User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Diffuse Pustular Eruption Following Computed Tomography
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
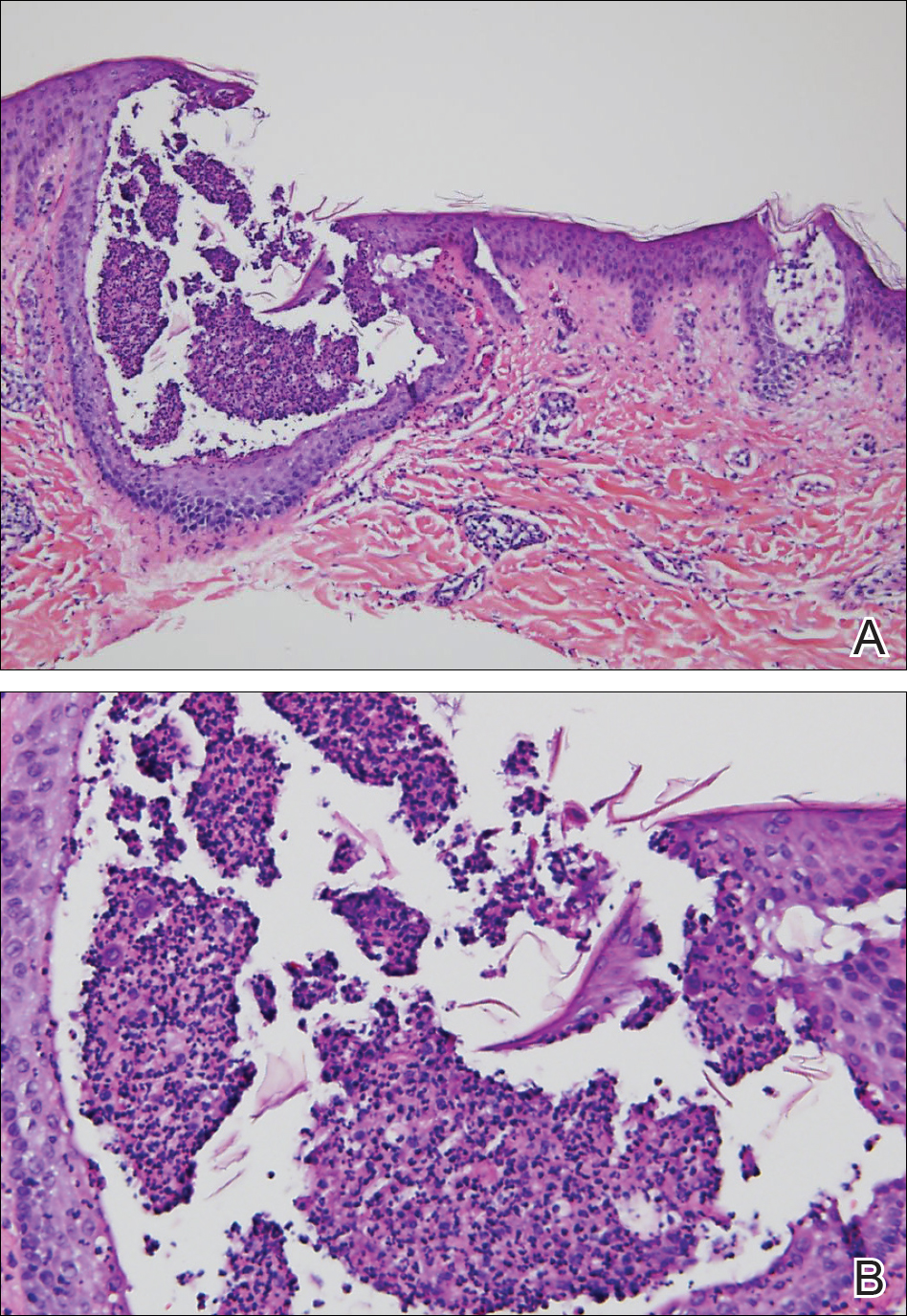
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
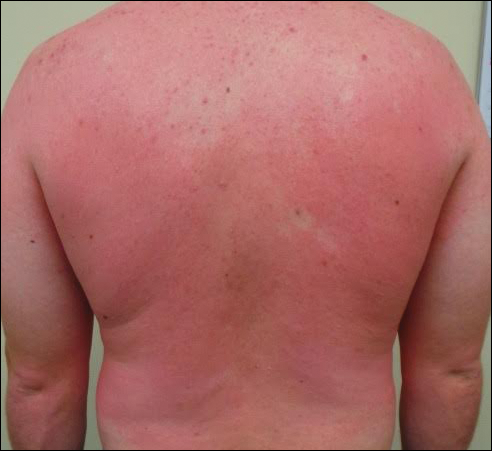
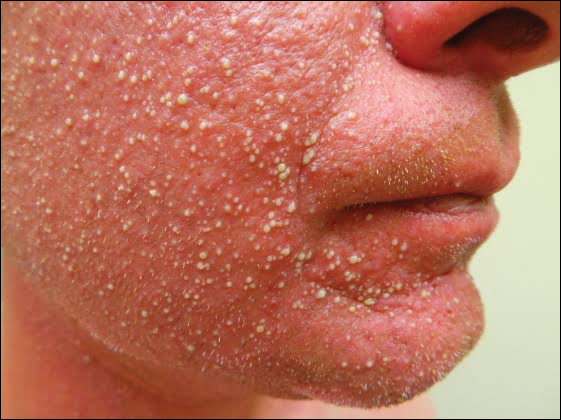
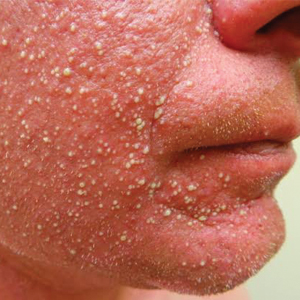
A 31-year-old man presented with a rapidly progressive, burning rash of 1 day's duration, along with malaise, nausea, and dizziness. At the time of presentation, he was hemodynamically stable and afebrile. Laboratory analysis revealed mild leukocytosis with neutrophilia. A complete metabolic panel was within normal limits. He had no chronic medical conditions and was taking no medications or supplements. One day prior to onset of the rash, he underwent contrast-enhanced (iopamidol) computed tomography of the abdomen. Physical examination revealed large edematous plaques on the face, neck, and trunk (top) that were studded with numerous pinpoint pustules (bottom). He also had subtle facial edema. There was relative sparing of the flexural sites and no involvement of the palms, soles, or mucous membranes. A shave biopsy was obtained from a pustular area on the neck.
Google Search Results for Diet and Psoriasis: Advice Patients Get on the Internet
Innovations in Dermatology: Sarecycline Approved for Acne


Inpatient Dermatology: Developing Standards of Care for Hospitalized Patients With Skin Disease


Treatment Options for Pilonidal Sinus
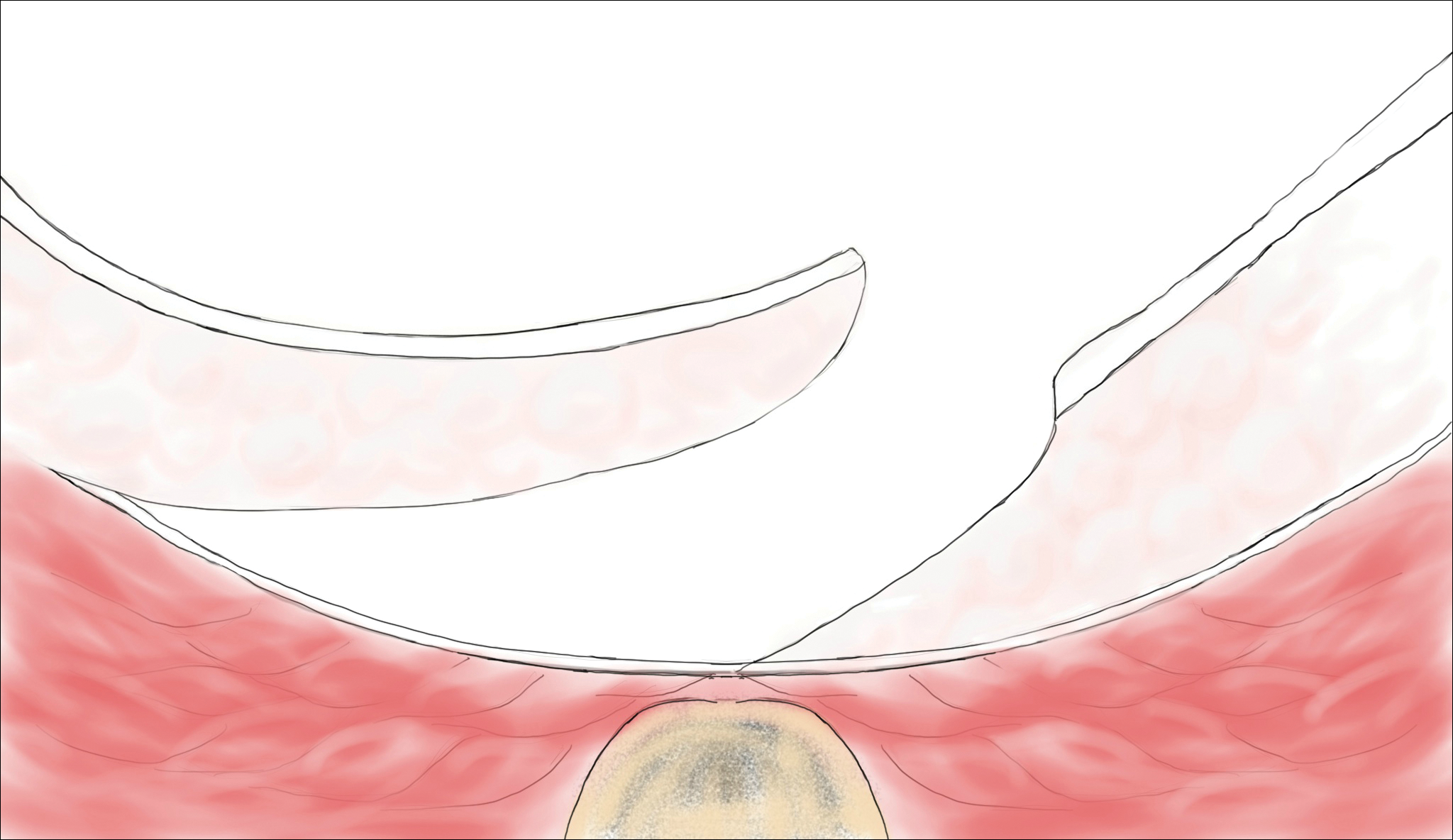
Pilonidal disease was first described by Mayo1 in 1833 who hypothesized that the underlying etiology is incomplete separation of the mesoderm and ectoderm layers during embryogenesis. In 1880, Hodges2 coined the term pilonidal sinus; he postulated that sinus formation was incited by hair.2 Today, Hodges theory is known as the acquired theory: hair induces a foreign body response in surrounding tissue, leading to sinus formation. Although pilonidal cysts can occur anywhere on the body, they most commonly extend cephalad in the sacrococcygeal and upper gluteal cleft (Figure 1).3,4 An acute pilonidal cyst typically presents with pain, tenderness, and swelling, similar to the presentation of a superficial abscess in other locations; however, a clue to the diagnosis is the presence of cutaneous pits along the midline of the gluteal cleft.5 Chronic pilonidal disease varies based on the extent of inflammation and scarring; the underlying cavity communicates with the overlying skin through sinuses and often drains with pressure.6
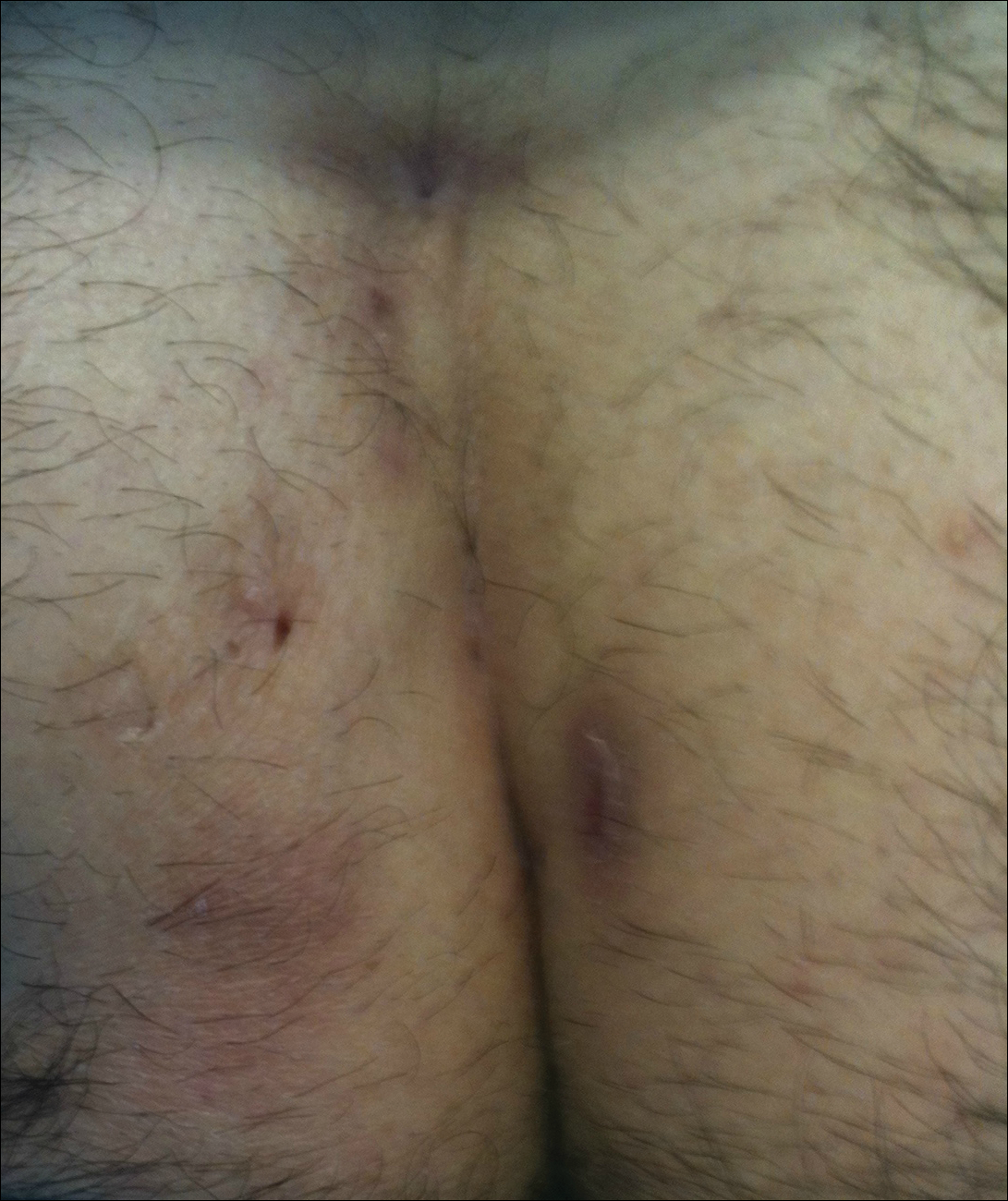
Pilonidal sinuses are rare before puberty or after 40 years of age7 and occur primarily in hirsute men. The ratio of men to women affected is between 3:1 and 4:1.8 Although pilonidal sinuses account for only 15% of anal suppurations, complications arising from pilonidal sinuses are a considerable cause of morbidity, resulting in loss of productivity in otherwise healthy individuals.9 Complications include chronic nonhealing wounds,10 as recurrent pilonidal sinuses tend to become colonized with gram-positive and facultative anaerobic bacteria, whereas primary pilonidal cysts more commonly become infected with anaerobic and gram-negative bacteria.11 Long-standing disease increases the risk of squamous cell carcinoma arising within sinus tracts.10,12
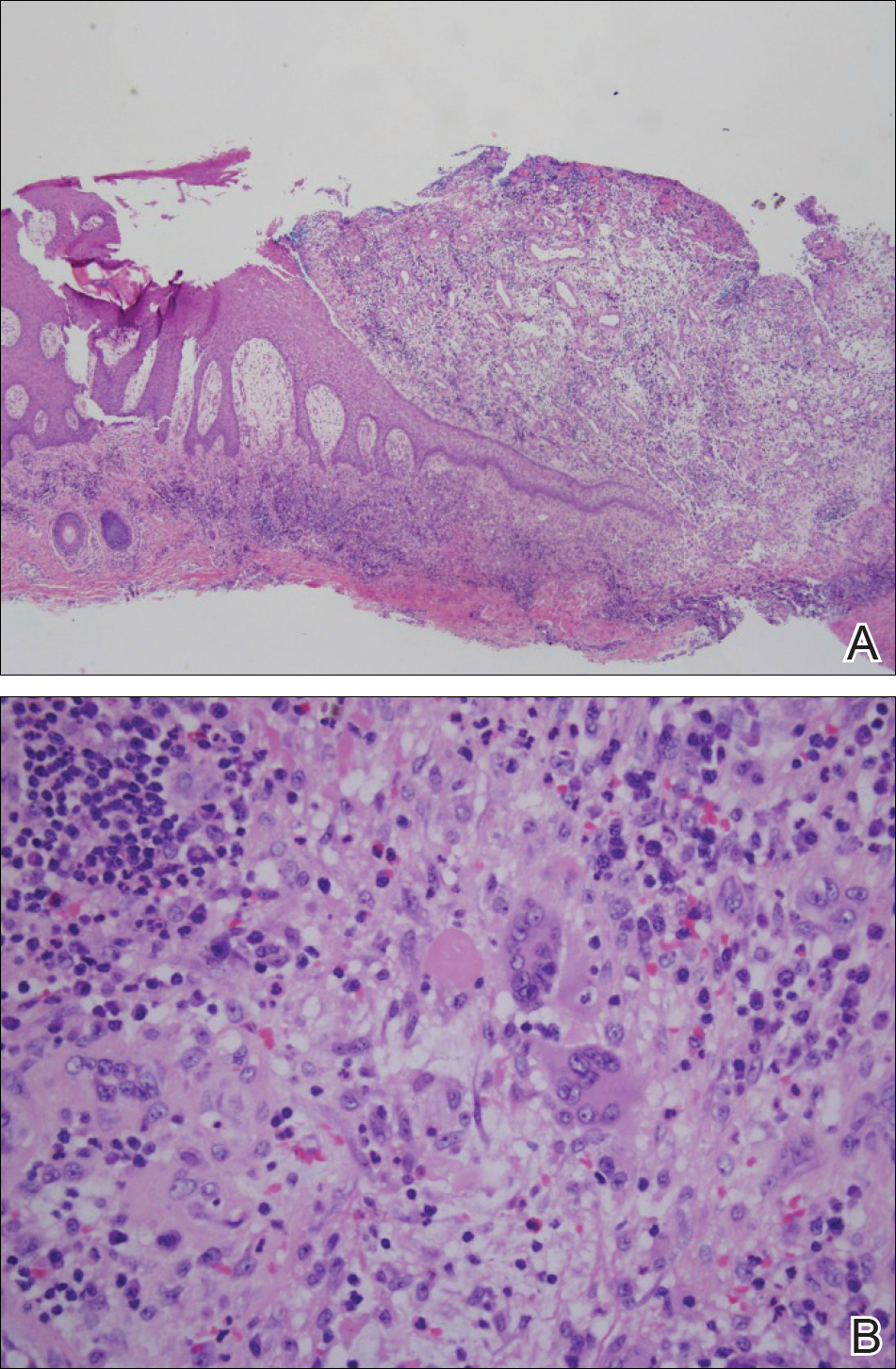
Histopathologically, pilonidal cysts are not true cysts because they lack an epithelial lining. Examination of the cavity commonly reveals hair, debris, and granulation tissue with surrounding foreign-body giant cells (Figure 2).5
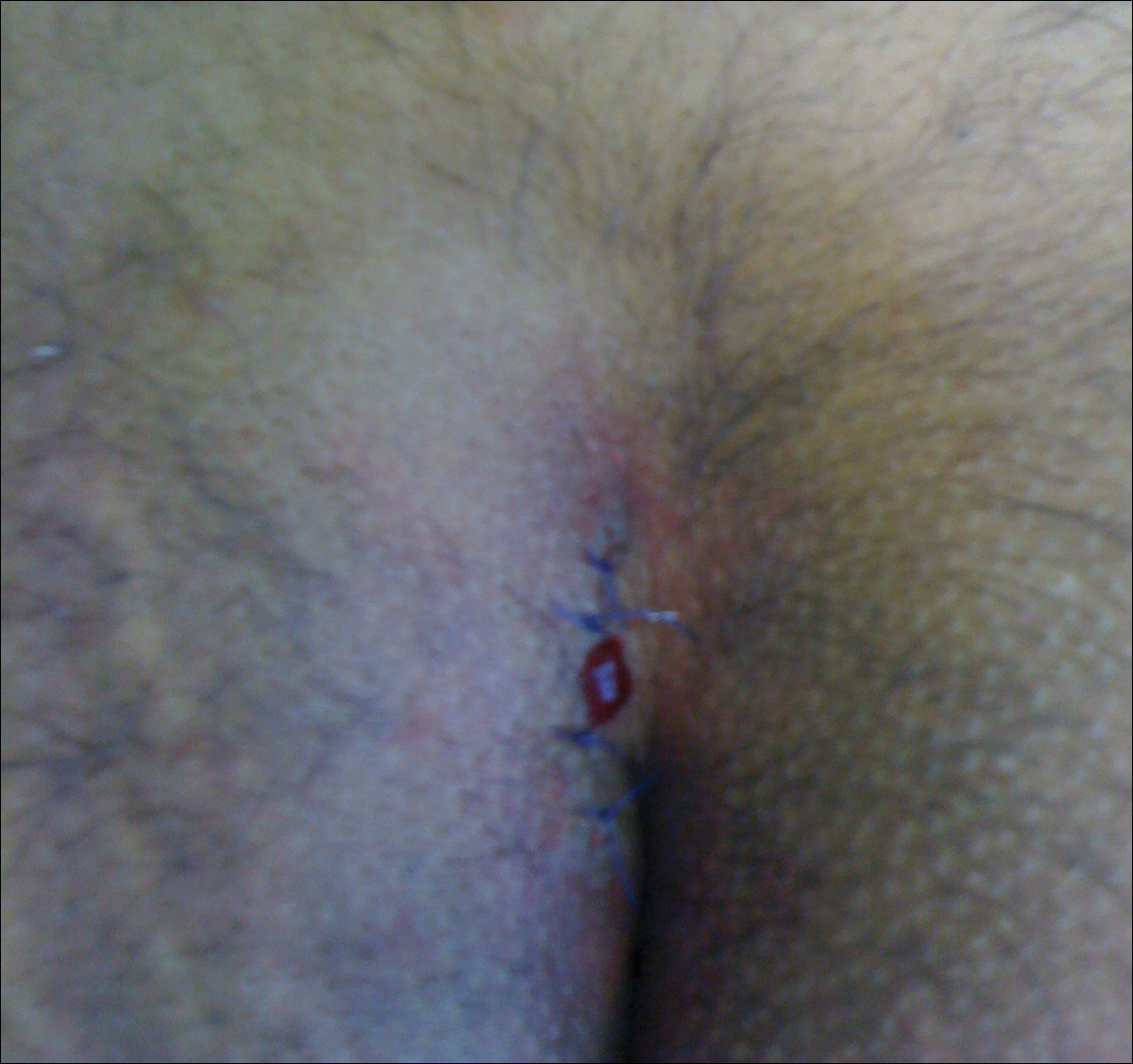
The preferred treatment of pilonidal cysts continues to be debated. In this article, we review evidence supporting current modalities including conservative and surgical techniques as well as novel laser therapy for the treatment of pilonidal disease.
Conservative Management Techniques
Phenol Injections
Liquid or crystallized phenol injections have been used for treatment of mild to moderate pilonidal cysts.13 Excess debris is removed by curettage, and phenol is administered through the existing orifices or pits without pressure. The phenol remains in the cavity for 1 to 3 minutes before aspiration. Remaining cyst contents are removed through tissue manipulation, and the sinus is washed with saline. Mean healing time is 20 days (range, +/−14 days).13
Classically, phenol injections have a failure rate of 30% to 40%, especially with multiple sinuses and suppurative disease6; however, the success rate improves with limited disease (ie, no more than 1–3 sinus pits).3 With multiple treatment sessions, a recurrence rate as low as 2% over 25 months has been reported.14 Phenol injection also has been proposed as an adjuvant therapy to pit excision to minimize the need for extensive surgery.15
Simple Incision and Drainage
Simple incision and drainage has a crucial role in the treatment of acute pilonidal disease to decrease pain and relieve tension. Off-midline incisions have been recommended for because the resulting closures fared better against sheer forces applied by the gluteal muscles on the cleft.6 Therefore, the incision often is made off-midline from the gluteal cleft even when the cyst lies directly on the gluteal cleft.
Rates of healing vary widely after incision and drainage, ranging from 45% to 82%.6 Primary pilonidal cysts may respond well, particularly if the cavity is abraded; in one series, 79% (58/73) of patients did not have a recurrence at the average follow-up of 60 months.16
Excision and Unroofing
Techniques for excision and unroofing without primary closure include 2 variants: wide and limited. The wide technique consists of an inwardly slanted excision that is deepest in the center of the cavity. The inward sloping angle of the incision aids in healing because it allows granulation to progress evenly from the base of the wound upward. The depth of the incision should spare the fascia and leave as much fatty tissue as possible while still resecting the entire cavity and associated pits.6 Limited incision techniques aim to shorten the healing period by making smaller incisions into the sinuses, pits, and secondary tracts, and they are frequently supplemented with curettage.6 Noteworthy disadvantages include prolonged healing time, need for professional wound management, and extended medical observation.5 The average duration of wound healing in a study of 300 patients was 5.4 weeks (range, +/−1.1 weeks),17 and the recurrence rate has ranged from 5% to 13%.18,19 Care must be taken to respond to numerous possible complications, including excessive exudation and granulation, superinfection, and walling off.6
