User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Ichthyosiform Sarcoidosis and Systemic Involvement
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
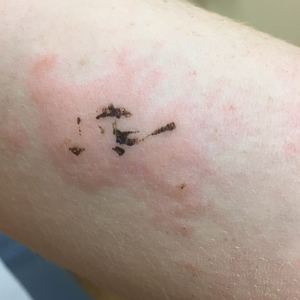
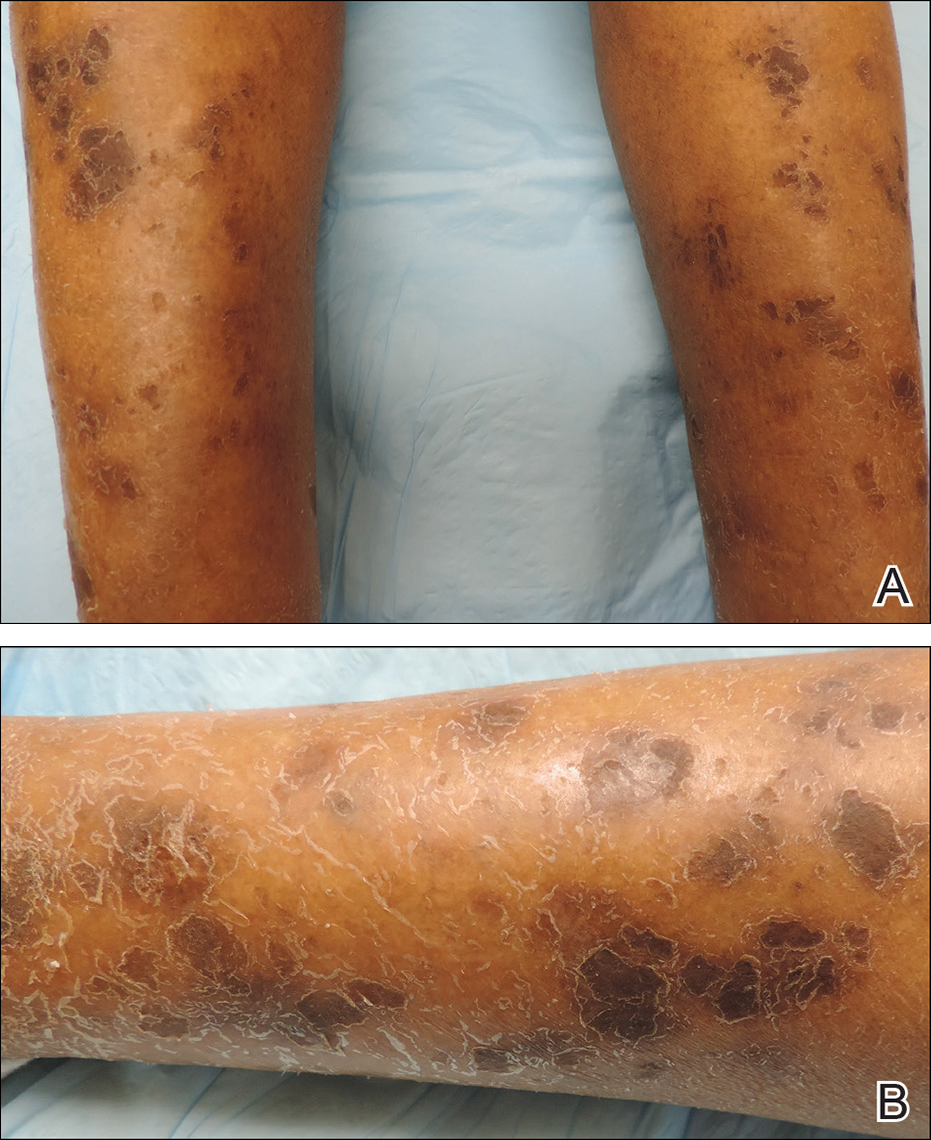
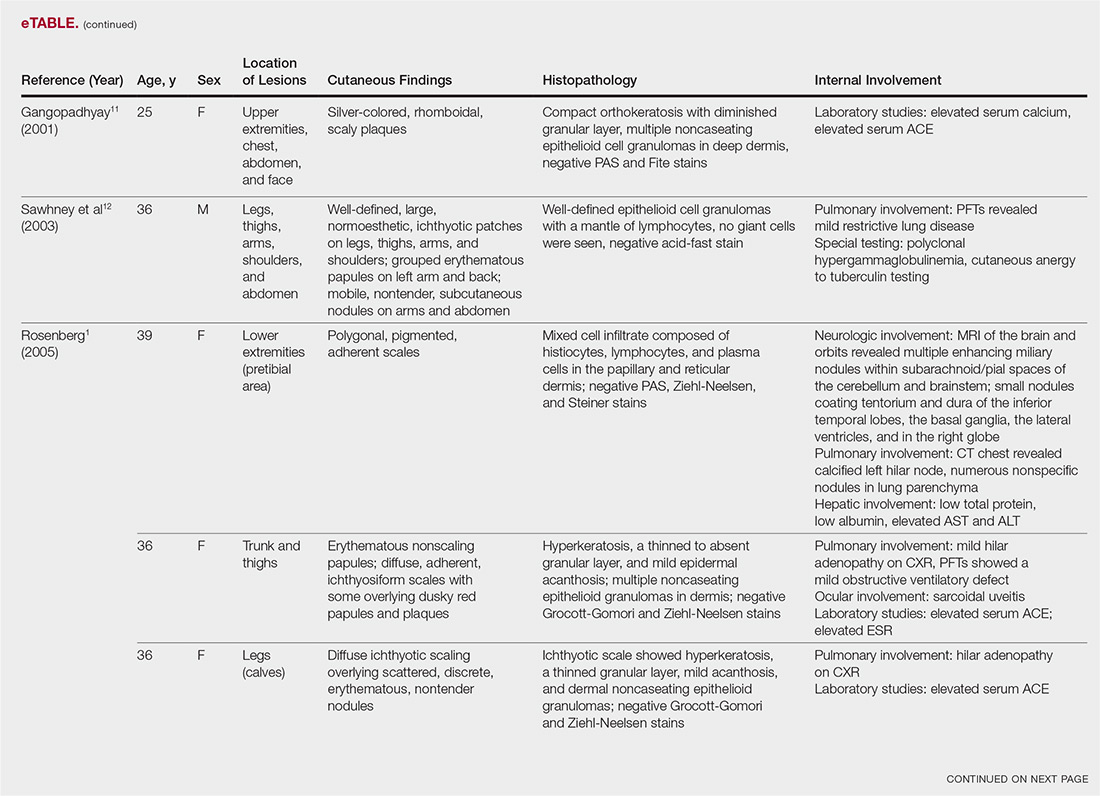
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
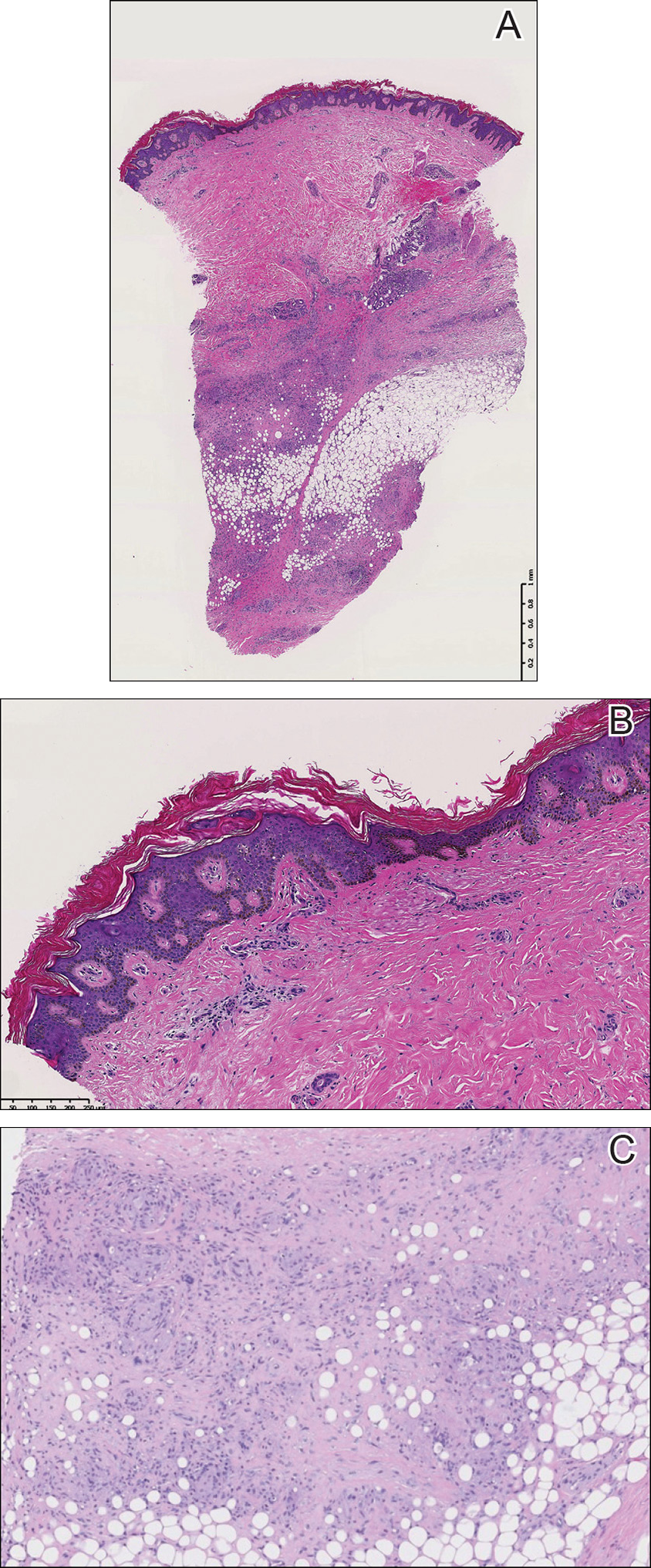
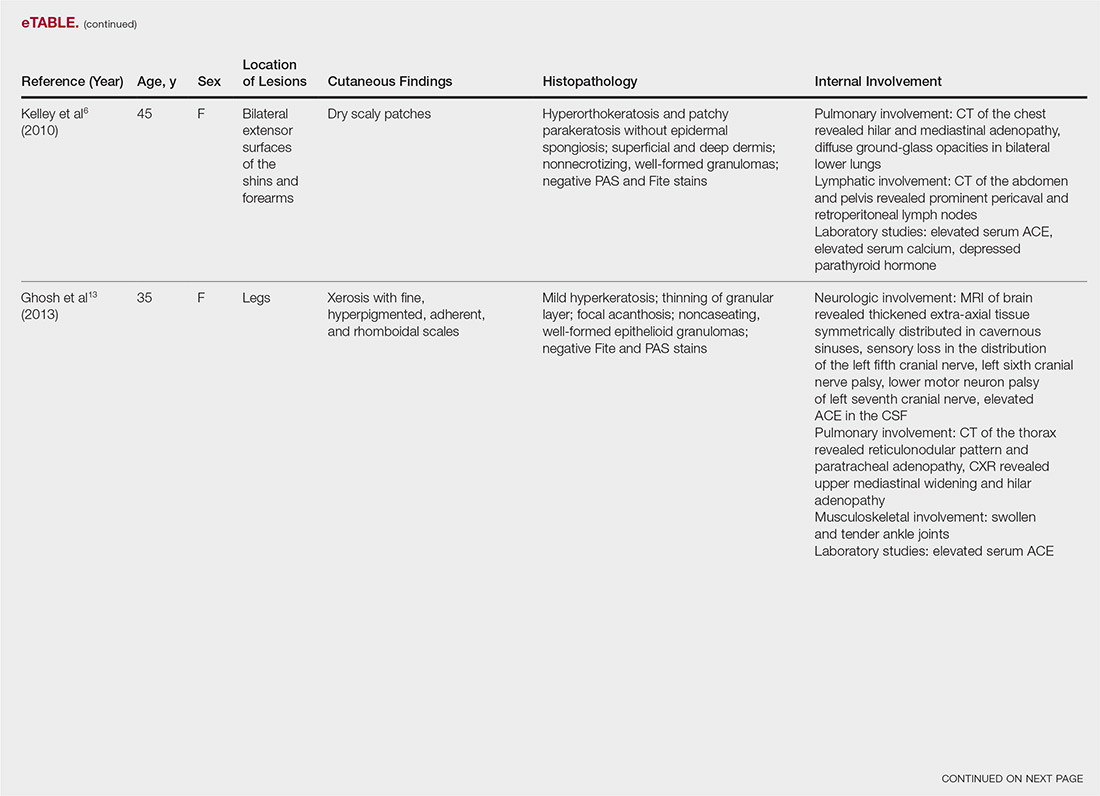
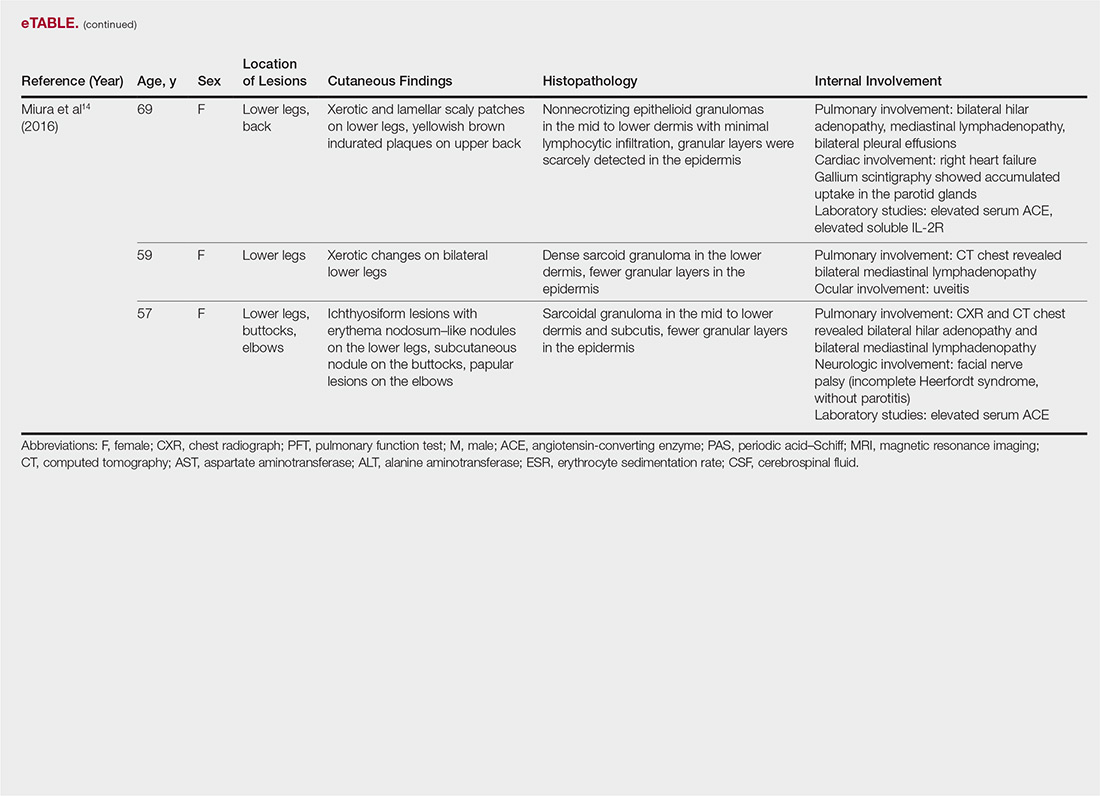
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
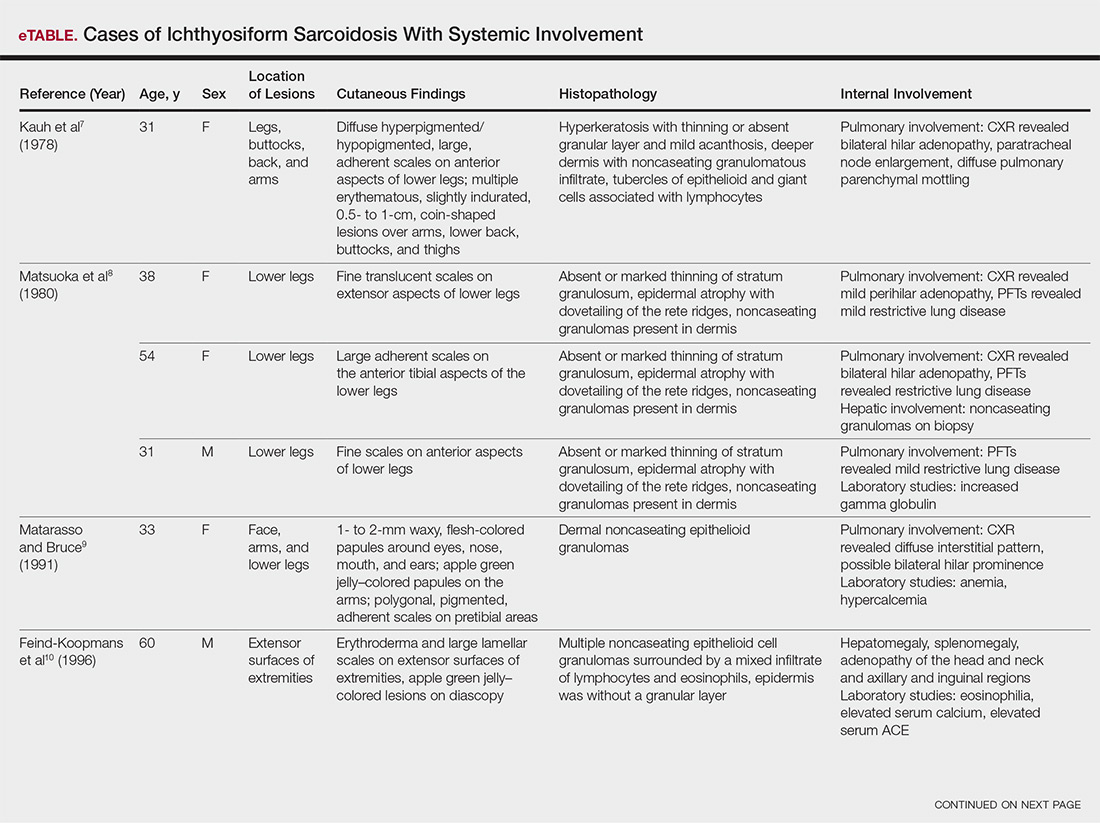
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
Sarcoidosis is a multiorgan, systemic, granulomatous disease that most commonly affects the cutaneous, pulmonary, ocular, and cardiac organ systems. Cutaneous involvement occurs in approximately 20% to 35% of patients, with approximately 25% of patients demonstrating only dermatologic findings.1 Cutaneous sarcoidosis can have a highly variable presentation. Ichthyosiform sarcoidosis (IS) is a rare form of this disease that has been described as presenting as polygonal adherent scales.2 It often is associated with internal organ involvement. We present a case of IS without any organ system involvement at the time of diagnosis. A review of the English-language literature was performed to ascertain the internal organ associations most commonly reported with IS.
Case Report
A 66-year-old black woman presented to dermatology with dark scaly patches noted by her primary care physician to be present on both of the lower extremities. The patient believed they were present for at least 4 years. She described dark spots confined to the lower legs that had gradually increased in size. Review of systems was negative for fever, chills, night sweats, weight loss, vision changes, cough, dyspnea, and joint pains, and there was no history of either personal or familial cutaneous diseases.
Physical examination revealed cutaneous patches of thin white scale with a sharp edge in arciform patterns on the lower extremities. Several of these patches were hyperpigmented and xerotic in appearance (Figure 1). The patches were limited to the lower legs, with no other lesions noted.
A punch biopsy of the skin on the right lower leg was performed. Histopathologic analysis showed epidermal compact hyperkeratosis with deep granulomatous infiltration into the subcutaneous tissue (Figures 2A and 2B). At high power, these granulomas were noted to be noncaseating naked granulomas composed of epithelioid histiocytes surrounded by sparse lymphocytic inflammation (Figure 2C). Special stains including acid-fast bacilli, Fite, and periodic acid–Schiff were negative. The diagnosis of IS was made based on clinical presentation and primarily by histopathologic analysis.
The patient’s cutaneous lesions were treated with fluocinonide ointment 0.05% twice daily. Although she did not notice a dramatic improvement in the plaques, they stabilized in size. Her primary care physician was notified and advised to begin a workup for involvement of other organ systems by sarcoidosis. Her initial evaluation, which included a chest radiograph and electrocardiogram, were unremarkable. Despite multiple attempts to persuade the patient to return for further follow-up, neither dermatology nor her primary care physician were able to complete a full workup.
Comment
Etiology
Although there are several theories regarding the etiology of sarcoidosis, the exact cause remains unknown. The body’s immune response, infectious agents, genetics, and the environment have all been thought to play a role. It has been well established that helper T cell (TH1) production of interferon and increased levels of tumor necrosis factor propagate the inflammatory response seen in sarcoidosis.3 More recently, TH17 cells have been found in cutaneous lesions, bronchoalveolar lavage samples, and the blood of patients with sarcoidosis, especially in those with active disease progression.3 Infectious agents such as mycobacteria and propionibacteria DNA or RNA also have been found in sarcoid samples.4 Several HLA-DRB1 variants have been associated with an increased incidence of sarcoidosis.5
Presentation
Characteristic dermatologic findings of sarcoidosis include macules, papules, nodules, and plaques located on the face, especially the nose, cheeks, and ears, and on the shins or ankles, as well as similar lesions around tattoos or scars. Sarcoid lesions also have been described as angiolupoid, lichenoid, annular, verrucous, ulcerative, and psoriasiform. Here we present an example of the uncommon type, ichthyosiform. Ichthyosiform sarcoidosis is a rare variant described primarily in dark-skinned individuals, a finding supported by both our case and prior reports. Most reported cases have described IS lesions as having a pasted-on appearance, with adherent centers on the extensor surfaces of the lower extremities, head, and/or neck.6 Our case follows this descriptive pattern previously reported with adherent patches limited to the lower extremities.
