User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Management of Facial Hair in Women
Facial hair growth in women is complex and multifaceted. It is not a disease but rather a part of normal anatomy or a symptom influenced by an underlying condition such as hypertrichosis, a hormonal imbalance (eg, hirsutism due to polycystic ovary syndrome [PCOS]), mechanical factors such as pseudofolliculitis barbae (PFB) from shaving, and perimenopausal and postmenopausal hormonal shifts. Additionally, normal facial hair patterns can vary substantially based on genetics, ethnicity, and cultural background. Some populations may naturally have more visible vellus or terminal hairs on the face, which are entirely physiologic rather than indicative of an underlying disorder. Despite this, societal expectations and beauty standards across many cultures dictate that facial hair in women is undesirable, often associating hair-free skin with femininity and attractiveness. This perception drives many women to seek treatment—not necessarily for medical reasons, but due to social pressure and aesthetic preferences.
Hypertrichosis, whether congenital or acquired, refers to excessive hair growth that is not androgen dependent and can appear on any site of the body. Causes include genetic predisposition, porphyria, thyroid disorders, internal malignancies, malnutrition, anorexia nervosa, or use of medications such as cyclosporine, prednisolone, and phenytoin.1 Hirsutism, by contrast, is characterized by the growth of terminal hairs in women at androgen-dependent sites such as the face, neck, and upper chest, where coarse hair typically grows in men.2 This condition often is associated with excess androgens produced by the ovaries or adrenal glands, most commonly due to PCOS although genetic factors may contribute.
Before initiating treatment, a thorough history and physical examination are essential to determine the underlying cause of conditions associated with facial hair growth in women. Clinicians should assess for signs of hyperandrogenism, menstrual irregularities, virilization, medication use, and family history. In cases of a suspected endocrine disorder, further laboratory evaluation may be warranted to guide appropriate management. While each cause of facial hair growth in women has unique management considerations, the shared impact on psychosocial well-being and adherence to grooming standards in the US military warrants an all-encompassing yet targeted approach. This comprehensive review discusses management options for women with facial hair in the military based on a review of PubMed articles indexed for MEDLINE conducted in November 2024 using combinations of the following search terms: hirsutism, facial hair, pseudofolliculitis barbae, women, female, military, grooming standards, hyperandrogenism, and hair removal.
Treatment Modalities
The available treatment modalities, including their mechanisms, potential risks, and considerations are summarized in the eTable.

Mechanical—Shaving remains one of the most widely utilized methods of hair removal in women due to its accessibility and ease of use. It does not disrupt the anagen phase of the hair growth cycle, making it a temporary method that requires frequent repetition (often daily), particularly for individuals with rapid hair growth. The belief that shaving causes hair to grow back thicker or faster is a common misconception. Shaving does not alter the thickness or growth rate of hair; instead, it leaves a blunt tip, making the hair feel coarser or appear thicker than uncut hair.3 Despite its relative convenience, shaving can lead to skin irritation due to mechanical trauma. Potential complications include PFB, superficial abrasions known more broadly as shaving irritation, and an increased risk for infections such as bacterial or fungal folliculitis.4
Chemical depilation, which uses thioglycolates mixed with alkali compounds, disrupts disulfide bonds in the hair, effectively breaking down the shaft without affecting the bulb. The depilatory requires application to the skin for approximately 3 to 15 minutes depending on the specific formulation and the thickness or texture of the hair. While it is a cost-effective option that easily can be done at home, the chemicals involved may trigger irritant contact dermatitis or folliculitis and produce an unpleasant odor from hydrogen disulfide gas.5 They also can lead to PFB.
Epilation removes the entire hair shaft and bulb, with results lasting approximately 6 weeks.6 Methods range from using tweezers to pluck single hairs and devices that simultaneously remove multiple hairs to hot or cold waxing, which use resin to grip and remove hair. Threading is a technique that uses twisted thread to remove the hair at the follicle level; this method may not alter hair growth unless performed during the anagen phase, during which repeated plucking can damage the matrix and potentially lead to permanent hair reduction.5 Common adverse effects include pain during removal, burns from waxing, folliculitis, PFB, postinflammatory hyperpigmentation, and scarring, particularly when multiple hairs are removed at once.
Pharmacologic—Pharmacologic therapy commonly is used to manage hirsutism and typically begins with a trial of combined oral contraceptives (COCs) containing estrogen and progestin, which are considered the first-line option unless contraindicated.7 If response to COC monotherapy is inadequate, an antiandrogen such as spironolactone may be added. Combination therapy with a COC and an antiandrogen generally is reserved for severe cases or patients who previously have shown suboptimal response to COCs alone.7 Patients should be counseled to discontinue antiandrogen therapy if they become pregnant due to the risk for fetal undervirilization observed in animal studies.8,9 Typical dosing of spironolactone, a competitive inhibitor of 5-α-reductase and androgen receptors, ranges from 100 mg to 200 mg daily.10 Reported adverse effects include polyuria, postural hypotension, menstrual irregularities, hyperkalemia, and potential liver dysfunction. Although spironolactone has demonstrated tumorigenic effects in animal studies, no such effects have been observed in humans.11
Eflornithine hydrochloride cream 13.9% is the first topical prescription medication approved by the US Food and Drug Administration for reduction of unwanted facial hair in women.12 It works by irreversibly blocking the activity of ornithine decarboxylase, an enzyme involved in the rate-limiting step of polyamine synthesis, which is essential for hair growth. In a randomized, double-blind clinical trial evaluating its effectiveness and safety, twice-daily application for 24 weeks resulted in a clinically meaningful reduction in hair length and density (measured as surface area) compared with the control group.13 When eflornithine hydrochloride cream 13.9% is discontinued, hair growth gradually returns to baseline. Studies have shown that hair regrowth typically begins within 8 weeks after treatment is stopped; within several months, hair returns to pretreatment levels.14 Adverse effects of eflornithine hydrochloride cream generally are mild and may include local irritation and acneform eruptions. In a randomized bilateral vehicle-controlled trial of 31 women, both eflornithine and vehicle creams were well tolerated, with 1 patient reporting mild tingling with eflornithine that resolved with continued use for 7 days.15
Procedural—Photoepilation therapies widely are considered by dermatologists to be among the most effective methods for reducing unwanted hair.16 Laser hair removal employs selective photothermolysis, a principle by which specific wavelengths of light target melanin in hair follicles. This method results in localized thermal damage, destroying hair follicles and reducing regrowth. Wavelengths between 600 and 1100 nm are most effective for hair removal; widely used devices include the ruby (694 nm), alexandrite (755 nm), diode (800-810 nm), and long-pulsed Nd:YAG lasers (1064 nm). Cooling mechanisms such as cryogen spray or contact cooling often are employed to minimize epidermal damage and lessen patient discomfort.
The hair matrix is most responsive to laser treatment during the anagen phase, necessitating multiple sessions to ensure all hairs are treated during this optimal growth stage. Generally, 4 to 6 sessions spaced at intervals of 4 to 6 weeks are required to achieve satisfactory results.17 Matching the laser wavelength to the absorption properties of melanin—the target chromophore—enables selective destruction of melanin-rich hair follicles while minimizing damage to surrounding skin.
The ideal laser wavelength primarily affects melanin concentrated in the hair bulb, leading to follicular destruction while reducing the risk for unintended depigmentation of the epidermis; however, competing structures in the skin (eg, epidermal pigment) also can absorb laser energy, diminishing treatment efficacy and increasing the risk for adverse effects. Shorter wavelengths are effective for lighter skin types, while longer wavelengths such as the Nd:YAG laser are safer for individuals with darker skin types as they bypass melanin in the epidermis.
It is important to note that laser hair removal is ineffective for white and gray hairs due to the lack of melanin. As a result, alternative methods such as electrolysis, which does not rely on pigment, may be more appropriate for permanent hair removal in individuals with nonpigmented hairs. Research indicates that combining topical eflornithine with alexandrite or Nd:YAG lasers improves outcomes for reducing unwanted facial hair.18
In military settings, laser hair removal is utilized for specific conditions such as PFB in male service members to assist with the reduction of hair and mitigation of symptoms.19 The majority of military dermatology clinics have devices for laser hair removal; however, dermatology services are not available at many military treatment facilities, and dermatologic care may be provided by the local civilian dermatologists. That said, laser therapy is covered in the civilian sector for active-duty service members with PFB of the face and neck under certain criteria. These include a documented safety risk in environments requiring respiratory protection, failure of conservative treatments, and evaluation by a military dermatologist who confirms the necessity of civilian-provided laser therapy when it is unavailable at a military facility.20 While such policies demonstrate the military’s recognition of laser therapy as a viable solution for certain grooming-related conditions, many are unaware that the existing laser hair removal policy also applies to women. Increasing awareness of this coverage could help female service members access treatment options that align with both medical and professional grooming needs.
Intense pulsed light (IPL) systems are nonlaser devices that emit broad-spectrum light in the 590- to 1200-nm range. They utilize a flash lamp to achieve thermal damage. Filters are used to narrow the wavelength range based on the specific target. Intense pulsed light devices are less precise than lasers but remain effective for hair reduction. In addition to hair removal, IPL devices are employed in the treatment of pigmented and vascular lesions. Common adverse effects of both laser and IPL hair removal include transient erythema, perifollicular edema, and pigmentary changes, especially in patients with darker skin types. Rare complications include blistering, scarring, and paradoxical hair stimulation in which untreated areas develop increased hair growth.
Electrolysis is recognized as the only method of truly permanent hair removal and is effective for all hair colors.21 However, the variability in technique among practitioners often leads to inconsistent results, with some patients experiencing hair regrowth. Galvanic electrolysis involves inserting a fine needle into the hair follicle and applying an electrical current to destroy the it and the rapidly dividing cells of the matrix.22 The introduction of thermolytic electrolysis, which uses a high-frequency alternating current (commonly 13.56 MHz or 27.12 MHz), has enhanced efficiency by creating heat at the needle tip to destroy the follicle. This approach is faster and now is commonly combined with galvanic electrolysis.23 While no controlled clinical trials directly compare these methods, many patients experience permanent hair removal, with approximately 15% to 25% regrowth within 6 months.22,24
Alternative Options—Home-use laser and light-based devices have become increasingly popular for managing unwanted hair due to their affordability and convenience, with most devices priced less than $1000.25 These devices utilize various technologies, including lasers (808 nm), IPL, or combinations of IPL and radiofrequency.26 Despite their accessibility, peer-reviewed research on their safety profile and effectiveness is limited, as existing data primarily come from industry-funded, uncontrolled studies with short follow-up durations—making it difficult to assess long-term outcomes.25
Psychosocial Impact
A 2023 study of active-duty female service members with PCOS highlighted the unique challenges they face while managing symptoms such as facial hair within the constraints of military service.27 Although the study focused on PCOS, the findings shed light on how facial hair specifically impacts the psychological well-being of servicewomen. Participants described facial hair as one of the most visible and stigmatizing symptoms, often leading to feelings of embarrassment and diminished confidence. Participants also highlighted the professional implications of facial hair, with some describing feelings of scrutiny and judgment from peers and leadership in public. These challenges can be more pronounced in deployments or field exercises where hygiene resources are limited. The lack of access not only affects self-perception but also can hinder the ability of servicewomen to meet implicit expectations for grooming and appearance.27 There is a notable gap in research examining the impact of facial hair on military servicewomen. Given the unique environmental challenges and professional expectations, further investigation is warranted to better understand how facial hair affects women and to optimize treatment approaches in this population.
Final Thoughts
Limited awareness and understanding of facial hair in woman contribute to stigma, often leaving affected individuals to navigate challenges in isolation. Given the impact on confidence, professional appearance, and adherence to military grooming standards, it is essential for health care practitioners to recognize and address facial hair in women. Importantly, laser hair removal is covered by TRICARE for active-duty female service members with PFB, yet many remain unaware of this benefit. Increased awareness of available mechanical, pharmacologic, and procedural treatment options allows for tailored management, ensuring that women receive appropriate medical care.
Wendelin DS, Pope DN, Mallory SB. Hypertrichosis. J Am Acad Dermatol. 2003;48:161-181. doi:10.1067/mjd.2003.100
Blume-Peytavi U, Hahn S. Medical treatment of hirsutism. Dermatol Ther. 2008;21:329-339. doi:10.1111/j.1529-8019.2008.00215.x
Kang CN, Shah M, Lynde C, et al. Hair removal practices: a literature review. Skin Therapy Lett. 2021;26:6-11.
Matheson E, Bain J. Hirsutism in women. Am Fam Physician. 2019;100:168-175.
Shenenberger DW, Utecht LM. Removal of unwanted facial hair. Am Fam Physician. 2002;66:1907-1911.
Johnson E, Ebling FJ. The effect of plucking hairs during different phases of the follicular cycle. J Embryol Exp Morphol. 1964;12:465-474.
Martin KA, Anderson RR, Chang RJ, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2018;103:1233-1257. doi:10.1210/jc.2018-00241
Barrionuevo P, Nabhan M, Altayar O, et al. Treatment options for hirsutism: a systematic review and network meta-analysis. J Clin Endocrinol Metab. 2018;103:1258-1264. doi:10.1210/jc.2017-02052
Alesi S, Forslund M, Melin J, et al. Efficacy and safety of anti-androgens in the management of polycystic ovary syndrome: a systematic review and meta-analysis of randomised controlled trials. EClinicalMedicine. Published online August 9, 2023. doi:10.1016/j.eclinm.2023.102162
Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement. Hum Reprod Update. 2012;18:146-170.
Hussein RS, Abdelbasset WK. Updates on hirsutism: a narrative review. Int J Biomedicine. 2022;12:193-198. doi:10.21103/Article12(2)_RA4
Shapiro J, Lui H. Vaniqa—eflornithine 13.9% cream. Skin Therapy Lett. 2001;6:1-5.
Wolf JE Jr, Shander D, Huber F, et al. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair. Int J Dermatol. 2007;46:94-98. doi:10.1111/j.1365-4632.2006.03079.x
Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol. 2001;2:197-202. doi:10.2165/00128071-200102030-00009
Hamzavi I, Tan E, Shapiro J, et al. A randomized bilateral vehicle-controlled study of eflornithine cream combined with laser treatment versus laser treatment alone for facial hirsutism in women. J Am Acad Dermatol. 2007;57:54-59. doi:10.1016/j.jaad.2006.09.025
Goldberg DJ. Laser hair removal. In: Goldberg DJ, ed. Laser Dermatology: Pearls and Problems. Blackwell; 2008.
Hussain M, Polnikorn N, Goldberg DJ. Laser-assisted hair removal in Asian skin: efficacy, complications, and the effect of single versus multiple treatments. Dermatol Surg. 2003;29:249-254. doi:10.1046/j.1524-4725.2003.29059.x
Smith SR, Piacquadio DJ, Beger B, et al. Eflornithine cream combined with laser therapy in the management of unwanted facial hair growth in women: a randomized trial. Dermatol Surg. 2006;32:1237-1243. doi:10.1111/j.1524-4725.2006.32282.x
Jung I, Lannan FM, Weiss A, et al. Treatment and current policies on pseudofolliculitis barbae in the US military. Cutis. 2023;112:299-302. doi:10.12788/cutis.0907
TRICARE Operations Manual 6010.59-M. Supplemental Health Care Program (SHCP)—Chapter 17. Contractor Responsibilities. Military Health System and Defense Health Agency website. Revised November 5, 2021. Accessed February 13, 2024. https://manuals.health.mil/pages/DisplayManualHtmlFile/2022-08-31/AsOf/TO15/C17S3.html
Yanes DA, Smith P, Avram MM. A review of best practices for gender-affirming laser hair removal. Dermatol Surg. 2024;50:S201-S204. doi:10.1097/DSS.0000000000004441
Wagner RF Jr, Tomich JM, Grande DJ. Electrolysis and thermolysis for permanent hair removal. J Am Acad Dermatol. 1985;12:441-449. doi:10.1016/s0190-9622(85)70062-x
Olsen EA. Methods of hair removal. J Am Acad Dermatol. 1999;40:143-157. doi:10.1016/s0190-9622(99)70181-7
Kligman AM, Peters L. Histologic changes of human hair follicles after electrolysis: a comparison of two methods. Cutis. 1984;34:169-176.
Hession MT, Markova A, Graber EM. A review of hand-held, home-use cosmetic laser and light devices. Dermatol Surg. 2015;41:307-320. doi:10.1097/DSS.0000000000000283
Wheeland RG. Permanent hair reduction with a home-use diode laser: safety and effectiveness 1 year after eight treatments. Lasers Surg Med. 2012;44:550-557. doi:10.1002/lsm.22051
Hopkins D, Walker SC, Wilson C, et al. The experience of living with polycystic ovary syndrome in the military. Mil Med. 2024;189:E188-E197. doi:10.1093/milmed/usad241
Facial hair growth in women is complex and multifaceted. It is not a disease but rather a part of normal anatomy or a symptom influenced by an underlying condition such as hypertrichosis, a hormonal imbalance (eg, hirsutism due to polycystic ovary syndrome [PCOS]), mechanical factors such as pseudofolliculitis barbae (PFB) from shaving, and perimenopausal and postmenopausal hormonal shifts. Additionally, normal facial hair patterns can vary substantially based on genetics, ethnicity, and cultural background. Some populations may naturally have more visible vellus or terminal hairs on the face, which are entirely physiologic rather than indicative of an underlying disorder. Despite this, societal expectations and beauty standards across many cultures dictate that facial hair in women is undesirable, often associating hair-free skin with femininity and attractiveness. This perception drives many women to seek treatment—not necessarily for medical reasons, but due to social pressure and aesthetic preferences.
Hypertrichosis, whether congenital or acquired, refers to excessive hair growth that is not androgen dependent and can appear on any site of the body. Causes include genetic predisposition, porphyria, thyroid disorders, internal malignancies, malnutrition, anorexia nervosa, or use of medications such as cyclosporine, prednisolone, and phenytoin.1 Hirsutism, by contrast, is characterized by the growth of terminal hairs in women at androgen-dependent sites such as the face, neck, and upper chest, where coarse hair typically grows in men.2 This condition often is associated with excess androgens produced by the ovaries or adrenal glands, most commonly due to PCOS although genetic factors may contribute.
Before initiating treatment, a thorough history and physical examination are essential to determine the underlying cause of conditions associated with facial hair growth in women. Clinicians should assess for signs of hyperandrogenism, menstrual irregularities, virilization, medication use, and family history. In cases of a suspected endocrine disorder, further laboratory evaluation may be warranted to guide appropriate management. While each cause of facial hair growth in women has unique management considerations, the shared impact on psychosocial well-being and adherence to grooming standards in the US military warrants an all-encompassing yet targeted approach. This comprehensive review discusses management options for women with facial hair in the military based on a review of PubMed articles indexed for MEDLINE conducted in November 2024 using combinations of the following search terms: hirsutism, facial hair, pseudofolliculitis barbae, women, female, military, grooming standards, hyperandrogenism, and hair removal.
Treatment Modalities
The available treatment modalities, including their mechanisms, potential risks, and considerations are summarized in the eTable.
Mechanical—Shaving remains one of the most widely utilized methods of hair removal in women due to its accessibility and ease of use. It does not disrupt the anagen phase of the hair growth cycle, making it a temporary method that requires frequent repetition (often daily), particularly for individuals with rapid hair growth. The belief that shaving causes hair to grow back thicker or faster is a common misconception. Shaving does not alter the thickness or growth rate of hair; instead, it leaves a blunt tip, making the hair feel coarser or appear thicker than uncut hair.3 Despite its relative convenience, shaving can lead to skin irritation due to mechanical trauma. Potential complications include PFB, superficial abrasions known more broadly as shaving irritation, and an increased risk for infections such as bacterial or fungal folliculitis.4
Chemical depilation, which uses thioglycolates mixed with alkali compounds, disrupts disulfide bonds in the hair, effectively breaking down the shaft without affecting the bulb. The depilatory requires application to the skin for approximately 3 to 15 minutes depending on the specific formulation and the thickness or texture of the hair. While it is a cost-effective option that easily can be done at home, the chemicals involved may trigger irritant contact dermatitis or folliculitis and produce an unpleasant odor from hydrogen disulfide gas.5 They also can lead to PFB.
Epilation removes the entire hair shaft and bulb, with results lasting approximately 6 weeks.6 Methods range from using tweezers to pluck single hairs and devices that simultaneously remove multiple hairs to hot or cold waxing, which use resin to grip and remove hair. Threading is a technique that uses twisted thread to remove the hair at the follicle level; this method may not alter hair growth unless performed during the anagen phase, during which repeated plucking can damage the matrix and potentially lead to permanent hair reduction.5 Common adverse effects include pain during removal, burns from waxing, folliculitis, PFB, postinflammatory hyperpigmentation, and scarring, particularly when multiple hairs are removed at once.
Pharmacologic—Pharmacologic therapy commonly is used to manage hirsutism and typically begins with a trial of combined oral contraceptives (COCs) containing estrogen and progestin, which are considered the first-line option unless contraindicated.7 If response to COC monotherapy is inadequate, an antiandrogen such as spironolactone may be added. Combination therapy with a COC and an antiandrogen generally is reserved for severe cases or patients who previously have shown suboptimal response to COCs alone.7 Patients should be counseled to discontinue antiandrogen therapy if they become pregnant due to the risk for fetal undervirilization observed in animal studies.8,9 Typical dosing of spironolactone, a competitive inhibitor of 5-α-reductase and androgen receptors, ranges from 100 mg to 200 mg daily.10 Reported adverse effects include polyuria, postural hypotension, menstrual irregularities, hyperkalemia, and potential liver dysfunction. Although spironolactone has demonstrated tumorigenic effects in animal studies, no such effects have been observed in humans.11
Eflornithine hydrochloride cream 13.9% is the first topical prescription medication approved by the US Food and Drug Administration for reduction of unwanted facial hair in women.12 It works by irreversibly blocking the activity of ornithine decarboxylase, an enzyme involved in the rate-limiting step of polyamine synthesis, which is essential for hair growth. In a randomized, double-blind clinical trial evaluating its effectiveness and safety, twice-daily application for 24 weeks resulted in a clinically meaningful reduction in hair length and density (measured as surface area) compared with the control group.13 When eflornithine hydrochloride cream 13.9% is discontinued, hair growth gradually returns to baseline. Studies have shown that hair regrowth typically begins within 8 weeks after treatment is stopped; within several months, hair returns to pretreatment levels.14 Adverse effects of eflornithine hydrochloride cream generally are mild and may include local irritation and acneform eruptions. In a randomized bilateral vehicle-controlled trial of 31 women, both eflornithine and vehicle creams were well tolerated, with 1 patient reporting mild tingling with eflornithine that resolved with continued use for 7 days.15
Procedural—Photoepilation therapies widely are considered by dermatologists to be among the most effective methods for reducing unwanted hair.16 Laser hair removal employs selective photothermolysis, a principle by which specific wavelengths of light target melanin in hair follicles. This method results in localized thermal damage, destroying hair follicles and reducing regrowth. Wavelengths between 600 and 1100 nm are most effective for hair removal; widely used devices include the ruby (694 nm), alexandrite (755 nm), diode (800-810 nm), and long-pulsed Nd:YAG lasers (1064 nm). Cooling mechanisms such as cryogen spray or contact cooling often are employed to minimize epidermal damage and lessen patient discomfort.
The hair matrix is most responsive to laser treatment during the anagen phase, necessitating multiple sessions to ensure all hairs are treated during this optimal growth stage. Generally, 4 to 6 sessions spaced at intervals of 4 to 6 weeks are required to achieve satisfactory results.17 Matching the laser wavelength to the absorption properties of melanin—the target chromophore—enables selective destruction of melanin-rich hair follicles while minimizing damage to surrounding skin.
The ideal laser wavelength primarily affects melanin concentrated in the hair bulb, leading to follicular destruction while reducing the risk for unintended depigmentation of the epidermis; however, competing structures in the skin (eg, epidermal pigment) also can absorb laser energy, diminishing treatment efficacy and increasing the risk for adverse effects. Shorter wavelengths are effective for lighter skin types, while longer wavelengths such as the Nd:YAG laser are safer for individuals with darker skin types as they bypass melanin in the epidermis.
It is important to note that laser hair removal is ineffective for white and gray hairs due to the lack of melanin. As a result, alternative methods such as electrolysis, which does not rely on pigment, may be more appropriate for permanent hair removal in individuals with nonpigmented hairs. Research indicates that combining topical eflornithine with alexandrite or Nd:YAG lasers improves outcomes for reducing unwanted facial hair.18
In military settings, laser hair removal is utilized for specific conditions such as PFB in male service members to assist with the reduction of hair and mitigation of symptoms.19 The majority of military dermatology clinics have devices for laser hair removal; however, dermatology services are not available at many military treatment facilities, and dermatologic care may be provided by the local civilian dermatologists. That said, laser therapy is covered in the civilian sector for active-duty service members with PFB of the face and neck under certain criteria. These include a documented safety risk in environments requiring respiratory protection, failure of conservative treatments, and evaluation by a military dermatologist who confirms the necessity of civilian-provided laser therapy when it is unavailable at a military facility.20 While such policies demonstrate the military’s recognition of laser therapy as a viable solution for certain grooming-related conditions, many are unaware that the existing laser hair removal policy also applies to women. Increasing awareness of this coverage could help female service members access treatment options that align with both medical and professional grooming needs.
Intense pulsed light (IPL) systems are nonlaser devices that emit broad-spectrum light in the 590- to 1200-nm range. They utilize a flash lamp to achieve thermal damage. Filters are used to narrow the wavelength range based on the specific target. Intense pulsed light devices are less precise than lasers but remain effective for hair reduction. In addition to hair removal, IPL devices are employed in the treatment of pigmented and vascular lesions. Common adverse effects of both laser and IPL hair removal include transient erythema, perifollicular edema, and pigmentary changes, especially in patients with darker skin types. Rare complications include blistering, scarring, and paradoxical hair stimulation in which untreated areas develop increased hair growth.
Electrolysis is recognized as the only method of truly permanent hair removal and is effective for all hair colors.21 However, the variability in technique among practitioners often leads to inconsistent results, with some patients experiencing hair regrowth. Galvanic electrolysis involves inserting a fine needle into the hair follicle and applying an electrical current to destroy the it and the rapidly dividing cells of the matrix.22 The introduction of thermolytic electrolysis, which uses a high-frequency alternating current (commonly 13.56 MHz or 27.12 MHz), has enhanced efficiency by creating heat at the needle tip to destroy the follicle. This approach is faster and now is commonly combined with galvanic electrolysis.23 While no controlled clinical trials directly compare these methods, many patients experience permanent hair removal, with approximately 15% to 25% regrowth within 6 months.22,24
Alternative Options—Home-use laser and light-based devices have become increasingly popular for managing unwanted hair due to their affordability and convenience, with most devices priced less than $1000.25 These devices utilize various technologies, including lasers (808 nm), IPL, or combinations of IPL and radiofrequency.26 Despite their accessibility, peer-reviewed research on their safety profile and effectiveness is limited, as existing data primarily come from industry-funded, uncontrolled studies with short follow-up durations—making it difficult to assess long-term outcomes.25
Psychosocial Impact
A 2023 study of active-duty female service members with PCOS highlighted the unique challenges they face while managing symptoms such as facial hair within the constraints of military service.27 Although the study focused on PCOS, the findings shed light on how facial hair specifically impacts the psychological well-being of servicewomen. Participants described facial hair as one of the most visible and stigmatizing symptoms, often leading to feelings of embarrassment and diminished confidence. Participants also highlighted the professional implications of facial hair, with some describing feelings of scrutiny and judgment from peers and leadership in public. These challenges can be more pronounced in deployments or field exercises where hygiene resources are limited. The lack of access not only affects self-perception but also can hinder the ability of servicewomen to meet implicit expectations for grooming and appearance.27 There is a notable gap in research examining the impact of facial hair on military servicewomen. Given the unique environmental challenges and professional expectations, further investigation is warranted to better understand how facial hair affects women and to optimize treatment approaches in this population.
Final Thoughts
Limited awareness and understanding of facial hair in woman contribute to stigma, often leaving affected individuals to navigate challenges in isolation. Given the impact on confidence, professional appearance, and adherence to military grooming standards, it is essential for health care practitioners to recognize and address facial hair in women. Importantly, laser hair removal is covered by TRICARE for active-duty female service members with PFB, yet many remain unaware of this benefit. Increased awareness of available mechanical, pharmacologic, and procedural treatment options allows for tailored management, ensuring that women receive appropriate medical care.
Facial hair growth in women is complex and multifaceted. It is not a disease but rather a part of normal anatomy or a symptom influenced by an underlying condition such as hypertrichosis, a hormonal imbalance (eg, hirsutism due to polycystic ovary syndrome [PCOS]), mechanical factors such as pseudofolliculitis barbae (PFB) from shaving, and perimenopausal and postmenopausal hormonal shifts. Additionally, normal facial hair patterns can vary substantially based on genetics, ethnicity, and cultural background. Some populations may naturally have more visible vellus or terminal hairs on the face, which are entirely physiologic rather than indicative of an underlying disorder. Despite this, societal expectations and beauty standards across many cultures dictate that facial hair in women is undesirable, often associating hair-free skin with femininity and attractiveness. This perception drives many women to seek treatment—not necessarily for medical reasons, but due to social pressure and aesthetic preferences.
