User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
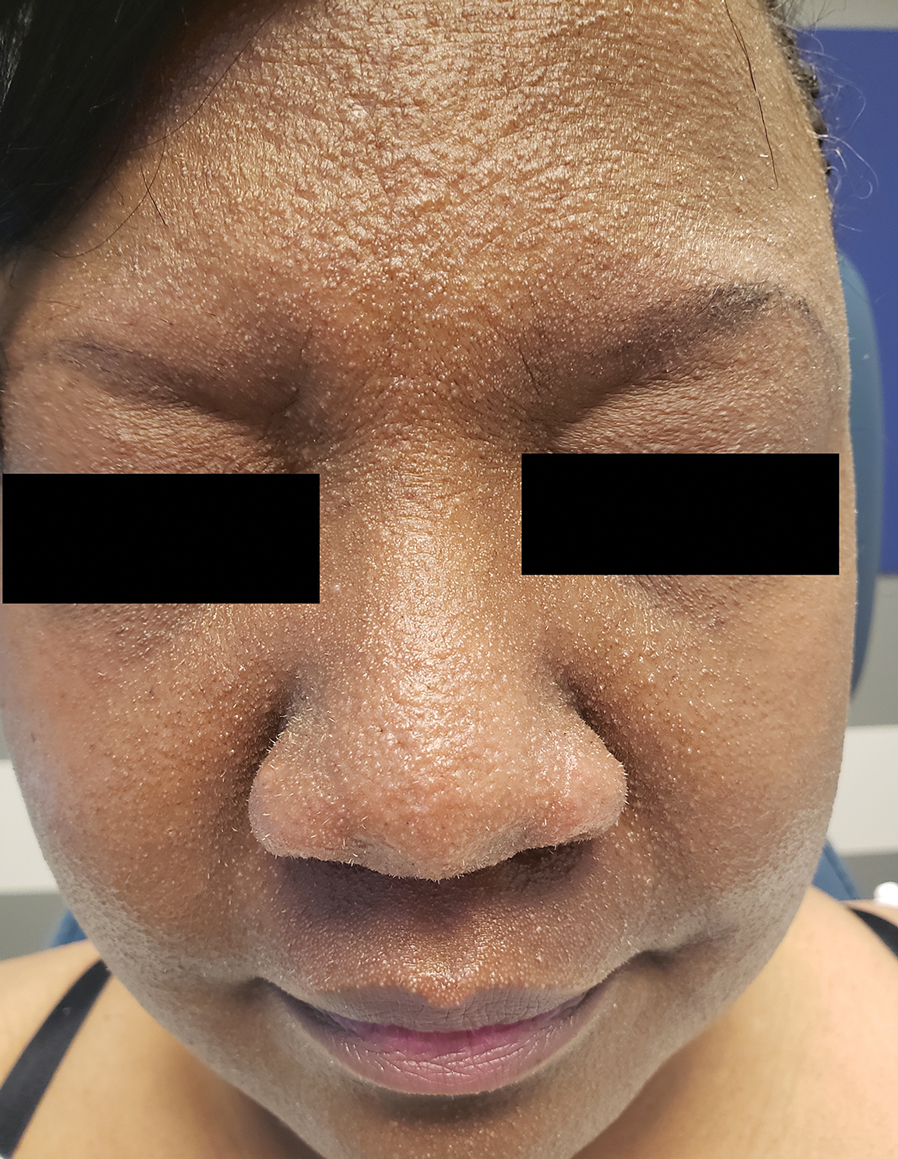
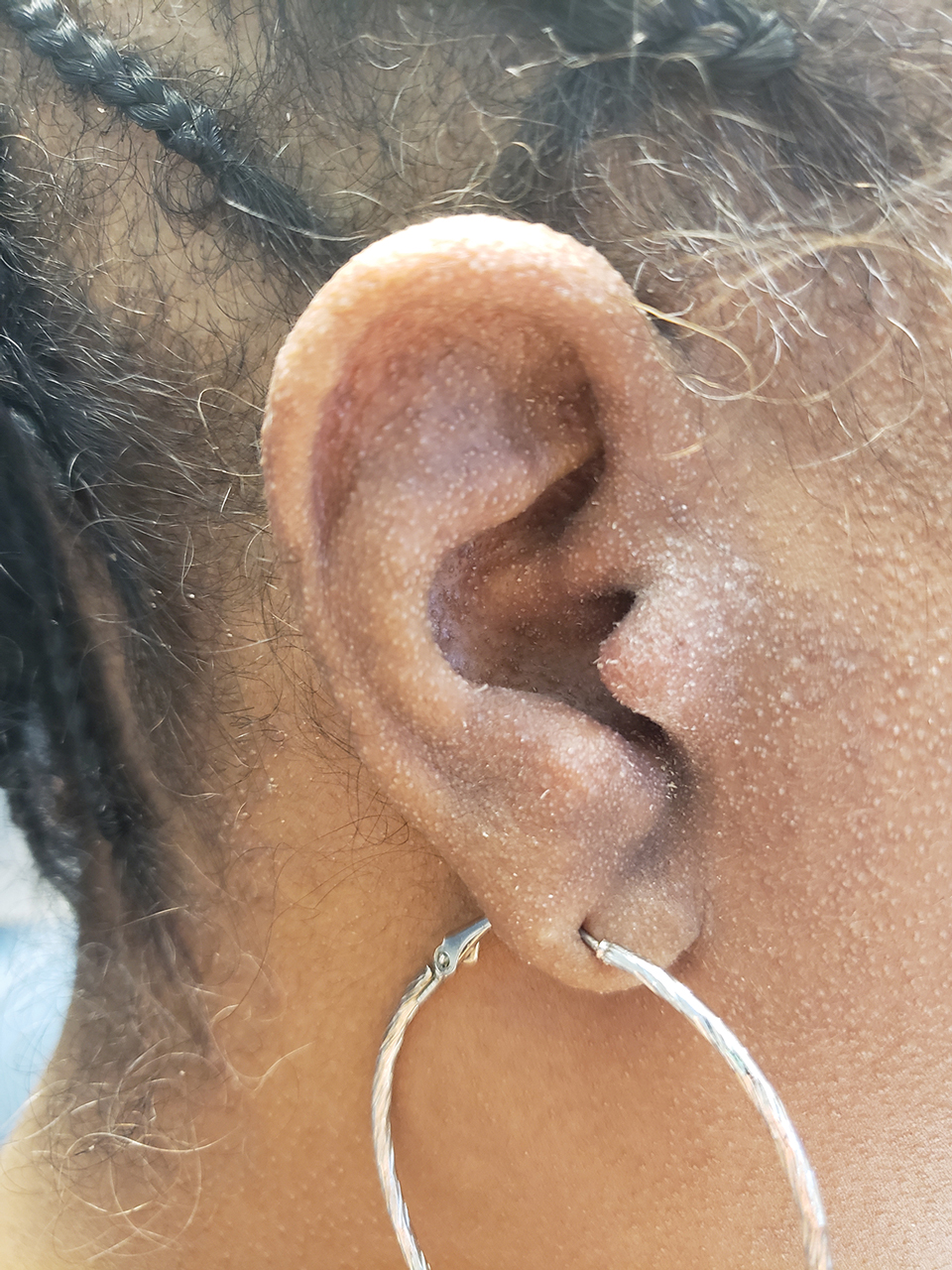
Flesh-Colored Pinpoint Papules With Fine White Spicules on the Upper Body
The Diagnosis: Trichodysplasia Spinulosa
A diagnosis of trichodysplasia spinulosa (TS) was rendered based on the clinical presentation— diffuse folliculocentric keratotic papules with spicules and leonine facies—coinciding with cyclosporine initiation. Biopsy was deferred given the classic presentation. The patient applied cidofovir cream 1% daily to lesions on the face. She was prescribed leflunomide 10 mg daily, which was later increased to 20 mg daily, for polyarthritis associated with systemic lupus erythematosus (SLE). Her transplant physician increased her cyclosporine dosage from 50 mg twice daily to 75 mg each morning and 50 mg each evening due to rising creatinine and donor-specific antibodies from the renal transplant. The patient’s TS eruption mildly improved 3 months after the cyclosporine dose was increased. To treat persistent lesions, oral valganciclovir was started at 450 mg once daily and later reduced to every other day due to leukopenia. After 3 months of taking valganciclovir 450 mg every other day, the patient’s TS rash resolved.
Trichodysplasia spinulosa is a rare condition caused by TS-associated polyomavirus1 that may arise in immunosuppressed patients, especially in solid organ transplant recipients.2 It is characterized by spiculated and folliculocentric papules, mainly on the face,1 and often is diagnosed clinically, but if the presentation is not classic, a skin biopsy can help to confirm the diagnosis. Because of its rarity, treatment options do not have well-established efficacy1 but include reducing immunosuppression and using the antivirals cidofovir1 or valganciclovir3 to treat the polyomavirus. Topical retinoids,3 photodynamic therapy, 4 and leflunomide5 also may be effective.
Although the typical approach to treating TS is to reduce immunosuppression, this was not an option for our patient, as she required increased immunosuppression for the treatment of active SLE. Leflunomide can be used for SLE, and in some reports it can be effective for BK viremia in kidney transplant recipients5 as well as for TS in solid organ transplant recipients.6 Our patient showed improvement of the TS, BK viremia, renal function, and SLE while taking leflunomide and valganciclovir.
The differential diagnosis includes keratosis pilaris, lichen nitidus, scleromyxedema, and trichostasis spinulosa. Keratosis pilaris is a benign skin disorder consisting of patches of keratotic papules with varying degrees of erythema and inflammation that are formed by dead keratinocytes plugging the hair follicles and often are seen on the extremities, face, and trunk.7 Our patient’s papules were flesh colored with no notable background erythema. Additionally, the presence of leonine facies was atypical for keratosis pilaris. Acids, steroids, and kinase inhibitors are the most frequently used treatments for keratosis pilaris.8
Lichen nitidus is a skin condition characterized by multiple shiny, dome-shaped, flesh-colored papules usually found on the flexor surfaces of the arms, anterior trunk, and genitalia. It is mostly asymptomatic, but patients may experience pruritus. Most cases occur in children and young adults, with no obvious racial or gender predilection. The diagnosis often is clinical, but biopsy shows downward enlargement of the epidermal rete ridges surrounding a focal inflammatory infiltrate, known as a ball-in-claw configuration.9-11 Lichen nitidus spontaneously resolves within a few years without treatment. Our patient did have flesh-colored papules on the arms and chest; however, major involvement of the face is not typical in lichen nitidus. Additionally, fine white spicules would not be seen in lichen nitidus. For severe generalized lichen nitidus, treatment options include topical corticosteroids, topical calcineurin inhibitors, oral antihistamines, or UV light to decrease inflammation.9-11
Scleromyxedema is a rare condition involving the deposition of mucinous material in the papillary dermis to cause the formation of infiltrative skin lesions.12 It is thought that immunoglobulins and cytokines secreted by inflammatory cells lead to the synthesis of glycosaminoglycans, which then causes deposition of mucin in the dermis.13 The classic cutaneous features of scleromyxedema include waxy indurated papules and plaques with skin thickening throughout the entire body.12 Our patient’s papules were not notably indurated and involved less than 50% of the total body surface area. An important diagnostic feature of scleromyxedema is monoclonal gammopathy, which our patient did not have. Intravenous immunoglobulin is the first-line treatment of scleromyxedema, and second-line treatments include systemic corticosteroids and thalidomide.14 Our patient also did not require treatment with intravenous immunoglobulin, as her rash improved with antiviral medication, which would not address the underlying inflammatory processes associated with scleromyxedema.
Trichostasis spinulosa is a rare hair follicle disorder consisting of dark, spiny, hyperkeratotic follicular papules that can be found on the extremities and face, especially the nose. The etiology is unknown, but risk factors include congenital dysplasia of hair follicles; exposure to UV light, dust, oil, or heat; chronic renal failure; Malassezia yeast; and Propionibacterium acnes. Adult women with darker skin types are most commonly affected by trichostasis spinulosa.15,16 Our patient fit the epidemiologic demographic of trichostasis spinulosa, including a history of chronic renal failure. Her rash covered the face, nose, and arms; however, the papules were flesh colored, whereas trichostasis spinulosa would appear as black papules. Furthermore, yeast and bacterial infections have been identified as potential agents associated with trichostasis spinulosa; therefore, antiviral agents would be ineffective. Viable treatments for trichostasis spinulosa include emollients, topical keratolytic agents, retinoic acids, and lasers to remove abnormal hair follicles.15,16
- Curman P, Näsman A, Brauner H. Trichodysplasia spinulosa: a comprehensive disease and its treatment. J Eur Acad Dermatol Venereol. 2021;35:1067-1076.
- Fischer MK, Kao GF, Nguyen HP, et al. Specific detection of trichodysplasia spinulosa-associated polyomavirus DNA in skin and renal allograft tissues in a patient with trichodysplasia spinulosa. Arch Dermatol. 2021;148:726-733.
- Shah PR, Esaa FS, Gupta P, et al. Trichodysplasia spinulosa successfully treated with adapalene 0.1% gel and oral valganciclovir in a renal transplant recipient. JAAD Case Rep. 2020;6:23-25.
- Liew YCC, Kee TYS, Kwek JL, et al. Photodynamic therapy for the treatment of trichodysplasia spinulosa in an Asian renal transplant recipient: a case report and review of the literature. JAAD Case Rep. 2021;7:74-83.
- Pierrotti LC, Urbano PRP, da Silva Nali LH, et al. Viremia and viuria of trichodysplasia spinulosa-associated polyomavirus before the development of clinical disease in a kidney transplant recipient. Transpl Infect Dis. 2019;21:E13133.
- Kassar R, Chang J, Chan AW, et al. Leflunomide for the treatment of trichodysplasia spinulosa in a liver transplant recipient. Transpl Infect Dis. 2017;19:E12702.
- Eckburg A, Kazemi T, Maguiness S. Keratosis pilaris rubra successfully treated with topical sirolimus: report of a case and review of the literature. Pediatr Dermatol. 2022;39:429-431.
- Reddy S, Brahmbhatt H. A narrative review on the role of acids, steroids, and kinase inhibitors in the treatment of keratosis pilaris. Cureus. 2021;13:E18917.
- Jordan AS, Green MC, Sulit DJ. Lichen nitidus. J Am Osteopath Assoc. 2019;119:704.
- Arizaga AT, Gaughan MD, Bang RH. Generalized lichen nitidus. Clin Exp Dermatol. 2002;27:115-117.
- Chu J, Lam JM. Lichen nitidus. CMAJ. 2014;186:E688.
- Haber R, Bachour J, El Gemayel M. Scleromyxedema treatment: a systematic review and update. Int J Dermatol. 2020;59:1191-1201.
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosis (LM) (discrete papular type). Dermatol Online J. 2017;23:8.
- Hoffman JHO, Enk AH. Scleromyxedema. J Dtsch Dermatol Ges. 2020;18:1449-1467.
- Kositkuljorn C, Suchonwanit P. Trichostasis spinulosa: a case report with an unusual presentation. Case Rep Dermatol. 2020;12:178-185.
- Ramteke MN, Bhide AA. Trichostasis spinulosa at an unusual site. Int J Trichology. 2016;8:78-80.
The Diagnosis: Trichodysplasia Spinulosa
A diagnosis of trichodysplasia spinulosa (TS) was rendered based on the clinical presentation— diffuse folliculocentric keratotic papules with spicules and leonine facies—coinciding with cyclosporine initiation. Biopsy was deferred given the classic presentation. The patient applied cidofovir cream 1% daily to lesions on the face. She was prescribed leflunomide 10 mg daily, which was later increased to 20 mg daily, for polyarthritis associated with systemic lupus erythematosus (SLE). Her transplant physician increased her cyclosporine dosage from 50 mg twice daily to 75 mg each morning and 50 mg each evening due to rising creatinine and donor-specific antibodies from the renal transplant. The patient’s TS eruption mildly improved 3 months after the cyclosporine dose was increased. To treat persistent lesions, oral valganciclovir was started at 450 mg once daily and later reduced to every other day due to leukopenia. After 3 months of taking valganciclovir 450 mg every other day, the patient’s TS rash resolved.
Trichodysplasia spinulosa is a rare condition caused by TS-associated polyomavirus1 that may arise in immunosuppressed patients, especially in solid organ transplant recipients.2 It is characterized by spiculated and folliculocentric papules, mainly on the face,1 and often is diagnosed clinically, but if the presentation is not classic, a skin biopsy can help to confirm the diagnosis. Because of its rarity, treatment options do not have well-established efficacy1 but include reducing immunosuppression and using the antivirals cidofovir1 or valganciclovir3 to treat the polyomavirus. Topical retinoids,3 photodynamic therapy, 4 and leflunomide5 also may be effective.
Although the typical approach to treating TS is to reduce immunosuppression, this was not an option for our patient, as she required increased immunosuppression for the treatment of active SLE. Leflunomide can be used for SLE, and in some reports it can be effective for BK viremia in kidney transplant recipients5 as well as for TS in solid organ transplant recipients.6 Our patient showed improvement of the TS, BK viremia, renal function, and SLE while taking leflunomide and valganciclovir.
The differential diagnosis includes keratosis pilaris, lichen nitidus, scleromyxedema, and trichostasis spinulosa. Keratosis pilaris is a benign skin disorder consisting of patches of keratotic papules with varying degrees of erythema and inflammation that are formed by dead keratinocytes plugging the hair follicles and often are seen on the extremities, face, and trunk.7 Our patient’s papules were flesh colored with no notable background erythema. Additionally, the presence of leonine facies was atypical for keratosis pilaris. Acids, steroids, and kinase inhibitors are the most frequently used treatments for keratosis pilaris.8
Lichen nitidus is a skin condition characterized by multiple shiny, dome-shaped, flesh-colored papules usually found on the flexor surfaces of the arms, anterior trunk, and genitalia. It is mostly asymptomatic, but patients may experience pruritus. Most cases occur in children and young adults, with no obvious racial or gender predilection. The diagnosis often is clinical, but biopsy shows downward enlargement of the epidermal rete ridges surrounding a focal inflammatory infiltrate, known as a ball-in-claw configuration.9-11 Lichen nitidus spontaneously resolves within a few years without treatment. Our patient did have flesh-colored papules on the arms and chest; however, major involvement of the face is not typical in lichen nitidus. Additionally, fine white spicules would not be seen in lichen nitidus. For severe generalized lichen nitidus, treatment options include topical corticosteroids, topical calcineurin inhibitors, oral antihistamines, or UV light to decrease inflammation.9-11
Scleromyxedema is a rare condition involving the deposition of mucinous material in the papillary dermis to cause the formation of infiltrative skin lesions.12 It is thought that immunoglobulins and cytokines secreted by inflammatory cells lead to the synthesis of glycosaminoglycans, which then causes deposition of mucin in the dermis.13 The classic cutaneous features of scleromyxedema include waxy indurated papules and plaques with skin thickening throughout the entire body.12 Our patient’s papules were not notably indurated and involved less than 50% of the total body surface area. An important diagnostic feature of scleromyxedema is monoclonal gammopathy, which our patient did not have. Intravenous immunoglobulin is the first-line treatment of scleromyxedema, and second-line treatments include systemic corticosteroids and thalidomide.14 Our patient also did not require treatment with intravenous immunoglobulin, as her rash improved with antiviral medication, which would not address the underlying inflammatory processes associated with scleromyxedema.
Trichostasis spinulosa is a rare hair follicle disorder consisting of dark, spiny, hyperkeratotic follicular papules that can be found on the extremities and face, especially the nose. The etiology is unknown, but risk factors include congenital dysplasia of hair follicles; exposure to UV light, dust, oil, or heat; chronic renal failure; Malassezia yeast; and Propionibacterium acnes. Adult women with darker skin types are most commonly affected by trichostasis spinulosa.15,16 Our patient fit the epidemiologic demographic of trichostasis spinulosa, including a history of chronic renal failure. Her rash covered the face, nose, and arms; however, the papules were flesh colored, whereas trichostasis spinulosa would appear as black papules. Furthermore, yeast and bacterial infections have been identified as potential agents associated with trichostasis spinulosa; therefore, antiviral agents would be ineffective. Viable treatments for trichostasis spinulosa include emollients, topical keratolytic agents, retinoic acids, and lasers to remove abnormal hair follicles.15,16
The Diagnosis: Trichodysplasia Spinulosa
A diagnosis of trichodysplasia spinulosa (TS) was rendered based on the clinical presentation— diffuse folliculocentric keratotic papules with spicules and leonine facies—coinciding with cyclosporine initiation. Biopsy was deferred given the classic presentation. The patient applied cidofovir cream 1% daily to lesions on the face. She was prescribed leflunomide 10 mg daily, which was later increased to 20 mg daily, for polyarthritis associated with systemic lupus erythematosus (SLE). Her transplant physician increased her cyclosporine dosage from 50 mg twice daily to 75 mg each morning and 50 mg each evening due to rising creatinine and donor-specific antibodies from the renal transplant. The patient’s TS eruption mildly improved 3 months after the cyclosporine dose was increased. To treat persistent lesions, oral valganciclovir was started at 450 mg once daily and later reduced to every other day due to leukopenia. After 3 months of taking valganciclovir 450 mg every other day, the patient’s TS rash resolved.
Trichodysplasia spinulosa is a rare condition caused by TS-associated polyomavirus1 that may arise in immunosuppressed patients, especially in solid organ transplant recipients.2 It is characterized by spiculated and folliculocentric papules, mainly on the face,1 and often is diagnosed clinically, but if the presentation is not classic, a skin biopsy can help to confirm the diagnosis. Because of its rarity, treatment options do not have well-established efficacy1 but include reducing immunosuppression and using the antivirals cidofovir1 or valganciclovir3 to treat the polyomavirus. Topical retinoids,3 photodynamic therapy, 4 and leflunomide5 also may be effective.
Although the typical approach to treating TS is to reduce immunosuppression, this was not an option for our patient, as she required increased immunosuppression for the treatment of active SLE. Leflunomide can be used for SLE, and in some reports it can be effective for BK viremia in kidney transplant recipients5 as well as for TS in solid organ transplant recipients.6 Our patient showed improvement of the TS, BK viremia, renal function, and SLE while taking leflunomide and valganciclovir.
The differential diagnosis includes keratosis pilaris, lichen nitidus, scleromyxedema, and trichostasis spinulosa. Keratosis pilaris is a benign skin disorder consisting of patches of keratotic papules with varying degrees of erythema and inflammation that are formed by dead keratinocytes plugging the hair follicles and often are seen on the extremities, face, and trunk.7 Our patient’s papules were flesh colored with no notable background erythema. Additionally, the presence of leonine facies was atypical for keratosis pilaris. Acids, steroids, and kinase inhibitors are the most frequently used treatments for keratosis pilaris.8
Lichen nitidus is a skin condition characterized by multiple shiny, dome-shaped, flesh-colored papules usually found on the flexor surfaces of the arms, anterior trunk, and genitalia. It is mostly asymptomatic, but patients may experience pruritus. Most cases occur in children and young adults, with no obvious racial or gender predilection. The diagnosis often is clinical, but biopsy shows downward enlargement of the epidermal rete ridges surrounding a focal inflammatory infiltrate, known as a ball-in-claw configuration.9-11 Lichen nitidus spontaneously resolves within a few years without treatment. Our patient did have flesh-colored papules on the arms and chest; however, major involvement of the face is not typical in lichen nitidus. Additionally, fine white spicules would not be seen in lichen nitidus. For severe generalized lichen nitidus, treatment options include topical corticosteroids, topical calcineurin inhibitors, oral antihistamines, or UV light to decrease inflammation.9-11
Scleromyxedema is a rare condition involving the deposition of mucinous material in the papillary dermis to cause the formation of infiltrative skin lesions.12 It is thought that immunoglobulins and cytokines secreted by inflammatory cells lead to the synthesis of glycosaminoglycans, which then causes deposition of mucin in the dermis.13 The classic cutaneous features of scleromyxedema include waxy indurated papules and plaques with skin thickening throughout the entire body.12 Our patient’s papules were not notably indurated and involved less than 50% of the total body surface area. An important diagnostic feature of scleromyxedema is monoclonal gammopathy, which our patient did not have. Intravenous immunoglobulin is the first-line treatment of scleromyxedema, and second-line treatments include systemic corticosteroids and thalidomide.14 Our patient also did not require treatment with intravenous immunoglobulin, as her rash improved with antiviral medication, which would not address the underlying inflammatory processes associated with scleromyxedema.
Trichostasis spinulosa is a rare hair follicle disorder consisting of dark, spiny, hyperkeratotic follicular papules that can be found on the extremities and face, especially the nose. The etiology is unknown, but risk factors include congenital dysplasia of hair follicles; exposure to UV light, dust, oil, or heat; chronic renal failure; Malassezia yeast; and Propionibacterium acnes. Adult women with darker skin types are most commonly affected by trichostasis spinulosa.15,16 Our patient fit the epidemiologic demographic of trichostasis spinulosa, including a history of chronic renal failure. Her rash covered the face, nose, and arms; however, the papules were flesh colored, whereas trichostasis spinulosa would appear as black papules. Furthermore, yeast and bacterial infections have been identified as potential agents associated with trichostasis spinulosa; therefore, antiviral agents would be ineffective. Viable treatments for trichostasis spinulosa include emollients, topical keratolytic agents, retinoic acids, and lasers to remove abnormal hair follicles.15,16
- Curman P, Näsman A, Brauner H. Trichodysplasia spinulosa: a comprehensive disease and its treatment. J Eur Acad Dermatol Venereol. 2021;35:1067-1076.
- Fischer MK, Kao GF, Nguyen HP, et al. Specific detection of trichodysplasia spinulosa-associated polyomavirus DNA in skin and renal allograft tissues in a patient with trichodysplasia spinulosa. Arch Dermatol. 2021;148:726-733.
- Shah PR, Esaa FS, Gupta P, et al. Trichodysplasia spinulosa successfully treated with adapalene 0.1% gel and oral valganciclovir in a renal transplant recipient. JAAD Case Rep. 2020;6:23-25.
- Liew YCC, Kee TYS, Kwek JL, et al. Photodynamic therapy for the treatment of trichodysplasia spinulosa in an Asian renal transplant recipient: a case report and review of the literature. JAAD Case Rep. 2021;7:74-83.
- Pierrotti LC, Urbano PRP, da Silva Nali LH, et al. Viremia and viuria of trichodysplasia spinulosa-associated polyomavirus before the development of clinical disease in a kidney transplant recipient. Transpl Infect Dis. 2019;21:E13133.
- Kassar R, Chang J, Chan AW, et al. Leflunomide for the treatment of trichodysplasia spinulosa in a liver transplant recipient. Transpl Infect Dis. 2017;19:E12702.
- Eckburg A, Kazemi T, Maguiness S. Keratosis pilaris rubra successfully treated with topical sirolimus: report of a case and review of the literature. Pediatr Dermatol. 2022;39:429-431.
- Reddy S, Brahmbhatt H. A narrative review on the role of acids, steroids, and kinase inhibitors in the treatment of keratosis pilaris. Cureus. 2021;13:E18917.
- Jordan AS, Green MC, Sulit DJ. Lichen nitidus. J Am Osteopath Assoc. 2019;119:704.
- Arizaga AT, Gaughan MD, Bang RH. Generalized lichen nitidus. Clin Exp Dermatol. 2002;27:115-117.
- Chu J, Lam JM. Lichen nitidus. CMAJ. 2014;186:E688.
- Haber R, Bachour J, El Gemayel M. Scleromyxedema treatment: a systematic review and update. Int J Dermatol. 2020;59:1191-1201.
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosis (LM) (discrete papular type). Dermatol Online J. 2017;23:8.
- Hoffman JHO, Enk AH. Scleromyxedema. J Dtsch Dermatol Ges. 2020;18:1449-1467.
- Kositkuljorn C, Suchonwanit P. Trichostasis spinulosa: a case report with an unusual presentation. Case Rep Dermatol. 2020;12:178-185.
- Ramteke MN, Bhide AA. Trichostasis spinulosa at an unusual site. Int J Trichology. 2016;8:78-80.
- Curman P, Näsman A, Brauner H. Trichodysplasia spinulosa: a comprehensive disease and its treatment. J Eur Acad Dermatol Venereol. 2021;35:1067-1076.
- Fischer MK, Kao GF, Nguyen HP, et al. Specific detection of trichodysplasia spinulosa-associated polyomavirus DNA in skin and renal allograft tissues in a patient with trichodysplasia spinulosa. Arch Dermatol. 2021;148:726-733.
- Shah PR, Esaa FS, Gupta P, et al. Trichodysplasia spinulosa successfully treated with adapalene 0.1% gel and oral valganciclovir in a renal transplant recipient. JAAD Case Rep. 2020;6:23-25.
- Liew YCC, Kee TYS, Kwek JL, et al. Photodynamic therapy for the treatment of trichodysplasia spinulosa in an Asian renal transplant recipient: a case report and review of the literature. JAAD Case Rep. 2021;7:74-83.
- Pierrotti LC, Urbano PRP, da Silva Nali LH, et al. Viremia and viuria of trichodysplasia spinulosa-associated polyomavirus before the development of clinical disease in a kidney transplant recipient. Transpl Infect Dis. 2019;21:E13133.
- Kassar R, Chang J, Chan AW, et al. Leflunomide for the treatment of trichodysplasia spinulosa in a liver transplant recipient. Transpl Infect Dis. 2017;19:E12702.
- Eckburg A, Kazemi T, Maguiness S. Keratosis pilaris rubra successfully treated with topical sirolimus: report of a case and review of the literature. Pediatr Dermatol. 2022;39:429-431.
- Reddy S, Brahmbhatt H. A narrative review on the role of acids, steroids, and kinase inhibitors in the treatment of keratosis pilaris. Cureus. 2021;13:E18917.
- Jordan AS, Green MC, Sulit DJ. Lichen nitidus. J Am Osteopath Assoc. 2019;119:704.
- Arizaga AT, Gaughan MD, Bang RH. Generalized lichen nitidus. Clin Exp Dermatol. 2002;27:115-117.
