User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
A Rare Association in Down Syndrome: Milialike Idiopathic Calcinosis Cutis and Palpebral Syringoma
To the Editor:
Down syndrome (DS) is associated with rare dermatological disorders, and the prevalence of some common dermatoses is greater in patients with DS. We report a case of milialike idiopathic calcinosis cutis (MICC) associated with syringomas in a patient with DS. We emphasize that MICC is one of the rare dermatoses associated with DS.
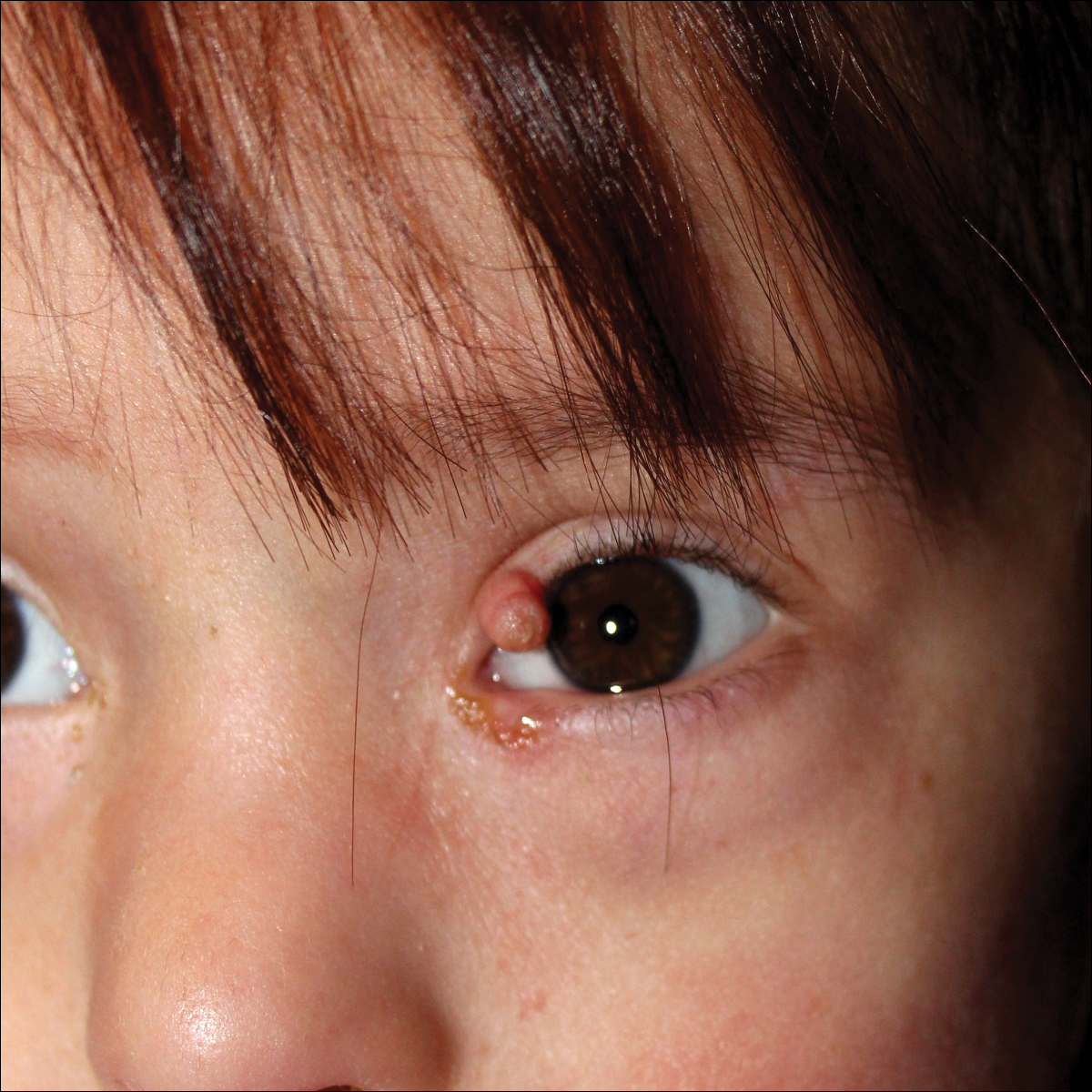
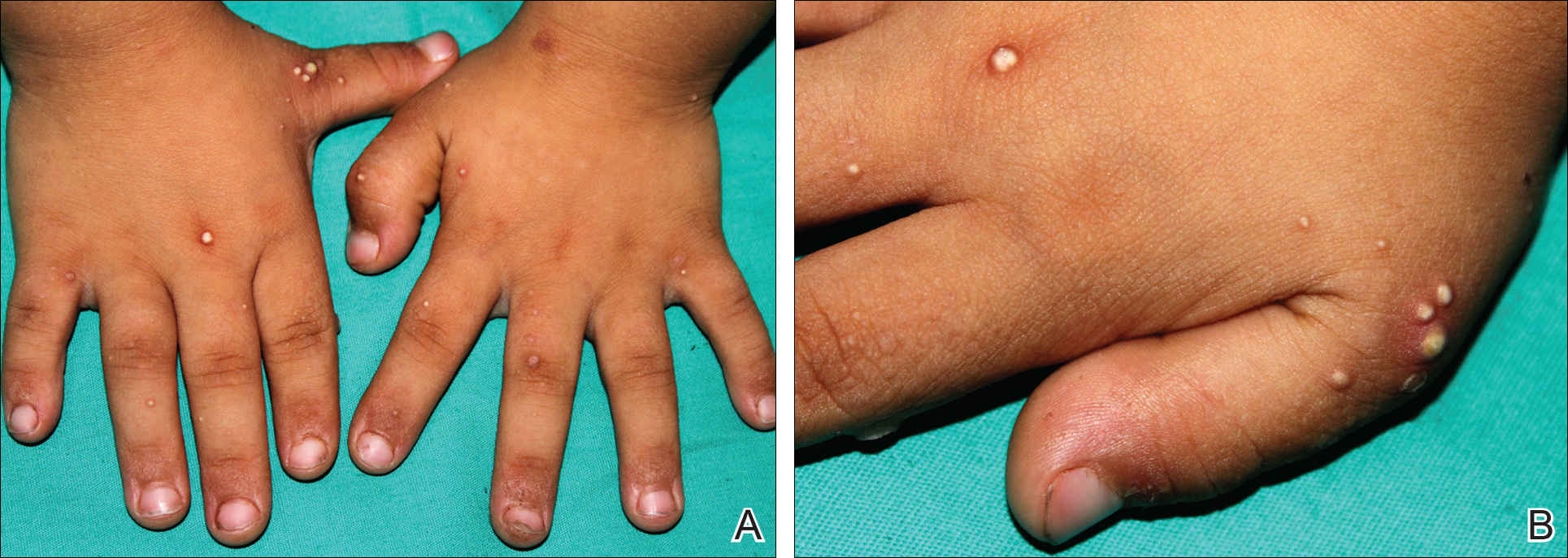
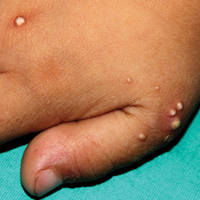
A 4-year-old girl with DS presented with a 4-mm, flesh-colored, regular-bordered, exophytic papular lesion on the left upper eyelid of 8 months' duration (Figure 1). It was clinically recognized as syringoma. On dermatologic examination of the patient, there also were 1- to 3-mm, round, whitish, painless, milialike papules on the dorsal surface of the hands and wrists (Figure 2). Some of these papules were surrounded by erythema. There was no sign of perforation. Her personal and family history were unremarkable.
Histopathologic examination of a biopsy from a milialike lesion on the hand showed a hyperkeratotic epidermis. In the dermis there was a roundish calcific nodule surrounded by a fibrovascular rim. The patient's guardians refused a biopsy from the lesion on the eyelid.
Laboratory tests including serum vitamin D, thyroid and parathyroid hormone, calcium, phosphorus, and urinary calcium levels, as well as renal function tests, were within reference range. On the basis of these clinical and histopathological findings, the patient was diagnosed with MICC and palpebral syringoma.
Many dermatoses associated with DS have been reported including elastosis perforans serpiginosa, alopecia areata, and syringomas.1-3 Sano et al4 first described MICC and syringomas in a patient with DS in 1978. Milialike idiopathic calcinosis cutis is characterized by asymptomatic, millimetric, firm, round, whitish papules that are sometimes surrounded by erythema. These papules may show perforation leading to transepidermal elimination of calcium, similar to the transdermal elimination of elastic fibrils in elastosis perforans serpiginosa. Although MICC usually is described in acral sites of children with DS, it also is reported in adults without DS and on other parts of the body.5-7
The cause of MICC is unknown. One hypothesis of the development of MICC is an increase of the calcium content in the sweat leading to calcification of the acrosyringium.8 Milia are small keratin cysts that usually develop by occlusion of the hair follicle, sweat duct, or sebaceous duct. However, milia also can occur from occlusion of the eccrine ducts where syringomas originate.9 Therefore, syringomas can be seen in association with milia and calcium deposits.5,9-11
We believe that MICC in DS may be more common than usually recognized, as the lesions often are asymptomatic. It is important to differentiate MICC from other dermatological diseases such as molluscum contagiosum, verruca plana, milia, and inclusion cysts. Histopathology and dermoscopy could aid in the accurate diagnosis of MICC.
- Dourmishev A, Miteva L, Mitev V, et al. Cutaneous aspects of Down syndrome. Cutis. 2000;66:420-424.
- Madan V, Williams J, Lear JT. Dermatological manifestations of Down's syndrome. Clin Exp Dermatol. 2006;31:623-629.
- Schepis C, Barone C, Siragusa M, et al. An updated survey on skin conditions in Down syndrome. Dermatology. 2002;205:234-238.
- Sano T, Tate S, Ishikawa C. A case of Down's syndrome associated with syringoma, milia, and subepidermal calcified nodule. Jpn J Dermatol. 1978;88:740.
- Schepis C, Siragusa M, Palazzo R, et al. Perforating milia-like idiopathic calcinosis cutis and periorbital syringomas in a girl with Down syndrome. Pediatr Dermatol. 1994;11:258-260.
- Schepis C, Siragusa M, Palazzo R, et al. Milia like idiopathic calcinosis cutis: an unusual dermatosis associated with Down syndrome. Br J Dermatol. 1996;134:143-146.
- Houtappel M, Leguit R, Sigurdsson V. Milia-like idiopathic calcinosis cutis in an adult without Down's syndrome. J Dermatol Case Rep. 2007;1:16-19.
- Eng AM, Mandrea E. Perforating calcinosis cutis presenting as milia. J Cutan Pathol. 1981;8:247-250.
- Wang KH, Chu JS, Lin YH, et al. Milium-like syringoma: a case study on histogenesis. J Cutan Pathol. 2004;31:336-340.
- Weiss E, Paez E, Greenberg AS, et al. Eruptive syringomas associated with milia. Int J Dermatol. 1995;34:193-195.
- Kim SJ, Won YH, Chun IK. Subepidermal calcified nodules and syringoma. J Eur Acad Dermatol Venereol. 1997;8:51-52.
To the Editor:
Down syndrome (DS) is associated with rare dermatological disorders, and the prevalence of some common dermatoses is greater in patients with DS. We report a case of milialike idiopathic calcinosis cutis (MICC) associated with syringomas in a patient with DS. We emphasize that MICC is one of the rare dermatoses associated with DS.
A 4-year-old girl with DS presented with a 4-mm, flesh-colored, regular-bordered, exophytic papular lesion on the left upper eyelid of 8 months' duration (Figure 1). It was clinically recognized as syringoma. On dermatologic examination of the patient, there also were 1- to 3-mm, round, whitish, painless, milialike papules on the dorsal surface of the hands and wrists (Figure 2). Some of these papules were surrounded by erythema. There was no sign of perforation. Her personal and family history were unremarkable.
Histopathologic examination of a biopsy from a milialike lesion on the hand showed a hyperkeratotic epidermis. In the dermis there was a roundish calcific nodule surrounded by a fibrovascular rim. The patient's guardians refused a biopsy from the lesion on the eyelid.
Laboratory tests including serum vitamin D, thyroid and parathyroid hormone, calcium, phosphorus, and urinary calcium levels, as well as renal function tests, were within reference range. On the basis of these clinical and histopathological findings, the patient was diagnosed with MICC and palpebral syringoma.
Many dermatoses associated with DS have been reported including elastosis perforans serpiginosa, alopecia areata, and syringomas.1-3 Sano et al4 first described MICC and syringomas in a patient with DS in 1978. Milialike idiopathic calcinosis cutis is characterized by asymptomatic, millimetric, firm, round, whitish papules that are sometimes surrounded by erythema. These papules may show perforation leading to transepidermal elimination of calcium, similar to the transdermal elimination of elastic fibrils in elastosis perforans serpiginosa. Although MICC usually is described in acral sites of children with DS, it also is reported in adults without DS and on other parts of the body.5-7
The cause of MICC is unknown. One hypothesis of the development of MICC is an increase of the calcium content in the sweat leading to calcification of the acrosyringium.8 Milia are small keratin cysts that usually develop by occlusion of the hair follicle, sweat duct, or sebaceous duct. However, milia also can occur from occlusion of the eccrine ducts where syringomas originate.9 Therefore, syringomas can be seen in association with milia and calcium deposits.5,9-11
We believe that MICC in DS may be more common than usually recognized, as the lesions often are asymptomatic. It is important to differentiate MICC from other dermatological diseases such as molluscum contagiosum, verruca plana, milia, and inclusion cysts. Histopathology and dermoscopy could aid in the accurate diagnosis of MICC.
To the Editor:
Down syndrome (DS) is associated with rare dermatological disorders, and the prevalence of some common dermatoses is greater in patients with DS. We report a case of milialike idiopathic calcinosis cutis (MICC) associated with syringomas in a patient with DS. We emphasize that MICC is one of the rare dermatoses associated with DS.
A 4-year-old girl with DS presented with a 4-mm, flesh-colored, regular-bordered, exophytic papular lesion on the left upper eyelid of 8 months' duration (Figure 1). It was clinically recognized as syringoma. On dermatologic examination of the patient, there also were 1- to 3-mm, round, whitish, painless, milialike papules on the dorsal surface of the hands and wrists (Figure 2). Some of these papules were surrounded by erythema. There was no sign of perforation. Her personal and family history were unremarkable.
Histopathologic examination of a biopsy from a milialike lesion on the hand showed a hyperkeratotic epidermis. In the dermis there was a roundish calcific nodule surrounded by a fibrovascular rim. The patient's guardians refused a biopsy from the lesion on the eyelid.
Laboratory tests including serum vitamin D, thyroid and parathyroid hormone, calcium, phosphorus, and urinary calcium levels, as well as renal function tests, were within reference range. On the basis of these clinical and histopathological findings, the patient was diagnosed with MICC and palpebral syringoma.
Many dermatoses associated with DS have been reported including elastosis perforans serpiginosa, alopecia areata, and syringomas.1-3 Sano et al4 first described MICC and syringomas in a patient with DS in 1978. Milialike idiopathic calcinosis cutis is characterized by asymptomatic, millimetric, firm, round, whitish papules that are sometimes surrounded by erythema. These papules may show perforation leading to transepidermal elimination of calcium, similar to the transdermal elimination of elastic fibrils in elastosis perforans serpiginosa. Although MICC usually is described in acral sites of children with DS, it also is reported in adults without DS and on other parts of the body.5-7
The cause of MICC is unknown. One hypothesis of the development of MICC is an increase of the calcium content in the sweat leading to calcification of the acrosyringium.8 Milia are small keratin cysts that usually develop by occlusion of the hair follicle, sweat duct, or sebaceous duct. However, milia also can occur from occlusion of the eccrine ducts where syringomas originate.9 Therefore, syringomas can be seen in association with milia and calcium deposits.5,9-11
We believe that MICC in DS may be more common than usually recognized, as the lesions often are asymptomatic. It is important to differentiate MICC from other dermatological diseases such as molluscum contagiosum, verruca plana, milia, and inclusion cysts. Histopathology and dermoscopy could aid in the accurate diagnosis of MICC.
- Dourmishev A, Miteva L, Mitev V, et al. Cutaneous aspects of Down syndrome. Cutis. 2000;66:420-424.
- Madan V, Williams J, Lear JT. Dermatological manifestations of Down's syndrome. Clin Exp Dermatol. 2006;31:623-629.
- Schepis C, Barone C, Siragusa M, et al. An updated survey on skin conditions in Down syndrome. Dermatology. 2002;205:234-238.
- Sano T, Tate S, Ishikawa C. A case of Down's syndrome associated with syringoma, milia, and subepidermal calcified nodule. Jpn J Dermatol. 1978;88:740.
- Schepis C, Siragusa M, Palazzo R, et al. Perforating milia-like idiopathic calcinosis cutis and periorbital syringomas in a girl with Down syndrome. Pediatr Dermatol. 1994;11:258-260.
- Schepis C, Siragusa M, Palazzo R, et al. Milia like idiopathic calcinosis cutis: an unusual dermatosis associated with Down syndrome. Br J Dermatol. 1996;134:143-146.
- Houtappel M, Leguit R, Sigurdsson V. Milia-like idiopathic calcinosis cutis in an adult without Down's syndrome. J Dermatol Case Rep. 2007;1:16-19.
- Eng AM, Mandrea E. Perforating calcinosis cutis presenting as milia. J Cutan Pathol. 1981;8:247-250.
- Wang KH, Chu JS, Lin YH, et al. Milium-like syringoma: a case study on histogenesis. J Cutan Pathol. 2004;31:336-340.
- Weiss E, Paez E, Greenberg AS, et al. Eruptive syringomas associated with milia. Int J Dermatol. 1995;34:193-195.
- Kim SJ, Won YH, Chun IK. Subepidermal calcified nodules and syringoma. J Eur Acad Dermatol Venereol. 1997;8:51-52.
- Dourmishev A, Miteva L, Mitev V, et al. Cutaneous aspects of Down syndrome. Cutis. 2000;66:420-424.
- Madan V, Williams J, Lear JT. Dermatological manifestations of Down's syndrome. Clin Exp Dermatol. 2006;31:623-629.
- Schepis C, Barone C, Siragusa M, et al. An updated survey on skin conditions in Down syndrome. Dermatology. 2002;205:234-238.
- Sano T, Tate S, Ishikawa C. A case of Down's syndrome associated with syringoma, milia, and subepidermal calcified nodule. Jpn J Dermatol. 1978;88:740.
- Schepis C, Siragusa M, Palazzo R, et al. Perforating milia-like idiopathic calcinosis cutis and periorbital syringomas in a girl with Down syndrome. Pediatr Dermatol. 1994;11:258-260.
- Schepis C, Siragusa M, Palazzo R, et al. Milia like idiopathic calcinosis cutis: an unusual dermatosis associated with Down syndrome. Br J Dermatol. 1996;134:143-146.
- Houtappel M, Leguit R, Sigurdsson V. Milia-like idiopathic calcinosis cutis in an adult without Down's syndrome. J Dermatol Case Rep. 2007;1:16-19.
- Eng AM, Mandrea E. Perforating calcinosis cutis presenting as milia. J Cutan Pathol. 1981;8:247-250.
- Wang KH, Chu JS, Lin YH, et al. Milium-like syringoma: a case study on histogenesis. J Cutan Pathol. 2004;31:336-340.
- Weiss E, Paez E, Greenberg AS, et al. Eruptive syringomas associated with milia. Int J Dermatol. 1995;34:193-195.
- Kim SJ, Won YH, Chun IK. Subepidermal calcified nodules and syringoma. J Eur Acad Dermatol Venereol. 1997;8:51-52.
Practice Points
- Down syndrome is associated with rare dermatological disorders and an increased prevalence of common dermatoses.
- It is important to differentiate milialike idiopathic calcinosis cutis from other dermatological diseases using histopathology and dermoscopy.
Nominate a Patient or Colleague for a NORD Rare Impact Award
Do you have a patient or colleague who has demonstrated extraordinary commitment to advancing understanding of rare diseases or improving the lives of those affected by rare diseases? January 13th is the deadline to nominate individuals for NORD’s Rare Impact Awards. Nominations can be submitted through the NORD website.
NORD’s Rare Impact Awards ceremony will take place on May 18, 2017, in the amphitheater of the Ronald Reagan Building and International Trade Center, the largest structure in Washington, DC, and the first and only federal building dedicated to both federal and private use. More than 500 distinguished guests are expected to attend. Registration will be available soon on the NORD website.
The Rare Impact Awards honor individuals and organizations for commitment to improving the lives of patients and families affected by rare diseases. Nominees may include patients, caregivers, clinicians, researchers, advocates, and others who in some way have contributed to the greater good of the community. Nominations are also being sought for organizations that have helped drive better understanding of rare diseases and/or improved care for those affected.
Do you have a patient or colleague who has demonstrated extraordinary commitment to advancing understanding of rare diseases or improving the lives of those affected by rare diseases? January 13th is the deadline to nominate individuals for NORD’s Rare Impact Awards. Nominations can be submitted through the NORD website.
NORD’s Rare Impact Awards ceremony will take place on May 18, 2017, in the amphitheater of the Ronald Reagan Building and International Trade Center, the largest structure in Washington, DC, and the first and only federal building dedicated to both federal and private use. More than 500 distinguished guests are expected to attend. Registration will be available soon on the NORD website.
The Rare Impact Awards honor individuals and organizations for commitment to improving the lives of patients and families affected by rare diseases. Nominees may include patients, caregivers, clinicians, researchers, advocates, and others who in some way have contributed to the greater good of the community. Nominations are also being sought for organizations that have helped drive better understanding of rare diseases and/or improved care for those affected.
Do you have a patient or colleague who has demonstrated extraordinary commitment to advancing understanding of rare diseases or improving the lives of those affected by rare diseases? January 13th is the deadline to nominate individuals for NORD’s Rare Impact Awards. Nominations can be submitted through the NORD website.
NORD’s Rare Impact Awards ceremony will take place on May 18, 2017, in the amphitheater of the Ronald Reagan Building and International Trade Center, the largest structure in Washington, DC, and the first and only federal building dedicated to both federal and private use. More than 500 distinguished guests are expected to attend. Registration will be available soon on the NORD website.
The Rare Impact Awards honor individuals and organizations for commitment to improving the lives of patients and families affected by rare diseases. Nominees may include patients, caregivers, clinicians, researchers, advocates, and others who in some way have contributed to the greater good of the community. Nominations are also being sought for organizations that have helped drive better understanding of rare diseases and/or improved care for those affected.
Transient Benign Neonatal Skin Findings
Review the PDF of the fact sheet on transient benign neonatal skin findings with board-relevant, easy-to-review material. This fact sheet lists benign findings that can be seen in neonates and infants.
Practice Questions
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Answers to practice questions provided on next page
Practice Question Answers
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Review the PDF of the fact sheet on transient benign neonatal skin findings with board-relevant, easy-to-review material. This fact sheet lists benign findings that can be seen in neonates and infants.
Practice Questions
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Answers to practice questions provided on next page
Practice Question Answers
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Review the PDF of the fact sheet on transient benign neonatal skin findings with board-relevant, easy-to-review material. This fact sheet lists benign findings that can be seen in neonates and infants.
Practice Questions
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis
Answers to practice questions provided on next page
Practice Question Answers
1. The parents of a 2-month-old infant present with their child. They are worried because the infant has “acne” that is not going away. Friends told them to try gentle cleansers and they have avoided using lotions or cream on her face. However, the bumps will not go away. On examination she has papules and pustules. Comedones cannot be identified. What are your next steps?
a. adapalene cream 0.1% every night at bedtime
b. benzoyl peroxide cream 4%
c. benzoyl peroxide wash 2.5%
d. erythromycin gel 2%
e. ketoconazole cream 2% twice daily
2. While in the newborn nursery prior to discharge, the attending pediatrician notices a rash on a 2-day-old neonate who is otherwise completely healthy. The pediatrician consults a dermatologist for his/her opinion. The dermatologist sees erythematous macules with central pustules located predominately on the trunk and proximal extremities. A pustule is unroofed with a blade, the contents smeared on a glass slide, and a Giemsa stain is performed. What is the predominant cell type you would expect to see on histological examination?
a. eosinophils
b. Langerhans cells
c. lymphocytes
d. neutrophils
e. no cells are visualized
3. Shortly after delivery, the pediatricians notice that the baby has numerous hyperpigmented macules on the back. No other primary lesions are seen. The neonate is otherwise normal in appearance and nontoxic appearing. A dermatologist is consulted for a recommendation for further workup or potential biopsy. The dermatologist examines the newborn. He is a well-appearing black boy with skin that is otherwise intact. A few pustules on the back are present that have a collarette of scale. The dermatologist reviews the mother’s prenatal history and the review shows that she was screened for syphilis and had a negative screening test with no other history of infectious diseases. What is the most appropriate next step to confirm your suspicions?