Hypertrichosis, whether congenital or acquired, refers to excessive hair growth that is not androgen dependent and can appear on any site of the body. Causes include genetic predisposition, porphyria, thyroid disorders, internal malignancies, malnutrition, anorexia nervosa, or use of medications such as cyclosporine, prednisolone, and phenytoin.1 Hirsutism, by contrast, is characterized by the growth of terminal hairs in women at androgen-dependent sites such as the face, neck, and upper chest, where coarse hair typically grows in men.2 This condition often is associated with excess androgens produced by the ovaries or adrenal glands, most commonly due to PCOS although genetic factors may contribute.
Before initiating treatment, a thorough history and physical examination are essential to determine the underlying cause of conditions associated with facial hair growth in women. Clinicians should assess for signs of hyperandrogenism, menstrual irregularities, virilization, medication use, and family history. In cases of a suspected endocrine disorder, further laboratory evaluation may be warranted to guide appropriate management. While each cause of facial hair growth in women has unique management considerations, the shared impact on psychosocial well-being and adherence to grooming standards in the US military warrants an all-encompassing yet targeted approach. This comprehensive review discusses management options for women with facial hair in the military based on a review of PubMed articles indexed for MEDLINE conducted in November 2024 using combinations of the following search terms: hirsutism, facial hair, pseudofolliculitis barbae, women, female, military, grooming standards, hyperandrogenism, and hair removal.
Treatment Modalities
The available treatment modalities, including their mechanisms, potential risks, and considerations are summarized in the eTable.
Mechanical—Shaving remains one of the most widely utilized methods of hair removal in women due to its accessibility and ease of use. It does not disrupt the anagen phase of the hair growth cycle, making it a temporary method that requires frequent repetition (often daily), particularly for individuals with rapid hair growth. The belief that shaving causes hair to grow back thicker or faster is a common misconception. Shaving does not alter the thickness or growth rate of hair; instead, it leaves a blunt tip, making the hair feel coarser or appear thicker than uncut hair.3 Despite its relative convenience, shaving can lead to skin irritation due to mechanical trauma. Potential complications include PFB, superficial abrasions known more broadly as shaving irritation, and an increased risk for infections such as bacterial or fungal folliculitis.4
Chemical depilation, which uses thioglycolates mixed with alkali compounds, disrupts disulfide bonds in the hair, effectively breaking down the shaft without affecting the bulb. The depilatory requires application to the skin for approximately 3 to 15 minutes depending on the specific formulation and the thickness or texture of the hair. While it is a cost-effective option that easily can be done at home, the chemicals involved may trigger irritant contact dermatitis or folliculitis and produce an unpleasant odor from hydrogen disulfide gas.5 They also can lead to PFB.
Epilation removes the entire hair shaft and bulb, with results lasting approximately 6 weeks.6 Methods range from using tweezers to pluck single hairs and devices that simultaneously remove multiple hairs to hot or cold waxing, which use resin to grip and remove hair. Threading is a technique that uses twisted thread to remove the hair at the follicle level; this method may not alter hair growth unless performed during the anagen phase, during which repeated plucking can damage the matrix and potentially lead to permanent hair reduction.5 Common adverse effects include pain during removal, burns from waxing, folliculitis, PFB, postinflammatory hyperpigmentation, and scarring, particularly when multiple hairs are removed at once.
Pharmacologic—Pharmacologic therapy commonly is used to manage hirsutism and typically begins with a trial of combined oral contraceptives (COCs) containing estrogen and progestin, which are considered the first-line option unless contraindicated.7 If response to COC monotherapy is inadequate, an antiandrogen such as spironolactone may be added. Combination therapy with a COC and an antiandrogen generally is reserved for severe cases or patients who previously have shown suboptimal response to COCs alone.7 Patients should be counseled to discontinue antiandrogen therapy if they become pregnant due to the risk for fetal undervirilization observed in animal studies.8,9 Typical dosing of spironolactone, a competitive inhibitor of 5-α-reductase and androgen receptors, ranges from 100 mg to 200 mg daily.10 Reported adverse effects include polyuria, postural hypotension, menstrual irregularities, hyperkalemia, and potential liver dysfunction. Although spironolactone has demonstrated tumorigenic effects in animal studies, no such effects have been observed in humans.11
Eflornithine hydrochloride cream 13.9% is the first topical prescription medication approved by the US Food and Drug Administration for reduction of unwanted facial hair in women.12 It works by irreversibly blocking the activity of ornithine decarboxylase, an enzyme involved in the rate-limiting step of polyamine synthesis, which is essential for hair growth. In a randomized, double-blind clinical trial evaluating its effectiveness and safety, twice-daily application for 24 weeks resulted in a clinically meaningful reduction in hair length and density (measured as surface area) compared with the control group.13 When eflornithine hydrochloride cream 13.9% is discontinued, hair growth gradually returns to baseline. Studies have shown that hair regrowth typically begins within 8 weeks after treatment is stopped; within several months, hair returns to pretreatment levels.14 Adverse effects of eflornithine hydrochloride cream generally are mild and may include local irritation and acneform eruptions. In a randomized bilateral vehicle-controlled trial of 31 women, both eflornithine and vehicle creams were well tolerated, with 1 patient reporting mild tingling with eflornithine that resolved with continued use for 7 days.15
Procedural—Photoepilation therapies widely are considered by dermatologists to be among the most effective methods for reducing unwanted hair.16 Laser hair removal employs selective photothermolysis, a principle by which specific wavelengths of light target melanin in hair follicles. This method results in localized thermal damage, destroying hair follicles and reducing regrowth. Wavelengths between 600 and 1100 nm are most effective for hair removal; widely used devices include the ruby (694 nm), alexandrite (755 nm), diode (800-810 nm), and long-pulsed Nd:YAG lasers (1064 nm). Cooling mechanisms such as cryogen spray or contact cooling often are employed to minimize epidermal damage and lessen patient discomfort.
The hair matrix is most responsive to laser treatment during the anagen phase, necessitating multiple sessions to ensure all hairs are treated during this optimal growth stage. Generally, 4 to 6 sessions spaced at intervals of 4 to 6 weeks are required to achieve satisfactory results.17 Matching the laser wavelength to the absorption properties of melanin—the target chromophore—enables selective destruction of melanin-rich hair follicles while minimizing damage to surrounding skin.
The ideal laser wavelength primarily affects melanin concentrated in the hair bulb, leading to follicular destruction while reducing the risk for unintended depigmentation of the epidermis; however, competing structures in the skin (eg, epidermal pigment) also can absorb laser energy, diminishing treatment efficacy and increasing the risk for adverse effects. Shorter wavelengths are effective for lighter skin types, while longer wavelengths such as the Nd:YAG laser are safer for individuals with darker skin types as they bypass melanin in the epidermis.
It is important to note that laser hair removal is ineffective for white and gray hairs due to the lack of melanin. As a result, alternative methods such as electrolysis, which does not rely on pigment, may be more appropriate for permanent hair removal in individuals with nonpigmented hairs. Research indicates that combining topical eflornithine with alexandrite or Nd:YAG lasers improves outcomes for reducing unwanted facial hair.18
In military settings, laser hair removal is utilized for specific conditions such as PFB in male service members to assist with the reduction of hair and mitigation of symptoms.19 The majority of military dermatology clinics have devices for laser hair removal; however, dermatology services are not available at many military treatment facilities, and dermatologic care may be provided by the local civilian dermatologists. That said, laser therapy is covered in the civilian sector for active-duty service members with PFB of the face and neck under certain criteria. These include a documented safety risk in environments requiring respiratory protection, failure of conservative treatments, and evaluation by a military dermatologist who confirms the necessity of civilian-provided laser therapy when it is unavailable at a military facility.20 While such policies demonstrate the military’s recognition of laser therapy as a viable solution for certain grooming-related conditions, many are unaware that the existing laser hair removal policy also applies to women. Increasing awareness of this coverage could help female service members access treatment options that align with both medical and professional grooming needs.
Intense pulsed light (IPL) systems are nonlaser devices that emit broad-spectrum light in the 590- to 1200-nm range. They utilize a flash lamp to achieve thermal damage. Filters are used to narrow the wavelength range based on the specific target. Intense pulsed light devices are less precise than lasers but remain effective for hair reduction. In addition to hair removal, IPL devices are employed in the treatment of pigmented and vascular lesions. Common adverse effects of both laser and IPL hair removal include transient erythema, perifollicular edema, and pigmentary changes, especially in patients with darker skin types. Rare complications include blistering, scarring, and paradoxical hair stimulation in which untreated areas develop increased hair growth.
Electrolysis is recognized as the only method of truly permanent hair removal and is effective for all hair colors.21 However, the variability in technique among practitioners often leads to inconsistent results, with some patients experiencing hair regrowth. Galvanic electrolysis involves inserting a fine needle into the hair follicle and applying an electrical current to destroy the it and the rapidly dividing cells of the matrix.22 The introduction of thermolytic electrolysis, which uses a high-frequency alternating current (commonly 13.56 MHz or 27.12 MHz), has enhanced efficiency by creating heat at the needle tip to destroy the follicle. This approach is faster and now is commonly combined with galvanic electrolysis.23 While no controlled clinical trials directly compare these methods, many patients experience permanent hair removal, with approximately 15% to 25% regrowth within 6 months.22,24
Alternative Options—Home-use laser and light-based devices have become increasingly popular for managing unwanted hair due to their affordability and convenience, with most devices priced less than $1000.25 These devices utilize various technologies, including lasers (808 nm), IPL, or combinations of IPL and radiofrequency.26 Despite their accessibility, peer-reviewed research on their safety profile and effectiveness is limited, as existing data primarily come from industry-funded, uncontrolled studies with short follow-up durations—making it difficult to assess long-term outcomes.25
Psychosocial Impact
A 2023 study of active-duty female service members with PCOS highlighted the unique challenges they face while managing symptoms such as facial hair within the constraints of military service.27 Although the study focused on PCOS, the findings shed light on how facial hair specifically impacts the psychological well-being of servicewomen. Participants described facial hair as one of the most visible and stigmatizing symptoms, often leading to feelings of embarrassment and diminished confidence. Participants also highlighted the professional implications of facial hair, with some describing feelings of scrutiny and judgment from peers and leadership in public. These challenges can be more pronounced in deployments or field exercises where hygiene resources are limited. The lack of access not only affects self-perception but also can hinder the ability of servicewomen to meet implicit expectations for grooming and appearance.27 There is a notable gap in research examining the impact of facial hair on military servicewomen. Given the unique environmental challenges and professional expectations, further investigation is warranted to better understand how facial hair affects women and to optimize treatment approaches in this population.
Final Thoughts
Limited awareness and understanding of facial hair in woman contribute to stigma, often leaving affected individuals to navigate challenges in isolation. Given the impact on confidence, professional appearance, and adherence to military grooming standards, it is essential for health care practitioners to recognize and address facial hair in women. Importantly, laser hair removal is covered by TRICARE for active-duty female service members with PFB, yet many remain unaware of this benefit. Increased awareness of available mechanical, pharmacologic, and procedural treatment options allows for tailored management, ensuring that women receive appropriate medical care.
Wendelin DS, Pope DN, Mallory SB. Hypertrichosis. J Am Acad Dermatol. 2003;48:161-181. doi:10.1067/mjd.2003.100
Blume-Peytavi U, Hahn S. Medical treatment of hirsutism. Dermatol Ther. 2008;21:329-339. doi:10.1111/j.1529-8019.2008.00215.x
Kang CN, Shah M, Lynde C, et al. Hair removal practices: a literature review. Skin Therapy Lett. 2021;26:6-11.
Matheson E, Bain J. Hirsutism in women. Am Fam Physician. 2019;100:168-175.
Shenenberger DW, Utecht LM. Removal of unwanted facial hair. Am Fam Physician. 2002;66:1907-1911.
Johnson E, Ebling FJ. The effect of plucking hairs during different phases of the follicular cycle. J Embryol Exp Morphol. 1964;12:465-474.
Martin KA, Anderson RR, Chang RJ, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2018;103:1233-1257. doi:10.1210/jc.2018-00241
Barrionuevo P, Nabhan M, Altayar O, et al. Treatment options for hirsutism: a systematic review and network meta-analysis. J Clin Endocrinol Metab. 2018;103:1258-1264. doi:10.1210/jc.2017-02052
Alesi S, Forslund M, Melin J, et al. Efficacy and safety of anti-androgens in the management of polycystic ovary syndrome: a systematic review and meta-analysis of randomised controlled trials. EClinicalMedicine. Published online August 9, 2023. doi:10.1016/j.eclinm.2023.102162
Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement. Hum Reprod Update. 2012;18:146-170.
Hussein RS, Abdelbasset WK. Updates on hirsutism: a narrative review. Int J Biomedicine. 2022;12:193-198. doi:10.21103/Article12(2)_RA4
Shapiro J, Lui H. Vaniqa—eflornithine 13.9% cream. Skin Therapy Lett. 2001;6:1-5.
Wolf JE Jr, Shander D, Huber F, et al. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair. Int J Dermatol. 2007;46:94-98. doi:10.1111/j.1365-4632.2006.03079.x
Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol. 2001;2:197-202. doi:10.2165/00128071-200102030-00009
Hamzavi I, Tan E, Shapiro J, et al. A randomized bilateral vehicle-controlled study of eflornithine cream combined with laser treatment versus laser treatment alone for facial hirsutism in women. J Am Acad Dermatol. 2007;57:54-59. doi:10.1016/j.jaad.2006.09.025
Goldberg DJ. Laser hair removal. In: Goldberg DJ, ed. Laser Dermatology: Pearls and Problems. Blackwell; 2008.
Hussain M, Polnikorn N, Goldberg DJ. Laser-assisted hair removal in Asian skin: efficacy, complications, and the effect of single versus multiple treatments. Dermatol Surg. 2003;29:249-254. doi:10.1046/j.1524-4725.2003.29059.x
Smith SR, Piacquadio DJ, Beger B, et al. Eflornithine cream combined with laser therapy in the management of unwanted facial hair growth in women: a randomized trial. Dermatol Surg. 2006;32:1237-1243. doi:10.1111/j.1524-4725.2006.32282.x
Jung I, Lannan FM, Weiss A, et al. Treatment and current policies on pseudofolliculitis barbae in the US military. Cutis. 2023;112:299-302. doi:10.12788/cutis.0907
TRICARE Operations Manual 6010.59-M. Supplemental Health Care Program (SHCP)—Chapter 17. Contractor Responsibilities. Military Health System and Defense Health Agency website. Revised November 5, 2021. Accessed February 13, 2024. https://manuals.health.mil/pages/DisplayManualHtmlFile/2022-08-31/AsOf/TO15/C17S3.html
Yanes DA, Smith P, Avram MM. A review of best practices for gender-affirming laser hair removal. Dermatol Surg. 2024;50:S201-S204. doi:10.1097/DSS.0000000000004441
Wagner RF Jr, Tomich JM, Grande DJ. Electrolysis and thermolysis for permanent hair removal. J Am Acad Dermatol. 1985;12:441-449. doi:10.1016/s0190-9622(85)70062-x
Olsen EA. Methods of hair removal. J Am Acad Dermatol. 1999;40:143-157. doi:10.1016/s0190-9622(99)70181-7
Kligman AM, Peters L. Histologic changes of human hair follicles after electrolysis: a comparison of two methods. Cutis. 1984;34:169-176.
Hession MT, Markova A, Graber EM. A review of hand-held, home-use cosmetic laser and light devices. Dermatol Surg. 2015;41:307-320. doi:10.1097/DSS.0000000000000283
Wheeland RG. Permanent hair reduction with a home-use diode laser: safety and effectiveness 1 year after eight treatments. Lasers Surg Med. 2012;44:550-557. doi:10.1002/lsm.22051
Hopkins D, Walker SC, Wilson C, et al. The experience of living with polycystic ovary syndrome in the military. Mil Med. 2024;189:E188-E197. doi:10.1093/milmed/usad241
Wendelin DS, Pope DN, Mallory SB. Hypertrichosis. J Am Acad Dermatol. 2003;48:161-181. doi:10.1067/mjd.2003.100
Blume-Peytavi U, Hahn S. Medical treatment of hirsutism. Dermatol Ther. 2008;21:329-339. doi:10.1111/j.1529-8019.2008.00215.x
Kang CN, Shah M, Lynde C, et al. Hair removal practices: a literature review. Skin Therapy Lett. 2021;26:6-11.
Matheson E, Bain J. Hirsutism in women. Am Fam Physician. 2019;100:168-175.
Shenenberger DW, Utecht LM. Removal of unwanted facial hair. Am Fam Physician. 2002;66:1907-1911.
Johnson E, Ebling FJ. The effect of plucking hairs during different phases of the follicular cycle. J Embryol Exp Morphol. 1964;12:465-474.
Martin KA, Anderson RR, Chang RJ, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2018;103:1233-1257. doi:10.1210/jc.2018-00241
Barrionuevo P, Nabhan M, Altayar O, et al. Treatment options for hirsutism: a systematic review and network meta-analysis. J Clin Endocrinol Metab. 2018;103:1258-1264. doi:10.1210/jc.2017-02052
Alesi S, Forslund M, Melin J, et al. Efficacy and safety of anti-androgens in the management of polycystic ovary syndrome: a systematic review and meta-analysis of randomised controlled trials. EClinicalMedicine. Published online August 9, 2023. doi:10.1016/j.eclinm.2023.102162
Escobar-Morreale HF, Carmina E, Dewailly D, et al. Epidemiology, diagnosis and management of hirsutism: a consensus statement. Hum Reprod Update. 2012;18:146-170.
Hussein RS, Abdelbasset WK. Updates on hirsutism: a narrative review. Int J Biomedicine. 2022;12:193-198. doi:10.21103/Article12(2)_RA4
Shapiro J, Lui H. Vaniqa—eflornithine 13.9% cream. Skin Therapy Lett. 2001;6:1-5.
Wolf JE Jr, Shander D, Huber F, et al. Randomized, double-blind clinical evaluation of the efficacy and safety of topical eflornithine HCl 13.9% cream in the treatment of women with facial hair. Int J Dermatol. 2007;46:94-98. doi:10.1111/j.1365-4632.2006.03079.x
Balfour JA, McClellan K. Topical eflornithine. Am J Clin Dermatol. 2001;2:197-202. doi:10.2165/00128071-200102030-00009
Hamzavi I, Tan E, Shapiro J, et al. A randomized bilateral vehicle-controlled study of eflornithine cream combined with laser treatment versus laser treatment alone for facial hirsutism in women. J Am Acad Dermatol. 2007;57:54-59. doi:10.1016/j.jaad.2006.09.025
Goldberg DJ. Laser hair removal. In: Goldberg DJ, ed. Laser Dermatology: Pearls and Problems. Blackwell; 2008.
Hussain M, Polnikorn N, Goldberg DJ. Laser-assisted hair removal in Asian skin: efficacy, complications, and the effect of single versus multiple treatments. Dermatol Surg. 2003;29:249-254. doi:10.1046/j.1524-4725.2003.29059.x
Smith SR, Piacquadio DJ, Beger B, et al. Eflornithine cream combined with laser therapy in the management of unwanted facial hair growth in women: a randomized trial. Dermatol Surg. 2006;32:1237-1243. doi:10.1111/j.1524-4725.2006.32282.x
Jung I, Lannan FM, Weiss A, et al. Treatment and current policies on pseudofolliculitis barbae in the US military. Cutis. 2023;112:299-302. doi:10.12788/cutis.0907
TRICARE Operations Manual 6010.59-M. Supplemental Health Care Program (SHCP)—Chapter 17. Contractor Responsibilities. Military Health System and Defense Health Agency website. Revised November 5, 2021. Accessed February 13, 2024. https://manuals.health.mil/pages/DisplayManualHtmlFile/2022-08-31/AsOf/TO15/C17S3.html
Yanes DA, Smith P, Avram MM. A review of best practices for gender-affirming laser hair removal. Dermatol Surg. 2024;50:S201-S204. doi:10.1097/DSS.0000000000004441
Wagner RF Jr, Tomich JM, Grande DJ. Electrolysis and thermolysis for permanent hair removal. J Am Acad Dermatol. 1985;12:441-449. doi:10.1016/s0190-9622(85)70062-x
Olsen EA. Methods of hair removal. J Am Acad Dermatol. 1999;40:143-157. doi:10.1016/s0190-9622(99)70181-7
Kligman AM, Peters L. Histologic changes of human hair follicles after electrolysis: a comparison of two methods. Cutis. 1984;34:169-176.
Hession MT, Markova A, Graber EM. A review of hand-held, home-use cosmetic laser and light devices. Dermatol Surg. 2015;41:307-320. doi:10.1097/DSS.0000000000000283
Wheeland RG. Permanent hair reduction with a home-use diode laser: safety and effectiveness 1 year after eight treatments. Lasers Surg Med. 2012;44:550-557. doi:10.1002/lsm.22051
Hopkins D, Walker SC, Wilson C, et al. The experience of living with polycystic ovary syndrome in the military. Mil Med. 2024;189:E188-E197. doi:10.1093/milmed/usad241

Millipede Burns: An Unusual Cause of Purplish Toes
To the Editor:
Millipedes do not have nearly as many feet as their name would suggest; most have fewer than 100.1 They are not actually insects; they are a wormlike arthropod in the Diplopoda class. Generally these harmless animals can be a welcome resident in gardens because they break down decaying plant material and rejuvenate the soil.1 However, they are less welcome in the home or underfoot because of what happens when these invertebrates are threatened or crushed.2
Millipedes, which typically have at least 30 pairs of legs, have 2 defense mechanisms: (1) body coiling to withstand external pressure, and (2) secretion of fluids with insecticidal properties from specialized glands distributed along their body.3 These secretions, which are used by the millipede to defend against predators, contain organic compounds including benzoquinone. When these secretions come into contact with skin, pigmentary changes resembling a burn or necrosis and irritation to the skin (pain, burning, itching) occur.4,5
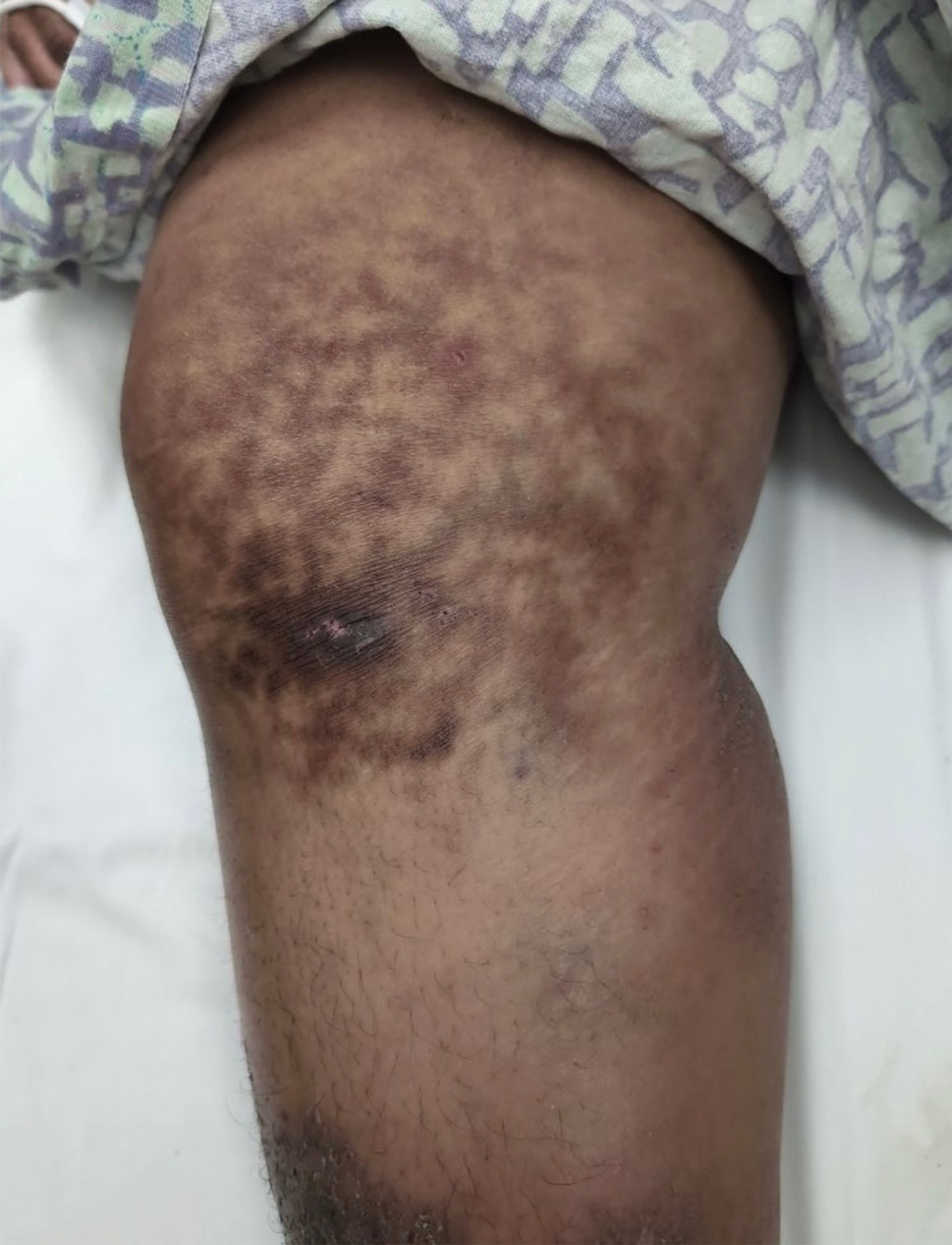
Millipedes typically are found in tropical and temperate regions worldwide, such as the Amazon rainforest, Southeast Asia, tropical areas of Africa, forests, grasslands, and gardens in North America and Europe.6 They also are found in every US state as well as Puerto Rico.1 Millipedes are nocturnal, favor dark places, and can make their way into residential areas, including homes, basements, gardens, and yards.2,6 Although millipede burns commonly are reported in tropical regions, we present a case in China.6A 33-year-old woman presented with purplish-red discoloration on all 5 toes on the left foot. The patient recounted that she discovered a millipede in her shoe earlier in the day, removed it, and crushed it with her bare foot. That night, while taking a bath, she noticed that the toes had turned purplish-red (Figure 1). The patient brought the crushed millipede with her to the emergency department where she sought treatment. The dermatologist confirmed that it was a millipede; however, the team was unable to determine the specific species because it had been crushed (Figure 2).
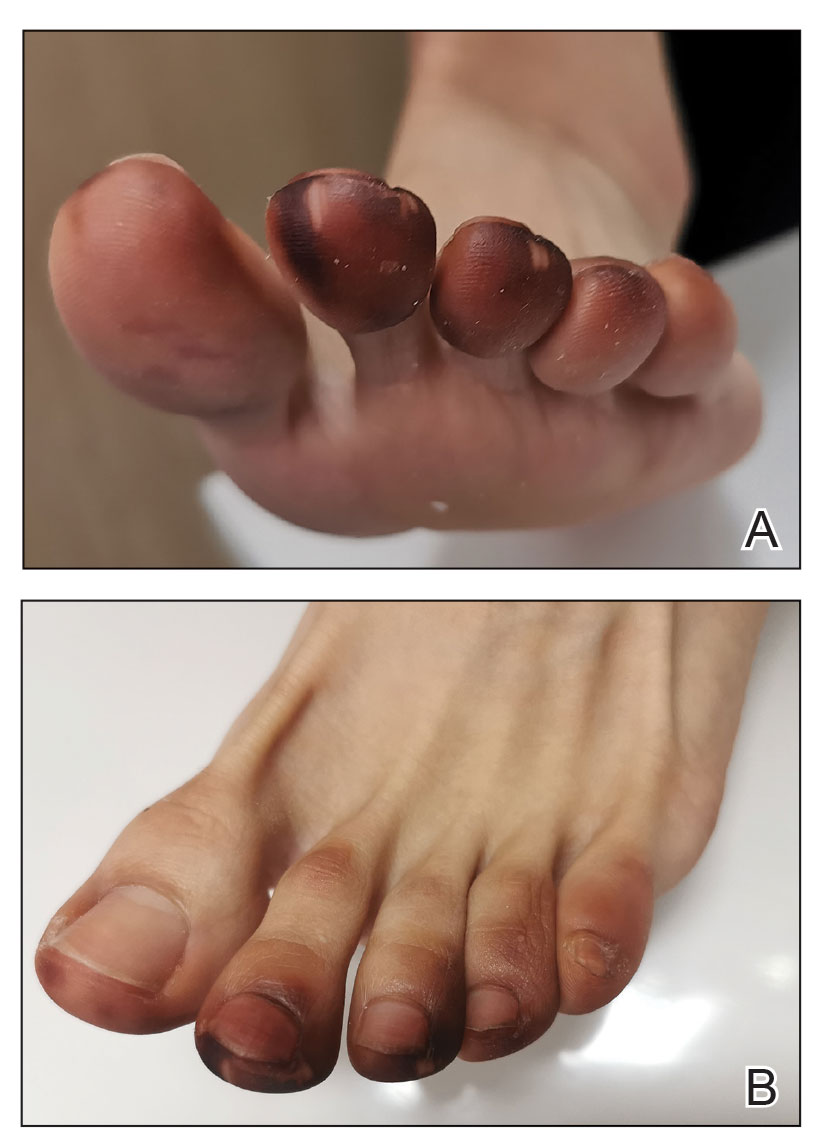

Physical examination of the affected toes showed a clear boundary and iodinelike staining. The patient did not report pain. The stained skin had a normal temperature, pulse, texture, and sensation. Dermoscopy revealed multiple black-brown patches on the toes (Figure 3). The pigmented area gradually faded over a 1-month period. Superficial damage to the toenail revealed evidence of black-brown pigmentation on both the nail and the skin underneath. The diagnosis in the dermoscopy report suggested exogenous pigmentation of the toes. The patient was advised that no treatment was needed and that the condition would resolve on its own. At 1-month follow-up, the patient’s toes had returned to their normal color (Figure 4).
