User login
Lichen Sclerosus: The Silent Genital Health Concern Often Missed
Ashley Winter, MD, remembers the first time she Googled the skin condition lichen sclerosus. Most of the websites listed the autoimmune condition as a rare disease.
In the realm of genital health, some conditions remain shrouded in silence and consequently are more likely to go undercounted and underdiagnosed, said Dr. Winter, a urologist based in Los Angeles.
“I truly believe that we just miss the diagnosis a vast majority of the time because there isn’t enough training on [detecting] it,” said Dr. Winter.
, according to the US National Institutes of Health. The condition also more commonly occurs among women, and symptoms include hypopigmentation, itching, pain, changes in skin appearance, and skin atrophy.
“Most cases [affect the] genital [area] only, so often patients don’t bring it up because they don’t want to be examined,” said Sarah Lonowski, MD, assistant professor of dermatology and codirector of the Multidisciplinary Autoimmune Skin Disease/Derm-Rheum Program at the University of Nebraska–Lincoln. “It’s a sensitive area, it’s an uncomfortable area to have examined, so it comes with a lot of emotional burden,” for patients, Dr. Lonowski said.
Receiving a lichen sclerosis diagnosis can take 5 years or longer, in part because the condition’s symptoms can lead clinicians to first make a diagnosis of a yeast infection or bacterial vaginosis, according to Christina Kraus, MD, assistant professor of dermatology at UCI Health in Irvine, California.
“There is still limited information on this condition in medical education, and it is not uncommon for clinicians who are not in dermatology or gynecology to be unfamiliar with this diagnosis,” Dr. Kraus said.
Because no medical tests are available to confirm lichen sclerosus, clinicians diagnose the condition based on the skin’s appearance and symptoms. In some cases, a skin biopsy may help differentiate it from similar rashes that occur in the genital area.
Prepubescent children and postmenopausal women are most likely to develop genital lichen sclerosis, so pediatricians and primary care physicians may be the first to see possible cases, Dr. Lonowski said.
Patients “may not mention it unless they’re asked,” Dr. Lonowski said, adding clinicians can inquire with patients about genital health, examine bothersome areas, “and refer if you’re not sure.”
Clinicians may also miss the condition during physical exams if they do not examine the vulvar skin, she said. The exact cause also remains elusive, but researchers believe genetic and hormonal factors, as well as an overactive immune response, may contribute to development of the condition.
Watch Out for Presentation
While lichen sclerosus more frequently occurs in women, men are also affected by the condition. Benjamin N. Breyer, MD, professor and chair of urology at the University of California San Francisco, said lichen sclerosus is one of the most common skin conditions he treats in his male patients.
“Advanced cases can cause urethral narrowing, which is a condition I treat commonly,” said Dr. Breyer. “Lichen sclerosus is often an underrecognized cause of pain or tearing with erections and sex in men.”
Similar to women, lichen sclerosus presents as white color changes on the skin. For men, the condition can also result in fusion of the shaft skin to the head of the penis and burying or concealment of the penis, Dr. Breyer said.
“This leads to challenges with intimacy and urination and can have extensive impacts on quality of life,” said Dr. Breyer.
For women, the skin changes often extend to the perianal area and can cause scarring, said Dr. Kraus.
“Early scarring may present as adherence of the labia minora to the labia majora or inability to fully retract the clitoral hood from the clitoris,” said Dr. Kraus.
In both men and women, lichen sclerosus and another autoimmune condition known as morphea, characterized by skin hardening and discoloration, often present together, said Dr. Lonowski.
“If you have a patient with known morphea, it’s important to ask about genital symptoms,” said Dr. Lonowski. “The association between the two is fairly strong.”
Circumcision is often the first step to help prevent chronic inflammation among male patients, said Dr. Breyer. Because lichen sclerosus is associated with an increased risk for penile cancer, “it is important to biopsy suspicious lesions,” Dr. Breyer added.
Increasing awareness of lichen sclerosus is crucial for early detection and timely intervention, said Dr. Lonowski. The first-line treatment of genital lichen sclerosus is strong topical steroid ointments to reduce inflammation. Clinicians might prescribe this treatment for use twice daily for 2-3 months and then assesses the patient on their response.
“It is fairly responsive to treatment in most cases,” said Dr. Lonowski.
Once symptoms have improved, Dr. Lonowski transitions patients to a maintenance regimen, which might include using the same steroid but only three times a week, switching to a topical with a less potent steroid dosage, or using a combination of a topical steroid and a nonsteroidal anti-inflammatory cream. Despite the prolonged use of the drug, she said patients with lichen sclerosus usually do not present with side effects like discoloration or thinning of skin.
“You may achieve control or remission, but we don’t stop treatment completely because we know the natural history of the disease is to have flares and recurrence.”
If left untreated, the condition can lead to atrophy, scarring, and distortion of the genital anatomy and, in some cases, develop into squamous cell carcinoma.
“The fact that you can do a topical cream intervention and prevent cancer is huge,” said Dr. Winter.
She said open discussions surrounding genital health led by primary care providers can destigmatize conditions like lichen sclerosus and promote early detection and management.
“We need to foster an environment where individuals feel comfortable discussing their symptoms openly,” Dr. Winter said. “Increased awareness can pave the way for early detection, which is crucial for managing the condition effectively.”
The experts included in the story reported no relevant disclosures.
A version of this article appeared on Medscape.com.
Ashley Winter, MD, remembers the first time she Googled the skin condition lichen sclerosus. Most of the websites listed the autoimmune condition as a rare disease.
In the realm of genital health, some conditions remain shrouded in silence and consequently are more likely to go undercounted and underdiagnosed, said Dr. Winter, a urologist based in Los Angeles.
“I truly believe that we just miss the diagnosis a vast majority of the time because there isn’t enough training on [detecting] it,” said Dr. Winter.
, according to the US National Institutes of Health. The condition also more commonly occurs among women, and symptoms include hypopigmentation, itching, pain, changes in skin appearance, and skin atrophy.
“Most cases [affect the] genital [area] only, so often patients don’t bring it up because they don’t want to be examined,” said Sarah Lonowski, MD, assistant professor of dermatology and codirector of the Multidisciplinary Autoimmune Skin Disease/Derm-Rheum Program at the University of Nebraska–Lincoln. “It’s a sensitive area, it’s an uncomfortable area to have examined, so it comes with a lot of emotional burden,” for patients, Dr. Lonowski said.
Receiving a lichen sclerosis diagnosis can take 5 years or longer, in part because the condition’s symptoms can lead clinicians to first make a diagnosis of a yeast infection or bacterial vaginosis, according to Christina Kraus, MD, assistant professor of dermatology at UCI Health in Irvine, California.
“There is still limited information on this condition in medical education, and it is not uncommon for clinicians who are not in dermatology or gynecology to be unfamiliar with this diagnosis,” Dr. Kraus said.
Because no medical tests are available to confirm lichen sclerosus, clinicians diagnose the condition based on the skin’s appearance and symptoms. In some cases, a skin biopsy may help differentiate it from similar rashes that occur in the genital area.
Prepubescent children and postmenopausal women are most likely to develop genital lichen sclerosis, so pediatricians and primary care physicians may be the first to see possible cases, Dr. Lonowski said.
Patients “may not mention it unless they’re asked,” Dr. Lonowski said, adding clinicians can inquire with patients about genital health, examine bothersome areas, “and refer if you’re not sure.”
Clinicians may also miss the condition during physical exams if they do not examine the vulvar skin, she said. The exact cause also remains elusive, but researchers believe genetic and hormonal factors, as well as an overactive immune response, may contribute to development of the condition.
Watch Out for Presentation
While lichen sclerosus more frequently occurs in women, men are also affected by the condition. Benjamin N. Breyer, MD, professor and chair of urology at the University of California San Francisco, said lichen sclerosus is one of the most common skin conditions he treats in his male patients.
“Advanced cases can cause urethral narrowing, which is a condition I treat commonly,” said Dr. Breyer. “Lichen sclerosus is often an underrecognized cause of pain or tearing with erections and sex in men.”
Similar to women, lichen sclerosus presents as white color changes on the skin. For men, the condition can also result in fusion of the shaft skin to the head of the penis and burying or concealment of the penis, Dr. Breyer said.
“This leads to challenges with intimacy and urination and can have extensive impacts on quality of life,” said Dr. Breyer.
For women, the skin changes often extend to the perianal area and can cause scarring, said Dr. Kraus.
“Early scarring may present as adherence of the labia minora to the labia majora or inability to fully retract the clitoral hood from the clitoris,” said Dr. Kraus.
In both men and women, lichen sclerosus and another autoimmune condition known as morphea, characterized by skin hardening and discoloration, often present together, said Dr. Lonowski.
“If you have a patient with known morphea, it’s important to ask about genital symptoms,” said Dr. Lonowski. “The association between the two is fairly strong.”
Circumcision is often the first step to help prevent chronic inflammation among male patients, said Dr. Breyer. Because lichen sclerosus is associated with an increased risk for penile cancer, “it is important to biopsy suspicious lesions,” Dr. Breyer added.
Increasing awareness of lichen sclerosus is crucial for early detection and timely intervention, said Dr. Lonowski. The first-line treatment of genital lichen sclerosus is strong topical steroid ointments to reduce inflammation. Clinicians might prescribe this treatment for use twice daily for 2-3 months and then assesses the patient on their response.
“It is fairly responsive to treatment in most cases,” said Dr. Lonowski.
Once symptoms have improved, Dr. Lonowski transitions patients to a maintenance regimen, which might include using the same steroid but only three times a week, switching to a topical with a less potent steroid dosage, or using a combination of a topical steroid and a nonsteroidal anti-inflammatory cream. Despite the prolonged use of the drug, she said patients with lichen sclerosus usually do not present with side effects like discoloration or thinning of skin.
“You may achieve control or remission, but we don’t stop treatment completely because we know the natural history of the disease is to have flares and recurrence.”
If left untreated, the condition can lead to atrophy, scarring, and distortion of the genital anatomy and, in some cases, develop into squamous cell carcinoma.
“The fact that you can do a topical cream intervention and prevent cancer is huge,” said Dr. Winter.
She said open discussions surrounding genital health led by primary care providers can destigmatize conditions like lichen sclerosus and promote early detection and management.
“We need to foster an environment where individuals feel comfortable discussing their symptoms openly,” Dr. Winter said. “Increased awareness can pave the way for early detection, which is crucial for managing the condition effectively.”
The experts included in the story reported no relevant disclosures.
A version of this article appeared on Medscape.com.
Ashley Winter, MD, remembers the first time she Googled the skin condition lichen sclerosus. Most of the websites listed the autoimmune condition as a rare disease.
In the realm of genital health, some conditions remain shrouded in silence and consequently are more likely to go undercounted and underdiagnosed, said Dr. Winter, a urologist based in Los Angeles.
“I truly believe that we just miss the diagnosis a vast majority of the time because there isn’t enough training on [detecting] it,” said Dr. Winter.
, according to the US National Institutes of Health. The condition also more commonly occurs among women, and symptoms include hypopigmentation, itching, pain, changes in skin appearance, and skin atrophy.
“Most cases [affect the] genital [area] only, so often patients don’t bring it up because they don’t want to be examined,” said Sarah Lonowski, MD, assistant professor of dermatology and codirector of the Multidisciplinary Autoimmune Skin Disease/Derm-Rheum Program at the University of Nebraska–Lincoln. “It’s a sensitive area, it’s an uncomfortable area to have examined, so it comes with a lot of emotional burden,” for patients, Dr. Lonowski said.
Receiving a lichen sclerosis diagnosis can take 5 years or longer, in part because the condition’s symptoms can lead clinicians to first make a diagnosis of a yeast infection or bacterial vaginosis, according to Christina Kraus, MD, assistant professor of dermatology at UCI Health in Irvine, California.
“There is still limited information on this condition in medical education, and it is not uncommon for clinicians who are not in dermatology or gynecology to be unfamiliar with this diagnosis,” Dr. Kraus said.
Because no medical tests are available to confirm lichen sclerosus, clinicians diagnose the condition based on the skin’s appearance and symptoms. In some cases, a skin biopsy may help differentiate it from similar rashes that occur in the genital area.
Prepubescent children and postmenopausal women are most likely to develop genital lichen sclerosis, so pediatricians and primary care physicians may be the first to see possible cases, Dr. Lonowski said.
Patients “may not mention it unless they’re asked,” Dr. Lonowski said, adding clinicians can inquire with patients about genital health, examine bothersome areas, “and refer if you’re not sure.”
Clinicians may also miss the condition during physical exams if they do not examine the vulvar skin, she said. The exact cause also remains elusive, but researchers believe genetic and hormonal factors, as well as an overactive immune response, may contribute to development of the condition.
Watch Out for Presentation
While lichen sclerosus more frequently occurs in women, men are also affected by the condition. Benjamin N. Breyer, MD, professor and chair of urology at the University of California San Francisco, said lichen sclerosus is one of the most common skin conditions he treats in his male patients.
“Advanced cases can cause urethral narrowing, which is a condition I treat commonly,” said Dr. Breyer. “Lichen sclerosus is often an underrecognized cause of pain or tearing with erections and sex in men.”
Similar to women, lichen sclerosus presents as white color changes on the skin. For men, the condition can also result in fusion of the shaft skin to the head of the penis and burying or concealment of the penis, Dr. Breyer said.
“This leads to challenges with intimacy and urination and can have extensive impacts on quality of life,” said Dr. Breyer.
For women, the skin changes often extend to the perianal area and can cause scarring, said Dr. Kraus.
“Early scarring may present as adherence of the labia minora to the labia majora or inability to fully retract the clitoral hood from the clitoris,” said Dr. Kraus.
In both men and women, lichen sclerosus and another autoimmune condition known as morphea, characterized by skin hardening and discoloration, often present together, said Dr. Lonowski.
“If you have a patient with known morphea, it’s important to ask about genital symptoms,” said Dr. Lonowski. “The association between the two is fairly strong.”
Circumcision is often the first step to help prevent chronic inflammation among male patients, said Dr. Breyer. Because lichen sclerosus is associated with an increased risk for penile cancer, “it is important to biopsy suspicious lesions,” Dr. Breyer added.
Increasing awareness of lichen sclerosus is crucial for early detection and timely intervention, said Dr. Lonowski. The first-line treatment of genital lichen sclerosus is strong topical steroid ointments to reduce inflammation. Clinicians might prescribe this treatment for use twice daily for 2-3 months and then assesses the patient on their response.
“It is fairly responsive to treatment in most cases,” said Dr. Lonowski.