Histopathology
The key histopathologic finding is the presence of noncaseating granulomas on biopsy. Sarcoid “specific” lesions rest on the identification of the noncaseating granulomas, while “nonspecific” lesions such as erythema nodosum fail to demonstrate this finding.1
Systemic Involvement
The IS type is believed to be an excellent marker for systemic disease, with approximately 95% of reported cases having some form of systemic illness.6 Acquired ichthyosis should warrant further investigation for systemic disease. Early recognition could be beneficial for the patient because the ichthyosiform type is believed to precede the diagnosis of systemic disease in most cases by a median of 3 months.6
The most common site of internal sarcoid involvement is the lungs, but the lymph nodes, eyes, liver, spleen, heart, and central nervous system also can be involved. Patients can present with nonspecific symptoms such as erythema nodosum in the skin, dyspnea, cough, chest pain, vision changes, enlarged lymph nodes, headaches, joint pain, fever, fatigue, weight loss, and malaise. According to a PubMed search of articles indexed for MEDLINE using the term ichthyosiform sarcoidosis, 16 cases have been reported in the English-language literature (eTable).1,6-14 Of these 16 cases, 3 involved men and 13 involved women. The median age of a patient diagnosed with IS was 37 years. The respiratory system was found to be the most common organ system involved (14 of 16 patients), with hilar adenopathy and restrictive lung disease being the most common findings. Neurologic findings and hepatic involvement also were seen in 3 and 3 patients, respectively. Eight of 16 cases had an elevated serum angiotensin-converting enzyme level. Details of systemic involvement in other cases of IS are listed in the eTable.
Management
Most patients are given topical corticosteroids for their cutaneous lesions, but patients with systemic involvement will likely need some type of systemic immunosuppressive therapy to control their disease. Systemic therapy often is warranted in IS because of reports of rapid progression. Our case differs from these prior reports in the relative stability of the disease at the last patient encounter. Systemic treatment commonly includes oral corticosteroids such as prednisone. Other options, such as hydroxychloroquine, methotrexate, azathioprine, pentoxifylline, thalidomide, cyclophosphamide, cyclosporine, and infliximab, can be considered if other treatments fail.13 Ichthyosiform sarcoidosis patients should continue to have regular follow-up to monitor for disease progression.
Differential
When evaluating an acquired ichthyosis, dermatologists can consider other associations such as Hodgkin disease, hypothyroidism, multiple myeloma, carcinomatosis, and chronic malnutrition.1 Skin biopsy demonstrating granuloma formation also is not specific for sarcoidosis. Other etiologies, such as autoimmune diseases, immunodeficiency disorders, infections, foreign body granulomas, neoplasms, and drug reactions, should be considered.15 All patients with acquired ichthyosis should undergo a thorough evaluation for internal involvement.
Conclusion
We presented a case of IS, a rare type of sarcoidosis commonly associated with further internal involvement of the respiratory, nervous, or hepatic organ systems. Recognition of an acquired form of ichthyosis and its potential disease associations, including sarcoidosis, is important to improve early detection of any internal disease, allowing prompt initiation of treatment.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
- Rosenberg B. Ichthyosiform sarcoidosis. Dermatol Online J. 2005;11:15.
- Banse-Kupin L, Pelachyk JM. Ichthyosiform sarcoidosis: report of two cases and review of the literature. J Am Acad Dermatol. 1987;17:616-620.
- Sanchez M, Haimovic A, Prystowsky S. Sarcoidosis. Dermatol Clin. 2015;33:389-416.
- Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36:561-568.
- Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36:569-584.
- Kelley BP, George DE, LeLeux TM, et al. Ichthyosiform sarcoidosis: a case report and review of the literature. Dermatol Online J. 2010;16:5.
- Kauh YC, Goody HE, Luscombe HA. Ichthyosiform sarcoidosis. Arch Dermatol. 1978;114:100-101.
- Matsuoka LY, LeVine M, Glasser S, et al. Ichthyosiform sarcoid. Cutis. 1980;25:188-189.
- Matarasso SL, Bruce S. Ichthyosiform sarcoidosis: report of a case. Cutis. 1991;47:405-408.
- Feind-Koopmans AG, Lucker GP, van de Kerkhof PC. Acquired ichthyosiform erythroderma and sarcoidosis. J Am Acad Dermatol. 1996;35:826-828.
- Gangopadhyay AK. Ichthyosiform sarcoidosis. Indian J Dermatol Venereol Leprol. 2001;67:91-92.
- Sawhney M, Sharma YK, Gera V, et al. Ichthyosiform sarcoidosis following chemotherapy of Hodgkin’s disease. Indian J Dermatol Venereol Leprol. 2003;69:220-222.
- Ghosh UC, Ghosh SK, Hazra K, et al. Ichthyosiform sarcoidosis revisited. Indian J Dermatol Venereol Leprol. 2013;79:795-798.
- Miura T, Kato Y, Yamamoto T. Ichthyosiform sarcoidosis: report of three cases from Japan and literature review. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:392-397.
- Fernandez-Faith E, McDonnell J. Cutaneous sarcoidosis: differential diagnosis. Clin Dermatol. 2007;25:276-287.
Practice Points
- Ichthyosiform sarcoidosis is a rare form of sarcoidosis that presents as polygonal adherent scales.
- Ichthyosiform sarcoidosis is commonly associated with pulmonary, neurologic, and hepatic involvement.
- Acquired ichthyosis should warrant further investigation for systemic disease.
Crizotinib-Induced Lichenoid Drug Eruption in a Patient With Lung Cancer
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
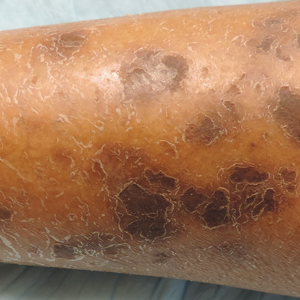
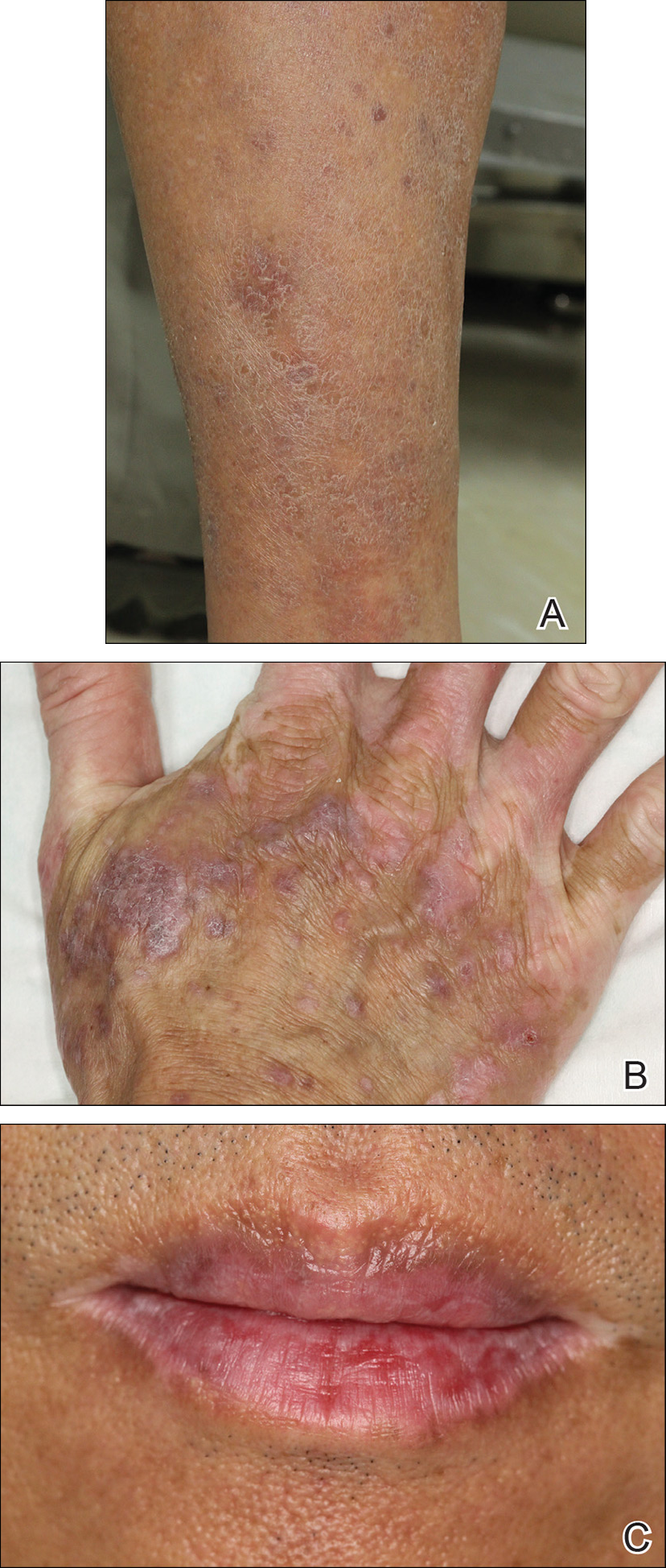
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
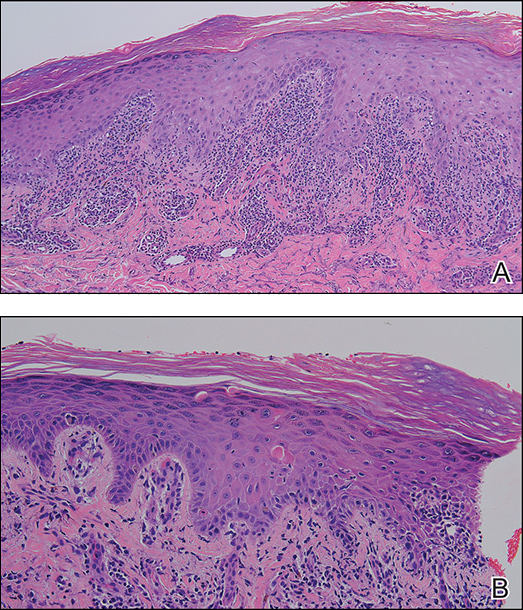
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
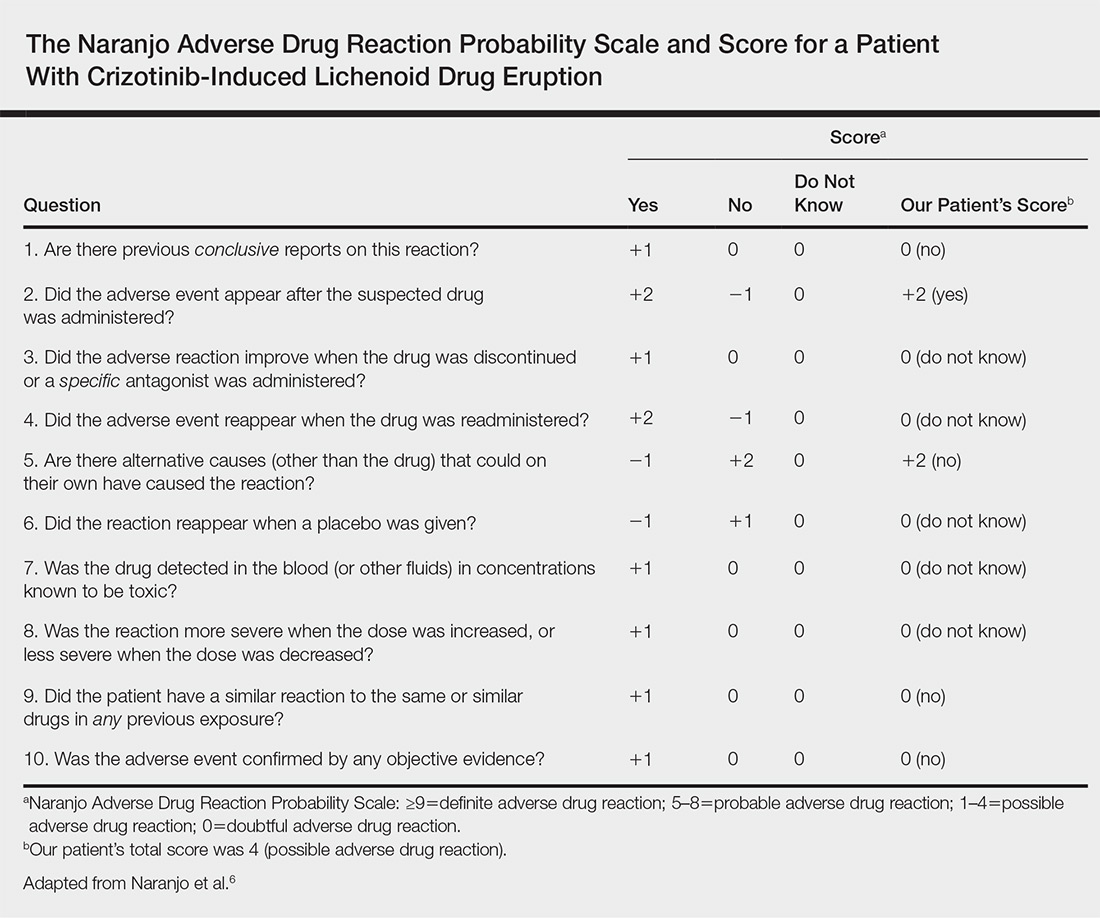
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
Crizotinib is a multitargeted tyrosine kinase inhibitor that blocks anaplastic lymphoma kinase (ALK), hepatocyte growth factor receptor (c-Met), and their oncogenic variants ALK fusion proteins or c-Met/hepatocyte growth factor receptor mutant variants.1 Additionally, crizotinib was approved by the US Food and Drug Administration in 2011 for the treatment of patients with non–small cell lung cancer (NSCLC) whose tumors are echinoderm microtubule-associated proteinlike 4 (EML4)/ALK or ROS1 positive.2,3 Among unselected populations of patients with NSCLC, the frequency of EML4/ALK rearrangements ranges from 1.5% to 6.7%.1 Crizotinib is superior to standard chemotherapy in patients with ALK-positive NSCLC.2
In clinical trials, adverse reactions (grades 1 to 4) to crizotinib occurring in at least 25% of patients included visual disturbances, gastrointestinal tract disorders, fatigue, and pitting edema.1,2,4 Adverse reactions (grades 3 and 4) occurring in more than 5% of patients included elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, dyspnea, pneumonia, and neutropenia.1,4 Although the incidence of dermatologic adverse reactions is approximately 11%, substantial progression of drug eruptions rarely has been reported.2,5 We describe a case of lichenoid drug eruption (LDE) that appeared 4 weeks after initiation of crizotinib treatment in a patient with ALK-positive metastatic lung adenocarcinoma.
Case Report
A 61-year-old man presented with a history of ALK-positive NSCLC with lung-to-lung metastasis and pleural seeding treated with a right lower lobectomy and chemotherapy 9 years prior. Chemotherapy was reattempted 5 years later. Targeted therapy with gefitinib was initiated following the lobectomy and 5 years later with erlotinib. The NSCLC was stable, as indicated by computed tomography performed once every 3 or 6 months. After 5 years of treatment, follow-up computed tomography showed slowly growing nodular shadows in the right middle and lower lung fields. Due to this disease progression, treatment with crizotinib (250 mg twice daily) was initiated. Four weeks after the initiation of crizotinib therapy, mild itchy skin eruptions developed on all extremities and the lower lip. He also reported that the skin lesions became more itchy and red with sun exposure. He had no history of drug allergies and denied taking any other medications.
Physical examination revealed multiple brown to violaceous, slightly scaly, flat-topped polygonal papules or plaques on both lower legs (Figure 1A), dorsal hands (Figure 1B), and extensor sites of the elbows, as well as lacelike fine white lines on the lower lip (Figure 1C). There were no nail lesions. The patient’s dermatologic history was unremarkable, except for a few vitiligo lesions on the dorsal hands, extensor sites of the elbows, and mouth angles diagnosed 20 years earlier.
A skin biopsy from the right dorsal hand revealed a lichenoid infiltrate in the superficial dermis composed of lymphocytes, histiocytes and scattered eosinophils, focal parakeratosis, focal hypergranulosis, mild acanthosis, and basal vacuolization (Figure 2A). In addition, some dyskeratotic keratinocytes in the stratum spinosum and granulosum were identified (Figure 2B). The histopathology was consistent with the diagnosis of an LDE. Direct immunofluorescence revealed no globular or cytoid body–like deposits of immunoglobulin, with IgM, IgA, IgG, or C3 in the epidermis, dermis, and basement membrane zone. Routine laboratory studies revealed elevated liver enzymes, including an ALT level of 115 U/L (reference range, 0–40 U/L) and AST level of 60 U/L (reference range, 5–45 U/L). Negative results for the serum hepatitis B surface antigen and anti– hepatitis C virus tests were recorded. The patient had no medical history of alcohol consumption or abnormal liver function tests. The skin lesions were treated with diflucortolone valerate fatty ointment 0.1% twice daily and abnormal liver functions were treated with silymarin (150 mg per cap twice daily). He experienced some improvement.
A causality assessment was performed using the Naranjo Adverse Drug Reaction Probability Scale,6,7 and we concluded that crizotinib was the possible cause (Naranjo score, 4) of this adverse drug reaction (Table). Because the skin reaction was tolerable and liver enzymes were mildly elevated (ALT, 50 U/L; AST, 48 U/L), the offending drug was continued to benefit the underlying disease. His NSCLC was stable on computed tomography 3 months later.
Comment
The number of indicated uses of crizotinib, an oral small-molecule ALK tyrosine kinase inhibitor for the treatment of NSCLC, has gradually increased, but only a few cases of cutaneous adverse reactions, such as erythema multiforme and severe photosensitivity dermatitis, have been reported.2,5 Skin toxicity is a common and well-known side effect of other small-molecule tyrosine kinase inhibitors, particularly epidermal growth factor receptor inhibitors.8 However, LDE is not commonly associated with small-molecule tyrosine kinase inhibitors, though it has been described in a few patients taking imatinib for chronic myelogenous leukemia and gastrointestinal tract stromal tumors.9,10
The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.10 Histologically, some findings are more common in LDE, including focal parakeratosis, cytoid bodies in the cornified and granular layers, and the presence of eosinophils.11
Our patient developed lichenoid rashes after 1 month of crizotinib therapy. The latency period for developing a medication-induced LDE varies from months to 1 year and is dependent on the dosage, host response, prior exposure, and concomitant drug administration. No additional medications had been added to our patient’s regimen after initiating crizotinib therapy, and he did not take any other known medications. Ultimately, based on the time-event relationship, morphology, distribution, and histopathologic findings, we concluded that our patient developed an LDE due to crizotinib.
Our patient also had a history of vitiligo affecting the hands, elbows, and mouth angles for 20 years. Although there are limited reports of a possible causal link between lichen planus or drug-induced lichen planus eruption and vitiligo,12-14 we do not think these conditions were associated in our case because the patient’s vitiligo lesions persisted for many years, did not progress, and remained inactive and stable, and there was a lack of co-localization of LDE and vitiligo.
Our patient reported that the skin eruptions worsened after sun exposure. Oser and Janne5 also reported a patient with ALK-positive metastatic lung adenocarcinoma who developed severe crizotinib-induced photosensitive rashes. Further accumulation of similar cases and pathophysiological studies will be necessary to clarify whether this photosensitivity dermatitis is caused by ALK inhibition itself or mediated through host-immune mechanisms.5
Conclusion
As crizotinib prescriptions for patients with NSCLC are increasing, clinicians should be aware of the possibility of cutaneous LDEs occurring as an adverse effect. Additionally, physicians should treat appropriately to avoid unnecessarily discontinuing a potentially life-saving medication and to improve quality of life for patients with NSCLC who are treated with crizotinib.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
- Malik SM, Maher VE, Bijwaard KE, et al. U.S. Food and Drug Administration approval: crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-2034.
- Sawamura S, Kajihara I, Ichihara A, et al. Crizotinib-associated erythema multiforme in a lung cancer patient. Drug Discov Ther. 2015;9:142-143.
- Liao BC, Lin CC, Shih JY, et al. Treating patients with ALK-positive non-small cell lung cancer: latest evidence and management strategy. Ther Adv Med Oncol. 2015;7:274-290.
- Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011-1019.
- Oser MG, Janne PA. A severe photosensitivity dermatitis caused by crizotinib. J Thorac Oncol. 2014;9:E51-E53.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Zaki SA. Adverse drug reaction and causality assessment scales. Lung India. 2011;28:152-153.
- Aw DC, Tan EH, Chin TM, et al. Management of epidermal growth factor receptor tyrosine kinase inhibitor-related cutaneous and gastrointestinal toxicities. Asia Pac J Clin Oncol. 2018;14:23-31.
- Penn EH, Chung HJ, Keller M. Imatinib mesylate-induced lichenoid drug eruption. Cutis. 2017;99:189-192.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Lage D, Juliano PB, Metze K, et al. Lichen planus and lichenoid drug-induced eruption: a histological and immunohistochemical study. Int J Dermatol. 2012;51:1199-1205.
- Veitch D, Kravvas G, Hughes S, et al. A rare colocalization of lichen planus and vitiligo. Case Rep Dermatol Med. 2015;2015:840193.
- Baghestani S, Moosavi A, Eftekhari T. Familial colocalization of lichen planus and vitiligo on sun exposed areas. Ann Dermatol. 2013;25:223-225.
- Chan WP, Mackey VT, Sun DK. Telmisartan-induced lichen planus eruption manifested on vitiliginous skin. Cutis. 2017;99:E16-E19.
Practice Points
- Cutaneous lichenoid drug eruptions (LDEs) and photosensitive rash may be caused by crizotinib.
- The clinical morphology of LDE may resemble lichen planus, but certain features, such as larger skin lesions, the absence of Wickham striae, and photodistribution, help to differentiate between the two.
