User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Innovations in Dermatology: Laboratory Monitoring With Isotretinoin

DRESS Syndrome Induced by Telaprevir: A Potentially Fatal Adverse Event in Chronic Hepatitis C Therapy
To the Editor:
A 58-year-old woman with a history of hyperprolactinemia and gastrointestinal angiodysplasia presented to the dermatology department with a generalized skin rash of 3 weeks’ duration. She did not have a history of toxic habits. She had a history of chronic hepatitis C virus (HCV) genotype 1b (IL-28B locus) with severe hepatic fibrosis (stage 4) as assessed by ultrasound-based elastography. Due to lack of response, plasma HCV RNA was still detectable at week 12 of pegylated interferon and ribavirin (RIB) therapy, and triple therapy with pegylated interferon, RIB, and telaprevir was initiated.
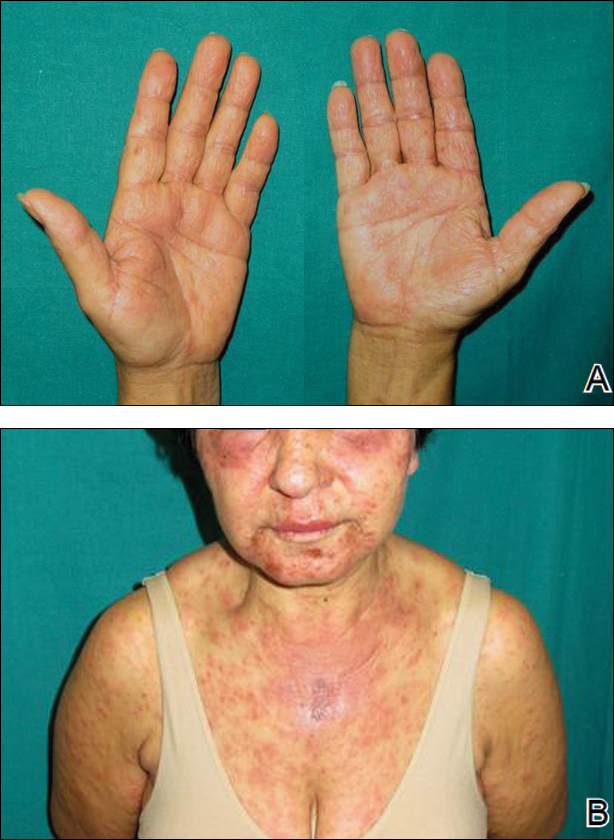
Two months later, she was admitted to the hospital after developing a generalized cutaneous rash that covered 90% of the body surface area (BSA) along with fever (temperature, 38.5°C). Laboratory blood tests showed an elevated absolute eosinophil count (2000 cells/µL [reference range, 0–500 cells/µL]), anemia (hemoglobin, 6.5 g/dL [reference range, 12–16 g/dL]), thrombocytopenia (26×103/µL [reference range, 150–400×103/µL]), and altered liver function tests (serum alanine aminotransferase, 60 U/L [reference range, 0–45 U/L]; aspartate aminotransferase, 80 U/L [reference range, 0–40 U/L]). Plasma HCV RNA was undetectable at this visit. On physical examination a generalized exanthema with coalescing plaques was observed, as well as crusted vesicles covering the arms, legs, chest, abdomen, and back. Palmoplantar papules (Figure, A) and facial swelling (Figure, B) also were present. A skin biopsy specimen taken from a papule on the left arm showed superficial perivascular lymphocytic infiltration with dermal edema. These findings were consistent with a diagnosis of DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome. Application of the Adverse Drug Reaction Probability Scale1 in our patient (total score of 5) suggested that DRESS syndrome was a moderate adverse event likely related to the use of telaprevir.
After diagnosis of DRESS syndrome, telaprevir was discontinued, and the doses of RIB and pegylated interferon were reduced to 200 mg and 180 µg weekly, respectively. Laboratory test values including liver function tests normalized within 3 weeks and remained normal on follow-up. Plasma HCV RNA continued to be undetectable.
Hepatitis C virus is relatively common with an incidence of 3% worldwide.2 It may present as an acute hepatitis or, more frequently, as asymptomatic chronic hepatitis. The acute process is self-limited and rarely causes hepatic failure. It usually leads to a chronic infection, which can result in cirrhosis, hepatocellular carcinoma, and the need for liver transplantation. The aim of treatment is eradication of HCV RNA, which is predicted by the attainment of a sustained virologic response. The latter is defined by the absence of HCV RNA by a polymerase chain reaction within 3 to 6 months after cessation of treatment.
Treatment of chronic HCV was based on the combination of pegylated interferon alfa-2a or -2b with RIB until 2015. Guidelines for the diagnosis and management of HCV infection have been published by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.2 These guidelines include new protease inhibitors, telaprevir and boceprevir, in the therapeutic approach of these patients. The main limitation of both drugs is the cutaneous toxicity.
Factors to be considered when treating HCV include viral genotype, if the patient is naïve or pretreated, the degree of fibrosis, established cirrhosis, and the treatment response. For patients with genotype 1,2 as in our case, combination therapy with 3 drugs is recommended: pegylated interferon 180 µg subcutaneous injection weekly, RIB 15 mg/kg daily, and telaprevir 2250 mg or boceprevir 2400 mg daily. Triple therapy has been shown to achieve a successful response in 75% of naïve patients and in 50% of patients refractory to standard therapy.3
Telaprevir is an NS3/4A protease inhibitor approved by the US Food and Drug Administration and the European Medicines Agency for treatment of chronic HCV infection in naïve patients and in those unresponsive to double therapy. In phase 2 clinical trials, 41% to 61% of patients treated with telaprevir developed cutaneous reactions, of which 5% to 8% required cessation of treatment.4 The predicting risk factors for developing a secondary rash to telaprevir include age older than 45 years, body mass index less than 30, Caucasian ethnicity, and receiving HCV therapy for the first time.4
This cutaneous side effect is managed depending on the extension of the lesions, the presence of systemic symptoms, and laboratory abnormalities.5 Therefore, the severity of the skin reaction can be divided into 4 stages4,5: (1) grade I or mild, defined as a localized rash with no systemic signs or mucosal involvement; (2) grade II or moderate, a maximum of 50% BSA involvement without epidermal detachment, and inflammation of the mucous membranes may be present without ulcers, as well as systemic symptoms such as fever, arthralgia, or eosinophilia; (3) grade III or severe, skin lesions affecting more than 50% BSA or less if any of the following lesions are present: vesicles or blisters, ulcers, epidermal detachment, palpable purpura, or erythema that does not blanch under pressure; (4) grade IV or life-threatening, when the clinical picture is consistent with acute generalized exanthematous pustulosis, DRESS syndrome, toxic epidermal necrolysis, or Stevens-Johnson syndrome.
DRESS syndrome is a condition clinically characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests. Cutaneous histopathologic examination may be unspecific, though atypical lymphocytes with a marked epidermotropism mimicking fungoid mycosis also have been described.6 In addition, human herpesvirus 6 serology may be negative, despite infection with this herpesvirus subtype having been associated with the development of DRESS syndrome. The pathophysiologic mechanism of DRESS syndrome is not completely understood; however, one theory ascribes an immunologic activation due to drug metabolite formation as the main mechanism.1
Eleven patients7 with possible DRESS syndrome have been reported in clinical trials (less than 5% of the total of patients), with an addition of 1 more by Montaudié et al.8 No notable differences were found between telaprevir levels in these patients with respect to those of the control group.
For the management of DRESS syndrome, the occurrence of early signs of a severe acute skin reaction requires the immediate cessation of the drug, telaprevir in this case. The withdrawal of the dual therapy will depend on the short-term clinical course, according to the general condition of the patient, as well as the analytical abnormalities observed.9
In conclusion, telaprevir is a promising novel therapy for the treatment of HCV infection, but its cutaneous side effects still need to be properly established.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharacol Ther. 1981;30:239-245.
- HCV guidance: recommendations for testing, managing, and treating hepatitis C. HCV Guidelines website. http://www.hcvguidelines.org. Accessed August 11, 2018.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405-2416.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Roujeau JC, Mockenhaupt M, Tahan SR, et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013;149:152-158.
- De Vriese AS, Philippe J, Van Renterghem DM, et al. Carbamazepine hypersensitivity syndrome: report of 4 cases and review of the literature. Medicine (Baltimore). 1995;74:144-151.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review [published online May 17, 2011]. Am J Med. 2011;124:588-597.
- Montaudié H, Passeron T, Cardot-Leccia N, et al. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303-305.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
To the Editor:
A 58-year-old woman with a history of hyperprolactinemia and gastrointestinal angiodysplasia presented to the dermatology department with a generalized skin rash of 3 weeks’ duration. She did not have a history of toxic habits. She had a history of chronic hepatitis C virus (HCV) genotype 1b (IL-28B locus) with severe hepatic fibrosis (stage 4) as assessed by ultrasound-based elastography. Due to lack of response, plasma HCV RNA was still detectable at week 12 of pegylated interferon and ribavirin (RIB) therapy, and triple therapy with pegylated interferon, RIB, and telaprevir was initiated.
Two months later, she was admitted to the hospital after developing a generalized cutaneous rash that covered 90% of the body surface area (BSA) along with fever (temperature, 38.5°C). Laboratory blood tests showed an elevated absolute eosinophil count (2000 cells/µL [reference range, 0–500 cells/µL]), anemia (hemoglobin, 6.5 g/dL [reference range, 12–16 g/dL]), thrombocytopenia (26×103/µL [reference range, 150–400×103/µL]), and altered liver function tests (serum alanine aminotransferase, 60 U/L [reference range, 0–45 U/L]; aspartate aminotransferase, 80 U/L [reference range, 0–40 U/L]). Plasma HCV RNA was undetectable at this visit. On physical examination a generalized exanthema with coalescing plaques was observed, as well as crusted vesicles covering the arms, legs, chest, abdomen, and back. Palmoplantar papules (Figure, A) and facial swelling (Figure, B) also were present. A skin biopsy specimen taken from a papule on the left arm showed superficial perivascular lymphocytic infiltration with dermal edema. These findings were consistent with a diagnosis of DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome. Application of the Adverse Drug Reaction Probability Scale1 in our patient (total score of 5) suggested that DRESS syndrome was a moderate adverse event likely related to the use of telaprevir.
After diagnosis of DRESS syndrome, telaprevir was discontinued, and the doses of RIB and pegylated interferon were reduced to 200 mg and 180 µg weekly, respectively. Laboratory test values including liver function tests normalized within 3 weeks and remained normal on follow-up. Plasma HCV RNA continued to be undetectable.
Hepatitis C virus is relatively common with an incidence of 3% worldwide.2 It may present as an acute hepatitis or, more frequently, as asymptomatic chronic hepatitis. The acute process is self-limited and rarely causes hepatic failure. It usually leads to a chronic infection, which can result in cirrhosis, hepatocellular carcinoma, and the need for liver transplantation. The aim of treatment is eradication of HCV RNA, which is predicted by the attainment of a sustained virologic response. The latter is defined by the absence of HCV RNA by a polymerase chain reaction within 3 to 6 months after cessation of treatment.
Treatment of chronic HCV was based on the combination of pegylated interferon alfa-2a or -2b with RIB until 2015. Guidelines for the diagnosis and management of HCV infection have been published by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.2 These guidelines include new protease inhibitors, telaprevir and boceprevir, in the therapeutic approach of these patients. The main limitation of both drugs is the cutaneous toxicity.
Factors to be considered when treating HCV include viral genotype, if the patient is naïve or pretreated, the degree of fibrosis, established cirrhosis, and the treatment response. For patients with genotype 1,2 as in our case, combination therapy with 3 drugs is recommended: pegylated interferon 180 µg subcutaneous injection weekly, RIB 15 mg/kg daily, and telaprevir 2250 mg or boceprevir 2400 mg daily. Triple therapy has been shown to achieve a successful response in 75% of naïve patients and in 50% of patients refractory to standard therapy.3
Telaprevir is an NS3/4A protease inhibitor approved by the US Food and Drug Administration and the European Medicines Agency for treatment of chronic HCV infection in naïve patients and in those unresponsive to double therapy. In phase 2 clinical trials, 41% to 61% of patients treated with telaprevir developed cutaneous reactions, of which 5% to 8% required cessation of treatment.4 The predicting risk factors for developing a secondary rash to telaprevir include age older than 45 years, body mass index less than 30, Caucasian ethnicity, and receiving HCV therapy for the first time.4
This cutaneous side effect is managed depending on the extension of the lesions, the presence of systemic symptoms, and laboratory abnormalities.5 Therefore, the severity of the skin reaction can be divided into 4 stages4,5: (1) grade I or mild, defined as a localized rash with no systemic signs or mucosal involvement; (2) grade II or moderate, a maximum of 50% BSA involvement without epidermal detachment, and inflammation of the mucous membranes may be present without ulcers, as well as systemic symptoms such as fever, arthralgia, or eosinophilia; (3) grade III or severe, skin lesions affecting more than 50% BSA or less if any of the following lesions are present: vesicles or blisters, ulcers, epidermal detachment, palpable purpura, or erythema that does not blanch under pressure; (4) grade IV or life-threatening, when the clinical picture is consistent with acute generalized exanthematous pustulosis, DRESS syndrome, toxic epidermal necrolysis, or Stevens-Johnson syndrome.
DRESS syndrome is a condition clinically characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests. Cutaneous histopathologic examination may be unspecific, though atypical lymphocytes with a marked epidermotropism mimicking fungoid mycosis also have been described.6 In addition, human herpesvirus 6 serology may be negative, despite infection with this herpesvirus subtype having been associated with the development of DRESS syndrome. The pathophysiologic mechanism of DRESS syndrome is not completely understood; however, one theory ascribes an immunologic activation due to drug metabolite formation as the main mechanism.1
Eleven patients7 with possible DRESS syndrome have been reported in clinical trials (less than 5% of the total of patients), with an addition of 1 more by Montaudié et al.8 No notable differences were found between telaprevir levels in these patients with respect to those of the control group.
For the management of DRESS syndrome, the occurrence of early signs of a severe acute skin reaction requires the immediate cessation of the drug, telaprevir in this case. The withdrawal of the dual therapy will depend on the short-term clinical course, according to the general condition of the patient, as well as the analytical abnormalities observed.9
In conclusion, telaprevir is a promising novel therapy for the treatment of HCV infection, but its cutaneous side effects still need to be properly established.
To the Editor:
A 58-year-old woman with a history of hyperprolactinemia and gastrointestinal angiodysplasia presented to the dermatology department with a generalized skin rash of 3 weeks’ duration. She did not have a history of toxic habits. She had a history of chronic hepatitis C virus (HCV) genotype 1b (IL-28B locus) with severe hepatic fibrosis (stage 4) as assessed by ultrasound-based elastography. Due to lack of response, plasma HCV RNA was still detectable at week 12 of pegylated interferon and ribavirin (RIB) therapy, and triple therapy with pegylated interferon, RIB, and telaprevir was initiated.
Two months later, she was admitted to the hospital after developing a generalized cutaneous rash that covered 90% of the body surface area (BSA) along with fever (temperature, 38.5°C). Laboratory blood tests showed an elevated absolute eosinophil count (2000 cells/µL [reference range, 0–500 cells/µL]), anemia (hemoglobin, 6.5 g/dL [reference range, 12–16 g/dL]), thrombocytopenia (26×103/µL [reference range, 150–400×103/µL]), and altered liver function tests (serum alanine aminotransferase, 60 U/L [reference range, 0–45 U/L]; aspartate aminotransferase, 80 U/L [reference range, 0–40 U/L]). Plasma HCV RNA was undetectable at this visit. On physical examination a generalized exanthema with coalescing plaques was observed, as well as crusted vesicles covering the arms, legs, chest, abdomen, and back. Palmoplantar papules (Figure, A) and facial swelling (Figure, B) also were present. A skin biopsy specimen taken from a papule on the left arm showed superficial perivascular lymphocytic infiltration with dermal edema. These findings were consistent with a diagnosis of DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome. Application of the Adverse Drug Reaction Probability Scale1 in our patient (total score of 5) suggested that DRESS syndrome was a moderate adverse event likely related to the use of telaprevir.
After diagnosis of DRESS syndrome, telaprevir was discontinued, and the doses of RIB and pegylated interferon were reduced to 200 mg and 180 µg weekly, respectively. Laboratory test values including liver function tests normalized within 3 weeks and remained normal on follow-up. Plasma HCV RNA continued to be undetectable.
Hepatitis C virus is relatively common with an incidence of 3% worldwide.2 It may present as an acute hepatitis or, more frequently, as asymptomatic chronic hepatitis. The acute process is self-limited and rarely causes hepatic failure. It usually leads to a chronic infection, which can result in cirrhosis, hepatocellular carcinoma, and the need for liver transplantation. The aim of treatment is eradication of HCV RNA, which is predicted by the attainment of a sustained virologic response. The latter is defined by the absence of HCV RNA by a polymerase chain reaction within 3 to 6 months after cessation of treatment.
Treatment of chronic HCV was based on the combination of pegylated interferon alfa-2a or -2b with RIB until 2015. Guidelines for the diagnosis and management of HCV infection have been published by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.2 These guidelines include new protease inhibitors, telaprevir and boceprevir, in the therapeutic approach of these patients. The main limitation of both drugs is the cutaneous toxicity.
Factors to be considered when treating HCV include viral genotype, if the patient is naïve or pretreated, the degree of fibrosis, established cirrhosis, and the treatment response. For patients with genotype 1,2 as in our case, combination therapy with 3 drugs is recommended: pegylated interferon 180 µg subcutaneous injection weekly, RIB 15 mg/kg daily, and telaprevir 2250 mg or boceprevir 2400 mg daily. Triple therapy has been shown to achieve a successful response in 75% of naïve patients and in 50% of patients refractory to standard therapy.3
Telaprevir is an NS3/4A protease inhibitor approved by the US Food and Drug Administration and the European Medicines Agency for treatment of chronic HCV infection in naïve patients and in those unresponsive to double therapy. In phase 2 clinical trials, 41% to 61% of patients treated with telaprevir developed cutaneous reactions, of which 5% to 8% required cessation of treatment.4 The predicting risk factors for developing a secondary rash to telaprevir include age older than 45 years, body mass index less than 30, Caucasian ethnicity, and receiving HCV therapy for the first time.4
This cutaneous side effect is managed depending on the extension of the lesions, the presence of systemic symptoms, and laboratory abnormalities.5 Therefore, the severity of the skin reaction can be divided into 4 stages4,5: (1) grade I or mild, defined as a localized rash with no systemic signs or mucosal involvement; (2) grade II or moderate, a maximum of 50% BSA involvement without epidermal detachment, and inflammation of the mucous membranes may be present without ulcers, as well as systemic symptoms such as fever, arthralgia, or eosinophilia; (3) grade III or severe, skin lesions affecting more than 50% BSA or less if any of the following lesions are present: vesicles or blisters, ulcers, epidermal detachment, palpable purpura, or erythema that does not blanch under pressure; (4) grade IV or life-threatening, when the clinical picture is consistent with acute generalized exanthematous pustulosis, DRESS syndrome, toxic epidermal necrolysis, or Stevens-Johnson syndrome.
DRESS syndrome is a condition clinically characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests. Cutaneous histopathologic examination may be unspecific, though atypical lymphocytes with a marked epidermotropism mimicking fungoid mycosis also have been described.6 In addition, human herpesvirus 6 serology may be negative, despite infection with this herpesvirus subtype having been associated with the development of DRESS syndrome. The pathophysiologic mechanism of DRESS syndrome is not completely understood; however, one theory ascribes an immunologic activation due to drug metabolite formation as the main mechanism.1
Eleven patients7 with possible DRESS syndrome have been reported in clinical trials (less than 5% of the total of patients), with an addition of 1 more by Montaudié et al.8 No notable differences were found between telaprevir levels in these patients with respect to those of the control group.
For the management of DRESS syndrome, the occurrence of early signs of a severe acute skin reaction requires the immediate cessation of the drug, telaprevir in this case. The withdrawal of the dual therapy will depend on the short-term clinical course, according to the general condition of the patient, as well as the analytical abnormalities observed.9
In conclusion, telaprevir is a promising novel therapy for the treatment of HCV infection, but its cutaneous side effects still need to be properly established.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharacol Ther. 1981;30:239-245.
- HCV guidance: recommendations for testing, managing, and treating hepatitis C. HCV Guidelines website. http://www.hcvguidelines.org. Accessed August 11, 2018.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405-2416.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Roujeau JC, Mockenhaupt M, Tahan SR, et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013;149:152-158.
- De Vriese AS, Philippe J, Van Renterghem DM, et al. Carbamazepine hypersensitivity syndrome: report of 4 cases and review of the literature. Medicine (Baltimore). 1995;74:144-151.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review [published online May 17, 2011]. Am J Med. 2011;124:588-597.
- Montaudié H, Passeron T, Cardot-Leccia N, et al. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303-305.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharacol Ther. 1981;30:239-245.
- HCV guidance: recommendations for testing, managing, and treating hepatitis C. HCV Guidelines website. http://www.hcvguidelines.org. Accessed August 11, 2018.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405-2416.
- Kardaun SH, Sidoroff A, Valeyrie-Allanore L, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156:609-611.
- Roujeau JC, Mockenhaupt M, Tahan SR, et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013;149:152-158.
- De Vriese AS, Philippe J, Van Renterghem DM, et al. Carbamazepine hypersensitivity syndrome: report of 4 cases and review of the literature. Medicine (Baltimore). 1995;74:144-151.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review [published online May 17, 2011]. Am J Med. 2011;124:588-597.
- Montaudié H, Passeron T, Cardot-Leccia N, et al. Drug rash with eosinophilia and systemic symptoms due to telaprevir. Dermatology. 2010;221:303-305.
- Tas S, Simonart T. Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update. Dermatology. 2003;206:353-356.
Practice Points
- DRESS syndrome is characterized by a generalized skin rash, facial angioedema, high fever, lymph node enlargement, and leukocytosis with eosinophilia or atypical lymphocytosis, along with abnormal renal and hepatic function tests.
- Severity of the skin reaction can be divided into 4 stages; in the third and fourth stages, adequate patient monitoring is necessary.
- Telaprevir is an NS3/4A protease inhibitor approved for treatment of chronic hepatitis C virus infection in naïve patients and in those unresponsive to double therapy. Its cutaneous side effects still need to be properly established.
Eumycetoma Pedis in an Albanian Farmer
To the Editor:
Mycetoma is a noncontagious chronic infection of the skin and subcutaneous tissue caused by exogenous fungi or bacteria that can involve deeper structures such as the fasciae, muscles, and bones. Clinically it is characterized by increased swelling of the affected area, fibrosis, nodules, tumefaction, formation of draining sinuses, and abscesses that drain pus-containing grains through fistulae.1 The initiation of the infection is related to local trauma and can involve muscle, underlying bone, and adjacent organs. The feet are the most commonly affected region, and the incubation period is variable. Patients rarely report prior trauma to the affected area and only seek medical consultation when the nodules and draining sinuses become evident. The etiopathogenesis of mycetoma is associated with aerobic actinomycetes (ie, Nocardia, Actinomadura, Streptomyces), known as actinomycetoma, and fungal infections, known as eumycetomas.1
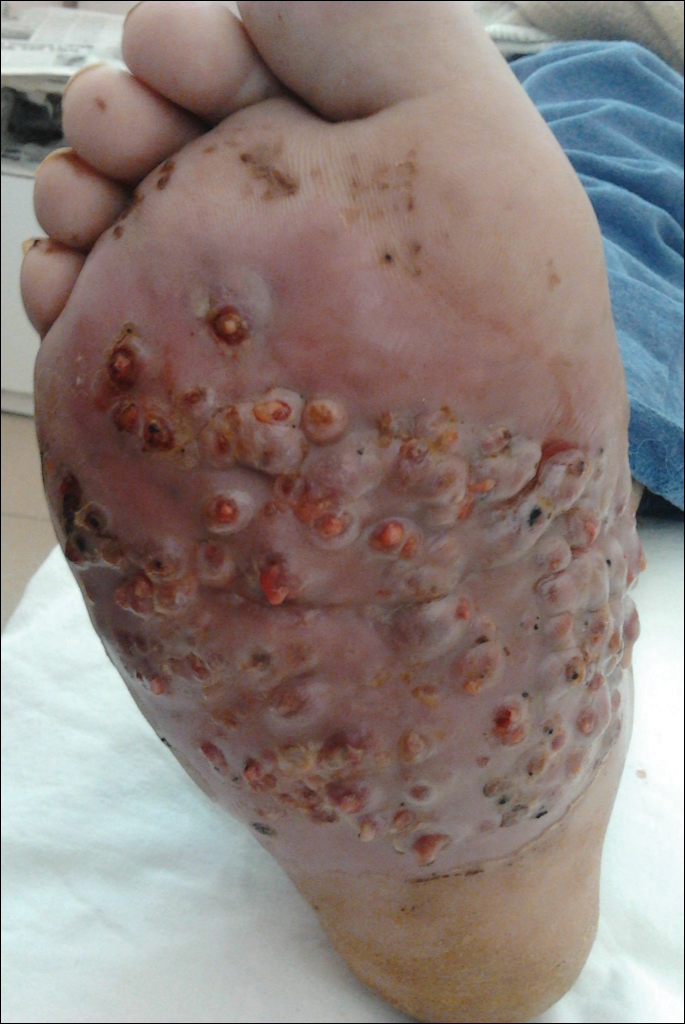
We report the case of a 57-year-old Albanian man who was referred to the outpatient clinic of our dermatology department for diagnosis and treatment of a chronic, suppurative, subcutaneous infection on the right foot presenting as abscesses and draining sinuses. The patient was a farmer and reported that the condition appeared 4 years prior following a laceration he sustained while at work. Dermatologic examination revealed local tumefaction, fistulated nodules, and abscesses discharging a serohemorrhagic fluid on the right foot (Figure 1). Perilesional erythema and subcutaneous swelling were evident. There was no regional lymphadenopathy. Standard laboratory examination was normal. Radiography of the right foot showed no osteolytic lesions or evidence of osteomyelitis.
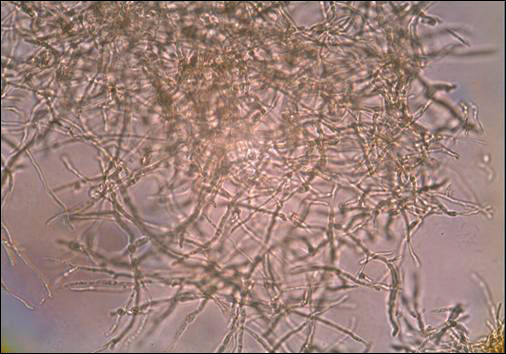
A skin biopsy from a lesion on the right foot was performed, and identification of the possible etiologic agent was based on direct microscopic examination of the granules, culture isolation of the agent, and fungal microscopic morphology.2 Granules were studied under direct examination with potassium hydroxide solution 20% and showed septate branching hyphae (Figure 2). The culture produced colonies that were white, yellow, and brown. Colonies were comprised of dense mycelium with melanin pigment and were grown at 37°C. A lactose tolerance test was positive.2 Therefore, the strain was identified as Madurella mycetomatis, and a diagnosis of eumycetoma pedis was made.
The patient was hospitalized for 2 weeks and treated with intravenous fluconazole, then treatment with oral itraconazole 200 mg once daily was initiated. At 4-month follow-up, he had self-discontinued treatment but demonstrated partial improvement of the tumefaction, healing of sinus tracts, and functional recovery of the right foot.
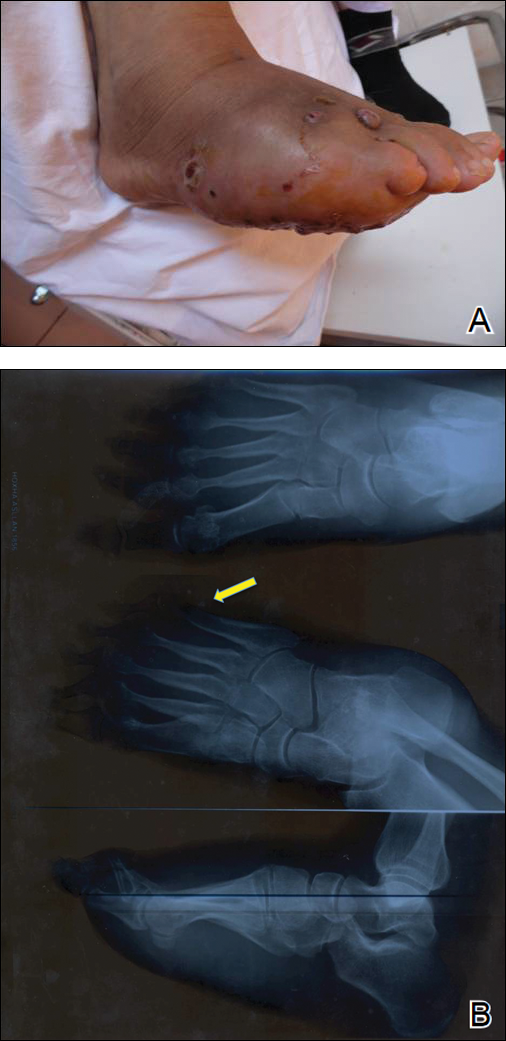
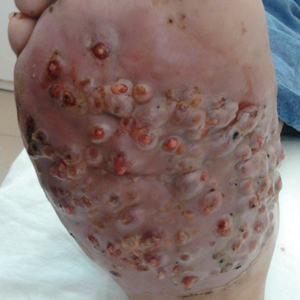
One year following the initial presentation, the patient’s clinical condition worsened (Figure 3A). Radiography of the right foot showed osteolytic lesions on bones in the right foot (Figure 3B), and a repeat culture showed the presence of Staphylococcus aureus; thus, treatment with itraconazole 200 mg once daily along with antibiotics (cefuroxime and gentamicin) was started immediately. Surgical treatment was recommended, but the patient refused treatment.