Although the cost of treatment varies by hospital, location, and a patient’s insurance coverage, patient reports to the Pilonidal Support Alliance indicate that the cost of conservative management ranges from $500 to $2000.20
Excision and Primary Closure
An elliptical excision that includes some of the lateral margin is excised down to the level of the fascia. Adjacent lateral tracts may be excised by expanding the incision. To close the wound, edges are approximated with placement of deep and superficial sutures. Wound healing typically occurs faster than secondary granulation, as seen in one randomized controlled trial with a mean of 10 days for primary closure compared to 13 weeks for secondary intention.21 However, as with any surgical procedure, postoperative complications can delay wound healing.19 The recurrence rate after primary closure varies considerably, ranging from 10% to 38%.18,21-23 The average cost of an excision ranges from $3000 to $6000.20
A
Surgical Techniques
For severe or recurrent pilonidal disease, skin flaps often are required. Several flaps have been developed, including advancement, Bascom cleft lift, Karydakis, and modified Limberg flap. Flaps require a vascular pedicle but allow for closure without tension.26 The cost of a flap procedure, ranging from $10,000 to $30,000, is greater than the cost of excision or other conservative therapy20; however, with a lower recurrence rate of pilonidal disease following flap procedures compared to other treatments, patients may save more on treatment over the long-term.
Advancement Flaps
The most commonly used advancement flaps are the V-Y advancement flap and Z-plasty. The V-Y advancement flap creates a full-thickness V-shaped incision down to gluteal fascia that is closed to form a postrepair suture line in the shape of a Y.5 Depending on the size of the defect, the flaps may be utilized unilaterally or bilaterally. A defect as large as 8 to 10 cm can be covered unilaterally; however, defects larger than 10 cm commonly require a bilateral flap.26 The V-Y advancement flap failed to show superiority to primary closure techniques based on complications, recurrence, and patient satisfaction in a large randomized controlled trial.27
Performing a Z-plasty requires excision of diseased tissue with recruitment of lateral flaps incised down to the level of the fascia. The lateral edges are transposed to increase transverse length.26 No statistically significant difference in infection or recurrence rates was noted between excision alone and excision plus Z-plasty; however, wounds were reported to heal faster in patients receiving excision plus Z-plasty (41 vs 15 days).28
Cleft Lift Closure
In 1987, Bascom29 introduced the cleft lift closure for recurrent pilonidal disease. This technique aims to reduce or eliminate lateral gluteal forces on the wounds by filling the gluteal cleft.5 The sinus tracts are excised and a full-thickness skin flap is extended across the cleft and closed off-midline. The adipose tissue fills in the previous space of the gluteal cleft. In the initial study, no recurrences were reported in 30 patients who underwent this procedure at 2-year follow-up; similarly, in another case series of 26 patients who underwent the procedure, no recurrences were noted at a median follow-up of 3 years.30 Compared to excision with secondary wound healing and primary closure on the midline, the Bascom cleft lift demonstrated a decrease in wound healing time (62, 52, and 29 days, respectively).31
The classic Karydakis flap consists of an oblique elliptical excision of diseased tissue with fixation of the flap base to the sacral fascia (Figures 4 and 5). The flap is closed by suturing the edge off-midline.32 This technique prevents a midline wound and aims to remodel and flatten the natal cleft. Karydakis33 performed the most important study for treatment of pilonidal disease with the Karydakis flap, which included more than 5000 patients. The results showed a 0.9% recurrence rate and an 8.5% wound complication rate over a 2- to 20-year follow-up.33 These results have been substantiated by more recent studies, which produced similar results: a 1.8% to 5.3% infection rate and a recurrence rate of 0.9% to 4.4%.34,35
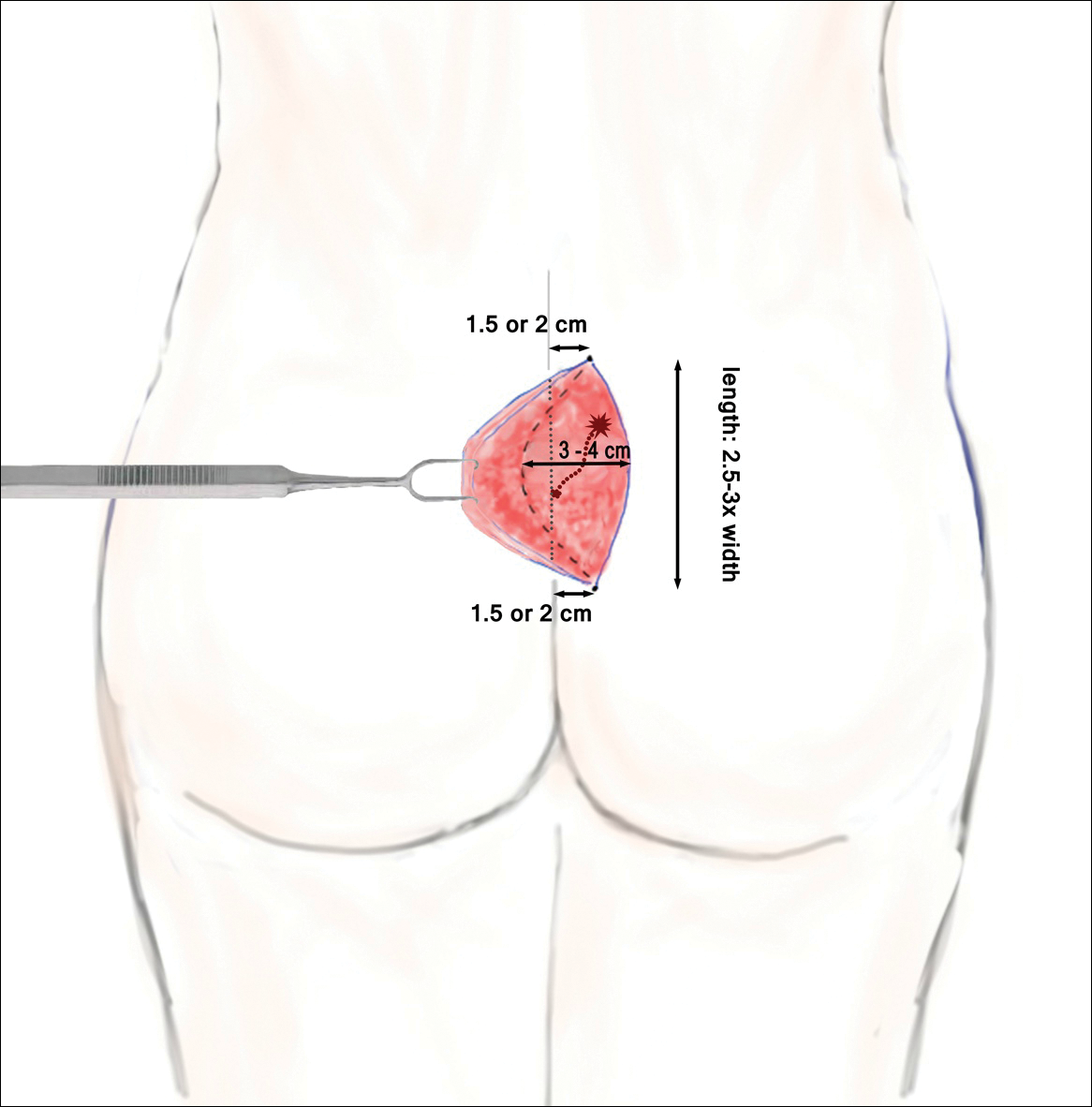
In the modified Karydakis flap, the same excision and closure is performed without tacking the flap to the sacral fascia, aiming to prevent formation of a new vulnerable raphe by flattening the natal cleft. The infection rate was similar to the classic Karydakis flap, and no recurrences were noted during a 20-month follow-up.36
Limberg Flap
The Limberg flap is derived from a rhomboid flap. In the classic Limberg flap, a midline rhomboid incision to the presacral fascia including the sinus is performed. The flap gains mobility by extending the excision laterally to the fascia of the gluteus maximus muscle. A variant of the original flap includes the modified Limberg flap, which lateralizes the midline sutures and flattens the intergluteal sulcus. Compared to the traditional Limberg approach, the modified Limberg flap was associated with a lower failure rate at both early and late time points and a lower rate of infection37,38; however, based on the data it is unclear when primary closure should be favored over a Limberg flap. Several studies show the recurrence rate to be identical; however, hospital stay and pain were reduced in the Limberg flap group compared to primary closure.39,40
Results from randomized controlled trials comparing the modified Limberg flap to the Karydakis flap vary. One of the largest prospective, randomized, controlled trials comparing the 2 flaps included 269 patients.Results showed a lower postoperative complication rate, lower pain scores, shorter operation time, and shorter hospital stay with the Karydakis flap compared to the Limberg flap, though no difference in recurrence was noted between the 2 groups.41
Tw
Overall, larger prospective trials are needed to clarify the differences in outcomes between flap techniques. In
Laser Therapy
Lasers are emerging as primary and adjuvant treatment options for pilonidal sinuses. Depilation with alexandrite, diode, and Nd:YAG lasers has demonstrated the most consistent evidence.50-54 Th
Large randomized controlled trials are needed to fully determine the utility of laser therapy as a primary or adjuvant treatment in pilonidal disease; however, given that laser therapies address the core pathogenesis of pilonidal disease and generally are well tolerated, their use may be strongly considered.
