demonstrated to carry over to HFrEF patients who also have type 2 diabetes mellitus (T2DM).
Now, a post-hoc analysis of a pivotal clinical trial reports that those benefits do extend to patients with HFrEF and T2DM.
“It’s really not about a Sophie’s choice of whether you give this or that drug in these patients,” said corresponding author Javed Butler, MD, MPH, MBA. “We really ought to be giving all of these drugs – the angiotensin receptor neprilysin inhibitors (ARNIs) and sodium-glucose transporter 2 (SGLT-2) inhibitors – to our patients for the best outcomes.”
The post-hoc analysis, published in JACC: Heart Failure, evaluated 361 patients with T2DM who were enrolled in the PROVE-HF trial of sac/val therapy for HF, published in JAMA.
PROVE-HF evaluated biomarkers, myocardial remodeling, and outcomes through a year of treatment with sac/val. The primary endpoint was the level of changes in natriuretic peptide (NT-proBNP) concentrations, left-ventricle ejection fraction (LVEF) and overall Kansas City Cardiomyopathy Questionnaire (KCCQ)-23 scores through 12 months of treatment.
The post hoc study reported that baseline NT-proBNP concentrations were higher in the DM patients (854 pg/mL vs. 706 pg/mL), but at 12 months those levels were 513 and 441 pg/mL, respectively.
LVEF changed similarly from baseline to 12 months in both groups: from 28.3% to 37% in the DM patients and from 28.1% to 38.34% in non-DM patients. Overall KCCQ-23 scores improved similarly in both groups, but longitudinal analyses found modestly higher gains in the T2DM group, 9.3 vs. 8.6 points (P = .07).
“The real reason I wanted to do this study is that I’m a huge fan of all the SGLT-2 inhibitors, and I’m very involved in those trials, and there is right now so much momentum behind SGLT-2 inhibitors that I don’t want people to forget that ARNI is still the base therapy for HF,” said Dr. Butler, chair of cardiovascular research and the department of medicine at the University of Mississippi in Jackson.
He noted that the size of the diabetes cohort in PROVE-HF “is a nonissue” for evaluating power of the post hoc analysis because it tracked key measures in the study population continuously at eight intervals over the 12 months.
The analysis further demonstrates the synergistic effects of ARNI and SGLT-2 inhibitors in patients with T2DM and HF that were also reported in the PARADIGM-HF study, Dr. Butler said.
“We have sort of moved on, saying that SGLT-2 inhibitors have a benefit on the heart, but the reverse is also true: ARNIs are still heart failure drugs, and we don’t think of them as diabetes drugs, but the PARADIGM-HF data showed that there was a substantial reduction in hemoglobin A1c in those who had diabetes,” he said.
The researchers noted that an absence of a control group may contribute to an overestimation of reverse cardiac remodeling in the T2DM patients, and that the PROVE-HF study wasn’t prospectively powered to delineate differences in how sac/val therapy affected patients with or without diabetes. “Future investigations seeking to evaluate differences by T2DM status after sacubitril/valsartan initiation may use our findings to plan prospective sample sizes,” the researchers wrote.
Dr. Butler disclosed financial relationships with Abbott, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, Eli Lilly, G3 Pharmaceutical, Impulse Dynamics, Innolife, Janssen, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana, StealthPeptide and Vifor. Lead author Muhammad Shahzeb Khan, MD, MSc, a professor at the University of Mississippi, has no relevant financial relationships to disclose.
SOURCE: Kahn MS et al. JACC: HF. 2020 Dec 9. doi: 10.1016/j.jchf.2020.09.014.
The beneficial effects of sacubitril/valsartan on reverse cardiac remodeling in patients with heart failure and reduced ejection fraction have been well established, but those benefits haven’t been as clearly demonstrated to carry over to HFrEF patients who also have type 2 diabetes mellitus (T2DM).
Now, a post-hoc analysis of a pivotal clinical trial reports that those benefits do extend to patients with HFrEF and T2DM.
“It’s really not about a Sophie’s choice of whether you give this or that drug in these patients,” said corresponding author Javed Butler, MD, MPH, MBA. “We really ought to be giving all of these drugs – the angiotensin receptor neprilysin inhibitors (ARNIs) and sodium-glucose transporter 2 (SGLT-2) inhibitors – to our patients for the best outcomes.”
The post-hoc analysis, published in JACC: Heart Failure, evaluated 361 patients with T2DM who were enrolled in the PROVE-HF trial of sac/val therapy for HF, published in JAMA.
PROVE-HF evaluated biomarkers, myocardial remodeling, and outcomes through a year of treatment with sac/val. The primary endpoint was the level of changes in natriuretic peptide (NT-proBNP) concentrations, left-ventricle ejection fraction (LVEF) and overall Kansas City Cardiomyopathy Questionnaire (KCCQ)-23 scores through 12 months of treatment.
The post hoc study reported that baseline NT-proBNP concentrations were higher in the DM patients (854 pg/mL vs. 706 pg/mL), but at 12 months those levels were 513 and 441 pg/mL, respectively.
LVEF changed similarly from baseline to 12 months in both groups: from 28.3% to 37% in the DM patients and from 28.1% to 38.34% in non-DM patients. Overall KCCQ-23 scores improved similarly in both groups, but longitudinal analyses found modestly higher gains in the T2DM group, 9.3 vs. 8.6 points (P = .07).
“The real reason I wanted to do this study is that I’m a huge fan of all the SGLT-2 inhibitors, and I’m very involved in those trials, and there is right now so much momentum behind SGLT-2 inhibitors that I don’t want people to forget that ARNI is still the base therapy for HF,” said Dr. Butler, chair of cardiovascular research and the department of medicine at the University of Mississippi in Jackson.
He noted that the size of the diabetes cohort in PROVE-HF “is a nonissue” for evaluating power of the post hoc analysis because it tracked key measures in the study population continuously at eight intervals over the 12 months.
The analysis further demonstrates the synergistic effects of ARNI and SGLT-2 inhibitors in patients with T2DM and HF that were also reported in the PARADIGM-HF study, Dr. Butler said.
“We have sort of moved on, saying that SGLT-2 inhibitors have a benefit on the heart, but the reverse is also true: ARNIs are still heart failure drugs, and we don’t think of them as diabetes drugs, but the PARADIGM-HF data showed that there was a substantial reduction in hemoglobin A1c in those who had diabetes,” he said.
The researchers noted that an absence of a control group may contribute to an overestimation of reverse cardiac remodeling in the T2DM patients, and that the PROVE-HF study wasn’t prospectively powered to delineate differences in how sac/val therapy affected patients with or without diabetes. “Future investigations seeking to evaluate differences by T2DM status after sacubitril/valsartan initiation may use our findings to plan prospective sample sizes,” the researchers wrote.
Dr. Butler disclosed financial relationships with Abbott, Amgen, Array, AstraZeneca, Bayer, Boehringer Ingelheim, CVRx, Eli Lilly, G3 Pharmaceutical, Impulse Dynamics, Innolife, Janssen, Luitpold, Medtronic, Merck, Novartis, Novo Nordisk, Relypsa, Sequana, StealthPeptide and Vifor. Lead author Muhammad Shahzeb Khan, MD, MSc, a professor at the University of Mississippi, has no relevant financial relationships to disclose.
SOURCE: Kahn MS et al. JACC: HF. 2020 Dec 9. doi: 10.1016/j.jchf.2020.09.014.
FROM JACC: HEART FAILURE
Understanding messenger RNA and other SARS-CoV-2 vaccines
In mid-November, Pfizer/BioNTech were the first with surprising positive protection interim data for their coronavirus vaccine, BNT162b2. A week later, Moderna released interim efficacy results showing its coronavirus vaccine, mRNA-1273, also protected patients from developing SARS-CoV-2 infections. Both studies included mostly healthy adults. A diverse ethnic and racial vaccinated population was included. A reasonable number of persons aged over 65 years, and persons with stable compromising medical conditions were included. Adolescents aged 16 years and over were included. Younger adolescents have been vaccinated or such studies are in the planning or early implementation stage as 2020 came to a close.
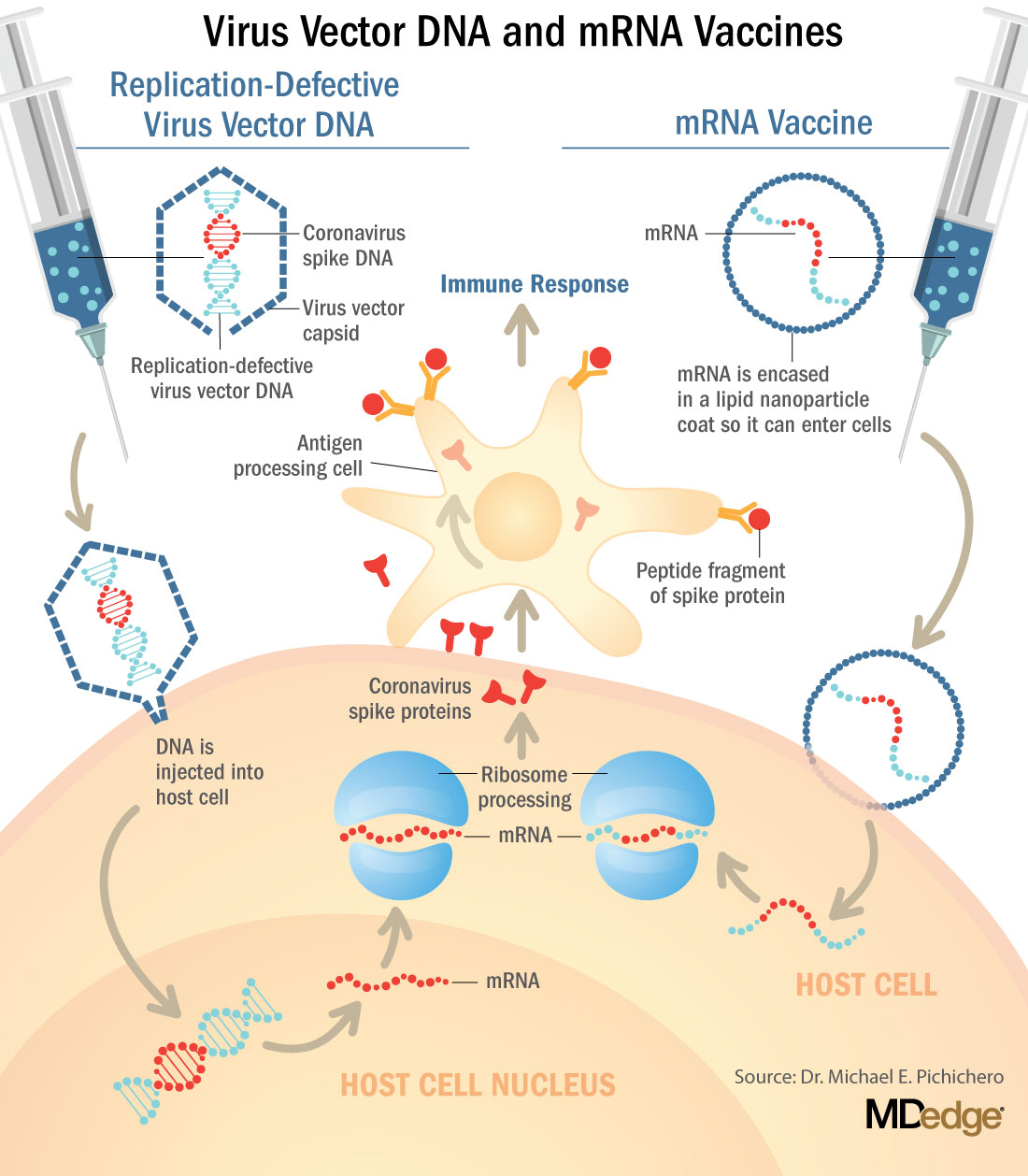
These are new and revolutionary vaccines, although the ability to inject mRNA into animals dates back to 1990, technological advances today make it a reality.1 Traditional vaccines typically involve injection with antigens such as purified proteins or polysaccharides or inactivated/attenuated viruses. In the case of Pfizer’s and Moderna’s vaccines, the mRNA provides the genetic information to synthesize the spike protein that the SARS-CoV-2 virus uses to attach to and infect human cells. Each type of vaccine is packaged in proprietary lipid nanoparticles to protect the mRNA from rapid degradation, and the nanoparticles serve as an adjuvant to attract immune cells to the site of injection. (The properties of the respective lipid nanoparticle packaging may be the factor that impacts storage requirements discussed below.) When injected into muscle (myocyte), the lipid nanoparticles containing the mRNA inside are taken into muscle cells, where the cytoplasmic ribosomes detect and decode the mRNA resulting in the production of the spike protein antigen. It should be noted that the mRNA does not enter the nucleus, where the genetic information (DNA) of a cell is located, and can’t be reproduced or integrated into the DNA. The antigen is exported to the myocyte cell surface where the immune system’s antigen presenting cells detect the protein, ingest it, and take it to regional lymph nodes where interactions with T cells and B cells results in antibodies, T cell–mediated immunity, and generation of immune memory T cells and B cells. A particular subset of T cells – cytotoxic or killer T cells – destroy cells that have been infected by a pathogen. The SARS-CoV-2 mRNA vaccine from Pfizer was reported to induce powerful cytotoxic T-cell responses. Results for Moderna’s vaccine had not been reported at the time this column was prepared, but I anticipate the same positive results.
The revolutionary aspect of mRNA vaccines is the speed at which they can be designed and produced. This is why they lead the pack among the SARS-CoV-2 vaccine candidates and why the National Institute of Allergy and Infectious Diseases provided financial, technical, and/or clinical support. Indeed, once the amino acid sequence of a protein can be determined (a relatively easy task these days) it’s straightforward to synthesize mRNA in the lab – and it can be done incredibly fast. It is reported that the mRNA code for the vaccine by Moderna was made in 2 days and production development was completed in about 2 months.2
A 2007 World Health Organization report noted that infectious diseases are emerging at “the historically unprecedented rate of one per year.”3 Severe acute respiratory syndrome (SARS), Zika, Ebola, and avian and swine flu are recent examples. For most vaccines against emerging diseases, the challenge is about speed: developing and manufacturing a vaccine and getting it to persons who need it as quickly as possible. The current seasonal flu vaccine takes about 6 months to develop; it takes years for most of the traditional vaccines. That’s why once the infrastructure is in place, mRNA vaccines may prove to offer a big advantage as vaccines against emerging pathogens.