- Chu J, Lam JM. Lichen nitidus. CMAJ. 2014;186:E688.
- Haber R, Bachour J, El Gemayel M. Scleromyxedema treatment: a systematic review and update. Int J Dermatol. 2020;59:1191-1201.
- Christman MP, Sukhdeo K, Kim RH, et al. Papular mucinosis, or localized lichen myxedematosis (LM) (discrete papular type). Dermatol Online J. 2017;23:8.
- Hoffman JHO, Enk AH. Scleromyxedema. J Dtsch Dermatol Ges. 2020;18:1449-1467.
- Kositkuljorn C, Suchonwanit P. Trichostasis spinulosa: a case report with an unusual presentation. Case Rep Dermatol. 2020;12:178-185.
- Ramteke MN, Bhide AA. Trichostasis spinulosa at an unusual site. Int J Trichology. 2016;8:78-80.
A 54-year-old Black woman presented with a rash that developed 6 months after a renal transplant due to a history of systemic lupus erythematosus with lupus nephritis. She was started on mycophenolate mofetil and tacrolimus after the transplant but was switched to cyclosporine because of BK viremia. The rash developed 1 week after cyclosporine was initiated and consisted of pruritic papules that started on the face and spread to the trunk and arms. Physical examination revealed innumerable follicular-based, keratotic, flesh-colored, pinpoint papules with fine white spicules on the face (top), neck, chest, arms, and back. Leonine facies was seen along the glabella with madarosis of the lateral eyebrows (top) and ears (bottom).
Inpatient Management of Hidradenitis Suppurativa: A Delphi Consensus Study
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition that affects approximately 0.1% of the US population.1,2 Severe disease or HS flares can lead patients to seek care through the emergency department (ED), with some requiring inpatient admission. 3 Inpatient hospitalization of patients with HS has increased over the last 2 decades, and patients with HS utilize emergency and inpatient care more frequently than those with other dermatologic conditions.4,5 Minority patients and those of lower socioeconomic status are more likely to present to the ED for HS management due to limited access to care and other existing comorbid conditions. 4 In a 2022 study of the Nationwide Readmissions Database, the authors looked at hospital readmission rates of patients with HS compared with those with heart failure—both patient populations with chronic debilitating conditions. Results indicated that the hospital readmission rates for patients with HS surpassed those of patients with heart failure for that year, highlighting the need for improved inpatient management of HS.6
Patients with HS present to the ED with severe pain, fever, wound care, or the need for surgical intervention. The ED and inpatient hospital setting are locations in which physicians may not be as familiar with the diagnosis or treatment of HS, specifically flares or severe disease. 7 The inpatient care setting provides access to certain resources that can be challenging to obtain in the outpatient clinical setting, such as social workers and pain specialists, but also can prove challenging in obtaining other resources for HS management, such as advanced medical therapies. Given the increase in hospital- based care for HS and lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial. In our study, we sought to generate a collection of expert consensus statements providers can refer to when managing patients with HS in the inpatient setting.
Methods
The study team at the Wake Forest University School of Medicine (Winston-Salem, North Carolina)(M.N., R.P., L.C.S.) developed an initial set of consensus statements based on current published HS treatment guidelines,8,9 publications on management of inpatient HS,3 published supportive care guidelines for Stevens-Johnson syndrome, 10 and personal clinical experience in managing inpatient HS, which resulted in 50 statements organized into the following categories: overall care, wound care, genital care, pain management, infection control, medical management, surgical management, nutrition, and transitional care guidelines. This study was approved by the Wake Forest University institutional review board (IRB00084257).
Participant Recruitment—Dermatologists were identified for participation in the study based on membership in the Society of Dermatology Hospitalists and the Hidradenitis Suppurativa Foundation or authorship of publications relevant to HS or inpatient dermatology. Dermatologists from larger academic institutions with HS specialty clinics and inpatient dermatology services also were identified. Participants were invited via email and could suggest other experts for inclusion. A total of 31 dermatologists were invited to participate in the study, with 26 agreeing to participate. All participating dermatologists were practicing in the United States.
Delphi Study—In the first round of the Delphi study, the participants were sent an online survey via REDCap in which they were asked to rank the appropriateness of each of the proposed 50 guideline statements on a scale of 1 (very inappropriate) to 9 (very appropriate). Participants also were able to provide commentary and feedback on each of the statements. Survey results were analyzed using the RAND/ UCLA Appropriateness Method.11 For each statement, the median rating for appropriateness, interpercentile range (IPR), IPR adjusted for symmetry, and disagreement index (DI) were calculated (DI=IPR/IPR adjusted for symmetry). The 30th and 70th percentiles were used in the DI calculation as the upper and lower limits, respectively. A median rating for appropriateness of 1.0 to 3.9 was considered “inappropriate,” 4.0 to 6.9 was considered “uncertain appropriateness,” and 7.0 to 9.0 was “appropriate.” A DI value greater than or equal to 1 indicated a lack of consensus regarding the appropriateness of the statement. Following each round, participants received a copy of their responses along with the group median rank of each statement. Statements that did not reach consensus in the first Delphi round were revised based on feedback received by the participants, and a second survey with 14 statements was sent via REDCap 2 weeks later. The RAND/UCLA Appropriateness Method also was applied to this second Delphi round. After the second survey, participants received a copy of anonymized comments regarding the consensus statements and were allowed to provide additional final commentary to be included in the discussion of these recommendations.
Results
Twenty-six dermatologists completed the first-round survey, and 24 participants completed the second-round survey. All participants self-identified as having expertise in either HS (n=22 [85%]) or inpatient dermatology (n=17 [65%]), and 13 (50%) participants self-identified as experts in both HS and inpatient dermatology. All participants, except 1, were affiliated with an academic health system with inpatient dermatology services. The average length of time in practice as a dermatologist was 10 years (median, 9 years [range, 3–27 years]).
Of the 50 initial proposed consensus statements, 26 (52%) achieved consensus after the first round; 21 statements revealed DI calculations that did not achieve consensus. Two statements achieved consensus but received median ratings for appropriateness, indicating uncertain appropriateness; because of this, 1 statement was removed and 1 was revised based on participant feedback, resulting in 13 revised statements (eTable 1). Controversial topics in the consensus process included obtaining wound cultures and meaningful culture data interpretation, use of specific biologic medications in the inpatient setting, and use of intravenous ertapenem. Participant responses to these topics are discussed in detail below. Of these secondround statements, all achieved consensus. The final set of consensus statements can be found in eTable 2.
Comment
Our Delphi consensus study combined the expertise of both dermatologists who care for patients with HS and those with inpatient dermatology experience to produce a set of recommendations for the management of HS in the hospital care setting. A strength of this study is inclusion of many national leaders in both HS and inpatient dermatology, with some participants having developed the previously published HS treatment guidelines and others having participated in inpatient dermatology Delphi studies.8-10 The expertise is further strengthened by the geographically diverse institutional representation within the United States.
The final consensus recommendations included 40 statements covering a range of patient care issues, including use of appropriate inpatient subspecialists (care team), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition back to outpatient management (transitional care). These recommendations are meant to serve as a resource for providers to consider when taking care of inpatient HS flares, recognizing that the complexity and individual circumstances of each patient are unique.
Delphi Consensus Recommendations Compared to Prior Guidelines—Several recommendations in the current study align with the previously published North American clinical management guidelines for HS.8,9 Our recommendations agree with prior guidelines on the importance of disease staging and pain assessment using validated assessment tools as well as screening for HS comorbidities. There also is agreement in the potential benefit of involving pain specialists in the development of a comprehensive pain management plan. The inpatient care setting provides a unique opportunity to engage multiple specialists and collaborate on patient care in a timely manner. Our recommendations regarding surgical care also align with established guidelines in recommending incision and drainage as an acute bedside procedure best utilized for symptom relief in inflamed abscesses and relegating most other surgical management to the outpatient setting. Wound care recommendations also are similar, with our expert participants agreeing on individualizing dressing choices based on wound characteristics. A benefit of inpatient wound care is access to skilled nursing for dressing changes and potentially improved access to more sophisticated dressing materials. Our recommendations differ from the prior guidelines in our focus on severe HS, HS flares, and HS complications, which constitute the majority of inpatient disease management. We provide additional guidance on management of secondary infections, perianal fistulous disease, and importantly transitional care to optimize discharge planning.
Differing Opinions in Our Analysis—Despite the success of our Delphi consensus process, there were some differing opinions regarding certain aspects of inpatient HS management, which is to be expected given the lack of strong evidence-based research to support some of the recommended practices. There were differing opinions on the utility of wound culture data, with some participants feeling culture data could help with antibiotic susceptibility and resistance patterns, while others felt wound cultures represent bacterial colonization or biofilm formation.
Initial consensus statements in the first Delphi round were created for individual biologic medications but did not achieve consensus, and feedback on the use of biologics in the inpatient environment was mixed, largely due to logistic and insurance issues. Many participants felt biologic medication cost, difficulty obtaining inpatient reimbursement, health care resource utilization, and availability of biologics in different hospital systems prevented recommending the use of specific biologics during hospitalization. The one exception was in the case of a hospitalized patient who was already receiving infliximab for HS: there was consensus on ensuring the patient dosing was maximized, if appropriate, to 10 mg/kg.12 Ertapenem use also was controversial, with some participants using it as a bridge therapy to either outpatient biologic use or surgery, while others felt it was onerous and difficult to establish reliable access to secure intravenous administration and regular dosing once the patient left the inpatient setting.13 Others said they have experienced objections from infectious disease colleagues on the use of intravenous antibiotics, citing antibiotic stewardship concerns.
Patient Care in the Inpatient Setting—Prior literature suggests patients admitted as inpatients for HS tend to be of lower socioeconomic status and are admitted to larger urban teaching hospitals.14,15 Patients with lower socioeconomic status have increased difficulty accessing health care resources; therefore, inpatient admission serves as an opportunity to provide a holistic HS assessment and coordinate resources for chronic outpatient management.
Study Limitations—This Delphi consensus study has some limitations. The existing literature on inpatient management of HS is limited, challenging our ability to assess the extent to which these published recommendations are already being implemented. Additionally, the study included HS and inpatient dermatology experts from the United States, which means the recommendations may not be generalizable to other countries. Most participants practiced dermatology at large tertiary care academic medical centers, which may limit the ability to implement recommendations in all US inpatient care settings such as small community-based hospitals; however, many of the supportive care guidelines such as pain control, wound care, nutritional support, and social work should be achievable in most inpatient care settings.
Conclusion
Given the increase in inpatient and ED health care utilization for HS, there is an urgent need for expert consensus recommendations on inpatient management of this unique patient population, which requires complex multidisciplinary care. Our recommendations are a resource for providers to utilize and potentially improve the standard of care we provide these patients.
Acknowledgment—We thank the Wake Forest University Clinical and Translational Science Institute (Winston- Salem, North Carolina) for providing statistical help.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764.
- Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183:990-998. doi:10.1111/bjd.19435
- Charrow A, Savage KT, Flood K, et al. Hidradenitis suppurativa for the dermatologic hospitalist. Cutis. 2019;104:276-280.
- Anzaldi L, Perkins JA, Byrd AS, et al. Characterizing inpatient hospitalizations for hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2020;82:510-513. doi:10.1016/j.jaad.2019.09.019
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614. doi:10.1016/j.jaad.2015.06.053
- Edigin E, Kaul S, Eseaton PO, et al. At 180 days hidradenitis suppurativa readmission rate is comparable to heart failure: analysis of the nationwide readmissions database. J Am Acad Dermatol. 2022;87:188-192. doi:10.1016/j.jaad.2021.06.894
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944. doi:10.1001/jamadermatol.2014.691
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j .jaad.2019.02.067
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101. doi:10.1016/j.jaad.2019.02.068
- Seminario-Vidal L, Kroshinsky D, Malachowski SJ, et al. Society of Dermatology Hospitalists supportive care guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults. J Am Acad Dermatol. 2020;82:1553-1567. doi:10.1016/j .jaad.2020.02.066
- Fitch K, Bernstein SJ, Burnand B, et al. The RAND/UCLA Appropriateness Method: User’s Manual. Rand; 2001.
- Oskardmay AN, Miles JA, Sayed CJ. Determining the optimal dose of infliximab for treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;81:702-708. doi:10.1016/j.jaad.2019.05.022
- Join-Lambert O, Coignard-Biehler H, Jais JP, et al. Efficacy of ertapenem in severe hidradenitis suppurativa: a pilot study in a cohort of 30 consecutive patients. J Antimicrob Chemother. 2016;71:513-520. doi:10.1093/jac/dkv361
- Khanna R, Whang KA, Huang AH, et al. Inpatient burden of hidradenitis suppurativa in the United States: analysis of the 2016 National Inpatient Sample. J Dermatolog Treat. 2022;33:1150-1152. doi:10.1080/09 546634.2020.1773380
- Patel A, Patel A, Solanki D, et al. Hidradenitis suppurativa in the United States: insights from the national inpatient sample (2008-2017) on contemporary trends in demographics, hospitalization rates, chronic comorbid conditions, and mortality. Cureus. 2022;14:E24755. doi:10.7759/cureus.24755
Practice Points
- Given the increase in hospital-based care for hidradenitis suppurativa (HS) and the lack of widespread inpatient access to dermatology and HS experts, consensus recommendations for management of HS in the acute hospital setting would be beneficial.
- Our Delphi study yielded 40 statements that reached consensus covering a range of patient care issues (eg, appropriate inpatient subspecialists [care team]), supportive care measures (wound care, pain control, genital care), disease-oriented treatment (medical management, surgical management), inpatient complications (infection control, nutrition), and successful transition to outpatient management (transitional care).
- These recommendations serve as an important resource for providers caring for inpatients with HS and represent a successful collaboration between inpatient dermatology and HS experts.
Reticulated Brownish Erythema on the Lower Back
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
A 42-year-old woman presented with an asymptomatic, erythematous, lacelike rash on the lower back of 8 months’ duration that was first noticed by her husband. The patient had a long-standing history of chronic fatigue and lower back pain treated with acetaminophen, diclofenac gel, and heating pads. Physical examination revealed reticulated brownish erythema confined to the lower back. Laboratory findings were unremarkable.
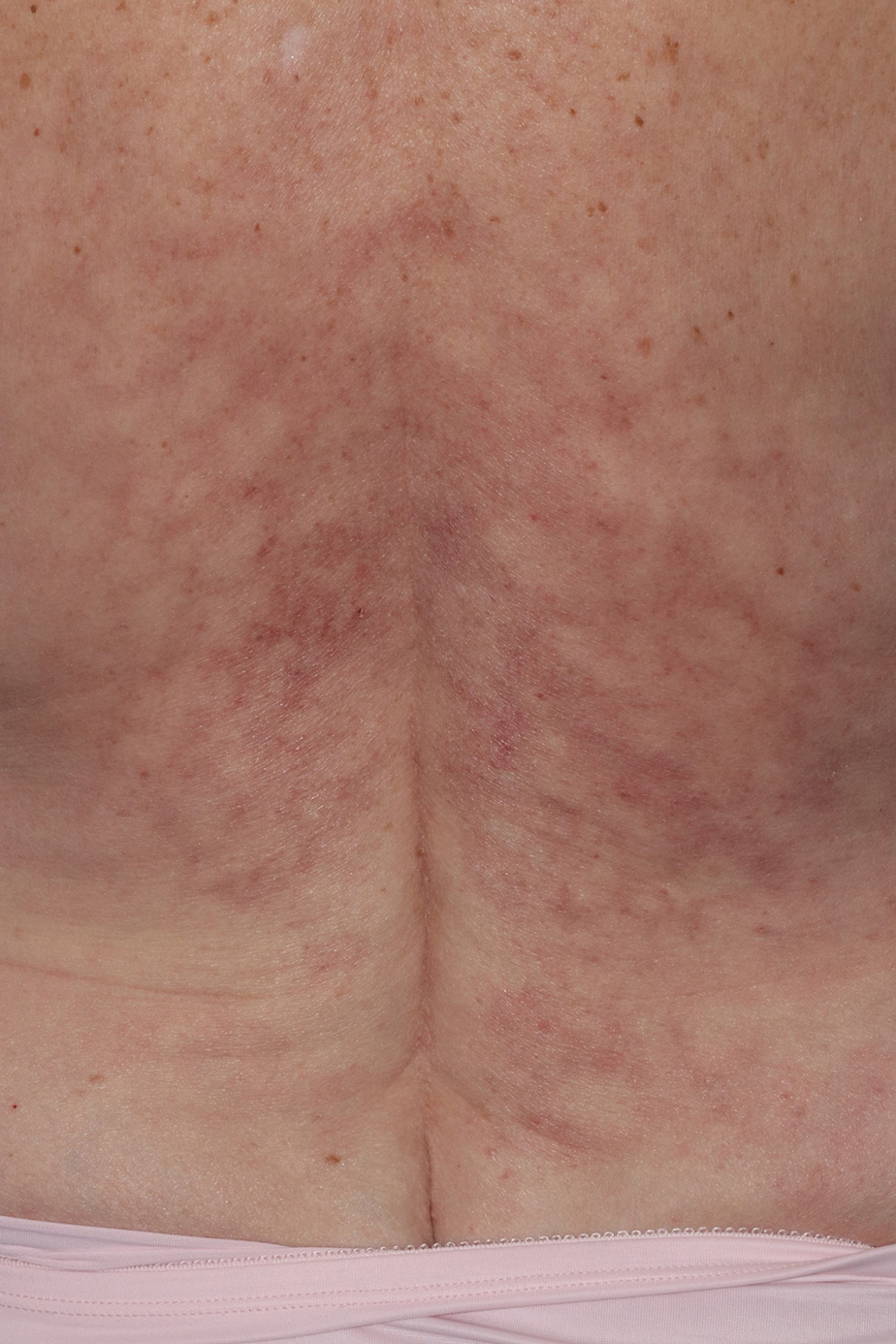
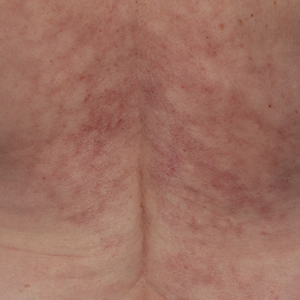
Plantar Hyperpigmentation
The Comparison
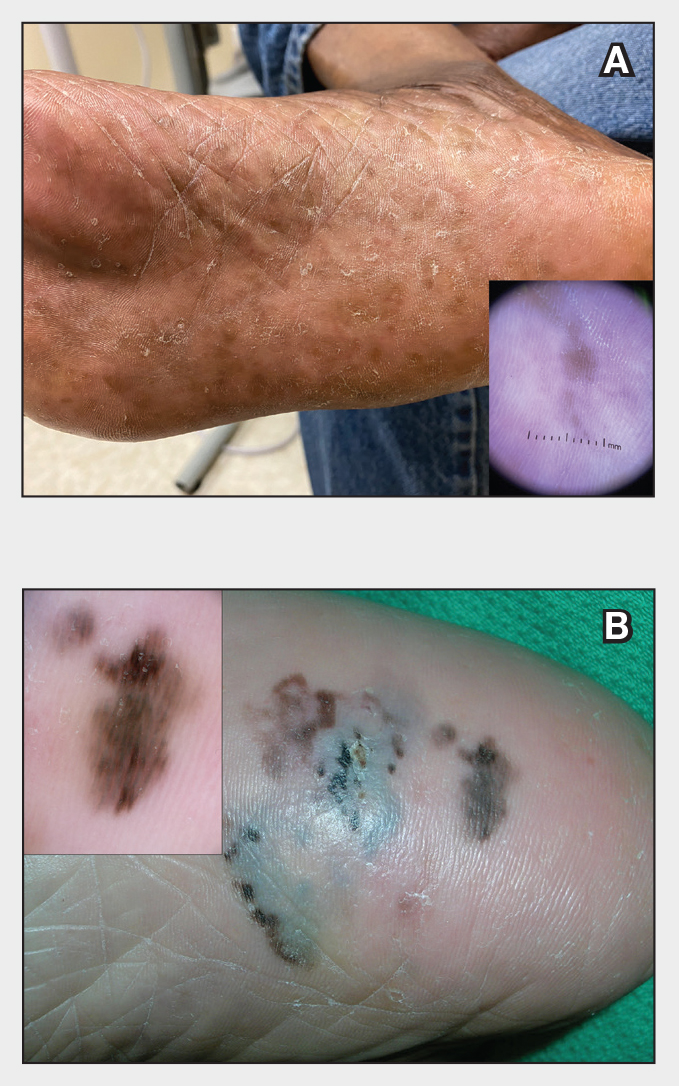
Plantar hyperpigmentation (also known as plantar melanosis [increased melanin], volar pigmented macules, benign racial melanosis, acral pigmentation, acral ethnic melanosis, or mottled hyperpigmentation of the plantar surface) is a benign finding in many individuals and is especially prevalent in those with darker skin tones. Acral refers to manifestation on the hands and feet, volar on the palms and soles, and plantar on the soles only. Here, we focus on plantar hyperpigmentation. We use the terms ethnic and racial interchangeably.
It is critically important to differentiate benign hyperpigmentation, which is common in patients with skin of color, from melanoma. Although rare, Black patients in the United States experience high morbidity and mortality from acral melanoma, which often is diagnosed late in the disease course.1
There are many causes of hyperpigmentation on the plantar surfaces, including benign ethnic melanosis, nevi, melanoma, infections such as syphilis and tinea nigra, conditions such as Peutz-Jeghers syndrome and Laugier-Hunziker syndrome, and postinflammatory hyperpigmentation secondary to atopic dermatitis and psoriasis. We focus on the most common causes, ethnic melanosis and nevi, as well as melanoma, which is the deadliest cause.
Epidemiology
In a 1980 study (N=251), Black Americans had a high incidence of plantar hyperpigmentation, with 52% of affected patients having dark brown skin and 31% having light brown skin.2
The epidemiology of melanoma varies by race/ethnicity. Melanoma in Black individuals is relatively rare, with an annual incidence of approximately 1 in 100,000 individuals.3 However, when individuals with skin of color develop melanoma, they are more likely than their White counterparts to have acral melanoma (acral lentiginous melanoma), one of the deadliest types.1 In a case series of Black patients with melanoma (N=48) from 2 tertiary care centers in Texas, 30 of 40 primary cutaneous melanomas (75%) were located on acral skin.4 Overall, 13 patients developed stage IV disease and 12 died due to disease progression. All patients who developed distant metastases or died of melanoma had acral melanoma.4 Individuals of Asian descent also have a high incidence of acral melanoma, as shown in research from Japan.5-9
Key clinical features in individuals with darker skin tones
Dermoscopy is an evidence-based clinical examination method for earlier diagnosis of cutaneous melanoma, including on acral skin.10,11 Benign nevi on the volar skin as well as the palms and soles tend to have one of these 3 dermoscopic patterns: parallel furrow, lattice, or irregular fibrillar. The pattern that is most predictive of volar melanoma is the parallel ridge pattern (PRP) (Figures A and B [insets]), which showed a high specificity (99.0%) and very high negative predictive value (97.7%) for malignant melanoma in a Japanese population.7 The PRP data from this study cannot be applied reliably to Black individuals, especially because benign ethnic melanosis and other benign conditions can demonstrate PRP.12 Reliance on the PRP as a diagnostic clue could result in unneccessary biopsies in as many as 50% of Black patients with benign plantar hyperpigmentation.2 Furthermore, biopsies of the plantar surface can be painful and cause pain while walking.
It has been suggested that PRP seen on dermoscopy in benign hyperpigmentation such as ethnic melanosis and nevi may preserve the acrosyringia (eccrine gland openings on the ridge), whereas PRP in melanoma may obliterate the acrosyringia.13 This observation is based on case reports only and needs further study. However, if validated, it could be a useful diagnostic clue.
Worth noting
In a retrospective cohort study of skin cancer in Black individuals (n=165) at a New York City–based cancer center from 2000 to 2020, 68% of patients were diagnosed with melanomas—80% were the acral subtype and 75% displayed a PRP. However, the surrounding uninvolved background skin, which was visible in most cases, also demonstrated a PRP.14 Because of the high morbidity and mortality rates of acral melanoma, clinicians should biopsy or immediately refer patients with concerning plantar hyperpigmentation to a dermatologist.
Health disparity highlight
The mortality rate for acral melanoma in Black patients is disproportionately high for the following reasons15,16:
- Patients and health care providers do not expect to see melanoma in Black patients (it truly is rare!), so screening and education on sun protection are limited.
- Benign ethnic melanosis makes it more difficult to distinguish between early acral melanoma and benign skin changes.
- Black patients and other US patient populations with skin of color may be less likely to have health insurance, which contributes to inequities in access to health care. As of 2022, the uninsured rates for nonelderly American Indian and Alaska Native, Hispanic, Native Hawaiian and Other Pacific Islander, Black, and White individuals were 19.1%, 18.0%, 12.7%, 10.0%, and 6.6%, respectively.17
Multi-institutional registries could improve understanding of acral melanoma in Black patients.4 More studies are needed to help differentiate between the dermoscopic finding of PRP in benign ethnic melanosis vs malignant melanoma.
- Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015: an analysis of the SEER registry. J Surg Res. 2020;251:329-339. doi:10.1016/j.jss.2020.02.010
- Coleman WP, Gately LE, Krementz AB, et al. Nevi, lentigines, and melanomas in blacks. Arch Dermatol. 1980;116:548-551.
- Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States: 2012-2016. USCS Data Brief, no. 9. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no9-melanoma-incidence-mortality-UnitedStates-2012-2016.htm
- Wix SN, Brown AB, Heberton M, et al. Clinical features and outcomes of black patients with melanoma. JAMA Dermatol. 2024;160:328-333. doi:10.1001/jamadermatol.2023.5789
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426. doi:10.1001/archderm.143.11.1423
- Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34. doi:10.1111/j.1346-8138.2010.01174.x
- Saida T, Miyazaki A, Oguchi S. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238. doi:10.1001/archderm.140.10.1233
- Saida T, Koga H, Uhara H. Dermoscopy for acral melanocytic lesions: revision of the 3-step algorithm and refined definition of the regular and irregular fibrillar pattern. Dermatol Pract Concept. 2022;12:e2022123. doi:10.5826/dpc.1203a123
- Heath CR, Usatine RP. Melanoma. Cutis. 2022;109:284-285.doi:10.12788/cutis.0513.
- Dinnes J, Deeks JJ, Chuchu N, et al; Cochrane Skin Cancer Diagnostic Test Accuracy Group. Visual inspection and dermoscopy, alone or in combination, for diagnosing keratinocyte skin cancers in adults. Cochrane Database Syst Rev. 2018; 12:CD011901. doi:10.1002/14651858.CD011901.pub2
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked-eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676. doi:10.1111/j.1365-2133.2008.08713.x
- Phan A, Dalle S, Marcilly MC, et al. Benign dermoscopic parallel ridge pattern variants. Arch Dermatol. 2011;147:634. doi:10.1001/archdermatol.2011.47
- Fracaroli TS, Lavorato FG, Maceira JP, et al. Parallel ridge pattern on dermoscopy: observation in non-melanoma cases. An Bras Dermatol. 2013;88:646-648. doi:10.1590/abd1806-4841.20132058
- Manci RN, Dauscher M, Marchetti MA, et al. Features of skin cancer in black individuals: a single-institution retrospective cohort study. Dermatol Pract Concept. 2022;12:e2022075. doi:10.5826/dpc.1202a75
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991. doi:10.1016/j.jaad.2016.06.006
- Ingrassia JP, Stein JA, Levine A, et al. Diagnosis and management of acral pigmented lesions. Dermatol Surg Off Publ Am Soc Dermatol Surg Al. 2023;49:926-931. doi:10.1097/DSS.0000000000003891
- Hill L, Artiga S, Damico A. Health coverage by race and ethnicity, 2010-2022. Kaiser Family Foundation. Published January 11, 2024. Accessed May 9, 2024. https://www.kff.org/racial-equity-and-health-policy/issue-brief/health-coverage-by-race-and-ethnicity
The Comparison
Plantar hyperpigmentation (also known as plantar melanosis [increased melanin], volar pigmented macules, benign racial melanosis, acral pigmentation, acral ethnic melanosis, or mottled hyperpigmentation of the plantar surface) is a benign finding in many individuals and is especially prevalent in those with darker skin tones. Acral refers to manifestation on the hands and feet, volar on the palms and soles, and plantar on the soles only. Here, we focus on plantar hyperpigmentation. We use the terms ethnic and racial interchangeably.
It is critically important to differentiate benign hyperpigmentation, which is common in patients with skin of color, from melanoma. Although rare, Black patients in the United States experience high morbidity and mortality from acral melanoma, which often is diagnosed late in the disease course.1
There are many causes of hyperpigmentation on the plantar surfaces, including benign ethnic melanosis, nevi, melanoma, infections such as syphilis and tinea nigra, conditions such as Peutz-Jeghers syndrome and Laugier-Hunziker syndrome, and postinflammatory hyperpigmentation secondary to atopic dermatitis and psoriasis. We focus on the most common causes, ethnic melanosis and nevi, as well as melanoma, which is the deadliest cause.
Epidemiology
In a 1980 study (N=251), Black Americans had a high incidence of plantar hyperpigmentation, with 52% of affected patients having dark brown skin and 31% having light brown skin.2
The epidemiology of melanoma varies by race/ethnicity. Melanoma in Black individuals is relatively rare, with an annual incidence of approximately 1 in 100,000 individuals.3 However, when individuals with skin of color develop melanoma, they are more likely than their White counterparts to have acral melanoma (acral lentiginous melanoma), one of the deadliest types.1 In a case series of Black patients with melanoma (N=48) from 2 tertiary care centers in Texas, 30 of 40 primary cutaneous melanomas (75%) were located on acral skin.4 Overall, 13 patients developed stage IV disease and 12 died due to disease progression. All patients who developed distant metastases or died of melanoma had acral melanoma.4 Individuals of Asian descent also have a high incidence of acral melanoma, as shown in research from Japan.5-9
Key clinical features in individuals with darker skin tones
Dermoscopy is an evidence-based clinical examination method for earlier diagnosis of cutaneous melanoma, including on acral skin.10,11 Benign nevi on the volar skin as well as the palms and soles tend to have one of these 3 dermoscopic patterns: parallel furrow, lattice, or irregular fibrillar. The pattern that is most predictive of volar melanoma is the parallel ridge pattern (PRP) (Figures A and B [insets]), which showed a high specificity (99.0%) and very high negative predictive value (97.7%) for malignant melanoma in a Japanese population.7 The PRP data from this study cannot be applied reliably to Black individuals, especially because benign ethnic melanosis and other benign conditions can demonstrate PRP.12 Reliance on the PRP as a diagnostic clue could result in unneccessary biopsies in as many as 50% of Black patients with benign plantar hyperpigmentation.2 Furthermore, biopsies of the plantar surface can be painful and cause pain while walking.