Conclusion
With mild pilonidal disease, more conservative measures can be employed; however, in cases of recurrent or suppurative disease or extensive scarring, excision with flap closure typically is required. Although no single surgical procedure has been identified as superior, one review demonstrated that off-midline procedures are statistically superior to midline closure in healing time, surgical site infection, and recurrence rate.24 Novel techniques continue to emerge in the management of pilonidal disease, including laser therapy. This modality shows promise as either a primary or adjuvant treatment; however, large randomized controlled trials are needed to confirm early findings.
Given that pilonidal disease most commonly occurs in the actively employed population, we recommend that dermatologic surgeons discuss treatment options with patients who have pilonidal disease, taking into consideration cost, length of hospital stay, and recovery time when deciding on a treatment course.
- Mayo OH. Observations on Injuries and Diseases of the Rectum. London, England: Burgess and Hill; 1833.
- Hodges RM. Pilonidal sinus. Boston Med Surg J. 1880;103:485-486.
- Eryilmaz R, Okan I, Ozkan OV, et al. Interdigital pilonidal sinus: a case report and literature review. Dermatol Surg. 2012;38:1400-1403.
- Stone MS. Cysts with a lining of stratified epithelium. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Amsterdam, Netherlands: Elsevier Limited; 2012:1917-1929.
- Khanna A, Rombeau JL. Pilonidal disease. Clin Colon Rectal Surg. 2011;24:46-53.
- de Parades V, Bouchard D, Janier M, et al. Pilonidal sinus disease. J Visc Surg. 2013;150:237-247.
- Harris CL, Laforet K, Sibbald RG, et al. Twelve common mistakes in pilonidal sinus care. Adv Skin Wound Care. 2012;25:325-332.
- Lindholt-Jensen C, Lindholt J, Beyer M, et al. Nd-YAG laser treatment of primary and recurrent pilonidal sinus. Lasers Med Sci. 2012;27:505-508.
- Oueidat D, Rizkallah A, Dirani M, et al. 25 years’ experience in the management of pilonidal sinus disease. Open J Gastro. 2014;4:1-5.
- Gordon P, Grant L, Irwin T. Recurrent pilonidal sepsis. Ulster Med J. 2014;83:10-12.
- Ardelt M, Dittmar Y, Kocijan R, et al. Microbiology of the infected recurrent sacrococcygeal pilonidal sinus. Int Wound J. 2016;13:231-237.
- Eryilmaz R, Bilecik T, Okan I, et al. Recurrent squamous cell carcinoma arising in a neglected pilonidal sinus: report of a case and literature review. Int J Clin Exp Med. 2014;7:446-450.
- Kayaalp C, Aydin C. Review of phenol treatment in sacrococcygeal pilonidal disease. Tech Coloproctol. 2009;13:189-193.
- Dag A, Colak T, Turkmenoglu O, et al. Phenol procedure for pilonidal sinus disease and risk factors for treatment failure. Surgery. 2012;151:113-117.
- Olmez A, Kayaalp C, Aydin C. Treatment of pilonidal disease by combination of pit excision and phenol application. Tech Coloproctol. 2013;17:201-206.
- Jensen SL, Harling H. Prognosis after simple incision and drainage for a first-episode acute pilonidal abscess. Br J Surg. 1988;75:60-61.
- Kepenekci I, Demirkan A, Celasin H, et al. Unroofing and curettage for the treatment of acute and chronic pilonidal disease. World J Surg. 2010;34:153-157.
- Søndenaa K, Nesvik I, Anderson E, et al. Recurrent pilonidal sinus after excision with closed or open treatment: final results of a randomized trial. Eur J Surg. 1996;162:237-240.
- Spivak H, Brooks VL, Nussbaum M, et al. Treatment of chronic pilonidal disease. Dis Colon Rectum. 1996;39:1136-1139.
- Pilonidal surgery costs. Pilonidal Support Alliance website. https://www.pilonidal.org/treatments/surgical-costs/. Updated January 30, 2016. Accessed October 14, 2018.21. al-Hassan HK, Francis IM, Neglén P. Primary closure or secondary granulation after excision of pilonidal sinus? Acta Chir Scand. 1990;156:695-699.
- Khaira HS, Brown JH. Excision and primary suture of pilonidal sinus. Ann R Coll Surg Engl. 1995;77:242-244.
- Clothier PR, Haywood IR. The natural history of the post anal (pilonidal) sinus. Ann R Coll Surg Engl. 1984;66:201-203.
- Al-Khamis A, McCallum I, King PM, et al. Healing by primary versus secondary intention after surgical treatment for pilonidal sinus. Cochrane Database Syst Rev. 2010;1:CD006213.
- McCallum I, King PM, Bruce J. Healing by primary closure versus open healing after surgery for pilonidal sinus: systematic review and meta-analysis. BMJ. 2008;336:868-871.
- Lee PJ, Raniga S, Biyani DK, et al. Sacrococcygeal pilonidal disease. Colorect Dis. 2008;10:639-650.
- Nursal TZ, Ezer A, Calişkan K, et al. Prospective randomized controlled trial comparing V-Y advancement flaps with primary suture methods in pilonidal disease. Am J Surg. 2010;199:170-177.
- Fazeli MS, Adel MG, Lebaschi AH. Comparison of outcomes in Z-plasty and delayed healing by secondary intention of the wound after excision in the sacral pilonidal sinus: results of a randomized, clinical trial. Dis Col Rectum. 2006;49:1831-1836.
- Bascom JU. Repeat pilonidal operations. Am J Surg. 1987;154:118-122.
- Nordon IM, Senapati A, Cripps NP. A prospective randomized controlled trial of simple Bascom’s technique versus Bascom’s cleft closure in the treatment of chronic pilonidal disease. Am J Surg. 2009;197:189-192.
- Dudnik R, Veldkamp J, Nienhujis S, et al. Secondary healing versus midline closure and modified Bascom natal cleft lift for pilonidal sinus disease. Scand J Surg. 2011;100:110-113.
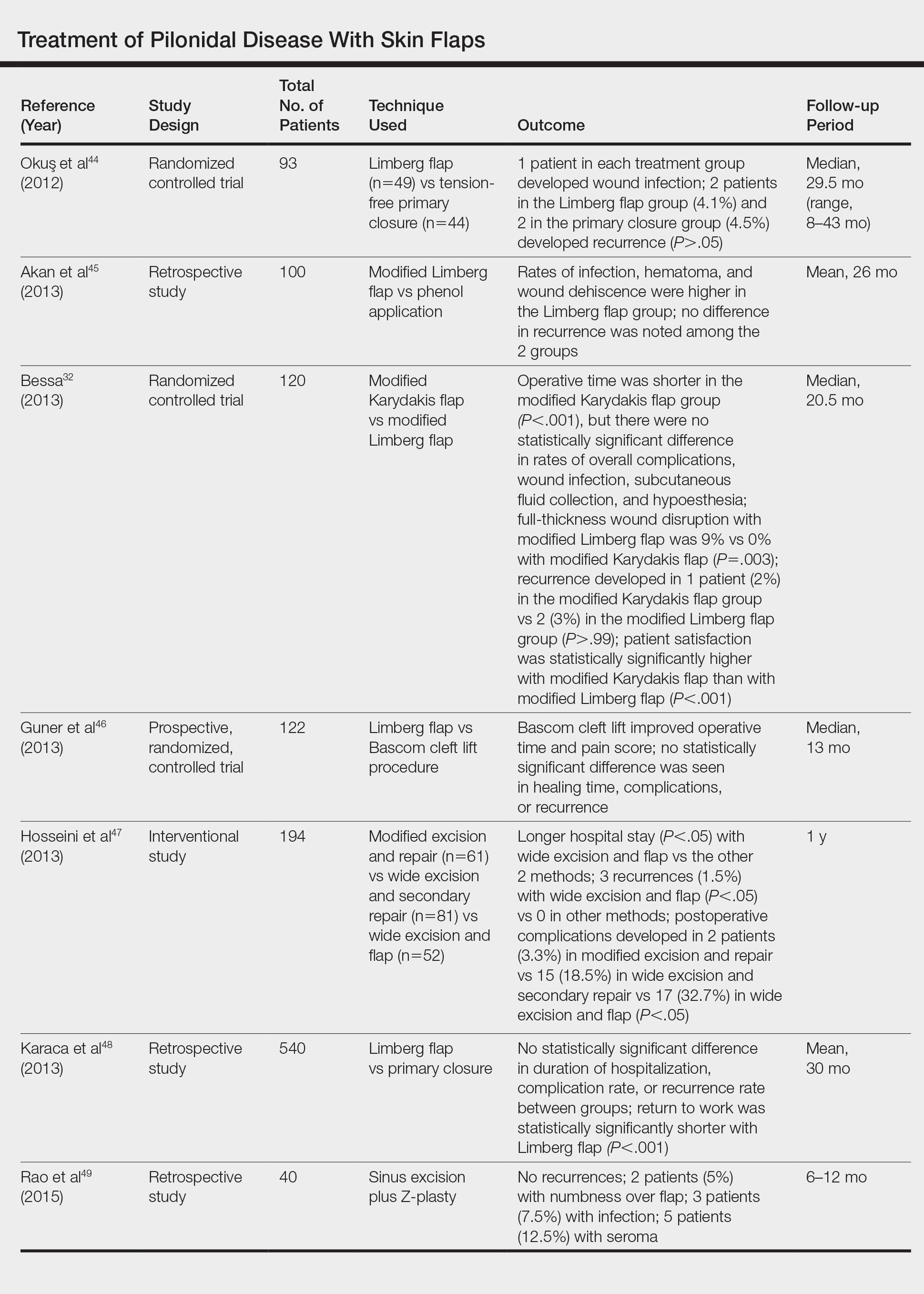
- Bessa SS. Comparison of short-term results between the modified Karydakis flap and the modified Limberg flap in the management of pilonidal sinus disease: a randomized controlled study. Dis Colon Rectum. 2013;56:491-498.
- Karydakis GE. Easy and successful treatment of pilonidal sinus after explanation of its causative process. Aust N Z J Surg. 1992;62:385-389.
- Kitchen PR. Pilonidal sinus: excision and primary closure with a lateralised wound - the Karydakis operation. Aust N Z J Surg. 1982;52:302-305.
- Akinci OF, Coskun A, Uzunköy A. Simple and effective surgical treatment of pilonidal sinus: asymmetric excision and primary closure using suction drain and subcuticular skin closure. Dis Colon Rectum. 2000;43:701-706.
- Bessa SS. Results of the lateral advancing flap operation (modified Karydakis procedure) for the management of pilonidal sinus disease. Dis Colon Rectum. 2007;50:1935-1940.
- Mentes BB, Leventoglu S, Chin A, et al. Modified Limberg transposition flap for sacrococcygeal pilonidal sinus. Surg Today. 2004;34:419-423.
- Cihan A, Ucan BH, Comert M, et al. Superiority of asymmetric modified Limberg flap for surgical treatment of pilonidal cyst disease. Dis Colon Rectum. 2006;49:244-249.
- Muzi MG, Milito G, Cadeddu F, et al. Randomized comparison of Limberg flap versus modified primary closure for treatment of pilonidal disease. Am J Surg. 2010;200:9-14.
- Tavassoli A, Noorshafiee S, Nazarzadeh R. Comparison of excision with primary repair versus Limberg flap. Int J Surg. 2011;9:343-346.
- Ates M, Dirican A, Sarac M, et al. Short and long-term results of the Karydakis flap versus the Limberg flap for treating pilonidal sinus disease: a prospective randomized study. Am J Surg. 2011;202:568-573.
- Can MF, Sevinc MM, Hancerliogullari O, et al. Multicenter prospective randomized trial comparing modified Limberg flap transposition and Karydakis flap reconstruction in patients with saccrococcygeal pilonidal disease. Am J Surg. 2010;200:318-327.
- Ersoy E, Devay AO, Aktimur R, et al. Comparison of short-term results after Limberg and Karydakis procedures for pilonidal disease: randomized prospective analysis of 100 patients. Colorectal Dis. 2009;11:705-710.
- Okuş A, Sevinç B, Karahan O, et al. Comparison of Limberg flap and tension-free primary closure during pilonidal sinus surgery. World J Surg. 2012;36:431-435.
- Akan K, Tihan D, Duman U, et al. Comparison of surgical Limberg flap technique and crystallized phenol application in the treatment of pilonidal sinus disease: a retrospective study. Ulus Cerrahi Derg. 2013;29:162-166.
- Guner A, Boz A, Ozkan OF, et al. Limberg flap versus Bascom cleft lift techniques for sacrococcygeal pilonidal sinus: prospective, randomized trial. World J Surg. 2013;37:2074-2080.
- Hosseini H, Heidari A, Jafarnejad B. Comparison of three surgical methods in treatment of patients with pilonidal sinus: modified excision and repair/wide excision/wide excision and flap in RASOUL, OMID and SADR hospitals (2004-2007). Indian J Surg. 2013;75:395-400.
- Karaca AS, Ali R, Capar M, et al. Comparison of Limberg flap and excision and primary closure of pilonidal sinus disease, in terms of quality of life and complications. J Korean Surg Soc. 2013;85:236-239.
- Rao J, Deora H, Mandia R. A retrospective study of 50 cases of pilonidal sinus with excision of tract and Z-plasty as treatment of choice for both primary and recurrent cases. Indian J Surg. 2015;77(suppl 2):691-693.
- Landa N, Aller O, Landa-Gundin N, et al. Successful treatment of recurrent pilonidal sinus with laser epilation. Dermatol Surg. 2005;31:726-728.
- Oram Y, Kahraman D, Karincaoğlu Y, et al. Evaluation of 60 patients with pilonidal sinus treated with laser epilation after surgery. Dermatol Surg. 2010;36:88-91.
- Benedetto AV, Lewis AT. Pilonidal sinus disease treated by depilation using an 800 nm diode laser and review of the literature. Dermatol Surg. 2005;31:587-591.
- Lindholt-Jensen CS, Lindholt JS, Beyer M, et al. Nd-YAG treatment of primary and recurrent pilonidal sinus. Lasers Med Sci. 2012;27:505-508.
- Jain V, Jain A. Use of lasers for the management of refractory cases of hidradenitis suppurativa and pilonidal sinus. J Cutan Aesthet. 2012;5:190-192.
Pilonidal disease was first described by Mayo1 in 1833 who hypothesized that the underlying etiology is incomplete separation of the mesoderm and ectoderm layers during embryogenesis. In 1880, Hodges2 coined the term pilonidal sinus; he postulated that sinus formation was incited by hair.2 Today, Hodges theory is known as the acquired theory: hair induces a foreign body response in surrounding tissue, leading to sinus formation. Although pilonidal cysts can occur anywhere on the body, they most commonly extend cephalad in the sacrococcygeal and upper gluteal cleft (Figure 1).3,4 An acute pilonidal cyst typically presents with pain, tenderness, and swelling, similar to the presentation of a superficial abscess in other locations; however, a clue to the diagnosis is the presence of cutaneous pits along the midline of the gluteal cleft.5 Chronic pilonidal disease varies based on the extent of inflammation and scarring; the underlying cavity communicates with the overlying skin through sinuses and often drains with pressure.6
Pilonidal sinuses are rare before puberty or after 40 years of age7 and occur primarily in hirsute men. The ratio of men to women affected is between 3:1 and 4:1.8 Although pilonidal sinuses account for only 15% of anal suppurations, complications arising from pilonidal sinuses are a considerable cause of morbidity, resulting in loss of productivity in otherwise healthy individuals.9 Complications include chronic nonhealing wounds,10 as recurrent pilonidal sinuses tend to become colonized with gram-positive and facultative anaerobic bacteria, whereas primary pilonidal cysts more commonly become infected with anaerobic and gram-negative bacteria.11 Long-standing disease increases the risk of squamous cell carcinoma arising within sinus tracts.10,12
Histopathologically, pilonidal cysts are not true cysts because they lack an epithelial lining. Examination of the cavity commonly reveals hair, debris, and granulation tissue with surrounding foreign-body giant cells (Figure 2).5
The preferred treatment of pilonidal cysts continues to be debated. In this article, we review evidence supporting current modalities including conservative and surgical techniques as well as novel laser therapy for the treatment of pilonidal disease.
