User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Relapsing Polychondritis With Meningoencephalitis
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
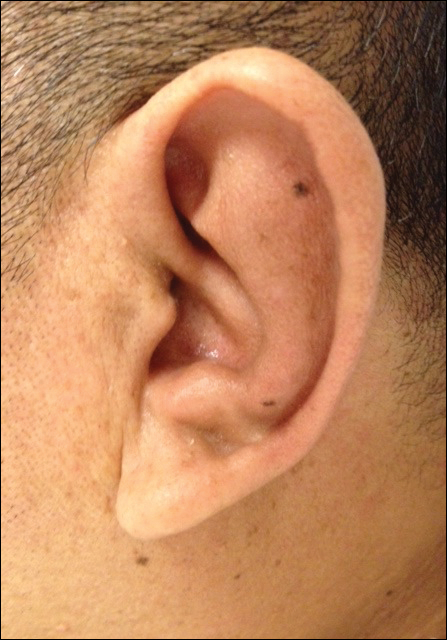
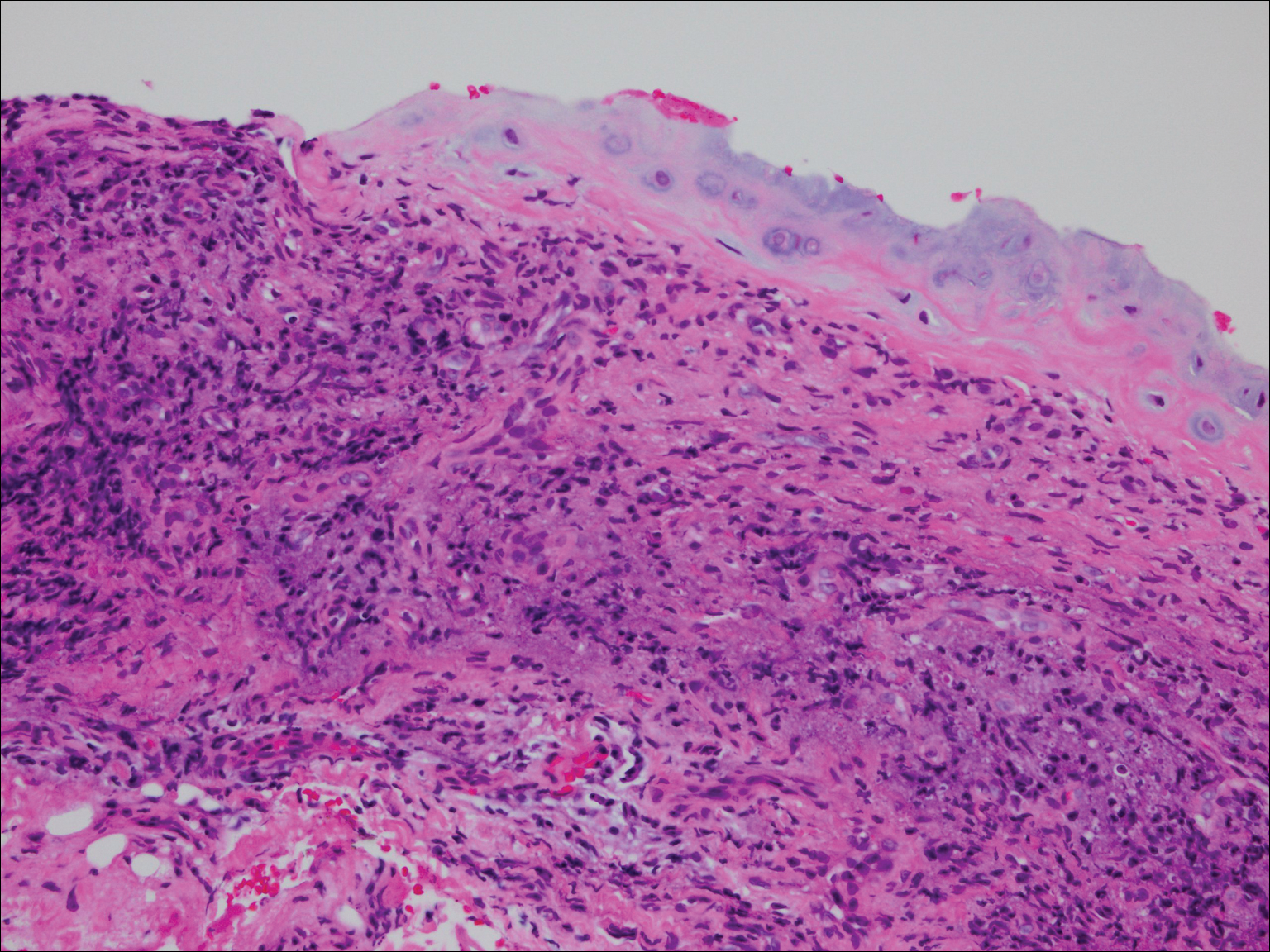
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
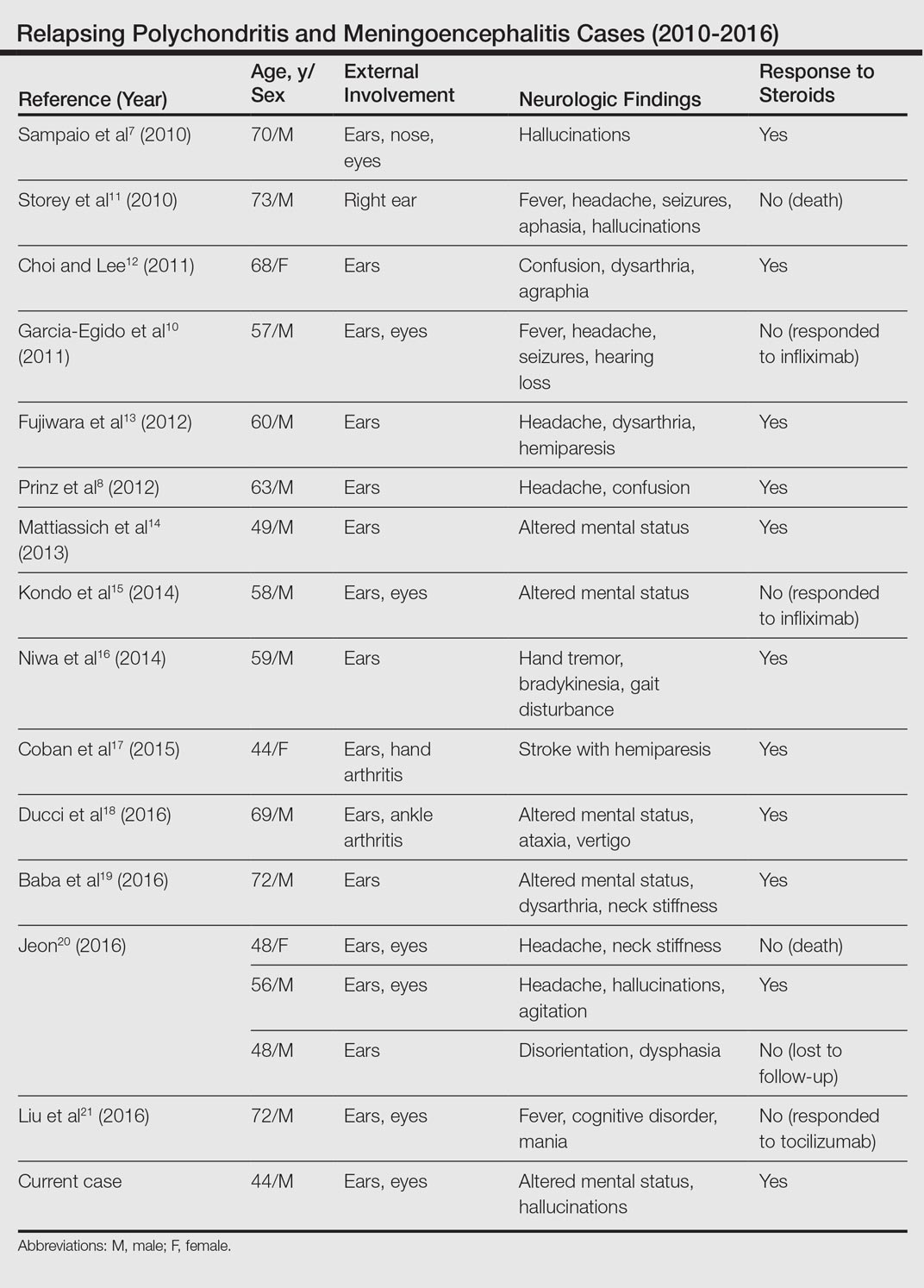
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
Relapsing polychondritis (RP) is an autoimmune disease affecting cartilaginous structures such as the ears, respiratory passages, joints, and cardiovascular system.1,2 In rare cases, the systemic effects of this autoimmune process can cause central nervous system (CNS) involvement such as meningoencephalitis (ME).3 In 2011, Wang et al4 described 4 cases of RP with ME and reviewed 24 cases from the literature. We present a case of a man with RP-associated ME that was responsive to steroid treatment. We also provide an updated review of the literature.
Case Report
A 44-year-old man developed gradually worsening bilateral ear pain, headaches, and seizures. He was briefly hospitalized and discharged with levetiracetam and quetiapine. However, his mental status continued to deteriorate and he was subsequently hospitalized 3 months later with confusion, hallucinations, and seizures.
On physical examination the patient was disoriented and unable to form cohesive sentences. He had bilateral tenderness, erythema, and edema of the auricles, which notably spared the lobules (Figure 1). The conjunctivae were injected bilaterally, and joint involvement included bilateral knee tenderness and swelling. Neurologic examination revealed questionable meningeal signs but no motor or sensory deficits. An extensive laboratory workup for the etiology of his altered mental status was unremarkable, except for a mildly elevated white blood cell count in the cerebrospinal fluid with predominantly lymphocytes. No infectious etiologies were identified on laboratory testing, and rheumatologic markers were negative including antinuclear antibody, rheumatoid factor, and anti–Sjögren syndrome antigen A/Sjögren syndrome antigen B. Magnetic resonance imaging revealed nonspecific findings of bilateral T2 hyperdensities in the subcortical white matter; however, cerebral angiography revealed no evidence of vasculitis. A biopsy of the right antihelix revealed prominent perichondritis and a neutrophilic inflammatory infiltrate with several lymphocytes and histiocytes (Figure 2). There was degeneration of the cartilaginous tissue with evidence of pyknotic nuclei, eosinophilia, and vacuolization of the chondrocytes. He was diagnosed with RP on the basis of clinical and histologic inflammation of the auricular cartilage, polyarthritis, and ocular inflammation.
The patient was treated with high-dose immunosuppression with methylprednisolone (1000 mg intravenous once daily for 5 days) and cyclophosphamide (one dose at 500 mg/m2), which resulted in remarkable improvement in his mental status, auricular inflammation, and knee pain. After 31 days of hospitalization the patient was discharged with a course of oral prednisone (starting at 60 mg/d, then tapered over the following 2 months) and monthly cyclophosphamide infusions (5 months total; starting at 500 mg/m2, then uptitrated to goal of 1000 mg/m2). Maintenance suppression was achieved with azathioprine (starting at 50 mg daily, then uptitrated to 100 mg daily), which was continued without any evidence of relapsed disease through his last outpatient visit 1 year after the diagnosis.
Comment
Auricular inflammation is a hallmark of RP and is present in 83% to 95% of patients.1,3 The affected ears can appear erythematous to violaceous with tender edema of the auricle that spares the lobules where no cartilage is present. The inflammation can extend into the ear canal and cause hearing loss, tinnitus, and vertigo. Histologically, RP can present with a nonspecific leukocytoclastic vasculitis and inflammatory destruction of the cartilage. Therefore, diagnosis of RP is reliant mainly on clinical characteristics rather than pathologic findings. In 1976, McAdam et al5 established diagnostic criteria for RP based on the presence of common clinical manifestations (eg, auricular chondritis, seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation). Michet et al6 later proposed major and minor criteria to classify and diagnose RP based on clinical manifestations. Diagnosis of our patient was confirmed by the presence of auricular chondritis, polyarthritis, and ocular inflammation. Diagnosing RP can be difficult because it has many systemic manifestations that can evoke a broad differential diagnosis. The time to diagnosis in our patient was 3 months, but the mean delay in diagnosis for patients with RP and ME is 2.9 years.4
The etiology of RP remains unclear, but current evidence supports an immune-mediated process directed toward proteins found in cartilage. Animal studies have suggested that RP may be driven by antibodies to matrillin 1 and type II collagen. There also may be a familial association with HLA-DR4 and genetic predisposition to autoimmune diseases in individuals affected by RP.1,3 The pathogenesis of CNS involvement in RP is thought to be due to a localized small vessel vasculitis.7,8 In our patient, however, cerebral angiography was negative for vasculitis, and thus our case may represent another mechanism for CNS involvement. There have been cases of encephalitis in RP caused by pathways other than CNS vasculitis. Kashihara et al9 reported a case of RP with encephalitis associated with antiglutamate receptor antibodies found in the cerebrospinal fluid and blood.
Treatment of RP has been based on pathophysiological considerations rather than empiric data due to its rarity. Relapsing polychondritis has been responsive to steroid treatment in reported cases as well as in our patient; however, in cases in which RP did not respond to steroids, infliximab may be effective for RP with ME.10 Further research regarding the treatment outcomes of RP with ME may be warranted.
Although rare, additional cases of RP with ME have been reported (Table). Wang et al4 described a series of 28 patients with RP and ME from 1960 to 2010. A PubMed search of articles indexed for MEDLINE that were published in the English-language literature from 2010 to 2016 was performed using the search terms relapsing polychondritis and nervous system. Including our patient, RP with ME was reported in 17 additional cases since Wang et al4 published their findings. These cases involved adults ranging in age from 44 to 73 years who were mainly men (14/17 [82%]). All of the patients presented with bilateral auricular chondritis, except for a case of unilateral ear involvement reported by Storey et al.11 Common neurologic manifestations included fever, headache, and altered mental status. Motor symptoms ranged from dysarthria and agraphia12 to hemiparesis.13 The mechanism of CNS involvement in RP was not identified in most cases; however, Mattiassich et al14 documented cerebral vasculitis in their patient, and Niwa et al16 found diffuse cerebral vasculitis on autopsy. Eleven of 17 (65%) cases responded to steroid treatment. Of the 6 cases in which RP did not respond to steroids, 2 patients died despite high-dose steroid treatment,11,20 2 responded to infliximab,10,15 1 responded to tocilizumab,21 and 1 was lost to follow-up after initial treatment failure.20
Conclusion
Although rare, RP should not be overlooked in the inpatient setting due to its potential for life-threatening systemic effects. Early diagnosis of this condition may be of benefit to this select population of patients, and further research regarding the prognosis, mechanisms, and treatment of RP may be necessary in the future.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: a 2013 update. Autoimmun Rev. 2014;13:90-95.
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-461.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Wang ZJ, Pu CQ, Wang ZJ, et al. Meningoencephalitis or meningitis in relapsing polychondritis: four case reports and a literature review. J Clin Neurosci. 2011;18:1608-1615.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet C, McKenna C, Luthra H, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Sampaio L, Silva L, Mariz E, et al. CNS involvement in relapsing polychondritis. Joint Bone Spine. 2010;77:619-620.
- Prinz S, Dafotakis M, Schneider RK, et al. The red puffy ear sign—a clinical sign to diagnose a rare cause of meningoencephalitis. Fortschr Neurol Psychiatr. 2012;80:463-467.
- Kashihara K, Kawada S, Takahashi Y. Autoantibodies to glutamate receptor GluR2 in a patient with limic encephalitis associated with relapsing polychondritis. J Neurol Sci. 2009;287:275-277.
- Garcia-Egido A, Gutierrez C, de la Fuente C, et al. Relapsing polychondritis-associated meningitis and encephalitis: response to infliximab. Rheumatology (Oxford). 2011;50:1721-1723.
- Storey K, Matej R, Rusina R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthemia: a case study. J Neurol. 2010;258:159-161.
- Choi HJ, Lee HJ. Relapsing polychondritis with encephalitis. J Clin Rheum. 2011;6:329-331.
- Fujiwara S, Zenke K, Iwata S, et al. Relapsing polychondritis presenting as encephalitis. No Shinkei Geka. 2012;40:247-253.
- Mattiassich G, Egger M, Semlitsch G, et al. Occurrence of relapsing polychondritis with a rising cANCA titre in a cANCA-positive systemic and cerebral vasculitis patient [published online February 5, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-008717.
- Kondo T, Fukuta M, Takemoto A, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. 2014;76:361-368.