It has been suggested that PRP seen on dermoscopy in benign hyperpigmentation such as ethnic melanosis and nevi may preserve the acrosyringia (eccrine gland openings on the ridge), whereas PRP in melanoma may obliterate the acrosyringia.13 This observation is based on case reports only and needs further study. However, if validated, it could be a useful diagnostic clue.
Worth noting
In a retrospective cohort study of skin cancer in Black individuals (n=165) at a New York City–based cancer center from 2000 to 2020, 68% of patients were diagnosed with melanomas—80% were the acral subtype and 75% displayed a PRP. However, the surrounding uninvolved background skin, which was visible in most cases, also demonstrated a PRP.14 Because of the high morbidity and mortality rates of acral melanoma, clinicians should biopsy or immediately refer patients with concerning plantar hyperpigmentation to a dermatologist.
Health disparity highlight
The mortality rate for acral melanoma in Black patients is disproportionately high for the following reasons15,16:
- Patients and health care providers do not expect to see melanoma in Black patients (it truly is rare!), so screening and education on sun protection are limited.
- Benign ethnic melanosis makes it more difficult to distinguish between early acral melanoma and benign skin changes.
- Black patients and other US patient populations with skin of color may be less likely to have health insurance, which contributes to inequities in access to health care. As of 2022, the uninsured rates for nonelderly American Indian and Alaska Native, Hispanic, Native Hawaiian and Other Pacific Islander, Black, and White individuals were 19.1%, 18.0%, 12.7%, 10.0%, and 6.6%, respectively.17
Multi-institutional registries could improve understanding of acral melanoma in Black patients.4 More studies are needed to help differentiate between the dermoscopic finding of PRP in benign ethnic melanosis vs malignant melanoma.
The Comparison
Plantar hyperpigmentation (also known as plantar melanosis [increased melanin], volar pigmented macules, benign racial melanosis, acral pigmentation, acral ethnic melanosis, or mottled hyperpigmentation of the plantar surface) is a benign finding in many individuals and is especially prevalent in those with darker skin tones. Acral refers to manifestation on the hands and feet, volar on the palms and soles, and plantar on the soles only. Here, we focus on plantar hyperpigmentation. We use the terms ethnic and racial interchangeably.
It is critically important to differentiate benign hyperpigmentation, which is common in patients with skin of color, from melanoma. Although rare, Black patients in the United States experience high morbidity and mortality from acral melanoma, which often is diagnosed late in the disease course.1
There are many causes of hyperpigmentation on the plantar surfaces, including benign ethnic melanosis, nevi, melanoma, infections such as syphilis and tinea nigra, conditions such as Peutz-Jeghers syndrome and Laugier-Hunziker syndrome, and postinflammatory hyperpigmentation secondary to atopic dermatitis and psoriasis. We focus on the most common causes, ethnic melanosis and nevi, as well as melanoma, which is the deadliest cause.
Epidemiology
In a 1980 study (N=251), Black Americans had a high incidence of plantar hyperpigmentation, with 52% of affected patients having dark brown skin and 31% having light brown skin.2
The epidemiology of melanoma varies by race/ethnicity. Melanoma in Black individuals is relatively rare, with an annual incidence of approximately 1 in 100,000 individuals.3 However, when individuals with skin of color develop melanoma, they are more likely than their White counterparts to have acral melanoma (acral lentiginous melanoma), one of the deadliest types.1 In a case series of Black patients with melanoma (N=48) from 2 tertiary care centers in Texas, 30 of 40 primary cutaneous melanomas (75%) were located on acral skin.4 Overall, 13 patients developed stage IV disease and 12 died due to disease progression. All patients who developed distant metastases or died of melanoma had acral melanoma.4 Individuals of Asian descent also have a high incidence of acral melanoma, as shown in research from Japan.5-9
Key clinical features in individuals with darker skin tones
Dermoscopy is an evidence-based clinical examination method for earlier diagnosis of cutaneous melanoma, including on acral skin.10,11 Benign nevi on the volar skin as well as the palms and soles tend to have one of these 3 dermoscopic patterns: parallel furrow, lattice, or irregular fibrillar. The pattern that is most predictive of volar melanoma is the parallel ridge pattern (PRP) (Figures A and B [insets]), which showed a high specificity (99.0%) and very high negative predictive value (97.7%) for malignant melanoma in a Japanese population.7 The PRP data from this study cannot be applied reliably to Black individuals, especially because benign ethnic melanosis and other benign conditions can demonstrate PRP.12 Reliance on the PRP as a diagnostic clue could result in unneccessary biopsies in as many as 50% of Black patients with benign plantar hyperpigmentation.2 Furthermore, biopsies of the plantar surface can be painful and cause pain while walking.
It has been suggested that PRP seen on dermoscopy in benign hyperpigmentation such as ethnic melanosis and nevi may preserve the acrosyringia (eccrine gland openings on the ridge), whereas PRP in melanoma may obliterate the acrosyringia.13 This observation is based on case reports only and needs further study. However, if validated, it could be a useful diagnostic clue.
Worth noting
In a retrospective cohort study of skin cancer in Black individuals (n=165) at a New York City–based cancer center from 2000 to 2020, 68% of patients were diagnosed with melanomas—80% were the acral subtype and 75% displayed a PRP. However, the surrounding uninvolved background skin, which was visible in most cases, also demonstrated a PRP.14 Because of the high morbidity and mortality rates of acral melanoma, clinicians should biopsy or immediately refer patients with concerning plantar hyperpigmentation to a dermatologist.
Health disparity highlight
The mortality rate for acral melanoma in Black patients is disproportionately high for the following reasons15,16:
- Patients and health care providers do not expect to see melanoma in Black patients (it truly is rare!), so screening and education on sun protection are limited.
- Benign ethnic melanosis makes it more difficult to distinguish between early acral melanoma and benign skin changes.
- Black patients and other US patient populations with skin of color may be less likely to have health insurance, which contributes to inequities in access to health care. As of 2022, the uninsured rates for nonelderly American Indian and Alaska Native, Hispanic, Native Hawaiian and Other Pacific Islander, Black, and White individuals were 19.1%, 18.0%, 12.7%, 10.0%, and 6.6%, respectively.17
Multi-institutional registries could improve understanding of acral melanoma in Black patients.4 More studies are needed to help differentiate between the dermoscopic finding of PRP in benign ethnic melanosis vs malignant melanoma.
- Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015: an analysis of the SEER registry. J Surg Res. 2020;251:329-339. doi:10.1016/j.jss.2020.02.010
- Coleman WP, Gately LE, Krementz AB, et al. Nevi, lentigines, and melanomas in blacks. Arch Dermatol. 1980;116:548-551.
- Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States: 2012-2016. USCS Data Brief, no. 9. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no9-melanoma-incidence-mortality-UnitedStates-2012-2016.htm
- Wix SN, Brown AB, Heberton M, et al. Clinical features and outcomes of black patients with melanoma. JAMA Dermatol. 2024;160:328-333. doi:10.1001/jamadermatol.2023.5789
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426. doi:10.1001/archderm.143.11.1423
- Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34. doi:10.1111/j.1346-8138.2010.01174.x
- Saida T, Miyazaki A, Oguchi S. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238. doi:10.1001/archderm.140.10.1233
- Saida T, Koga H, Uhara H. Dermoscopy for acral melanocytic lesions: revision of the 3-step algorithm and refined definition of the regular and irregular fibrillar pattern. Dermatol Pract Concept. 2022;12:e2022123. doi:10.5826/dpc.1203a123
- Heath CR, Usatine RP. Melanoma. Cutis. 2022;109:284-285.doi:10.12788/cutis.0513.
- Dinnes J, Deeks JJ, Chuchu N, et al; Cochrane Skin Cancer Diagnostic Test Accuracy Group. Visual inspection and dermoscopy, alone or in combination, for diagnosing keratinocyte skin cancers in adults. Cochrane Database Syst Rev. 2018; 12:CD011901. doi:10.1002/14651858.CD011901.pub2
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked-eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676. doi:10.1111/j.1365-2133.2008.08713.x
- Phan A, Dalle S, Marcilly MC, et al. Benign dermoscopic parallel ridge pattern variants. Arch Dermatol. 2011;147:634. doi:10.1001/archdermatol.2011.47
- Fracaroli TS, Lavorato FG, Maceira JP, et al. Parallel ridge pattern on dermoscopy: observation in non-melanoma cases. An Bras Dermatol. 2013;88:646-648. doi:10.1590/abd1806-4841.20132058
- Manci RN, Dauscher M, Marchetti MA, et al. Features of skin cancer in black individuals: a single-institution retrospective cohort study. Dermatol Pract Concept. 2022;12:e2022075. doi:10.5826/dpc.1202a75
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991. doi:10.1016/j.jaad.2016.06.006
- Ingrassia JP, Stein JA, Levine A, et al. Diagnosis and management of acral pigmented lesions. Dermatol Surg Off Publ Am Soc Dermatol Surg Al. 2023;49:926-931. doi:10.1097/DSS.0000000000003891
- Hill L, Artiga S, Damico A. Health coverage by race and ethnicity, 2010-2022. Kaiser Family Foundation. Published January 11, 2024. Accessed May 9, 2024. https://www.kff.org/racial-equity-and-health-policy/issue-brief/health-coverage-by-race-and-ethnicity
- Huang K, Fan J, Misra S. Acral lentiginous melanoma: incidence and survival in the United States, 2006-2015: an analysis of the SEER registry. J Surg Res. 2020;251:329-339. doi:10.1016/j.jss.2020.02.010
- Coleman WP, Gately LE, Krementz AB, et al. Nevi, lentigines, and melanomas in blacks. Arch Dermatol. 1980;116:548-551.
- Centers for Disease Control and Prevention. Melanoma Incidence and Mortality, United States: 2012-2016. USCS Data Brief, no. 9. Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019. https://www.cdc.gov/cancer/uscs/about/data-briefs/no9-melanoma-incidence-mortality-UnitedStates-2012-2016.htm
- Wix SN, Brown AB, Heberton M, et al. Clinical features and outcomes of black patients with melanoma. JAMA Dermatol. 2024;160:328-333. doi:10.1001/jamadermatol.2023.5789
- Saida T, Koga H. Dermoscopic patterns of acral melanocytic nevi: their variations, changes, and significance. Arch Dermatol. 2007;143:1423-1426. doi:10.1001/archderm.143.11.1423
- Saida T, Koga H, Uhara H. Key points in dermoscopic differentiation between early acral melanoma and acral nevus. J Dermatol. 2011;38:25-34. doi:10.1111/j.1346-8138.2010.01174.x
- Saida T, Miyazaki A, Oguchi S. Significance of dermoscopic patterns in detecting malignant melanoma on acral volar skin: results of a multicenter study in Japan. Arch Dermatol. 2004;140:1233-1238. doi:10.1001/archderm.140.10.1233
- Saida T, Koga H, Uhara H. Dermoscopy for acral melanocytic lesions: revision of the 3-step algorithm and refined definition of the regular and irregular fibrillar pattern. Dermatol Pract Concept. 2022;12:e2022123. doi:10.5826/dpc.1203a123
- Heath CR, Usatine RP. Melanoma. Cutis. 2022;109:284-285.doi:10.12788/cutis.0513.
- Dinnes J, Deeks JJ, Chuchu N, et al; Cochrane Skin Cancer Diagnostic Test Accuracy Group. Visual inspection and dermoscopy, alone or in combination, for diagnosing keratinocyte skin cancers in adults. Cochrane Database Syst Rev. 2018; 12:CD011901. doi:10.1002/14651858.CD011901.pub2
- Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked-eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159:669-676. doi:10.1111/j.1365-2133.2008.08713.x
- Phan A, Dalle S, Marcilly MC, et al. Benign dermoscopic parallel ridge pattern variants. Arch Dermatol. 2011;147:634. doi:10.1001/archdermatol.2011.47
- Fracaroli TS, Lavorato FG, Maceira JP, et al. Parallel ridge pattern on dermoscopy: observation in non-melanoma cases. An Bras Dermatol. 2013;88:646-648. doi:10.1590/abd1806-4841.20132058
- Manci RN, Dauscher M, Marchetti MA, et al. Features of skin cancer in black individuals: a single-institution retrospective cohort study. Dermatol Pract Concept. 2022;12:e2022075. doi:10.5826/dpc.1202a75
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991. doi:10.1016/j.jaad.2016.06.006
- Ingrassia JP, Stein JA, Levine A, et al. Diagnosis and management of acral pigmented lesions. Dermatol Surg Off Publ Am Soc Dermatol Surg Al. 2023;49:926-931. doi:10.1097/DSS.0000000000003891
- Hill L, Artiga S, Damico A. Health coverage by race and ethnicity, 2010-2022. Kaiser Family Foundation. Published January 11, 2024. Accessed May 9, 2024. https://www.kff.org/racial-equity-and-health-policy/issue-brief/health-coverage-by-race-and-ethnicity
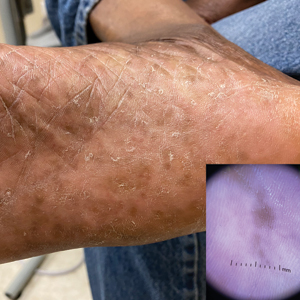
How Media Coverage of Oral Minoxidil for Hair Loss Has Impacted Prescribing Habits
Minoxidil, a potent vasodilator, was approved by the US Food and Drug Administration (FDA) in 1963 to treat high blood pressure. Its application as a hair loss treatment was discovered by accident—patients taking oral minoxidil for blood pressure noticed hair growth on their bodies as a side effect of the medication. In 1988, topical minoxidil (Rogaine [Johnson & Johnson Consumer Inc]) was approved by the FDA for the treatment of androgenetic alopecia in men, and then it was approved for the same indication in women in 1991. The mechanism of action by which minoxidil increases hair growth still has not been fully elucidated. When applied topically, it is thought to extend the anagen phase (or growth phase) of the hair cycle and increase hair follicle size. It also increases oxygen to the hair follicle through vasodilation and stimulates the production of vascular endothelial growth factor, which is thought to promote hair growth.1 Since its approval, topical minoxidil has become a first-line treatment of androgenetic alopecia in men and women.
In August 2022, The New York Times (NYT) published an article on dermatologists’ use of oral minoxidil at a fraction of the dose prescribed for blood pressure with profound results in hair regrowth.2 Several dermatologists quoted in the article endorsed that the decreased dose minimizes unwanted side effects such as hypertrichosis, hypotension, and other cardiac issues while still being effective for hair loss. Also, compared to topical minoxidil, low-dose oral minoxidil (LDOM) is relatively cheaper and easier to use; topicals are more cumbersome to apply and often leave the hair and scalp sticky, leading to noncompliance among patients.2 Currently, oral minoxidil is not approved by the FDA for use in hair loss, making it an off-label use.
Since the NYT article was published, we have observed an increase in patient questions and requests for LDOM as well as heightened use by fellow dermatologists in our community. As of November 2022, the NYT had approximately 9,330,000 total subscribers, solidifying its place as a newspaper of record in the United States and across the world.3 In April 2023, we conducted a survey of US-based board-certified dermatologists to investigate the impact of the NYT article on prescribing practices of LDOM for alopecia. The survey was conducted as a poll in a Facebook group for board-certified dermatologists and asked, “How did the NYT article on oral minoxidil for alopecia change your utilization of LDOM (low-dose oral minoxidil) for alopecia?” Three answer choices were given: (1) I started Rx’ing LDOM or increased the number of patients I manage with LDOM; (2) No change. I never Rx’d LDOM and/or no increase in utilization; and (3) I was already prescribing LDOM.
Of the 65 total respondents, 27 (42%) reported that the NYT article influenced their decision to start prescribing LDOM for alopecia. Nine respondents (14%) reported that the article did not influence their prescribing habits, and 27 (42%) responded that they were already prescribing the medication prior to the article’s publication.
Data from Epiphany Dermatology, a practice with more than 70 locations throughout the United States, showed that oral minoxidil was prescribed for alopecia 107 times in 2020 and 672 times in 2021 (Amy Hadley, Epiphany Dermatology, written communication, March 24, 2023). In 2022, prescriptions increased exponentially to 1626, and in the period of January 2023 to March 2023 alone, oral minoxidil was prescribed 510 times. Following publication of the NYT article in August 2022, LDOM was prescribed a total of 1377 times in the next 8 months.
Moreover, data from Summit Pharmacy, a retail pharmacy in Centennial, Colorado, showed an 1800% increase in LDOM prescriptions in the 7 months following the NYT article’s publication (August 2022 to March 2023) compared with the 7 months prior (January 2022 to August 2022)(Brandon Johnson, Summit Pharmacy, written communication, March 30, 2023). These data provide evidence for the influence of the NYT article on prescribing habits of dermatology providers in the United States.
The safety of oral minoxidil for use in hair loss has been established through several studies in the literature.4,5 These results show that LDOM may be a safe, readily accessible, and revolutionary treatment for hair loss. A retrospective multicenter study of 1404 patients treated with LDOM for any type of alopecia found that side effects were infrequent, and only 1.7% of patients discontinued treatment due to adverse effects. The most frequent adverse effect was hypertrichosis, occurring in 15.1% of patients but leading to treatment withdrawal in only 0.5% of patients.4 Similarly, Randolph and Tosti5 found that hypertrichosis of the face and body was the most common adverse effect observed, though it rarely resulted in discontinuation and likely was dose dependent: less than 10% of patients receiving 0.25 mg/d experienced hypertrichosis compared with more than 50% of those receiving 5 mg/d (N=634). They also described patients in whom topical minoxidil, though effective, posed major barriers to compliance due to the twice-daily application, changes to hair texture from the medication, and scalp irritation. A literature review of 17 studies with 634 patients on LDOM as a primary treatment for hair loss found that it was an effective, well-tolerated treatment and should be considered for healthy patients who have difficulty with topical formulations.5
In the age of media with data constantly at users’ fingertips, the art of practicing medicine also has changed. Although physicians pride themselves on evidence-based medicine, it appears that an NYT article had an impact on how physicians, particularly dermatologists, prescribe oral minoxidil. However, it is difficult to know if the article exposed dermatologists to another treatment in their armamentarium for hair loss or if it influenced patients to ask their health care provider about LDOM for hair loss. One thing is clear—since the article’s publication, the off-label use of LDOM for alopecia has produced what many may call “miracles” for patients with hair loss.5
- Messenger AG, Rundegren J. Minoxidil: mechanisms of action on hair growth. Br J Dermatol. 2004;150:186-194. doi:10.1111/j.1365-2133.2004.05785.x
- Kolata G. An old medicine grows new hair for pennies a day, doctors say. The New York Times. August 18, 2022. Accessed May 20, 2024. https://www.nytimes.com/2022/08/18/health/minoxidil-hair-loss-pills.html
- The New York Times Company reports third-quarter 2022 results. Press release. The New York Times Company; November 2, 2022. Accessed May 20, 2024. https://nytco-assets.nytimes.com/2022/11/NYT-Press-Release-Q3-2022-Final-nM7GzWGr.pdf
- Vañó-Galván S, Pirmez R, Hermosa-Gelbard A, et al. Safety of low-dose oral minoxidil for hair loss: a multicenter study of 1404 patients. J Am Acad Dermatol. 2021;84:1644-1651. doi:10.1016/j.jaad.2021.02.054
- Randolph M, Tosti A. Oral minoxidil treatment for hair loss: a review of efficacy and safety. J Am Acad Dermatol. 2021;84:737-746. doi:10.1016/j.jaad.2020.06.1009
Minoxidil, a potent vasodilator, was approved by the US Food and Drug Administration (FDA) in 1963 to treat high blood pressure. Its application as a hair loss treatment was discovered by accident—patients taking oral minoxidil for blood pressure noticed hair growth on their bodies as a side effect of the medication. In 1988, topical minoxidil (Rogaine [Johnson & Johnson Consumer Inc]) was approved by the FDA for the treatment of androgenetic alopecia in men, and then it was approved for the same indication in women in 1991. The mechanism of action by which minoxidil increases hair growth still has not been fully elucidated. When applied topically, it is thought to extend the anagen phase (or growth phase) of the hair cycle and increase hair follicle size. It also increases oxygen to the hair follicle through vasodilation and stimulates the production of vascular endothelial growth factor, which is thought to promote hair growth.1 Since its approval, topical minoxidil has become a first-line treatment of androgenetic alopecia in men and women.
In August 2022, The New York Times (NYT) published an article on dermatologists’ use of oral minoxidil at a fraction of the dose prescribed for blood pressure with profound results in hair regrowth.2 Several dermatologists quoted in the article endorsed that the decreased dose minimizes unwanted side effects such as hypertrichosis, hypotension, and other cardiac issues while still being effective for hair loss. Also, compared to topical minoxidil, low-dose oral minoxidil (LDOM) is relatively cheaper and easier to use; topicals are more cumbersome to apply and often leave the hair and scalp sticky, leading to noncompliance among patients.2 Currently, oral minoxidil is not approved by the FDA for use in hair loss, making it an off-label use.
Since the NYT article was published, we have observed an increase in patient questions and requests for LDOM as well as heightened use by fellow dermatologists in our community. As of November 2022, the NYT had approximately 9,330,000 total subscribers, solidifying its place as a newspaper of record in the United States and across the world.3 In April 2023, we conducted a survey of US-based board-certified dermatologists to investigate the impact of the NYT article on prescribing practices of LDOM for alopecia. The survey was conducted as a poll in a Facebook group for board-certified dermatologists and asked, “How did the NYT article on oral minoxidil for alopecia change your utilization of LDOM (low-dose oral minoxidil) for alopecia?” Three answer choices were given: (1) I started Rx’ing LDOM or increased the number of patients I manage with LDOM; (2) No change. I never Rx’d LDOM and/or no increase in utilization; and (3) I was already prescribing LDOM.
Of the 65 total respondents, 27 (42%) reported that the NYT article influenced their decision to start prescribing LDOM for alopecia. Nine respondents (14%) reported that the article did not influence their prescribing habits, and 27 (42%) responded that they were already prescribing the medication prior to the article’s publication.
Data from Epiphany Dermatology, a practice with more than 70 locations throughout the United States, showed that oral minoxidil was prescribed for alopecia 107 times in 2020 and 672 times in 2021 (Amy Hadley, Epiphany Dermatology, written communication, March 24, 2023). In 2022, prescriptions increased exponentially to 1626, and in the period of January 2023 to March 2023 alone, oral minoxidil was prescribed 510 times. Following publication of the NYT article in August 2022, LDOM was prescribed a total of 1377 times in the next 8 months.
Moreover, data from Summit Pharmacy, a retail pharmacy in Centennial, Colorado, showed an 1800% increase in LDOM prescriptions in the 7 months following the NYT article’s publication (August 2022 to March 2023) compared with the 7 months prior (January 2022 to August 2022)(Brandon Johnson, Summit Pharmacy, written communication, March 30, 2023). These data provide evidence for the influence of the NYT article on prescribing habits of dermatology providers in the United States.
The safety of oral minoxidil for use in hair loss has been established through several studies in the literature.4,5 These results show that LDOM may be a safe, readily accessible, and revolutionary treatment for hair loss. A retrospective multicenter study of 1404 patients treated with LDOM for any type of alopecia found that side effects were infrequent, and only 1.7% of patients discontinued treatment due to adverse effects. The most frequent adverse effect was hypertrichosis, occurring in 15.1% of patients but leading to treatment withdrawal in only 0.5% of patients.4 Similarly, Randolph and Tosti5 found that hypertrichosis of the face and body was the most common adverse effect observed, though it rarely resulted in discontinuation and likely was dose dependent: less than 10% of patients receiving 0.25 mg/d experienced hypertrichosis compared with more than 50% of those receiving 5 mg/d (N=634). They also described patients in whom topical minoxidil, though effective, posed major barriers to compliance due to the twice-daily application, changes to hair texture from the medication, and scalp irritation. A literature review of 17 studies with 634 patients on LDOM as a primary treatment for hair loss found that it was an effective, well-tolerated treatment and should be considered for healthy patients who have difficulty with topical formulations.5
In the age of media with data constantly at users’ fingertips, the art of practicing medicine also has changed. Although physicians pride themselves on evidence-based medicine, it appears that an NYT article had an impact on how physicians, particularly dermatologists, prescribe oral minoxidil. However, it is difficult to know if the article exposed dermatologists to another treatment in their armamentarium for hair loss or if it influenced patients to ask their health care provider about LDOM for hair loss. One thing is clear—since the article’s publication, the off-label use of LDOM for alopecia has produced what many may call “miracles” for patients with hair loss.5
Minoxidil, a potent vasodilator, was approved by the US Food and Drug Administration (FDA) in 1963 to treat high blood pressure. Its application as a hair loss treatment was discovered by accident—patients taking oral minoxidil for blood pressure noticed hair growth on their bodies as a side effect of the medication. In 1988, topical minoxidil (Rogaine [Johnson & Johnson Consumer Inc]) was approved by the FDA for the treatment of androgenetic alopecia in men, and then it was approved for the same indication in women in 1991. The mechanism of action by which minoxidil increases hair growth still has not been fully elucidated. When applied topically, it is thought to extend the anagen phase (or growth phase) of the hair cycle and increase hair follicle size. It also increases oxygen to the hair follicle through vasodilation and stimulates the production of vascular endothelial growth factor, which is thought to promote hair growth.1 Since its approval, topical minoxidil has become a first-line treatment of androgenetic alopecia in men and women.
In August 2022, The New York Times (NYT) published an article on dermatologists’ use of oral minoxidil at a fraction of the dose prescribed for blood pressure with profound results in hair regrowth.2 Several dermatologists quoted in the article endorsed that the decreased dose minimizes unwanted side effects such as hypertrichosis, hypotension, and other cardiac issues while still being effective for hair loss. Also, compared to topical minoxidil, low-dose oral minoxidil (LDOM) is relatively cheaper and easier to use; topicals are more cumbersome to apply and often leave the hair and scalp sticky, leading to noncompliance among patients.2 Currently, oral minoxidil is not approved by the FDA for use in hair loss, making it an off-label use.
Since the NYT article was published, we have observed an increase in patient questions and requests for LDOM as well as heightened use by fellow dermatologists in our community. As of November 2022, the NYT had approximately 9,330,000 total subscribers, solidifying its place as a newspaper of record in the United States and across the world.3 In April 2023, we conducted a survey of US-based board-certified dermatologists to investigate the impact of the NYT article on prescribing practices of LDOM for alopecia. The survey was conducted as a poll in a Facebook group for board-certified dermatologists and asked, “How did the NYT article on oral minoxidil for alopecia change your utilization of LDOM (low-dose oral minoxidil) for alopecia?” Three answer choices were given: (1) I started Rx’ing LDOM or increased the number of patients I manage with LDOM; (2) No change. I never Rx’d LDOM and/or no increase in utilization; and (3) I was already prescribing LDOM.
Of the 65 total respondents, 27 (42%) reported that the NYT article influenced their decision to start prescribing LDOM for alopecia. Nine respondents (14%) reported that the article did not influence their prescribing habits, and 27 (42%) responded that they were already prescribing the medication prior to the article’s publication.
Data from Epiphany Dermatology, a practice with more than 70 locations throughout the United States, showed that oral minoxidil was prescribed for alopecia 107 times in 2020 and 672 times in 2021 (Amy Hadley, Epiphany Dermatology, written communication, March 24, 2023). In 2022, prescriptions increased exponentially to 1626, and in the period of January 2023 to March 2023 alone, oral minoxidil was prescribed 510 times. Following publication of the NYT article in August 2022, LDOM was prescribed a total of 1377 times in the next 8 months.
Moreover, data from Summit Pharmacy, a retail pharmacy in Centennial, Colorado, showed an 1800% increase in LDOM prescriptions in the 7 months following the NYT article’s publication (August 2022 to March 2023) compared with the 7 months prior (January 2022 to August 2022)(Brandon Johnson, Summit Pharmacy, written communication, March 30, 2023). These data provide evidence for the influence of the NYT article on prescribing habits of dermatology providers in the United States.