Once symptoms have improved, Dr. Lonowski transitions patients to a maintenance regimen, which might include using the same steroid but only three times a week, switching to a topical with a less potent steroid dosage, or using a combination of a topical steroid and a nonsteroidal anti-inflammatory cream. Despite the prolonged use of the drug, she said patients with lichen sclerosus usually do not present with side effects like discoloration or thinning of skin.
“You may achieve control or remission, but we don’t stop treatment completely because we know the natural history of the disease is to have flares and recurrence.”
If left untreated, the condition can lead to atrophy, scarring, and distortion of the genital anatomy and, in some cases, develop into squamous cell carcinoma.
“The fact that you can do a topical cream intervention and prevent cancer is huge,” said Dr. Winter.
She said open discussions surrounding genital health led by primary care providers can destigmatize conditions like lichen sclerosus and promote early detection and management.
“We need to foster an environment where individuals feel comfortable discussing their symptoms openly,” Dr. Winter said. “Increased awareness can pave the way for early detection, which is crucial for managing the condition effectively.”
The experts included in the story reported no relevant disclosures.
A version of this article appeared on Medscape.com.
Study: Lifetime Cost of Vyjuvek Gene Therapy for DEB Could Be $15-$22 Million
according to the authors of a new study.
The US Food and Drug Administration (FDA) approved Vyjuvek (Krystal Biotech) in May 2023 for the treatment of wounds in patients ages 6 months and older with dystrophic epidermolysis bullosa (DEB), which includes two types, the most severe form (autosomal recessive, or RDEB) and the autosomal dominant form of DEB (DDEB), which tends to be milder.

Treatment with Vyjuvek “represents an important advance in the treatment of RDEB,” wrote Adam J.N. Raymakers, PhD, and colleagues at the Program on Regulation, Therapeutics, and Law; the Department of Dermatology; and the Division of Pulmonary and Critical Care Medicine at Brigham & Women’s Hospital in Boston, Massachusetts, in their paper, published in JAMA Dermatology. But the price “will be high, potentially limiting patients’ access to it,” they added. Evidence to support it in DDEB “is less conclusive,” they wrote, noting that the pivotal phase 3 study that led to approval included one patient with DDEB.
“The wider indication granted by the FDA may lead to friction between payers on the one hand and patients and physicians on the other,” they wrote, noting a potential minimum price of $300,000 per patient a year, which was based on Krystal’s regulatory filings.
There is no cure for DEB. Vyjuvek, applied as a gel on an ongoing basis, uses a nonreplicating herpes simplex virus type 1 vector to deliver the COL7A1 gene directly to skin cells, restoring the COL7 protein fibrils that stabilize skin structure.
The authors estimated that in the United States, 894 individuals – largely children – with both forms of the disease would be eligible for Vyjuvek treatment in the first year. Based on the $300,000 price, spending on gene therapy could range from $179 million to $357 million for those 894 patients, they reported in the study.
Over the first 3 years, spending could range as high as $1 billion, the authors estimated. Even if patients with only the most severe disease (RDEB) — an estimated 442 patients — received treatment, spending could be $132 million and up to $400 million or more over the first 3 years, they wrote.
Some media outlets have reported that Vyjuvek could cost as much as $600,000, said Dr. Raymakers, a research fellow. That price “would double all of our estimates,” he told this news organization.
The study assumed that patients with RDEB would only live to age 50, which led to a lifetime cost estimate of $15 million. But that is likely a conservative estimate, he and his coauthors wrote, noting that many patients with RDEB die from squamous cell carcinoma, but that Vyjuvek could, by attenuating skin damage, also potentially prevent skin cancer.
Dr. Raymakers said he and his colleagues began their study when Vyjuvek was approved, and thus they did not have any real-world data on the price or payer responses. Their estimates also did not include differing dosing regimens, which also could change their spending figures.
Krystal Biotech recently reported that in its third quarter of 2023 – representing just 1 month of Vyjuvek availability – it received requests to begin treatment for 284 patients from 136 unique clinicians. Twenty percent of the start requests were for patients with the milder form (DDEB), and a third of all the requests were for patients 10 years of age or younger. The company also said that it had “received positive coverage determinations from all major commercial national health plans” and that it was on track to receive approval from most state Medicaid plans.
In 1 month, Krystal reported net Vyjuvek revenues of $8.6 million.
The authors suggested that one way to evaluate Vyjuvek’s value — especially for those with DDEB — would be through a cost-effectiveness study. While important, a cost-effectiveness study would not get at the impact on a payer, said Dr. Raymakers. “Something can be cost-effective but unaffordable to the system,” he said.
“When there’s one of these very expensive therapies, that’s one thing,” he said. “But when there’s more and more coming to market, you wonder how much can be tolerated,” said Dr. Raymakers.
CMS Launching Gene Therapy Program
The Biden administration recently announced that it was launching a program aimed at increasing access, curbing costs, and ensuring value of gene therapies, starting with sickle cell disease. The program will begin in early 2025. Among other aspects, the federal government will negotiate the price of the product with the manufacturer.
“The goal of the Cell and Gene Therapy Access Model is to increase access to innovative cell and gene therapies for people with Medicaid by making it easier for states to pay for these therapies,” said Liz Fowler, CMS Deputy Administrator and Director of the CMS Innovation Center, in a statement announcing the program.
Whether the new program takes a look at Vyjuvek – and when – is not clear.
But the authors of the study noted that the lifetime costs of treating a patient with Vyjuvek “exceed the costs of all other one-time gene therapies for other diseases.” And they wrote, even at the most conservative estimates, Vyjuvek “will be the most expensive gene therapy currently marketed in the US.”
The study was funded by a grant from Arnold Ventures, grants from the Kaiser Permanente Institute for Health Policy, the Commonwealth Fund, and the National Heart, Lung, and Blood Institute. Dr. Raymakers and co-authors reported no financial relationships relevant to the work.
according to the authors of a new study.
The US Food and Drug Administration (FDA) approved Vyjuvek (Krystal Biotech) in May 2023 for the treatment of wounds in patients ages 6 months and older with dystrophic epidermolysis bullosa (DEB), which includes two types, the most severe form (autosomal recessive, or RDEB) and the autosomal dominant form of DEB (DDEB), which tends to be milder.
Treatment with Vyjuvek “represents an important advance in the treatment of RDEB,” wrote Adam J.N. Raymakers, PhD, and colleagues at the Program on Regulation, Therapeutics, and Law; the Department of Dermatology; and the Division of Pulmonary and Critical Care Medicine at Brigham & Women’s Hospital in Boston, Massachusetts, in their paper, published in JAMA Dermatology. But the price “will be high, potentially limiting patients’ access to it,” they added. Evidence to support it in DDEB “is less conclusive,” they wrote, noting that the pivotal phase 3 study that led to approval included one patient with DDEB.
“The wider indication granted by the FDA may lead to friction between payers on the one hand and patients and physicians on the other,” they wrote, noting a potential minimum price of $300,000 per patient a year, which was based on Krystal’s regulatory filings.
There is no cure for DEB. Vyjuvek, applied as a gel on an ongoing basis, uses a nonreplicating herpes simplex virus type 1 vector to deliver the COL7A1 gene directly to skin cells, restoring the COL7 protein fibrils that stabilize skin structure.
The authors estimated that in the United States, 894 individuals – largely children – with both forms of the disease would be eligible for Vyjuvek treatment in the first year. Based on the $300,000 price, spending on gene therapy could range from $179 million to $357 million for those 894 patients, they reported in the study.
Over the first 3 years, spending could range as high as $1 billion, the authors estimated. Even if patients with only the most severe disease (RDEB) — an estimated 442 patients — received treatment, spending could be $132 million and up to $400 million or more over the first 3 years, they wrote.
Some media outlets have reported that Vyjuvek could cost as much as $600,000, said Dr. Raymakers, a research fellow. That price “would double all of our estimates,” he told this news organization.
The study assumed that patients with RDEB would only live to age 50, which led to a lifetime cost estimate of $15 million. But that is likely a conservative estimate, he and his coauthors wrote, noting that many patients with RDEB die from squamous cell carcinoma, but that Vyjuvek could, by attenuating skin damage, also potentially prevent skin cancer.
Dr. Raymakers said he and his colleagues began their study when Vyjuvek was approved, and thus they did not have any real-world data on the price or payer responses. Their estimates also did not include differing dosing regimens, which also could change their spending figures.
Krystal Biotech recently reported that in its third quarter of 2023 – representing just 1 month of Vyjuvek availability – it received requests to begin treatment for 284 patients from 136 unique clinicians. Twenty percent of the start requests were for patients with the milder form (DDEB), and a third of all the requests were for patients 10 years of age or younger. The company also said that it had “received positive coverage determinations from all major commercial national health plans” and that it was on track to receive approval from most state Medicaid plans.
In 1 month, Krystal reported net Vyjuvek revenues of $8.6 million.
The authors suggested that one way to evaluate Vyjuvek’s value — especially for those with DDEB — would be through a cost-effectiveness study. While important, a cost-effectiveness study would not get at the impact on a payer, said Dr. Raymakers. “Something can be cost-effective but unaffordable to the system,” he said.
“When there’s one of these very expensive therapies, that’s one thing,” he said. “But when there’s more and more coming to market, you wonder how much can be tolerated,” said Dr. Raymakers.
CMS Launching Gene Therapy Program
The Biden administration recently announced that it was launching a program aimed at increasing access, curbing costs, and ensuring value of gene therapies, starting with sickle cell disease. The program will begin in early 2025. Among other aspects, the federal government will negotiate the price of the product with the manufacturer.
“The goal of the Cell and Gene Therapy Access Model is to increase access to innovative cell and gene therapies for people with Medicaid by making it easier for states to pay for these therapies,” said Liz Fowler, CMS Deputy Administrator and Director of the CMS Innovation Center, in a statement announcing the program.
Whether the new program takes a look at Vyjuvek – and when – is not clear.
But the authors of the study noted that the lifetime costs of treating a patient with Vyjuvek “exceed the costs of all other one-time gene therapies for other diseases.” And they wrote, even at the most conservative estimates, Vyjuvek “will be the most expensive gene therapy currently marketed in the US.”
The study was funded by a grant from Arnold Ventures, grants from the Kaiser Permanente Institute for Health Policy, the Commonwealth Fund, and the National Heart, Lung, and Blood Institute. Dr. Raymakers and co-authors reported no financial relationships relevant to the work.
according to the authors of a new study.
The US Food and Drug Administration (FDA) approved Vyjuvek (Krystal Biotech) in May 2023 for the treatment of wounds in patients ages 6 months and older with dystrophic epidermolysis bullosa (DEB), which includes two types, the most severe form (autosomal recessive, or RDEB) and the autosomal dominant form of DEB (DDEB), which tends to be milder.
Treatment with Vyjuvek “represents an important advance in the treatment of RDEB,” wrote Adam J.N. Raymakers, PhD, and colleagues at the Program on Regulation, Therapeutics, and Law; the Department of Dermatology; and the Division of Pulmonary and Critical Care Medicine at Brigham & Women’s Hospital in Boston, Massachusetts, in their paper, published in JAMA Dermatology. But the price “will be high, potentially limiting patients’ access to it,” they added. Evidence to support it in DDEB “is less conclusive,” they wrote, noting that the pivotal phase 3 study that led to approval included one patient with DDEB.
“The wider indication granted by the FDA may lead to friction between payers on the one hand and patients and physicians on the other,” they wrote, noting a potential minimum price of $300,000 per patient a year, which was based on Krystal’s regulatory filings.
There is no cure for DEB. Vyjuvek, applied as a gel on an ongoing basis, uses a nonreplicating herpes simplex virus type 1 vector to deliver the COL7A1 gene directly to skin cells, restoring the COL7 protein fibrils that stabilize skin structure.
The authors estimated that in the United States, 894 individuals – largely children – with both forms of the disease would be eligible for Vyjuvek treatment in the first year. Based on the $300,000 price, spending on gene therapy could range from $179 million to $357 million for those 894 patients, they reported in the study.
Over the first 3 years, spending could range as high as $1 billion, the authors estimated. Even if patients with only the most severe disease (RDEB) — an estimated 442 patients — received treatment, spending could be $132 million and up to $400 million or more over the first 3 years, they wrote.
Some media outlets have reported that Vyjuvek could cost as much as $600,000, said Dr. Raymakers, a research fellow. That price “would double all of our estimates,” he told this news organization.
The study assumed that patients with RDEB would only live to age 50, which led to a lifetime cost estimate of $15 million. But that is likely a conservative estimate, he and his coauthors wrote, noting that many patients with RDEB die from squamous cell carcinoma, but that Vyjuvek could, by attenuating skin damage, also potentially prevent skin cancer.
Dr. Raymakers said he and his colleagues began their study when Vyjuvek was approved, and thus they did not have any real-world data on the price or payer responses. Their estimates also did not include differing dosing regimens, which also could change their spending figures.
Krystal Biotech recently reported that in its third quarter of 2023 – representing just 1 month of Vyjuvek availability – it received requests to begin treatment for 284 patients from 136 unique clinicians. Twenty percent of the start requests were for patients with the milder form (DDEB), and a third of all the requests were for patients 10 years of age or younger. The company also said that it had “received positive coverage determinations from all major commercial national health plans” and that it was on track to receive approval from most state Medicaid plans.
In 1 month, Krystal reported net Vyjuvek revenues of $8.6 million.
The authors suggested that one way to evaluate Vyjuvek’s value — especially for those with DDEB — would be through a cost-effectiveness study. While important, a cost-effectiveness study would not get at the impact on a payer, said Dr. Raymakers. “Something can be cost-effective but unaffordable to the system,” he said.
“When there’s one of these very expensive therapies, that’s one thing,” he said. “But when there’s more and more coming to market, you wonder how much can be tolerated,” said Dr. Raymakers.
CMS Launching Gene Therapy Program
The Biden administration recently announced that it was launching a program aimed at increasing access, curbing costs, and ensuring value of gene therapies, starting with sickle cell disease. The program will begin in early 2025. Among other aspects, the federal government will negotiate the price of the product with the manufacturer.
“The goal of the Cell and Gene Therapy Access Model is to increase access to innovative cell and gene therapies for people with Medicaid by making it easier for states to pay for these therapies,” said Liz Fowler, CMS Deputy Administrator and Director of the CMS Innovation Center, in a statement announcing the program.
Whether the new program takes a look at Vyjuvek – and when – is not clear.
But the authors of the study noted that the lifetime costs of treating a patient with Vyjuvek “exceed the costs of all other one-time gene therapies for other diseases.” And they wrote, even at the most conservative estimates, Vyjuvek “will be the most expensive gene therapy currently marketed in the US.”