Coalescing Hyperkeratotic Plaques and Papules
The Diagnosis: X-Linked Ichthyosis
Immunohistochemical staining of a punch biopsy specimen from the left foot with cytokeratin markers AE1/3, 5/6, and 19 showed normal positive uptake. Further workup was recommended, and the patient was referred to genetics for an ichthyosis gene panel. DNA sequencing revealed a c.1121G>A transition in exon 10 of the steroid sulfatase gene, STS, consistent with X-linked ichthyosis (XLI).
X-linked ichthyosis, also known as steroid sulfatase deficiency and X-linked recessive ichthyosis, is a congenital skin disorder classified in 1965 by Wells and Kerr.1 Ichthyoses are a heterogenous group of acquired and congenital disorders of keratinization that manifest with xerosis, hyperkeratosis, and scaling.2 Of more than 20 ichthyoses, XLI is the second most common ichthyosis, with a prevalence of 1 in 6000 males.3 X-linked ichthyosis occurs almost exclusively in males, and although females can be carriers, they rarely exhibit skin manifestations.4
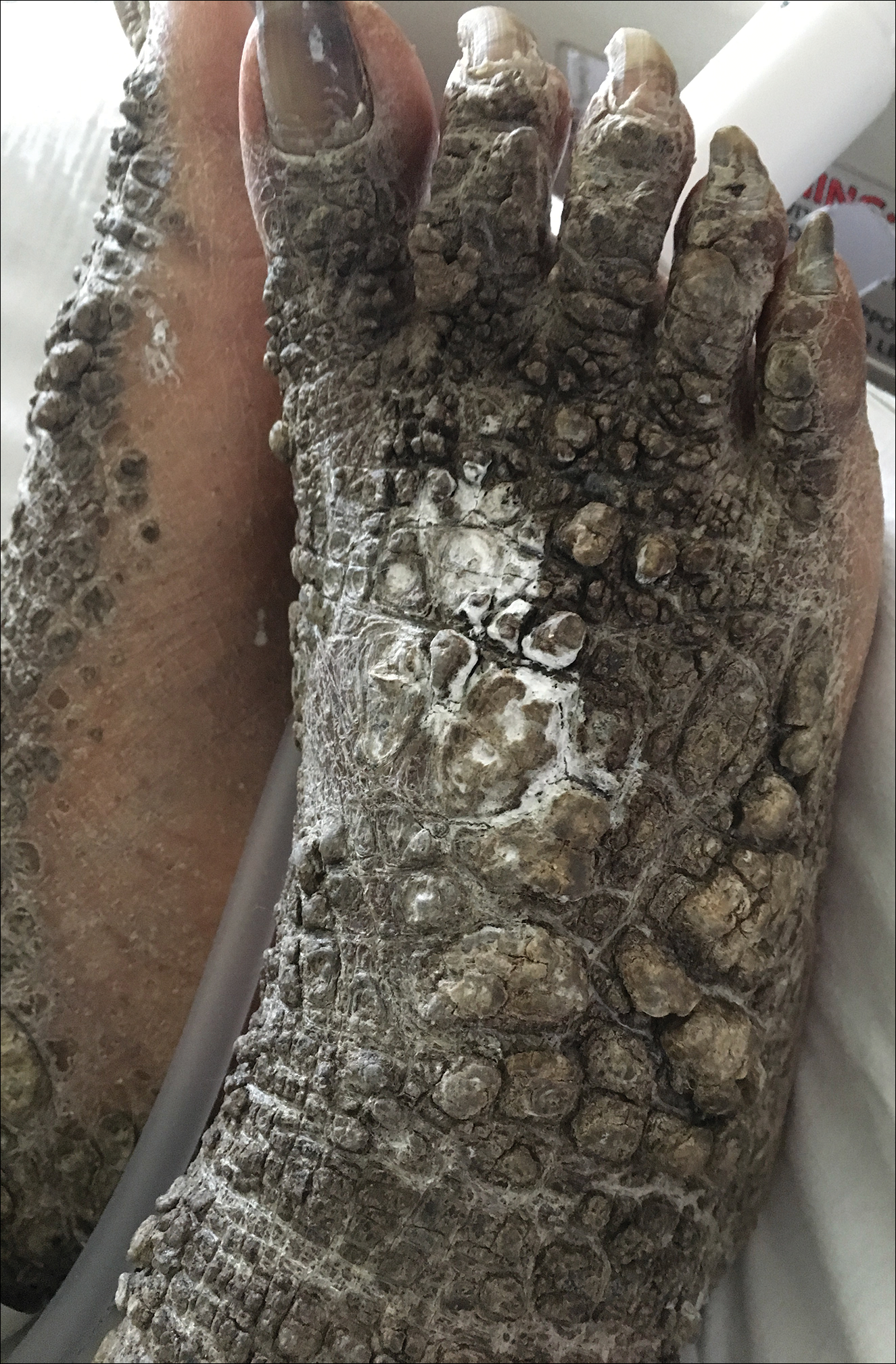
X-linked ichthyosis is caused by either a partial or full deletion or mutation in the STS gene on the X chromosome.2 The absence of STS activity results in the accumulation of cholesterol sulfate in the stratum corneum, leading to corneocyte cohesion, hyperkeratosis, and impaired skin permeability. The most common clinical phenotype is characterized by polygonal scales concentrated on the upper and lower extremities as well as the trunk (Figure), consistent with our patient's clinical presentation.5
anterior knees (A) as well as large exophytic papules on the upper
chest and neck (B).
X-linked ichthyosis typically presents in the first 6 months of life as generalized desquamation and xerosis that progresses to fine scaling on the trunk and extremities, more commonly and heavily involving the legs; however, the extensor surfaces of the arms also may be affected.6 After the neonatal period, fine scaling persists on the trunk and extremities, but scales often become coarser and darker over time. Although scaling is generalized, it typically spares the antecubital and popliteal fossae, palms, soles, and midface. The lateral face, axillae, and neck always remain involved.4 The most common extracutaneous manifestations of XLI affect the ocular, genitourinary, and cognitive/behavioral systems. Patients can develop corneal comma-shaped opacities, hypogonadism, cryptorchidism, and an increased risk for testicular cancer. Female carriers may have prolonged delivery of affected neonates.2,5,7-9 Given the unrelated debilitating neurologic consequences of our patient's presenting subarachnoid hemorrhage, further workup was not pursued into these associations.
Although XLI is most commonly diagnosed in early childhood, it also must be considered in adult patients presenting with severe scaling of the trunk, arms, and legs who have not had prior dermatologic workup. Given the similarity of XLI presentation to other ichthyoses, particularly ichthyosis vulgaris, lamellar ichthyosis, and ichthyosis bullosa of Siemens, genetic analysis is the most accurate diagnostic tool and should be considered in patients with an atypical presentation. Rupioid psoriasis also may be considered and can be confirmed on biopsy. Diagnosis of XLI should prompt symptomatic treatment, genetic counseling, and workup for extracutaneous manifestations.
- Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol. 1965;92:1-6.
- Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62:480-485.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141:617-627.
- Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis [published online November 27, 2013]. Biochim Biophys Acta. 2014;1841:353-361.
- Wu B, Paller AS. Ichthyosis, X-Linked. Treasure Island, FL: StatPearls Publishing LLC; 2019.
- Marukian NV, Choate KA. Recent advances in understanding ichthyosis pathogenesis. F1000Res. 2016;5. doi:10.12688/f1000research.8584.1.
- Baek WS, Aypar U. Case report neurological manifestations of X-linked ichthyosis: case report and review of the literature [published online August 13, 2017]. 2017;2017:9086408.
- Brookes KJ, Hawi Z, Park J, et al. Polymorphisms of the steroid sulfatase (STS) gene are associated with attention deficit hyperactivity disorder and influence brain tissue mRNA expression. Am J Med Genet Part B Neuropsychiatr Genet. 2010;153:1417-1424.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519-524.
The Diagnosis: X-Linked Ichthyosis
Immunohistochemical staining of a punch biopsy specimen from the left foot with cytokeratin markers AE1/3, 5/6, and 19 showed normal positive uptake. Further workup was recommended, and the patient was referred to genetics for an ichthyosis gene panel. DNA sequencing revealed a c.1121G>A transition in exon 10 of the steroid sulfatase gene, STS, consistent with X-linked ichthyosis (XLI).
X-linked ichthyosis, also known as steroid sulfatase deficiency and X-linked recessive ichthyosis, is a congenital skin disorder classified in 1965 by Wells and Kerr.1 Ichthyoses are a heterogenous group of acquired and congenital disorders of keratinization that manifest with xerosis, hyperkeratosis, and scaling.2 Of more than 20 ichthyoses, XLI is the second most common ichthyosis, with a prevalence of 1 in 6000 males.3 X-linked ichthyosis occurs almost exclusively in males, and although females can be carriers, they rarely exhibit skin manifestations.4
X-linked ichthyosis is caused by either a partial or full deletion or mutation in the STS gene on the X chromosome.2 The absence of STS activity results in the accumulation of cholesterol sulfate in the stratum corneum, leading to corneocyte cohesion, hyperkeratosis, and impaired skin permeability. The most common clinical phenotype is characterized by polygonal scales concentrated on the upper and lower extremities as well as the trunk (Figure), consistent with our patient's clinical presentation.5
anterior knees (A) as well as large exophytic papules on the upper
chest and neck (B).
X-linked ichthyosis typically presents in the first 6 months of life as generalized desquamation and xerosis that progresses to fine scaling on the trunk and extremities, more commonly and heavily involving the legs; however, the extensor surfaces of the arms also may be affected.6 After the neonatal period, fine scaling persists on the trunk and extremities, but scales often become coarser and darker over time. Although scaling is generalized, it typically spares the antecubital and popliteal fossae, palms, soles, and midface. The lateral face, axillae, and neck always remain involved.4 The most common extracutaneous manifestations of XLI affect the ocular, genitourinary, and cognitive/behavioral systems. Patients can develop corneal comma-shaped opacities, hypogonadism, cryptorchidism, and an increased risk for testicular cancer. Female carriers may have prolonged delivery of affected neonates.2,5,7-9 Given the unrelated debilitating neurologic consequences of our patient's presenting subarachnoid hemorrhage, further workup was not pursued into these associations.
Although XLI is most commonly diagnosed in early childhood, it also must be considered in adult patients presenting with severe scaling of the trunk, arms, and legs who have not had prior dermatologic workup. Given the similarity of XLI presentation to other ichthyoses, particularly ichthyosis vulgaris, lamellar ichthyosis, and ichthyosis bullosa of Siemens, genetic analysis is the most accurate diagnostic tool and should be considered in patients with an atypical presentation. Rupioid psoriasis also may be considered and can be confirmed on biopsy. Diagnosis of XLI should prompt symptomatic treatment, genetic counseling, and workup for extracutaneous manifestations.
The Diagnosis: X-Linked Ichthyosis
Immunohistochemical staining of a punch biopsy specimen from the left foot with cytokeratin markers AE1/3, 5/6, and 19 showed normal positive uptake. Further workup was recommended, and the patient was referred to genetics for an ichthyosis gene panel. DNA sequencing revealed a c.1121G>A transition in exon 10 of the steroid sulfatase gene, STS, consistent with X-linked ichthyosis (XLI).
X-linked ichthyosis, also known as steroid sulfatase deficiency and X-linked recessive ichthyosis, is a congenital skin disorder classified in 1965 by Wells and Kerr.1 Ichthyoses are a heterogenous group of acquired and congenital disorders of keratinization that manifest with xerosis, hyperkeratosis, and scaling.2 Of more than 20 ichthyoses, XLI is the second most common ichthyosis, with a prevalence of 1 in 6000 males.3 X-linked ichthyosis occurs almost exclusively in males, and although females can be carriers, they rarely exhibit skin manifestations.4
X-linked ichthyosis is caused by either a partial or full deletion or mutation in the STS gene on the X chromosome.2 The absence of STS activity results in the accumulation of cholesterol sulfate in the stratum corneum, leading to corneocyte cohesion, hyperkeratosis, and impaired skin permeability. The most common clinical phenotype is characterized by polygonal scales concentrated on the upper and lower extremities as well as the trunk (Figure), consistent with our patient's clinical presentation.5
anterior knees (A) as well as large exophytic papules on the upper
chest and neck (B).
X-linked ichthyosis typically presents in the first 6 months of life as generalized desquamation and xerosis that progresses to fine scaling on the trunk and extremities, more commonly and heavily involving the legs; however, the extensor surfaces of the arms also may be affected.6 After the neonatal period, fine scaling persists on the trunk and extremities, but scales often become coarser and darker over time. Although scaling is generalized, it typically spares the antecubital and popliteal fossae, palms, soles, and midface. The lateral face, axillae, and neck always remain involved.4 The most common extracutaneous manifestations of XLI affect the ocular, genitourinary, and cognitive/behavioral systems. Patients can develop corneal comma-shaped opacities, hypogonadism, cryptorchidism, and an increased risk for testicular cancer. Female carriers may have prolonged delivery of affected neonates.2,5,7-9 Given the unrelated debilitating neurologic consequences of our patient's presenting subarachnoid hemorrhage, further workup was not pursued into these associations.
Although XLI is most commonly diagnosed in early childhood, it also must be considered in adult patients presenting with severe scaling of the trunk, arms, and legs who have not had prior dermatologic workup. Given the similarity of XLI presentation to other ichthyoses, particularly ichthyosis vulgaris, lamellar ichthyosis, and ichthyosis bullosa of Siemens, genetic analysis is the most accurate diagnostic tool and should be considered in patients with an atypical presentation. Rupioid psoriasis also may be considered and can be confirmed on biopsy. Diagnosis of XLI should prompt symptomatic treatment, genetic counseling, and workup for extracutaneous manifestations.
- Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol. 1965;92:1-6.
- Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62:480-485.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141:617-627.
- Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis [published online November 27, 2013]. Biochim Biophys Acta. 2014;1841:353-361.
- Wu B, Paller AS. Ichthyosis, X-Linked. Treasure Island, FL: StatPearls Publishing LLC; 2019.
- Marukian NV, Choate KA. Recent advances in understanding ichthyosis pathogenesis. F1000Res. 2016;5. doi:10.12688/f1000research.8584.1.
- Baek WS, Aypar U. Case report neurological manifestations of X-linked ichthyosis: case report and review of the literature [published online August 13, 2017]. 2017;2017:9086408.
- Brookes KJ, Hawi Z, Park J, et al. Polymorphisms of the steroid sulfatase (STS) gene are associated with attention deficit hyperactivity disorder and influence brain tissue mRNA expression. Am J Med Genet Part B Neuropsychiatr Genet. 2010;153:1417-1424.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519-524.
- Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol. 1965;92:1-6.
- Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62:480-485.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141:617-627.
- Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis [published online November 27, 2013]. Biochim Biophys Acta. 2014;1841:353-361.
- Wu B, Paller AS. Ichthyosis, X-Linked. Treasure Island, FL: StatPearls Publishing LLC; 2019.
- Marukian NV, Choate KA. Recent advances in understanding ichthyosis pathogenesis. F1000Res. 2016;5. doi:10.12688/f1000research.8584.1.
- Baek WS, Aypar U. Case report neurological manifestations of X-linked ichthyosis: case report and review of the literature [published online August 13, 2017]. 2017;2017:9086408.
- Brookes KJ, Hawi Z, Park J, et al. Polymorphisms of the steroid sulfatase (STS) gene are associated with attention deficit hyperactivity disorder and influence brain tissue mRNA expression. Am J Med Genet Part B Neuropsychiatr Genet. 2010;153:1417-1424.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519-524.
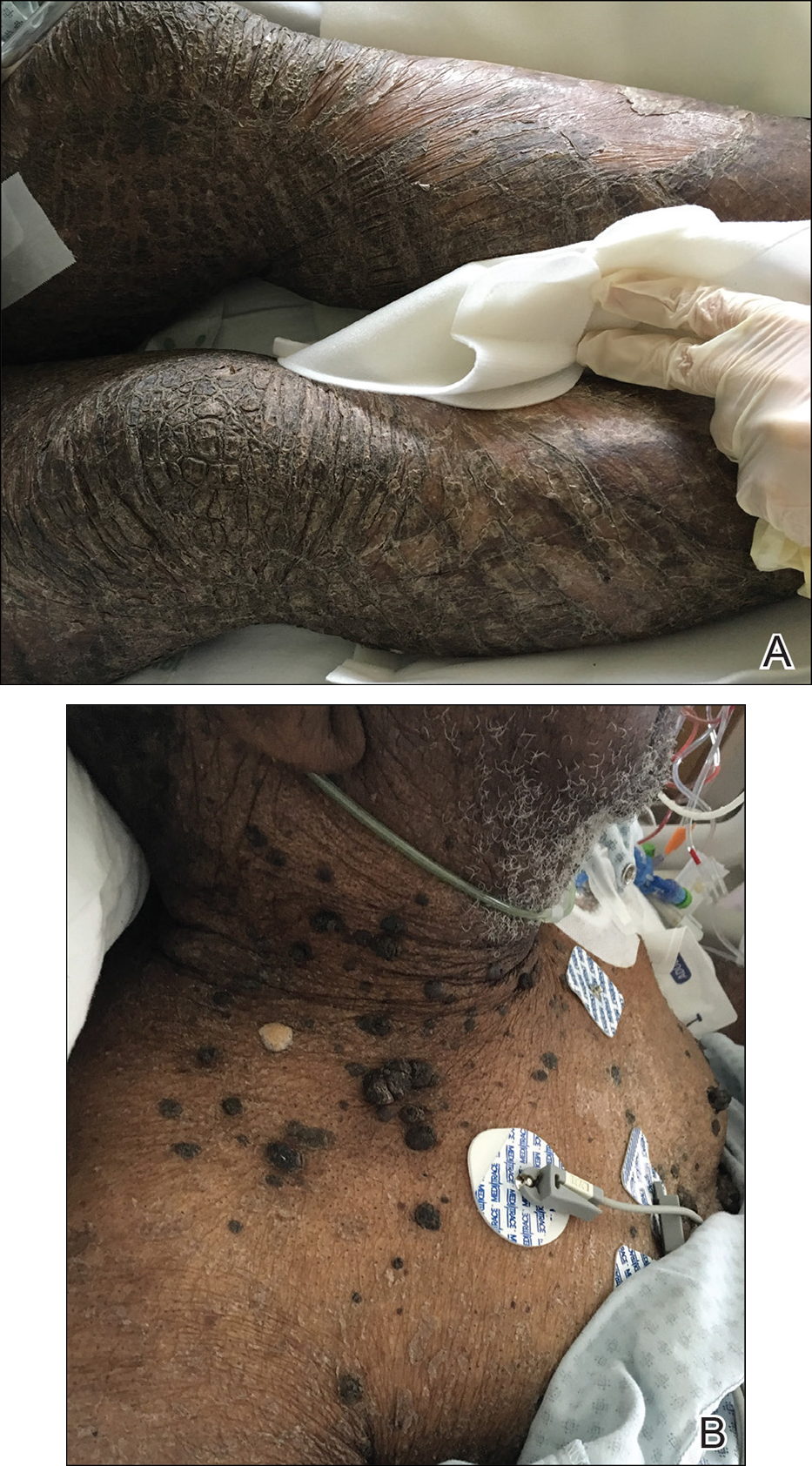
A 67-year-old man with a history of congestive heart failure, type 2 diabetes mellitus, hypertension, and schizophrenia was admitted to the hospital for subarachnoid hemorrhage and was noted to have heavy scaling on the bilateral legs. Given recent medical events, the patient was nonconversant at the time of consultation, but his daughter provided his medical history at bedside. The patient usually wore long-sleeved clothing and pants, thus no one had seen his skin in many years, and it was unclear how long the scaling had been present. His family history was notable for eczema in distant relatives but negative for comparable conditions. Physical examination revealed thick lichenified skin with many large, exophytic, brown papules (largest measured 1.5×1×1 cm) and platelike scaling on the anterior chest, abdomen, lateral arms, and forearms. Extensive coalescing hyperkeratotic plaques and papules (up to 1 cm in thickness) were present on the anterior legs and feet, and scattered verrucous brown papules were noted on the plantar aspects of the bilateral feet. A punch biopsy of the left foot revealed extensive, dense, compact, orthokeratotic hyperkeratosis with a preserved granular layer with no epidermolysis.
Update on Calciphylaxis Etiopathogenesis, Diagnosis, and Management
Calciphylaxis, also known as calcific uremic arteriolopathy, is a painful skin condition classically seen in patients with end-stage renal disease (ESRD), particularly those on chronic dialysis.1,2 It also has increasingly been reported in patients with normal renal function and calcium and phosphate homeostasis.3,4 Effective diagnosis and management of calciphylaxis remains challenging for physicians.2,5 The condition is characterized by tissue ischemia caused by calcification of cutaneous arteriolar vessels. As a result, calciphylaxis is associated with high mortality rates, ranging from 60% to 80%.5,6 Excruciating pain and nonhealing ulcers often lead to recurrent hospitalizations and infectious complications,7 and poor nutritional status, chronic pain, depression, and insomnia can further complicate recovery and lead to poor quality of life.8
We provide an update on calciphylaxis etiopathogenesis, diagnosis, and management. We also highlight some challenges faced in managing this potentially fatal condition.