The safety of oral minoxidil for use in hair loss has been established through several studies in the literature.4,5 These results show that LDOM may be a safe, readily accessible, and revolutionary treatment for hair loss. A retrospective multicenter study of 1404 patients treated with LDOM for any type of alopecia found that side effects were infrequent, and only 1.7% of patients discontinued treatment due to adverse effects. The most frequent adverse effect was hypertrichosis, occurring in 15.1% of patients but leading to treatment withdrawal in only 0.5% of patients.4 Similarly, Randolph and Tosti5 found that hypertrichosis of the face and body was the most common adverse effect observed, though it rarely resulted in discontinuation and likely was dose dependent: less than 10% of patients receiving 0.25 mg/d experienced hypertrichosis compared with more than 50% of those receiving 5 mg/d (N=634). They also described patients in whom topical minoxidil, though effective, posed major barriers to compliance due to the twice-daily application, changes to hair texture from the medication, and scalp irritation. A literature review of 17 studies with 634 patients on LDOM as a primary treatment for hair loss found that it was an effective, well-tolerated treatment and should be considered for healthy patients who have difficulty with topical formulations.5
In the age of media with data constantly at users’ fingertips, the art of practicing medicine also has changed. Although physicians pride themselves on evidence-based medicine, it appears that an NYT article had an impact on how physicians, particularly dermatologists, prescribe oral minoxidil. However, it is difficult to know if the article exposed dermatologists to another treatment in their armamentarium for hair loss or if it influenced patients to ask their health care provider about LDOM for hair loss. One thing is clear—since the article’s publication, the off-label use of LDOM for alopecia has produced what many may call “miracles” for patients with hair loss.5
- Messenger AG, Rundegren J. Minoxidil: mechanisms of action on hair growth. Br J Dermatol. 2004;150:186-194. doi:10.1111/j.1365-2133.2004.05785.x
- Kolata G. An old medicine grows new hair for pennies a day, doctors say. The New York Times. August 18, 2022. Accessed May 20, 2024. https://www.nytimes.com/2022/08/18/health/minoxidil-hair-loss-pills.html
- The New York Times Company reports third-quarter 2022 results. Press release. The New York Times Company; November 2, 2022. Accessed May 20, 2024. https://nytco-assets.nytimes.com/2022/11/NYT-Press-Release-Q3-2022-Final-nM7GzWGr.pdf
- Vañó-Galván S, Pirmez R, Hermosa-Gelbard A, et al. Safety of low-dose oral minoxidil for hair loss: a multicenter study of 1404 patients. J Am Acad Dermatol. 2021;84:1644-1651. doi:10.1016/j.jaad.2021.02.054
- Randolph M, Tosti A. Oral minoxidil treatment for hair loss: a review of efficacy and safety. J Am Acad Dermatol. 2021;84:737-746. doi:10.1016/j.jaad.2020.06.1009
- Messenger AG, Rundegren J. Minoxidil: mechanisms of action on hair growth. Br J Dermatol. 2004;150:186-194. doi:10.1111/j.1365-2133.2004.05785.x
- Kolata G. An old medicine grows new hair for pennies a day, doctors say. The New York Times. August 18, 2022. Accessed May 20, 2024. https://www.nytimes.com/2022/08/18/health/minoxidil-hair-loss-pills.html
- The New York Times Company reports third-quarter 2022 results. Press release. The New York Times Company; November 2, 2022. Accessed May 20, 2024. https://nytco-assets.nytimes.com/2022/11/NYT-Press-Release-Q3-2022-Final-nM7GzWGr.pdf
- Vañó-Galván S, Pirmez R, Hermosa-Gelbard A, et al. Safety of low-dose oral minoxidil for hair loss: a multicenter study of 1404 patients. J Am Acad Dermatol. 2021;84:1644-1651. doi:10.1016/j.jaad.2021.02.054
- Randolph M, Tosti A. Oral minoxidil treatment for hair loss: a review of efficacy and safety. J Am Acad Dermatol. 2021;84:737-746. doi:10.1016/j.jaad.2020.06.1009
Practice Points
- Low-dose oral minoxidil (LDOM) prescriptions have increased due to rising attention to its efficacy and safety.
- Media outlets can have a powerful effect on prescribing habits of physicians.
- Physicians should be aware of media trends to help direct patient education.
Aquatic Antagonists: Dermatologic Injuries From Sea Urchins (Echinoidea)
Sea urchins—members of the phylum Echinodermata and the class Echinoidea—are spiny marine invertebrates. Their consumption of fleshy algae makes them essential players in maintaining reef ecosystems.1,2 Echinoids, a class that includes heart urchins and sand dollars, are ubiquitous in benthic marine environments, both free floating and rock boring, and inhabit a wide range of latitudes spanning from polar oceans to warm seas.3 Despite their immobility and nonaggression, sea urchin puncture wounds are common among divers, snorkelers, swimmers, surfers, and fishers who accidentally come into contact with their sharp spines. Although the epidemiology of sea urchin exposure and injury is difficult to assess, the American Association of Poison Control Centers’ most recent annual report in 2022 documents approximately 1426 annual aquatic bites and/or envenomations.4
Sea Urchin Morphology and Toxicity
Echinoderms (a term of Greek origin meaning spiny skin) share a radially symmetric calcium carbonate skeleton (termed stereom) that is supported by collagenous ligaments.1 Sea urchins possess spines composed of calcite crystals, which radiate from their body and play a role in locomotion and defense against predators—namely sea otters, starfish/sea stars, wolf eels, and triggerfish, among others (Figure).5 These brittle spines can easily penetrate human skin and subsequently break off the sea urchin body. Most species of sea urchins possess solid spines, but a small percentage (80 of approximately 700 extant species) have hollow spines containing various toxic substances.6 Penetration and systemic absorption of the toxins within these spines can generate severe systemic responses.
The venomous flower urchin (Toxopneustes pileolus), found in the Indian and Pacific oceans, is one of the more common species known to produce a systemic reaction involving neuromuscular blockage.7-9 The most common species harvested off the Pacific coast of the United States—Strongylocentrotus purpuratus (purple sea urchin) and Strongylocentrotus franciscanus (red sea urchins)—are not inherently venomous.8

Both the sea urchin body and spines are covered in a unique epithelium thought to be responsible for the majority of their proinflammatory and pronociceptive properties. Epithelial compounds identified include serotonin, histamines, steroids, glycosides, hemolysins, proteases, and bradykininlike and cholinergic substances.5,7 Additionally, certain sea urchin species possess 3-pronged pincerlike organs at the base of spines called pedicellariae, which are used in feeding.10 Skin penetration by the pedicellariae is especially dangerous, as they tightly adhere to wounds and contain venom-producing organs that allow them to continue injecting toxins after their detachment from the sea urchin body.11
Presentation and Diagnosis of Sea Urchin Injuries
Sea urchin injuries have a wide range of manifestations depending on the number of spines involved, the presence of venom, the depth and location of spine penetration, the duration of spine retention in the skin, and the time before treatment initiation. The most common site of sea urchin injury unsurprisingly is the lower extremities and feet, often in the context of divers and swimmers walking across the sea floor. The hands are another frequently injured site, along with the legs, arms, back, scalp, and even oral mucosa.11
Although clinical history and presentation frequently reveal the mechanism of aquatic injury, patients often are unsure of the agent to which they were exposed and may be unaware of retained foreign bodies. Dermoscopy can distinguish the distinct lines radiating from the core of sea urchin spines from other foreign bodies lodged within the skin.6 It also can be used to locate spines for removal or for their analysis following punch biopsy.6,12 The radiopaque nature of sea urchin spines makes radiography and magnetic resonance imaging useful tools in assessment of periarticular soft-tissue damage and spine removal.8,11,13 Ultrasonography can reveal spines that no longer appear on radiography due to absorption by human tissue.14
Immediate Dermatologic Effects
Sea urchin injuries can be broadly categorized into immediate and delayed reactions. Immediate manifestations of contact with sea urchin spines include localized pain, bleeding, erythema, myalgia, and edema at the site of injury that can last from a few hours to 1 week without proper wound care and spine removal.5 Systemic symptoms ranging from dizziness, lightheadedness, paresthesia, aphonia, paralysis, coma, and death generally are only seen following injuries from venomous species, attachment of pedicellariae, injuries involving neurovascular structures, or penetration by more than 15 spines.7,11
Initial treatment includes soaking the wound in hot water (113 °F [45 °C]) for 30 to 90 minutes and subsequently removing spines and pedicellariae to prevent development of delayed reactions.5,15,16 The compounds in the sea urchin epithelium are heat labile and will be inactivated upon soaking in hot water.16 Extraction of spines can be difficult, as they are brittle and easily break in the skin. Successful removal has been reported using forceps and a hypodermic needle as well as excision; both approaches may require local anesthesia.8,17 Another technique involves freezing the localized area with liquid nitrogen to allow easier removal upon skin blistering.18 Punch biopsy also has been utilized as an effective means of ensuring all spiny fragments are removed.9,19,20 These spines often cause black or purple tattoolike staining at the puncture site, which can persist for a few days after spine extraction.8 Ablation using the erbium-doped:YAG laser may be helpful for removal of associated pigment.21,22
Delayed Dermatologic Effects
Delayed reactions to sea urchin injuries often are attributable to prolonged retention of spines in the skin. Granulomatous reactions typically manifest 2 weeks after injury as firm nonsuppurative nodules with central umbilication and a hyperkeratotic surface.7 These nodules may or may not be painful. Histopathology most often reveals foreign body and sarcoidal-type granulomatous reactions. However, tuberculoid, necrobiotic, and suppurative granulomas also may develop.13 Other microscopic features include inflammatory reactions, suppurative dermatitis, focal necrosis, and microabscesses.23 Wounds with progression to granulomatous disease often require surgical debridement.
Other more serious sequalae can result from involvement of joint capsules, especially in the hands and feet. Sea urchin injury involving joint spaces should be treated aggressively, as progression to inflammatory or infectious synovitis and tenosynovitis can cause irreversible loss of joint function. Inflammatory synovitis occurs 1 to 2 months on average after injury following a period of minimal symptoms and begins as a gradual increase in joint swelling and decrease in range of motion.8 Infectious tenosynovitis manifests quite similarly. Although suppurative etiologies generally progress with a more acute onset, certain infectious organisms (eg, Mycobacterium) take on an indolent course and should not be overlooked as a cause of delayed symptoms.8 The Kavanel cardinal signs are a sensitive tool used in the diagnosis of infectious flexor sheath tenosynovitis.8,24 If suspicion for joint infection is high, emergency referral should be made for debridement and culture-guided antibiotic therapy. Left untreated, infectious tenosynovitis can result in tendon necrosis or rupture, digit necrosis, and systemic infection.24 Patients with joint involvement should be referred to specialty care (eg, hand surgeon), as they often require synovectomy and surgical removal of foreign material.8
From 1 month to 1 year after injury, prolonged granulomatous synovitis of the hand may eventually lead to joint destruction known as “sea urchin arthritis.” These patients present with decreased range of motion and numerous nodules on the hand with a hyperkeratotic surface. Radiography reveals joint space narrowing, osteolysis, subchondral sclerosis, and periosteal reaction. Synovectomy and debridement are necessary to prevent irreversible joint damage or the need for arthrodesis and bone grafting.24
Other Treatment Considerations
Other important considerations in the care of sea urchin spine injuries include assessment of tetanus immunization status and administration of necessary prophylaxis as soon as possible, even in delayed presentations (Table).16,25 Cultures should be taken only if infection is suspected. Prophylactic antibiotics are not recommended unless the patient is immunocompromised or otherwise has impaired wound healing. If a patient presents with systemic symptoms, they should be referred to an emergency care facility for further management.
Final Thoughts
Sea urchin injuries can lead to serious complications if not diagnosed quickly and treated properly. Retention of sea urchin spines in the deep tissues and joint spaces may lead to granulomas, inflammatory and infectious tenosynovitis (including mycobacterial infection), and sea urchin arthritis requiring surgical debridement and possible irreversible joint damage, up to a year after initial injury. Patients should be educated on the possibility of developing these delayed reactions and instructed to seek immediate care. Joint deformities, range-of-motion deficits, and involvement of neurovascular structures should be considered emergent and referred for proper management. Shoes and diving gear offer some protection but are easily penetrable by sharp sea urchin spines. Preventive focus should be aimed at educating patients and providers on the importance of prompt spine removal upon injury. Although dermatologic and systemic manifestations vary widely, a thorough history, physical examination, and appropriate use of imaging modalities can facilitate accurate diagnosis and guide treatment.

- Amemiya CT, Miyake T, Rast JP. Echinoderms. Curr Biol. 2005;15:R944-R946. doi:10.1016/j.cub.2005.11.026
- Koch NM, Coppard SE, Lessios HA, et al. A phylogenomic resolution of the sea urchin tree of life. BMC Evol Biol. 2018;18:189. doi:10.1186/s12862-018-1300-4
- Amir Y, Insler M, Giller A, et al. Senescence and longevity of sea urchins. Genes (Basel). 2020;11:573. doi:10.3390/genes11050573
- Gummin DD, Mowry JB, Beuhler MC, et al. 2022 Annual Report of the National Poison Data System® (NPDS) from America's Poison Centers®: 40th annual report. Clin Toxicol (Phila). 2023;61:717-939. doi:10.1080/15563650.2023.2268981
- Gelman Y, Kong EL, Murphy-Lavoie HM. Sea urchin toxicity. In: StatPearls [Internet]. StatPearls Publishing; 2021.
- Suarez-Conde MF, Vallone MG, González VM, et al. Sea urchin skin lesions: a case report. Dermatol Pract Concept. 2021;11:E2021009. doi:10.5826/dpc.1102a09
- Al-Kathiri L, Al-Najjar T, Sulaiman I. Sea urchin granuloma of the hands: a case report. Oman Med J. 2019;34:350-353. doi:10.5001/omj.2019.68
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Hatakeyama T, Ichise A, Unno H, et al. Carbohydrate recognition by the rhamnose-binding lectin SUL-I with a novel three-domain structure isolated from the venom of globiferous pedicellariae of the flower sea urchin Toxopneustes pileolus. Protein Sci. 2017;26:1574-1583. doi:10.1002/pro.3185
- Balhara KS, Stolbach A. Marine envenomations. Emerg Med Clin North Am. 2014;32:223-243. doi:10.1016/j.emc.2013.09.009
- Schwartz Z, Cohen M, Lipner SR. Sea urchin injuries: a review and clinical approach algorithm. J Dermatolog Treat. 2021;32:150-156. doi:10.1080/09546634.2019.1638884
- Park SJ, Park JW, Choi SY, et al. Use of dermoscopy after punch removal of a veiled sea urchin spine. Dermatol Ther. 2021;34:E14947. doi:10.1111/dth.14947
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg Am. 2008;33:398-401. doi:10.1016/j.jhsa.2007.11.016
- Groleau S, Chhem RK, Younge D, et al. Ultrasonography of foreign-body tenosynovitis. Can Assoc Radiol J. 1992;43:454-456.
- Hornbeak KB, Auerbach PS. Marine envenomation. Emerg Med Clin North Am. 2017;35:321-337. doi:10.1016/j.emc.2016.12.004
- Noonburg GE. Management of extremity trauma and related infections occurring in the aquatic environment. J Am Acad Orthop Surg. 2005;13:243-253. doi:10.5435/00124635-200507000-00004
- Haddad Junior V. Observation of initial clinical manifestations and repercussions from the treatment of 314 human injuries caused by black sea urchins (Echinometra lucunter) on the southeastern Brazilian coast. Rev Soc Bras Med Trop. 2012;45:390-392. doi:10.1590/s0037-86822012000300021
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868. doi:10.1056/NEJMc1209382
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383. doi:10.1111/j.1445-2197.2010.05296.x
- Cardenas-de la Garza JA, Cuellar-Barboza A, Ancer-Arellano J, et al. Classic dermatological tools: foreign body removal with punch biopsy.J Am Acad Dermatol. 2019;81:E93-E94. doi:10.1016/j.jaad.2018.10.038
- Gungor S, Tarikçi N, Gokdemir G. Removal of sea urchin spines using erbium-doped yttrium aluminum garnet ablation. Dermatol Surg. 2012;38:508-510. doi:10.1111/j.1524-4725.2011.02259.x
- Böer A, Ochsendorf FR, Beier C, et al. Effective removal of sea-urchin spines by erbium:YAG laser ablation. Br J Dermatol. 2001;145:169-170. doi:10.1046/j.1365-2133.2001.04306.x
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228. doi:10.1034/j.1600-0560.2001.028005223.x
- Yi A, Kennedy C, Chia B, et al. Radiographic soft tissue thickness differentiating pyogenic flexor tenosynovitis from other finger infections. J Hand Surg Am. 2019;44:394-399. doi:10.1016/j.jhsa.2019.01.013
- Callison C, Nguyen H. Tetanus prophylaxis. In: StatPearls [Internet]. StatPearls Publishing; 2022.
Sea urchins—members of the phylum Echinodermata and the class Echinoidea—are spiny marine invertebrates. Their consumption of fleshy algae makes them essential players in maintaining reef ecosystems.1,2 Echinoids, a class that includes heart urchins and sand dollars, are ubiquitous in benthic marine environments, both free floating and rock boring, and inhabit a wide range of latitudes spanning from polar oceans to warm seas.3 Despite their immobility and nonaggression, sea urchin puncture wounds are common among divers, snorkelers, swimmers, surfers, and fishers who accidentally come into contact with their sharp spines. Although the epidemiology of sea urchin exposure and injury is difficult to assess, the American Association of Poison Control Centers’ most recent annual report in 2022 documents approximately 1426 annual aquatic bites and/or envenomations.4
Sea Urchin Morphology and Toxicity
Echinoderms (a term of Greek origin meaning spiny skin) share a radially symmetric calcium carbonate skeleton (termed stereom) that is supported by collagenous ligaments.1 Sea urchins possess spines composed of calcite crystals, which radiate from their body and play a role in locomotion and defense against predators—namely sea otters, starfish/sea stars, wolf eels, and triggerfish, among others (Figure).5 These brittle spines can easily penetrate human skin and subsequently break off the sea urchin body. Most species of sea urchins possess solid spines, but a small percentage (80 of approximately 700 extant species) have hollow spines containing various toxic substances.6 Penetration and systemic absorption of the toxins within these spines can generate severe systemic responses.
The venomous flower urchin (Toxopneustes pileolus), found in the Indian and Pacific oceans, is one of the more common species known to produce a systemic reaction involving neuromuscular blockage.7-9 The most common species harvested off the Pacific coast of the United States—Strongylocentrotus purpuratus (purple sea urchin) and Strongylocentrotus franciscanus (red sea urchins)—are not inherently venomous.8
Both the sea urchin body and spines are covered in a unique epithelium thought to be responsible for the majority of their proinflammatory and pronociceptive properties. Epithelial compounds identified include serotonin, histamines, steroids, glycosides, hemolysins, proteases, and bradykininlike and cholinergic substances.5,7 Additionally, certain sea urchin species possess 3-pronged pincerlike organs at the base of spines called pedicellariae, which are used in feeding.10 Skin penetration by the pedicellariae is especially dangerous, as they tightly adhere to wounds and contain venom-producing organs that allow them to continue injecting toxins after their detachment from the sea urchin body.11
Presentation and Diagnosis of Sea Urchin Injuries
Sea urchin injuries have a wide range of manifestations depending on the number of spines involved, the presence of venom, the depth and location of spine penetration, the duration of spine retention in the skin, and the time before treatment initiation. The most common site of sea urchin injury unsurprisingly is the lower extremities and feet, often in the context of divers and swimmers walking across the sea floor. The hands are another frequently injured site, along with the legs, arms, back, scalp, and even oral mucosa.11
Although clinical history and presentation frequently reveal the mechanism of aquatic injury, patients often are unsure of the agent to which they were exposed and may be unaware of retained foreign bodies. Dermoscopy can distinguish the distinct lines radiating from the core of sea urchin spines from other foreign bodies lodged within the skin.6 It also can be used to locate spines for removal or for their analysis following punch biopsy.6,12 The radiopaque nature of sea urchin spines makes radiography and magnetic resonance imaging useful tools in assessment of periarticular soft-tissue damage and spine removal.8,11,13 Ultrasonography can reveal spines that no longer appear on radiography due to absorption by human tissue.14
Immediate Dermatologic Effects
Sea urchin injuries can be broadly categorized into immediate and delayed reactions. Immediate manifestations of contact with sea urchin spines include localized pain, bleeding, erythema, myalgia, and edema at the site of injury that can last from a few hours to 1 week without proper wound care and spine removal.5 Systemic symptoms ranging from dizziness, lightheadedness, paresthesia, aphonia, paralysis, coma, and death generally are only seen following injuries from venomous species, attachment of pedicellariae, injuries involving neurovascular structures, or penetration by more than 15 spines.7,11
Initial treatment includes soaking the wound in hot water (113 °F [45 °C]) for 30 to 90 minutes and subsequently removing spines and pedicellariae to prevent development of delayed reactions.5,15,16 The compounds in the sea urchin epithelium are heat labile and will be inactivated upon soaking in hot water.16 Extraction of spines can be difficult, as they are brittle and easily break in the skin. Successful removal has been reported using forceps and a hypodermic needle as well as excision; both approaches may require local anesthesia.8,17 Another technique involves freezing the localized area with liquid nitrogen to allow easier removal upon skin blistering.18 Punch biopsy also has been utilized as an effective means of ensuring all spiny fragments are removed.9,19,20 These spines often cause black or purple tattoolike staining at the puncture site, which can persist for a few days after spine extraction.8 Ablation using the erbium-doped:YAG laser may be helpful for removal of associated pigment.21,22
Delayed Dermatologic Effects
Delayed reactions to sea urchin injuries often are attributable to prolonged retention of spines in the skin. Granulomatous reactions typically manifest 2 weeks after injury as firm nonsuppurative nodules with central umbilication and a hyperkeratotic surface.7 These nodules may or may not be painful. Histopathology most often reveals foreign body and sarcoidal-type granulomatous reactions. However, tuberculoid, necrobiotic, and suppurative granulomas also may develop.13 Other microscopic features include inflammatory reactions, suppurative dermatitis, focal necrosis, and microabscesses.23 Wounds with progression to granulomatous disease often require surgical debridement.
Other more serious sequalae can result from involvement of joint capsules, especially in the hands and feet. Sea urchin injury involving joint spaces should be treated aggressively, as progression to inflammatory or infectious synovitis and tenosynovitis can cause irreversible loss of joint function. Inflammatory synovitis occurs 1 to 2 months on average after injury following a period of minimal symptoms and begins as a gradual increase in joint swelling and decrease in range of motion.8 Infectious tenosynovitis manifests quite similarly. Although suppurative etiologies generally progress with a more acute onset, certain infectious organisms (eg, Mycobacterium) take on an indolent course and should not be overlooked as a cause of delayed symptoms.8 The Kavanel cardinal signs are a sensitive tool used in the diagnosis of infectious flexor sheath tenosynovitis.8,24 If suspicion for joint infection is high, emergency referral should be made for debridement and culture-guided antibiotic therapy. Left untreated, infectious tenosynovitis can result in tendon necrosis or rupture, digit necrosis, and systemic infection.24 Patients with joint involvement should be referred to specialty care (eg, hand surgeon), as they often require synovectomy and surgical removal of foreign material.8
From 1 month to 1 year after injury, prolonged granulomatous synovitis of the hand may eventually lead to joint destruction known as “sea urchin arthritis.” These patients present with decreased range of motion and numerous nodules on the hand with a hyperkeratotic surface. Radiography reveals joint space narrowing, osteolysis, subchondral sclerosis, and periosteal reaction. Synovectomy and debridement are necessary to prevent irreversible joint damage or the need for arthrodesis and bone grafting.24
Other Treatment Considerations
Other important considerations in the care of sea urchin spine injuries include assessment of tetanus immunization status and administration of necessary prophylaxis as soon as possible, even in delayed presentations (Table).16,25 Cultures should be taken only if infection is suspected. Prophylactic antibiotics are not recommended unless the patient is immunocompromised or otherwise has impaired wound healing. If a patient presents with systemic symptoms, they should be referred to an emergency care facility for further management.
Final Thoughts
Sea urchin injuries can lead to serious complications if not diagnosed quickly and treated properly. Retention of sea urchin spines in the deep tissues and joint spaces may lead to granulomas, inflammatory and infectious tenosynovitis (including mycobacterial infection), and sea urchin arthritis requiring surgical debridement and possible irreversible joint damage, up to a year after initial injury. Patients should be educated on the possibility of developing these delayed reactions and instructed to seek immediate care. Joint deformities, range-of-motion deficits, and involvement of neurovascular structures should be considered emergent and referred for proper management. Shoes and diving gear offer some protection but are easily penetrable by sharp sea urchin spines. Preventive focus should be aimed at educating patients and providers on the importance of prompt spine removal upon injury. Although dermatologic and systemic manifestations vary widely, a thorough history, physical examination, and appropriate use of imaging modalities can facilitate accurate diagnosis and guide treatment.
Sea urchins—members of the phylum Echinodermata and the class Echinoidea—are spiny marine invertebrates. Their consumption of fleshy algae makes them essential players in maintaining reef ecosystems.1,2 Echinoids, a class that includes heart urchins and sand dollars, are ubiquitous in benthic marine environments, both free floating and rock boring, and inhabit a wide range of latitudes spanning from polar oceans to warm seas.3 Despite their immobility and nonaggression, sea urchin puncture wounds are common among divers, snorkelers, swimmers, surfers, and fishers who accidentally come into contact with their sharp spines. Although the epidemiology of sea urchin exposure and injury is difficult to assess, the American Association of Poison Control Centers’ most recent annual report in 2022 documents approximately 1426 annual aquatic bites and/or envenomations.4
Sea Urchin Morphology and Toxicity
Echinoderms (a term of Greek origin meaning spiny skin) share a radially symmetric calcium carbonate skeleton (termed stereom) that is supported by collagenous ligaments.1 Sea urchins possess spines composed of calcite crystals, which radiate from their body and play a role in locomotion and defense against predators—namely sea otters, starfish/sea stars, wolf eels, and triggerfish, among others (Figure).5 These brittle spines can easily penetrate human skin and subsequently break off the sea urchin body. Most species of sea urchins possess solid spines, but a small percentage (80 of approximately 700 extant species) have hollow spines containing various toxic substances.6 Penetration and systemic absorption of the toxins within these spines can generate severe systemic responses.
The venomous flower urchin (Toxopneustes pileolus), found in the Indian and Pacific oceans, is one of the more common species known to produce a systemic reaction involving neuromuscular blockage.7-9 The most common species harvested off the Pacific coast of the United States—Strongylocentrotus purpuratus (purple sea urchin) and Strongylocentrotus franciscanus (red sea urchins)—are not inherently venomous.8
Both the sea urchin body and spines are covered in a unique epithelium thought to be responsible for the majority of their proinflammatory and pronociceptive properties. Epithelial compounds identified include serotonin, histamines, steroids, glycosides, hemolysins, proteases, and bradykininlike and cholinergic substances.5,7 Additionally, certain sea urchin species possess 3-pronged pincerlike organs at the base of spines called pedicellariae, which are used in feeding.10 Skin penetration by the pedicellariae is especially dangerous, as they tightly adhere to wounds and contain venom-producing organs that allow them to continue injecting toxins after their detachment from the sea urchin body.11
Presentation and Diagnosis of Sea Urchin Injuries
Sea urchin injuries have a wide range of manifestations depending on the number of spines involved, the presence of venom, the depth and location of spine penetration, the duration of spine retention in the skin, and the time before treatment initiation. The most common site of sea urchin injury unsurprisingly is the lower extremities and feet, often in the context of divers and swimmers walking across the sea floor. The hands are another frequently injured site, along with the legs, arms, back, scalp, and even oral mucosa.11
Although clinical history and presentation frequently reveal the mechanism of aquatic injury, patients often are unsure of the agent to which they were exposed and may be unaware of retained foreign bodies. Dermoscopy can distinguish the distinct lines radiating from the core of sea urchin spines from other foreign bodies lodged within the skin.6 It also can be used to locate spines for removal or for their analysis following punch biopsy.6,12 The radiopaque nature of sea urchin spines makes radiography and magnetic resonance imaging useful tools in assessment of periarticular soft-tissue damage and spine removal.8,11,13 Ultrasonography can reveal spines that no longer appear on radiography due to absorption by human tissue.14
Immediate Dermatologic Effects
Sea urchin injuries can be broadly categorized into immediate and delayed reactions. Immediate manifestations of contact with sea urchin spines include localized pain, bleeding, erythema, myalgia, and edema at the site of injury that can last from a few hours to 1 week without proper wound care and spine removal.5 Systemic symptoms ranging from dizziness, lightheadedness, paresthesia, aphonia, paralysis, coma, and death generally are only seen following injuries from venomous species, attachment of pedicellariae, injuries involving neurovascular structures, or penetration by more than 15 spines.7,11
Initial treatment includes soaking the wound in hot water (113 °F [45 °C]) for 30 to 90 minutes and subsequently removing spines and pedicellariae to prevent development of delayed reactions.5,15,16 The compounds in the sea urchin epithelium are heat labile and will be inactivated upon soaking in hot water.16 Extraction of spines can be difficult, as they are brittle and easily break in the skin. Successful removal has been reported using forceps and a hypodermic needle as well as excision; both approaches may require local anesthesia.8,17 Another technique involves freezing the localized area with liquid nitrogen to allow easier removal upon skin blistering.18 Punch biopsy also has been utilized as an effective means of ensuring all spiny fragments are removed.9,19,20 These spines often cause black or purple tattoolike staining at the puncture site, which can persist for a few days after spine extraction.8 Ablation using the erbium-doped:YAG laser may be helpful for removal of associated pigment.21,22
Delayed Dermatologic Effects
Delayed reactions to sea urchin injuries often are attributable to prolonged retention of spines in the skin. Granulomatous reactions typically manifest 2 weeks after injury as firm nonsuppurative nodules with central umbilication and a hyperkeratotic surface.7 These nodules may or may not be painful. Histopathology most often reveals foreign body and sarcoidal-type granulomatous reactions. However, tuberculoid, necrobiotic, and suppurative granulomas also may develop.13 Other microscopic features include inflammatory reactions, suppurative dermatitis, focal necrosis, and microabscesses.23 Wounds with progression to granulomatous disease often require surgical debridement.