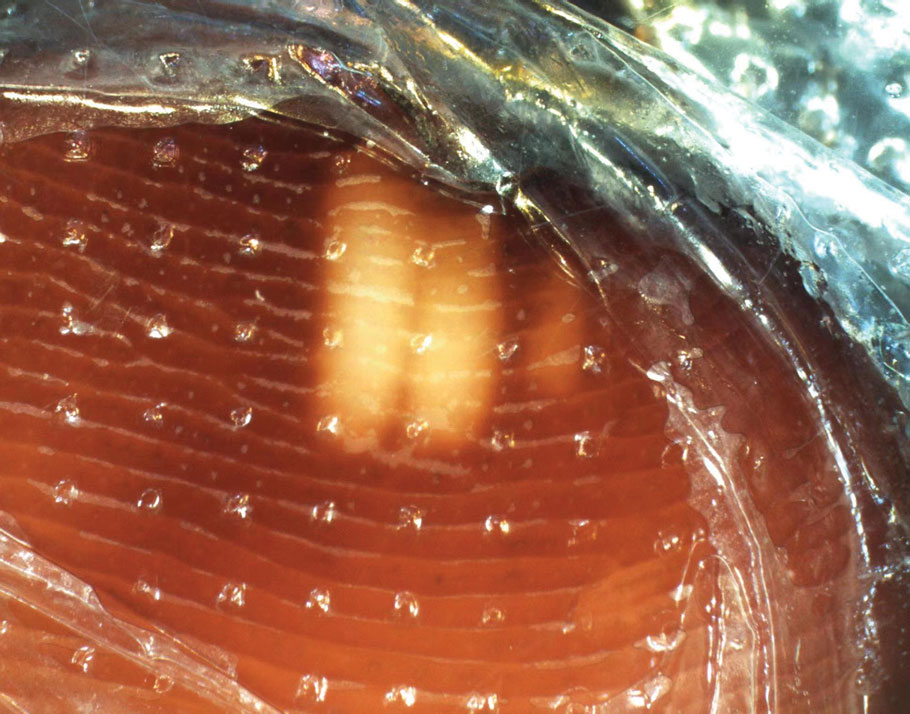
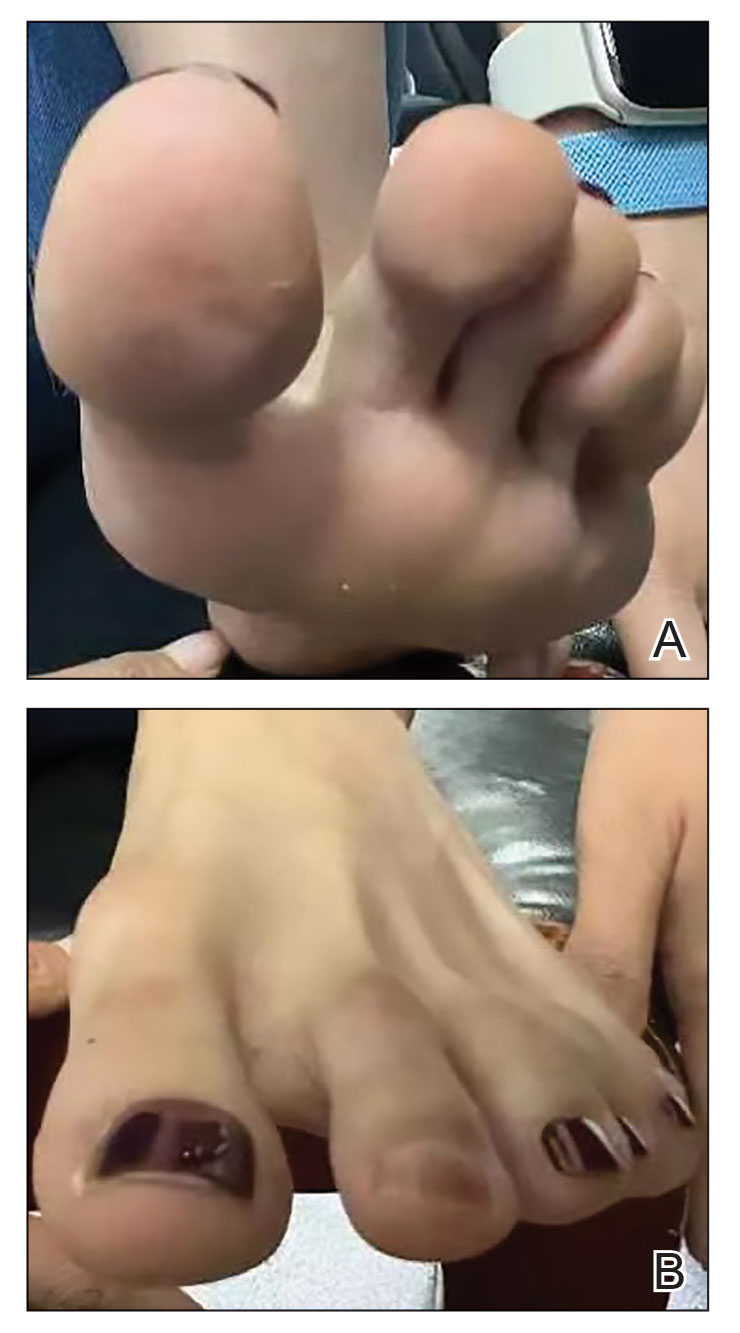
The feet are common sites of millipede burns; other exposed areas, such as the arms, face, and eyes, also are potential sites of involvement.5 The cutaneous pigmentary changes seen on our patient’s foot were a result of the millipede’s defense mechanism—secreted toxic chemicals that stained the foot. It is important to note that the pigmentation was not associated with the death of the millipede, as the millipede was still alive upon initial contact with the patient’s foot in her shoe.
When a patient presents with pigmentary changes, several conditions must be ruled out—notably acute arterial thrombosis. Patients with this condition will describe acute pain and weakness in the area of involvement. Physicians inspecting the area will note coldness and pallor in the affected limb as well as a diminished or absent pulse. In severe cases, the skin may exhibit a purplish-red appearance.5 Millipede burns also should be distinguished from bacterial endocarditis and cryoglobulinemia.7 All 3 conditions can manifest with redness, swelling, blisters, and purpuralike changes. Positive blood culture is an important diagnostic basis for bacterial endocarditis; in addition, routine blood tests will demonstrate a decrease in red blood cells and hemoglobin, and routine urinalysis may show proteinuria and microscopic hematuria. Patients with cryoglobulinemia will have a positive cryoglobulin assay, increased IgM, and often decreased complement.7 It also is worth noting that millipede burns might resemble child abuse in pediatric patients, necessitating further evaluation.5
It is unusual to see a millipede burn in nontropical regions. Therefore, the identification of our patient’s millipede burn was notable and serves as a reminder to keep this diagnosis in the differential when caring for patients with pigmentary changes. An accurate diagnosis hinges on being alert to a millipede exposure history and recognizing the clinical manifestations. For affected patients, it may be beneficial to recommend they advise friends and relatives to avoid skin contact with millipedes and most importantly to avoid stepping on them with bare feet.
Millipedes. National Wildlife Federation. Accessed October 15, 2025. https://www.nwf.org/Educational-Resources/Wildlife-Guide/Invertebrates/Millipedes
Pennini SN, Rebello PFB, Guerra MdGVB, et al. Millipede accident with unusual dermatological lesion. An Bras Dermatol. 2019;94:765-767. doi:10.1016/j.abd.2019.10.003
Lima CAJ, Cardoso JLC, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda Class (“millipedes“). An Bras Dermatol. 2010;85:391-392. doi:10.1590/s0365-05962910000300018
De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190. doi:10.3109/15563650.2011.560855
Lacy FA, Elston DM. What’s eating you? millipede burns. Cutis. 2019;103:195-196.
Neto ASH, Filho FB, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258. doi:10.1590/0037-8682-0212-2013
Sampaio FMS, Valviesse VRGdA, Lyra-da-Silva JO, et al. Pain and hyperpigmentation of the toes: a quiz. hyperpigmentation of the toes caused by millipedes. Acta Derm Venereol. 2014;94:253-254. doi:10.2340/00015555-1645
To the Editor:
Millipedes do not have nearly as many feet as their name would suggest; most have fewer than 100.1 They are not actually insects; they are a wormlike arthropod in the Diplopoda class. Generally these harmless animals can be a welcome resident in gardens because they break down decaying plant material and rejuvenate the soil.1 However, they are less welcome in the home or underfoot because of what happens when these invertebrates are threatened or crushed.2
Millipedes, which typically have at least 30 pairs of legs, have 2 defense mechanisms: (1) body coiling to withstand external pressure, and (2) secretion of fluids with insecticidal properties from specialized glands distributed along their body.3 These secretions, which are used by the millipede to defend against predators, contain organic compounds including benzoquinone. When these secretions come into contact with skin, pigmentary changes resembling a burn or necrosis and irritation to the skin (pain, burning, itching) occur.4,5
Millipedes typically are found in tropical and temperate regions worldwide, such as the Amazon rainforest, Southeast Asia, tropical areas of Africa, forests, grasslands, and gardens in North America and Europe.6 They also are found in every US state as well as Puerto Rico.1 Millipedes are nocturnal, favor dark places, and can make their way into residential areas, including homes, basements, gardens, and yards.2,6 Although millipede burns commonly are reported in tropical regions, we present a case in China.6A 33-year-old woman presented with purplish-red discoloration on all 5 toes on the left foot. The patient recounted that she discovered a millipede in her shoe earlier in the day, removed it, and crushed it with her bare foot. That night, while taking a bath, she noticed that the toes had turned purplish-red (Figure 1). The patient brought the crushed millipede with her to the emergency department where she sought treatment. The dermatologist confirmed that it was a millipede; however, the team was unable to determine the specific species because it had been crushed (Figure 2).
Physical examination of the affected toes showed a clear boundary and iodinelike staining. The patient did not report pain. The stained skin had a normal temperature, pulse, texture, and sensation. Dermoscopy revealed multiple black-brown patches on the toes (Figure 3). The pigmented area gradually faded over a 1-month period. Superficial damage to the toenail revealed evidence of black-brown pigmentation on both the nail and the skin underneath. The diagnosis in the dermoscopy report suggested exogenous pigmentation of the toes. The patient was advised that no treatment was needed and that the condition would resolve on its own. At 1-month follow-up, the patient’s toes had returned to their normal color (Figure 4).
The feet are common sites of millipede burns; other exposed areas, such as the arms, face, and eyes, also are potential sites of involvement.5 The cutaneous pigmentary changes seen on our patient’s foot were a result of the millipede’s defense mechanism—secreted toxic chemicals that stained the foot. It is important to note that the pigmentation was not associated with the death of the millipede, as the millipede was still alive upon initial contact with the patient’s foot in her shoe.
When a patient presents with pigmentary changes, several conditions must be ruled out—notably acute arterial thrombosis. Patients with this condition will describe acute pain and weakness in the area of involvement. Physicians inspecting the area will note coldness and pallor in the affected limb as well as a diminished or absent pulse. In severe cases, the skin may exhibit a purplish-red appearance.5 Millipede burns also should be distinguished from bacterial endocarditis and cryoglobulinemia.7 All 3 conditions can manifest with redness, swelling, blisters, and purpuralike changes. Positive blood culture is an important diagnostic basis for bacterial endocarditis; in addition, routine blood tests will demonstrate a decrease in red blood cells and hemoglobin, and routine urinalysis may show proteinuria and microscopic hematuria. Patients with cryoglobulinemia will have a positive cryoglobulin assay, increased IgM, and often decreased complement.7 It also is worth noting that millipede burns might resemble child abuse in pediatric patients, necessitating further evaluation.5
It is unusual to see a millipede burn in nontropical regions. Therefore, the identification of our patient’s millipede burn was notable and serves as a reminder to keep this diagnosis in the differential when caring for patients with pigmentary changes. An accurate diagnosis hinges on being alert to a millipede exposure history and recognizing the clinical manifestations. For affected patients, it may be beneficial to recommend they advise friends and relatives to avoid skin contact with millipedes and most importantly to avoid stepping on them with bare feet.
To the Editor:
Millipedes do not have nearly as many feet as their name would suggest; most have fewer than 100.1 They are not actually insects; they are a wormlike arthropod in the Diplopoda class. Generally these harmless animals can be a welcome resident in gardens because they break down decaying plant material and rejuvenate the soil.1 However, they are less welcome in the home or underfoot because of what happens when these invertebrates are threatened or crushed.2
Millipedes, which typically have at least 30 pairs of legs, have 2 defense mechanisms: (1) body coiling to withstand external pressure, and (2) secretion of fluids with insecticidal properties from specialized glands distributed along their body.3 These secretions, which are used by the millipede to defend against predators, contain organic compounds including benzoquinone. When these secretions come into contact with skin, pigmentary changes resembling a burn or necrosis and irritation to the skin (pain, burning, itching) occur.4,5
Millipedes typically are found in tropical and temperate regions worldwide, such as the Amazon rainforest, Southeast Asia, tropical areas of Africa, forests, grasslands, and gardens in North America and Europe.6 They also are found in every US state as well as Puerto Rico.1 Millipedes are nocturnal, favor dark places, and can make their way into residential areas, including homes, basements, gardens, and yards.2,6 Although millipede burns commonly are reported in tropical regions, we present a case in China.6A 33-year-old woman presented with purplish-red discoloration on all 5 toes on the left foot. The patient recounted that she discovered a millipede in her shoe earlier in the day, removed it, and crushed it with her bare foot. That night, while taking a bath, she noticed that the toes had turned purplish-red (Figure 1). The patient brought the crushed millipede with her to the emergency department where she sought treatment. The dermatologist confirmed that it was a millipede; however, the team was unable to determine the specific species because it had been crushed (Figure 2).
Physical examination of the affected toes showed a clear boundary and iodinelike staining. The patient did not report pain. The stained skin had a normal temperature, pulse, texture, and sensation. Dermoscopy revealed multiple black-brown patches on the toes (Figure 3). The pigmented area gradually faded over a 1-month period. Superficial damage to the toenail revealed evidence of black-brown pigmentation on both the nail and the skin underneath. The diagnosis in the dermoscopy report suggested exogenous pigmentation of the toes. The patient was advised that no treatment was needed and that the condition would resolve on its own. At 1-month follow-up, the patient’s toes had returned to their normal color (Figure 4).
The feet are common sites of millipede burns; other exposed areas, such as the arms, face, and eyes, also are potential sites of involvement.5 The cutaneous pigmentary changes seen on our patient’s foot were a result of the millipede’s defense mechanism—secreted toxic chemicals that stained the foot. It is important to note that the pigmentation was not associated with the death of the millipede, as the millipede was still alive upon initial contact with the patient’s foot in her shoe.
When a patient presents with pigmentary changes, several conditions must be ruled out—notably acute arterial thrombosis. Patients with this condition will describe acute pain and weakness in the area of involvement. Physicians inspecting the area will note coldness and pallor in the affected limb as well as a diminished or absent pulse. In severe cases, the skin may exhibit a purplish-red appearance.5 Millipede burns also should be distinguished from bacterial endocarditis and cryoglobulinemia.7 All 3 conditions can manifest with redness, swelling, blisters, and purpuralike changes. Positive blood culture is an important diagnostic basis for bacterial endocarditis; in addition, routine blood tests will demonstrate a decrease in red blood cells and hemoglobin, and routine urinalysis may show proteinuria and microscopic hematuria. Patients with cryoglobulinemia will have a positive cryoglobulin assay, increased IgM, and often decreased complement.7 It also is worth noting that millipede burns might resemble child abuse in pediatric patients, necessitating further evaluation.5
It is unusual to see a millipede burn in nontropical regions. Therefore, the identification of our patient’s millipede burn was notable and serves as a reminder to keep this diagnosis in the differential when caring for patients with pigmentary changes. An accurate diagnosis hinges on being alert to a millipede exposure history and recognizing the clinical manifestations. For affected patients, it may be beneficial to recommend they advise friends and relatives to avoid skin contact with millipedes and most importantly to avoid stepping on them with bare feet.
Millipedes. National Wildlife Federation. Accessed October 15, 2025. https://www.nwf.org/Educational-Resources/Wildlife-Guide/Invertebrates/Millipedes
Pennini SN, Rebello PFB, Guerra MdGVB, et al. Millipede accident with unusual dermatological lesion. An Bras Dermatol. 2019;94:765-767. doi:10.1016/j.abd.2019.10.003
Lima CAJ, Cardoso JLC, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda Class (“millipedes“). An Bras Dermatol. 2010;85:391-392. doi:10.1590/s0365-05962910000300018
De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190. doi:10.3109/15563650.2011.560855
Lacy FA, Elston DM. What’s eating you? millipede burns. Cutis. 2019;103:195-196.
Neto ASH, Filho FB, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258. doi:10.1590/0037-8682-0212-2013
Sampaio FMS, Valviesse VRGdA, Lyra-da-Silva JO, et al. Pain and hyperpigmentation of the toes: a quiz. hyperpigmentation of the toes caused by millipedes. Acta Derm Venereol. 2014;94:253-254. doi:10.2340/00015555-1645
Millipedes. National Wildlife Federation. Accessed October 15, 2025. https://www.nwf.org/Educational-Resources/Wildlife-Guide/Invertebrates/Millipedes
Pennini SN, Rebello PFB, Guerra MdGVB, et al. Millipede accident with unusual dermatological lesion. An Bras Dermatol. 2019;94:765-767. doi:10.1016/j.abd.2019.10.003
Lima CAJ, Cardoso JLC, Magela A, et al. Exogenous pigmentation in toes feigning ischemia of the extremities: a diagnostic challenge brought by arthropods of the Diplopoda Class (“millipedes“). An Bras Dermatol. 2010;85:391-392. doi:10.1590/s0365-05962910000300018
De Capitani EM, Vieira RJ, Bucaretchi F, et al. Human accidents involving Rhinocricus spp., a common millipede genus observed in urban areas of Brazil. Clin Toxicol (Phila). 2011;49:187-190. doi:10.3109/15563650.2011.560855
Lacy FA, Elston DM. What’s eating you? millipede burns. Cutis. 2019;103:195-196.
Neto ASH, Filho FB, Martins G. Skin lesions simulating blue toe syndrome caused by prolonged contact with a millipede. Rev Soc Bras Med Trop. 2014;47:257-258. doi:10.1590/0037-8682-0212-2013
Sampaio FMS, Valviesse VRGdA, Lyra-da-Silva JO, et al. Pain and hyperpigmentation of the toes: a quiz. hyperpigmentation of the toes caused by millipedes. Acta Derm Venereol. 2014;94:253-254. doi:10.2340/00015555-1645
PRACTICE POINTS
- Millipede burns can resemble ischemia. The most common site of a millipede burn is the feet.
- Diagnosing a millipede burn hinges on obtaining a detailed history, viewing the site under a dermatoscope, and carefully assessing the temperature and pulse of the affected area.

Growing Nodule on the Parietal Scalp
Growing Nodule on the Parietal Scalp
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Biopsy revealed a squamous epithelium with cystic changes, trichilemmal differentiation, squamous eddy formation, keratinocyte atypia, focal necrotic changes, and a focus of atypical keratinocytes invading the dermis (Figure 1). Based on these findings, a diagnosis of malignant proliferating trichilemmal tumor (MPTT) was made.

Malignant proliferating trichilemmal tumor is a rare adnexal tumor that develops from the outer root sheath of the hair follicle. It often arises due to malignant transformation of pre-existing trichilemmal cysts, but some cases occur de novo.1 Malignant transformation is thought to start from a trichilemmal cyst in an adenomatous histologic stage, progressing to a proliferating trichilemmal cyst (PTC) in an epitheliomatous phase, ultimately becoming carcinomatous with MPTT.2-4 This transformation has been categorized into 3 morphologic groups to predict tumor behavior, including benign PTCs (curable by excision), low-grade malignant PTCs (minor risk for local recurrence), and high-grade malignant PTCs (risk for regional spread and metastasis with cytologic atypical features and potential for aggressive growth).1
More commonly observed in women in the fourth to eighth decades of life, MPTT may manifest as a fast- growing, painless, solitary nodule or as a progressively enlarging nodule at the site of a previously stable, long-standing lesion. Malignant proliferating trichilemmal tumor manifests frequently on the scalp, face, or neck, but there are reports of MPTT manifesting on the trunk and even as multiple concurrent lesions.1-4 The variability in clinical presentation and the potential to be mistaken for benign conditions makes excisional biopsy essential for diagnosis of MPTT. Histopathology classically demonstrates trichilemmal keratinization, a high mitotic index, and cellular atypia with invasion into the dermis.4 Malignant transformation frequently follows a prior history of trauma to the area or local inflammation.
Given the locally aggressive nature of MPTT, our patient was referred to a Mohs micrographic surgeon. While both wide excision with tumor-free margins and Mohs micrographic surgery are accepted surgical procedures for MPTT, there is no consensus in the literature on a standard treatment recommendation. Following surgery, close monitoring is needed for potential recurrence and metastases intracranially to the dura and muscles,5 as well as to the lungs.6 Further imaging using computed tomography or positron emission tomography can be ordered to rule out metastatic disease.4
Pilomatrixomas are benign neoplasms that arise from hair matrix cells and have been associated with catenin beta-1 gene mutations, as well as genetic syndromes and trauma.7 Clinically, pilomatrixomas manifest as solitary, firm, painless, slow-growing nodules that commonly are found in the head and neck region. This tumor has a slight predominance in women and occurs frequently in adolescent years. The overlying skin may appear normal or show grey-bluish discoloration.8 Histopathology shows basaloid cells resembling primitive hair matrix cells with an abrupt transition to shadow cells composed of transformed keratinocytes without nuclei and calcification.7-8 This tumor can be differentiated by the presence of basaloid and shadow cells with calcification on histopathology, while MPTT will show atypical, mitotically active squamous cells with trichilemmal keratinization (Figure 2).

Proliferating trichilemmal cyst is a variant of trichilemmal cyst (TC) arising from the outer root sheath cells of the hair follicle. While TCs usually are slow growing and benign, the proliferating variant can be more aggressive with malignant potential. Patients often present with a solitary, well-circumscribed, rapidly growing nodule on the scalp. The lesion may be painful, and ulceration can occur, exposing the cystic contents. Histopathologically, PTCs resemble TCs with trichilemmal keratinization but also exhibit notable epithelial proliferation within the cystic space.9 While there can be considerable histopathologic overlap between PTC and MPTT—including extensive trichilemmal keratinization, variable atypia, and mitotic activity—PTC typically should not demonstrate invasion into the surrounding soft tissue or the degree of high-grade atypia, brisk mitoses, or necrosis seen in MPTT (eFigure 1).1 Immunohistochemistry may help distinguish PTC from MPTT and squamous cell carcinoma (SCC).10-11 The pattern of Ki-67 and p53 expression may be helpful with classification of PTC/MPTT into the 3 groups (benign, low-grade malignant, and high-grade malignant) proposed by Ye et al.1 Other investigators have suggested that Ki-67 expression may correlate potential for recurrence and clinical prognosis.12 Expression of CD34 (a marker that supports outer root sheath origin) might favor PTC/MPTT over SCC; however, cases of CD34- negative MPTT have been reported, particularly those with poorly differentiated histopathology.
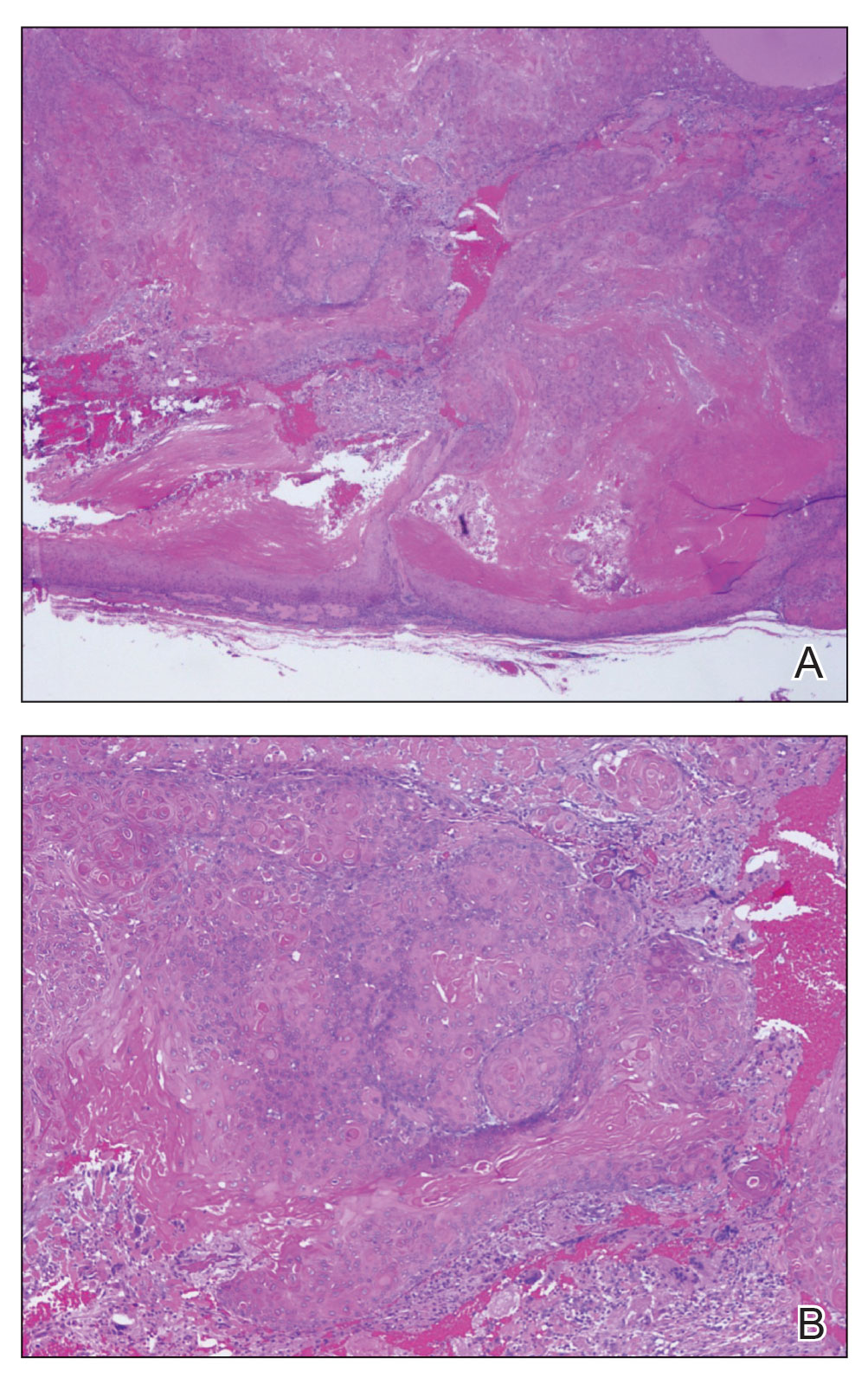
Squamous cell carcinoma with cystic features is a histologic variant of SCC characterized by cystlike spaces containing malignant squamous epithelial cells.13 Squamous cell carcinoma with cystic features can manifest as a firm nodule with ulceration similar to MPTT or PTC but also can mimic a benign cyst.14 The diagnosis of invasive SCC with cystic features typically is straightforward and characterized by cords and nests of atypical keratinocytes extending into the dermis with areas of cystic architecture (eFigure 2). While both SCC with cystic features and MPTT may show cystic histopathologic architecture, MPTT typically shows areas of PTC, whereas SCC with cystic features lacks such areas.