Mycetomas are rare in Albania but are common in countries of tropical and subtropical regions. K
Clinical features of eumycetoma include lesions with clear margins, few sinuses, black grains, slow progression, and long-term involvement of bone. The grains represent an aggregate of hyphae produced by fungi; thus, the characteristic feature of eumycetoma is the formation of large granules that can involve bone.1 A critical diagnostic step is to distinguish between eumycetoma and actinomycetoma. If possible, it is important to culture the organism because treatment varies depending on the cause of the infection.
Fungal identification is crucial in the diagnosis of mycetoma. In our case, diagnosis of eumycetoma pedis was based on clinical examination and detection of fungal species by microscopic examination and culture. The color of small granules (black grains) is a parameter used to identify different pathogens on histology but is not sufficient for diagnosis.5 The examination by potassium hydroxide preparation is helpful to identify the hyphae; however, culture is necessary.2
Therapeutic management of eumycetoma needs a combined strategy that includes systemic treatment and surgical therapy. Eumycetomas generally are more difficult to treat then actinomycetomas. Some authors recommend a high dose of amphotericin B as the treatment of choice for eumycetoma,6,7 but there are some that emphasize that amphotericin B is partially effective.8,9 There also is evidence in the literature of resistance of eumycetoma to ketoconazole treatment10,11 and successful treatment with fluconazole and itraconazole.10-13 For this reason, we treated our patient with the latter agents. In cases of osteolysis, amputation often is required.
In conclusion, eumycetoma pedis is a rare deep fungal infection that can cause considerable morbidity. P
- Rook A, Burns T. Rook’s Textbook of Dermatology. 8th ed. West Sussex, UK; Hoboken, NJ: Wiley-Blackwell; 2010.
- Balows A, Hausler WJ, eds. Manual of Clinical Microbiology. 5th ed. Washington, DC: American Society for Microbiology; 1991.
- Carter HV. On a new striking form of fungus disease principally affecting the foot and prevailing endemically in many parts of India. Trans Med Phys Soc Bombay. 1860;6:104-142.
- Kwon-Chung KJ, Bennet JE. Medical Mycology. Philadelphia, PA: Lea & Febiger; 1992.
- Venugopal PV, Venugopal TV. Pale grain eumycetomas in Madras. Australas J Dermatol. 1995;36:149-151.
- Guarro J, Gams W, Pujol I, et al. Acremonium species: new emerging fungal opportunists—in vitro antifungal susceptibilities and review. Clin Infec Dis. 1997;25:1222-1229.
- Lau YL, Yuen KY, Lee CW, et al. Invasive Acremonium falciforme infection in a patient with severe combined immunodeficiency. Clin Infect Dis. 1995;20:197-198.
- Fincher RM, Fisher JF, Lovell RD, et al. Infection due to the fungus Acremonium (cephalosporium). Medicine (Baltimore). 1991;70:398-409.
- Milburn PB, Papayanopulos DM, Pomerantz BM. Mycetoma due to Acremonium falciforme. Int J Dermatol. 1988;27:408-410.
- Welsh O, Salinas MC, Rodriguez MA. Treatment of eumycetoma and actinomycetoma. Cur Top Med Mycol. 1995;6:47-71.
- Restrepo A. Treatment of tropical mycoses. J Am Acad Dermatol. 1994;31:S91-S102.
- Gugnani HC, Ezeanolue BC, Khalil M, et al. Fluconazole in the therapy of tropical deep mycoses. Mycoses. 1995;38:485-488.
- Welsh O. Mycetoma. current concepts in treatment. Int J Dermatol. 1991;30:387-398.
To the Editor:
Mycetoma is a noncontagious chronic infection of the skin and subcutaneous tissue caused by exogenous fungi or bacteria that can involve deeper structures such as the fasciae, muscles, and bones. Clinically it is characterized by increased swelling of the affected area, fibrosis, nodules, tumefaction, formation of draining sinuses, and abscesses that drain pus-containing grains through fistulae.1 The initiation of the infection is related to local trauma and can involve muscle, underlying bone, and adjacent organs. The feet are the most commonly affected region, and the incubation period is variable. Patients rarely report prior trauma to the affected area and only seek medical consultation when the nodules and draining sinuses become evident. The etiopathogenesis of mycetoma is associated with aerobic actinomycetes (ie, Nocardia, Actinomadura, Streptomyces), known as actinomycetoma, and fungal infections, known as eumycetomas.1
We report the case of a 57-year-old Albanian man who was referred to the outpatient clinic of our dermatology department for diagnosis and treatment of a chronic, suppurative, subcutaneous infection on the right foot presenting as abscesses and draining sinuses. The patient was a farmer and reported that the condition appeared 4 years prior following a laceration he sustained while at work. Dermatologic examination revealed local tumefaction, fistulated nodules, and abscesses discharging a serohemorrhagic fluid on the right foot (Figure 1). Perilesional erythema and subcutaneous swelling were evident. There was no regional lymphadenopathy. Standard laboratory examination was normal. Radiography of the right foot showed no osteolytic lesions or evidence of osteomyelitis.
A skin biopsy from a lesion on the right foot was performed, and identification of the possible etiologic agent was based on direct microscopic examination of the granules, culture isolation of the agent, and fungal microscopic morphology.2 Granules were studied under direct examination with potassium hydroxide solution 20% and showed septate branching hyphae (Figure 2). The culture produced colonies that were white, yellow, and brown. Colonies were comprised of dense mycelium with melanin pigment and were grown at 37°C. A lactose tolerance test was positive.2 Therefore, the strain was identified as Madurella mycetomatis, and a diagnosis of eumycetoma pedis was made.
The patient was hospitalized for 2 weeks and treated with intravenous fluconazole, then treatment with oral itraconazole 200 mg once daily was initiated. At 4-month follow-up, he had self-discontinued treatment but demonstrated partial improvement of the tumefaction, healing of sinus tracts, and functional recovery of the right foot.
One year following the initial presentation, the patient’s clinical condition worsened (Figure 3A). Radiography of the right foot showed osteolytic lesions on bones in the right foot (Figure 3B), and a repeat culture showed the presence of Staphylococcus aureus; thus, treatment with itraconazole 200 mg once daily along with antibiotics (cefuroxime and gentamicin) was started immediately. Surgical treatment was recommended, but the patient refused treatment.
Mycetomas are rare in Albania but are common in countries of tropical and subtropical regions. K
Clinical features of eumycetoma include lesions with clear margins, few sinuses, black grains, slow progression, and long-term involvement of bone. The grains represent an aggregate of hyphae produced by fungi; thus, the characteristic feature of eumycetoma is the formation of large granules that can involve bone.1 A critical diagnostic step is to distinguish between eumycetoma and actinomycetoma. If possible, it is important to culture the organism because treatment varies depending on the cause of the infection.
Fungal identification is crucial in the diagnosis of mycetoma. In our case, diagnosis of eumycetoma pedis was based on clinical examination and detection of fungal species by microscopic examination and culture. The color of small granules (black grains) is a parameter used to identify different pathogens on histology but is not sufficient for diagnosis.5 The examination by potassium hydroxide preparation is helpful to identify the hyphae; however, culture is necessary.2
Therapeutic management of eumycetoma needs a combined strategy that includes systemic treatment and surgical therapy. Eumycetomas generally are more difficult to treat then actinomycetomas. Some authors recommend a high dose of amphotericin B as the treatment of choice for eumycetoma,6,7 but there are some that emphasize that amphotericin B is partially effective.8,9 There also is evidence in the literature of resistance of eumycetoma to ketoconazole treatment10,11 and successful treatment with fluconazole and itraconazole.10-13 For this reason, we treated our patient with the latter agents. In cases of osteolysis, amputation often is required.
In conclusion, eumycetoma pedis is a rare deep fungal infection that can cause considerable morbidity. P
To the Editor:
Mycetoma is a noncontagious chronic infection of the skin and subcutaneous tissue caused by exogenous fungi or bacteria that can involve deeper structures such as the fasciae, muscles, and bones. Clinically it is characterized by increased swelling of the affected area, fibrosis, nodules, tumefaction, formation of draining sinuses, and abscesses that drain pus-containing grains through fistulae.1 The initiation of the infection is related to local trauma and can involve muscle, underlying bone, and adjacent organs. The feet are the most commonly affected region, and the incubation period is variable. Patients rarely report prior trauma to the affected area and only seek medical consultation when the nodules and draining sinuses become evident. The etiopathogenesis of mycetoma is associated with aerobic actinomycetes (ie, Nocardia, Actinomadura, Streptomyces), known as actinomycetoma, and fungal infections, known as eumycetomas.1
We report the case of a 57-year-old Albanian man who was referred to the outpatient clinic of our dermatology department for diagnosis and treatment of a chronic, suppurative, subcutaneous infection on the right foot presenting as abscesses and draining sinuses. The patient was a farmer and reported that the condition appeared 4 years prior following a laceration he sustained while at work. Dermatologic examination revealed local tumefaction, fistulated nodules, and abscesses discharging a serohemorrhagic fluid on the right foot (Figure 1). Perilesional erythema and subcutaneous swelling were evident. There was no regional lymphadenopathy. Standard laboratory examination was normal. Radiography of the right foot showed no osteolytic lesions or evidence of osteomyelitis.
A skin biopsy from a lesion on the right foot was performed, and identification of the possible etiologic agent was based on direct microscopic examination of the granules, culture isolation of the agent, and fungal microscopic morphology.2 Granules were studied under direct examination with potassium hydroxide solution 20% and showed septate branching hyphae (Figure 2). The culture produced colonies that were white, yellow, and brown. Colonies were comprised of dense mycelium with melanin pigment and were grown at 37°C. A lactose tolerance test was positive.2 Therefore, the strain was identified as Madurella mycetomatis, and a diagnosis of eumycetoma pedis was made.
The patient was hospitalized for 2 weeks and treated with intravenous fluconazole, then treatment with oral itraconazole 200 mg once daily was initiated. At 4-month follow-up, he had self-discontinued treatment but demonstrated partial improvement of the tumefaction, healing of sinus tracts, and functional recovery of the right foot.
One year following the initial presentation, the patient’s clinical condition worsened (Figure 3A). Radiography of the right foot showed osteolytic lesions on bones in the right foot (Figure 3B), and a repeat culture showed the presence of Staphylococcus aureus; thus, treatment with itraconazole 200 mg once daily along with antibiotics (cefuroxime and gentamicin) was started immediately. Surgical treatment was recommended, but the patient refused treatment.
Mycetomas are rare in Albania but are common in countries of tropical and subtropical regions. K
Clinical features of eumycetoma include lesions with clear margins, few sinuses, black grains, slow progression, and long-term involvement of bone. The grains represent an aggregate of hyphae produced by fungi; thus, the characteristic feature of eumycetoma is the formation of large granules that can involve bone.1 A critical diagnostic step is to distinguish between eumycetoma and actinomycetoma. If possible, it is important to culture the organism because treatment varies depending on the cause of the infection.
Fungal identification is crucial in the diagnosis of mycetoma. In our case, diagnosis of eumycetoma pedis was based on clinical examination and detection of fungal species by microscopic examination and culture. The color of small granules (black grains) is a parameter used to identify different pathogens on histology but is not sufficient for diagnosis.5 The examination by potassium hydroxide preparation is helpful to identify the hyphae; however, culture is necessary.2
Therapeutic management of eumycetoma needs a combined strategy that includes systemic treatment and surgical therapy. Eumycetomas generally are more difficult to treat then actinomycetomas. Some authors recommend a high dose of amphotericin B as the treatment of choice for eumycetoma,6,7 but there are some that emphasize that amphotericin B is partially effective.8,9 There also is evidence in the literature of resistance of eumycetoma to ketoconazole treatment10,11 and successful treatment with fluconazole and itraconazole.10-13 For this reason, we treated our patient with the latter agents. In cases of osteolysis, amputation often is required.
In conclusion, eumycetoma pedis is a rare deep fungal infection that can cause considerable morbidity. P
- Rook A, Burns T. Rook’s Textbook of Dermatology. 8th ed. West Sussex, UK; Hoboken, NJ: Wiley-Blackwell; 2010.
- Balows A, Hausler WJ, eds. Manual of Clinical Microbiology. 5th ed. Washington, DC: American Society for Microbiology; 1991.
- Carter HV. On a new striking form of fungus disease principally affecting the foot and prevailing endemically in many parts of India. Trans Med Phys Soc Bombay. 1860;6:104-142.
- Kwon-Chung KJ, Bennet JE. Medical Mycology. Philadelphia, PA: Lea & Febiger; 1992.
- Venugopal PV, Venugopal TV. Pale grain eumycetomas in Madras. Australas J Dermatol. 1995;36:149-151.
- Guarro J, Gams W, Pujol I, et al. Acremonium species: new emerging fungal opportunists—in vitro antifungal susceptibilities and review. Clin Infec Dis. 1997;25:1222-1229.
- Lau YL, Yuen KY, Lee CW, et al. Invasive Acremonium falciforme infection in a patient with severe combined immunodeficiency. Clin Infect Dis. 1995;20:197-198.
- Fincher RM, Fisher JF, Lovell RD, et al. Infection due to the fungus Acremonium (cephalosporium). Medicine (Baltimore). 1991;70:398-409.
- Milburn PB, Papayanopulos DM, Pomerantz BM. Mycetoma due to Acremonium falciforme. Int J Dermatol. 1988;27:408-410.
- Welsh O, Salinas MC, Rodriguez MA. Treatment of eumycetoma and actinomycetoma. Cur Top Med Mycol. 1995;6:47-71.
- Restrepo A. Treatment of tropical mycoses. J Am Acad Dermatol. 1994;31:S91-S102.
- Gugnani HC, Ezeanolue BC, Khalil M, et al. Fluconazole in the therapy of tropical deep mycoses. Mycoses. 1995;38:485-488.
- Welsh O. Mycetoma. current concepts in treatment. Int J Dermatol. 1991;30:387-398.
- Rook A, Burns T. Rook’s Textbook of Dermatology. 8th ed. West Sussex, UK; Hoboken, NJ: Wiley-Blackwell; 2010.
- Balows A, Hausler WJ, eds. Manual of Clinical Microbiology. 5th ed. Washington, DC: American Society for Microbiology; 1991.
- Carter HV. On a new striking form of fungus disease principally affecting the foot and prevailing endemically in many parts of India. Trans Med Phys Soc Bombay. 1860;6:104-142.
- Kwon-Chung KJ, Bennet JE. Medical Mycology. Philadelphia, PA: Lea & Febiger; 1992.
- Venugopal PV, Venugopal TV. Pale grain eumycetomas in Madras. Australas J Dermatol. 1995;36:149-151.
- Guarro J, Gams W, Pujol I, et al. Acremonium species: new emerging fungal opportunists—in vitro antifungal susceptibilities and review. Clin Infec Dis. 1997;25:1222-1229.
- Lau YL, Yuen KY, Lee CW, et al. Invasive Acremonium falciforme infection in a patient with severe combined immunodeficiency. Clin Infect Dis. 1995;20:197-198.
- Fincher RM, Fisher JF, Lovell RD, et al. Infection due to the fungus Acremonium (cephalosporium). Medicine (Baltimore). 1991;70:398-409.
- Milburn PB, Papayanopulos DM, Pomerantz BM. Mycetoma due to Acremonium falciforme. Int J Dermatol. 1988;27:408-410.
- Welsh O, Salinas MC, Rodriguez MA. Treatment of eumycetoma and actinomycetoma. Cur Top Med Mycol. 1995;6:47-71.
- Restrepo A. Treatment of tropical mycoses. J Am Acad Dermatol. 1994;31:S91-S102.
- Gugnani HC, Ezeanolue BC, Khalil M, et al. Fluconazole in the therapy of tropical deep mycoses. Mycoses. 1995;38:485-488.
- Welsh O. Mycetoma. current concepts in treatment. Int J Dermatol. 1991;30:387-398.
Practice Points
- A critical step in the diagnosis of mycetomas is to distinguish between eumycetoma and actinomycetoma.
- Potassium hydroxide preparation is helpful to identify fungal infection.
- Eumycetomas generally are more difficult to treat and require a combined strategy including systemic treatment and surgical therapy.
Postirradiation Morphea: Unique Presentation on the Breast
To the Editor:
Postirradiation morphea (PIM) is a rare but well-documented phenomenon that primarily occurs in breast cancer patients who have received radiation therapy; however, it also has been reported in patients who have received radiation therapy for lymphoma as well as endocervical, endometrial, and gastric carcinomas.1 Importantly, clinicians must be able to recognize and differentiate this condition from other causes of new-onset induration and erythema of the breast, such as cancer recurrence, a new primary malignancy, or inflammatory etiologies (eg, radiation or contact dermatitis). Typically, PIM presents months to years after radiation therapy as an erythematous patch within the irradiated area that progressively becomes indurated. We report an unusual case of PIM with a reticulated appearance occurring 3 weeks after radiotherapy, chemotherapy, and surgery for an infiltrating ductal carcinoma of the left breast.
A 62-year-old woman presented to the dermatology department with a stage IIA, lymph node–negative, estrogen and progesterone receptor–negative, human epidermal growth factor receptor 2–negative infiltrating ductal carcinoma of the left breast. She was treated with a partial mastectomy of the left breast followed by external beam radiotherapy to the entire left breast in combination with chemotherapy (doxorubicin, cyclophosphamide, paclitaxel). The patient received 15 fractions of 270 cGy (4050 cGy total) with a weekly 600-cGy boost over 21 days without any complications.
Three weeks after finishing radiation therapy, the patient developed redness and swelling of the left breast that did not encompass the entire radiation field. There was no associated pain or pruritus. She was treated by her surgical oncologist with topical calendula and 3 courses of cephalexin for suspected mastitis with only modest improvement, then was referred to dermatology 3 months later.
At the initial dermatology evaluation, the patient reported little improvement after antibiotics and topical calendula. On physical examination, there were erythematous, reticulated, dusky, indurated patches on the entire left breast. The area of most pronounced induration surrounded the surgical scar on the left superior breast. Punch biopsy for hematoxylin and eosin staining and tissue cultures was obtained at this appointment. The patient was started on doxycycline 100 mg twice daily and was instructed to apply triamcinolone ointment 0.1% twice daily to the affected area. After 1 month of therapy, she reported slight improvement in the degree of erythema with this regimen, but the involved area continued to extend outside of the radiation field to the central chest wall and medial right breast (Figure 1). Two additional biopsies—one from the central chest and another from the right breast—were then taken over the course of 4 months, given the consistently inconclusive clinicopathologic nature and failure of the eruption to respond to antibiotics plus topical corticosteroids.
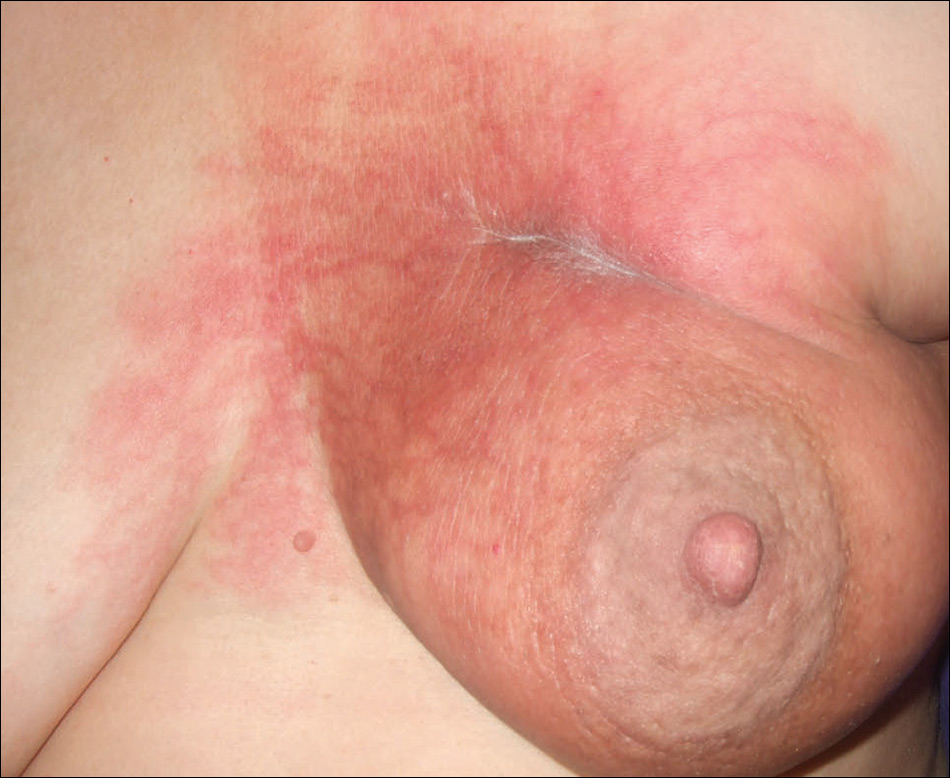
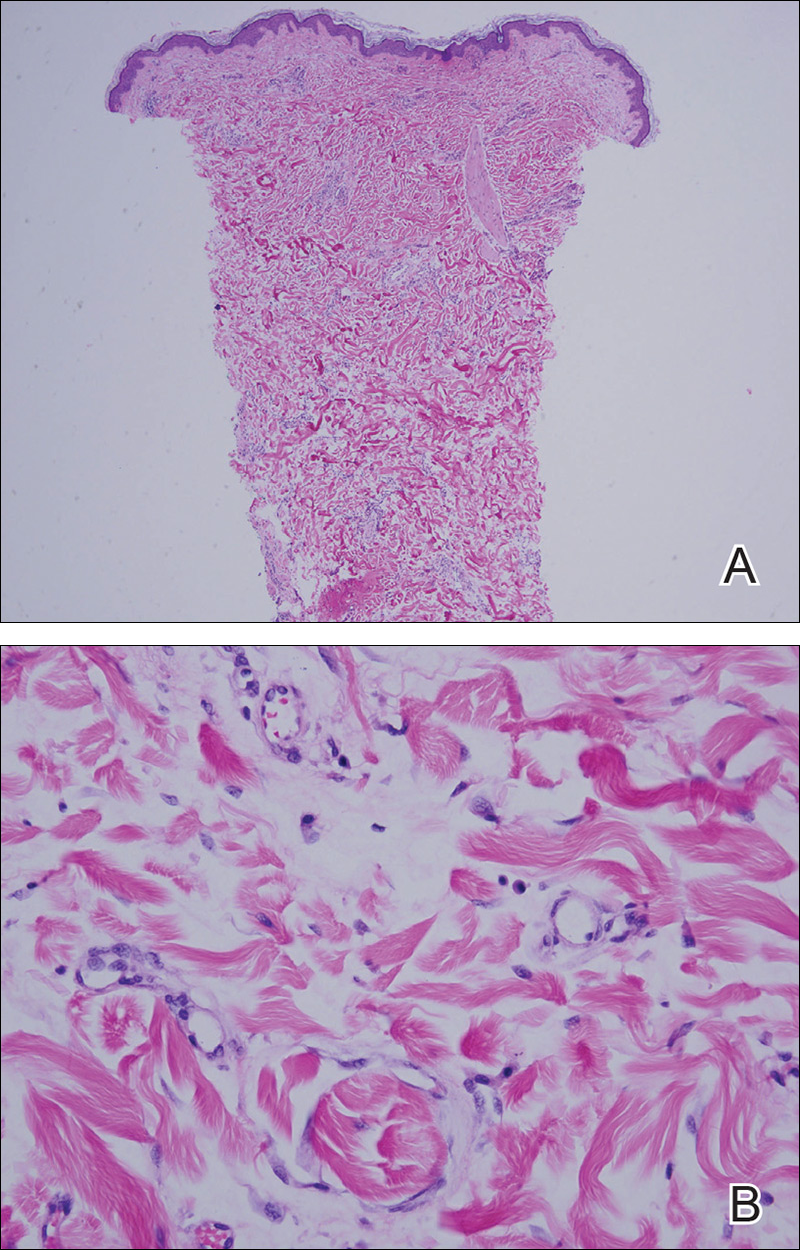
Punch biopsy from the central chest revealed a sparse perivascular infiltrate comprised predominantly of lymphocytes with occasional eosinophils (Figure 2). There were foci suggestive of early dermal sclerosis, an increased number of small blood vessels in the dermis, and scattered enlarged fibroblasts. Metastatic carcinoma was not identified. Although the histologic findings were not entirely specific, the changes were most suggestive of PIM, for which the patient was started on pentoxifylline (400 mg 3 times daily) and oral vitamin E supplementation (400 IU daily). At subsequent follow-up appointments, she showed markedly decreased skin erythema and induration.
Morphea, also known as localized scleroderma, is an inflammatory skin condition characterized by sclerosis of the dermis and subcutis leading to scarlike tissue formation. Worldwide incidence ranges from 0.4 to 2.7 cases per 100,000 individuals with a predilection for white women.2 Unlike systemic scleroderma, morphea patients lack Raynaud phenomenon and visceral involvement.3,4
There are several clinical subtypes of morphea, including plaque, linear, generalized, and pansclerotic morphea. Lesions may vary in appearance based on configuration, stage of development, and depth of involvement.4 During the earliest phases, morphea lesions are asymptomatic, asymmetrically distributed, erythematous to violaceous patches or subtly indurated plaques expanding centrifugally with a lilac ring. Central sclerosis with loss of follicles and sweat glands is a later finding associated with advanced disease. Moreover, some reports of early-stage morphea have suggested a reticulated or geographic vascular morphology that may be misdiagnosed for other conditions such as a port-wine stain.5
Local skin exposures have long been hypothesized to contribute to development of morphea, including infection, especially Borrelia burgdorferi; trauma; chronic venous insufficiency; cosmetic surgery; medications; and exposure to toxic cooking oils, silicones, silica, pesticides, organic solvents, and vinyl chloride.2,6,7
Radiation therapy is an often overlooked cause of morphea. It was first described in 1905 but then rarely discussed until a 1989 case series of 9 patients, 7 of whom had received irradiation for breast cancer.8,9 Today, the increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in PIM incidence.10
In contrast to other radiation-induced skin conditions, development of PIM is independent of the presence or absence of adjuvant chemotherapy, type of radiation therapy, or the total radiation dose or fractionation number, with reported doses ranging from less than 20.0 Gy to up to 59.4 Gy and dose fractions ranging from 10 to 30. In 20% to 30% of cases, PIM extends beyond the radiation field, sometimes involving distant sites never exposed to high-energy rays.1,10,11 This observation suggests a mechanism reliant on more widespread cascade rather than solely local tissue damage.
Prominent culture-negative, lymphoplasmacytic inflammation is another important diagnostic clue. Radiation dermatitis and fibrosis do not have the marked erythematous to violaceous hue seen in early morphea plaques. This color seen in early morphea plaques may be intense enough and in a geographic pattern, emulating a vascular lesion.
There is no standardized treatment of PIM, but traditional therapies for morphea may provide some benefit. Several randomized controlled clinical trials have shown success with pentoxifylline and oral vitamin E supplementation to treat or prevent radiation-induced breast fibrosis.12 Extrapolating from this data, our patient was started on this combination therapy and showed marked improvement in skin color and texture.
- Morganroth PA, Dehoratius D, Curry H, et al. Postirradiation morphea: a case report with a review of the literature and summary of the clinicopathologic differential diagnosis [published online October 4, 2013]. Am J
Dermatopathol. doi:10.1097/DAD.0b013e3181cb3fdd. - Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228; quiz 229-230.
- Noh JW, Kim J, Kim JW. Localized scleroderma: a clinical study at a single center in Korea. Int J Rheum Dis. 2013;16:437-441.
- Vasquez R, Sendejo C, Jacobe H. Morphea and other localized forms of scleroderma. Curr Opin Rheumatol. 2012;24:685-693.
- Nijhawan RI, Bard S, Blyumin M, et al. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64:779-782.
- Haustein UF, Ziegler V. Environmentally induced systemic sclerosis-like disorders. Int J Dermatol. 1985;24:147-151.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol. 2009;36:2383-2396.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol. 1989;120:831-835.
- Crocker HR. Diseases of the Skin: Their Description, Pathology, Diagnosis, and Treatment. Philadelphia, PA: P. Blakiston Son & Co; 1905.
- Laetsch B, Hofer T, Lombriser N, et al. Irradiation-induced morphea: x-rays as triggers of autoimmunity. Dermatology. 2011;223:9-12.
- Shetty G, Lewis F, Thrush S. Morphea of the breast: case reports and review of literature. Breast J. 2007;13:302-304.
- Jacobson G, Bhatia S, Smith BJ, et al. Randomized trial of pentoxifylline and vitamin E vs standard follow-up after breast irradiation to prevent breast fibrosis, evaluated by tissue compliance meter. Int J Radiat Oncol Biol Phys. 2013;85:604-608.
To the Editor:
Postirradiation morphea (PIM) is a rare but well-documented phenomenon that primarily occurs in breast cancer patients who have received radiation therapy; however, it also has been reported in patients who have received radiation therapy for lymphoma as well as endocervical, endometrial, and gastric carcinomas.1 Importantly, clinicians must be able to recognize and differentiate this condition from other causes of new-onset induration and erythema of the breast, such as cancer recurrence, a new primary malignancy, or inflammatory etiologies (eg, radiation or contact dermatitis). Typically, PIM presents months to years after radiation therapy as an erythematous patch within the irradiated area that progressively becomes indurated. We report an unusual case of PIM with a reticulated appearance occurring 3 weeks after radiotherapy, chemotherapy, and surgery for an infiltrating ductal carcinoma of the left breast.