Conservative Management Techniques
Phenol Injections
Liquid or crystallized phenol injections have been used for treatment of mild to moderate pilonidal cysts.13 Excess debris is removed by curettage, and phenol is administered through the existing orifices or pits without pressure. The phenol remains in the cavity for 1 to 3 minutes before aspiration. Remaining cyst contents are removed through tissue manipulation, and the sinus is washed with saline. Mean healing time is 20 days (range, +/−14 days).13
Classically, phenol injections have a failure rate of 30% to 40%, especially with multiple sinuses and suppurative disease6; however, the success rate improves with limited disease (ie, no more than 1–3 sinus pits).3 With multiple treatment sessions, a recurrence rate as low as 2% over 25 months has been reported.14 Phenol injection also has been proposed as an adjuvant therapy to pit excision to minimize the need for extensive surgery.15
Simple Incision and Drainage
Simple incision and drainage has a crucial role in the treatment of acute pilonidal disease to decrease pain and relieve tension. Off-midline incisions have been recommended for because the resulting closures fared better against sheer forces applied by the gluteal muscles on the cleft.6 Therefore, the incision often is made off-midline from the gluteal cleft even when the cyst lies directly on the gluteal cleft.
Rates of healing vary widely after incision and drainage, ranging from 45% to 82%.6 Primary pilonidal cysts may respond well, particularly if the cavity is abraded; in one series, 79% (58/73) of patients did not have a recurrence at the average follow-up of 60 months.16
Excision and Unroofing
Techniques for excision and unroofing without primary closure include 2 variants: wide and limited. The wide technique consists of an inwardly slanted excision that is deepest in the center of the cavity. The inward sloping angle of the incision aids in healing because it allows granulation to progress evenly from the base of the wound upward. The depth of the incision should spare the fascia and leave as much fatty tissue as possible while still resecting the entire cavity and associated pits.6 Limited incision techniques aim to shorten the healing period by making smaller incisions into the sinuses, pits, and secondary tracts, and they are frequently supplemented with curettage.6 Noteworthy disadvantages include prolonged healing time, need for professional wound management, and extended medical observation.5 The average duration of wound healing in a study of 300 patients was 5.4 weeks (range, +/−1.1 weeks),17 and the recurrence rate has ranged from 5% to 13%.18,19 Care must be taken to respond to numerous possible complications, including excessive exudation and granulation, superinfection, and walling off.6
Although the cost of treatment varies by hospital, location, and a patient’s insurance coverage, patient reports to the Pilonidal Support Alliance indicate that the cost of conservative management ranges from $500 to $2000.20
Excision and Primary Closure
An elliptical excision that includes some of the lateral margin is excised down to the level of the fascia. Adjacent lateral tracts may be excised by expanding the incision. To close the wound, edges are approximated with placement of deep and superficial sutures. Wound healing typically occurs faster than secondary granulation, as seen in one randomized controlled trial with a mean of 10 days for primary closure compared to 13 weeks for secondary intention.21 However, as with any surgical procedure, postoperative complications can delay wound healing.19 The recurrence rate after primary closure varies considerably, ranging from 10% to 38%.18,21-23 The average cost of an excision ranges from $3000 to $6000.20
A
Surgical Techniques
For severe or recurrent pilonidal disease, skin flaps often are required. Several flaps have been developed, including advancement, Bascom cleft lift, Karydakis, and modified Limberg flap. Flaps require a vascular pedicle but allow for closure without tension.26 The cost of a flap procedure, ranging from $10,000 to $30,000, is greater than the cost of excision or other conservative therapy20; however, with a lower recurrence rate of pilonidal disease following flap procedures compared to other treatments, patients may save more on treatment over the long-term.
Advancement Flaps
The most commonly used advancement flaps are the V-Y advancement flap and Z-plasty. The V-Y advancement flap creates a full-thickness V-shaped incision down to gluteal fascia that is closed to form a postrepair suture line in the shape of a Y.5 Depending on the size of the defect, the flaps may be utilized unilaterally or bilaterally. A defect as large as 8 to 10 cm can be covered unilaterally; however, defects larger than 10 cm commonly require a bilateral flap.26 The V-Y advancement flap failed to show superiority to primary closure techniques based on complications, recurrence, and patient satisfaction in a large randomized controlled trial.27
Performing a Z-plasty requires excision of diseased tissue with recruitment of lateral flaps incised down to the level of the fascia. The lateral edges are transposed to increase transverse length.26 No statistically significant difference in infection or recurrence rates was noted between excision alone and excision plus Z-plasty; however, wounds were reported to heal faster in patients receiving excision plus Z-plasty (41 vs 15 days).28
Cleft Lift Closure
In 1987, Bascom29 introduced the cleft lift closure for recurrent pilonidal disease. This technique aims to reduce or eliminate lateral gluteal forces on the wounds by filling the gluteal cleft.5 The sinus tracts are excised and a full-thickness skin flap is extended across the cleft and closed off-midline. The adipose tissue fills in the previous space of the gluteal cleft. In the initial study, no recurrences were reported in 30 patients who underwent this procedure at 2-year follow-up; similarly, in another case series of 26 patients who underwent the procedure, no recurrences were noted at a median follow-up of 3 years.30 Compared to excision with secondary wound healing and primary closure on the midline, the Bascom cleft lift demonstrated a decrease in wound healing time (62, 52, and 29 days, respectively).31
The classic Karydakis flap consists of an oblique elliptical excision of diseased tissue with fixation of the flap base to the sacral fascia (Figures 4 and 5). The flap is closed by suturing the edge off-midline.32 This technique prevents a midline wound and aims to remodel and flatten the natal cleft. Karydakis33 performed the most important study for treatment of pilonidal disease with the Karydakis flap, which included more than 5000 patients. The results showed a 0.9% recurrence rate and an 8.5% wound complication rate over a 2- to 20-year follow-up.33 These results have been substantiated by more recent studies, which produced similar results: a 1.8% to 5.3% infection rate and a recurrence rate of 0.9% to 4.4%.34,35
In the modified Karydakis flap, the same excision and closure is performed without tacking the flap to the sacral fascia, aiming to prevent formation of a new vulnerable raphe by flattening the natal cleft. The infection rate was similar to the classic Karydakis flap, and no recurrences were noted during a 20-month follow-up.36
Limberg Flap
The Limberg flap is derived from a rhomboid flap. In the classic Limberg flap, a midline rhomboid incision to the presacral fascia including the sinus is performed. The flap gains mobility by extending the excision laterally to the fascia of the gluteus maximus muscle. A variant of the original flap includes the modified Limberg flap, which lateralizes the midline sutures and flattens the intergluteal sulcus. Compared to the traditional Limberg approach, the modified Limberg flap was associated with a lower failure rate at both early and late time points and a lower rate of infection37,38; however, based on the data it is unclear when primary closure should be favored over a Limberg flap. Several studies show the recurrence rate to be identical; however, hospital stay and pain were reduced in the Limberg flap group compared to primary closure.39,40
Results from randomized controlled trials comparing the modified Limberg flap to the Karydakis flap vary. One of the largest prospective, randomized, controlled trials comparing the 2 flaps included 269 patients.Results showed a lower postoperative complication rate, lower pain scores, shorter operation time, and shorter hospital stay with the Karydakis flap compared to the Limberg flap, though no difference in recurrence was noted between the 2 groups.41
Tw
Overall, larger prospective trials are needed to clarify the differences in outcomes between flap techniques. In
Laser Therapy
Lasers are emerging as primary and adjuvant treatment options for pilonidal sinuses. Depilation with alexandrite, diode, and Nd:YAG lasers has demonstrated the most consistent evidence.50-54 Th
Large randomized controlled trials are needed to fully determine the utility of laser therapy as a primary or adjuvant treatment in pilonidal disease; however, given that laser therapies address the core pathogenesis of pilonidal disease and generally are well tolerated, their use may be strongly considered.
Conclusion
With mild pilonidal disease, more conservative measures can be employed; however, in cases of recurrent or suppurative disease or extensive scarring, excision with flap closure typically is required. Although no single surgical procedure has been identified as superior, one review demonstrated that off-midline procedures are statistically superior to midline closure in healing time, surgical site infection, and recurrence rate.24 Novel techniques continue to emerge in the management of pilonidal disease, including laser therapy. This modality shows promise as either a primary or adjuvant treatment; however, large randomized controlled trials are needed to confirm early findings.
Given that pilonidal disease most commonly occurs in the actively employed population, we recommend that dermatologic surgeons discuss treatment options with patients who have pilonidal disease, taking into consideration cost, length of hospital stay, and recovery time when deciding on a treatment course.
Pilonidal disease was first described by Mayo1 in 1833 who hypothesized that the underlying etiology is incomplete separation of the mesoderm and ectoderm layers during embryogenesis. In 1880, Hodges2 coined the term pilonidal sinus; he postulated that sinus formation was incited by hair.2 Today, Hodges theory is known as the acquired theory: hair induces a foreign body response in surrounding tissue, leading to sinus formation. Although pilonidal cysts can occur anywhere on the body, they most commonly extend cephalad in the sacrococcygeal and upper gluteal cleft (Figure 1).3,4 An acute pilonidal cyst typically presents with pain, tenderness, and swelling, similar to the presentation of a superficial abscess in other locations; however, a clue to the diagnosis is the presence of cutaneous pits along the midline of the gluteal cleft.5 Chronic pilonidal disease varies based on the extent of inflammation and scarring; the underlying cavity communicates with the overlying skin through sinuses and often drains with pressure.6
Pilonidal sinuses are rare before puberty or after 40 years of age7 and occur primarily in hirsute men. The ratio of men to women affected is between 3:1 and 4:1.8 Although pilonidal sinuses account for only 15% of anal suppurations, complications arising from pilonidal sinuses are a considerable cause of morbidity, resulting in loss of productivity in otherwise healthy individuals.9 Complications include chronic nonhealing wounds,10 as recurrent pilonidal sinuses tend to become colonized with gram-positive and facultative anaerobic bacteria, whereas primary pilonidal cysts more commonly become infected with anaerobic and gram-negative bacteria.11 Long-standing disease increases the risk of squamous cell carcinoma arising within sinus tracts.10,12
Histopathologically, pilonidal cysts are not true cysts because they lack an epithelial lining. Examination of the cavity commonly reveals hair, debris, and granulation tissue with surrounding foreign-body giant cells (Figure 2).5
The preferred treatment of pilonidal cysts continues to be debated. In this article, we review evidence supporting current modalities including conservative and surgical techniques as well as novel laser therapy for the treatment of pilonidal disease.
Conservative Management Techniques
Phenol Injections
Liquid or crystallized phenol injections have been used for treatment of mild to moderate pilonidal cysts.13 Excess debris is removed by curettage, and phenol is administered through the existing orifices or pits without pressure. The phenol remains in the cavity for 1 to 3 minutes before aspiration. Remaining cyst contents are removed through tissue manipulation, and the sinus is washed with saline. Mean healing time is 20 days (range, +/−14 days).13
Classically, phenol injections have a failure rate of 30% to 40%, especially with multiple sinuses and suppurative disease6; however, the success rate improves with limited disease (ie, no more than 1–3 sinus pits).3 With multiple treatment sessions, a recurrence rate as low as 2% over 25 months has been reported.14 Phenol injection also has been proposed as an adjuvant therapy to pit excision to minimize the need for extensive surgery.15
Simple Incision and Drainage
Simple incision and drainage has a crucial role in the treatment of acute pilonidal disease to decrease pain and relieve tension. Off-midline incisions have been recommended for because the resulting closures fared better against sheer forces applied by the gluteal muscles on the cleft.6 Therefore, the incision often is made off-midline from the gluteal cleft even when the cyst lies directly on the gluteal cleft.
Rates of healing vary widely after incision and drainage, ranging from 45% to 82%.6 Primary pilonidal cysts may respond well, particularly if the cavity is abraded; in one series, 79% (58/73) of patients did not have a recurrence at the average follow-up of 60 months.16
Excision and Unroofing
Techniques for excision and unroofing without primary closure include 2 variants: wide and limited. The wide technique consists of an inwardly slanted excision that is deepest in the center of the cavity. The inward sloping angle of the incision aids in healing because it allows granulation to progress evenly from the base of the wound upward. The depth of the incision should spare the fascia and leave as much fatty tissue as possible while still resecting the entire cavity and associated pits.6 Limited incision techniques aim to shorten the healing period by making smaller incisions into the sinuses, pits, and secondary tracts, and they are frequently supplemented with curettage.6 Noteworthy disadvantages include prolonged healing time, need for professional wound management, and extended medical observation.5 The average duration of wound healing in a study of 300 patients was 5.4 weeks (range, +/−1.1 weeks),17 and the recurrence rate has ranged from 5% to 13%.18,19 Care must be taken to respond to numerous possible complications, including excessive exudation and granulation, superinfection, and walling off.6
Although the cost of treatment varies by hospital, location, and a patient’s insurance coverage, patient reports to the Pilonidal Support Alliance indicate that the cost of conservative management ranges from $500 to $2000.20
Excision and Primary Closure
An elliptical excision that includes some of the lateral margin is excised down to the level of the fascia. Adjacent lateral tracts may be excised by expanding the incision. To close the wound, edges are approximated with placement of deep and superficial sutures. Wound healing typically occurs faster than secondary granulation, as seen in one randomized controlled trial with a mean of 10 days for primary closure compared to 13 weeks for secondary intention.21 However, as with any surgical procedure, postoperative complications can delay wound healing.19 The recurrence rate after primary closure varies considerably, ranging from 10% to 38%.18,21-23 The average cost of an excision ranges from $3000 to $6000.20
A
Surgical Techniques
For severe or recurrent pilonidal disease, skin flaps often are required. Several flaps have been developed, including advancement, Bascom cleft lift, Karydakis, and modified Limberg flap. Flaps require a vascular pedicle but allow for closure without tension.26 The cost of a flap procedure, ranging from $10,000 to $30,000, is greater than the cost of excision or other conservative therapy20; however, with a lower recurrence rate of pilonidal disease following flap procedures compared to other treatments, patients may save more on treatment over the long-term.
Advancement Flaps
The most commonly used advancement flaps are the V-Y advancement flap and Z-plasty. The V-Y advancement flap creates a full-thickness V-shaped incision down to gluteal fascia that is closed to form a postrepair suture line in the shape of a Y.5 Depending on the size of the defect, the flaps may be utilized unilaterally or bilaterally. A defect as large as 8 to 10 cm can be covered unilaterally; however, defects larger than 10 cm commonly require a bilateral flap.26 The V-Y advancement flap failed to show superiority to primary closure techniques based on complications, recurrence, and patient satisfaction in a large randomized controlled trial.27
Performing a Z-plasty requires excision of diseased tissue with recruitment of lateral flaps incised down to the level of the fascia. The lateral edges are transposed to increase transverse length.26 No statistically significant difference in infection or recurrence rates was noted between excision alone and excision plus Z-plasty; however, wounds were reported to heal faster in patients receiving excision plus Z-plasty (41 vs 15 days).28
Cleft Lift Closure
In 1987, Bascom29 introduced the cleft lift closure for recurrent pilonidal disease. This technique aims to reduce or eliminate lateral gluteal forces on the wounds by filling the gluteal cleft.5 The sinus tracts are excised and a full-thickness skin flap is extended across the cleft and closed off-midline. The adipose tissue fills in the previous space of the gluteal cleft. In the initial study, no recurrences were reported in 30 patients who underwent this procedure at 2-year follow-up; similarly, in another case series of 26 patients who underwent the procedure, no recurrences were noted at a median follow-up of 3 years.30 Compared to excision with secondary wound healing and primary closure on the midline, the Bascom cleft lift demonstrated a decrease in wound healing time (62, 52, and 29 days, respectively).31
The classic Karydakis flap consists of an oblique elliptical excision of diseased tissue with fixation of the flap base to the sacral fascia (Figures 4 and 5). The flap is closed by suturing the edge off-midline.32 This technique prevents a midline wound and aims to remodel and flatten the natal cleft. Karydakis33 performed the most important study for treatment of pilonidal disease with the Karydakis flap, which included more than 5000 patients. The results showed a 0.9% recurrence rate and an 8.5% wound complication rate over a 2- to 20-year follow-up.33 These results have been substantiated by more recent studies, which produced similar results: a 1.8% to 5.3% infection rate and a recurrence rate of 0.9% to 4.4%.34,35
In the modified Karydakis flap, the same excision and closure is performed without tacking the flap to the sacral fascia, aiming to prevent formation of a new vulnerable raphe by flattening the natal cleft. The infection rate was similar to the classic Karydakis flap, and no recurrences were noted during a 20-month follow-up.36
Limberg Flap
The Limberg flap is derived from a rhomboid flap. In the classic Limberg flap, a midline rhomboid incision to the presacral fascia including the sinus is performed. The flap gains mobility by extending the excision laterally to the fascia of the gluteus maximus muscle. A variant of the original flap includes the modified Limberg flap, which lateralizes the midline sutures and flattens the intergluteal sulcus. Compared to the traditional Limberg approach, the modified Limberg flap was associated with a lower failure rate at both early and late time points and a lower rate of infection37,38; however, based on the data it is unclear when primary closure should be favored over a Limberg flap. Several studies show the recurrence rate to be identical; however, hospital stay and pain were reduced in the Limberg flap group compared to primary closure.39,40
Results from randomized controlled trials comparing the modified Limberg flap to the Karydakis flap vary. One of the largest prospective, randomized, controlled trials comparing the 2 flaps included 269 patients.Results showed a lower postoperative complication rate, lower pain scores, shorter operation time, and shorter hospital stay with the Karydakis flap compared to the Limberg flap, though no difference in recurrence was noted between the 2 groups.41
Tw
Overall, larger prospective trials are needed to clarify the differences in outcomes between flap techniques. In
Laser Therapy
Lasers are emerging as primary and adjuvant treatment options for pilonidal sinuses. Depilation with alexandrite, diode, and Nd:YAG lasers has demonstrated the most consistent evidence.50-54 Th
Large randomized controlled trials are needed to fully determine the utility of laser therapy as a primary or adjuvant treatment in pilonidal disease; however, given that laser therapies address the core pathogenesis of pilonidal disease and generally are well tolerated, their use may be strongly considered.