a. do a swab of a pustule and send it for viral culture
b. have his blood drawn and check for signs of neonatal herpes simplex virus infection
c. perform a biopsy of a pustule
d. perform a Giemsa stain on a smear of the pustule
e. start treatment with permethrin
4. Which intraoral cysts occur on the alveolar ridge of a neonate?
a. Bohn nodule
b. branchial cleft cyst
c. Epstein pearls
d. median raphe cyst
e. palatal cysts of the newborn
5. Miliaria rubra is associated with inflammation of the sweat glands in what portion of the skin?
a. basement membrane zone
b. dermis
c. dermoepidermal junction
d. intraepidermal
e. subcutis

Red-Blue Nodule on the Scalp
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
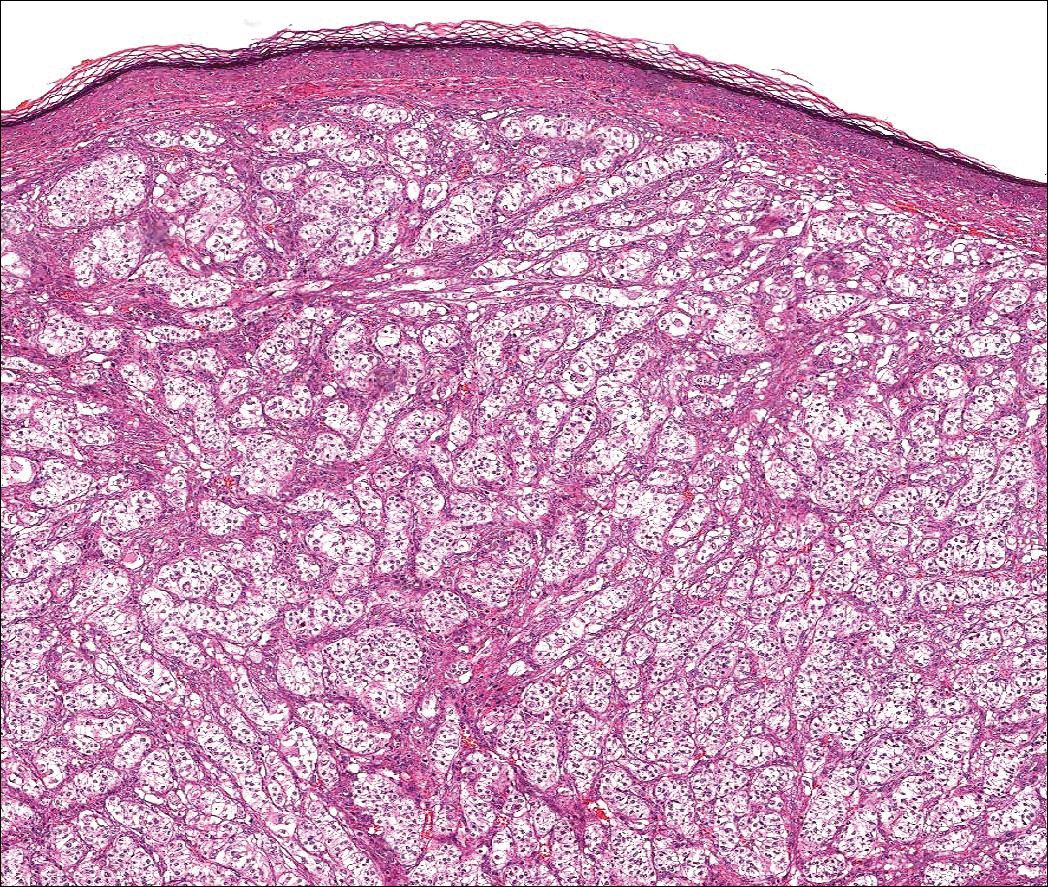
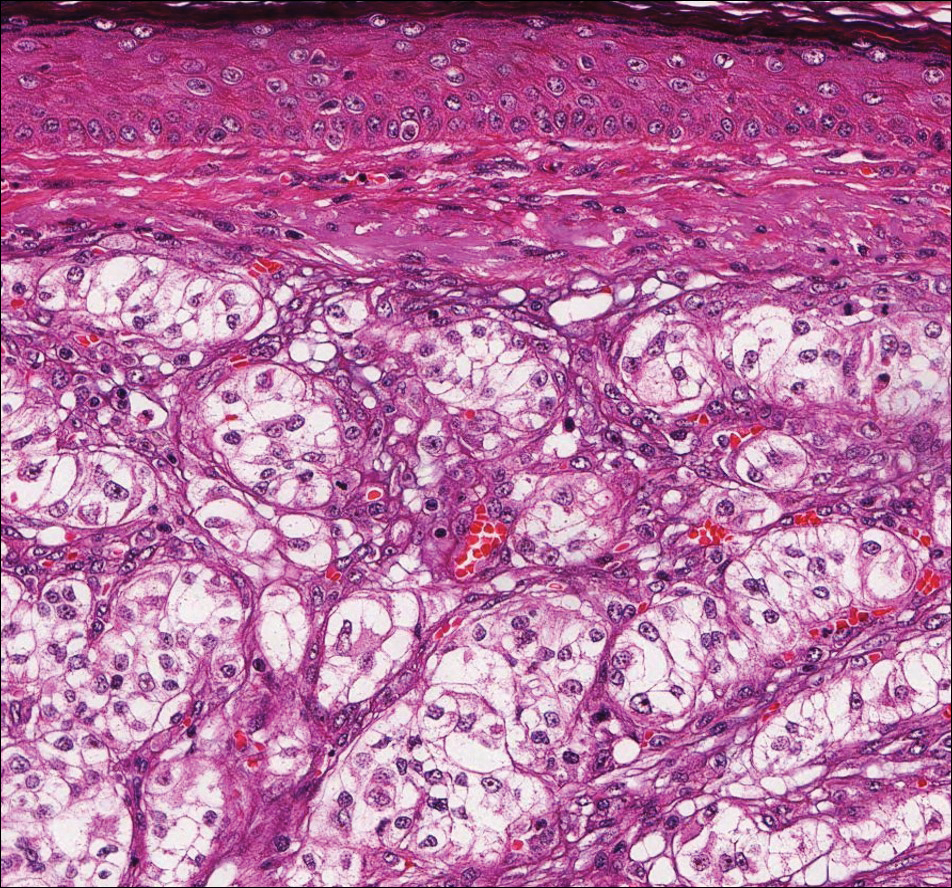
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
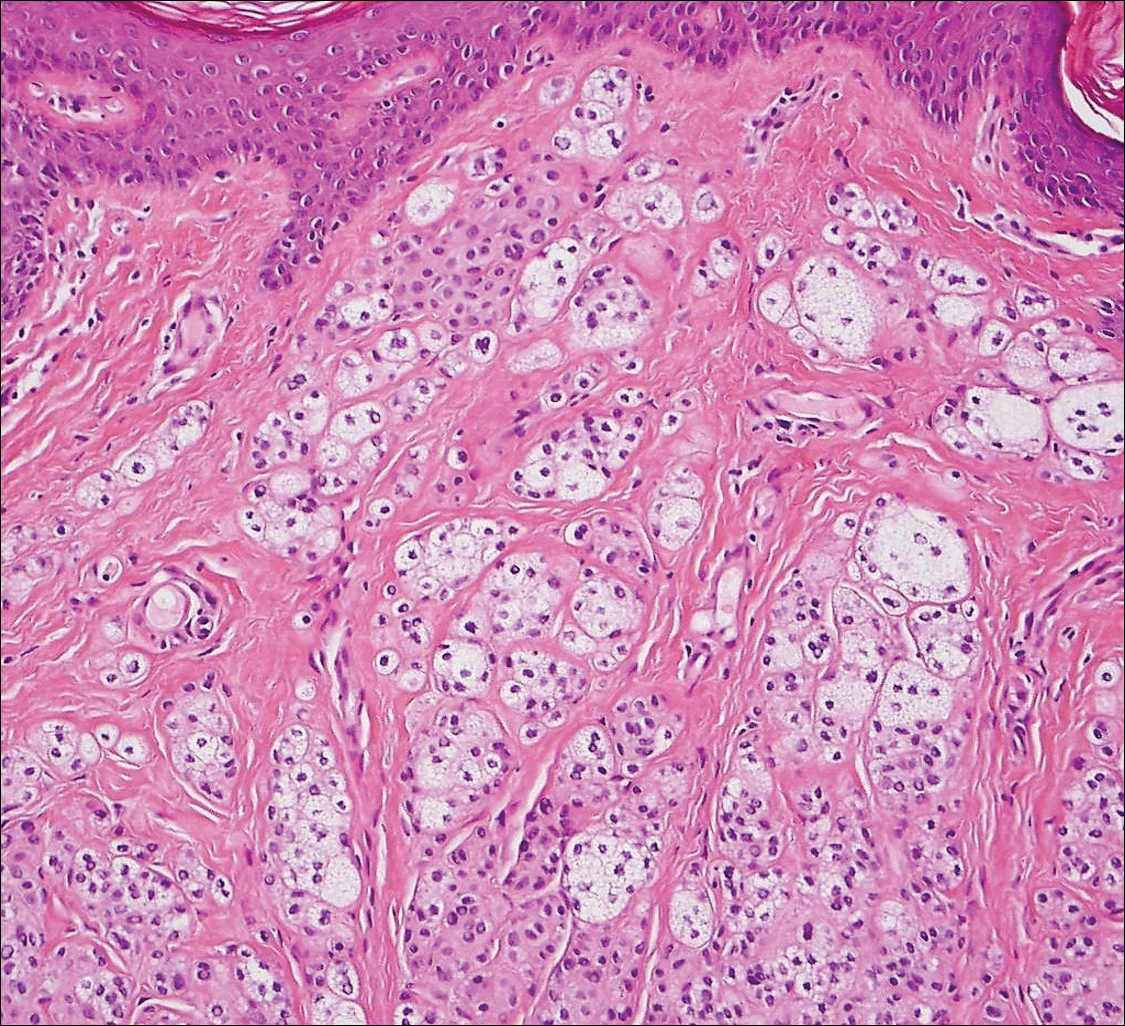
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
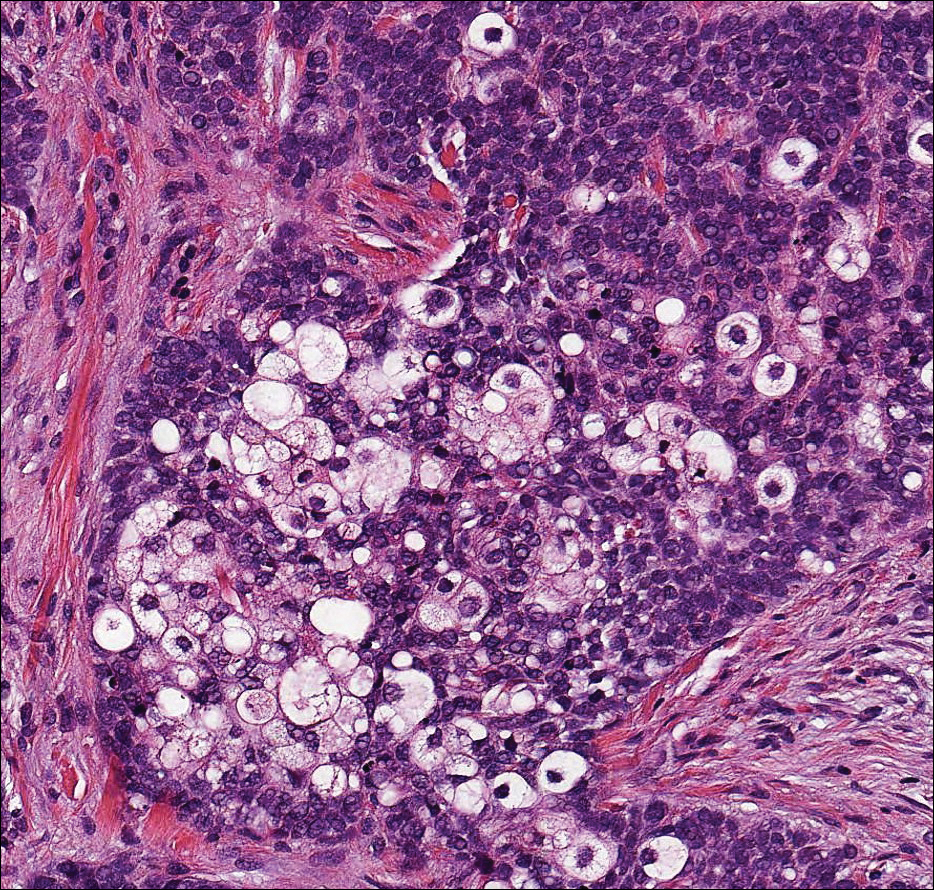
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6

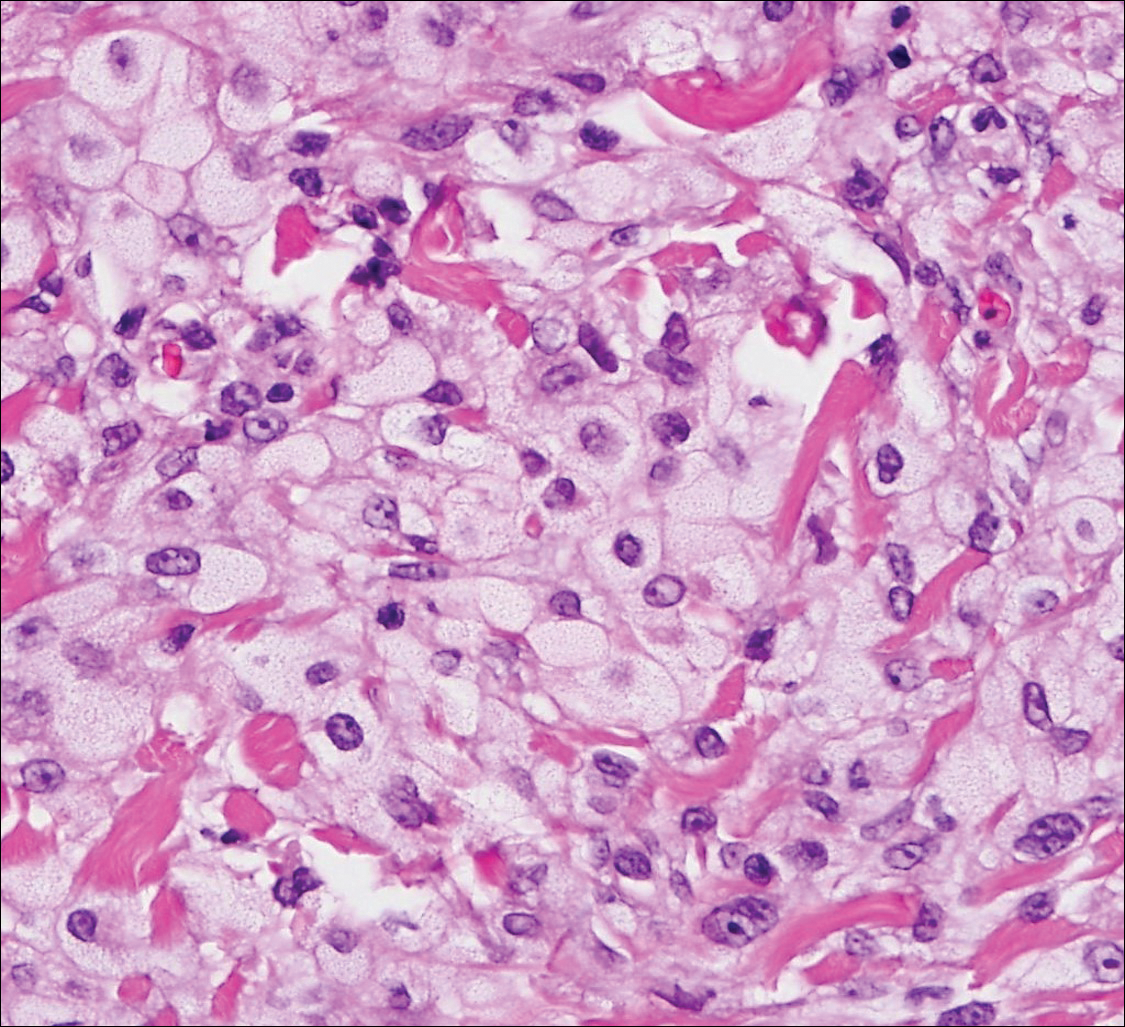
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
Metastatic Clear Cell Renal Cell Carcinoma
The differential diagnosis of cutaneous neoplasms with clear cells is broad. Clear cell features can be seen in primary tumors arising from the epidermis and cutaneous adnexa as well as in mesenchymal and melanocytic neoplasms. Furthermore, metastatic disease should be considered in the histologic differential diagnosis, as many visceral malignancies have clear cell features. This patient was subsequently found to have a large renal mass with metastasis to the lungs, spleen, and bone. The histologic findings support the diagnosis of metastatic clear cell renal cell carcinoma (RCC) to the skin.
Approximately 30% of patients with clear cell RCC present with metastatic disease with approximately 8% of those involving the skin.1,2 Cutaneous RCC metastases show a predilection for the head, especially the scalp. The clinical presentation is variable, but there often is a history of a rapidly growing brown, black, or purple nodule or plaque. A thorough review of the patient's history should be conducted if metastatic RCC is in the differential diagnosis, as it has been reported to occur up to 20 years after initial diagnosis.3
Histologically, clear cell RCC (quiz image) is composed of nests of tumor cells with clear cytoplasm and centrally located nuclei with prominent nucleoli. The clear cell features result from abundant cytoplasmic glycogen and lipid but may not be present in every case. One of the most important histologic features is the presence of delicate branching blood vessels (Figure 1). Numerous extravasated red blood cells also may be present. Positive immunohistochemical staining for PAX8, CD10, and RCC antigens support the diagnosis.4
Balloon cell nevi (Figure 2) most commonly occur on the head and neck in adolescents and young adults but clinically are indistinguishable from other banal nevi. The nevus cells are large with foamy to finely vacuolated cytoplasm and lack atypia. The clear cell change is the result of melanosome degeneration and may be extensive. The presence of melanin pigment, nests of typical nevus cells, and positive staining with MART-1 can help distinguish the tumor from xanthomas and RCC.5
Clear cell hidradenoma (Figure 3) is a well-circumscribed tumor of sweat gland origin that arises in the dermis. The architecture usually is solid, cystic, or a combination of both. The cytology is classically bland with poroid, squamoid, or clear cell morphology. Clear cells that are positive on periodic acid-Schiff staining predominate in up to one-third of cases. Carcinoembryonic antigen and epithelial membrane antigen can be used to highlight the eosinophilic cuticles of ducts within solid areas.6
Sebaceous carcinoma (Figure 4) most frequently arises in a periorbital distribution, although extraocular lesions are known to occur. Histologically, there is a proliferation of both mature sebocytes and basaloid cells in the dermis, occasionally involving the epidermis. The mature sebocytes demonstrate clear cell features with foamy to vacuolated cytoplasm and large nuclei with scalloped borders. The clear cells may vary greatly in number and often are sparse in poorly differentiated tumors in which pleomorphic basaloid cells may predominate. The basaloid cells may resemble those of squamous or basal cell carcinoma, leading to a diagnostic dilemma in some cases. Special staining with Sudan black B and oil red O highlights the cytoplasmic lipid but must be performed on frozen section specimens. Although not entirely specific, immunohistochemical expression of epithelial membrane antigen, androgen receptor, and membranous vesicular adipophilin staining in sebaceous carcinoma can assist in the diagnosis.7
Cutaneous xanthomas (Figure 5) may arise in patients of any age and represent deposition of lipid-laden macrophages. Classification often is dependent on the clinical presentation; however, some subtypes demonstrate unique morphologic features (eg, verruciform xanthomas). Xanthomas classically arise in association with elevated serum lipids, but they also may occur in normolipemic patients. Individuals with Erdheim-Chester disease have an increased propensity to develop xanthelasma. Similarly, plane xanthomas have been associated with monoclonal gammopathy. Histologically, xanthomas are characterized by sheets of foamy macrophages within the dermis and subcutis. Positive immunohistochemical staining for CD68 highlighting the histiocytic nature of the cells and the absence of a delicate vascular network aid in the differentiation from RCC.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Alcaraz I, Cerroni L, Rutten A, et al. Cutaneous metastases from internal malignancies: a clinicopathologic and immunohistochemical review. Am J Dermatopathol. 2012;34:347-393.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Lin F, Prichard J. Handbook of Practical Immunohistochemistry: Frequently Asked Questions. 2nd ed. New York, NY: Springer; 2015.
- McKee PH, Calonje E. Diagnostic Atlas of Melanocytic Pathology. Edinburgh, Scotland: Mosby/Elsevier; 2009.
- Elston DM, Ferringer T, Ko CJ. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Ansai S, Takeichi H, Arase S, et al. Sebaceous carcinoma: an immunohistochemical reappraisal. Am J Dermatopathol. 2011;33:579-587.
A 59-year-old man presented with a 1.5×1.0-cm asymptomatic, smooth, red-blue nodule on the left parietal scalp. The nodule had been rapidly enlarging over the last 3 weeks. After resection, the cut surface was golden yellow and focally hemorrhagic.
Superficial Ulceration on the Vulva
Foscarnet-Induced Ulceration
Viral swabs were negative for herpes simplex virus. The diagnosis of foscarnet-induced ulceration was reached and the drug was discontinued. Symptomatic treatment with soap substitutes and lidocaine ointment was used.
Foscarnet is an antiviral agent used when resistance develops to first-line therapies.1 It is a pyrophosphate analogue that inhibits viral DNA polymerase, thereby preventing viral replication. It is used in treating cytomegalovirus and herpes simplex virus, which are resistant to first-line therapies, or patients who develop hematologic toxicity from antivirals. The main side effects of foscarnet include nephrotoxicity, alteration of calcium homeostasis, and malaise. Genital ulceration is a known side effect of therapy, though it is rare and more commonly seen in uncircumcised males. Approximately 94% of the drug is excreted unchanged in the urine, which causes an irritant dermatitis that is more pronounced in males as the urine stays in the subpreputial area.1
Vulval ulceration2 and penile ulceration3 has been reported in AIDS patients treated with foscarnet. In these patients, the onset of ulceration is temporally related to foscarnet therapy, occurring at approximately day 7 to 24 of treatment and resolving after discontinuation of therapy.
- Wagstaff AJ, Bryson HM. Foscarnet. a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs. 1994;48:199-226.
- Lacey HB, Ness A, Mandal BK. Vulval ulceration associated with foscarnet. Genitourin Med. 1992;68:182.
- Moyle G, Barton S, Gazzard BG. Penile ulceration with foscarnet therapy. AIDS. 1993;7:140-141.
Foscarnet-Induced Ulceration
Viral swabs were negative for herpes simplex virus. The diagnosis of foscarnet-induced ulceration was reached and the drug was discontinued. Symptomatic treatment with soap substitutes and lidocaine ointment was used.
Foscarnet is an antiviral agent used when resistance develops to first-line therapies.1 It is a pyrophosphate analogue that inhibits viral DNA polymerase, thereby preventing viral replication. It is used in treating cytomegalovirus and herpes simplex virus, which are resistant to first-line therapies, or patients who develop hematologic toxicity from antivirals. The main side effects of foscarnet include nephrotoxicity, alteration of calcium homeostasis, and malaise. Genital ulceration is a known side effect of therapy, though it is rare and more commonly seen in uncircumcised males. Approximately 94% of the drug is excreted unchanged in the urine, which causes an irritant dermatitis that is more pronounced in males as the urine stays in the subpreputial area.1
Vulval ulceration2 and penile ulceration3 has been reported in AIDS patients treated with foscarnet. In these patients, the onset of ulceration is temporally related to foscarnet therapy, occurring at approximately day 7 to 24 of treatment and resolving after discontinuation of therapy.
Foscarnet-Induced Ulceration
Viral swabs were negative for herpes simplex virus. The diagnosis of foscarnet-induced ulceration was reached and the drug was discontinued. Symptomatic treatment with soap substitutes and lidocaine ointment was used.
Foscarnet is an antiviral agent used when resistance develops to first-line therapies.1 It is a pyrophosphate analogue that inhibits viral DNA polymerase, thereby preventing viral replication. It is used in treating cytomegalovirus and herpes simplex virus, which are resistant to first-line therapies, or patients who develop hematologic toxicity from antivirals. The main side effects of foscarnet include nephrotoxicity, alteration of calcium homeostasis, and malaise. Genital ulceration is a known side effect of therapy, though it is rare and more commonly seen in uncircumcised males. Approximately 94% of the drug is excreted unchanged in the urine, which causes an irritant dermatitis that is more pronounced in males as the urine stays in the subpreputial area.1
Vulval ulceration2 and penile ulceration3 has been reported in AIDS patients treated with foscarnet. In these patients, the onset of ulceration is temporally related to foscarnet therapy, occurring at approximately day 7 to 24 of treatment and resolving after discontinuation of therapy.
- Wagstaff AJ, Bryson HM. Foscarnet. a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs. 1994;48:199-226.
- Lacey HB, Ness A, Mandal BK. Vulval ulceration associated with foscarnet. Genitourin Med. 1992;68:182.
- Moyle G, Barton S, Gazzard BG. Penile ulceration with foscarnet therapy. AIDS. 1993;7:140-141.
- Wagstaff AJ, Bryson HM. Foscarnet. a reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs. 1994;48:199-226.
- Lacey HB, Ness A, Mandal BK. Vulval ulceration associated with foscarnet. Genitourin Med. 1992;68:182.
- Moyle G, Barton S, Gazzard BG. Penile ulceration with foscarnet therapy. AIDS. 1993;7:140-141.
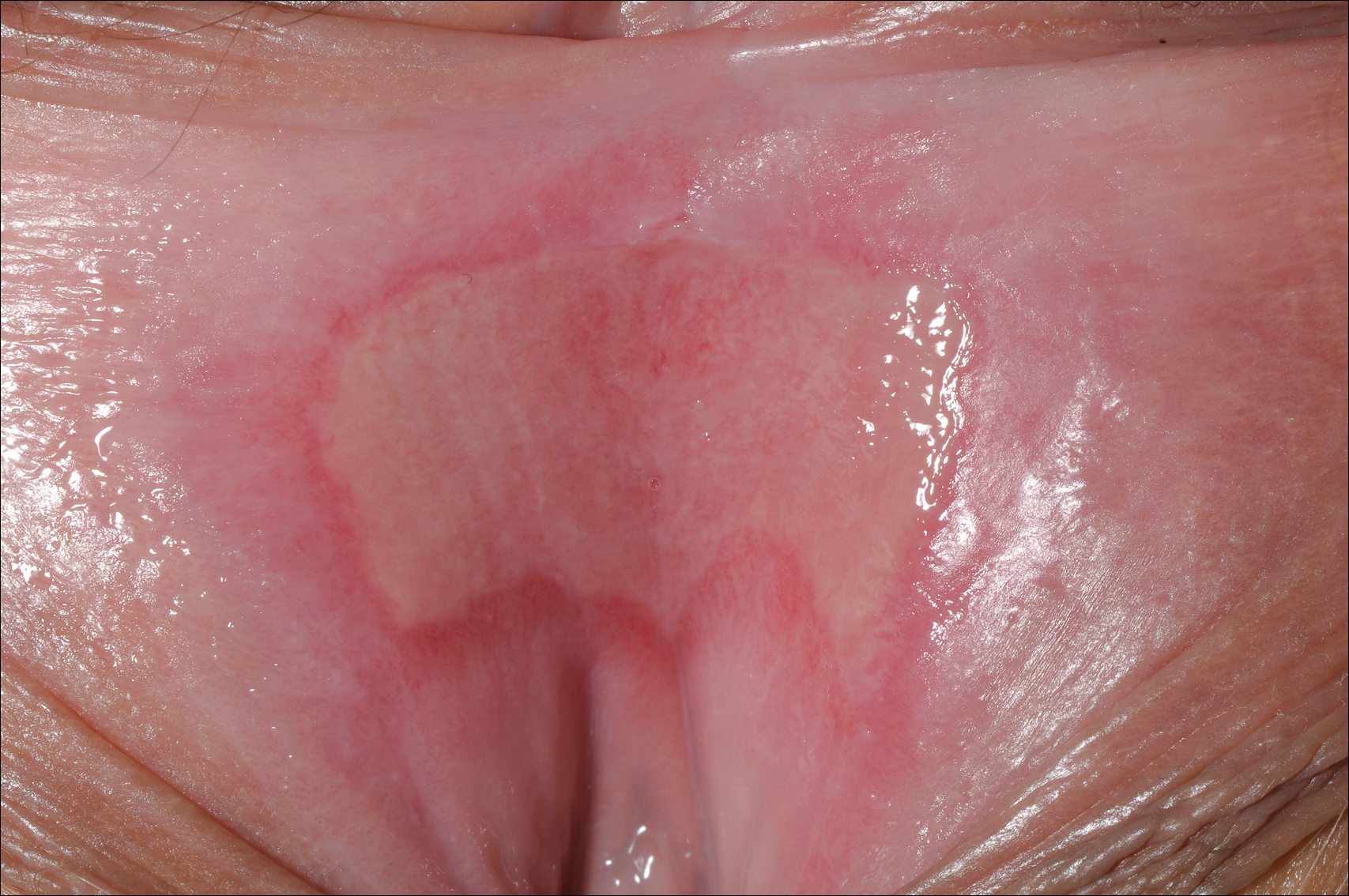
A 23-year-old woman who was immunosuppressed secondary to cyclophosphamide and prednisolone treatment of autoimmune panniculitis was admitted to intensive care with dyspnea. Cytomegalovirus and Pneumocystis jiroveci pneumonia were diagnosed on bronchoscopy and bronchial washings. Management with valganciclovir was started but worsened the patient's pancytopenia. She was started on intravenous foscarnet. After a week of therapy, the patient reported vulval soreness and painful micturition. On examination there was superficial ulceration of the labia minora. The affected area was symmetrical, and there was some extension into the vestibule. There were no vesicles or lesions on the cutaneous skin.
Shedding Light on Onychomadesis
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23

Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
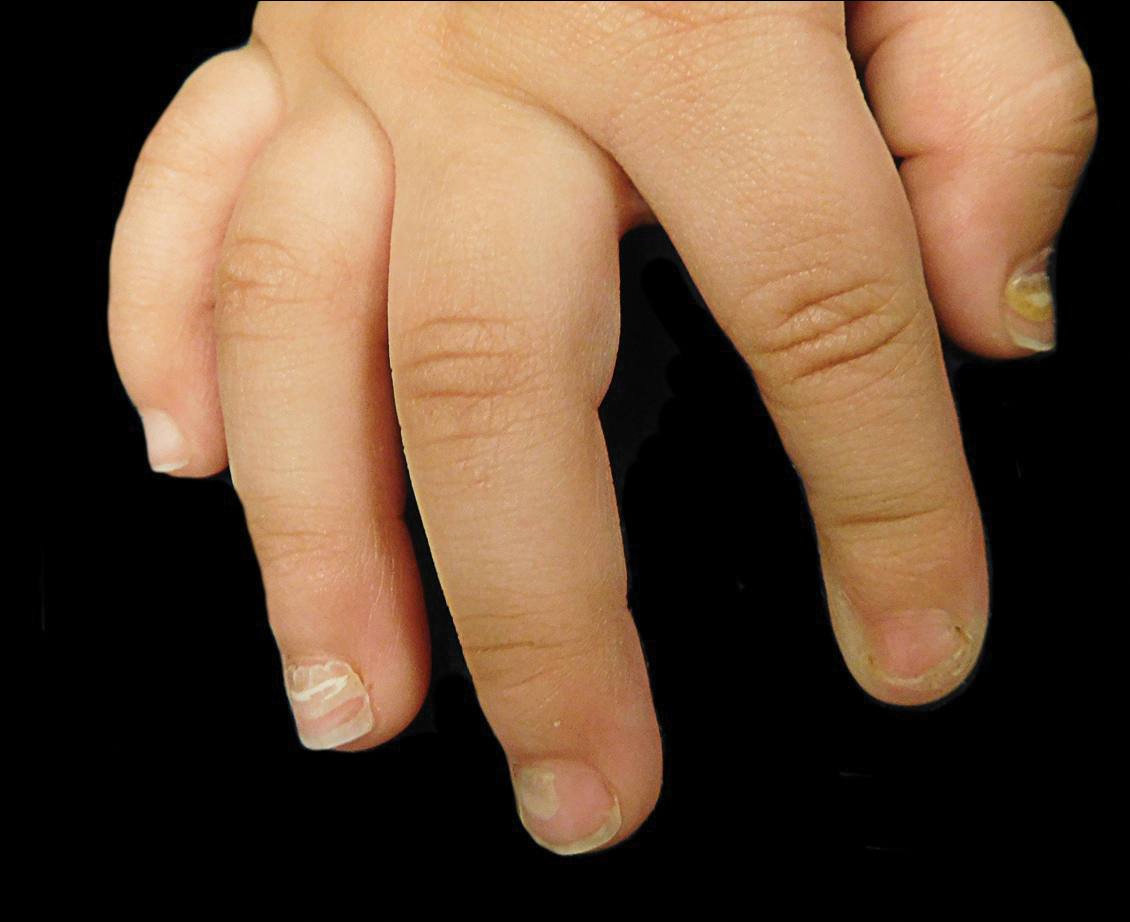
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Practice Points
- Onychomadesis in a child may be a cutaneous sign of systemic disease.