A 62-year-old woman presented to the dermatology department with a stage IIA, lymph node–negative, estrogen and progesterone receptor–negative, human epidermal growth factor receptor 2–negative infiltrating ductal carcinoma of the left breast. She was treated with a partial mastectomy of the left breast followed by external beam radiotherapy to the entire left breast in combination with chemotherapy (doxorubicin, cyclophosphamide, paclitaxel). The patient received 15 fractions of 270 cGy (4050 cGy total) with a weekly 600-cGy boost over 21 days without any complications.
Three weeks after finishing radiation therapy, the patient developed redness and swelling of the left breast that did not encompass the entire radiation field. There was no associated pain or pruritus. She was treated by her surgical oncologist with topical calendula and 3 courses of cephalexin for suspected mastitis with only modest improvement, then was referred to dermatology 3 months later.
At the initial dermatology evaluation, the patient reported little improvement after antibiotics and topical calendula. On physical examination, there were erythematous, reticulated, dusky, indurated patches on the entire left breast. The area of most pronounced induration surrounded the surgical scar on the left superior breast. Punch biopsy for hematoxylin and eosin staining and tissue cultures was obtained at this appointment. The patient was started on doxycycline 100 mg twice daily and was instructed to apply triamcinolone ointment 0.1% twice daily to the affected area. After 1 month of therapy, she reported slight improvement in the degree of erythema with this regimen, but the involved area continued to extend outside of the radiation field to the central chest wall and medial right breast (Figure 1). Two additional biopsies—one from the central chest and another from the right breast—were then taken over the course of 4 months, given the consistently inconclusive clinicopathologic nature and failure of the eruption to respond to antibiotics plus topical corticosteroids.
Punch biopsy from the central chest revealed a sparse perivascular infiltrate comprised predominantly of lymphocytes with occasional eosinophils (Figure 2). There were foci suggestive of early dermal sclerosis, an increased number of small blood vessels in the dermis, and scattered enlarged fibroblasts. Metastatic carcinoma was not identified. Although the histologic findings were not entirely specific, the changes were most suggestive of PIM, for which the patient was started on pentoxifylline (400 mg 3 times daily) and oral vitamin E supplementation (400 IU daily). At subsequent follow-up appointments, she showed markedly decreased skin erythema and induration.
Morphea, also known as localized scleroderma, is an inflammatory skin condition characterized by sclerosis of the dermis and subcutis leading to scarlike tissue formation. Worldwide incidence ranges from 0.4 to 2.7 cases per 100,000 individuals with a predilection for white women.2 Unlike systemic scleroderma, morphea patients lack Raynaud phenomenon and visceral involvement.3,4
There are several clinical subtypes of morphea, including plaque, linear, generalized, and pansclerotic morphea. Lesions may vary in appearance based on configuration, stage of development, and depth of involvement.4 During the earliest phases, morphea lesions are asymptomatic, asymmetrically distributed, erythematous to violaceous patches or subtly indurated plaques expanding centrifugally with a lilac ring. Central sclerosis with loss of follicles and sweat glands is a later finding associated with advanced disease. Moreover, some reports of early-stage morphea have suggested a reticulated or geographic vascular morphology that may be misdiagnosed for other conditions such as a port-wine stain.5
Local skin exposures have long been hypothesized to contribute to development of morphea, including infection, especially Borrelia burgdorferi; trauma; chronic venous insufficiency; cosmetic surgery; medications; and exposure to toxic cooking oils, silicones, silica, pesticides, organic solvents, and vinyl chloride.2,6,7
Radiation therapy is an often overlooked cause of morphea. It was first described in 1905 but then rarely discussed until a 1989 case series of 9 patients, 7 of whom had received irradiation for breast cancer.8,9 Today, the increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in PIM incidence.10
In contrast to other radiation-induced skin conditions, development of PIM is independent of the presence or absence of adjuvant chemotherapy, type of radiation therapy, or the total radiation dose or fractionation number, with reported doses ranging from less than 20.0 Gy to up to 59.4 Gy and dose fractions ranging from 10 to 30. In 20% to 30% of cases, PIM extends beyond the radiation field, sometimes involving distant sites never exposed to high-energy rays.1,10,11 This observation suggests a mechanism reliant on more widespread cascade rather than solely local tissue damage.
Prominent culture-negative, lymphoplasmacytic inflammation is another important diagnostic clue. Radiation dermatitis and fibrosis do not have the marked erythematous to violaceous hue seen in early morphea plaques. This color seen in early morphea plaques may be intense enough and in a geographic pattern, emulating a vascular lesion.
There is no standardized treatment of PIM, but traditional therapies for morphea may provide some benefit. Several randomized controlled clinical trials have shown success with pentoxifylline and oral vitamin E supplementation to treat or prevent radiation-induced breast fibrosis.12 Extrapolating from this data, our patient was started on this combination therapy and showed marked improvement in skin color and texture.
To the Editor:
Postirradiation morphea (PIM) is a rare but well-documented phenomenon that primarily occurs in breast cancer patients who have received radiation therapy; however, it also has been reported in patients who have received radiation therapy for lymphoma as well as endocervical, endometrial, and gastric carcinomas.1 Importantly, clinicians must be able to recognize and differentiate this condition from other causes of new-onset induration and erythema of the breast, such as cancer recurrence, a new primary malignancy, or inflammatory etiologies (eg, radiation or contact dermatitis). Typically, PIM presents months to years after radiation therapy as an erythematous patch within the irradiated area that progressively becomes indurated. We report an unusual case of PIM with a reticulated appearance occurring 3 weeks after radiotherapy, chemotherapy, and surgery for an infiltrating ductal carcinoma of the left breast.
A 62-year-old woman presented to the dermatology department with a stage IIA, lymph node–negative, estrogen and progesterone receptor–negative, human epidermal growth factor receptor 2–negative infiltrating ductal carcinoma of the left breast. She was treated with a partial mastectomy of the left breast followed by external beam radiotherapy to the entire left breast in combination with chemotherapy (doxorubicin, cyclophosphamide, paclitaxel). The patient received 15 fractions of 270 cGy (4050 cGy total) with a weekly 600-cGy boost over 21 days without any complications.
Three weeks after finishing radiation therapy, the patient developed redness and swelling of the left breast that did not encompass the entire radiation field. There was no associated pain or pruritus. She was treated by her surgical oncologist with topical calendula and 3 courses of cephalexin for suspected mastitis with only modest improvement, then was referred to dermatology 3 months later.
At the initial dermatology evaluation, the patient reported little improvement after antibiotics and topical calendula. On physical examination, there were erythematous, reticulated, dusky, indurated patches on the entire left breast. The area of most pronounced induration surrounded the surgical scar on the left superior breast. Punch biopsy for hematoxylin and eosin staining and tissue cultures was obtained at this appointment. The patient was started on doxycycline 100 mg twice daily and was instructed to apply triamcinolone ointment 0.1% twice daily to the affected area. After 1 month of therapy, she reported slight improvement in the degree of erythema with this regimen, but the involved area continued to extend outside of the radiation field to the central chest wall and medial right breast (Figure 1). Two additional biopsies—one from the central chest and another from the right breast—were then taken over the course of 4 months, given the consistently inconclusive clinicopathologic nature and failure of the eruption to respond to antibiotics plus topical corticosteroids.
Punch biopsy from the central chest revealed a sparse perivascular infiltrate comprised predominantly of lymphocytes with occasional eosinophils (Figure 2). There were foci suggestive of early dermal sclerosis, an increased number of small blood vessels in the dermis, and scattered enlarged fibroblasts. Metastatic carcinoma was not identified. Although the histologic findings were not entirely specific, the changes were most suggestive of PIM, for which the patient was started on pentoxifylline (400 mg 3 times daily) and oral vitamin E supplementation (400 IU daily). At subsequent follow-up appointments, she showed markedly decreased skin erythema and induration.
Morphea, also known as localized scleroderma, is an inflammatory skin condition characterized by sclerosis of the dermis and subcutis leading to scarlike tissue formation. Worldwide incidence ranges from 0.4 to 2.7 cases per 100,000 individuals with a predilection for white women.2 Unlike systemic scleroderma, morphea patients lack Raynaud phenomenon and visceral involvement.3,4
There are several clinical subtypes of morphea, including plaque, linear, generalized, and pansclerotic morphea. Lesions may vary in appearance based on configuration, stage of development, and depth of involvement.4 During the earliest phases, morphea lesions are asymptomatic, asymmetrically distributed, erythematous to violaceous patches or subtly indurated plaques expanding centrifugally with a lilac ring. Central sclerosis with loss of follicles and sweat glands is a later finding associated with advanced disease. Moreover, some reports of early-stage morphea have suggested a reticulated or geographic vascular morphology that may be misdiagnosed for other conditions such as a port-wine stain.5
Local skin exposures have long been hypothesized to contribute to development of morphea, including infection, especially Borrelia burgdorferi; trauma; chronic venous insufficiency; cosmetic surgery; medications; and exposure to toxic cooking oils, silicones, silica, pesticides, organic solvents, and vinyl chloride.2,6,7
Radiation therapy is an often overlooked cause of morphea. It was first described in 1905 but then rarely discussed until a 1989 case series of 9 patients, 7 of whom had received irradiation for breast cancer.8,9 Today, the increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in PIM incidence.10
In contrast to other radiation-induced skin conditions, development of PIM is independent of the presence or absence of adjuvant chemotherapy, type of radiation therapy, or the total radiation dose or fractionation number, with reported doses ranging from less than 20.0 Gy to up to 59.4 Gy and dose fractions ranging from 10 to 30. In 20% to 30% of cases, PIM extends beyond the radiation field, sometimes involving distant sites never exposed to high-energy rays.1,10,11 This observation suggests a mechanism reliant on more widespread cascade rather than solely local tissue damage.
Prominent culture-negative, lymphoplasmacytic inflammation is another important diagnostic clue. Radiation dermatitis and fibrosis do not have the marked erythematous to violaceous hue seen in early morphea plaques. This color seen in early morphea plaques may be intense enough and in a geographic pattern, emulating a vascular lesion.
There is no standardized treatment of PIM, but traditional therapies for morphea may provide some benefit. Several randomized controlled clinical trials have shown success with pentoxifylline and oral vitamin E supplementation to treat or prevent radiation-induced breast fibrosis.12 Extrapolating from this data, our patient was started on this combination therapy and showed marked improvement in skin color and texture.
- Morganroth PA, Dehoratius D, Curry H, et al. Postirradiation morphea: a case report with a review of the literature and summary of the clinicopathologic differential diagnosis [published online October 4, 2013]. Am J
Dermatopathol. doi:10.1097/DAD.0b013e3181cb3fdd. - Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228; quiz 229-230.
- Noh JW, Kim J, Kim JW. Localized scleroderma: a clinical study at a single center in Korea. Int J Rheum Dis. 2013;16:437-441.
- Vasquez R, Sendejo C, Jacobe H. Morphea and other localized forms of scleroderma. Curr Opin Rheumatol. 2012;24:685-693.
- Nijhawan RI, Bard S, Blyumin M, et al. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64:779-782.
- Haustein UF, Ziegler V. Environmentally induced systemic sclerosis-like disorders. Int J Dermatol. 1985;24:147-151.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol. 2009;36:2383-2396.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol. 1989;120:831-835.
- Crocker HR. Diseases of the Skin: Their Description, Pathology, Diagnosis, and Treatment. Philadelphia, PA: P. Blakiston Son & Co; 1905.
- Laetsch B, Hofer T, Lombriser N, et al. Irradiation-induced morphea: x-rays as triggers of autoimmunity. Dermatology. 2011;223:9-12.
- Shetty G, Lewis F, Thrush S. Morphea of the breast: case reports and review of literature. Breast J. 2007;13:302-304.
- Jacobson G, Bhatia S, Smith BJ, et al. Randomized trial of pentoxifylline and vitamin E vs standard follow-up after breast irradiation to prevent breast fibrosis, evaluated by tissue compliance meter. Int J Radiat Oncol Biol Phys. 2013;85:604-608.
- Morganroth PA, Dehoratius D, Curry H, et al. Postirradiation morphea: a case report with a review of the literature and summary of the clinicopathologic differential diagnosis [published online October 4, 2013]. Am J
Dermatopathol. doi:10.1097/DAD.0b013e3181cb3fdd. - Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228; quiz 229-230.
- Noh JW, Kim J, Kim JW. Localized scleroderma: a clinical study at a single center in Korea. Int J Rheum Dis. 2013;16:437-441.
- Vasquez R, Sendejo C, Jacobe H. Morphea and other localized forms of scleroderma. Curr Opin Rheumatol. 2012;24:685-693.
- Nijhawan RI, Bard S, Blyumin M, et al. Early localized morphea mimicking an acquired port-wine stain. J Am Acad Dermatol. 2011;64:779-782.
- Haustein UF, Ziegler V. Environmentally induced systemic sclerosis-like disorders. Int J Dermatol. 1985;24:147-151.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol. 2009;36:2383-2396.
- Colver GB, Rodger A, Mortimer PS, et al. Post-irradiation morphoea. Br J Dermatol. 1989;120:831-835.
- Crocker HR. Diseases of the Skin: Their Description, Pathology, Diagnosis, and Treatment. Philadelphia, PA: P. Blakiston Son & Co; 1905.
- Laetsch B, Hofer T, Lombriser N, et al. Irradiation-induced morphea: x-rays as triggers of autoimmunity. Dermatology. 2011;223:9-12.
- Shetty G, Lewis F, Thrush S. Morphea of the breast: case reports and review of literature. Breast J. 2007;13:302-304.
- Jacobson G, Bhatia S, Smith BJ, et al. Randomized trial of pentoxifylline and vitamin E vs standard follow-up after breast irradiation to prevent breast fibrosis, evaluated by tissue compliance meter. Int J Radiat Oncol Biol Phys. 2013;85:604-608.
Practice Points
- Radiation therapy is an often overlooked cause of morphea.
- The increasing popularity of lumpectomy plus radiation therapy for treatment of early-stage breast cancer has led to a rise in postirradiation morphea incidence.
- Tissue changes occur as early as weeks or as late as 32 years after radiation treatment.
- Postirradiation morphea may extend beyond the radiation field.
Allergy Testing in Dermatology and Beyond
Allergy testing typically refers to evaluation of a patient for suspected type I or type IV hypersensitivity.1,2 The possibility of type I hypersensitivity is raised in patients presenting with food allergies, allergic rhinitis, asthma, and immediate adverse reactions to medications, whereas type IV hypersensitivity is suspected in patients with eczematous eruptions, delayed adverse cutaneous reactions to medications, and failure of metallic implants (eg, metal joint replacements, cardiac stents) in conjunction with overlying skin rashes (Table 1).1-5 Type II (eg, pemphigus vulgaris) and type III (eg, IgA vasculitis) hypersensitivities are not evaluated with screening allergy tests.

Type I Sensitization
Type I hypersensitivity is an immediate hypersensitivity mediated predominantly by IgE activation of mast cells in the skin as well as the respiratory and gastric mucosa.1 Sensitization of an individual patient occurs when antigen-presenting cells induce a helper T cell (TH2) cytokine response leading to B-cell class switching and allergen-specific IgE production. Upon repeat exposure to the allergen, circulating antibodies then bind to high-affinity receptors on mast cells and basophils and initiate an allergic inflammatory response, leading to a clinical presentation of allergic rhinitis, urticaria, or immediate drug reactions. Confirming type I sensitization may be performed via serologic (in vitro) or skin testing (in vivo).5,6
Serologic Testing (In Vitro)
Serologic testing is a blood test that detects circulating IgE levels against specific allergens.5 The first such test, the radioallergosorbent test, was introduced in the 1970s but is not quantitative and is no longer used. Although common, it is inaccurate to describe current serum IgE (s-IgE) testing as radioallergosorbent testing. There are several US Food and Drug Administration-approved s-IgE assays in common use, and these tests may be helpful in elucidating relevant allergens and for tailoring therapy appropriately, which may consist of avoidance of certain foods or environmental agents and/or allergen immunotherapy.
Skin Testing (In Vivo)
Skin testing can be performed percutaneously (eg, percutaneous skin testing) or intradermally (eg, intradermal testing).6 Percutaneous skin testing is performed by placing a drop of allergen extract on the skin, after which a lancet is used to lightly scratch the skin; intradermal testing is performed by injecting a small amount of allergen extract into the dermis. In both cases, the skin is evaluated after 15 to 20 minutes for the presence and size of a cutaneous wheal. Medications with antihistaminergic activity must be discontinued prior to testing. Both s-IgE and skin testing assess for type I hypersensitivity, and factors such as extensive rash, concern for anaphylaxis, or inability to discontinue antihistamines may favor s-IgE testing versus skin testing. False-positive results can occur with both tests, and for this reason, test results should always be interpreted in conjunction with clinical examination and patient history to determine relevant allergies.
Type IV Sensitization
Type IV hypersensitivity is a delayed hypersensitivity mediated primarily by lymphocytes.2 Sensitization occurs when haptens bind to host proteins and are presented by epidermal and dermal dendritic cells to T lymphocytes in the skin. These lymphocytes then migrate to regional lymph nodes where antigen-specific T lymphocytes are produced and home back to the skin. Upon reexposure to the allergen, these memory T lymphocytes become activated and incite a delayed allergic response. Confirming type IV hypersensitivity primarily is accomplished via patch testing, though other testing modalities exist.
Skin Biopsy
Biopsy is sometimes performed in the workup of an individual presenting with allergic contact dermatitis (ACD) and typically will show spongiosis with normal stratum corneum and epidermal thickness in the setting of acute ACD and mild to marked acanthosis and parakeratosis in chronic ACD.7 The findings, however, are nonspecific and the differential of these histopathologic findings encompasses nummular dermatitis, atopic dermatitis, irritant contact dermatitis, and dyshidrotic eczema, among others. The presence of eosinophils and Langerhans cell microabscesses may provide supportive evidence for ACD over the other spongiotic dermatitides.7,8
Patch Testing
Patch testing is the gold standard in diagnosing type IV hypersensitivities resulting in a clinical presentation of ACD. Hundreds of allergens are commercially available for patch testing, and more commonly tested allergens fall into one of several categories, such as cosmetic preservatives, rubbers, metals, textiles, fragrances, adhesives, antibiotics, plants, and even corticosteroids. Of note, a common misconception is that ACD must result from new exposures; however, patients may develop ACD secondary to an exposure or product they have been using for many years without a problem.
Three commonly used screening series are the thin-layer rapid use epicutaneous (T.R.U.E.) test (SmartPractice), North American Contact Dermatitis Group screening series, and American Contact Dermatitis Society Core 80 allergen series, which have some variation in the type and number of allergens included (Table 2). The T.R.U.E. test will miss a notable number of clinically relevant allergens in comparison to the North American Contact Dermatitis Group and American Contact Dermatitis Society Core series, and it may be of particularly low utility in identifying fragrance or preservative ACD.9
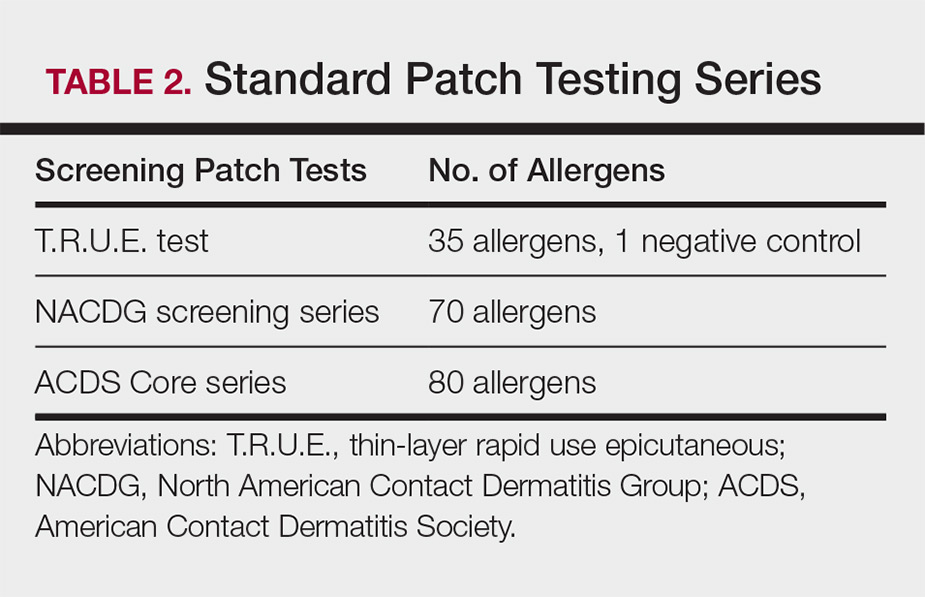
Allergens are placed on the back in chambers in a petrolatum or aqueous medium. The patches remain affixed for 48 hours, during which time the patient is asked to refrain from showering or exercising to prevent loss of patches. The patient's skin is then evaluated for reactions to allergens on 2 separate occasions: at the time of patch removal 48 hours after initial placement, then the areas of patches are marked for delayed readings at day 4 to day 7 after initial patch placement. Results are scored based on the degree of the inflammatory reaction (Table 3). Delayed readings beyond day 7 may be necessary for metals, specific preservatives (eg, dodecyl gallate, propolis), and neomycin.10
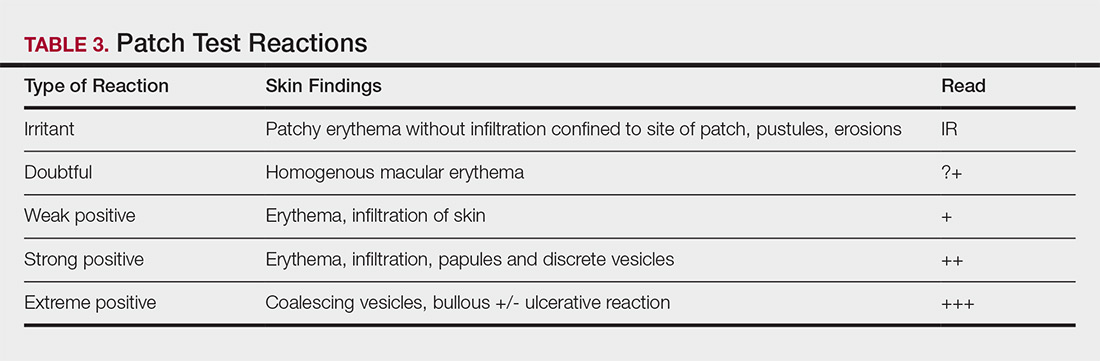
There is a wide spectrum of cutaneous disease that should prompt consideration of patch testing, including well-circumscribed eczematous dermatitis (eg, recurrent lip, hand, and foot dermatitis); patchy or diffuse eczema, especially if recently worsened and/or unresponsive to topical steroids; lichenoid eruptions, particularly of mucosal surfaces; mucous membrane eruptions (eg, stomatitis, vulvitis); and eczematous presentations that raise concern for airborne (photodistributed) or systemic contact dermatitis.11-13 Although further studies of efficacy and safety are ongoing, patch testing also may be useful in the diagnosis of nonimmediate cutaneous adverse drug reactions, especially fixed drug eruptions, acute generalized exanthematous pustulosis, systemic contact dermatitis from medications, and drug-induced hypersensitivity syndrome.3 Lastly, patients with type IV hypersensitivity to metals, adhesives, or antibiotics used in metallic orthopedic or cardiac implants may experience implant failure, regional contact dermatitis, or both, and benefit from patch testing prior to implant replacement to assess for potential allergens. Of the joints that fail, it is estimated that up to 5% are due to metal hypersensitivity.4
Throughout patch testing, patients may continue to manage their skin condition with oral antihistamines and topical steroids, though application to the site at which the patches are applied should be avoided throughout patch testing and during the week prior. According to expert consensus, immunosuppressive medications that are less likely to impact patch testing and therefore may be continued include low-dose methotrexate, oral prednisone less than 10 mg daily, biologic therapy, and low-dose cyclosporine (<2 mg/kg daily). Therapeutic interventions that are more likely to impact patch testing and should be avoided include phototherapy or extensive sun exposure within a week prior to testing, oral prednisone more than 10 mg daily, intramuscular triamcinolone within the preceding month, and high-dose cyclosporine (>2 mg/kg daily).14
An important component to successful patch testing is posttest patient counseling. Providers can create a safe list of products for patients by logging onto the American Contact Dermatitis Society website and accessing the Contact Allergen Management Program (CAMP).15 All relevant allergens found on patch testing may be selected and patient-specific identification codes generated. Once these codes are entered into the CAMP app on the patient's cellular device, a personalized, regularly updated list of safe products appears for many categories of products, including shampoos, sunscreens, moisturizers, cosmetic products, and laundry or dish detergents, among others. Of note, this app is not helpful for avoidance in patients with textile allergies. Patients should be counseled that improvement occurs with avoidance, which usually occurs within weeks but may slowly occur over time in some cases.
Lymphocyte Transformation Test (In Vitro)
The lymphocyte transformation test is an experimental in vitro test for type IV hypersensitivity. This serologic test utilizes allergens to stimulate memory T lymphocytes in vitro and measures the degree of response to the allergen. Although this test has generated excitement, particularly for the potential to safely evaluate for severe adverse cutaneous drug reactions, it currently is not the standard of care and is not utilized in the United States.16
Conclusion
Dermatologists play a vital role in the workup of suspected type IV hypersensitivities. Patch testing is an important but underutilized tool in the arsenal of allergy testing and may be indicated in a wide variety of cutaneous presentations, adverse reactions to medications, and implanted device failures. Identification and avoidance of a culprit allergen has the potential to lead to complete resolution of disease and notable improvement in quality of life for patients.
Acknowledgments
The author thanks Nina Botto, MD (San Francisco, California), for her mentorship in the arena of ACD as well as the Women's Dermatologic Society for the support they provided through the mentorship program.
- Oettgen H, Broide DH. Introduction to the mechanisms of allergic disease. In: Holgate ST, Church MK, Broide DH, et al, eds. Allergy. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:1-32.
- Werfel T, Kapp A. Atopic dermatitis and allergic contact dermatitis. In: Holgate ST, Church MK, Broide DH, et al, eds. Allergy. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:263-286.
- Zinn A, Gayam S, Chelliah MP, et al. Patch testing for nonimmediate cutaneous adverse drug reactions. J Am Acad Dermatol. 2018;78:421-423.
- Thyssen JP, Menne T, Schalock PC, et al. Pragmatic approach to the clinical work-up of patients with putative allergic disease to metallic orthopaedic implants before and after surgery. Br J Dermatol. 2011;164:473-478.
- Cox L. Overview of serological-specific IgE antibody testing in children. Curr Allergy Asthma Rep. 2011;11:447-453.
- Dolen WK. Skin testing and immunoassays for allergen-specific IgE. Clin Rev Allergy Immunol. 2001;21:229-239.
- Keeling BH, Gavino AC, Gavino AC. Skin biopsy, the allergists' tool: how to interpret a report. Curr Allergy Asthma Rep. 2015;15:62.
- Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504.
- DeKoven JG, Warshaw EM, Belsito DV. North American Contact Dermatitis Group patch test results 2013-2014. Dermatitis. 2017;28:33-46.
- Davis MD, Bhate K, Rohlinger AL, et al. Delayed patch test reading after 5 days: the Mayo Clinic experience. J Am Acad Dermatol. 2008;59:225-233.
- Rajagopalan R, Anderson RT. The profile of a patient with contact dermatitis and a suspicion of contact allergy (history, physical characteristics, and dermatology-specific quality of life). Am J Contact Dermat. 1997;8:26-31.
- Huygens S, Goossens A. An update on airborne contact dermatitis. Contact Dermatitis. 2001;44:1-6.
- Salam TN, Fowler JF. Balsam-related systemic contact dermatitis. J Am Acad Dermatol. 2001;45:377-381.
- Fowler JF, Maibach HI, Zirwas M, et al. Effects of immunomodulatory agents on patch testing: expert opinion 2012. Dermatitis. 2012;23:301-303.
- ACDS CAMP. American Contact Dermatitis Society website. https://www.contactderm.org/i4a/pages/index.cfm?pageid=3489. Accessed November 14, 2018.
- Popple A, Williams J, Maxwell G, et al. The lymphocyte transformation test in allergic contact dermatitis: new opportunities. J Immunotoxicol. 2016;13:84-91.
Allergy testing typically refers to evaluation of a patient for suspected type I or type IV hypersensitivity.1,2 The possibility of type I hypersensitivity is raised in patients presenting with food allergies, allergic rhinitis, asthma, and immediate adverse reactions to medications, whereas type IV hypersensitivity is suspected in patients with eczematous eruptions, delayed adverse cutaneous reactions to medications, and failure of metallic implants (eg, metal joint replacements, cardiac stents) in conjunction with overlying skin rashes (Table 1).1-5 Type II (eg, pemphigus vulgaris) and type III (eg, IgA vasculitis) hypersensitivities are not evaluated with screening allergy tests.
Type I Sensitization
Type I hypersensitivity is an immediate hypersensitivity mediated predominantly by IgE activation of mast cells in the skin as well as the respiratory and gastric mucosa.1 Sensitization of an individual patient occurs when antigen-presenting cells induce a helper T cell (TH2) cytokine response leading to B-cell class switching and allergen-specific IgE production. Upon repeat exposure to the allergen, circulating antibodies then bind to high-affinity receptors on mast cells and basophils and initiate an allergic inflammatory response, leading to a clinical presentation of allergic rhinitis, urticaria, or immediate drug reactions. Confirming type I sensitization may be performed via serologic (in vitro) or skin testing (in vivo).5,6
Serologic Testing (In Vitro)
Serologic testing is a blood test that detects circulating IgE levels against specific allergens.5 The first such test, the radioallergosorbent test, was introduced in the 1970s but is not quantitative and is no longer used. Although common, it is inaccurate to describe current serum IgE (s-IgE) testing as radioallergosorbent testing. There are several US Food and Drug Administration-approved s-IgE assays in common use, and these tests may be helpful in elucidating relevant allergens and for tailoring therapy appropriately, which may consist of avoidance of certain foods or environmental agents and/or allergen immunotherapy.