- Niwa A, Okamoto Y, Kondo T, et al. Perivasculitic pancencephalitis with relapsing polychondritis: an autopsy case report and review of previous cases. Intern Med. 2014;53:1191-1195.
- Coban EK, Xanmemmedoy E, Colak M, et al. A rare complication of a rare disease; stroke due to relapsing polychondritis. Ideggyogy Sz. 2015;68:429-432.
- Ducci R, Germiniani F, Czecko L, et al. Relapsing polychondritis and lymphocytic meningitis with varied neurological symptoms [published online February 5, 2016]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.09.005.
- Baba T, Kanno S, Shijo T, et al. Callosal disconnection syndrome associated with relapsing polychondritis. Intern Med. 2016;55:1191-1193.
- Jeon C. Relapsing polychondritis with central nervous system involvement: experience of three different cases in a single center. J Korean Med. 2016;31:1846-1850.
- Liu L, Liu S, Guan W, et al. Efficacy of tocilizumab for psychiatric symptoms associated with relapsing polychondritis: the first case report and review of the literature. Rheumatol Int. 2016;36:1185-1189.
Practice Points
- Meningoencephalitis (ME) is a potentially rare complication of relapsing polychondritis (RP).
- Treatment of ME due to RP can include high-dose steroids and biologics.
Clinicians Should Retain the Ability to Choose a Pathologist
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
As employers search for ways to reduce the cost of providing health care to their employees, there is a growing trend toward narrowed provider networks and exclusive laboratory contracts. In the case of clinical pathology, some of these choices make sense from the employer’s perspective. A complete blood cell count or comprehensive metabolic panel is done on a machine and the result is much the same regardless of the laboratory. So why not have all laboratory tests performed by the lowest bidder?
Laboratories vary in quality and anatomic pathology services are different from blood tests. Each slide must be interpreted by a physician and skill in the interpretation of skin specimens varies widely. Dermatopathology was one of the first subspecialties to be recognized within pathology, as it requires a high level of expertise. Clinicopathological correlation often is key to the accurate interpretation of a specimen. The stakes are high, and a delay in diagnosis of melanoma remains one of the most serious errors in medicine and one of the most common causes for litigation in dermatology.
The accurate interpretation of skin biopsy specimens becomes especially difficult when inadequate or misleading clinical information accompanies the specimen. A study of 589 biopsies submitted by primary care physicians and reported by general pathologists demonstrated a 6.5% error rate. False-negative errors were the most common, but false-positives also were observed.1 A study of pigmented lesions referred to the University of California, San Francisco, demonstrated a discordance rate of 14.3%.2 The degree of discordance would be expected to vary based on the range of diagnoses included in each study.
Board-certified dermatopathologists have varying areas of expertise and there is notable subjectivity in the interpretation of biopsy specimens. In the case of problematic pigmented lesions such as atypical Spitz nevi, there can be low interobserver agreement even among the experts in categorizing lesions as malignant versus nonmalignant (κ=0.30).3 The low concordance among expert dermatopathologists demonstrates that light microscopic features alone often are inadequate for diagnosis. Advanced studies, including immunohistochemical stains, can help to clarify the diagnosis. In the case of atypical Spitz tumors, the contribution of special stains to the final diagnosis is statistically similar to that of hematoxylin and eosin sections and age, suggesting that nothing alone is sufficiently reliable to establish a definitive diagnosis in every case.4 Although helpful, these studies are costly, and savings obtained by sending cases to the lowest bidder can evaporate quickly. Costs are higher when factoring in molecular studies, which can run upwards of $3000 per slide; the cost of litigation related to incorrect diagnoses; or the human costs of an incorrect diagnosis.
As a group, dermatopathologists are highly skilled in the interpretation of skin specimens, but challenging lesions are common in the routine practice of dermatopathology. A study of 1249 pigmented melanocytic lesions demonstrated substantial agreement among expert dermatopathologists for less problematic lesions, though agreement was greater for patients 40 years and older (κ=0.67) than for younger patients (κ=0.49). Agreement was lower for patients with atypical mole syndrome (κ=0.31).5 These discrepancies occur despite the fact that there is good interobserver reproducibility for grading of individual histological features such as asymmetry, circumscription, irregular confluent nests, single melanocytes predominating, absence of maturation, suprabasal melanocytes, symmetrical melanin, deep melanin, cytological atypia, mitoses, dermal lymphocytic infiltrate, and necrosis.6 These results indicate that accurate diagnoses cannot be reliably established simply by grading a list of histological features. Accurate diagnosis requires complex pattern recognition and integration of findings. Conflicting criteria often are present and an accurate interpretation requires considerable judgment as to which features are significant and which are not.
Separation of sebaceous adenoma, sebaceoma, and well-differentiated sebaceous carcinoma is another challenging area, and interobserver consensus can be as low as 11%,7 which suggests notable subjectivity in the criteria for diagnosis of nonmelanocytic tumors and emphasizes the importance of communication between the dermatopathologist and clinician when determining how to manage an ambiguous lesion. The interpretation of inflammatory skin diseases, alopecia, and lymphoid proliferations also can be problematic, and expert consultation often is required.
All dermatologists receive substantial training in dermatopathology, which puts them in an excellent position to interpret ambiguous findings in the context of the clinical presentation. Sometimes the dermatologist who has seen the clinical presentation can be in the best position to make the diagnosis. All clinicians must be wary of bias and an objective set of eyes often can be helpful. Communication is crucial to ensure appropriate care for each patient, and policies that restrict the choice of pathologist can be damaging.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.
- Trotter MJ, Bruecks AK. Interpretation of skin biopsies by general pathologists: diagnostic discrepancy rate measured by blinded review. Arch Pathol Lab Med. 2003;127:1489-1492.
- Shoo BA, Sagebiel RW, Kashani-Sabet M. Discordance in the histopathologic diagnosis of melanoma at a melanoma referral center [published online March 19, 2010]. J Am Acad Dermatol. 2010;62:751-756.
- Gerami P, Busam K, Cochran A, et al. Histomorphologic assessment and interobserver diagnostic reproducibility of atypical spitzoid melanocytic neoplasms with long-term follow-up. Am J Surg Pathol. 2014;38:934-940.
- Puri PK, Ferringer TC, Tyler WB, et al. Statistical analysis of the concordance of immunohistochemical stains with the final diagnosis in spitzoid neoplasms. Am J Dermatopathol. 2011;33:72-77.
- Braun RP, Gutkowicz-Krusin D, Rabinovitz H, et al. Agreement of dermatopathologists in the evaluation of clinically difficult melanocytic lesions: how golden is the ‘gold standard’? Dermatology. 2012;224:51-58.
- Urso C, Rongioletti F, Innocenzi D, et al. Interobserver reproducibility of histological features in cutaneous malignant melanoma. J Clin Pathol. 2005;58:1194-1198.
- Harvey NT, Budgeon CA, Leecy T, et al. Interobserver variability in the diagnosis of circumscribed sebaceous neoplasms of the skin. Pathology. 2013;45:581-586.

Papillary Transitional Cell Bladder Carcinoma and Systematized Epidermal Nevus Syndrome
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
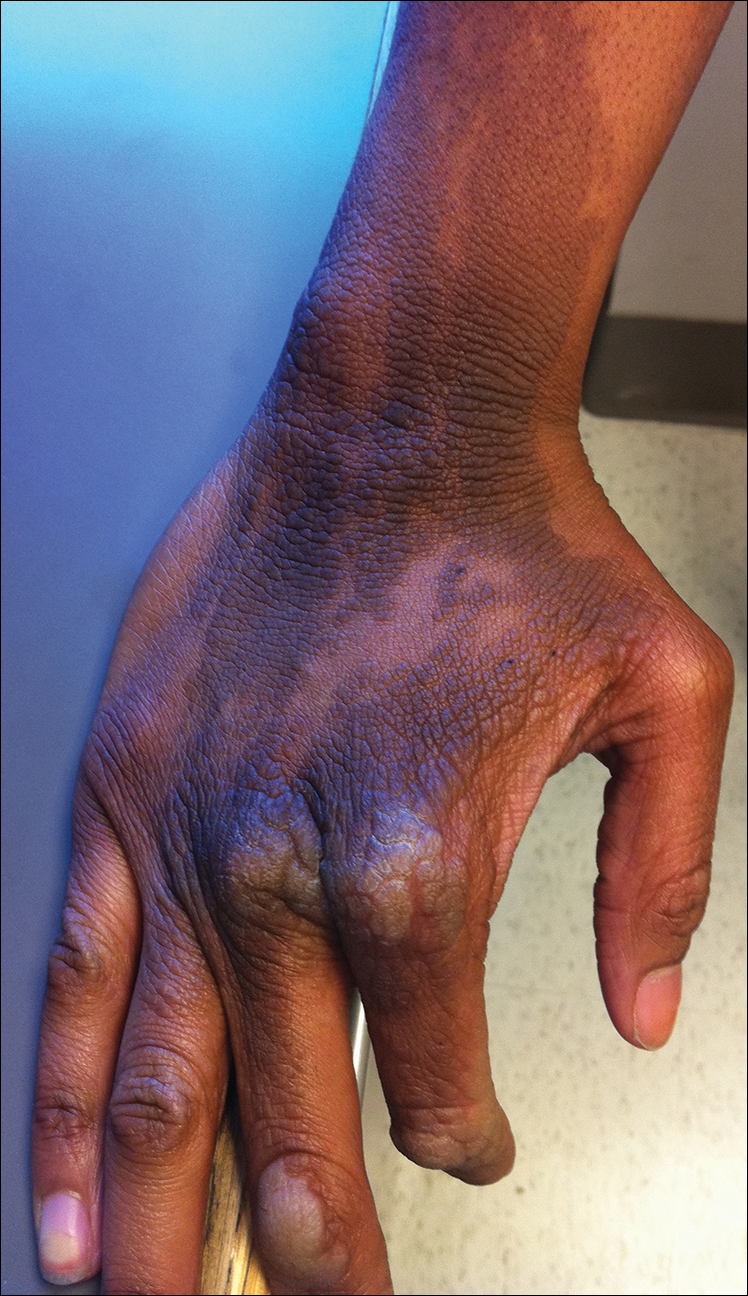
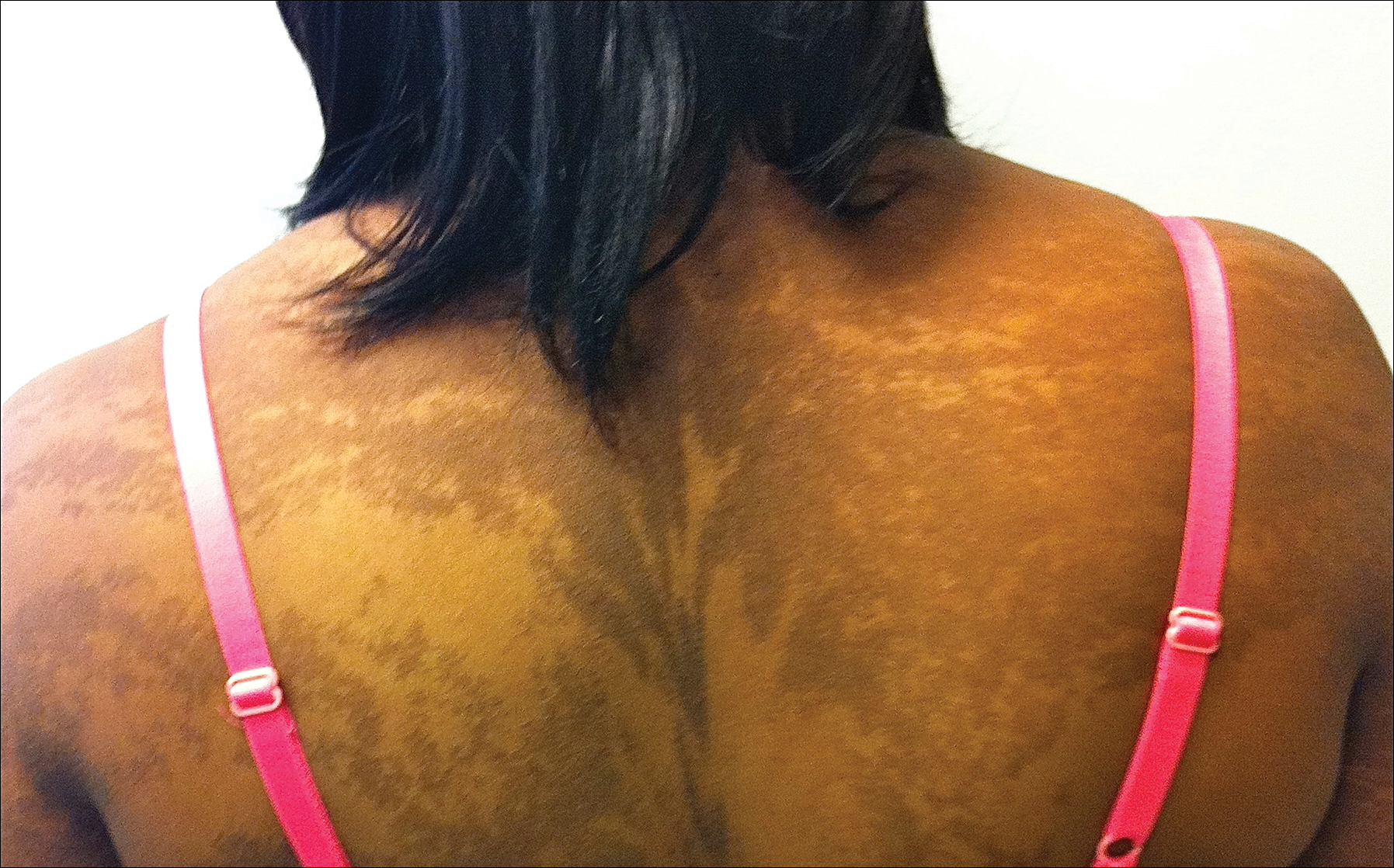
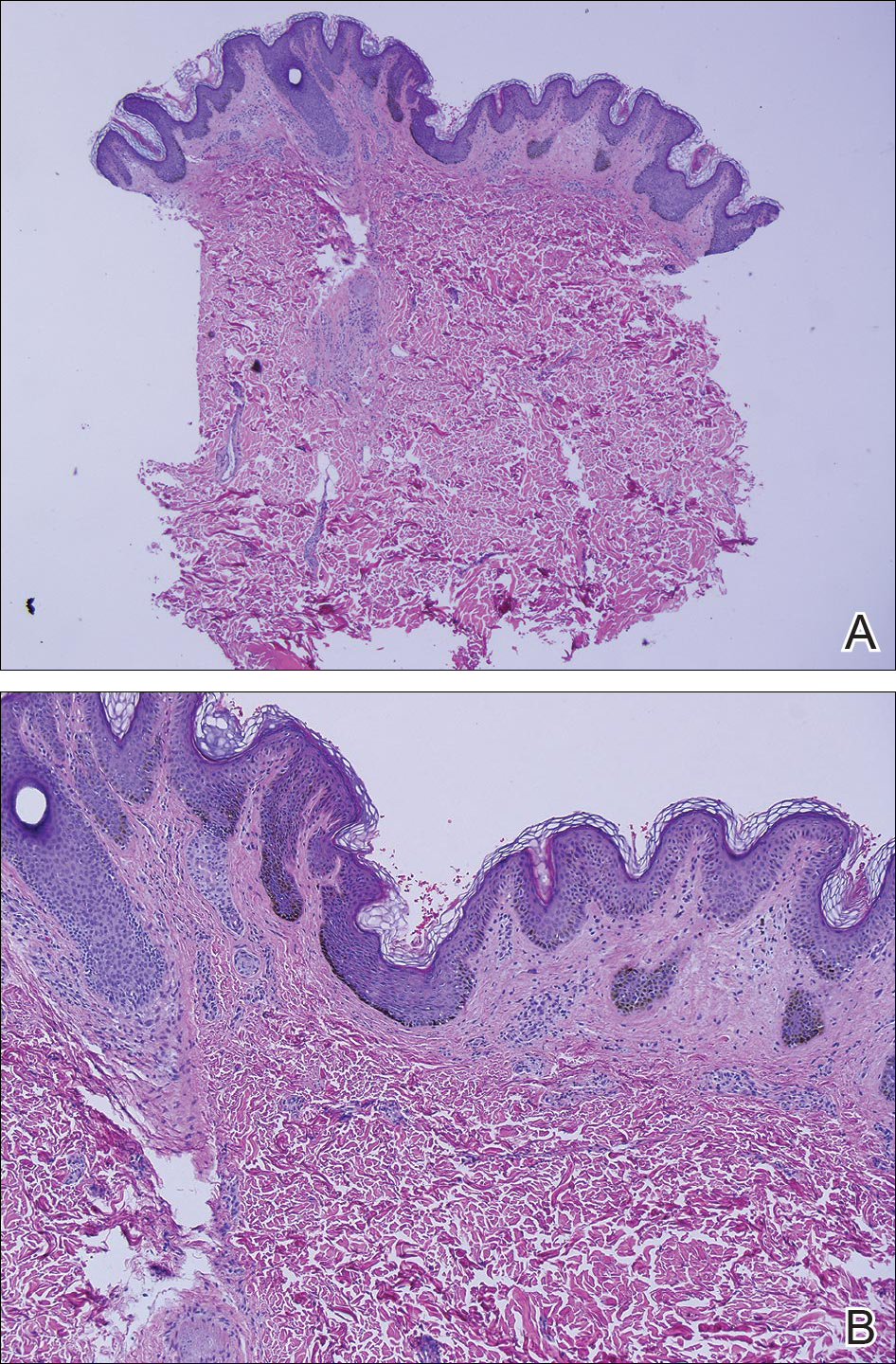
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
Epidermal nevi can occur in isolation or in association with internal abnormalities. Epidermal nevus syndrome (ENS) is a heterogeneous group of neurocutaneous disorders characterized by mosaicism and epidermal nevi found in association with various systemic abnormalities.1-4 There are many possible associated systemic findings, including abnormalities of the central nervous, musculoskeletal, renal, and hematologic systems. Epidermal nevi have been associated with internal malignancies. We present the case of a patient with epidermal nevi associated with papillary transitional cell bladder carcinoma. According to a PubMed search of articles indexed for MEDLINE using the search terms transitional cell bladder carcinoma and epidermal nevus, there have only been 4 other cases of transitional cell bladder carcinoma and ENS reported in the literature,5-8 2 of which were reports of papillary transitional cell bladder carcinoma.5,6
Case Report
A 29-year-old woman presented to our clinic with a rash that had been present since 3 years of age. The emergency department consulted dermatology for evaluation of what was believed to be contact dermatitis; however, upon questioning the patient, it was revealed that the rash was chronic and persistent.