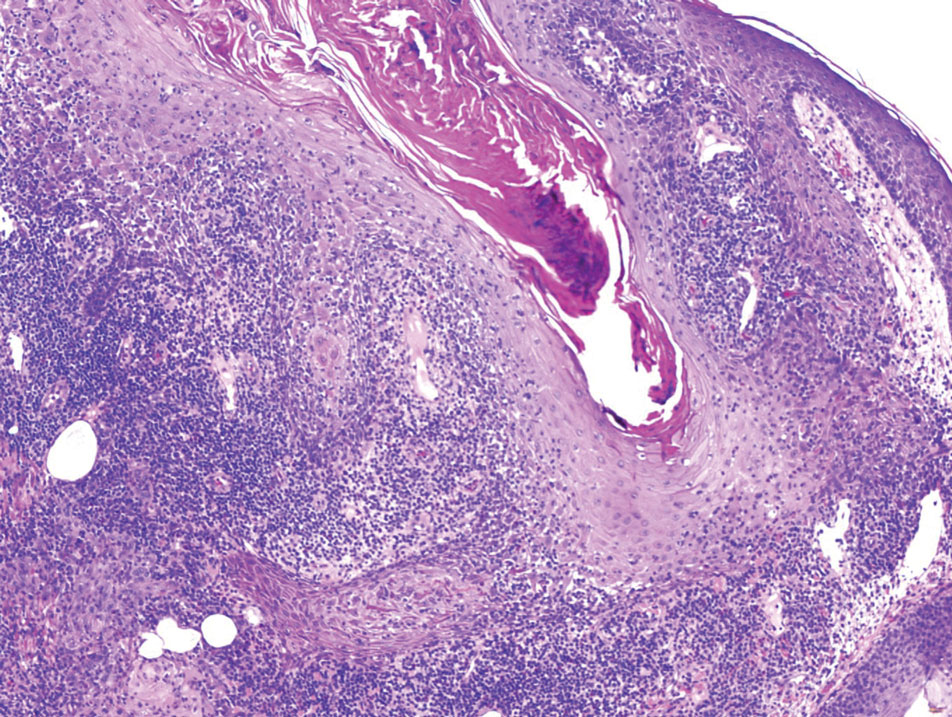
Verrucous cysts refer to infundibular cysts or less commonly pilar cysts or hybrid pilar-epidermoid cysts that exhibit superimposed human papillomavirus (HPV) cytopathic changes. Clinically, a verrucous cyst manifests as a single, asymptomatic, slow-growing, firm lesion most commonly manifesting on the face and back. Histopathologically, the cyst wall may show acanthosis, papillomatosis, hypergranulosis with coarse keratohyalin granules, and koilocytic changes (eFigure 3). These histopathologic features are believed to be induced by secondary HPV infection. While HPV-related change, characterized by koilocytic alteration, papillomatosis, and verruciform hyperplasia, more commonly affects epidermal cysts, occasionally trichilemmal (pilar) cysts are involved. In these cases, verrucous cysts should be distinguished from MPTT. Verrucous cysts may contain rare normal mitotic figures, but do not contain atypical mitosis, marked cellular pleomorphism, or an infiltrating pattern similar to MPTT.15
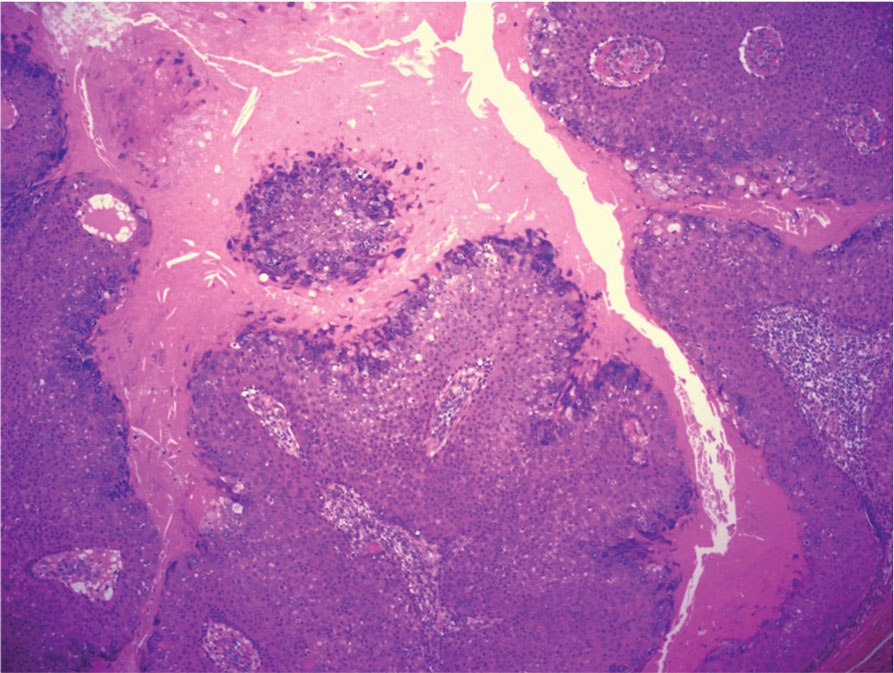
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumors: a clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574. doi:10.1309/0XLEGFQ64XYJU4G6
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208. doi:10.1159/000249868
- Rao S, Ramakrishnan R, Kamakshi D, et al. Malignant proliferating trichilemmal tumour presenting early in life: an uncommon feature. J Cutan Aesthet Surg. 2011;4:51-55. doi:10.4103/0974-2077.79196
- Kearns-Turcotte S, Thériault M, Blouin MM. Malignant proliferating trichilemmal tumors arising in patients with multiple trichilemmal cysts: a case series. JAAD Case Rep. 2022;22:42-46. doi:10.1016
- Karamese M, Akatekin A, Abaci M, et al. Unusual invasion of trichilemmal tumors: two case reports. Modern Plastic Surg. 2012; 2:54-57. doi:10.4236/MPS.2012.23014 /j.jdcr.2022.01.033
- Lobo L, Amonkar AD, Dontamsetty VV. Malignant proliferating trichilemmal tumour of the scalp with intra-cranial extension and lung metastasis-a case report. Indian J Surg. 2016;78:493-495. doi:10.1007/s12262-015-1427-0
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018;40:631-641. doi:10.1097/DAD.0000000000001118
- Sharma D, Agarwal S, Jain LS, et al. Pilomatrixoma masquerading as metastatic adenocarcinoma. A diagnostic pitfall on cytology. J Clin Diagn Res. 2014;8:FD13-FD14. doi:10.7860/JCDR/2014/9696.5064
- Valerio E, Parro FHS, Macedo MP, et al. Proliferating trichilemmal cyst with clinical, radiological, macroscopic, and microscopic orrelation. An Bras Dermatol. 2019;94:452-454. doi:10.1590 /abd1806-4841.20198199
- Joshi TP, Marchand S, Tschen J. Malignant proliferating trichilemmal tumor: a subtle presentation in an African American woman and review of immunohistochemical markers for this rare condition. Cureus. 2021;13:E17289. doi:10.7759/cureus.17289
- Gulati HK, Deshmukh SD, Anand M, et al. Low-grade malignant proliferating pilar tumor simulating a squamous-cell carcinoma in an elderly female: a case report and immunohistochemical study. Int J Trichology. 2011;3:98-101. doi:10.4103/0974-7753.90818
- Rangel-Gamboa L, Reyes-Castro M, Dominguez-Cherit J, et al. Proliferating trichilemmal cyst: the value of ki67 immunostaining. Int J Trichology. 2013;5:115-117. doi:10.4103/0974-7753.125599
- Asad U, Alkul S, Shimizu I, et al. Squamous cell carcinoma with unusual benign-appearing cystic features on histology. Cureus. 2023;15:E33610. doi:10.7759/cureus.33610
- Alkul S, Nguyen CN, Ramani NS, et al. Squamous cell carcinoma arising in an epidermal inclusion cyst. Baylor Univ Med Cent Proc. 2022;35:688-690. doi:10.1080/08998280.2022.207760
- Nanes BA, Laknezhad S, Chamseddin B, et al. Verrucous pilar cysts infected with beta human papillomavirus. J Cutan Pathol. 2020;47:381-386. doi:10.1111/cup.13599
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Biopsy revealed a squamous epithelium with cystic changes, trichilemmal differentiation, squamous eddy formation, keratinocyte atypia, focal necrotic changes, and a focus of atypical keratinocytes invading the dermis (Figure 1). Based on these findings, a diagnosis of malignant proliferating trichilemmal tumor (MPTT) was made.
Malignant proliferating trichilemmal tumor is a rare adnexal tumor that develops from the outer root sheath of the hair follicle. It often arises due to malignant transformation of pre-existing trichilemmal cysts, but some cases occur de novo.1 Malignant transformation is thought to start from a trichilemmal cyst in an adenomatous histologic stage, progressing to a proliferating trichilemmal cyst (PTC) in an epitheliomatous phase, ultimately becoming carcinomatous with MPTT.2-4 This transformation has been categorized into 3 morphologic groups to predict tumor behavior, including benign PTCs (curable by excision), low-grade malignant PTCs (minor risk for local recurrence), and high-grade malignant PTCs (risk for regional spread and metastasis with cytologic atypical features and potential for aggressive growth).1
More commonly observed in women in the fourth to eighth decades of life, MPTT may manifest as a fast- growing, painless, solitary nodule or as a progressively enlarging nodule at the site of a previously stable, long-standing lesion. Malignant proliferating trichilemmal tumor manifests frequently on the scalp, face, or neck, but there are reports of MPTT manifesting on the trunk and even as multiple concurrent lesions.1-4 The variability in clinical presentation and the potential to be mistaken for benign conditions makes excisional biopsy essential for diagnosis of MPTT. Histopathology classically demonstrates trichilemmal keratinization, a high mitotic index, and cellular atypia with invasion into the dermis.4 Malignant transformation frequently follows a prior history of trauma to the area or local inflammation.
Given the locally aggressive nature of MPTT, our patient was referred to a Mohs micrographic surgeon. While both wide excision with tumor-free margins and Mohs micrographic surgery are accepted surgical procedures for MPTT, there is no consensus in the literature on a standard treatment recommendation. Following surgery, close monitoring is needed for potential recurrence and metastases intracranially to the dura and muscles,5 as well as to the lungs.6 Further imaging using computed tomography or positron emission tomography can be ordered to rule out metastatic disease.4
Pilomatrixomas are benign neoplasms that arise from hair matrix cells and have been associated with catenin beta-1 gene mutations, as well as genetic syndromes and trauma.7 Clinically, pilomatrixomas manifest as solitary, firm, painless, slow-growing nodules that commonly are found in the head and neck region. This tumor has a slight predominance in women and occurs frequently in adolescent years. The overlying skin may appear normal or show grey-bluish discoloration.8 Histopathology shows basaloid cells resembling primitive hair matrix cells with an abrupt transition to shadow cells composed of transformed keratinocytes without nuclei and calcification.7-8 This tumor can be differentiated by the presence of basaloid and shadow cells with calcification on histopathology, while MPTT will show atypical, mitotically active squamous cells with trichilemmal keratinization (Figure 2).
Proliferating trichilemmal cyst is a variant of trichilemmal cyst (TC) arising from the outer root sheath cells of the hair follicle. While TCs usually are slow growing and benign, the proliferating variant can be more aggressive with malignant potential. Patients often present with a solitary, well-circumscribed, rapidly growing nodule on the scalp. The lesion may be painful, and ulceration can occur, exposing the cystic contents. Histopathologically, PTCs resemble TCs with trichilemmal keratinization but also exhibit notable epithelial proliferation within the cystic space.9 While there can be considerable histopathologic overlap between PTC and MPTT—including extensive trichilemmal keratinization, variable atypia, and mitotic activity—PTC typically should not demonstrate invasion into the surrounding soft tissue or the degree of high-grade atypia, brisk mitoses, or necrosis seen in MPTT (eFigure 1).1 Immunohistochemistry may help distinguish PTC from MPTT and squamous cell carcinoma (SCC).10-11 The pattern of Ki-67 and p53 expression may be helpful with classification of PTC/MPTT into the 3 groups (benign, low-grade malignant, and high-grade malignant) proposed by Ye et al.1 Other investigators have suggested that Ki-67 expression may correlate potential for recurrence and clinical prognosis.12 Expression of CD34 (a marker that supports outer root sheath origin) might favor PTC/MPTT over SCC; however, cases of CD34- negative MPTT have been reported, particularly those with poorly differentiated histopathology.
Squamous cell carcinoma with cystic features is a histologic variant of SCC characterized by cystlike spaces containing malignant squamous epithelial cells.13 Squamous cell carcinoma with cystic features can manifest as a firm nodule with ulceration similar to MPTT or PTC but also can mimic a benign cyst.14 The diagnosis of invasive SCC with cystic features typically is straightforward and characterized by cords and nests of atypical keratinocytes extending into the dermis with areas of cystic architecture (eFigure 2). While both SCC with cystic features and MPTT may show cystic histopathologic architecture, MPTT typically shows areas of PTC, whereas SCC with cystic features lacks such areas.
Verrucous cysts refer to infundibular cysts or less commonly pilar cysts or hybrid pilar-epidermoid cysts that exhibit superimposed human papillomavirus (HPV) cytopathic changes. Clinically, a verrucous cyst manifests as a single, asymptomatic, slow-growing, firm lesion most commonly manifesting on the face and back. Histopathologically, the cyst wall may show acanthosis, papillomatosis, hypergranulosis with coarse keratohyalin granules, and koilocytic changes (eFigure 3). These histopathologic features are believed to be induced by secondary HPV infection. While HPV-related change, characterized by koilocytic alteration, papillomatosis, and verruciform hyperplasia, more commonly affects epidermal cysts, occasionally trichilemmal (pilar) cysts are involved. In these cases, verrucous cysts should be distinguished from MPTT. Verrucous cysts may contain rare normal mitotic figures, but do not contain atypical mitosis, marked cellular pleomorphism, or an infiltrating pattern similar to MPTT.15
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Biopsy revealed a squamous epithelium with cystic changes, trichilemmal differentiation, squamous eddy formation, keratinocyte atypia, focal necrotic changes, and a focus of atypical keratinocytes invading the dermis (Figure 1). Based on these findings, a diagnosis of malignant proliferating trichilemmal tumor (MPTT) was made.
Malignant proliferating trichilemmal tumor is a rare adnexal tumor that develops from the outer root sheath of the hair follicle. It often arises due to malignant transformation of pre-existing trichilemmal cysts, but some cases occur de novo.1 Malignant transformation is thought to start from a trichilemmal cyst in an adenomatous histologic stage, progressing to a proliferating trichilemmal cyst (PTC) in an epitheliomatous phase, ultimately becoming carcinomatous with MPTT.2-4 This transformation has been categorized into 3 morphologic groups to predict tumor behavior, including benign PTCs (curable by excision), low-grade malignant PTCs (minor risk for local recurrence), and high-grade malignant PTCs (risk for regional spread and metastasis with cytologic atypical features and potential for aggressive growth).1
More commonly observed in women in the fourth to eighth decades of life, MPTT may manifest as a fast- growing, painless, solitary nodule or as a progressively enlarging nodule at the site of a previously stable, long-standing lesion. Malignant proliferating trichilemmal tumor manifests frequently on the scalp, face, or neck, but there are reports of MPTT manifesting on the trunk and even as multiple concurrent lesions.1-4 The variability in clinical presentation and the potential to be mistaken for benign conditions makes excisional biopsy essential for diagnosis of MPTT. Histopathology classically demonstrates trichilemmal keratinization, a high mitotic index, and cellular atypia with invasion into the dermis.4 Malignant transformation frequently follows a prior history of trauma to the area or local inflammation.
Given the locally aggressive nature of MPTT, our patient was referred to a Mohs micrographic surgeon. While both wide excision with tumor-free margins and Mohs micrographic surgery are accepted surgical procedures for MPTT, there is no consensus in the literature on a standard treatment recommendation. Following surgery, close monitoring is needed for potential recurrence and metastases intracranially to the dura and muscles,5 as well as to the lungs.6 Further imaging using computed tomography or positron emission tomography can be ordered to rule out metastatic disease.4
Pilomatrixomas are benign neoplasms that arise from hair matrix cells and have been associated with catenin beta-1 gene mutations, as well as genetic syndromes and trauma.7 Clinically, pilomatrixomas manifest as solitary, firm, painless, slow-growing nodules that commonly are found in the head and neck region. This tumor has a slight predominance in women and occurs frequently in adolescent years. The overlying skin may appear normal or show grey-bluish discoloration.8 Histopathology shows basaloid cells resembling primitive hair matrix cells with an abrupt transition to shadow cells composed of transformed keratinocytes without nuclei and calcification.7-8 This tumor can be differentiated by the presence of basaloid and shadow cells with calcification on histopathology, while MPTT will show atypical, mitotically active squamous cells with trichilemmal keratinization (Figure 2).
Proliferating trichilemmal cyst is a variant of trichilemmal cyst (TC) arising from the outer root sheath cells of the hair follicle. While TCs usually are slow growing and benign, the proliferating variant can be more aggressive with malignant potential. Patients often present with a solitary, well-circumscribed, rapidly growing nodule on the scalp. The lesion may be painful, and ulceration can occur, exposing the cystic contents. Histopathologically, PTCs resemble TCs with trichilemmal keratinization but also exhibit notable epithelial proliferation within the cystic space.9 While there can be considerable histopathologic overlap between PTC and MPTT—including extensive trichilemmal keratinization, variable atypia, and mitotic activity—PTC typically should not demonstrate invasion into the surrounding soft tissue or the degree of high-grade atypia, brisk mitoses, or necrosis seen in MPTT (eFigure 1).1 Immunohistochemistry may help distinguish PTC from MPTT and squamous cell carcinoma (SCC).10-11 The pattern of Ki-67 and p53 expression may be helpful with classification of PTC/MPTT into the 3 groups (benign, low-grade malignant, and high-grade malignant) proposed by Ye et al.1 Other investigators have suggested that Ki-67 expression may correlate potential for recurrence and clinical prognosis.12 Expression of CD34 (a marker that supports outer root sheath origin) might favor PTC/MPTT over SCC; however, cases of CD34- negative MPTT have been reported, particularly those with poorly differentiated histopathology.
Squamous cell carcinoma with cystic features is a histologic variant of SCC characterized by cystlike spaces containing malignant squamous epithelial cells.13 Squamous cell carcinoma with cystic features can manifest as a firm nodule with ulceration similar to MPTT or PTC but also can mimic a benign cyst.14 The diagnosis of invasive SCC with cystic features typically is straightforward and characterized by cords and nests of atypical keratinocytes extending into the dermis with areas of cystic architecture (eFigure 2). While both SCC with cystic features and MPTT may show cystic histopathologic architecture, MPTT typically shows areas of PTC, whereas SCC with cystic features lacks such areas.
Verrucous cysts refer to infundibular cysts or less commonly pilar cysts or hybrid pilar-epidermoid cysts that exhibit superimposed human papillomavirus (HPV) cytopathic changes. Clinically, a verrucous cyst manifests as a single, asymptomatic, slow-growing, firm lesion most commonly manifesting on the face and back. Histopathologically, the cyst wall may show acanthosis, papillomatosis, hypergranulosis with coarse keratohyalin granules, and koilocytic changes (eFigure 3). These histopathologic features are believed to be induced by secondary HPV infection. While HPV-related change, characterized by koilocytic alteration, papillomatosis, and verruciform hyperplasia, more commonly affects epidermal cysts, occasionally trichilemmal (pilar) cysts are involved. In these cases, verrucous cysts should be distinguished from MPTT. Verrucous cysts may contain rare normal mitotic figures, but do not contain atypical mitosis, marked cellular pleomorphism, or an infiltrating pattern similar to MPTT.15
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumors: a clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574. doi:10.1309/0XLEGFQ64XYJU4G6
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208. doi:10.1159/000249868
- Rao S, Ramakrishnan R, Kamakshi D, et al. Malignant proliferating trichilemmal tumour presenting early in life: an uncommon feature. J Cutan Aesthet Surg. 2011;4:51-55. doi:10.4103/0974-2077.79196
- Kearns-Turcotte S, Thériault M, Blouin MM. Malignant proliferating trichilemmal tumors arising in patients with multiple trichilemmal cysts: a case series. JAAD Case Rep. 2022;22:42-46. doi:10.1016
- Karamese M, Akatekin A, Abaci M, et al. Unusual invasion of trichilemmal tumors: two case reports. Modern Plastic Surg. 2012; 2:54-57. doi:10.4236/MPS.2012.23014 /j.jdcr.2022.01.033
- Lobo L, Amonkar AD, Dontamsetty VV. Malignant proliferating trichilemmal tumour of the scalp with intra-cranial extension and lung metastasis-a case report. Indian J Surg. 2016;78:493-495. doi:10.1007/s12262-015-1427-0
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018;40:631-641. doi:10.1097/DAD.0000000000001118
- Sharma D, Agarwal S, Jain LS, et al. Pilomatrixoma masquerading as metastatic adenocarcinoma. A diagnostic pitfall on cytology. J Clin Diagn Res. 2014;8:FD13-FD14. doi:10.7860/JCDR/2014/9696.5064
- Valerio E, Parro FHS, Macedo MP, et al. Proliferating trichilemmal cyst with clinical, radiological, macroscopic, and microscopic orrelation. An Bras Dermatol. 2019;94:452-454. doi:10.1590 /abd1806-4841.20198199
- Joshi TP, Marchand S, Tschen J. Malignant proliferating trichilemmal tumor: a subtle presentation in an African American woman and review of immunohistochemical markers for this rare condition. Cureus. 2021;13:E17289. doi:10.7759/cureus.17289
- Gulati HK, Deshmukh SD, Anand M, et al. Low-grade malignant proliferating pilar tumor simulating a squamous-cell carcinoma in an elderly female: a case report and immunohistochemical study. Int J Trichology. 2011;3:98-101. doi:10.4103/0974-7753.90818
- Rangel-Gamboa L, Reyes-Castro M, Dominguez-Cherit J, et al. Proliferating trichilemmal cyst: the value of ki67 immunostaining. Int J Trichology. 2013;5:115-117. doi:10.4103/0974-7753.125599
- Asad U, Alkul S, Shimizu I, et al. Squamous cell carcinoma with unusual benign-appearing cystic features on histology. Cureus. 2023;15:E33610. doi:10.7759/cureus.33610
- Alkul S, Nguyen CN, Ramani NS, et al. Squamous cell carcinoma arising in an epidermal inclusion cyst. Baylor Univ Med Cent Proc. 2022;35:688-690. doi:10.1080/08998280.2022.207760
- Nanes BA, Laknezhad S, Chamseddin B, et al. Verrucous pilar cysts infected with beta human papillomavirus. J Cutan Pathol. 2020;47:381-386. doi:10.1111/cup.13599
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumors: a clinicopathologic study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574. doi:10.1309/0XLEGFQ64XYJU4G6
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208. doi:10.1159/000249868
- Rao S, Ramakrishnan R, Kamakshi D, et al. Malignant proliferating trichilemmal tumour presenting early in life: an uncommon feature. J Cutan Aesthet Surg. 2011;4:51-55. doi:10.4103/0974-2077.79196
- Kearns-Turcotte S, Thériault M, Blouin MM. Malignant proliferating trichilemmal tumors arising in patients with multiple trichilemmal cysts: a case series. JAAD Case Rep. 2022;22:42-46. doi:10.1016
- Karamese M, Akatekin A, Abaci M, et al. Unusual invasion of trichilemmal tumors: two case reports. Modern Plastic Surg. 2012; 2:54-57. doi:10.4236/MPS.2012.23014 /j.jdcr.2022.01.033
- Lobo L, Amonkar AD, Dontamsetty VV. Malignant proliferating trichilemmal tumour of the scalp with intra-cranial extension and lung metastasis-a case report. Indian J Surg. 2016;78:493-495. doi:10.1007/s12262-015-1427-0
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018;40:631-641. doi:10.1097/DAD.0000000000001118
- Sharma D, Agarwal S, Jain LS, et al. Pilomatrixoma masquerading as metastatic adenocarcinoma. A diagnostic pitfall on cytology. J Clin Diagn Res. 2014;8:FD13-FD14. doi:10.7860/JCDR/2014/9696.5064
- Valerio E, Parro FHS, Macedo MP, et al. Proliferating trichilemmal cyst with clinical, radiological, macroscopic, and microscopic orrelation. An Bras Dermatol. 2019;94:452-454. doi:10.1590 /abd1806-4841.20198199
- Joshi TP, Marchand S, Tschen J. Malignant proliferating trichilemmal tumor: a subtle presentation in an African American woman and review of immunohistochemical markers for this rare condition. Cureus. 2021;13:E17289. doi:10.7759/cureus.17289
- Gulati HK, Deshmukh SD, Anand M, et al. Low-grade malignant proliferating pilar tumor simulating a squamous-cell carcinoma in an elderly female: a case report and immunohistochemical study. Int J Trichology. 2011;3:98-101. doi:10.4103/0974-7753.90818
- Rangel-Gamboa L, Reyes-Castro M, Dominguez-Cherit J, et al. Proliferating trichilemmal cyst: the value of ki67 immunostaining. Int J Trichology. 2013;5:115-117. doi:10.4103/0974-7753.125599
- Asad U, Alkul S, Shimizu I, et al. Squamous cell carcinoma with unusual benign-appearing cystic features on histology. Cureus. 2023;15:E33610. doi:10.7759/cureus.33610
- Alkul S, Nguyen CN, Ramani NS, et al. Squamous cell carcinoma arising in an epidermal inclusion cyst. Baylor Univ Med Cent Proc. 2022;35:688-690. doi:10.1080/08998280.2022.207760
- Nanes BA, Laknezhad S, Chamseddin B, et al. Verrucous pilar cysts infected with beta human papillomavirus. J Cutan Pathol. 2020;47:381-386. doi:10.1111/cup.13599
Growing Nodule on the Parietal Scalp
Growing Nodule on the Parietal Scalp
A 38-year-old woman with no notable medical history presented to the dermatology department with a firm enlarging nodule on the scalp of many years’ duration. The patient noted there was no drainage or bleeding. Physical examination revealed a mobile, 2.5-cm, subcutaneous nodule on the right parietal medial scalp. An excisional biopsy was performed.
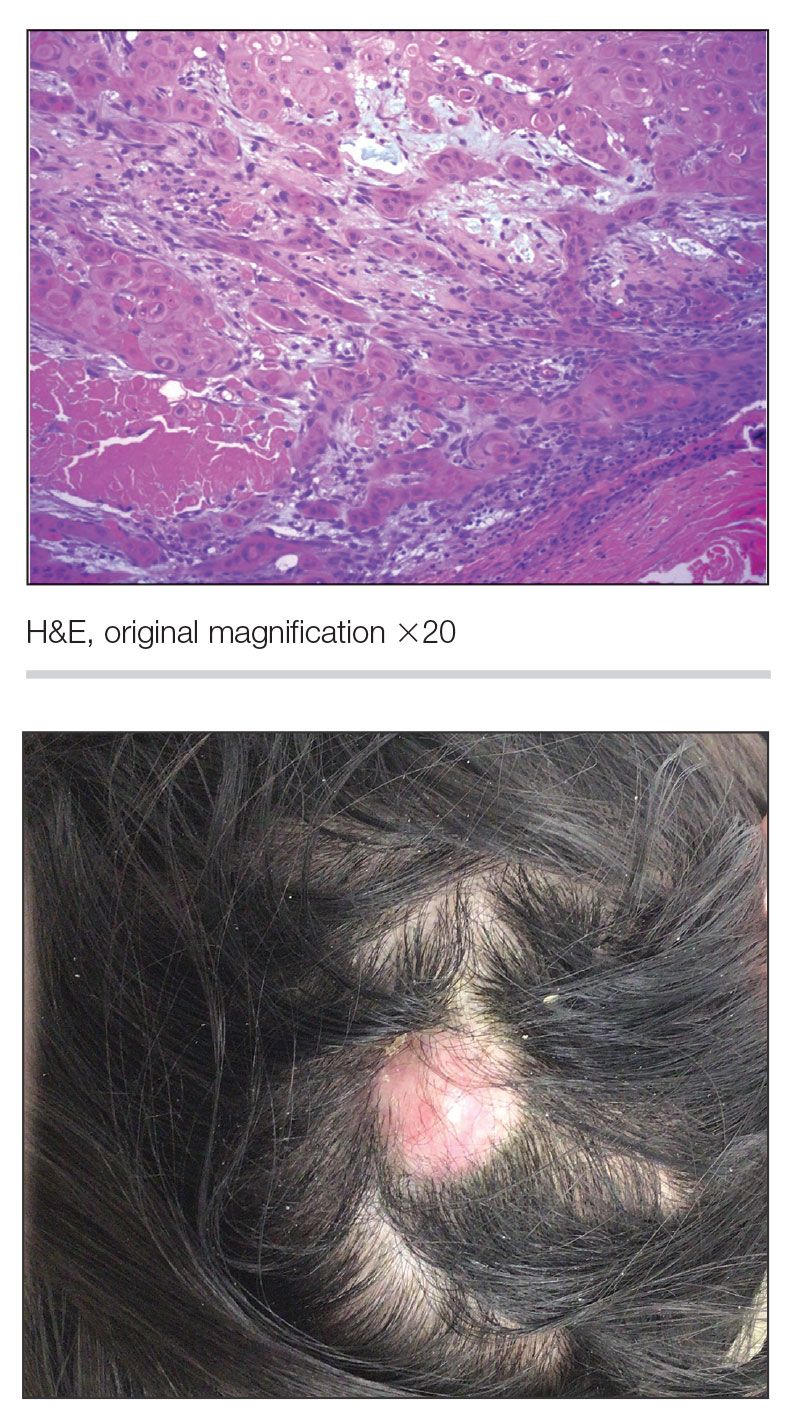

Path of Least Resistance: Guidance for Antibiotic Stewardship in Acne
Path of Least Resistance: Guidance for Antibiotic Stewardship in Acne
Dermatologists have long relied on oral antibiotics to manage moderate to severe acne1-4; however, it is critical to reassess how these medications are used in clinical practice as concerns about antibiotic resistance grow.5 The question is not whether antibiotics are effective for acne treatment—we know they are—but how to optimize their use to balance clinical benefit with responsible prescribing. Resistance in Cutibacterium acnes has been well documented in laboratory settings, but clinical treatment failure due to resistance remains rare and difficult to quantify.6,7 Still, minimizing unnecessary exposure is good clinical practice. Whether antibiotic resistance ultimately proves to drive clinical failure or remains largely theoretical, stewardship safeguards future treatment options.