Early efficacy results have been surprising
Both vaccines were reported to produce about 95% efficacy in the final analysis. That was unexpectedly high because most vaccines for respiratory illness achieve efficacy of 60%-80%, e.g., flu vaccines. However, the efficacy rate may drop as time goes by because stimulation of short-term immunity would be in the earliest reported results.

Preventing SARS-CoV-2 cases is an important aspect of a coronavirus vaccine, but preventing severe illness is especially important considering that severe cases can result in prolonged intubation/artificial ventilation, prolonged disability and death. Pfizer/BioNTech had not released any data on the breakdown of severe cases as this column was finalized. In Moderna’s clinical trial, a secondary endpoint analyzed severe cases of COVID-19 and included 30 severe cases (as defined in the study protocol) in this analysis. All 30 cases occurred in the placebo group and none in the mRNA-1273–vaccinated group. In the Pfizer/BioNTech trial there were too few cases of severe illness to calculate efficacy.
Duration of immunity and need to revaccinate after initial primary vaccination are unknowns. Study of induction of B- and T-cell memory and levels of long-term protection have not been reported thus far.
Could mRNA COVID-19 vaccines be dangerous in the long term?
These will be the first-ever mRNA vaccines brought to market for humans. In order to receive Food and Drug Administration approval, the companies had to prove there were no immediate or short-term negative adverse effects from the vaccines. The companies reported that their independent data-monitoring committees hadn’t “reported any serious safety concerns.” However, fairly significant local reactions at the site of injection, fever, malaise, and fatigue occur with modest frequency following vaccinations with these products, reportedly in 10%-15% of vaccinees. Overall, the immediate reaction profile appears to be more severe than what occurs following seasonal influenza vaccination. When mass inoculations with these completely new and revolutionary vaccines begins, we will know virtually nothing about their long-term side effects. The possibility of systemic inflammatory responses that could lead to autoimmune conditions, persistence of the induced immunogen expression, development of autoreactive antibodies, and toxic effects of delivery components have been raised as theoretical concerns.4-6 None of these theoretical risks have been observed to date and postmarketing phase 4 safety monitoring studies are in place from the Centers for Disease Control and Prevention and the companies that produce the vaccines. This is a risk public health authorities are willing to take because the risk to benefit calculation strongly favors taking theoretical risks, compared with clear benefits in preventing severe illnesses and death.
What about availability?
Pfizer/BioNTech expects to be able to produce up to 50 million vaccine doses in 2020 and up to 1.3 billion doses in 2021. Moderna expects to produce 20 million doses by the end of 2020, and 500 million to 1 billion doses in 2021. Storage requirements are inherent to the composition of the vaccines with their differing lipid nanoparticle delivery systems. Pfizer/BioNTech’s BNT162b2 has to be stored and transported at –80° C, which requires specialized freezers, which most doctors’ offices and pharmacies are unlikely to have on site, or dry ice containers. Once the vaccine is thawed, it can only remain in the refrigerator for 24 hours. Moderna’s mRNA-1273 will be much easier to distribute. The vaccine is stable in a standard freezer at –20° C for up to 6 months, in a refrigerator for up to 30 days within that 6-month shelf life, and at room temperature for up to 12 hours.
Timelines and testing other vaccines
Strong efficacy data from the two leading SARS-CoV-2 vaccines and emergency-use authorization Food and Drug Administration approval suggest the window for testing additional vaccine candidates in the United States could soon start to close. Of the more than 200 vaccines in development for SARS-CoV-2, at least 7 have a chance of gathering pivotal data before the front-runners become broadly available.
Testing diverse vaccine candidates, based on different technologies, is important for ensuring sufficient supply and could lead to products with tolerability and safety profiles that make them better suited, or more attractive, to subsets of the population. Different vaccine antigens and technologies also may yield different durations of protection, a question that will not be answered until long after the first products are on the market.
AstraZeneca enrolled about 23,000 subjects into its two phase 3 trials of AZD1222 (ChAdOx1 nCoV-19): a 40,000-subject U.S. trial and a 10,000-subject study in Brazil. AstraZeneca’s AZD1222, developed with the University of Oxford (England), uses a replication defective simian adenovirus vector called ChAdOx1.AZD1222 which encodes the SARS-CoV-2 spike protein. After injection, the viral vector delivers recombinant DNA that is decoded to mRNA, followed by mRNA decoding to become a protein. A serendipitous manufacturing error for the first 3,000 doses resulted in a half dose for those subjects before the error was discovered. Full doses were given to those subjects on second injections and those subjects showed 90% efficacy. Subjects who received 2 full doses showed 62% efficacy. A vaccine cannot be licensed based on 3,000 subjects so AstraZeneca has started a new phase 3 trial involving many more subjects to receive the combination lower dose followed by the full dose.
Johnson and Johnson (J&J) started its phase 3 trial evaluating a single dose of JNJ-78436735 in September. Phase 3 data may be reported by the end of2020. In November, J&J announced it was starting a second phase 3 trial to test two doses of the candidate. J&J’s JNJ-78436735 encodes the SARS-CoV-2 spike protein in an adenovirus serotype 26 (Ad26) vector, which is one of the two adenovirus vectors used in Sputnik V, the Russian vaccine reported to have 90% efficacy at an early interim analysis.
Sanofi and Novavax are both developing protein-based vaccines, a proven modality. Sanofi, in partnership with GlaxoSmithKline started a phase 1/2 clinical trial in the Fall 2020 with plans to commence a phase 3 trial in late December. Sanofi developed the protein ingredients and GlaxoSmithKline added one of their novel adjuvants. Novavax expects data from a U.K. phase 3 trial of NVX-CoV2373 in early 2021 and began a U.S. phase 3 study in late November. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike protein and contains Novavax’s patented saponin-based Matrix-M adjuvant.
Inovio Pharmaceuticals was gearing up to start a U.S. phase 2/3 trial of DNA vaccine INO-4800 by the end of 2020.
After Moderna and Pfizer-BioNTech, CureVac has the next most advanced mRNA vaccine. It was planned that a phase 2b/3 trial of CVnCoV would be conducted in Europe, Latin America, Africa, and Asia. Sanofi is also developing a mRNA vaccine as a second product in addition to its protein vaccine.
Vaxxinity planned to begin phase 3 testing of UB-612, a multitope peptide–based vaccine, in Brazil by the end of 2020.
However, emergency-use authorizations for the Pfizer and Moderna vaccines could hinder trial recruitment in at least two ways. Given the gravity of the pandemic, some stakeholders believe it would be ethical to unblind ongoing trials to give subjects the opportunity to switch to a vaccine proven to be effective. Even if unblinding doesn’t occur, as the two authorized vaccines start to become widely available, volunteering for clinical trials may become less attractive.
Dr. Pichichero is a specialist in pediatric infectious diseases, and director of the Research Institute at Rochester (N.Y.) General Hospital. He said he has no relevant financial disclosures. Email Dr. Pichichero at [email protected].
References
1. Wolff JA et al. Science. 1990 Mar 23. doi: 10.1126/science.1690918.
2. Jackson LA et al. N Engl J Med. 2020 Nov 12. doi: 10.1056/NEJMoa2022483.
3. Prentice T and Reinders LT. The world health report 2007. (Geneva Switzerland: World Health Organization, 2007).
4. Peck KM and Lauring AS. J Virol. 2018. doi: 10.1128/JVI.01031-17.
5. Pepini T et al. J Immunol. 2017 May 15. doi: 10.4049/jimmunol.1601877.
6. Theofilopoulos AN et al. Annu Rev Immunol. 2005. doi: 10.1146/annurev.immunol.23.021704.115843.
In mid-November, Pfizer/BioNTech were the first with surprising positive protection interim data for their coronavirus vaccine, BNT162b2. A week later, Moderna released interim efficacy results showing its coronavirus vaccine, mRNA-1273, also protected patients from developing SARS-CoV-2 infections. Both studies included mostly healthy adults. A diverse ethnic and racial vaccinated population was included. A reasonable number of persons aged over 65 years, and persons with stable compromising medical conditions were included. Adolescents aged 16 years and over were included. Younger adolescents have been vaccinated or such studies are in the planning or early implementation stage as 2020 came to a close.
These are new and revolutionary vaccines, although the ability to inject mRNA into animals dates back to 1990, technological advances today make it a reality.1 Traditional vaccines typically involve injection with antigens such as purified proteins or polysaccharides or inactivated/attenuated viruses. In the case of Pfizer’s and Moderna’s vaccines, the mRNA provides the genetic information to synthesize the spike protein that the SARS-CoV-2 virus uses to attach to and infect human cells. Each type of vaccine is packaged in proprietary lipid nanoparticles to protect the mRNA from rapid degradation, and the nanoparticles serve as an adjuvant to attract immune cells to the site of injection. (The properties of the respective lipid nanoparticle packaging may be the factor that impacts storage requirements discussed below.) When injected into muscle (myocyte), the lipid nanoparticles containing the mRNA inside are taken into muscle cells, where the cytoplasmic ribosomes detect and decode the mRNA resulting in the production of the spike protein antigen. It should be noted that the mRNA does not enter the nucleus, where the genetic information (DNA) of a cell is located, and can’t be reproduced or integrated into the DNA. The antigen is exported to the myocyte cell surface where the immune system’s antigen presenting cells detect the protein, ingest it, and take it to regional lymph nodes where interactions with T cells and B cells results in antibodies, T cell–mediated immunity, and generation of immune memory T cells and B cells. A particular subset of T cells – cytotoxic or killer T cells – destroy cells that have been infected by a pathogen. The SARS-CoV-2 mRNA vaccine from Pfizer was reported to induce powerful cytotoxic T-cell responses. Results for Moderna’s vaccine had not been reported at the time this column was prepared, but I anticipate the same positive results.
The revolutionary aspect of mRNA vaccines is the speed at which they can be designed and produced. This is why they lead the pack among the SARS-CoV-2 vaccine candidates and why the National Institute of Allergy and Infectious Diseases provided financial, technical, and/or clinical support. Indeed, once the amino acid sequence of a protein can be determined (a relatively easy task these days) it’s straightforward to synthesize mRNA in the lab – and it can be done incredibly fast. It is reported that the mRNA code for the vaccine by Moderna was made in 2 days and production development was completed in about 2 months.2
A 2007 World Health Organization report noted that infectious diseases are emerging at “the historically unprecedented rate of one per year.”3 Severe acute respiratory syndrome (SARS), Zika, Ebola, and avian and swine flu are recent examples. For most vaccines against emerging diseases, the challenge is about speed: developing and manufacturing a vaccine and getting it to persons who need it as quickly as possible. The current seasonal flu vaccine takes about 6 months to develop; it takes years for most of the traditional vaccines. That’s why once the infrastructure is in place, mRNA vaccines may prove to offer a big advantage as vaccines against emerging pathogens.
Early efficacy results have been surprising
Both vaccines were reported to produce about 95% efficacy in the final analysis. That was unexpectedly high because most vaccines for respiratory illness achieve efficacy of 60%-80%, e.g., flu vaccines. However, the efficacy rate may drop as time goes by because stimulation of short-term immunity would be in the earliest reported results.
Preventing SARS-CoV-2 cases is an important aspect of a coronavirus vaccine, but preventing severe illness is especially important considering that severe cases can result in prolonged intubation/artificial ventilation, prolonged disability and death. Pfizer/BioNTech had not released any data on the breakdown of severe cases as this column was finalized. In Moderna’s clinical trial, a secondary endpoint analyzed severe cases of COVID-19 and included 30 severe cases (as defined in the study protocol) in this analysis. All 30 cases occurred in the placebo group and none in the mRNA-1273–vaccinated group. In the Pfizer/BioNTech trial there were too few cases of severe illness to calculate efficacy.
Duration of immunity and need to revaccinate after initial primary vaccination are unknowns. Study of induction of B- and T-cell memory and levels of long-term protection have not been reported thus far.
Could mRNA COVID-19 vaccines be dangerous in the long term?
These will be the first-ever mRNA vaccines brought to market for humans. In order to receive Food and Drug Administration approval, the companies had to prove there were no immediate or short-term negative adverse effects from the vaccines. The companies reported that their independent data-monitoring committees hadn’t “reported any serious safety concerns.” However, fairly significant local reactions at the site of injection, fever, malaise, and fatigue occur with modest frequency following vaccinations with these products, reportedly in 10%-15% of vaccinees. Overall, the immediate reaction profile appears to be more severe than what occurs following seasonal influenza vaccination. When mass inoculations with these completely new and revolutionary vaccines begins, we will know virtually nothing about their long-term side effects. The possibility of systemic inflammatory responses that could lead to autoimmune conditions, persistence of the induced immunogen expression, development of autoreactive antibodies, and toxic effects of delivery components have been raised as theoretical concerns.4-6 None of these theoretical risks have been observed to date and postmarketing phase 4 safety monitoring studies are in place from the Centers for Disease Control and Prevention and the companies that produce the vaccines. This is a risk public health authorities are willing to take because the risk to benefit calculation strongly favors taking theoretical risks, compared with clear benefits in preventing severe illnesses and death.
What about availability?