The rash was nonpruritic and was located on the face, hands (Figure 1), chest, buttocks, thighs, legs, and back (Figure 2). Although asymptomatic, the appearance of the skin caused the patient some emotional distress. As a child she had been evaluated by a dermatologist and a biopsy was performed, but she did not recall the results or have any records. She had been prescribed an oral medication by the dermatologist, but treatment was terminated early due to nausea. The skin lesions did not improve with the short course of treatment.
Eighteen months prior to presentation to our clinic, the patient was discovered to have hematuria on routine examination by her primary care physician. At that time, the patient underwent a workup for hematuria and a mass was discovered in the bladder via cystoscopy. A diagnosis of low-grade papillary transitional cell bladder carcinoma was made, and she underwent a partial cystectomy. No radiation or chemotherapy was required. The remainder of her medical history was only remarkable for asthma, which was well controlled with albuterol. On examination, generalized, hyperpigmented, reticulated patches, macules, and hyperpigmented verrucous plaques were distributed along the Blaschko lines, sparing the face. No limb abnormalities or dental or nail abnormalities were noted. Examination of the axillary and cervical lymph nodes was unremarkable, and no neurological abnormalities were noted. A 3-mm punch biopsy of the mid upper back was performed. Histopathology revealed papillomatous, nonorganoid, nonepidermolytic hyperplasia of the epidermis with elongated rete ridges (Figure 3), which was diagnosed as a nonorganoid nonepidermolytic epidermal nevus.
Comment
Epidermal nevus syndrome is a group of disorders characterized by both local or systematized epidermal nevi and systemic findings. Solomon et al4 first coined the term epidermal nevus syndrome more than 40 years ago; however, since then there has been confusion about how to define ENS. Epidermal nevus syndrome has been considered an umbrella term that includes more specific syndromes involving epidermal nevi, such as Proteus syndrome and Schimmelpenning syndrome; conversely, it also has been considered a term for those who do not meet the criteria for more specific syndromes.1,9 Happle1 discussed that the genetic variations found in ENS warrant recognition. Simply put, ENS is a heterogeneous group of syndromes that are similar in that they involve epidermal nevi and internal abnormalities but are genetically distinct. The list of definitive ENSs, as suggested by Happle1 and others, will likely continue to grow.3,5
The exact pathomechanism of ENS is unknown, but the clinical presentation most likely represents a lethal disorder mitigated by mosaicism.2,9 Gene defects vary depending on the specific ENS. For instance, the phosphatase and tensin homolog gene, PTEN, mutations have been associated with type 2 segmental Cowden disease. Fibroblast growth factor receptor 3, FGFR3, mutations have been linked to Garcia-Hafner-Happle syndrome.3FGFR3 mutations have been found in nonepidermolytic epidermal nevi, and some suggest that the majority of epidermal nevi exhibit mutations in FGFR3.5,10,11 On the other hand, other gene defects have not been elucidated, such as in Schimmelpenning syndrome.3
Clinically, ENS may involve nonepidermolytic verrucous nevi, sebaceous nevi, organoid nevi, linear Cowden nevi, and woolly hair nevi. Lesions may be flesh-colored, pink, yellow, or hyperpigmented plaques in a blaschkoid distribution and may be localized or systematized. Nevi typically are present at birth or develop within the first year of life.9,12,13 Other cutaneous findings may be noted apart from epidermal nevi, including melanocytic nevi, aplasia cutis congenita, and hemangiomas.13,14
Extracutaneous findings include central nervous system, skeletal, ocular, cardiac, and genitourinary defects, which are often observed in these patients.3,9,13,14 Central nervous system findings are seen in 50% to 70% of cases, with seizures and mental retardation among the most common.13-15 Genitourinary abnormalities associated with epidermal nevi, including horseshoe kidney, cystic kidney, duplicated collecting system, testicular and paratesticular tumors, and hypospadias have been documented in the literature.16 Our patient had a history of papillary transitional cell bladder carcinoma, which is rare for a patient younger than 30 years. The overall median age of diagnosis of bladder cancer is 65 years, and it is more common in men than in women.17 Transitional cell carcinomas account for approximately 90% of all bladder cancers in the United States. Other common types of bladder cancer include squamous cell carcinoma, adenocarcinoma, and rhabdomyosarcoma.16 Typically, transitional cell carcinoma is associated with smoking, exposure to aniline dyes, cyclophosphamide, and living in industrialized areas.16,17 Individuals who work with textiles, dyes, leather, tires, rubber, and/or petroleum; painters; truck drivers; drill press operators; and hairdressers are at an increased risk for development of bladder cancer.16
Interestingly, it has been shown in some studies that papillary transitional cell bladder carcinoma frequently is associated with FGFR3 mutations, which may be the missing link in the rare finding of papillary transitional cell bladder carcinoma and epidermal nevi.5,18,19 In addition, PTEN mutations also have been identified in low-grade papillary transitional cell carcinomas of the bladder, another gene linked to an ENS with type 2 segmental Cowden disease.3,20
Histopathologically, epidermal nevi have 10 different descriptions. Our patient had a nonorganoid nonepidermolytic epidermal nevus characterized by hyperkeratosis, acanthosis, papillomatosis, and elongated rete ridges. Focal acantholysis and epidermolytic hyperkeratosis also is seen in some epidermal nevi but was not seen in this case.9,21
Simple epidermal nevi occur in approximately 1 in 1000 newborns; however, when a child presents with multiple or systematized epidermal nevi, investigation should be undertaken for other possible associations.13,14 Of note, there have been several cases of squamous cell, verrucous, basal cell, and adnexal carcinomas arising in linear epidermal nevi.22-24
Epidermal nevi can be difficult to treat. Some patients are troubled by the appearance of these nevi, especially those with systematized disease. Unfortunately, for patients with multiple nevi or systematized disease, there are no consistently effective treatment options; however, there are case reports25,26 in the literature citing improvement or cure of epidermal nevi with full-thickness excision, continuous and pulsed CO2 laser, pulsed dye laser, and erbium-doped YAG laser.25 Other therapies that have been purported to help improve epidermal nevi are topical and oral retinoids, corticosteroids, topical 5-fluorouracil, anthralin, and podophyllin.26
Conclusion
Transitional cell bladder carcinoma is rare in patients in the third decade of life and younger. Given the age of our patient and her concomitant lack of risk factors, such as older age, history of smoking, and exposure to certain chemicals (eg, aniline dyes) and medications (eg, cyclophosphamide), it is more likely that the finding of papillary transitional cell bladder carcinoma and ENS are related. A clear genetic link between ENS and transitional cell papillary bladder carcinoma has yet to be elucidated, but the FGFR3 gene is promising.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
- Happle R. What is a nevus? a proposed definition of a common medical term. Dermatology. 1995;191:1-5.
- Gonzalez ME, Jabbari A, Tlougan BE, et al. Epidermal nevus. Dermatol Online J. 2010;16:12.
- Happle R. The group of epidermal nevus syndromes. part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22.
- Solomon LM, Fretzin DF, Dewald RL. The epidermal nevus syndrome. Arch Dermatol. 1968;97:273-285.
- Flosadottir E, Bjarnason B. A non-epidermolytic epidermal nevus of a soft, papillomatous type with transitional cell cancer of the bladder: a case report and review of non-cutaneous cancers associated with epidermal naevi. Acta Derm Venerol. 2008;88:173-175.
- Rosenthal D, Fretzin DF. Epidermal nevus syndrome: report of association with transitional cell carcinoma of the bladder. Pediatr Dermatol. 1986;3:455-458.
- Garcia de Jalon A, Azua-Romea J, Trivez MA, et al. Epidermal naevus syndrome (Solomon’s syndrome) associated with bladder cancer in a 20-year-old female. Scand J Urol Nephrol. 2004;38:85-87.
- Rongioletti F, Rebora A. Epidermal nevus with transitional cell carcinomas of the urinary tract. J Am Acad Dermatol. 1991;25:856-858.
- Moss C. Mosacism and linear lesions. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:943-962.
- Hafner C, van Oers JM, Vogt T, et al. Mosaicisim of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201-2207.
- Bygum A, Fagerberg CR, Clemmensen OJ, et al. Systemic epidermal nevus with involvement of the oral mucosa due to FGFR3 mutation. BMC Med Genet. 2011;12:79.
- Happle R. Linear Cowden nevus: a new distinct epidermal nevus. Eur J Dermatol. 2007;17:133-136.
- Vujevich JJ, Mancini AJ. The epidermal nevus syndromes: multisystem disorders. J Am Acad Dermatol. 2004;50:957-961.
- Solomon L, Esterly N. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6:1-56.
- Grebe TA, Rimsa ME, Richter SF, et al. Further delineation of the epidermal nevus syndrome: two cases with new findings and literature review. Am J Med Genet. 1993;47:24-30.
- Lamm DL, Torti FM. Bladder cancer, 1996. Ca Cancer J Clin. 1996;46:93-112.
- Metts MC, Metts JC, Milito SJ, et al. Bladder cancer: a review of diagnosis and management. J Natl Med Assoc. 2000;92:285-294.
- Kimura T, Suzuki H, Ohashi T, et al. The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas. Cancer. 2001;92:2555-2561.
- Cappellen D, DeOliveira C, Ricol D, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18-20.
- Knowles MA, Platt FM, Ross RL, et al. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28:305-316.
- Luzar B, Calonje E, Bastian B. Tumors of the surface epithelium. In: Calonje JE, Breen T, McKee PH, eds. McKee’s Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012:1076-1149.
- Masood Q, Narayan D. Squamous cell carcinoma in a linear epidermal nevus. J Plast Reconstr Aesthet Surg. 2009;62:693-694.
- Cramer SF, Mandel MA, Hauler R, et al. Squamous cell carcinoma arising in a linear epidermal nevus. Arch Dermatol. 1981;117:222-224.
- Affleck AG, Leach IJ, Varma S. Two squamous cell carcinomas arising in a linear epidermal nevus in a 28-year-old female. Clin Exp Dermatol. 2005;30:382-384.
- Alam M, Arndt KA. A method for pulsed carbon dioxide laser treatment of epidermal nevi. J Am Acad Dermatol. 2002;46:554-556.
- Requena L, Requena C, Cockerell CJ. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012:1809-1810.
Practice Points
- Epidermal nevi are common benign cutaneous neoplasms.
- Extensive systematized epidermal nevi can be a sign of internal disease.
Cutaneous Adnexal Carcinoma With Apocrine Differentiation
Differentiation between a primary adnexal carcinoma and a metastatic carcinoma to the skin is a challenging yet critical task for dermatologists and pathologists. Carcinomas that have metastasized to the skin are a sign of widespread systemic involvement and poor prognosis, while primary adnexal carcinomas tend to progress with an indolent clinical course. Although many patients with cutaneous metastases from an internal primary neoplasm can expect a median survival of no more than 12 months,1 patients with primary adnexal carcinomas are reported to have a 5-year survival rate of 95.5% for localized disease and 85% with spread to regional lymph nodes.2 We report a case of multiple cutaneous neoplasms of unknown primary origin in a 71-year-old man and describe our approach to identification of the possible primary site as well as management of the disease.
Case Report
A 71-year-old man initially presented to his primary physician for evaluation of a mass on the left side of the neck of 3 months' duration. On physical examination, a firm 2.5×3.0-cm nodule was noted at the anterior border of the trapezius muscle. Palpation of the thyroid revealed an additional right-sided nodule. The submandibular and parotid glands were unremarkable to palpation. The patient was referred to general surgery for biopsy, which revealed an infiltrating, moderately differentiated adenocarcinoma with extensive lymphatic permeation. Immunohistochemical staining for cytokeratin (CK) 7 was positive, while CK20 and thyroid transcription factor 1 were negative. A positron emission tomography/computed tomography (CT) fusion scan demonstrated 3 areas of enhanced uptake: one in the right side of the thyroid, a second corresponding to the mass on the left side of the neck at the level of the trapezius muscle, and a third in the left masseter muscle. Surgical excision with negative margins with possible chemotherapy was recommended; however, the patient declined treatment and was lost to follow-up until 2 years later when he presented to his primary physician with an additional lesion on his scalp.
Four years after the biopsy, the patient presented to the dermatology department with additional tumor nodules including a 4-cm, annular, indurated, focally eroded plaque on the left side of the lateral neck (Figure 1); 3 separate 1-cm nodules on the right side of the lateral neck; and an ulcerated, crusted, 10×8-cm plaque on the posterior aspect of the scalp. Despite the extensive lesions, the patient remained in good health and reported no recent weight loss or signs or symptoms of systemic involvement. The posterior scalp lesion, which developed 2 years after the initial appearance of the mass on the neck and was thought to represent a possible metastasis of the tumor, was biopsied and showed diffuse infiltration of the dermis by poorly differentiated tumor cells with vacuolated cytoplasm arranged in nests and cords and sometimes in a single-file arrangement (Figure 2). A CT scan demonstrated pretracheal lymphadenopathy as well as small intraparenchymal and subpleural pulmonary nodules throughout both lung fields.