The study was funded by a grant from Arnold Ventures, grants from the Kaiser Permanente Institute for Health Policy, the Commonwealth Fund, and the National Heart, Lung, and Blood Institute. Dr. Raymakers and co-authors reported no financial relationships relevant to the work.
FROM JAMA DERMATOLOGY
Success with Sirolimus in Treating Skin Sarcoidosis Could Spur Studies in Other Organs
Sirolimus may be an effective treatment for patients with persistent cutaneous sarcoidosis.
In a small clinical trial, 7 of 10 patients treated with sirolimus via oral solution had improvements in skin lesions after 4 months, which was sustained for up to 2 years after the study concluded.
The results suggested that mechanistic target of rapamycin (mTOR) inhibition is a potential therapeutic avenue for sarcoidosis, which the authors said should be explored in larger clinical trials.
In the past decade, there has been a growing amount of evidence suggesting mTOR’s role in sarcoidosis. In 2017, researchers showed that activation of mTOR in macrophages could cause progressive sarcoidosis in mice. In additional studies, high levels of mTOR activity were detected in human sarcoidosis granulomas in various organs, including the skin, lung, and heart.
Three case reports also documented using the mTOR inhibitor sirolimus to effectively treat systemic sarcoidosis.
“Although all reports observed improvement of the disease following the treatment, no clinical trial investigating the efficacy and safety of sirolimus in patients with sarcoidosis had been published” prior to this study, wrote senior author Georg Stary, MD, of the Medical University of Vienna and the Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria, and colleagues.
The findings were published in the The Lancet Rheumatology.
For the study, researchers recruited 16 individuals with persistent and glucocorticoid-refractory cutaneous sarcoidosis between September 2019 and June 2021. A total of 14 participants were randomly assigned to the topical phase of the study, whereas two immediately received systemic treatment. All treatment was conducted at Vienna General Hospital.
In the placebo-controlled, double-blinded topical treatment arm, patients received either 0.1% topical sirolimus in Vaseline or Vaseline alone (placebo) twice daily for 2 months. After a 1-month washout period, participants were switched to the alternate treatment arm for an additional 2 months.
Following this topical phase and an additional 1-month washout period, all remaining participants received systemic sirolimus via a 1-mg/mL solution, starting with a 6-mg loading dose and continuing with 2 mg once daily for 4 months. The primary outcome was change in Cutaneous Sarcoidosis Activity and Morphology Index (CSAMI) from baseline, with decrease of more than five points representing a response to treatment.
A total of 10 patients completed the trial.
There was no change in CSAMI in either topical treatment groups. In the systemic group, 70% of patients had clinical improvement in skin lesions, with three responders in this group having complete resolution of skin lesions. The median change in CSAMI was −7.0 points (P = .018).
This improvement persisted for 2 months following study conclusion, with more pronounced improvement from baseline after 2 years of drug-free follow-up (−11.5 points).
There were no serious adverse events reported during the study, but 42% of patients treated with systemic sirolimus reported mild skin reactions, such as acne and eczema. Other related adverse events were hypertriglyceridemia (17%), hyperglycemia (17%), and proteinuria (8%).
Compared with clinical outcomes with tofacitinib and tumor necrosis factor (TNF) inhibitors, “the strength of our study lies in the sustained treatment effect after drug withdrawal among all responders. This prolonged effect has not yet been explored with tofacitinib, whereas with TNF inhibitors disease relapse was seen in more than 50% of patients at 3-8 months,” the authors wrote.
The researchers also analyzed participants’ skin biopsies to gain a better understanding of how mTOR inhibition affected granuloma structures. They found that, at baseline, mTOR activity was significantly lower in the fibroblasts of treatment nonresponders than in responders. They speculated that lower expression of mTOR could make these granuloma-associated cells resistant to systemic sirolimus.
These promising findings combine “clinical response with a molecular analysis,” Avrom Caplan, MD, co-director of the Sarcoidosis Program at NYU Langone in New York City, told this news organization. He was not involved with the research. Adding molecular information to clinical outcome data “helps solidify that [the mTOR] pathway has relevance in the sarcoid granuloma formation.”
The study had a limited sample size — a challenge for many clinical trials of rare diseases, Dr. Caplan said. Larger clinical trials are necessary to explore mTOR inhibition in sarcoidosis, both he and the authors agreed. A larger trial could also include greater heterogeneity of patients, including varied sarcoid presentation and demographics, Dr. Caplan noted. In this study, all but one participants were White individuals, and 63% of participants were female.
Larger studies could also address important questions on ideal length of therapy, dosing, and where this therapy “would fall within the therapeutic step ladder,” Dr. Caplan continued.
Whether mTOR inhibition could be effective at treating individuals with sarcoidosis in other organs beyond the skin is also unknown.
“If the pathogenesis of sarcoid granuloma formation does include mTOR upregulation, which they are showing here…then you could hypothesize that, yes, using this therapy could benefit other organs,” he said. “But that has to be investigated in larger trials.”
The study was funded in part by a Vienna Science and Technology Fund project. Several authors report receiving grants from the Austrian Science Fund and one from the Ann Theodore Foundation Breakthrough Sarcoidosis Initiative. Dr. Caplan reported no relevant financial relationships.
A version of this article appeared on Medscape.com .
Sirolimus may be an effective treatment for patients with persistent cutaneous sarcoidosis.
In a small clinical trial, 7 of 10 patients treated with sirolimus via oral solution had improvements in skin lesions after 4 months, which was sustained for up to 2 years after the study concluded.
The results suggested that mechanistic target of rapamycin (mTOR) inhibition is a potential therapeutic avenue for sarcoidosis, which the authors said should be explored in larger clinical trials.
In the past decade, there has been a growing amount of evidence suggesting mTOR’s role in sarcoidosis. In 2017, researchers showed that activation of mTOR in macrophages could cause progressive sarcoidosis in mice. In additional studies, high levels of mTOR activity were detected in human sarcoidosis granulomas in various organs, including the skin, lung, and heart.
Three case reports also documented using the mTOR inhibitor sirolimus to effectively treat systemic sarcoidosis.
“Although all reports observed improvement of the disease following the treatment, no clinical trial investigating the efficacy and safety of sirolimus in patients with sarcoidosis had been published” prior to this study, wrote senior author Georg Stary, MD, of the Medical University of Vienna and the Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria, and colleagues.
The findings were published in the The Lancet Rheumatology.
For the study, researchers recruited 16 individuals with persistent and glucocorticoid-refractory cutaneous sarcoidosis between September 2019 and June 2021. A total of 14 participants were randomly assigned to the topical phase of the study, whereas two immediately received systemic treatment. All treatment was conducted at Vienna General Hospital.
In the placebo-controlled, double-blinded topical treatment arm, patients received either 0.1% topical sirolimus in Vaseline or Vaseline alone (placebo) twice daily for 2 months. After a 1-month washout period, participants were switched to the alternate treatment arm for an additional 2 months.
Following this topical phase and an additional 1-month washout period, all remaining participants received systemic sirolimus via a 1-mg/mL solution, starting with a 6-mg loading dose and continuing with 2 mg once daily for 4 months. The primary outcome was change in Cutaneous Sarcoidosis Activity and Morphology Index (CSAMI) from baseline, with decrease of more than five points representing a response to treatment.
A total of 10 patients completed the trial.
There was no change in CSAMI in either topical treatment groups. In the systemic group, 70% of patients had clinical improvement in skin lesions, with three responders in this group having complete resolution of skin lesions. The median change in CSAMI was −7.0 points (P = .018).
This improvement persisted for 2 months following study conclusion, with more pronounced improvement from baseline after 2 years of drug-free follow-up (−11.5 points).
There were no serious adverse events reported during the study, but 42% of patients treated with systemic sirolimus reported mild skin reactions, such as acne and eczema. Other related adverse events were hypertriglyceridemia (17%), hyperglycemia (17%), and proteinuria (8%).
Compared with clinical outcomes with tofacitinib and tumor necrosis factor (TNF) inhibitors, “the strength of our study lies in the sustained treatment effect after drug withdrawal among all responders. This prolonged effect has not yet been explored with tofacitinib, whereas with TNF inhibitors disease relapse was seen in more than 50% of patients at 3-8 months,” the authors wrote.
The researchers also analyzed participants’ skin biopsies to gain a better understanding of how mTOR inhibition affected granuloma structures. They found that, at baseline, mTOR activity was significantly lower in the fibroblasts of treatment nonresponders than in responders. They speculated that lower expression of mTOR could make these granuloma-associated cells resistant to systemic sirolimus.
These promising findings combine “clinical response with a molecular analysis,” Avrom Caplan, MD, co-director of the Sarcoidosis Program at NYU Langone in New York City, told this news organization. He was not involved with the research. Adding molecular information to clinical outcome data “helps solidify that [the mTOR] pathway has relevance in the sarcoid granuloma formation.”
The study had a limited sample size — a challenge for many clinical trials of rare diseases, Dr. Caplan said. Larger clinical trials are necessary to explore mTOR inhibition in sarcoidosis, both he and the authors agreed. A larger trial could also include greater heterogeneity of patients, including varied sarcoid presentation and demographics, Dr. Caplan noted. In this study, all but one participants were White individuals, and 63% of participants were female.
Larger studies could also address important questions on ideal length of therapy, dosing, and where this therapy “would fall within the therapeutic step ladder,” Dr. Caplan continued.
Whether mTOR inhibition could be effective at treating individuals with sarcoidosis in other organs beyond the skin is also unknown.
“If the pathogenesis of sarcoid granuloma formation does include mTOR upregulation, which they are showing here…then you could hypothesize that, yes, using this therapy could benefit other organs,” he said. “But that has to be investigated in larger trials.”
The study was funded in part by a Vienna Science and Technology Fund project. Several authors report receiving grants from the Austrian Science Fund and one from the Ann Theodore Foundation Breakthrough Sarcoidosis Initiative. Dr. Caplan reported no relevant financial relationships.
A version of this article appeared on Medscape.com .
Sirolimus may be an effective treatment for patients with persistent cutaneous sarcoidosis.
In a small clinical trial, 7 of 10 patients treated with sirolimus via oral solution had improvements in skin lesions after 4 months, which was sustained for up to 2 years after the study concluded.
The results suggested that mechanistic target of rapamycin (mTOR) inhibition is a potential therapeutic avenue for sarcoidosis, which the authors said should be explored in larger clinical trials.
In the past decade, there has been a growing amount of evidence suggesting mTOR’s role in sarcoidosis. In 2017, researchers showed that activation of mTOR in macrophages could cause progressive sarcoidosis in mice. In additional studies, high levels of mTOR activity were detected in human sarcoidosis granulomas in various organs, including the skin, lung, and heart.
Three case reports also documented using the mTOR inhibitor sirolimus to effectively treat systemic sarcoidosis.
“Although all reports observed improvement of the disease following the treatment, no clinical trial investigating the efficacy and safety of sirolimus in patients with sarcoidosis had been published” prior to this study, wrote senior author Georg Stary, MD, of the Medical University of Vienna and the Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria, and colleagues.
The findings were published in the The Lancet Rheumatology.
For the study, researchers recruited 16 individuals with persistent and glucocorticoid-refractory cutaneous sarcoidosis between September 2019 and June 2021. A total of 14 participants were randomly assigned to the topical phase of the study, whereas two immediately received systemic treatment. All treatment was conducted at Vienna General Hospital.
In the placebo-controlled, double-blinded topical treatment arm, patients received either 0.1% topical sirolimus in Vaseline or Vaseline alone (placebo) twice daily for 2 months. After a 1-month washout period, participants were switched to the alternate treatment arm for an additional 2 months.
Following this topical phase and an additional 1-month washout period, all remaining participants received systemic sirolimus via a 1-mg/mL solution, starting with a 6-mg loading dose and continuing with 2 mg once daily for 4 months. The primary outcome was change in Cutaneous Sarcoidosis Activity and Morphology Index (CSAMI) from baseline, with decrease of more than five points representing a response to treatment.
A total of 10 patients completed the trial.
There was no change in CSAMI in either topical treatment groups. In the systemic group, 70% of patients had clinical improvement in skin lesions, with three responders in this group having complete resolution of skin lesions. The median change in CSAMI was −7.0 points (P = .018).
This improvement persisted for 2 months following study conclusion, with more pronounced improvement from baseline after 2 years of drug-free follow-up (−11.5 points).
There were no serious adverse events reported during the study, but 42% of patients treated with systemic sirolimus reported mild skin reactions, such as acne and eczema. Other related adverse events were hypertriglyceridemia (17%), hyperglycemia (17%), and proteinuria (8%).
Compared with clinical outcomes with tofacitinib and tumor necrosis factor (TNF) inhibitors, “the strength of our study lies in the sustained treatment effect after drug withdrawal among all responders. This prolonged effect has not yet been explored with tofacitinib, whereas with TNF inhibitors disease relapse was seen in more than 50% of patients at 3-8 months,” the authors wrote.
The researchers also analyzed participants’ skin biopsies to gain a better understanding of how mTOR inhibition affected granuloma structures. They found that, at baseline, mTOR activity was significantly lower in the fibroblasts of treatment nonresponders than in responders. They speculated that lower expression of mTOR could make these granuloma-associated cells resistant to systemic sirolimus.
These promising findings combine “clinical response with a molecular analysis,” Avrom Caplan, MD, co-director of the Sarcoidosis Program at NYU Langone in New York City, told this news organization. He was not involved with the research. Adding molecular information to clinical outcome data “helps solidify that [the mTOR] pathway has relevance in the sarcoid granuloma formation.”
The study had a limited sample size — a challenge for many clinical trials of rare diseases, Dr. Caplan said. Larger clinical trials are necessary to explore mTOR inhibition in sarcoidosis, both he and the authors agreed. A larger trial could also include greater heterogeneity of patients, including varied sarcoid presentation and demographics, Dr. Caplan noted. In this study, all but one participants were White individuals, and 63% of participants were female.
Larger studies could also address important questions on ideal length of therapy, dosing, and where this therapy “would fall within the therapeutic step ladder,” Dr. Caplan continued.
Whether mTOR inhibition could be effective at treating individuals with sarcoidosis in other organs beyond the skin is also unknown.
“If the pathogenesis of sarcoid granuloma formation does include mTOR upregulation, which they are showing here…then you could hypothesize that, yes, using this therapy could benefit other organs,” he said. “But that has to be investigated in larger trials.”
The study was funded in part by a Vienna Science and Technology Fund project. Several authors report receiving grants from the Austrian Science Fund and one from the Ann Theodore Foundation Breakthrough Sarcoidosis Initiative. Dr. Caplan reported no relevant financial relationships.
A version of this article appeared on Medscape.com .