In this article, we present a practical, expert-based framework aligned with American Academy of Dermatology (AAD) guidelines to support responsible antibiotic use in acne management. Seven prescribing principles are outlined to help clinicians maintain efficacy while minimizing resistance risk. Mechanisms of resistance in C acnes and broader microbiome impacts also are discussed.
MECHANISMS OF RESISTANCE IN ACNE THERAPY
Antibiotic resistance in acne primarily involves C acnes and arises through selective pressure from prolonged or subtherapeutic antibiotic exposure. Resistance mechanisms include point mutations in ribosomal binding sites, leading to decreased binding affinity for tetracyclines and macrolides as well as efflux pump activation and biofilm formation.8,9 Over time, resistant strains may proliferate and outcompete susceptible populations, potentially contributing to reduced clinical efficacy. Importantly, the use of broad-spectrum antibiotics may disrupt the skin and gut microbiota, promoting resistance among nontarget organisms.5 These concerns underscore the importance of limiting antibiotic use to appropriate indications, combining antibiotics with adjunctive nonantibiotic therapies, and avoiding monotherapy.
PRESCRIBING PRINCIPLES FOR RESPONSIBLE ORAL ANTIBIOTIC USE IN ACNE
The following principles are derived from our clinical experience and are aligned with AAD guidelines on acne treatment.10 This practical framework supports safe, effective, and streamlined prescribing.
Reserve Oral Antibiotics for Appropriate Cases
Oral antibiotics should be considered for patients with moderate to severe inflammatory acne when rapid anti-inflammatory control is needed. They are not indicated for comedonal or mild papulopustular acne. Before initiating treatment, clinicians should weigh the potential benefits against the risks associated with antibiotic exposure, including resistance and microbiome disruption.
Combine Oral Antibiotics With Topical Retinoids
Oral antibiotics should not be used as monotherapy. Topical retinoids should be initiated concurrently with oral antibiotics to maximize anti-inflammatory benefit, support transition to maintenance therapy, and reduce risk for resistance.
Consider Adding an Adjunctive Topical Antimicrobial Agent
Adjunctive topical antimicrobials can help reduce bacterial load. Benzoyl peroxide remains a first-line option due to its bactericidal activity and lack of resistance induction; however, recent product recalls involving benzene contamination may have raised safety concerns among some clinicians and patients.11,12 While no definitive harm has been established, alternative topical agents approved by the US Food and Drug Administration (eg, azelaic acid) may be used based on shared decision-making, tolerability, cost, access, and patient preference. Use of topical antibiotics (eg, clindamycin, erythromycin) as monotherapy is discouraged due to their higher resistance potential, which is consistent with AAD guidance.
Limit Treatment Duration to 12 Weeks or Less
Antibiotic use should be time limited, with discontinuation ideally within 8 to 12 weeks as clinical improvement is demonstrated. Repeated or prolonged courses should be avoided to minimize risk for resistance.
Simplify Treatment Regimens to Enhance Adherence
Regimen simplicity improves adherence, especially in adolescents. A two-agent regimen of an oral antibiotic and a topical retinoid typically is sufficient during the induction phase.13,14
Select Narrower-Spectrum Antibiotics When Feasible
Using a narrower-spectrum antibiotic may help minimize disruption to nontarget microbiota.15,16 Sarecycline has shown narrower in vitro activity within the tetracycline class,17,18 though clinical decisions should be informed by access, availability, and cost. Regardless of the agent used (eg, doxycycline, minocycline, or sarecycline), all antibiotics should be used judiciously and for the shortest effective duration.
Use Systemic Nonantibiotic Therapies When Appropriate
If there is inadequate response to oral antibiotic therapy, consider switching to systemic nonantibiotic options. Hormonal therapy may be appropriate for select female patients. Oral isotretinoin should be considered for patients with severe, recalcitrant, or scarring acne. Cycling between antibiotic classes without clear benefit is discouraged.
FINAL THOUGHTS
Oral antibiotics remain a foundational component in the management of moderate to severe acne; however, their use must be intentional, time limited, and guided by best practices to minimize the emergence of antimicrobial resistance. By adhering to the prescribing principles we have outlined here, which are rooted in clinical expertise and consistent with AAD guidelines, dermatologists can preserve antibiotic efficacy, optimize patient outcomes, and reduce long-term microbiologic risks. Stewardship is not about withholding treatment; it is about optimizing care today to protect treatment options for tomorrow.
- Bhate K, Williams H. Epidemiology of acne vulgaris. Br J Dermatol. 2013;168:474-485.
- Barbieri JS, Bhate K, Hartnett KP, et al. Trends in oral antibiotic prescription in dermatology, 2008 to 2016. JAMA Dermatol. 2019;155:290-297.
- Grada A, Armstrong A, Bunick C, et al. Trends in oral antibiotic use for acne treatment: a retrospective, population-based study in the United States, 2014 to 2016. J Drugs Dermatol. 2023;22:265-270.
- Perche PO, Peck GM, Robinson L, et al. Prescribing trends for acne vulgaris visits in the United States. Antibiotics. 2023;12:269.
- Karadag A, Aslan Kayıran M, Wu CY, et al. Antibiotic resistance in acne: changes, consequences and concerns. J Eur Acad Dermatol Venereol. 2021;35:73-78.
- Eady AE, Cove JH, Layton AM. Is antibiotic resistance in cutaneous propionibacteria clinically relevant? implications of resistance for acne patients and prescribers. Am J Clin Dermatol. 2003;4:813-831.
- Eady EA, Cove J, Holland K, et al. Erythromycin resistant propionibacteria in antibiotic treated acne patients: association with therapeutic failure. Br J Dermatol. 1989;121:51-57.
- Grossman TH. Tetracycline antibiotics and resistance. Cold Spring Harb Perspect Med. 2016;6:a025387.
- Kayiran M AS, Karadag AS, Al-Khuzaei S, et al. Antibiotic resistance in acne: mechanisms, complications and management. Am J Clin Dermatol. 2020;21:813-819.
- Reynolds RV, Yeung H, Cheng CE, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2024;90:1006-1035.
- Kucera K, Zenzola N, Hudspeth A, et al. Benzoyl peroxide drug products form benzene. Environ Health Perspect. 2024;132:037702.
- Kucera K, Zenzola N, Hudspeth A, et al. Evaluation of benzene presence and formation in benzoyl peroxide drug products. J Invest Dermatol. 2025;145:1147-1154.E11.
- Grada A, Perche P, Feldman S. Adherence and persistence to acne medications: a population-based claims database analysis. J Drugs Dermatol. 2022;21:758-764.<.li>
- Anderson KL, Dothard EH, Huang KE, et al. Frequency of primary nonadherence to acne treatment. JAMA Dermatol. 2015;151:623-626.
- Grada A, Bunick CG. Spectrum of antibiotic activity and its relevance to the microbiome. JAMA Netw Open. 2021;4:E215357-E215357.
- Francino M. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol. 2016;6:164577.
- Moura IB, Grada A, Spittal W, et al. Profiling the effects of systemic antibiotics for acne, including the narrow-spectrum antibiotic sarecycline, on the human gut microbiota. Front Microbiol. 2022;13:901911.
- Zhanel G, Critchley I, Lin L-Y, et al. Microbiological profile of sarecycline, a novel targeted spectrum tetracycline for the treatment of acne vulgaris. Antimicrob Agents Chemother. 2019;63:1297-1318.
Dermatologists have long relied on oral antibiotics to manage moderate to severe acne1-4; however, it is critical to reassess how these medications are used in clinical practice as concerns about antibiotic resistance grow.5 The question is not whether antibiotics are effective for acne treatment—we know they are—but how to optimize their use to balance clinical benefit with responsible prescribing. Resistance in Cutibacterium acnes has been well documented in laboratory settings, but clinical treatment failure due to resistance remains rare and difficult to quantify.6,7 Still, minimizing unnecessary exposure is good clinical practice. Whether antibiotic resistance ultimately proves to drive clinical failure or remains largely theoretical, stewardship safeguards future treatment options.
In this article, we present a practical, expert-based framework aligned with American Academy of Dermatology (AAD) guidelines to support responsible antibiotic use in acne management. Seven prescribing principles are outlined to help clinicians maintain efficacy while minimizing resistance risk. Mechanisms of resistance in C acnes and broader microbiome impacts also are discussed.
MECHANISMS OF RESISTANCE IN ACNE THERAPY
Antibiotic resistance in acne primarily involves C acnes and arises through selective pressure from prolonged or subtherapeutic antibiotic exposure. Resistance mechanisms include point mutations in ribosomal binding sites, leading to decreased binding affinity for tetracyclines and macrolides as well as efflux pump activation and biofilm formation.8,9 Over time, resistant strains may proliferate and outcompete susceptible populations, potentially contributing to reduced clinical efficacy. Importantly, the use of broad-spectrum antibiotics may disrupt the skin and gut microbiota, promoting resistance among nontarget organisms.5 These concerns underscore the importance of limiting antibiotic use to appropriate indications, combining antibiotics with adjunctive nonantibiotic therapies, and avoiding monotherapy.
PRESCRIBING PRINCIPLES FOR RESPONSIBLE ORAL ANTIBIOTIC USE IN ACNE
The following principles are derived from our clinical experience and are aligned with AAD guidelines on acne treatment.10 This practical framework supports safe, effective, and streamlined prescribing.
Reserve Oral Antibiotics for Appropriate Cases
Oral antibiotics should be considered for patients with moderate to severe inflammatory acne when rapid anti-inflammatory control is needed. They are not indicated for comedonal or mild papulopustular acne. Before initiating treatment, clinicians should weigh the potential benefits against the risks associated with antibiotic exposure, including resistance and microbiome disruption.
Combine Oral Antibiotics With Topical Retinoids
Oral antibiotics should not be used as monotherapy. Topical retinoids should be initiated concurrently with oral antibiotics to maximize anti-inflammatory benefit, support transition to maintenance therapy, and reduce risk for resistance.
Consider Adding an Adjunctive Topical Antimicrobial Agent
Adjunctive topical antimicrobials can help reduce bacterial load. Benzoyl peroxide remains a first-line option due to its bactericidal activity and lack of resistance induction; however, recent product recalls involving benzene contamination may have raised safety concerns among some clinicians and patients.11,12 While no definitive harm has been established, alternative topical agents approved by the US Food and Drug Administration (eg, azelaic acid) may be used based on shared decision-making, tolerability, cost, access, and patient preference. Use of topical antibiotics (eg, clindamycin, erythromycin) as monotherapy is discouraged due to their higher resistance potential, which is consistent with AAD guidance.
Limit Treatment Duration to 12 Weeks or Less
Antibiotic use should be time limited, with discontinuation ideally within 8 to 12 weeks as clinical improvement is demonstrated. Repeated or prolonged courses should be avoided to minimize risk for resistance.
Simplify Treatment Regimens to Enhance Adherence
Regimen simplicity improves adherence, especially in adolescents. A two-agent regimen of an oral antibiotic and a topical retinoid typically is sufficient during the induction phase.13,14
Select Narrower-Spectrum Antibiotics When Feasible
Using a narrower-spectrum antibiotic may help minimize disruption to nontarget microbiota.15,16 Sarecycline has shown narrower in vitro activity within the tetracycline class,17,18 though clinical decisions should be informed by access, availability, and cost. Regardless of the agent used (eg, doxycycline, minocycline, or sarecycline), all antibiotics should be used judiciously and for the shortest effective duration.
Use Systemic Nonantibiotic Therapies When Appropriate
If there is inadequate response to oral antibiotic therapy, consider switching to systemic nonantibiotic options. Hormonal therapy may be appropriate for select female patients. Oral isotretinoin should be considered for patients with severe, recalcitrant, or scarring acne. Cycling between antibiotic classes without clear benefit is discouraged.
FINAL THOUGHTS
Oral antibiotics remain a foundational component in the management of moderate to severe acne; however, their use must be intentional, time limited, and guided by best practices to minimize the emergence of antimicrobial resistance. By adhering to the prescribing principles we have outlined here, which are rooted in clinical expertise and consistent with AAD guidelines, dermatologists can preserve antibiotic efficacy, optimize patient outcomes, and reduce long-term microbiologic risks. Stewardship is not about withholding treatment; it is about optimizing care today to protect treatment options for tomorrow.
Dermatologists have long relied on oral antibiotics to manage moderate to severe acne1-4; however, it is critical to reassess how these medications are used in clinical practice as concerns about antibiotic resistance grow.5 The question is not whether antibiotics are effective for acne treatment—we know they are—but how to optimize their use to balance clinical benefit with responsible prescribing. Resistance in Cutibacterium acnes has been well documented in laboratory settings, but clinical treatment failure due to resistance remains rare and difficult to quantify.6,7 Still, minimizing unnecessary exposure is good clinical practice. Whether antibiotic resistance ultimately proves to drive clinical failure or remains largely theoretical, stewardship safeguards future treatment options.
In this article, we present a practical, expert-based framework aligned with American Academy of Dermatology (AAD) guidelines to support responsible antibiotic use in acne management. Seven prescribing principles are outlined to help clinicians maintain efficacy while minimizing resistance risk. Mechanisms of resistance in C acnes and broader microbiome impacts also are discussed.
MECHANISMS OF RESISTANCE IN ACNE THERAPY
Antibiotic resistance in acne primarily involves C acnes and arises through selective pressure from prolonged or subtherapeutic antibiotic exposure. Resistance mechanisms include point mutations in ribosomal binding sites, leading to decreased binding affinity for tetracyclines and macrolides as well as efflux pump activation and biofilm formation.8,9 Over time, resistant strains may proliferate and outcompete susceptible populations, potentially contributing to reduced clinical efficacy. Importantly, the use of broad-spectrum antibiotics may disrupt the skin and gut microbiota, promoting resistance among nontarget organisms.5 These concerns underscore the importance of limiting antibiotic use to appropriate indications, combining antibiotics with adjunctive nonantibiotic therapies, and avoiding monotherapy.
PRESCRIBING PRINCIPLES FOR RESPONSIBLE ORAL ANTIBIOTIC USE IN ACNE
The following principles are derived from our clinical experience and are aligned with AAD guidelines on acne treatment.10 This practical framework supports safe, effective, and streamlined prescribing.
Reserve Oral Antibiotics for Appropriate Cases
Oral antibiotics should be considered for patients with moderate to severe inflammatory acne when rapid anti-inflammatory control is needed. They are not indicated for comedonal or mild papulopustular acne. Before initiating treatment, clinicians should weigh the potential benefits against the risks associated with antibiotic exposure, including resistance and microbiome disruption.
Combine Oral Antibiotics With Topical Retinoids
Oral antibiotics should not be used as monotherapy. Topical retinoids should be initiated concurrently with oral antibiotics to maximize anti-inflammatory benefit, support transition to maintenance therapy, and reduce risk for resistance.
Consider Adding an Adjunctive Topical Antimicrobial Agent
Adjunctive topical antimicrobials can help reduce bacterial load. Benzoyl peroxide remains a first-line option due to its bactericidal activity and lack of resistance induction; however, recent product recalls involving benzene contamination may have raised safety concerns among some clinicians and patients.11,12 While no definitive harm has been established, alternative topical agents approved by the US Food and Drug Administration (eg, azelaic acid) may be used based on shared decision-making, tolerability, cost, access, and patient preference. Use of topical antibiotics (eg, clindamycin, erythromycin) as monotherapy is discouraged due to their higher resistance potential, which is consistent with AAD guidance.
Limit Treatment Duration to 12 Weeks or Less
Antibiotic use should be time limited, with discontinuation ideally within 8 to 12 weeks as clinical improvement is demonstrated. Repeated or prolonged courses should be avoided to minimize risk for resistance.
Simplify Treatment Regimens to Enhance Adherence
Regimen simplicity improves adherence, especially in adolescents. A two-agent regimen of an oral antibiotic and a topical retinoid typically is sufficient during the induction phase.13,14
Select Narrower-Spectrum Antibiotics When Feasible
Using a narrower-spectrum antibiotic may help minimize disruption to nontarget microbiota.15,16 Sarecycline has shown narrower in vitro activity within the tetracycline class,17,18 though clinical decisions should be informed by access, availability, and cost. Regardless of the agent used (eg, doxycycline, minocycline, or sarecycline), all antibiotics should be used judiciously and for the shortest effective duration.
Use Systemic Nonantibiotic Therapies When Appropriate
If there is inadequate response to oral antibiotic therapy, consider switching to systemic nonantibiotic options. Hormonal therapy may be appropriate for select female patients. Oral isotretinoin should be considered for patients with severe, recalcitrant, or scarring acne. Cycling between antibiotic classes without clear benefit is discouraged.
FINAL THOUGHTS
Oral antibiotics remain a foundational component in the management of moderate to severe acne; however, their use must be intentional, time limited, and guided by best practices to minimize the emergence of antimicrobial resistance. By adhering to the prescribing principles we have outlined here, which are rooted in clinical expertise and consistent with AAD guidelines, dermatologists can preserve antibiotic efficacy, optimize patient outcomes, and reduce long-term microbiologic risks. Stewardship is not about withholding treatment; it is about optimizing care today to protect treatment options for tomorrow.
- Bhate K, Williams H. Epidemiology of acne vulgaris. Br J Dermatol. 2013;168:474-485.
- Barbieri JS, Bhate K, Hartnett KP, et al. Trends in oral antibiotic prescription in dermatology, 2008 to 2016. JAMA Dermatol. 2019;155:290-297.
- Grada A, Armstrong A, Bunick C, et al. Trends in oral antibiotic use for acne treatment: a retrospective, population-based study in the United States, 2014 to 2016. J Drugs Dermatol. 2023;22:265-270.
- Perche PO, Peck GM, Robinson L, et al. Prescribing trends for acne vulgaris visits in the United States. Antibiotics. 2023;12:269.
- Karadag A, Aslan Kayıran M, Wu CY, et al. Antibiotic resistance in acne: changes, consequences and concerns. J Eur Acad Dermatol Venereol. 2021;35:73-78.
- Eady AE, Cove JH, Layton AM. Is antibiotic resistance in cutaneous propionibacteria clinically relevant? implications of resistance for acne patients and prescribers. Am J Clin Dermatol. 2003;4:813-831.
- Eady EA, Cove J, Holland K, et al. Erythromycin resistant propionibacteria in antibiotic treated acne patients: association with therapeutic failure. Br J Dermatol. 1989;121:51-57.
- Grossman TH. Tetracycline antibiotics and resistance. Cold Spring Harb Perspect Med. 2016;6:a025387.
- Kayiran M AS, Karadag AS, Al-Khuzaei S, et al. Antibiotic resistance in acne: mechanisms, complications and management. Am J Clin Dermatol. 2020;21:813-819.
- Reynolds RV, Yeung H, Cheng CE, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2024;90:1006-1035.
- Kucera K, Zenzola N, Hudspeth A, et al. Benzoyl peroxide drug products form benzene. Environ Health Perspect. 2024;132:037702.
- Kucera K, Zenzola N, Hudspeth A, et al. Evaluation of benzene presence and formation in benzoyl peroxide drug products. J Invest Dermatol. 2025;145:1147-1154.E11.
- Grada A, Perche P, Feldman S. Adherence and persistence to acne medications: a population-based claims database analysis. J Drugs Dermatol. 2022;21:758-764.<.li>
- Anderson KL, Dothard EH, Huang KE, et al. Frequency of primary nonadherence to acne treatment. JAMA Dermatol. 2015;151:623-626.
- Grada A, Bunick CG. Spectrum of antibiotic activity and its relevance to the microbiome. JAMA Netw Open. 2021;4:E215357-E215357.
- Francino M. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol. 2016;6:164577.
- Moura IB, Grada A, Spittal W, et al. Profiling the effects of systemic antibiotics for acne, including the narrow-spectrum antibiotic sarecycline, on the human gut microbiota. Front Microbiol. 2022;13:901911.
- Zhanel G, Critchley I, Lin L-Y, et al. Microbiological profile of sarecycline, a novel targeted spectrum tetracycline for the treatment of acne vulgaris. Antimicrob Agents Chemother. 2019;63:1297-1318.
- Bhate K, Williams H. Epidemiology of acne vulgaris. Br J Dermatol. 2013;168:474-485.
- Barbieri JS, Bhate K, Hartnett KP, et al. Trends in oral antibiotic prescription in dermatology, 2008 to 2016. JAMA Dermatol. 2019;155:290-297.
- Grada A, Armstrong A, Bunick C, et al. Trends in oral antibiotic use for acne treatment: a retrospective, population-based study in the United States, 2014 to 2016. J Drugs Dermatol. 2023;22:265-270.
- Perche PO, Peck GM, Robinson L, et al. Prescribing trends for acne vulgaris visits in the United States. Antibiotics. 2023;12:269.
- Karadag A, Aslan Kayıran M, Wu CY, et al. Antibiotic resistance in acne: changes, consequences and concerns. J Eur Acad Dermatol Venereol. 2021;35:73-78.
- Eady AE, Cove JH, Layton AM. Is antibiotic resistance in cutaneous propionibacteria clinically relevant? implications of resistance for acne patients and prescribers. Am J Clin Dermatol. 2003;4:813-831.
- Eady EA, Cove J, Holland K, et al. Erythromycin resistant propionibacteria in antibiotic treated acne patients: association with therapeutic failure. Br J Dermatol. 1989;121:51-57.
- Grossman TH. Tetracycline antibiotics and resistance. Cold Spring Harb Perspect Med. 2016;6:a025387.
- Kayiran M AS, Karadag AS, Al-Khuzaei S, et al. Antibiotic resistance in acne: mechanisms, complications and management. Am J Clin Dermatol. 2020;21:813-819.
- Reynolds RV, Yeung H, Cheng CE, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2024;90:1006-1035.
- Kucera K, Zenzola N, Hudspeth A, et al. Benzoyl peroxide drug products form benzene. Environ Health Perspect. 2024;132:037702.
- Kucera K, Zenzola N, Hudspeth A, et al. Evaluation of benzene presence and formation in benzoyl peroxide drug products. J Invest Dermatol. 2025;145:1147-1154.E11.
- Grada A, Perche P, Feldman S. Adherence and persistence to acne medications: a population-based claims database analysis. J Drugs Dermatol. 2022;21:758-764.<.li>
- Anderson KL, Dothard EH, Huang KE, et al. Frequency of primary nonadherence to acne treatment. JAMA Dermatol. 2015;151:623-626.
- Grada A, Bunick CG. Spectrum of antibiotic activity and its relevance to the microbiome. JAMA Netw Open. 2021;4:E215357-E215357.
- Francino M. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol. 2016;6:164577.
- Moura IB, Grada A, Spittal W, et al. Profiling the effects of systemic antibiotics for acne, including the narrow-spectrum antibiotic sarecycline, on the human gut microbiota. Front Microbiol. 2022;13:901911.
- Zhanel G, Critchley I, Lin L-Y, et al. Microbiological profile of sarecycline, a novel targeted spectrum tetracycline for the treatment of acne vulgaris. Antimicrob Agents Chemother. 2019;63:1297-1318.
Path of Least Resistance: Guidance for Antibiotic Stewardship in Acne
Path of Least Resistance: Guidance for Antibiotic Stewardship in Acne
Practice Point
- Oral antibiotics remain a cornerstone in the treatment of moderate to severe acne, but growing concerns about antibiotic resistance necessitate more intentional prescribing.

Poly-L-Lactic Acid Reconstitution Technique to Reduce Needle Obstruction
Poly-L-Lactic Acid Reconstitution Technique to Reduce Needle Obstruction
Practice Gap
Poly-L-lactic acid is approved by the US Food and Drug Administration for addressing fat loss due to HAART in patients with HIV.2,3 When used as a dermal filler for correction of facial lipoatrophy, PLLA is well tolerated and has been shown to improve quality of life.2,3 Poly-L-lactic acid is available for clinical use as microparticles of lyophilized alpha hydroxy acid polymers. Once injected (after the carrier substance is absorbed), PLLA induces an inflammatory response that ultimately leads to the production of new collagen.3 Unfortunately, PLLA microparticles often obstruct needles and make the product difficult to use, potentially hindering effective injection; thus, it is in the best interest of the patient to mitigate needle obstruction during this procedure. In this article, we describe a simple and effective way to mitigate this problem by utilizing a water bath to warm the filler prior to injection.
Technique
The required supplies include a thermostatic water bath, reconstituted PLLA, a syringe, and a 26-gauge injection needle. Because laboratory-grade heated water baths typically cost between $300 and $3000,4 we recommend using a more affordable, commercially available thermostatic water bath (eg, baby bottle warmer)(Figure 1) to warm the filler prior to injection, as the optimal temperature for this technique can still be achieved while remaining cost effective. Vials of PLLA reconstituted with 7 mL of sterile water and 2 mL lidocaine hydrochloride 1% should be labeled with the date of reconstitution and manually agitated for 30 seconds. The reconstituted product should be stored for 24 hours to ensure even suspension and powder saturation.5 On the day of the procedure, the vial should be placed into the water bath (heated to 100 °C) for 10 minutes prior to injection (Figure 2) and agitated again immediately before withdrawal into the syringe. The clinician then should sterilize the rubber top and draw the product from the warmed vial using the same size needle that will be used for injection. Although a larger gauge needle may make drawing up the product easier in typical practice, drawing and injecting with the same gauge needle helps prevent larger particles from clogging a smaller injection needle. Using a 26-gauge injection needle for withdrawal further reduces clogging by serving as a filter to prevent larger product particles from entering the injection syringe. The vials of PLLA can be kept in the water bath throughout the procedure between uses to keep the filler at a consistent temperature.

Practice Implications
Although many clinicians reduce needle obstructions by warming PLLA before injection, a published protocol currently is not available. One consideration when utilizing this technique is the limited data on the clinical stability and efficacy of PLLA at varying temperatures. Two studies recommend bringing the reconstituted vial to room temperature prior to injection, while others have documented an endothermic melting point in the range of 120 °C to 180
- James J, Carruthers A, Carruthers J. HIV-associated facial lipoatrophy. Dermatol Surg. 2002;28:979-986. doi:10.1046/j.1524-4725.2002.02099.x
- Duracinsky M, Leclercq P, Herrmann S, et al. Safety of poly-L-lactic acid (New-Fill®) in the treatment of facial lipoatrophy: a large observational study among HIV-positive patients. BMC Infect Dis. 2014;14:474. doi:10.1186/1471-2334-14-474
- Sickles CK, Nassereddin A, Patel P, et al. Poly-L-lactic acid. StatPearls [Internet]. Updated February 28, 2024. Accessed October 31, 2025. https://www.ncbi.nlm.nih.gov/books/NBK507871/
- Laboratory equipment: Water bath. Global Lab Supply. (n.d.). http://www.globallabsupply.com/Water-Bath-s/2122.htm
- Lin MJ, Dubin DP, Goldberg DJ, et al. Practices in the usage and reconstitution of poly-L-lactic acid. J Drugs Dermatol. 2019;18:880-886.
- Vleggaar D, Fitzgerald R, Lorenc ZP, et al. Consensus recommendations on the use of injectable poly-L-lactic acid for facial and nonfacial volumization. J Drugs Dermatol. 2014;13:s44-51.
- Sedush NG, Kalinin KT, Azarkevich PN, et al. Physicochemical characteristics and hydrolytic degradation of polylactic acid dermal fillers: a comparative study. Cosmetics. 2023;10:110. doi:10.3390/cosmetics10040110
Practice Gap
Poly-L-lactic acid is approved by the US Food and Drug Administration for addressing fat loss due to HAART in patients with HIV.2,3 When used as a dermal filler for correction of facial lipoatrophy, PLLA is well tolerated and has been shown to improve quality of life.2,3 Poly-L-lactic acid is available for clinical use as microparticles of lyophilized alpha hydroxy acid polymers. Once injected (after the carrier substance is absorbed), PLLA induces an inflammatory response that ultimately leads to the production of new collagen.3 Unfortunately, PLLA microparticles often obstruct needles and make the product difficult to use, potentially hindering effective injection; thus, it is in the best interest of the patient to mitigate needle obstruction during this procedure. In this article, we describe a simple and effective way to mitigate this problem by utilizing a water bath to warm the filler prior to injection.