Epidemiology
Calciphylaxis is considered a rare dermatosis with an estimated annual incidence of 1% to 4% in ESRD patients on dialysis. Recent data suggest that incidence of calciphylaxis is rising,5,7,9 which may stem from an increased use of calcium-based phosphate binders, an actual rise in disease incidence, and/or increased recognition of the disease.5 It is difficult to estimate the exact disease burden of calciphylaxis because the diagnostic criteria are not well defined, often leading to missed or delayed diagnosis.3,10 Furthermore, there is no centralized registry for calciphylaxis cases.3
Etiology and Pathogenesis
Calciphylaxis is thought to have a multifactorial etiology with the exact cause or trigger unknown.7 A long list of risk factors and triggers is associated with the condition (Table 1). Calciphylaxis primarily affects small arteries (40–600 μm in diameter) that become calcified due to an imbalance between inhibitors and promoters of calcification.2,11 Fetuin-A and matrix Gla protein inhibit vascular calcification and are downregulated in calciphylaxis.12,13 Dysfunctional calcium, phosphate, and parathyroid hormone regulatory pathways provide an increased substrate for the process of calcification, which causes endothelial damage and microthrombosis, resulting in tissue ischemia and infarction.14,15 Notably, there is growing interest in the role of vitamin K in the pathogenesis of calciphylaxis. Vitamin K inhibits vascular calcification, possibly by increasing the circulating levels of carboxylated matrix Gla protein.16
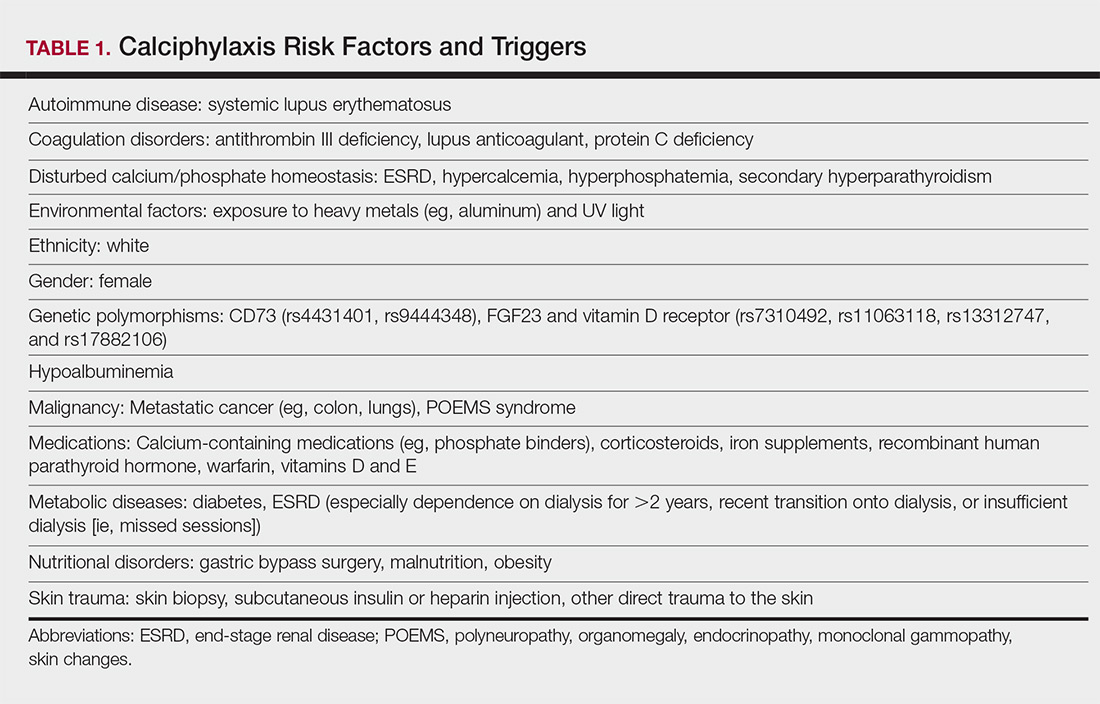
Clinical Features
Calciphylaxis is most commonly seen on the legs, abdomen, and buttocks.2 Patients with ESRD commonly develop proximal lesions affecting adipose-rich sites and have a poor prognosis. Distal lesions are more common in patients with nonuremic calciphylaxis, and mortality rates are lower in this population.2
Early lesions present as painful skin nodules or indurated plaques that often are rock-hard or firm to palpation with overlying mottling or a livedoid pattern (Figure, A). Early lesions progress from livedo reticularis to livedo racemosa and then to retiform purpura (Figure, B). Purpuric lesions later evolve into black eschars (Figure, C), then to necrotic, ulcerated, malodorous plaques or nodules in later stages of the disease (Figure, D). Lesions also may develop a gangrenous sclerotic appearance.2,5
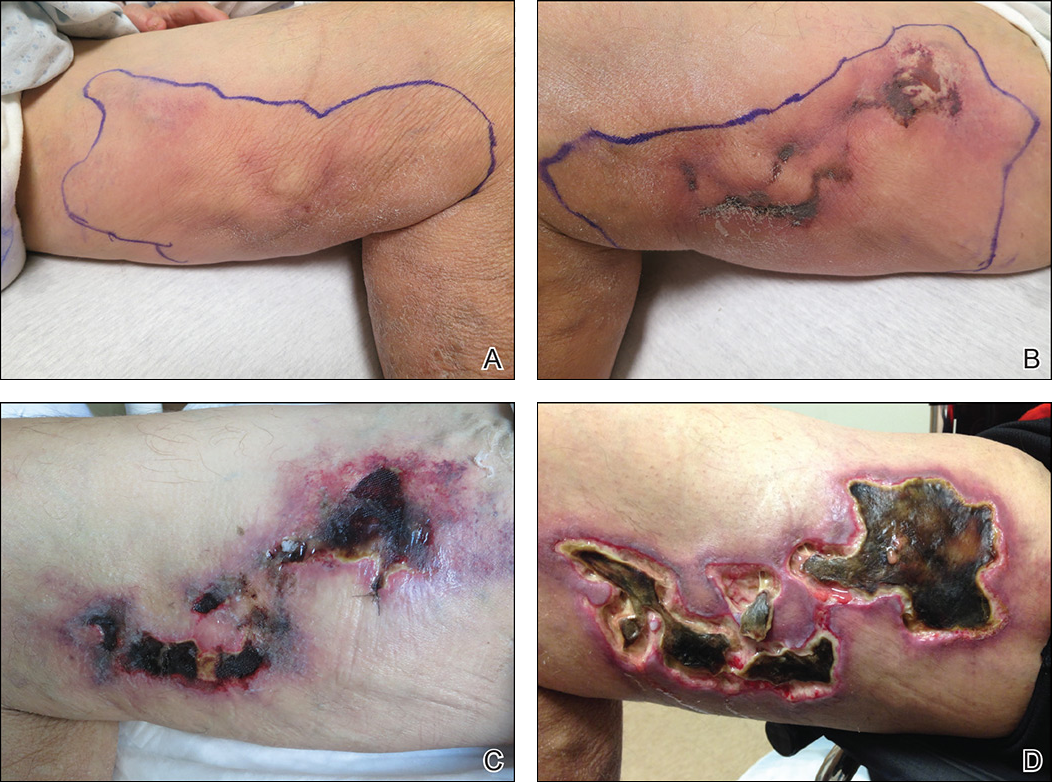
Although most patients with calciphylaxis have ESRD, nonuremic patients also can develop the disease. Those with calciphylaxis who do not have renal dysfunction frequently have other risk factors for the disease and often report another notable health problem in the weeks or months prior to presentation.4 More than half of patients with calciphylaxis become bedridden or require use of a wheelchair.17 Pain is characteristically severe throughout the course of the disease; it may even precede the appearance of the skin lesions.18 Because the pain is associated with ischemia, it tends to be relatively refractory to treatment with opioids. Rare extracutaneous vascular calcifications may lead to visual impairment, gastrointestinal tract bleeding, and myopathy.5,9,19,20
Diagnosis
Considering the high morbidity and mortality associated with calciphylaxis, it is important to provide accurate and timely diagnosis; however, there currently are no validated diagnostic criteria for calciphylaxis. Careful correlation of clinical and histologic findings is required. Calciphylaxis biopsies have demonstrated medial calcification and proliferation of the intima of small- to medium-sized arteries.21 Lobular and septal panniculitis and extravascular soft-tissue calcification, particularly stippled calcification of the eccrine sweat glands, also has been seen.2,22 Special calcium stains (eg, von Kossa, Alizarin red) increase the sensitivity of biopsy by highlighting subtle areas of intravascular and extravascular calcification.5,23 Sufficient sampling of subcutaneous tissue and specimen evaluation by an experienced dermatopathologist are necessary to ensure proper interpretation of the histologic findings.
Despite these measures, skin biopsies may be nondiagnostic or falsely negative; therefore, when there is high clinical suspicion, it may be appropriate to move forward with a presumptive diagnosis of calciphylaxis even if the histologic findings are nondiagnostic.1,9,24 It also is worth noting that localized progression and ulceration may occur following skin biopsy, such that biopsy may even be contraindicated in certain cases (eg, penile calciphylaxis).
Standard laboratory workup for calciphylaxis includes evaluation for associated risk factors as well as exclusion of other conditions in the differential diagnosis (Table 2). Blood tests to evaluate for risk factors include liver and renal function tests, a complete metabolic panel, parathyroid hormone level, and serum albumin level.5 Elevated calcium and phosphate levels may signal disturbed calcium and phosphate homeostasis but are neither sensitive nor specific for the diagnosis.25 Complete blood cell count, blood cultures, thorough hypercoagulability workup (including but not limited to antiphospholipid antibodies, proteins C and S, factor V Leiden, antithrombin III, homocysteine, methylenetetrahydrofolate reductase mutation, and cryoglobulins), rheumatoid factor, antineutrophil cytoplasmic antibodies, and antinuclear antibody testing may be relevant to help identify contributing factors or mimickers of calciphylaxis.5 Various imaging modalities also have been used to evaluate for the presence of soft-tissue calcification in areas of suspected calciphylaxis, including radiography, mammography, computed tomography, ultrasonography, nuclear bone scintigraphy, and spectroscopy.2,26,27 Unfortunately, there currently is no standardized reproducible imaging modality for reliable diagnosis of calciphylaxis. Ultimately, histologic and radiographic findings should always be interpreted in the context of relevant clinical findings.2,9

Prevention
Reduction of the net calcium phosphorus product may help reduce the risk of calciphylaxis in ESRD patients, which can be accomplished by using non–calcium-phosphate binders, adequate dialysis, and restricting use of vitamin D and vitamin K antagonists.2,5 There are limited data regarding the benefits of using bisphosphonates and cinacalcet in ESRD patients on dialysis to prevent calciphylaxis.28,29
Management
Management of calciphylaxis is multifactorial. Besides dermatology and nephrology, specialists in pain management, wound care, plastic surgery, and nutrition are critical partners in management.1,5,9,30 Nephrologists can help optimize calcium and phosphate balance and ensure adequate dialysis. Pain specialists can aid in creating aggressive multiagent pain regimens that target the neuropathic/ischemic and physical aspects of calciphylaxis pain. When appropriate, nutrition specialists can help establish high-protein, low-phosphorus diets, and wound specialists can provide access to advanced wound dressings and adjunctive hyperbaric oxygen therapy. Plastic surgeons can provide conservative debridement procedures in a subset of patients, usually those with distal stable disease.
The limited understanding of the etiopathogenesis of calciphylaxis and the lack of data on its management are reflected in the limited treatment options for the disease (Table 3).2,5,9 There are no formal algorithms for the treatment of calciphylaxis. Therapeutic trials are scarce, and most of the current treatment recommendations are based on small retrospective reports or case series. Sodium thiosulfate has been the most widely used treatment option since 2004, when its use in calciphylaxis was first reported.31 Sodium thiosulfate chelates calcium and is thought to have antioxidant and vasodilatory properties.32 There are a few promising clinical trials and large-scale studies (Table 4) that aim to evaluate the efficacy of existing treatments (eg, sodium thiosulfate) as well as novel treatment options such as lanthanum carbonate, SNF472 (hexasodium phytate), and vitamin K.33-36

Prognosis
Calciphylaxis is a potentially fatal condition with a poor prognosis and a median survival rate of approximately 1 year following the appearance of skin lesions.37-39 Patients with proximal lesions and those on peritoneal dialysis (as opposed to hemodialysis) have a worse prognosis.40 Mortality rates are estimated to be 30% at 6 months, 50% at 12 months, and 80% at 2 years, with sepsis secondary to infection of cutaneous ulcers being the leading cause of death.37-39 The impact of calciphylaxis on patient quality of life and activities of daily living is severe.8,17
Future Directions
Multi-institution cohort studies and collaborative registries are needed to provide updated information related to the epidemiology, diagnosis, treatment, morbidity, and mortality associated with calciphylaxis and to help formulate evidence-based diagnostic criteria. Radiographic and histologic studies, as well as other tools for early and accurate diagnosis of calciphylaxis, should be studied for feasibility, accuracy, and reproducibility. The incidence of nonuremic calciphylaxis points toward pathogenic pathways besides those based on the bone-mineral axis. Basic science research directed at improving understanding of the pathophysiology of calciphylaxis would be helpful in devising new treatment strategies targeting these pathways. Establishment of a collaborative, multi-institutional calciphylaxis working group would enable experts to formulate therapeutic guidelines based on current evidence. Such a group could facilitate initiation of large prospective studies to establish the efficacy of existing and new treatment modalities for calciphylaxis. A working group within the Society for Dermatology Hospitalists has been tasked with addressing these issues and is currently establishing a multicenter calciphylaxis database.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Nigwekar SU, Thadhani RI, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Davis JM. The relationship between obesity and calciphylaxis: a review of the literature. Ostomy Wound Manage. 2016;62:12-18.
- Bajaj R, Courbebaisse M, Kroshinsky D, et al. Calciphylaxis in patients with normal renal function: a case series and systematic review. Mayo Clin Proc. 2018;93:1202-1212.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Westphal SG, Plumb T. Calciphylaxis. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2018. https://www.ncbi.nlm.nih.gov/books/NBK519020. Accessed November 12, 2018.
- Riemer CA, El-Azhary RA, Wu KL, et al. Underreported use of palliative care and patient-reported outcome measures to address reduced quality of life in patients with calciphylaxis: a systematic review. Br J Dermatol. 2017;177:1510-1518.
- Nigwekar SU. Calciphylaxis. Curr Opin Nephrol Hypertens. 2017;26:276-281.
- Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Perit Dial Int. 2008;28:268-270.
- Lin WT, Chao CM. Tumoral calcinosis in renal failure. QJM. 2014;107:387.
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Bleyer AJ, Choi M, Igwemezie B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376-383.
- Ahmed S, O’Neill KD, Hood AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:267-276.
- Nigwekar SU, Bloch DB, Nazarian RM, et al. Vitamin K-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717-1722.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Polizzotto MN, Bryan T, Ashby MA, et al. Symptomatic management of calciphylaxis: a case series and review of the literature. J Pain Symptom Manage. 2006;32:186-190.
- Gupta N, Haq KF, Mahajan S, et al. Gastrointestinal bleeding secondary to calciphylaxis. Am J Case Rep. 2015;16:818-822.
- Edelstein CL, Wickham MK, Kirby PA. Systemic calciphylaxis presenting as a painful, proximal myopathy. Postgrad Med J. 1992;68:209-211.
- Mochel MC, Arakari RY, Wang G, et al. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol. 2013;35:582-586.
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802.
- Cassius C, Moguelet P, Monfort JB, et al. Calciphylaxis in haemodialysed patients: diagnostic value of calcifications in cutaneous biopsy. Br J Dermatol. 2018;178:292-293.
- Sreedhar A, Sheikh HA, Scagliotti CJ, et al. Advanced-stage calciphylaxis: think before you punch. Cleve Clin J Med. 2016;83:562-564.
- Brandenburg VM, Kramann R, Rothe H, et al. Calcific uraemic arteriolopathy (calciphylaxis): data from a large nation-wide registry. Nephrol Dial Transplant. 2017;32:126-132.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017;153:101-103.
- Shmidt E, Murthy NS, Knudsen JM, et al. Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol. 2012;67:1296-1301.
- EVOLVE Trial Investigators; Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482-2494.
- Rogers NM, Teubner DJO, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150-157.
- Nigwekar SU. Multidisciplinary approach to calcific uremic arteriolopathy. Curr Opin Nephrol Hypertens. 2015;24:531-537.
- Cicone JS, Petronis JB, Embert CD, et al. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104-1108.
- Chen NX, O’Neill K, Akl NK, et al. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151-156.
- Chan MR, Ghandour F, Murali NS, et al. Pilot study of the effect of lanthanum carbonate in patients with calciphylaxis: a Wisconsin Network for Health Research (WiNHR) study. J Nephrol Ther. 2014;4:1000162.
- Perelló J, Gómez M, Ferrer MD, et al. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J Nephrol. 2018;31:287-296.
- Christiadi D, Singer RF. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin Kidney J. 2018;11:528-529.
- Caluwe R, Vandecasteele S, Van Vlem B, et al. Vitamin K2 supplementation in haemodialysis patients: a randomized dose-finding study. Nephrol Dial Transplant. 2014;29:1385-1390.
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429.
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241.
Calciphylaxis, also known as calcific uremic arteriolopathy, is a painful skin condition classically seen in patients with end-stage renal disease (ESRD), particularly those on chronic dialysis.1,2 It also has increasingly been reported in patients with normal renal function and calcium and phosphate homeostasis.3,4 Effective diagnosis and management of calciphylaxis remains challenging for physicians.2,5 The condition is characterized by tissue ischemia caused by calcification of cutaneous arteriolar vessels. As a result, calciphylaxis is associated with high mortality rates, ranging from 60% to 80%.5,6 Excruciating pain and nonhealing ulcers often lead to recurrent hospitalizations and infectious complications,7 and poor nutritional status, chronic pain, depression, and insomnia can further complicate recovery and lead to poor quality of life.8
We provide an update on calciphylaxis etiopathogenesis, diagnosis, and management. We also highlight some challenges faced in managing this potentially fatal condition.
Epidemiology
Calciphylaxis is considered a rare dermatosis with an estimated annual incidence of 1% to 4% in ESRD patients on dialysis. Recent data suggest that incidence of calciphylaxis is rising,5,7,9 which may stem from an increased use of calcium-based phosphate binders, an actual rise in disease incidence, and/or increased recognition of the disease.5 It is difficult to estimate the exact disease burden of calciphylaxis because the diagnostic criteria are not well defined, often leading to missed or delayed diagnosis.3,10 Furthermore, there is no centralized registry for calciphylaxis cases.3
Etiology and Pathogenesis
Calciphylaxis is thought to have a multifactorial etiology with the exact cause or trigger unknown.7 A long list of risk factors and triggers is associated with the condition (Table 1). Calciphylaxis primarily affects small arteries (40–600 μm in diameter) that become calcified due to an imbalance between inhibitors and promoters of calcification.2,11 Fetuin-A and matrix Gla protein inhibit vascular calcification and are downregulated in calciphylaxis.12,13 Dysfunctional calcium, phosphate, and parathyroid hormone regulatory pathways provide an increased substrate for the process of calcification, which causes endothelial damage and microthrombosis, resulting in tissue ischemia and infarction.14,15 Notably, there is growing interest in the role of vitamin K in the pathogenesis of calciphylaxis. Vitamin K inhibits vascular calcification, possibly by increasing the circulating levels of carboxylated matrix Gla protein.16
Clinical Features
Calciphylaxis is most commonly seen on the legs, abdomen, and buttocks.2 Patients with ESRD commonly develop proximal lesions affecting adipose-rich sites and have a poor prognosis. Distal lesions are more common in patients with nonuremic calciphylaxis, and mortality rates are lower in this population.2
Early lesions present as painful skin nodules or indurated plaques that often are rock-hard or firm to palpation with overlying mottling or a livedoid pattern (Figure, A). Early lesions progress from livedo reticularis to livedo racemosa and then to retiform purpura (Figure, B). Purpuric lesions later evolve into black eschars (Figure, C), then to necrotic, ulcerated, malodorous plaques or nodules in later stages of the disease (Figure, D). Lesions also may develop a gangrenous sclerotic appearance.2,5
Although most patients with calciphylaxis have ESRD, nonuremic patients also can develop the disease. Those with calciphylaxis who do not have renal dysfunction frequently have other risk factors for the disease and often report another notable health problem in the weeks or months prior to presentation.4 More than half of patients with calciphylaxis become bedridden or require use of a wheelchair.17 Pain is characteristically severe throughout the course of the disease; it may even precede the appearance of the skin lesions.18 Because the pain is associated with ischemia, it tends to be relatively refractory to treatment with opioids. Rare extracutaneous vascular calcifications may lead to visual impairment, gastrointestinal tract bleeding, and myopathy.5,9,19,20
Diagnosis
Considering the high morbidity and mortality associated with calciphylaxis, it is important to provide accurate and timely diagnosis; however, there currently are no validated diagnostic criteria for calciphylaxis. Careful correlation of clinical and histologic findings is required. Calciphylaxis biopsies have demonstrated medial calcification and proliferation of the intima of small- to medium-sized arteries.21 Lobular and septal panniculitis and extravascular soft-tissue calcification, particularly stippled calcification of the eccrine sweat glands, also has been seen.2,22 Special calcium stains (eg, von Kossa, Alizarin red) increase the sensitivity of biopsy by highlighting subtle areas of intravascular and extravascular calcification.5,23 Sufficient sampling of subcutaneous tissue and specimen evaluation by an experienced dermatopathologist are necessary to ensure proper interpretation of the histologic findings.
Despite these measures, skin biopsies may be nondiagnostic or falsely negative; therefore, when there is high clinical suspicion, it may be appropriate to move forward with a presumptive diagnosis of calciphylaxis even if the histologic findings are nondiagnostic.1,9,24 It also is worth noting that localized progression and ulceration may occur following skin biopsy, such that biopsy may even be contraindicated in certain cases (eg, penile calciphylaxis).