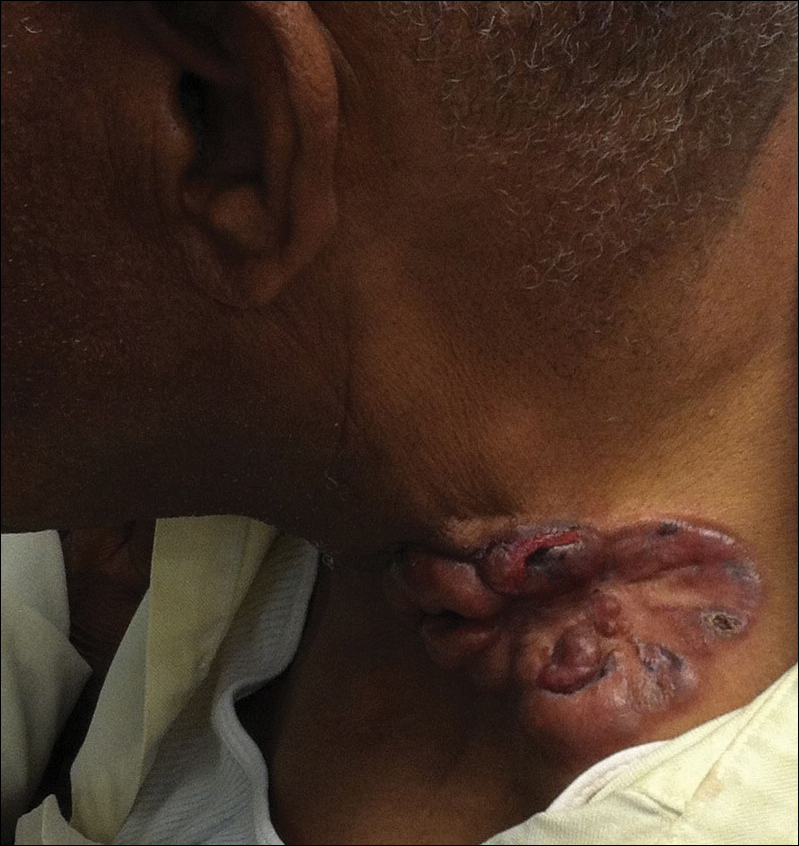
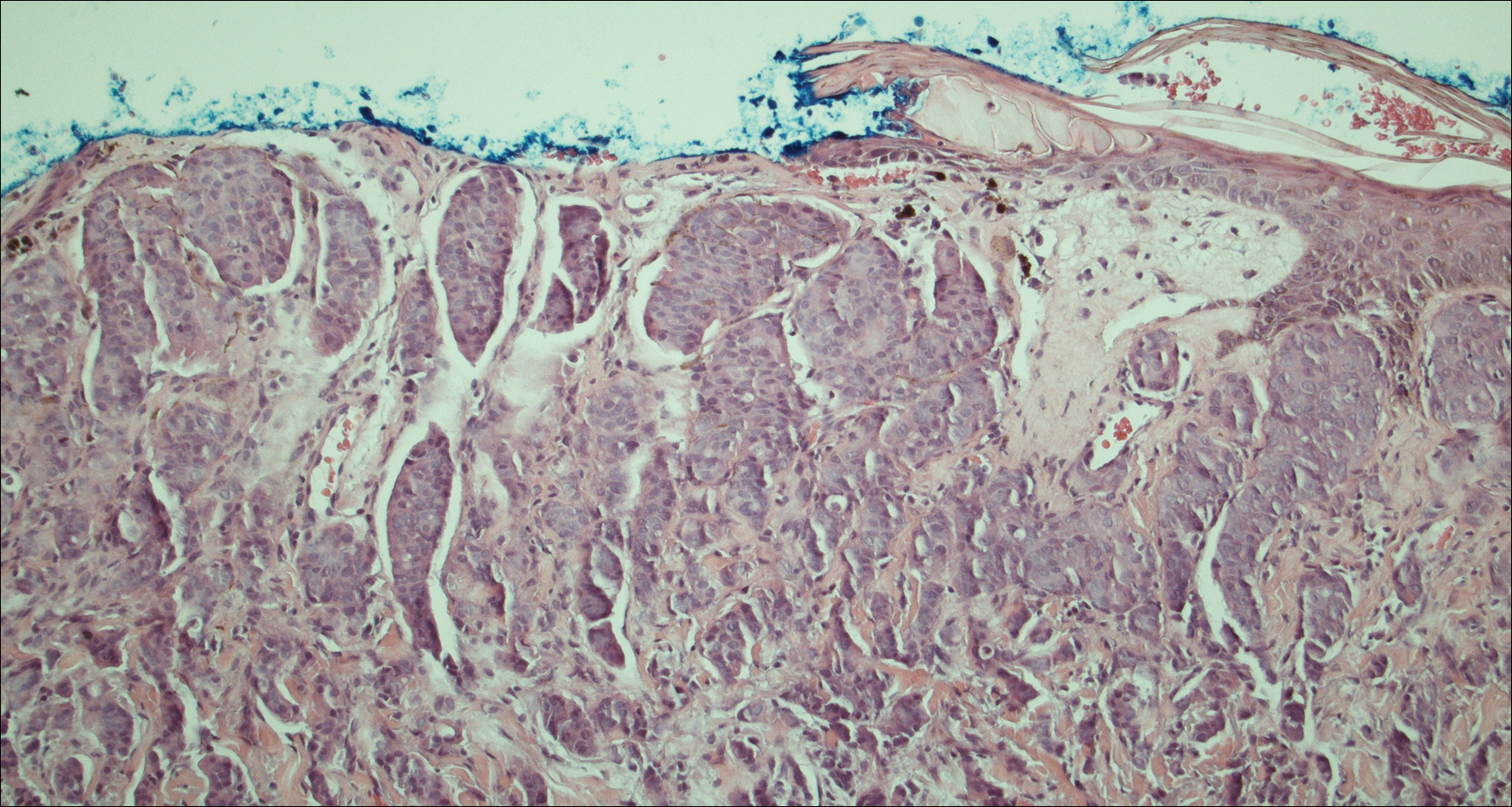
Another scalp biopsy was taken. Tumor cells were negative on mucicarmine staining. Additional immunohistochemical staining, including a periodic acid-Schiff stain with diastase digestion for epithelial mucin revealed minimal luminal positivity. Immunostaining was positive for CK7, carcinoembryonic antigen, CD15, estrogen receptor, progesterone receptor, gross cystic disease fluid protein 15 (GCDFP-15), and mammaglobin, and negative for CK20, podoplanin, thyroid transcription factor 1, S-100 protein, p63, and prostate specific antigen. ERBB2 (formerly HER2/neu) staining was negative according to fluorescence in situ hybridization analysis. Tumor cells showed a Ki-67 nuclear proliferation index of greater than 50%, indicating progression to aggressive carcinoma.
Based on the histological and immunochemical studies, the differential diagnosis included primary cutaneous apocrine carcinoma versus breast carcinoma; however, the prolonged clinical progression of these lesions favored a primary cutaneous adnexal tumor over a metastatic adenocarcinoma. Nevertheless, despite the initially indolent growth of the lesions over the first 5 years, the Ki-67 proliferation index and presence of widespread metastases on the posterior scalp indicated progression to an aggressive carcinoma. Chemotherapy was recommended as the treatment of choice. At his most recent follow-up visit 4 months later, the patient chose to begin treatment with tamoxifen and refused other treatment options.
Comment
The distinction between primary adnexal and metastatic adenocarcinomas of the skin is challenging both clinically and histologically. Some pathologists have argued that metastatic breast carcinomas and primary cutaneous apocrine carcinomas are essentially indistinguishable.3 Patients with cutaneous metastases, which occur in approximately 5.3% of all malignancies,4 typically can expect survival of no more than 12 months from the time of detection.1 In contrast, primary apocrine carcinomas of the skin, though much less common, carry a remarkably better prognosis, with 5-year relative survival rates of 95.5% and 85.5% reported for patients with localized disease and spread to regional lymph nodes, respectively.2
Fewer than 100 cases of primary cutaneous adnexal (apocrine) carcinomas have been reported overall, with the earliest known report dating back to 1944.5 According to the literature, primary apocrine carcinomas were diagnosed at a median age of 66 years and were slightly more common in females than males.2,6 Apocrine carcinomas were seen most frequently on the head, neck, and trunk,2 generally presenting in the form of asymptomatic nodules or plaques of 2 to 3 cm in size, with gradual progression occurring over months to years.6 Approximately 40% of patients have been reported with positive regional lymph nodes at diagnosis. Treatment of apocrine carcinoma typically has involved local excision with clear margins with or without lymph node dissection. Chemotherapy and radiation therapy have shown no proven benefit.7
Currently, there is no standardized approach to evaluating patients with possible cutaneous metastasis versus primary cutaneous adnexal carcinomas. Imaging studies such as mammography and abdominal CT typically reveal an internal primary cancer in one-third of patients. However, additional studies such as gastrointestinal radiography, chest and pelvic CT, barium enema, and intravenous pyelogram have shown to be of limited value.8 Although specificity and sensitivity of immunohistochemistry is limited, a number of immunomarkers, including CK7 and CK20, are routinely studied to narrow the differential diagnosis of a cutaneous neoplasm of unclear origin. Urothelial, gastric, colorectal, and pancreatic carcinomas generally are positive for CK20; CK7-positive adenocarcinomas include salivary, non-small cell lung, breast, ovarian, pancreatic, endometrial, and transitional cell adenocarcinomas. Carcinomas negative for both CK7 and CK20 include colorectal, hepatocellular, renal cell, prostate, and squamous cell carcinoma of the lung.
The presence of positive staining for estrogen and progesterone receptors as well as GCDFP-15 and mammaglobin raised the possibility of primary breast adenocarcinoma in our patient, but given that these markers can be positive in primary cutaneous adnexal tumors, immunohistochemistry results were not able to provide a definitive primary site. The overall staining pattern was nearly identical to 26 cases of primary cutaneous cribriform apocrine carcinoma, which was found to be positive for CK7 and carcinoembryonic antigen, and negative for CK20 and S-100. The only difference was in GCDFP-15 staining, which was positive in our case and negative in the cases of cribriform apocrine carcinoma.9 Histologic features favoring a primary apocrine origin include normal apocrine glands in the vicinity, glandular structures with decapitation secretion high in the dermis, and intracytoplasmic iron granules.10 Additionally, positive estrogen receptor staining appears to be much more common in apocrine carcinomas (5/10) than in eccrine carcinomas (1/7).11
A number of other markers have been investigated for possible diagnostic utility for distinction between primary adnexal carcinomas and metastatic adenocarcinomas. The nuclear transcription factor p63, which plays a role in keratinocyte differentiation, is preferentially expressed in a number of primary adnexal carcinomas and is purported to be the most sensitive marker overall, with a sensitivity of 78% to 91%.12-14 However, p63 has shown incomplete specificity for primary adnexal neoplasms, having been reported as positive in 11% to 22% of adenocarcinomas metastatic to skin.15-18 Nestin and CK15, which are expressed in hair follicle progenitor cells, also are potential specific markers for some primary adnexal lesions, specifically eccrine carcinoma, porocarcinoma, hidradenocarcinoma, and microcystic adnexal carcinoma; however, in one report, none of the apocrine carcinomas were positive for p63, cytokeratin 15, or D2-40.19 Thus, while markers for some primary adnexal neoplasms are emerging, specific tests at the immunohistochemical level for the apocrine carcinoma subgroup are still lacking.
Conclusion
In summary, a conclusive distinction between primary cutaneous apocrine carcinoma and metastatic adenocarcinoma to the skin remains challenging. Although new markers provide more specificity and sensitivity for neoplasms of eccrine origin, these markers do not appear to differentiate between primary apocrine carcinoma and metastatic breast carcinoma. In this case, as in other recent reports, diagnosis remained dependent on the clinical course of the patient. Although considerable progress has been made regarding immunohistochemical analysis of these cases, additional markers, especially ones more specific for primary skin cancers with apocrine differentiation, are still needed.
- Nashan D, Müller ML, Braun-Falco M, et al. Cutaneous metastases of visceral tumours: a review. J Cancer Res Clin Oncol. 2009;135:1-14.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:141-142.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Horn RC. Malignant papillary cystadenoma of sweat glands with metastases to the regional lymph nodes. Surgery. 1944;16:348-355.
- Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
- Vasilakaki T, Skafida E, Moustou E, et al. Primary cutaneous apocrine carcinoma of sweat glands: a rare case report [published online December 17, 2011]. Case Rep Oncol. 2011;4:597-601.
- Hainsworth JD, Greco FA. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257-263.
- Rutten A, Kutzner H, Mentzel T, et al. Primary cutaneous cribriform apocrine carcinoma: a clinicopathologic and immunohistochemical study of 26 cases of an under-recognized cutaneous adnexal neoplasm. J Am Acad Dermatol. 2009;61:644-651.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever's Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2009.
- Le LP, Dias-Santagata D, Pawlak AC, et al. Apocrine-eccrine carcinomas: molecular and immunohistochemical analyses. PLoS One. 2012;7:e47290.
- Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113:1661-1670.
- Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156-3161.
- Reis-Filho JS, Torio B, Albergaria A, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517-523.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Mahalingam M, Nguyen LP, Richards JE, et al. The diagnostic utility of immunohistochemistry in distinguishing primary skin adnexal carcinomas from metastatic adenocarcinoma to skin: an immunohistochemical reappraisal using cytokeratin 15, nestin, p63, D2-40, and calretinin. Mod Pathol. 2010;23:713-719.
Differentiation between a primary adnexal carcinoma and a metastatic carcinoma to the skin is a challenging yet critical task for dermatologists and pathologists. Carcinomas that have metastasized to the skin are a sign of widespread systemic involvement and poor prognosis, while primary adnexal carcinomas tend to progress with an indolent clinical course. Although many patients with cutaneous metastases from an internal primary neoplasm can expect a median survival of no more than 12 months,1 patients with primary adnexal carcinomas are reported to have a 5-year survival rate of 95.5% for localized disease and 85% with spread to regional lymph nodes.2 We report a case of multiple cutaneous neoplasms of unknown primary origin in a 71-year-old man and describe our approach to identification of the possible primary site as well as management of the disease.
Case Report
A 71-year-old man initially presented to his primary physician for evaluation of a mass on the left side of the neck of 3 months' duration. On physical examination, a firm 2.5×3.0-cm nodule was noted at the anterior border of the trapezius muscle. Palpation of the thyroid revealed an additional right-sided nodule. The submandibular and parotid glands were unremarkable to palpation. The patient was referred to general surgery for biopsy, which revealed an infiltrating, moderately differentiated adenocarcinoma with extensive lymphatic permeation. Immunohistochemical staining for cytokeratin (CK) 7 was positive, while CK20 and thyroid transcription factor 1 were negative. A positron emission tomography/computed tomography (CT) fusion scan demonstrated 3 areas of enhanced uptake: one in the right side of the thyroid, a second corresponding to the mass on the left side of the neck at the level of the trapezius muscle, and a third in the left masseter muscle. Surgical excision with negative margins with possible chemotherapy was recommended; however, the patient declined treatment and was lost to follow-up until 2 years later when he presented to his primary physician with an additional lesion on his scalp.
Four years after the biopsy, the patient presented to the dermatology department with additional tumor nodules including a 4-cm, annular, indurated, focally eroded plaque on the left side of the lateral neck (Figure 1); 3 separate 1-cm nodules on the right side of the lateral neck; and an ulcerated, crusted, 10×8-cm plaque on the posterior aspect of the scalp. Despite the extensive lesions, the patient remained in good health and reported no recent weight loss or signs or symptoms of systemic involvement. The posterior scalp lesion, which developed 2 years after the initial appearance of the mass on the neck and was thought to represent a possible metastasis of the tumor, was biopsied and showed diffuse infiltration of the dermis by poorly differentiated tumor cells with vacuolated cytoplasm arranged in nests and cords and sometimes in a single-file arrangement (Figure 2). A CT scan demonstrated pretracheal lymphadenopathy as well as small intraparenchymal and subpleural pulmonary nodules throughout both lung fields.
Another scalp biopsy was taken. Tumor cells were negative on mucicarmine staining. Additional immunohistochemical staining, including a periodic acid-Schiff stain with diastase digestion for epithelial mucin revealed minimal luminal positivity. Immunostaining was positive for CK7, carcinoembryonic antigen, CD15, estrogen receptor, progesterone receptor, gross cystic disease fluid protein 15 (GCDFP-15), and mammaglobin, and negative for CK20, podoplanin, thyroid transcription factor 1, S-100 protein, p63, and prostate specific antigen. ERBB2 (formerly HER2/neu) staining was negative according to fluorescence in situ hybridization analysis. Tumor cells showed a Ki-67 nuclear proliferation index of greater than 50%, indicating progression to aggressive carcinoma.
Based on the histological and immunochemical studies, the differential diagnosis included primary cutaneous apocrine carcinoma versus breast carcinoma; however, the prolonged clinical progression of these lesions favored a primary cutaneous adnexal tumor over a metastatic adenocarcinoma. Nevertheless, despite the initially indolent growth of the lesions over the first 5 years, the Ki-67 proliferation index and presence of widespread metastases on the posterior scalp indicated progression to an aggressive carcinoma. Chemotherapy was recommended as the treatment of choice. At his most recent follow-up visit 4 months later, the patient chose to begin treatment with tamoxifen and refused other treatment options.