Pfizer/BioNTech expects to be able to produce up to 50 million vaccine doses in 2020 and up to 1.3 billion doses in 2021. Moderna expects to produce 20 million doses by the end of 2020, and 500 million to 1 billion doses in 2021. Storage requirements are inherent to the composition of the vaccines with their differing lipid nanoparticle delivery systems. Pfizer/BioNTech’s BNT162b2 has to be stored and transported at –80° C, which requires specialized freezers, which most doctors’ offices and pharmacies are unlikely to have on site, or dry ice containers. Once the vaccine is thawed, it can only remain in the refrigerator for 24 hours. Moderna’s mRNA-1273 will be much easier to distribute. The vaccine is stable in a standard freezer at –20° C for up to 6 months, in a refrigerator for up to 30 days within that 6-month shelf life, and at room temperature for up to 12 hours.
Timelines and testing other vaccines
Strong efficacy data from the two leading SARS-CoV-2 vaccines and emergency-use authorization Food and Drug Administration approval suggest the window for testing additional vaccine candidates in the United States could soon start to close. Of the more than 200 vaccines in development for SARS-CoV-2, at least 7 have a chance of gathering pivotal data before the front-runners become broadly available.
Testing diverse vaccine candidates, based on different technologies, is important for ensuring sufficient supply and could lead to products with tolerability and safety profiles that make them better suited, or more attractive, to subsets of the population. Different vaccine antigens and technologies also may yield different durations of protection, a question that will not be answered until long after the first products are on the market.
AstraZeneca enrolled about 23,000 subjects into its two phase 3 trials of AZD1222 (ChAdOx1 nCoV-19): a 40,000-subject U.S. trial and a 10,000-subject study in Brazil. AstraZeneca’s AZD1222, developed with the University of Oxford (England), uses a replication defective simian adenovirus vector called ChAdOx1.AZD1222 which encodes the SARS-CoV-2 spike protein. After injection, the viral vector delivers recombinant DNA that is decoded to mRNA, followed by mRNA decoding to become a protein. A serendipitous manufacturing error for the first 3,000 doses resulted in a half dose for those subjects before the error was discovered. Full doses were given to those subjects on second injections and those subjects showed 90% efficacy. Subjects who received 2 full doses showed 62% efficacy. A vaccine cannot be licensed based on 3,000 subjects so AstraZeneca has started a new phase 3 trial involving many more subjects to receive the combination lower dose followed by the full dose.
Johnson and Johnson (J&J) started its phase 3 trial evaluating a single dose of JNJ-78436735 in September. Phase 3 data may be reported by the end of2020. In November, J&J announced it was starting a second phase 3 trial to test two doses of the candidate. J&J’s JNJ-78436735 encodes the SARS-CoV-2 spike protein in an adenovirus serotype 26 (Ad26) vector, which is one of the two adenovirus vectors used in Sputnik V, the Russian vaccine reported to have 90% efficacy at an early interim analysis.
Sanofi and Novavax are both developing protein-based vaccines, a proven modality. Sanofi, in partnership with GlaxoSmithKline started a phase 1/2 clinical trial in the Fall 2020 with plans to commence a phase 3 trial in late December. Sanofi developed the protein ingredients and GlaxoSmithKline added one of their novel adjuvants. Novavax expects data from a U.K. phase 3 trial of NVX-CoV2373 in early 2021 and began a U.S. phase 3 study in late November. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike protein and contains Novavax’s patented saponin-based Matrix-M adjuvant.
Inovio Pharmaceuticals was gearing up to start a U.S. phase 2/3 trial of DNA vaccine INO-4800 by the end of 2020.
After Moderna and Pfizer-BioNTech, CureVac has the next most advanced mRNA vaccine. It was planned that a phase 2b/3 trial of CVnCoV would be conducted in Europe, Latin America, Africa, and Asia. Sanofi is also developing a mRNA vaccine as a second product in addition to its protein vaccine.
Vaxxinity planned to begin phase 3 testing of UB-612, a multitope peptide–based vaccine, in Brazil by the end of 2020.
However, emergency-use authorizations for the Pfizer and Moderna vaccines could hinder trial recruitment in at least two ways. Given the gravity of the pandemic, some stakeholders believe it would be ethical to unblind ongoing trials to give subjects the opportunity to switch to a vaccine proven to be effective. Even if unblinding doesn’t occur, as the two authorized vaccines start to become widely available, volunteering for clinical trials may become less attractive.
Dr. Pichichero is a specialist in pediatric infectious diseases, and director of the Research Institute at Rochester (N.Y.) General Hospital. He said he has no relevant financial disclosures. Email Dr. Pichichero at [email protected].
References
1. Wolff JA et al. Science. 1990 Mar 23. doi: 10.1126/science.1690918.
2. Jackson LA et al. N Engl J Med. 2020 Nov 12. doi: 10.1056/NEJMoa2022483.
3. Prentice T and Reinders LT. The world health report 2007. (Geneva Switzerland: World Health Organization, 2007).
4. Peck KM and Lauring AS. J Virol. 2018. doi: 10.1128/JVI.01031-17.
5. Pepini T et al. J Immunol. 2017 May 15. doi: 10.4049/jimmunol.1601877.
6. Theofilopoulos AN et al. Annu Rev Immunol. 2005. doi: 10.1146/annurev.immunol.23.021704.115843.
In mid-November, Pfizer/BioNTech were the first with surprising positive protection interim data for their coronavirus vaccine, BNT162b2. A week later, Moderna released interim efficacy results showing its coronavirus vaccine, mRNA-1273, also protected patients from developing SARS-CoV-2 infections. Both studies included mostly healthy adults. A diverse ethnic and racial vaccinated population was included. A reasonable number of persons aged over 65 years, and persons with stable compromising medical conditions were included. Adolescents aged 16 years and over were included. Younger adolescents have been vaccinated or such studies are in the planning or early implementation stage as 2020 came to a close.
These are new and revolutionary vaccines, although the ability to inject mRNA into animals dates back to 1990, technological advances today make it a reality.1 Traditional vaccines typically involve injection with antigens such as purified proteins or polysaccharides or inactivated/attenuated viruses. In the case of Pfizer’s and Moderna’s vaccines, the mRNA provides the genetic information to synthesize the spike protein that the SARS-CoV-2 virus uses to attach to and infect human cells. Each type of vaccine is packaged in proprietary lipid nanoparticles to protect the mRNA from rapid degradation, and the nanoparticles serve as an adjuvant to attract immune cells to the site of injection. (The properties of the respective lipid nanoparticle packaging may be the factor that impacts storage requirements discussed below.) When injected into muscle (myocyte), the lipid nanoparticles containing the mRNA inside are taken into muscle cells, where the cytoplasmic ribosomes detect and decode the mRNA resulting in the production of the spike protein antigen. It should be noted that the mRNA does not enter the nucleus, where the genetic information (DNA) of a cell is located, and can’t be reproduced or integrated into the DNA. The antigen is exported to the myocyte cell surface where the immune system’s antigen presenting cells detect the protein, ingest it, and take it to regional lymph nodes where interactions with T cells and B cells results in antibodies, T cell–mediated immunity, and generation of immune memory T cells and B cells. A particular subset of T cells – cytotoxic or killer T cells – destroy cells that have been infected by a pathogen. The SARS-CoV-2 mRNA vaccine from Pfizer was reported to induce powerful cytotoxic T-cell responses. Results for Moderna’s vaccine had not been reported at the time this column was prepared, but I anticipate the same positive results.
The revolutionary aspect of mRNA vaccines is the speed at which they can be designed and produced. This is why they lead the pack among the SARS-CoV-2 vaccine candidates and why the National Institute of Allergy and Infectious Diseases provided financial, technical, and/or clinical support. Indeed, once the amino acid sequence of a protein can be determined (a relatively easy task these days) it’s straightforward to synthesize mRNA in the lab – and it can be done incredibly fast. It is reported that the mRNA code for the vaccine by Moderna was made in 2 days and production development was completed in about 2 months.2
A 2007 World Health Organization report noted that infectious diseases are emerging at “the historically unprecedented rate of one per year.”3 Severe acute respiratory syndrome (SARS), Zika, Ebola, and avian and swine flu are recent examples. For most vaccines against emerging diseases, the challenge is about speed: developing and manufacturing a vaccine and getting it to persons who need it as quickly as possible. The current seasonal flu vaccine takes about 6 months to develop; it takes years for most of the traditional vaccines. That’s why once the infrastructure is in place, mRNA vaccines may prove to offer a big advantage as vaccines against emerging pathogens.
Early efficacy results have been surprising
Both vaccines were reported to produce about 95% efficacy in the final analysis. That was unexpectedly high because most vaccines for respiratory illness achieve efficacy of 60%-80%, e.g., flu vaccines. However, the efficacy rate may drop as time goes by because stimulation of short-term immunity would be in the earliest reported results.
Preventing SARS-CoV-2 cases is an important aspect of a coronavirus vaccine, but preventing severe illness is especially important considering that severe cases can result in prolonged intubation/artificial ventilation, prolonged disability and death. Pfizer/BioNTech had not released any data on the breakdown of severe cases as this column was finalized. In Moderna’s clinical trial, a secondary endpoint analyzed severe cases of COVID-19 and included 30 severe cases (as defined in the study protocol) in this analysis. All 30 cases occurred in the placebo group and none in the mRNA-1273–vaccinated group. In the Pfizer/BioNTech trial there were too few cases of severe illness to calculate efficacy.
Duration of immunity and need to revaccinate after initial primary vaccination are unknowns. Study of induction of B- and T-cell memory and levels of long-term protection have not been reported thus far.
Could mRNA COVID-19 vaccines be dangerous in the long term?
These will be the first-ever mRNA vaccines brought to market for humans. In order to receive Food and Drug Administration approval, the companies had to prove there were no immediate or short-term negative adverse effects from the vaccines. The companies reported that their independent data-monitoring committees hadn’t “reported any serious safety concerns.” However, fairly significant local reactions at the site of injection, fever, malaise, and fatigue occur with modest frequency following vaccinations with these products, reportedly in 10%-15% of vaccinees. Overall, the immediate reaction profile appears to be more severe than what occurs following seasonal influenza vaccination. When mass inoculations with these completely new and revolutionary vaccines begins, we will know virtually nothing about their long-term side effects. The possibility of systemic inflammatory responses that could lead to autoimmune conditions, persistence of the induced immunogen expression, development of autoreactive antibodies, and toxic effects of delivery components have been raised as theoretical concerns.4-6 None of these theoretical risks have been observed to date and postmarketing phase 4 safety monitoring studies are in place from the Centers for Disease Control and Prevention and the companies that produce the vaccines. This is a risk public health authorities are willing to take because the risk to benefit calculation strongly favors taking theoretical risks, compared with clear benefits in preventing severe illnesses and death.
What about availability?
Pfizer/BioNTech expects to be able to produce up to 50 million vaccine doses in 2020 and up to 1.3 billion doses in 2021. Moderna expects to produce 20 million doses by the end of 2020, and 500 million to 1 billion doses in 2021. Storage requirements are inherent to the composition of the vaccines with their differing lipid nanoparticle delivery systems. Pfizer/BioNTech’s BNT162b2 has to be stored and transported at –80° C, which requires specialized freezers, which most doctors’ offices and pharmacies are unlikely to have on site, or dry ice containers. Once the vaccine is thawed, it can only remain in the refrigerator for 24 hours. Moderna’s mRNA-1273 will be much easier to distribute. The vaccine is stable in a standard freezer at –20° C for up to 6 months, in a refrigerator for up to 30 days within that 6-month shelf life, and at room temperature for up to 12 hours.
Timelines and testing other vaccines
Strong efficacy data from the two leading SARS-CoV-2 vaccines and emergency-use authorization Food and Drug Administration approval suggest the window for testing additional vaccine candidates in the United States could soon start to close. Of the more than 200 vaccines in development for SARS-CoV-2, at least 7 have a chance of gathering pivotal data before the front-runners become broadly available.
Testing diverse vaccine candidates, based on different technologies, is important for ensuring sufficient supply and could lead to products with tolerability and safety profiles that make them better suited, or more attractive, to subsets of the population. Different vaccine antigens and technologies also may yield different durations of protection, a question that will not be answered until long after the first products are on the market.
AstraZeneca enrolled about 23,000 subjects into its two phase 3 trials of AZD1222 (ChAdOx1 nCoV-19): a 40,000-subject U.S. trial and a 10,000-subject study in Brazil. AstraZeneca’s AZD1222, developed with the University of Oxford (England), uses a replication defective simian adenovirus vector called ChAdOx1.AZD1222 which encodes the SARS-CoV-2 spike protein. After injection, the viral vector delivers recombinant DNA that is decoded to mRNA, followed by mRNA decoding to become a protein. A serendipitous manufacturing error for the first 3,000 doses resulted in a half dose for those subjects before the error was discovered. Full doses were given to those subjects on second injections and those subjects showed 90% efficacy. Subjects who received 2 full doses showed 62% efficacy. A vaccine cannot be licensed based on 3,000 subjects so AstraZeneca has started a new phase 3 trial involving many more subjects to receive the combination lower dose followed by the full dose.
Johnson and Johnson (J&J) started its phase 3 trial evaluating a single dose of JNJ-78436735 in September. Phase 3 data may be reported by the end of2020. In November, J&J announced it was starting a second phase 3 trial to test two doses of the candidate. J&J’s JNJ-78436735 encodes the SARS-CoV-2 spike protein in an adenovirus serotype 26 (Ad26) vector, which is one of the two adenovirus vectors used in Sputnik V, the Russian vaccine reported to have 90% efficacy at an early interim analysis.
Sanofi and Novavax are both developing protein-based vaccines, a proven modality. Sanofi, in partnership with GlaxoSmithKline started a phase 1/2 clinical trial in the Fall 2020 with plans to commence a phase 3 trial in late December. Sanofi developed the protein ingredients and GlaxoSmithKline added one of their novel adjuvants. Novavax expects data from a U.K. phase 3 trial of NVX-CoV2373 in early 2021 and began a U.S. phase 3 study in late November. NVX-CoV2373 was created using Novavax’ recombinant nanoparticle technology to generate antigen derived from the coronavirus spike protein and contains Novavax’s patented saponin-based Matrix-M adjuvant.