Conclusion
With mild pilonidal disease, more conservative measures can be employed; however, in cases of recurrent or suppurative disease or extensive scarring, excision with flap closure typically is required. Although no single surgical procedure has been identified as superior, one review demonstrated that off-midline procedures are statistically superior to midline closure in healing time, surgical site infection, and recurrence rate.24 Novel techniques continue to emerge in the management of pilonidal disease, including laser therapy. This modality shows promise as either a primary or adjuvant treatment; however, large randomized controlled trials are needed to confirm early findings.
Given that pilonidal disease most commonly occurs in the actively employed population, we recommend that dermatologic surgeons discuss treatment options with patients who have pilonidal disease, taking into consideration cost, length of hospital stay, and recovery time when deciding on a treatment course.
- Mayo OH. Observations on Injuries and Diseases of the Rectum. London, England: Burgess and Hill; 1833.
- Hodges RM. Pilonidal sinus. Boston Med Surg J. 1880;103:485-486.
- Eryilmaz R, Okan I, Ozkan OV, et al. Interdigital pilonidal sinus: a case report and literature review. Dermatol Surg. 2012;38:1400-1403.
- Stone MS. Cysts with a lining of stratified epithelium. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Amsterdam, Netherlands: Elsevier Limited; 2012:1917-1929.
- Khanna A, Rombeau JL. Pilonidal disease. Clin Colon Rectal Surg. 2011;24:46-53.
- de Parades V, Bouchard D, Janier M, et al. Pilonidal sinus disease. J Visc Surg. 2013;150:237-247.
- Harris CL, Laforet K, Sibbald RG, et al. Twelve common mistakes in pilonidal sinus care. Adv Skin Wound Care. 2012;25:325-332.
- Lindholt-Jensen C, Lindholt J, Beyer M, et al. Nd-YAG laser treatment of primary and recurrent pilonidal sinus. Lasers Med Sci. 2012;27:505-508.
- Oueidat D, Rizkallah A, Dirani M, et al. 25 years’ experience in the management of pilonidal sinus disease. Open J Gastro. 2014;4:1-5.
- Gordon P, Grant L, Irwin T. Recurrent pilonidal sepsis. Ulster Med J. 2014;83:10-12.
- Ardelt M, Dittmar Y, Kocijan R, et al. Microbiology of the infected recurrent sacrococcygeal pilonidal sinus. Int Wound J. 2016;13:231-237.
- Eryilmaz R, Bilecik T, Okan I, et al. Recurrent squamous cell carcinoma arising in a neglected pilonidal sinus: report of a case and literature review. Int J Clin Exp Med. 2014;7:446-450.
- Kayaalp C, Aydin C. Review of phenol treatment in sacrococcygeal pilonidal disease. Tech Coloproctol. 2009;13:189-193.
- Dag A, Colak T, Turkmenoglu O, et al. Phenol procedure for pilonidal sinus disease and risk factors for treatment failure. Surgery. 2012;151:113-117.
- Olmez A, Kayaalp C, Aydin C. Treatment of pilonidal disease by combination of pit excision and phenol application. Tech Coloproctol. 2013;17:201-206.
- Jensen SL, Harling H. Prognosis after simple incision and drainage for a first-episode acute pilonidal abscess. Br J Surg. 1988;75:60-61.
- Kepenekci I, Demirkan A, Celasin H, et al. Unroofing and curettage for the treatment of acute and chronic pilonidal disease. World J Surg. 2010;34:153-157.
- Søndenaa K, Nesvik I, Anderson E, et al. Recurrent pilonidal sinus after excision with closed or open treatment: final results of a randomized trial. Eur J Surg. 1996;162:237-240.
- Spivak H, Brooks VL, Nussbaum M, et al. Treatment of chronic pilonidal disease. Dis Colon Rectum. 1996;39:1136-1139.
- Pilonidal surgery costs. Pilonidal Support Alliance website. https://www.pilonidal.org/treatments/surgical-costs/. Updated January 30, 2016. Accessed October 14, 2018.21. al-Hassan HK, Francis IM, Neglén P. Primary closure or secondary granulation after excision of pilonidal sinus? Acta Chir Scand. 1990;156:695-699.
- Khaira HS, Brown JH. Excision and primary suture of pilonidal sinus. Ann R Coll Surg Engl. 1995;77:242-244.
- Clothier PR, Haywood IR. The natural history of the post anal (pilonidal) sinus. Ann R Coll Surg Engl. 1984;66:201-203.
- Al-Khamis A, McCallum I, King PM, et al. Healing by primary versus secondary intention after surgical treatment for pilonidal sinus. Cochrane Database Syst Rev. 2010;1:CD006213.
- McCallum I, King PM, Bruce J. Healing by primary closure versus open healing after surgery for pilonidal sinus: systematic review and meta-analysis. BMJ. 2008;336:868-871.
- Lee PJ, Raniga S, Biyani DK, et al. Sacrococcygeal pilonidal disease. Colorect Dis. 2008;10:639-650.
- Nursal TZ, Ezer A, Calişkan K, et al. Prospective randomized controlled trial comparing V-Y advancement flaps with primary suture methods in pilonidal disease. Am J Surg. 2010;199:170-177.
- Fazeli MS, Adel MG, Lebaschi AH. Comparison of outcomes in Z-plasty and delayed healing by secondary intention of the wound after excision in the sacral pilonidal sinus: results of a randomized, clinical trial. Dis Col Rectum. 2006;49:1831-1836.
- Bascom JU. Repeat pilonidal operations. Am J Surg. 1987;154:118-122.
- Nordon IM, Senapati A, Cripps NP. A prospective randomized controlled trial of simple Bascom’s technique versus Bascom’s cleft closure in the treatment of chronic pilonidal disease. Am J Surg. 2009;197:189-192.
- Dudnik R, Veldkamp J, Nienhujis S, et al. Secondary healing versus midline closure and modified Bascom natal cleft lift for pilonidal sinus disease. Scand J Surg. 2011;100:110-113.
- Bessa SS. Comparison of short-term results between the modified Karydakis flap and the modified Limberg flap in the management of pilonidal sinus disease: a randomized controlled study. Dis Colon Rectum. 2013;56:491-498.
- Karydakis GE. Easy and successful treatment of pilonidal sinus after explanation of its causative process. Aust N Z J Surg. 1992;62:385-389.
- Kitchen PR. Pilonidal sinus: excision and primary closure with a lateralised wound - the Karydakis operation. Aust N Z J Surg. 1982;52:302-305.
- Akinci OF, Coskun A, Uzunköy A. Simple and effective surgical treatment of pilonidal sinus: asymmetric excision and primary closure using suction drain and subcuticular skin closure. Dis Colon Rectum. 2000;43:701-706.
- Bessa SS. Results of the lateral advancing flap operation (modified Karydakis procedure) for the management of pilonidal sinus disease. Dis Colon Rectum. 2007;50:1935-1940.
- Mentes BB, Leventoglu S, Chin A, et al. Modified Limberg transposition flap for sacrococcygeal pilonidal sinus. Surg Today. 2004;34:419-423.
- Cihan A, Ucan BH, Comert M, et al. Superiority of asymmetric modified Limberg flap for surgical treatment of pilonidal cyst disease. Dis Colon Rectum. 2006;49:244-249.
- Muzi MG, Milito G, Cadeddu F, et al. Randomized comparison of Limberg flap versus modified primary closure for treatment of pilonidal disease. Am J Surg. 2010;200:9-14.
- Tavassoli A, Noorshafiee S, Nazarzadeh R. Comparison of excision with primary repair versus Limberg flap. Int J Surg. 2011;9:343-346.
- Ates M, Dirican A, Sarac M, et al. Short and long-term results of the Karydakis flap versus the Limberg flap for treating pilonidal sinus disease: a prospective randomized study. Am J Surg. 2011;202:568-573.
- Can MF, Sevinc MM, Hancerliogullari O, et al. Multicenter prospective randomized trial comparing modified Limberg flap transposition and Karydakis flap reconstruction in patients with saccrococcygeal pilonidal disease. Am J Surg. 2010;200:318-327.
- Ersoy E, Devay AO, Aktimur R, et al. Comparison of short-term results after Limberg and Karydakis procedures for pilonidal disease: randomized prospective analysis of 100 patients. Colorectal Dis. 2009;11:705-710.
- Okuş A, Sevinç B, Karahan O, et al. Comparison of Limberg flap and tension-free primary closure during pilonidal sinus surgery. World J Surg. 2012;36:431-435.
- Akan K, Tihan D, Duman U, et al. Comparison of surgical Limberg flap technique and crystallized phenol application in the treatment of pilonidal sinus disease: a retrospective study. Ulus Cerrahi Derg. 2013;29:162-166.
- Guner A, Boz A, Ozkan OF, et al. Limberg flap versus Bascom cleft lift techniques for sacrococcygeal pilonidal sinus: prospective, randomized trial. World J Surg. 2013;37:2074-2080.
- Hosseini H, Heidari A, Jafarnejad B. Comparison of three surgical methods in treatment of patients with pilonidal sinus: modified excision and repair/wide excision/wide excision and flap in RASOUL, OMID and SADR hospitals (2004-2007). Indian J Surg. 2013;75:395-400.
- Karaca AS, Ali R, Capar M, et al. Comparison of Limberg flap and excision and primary closure of pilonidal sinus disease, in terms of quality of life and complications. J Korean Surg Soc. 2013;85:236-239.
- Rao J, Deora H, Mandia R. A retrospective study of 50 cases of pilonidal sinus with excision of tract and Z-plasty as treatment of choice for both primary and recurrent cases. Indian J Surg. 2015;77(suppl 2):691-693.
- Landa N, Aller O, Landa-Gundin N, et al. Successful treatment of recurrent pilonidal sinus with laser epilation. Dermatol Surg. 2005;31:726-728.
- Oram Y, Kahraman D, Karincaoğlu Y, et al. Evaluation of 60 patients with pilonidal sinus treated with laser epilation after surgery. Dermatol Surg. 2010;36:88-91.
- Benedetto AV, Lewis AT. Pilonidal sinus disease treated by depilation using an 800 nm diode laser and review of the literature. Dermatol Surg. 2005;31:587-591.
- Lindholt-Jensen CS, Lindholt JS, Beyer M, et al. Nd-YAG treatment of primary and recurrent pilonidal sinus. Lasers Med Sci. 2012;27:505-508.
- Jain V, Jain A. Use of lasers for the management of refractory cases of hidradenitis suppurativa and pilonidal sinus. J Cutan Aesthet. 2012;5:190-192.
- Mayo OH. Observations on Injuries and Diseases of the Rectum. London, England: Burgess and Hill; 1833.
- Hodges RM. Pilonidal sinus. Boston Med Surg J. 1880;103:485-486.
- Eryilmaz R, Okan I, Ozkan OV, et al. Interdigital pilonidal sinus: a case report and literature review. Dermatol Surg. 2012;38:1400-1403.
- Stone MS. Cysts with a lining of stratified epithelium. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Amsterdam, Netherlands: Elsevier Limited; 2012:1917-1929.
- Khanna A, Rombeau JL. Pilonidal disease. Clin Colon Rectal Surg. 2011;24:46-53.
- de Parades V, Bouchard D, Janier M, et al. Pilonidal sinus disease. J Visc Surg. 2013;150:237-247.
- Harris CL, Laforet K, Sibbald RG, et al. Twelve common mistakes in pilonidal sinus care. Adv Skin Wound Care. 2012;25:325-332.
- Lindholt-Jensen C, Lindholt J, Beyer M, et al. Nd-YAG laser treatment of primary and recurrent pilonidal sinus. Lasers Med Sci. 2012;27:505-508.
- Oueidat D, Rizkallah A, Dirani M, et al. 25 years’ experience in the management of pilonidal sinus disease. Open J Gastro. 2014;4:1-5.
- Gordon P, Grant L, Irwin T. Recurrent pilonidal sepsis. Ulster Med J. 2014;83:10-12.
- Ardelt M, Dittmar Y, Kocijan R, et al. Microbiology of the infected recurrent sacrococcygeal pilonidal sinus. Int Wound J. 2016;13:231-237.
- Eryilmaz R, Bilecik T, Okan I, et al. Recurrent squamous cell carcinoma arising in a neglected pilonidal sinus: report of a case and literature review. Int J Clin Exp Med. 2014;7:446-450.
- Kayaalp C, Aydin C. Review of phenol treatment in sacrococcygeal pilonidal disease. Tech Coloproctol. 2009;13:189-193.
- Dag A, Colak T, Turkmenoglu O, et al. Phenol procedure for pilonidal sinus disease and risk factors for treatment failure. Surgery. 2012;151:113-117.
- Olmez A, Kayaalp C, Aydin C. Treatment of pilonidal disease by combination of pit excision and phenol application. Tech Coloproctol. 2013;17:201-206.
- Jensen SL, Harling H. Prognosis after simple incision and drainage for a first-episode acute pilonidal abscess. Br J Surg. 1988;75:60-61.
- Kepenekci I, Demirkan A, Celasin H, et al. Unroofing and curettage for the treatment of acute and chronic pilonidal disease. World J Surg. 2010;34:153-157.
- Søndenaa K, Nesvik I, Anderson E, et al. Recurrent pilonidal sinus after excision with closed or open treatment: final results of a randomized trial. Eur J Surg. 1996;162:237-240.
- Spivak H, Brooks VL, Nussbaum M, et al. Treatment of chronic pilonidal disease. Dis Colon Rectum. 1996;39:1136-1139.
- Pilonidal surgery costs. Pilonidal Support Alliance website. https://www.pilonidal.org/treatments/surgical-costs/. Updated January 30, 2016. Accessed October 14, 2018.21. al-Hassan HK, Francis IM, Neglén P. Primary closure or secondary granulation after excision of pilonidal sinus? Acta Chir Scand. 1990;156:695-699.
- Khaira HS, Brown JH. Excision and primary suture of pilonidal sinus. Ann R Coll Surg Engl. 1995;77:242-244.
- Clothier PR, Haywood IR. The natural history of the post anal (pilonidal) sinus. Ann R Coll Surg Engl. 1984;66:201-203.
- Al-Khamis A, McCallum I, King PM, et al. Healing by primary versus secondary intention after surgical treatment for pilonidal sinus. Cochrane Database Syst Rev. 2010;1:CD006213.
- McCallum I, King PM, Bruce J. Healing by primary closure versus open healing after surgery for pilonidal sinus: systematic review and meta-analysis. BMJ. 2008;336:868-871.
- Lee PJ, Raniga S, Biyani DK, et al. Sacrococcygeal pilonidal disease. Colorect Dis. 2008;10:639-650.
- Nursal TZ, Ezer A, Calişkan K, et al. Prospective randomized controlled trial comparing V-Y advancement flaps with primary suture methods in pilonidal disease. Am J Surg. 2010;199:170-177.
- Fazeli MS, Adel MG, Lebaschi AH. Comparison of outcomes in Z-plasty and delayed healing by secondary intention of the wound after excision in the sacral pilonidal sinus: results of a randomized, clinical trial. Dis Col Rectum. 2006;49:1831-1836.
- Bascom JU. Repeat pilonidal operations. Am J Surg. 1987;154:118-122.
- Nordon IM, Senapati A, Cripps NP. A prospective randomized controlled trial of simple Bascom’s technique versus Bascom’s cleft closure in the treatment of chronic pilonidal disease. Am J Surg. 2009;197:189-192.
- Dudnik R, Veldkamp J, Nienhujis S, et al. Secondary healing versus midline closure and modified Bascom natal cleft lift for pilonidal sinus disease. Scand J Surg. 2011;100:110-113.
- Bessa SS. Comparison of short-term results between the modified Karydakis flap and the modified Limberg flap in the management of pilonidal sinus disease: a randomized controlled study. Dis Colon Rectum. 2013;56:491-498.
- Karydakis GE. Easy and successful treatment of pilonidal sinus after explanation of its causative process. Aust N Z J Surg. 1992;62:385-389.