Other more serious sequalae can result from involvement of joint capsules, especially in the hands and feet. Sea urchin injury involving joint spaces should be treated aggressively, as progression to inflammatory or infectious synovitis and tenosynovitis can cause irreversible loss of joint function. Inflammatory synovitis occurs 1 to 2 months on average after injury following a period of minimal symptoms and begins as a gradual increase in joint swelling and decrease in range of motion.8 Infectious tenosynovitis manifests quite similarly. Although suppurative etiologies generally progress with a more acute onset, certain infectious organisms (eg, Mycobacterium) take on an indolent course and should not be overlooked as a cause of delayed symptoms.8 The Kavanel cardinal signs are a sensitive tool used in the diagnosis of infectious flexor sheath tenosynovitis.8,24 If suspicion for joint infection is high, emergency referral should be made for debridement and culture-guided antibiotic therapy. Left untreated, infectious tenosynovitis can result in tendon necrosis or rupture, digit necrosis, and systemic infection.24 Patients with joint involvement should be referred to specialty care (eg, hand surgeon), as they often require synovectomy and surgical removal of foreign material.8
From 1 month to 1 year after injury, prolonged granulomatous synovitis of the hand may eventually lead to joint destruction known as “sea urchin arthritis.” These patients present with decreased range of motion and numerous nodules on the hand with a hyperkeratotic surface. Radiography reveals joint space narrowing, osteolysis, subchondral sclerosis, and periosteal reaction. Synovectomy and debridement are necessary to prevent irreversible joint damage or the need for arthrodesis and bone grafting.24
Other Treatment Considerations
Other important considerations in the care of sea urchin spine injuries include assessment of tetanus immunization status and administration of necessary prophylaxis as soon as possible, even in delayed presentations (Table).16,25 Cultures should be taken only if infection is suspected. Prophylactic antibiotics are not recommended unless the patient is immunocompromised or otherwise has impaired wound healing. If a patient presents with systemic symptoms, they should be referred to an emergency care facility for further management.
Final Thoughts
Sea urchin injuries can lead to serious complications if not diagnosed quickly and treated properly. Retention of sea urchin spines in the deep tissues and joint spaces may lead to granulomas, inflammatory and infectious tenosynovitis (including mycobacterial infection), and sea urchin arthritis requiring surgical debridement and possible irreversible joint damage, up to a year after initial injury. Patients should be educated on the possibility of developing these delayed reactions and instructed to seek immediate care. Joint deformities, range-of-motion deficits, and involvement of neurovascular structures should be considered emergent and referred for proper management. Shoes and diving gear offer some protection but are easily penetrable by sharp sea urchin spines. Preventive focus should be aimed at educating patients and providers on the importance of prompt spine removal upon injury. Although dermatologic and systemic manifestations vary widely, a thorough history, physical examination, and appropriate use of imaging modalities can facilitate accurate diagnosis and guide treatment.
- Amemiya CT, Miyake T, Rast JP. Echinoderms. Curr Biol. 2005;15:R944-R946. doi:10.1016/j.cub.2005.11.026
- Koch NM, Coppard SE, Lessios HA, et al. A phylogenomic resolution of the sea urchin tree of life. BMC Evol Biol. 2018;18:189. doi:10.1186/s12862-018-1300-4
- Amir Y, Insler M, Giller A, et al. Senescence and longevity of sea urchins. Genes (Basel). 2020;11:573. doi:10.3390/genes11050573
- Gummin DD, Mowry JB, Beuhler MC, et al. 2022 Annual Report of the National Poison Data System® (NPDS) from America's Poison Centers®: 40th annual report. Clin Toxicol (Phila). 2023;61:717-939. doi:10.1080/15563650.2023.2268981
- Gelman Y, Kong EL, Murphy-Lavoie HM. Sea urchin toxicity. In: StatPearls [Internet]. StatPearls Publishing; 2021.
- Suarez-Conde MF, Vallone MG, González VM, et al. Sea urchin skin lesions: a case report. Dermatol Pract Concept. 2021;11:E2021009. doi:10.5826/dpc.1102a09
- Al-Kathiri L, Al-Najjar T, Sulaiman I. Sea urchin granuloma of the hands: a case report. Oman Med J. 2019;34:350-353. doi:10.5001/omj.2019.68
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Hatakeyama T, Ichise A, Unno H, et al. Carbohydrate recognition by the rhamnose-binding lectin SUL-I with a novel three-domain structure isolated from the venom of globiferous pedicellariae of the flower sea urchin Toxopneustes pileolus. Protein Sci. 2017;26:1574-1583. doi:10.1002/pro.3185
- Balhara KS, Stolbach A. Marine envenomations. Emerg Med Clin North Am. 2014;32:223-243. doi:10.1016/j.emc.2013.09.009
- Schwartz Z, Cohen M, Lipner SR. Sea urchin injuries: a review and clinical approach algorithm. J Dermatolog Treat. 2021;32:150-156. doi:10.1080/09546634.2019.1638884
- Park SJ, Park JW, Choi SY, et al. Use of dermoscopy after punch removal of a veiled sea urchin spine. Dermatol Ther. 2021;34:E14947. doi:10.1111/dth.14947
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg Am. 2008;33:398-401. doi:10.1016/j.jhsa.2007.11.016
- Groleau S, Chhem RK, Younge D, et al. Ultrasonography of foreign-body tenosynovitis. Can Assoc Radiol J. 1992;43:454-456.
- Hornbeak KB, Auerbach PS. Marine envenomation. Emerg Med Clin North Am. 2017;35:321-337. doi:10.1016/j.emc.2016.12.004
- Noonburg GE. Management of extremity trauma and related infections occurring in the aquatic environment. J Am Acad Orthop Surg. 2005;13:243-253. doi:10.5435/00124635-200507000-00004
- Haddad Junior V. Observation of initial clinical manifestations and repercussions from the treatment of 314 human injuries caused by black sea urchins (Echinometra lucunter) on the southeastern Brazilian coast. Rev Soc Bras Med Trop. 2012;45:390-392. doi:10.1590/s0037-86822012000300021
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868. doi:10.1056/NEJMc1209382
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383. doi:10.1111/j.1445-2197.2010.05296.x
- Cardenas-de la Garza JA, Cuellar-Barboza A, Ancer-Arellano J, et al. Classic dermatological tools: foreign body removal with punch biopsy.J Am Acad Dermatol. 2019;81:E93-E94. doi:10.1016/j.jaad.2018.10.038
- Gungor S, Tarikçi N, Gokdemir G. Removal of sea urchin spines using erbium-doped yttrium aluminum garnet ablation. Dermatol Surg. 2012;38:508-510. doi:10.1111/j.1524-4725.2011.02259.x
- Böer A, Ochsendorf FR, Beier C, et al. Effective removal of sea-urchin spines by erbium:YAG laser ablation. Br J Dermatol. 2001;145:169-170. doi:10.1046/j.1365-2133.2001.04306.x
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228. doi:10.1034/j.1600-0560.2001.028005223.x
- Yi A, Kennedy C, Chia B, et al. Radiographic soft tissue thickness differentiating pyogenic flexor tenosynovitis from other finger infections. J Hand Surg Am. 2019;44:394-399. doi:10.1016/j.jhsa.2019.01.013
- Callison C, Nguyen H. Tetanus prophylaxis. In: StatPearls [Internet]. StatPearls Publishing; 2022.
- Amemiya CT, Miyake T, Rast JP. Echinoderms. Curr Biol. 2005;15:R944-R946. doi:10.1016/j.cub.2005.11.026
- Koch NM, Coppard SE, Lessios HA, et al. A phylogenomic resolution of the sea urchin tree of life. BMC Evol Biol. 2018;18:189. doi:10.1186/s12862-018-1300-4
- Amir Y, Insler M, Giller A, et al. Senescence and longevity of sea urchins. Genes (Basel). 2020;11:573. doi:10.3390/genes11050573
- Gummin DD, Mowry JB, Beuhler MC, et al. 2022 Annual Report of the National Poison Data System® (NPDS) from America's Poison Centers®: 40th annual report. Clin Toxicol (Phila). 2023;61:717-939. doi:10.1080/15563650.2023.2268981
- Gelman Y, Kong EL, Murphy-Lavoie HM. Sea urchin toxicity. In: StatPearls [Internet]. StatPearls Publishing; 2021.
- Suarez-Conde MF, Vallone MG, González VM, et al. Sea urchin skin lesions: a case report. Dermatol Pract Concept. 2021;11:E2021009. doi:10.5826/dpc.1102a09
- Al-Kathiri L, Al-Najjar T, Sulaiman I. Sea urchin granuloma of the hands: a case report. Oman Med J. 2019;34:350-353. doi:10.5001/omj.2019.68
- Dahl WJ, Jebson P, Louis DS. Sea urchin injuries to the hand: a case report and review of the literature. Iowa Orthop J. 2010;30:153-156.
- Hatakeyama T, Ichise A, Unno H, et al. Carbohydrate recognition by the rhamnose-binding lectin SUL-I with a novel three-domain structure isolated from the venom of globiferous pedicellariae of the flower sea urchin Toxopneustes pileolus. Protein Sci. 2017;26:1574-1583. doi:10.1002/pro.3185
- Balhara KS, Stolbach A. Marine envenomations. Emerg Med Clin North Am. 2014;32:223-243. doi:10.1016/j.emc.2013.09.009
- Schwartz Z, Cohen M, Lipner SR. Sea urchin injuries: a review and clinical approach algorithm. J Dermatolog Treat. 2021;32:150-156. doi:10.1080/09546634.2019.1638884
- Park SJ, Park JW, Choi SY, et al. Use of dermoscopy after punch removal of a veiled sea urchin spine. Dermatol Ther. 2021;34:E14947. doi:10.1111/dth.14947
- Wada T, Soma T, Gaman K, et al. Sea urchin spine arthritis of the hand. J Hand Surg Am. 2008;33:398-401. doi:10.1016/j.jhsa.2007.11.016
- Groleau S, Chhem RK, Younge D, et al. Ultrasonography of foreign-body tenosynovitis. Can Assoc Radiol J. 1992;43:454-456.
- Hornbeak KB, Auerbach PS. Marine envenomation. Emerg Med Clin North Am. 2017;35:321-337. doi:10.1016/j.emc.2016.12.004
- Noonburg GE. Management of extremity trauma and related infections occurring in the aquatic environment. J Am Acad Orthop Surg. 2005;13:243-253. doi:10.5435/00124635-200507000-00004
- Haddad Junior V. Observation of initial clinical manifestations and repercussions from the treatment of 314 human injuries caused by black sea urchins (Echinometra lucunter) on the southeastern Brazilian coast. Rev Soc Bras Med Trop. 2012;45:390-392. doi:10.1590/s0037-86822012000300021
- Gargus MD, Morohashi DK. A sea-urchin spine chilling remedy. N Engl J Med. 2012;367:1867-1868. doi:10.1056/NEJMc1209382
- Sjøberg T, de Weerd L. The usefulness of a skin biopsy punch to remove sea urchin spines. ANZ J Surg. 2010;80:383. doi:10.1111/j.1445-2197.2010.05296.x
- Cardenas-de la Garza JA, Cuellar-Barboza A, Ancer-Arellano J, et al. Classic dermatological tools: foreign body removal with punch biopsy.J Am Acad Dermatol. 2019;81:E93-E94. doi:10.1016/j.jaad.2018.10.038
- Gungor S, Tarikçi N, Gokdemir G. Removal of sea urchin spines using erbium-doped yttrium aluminum garnet ablation. Dermatol Surg. 2012;38:508-510. doi:10.1111/j.1524-4725.2011.02259.x
- Böer A, Ochsendorf FR, Beier C, et al. Effective removal of sea-urchin spines by erbium:YAG laser ablation. Br J Dermatol. 2001;145:169-170. doi:10.1046/j.1365-2133.2001.04306.x
- De La Torre C, Toribio J. Sea-urchin granuloma: histologic profile. a pathologic study of 50 biopsies. J Cutan Pathol. 2001;28:223-228. doi:10.1034/j.1600-0560.2001.028005223.x
- Yi A, Kennedy C, Chia B, et al. Radiographic soft tissue thickness differentiating pyogenic flexor tenosynovitis from other finger infections. J Hand Surg Am. 2019;44:394-399. doi:10.1016/j.jhsa.2019.01.013
- Callison C, Nguyen H. Tetanus prophylaxis. In: StatPearls [Internet]. StatPearls Publishing; 2022.
Practice Points
- Sea urchin spines easily become embedded in human skin upon contact and cause localized pain, edema, and black or purple pinpoint markings.
- Immediate treatment includes soaking in hot water (113 12°F [45 12°C]) for 30 to 90 minutes to inactivate proinflammatory compounds, followed by extraction of the spines.
- Successful methods of spine removal include the use of forceps and a hypodermic needle, as well as excision, liquid nitrogen, and punch biopsy.
- Prompt removal of the spines can reduce the incidence of delayed granulomatous reactions, synovitis, and sea urchin arthritis.

Central Centrifugal Cicatricial Alopecia in Males: Analysis of Time to Diagnosis and Disease Severity
To the Editor:
Central centrifugal cicatricial alopecia (CCCA) is a chronic progressive type of scarring alopecia that primarily affects women of African descent.1 The disorder rarely is reported in men, which may be due to misdiagnosis or delayed diagnosis. Early diagnosis and treatment are the cornerstones to slow or halt disease progression and prevent permanent damage to hair follicles. This study aimed to investigate the time to diagnosis and disease severity among males with CCCA.
We conducted a retrospective chart review of male patients older than 18 years seen in outpatient clinics at an academic dermatology department (Philadelphia, Pennsylvania) between January 2012 and December 2022. An electronic query using the International Classification of Diseases, Ninth and Tenth Revisions, code L66.9 (cicatricial alopecia, unspecified) was performed. Patients were included if they had a clinical diagnosis of CCCA, histologic evidence of CCCA, and scalp photographs from the initial dermatology visit. Patients with folliculitis decalvans, scalp biopsy features that limited characterization, or no scalp biopsy were excluded from the study. Onset of CCCA was defined as the patient-reported start time of hair loss and/or scalp symptoms. To determine alopecia severity, the degree of central scalp hair loss was independently assessed by 2 dermatologists (S.C.T., T.O.) using the central scalp alopecia photographic scale in African American women.2,3 This 6-point photographic scale displays images with grades ranging from 0 (normal) to 5 (bald scalp); higher grades indicate probable and more severe CCCA. The scale also divides the central hair loss in a frontal-accentuation or vertex-predominant pattern, which corresponds to the A or B designations, respectively; thus, a score of 5A indicates probable severe CCCA with a frontal accentuation pattern, while 5B indicates probable severe CCCA with hair loss focused on the vertex scalp. This study was approved by the University of Pennsylvania institutional review board (approval #850730).
Of 108 male patients, 12 met the eligibility criteria. Nearly all patients (91.7% [11/12]) had a CCCA severity grade of 3 or higher at the initial dermatology visit, indicating extensive hair loss (Table). The clinical appearance of severity grades 2 through 5 is demonstrated in the Figure. Among patients with a known disease duration prior to diagnosis, 72.7% (8/11) were diagnosed more than 1 year after onset of CCCA, and 45.4% (5/11) were diagnosed more than 5 years after onset. On average (SD), it took 6.4 (5.9) years for patients to receive a diagnosis of CCCA after the onset of scalp symptoms and/or hair loss.
Randomized controlled trials evaluating treatment of CCCA are lacking, and anecdotal evidence posits a better treatment response in early CCCA; however, our results suggest that most male patients present with advanced CCCA and receive a diagnosis years after disease onset. Similar research in alopecia areata has shown that 72.4% (105/145) of patients received their diagnosis within a year after onset of symptoms, and the mean time from onset of symptoms to diagnosis was 1 year.4 In contrast, male patients with CCCA experience considerable diagnostic delays. This disparity indicates the need for clinicians to increase recognition of CCCA in men and quickly refer them to a dermatologist for prompt treatment.
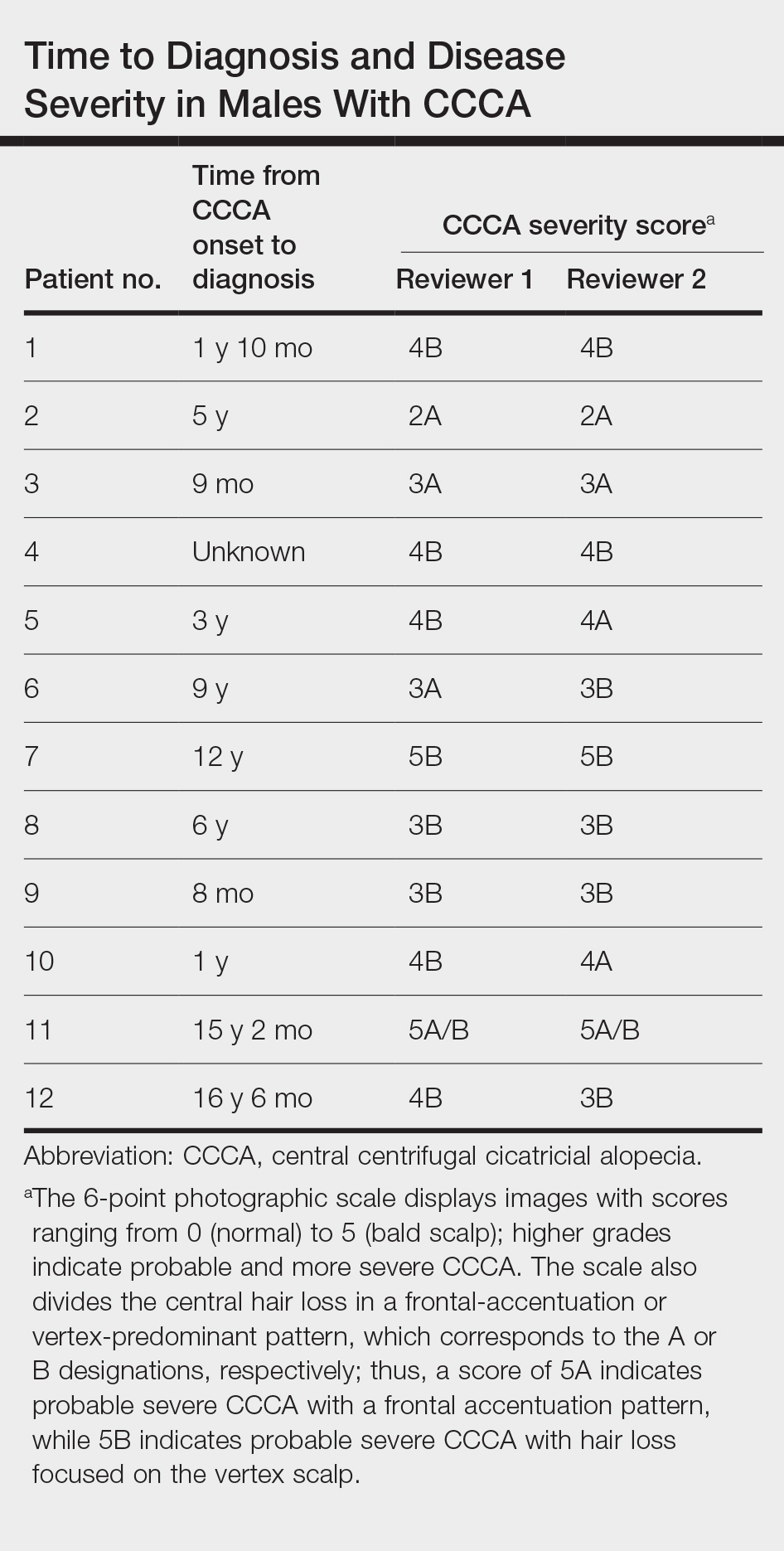
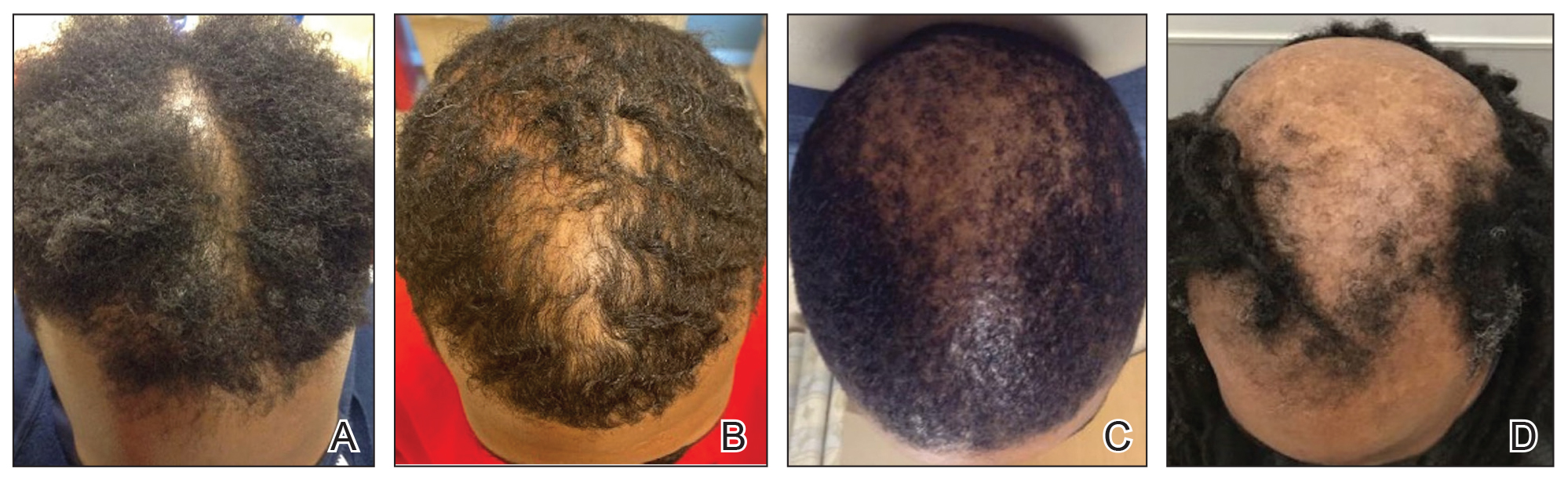
Androgenetic alopecia (AGA) commonly is at the top of the differential diagnosis for hair loss on the vertex of the scalp in males, but clinicians should maintain a high index of suspicion for CCCA, especially when scalp symptoms or atypical features of AGA are present.5 Androgenetic alopecia typically is asymptomatic, whereas the symptoms of CCCA may include itching, tenderness, and/or burning.6,7 Trichoscopy is useful to evaluate for scarring, and a scalp biopsy may reveal other features to lower AGA on the differential. Educating patients, barbers, and hairstylists about the importance of early intervention also may encourage earlier visits before the scarring process is advanced. Further exploration into factors impacting diagnosis and CCCA severity may uncover implications for prognosis and treatment.
This study was limited by a small sample size, retrospective design, and single-center analysis. Some patients had comorbid hair loss conditions, which could affect disease severity. Moreover, the central scalp alopecia photographic scale2 was not validated in men or designed for assessment of the nonclassical hair loss distributions noted in some of our patients. Nonetheless, we hope these data will support clinicians in efforts to advocate for early diagnosis and treatment in patients with CCCA to ultimately help improve outcomes.
- Ogunleye TA, McMichael A, Olsen EA. Central centrifugal cicatricial alopecia: what has been achieved, current clues for future research. Dermatol Clin. 2014;32:173-181. doi:10.1016/j.det.2013.12.005
- Olsen EA, Callender V, McMichael A, et al. Central hair loss in African American women: incidence and potential risk factors. J Am Acad Dermatol. 2011;64:245-252. doi:10.1016/j.jaad.2009.11.693
- Olsen EA, Callendar V, Sperling L, et al. Central scalp alopecia photographic scale in African American women. Dermatol Ther. 2008;21:264-267. doi:10.1111/j.1529-8019.2008.00208.x
- Andersen YMF, Nymand L, DeLozier AM, et al. Patient characteristics and disease burden of alopecia areata in the Danish Skin Cohort. BMJ Open. 2022;12:E053137. doi:10.1136/bmjopen-2021-053137
- Davis EC, Reid SD, Callender VD, et al. Differentiating central centrifugal cicatricial alopecia and androgenetic alopecia in African American men. J Clin Aesthetic Dermatol. 2012;5:37-40.
- Jackson TK, Sow Y, Ayoade KO, et al. Central centrifugal cicatricial alopecia in males. J Am Acad Dermatol. 2023;89:1136-1140. doi:10.1016/j.jaad.2023.07.1011
- Lawson CN, Bakayoko A, Callender VD. Central centrifugal cicatricial alopecia: challenges and treatments. Dermatol Clin. 2021;39:389-405. doi:10.1016/j.det.2021.03.004
To the Editor:
Central centrifugal cicatricial alopecia (CCCA) is a chronic progressive type of scarring alopecia that primarily affects women of African descent.1 The disorder rarely is reported in men, which may be due to misdiagnosis or delayed diagnosis. Early diagnosis and treatment are the cornerstones to slow or halt disease progression and prevent permanent damage to hair follicles. This study aimed to investigate the time to diagnosis and disease severity among males with CCCA.
We conducted a retrospective chart review of male patients older than 18 years seen in outpatient clinics at an academic dermatology department (Philadelphia, Pennsylvania) between January 2012 and December 2022. An electronic query using the International Classification of Diseases, Ninth and Tenth Revisions, code L66.9 (cicatricial alopecia, unspecified) was performed. Patients were included if they had a clinical diagnosis of CCCA, histologic evidence of CCCA, and scalp photographs from the initial dermatology visit. Patients with folliculitis decalvans, scalp biopsy features that limited characterization, or no scalp biopsy were excluded from the study. Onset of CCCA was defined as the patient-reported start time of hair loss and/or scalp symptoms. To determine alopecia severity, the degree of central scalp hair loss was independently assessed by 2 dermatologists (S.C.T., T.O.) using the central scalp alopecia photographic scale in African American women.2,3 This 6-point photographic scale displays images with grades ranging from 0 (normal) to 5 (bald scalp); higher grades indicate probable and more severe CCCA. The scale also divides the central hair loss in a frontal-accentuation or vertex-predominant pattern, which corresponds to the A or B designations, respectively; thus, a score of 5A indicates probable severe CCCA with a frontal accentuation pattern, while 5B indicates probable severe CCCA with hair loss focused on the vertex scalp. This study was approved by the University of Pennsylvania institutional review board (approval #850730).
Of 108 male patients, 12 met the eligibility criteria. Nearly all patients (91.7% [11/12]) had a CCCA severity grade of 3 or higher at the initial dermatology visit, indicating extensive hair loss (Table). The clinical appearance of severity grades 2 through 5 is demonstrated in the Figure. Among patients with a known disease duration prior to diagnosis, 72.7% (8/11) were diagnosed more than 1 year after onset of CCCA, and 45.4% (5/11) were diagnosed more than 5 years after onset. On average (SD), it took 6.4 (5.9) years for patients to receive a diagnosis of CCCA after the onset of scalp symptoms and/or hair loss.
Randomized controlled trials evaluating treatment of CCCA are lacking, and anecdotal evidence posits a better treatment response in early CCCA; however, our results suggest that most male patients present with advanced CCCA and receive a diagnosis years after disease onset. Similar research in alopecia areata has shown that 72.4% (105/145) of patients received their diagnosis within a year after onset of symptoms, and the mean time from onset of symptoms to diagnosis was 1 year.4 In contrast, male patients with CCCA experience considerable diagnostic delays. This disparity indicates the need for clinicians to increase recognition of CCCA in men and quickly refer them to a dermatologist for prompt treatment.
Androgenetic alopecia (AGA) commonly is at the top of the differential diagnosis for hair loss on the vertex of the scalp in males, but clinicians should maintain a high index of suspicion for CCCA, especially when scalp symptoms or atypical features of AGA are present.5 Androgenetic alopecia typically is asymptomatic, whereas the symptoms of CCCA may include itching, tenderness, and/or burning.6,7 Trichoscopy is useful to evaluate for scarring, and a scalp biopsy may reveal other features to lower AGA on the differential. Educating patients, barbers, and hairstylists about the importance of early intervention also may encourage earlier visits before the scarring process is advanced. Further exploration into factors impacting diagnosis and CCCA severity may uncover implications for prognosis and treatment.
This study was limited by a small sample size, retrospective design, and single-center analysis. Some patients had comorbid hair loss conditions, which could affect disease severity. Moreover, the central scalp alopecia photographic scale2 was not validated in men or designed for assessment of the nonclassical hair loss distributions noted in some of our patients. Nonetheless, we hope these data will support clinicians in efforts to advocate for early diagnosis and treatment in patients with CCCA to ultimately help improve outcomes.
To the Editor:
Central centrifugal cicatricial alopecia (CCCA) is a chronic progressive type of scarring alopecia that primarily affects women of African descent.1 The disorder rarely is reported in men, which may be due to misdiagnosis or delayed diagnosis. Early diagnosis and treatment are the cornerstones to slow or halt disease progression and prevent permanent damage to hair follicles. This study aimed to investigate the time to diagnosis and disease severity among males with CCCA.
We conducted a retrospective chart review of male patients older than 18 years seen in outpatient clinics at an academic dermatology department (Philadelphia, Pennsylvania) between January 2012 and December 2022. An electronic query using the International Classification of Diseases, Ninth and Tenth Revisions, code L66.9 (cicatricial alopecia, unspecified) was performed. Patients were included if they had a clinical diagnosis of CCCA, histologic evidence of CCCA, and scalp photographs from the initial dermatology visit. Patients with folliculitis decalvans, scalp biopsy features that limited characterization, or no scalp biopsy were excluded from the study. Onset of CCCA was defined as the patient-reported start time of hair loss and/or scalp symptoms. To determine alopecia severity, the degree of central scalp hair loss was independently assessed by 2 dermatologists (S.C.T., T.O.) using the central scalp alopecia photographic scale in African American women.2,3 This 6-point photographic scale displays images with grades ranging from 0 (normal) to 5 (bald scalp); higher grades indicate probable and more severe CCCA. The scale also divides the central hair loss in a frontal-accentuation or vertex-predominant pattern, which corresponds to the A or B designations, respectively; thus, a score of 5A indicates probable severe CCCA with a frontal accentuation pattern, while 5B indicates probable severe CCCA with hair loss focused on the vertex scalp. This study was approved by the University of Pennsylvania institutional review board (approval #850730).
Of 108 male patients, 12 met the eligibility criteria. Nearly all patients (91.7% [11/12]) had a CCCA severity grade of 3 or higher at the initial dermatology visit, indicating extensive hair loss (Table). The clinical appearance of severity grades 2 through 5 is demonstrated in the Figure. Among patients with a known disease duration prior to diagnosis, 72.7% (8/11) were diagnosed more than 1 year after onset of CCCA, and 45.4% (5/11) were diagnosed more than 5 years after onset. On average (SD), it took 6.4 (5.9) years for patients to receive a diagnosis of CCCA after the onset of scalp symptoms and/or hair loss.