- In childhood, onychomadesis is sometimes linked with hand-foot-and-mouth disease.
- Spontaneous nail regrowth usually occurs within 12 weeks but may occur faster in children.
Product News: January 2017
Enbrel
Amgen Inc announces that the US Food and Drug Administration has approved the supplemental Biologics License Application for the expanded use of Enbrel (etanercept) for the treatment of moderate to severe plaque psoriasis in pediatric patients (aged 4–17 years). Enbrel, a tumor necrosis factor blocker, was approved for the treatment of moderate to severe plaque psoriasis in adults in 2004. For more information, visit www.enbrel.com.
Eucrisa
Pfizer Inc announces US Food and Drug Administration approval of Eucrisa (crisaborole) ointment 2% for the treatment of mild to moderate atopic dermatitis in patients 2 years and older. Eucrisa is a nonsteroidal topical phosphodiesterase 4 inhibitor and is applied twice daily. This approval provides patients with atopic dermatitis another treatment alternative, as this community has not had a new prescription treatment for more than 10 years. For more information, visit www.pfizer.com.
Isdinceutics
Isdin based in Spain has launched the Isdinceutics line of physician-dispensed cosmeceuticals to the US market, which focuses on vitamins and hydrators rather than chemicals to rejuvenate the skin. Isdinceutics features a daily antioxidant routine with Flavo-C Ultraglican and Flavo-C Serum to reduce the appearance of microwrinkles and elevate the skin’s natural moisture production. Products to correct pigmentation problems as well as undereye circles and puffiness also are
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Enbrel
Amgen Inc announces that the US Food and Drug Administration has approved the supplemental Biologics License Application for the expanded use of Enbrel (etanercept) for the treatment of moderate to severe plaque psoriasis in pediatric patients (aged 4–17 years). Enbrel, a tumor necrosis factor blocker, was approved for the treatment of moderate to severe plaque psoriasis in adults in 2004. For more information, visit www.enbrel.com.
Eucrisa
Pfizer Inc announces US Food and Drug Administration approval of Eucrisa (crisaborole) ointment 2% for the treatment of mild to moderate atopic dermatitis in patients 2 years and older. Eucrisa is a nonsteroidal topical phosphodiesterase 4 inhibitor and is applied twice daily. This approval provides patients with atopic dermatitis another treatment alternative, as this community has not had a new prescription treatment for more than 10 years. For more information, visit www.pfizer.com.
Isdinceutics
Isdin based in Spain has launched the Isdinceutics line of physician-dispensed cosmeceuticals to the US market, which focuses on vitamins and hydrators rather than chemicals to rejuvenate the skin. Isdinceutics features a daily antioxidant routine with Flavo-C Ultraglican and Flavo-C Serum to reduce the appearance of microwrinkles and elevate the skin’s natural moisture production. Products to correct pigmentation problems as well as undereye circles and puffiness also are
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Enbrel
Amgen Inc announces that the US Food and Drug Administration has approved the supplemental Biologics License Application for the expanded use of Enbrel (etanercept) for the treatment of moderate to severe plaque psoriasis in pediatric patients (aged 4–17 years). Enbrel, a tumor necrosis factor blocker, was approved for the treatment of moderate to severe plaque psoriasis in adults in 2004. For more information, visit www.enbrel.com.
Eucrisa
Pfizer Inc announces US Food and Drug Administration approval of Eucrisa (crisaborole) ointment 2% for the treatment of mild to moderate atopic dermatitis in patients 2 years and older. Eucrisa is a nonsteroidal topical phosphodiesterase 4 inhibitor and is applied twice daily. This approval provides patients with atopic dermatitis another treatment alternative, as this community has not had a new prescription treatment for more than 10 years. For more information, visit www.pfizer.com.
Isdinceutics
Isdin based in Spain has launched the Isdinceutics line of physician-dispensed cosmeceuticals to the US market, which focuses on vitamins and hydrators rather than chemicals to rejuvenate the skin. Isdinceutics features a daily antioxidant routine with Flavo-C Ultraglican and Flavo-C Serum to reduce the appearance of microwrinkles and elevate the skin’s natural moisture production. Products to correct pigmentation problems as well as undereye circles and puffiness also are
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].

Advances in Minimally Invasive and Noninvasive Treatments for Submental Fat
Submental fat (SMF) accumulation within the subcutaneous (preplatysmal) or subplatysmal fat compartment of the cervical anatomy results in an obtuse cervicomental angle and loss of mandibular and cervical contours. It is a common cosmetic concern due to its aesthetic association with weight gain and aging.1 Minimally invasive or noninvasive submental lipolytic agents and techniques are sought for patients who are not candidates for surgery or prefer more conservative cosmetic treatments. These methods typically are only effective in addressing preplatysmal SMF, as subplatysmal SMF requires more surgical methods due to its less-accessible location. The pathology of SMF should initially be assessed by clinical examination or ultrasonography. In this article, we review the most relevant clinical and safety data on minimally invasive and noninvasive treatments for SMF, including laser-assisted lipolysis (LAL), radiofrequency (RF)–assisted lipolysis, deoxycholic acid (DCA), and cryolipolysis.
MINIMALLY INVASIVE MODALITIES
Traditional, or tumescent, liposuction is still widely considered the most effective method for removal of large masses of adiposity. Laser- and RF-assisted adjuncts have been more recently developed to improve patient side effects and recovery time and reduce the manual effort of surgeons. Of note, these adjuncts, with some exceptions, still require the same invasiveness as traditional liposuction, involving submental stab incisions of up to 2.4 mm.
Laser-Assisted Lipolysis
Laser-assisted lipolysis produces a similar effect as suction-assisted lipoplasty by focusing pulses of laser energy through a 1-mm wide fiber optic cannula and inducing thermally mediated adipolysis. The directed laser results in adipocyte rupturing with added benefits of skin retraction and small vessel coagulation, thus lessening intraoperative blood loss.2 This technique typically requires smaller incisions than traditional liposuction. The most common laser lipolysis systems used in cosmetic dermatology are the 920- to 980-nm diode lasers and 1064- to 1440-nm Nd:YAG lasers. The 924-nm diode, 1064-nm Nd:YAG, and 1064/1320-nm Nd:YAG have been best characterized in clinical trials, as reviewed by Fakhouri et al,3 with demonstrated efficacy in reducing SMF density.
The first randomized prospective trial comparing LAL (using 1064-nm Nd:YAG) and traditional liposuction in various anatomical areas on 25 patients showed no difference in cosmetic results, ecchymoses, edema, or retraction, and significantly lower postoperative pain ratings (P<.0001) in LAL.4 A more recent prospective randomized comparison of LAL (980-nm diode laser; 6–8 W) and traditional liposuction of the submental area in 40 female patients showed greater reduction in SMF thickness in the LAL group compared to the liposuction group at 2-month follow-up (6.2 vs 8.22 unspecified units; P<.001) with significant improvement from baseline in both groups (P<.001).5 However, the cosmetic benefit of LAL over traditional liposuction remains controversial and has not been unequivocally established in the literature.
Common adverse events (AEs) are postoperative swelling, ecchymoses, and pain, and complications of interest are nodularity, skin infections, burns, and nerve damage.6 In one retrospective investigation (N=537), these complications occurred at a rate of less than 1% (4 burns and 1 skin infection).6 Patients treated with LAL may report fewer AEs, especially pain and bleeding, compared to liposuction-treated patients.3
RF-Assisted Lipolysis
Radiofrequency-assisted lipolysis is one of the newest technologies in lipocontouring. NeckTite (Invasix Aesthetic Solutions) is effective for treatment of preplatysmal adiposity and cervicomental lipocontouring; a 2.4-mm bipolar probe that is inserted into the subdermal space and connected with an external electrode emits RF energy and simultaneously coagulates and aspirates adipose tissue. NeckTite also may be used in conjunction with FaceTite (Invasix Aesthetic Solutions), which promotes fibroseptal network remodeling and dermal contraction.2
In the first published investigation of the efficacy and safety of NeckTite, 47 of 55 patients received treatment of slight to moderate SMF (average body mass index [BMI], 25 kg/m2) with NeckTite and FaceTite or NeckTite alone.7 At 6-month follow-up, 87% (48/55) of patients subjectively rated treatment efficacy as satisfactory, and 2 independent physicians rated the improvement between before-and-after frontal and lateral photographs of the submental area as moderate to excellent in 95% (52/55) of all cases. Reported complications in this study were full-thickness burns resulting in minor scarring (2/55 [4%]), neck tissue hardness that resolved with daily massage after 3 months (5/55 [9%]), and transient facial nerve paresis of the mandibular branch that resolved after 2 months (1/55 [2%]).7
NONINVASIVE MODALITIES
RF-Assisted Contouring
Another exciting development in RF technology is truSculpt (Cutera), a noninvasive contouring device that is placed over the epidermis and emits RF energy that preferentially heats fat more than other tissue types. In a single-center prospective trial of efficacy and safety in the treatment of SMF, 17 patients received 2 treatments with truSculpt administered 1 month apart.8 At 1- and 6-month follow-up, 82.3% (14/17) and 52.9% (9/17) of patients showed improvement on physician assessment. Submental circumference and ultrasonographic fat thickness reductions at 1-month follow-up were 1.4 cm (5.7% of pretreatment circumference [P<.001]) and 5.4 mm (9.7% of pretreatment fat thickness [P=.005]), respectively. At further longer-term follow-up to 6 months, submental circumference was 0.9 cm (3.8% of pretreatment circumference [P<.001]) and ultrasonographic fat reduction was 6.8 mm (10.5% of pretreatment fat thickness [P=.006]). Commonly reported AEs were pain (rate not given), erythema (8/17 [47%]), edema (1/17 [6%]), and vesicle formation (1/17 [6%]); all were self-resolving. Erythema usually subsided within 6 hours posttreatment. No other AEs or complications were reported.8
Deoxycholic Acid
Deoxycholic acid (DCA)(formerly ATX-101) is an injectable liquid formulation of synthetic DCA that was approved by the US Food and Drug Administration (FDA) in 2015 for moderate to severe SMF. Deoxycholic acid exists endogenously as a bile salt emulsifier and has been shown to cause dose-dependent adipocyte lysis, necrosis, disruption and dissolution of fat architecture, and inflammatory targeting of adipocytes by immune cells.9,10 Thus, DCA causes targeted adipocytolysis and is a novel medical agent in the treatment of SMF. Supplied in 2-mL vials, clinicians may inject 10 mL at each treatment for up to 6 treatments administered 1 month apart.11
Efficacy
REFINE-1, a pivotal North American–based phase 3 trial, investigated the efficacy and safety of DCA.12 A total of 506 participants with scores of 2 (moderate) or 3 (severe) on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS) and a mean BMI of 29 kg/m2 were randomized to receive preplatysmal fat injections of 2 mg/cm2 of DCA (n=256) or placebo (n=250). Participants received up to 10 mL of product (mean total of 25 mL of DCA across all visits) at each treatment session for up to 6 sessions depending on individual efficacy, with approximately 28 days between sessions. Sixty-four percent of the treatment group received all 6 treatments. At 12-week follow-up after the last treatment session, 70% of DCA-treated participants versus 18.6% of placebo-treated participants (P<.001) improved by 1 grade or more on the CR-SMFRS and 13.4% versus 0% (P<.001) improved by 2 grades or more. Skin laxity was unchanged or improved in 92.7% of the DCA group and 87.6% of the placebo group.12
REFINE-2, the second of the North American phase 3 trials, had parallel inclusionary criteria and study design and established efficacy of 2 mg/cm2 DCA over placebo in 516 participants (randomized 1:1).13 At 12 weeks posttreatment, 66.5% of DCA-treated participants versus 22.2% of placebo-treated participants improved by 1 grade or more according to the CR-SMFRS (P<.001) and 18.6% versus 3% improved by 2 grades or more in SMF (P<.001). Magnetic resonance imaging analysis of participants in the DCA (n=113) and placebo groups (n=112) showed that 40.2% versus 5.2% (P<.001) exhibited 10% or more reduction in submental volume, with similar comparative rates of SMF thickness reduction via caliper measurements.13
Safety
Safety data from REFINE-1 showed higher rates of treatment-related AEs in DCA-treated participants compared to placebo, including hematoma (70% vs 67.3%), anesthesia (66.9% vs 4.4%), pain (65.4% vs 23.4%), edema (52.9% vs 21.8%), induration (18.3% vs 1.6%), paresthesia (12.8% vs 3.2%), nodule formation (12.5% vs 0.8%), and pruritus (8.6% vs 3.6%).12 In this trial, 11 of 258 cases (4.3%) of marginal mandibular nerve paresis and asymmetric smile occurred, all in DCA-treated participants and with a median duration of 31 days. Dysphagia resolving in a median duration of 4 days occurred in 1.6% (4/258) of DCA-treated participants.12 REFINE-2 exhibited similar rates of common AEs. Complications of note were 14 cases of marginal mandibular nerve paresis (11 in DCA group, 3 in placebo group) attributed to injection technique, 1 case of skin ulceration possibly related to accidental injection into dermis, and 6 cases of dysphagia in DCA participants attributed to higher volume treatment sessions and postinjection swelling. Dysphagia lasted a median of 2.5 days and resolved without sequelae.13
Overall, DCA demonstrated high rates of minor injection-site AEs that resolved without sequelae and could be mitigated by comfort therapies (eg, lidocaine, nonsteroidal anti-inflammatory drugs) as well as understanding the anatomy of the submental region. Adverse effects of particular interest included marginal mandibular nerve palsy, skin ulceration, and dysphagia.12,13
Cryolipolysis
Cryolipolysis is an advancement that utilizes the application of noninvasive cooling temperatures to the skin’s surface to destroy underlying adipocytes based on the concept that lipid-filled cells are more susceptible to cold-induced injury than water-filled cells. Thus, cryolipolysis selectively targets adipose tissue, leading to cell death without harm to surrounding cells and without the need for surgery or injections.14
Cryolipolysis typically is delivered via a vacuum applicator (CoolMini, Zeltiq Aesthetics Inc), which applies temperatures of –10°C (14°F) to the skin in cycles of 60 minutes each. Initially approved by the FDA for treatment of flank adiposity in 2010, cryolipolysis has since been approved for treatment of the abdomen, thighs, and submental area.14 An advantage of cryolipolysis is that it does not require frequent treatment sessions for maximal efficacy.
Efficacy
The efficacy of cryolipolysis in the treatment of SMF was established in a multicenter device investigation resulting in its FDA approval for the submental region.15 Sixty participants with a mean BMI of 31.8 kg/m2 received 1 (1/60) or 2 (59/60) treatment sessions of the submental area administered 6 weeks apart. Primary efficacy assessments included analysis by 3 blinded reviewers who viewed photographs of each participant at baseline, immediately posttreatment, 6 weeks posttreatment, and 12 weeks posttreatment; ultrasonographic measurements of SMF thickness; and a 12-point patient satisfaction questionnaire. Blinded reviewers correctly identified baseline images in 91.4% (55/60) of cases. Ultrasonography confirmed a mean reduction in SMF of 2 mm (P<.0001) or 20% of fat thickness at 12 weeks posttreatment. On subjective patient satisfaction surveys, 83% (50/60) of participants were satisfied with the procedure and 77% (46/60) reported a visible reduction in fat and perceived an improvement in appearance.15
Safety
The most common immediate posttreatment AEs were erythema/purpura (100%), numbness (90%), edema (62%), tingling (30%), blanching (25%), and bruising (3%) at the site of cryolipolysis with resolution within 1 week posttreatment, except for numbness.15 At 6-week follow-up, all AEs had resolved, except continued numbness in 4 participants that resolved by 12-week follow-up. A further event of note was fullness in the throat in 1 participant that was attributed to swelling and resolved at 40 days posttreatment without incident. No serious AEs were reported in this trial.15
A particularly concerning but rare complication that is increasing in awareness is paradoxical adipose hyperplasia following cryolipolysis. Patients may develop firm painless areas of soft tissue enlargements in the area of cryolipolysis typically 3 to 6 months posttreatment.16 The largest published report recorded an incidence rate of 0.46% (n=2, all males) at a single-center institution of 422 cryolipolysis treatments.16 Other incidence rates reported are 0.0051% and 0.78%.17 Causes and associations are not known, though male gender is speculated to increase risk.
Conclusion
This article highlights the available information on advances in minimally invasive and noninvasive treatments for SMF accumulation. The efficacy and safety trials varied in quality and in different methods of end point analysis of SMF reduction. Further, few trials have featured head-to-head comparisons of treatments.
Although liposuction and adjuncts remain the gold standard in large-mass lipid removal, these procedures are invasive and exhibit typical risks of surgery. Given its sensitive location, the submental area may require the use of more delicate therapeutic methods, including completely noninvasive devices such as truSculpt and cryolipolysis. Regardless of the chosen treatment, the most important factors in yielding patient satisfaction and SMF improvement are proper patient selection and an understanding of the anatomical source of adiposity to be addressed with the therapeutic modalities.
[polldaddy:9711250]
- Hatef DA, Koshy JC, Sandoval SE, et al. The submental fat compartment of the neck. Semin Plast Surg. 2009;23:288-291.
- Mulholland RS. Nonexcisional, minimally invasive rejuvenation of the neck. Clin Plast Surg. 2014;41:11-31.
- Fakhouri TM, El Tal AK, Abrou AE, et al. Laser-assisted lipolysis: a review. Dermatol Surg. 2012;38:155-169.
- Prado A, Andrades P, Danilla S, et al. A prospective, randomized, double-blind, controlled clinical trial comparing laser-assisted lipoplasty with suction-assisted lipoplasty. Plast Reconstr Surg. 2006;118:1032-1045.
- Valizadeh N, Jalaly NY, Zarghampour M, et al. Evaluation of safety and efficacy of 980-nm diode laser-assisted lipolysis versus traditional liposuction for submental rejuvenation: a randomized clinical trial. J Cosmet Laser Ther. 2016;18:41-45.
- Katz B, McBean J. Laser-assisted lipolysis: a report on complications. J Cosmet Laser Ther. 2008;10:231-233.
- Keramidas E, Rodopoulou S. Radiofrequency-assisted liposuction for neck and lower face adipodermal remodeling and contouring. Plast Reconstr Surg Glob Open. 2016;4:e850.
- Park JH, Kim JI, Park HJ, et al. Evaluation of safety and efficacy of noninvasive radiofrequency technology for submental rejuvenation [published online July 12, 2016]. Lasers Med Sci. 2016;31:1599-1605.
- Yagima Odo ME, Cucé LC, Odo LM, et al. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178-188; discussion 188-189.
- Rotunda AM, Suzuki H, Moy RL, et al. Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution. Dermatol Surg. 2004;30:1001-1008.
- Kybella [package insert]. Westlake Village, CA: Kythera Biopharmaceuticals, Inc; 2015.
- Jones DH, Carruthers J, Joseph JH, et al. REFINE-1, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial with ATX-101, an injectable drug for submental fat reduction. Dermatol Surg. 2016;42:38-49.
- Humphrey S, Sykes J, Kantor J, et al. ATX-101 for reduction of submental fat: a phase III randomized controlled trial [published online July 16, 2016]. J Am Acad Dermatol. 2016;75:788-797.e7.
- Manstein D, Laubach H, Watanabe K, et al. Selective cryolysis: a novel method of non-invasive fat removal. Lasers Surg Med. 2008;40:595-604.
- Kilmer SL, Burns AJ, Zelickson BD. Safety and efficacy of cryolipolysis for non-invasive reduction of submental fat. Lasers Surg Med. 2016;48:3-13.
- Singh SM, Geddes ER, Boutrous SG, et al. Paradoxical adipose hyperplasia secondary to cryolipolysis: an underreported entity? Lasers Surg Med. 2015;47:476-478.
- Kelly E, Rodriguez-Feliz J, Kelly ME. Paradoxical adipose hyperplasia after cryolipolysis: a report on incidence and common factors identified in 510 patients. Plast Reconst Surg. 2016;137:639e-640e.
Submental fat (SMF) accumulation within the subcutaneous (preplatysmal) or subplatysmal fat compartment of the cervical anatomy results in an obtuse cervicomental angle and loss of mandibular and cervical contours. It is a common cosmetic concern due to its aesthetic association with weight gain and aging.1 Minimally invasive or noninvasive submental lipolytic agents and techniques are sought for patients who are not candidates for surgery or prefer more conservative cosmetic treatments. These methods typically are only effective in addressing preplatysmal SMF, as subplatysmal SMF requires more surgical methods due to its less-accessible location. The pathology of SMF should initially be assessed by clinical examination or ultrasonography. In this article, we review the most relevant clinical and safety data on minimally invasive and noninvasive treatments for SMF, including laser-assisted lipolysis (LAL), radiofrequency (RF)–assisted lipolysis, deoxycholic acid (DCA), and cryolipolysis.
MINIMALLY INVASIVE MODALITIES
Traditional, or tumescent, liposuction is still widely considered the most effective method for removal of large masses of adiposity. Laser- and RF-assisted adjuncts have been more recently developed to improve patient side effects and recovery time and reduce the manual effort of surgeons. Of note, these adjuncts, with some exceptions, still require the same invasiveness as traditional liposuction, involving submental stab incisions of up to 2.4 mm.
Laser-Assisted Lipolysis
Laser-assisted lipolysis produces a similar effect as suction-assisted lipoplasty by focusing pulses of laser energy through a 1-mm wide fiber optic cannula and inducing thermally mediated adipolysis. The directed laser results in adipocyte rupturing with added benefits of skin retraction and small vessel coagulation, thus lessening intraoperative blood loss.2 This technique typically requires smaller incisions than traditional liposuction. The most common laser lipolysis systems used in cosmetic dermatology are the 920- to 980-nm diode lasers and 1064- to 1440-nm Nd:YAG lasers. The 924-nm diode, 1064-nm Nd:YAG, and 1064/1320-nm Nd:YAG have been best characterized in clinical trials, as reviewed by Fakhouri et al,3 with demonstrated efficacy in reducing SMF density.
The first randomized prospective trial comparing LAL (using 1064-nm Nd:YAG) and traditional liposuction in various anatomical areas on 25 patients showed no difference in cosmetic results, ecchymoses, edema, or retraction, and significantly lower postoperative pain ratings (P<.0001) in LAL.4 A more recent prospective randomized comparison of LAL (980-nm diode laser; 6–8 W) and traditional liposuction of the submental area in 40 female patients showed greater reduction in SMF thickness in the LAL group compared to the liposuction group at 2-month follow-up (6.2 vs 8.22 unspecified units; P<.001) with significant improvement from baseline in both groups (P<.001).5 However, the cosmetic benefit of LAL over traditional liposuction remains controversial and has not been unequivocally established in the literature.
Common adverse events (AEs) are postoperative swelling, ecchymoses, and pain, and complications of interest are nodularity, skin infections, burns, and nerve damage.6 In one retrospective investigation (N=537), these complications occurred at a rate of less than 1% (4 burns and 1 skin infection).6 Patients treated with LAL may report fewer AEs, especially pain and bleeding, compared to liposuction-treated patients.3
RF-Assisted Lipolysis
Radiofrequency-assisted lipolysis is one of the newest technologies in lipocontouring. NeckTite (Invasix Aesthetic Solutions) is effective for treatment of preplatysmal adiposity and cervicomental lipocontouring; a 2.4-mm bipolar probe that is inserted into the subdermal space and connected with an external electrode emits RF energy and simultaneously coagulates and aspirates adipose tissue. NeckTite also may be used in conjunction with FaceTite (Invasix Aesthetic Solutions), which promotes fibroseptal network remodeling and dermal contraction.2
In the first published investigation of the efficacy and safety of NeckTite, 47 of 55 patients received treatment of slight to moderate SMF (average body mass index [BMI], 25 kg/m2) with NeckTite and FaceTite or NeckTite alone.7 At 6-month follow-up, 87% (48/55) of patients subjectively rated treatment efficacy as satisfactory, and 2 independent physicians rated the improvement between before-and-after frontal and lateral photographs of the submental area as moderate to excellent in 95% (52/55) of all cases. Reported complications in this study were full-thickness burns resulting in minor scarring (2/55 [4%]), neck tissue hardness that resolved with daily massage after 3 months (5/55 [9%]), and transient facial nerve paresis of the mandibular branch that resolved after 2 months (1/55 [2%]).7
NONINVASIVE MODALITIES
RF-Assisted Contouring
Another exciting development in RF technology is truSculpt (Cutera), a noninvasive contouring device that is placed over the epidermis and emits RF energy that preferentially heats fat more than other tissue types. In a single-center prospective trial of efficacy and safety in the treatment of SMF, 17 patients received 2 treatments with truSculpt administered 1 month apart.8 At 1- and 6-month follow-up, 82.3% (14/17) and 52.9% (9/17) of patients showed improvement on physician assessment. Submental circumference and ultrasonographic fat thickness reductions at 1-month follow-up were 1.4 cm (5.7% of pretreatment circumference [P<.001]) and 5.4 mm (9.7% of pretreatment fat thickness [P=.005]), respectively. At further longer-term follow-up to 6 months, submental circumference was 0.9 cm (3.8% of pretreatment circumference [P<.001]) and ultrasonographic fat reduction was 6.8 mm (10.5% of pretreatment fat thickness [P=.006]). Commonly reported AEs were pain (rate not given), erythema (8/17 [47%]), edema (1/17 [6%]), and vesicle formation (1/17 [6%]); all were self-resolving. Erythema usually subsided within 6 hours posttreatment. No other AEs or complications were reported.8
Deoxycholic Acid
Deoxycholic acid (DCA)(formerly ATX-101) is an injectable liquid formulation of synthetic DCA that was approved by the US Food and Drug Administration (FDA) in 2015 for moderate to severe SMF. Deoxycholic acid exists endogenously as a bile salt emulsifier and has been shown to cause dose-dependent adipocyte lysis, necrosis, disruption and dissolution of fat architecture, and inflammatory targeting of adipocytes by immune cells.9,10 Thus, DCA causes targeted adipocytolysis and is a novel medical agent in the treatment of SMF. Supplied in 2-mL vials, clinicians may inject 10 mL at each treatment for up to 6 treatments administered 1 month apart.11
Efficacy
REFINE-1, a pivotal North American–based phase 3 trial, investigated the efficacy and safety of DCA.12 A total of 506 participants with scores of 2 (moderate) or 3 (severe) on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS) and a mean BMI of 29 kg/m2 were randomized to receive preplatysmal fat injections of 2 mg/cm2 of DCA (n=256) or placebo (n=250). Participants received up to 10 mL of product (mean total of 25 mL of DCA across all visits) at each treatment session for up to 6 sessions depending on individual efficacy, with approximately 28 days between sessions. Sixty-four percent of the treatment group received all 6 treatments. At 12-week follow-up after the last treatment session, 70% of DCA-treated participants versus 18.6% of placebo-treated participants (P<.001) improved by 1 grade or more on the CR-SMFRS and 13.4% versus 0% (P<.001) improved by 2 grades or more. Skin laxity was unchanged or improved in 92.7% of the DCA group and 87.6% of the placebo group.12
REFINE-2, the second of the North American phase 3 trials, had parallel inclusionary criteria and study design and established efficacy of 2 mg/cm2 DCA over placebo in 516 participants (randomized 1:1).13 At 12 weeks posttreatment, 66.5% of DCA-treated participants versus 22.2% of placebo-treated participants improved by 1 grade or more according to the CR-SMFRS (P<.001) and 18.6% versus 3% improved by 2 grades or more in SMF (P<.001). Magnetic resonance imaging analysis of participants in the DCA (n=113) and placebo groups (n=112) showed that 40.2% versus 5.2% (P<.001) exhibited 10% or more reduction in submental volume, with similar comparative rates of SMF thickness reduction via caliper measurements.13
Safety
Safety data from REFINE-1 showed higher rates of treatment-related AEs in DCA-treated participants compared to placebo, including hematoma (70% vs 67.3%), anesthesia (66.9% vs 4.4%), pain (65.4% vs 23.4%), edema (52.9% vs 21.8%), induration (18.3% vs 1.6%), paresthesia (12.8% vs 3.2%), nodule formation (12.5% vs 0.8%), and pruritus (8.6% vs 3.6%).12 In this trial, 11 of 258 cases (4.3%) of marginal mandibular nerve paresis and asymmetric smile occurred, all in DCA-treated participants and with a median duration of 31 days. Dysphagia resolving in a median duration of 4 days occurred in 1.6% (4/258) of DCA-treated participants.12 REFINE-2 exhibited similar rates of common AEs. Complications of note were 14 cases of marginal mandibular nerve paresis (11 in DCA group, 3 in placebo group) attributed to injection technique, 1 case of skin ulceration possibly related to accidental injection into dermis, and 6 cases of dysphagia in DCA participants attributed to higher volume treatment sessions and postinjection swelling. Dysphagia lasted a median of 2.5 days and resolved without sequelae.13
Overall, DCA demonstrated high rates of minor injection-site AEs that resolved without sequelae and could be mitigated by comfort therapies (eg, lidocaine, nonsteroidal anti-inflammatory drugs) as well as understanding the anatomy of the submental region. Adverse effects of particular interest included marginal mandibular nerve palsy, skin ulceration, and dysphagia.12,13
Cryolipolysis
Cryolipolysis is an advancement that utilizes the application of noninvasive cooling temperatures to the skin’s surface to destroy underlying adipocytes based on the concept that lipid-filled cells are more susceptible to cold-induced injury than water-filled cells. Thus, cryolipolysis selectively targets adipose tissue, leading to cell death without harm to surrounding cells and without the need for surgery or injections.14
Cryolipolysis typically is delivered via a vacuum applicator (CoolMini, Zeltiq Aesthetics Inc), which applies temperatures of –10°C (14°F) to the skin in cycles of 60 minutes each. Initially approved by the FDA for treatment of flank adiposity in 2010, cryolipolysis has since been approved for treatment of the abdomen, thighs, and submental area.14 An advantage of cryolipolysis is that it does not require frequent treatment sessions for maximal efficacy.