Inovio Pharmaceuticals was gearing up to start a U.S. phase 2/3 trial of DNA vaccine INO-4800 by the end of 2020.
After Moderna and Pfizer-BioNTech, CureVac has the next most advanced mRNA vaccine. It was planned that a phase 2b/3 trial of CVnCoV would be conducted in Europe, Latin America, Africa, and Asia. Sanofi is also developing a mRNA vaccine as a second product in addition to its protein vaccine.
Vaxxinity planned to begin phase 3 testing of UB-612, a multitope peptide–based vaccine, in Brazil by the end of 2020.
However, emergency-use authorizations for the Pfizer and Moderna vaccines could hinder trial recruitment in at least two ways. Given the gravity of the pandemic, some stakeholders believe it would be ethical to unblind ongoing trials to give subjects the opportunity to switch to a vaccine proven to be effective. Even if unblinding doesn’t occur, as the two authorized vaccines start to become widely available, volunteering for clinical trials may become less attractive.
Dr. Pichichero is a specialist in pediatric infectious diseases, and director of the Research Institute at Rochester (N.Y.) General Hospital. He said he has no relevant financial disclosures. Email Dr. Pichichero at [email protected].
References
1. Wolff JA et al. Science. 1990 Mar 23. doi: 10.1126/science.1690918.
2. Jackson LA et al. N Engl J Med. 2020 Nov 12. doi: 10.1056/NEJMoa2022483.
3. Prentice T and Reinders LT. The world health report 2007. (Geneva Switzerland: World Health Organization, 2007).
4. Peck KM and Lauring AS. J Virol. 2018. doi: 10.1128/JVI.01031-17.
5. Pepini T et al. J Immunol. 2017 May 15. doi: 10.4049/jimmunol.1601877.
6. Theofilopoulos AN et al. Annu Rev Immunol. 2005. doi: 10.1146/annurev.immunol.23.021704.115843.
COVID-19 neurologic fallout not limited to the severely ill
Serious neurologic complications in patients with COVID-19 are not limited to the severely ill, new research confirms.
“We found a range of neurologic diagnoses, including stroke and seizures, among hospitalized patients with COVID-19 and the majority were not critically ill, suggesting that these complications are not limited just to those patients who require ICU care or a ventilator,” study investigator Pria Anand, MD, division of neuro-infectious diseases, Boston University, said in an interview.
The study was published online Dec. 9 in Neurology Clinical Practice.
‘Moderately severe’ disability
For the study, the investigators reviewed the medical records of 74 adults (mean age, 64 years) who were hospitalized with COVID-19 and evaluated for neurologic conditions at Boston Medical Center, a safety-net hospital caring primarily for underserved, low-income, racial and ethnic minority populations.
The most common COVID-19 symptoms on arrival to the hospital were cough (39%), dyspnea (36%), and fever (34%). Eleven patients required intubation (15%) and 28 required some form of supplemental oxygen (38%). Thirty-four patients required intensive care (46%).
The most common neurologic COVID-19 symptoms at presentation were altered mental status (53%), myalgia (24%), fatigue (24%), and headache (18%).
After neurologic assessment, the most common final neurologic diagnosis was multifactorial or toxic-metabolic encephalopathy (35%), followed by seizure (20%), ischemic stroke (20%), primary movement disorder (9%), peripheral neuropathy (8%), and hemorrhagic stroke (4%).
Three patients (4%) suffered traumatic brain injuries after falling in their homes after developing COVID-19.
Ten (14%) patients died in the hospital. Survivors had “moderately severe” disability at discharge (median modified Rankin Scale score of 4 from a preadmission mRS score of 2) and many were discharged to nursing facilities or rehabilitation hospitals.
“Although we do not have data on their posthospital course, this suggests that patients with neurologic complications of COVID-19 are likely to require ongoing rehabilitation, even after they leave the hospital,” Dr. Anand, a member of the American Academy of Neurology, said in an interview.
“There are a diverse range of mechanisms by which COVID-19 can cause neurologic complications,” Dr. Anand said.
“These complications can result from the body’s immunological response to the virus (e.g., Guillain-Barré syndrome, an autoimmune disorder affecting the nerves), from having a systemic severe illness (e.g., brain injury as a result of insufficient oxygenation), from the increased tendency to form blood clots (e.g., stroke), from worsening of preexisting neurologic disorders, and possibly from involvement of the nervous system by the virus itself,” she explained.
The researchers said more study is needed to characterize the infectious and postinfectious neurologic complications of COVID-19 in diverse patient populations.
Lingering issues
In an interview, Kenneth L. Tyler, MD, chair of neurology, University of Colorado, Denver, noted that this is one of the larger series published to date of the neurologic complications associated with COVID-19, and the first to come from a U.S. safety-net hospital in a large metropolitan area.
“Overall, the types and categories of neurological complications reported including encephalopathy (35%) and acute cerebrovascular events (~20%) are similar to those reported elsewhere,” said Dr. Tyler.
However, the frequency of stroke (~20%) is higher than in some other reports, “likely reflecting the comorbidities such as diabetes, hypertension, limited access to care [that are] present in this population,” he said.
Dr. Tyler also noted that the “relatively high frequency” of primary movement disorders, notably myoclonus, “hasn’t been particularly well recognized or described, although one of the authors has written on this in COVID-19, so perhaps there is a bit of an ‘ascertainment bias’ – as they were looking harder for it?”
Finally, he noted, it’s important to understand that all the published studies “vary tremendously in the populations they examine, so direct comparisons can be difficult.”
Also weighing in on the report in an interview, Richard Temes, MD, director, Northwell Health’s Center for Neurocritical Care in Manhasset, N.Y., said neurologic problems have been noted since the start of COVID-19 and have been well described.
“It’s common for patients to present with very nonspecific neurological complaints like confusion, disorientation, altered mental status, lethargy, but also neurological disease such as strokes, brain hemorrhages, and seizures are quite common as well,” said Dr. Temes.
He also noted that a number of patients with COVID-19 will have “lingering effects, especially patients who are hospitalized, that can range from memory deficit, cognitive slowing, and trouble with activities of daily living and depression.
“These effects can occur with any patient who is hospitalized for a [significant] period of time, especially in the intensive care unit, so it’s hard to tease out whether or not this is truly from COVID itself or if it’s just being a survivor from a very severe, critical illness. We don’t know yet. We need more data on that,” he cautioned.
A version of this article originally appeared on Medscape.com.
Serious neurologic complications in patients with COVID-19 are not limited to the severely ill, new research confirms.
“We found a range of neurologic diagnoses, including stroke and seizures, among hospitalized patients with COVID-19 and the majority were not critically ill, suggesting that these complications are not limited just to those patients who require ICU care or a ventilator,” study investigator Pria Anand, MD, division of neuro-infectious diseases, Boston University, said in an interview.
The study was published online Dec. 9 in Neurology Clinical Practice.
‘Moderately severe’ disability
For the study, the investigators reviewed the medical records of 74 adults (mean age, 64 years) who were hospitalized with COVID-19 and evaluated for neurologic conditions at Boston Medical Center, a safety-net hospital caring primarily for underserved, low-income, racial and ethnic minority populations.
The most common COVID-19 symptoms on arrival to the hospital were cough (39%), dyspnea (36%), and fever (34%). Eleven patients required intubation (15%) and 28 required some form of supplemental oxygen (38%). Thirty-four patients required intensive care (46%).
The most common neurologic COVID-19 symptoms at presentation were altered mental status (53%), myalgia (24%), fatigue (24%), and headache (18%).
After neurologic assessment, the most common final neurologic diagnosis was multifactorial or toxic-metabolic encephalopathy (35%), followed by seizure (20%), ischemic stroke (20%), primary movement disorder (9%), peripheral neuropathy (8%), and hemorrhagic stroke (4%).
Three patients (4%) suffered traumatic brain injuries after falling in their homes after developing COVID-19.
Ten (14%) patients died in the hospital. Survivors had “moderately severe” disability at discharge (median modified Rankin Scale score of 4 from a preadmission mRS score of 2) and many were discharged to nursing facilities or rehabilitation hospitals.
“Although we do not have data on their posthospital course, this suggests that patients with neurologic complications of COVID-19 are likely to require ongoing rehabilitation, even after they leave the hospital,” Dr. Anand, a member of the American Academy of Neurology, said in an interview.
“There are a diverse range of mechanisms by which COVID-19 can cause neurologic complications,” Dr. Anand said.
“These complications can result from the body’s immunological response to the virus (e.g., Guillain-Barré syndrome, an autoimmune disorder affecting the nerves), from having a systemic severe illness (e.g., brain injury as a result of insufficient oxygenation), from the increased tendency to form blood clots (e.g., stroke), from worsening of preexisting neurologic disorders, and possibly from involvement of the nervous system by the virus itself,” she explained.
The researchers said more study is needed to characterize the infectious and postinfectious neurologic complications of COVID-19 in diverse patient populations.
Lingering issues
In an interview, Kenneth L. Tyler, MD, chair of neurology, University of Colorado, Denver, noted that this is one of the larger series published to date of the neurologic complications associated with COVID-19, and the first to come from a U.S. safety-net hospital in a large metropolitan area.
“Overall, the types and categories of neurological complications reported including encephalopathy (35%) and acute cerebrovascular events (~20%) are similar to those reported elsewhere,” said Dr. Tyler.
However, the frequency of stroke (~20%) is higher than in some other reports, “likely reflecting the comorbidities such as diabetes, hypertension, limited access to care [that are] present in this population,” he said.
Dr. Tyler also noted that the “relatively high frequency” of primary movement disorders, notably myoclonus, “hasn’t been particularly well recognized or described, although one of the authors has written on this in COVID-19, so perhaps there is a bit of an ‘ascertainment bias’ – as they were looking harder for it?”
Finally, he noted, it’s important to understand that all the published studies “vary tremendously in the populations they examine, so direct comparisons can be difficult.”
Also weighing in on the report in an interview, Richard Temes, MD, director, Northwell Health’s Center for Neurocritical Care in Manhasset, N.Y., said neurologic problems have been noted since the start of COVID-19 and have been well described.
“It’s common for patients to present with very nonspecific neurological complaints like confusion, disorientation, altered mental status, lethargy, but also neurological disease such as strokes, brain hemorrhages, and seizures are quite common as well,” said Dr. Temes.
He also noted that a number of patients with COVID-19 will have “lingering effects, especially patients who are hospitalized, that can range from memory deficit, cognitive slowing, and trouble with activities of daily living and depression.
“These effects can occur with any patient who is hospitalized for a [significant] period of time, especially in the intensive care unit, so it’s hard to tease out whether or not this is truly from COVID itself or if it’s just being a survivor from a very severe, critical illness. We don’t know yet. We need more data on that,” he cautioned.
A version of this article originally appeared on Medscape.com.
Serious neurologic complications in patients with COVID-19 are not limited to the severely ill, new research confirms.
“We found a range of neurologic diagnoses, including stroke and seizures, among hospitalized patients with COVID-19 and the majority were not critically ill, suggesting that these complications are not limited just to those patients who require ICU care or a ventilator,” study investigator Pria Anand, MD, division of neuro-infectious diseases, Boston University, said in an interview.
The study was published online Dec. 9 in Neurology Clinical Practice.
‘Moderately severe’ disability
For the study, the investigators reviewed the medical records of 74 adults (mean age, 64 years) who were hospitalized with COVID-19 and evaluated for neurologic conditions at Boston Medical Center, a safety-net hospital caring primarily for underserved, low-income, racial and ethnic minority populations.
The most common COVID-19 symptoms on arrival to the hospital were cough (39%), dyspnea (36%), and fever (34%). Eleven patients required intubation (15%) and 28 required some form of supplemental oxygen (38%). Thirty-four patients required intensive care (46%).
The most common neurologic COVID-19 symptoms at presentation were altered mental status (53%), myalgia (24%), fatigue (24%), and headache (18%).
After neurologic assessment, the most common final neurologic diagnosis was multifactorial or toxic-metabolic encephalopathy (35%), followed by seizure (20%), ischemic stroke (20%), primary movement disorder (9%), peripheral neuropathy (8%), and hemorrhagic stroke (4%).
Three patients (4%) suffered traumatic brain injuries after falling in their homes after developing COVID-19.
Ten (14%) patients died in the hospital. Survivors had “moderately severe” disability at discharge (median modified Rankin Scale score of 4 from a preadmission mRS score of 2) and many were discharged to nursing facilities or rehabilitation hospitals.
“Although we do not have data on their posthospital course, this suggests that patients with neurologic complications of COVID-19 are likely to require ongoing rehabilitation, even after they leave the hospital,” Dr. Anand, a member of the American Academy of Neurology, said in an interview.
“There are a diverse range of mechanisms by which COVID-19 can cause neurologic complications,” Dr. Anand said.
“These complications can result from the body’s immunological response to the virus (e.g., Guillain-Barré syndrome, an autoimmune disorder affecting the nerves), from having a systemic severe illness (e.g., brain injury as a result of insufficient oxygenation), from the increased tendency to form blood clots (e.g., stroke), from worsening of preexisting neurologic disorders, and possibly from involvement of the nervous system by the virus itself,” she explained.
The researchers said more study is needed to characterize the infectious and postinfectious neurologic complications of COVID-19 in diverse patient populations.
Lingering issues
In an interview, Kenneth L. Tyler, MD, chair of neurology, University of Colorado, Denver, noted that this is one of the larger series published to date of the neurologic complications associated with COVID-19, and the first to come from a U.S. safety-net hospital in a large metropolitan area.
“Overall, the types and categories of neurological complications reported including encephalopathy (35%) and acute cerebrovascular events (~20%) are similar to those reported elsewhere,” said Dr. Tyler.
However, the frequency of stroke (~20%) is higher than in some other reports, “likely reflecting the comorbidities such as diabetes, hypertension, limited access to care [that are] present in this population,” he said.
Dr. Tyler also noted that the “relatively high frequency” of primary movement disorders, notably myoclonus, “hasn’t been particularly well recognized or described, although one of the authors has written on this in COVID-19, so perhaps there is a bit of an ‘ascertainment bias’ – as they were looking harder for it?”