Randomized controlled trials evaluating treatment of CCCA are lacking, and anecdotal evidence posits a better treatment response in early CCCA; however, our results suggest that most male patients present with advanced CCCA and receive a diagnosis years after disease onset. Similar research in alopecia areata has shown that 72.4% (105/145) of patients received their diagnosis within a year after onset of symptoms, and the mean time from onset of symptoms to diagnosis was 1 year.4 In contrast, male patients with CCCA experience considerable diagnostic delays. This disparity indicates the need for clinicians to increase recognition of CCCA in men and quickly refer them to a dermatologist for prompt treatment.
Androgenetic alopecia (AGA) commonly is at the top of the differential diagnosis for hair loss on the vertex of the scalp in males, but clinicians should maintain a high index of suspicion for CCCA, especially when scalp symptoms or atypical features of AGA are present.5 Androgenetic alopecia typically is asymptomatic, whereas the symptoms of CCCA may include itching, tenderness, and/or burning.6,7 Trichoscopy is useful to evaluate for scarring, and a scalp biopsy may reveal other features to lower AGA on the differential. Educating patients, barbers, and hairstylists about the importance of early intervention also may encourage earlier visits before the scarring process is advanced. Further exploration into factors impacting diagnosis and CCCA severity may uncover implications for prognosis and treatment.
This study was limited by a small sample size, retrospective design, and single-center analysis. Some patients had comorbid hair loss conditions, which could affect disease severity. Moreover, the central scalp alopecia photographic scale2 was not validated in men or designed for assessment of the nonclassical hair loss distributions noted in some of our patients. Nonetheless, we hope these data will support clinicians in efforts to advocate for early diagnosis and treatment in patients with CCCA to ultimately help improve outcomes.
- Ogunleye TA, McMichael A, Olsen EA. Central centrifugal cicatricial alopecia: what has been achieved, current clues for future research. Dermatol Clin. 2014;32:173-181. doi:10.1016/j.det.2013.12.005
- Olsen EA, Callender V, McMichael A, et al. Central hair loss in African American women: incidence and potential risk factors. J Am Acad Dermatol. 2011;64:245-252. doi:10.1016/j.jaad.2009.11.693
- Olsen EA, Callendar V, Sperling L, et al. Central scalp alopecia photographic scale in African American women. Dermatol Ther. 2008;21:264-267. doi:10.1111/j.1529-8019.2008.00208.x
- Andersen YMF, Nymand L, DeLozier AM, et al. Patient characteristics and disease burden of alopecia areata in the Danish Skin Cohort. BMJ Open. 2022;12:E053137. doi:10.1136/bmjopen-2021-053137
- Davis EC, Reid SD, Callender VD, et al. Differentiating central centrifugal cicatricial alopecia and androgenetic alopecia in African American men. J Clin Aesthetic Dermatol. 2012;5:37-40.
- Jackson TK, Sow Y, Ayoade KO, et al. Central centrifugal cicatricial alopecia in males. J Am Acad Dermatol. 2023;89:1136-1140. doi:10.1016/j.jaad.2023.07.1011
- Lawson CN, Bakayoko A, Callender VD. Central centrifugal cicatricial alopecia: challenges and treatments. Dermatol Clin. 2021;39:389-405. doi:10.1016/j.det.2021.03.004
- Ogunleye TA, McMichael A, Olsen EA. Central centrifugal cicatricial alopecia: what has been achieved, current clues for future research. Dermatol Clin. 2014;32:173-181. doi:10.1016/j.det.2013.12.005
- Olsen EA, Callender V, McMichael A, et al. Central hair loss in African American women: incidence and potential risk factors. J Am Acad Dermatol. 2011;64:245-252. doi:10.1016/j.jaad.2009.11.693
- Olsen EA, Callendar V, Sperling L, et al. Central scalp alopecia photographic scale in African American women. Dermatol Ther. 2008;21:264-267. doi:10.1111/j.1529-8019.2008.00208.x
- Andersen YMF, Nymand L, DeLozier AM, et al. Patient characteristics and disease burden of alopecia areata in the Danish Skin Cohort. BMJ Open. 2022;12:E053137. doi:10.1136/bmjopen-2021-053137
- Davis EC, Reid SD, Callender VD, et al. Differentiating central centrifugal cicatricial alopecia and androgenetic alopecia in African American men. J Clin Aesthetic Dermatol. 2012;5:37-40.
- Jackson TK, Sow Y, Ayoade KO, et al. Central centrifugal cicatricial alopecia in males. J Am Acad Dermatol. 2023;89:1136-1140. doi:10.1016/j.jaad.2023.07.1011
- Lawson CN, Bakayoko A, Callender VD. Central centrifugal cicatricial alopecia: challenges and treatments. Dermatol Clin. 2021;39:389-405. doi:10.1016/j.det.2021.03.004
Practice Points
- Most males with central centrifugal cicatricial alopecia (CCCA) experience considerable diagnostic delays and typically present to dermatology with late-stage disease.
- Dermatologists should consider CCCA in the differential diagnosis for adult Black males with alopecia.
- More research is needed to explore advanced CCCA in males, including factors limiting timely diagnosis and the impact on quality of life in this population.
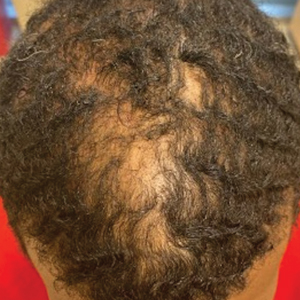
The Impact of the Recent Supreme Court Ruling on the Dermatology Recruitment Pipeline
The ruling by the Supreme Court of the United States (SCOTUS) in 20231,2 on the use of race-based criteria in college admissions was met with a range of reactions across the country. Given the implications of this decision on the future makeup of higher education, the downstream effects on medical school admissions, and the possible further impact on graduate medical education programs, we sought to explore the potential impact of the landmark decision from the perspective of dermatology residency program directors and offer insights on this pivotal judgment.
Background on the SCOTUS Ruling
In June 2023, SCOTUS issued its formal decision on 2 court cases brought by the organization Students for Fair Admissions (SFFA) against the University of North Carolina at Chapel Hill1 and Harvard University (Cambridge, Massachusetts)2 that addressed college admissions practices dealing with the use of race as a selection criterion in the application process. The cases alleged that these universities had overly emphasized race in the admissions process and thus were in violation of the Civil Rights Act of 1964 as well as the 14th Amendment.1,2
The SCOTUS justices voted 6 to 3 in favor of the argument presented by the SFFA, determining that the use of race in the college admissions process essentially constituted a form of racial discrimination. The ruling was in contrast to a prior decision in 2003 that centered on law school admissions at the University of Michigan (Ann Arbor, Michigan) in which SCOTUS previously had determined that race could be used as one factor amongst other criteria in the higher education selection process.3 In the 2023 decision siding with SFFA, SCOTUS did acknowledge that it was still acceptable for selection processes to consider “an applicant’s discussion of how race affected his or her life, be it through discrimination, inspiration, or otherwise.”2
Effect on Undergraduate Admissions
Prior to the 2023 ruling, several states had already passed independent laws against the use of affirmative action or race-based selection criteria in the admissions process at public colleges and universities.4 As a result, these institutions would already be conforming to the principles set forth in the SCOTUS ruling and major changes to their undergraduate admissions policies would not be expected; however, a considerable number of colleges and universities—particularly those considered highly selective with applicant acceptance rates that are well below the national average—reported the use of race as a factor in their admissions processes in standardized reporting surveys.5 For these institutions, it is no longer considered acceptable (based on the SCOTUS decision) to use race as a singular factor in admissions or to implement race-conscious decision-making—in which individuals are considered differently based solely on their race—as part of the undergraduate selection process.
In light of these rulings, many institutions have explicitly committed to upholding principles of diversity in their recruitment processes, acknowledging the multifaceted nature of diversity beyond strictly racial terms—including but not limited to socioeconomic diversity, religious diversity, or gender diversity—which is in compliance with the interpretation ruling by the US Department of Education and the US Department of Justice.6 Additionally, select institutions have taken approaches to explicitly include questions on ways in which applicants have overcome obstacles or challenges, allowing an opportunity for individuals who have had such experiences related to race an opportunity to incorporate these elements into their applications. Finally, some institutions have taken a more limited approach, eliminating ways in which race is explicitly addressed in the application and focusing on race-neutral elements of the application in their approach to selection.7
Because the first college admission cycle since the 2023 SCOTUS ruling is still underway, we have yet to witness the full impact of this decision on the current undergraduate admissions landscape.
Effect on Medical School Admissions and Rotations
Although SCOTUS specifically examined the undergraduate admissions process, the ruling on race-conscious admissions also had a profound impact on graduate school admissions including medical school admission processes.1,2,8,9 This is because the language of the majority opinion refers to “university programs” in its ruling, which also has been broadly interpreted to include graduate school programs. As with undergraduate admissions, it has been interpreted by national medical education organizations and institutions that medical schools also cannot consider an applicant’s race or ethnicity as a specific factor in the admissions process.1,2,8,9
Lived individual experiences, including essays that speak to an applicant’s lived experiences and career aspirations related to race, still can be taken into account. In particular, holistic review still can be utilized to evaluate medical school candidates and may play a more integral role in the medical school admissions process now than in the past.8,10,11 After the ruling, Justice Sonia Sotomayor noted that “today’s decision leaves intact holistic college admissions and recruitment efforts that seek to enroll diverse classes without using racial classifications.”1
The ruling asserted that universities may define their mission as they see fit. As a result, the ruling did not affect medical school missions or strategic plans, including those that may aim to diversify the health care workforce.8,10,11 The ruling also did not affect the ability to utilize pathway programs to encourage a career in medicine or recruitment relationships with diverse undergraduate or community-based organizations. Student interest groups also can be involved in the relationship-building or recruitment activities for medical schools.8,10,11 Guidance from the US Department of Education and US Department of Justice noted that institutions may consider race in identifying prospective applicants through recruitment and outreach, “provided that their outreach and recruitment programs do not provide targeted groups of prospective students preference in the admissions process, and provided that all students—whether part of a specifically targeted group or not—enjoy the same opportunity to apply and compete for admission.”12
In regard to pathways programs, slots cannot be reserved and preference cannot be given to applicants who participated in these programs if race was a factor in selecting participants.8 Similarly, medical school away electives related to diversity cannot be reserved for those of a specific race or ethnicity; however, these electives can utilize commitment to stated aims and missions of the rotation, such as a commitment to diversity within medicine, as a basis to selecting candidates.8
The ruling did not address how race or ethnicity is factored into financial aid or scholarship determination. There has been concern in higher education that the legal framework utilized in the SCOTUS decision could affect financial aid and scholarship decisions; therefore, many institutions are proceeding with caution in their approach.8
Effect on Residency Selection
Because the SCOTUS ruling references colleges and universities, not health care employers, it should not affect the residency selection process; however, there is variability in how health care institutions are interpreting the impact of the ruling on residency selection, with some taking a more prescriptive and cautious view on the matter. Additionally, with that said, residency selection is considered an employment practice covered by Title VII of the Civil Rights Act of 1964,13 which already prohibits the consideration of race in hiring decisions.7 Under Title VII, it is unlawful for employers to discriminate against someone because of race, color, religion, sex, or national origin, and it is “unlawful to use policies or practices that seem neutral but have the effect of discriminating against people because of their race, color, religion, sex … or national origin.” Title VII also states that employers cannot “make employment decisions based on stereotypes or assumptions about a person’s abilities, traits, or performance because of their race, color, religion, sex … or national origin.”13
Importantly, Title VII does not imply that employers need to abandon their diversity, equity, or inclusion initiatives, and it does not imply that employers must revoke their mission to improve diversity in the workforce. Title VII does not state that racial information cannot be available. It would be permissible to use racial data to assess recruitment trends, identify inequities, and create programs to eliminate barriers and decrease bias14; for example, if a program identified that, based on their current review system, students who are underrepresented in medicine were disproportionately screened out of the applicant pool or interview group, they may wish to revisit their review process to identify and eliminate possible biases. Programs also may wish to adopt educational programs for reviewers (eg, implicit bias training) or educational content on the potential for bias in commonly used review criteria, such as the US Medical Licensing Examination, clerkship grades, and the Medical Student Performance Evaluation.15 Reviewers can and should consider applications in an individualized and holistic manner in which experiences, traits, skills, and academic metrics are assessed together for compatibility with the values and mission of the training program.16
Future Directions for Dermatology
Beyond the SCOTUS ruling, there have been other shifts in the dermatology residency application process that have affected candidate review. Dermatology programs recently have adopted the use of preference signaling in residency applications. Preliminary data from the Association of American Medical Colleges for the 2024 application cycle indicated that of the 81 programs analyzed, there was a nearly 0% chance of an applicant receiving an interview invitation from a program that they did not signal. The median signal-to-interview conversion rate for the 81 dermatology programs analyzed was 55% for gold signals and 15% for silver signals.17 It can be inferred from these data that programs are using preference signaling as important criteria for consideration of interview invitation. Programs may choose to focus most of their attention on the applicant pool who has signaled them. Because the number and type of signals available is equal among all applicants, we hope that this provides an equitable way for all applicants to garner holistic review from programs that interested them. In addition, there has been a 30% decrease in average applications submitted per dermatology applicant.18 With a substantial decline in applications to dermatology, we hope that reviewers are able to spend more time devoted to comprehensive holistic review.
Although signals are equitable for applicants, their distribution among programs may not be; for example, in a given year, a program might find that all their gold signals came from non–underrepresented in medicine students. We encourage programs to carefully review applicant data to ensure their recruitment process is not inadvertently discriminatory and is in alignment with their goals and mission.
- Students for Fair Admissions, Inc. v University of North Carolina, 567 F. Supp. 3d 580 (M.D.N.C. 2021).
- Students for Fair Admissions, Inc. v President and Fellows of Harvard College, 600 US ___ (2023).
- Grutter v Bollinger, 539 US 306 (2003).
- Saul S. 9 states have banned affirmative action. here’s what that looks like. The New York Times. October 31, 2022. https://www.nytimes.com/2022/10/31/us/politics/affirmative-action-ban-states.html
- Desilver D. Private, selective colleges are most likely to use race, ethnicity as a factor in admissions decisions. Pew Research Center. July 14, 2023. Accessed May 29, 2024. https://www.pewresearch.org/short-reads/2023/07/14/private-selective-colleges-are-most-likely-to-use-race-ethnicity-as-a-factor-in-admissions-decisions/
- US Department of Education. Justice and education departments release resources to advance diversity and opportunity in higher education. August 14, 2023. Accessed May 17, 2024. https://www.ed.gov/news/press-releases/advance-diversity-and-opportunity-higher-education-justice-and-education-departments-release-resources-advance-diversity-and-opportunity-higher-education
- Amponsah MN, Hamid RD. Harvard overhauls college application in wake of affirmative action decision. The Harvard Crimson. August 3, 2023. Accessed May 17, 2024. https://www.thecrimson.com/article/2023/8/3/harvard-admission-essay-change/
- Association of American Medical Colleges. Frequently asked questions: what does the Harvard and UNC decision mean for medical education? August 24, 2023. Accessed May 17, 2024. https://www.aamc.org/media/68771/download?attachment%3Fattachment
- American Medical Association. Affirmative action ends: how Supreme Court ruling impacts medical schools & the health care workforce. July 7, 2023. Accessed May 17, 2024. https://www.ama-assn.org/medical-students/medical-school-life/affirmative-action-ends-how-supreme-court-ruling-impacts
- Association of American Medical Colleges. How can medical schools boost racial diversity in the wake of the recent Supreme Court ruling? July 27, 2023. Accessed May 17, 2024. https://www.aamc.org/news/how-can-medical-schools-boost-racial-diversity-wake-recent-supreme-court-ruling
- Association of American Medical Colleges. Diversity in medical school admissions. Updated March 18, 2024. Accessed May 17, 2024. https://www.aamc.org/about-us/mission-areas/medical-education/diversity-medical-school-admissions
- United States Department of Justice. Questions and answers regarding the Supreme Court’s decision in Students For Fair Admissions, Inc. v. Harvard College and University of North Carolina. August 14, 2023. Accessed May 29, 2024. https://www.justice.gov/d9/2023-08/post-sffa_resource_faq_final_508.pdf
- US Department of Justice. Title VII of the Civil Rights Act of 1964. Accessed May 17, 2024. https://www.justice.gov/crt/laws-we-enforce
- Zheng L. How to effectively—and legally—use racial data for DEI. Harvard Business Review. July 24, 2023. Accessed May 17, 2024. https://hbr.org/2023/07/how-to-effectively-and-legally-use-racial-data-for-dei
- Crites K, Johnson J, Scott N, et al. Increasing diversity in residency training programs. Cureus. 2022;14:E25962. doi:10.7759/cureus.25962
- Association of American Medical Colleges. Holistic principles in resident selection: an introduction. Accessed May 17, 2024. https://www.aamc.org/media/44586/download?attachment
- Association of American Medical Colleges. Exploring the relationship between program signaling & interview invitations across specialties 2024 ERAS® preliminary analysis. December 29, 2023. Accessed May 17, 2024. https://www.aamc.org/media/74811/download?attachment
- Association of American Medical Colleges. Preliminary program signaling data and their impact on residency selection. October 24, 2023. Accessed May 17, 2024. https://www.aamc.org/services/eras-institutions/program-signaling-data#:~:text=Preliminary%20Program%20Signaling%20Data%20and%20Their%20Impact%20on%20Residency%20Selection,-Oct.&text=Program%20signals%20are%20a%20mechanism,whom%20to%20invite%20for%20interview
The ruling by the Supreme Court of the United States (SCOTUS) in 20231,2 on the use of race-based criteria in college admissions was met with a range of reactions across the country. Given the implications of this decision on the future makeup of higher education, the downstream effects on medical school admissions, and the possible further impact on graduate medical education programs, we sought to explore the potential impact of the landmark decision from the perspective of dermatology residency program directors and offer insights on this pivotal judgment.
Background on the SCOTUS Ruling
In June 2023, SCOTUS issued its formal decision on 2 court cases brought by the organization Students for Fair Admissions (SFFA) against the University of North Carolina at Chapel Hill1 and Harvard University (Cambridge, Massachusetts)2 that addressed college admissions practices dealing with the use of race as a selection criterion in the application process. The cases alleged that these universities had overly emphasized race in the admissions process and thus were in violation of the Civil Rights Act of 1964 as well as the 14th Amendment.1,2
The SCOTUS justices voted 6 to 3 in favor of the argument presented by the SFFA, determining that the use of race in the college admissions process essentially constituted a form of racial discrimination. The ruling was in contrast to a prior decision in 2003 that centered on law school admissions at the University of Michigan (Ann Arbor, Michigan) in which SCOTUS previously had determined that race could be used as one factor amongst other criteria in the higher education selection process.3 In the 2023 decision siding with SFFA, SCOTUS did acknowledge that it was still acceptable for selection processes to consider “an applicant’s discussion of how race affected his or her life, be it through discrimination, inspiration, or otherwise.”2
Effect on Undergraduate Admissions
Prior to the 2023 ruling, several states had already passed independent laws against the use of affirmative action or race-based selection criteria in the admissions process at public colleges and universities.4 As a result, these institutions would already be conforming to the principles set forth in the SCOTUS ruling and major changes to their undergraduate admissions policies would not be expected; however, a considerable number of colleges and universities—particularly those considered highly selective with applicant acceptance rates that are well below the national average—reported the use of race as a factor in their admissions processes in standardized reporting surveys.5 For these institutions, it is no longer considered acceptable (based on the SCOTUS decision) to use race as a singular factor in admissions or to implement race-conscious decision-making—in which individuals are considered differently based solely on their race—as part of the undergraduate selection process.
In light of these rulings, many institutions have explicitly committed to upholding principles of diversity in their recruitment processes, acknowledging the multifaceted nature of diversity beyond strictly racial terms—including but not limited to socioeconomic diversity, religious diversity, or gender diversity—which is in compliance with the interpretation ruling by the US Department of Education and the US Department of Justice.6 Additionally, select institutions have taken approaches to explicitly include questions on ways in which applicants have overcome obstacles or challenges, allowing an opportunity for individuals who have had such experiences related to race an opportunity to incorporate these elements into their applications. Finally, some institutions have taken a more limited approach, eliminating ways in which race is explicitly addressed in the application and focusing on race-neutral elements of the application in their approach to selection.7
Because the first college admission cycle since the 2023 SCOTUS ruling is still underway, we have yet to witness the full impact of this decision on the current undergraduate admissions landscape.
Effect on Medical School Admissions and Rotations
Although SCOTUS specifically examined the undergraduate admissions process, the ruling on race-conscious admissions also had a profound impact on graduate school admissions including medical school admission processes.1,2,8,9 This is because the language of the majority opinion refers to “university programs” in its ruling, which also has been broadly interpreted to include graduate school programs. As with undergraduate admissions, it has been interpreted by national medical education organizations and institutions that medical schools also cannot consider an applicant’s race or ethnicity as a specific factor in the admissions process.1,2,8,9
Lived individual experiences, including essays that speak to an applicant’s lived experiences and career aspirations related to race, still can be taken into account. In particular, holistic review still can be utilized to evaluate medical school candidates and may play a more integral role in the medical school admissions process now than in the past.8,10,11 After the ruling, Justice Sonia Sotomayor noted that “today’s decision leaves intact holistic college admissions and recruitment efforts that seek to enroll diverse classes without using racial classifications.”1
The ruling asserted that universities may define their mission as they see fit. As a result, the ruling did not affect medical school missions or strategic plans, including those that may aim to diversify the health care workforce.8,10,11 The ruling also did not affect the ability to utilize pathway programs to encourage a career in medicine or recruitment relationships with diverse undergraduate or community-based organizations. Student interest groups also can be involved in the relationship-building or recruitment activities for medical schools.8,10,11 Guidance from the US Department of Education and US Department of Justice noted that institutions may consider race in identifying prospective applicants through recruitment and outreach, “provided that their outreach and recruitment programs do not provide targeted groups of prospective students preference in the admissions process, and provided that all students—whether part of a specifically targeted group or not—enjoy the same opportunity to apply and compete for admission.”12
In regard to pathways programs, slots cannot be reserved and preference cannot be given to applicants who participated in these programs if race was a factor in selecting participants.8 Similarly, medical school away electives related to diversity cannot be reserved for those of a specific race or ethnicity; however, these electives can utilize commitment to stated aims and missions of the rotation, such as a commitment to diversity within medicine, as a basis to selecting candidates.8
The ruling did not address how race or ethnicity is factored into financial aid or scholarship determination. There has been concern in higher education that the legal framework utilized in the SCOTUS decision could affect financial aid and scholarship decisions; therefore, many institutions are proceeding with caution in their approach.8
Effect on Residency Selection
Because the SCOTUS ruling references colleges and universities, not health care employers, it should not affect the residency selection process; however, there is variability in how health care institutions are interpreting the impact of the ruling on residency selection, with some taking a more prescriptive and cautious view on the matter. Additionally, with that said, residency selection is considered an employment practice covered by Title VII of the Civil Rights Act of 1964,13 which already prohibits the consideration of race in hiring decisions.7 Under Title VII, it is unlawful for employers to discriminate against someone because of race, color, religion, sex, or national origin, and it is “unlawful to use policies or practices that seem neutral but have the effect of discriminating against people because of their race, color, religion, sex … or national origin.” Title VII also states that employers cannot “make employment decisions based on stereotypes or assumptions about a person’s abilities, traits, or performance because of their race, color, religion, sex … or national origin.”13
Importantly, Title VII does not imply that employers need to abandon their diversity, equity, or inclusion initiatives, and it does not imply that employers must revoke their mission to improve diversity in the workforce. Title VII does not state that racial information cannot be available. It would be permissible to use racial data to assess recruitment trends, identify inequities, and create programs to eliminate barriers and decrease bias14; for example, if a program identified that, based on their current review system, students who are underrepresented in medicine were disproportionately screened out of the applicant pool or interview group, they may wish to revisit their review process to identify and eliminate possible biases. Programs also may wish to adopt educational programs for reviewers (eg, implicit bias training) or educational content on the potential for bias in commonly used review criteria, such as the US Medical Licensing Examination, clerkship grades, and the Medical Student Performance Evaluation.15 Reviewers can and should consider applications in an individualized and holistic manner in which experiences, traits, skills, and academic metrics are assessed together for compatibility with the values and mission of the training program.16
Future Directions for Dermatology
Beyond the SCOTUS ruling, there have been other shifts in the dermatology residency application process that have affected candidate review. Dermatology programs recently have adopted the use of preference signaling in residency applications. Preliminary data from the Association of American Medical Colleges for the 2024 application cycle indicated that of the 81 programs analyzed, there was a nearly 0% chance of an applicant receiving an interview invitation from a program that they did not signal. The median signal-to-interview conversion rate for the 81 dermatology programs analyzed was 55% for gold signals and 15% for silver signals.17 It can be inferred from these data that programs are using preference signaling as important criteria for consideration of interview invitation. Programs may choose to focus most of their attention on the applicant pool who has signaled them. Because the number and type of signals available is equal among all applicants, we hope that this provides an equitable way for all applicants to garner holistic review from programs that interested them. In addition, there has been a 30% decrease in average applications submitted per dermatology applicant.18 With a substantial decline in applications to dermatology, we hope that reviewers are able to spend more time devoted to comprehensive holistic review.
Although signals are equitable for applicants, their distribution among programs may not be; for example, in a given year, a program might find that all their gold signals came from non–underrepresented in medicine students. We encourage programs to carefully review applicant data to ensure their recruitment process is not inadvertently discriminatory and is in alignment with their goals and mission.
The ruling by the Supreme Court of the United States (SCOTUS) in 20231,2 on the use of race-based criteria in college admissions was met with a range of reactions across the country. Given the implications of this decision on the future makeup of higher education, the downstream effects on medical school admissions, and the possible further impact on graduate medical education programs, we sought to explore the potential impact of the landmark decision from the perspective of dermatology residency program directors and offer insights on this pivotal judgment.
Background on the SCOTUS Ruling
In June 2023, SCOTUS issued its formal decision on 2 court cases brought by the organization Students for Fair Admissions (SFFA) against the University of North Carolina at Chapel Hill1 and Harvard University (Cambridge, Massachusetts)2 that addressed college admissions practices dealing with the use of race as a selection criterion in the application process. The cases alleged that these universities had overly emphasized race in the admissions process and thus were in violation of the Civil Rights Act of 1964 as well as the 14th Amendment.1,2
The SCOTUS justices voted 6 to 3 in favor of the argument presented by the SFFA, determining that the use of race in the college admissions process essentially constituted a form of racial discrimination. The ruling was in contrast to a prior decision in 2003 that centered on law school admissions at the University of Michigan (Ann Arbor, Michigan) in which SCOTUS previously had determined that race could be used as one factor amongst other criteria in the higher education selection process.3 In the 2023 decision siding with SFFA, SCOTUS did acknowledge that it was still acceptable for selection processes to consider “an applicant’s discussion of how race affected his or her life, be it through discrimination, inspiration, or otherwise.”2
Effect on Undergraduate Admissions
Prior to the 2023 ruling, several states had already passed independent laws against the use of affirmative action or race-based selection criteria in the admissions process at public colleges and universities.4 As a result, these institutions would already be conforming to the principles set forth in the SCOTUS ruling and major changes to their undergraduate admissions policies would not be expected; however, a considerable number of colleges and universities—particularly those considered highly selective with applicant acceptance rates that are well below the national average—reported the use of race as a factor in their admissions processes in standardized reporting surveys.5 For these institutions, it is no longer considered acceptable (based on the SCOTUS decision) to use race as a singular factor in admissions or to implement race-conscious decision-making—in which individuals are considered differently based solely on their race—as part of the undergraduate selection process.
In light of these rulings, many institutions have explicitly committed to upholding principles of diversity in their recruitment processes, acknowledging the multifaceted nature of diversity beyond strictly racial terms—including but not limited to socioeconomic diversity, religious diversity, or gender diversity—which is in compliance with the interpretation ruling by the US Department of Education and the US Department of Justice.6 Additionally, select institutions have taken approaches to explicitly include questions on ways in which applicants have overcome obstacles or challenges, allowing an opportunity for individuals who have had such experiences related to race an opportunity to incorporate these elements into their applications. Finally, some institutions have taken a more limited approach, eliminating ways in which race is explicitly addressed in the application and focusing on race-neutral elements of the application in their approach to selection.7
Because the first college admission cycle since the 2023 SCOTUS ruling is still underway, we have yet to witness the full impact of this decision on the current undergraduate admissions landscape.