Skin Testing (In Vivo)
Skin testing can be performed percutaneously (eg, percutaneous skin testing) or intradermally (eg, intradermal testing).6 Percutaneous skin testing is performed by placing a drop of allergen extract on the skin, after which a lancet is used to lightly scratch the skin; intradermal testing is performed by injecting a small amount of allergen extract into the dermis. In both cases, the skin is evaluated after 15 to 20 minutes for the presence and size of a cutaneous wheal. Medications with antihistaminergic activity must be discontinued prior to testing. Both s-IgE and skin testing assess for type I hypersensitivity, and factors such as extensive rash, concern for anaphylaxis, or inability to discontinue antihistamines may favor s-IgE testing versus skin testing. False-positive results can occur with both tests, and for this reason, test results should always be interpreted in conjunction with clinical examination and patient history to determine relevant allergies.
Type IV Sensitization
Type IV hypersensitivity is a delayed hypersensitivity mediated primarily by lymphocytes.2 Sensitization occurs when haptens bind to host proteins and are presented by epidermal and dermal dendritic cells to T lymphocytes in the skin. These lymphocytes then migrate to regional lymph nodes where antigen-specific T lymphocytes are produced and home back to the skin. Upon reexposure to the allergen, these memory T lymphocytes become activated and incite a delayed allergic response. Confirming type IV hypersensitivity primarily is accomplished via patch testing, though other testing modalities exist.
Skin Biopsy
Biopsy is sometimes performed in the workup of an individual presenting with allergic contact dermatitis (ACD) and typically will show spongiosis with normal stratum corneum and epidermal thickness in the setting of acute ACD and mild to marked acanthosis and parakeratosis in chronic ACD.7 The findings, however, are nonspecific and the differential of these histopathologic findings encompasses nummular dermatitis, atopic dermatitis, irritant contact dermatitis, and dyshidrotic eczema, among others. The presence of eosinophils and Langerhans cell microabscesses may provide supportive evidence for ACD over the other spongiotic dermatitides.7,8
Patch Testing
Patch testing is the gold standard in diagnosing type IV hypersensitivities resulting in a clinical presentation of ACD. Hundreds of allergens are commercially available for patch testing, and more commonly tested allergens fall into one of several categories, such as cosmetic preservatives, rubbers, metals, textiles, fragrances, adhesives, antibiotics, plants, and even corticosteroids. Of note, a common misconception is that ACD must result from new exposures; however, patients may develop ACD secondary to an exposure or product they have been using for many years without a problem.
Three commonly used screening series are the thin-layer rapid use epicutaneous (T.R.U.E.) test (SmartPractice), North American Contact Dermatitis Group screening series, and American Contact Dermatitis Society Core 80 allergen series, which have some variation in the type and number of allergens included (Table 2). The T.R.U.E. test will miss a notable number of clinically relevant allergens in comparison to the North American Contact Dermatitis Group and American Contact Dermatitis Society Core series, and it may be of particularly low utility in identifying fragrance or preservative ACD.9
Allergens are placed on the back in chambers in a petrolatum or aqueous medium. The patches remain affixed for 48 hours, during which time the patient is asked to refrain from showering or exercising to prevent loss of patches. The patient's skin is then evaluated for reactions to allergens on 2 separate occasions: at the time of patch removal 48 hours after initial placement, then the areas of patches are marked for delayed readings at day 4 to day 7 after initial patch placement. Results are scored based on the degree of the inflammatory reaction (Table 3). Delayed readings beyond day 7 may be necessary for metals, specific preservatives (eg, dodecyl gallate, propolis), and neomycin.10
There is a wide spectrum of cutaneous disease that should prompt consideration of patch testing, including well-circumscribed eczematous dermatitis (eg, recurrent lip, hand, and foot dermatitis); patchy or diffuse eczema, especially if recently worsened and/or unresponsive to topical steroids; lichenoid eruptions, particularly of mucosal surfaces; mucous membrane eruptions (eg, stomatitis, vulvitis); and eczematous presentations that raise concern for airborne (photodistributed) or systemic contact dermatitis.11-13 Although further studies of efficacy and safety are ongoing, patch testing also may be useful in the diagnosis of nonimmediate cutaneous adverse drug reactions, especially fixed drug eruptions, acute generalized exanthematous pustulosis, systemic contact dermatitis from medications, and drug-induced hypersensitivity syndrome.3 Lastly, patients with type IV hypersensitivity to metals, adhesives, or antibiotics used in metallic orthopedic or cardiac implants may experience implant failure, regional contact dermatitis, or both, and benefit from patch testing prior to implant replacement to assess for potential allergens. Of the joints that fail, it is estimated that up to 5% are due to metal hypersensitivity.4
Throughout patch testing, patients may continue to manage their skin condition with oral antihistamines and topical steroids, though application to the site at which the patches are applied should be avoided throughout patch testing and during the week prior. According to expert consensus, immunosuppressive medications that are less likely to impact patch testing and therefore may be continued include low-dose methotrexate, oral prednisone less than 10 mg daily, biologic therapy, and low-dose cyclosporine (<2 mg/kg daily). Therapeutic interventions that are more likely to impact patch testing and should be avoided include phototherapy or extensive sun exposure within a week prior to testing, oral prednisone more than 10 mg daily, intramuscular triamcinolone within the preceding month, and high-dose cyclosporine (>2 mg/kg daily).14
An important component to successful patch testing is posttest patient counseling. Providers can create a safe list of products for patients by logging onto the American Contact Dermatitis Society website and accessing the Contact Allergen Management Program (CAMP).15 All relevant allergens found on patch testing may be selected and patient-specific identification codes generated. Once these codes are entered into the CAMP app on the patient's cellular device, a personalized, regularly updated list of safe products appears for many categories of products, including shampoos, sunscreens, moisturizers, cosmetic products, and laundry or dish detergents, among others. Of note, this app is not helpful for avoidance in patients with textile allergies. Patients should be counseled that improvement occurs with avoidance, which usually occurs within weeks but may slowly occur over time in some cases.
Lymphocyte Transformation Test (In Vitro)
The lymphocyte transformation test is an experimental in vitro test for type IV hypersensitivity. This serologic test utilizes allergens to stimulate memory T lymphocytes in vitro and measures the degree of response to the allergen. Although this test has generated excitement, particularly for the potential to safely evaluate for severe adverse cutaneous drug reactions, it currently is not the standard of care and is not utilized in the United States.16
Conclusion
Dermatologists play a vital role in the workup of suspected type IV hypersensitivities. Patch testing is an important but underutilized tool in the arsenal of allergy testing and may be indicated in a wide variety of cutaneous presentations, adverse reactions to medications, and implanted device failures. Identification and avoidance of a culprit allergen has the potential to lead to complete resolution of disease and notable improvement in quality of life for patients.
Acknowledgments
The author thanks Nina Botto, MD (San Francisco, California), for her mentorship in the arena of ACD as well as the Women's Dermatologic Society for the support they provided through the mentorship program.
Allergy testing typically refers to evaluation of a patient for suspected type I or type IV hypersensitivity.1,2 The possibility of type I hypersensitivity is raised in patients presenting with food allergies, allergic rhinitis, asthma, and immediate adverse reactions to medications, whereas type IV hypersensitivity is suspected in patients with eczematous eruptions, delayed adverse cutaneous reactions to medications, and failure of metallic implants (eg, metal joint replacements, cardiac stents) in conjunction with overlying skin rashes (Table 1).1-5 Type II (eg, pemphigus vulgaris) and type III (eg, IgA vasculitis) hypersensitivities are not evaluated with screening allergy tests.
Type I Sensitization
Type I hypersensitivity is an immediate hypersensitivity mediated predominantly by IgE activation of mast cells in the skin as well as the respiratory and gastric mucosa.1 Sensitization of an individual patient occurs when antigen-presenting cells induce a helper T cell (TH2) cytokine response leading to B-cell class switching and allergen-specific IgE production. Upon repeat exposure to the allergen, circulating antibodies then bind to high-affinity receptors on mast cells and basophils and initiate an allergic inflammatory response, leading to a clinical presentation of allergic rhinitis, urticaria, or immediate drug reactions. Confirming type I sensitization may be performed via serologic (in vitro) or skin testing (in vivo).5,6
Serologic Testing (In Vitro)
Serologic testing is a blood test that detects circulating IgE levels against specific allergens.5 The first such test, the radioallergosorbent test, was introduced in the 1970s but is not quantitative and is no longer used. Although common, it is inaccurate to describe current serum IgE (s-IgE) testing as radioallergosorbent testing. There are several US Food and Drug Administration-approved s-IgE assays in common use, and these tests may be helpful in elucidating relevant allergens and for tailoring therapy appropriately, which may consist of avoidance of certain foods or environmental agents and/or allergen immunotherapy.
Skin Testing (In Vivo)
Skin testing can be performed percutaneously (eg, percutaneous skin testing) or intradermally (eg, intradermal testing).6 Percutaneous skin testing is performed by placing a drop of allergen extract on the skin, after which a lancet is used to lightly scratch the skin; intradermal testing is performed by injecting a small amount of allergen extract into the dermis. In both cases, the skin is evaluated after 15 to 20 minutes for the presence and size of a cutaneous wheal. Medications with antihistaminergic activity must be discontinued prior to testing. Both s-IgE and skin testing assess for type I hypersensitivity, and factors such as extensive rash, concern for anaphylaxis, or inability to discontinue antihistamines may favor s-IgE testing versus skin testing. False-positive results can occur with both tests, and for this reason, test results should always be interpreted in conjunction with clinical examination and patient history to determine relevant allergies.
Type IV Sensitization
Type IV hypersensitivity is a delayed hypersensitivity mediated primarily by lymphocytes.2 Sensitization occurs when haptens bind to host proteins and are presented by epidermal and dermal dendritic cells to T lymphocytes in the skin. These lymphocytes then migrate to regional lymph nodes where antigen-specific T lymphocytes are produced and home back to the skin. Upon reexposure to the allergen, these memory T lymphocytes become activated and incite a delayed allergic response. Confirming type IV hypersensitivity primarily is accomplished via patch testing, though other testing modalities exist.
Skin Biopsy
Biopsy is sometimes performed in the workup of an individual presenting with allergic contact dermatitis (ACD) and typically will show spongiosis with normal stratum corneum and epidermal thickness in the setting of acute ACD and mild to marked acanthosis and parakeratosis in chronic ACD.7 The findings, however, are nonspecific and the differential of these histopathologic findings encompasses nummular dermatitis, atopic dermatitis, irritant contact dermatitis, and dyshidrotic eczema, among others. The presence of eosinophils and Langerhans cell microabscesses may provide supportive evidence for ACD over the other spongiotic dermatitides.7,8
Patch Testing
Patch testing is the gold standard in diagnosing type IV hypersensitivities resulting in a clinical presentation of ACD. Hundreds of allergens are commercially available for patch testing, and more commonly tested allergens fall into one of several categories, such as cosmetic preservatives, rubbers, metals, textiles, fragrances, adhesives, antibiotics, plants, and even corticosteroids. Of note, a common misconception is that ACD must result from new exposures; however, patients may develop ACD secondary to an exposure or product they have been using for many years without a problem.
Three commonly used screening series are the thin-layer rapid use epicutaneous (T.R.U.E.) test (SmartPractice), North American Contact Dermatitis Group screening series, and American Contact Dermatitis Society Core 80 allergen series, which have some variation in the type and number of allergens included (Table 2). The T.R.U.E. test will miss a notable number of clinically relevant allergens in comparison to the North American Contact Dermatitis Group and American Contact Dermatitis Society Core series, and it may be of particularly low utility in identifying fragrance or preservative ACD.9
Allergens are placed on the back in chambers in a petrolatum or aqueous medium. The patches remain affixed for 48 hours, during which time the patient is asked to refrain from showering or exercising to prevent loss of patches. The patient's skin is then evaluated for reactions to allergens on 2 separate occasions: at the time of patch removal 48 hours after initial placement, then the areas of patches are marked for delayed readings at day 4 to day 7 after initial patch placement. Results are scored based on the degree of the inflammatory reaction (Table 3). Delayed readings beyond day 7 may be necessary for metals, specific preservatives (eg, dodecyl gallate, propolis), and neomycin.10
There is a wide spectrum of cutaneous disease that should prompt consideration of patch testing, including well-circumscribed eczematous dermatitis (eg, recurrent lip, hand, and foot dermatitis); patchy or diffuse eczema, especially if recently worsened and/or unresponsive to topical steroids; lichenoid eruptions, particularly of mucosal surfaces; mucous membrane eruptions (eg, stomatitis, vulvitis); and eczematous presentations that raise concern for airborne (photodistributed) or systemic contact dermatitis.11-13 Although further studies of efficacy and safety are ongoing, patch testing also may be useful in the diagnosis of nonimmediate cutaneous adverse drug reactions, especially fixed drug eruptions, acute generalized exanthematous pustulosis, systemic contact dermatitis from medications, and drug-induced hypersensitivity syndrome.3 Lastly, patients with type IV hypersensitivity to metals, adhesives, or antibiotics used in metallic orthopedic or cardiac implants may experience implant failure, regional contact dermatitis, or both, and benefit from patch testing prior to implant replacement to assess for potential allergens. Of the joints that fail, it is estimated that up to 5% are due to metal hypersensitivity.4
Throughout patch testing, patients may continue to manage their skin condition with oral antihistamines and topical steroids, though application to the site at which the patches are applied should be avoided throughout patch testing and during the week prior. According to expert consensus, immunosuppressive medications that are less likely to impact patch testing and therefore may be continued include low-dose methotrexate, oral prednisone less than 10 mg daily, biologic therapy, and low-dose cyclosporine (<2 mg/kg daily). Therapeutic interventions that are more likely to impact patch testing and should be avoided include phototherapy or extensive sun exposure within a week prior to testing, oral prednisone more than 10 mg daily, intramuscular triamcinolone within the preceding month, and high-dose cyclosporine (>2 mg/kg daily).14
An important component to successful patch testing is posttest patient counseling. Providers can create a safe list of products for patients by logging onto the American Contact Dermatitis Society website and accessing the Contact Allergen Management Program (CAMP).15 All relevant allergens found on patch testing may be selected and patient-specific identification codes generated. Once these codes are entered into the CAMP app on the patient's cellular device, a personalized, regularly updated list of safe products appears for many categories of products, including shampoos, sunscreens, moisturizers, cosmetic products, and laundry or dish detergents, among others. Of note, this app is not helpful for avoidance in patients with textile allergies. Patients should be counseled that improvement occurs with avoidance, which usually occurs within weeks but may slowly occur over time in some cases.
Lymphocyte Transformation Test (In Vitro)
The lymphocyte transformation test is an experimental in vitro test for type IV hypersensitivity. This serologic test utilizes allergens to stimulate memory T lymphocytes in vitro and measures the degree of response to the allergen. Although this test has generated excitement, particularly for the potential to safely evaluate for severe adverse cutaneous drug reactions, it currently is not the standard of care and is not utilized in the United States.16
Conclusion
Dermatologists play a vital role in the workup of suspected type IV hypersensitivities. Patch testing is an important but underutilized tool in the arsenal of allergy testing and may be indicated in a wide variety of cutaneous presentations, adverse reactions to medications, and implanted device failures. Identification and avoidance of a culprit allergen has the potential to lead to complete resolution of disease and notable improvement in quality of life for patients.
Acknowledgments
The author thanks Nina Botto, MD (San Francisco, California), for her mentorship in the arena of ACD as well as the Women's Dermatologic Society for the support they provided through the mentorship program.
- Oettgen H, Broide DH. Introduction to the mechanisms of allergic disease. In: Holgate ST, Church MK, Broide DH, et al, eds. Allergy. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:1-32.
- Werfel T, Kapp A. Atopic dermatitis and allergic contact dermatitis. In: Holgate ST, Church MK, Broide DH, et al, eds. Allergy. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:263-286.
- Zinn A, Gayam S, Chelliah MP, et al. Patch testing for nonimmediate cutaneous adverse drug reactions. J Am Acad Dermatol. 2018;78:421-423.
- Thyssen JP, Menne T, Schalock PC, et al. Pragmatic approach to the clinical work-up of patients with putative allergic disease to metallic orthopaedic implants before and after surgery. Br J Dermatol. 2011;164:473-478.
- Cox L. Overview of serological-specific IgE antibody testing in children. Curr Allergy Asthma Rep. 2011;11:447-453.
- Dolen WK. Skin testing and immunoassays for allergen-specific IgE. Clin Rev Allergy Immunol. 2001;21:229-239.
- Keeling BH, Gavino AC, Gavino AC. Skin biopsy, the allergists' tool: how to interpret a report. Curr Allergy Asthma Rep. 2015;15:62.
- Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504.
- DeKoven JG, Warshaw EM, Belsito DV. North American Contact Dermatitis Group patch test results 2013-2014. Dermatitis. 2017;28:33-46.
- Davis MD, Bhate K, Rohlinger AL, et al. Delayed patch test reading after 5 days: the Mayo Clinic experience. J Am Acad Dermatol. 2008;59:225-233.
- Rajagopalan R, Anderson RT. The profile of a patient with contact dermatitis and a suspicion of contact allergy (history, physical characteristics, and dermatology-specific quality of life). Am J Contact Dermat. 1997;8:26-31.
- Huygens S, Goossens A. An update on airborne contact dermatitis. Contact Dermatitis. 2001;44:1-6.
- Salam TN, Fowler JF. Balsam-related systemic contact dermatitis. J Am Acad Dermatol. 2001;45:377-381.
- Fowler JF, Maibach HI, Zirwas M, et al. Effects of immunomodulatory agents on patch testing: expert opinion 2012. Dermatitis. 2012;23:301-303.
- ACDS CAMP. American Contact Dermatitis Society website. https://www.contactderm.org/i4a/pages/index.cfm?pageid=3489. Accessed November 14, 2018.
- Popple A, Williams J, Maxwell G, et al. The lymphocyte transformation test in allergic contact dermatitis: new opportunities. J Immunotoxicol. 2016;13:84-91.
- Oettgen H, Broide DH. Introduction to the mechanisms of allergic disease. In: Holgate ST, Church MK, Broide DH, et al, eds. Allergy. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:1-32.
- Werfel T, Kapp A. Atopic dermatitis and allergic contact dermatitis. In: Holgate ST, Church MK, Broide DH, et al, eds. Allergy. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:263-286.
- Zinn A, Gayam S, Chelliah MP, et al. Patch testing for nonimmediate cutaneous adverse drug reactions. J Am Acad Dermatol. 2018;78:421-423.
- Thyssen JP, Menne T, Schalock PC, et al. Pragmatic approach to the clinical work-up of patients with putative allergic disease to metallic orthopaedic implants before and after surgery. Br J Dermatol. 2011;164:473-478.
- Cox L. Overview of serological-specific IgE antibody testing in children. Curr Allergy Asthma Rep. 2011;11:447-453.
- Dolen WK. Skin testing and immunoassays for allergen-specific IgE. Clin Rev Allergy Immunol. 2001;21:229-239.
- Keeling BH, Gavino AC, Gavino AC. Skin biopsy, the allergists' tool: how to interpret a report. Curr Allergy Asthma Rep. 2015;15:62.
- Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504.
- DeKoven JG, Warshaw EM, Belsito DV. North American Contact Dermatitis Group patch test results 2013-2014. Dermatitis. 2017;28:33-46.
- Davis MD, Bhate K, Rohlinger AL, et al. Delayed patch test reading after 5 days: the Mayo Clinic experience. J Am Acad Dermatol. 2008;59:225-233.
- Rajagopalan R, Anderson RT. The profile of a patient with contact dermatitis and a suspicion of contact allergy (history, physical characteristics, and dermatology-specific quality of life). Am J Contact Dermat. 1997;8:26-31.
- Huygens S, Goossens A. An update on airborne contact dermatitis. Contact Dermatitis. 2001;44:1-6.
- Salam TN, Fowler JF. Balsam-related systemic contact dermatitis. J Am Acad Dermatol. 2001;45:377-381.
- Fowler JF, Maibach HI, Zirwas M, et al. Effects of immunomodulatory agents on patch testing: expert opinion 2012. Dermatitis. 2012;23:301-303.
- ACDS CAMP. American Contact Dermatitis Society website. https://www.contactderm.org/i4a/pages/index.cfm?pageid=3489. Accessed November 14, 2018.
- Popple A, Williams J, Maxwell G, et al. The lymphocyte transformation test in allergic contact dermatitis: new opportunities. J Immunotoxicol. 2016;13:84-91.

Health Care Barriers and Quality of Life in Central Centrifugal Cicatricial Alopecia Patients
The etiology of central centrifugal cicatricial alopecia (CCCA), a clinical and histological pattern of hair loss on the central scalp, has been well studied. This disease is chronic and progressive, with extensive follicular destruction and eventual burnout.1,2 Central centrifugal cicatricial alopecia is most commonly seen in patients of African descent and has been shown to be 1 of the 5 most common dermatologic diagnoses in black patients.3,4 The top 5 dermatologic diagnoses within this population include acne vulgaris (28.4%), dyschromia (19.9%), eczema (9.1%), alopecia (8.3%), and seborrheic dermatitis (6.7%).4 The incidence rate of CCCA is estimated to be 5.6%.3,5 Most patients are women, with onset between the second and fourth decades of life.6
Central centrifugal cicatricial alopecia treatment efficacy is inversely correlated with disease duration. The primary goal of treatment is to prevent progression. Efforts are made to stimulate regrowth in areas that are not permanently scarred. When patients present with a substantial amount of scarring hair loss, dermatologists often are limited in their ability to achieve a cosmetically acceptable pattern of growth. Generally, hair is connected to a sense of self-worth in black women, and any type of hair loss has been shown to lead to frustration and decreased self-esteem.7 A 1994 study showed that 75% (44/58) of women with androgenetic alopecia had decreased self-esteem and 50% (29/58) had social challenges.8
The purpose of this pilot study was to determine the personal, historical, logistical, or environmental factors that preclude women from obtaining medical care for CCCA and to investigate how CCCA affects quality of life (QOL) and psychological well-being.
Methods
The investigators designed a survey study of adult, English-speaking, black women diagnosed with CCCA at the Northwestern University Department of Dermatology (Chicago, Illinois) between 2011 and 2017. Patients were selected from the electronic data warehouse compiled by the Department of Dermatology and were included if they fulfilled the following criteria: evaluated in the dermatology department between September 1, 2011, and September 30, 2017, by any faculty physician; diagnosed with CCCA; and aged 18 years or older. Patients were excluded if they did not speak English, as interpreters were not available. All patients who fulfilled the inclusion criteria provided signed informed consent prior to participation. All surveys were disseminated in the office or via telephone from fall 2016 to spring 2017 and took 10 to 15 minutes to complete. The research was approved by the authors’ institutional review board (IRB ID STU00203449).
Survey Instrument
The
Data Analysis
Analyses were completed using data analysis software JMP Pro 13 from SAS and a Microsoft Excel spreadsheet. Continuous data were presented as mean, SD, median, minimum, and maximum. Categorical data were presented as counts and percentages. Nine QOL items were aggregated into a self-esteem category (questions 30–38).
Cronbach α, a statistical measure of internal consistency and how closely related items are in a group, was used to evaluate internal consistency reliability; values of 0.70 or greater indicate acceptable reliability.
Results
Of 501 individuals contacted, 34 completed the survey (7% completion rate). Nonrespondents included 7 who refused to participate and 460 who could not be contacted. All respondents self-identified as black women. Median age at time of survey administration was 46 years (range, 28–79 years); median age at CCCA diagnosis was 42 years (range, 15–73 years). Respondents did not significantly differ in age from nonrespondents (P=.46). The majority of respondents had an associate’s degree, bachelor’s degree, or advanced degree of education (master of arts, doctor of medicine, doctor of jurisprudence, doctor of philosophy); however, 8 women reported completing some college, 1 reported completing high school, and 1 reported no schooling. Three respondents had no health insurance.
Initial Hair Loss Discovery
The majority of respondents (22/34 [65%]) were first to notice their hair loss, while 5 (15%) reported hairstylists as the initial observers. Twelve respondents (35%) initially went to a physician to learn why they were losing hair; 6 (18%) instead utilized hairstylists or the Internet. Fifteen women (44%) waited more than 1 month up to 6 months after noticing hair loss before seeing a physician instead of going immediately within a 4-week period, and 16 (47%) waited 1 year or more.
Nondermatologist Consultation
Almost half (16/34 [47%]) of the women went to a nondermatologist physician regarding their hair loss; of them, half (8/16 [50%]) reported their physician did not examine the scalp, 3 (19%) reported their physician offered a biopsy, and none of them reported that their physician diagnosed them with CCCA. The median patient rating of their nondermatologist physician interactions was good (3 on a 5-point scale). Table 1 and Figure 1 show responses to individual items.
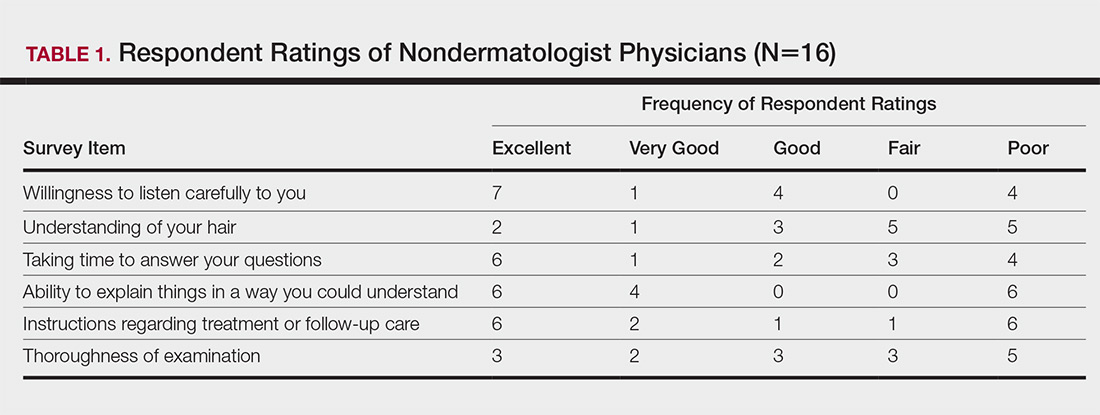
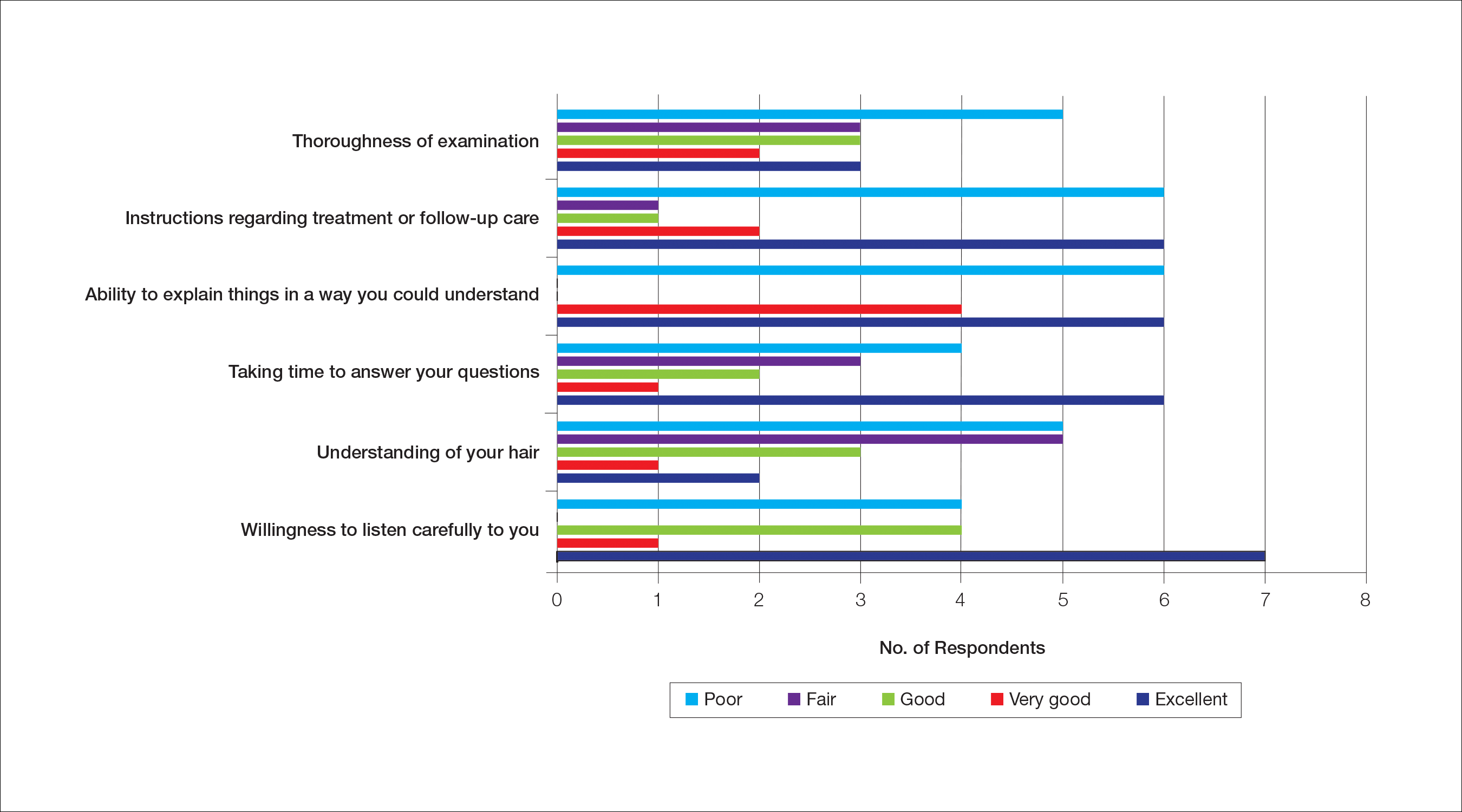
Dermatologist Consultation
All 34 respondents presented to a dermatologist. The majority of respondents (22/34 [65%]) saw either 1 or 2 dermatologists for their hair loss. Three (9%) reported their dermatologist did not examine their scalp. Twelve respondents (35%) reported their dermatologist did not offer a biopsy. Twenty-one respondents (62%) reported a CCCA diagnosis from the first dermatologist they saw. Twenty-three respondents (68%) were diagnosed by dermatologists with expertise in hair disorders. Sixteen (47%) were diagnosed by dermatologists within a skin-of-color center. Fourteen (41%) initial dermatology consultations were race concordant.