Efficacy
The efficacy of cryolipolysis in the treatment of SMF was established in a multicenter device investigation resulting in its FDA approval for the submental region.15 Sixty participants with a mean BMI of 31.8 kg/m2 received 1 (1/60) or 2 (59/60) treatment sessions of the submental area administered 6 weeks apart. Primary efficacy assessments included analysis by 3 blinded reviewers who viewed photographs of each participant at baseline, immediately posttreatment, 6 weeks posttreatment, and 12 weeks posttreatment; ultrasonographic measurements of SMF thickness; and a 12-point patient satisfaction questionnaire. Blinded reviewers correctly identified baseline images in 91.4% (55/60) of cases. Ultrasonography confirmed a mean reduction in SMF of 2 mm (P<.0001) or 20% of fat thickness at 12 weeks posttreatment. On subjective patient satisfaction surveys, 83% (50/60) of participants were satisfied with the procedure and 77% (46/60) reported a visible reduction in fat and perceived an improvement in appearance.15
Safety
The most common immediate posttreatment AEs were erythema/purpura (100%), numbness (90%), edema (62%), tingling (30%), blanching (25%), and bruising (3%) at the site of cryolipolysis with resolution within 1 week posttreatment, except for numbness.15 At 6-week follow-up, all AEs had resolved, except continued numbness in 4 participants that resolved by 12-week follow-up. A further event of note was fullness in the throat in 1 participant that was attributed to swelling and resolved at 40 days posttreatment without incident. No serious AEs were reported in this trial.15
A particularly concerning but rare complication that is increasing in awareness is paradoxical adipose hyperplasia following cryolipolysis. Patients may develop firm painless areas of soft tissue enlargements in the area of cryolipolysis typically 3 to 6 months posttreatment.16 The largest published report recorded an incidence rate of 0.46% (n=2, all males) at a single-center institution of 422 cryolipolysis treatments.16 Other incidence rates reported are 0.0051% and 0.78%.17 Causes and associations are not known, though male gender is speculated to increase risk.
Conclusion
This article highlights the available information on advances in minimally invasive and noninvasive treatments for SMF accumulation. The efficacy and safety trials varied in quality and in different methods of end point analysis of SMF reduction. Further, few trials have featured head-to-head comparisons of treatments.
Although liposuction and adjuncts remain the gold standard in large-mass lipid removal, these procedures are invasive and exhibit typical risks of surgery. Given its sensitive location, the submental area may require the use of more delicate therapeutic methods, including completely noninvasive devices such as truSculpt and cryolipolysis. Regardless of the chosen treatment, the most important factors in yielding patient satisfaction and SMF improvement are proper patient selection and an understanding of the anatomical source of adiposity to be addressed with the therapeutic modalities.
[polldaddy:9711250]
Submental fat (SMF) accumulation within the subcutaneous (preplatysmal) or subplatysmal fat compartment of the cervical anatomy results in an obtuse cervicomental angle and loss of mandibular and cervical contours. It is a common cosmetic concern due to its aesthetic association with weight gain and aging.1 Minimally invasive or noninvasive submental lipolytic agents and techniques are sought for patients who are not candidates for surgery or prefer more conservative cosmetic treatments. These methods typically are only effective in addressing preplatysmal SMF, as subplatysmal SMF requires more surgical methods due to its less-accessible location. The pathology of SMF should initially be assessed by clinical examination or ultrasonography. In this article, we review the most relevant clinical and safety data on minimally invasive and noninvasive treatments for SMF, including laser-assisted lipolysis (LAL), radiofrequency (RF)–assisted lipolysis, deoxycholic acid (DCA), and cryolipolysis.
MINIMALLY INVASIVE MODALITIES
Traditional, or tumescent, liposuction is still widely considered the most effective method for removal of large masses of adiposity. Laser- and RF-assisted adjuncts have been more recently developed to improve patient side effects and recovery time and reduce the manual effort of surgeons. Of note, these adjuncts, with some exceptions, still require the same invasiveness as traditional liposuction, involving submental stab incisions of up to 2.4 mm.
Laser-Assisted Lipolysis
Laser-assisted lipolysis produces a similar effect as suction-assisted lipoplasty by focusing pulses of laser energy through a 1-mm wide fiber optic cannula and inducing thermally mediated adipolysis. The directed laser results in adipocyte rupturing with added benefits of skin retraction and small vessel coagulation, thus lessening intraoperative blood loss.2 This technique typically requires smaller incisions than traditional liposuction. The most common laser lipolysis systems used in cosmetic dermatology are the 920- to 980-nm diode lasers and 1064- to 1440-nm Nd:YAG lasers. The 924-nm diode, 1064-nm Nd:YAG, and 1064/1320-nm Nd:YAG have been best characterized in clinical trials, as reviewed by Fakhouri et al,3 with demonstrated efficacy in reducing SMF density.
The first randomized prospective trial comparing LAL (using 1064-nm Nd:YAG) and traditional liposuction in various anatomical areas on 25 patients showed no difference in cosmetic results, ecchymoses, edema, or retraction, and significantly lower postoperative pain ratings (P<.0001) in LAL.4 A more recent prospective randomized comparison of LAL (980-nm diode laser; 6–8 W) and traditional liposuction of the submental area in 40 female patients showed greater reduction in SMF thickness in the LAL group compared to the liposuction group at 2-month follow-up (6.2 vs 8.22 unspecified units; P<.001) with significant improvement from baseline in both groups (P<.001).5 However, the cosmetic benefit of LAL over traditional liposuction remains controversial and has not been unequivocally established in the literature.
Common adverse events (AEs) are postoperative swelling, ecchymoses, and pain, and complications of interest are nodularity, skin infections, burns, and nerve damage.6 In one retrospective investigation (N=537), these complications occurred at a rate of less than 1% (4 burns and 1 skin infection).6 Patients treated with LAL may report fewer AEs, especially pain and bleeding, compared to liposuction-treated patients.3
RF-Assisted Lipolysis
Radiofrequency-assisted lipolysis is one of the newest technologies in lipocontouring. NeckTite (Invasix Aesthetic Solutions) is effective for treatment of preplatysmal adiposity and cervicomental lipocontouring; a 2.4-mm bipolar probe that is inserted into the subdermal space and connected with an external electrode emits RF energy and simultaneously coagulates and aspirates adipose tissue. NeckTite also may be used in conjunction with FaceTite (Invasix Aesthetic Solutions), which promotes fibroseptal network remodeling and dermal contraction.2
In the first published investigation of the efficacy and safety of NeckTite, 47 of 55 patients received treatment of slight to moderate SMF (average body mass index [BMI], 25 kg/m2) with NeckTite and FaceTite or NeckTite alone.7 At 6-month follow-up, 87% (48/55) of patients subjectively rated treatment efficacy as satisfactory, and 2 independent physicians rated the improvement between before-and-after frontal and lateral photographs of the submental area as moderate to excellent in 95% (52/55) of all cases. Reported complications in this study were full-thickness burns resulting in minor scarring (2/55 [4%]), neck tissue hardness that resolved with daily massage after 3 months (5/55 [9%]), and transient facial nerve paresis of the mandibular branch that resolved after 2 months (1/55 [2%]).7
NONINVASIVE MODALITIES
RF-Assisted Contouring
Another exciting development in RF technology is truSculpt (Cutera), a noninvasive contouring device that is placed over the epidermis and emits RF energy that preferentially heats fat more than other tissue types. In a single-center prospective trial of efficacy and safety in the treatment of SMF, 17 patients received 2 treatments with truSculpt administered 1 month apart.8 At 1- and 6-month follow-up, 82.3% (14/17) and 52.9% (9/17) of patients showed improvement on physician assessment. Submental circumference and ultrasonographic fat thickness reductions at 1-month follow-up were 1.4 cm (5.7% of pretreatment circumference [P<.001]) and 5.4 mm (9.7% of pretreatment fat thickness [P=.005]), respectively. At further longer-term follow-up to 6 months, submental circumference was 0.9 cm (3.8% of pretreatment circumference [P<.001]) and ultrasonographic fat reduction was 6.8 mm (10.5% of pretreatment fat thickness [P=.006]). Commonly reported AEs were pain (rate not given), erythema (8/17 [47%]), edema (1/17 [6%]), and vesicle formation (1/17 [6%]); all were self-resolving. Erythema usually subsided within 6 hours posttreatment. No other AEs or complications were reported.8
Deoxycholic Acid
Deoxycholic acid (DCA)(formerly ATX-101) is an injectable liquid formulation of synthetic DCA that was approved by the US Food and Drug Administration (FDA) in 2015 for moderate to severe SMF. Deoxycholic acid exists endogenously as a bile salt emulsifier and has been shown to cause dose-dependent adipocyte lysis, necrosis, disruption and dissolution of fat architecture, and inflammatory targeting of adipocytes by immune cells.9,10 Thus, DCA causes targeted adipocytolysis and is a novel medical agent in the treatment of SMF. Supplied in 2-mL vials, clinicians may inject 10 mL at each treatment for up to 6 treatments administered 1 month apart.11
Efficacy
REFINE-1, a pivotal North American–based phase 3 trial, investigated the efficacy and safety of DCA.12 A total of 506 participants with scores of 2 (moderate) or 3 (severe) on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS) and a mean BMI of 29 kg/m2 were randomized to receive preplatysmal fat injections of 2 mg/cm2 of DCA (n=256) or placebo (n=250). Participants received up to 10 mL of product (mean total of 25 mL of DCA across all visits) at each treatment session for up to 6 sessions depending on individual efficacy, with approximately 28 days between sessions. Sixty-four percent of the treatment group received all 6 treatments. At 12-week follow-up after the last treatment session, 70% of DCA-treated participants versus 18.6% of placebo-treated participants (P<.001) improved by 1 grade or more on the CR-SMFRS and 13.4% versus 0% (P<.001) improved by 2 grades or more. Skin laxity was unchanged or improved in 92.7% of the DCA group and 87.6% of the placebo group.12
REFINE-2, the second of the North American phase 3 trials, had parallel inclusionary criteria and study design and established efficacy of 2 mg/cm2 DCA over placebo in 516 participants (randomized 1:1).13 At 12 weeks posttreatment, 66.5% of DCA-treated participants versus 22.2% of placebo-treated participants improved by 1 grade or more according to the CR-SMFRS (P<.001) and 18.6% versus 3% improved by 2 grades or more in SMF (P<.001). Magnetic resonance imaging analysis of participants in the DCA (n=113) and placebo groups (n=112) showed that 40.2% versus 5.2% (P<.001) exhibited 10% or more reduction in submental volume, with similar comparative rates of SMF thickness reduction via caliper measurements.13
Safety
Safety data from REFINE-1 showed higher rates of treatment-related AEs in DCA-treated participants compared to placebo, including hematoma (70% vs 67.3%), anesthesia (66.9% vs 4.4%), pain (65.4% vs 23.4%), edema (52.9% vs 21.8%), induration (18.3% vs 1.6%), paresthesia (12.8% vs 3.2%), nodule formation (12.5% vs 0.8%), and pruritus (8.6% vs 3.6%).12 In this trial, 11 of 258 cases (4.3%) of marginal mandibular nerve paresis and asymmetric smile occurred, all in DCA-treated participants and with a median duration of 31 days. Dysphagia resolving in a median duration of 4 days occurred in 1.6% (4/258) of DCA-treated participants.12 REFINE-2 exhibited similar rates of common AEs. Complications of note were 14 cases of marginal mandibular nerve paresis (11 in DCA group, 3 in placebo group) attributed to injection technique, 1 case of skin ulceration possibly related to accidental injection into dermis, and 6 cases of dysphagia in DCA participants attributed to higher volume treatment sessions and postinjection swelling. Dysphagia lasted a median of 2.5 days and resolved without sequelae.13
Overall, DCA demonstrated high rates of minor injection-site AEs that resolved without sequelae and could be mitigated by comfort therapies (eg, lidocaine, nonsteroidal anti-inflammatory drugs) as well as understanding the anatomy of the submental region. Adverse effects of particular interest included marginal mandibular nerve palsy, skin ulceration, and dysphagia.12,13
Cryolipolysis
Cryolipolysis is an advancement that utilizes the application of noninvasive cooling temperatures to the skin’s surface to destroy underlying adipocytes based on the concept that lipid-filled cells are more susceptible to cold-induced injury than water-filled cells. Thus, cryolipolysis selectively targets adipose tissue, leading to cell death without harm to surrounding cells and without the need for surgery or injections.14
Cryolipolysis typically is delivered via a vacuum applicator (CoolMini, Zeltiq Aesthetics Inc), which applies temperatures of –10°C (14°F) to the skin in cycles of 60 minutes each. Initially approved by the FDA for treatment of flank adiposity in 2010, cryolipolysis has since been approved for treatment of the abdomen, thighs, and submental area.14 An advantage of cryolipolysis is that it does not require frequent treatment sessions for maximal efficacy.
Efficacy
The efficacy of cryolipolysis in the treatment of SMF was established in a multicenter device investigation resulting in its FDA approval for the submental region.15 Sixty participants with a mean BMI of 31.8 kg/m2 received 1 (1/60) or 2 (59/60) treatment sessions of the submental area administered 6 weeks apart. Primary efficacy assessments included analysis by 3 blinded reviewers who viewed photographs of each participant at baseline, immediately posttreatment, 6 weeks posttreatment, and 12 weeks posttreatment; ultrasonographic measurements of SMF thickness; and a 12-point patient satisfaction questionnaire. Blinded reviewers correctly identified baseline images in 91.4% (55/60) of cases. Ultrasonography confirmed a mean reduction in SMF of 2 mm (P<.0001) or 20% of fat thickness at 12 weeks posttreatment. On subjective patient satisfaction surveys, 83% (50/60) of participants were satisfied with the procedure and 77% (46/60) reported a visible reduction in fat and perceived an improvement in appearance.15
Safety
The most common immediate posttreatment AEs were erythema/purpura (100%), numbness (90%), edema (62%), tingling (30%), blanching (25%), and bruising (3%) at the site of cryolipolysis with resolution within 1 week posttreatment, except for numbness.15 At 6-week follow-up, all AEs had resolved, except continued numbness in 4 participants that resolved by 12-week follow-up. A further event of note was fullness in the throat in 1 participant that was attributed to swelling and resolved at 40 days posttreatment without incident. No serious AEs were reported in this trial.15
A particularly concerning but rare complication that is increasing in awareness is paradoxical adipose hyperplasia following cryolipolysis. Patients may develop firm painless areas of soft tissue enlargements in the area of cryolipolysis typically 3 to 6 months posttreatment.16 The largest published report recorded an incidence rate of 0.46% (n=2, all males) at a single-center institution of 422 cryolipolysis treatments.16 Other incidence rates reported are 0.0051% and 0.78%.17 Causes and associations are not known, though male gender is speculated to increase risk.
Conclusion
This article highlights the available information on advances in minimally invasive and noninvasive treatments for SMF accumulation. The efficacy and safety trials varied in quality and in different methods of end point analysis of SMF reduction. Further, few trials have featured head-to-head comparisons of treatments.
Although liposuction and adjuncts remain the gold standard in large-mass lipid removal, these procedures are invasive and exhibit typical risks of surgery. Given its sensitive location, the submental area may require the use of more delicate therapeutic methods, including completely noninvasive devices such as truSculpt and cryolipolysis. Regardless of the chosen treatment, the most important factors in yielding patient satisfaction and SMF improvement are proper patient selection and an understanding of the anatomical source of adiposity to be addressed with the therapeutic modalities.
[polldaddy:9711250]
- Hatef DA, Koshy JC, Sandoval SE, et al. The submental fat compartment of the neck. Semin Plast Surg. 2009;23:288-291.
- Mulholland RS. Nonexcisional, minimally invasive rejuvenation of the neck. Clin Plast Surg. 2014;41:11-31.
- Fakhouri TM, El Tal AK, Abrou AE, et al. Laser-assisted lipolysis: a review. Dermatol Surg. 2012;38:155-169.
- Prado A, Andrades P, Danilla S, et al. A prospective, randomized, double-blind, controlled clinical trial comparing laser-assisted lipoplasty with suction-assisted lipoplasty. Plast Reconstr Surg. 2006;118:1032-1045.
- Valizadeh N, Jalaly NY, Zarghampour M, et al. Evaluation of safety and efficacy of 980-nm diode laser-assisted lipolysis versus traditional liposuction for submental rejuvenation: a randomized clinical trial. J Cosmet Laser Ther. 2016;18:41-45.
- Katz B, McBean J. Laser-assisted lipolysis: a report on complications. J Cosmet Laser Ther. 2008;10:231-233.
- Keramidas E, Rodopoulou S. Radiofrequency-assisted liposuction for neck and lower face adipodermal remodeling and contouring. Plast Reconstr Surg Glob Open. 2016;4:e850.
- Park JH, Kim JI, Park HJ, et al. Evaluation of safety and efficacy of noninvasive radiofrequency technology for submental rejuvenation [published online July 12, 2016]. Lasers Med Sci. 2016;31:1599-1605.
- Yagima Odo ME, Cucé LC, Odo LM, et al. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178-188; discussion 188-189.
- Rotunda AM, Suzuki H, Moy RL, et al. Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution. Dermatol Surg. 2004;30:1001-1008.
- Kybella [package insert]. Westlake Village, CA: Kythera Biopharmaceuticals, Inc; 2015.
- Jones DH, Carruthers J, Joseph JH, et al. REFINE-1, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial with ATX-101, an injectable drug for submental fat reduction. Dermatol Surg. 2016;42:38-49.
- Humphrey S, Sykes J, Kantor J, et al. ATX-101 for reduction of submental fat: a phase III randomized controlled trial [published online July 16, 2016]. J Am Acad Dermatol. 2016;75:788-797.e7.
- Manstein D, Laubach H, Watanabe K, et al. Selective cryolysis: a novel method of non-invasive fat removal. Lasers Surg Med. 2008;40:595-604.
- Kilmer SL, Burns AJ, Zelickson BD. Safety and efficacy of cryolipolysis for non-invasive reduction of submental fat. Lasers Surg Med. 2016;48:3-13.
- Singh SM, Geddes ER, Boutrous SG, et al. Paradoxical adipose hyperplasia secondary to cryolipolysis: an underreported entity? Lasers Surg Med. 2015;47:476-478.
- Kelly E, Rodriguez-Feliz J, Kelly ME. Paradoxical adipose hyperplasia after cryolipolysis: a report on incidence and common factors identified in 510 patients. Plast Reconst Surg. 2016;137:639e-640e.
- Hatef DA, Koshy JC, Sandoval SE, et al. The submental fat compartment of the neck. Semin Plast Surg. 2009;23:288-291.
- Mulholland RS. Nonexcisional, minimally invasive rejuvenation of the neck. Clin Plast Surg. 2014;41:11-31.
- Fakhouri TM, El Tal AK, Abrou AE, et al. Laser-assisted lipolysis: a review. Dermatol Surg. 2012;38:155-169.
- Prado A, Andrades P, Danilla S, et al. A prospective, randomized, double-blind, controlled clinical trial comparing laser-assisted lipoplasty with suction-assisted lipoplasty. Plast Reconstr Surg. 2006;118:1032-1045.
- Valizadeh N, Jalaly NY, Zarghampour M, et al. Evaluation of safety and efficacy of 980-nm diode laser-assisted lipolysis versus traditional liposuction for submental rejuvenation: a randomized clinical trial. J Cosmet Laser Ther. 2016;18:41-45.
- Katz B, McBean J. Laser-assisted lipolysis: a report on complications. J Cosmet Laser Ther. 2008;10:231-233.
- Keramidas E, Rodopoulou S. Radiofrequency-assisted liposuction for neck and lower face adipodermal remodeling and contouring. Plast Reconstr Surg Glob Open. 2016;4:e850.
- Park JH, Kim JI, Park HJ, et al. Evaluation of safety and efficacy of noninvasive radiofrequency technology for submental rejuvenation [published online July 12, 2016]. Lasers Med Sci. 2016;31:1599-1605.
- Yagima Odo ME, Cucé LC, Odo LM, et al. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178-188; discussion 188-189.
- Rotunda AM, Suzuki H, Moy RL, et al. Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution. Dermatol Surg. 2004;30:1001-1008.
- Kybella [package insert]. Westlake Village, CA: Kythera Biopharmaceuticals, Inc; 2015.
- Jones DH, Carruthers J, Joseph JH, et al. REFINE-1, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial with ATX-101, an injectable drug for submental fat reduction. Dermatol Surg. 2016;42:38-49.
- Humphrey S, Sykes J, Kantor J, et al. ATX-101 for reduction of submental fat: a phase III randomized controlled trial [published online July 16, 2016]. J Am Acad Dermatol. 2016;75:788-797.e7.
- Manstein D, Laubach H, Watanabe K, et al. Selective cryolysis: a novel method of non-invasive fat removal. Lasers Surg Med. 2008;40:595-604.
- Kilmer SL, Burns AJ, Zelickson BD. Safety and efficacy of cryolipolysis for non-invasive reduction of submental fat. Lasers Surg Med. 2016;48:3-13.
- Singh SM, Geddes ER, Boutrous SG, et al. Paradoxical adipose hyperplasia secondary to cryolipolysis: an underreported entity? Lasers Surg Med. 2015;47:476-478.
- Kelly E, Rodriguez-Feliz J, Kelly ME. Paradoxical adipose hyperplasia after cryolipolysis: a report on incidence and common factors identified in 510 patients. Plast Reconst Surg. 2016;137:639e-640e.
Practice Points
- New developments in minimally invasive techniques for treating submental adiposity include laser-assisted and radiofrequency-assisted lipoplasty with demonstrated clinical benefit and acceptable safety.
- Noninvasive treatments for submental adiposity include radiofrequency-assisted contouring devices, deoxycholic acid, and cryolipolysis, which offer an alternative to more invasive procedures such as lipoplasty.
- There are no comparative studies to date to suggest noninferiority of these noninvasive treatments compared to lipoplasty.
Basal Cell Carcinoma Arising in Outdoor Workers Versus Indoor Workers: A Retrospective Study
Basal cell carcinoma (BCC) is the most prevalent malignancy in white individuals and its incidence is rapidly increasing. Despite its low mortality rate, BCC can cause severe morbidity and remains a serious health problem with a high economic burden for health care systems. The incidence of BCC is higher in individuals who have red or blonde hair, light eye color, and/or Fitzpatrick skin types I and II. The risk for developing BCC also increases with age, and men are more frequently affected than women.1,2 Although several factors have been implicated in the etiology of this condition, such as exposure to ionizing radiation, trauma, chemical carcinogenesis, immunosuppression, predisposing syndromes, and host factors (eg, traits that affect susceptibility to disease),3-5 exposure to UV radiation is considered to be a major risk factor, with most BCCs presenting in sun-exposed areas of the body (eg, face, neck). Prolongate suberythrodermal UV doses, which do not burn the skin but cause erythema in the histological level, can lead to formation of pyrimidine dimers in the dermal and epidermal tissues and cause DNA mutation with potential carcinogenic effects. Due to a large number of outdoor occupations, it is likely that outdoor workers (OWs) with a history of UV exposure may develop BCCs with different features than those seen in indoor workers (IWs). However, there has been debate about the relevance of occupational UV exposure as a risk factor for BCC development.6,7 The aim of this study was to compare the clinical and histological features of BCCs in OWs versus IWs at a referral hospital in southern Spain.
Methods
Using the electronic pathology records at a referral hospital in southern Spain, we identified medical records between May 1, 2010, and May 1, 2011, of specimens containing the term skin in the specimen box and basal cell carcinoma in the diagnosis box. We excluded patients with a history of or concomitant squamous cell carcinoma. Reexcision of incompletely excised lesions; punch, shave or incisional biopsies; and palliative excisions also were excluded. The specimens were reviewed and classified according to the differentiation pattern of BCC (ie, nodular, superficial, morpheic, micronodular). Basal cell carcinomas with mixed features were classified according to the most predominant subtype.
We also gathered information regarding the patients’ work history (ie, any job held during their lifetime with a minimum duration of 6 months). Patients were asked about the type of work and start/end dates. In patients who performed OW, we evaluated hours per day and months as well as the type of clothing worn (eg, head covering, socks/stockings during work in the summer months).
Each patient was classified as an OW or IW based on his/her stated occupation. The OWs included those who performed all or most of their work (≥6 hours per day for at least 6 months) outdoors in direct sunlight. Most patients in this group included farmers and fishermen. Indoor workers were those who performed most of their work in an indoor environment (eg, shop, factory, office, hospital, library, bank, school, laboratory). Most patients in this group included mechanics and shop assistants. A small group of individuals could not be classified as OWs or IWs and therefore were excluded from the study. Individuals with a history of exposure to ionizing radiation, chemical carcinogenesis, immunosuppression, or predisposing syndromes also were excluded.
We included variables that could be considered independent risk factors for BCC, including age, sex, eye color, natural hair color, Fitzpatrick skin type, history of sunburns, and family history. All data were collected via a personal interview performed by a single dermatologist (H.H-E.) during the follow-up with the patients conducted after obtaining all medical records and contacting eligible patients; none of the patients were lost on follow-up.
The study was approved by the hospital’s ethics committee and written consent was obtained from all recruited patients for analyzing the data acquired and accessing the relevant diagnostic documents (eg, pathology reports).
The cohorts were compared by a χ2 test and Student t test, which were performed using the SPSS software version 15. Statistical significance was determined using α=.05, and all tests were 2-sided.
Results
A total of 308 patients were included in the study, comprising 178 (58%) OWs and 130 (42%) IWs. Table 1 summarizes the characteristics of each cohort with the statistical outcomes.