Effect on Medical School Admissions and Rotations
Although SCOTUS specifically examined the undergraduate admissions process, the ruling on race-conscious admissions also had a profound impact on graduate school admissions including medical school admission processes.1,2,8,9 This is because the language of the majority opinion refers to “university programs” in its ruling, which also has been broadly interpreted to include graduate school programs. As with undergraduate admissions, it has been interpreted by national medical education organizations and institutions that medical schools also cannot consider an applicant’s race or ethnicity as a specific factor in the admissions process.1,2,8,9
Lived individual experiences, including essays that speak to an applicant’s lived experiences and career aspirations related to race, still can be taken into account. In particular, holistic review still can be utilized to evaluate medical school candidates and may play a more integral role in the medical school admissions process now than in the past.8,10,11 After the ruling, Justice Sonia Sotomayor noted that “today’s decision leaves intact holistic college admissions and recruitment efforts that seek to enroll diverse classes without using racial classifications.”1
The ruling asserted that universities may define their mission as they see fit. As a result, the ruling did not affect medical school missions or strategic plans, including those that may aim to diversify the health care workforce.8,10,11 The ruling also did not affect the ability to utilize pathway programs to encourage a career in medicine or recruitment relationships with diverse undergraduate or community-based organizations. Student interest groups also can be involved in the relationship-building or recruitment activities for medical schools.8,10,11 Guidance from the US Department of Education and US Department of Justice noted that institutions may consider race in identifying prospective applicants through recruitment and outreach, “provided that their outreach and recruitment programs do not provide targeted groups of prospective students preference in the admissions process, and provided that all students—whether part of a specifically targeted group or not—enjoy the same opportunity to apply and compete for admission.”12
In regard to pathways programs, slots cannot be reserved and preference cannot be given to applicants who participated in these programs if race was a factor in selecting participants.8 Similarly, medical school away electives related to diversity cannot be reserved for those of a specific race or ethnicity; however, these electives can utilize commitment to stated aims and missions of the rotation, such as a commitment to diversity within medicine, as a basis to selecting candidates.8
The ruling did not address how race or ethnicity is factored into financial aid or scholarship determination. There has been concern in higher education that the legal framework utilized in the SCOTUS decision could affect financial aid and scholarship decisions; therefore, many institutions are proceeding with caution in their approach.8
Effect on Residency Selection
Because the SCOTUS ruling references colleges and universities, not health care employers, it should not affect the residency selection process; however, there is variability in how health care institutions are interpreting the impact of the ruling on residency selection, with some taking a more prescriptive and cautious view on the matter. Additionally, with that said, residency selection is considered an employment practice covered by Title VII of the Civil Rights Act of 1964,13 which already prohibits the consideration of race in hiring decisions.7 Under Title VII, it is unlawful for employers to discriminate against someone because of race, color, religion, sex, or national origin, and it is “unlawful to use policies or practices that seem neutral but have the effect of discriminating against people because of their race, color, religion, sex … or national origin.” Title VII also states that employers cannot “make employment decisions based on stereotypes or assumptions about a person’s abilities, traits, or performance because of their race, color, religion, sex … or national origin.”13
Importantly, Title VII does not imply that employers need to abandon their diversity, equity, or inclusion initiatives, and it does not imply that employers must revoke their mission to improve diversity in the workforce. Title VII does not state that racial information cannot be available. It would be permissible to use racial data to assess recruitment trends, identify inequities, and create programs to eliminate barriers and decrease bias14; for example, if a program identified that, based on their current review system, students who are underrepresented in medicine were disproportionately screened out of the applicant pool or interview group, they may wish to revisit their review process to identify and eliminate possible biases. Programs also may wish to adopt educational programs for reviewers (eg, implicit bias training) or educational content on the potential for bias in commonly used review criteria, such as the US Medical Licensing Examination, clerkship grades, and the Medical Student Performance Evaluation.15 Reviewers can and should consider applications in an individualized and holistic manner in which experiences, traits, skills, and academic metrics are assessed together for compatibility with the values and mission of the training program.16
Future Directions for Dermatology
Beyond the SCOTUS ruling, there have been other shifts in the dermatology residency application process that have affected candidate review. Dermatology programs recently have adopted the use of preference signaling in residency applications. Preliminary data from the Association of American Medical Colleges for the 2024 application cycle indicated that of the 81 programs analyzed, there was a nearly 0% chance of an applicant receiving an interview invitation from a program that they did not signal. The median signal-to-interview conversion rate for the 81 dermatology programs analyzed was 55% for gold signals and 15% for silver signals.17 It can be inferred from these data that programs are using preference signaling as important criteria for consideration of interview invitation. Programs may choose to focus most of their attention on the applicant pool who has signaled them. Because the number and type of signals available is equal among all applicants, we hope that this provides an equitable way for all applicants to garner holistic review from programs that interested them. In addition, there has been a 30% decrease in average applications submitted per dermatology applicant.18 With a substantial decline in applications to dermatology, we hope that reviewers are able to spend more time devoted to comprehensive holistic review.
Although signals are equitable for applicants, their distribution among programs may not be; for example, in a given year, a program might find that all their gold signals came from non–underrepresented in medicine students. We encourage programs to carefully review applicant data to ensure their recruitment process is not inadvertently discriminatory and is in alignment with their goals and mission.
- Students for Fair Admissions, Inc. v University of North Carolina, 567 F. Supp. 3d 580 (M.D.N.C. 2021).
- Students for Fair Admissions, Inc. v President and Fellows of Harvard College, 600 US ___ (2023).
- Grutter v Bollinger, 539 US 306 (2003).
- Saul S. 9 states have banned affirmative action. here’s what that looks like. The New York Times. October 31, 2022. https://www.nytimes.com/2022/10/31/us/politics/affirmative-action-ban-states.html
- Desilver D. Private, selective colleges are most likely to use race, ethnicity as a factor in admissions decisions. Pew Research Center. July 14, 2023. Accessed May 29, 2024. https://www.pewresearch.org/short-reads/2023/07/14/private-selective-colleges-are-most-likely-to-use-race-ethnicity-as-a-factor-in-admissions-decisions/
- US Department of Education. Justice and education departments release resources to advance diversity and opportunity in higher education. August 14, 2023. Accessed May 17, 2024. https://www.ed.gov/news/press-releases/advance-diversity-and-opportunity-higher-education-justice-and-education-departments-release-resources-advance-diversity-and-opportunity-higher-education
- Amponsah MN, Hamid RD. Harvard overhauls college application in wake of affirmative action decision. The Harvard Crimson. August 3, 2023. Accessed May 17, 2024. https://www.thecrimson.com/article/2023/8/3/harvard-admission-essay-change/
- Association of American Medical Colleges. Frequently asked questions: what does the Harvard and UNC decision mean for medical education? August 24, 2023. Accessed May 17, 2024. https://www.aamc.org/media/68771/download?attachment%3Fattachment
- American Medical Association. Affirmative action ends: how Supreme Court ruling impacts medical schools & the health care workforce. July 7, 2023. Accessed May 17, 2024. https://www.ama-assn.org/medical-students/medical-school-life/affirmative-action-ends-how-supreme-court-ruling-impacts
- Association of American Medical Colleges. How can medical schools boost racial diversity in the wake of the recent Supreme Court ruling? July 27, 2023. Accessed May 17, 2024. https://www.aamc.org/news/how-can-medical-schools-boost-racial-diversity-wake-recent-supreme-court-ruling
- Association of American Medical Colleges. Diversity in medical school admissions. Updated March 18, 2024. Accessed May 17, 2024. https://www.aamc.org/about-us/mission-areas/medical-education/diversity-medical-school-admissions
- United States Department of Justice. Questions and answers regarding the Supreme Court’s decision in Students For Fair Admissions, Inc. v. Harvard College and University of North Carolina. August 14, 2023. Accessed May 29, 2024. https://www.justice.gov/d9/2023-08/post-sffa_resource_faq_final_508.pdf
- US Department of Justice. Title VII of the Civil Rights Act of 1964. Accessed May 17, 2024. https://www.justice.gov/crt/laws-we-enforce
- Zheng L. How to effectively—and legally—use racial data for DEI. Harvard Business Review. July 24, 2023. Accessed May 17, 2024. https://hbr.org/2023/07/how-to-effectively-and-legally-use-racial-data-for-dei
- Crites K, Johnson J, Scott N, et al. Increasing diversity in residency training programs. Cureus. 2022;14:E25962. doi:10.7759/cureus.25962
- Association of American Medical Colleges. Holistic principles in resident selection: an introduction. Accessed May 17, 2024. https://www.aamc.org/media/44586/download?attachment
- Association of American Medical Colleges. Exploring the relationship between program signaling & interview invitations across specialties 2024 ERAS® preliminary analysis. December 29, 2023. Accessed May 17, 2024. https://www.aamc.org/media/74811/download?attachment
- Association of American Medical Colleges. Preliminary program signaling data and their impact on residency selection. October 24, 2023. Accessed May 17, 2024. https://www.aamc.org/services/eras-institutions/program-signaling-data#:~:text=Preliminary%20Program%20Signaling%20Data%20and%20Their%20Impact%20on%20Residency%20Selection,-Oct.&text=Program%20signals%20are%20a%20mechanism,whom%20to%20invite%20for%20interview
- Students for Fair Admissions, Inc. v University of North Carolina, 567 F. Supp. 3d 580 (M.D.N.C. 2021).
- Students for Fair Admissions, Inc. v President and Fellows of Harvard College, 600 US ___ (2023).
- Grutter v Bollinger, 539 US 306 (2003).
- Saul S. 9 states have banned affirmative action. here’s what that looks like. The New York Times. October 31, 2022. https://www.nytimes.com/2022/10/31/us/politics/affirmative-action-ban-states.html
- Desilver D. Private, selective colleges are most likely to use race, ethnicity as a factor in admissions decisions. Pew Research Center. July 14, 2023. Accessed May 29, 2024. https://www.pewresearch.org/short-reads/2023/07/14/private-selective-colleges-are-most-likely-to-use-race-ethnicity-as-a-factor-in-admissions-decisions/
- US Department of Education. Justice and education departments release resources to advance diversity and opportunity in higher education. August 14, 2023. Accessed May 17, 2024. https://www.ed.gov/news/press-releases/advance-diversity-and-opportunity-higher-education-justice-and-education-departments-release-resources-advance-diversity-and-opportunity-higher-education
- Amponsah MN, Hamid RD. Harvard overhauls college application in wake of affirmative action decision. The Harvard Crimson. August 3, 2023. Accessed May 17, 2024. https://www.thecrimson.com/article/2023/8/3/harvard-admission-essay-change/
- Association of American Medical Colleges. Frequently asked questions: what does the Harvard and UNC decision mean for medical education? August 24, 2023. Accessed May 17, 2024. https://www.aamc.org/media/68771/download?attachment%3Fattachment
- American Medical Association. Affirmative action ends: how Supreme Court ruling impacts medical schools & the health care workforce. July 7, 2023. Accessed May 17, 2024. https://www.ama-assn.org/medical-students/medical-school-life/affirmative-action-ends-how-supreme-court-ruling-impacts
- Association of American Medical Colleges. How can medical schools boost racial diversity in the wake of the recent Supreme Court ruling? July 27, 2023. Accessed May 17, 2024. https://www.aamc.org/news/how-can-medical-schools-boost-racial-diversity-wake-recent-supreme-court-ruling
- Association of American Medical Colleges. Diversity in medical school admissions. Updated March 18, 2024. Accessed May 17, 2024. https://www.aamc.org/about-us/mission-areas/medical-education/diversity-medical-school-admissions
- United States Department of Justice. Questions and answers regarding the Supreme Court’s decision in Students For Fair Admissions, Inc. v. Harvard College and University of North Carolina. August 14, 2023. Accessed May 29, 2024. https://www.justice.gov/d9/2023-08/post-sffa_resource_faq_final_508.pdf
- US Department of Justice. Title VII of the Civil Rights Act of 1964. Accessed May 17, 2024. https://www.justice.gov/crt/laws-we-enforce
- Zheng L. How to effectively—and legally—use racial data for DEI. Harvard Business Review. July 24, 2023. Accessed May 17, 2024. https://hbr.org/2023/07/how-to-effectively-and-legally-use-racial-data-for-dei
- Crites K, Johnson J, Scott N, et al. Increasing diversity in residency training programs. Cureus. 2022;14:E25962. doi:10.7759/cureus.25962
- Association of American Medical Colleges. Holistic principles in resident selection: an introduction. Accessed May 17, 2024. https://www.aamc.org/media/44586/download?attachment
- Association of American Medical Colleges. Exploring the relationship between program signaling & interview invitations across specialties 2024 ERAS® preliminary analysis. December 29, 2023. Accessed May 17, 2024. https://www.aamc.org/media/74811/download?attachment
- Association of American Medical Colleges. Preliminary program signaling data and their impact on residency selection. October 24, 2023. Accessed May 17, 2024. https://www.aamc.org/services/eras-institutions/program-signaling-data#:~:text=Preliminary%20Program%20Signaling%20Data%20and%20Their%20Impact%20on%20Residency%20Selection,-Oct.&text=Program%20signals%20are%20a%20mechanism,whom%20to%20invite%20for%20interview
Practice Points
- The 2023 ruling by the Supreme Court of the United States on the use of race-based criteria in college admissions may have implications for the selection of individuals into the dermatology workforce.
- We highlight the impacts of these decisions at the college, medical school, and dermatology residency levels and provide context for future directions in the selection processes for practicing dermatologists.
Erythematous Flaky Rash on the Toe
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
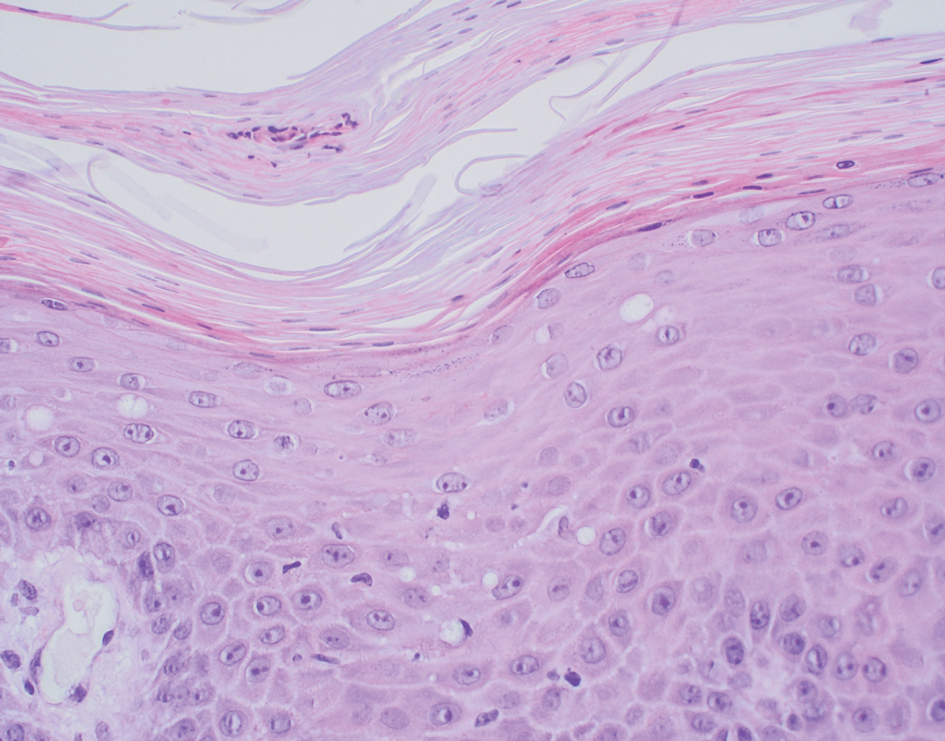
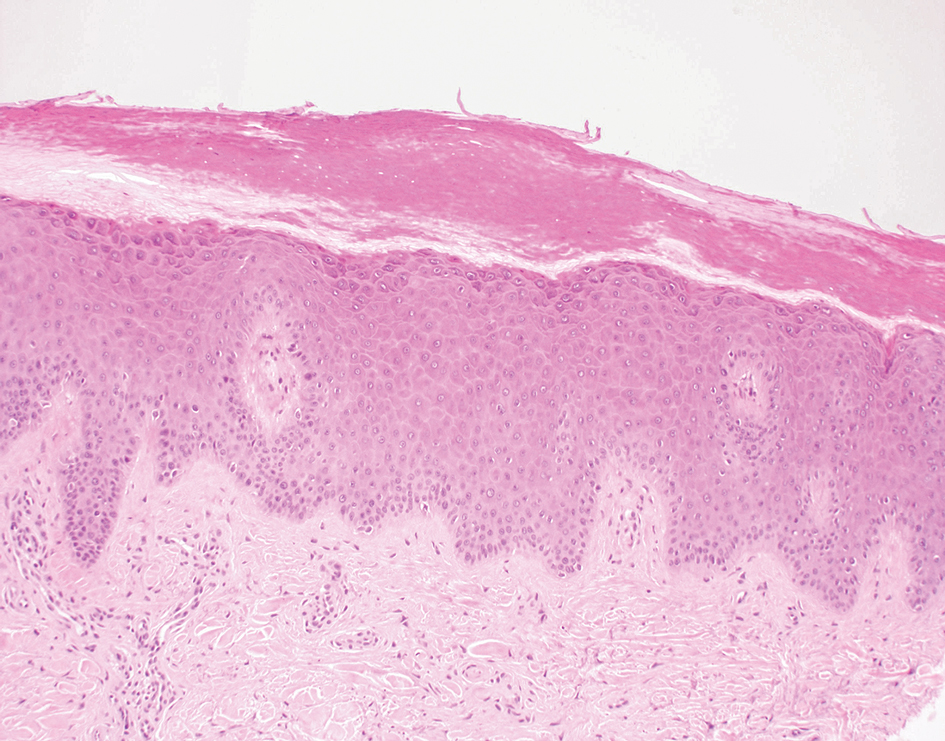
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
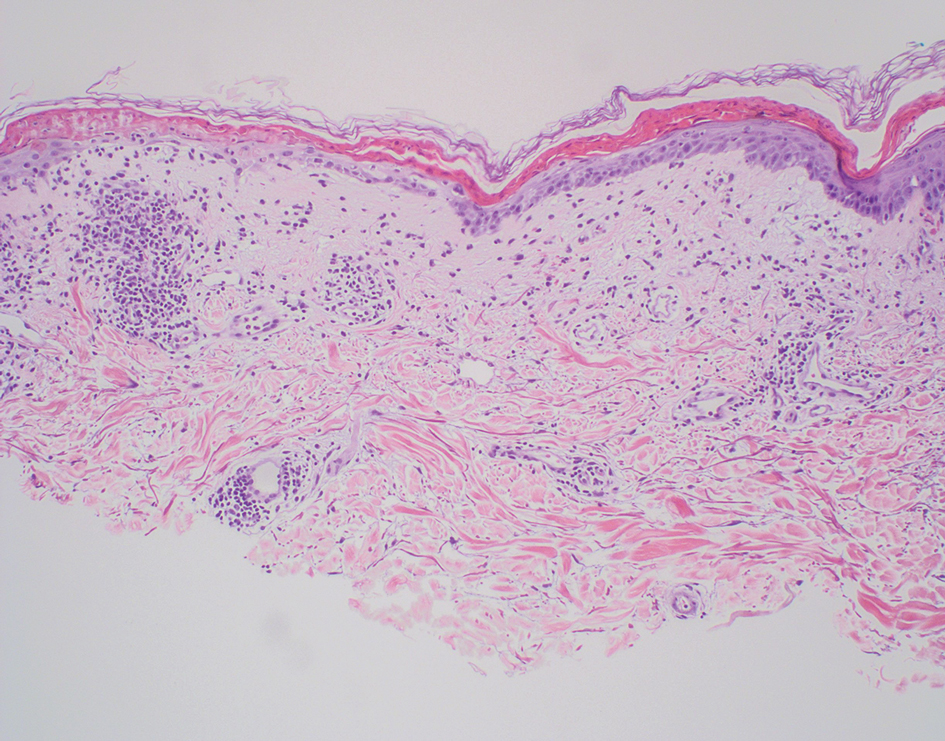
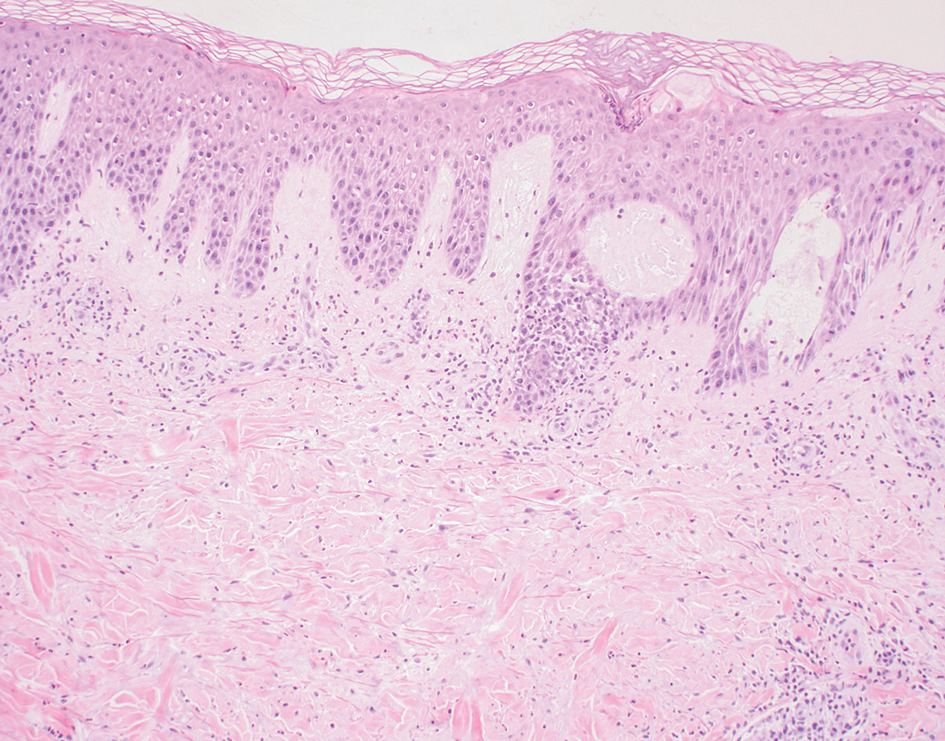
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
The Diagnosis: Necrolytic Migratory Erythema
Necrolytic migratory erythema (NME) is a waxing and waning rash associated with rare pancreatic neuroendocrine tumors called glucagonomas. It is characterized by pruritic and painful, well-demarcated, erythematous plaques that manifest in the intertriginous areas and on the perineum and buttocks.1 Due to the evolving nature of the rash, the histopathologic findings in NME vary depending on the stage of the cutaneous lesions at the time of biopsy.2 Multiple dyskeratotic keratinocytes spanning all epidermal layers may be a diagnostic clue in early lesions of NME.3 Typical features of longstanding lesions include confluent parakeratosis, psoriasiform hyperplasia with mild or absent spongiosis, and upper epidermal necrosis with keratinocyte vacuolization and pallor.4 Morphologic features that are present prior to the development of epidermal vacuolation and necrosis frequently are misattributed to psoriasis or eczema. Long-standing lesions also may develop a neutrophilic infiltrate with subcorneal and intraepidermal pustules.2 Other common features include a discrete perivascular lymphocytic infiltrate and an erosive or encrusted epidermis.5 Although direct immunofluorescence typically is negative, nonspecific findings can be seen, including apoptotic keratinocytes labeling with fibrinogen and C3, as well as scattered, clumped, IgM-positive cytoid bodies present at the dermal-epidermal junction (DEJ).6 Biopsies also have shown scattered, clumped, IgM-positive cytoid bodies present at the DEJ.5
Psoriasis is a chronic relapsing papulosquamous disorder characterized by scaly erythematous plaques often overlying the extensor surfaces of the extremities. Histopathology shows a psoriasiform pattern of inflammation with thinning of the suprapapillary plates and elongation of the rete ridges. Further diagnostic clues of psoriasis include regular acanthosis, characteristic Munro microabscesses with neutrophils in a hyperkeratotic stratum corneum (Figure 1), hypogranulosis, and neutrophilic spongiform pustules of Kogoj in the stratum spinosum. Generally, there is a lack of the epidermal necrosis seen with NME.7,8
Lichen simplex chronicus manifests as pruritic, often hyperpigmented, well-defined, lichenified plaques with excoriation following repetitive mechanical trauma, commonly on the lower lateral legs, posterior neck, and flexural areas.9 The histologic landscape is marked by well-developed lesions evolving to show compact orthokeratosis, hypergranulosis, irregularly elongated rete ridges (ie, irregular acanthosis), and papillary dermal fibrosis with vertical streaking of collagen (Figure 2).9,10
Subacute cutaneous lupus erythematosus (SCLE) is recognized clinically by scaly/psoriasiform and annular lesions with mild or absent systemic involvement. Common histopathologic findings include epidermal atrophy, vacuolar interface dermatitis with hydropic degeneration of the basal layer, a subepidermal lymphocytic infiltrate, and a periadnexal and perivascular infiltrate (Figure 3).11 Upper dermal edema, spotty necrosis of individual cells in the epidermis, dermal-epidermal separation caused by prominent basal cell degeneration, and accumulation of acid mucopolysaccharides (mucin) are other histologic features associated with SCLE.12,13
The immunofluorescence pattern in SCLE features dustlike particles of IgG deposition in the epidermis, subepidermal region, and dermal cellular infiltrate. Lesions also may have granular deposition of immunoreactions at the DEJ.11,13
The manifestation of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome (also known as drug-induced hypersensitivity syndrome) is variable, with a morbilliform rash that spreads from the face to the entire body, urticaria, atypical target lesions, purpuriform lesions, lymphadenopathy, and exfoliative dermatitis.14 The nonspecific morphologic features of DRESS syndrome lesions are associated with variable histologic features, which include focal interface changes with vacuolar alteration of the basal layer; atypical lymphocytes with hyperchromic nuclei; and a superficial, inconsistently dense, perivascular lymphocytic infiltrate. Other relatively common histopathologic patterns include an upper dermis with dilated blood vessels, spongiosis with exocytosis of lymphocytes (Figure 4), and necrotic keratinocytes. Although peripheral eosinophilia is an important diagnostic criterion and is observed consistently, eosinophils are variably present on skin biopsy.15,16 Given the histopathologic variability and nonspecific findings, clinical correlation is required when diagnosing DRESS syndrome.
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
- Halvorson SA, Gilbert E, Hopkins RS, et al. Putting the pieces together: necrolytic migratory erythema and the glucagonoma syndrome. J Gen Intern Med. 2013;28:1525-1529. doi:10.1007 /s11606-013-2490-5
- Toberer F, Hartschuh W, Wiedemeyer K. Glucagonoma-associated necrolytic migratory erythema: the broad spectrum of the clinical and histopathological findings and clues to the diagnosis. Am J Dermatopathol. 2019;41:E29-E32. doi:10.1097DAD .0000000000001219
- Hunt SJ, Narus VT, Abell E. Necrolytic migratory erythema: dyskeratotic dermatitis, a clue to early diagnosis. J Am Acad Dermatol. 1991; 24:473-477. doi:10.1016/0190-9622(91)70076-e
- van Beek AP, de Haas ER, van Vloten WA, et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol. 2004;151:531-537. doi:10.1530/eje.0.1510531
- Pujol RM, Wang C-Y E, el-Azhary RA, et al. Necrolytic migratory erythema: clinicopathologic study of 13 cases. Int J Dermatol. 2004;43:12- 18. doi:10.1111/j.1365-4632.2004.01844.x
- Johnson SM, Smoller BR, Lamps LW, et al. Necrolytic migratory erythema as the only presenting sign of a glucagonoma. J Am Acad Dermatol. 2003;49:325-328. doi:10.1067/s0190-9622(02)61774-8
- De Rosa G, Mignogna C. The histopathology of psoriasis. Reumatismo. 2007;59(suppl 1):46-48. doi:10.4081/reumatismo.2007.1s.46
- Kimmel GW, Lebwohl M. Psoriasis: overview and diagnosis. In: Bhutani T, Liao W, Nakamura M, eds. Evidence-Based Psoriasis. Springer; 2018:1-16. doi:10.1007/978-3-319-90107-7_1
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68. doi:10.22551/2019.24.0603.10155
- O’Keefe RJ, Scurry JP, Dennerstein G, et al. Audit of 114 nonneoplastic vulvar biopsies. Br J Obstet Gynaecol. 1995;102:780-786. doi:10.1111/j.1471-0528.1995.tb10842.x
- Parodi A, Caproni M, Cardinali C, et al P. Clinical, histological and immunopathological features of 58 patients with subacute cutaneous lupus erythematosus. Dermatology. 2000;200:6-10. doi:10.1159/000018307
- Lyon CC, Blewitt R, Harrison PV. Subacute cutaneous lupus erythematosus: two cases of delayed diagnosis. Acta Derm Venereol. 1998;78:57-59. doi:10.1080/00015559850135869
- David-Bajar KM. Subacute cutaneous lupus erythematosus. J Invest Dermatol. 1993;100:2S-8S. doi:10.1111/1523-1747.ep12355164
- Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625-643. doi:10.1111/ddg.12747
- Borroni G, Torti S, Pezzini C, et al. Histopathologic spectrum of drug reaction with eosinophilia and systemic symptoms (DRESS): a diagnosis that needs clinico-pathological correlation. G Ital Dermatol Venereol. 2014;149:291-300.
- Ortonne N, Valeyrie-Allanore L, Bastuji-Garin S, et al. Histopathology of drug rash with eosinophilia and systemic symptoms syndrome: a morphological and phenotypical study. Br J Dermatol. 2015;173:50-58. doi:10.1111/bjd.13683
A 62-year-old man presented with an erythematous flaky rash associated with burning pain on the right medial second toe that persisted for several months. Prior treatment with econazole, ciclopirox, and oral amoxicillin had failed. A shave biopsy was performed.