Standard laboratory workup for calciphylaxis includes evaluation for associated risk factors as well as exclusion of other conditions in the differential diagnosis (Table 2). Blood tests to evaluate for risk factors include liver and renal function tests, a complete metabolic panel, parathyroid hormone level, and serum albumin level.5 Elevated calcium and phosphate levels may signal disturbed calcium and phosphate homeostasis but are neither sensitive nor specific for the diagnosis.25 Complete blood cell count, blood cultures, thorough hypercoagulability workup (including but not limited to antiphospholipid antibodies, proteins C and S, factor V Leiden, antithrombin III, homocysteine, methylenetetrahydrofolate reductase mutation, and cryoglobulins), rheumatoid factor, antineutrophil cytoplasmic antibodies, and antinuclear antibody testing may be relevant to help identify contributing factors or mimickers of calciphylaxis.5 Various imaging modalities also have been used to evaluate for the presence of soft-tissue calcification in areas of suspected calciphylaxis, including radiography, mammography, computed tomography, ultrasonography, nuclear bone scintigraphy, and spectroscopy.2,26,27 Unfortunately, there currently is no standardized reproducible imaging modality for reliable diagnosis of calciphylaxis. Ultimately, histologic and radiographic findings should always be interpreted in the context of relevant clinical findings.2,9
Prevention
Reduction of the net calcium phosphorus product may help reduce the risk of calciphylaxis in ESRD patients, which can be accomplished by using non–calcium-phosphate binders, adequate dialysis, and restricting use of vitamin D and vitamin K antagonists.2,5 There are limited data regarding the benefits of using bisphosphonates and cinacalcet in ESRD patients on dialysis to prevent calciphylaxis.28,29
Management
Management of calciphylaxis is multifactorial. Besides dermatology and nephrology, specialists in pain management, wound care, plastic surgery, and nutrition are critical partners in management.1,5,9,30 Nephrologists can help optimize calcium and phosphate balance and ensure adequate dialysis. Pain specialists can aid in creating aggressive multiagent pain regimens that target the neuropathic/ischemic and physical aspects of calciphylaxis pain. When appropriate, nutrition specialists can help establish high-protein, low-phosphorus diets, and wound specialists can provide access to advanced wound dressings and adjunctive hyperbaric oxygen therapy. Plastic surgeons can provide conservative debridement procedures in a subset of patients, usually those with distal stable disease.
The limited understanding of the etiopathogenesis of calciphylaxis and the lack of data on its management are reflected in the limited treatment options for the disease (Table 3).2,5,9 There are no formal algorithms for the treatment of calciphylaxis. Therapeutic trials are scarce, and most of the current treatment recommendations are based on small retrospective reports or case series. Sodium thiosulfate has been the most widely used treatment option since 2004, when its use in calciphylaxis was first reported.31 Sodium thiosulfate chelates calcium and is thought to have antioxidant and vasodilatory properties.32 There are a few promising clinical trials and large-scale studies (Table 4) that aim to evaluate the efficacy of existing treatments (eg, sodium thiosulfate) as well as novel treatment options such as lanthanum carbonate, SNF472 (hexasodium phytate), and vitamin K.33-36
Prognosis
Calciphylaxis is a potentially fatal condition with a poor prognosis and a median survival rate of approximately 1 year following the appearance of skin lesions.37-39 Patients with proximal lesions and those on peritoneal dialysis (as opposed to hemodialysis) have a worse prognosis.40 Mortality rates are estimated to be 30% at 6 months, 50% at 12 months, and 80% at 2 years, with sepsis secondary to infection of cutaneous ulcers being the leading cause of death.37-39 The impact of calciphylaxis on patient quality of life and activities of daily living is severe.8,17
Future Directions
Multi-institution cohort studies and collaborative registries are needed to provide updated information related to the epidemiology, diagnosis, treatment, morbidity, and mortality associated with calciphylaxis and to help formulate evidence-based diagnostic criteria. Radiographic and histologic studies, as well as other tools for early and accurate diagnosis of calciphylaxis, should be studied for feasibility, accuracy, and reproducibility. The incidence of nonuremic calciphylaxis points toward pathogenic pathways besides those based on the bone-mineral axis. Basic science research directed at improving understanding of the pathophysiology of calciphylaxis would be helpful in devising new treatment strategies targeting these pathways. Establishment of a collaborative, multi-institutional calciphylaxis working group would enable experts to formulate therapeutic guidelines based on current evidence. Such a group could facilitate initiation of large prospective studies to establish the efficacy of existing and new treatment modalities for calciphylaxis. A working group within the Society for Dermatology Hospitalists has been tasked with addressing these issues and is currently establishing a multicenter calciphylaxis database.
Calciphylaxis, also known as calcific uremic arteriolopathy, is a painful skin condition classically seen in patients with end-stage renal disease (ESRD), particularly those on chronic dialysis.1,2 It also has increasingly been reported in patients with normal renal function and calcium and phosphate homeostasis.3,4 Effective diagnosis and management of calciphylaxis remains challenging for physicians.2,5 The condition is characterized by tissue ischemia caused by calcification of cutaneous arteriolar vessels. As a result, calciphylaxis is associated with high mortality rates, ranging from 60% to 80%.5,6 Excruciating pain and nonhealing ulcers often lead to recurrent hospitalizations and infectious complications,7 and poor nutritional status, chronic pain, depression, and insomnia can further complicate recovery and lead to poor quality of life.8
We provide an update on calciphylaxis etiopathogenesis, diagnosis, and management. We also highlight some challenges faced in managing this potentially fatal condition.
Epidemiology
Calciphylaxis is considered a rare dermatosis with an estimated annual incidence of 1% to 4% in ESRD patients on dialysis. Recent data suggest that incidence of calciphylaxis is rising,5,7,9 which may stem from an increased use of calcium-based phosphate binders, an actual rise in disease incidence, and/or increased recognition of the disease.5 It is difficult to estimate the exact disease burden of calciphylaxis because the diagnostic criteria are not well defined, often leading to missed or delayed diagnosis.3,10 Furthermore, there is no centralized registry for calciphylaxis cases.3
Etiology and Pathogenesis
Calciphylaxis is thought to have a multifactorial etiology with the exact cause or trigger unknown.7 A long list of risk factors and triggers is associated with the condition (Table 1). Calciphylaxis primarily affects small arteries (40–600 μm in diameter) that become calcified due to an imbalance between inhibitors and promoters of calcification.2,11 Fetuin-A and matrix Gla protein inhibit vascular calcification and are downregulated in calciphylaxis.12,13 Dysfunctional calcium, phosphate, and parathyroid hormone regulatory pathways provide an increased substrate for the process of calcification, which causes endothelial damage and microthrombosis, resulting in tissue ischemia and infarction.14,15 Notably, there is growing interest in the role of vitamin K in the pathogenesis of calciphylaxis. Vitamin K inhibits vascular calcification, possibly by increasing the circulating levels of carboxylated matrix Gla protein.16
Clinical Features
Calciphylaxis is most commonly seen on the legs, abdomen, and buttocks.2 Patients with ESRD commonly develop proximal lesions affecting adipose-rich sites and have a poor prognosis. Distal lesions are more common in patients with nonuremic calciphylaxis, and mortality rates are lower in this population.2
Early lesions present as painful skin nodules or indurated plaques that often are rock-hard or firm to palpation with overlying mottling or a livedoid pattern (Figure, A). Early lesions progress from livedo reticularis to livedo racemosa and then to retiform purpura (Figure, B). Purpuric lesions later evolve into black eschars (Figure, C), then to necrotic, ulcerated, malodorous plaques or nodules in later stages of the disease (Figure, D). Lesions also may develop a gangrenous sclerotic appearance.2,5
Although most patients with calciphylaxis have ESRD, nonuremic patients also can develop the disease. Those with calciphylaxis who do not have renal dysfunction frequently have other risk factors for the disease and often report another notable health problem in the weeks or months prior to presentation.4 More than half of patients with calciphylaxis become bedridden or require use of a wheelchair.17 Pain is characteristically severe throughout the course of the disease; it may even precede the appearance of the skin lesions.18 Because the pain is associated with ischemia, it tends to be relatively refractory to treatment with opioids. Rare extracutaneous vascular calcifications may lead to visual impairment, gastrointestinal tract bleeding, and myopathy.5,9,19,20
Diagnosis
Considering the high morbidity and mortality associated with calciphylaxis, it is important to provide accurate and timely diagnosis; however, there currently are no validated diagnostic criteria for calciphylaxis. Careful correlation of clinical and histologic findings is required. Calciphylaxis biopsies have demonstrated medial calcification and proliferation of the intima of small- to medium-sized arteries.21 Lobular and septal panniculitis and extravascular soft-tissue calcification, particularly stippled calcification of the eccrine sweat glands, also has been seen.2,22 Special calcium stains (eg, von Kossa, Alizarin red) increase the sensitivity of biopsy by highlighting subtle areas of intravascular and extravascular calcification.5,23 Sufficient sampling of subcutaneous tissue and specimen evaluation by an experienced dermatopathologist are necessary to ensure proper interpretation of the histologic findings.
Despite these measures, skin biopsies may be nondiagnostic or falsely negative; therefore, when there is high clinical suspicion, it may be appropriate to move forward with a presumptive diagnosis of calciphylaxis even if the histologic findings are nondiagnostic.1,9,24 It also is worth noting that localized progression and ulceration may occur following skin biopsy, such that biopsy may even be contraindicated in certain cases (eg, penile calciphylaxis).
Standard laboratory workup for calciphylaxis includes evaluation for associated risk factors as well as exclusion of other conditions in the differential diagnosis (Table 2). Blood tests to evaluate for risk factors include liver and renal function tests, a complete metabolic panel, parathyroid hormone level, and serum albumin level.5 Elevated calcium and phosphate levels may signal disturbed calcium and phosphate homeostasis but are neither sensitive nor specific for the diagnosis.25 Complete blood cell count, blood cultures, thorough hypercoagulability workup (including but not limited to antiphospholipid antibodies, proteins C and S, factor V Leiden, antithrombin III, homocysteine, methylenetetrahydrofolate reductase mutation, and cryoglobulins), rheumatoid factor, antineutrophil cytoplasmic antibodies, and antinuclear antibody testing may be relevant to help identify contributing factors or mimickers of calciphylaxis.5 Various imaging modalities also have been used to evaluate for the presence of soft-tissue calcification in areas of suspected calciphylaxis, including radiography, mammography, computed tomography, ultrasonography, nuclear bone scintigraphy, and spectroscopy.2,26,27 Unfortunately, there currently is no standardized reproducible imaging modality for reliable diagnosis of calciphylaxis. Ultimately, histologic and radiographic findings should always be interpreted in the context of relevant clinical findings.2,9
Prevention
Reduction of the net calcium phosphorus product may help reduce the risk of calciphylaxis in ESRD patients, which can be accomplished by using non–calcium-phosphate binders, adequate dialysis, and restricting use of vitamin D and vitamin K antagonists.2,5 There are limited data regarding the benefits of using bisphosphonates and cinacalcet in ESRD patients on dialysis to prevent calciphylaxis.28,29
Management
Management of calciphylaxis is multifactorial. Besides dermatology and nephrology, specialists in pain management, wound care, plastic surgery, and nutrition are critical partners in management.1,5,9,30 Nephrologists can help optimize calcium and phosphate balance and ensure adequate dialysis. Pain specialists can aid in creating aggressive multiagent pain regimens that target the neuropathic/ischemic and physical aspects of calciphylaxis pain. When appropriate, nutrition specialists can help establish high-protein, low-phosphorus diets, and wound specialists can provide access to advanced wound dressings and adjunctive hyperbaric oxygen therapy. Plastic surgeons can provide conservative debridement procedures in a subset of patients, usually those with distal stable disease.
The limited understanding of the etiopathogenesis of calciphylaxis and the lack of data on its management are reflected in the limited treatment options for the disease (Table 3).2,5,9 There are no formal algorithms for the treatment of calciphylaxis. Therapeutic trials are scarce, and most of the current treatment recommendations are based on small retrospective reports or case series. Sodium thiosulfate has been the most widely used treatment option since 2004, when its use in calciphylaxis was first reported.31 Sodium thiosulfate chelates calcium and is thought to have antioxidant and vasodilatory properties.32 There are a few promising clinical trials and large-scale studies (Table 4) that aim to evaluate the efficacy of existing treatments (eg, sodium thiosulfate) as well as novel treatment options such as lanthanum carbonate, SNF472 (hexasodium phytate), and vitamin K.33-36
Prognosis
Calciphylaxis is a potentially fatal condition with a poor prognosis and a median survival rate of approximately 1 year following the appearance of skin lesions.37-39 Patients with proximal lesions and those on peritoneal dialysis (as opposed to hemodialysis) have a worse prognosis.40 Mortality rates are estimated to be 30% at 6 months, 50% at 12 months, and 80% at 2 years, with sepsis secondary to infection of cutaneous ulcers being the leading cause of death.37-39 The impact of calciphylaxis on patient quality of life and activities of daily living is severe.8,17
Future Directions
Multi-institution cohort studies and collaborative registries are needed to provide updated information related to the epidemiology, diagnosis, treatment, morbidity, and mortality associated with calciphylaxis and to help formulate evidence-based diagnostic criteria. Radiographic and histologic studies, as well as other tools for early and accurate diagnosis of calciphylaxis, should be studied for feasibility, accuracy, and reproducibility. The incidence of nonuremic calciphylaxis points toward pathogenic pathways besides those based on the bone-mineral axis. Basic science research directed at improving understanding of the pathophysiology of calciphylaxis would be helpful in devising new treatment strategies targeting these pathways. Establishment of a collaborative, multi-institutional calciphylaxis working group would enable experts to formulate therapeutic guidelines based on current evidence. Such a group could facilitate initiation of large prospective studies to establish the efficacy of existing and new treatment modalities for calciphylaxis. A working group within the Society for Dermatology Hospitalists has been tasked with addressing these issues and is currently establishing a multicenter calciphylaxis database.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Nigwekar SU, Thadhani RI, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Davis JM. The relationship between obesity and calciphylaxis: a review of the literature. Ostomy Wound Manage. 2016;62:12-18.
- Bajaj R, Courbebaisse M, Kroshinsky D, et al. Calciphylaxis in patients with normal renal function: a case series and systematic review. Mayo Clin Proc. 2018;93:1202-1212.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Westphal SG, Plumb T. Calciphylaxis. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2018. https://www.ncbi.nlm.nih.gov/books/NBK519020. Accessed November 12, 2018.
- Riemer CA, El-Azhary RA, Wu KL, et al. Underreported use of palliative care and patient-reported outcome measures to address reduced quality of life in patients with calciphylaxis: a systematic review. Br J Dermatol. 2017;177:1510-1518.
- Nigwekar SU. Calciphylaxis. Curr Opin Nephrol Hypertens. 2017;26:276-281.
- Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Perit Dial Int. 2008;28:268-270.
- Lin WT, Chao CM. Tumoral calcinosis in renal failure. QJM. 2014;107:387.
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Bleyer AJ, Choi M, Igwemezie B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376-383.
- Ahmed S, O’Neill KD, Hood AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:267-276.
- Nigwekar SU, Bloch DB, Nazarian RM, et al. Vitamin K-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717-1722.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Polizzotto MN, Bryan T, Ashby MA, et al. Symptomatic management of calciphylaxis: a case series and review of the literature. J Pain Symptom Manage. 2006;32:186-190.
- Gupta N, Haq KF, Mahajan S, et al. Gastrointestinal bleeding secondary to calciphylaxis. Am J Case Rep. 2015;16:818-822.
- Edelstein CL, Wickham MK, Kirby PA. Systemic calciphylaxis presenting as a painful, proximal myopathy. Postgrad Med J. 1992;68:209-211.
- Mochel MC, Arakari RY, Wang G, et al. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol. 2013;35:582-586.
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802.
- Cassius C, Moguelet P, Monfort JB, et al. Calciphylaxis in haemodialysed patients: diagnostic value of calcifications in cutaneous biopsy. Br J Dermatol. 2018;178:292-293.
- Sreedhar A, Sheikh HA, Scagliotti CJ, et al. Advanced-stage calciphylaxis: think before you punch. Cleve Clin J Med. 2016;83:562-564.
- Brandenburg VM, Kramann R, Rothe H, et al. Calcific uraemic arteriolopathy (calciphylaxis): data from a large nation-wide registry. Nephrol Dial Transplant. 2017;32:126-132.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017;153:101-103.
- Shmidt E, Murthy NS, Knudsen JM, et al. Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol. 2012;67:1296-1301.
- EVOLVE Trial Investigators; Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482-2494.
- Rogers NM, Teubner DJO, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150-157.
- Nigwekar SU. Multidisciplinary approach to calcific uremic arteriolopathy. Curr Opin Nephrol Hypertens. 2015;24:531-537.
- Cicone JS, Petronis JB, Embert CD, et al. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104-1108.
- Chen NX, O’Neill K, Akl NK, et al. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151-156.
- Chan MR, Ghandour F, Murali NS, et al. Pilot study of the effect of lanthanum carbonate in patients with calciphylaxis: a Wisconsin Network for Health Research (WiNHR) study. J Nephrol Ther. 2014;4:1000162.
- Perelló J, Gómez M, Ferrer MD, et al. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J Nephrol. 2018;31:287-296.
- Christiadi D, Singer RF. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin Kidney J. 2018;11:528-529.
- Caluwe R, Vandecasteele S, Van Vlem B, et al. Vitamin K2 supplementation in haemodialysis patients: a randomized dose-finding study. Nephrol Dial Transplant. 2014;29:1385-1390.
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429.
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Nigwekar SU, Thadhani RI, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Davis JM. The relationship between obesity and calciphylaxis: a review of the literature. Ostomy Wound Manage. 2016;62:12-18.
- Bajaj R, Courbebaisse M, Kroshinsky D, et al. Calciphylaxis in patients with normal renal function: a case series and systematic review. Mayo Clin Proc. 2018;93:1202-1212.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Westphal SG, Plumb T. Calciphylaxis. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2018. https://www.ncbi.nlm.nih.gov/books/NBK519020. Accessed November 12, 2018.
- Riemer CA, El-Azhary RA, Wu KL, et al. Underreported use of palliative care and patient-reported outcome measures to address reduced quality of life in patients with calciphylaxis: a systematic review. Br J Dermatol. 2017;177:1510-1518.
- Nigwekar SU. Calciphylaxis. Curr Opin Nephrol Hypertens. 2017;26:276-281.
- Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Perit Dial Int. 2008;28:268-270.
- Lin WT, Chao CM. Tumoral calcinosis in renal failure. QJM. 2014;107:387.
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Bleyer AJ, Choi M, Igwemezie B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376-383.
- Ahmed S, O’Neill KD, Hood AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:267-276.
- Nigwekar SU, Bloch DB, Nazarian RM, et al. Vitamin K-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717-1722.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Polizzotto MN, Bryan T, Ashby MA, et al. Symptomatic management of calciphylaxis: a case series and review of the literature. J Pain Symptom Manage. 2006;32:186-190.
- Gupta N, Haq KF, Mahajan S, et al. Gastrointestinal bleeding secondary to calciphylaxis. Am J Case Rep. 2015;16:818-822.
- Edelstein CL, Wickham MK, Kirby PA. Systemic calciphylaxis presenting as a painful, proximal myopathy. Postgrad Med J. 1992;68:209-211.
- Mochel MC, Arakari RY, Wang G, et al. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol. 2013;35:582-586.
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802.
- Cassius C, Moguelet P, Monfort JB, et al. Calciphylaxis in haemodialysed patients: diagnostic value of calcifications in cutaneous biopsy. Br J Dermatol. 2018;178:292-293.
- Sreedhar A, Sheikh HA, Scagliotti CJ, et al. Advanced-stage calciphylaxis: think before you punch. Cleve Clin J Med. 2016;83:562-564.
- Brandenburg VM, Kramann R, Rothe H, et al. Calcific uraemic arteriolopathy (calciphylaxis): data from a large nation-wide registry. Nephrol Dial Transplant. 2017;32:126-132.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017;153:101-103.
- Shmidt E, Murthy NS, Knudsen JM, et al. Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol. 2012;67:1296-1301.
- EVOLVE Trial Investigators; Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482-2494.
- Rogers NM, Teubner DJO, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150-157.
- Nigwekar SU. Multidisciplinary approach to calcific uremic arteriolopathy. Curr Opin Nephrol Hypertens. 2015;24:531-537.
- Cicone JS, Petronis JB, Embert CD, et al. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104-1108.
- Chen NX, O’Neill K, Akl NK, et al. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151-156.
- Chan MR, Ghandour F, Murali NS, et al. Pilot study of the effect of lanthanum carbonate in patients with calciphylaxis: a Wisconsin Network for Health Research (WiNHR) study. J Nephrol Ther. 2014;4:1000162.
- Perelló J, Gómez M, Ferrer MD, et al. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J Nephrol. 2018;31:287-296.
- Christiadi D, Singer RF. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin Kidney J. 2018;11:528-529.
- Caluwe R, Vandecasteele S, Van Vlem B, et al. Vitamin K2 supplementation in haemodialysis patients: a randomized dose-finding study. Nephrol Dial Transplant. 2014;29:1385-1390.
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429.
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241.
Practice Points
- Maintain a high index of suspicion for calciphylaxis in patients with end-stage renal disease on chronic dialysis presenting with severely painful livedoid plaques or retiform purpura, particularly in fat-rich body sites.
- Skin biopsies may be limited by biopsy site, inadequate biopsy depth, missed areas of microcalcification, and absence of definitive histologic criteria. Special calcium stains and review by an experienced dermatopathologist may lower the rate of false-negative biopsies.
- In cases where the most likely clinical diagnosis is calciphylaxis, treatment should be initiated even if definitive histopathology findings are lacking.
- Treatment should be multimodal, including elimination of risk factors, intravenous sodium thiosulfate, agents addressing calcium-phosphate metabolism, and surgical debridement, if indicated.
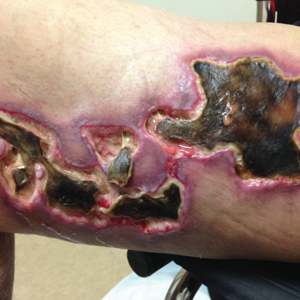
Optimizing Topical Therapy for Onychomycosis: The Importance of Patient Education
Onychomycosis is a fungal infection of the nail unit due to dermatophytes, yeasts, and nondermatophyte molds (NDMs). It accounts for approximately 50% of all nail disorders seen in clinical practice and is estimated to affect 10% to 12% of the US population.1,2 Oral medications approved by the US Food and Drug Administration (FDA) include terbinafine and itraconazole, which have demonstrated good efficacy in treating onychomycosis but are associated with potential drug-drug interactions and systemic side effects.3,4 Although liver failure associated with these drugs is rare,5 many patients are anxious about systemic adverse events and therefore prefer to use topical therapies for onychomycosis.