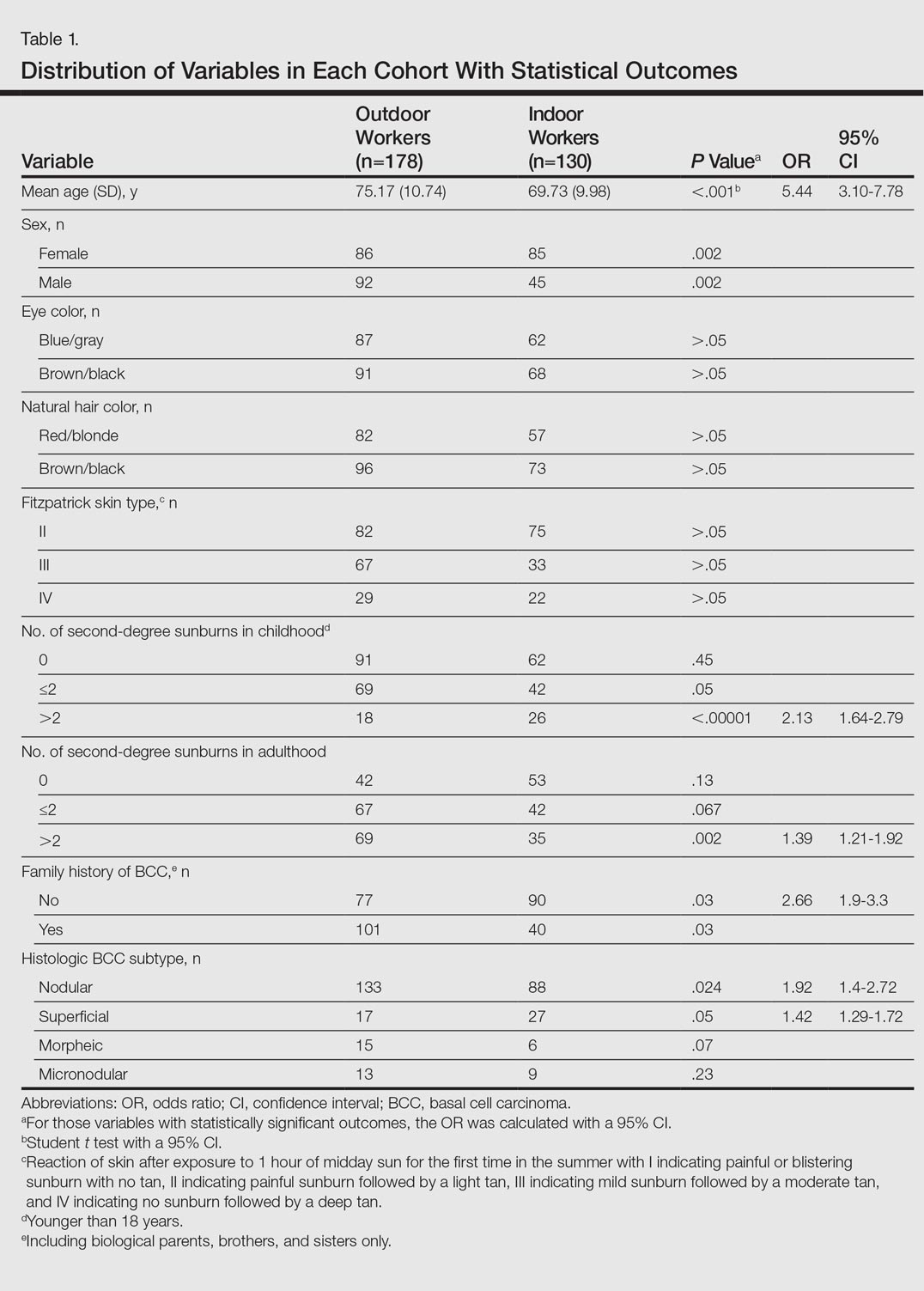
The mean age (SD) of the OWs was significantly higher than the IWs (75.17 [10.74] vs 69.73 [9.98] years; P<.001). The sex distribution among the 2 cohorts was significantly different (P=.002); the OW group featured a slightly higher proportion of men than women (92 [52%] vs 86 [48%]), whereas women were clearly more prevalent in the IW group than men (85 [65%] vs 45 [35%]).
No significant differences regarding eye color (blue/gray vs brown/black) between the 2 cohorts were found (P>.05). In the same way, the 2 cohorts did not show differences in the natural hair color (red/blonde vs brown/black)(P>.05).
Fitzpatrick skin type II was the most common between both cohorts (82 [46%] OWs and 75 [58%] IWs), but no statistical differences regarding the proportions of each skin type were found (P>.05).
History of sunburns (>2 episodes) was significantly different between the 2 cohorts. The incidence of second-degree sunburns in childhood was higher in IWs (P<.00001), while the incidence of second-degree sunburns in adulthood was higher in OWs (P=.002).
Most OWs had a positive family history of BCC (101 [57%]), while the majority of IWs had a negative family history of BCC (90 [69%]). This difference was statistically significant (P=.03).
Table 2 shows the distribution of anatomic sites of BCCs in OWs and IWs. The nose was the most frequently affected area in OWs (35 cases [20%]), while the cheek was the most common location (23 [18%]) in IWs. Comparison of the frequency of BCC incidence for each anatomic location revealed that only the rate for truncal BCC was significantly different; IWs had a higher incidence of truncal BCCs than OWs (P=.0035). Although the differences between groups were not statistically significant, there was a trend toward a higher incidence of BCCs on the forehead in OW (P=.06).
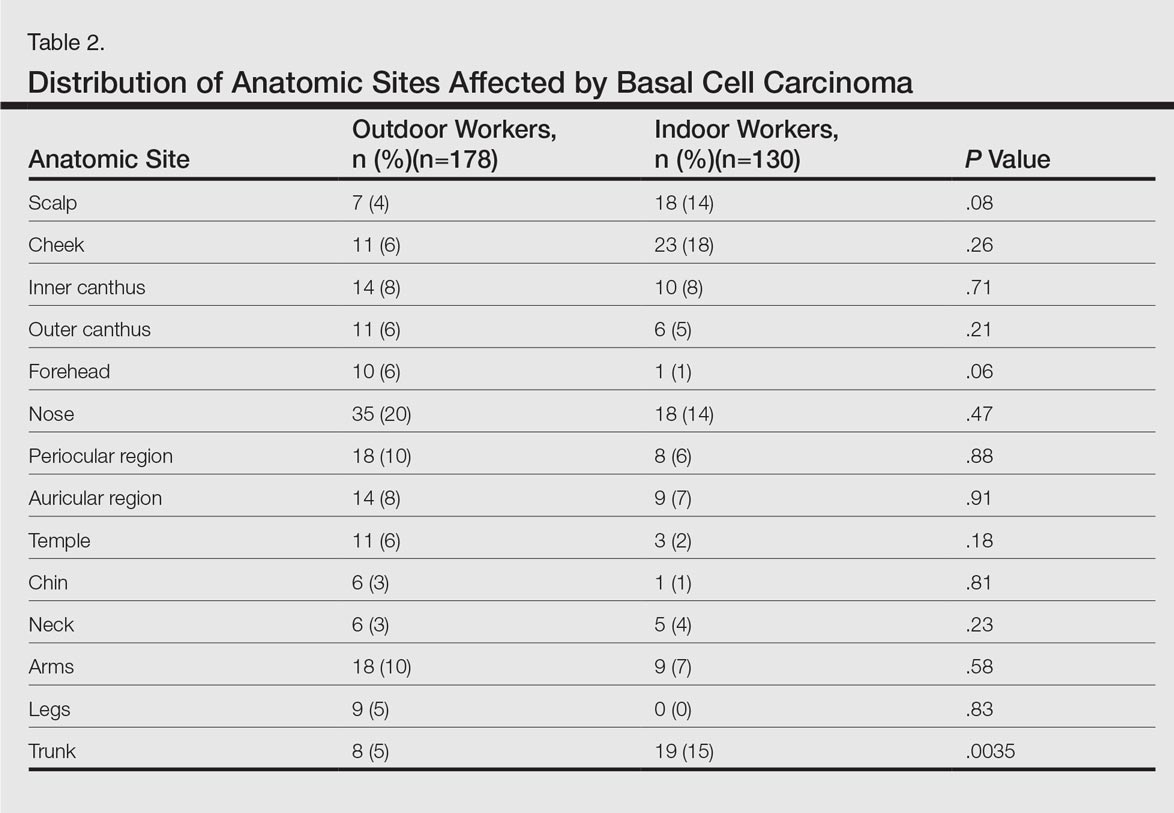
In both cohorts, the most prevalent histologic subtype was nodular BCC (133 [75%] OWs and 88 [68%] IWs), followed by superficial BCC (17 [10%] OWs and 27 [21%] IWs). The incidence rate of nodular BCCs was statistically different between the 2 cohorts, with OWs showing a higher incidence compared to IWs (P=.024). Regarding the superficial subtype, the opposite was observed: IWs had significantly increased risk compared to OWs (P=.05). There was a trend toward a higher incidence of morpheic BCCs in OWs than IWs, but the difference was not statistically significant (P=.07).
Comment
Skin cancer due to occupational UV exposure is more common than is generally recognized,6,7 but occupational UV exposure as a risk factor for BCC is still an ongoing debate. In this study, we analyzed the different clinical and histological features of BCC in OWs versus IWs.
The geographic area where this study was performed is characterized by a subtropical Mediterranean climate with irregular rainfall; a short, cool to mild winter; and long, dry, hot summers. Summer temperatures usually are hot and regularly exceed 35°C (95°F). UV index (UVI) is a measure of the amount of skin-damaging UV radiation expected to reach the earth’s surface when the sun is highest in the sky (around midday) and ranges from 1 (low risk) to 10 (maximum risk). In southern Spain, the mean UVI is approximately 6 and can reach up to 9 or sometimes 10 in the summer months. Although Fitzpatrick skin types II and III are most common, the elevated UVI indicates that the general population in southern Spain is at a high risk for developing skin cancer.
In our study the mean age of IWs was lower than OWs, which suggests that IWs may develop BCC at a younger age than OWs. This finding is consistent with studies showing that cumulative occupational UV exposure has been associated with development of BCCs in older age groups, while acute intermittent recreational sun exposure, particularly sustained in childhood and adolescence, is linked with BCC in younger patients.6
The role of sex as a risk factor for BCC remains unclear. Some reports show that BCC is more common in men than in women.8-10 In our study, sex distribution was statistically significant (P=.002); there were more women in the IW cohort and more men in the OW cohort. These differences may be explained by cultural and lifestyle patterns, as women who are IWs tend to have office jobs in urban settings and wear modern fashion clothes at work and for recreation. In rural settings, women have agricultural jobs and tend to wear more traditional clothes that offer sun protection.
Positive family history has been suggested to be a constitutional risk factor for BCC development.8,11,12 In our study, we observed that positive family history was more common in OWs, while most IWs had a negative family history. These differences were significant (P=.03), and OWs had a 2.6-fold increased likelihood of having a positive family history of BCC compared to IWs. Cultural and lifestyle patterns may partially explain this finding. In rural settings, workers tend to have the same job as their parents as a traditional way of life and therefore have similar patterns of UV exposure; in urban settings, individuals may have different jobs than their parents and therefore the pattern of UV exposure may be different. However, a genetic predisposition for developing BCC cannot be excluded. In addition, we have to consider that the information on family history of BCC in the patients was self-reported and not validated, which may limit the results.
The difference in history of second-degree sunburn in childhood was significantly higher in IWs than in OWs (P<.00001). The OW group had a significant rate of sunburns in adulthood (P=.002). The relationship between UV radiation and BCC is complex, and the patterns of sun exposure and their occurrence in different periods of lifetime (ie, childhood vs adulthood) remain controversial.13 The overall history of severe sunburns seems to be more important than simply the tendency to burn or tan,14,15 and a history of sunburns in childhood and adolescence has been associated with early-onset BCC.6 Our findings were consistent in that the age of onset of BCCs was lower in IWs who had a history of sunburns in childhood. Basal cell carcinomas developed at older ages in OWs who had a higher incidence of sunburns in adulthood. However, we have to consider that the retrospective nature of the data collection on sunburns in childhood and adulthood was potentially limited, as the information was based on the patients’ memory. Additionally, other non-UV risk factors for BCC, such as ionizing radiation exposure, were not analyzed.
The majority of BCCs developed in sun-exposed areas of the head and neck in both cohorts, and only 35 (20%) and 28 (22%) BCCs were located on the trunk, arms, or legs in OWs and IWs, respectively. In our study, the rate of BCCs on the trunk was significantly lower in OWs than in IWs (P=.0035). Basal cell carcinomas on the trunk have been suggested to be linked to genetic susceptibility16,17 and reduced DNA repair capacity18 rather than sun exposure. Our findings support this hypothesis and suggest that occupational sun exposure has no direct relation with truncal BCC. This outcome is consistent with the result of a case-control study conducted by Pelucchi et al19 (N=1040). The authors concluded that occupational UV exposure was not associated with truncal BCC development but with head/neck BCC, indicating that there may be different etiological mechanisms between truncal and head/neck BCC.19 In the largest BCC case series published in the literature with 13,457 specimens, the authors stated that tumors on the trunk may represent a particular variant of BCC, in which the theory of chronic versus intermittent UV exposure cannot be simply extrapolated as it is for the rest of BCC sites. Other factors such as genetic predisposition could be involved in the development of truncal BCC.20 Similarly, Ramos et al21 suggested that nonmelanoma skin cancers in sun-protected anatomic sites may occur in individuals with impairment in the DNA repair process.
The classification of histological subtypes of BCC helps to predict tumor behavior,22 which can impact the prognosis. In our study, nodular BCC was the most common subtype in both cohorts, followed by superficial BCC. The nodular subtype was increased in OWs compared to IWs, while the superficial subtype was most common in IWs. Bastiaens et al23 and McCormack et al24 have suggested that the most frequent subtypes of BCC (nodular and superficial) may represent different tumors with distinct causal factors. According to these authors, nodular subtypes are associated with cumulative UV exposure, while superficial subtypes are associated with more intense and intermittent UV exposure. The results of the current study support this hypothesis, as the OW cohort with cumulative UV exposure showed more incidence of nodular BCC than IWs, while the patients with intense and intermittent sun exposure (the IWs) showed more risk of superficial BCC.
The importance of occupational UV exposure in OWs as a risk factor for BCC is still an ongoing discussion. Our data show that occupational UV exposure may be considered an etiological factor for BCC according to histological subtype and anatomic site. Our study is limited by the retrospective nature of the data collection regarding occupation and childhood sunburns, which were based on the patients’ memory and therefore potentially biased. Data regarding family history of BCC also was self-reported and not validated. Another limiting factor was that other non-UV risk factors for BCC, such as ionizing radiation exposure, were not considered. The limited sample size also may have impacted the study results. Among the strengths of the study are the complete response rate, the similar catchment area of OWs and IWs, the common hospital setting of the 2 cohorts, and the similar attention to medical history. All patients were obtained from the practice of a single referral dermatologist and are felt to be representative of our working area. The use of a single dermatologist reduces provider-associated variability.
Conclusion
According to the results of this study, OWs are more likely to develop nodular BCCs with no increased risk for superficial BCCs. The age of onset in OWs is older than in IWs. Some anatomical sites such as the trunk are more commonly affected in IWs. Truncal BCCs may have etiological factors other than UV exposure, such as a genetic predisposition. This study is useful to occupational safety representatives and physicians to stimulate the implementation of prevention strategies for this easily preventable malignancy and may encourage further research.
- de Vries E, van de Poll-Franse LV, Louwman WJ, et al. Predictions of skin cancer incidence in the Netherlands up to 2015. Br J Dermatol. 2005;152:481-488.
- Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
- Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(suppl 61):1-6.
- Netscher DT, Spira M. Basal cell carcinoma: an overview of tumor biology and treatment. Plast Reconstr Surg. 2004;113:e74-e94.
- Miller SJ. Etiology and pathogenesis of basal cell carcinoma. Clin Dermatol. 1995;13:527-536.
- Dessinioti C, Tzannis K, Sypsa V, et al. Epidemiologic risk factors of basal cell carcinoma development and age at onset in a Southern European population from Greece. Exp Dermatol. 2011;20:622-626.
- Bauer A, Diepgen TL, Schmitt J. Is occupational solar UV-irradiation a relevant risk factor for basal cell carcinoma? a systematic review and meta-analysis of the epidemiologic literature. Br J Dermatol. 2011;165:612-625.
- Tran H, Chen K, Shumack S. Epidemiology and aetiology of basal cell carcinoma. Br J Dermatol. 2003;149(suppl 66):50-52.
- Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18.
- Stern RS. The mysteries of geographic variability in nonmelanoma skin cancer incidence. Arch Dermatol. 1999;135:843-844.
- Chinem VP, Miot HA. Epidemiology of basal cell carcinoma. An Bras Dermatol. 2011;86:292-305.
- Wong CS, Strange RC, Lear JT. Basal cell carcinoma. Br Med J. 2003;327:794-798.
- Dessinioti C, Antoniou C, Katsambas AD, et al. Basal cell carcinoma: what’s new under the sun. Photochem Photobiol. 2010;86:481-491.
- Van Dam RM, Huang Z, Rimm EB, et al. Risk factors for basal cell carcinoma of the skin in men: results from the health professionals follow-up study. Am J Epidemiol. 1999;150:459-468.
- Hunter DJ, Colditz GA, Stampfer MJ, et al. Risk factors for basal cell carcinoma in a prospective cohort of women. Ann Epidemiol. 1990;1:13-23.
- Ramachandran S, Fryer AA, Smith A, et al. Cutaneous basal cell carcinomas: distinct host factors are associated with the development of tumors on the trunk and on the head and neck. Cancer. 2001;92:354-358.
- Ramachandran S, Lear JT, Ramsay H, et al. Presentation with multiple cutaneous basal cell carcinomas: association of glutathione S-transferase and cytochrome P450 genotypes with clinical phenotype. Cancer Epidemiol Biomarkers Prev. 1999;8:61-67.
- Wei Q, Matanoski GM, Farmer ER, et al. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proc Natl Acad Sci USA. 1993;90:1614-1618.
- Pelucchi C, Di Landro A, Naldi L, et al. Risk factors for histological types and anatomic sites of cutaneous basal-cell carcinoma: an Italian case-control study [published online ahead of print Oct 19, 2006]. J Invest Dermatol. 2007;127:935-944.
- Scrivener Y, Grosshans E, Cribier B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br J Dermatol. 2002;147:41-47.
- Ramos J, Villa J, Ruiz A, et al. UV dose determines key characteristics of nonmelanoma skin cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:2006-2011.
- Rippey JJ. Why classify basal cell carcinomas? Histopathology. 1998;32:393-398.
- Bastiaens MT, Hoefnagel JJ, Bruijn JA, et al. Differences in age, site distribution and sex between nodular and superficial basal cell carcinomas indicate different type of tumors. J Invest Dermatol. 1998;110:880-884.
- McCormack CJ, Kelly JW, Dorevitch AP. Differences in age and body site distribution of histological subtypes of basal cell carcinoma. a possible indicator of different causes. Arch Dermatol. 1997;133:593-596.
Basal cell carcinoma (BCC) is the most prevalent malignancy in white individuals and its incidence is rapidly increasing. Despite its low mortality rate, BCC can cause severe morbidity and remains a serious health problem with a high economic burden for health care systems. The incidence of BCC is higher in individuals who have red or blonde hair, light eye color, and/or Fitzpatrick skin types I and II. The risk for developing BCC also increases with age, and men are more frequently affected than women.1,2 Although several factors have been implicated in the etiology of this condition, such as exposure to ionizing radiation, trauma, chemical carcinogenesis, immunosuppression, predisposing syndromes, and host factors (eg, traits that affect susceptibility to disease),3-5 exposure to UV radiation is considered to be a major risk factor, with most BCCs presenting in sun-exposed areas of the body (eg, face, neck). Prolongate suberythrodermal UV doses, which do not burn the skin but cause erythema in the histological level, can lead to formation of pyrimidine dimers in the dermal and epidermal tissues and cause DNA mutation with potential carcinogenic effects. Due to a large number of outdoor occupations, it is likely that outdoor workers (OWs) with a history of UV exposure may develop BCCs with different features than those seen in indoor workers (IWs). However, there has been debate about the relevance of occupational UV exposure as a risk factor for BCC development.6,7 The aim of this study was to compare the clinical and histological features of BCCs in OWs versus IWs at a referral hospital in southern Spain.
Methods
Using the electronic pathology records at a referral hospital in southern Spain, we identified medical records between May 1, 2010, and May 1, 2011, of specimens containing the term skin in the specimen box and basal cell carcinoma in the diagnosis box. We excluded patients with a history of or concomitant squamous cell carcinoma. Reexcision of incompletely excised lesions; punch, shave or incisional biopsies; and palliative excisions also were excluded. The specimens were reviewed and classified according to the differentiation pattern of BCC (ie, nodular, superficial, morpheic, micronodular). Basal cell carcinomas with mixed features were classified according to the most predominant subtype.
We also gathered information regarding the patients’ work history (ie, any job held during their lifetime with a minimum duration of 6 months). Patients were asked about the type of work and start/end dates. In patients who performed OW, we evaluated hours per day and months as well as the type of clothing worn (eg, head covering, socks/stockings during work in the summer months).
Each patient was classified as an OW or IW based on his/her stated occupation. The OWs included those who performed all or most of their work (≥6 hours per day for at least 6 months) outdoors in direct sunlight. Most patients in this group included farmers and fishermen. Indoor workers were those who performed most of their work in an indoor environment (eg, shop, factory, office, hospital, library, bank, school, laboratory). Most patients in this group included mechanics and shop assistants. A small group of individuals could not be classified as OWs or IWs and therefore were excluded from the study. Individuals with a history of exposure to ionizing radiation, chemical carcinogenesis, immunosuppression, or predisposing syndromes also were excluded.
We included variables that could be considered independent risk factors for BCC, including age, sex, eye color, natural hair color, Fitzpatrick skin type, history of sunburns, and family history. All data were collected via a personal interview performed by a single dermatologist (H.H-E.) during the follow-up with the patients conducted after obtaining all medical records and contacting eligible patients; none of the patients were lost on follow-up.
The study was approved by the hospital’s ethics committee and written consent was obtained from all recruited patients for analyzing the data acquired and accessing the relevant diagnostic documents (eg, pathology reports).
The cohorts were compared by a χ2 test and Student t test, which were performed using the SPSS software version 15. Statistical significance was determined using α=.05, and all tests were 2-sided.
Results
A total of 308 patients were included in the study, comprising 178 (58%) OWs and 130 (42%) IWs. Table 1 summarizes the characteristics of each cohort with the statistical outcomes.
The mean age (SD) of the OWs was significantly higher than the IWs (75.17 [10.74] vs 69.73 [9.98] years; P<.001). The sex distribution among the 2 cohorts was significantly different (P=.002); the OW group featured a slightly higher proportion of men than women (92 [52%] vs 86 [48%]), whereas women were clearly more prevalent in the IW group than men (85 [65%] vs 45 [35%]).
No significant differences regarding eye color (blue/gray vs brown/black) between the 2 cohorts were found (P>.05). In the same way, the 2 cohorts did not show differences in the natural hair color (red/blonde vs brown/black)(P>.05).
Fitzpatrick skin type II was the most common between both cohorts (82 [46%] OWs and 75 [58%] IWs), but no statistical differences regarding the proportions of each skin type were found (P>.05).
History of sunburns (>2 episodes) was significantly different between the 2 cohorts. The incidence of second-degree sunburns in childhood was higher in IWs (P<.00001), while the incidence of second-degree sunburns in adulthood was higher in OWs (P=.002).
Most OWs had a positive family history of BCC (101 [57%]), while the majority of IWs had a negative family history of BCC (90 [69%]). This difference was statistically significant (P=.03).
Table 2 shows the distribution of anatomic sites of BCCs in OWs and IWs. The nose was the most frequently affected area in OWs (35 cases [20%]), while the cheek was the most common location (23 [18%]) in IWs. Comparison of the frequency of BCC incidence for each anatomic location revealed that only the rate for truncal BCC was significantly different; IWs had a higher incidence of truncal BCCs than OWs (P=.0035). Although the differences between groups were not statistically significant, there was a trend toward a higher incidence of BCCs on the forehead in OW (P=.06).
In both cohorts, the most prevalent histologic subtype was nodular BCC (133 [75%] OWs and 88 [68%] IWs), followed by superficial BCC (17 [10%] OWs and 27 [21%] IWs). The incidence rate of nodular BCCs was statistically different between the 2 cohorts, with OWs showing a higher incidence compared to IWs (P=.024). Regarding the superficial subtype, the opposite was observed: IWs had significantly increased risk compared to OWs (P=.05). There was a trend toward a higher incidence of morpheic BCCs in OWs than IWs, but the difference was not statistically significant (P=.07).
Comment
Skin cancer due to occupational UV exposure is more common than is generally recognized,6,7 but occupational UV exposure as a risk factor for BCC is still an ongoing debate. In this study, we analyzed the different clinical and histological features of BCC in OWs versus IWs.
The geographic area where this study was performed is characterized by a subtropical Mediterranean climate with irregular rainfall; a short, cool to mild winter; and long, dry, hot summers. Summer temperatures usually are hot and regularly exceed 35°C (95°F). UV index (UVI) is a measure of the amount of skin-damaging UV radiation expected to reach the earth’s surface when the sun is highest in the sky (around midday) and ranges from 1 (low risk) to 10 (maximum risk). In southern Spain, the mean UVI is approximately 6 and can reach up to 9 or sometimes 10 in the summer months. Although Fitzpatrick skin types II and III are most common, the elevated UVI indicates that the general population in southern Spain is at a high risk for developing skin cancer.
In our study the mean age of IWs was lower than OWs, which suggests that IWs may develop BCC at a younger age than OWs. This finding is consistent with studies showing that cumulative occupational UV exposure has been associated with development of BCCs in older age groups, while acute intermittent recreational sun exposure, particularly sustained in childhood and adolescence, is linked with BCC in younger patients.6
The role of sex as a risk factor for BCC remains unclear. Some reports show that BCC is more common in men than in women.8-10 In our study, sex distribution was statistically significant (P=.002); there were more women in the IW cohort and more men in the OW cohort. These differences may be explained by cultural and lifestyle patterns, as women who are IWs tend to have office jobs in urban settings and wear modern fashion clothes at work and for recreation. In rural settings, women have agricultural jobs and tend to wear more traditional clothes that offer sun protection.
Positive family history has been suggested to be a constitutional risk factor for BCC development.8,11,12 In our study, we observed that positive family history was more common in OWs, while most IWs had a negative family history. These differences were significant (P=.03), and OWs had a 2.6-fold increased likelihood of having a positive family history of BCC compared to IWs. Cultural and lifestyle patterns may partially explain this finding. In rural settings, workers tend to have the same job as their parents as a traditional way of life and therefore have similar patterns of UV exposure; in urban settings, individuals may have different jobs than their parents and therefore the pattern of UV exposure may be different. However, a genetic predisposition for developing BCC cannot be excluded. In addition, we have to consider that the information on family history of BCC in the patients was self-reported and not validated, which may limit the results.
The difference in history of second-degree sunburn in childhood was significantly higher in IWs than in OWs (P<.00001). The OW group had a significant rate of sunburns in adulthood (P=.002). The relationship between UV radiation and BCC is complex, and the patterns of sun exposure and their occurrence in different periods of lifetime (ie, childhood vs adulthood) remain controversial.13 The overall history of severe sunburns seems to be more important than simply the tendency to burn or tan,14,15 and a history of sunburns in childhood and adolescence has been associated with early-onset BCC.6 Our findings were consistent in that the age of onset of BCCs was lower in IWs who had a history of sunburns in childhood. Basal cell carcinomas developed at older ages in OWs who had a higher incidence of sunburns in adulthood. However, we have to consider that the retrospective nature of the data collection on sunburns in childhood and adulthood was potentially limited, as the information was based on the patients’ memory. Additionally, other non-UV risk factors for BCC, such as ionizing radiation exposure, were not analyzed.
The majority of BCCs developed in sun-exposed areas of the head and neck in both cohorts, and only 35 (20%) and 28 (22%) BCCs were located on the trunk, arms, or legs in OWs and IWs, respectively. In our study, the rate of BCCs on the trunk was significantly lower in OWs than in IWs (P=.0035). Basal cell carcinomas on the trunk have been suggested to be linked to genetic susceptibility16,17 and reduced DNA repair capacity18 rather than sun exposure. Our findings support this hypothesis and suggest that occupational sun exposure has no direct relation with truncal BCC. This outcome is consistent with the result of a case-control study conducted by Pelucchi et al19 (N=1040). The authors concluded that occupational UV exposure was not associated with truncal BCC development but with head/neck BCC, indicating that there may be different etiological mechanisms between truncal and head/neck BCC.19 In the largest BCC case series published in the literature with 13,457 specimens, the authors stated that tumors on the trunk may represent a particular variant of BCC, in which the theory of chronic versus intermittent UV exposure cannot be simply extrapolated as it is for the rest of BCC sites. Other factors such as genetic predisposition could be involved in the development of truncal BCC.20 Similarly, Ramos et al21 suggested that nonmelanoma skin cancers in sun-protected anatomic sites may occur in individuals with impairment in the DNA repair process.