Comment
The distinction between primary adnexal and metastatic adenocarcinomas of the skin is challenging both clinically and histologically. Some pathologists have argued that metastatic breast carcinomas and primary cutaneous apocrine carcinomas are essentially indistinguishable.3 Patients with cutaneous metastases, which occur in approximately 5.3% of all malignancies,4 typically can expect survival of no more than 12 months from the time of detection.1 In contrast, primary apocrine carcinomas of the skin, though much less common, carry a remarkably better prognosis, with 5-year relative survival rates of 95.5% and 85.5% reported for patients with localized disease and spread to regional lymph nodes, respectively.2
Fewer than 100 cases of primary cutaneous adnexal (apocrine) carcinomas have been reported overall, with the earliest known report dating back to 1944.5 According to the literature, primary apocrine carcinomas were diagnosed at a median age of 66 years and were slightly more common in females than males.2,6 Apocrine carcinomas were seen most frequently on the head, neck, and trunk,2 generally presenting in the form of asymptomatic nodules or plaques of 2 to 3 cm in size, with gradual progression occurring over months to years.6 Approximately 40% of patients have been reported with positive regional lymph nodes at diagnosis. Treatment of apocrine carcinoma typically has involved local excision with clear margins with or without lymph node dissection. Chemotherapy and radiation therapy have shown no proven benefit.7
Currently, there is no standardized approach to evaluating patients with possible cutaneous metastasis versus primary cutaneous adnexal carcinomas. Imaging studies such as mammography and abdominal CT typically reveal an internal primary cancer in one-third of patients. However, additional studies such as gastrointestinal radiography, chest and pelvic CT, barium enema, and intravenous pyelogram have shown to be of limited value.8 Although specificity and sensitivity of immunohistochemistry is limited, a number of immunomarkers, including CK7 and CK20, are routinely studied to narrow the differential diagnosis of a cutaneous neoplasm of unclear origin. Urothelial, gastric, colorectal, and pancreatic carcinomas generally are positive for CK20; CK7-positive adenocarcinomas include salivary, non-small cell lung, breast, ovarian, pancreatic, endometrial, and transitional cell adenocarcinomas. Carcinomas negative for both CK7 and CK20 include colorectal, hepatocellular, renal cell, prostate, and squamous cell carcinoma of the lung.
The presence of positive staining for estrogen and progesterone receptors as well as GCDFP-15 and mammaglobin raised the possibility of primary breast adenocarcinoma in our patient, but given that these markers can be positive in primary cutaneous adnexal tumors, immunohistochemistry results were not able to provide a definitive primary site. The overall staining pattern was nearly identical to 26 cases of primary cutaneous cribriform apocrine carcinoma, which was found to be positive for CK7 and carcinoembryonic antigen, and negative for CK20 and S-100. The only difference was in GCDFP-15 staining, which was positive in our case and negative in the cases of cribriform apocrine carcinoma.9 Histologic features favoring a primary apocrine origin include normal apocrine glands in the vicinity, glandular structures with decapitation secretion high in the dermis, and intracytoplasmic iron granules.10 Additionally, positive estrogen receptor staining appears to be much more common in apocrine carcinomas (5/10) than in eccrine carcinomas (1/7).11
A number of other markers have been investigated for possible diagnostic utility for distinction between primary adnexal carcinomas and metastatic adenocarcinomas. The nuclear transcription factor p63, which plays a role in keratinocyte differentiation, is preferentially expressed in a number of primary adnexal carcinomas and is purported to be the most sensitive marker overall, with a sensitivity of 78% to 91%.12-14 However, p63 has shown incomplete specificity for primary adnexal neoplasms, having been reported as positive in 11% to 22% of adenocarcinomas metastatic to skin.15-18 Nestin and CK15, which are expressed in hair follicle progenitor cells, also are potential specific markers for some primary adnexal lesions, specifically eccrine carcinoma, porocarcinoma, hidradenocarcinoma, and microcystic adnexal carcinoma; however, in one report, none of the apocrine carcinomas were positive for p63, cytokeratin 15, or D2-40.19 Thus, while markers for some primary adnexal neoplasms are emerging, specific tests at the immunohistochemical level for the apocrine carcinoma subgroup are still lacking.
Conclusion
In summary, a conclusive distinction between primary cutaneous apocrine carcinoma and metastatic adenocarcinoma to the skin remains challenging. Although new markers provide more specificity and sensitivity for neoplasms of eccrine origin, these markers do not appear to differentiate between primary apocrine carcinoma and metastatic breast carcinoma. In this case, as in other recent reports, diagnosis remained dependent on the clinical course of the patient. Although considerable progress has been made regarding immunohistochemical analysis of these cases, additional markers, especially ones more specific for primary skin cancers with apocrine differentiation, are still needed.
Differentiation between a primary adnexal carcinoma and a metastatic carcinoma to the skin is a challenging yet critical task for dermatologists and pathologists. Carcinomas that have metastasized to the skin are a sign of widespread systemic involvement and poor prognosis, while primary adnexal carcinomas tend to progress with an indolent clinical course. Although many patients with cutaneous metastases from an internal primary neoplasm can expect a median survival of no more than 12 months,1 patients with primary adnexal carcinomas are reported to have a 5-year survival rate of 95.5% for localized disease and 85% with spread to regional lymph nodes.2 We report a case of multiple cutaneous neoplasms of unknown primary origin in a 71-year-old man and describe our approach to identification of the possible primary site as well as management of the disease.
Case Report
A 71-year-old man initially presented to his primary physician for evaluation of a mass on the left side of the neck of 3 months' duration. On physical examination, a firm 2.5×3.0-cm nodule was noted at the anterior border of the trapezius muscle. Palpation of the thyroid revealed an additional right-sided nodule. The submandibular and parotid glands were unremarkable to palpation. The patient was referred to general surgery for biopsy, which revealed an infiltrating, moderately differentiated adenocarcinoma with extensive lymphatic permeation. Immunohistochemical staining for cytokeratin (CK) 7 was positive, while CK20 and thyroid transcription factor 1 were negative. A positron emission tomography/computed tomography (CT) fusion scan demonstrated 3 areas of enhanced uptake: one in the right side of the thyroid, a second corresponding to the mass on the left side of the neck at the level of the trapezius muscle, and a third in the left masseter muscle. Surgical excision with negative margins with possible chemotherapy was recommended; however, the patient declined treatment and was lost to follow-up until 2 years later when he presented to his primary physician with an additional lesion on his scalp.
Four years after the biopsy, the patient presented to the dermatology department with additional tumor nodules including a 4-cm, annular, indurated, focally eroded plaque on the left side of the lateral neck (Figure 1); 3 separate 1-cm nodules on the right side of the lateral neck; and an ulcerated, crusted, 10×8-cm plaque on the posterior aspect of the scalp. Despite the extensive lesions, the patient remained in good health and reported no recent weight loss or signs or symptoms of systemic involvement. The posterior scalp lesion, which developed 2 years after the initial appearance of the mass on the neck and was thought to represent a possible metastasis of the tumor, was biopsied and showed diffuse infiltration of the dermis by poorly differentiated tumor cells with vacuolated cytoplasm arranged in nests and cords and sometimes in a single-file arrangement (Figure 2). A CT scan demonstrated pretracheal lymphadenopathy as well as small intraparenchymal and subpleural pulmonary nodules throughout both lung fields.
Another scalp biopsy was taken. Tumor cells were negative on mucicarmine staining. Additional immunohistochemical staining, including a periodic acid-Schiff stain with diastase digestion for epithelial mucin revealed minimal luminal positivity. Immunostaining was positive for CK7, carcinoembryonic antigen, CD15, estrogen receptor, progesterone receptor, gross cystic disease fluid protein 15 (GCDFP-15), and mammaglobin, and negative for CK20, podoplanin, thyroid transcription factor 1, S-100 protein, p63, and prostate specific antigen. ERBB2 (formerly HER2/neu) staining was negative according to fluorescence in situ hybridization analysis. Tumor cells showed a Ki-67 nuclear proliferation index of greater than 50%, indicating progression to aggressive carcinoma.
Based on the histological and immunochemical studies, the differential diagnosis included primary cutaneous apocrine carcinoma versus breast carcinoma; however, the prolonged clinical progression of these lesions favored a primary cutaneous adnexal tumor over a metastatic adenocarcinoma. Nevertheless, despite the initially indolent growth of the lesions over the first 5 years, the Ki-67 proliferation index and presence of widespread metastases on the posterior scalp indicated progression to an aggressive carcinoma. Chemotherapy was recommended as the treatment of choice. At his most recent follow-up visit 4 months later, the patient chose to begin treatment with tamoxifen and refused other treatment options.
Comment
The distinction between primary adnexal and metastatic adenocarcinomas of the skin is challenging both clinically and histologically. Some pathologists have argued that metastatic breast carcinomas and primary cutaneous apocrine carcinomas are essentially indistinguishable.3 Patients with cutaneous metastases, which occur in approximately 5.3% of all malignancies,4 typically can expect survival of no more than 12 months from the time of detection.1 In contrast, primary apocrine carcinomas of the skin, though much less common, carry a remarkably better prognosis, with 5-year relative survival rates of 95.5% and 85.5% reported for patients with localized disease and spread to regional lymph nodes, respectively.2
Fewer than 100 cases of primary cutaneous adnexal (apocrine) carcinomas have been reported overall, with the earliest known report dating back to 1944.5 According to the literature, primary apocrine carcinomas were diagnosed at a median age of 66 years and were slightly more common in females than males.2,6 Apocrine carcinomas were seen most frequently on the head, neck, and trunk,2 generally presenting in the form of asymptomatic nodules or plaques of 2 to 3 cm in size, with gradual progression occurring over months to years.6 Approximately 40% of patients have been reported with positive regional lymph nodes at diagnosis. Treatment of apocrine carcinoma typically has involved local excision with clear margins with or without lymph node dissection. Chemotherapy and radiation therapy have shown no proven benefit.7
Currently, there is no standardized approach to evaluating patients with possible cutaneous metastasis versus primary cutaneous adnexal carcinomas. Imaging studies such as mammography and abdominal CT typically reveal an internal primary cancer in one-third of patients. However, additional studies such as gastrointestinal radiography, chest and pelvic CT, barium enema, and intravenous pyelogram have shown to be of limited value.8 Although specificity and sensitivity of immunohistochemistry is limited, a number of immunomarkers, including CK7 and CK20, are routinely studied to narrow the differential diagnosis of a cutaneous neoplasm of unclear origin. Urothelial, gastric, colorectal, and pancreatic carcinomas generally are positive for CK20; CK7-positive adenocarcinomas include salivary, non-small cell lung, breast, ovarian, pancreatic, endometrial, and transitional cell adenocarcinomas. Carcinomas negative for both CK7 and CK20 include colorectal, hepatocellular, renal cell, prostate, and squamous cell carcinoma of the lung.
The presence of positive staining for estrogen and progesterone receptors as well as GCDFP-15 and mammaglobin raised the possibility of primary breast adenocarcinoma in our patient, but given that these markers can be positive in primary cutaneous adnexal tumors, immunohistochemistry results were not able to provide a definitive primary site. The overall staining pattern was nearly identical to 26 cases of primary cutaneous cribriform apocrine carcinoma, which was found to be positive for CK7 and carcinoembryonic antigen, and negative for CK20 and S-100. The only difference was in GCDFP-15 staining, which was positive in our case and negative in the cases of cribriform apocrine carcinoma.9 Histologic features favoring a primary apocrine origin include normal apocrine glands in the vicinity, glandular structures with decapitation secretion high in the dermis, and intracytoplasmic iron granules.10 Additionally, positive estrogen receptor staining appears to be much more common in apocrine carcinomas (5/10) than in eccrine carcinomas (1/7).11
A number of other markers have been investigated for possible diagnostic utility for distinction between primary adnexal carcinomas and metastatic adenocarcinomas. The nuclear transcription factor p63, which plays a role in keratinocyte differentiation, is preferentially expressed in a number of primary adnexal carcinomas and is purported to be the most sensitive marker overall, with a sensitivity of 78% to 91%.12-14 However, p63 has shown incomplete specificity for primary adnexal neoplasms, having been reported as positive in 11% to 22% of adenocarcinomas metastatic to skin.15-18 Nestin and CK15, which are expressed in hair follicle progenitor cells, also are potential specific markers for some primary adnexal lesions, specifically eccrine carcinoma, porocarcinoma, hidradenocarcinoma, and microcystic adnexal carcinoma; however, in one report, none of the apocrine carcinomas were positive for p63, cytokeratin 15, or D2-40.19 Thus, while markers for some primary adnexal neoplasms are emerging, specific tests at the immunohistochemical level for the apocrine carcinoma subgroup are still lacking.
Conclusion
In summary, a conclusive distinction between primary cutaneous apocrine carcinoma and metastatic adenocarcinoma to the skin remains challenging. Although new markers provide more specificity and sensitivity for neoplasms of eccrine origin, these markers do not appear to differentiate between primary apocrine carcinoma and metastatic breast carcinoma. In this case, as in other recent reports, diagnosis remained dependent on the clinical course of the patient. Although considerable progress has been made regarding immunohistochemical analysis of these cases, additional markers, especially ones more specific for primary skin cancers with apocrine differentiation, are still needed.
- Nashan D, Müller ML, Braun-Falco M, et al. Cutaneous metastases of visceral tumours: a review. J Cancer Res Clin Oncol. 2009;135:1-14.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:141-142.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Horn RC. Malignant papillary cystadenoma of sweat glands with metastases to the regional lymph nodes. Surgery. 1944;16:348-355.
- Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
- Vasilakaki T, Skafida E, Moustou E, et al. Primary cutaneous apocrine carcinoma of sweat glands: a rare case report [published online December 17, 2011]. Case Rep Oncol. 2011;4:597-601.
- Hainsworth JD, Greco FA. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257-263.
- Rutten A, Kutzner H, Mentzel T, et al. Primary cutaneous cribriform apocrine carcinoma: a clinicopathologic and immunohistochemical study of 26 cases of an under-recognized cutaneous adnexal neoplasm. J Am Acad Dermatol. 2009;61:644-651.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever's Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2009.
- Le LP, Dias-Santagata D, Pawlak AC, et al. Apocrine-eccrine carcinomas: molecular and immunohistochemical analyses. PLoS One. 2012;7:e47290.
- Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113:1661-1670.
- Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156-3161.
- Reis-Filho JS, Torio B, Albergaria A, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517-523.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Mahalingam M, Nguyen LP, Richards JE, et al. The diagnostic utility of immunohistochemistry in distinguishing primary skin adnexal carcinomas from metastatic adenocarcinoma to skin: an immunohistochemical reappraisal using cytokeratin 15, nestin, p63, D2-40, and calretinin. Mod Pathol. 2010;23:713-719.
- Nashan D, Müller ML, Braun-Falco M, et al. Cutaneous metastases of visceral tumours: a review. J Cancer Res Clin Oncol. 2009;135:1-14.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Fernandez-Flores A. The elusive differential diagnosis of cutaneous apocrine adenocarcinoma vs. metastasis: the current role of clinical correlation. Acta Dermatovenerol Alp Pannonica Adriat. 2009;18:141-142.
- Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7316 cancer patients. J Am Acad Dermatol. 1990;22:19-26.
- Horn RC. Malignant papillary cystadenoma of sweat glands with metastases to the regional lymph nodes. Surgery. 1944;16:348-355.
- Pucevich B, Catinchi-Jaime S, Ho J, et al. Invasive primary ductal apocrine adenocarcinoma of axilla: a case report with immunohistochemical profiling and a review of literature. Dermatol Online J. 2008;14:5.
- Vasilakaki T, Skafida E, Moustou E, et al. Primary cutaneous apocrine carcinoma of sweat glands: a rare case report [published online December 17, 2011]. Case Rep Oncol. 2011;4:597-601.
- Hainsworth JD, Greco FA. Treatment of patients with cancer of an unknown primary site. N Engl J Med. 1993;329:257-263.
- Rutten A, Kutzner H, Mentzel T, et al. Primary cutaneous cribriform apocrine carcinoma: a clinicopathologic and immunohistochemical study of 26 cases of an under-recognized cutaneous adnexal neoplasm. J Am Acad Dermatol. 2009;61:644-651.
- Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever's Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2009.
- Le LP, Dias-Santagata D, Pawlak AC, et al. Apocrine-eccrine carcinomas: molecular and immunohistochemical analyses. PLoS One. 2012;7:e47290.
- Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113:1661-1670.
- Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156-3161.
- Reis-Filho JS, Torio B, Albergaria A, et al. p63 expression in normal skin and usual cutaneous carcinomas. J Cutan Pathol. 2002;29:517-523.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Mahalingam M, Nguyen LP, Richards JE, et al. The diagnostic utility of immunohistochemistry in distinguishing primary skin adnexal carcinomas from metastatic adenocarcinoma to skin: an immunohistochemical reappraisal using cytokeratin 15, nestin, p63, D2-40, and calretinin. Mod Pathol. 2010;23:713-719.
Practice Points
- Despite advances in immunohistochemical analysis, differentiating between primary apocrine carcinoma and metastatic breast carcinoma remains largely dependent on the clinical course of the patient.