The median patient rating of their dermatologist interactions was excellent (5 on a 5-point scale). Table 2 and Figure 2 show responses to individual items. Respondents saw an average of 3 different providers, both dermatologists and otherwise.
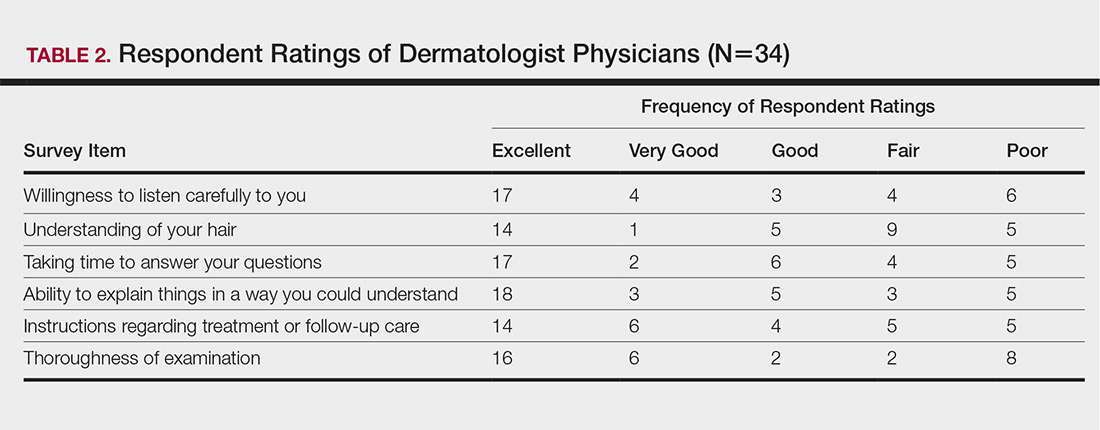
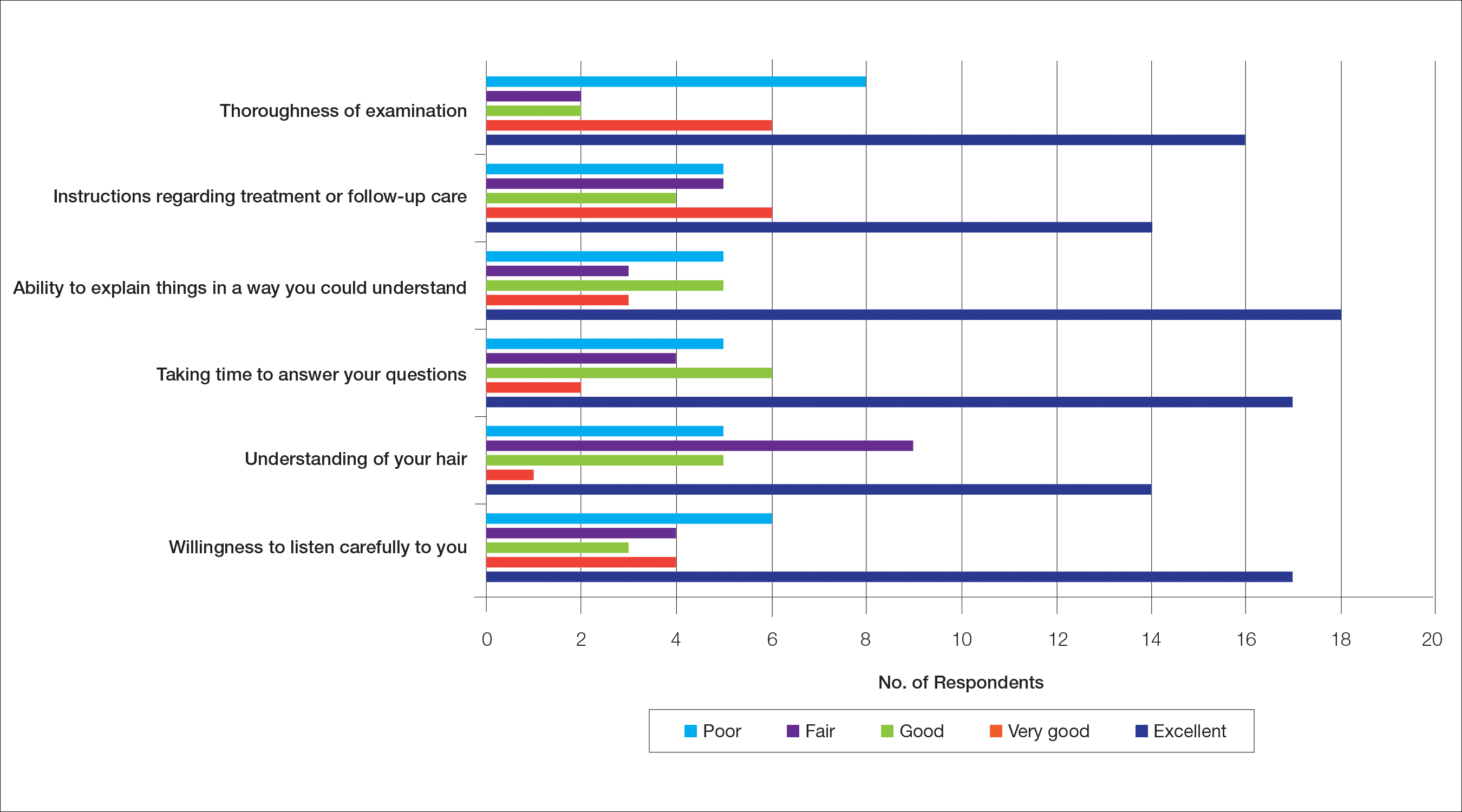
Waiting to See a Dermatologist
Nearly all respondents (31/34 [91%]) recommended that other women with hair loss immediately go see a dermatologist.
Barriers to Care
The top 5 factors reported as most important when initially seeking care included the physician’s experience with black hair and CCCA, the patient’s personal hairstyling practices, the physician’s ethnicity, availability of effective treatment options, and treatment cost. Table 3 shows frequency counts for these freely reported factors.
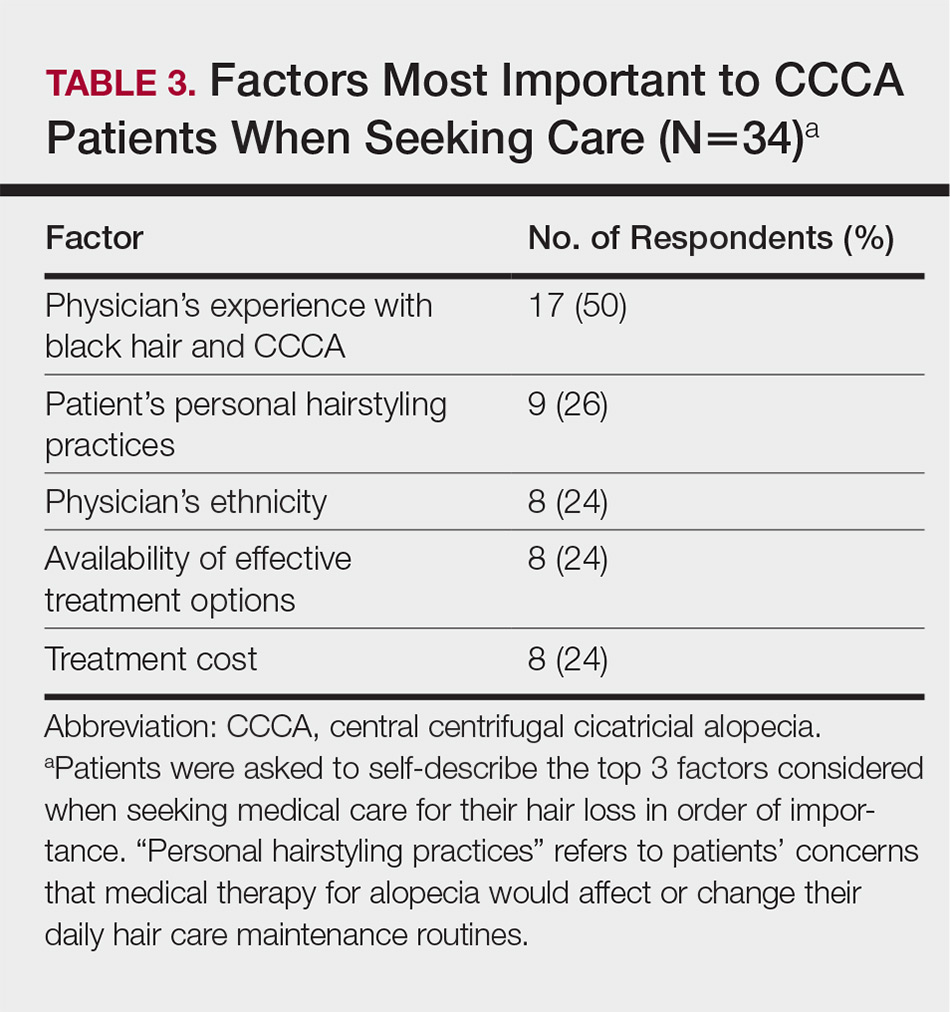
Quality of Life
The median score on 9 aggregated self-esteem items was 4 on a 5-point scale, representing an agree response to statements such as “I feel embarrassed, self-conscious, or frustrated about my hair loss” (28/34 [82%]) and “My hair loss bothers me” (28/34 [82%])(Table 4). Cronbach α for self-esteem survey items was 0.7826.

For the nonaggregated items, many respondents strongly disagreed with statements pertaining to activities of daily living, including “I take care of where I sit or stand at social gatherings due to my hair loss” (18/34 [53%]), “My hair loss makes it difficult for me to go to the grocery store” (29/34 [85%]), “My hair loss makes it difficult for me to attend faith-based activities” (30/34 [88%]), “My hair loss makes it difficult for me to exercise” (23/34 [68%]), “My hair loss makes it difficult for me to go to work and/or school” (24/34 [71%]), “My hair loss makes it difficult for me to go out with a significant other” (24/34 [71%]), “My hair loss makes it difficult for me to spend time with family” (27/34 [79%]), and “My hair loss makes it difficult for me to go to a hairstylist” (16/34 [47%]).
Comment
The majority of respondents were first to discover their hair loss. Harbingers of CCCA hair loss include paresthesia, tenderness, and itch,6 symptoms that are hard to ignore. Unfortunately, many patients notice hair thinning years after the scarring process has begun and a notable amount of hair has already been lost.6,9
Fifteen percent of respondents learned about their hair loss from their hairstylist. Women of African descent often maintain hairstyles that require frequent interactions with a hair care professional.7,10 As a result, hairstylists are at the forefront of early alopecia detection and are a valued resource in the black community. Open dialogue between dermatologists and hair care professionals could funnel women with hair loss into treatment before extensive damage.
Fifteen women (44%) recalled a waiting period of several months before seeking medical assistance, and 16 (47%) reported waiting 1 year or more. However, 91% of respondents indicated that women with hair loss should immediately see a physician for evaluation, thus patient experiences underscore the importance of early treatment. In our experience, many patients wait years before presenting to a physician. Some work with their hairstylists first to address the issue, while others do not realize how notable the loss has become. Some have a negative experience with one provider or are told there is nothing that can be done and then wait many years to see a second provider. Proper education of patients, physicians, and hairstylists is important in the identification and prompt treatment of this condition.
It is perhaps to be expected that patients rated interactions with dermatologists as excellent and very good more frequently than interactions with nondermatologists, which may be due to an absence of thorough hair evaluation with nondermatologists. Respondents reported that only half of nondermatologist providers actually examined their scalp during an initial encounter. However, both physician groups had the lowest frequencies of excellent and very good ratings on “understanding of your hair” (Tables 1 and 2). Patients with hair loss seek immediate answers, and often it is the specialist that can give them a firm diagnosis as opposed to a primary care provider. The fact that dermatologists and nondermatologists alike scored poorly on patient-perceived understanding of CCCA indicates an area for improvement within patient-physician interactions and physician knowledge.
The top 5 factors important to respondents when obtaining medical care included the physician’s experience with black hair and CCCA, the patient’s personal hairstyling practices, the physician’s ethnicity, availability of effective treatment options, and treatment cost. Patients with CCCA seeing dermatologists may discern a lack of experience with ethnic hair that leads patients to doubt their physicians’ ability to provide adequate care and decreased shared decision-making.11,12 These patient perceptions are not unfounded; a 2008 study showed that dermatology residents are not uniformly trained in diseases pertaining to patients with skin of color.13 Thus, incorporation of education on skin of color in dermatology training programs is critical.
Finally, hair loss patients often have concerns regarding how medical therapeutics could adversely affect personal hair care regimens, including washing and hairstyling practices. Current research demonstrates that patients consider treatment effectiveness and ability to be integrated into daily routines after establishing medical care.14 The present study shows that some CCCA patients contemplate how well a therapy will work before seeking medical care, demonstrating that patients continue to have these concerns after establishing medical care. Consideration of treatment effectiveness is important for both patients and providers, as there is minimal evidence behind current CCCA management practices. The ability for treatments to be easily integrated into daily hair care habits is important to maintain patient compliance.
Participants’ median self-esteem scores indicate the effect of CCCA on morale and self-perception. Items scrutinizing this construct had acceptable internal consistency reliability. It is interesting to note that activities of daily living were not impacted by hair loss. Examination of self-esteem is important in the alopecia population because the effect of hair loss on mental status is well documented.15-17 Low self-esteem has been reported as a prospective risk factor for clinical depression.18-20 In black patients, clinical depression rates surpass those of Hispanics and non-Hispanic white individuals.21 Dermatologists must consider the psychological status of all patients, particularly populations at risk for severe disease.
Limitations of this study include the small (34 participants) and mostly highly educated sample size, limited survey validity, and potential patient bias. Because many patients changed their address and/or telephone number in the time between CCCA diagnosis and the present study, we were left with a small pilot study, which minimizes the impact of our findings. Furthermore, our survey was created by a single expert’s opinion and modeling from preexisting alopecia questionnaires16; full validity procedures analyzing face, content, and criterion validity were not undertaken. Finally, the majority of respondents were patients of one of the study’s authors (S.S.L.P.), which could influence survey responses. The fact that some providers were hair experts and some were race concordant with their patients also could potentially affect the responses received, which was not analyzed in the present study. Future studies with more respondents from multiple providers would help clarify our preliminary findings.
Conclusion
Analysis of barriers to care and QOL in patients with skin of color is an essential addition to dermatologic discourse. Alopecia is particularly important to investigate, as prior research has found it to be one of the top 5 diagnoses made in patients with skin of color.3,4 Alopecia has been shown to negatively affect QOL.15,22,23 This study, although limited by small sample size, suggests CCCA also is a contributor to self-esteem challenges, similar to other forms of hair loss. Patient-physician interactions and personal hairstyling practices are prominent barriers to care for CCCA patients, demonstrating the need for quality education on skin of color and cultural competency in dermatology residencies across the country.
- Ogunleye TA, McMichael A, Olsen EA. Central centrifugal cicatricial alopecia: what has been achieved, current clues for future research. Dermatol Clin. 2014;32:173-181.
- Sperling LC. Scarring alopecia and the dermatopathologist. J Cutan Pathol. 2001;28:333-342.
- Halder RM, Grimes PE, McLaurin CI, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Olsen EA, Callender V, McMichael A, et al. Central hair loss in African American women: incidence and potential risk factors. J Am Acad Dermatol. 2011;64:245-252.
- Gathers RC, Lim HW. Central centrifugal cicatricial alopecia: past, present, and future. J Am Acad Dermatol. 2009;60:660-668.
- Gathers RC, Mahan MG. African american women, hair care, and health barriers. J Clin Aesthet Dermatol. 2014;7:26-29.
- Van Der Donk J, Hunfeld JA, Passchier J, et al. Quality of life and maladjustment associated with hair loss in women with alopecia androgenetica. Social Sci Med. 1994;38:159-163.
- Sperling LC, Sau P. The follicular degeneration syndrome in black patients. ‘hot comb alopecia’ revisited and revised. Arch Dermatol. 1992;128:68-74.
- Gathers RC, Jankowski M, Eide M, et al. Hair grooming practices and central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2009;60:574-578.
- Harvey VM, Ozoemena U, Paul J, et al. Patient-provider communication, concordance, and ratings of care in dermatology: results of a cross-sectional study. Dermatol Online J. 2016;22. pii: 13030/qt06j6p7gh.
- Laveist TA, Nuru-Jeter A. Is doctor-patient race concordance associated with greater satisfaction with care? J Health Soc Behav. 2002;43:296-306.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Suchonwanit P, Hector CE, Bin Saif GA, et al. Factors affecting the severity of central centrifugal cicatricial alopecia. Int J Dermatol. 2016;55:E338-E343.
- Williamson D, Gonzalez M, Finlay AY. The effect of hair loss on quality of life. J Eur Acad Dermatol Venereol. 2001;15:137-139.
- Fabbrocini G, Panariello L, De Vita V, et al. Quality of life in alopecia areata: a disease-specific questionnaire. J Eur Acad Dermatol Venereol. 2013;27:E276-E281.
- Ramos PM, Miot HA. Female pattern hair loss: a clinical and pathophysiological review. An Bras Dermatol. 2015;90:529-543.
- Sowislo JF, Orth U. Does low self-esteem predict depression and anxiety? a meta-analysis of longitudinal studies. Psychol Bull. 2013;139:213-240.
- Steiger AE, Allemand M, Robins RW, et al. Low and decreasing self-esteem during adolescence predict adult depression two decades later. J Pers Soc Psychol. 2014;106:325-338.
- Wegener I, Geiser F, Alfter S, et al. Changes of explicitly and implicitly measured self-esteem in the treatment of major depression: evidence for implicit self-esteem compensation. Compr Psychiatry. 2015;58:57-67.
- Pratt LAB, Brody DJ. Depression in the U.S. Household Population, 2009-2012. Hyattsville, MD: National Center for Health Statistics; 2014. NCHS Data Brief, No. 172. https://www.cdc.gov/nchs/data/databriefs/db172.pdf. Published December 2014. Accessed November 19, 2018.
- Schmidt S, Fischer TW, Chren MM, et al. Strategies of coping and quality of life in women with alopecia. Br J Dermatol. 2001;144:1038-1043.
- Hunt N, McHale S. The psychological impact of alopecia. Br Med J. 2005;331:951-953.
The etiology of central centrifugal cicatricial alopecia (CCCA), a clinical and histological pattern of hair loss on the central scalp, has been well studied. This disease is chronic and progressive, with extensive follicular destruction and eventual burnout.1,2 Central centrifugal cicatricial alopecia is most commonly seen in patients of African descent and has been shown to be 1 of the 5 most common dermatologic diagnoses in black patients.3,4 The top 5 dermatologic diagnoses within this population include acne vulgaris (28.4%), dyschromia (19.9%), eczema (9.1%), alopecia (8.3%), and seborrheic dermatitis (6.7%).4 The incidence rate of CCCA is estimated to be 5.6%.3,5 Most patients are women, with onset between the second and fourth decades of life.6
Central centrifugal cicatricial alopecia treatment efficacy is inversely correlated with disease duration. The primary goal of treatment is to prevent progression. Efforts are made to stimulate regrowth in areas that are not permanently scarred. When patients present with a substantial amount of scarring hair loss, dermatologists often are limited in their ability to achieve a cosmetically acceptable pattern of growth. Generally, hair is connected to a sense of self-worth in black women, and any type of hair loss has been shown to lead to frustration and decreased self-esteem.7 A 1994 study showed that 75% (44/58) of women with androgenetic alopecia had decreased self-esteem and 50% (29/58) had social challenges.8
The purpose of this pilot study was to determine the personal, historical, logistical, or environmental factors that preclude women from obtaining medical care for CCCA and to investigate how CCCA affects quality of life (QOL) and psychological well-being.
Methods
The investigators designed a survey study of adult, English-speaking, black women diagnosed with CCCA at the Northwestern University Department of Dermatology (Chicago, Illinois) between 2011 and 2017. Patients were selected from the electronic data warehouse compiled by the Department of Dermatology and were included if they fulfilled the following criteria: evaluated in the dermatology department between September 1, 2011, and September 30, 2017, by any faculty physician; diagnosed with CCCA; and aged 18 years or older. Patients were excluded if they did not speak English, as interpreters were not available. All patients who fulfilled the inclusion criteria provided signed informed consent prior to participation. All surveys were disseminated in the office or via telephone from fall 2016 to spring 2017 and took 10 to 15 minutes to complete. The research was approved by the authors’ institutional review board (IRB ID STU00203449).
Survey Instrument
The
Data Analysis
Analyses were completed using data analysis software JMP Pro 13 from SAS and a Microsoft Excel spreadsheet. Continuous data were presented as mean, SD, median, minimum, and maximum. Categorical data were presented as counts and percentages. Nine QOL items were aggregated into a self-esteem category (questions 30–38).
Cronbach α, a statistical measure of internal consistency and how closely related items are in a group, was used to evaluate internal consistency reliability; values of 0.70 or greater indicate acceptable reliability.
Results
Of 501 individuals contacted, 34 completed the survey (7% completion rate). Nonrespondents included 7 who refused to participate and 460 who could not be contacted. All respondents self-identified as black women. Median age at time of survey administration was 46 years (range, 28–79 years); median age at CCCA diagnosis was 42 years (range, 15–73 years). Respondents did not significantly differ in age from nonrespondents (P=.46). The majority of respondents had an associate’s degree, bachelor’s degree, or advanced degree of education (master of arts, doctor of medicine, doctor of jurisprudence, doctor of philosophy); however, 8 women reported completing some college, 1 reported completing high school, and 1 reported no schooling. Three respondents had no health insurance.
Initial Hair Loss Discovery
The majority of respondents (22/34 [65%]) were first to notice their hair loss, while 5 (15%) reported hairstylists as the initial observers. Twelve respondents (35%) initially went to a physician to learn why they were losing hair; 6 (18%) instead utilized hairstylists or the Internet. Fifteen women (44%) waited more than 1 month up to 6 months after noticing hair loss before seeing a physician instead of going immediately within a 4-week period, and 16 (47%) waited 1 year or more.
Nondermatologist Consultation
Almost half (16/34 [47%]) of the women went to a nondermatologist physician regarding their hair loss; of them, half (8/16 [50%]) reported their physician did not examine the scalp, 3 (19%) reported their physician offered a biopsy, and none of them reported that their physician diagnosed them with CCCA. The median patient rating of their nondermatologist physician interactions was good (3 on a 5-point scale). Table 1 and Figure 1 show responses to individual items.
Dermatologist Consultation
All 34 respondents presented to a dermatologist. The majority of respondents (22/34 [65%]) saw either 1 or 2 dermatologists for their hair loss. Three (9%) reported their dermatologist did not examine their scalp. Twelve respondents (35%) reported their dermatologist did not offer a biopsy. Twenty-one respondents (62%) reported a CCCA diagnosis from the first dermatologist they saw. Twenty-three respondents (68%) were diagnosed by dermatologists with expertise in hair disorders. Sixteen (47%) were diagnosed by dermatologists within a skin-of-color center. Fourteen (41%) initial dermatology consultations were race concordant.
The median patient rating of their dermatologist interactions was excellent (5 on a 5-point scale). Table 2 and Figure 2 show responses to individual items. Respondents saw an average of 3 different providers, both dermatologists and otherwise.
Waiting to See a Dermatologist
Nearly all respondents (31/34 [91%]) recommended that other women with hair loss immediately go see a dermatologist.
Barriers to Care
The top 5 factors reported as most important when initially seeking care included the physician’s experience with black hair and CCCA, the patient’s personal hairstyling practices, the physician’s ethnicity, availability of effective treatment options, and treatment cost. Table 3 shows frequency counts for these freely reported factors.
Quality of Life
The median score on 9 aggregated self-esteem items was 4 on a 5-point scale, representing an agree response to statements such as “I feel embarrassed, self-conscious, or frustrated about my hair loss” (28/34 [82%]) and “My hair loss bothers me” (28/34 [82%])(Table 4). Cronbach α for self-esteem survey items was 0.7826.
For the nonaggregated items, many respondents strongly disagreed with statements pertaining to activities of daily living, including “I take care of where I sit or stand at social gatherings due to my hair loss” (18/34 [53%]), “My hair loss makes it difficult for me to go to the grocery store” (29/34 [85%]), “My hair loss makes it difficult for me to attend faith-based activities” (30/34 [88%]), “My hair loss makes it difficult for me to exercise” (23/34 [68%]), “My hair loss makes it difficult for me to go to work and/or school” (24/34 [71%]), “My hair loss makes it difficult for me to go out with a significant other” (24/34 [71%]), “My hair loss makes it difficult for me to spend time with family” (27/34 [79%]), and “My hair loss makes it difficult for me to go to a hairstylist” (16/34 [47%]).
Comment
The majority of respondents were first to discover their hair loss. Harbingers of CCCA hair loss include paresthesia, tenderness, and itch,6 symptoms that are hard to ignore. Unfortunately, many patients notice hair thinning years after the scarring process has begun and a notable amount of hair has already been lost.6,9
Fifteen percent of respondents learned about their hair loss from their hairstylist. Women of African descent often maintain hairstyles that require frequent interactions with a hair care professional.7,10 As a result, hairstylists are at the forefront of early alopecia detection and are a valued resource in the black community. Open dialogue between dermatologists and hair care professionals could funnel women with hair loss into treatment before extensive damage.
Fifteen women (44%) recalled a waiting period of several months before seeking medical assistance, and 16 (47%) reported waiting 1 year or more. However, 91% of respondents indicated that women with hair loss should immediately see a physician for evaluation, thus patient experiences underscore the importance of early treatment. In our experience, many patients wait years before presenting to a physician. Some work with their hairstylists first to address the issue, while others do not realize how notable the loss has become. Some have a negative experience with one provider or are told there is nothing that can be done and then wait many years to see a second provider. Proper education of patients, physicians, and hairstylists is important in the identification and prompt treatment of this condition.
It is perhaps to be expected that patients rated interactions with dermatologists as excellent and very good more frequently than interactions with nondermatologists, which may be due to an absence of thorough hair evaluation with nondermatologists. Respondents reported that only half of nondermatologist providers actually examined their scalp during an initial encounter. However, both physician groups had the lowest frequencies of excellent and very good ratings on “understanding of your hair” (Tables 1 and 2). Patients with hair loss seek immediate answers, and often it is the specialist that can give them a firm diagnosis as opposed to a primary care provider. The fact that dermatologists and nondermatologists alike scored poorly on patient-perceived understanding of CCCA indicates an area for improvement within patient-physician interactions and physician knowledge.
The top 5 factors important to respondents when obtaining medical care included the physician’s experience with black hair and CCCA, the patient’s personal hairstyling practices, the physician’s ethnicity, availability of effective treatment options, and treatment cost. Patients with CCCA seeing dermatologists may discern a lack of experience with ethnic hair that leads patients to doubt their physicians’ ability to provide adequate care and decreased shared decision-making.11,12 These patient perceptions are not unfounded; a 2008 study showed that dermatology residents are not uniformly trained in diseases pertaining to patients with skin of color.13 Thus, incorporation of education on skin of color in dermatology training programs is critical.
Finally, hair loss patients often have concerns regarding how medical therapeutics could adversely affect personal hair care regimens, including washing and hairstyling practices. Current research demonstrates that patients consider treatment effectiveness and ability to be integrated into daily routines after establishing medical care.14 The present study shows that some CCCA patients contemplate how well a therapy will work before seeking medical care, demonstrating that patients continue to have these concerns after establishing medical care. Consideration of treatment effectiveness is important for both patients and providers, as there is minimal evidence behind current CCCA management practices. The ability for treatments to be easily integrated into daily hair care habits is important to maintain patient compliance.
Participants’ median self-esteem scores indicate the effect of CCCA on morale and self-perception. Items scrutinizing this construct had acceptable internal consistency reliability. It is interesting to note that activities of daily living were not impacted by hair loss. Examination of self-esteem is important in the alopecia population because the effect of hair loss on mental status is well documented.15-17 Low self-esteem has been reported as a prospective risk factor for clinical depression.18-20 In black patients, clinical depression rates surpass those of Hispanics and non-Hispanic white individuals.21 Dermatologists must consider the psychological status of all patients, particularly populations at risk for severe disease.
Limitations of this study include the small (34 participants) and mostly highly educated sample size, limited survey validity, and potential patient bias. Because many patients changed their address and/or telephone number in the time between CCCA diagnosis and the present study, we were left with a small pilot study, which minimizes the impact of our findings. Furthermore, our survey was created by a single expert’s opinion and modeling from preexisting alopecia questionnaires16; full validity procedures analyzing face, content, and criterion validity were not undertaken. Finally, the majority of respondents were patients of one of the study’s authors (S.S.L.P.), which could influence survey responses. The fact that some providers were hair experts and some were race concordant with their patients also could potentially affect the responses received, which was not analyzed in the present study. Future studies with more respondents from multiple providers would help clarify our preliminary findings.