Technique
The required supplies include a thermostatic water bath, reconstituted PLLA, a syringe, and a 26-gauge injection needle. Because laboratory-grade heated water baths typically cost between $300 and $3000,4 we recommend using a more affordable, commercially available thermostatic water bath (eg, baby bottle warmer)(Figure 1) to warm the filler prior to injection, as the optimal temperature for this technique can still be achieved while remaining cost effective. Vials of PLLA reconstituted with 7 mL of sterile water and 2 mL lidocaine hydrochloride 1% should be labeled with the date of reconstitution and manually agitated for 30 seconds. The reconstituted product should be stored for 24 hours to ensure even suspension and powder saturation.5 On the day of the procedure, the vial should be placed into the water bath (heated to 100 °C) for 10 minutes prior to injection (Figure 2) and agitated again immediately before withdrawal into the syringe. The clinician then should sterilize the rubber top and draw the product from the warmed vial using the same size needle that will be used for injection. Although a larger gauge needle may make drawing up the product easier in typical practice, drawing and injecting with the same gauge needle helps prevent larger particles from clogging a smaller injection needle. Using a 26-gauge injection needle for withdrawal further reduces clogging by serving as a filter to prevent larger product particles from entering the injection syringe. The vials of PLLA can be kept in the water bath throughout the procedure between uses to keep the filler at a consistent temperature.
Practice Implications
Although many clinicians reduce needle obstructions by warming PLLA before injection, a published protocol currently is not available. One consideration when utilizing this technique is the limited data on the clinical stability and efficacy of PLLA at varying temperatures. Two studies recommend bringing the reconstituted vial to room temperature prior to injection, while others have documented an endothermic melting point in the range of 120 °C to 180
Practice Gap
Poly-L-lactic acid is approved by the US Food and Drug Administration for addressing fat loss due to HAART in patients with HIV.2,3 When used as a dermal filler for correction of facial lipoatrophy, PLLA is well tolerated and has been shown to improve quality of life.2,3 Poly-L-lactic acid is available for clinical use as microparticles of lyophilized alpha hydroxy acid polymers. Once injected (after the carrier substance is absorbed), PLLA induces an inflammatory response that ultimately leads to the production of new collagen.3 Unfortunately, PLLA microparticles often obstruct needles and make the product difficult to use, potentially hindering effective injection; thus, it is in the best interest of the patient to mitigate needle obstruction during this procedure. In this article, we describe a simple and effective way to mitigate this problem by utilizing a water bath to warm the filler prior to injection.
Technique
The required supplies include a thermostatic water bath, reconstituted PLLA, a syringe, and a 26-gauge injection needle. Because laboratory-grade heated water baths typically cost between $300 and $3000,4 we recommend using a more affordable, commercially available thermostatic water bath (eg, baby bottle warmer)(Figure 1) to warm the filler prior to injection, as the optimal temperature for this technique can still be achieved while remaining cost effective. Vials of PLLA reconstituted with 7 mL of sterile water and 2 mL lidocaine hydrochloride 1% should be labeled with the date of reconstitution and manually agitated for 30 seconds. The reconstituted product should be stored for 24 hours to ensure even suspension and powder saturation.5 On the day of the procedure, the vial should be placed into the water bath (heated to 100 °C) for 10 minutes prior to injection (Figure 2) and agitated again immediately before withdrawal into the syringe. The clinician then should sterilize the rubber top and draw the product from the warmed vial using the same size needle that will be used for injection. Although a larger gauge needle may make drawing up the product easier in typical practice, drawing and injecting with the same gauge needle helps prevent larger particles from clogging a smaller injection needle. Using a 26-gauge injection needle for withdrawal further reduces clogging by serving as a filter to prevent larger product particles from entering the injection syringe. The vials of PLLA can be kept in the water bath throughout the procedure between uses to keep the filler at a consistent temperature.
Practice Implications
Although many clinicians reduce needle obstructions by warming PLLA before injection, a published protocol currently is not available. One consideration when utilizing this technique is the limited data on the clinical stability and efficacy of PLLA at varying temperatures. Two studies recommend bringing the reconstituted vial to room temperature prior to injection, while others have documented an endothermic melting point in the range of 120 °C to 180
- James J, Carruthers A, Carruthers J. HIV-associated facial lipoatrophy. Dermatol Surg. 2002;28:979-986. doi:10.1046/j.1524-4725.2002.02099.x
- Duracinsky M, Leclercq P, Herrmann S, et al. Safety of poly-L-lactic acid (New-Fill®) in the treatment of facial lipoatrophy: a large observational study among HIV-positive patients. BMC Infect Dis. 2014;14:474. doi:10.1186/1471-2334-14-474
- Sickles CK, Nassereddin A, Patel P, et al. Poly-L-lactic acid. StatPearls [Internet]. Updated February 28, 2024. Accessed October 31, 2025. https://www.ncbi.nlm.nih.gov/books/NBK507871/
- Laboratory equipment: Water bath. Global Lab Supply. (n.d.). http://www.globallabsupply.com/Water-Bath-s/2122.htm
- Lin MJ, Dubin DP, Goldberg DJ, et al. Practices in the usage and reconstitution of poly-L-lactic acid. J Drugs Dermatol. 2019;18:880-886.
- Vleggaar D, Fitzgerald R, Lorenc ZP, et al. Consensus recommendations on the use of injectable poly-L-lactic acid for facial and nonfacial volumization. J Drugs Dermatol. 2014;13:s44-51.
- Sedush NG, Kalinin KT, Azarkevich PN, et al. Physicochemical characteristics and hydrolytic degradation of polylactic acid dermal fillers: a comparative study. Cosmetics. 2023;10:110. doi:10.3390/cosmetics10040110
- James J, Carruthers A, Carruthers J. HIV-associated facial lipoatrophy. Dermatol Surg. 2002;28:979-986. doi:10.1046/j.1524-4725.2002.02099.x
- Duracinsky M, Leclercq P, Herrmann S, et al. Safety of poly-L-lactic acid (New-Fill®) in the treatment of facial lipoatrophy: a large observational study among HIV-positive patients. BMC Infect Dis. 2014;14:474. doi:10.1186/1471-2334-14-474
- Sickles CK, Nassereddin A, Patel P, et al. Poly-L-lactic acid. StatPearls [Internet]. Updated February 28, 2024. Accessed October 31, 2025. https://www.ncbi.nlm.nih.gov/books/NBK507871/
- Laboratory equipment: Water bath. Global Lab Supply. (n.d.). http://www.globallabsupply.com/Water-Bath-s/2122.htm
- Lin MJ, Dubin DP, Goldberg DJ, et al. Practices in the usage and reconstitution of poly-L-lactic acid. J Drugs Dermatol. 2019;18:880-886.
- Vleggaar D, Fitzgerald R, Lorenc ZP, et al. Consensus recommendations on the use of injectable poly-L-lactic acid for facial and nonfacial volumization. J Drugs Dermatol. 2014;13:s44-51.
- Sedush NG, Kalinin KT, Azarkevich PN, et al. Physicochemical characteristics and hydrolytic degradation of polylactic acid dermal fillers: a comparative study. Cosmetics. 2023;10:110. doi:10.3390/cosmetics10040110
Poly-L-Lactic Acid Reconstitution Technique to Reduce Needle Obstruction
Poly-L-Lactic Acid Reconstitution Technique to Reduce Needle Obstruction

Early Infantile Hemangioma Diagnosis Is Key in Skin of Color
Early Infantile Hemangioma Diagnosis Is Key in Skin of Color
Infantile hemangioma (IH) is the most common vascular tumor of infancy, appearing within the first few weeks of life and typically reaching peak size by age 3 to 5 months.1 It classically manifests as a raised or flat bright-red lesion in the upper dermis of the skin and/or subcutaneous tissue and can vary in number, size, shape, and location.2 It is characterized by a rapid proliferative phase, especially between 5 and 8 weeks of age, followed by gradual spontaneous regression over 1 to 10 years.1-3
Infantile hemangiomas are categorized based on depth (superficial, deep, or mixed) and distribution pattern (focal, multifocal, segmental, or indeterminate).4 In most cases, complete regression occurs by age 4 years, but there can be residual telangiectasia, fibrofatty tissue, and/or scarring.1,4 About 10% to 15% of IHs result in complications that require medical intervention (eg, visual, airway, or auditory compromise; ulceration; disfigurement); ideally, these patients should be referred to a specialist by 5 weeks of age.4 Prompt assessment of IH severity is essential to prevent or mitigate potential complications and ultimately improve outcomes.3 Social drivers of health contribute to delayed diagnosis and management of hemangiomas, leading to increased complications in some patient populations.5-7
Epidemiology
Infantile hemangiomas are estimated to manifest in 4.5% of infants in the United States.1 The most common type is superficial IH, typically found on the head or neck.5 Risk factors in infants include female sex, White race, premature birth, and low birth weight (<1000 g).1,3 Maternal risk factors include advanced gestational age (ie, >35 years), multiple gestations, family history of IH, tobacco use, use of progesterone therapy during pregnancy, and pre-eclampsia.1,3
Focal IH typically manifests as a single localized lesion that can occur anywhere on the body.2,3 In contrast, segmental IH manifests in a linear pattern and/or is distributed on a large anatomic area, most commonly on the face and less frequently the extremities and trunk.
Key Clinical Features
Superficial IH in patients with darker skin tones may appear as a dark-red or violaceous papule or plaque compared to bright red in lighter skin tones.5 Deep IH may appear as a soft, round, flesh-colored or blue-hued subcutaneous mass, the color of which may be harder to appreciate in those with darker skin tones.5
Worth Noting
Complications from IH may require imaging, close follow-up, systemic therapy, multidisciplinary care, and advanced health literacy and patient/family navigation. Multifocal IHs (≥5 lesions) are more likely to be associated with infantile hepatic hemangiomas.2,3 Large (>5 cm) segmental IHs on the face and lumbosacral area require further evaluation for PHACES (posterior fossa malformation, hemangiomas, arterial anomalies, cardiac defects, eye anomalies, and sternal raphe/cleft defects) and LUMBAR (lower-body segmental IH; urogenital anomalies and ulceration; myelopathy; bony deformities; anorectal malformations and arterial anomalies; and renal anomalies) syndromes, which are more common in patients of Hispanic ethnicity.2,3
The Infantile Hemangioma Referral Score is a recently validated tool that can assist primary care physicians in timely referral of IHs requiring early specialist intervention.4,9 It takes into account the location, number, and size of the lesions and the age of the patient; these factors help to determine which IHs may be managed conservatively vs those that may require treatment to prevent life-threatening complications.1-3
Systemic corticosteroids historically have been the primary treatment for IH; however, in the past decade, propranolol oral solution (4.28 mg/mL) has become the first-line therapy for most infants requiring systemic management.10 It is the only medication approved by the US Food and Drug Administration for proliferating IH, with treatment initiation as young as 5 weeks corrected age.11 As a nonselective beta-blocker, propranolol is believed to reduce IHs through vasoconstriction or by inhibition of angiogenesis.1,4,10
For small superficial IHs, treatment options include timolol maleate ophthalmic solution 0.5% (one drop applied twice daily to the IH) or pulsed dye laser therapy.4,10 Surgical excision typically is avoided during infancy due to concerns about anesthetic risks and potential blood loss.4,10 Surgery is reserved for cases involving residual fibrofatty tissue, postinvolution scarring, obstruction of vital structures, or lesions in aesthetically sensitive areas as well as when propranolol is contraindicated.4,10
Health Disparity Highlight
Infants with skin of color and those of lower socioeconomic status (SES) face a heightened risk for delayed diagnosis and more advanced disease at the initial evaluation for IH.5,7 Access barriers such as geographic limitations to specialty services, lack of insurance, underinsurance, and language differences impact timely diagnosis and treatment.5,6 Implementation of telemedicine services in areas with limited access to specialists can facilitate early evaluation and risk stratification for IH.12
A retrospective cohort study of 804 children seen at a large academic hospital found that those of lower SES were more likely to seek care after 3 months of age than their higher-SES counterparts.6 Those who presented after 6 months of age also had higher IH severity scores compared to their counterparts with higher SES.6 Delayed access to care may cause children to miss the critical treatment window during the rapid proliferative growth phase.6,12 However, children insured through Medicaid or the Children’s Health Insurance Program who participated in institutional care management programs (which assist in scheduling specialty care appointments within the institution) sought treatment earlier regardless of their SES, suggesting that such programs may help reduce disparities in timely access for children of lower SES.6
An epidemiologic study analyzing the demographics of children hospitalized across the United States demonstrated that Black infants with IH were more likely to belong to the lowest income quartile compared with White infants or those of other races. They also were 2 times older on average at initial presentation (1.8 vs 1.0 years), experienced longer hospitalizations (16.4 vs 13.8 days), and underwent more IH-related procedures than White infants and infants of other races (2.4, 1.9, and 2.1, respectively).7
These and other factors may contribute to missed windows of opportunity for timely treatment of high-risk IHs in patients with darker skin tones and/or those facing challenges stemming from social drivers of health.
- Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390:85-94.
- Mitra R, Fitzsimons HL, Hale T, et al. Recent advances in understanding the molecular basis of infantile haemangioma development. Br J Dermatol. 2024;191:661-669.
- Rodríguez Bandera AI, Sebaratnam DF, Wargon O, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392.
- Sebaratnam DF, Rodríguez Bandera AL, Wong LCF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85:1395-1404.
- Taye ME, Shah J, Seiverling EV, et al. Diagnosis of vascular anomalies in patients with skin of color. J Clin Aesthet Dermatol. 2024;17:54-62.
- Lie E, Psoter KJ, Püttgen KB. Lower socioeconomic status is associated with delayed access to care for infantile hemangioma: a cohort study. J Am Acad Dermatol. 2023;88:E221-E230.
- Kumar KD, Desai AD, Shah VP, et al. Racial discrepancies in presentation of hospitalized infantile hemangioma cases using the Kids’ Inpatient Database. Health Sci Rep. 2023;6:E1092.
- Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138:1567.
- Léauté-Labrèze C, Baselga Torres E, Weibel L, et al. The infantile hemangioma referral score: a validated tool for physicians. Pediatrics. 2020;145:E20191628.
- Macca L, Altavilla D, Di Bartolomeo L, et al. Update on treatment of infantile hemangiomas: what’s new in the last five years? Front Pharmacol. 2022;13:879602.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475.
- Frieden IJ, Püttgen KB, Drolet BA, et al. Management of infantile hemangiomas during the COVID pandemic. Pediatr Dermatol. 2020;37:412-418.
Infantile hemangioma (IH) is the most common vascular tumor of infancy, appearing within the first few weeks of life and typically reaching peak size by age 3 to 5 months.1 It classically manifests as a raised or flat bright-red lesion in the upper dermis of the skin and/or subcutaneous tissue and can vary in number, size, shape, and location.2 It is characterized by a rapid proliferative phase, especially between 5 and 8 weeks of age, followed by gradual spontaneous regression over 1 to 10 years.1-3
Infantile hemangiomas are categorized based on depth (superficial, deep, or mixed) and distribution pattern (focal, multifocal, segmental, or indeterminate).4 In most cases, complete regression occurs by age 4 years, but there can be residual telangiectasia, fibrofatty tissue, and/or scarring.1,4 About 10% to 15% of IHs result in complications that require medical intervention (eg, visual, airway, or auditory compromise; ulceration; disfigurement); ideally, these patients should be referred to a specialist by 5 weeks of age.4 Prompt assessment of IH severity is essential to prevent or mitigate potential complications and ultimately improve outcomes.3 Social drivers of health contribute to delayed diagnosis and management of hemangiomas, leading to increased complications in some patient populations.5-7
Epidemiology
Infantile hemangiomas are estimated to manifest in 4.5% of infants in the United States.1 The most common type is superficial IH, typically found on the head or neck.5 Risk factors in infants include female sex, White race, premature birth, and low birth weight (<1000 g).1,3 Maternal risk factors include advanced gestational age (ie, >35 years), multiple gestations, family history of IH, tobacco use, use of progesterone therapy during pregnancy, and pre-eclampsia.1,3
Focal IH typically manifests as a single localized lesion that can occur anywhere on the body.2,3 In contrast, segmental IH manifests in a linear pattern and/or is distributed on a large anatomic area, most commonly on the face and less frequently the extremities and trunk.
Key Clinical Features
Superficial IH in patients with darker skin tones may appear as a dark-red or violaceous papule or plaque compared to bright red in lighter skin tones.5 Deep IH may appear as a soft, round, flesh-colored or blue-hued subcutaneous mass, the color of which may be harder to appreciate in those with darker skin tones.5
Worth Noting
Complications from IH may require imaging, close follow-up, systemic therapy, multidisciplinary care, and advanced health literacy and patient/family navigation. Multifocal IHs (≥5 lesions) are more likely to be associated with infantile hepatic hemangiomas.2,3 Large (>5 cm) segmental IHs on the face and lumbosacral area require further evaluation for PHACES (posterior fossa malformation, hemangiomas, arterial anomalies, cardiac defects, eye anomalies, and sternal raphe/cleft defects) and LUMBAR (lower-body segmental IH; urogenital anomalies and ulceration; myelopathy; bony deformities; anorectal malformations and arterial anomalies; and renal anomalies) syndromes, which are more common in patients of Hispanic ethnicity.2,3
The Infantile Hemangioma Referral Score is a recently validated tool that can assist primary care physicians in timely referral of IHs requiring early specialist intervention.4,9 It takes into account the location, number, and size of the lesions and the age of the patient; these factors help to determine which IHs may be managed conservatively vs those that may require treatment to prevent life-threatening complications.1-3
Systemic corticosteroids historically have been the primary treatment for IH; however, in the past decade, propranolol oral solution (4.28 mg/mL) has become the first-line therapy for most infants requiring systemic management.10 It is the only medication approved by the US Food and Drug Administration for proliferating IH, with treatment initiation as young as 5 weeks corrected age.11 As a nonselective beta-blocker, propranolol is believed to reduce IHs through vasoconstriction or by inhibition of angiogenesis.1,4,10
For small superficial IHs, treatment options include timolol maleate ophthalmic solution 0.5% (one drop applied twice daily to the IH) or pulsed dye laser therapy.4,10 Surgical excision typically is avoided during infancy due to concerns about anesthetic risks and potential blood loss.4,10 Surgery is reserved for cases involving residual fibrofatty tissue, postinvolution scarring, obstruction of vital structures, or lesions in aesthetically sensitive areas as well as when propranolol is contraindicated.4,10
Health Disparity Highlight
Infants with skin of color and those of lower socioeconomic status (SES) face a heightened risk for delayed diagnosis and more advanced disease at the initial evaluation for IH.5,7 Access barriers such as geographic limitations to specialty services, lack of insurance, underinsurance, and language differences impact timely diagnosis and treatment.5,6 Implementation of telemedicine services in areas with limited access to specialists can facilitate early evaluation and risk stratification for IH.12
A retrospective cohort study of 804 children seen at a large academic hospital found that those of lower SES were more likely to seek care after 3 months of age than their higher-SES counterparts.6 Those who presented after 6 months of age also had higher IH severity scores compared to their counterparts with higher SES.6 Delayed access to care may cause children to miss the critical treatment window during the rapid proliferative growth phase.6,12 However, children insured through Medicaid or the Children’s Health Insurance Program who participated in institutional care management programs (which assist in scheduling specialty care appointments within the institution) sought treatment earlier regardless of their SES, suggesting that such programs may help reduce disparities in timely access for children of lower SES.6
An epidemiologic study analyzing the demographics of children hospitalized across the United States demonstrated that Black infants with IH were more likely to belong to the lowest income quartile compared with White infants or those of other races. They also were 2 times older on average at initial presentation (1.8 vs 1.0 years), experienced longer hospitalizations (16.4 vs 13.8 days), and underwent more IH-related procedures than White infants and infants of other races (2.4, 1.9, and 2.1, respectively).7
These and other factors may contribute to missed windows of opportunity for timely treatment of high-risk IHs in patients with darker skin tones and/or those facing challenges stemming from social drivers of health.
Infantile hemangioma (IH) is the most common vascular tumor of infancy, appearing within the first few weeks of life and typically reaching peak size by age 3 to 5 months.1 It classically manifests as a raised or flat bright-red lesion in the upper dermis of the skin and/or subcutaneous tissue and can vary in number, size, shape, and location.2 It is characterized by a rapid proliferative phase, especially between 5 and 8 weeks of age, followed by gradual spontaneous regression over 1 to 10 years.1-3
Infantile hemangiomas are categorized based on depth (superficial, deep, or mixed) and distribution pattern (focal, multifocal, segmental, or indeterminate).4 In most cases, complete regression occurs by age 4 years, but there can be residual telangiectasia, fibrofatty tissue, and/or scarring.1,4 About 10% to 15% of IHs result in complications that require medical intervention (eg, visual, airway, or auditory compromise; ulceration; disfigurement); ideally, these patients should be referred to a specialist by 5 weeks of age.4 Prompt assessment of IH severity is essential to prevent or mitigate potential complications and ultimately improve outcomes.3 Social drivers of health contribute to delayed diagnosis and management of hemangiomas, leading to increased complications in some patient populations.5-7
Epidemiology
Infantile hemangiomas are estimated to manifest in 4.5% of infants in the United States.1 The most common type is superficial IH, typically found on the head or neck.5 Risk factors in infants include female sex, White race, premature birth, and low birth weight (<1000 g).1,3 Maternal risk factors include advanced gestational age (ie, >35 years), multiple gestations, family history of IH, tobacco use, use of progesterone therapy during pregnancy, and pre-eclampsia.1,3
Focal IH typically manifests as a single localized lesion that can occur anywhere on the body.2,3 In contrast, segmental IH manifests in a linear pattern and/or is distributed on a large anatomic area, most commonly on the face and less frequently the extremities and trunk.
Key Clinical Features
Superficial IH in patients with darker skin tones may appear as a dark-red or violaceous papule or plaque compared to bright red in lighter skin tones.5 Deep IH may appear as a soft, round, flesh-colored or blue-hued subcutaneous mass, the color of which may be harder to appreciate in those with darker skin tones.5
Worth Noting
Complications from IH may require imaging, close follow-up, systemic therapy, multidisciplinary care, and advanced health literacy and patient/family navigation. Multifocal IHs (≥5 lesions) are more likely to be associated with infantile hepatic hemangiomas.2,3 Large (>5 cm) segmental IHs on the face and lumbosacral area require further evaluation for PHACES (posterior fossa malformation, hemangiomas, arterial anomalies, cardiac defects, eye anomalies, and sternal raphe/cleft defects) and LUMBAR (lower-body segmental IH; urogenital anomalies and ulceration; myelopathy; bony deformities; anorectal malformations and arterial anomalies; and renal anomalies) syndromes, which are more common in patients of Hispanic ethnicity.2,3
The Infantile Hemangioma Referral Score is a recently validated tool that can assist primary care physicians in timely referral of IHs requiring early specialist intervention.4,9 It takes into account the location, number, and size of the lesions and the age of the patient; these factors help to determine which IHs may be managed conservatively vs those that may require treatment to prevent life-threatening complications.1-3
Systemic corticosteroids historically have been the primary treatment for IH; however, in the past decade, propranolol oral solution (4.28 mg/mL) has become the first-line therapy for most infants requiring systemic management.10 It is the only medication approved by the US Food and Drug Administration for proliferating IH, with treatment initiation as young as 5 weeks corrected age.11 As a nonselective beta-blocker, propranolol is believed to reduce IHs through vasoconstriction or by inhibition of angiogenesis.1,4,10
For small superficial IHs, treatment options include timolol maleate ophthalmic solution 0.5% (one drop applied twice daily to the IH) or pulsed dye laser therapy.4,10 Surgical excision typically is avoided during infancy due to concerns about anesthetic risks and potential blood loss.4,10 Surgery is reserved for cases involving residual fibrofatty tissue, postinvolution scarring, obstruction of vital structures, or lesions in aesthetically sensitive areas as well as when propranolol is contraindicated.4,10
Health Disparity Highlight
Infants with skin of color and those of lower socioeconomic status (SES) face a heightened risk for delayed diagnosis and more advanced disease at the initial evaluation for IH.5,7 Access barriers such as geographic limitations to specialty services, lack of insurance, underinsurance, and language differences impact timely diagnosis and treatment.5,6 Implementation of telemedicine services in areas with limited access to specialists can facilitate early evaluation and risk stratification for IH.12
A retrospective cohort study of 804 children seen at a large academic hospital found that those of lower SES were more likely to seek care after 3 months of age than their higher-SES counterparts.6 Those who presented after 6 months of age also had higher IH severity scores compared to their counterparts with higher SES.6 Delayed access to care may cause children to miss the critical treatment window during the rapid proliferative growth phase.6,12 However, children insured through Medicaid or the Children’s Health Insurance Program who participated in institutional care management programs (which assist in scheduling specialty care appointments within the institution) sought treatment earlier regardless of their SES, suggesting that such programs may help reduce disparities in timely access for children of lower SES.6
An epidemiologic study analyzing the demographics of children hospitalized across the United States demonstrated that Black infants with IH were more likely to belong to the lowest income quartile compared with White infants or those of other races. They also were 2 times older on average at initial presentation (1.8 vs 1.0 years), experienced longer hospitalizations (16.4 vs 13.8 days), and underwent more IH-related procedures than White infants and infants of other races (2.4, 1.9, and 2.1, respectively).7
These and other factors may contribute to missed windows of opportunity for timely treatment of high-risk IHs in patients with darker skin tones and/or those facing challenges stemming from social drivers of health.
- Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390:85-94.
- Mitra R, Fitzsimons HL, Hale T, et al. Recent advances in understanding the molecular basis of infantile haemangioma development. Br J Dermatol. 2024;191:661-669.
- Rodríguez Bandera AI, Sebaratnam DF, Wargon O, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392.
- Sebaratnam DF, Rodríguez Bandera AL, Wong LCF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85:1395-1404.
- Taye ME, Shah J, Seiverling EV, et al. Diagnosis of vascular anomalies in patients with skin of color. J Clin Aesthet Dermatol. 2024;17:54-62.
- Lie E, Psoter KJ, Püttgen KB. Lower socioeconomic status is associated with delayed access to care for infantile hemangioma: a cohort study. J Am Acad Dermatol. 2023;88:E221-E230.
- Kumar KD, Desai AD, Shah VP, et al. Racial discrepancies in presentation of hospitalized infantile hemangioma cases using the Kids’ Inpatient Database. Health Sci Rep. 2023;6:E1092.
- Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138:1567.
- Léauté-Labrèze C, Baselga Torres E, Weibel L, et al. The infantile hemangioma referral score: a validated tool for physicians. Pediatrics. 2020;145:E20191628.
- Macca L, Altavilla D, Di Bartolomeo L, et al. Update on treatment of infantile hemangiomas: what’s new in the last five years? Front Pharmacol. 2022;13:879602.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475.
- Frieden IJ, Püttgen KB, Drolet BA, et al. Management of infantile hemangiomas during the COVID pandemic. Pediatr Dermatol. 2020;37:412-418.
- Léauté-Labrèze C, Harper JI, Hoeger PH. Infantile haemangioma. Lancet. 2017;390:85-94.
- Mitra R, Fitzsimons HL, Hale T, et al. Recent advances in understanding the molecular basis of infantile haemangioma development. Br J Dermatol. 2024;191:661-669.
- Rodríguez Bandera AI, Sebaratnam DF, Wargon O, et al. Infantile hemangioma. part 1: epidemiology, pathogenesis, clinical presentation and assessment. J Am Acad Dermatol. 2021;85:1379-1392.
- Sebaratnam DF, Rodríguez Bandera AL, Wong LCF, et al. Infantile hemangioma. part 2: management. J Am Acad Dermatol. 2021;85:1395-1404.
- Taye ME, Shah J, Seiverling EV, et al. Diagnosis of vascular anomalies in patients with skin of color. J Clin Aesthet Dermatol. 2024;17:54-62.
- Lie E, Psoter KJ, Püttgen KB. Lower socioeconomic status is associated with delayed access to care for infantile hemangioma: a cohort study. J Am Acad Dermatol. 2023;88:E221-E230.
- Kumar KD, Desai AD, Shah VP, et al. Racial discrepancies in presentation of hospitalized infantile hemangioma cases using the Kids’ Inpatient Database. Health Sci Rep. 2023;6:E1092.
- Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138:1567.
- Léauté-Labrèze C, Baselga Torres E, Weibel L, et al. The infantile hemangioma referral score: a validated tool for physicians. Pediatrics. 2020;145:E20191628.
- Macca L, Altavilla D, Di Bartolomeo L, et al. Update on treatment of infantile hemangiomas: what’s new in the last five years? Front Pharmacol. 2022;13:879602.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:E20183475.
- Frieden IJ, Püttgen KB, Drolet BA, et al. Management of infantile hemangiomas during the COVID pandemic. Pediatr Dermatol. 2020;37:412-418.