- Kitchen PR. Pilonidal sinus: excision and primary closure with a lateralised wound - the Karydakis operation. Aust N Z J Surg. 1982;52:302-305.
- Akinci OF, Coskun A, Uzunköy A. Simple and effective surgical treatment of pilonidal sinus: asymmetric excision and primary closure using suction drain and subcuticular skin closure. Dis Colon Rectum. 2000;43:701-706.
- Bessa SS. Results of the lateral advancing flap operation (modified Karydakis procedure) for the management of pilonidal sinus disease. Dis Colon Rectum. 2007;50:1935-1940.
- Mentes BB, Leventoglu S, Chin A, et al. Modified Limberg transposition flap for sacrococcygeal pilonidal sinus. Surg Today. 2004;34:419-423.
- Cihan A, Ucan BH, Comert M, et al. Superiority of asymmetric modified Limberg flap for surgical treatment of pilonidal cyst disease. Dis Colon Rectum. 2006;49:244-249.
- Muzi MG, Milito G, Cadeddu F, et al. Randomized comparison of Limberg flap versus modified primary closure for treatment of pilonidal disease. Am J Surg. 2010;200:9-14.
- Tavassoli A, Noorshafiee S, Nazarzadeh R. Comparison of excision with primary repair versus Limberg flap. Int J Surg. 2011;9:343-346.
- Ates M, Dirican A, Sarac M, et al. Short and long-term results of the Karydakis flap versus the Limberg flap for treating pilonidal sinus disease: a prospective randomized study. Am J Surg. 2011;202:568-573.
- Can MF, Sevinc MM, Hancerliogullari O, et al. Multicenter prospective randomized trial comparing modified Limberg flap transposition and Karydakis flap reconstruction in patients with saccrococcygeal pilonidal disease. Am J Surg. 2010;200:318-327.
- Ersoy E, Devay AO, Aktimur R, et al. Comparison of short-term results after Limberg and Karydakis procedures for pilonidal disease: randomized prospective analysis of 100 patients. Colorectal Dis. 2009;11:705-710.
- Okuş A, Sevinç B, Karahan O, et al. Comparison of Limberg flap and tension-free primary closure during pilonidal sinus surgery. World J Surg. 2012;36:431-435.
- Akan K, Tihan D, Duman U, et al. Comparison of surgical Limberg flap technique and crystallized phenol application in the treatment of pilonidal sinus disease: a retrospective study. Ulus Cerrahi Derg. 2013;29:162-166.
- Guner A, Boz A, Ozkan OF, et al. Limberg flap versus Bascom cleft lift techniques for sacrococcygeal pilonidal sinus: prospective, randomized trial. World J Surg. 2013;37:2074-2080.
- Hosseini H, Heidari A, Jafarnejad B. Comparison of three surgical methods in treatment of patients with pilonidal sinus: modified excision and repair/wide excision/wide excision and flap in RASOUL, OMID and SADR hospitals (2004-2007). Indian J Surg. 2013;75:395-400.
- Karaca AS, Ali R, Capar M, et al. Comparison of Limberg flap and excision and primary closure of pilonidal sinus disease, in terms of quality of life and complications. J Korean Surg Soc. 2013;85:236-239.
- Rao J, Deora H, Mandia R. A retrospective study of 50 cases of pilonidal sinus with excision of tract and Z-plasty as treatment of choice for both primary and recurrent cases. Indian J Surg. 2015;77(suppl 2):691-693.
- Landa N, Aller O, Landa-Gundin N, et al. Successful treatment of recurrent pilonidal sinus with laser epilation. Dermatol Surg. 2005;31:726-728.
- Oram Y, Kahraman D, Karincaoğlu Y, et al. Evaluation of 60 patients with pilonidal sinus treated with laser epilation after surgery. Dermatol Surg. 2010;36:88-91.
- Benedetto AV, Lewis AT. Pilonidal sinus disease treated by depilation using an 800 nm diode laser and review of the literature. Dermatol Surg. 2005;31:587-591.
- Lindholt-Jensen CS, Lindholt JS, Beyer M, et al. Nd-YAG treatment of primary and recurrent pilonidal sinus. Lasers Med Sci. 2012;27:505-508.
- Jain V, Jain A. Use of lasers for the management of refractory cases of hidradenitis suppurativa and pilonidal sinus. J Cutan Aesthet. 2012;5:190-192.
Practice Points
- Mild pilonidal disease can be treated with conservative measures, including phenol injection and simple excision and drainage. Recurrent disease or the presence of extensive scarring or suppurative disease typically necessitates excision with flap closure.
- Off-midline procedures have been shown to be statistically superior to midline closure with regard to healing time, infection at the surgical site, and rate of recurrence.
- Laser excision holds promise as a primary or adjuvant treatment of pilonidal disease; however, large randomized controlled trials are needed to confirm early findings.
Concomitant Fibrofolliculoma and Trichodiscoma on the Abdomen
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
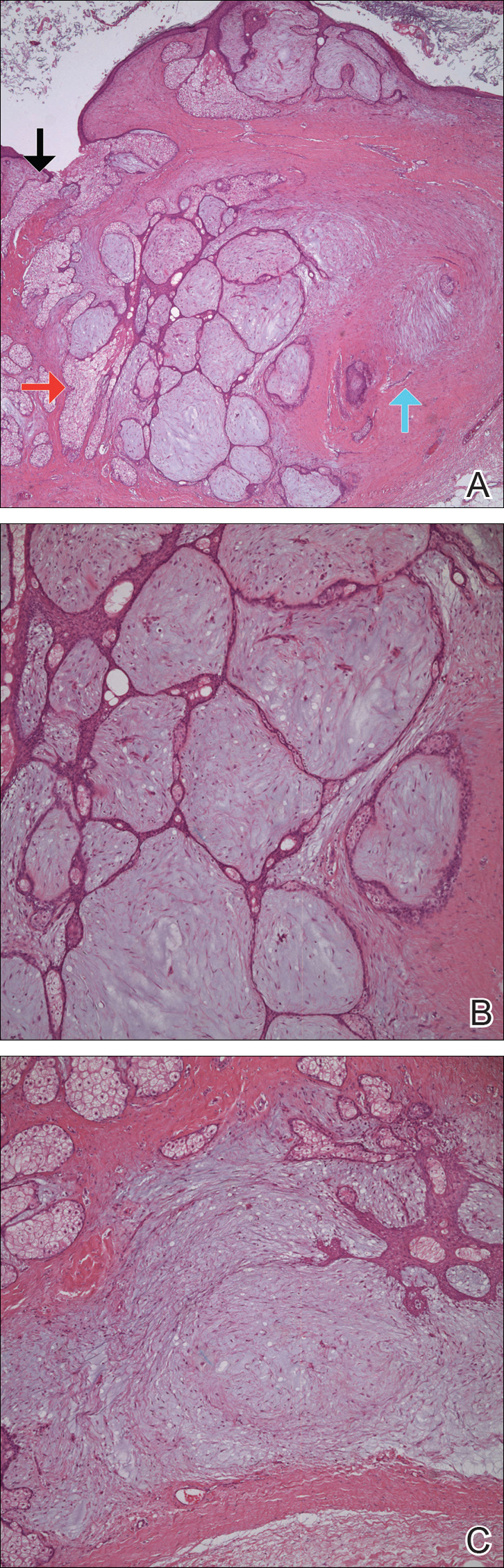
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.

Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
Fibrofolliculomas and trichodiscomas typically present on the head or neck as smooth, flesh-colored, dome-shaped papules. These two entities are considered to constitute two separate time points on a spectrum of histopathologic changes in mantleoma differentiation.1 Histologically, both are benign hamartomas of the pilosebaceous subunit and collectively are known as mantleomas. We present an unusual case of a concomitant fibrofolliculoma and trichodiscoma on the abdomen.
Case Report
An asymptomatic 54-year-old man presented for a routine full-body skin examination. A solitary, 2×1-cm, subcutaneous, doughy, mobile nodule was found on the left side of the abdomen with an overlying 2-mm yellow fleshy papule. The patient declined excision of the lesion, and it was recommended that he return for follow-up 3 months later.
The patient did not present for follow-up until 4.5 years later, at which point the lesion had grown to 3.0×2.5 cm in size. An excision was performed, at which time the lesion was noted to be cystic, extruding an oily, yellow-white liquid. Bacterial culture was negative. Histopathologic sections showed a dome-shaped papule with connection to the overlying epidermis. Epithelial extensions from the infundibular epithelium formed a fenestrated pattern surrounding a fibrous and mucinous stroma (Figure, A and B). The differential diagnosis at this time included an epidermal inclusion cyst, fibroma, intradermal nevus, verruca, hemangioma, angiofibroma, and lipoma.2-4
The same lesion cut in a different plane of sectioning showed an expansile dermal nodule comprising clusters of sebaceous lobules surrounding a fibrous and mucinous stroma. Within the second lesion, fibrous and stromal components predominated over epithelial components (Figure, C). A diagnosis of fibrofolliculoma showing features of a trichodiscoma arising in the unusual location of the abdomen was made.
Comment
Solitary fibrofolliculomas and trichodiscomas are flesh-colored, dome-shaped papules that generally present on the face, specifically on the chin, nose, cheeks, ears, and eyebrows without considerable symptoms.2,4,5 Clinically, fibrofolliculomas are indistinguishable from trichodiscomas but demonstrate different features on biopsy.1,5
Fibrofolliculomas and trichodiscomas are well known for their association with Birt-Hogg-Dubé (BHD) syndrome when they present concomitantly and typically arise earlier in the third decade of life than solitary fibrofolliculomas; however, there have been reports of solitary fibrofolliculomas in patients aged 1 to 36 years.4,6 The triad of BHD syndrome consists of multiple fibrofolliculomas, trichodiscomas, and acrochordons, and it is acquired in an autosomal-dominant manner, unlike solitary fibrofolliculomas, which typically are not inherited. Birt-Hogg-Dubé syndrome is caused by a mutation in the FLCN gene that codes for the tumor-suppressor protein folliculin, which when mutated can cause unregulated proliferation of cells.7 Solitary fibrofolliculomas and the multiple fibrofolliculomas seen in BHD syndrome are histologically similar.
Fibrofolliculoma can be clinically indistinguishable from fibroepithelioma of Pinkus, perifollicular fibroma, trichilemmoma, trichodiscoma, trichoepithelioma, and trichofolliculoma. All typically present clinically as flesh-colored papules,1 although histologic distinction can be made (Table).5,8-13
Fibrofolliculoma is a benign hamartoma that arises from the pilosebaceous follicle and consists of an expansion of the fibrous root sheath, which typically surrounds the hair follicle along with proliferating bands or ribbons of perifollicular connective tissue. As such, the hair follicle may be dilated and filled with keratin in the expanded infundibulum.8 Follicles also may be surrounded by a myxoid stroma.2 In contrast, trichodiscoma is characterized by connective tissue with mature sebaceous lobules in the periphery. It has a myxoid stroma, as opposed to the more fibrous stroma seen in fibrofolliculomas.
Reports have examined the staining patterns of fibrofolliculomas, which show characteristics similar to those of other hair follicle hamartomas, including trichodiscomas.10 The connective tissue and epithelial components that constitute a fibrofolliculoma show different staining patterns. The connective tissue component stains positive for CD34 spindle cells, factor XIIIa, and nestin (a marker of angiogenesis). CD117 (c-kit) expression in the stroma, a marker of fibrocytes, is a feature of both fibrofolliculoma and perifollicular fibromas. The epithelial component, consisting of the hair follicle itself, stains positive for CK15. CK15 expression has been reported in undifferentiated sebocytes of the mantle and in the hair follicle.10 Immunohistochemical staining supports the notion that fibrofolliculomas contain connective tissue and epithelial components and helps to compare and contrast them to those of other hair follicle hamartomas.
Ackerman et al1 considered both fibrofolliculomas and trichodiscomas to be hamartomas of the epithelial hair follicle. The exact etiology of each of these hamartomas is unknown, but the undifferentiated epithelial strands protruding from the hair follicle in a fibrofolliculoma lie in close proximity to sebaceous glands. Furthermore, the authors postulated that fibrofolliculomas and trichodiscomas constitute a spectrum that encompasses the differentiation process of a mantleoma, with fibrofolliculoma representing the beginning of mantleoma differentiation and trichodiscoma representing the end. This end stage of follicular differentiation is one in which there is a predominant stroma and the previously undifferentiated epithelium has formed into sebaceous ducts and lobules in the stroma.1
Most cases of fibrofolliculoma and/or trichodiscoma arise in areas of dense sebaceous follicle concentration (eg, face), further supporting the hypothesis that sebaceous gland proliferation contributes to fibrofolliculoma.14 The case described here, with the fibrofolliculoma arising on the abdomen in conjunction with a trichodiscoma, is therefore worth noting because its location differs from what has been observed in previously reported cases.4
There are both surgical and medical options for treatment of fibrofolliculoma. Although surgical excision is an option for a single lesion, patients with multiple fibrofolliculomas or BHD may prefer removal with the combined CO2 laser and erbium-doped YAG laser.15
Conclusion
We present a rare case of concomitant fibrofolliculoma and trichodiscoma arising on the unusual location of the abdomen. This report highlights the histopathologic features of multiple adnexal tumors and emphasizes the importance of biopsy for differentiating fibrofolliculoma and trichodiscoma.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
- Ackerman AB, Chongchitnant N, DeViragh P. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
- Scully K, Bargman H, Assaad D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11:361-363.
- Chang JK, Lee DC, Chang MH. A solitary fibrofolliculoma in the eyelid. Korean J Ophthalmol. 2007;21:169-171.
- Starink TM, Brownstein MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17:493-496.
- Cho EU, Lee JD, Cho SH. A solitary fibrofolliculoma on the concha of the ear. Int J Dermatol. 2012;51:616-628.
- Mo HJ, Park CK, Yi JY. A case of solitary fibrofolliculoma. Korean J Dermatol. 2001;39:602-604.
- Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell. 2002;2:157-164.
- Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. 1977;113:1674-1677.
- Foucar K, Rosen TH, Foucar E, et al. Fibrofolliculoma: a clinicopathologic study. Cutis. 1981;28:429-432.
- Misago NO, Kimura TE, Narisawa YU. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36:943-951.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol. 2005;53(2 suppl 1):S108-S111.
- Lee Y, Su H, Chen H. Fibroepithelioma of Pinkus. a case report. Dermatologica Sinica. 2002;20:142-146.
- Nam JH, Min JH, Lee GY, et al. A case of perifollicular fibroma. Ann Dermatol. 2011:23:236-238.
- Vernooij M, Claessens T, Luijten M, et al. Birt-Hogg-Dubé syndrome and the skin. Fam Cancer. 2013;12:381-385.
- Jacob CI, Dover JS. Birt-Hogg-Dubé syndrome: treatment of cutaneous manifestations with laser skin resurfacing. Arch Dermatol. 2001;137:98-99.
Practice Points
- Fibrofolliculoma and trichodiscoma are flesh-colored adnexal tumors that arise from or around hair follicles.
- It is important to recognize these entities, as they can be related to Birt-Hogg-Dubé syndrome.
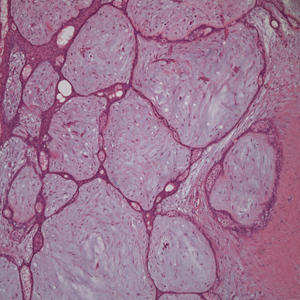
Debunking Psoriasis Myths: Remove Psoriasis Scales Gently
Myth: Pick Psoriasis Scales to Remove Them
Patients may be inclined to pick psoriasis scales that appear in noticeable areas or on the scalp. However, they should be counseled to avoid this practice, which could cause an infection. Instead, Dr. Steven Feldman (Winston-Salem, North Carolina) suggests putting on an ointment or oil-like medication to soften the scale. “Almost any kind of moisturizer will change the reflective properties of the scale so that you don’t see the scale,” he advised. He also suggested descaling agents such as topical salicylic acid or lactic acid. His patient education video is available on the American Academy of Dermatology website should you wish to direct your patients to it.
Because salicylic acid is a keratolytic (or peeling agent), it works by causing the outer layer of skin to shed. When applied topically, it helps to soften and lift psoriasis scales. Coal tar over-the-counter products also can be used for the same purpose. The over-the-counter product guide from the National Psoriasis Foundation is a valuable resource to share with patients.
Expert Commentary
I agree that it is very important to treat scale very gently. In addition to risk for infection, picking and traumatizing scale can lead to worsening of the psoriasis. This is known as the Koebner phenomenon. The phenomenon was first described by Heinrich Koebner in 1876 as the formation of psoriatic lesions in uninvolved skin of patients with psoriasis after cutaneous trauma. This isomorphic phenomenon is now known to involve numerous diseases, among them vitiligo, lichen planus, and Darier disease.
—Jeffrey M. Weinberg, MD (New York, New York)
Feldman S. How should I remove psoriasis scale? American Academy of Dermatology website. https://www.aad.org/public/diseases/scaly-skin/psoriasis/tips-for-managing-psoriasis/how-should-i-remove-psoriasis-scale. Accessed October 31, 2018.