FROM THE LANCET RHEUMATOLOGY
Cutaneous lupus, dermatomyositis: Excitement growing around emerging therapies
ORLANDO, FLORIDA — Advances in treating medical conditions rarely emerge in a straight line. Oftentimes, progress comes in fits and starts, and therapies to treat cutaneous lupus erythematosus (CLE) and dermatomyositis are no exception.
Beyond approved treatments that deserve more attention, like belimumab, approved by the Food and Drug Administration (FDA) for systemic lupus erythematosus (SLE) in 2011, and Octagam 10%, an intravenous immune globulin (IVIG) preparation approved for dermatomyositis in 2021, anticipation is growing for emerging therapies and their potential to provide relief to patients, Anthony Fernandez, MD, PhD, said at the ODAC Dermatology, Aesthetic & Surgical Conference. The tyrosine kinase 2 (TYK2) inhibitor deucravacitinib, Janus kinase (JAK) inhibitors brepocitinib and baricitinib, and the monoclonal antibody anifrolumab, he noted, are prime examples.
“. In my opinion, this is the start of what will be the most exciting decade in the history of these two diseases,” said Dr. Fernandez, director of medical dermatology at the Cleveland Clinic.
Emerging Treatments for Cutaneous Lupus
Although SLE can involve many organ systems, the skin is one of the most affected. There are specific cutaneous lesions categorized as either acute cutaneous lupus, subacute cutaneous lupus, or chronic cutaneous lupus.
The oral TYK2 inhibitor deucravacitinib, for example, should be able to dampen interleukin responses in people with CLE, Dr. Fernandez said. Deucravacitinib was approved by the FDA to treat psoriasis in September 2022.

A phase 2 study published in 2023 focused on this agent for relief of systemic lupus. Improvements in cutaneous disease were a secondary endpoint. The trial demonstrated that the patients treated with deucravacitinib achieved a 56%-70% CLASI-50 response, depending on dosing, compared with a 17% response among those on placebo at week 48.
Based on the trial results, recruitment has begun for a phase 2 trial to evaluate deucravacitinib, compared with placebo, in patients with discoid and/or subacute cutaneous lupus. “This may be another medicine we have available to give to any of our patients with cutaneous lupus,” Dr. Fernandez said.
Anifrolumab Appears Promising
The FDA approval of anifrolumab, a type I interferon (IFN) receptor antagonist, for treating moderate to severe SLE in July 2021, for example, is good news for dermatologists and their patients, added Dr. Fernandez. “Almost immediately after approval, case studies showed marked improvement in patients with refractory cutaneous lupus.” While the therapy was approved for treating systemic lupus, it allows for off-label treatment of the cutaneous predominant form of the disease, he said.
Furthermore, the manufacturer of anifrolumab, AstraZeneca, is launching the LAVENDER clinical trial to assess the monoclonal antibody specifically for treating CLE. “This is a big deal because we may be able to prescribe anifrolumab for our cutaneous lupus patients who don’t have systemic lupus,” Dr. Fernandez said.
Phase 3 data supported use the of anifrolumab in systemic lupus, including the TULIP-2 trial, which demonstrated its superiority to placebo for reducing severity of systemic disease and lowering corticosteroid use. A study published in March 2023 of 11 patients showed that they had a “very fast response” to the agent, Dr. Fernandez said, with a 50% or greater improvement in the Cutaneous Lupus Erythematosus Disease Area and Severity Index activity score reached by all participants at week 16. Improvements of 50% or more in this scoring system are considered clinically meaningful, he added.
Upcoming Dermatomyositis Treatments
Why highlight emerging therapies for CLE and dermatomyositis in the same ODAC presentation? Although distinct conditions, these autoimmune conditions are both mediated by type 1 IFN inflammation.
Dermatomyositis is a relatively rare immune-mediated disease that most commonly affects the skin and muscle. Doctors score disease presentation, activity, and clinical improvements on a scale similar to CLASI for cutaneous lupus, the CDASI or Cutaneous Dermatomyositis Disease Area and Severity Index. Among people with CDASI activity scores of at least 14, which is the threshold for moderate to severe disease, a 20% improvement is clinically meaningful, Dr. Fernandez said. In addition, a 40% or greater improvement correlates with significant improvements in quality of life.
There is now more evidence for the use of IVIG to treat dermatomyositis. “Among those of us who treat dermatomyositis on a regular basis, we believe IVIG is the most potent treatment. We’ve known that for a long time,” Dr. Fernandez said.
Despite this tenet, for years, there was only one placebo-controlled trial, published in 1993, that evaluated IVIG treatment for dermatomyositis, and it included only 15 participants. That was until October 2022, he said, when the New England Journal of Medicine published a study comparing a specific brand of IVIG (Octagam) with placebo in 95 people with dermatomyositis.
In the study, 79% of participants treated with IVIG had a total improvement score of at least 20 (minimal improvement), the primary endpoint, at 16 weeks, compared with 44% of those receiving a placebo. Those treated with IVIG also had significant improvements in the CDASI score, a secondary endpoint, compared with those on placebo, he said.
Based on results of this trial, the FDA approved Octagam 10% for dermatomyositis in adults. Dr. Fernandez noted the approval is restricted to the brand of IVIG in the trial, not all IVIG products. However, “the FDA approval is most important to us because it gives us ammunition to fight for insurers to approve IVIG when we feel our patients with dermatomyositis need it,” regardless of the brand.
The Potential of JAK1 Inhibitors
An open-label study of the JAK inhibitor tofacitinib, published in December 2020, showed that mean changes in CDASI activity scores at 12 weeks were statistically significant compared with baseline in 10 people with dermatomyositis. “The importance of this study is that it is proof of concept that JAK inhibition can be effective for treating dermatomyositis, especially with active skin disease,” Dr. Fernandez said.
In addition, two large phase 3 trials are evaluating JAK inhibitor safety and efficacy for treating dermatomyositis. One is the VALOR trial, currently recruiting people with recalcitrant dermatomyositis to evaluate treatment with brepocitinib. Researchers in France are looking at another JAK inhibitor, baricitinib, for treating relapsing or treatment-naive dermatomyositis. Recruitment for the BIRD clinical trial is ongoing.
Monoclonal Antibody Showing Promise
“When it comes to looking specifically at dermatomyositis cutaneous disease, it’s been found that the levels of IFN beta correlate best with not only lesional skin type 1 IFN inflammatory signatures but also overall clinical disease activity,” Dr. Fernandez said. This correlation is stronger than for any other IFN-1-type cytokine active in the disorder.
“Perhaps blocking IFN beta might be best way to get control of dermatomyositis activity,” he added.
With that in mind, a phase 2 trial of dazukibart presented at the American Academy of Dermatology 2023 annual meeting highlighted the promise of this agent that targets type 1 IFN beta.
The primary endpoint was improvement in CDASI at 12 weeks. “This medication has remarkable efficacy,” Dr. Fernandez said. “We were one of the sites for this trial. Despite being blinded, there was no question about who was receiving drug and who was receiving placebo.”
“A minimal clinical improvement in disease activity was seen in more than 90%, so almost every patient who received this medication had meaningful improvement,” he added.
Based on the results, the manufacturer, Pfizer, is recruiting participants for a phase 3 trial to further assess dazukibart in dermatomyositis and polymyositis. Dr. Fernandez said, “This is a story you should pay attention to if you treat any dermatomyositis patients at all.”
Regarding these emerging therapies for CLE and dermatomyositis, “This looks very much like the early days of psoriasis, in the early 2000s, when there was a lot of activity developing treatments,” Dr. Fernandez said. “I will predict that within 10 years, we will have multiple novel agents available that will probably work better than anything we have today.”
Dr. Fernandez reported receiving grant and/or research support from Alexion, Incyte, Mallinckrodt Pharmaceuticals, Novartis, Pfizer, and Priovant Therapeutics; acting as a consultant or advisory board member for AbbVie, Biogen, Mallinckrodt Pharmaceuticals; and being a member of the speaker bureau or receiving honoraria for non-CME from AbbVie, Kyowa Kirin, and Mallinckrodt Pharmaceuticals.
A version of this article appeared on Medscape.com.
ORLANDO, FLORIDA — Advances in treating medical conditions rarely emerge in a straight line. Oftentimes, progress comes in fits and starts, and therapies to treat cutaneous lupus erythematosus (CLE) and dermatomyositis are no exception.
Beyond approved treatments that deserve more attention, like belimumab, approved by the Food and Drug Administration (FDA) for systemic lupus erythematosus (SLE) in 2011, and Octagam 10%, an intravenous immune globulin (IVIG) preparation approved for dermatomyositis in 2021, anticipation is growing for emerging therapies and their potential to provide relief to patients, Anthony Fernandez, MD, PhD, said at the ODAC Dermatology, Aesthetic & Surgical Conference. The tyrosine kinase 2 (TYK2) inhibitor deucravacitinib, Janus kinase (JAK) inhibitors brepocitinib and baricitinib, and the monoclonal antibody anifrolumab, he noted, are prime examples.
“. In my opinion, this is the start of what will be the most exciting decade in the history of these two diseases,” said Dr. Fernandez, director of medical dermatology at the Cleveland Clinic.
Emerging Treatments for Cutaneous Lupus
Although SLE can involve many organ systems, the skin is one of the most affected. There are specific cutaneous lesions categorized as either acute cutaneous lupus, subacute cutaneous lupus, or chronic cutaneous lupus.
The oral TYK2 inhibitor deucravacitinib, for example, should be able to dampen interleukin responses in people with CLE, Dr. Fernandez said. Deucravacitinib was approved by the FDA to treat psoriasis in September 2022.
A phase 2 study published in 2023 focused on this agent for relief of systemic lupus. Improvements in cutaneous disease were a secondary endpoint. The trial demonstrated that the patients treated with deucravacitinib achieved a 56%-70% CLASI-50 response, depending on dosing, compared with a 17% response among those on placebo at week 48.
Based on the trial results, recruitment has begun for a phase 2 trial to evaluate deucravacitinib, compared with placebo, in patients with discoid and/or subacute cutaneous lupus. “This may be another medicine we have available to give to any of our patients with cutaneous lupus,” Dr. Fernandez said.
Anifrolumab Appears Promising
The FDA approval of anifrolumab, a type I interferon (IFN) receptor antagonist, for treating moderate to severe SLE in July 2021, for example, is good news for dermatologists and their patients, added Dr. Fernandez. “Almost immediately after approval, case studies showed marked improvement in patients with refractory cutaneous lupus.” While the therapy was approved for treating systemic lupus, it allows for off-label treatment of the cutaneous predominant form of the disease, he said.
Furthermore, the manufacturer of anifrolumab, AstraZeneca, is launching the LAVENDER clinical trial to assess the monoclonal antibody specifically for treating CLE. “This is a big deal because we may be able to prescribe anifrolumab for our cutaneous lupus patients who don’t have systemic lupus,” Dr. Fernandez said.
Phase 3 data supported use the of anifrolumab in systemic lupus, including the TULIP-2 trial, which demonstrated its superiority to placebo for reducing severity of systemic disease and lowering corticosteroid use. A study published in March 2023 of 11 patients showed that they had a “very fast response” to the agent, Dr. Fernandez said, with a 50% or greater improvement in the Cutaneous Lupus Erythematosus Disease Area and Severity Index activity score reached by all participants at week 16. Improvements of 50% or more in this scoring system are considered clinically meaningful, he added.
Upcoming Dermatomyositis Treatments
Why highlight emerging therapies for CLE and dermatomyositis in the same ODAC presentation? Although distinct conditions, these autoimmune conditions are both mediated by type 1 IFN inflammation.
Dermatomyositis is a relatively rare immune-mediated disease that most commonly affects the skin and muscle. Doctors score disease presentation, activity, and clinical improvements on a scale similar to CLASI for cutaneous lupus, the CDASI or Cutaneous Dermatomyositis Disease Area and Severity Index. Among people with CDASI activity scores of at least 14, which is the threshold for moderate to severe disease, a 20% improvement is clinically meaningful, Dr. Fernandez said. In addition, a 40% or greater improvement correlates with significant improvements in quality of life.
There is now more evidence for the use of IVIG to treat dermatomyositis. “Among those of us who treat dermatomyositis on a regular basis, we believe IVIG is the most potent treatment. We’ve known that for a long time,” Dr. Fernandez said.
Despite this tenet, for years, there was only one placebo-controlled trial, published in 1993, that evaluated IVIG treatment for dermatomyositis, and it included only 15 participants. That was until October 2022, he said, when the New England Journal of Medicine published a study comparing a specific brand of IVIG (Octagam) with placebo in 95 people with dermatomyositis.
In the study, 79% of participants treated with IVIG had a total improvement score of at least 20 (minimal improvement), the primary endpoint, at 16 weeks, compared with 44% of those receiving a placebo. Those treated with IVIG also had significant improvements in the CDASI score, a secondary endpoint, compared with those on placebo, he said.
Based on results of this trial, the FDA approved Octagam 10% for dermatomyositis in adults. Dr. Fernandez noted the approval is restricted to the brand of IVIG in the trial, not all IVIG products. However, “the FDA approval is most important to us because it gives us ammunition to fight for insurers to approve IVIG when we feel our patients with dermatomyositis need it,” regardless of the brand.
The Potential of JAK1 Inhibitors
An open-label study of the JAK inhibitor tofacitinib, published in December 2020, showed that mean changes in CDASI activity scores at 12 weeks were statistically significant compared with baseline in 10 people with dermatomyositis. “The importance of this study is that it is proof of concept that JAK inhibition can be effective for treating dermatomyositis, especially with active skin disease,” Dr. Fernandez said.
In addition, two large phase 3 trials are evaluating JAK inhibitor safety and efficacy for treating dermatomyositis. One is the VALOR trial, currently recruiting people with recalcitrant dermatomyositis to evaluate treatment with brepocitinib. Researchers in France are looking at another JAK inhibitor, baricitinib, for treating relapsing or treatment-naive dermatomyositis. Recruitment for the BIRD clinical trial is ongoing.
Monoclonal Antibody Showing Promise
“When it comes to looking specifically at dermatomyositis cutaneous disease, it’s been found that the levels of IFN beta correlate best with not only lesional skin type 1 IFN inflammatory signatures but also overall clinical disease activity,” Dr. Fernandez said. This correlation is stronger than for any other IFN-1-type cytokine active in the disorder.
“Perhaps blocking IFN beta might be best way to get control of dermatomyositis activity,” he added.
With that in mind, a phase 2 trial of dazukibart presented at the American Academy of Dermatology 2023 annual meeting highlighted the promise of this agent that targets type 1 IFN beta.