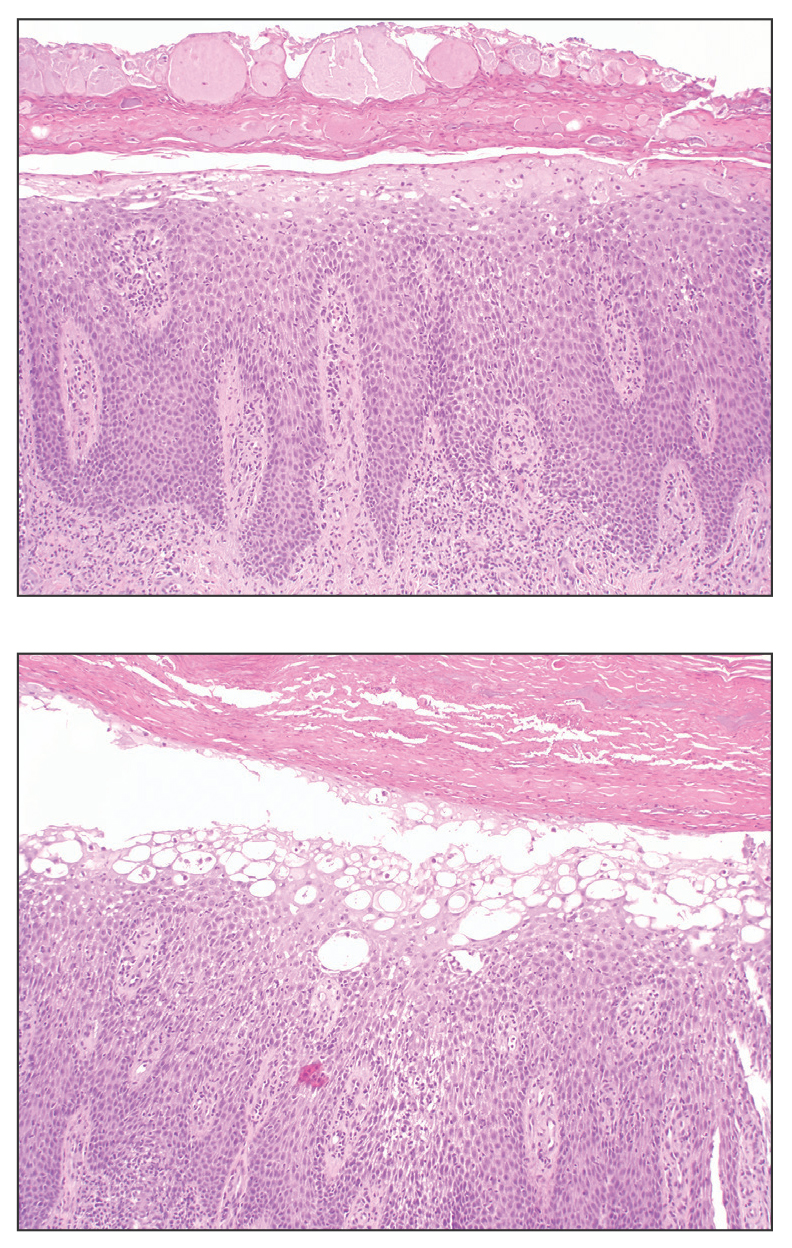
Latest Breakthroughs in Molluscum Contagiosum Therapy
Molluscum contagiosum (ie, molluscum) is a ubiquitous infection caused by the poxvirus molluscum contagiosum virus (MCV). Although skin deep, molluscum shares many factors with the more virulent poxviridae. Moisture and trauma can cause viral material to be released from the pearly papules through a small opening, which also allows entry of bacteria and medications into the lesion. The MCV is transmitted by direct contact with skin or via fomites.1
Molluscum can affect children of any age, with MCV type 1 peaking in toddlers and school-aged children and MCV type 2 after the sexual debut. The prevalence of molluscum has increased since the 1980s. It is stressful for children and caregivers and poses challenges in schools as well as sports such as swimming, wrestling, and karate.1,2
For the first time, we have US Food and Drug Administration (FDA)–approved products to treat MCV infections. Previously, only off-label agents were used. Therefore, we have to contemplate why treatment is important to our patients.
What type of care is required for molluscum?
Counseling is the first and only mandatory treatment, which consists of 3 parts: natural history, risk factors for spread, and options for therapy. The natural history of molluscum in children is early spread, contagion to oneself and others (as high as 60% of sibling co-bathers3), triggering of dermatitis, eventual onset of the beginning-of-the-end (BOTE) sign, and eventually clearance. The natural history in adults is poorly understood.
Early clearance is uncommon; reports have suggested 45.6% to 48.4% of affected patients are clear at 1 year and 69.5% to 72.6% at 1.5 years.4 For many children, especially those with atopic dermatitis (AD), lesions linger and often spread, with many experiencing disease for 3 to 4 years. Fomites such as towels, washcloths, and sponges can transfer the virus and spread lesions; therefore, I advise patients to gently pat their skin dry, wash towels frequently, and avoid sharing bathing equipment.1,3,5 Children and adults with immunosuppression may have a greater number of lesions and more prolonged course of disease, including those with HIV as well as DOC8 and CARD11 mutations.6 The American Academy of Pediatrics (AAP) emphasizes that children should not be excluded from attending child care/school or from swimming in public pools but lesions should be covered.6 Lesions, especially those in the antecubital region, can trigger new-onset AD or AD flares.3 In response, gentle skin care including fragrance-free cleansers and periodic application of moisturizers may ward off AD. Topical corticosteroids are preferred.
Dermatitis in MCV is a great mimicker and can resemble erythema multiforme, Gianotti-Crosti syndrome, impetigo, and AD.1 Superinfection recently has been reported; however, in a retrospective analysis of 56 patients with inflamed lesions secondary to molluscum infection, only 7 had positive bacterial cultures, which supports the idea of the swelling and redness of inflammation as a mimic for infection.7 When true infection does occur, tender, swollen, pus-filled lesions should be lanced and cultured.1,7,8
When should we consider therapy?
Therapy is highly dependent on the child, the caregiver, and the social circumstances.1 More than 80% of parents are anxious about molluscum, and countless children are embarrassed or ashamed.1 Ultimately, an unhappy child merits care. The AAP cites the following as reasons to treat: “(1) alleviate discomfort, including itching; (2) reduce autoinoculation; (3) limit transmission of the virus to close contacts; (4) reduce cosmetic concerns; and (5) prevent secondary infection.”6 For adults, we should consider limitations to intimacy and reduction of sexual transmission risk.6
Treatment can be based on the number of lesions. With a few lesions (<3), therapy is worthwhile if they are unsightly; appear on exposed skin causing embarrassment; and/or are itchy, uncomfortable, or large. In a report of 300 children with molluscum treated with cantharidin, most patients choosing therapy had 10 to 20 lesions, but this was over multiple visits.8 Looking at a 2018 data set of 50 patients (all-comers) with molluscum,3 the mean number of lesions was 10 (median, 7); 3 lesions were 1 SD below, while 14, 17, and 45 were 1, 2, and 3 SDs above, respectively. This data set shows that patients can develop more lesions rapidly, and most children have many visible lesions (N.B. Silverberg, MD, unpublished data).
Because each lesion contains infectious viral particles and patients scratch, more lesions are equated to greater autoinoculation and contagion. In addition to the AAP criteria, treatment can be considered for households with immunocompromised individuals, children at risk for new-onset AD, or those with AD at risk for flare. For patients with 45 lesions or more (3 SDs), clearance is harder to achieve with 2 sessions of in-office therapy, and multiple methods or the addition of immunomodulatory therapeutics should be considered.
Do we have to clear every lesion?
New molluscum lesions may arise until a patient achieves immunity, and they may appear more than a month after inoculation, making it difficult to keep up with the rapid spread. Latency between exposure and lesion development usually is 2 to 7 weeks but may be as long as 6 months, making it difficult to prevent spread.6 Therefore, when we treat, we should not promise full clearance to patients and parents. Rather, we should inform them that new lesions may develop later, and therapy is only effective on visible lesions. In a recent study, a 50% clearance of lesions was the satisfactory threshold for parents, demonstrating that satisfaction is possible with partial clearance.9
What is new in therapeutics for molluscum?
Molluscum therapies are either destructive, immunomodulatory, or antiviral. Two agents now are approved by the FDA for the treatment of molluscum infections.
Berdazimer gel 10.3% is approved for patients 1 year or older, but it is not yet available. This agent has both immunomodulatory and antiviral properties.10 It features a home therapy that is mixed on a small palette, then painted on by the patient or parent once daily for 12 weeks. Study outcomes demonstrated more than 50% lesional clearance.11,12 Complete clearance was achieved in at least 30% of patients.12A proprietary topical version of cantharidin 0.7% in flexible collodion is now FDA approved for patients 2 years and older. This vesicant-triggering iatrogenic is targeted at creating blisters overlying molluscum lesions. It is conceptually similar to older versions but with some enhanced features.5,13,14 This version was used for therapy every 3 weeks for up to 4 sessions in clinical trials. Safety is similar across all body sites treated (nonmucosal and not near the mucosal surfaces) but not for mucosa, the mid face, or eyelids.13 Complete lesion clearance was 46.3% to 54% and statistically greater than placebo (P<.001).14Both agents are well tolerated in children with AD; adverse effects include blistering with cantharidin and dermatitislike symptoms with berdazimer.15,16 These therapies have the advantage of being easy to use.
Final Thoughts
We have entered an era of high-quality molluscum therapy. Patient care involves developing a good knowledge of the agents, incorporating shared decision-making with patients and caregivers, and addressing therapy in the context of comorbid diseases such as AD.
- Silverberg NB. Pediatric molluscum: an update. Cutis. 2019;104:301-305, E1-E2.
- Thompson AJ, Matinpour K, Hardin J, et al. Molluscum gladiatorum. Dermatol Online J. 2014;20:13030/qt0nj121n1.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357. doi:10.1111/pde.12504
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Molluscum contagiosum. In: Kimberlin DW, Lynfield R, Barnett ED, et al (eds). Red Book: 2021–2024 Report of the Committee on Infectious Diseases. 32nd edition. American Academy of Pediatrics. May 26, 2021. Accessed May 20, 2024. https://publications.aap.org/redbook/book/347/chapter/5754264/Molluscum-Contagiosum
- Gross I, Ben Nachum N, Molho-Pessach V, et al. The molluscum contagiosum BOTE sign—infected or inflamed? Pediatr Dermatol. 2020;37:476-479. doi:10.1111/pde.14124
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507. doi:10.1067/mjd.2000.106370
- Maeda-Chubachi T, McLeod L, Enloe C, et al. Defining clinically meaningful improvement in molluscum contagiosum. J Am Acad Dermatol. 2024;90:443-445. doi:10.1016/j.jaad.2023.10.033
- Guttman-Yassky E, Gallo RL, Pavel AB, et al. A nitric oxide-releasing topical medication as a potential treatment option for atopic dermatitis through antimicrobial and anti-inflammatory activity. J Invest Dermatol. 2020;140:2531-2535.e2. doi:10.1016/j.jid.2020.04.013
- Browning JC, Cartwright M, Thorla I Jr, et al. A patient-centered perspective of molluscum contagiosum as reported by B-SIMPLE4 Clinical Trial patients and caregivers: Global Impression of Change and Exit Interview substudy results. Am J Clin Dermatol. 2023;24:119-133. doi:10.1007/s40257-022-00733-9
- Sugarman JL, Hebert A, Browning JC, et al. Berdazimer gel for molluscum contagiosum: an integrated analysis of 3 randomized controlled trials. J Am Acad Dermatol. 2024;90:299-308. doi:10.1016/j.jaad.2023.09.066
- Eichenfield LF, Kwong P, Gonzalez ME, et al. Safety and efficacy of VP-102 (cantharidin, 0.7% w/v) in molluscum contagiosum by body region: post hoc pooled analyses from two phase III randomized trials. J Clin Aesthet Dermatol. 2021;14:42-47.
- Eichenfield LF, McFalda W, Brabec B, et al. Safety and efficacy of VP-102, a proprietary, drug-device combination product containing cantharidin, 0.7% (w/v), in children and adults with molluscum contagiosum: two phase 3 randomized clinical trials. JAMA Dermatol. 2020;156:1315-1323. doi:10.1001/jamadermatol.2020.3238
- Paller AS, Green LJ, Silverberg N, et al. Berdazimer gel for molluscum contagiosum in patients with atopic dermatitis. Pediatr Dermatol.Published online February 27, 2024. doi:10.1111/pde.15575
- Eichenfield L, Hebert A, Mancini A, et al. Therapeutic approaches and special considerations for treating molluscum contagiosum. J Drugs Dermatol. 2021;20:1185-1190. doi:10.36849/jdd.6383
Molluscum contagiosum (ie, molluscum) is a ubiquitous infection caused by the poxvirus molluscum contagiosum virus (MCV). Although skin deep, molluscum shares many factors with the more virulent poxviridae. Moisture and trauma can cause viral material to be released from the pearly papules through a small opening, which also allows entry of bacteria and medications into the lesion. The MCV is transmitted by direct contact with skin or via fomites.1
Molluscum can affect children of any age, with MCV type 1 peaking in toddlers and school-aged children and MCV type 2 after the sexual debut. The prevalence of molluscum has increased since the 1980s. It is stressful for children and caregivers and poses challenges in schools as well as sports such as swimming, wrestling, and karate.1,2
For the first time, we have US Food and Drug Administration (FDA)–approved products to treat MCV infections. Previously, only off-label agents were used. Therefore, we have to contemplate why treatment is important to our patients.
What type of care is required for molluscum?
Counseling is the first and only mandatory treatment, which consists of 3 parts: natural history, risk factors for spread, and options for therapy. The natural history of molluscum in children is early spread, contagion to oneself and others (as high as 60% of sibling co-bathers3), triggering of dermatitis, eventual onset of the beginning-of-the-end (BOTE) sign, and eventually clearance. The natural history in adults is poorly understood.
Early clearance is uncommon; reports have suggested 45.6% to 48.4% of affected patients are clear at 1 year and 69.5% to 72.6% at 1.5 years.4 For many children, especially those with atopic dermatitis (AD), lesions linger and often spread, with many experiencing disease for 3 to 4 years. Fomites such as towels, washcloths, and sponges can transfer the virus and spread lesions; therefore, I advise patients to gently pat their skin dry, wash towels frequently, and avoid sharing bathing equipment.1,3,5 Children and adults with immunosuppression may have a greater number of lesions and more prolonged course of disease, including those with HIV as well as DOC8 and CARD11 mutations.6 The American Academy of Pediatrics (AAP) emphasizes that children should not be excluded from attending child care/school or from swimming in public pools but lesions should be covered.6 Lesions, especially those in the antecubital region, can trigger new-onset AD or AD flares.3 In response, gentle skin care including fragrance-free cleansers and periodic application of moisturizers may ward off AD. Topical corticosteroids are preferred.
Dermatitis in MCV is a great mimicker and can resemble erythema multiforme, Gianotti-Crosti syndrome, impetigo, and AD.1 Superinfection recently has been reported; however, in a retrospective analysis of 56 patients with inflamed lesions secondary to molluscum infection, only 7 had positive bacterial cultures, which supports the idea of the swelling and redness of inflammation as a mimic for infection.7 When true infection does occur, tender, swollen, pus-filled lesions should be lanced and cultured.1,7,8
When should we consider therapy?
Therapy is highly dependent on the child, the caregiver, and the social circumstances.1 More than 80% of parents are anxious about molluscum, and countless children are embarrassed or ashamed.1 Ultimately, an unhappy child merits care. The AAP cites the following as reasons to treat: “(1) alleviate discomfort, including itching; (2) reduce autoinoculation; (3) limit transmission of the virus to close contacts; (4) reduce cosmetic concerns; and (5) prevent secondary infection.”6 For adults, we should consider limitations to intimacy and reduction of sexual transmission risk.6
Treatment can be based on the number of lesions. With a few lesions (<3), therapy is worthwhile if they are unsightly; appear on exposed skin causing embarrassment; and/or are itchy, uncomfortable, or large. In a report of 300 children with molluscum treated with cantharidin, most patients choosing therapy had 10 to 20 lesions, but this was over multiple visits.8 Looking at a 2018 data set of 50 patients (all-comers) with molluscum,3 the mean number of lesions was 10 (median, 7); 3 lesions were 1 SD below, while 14, 17, and 45 were 1, 2, and 3 SDs above, respectively. This data set shows that patients can develop more lesions rapidly, and most children have many visible lesions (N.B. Silverberg, MD, unpublished data).
Because each lesion contains infectious viral particles and patients scratch, more lesions are equated to greater autoinoculation and contagion. In addition to the AAP criteria, treatment can be considered for households with immunocompromised individuals, children at risk for new-onset AD, or those with AD at risk for flare. For patients with 45 lesions or more (3 SDs), clearance is harder to achieve with 2 sessions of in-office therapy, and multiple methods or the addition of immunomodulatory therapeutics should be considered.
Do we have to clear every lesion?
New molluscum lesions may arise until a patient achieves immunity, and they may appear more than a month after inoculation, making it difficult to keep up with the rapid spread. Latency between exposure and lesion development usually is 2 to 7 weeks but may be as long as 6 months, making it difficult to prevent spread.6 Therefore, when we treat, we should not promise full clearance to patients and parents. Rather, we should inform them that new lesions may develop later, and therapy is only effective on visible lesions. In a recent study, a 50% clearance of lesions was the satisfactory threshold for parents, demonstrating that satisfaction is possible with partial clearance.9
What is new in therapeutics for molluscum?
Molluscum therapies are either destructive, immunomodulatory, or antiviral. Two agents now are approved by the FDA for the treatment of molluscum infections.
Berdazimer gel 10.3% is approved for patients 1 year or older, but it is not yet available. This agent has both immunomodulatory and antiviral properties.10 It features a home therapy that is mixed on a small palette, then painted on by the patient or parent once daily for 12 weeks. Study outcomes demonstrated more than 50% lesional clearance.11,12 Complete clearance was achieved in at least 30% of patients.12A proprietary topical version of cantharidin 0.7% in flexible collodion is now FDA approved for patients 2 years and older. This vesicant-triggering iatrogenic is targeted at creating blisters overlying molluscum lesions. It is conceptually similar to older versions but with some enhanced features.5,13,14 This version was used for therapy every 3 weeks for up to 4 sessions in clinical trials. Safety is similar across all body sites treated (nonmucosal and not near the mucosal surfaces) but not for mucosa, the mid face, or eyelids.13 Complete lesion clearance was 46.3% to 54% and statistically greater than placebo (P<.001).14Both agents are well tolerated in children with AD; adverse effects include blistering with cantharidin and dermatitislike symptoms with berdazimer.15,16 These therapies have the advantage of being easy to use.
Final Thoughts
We have entered an era of high-quality molluscum therapy. Patient care involves developing a good knowledge of the agents, incorporating shared decision-making with patients and caregivers, and addressing therapy in the context of comorbid diseases such as AD.
Molluscum contagiosum (ie, molluscum) is a ubiquitous infection caused by the poxvirus molluscum contagiosum virus (MCV). Although skin deep, molluscum shares many factors with the more virulent poxviridae. Moisture and trauma can cause viral material to be released from the pearly papules through a small opening, which also allows entry of bacteria and medications into the lesion. The MCV is transmitted by direct contact with skin or via fomites.1
Molluscum can affect children of any age, with MCV type 1 peaking in toddlers and school-aged children and MCV type 2 after the sexual debut. The prevalence of molluscum has increased since the 1980s. It is stressful for children and caregivers and poses challenges in schools as well as sports such as swimming, wrestling, and karate.1,2
For the first time, we have US Food and Drug Administration (FDA)–approved products to treat MCV infections. Previously, only off-label agents were used. Therefore, we have to contemplate why treatment is important to our patients.
What type of care is required for molluscum?
Counseling is the first and only mandatory treatment, which consists of 3 parts: natural history, risk factors for spread, and options for therapy. The natural history of molluscum in children is early spread, contagion to oneself and others (as high as 60% of sibling co-bathers3), triggering of dermatitis, eventual onset of the beginning-of-the-end (BOTE) sign, and eventually clearance. The natural history in adults is poorly understood.
Early clearance is uncommon; reports have suggested 45.6% to 48.4% of affected patients are clear at 1 year and 69.5% to 72.6% at 1.5 years.4 For many children, especially those with atopic dermatitis (AD), lesions linger and often spread, with many experiencing disease for 3 to 4 years. Fomites such as towels, washcloths, and sponges can transfer the virus and spread lesions; therefore, I advise patients to gently pat their skin dry, wash towels frequently, and avoid sharing bathing equipment.1,3,5 Children and adults with immunosuppression may have a greater number of lesions and more prolonged course of disease, including those with HIV as well as DOC8 and CARD11 mutations.6 The American Academy of Pediatrics (AAP) emphasizes that children should not be excluded from attending child care/school or from swimming in public pools but lesions should be covered.6 Lesions, especially those in the antecubital region, can trigger new-onset AD or AD flares.3 In response, gentle skin care including fragrance-free cleansers and periodic application of moisturizers may ward off AD. Topical corticosteroids are preferred.
Dermatitis in MCV is a great mimicker and can resemble erythema multiforme, Gianotti-Crosti syndrome, impetigo, and AD.1 Superinfection recently has been reported; however, in a retrospective analysis of 56 patients with inflamed lesions secondary to molluscum infection, only 7 had positive bacterial cultures, which supports the idea of the swelling and redness of inflammation as a mimic for infection.7 When true infection does occur, tender, swollen, pus-filled lesions should be lanced and cultured.1,7,8
When should we consider therapy?
Therapy is highly dependent on the child, the caregiver, and the social circumstances.1 More than 80% of parents are anxious about molluscum, and countless children are embarrassed or ashamed.1 Ultimately, an unhappy child merits care. The AAP cites the following as reasons to treat: “(1) alleviate discomfort, including itching; (2) reduce autoinoculation; (3) limit transmission of the virus to close contacts; (4) reduce cosmetic concerns; and (5) prevent secondary infection.”6 For adults, we should consider limitations to intimacy and reduction of sexual transmission risk.6
Treatment can be based on the number of lesions. With a few lesions (<3), therapy is worthwhile if they are unsightly; appear on exposed skin causing embarrassment; and/or are itchy, uncomfortable, or large. In a report of 300 children with molluscum treated with cantharidin, most patients choosing therapy had 10 to 20 lesions, but this was over multiple visits.8 Looking at a 2018 data set of 50 patients (all-comers) with molluscum,3 the mean number of lesions was 10 (median, 7); 3 lesions were 1 SD below, while 14, 17, and 45 were 1, 2, and 3 SDs above, respectively. This data set shows that patients can develop more lesions rapidly, and most children have many visible lesions (N.B. Silverberg, MD, unpublished data).
Because each lesion contains infectious viral particles and patients scratch, more lesions are equated to greater autoinoculation and contagion. In addition to the AAP criteria, treatment can be considered for households with immunocompromised individuals, children at risk for new-onset AD, or those with AD at risk for flare. For patients with 45 lesions or more (3 SDs), clearance is harder to achieve with 2 sessions of in-office therapy, and multiple methods or the addition of immunomodulatory therapeutics should be considered.
Do we have to clear every lesion?
New molluscum lesions may arise until a patient achieves immunity, and they may appear more than a month after inoculation, making it difficult to keep up with the rapid spread. Latency between exposure and lesion development usually is 2 to 7 weeks but may be as long as 6 months, making it difficult to prevent spread.6 Therefore, when we treat, we should not promise full clearance to patients and parents. Rather, we should inform them that new lesions may develop later, and therapy is only effective on visible lesions. In a recent study, a 50% clearance of lesions was the satisfactory threshold for parents, demonstrating that satisfaction is possible with partial clearance.9
What is new in therapeutics for molluscum?
Molluscum therapies are either destructive, immunomodulatory, or antiviral. Two agents now are approved by the FDA for the treatment of molluscum infections.
Berdazimer gel 10.3% is approved for patients 1 year or older, but it is not yet available. This agent has both immunomodulatory and antiviral properties.10 It features a home therapy that is mixed on a small palette, then painted on by the patient or parent once daily for 12 weeks. Study outcomes demonstrated more than 50% lesional clearance.11,12 Complete clearance was achieved in at least 30% of patients.12A proprietary topical version of cantharidin 0.7% in flexible collodion is now FDA approved for patients 2 years and older. This vesicant-triggering iatrogenic is targeted at creating blisters overlying molluscum lesions. It is conceptually similar to older versions but with some enhanced features.5,13,14 This version was used for therapy every 3 weeks for up to 4 sessions in clinical trials. Safety is similar across all body sites treated (nonmucosal and not near the mucosal surfaces) but not for mucosa, the mid face, or eyelids.13 Complete lesion clearance was 46.3% to 54% and statistically greater than placebo (P<.001).14Both agents are well tolerated in children with AD; adverse effects include blistering with cantharidin and dermatitislike symptoms with berdazimer.15,16 These therapies have the advantage of being easy to use.
Final Thoughts
We have entered an era of high-quality molluscum therapy. Patient care involves developing a good knowledge of the agents, incorporating shared decision-making with patients and caregivers, and addressing therapy in the context of comorbid diseases such as AD.
- Silverberg NB. Pediatric molluscum: an update. Cutis. 2019;104:301-305, E1-E2.
- Thompson AJ, Matinpour K, Hardin J, et al. Molluscum gladiatorum. Dermatol Online J. 2014;20:13030/qt0nj121n1.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357. doi:10.1111/pde.12504
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Molluscum contagiosum. In: Kimberlin DW, Lynfield R, Barnett ED, et al (eds). Red Book: 2021–2024 Report of the Committee on Infectious Diseases. 32nd edition. American Academy of Pediatrics. May 26, 2021. Accessed May 20, 2024. https://publications.aap.org/redbook/book/347/chapter/5754264/Molluscum-Contagiosum
- Gross I, Ben Nachum N, Molho-Pessach V, et al. The molluscum contagiosum BOTE sign—infected or inflamed? Pediatr Dermatol. 2020;37:476-479. doi:10.1111/pde.14124
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507. doi:10.1067/mjd.2000.106370
- Maeda-Chubachi T, McLeod L, Enloe C, et al. Defining clinically meaningful improvement in molluscum contagiosum. J Am Acad Dermatol. 2024;90:443-445. doi:10.1016/j.jaad.2023.10.033
- Guttman-Yassky E, Gallo RL, Pavel AB, et al. A nitric oxide-releasing topical medication as a potential treatment option for atopic dermatitis through antimicrobial and anti-inflammatory activity. J Invest Dermatol. 2020;140:2531-2535.e2. doi:10.1016/j.jid.2020.04.013
- Browning JC, Cartwright M, Thorla I Jr, et al. A patient-centered perspective of molluscum contagiosum as reported by B-SIMPLE4 Clinical Trial patients and caregivers: Global Impression of Change and Exit Interview substudy results. Am J Clin Dermatol. 2023;24:119-133. doi:10.1007/s40257-022-00733-9
- Sugarman JL, Hebert A, Browning JC, et al. Berdazimer gel for molluscum contagiosum: an integrated analysis of 3 randomized controlled trials. J Am Acad Dermatol. 2024;90:299-308. doi:10.1016/j.jaad.2023.09.066
- Eichenfield LF, Kwong P, Gonzalez ME, et al. Safety and efficacy of VP-102 (cantharidin, 0.7% w/v) in molluscum contagiosum by body region: post hoc pooled analyses from two phase III randomized trials. J Clin Aesthet Dermatol. 2021;14:42-47.
- Eichenfield LF, McFalda W, Brabec B, et al. Safety and efficacy of VP-102, a proprietary, drug-device combination product containing cantharidin, 0.7% (w/v), in children and adults with molluscum contagiosum: two phase 3 randomized clinical trials. JAMA Dermatol. 2020;156:1315-1323. doi:10.1001/jamadermatol.2020.3238
- Paller AS, Green LJ, Silverberg N, et al. Berdazimer gel for molluscum contagiosum in patients with atopic dermatitis. Pediatr Dermatol.Published online February 27, 2024. doi:10.1111/pde.15575
- Eichenfield L, Hebert A, Mancini A, et al. Therapeutic approaches and special considerations for treating molluscum contagiosum. J Drugs Dermatol. 2021;20:1185-1190. doi:10.36849/jdd.6383
- Silverberg NB. Pediatric molluscum: an update. Cutis. 2019;104:301-305, E1-E2.
- Thompson AJ, Matinpour K, Hardin J, et al. Molluscum gladiatorum. Dermatol Online J. 2014;20:13030/qt0nj121n1.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357. doi:10.1111/pde.12504
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Molluscum contagiosum. In: Kimberlin DW, Lynfield R, Barnett ED, et al (eds). Red Book: 2021–2024 Report of the Committee on Infectious Diseases. 32nd edition. American Academy of Pediatrics. May 26, 2021. Accessed May 20, 2024. https://publications.aap.org/redbook/book/347/chapter/5754264/Molluscum-Contagiosum
- Gross I, Ben Nachum N, Molho-Pessach V, et al. The molluscum contagiosum BOTE sign—infected or inflamed? Pediatr Dermatol. 2020;37:476-479. doi:10.1111/pde.14124
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507. doi:10.1067/mjd.2000.106370
- Maeda-Chubachi T, McLeod L, Enloe C, et al. Defining clinically meaningful improvement in molluscum contagiosum. J Am Acad Dermatol. 2024;90:443-445. doi:10.1016/j.jaad.2023.10.033
- Guttman-Yassky E, Gallo RL, Pavel AB, et al. A nitric oxide-releasing topical medication as a potential treatment option for atopic dermatitis through antimicrobial and anti-inflammatory activity. J Invest Dermatol. 2020;140:2531-2535.e2. doi:10.1016/j.jid.2020.04.013
- Browning JC, Cartwright M, Thorla I Jr, et al. A patient-centered perspective of molluscum contagiosum as reported by B-SIMPLE4 Clinical Trial patients and caregivers: Global Impression of Change and Exit Interview substudy results. Am J Clin Dermatol. 2023;24:119-133. doi:10.1007/s40257-022-00733-9
- Sugarman JL, Hebert A, Browning JC, et al. Berdazimer gel for molluscum contagiosum: an integrated analysis of 3 randomized controlled trials. J Am Acad Dermatol. 2024;90:299-308. doi:10.1016/j.jaad.2023.09.066
- Eichenfield LF, Kwong P, Gonzalez ME, et al. Safety and efficacy of VP-102 (cantharidin, 0.7% w/v) in molluscum contagiosum by body region: post hoc pooled analyses from two phase III randomized trials. J Clin Aesthet Dermatol. 2021;14:42-47.
- Eichenfield LF, McFalda W, Brabec B, et al. Safety and efficacy of VP-102, a proprietary, drug-device combination product containing cantharidin, 0.7% (w/v), in children and adults with molluscum contagiosum: two phase 3 randomized clinical trials. JAMA Dermatol. 2020;156:1315-1323. doi:10.1001/jamadermatol.2020.3238
- Paller AS, Green LJ, Silverberg N, et al. Berdazimer gel for molluscum contagiosum in patients with atopic dermatitis. Pediatr Dermatol.Published online February 27, 2024. doi:10.1111/pde.15575
- Eichenfield L, Hebert A, Mancini A, et al. Therapeutic approaches and special considerations for treating molluscum contagiosum. J Drugs Dermatol. 2021;20:1185-1190. doi:10.36849/jdd.6383