National Psoriasis Foundation. Over-the-counter products. https://www.aad.org/public/diseases/scaly-skin/psoriasis/tips-for-managing-psoriasis/how-should-i-remove-psoriasis-scale. Published June 2017. Accessed October 31, 2018.
Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
Myth: Pick Psoriasis Scales to Remove Them
Patients may be inclined to pick psoriasis scales that appear in noticeable areas or on the scalp. However, they should be counseled to avoid this practice, which could cause an infection. Instead, Dr. Steven Feldman (Winston-Salem, North Carolina) suggests putting on an ointment or oil-like medication to soften the scale. “Almost any kind of moisturizer will change the reflective properties of the scale so that you don’t see the scale,” he advised. He also suggested descaling agents such as topical salicylic acid or lactic acid. His patient education video is available on the American Academy of Dermatology website should you wish to direct your patients to it.
Because salicylic acid is a keratolytic (or peeling agent), it works by causing the outer layer of skin to shed. When applied topically, it helps to soften and lift psoriasis scales. Coal tar over-the-counter products also can be used for the same purpose. The over-the-counter product guide from the National Psoriasis Foundation is a valuable resource to share with patients.
Expert Commentary
I agree that it is very important to treat scale very gently. In addition to risk for infection, picking and traumatizing scale can lead to worsening of the psoriasis. This is known as the Koebner phenomenon. The phenomenon was first described by Heinrich Koebner in 1876 as the formation of psoriatic lesions in uninvolved skin of patients with psoriasis after cutaneous trauma. This isomorphic phenomenon is now known to involve numerous diseases, among them vitiligo, lichen planus, and Darier disease.
—Jeffrey M. Weinberg, MD (New York, New York)
Myth: Pick Psoriasis Scales to Remove Them
Patients may be inclined to pick psoriasis scales that appear in noticeable areas or on the scalp. However, they should be counseled to avoid this practice, which could cause an infection. Instead, Dr. Steven Feldman (Winston-Salem, North Carolina) suggests putting on an ointment or oil-like medication to soften the scale. “Almost any kind of moisturizer will change the reflective properties of the scale so that you don’t see the scale,” he advised. He also suggested descaling agents such as topical salicylic acid or lactic acid. His patient education video is available on the American Academy of Dermatology website should you wish to direct your patients to it.
Because salicylic acid is a keratolytic (or peeling agent), it works by causing the outer layer of skin to shed. When applied topically, it helps to soften and lift psoriasis scales. Coal tar over-the-counter products also can be used for the same purpose. The over-the-counter product guide from the National Psoriasis Foundation is a valuable resource to share with patients.
Expert Commentary
I agree that it is very important to treat scale very gently. In addition to risk for infection, picking and traumatizing scale can lead to worsening of the psoriasis. This is known as the Koebner phenomenon. The phenomenon was first described by Heinrich Koebner in 1876 as the formation of psoriatic lesions in uninvolved skin of patients with psoriasis after cutaneous trauma. This isomorphic phenomenon is now known to involve numerous diseases, among them vitiligo, lichen planus, and Darier disease.
—Jeffrey M. Weinberg, MD (New York, New York)
Feldman S. How should I remove psoriasis scale? American Academy of Dermatology website. https://www.aad.org/public/diseases/scaly-skin/psoriasis/tips-for-managing-psoriasis/how-should-i-remove-psoriasis-scale. Accessed October 31, 2018.
National Psoriasis Foundation. Over-the-counter products. https://www.aad.org/public/diseases/scaly-skin/psoriasis/tips-for-managing-psoriasis/how-should-i-remove-psoriasis-scale. Published June 2017. Accessed October 31, 2018.
Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
Feldman S. How should I remove psoriasis scale? American Academy of Dermatology website. https://www.aad.org/public/diseases/scaly-skin/psoriasis/tips-for-managing-psoriasis/how-should-i-remove-psoriasis-scale. Accessed October 31, 2018.
National Psoriasis Foundation. Over-the-counter products. https://www.aad.org/public/diseases/scaly-skin/psoriasis/tips-for-managing-psoriasis/how-should-i-remove-psoriasis-scale. Published June 2017. Accessed October 31, 2018.
Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.

Autoimmune Progesterone Dermatitis
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
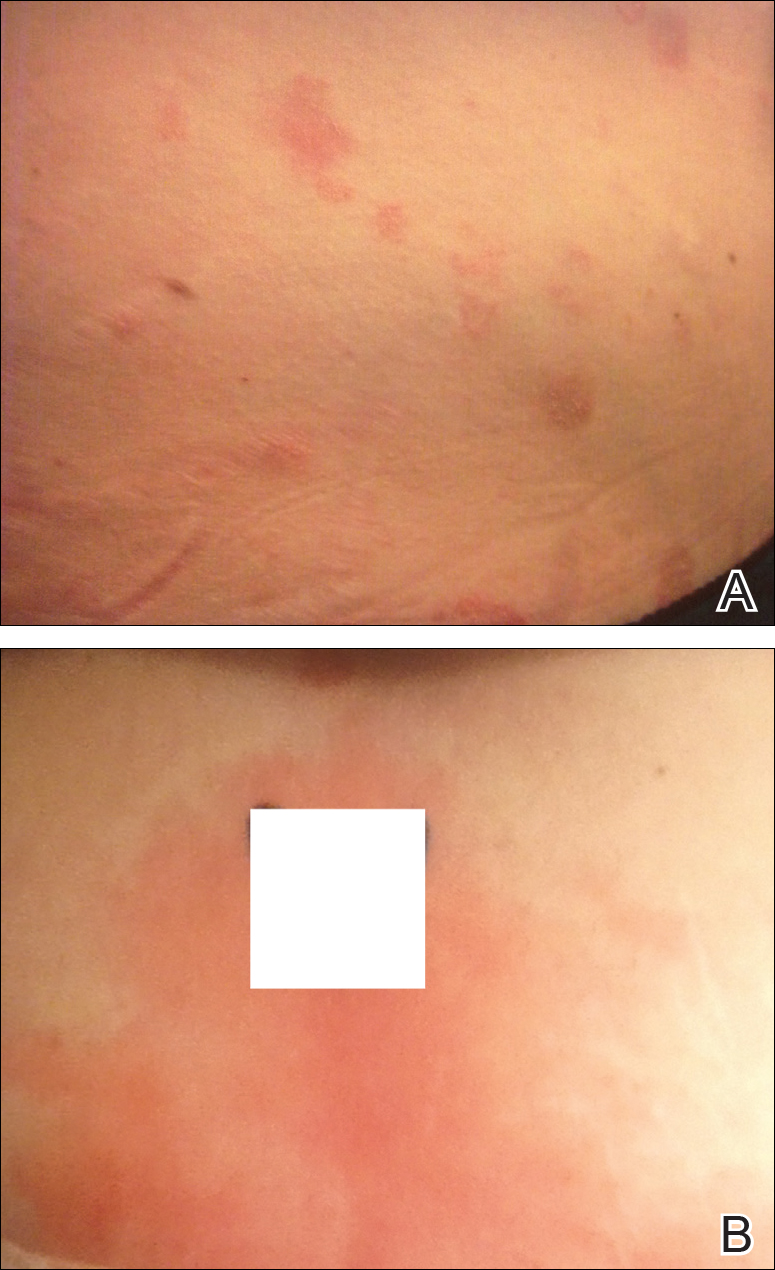
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
To the Editor:
Autoimmune progesterone dermatitis (APD) is a rare dermatologic condition that can be challenging to diagnose. The associated skin lesions are not only variable in physical presentation but also in the timing of the outbreak. The skin disorder stems from an internal reaction to elevated levels of progesterone during the luteal phase of the menstrual cycle. Autoimmune progesterone dermatitis can be difficult to detect; although the typical menstrual cycle is 28 days, many women have longer or shorter hormonal phases, leading to cyclical irregularity that can cause the lesions to appear sporadic in nature when in fact they are not.1
A 34-year-old woman with a history of endometriosis, psoriasis, and malignant melanoma presented to our dermatology clinic 2 days after a brief hospitalization during which she was diagnosed with a hypersensitivity reaction. Two days prior to her hospital admission, the patient developed a rash on the lower back with associated myalgia. The rash progressively worsened, spreading laterally to the flanks, which prompted her to seek medical attention. Blood work included a complete blood cell count with differential, complete metabolic panel, antinuclear antibody test, and erythrocyte sedimentation rate, which all were within reference range. A 4-mm punch biopsy from the left lateral flank was performed and was consistent with a neutrophilic dermatosis. The patient’s symptoms diminished and she was discharged the next day with instructions to follow up with a dermatologist.
Physical examination at our clinic revealed multiple minimally indurated, erythematous plaques with superficial scaling along the left lower back and upper buttock (Figure 1). No other skin lesions were present, and palpation of the cervical, axillary, and inguinal lymph nodes was unremarkable. A repeat 6-mm punch biopsy was performed and she was sent for fasting blood work.
Histologic examination of the punch biopsy revealed a superficial and deep perivascular and interstitial dermatitis with scattered neutrophils and eosinophils. Findings were described as nonspecific, possibly representing a dermal hypersensitivity or urticarial reaction.
Glucose-6-phosphate dehydrogenase testing was within reference range, and therapy was initiated with oral dapsone 50 mg once daily as well as fexofenadine 180 mg once daily. The patient initially responded well to the oral therapy, but she experienced recurrence of the skin eruption at infrequent intervals over the next few months, requiring escalating doses of dapsone to control the symptoms. After further questioning at a subsequent visit a few months later, it was discovered that the eruption occurred near the onset of the patient’s irregular menstrual cycle.
Approximately 1 year after her initial presentation, the patient returned for intradermal hormone injections to test for hormonally induced hypersensitivities. An injection of0.1 mL of a 50-mg/mL progesterone solution was administered in the right forearm as well as 0.1 mL of a 5-mg/mL estradiol solution and 0.1 mL of saline in the left forearm as a control. One hour after the injections, a strong positive reaction consisting of a 15-mm indurated plaque with surrounding wheal was noted at the site of the progesterone injection. The estradiol and saline control sites were clear of any dermal reaction (Figure 2). A diagnosis of APD was established, and the patient was referred to her gynecologist for treatment.
Due to the aggressive nature of her endometriosis, the gonadotropin-releasing hormone agonist leuprolide acetate was the first-line treatment prescribed by her gynecologist; however, after 8 months of therapy with leuprolide acetate, she was still experiencing breakthrough myalgia with her menstrual cycle and opted for a hysterectomy with a bilateral salpingo-oophorectomy. Within weeks of surgery, the myalgia ceased and the patient was completely asymptomatic.
Autoimmune progesterone dermatitis was first described in 1921.2 In affected women, the body reacts to the progesterone hormone surge during the luteal phase of the menstrual cycle. Symptoms begin approximately 3 to 4 days prior to menses and resolve 2 to 3 days after onset of flow. These progesterone hypersensitivity reactions can present within a spectrum of morphologies and severities. The lesions can appear eczematous, urticarial, as an angioedemalike reaction, as an erythema multiforme–like reaction with targetoid lesions, or in other nonspecific ways.1,3 Some patients experience a very mild, almost asymptomatic reaction, while others have a profound reaction progressing to anaphylaxis. Originally it was thought that exogenous exposure to progesterone led to a cross-reaction or hypersensitivity to the hormone; however, there have been cases reported in females as young as 12 years of age with no prior exposure.3,4 Reactions also can vary during pregnancy. There have been reports of spontaneous abortion in some affected females, but symptoms may dissipate in others, possibly due to a slow rise in progesterone causing a desensitization reaction.3,5
According to Bandino et al,6 there are 3 criteria for diagnosis of APD: (1) skin lesions related to the menstrual cycle, (2) positive response to intradermal testing with progesterone, and (3) symptomatic improvement after inhibiting progesterone secretions by suppressing ovulation.Areas checked with intradermal testing need to be evaluated 24 and 48 hours later for possible immediate or delayed-type hypersensitivity reactions. Biopsy typically is not helpful in this diagnosis because results usually are nonspecific.
Treatment of APD is targeted toward suppressing the internal hormonal surge. By suppressing the progesterone hormone, the symptoms are alleviated. The discomfort from the skin reaction typically is unresponsive to steroids or antihistamines. Oral contraceptives are first line in most cases because they suppress ovulation. Gonadotropin-releasing hormone analogues and tamoxifen also have been successful. For patients with severe disease that is recalcitrant to standard therapy or those who are postmenopausal, an oophorectemy is a curative option.2,4,5,7
Autoimmune progesterone dermatitis is a rare cyclical dermatologic condition in which the body responds to a surge of the patient’s own progesterone hormone. The disorder is difficult to diagnose because it can present with differing morphologies and biopsy is nonspecific. It also can be increasingly difficult to diagnose in women who do not have a typical 28-day menstrual cycle. In our patient, her irregular menstrual cycle may have caused a delay in diagnosis. Although the condition is rare, APD should be included in the differential diagnosis in females with a recurrent, cyclical, or recalcitrant cutaneous eruption.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
- Wojnarowska F, Greaves MW, Peachey RD, et al. Progesterone-induced erythema multiforme. J R Soc Med. 1985;78:407-408.
- Lee MK, Lee WY, Yong SJ, et al. A case of autoimmune progesterone dermatitis misdiagnosed as allergic contact dermatitis [published online February 9, 2011]. Allergy Asthma Immunol Res. 2011;3:141-144.
- Baptist AP, Baldwin JL. Autoimmune progesterone dermatitis in a patient with endometriosis: a case report and review of the literature. Clin Mol Allergy. 2004;2:10.
- Baççıoğlu A, Kocak M, Bozdag O, et al. An unusual form of autoimmune progesterone dermatitis (ADP): the role of diagnostic challenge test. World Allergy Organ J. 2007;10:S52.
- George R, Badawy SZ. Autoimmune progesterone dermatitis: a case report [published online August 9, 2012]. Case Rep Obstet Gynecol. doi:10.1155/2012/757854.
- Bandino JP, Thoppil J, Kennedy JS, et al. Iatrogenic autoimmune progesterone dermatitis causes by 17α-hydroxyprogesterone caproate for preterm labor prevention. Cutis. 2011;88:241-243.
- Magen E, Feldman V. Autoimmune progesterone anaphylaxis in a 24-year-old woman. Isr Med Assoc J. 2012;14:518-519.
Practice Points
- Autoimmune progesterone dermatitis (APD) is a hypersensitivity reaction to the progesterone surge during a woman’s menstrual cycle.
- Patients with APD often are misdiagnosed for years due to the variability of each woman’s menstrual cycle, making the correlation difficult.
- It is important to keep APD in mind for any recalcitrant or recurrent rash in females. A thorough history is critical when formulating a diagnosis.
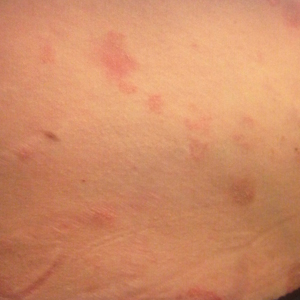
Mohs Micrographic Surgery Overlying a Pacemaker
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.

Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
To the Editor:
Pacemakers and defibrillators are common in patients presenting for cutaneous surgery. The use and application of electrosurgery in this patient population has been reviewed extensively.1 The presence of a cardiac device immediately below a cutaneous surgical site presents as a potentially more complex surgical procedure. Damage to and/or manipulation of the cardiac device could activate the device and/or require subsequent repair of the unit. We present the case of a basal cell carcinoma (BCC) overlying a pacemaker along with a brief review of the literature.
An 89-year-old man presented to our Mohs surgical unit for treatment of a long-standing BCC on the left upper chest (Figure, A) via Mohs micrographic surgery (MMS), which was utilized due to the infiltrative nature of the tumor and its close proximity to the cardiac device. He had a history of heart disease including paroxysmal atrial fibrillation, first-degree atrioventricular block, and sick sinus syndrome, and a pacemaker had been placed 5 years prior. The tumor was located on the skin directly above the pacemaker. The pacemaker and associated lead wires were easily palpable to touch. Prior to the procedure, treatment options were discussed with the patient’s cardiologist. Due to the size of the tumor (21×22 mm) and more importantly its location directly above the pacemaker, the BCC was treated with a single stage of MMS (Figure, B). In an effort to minimize potential exposure of the pacemaker, the surgical site was infiltrated with additional local anesthesia, which created a temporary edematous thickening to provide an increased barrier between the surgical site and pacemaker. Hemostasis was achieved with thermocautery, and a fusiform repair was completed without consequence (Figure, C). There were no postoperative changes or concerns, and preoperative and postoperative electrocardiograms reviewed by the patient’s cardiologist revealed no change.