The primary endpoint was improvement in CDASI at 12 weeks. “This medication has remarkable efficacy,” Dr. Fernandez said. “We were one of the sites for this trial. Despite being blinded, there was no question about who was receiving drug and who was receiving placebo.”
“A minimal clinical improvement in disease activity was seen in more than 90%, so almost every patient who received this medication had meaningful improvement,” he added.
Based on the results, the manufacturer, Pfizer, is recruiting participants for a phase 3 trial to further assess dazukibart in dermatomyositis and polymyositis. Dr. Fernandez said, “This is a story you should pay attention to if you treat any dermatomyositis patients at all.”
Regarding these emerging therapies for CLE and dermatomyositis, “This looks very much like the early days of psoriasis, in the early 2000s, when there was a lot of activity developing treatments,” Dr. Fernandez said. “I will predict that within 10 years, we will have multiple novel agents available that will probably work better than anything we have today.”
Dr. Fernandez reported receiving grant and/or research support from Alexion, Incyte, Mallinckrodt Pharmaceuticals, Novartis, Pfizer, and Priovant Therapeutics; acting as a consultant or advisory board member for AbbVie, Biogen, Mallinckrodt Pharmaceuticals; and being a member of the speaker bureau or receiving honoraria for non-CME from AbbVie, Kyowa Kirin, and Mallinckrodt Pharmaceuticals.
A version of this article appeared on Medscape.com.
ORLANDO, FLORIDA — Advances in treating medical conditions rarely emerge in a straight line. Oftentimes, progress comes in fits and starts, and therapies to treat cutaneous lupus erythematosus (CLE) and dermatomyositis are no exception.
Beyond approved treatments that deserve more attention, like belimumab, approved by the Food and Drug Administration (FDA) for systemic lupus erythematosus (SLE) in 2011, and Octagam 10%, an intravenous immune globulin (IVIG) preparation approved for dermatomyositis in 2021, anticipation is growing for emerging therapies and their potential to provide relief to patients, Anthony Fernandez, MD, PhD, said at the ODAC Dermatology, Aesthetic & Surgical Conference. The tyrosine kinase 2 (TYK2) inhibitor deucravacitinib, Janus kinase (JAK) inhibitors brepocitinib and baricitinib, and the monoclonal antibody anifrolumab, he noted, are prime examples.
“. In my opinion, this is the start of what will be the most exciting decade in the history of these two diseases,” said Dr. Fernandez, director of medical dermatology at the Cleveland Clinic.
Emerging Treatments for Cutaneous Lupus
Although SLE can involve many organ systems, the skin is one of the most affected. There are specific cutaneous lesions categorized as either acute cutaneous lupus, subacute cutaneous lupus, or chronic cutaneous lupus.
The oral TYK2 inhibitor deucravacitinib, for example, should be able to dampen interleukin responses in people with CLE, Dr. Fernandez said. Deucravacitinib was approved by the FDA to treat psoriasis in September 2022.
A phase 2 study published in 2023 focused on this agent for relief of systemic lupus. Improvements in cutaneous disease were a secondary endpoint. The trial demonstrated that the patients treated with deucravacitinib achieved a 56%-70% CLASI-50 response, depending on dosing, compared with a 17% response among those on placebo at week 48.
Based on the trial results, recruitment has begun for a phase 2 trial to evaluate deucravacitinib, compared with placebo, in patients with discoid and/or subacute cutaneous lupus. “This may be another medicine we have available to give to any of our patients with cutaneous lupus,” Dr. Fernandez said.
Anifrolumab Appears Promising
The FDA approval of anifrolumab, a type I interferon (IFN) receptor antagonist, for treating moderate to severe SLE in July 2021, for example, is good news for dermatologists and their patients, added Dr. Fernandez. “Almost immediately after approval, case studies showed marked improvement in patients with refractory cutaneous lupus.” While the therapy was approved for treating systemic lupus, it allows for off-label treatment of the cutaneous predominant form of the disease, he said.
Furthermore, the manufacturer of anifrolumab, AstraZeneca, is launching the LAVENDER clinical trial to assess the monoclonal antibody specifically for treating CLE. “This is a big deal because we may be able to prescribe anifrolumab for our cutaneous lupus patients who don’t have systemic lupus,” Dr. Fernandez said.
Phase 3 data supported use the of anifrolumab in systemic lupus, including the TULIP-2 trial, which demonstrated its superiority to placebo for reducing severity of systemic disease and lowering corticosteroid use. A study published in March 2023 of 11 patients showed that they had a “very fast response” to the agent, Dr. Fernandez said, with a 50% or greater improvement in the Cutaneous Lupus Erythematosus Disease Area and Severity Index activity score reached by all participants at week 16. Improvements of 50% or more in this scoring system are considered clinically meaningful, he added.
Upcoming Dermatomyositis Treatments
Why highlight emerging therapies for CLE and dermatomyositis in the same ODAC presentation? Although distinct conditions, these autoimmune conditions are both mediated by type 1 IFN inflammation.
Dermatomyositis is a relatively rare immune-mediated disease that most commonly affects the skin and muscle. Doctors score disease presentation, activity, and clinical improvements on a scale similar to CLASI for cutaneous lupus, the CDASI or Cutaneous Dermatomyositis Disease Area and Severity Index. Among people with CDASI activity scores of at least 14, which is the threshold for moderate to severe disease, a 20% improvement is clinically meaningful, Dr. Fernandez said. In addition, a 40% or greater improvement correlates with significant improvements in quality of life.
There is now more evidence for the use of IVIG to treat dermatomyositis. “Among those of us who treat dermatomyositis on a regular basis, we believe IVIG is the most potent treatment. We’ve known that for a long time,” Dr. Fernandez said.
Despite this tenet, for years, there was only one placebo-controlled trial, published in 1993, that evaluated IVIG treatment for dermatomyositis, and it included only 15 participants. That was until October 2022, he said, when the New England Journal of Medicine published a study comparing a specific brand of IVIG (Octagam) with placebo in 95 people with dermatomyositis.
In the study, 79% of participants treated with IVIG had a total improvement score of at least 20 (minimal improvement), the primary endpoint, at 16 weeks, compared with 44% of those receiving a placebo. Those treated with IVIG also had significant improvements in the CDASI score, a secondary endpoint, compared with those on placebo, he said.
Based on results of this trial, the FDA approved Octagam 10% for dermatomyositis in adults. Dr. Fernandez noted the approval is restricted to the brand of IVIG in the trial, not all IVIG products. However, “the FDA approval is most important to us because it gives us ammunition to fight for insurers to approve IVIG when we feel our patients with dermatomyositis need it,” regardless of the brand.
The Potential of JAK1 Inhibitors
An open-label study of the JAK inhibitor tofacitinib, published in December 2020, showed that mean changes in CDASI activity scores at 12 weeks were statistically significant compared with baseline in 10 people with dermatomyositis. “The importance of this study is that it is proof of concept that JAK inhibition can be effective for treating dermatomyositis, especially with active skin disease,” Dr. Fernandez said.
In addition, two large phase 3 trials are evaluating JAK inhibitor safety and efficacy for treating dermatomyositis. One is the VALOR trial, currently recruiting people with recalcitrant dermatomyositis to evaluate treatment with brepocitinib. Researchers in France are looking at another JAK inhibitor, baricitinib, for treating relapsing or treatment-naive dermatomyositis. Recruitment for the BIRD clinical trial is ongoing.
Monoclonal Antibody Showing Promise
“When it comes to looking specifically at dermatomyositis cutaneous disease, it’s been found that the levels of IFN beta correlate best with not only lesional skin type 1 IFN inflammatory signatures but also overall clinical disease activity,” Dr. Fernandez said. This correlation is stronger than for any other IFN-1-type cytokine active in the disorder.
“Perhaps blocking IFN beta might be best way to get control of dermatomyositis activity,” he added.
With that in mind, a phase 2 trial of dazukibart presented at the American Academy of Dermatology 2023 annual meeting highlighted the promise of this agent that targets type 1 IFN beta.
The primary endpoint was improvement in CDASI at 12 weeks. “This medication has remarkable efficacy,” Dr. Fernandez said. “We were one of the sites for this trial. Despite being blinded, there was no question about who was receiving drug and who was receiving placebo.”
“A minimal clinical improvement in disease activity was seen in more than 90%, so almost every patient who received this medication had meaningful improvement,” he added.
Based on the results, the manufacturer, Pfizer, is recruiting participants for a phase 3 trial to further assess dazukibart in dermatomyositis and polymyositis. Dr. Fernandez said, “This is a story you should pay attention to if you treat any dermatomyositis patients at all.”
Regarding these emerging therapies for CLE and dermatomyositis, “This looks very much like the early days of psoriasis, in the early 2000s, when there was a lot of activity developing treatments,” Dr. Fernandez said. “I will predict that within 10 years, we will have multiple novel agents available that will probably work better than anything we have today.”
Dr. Fernandez reported receiving grant and/or research support from Alexion, Incyte, Mallinckrodt Pharmaceuticals, Novartis, Pfizer, and Priovant Therapeutics; acting as a consultant or advisory board member for AbbVie, Biogen, Mallinckrodt Pharmaceuticals; and being a member of the speaker bureau or receiving honoraria for non-CME from AbbVie, Kyowa Kirin, and Mallinckrodt Pharmaceuticals.
A version of this article appeared on Medscape.com.
FROM ODAC 2024
Hair Loss in Children: How to Spot and Treat Different Causes
ORLANDO, FLORIDA — There are subtleties and nuances to diagnosing, treating, and monitoring the progress of treatment of hair loss in children. Moreover, hair loss in children can be challenging because it can be caused by a range of conditions, some common and others relatively rare.
Michelle Oboite, MD, shared tips on how to distinguish types of hair loss, when to treat with medications such as topical corticosteroids or Janus kinase (JAK) inhibitors, and why shared decision-making is important, at the ODAC Dermatology, Aesthetic & Surgical Conference.
What these conditions share is that they can negatively affect the quality of life for a child or teenager when the condition leads to anxiety, teasing, or bullying. “It is very isolating to have this condition that everyone in the world can see that you have and judge you for it,” said Dr. Oboite, an attending physician in the dermatology section of Children’s Hospital of Philadelphia.
Others are lichen planopilaris and genetic conditions, including loose anagen syndrome, uncombable hair syndrome, and “something so rare” — it has no acronym — autosomal recessive hypotrichosis with recurrent skin vesicles, Dr. Oboite said.
Alopecia Areata
Alopecia areata can differ from child to child and can appear in different stages: A localized patch stage, a diffuse patchy stage, or alopecia universalis. In this last stage, the child has already lost most or all the hair on the scalp and eyebrows, as well as the eyelashes.
The decision to treat or not to treat, particularly in younger children, should be on the basis of shared decision-making between a healthcare provider and caregiver, said Dr. Oboite, who is also an assistant professor of clinical dermatology at the University of Pennsylvania, Philadelphia.
Some younger children may not experience any negative impact from the condition, so waiting until they are older is an option.
Also, consider the impact of treatment on a child. Some therapies require frequent blood draws for monitoring, and some topical therapies that are applied multiple times a day “can be very overwhelming” for young children, Dr. Oboite said.
Most children with alopecia areata are healthy and do not need extensive screening laboratory testing. However, one exception is if thyroid dysfunction, commonly associated with alopecia areata, is suspected.
For alopecia areata, Dr. Oboite recommends starting with topical therapies, either topical corticosteroids (as first line) or topical JAK inhibitors (either topical ruxolitinib or compounded topical tofacitinib, both off-label for this indication).
Topical corticosteroids can be effective, but “you want to be thoughtful of the strength you’re using, the application frequency, and then the total amount of surface area that you’re treating,” Dr. Oboite said. Too potent or too much of a topical corticosteroid increases the risk for atrophy and systemic absorption, respectively. To reduce the risk, she reserves the use of ultrahigh-potency topical corticosteroids, such as clobetasol, for children ages 10 years or older. For children younger than 10 years, she recommends using mid-high-potency topical corticosteroids instead.
She recommends once-a-day application around bedtime 5 days a week, generally Monday through Friday to make it easier to remember.
“For children who have over 50% of the scalp involved, I do consider systemic therapy,” Dr. Oboite said. This can include oral steroids such as dexamethasone, prednisone, or prednisolone. For children with recalcitrant disease, she is more likely to use the oral JAK inhibitor ritlecitinib because it was recently approved by the Food and Drug Administration for treating severe alopecia areata in children 12 years and older and in adults.
Another strategy Dr. Oboite uses is to add low-dose oral minoxidil as an adjuvant to other systemic therapy. “I find that it helps with faster hair regrowth,” she said.
Tinea Capitis
Oral treatment is indicated for tinea capitis. “Topicals just don’t really clear this,” Dr. Oboite said. Also, talk to patients and families about preventing reinfection with the dermatophyte that causes this condition. “Make sure we’re cleaning hats, combs, brushes, and pillowcases. That is really important.”
Some patients can develop a widespread rash while on treatment. But in most cases, it’s not an adverse reaction to the medication but rather an indication that the body’s response is revving up, she noted.
Griseofulvin 20 mg/kg/d is one treatment option. Another is terbinafine (using weight-based dosing). A tip with terbinafine is that because the tablet needs to be crushed for a young child, “you can put it in anything, besides applesauce or yogurt with fruit on the bottom, which can be acidic and reduce the effectiveness of the medication,” Dr. Oboite said.
For cases of severe, inflammatory tinea capitis such as a kerion, “I will say you have to hold the hands of these patients, the journey can be long,” she added.
Trichotillomania
Trichotillomania occurs when someone cannot stop pulling their own hair, and in the early phases, it can be confused with alopecia areata. A thorough history and examination of the patient can help distinguish the two conditions. Sometimes a child or teen has a history of anxiety-related behaviors like nail biting that points to trichotillomania. Another tip is to use a dermatoscope to help distinguish hair loss conditions because it avoids having to do as many biopsies in children.
Redirection therapy can work for younger children, and cognitive behavioral therapy (CBT) can help older children with trichotillomania. In response to a question during the Q&A period, Dr. Oboite said psychiatrists or psychologists can perform CBT. If it takes time to get an appointment, there are some CBT apps that can help in the meantime, she said.
“One thing really important is to not blame the child,” Dr. Oboite said. “Most children don’t even know that they’re doing this. This is often not a behavior that is being done on purpose.”
Androgenetic Alopecia
Rarely, children and teenagers can also present with androgenetic alopecia, which Dr. Oboite has successfully treated with topical minoxidil, applied once a day before increasing to twice a day if tolerated. “I will tell them that when they pick it up, it will say ‘you should not use in children.’ But it actually can be used in children safely.”
Low-dose oral minoxidil is another option. Both treatments require a commitment by patients and parents because they are “taking this for a long time.”