- Treatment of apocrine carcinoma typically involves local excision with clear margins with or without lymph node dissection.
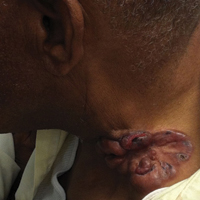
Teledermatology in Tijuana, Mexico
The Health Frontiers in Tijuana (HFiT) clinic is a binational partnership between the University of California, San Diego School of Medicine (San Diego, California); the Universidad Autónoma de Baja California School of Medicine (Tijuana, Mexico); and Desayunador Salesiano Padre Chava, a community grassroots organization in Tijuana, Mexico. Health Frontiers in Tijuana provides accessible quality health care for the underserved in Tijuana's Zona Norte.1 This article is a narrative meant to share my clinical experience as a dermatology resident who worked with HFiT to establish teledermatology services at this clinic.
Teledermatology in Tijuana
The patient population served by the HFiT clinic includes substance users, sex workers, the homeless, deportees, indigent patients, and recently Haitian immigrants.1 We established teledermatology services under the faculty leadership of Casey Carlos, MD, who was awarded a SkinCare for Developing Countries grant from the American Academy of Dermatology in April 2015 to address the need for teledermatology support for the clinic.2
Over the last 2 years, we have worked closely with 2 medical students from the University of California, San Diego--Nicole Herrick, BS, and Nicole DeMartinis, BA--to apply for the grant and create a system whereby volunteer residents and faculty consultants at the University of California, San Diego, can provide teledermatology services on a weekly basis to support the HFiT staff as they see patients with dermatologic conditions. Initially, we purchased touch screen tablets to use the Africa Teledermatology Project (africa.telederm.org) web-based program. The clinic was already functioning with electronic medical records with volunteers who carried tablets and scribed for the providers as they saw patients. We felt this method would be a great way to incorporate teledermatology into the clinic, and it functioned moderately well for several weeks but was very labor intensive on our part, as we frequently had to travel to Tijuana to retrain rotating clinic volunteers on how to use the program. Often, the Internet connection was slow, which made pulling up the Africa Teledermatology Project website difficult, and photographs also would take too long to upload in the middle of a busy clinic.
We are now exploring how to use a more simple email format to send the teledermatology consultations while still being compliant with the Health Insurance Portability and Accountability Act. We currently use secure university email accounts. Although we are still working out the details, this email-based method seems to work well. It has been a simple solution to accommodate a slow Internet connection and many rotating volunteers without requiring additional training. The email format also allows the photographs to be saved in draft messages, even if the Internet connection times out.
Once the teledermatology consultation is sent, the medical students and I review them and then get an attending physician's input on our proposed working diagnosis and plan. We work to have this process complete within several days to return the answered consultation to the requesting provider.
Final Thoughts
The HFiT providers have shared a lot of positive verbal feedback about this project. One frequent comment is how helpful it is to have access to a dermatologist for challenging cases. We also have heard many times that this project has inspired medical students and volunteers to expand their knowledge of dermatology. We are continuing to form new collaborative relationships with physicians in Tijuana. We will soon have the ability to train primary care providers at HFiT on performing simple skin biopsies and managing basic dermatologic conditions. Through our support of these providers, we are creating a sustainable partnership that is mutually beneficial to the patients in Tijuana as well as the medical students and residents in the United States. It is highly rewarding to all those involved with this project, and I am excited to see what challenges this next year will bring as we welcome many new patients from Haiti into the HFiT patient population.
- About Health Frontiers in Tijuana. University of California, San Diego School of Medicine website. https://meded.ucsd.edu/index.cfm/groups/hfit/about/. Accessed November 29, 2016.
- SkinCare for developing countries. American Academy of Dermatology website. https://www.aad.org/members/awards/skincare-for-developing-countries#undefined. Accessed November 29, 2016.
The Health Frontiers in Tijuana (HFiT) clinic is a binational partnership between the University of California, San Diego School of Medicine (San Diego, California); the Universidad Autónoma de Baja California School of Medicine (Tijuana, Mexico); and Desayunador Salesiano Padre Chava, a community grassroots organization in Tijuana, Mexico. Health Frontiers in Tijuana provides accessible quality health care for the underserved in Tijuana's Zona Norte.1 This article is a narrative meant to share my clinical experience as a dermatology resident who worked with HFiT to establish teledermatology services at this clinic.
Teledermatology in Tijuana
The patient population served by the HFiT clinic includes substance users, sex workers, the homeless, deportees, indigent patients, and recently Haitian immigrants.1 We established teledermatology services under the faculty leadership of Casey Carlos, MD, who was awarded a SkinCare for Developing Countries grant from the American Academy of Dermatology in April 2015 to address the need for teledermatology support for the clinic.2
Over the last 2 years, we have worked closely with 2 medical students from the University of California, San Diego--Nicole Herrick, BS, and Nicole DeMartinis, BA--to apply for the grant and create a system whereby volunteer residents and faculty consultants at the University of California, San Diego, can provide teledermatology services on a weekly basis to support the HFiT staff as they see patients with dermatologic conditions. Initially, we purchased touch screen tablets to use the Africa Teledermatology Project (africa.telederm.org) web-based program. The clinic was already functioning with electronic medical records with volunteers who carried tablets and scribed for the providers as they saw patients. We felt this method would be a great way to incorporate teledermatology into the clinic, and it functioned moderately well for several weeks but was very labor intensive on our part, as we frequently had to travel to Tijuana to retrain rotating clinic volunteers on how to use the program. Often, the Internet connection was slow, which made pulling up the Africa Teledermatology Project website difficult, and photographs also would take too long to upload in the middle of a busy clinic.
We are now exploring how to use a more simple email format to send the teledermatology consultations while still being compliant with the Health Insurance Portability and Accountability Act. We currently use secure university email accounts. Although we are still working out the details, this email-based method seems to work well. It has been a simple solution to accommodate a slow Internet connection and many rotating volunteers without requiring additional training. The email format also allows the photographs to be saved in draft messages, even if the Internet connection times out.
Once the teledermatology consultation is sent, the medical students and I review them and then get an attending physician's input on our proposed working diagnosis and plan. We work to have this process complete within several days to return the answered consultation to the requesting provider.
Final Thoughts
The HFiT providers have shared a lot of positive verbal feedback about this project. One frequent comment is how helpful it is to have access to a dermatologist for challenging cases. We also have heard many times that this project has inspired medical students and volunteers to expand their knowledge of dermatology. We are continuing to form new collaborative relationships with physicians in Tijuana. We will soon have the ability to train primary care providers at HFiT on performing simple skin biopsies and managing basic dermatologic conditions. Through our support of these providers, we are creating a sustainable partnership that is mutually beneficial to the patients in Tijuana as well as the medical students and residents in the United States. It is highly rewarding to all those involved with this project, and I am excited to see what challenges this next year will bring as we welcome many new patients from Haiti into the HFiT patient population.
The Health Frontiers in Tijuana (HFiT) clinic is a binational partnership between the University of California, San Diego School of Medicine (San Diego, California); the Universidad Autónoma de Baja California School of Medicine (Tijuana, Mexico); and Desayunador Salesiano Padre Chava, a community grassroots organization in Tijuana, Mexico. Health Frontiers in Tijuana provides accessible quality health care for the underserved in Tijuana's Zona Norte.1 This article is a narrative meant to share my clinical experience as a dermatology resident who worked with HFiT to establish teledermatology services at this clinic.
Teledermatology in Tijuana
The patient population served by the HFiT clinic includes substance users, sex workers, the homeless, deportees, indigent patients, and recently Haitian immigrants.1 We established teledermatology services under the faculty leadership of Casey Carlos, MD, who was awarded a SkinCare for Developing Countries grant from the American Academy of Dermatology in April 2015 to address the need for teledermatology support for the clinic.2
Over the last 2 years, we have worked closely with 2 medical students from the University of California, San Diego--Nicole Herrick, BS, and Nicole DeMartinis, BA--to apply for the grant and create a system whereby volunteer residents and faculty consultants at the University of California, San Diego, can provide teledermatology services on a weekly basis to support the HFiT staff as they see patients with dermatologic conditions. Initially, we purchased touch screen tablets to use the Africa Teledermatology Project (africa.telederm.org) web-based program. The clinic was already functioning with electronic medical records with volunteers who carried tablets and scribed for the providers as they saw patients. We felt this method would be a great way to incorporate teledermatology into the clinic, and it functioned moderately well for several weeks but was very labor intensive on our part, as we frequently had to travel to Tijuana to retrain rotating clinic volunteers on how to use the program. Often, the Internet connection was slow, which made pulling up the Africa Teledermatology Project website difficult, and photographs also would take too long to upload in the middle of a busy clinic.
We are now exploring how to use a more simple email format to send the teledermatology consultations while still being compliant with the Health Insurance Portability and Accountability Act. We currently use secure university email accounts. Although we are still working out the details, this email-based method seems to work well. It has been a simple solution to accommodate a slow Internet connection and many rotating volunteers without requiring additional training. The email format also allows the photographs to be saved in draft messages, even if the Internet connection times out.
Once the teledermatology consultation is sent, the medical students and I review them and then get an attending physician's input on our proposed working diagnosis and plan. We work to have this process complete within several days to return the answered consultation to the requesting provider.
Final Thoughts
The HFiT providers have shared a lot of positive verbal feedback about this project. One frequent comment is how helpful it is to have access to a dermatologist for challenging cases. We also have heard many times that this project has inspired medical students and volunteers to expand their knowledge of dermatology. We are continuing to form new collaborative relationships with physicians in Tijuana. We will soon have the ability to train primary care providers at HFiT on performing simple skin biopsies and managing basic dermatologic conditions. Through our support of these providers, we are creating a sustainable partnership that is mutually beneficial to the patients in Tijuana as well as the medical students and residents in the United States. It is highly rewarding to all those involved with this project, and I am excited to see what challenges this next year will bring as we welcome many new patients from Haiti into the HFiT patient population.
- About Health Frontiers in Tijuana. University of California, San Diego School of Medicine website. https://meded.ucsd.edu/index.cfm/groups/hfit/about/. Accessed November 29, 2016.
- SkinCare for developing countries. American Academy of Dermatology website. https://www.aad.org/members/awards/skincare-for-developing-countries#undefined. Accessed November 29, 2016.
- About Health Frontiers in Tijuana. University of California, San Diego School of Medicine website. https://meded.ucsd.edu/index.cfm/groups/hfit/about/. Accessed November 29, 2016.
- SkinCare for developing countries. American Academy of Dermatology website. https://www.aad.org/members/awards/skincare-for-developing-countries#undefined. Accessed November 29, 2016.

Facial Rejuvenation With Fractional Laser Resurfacing

Pulley Suture for Wound Closure

Addressing Patient Concerns About Treatment Safety Data


Multiple Keratoacanthomas Occurring in Surgical Margins and De Novo Treated With Intralesional Methotrexate
Keratoacanthomas (KAs) are rapidly growing tumors most prominently found on sun-exposed areas of the skin. The normal progression of a KA is to show rapid growth followed by spontaneous resolution.1 Most KAs are solitary; however, there are several variants of multiple KAs including the familial Ferguson-Smith type, Gryzbowski syndrome (generalized eruptive KAs), KA centrifugum marginatum, Muir-Torre syndrome, and xeroderma pigmentosum.2-4 Keratoacanthomas also may develop in areas of trauma, including burns, laser treatment, radiation, and surgical margins from excisional biopsies or skin grafting.5 Treatment of multiple KAs can be difficult due to a potentially large field size and number of lesions.6 We present a case of multiple KAs developing both in the surgical margins and de novo that responded dramatically to treatment with intralesional methotrexate (MTX).
Case Report
A 55-year-old man with a history of a surgically treated squamous cell carcinoma (SCC) on the anterior aspect of the right leg developed multiple nodules involving the surgical scar. He previously underwent Mohs micrographic surgery (MMS); within a month after the second surgery the patient noticed increased pruritus along with scaly pink changes at the site of the surgical scar.
One month prior to presentation, biopsies from the anterior aspect of the right leg demonstrated well-differentiated SCC and he was subsequently treated with MMS; however, examination 1 month after MMS revealed an 11×7-cm indurated plaque with multiple nodules ranging from 1 to 2 cm near the periphery of the plaque with central atrophy and scarring, reminiscent of KA centrifugum marginatum (Figure, A). In a similar fashion, an 8×5-cm plaque composed of 7 nodular areas was noted on the posterior aspect of the right leg (Figure, B). The patient denied any history of trauma to this area. There was no palpable regional lymphadenopathy and the remainder of the skin examination was normal, except for signs of venous stasis in both legs.
Based on the location and morphology of the lesions, the clinical presentation was consistent with multiple KAs. Histologic examination from punch biopsies taken from the plaque's periphery demonstrated well-differentiated SCC (KA type), as well as a lichenoid inflammatory process, epidermal hyperplasia, and cystic and endophytic squamous proliferation suggestive of hypertrophic lichen planus (HLP).
In consideration of the size and number of the lesions as well as the prolonged wound healing with prior surgery, the patient consented to treatment with intralesional MTX (1 mL of 12.5 mg/mL every 2 weeks) rather than undergoing further surgery. The MTX injection was distributed between the lesions on the anterior and posterior aspects of the lower right leg. At each injection session, the size, thickness, and nodularity of the tumor decreased with markedly less pruritus and symptomatic relief was achieved. After 3 injection sessions, resulting in a total of 3 mL of 12.5 mg/mL of MTX, biopsies were taken from the residual atrophic scar on the anterior aspect of the right leg and the remaining 3 papules on the posterior aspect of the right leg to rule out HLP and invasive SCC. The pathology report commented on the presence of prurigo nodules without any evidence of SCC.
At 3-month follow-up, the patient demonstrated no new lesions or recurrence (Figure, C and D). The right leg continued to heal with scarring and postinflammatory pigmentary changes. The patient was monitored for recurrence and to determine the diagnosis of HLP.
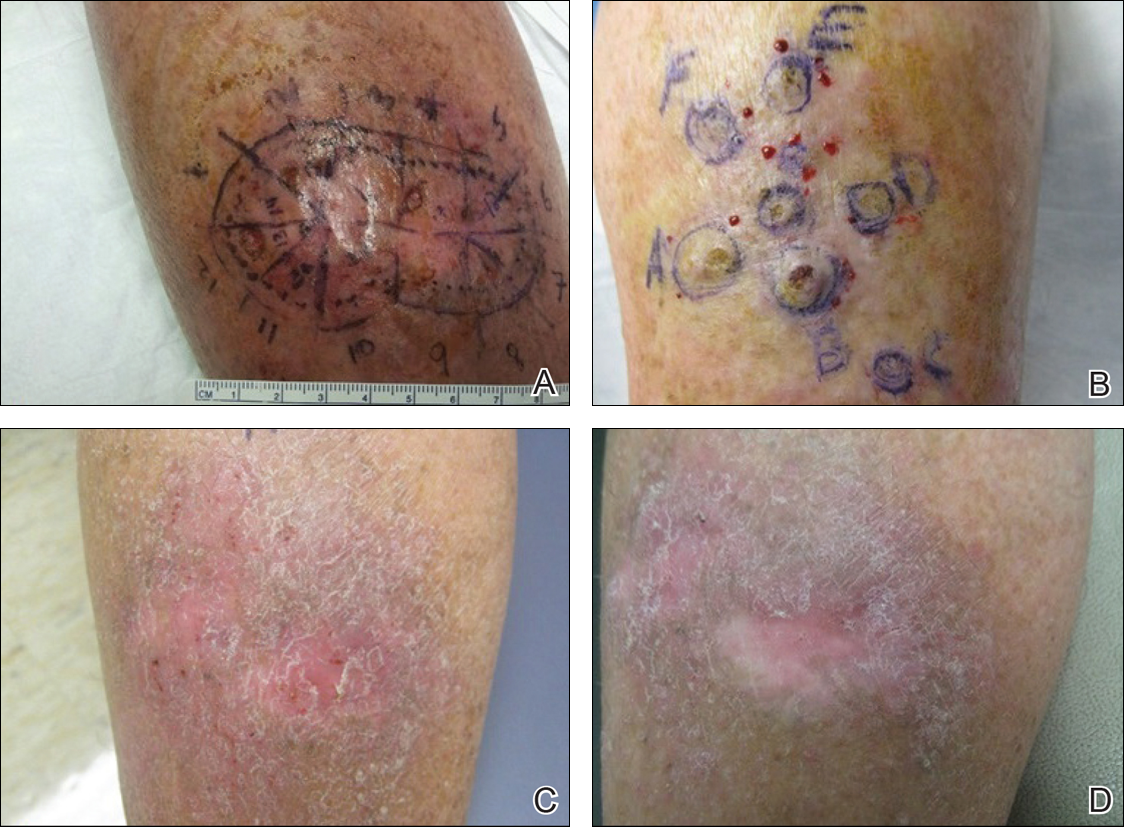
Comment
We report the development of multiple KAs arising both from within surgical margins and de novo, and resolution with intralesional MTX. Keratoacanthomas, especially various KA types, have been observed to develop due to various types of trauma, including sites of surgical scars, lichen planus, tattoos, thermal burns, radiation, and discoid lupus erythematosus, and within skin grafts and donor sites.5-19
Hypertrophic lichen planus is a chronic variant of lichen planus that often is found on the pretibial areas of the lower legs.13 Both SCC and reactive KAs have been observed to develop within lesions of HLP.14 Our pathologist commented on the presence of a lichenoid infiltrate with necrotic keratinocytes and epidermal hyperplasia suspicious for HLP, with a small focus of cystic and endophytic squamous proliferation. The latter lacked notable atypia or an invasive component and could represent an irritated infundibular cyst versus an early evolving KA.