Many patients desire topical therapy but not every patient is an appropriate candidate. Patients who will likely respond well to topical therapy include those with superficial onychomycosis, distal lateral subungual onychomycosis that involves less than 50% of the nail plate surface area (without matrix involvement and a nail plate thickness less than 2 mm), and only up to 3 or 4 nails affected.6 In patients who have contraindications to oral therapy, topical therapy may be the only treatment option. To maximize efficacy of FDA-approved topical agents for onychomycosis therapy, patient education is of utmost importance. Failure to properly counsel the patient on proper medication application may result in decreased antifungal efficacy; poor patient compliance due to lack of improvement; and progression of disease, leading to increased onychodystrophy and pain.
Before initiating therapy, patients should be counseled that treatment with topical drugs is long, requiring daily application of the medication for 6 months for fingernails and 12 months for toenails, based on average nail growth rates (2–3 mm per month for fingernails; 1 mm per month for toenails).7 Patients also are advised to avoid nail polish application during the course of therapy, as clinical trials were performed without nail polish and the true efficacy with nail polish is unknown.8-10 Because patients who have had onychomycosis for shorter durations generally have better cure rates than those who have disease for longer durations, it is prudent to initiate topical therapy as early as possible.11,12 Treating the feet with an antifungal while treating the nails for onychomycosis further enhances efficacy.13,14 There are 3 FDA-approved topical therapies for onychomycosis: ciclopirox nail lacquer 8%, efinaconazole solution 10%, and tavaborole solution 5%.15-17
Ciclopirox is a hydroxypyridone with broad-spectrum antimicrobial activity against dermatophytes, NDMs, yeasts, and bacteria. Its mechanism of action is to chelate polyvalent cations, such as Fe3+, and inhibit fungal metal-dependent enzymes responsible for the degradation of toxic metabolites.15 Ciclopirox nail lacquer 8% was FDA approved for the treatment of onychomycosis in 1999, making it the first topical approved for this purpose. Its indication is for immunocompetent patients with mild to moderate onychomycosis (Trichophyton rubrum) without lunula involvement, with mycologic cure rates of 29% to 36% and complete cure rates of 5.5% to 8.5% (toenails).15 It is the only FDA-approved topical treatment for both fingernails and toenails. Using a brush applicator, it is applied daily to the nail plate and its undersurface, hyponychium, and 5 mm of the surrounding skin. It is important to counsel the patient to remove the lacquer from the nail plate weekly because failure to do so will result in accumulation of numerous layers of medication, such that the active drug cannot reach the site of infection (Figures 1 and 2). The nail plate also should be trimmed and filed weekly by the patient, with monthly clipping/debridement by a physician recommended.6,15
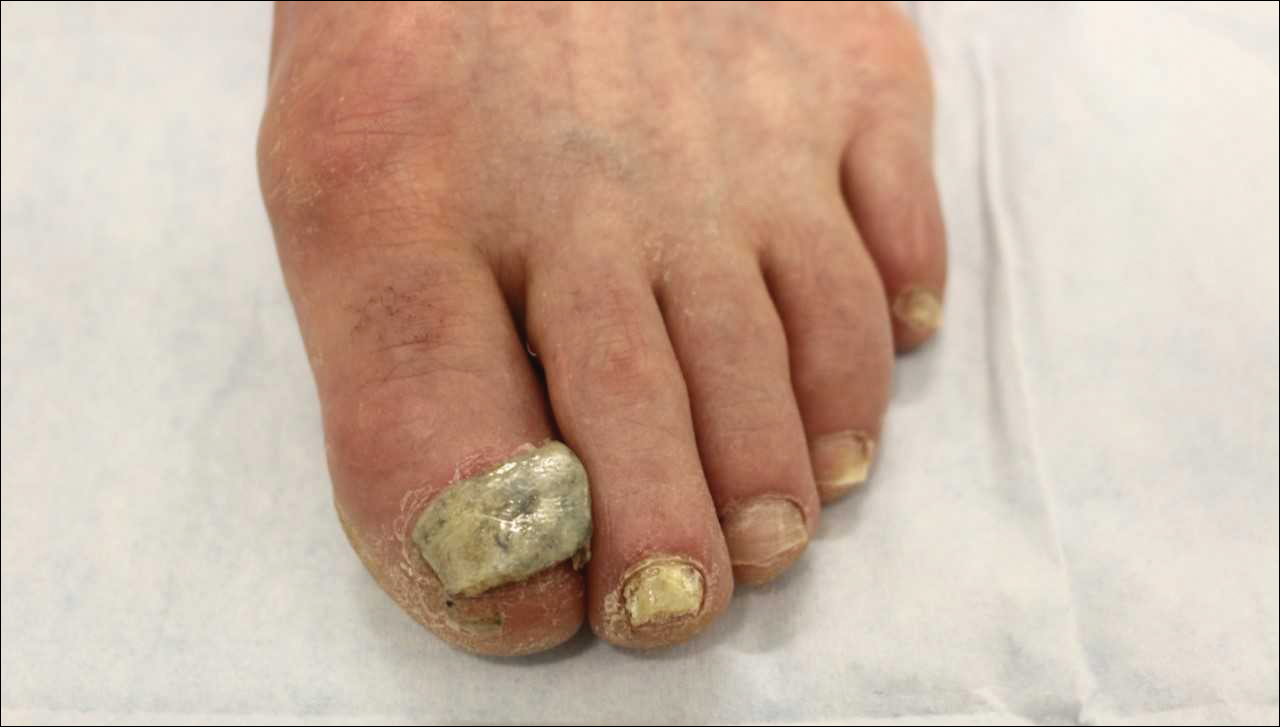
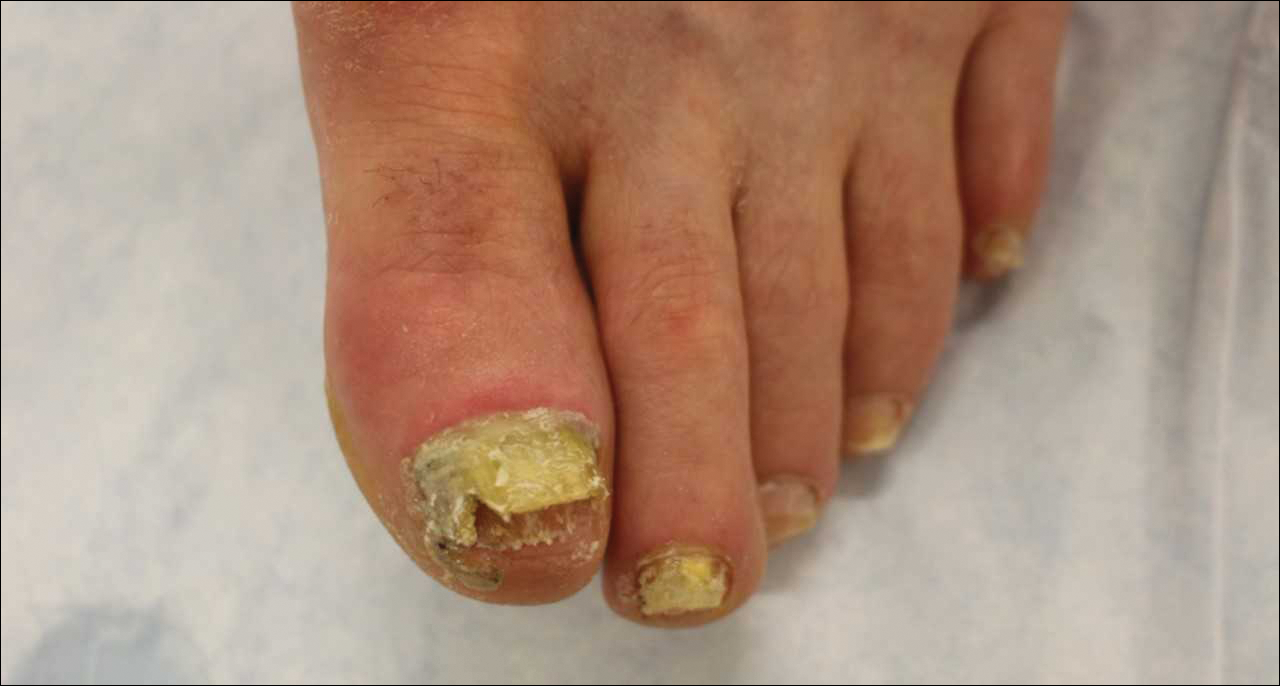
Efinaconazole is a triazole with antifungal activity against dermatophytes, NDMs, and Candida species. Its mechanism of action is inhibition of lanosterol 14α-demethylase, an enzyme involved in the biosynthesis of ergosterol, which is a component of the fungal cell membrane. Efinaconazole solution 10% was FDA approved in June 2014 for the treatment of toenail onychomycosis due to T rubrum and Trichophyton mentagrophytes, with package insert mycologic cure rates of 53.4% to 55.2% and complete cure rates of 15.2% to 17.8%.9,16 It is applied with a brush applicator to the nail plate, as well as its undersurface, nail folds, and hyponychium. Two drops are recommended for the great toenail and one drop for all other toenails, and no removal of the solution or debridement is required.6,16
Tavaborole is a benzoxaborole with antifungal activity against dermatophytes, NDMs, and yeasts. Its mechanism of action is inhibition of fungal aminoacyl transfer RNA synthetase, thus impeding protein synthesis.18 Tavaborole solution 5% was FDA approved in July 2014 for the treatment of toenail onychomycosis due to T rubrum and T mentagrophytes, with mycologic cure rates of 31.1% and 35.9% and complete cure rates of 6.5% and 9.1%, respectively.11,17 It is applied with a glass pointed-tip dropper to the nail plate, such that the entire nail is covered as well as under the nail tip. No removal of the solution or debridement is required.17
Topical therapies for onychomycosis require long treatment durations, thus excellent compliance and adherence to the treatment protocol are vital to maximize efficacy. Dermatologists who prescribe ciclopirox nail lacquer 8% should counsel patients to remove the lacquer with alcohol weekly, such that the antifungal penetrates the nail plate to reach the site of infection. Monthly debridement also must be clarified before initiating therapy. With all topical therapy for onychomycosis, it is important to treat early, treat concurrently for tinea pedis, and avoid use of nail polish so that patients have the best possible cure rates.
- Lipner SR, Scher RK. Onychomycosis: diagnosis and therapy. In: Razzaghi-Abyaneh M, Shams-Ghahfarokhi M, Rai M, eds. Medical Mycology: Current Trends and Future Prospects. Boca Raton, FL: CRC Press; 2015:28.
- Scher RK, Rich P, Pariser D, et al. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg. 2013;32(2 suppl 1):S2-S4.
- Lamisil [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 1997.
- Sporanox [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2001.
- National Institutes of Health. Terbinafine. LiverTox website. https://livertox.nlm.nih.gov/Terbinafine.htm. Accessed November 7, 2018.
- Lipner SR, Scher RK. Onychomycosis: topical therapy and devices. In: Rubin AI, Jellinek NJ, Daniel CR III, et al, eds. Scher and Daniel’s Nails: Diagnosis, Surgery, Therapy. 4th ed. Cham, Switzerland: Springer International Publishing; 2018:173-184.
- Lipner SR, Scher RK. Nail growth evaluation and factors affecting nail growth. In: Humbert P, Fanian F, Maibach HI, et al, eds. Agache’s Measuring the Skin. 2nd ed. Berlin, Germany: Springer; 2017:867-881.
- Gupta AK, Elewski BE, Sugarman JL, et al. The efficacy and safety of efinaconazole 10% solution for treatment of mild to moderate onychomycosis: a pooled analysis of two phase 3 randomized trials. J Drugs Dermatol. 2014;13:815-820.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol. 2015;73:62-69.
- Rich P. Efinaconazole topical solution, 10%: the benefits of treating onychomycosis early. J Drugs Dermatol. 2015;14:58-62.
- Lipner SR, Scher RK. Efinaconazole 10% topical solution for the topical treatment of onychomycosis of the toenail. Expert Rev Clin Pharmacol. 2015;8:719-731.
- Del Rosso JQ. Onychomycosis of toenails and post-hoc analyses with efinaconazole 10% solution once-daily treatment: impact of disease severity and other concomitant associated factors on selection of therapy and therapeutic outcomes. J Clin Aesthet Dermatol. 2016;9:42.
- Lipner SR, Scher RK. Management of onychomycosis and co-existing tinea pedis. J Drugs Dermatol. 2015;14:492-494.
- Penlac [package insert]. Berwyn, PA: Dermik Laboratories; 2004.
- Jublia [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals, LLC; 2014.
- Kerydin [package insert]. Palo Alto, CA: Anacor Pharmaceuticals, Inc; 2014.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
Onychomycosis is a fungal infection of the nail unit due to dermatophytes, yeasts, and nondermatophyte molds (NDMs). It accounts for approximately 50% of all nail disorders seen in clinical practice and is estimated to affect 10% to 12% of the US population.1,2 Oral medications approved by the US Food and Drug Administration (FDA) include terbinafine and itraconazole, which have demonstrated good efficacy in treating onychomycosis but are associated with potential drug-drug interactions and systemic side effects.3,4 Although liver failure associated with these drugs is rare,5 many patients are anxious about systemic adverse events and therefore prefer to use topical therapies for onychomycosis.
Many patients desire topical therapy but not every patient is an appropriate candidate. Patients who will likely respond well to topical therapy include those with superficial onychomycosis, distal lateral subungual onychomycosis that involves less than 50% of the nail plate surface area (without matrix involvement and a nail plate thickness less than 2 mm), and only up to 3 or 4 nails affected.6 In patients who have contraindications to oral therapy, topical therapy may be the only treatment option. To maximize efficacy of FDA-approved topical agents for onychomycosis therapy, patient education is of utmost importance. Failure to properly counsel the patient on proper medication application may result in decreased antifungal efficacy; poor patient compliance due to lack of improvement; and progression of disease, leading to increased onychodystrophy and pain.
Before initiating therapy, patients should be counseled that treatment with topical drugs is long, requiring daily application of the medication for 6 months for fingernails and 12 months for toenails, based on average nail growth rates (2–3 mm per month for fingernails; 1 mm per month for toenails).7 Patients also are advised to avoid nail polish application during the course of therapy, as clinical trials were performed without nail polish and the true efficacy with nail polish is unknown.8-10 Because patients who have had onychomycosis for shorter durations generally have better cure rates than those who have disease for longer durations, it is prudent to initiate topical therapy as early as possible.11,12 Treating the feet with an antifungal while treating the nails for onychomycosis further enhances efficacy.13,14 There are 3 FDA-approved topical therapies for onychomycosis: ciclopirox nail lacquer 8%, efinaconazole solution 10%, and tavaborole solution 5%.15-17
Ciclopirox is a hydroxypyridone with broad-spectrum antimicrobial activity against dermatophytes, NDMs, yeasts, and bacteria. Its mechanism of action is to chelate polyvalent cations, such as Fe3+, and inhibit fungal metal-dependent enzymes responsible for the degradation of toxic metabolites.15 Ciclopirox nail lacquer 8% was FDA approved for the treatment of onychomycosis in 1999, making it the first topical approved for this purpose. Its indication is for immunocompetent patients with mild to moderate onychomycosis (Trichophyton rubrum) without lunula involvement, with mycologic cure rates of 29% to 36% and complete cure rates of 5.5% to 8.5% (toenails).15 It is the only FDA-approved topical treatment for both fingernails and toenails. Using a brush applicator, it is applied daily to the nail plate and its undersurface, hyponychium, and 5 mm of the surrounding skin. It is important to counsel the patient to remove the lacquer from the nail plate weekly because failure to do so will result in accumulation of numerous layers of medication, such that the active drug cannot reach the site of infection (Figures 1 and 2). The nail plate also should be trimmed and filed weekly by the patient, with monthly clipping/debridement by a physician recommended.6,15
Efinaconazole is a triazole with antifungal activity against dermatophytes, NDMs, and Candida species. Its mechanism of action is inhibition of lanosterol 14α-demethylase, an enzyme involved in the biosynthesis of ergosterol, which is a component of the fungal cell membrane. Efinaconazole solution 10% was FDA approved in June 2014 for the treatment of toenail onychomycosis due to T rubrum and Trichophyton mentagrophytes, with package insert mycologic cure rates of 53.4% to 55.2% and complete cure rates of 15.2% to 17.8%.9,16 It is applied with a brush applicator to the nail plate, as well as its undersurface, nail folds, and hyponychium. Two drops are recommended for the great toenail and one drop for all other toenails, and no removal of the solution or debridement is required.6,16
Tavaborole is a benzoxaborole with antifungal activity against dermatophytes, NDMs, and yeasts. Its mechanism of action is inhibition of fungal aminoacyl transfer RNA synthetase, thus impeding protein synthesis.18 Tavaborole solution 5% was FDA approved in July 2014 for the treatment of toenail onychomycosis due to T rubrum and T mentagrophytes, with mycologic cure rates of 31.1% and 35.9% and complete cure rates of 6.5% and 9.1%, respectively.11,17 It is applied with a glass pointed-tip dropper to the nail plate, such that the entire nail is covered as well as under the nail tip. No removal of the solution or debridement is required.17
Topical therapies for onychomycosis require long treatment durations, thus excellent compliance and adherence to the treatment protocol are vital to maximize efficacy. Dermatologists who prescribe ciclopirox nail lacquer 8% should counsel patients to remove the lacquer with alcohol weekly, such that the antifungal penetrates the nail plate to reach the site of infection. Monthly debridement also must be clarified before initiating therapy. With all topical therapy for onychomycosis, it is important to treat early, treat concurrently for tinea pedis, and avoid use of nail polish so that patients have the best possible cure rates.
Onychomycosis is a fungal infection of the nail unit due to dermatophytes, yeasts, and nondermatophyte molds (NDMs). It accounts for approximately 50% of all nail disorders seen in clinical practice and is estimated to affect 10% to 12% of the US population.1,2 Oral medications approved by the US Food and Drug Administration (FDA) include terbinafine and itraconazole, which have demonstrated good efficacy in treating onychomycosis but are associated with potential drug-drug interactions and systemic side effects.3,4 Although liver failure associated with these drugs is rare,5 many patients are anxious about systemic adverse events and therefore prefer to use topical therapies for onychomycosis.
Many patients desire topical therapy but not every patient is an appropriate candidate. Patients who will likely respond well to topical therapy include those with superficial onychomycosis, distal lateral subungual onychomycosis that involves less than 50% of the nail plate surface area (without matrix involvement and a nail plate thickness less than 2 mm), and only up to 3 or 4 nails affected.6 In patients who have contraindications to oral therapy, topical therapy may be the only treatment option. To maximize efficacy of FDA-approved topical agents for onychomycosis therapy, patient education is of utmost importance. Failure to properly counsel the patient on proper medication application may result in decreased antifungal efficacy; poor patient compliance due to lack of improvement; and progression of disease, leading to increased onychodystrophy and pain.
Before initiating therapy, patients should be counseled that treatment with topical drugs is long, requiring daily application of the medication for 6 months for fingernails and 12 months for toenails, based on average nail growth rates (2–3 mm per month for fingernails; 1 mm per month for toenails).7 Patients also are advised to avoid nail polish application during the course of therapy, as clinical trials were performed without nail polish and the true efficacy with nail polish is unknown.8-10 Because patients who have had onychomycosis for shorter durations generally have better cure rates than those who have disease for longer durations, it is prudent to initiate topical therapy as early as possible.11,12 Treating the feet with an antifungal while treating the nails for onychomycosis further enhances efficacy.13,14 There are 3 FDA-approved topical therapies for onychomycosis: ciclopirox nail lacquer 8%, efinaconazole solution 10%, and tavaborole solution 5%.15-17
Ciclopirox is a hydroxypyridone with broad-spectrum antimicrobial activity against dermatophytes, NDMs, yeasts, and bacteria. Its mechanism of action is to chelate polyvalent cations, such as Fe3+, and inhibit fungal metal-dependent enzymes responsible for the degradation of toxic metabolites.15 Ciclopirox nail lacquer 8% was FDA approved for the treatment of onychomycosis in 1999, making it the first topical approved for this purpose. Its indication is for immunocompetent patients with mild to moderate onychomycosis (Trichophyton rubrum) without lunula involvement, with mycologic cure rates of 29% to 36% and complete cure rates of 5.5% to 8.5% (toenails).15 It is the only FDA-approved topical treatment for both fingernails and toenails. Using a brush applicator, it is applied daily to the nail plate and its undersurface, hyponychium, and 5 mm of the surrounding skin. It is important to counsel the patient to remove the lacquer from the nail plate weekly because failure to do so will result in accumulation of numerous layers of medication, such that the active drug cannot reach the site of infection (Figures 1 and 2). The nail plate also should be trimmed and filed weekly by the patient, with monthly clipping/debridement by a physician recommended.6,15
Efinaconazole is a triazole with antifungal activity against dermatophytes, NDMs, and Candida species. Its mechanism of action is inhibition of lanosterol 14α-demethylase, an enzyme involved in the biosynthesis of ergosterol, which is a component of the fungal cell membrane. Efinaconazole solution 10% was FDA approved in June 2014 for the treatment of toenail onychomycosis due to T rubrum and Trichophyton mentagrophytes, with package insert mycologic cure rates of 53.4% to 55.2% and complete cure rates of 15.2% to 17.8%.9,16 It is applied with a brush applicator to the nail plate, as well as its undersurface, nail folds, and hyponychium. Two drops are recommended for the great toenail and one drop for all other toenails, and no removal of the solution or debridement is required.6,16
Tavaborole is a benzoxaborole with antifungal activity against dermatophytes, NDMs, and yeasts. Its mechanism of action is inhibition of fungal aminoacyl transfer RNA synthetase, thus impeding protein synthesis.18 Tavaborole solution 5% was FDA approved in July 2014 for the treatment of toenail onychomycosis due to T rubrum and T mentagrophytes, with mycologic cure rates of 31.1% and 35.9% and complete cure rates of 6.5% and 9.1%, respectively.11,17 It is applied with a glass pointed-tip dropper to the nail plate, such that the entire nail is covered as well as under the nail tip. No removal of the solution or debridement is required.17
Topical therapies for onychomycosis require long treatment durations, thus excellent compliance and adherence to the treatment protocol are vital to maximize efficacy. Dermatologists who prescribe ciclopirox nail lacquer 8% should counsel patients to remove the lacquer with alcohol weekly, such that the antifungal penetrates the nail plate to reach the site of infection. Monthly debridement also must be clarified before initiating therapy. With all topical therapy for onychomycosis, it is important to treat early, treat concurrently for tinea pedis, and avoid use of nail polish so that patients have the best possible cure rates.