Loose Anagen and Uncombable Hair Syndromes
A rare genetic form of hair loss is called loose anagen syndrome. Children with this disorder will have thin hair that is easily pulled out without a lot of force. Their hair appears to typically only grow to a certain length (such as to the nape of the neck) and then stops.
Another genetic hair loss condition is uncombable hair syndrome. It can cause hair to grow out of the scalp in all directions, and as the name suggests, it is almost impossible to comb or brush down. Along with loose anagen syndrome, uncombable hair syndrome tends to improve as the child gets older. “The key point here is telling parents that it can get better with time,” Dr. Oboite said.
A Condition With No Well-Known Acronym
She described a child she treated who had hair that never grew and was easily broken. The patient’s skin was prone to bruising, and her fingernails would easily fall off after trauma; her dentist noted that she had no buds for adult teeth on x-rays. These different presentations are important because hair, teeth, and nails all come from the same ectoderm germ line in embryo development, Dr. Oboite said.
Exome sequencing revealed the girl had a very rare diagnosis called autosomal recessive hypotrichosis with recurrent skin vesicles. “So, it is really important to recognize that children who are presenting with hair issues can have a genetic, underlying condition,” she said. Examining the skin, nails, and teeth, in addition to the hair, can be clues to these very rare diagnoses.
Some of these hair loss conditions in children can be challenging to diagnose and manage, Dr. Oboite said. “So don’t be afraid to ask for help on complex or rare cases.” Pediatric dermatologists “are always happy to help you. Hair loss is daunting, and hair loss in children can be even more daunting,” but the rewards of accurate diagnosis and successful treatment can be great, she said.
Dr. Oboite reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
ORLANDO, FLORIDA — There are subtleties and nuances to diagnosing, treating, and monitoring the progress of treatment of hair loss in children. Moreover, hair loss in children can be challenging because it can be caused by a range of conditions, some common and others relatively rare.
Michelle Oboite, MD, shared tips on how to distinguish types of hair loss, when to treat with medications such as topical corticosteroids or Janus kinase (JAK) inhibitors, and why shared decision-making is important, at the ODAC Dermatology, Aesthetic & Surgical Conference.
What these conditions share is that they can negatively affect the quality of life for a child or teenager when the condition leads to anxiety, teasing, or bullying. “It is very isolating to have this condition that everyone in the world can see that you have and judge you for it,” said Dr. Oboite, an attending physician in the dermatology section of Children’s Hospital of Philadelphia.
Others are lichen planopilaris and genetic conditions, including loose anagen syndrome, uncombable hair syndrome, and “something so rare” — it has no acronym — autosomal recessive hypotrichosis with recurrent skin vesicles, Dr. Oboite said.
Alopecia Areata
Alopecia areata can differ from child to child and can appear in different stages: A localized patch stage, a diffuse patchy stage, or alopecia universalis. In this last stage, the child has already lost most or all the hair on the scalp and eyebrows, as well as the eyelashes.
The decision to treat or not to treat, particularly in younger children, should be on the basis of shared decision-making between a healthcare provider and caregiver, said Dr. Oboite, who is also an assistant professor of clinical dermatology at the University of Pennsylvania, Philadelphia.
Some younger children may not experience any negative impact from the condition, so waiting until they are older is an option.
Also, consider the impact of treatment on a child. Some therapies require frequent blood draws for monitoring, and some topical therapies that are applied multiple times a day “can be very overwhelming” for young children, Dr. Oboite said.
Most children with alopecia areata are healthy and do not need extensive screening laboratory testing. However, one exception is if thyroid dysfunction, commonly associated with alopecia areata, is suspected.
For alopecia areata, Dr. Oboite recommends starting with topical therapies, either topical corticosteroids (as first line) or topical JAK inhibitors (either topical ruxolitinib or compounded topical tofacitinib, both off-label for this indication).
Topical corticosteroids can be effective, but “you want to be thoughtful of the strength you’re using, the application frequency, and then the total amount of surface area that you’re treating,” Dr. Oboite said. Too potent or too much of a topical corticosteroid increases the risk for atrophy and systemic absorption, respectively. To reduce the risk, she reserves the use of ultrahigh-potency topical corticosteroids, such as clobetasol, for children ages 10 years or older. For children younger than 10 years, she recommends using mid-high-potency topical corticosteroids instead.
She recommends once-a-day application around bedtime 5 days a week, generally Monday through Friday to make it easier to remember.
“For children who have over 50% of the scalp involved, I do consider systemic therapy,” Dr. Oboite said. This can include oral steroids such as dexamethasone, prednisone, or prednisolone. For children with recalcitrant disease, she is more likely to use the oral JAK inhibitor ritlecitinib because it was recently approved by the Food and Drug Administration for treating severe alopecia areata in children 12 years and older and in adults.
Another strategy Dr. Oboite uses is to add low-dose oral minoxidil as an adjuvant to other systemic therapy. “I find that it helps with faster hair regrowth,” she said.
Tinea Capitis
Oral treatment is indicated for tinea capitis. “Topicals just don’t really clear this,” Dr. Oboite said. Also, talk to patients and families about preventing reinfection with the dermatophyte that causes this condition. “Make sure we’re cleaning hats, combs, brushes, and pillowcases. That is really important.”
Some patients can develop a widespread rash while on treatment. But in most cases, it’s not an adverse reaction to the medication but rather an indication that the body’s response is revving up, she noted.
Griseofulvin 20 mg/kg/d is one treatment option. Another is terbinafine (using weight-based dosing). A tip with terbinafine is that because the tablet needs to be crushed for a young child, “you can put it in anything, besides applesauce or yogurt with fruit on the bottom, which can be acidic and reduce the effectiveness of the medication,” Dr. Oboite said.
For cases of severe, inflammatory tinea capitis such as a kerion, “I will say you have to hold the hands of these patients, the journey can be long,” she added.
Trichotillomania
Trichotillomania occurs when someone cannot stop pulling their own hair, and in the early phases, it can be confused with alopecia areata. A thorough history and examination of the patient can help distinguish the two conditions. Sometimes a child or teen has a history of anxiety-related behaviors like nail biting that points to trichotillomania. Another tip is to use a dermatoscope to help distinguish hair loss conditions because it avoids having to do as many biopsies in children.
Redirection therapy can work for younger children, and cognitive behavioral therapy (CBT) can help older children with trichotillomania. In response to a question during the Q&A period, Dr. Oboite said psychiatrists or psychologists can perform CBT. If it takes time to get an appointment, there are some CBT apps that can help in the meantime, she said.
“One thing really important is to not blame the child,” Dr. Oboite said. “Most children don’t even know that they’re doing this. This is often not a behavior that is being done on purpose.”
Androgenetic Alopecia
Rarely, children and teenagers can also present with androgenetic alopecia, which Dr. Oboite has successfully treated with topical minoxidil, applied once a day before increasing to twice a day if tolerated. “I will tell them that when they pick it up, it will say ‘you should not use in children.’ But it actually can be used in children safely.”
Low-dose oral minoxidil is another option. Both treatments require a commitment by patients and parents because they are “taking this for a long time.”
Loose Anagen and Uncombable Hair Syndromes
A rare genetic form of hair loss is called loose anagen syndrome. Children with this disorder will have thin hair that is easily pulled out without a lot of force. Their hair appears to typically only grow to a certain length (such as to the nape of the neck) and then stops.
Another genetic hair loss condition is uncombable hair syndrome. It can cause hair to grow out of the scalp in all directions, and as the name suggests, it is almost impossible to comb or brush down. Along with loose anagen syndrome, uncombable hair syndrome tends to improve as the child gets older. “The key point here is telling parents that it can get better with time,” Dr. Oboite said.
A Condition With No Well-Known Acronym
She described a child she treated who had hair that never grew and was easily broken. The patient’s skin was prone to bruising, and her fingernails would easily fall off after trauma; her dentist noted that she had no buds for adult teeth on x-rays. These different presentations are important because hair, teeth, and nails all come from the same ectoderm germ line in embryo development, Dr. Oboite said.
Exome sequencing revealed the girl had a very rare diagnosis called autosomal recessive hypotrichosis with recurrent skin vesicles. “So, it is really important to recognize that children who are presenting with hair issues can have a genetic, underlying condition,” she said. Examining the skin, nails, and teeth, in addition to the hair, can be clues to these very rare diagnoses.
Some of these hair loss conditions in children can be challenging to diagnose and manage, Dr. Oboite said. “So don’t be afraid to ask for help on complex or rare cases.” Pediatric dermatologists “are always happy to help you. Hair loss is daunting, and hair loss in children can be even more daunting,” but the rewards of accurate diagnosis and successful treatment can be great, she said.
Dr. Oboite reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
ORLANDO, FLORIDA — There are subtleties and nuances to diagnosing, treating, and monitoring the progress of treatment of hair loss in children. Moreover, hair loss in children can be challenging because it can be caused by a range of conditions, some common and others relatively rare.
Michelle Oboite, MD, shared tips on how to distinguish types of hair loss, when to treat with medications such as topical corticosteroids or Janus kinase (JAK) inhibitors, and why shared decision-making is important, at the ODAC Dermatology, Aesthetic & Surgical Conference.
What these conditions share is that they can negatively affect the quality of life for a child or teenager when the condition leads to anxiety, teasing, or bullying. “It is very isolating to have this condition that everyone in the world can see that you have and judge you for it,” said Dr. Oboite, an attending physician in the dermatology section of Children’s Hospital of Philadelphia.
Others are lichen planopilaris and genetic conditions, including loose anagen syndrome, uncombable hair syndrome, and “something so rare” — it has no acronym — autosomal recessive hypotrichosis with recurrent skin vesicles, Dr. Oboite said.
Alopecia Areata
Alopecia areata can differ from child to child and can appear in different stages: A localized patch stage, a diffuse patchy stage, or alopecia universalis. In this last stage, the child has already lost most or all the hair on the scalp and eyebrows, as well as the eyelashes.
The decision to treat or not to treat, particularly in younger children, should be on the basis of shared decision-making between a healthcare provider and caregiver, said Dr. Oboite, who is also an assistant professor of clinical dermatology at the University of Pennsylvania, Philadelphia.
Some younger children may not experience any negative impact from the condition, so waiting until they are older is an option.
Also, consider the impact of treatment on a child. Some therapies require frequent blood draws for monitoring, and some topical therapies that are applied multiple times a day “can be very overwhelming” for young children, Dr. Oboite said.
Most children with alopecia areata are healthy and do not need extensive screening laboratory testing. However, one exception is if thyroid dysfunction, commonly associated with alopecia areata, is suspected.
For alopecia areata, Dr. Oboite recommends starting with topical therapies, either topical corticosteroids (as first line) or topical JAK inhibitors (either topical ruxolitinib or compounded topical tofacitinib, both off-label for this indication).
Topical corticosteroids can be effective, but “you want to be thoughtful of the strength you’re using, the application frequency, and then the total amount of surface area that you’re treating,” Dr. Oboite said. Too potent or too much of a topical corticosteroid increases the risk for atrophy and systemic absorption, respectively. To reduce the risk, she reserves the use of ultrahigh-potency topical corticosteroids, such as clobetasol, for children ages 10 years or older. For children younger than 10 years, she recommends using mid-high-potency topical corticosteroids instead.
She recommends once-a-day application around bedtime 5 days a week, generally Monday through Friday to make it easier to remember.
“For children who have over 50% of the scalp involved, I do consider systemic therapy,” Dr. Oboite said. This can include oral steroids such as dexamethasone, prednisone, or prednisolone. For children with recalcitrant disease, she is more likely to use the oral JAK inhibitor ritlecitinib because it was recently approved by the Food and Drug Administration for treating severe alopecia areata in children 12 years and older and in adults.
Another strategy Dr. Oboite uses is to add low-dose oral minoxidil as an adjuvant to other systemic therapy. “I find that it helps with faster hair regrowth,” she said.
Tinea Capitis
Oral treatment is indicated for tinea capitis. “Topicals just don’t really clear this,” Dr. Oboite said. Also, talk to patients and families about preventing reinfection with the dermatophyte that causes this condition. “Make sure we’re cleaning hats, combs, brushes, and pillowcases. That is really important.”
Some patients can develop a widespread rash while on treatment. But in most cases, it’s not an adverse reaction to the medication but rather an indication that the body’s response is revving up, she noted.
Griseofulvin 20 mg/kg/d is one treatment option. Another is terbinafine (using weight-based dosing). A tip with terbinafine is that because the tablet needs to be crushed for a young child, “you can put it in anything, besides applesauce or yogurt with fruit on the bottom, which can be acidic and reduce the effectiveness of the medication,” Dr. Oboite said.
For cases of severe, inflammatory tinea capitis such as a kerion, “I will say you have to hold the hands of these patients, the journey can be long,” she added.
Trichotillomania
Trichotillomania occurs when someone cannot stop pulling their own hair, and in the early phases, it can be confused with alopecia areata. A thorough history and examination of the patient can help distinguish the two conditions. Sometimes a child or teen has a history of anxiety-related behaviors like nail biting that points to trichotillomania. Another tip is to use a dermatoscope to help distinguish hair loss conditions because it avoids having to do as many biopsies in children.
Redirection therapy can work for younger children, and cognitive behavioral therapy (CBT) can help older children with trichotillomania. In response to a question during the Q&A period, Dr. Oboite said psychiatrists or psychologists can perform CBT. If it takes time to get an appointment, there are some CBT apps that can help in the meantime, she said.
“One thing really important is to not blame the child,” Dr. Oboite said. “Most children don’t even know that they’re doing this. This is often not a behavior that is being done on purpose.”
Androgenetic Alopecia
Rarely, children and teenagers can also present with androgenetic alopecia, which Dr. Oboite has successfully treated with topical minoxidil, applied once a day before increasing to twice a day if tolerated. “I will tell them that when they pick it up, it will say ‘you should not use in children.’ But it actually can be used in children safely.”
Low-dose oral minoxidil is another option. Both treatments require a commitment by patients and parents because they are “taking this for a long time.”
Loose Anagen and Uncombable Hair Syndromes
A rare genetic form of hair loss is called loose anagen syndrome. Children with this disorder will have thin hair that is easily pulled out without a lot of force. Their hair appears to typically only grow to a certain length (such as to the nape of the neck) and then stops.
Another genetic hair loss condition is uncombable hair syndrome. It can cause hair to grow out of the scalp in all directions, and as the name suggests, it is almost impossible to comb or brush down. Along with loose anagen syndrome, uncombable hair syndrome tends to improve as the child gets older. “The key point here is telling parents that it can get better with time,” Dr. Oboite said.