The classification of histological subtypes of BCC helps to predict tumor behavior,22 which can impact the prognosis. In our study, nodular BCC was the most common subtype in both cohorts, followed by superficial BCC. The nodular subtype was increased in OWs compared to IWs, while the superficial subtype was most common in IWs. Bastiaens et al23 and McCormack et al24 have suggested that the most frequent subtypes of BCC (nodular and superficial) may represent different tumors with distinct causal factors. According to these authors, nodular subtypes are associated with cumulative UV exposure, while superficial subtypes are associated with more intense and intermittent UV exposure. The results of the current study support this hypothesis, as the OW cohort with cumulative UV exposure showed more incidence of nodular BCC than IWs, while the patients with intense and intermittent sun exposure (the IWs) showed more risk of superficial BCC.
The importance of occupational UV exposure in OWs as a risk factor for BCC is still an ongoing discussion. Our data show that occupational UV exposure may be considered an etiological factor for BCC according to histological subtype and anatomic site. Our study is limited by the retrospective nature of the data collection regarding occupation and childhood sunburns, which were based on the patients’ memory and therefore potentially biased. Data regarding family history of BCC also was self-reported and not validated. Another limiting factor was that other non-UV risk factors for BCC, such as ionizing radiation exposure, were not considered. The limited sample size also may have impacted the study results. Among the strengths of the study are the complete response rate, the similar catchment area of OWs and IWs, the common hospital setting of the 2 cohorts, and the similar attention to medical history. All patients were obtained from the practice of a single referral dermatologist and are felt to be representative of our working area. The use of a single dermatologist reduces provider-associated variability.
Conclusion
According to the results of this study, OWs are more likely to develop nodular BCCs with no increased risk for superficial BCCs. The age of onset in OWs is older than in IWs. Some anatomical sites such as the trunk are more commonly affected in IWs. Truncal BCCs may have etiological factors other than UV exposure, such as a genetic predisposition. This study is useful to occupational safety representatives and physicians to stimulate the implementation of prevention strategies for this easily preventable malignancy and may encourage further research.
Basal cell carcinoma (BCC) is the most prevalent malignancy in white individuals and its incidence is rapidly increasing. Despite its low mortality rate, BCC can cause severe morbidity and remains a serious health problem with a high economic burden for health care systems. The incidence of BCC is higher in individuals who have red or blonde hair, light eye color, and/or Fitzpatrick skin types I and II. The risk for developing BCC also increases with age, and men are more frequently affected than women.1,2 Although several factors have been implicated in the etiology of this condition, such as exposure to ionizing radiation, trauma, chemical carcinogenesis, immunosuppression, predisposing syndromes, and host factors (eg, traits that affect susceptibility to disease),3-5 exposure to UV radiation is considered to be a major risk factor, with most BCCs presenting in sun-exposed areas of the body (eg, face, neck). Prolongate suberythrodermal UV doses, which do not burn the skin but cause erythema in the histological level, can lead to formation of pyrimidine dimers in the dermal and epidermal tissues and cause DNA mutation with potential carcinogenic effects. Due to a large number of outdoor occupations, it is likely that outdoor workers (OWs) with a history of UV exposure may develop BCCs with different features than those seen in indoor workers (IWs). However, there has been debate about the relevance of occupational UV exposure as a risk factor for BCC development.6,7 The aim of this study was to compare the clinical and histological features of BCCs in OWs versus IWs at a referral hospital in southern Spain.
Methods
Using the electronic pathology records at a referral hospital in southern Spain, we identified medical records between May 1, 2010, and May 1, 2011, of specimens containing the term skin in the specimen box and basal cell carcinoma in the diagnosis box. We excluded patients with a history of or concomitant squamous cell carcinoma. Reexcision of incompletely excised lesions; punch, shave or incisional biopsies; and palliative excisions also were excluded. The specimens were reviewed and classified according to the differentiation pattern of BCC (ie, nodular, superficial, morpheic, micronodular). Basal cell carcinomas with mixed features were classified according to the most predominant subtype.
We also gathered information regarding the patients’ work history (ie, any job held during their lifetime with a minimum duration of 6 months). Patients were asked about the type of work and start/end dates. In patients who performed OW, we evaluated hours per day and months as well as the type of clothing worn (eg, head covering, socks/stockings during work in the summer months).
Each patient was classified as an OW or IW based on his/her stated occupation. The OWs included those who performed all or most of their work (≥6 hours per day for at least 6 months) outdoors in direct sunlight. Most patients in this group included farmers and fishermen. Indoor workers were those who performed most of their work in an indoor environment (eg, shop, factory, office, hospital, library, bank, school, laboratory). Most patients in this group included mechanics and shop assistants. A small group of individuals could not be classified as OWs or IWs and therefore were excluded from the study. Individuals with a history of exposure to ionizing radiation, chemical carcinogenesis, immunosuppression, or predisposing syndromes also were excluded.
We included variables that could be considered independent risk factors for BCC, including age, sex, eye color, natural hair color, Fitzpatrick skin type, history of sunburns, and family history. All data were collected via a personal interview performed by a single dermatologist (H.H-E.) during the follow-up with the patients conducted after obtaining all medical records and contacting eligible patients; none of the patients were lost on follow-up.
The study was approved by the hospital’s ethics committee and written consent was obtained from all recruited patients for analyzing the data acquired and accessing the relevant diagnostic documents (eg, pathology reports).
The cohorts were compared by a χ2 test and Student t test, which were performed using the SPSS software version 15. Statistical significance was determined using α=.05, and all tests were 2-sided.
Results
A total of 308 patients were included in the study, comprising 178 (58%) OWs and 130 (42%) IWs. Table 1 summarizes the characteristics of each cohort with the statistical outcomes.
The mean age (SD) of the OWs was significantly higher than the IWs (75.17 [10.74] vs 69.73 [9.98] years; P<.001). The sex distribution among the 2 cohorts was significantly different (P=.002); the OW group featured a slightly higher proportion of men than women (92 [52%] vs 86 [48%]), whereas women were clearly more prevalent in the IW group than men (85 [65%] vs 45 [35%]).
No significant differences regarding eye color (blue/gray vs brown/black) between the 2 cohorts were found (P>.05). In the same way, the 2 cohorts did not show differences in the natural hair color (red/blonde vs brown/black)(P>.05).
Fitzpatrick skin type II was the most common between both cohorts (82 [46%] OWs and 75 [58%] IWs), but no statistical differences regarding the proportions of each skin type were found (P>.05).
History of sunburns (>2 episodes) was significantly different between the 2 cohorts. The incidence of second-degree sunburns in childhood was higher in IWs (P<.00001), while the incidence of second-degree sunburns in adulthood was higher in OWs (P=.002).
Most OWs had a positive family history of BCC (101 [57%]), while the majority of IWs had a negative family history of BCC (90 [69%]). This difference was statistically significant (P=.03).
Table 2 shows the distribution of anatomic sites of BCCs in OWs and IWs. The nose was the most frequently affected area in OWs (35 cases [20%]), while the cheek was the most common location (23 [18%]) in IWs. Comparison of the frequency of BCC incidence for each anatomic location revealed that only the rate for truncal BCC was significantly different; IWs had a higher incidence of truncal BCCs than OWs (P=.0035). Although the differences between groups were not statistically significant, there was a trend toward a higher incidence of BCCs on the forehead in OW (P=.06).
In both cohorts, the most prevalent histologic subtype was nodular BCC (133 [75%] OWs and 88 [68%] IWs), followed by superficial BCC (17 [10%] OWs and 27 [21%] IWs). The incidence rate of nodular BCCs was statistically different between the 2 cohorts, with OWs showing a higher incidence compared to IWs (P=.024). Regarding the superficial subtype, the opposite was observed: IWs had significantly increased risk compared to OWs (P=.05). There was a trend toward a higher incidence of morpheic BCCs in OWs than IWs, but the difference was not statistically significant (P=.07).
Comment
Skin cancer due to occupational UV exposure is more common than is generally recognized,6,7 but occupational UV exposure as a risk factor for BCC is still an ongoing debate. In this study, we analyzed the different clinical and histological features of BCC in OWs versus IWs.
The geographic area where this study was performed is characterized by a subtropical Mediterranean climate with irregular rainfall; a short, cool to mild winter; and long, dry, hot summers. Summer temperatures usually are hot and regularly exceed 35°C (95°F). UV index (UVI) is a measure of the amount of skin-damaging UV radiation expected to reach the earth’s surface when the sun is highest in the sky (around midday) and ranges from 1 (low risk) to 10 (maximum risk). In southern Spain, the mean UVI is approximately 6 and can reach up to 9 or sometimes 10 in the summer months. Although Fitzpatrick skin types II and III are most common, the elevated UVI indicates that the general population in southern Spain is at a high risk for developing skin cancer.
In our study the mean age of IWs was lower than OWs, which suggests that IWs may develop BCC at a younger age than OWs. This finding is consistent with studies showing that cumulative occupational UV exposure has been associated with development of BCCs in older age groups, while acute intermittent recreational sun exposure, particularly sustained in childhood and adolescence, is linked with BCC in younger patients.6
The role of sex as a risk factor for BCC remains unclear. Some reports show that BCC is more common in men than in women.8-10 In our study, sex distribution was statistically significant (P=.002); there were more women in the IW cohort and more men in the OW cohort. These differences may be explained by cultural and lifestyle patterns, as women who are IWs tend to have office jobs in urban settings and wear modern fashion clothes at work and for recreation. In rural settings, women have agricultural jobs and tend to wear more traditional clothes that offer sun protection.
Positive family history has been suggested to be a constitutional risk factor for BCC development.8,11,12 In our study, we observed that positive family history was more common in OWs, while most IWs had a negative family history. These differences were significant (P=.03), and OWs had a 2.6-fold increased likelihood of having a positive family history of BCC compared to IWs. Cultural and lifestyle patterns may partially explain this finding. In rural settings, workers tend to have the same job as their parents as a traditional way of life and therefore have similar patterns of UV exposure; in urban settings, individuals may have different jobs than their parents and therefore the pattern of UV exposure may be different. However, a genetic predisposition for developing BCC cannot be excluded. In addition, we have to consider that the information on family history of BCC in the patients was self-reported and not validated, which may limit the results.
The difference in history of second-degree sunburn in childhood was significantly higher in IWs than in OWs (P<.00001). The OW group had a significant rate of sunburns in adulthood (P=.002). The relationship between UV radiation and BCC is complex, and the patterns of sun exposure and their occurrence in different periods of lifetime (ie, childhood vs adulthood) remain controversial.13 The overall history of severe sunburns seems to be more important than simply the tendency to burn or tan,14,15 and a history of sunburns in childhood and adolescence has been associated with early-onset BCC.6 Our findings were consistent in that the age of onset of BCCs was lower in IWs who had a history of sunburns in childhood. Basal cell carcinomas developed at older ages in OWs who had a higher incidence of sunburns in adulthood. However, we have to consider that the retrospective nature of the data collection on sunburns in childhood and adulthood was potentially limited, as the information was based on the patients’ memory. Additionally, other non-UV risk factors for BCC, such as ionizing radiation exposure, were not analyzed.
The majority of BCCs developed in sun-exposed areas of the head and neck in both cohorts, and only 35 (20%) and 28 (22%) BCCs were located on the trunk, arms, or legs in OWs and IWs, respectively. In our study, the rate of BCCs on the trunk was significantly lower in OWs than in IWs (P=.0035). Basal cell carcinomas on the trunk have been suggested to be linked to genetic susceptibility16,17 and reduced DNA repair capacity18 rather than sun exposure. Our findings support this hypothesis and suggest that occupational sun exposure has no direct relation with truncal BCC. This outcome is consistent with the result of a case-control study conducted by Pelucchi et al19 (N=1040). The authors concluded that occupational UV exposure was not associated with truncal BCC development but with head/neck BCC, indicating that there may be different etiological mechanisms between truncal and head/neck BCC.19 In the largest BCC case series published in the literature with 13,457 specimens, the authors stated that tumors on the trunk may represent a particular variant of BCC, in which the theory of chronic versus intermittent UV exposure cannot be simply extrapolated as it is for the rest of BCC sites. Other factors such as genetic predisposition could be involved in the development of truncal BCC.20 Similarly, Ramos et al21 suggested that nonmelanoma skin cancers in sun-protected anatomic sites may occur in individuals with impairment in the DNA repair process.
The classification of histological subtypes of BCC helps to predict tumor behavior,22 which can impact the prognosis. In our study, nodular BCC was the most common subtype in both cohorts, followed by superficial BCC. The nodular subtype was increased in OWs compared to IWs, while the superficial subtype was most common in IWs. Bastiaens et al23 and McCormack et al24 have suggested that the most frequent subtypes of BCC (nodular and superficial) may represent different tumors with distinct causal factors. According to these authors, nodular subtypes are associated with cumulative UV exposure, while superficial subtypes are associated with more intense and intermittent UV exposure. The results of the current study support this hypothesis, as the OW cohort with cumulative UV exposure showed more incidence of nodular BCC than IWs, while the patients with intense and intermittent sun exposure (the IWs) showed more risk of superficial BCC.
The importance of occupational UV exposure in OWs as a risk factor for BCC is still an ongoing discussion. Our data show that occupational UV exposure may be considered an etiological factor for BCC according to histological subtype and anatomic site. Our study is limited by the retrospective nature of the data collection regarding occupation and childhood sunburns, which were based on the patients’ memory and therefore potentially biased. Data regarding family history of BCC also was self-reported and not validated. Another limiting factor was that other non-UV risk factors for BCC, such as ionizing radiation exposure, were not considered. The limited sample size also may have impacted the study results. Among the strengths of the study are the complete response rate, the similar catchment area of OWs and IWs, the common hospital setting of the 2 cohorts, and the similar attention to medical history. All patients were obtained from the practice of a single referral dermatologist and are felt to be representative of our working area. The use of a single dermatologist reduces provider-associated variability.
Conclusion
According to the results of this study, OWs are more likely to develop nodular BCCs with no increased risk for superficial BCCs. The age of onset in OWs is older than in IWs. Some anatomical sites such as the trunk are more commonly affected in IWs. Truncal BCCs may have etiological factors other than UV exposure, such as a genetic predisposition. This study is useful to occupational safety representatives and physicians to stimulate the implementation of prevention strategies for this easily preventable malignancy and may encourage further research.
- de Vries E, van de Poll-Franse LV, Louwman WJ, et al. Predictions of skin cancer incidence in the Netherlands up to 2015. Br J Dermatol. 2005;152:481-488.
- Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
- Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(suppl 61):1-6.
- Netscher DT, Spira M. Basal cell carcinoma: an overview of tumor biology and treatment. Plast Reconstr Surg. 2004;113:e74-e94.
- Miller SJ. Etiology and pathogenesis of basal cell carcinoma. Clin Dermatol. 1995;13:527-536.
- Dessinioti C, Tzannis K, Sypsa V, et al. Epidemiologic risk factors of basal cell carcinoma development and age at onset in a Southern European population from Greece. Exp Dermatol. 2011;20:622-626.
- Bauer A, Diepgen TL, Schmitt J. Is occupational solar UV-irradiation a relevant risk factor for basal cell carcinoma? a systematic review and meta-analysis of the epidemiologic literature. Br J Dermatol. 2011;165:612-625.
- Tran H, Chen K, Shumack S. Epidemiology and aetiology of basal cell carcinoma. Br J Dermatol. 2003;149(suppl 66):50-52.
- Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18.
- Stern RS. The mysteries of geographic variability in nonmelanoma skin cancer incidence. Arch Dermatol. 1999;135:843-844.
- Chinem VP, Miot HA. Epidemiology of basal cell carcinoma. An Bras Dermatol. 2011;86:292-305.
- Wong CS, Strange RC, Lear JT. Basal cell carcinoma. Br Med J. 2003;327:794-798.
- Dessinioti C, Antoniou C, Katsambas AD, et al. Basal cell carcinoma: what’s new under the sun. Photochem Photobiol. 2010;86:481-491.
- Van Dam RM, Huang Z, Rimm EB, et al. Risk factors for basal cell carcinoma of the skin in men: results from the health professionals follow-up study. Am J Epidemiol. 1999;150:459-468.
- Hunter DJ, Colditz GA, Stampfer MJ, et al. Risk factors for basal cell carcinoma in a prospective cohort of women. Ann Epidemiol. 1990;1:13-23.
- Ramachandran S, Fryer AA, Smith A, et al. Cutaneous basal cell carcinomas: distinct host factors are associated with the development of tumors on the trunk and on the head and neck. Cancer. 2001;92:354-358.
- Ramachandran S, Lear JT, Ramsay H, et al. Presentation with multiple cutaneous basal cell carcinomas: association of glutathione S-transferase and cytochrome P450 genotypes with clinical phenotype. Cancer Epidemiol Biomarkers Prev. 1999;8:61-67.
- Wei Q, Matanoski GM, Farmer ER, et al. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proc Natl Acad Sci USA. 1993;90:1614-1618.
- Pelucchi C, Di Landro A, Naldi L, et al. Risk factors for histological types and anatomic sites of cutaneous basal-cell carcinoma: an Italian case-control study [published online ahead of print Oct 19, 2006]. J Invest Dermatol. 2007;127:935-944.
- Scrivener Y, Grosshans E, Cribier B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br J Dermatol. 2002;147:41-47.
- Ramos J, Villa J, Ruiz A, et al. UV dose determines key characteristics of nonmelanoma skin cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:2006-2011.
- Rippey JJ. Why classify basal cell carcinomas? Histopathology. 1998;32:393-398.
- Bastiaens MT, Hoefnagel JJ, Bruijn JA, et al. Differences in age, site distribution and sex between nodular and superficial basal cell carcinomas indicate different type of tumors. J Invest Dermatol. 1998;110:880-884.
- McCormack CJ, Kelly JW, Dorevitch AP. Differences in age and body site distribution of histological subtypes of basal cell carcinoma. a possible indicator of different causes. Arch Dermatol. 1997;133:593-596.
- de Vries E, van de Poll-Franse LV, Louwman WJ, et al. Predictions of skin cancer incidence in the Netherlands up to 2015. Br J Dermatol. 2005;152:481-488.
- Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30:774-778.
- Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(suppl 61):1-6.
- Netscher DT, Spira M. Basal cell carcinoma: an overview of tumor biology and treatment. Plast Reconstr Surg. 2004;113:e74-e94.
- Miller SJ. Etiology and pathogenesis of basal cell carcinoma. Clin Dermatol. 1995;13:527-536.
- Dessinioti C, Tzannis K, Sypsa V, et al. Epidemiologic risk factors of basal cell carcinoma development and age at onset in a Southern European population from Greece. Exp Dermatol. 2011;20:622-626.
- Bauer A, Diepgen TL, Schmitt J. Is occupational solar UV-irradiation a relevant risk factor for basal cell carcinoma? a systematic review and meta-analysis of the epidemiologic literature. Br J Dermatol. 2011;165:612-625.
- Tran H, Chen K, Shumack S. Epidemiology and aetiology of basal cell carcinoma. Br J Dermatol. 2003;149(suppl 66):50-52.
- Armstrong BK, Kricker A. The epidemiology of UV induced skin cancer. J Photochem Photobiol B. 2001;63:8-18.
- Stern RS. The mysteries of geographic variability in nonmelanoma skin cancer incidence. Arch Dermatol. 1999;135:843-844.
- Chinem VP, Miot HA. Epidemiology of basal cell carcinoma. An Bras Dermatol. 2011;86:292-305.
- Wong CS, Strange RC, Lear JT. Basal cell carcinoma. Br Med J. 2003;327:794-798.
- Dessinioti C, Antoniou C, Katsambas AD, et al. Basal cell carcinoma: what’s new under the sun. Photochem Photobiol. 2010;86:481-491.
- Van Dam RM, Huang Z, Rimm EB, et al. Risk factors for basal cell carcinoma of the skin in men: results from the health professionals follow-up study. Am J Epidemiol. 1999;150:459-468.
- Hunter DJ, Colditz GA, Stampfer MJ, et al. Risk factors for basal cell carcinoma in a prospective cohort of women. Ann Epidemiol. 1990;1:13-23.
- Ramachandran S, Fryer AA, Smith A, et al. Cutaneous basal cell carcinomas: distinct host factors are associated with the development of tumors on the trunk and on the head and neck. Cancer. 2001;92:354-358.
- Ramachandran S, Lear JT, Ramsay H, et al. Presentation with multiple cutaneous basal cell carcinomas: association of glutathione S-transferase and cytochrome P450 genotypes with clinical phenotype. Cancer Epidemiol Biomarkers Prev. 1999;8:61-67.
- Wei Q, Matanoski GM, Farmer ER, et al. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proc Natl Acad Sci USA. 1993;90:1614-1618.
- Pelucchi C, Di Landro A, Naldi L, et al. Risk factors for histological types and anatomic sites of cutaneous basal-cell carcinoma: an Italian case-control study [published online ahead of print Oct 19, 2006]. J Invest Dermatol. 2007;127:935-944.
- Scrivener Y, Grosshans E, Cribier B. Variations of basal cell carcinomas according to gender, age, location and histopathological subtype. Br J Dermatol. 2002;147:41-47.
- Ramos J, Villa J, Ruiz A, et al. UV dose determines key characteristics of nonmelanoma skin cancer. Cancer Epidemiol Biomarkers Prev. 2004;13:2006-2011.
- Rippey JJ. Why classify basal cell carcinomas? Histopathology. 1998;32:393-398.
- Bastiaens MT, Hoefnagel JJ, Bruijn JA, et al. Differences in age, site distribution and sex between nodular and superficial basal cell carcinomas indicate different type of tumors. J Invest Dermatol. 1998;110:880-884.
- McCormack CJ, Kelly JW, Dorevitch AP. Differences in age and body site distribution of histological subtypes of basal cell carcinoma. a possible indicator of different causes. Arch Dermatol. 1997;133:593-596.
Practice Points
- Basal cell carcinoma (BCC) is the most common cancer in white individuals with rapidly increasing incidence rates and a high economic burden.
- Despite a large number of epidemiologic studies and the known importance of UV exposure in BCC carcinogenesis, there are no clear conclusions regarding the role of chronic and acute sun exposure related to BCC subtypes.
- It is reasonable to assume that outdoor workers with a history of UV exposure may develop BCCs with different features than those observed in indoor workers.
Patch Testing for Adverse Drug Reactions
Adverse drug reactions account for 3% to 6% of hospital admissions in the United States and occur in 10% to 15% of hospitalized patients.1,2 The most common culprits are antibiotics and nonsteroidal anti-inflammatory drugs (NSAIDs).3-12 In most cases, diagnoses are made clinically without diagnostic testing. To identify drug allergies associated with diagnostic testing, one center selected patients with suspected cutaneous drug reactions (2006-2010) for further evaluation.13 Of 612 patients who were evaluated, 141 had a high suspicion of drug allergy and were included in the analysis. The excluded patients had pseudoallergic reactions, reactive exanthemas due to infection, histopathologic exclusion of drug allergy, angioedema, or other dermatological conditions such as contact dermatitis and eczema. Of the included patients, 107 were diagnosed with drug reactions, while the remainder had non–drug-related exanthemas or unknown etiology after testing. Identified culprit drugs were predominantly antibiotics (39.8%) and NSAIDs (21.2%); contrast media, anticoagulants, anticonvulsants, antimalarials, antifungals, glucocorticoids, antihypertensives, and proton pump inhibitors also were implicated. They were identified with skin prick, intradermal, and patch tests (62.6%); lymphocyte transformation test (17.7%); oral rechallenge (5.6%); or without skin testing (6.5%). One quarter of patients with a high suspicion for drug allergy did not have a confirmed drug eruption in this study. Another study found that 10% to 20% of patients with reported penicillin allergy had confirmation via skin prick testing.14 These findings suggest that confirmation of suspected drug allergy may require more than one diagnostic test.
Tests for Adverse Drug Reactions
The following tests have been shown to aid in the identification of cutaneous drug eruptions: (1) patch tests15-21; (2) intradermal tests14,15,19,20; (3) drug provocation tests15,20; and (4) lymphocyte transformation tests.20 Intradermal or skin prick tests are most useful in urticarial eruptions but can be considered in nonurticarial eruptions with delayed inspection of test sites up to 1 week after testing. Drug provocation tests are considered the gold standard but involve patient risk. Lymphocyte transformation tests use the principle that T lymphocytes proliferate in the presence of drugs to which the patient is sensitized. Patch tests will be discussed in greater detail below. Immunohistochemistry can determine immunologic mechanisms of eruptions but cannot identify causative agents.16,17,22
A retrospective study of patients referred for evaluation of adverse drug reactions between 1996 and 2006 found the collective negative predictive value (NPV)—the percentage of truly negative skin tests based on provocation or substitution testing—of cutaneous drug tests including patch, prick, and intradermal tests to be 89.6% (95% confidence interval, 85.9%-93.3%).23 The NPVs of each test were not reported. Patients with negative cutaneous tests had subsequent oral rechallenge or substitution testing with medication from the same drug class.23 Another study16 found the NPV of patch testing to be at least 79% after review of data from other studies using patch and provocation testing.16,24 These studies suggest that cutaneous testing can be useful, albeit imperfect, in the evaluation and diagnosis of drug allergy.
Review of the Patch Test
Patch tests can be helpful in diagnosis of delayed hypersensitivities.18 Patch testing is most commonly and effectively used to diagnose allergic contact dermatitis, but its utility in other applications, such as diagnosis of cutaneous drug eruptions, has not been extensively studied.
The development of patch tests to diagnose systemic drug allergies is inhibited by the uncertainty of percutaneous drug penetration, a dearth of studies to determine the best test concentrations of active drug in the patch test, and the potential for nonimmunologic contact urticaria upon skin exposure. Furthermore, cutaneous metabolism of many antigens is well documented, but correlation to systemic metabolism often is unknown, which can confound patch test results and lead to false-negative results when the skin’s metabolic capacity does not match the body’s capacity to generate antigens capable of eliciting immunogenic responses.21 Additionally, the method used to suspend and disperse drugs in patch test vehicles is unfamiliar to most pharmacists, and standardized concentrations and vehicles are available only for some medications.25 Studies sufficient to obtain US Food and Drug Administration approval of patch tests for systemic drug eruptions would be costly and therefore prohibitive to investigators. The majority of the literature consists of case reports and data extrapolated from reviews. Patch test results of many drugs have been reported in the literature, with the highest frequencies of positive results associated with anticonvulsants,26 antibiotics, corticosteroids, calcium channel blockers, and benzodiazepines.21
Patch test placement affects the diagnostic value of the test. Placing patch tests on previously involved sites of fixed drug eruptions improves yield over placement on uninvolved skin.27 Placing patch tests on previously involved sites of other drug eruptions such as toxic epidermal necrolysis also may aid in diagnosis, though the literature is sparse.25,26,28
Patch Testing in Drug Eruptions
Morbilliform eruptions account for 48% to 91% of patients with adverse drug reactions.4-6 Other drug eruptions include urticarial eruptions, acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, toxic epidermal necrolysis, Stevens-Johnson syndrome, lichenoid drug eruption, symmetric drug-related intertriginous and flexural exanthema (SDRIFE), erythema multiforme (EM), and systemic contact dermatitis. The Table summarizes reports of positive patch tests with various medications for these drug eruptions.
In general, antimicrobials and NSAIDs were the most implicated drugs with positive patch test results in AGEP, DRESS syndrome, EM, fixed drug eruptions, and morbilliform eruptions. In AGEP, positive results also were reported for other drugs, including terbinafine and morphine.29-38 In fixed drug eruptions, patch testing on involved skin showed positive results to NSAIDs, analgesics, platelet inhibitors, and antimicrobials.27,52-55 Patch testing in DRESS syndrome has shown many positive reactions to antiepileptics and antipsychotics.39-43 One study used patch tests in SDRIFE, reporting positive results with antimicrobials, antineoplastics, decongestants, and glucocorticoids.61 Nonsteroidal anti-inflammatory drugs, antimicrobials, calcium channel blockers, and histamine antagonists were implicated in EM.47-51 Positive patch tests were seen in morbilliform eruptions with selective serotonin reuptake inhibitors, antiepileptics/benzodiazepines, NSAIDs, and antimicrobials.28,57-60 In toxic epidermal necrolysis, diagnosis with patch testing was made using patches placed on previously involved skin with sulfamethoxazole.62
Systemic Contact Dermatitis
Drugs historically recognized as causing allergic contact dermatitis (eg, topical gentamycin) can cause systemic contact dermatitis, which can be patch tested. In these situations, systemic contact dermatitis may be due to either the active drug or excipients in the medication formulation. Excipients are inactive ingredients in medications that provide a suitable consistency, appearance, or form. Often overlooked as culprits of drug hypersensitivity because they are theoretically inert, excipients are increasingly implicated in drug allergy. Swerlick and Campbell63 described 11 cases in which chronic unexplained pruritus responded to medication changes to avoid coloring agents. The most common culprits were FD&C Blue No. 1 and FD&C Blue No. 2. Patch testing for allergies to dyes can be clinically useful, though a lack of commercially available patch tests makes diagnosis difficult.64
Other excipients can cause cutaneous reactions. Propylene glycol, commonly implicated in allergic contact dermatitis, also can cause cutaneous eruptions upon systemic exposure.65 Corticosteroid-induced systemic contact dermatitis has been reported, though it is less prevalent than allergic contact dermatitis.66 These reactions usually are due to nonmethylated and nonhalogenated corticosteroids including budesonide, cortisone, hydrocortisone, prednisolone, and methylprednisolone.67,68 Patch testing in these situations is complicated by the possibility of false-negative results due to the anti-inflammatory effects of the corticosteroids. Therefore, patch testing should be performed using standardized and not treatment concentrations.