Conclusion
Analysis of barriers to care and QOL in patients with skin of color is an essential addition to dermatologic discourse. Alopecia is particularly important to investigate, as prior research has found it to be one of the top 5 diagnoses made in patients with skin of color.3,4 Alopecia has been shown to negatively affect QOL.15,22,23 This study, although limited by small sample size, suggests CCCA also is a contributor to self-esteem challenges, similar to other forms of hair loss. Patient-physician interactions and personal hairstyling practices are prominent barriers to care for CCCA patients, demonstrating the need for quality education on skin of color and cultural competency in dermatology residencies across the country.
The etiology of central centrifugal cicatricial alopecia (CCCA), a clinical and histological pattern of hair loss on the central scalp, has been well studied. This disease is chronic and progressive, with extensive follicular destruction and eventual burnout.1,2 Central centrifugal cicatricial alopecia is most commonly seen in patients of African descent and has been shown to be 1 of the 5 most common dermatologic diagnoses in black patients.3,4 The top 5 dermatologic diagnoses within this population include acne vulgaris (28.4%), dyschromia (19.9%), eczema (9.1%), alopecia (8.3%), and seborrheic dermatitis (6.7%).4 The incidence rate of CCCA is estimated to be 5.6%.3,5 Most patients are women, with onset between the second and fourth decades of life.6
Central centrifugal cicatricial alopecia treatment efficacy is inversely correlated with disease duration. The primary goal of treatment is to prevent progression. Efforts are made to stimulate regrowth in areas that are not permanently scarred. When patients present with a substantial amount of scarring hair loss, dermatologists often are limited in their ability to achieve a cosmetically acceptable pattern of growth. Generally, hair is connected to a sense of self-worth in black women, and any type of hair loss has been shown to lead to frustration and decreased self-esteem.7 A 1994 study showed that 75% (44/58) of women with androgenetic alopecia had decreased self-esteem and 50% (29/58) had social challenges.8
The purpose of this pilot study was to determine the personal, historical, logistical, or environmental factors that preclude women from obtaining medical care for CCCA and to investigate how CCCA affects quality of life (QOL) and psychological well-being.
Methods
The investigators designed a survey study of adult, English-speaking, black women diagnosed with CCCA at the Northwestern University Department of Dermatology (Chicago, Illinois) between 2011 and 2017. Patients were selected from the electronic data warehouse compiled by the Department of Dermatology and were included if they fulfilled the following criteria: evaluated in the dermatology department between September 1, 2011, and September 30, 2017, by any faculty physician; diagnosed with CCCA; and aged 18 years or older. Patients were excluded if they did not speak English, as interpreters were not available. All patients who fulfilled the inclusion criteria provided signed informed consent prior to participation. All surveys were disseminated in the office or via telephone from fall 2016 to spring 2017 and took 10 to 15 minutes to complete. The research was approved by the authors’ institutional review board (IRB ID STU00203449).
Survey Instrument
The
Data Analysis
Analyses were completed using data analysis software JMP Pro 13 from SAS and a Microsoft Excel spreadsheet. Continuous data were presented as mean, SD, median, minimum, and maximum. Categorical data were presented as counts and percentages. Nine QOL items were aggregated into a self-esteem category (questions 30–38).
Cronbach α, a statistical measure of internal consistency and how closely related items are in a group, was used to evaluate internal consistency reliability; values of 0.70 or greater indicate acceptable reliability.
Results
Of 501 individuals contacted, 34 completed the survey (7% completion rate). Nonrespondents included 7 who refused to participate and 460 who could not be contacted. All respondents self-identified as black women. Median age at time of survey administration was 46 years (range, 28–79 years); median age at CCCA diagnosis was 42 years (range, 15–73 years). Respondents did not significantly differ in age from nonrespondents (P=.46). The majority of respondents had an associate’s degree, bachelor’s degree, or advanced degree of education (master of arts, doctor of medicine, doctor of jurisprudence, doctor of philosophy); however, 8 women reported completing some college, 1 reported completing high school, and 1 reported no schooling. Three respondents had no health insurance.
Initial Hair Loss Discovery
The majority of respondents (22/34 [65%]) were first to notice their hair loss, while 5 (15%) reported hairstylists as the initial observers. Twelve respondents (35%) initially went to a physician to learn why they were losing hair; 6 (18%) instead utilized hairstylists or the Internet. Fifteen women (44%) waited more than 1 month up to 6 months after noticing hair loss before seeing a physician instead of going immediately within a 4-week period, and 16 (47%) waited 1 year or more.
Nondermatologist Consultation
Almost half (16/34 [47%]) of the women went to a nondermatologist physician regarding their hair loss; of them, half (8/16 [50%]) reported their physician did not examine the scalp, 3 (19%) reported their physician offered a biopsy, and none of them reported that their physician diagnosed them with CCCA. The median patient rating of their nondermatologist physician interactions was good (3 on a 5-point scale). Table 1 and Figure 1 show responses to individual items.
Dermatologist Consultation
All 34 respondents presented to a dermatologist. The majority of respondents (22/34 [65%]) saw either 1 or 2 dermatologists for their hair loss. Three (9%) reported their dermatologist did not examine their scalp. Twelve respondents (35%) reported their dermatologist did not offer a biopsy. Twenty-one respondents (62%) reported a CCCA diagnosis from the first dermatologist they saw. Twenty-three respondents (68%) were diagnosed by dermatologists with expertise in hair disorders. Sixteen (47%) were diagnosed by dermatologists within a skin-of-color center. Fourteen (41%) initial dermatology consultations were race concordant.
The median patient rating of their dermatologist interactions was excellent (5 on a 5-point scale). Table 2 and Figure 2 show responses to individual items. Respondents saw an average of 3 different providers, both dermatologists and otherwise.
Waiting to See a Dermatologist
Nearly all respondents (31/34 [91%]) recommended that other women with hair loss immediately go see a dermatologist.
Barriers to Care
The top 5 factors reported as most important when initially seeking care included the physician’s experience with black hair and CCCA, the patient’s personal hairstyling practices, the physician’s ethnicity, availability of effective treatment options, and treatment cost. Table 3 shows frequency counts for these freely reported factors.
Quality of Life
The median score on 9 aggregated self-esteem items was 4 on a 5-point scale, representing an agree response to statements such as “I feel embarrassed, self-conscious, or frustrated about my hair loss” (28/34 [82%]) and “My hair loss bothers me” (28/34 [82%])(Table 4). Cronbach α for self-esteem survey items was 0.7826.
For the nonaggregated items, many respondents strongly disagreed with statements pertaining to activities of daily living, including “I take care of where I sit or stand at social gatherings due to my hair loss” (18/34 [53%]), “My hair loss makes it difficult for me to go to the grocery store” (29/34 [85%]), “My hair loss makes it difficult for me to attend faith-based activities” (30/34 [88%]), “My hair loss makes it difficult for me to exercise” (23/34 [68%]), “My hair loss makes it difficult for me to go to work and/or school” (24/34 [71%]), “My hair loss makes it difficult for me to go out with a significant other” (24/34 [71%]), “My hair loss makes it difficult for me to spend time with family” (27/34 [79%]), and “My hair loss makes it difficult for me to go to a hairstylist” (16/34 [47%]).
Comment
The majority of respondents were first to discover their hair loss. Harbingers of CCCA hair loss include paresthesia, tenderness, and itch,6 symptoms that are hard to ignore. Unfortunately, many patients notice hair thinning years after the scarring process has begun and a notable amount of hair has already been lost.6,9
Fifteen percent of respondents learned about their hair loss from their hairstylist. Women of African descent often maintain hairstyles that require frequent interactions with a hair care professional.7,10 As a result, hairstylists are at the forefront of early alopecia detection and are a valued resource in the black community. Open dialogue between dermatologists and hair care professionals could funnel women with hair loss into treatment before extensive damage.
Fifteen women (44%) recalled a waiting period of several months before seeking medical assistance, and 16 (47%) reported waiting 1 year or more. However, 91% of respondents indicated that women with hair loss should immediately see a physician for evaluation, thus patient experiences underscore the importance of early treatment. In our experience, many patients wait years before presenting to a physician. Some work with their hairstylists first to address the issue, while others do not realize how notable the loss has become. Some have a negative experience with one provider or are told there is nothing that can be done and then wait many years to see a second provider. Proper education of patients, physicians, and hairstylists is important in the identification and prompt treatment of this condition.
It is perhaps to be expected that patients rated interactions with dermatologists as excellent and very good more frequently than interactions with nondermatologists, which may be due to an absence of thorough hair evaluation with nondermatologists. Respondents reported that only half of nondermatologist providers actually examined their scalp during an initial encounter. However, both physician groups had the lowest frequencies of excellent and very good ratings on “understanding of your hair” (Tables 1 and 2). Patients with hair loss seek immediate answers, and often it is the specialist that can give them a firm diagnosis as opposed to a primary care provider. The fact that dermatologists and nondermatologists alike scored poorly on patient-perceived understanding of CCCA indicates an area for improvement within patient-physician interactions and physician knowledge.
The top 5 factors important to respondents when obtaining medical care included the physician’s experience with black hair and CCCA, the patient’s personal hairstyling practices, the physician’s ethnicity, availability of effective treatment options, and treatment cost. Patients with CCCA seeing dermatologists may discern a lack of experience with ethnic hair that leads patients to doubt their physicians’ ability to provide adequate care and decreased shared decision-making.11,12 These patient perceptions are not unfounded; a 2008 study showed that dermatology residents are not uniformly trained in diseases pertaining to patients with skin of color.13 Thus, incorporation of education on skin of color in dermatology training programs is critical.
Finally, hair loss patients often have concerns regarding how medical therapeutics could adversely affect personal hair care regimens, including washing and hairstyling practices. Current research demonstrates that patients consider treatment effectiveness and ability to be integrated into daily routines after establishing medical care.14 The present study shows that some CCCA patients contemplate how well a therapy will work before seeking medical care, demonstrating that patients continue to have these concerns after establishing medical care. Consideration of treatment effectiveness is important for both patients and providers, as there is minimal evidence behind current CCCA management practices. The ability for treatments to be easily integrated into daily hair care habits is important to maintain patient compliance.
Participants’ median self-esteem scores indicate the effect of CCCA on morale and self-perception. Items scrutinizing this construct had acceptable internal consistency reliability. It is interesting to note that activities of daily living were not impacted by hair loss. Examination of self-esteem is important in the alopecia population because the effect of hair loss on mental status is well documented.15-17 Low self-esteem has been reported as a prospective risk factor for clinical depression.18-20 In black patients, clinical depression rates surpass those of Hispanics and non-Hispanic white individuals.21 Dermatologists must consider the psychological status of all patients, particularly populations at risk for severe disease.
Limitations of this study include the small (34 participants) and mostly highly educated sample size, limited survey validity, and potential patient bias. Because many patients changed their address and/or telephone number in the time between CCCA diagnosis and the present study, we were left with a small pilot study, which minimizes the impact of our findings. Furthermore, our survey was created by a single expert’s opinion and modeling from preexisting alopecia questionnaires16; full validity procedures analyzing face, content, and criterion validity were not undertaken. Finally, the majority of respondents were patients of one of the study’s authors (S.S.L.P.), which could influence survey responses. The fact that some providers were hair experts and some were race concordant with their patients also could potentially affect the responses received, which was not analyzed in the present study. Future studies with more respondents from multiple providers would help clarify our preliminary findings.
Conclusion
Analysis of barriers to care and QOL in patients with skin of color is an essential addition to dermatologic discourse. Alopecia is particularly important to investigate, as prior research has found it to be one of the top 5 diagnoses made in patients with skin of color.3,4 Alopecia has been shown to negatively affect QOL.15,22,23 This study, although limited by small sample size, suggests CCCA also is a contributor to self-esteem challenges, similar to other forms of hair loss. Patient-physician interactions and personal hairstyling practices are prominent barriers to care for CCCA patients, demonstrating the need for quality education on skin of color and cultural competency in dermatology residencies across the country.
- Ogunleye TA, McMichael A, Olsen EA. Central centrifugal cicatricial alopecia: what has been achieved, current clues for future research. Dermatol Clin. 2014;32:173-181.
- Sperling LC. Scarring alopecia and the dermatopathologist. J Cutan Pathol. 2001;28:333-342.
- Halder RM, Grimes PE, McLaurin CI, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Olsen EA, Callender V, McMichael A, et al. Central hair loss in African American women: incidence and potential risk factors. J Am Acad Dermatol. 2011;64:245-252.
- Gathers RC, Lim HW. Central centrifugal cicatricial alopecia: past, present, and future. J Am Acad Dermatol. 2009;60:660-668.
- Gathers RC, Mahan MG. African american women, hair care, and health barriers. J Clin Aesthet Dermatol. 2014;7:26-29.
- Van Der Donk J, Hunfeld JA, Passchier J, et al. Quality of life and maladjustment associated with hair loss in women with alopecia androgenetica. Social Sci Med. 1994;38:159-163.
- Sperling LC, Sau P. The follicular degeneration syndrome in black patients. ‘hot comb alopecia’ revisited and revised. Arch Dermatol. 1992;128:68-74.
- Gathers RC, Jankowski M, Eide M, et al. Hair grooming practices and central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2009;60:574-578.
- Harvey VM, Ozoemena U, Paul J, et al. Patient-provider communication, concordance, and ratings of care in dermatology: results of a cross-sectional study. Dermatol Online J. 2016;22. pii: 13030/qt06j6p7gh.
- Laveist TA, Nuru-Jeter A. Is doctor-patient race concordance associated with greater satisfaction with care? J Health Soc Behav. 2002;43:296-306.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Suchonwanit P, Hector CE, Bin Saif GA, et al. Factors affecting the severity of central centrifugal cicatricial alopecia. Int J Dermatol. 2016;55:E338-E343.
- Williamson D, Gonzalez M, Finlay AY. The effect of hair loss on quality of life. J Eur Acad Dermatol Venereol. 2001;15:137-139.
- Fabbrocini G, Panariello L, De Vita V, et al. Quality of life in alopecia areata: a disease-specific questionnaire. J Eur Acad Dermatol Venereol. 2013;27:E276-E281.
- Ramos PM, Miot HA. Female pattern hair loss: a clinical and pathophysiological review. An Bras Dermatol. 2015;90:529-543.
- Sowislo JF, Orth U. Does low self-esteem predict depression and anxiety? a meta-analysis of longitudinal studies. Psychol Bull. 2013;139:213-240.
- Steiger AE, Allemand M, Robins RW, et al. Low and decreasing self-esteem during adolescence predict adult depression two decades later. J Pers Soc Psychol. 2014;106:325-338.
- Wegener I, Geiser F, Alfter S, et al. Changes of explicitly and implicitly measured self-esteem in the treatment of major depression: evidence for implicit self-esteem compensation. Compr Psychiatry. 2015;58:57-67.
- Pratt LAB, Brody DJ. Depression in the U.S. Household Population, 2009-2012. Hyattsville, MD: National Center for Health Statistics; 2014. NCHS Data Brief, No. 172. https://www.cdc.gov/nchs/data/databriefs/db172.pdf. Published December 2014. Accessed November 19, 2018.
- Schmidt S, Fischer TW, Chren MM, et al. Strategies of coping and quality of life in women with alopecia. Br J Dermatol. 2001;144:1038-1043.
- Hunt N, McHale S. The psychological impact of alopecia. Br Med J. 2005;331:951-953.
- Ogunleye TA, McMichael A, Olsen EA. Central centrifugal cicatricial alopecia: what has been achieved, current clues for future research. Dermatol Clin. 2014;32:173-181.
- Sperling LC. Scarring alopecia and the dermatopathologist. J Cutan Pathol. 2001;28:333-342.
- Halder RM, Grimes PE, McLaurin CI, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Olsen EA, Callender V, McMichael A, et al. Central hair loss in African American women: incidence and potential risk factors. J Am Acad Dermatol. 2011;64:245-252.
- Gathers RC, Lim HW. Central centrifugal cicatricial alopecia: past, present, and future. J Am Acad Dermatol. 2009;60:660-668.
- Gathers RC, Mahan MG. African american women, hair care, and health barriers. J Clin Aesthet Dermatol. 2014;7:26-29.
- Van Der Donk J, Hunfeld JA, Passchier J, et al. Quality of life and maladjustment associated with hair loss in women with alopecia androgenetica. Social Sci Med. 1994;38:159-163.
- Sperling LC, Sau P. The follicular degeneration syndrome in black patients. ‘hot comb alopecia’ revisited and revised. Arch Dermatol. 1992;128:68-74.
- Gathers RC, Jankowski M, Eide M, et al. Hair grooming practices and central centrifugal cicatricial alopecia. J Am Acad Dermatol. 2009;60:574-578.
- Harvey VM, Ozoemena U, Paul J, et al. Patient-provider communication, concordance, and ratings of care in dermatology: results of a cross-sectional study. Dermatol Online J. 2016;22. pii: 13030/qt06j6p7gh.
- Laveist TA, Nuru-Jeter A. Is doctor-patient race concordance associated with greater satisfaction with care? J Health Soc Behav. 2002;43:296-306.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Suchonwanit P, Hector CE, Bin Saif GA, et al. Factors affecting the severity of central centrifugal cicatricial alopecia. Int J Dermatol. 2016;55:E338-E343.
- Williamson D, Gonzalez M, Finlay AY. The effect of hair loss on quality of life. J Eur Acad Dermatol Venereol. 2001;15:137-139.
- Fabbrocini G, Panariello L, De Vita V, et al. Quality of life in alopecia areata: a disease-specific questionnaire. J Eur Acad Dermatol Venereol. 2013;27:E276-E281.
- Ramos PM, Miot HA. Female pattern hair loss: a clinical and pathophysiological review. An Bras Dermatol. 2015;90:529-543.
- Sowislo JF, Orth U. Does low self-esteem predict depression and anxiety? a meta-analysis of longitudinal studies. Psychol Bull. 2013;139:213-240.
- Steiger AE, Allemand M, Robins RW, et al. Low and decreasing self-esteem during adolescence predict adult depression two decades later. J Pers Soc Psychol. 2014;106:325-338.
- Wegener I, Geiser F, Alfter S, et al. Changes of explicitly and implicitly measured self-esteem in the treatment of major depression: evidence for implicit self-esteem compensation. Compr Psychiatry. 2015;58:57-67.
- Pratt LAB, Brody DJ. Depression in the U.S. Household Population, 2009-2012. Hyattsville, MD: National Center for Health Statistics; 2014. NCHS Data Brief, No. 172. https://www.cdc.gov/nchs/data/databriefs/db172.pdf. Published December 2014. Accessed November 19, 2018.
- Schmidt S, Fischer TW, Chren MM, et al. Strategies of coping and quality of life in women with alopecia. Br J Dermatol. 2001;144:1038-1043.
- Hunt N, McHale S. The psychological impact of alopecia. Br Med J. 2005;331:951-953.
Practice Points
- Central centrifugal cicatricial alopecia (CCCA) presents a unique set of challenges for both patients and providers.
- Lack of physician experience with black hair/CCCA and the potential impact of care on personal hairstyling practices are 2 barriers to care for many patients with this disease.
- There is a need for improved patient-provider communication strategies, quality education on hair in skin of color patients, and cultural competency training in dermatology residencies across the country.
Product News: 12 2018
Altreno Lotion Now Available for Acne Vulgaris
Ortho Dermatologics launches Altreno (tretinoin) Lotion 0.05% for the treatment of acne vulgaris in patients 9 years and older. It was approved by the US Food and Drug Administration in August 2018, providing patients with the efficacy of tretinoin and the tolerability of a lotion formulation containing hyaluronic acid, glycerin, and collagen to help hydrate and moisturize the skin. For more information, visit www.ortho-dermatologics.com.
Bryhali Approved for Plaque Psoriasis in Adults
Ortho Dermatologics announces US Food and Drug Administration approval of Bryhali (halobetasol propionate) Lotion 0.01% for the treatment of plaque psoriasis in adults. Bryhali is a corticosteroid in a novel vehicle lotion with safety established for dosing up to 8 weeks, offering patients a longer duration of use than other topical steroids. For more information, visit www.bryhali.com.
CoolSculpting Cleared for the Submandibular Area
Zeltiq Aesthetics, Inc, an Allergan affiliate, announces US Food and Drug Administration (FDA) clearance of CoolSculpting to treat the submandibular area. The FDA clearance also was expanded to include patients with a body mass index of up to 46.2 when treating the submental and submandibular areas. CoolSculpting is a nonsurgical treatment that works by gently cooling targeted fat cells in the body to induce natural controlled elimination of fat cells without affecting surrounding tissue. CoolSculpting also is cleared for treatment of visible fat bulges on the thighs, abdomen, and flanks. For more information, visit www.coolsculpting.com.
Glytone Age-Defying Vitamin C+E Serum Reduces Signs of Aging
Pierre Fabre Dermo-Cosmetique introduces Glytone Age-Defying Vitamin C+E Serum, a layering serum with time-released, high concentrations of stabilized vitamins C and E combined with red tea flavonoids to deliver antioxidant protection and antiaging benefits. It is indicated for premature aging caused by environmental damage and oxidative stress as well as postprocedure relief. For more information, visit www.glytone-usa.com.
Hyrimoz Biosimilar Approved for Psoriasis
Sandoz, a Novartis Division, announces US Food and Drug Administration (FDA) approval of Hyrimoz (adalimumab-adaz), a biosimilar indicated for the treatment of psoriatic arthritis and plaque psoriasis, as well as rheumatoid arthritis, juvenile idiopathic arthritis in patients 4 years and older, ankylosing spondylitis, adult Crohn disease, and ulcerative colitis. Hyrimoz is the third FDA-approved biosimilar from Sandoz. For more information, visit www.sandoz.com.
Libtayo Approved for SCC
Regeneron Pharmaceuticals, Inc, and sanofi-aventis US LLC, announce US Food and Drug Administration (FDA) approval of Libtayo (cemiplimab-rwlc) for the treatment of patients with metastatic cutaneous squamous cell carcinoma (SCC) or locally advanced cutaneous SCC who are not candidates for curative surgery or radiation. Libtayo is a monoclonal antibody targeting the programmed death receptor 1.
Restylane Lyft Now Approved for Hand Rejuvenation
Nestlé Skin Health announces US Food and Drug Administration approval of Restylane Lyft with Lidocaine, a hyaluronic acid dermal filler, for the correction of age-related volume loss in the back of the hands for patients older than 21 years. Restylane Lyft with Lidocaine also is indicated for correction of moderate to severe facial wrinkles and folds such as the nasolabial folds, cheek augmentation, and age-related midface contour deficiencies. For more information, visit www.RestylaneUSA.com.
Xepi Launches for Impetigo
Cutanea Life Sciences launches Xepi (ozenoxacin) Cream 1%, a quinolone antimicrobial, for the treatment of impetigo in adult and pediatric patients 2 months or older. Xepi is applied twice daily for 5 days and has been shown to be active against most isolates of Staphylococcus aureus (including methicillin-resistant isolates) and Streptococcus pyogenes, both in vitro and in clinical infections. For more information, visit www.XepiCream.com.
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Altreno Lotion Now Available for Acne Vulgaris
Ortho Dermatologics launches Altreno (tretinoin) Lotion 0.05% for the treatment of acne vulgaris in patients 9 years and older. It was approved by the US Food and Drug Administration in August 2018, providing patients with the efficacy of tretinoin and the tolerability of a lotion formulation containing hyaluronic acid, glycerin, and collagen to help hydrate and moisturize the skin. For more information, visit www.ortho-dermatologics.com.
Bryhali Approved for Plaque Psoriasis in Adults
Ortho Dermatologics announces US Food and Drug Administration approval of Bryhali (halobetasol propionate) Lotion 0.01% for the treatment of plaque psoriasis in adults. Bryhali is a corticosteroid in a novel vehicle lotion with safety established for dosing up to 8 weeks, offering patients a longer duration of use than other topical steroids. For more information, visit www.bryhali.com.
CoolSculpting Cleared for the Submandibular Area
Zeltiq Aesthetics, Inc, an Allergan affiliate, announces US Food and Drug Administration (FDA) clearance of CoolSculpting to treat the submandibular area. The FDA clearance also was expanded to include patients with a body mass index of up to 46.2 when treating the submental and submandibular areas. CoolSculpting is a nonsurgical treatment that works by gently cooling targeted fat cells in the body to induce natural controlled elimination of fat cells without affecting surrounding tissue. CoolSculpting also is cleared for treatment of visible fat bulges on the thighs, abdomen, and flanks. For more information, visit www.coolsculpting.com.
Glytone Age-Defying Vitamin C+E Serum Reduces Signs of Aging
Pierre Fabre Dermo-Cosmetique introduces Glytone Age-Defying Vitamin C+E Serum, a layering serum with time-released, high concentrations of stabilized vitamins C and E combined with red tea flavonoids to deliver antioxidant protection and antiaging benefits. It is indicated for premature aging caused by environmental damage and oxidative stress as well as postprocedure relief. For more information, visit www.glytone-usa.com.
Hyrimoz Biosimilar Approved for Psoriasis
Sandoz, a Novartis Division, announces US Food and Drug Administration (FDA) approval of Hyrimoz (adalimumab-adaz), a biosimilar indicated for the treatment of psoriatic arthritis and plaque psoriasis, as well as rheumatoid arthritis, juvenile idiopathic arthritis in patients 4 years and older, ankylosing spondylitis, adult Crohn disease, and ulcerative colitis. Hyrimoz is the third FDA-approved biosimilar from Sandoz. For more information, visit www.sandoz.com.
Libtayo Approved for SCC
Regeneron Pharmaceuticals, Inc, and sanofi-aventis US LLC, announce US Food and Drug Administration (FDA) approval of Libtayo (cemiplimab-rwlc) for the treatment of patients with metastatic cutaneous squamous cell carcinoma (SCC) or locally advanced cutaneous SCC who are not candidates for curative surgery or radiation. Libtayo is a monoclonal antibody targeting the programmed death receptor 1.
Restylane Lyft Now Approved for Hand Rejuvenation
Nestlé Skin Health announces US Food and Drug Administration approval of Restylane Lyft with Lidocaine, a hyaluronic acid dermal filler, for the correction of age-related volume loss in the back of the hands for patients older than 21 years. Restylane Lyft with Lidocaine also is indicated for correction of moderate to severe facial wrinkles and folds such as the nasolabial folds, cheek augmentation, and age-related midface contour deficiencies. For more information, visit www.RestylaneUSA.com.
Xepi Launches for Impetigo
Cutanea Life Sciences launches Xepi (ozenoxacin) Cream 1%, a quinolone antimicrobial, for the treatment of impetigo in adult and pediatric patients 2 months or older. Xepi is applied twice daily for 5 days and has been shown to be active against most isolates of Staphylococcus aureus (including methicillin-resistant isolates) and Streptococcus pyogenes, both in vitro and in clinical infections. For more information, visit www.XepiCream.com.
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].
Altreno Lotion Now Available for Acne Vulgaris
Ortho Dermatologics launches Altreno (tretinoin) Lotion 0.05% for the treatment of acne vulgaris in patients 9 years and older. It was approved by the US Food and Drug Administration in August 2018, providing patients with the efficacy of tretinoin and the tolerability of a lotion formulation containing hyaluronic acid, glycerin, and collagen to help hydrate and moisturize the skin. For more information, visit www.ortho-dermatologics.com.
Bryhali Approved for Plaque Psoriasis in Adults
Ortho Dermatologics announces US Food and Drug Administration approval of Bryhali (halobetasol propionate) Lotion 0.01% for the treatment of plaque psoriasis in adults. Bryhali is a corticosteroid in a novel vehicle lotion with safety established for dosing up to 8 weeks, offering patients a longer duration of use than other topical steroids. For more information, visit www.bryhali.com.
CoolSculpting Cleared for the Submandibular Area
Zeltiq Aesthetics, Inc, an Allergan affiliate, announces US Food and Drug Administration (FDA) clearance of CoolSculpting to treat the submandibular area. The FDA clearance also was expanded to include patients with a body mass index of up to 46.2 when treating the submental and submandibular areas. CoolSculpting is a nonsurgical treatment that works by gently cooling targeted fat cells in the body to induce natural controlled elimination of fat cells without affecting surrounding tissue. CoolSculpting also is cleared for treatment of visible fat bulges on the thighs, abdomen, and flanks. For more information, visit www.coolsculpting.com.
Glytone Age-Defying Vitamin C+E Serum Reduces Signs of Aging
Pierre Fabre Dermo-Cosmetique introduces Glytone Age-Defying Vitamin C+E Serum, a layering serum with time-released, high concentrations of stabilized vitamins C and E combined with red tea flavonoids to deliver antioxidant protection and antiaging benefits. It is indicated for premature aging caused by environmental damage and oxidative stress as well as postprocedure relief. For more information, visit www.glytone-usa.com.
Hyrimoz Biosimilar Approved for Psoriasis
Sandoz, a Novartis Division, announces US Food and Drug Administration (FDA) approval of Hyrimoz (adalimumab-adaz), a biosimilar indicated for the treatment of psoriatic arthritis and plaque psoriasis, as well as rheumatoid arthritis, juvenile idiopathic arthritis in patients 4 years and older, ankylosing spondylitis, adult Crohn disease, and ulcerative colitis. Hyrimoz is the third FDA-approved biosimilar from Sandoz. For more information, visit www.sandoz.com.
Libtayo Approved for SCC
Regeneron Pharmaceuticals, Inc, and sanofi-aventis US LLC, announce US Food and Drug Administration (FDA) approval of Libtayo (cemiplimab-rwlc) for the treatment of patients with metastatic cutaneous squamous cell carcinoma (SCC) or locally advanced cutaneous SCC who are not candidates for curative surgery or radiation. Libtayo is a monoclonal antibody targeting the programmed death receptor 1.