Finally, he noted, it’s important to understand that all the published studies “vary tremendously in the populations they examine, so direct comparisons can be difficult.”
Also weighing in on the report in an interview, Richard Temes, MD, director, Northwell Health’s Center for Neurocritical Care in Manhasset, N.Y., said neurologic problems have been noted since the start of COVID-19 and have been well described.
“It’s common for patients to present with very nonspecific neurological complaints like confusion, disorientation, altered mental status, lethargy, but also neurological disease such as strokes, brain hemorrhages, and seizures are quite common as well,” said Dr. Temes.
He also noted that a number of patients with COVID-19 will have “lingering effects, especially patients who are hospitalized, that can range from memory deficit, cognitive slowing, and trouble with activities of daily living and depression.
“These effects can occur with any patient who is hospitalized for a [significant] period of time, especially in the intensive care unit, so it’s hard to tease out whether or not this is truly from COVID itself or if it’s just being a survivor from a very severe, critical illness. We don’t know yet. We need more data on that,” he cautioned.
A version of this article originally appeared on Medscape.com.
Twincretin ‘impressive’: Topline data from phase 3 trial in diabetes
Tirzepatide, a novel subcutaneously injected drug that acts via two related but separate pathways of glucose control, produced strikingly positive effects in top-line results from the phase 3, placebo-controlled study SURPASS-1 in 478 adults with type 2 diabetes, according to a Dec. 9 press release from the manufacturer, Lilly.
The tirzepatide molecule exerts agonist effects at both the glucose-dependent insulinotropic polypeptide (GIP) receptor and the glucagon-like peptide-1 (GLP-1) receptor, and has been called a “twincretin” for its activity encompassing two different incretins. Phase 2 trial results caused excitement, with one physician calling the data “unbelievable” when reported in 2018.
SURPASS-1 enrolled patients who were very early in the course of their disease, had on average relatively mild elevation in glucose levels, and few metabolic comorbidities. They took one of three doses of the agent (5, 10, or 15 mg) as monotherapy or placebo for 40 weeks.
Julio Rosenstock, MD, said in the Lilly statement: “The study took a bold approach in assessing A1c targets. Not only did nearly 90% of all participants taking tirzepatide meet the standard A1c goal of less than 7%, more than half taking the highest dose also achieved an A1c less than 5.7%, the level seen in people without diabetes.”
Dr. Rosenstock is principal investigator of SURPASS-1 and director of the Dallas Diabetes Research Center in Texas.
The discontinuation rate in the high-dose group was 21.5% compared with less than 10% in the two lower-dose cohorts. Lilly said most of the dropouts “were due to the pandemic and family or work reasons.” The dropout rate in the placebo group was 14.8%.
These data were not included in the efficacy analysis, however, which “muddied” the analysis somewhat, one pharma analyst told BioPharma Dive.
Commenting on the new trial data, Ildiko Lingvay, MD, said in an interview: “I am very impressed with these results,” which are “unprecedented for any glucose-lowering medication that has ever been tested.”
Dr. Lingvay, of the department of internal medicine/endocrinology, and medical director, office of clinical trials management at UT Southwestern Medical Center, Dallas, was not involved in the study.
She added that the weight loss seen with tirzepatide “is equally impressive with greater than 10% of body weight loss above placebo achieved within 40 weeks of treatment and without any directed weight loss efforts.”
If the agent is eventually approved, “I am enthusiastic about the prospect of having another very powerful tool to address both diabetes and obesity,” she added.
The full results of SURPASS-1 will be presented at the American Diabetes Association 81st Scientific Sessions and published in a peer-reviewed journal in 2021.
SURPASS-1 is one of eight phase 3 studies of the drug, including five registration studies and one large 12,500-patient cardiovascular outcomes trial.
Tirzepatide patients lost up to 20 lb, side effect profile ‘reassuring’
In the study, patients had been recently diagnosed with type 2 diabetes (average duration, 4.8 years) and 54% were treatment-naive. Average baseline hemoglobin A1c was 7.9% and mean weight was 85.9 kg (189 pounds).
Patients started on a subcutaneous injectable dose of tirzepatide of 2.5 mg per week, which was titrated up to the final dose – 5, 10, or 15 mg – in 2.5-mg increments given as monotherapy for 40 weeks and compared with placebo.
Treatment with tirzepatide resulted in average reductions in A1c from baseline that ranged from 1.87% to 2.07%, depending on the dose, and were all significant compared with an increase of 0.4% with placebo.
The percentage of patients whose A1c fell to normal levels (less than 5.7%) ranged from 30.5% to 51.7%, compared with 0.9% among controls, and again, was significant for all doses.
Patients treated with tirzepatide also lost weight. Average weight reductions after 40 weeks were significant and ranged from 7.0 to 9.5 kg (15-21 pounds) compared with an average loss of 0.7 kg (1.5 pounds) among patients who received placebo.
The most common adverse events were gastrointestinal-related and mild to moderate in severity, and usually occurred during dose escalation.
Dr. Lingvay said the safety data reported are “reassuring, with side effects in the anticipated range and comparable with other medications in the GLP-1 agonist class.”
And no hypoglycemic (level 2, < 54 mg/dL) events were reported, “which is impressive considering the overall glucose level achieved,” she noted.
“I am eagerly awaiting the results of the other studies within the SURPASS program and hope those will confirm these initial findings and provide additional safety and efficacy information in a wider range of patients with type 2 diabetes,” she concluded.
Dr. Lingvay has reported receiving research funding, advisory/consulting fees, and/or other support from Novo Nordisk, Eli Lilly, Sanofi, AstraZeneca, Boehringer Ingelheim, Janssen, Intercept, Intarcia, Target Pharma, Merck, Pfizer, Novartis, GI Dynamics, Mylan, MannKind, Valeritas, Bayer, and Zealand Pharma.
A version of this article originally appeared on Medscape.com.
Tirzepatide, a novel subcutaneously injected drug that acts via two related but separate pathways of glucose control, produced strikingly positive effects in top-line results from the phase 3, placebo-controlled study SURPASS-1 in 478 adults with type 2 diabetes, according to a Dec. 9 press release from the manufacturer, Lilly.
The tirzepatide molecule exerts agonist effects at both the glucose-dependent insulinotropic polypeptide (GIP) receptor and the glucagon-like peptide-1 (GLP-1) receptor, and has been called a “twincretin” for its activity encompassing two different incretins. Phase 2 trial results caused excitement, with one physician calling the data “unbelievable” when reported in 2018.
SURPASS-1 enrolled patients who were very early in the course of their disease, had on average relatively mild elevation in glucose levels, and few metabolic comorbidities. They took one of three doses of the agent (5, 10, or 15 mg) as monotherapy or placebo for 40 weeks.
Julio Rosenstock, MD, said in the Lilly statement: “The study took a bold approach in assessing A1c targets. Not only did nearly 90% of all participants taking tirzepatide meet the standard A1c goal of less than 7%, more than half taking the highest dose also achieved an A1c less than 5.7%, the level seen in people without diabetes.”
Dr. Rosenstock is principal investigator of SURPASS-1 and director of the Dallas Diabetes Research Center in Texas.
The discontinuation rate in the high-dose group was 21.5% compared with less than 10% in the two lower-dose cohorts. Lilly said most of the dropouts “were due to the pandemic and family or work reasons.” The dropout rate in the placebo group was 14.8%.
These data were not included in the efficacy analysis, however, which “muddied” the analysis somewhat, one pharma analyst told BioPharma Dive.
Commenting on the new trial data, Ildiko Lingvay, MD, said in an interview: “I am very impressed with these results,” which are “unprecedented for any glucose-lowering medication that has ever been tested.”
Dr. Lingvay, of the department of internal medicine/endocrinology, and medical director, office of clinical trials management at UT Southwestern Medical Center, Dallas, was not involved in the study.
She added that the weight loss seen with tirzepatide “is equally impressive with greater than 10% of body weight loss above placebo achieved within 40 weeks of treatment and without any directed weight loss efforts.”
If the agent is eventually approved, “I am enthusiastic about the prospect of having another very powerful tool to address both diabetes and obesity,” she added.
The full results of SURPASS-1 will be presented at the American Diabetes Association 81st Scientific Sessions and published in a peer-reviewed journal in 2021.
SURPASS-1 is one of eight phase 3 studies of the drug, including five registration studies and one large 12,500-patient cardiovascular outcomes trial.
Tirzepatide patients lost up to 20 lb, side effect profile ‘reassuring’
In the study, patients had been recently diagnosed with type 2 diabetes (average duration, 4.8 years) and 54% were treatment-naive. Average baseline hemoglobin A1c was 7.9% and mean weight was 85.9 kg (189 pounds).
Patients started on a subcutaneous injectable dose of tirzepatide of 2.5 mg per week, which was titrated up to the final dose – 5, 10, or 15 mg – in 2.5-mg increments given as monotherapy for 40 weeks and compared with placebo.
Treatment with tirzepatide resulted in average reductions in A1c from baseline that ranged from 1.87% to 2.07%, depending on the dose, and were all significant compared with an increase of 0.4% with placebo.
The percentage of patients whose A1c fell to normal levels (less than 5.7%) ranged from 30.5% to 51.7%, compared with 0.9% among controls, and again, was significant for all doses.
Patients treated with tirzepatide also lost weight. Average weight reductions after 40 weeks were significant and ranged from 7.0 to 9.5 kg (15-21 pounds) compared with an average loss of 0.7 kg (1.5 pounds) among patients who received placebo.
The most common adverse events were gastrointestinal-related and mild to moderate in severity, and usually occurred during dose escalation.
Dr. Lingvay said the safety data reported are “reassuring, with side effects in the anticipated range and comparable with other medications in the GLP-1 agonist class.”
And no hypoglycemic (level 2, < 54 mg/dL) events were reported, “which is impressive considering the overall glucose level achieved,” she noted.
“I am eagerly awaiting the results of the other studies within the SURPASS program and hope those will confirm these initial findings and provide additional safety and efficacy information in a wider range of patients with type 2 diabetes,” she concluded.
Dr. Lingvay has reported receiving research funding, advisory/consulting fees, and/or other support from Novo Nordisk, Eli Lilly, Sanofi, AstraZeneca, Boehringer Ingelheim, Janssen, Intercept, Intarcia, Target Pharma, Merck, Pfizer, Novartis, GI Dynamics, Mylan, MannKind, Valeritas, Bayer, and Zealand Pharma.
A version of this article originally appeared on Medscape.com.
Tirzepatide, a novel subcutaneously injected drug that acts via two related but separate pathways of glucose control, produced strikingly positive effects in top-line results from the phase 3, placebo-controlled study SURPASS-1 in 478 adults with type 2 diabetes, according to a Dec. 9 press release from the manufacturer, Lilly.
The tirzepatide molecule exerts agonist effects at both the glucose-dependent insulinotropic polypeptide (GIP) receptor and the glucagon-like peptide-1 (GLP-1) receptor, and has been called a “twincretin” for its activity encompassing two different incretins. Phase 2 trial results caused excitement, with one physician calling the data “unbelievable” when reported in 2018.
SURPASS-1 enrolled patients who were very early in the course of their disease, had on average relatively mild elevation in glucose levels, and few metabolic comorbidities. They took one of three doses of the agent (5, 10, or 15 mg) as monotherapy or placebo for 40 weeks.
Julio Rosenstock, MD, said in the Lilly statement: “The study took a bold approach in assessing A1c targets. Not only did nearly 90% of all participants taking tirzepatide meet the standard A1c goal of less than 7%, more than half taking the highest dose also achieved an A1c less than 5.7%, the level seen in people without diabetes.”
Dr. Rosenstock is principal investigator of SURPASS-1 and director of the Dallas Diabetes Research Center in Texas.
The discontinuation rate in the high-dose group was 21.5% compared with less than 10% in the two lower-dose cohorts. Lilly said most of the dropouts “were due to the pandemic and family or work reasons.” The dropout rate in the placebo group was 14.8%.
These data were not included in the efficacy analysis, however, which “muddied” the analysis somewhat, one pharma analyst told BioPharma Dive.
Commenting on the new trial data, Ildiko Lingvay, MD, said in an interview: “I am very impressed with these results,” which are “unprecedented for any glucose-lowering medication that has ever been tested.”
Dr. Lingvay, of the department of internal medicine/endocrinology, and medical director, office of clinical trials management at UT Southwestern Medical Center, Dallas, was not involved in the study.
She added that the weight loss seen with tirzepatide “is equally impressive with greater than 10% of body weight loss above placebo achieved within 40 weeks of treatment and without any directed weight loss efforts.”
If the agent is eventually approved, “I am enthusiastic about the prospect of having another very powerful tool to address both diabetes and obesity,” she added.
The full results of SURPASS-1 will be presented at the American Diabetes Association 81st Scientific Sessions and published in a peer-reviewed journal in 2021.
SURPASS-1 is one of eight phase 3 studies of the drug, including five registration studies and one large 12,500-patient cardiovascular outcomes trial.
Tirzepatide patients lost up to 20 lb, side effect profile ‘reassuring’
In the study, patients had been recently diagnosed with type 2 diabetes (average duration, 4.8 years) and 54% were treatment-naive. Average baseline hemoglobin A1c was 7.9% and mean weight was 85.9 kg (189 pounds).
Patients started on a subcutaneous injectable dose of tirzepatide of 2.5 mg per week, which was titrated up to the final dose – 5, 10, or 15 mg – in 2.5-mg increments given as monotherapy for 40 weeks and compared with placebo.
Treatment with tirzepatide resulted in average reductions in A1c from baseline that ranged from 1.87% to 2.07%, depending on the dose, and were all significant compared with an increase of 0.4% with placebo.
The percentage of patients whose A1c fell to normal levels (less than 5.7%) ranged from 30.5% to 51.7%, compared with 0.9% among controls, and again, was significant for all doses.