Early Infantile Hemangioma Diagnosis Is Key in Skin of Color
Early Infantile Hemangioma Diagnosis Is Key in Skin of Color

Cost Analysis of Dermatology Residency Applications From 2021 to 2024 Using the Texas Seeking Transparency in Application to Residency Database
Cost Analysis of Dermatology Residency Applications From 2021 to 2024 Using the Texas Seeking Transparency in Application to Residency Database
To the Editor:
Residency applicants, especially in competitive specialties such as dermatology, face major financial barriers due to the high costs of applications, interviews, and away rotations.1 While several studies have examined application costs of other specialties, few have analyzed expenses associated with dermatology applications.1,2 There are no data examining costs following the start of the COVID-19 pandemic in 2020; thus, our study evaluated dermatology application cost trends from 2021 to 2024 and compared them to other specialties to identify strategies to reduce the financial burden on applicants.
Self-reported total application costs, application fees, interview expenses, and away rotation costs from 2021 to 2024 were collected from the Texas Seeking Transparency in Application to Residency (STAR) database powered by the UT Southwestern Medical Center (Dallas, Texas).3 The mean total application expenses per year were compared among specialties, and an analysis of variance was used to determine if the differences were statistically significant.
The number of applicants who recorded information in the Texas STAR database was 110 in 2021, 163 in 2022, 136 in 2023, and 129 in 2024.3 The total dermatology application expenses increased from $2805 in 2021 to $6231 in 2024; interview costs increased from $404 in 2021 to $911 in 2024; and away rotation costs increased from $850 in 2021 to $3812 in 2024 (all P<.05)(Table). There was no significant change in application fees during the study period ($2176 in 2021 to $2125 in 2024 [P=.58]). Dermatology had the fourth highest average total cost over the study period compared to all other specialties, increasing from $2250 in 2021 to $5250 in 2024, following orthopedic surgery ($2250 in 2021 to $6750 in 2024), plastic surgery ($2250 in 2021 to $9750 in 2024), and neurosurgery ($1750 in 2021 to $11,250 in 2024).

Our study found that dermatology residency application costs have increased significantly from 2021 to 2024, primarily driven by rising interview and away rotation expenses (both P<.05). This trend places dermatology among the most expensive fields to apply to for residency. A cross-sectional survey of dermatology residency program directors identified away rotations as one of the top 5 selection criteria, underscoring their importance in the matching process.4 In addition, a cross-sectional analysis of 345 dermatology residents found that 26.2% matched at institutions where they had mentors, including those they connected with through away rotations.5,6 Overall, the high cost of away rotations partially may reflect the competitive nature of the specialty, as building connections at programs may enhance the chances of matching. These costs also can vary based on geography, as rotating in high-cost urban centers can be more expensive than in rural areas; however, rural rotations may be less common due to limited program availability and applicant preferences. For example, nearly 50% of 2024 Electronic Residency Application Service applicants indicated a preference for urban settings, while fewer than 5% selected rural settings.7 Additionally, the high costs associated with applying to residency programs and completing away rotations can disproportionately impact students from rural backgrounds and underrepresented minorities, who may have fewer financial resources.
In our study, the lower application-related expenses in 2021 (during the pandemic) compared to those of 2024 (postpandemic) likely stem from the Association of American Medical Colleges’ recommendation to conduct virtual interviews during the pandemic.8 In 2024, some dermatology programs returned to in-person interviews, with some applicants consequently incurring higher costs related to travel, lodging, and other associated expenses.8 A cost-analysis study of 4153 dermatology applicants from 2016 to 2021 found that the average application costs were $1759 per applicant during the pandemic, when virtual interviews replaced in-person ones, whereas costs were $8476 per applicant during periods with in-person interviews and no COVID-19 restrictions.2 However, we did not observe a significant change in application fees over our study period, likely because the pandemic did not affect application numbers. A cross-sectional analysis of dermatology applicants during the pandemic similarly reported reductions in application-related expenses during the period when interviews were conducted virtually,9 supporting the trend observed in our study. Overall, our findings taken together with other studies highlight the pandemic’s role in reducing expenses and underscore the potential for exploring additional cost-saving measures.
Implementing strategies to reduce these financial burdens—including virtual interviews, increasing student funding for away rotations, and limiting the number of applications individual students can submit—could help alleviate socioeconomic disparities. The new signaling system for residency programs aims to reduce the number of applications submitted, as applicants typically receive interviews only from the limited number of programs they signal, reducing overall application costs. However, our data from the Texas STAR database suggest that application numbers remained relatively stable from 2021 to 2024, indicating that, despite signaling, many applicants still may apply broadly in hopes of improving their chances in an increasingly competitive field. Although a definitive solution to reducing the financial burden on dermatology applicants remains elusive, these strategies can raise awareness and encourage important dialogues.
Limitations of our study include the voluntary nature of the Texas STAR survey, leading to potential voluntary response bias, as well as the small sample size. Students who choose to submit cost data may differ systematically from those who do not; for example, students who match may be more likely to report their outcomes, while those who do not match may be less likely to participate, potentially introducing selection bias. In addition, general awareness of the Texas STAR survey may vary across institutions and among students, further limiting the number of students who participate. Additionally, 2021 was the only presignaling year included, making it difficult to assess longer-term trends. Despite these limitations, the Texas STAR database remains a valuable resource for analyzing general residency application expenses and trends, as it offers comprehensive data from more than 100 medical schools and includes many variables.3
In conclusion, our study found that total dermatology residency application costs have increased significantly from 2021 to 2024 (all P<.05), making dermatology among the most expensive specialties for applying. This study sets the foundation for future survey-based research for applicants and program directors on strategies to alleviate financial burdens.
- Mansouri B, Walker GD, Mitchell J, et al. The cost of applying to dermatology residency: 2014 data estimates. J Am Acad Dermatol. 2016;74:754-756. doi:10.1016/j.jaad.2015.10.049
- Gorgy M, Shah S, Arbuiso S, et al. Comparison of cost changes due to the COVID-19 pandemic for dermatology residency applications in the USA. Clin Exp Dermatol. 2022;47:600-602. doi:10.1111/ced.15001<.li>
- UT Southwestern. Texas STAR. 2024. Accessed November 5, 2025. https://www.utsouthwestern.edu/education/medical-school/about-the-school/student-affairs/texas-star.html
- Baldwin K, Weidner Z, Ahn J, et al. Are away rotations critical for a successful match in orthopaedic surgery? Clin Orthop Relat Res. 2009;467:3340-3345. doi:10.1007/s11999-009-0920-9
- Yeh C, Desai AD, Wilson BN, et al. Cross-sectional analysis of scholarly work and mentor relationships in matched dermatology residency applicants. J Am Acad Dermatol. 2022;86:1437-1439. doi:10.1016/j.jaad.2021.06.861
- Gorouhi F, Alikhan A, Rezaei A, et al. Dermatology residency selection criteria with an emphasis on program characteristics: a national program director survey. Dermatol Res Pract. 2014;2014:692760. doi:10.1155/2014/692760
- Association of American Medical Colleges. Decoding geographic and setting preferences in residency selection. January 18, 2024. Accessed October 27, 2025. https://www.aamc.org/services/eras-institutions/geographic-preferences
- Association of American Medical Colleges. Virtual interviews: tips for program directors. Updated May 14, 2020. https://med.stanford.edu/content/dam/sm/gme/program_portal/pd/pd_meet/2019-2020/8-6-20-Virtual_Interview_Tips_for_Program_Directors_05142020.pdf
- Williams GE, Zimmerman JM, Wiggins CJ, et al. The indelible marks on dermatology: impacts of COVID-19 on dermatology residency match using the Texas STAR database. Clin Dermatol. 2023;41:215-218. doi:10.1016/j.clindermatol.2022.12.001
To the Editor:
Residency applicants, especially in competitive specialties such as dermatology, face major financial barriers due to the high costs of applications, interviews, and away rotations.1 While several studies have examined application costs of other specialties, few have analyzed expenses associated with dermatology applications.1,2 There are no data examining costs following the start of the COVID-19 pandemic in 2020; thus, our study evaluated dermatology application cost trends from 2021 to 2024 and compared them to other specialties to identify strategies to reduce the financial burden on applicants.
Self-reported total application costs, application fees, interview expenses, and away rotation costs from 2021 to 2024 were collected from the Texas Seeking Transparency in Application to Residency (STAR) database powered by the UT Southwestern Medical Center (Dallas, Texas).3 The mean total application expenses per year were compared among specialties, and an analysis of variance was used to determine if the differences were statistically significant.
The number of applicants who recorded information in the Texas STAR database was 110 in 2021, 163 in 2022, 136 in 2023, and 129 in 2024.3 The total dermatology application expenses increased from $2805 in 2021 to $6231 in 2024; interview costs increased from $404 in 2021 to $911 in 2024; and away rotation costs increased from $850 in 2021 to $3812 in 2024 (all P<.05)(Table). There was no significant change in application fees during the study period ($2176 in 2021 to $2125 in 2024 [P=.58]). Dermatology had the fourth highest average total cost over the study period compared to all other specialties, increasing from $2250 in 2021 to $5250 in 2024, following orthopedic surgery ($2250 in 2021 to $6750 in 2024), plastic surgery ($2250 in 2021 to $9750 in 2024), and neurosurgery ($1750 in 2021 to $11,250 in 2024).
Our study found that dermatology residency application costs have increased significantly from 2021 to 2024, primarily driven by rising interview and away rotation expenses (both P<.05). This trend places dermatology among the most expensive fields to apply to for residency. A cross-sectional survey of dermatology residency program directors identified away rotations as one of the top 5 selection criteria, underscoring their importance in the matching process.4 In addition, a cross-sectional analysis of 345 dermatology residents found that 26.2% matched at institutions where they had mentors, including those they connected with through away rotations.5,6 Overall, the high cost of away rotations partially may reflect the competitive nature of the specialty, as building connections at programs may enhance the chances of matching. These costs also can vary based on geography, as rotating in high-cost urban centers can be more expensive than in rural areas; however, rural rotations may be less common due to limited program availability and applicant preferences. For example, nearly 50% of 2024 Electronic Residency Application Service applicants indicated a preference for urban settings, while fewer than 5% selected rural settings.7 Additionally, the high costs associated with applying to residency programs and completing away rotations can disproportionately impact students from rural backgrounds and underrepresented minorities, who may have fewer financial resources.
In our study, the lower application-related expenses in 2021 (during the pandemic) compared to those of 2024 (postpandemic) likely stem from the Association of American Medical Colleges’ recommendation to conduct virtual interviews during the pandemic.8 In 2024, some dermatology programs returned to in-person interviews, with some applicants consequently incurring higher costs related to travel, lodging, and other associated expenses.8 A cost-analysis study of 4153 dermatology applicants from 2016 to 2021 found that the average application costs were $1759 per applicant during the pandemic, when virtual interviews replaced in-person ones, whereas costs were $8476 per applicant during periods with in-person interviews and no COVID-19 restrictions.2 However, we did not observe a significant change in application fees over our study period, likely because the pandemic did not affect application numbers. A cross-sectional analysis of dermatology applicants during the pandemic similarly reported reductions in application-related expenses during the period when interviews were conducted virtually,9 supporting the trend observed in our study. Overall, our findings taken together with other studies highlight the pandemic’s role in reducing expenses and underscore the potential for exploring additional cost-saving measures.
Implementing strategies to reduce these financial burdens—including virtual interviews, increasing student funding for away rotations, and limiting the number of applications individual students can submit—could help alleviate socioeconomic disparities. The new signaling system for residency programs aims to reduce the number of applications submitted, as applicants typically receive interviews only from the limited number of programs they signal, reducing overall application costs. However, our data from the Texas STAR database suggest that application numbers remained relatively stable from 2021 to 2024, indicating that, despite signaling, many applicants still may apply broadly in hopes of improving their chances in an increasingly competitive field. Although a definitive solution to reducing the financial burden on dermatology applicants remains elusive, these strategies can raise awareness and encourage important dialogues.
Limitations of our study include the voluntary nature of the Texas STAR survey, leading to potential voluntary response bias, as well as the small sample size. Students who choose to submit cost data may differ systematically from those who do not; for example, students who match may be more likely to report their outcomes, while those who do not match may be less likely to participate, potentially introducing selection bias. In addition, general awareness of the Texas STAR survey may vary across institutions and among students, further limiting the number of students who participate. Additionally, 2021 was the only presignaling year included, making it difficult to assess longer-term trends. Despite these limitations, the Texas STAR database remains a valuable resource for analyzing general residency application expenses and trends, as it offers comprehensive data from more than 100 medical schools and includes many variables.3
In conclusion, our study found that total dermatology residency application costs have increased significantly from 2021 to 2024 (all P<.05), making dermatology among the most expensive specialties for applying. This study sets the foundation for future survey-based research for applicants and program directors on strategies to alleviate financial burdens.
To the Editor:
Residency applicants, especially in competitive specialties such as dermatology, face major financial barriers due to the high costs of applications, interviews, and away rotations.1 While several studies have examined application costs of other specialties, few have analyzed expenses associated with dermatology applications.1,2 There are no data examining costs following the start of the COVID-19 pandemic in 2020; thus, our study evaluated dermatology application cost trends from 2021 to 2024 and compared them to other specialties to identify strategies to reduce the financial burden on applicants.
Self-reported total application costs, application fees, interview expenses, and away rotation costs from 2021 to 2024 were collected from the Texas Seeking Transparency in Application to Residency (STAR) database powered by the UT Southwestern Medical Center (Dallas, Texas).3 The mean total application expenses per year were compared among specialties, and an analysis of variance was used to determine if the differences were statistically significant.
The number of applicants who recorded information in the Texas STAR database was 110 in 2021, 163 in 2022, 136 in 2023, and 129 in 2024.3 The total dermatology application expenses increased from $2805 in 2021 to $6231 in 2024; interview costs increased from $404 in 2021 to $911 in 2024; and away rotation costs increased from $850 in 2021 to $3812 in 2024 (all P<.05)(Table). There was no significant change in application fees during the study period ($2176 in 2021 to $2125 in 2024 [P=.58]). Dermatology had the fourth highest average total cost over the study period compared to all other specialties, increasing from $2250 in 2021 to $5250 in 2024, following orthopedic surgery ($2250 in 2021 to $6750 in 2024), plastic surgery ($2250 in 2021 to $9750 in 2024), and neurosurgery ($1750 in 2021 to $11,250 in 2024).
Our study found that dermatology residency application costs have increased significantly from 2021 to 2024, primarily driven by rising interview and away rotation expenses (both P<.05). This trend places dermatology among the most expensive fields to apply to for residency. A cross-sectional survey of dermatology residency program directors identified away rotations as one of the top 5 selection criteria, underscoring their importance in the matching process.4 In addition, a cross-sectional analysis of 345 dermatology residents found that 26.2% matched at institutions where they had mentors, including those they connected with through away rotations.5,6 Overall, the high cost of away rotations partially may reflect the competitive nature of the specialty, as building connections at programs may enhance the chances of matching. These costs also can vary based on geography, as rotating in high-cost urban centers can be more expensive than in rural areas; however, rural rotations may be less common due to limited program availability and applicant preferences. For example, nearly 50% of 2024 Electronic Residency Application Service applicants indicated a preference for urban settings, while fewer than 5% selected rural settings.7 Additionally, the high costs associated with applying to residency programs and completing away rotations can disproportionately impact students from rural backgrounds and underrepresented minorities, who may have fewer financial resources.
In our study, the lower application-related expenses in 2021 (during the pandemic) compared to those of 2024 (postpandemic) likely stem from the Association of American Medical Colleges’ recommendation to conduct virtual interviews during the pandemic.8 In 2024, some dermatology programs returned to in-person interviews, with some applicants consequently incurring higher costs related to travel, lodging, and other associated expenses.8 A cost-analysis study of 4153 dermatology applicants from 2016 to 2021 found that the average application costs were $1759 per applicant during the pandemic, when virtual interviews replaced in-person ones, whereas costs were $8476 per applicant during periods with in-person interviews and no COVID-19 restrictions.2 However, we did not observe a significant change in application fees over our study period, likely because the pandemic did not affect application numbers. A cross-sectional analysis of dermatology applicants during the pandemic similarly reported reductions in application-related expenses during the period when interviews were conducted virtually,9 supporting the trend observed in our study. Overall, our findings taken together with other studies highlight the pandemic’s role in reducing expenses and underscore the potential for exploring additional cost-saving measures.
Implementing strategies to reduce these financial burdens—including virtual interviews, increasing student funding for away rotations, and limiting the number of applications individual students can submit—could help alleviate socioeconomic disparities. The new signaling system for residency programs aims to reduce the number of applications submitted, as applicants typically receive interviews only from the limited number of programs they signal, reducing overall application costs. However, our data from the Texas STAR database suggest that application numbers remained relatively stable from 2021 to 2024, indicating that, despite signaling, many applicants still may apply broadly in hopes of improving their chances in an increasingly competitive field. Although a definitive solution to reducing the financial burden on dermatology applicants remains elusive, these strategies can raise awareness and encourage important dialogues.
Limitations of our study include the voluntary nature of the Texas STAR survey, leading to potential voluntary response bias, as well as the small sample size. Students who choose to submit cost data may differ systematically from those who do not; for example, students who match may be more likely to report their outcomes, while those who do not match may be less likely to participate, potentially introducing selection bias. In addition, general awareness of the Texas STAR survey may vary across institutions and among students, further limiting the number of students who participate. Additionally, 2021 was the only presignaling year included, making it difficult to assess longer-term trends. Despite these limitations, the Texas STAR database remains a valuable resource for analyzing general residency application expenses and trends, as it offers comprehensive data from more than 100 medical schools and includes many variables.3
In conclusion, our study found that total dermatology residency application costs have increased significantly from 2021 to 2024 (all P<.05), making dermatology among the most expensive specialties for applying. This study sets the foundation for future survey-based research for applicants and program directors on strategies to alleviate financial burdens.
- Mansouri B, Walker GD, Mitchell J, et al. The cost of applying to dermatology residency: 2014 data estimates. J Am Acad Dermatol. 2016;74:754-756. doi:10.1016/j.jaad.2015.10.049
- Gorgy M, Shah S, Arbuiso S, et al. Comparison of cost changes due to the COVID-19 pandemic for dermatology residency applications in the USA. Clin Exp Dermatol. 2022;47:600-602. doi:10.1111/ced.15001<.li>
- UT Southwestern. Texas STAR. 2024. Accessed November 5, 2025. https://www.utsouthwestern.edu/education/medical-school/about-the-school/student-affairs/texas-star.html
- Baldwin K, Weidner Z, Ahn J, et al. Are away rotations critical for a successful match in orthopaedic surgery? Clin Orthop Relat Res. 2009;467:3340-3345. doi:10.1007/s11999-009-0920-9
- Yeh C, Desai AD, Wilson BN, et al. Cross-sectional analysis of scholarly work and mentor relationships in matched dermatology residency applicants. J Am Acad Dermatol. 2022;86:1437-1439. doi:10.1016/j.jaad.2021.06.861
- Gorouhi F, Alikhan A, Rezaei A, et al. Dermatology residency selection criteria with an emphasis on program characteristics: a national program director survey. Dermatol Res Pract. 2014;2014:692760. doi:10.1155/2014/692760
- Association of American Medical Colleges. Decoding geographic and setting preferences in residency selection. January 18, 2024. Accessed October 27, 2025. https://www.aamc.org/services/eras-institutions/geographic-preferences
- Association of American Medical Colleges. Virtual interviews: tips for program directors. Updated May 14, 2020. https://med.stanford.edu/content/dam/sm/gme/program_portal/pd/pd_meet/2019-2020/8-6-20-Virtual_Interview_Tips_for_Program_Directors_05142020.pdf
- Williams GE, Zimmerman JM, Wiggins CJ, et al. The indelible marks on dermatology: impacts of COVID-19 on dermatology residency match using the Texas STAR database. Clin Dermatol. 2023;41:215-218. doi:10.1016/j.clindermatol.2022.12.001
- Mansouri B, Walker GD, Mitchell J, et al. The cost of applying to dermatology residency: 2014 data estimates. J Am Acad Dermatol. 2016;74:754-756. doi:10.1016/j.jaad.2015.10.049
- Gorgy M, Shah S, Arbuiso S, et al. Comparison of cost changes due to the COVID-19 pandemic for dermatology residency applications in the USA. Clin Exp Dermatol. 2022;47:600-602. doi:10.1111/ced.15001<.li>
- UT Southwestern. Texas STAR. 2024. Accessed November 5, 2025. https://www.utsouthwestern.edu/education/medical-school/about-the-school/student-affairs/texas-star.html
- Baldwin K, Weidner Z, Ahn J, et al. Are away rotations critical for a successful match in orthopaedic surgery? Clin Orthop Relat Res. 2009;467:3340-3345. doi:10.1007/s11999-009-0920-9
- Yeh C, Desai AD, Wilson BN, et al. Cross-sectional analysis of scholarly work and mentor relationships in matched dermatology residency applicants. J Am Acad Dermatol. 2022;86:1437-1439. doi:10.1016/j.jaad.2021.06.861
- Gorouhi F, Alikhan A, Rezaei A, et al. Dermatology residency selection criteria with an emphasis on program characteristics: a national program director survey. Dermatol Res Pract. 2014;2014:692760. doi:10.1155/2014/692760
- Association of American Medical Colleges. Decoding geographic and setting preferences in residency selection. January 18, 2024. Accessed October 27, 2025. https://www.aamc.org/services/eras-institutions/geographic-preferences
- Association of American Medical Colleges. Virtual interviews: tips for program directors. Updated May 14, 2020. https://med.stanford.edu/content/dam/sm/gme/program_portal/pd/pd_meet/2019-2020/8-6-20-Virtual_Interview_Tips_for_Program_Directors_05142020.pdf
- Williams GE, Zimmerman JM, Wiggins CJ, et al. The indelible marks on dermatology: impacts of COVID-19 on dermatology residency match using the Texas STAR database. Clin Dermatol. 2023;41:215-218. doi:10.1016/j.clindermatol.2022.12.001
Cost Analysis of Dermatology Residency Applications From 2021 to 2024 Using the Texas Seeking Transparency in Application to Residency Database
Cost Analysis of Dermatology Residency Applications From 2021 to 2024 Using the Texas Seeking Transparency in Application to Residency Database
PRACTICE POINTS
- Dermatology application costs increased from 2021 to 2024, largely due to expenses related to away rotations and, in some cases, a return to in-person interviews.
- Away rotations play a critical role in the dermatology match; however, they also contribute substantially to financial burden.
- The cost-saving impact of virtual interviews during the COVID-19 pandemic highlights a meaningful opportunity for future cost reduction.
- Further interventions are needed to meaningfully reduce financial burden and promote equity.

Therapeutic Approaches for Alopecia Areata in Children Aged 6 to 11 Years
Therapeutic Approaches for Alopecia Areata in Children Aged 6 to 11 Years
Pediatric alopecia areata (AA) is a chronic autoimmune disease of the hair follicles characterized by nonscarring hair loss. Its incidence in children in the United States ranges from 13.6 to 33.5 per 100,000 person-years, with a prevalence of 0.04% to 0.11%.1 Alopecia areata has important effects on quality of life, particularly in children. Hair loss at an early age can decrease participation in school, sports, and extracurricular activities2 and is associated with increased rates of comorbid anxiety and depression.3 Families also experience psychosocial stress, often comparable to other chronic pediatric illnesses.4 Thus, management requires not only medical therapy but also psychosocial support and school-based accommodations.
Systemic therapies for treatment of AA in adolescents and adults are increasingly available, including US Food and Drug Administration (FDA)–approved Janus kinase (JAK) inhibitors such as baricitinib, deuruxolitinib (for adults), and ritlecitinib (for adolescents and adults); however, no systemic therapies have been approved by the FDA for children younger than 12 years. The therapeutic gap is most acute for those aged 6 to 11 years, for whom the psychosocial burden is high but treatment options are limited.3
This article highlights options and strategies for managing AA in children aged 6 to 11 years, emphasizing supportive and psychosocial care (including camouflage techniques), topical therapies, and off-label systemic approaches.
Supportive and Psychosocial Care
Treatment of AA in children extends beyond the affected child to include parents, caregivers, and even school staff (eg, teachers, principals, nurses).4 Disease-specific organizations such as the National Alopecia Areata Foundation (naaf.org) and the Children’s Alopecia Project (childrensalopeciaproject.org) provide education, support groups, and advocacy resources. These organizations assist families in navigating school accommodations, including Section 504 plans that may allow children with AA to wear hats in school to mitigate stigma. Additional resources include handouts for teachers and school nurses developed by the Society for Pediatric Dermatology.5
Psychological support for these patients is critical. Many children benefit from seeing a psychologist, particularly if anxiety, depression, and/or bullying is present.3 In clinics without embedded psychology services, dermatologists should maintain referral lists or encourage families to seek guidance from their pediatrician.
Camouflage techniques can help children cope with visible hair loss. Wigs and hairpieces are available free of charge through charitable organizations for patients younger than 17; however, young children often find adhesives uncomfortable, and they will not wear nonadherent wigs for long periods of time. Alternatives include soft hats, bonnets, scarves, and beanies. For partial hair loss, root concealers, scalp powders, or hair mascara can be useful. Temporary eyebrow tattoos are a good cosmetic approach, whereas microblading generally is not advised in children younger than 12 due to procedural risks including pain.
Topical Therapies
Topical agents remain the mainstay of treatment for AA in children aged 6 to 11 years. Potent class 1 or class 2 topical corticosteroids commonly are used, sometimes in combination with calcineurin inhibitors or topical minoxidil. Off-label compounded topical JAK inhibitors also have been tried in this population and may be helpful for eyebrow hair loss,6 though data on their efficacy for scalp AA are mixed.7 Intralesional corticosteroid injections, effective in adolescents and adults, generally are poorly tolerated by younger children and may cause considerable distress. Contact immunotherapy with squaric acid dibutyl ester or anthralin can be considered, but these agents are designed to elicit irritation, which may be intolerable for young children.8 Shared decision-making with families is essential to balance efficacy, tolerability, and treatment burden.
Systemic Therapies
Systemic therapy generally is reserved for children with extensive or refractory AA. Low-dose oral minoxidil is emerging as an off-label option. One systematic review reported that low-dose oral minoxidil was well tolerated in pediatric patients with minimal adverse effects.9 Doses of 0.01 to 0.02 mg/kg/d are reasonable starting points, achieved by cutting tablets or compounding oral solutions.10
In children with AA and concurrent atopic dermatitis, dupilumab may offer dual benefit. A real-world observational study demonstrated hair regrowth in pediatric patients with AA treated with dupilumab.11 Immunosuppressive options such as low-dose methotrexate or pulse corticosteroids (dexamethasone or prednisolone) also may be considered, although use of these agents requires careful monitoring due to increased risk for infection, clinically significant blood count and liver enzyme changes, and metabolic adverse effects related to long-term use of corticosteroids.
Clinical trials of JAK inhibitors in children aged 6 to 11 years are anticipated to begin in late 2025. Until then, off-label use of ritlecitinib, baricitinib, tofacitinib, or other JAK inhibitors may be considered in select cases with considerable disease burden and quality-of-life impairment following thorough discussion with the patient and their caregivers. Currently available pediatric data show few serious adverse events in children—the most common included upper respiratory infections (nasopharyngitis), acne, and headaches—but long-term risks remain unknown. Dosing challenges also exist for children who cannot swallow pills; currently ritlecitinib is available only as a capsule that cannot be opened while other JAK inhibitors are available in more accessible forms (baricitinib can be crushed and dissolved, and tofacitinib is available in liquid formulation for other pediatric indications). Insurance coverage is a major barrier, as these therapies are not FDA approved for AA in this age group.
Final Thoughts
Alopecia areata in children aged 6 to 11 years presents unique therapeutic challenges. While highly effective systemic therapies exist for older patients, younger children have limited options. For the 6-to-11 age group, management strategies should prioritize psychosocial support, topical therapy, and low-burden systemic alternatives such as low-dose oral minoxidil. Family education, school-based accommodations, and access to camouflage techniques are integral to holistic care. The commencement of pediatric clinical trials for JAK inhibitors offers hope for more robust treatment strategies in the near future. In the meantime, clinicians must engage in shared decision-making, tailoring therapy to the child’s disease severity, emotional well-being, and family priorities.
- Adhanom R, Ansbro B, Castelo-Soccio L. Epidemiology of pediatric alopecia areata. Pediatr Dermatol. 2025;42(suppl 1):12-23. doi:10.1111/pde.15803
- Paller AS, Rangel SM, Chamlin SL, et al; Pediatric Dermatology Research Alliance. Stigmatization and mental health impact of chronic pediatric skin disorders. JAMA Dermatol. 2024;160:621-630.
- van Dalen M, Muller KS, Kasperkovitz-Oosterloo JM, et al. Anxiety, depression, and quality of life in children and adults with alopecia areata: systematic review and meta-analysis. Front Med (Lausanne). 2022;9:1054898.
- Yücesoy SN, Uzunçakmak TK, Selçukog?lu Ö, et al. Evaluation of quality of life scores and family impact scales in pediatric patients with alopecia areata: a cross-sectional cohort study. Int J Dermatol. 2024;63:1414-1420.