Restylane Lyft Now Approved for Hand Rejuvenation
Nestlé Skin Health announces US Food and Drug Administration approval of Restylane Lyft with Lidocaine, a hyaluronic acid dermal filler, for the correction of age-related volume loss in the back of the hands for patients older than 21 years. Restylane Lyft with Lidocaine also is indicated for correction of moderate to severe facial wrinkles and folds such as the nasolabial folds, cheek augmentation, and age-related midface contour deficiencies. For more information, visit www.RestylaneUSA.com.
Xepi Launches for Impetigo
Cutanea Life Sciences launches Xepi (ozenoxacin) Cream 1%, a quinolone antimicrobial, for the treatment of impetigo in adult and pediatric patients 2 months or older. Xepi is applied twice daily for 5 days and has been shown to be active against most isolates of Staphylococcus aureus (including methicillin-resistant isolates) and Streptococcus pyogenes, both in vitro and in clinical infections. For more information, visit www.XepiCream.com.
If you would like your product included in Product News, please email a press release to the Editorial Office at [email protected].

Primary Cutaneous Epstein-Barr Virus–Positive Diffuse Large B-Cell Lymphoma: A Rare and Aggressive Cutaneous Lymphoma
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
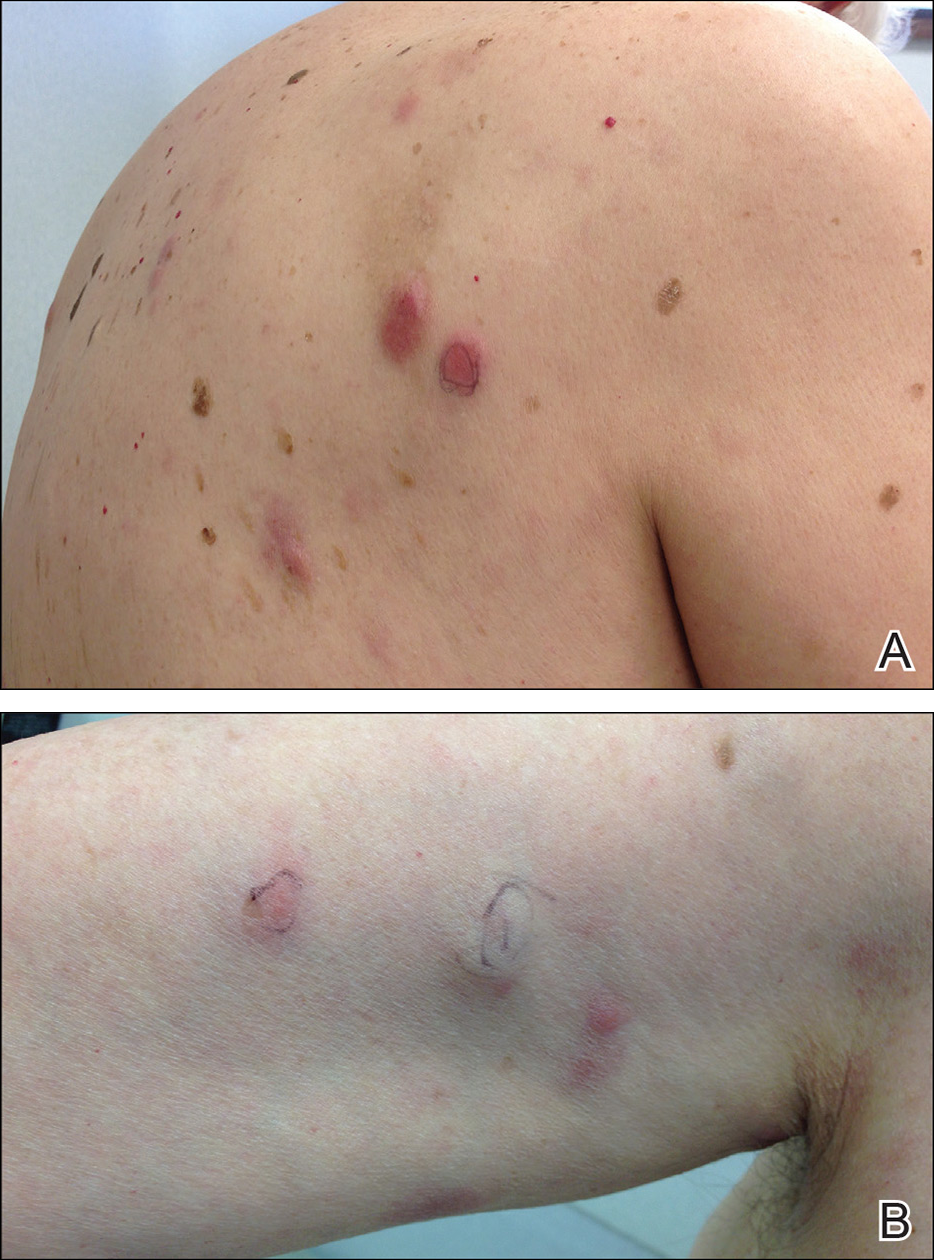
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
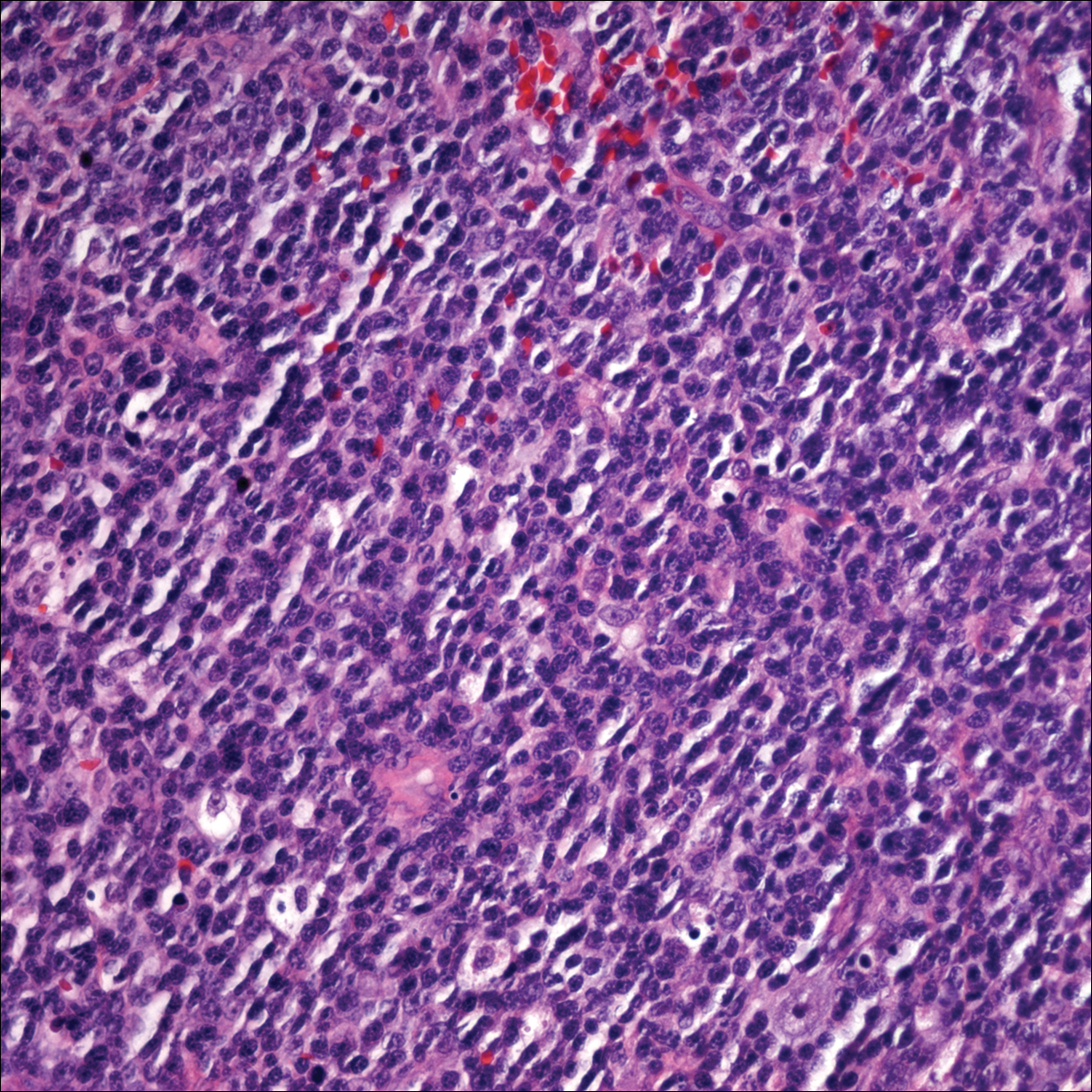
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
Cutaneous B-cell lymphomas represent a group of lymphomas derived from B lymphocytes in various stages of differentiation. The skin can be the site of primary or secondary involvement of any of the B-cell lymphomas. Primary cutaneous B-cell lymphomas present in the skin without evidence of extracutaneous disease at the time of diagnosis.1 The World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues recognizes 5 distinct primary cutaneous B-cell lymphoma subtypes: primary cutaneous follicle center lymphoma; primary cutaneous marginal zone lymphoma; primary cutaneous diffuse large B-cell lymphoma (DLBCL), leg type; DLBCL, not otherwise specified; and intravascular DLBCL.1-3 The DLBCL, not otherwise specified, category includes less common provisional entities with insufficient evidence to be recognized as distinct diseases. Epstein-Barr virus (EBV)–positive DLBCL is a rare subtype in this group.4
This article reviews the different clinicopathologic subtypes of primary cutaneous B-cell lymphoma. It also serves to help dermatologists recognize primary cutaneous EBV-positive DLBCL as a rare and aggressive form of this disease.
Case Report
An 84-year-old white man presented with a pruritic eruption on the arms, legs, back, neck, and face of 5 months’ duration. His medical history was notable for prostate cancer that was successfully treated with radiation therapy 6 years prior. The patient denied any constitutional symptoms such as fever, chills, night sweats, or weight loss, and review of systems was negative. The patient was taking prednisone, which alleviated the pruritus, but the lesions persisted.
Physical examination revealed multiple pink to erythematous papules and subcutaneous nodules involving the face, neck, back, arms, and legs (Figure 1). No scale, crust, or ulceration was present. Palpation of the cervical, supraclavicular, axillary, and inguinal lymph nodes was negative for lymphadenopathy.
Punch biopsies of representative lesions on the upper back and right arm revealed diffuse and nodular infiltrates of large atypical lymphoid cells with scattered centroblasts and immunoblasts (Figures 2 and 3). Immunohistochemical staining demonstrated CD79, MUM-1, and EBV-encoded RNA positivity among the neoplastic cells. The Ki-67 proliferative index was greater than 90%. The neoplastic cells were negative for CD5, CD10, CD20, CD21, CD30, CD56, CD123, CD138, PAX5, C-MYC, BCL-2, BCL-6, cyclin D1, TCL-1A, and terminal deoxynucleotidyl transferase
A peripheral blood smear did not show evidence of a B-cell lymphoproliferative process. A bone marrow biopsy was performed and did not show evidence of B-cell lymphoid neoplasia but did show reactive lymphoid aggregates composed of CD4+ and CD10+ T cells. Peripheral blood T-cell rearrangement and JAK2 were negative.
Based on clinical and histologic findings, the patient was diagnosed with primary cutaneous EBV-positive DLBCL. The patient was started on CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy for treatment of this aggressive cutaneous lymphoma, which initially resulted in clinical improvement of the lesions and complete involution of the subcutaneous nodules. After the sixth cycle of CHOP, he developed faintly erythematous indurated papules on the upper arms, chest, and back. Biopsy confirmed recurrence of the EBV-positive cutaneous lymphoma, and he started salvage chemotherapy with gemcitabine, oxaliplatin, and rituximab every 2 weeks; however, 4 months later (9 months after the initial presentation) he died from complications of the disease.
Comment
Etiology
Epstein-Barr virus–positive DLBCL, also called EBV-positive DLBCL of the elderly, was initially described in 2003 by Oyama et al5 and was included as a provisional entity in the 2008 World Health Organization classification system as a rare subtype of the DLBCL, not otherwise specified, category.2 It is defined as an EBV-positive monoclonal large B-cell proliferation that occurs in immunocompetent patients older than 50 years.6 Epstein-Barr virus is a human herpesvirus that demonstrates tropism for lymphocytes and survives in human hosts by establishing latency in B cells. Under normal immune conditions, the proliferation of EBV-infected B cells is prevented by cytotoxic T cells.7 It is important to recognize that patients with EBV-positive DLBCL do not have a known immunodeficiency state; therefore, it has been postulated that EBV-positive DLBCL might be caused by age-related senescence of the immune system.4,8
Epidemiology and Clinical Features
Epstein-Barr virus–positive DLBCL is more common in Asian countries than in Western countries, and there is a slight male predominance.6 A majority of patients present with extranodal disease at the time of diagnosis, and the skin is the most common extranodal site of involvement.6,9 Rare cases of primary cutaneous involvement also have been described.7,9,10 Cutaneous manifestations include erythematous papules and subcutaneous nodules. Other sites of extranodal involvement include the lungs, oral cavity, pharynx, gastrointestinal tract, and bone marrow.8,9 However, EBV-positive DLBCL is an aggressive lymphoma and prognosis is poor irrespective of the primary site of involvement.
Histopathology
Two morphologic subtypes can be seen on histology. The polymorphic pattern is characterized by a broad range of B-cell maturation with admixed reactive cells (eg, lymphocytes, histiocytes, plasma cells). The monomorphic or large-cell pattern is characterized by monotonous sheets of large transformed B cells.4,11 Many cases show both histologic patterns, and these morphologic variants do not impart any clinical or prognostic significance. Regardless of the histologic subtype, the neoplastic cells express pan B-cell antigens (eg, CD19, CD20, CD79a, PAX5), as well as MUM-1, BCL-2, and EBV-encoded RNA.4 Cases with plasmablastic features, as in our patient, may show weak or absent CD20 staining.12 Detection of EBV by in situ hybridization is required for the diagnosis.
Diagnosis
Workup for a suspected cutaneous lymphoma should include a complete history and physical examination; laboratory studies; and relevant imaging evaluation such as computed tomography of the chest, abdomen, and pelvis with or without whole-body positron emission tomography. A bone marrow biopsy and aspirate also should be performed in all cutaneous lymphomas with intermediate to aggressive clinical behavior. Accurate staging evaluation is integral to confirm the absence of extracutaneous involvement and to provide prognostic and anatomic information for the appropriate selection of treatment.13
Prognosis and Management
Primary cutaneous lymphomas tend to have different clinical behaviors and prognoses compared to histologically similar systemic lymphomas; therefore, different therapeutic strategies are warranted.14 Epstein-Barr virus–positive DLBCL has an aggressive clinical course with a median survival of 2 years.8 Patients with EBV-positive DLBCL have a poorer overall survival and treatment response when compared to patients with EBV-negative DLBCLs.4 Primary cutaneous B-cell lymphomas with indolent behavior, such as primary cutaneous marginal zone lymphoma and primary cutaneous follicle center lymphoma, can be treated with surgical excision, radiation therapy, or observation.15 No standard treatment exists for EBV-positive DLBCL, but R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone), which is the standard treatment of primary cutaneous DLBCL, leg type, may provide a survival benefit.13,15 Further studies are required to determine optimal treatment strategies.
Conclusion
Although rare, EBV-positive DLBCL is an important entity to consider when evaluating a patient with a suspected primary cutaneous lymphoma. Workup to rule out an underlying systemic lymphoma with relevant laboratory evaluation, imaging studies, and bone marrow biopsy is critical. Prognosis is poor and treatment is difficult, as standard treatment protocols have yet to be determined.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nakmura S, Jaffe ES, Swerdlow SH. EBV positive diffuse large B-cell lymphoma of the elderly. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2008:243-244.
- Kempf W, Sander CA. Classification of cutaneous lymphomas—an update. Histopathology. 2010;56:57-70.
- Castillo JJ, Beltran BE, Miranda RN, et al. Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly: what we know so far. Oncologist. 2011;16:87-96.
- Oyama T, Ichimura K, Suzuki R, et al. Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients. Am J Surg Pathol. 2003;27:16-26.
- Ok CY, Papathomas TG, Medeiros LJ, et al. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood. 2013;122:328-340.
- Tokuda Y, Fukushima M, Nakazawa K, et al. A case of primary Epstein-Barr virus-associated cutaneous diffuse large B-cell lymphoma unassociated with iatrogenic or endogenous immune dysregulation. J Cutan Pathol. 2008;35:666-671.
- Oyama T, Yamamoto K, Asano N, et al. Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients. Clin Cancer Res. 2007;13:5124-5132.
- Eminger LA, Hall LD, Hesterman KS, et al. Epstein-Barr virus: dermatologic associations and implications. J Am Acad Dermatol. 2015;72:21-34.
- Martin B, Whittaker S, Morris S, et al. A case of primary cutaneous senile EBV-related diffuse large B-cell lymphoma. Am J Dermatopathol. 2010;32:190-193.
- Gibson SE, Hsi ED. Epstein-Barr virus-positive B-cell lymphoma of the elderly at a United States tertiary medical center: an uncommon aggressive lymphoma with a nongerminal center B-cell phenotype. Hum Pathol. 2009;40:653-661.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:479-484.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:329.e1-329.e13; quiz 341-342.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:343.e1-343.e11; quiz 355-356.
Practice Points
- Primary cutaneous lymphomas are malignant lymphomas confined to the skin.
- Complete staging workup is necessary to rule out secondary involvement of the skin from a nodal lymphoma.
- Epstein-Barr virus-positive diffuse large B-cell lymphoma is a rare and aggressive primary cutaneous lymphoma.
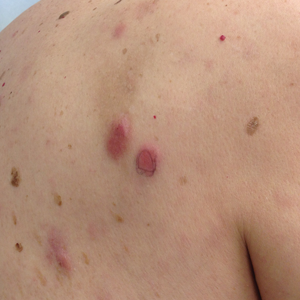
Automobile Injury: A Common Familiar Risk for Presenting and Comparing Risks in Dermatology
Numerous highly efficacious treatment modalities exist in dermatology, yet patients may be highly wary of their possible adverse events, even when those risks are rare.1,2 Such fears can lead to poor medication adherence and treatment refusal. A key determinant in successful patient-provider care is to effectively communicate risk. The communication of risk is hampered by the lack of any common currency for comparing risks. The development of a standardized unit of risk could help facilitate risk comparisons, allowing physicians and patients to put risk levels into better perspective.
One easily relatable event is the risk of injury in an automobile crash. Driving, whether to the dermatology clinic for a monitoring visit or to the supermarket for weekly groceries, is associated with risk of injury and death. The risk of automobile-related injury warranting a visit to the emergency department could provide a comparator that physicians can use to give patients a more objective sense of treatment risks or to introduce the justification of a monitoring visit. The objective of this study was to develop a standard risk unit based on the lifetime risk (LTR) of automobile injury and to compare this unit of risk to various risks of dermatologic treatments.
Methods
Literature Review
We first identified common risks in dermatology that would be illustrative and then identified keywords. PubMed searches for articles indexed for MEDLINE from November 1996 to February 2017 were performed combining the following terms: (relative risk, odds ratio, lifetime risk) and (isotretinoin, IBD; melanoma, SCC, transplantation; indoor tanning, BCC, SCC; transplant and SCC; biologics and tuberculosis; hydroxychloroquine retinal toxicity; psoriasis and psoriatic arthritis). An additional search was performed in June 2018 including the term blindness and injectable fillers. Our search combined these terms in numerous ways. Results were focused on meta-analyses and observational studies.
The references of relevant studies were included. Articles not focused on meta-analyses but rather on observational studies were individually analyzed for quality and bias using the 9-point Newcastle-Ottawa Scale, with a score of 7 or more as a cutoff for inclusion.
Determination of Risk Comparators
Data from the 2016 National Safety Council’s Injury Facts report were searched for nonmedical-related risk comparators, such as the risk of death by dog attack, by lightning, and by fire or smoke.3 Data from the 2015 US Department of Transportation Traffic Safety Facts were searched for relatable risk comparators, such as the LTR of automobile death and injury.4
Definitions
Automobile injury was defined as an injury warranting a visit to the emergency department.5 Automobile was defined as a road vehicle with 4 wheels and powered by an internal combustion engine or electric motor.6 This definition excluded light trucks, large trucks, and motorcycles.
LTR Calculation
Lifetime risk was used as the comparative measure. Lifetime risk is a type of absolute risk that depicts the probability that a specific disease or event will occur in an individual’s lifespan. The LTRof developing a disease or adverse event due to a dermatologic therapy or interventionwas denoted as LTRadverse event and calculated by the following equation7,8:

In this equation, LTRgeneral population is the LTR of developing the disease or adverse event without being subject to the therapy or intervention, and RRintervention is the relative risk (RR) from previously published RR data (relating to the development of the disease in question or an adverse event of the intervention). The use of equation (1) holds true only when the absolute risk of developing the disease or adverse event (LTRgeneral population) is low.7 Although the calculation of an LTR using a constant lifetime RR may require major approximations, studies evaluating the variation of RR over time are sparse.7,9 The Newcastle-Ottawa Scale was used to control such variance; only high-quality, nonrandomized studies were included. Although the use of residual LTR would be preferable, as LTR depends on age, such epidemiological data do not exist for complex diseases.
When not available, the LTRgeneral population was calculated from the rate of disease (cases per 100,000 individuals per year) multiplied by the average lifespan of an American (78.8 years)10:
When an odds ratio (OR) was presented, its conversion to RR followed11:
In this equation, RC is the absolute risk in the unexposed group. If the prevalence of the disease was considered low, the rare disease assumption was implemented as the following11,12:

The use of this approximation overestimates the LTR of an event. From a patient perspective, this approach is conservative. If prior LTR values were available, such as the LTR of automobile injury, automobile death, or other intervention, they were used without the need for calculation.
Unit Comparator
The LTRs of all adverse events were normalized to a unit comparator, using the LTR of an automobile injury as reference point, denoted as 1 risk unit (RU):

This equation allows for quick comparison of the magnitude of LTRs between events. Events with an RU less than 1 are less likely to occur than the risk of automobile injury; events with an RU greater than 1 are more likely than the risk of automobile injury. All RR, LTR, and unit comparators were presented as a single pooled estimate of their respective upper-limit CIs. The use of the upper-limit CI conservatively overestimates the LTR of an event.
Results
Ten dermatologic interventions were identified as illustrative, to be presented alongside the risk of automobile injury and death. The LTR of automobile injury was 32%, defined as 1.0 RU. The LTR of automobile death was 0.89% (1/36 RU).
Two events had LTRs roughly similar to automobile injury: development of a subsequent basal cell carcinoma within 3 years (1.4 RU) and development of a squamous cell carcinoma (SCC) secondary to indoor tanning (1.6 RU). Development of SCC following organ transplantation (34 RU) was considerably more likely than automobile injury. All other identified events had lower RUs than automobile injury (Table). Three events with small RUs included tuberculosis development with a tumor necrosis factor α inhibitor (1/32 RU), Crohn disease development with isotretinoin (1/41 RU), and blindness following facial hyaluronic acid injection (1/80 RU). The LTR of death by dog attack (1/42,436 RU) and death by lightning strike (1/36,542 RU) also had small RUs.
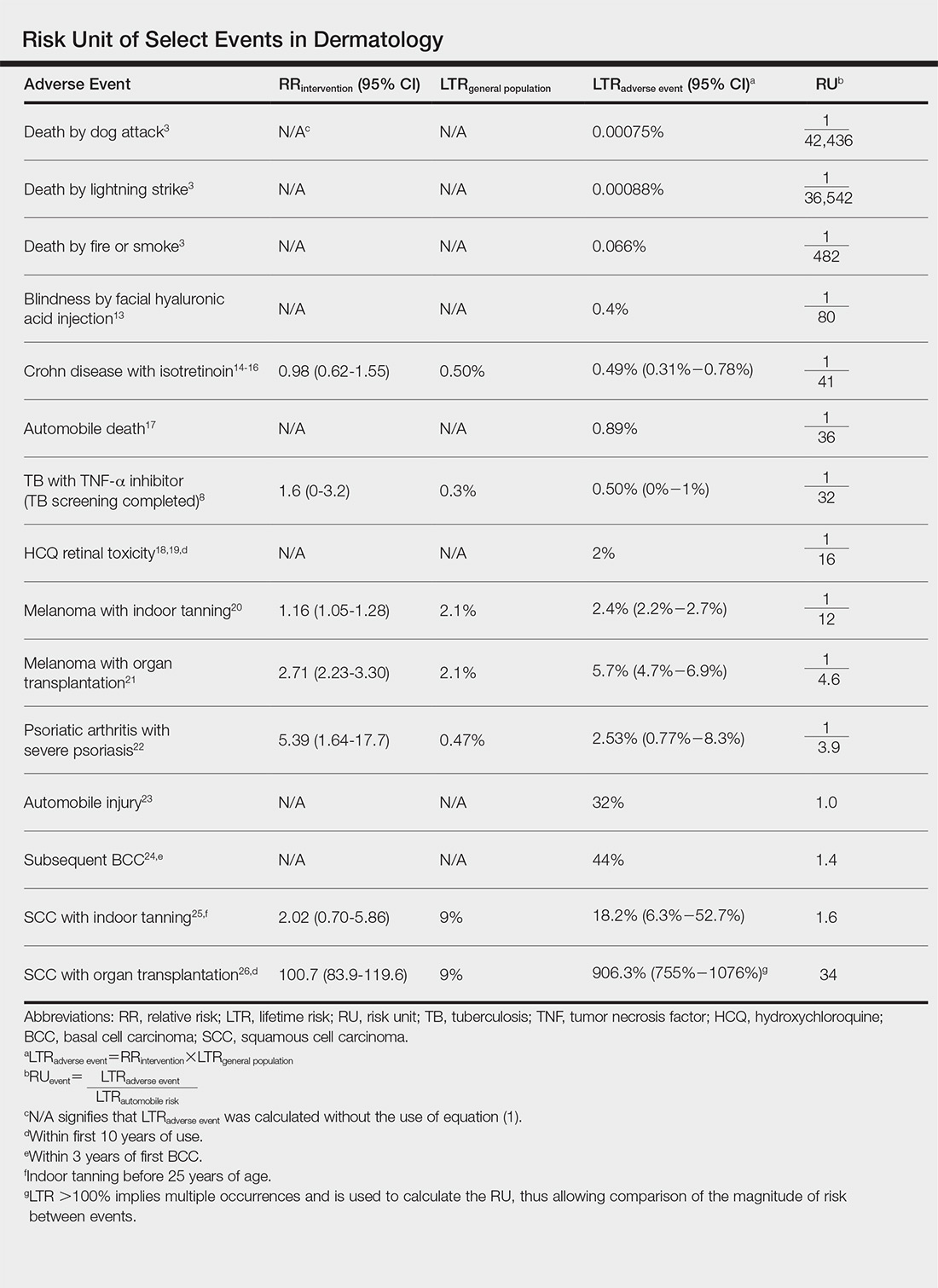
The unit comparators from the Table were adapted into graphic form to depict risk relative to the risk of automobile injury (Figure).
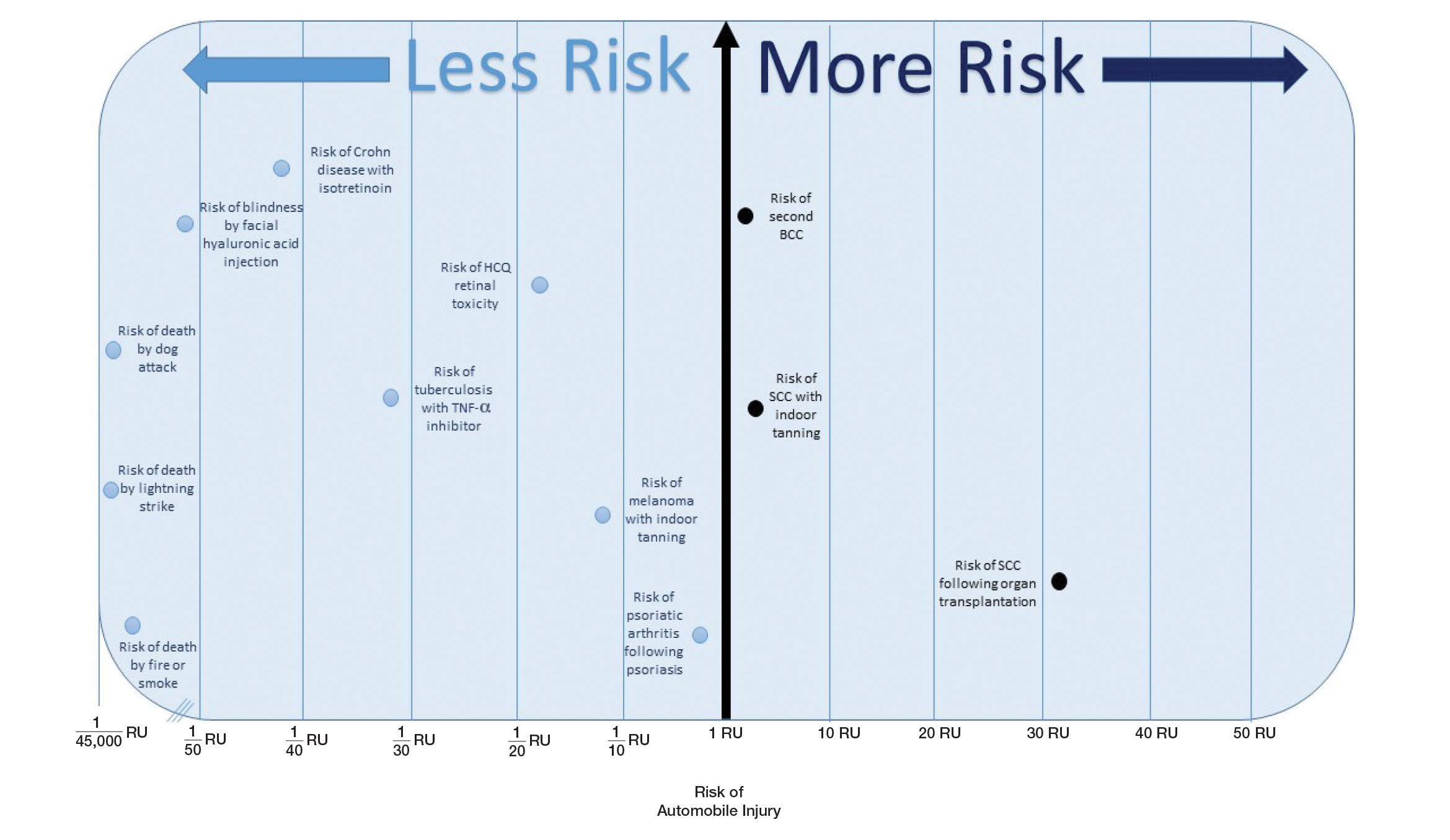
Comment
Numerous interventions in dermatology offer much less risk of an adverse event than the LTR of automobile injury. However, this concept of risk includes only the likelihood of development of an event, not the severity of the measured event, as our numerical and visual tool objectively captures the related risks using an RU comparator. Such use of a standardized RU demonstrates the essence of risk; “zero risk” does not exist, and each intervention or treatment, albeit how small, must be justified in concordance with other types of risk, such as the automobile.