Patients treated with tirzepatide also lost weight. Average weight reductions after 40 weeks were significant and ranged from 7.0 to 9.5 kg (15-21 pounds) compared with an average loss of 0.7 kg (1.5 pounds) among patients who received placebo.
The most common adverse events were gastrointestinal-related and mild to moderate in severity, and usually occurred during dose escalation.
Dr. Lingvay said the safety data reported are “reassuring, with side effects in the anticipated range and comparable with other medications in the GLP-1 agonist class.”
And no hypoglycemic (level 2, < 54 mg/dL) events were reported, “which is impressive considering the overall glucose level achieved,” she noted.
“I am eagerly awaiting the results of the other studies within the SURPASS program and hope those will confirm these initial findings and provide additional safety and efficacy information in a wider range of patients with type 2 diabetes,” she concluded.
Dr. Lingvay has reported receiving research funding, advisory/consulting fees, and/or other support from Novo Nordisk, Eli Lilly, Sanofi, AstraZeneca, Boehringer Ingelheim, Janssen, Intercept, Intarcia, Target Pharma, Merck, Pfizer, Novartis, GI Dynamics, Mylan, MannKind, Valeritas, Bayer, and Zealand Pharma.
A version of this article originally appeared on Medscape.com.
ACC/AHA update two atrial fibrillation performance measures
The American College of Cardiology and American Heart Association Task Force on Performance Measures have made two changes to performance measures for adults with atrial fibrillation or atrial flutter.

The 2020 Update to the 2016 ACC/AHA Clinical Performance and Quality Measures for Adults With Atrial Fibrillation or Atrial Flutter was published online Dec. 7 in the Journal of the American College of Cardiology and Circulation: Cardiovascular Quality and Outcomes. It was developed in collaboration with the Heart Rhythm Society.
Both performance measure changes were prompted by, and are in accordance with, the 2019 ACC/AHA/Heart Rhythm Society atrial fibrillation guideline focused update issued in January 2019, and reported by this news organization at that time.
The first change is the clarification that valvular atrial fibrillation is atrial fibrillation with either moderate or severe mitral stenosis or a mechanical heart valve. This change is incorporated into all the performance measures.
The second change, which only applies to the performance measure of anticoagulation prescribed, is the separation of a male and female threshold for the CHA2DS2-VASc score.
This threshold is now a score higher than 1 for men and higher than 2 for women, further demonstrating that the risk for stroke differs for men and women with atrial fibrillation or atrial flutter, the ACC/AHA noted in a press release.
“Successful implementation of these updated performance measures by clinicians and healthcare organizations will lead to quality improvement for adult patients with atrial fibrillation or atrial flutter,” they said.
A version of this article originally appeared on Medscape.com.
The American College of Cardiology and American Heart Association Task Force on Performance Measures have made two changes to performance measures for adults with atrial fibrillation or atrial flutter.
The 2020 Update to the 2016 ACC/AHA Clinical Performance and Quality Measures for Adults With Atrial Fibrillation or Atrial Flutter was published online Dec. 7 in the Journal of the American College of Cardiology and Circulation: Cardiovascular Quality and Outcomes. It was developed in collaboration with the Heart Rhythm Society.
Both performance measure changes were prompted by, and are in accordance with, the 2019 ACC/AHA/Heart Rhythm Society atrial fibrillation guideline focused update issued in January 2019, and reported by this news organization at that time.
The first change is the clarification that valvular atrial fibrillation is atrial fibrillation with either moderate or severe mitral stenosis or a mechanical heart valve. This change is incorporated into all the performance measures.
The second change, which only applies to the performance measure of anticoagulation prescribed, is the separation of a male and female threshold for the CHA2DS2-VASc score.
This threshold is now a score higher than 1 for men and higher than 2 for women, further demonstrating that the risk for stroke differs for men and women with atrial fibrillation or atrial flutter, the ACC/AHA noted in a press release.
“Successful implementation of these updated performance measures by clinicians and healthcare organizations will lead to quality improvement for adult patients with atrial fibrillation or atrial flutter,” they said.
A version of this article originally appeared on Medscape.com.
The American College of Cardiology and American Heart Association Task Force on Performance Measures have made two changes to performance measures for adults with atrial fibrillation or atrial flutter.
The 2020 Update to the 2016 ACC/AHA Clinical Performance and Quality Measures for Adults With Atrial Fibrillation or Atrial Flutter was published online Dec. 7 in the Journal of the American College of Cardiology and Circulation: Cardiovascular Quality and Outcomes. It was developed in collaboration with the Heart Rhythm Society.
Both performance measure changes were prompted by, and are in accordance with, the 2019 ACC/AHA/Heart Rhythm Society atrial fibrillation guideline focused update issued in January 2019, and reported by this news organization at that time.
The first change is the clarification that valvular atrial fibrillation is atrial fibrillation with either moderate or severe mitral stenosis or a mechanical heart valve. This change is incorporated into all the performance measures.
The second change, which only applies to the performance measure of anticoagulation prescribed, is the separation of a male and female threshold for the CHA2DS2-VASc score.
This threshold is now a score higher than 1 for men and higher than 2 for women, further demonstrating that the risk for stroke differs for men and women with atrial fibrillation or atrial flutter, the ACC/AHA noted in a press release.
“Successful implementation of these updated performance measures by clinicians and healthcare organizations will lead to quality improvement for adult patients with atrial fibrillation or atrial flutter,” they said.
A version of this article originally appeared on Medscape.com.
CDC panel recommends Pfizer’s COVID-19 vaccine for people 16 and over
stating they found it was safe and effective.
The agency said it will quickly issue guidance to clinicians so they can determine when and when not to give the vaccine, and to help them communicate the risks and benefits to patients.
CDC staff gave a preview of those clinical considerations at the agency’s Advisory Committee on Immunization Practices (ACIP) meeting on December 12 and said it would be holding calls with clinicians on December 13 and 14.
The CDC will also issue guidance December 13 on how organizations can handle the workforce problems that might arise as health care workers experience side effects from vaccination.
ACIP voted 11-0, with three recusals, to recommend use of the Pfizer-BioNTech mRNA vaccine in individuals 16 years or older according to the guidelines of the Food and Drug Administration’s (FDA’s) emergency use authorization issued December 11.
The panel also voted unanimously to include the vaccine in 2021 immunization schedules. All panel members said the recommendation should go hand-in-hand with ACIP’s previous recommendation on December 1 that allocation of the vaccine be phased-in, with health care workers and residents and staff of long-term care facilities in phase 1a.
Allergies, pregnant women?
ACIP panelists said clinicians need more guidance on whether to use the vaccine in pregnant or breastfeeding women, the immunocompromised, or those who have a history of allergies.
The FDA health care provider information sheet said there is not enough data to recommend vaccinating those women or the immunocompromised, and also advises against giving the vaccine to individuals who have a history of serious allergic reaction to any component of the vaccine.
Peter Marks, MD, PhD, director of the FDA’s Center for Biologic Evaluation and Research (CBER) clarified this in a briefing on December 12, noting that women who are pregnant or lactating can make the decision in consultation with their physician. And, he said, patients with any other history of allergy should be able to safely get the vaccine.
The CDC — in its soon-to-be-released guidance — will make the same recommendations. For any woman considering vaccination, she should consider the level of COVID-19 in the community, her personal risk of contracting the virus, the risks to her or her fetus of developing the disease, and the vaccine’s known side effects, Sarah Mbaeyi, MD, MPH, a medical officer at the agency, said during the panel meeting December 12.
She added that the CDC will also urge physicians to advise women to take acetaminophen if they develop a fever after vaccination — to protect the developing fetus from fever.
Sandra Fryhofer, MD, representing the American Medical Association, commended the CDC for these recommendations. But she also called on Pfizer, the FDA, and the CDC to make data from the developmental and reproductive toxicity (DART) studies public as soon as possible.
“We really need to put those results on warp speed and get them out there to give our physicians and pregnant women more information,” said Fryhofer, an adjunct associate professor of medicine at Emory University School of Medicine in Atlanta, Georgia.
The American College of Obstetricians and Gynecologists (ACOG) will also soon release guidance for vaccinating pregnant and breastfeeding women, said Linda Eckert, MD, FACOG, an ACOG representative on the panel.
ACOG and the CDC met the morning of December 12 to discuss risks and benefits with experts in immunology, placental pathology, and vaccine kinetics, she said.
“The overall complete consensus was that we don’t see biological plausibility at this time for placental transfer of the mRNA and that we see that direct fetal exposure or the possibility of fetal inflammatory response is extremely unlikely,” said Eckert, professor of obstetrics and gynecology at the University of Washington, Seattle. “Clearly we are waiting on the data.”
A Pfizer official told the ACIP panel that preliminary data “show no indication of either developmental or reproductive toxicity,” and that the company plans to send the final DART data to the FDA at the end of December.
On the potential for allergic reactions, the CDC concurred with the FDA that the vaccine should not be given to people with a history of serious reactions. The agency added that the category should include anyone who has had a reaction to any vaccine or injectable drug product because injectables may contain the same ingredients as the Pfizer vaccine, said Mbaeyi.
The CDC will also urge clinicians to observe patients with a history of anaphylaxis for 30 minutes after vaccination and all patients for at least 15 minutes afterward.
Should teens be a special population?
At least one ACIP panel member — Henry Bernstein, DO, MHCM, FAAP — said he was concerned that backing use of the vaccine in 16- and 17-year-olds was a leap of faith, given that Pfizer had extremely limited data on this cohort.
Bernstein, professor of pediatrics at the Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, also said that systemic reactions were more common in that age group.
He argued for making the 16- and 17-year-olds a “special population” that would get specific attention and guidance for vaccination from the federal agencies and professional societies.
Bernstein said he did not want to sow any more doubts in parents’ minds about vaccination, noting that hesitancy was a growing concern. “A successful pediatric vaccination program depends on creating and sustaining parental confidence in both the safety and effectiveness of this vaccine,” he said.
Many panelists, however, noted that there has been no evidence to suggest that the vaccine is not safe or less effective in that younger age group.
Yvonne Maldonado, MD, the American Academy of Pediatrics representative on the panel, said that this age group should not be denied the vaccine as they often have essential or front-line jobs that put them at higher risk for infection.
“I am very concerned about this message being sent out that this vaccine will not be safe in children,” said Maldonado, professor of pediatrics and health research and policy at Stanford University School of Medicine in California.
“We currently have no evidence that that is the case,” she said, adding there is also no indication younger children are biologically or physiologically different in their response or safety risk than 18-year-olds.
Vaccine = hope
Committee members breathed a sigh of relief at the end of the 2-day meeting, saying that although the Pfizer vaccine is not perfect, it represents a scientific milestone and a significant advance against the continuing march of the SARS-CoV-2 pandemic.
“This vaccine and future vaccines do provide a promise for a lot of progress in the future,” said panelist Beth P. Bell, MD, MPH, clinical professor of global health at the University of Washington School of Public Health in Seattle.
Peter Szilagyi, MD, MPH, executive vice-chair and vice-chair for research at the University of California, Los Angeles pediatrics department, said, “I’m really hopeful that this is the beginning of the end of the coronavirus pandemic.”
“The need for this vaccine is profound,” said Veronica McNally, president and CEO of the Franny Strong Foundation in West Bloomfield, Michigan.
The ACIP panel also made the argument that while the at least $10 billion spent on vaccine development by the federal government’s Operation Warp Speed alone has been a good investment, more spending is needed to actually get Americans vaccinated.
The imbalance between the two is “shocking and needs to be corrected,” said Bell. “We are not going to be able to protect the American public if we don’t have a way to deliver the vaccine to them.”
This article first appeared on Medscape.com.
stating they found it was safe and effective.
The agency said it will quickly issue guidance to clinicians so they can determine when and when not to give the vaccine, and to help them communicate the risks and benefits to patients.
CDC staff gave a preview of those clinical considerations at the agency’s Advisory Committee on Immunization Practices (ACIP) meeting on December 12 and said it would be holding calls with clinicians on December 13 and 14.
The CDC will also issue guidance December 13 on how organizations can handle the workforce problems that might arise as health care workers experience side effects from vaccination.
ACIP voted 11-0, with three recusals, to recommend use of the Pfizer-BioNTech mRNA vaccine in individuals 16 years or older according to the guidelines of the Food and Drug Administration’s (FDA’s) emergency use authorization issued December 11.
The panel also voted unanimously to include the vaccine in 2021 immunization schedules. All panel members said the recommendation should go hand-in-hand with ACIP’s previous recommendation on December 1 that allocation of the vaccine be phased-in, with health care workers and residents and staff of long-term care facilities in phase 1a.
Allergies, pregnant women?
ACIP panelists said clinicians need more guidance on whether to use the vaccine in pregnant or breastfeeding women, the immunocompromised, or those who have a history of allergies.
The FDA health care provider information sheet said there is not enough data to recommend vaccinating those women or the immunocompromised, and also advises against giving the vaccine to individuals who have a history of serious allergic reaction to any component of the vaccine.
Peter Marks, MD, PhD, director of the FDA’s Center for Biologic Evaluation and Research (CBER) clarified this in a briefing on December 12, noting that women who are pregnant or lactating can make the decision in consultation with their physician. And, he said, patients with any other history of allergy should be able to safely get the vaccine.
The CDC — in its soon-to-be-released guidance — will make the same recommendations. For any woman considering vaccination, she should consider the level of COVID-19 in the community, her personal risk of contracting the virus, the risks to her or her fetus of developing the disease, and the vaccine’s known side effects, Sarah Mbaeyi, MD, MPH, a medical officer at the agency, said during the panel meeting December 12.
She added that the CDC will also urge physicians to advise women to take acetaminophen if they develop a fever after vaccination — to protect the developing fetus from fever.
Sandra Fryhofer, MD, representing the American Medical Association, commended the CDC for these recommendations. But she also called on Pfizer, the FDA, and the CDC to make data from the developmental and reproductive toxicity (DART) studies public as soon as possible.