- Alopecia areata. Society for Pediatric Dermatology. Accessed November 17, 2025. https://pedsderm.net/site/assets/files/18580/spd_school_handout_1_alopecia.pdf
- Liu LY, King BA. Response to tofacitinib therapy of eyebrows and eyelashes in alopecia areata. J Am Acad Dermatol. 2019;80:1778-1779.
- Bokhari L, Sinclair R. Treatment of alopecia universalis with topical Janus kinase inhibitors—a double blind, placebo, and active controlled pilot study. Int J Dermatol. 2018;57:1464-1470.
- Hill ND, Bunata K, Hebert AA. Treatment of alopecia areata with squaric acid dibutylester. Clin Dermatol. 2015;33:300-304.
- Williams KN, Olukoga CTY, Tosti A. Evaluation of the safety and effectiveness of oral minoxidil in children: a systematic review. Dermatol Ther (Heidelb). 2024;14:1709-1727.
- Lemes LR, Melo DF, de Oliveira DS, et al. Topical and oral minoxidil for hair disorders in pediatric patients: what do we know so far? Dermatol Ther. 2020;33:E13950.
- David E, Shokrian N, Del Duca E, et al. Dupilumab induces hair regrowth in pediatric alopecia areata: a real-world, single-center observational study. Arch Dermatol Res. 2024;316:487.
Pediatric alopecia areata (AA) is a chronic autoimmune disease of the hair follicles characterized by nonscarring hair loss. Its incidence in children in the United States ranges from 13.6 to 33.5 per 100,000 person-years, with a prevalence of 0.04% to 0.11%.1 Alopecia areata has important effects on quality of life, particularly in children. Hair loss at an early age can decrease participation in school, sports, and extracurricular activities2 and is associated with increased rates of comorbid anxiety and depression.3 Families also experience psychosocial stress, often comparable to other chronic pediatric illnesses.4 Thus, management requires not only medical therapy but also psychosocial support and school-based accommodations.
Systemic therapies for treatment of AA in adolescents and adults are increasingly available, including US Food and Drug Administration (FDA)–approved Janus kinase (JAK) inhibitors such as baricitinib, deuruxolitinib (for adults), and ritlecitinib (for adolescents and adults); however, no systemic therapies have been approved by the FDA for children younger than 12 years. The therapeutic gap is most acute for those aged 6 to 11 years, for whom the psychosocial burden is high but treatment options are limited.3
This article highlights options and strategies for managing AA in children aged 6 to 11 years, emphasizing supportive and psychosocial care (including camouflage techniques), topical therapies, and off-label systemic approaches.
Supportive and Psychosocial Care
Treatment of AA in children extends beyond the affected child to include parents, caregivers, and even school staff (eg, teachers, principals, nurses).4 Disease-specific organizations such as the National Alopecia Areata Foundation (naaf.org) and the Children’s Alopecia Project (childrensalopeciaproject.org) provide education, support groups, and advocacy resources. These organizations assist families in navigating school accommodations, including Section 504 plans that may allow children with AA to wear hats in school to mitigate stigma. Additional resources include handouts for teachers and school nurses developed by the Society for Pediatric Dermatology.5
Psychological support for these patients is critical. Many children benefit from seeing a psychologist, particularly if anxiety, depression, and/or bullying is present.3 In clinics without embedded psychology services, dermatologists should maintain referral lists or encourage families to seek guidance from their pediatrician.
Camouflage techniques can help children cope with visible hair loss. Wigs and hairpieces are available free of charge through charitable organizations for patients younger than 17; however, young children often find adhesives uncomfortable, and they will not wear nonadherent wigs for long periods of time. Alternatives include soft hats, bonnets, scarves, and beanies. For partial hair loss, root concealers, scalp powders, or hair mascara can be useful. Temporary eyebrow tattoos are a good cosmetic approach, whereas microblading generally is not advised in children younger than 12 due to procedural risks including pain.
Topical Therapies
Topical agents remain the mainstay of treatment for AA in children aged 6 to 11 years. Potent class 1 or class 2 topical corticosteroids commonly are used, sometimes in combination with calcineurin inhibitors or topical minoxidil. Off-label compounded topical JAK inhibitors also have been tried in this population and may be helpful for eyebrow hair loss,6 though data on their efficacy for scalp AA are mixed.7 Intralesional corticosteroid injections, effective in adolescents and adults, generally are poorly tolerated by younger children and may cause considerable distress. Contact immunotherapy with squaric acid dibutyl ester or anthralin can be considered, but these agents are designed to elicit irritation, which may be intolerable for young children.8 Shared decision-making with families is essential to balance efficacy, tolerability, and treatment burden.
Systemic Therapies
Systemic therapy generally is reserved for children with extensive or refractory AA. Low-dose oral minoxidil is emerging as an off-label option. One systematic review reported that low-dose oral minoxidil was well tolerated in pediatric patients with minimal adverse effects.9 Doses of 0.01 to 0.02 mg/kg/d are reasonable starting points, achieved by cutting tablets or compounding oral solutions.10
In children with AA and concurrent atopic dermatitis, dupilumab may offer dual benefit. A real-world observational study demonstrated hair regrowth in pediatric patients with AA treated with dupilumab.11 Immunosuppressive options such as low-dose methotrexate or pulse corticosteroids (dexamethasone or prednisolone) also may be considered, although use of these agents requires careful monitoring due to increased risk for infection, clinically significant blood count and liver enzyme changes, and metabolic adverse effects related to long-term use of corticosteroids.
Clinical trials of JAK inhibitors in children aged 6 to 11 years are anticipated to begin in late 2025. Until then, off-label use of ritlecitinib, baricitinib, tofacitinib, or other JAK inhibitors may be considered in select cases with considerable disease burden and quality-of-life impairment following thorough discussion with the patient and their caregivers. Currently available pediatric data show few serious adverse events in children—the most common included upper respiratory infections (nasopharyngitis), acne, and headaches—but long-term risks remain unknown. Dosing challenges also exist for children who cannot swallow pills; currently ritlecitinib is available only as a capsule that cannot be opened while other JAK inhibitors are available in more accessible forms (baricitinib can be crushed and dissolved, and tofacitinib is available in liquid formulation for other pediatric indications). Insurance coverage is a major barrier, as these therapies are not FDA approved for AA in this age group.
Final Thoughts
Alopecia areata in children aged 6 to 11 years presents unique therapeutic challenges. While highly effective systemic therapies exist for older patients, younger children have limited options. For the 6-to-11 age group, management strategies should prioritize psychosocial support, topical therapy, and low-burden systemic alternatives such as low-dose oral minoxidil. Family education, school-based accommodations, and access to camouflage techniques are integral to holistic care. The commencement of pediatric clinical trials for JAK inhibitors offers hope for more robust treatment strategies in the near future. In the meantime, clinicians must engage in shared decision-making, tailoring therapy to the child’s disease severity, emotional well-being, and family priorities.
Pediatric alopecia areata (AA) is a chronic autoimmune disease of the hair follicles characterized by nonscarring hair loss. Its incidence in children in the United States ranges from 13.6 to 33.5 per 100,000 person-years, with a prevalence of 0.04% to 0.11%.1 Alopecia areata has important effects on quality of life, particularly in children. Hair loss at an early age can decrease participation in school, sports, and extracurricular activities2 and is associated with increased rates of comorbid anxiety and depression.3 Families also experience psychosocial stress, often comparable to other chronic pediatric illnesses.4 Thus, management requires not only medical therapy but also psychosocial support and school-based accommodations.
Systemic therapies for treatment of AA in adolescents and adults are increasingly available, including US Food and Drug Administration (FDA)–approved Janus kinase (JAK) inhibitors such as baricitinib, deuruxolitinib (for adults), and ritlecitinib (for adolescents and adults); however, no systemic therapies have been approved by the FDA for children younger than 12 years. The therapeutic gap is most acute for those aged 6 to 11 years, for whom the psychosocial burden is high but treatment options are limited.3
This article highlights options and strategies for managing AA in children aged 6 to 11 years, emphasizing supportive and psychosocial care (including camouflage techniques), topical therapies, and off-label systemic approaches.
Supportive and Psychosocial Care
Treatment of AA in children extends beyond the affected child to include parents, caregivers, and even school staff (eg, teachers, principals, nurses).4 Disease-specific organizations such as the National Alopecia Areata Foundation (naaf.org) and the Children’s Alopecia Project (childrensalopeciaproject.org) provide education, support groups, and advocacy resources. These organizations assist families in navigating school accommodations, including Section 504 plans that may allow children with AA to wear hats in school to mitigate stigma. Additional resources include handouts for teachers and school nurses developed by the Society for Pediatric Dermatology.5
Psychological support for these patients is critical. Many children benefit from seeing a psychologist, particularly if anxiety, depression, and/or bullying is present.3 In clinics without embedded psychology services, dermatologists should maintain referral lists or encourage families to seek guidance from their pediatrician.
Camouflage techniques can help children cope with visible hair loss. Wigs and hairpieces are available free of charge through charitable organizations for patients younger than 17; however, young children often find adhesives uncomfortable, and they will not wear nonadherent wigs for long periods of time. Alternatives include soft hats, bonnets, scarves, and beanies. For partial hair loss, root concealers, scalp powders, or hair mascara can be useful. Temporary eyebrow tattoos are a good cosmetic approach, whereas microblading generally is not advised in children younger than 12 due to procedural risks including pain.
Topical Therapies
Topical agents remain the mainstay of treatment for AA in children aged 6 to 11 years. Potent class 1 or class 2 topical corticosteroids commonly are used, sometimes in combination with calcineurin inhibitors or topical minoxidil. Off-label compounded topical JAK inhibitors also have been tried in this population and may be helpful for eyebrow hair loss,6 though data on their efficacy for scalp AA are mixed.7 Intralesional corticosteroid injections, effective in adolescents and adults, generally are poorly tolerated by younger children and may cause considerable distress. Contact immunotherapy with squaric acid dibutyl ester or anthralin can be considered, but these agents are designed to elicit irritation, which may be intolerable for young children.8 Shared decision-making with families is essential to balance efficacy, tolerability, and treatment burden.
Systemic Therapies
Systemic therapy generally is reserved for children with extensive or refractory AA. Low-dose oral minoxidil is emerging as an off-label option. One systematic review reported that low-dose oral minoxidil was well tolerated in pediatric patients with minimal adverse effects.9 Doses of 0.01 to 0.02 mg/kg/d are reasonable starting points, achieved by cutting tablets or compounding oral solutions.10
In children with AA and concurrent atopic dermatitis, dupilumab may offer dual benefit. A real-world observational study demonstrated hair regrowth in pediatric patients with AA treated with dupilumab.11 Immunosuppressive options such as low-dose methotrexate or pulse corticosteroids (dexamethasone or prednisolone) also may be considered, although use of these agents requires careful monitoring due to increased risk for infection, clinically significant blood count and liver enzyme changes, and metabolic adverse effects related to long-term use of corticosteroids.
Clinical trials of JAK inhibitors in children aged 6 to 11 years are anticipated to begin in late 2025. Until then, off-label use of ritlecitinib, baricitinib, tofacitinib, or other JAK inhibitors may be considered in select cases with considerable disease burden and quality-of-life impairment following thorough discussion with the patient and their caregivers. Currently available pediatric data show few serious adverse events in children—the most common included upper respiratory infections (nasopharyngitis), acne, and headaches—but long-term risks remain unknown. Dosing challenges also exist for children who cannot swallow pills; currently ritlecitinib is available only as a capsule that cannot be opened while other JAK inhibitors are available in more accessible forms (baricitinib can be crushed and dissolved, and tofacitinib is available in liquid formulation for other pediatric indications). Insurance coverage is a major barrier, as these therapies are not FDA approved for AA in this age group.
Final Thoughts
Alopecia areata in children aged 6 to 11 years presents unique therapeutic challenges. While highly effective systemic therapies exist for older patients, younger children have limited options. For the 6-to-11 age group, management strategies should prioritize psychosocial support, topical therapy, and low-burden systemic alternatives such as low-dose oral minoxidil. Family education, school-based accommodations, and access to camouflage techniques are integral to holistic care. The commencement of pediatric clinical trials for JAK inhibitors offers hope for more robust treatment strategies in the near future. In the meantime, clinicians must engage in shared decision-making, tailoring therapy to the child’s disease severity, emotional well-being, and family priorities.
- Adhanom R, Ansbro B, Castelo-Soccio L. Epidemiology of pediatric alopecia areata. Pediatr Dermatol. 2025;42(suppl 1):12-23. doi:10.1111/pde.15803
- Paller AS, Rangel SM, Chamlin SL, et al; Pediatric Dermatology Research Alliance. Stigmatization and mental health impact of chronic pediatric skin disorders. JAMA Dermatol. 2024;160:621-630.
- van Dalen M, Muller KS, Kasperkovitz-Oosterloo JM, et al. Anxiety, depression, and quality of life in children and adults with alopecia areata: systematic review and meta-analysis. Front Med (Lausanne). 2022;9:1054898.
- Yücesoy SN, Uzunçakmak TK, Selçukog?lu Ö, et al. Evaluation of quality of life scores and family impact scales in pediatric patients with alopecia areata: a cross-sectional cohort study. Int J Dermatol. 2024;63:1414-1420.
- Alopecia areata. Society for Pediatric Dermatology. Accessed November 17, 2025. https://pedsderm.net/site/assets/files/18580/spd_school_handout_1_alopecia.pdf
- Liu LY, King BA. Response to tofacitinib therapy of eyebrows and eyelashes in alopecia areata. J Am Acad Dermatol. 2019;80:1778-1779.
- Bokhari L, Sinclair R. Treatment of alopecia universalis with topical Janus kinase inhibitors—a double blind, placebo, and active controlled pilot study. Int J Dermatol. 2018;57:1464-1470.
- Hill ND, Bunata K, Hebert AA. Treatment of alopecia areata with squaric acid dibutylester. Clin Dermatol. 2015;33:300-304.
- Williams KN, Olukoga CTY, Tosti A. Evaluation of the safety and effectiveness of oral minoxidil in children: a systematic review. Dermatol Ther (Heidelb). 2024;14:1709-1727.
- Lemes LR, Melo DF, de Oliveira DS, et al. Topical and oral minoxidil for hair disorders in pediatric patients: what do we know so far? Dermatol Ther. 2020;33:E13950.
- David E, Shokrian N, Del Duca E, et al. Dupilumab induces hair regrowth in pediatric alopecia areata: a real-world, single-center observational study. Arch Dermatol Res. 2024;316:487.
- Adhanom R, Ansbro B, Castelo-Soccio L. Epidemiology of pediatric alopecia areata. Pediatr Dermatol. 2025;42(suppl 1):12-23. doi:10.1111/pde.15803
- Paller AS, Rangel SM, Chamlin SL, et al; Pediatric Dermatology Research Alliance. Stigmatization and mental health impact of chronic pediatric skin disorders. JAMA Dermatol. 2024;160:621-630.
- van Dalen M, Muller KS, Kasperkovitz-Oosterloo JM, et al. Anxiety, depression, and quality of life in children and adults with alopecia areata: systematic review and meta-analysis. Front Med (Lausanne). 2022;9:1054898.
- Yücesoy SN, Uzunçakmak TK, Selçukog?lu Ö, et al. Evaluation of quality of life scores and family impact scales in pediatric patients with alopecia areata: a cross-sectional cohort study. Int J Dermatol. 2024;63:1414-1420.
- Alopecia areata. Society for Pediatric Dermatology. Accessed November 17, 2025. https://pedsderm.net/site/assets/files/18580/spd_school_handout_1_alopecia.pdf
- Liu LY, King BA. Response to tofacitinib therapy of eyebrows and eyelashes in alopecia areata. J Am Acad Dermatol. 2019;80:1778-1779.
- Bokhari L, Sinclair R. Treatment of alopecia universalis with topical Janus kinase inhibitors—a double blind, placebo, and active controlled pilot study. Int J Dermatol. 2018;57:1464-1470.
- Hill ND, Bunata K, Hebert AA. Treatment of alopecia areata with squaric acid dibutylester. Clin Dermatol. 2015;33:300-304.
- Williams KN, Olukoga CTY, Tosti A. Evaluation of the safety and effectiveness of oral minoxidil in children: a systematic review. Dermatol Ther (Heidelb). 2024;14:1709-1727.
- Lemes LR, Melo DF, de Oliveira DS, et al. Topical and oral minoxidil for hair disorders in pediatric patients: what do we know so far? Dermatol Ther. 2020;33:E13950.
- David E, Shokrian N, Del Duca E, et al. Dupilumab induces hair regrowth in pediatric alopecia areata: a real-world, single-center observational study. Arch Dermatol Res. 2024;316:487.
Therapeutic Approaches for Alopecia Areata in Children Aged 6 to 11 Years
Therapeutic Approaches for Alopecia Areata in Children Aged 6 to 11 Years

Solitary Yellow Papule on the Upper Back in an Infant
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
A 6-month-old male infant with a history of cradle cap and an infantile hemangioma on the left shoulder presented to the dermatology clinic for evaluation of a slow-growing yellow papule on the upper back of 3 months’ duration. The lesion initially was noted 2 months prior to the current presentation by the patient’s pediatrician, who recommended follow-up with dermatology after an unsuccessful attempt at incision and drainage. Physical examination revealed a 7-mm, yellow, dome-shaped papule with a red collarette on the right upper back. No axillary freckling, ocular findings, or other skin findings were found. The patient was born at term with no complications, and his mother reported that he was otherwise healthy. There were no developmental concerns or known allergies, and his family history was negative for any similar lesions. Dermoscopic examination of the lesion revealed a well-circumscribed, circular, yellow-orange papule with an erythematous border and setting-sun appearance.
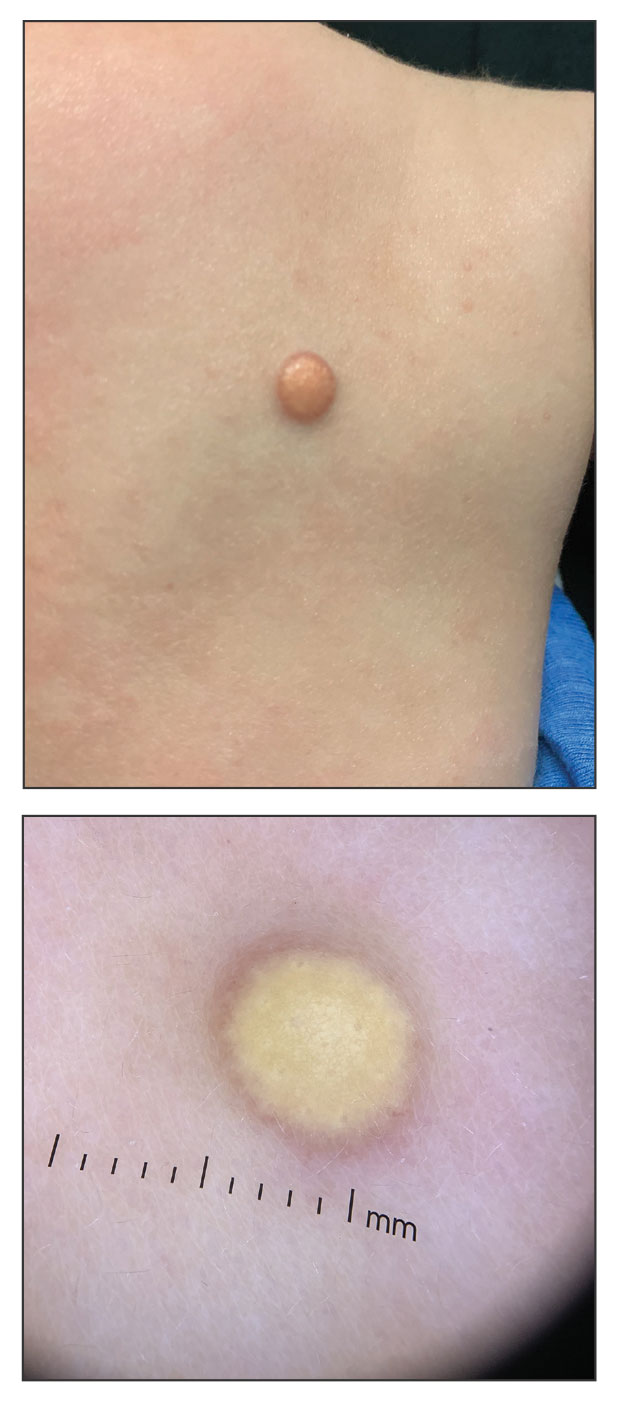
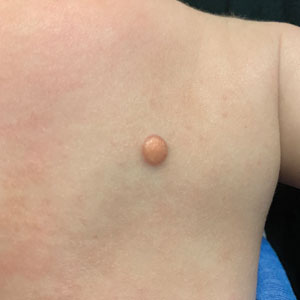
Reticulated Hyperpigmentation on the Knee and Thigh
Reticulated Hyperpigmentation on the Knee and Thigh
The patient was diagnosed with erythema ab igne based on characteristic skin findings on physical examination along with a convincing history of chronic localized heat exposure. Erythema ab igne manifests as a persistent reticulated, erythematous, or hyperpigmented rash at sites of chronic heat exposure.1 Commonplace items that emit heat such as electric heaters, car heaters, heating pads, hot water bottles, and, in our case, laptops also emit infrared radiation, which can lead to changes in the skin with long-term exposure.2 Because exposure to these sources often is limited to one area of the body, erythema ab igne usually manifests locally, as exemplified in this case. Chronic heat exposure and infrared radiation from these sources are thought to induce hyperthermia below the threshold for a thermal burn, and the cutaneous findings correspond with the dermal venous plexus.3
Diagnosis of erythema ab igne primarily is made clinically based on characteristic skin findings and exposure history. Relevant history may include occupations with prolonged heat exposure, such as baking, silversmithing, or foundry work. Heat exposure also may result from cultural practices such as cupping with moxibustion.4 Additionally, repeated use of heating pads or hot water bottles for pain relief by patients diagnosed with chronic pain or an underlying illness may contribute to development of erythema ab igne.1,4
Biopsy was not needed for diagnosis of this patient, but if the presentation is equivocal and history of potential exposures is unclear, a biopsy may be taken. A hematoxylin and eosin stain would reveal dilation of small vascular channels in the superficial dermis, contributing to the classic reticulated appearance. Biopsy findings also would reveal either an interface dermatitis or pigment incontinence containing melanin-laden macrophages correlating to either the erythema or hyperpigmentation, respectively.4
The prognosis for erythema ab igne is excellent, especially if diagnosed early. Treatment involves removal of the inciting heat source.1 The discoloration may resolve within a few months to years or may persist. If the hyperpigmentation is persistent, patients may consider laser treatments or lightening agents such as topical hydroquinone or topical tretinoin.4 However, if undiagnosed, patients may be at risk for development of a cutaneous malignancy, such as squamous cell carcinoma, Merkel cell carcinoma, poorly differentiated carcinoma, or cutaneous marginal zone lymphoma.2,4 Malignant transformation has been reported to occur decades after the initial skin eruption, although the risk is rare5; however, due to this risk, patients with erythema ab igne should be followed regularly and screened for new lesions in the affected areas.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kesty K, Feldman SR. Erythema ab igne: evolving technology, evolving presentation. Dermatol Online J. 2014;20:13030.
- Harview CL, Krenitsky A. Erythema ab igne: a clinical review. Cutis. 2023;111:E33-E38. doi:10.12788/cutis.0771
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
The patient was diagnosed with erythema ab igne based on characteristic skin findings on physical examination along with a convincing history of chronic localized heat exposure. Erythema ab igne manifests as a persistent reticulated, erythematous, or hyperpigmented rash at sites of chronic heat exposure.1 Commonplace items that emit heat such as electric heaters, car heaters, heating pads, hot water bottles, and, in our case, laptops also emit infrared radiation, which can lead to changes in the skin with long-term exposure.2 Because exposure to these sources often is limited to one area of the body, erythema ab igne usually manifests locally, as exemplified in this case. Chronic heat exposure and infrared radiation from these sources are thought to induce hyperthermia below the threshold for a thermal burn, and the cutaneous findings correspond with the dermal venous plexus.3
Diagnosis of erythema ab igne primarily is made clinically based on characteristic skin findings and exposure history. Relevant history may include occupations with prolonged heat exposure, such as baking, silversmithing, or foundry work. Heat exposure also may result from cultural practices such as cupping with moxibustion.4 Additionally, repeated use of heating pads or hot water bottles for pain relief by patients diagnosed with chronic pain or an underlying illness may contribute to development of erythema ab igne.1,4
Biopsy was not needed for diagnosis of this patient, but if the presentation is equivocal and history of potential exposures is unclear, a biopsy may be taken. A hematoxylin and eosin stain would reveal dilation of small vascular channels in the superficial dermis, contributing to the classic reticulated appearance. Biopsy findings also would reveal either an interface dermatitis or pigment incontinence containing melanin-laden macrophages correlating to either the erythema or hyperpigmentation, respectively.4
The prognosis for erythema ab igne is excellent, especially if diagnosed early. Treatment involves removal of the inciting heat source.1 The discoloration may resolve within a few months to years or may persist. If the hyperpigmentation is persistent, patients may consider laser treatments or lightening agents such as topical hydroquinone or topical tretinoin.4 However, if undiagnosed, patients may be at risk for development of a cutaneous malignancy, such as squamous cell carcinoma, Merkel cell carcinoma, poorly differentiated carcinoma, or cutaneous marginal zone lymphoma.2,4 Malignant transformation has been reported to occur decades after the initial skin eruption, although the risk is rare5; however, due to this risk, patients with erythema ab igne should be followed regularly and screened for new lesions in the affected areas.
The patient was diagnosed with erythema ab igne based on characteristic skin findings on physical examination along with a convincing history of chronic localized heat exposure. Erythema ab igne manifests as a persistent reticulated, erythematous, or hyperpigmented rash at sites of chronic heat exposure.1 Commonplace items that emit heat such as electric heaters, car heaters, heating pads, hot water bottles, and, in our case, laptops also emit infrared radiation, which can lead to changes in the skin with long-term exposure.2 Because exposure to these sources often is limited to one area of the body, erythema ab igne usually manifests locally, as exemplified in this case. Chronic heat exposure and infrared radiation from these sources are thought to induce hyperthermia below the threshold for a thermal burn, and the cutaneous findings correspond with the dermal venous plexus.3
Diagnosis of erythema ab igne primarily is made clinically based on characteristic skin findings and exposure history. Relevant history may include occupations with prolonged heat exposure, such as baking, silversmithing, or foundry work. Heat exposure also may result from cultural practices such as cupping with moxibustion.4 Additionally, repeated use of heating pads or hot water bottles for pain relief by patients diagnosed with chronic pain or an underlying illness may contribute to development of erythema ab igne.1,4
Biopsy was not needed for diagnosis of this patient, but if the presentation is equivocal and history of potential exposures is unclear, a biopsy may be taken. A hematoxylin and eosin stain would reveal dilation of small vascular channels in the superficial dermis, contributing to the classic reticulated appearance. Biopsy findings also would reveal either an interface dermatitis or pigment incontinence containing melanin-laden macrophages correlating to either the erythema or hyperpigmentation, respectively.4
The prognosis for erythema ab igne is excellent, especially if diagnosed early. Treatment involves removal of the inciting heat source.1 The discoloration may resolve within a few months to years or may persist. If the hyperpigmentation is persistent, patients may consider laser treatments or lightening agents such as topical hydroquinone or topical tretinoin.4 However, if undiagnosed, patients may be at risk for development of a cutaneous malignancy, such as squamous cell carcinoma, Merkel cell carcinoma, poorly differentiated carcinoma, or cutaneous marginal zone lymphoma.2,4 Malignant transformation has been reported to occur decades after the initial skin eruption, although the risk is rare5; however, due to this risk, patients with erythema ab igne should be followed regularly and screened for new lesions in the affected areas.
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kesty K, Feldman SR. Erythema ab igne: evolving technology, evolving presentation. Dermatol Online J. 2014;20:13030.
- Harview CL, Krenitsky A. Erythema ab igne: a clinical review. Cutis. 2023;111:E33-E38. doi:10.12788/cutis.0771
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Tan S, Bertucci V. Erythema ab igne: an old condition new again. CMAJ. 2000;162:77-78.
- Miller K, Hunt R, Chu J, et al. Erythema ab igne. Dermatol Online J. 2011;17:28.
- Kesty K, Feldman SR. Erythema ab igne: evolving technology, evolving presentation. Dermatol Online J. 2014;20:13030.
- Harview CL, Krenitsky A. Erythema ab igne: a clinical review. Cutis. 2023;111:E33-E38. doi:10.12788/cutis.0771
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
Reticulated Hyperpigmentation on the Knee and Thigh
Reticulated Hyperpigmentation on the Knee and Thigh
A 25-year-old woman with an unremarkable medical history presented to the dermatology clinic for evaluation of a persistent rash on the right knee and distal thigh of several months’ duration. The patient noted that the rash had been asymptomatic, and she denied any history of trauma to the area. She reported that she worked as a teacher and had repeatedly stayed up late using her laptop for months. Rather than use a desk, she often would work sitting with her laptop in her lap.