A Condition With No Well-Known Acronym
She described a child she treated who had hair that never grew and was easily broken. The patient’s skin was prone to bruising, and her fingernails would easily fall off after trauma; her dentist noted that she had no buds for adult teeth on x-rays. These different presentations are important because hair, teeth, and nails all come from the same ectoderm germ line in embryo development, Dr. Oboite said.
Exome sequencing revealed the girl had a very rare diagnosis called autosomal recessive hypotrichosis with recurrent skin vesicles. “So, it is really important to recognize that children who are presenting with hair issues can have a genetic, underlying condition,” she said. Examining the skin, nails, and teeth, in addition to the hair, can be clues to these very rare diagnoses.
Some of these hair loss conditions in children can be challenging to diagnose and manage, Dr. Oboite said. “So don’t be afraid to ask for help on complex or rare cases.” Pediatric dermatologists “are always happy to help you. Hair loss is daunting, and hair loss in children can be even more daunting,” but the rewards of accurate diagnosis and successful treatment can be great, she said.
Dr. Oboite reported no relevant financial relationships.
A version of this article appeared on Medscape.com.
AT ODAC 2024
FDA Approves Topical Gel For Wounds Associated With JEB and DEB
The FDA has approved a topical gel containing birch triterpenes for the treatment of partial thickness wounds in patients 6 months and older with junctional epidermolysis bullosa (JEB) and dystrophic epidermolysis bullosa (DEB).
The gel is marketed under the name Filsuvez. It is the first approved treatment for wounds associated with JEB and the second for patients with DEB, following the approval of Vyjuvek (Krystal Biotech), a topical gene therapy gel, in May 2023.
First developed by Amryt Pharma and intended for home use, Filsuvez is now marketed by Chiesi Global Rare Diseases, which acquired Amryt in January 2023. The gel is applied topically to the wound at each dressing change.
The approval of Filsuvez is based on results from the Efficacy and Safety Study of Oleogel-S10 in Epidermolysis Bullosa (EASE), a randomized, placebo-controlled study of 223 people, the largest-ever phase 3 clinical trial for the treatment of EB, according to the Chiesi news release. The gel was well tolerated and met the primary endpoint with statistical significance, with 41.3% of patients achieving first complete target wound closure within 45 days (compared with 28.9% on placebo).
“I am so excited to say that this is another hurdle cleared and milestone achieved for the EB Community,” Brett Kopelan, executive director at debra of America said in a blog post. “We are now on the road to being able to treat EB more effectively, and to make the worst disease you’ve never heard of chronic, but livable, by making use of multiple therapeutic options in conjunction with each other.”
The FDA has approved a topical gel containing birch triterpenes for the treatment of partial thickness wounds in patients 6 months and older with junctional epidermolysis bullosa (JEB) and dystrophic epidermolysis bullosa (DEB).
The gel is marketed under the name Filsuvez. It is the first approved treatment for wounds associated with JEB and the second for patients with DEB, following the approval of Vyjuvek (Krystal Biotech), a topical gene therapy gel, in May 2023.
First developed by Amryt Pharma and intended for home use, Filsuvez is now marketed by Chiesi Global Rare Diseases, which acquired Amryt in January 2023. The gel is applied topically to the wound at each dressing change.
The approval of Filsuvez is based on results from the Efficacy and Safety Study of Oleogel-S10 in Epidermolysis Bullosa (EASE), a randomized, placebo-controlled study of 223 people, the largest-ever phase 3 clinical trial for the treatment of EB, according to the Chiesi news release. The gel was well tolerated and met the primary endpoint with statistical significance, with 41.3% of patients achieving first complete target wound closure within 45 days (compared with 28.9% on placebo).
“I am so excited to say that this is another hurdle cleared and milestone achieved for the EB Community,” Brett Kopelan, executive director at debra of America said in a blog post. “We are now on the road to being able to treat EB more effectively, and to make the worst disease you’ve never heard of chronic, but livable, by making use of multiple therapeutic options in conjunction with each other.”
The FDA has approved a topical gel containing birch triterpenes for the treatment of partial thickness wounds in patients 6 months and older with junctional epidermolysis bullosa (JEB) and dystrophic epidermolysis bullosa (DEB).
The gel is marketed under the name Filsuvez. It is the first approved treatment for wounds associated with JEB and the second for patients with DEB, following the approval of Vyjuvek (Krystal Biotech), a topical gene therapy gel, in May 2023.
First developed by Amryt Pharma and intended for home use, Filsuvez is now marketed by Chiesi Global Rare Diseases, which acquired Amryt in January 2023. The gel is applied topically to the wound at each dressing change.
The approval of Filsuvez is based on results from the Efficacy and Safety Study of Oleogel-S10 in Epidermolysis Bullosa (EASE), a randomized, placebo-controlled study of 223 people, the largest-ever phase 3 clinical trial for the treatment of EB, according to the Chiesi news release. The gel was well tolerated and met the primary endpoint with statistical significance, with 41.3% of patients achieving first complete target wound closure within 45 days (compared with 28.9% on placebo).
“I am so excited to say that this is another hurdle cleared and milestone achieved for the EB Community,” Brett Kopelan, executive director at debra of America said in a blog post. “We are now on the road to being able to treat EB more effectively, and to make the worst disease you’ve never heard of chronic, but livable, by making use of multiple therapeutic options in conjunction with each other.”
Teen and young adult rheumatology patients report gaps in sexual health counseling
SAN DIEGO — Only half of teens and young adults on teratogenic medication report being asked about sexual activity by their rheumatologist, and 38% did not know that their medication would be harmful to a fetus, according to a new survey.
While pediatric rheumatology providers may think that health screenings and contraceptive counseling are happening elsewhere, “this study suggests that a lot of patients are being missed, including those on teratogens,” noted Brittany M. Huynh, MD, MPH, a pediatric rheumatology fellow at the Indiana University School of Medicine in Indianapolis. She led the study and presented the findings at the American College of Rheumatology annual meeting.

For the study, Dr. Huynh and colleagues recruited patients aged 14-23 years who were assigned female at birth and were followed at pediatric rheumatology clinics affiliated with Indiana University. Participants completed a one-time survey between October 2020 and July 2022 and were asked about their sexual reproductive health experience and knowledge. Notably, all but four surveys were completed prior to the US Supreme Court Dobbs decision overturning Roe v. Wade.
Of responses from 108 participants, the most common diagnoses were juvenile idiopathic arthritis (52%) and systemic lupus erythematosus (16%). About one third (36%) of patients were on teratogenic medication, with the most common being methotrexate. About three fourths (76%) were White, and the average age of respondents was 16.7.
Most participants (82%) said they had been asked about sexual activity by a health care provider, but only 38% said their pediatric rheumatologist discussed this topic with them. Of the 39 patients on teratogenic medication, 54% said they had been asked about sexual activity by their pediatric rheumatologist, and only 51% said they had received teratogenicity counseling.
A larger percentage (85%) of this group reported receiving sexual activity screenings by any provider, but there was little difference in counseling about teratogenic medication.
This suggests that this type of risk counseling “is almost exclusively done by (pediatric rheumatologists), if at all,” Dr. Huynh noted during her presentation.
In total, 56% of all patients said a provider had talked to them about how to prevent pregnancy, and 20% said they had been counseled about how to get and use emergency contraception. Only 6% of patients said their pediatric rheumatologist had discussed emergency contraception during appointments.
Although sexual activity screenings were associated with current teratogen use, pregnancy prevention counseling and emergency contraceptive counseling were not associated with teratogen use or reported sexual activity.
The survey also revealed that there were gaps in knowledge about the health effects of rheumatic medication. Of the patients on teratogens, 38% did not know that their medication could harm a fetus if they became pregnant. Only 9% of patients not on teratogens correctly answered that their medication would not harm a fetus.
Previous studies have also shown that rheumatology patients do not know that their medications can be teratogenic, noted Cuoghi Edens, MD, a rheumatologist at the University of Chicago, who sees both adult and pediatric patients. She was not involved with the study. The larger challenge is how to best educate patients, she said.
While hopefully a patient’s primary care provider is discussing these issues with them, these patients often see their rheumatologist more frequently and more consistently than other providers, Dr. Edens said.

“We are sometimes the continuity of care for the patient versus their primary care, even though it should be a group effort of trying to some of these questions,” she said.
Conducting reproductive health screenings in pediatric rheumatology clinics can be difficult though, Dr. Edens noted, not only because of time constraints but also because parents often attend appointments with their child and likely have been for years. These screenings are most accurate when done one-on-one, so pivoting and removing the parents from the room can be awkward for providers, Dr. Edens said.
She advised that starting these conversations early on can be one way to ease into talking about reproductive health. In her own practice, Dr. Huynh sets aside time during appointments to speak with adolescent patients privately.
“We always discuss teratogenic medication. I always talk to them about the fact that I’m going to be doing pregnancy testing with their other screening labs because of the risks associated,” she said. “I also specifically set time aside for patients on teratogens to talk about emergency contraception and offer a prescription, if they’re interested.”
Dr. Huynh emphasized that providing easy access to emergency contraception is key. The ACR reproductive health guidelines — although geared toward adults — recommend discussing emergency contraception with patients, and Dr. Huynh advocates writing prescriptions for interested patients.
“They can fill it and have it easily accessible, so that there are no additional barriers, particularly for people who have these higher risks,” she said.
While emergency contraceptives are also available over the counter, it can be awkward for young people to ask for them, she said, and they can be expensive if not covered under insurance. Providing a prescription is one way to avoid those issues, Dr. Huynh said.
“Certainly, you have to have some parent buy-in, because if there is going to be a script, it’s probably going to be under insurance,” she said. “But in my experience, parents are happy to have it around as long as you’re talking it through with them as well as the young person.”
Dr. Huynh and Dr. Edens had no disclosures.
A version of this article appeared on Medscape.com.
SAN DIEGO — Only half of teens and young adults on teratogenic medication report being asked about sexual activity by their rheumatologist, and 38% did not know that their medication would be harmful to a fetus, according to a new survey.
While pediatric rheumatology providers may think that health screenings and contraceptive counseling are happening elsewhere, “this study suggests that a lot of patients are being missed, including those on teratogens,” noted Brittany M. Huynh, MD, MPH, a pediatric rheumatology fellow at the Indiana University School of Medicine in Indianapolis. She led the study and presented the findings at the American College of Rheumatology annual meeting.
For the study, Dr. Huynh and colleagues recruited patients aged 14-23 years who were assigned female at birth and were followed at pediatric rheumatology clinics affiliated with Indiana University. Participants completed a one-time survey between October 2020 and July 2022 and were asked about their sexual reproductive health experience and knowledge. Notably, all but four surveys were completed prior to the US Supreme Court Dobbs decision overturning Roe v. Wade.
Of responses from 108 participants, the most common diagnoses were juvenile idiopathic arthritis (52%) and systemic lupus erythematosus (16%). About one third (36%) of patients were on teratogenic medication, with the most common being methotrexate. About three fourths (76%) were White, and the average age of respondents was 16.7.
Most participants (82%) said they had been asked about sexual activity by a health care provider, but only 38% said their pediatric rheumatologist discussed this topic with them. Of the 39 patients on teratogenic medication, 54% said they had been asked about sexual activity by their pediatric rheumatologist, and only 51% said they had received teratogenicity counseling.
A larger percentage (85%) of this group reported receiving sexual activity screenings by any provider, but there was little difference in counseling about teratogenic medication.
This suggests that this type of risk counseling “is almost exclusively done by (pediatric rheumatologists), if at all,” Dr. Huynh noted during her presentation.
In total, 56% of all patients said a provider had talked to them about how to prevent pregnancy, and 20% said they had been counseled about how to get and use emergency contraception. Only 6% of patients said their pediatric rheumatologist had discussed emergency contraception during appointments.
Although sexual activity screenings were associated with current teratogen use, pregnancy prevention counseling and emergency contraceptive counseling were not associated with teratogen use or reported sexual activity.
The survey also revealed that there were gaps in knowledge about the health effects of rheumatic medication. Of the patients on teratogens, 38% did not know that their medication could harm a fetus if they became pregnant. Only 9% of patients not on teratogens correctly answered that their medication would not harm a fetus.
Previous studies have also shown that rheumatology patients do not know that their medications can be teratogenic, noted Cuoghi Edens, MD, a rheumatologist at the University of Chicago, who sees both adult and pediatric patients. She was not involved with the study. The larger challenge is how to best educate patients, she said.
While hopefully a patient’s primary care provider is discussing these issues with them, these patients often see their rheumatologist more frequently and more consistently than other providers, Dr. Edens said.
“We are sometimes the continuity of care for the patient versus their primary care, even though it should be a group effort of trying to some of these questions,” she said.
Conducting reproductive health screenings in pediatric rheumatology clinics can be difficult though, Dr. Edens noted, not only because of time constraints but also because parents often attend appointments with their child and likely have been for years. These screenings are most accurate when done one-on-one, so pivoting and removing the parents from the room can be awkward for providers, Dr. Edens said.
She advised that starting these conversations early on can be one way to ease into talking about reproductive health. In her own practice, Dr. Huynh sets aside time during appointments to speak with adolescent patients privately.
“We always discuss teratogenic medication. I always talk to them about the fact that I’m going to be doing pregnancy testing with their other screening labs because of the risks associated,” she said. “I also specifically set time aside for patients on teratogens to talk about emergency contraception and offer a prescription, if they’re interested.”
Dr. Huynh emphasized that providing easy access to emergency contraception is key. The ACR reproductive health guidelines — although geared toward adults — recommend discussing emergency contraception with patients, and Dr. Huynh advocates writing prescriptions for interested patients.
“They can fill it and have it easily accessible, so that there are no additional barriers, particularly for people who have these higher risks,” she said.
While emergency contraceptives are also available over the counter, it can be awkward for young people to ask for them, she said, and they can be expensive if not covered under insurance. Providing a prescription is one way to avoid those issues, Dr. Huynh said.
“Certainly, you have to have some parent buy-in, because if there is going to be a script, it’s probably going to be under insurance,” she said. “But in my experience, parents are happy to have it around as long as you’re talking it through with them as well as the young person.”
Dr. Huynh and Dr. Edens had no disclosures.
A version of this article appeared on Medscape.com.
SAN DIEGO — Only half of teens and young adults on teratogenic medication report being asked about sexual activity by their rheumatologist, and 38% did not know that their medication would be harmful to a fetus, according to a new survey.
While pediatric rheumatology providers may think that health screenings and contraceptive counseling are happening elsewhere, “this study suggests that a lot of patients are being missed, including those on teratogens,” noted Brittany M. Huynh, MD, MPH, a pediatric rheumatology fellow at the Indiana University School of Medicine in Indianapolis. She led the study and presented the findings at the American College of Rheumatology annual meeting.