“We really need to put those results on warp speed and get them out there to give our physicians and pregnant women more information,” said Fryhofer, an adjunct associate professor of medicine at Emory University School of Medicine in Atlanta, Georgia.
The American College of Obstetricians and Gynecologists (ACOG) will also soon release guidance for vaccinating pregnant and breastfeeding women, said Linda Eckert, MD, FACOG, an ACOG representative on the panel.
ACOG and the CDC met the morning of December 12 to discuss risks and benefits with experts in immunology, placental pathology, and vaccine kinetics, she said.
“The overall complete consensus was that we don’t see biological plausibility at this time for placental transfer of the mRNA and that we see that direct fetal exposure or the possibility of fetal inflammatory response is extremely unlikely,” said Eckert, professor of obstetrics and gynecology at the University of Washington, Seattle. “Clearly we are waiting on the data.”
A Pfizer official told the ACIP panel that preliminary data “show no indication of either developmental or reproductive toxicity,” and that the company plans to send the final DART data to the FDA at the end of December.
On the potential for allergic reactions, the CDC concurred with the FDA that the vaccine should not be given to people with a history of serious reactions. The agency added that the category should include anyone who has had a reaction to any vaccine or injectable drug product because injectables may contain the same ingredients as the Pfizer vaccine, said Mbaeyi.
The CDC will also urge clinicians to observe patients with a history of anaphylaxis for 30 minutes after vaccination and all patients for at least 15 minutes afterward.
Should teens be a special population?
At least one ACIP panel member — Henry Bernstein, DO, MHCM, FAAP — said he was concerned that backing use of the vaccine in 16- and 17-year-olds was a leap of faith, given that Pfizer had extremely limited data on this cohort.
Bernstein, professor of pediatrics at the Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, also said that systemic reactions were more common in that age group.
He argued for making the 16- and 17-year-olds a “special population” that would get specific attention and guidance for vaccination from the federal agencies and professional societies.
Bernstein said he did not want to sow any more doubts in parents’ minds about vaccination, noting that hesitancy was a growing concern. “A successful pediatric vaccination program depends on creating and sustaining parental confidence in both the safety and effectiveness of this vaccine,” he said.
Many panelists, however, noted that there has been no evidence to suggest that the vaccine is not safe or less effective in that younger age group.
Yvonne Maldonado, MD, the American Academy of Pediatrics representative on the panel, said that this age group should not be denied the vaccine as they often have essential or front-line jobs that put them at higher risk for infection.
“I am very concerned about this message being sent out that this vaccine will not be safe in children,” said Maldonado, professor of pediatrics and health research and policy at Stanford University School of Medicine in California.
“We currently have no evidence that that is the case,” she said, adding there is also no indication younger children are biologically or physiologically different in their response or safety risk than 18-year-olds.
Vaccine = hope
Committee members breathed a sigh of relief at the end of the 2-day meeting, saying that although the Pfizer vaccine is not perfect, it represents a scientific milestone and a significant advance against the continuing march of the SARS-CoV-2 pandemic.
“This vaccine and future vaccines do provide a promise for a lot of progress in the future,” said panelist Beth P. Bell, MD, MPH, clinical professor of global health at the University of Washington School of Public Health in Seattle.
Peter Szilagyi, MD, MPH, executive vice-chair and vice-chair for research at the University of California, Los Angeles pediatrics department, said, “I’m really hopeful that this is the beginning of the end of the coronavirus pandemic.”
“The need for this vaccine is profound,” said Veronica McNally, president and CEO of the Franny Strong Foundation in West Bloomfield, Michigan.
The ACIP panel also made the argument that while the at least $10 billion spent on vaccine development by the federal government’s Operation Warp Speed alone has been a good investment, more spending is needed to actually get Americans vaccinated.
The imbalance between the two is “shocking and needs to be corrected,” said Bell. “We are not going to be able to protect the American public if we don’t have a way to deliver the vaccine to them.”
This article first appeared on Medscape.com.
stating they found it was safe and effective.
The agency said it will quickly issue guidance to clinicians so they can determine when and when not to give the vaccine, and to help them communicate the risks and benefits to patients.
CDC staff gave a preview of those clinical considerations at the agency’s Advisory Committee on Immunization Practices (ACIP) meeting on December 12 and said it would be holding calls with clinicians on December 13 and 14.
The CDC will also issue guidance December 13 on how organizations can handle the workforce problems that might arise as health care workers experience side effects from vaccination.
ACIP voted 11-0, with three recusals, to recommend use of the Pfizer-BioNTech mRNA vaccine in individuals 16 years or older according to the guidelines of the Food and Drug Administration’s (FDA’s) emergency use authorization issued December 11.
The panel also voted unanimously to include the vaccine in 2021 immunization schedules. All panel members said the recommendation should go hand-in-hand with ACIP’s previous recommendation on December 1 that allocation of the vaccine be phased-in, with health care workers and residents and staff of long-term care facilities in phase 1a.
Allergies, pregnant women?
ACIP panelists said clinicians need more guidance on whether to use the vaccine in pregnant or breastfeeding women, the immunocompromised, or those who have a history of allergies.
The FDA health care provider information sheet said there is not enough data to recommend vaccinating those women or the immunocompromised, and also advises against giving the vaccine to individuals who have a history of serious allergic reaction to any component of the vaccine.
Peter Marks, MD, PhD, director of the FDA’s Center for Biologic Evaluation and Research (CBER) clarified this in a briefing on December 12, noting that women who are pregnant or lactating can make the decision in consultation with their physician. And, he said, patients with any other history of allergy should be able to safely get the vaccine.
The CDC — in its soon-to-be-released guidance — will make the same recommendations. For any woman considering vaccination, she should consider the level of COVID-19 in the community, her personal risk of contracting the virus, the risks to her or her fetus of developing the disease, and the vaccine’s known side effects, Sarah Mbaeyi, MD, MPH, a medical officer at the agency, said during the panel meeting December 12.
She added that the CDC will also urge physicians to advise women to take acetaminophen if they develop a fever after vaccination — to protect the developing fetus from fever.
Sandra Fryhofer, MD, representing the American Medical Association, commended the CDC for these recommendations. But she also called on Pfizer, the FDA, and the CDC to make data from the developmental and reproductive toxicity (DART) studies public as soon as possible.
“We really need to put those results on warp speed and get them out there to give our physicians and pregnant women more information,” said Fryhofer, an adjunct associate professor of medicine at Emory University School of Medicine in Atlanta, Georgia.
The American College of Obstetricians and Gynecologists (ACOG) will also soon release guidance for vaccinating pregnant and breastfeeding women, said Linda Eckert, MD, FACOG, an ACOG representative on the panel.
ACOG and the CDC met the morning of December 12 to discuss risks and benefits with experts in immunology, placental pathology, and vaccine kinetics, she said.
“The overall complete consensus was that we don’t see biological plausibility at this time for placental transfer of the mRNA and that we see that direct fetal exposure or the possibility of fetal inflammatory response is extremely unlikely,” said Eckert, professor of obstetrics and gynecology at the University of Washington, Seattle. “Clearly we are waiting on the data.”
A Pfizer official told the ACIP panel that preliminary data “show no indication of either developmental or reproductive toxicity,” and that the company plans to send the final DART data to the FDA at the end of December.
On the potential for allergic reactions, the CDC concurred with the FDA that the vaccine should not be given to people with a history of serious reactions. The agency added that the category should include anyone who has had a reaction to any vaccine or injectable drug product because injectables may contain the same ingredients as the Pfizer vaccine, said Mbaeyi.
The CDC will also urge clinicians to observe patients with a history of anaphylaxis for 30 minutes after vaccination and all patients for at least 15 minutes afterward.
Should teens be a special population?
At least one ACIP panel member — Henry Bernstein, DO, MHCM, FAAP — said he was concerned that backing use of the vaccine in 16- and 17-year-olds was a leap of faith, given that Pfizer had extremely limited data on this cohort.
Bernstein, professor of pediatrics at the Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York, also said that systemic reactions were more common in that age group.
He argued for making the 16- and 17-year-olds a “special population” that would get specific attention and guidance for vaccination from the federal agencies and professional societies.
Bernstein said he did not want to sow any more doubts in parents’ minds about vaccination, noting that hesitancy was a growing concern. “A successful pediatric vaccination program depends on creating and sustaining parental confidence in both the safety and effectiveness of this vaccine,” he said.
Many panelists, however, noted that there has been no evidence to suggest that the vaccine is not safe or less effective in that younger age group.
Yvonne Maldonado, MD, the American Academy of Pediatrics representative on the panel, said that this age group should not be denied the vaccine as they often have essential or front-line jobs that put them at higher risk for infection.
“I am very concerned about this message being sent out that this vaccine will not be safe in children,” said Maldonado, professor of pediatrics and health research and policy at Stanford University School of Medicine in California.
“We currently have no evidence that that is the case,” she said, adding there is also no indication younger children are biologically or physiologically different in their response or safety risk than 18-year-olds.
Vaccine = hope
Committee members breathed a sigh of relief at the end of the 2-day meeting, saying that although the Pfizer vaccine is not perfect, it represents a scientific milestone and a significant advance against the continuing march of the SARS-CoV-2 pandemic.
“This vaccine and future vaccines do provide a promise for a lot of progress in the future,” said panelist Beth P. Bell, MD, MPH, clinical professor of global health at the University of Washington School of Public Health in Seattle.
Peter Szilagyi, MD, MPH, executive vice-chair and vice-chair for research at the University of California, Los Angeles pediatrics department, said, “I’m really hopeful that this is the beginning of the end of the coronavirus pandemic.”
“The need for this vaccine is profound,” said Veronica McNally, president and CEO of the Franny Strong Foundation in West Bloomfield, Michigan.
The ACIP panel also made the argument that while the at least $10 billion spent on vaccine development by the federal government’s Operation Warp Speed alone has been a good investment, more spending is needed to actually get Americans vaccinated.
The imbalance between the two is “shocking and needs to be corrected,” said Bell. “We are not going to be able to protect the American public if we don’t have a way to deliver the vaccine to them.”
This article first appeared on Medscape.com.
FDA OKs emergency use of Pfizer COVID-19 vaccine
The much-anticipated emergency use authorization (EUA) of this vaccine — the first such approval in the United States — was greeted with optimism by infectious disease and pulmonary experts, although unanswered questions remain regarding use in people with allergic hypersensitivity, safety in pregnant women, and how smooth distribution will be.
“I am delighted. This is a first, firm step on a long path to getting this COVID pandemic under control,” William Schaffner, MD, professor of infectious diseases at the Vanderbilt University School of Medicine in Nashville, Tennessee, said in an interview.
The FDA gave the green light after the December 10 recommendation from the agency’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) meeting. The committee voted 17-4 in favor of the emergency authorization.

The COVID-19 vaccine is “going to have a major impact here in the US. I’m very optimistic about it,” Dial Hewlett, MD, a spokesperson for the Infectious Diseases Society of American (IDSA), told this news organization.
Daniel Culver, DO, chair of medicine at the Cleveland Clinic in Ohio, is likewise hopeful. “My understanding is that supplies of the vaccine are already in place in hubs and will be shipped relatively quickly. The hope would be we can start vaccinating people as early as next week.”
Allergic reactions reported in the UK
After vaccinations with the Pfizer vaccine began in the UK on December 8, reports surfaced of two healthcare workers who experienced allergic reactions. They have since recovered, but officials warned that people with a history of severe allergic reactions should not receive the Pfizer vaccine at this time.
“For the moment, they are asking people who have had notable allergic reactions to step aside while this is investigated. It shows you that the system is working,” Schaffner said.
Both vaccine recipients who experienced anaphylaxis carried EpiPens, as they were at high risk for allergic reactions, Hewlett said. Also, if other COVID-19 vaccines are approved for use in the future, people allergic to the Pfizer vaccine might have another option, he added.
Reassuring role models
Schaffner supports the CDC Advisory Committee on Immunization Practices (ACIP) decision to start vaccinations with healthcare workers and residents of long-term care facilities.
“Vaccinating healthcare workers, in particular, will be a model for the general public,” said Schaffner, who is also a former member of the IDSA board of directors. “If they see those of us in white coats and blue scrubs lining up for the vaccine, that will provide confidence.”
To further increase acceptance of the COVID-19 vaccine, public health officials need to provide information and reassure the general public, Schaffner said.
Hewlett agreed. “I know there are a lot of people in the population who are very hesitant about vaccines. As infection disease specialists and people in public health, we are trying to allay a lot of concerns people have.”
Reassurance will be especially important in minority communities. “They have been disproportionately affected by the virus, and they have a traditional history of not being optimally vaccinated,” Schaffner said. “We need to reach them in particular with good information and reassurance…so they can make good decisions for themselves and their families.”
No vaccine is 100% effective or completely free of side effects. “There is always a chance there can be adverse reactions, but we think for the most part this is going to be a safe and effective vaccine,” said Hewlett, medical director at the Division of Disease Control and deputy to commissioner of health at the Westchester County Department of Health in White Plains, New York.
Distribution: Smooth or full of strife?
In addition to the concern that some people will not take advantage of vaccination against COVID-19, there could be vaccine supply issues down the road, Schaffner said.
Culver agreed. “In the early phases, I expect that there will be some kinks to work out, but because the numbers are relatively small, this should be okay,” he said.
“I think when we start to get into larger-scale vaccination programs — the supply chain, transport, and storage will be a Herculean undertaking,” Culver added. “It will take careful coordination between healthcare providers, distributors, suppliers, and public health officials to pull this off.”
Planning and distribution also should focus beyond US borders. Any issues in vaccine distribution or administration in the United States “will only be multiplied in several other parts of the world,” Culver said. Because COVID-19 is a pandemic, “we need to think about vaccinating globally.”
Investigating adverse events
Adverse events common to vaccinations in general — injection site pain, headaches, and fever — would not be unexpected with the COVID-19 vaccines. However, experts remain concerned that other, unrelated adverse events might be erroneously attributed to vaccination. For example, if a fall, heart attack, or death occurs within days of immunization, some might immediately blame the vaccine product.