The development of adverse events secondary to dermatologic intervention or therapy, for which monitoring visits are utilized, were used as important comparators to the risk of automobile injury. The continuous practice of monitoring visits may increase patient’s fears regarding possible adverse events secondary to therapy. Hydroxychloroquine retinal toxicity (1/16 RU) and psoriatic arthritis development following severe psoriasis (1/3.9 RU) were less likely to occur than automobile injury. The development of abnormal blood counts or blood tests secondary to therapy or intervention could not be formatted into an RU. The use of equation (1) for the calculation of LTRadverse eventholds true only when the absolute risk of developing the adverse event in the general population—in this case, abnormal blood counts or blood tests—is low.7
Although the unit comparator allows for the comparison of different dermatologic risk, a limitation of the RU model and its visual tool are a dependence on RR, a value that changes following publication of new studies. A solution was the use of a single pooled estimate to represent the upper-limit CIs of LTR. This practice overestimates risk. As with RR, new automobile injury rates are published annually.10 In the last 5 years, the LTR of automobile injury has stayed relatively constant: between 32% and 33%.4 Although the RU calculations and Figure included a wide variety of interventions in dermatology, select clinical situations were not included. It is beyond the scope of this article to systematically review all risk in dermatology but rather introduce the concept of the RU founded on automobile-associated risks. With the introduction of a methodical framework, the reader is invited to calculate RUs pertinent to their clinical interests.
Any intervention or treatment in dermatology is accompanied by risk. The use of a unit comparator using an easily relatable event—the LTR of automobile injury—allows the patient to easily compare risk and internally justify the practice of monitoring visits. Inclusion of a visual tool, such as the Figure, might alleviate many irrational fears that accompany some of the highly effective treatments and interventions used in dermatology and thus lead to better patient outcomes.
Acknowledgment
We thank Taranjeet Singh, MS (Dunn, North Carolina), for her comments on an earlier version of the manuscript.
- Rosen AB, Tsai JS, Downs SM. Variations in risk attitude across race, gender, and education. Med Decis Making. 2003;23:511-517.
- Sandoval LF, Pierce A, Feldman SR. Systemic therapies for psoriasis: an evidence-based update. Am J Clin Dermatol. 2014;15:165-180.
- National Safety Council. Odds of dying. Injury Facts website. http://injuryfacts.nsc.org/all-injuries/preventable-death-overview/odds-of-dying/. Accessed November 4, 2018.
- National Center for Statistics and Analysis (NCSA) motor vehicle traffic crash data resource page. National Highway Traffic Safety Administration website. https://crashstats.nhtsa.dot.gov/#/. Accessed November 4, 2018.
- CDC report shows motor vehicle crash injuries are frequent and costly. Centers for Disease Control and Prevention website. http://www.cdc.gov/media/releases/2014/p1007-crash-injuries.html. Published October 7, 2014. Accessed November 4, 2018.
- Automobile. Business Dictionary website. http://www.businessdictionary.com/definition/automobile.html. Accessed November 4, 2018.
- Dupont WD, Plummer WD Jr. Understanding the relationship between relative and absolute risk. Cancer. 1996;77:2193-2199.
- Kaminska E, Patel I, Dabade TS, et al. Comparing the lifetime risks of TNF-alpha inhibitor use to common benchmarks of risk. J Dermatolog Treat. 2011;24:101-106.
- Dupont WD. Converting relative risks to absolute risks: a graphical approach. Stat Med. 1989;8:641-651.
- Kochanek KD, Murphy SL, Xu J, et al. Deaths: final data for 2014. Natl Vital Stat Rep. 2016;65:1-122.
- Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0. The Cochrane Collaboration website. http://handbook.cochrane.org. Updated March 2011. Accessed November 15, 2018.
- Katz KA. The (relative) risks of using odds ratios. Arch Dermatol. 2006;142:761-764.
- Rayess HM, Svider PF, Hanba C, et al. A cross-sectional analysis of adverse events and litigation for injectable fillers. JAMA Facial Plast Surg. 2018;20:207-214.
- Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol. 2007;5:1424-1429.
- Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504-1517.
- Lee SY, Jamal MM, Nguyen ET, et al. Does exposure to isotretinoin increase the risk for the development of inflammatory bowel disease? A meta-analysis. Eur J Gastroenterol Hepatol. 2016;28:210-216.
- Injury Facts, 2017. Itasca, IL: National Safety Council; 2017.
- Marmor MF, Kellner U, Lai TY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016;123:1386-1394.
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol. 2014;132:1453-1460.
- Colantonio S, Bracken MB, Beecker J. The association of indoor tanning and melanoma in adults: systematic review and meta-analysis. J Am Acad Dermatol. 2014;70:847-857.e1-18.
- Green AC, Olsen CM. Increased risk of melanoma in organ transplant recipients: systematic review and meta-analysis of cohort studies. Acta Derm Venereol. 2015;95:923-927.
- Eder L, Haddad A, Rosen CF, et al. The incidence and risk factors for psoriatic arthritis in patients with psoriasis: a prospective cohort study. Arthritis Rheumatol. 2016;68:915-923.
- National Highway Traffic Safety Administration (NHTSA). Traffic Safety Facts 2015. Washington, DC: US Department of Transportation; 2015.
- Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch Dermatol. 2000;136:1524-1530.
- Wehner MR, Shive ML, Chren MM, et al. Indoor tanning and non-melanoma skin cancer: systematic review and meta-analysis. BMJ. 2012;345:E5909.
- Lindelöf B, Sigurgeirsson B, Gäbel H, et al. Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol. 2000;143:513-519.
Numerous highly efficacious treatment modalities exist in dermatology, yet patients may be highly wary of their possible adverse events, even when those risks are rare.1,2 Such fears can lead to poor medication adherence and treatment refusal. A key determinant in successful patient-provider care is to effectively communicate risk. The communication of risk is hampered by the lack of any common currency for comparing risks. The development of a standardized unit of risk could help facilitate risk comparisons, allowing physicians and patients to put risk levels into better perspective.
One easily relatable event is the risk of injury in an automobile crash. Driving, whether to the dermatology clinic for a monitoring visit or to the supermarket for weekly groceries, is associated with risk of injury and death. The risk of automobile-related injury warranting a visit to the emergency department could provide a comparator that physicians can use to give patients a more objective sense of treatment risks or to introduce the justification of a monitoring visit. The objective of this study was to develop a standard risk unit based on the lifetime risk (LTR) of automobile injury and to compare this unit of risk to various risks of dermatologic treatments.
Methods
Literature Review
We first identified common risks in dermatology that would be illustrative and then identified keywords. PubMed searches for articles indexed for MEDLINE from November 1996 to February 2017 were performed combining the following terms: (relative risk, odds ratio, lifetime risk) and (isotretinoin, IBD; melanoma, SCC, transplantation; indoor tanning, BCC, SCC; transplant and SCC; biologics and tuberculosis; hydroxychloroquine retinal toxicity; psoriasis and psoriatic arthritis). An additional search was performed in June 2018 including the term blindness and injectable fillers. Our search combined these terms in numerous ways. Results were focused on meta-analyses and observational studies.
The references of relevant studies were included. Articles not focused on meta-analyses but rather on observational studies were individually analyzed for quality and bias using the 9-point Newcastle-Ottawa Scale, with a score of 7 or more as a cutoff for inclusion.
Determination of Risk Comparators
Data from the 2016 National Safety Council’s Injury Facts report were searched for nonmedical-related risk comparators, such as the risk of death by dog attack, by lightning, and by fire or smoke.3 Data from the 2015 US Department of Transportation Traffic Safety Facts were searched for relatable risk comparators, such as the LTR of automobile death and injury.4
Definitions
Automobile injury was defined as an injury warranting a visit to the emergency department.5 Automobile was defined as a road vehicle with 4 wheels and powered by an internal combustion engine or electric motor.6 This definition excluded light trucks, large trucks, and motorcycles.
LTR Calculation
Lifetime risk was used as the comparative measure. Lifetime risk is a type of absolute risk that depicts the probability that a specific disease or event will occur in an individual’s lifespan. The LTRof developing a disease or adverse event due to a dermatologic therapy or interventionwas denoted as LTRadverse event and calculated by the following equation7,8:
In this equation, LTRgeneral population is the LTR of developing the disease or adverse event without being subject to the therapy or intervention, and RRintervention is the relative risk (RR) from previously published RR data (relating to the development of the disease in question or an adverse event of the intervention). The use of equation (1) holds true only when the absolute risk of developing the disease or adverse event (LTRgeneral population) is low.7 Although the calculation of an LTR using a constant lifetime RR may require major approximations, studies evaluating the variation of RR over time are sparse.7,9 The Newcastle-Ottawa Scale was used to control such variance; only high-quality, nonrandomized studies were included. Although the use of residual LTR would be preferable, as LTR depends on age, such epidemiological data do not exist for complex diseases.
When not available, the LTRgeneral population was calculated from the rate of disease (cases per 100,000 individuals per year) multiplied by the average lifespan of an American (78.8 years)10:
When an odds ratio (OR) was presented, its conversion to RR followed11:
In this equation, RC is the absolute risk in the unexposed group. If the prevalence of the disease was considered low, the rare disease assumption was implemented as the following11,12:
The use of this approximation overestimates the LTR of an event. From a patient perspective, this approach is conservative. If prior LTR values were available, such as the LTR of automobile injury, automobile death, or other intervention, they were used without the need for calculation.
Unit Comparator
The LTRs of all adverse events were normalized to a unit comparator, using the LTR of an automobile injury as reference point, denoted as 1 risk unit (RU):
This equation allows for quick comparison of the magnitude of LTRs between events. Events with an RU less than 1 are less likely to occur than the risk of automobile injury; events with an RU greater than 1 are more likely than the risk of automobile injury. All RR, LTR, and unit comparators were presented as a single pooled estimate of their respective upper-limit CIs. The use of the upper-limit CI conservatively overestimates the LTR of an event.
Results
Ten dermatologic interventions were identified as illustrative, to be presented alongside the risk of automobile injury and death. The LTR of automobile injury was 32%, defined as 1.0 RU. The LTR of automobile death was 0.89% (1/36 RU).
Two events had LTRs roughly similar to automobile injury: development of a subsequent basal cell carcinoma within 3 years (1.4 RU) and development of a squamous cell carcinoma (SCC) secondary to indoor tanning (1.6 RU). Development of SCC following organ transplantation (34 RU) was considerably more likely than automobile injury. All other identified events had lower RUs than automobile injury (Table). Three events with small RUs included tuberculosis development with a tumor necrosis factor α inhibitor (1/32 RU), Crohn disease development with isotretinoin (1/41 RU), and blindness following facial hyaluronic acid injection (1/80 RU). The LTR of death by dog attack (1/42,436 RU) and death by lightning strike (1/36,542 RU) also had small RUs.
The unit comparators from the Table were adapted into graphic form to depict risk relative to the risk of automobile injury (Figure).
Comment
Numerous interventions in dermatology offer much less risk of an adverse event than the LTR of automobile injury. However, this concept of risk includes only the likelihood of development of an event, not the severity of the measured event, as our numerical and visual tool objectively captures the related risks using an RU comparator. Such use of a standardized RU demonstrates the essence of risk; “zero risk” does not exist, and each intervention or treatment, albeit how small, must be justified in concordance with other types of risk, such as the automobile.
The development of adverse events secondary to dermatologic intervention or therapy, for which monitoring visits are utilized, were used as important comparators to the risk of automobile injury. The continuous practice of monitoring visits may increase patient’s fears regarding possible adverse events secondary to therapy. Hydroxychloroquine retinal toxicity (1/16 RU) and psoriatic arthritis development following severe psoriasis (1/3.9 RU) were less likely to occur than automobile injury. The development of abnormal blood counts or blood tests secondary to therapy or intervention could not be formatted into an RU. The use of equation (1) for the calculation of LTRadverse eventholds true only when the absolute risk of developing the adverse event in the general population—in this case, abnormal blood counts or blood tests—is low.7
Although the unit comparator allows for the comparison of different dermatologic risk, a limitation of the RU model and its visual tool are a dependence on RR, a value that changes following publication of new studies. A solution was the use of a single pooled estimate to represent the upper-limit CIs of LTR. This practice overestimates risk. As with RR, new automobile injury rates are published annually.10 In the last 5 years, the LTR of automobile injury has stayed relatively constant: between 32% and 33%.4 Although the RU calculations and Figure included a wide variety of interventions in dermatology, select clinical situations were not included. It is beyond the scope of this article to systematically review all risk in dermatology but rather introduce the concept of the RU founded on automobile-associated risks. With the introduction of a methodical framework, the reader is invited to calculate RUs pertinent to their clinical interests.
Any intervention or treatment in dermatology is accompanied by risk. The use of a unit comparator using an easily relatable event—the LTR of automobile injury—allows the patient to easily compare risk and internally justify the practice of monitoring visits. Inclusion of a visual tool, such as the Figure, might alleviate many irrational fears that accompany some of the highly effective treatments and interventions used in dermatology and thus lead to better patient outcomes.
Acknowledgment
We thank Taranjeet Singh, MS (Dunn, North Carolina), for her comments on an earlier version of the manuscript.
Numerous highly efficacious treatment modalities exist in dermatology, yet patients may be highly wary of their possible adverse events, even when those risks are rare.1,2 Such fears can lead to poor medication adherence and treatment refusal. A key determinant in successful patient-provider care is to effectively communicate risk. The communication of risk is hampered by the lack of any common currency for comparing risks. The development of a standardized unit of risk could help facilitate risk comparisons, allowing physicians and patients to put risk levels into better perspective.
One easily relatable event is the risk of injury in an automobile crash. Driving, whether to the dermatology clinic for a monitoring visit or to the supermarket for weekly groceries, is associated with risk of injury and death. The risk of automobile-related injury warranting a visit to the emergency department could provide a comparator that physicians can use to give patients a more objective sense of treatment risks or to introduce the justification of a monitoring visit. The objective of this study was to develop a standard risk unit based on the lifetime risk (LTR) of automobile injury and to compare this unit of risk to various risks of dermatologic treatments.
Methods
Literature Review
We first identified common risks in dermatology that would be illustrative and then identified keywords. PubMed searches for articles indexed for MEDLINE from November 1996 to February 2017 were performed combining the following terms: (relative risk, odds ratio, lifetime risk) and (isotretinoin, IBD; melanoma, SCC, transplantation; indoor tanning, BCC, SCC; transplant and SCC; biologics and tuberculosis; hydroxychloroquine retinal toxicity; psoriasis and psoriatic arthritis). An additional search was performed in June 2018 including the term blindness and injectable fillers. Our search combined these terms in numerous ways. Results were focused on meta-analyses and observational studies.
The references of relevant studies were included. Articles not focused on meta-analyses but rather on observational studies were individually analyzed for quality and bias using the 9-point Newcastle-Ottawa Scale, with a score of 7 or more as a cutoff for inclusion.
Determination of Risk Comparators
Data from the 2016 National Safety Council’s Injury Facts report were searched for nonmedical-related risk comparators, such as the risk of death by dog attack, by lightning, and by fire or smoke.3 Data from the 2015 US Department of Transportation Traffic Safety Facts were searched for relatable risk comparators, such as the LTR of automobile death and injury.4
Definitions
Automobile injury was defined as an injury warranting a visit to the emergency department.5 Automobile was defined as a road vehicle with 4 wheels and powered by an internal combustion engine or electric motor.6 This definition excluded light trucks, large trucks, and motorcycles.
LTR Calculation
Lifetime risk was used as the comparative measure. Lifetime risk is a type of absolute risk that depicts the probability that a specific disease or event will occur in an individual’s lifespan. The LTRof developing a disease or adverse event due to a dermatologic therapy or interventionwas denoted as LTRadverse event and calculated by the following equation7,8:
In this equation, LTRgeneral population is the LTR of developing the disease or adverse event without being subject to the therapy or intervention, and RRintervention is the relative risk (RR) from previously published RR data (relating to the development of the disease in question or an adverse event of the intervention). The use of equation (1) holds true only when the absolute risk of developing the disease or adverse event (LTRgeneral population) is low.7 Although the calculation of an LTR using a constant lifetime RR may require major approximations, studies evaluating the variation of RR over time are sparse.7,9 The Newcastle-Ottawa Scale was used to control such variance; only high-quality, nonrandomized studies were included. Although the use of residual LTR would be preferable, as LTR depends on age, such epidemiological data do not exist for complex diseases.
When not available, the LTRgeneral population was calculated from the rate of disease (cases per 100,000 individuals per year) multiplied by the average lifespan of an American (78.8 years)10:
When an odds ratio (OR) was presented, its conversion to RR followed11:
In this equation, RC is the absolute risk in the unexposed group. If the prevalence of the disease was considered low, the rare disease assumption was implemented as the following11,12:
The use of this approximation overestimates the LTR of an event. From a patient perspective, this approach is conservative. If prior LTR values were available, such as the LTR of automobile injury, automobile death, or other intervention, they were used without the need for calculation.
Unit Comparator
The LTRs of all adverse events were normalized to a unit comparator, using the LTR of an automobile injury as reference point, denoted as 1 risk unit (RU):
This equation allows for quick comparison of the magnitude of LTRs between events. Events with an RU less than 1 are less likely to occur than the risk of automobile injury; events with an RU greater than 1 are more likely than the risk of automobile injury. All RR, LTR, and unit comparators were presented as a single pooled estimate of their respective upper-limit CIs. The use of the upper-limit CI conservatively overestimates the LTR of an event.
Results
Ten dermatologic interventions were identified as illustrative, to be presented alongside the risk of automobile injury and death. The LTR of automobile injury was 32%, defined as 1.0 RU. The LTR of automobile death was 0.89% (1/36 RU).
Two events had LTRs roughly similar to automobile injury: development of a subsequent basal cell carcinoma within 3 years (1.4 RU) and development of a squamous cell carcinoma (SCC) secondary to indoor tanning (1.6 RU). Development of SCC following organ transplantation (34 RU) was considerably more likely than automobile injury. All other identified events had lower RUs than automobile injury (Table). Three events with small RUs included tuberculosis development with a tumor necrosis factor α inhibitor (1/32 RU), Crohn disease development with isotretinoin (1/41 RU), and blindness following facial hyaluronic acid injection (1/80 RU). The LTR of death by dog attack (1/42,436 RU) and death by lightning strike (1/36,542 RU) also had small RUs.
The unit comparators from the Table were adapted into graphic form to depict risk relative to the risk of automobile injury (Figure).
Comment
Numerous interventions in dermatology offer much less risk of an adverse event than the LTR of automobile injury. However, this concept of risk includes only the likelihood of development of an event, not the severity of the measured event, as our numerical and visual tool objectively captures the related risks using an RU comparator. Such use of a standardized RU demonstrates the essence of risk; “zero risk” does not exist, and each intervention or treatment, albeit how small, must be justified in concordance with other types of risk, such as the automobile.
The development of adverse events secondary to dermatologic intervention or therapy, for which monitoring visits are utilized, were used as important comparators to the risk of automobile injury. The continuous practice of monitoring visits may increase patient’s fears regarding possible adverse events secondary to therapy. Hydroxychloroquine retinal toxicity (1/16 RU) and psoriatic arthritis development following severe psoriasis (1/3.9 RU) were less likely to occur than automobile injury. The development of abnormal blood counts or blood tests secondary to therapy or intervention could not be formatted into an RU. The use of equation (1) for the calculation of LTRadverse eventholds true only when the absolute risk of developing the adverse event in the general population—in this case, abnormal blood counts or blood tests—is low.7
Although the unit comparator allows for the comparison of different dermatologic risk, a limitation of the RU model and its visual tool are a dependence on RR, a value that changes following publication of new studies. A solution was the use of a single pooled estimate to represent the upper-limit CIs of LTR. This practice overestimates risk. As with RR, new automobile injury rates are published annually.10 In the last 5 years, the LTR of automobile injury has stayed relatively constant: between 32% and 33%.4 Although the RU calculations and Figure included a wide variety of interventions in dermatology, select clinical situations were not included. It is beyond the scope of this article to systematically review all risk in dermatology but rather introduce the concept of the RU founded on automobile-associated risks. With the introduction of a methodical framework, the reader is invited to calculate RUs pertinent to their clinical interests.
Any intervention or treatment in dermatology is accompanied by risk. The use of a unit comparator using an easily relatable event—the LTR of automobile injury—allows the patient to easily compare risk and internally justify the practice of monitoring visits. Inclusion of a visual tool, such as the Figure, might alleviate many irrational fears that accompany some of the highly effective treatments and interventions used in dermatology and thus lead to better patient outcomes.
Acknowledgment
We thank Taranjeet Singh, MS (Dunn, North Carolina), for her comments on an earlier version of the manuscript.
- Rosen AB, Tsai JS, Downs SM. Variations in risk attitude across race, gender, and education. Med Decis Making. 2003;23:511-517.
- Sandoval LF, Pierce A, Feldman SR. Systemic therapies for psoriasis: an evidence-based update. Am J Clin Dermatol. 2014;15:165-180.
- National Safety Council. Odds of dying. Injury Facts website. http://injuryfacts.nsc.org/all-injuries/preventable-death-overview/odds-of-dying/. Accessed November 4, 2018.
- National Center for Statistics and Analysis (NCSA) motor vehicle traffic crash data resource page. National Highway Traffic Safety Administration website. https://crashstats.nhtsa.dot.gov/#/. Accessed November 4, 2018.
- CDC report shows motor vehicle crash injuries are frequent and costly. Centers for Disease Control and Prevention website. http://www.cdc.gov/media/releases/2014/p1007-crash-injuries.html. Published October 7, 2014. Accessed November 4, 2018.
- Automobile. Business Dictionary website. http://www.businessdictionary.com/definition/automobile.html. Accessed November 4, 2018.
- Dupont WD, Plummer WD Jr. Understanding the relationship between relative and absolute risk. Cancer. 1996;77:2193-2199.
- Kaminska E, Patel I, Dabade TS, et al. Comparing the lifetime risks of TNF-alpha inhibitor use to common benchmarks of risk. J Dermatolog Treat. 2011;24:101-106.
- Dupont WD. Converting relative risks to absolute risks: a graphical approach. Stat Med. 1989;8:641-651.
- Kochanek KD, Murphy SL, Xu J, et al. Deaths: final data for 2014. Natl Vital Stat Rep. 2016;65:1-122.
- Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0. The Cochrane Collaboration website. http://handbook.cochrane.org. Updated March 2011. Accessed November 15, 2018.
- Katz KA. The (relative) risks of using odds ratios. Arch Dermatol. 2006;142:761-764.
- Rayess HM, Svider PF, Hanba C, et al. A cross-sectional analysis of adverse events and litigation for injectable fillers. JAMA Facial Plast Surg. 2018;20:207-214.
- Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol. 2007;5:1424-1429.
- Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504-1517.
- Lee SY, Jamal MM, Nguyen ET, et al. Does exposure to isotretinoin increase the risk for the development of inflammatory bowel disease? A meta-analysis. Eur J Gastroenterol Hepatol. 2016;28:210-216.
- Injury Facts, 2017. Itasca, IL: National Safety Council; 2017.
- Marmor MF, Kellner U, Lai TY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016;123:1386-1394.
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol. 2014;132:1453-1460.
- Colantonio S, Bracken MB, Beecker J. The association of indoor tanning and melanoma in adults: systematic review and meta-analysis. J Am Acad Dermatol. 2014;70:847-857.e1-18.
- Green AC, Olsen CM. Increased risk of melanoma in organ transplant recipients: systematic review and meta-analysis of cohort studies. Acta Derm Venereol. 2015;95:923-927.
- Eder L, Haddad A, Rosen CF, et al. The incidence and risk factors for psoriatic arthritis in patients with psoriasis: a prospective cohort study. Arthritis Rheumatol. 2016;68:915-923.
- National Highway Traffic Safety Administration (NHTSA). Traffic Safety Facts 2015. Washington, DC: US Department of Transportation; 2015.
- Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch Dermatol. 2000;136:1524-1530.
- Wehner MR, Shive ML, Chren MM, et al. Indoor tanning and non-melanoma skin cancer: systematic review and meta-analysis. BMJ. 2012;345:E5909.
- Lindelöf B, Sigurgeirsson B, Gäbel H, et al. Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol. 2000;143:513-519.
- Rosen AB, Tsai JS, Downs SM. Variations in risk attitude across race, gender, and education. Med Decis Making. 2003;23:511-517.
- Sandoval LF, Pierce A, Feldman SR. Systemic therapies for psoriasis: an evidence-based update. Am J Clin Dermatol. 2014;15:165-180.
- National Safety Council. Odds of dying. Injury Facts website. http://injuryfacts.nsc.org/all-injuries/preventable-death-overview/odds-of-dying/. Accessed November 4, 2018.
- National Center for Statistics and Analysis (NCSA) motor vehicle traffic crash data resource page. National Highway Traffic Safety Administration website. https://crashstats.nhtsa.dot.gov/#/. Accessed November 4, 2018.
- CDC report shows motor vehicle crash injuries are frequent and costly. Centers for Disease Control and Prevention website. http://www.cdc.gov/media/releases/2014/p1007-crash-injuries.html. Published October 7, 2014. Accessed November 4, 2018.
- Automobile. Business Dictionary website. http://www.businessdictionary.com/definition/automobile.html. Accessed November 4, 2018.
- Dupont WD, Plummer WD Jr. Understanding the relationship between relative and absolute risk. Cancer. 1996;77:2193-2199.
- Kaminska E, Patel I, Dabade TS, et al. Comparing the lifetime risks of TNF-alpha inhibitor use to common benchmarks of risk. J Dermatolog Treat. 2011;24:101-106.
- Dupont WD. Converting relative risks to absolute risks: a graphical approach. Stat Med. 1989;8:641-651.
- Kochanek KD, Murphy SL, Xu J, et al. Deaths: final data for 2014. Natl Vital Stat Rep. 2016;65:1-122.
- Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0. The Cochrane Collaboration website. http://handbook.cochrane.org. Updated March 2011. Accessed November 15, 2018.
- Katz KA. The (relative) risks of using odds ratios. Arch Dermatol. 2006;142:761-764.
- Rayess HM, Svider PF, Hanba C, et al. A cross-sectional analysis of adverse events and litigation for injectable fillers. JAMA Facial Plast Surg. 2018;20:207-214.
- Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol. 2007;5:1424-1429.
- Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504-1517.
- Lee SY, Jamal MM, Nguyen ET, et al. Does exposure to isotretinoin increase the risk for the development of inflammatory bowel disease? A meta-analysis. Eur J Gastroenterol Hepatol. 2016;28:210-216.
- Injury Facts, 2017. Itasca, IL: National Safety Council; 2017.
- Marmor MF, Kellner U, Lai TY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016;123:1386-1394.
- Melles RB, Marmor MF. The risk of toxic retinopathy in patients on long-term hydroxychloroquine therapy. JAMA Ophthalmol. 2014;132:1453-1460.
- Colantonio S, Bracken MB, Beecker J. The association of indoor tanning and melanoma in adults: systematic review and meta-analysis. J Am Acad Dermatol. 2014;70:847-857.e1-18.
- Green AC, Olsen CM. Increased risk of melanoma in organ transplant recipients: systematic review and meta-analysis of cohort studies. Acta Derm Venereol. 2015;95:923-927.
- Eder L, Haddad A, Rosen CF, et al. The incidence and risk factors for psoriatic arthritis in patients with psoriasis: a prospective cohort study. Arthritis Rheumatol. 2016;68:915-923.
- National Highway Traffic Safety Administration (NHTSA). Traffic Safety Facts 2015. Washington, DC: US Department of Transportation; 2015.
- Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch Dermatol. 2000;136:1524-1530.
- Wehner MR, Shive ML, Chren MM, et al. Indoor tanning and non-melanoma skin cancer: systematic review and meta-analysis. BMJ. 2012;345:E5909.
- Lindelöf B, Sigurgeirsson B, Gäbel H, et al. Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol. 2000;143:513-519.
Practice Points
- Using common identifiable risks may help patients put the risk of certain dermatologic interventions into perspective.
- Numerous interventions in dermatology offer much less risk of an adverse event than the lifetime risk of automobile injury.