- Lipner SR, Scher RK. Onychomycosis: diagnosis and therapy. In: Razzaghi-Abyaneh M, Shams-Ghahfarokhi M, Rai M, eds. Medical Mycology: Current Trends and Future Prospects. Boca Raton, FL: CRC Press; 2015:28.
- Scher RK, Rich P, Pariser D, et al. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg. 2013;32(2 suppl 1):S2-S4.
- Lamisil [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 1997.
- Sporanox [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2001.
- National Institutes of Health. Terbinafine. LiverTox website. https://livertox.nlm.nih.gov/Terbinafine.htm. Accessed November 7, 2018.
- Lipner SR, Scher RK. Onychomycosis: topical therapy and devices. In: Rubin AI, Jellinek NJ, Daniel CR III, et al, eds. Scher and Daniel’s Nails: Diagnosis, Surgery, Therapy. 4th ed. Cham, Switzerland: Springer International Publishing; 2018:173-184.
- Lipner SR, Scher RK. Nail growth evaluation and factors affecting nail growth. In: Humbert P, Fanian F, Maibach HI, et al, eds. Agache’s Measuring the Skin. 2nd ed. Berlin, Germany: Springer; 2017:867-881.
- Gupta AK, Elewski BE, Sugarman JL, et al. The efficacy and safety of efinaconazole 10% solution for treatment of mild to moderate onychomycosis: a pooled analysis of two phase 3 randomized trials. J Drugs Dermatol. 2014;13:815-820.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol. 2015;73:62-69.
- Rich P. Efinaconazole topical solution, 10%: the benefits of treating onychomycosis early. J Drugs Dermatol. 2015;14:58-62.
- Lipner SR, Scher RK. Efinaconazole 10% topical solution for the topical treatment of onychomycosis of the toenail. Expert Rev Clin Pharmacol. 2015;8:719-731.
- Del Rosso JQ. Onychomycosis of toenails and post-hoc analyses with efinaconazole 10% solution once-daily treatment: impact of disease severity and other concomitant associated factors on selection of therapy and therapeutic outcomes. J Clin Aesthet Dermatol. 2016;9:42.
- Lipner SR, Scher RK. Management of onychomycosis and co-existing tinea pedis. J Drugs Dermatol. 2015;14:492-494.
- Penlac [package insert]. Berwyn, PA: Dermik Laboratories; 2004.
- Jublia [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals, LLC; 2014.
- Kerydin [package insert]. Palo Alto, CA: Anacor Pharmaceuticals, Inc; 2014.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
- Lipner SR, Scher RK. Onychomycosis: diagnosis and therapy. In: Razzaghi-Abyaneh M, Shams-Ghahfarokhi M, Rai M, eds. Medical Mycology: Current Trends and Future Prospects. Boca Raton, FL: CRC Press; 2015:28.
- Scher RK, Rich P, Pariser D, et al. The epidemiology, etiology, and pathophysiology of onychomycosis. Semin Cutan Med Surg. 2013;32(2 suppl 1):S2-S4.
- Lamisil [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 1997.
- Sporanox [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2001.
- National Institutes of Health. Terbinafine. LiverTox website. https://livertox.nlm.nih.gov/Terbinafine.htm. Accessed November 7, 2018.
- Lipner SR, Scher RK. Onychomycosis: topical therapy and devices. In: Rubin AI, Jellinek NJ, Daniel CR III, et al, eds. Scher and Daniel’s Nails: Diagnosis, Surgery, Therapy. 4th ed. Cham, Switzerland: Springer International Publishing; 2018:173-184.
- Lipner SR, Scher RK. Nail growth evaluation and factors affecting nail growth. In: Humbert P, Fanian F, Maibach HI, et al, eds. Agache’s Measuring the Skin. 2nd ed. Berlin, Germany: Springer; 2017:867-881.
- Gupta AK, Elewski BE, Sugarman JL, et al. The efficacy and safety of efinaconazole 10% solution for treatment of mild to moderate onychomycosis: a pooled analysis of two phase 3 randomized trials. J Drugs Dermatol. 2014;13:815-820.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol. 2015;73:62-69.
- Rich P. Efinaconazole topical solution, 10%: the benefits of treating onychomycosis early. J Drugs Dermatol. 2015;14:58-62.
- Lipner SR, Scher RK. Efinaconazole 10% topical solution for the topical treatment of onychomycosis of the toenail. Expert Rev Clin Pharmacol. 2015;8:719-731.
- Del Rosso JQ. Onychomycosis of toenails and post-hoc analyses with efinaconazole 10% solution once-daily treatment: impact of disease severity and other concomitant associated factors on selection of therapy and therapeutic outcomes. J Clin Aesthet Dermatol. 2016;9:42.
- Lipner SR, Scher RK. Management of onychomycosis and co-existing tinea pedis. J Drugs Dermatol. 2015;14:492-494.
- Penlac [package insert]. Berwyn, PA: Dermik Laboratories; 2004.
- Jublia [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals, LLC; 2014.
- Kerydin [package insert]. Palo Alto, CA: Anacor Pharmaceuticals, Inc; 2014.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
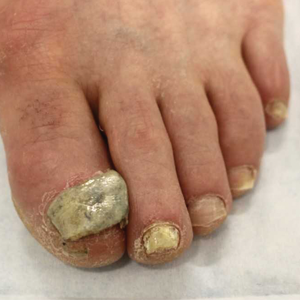
Strategies to Reduce Youth Indoor Tanning Injuries
Perusal of any lifestyle magazine reveals photographs of movie stars with sun-kissed skin. One can imagine their carefree lives afford ample time outdoors, a vast departure from the pasty masses trapped in their office cubicles. Our cultural norms dictate that a glowing look is a sign of health and attractiveness. Light-skinned individuals must receive regular exposure to sunlight to maintain their bronzed color. Over the last century, the indoor tanning industry has expanded to fill the niche created by the ceaseless pursuit of the ideal complexion.
Indoor tanning use causes up to 170,000 cases of skin cancer per year worldwide.1 Accumulating sunburns early in life is a leading risk factor for melanoma, the deadliest form of skin cancer. Campaigns to spread awareness about the link between UV radiation and skin cancer are ubiquitous. The US Food and Drug Administration (FDA) recommends against the use of tanning beds by minors, and several states have passed laws restricting their access. However, adolescents continue to engage. White female high school students remain frequenters of this practice, with more than 15% reporting current use of indoor tanning facilities.2 It seems targeted outreach and media campaigns are unsuccessful in influencing their behavior, and new approaches are needed.
Tanning-Related Injuries
Concentrated exposure to UV radiation during indoor tanning sessions carries the potential for immediate harm. Public health campaigns have focused on long-term skin cancer risk while overlooking thousands of injuries that occur annually at tanning salons across the country. The US Consumer Product Safety Commission first noted injuries associated with the largely unregulated tanning industry in 1974.3 In response, the FDA limited radiation levels, required indoor tanning devices to have timers and manual off switches, and mandated the use of protective eyewear. These changes sparked industry backlash due to the cost of compliance. The Indoor Tanning Association (no longer in operation) hired a lobbying firm in 2009 that successfully fought to resist further regulation.3
More than 3000 indoor tanning–related injuries are treated in emergency departments annually.4 White women aged 18 to 24 years who visit tanning salons are most likely to sustain injuries. In one study, severe skin burns accounted for 80% of emergency department visits, while the rest were due to fainting, eye injuries, and infections from unsanitary equipment.Timer malfunctions may play a role in tanning bed injuries, as several injured patients have reported falling asleep while tanning.4 Only 5% of tanning salons in North Carolina complied with FDA-recommended exposure schedules in 2003, suggesting that neglect or deliberate override of safety features also may contribute to injury.5
Challenges in Changing Tanning Behaviors
Use of indoor tanning facilities by adolescents is boosted by their misperceptions of peer engagement. Many teenagers overestimate the number of their peers who tan, which influences their own behavior.6 This phenomenon illustrates the importance of perceived social norms in this demographic group. Motivating adolescents to take actions that violate these norms poses a considerable challenge.
To teenagers, the perceived immediate benefits of indoor tanning far outweigh perceived costs. The immediate benefit of indoor tanning is having attractive skin, which may improve social standing and perceived self-worth. When adolescents weigh costs and benefits at different points in time, the present value of future events is discounted when compared to current events. For example, an immediate loss of $1000 is more impactful than losing $1000 ten years down the road. Adolescents are motivated to succeed in the short-term and may heavily discount future adverse effects such as the risk for developing cancer or premature aging of the skin. Therefore, getting a tan may be the “rational” decision even if there is an increased risk of future skin cancer.7
The addiction theory of tanning seeks to explain why individuals continue to tan despite knowledge of the associated risks. Exposure to UV radiation releases endorphins, producing a natural narcotic effect.8 The relaxing feeling sunbathers experience may lead to a phenomenon similar to addictions to opioids, alcohol, tobacco, and sugar. Behavior change is a process that unfolds over time. The 5 stages are precontemplation, contemplation, preparation, action, and maintenance.9 Education falls on deaf ears when the recipients are not ready to consider change. Individuals who are already thinking about cutting back on tanning fall into the category of contemplators and are the most open to educational techniques.9
Potential Solutions
Despite the dire long-term consequences of melanoma, warning adolescents of the increased cancer risk from tanning is an ineffective dissuasion strategy.10 Solutions that aim to limit tanning behaviors in this population should instead center on decreasing the present utility of a tan. Emphasis on the risk of immediate injury may be one effective route. The costs of potential damage to current appearance, vision, and overall health are not readily discounted by adolescents. Teens who devote time and money to the pursuit of a golden glow place high value on attractiveness. Such individuals respond best to loss-framed messages that focus on the impact of UV exposure on appearance, not just their health.11 However, appearance-motivated individuals may feel threatened by interventions that aim to reduce their decision freedom and display high reactance, leading them to reassert their freedom by resisting antitanning messages.12 Another strategy is altering media messaging to support a wider swathe of skin tones, reducing the social benefits of a tan. To swing the needle on our cultural norms, this intervention will require an enduring effort with backing from media outlets and celebrities.
Taxes on tanning salons and devices provide a basic economic disincentive to adolescents who typically have limited funds. State cigarette tax increases successfully reduced youth consumption of tobacco in the 1990s.13 A provision of the Patient Protection and Affordable Care Act levied a 10% excise tax on tanning salons with promising early results.14 Further taxation may generate revenue for educational campaigns on the injury risks of tanning. Continued safety improvements that limit user exposure to UV radiation and enforcement of FDA regulations also will decrease injury rates. Minimizing the UV output of tanning beds and designing protective equipment for tanners are 2 potential objectives. Improvement of over-the-counter sunless tanning agents also will provide alternatives to catching rays for adolescents who wish to attain a bronzed complexion.
Final Thoughts
Health care providers must assess a patient’s readiness for change and tailor interventions accordingly. Regardless of the method, new approaches to combat adolescent tanning injuries may reduce health care costs and minimize serious public health concerns for the next generation.
- Firger J. Indoor tanning injuries send thousands to the ER each year. CBS News. December 16, 2014. https://www.cbsnews.com/news/skin-cancer-burns-indoor-tanning-salon-injuries/. Accessed November 7, 2018.
- Guy GP, Berkowitz Z, Everett Jones S, et al. Prevalence of indoor tanning and association with sunburn among youth in the United States. JAMA Dermatol. 2017;153:387-390.
- Pulley MK. Government tan lines: examining the reach and effectiveness of federal and state efforts to protect consumers from the dangers of indoor tanning. Pepperdine Law Review. 2009;36:1163-1181.
- Guy GP Jr, Watson M, Haileyesus T, et al. Indoor tanning–related injuries treated in a national sample of US hospital emergency departments. JAMA Intern Med. 2015;175:309-311.
- Hornung RL, Magee KH, Lee WJ, et al. Tanning facility use: are we exceeding Food and Drug Administration limits? J Am Acad Dermatol. 2003;49:655-661.
- Hoerster KD, Mayer JA, Woodruff SI, et al. The influence of parents and peers on adolescent indoor tanning behavior: findings from a multi-city sample. J Am Acad Dermatol. 2007;57:990-997.
- Feldman SR, Dempsey JR, Grummer S, et al. Implications of a utility model for ultraviolet exposure behavior. J Am Acad Dermatol. 2001;45:718-722.
- Okhovat J, Feldman SR. Tanning: an addiction? The Melanoma Letter. 2013 Winter;31:5-7. https://www.skincancer.org/Media/Default/File/File/SCF_ML_31-3.pdf. Accessed November 11, 2017.
- Prochaska JO, DiClemente CC, Norcross JC. In search of how people change. applications to addictive behaviors. Am Psychol. 1992;47:1102-1114.
- Baker MK. Preventing Skin Cancer in Adolescent Girls Through Intervention With Their Mothers [dissertation]. Johnson City, TN: East Tennessee State University; 2013.
- Thomas K, Hevey D, Pertl M, et al. Appearance matters: the frame and focus of health messages influences beliefs about skin cancer. Br J Health Psychol. 2011;16(pt 2):418-429.
- Jones JL, Leary MR. Effects of appearance-based admonitions against sun exposure on tanning intentions in young adults. Health Psychol. 1994;13:86-90.
- Carpenter C, Cook PJ. Cigarette taxes and youth smoking: new evidence from national, state, and local youth risk behavior surveys. J Health Econ. 2008;27:287-99.
- Ryan E. The ‘tanning tax’ is a public health success story. Health Affairs website. https://www.healthaffairs.org/do/10.1377/hblog20170815.061547/full/. Published August 15, 2017. Accessed November 7, 2018.
Perusal of any lifestyle magazine reveals photographs of movie stars with sun-kissed skin. One can imagine their carefree lives afford ample time outdoors, a vast departure from the pasty masses trapped in their office cubicles. Our cultural norms dictate that a glowing look is a sign of health and attractiveness. Light-skinned individuals must receive regular exposure to sunlight to maintain their bronzed color. Over the last century, the indoor tanning industry has expanded to fill the niche created by the ceaseless pursuit of the ideal complexion.
Indoor tanning use causes up to 170,000 cases of skin cancer per year worldwide.1 Accumulating sunburns early in life is a leading risk factor for melanoma, the deadliest form of skin cancer. Campaigns to spread awareness about the link between UV radiation and skin cancer are ubiquitous. The US Food and Drug Administration (FDA) recommends against the use of tanning beds by minors, and several states have passed laws restricting their access. However, adolescents continue to engage. White female high school students remain frequenters of this practice, with more than 15% reporting current use of indoor tanning facilities.2 It seems targeted outreach and media campaigns are unsuccessful in influencing their behavior, and new approaches are needed.
Tanning-Related Injuries
Concentrated exposure to UV radiation during indoor tanning sessions carries the potential for immediate harm. Public health campaigns have focused on long-term skin cancer risk while overlooking thousands of injuries that occur annually at tanning salons across the country. The US Consumer Product Safety Commission first noted injuries associated with the largely unregulated tanning industry in 1974.3 In response, the FDA limited radiation levels, required indoor tanning devices to have timers and manual off switches, and mandated the use of protective eyewear. These changes sparked industry backlash due to the cost of compliance. The Indoor Tanning Association (no longer in operation) hired a lobbying firm in 2009 that successfully fought to resist further regulation.3
More than 3000 indoor tanning–related injuries are treated in emergency departments annually.4 White women aged 18 to 24 years who visit tanning salons are most likely to sustain injuries. In one study, severe skin burns accounted for 80% of emergency department visits, while the rest were due to fainting, eye injuries, and infections from unsanitary equipment.Timer malfunctions may play a role in tanning bed injuries, as several injured patients have reported falling asleep while tanning.4 Only 5% of tanning salons in North Carolina complied with FDA-recommended exposure schedules in 2003, suggesting that neglect or deliberate override of safety features also may contribute to injury.5
Challenges in Changing Tanning Behaviors
Use of indoor tanning facilities by adolescents is boosted by their misperceptions of peer engagement. Many teenagers overestimate the number of their peers who tan, which influences their own behavior.6 This phenomenon illustrates the importance of perceived social norms in this demographic group. Motivating adolescents to take actions that violate these norms poses a considerable challenge.
To teenagers, the perceived immediate benefits of indoor tanning far outweigh perceived costs. The immediate benefit of indoor tanning is having attractive skin, which may improve social standing and perceived self-worth. When adolescents weigh costs and benefits at different points in time, the present value of future events is discounted when compared to current events. For example, an immediate loss of $1000 is more impactful than losing $1000 ten years down the road. Adolescents are motivated to succeed in the short-term and may heavily discount future adverse effects such as the risk for developing cancer or premature aging of the skin. Therefore, getting a tan may be the “rational” decision even if there is an increased risk of future skin cancer.7
The addiction theory of tanning seeks to explain why individuals continue to tan despite knowledge of the associated risks. Exposure to UV radiation releases endorphins, producing a natural narcotic effect.8 The relaxing feeling sunbathers experience may lead to a phenomenon similar to addictions to opioids, alcohol, tobacco, and sugar. Behavior change is a process that unfolds over time. The 5 stages are precontemplation, contemplation, preparation, action, and maintenance.9 Education falls on deaf ears when the recipients are not ready to consider change. Individuals who are already thinking about cutting back on tanning fall into the category of contemplators and are the most open to educational techniques.9
Potential Solutions
Despite the dire long-term consequences of melanoma, warning adolescents of the increased cancer risk from tanning is an ineffective dissuasion strategy.10 Solutions that aim to limit tanning behaviors in this population should instead center on decreasing the present utility of a tan. Emphasis on the risk of immediate injury may be one effective route. The costs of potential damage to current appearance, vision, and overall health are not readily discounted by adolescents. Teens who devote time and money to the pursuit of a golden glow place high value on attractiveness. Such individuals respond best to loss-framed messages that focus on the impact of UV exposure on appearance, not just their health.11 However, appearance-motivated individuals may feel threatened by interventions that aim to reduce their decision freedom and display high reactance, leading them to reassert their freedom by resisting antitanning messages.12 Another strategy is altering media messaging to support a wider swathe of skin tones, reducing the social benefits of a tan. To swing the needle on our cultural norms, this intervention will require an enduring effort with backing from media outlets and celebrities.
Taxes on tanning salons and devices provide a basic economic disincentive to adolescents who typically have limited funds. State cigarette tax increases successfully reduced youth consumption of tobacco in the 1990s.13 A provision of the Patient Protection and Affordable Care Act levied a 10% excise tax on tanning salons with promising early results.14 Further taxation may generate revenue for educational campaigns on the injury risks of tanning. Continued safety improvements that limit user exposure to UV radiation and enforcement of FDA regulations also will decrease injury rates. Minimizing the UV output of tanning beds and designing protective equipment for tanners are 2 potential objectives. Improvement of over-the-counter sunless tanning agents also will provide alternatives to catching rays for adolescents who wish to attain a bronzed complexion.
Final Thoughts
Health care providers must assess a patient’s readiness for change and tailor interventions accordingly. Regardless of the method, new approaches to combat adolescent tanning injuries may reduce health care costs and minimize serious public health concerns for the next generation.
Perusal of any lifestyle magazine reveals photographs of movie stars with sun-kissed skin. One can imagine their carefree lives afford ample time outdoors, a vast departure from the pasty masses trapped in their office cubicles. Our cultural norms dictate that a glowing look is a sign of health and attractiveness. Light-skinned individuals must receive regular exposure to sunlight to maintain their bronzed color. Over the last century, the indoor tanning industry has expanded to fill the niche created by the ceaseless pursuit of the ideal complexion.
Indoor tanning use causes up to 170,000 cases of skin cancer per year worldwide.1 Accumulating sunburns early in life is a leading risk factor for melanoma, the deadliest form of skin cancer. Campaigns to spread awareness about the link between UV radiation and skin cancer are ubiquitous. The US Food and Drug Administration (FDA) recommends against the use of tanning beds by minors, and several states have passed laws restricting their access. However, adolescents continue to engage. White female high school students remain frequenters of this practice, with more than 15% reporting current use of indoor tanning facilities.2 It seems targeted outreach and media campaigns are unsuccessful in influencing their behavior, and new approaches are needed.
Tanning-Related Injuries
Concentrated exposure to UV radiation during indoor tanning sessions carries the potential for immediate harm. Public health campaigns have focused on long-term skin cancer risk while overlooking thousands of injuries that occur annually at tanning salons across the country. The US Consumer Product Safety Commission first noted injuries associated with the largely unregulated tanning industry in 1974.3 In response, the FDA limited radiation levels, required indoor tanning devices to have timers and manual off switches, and mandated the use of protective eyewear. These changes sparked industry backlash due to the cost of compliance. The Indoor Tanning Association (no longer in operation) hired a lobbying firm in 2009 that successfully fought to resist further regulation.3
More than 3000 indoor tanning–related injuries are treated in emergency departments annually.4 White women aged 18 to 24 years who visit tanning salons are most likely to sustain injuries. In one study, severe skin burns accounted for 80% of emergency department visits, while the rest were due to fainting, eye injuries, and infections from unsanitary equipment.Timer malfunctions may play a role in tanning bed injuries, as several injured patients have reported falling asleep while tanning.4 Only 5% of tanning salons in North Carolina complied with FDA-recommended exposure schedules in 2003, suggesting that neglect or deliberate override of safety features also may contribute to injury.5
Challenges in Changing Tanning Behaviors
Use of indoor tanning facilities by adolescents is boosted by their misperceptions of peer engagement. Many teenagers overestimate the number of their peers who tan, which influences their own behavior.6 This phenomenon illustrates the importance of perceived social norms in this demographic group. Motivating adolescents to take actions that violate these norms poses a considerable challenge.