“It’s important to remember that any new, highly touted medical therapy like this will receive a lot of scrutiny, so it would be unusual not to hear about something happening to somebody,” Culver said. Vaccine companies and health agencies will be carefully evaluating any reported adverse events to ensure no safety signal was missed in the trials.
“Fortunately, there are systems in place to investigate these events immediately,” Schaffner said.
Pregnancy recommendations pending
One question still looms: Is the COVID-19 vaccination safe for pregnant women? This isn’t just a question for the general public, either, Schaffner said. He estimated that about 70 percent of healthcare workers are women, and data suggests about 300,000 of these healthcare workers are pregnant.
“The CDC’s Advisory Committee on Immunization Practices will speak to that just as soon as the EUA is issued,” he added.
Patients are asking Culver about the priority order for vaccination. He said it’s difficult to provide firm guidance at this point.
People also have “lingering skepticism” about whether vaccine development was done in a prudent way, Culver said. Some people question whether the Pfizer vaccine and others were rushed to market. “So we try to spend time with the patients, reassuring them that all the usual safety evaluations were carefully done,” he said.
Another concern is whether mRNA vaccines can interact with human DNA. “The quick, short, and definitive answer is no,” Schaffner said. The m stands for messenger — the vaccines transmit information. "Once it gets into a cell, the mRNA does not go anywhere near the DNA, and once it transmits its information to the cell appropriately, it gets metabolized, and we excrete all the remnants."
Hewlett pointed out that investigations and surveillance will continue. Because this is an EUA and not full approval, “that essentially means they will still be obligated to collect a lot more data than they would ordinarily,” he said.
How long immunoprotection will last also remains an unknown. “The big question left on the table now is the durability,” Culver said. “Of course, we won’t know the answer to that for quite some time.”
Schaffner and Culver have disclosed no relevant financial relationships. Hewlett was an employee of Pfizer until mid-2019. His previous work as Pfizer’s senior medical director of global medical product evaluation was not associated with development of the COVID-19 vaccine.
This article first appeared on Medscape.com.
The much-anticipated emergency use authorization (EUA) of this vaccine — the first such approval in the United States — was greeted with optimism by infectious disease and pulmonary experts, although unanswered questions remain regarding use in people with allergic hypersensitivity, safety in pregnant women, and how smooth distribution will be.
“I am delighted. This is a first, firm step on a long path to getting this COVID pandemic under control,” William Schaffner, MD, professor of infectious diseases at the Vanderbilt University School of Medicine in Nashville, Tennessee, said in an interview.
The FDA gave the green light after the December 10 recommendation from the agency’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) meeting. The committee voted 17-4 in favor of the emergency authorization.
The COVID-19 vaccine is “going to have a major impact here in the US. I’m very optimistic about it,” Dial Hewlett, MD, a spokesperson for the Infectious Diseases Society of American (IDSA), told this news organization.
Daniel Culver, DO, chair of medicine at the Cleveland Clinic in Ohio, is likewise hopeful. “My understanding is that supplies of the vaccine are already in place in hubs and will be shipped relatively quickly. The hope would be we can start vaccinating people as early as next week.”
Allergic reactions reported in the UK
After vaccinations with the Pfizer vaccine began in the UK on December 8, reports surfaced of two healthcare workers who experienced allergic reactions. They have since recovered, but officials warned that people with a history of severe allergic reactions should not receive the Pfizer vaccine at this time.
“For the moment, they are asking people who have had notable allergic reactions to step aside while this is investigated. It shows you that the system is working,” Schaffner said.
Both vaccine recipients who experienced anaphylaxis carried EpiPens, as they were at high risk for allergic reactions, Hewlett said. Also, if other COVID-19 vaccines are approved for use in the future, people allergic to the Pfizer vaccine might have another option, he added.
Reassuring role models
Schaffner supports the CDC Advisory Committee on Immunization Practices (ACIP) decision to start vaccinations with healthcare workers and residents of long-term care facilities.
“Vaccinating healthcare workers, in particular, will be a model for the general public,” said Schaffner, who is also a former member of the IDSA board of directors. “If they see those of us in white coats and blue scrubs lining up for the vaccine, that will provide confidence.”
To further increase acceptance of the COVID-19 vaccine, public health officials need to provide information and reassure the general public, Schaffner said.
Hewlett agreed. “I know there are a lot of people in the population who are very hesitant about vaccines. As infection disease specialists and people in public health, we are trying to allay a lot of concerns people have.”
Reassurance will be especially important in minority communities. “They have been disproportionately affected by the virus, and they have a traditional history of not being optimally vaccinated,” Schaffner said. “We need to reach them in particular with good information and reassurance…so they can make good decisions for themselves and their families.”
No vaccine is 100% effective or completely free of side effects. “There is always a chance there can be adverse reactions, but we think for the most part this is going to be a safe and effective vaccine,” said Hewlett, medical director at the Division of Disease Control and deputy to commissioner of health at the Westchester County Department of Health in White Plains, New York.
Distribution: Smooth or full of strife?
In addition to the concern that some people will not take advantage of vaccination against COVID-19, there could be vaccine supply issues down the road, Schaffner said.
Culver agreed. “In the early phases, I expect that there will be some kinks to work out, but because the numbers are relatively small, this should be okay,” he said.
“I think when we start to get into larger-scale vaccination programs — the supply chain, transport, and storage will be a Herculean undertaking,” Culver added. “It will take careful coordination between healthcare providers, distributors, suppliers, and public health officials to pull this off.”
Planning and distribution also should focus beyond US borders. Any issues in vaccine distribution or administration in the United States “will only be multiplied in several other parts of the world,” Culver said. Because COVID-19 is a pandemic, “we need to think about vaccinating globally.”
Investigating adverse events
Adverse events common to vaccinations in general — injection site pain, headaches, and fever — would not be unexpected with the COVID-19 vaccines. However, experts remain concerned that other, unrelated adverse events might be erroneously attributed to vaccination. For example, if a fall, heart attack, or death occurs within days of immunization, some might immediately blame the vaccine product.
“It’s important to remember that any new, highly touted medical therapy like this will receive a lot of scrutiny, so it would be unusual not to hear about something happening to somebody,” Culver said. Vaccine companies and health agencies will be carefully evaluating any reported adverse events to ensure no safety signal was missed in the trials.
“Fortunately, there are systems in place to investigate these events immediately,” Schaffner said.
Pregnancy recommendations pending
One question still looms: Is the COVID-19 vaccination safe for pregnant women? This isn’t just a question for the general public, either, Schaffner said. He estimated that about 70 percent of healthcare workers are women, and data suggests about 300,000 of these healthcare workers are pregnant.
“The CDC’s Advisory Committee on Immunization Practices will speak to that just as soon as the EUA is issued,” he added.
Patients are asking Culver about the priority order for vaccination. He said it’s difficult to provide firm guidance at this point.
People also have “lingering skepticism” about whether vaccine development was done in a prudent way, Culver said. Some people question whether the Pfizer vaccine and others were rushed to market. “So we try to spend time with the patients, reassuring them that all the usual safety evaluations were carefully done,” he said.
Another concern is whether mRNA vaccines can interact with human DNA. “The quick, short, and definitive answer is no,” Schaffner said. The m stands for messenger — the vaccines transmit information. "Once it gets into a cell, the mRNA does not go anywhere near the DNA, and once it transmits its information to the cell appropriately, it gets metabolized, and we excrete all the remnants."
Hewlett pointed out that investigations and surveillance will continue. Because this is an EUA and not full approval, “that essentially means they will still be obligated to collect a lot more data than they would ordinarily,” he said.
How long immunoprotection will last also remains an unknown. “The big question left on the table now is the durability,” Culver said. “Of course, we won’t know the answer to that for quite some time.”
Schaffner and Culver have disclosed no relevant financial relationships. Hewlett was an employee of Pfizer until mid-2019. His previous work as Pfizer’s senior medical director of global medical product evaluation was not associated with development of the COVID-19 vaccine.
This article first appeared on Medscape.com.
The much-anticipated emergency use authorization (EUA) of this vaccine — the first such approval in the United States — was greeted with optimism by infectious disease and pulmonary experts, although unanswered questions remain regarding use in people with allergic hypersensitivity, safety in pregnant women, and how smooth distribution will be.
“I am delighted. This is a first, firm step on a long path to getting this COVID pandemic under control,” William Schaffner, MD, professor of infectious diseases at the Vanderbilt University School of Medicine in Nashville, Tennessee, said in an interview.
The FDA gave the green light after the December 10 recommendation from the agency’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) meeting. The committee voted 17-4 in favor of the emergency authorization.
The COVID-19 vaccine is “going to have a major impact here in the US. I’m very optimistic about it,” Dial Hewlett, MD, a spokesperson for the Infectious Diseases Society of American (IDSA), told this news organization.
Daniel Culver, DO, chair of medicine at the Cleveland Clinic in Ohio, is likewise hopeful. “My understanding is that supplies of the vaccine are already in place in hubs and will be shipped relatively quickly. The hope would be we can start vaccinating people as early as next week.”
Allergic reactions reported in the UK
After vaccinations with the Pfizer vaccine began in the UK on December 8, reports surfaced of two healthcare workers who experienced allergic reactions. They have since recovered, but officials warned that people with a history of severe allergic reactions should not receive the Pfizer vaccine at this time.
“For the moment, they are asking people who have had notable allergic reactions to step aside while this is investigated. It shows you that the system is working,” Schaffner said.
Both vaccine recipients who experienced anaphylaxis carried EpiPens, as they were at high risk for allergic reactions, Hewlett said. Also, if other COVID-19 vaccines are approved for use in the future, people allergic to the Pfizer vaccine might have another option, he added.
Reassuring role models
Schaffner supports the CDC Advisory Committee on Immunization Practices (ACIP) decision to start vaccinations with healthcare workers and residents of long-term care facilities.
“Vaccinating healthcare workers, in particular, will be a model for the general public,” said Schaffner, who is also a former member of the IDSA board of directors. “If they see those of us in white coats and blue scrubs lining up for the vaccine, that will provide confidence.”
To further increase acceptance of the COVID-19 vaccine, public health officials need to provide information and reassure the general public, Schaffner said.
Hewlett agreed. “I know there are a lot of people in the population who are very hesitant about vaccines. As infection disease specialists and people in public health, we are trying to allay a lot of concerns people have.”
Reassurance will be especially important in minority communities. “They have been disproportionately affected by the virus, and they have a traditional history of not being optimally vaccinated,” Schaffner said. “We need to reach them in particular with good information and reassurance…so they can make good decisions for themselves and their families.”
No vaccine is 100% effective or completely free of side effects. “There is always a chance there can be adverse reactions, but we think for the most part this is going to be a safe and effective vaccine,” said Hewlett, medical director at the Division of Disease Control and deputy to commissioner of health at the Westchester County Department of Health in White Plains, New York.
Distribution: Smooth or full of strife?
In addition to the concern that some people will not take advantage of vaccination against COVID-19, there could be vaccine supply issues down the road, Schaffner said.
Culver agreed. “In the early phases, I expect that there will be some kinks to work out, but because the numbers are relatively small, this should be okay,” he said.
“I think when we start to get into larger-scale vaccination programs — the supply chain, transport, and storage will be a Herculean undertaking,” Culver added. “It will take careful coordination between healthcare providers, distributors, suppliers, and public health officials to pull this off.”
Planning and distribution also should focus beyond US borders. Any issues in vaccine distribution or administration in the United States “will only be multiplied in several other parts of the world,” Culver said. Because COVID-19 is a pandemic, “we need to think about vaccinating globally.”
Investigating adverse events
Adverse events common to vaccinations in general — injection site pain, headaches, and fever — would not be unexpected with the COVID-19 vaccines. However, experts remain concerned that other, unrelated adverse events might be erroneously attributed to vaccination. For example, if a fall, heart attack, or death occurs within days of immunization, some might immediately blame the vaccine product.
“It’s important to remember that any new, highly touted medical therapy like this will receive a lot of scrutiny, so it would be unusual not to hear about something happening to somebody,” Culver said. Vaccine companies and health agencies will be carefully evaluating any reported adverse events to ensure no safety signal was missed in the trials.
“Fortunately, there are systems in place to investigate these events immediately,” Schaffner said.
Pregnancy recommendations pending
One question still looms: Is the COVID-19 vaccination safe for pregnant women? This isn’t just a question for the general public, either, Schaffner said. He estimated that about 70 percent of healthcare workers are women, and data suggests about 300,000 of these healthcare workers are pregnant.
“The CDC’s Advisory Committee on Immunization Practices will speak to that just as soon as the EUA is issued,” he added.
Patients are asking Culver about the priority order for vaccination. He said it’s difficult to provide firm guidance at this point.
People also have “lingering skepticism” about whether vaccine development was done in a prudent way, Culver said. Some people question whether the Pfizer vaccine and others were rushed to market. “So we try to spend time with the patients, reassuring them that all the usual safety evaluations were carefully done,” he said.
Another concern is whether mRNA vaccines can interact with human DNA. “The quick, short, and definitive answer is no,” Schaffner said. The m stands for messenger — the vaccines transmit information. "Once it gets into a cell, the mRNA does not go anywhere near the DNA, and once it transmits its information to the cell appropriately, it gets metabolized, and we excrete all the remnants."
Hewlett pointed out that investigations and surveillance will continue. Because this is an EUA and not full approval, “that essentially means they will still be obligated to collect a lot more data than they would ordinarily,” he said.
How long immunoprotection will last also remains an unknown. “The big question left on the table now is the durability,” Culver said. “Of course, we won’t know the answer to that for quite some time.”
Schaffner and Culver have disclosed no relevant financial relationships. Hewlett was an employee of Pfizer until mid-2019. His previous work as Pfizer’s senior medical director of global medical product evaluation was not associated with development of the COVID-19 vaccine.
This article first appeared on Medscape.com.