Treatment of cutaneous lesions near pacemakers or defibrillators requires caution, both in avoidance of the device itself as well as electrocautery interference.1-4 There are multiple treatment options available, including MMS, excision, curettage and desiccation, topical therapies, and radiation therapy. The benefits of MMS for cutaneous tumors overlying cardiac devices include decreased risk of damaging the underlying pacemaker by minimizing surgical depth of the defect, minimizing the risk of recurrence and hence any additional procedures, and minimizing the risk of surgical complications via a smaller surgical defect.4 Monopolar electrosurgery is associated with the risk of interfering with pacemaker function; however, the use of bipolar electrocoagulation has been shown to be safer.1,3,4 Additionally, thermocautery carries the least risk because it involves heat only.2,5
Awareness of the cardiac device location, communication with the patient’s cardiologist, use of local anesthesia infiltrates to maximize distance between the surgical site and cardiac device, and appropriate hemostasis methods offer the most effective and safest means for surgical removal of tumors overlying cardiac devices.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
- El-Gamal HM, Dufresne RG, Saddler K. Electrosurgery, pacemakers and ICDs: a survey of precautions and complications experienced by cutaneous surgeons. Dermatol Surg. 2001;27:385-390.
- Chapas AM, Lee D, Rogers GS. Excision of malignant melanoma overlying a pacemaker. Dermatol Surg. 2005;31:112-114.
- Matzke TJ, Christenson LJ, Christenson SD, et al. Pacemakers and implantable cardiac defibrillators in dermatologic surgery. Dermatol Surg. 2006;32:1155-1162.
- Herrmann JL, Mishra V, Greenway HT. Basal cell carcinoma overlying a cardiac pacemaker successfully treated using Mohs micrographic surgery. 2014;4:474-477.
- Lane JE, O’Brien EM, Kent DE. Optimization of thermocautery in excisional dermatologic surgery. Dermatol Surg. 2006;32:669-675.
Practice Points
- Surgical treatment of a cutaneous lesion overlying a cardiac device requires caution, both in avoidance of the device itself as well as electrocautery interference.
- Local anesthesia infiltrates can be used to create a temporary edematous thickening to minimize potential exposure of the device during the procedure.
Military Grooming Standards and Their Impact on Skin Diseases of the Head and Neck
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
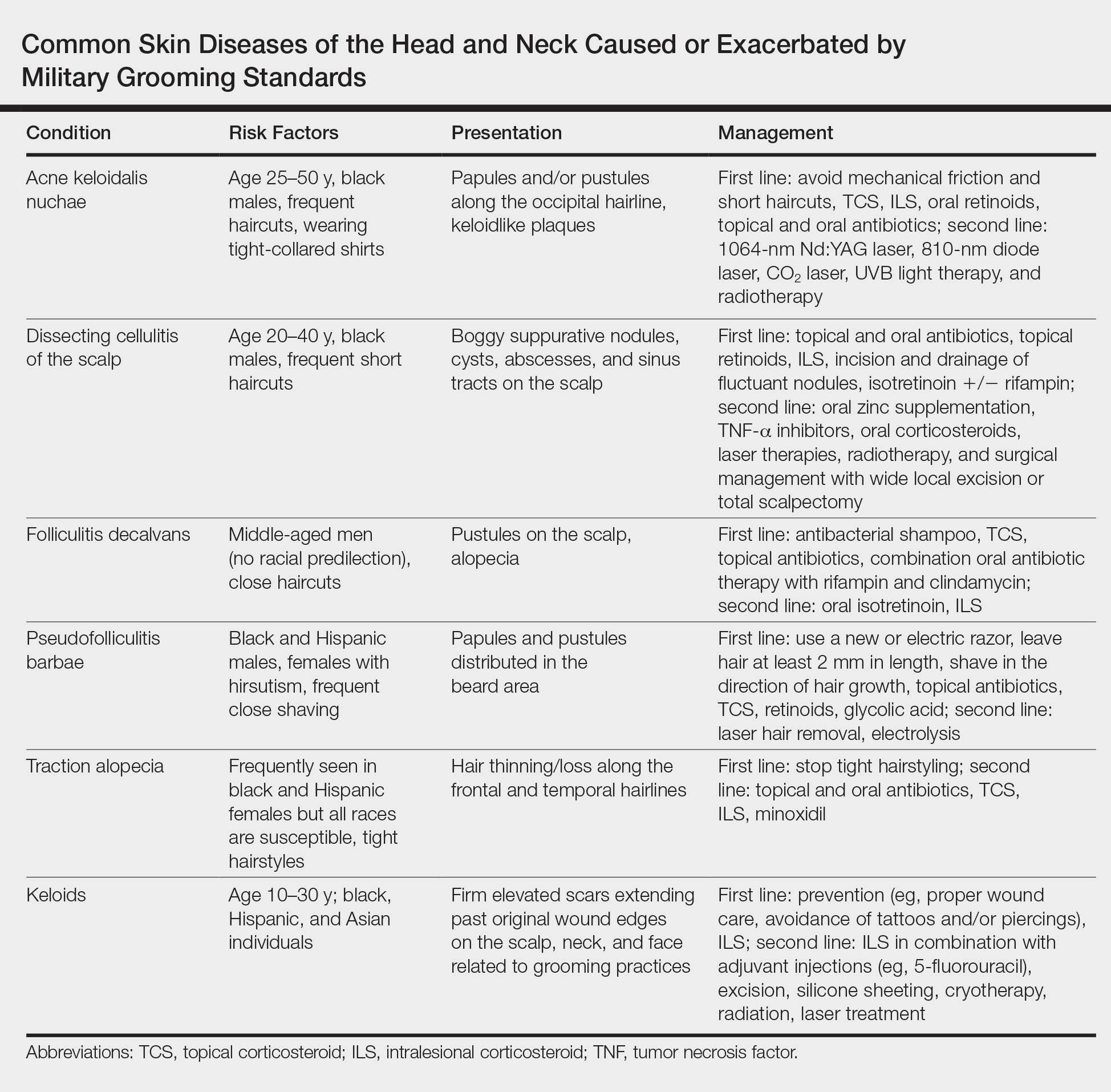
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
The US military enforces grooming standards to ensure the professional appearance and serviceability of soldiers in all operational settings. Although most individuals are able to uphold these regulations without incident, there is a growing cohort of servicemembers with skin diseases that were exacerbated or even initiated by haircuts, hairstyling, and shaving required to conform to these grooming standards. These skin diseases, which can affect both sexes and may not be appreciated until years into a soldier's service commitment, can have consequences related to individual morbidity and medical readiness for deployment, making it an important issue for medical practitioners to recognize and manage in servicemembers.
This review highlights several disorders of the pilosebaceous unit of the head and neck that can be caused or exacerbated by military grooming standards, including inflammatory hair disorders, traction alopecia, and pseudofolliculitis barbae. Discussion of each entity will include a review of susceptibility and causality as well as initial treatment options to consider (Table).
Inflammatory Hair Disorders
The proper appearance of servicemembers in uniform represents self-discipline and conformity to the high standards of the military. This transition occurs as a rite of passage for many new male recruits who receive shaved haircuts during their first days of basic training. Thereafter, male servicemembers are required to maintain a tapered appearance of the hair per military regulations.1 Clipping hair closely to the scalp or shaving the head entirely are authorized and often encouraged; therefore, high and tight haircuts and buzz cuts are popular among male soldiers due to the general ease of care and ability to maintain the haircut themselves. Conversely, these styles require servicemembers to get weekly or biweekly haircuts that in turn can lead to chronic trauma and irritation. In more susceptible populations, inflammatory hair disorders such as acne keloidalis nuchae (AKN), dissecting cellulitis of the scalp, and folliculitis decalvans may be incited.
Acne Keloidalis Nuchae
Acne keloidalis nuchae, also called folliculitis keloidalis, is a chronic scarring folliculitis presenting with papules and plaques on the occiput and nape of the neck that may merge to form hypertrophic scars or keloids. This disorder most commonly develops in young black men but also can be seen in black females and white patients of both sexes.2 Acne keloidalis nuchae shares many histologic features with central centrifugal cicatricial alopecia, which may suggest a similar pathogenesis. Apart from frequent haircuts, tight-collared shirts, such as those on military service uniforms, also have been associated with AKN. Because of these suspected etiologies, first-line treatment focuses on preventing further trauma by avoiding mechanical irritation and short haircuts, which may be difficult in the military setting. For earlier disease stages, topical and intralesional corticosteroids, oral retinoids, and topical and oral antibiotics are used for their anti-inflammatory properties.3 In refractory cases, surgical excision with healing by secondary intention may be attempted.4 Additional treatment options include the 1064-nm Nd:YAG and 810-nm diode lasers,3 UVB light therapy, CO2 laser, and radiotherapy.
Dissecting Cellulitis of the Scalp
Similar to AKN, dissecting cellulitis of the scalp is another inflammatory hair disorder that is worsened by frequent short haircuts.5 Dissecting cellulitis of the scalp is a primary cicatricial alopecia proposed to be secondary to follicular occlusion. It often is seen in black males aged 20 to 40 years and is characterized by boggy suppurative nodules and cysts with draining sinus tracts, abscesses, and resultant scarring alopecia. Dissecting cellulitis of the scalp is part of the follicular occlusion tetrad, which also includes hidradenitis suppurativa, acne conglobata, and pilonidal cysts. First-line therapies include topical and oral antibiotics, topical retinoids, intralesional corticosteroids, incision and drainage of fluctuant nodules, and oral isotretinoin with or without rifampin. Alternative treatments include oral zinc supplementation, oral corticosteroids, tumor necrosis factor α inhibitors, laser therapies, radiotherapy, and surgical management with wide local excision or total scalpectomy.6,7
Folliculitis Decalvans
Folliculitis decalvans is a primary cicatricial alopecia of the scalp that most commonly presents in middle-aged men without racial predilection.8 Folliculitis decalvans presents with multiple pustules, crusts, tufted hairs, and perifollicular hyperkeratosis, leading to scarring of the scalp, which often is most severe on the posterior vertex. Staphylococcus aureus is a presumed player in the pathogenesis of folliculitis decalvans with superantigens causing release of cytokines stimulating follicular destruction. Close haircuts in conformation with military grooming standards can contribute to this condition due to mechanical trauma and subsequent inflammation. It typically is diagnosed clinically, but if histologic confirmation is desired, a sample from the periphery of early lesions is preferred.9 Initial treatment consists of antibacterial shampoos, topical corticosteroids, topical antibiotics, and combination oral antibiotic therapy with rifampin and clindamycin. Studies using oral isotretinoin have shown variable results,10,11 and the most effective treatment of recalcitrant lesions appears to be intralesional corticosteroids.12
Follicular and Scarring Disorders
In addition to inflammatory hair disorders, military grooming standards have been linked to the pathogenesis of diseases such as pseudofolliculitis barbae, traction alopecia, and keloids, specifically through irritation of the face, neck, and scalp, as well as damage to the follicular unit.5 These conditions develop because grooming regulations necessitate certain hair practices such as close shaving of facial and neck hair and keeping long hair secured relatively tightly to the scalp.
Pseudofolliculitis Barbae
Males in the military are obligated to keep their faces clean-shaven.1 They may acquire a medical waiver for a specified beard length if deemed appropriate by the treating physician,1 which often leads to the need for continual waiver renewal and also may warrant possible negative perception from peers, subordinates, and leadership. One of the most prevalent conditions that is closely associated with shaving is pseudofolliculitis barbae. The combination of close shaving and tightly coiled hairs causes the hairs to grow toward and penetrate the skin, particularly on the neck.13 In some cases, the hairs never actually exit the skin and simply curl within the superficial epidermis. A foreign body reaction often arises, leading to inflamed follicular papules and pustules. Affected individuals may experience pain, pruritus, and secondary infections. Postinflammatory hyperpigmentation, hypertrophic scarring, and keloid formation are common sequelae in cases of untreated disease. Pseudofolliculitis barbae also is exacerbated by pulling the skin taut and shaving against the grain, making behavioral interventions a key component in management of this condition. Preliminary recommendations include using a new or electric razor, leaving hair at least 2 mm in length, and shaving in the direction of hair growth. Other treatment options with varying effectiveness include daily alternation of a mild topical corticosteroid and one of the following: a topical retinoid, topical antibiotics, or glycolic acid. The only treatments that approach definitive cure are laser hair removal and electrolysis for which patient skin type plays an important role in laser selection.5
Traction Alopecia
Similar to their male counterparts, female military members must also present a conservative professional appearance, including hair that is neatly groomed.1 If the length of the hair extends beyond the uniform collar, it must be inconspicuously fastened or pinned above the collar. As a result, loosely tied hair is unauthorized, and females with long hair must secure their hair tightly on a daily basis. Traction alopecia results from tight hairstyling over a prolonged period and commonly affects female soldiers. The etiology is presumed to be mechanical loosening of hair within the follicles, leading to inflammation. Although traditionally seen in black women along the frontal and temporal hairlines, traction alopecia has been identified in individuals of all races and can occur anywhere on the scalp.5 Perifollicular erythema may be the first sign, and papules and pustules may be visible. Although the hair loss in traction alopecia usually is reversible if the traction is ceased, end-stage disease may be permanent.6 Halting traction-inducing practices is paramount, and other treatment options that may slow progression include topical or oral antibiotics and topical or intralesional corticosteroids. Recovery of hair loss also may be aided by topical minoxidil.5
Keloids
Keloid formation is an important pathology to address, as it may result from several of the aforementioned conditions. Keloids are most commonly seen in black individuals but also can occur in Hispanic and Asian patients. The cause has not been fully elucidated but is thought to be a combination of dysfunctional fibroblasts with a genetic component based on racial predilection and twin concordance studies.5 The chest, shoulders, upper back, neck, and earlobes are particularly susceptible to keloid formation, which can appear from 1 to 24 years following dermal trauma.5 Unlike hypertrophic scars, keloids generally do not regress and frequently cause discomfort, pruritus, and emotional distress. They also can hinder wearing a military uniform. Sustained remission is problematic, making prevention a first-line approach, including proper care of wounds when they occur and avoiding elective procedures such as piercings and tattoos. Intralesional corticosteroids, adjuvant injections (eg, 5-fluorouracil), silicone sheeting, cryotherapy, radiation, laser therapy, and excision are some of the treatment options when keloids have formed.5
Final Comment
It is important to recognize military grooming standards as a cause or contributor to several diseases of the head and neck in military servicemembers. Specifically, frequent haircuts in male soldiers are associated with several inflammatory hair disorders, including AKN, dissecting cellulitis of the scalp, and folliculitis decalvans, while daily shaving predisposes individuals to pseudofolliculitis barbae with possible keloid formation. Females may develop traction alopecia from chronically tight, pulled back hairstyles. All of these conditions have health implications for the affected individuals and can compromise the military mission. Awareness, prevention, and recognition are key along with the knowledge base to provide anticipatory avoidance and initiate appropriate treatments, thereby mitigating these potential consequences.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
- US Department of the Army. Wear and Appearance of Army Uniforms and Insignia: Army Regulation 670-1. Washington, DC: Department of the Army; 2017. https://history.army.mil/html/forcestruc/docs/AR670-1.pdf. Accessed October 11, 2018.
- East-Innis AD, Stylianou K, Paolino A, et al. Acne keloidalis nuchae: risk factors and associated disorders--a retrospective study. Int J Dermatol. 2017;56:828-832.
- Maranda EL, Simmons BJ, Nguyen AH, et al. Treatment of acne keloidalis nuchae: a systematic review of the literature. Dermatol Ther (Heidelb). 2016;6:363-378.
- Glenn MJ, Bennett RG, Kelly AP. Acne keloidalis nuchae: treatment with excision and second-intention healing. J Am Acad Dermatol. 1995;33:243-246.
- Madu P, Kundu RV. Follicular and scarring disorders in skin of color: presentation and management. Am J Clin Dermatol. 2014;15:307-321.
- Rodney IJ, Onwudiwe OC. Hair and scalp disorders in ethnic populations. J Drugs Dermatol. 2013;12:420-427.
- Lindsey SF, Tosti A. Ethnic hair disorders. Curr Probl Dermatol. 2015;47:139-148.
- Whiting DA. Cicatricial alopecia: clinico-pathological findings and treatment. Clin Dermatol. 2001;19:211-225.
- Sperling LC, Cowper SE, Knopp EA. An Atlas of Hair Pathology with Clinical Correlations. 2nd ed. Boca Raton, FL: CRC Press; 2012.
- Gemmeke A, Wollina U. Folliculitis decalvans of the scalp: response to triple therapy with isotretinoin, clindamycin, and prednisolone. Acta Dermatovenerol Alp Pannonica Adriat. 2006;15:184-186.
- Hallai N, Thompson I, Williams P, et al. Folliculitis spinulosa decalvans: failure to respond to oral isotretinoin. J Eur Acad Dermatol Venereol. 2006;20:223-224.
- Bolduc C, Sperling LC, Shapiro J. Primary cicatricial alopecia. J Am Acad Dermatol. 2016;75:101-117.
- Perry PK, Cook-Bolden FE, Rahman Z, et al. Defining pseudofolliculitis barbae in 2001: a review of the literature and current trends. J Am Acad Dermatol. 2002;46(2 suppl):S113-S119.
Practice Points
- The short frequent haircuts required to maintain a tapered appearance of the hair per US military regulations may lead to inflammatory hair disorders such as acne keloidalis nuchae, dissecting cellulitis of the scalp, and folliculitis decalvans.
- The mainstay of prevention for these conditions is avoidance of inciting factors such as short haircuts, tight-collared shirts, frequent shaving, or tight hairstyles.
- Early identification and treatment of inflammatory follicular and scarring disorders can prevent further scarring, pigmentation changes, and/or disfigurement.