In our clinic, we have anecdotally observed several patients with chronic dermatitis and suspected NSAID allergies have positive patch test results with propylene glycol and not the suspected drug. Excipients encountered in multiple drugs and foods are more likely to present as chronic dermatitis, while active drug ingredients started in hospital settings more often present as acute dermatitis.

Our Experience
We have patch tested a handful of patients with suspected drug eruptions (University Hospitals Cleveland Medical Center institutional review board #07-12-27). Medications, excipients, and their concentrations (in % weight per weight) and vehicles that were tested include ibuprofen (10% petrolatum), aspirin (10% petrolatum), hydrochlorothiazide (10% petrolatum), captopril (5% petrolatum), and propylene glycol (30% water or 5% petrolatum). Patch tests were read at 48 and 72 hours and scored according to the International Contact Dermatitis Research Group patch test scoring guidelines.69 Two patients tested for ibuprofen reacted positively only to propylene glycol; the 3 other patients did not react to aspirin, hydrochlorothiazide, and captopril. Overall, we observed no positive patch tests to medications and 2 positive tests to propylene glycol in 5 patients tested (unpublished data).
Areas of Uncertainty
Although tests for immediate-type hypersensitivity reactions to drugs exist as skin prick tests, diagnostic testing for the majority of drug reactions does not exist. Drug allergy diagnosis is made with history and temporality, potentially resulting in unnecessary avoidance of helpful medications. Ideal patch test concentrations and vehicles as well as the sensitivity and specificity of these tests are unknown.
Guidelines From Professional Societies
Drug allergy testing guidelines are available from the British Society for Allergy and Clinical Immunology70 and American Academy of Allergy, Asthma and Immunology.71 The guidelines recommend diagnosis by history and temporality, and it is stated that patch testing is potentially useful in maculopapular rashes, AGEP, fixed drug eruptions, and DRESS syndrome.
Conclusion
Case reports in the literature suggest the utility of patch testing in some drug allergies. We suggest testing excipients such as propylene glycol and benzoic acid to rule out systemic contact dermatitis when patch testing with active drugs to confirm cause of suspected adverse cutaneous reactions to medications.
- Arndt KA, Jick H. Rates of cutaneous reactions to drugs. a report from the Boston Collaborative Drug Surveillance Program. JAMA. 1976;235:918-922.
- Bigby M, Jick S, Jick H, et al. Drug-induced cutaneous reactions. a report from the Boston Collaborative Drug Surveillance Program on 15,483 consecutive inpatients, 1975 to 1982. JAMA. 1986;256:3358-3363.
- Fiszenson-Albala F, Auzerie V, Mahe E, et al. A 6-month prospective survey of cutaneous drug reactions in a hospital setting. Br J Dermatol. 2003;149:1018-1022.
- Thong BY, Leong KP, Tang CY, et al. Drug allergy in a general hospital: results of a novel prospective inpatient reporting system. Ann Allergy Asthma Immunol. 2003;90:342-347.
- Hunziker T, Kunzi UP, Braunschweig S, et al. Comprehensive hospital drug monitoring (CHDM): adverse skin reactions, a 20-year survey. Allergy. 1997;52:388-393.
- Swanbeck G, Dahlberg E. Cutaneous drug reactions. an attempt to quantitative estimation. Arch Dermatol Res. 1992;284:215-218.
- Naldi L, Conforti A, Venegoni M, et al. Cutaneous reactions to drugs. an analysis of spontaneous reports in four Italian regions. Br J Clin Pharmacol. 1999;48:839-846.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis. In: Bolognia, JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:319-333.
- Vasconcelos C, Magina S, Quirino P, et al. Cutaneous drug reactions to piroxicam. Contact Dermatitis. 1998;39:145.
- Gerber D. Adverse reactions of piroxicam. Drug Intell Clin Pharm. 1987;21:707-710.
- Revuz J, Valeyrie-Allanore L. Drug reactions. In: Bolognia, JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:335-356.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-709.e9; quiz 718-720.
- Heinzerling LM, Tomsitz D, Anliker MD. Is drug allergy less prevalent than previously assumed? a 5-year analysis. Br J Dermatol. 2012;166:107-114.
- Salkind AR, Cuddy PG. Is this patient allergic to penicillin?: an evidence-based analysis of the likelihood of penicillin allergy. JAMA. 2001;285:2498-2505.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Cham PM, Warshaw EM. Patch testing for evaluating drug reactions due to systemic antibiotics. Dermatitis. 2007;18:63-77.
- Andrade P, Brinca A, Gonçalo M. Patch testing in fixed drug eruptions—a 20-year review. Contact Dermatitis. 2011;65:195-201.
- Romano A, Viola M, Gaeta F, et al. Patch testing in non-immediate drug eruptions. Allergy Asthma Clin Immunol. 2008;4:66-74.
- Rosso R, Mattiacci G, Bernardi ML, et al. Very delayed reactions to beta-lactam antibiotics. Contact Dermatitis. 2000;42:293-295.
- Romano A, Torres MJ, Castells M, et al. Diagnosis and management of drug hypersensitivity reactions. J Allergy Clin Immunol. 2011;127(3 suppl):S67-S73.
- Friedmann PS, Ardern-Jones M. Patch testing in drug allergy. Curr Opin Allergy Clin Immunol. 2010;10:291-296.
- Torres MJ, Mayorga C, Blanca M. Nonimmediate allergic reactions induced by drugs: pathogenesis and diagnostic tests. J Investig Allergol Clin Immunol. 2009;19:80-90.
- Waton J, Tréchot P, Loss-Ayay C, et al. Negative predictive value of drug skin tests in investigating cutaneous adverse drug reactions. Br J Dermatol. 2009;160:786-794.
- Romano A, Viola M, Mondino C, et al. Diagnosing nonimmediate reactions to penicillins by in vivo tests. Int Arch Allergy Immunol. 2002;129:169-174.
- De Groot AC. Patch Testing. Test Concentrations and Vehicles for 4350 Chemicals. 3rd ed. Wapserveen, Netherlands: acdegroot publishing; 2008.
- Elzagallaai AA, Knowles SR, Rieder MJ, et al. Patch testing for the diagnosis of anticonvulsant hypersensitivity syndrome: a systematic review. Drug Saf. 2009;32:391-408.
- Andrade P, Gonçalo M. Fixed drug eruption caused by etoricoxib—2 cases confirmed by patch testing. Contact Dermatitis. 2011;64:118-120.
- Barbaud A, Reichert-Penetrat S, Tréchot P, et al. The use of skin testing in the investigation of cutaneous adverse drug reactions. Br J Dermatol. 1998;139:49-58.
- Wolkenstein P, Chosidow O, Fléchet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
- Harries MJ, McIntyre SJ, Kingston TP. Co-amoxiclav-induced acute generalized exanthematous pustulosis confirmed by patch testing. Contact Dermatitis. 2006;55:372.
- Matsumoto Y, Okubo Y, Yamamoto T, et al. Case of acute generalized exanthematous pustulosis caused by ampicillin/cloxacillin sodium in a pregnant woman. J Dermatol. 2008;35:362-364.
- Chaabane A, Aouam K, Gassab L, et al. Acute generalized exanthematous pustulosis (AGEP) induced by cefotaxime. Fundam Clin Pharmacol. 2010;24:429-432.
- Hausermann P, Scherer K, Weber M, et al. Ciprofloxacin-induced acute generalized exanthematous pustulosis mimicking bullous drug eruption confirmed by a positive patch test. Dermatology. 2005;211:277-280.
- Moreau A, Dompmartin A, Castel B, et al. Drug-induced acute generalized exanthematous pustulosis with positive patch tests. Int J Dermatol. 1995;34:263-266.
- Kempinaire A, De Raevea L, Merckx M, et al. Terbinafine-induced acute generalized exanthematous pustulosis confirmed by a positive patch-test result. J Am Acad Dermatol. 1997;37:653-655.
- Mäkelä L, Lammintausta K. Etoricoxib-induced acute generalized exanthematous pustulosis. Acta Derm Venereol. 2008;88:200-201.
- Yang CC, Lee JY, Chen WC. Acute generalized exanthematous pustulosis caused by celecoxib. J Formos Med Assoc. 2004;103:555-557.
- Kardaun SH, de Monchy JG. Acute generalized exanthematous pustulosis caused by morphine, confirmed by positive patch test and lymphocyte transformation test. J Am Acad Dermatol. 2006;55(2 suppl):S21-S23.
- Inadomi T. Drug rash with eosinophilia and systemic symptoms (DRESS): changing carbamazepine to phenobarbital controlled epilepsy without the recurrence of DRESS. Eur J Dermatol. 2010;20:220-222.
- Buyuktiryaki AB, Bezirganoglu H, Sahiner UM, et al. Patch testing is an effective method for the diagnosis of carbamazepine-induced drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome in an 8-year-old girl. Australas J Dermatol. 2012;53:274-277.
- Aouam K, Ben Romdhane F, Loussaief C, et al. Hypersensitivity syndrome induced by anticonvulsants: possible cross-reactivity between carbamazepine and lamotrigine. J Clin Pharmacol. 2009;49:1488-1491.
- Santiago F, Gonçalo M, Vieira R, et al. Epicutaneous patch testing in drug hypersensitivity syndrome (DRESS). Contact Dermatitis. 2010;62:47-53.
- Prevost P, Bédry R, Lacoste D, et al. Hypersensitivity syndrome with olanzapine confirmed by patch tests. Eur J Dermatol. 2012;22:126-127.
- Hubiche T, Milpied B, Cazeau C, et al. Association of immunologically confirmed delayed drug reaction and human herpesvirus 6 viremia in a pediatric case of drug-induced hypersensitivity syndrome. Dermatology. 2011;222:140-141.
- Song WJ, Shim EJ, Kang MG, et al. Severe drug hypersensitivity induced by erdosteine and doxofylline as confirmed by patch and lymphocyte transformation tests: a case report. J Investig Allergol Clin Immunol. 2012;22:230-232.
- Lee JH, Park HK, Heo J, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23:521-525.
- González-Delgado P, Blanes M, Soriano V, et al. Erythema multiforme to amoxicillin with concurrent infection by Epstein-Barr virus. Allergol Immunopathol. 2006;34:76-78.
- Gonzalo Garijo MA, Pérez Calderón R, de Argila Fernández-Durán D, et al. Cutaneous reactions due to diltiazem and cross reactivity with other calcium channel blockers. Allergol Immunopathol (Madr). 2005;33:238-240.
- Peña AL, Henriquezsantana A, Gonzalez-Seco E, et al. Exudative erythema multiforme induced by hydroxyzine. Eur J Dermatol. 2008;18:194-195.
- Arakawa Y, Nakai N, Katoh N. Celecoxib-induced erythema multiforme-type drug eruption with a positive patch test. J Dermatol. 2011;38:1185-1188.
- Prieto A, De barrio M, Pérez C, et al. Piroxicam-induced erythema multiforme. Contact Dermatitis. 2004;50:263.
- Dalmau J, Serra-baldrich E, Roé E, et al. Use of patch test in fixed drug eruption due to metamizole (Nolotil). Contact Dermatitis. 2006;54:127-128.
- Gastaminza G, Anda M, Audicana MT, et al. Fixed-drug eruption due to metronidazole with positive topical provocation. Contact Dermatitis. 2001;44:36.
- Bellini V, Stingeni L, Lisi P. Multifocal fixed drug eruption due to celecoxib. Dermatitis. 2009;20:174-176.
- García CM, Carmena R, García R, et al. Fixed drug eruption from ticlopidine, with positive lesional patch test. Contact Dermatitis. 2001;44:40-41.
- Cruz MJ, Duarte AF, Baudrier T, et al. Lichenoid drug eruption induced by misoprostol. Contact Dermatitis. 2009;61:240-242.
- Alanko K. Patch testing in cutaneous reactions caused by carbamazepine. Contact Dermatitis. 1993;29:254-257.
- Grob M, Scheidegger P, Wüthrich B. Allergic skin reaction to celecoxib. Dermatology. 2000;201:383.
- Alonso JC, Ortega JD, Gonzalo MJ. Cutaneous reaction to oral celecoxib with positive patch test. Contact Dermatitis. 2004;50:48-49.
- Fernandes B, Brites M, Gonçalo M, et al. Maculopapular eruption from sertraline with positive patch tests. Contact Dermatitis. 2000;42:287.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Klein CE, Trautmann A, Zillikens D, et al. Patch testing in an unusual case of toxic epidermal necrolysis. Contact Dermatitis. 1996;35:175-176.
- Swerlick RA, Campbell CF. Medication dyes as a source of drug allergy. J Drugs Dermatol. 2013;12:99-102.
- Guin JD. Patch testing to FD&C and D&C dyes. Contact Dermatitis. 2003;49:217-218.
- Lowther A, McCormick T, Nedorost S. Systemic contact dermatitis from propylene glycol. Dermatitis. 2008;19:105-108.
- Baeck M, Goossens A. Systemic contact dermatitis to corticosteroids. Allergy. 2012;67:1580-1585.
- Baeck M, Goossens A. Immediate and delayed allergic hypersensitivity to corticosteroids: practical guidelines. Contact Dermatitis. 2012;66:38-45.
- Basedow S, Eigelshoven S, Homey B. Immediate and delayed hypersensitivity to corticosteroids. J Dtsch Dermatol Ges. 2011;9:885-888.
- Johansen JD, Aalto-korte K, Agner T, et al. European Society of Contact Dermatitis guideline for diagnostic patch testing—recommendations on best practice. Contact Dermatitis. 2015;73:195-221.
- Mirakian R, Ewan PW, Durham SR, et al. BSACI guidelines for the management of drug allergy. Clin Exp Allergy. 2009;39:43-61.
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259-273.
Adverse drug reactions account for 3% to 6% of hospital admissions in the United States and occur in 10% to 15% of hospitalized patients.1,2 The most common culprits are antibiotics and nonsteroidal anti-inflammatory drugs (NSAIDs).3-12 In most cases, diagnoses are made clinically without diagnostic testing. To identify drug allergies associated with diagnostic testing, one center selected patients with suspected cutaneous drug reactions (2006-2010) for further evaluation.13 Of 612 patients who were evaluated, 141 had a high suspicion of drug allergy and were included in the analysis. The excluded patients had pseudoallergic reactions, reactive exanthemas due to infection, histopathologic exclusion of drug allergy, angioedema, or other dermatological conditions such as contact dermatitis and eczema. Of the included patients, 107 were diagnosed with drug reactions, while the remainder had non–drug-related exanthemas or unknown etiology after testing. Identified culprit drugs were predominantly antibiotics (39.8%) and NSAIDs (21.2%); contrast media, anticoagulants, anticonvulsants, antimalarials, antifungals, glucocorticoids, antihypertensives, and proton pump inhibitors also were implicated. They were identified with skin prick, intradermal, and patch tests (62.6%); lymphocyte transformation test (17.7%); oral rechallenge (5.6%); or without skin testing (6.5%). One quarter of patients with a high suspicion for drug allergy did not have a confirmed drug eruption in this study. Another study found that 10% to 20% of patients with reported penicillin allergy had confirmation via skin prick testing.14 These findings suggest that confirmation of suspected drug allergy may require more than one diagnostic test.
Tests for Adverse Drug Reactions
The following tests have been shown to aid in the identification of cutaneous drug eruptions: (1) patch tests15-21; (2) intradermal tests14,15,19,20; (3) drug provocation tests15,20; and (4) lymphocyte transformation tests.20 Intradermal or skin prick tests are most useful in urticarial eruptions but can be considered in nonurticarial eruptions with delayed inspection of test sites up to 1 week after testing. Drug provocation tests are considered the gold standard but involve patient risk. Lymphocyte transformation tests use the principle that T lymphocytes proliferate in the presence of drugs to which the patient is sensitized. Patch tests will be discussed in greater detail below. Immunohistochemistry can determine immunologic mechanisms of eruptions but cannot identify causative agents.16,17,22
A retrospective study of patients referred for evaluation of adverse drug reactions between 1996 and 2006 found the collective negative predictive value (NPV)—the percentage of truly negative skin tests based on provocation or substitution testing—of cutaneous drug tests including patch, prick, and intradermal tests to be 89.6% (95% confidence interval, 85.9%-93.3%).23 The NPVs of each test were not reported. Patients with negative cutaneous tests had subsequent oral rechallenge or substitution testing with medication from the same drug class.23 Another study16 found the NPV of patch testing to be at least 79% after review of data from other studies using patch and provocation testing.16,24 These studies suggest that cutaneous testing can be useful, albeit imperfect, in the evaluation and diagnosis of drug allergy.
Review of the Patch Test
Patch tests can be helpful in diagnosis of delayed hypersensitivities.18 Patch testing is most commonly and effectively used to diagnose allergic contact dermatitis, but its utility in other applications, such as diagnosis of cutaneous drug eruptions, has not been extensively studied.
The development of patch tests to diagnose systemic drug allergies is inhibited by the uncertainty of percutaneous drug penetration, a dearth of studies to determine the best test concentrations of active drug in the patch test, and the potential for nonimmunologic contact urticaria upon skin exposure. Furthermore, cutaneous metabolism of many antigens is well documented, but correlation to systemic metabolism often is unknown, which can confound patch test results and lead to false-negative results when the skin’s metabolic capacity does not match the body’s capacity to generate antigens capable of eliciting immunogenic responses.21 Additionally, the method used to suspend and disperse drugs in patch test vehicles is unfamiliar to most pharmacists, and standardized concentrations and vehicles are available only for some medications.25 Studies sufficient to obtain US Food and Drug Administration approval of patch tests for systemic drug eruptions would be costly and therefore prohibitive to investigators. The majority of the literature consists of case reports and data extrapolated from reviews. Patch test results of many drugs have been reported in the literature, with the highest frequencies of positive results associated with anticonvulsants,26 antibiotics, corticosteroids, calcium channel blockers, and benzodiazepines.21
Patch test placement affects the diagnostic value of the test. Placing patch tests on previously involved sites of fixed drug eruptions improves yield over placement on uninvolved skin.27 Placing patch tests on previously involved sites of other drug eruptions such as toxic epidermal necrolysis also may aid in diagnosis, though the literature is sparse.25,26,28
Patch Testing in Drug Eruptions
Morbilliform eruptions account for 48% to 91% of patients with adverse drug reactions.4-6 Other drug eruptions include urticarial eruptions, acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, toxic epidermal necrolysis, Stevens-Johnson syndrome, lichenoid drug eruption, symmetric drug-related intertriginous and flexural exanthema (SDRIFE), erythema multiforme (EM), and systemic contact dermatitis. The Table summarizes reports of positive patch tests with various medications for these drug eruptions.
In general, antimicrobials and NSAIDs were the most implicated drugs with positive patch test results in AGEP, DRESS syndrome, EM, fixed drug eruptions, and morbilliform eruptions. In AGEP, positive results also were reported for other drugs, including terbinafine and morphine.29-38 In fixed drug eruptions, patch testing on involved skin showed positive results to NSAIDs, analgesics, platelet inhibitors, and antimicrobials.27,52-55 Patch testing in DRESS syndrome has shown many positive reactions to antiepileptics and antipsychotics.39-43 One study used patch tests in SDRIFE, reporting positive results with antimicrobials, antineoplastics, decongestants, and glucocorticoids.61 Nonsteroidal anti-inflammatory drugs, antimicrobials, calcium channel blockers, and histamine antagonists were implicated in EM.47-51 Positive patch tests were seen in morbilliform eruptions with selective serotonin reuptake inhibitors, antiepileptics/benzodiazepines, NSAIDs, and antimicrobials.28,57-60 In toxic epidermal necrolysis, diagnosis with patch testing was made using patches placed on previously involved skin with sulfamethoxazole.62
Systemic Contact Dermatitis
Drugs historically recognized as causing allergic contact dermatitis (eg, topical gentamycin) can cause systemic contact dermatitis, which can be patch tested. In these situations, systemic contact dermatitis may be due to either the active drug or excipients in the medication formulation. Excipients are inactive ingredients in medications that provide a suitable consistency, appearance, or form. Often overlooked as culprits of drug hypersensitivity because they are theoretically inert, excipients are increasingly implicated in drug allergy. Swerlick and Campbell63 described 11 cases in which chronic unexplained pruritus responded to medication changes to avoid coloring agents. The most common culprits were FD&C Blue No. 1 and FD&C Blue No. 2. Patch testing for allergies to dyes can be clinically useful, though a lack of commercially available patch tests makes diagnosis difficult.64
Other excipients can cause cutaneous reactions. Propylene glycol, commonly implicated in allergic contact dermatitis, also can cause cutaneous eruptions upon systemic exposure.65 Corticosteroid-induced systemic contact dermatitis has been reported, though it is less prevalent than allergic contact dermatitis.66 These reactions usually are due to nonmethylated and nonhalogenated corticosteroids including budesonide, cortisone, hydrocortisone, prednisolone, and methylprednisolone.67,68 Patch testing in these situations is complicated by the possibility of false-negative results due to the anti-inflammatory effects of the corticosteroids. Therefore, patch testing should be performed using standardized and not treatment concentrations.
In our clinic, we have anecdotally observed several patients with chronic dermatitis and suspected NSAID allergies have positive patch test results with propylene glycol and not the suspected drug. Excipients encountered in multiple drugs and foods are more likely to present as chronic dermatitis, while active drug ingredients started in hospital settings more often present as acute dermatitis.
Our Experience
We have patch tested a handful of patients with suspected drug eruptions (University Hospitals Cleveland Medical Center institutional review board #07-12-27). Medications, excipients, and their concentrations (in % weight per weight) and vehicles that were tested include ibuprofen (10% petrolatum), aspirin (10% petrolatum), hydrochlorothiazide (10% petrolatum), captopril (5% petrolatum), and propylene glycol (30% water or 5% petrolatum). Patch tests were read at 48 and 72 hours and scored according to the International Contact Dermatitis Research Group patch test scoring guidelines.69 Two patients tested for ibuprofen reacted positively only to propylene glycol; the 3 other patients did not react to aspirin, hydrochlorothiazide, and captopril. Overall, we observed no positive patch tests to medications and 2 positive tests to propylene glycol in 5 patients tested (unpublished data).
Areas of Uncertainty
Although tests for immediate-type hypersensitivity reactions to drugs exist as skin prick tests, diagnostic testing for the majority of drug reactions does not exist. Drug allergy diagnosis is made with history and temporality, potentially resulting in unnecessary avoidance of helpful medications. Ideal patch test concentrations and vehicles as well as the sensitivity and specificity of these tests are unknown.
Guidelines From Professional Societies
Drug allergy testing guidelines are available from the British Society for Allergy and Clinical Immunology70 and American Academy of Allergy, Asthma and Immunology.71 The guidelines recommend diagnosis by history and temporality, and it is stated that patch testing is potentially useful in maculopapular rashes, AGEP, fixed drug eruptions, and DRESS syndrome.
Conclusion
Case reports in the literature suggest the utility of patch testing in some drug allergies. We suggest testing excipients such as propylene glycol and benzoic acid to rule out systemic contact dermatitis when patch testing with active drugs to confirm cause of suspected adverse cutaneous reactions to medications.
Adverse drug reactions account for 3% to 6% of hospital admissions in the United States and occur in 10% to 15% of hospitalized patients.1,2 The most common culprits are antibiotics and nonsteroidal anti-inflammatory drugs (NSAIDs).3-12 In most cases, diagnoses are made clinically without diagnostic testing. To identify drug allergies associated with diagnostic testing, one center selected patients with suspected cutaneous drug reactions (2006-2010) for further evaluation.13 Of 612 patients who were evaluated, 141 had a high suspicion of drug allergy and were included in the analysis. The excluded patients had pseudoallergic reactions, reactive exanthemas due to infection, histopathologic exclusion of drug allergy, angioedema, or other dermatological conditions such as contact dermatitis and eczema. Of the included patients, 107 were diagnosed with drug reactions, while the remainder had non–drug-related exanthemas or unknown etiology after testing. Identified culprit drugs were predominantly antibiotics (39.8%) and NSAIDs (21.2%); contrast media, anticoagulants, anticonvulsants, antimalarials, antifungals, glucocorticoids, antihypertensives, and proton pump inhibitors also were implicated. They were identified with skin prick, intradermal, and patch tests (62.6%); lymphocyte transformation test (17.7%); oral rechallenge (5.6%); or without skin testing (6.5%). One quarter of patients with a high suspicion for drug allergy did not have a confirmed drug eruption in this study. Another study found that 10% to 20% of patients with reported penicillin allergy had confirmation via skin prick testing.14 These findings suggest that confirmation of suspected drug allergy may require more than one diagnostic test.
Tests for Adverse Drug Reactions
The following tests have been shown to aid in the identification of cutaneous drug eruptions: (1) patch tests15-21; (2) intradermal tests14,15,19,20; (3) drug provocation tests15,20; and (4) lymphocyte transformation tests.20 Intradermal or skin prick tests are most useful in urticarial eruptions but can be considered in nonurticarial eruptions with delayed inspection of test sites up to 1 week after testing. Drug provocation tests are considered the gold standard but involve patient risk. Lymphocyte transformation tests use the principle that T lymphocytes proliferate in the presence of drugs to which the patient is sensitized. Patch tests will be discussed in greater detail below. Immunohistochemistry can determine immunologic mechanisms of eruptions but cannot identify causative agents.16,17,22
A retrospective study of patients referred for evaluation of adverse drug reactions between 1996 and 2006 found the collective negative predictive value (NPV)—the percentage of truly negative skin tests based on provocation or substitution testing—of cutaneous drug tests including patch, prick, and intradermal tests to be 89.6% (95% confidence interval, 85.9%-93.3%).23 The NPVs of each test were not reported. Patients with negative cutaneous tests had subsequent oral rechallenge or substitution testing with medication from the same drug class.23 Another study16 found the NPV of patch testing to be at least 79% after review of data from other studies using patch and provocation testing.16,24 These studies suggest that cutaneous testing can be useful, albeit imperfect, in the evaluation and diagnosis of drug allergy.
Review of the Patch Test
Patch tests can be helpful in diagnosis of delayed hypersensitivities.18 Patch testing is most commonly and effectively used to diagnose allergic contact dermatitis, but its utility in other applications, such as diagnosis of cutaneous drug eruptions, has not been extensively studied.
The development of patch tests to diagnose systemic drug allergies is inhibited by the uncertainty of percutaneous drug penetration, a dearth of studies to determine the best test concentrations of active drug in the patch test, and the potential for nonimmunologic contact urticaria upon skin exposure. Furthermore, cutaneous metabolism of many antigens is well documented, but correlation to systemic metabolism often is unknown, which can confound patch test results and lead to false-negative results when the skin’s metabolic capacity does not match the body’s capacity to generate antigens capable of eliciting immunogenic responses.21 Additionally, the method used to suspend and disperse drugs in patch test vehicles is unfamiliar to most pharmacists, and standardized concentrations and vehicles are available only for some medications.25 Studies sufficient to obtain US Food and Drug Administration approval of patch tests for systemic drug eruptions would be costly and therefore prohibitive to investigators. The majority of the literature consists of case reports and data extrapolated from reviews. Patch test results of many drugs have been reported in the literature, with the highest frequencies of positive results associated with anticonvulsants,26 antibiotics, corticosteroids, calcium channel blockers, and benzodiazepines.21
Patch test placement affects the diagnostic value of the test. Placing patch tests on previously involved sites of fixed drug eruptions improves yield over placement on uninvolved skin.27 Placing patch tests on previously involved sites of other drug eruptions such as toxic epidermal necrolysis also may aid in diagnosis, though the literature is sparse.25,26,28
Patch Testing in Drug Eruptions
Morbilliform eruptions account for 48% to 91% of patients with adverse drug reactions.4-6 Other drug eruptions include urticarial eruptions, acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, toxic epidermal necrolysis, Stevens-Johnson syndrome, lichenoid drug eruption, symmetric drug-related intertriginous and flexural exanthema (SDRIFE), erythema multiforme (EM), and systemic contact dermatitis. The Table summarizes reports of positive patch tests with various medications for these drug eruptions.