The lichenoid inflammation is suspicious for HLP, which has been associated with eruptive KAs13-16 and may have contributed to the development of persistent KAs in our patient, both in sites of surgical scars (the anterior aspect of the leg) and in uninvolved skin (the posterior aspect of the leg). Trauma from the prior surgery may have stimulated a local inflammatory response and, if coupled with a preexisting underlying chronic inflammatory condition such as HLP, may have triggered the development of new lesions on the posterior leg. Skin pathergy reactions also are caused by an upregulated inflammatory response, which is reduced with immunosuppressive agents such as MTX.12
In our patient, there was both an isotopic and isomorphic response. The term isotopic response refers to the occurrence of a new skin disorder at the site of another unrelated and already healed skin disease. It was first defined by Wolf and Wolf20 in 1985 and hence is also known as Wolf isotopic response. The isotopic response in our patient occurred in the setting of lichen planus. The isomorphic response indicates the appearance of typical skin lesions of an existing dermatosis at sites of other skin injuries.
Initially, we thought the patient had recurrence of SCC, but with the rapid development of multiple lesions, the diagnosis of multiple KAs was more likely. Kimyai-Asadi et al8 demonstrated that surgical trauma can precede the development of KAs, as they reported a patient who developed a KA at an excision site. Tamir et al7 reported the simultaneous appearance of KAs in burn scars and skin graft donor sites 4 months after a 40% total body surface area burn. Hamilton et al11 described surgical trauma from a split-skin graft donor site as a trigger for the onset of a KA.
Multiple treatment alternatives exist for KAs, with the standard of care for large or high-risk KAs being excisional surgery21,22; however, other approaches may need to be considered in certain cases, such as with multiple KAs in which lesions may be large and extensive, thereby yielding poor cosmetic outcomes, or with increased surgical risk.23 Furthermore, multiple KAs that develop in the setting of surgical scars require special consideration. Topical 5-fluorouracil, various systemic and intralesional agents (eg, retinoids, interferon, bleomycin, MTX), laser therapy, electrodesiccation and curettage, radiotherapy, and photodynamic therapy all have been reported as methods employed for the treatment of KA.23-27 Goldberg et al5 reported cases of resolution of eruptive KAs arising in both surgical and nonsurgical sites with a combination of deep shave excision, MMS, curettage and desiccation, and oral isotretinoin.
For our patient, we opted for treatment with intralesional MTX, both due to its effectiveness for solitary KAs and reasonably decreased risk of morbidity compared to surgical excision of regions of the pretibial calves. Treatment with MTX would not have been attempted if there was any clinical doubt that the lesions were not the well-differentiated KA type. Also, we had a low threshold for discontinuing therapy and reverting to MMS treatment if any of the lesions displayed a paradoxical growth post-MTX treatment or failed to respond after 3 treatments. Intralesional MTX is less invasive, relatively inexpensive, and a treatment modality with decreased morbidity for KAs, especially for multiple KAs. It should be considered as a potential alternative to surgery in such cases.23-27
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Feldman RJ, Maize JC. Multiple keratoacanthomas in a young woman: report of a case emphasizing medical management and a review of the spectrum of multiple keratoacanthomas. Int J Dermatol. 2007;46:77-79.
- Ereaux LP, Schopflocher P, Fornier CJ. Keratoacanthoma. Arch Dermatol. 1955;71:73-83.
- Lloyd KM, Madsen DK, Lin PY. Grzybowski's eruptive keratoacanthoma. J Am Acad Dermatol. 1989;21(5, pt 1):1023-1024.
- Goldberg LH, Silapunt S, Beyrau KK, et al. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
- Pillsbury DM, Beerman H. Multiple keratoacanthoma. Am J Med Sci. 1958;236:614-623.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;400(5, pt 2):870-871.
- Kimyai-Asadi A, Shaffer C, Levine VJ, et al. Keratoacanthomas arising from an excisional surgery scar. J Drugs Dermatol. 2004;3:193-194.
- Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(suppl 2):S35-S38.
- Hendricks WM. Sudden appearance of multiple keratoacanthomas three weeks after thermal burns. Cutis. 1991;47:410-412.
- Hamilton SA, Dickson WA, O'Brien CJ. Keratoacanthoma developing in a split skin graft donor site. Br J Plast Surg. 1997;50:560-561.
- Bangash SJ, Green WH, Dolson DJ, et al. Eruptive postoperative squamous cell carcinomas exhibiting a pathergy-like reaction around surgical wound sites. J Am Acad Dermatol. 2009;61:892-897.
- Badell A, Marcoval J, Gallego I, et al. Keratoacanthomas arising in hypertrophic lichen planus. Br J Dermatol. 2000;142:370-393.
- Chave TA, Graham-Brown RAC. Keratoacanthoma developing in hypertrophic lichen planus. Br J Dermatol. 2003;148:592.
- Epstein R. Treatment of keratoacanthoma arising from hypertrophic lichen planus. J Am Acad Dermatol. 2010;62(3, suppl 1):AB28.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Toll A, Salgado R, Espinet B, et al. "Eruptive postoperative squamous cell carcinomas" or "Hypertrophic lichen planus-like reactions combined with infundibulocystic hyperplasia"? J Am Acad Dermatol. 2010;63:910-911.
- Fanti PA, Tosti A, Peluso AM, et al. Multiple keratoacanthoma in discoid lupus erythematosus. J Am Acad Dermatol. 1989;21(4, pt 1):809-810.
- Kossard S, Thompson C, Duncan GM. Hypertrophic lichen planus-like reactions combined with infundibulocystic hyperplasia: pathway to neoplasia. Arch Dermatol. 2004;140:1262-1267.
- Wolf R, Wolf D. Tinea in a site of healed herpes zoster (Isoloci response). Int J Dermatol. 1985;24:539.
- Larson PO. Keratoacanthomas treated with Mohs' micrographic surgery (chemosurgery): a review of forty-three cases. J Am Acad Dermatol. 1987;16:1040-1044.
- Benest L, Kaplan RP, Salit R, et al. Keratoacanthoma centrifugum marginatum of the lower extremity treated with Mohs micrographic surgery. J Am Acad Dermatol. 1994;31:501-502.
- Remling R, Mempel M, Schnopp N, et al. Intralesional methotrexate injection: an effective time and cost saving therapy alternative in keratoacanthomas that are difficult to treat surgically. Hautarzt. 2000;51:612-614.
- Annest NM, VanBeek MJ, Arpey CJ, et al. Intralesional methotrexate treatment for keratoacanthoma tumors: a retrospective study and review of the literature. J Am Acad Dermatol. 2007;56:989-993.
- Melton JL, Nelson BR, Stough DB, et al. Treatment of keratoacanthoma with intralesional methotrexate. J Am Acad Dermatol. 1991;25:1017-1023.
- Cuesta-Romero C, de Grado-Pena J. Intralesional methotrexate in solitary keratoacanthoma. Arch Dermatol. 1998;134:513-514.
- Richard MA, Gachon J, Choux R, et al. Treatment of keratoacanthoma with intralesional methotrexate injections. An Dermatol Venereol. 2000;127:1097.
Keratoacanthomas (KAs) are rapidly growing tumors most prominently found on sun-exposed areas of the skin. The normal progression of a KA is to show rapid growth followed by spontaneous resolution.1 Most KAs are solitary; however, there are several variants of multiple KAs including the familial Ferguson-Smith type, Gryzbowski syndrome (generalized eruptive KAs), KA centrifugum marginatum, Muir-Torre syndrome, and xeroderma pigmentosum.2-4 Keratoacanthomas also may develop in areas of trauma, including burns, laser treatment, radiation, and surgical margins from excisional biopsies or skin grafting.5 Treatment of multiple KAs can be difficult due to a potentially large field size and number of lesions.6 We present a case of multiple KAs developing both in the surgical margins and de novo that responded dramatically to treatment with intralesional methotrexate (MTX).
Case Report
A 55-year-old man with a history of a surgically treated squamous cell carcinoma (SCC) on the anterior aspect of the right leg developed multiple nodules involving the surgical scar. He previously underwent Mohs micrographic surgery (MMS); within a month after the second surgery the patient noticed increased pruritus along with scaly pink changes at the site of the surgical scar.
One month prior to presentation, biopsies from the anterior aspect of the right leg demonstrated well-differentiated SCC and he was subsequently treated with MMS; however, examination 1 month after MMS revealed an 11×7-cm indurated plaque with multiple nodules ranging from 1 to 2 cm near the periphery of the plaque with central atrophy and scarring, reminiscent of KA centrifugum marginatum (Figure, A). In a similar fashion, an 8×5-cm plaque composed of 7 nodular areas was noted on the posterior aspect of the right leg (Figure, B). The patient denied any history of trauma to this area. There was no palpable regional lymphadenopathy and the remainder of the skin examination was normal, except for signs of venous stasis in both legs.
Based on the location and morphology of the lesions, the clinical presentation was consistent with multiple KAs. Histologic examination from punch biopsies taken from the plaque's periphery demonstrated well-differentiated SCC (KA type), as well as a lichenoid inflammatory process, epidermal hyperplasia, and cystic and endophytic squamous proliferation suggestive of hypertrophic lichen planus (HLP).
In consideration of the size and number of the lesions as well as the prolonged wound healing with prior surgery, the patient consented to treatment with intralesional MTX (1 mL of 12.5 mg/mL every 2 weeks) rather than undergoing further surgery. The MTX injection was distributed between the lesions on the anterior and posterior aspects of the lower right leg. At each injection session, the size, thickness, and nodularity of the tumor decreased with markedly less pruritus and symptomatic relief was achieved. After 3 injection sessions, resulting in a total of 3 mL of 12.5 mg/mL of MTX, biopsies were taken from the residual atrophic scar on the anterior aspect of the right leg and the remaining 3 papules on the posterior aspect of the right leg to rule out HLP and invasive SCC. The pathology report commented on the presence of prurigo nodules without any evidence of SCC.
At 3-month follow-up, the patient demonstrated no new lesions or recurrence (Figure, C and D). The right leg continued to heal with scarring and postinflammatory pigmentary changes. The patient was monitored for recurrence and to determine the diagnosis of HLP.
Comment
We report the development of multiple KAs arising both from within surgical margins and de novo, and resolution with intralesional MTX. Keratoacanthomas, especially various KA types, have been observed to develop due to various types of trauma, including sites of surgical scars, lichen planus, tattoos, thermal burns, radiation, and discoid lupus erythematosus, and within skin grafts and donor sites.5-19
Hypertrophic lichen planus is a chronic variant of lichen planus that often is found on the pretibial areas of the lower legs.13 Both SCC and reactive KAs have been observed to develop within lesions of HLP.14 Our pathologist commented on the presence of a lichenoid infiltrate with necrotic keratinocytes and epidermal hyperplasia suspicious for HLP, with a small focus of cystic and endophytic squamous proliferation. The latter lacked notable atypia or an invasive component and could represent an irritated infundibular cyst versus an early evolving KA.
The lichenoid inflammation is suspicious for HLP, which has been associated with eruptive KAs13-16 and may have contributed to the development of persistent KAs in our patient, both in sites of surgical scars (the anterior aspect of the leg) and in uninvolved skin (the posterior aspect of the leg). Trauma from the prior surgery may have stimulated a local inflammatory response and, if coupled with a preexisting underlying chronic inflammatory condition such as HLP, may have triggered the development of new lesions on the posterior leg. Skin pathergy reactions also are caused by an upregulated inflammatory response, which is reduced with immunosuppressive agents such as MTX.12
In our patient, there was both an isotopic and isomorphic response. The term isotopic response refers to the occurrence of a new skin disorder at the site of another unrelated and already healed skin disease. It was first defined by Wolf and Wolf20 in 1985 and hence is also known as Wolf isotopic response. The isotopic response in our patient occurred in the setting of lichen planus. The isomorphic response indicates the appearance of typical skin lesions of an existing dermatosis at sites of other skin injuries.
Initially, we thought the patient had recurrence of SCC, but with the rapid development of multiple lesions, the diagnosis of multiple KAs was more likely. Kimyai-Asadi et al8 demonstrated that surgical trauma can precede the development of KAs, as they reported a patient who developed a KA at an excision site. Tamir et al7 reported the simultaneous appearance of KAs in burn scars and skin graft donor sites 4 months after a 40% total body surface area burn. Hamilton et al11 described surgical trauma from a split-skin graft donor site as a trigger for the onset of a KA.
Multiple treatment alternatives exist for KAs, with the standard of care for large or high-risk KAs being excisional surgery21,22; however, other approaches may need to be considered in certain cases, such as with multiple KAs in which lesions may be large and extensive, thereby yielding poor cosmetic outcomes, or with increased surgical risk.23 Furthermore, multiple KAs that develop in the setting of surgical scars require special consideration. Topical 5-fluorouracil, various systemic and intralesional agents (eg, retinoids, interferon, bleomycin, MTX), laser therapy, electrodesiccation and curettage, radiotherapy, and photodynamic therapy all have been reported as methods employed for the treatment of KA.23-27 Goldberg et al5 reported cases of resolution of eruptive KAs arising in both surgical and nonsurgical sites with a combination of deep shave excision, MMS, curettage and desiccation, and oral isotretinoin.
For our patient, we opted for treatment with intralesional MTX, both due to its effectiveness for solitary KAs and reasonably decreased risk of morbidity compared to surgical excision of regions of the pretibial calves. Treatment with MTX would not have been attempted if there was any clinical doubt that the lesions were not the well-differentiated KA type. Also, we had a low threshold for discontinuing therapy and reverting to MMS treatment if any of the lesions displayed a paradoxical growth post-MTX treatment or failed to respond after 3 treatments. Intralesional MTX is less invasive, relatively inexpensive, and a treatment modality with decreased morbidity for KAs, especially for multiple KAs. It should be considered as a potential alternative to surgery in such cases.23-27
Keratoacanthomas (KAs) are rapidly growing tumors most prominently found on sun-exposed areas of the skin. The normal progression of a KA is to show rapid growth followed by spontaneous resolution.1 Most KAs are solitary; however, there are several variants of multiple KAs including the familial Ferguson-Smith type, Gryzbowski syndrome (generalized eruptive KAs), KA centrifugum marginatum, Muir-Torre syndrome, and xeroderma pigmentosum.2-4 Keratoacanthomas also may develop in areas of trauma, including burns, laser treatment, radiation, and surgical margins from excisional biopsies or skin grafting.5 Treatment of multiple KAs can be difficult due to a potentially large field size and number of lesions.6 We present a case of multiple KAs developing both in the surgical margins and de novo that responded dramatically to treatment with intralesional methotrexate (MTX).
Case Report
A 55-year-old man with a history of a surgically treated squamous cell carcinoma (SCC) on the anterior aspect of the right leg developed multiple nodules involving the surgical scar. He previously underwent Mohs micrographic surgery (MMS); within a month after the second surgery the patient noticed increased pruritus along with scaly pink changes at the site of the surgical scar.
One month prior to presentation, biopsies from the anterior aspect of the right leg demonstrated well-differentiated SCC and he was subsequently treated with MMS; however, examination 1 month after MMS revealed an 11×7-cm indurated plaque with multiple nodules ranging from 1 to 2 cm near the periphery of the plaque with central atrophy and scarring, reminiscent of KA centrifugum marginatum (Figure, A). In a similar fashion, an 8×5-cm plaque composed of 7 nodular areas was noted on the posterior aspect of the right leg (Figure, B). The patient denied any history of trauma to this area. There was no palpable regional lymphadenopathy and the remainder of the skin examination was normal, except for signs of venous stasis in both legs.