To teenagers, the perceived immediate benefits of indoor tanning far outweigh perceived costs. The immediate benefit of indoor tanning is having attractive skin, which may improve social standing and perceived self-worth. When adolescents weigh costs and benefits at different points in time, the present value of future events is discounted when compared to current events. For example, an immediate loss of $1000 is more impactful than losing $1000 ten years down the road. Adolescents are motivated to succeed in the short-term and may heavily discount future adverse effects such as the risk for developing cancer or premature aging of the skin. Therefore, getting a tan may be the “rational” decision even if there is an increased risk of future skin cancer.7
The addiction theory of tanning seeks to explain why individuals continue to tan despite knowledge of the associated risks. Exposure to UV radiation releases endorphins, producing a natural narcotic effect.8 The relaxing feeling sunbathers experience may lead to a phenomenon similar to addictions to opioids, alcohol, tobacco, and sugar. Behavior change is a process that unfolds over time. The 5 stages are precontemplation, contemplation, preparation, action, and maintenance.9 Education falls on deaf ears when the recipients are not ready to consider change. Individuals who are already thinking about cutting back on tanning fall into the category of contemplators and are the most open to educational techniques.9
Potential Solutions
Despite the dire long-term consequences of melanoma, warning adolescents of the increased cancer risk from tanning is an ineffective dissuasion strategy.10 Solutions that aim to limit tanning behaviors in this population should instead center on decreasing the present utility of a tan. Emphasis on the risk of immediate injury may be one effective route. The costs of potential damage to current appearance, vision, and overall health are not readily discounted by adolescents. Teens who devote time and money to the pursuit of a golden glow place high value on attractiveness. Such individuals respond best to loss-framed messages that focus on the impact of UV exposure on appearance, not just their health.11 However, appearance-motivated individuals may feel threatened by interventions that aim to reduce their decision freedom and display high reactance, leading them to reassert their freedom by resisting antitanning messages.12 Another strategy is altering media messaging to support a wider swathe of skin tones, reducing the social benefits of a tan. To swing the needle on our cultural norms, this intervention will require an enduring effort with backing from media outlets and celebrities.
Taxes on tanning salons and devices provide a basic economic disincentive to adolescents who typically have limited funds. State cigarette tax increases successfully reduced youth consumption of tobacco in the 1990s.13 A provision of the Patient Protection and Affordable Care Act levied a 10% excise tax on tanning salons with promising early results.14 Further taxation may generate revenue for educational campaigns on the injury risks of tanning. Continued safety improvements that limit user exposure to UV radiation and enforcement of FDA regulations also will decrease injury rates. Minimizing the UV output of tanning beds and designing protective equipment for tanners are 2 potential objectives. Improvement of over-the-counter sunless tanning agents also will provide alternatives to catching rays for adolescents who wish to attain a bronzed complexion.
Final Thoughts
Health care providers must assess a patient’s readiness for change and tailor interventions accordingly. Regardless of the method, new approaches to combat adolescent tanning injuries may reduce health care costs and minimize serious public health concerns for the next generation.
- Firger J. Indoor tanning injuries send thousands to the ER each year. CBS News. December 16, 2014. https://www.cbsnews.com/news/skin-cancer-burns-indoor-tanning-salon-injuries/. Accessed November 7, 2018.
- Guy GP, Berkowitz Z, Everett Jones S, et al. Prevalence of indoor tanning and association with sunburn among youth in the United States. JAMA Dermatol. 2017;153:387-390.
- Pulley MK. Government tan lines: examining the reach and effectiveness of federal and state efforts to protect consumers from the dangers of indoor tanning. Pepperdine Law Review. 2009;36:1163-1181.
- Guy GP Jr, Watson M, Haileyesus T, et al. Indoor tanning–related injuries treated in a national sample of US hospital emergency departments. JAMA Intern Med. 2015;175:309-311.
- Hornung RL, Magee KH, Lee WJ, et al. Tanning facility use: are we exceeding Food and Drug Administration limits? J Am Acad Dermatol. 2003;49:655-661.
- Hoerster KD, Mayer JA, Woodruff SI, et al. The influence of parents and peers on adolescent indoor tanning behavior: findings from a multi-city sample. J Am Acad Dermatol. 2007;57:990-997.
- Feldman SR, Dempsey JR, Grummer S, et al. Implications of a utility model for ultraviolet exposure behavior. J Am Acad Dermatol. 2001;45:718-722.
- Okhovat J, Feldman SR. Tanning: an addiction? The Melanoma Letter. 2013 Winter;31:5-7. https://www.skincancer.org/Media/Default/File/File/SCF_ML_31-3.pdf. Accessed November 11, 2017.
- Prochaska JO, DiClemente CC, Norcross JC. In search of how people change. applications to addictive behaviors. Am Psychol. 1992;47:1102-1114.
- Baker MK. Preventing Skin Cancer in Adolescent Girls Through Intervention With Their Mothers [dissertation]. Johnson City, TN: East Tennessee State University; 2013.
- Thomas K, Hevey D, Pertl M, et al. Appearance matters: the frame and focus of health messages influences beliefs about skin cancer. Br J Health Psychol. 2011;16(pt 2):418-429.
- Jones JL, Leary MR. Effects of appearance-based admonitions against sun exposure on tanning intentions in young adults. Health Psychol. 1994;13:86-90.
- Carpenter C, Cook PJ. Cigarette taxes and youth smoking: new evidence from national, state, and local youth risk behavior surveys. J Health Econ. 2008;27:287-99.
- Ryan E. The ‘tanning tax’ is a public health success story. Health Affairs website. https://www.healthaffairs.org/do/10.1377/hblog20170815.061547/full/. Published August 15, 2017. Accessed November 7, 2018.
- Firger J. Indoor tanning injuries send thousands to the ER each year. CBS News. December 16, 2014. https://www.cbsnews.com/news/skin-cancer-burns-indoor-tanning-salon-injuries/. Accessed November 7, 2018.
- Guy GP, Berkowitz Z, Everett Jones S, et al. Prevalence of indoor tanning and association with sunburn among youth in the United States. JAMA Dermatol. 2017;153:387-390.
- Pulley MK. Government tan lines: examining the reach and effectiveness of federal and state efforts to protect consumers from the dangers of indoor tanning. Pepperdine Law Review. 2009;36:1163-1181.
- Guy GP Jr, Watson M, Haileyesus T, et al. Indoor tanning–related injuries treated in a national sample of US hospital emergency departments. JAMA Intern Med. 2015;175:309-311.
- Hornung RL, Magee KH, Lee WJ, et al. Tanning facility use: are we exceeding Food and Drug Administration limits? J Am Acad Dermatol. 2003;49:655-661.
- Hoerster KD, Mayer JA, Woodruff SI, et al. The influence of parents and peers on adolescent indoor tanning behavior: findings from a multi-city sample. J Am Acad Dermatol. 2007;57:990-997.
- Feldman SR, Dempsey JR, Grummer S, et al. Implications of a utility model for ultraviolet exposure behavior. J Am Acad Dermatol. 2001;45:718-722.
- Okhovat J, Feldman SR. Tanning: an addiction? The Melanoma Letter. 2013 Winter;31:5-7. https://www.skincancer.org/Media/Default/File/File/SCF_ML_31-3.pdf. Accessed November 11, 2017.
- Prochaska JO, DiClemente CC, Norcross JC. In search of how people change. applications to addictive behaviors. Am Psychol. 1992;47:1102-1114.
- Baker MK. Preventing Skin Cancer in Adolescent Girls Through Intervention With Their Mothers [dissertation]. Johnson City, TN: East Tennessee State University; 2013.
- Thomas K, Hevey D, Pertl M, et al. Appearance matters: the frame and focus of health messages influences beliefs about skin cancer. Br J Health Psychol. 2011;16(pt 2):418-429.
- Jones JL, Leary MR. Effects of appearance-based admonitions against sun exposure on tanning intentions in young adults. Health Psychol. 1994;13:86-90.
- Carpenter C, Cook PJ. Cigarette taxes and youth smoking: new evidence from national, state, and local youth risk behavior surveys. J Health Econ. 2008;27:287-99.
- Ryan E. The ‘tanning tax’ is a public health success story. Health Affairs website. https://www.healthaffairs.org/do/10.1377/hblog20170815.061547/full/. Published August 15, 2017. Accessed November 7, 2018.

DRESS Syndrome: How to Approach Systemic Workup, Treatment, and Long-term Follow-up


Lesions With a Distinct Black Pigment
The Diagnosis: Black-Spot Poison Ivy
Due to the detailed account of the patient's history including acuity of current presentation, history of recent activities, travel history, and recent exposures, as well as a thorough skin examination, a diagnosis of black-spot poison ivy was made. In this case, the linear distribution of the lesions with overlying black pigment that could not be removed (Figures 1 and 2) provided important clues to diagnosis.
Poison ivy is an allergic contact dermatitis that affects an estimated 25 to 40 million Americans annually who are exposed to its resin. Poison ivy is a plant from the Toxicodendron genus, and an estimated 85% of the North American population report sensitivity to these plants, of which poison ivy (Toxicodendron radicans) is the most common.1 Other related plants include poison sumac and poison oak. Poison ivy and other Toxicodendron plants produce urushiol, the oleoresin responsible for one of the most common allergic contact dermatitides in the United States.2 Black-spot poison ivy is an uncommon presentation following exposure to urushiol or oleoresin,3 as sufficient concentration of urushiol on the skin rarely is achieved.3,4 This plant's resin oxidizes and turns coal black when exposed to air.5 Contact with enough of this oleoresin will produce black-spot poison ivy.6 Patients with sufficient concentrations of oleoresin on their skin to cause this black oxidation usually have similar black spots on their clothing.7 Interestingly, some Toxicodendron species, such as the Japanese lacquer tree, Toxicodendron vernicifluum, have a black lacquer sap that was historically used as ink.8 This ink was used on Chinese and Japanese jars and has caused contact dermatitis hundreds of years after they were created.7
Poison ivy is characterized by a generalized, pruritic, erythematous rash with vesicles and papules in a linear distribution.9 Black-spot poison ivy presents the same with the addition of black lacquer-like macules with surrounding erythema.10 The skin lesions usually appear on exposed areas 24 to 48 hours after contact.11 Histology of black-spot poison ivy lesions should reveal yellow material in the stratum corneum with epidermal necrosis, in addition to classic features of acute allergic contact dermatitis.3 Interestingly, because these lesions occur with the first exposure to poison ivy, a patient may not develop the typical itchy eczematous eruption characteristic of poison ivy dermatitis. Differential diagnosis includes superficial purpura; exogenous pigment such as marker, ink, or tattoo pigment; tinea nigra; purpuric allergic contact dermatitis to resins or dyes; arthropod assault; irritant contact dermatitis; and infectious and noninfectious vasculitis.11
Similar to poison ivy, treatment of black-spot poison ivy involves oral and topical steroids combined with antihistamines if the patient continues to experience pruritus.6,12 It was recommended to our patient to apply cool compresses with water or Burow solution to alleviate itching and promote drying of the lesions. Calamine lotion can provide similar outcomes.13 Once the oleoresin is oxidized and bound to skin, the black spots cannot be removed with soap, water, or alcohol. The black spots gradually desquamate 1 to 2 weeks after formation without scarring,11 and patients do not require further monitoring.1 Patients should clean or discard clothing and evaluate for possible sources of poison ivy exposure. Because this type of poison ivy dermatitis is rare, most health care workers likely have never seen black-spot poison ivy, and it is an important diagnosis to consider.13
- Baer RL. Poison ivy dermatitis. Cutis. 1990;46:34-36.
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Hurwitz RM, Rivera HP, Guin JD. Black-spot poison ivy dermatitis. an acute irritant contact dermatitis superimposed upon an allergic contact dermatitis. Am J Dermatopathol. 1984;6:319-322.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Mallory SB, Hurwitz RM. Black-spot poison-ivy dermatitis. Clin Dermatol. 1986;4:149-151.
- Mallory SB, Miller OF, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Rietschel R, Fowler J. Toxicodendron plants and species. Fisher's Contact Dermatitis. 4th ed. Baltimore, MD: Williams & Wilkins; 1995:469-472.
- Fisher AA. Poison ivy/oak dermatitis. part I: prevention--soap and water, topical barriers, hyposensitization. Cutis. 1996;57:384-386.
- McClanahan C, Asarch A, Swick BL. Black spot poison ivy. Int J Dermatol. 2014;53:752-753.
- Mu EW, Capell BC, Castelo-Soccio L. Black spots on a toddler's skin. Contemp Pediatr. 2013;30:31-32.
- Schram SE, Willey A, Lee PK, et al. Black-spot poison ivy. Dermatitis. 2008;19:48-51.
- Paniagua CT, Bean AS. Black-spot poison ivy: a rare phenomenon. J Am Acad Nurse Pract. 2011;23:275-277.
The Diagnosis: Black-Spot Poison Ivy
Due to the detailed account of the patient's history including acuity of current presentation, history of recent activities, travel history, and recent exposures, as well as a thorough skin examination, a diagnosis of black-spot poison ivy was made. In this case, the linear distribution of the lesions with overlying black pigment that could not be removed (Figures 1 and 2) provided important clues to diagnosis.
Poison ivy is an allergic contact dermatitis that affects an estimated 25 to 40 million Americans annually who are exposed to its resin. Poison ivy is a plant from the Toxicodendron genus, and an estimated 85% of the North American population report sensitivity to these plants, of which poison ivy (Toxicodendron radicans) is the most common.1 Other related plants include poison sumac and poison oak. Poison ivy and other Toxicodendron plants produce urushiol, the oleoresin responsible for one of the most common allergic contact dermatitides in the United States.2 Black-spot poison ivy is an uncommon presentation following exposure to urushiol or oleoresin,3 as sufficient concentration of urushiol on the skin rarely is achieved.3,4 This plant's resin oxidizes and turns coal black when exposed to air.5 Contact with enough of this oleoresin will produce black-spot poison ivy.6 Patients with sufficient concentrations of oleoresin on their skin to cause this black oxidation usually have similar black spots on their clothing.7 Interestingly, some Toxicodendron species, such as the Japanese lacquer tree, Toxicodendron vernicifluum, have a black lacquer sap that was historically used as ink.8 This ink was used on Chinese and Japanese jars and has caused contact dermatitis hundreds of years after they were created.7
Poison ivy is characterized by a generalized, pruritic, erythematous rash with vesicles and papules in a linear distribution.9 Black-spot poison ivy presents the same with the addition of black lacquer-like macules with surrounding erythema.10 The skin lesions usually appear on exposed areas 24 to 48 hours after contact.11 Histology of black-spot poison ivy lesions should reveal yellow material in the stratum corneum with epidermal necrosis, in addition to classic features of acute allergic contact dermatitis.3 Interestingly, because these lesions occur with the first exposure to poison ivy, a patient may not develop the typical itchy eczematous eruption characteristic of poison ivy dermatitis. Differential diagnosis includes superficial purpura; exogenous pigment such as marker, ink, or tattoo pigment; tinea nigra; purpuric allergic contact dermatitis to resins or dyes; arthropod assault; irritant contact dermatitis; and infectious and noninfectious vasculitis.11
Similar to poison ivy, treatment of black-spot poison ivy involves oral and topical steroids combined with antihistamines if the patient continues to experience pruritus.6,12 It was recommended to our patient to apply cool compresses with water or Burow solution to alleviate itching and promote drying of the lesions. Calamine lotion can provide similar outcomes.13 Once the oleoresin is oxidized and bound to skin, the black spots cannot be removed with soap, water, or alcohol. The black spots gradually desquamate 1 to 2 weeks after formation without scarring,11 and patients do not require further monitoring.1 Patients should clean or discard clothing and evaluate for possible sources of poison ivy exposure. Because this type of poison ivy dermatitis is rare, most health care workers likely have never seen black-spot poison ivy, and it is an important diagnosis to consider.13
The Diagnosis: Black-Spot Poison Ivy
Due to the detailed account of the patient's history including acuity of current presentation, history of recent activities, travel history, and recent exposures, as well as a thorough skin examination, a diagnosis of black-spot poison ivy was made. In this case, the linear distribution of the lesions with overlying black pigment that could not be removed (Figures 1 and 2) provided important clues to diagnosis.
Poison ivy is an allergic contact dermatitis that affects an estimated 25 to 40 million Americans annually who are exposed to its resin. Poison ivy is a plant from the Toxicodendron genus, and an estimated 85% of the North American population report sensitivity to these plants, of which poison ivy (Toxicodendron radicans) is the most common.1 Other related plants include poison sumac and poison oak. Poison ivy and other Toxicodendron plants produce urushiol, the oleoresin responsible for one of the most common allergic contact dermatitides in the United States.2 Black-spot poison ivy is an uncommon presentation following exposure to urushiol or oleoresin,3 as sufficient concentration of urushiol on the skin rarely is achieved.3,4 This plant's resin oxidizes and turns coal black when exposed to air.5 Contact with enough of this oleoresin will produce black-spot poison ivy.6 Patients with sufficient concentrations of oleoresin on their skin to cause this black oxidation usually have similar black spots on their clothing.7 Interestingly, some Toxicodendron species, such as the Japanese lacquer tree, Toxicodendron vernicifluum, have a black lacquer sap that was historically used as ink.8 This ink was used on Chinese and Japanese jars and has caused contact dermatitis hundreds of years after they were created.7
Poison ivy is characterized by a generalized, pruritic, erythematous rash with vesicles and papules in a linear distribution.9 Black-spot poison ivy presents the same with the addition of black lacquer-like macules with surrounding erythema.10 The skin lesions usually appear on exposed areas 24 to 48 hours after contact.11 Histology of black-spot poison ivy lesions should reveal yellow material in the stratum corneum with epidermal necrosis, in addition to classic features of acute allergic contact dermatitis.3 Interestingly, because these lesions occur with the first exposure to poison ivy, a patient may not develop the typical itchy eczematous eruption characteristic of poison ivy dermatitis. Differential diagnosis includes superficial purpura; exogenous pigment such as marker, ink, or tattoo pigment; tinea nigra; purpuric allergic contact dermatitis to resins or dyes; arthropod assault; irritant contact dermatitis; and infectious and noninfectious vasculitis.11
Similar to poison ivy, treatment of black-spot poison ivy involves oral and topical steroids combined with antihistamines if the patient continues to experience pruritus.6,12 It was recommended to our patient to apply cool compresses with water or Burow solution to alleviate itching and promote drying of the lesions. Calamine lotion can provide similar outcomes.13 Once the oleoresin is oxidized and bound to skin, the black spots cannot be removed with soap, water, or alcohol. The black spots gradually desquamate 1 to 2 weeks after formation without scarring,11 and patients do not require further monitoring.1 Patients should clean or discard clothing and evaluate for possible sources of poison ivy exposure. Because this type of poison ivy dermatitis is rare, most health care workers likely have never seen black-spot poison ivy, and it is an important diagnosis to consider.13
- Baer RL. Poison ivy dermatitis. Cutis. 1990;46:34-36.
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Hurwitz RM, Rivera HP, Guin JD. Black-spot poison ivy dermatitis. an acute irritant contact dermatitis superimposed upon an allergic contact dermatitis. Am J Dermatopathol. 1984;6:319-322.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Mallory SB, Hurwitz RM. Black-spot poison-ivy dermatitis. Clin Dermatol. 1986;4:149-151.
- Mallory SB, Miller OF, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Rietschel R, Fowler J. Toxicodendron plants and species. Fisher's Contact Dermatitis. 4th ed. Baltimore, MD: Williams & Wilkins; 1995:469-472.
- Fisher AA. Poison ivy/oak dermatitis. part I: prevention--soap and water, topical barriers, hyposensitization. Cutis. 1996;57:384-386.
- McClanahan C, Asarch A, Swick BL. Black spot poison ivy. Int J Dermatol. 2014;53:752-753.
- Mu EW, Capell BC, Castelo-Soccio L. Black spots on a toddler's skin. Contemp Pediatr. 2013;30:31-32.
- Schram SE, Willey A, Lee PK, et al. Black-spot poison ivy. Dermatitis. 2008;19:48-51.
- Paniagua CT, Bean AS. Black-spot poison ivy: a rare phenomenon. J Am Acad Nurse Pract. 2011;23:275-277.
- Baer RL. Poison ivy dermatitis. Cutis. 1990;46:34-36.
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Hurwitz RM, Rivera HP, Guin JD. Black-spot poison ivy dermatitis. an acute irritant contact dermatitis superimposed upon an allergic contact dermatitis. Am J Dermatopathol. 1984;6:319-322.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Mallory SB, Hurwitz RM. Black-spot poison-ivy dermatitis. Clin Dermatol. 1986;4:149-151.
- Mallory SB, Miller OF, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Rietschel R, Fowler J. Toxicodendron plants and species. Fisher's Contact Dermatitis. 4th ed. Baltimore, MD: Williams & Wilkins; 1995:469-472.
- Fisher AA. Poison ivy/oak dermatitis. part I: prevention--soap and water, topical barriers, hyposensitization. Cutis. 1996;57:384-386.
- McClanahan C, Asarch A, Swick BL. Black spot poison ivy. Int J Dermatol. 2014;53:752-753.
- Mu EW, Capell BC, Castelo-Soccio L. Black spots on a toddler's skin. Contemp Pediatr. 2013;30:31-32.
- Schram SE, Willey A, Lee PK, et al. Black-spot poison ivy. Dermatitis. 2008;19:48-51.
- Paniagua CT, Bean AS. Black-spot poison ivy: a rare phenomenon. J Am Acad Nurse Pract. 2011;23:275-277.
A 17-year-old adolescent boy presented to urgent care with a pruritic eruption on the bilateral arms of 1 day's duration. He was camping in the woods the night prior to presentation. On physical examination linear, erythematous, edematous plaques were observed bilaterally with overlying brown and black pigment on the arms. The pigment could not be removed with alcohol or vigorous scrubbing. The patient's condition improved with prednisone.