In general, antimicrobials and NSAIDs were the most implicated drugs with positive patch test results in AGEP, DRESS syndrome, EM, fixed drug eruptions, and morbilliform eruptions. In AGEP, positive results also were reported for other drugs, including terbinafine and morphine.29-38 In fixed drug eruptions, patch testing on involved skin showed positive results to NSAIDs, analgesics, platelet inhibitors, and antimicrobials.27,52-55 Patch testing in DRESS syndrome has shown many positive reactions to antiepileptics and antipsychotics.39-43 One study used patch tests in SDRIFE, reporting positive results with antimicrobials, antineoplastics, decongestants, and glucocorticoids.61 Nonsteroidal anti-inflammatory drugs, antimicrobials, calcium channel blockers, and histamine antagonists were implicated in EM.47-51 Positive patch tests were seen in morbilliform eruptions with selective serotonin reuptake inhibitors, antiepileptics/benzodiazepines, NSAIDs, and antimicrobials.28,57-60 In toxic epidermal necrolysis, diagnosis with patch testing was made using patches placed on previously involved skin with sulfamethoxazole.62
Systemic Contact Dermatitis
Drugs historically recognized as causing allergic contact dermatitis (eg, topical gentamycin) can cause systemic contact dermatitis, which can be patch tested. In these situations, systemic contact dermatitis may be due to either the active drug or excipients in the medication formulation. Excipients are inactive ingredients in medications that provide a suitable consistency, appearance, or form. Often overlooked as culprits of drug hypersensitivity because they are theoretically inert, excipients are increasingly implicated in drug allergy. Swerlick and Campbell63 described 11 cases in which chronic unexplained pruritus responded to medication changes to avoid coloring agents. The most common culprits were FD&C Blue No. 1 and FD&C Blue No. 2. Patch testing for allergies to dyes can be clinically useful, though a lack of commercially available patch tests makes diagnosis difficult.64
Other excipients can cause cutaneous reactions. Propylene glycol, commonly implicated in allergic contact dermatitis, also can cause cutaneous eruptions upon systemic exposure.65 Corticosteroid-induced systemic contact dermatitis has been reported, though it is less prevalent than allergic contact dermatitis.66 These reactions usually are due to nonmethylated and nonhalogenated corticosteroids including budesonide, cortisone, hydrocortisone, prednisolone, and methylprednisolone.67,68 Patch testing in these situations is complicated by the possibility of false-negative results due to the anti-inflammatory effects of the corticosteroids. Therefore, patch testing should be performed using standardized and not treatment concentrations.
In our clinic, we have anecdotally observed several patients with chronic dermatitis and suspected NSAID allergies have positive patch test results with propylene glycol and not the suspected drug. Excipients encountered in multiple drugs and foods are more likely to present as chronic dermatitis, while active drug ingredients started in hospital settings more often present as acute dermatitis.
Our Experience
We have patch tested a handful of patients with suspected drug eruptions (University Hospitals Cleveland Medical Center institutional review board #07-12-27). Medications, excipients, and their concentrations (in % weight per weight) and vehicles that were tested include ibuprofen (10% petrolatum), aspirin (10% petrolatum), hydrochlorothiazide (10% petrolatum), captopril (5% petrolatum), and propylene glycol (30% water or 5% petrolatum). Patch tests were read at 48 and 72 hours and scored according to the International Contact Dermatitis Research Group patch test scoring guidelines.69 Two patients tested for ibuprofen reacted positively only to propylene glycol; the 3 other patients did not react to aspirin, hydrochlorothiazide, and captopril. Overall, we observed no positive patch tests to medications and 2 positive tests to propylene glycol in 5 patients tested (unpublished data).
Areas of Uncertainty
Although tests for immediate-type hypersensitivity reactions to drugs exist as skin prick tests, diagnostic testing for the majority of drug reactions does not exist. Drug allergy diagnosis is made with history and temporality, potentially resulting in unnecessary avoidance of helpful medications. Ideal patch test concentrations and vehicles as well as the sensitivity and specificity of these tests are unknown.
Guidelines From Professional Societies
Drug allergy testing guidelines are available from the British Society for Allergy and Clinical Immunology70 and American Academy of Allergy, Asthma and Immunology.71 The guidelines recommend diagnosis by history and temporality, and it is stated that patch testing is potentially useful in maculopapular rashes, AGEP, fixed drug eruptions, and DRESS syndrome.
Conclusion
Case reports in the literature suggest the utility of patch testing in some drug allergies. We suggest testing excipients such as propylene glycol and benzoic acid to rule out systemic contact dermatitis when patch testing with active drugs to confirm cause of suspected adverse cutaneous reactions to medications.
- Arndt KA, Jick H. Rates of cutaneous reactions to drugs. a report from the Boston Collaborative Drug Surveillance Program. JAMA. 1976;235:918-922.
- Bigby M, Jick S, Jick H, et al. Drug-induced cutaneous reactions. a report from the Boston Collaborative Drug Surveillance Program on 15,483 consecutive inpatients, 1975 to 1982. JAMA. 1986;256:3358-3363.
- Fiszenson-Albala F, Auzerie V, Mahe E, et al. A 6-month prospective survey of cutaneous drug reactions in a hospital setting. Br J Dermatol. 2003;149:1018-1022.
- Thong BY, Leong KP, Tang CY, et al. Drug allergy in a general hospital: results of a novel prospective inpatient reporting system. Ann Allergy Asthma Immunol. 2003;90:342-347.
- Hunziker T, Kunzi UP, Braunschweig S, et al. Comprehensive hospital drug monitoring (CHDM): adverse skin reactions, a 20-year survey. Allergy. 1997;52:388-393.
- Swanbeck G, Dahlberg E. Cutaneous drug reactions. an attempt to quantitative estimation. Arch Dermatol Res. 1992;284:215-218.
- Naldi L, Conforti A, Venegoni M, et al. Cutaneous reactions to drugs. an analysis of spontaneous reports in four Italian regions. Br J Clin Pharmacol. 1999;48:839-846.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis. In: Bolognia, JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:319-333.
- Vasconcelos C, Magina S, Quirino P, et al. Cutaneous drug reactions to piroxicam. Contact Dermatitis. 1998;39:145.
- Gerber D. Adverse reactions of piroxicam. Drug Intell Clin Pharm. 1987;21:707-710.
- Revuz J, Valeyrie-Allanore L. Drug reactions. In: Bolognia, JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:335-356.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-709.e9; quiz 718-720.
- Heinzerling LM, Tomsitz D, Anliker MD. Is drug allergy less prevalent than previously assumed? a 5-year analysis. Br J Dermatol. 2012;166:107-114.
- Salkind AR, Cuddy PG. Is this patient allergic to penicillin?: an evidence-based analysis of the likelihood of penicillin allergy. JAMA. 2001;285:2498-2505.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Cham PM, Warshaw EM. Patch testing for evaluating drug reactions due to systemic antibiotics. Dermatitis. 2007;18:63-77.
- Andrade P, Brinca A, Gonçalo M. Patch testing in fixed drug eruptions—a 20-year review. Contact Dermatitis. 2011;65:195-201.
- Romano A, Viola M, Gaeta F, et al. Patch testing in non-immediate drug eruptions. Allergy Asthma Clin Immunol. 2008;4:66-74.
- Rosso R, Mattiacci G, Bernardi ML, et al. Very delayed reactions to beta-lactam antibiotics. Contact Dermatitis. 2000;42:293-295.
- Romano A, Torres MJ, Castells M, et al. Diagnosis and management of drug hypersensitivity reactions. J Allergy Clin Immunol. 2011;127(3 suppl):S67-S73.
- Friedmann PS, Ardern-Jones M. Patch testing in drug allergy. Curr Opin Allergy Clin Immunol. 2010;10:291-296.
- Torres MJ, Mayorga C, Blanca M. Nonimmediate allergic reactions induced by drugs: pathogenesis and diagnostic tests. J Investig Allergol Clin Immunol. 2009;19:80-90.
- Waton J, Tréchot P, Loss-Ayay C, et al. Negative predictive value of drug skin tests in investigating cutaneous adverse drug reactions. Br J Dermatol. 2009;160:786-794.
- Romano A, Viola M, Mondino C, et al. Diagnosing nonimmediate reactions to penicillins by in vivo tests. Int Arch Allergy Immunol. 2002;129:169-174.
- De Groot AC. Patch Testing. Test Concentrations and Vehicles for 4350 Chemicals. 3rd ed. Wapserveen, Netherlands: acdegroot publishing; 2008.
- Elzagallaai AA, Knowles SR, Rieder MJ, et al. Patch testing for the diagnosis of anticonvulsant hypersensitivity syndrome: a systematic review. Drug Saf. 2009;32:391-408.
- Andrade P, Gonçalo M. Fixed drug eruption caused by etoricoxib—2 cases confirmed by patch testing. Contact Dermatitis. 2011;64:118-120.
- Barbaud A, Reichert-Penetrat S, Tréchot P, et al. The use of skin testing in the investigation of cutaneous adverse drug reactions. Br J Dermatol. 1998;139:49-58.
- Wolkenstein P, Chosidow O, Fléchet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
- Harries MJ, McIntyre SJ, Kingston TP. Co-amoxiclav-induced acute generalized exanthematous pustulosis confirmed by patch testing. Contact Dermatitis. 2006;55:372.
- Matsumoto Y, Okubo Y, Yamamoto T, et al. Case of acute generalized exanthematous pustulosis caused by ampicillin/cloxacillin sodium in a pregnant woman. J Dermatol. 2008;35:362-364.
- Chaabane A, Aouam K, Gassab L, et al. Acute generalized exanthematous pustulosis (AGEP) induced by cefotaxime. Fundam Clin Pharmacol. 2010;24:429-432.
- Hausermann P, Scherer K, Weber M, et al. Ciprofloxacin-induced acute generalized exanthematous pustulosis mimicking bullous drug eruption confirmed by a positive patch test. Dermatology. 2005;211:277-280.
- Moreau A, Dompmartin A, Castel B, et al. Drug-induced acute generalized exanthematous pustulosis with positive patch tests. Int J Dermatol. 1995;34:263-266.
- Kempinaire A, De Raevea L, Merckx M, et al. Terbinafine-induced acute generalized exanthematous pustulosis confirmed by a positive patch-test result. J Am Acad Dermatol. 1997;37:653-655.
- Mäkelä L, Lammintausta K. Etoricoxib-induced acute generalized exanthematous pustulosis. Acta Derm Venereol. 2008;88:200-201.
- Yang CC, Lee JY, Chen WC. Acute generalized exanthematous pustulosis caused by celecoxib. J Formos Med Assoc. 2004;103:555-557.
- Kardaun SH, de Monchy JG. Acute generalized exanthematous pustulosis caused by morphine, confirmed by positive patch test and lymphocyte transformation test. J Am Acad Dermatol. 2006;55(2 suppl):S21-S23.
- Inadomi T. Drug rash with eosinophilia and systemic symptoms (DRESS): changing carbamazepine to phenobarbital controlled epilepsy without the recurrence of DRESS. Eur J Dermatol. 2010;20:220-222.
- Buyuktiryaki AB, Bezirganoglu H, Sahiner UM, et al. Patch testing is an effective method for the diagnosis of carbamazepine-induced drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome in an 8-year-old girl. Australas J Dermatol. 2012;53:274-277.
- Aouam K, Ben Romdhane F, Loussaief C, et al. Hypersensitivity syndrome induced by anticonvulsants: possible cross-reactivity between carbamazepine and lamotrigine. J Clin Pharmacol. 2009;49:1488-1491.
- Santiago F, Gonçalo M, Vieira R, et al. Epicutaneous patch testing in drug hypersensitivity syndrome (DRESS). Contact Dermatitis. 2010;62:47-53.
- Prevost P, Bédry R, Lacoste D, et al. Hypersensitivity syndrome with olanzapine confirmed by patch tests. Eur J Dermatol. 2012;22:126-127.
- Hubiche T, Milpied B, Cazeau C, et al. Association of immunologically confirmed delayed drug reaction and human herpesvirus 6 viremia in a pediatric case of drug-induced hypersensitivity syndrome. Dermatology. 2011;222:140-141.
- Song WJ, Shim EJ, Kang MG, et al. Severe drug hypersensitivity induced by erdosteine and doxofylline as confirmed by patch and lymphocyte transformation tests: a case report. J Investig Allergol Clin Immunol. 2012;22:230-232.
- Lee JH, Park HK, Heo J, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23:521-525.
- González-Delgado P, Blanes M, Soriano V, et al. Erythema multiforme to amoxicillin with concurrent infection by Epstein-Barr virus. Allergol Immunopathol. 2006;34:76-78.
- Gonzalo Garijo MA, Pérez Calderón R, de Argila Fernández-Durán D, et al. Cutaneous reactions due to diltiazem and cross reactivity with other calcium channel blockers. Allergol Immunopathol (Madr). 2005;33:238-240.
- Peña AL, Henriquezsantana A, Gonzalez-Seco E, et al. Exudative erythema multiforme induced by hydroxyzine. Eur J Dermatol. 2008;18:194-195.
- Arakawa Y, Nakai N, Katoh N. Celecoxib-induced erythema multiforme-type drug eruption with a positive patch test. J Dermatol. 2011;38:1185-1188.
- Prieto A, De barrio M, Pérez C, et al. Piroxicam-induced erythema multiforme. Contact Dermatitis. 2004;50:263.
- Dalmau J, Serra-baldrich E, Roé E, et al. Use of patch test in fixed drug eruption due to metamizole (Nolotil). Contact Dermatitis. 2006;54:127-128.
- Gastaminza G, Anda M, Audicana MT, et al. Fixed-drug eruption due to metronidazole with positive topical provocation. Contact Dermatitis. 2001;44:36.
- Bellini V, Stingeni L, Lisi P. Multifocal fixed drug eruption due to celecoxib. Dermatitis. 2009;20:174-176.
- García CM, Carmena R, García R, et al. Fixed drug eruption from ticlopidine, with positive lesional patch test. Contact Dermatitis. 2001;44:40-41.
- Cruz MJ, Duarte AF, Baudrier T, et al. Lichenoid drug eruption induced by misoprostol. Contact Dermatitis. 2009;61:240-242.
- Alanko K. Patch testing in cutaneous reactions caused by carbamazepine. Contact Dermatitis. 1993;29:254-257.
- Grob M, Scheidegger P, Wüthrich B. Allergic skin reaction to celecoxib. Dermatology. 2000;201:383.
- Alonso JC, Ortega JD, Gonzalo MJ. Cutaneous reaction to oral celecoxib with positive patch test. Contact Dermatitis. 2004;50:48-49.
- Fernandes B, Brites M, Gonçalo M, et al. Maculopapular eruption from sertraline with positive patch tests. Contact Dermatitis. 2000;42:287.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Klein CE, Trautmann A, Zillikens D, et al. Patch testing in an unusual case of toxic epidermal necrolysis. Contact Dermatitis. 1996;35:175-176.
- Swerlick RA, Campbell CF. Medication dyes as a source of drug allergy. J Drugs Dermatol. 2013;12:99-102.
- Guin JD. Patch testing to FD&C and D&C dyes. Contact Dermatitis. 2003;49:217-218.
- Lowther A, McCormick T, Nedorost S. Systemic contact dermatitis from propylene glycol. Dermatitis. 2008;19:105-108.
- Baeck M, Goossens A. Systemic contact dermatitis to corticosteroids. Allergy. 2012;67:1580-1585.
- Baeck M, Goossens A. Immediate and delayed allergic hypersensitivity to corticosteroids: practical guidelines. Contact Dermatitis. 2012;66:38-45.
- Basedow S, Eigelshoven S, Homey B. Immediate and delayed hypersensitivity to corticosteroids. J Dtsch Dermatol Ges. 2011;9:885-888.
- Johansen JD, Aalto-korte K, Agner T, et al. European Society of Contact Dermatitis guideline for diagnostic patch testing—recommendations on best practice. Contact Dermatitis. 2015;73:195-221.
- Mirakian R, Ewan PW, Durham SR, et al. BSACI guidelines for the management of drug allergy. Clin Exp Allergy. 2009;39:43-61.
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259-273.
- Arndt KA, Jick H. Rates of cutaneous reactions to drugs. a report from the Boston Collaborative Drug Surveillance Program. JAMA. 1976;235:918-922.
- Bigby M, Jick S, Jick H, et al. Drug-induced cutaneous reactions. a report from the Boston Collaborative Drug Surveillance Program on 15,483 consecutive inpatients, 1975 to 1982. JAMA. 1986;256:3358-3363.
- Fiszenson-Albala F, Auzerie V, Mahe E, et al. A 6-month prospective survey of cutaneous drug reactions in a hospital setting. Br J Dermatol. 2003;149:1018-1022.
- Thong BY, Leong KP, Tang CY, et al. Drug allergy in a general hospital: results of a novel prospective inpatient reporting system. Ann Allergy Asthma Immunol. 2003;90:342-347.
- Hunziker T, Kunzi UP, Braunschweig S, et al. Comprehensive hospital drug monitoring (CHDM): adverse skin reactions, a 20-year survey. Allergy. 1997;52:388-393.
- Swanbeck G, Dahlberg E. Cutaneous drug reactions. an attempt to quantitative estimation. Arch Dermatol Res. 1992;284:215-218.
- Naldi L, Conforti A, Venegoni M, et al. Cutaneous reactions to drugs. an analysis of spontaneous reports in four Italian regions. Br J Clin Pharmacol. 1999;48:839-846.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis. In: Bolognia, JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:319-333.
- Vasconcelos C, Magina S, Quirino P, et al. Cutaneous drug reactions to piroxicam. Contact Dermatitis. 1998;39:145.
- Gerber D. Adverse reactions of piroxicam. Drug Intell Clin Pharm. 1987;21:707-710.
- Revuz J, Valeyrie-Allanore L. Drug reactions. In: Bolognia, JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012:335-356.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-709.e9; quiz 718-720.
- Heinzerling LM, Tomsitz D, Anliker MD. Is drug allergy less prevalent than previously assumed? a 5-year analysis. Br J Dermatol. 2012;166:107-114.
- Salkind AR, Cuddy PG. Is this patient allergic to penicillin?: an evidence-based analysis of the likelihood of penicillin allergy. JAMA. 2001;285:2498-2505.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Cham PM, Warshaw EM. Patch testing for evaluating drug reactions due to systemic antibiotics. Dermatitis. 2007;18:63-77.
- Andrade P, Brinca A, Gonçalo M. Patch testing in fixed drug eruptions—a 20-year review. Contact Dermatitis. 2011;65:195-201.
- Romano A, Viola M, Gaeta F, et al. Patch testing in non-immediate drug eruptions. Allergy Asthma Clin Immunol. 2008;4:66-74.
- Rosso R, Mattiacci G, Bernardi ML, et al. Very delayed reactions to beta-lactam antibiotics. Contact Dermatitis. 2000;42:293-295.
- Romano A, Torres MJ, Castells M, et al. Diagnosis and management of drug hypersensitivity reactions. J Allergy Clin Immunol. 2011;127(3 suppl):S67-S73.
- Friedmann PS, Ardern-Jones M. Patch testing in drug allergy. Curr Opin Allergy Clin Immunol. 2010;10:291-296.
- Torres MJ, Mayorga C, Blanca M. Nonimmediate allergic reactions induced by drugs: pathogenesis and diagnostic tests. J Investig Allergol Clin Immunol. 2009;19:80-90.
- Waton J, Tréchot P, Loss-Ayay C, et al. Negative predictive value of drug skin tests in investigating cutaneous adverse drug reactions. Br J Dermatol. 2009;160:786-794.
- Romano A, Viola M, Mondino C, et al. Diagnosing nonimmediate reactions to penicillins by in vivo tests. Int Arch Allergy Immunol. 2002;129:169-174.
- De Groot AC. Patch Testing. Test Concentrations and Vehicles for 4350 Chemicals. 3rd ed. Wapserveen, Netherlands: acdegroot publishing; 2008.
- Elzagallaai AA, Knowles SR, Rieder MJ, et al. Patch testing for the diagnosis of anticonvulsant hypersensitivity syndrome: a systematic review. Drug Saf. 2009;32:391-408.
- Andrade P, Gonçalo M. Fixed drug eruption caused by etoricoxib—2 cases confirmed by patch testing. Contact Dermatitis. 2011;64:118-120.
- Barbaud A, Reichert-Penetrat S, Tréchot P, et al. The use of skin testing in the investigation of cutaneous adverse drug reactions. Br J Dermatol. 1998;139:49-58.
- Wolkenstein P, Chosidow O, Fléchet ML, et al. Patch testing in severe cutaneous adverse drug reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Contact Dermatitis. 1996;35:234-236.
- Harries MJ, McIntyre SJ, Kingston TP. Co-amoxiclav-induced acute generalized exanthematous pustulosis confirmed by patch testing. Contact Dermatitis. 2006;55:372.
- Matsumoto Y, Okubo Y, Yamamoto T, et al. Case of acute generalized exanthematous pustulosis caused by ampicillin/cloxacillin sodium in a pregnant woman. J Dermatol. 2008;35:362-364.
- Chaabane A, Aouam K, Gassab L, et al. Acute generalized exanthematous pustulosis (AGEP) induced by cefotaxime. Fundam Clin Pharmacol. 2010;24:429-432.
- Hausermann P, Scherer K, Weber M, et al. Ciprofloxacin-induced acute generalized exanthematous pustulosis mimicking bullous drug eruption confirmed by a positive patch test. Dermatology. 2005;211:277-280.
- Moreau A, Dompmartin A, Castel B, et al. Drug-induced acute generalized exanthematous pustulosis with positive patch tests. Int J Dermatol. 1995;34:263-266.
- Kempinaire A, De Raevea L, Merckx M, et al. Terbinafine-induced acute generalized exanthematous pustulosis confirmed by a positive patch-test result. J Am Acad Dermatol. 1997;37:653-655.
- Mäkelä L, Lammintausta K. Etoricoxib-induced acute generalized exanthematous pustulosis. Acta Derm Venereol. 2008;88:200-201.
- Yang CC, Lee JY, Chen WC. Acute generalized exanthematous pustulosis caused by celecoxib. J Formos Med Assoc. 2004;103:555-557.
- Kardaun SH, de Monchy JG. Acute generalized exanthematous pustulosis caused by morphine, confirmed by positive patch test and lymphocyte transformation test. J Am Acad Dermatol. 2006;55(2 suppl):S21-S23.
- Inadomi T. Drug rash with eosinophilia and systemic symptoms (DRESS): changing carbamazepine to phenobarbital controlled epilepsy without the recurrence of DRESS. Eur J Dermatol. 2010;20:220-222.
- Buyuktiryaki AB, Bezirganoglu H, Sahiner UM, et al. Patch testing is an effective method for the diagnosis of carbamazepine-induced drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome in an 8-year-old girl. Australas J Dermatol. 2012;53:274-277.
- Aouam K, Ben Romdhane F, Loussaief C, et al. Hypersensitivity syndrome induced by anticonvulsants: possible cross-reactivity between carbamazepine and lamotrigine. J Clin Pharmacol. 2009;49:1488-1491.
- Santiago F, Gonçalo M, Vieira R, et al. Epicutaneous patch testing in drug hypersensitivity syndrome (DRESS). Contact Dermatitis. 2010;62:47-53.
- Prevost P, Bédry R, Lacoste D, et al. Hypersensitivity syndrome with olanzapine confirmed by patch tests. Eur J Dermatol. 2012;22:126-127.
- Hubiche T, Milpied B, Cazeau C, et al. Association of immunologically confirmed delayed drug reaction and human herpesvirus 6 viremia in a pediatric case of drug-induced hypersensitivity syndrome. Dermatology. 2011;222:140-141.
- Song WJ, Shim EJ, Kang MG, et al. Severe drug hypersensitivity induced by erdosteine and doxofylline as confirmed by patch and lymphocyte transformation tests: a case report. J Investig Allergol Clin Immunol. 2012;22:230-232.
- Lee JH, Park HK, Heo J, et al. Drug rash with eosinophilia and systemic symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23:521-525.
- González-Delgado P, Blanes M, Soriano V, et al. Erythema multiforme to amoxicillin with concurrent infection by Epstein-Barr virus. Allergol Immunopathol. 2006;34:76-78.
- Gonzalo Garijo MA, Pérez Calderón R, de Argila Fernández-Durán D, et al. Cutaneous reactions due to diltiazem and cross reactivity with other calcium channel blockers. Allergol Immunopathol (Madr). 2005;33:238-240.
- Peña AL, Henriquezsantana A, Gonzalez-Seco E, et al. Exudative erythema multiforme induced by hydroxyzine. Eur J Dermatol. 2008;18:194-195.
- Arakawa Y, Nakai N, Katoh N. Celecoxib-induced erythema multiforme-type drug eruption with a positive patch test. J Dermatol. 2011;38:1185-1188.
- Prieto A, De barrio M, Pérez C, et al. Piroxicam-induced erythema multiforme. Contact Dermatitis. 2004;50:263.
- Dalmau J, Serra-baldrich E, Roé E, et al. Use of patch test in fixed drug eruption due to metamizole (Nolotil). Contact Dermatitis. 2006;54:127-128.
- Gastaminza G, Anda M, Audicana MT, et al. Fixed-drug eruption due to metronidazole with positive topical provocation. Contact Dermatitis. 2001;44:36.
- Bellini V, Stingeni L, Lisi P. Multifocal fixed drug eruption due to celecoxib. Dermatitis. 2009;20:174-176.
- García CM, Carmena R, García R, et al. Fixed drug eruption from ticlopidine, with positive lesional patch test. Contact Dermatitis. 2001;44:40-41.
- Cruz MJ, Duarte AF, Baudrier T, et al. Lichenoid drug eruption induced by misoprostol. Contact Dermatitis. 2009;61:240-242.
- Alanko K. Patch testing in cutaneous reactions caused by carbamazepine. Contact Dermatitis. 1993;29:254-257.
- Grob M, Scheidegger P, Wüthrich B. Allergic skin reaction to celecoxib. Dermatology. 2000;201:383.
- Alonso JC, Ortega JD, Gonzalo MJ. Cutaneous reaction to oral celecoxib with positive patch test. Contact Dermatitis. 2004;50:48-49.
- Fernandes B, Brites M, Gonçalo M, et al. Maculopapular eruption from sertraline with positive patch tests. Contact Dermatitis. 2000;42:287.
- Häusermann P, Harr T, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Klein CE, Trautmann A, Zillikens D, et al. Patch testing in an unusual case of toxic epidermal necrolysis. Contact Dermatitis. 1996;35:175-176.
- Swerlick RA, Campbell CF. Medication dyes as a source of drug allergy. J Drugs Dermatol. 2013;12:99-102.
- Guin JD. Patch testing to FD&C and D&C dyes. Contact Dermatitis. 2003;49:217-218.
- Lowther A, McCormick T, Nedorost S. Systemic contact dermatitis from propylene glycol. Dermatitis. 2008;19:105-108.
- Baeck M, Goossens A. Systemic contact dermatitis to corticosteroids. Allergy. 2012;67:1580-1585.
- Baeck M, Goossens A. Immediate and delayed allergic hypersensitivity to corticosteroids: practical guidelines. Contact Dermatitis. 2012;66:38-45.
- Basedow S, Eigelshoven S, Homey B. Immediate and delayed hypersensitivity to corticosteroids. J Dtsch Dermatol Ges. 2011;9:885-888.
- Johansen JD, Aalto-korte K, Agner T, et al. European Society of Contact Dermatitis guideline for diagnostic patch testing—recommendations on best practice. Contact Dermatitis. 2015;73:195-221.
- Mirakian R, Ewan PW, Durham SR, et al. BSACI guidelines for the management of drug allergy. Clin Exp Allergy. 2009;39:43-61.
- Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. Drug allergy: an updated practice parameter. Ann Allergy Asthma Immunol. 2010;105:259-273.
Practice Points
- Consider patch testing in suspected eczematous drug rashes and fixed drug eruption.
- Patch test to inactive excipients as well as active ingredients.
- Caution patients that sensitivity of patch testing for systemic drug reactions is unknown and likely lower than specificity.