Based on the location and morphology of the lesions, the clinical presentation was consistent with multiple KAs. Histologic examination from punch biopsies taken from the plaque's periphery demonstrated well-differentiated SCC (KA type), as well as a lichenoid inflammatory process, epidermal hyperplasia, and cystic and endophytic squamous proliferation suggestive of hypertrophic lichen planus (HLP).
In consideration of the size and number of the lesions as well as the prolonged wound healing with prior surgery, the patient consented to treatment with intralesional MTX (1 mL of 12.5 mg/mL every 2 weeks) rather than undergoing further surgery. The MTX injection was distributed between the lesions on the anterior and posterior aspects of the lower right leg. At each injection session, the size, thickness, and nodularity of the tumor decreased with markedly less pruritus and symptomatic relief was achieved. After 3 injection sessions, resulting in a total of 3 mL of 12.5 mg/mL of MTX, biopsies were taken from the residual atrophic scar on the anterior aspect of the right leg and the remaining 3 papules on the posterior aspect of the right leg to rule out HLP and invasive SCC. The pathology report commented on the presence of prurigo nodules without any evidence of SCC.
At 3-month follow-up, the patient demonstrated no new lesions or recurrence (Figure, C and D). The right leg continued to heal with scarring and postinflammatory pigmentary changes. The patient was monitored for recurrence and to determine the diagnosis of HLP.
Comment
We report the development of multiple KAs arising both from within surgical margins and de novo, and resolution with intralesional MTX. Keratoacanthomas, especially various KA types, have been observed to develop due to various types of trauma, including sites of surgical scars, lichen planus, tattoos, thermal burns, radiation, and discoid lupus erythematosus, and within skin grafts and donor sites.5-19
Hypertrophic lichen planus is a chronic variant of lichen planus that often is found on the pretibial areas of the lower legs.13 Both SCC and reactive KAs have been observed to develop within lesions of HLP.14 Our pathologist commented on the presence of a lichenoid infiltrate with necrotic keratinocytes and epidermal hyperplasia suspicious for HLP, with a small focus of cystic and endophytic squamous proliferation. The latter lacked notable atypia or an invasive component and could represent an irritated infundibular cyst versus an early evolving KA.
The lichenoid inflammation is suspicious for HLP, which has been associated with eruptive KAs13-16 and may have contributed to the development of persistent KAs in our patient, both in sites of surgical scars (the anterior aspect of the leg) and in uninvolved skin (the posterior aspect of the leg). Trauma from the prior surgery may have stimulated a local inflammatory response and, if coupled with a preexisting underlying chronic inflammatory condition such as HLP, may have triggered the development of new lesions on the posterior leg. Skin pathergy reactions also are caused by an upregulated inflammatory response, which is reduced with immunosuppressive agents such as MTX.12
In our patient, there was both an isotopic and isomorphic response. The term isotopic response refers to the occurrence of a new skin disorder at the site of another unrelated and already healed skin disease. It was first defined by Wolf and Wolf20 in 1985 and hence is also known as Wolf isotopic response. The isotopic response in our patient occurred in the setting of lichen planus. The isomorphic response indicates the appearance of typical skin lesions of an existing dermatosis at sites of other skin injuries.
Initially, we thought the patient had recurrence of SCC, but with the rapid development of multiple lesions, the diagnosis of multiple KAs was more likely. Kimyai-Asadi et al8 demonstrated that surgical trauma can precede the development of KAs, as they reported a patient who developed a KA at an excision site. Tamir et al7 reported the simultaneous appearance of KAs in burn scars and skin graft donor sites 4 months after a 40% total body surface area burn. Hamilton et al11 described surgical trauma from a split-skin graft donor site as a trigger for the onset of a KA.
Multiple treatment alternatives exist for KAs, with the standard of care for large or high-risk KAs being excisional surgery21,22; however, other approaches may need to be considered in certain cases, such as with multiple KAs in which lesions may be large and extensive, thereby yielding poor cosmetic outcomes, or with increased surgical risk.23 Furthermore, multiple KAs that develop in the setting of surgical scars require special consideration. Topical 5-fluorouracil, various systemic and intralesional agents (eg, retinoids, interferon, bleomycin, MTX), laser therapy, electrodesiccation and curettage, radiotherapy, and photodynamic therapy all have been reported as methods employed for the treatment of KA.23-27 Goldberg et al5 reported cases of resolution of eruptive KAs arising in both surgical and nonsurgical sites with a combination of deep shave excision, MMS, curettage and desiccation, and oral isotretinoin.
For our patient, we opted for treatment with intralesional MTX, both due to its effectiveness for solitary KAs and reasonably decreased risk of morbidity compared to surgical excision of regions of the pretibial calves. Treatment with MTX would not have been attempted if there was any clinical doubt that the lesions were not the well-differentiated KA type. Also, we had a low threshold for discontinuing therapy and reverting to MMS treatment if any of the lesions displayed a paradoxical growth post-MTX treatment or failed to respond after 3 treatments. Intralesional MTX is less invasive, relatively inexpensive, and a treatment modality with decreased morbidity for KAs, especially for multiple KAs. It should be considered as a potential alternative to surgery in such cases.23-27
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Feldman RJ, Maize JC. Multiple keratoacanthomas in a young woman: report of a case emphasizing medical management and a review of the spectrum of multiple keratoacanthomas. Int J Dermatol. 2007;46:77-79.
- Ereaux LP, Schopflocher P, Fornier CJ. Keratoacanthoma. Arch Dermatol. 1955;71:73-83.
- Lloyd KM, Madsen DK, Lin PY. Grzybowski's eruptive keratoacanthoma. J Am Acad Dermatol. 1989;21(5, pt 1):1023-1024.
- Goldberg LH, Silapunt S, Beyrau KK, et al. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
- Pillsbury DM, Beerman H. Multiple keratoacanthoma. Am J Med Sci. 1958;236:614-623.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;400(5, pt 2):870-871.
- Kimyai-Asadi A, Shaffer C, Levine VJ, et al. Keratoacanthomas arising from an excisional surgery scar. J Drugs Dermatol. 2004;3:193-194.
- Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(suppl 2):S35-S38.
- Hendricks WM. Sudden appearance of multiple keratoacanthomas three weeks after thermal burns. Cutis. 1991;47:410-412.
- Hamilton SA, Dickson WA, O'Brien CJ. Keratoacanthoma developing in a split skin graft donor site. Br J Plast Surg. 1997;50:560-561.
- Bangash SJ, Green WH, Dolson DJ, et al. Eruptive postoperative squamous cell carcinomas exhibiting a pathergy-like reaction around surgical wound sites. J Am Acad Dermatol. 2009;61:892-897.
- Badell A, Marcoval J, Gallego I, et al. Keratoacanthomas arising in hypertrophic lichen planus. Br J Dermatol. 2000;142:370-393.
- Chave TA, Graham-Brown RAC. Keratoacanthoma developing in hypertrophic lichen planus. Br J Dermatol. 2003;148:592.
- Epstein R. Treatment of keratoacanthoma arising from hypertrophic lichen planus. J Am Acad Dermatol. 2010;62(3, suppl 1):AB28.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Toll A, Salgado R, Espinet B, et al. "Eruptive postoperative squamous cell carcinomas" or "Hypertrophic lichen planus-like reactions combined with infundibulocystic hyperplasia"? J Am Acad Dermatol. 2010;63:910-911.
- Fanti PA, Tosti A, Peluso AM, et al. Multiple keratoacanthoma in discoid lupus erythematosus. J Am Acad Dermatol. 1989;21(4, pt 1):809-810.
- Kossard S, Thompson C, Duncan GM. Hypertrophic lichen planus-like reactions combined with infundibulocystic hyperplasia: pathway to neoplasia. Arch Dermatol. 2004;140:1262-1267.
- Wolf R, Wolf D. Tinea in a site of healed herpes zoster (Isoloci response). Int J Dermatol. 1985;24:539.
- Larson PO. Keratoacanthomas treated with Mohs' micrographic surgery (chemosurgery): a review of forty-three cases. J Am Acad Dermatol. 1987;16:1040-1044.
- Benest L, Kaplan RP, Salit R, et al. Keratoacanthoma centrifugum marginatum of the lower extremity treated with Mohs micrographic surgery. J Am Acad Dermatol. 1994;31:501-502.
- Remling R, Mempel M, Schnopp N, et al. Intralesional methotrexate injection: an effective time and cost saving therapy alternative in keratoacanthomas that are difficult to treat surgically. Hautarzt. 2000;51:612-614.
- Annest NM, VanBeek MJ, Arpey CJ, et al. Intralesional methotrexate treatment for keratoacanthoma tumors: a retrospective study and review of the literature. J Am Acad Dermatol. 2007;56:989-993.
- Melton JL, Nelson BR, Stough DB, et al. Treatment of keratoacanthoma with intralesional methotrexate. J Am Acad Dermatol. 1991;25:1017-1023.
- Cuesta-Romero C, de Grado-Pena J. Intralesional methotrexate in solitary keratoacanthoma. Arch Dermatol. 1998;134:513-514.
- Richard MA, Gachon J, Choux R, et al. Treatment of keratoacanthoma with intralesional methotrexate injections. An Dermatol Venereol. 2000;127:1097.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Feldman RJ, Maize JC. Multiple keratoacanthomas in a young woman: report of a case emphasizing medical management and a review of the spectrum of multiple keratoacanthomas. Int J Dermatol. 2007;46:77-79.
- Ereaux LP, Schopflocher P, Fornier CJ. Keratoacanthoma. Arch Dermatol. 1955;71:73-83.
- Lloyd KM, Madsen DK, Lin PY. Grzybowski's eruptive keratoacanthoma. J Am Acad Dermatol. 1989;21(5, pt 1):1023-1024.
- Goldberg LH, Silapunt S, Beyrau KK, et al. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
- Pillsbury DM, Beerman H. Multiple keratoacanthoma. Am J Med Sci. 1958;236:614-623.
- Tamir G, Morgenstern S, Ben-Amitay D, et al. Synchronous appearance of keratoacanthomas in burn scar and skin graft donor site shortly after injury. J Am Acad Dermatol. 1999;400(5, pt 2):870-871.
- Kimyai-Asadi A, Shaffer C, Levine VJ, et al. Keratoacanthomas arising from an excisional surgery scar. J Drugs Dermatol. 2004;3:193-194.
- Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(suppl 2):S35-S38.
- Hendricks WM. Sudden appearance of multiple keratoacanthomas three weeks after thermal burns. Cutis. 1991;47:410-412.
- Hamilton SA, Dickson WA, O'Brien CJ. Keratoacanthoma developing in a split skin graft donor site. Br J Plast Surg. 1997;50:560-561.
- Bangash SJ, Green WH, Dolson DJ, et al. Eruptive postoperative squamous cell carcinomas exhibiting a pathergy-like reaction around surgical wound sites. J Am Acad Dermatol. 2009;61:892-897.
- Badell A, Marcoval J, Gallego I, et al. Keratoacanthomas arising in hypertrophic lichen planus. Br J Dermatol. 2000;142:370-393.
- Chave TA, Graham-Brown RAC. Keratoacanthoma developing in hypertrophic lichen planus. Br J Dermatol. 2003;148:592.
- Epstein R. Treatment of keratoacanthoma arising from hypertrophic lichen planus. J Am Acad Dermatol. 2010;62(3, suppl 1):AB28.
- Giesecke LM, Reid CM, James CL, et al. Giant keratoacanthoma arising in hypertrophic lichen planus. Australas J Dermatol. 2003;44:267-269.
- Toll A, Salgado R, Espinet B, et al. "Eruptive postoperative squamous cell carcinomas" or "Hypertrophic lichen planus-like reactions combined with infundibulocystic hyperplasia"? J Am Acad Dermatol. 2010;63:910-911.
- Fanti PA, Tosti A, Peluso AM, et al. Multiple keratoacanthoma in discoid lupus erythematosus. J Am Acad Dermatol. 1989;21(4, pt 1):809-810.
- Kossard S, Thompson C, Duncan GM. Hypertrophic lichen planus-like reactions combined with infundibulocystic hyperplasia: pathway to neoplasia. Arch Dermatol. 2004;140:1262-1267.
- Wolf R, Wolf D. Tinea in a site of healed herpes zoster (Isoloci response). Int J Dermatol. 1985;24:539.
- Larson PO. Keratoacanthomas treated with Mohs' micrographic surgery (chemosurgery): a review of forty-three cases. J Am Acad Dermatol. 1987;16:1040-1044.
- Benest L, Kaplan RP, Salit R, et al. Keratoacanthoma centrifugum marginatum of the lower extremity treated with Mohs micrographic surgery. J Am Acad Dermatol. 1994;31:501-502.
- Remling R, Mempel M, Schnopp N, et al. Intralesional methotrexate injection: an effective time and cost saving therapy alternative in keratoacanthomas that are difficult to treat surgically. Hautarzt. 2000;51:612-614.
- Annest NM, VanBeek MJ, Arpey CJ, et al. Intralesional methotrexate treatment for keratoacanthoma tumors: a retrospective study and review of the literature. J Am Acad Dermatol. 2007;56:989-993.
- Melton JL, Nelson BR, Stough DB, et al. Treatment of keratoacanthoma with intralesional methotrexate. J Am Acad Dermatol. 1991;25:1017-1023.
- Cuesta-Romero C, de Grado-Pena J. Intralesional methotrexate in solitary keratoacanthoma. Arch Dermatol. 1998;134:513-514.
- Richard MA, Gachon J, Choux R, et al. Treatment of keratoacanthoma with intralesional methotrexate injections. An Dermatol Venereol. 2000;127:1097.
Practice Points
- Keratoacanthomas (KAs) are rapidly growing tumors most prominently found on sun-exposed areas but also may develop in areas of trauma including burns, laser treatment, radiation, and surgical margins from excisional biopsies or skin grafting.
- Intralesional methotrexate is a potential alternative to surgical treatment of KAs as a less invasive and less costly treatment modality with decreased morbidity for multiple KAs.
- Isotopic response refers to the occurrence of a new skin disorder arising at the site of another unrelated and already healed skin disease. Isomorphic response indicates the appearance of typical skin lesions of an existing dermatosis at sites of injuries.
Cosmetic Corner: Dermatologists Weigh in on Self-tanners
To improve patient care and outcomes, leading dermatologists offered their recommendations on self-tanners. Consideration must be given to:
- Anthelios 50 Mineral Tinted
La Roche-Posay Laboratoire Dermatologique
Recommended by Gary Goldenberg, MD, New York, New York
- St. Tropez Self Tan products
PZ Cussons Beauty LLP
“It helps to produce an even and natural-looking skin tone.”—Anthony M. Rossi, MD, New York, New York
- Sun-Free Self-Tanning Formula
Kiehl’s
Recommended by Gary Goldenberg, MD, New York, New York
- Sunless Tanning Towelette
Sun Bum
“This product is easy to use. Make sure to use it in conjunction with a broad-spectrum sunscreen.”—Shari Lipner, MD, PhD, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, skin-lightening products, and athlete’s foot treatments will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on self-tanners. Consideration must be given to:
- Anthelios 50 Mineral Tinted
La Roche-Posay Laboratoire Dermatologique
Recommended by Gary Goldenberg, MD, New York, New York
- St. Tropez Self Tan products
PZ Cussons Beauty LLP
“It helps to produce an even and natural-looking skin tone.”—Anthony M. Rossi, MD, New York, New York
- Sun-Free Self-Tanning Formula
Kiehl’s
Recommended by Gary Goldenberg, MD, New York, New York
- Sunless Tanning Towelette
Sun Bum
“This product is easy to use. Make sure to use it in conjunction with a broad-spectrum sunscreen.”—Shari Lipner, MD, PhD, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, skin-lightening products, and athlete’s foot treatments will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on self-tanners. Consideration must be given to:
- Anthelios 50 Mineral Tinted
La Roche-Posay Laboratoire Dermatologique
Recommended by Gary Goldenberg, MD, New York, New York
- St. Tropez Self Tan products
PZ Cussons Beauty LLP
“It helps to produce an even and natural-looking skin tone.”—Anthony M. Rossi, MD, New York, New York
- Sun-Free Self-Tanning Formula
Kiehl’s
Recommended by Gary Goldenberg, MD, New York, New York
- Sunless Tanning Towelette
Sun Bum
“This product is easy to use. Make sure to use it in conjunction with a broad-spectrum sunscreen.”—Shari Lipner, MD, PhD, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, skin-lightening products, and athlete’s foot treatments will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