For the study, Dr. Huynh and colleagues recruited patients aged 14-23 years who were assigned female at birth and were followed at pediatric rheumatology clinics affiliated with Indiana University. Participants completed a one-time survey between October 2020 and July 2022 and were asked about their sexual reproductive health experience and knowledge. Notably, all but four surveys were completed prior to the US Supreme Court Dobbs decision overturning Roe v. Wade.
Of responses from 108 participants, the most common diagnoses were juvenile idiopathic arthritis (52%) and systemic lupus erythematosus (16%). About one third (36%) of patients were on teratogenic medication, with the most common being methotrexate. About three fourths (76%) were White, and the average age of respondents was 16.7.
Most participants (82%) said they had been asked about sexual activity by a health care provider, but only 38% said their pediatric rheumatologist discussed this topic with them. Of the 39 patients on teratogenic medication, 54% said they had been asked about sexual activity by their pediatric rheumatologist, and only 51% said they had received teratogenicity counseling.
A larger percentage (85%) of this group reported receiving sexual activity screenings by any provider, but there was little difference in counseling about teratogenic medication.
This suggests that this type of risk counseling “is almost exclusively done by (pediatric rheumatologists), if at all,” Dr. Huynh noted during her presentation.
In total, 56% of all patients said a provider had talked to them about how to prevent pregnancy, and 20% said they had been counseled about how to get and use emergency contraception. Only 6% of patients said their pediatric rheumatologist had discussed emergency contraception during appointments.
Although sexual activity screenings were associated with current teratogen use, pregnancy prevention counseling and emergency contraceptive counseling were not associated with teratogen use or reported sexual activity.
The survey also revealed that there were gaps in knowledge about the health effects of rheumatic medication. Of the patients on teratogens, 38% did not know that their medication could harm a fetus if they became pregnant. Only 9% of patients not on teratogens correctly answered that their medication would not harm a fetus.
Previous studies have also shown that rheumatology patients do not know that their medications can be teratogenic, noted Cuoghi Edens, MD, a rheumatologist at the University of Chicago, who sees both adult and pediatric patients. She was not involved with the study. The larger challenge is how to best educate patients, she said.
While hopefully a patient’s primary care provider is discussing these issues with them, these patients often see their rheumatologist more frequently and more consistently than other providers, Dr. Edens said.
“We are sometimes the continuity of care for the patient versus their primary care, even though it should be a group effort of trying to some of these questions,” she said.
Conducting reproductive health screenings in pediatric rheumatology clinics can be difficult though, Dr. Edens noted, not only because of time constraints but also because parents often attend appointments with their child and likely have been for years. These screenings are most accurate when done one-on-one, so pivoting and removing the parents from the room can be awkward for providers, Dr. Edens said.
She advised that starting these conversations early on can be one way to ease into talking about reproductive health. In her own practice, Dr. Huynh sets aside time during appointments to speak with adolescent patients privately.
“We always discuss teratogenic medication. I always talk to them about the fact that I’m going to be doing pregnancy testing with their other screening labs because of the risks associated,” she said. “I also specifically set time aside for patients on teratogens to talk about emergency contraception and offer a prescription, if they’re interested.”
Dr. Huynh emphasized that providing easy access to emergency contraception is key. The ACR reproductive health guidelines — although geared toward adults — recommend discussing emergency contraception with patients, and Dr. Huynh advocates writing prescriptions for interested patients.
“They can fill it and have it easily accessible, so that there are no additional barriers, particularly for people who have these higher risks,” she said.
While emergency contraceptives are also available over the counter, it can be awkward for young people to ask for them, she said, and they can be expensive if not covered under insurance. Providing a prescription is one way to avoid those issues, Dr. Huynh said.
“Certainly, you have to have some parent buy-in, because if there is going to be a script, it’s probably going to be under insurance,” she said. “But in my experience, parents are happy to have it around as long as you’re talking it through with them as well as the young person.”
Dr. Huynh and Dr. Edens had no disclosures.
A version of this article appeared on Medscape.com.
FROM ACR 2023
Why Are Prion Diseases on the Rise?
This transcript has been edited for clarity.
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here. 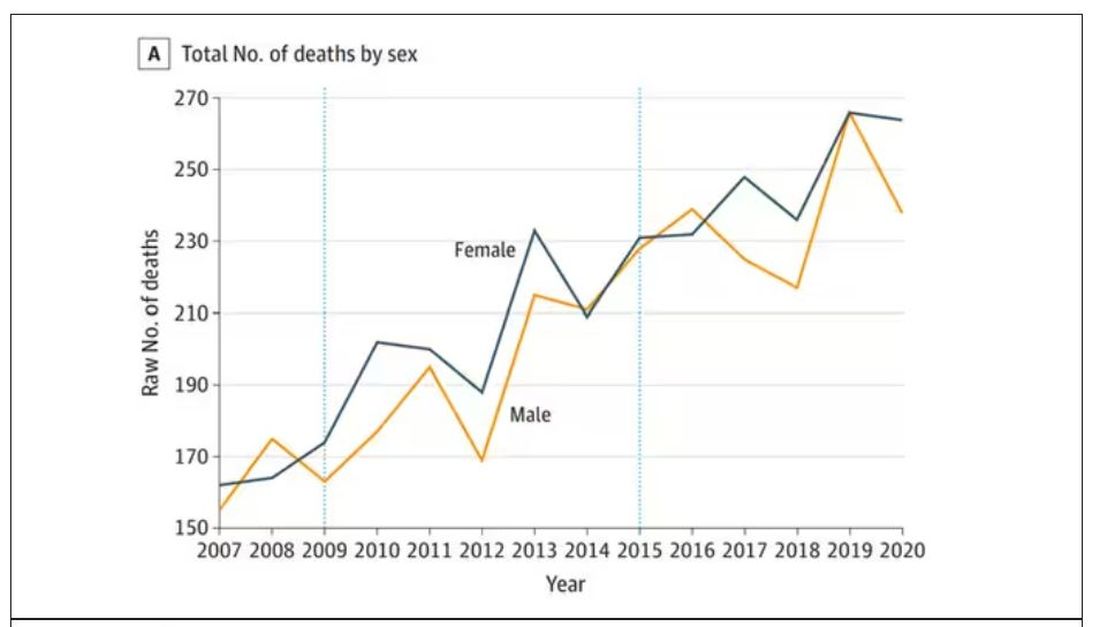
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.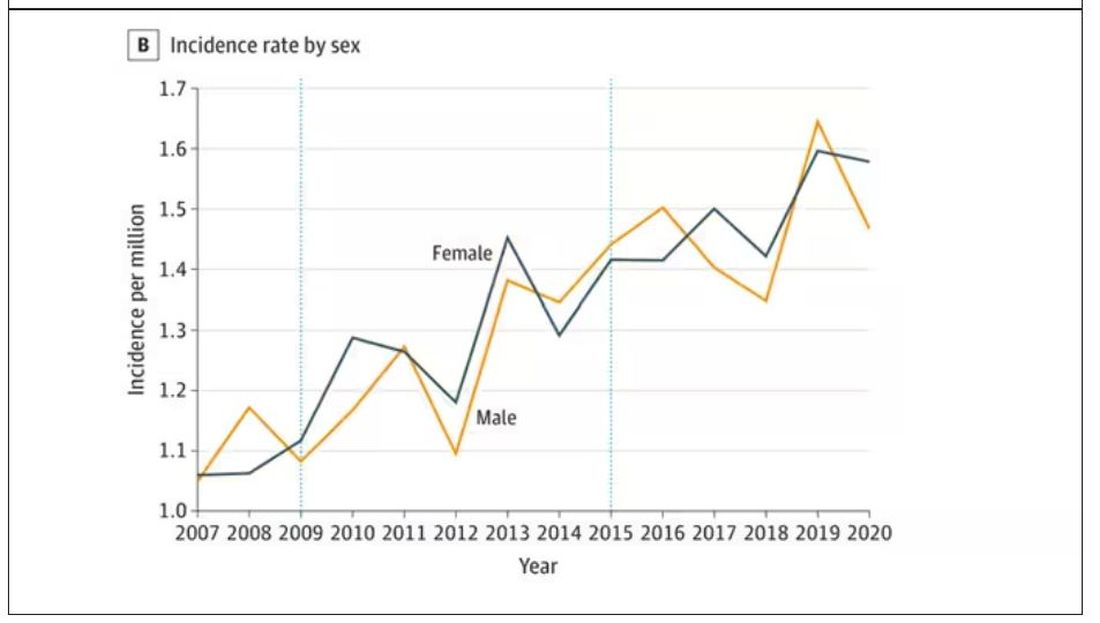
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here.
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
In 1986, in Britain, cattle started dying.
The condition, quickly nicknamed “mad cow disease,” was clearly infectious, but the particular pathogen was difficult to identify. By 1993, 120,000 cattle in Britain were identified as being infected. As yet, no human cases had occurred and the UK government insisted that cattle were a dead-end host for the pathogen. By the mid-1990s, however, multiple human cases, attributable to ingestion of meat and organs from infected cattle, were discovered. In humans, variant Creutzfeldt-Jakob disease (CJD) was a media sensation — a nearly uniformly fatal, untreatable condition with a rapid onset of dementia, mobility issues characterized by jerky movements, and autopsy reports finding that the brain itself had turned into a spongy mess.
The United States banned UK beef imports in 1996 and only lifted the ban in 2020.
The disease was made all the more mysterious because the pathogen involved was not a bacterium, parasite, or virus, but a protein — or a proteinaceous infectious particle, shortened to “prion.”
Prions are misfolded proteins that aggregate in cells — in this case, in nerve cells. But what makes prions different from other misfolded proteins is that the misfolded protein catalyzes the conversion of its non-misfolded counterpart into the misfolded configuration. It creates a chain reaction, leading to rapid accumulation of misfolded proteins and cell death.
And, like a time bomb, we all have prion protein inside us. In its normally folded state, the function of prion protein remains unclear — knockout mice do okay without it — but it is also highly conserved across mammalian species, so it probably does something worthwhile, perhaps protecting nerve fibers.
Far more common than humans contracting mad cow disease is the condition known as sporadic CJD, responsible for 85% of all cases of prion-induced brain disease. The cause of sporadic CJD is unknown.
But one thing is known: Cases are increasing.
I don’t want you to freak out; we are not in the midst of a CJD epidemic. But it’s been a while since I’ve seen people discussing the condition — which remains as horrible as it was in the 1990s — and a new research letter appearing in JAMA Neurology brought it back to the top of my mind.
Researchers, led by Matthew Crane at Hopkins, used the CDC’s WONDER cause-of-death database, which pulls diagnoses from death certificates. Normally, I’m not a fan of using death certificates for cause-of-death analyses, but in this case I’ll give it a pass. Assuming that the diagnosis of CJD is made, it would be really unlikely for it not to appear on a death certificate.
The main findings are seen here.
Note that we can’t tell whether these are sporadic CJD cases or variant CJD cases or even familial CJD cases; however, unless there has been a dramatic change in epidemiology, the vast majority of these will be sporadic.
The question is, why are there more cases?
Whenever this type of question comes up with any disease, there are basically three possibilities:
First, there may be an increase in the susceptible, or at-risk, population. In this case, we know that older people are at higher risk of developing sporadic CJD, and over time, the population has aged. To be fair, the authors adjusted for this and still saw an increase, though it was attenuated.
Second, we might be better at diagnosing the condition. A lot has happened since the mid-1990s, when the diagnosis was based more or less on symptoms. The advent of more sophisticated MRI protocols as well as a new diagnostic test called “real-time quaking-induced conversion testing” may mean we are just better at detecting people with this disease.
Third (and most concerning), a new exposure has occurred. What that exposure might be, where it might come from, is anyone’s guess. It’s hard to do broad-scale epidemiology on very rare diseases.
But given these findings, it seems that a bit more surveillance for this rare but devastating condition is well merited.
F. Perry Wilson, MD, MSCE, is an associate professor of medicine and public health and director of Yale’s Clinical and Translational Research Accelerator. His science communication work can be found in the Huffington Post, on NPR, and here on Medscape. He tweets @fperrywilson and his new book, How Medicine Works and When It Doesn’t, is available now.
F. Perry Wilson, MD, MSCE, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Hyperpigmented Flexural Plaques, Hypohidrosis, and Hypotrichosis
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
. B, Grocott-Gomori methenamine-silver staining showed numerous fungal elements")
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
The Diagnosis: Lelis Syndrome
Histopathology revealed spongiotic dermatitis with marked acanthosis and hyperkeratosis (Figure, A) with fungal colonization of the stratum corneum (Figure, B). Our patient was diagnosed with Lelis syndrome (also referred to as ectodermal dysplasia with acanthosis nigricans syndrome), a rare condition with hypotrichosis and hypohidrosis resulting from ectodermal dysplasia.1,2 The pruritic rash was diagnosed as chronic dermatitis due to fungal colonization in the setting of acanthosis nigricans. The fungal infection was treated with a 4-week course of oral fluconazole 200 mg/wk, ketoconazole cream 2% twice daily, and discontinuation of topical steroids, resulting in the thinning of the plaques on the neck and antecubital fossae as well as resolution of the pruritus. Following antifungal treatment, our patient was started on tazarotene cream 0.1% for acanthosis nigricans.
Ectodermal dysplasias are inherited disorders with abnormalities of the skin, hair, sweat glands, nails, teeth, and sometimes internal organs.3 Patients with Lelis syndrome may have other manifestations of ectodermal dysplasia in addition to hypohidrosis and hypotrichosis, including deafness and abnormal dentition,1,3 as seen in our patient. Intellectual disability has been described in many types of ectodermal dysplasia, including Lelis syndrome, but the association may be obscured by neurologic damage after repeat episodes of hyperthermia in infancy due to anhidrosis or hypohidrosis.4
When evaluating the differential diagnoses, the presence of hypotrichosis and hypohidrosis indicating ectodermal dysplasia is key. Confluent and reticulated papillomatosis presents with hyperkeratosis, papillomatosis, and focal acanthosis on histopathology. It can present on the neck and antecubital fossae; however, it is not associated with hypohidrosis and hypotrichosis.5 Although activating fibroblast growth factor receptor, FGFR, mutations have been implicated in the development of acanthosis nigricans in a variety of syndromes, these diagnoses are associated with abnormalities in skeletal development such as craniosynostosis and short stature; hypotrichosis and hypohidrosis are not seen.6,7 HAIR-AN (hyperandrogenism, insulin resistance, and acanthosis nigricans) syndrome typically presents in the prepubertal period with obesity and insulin resistance; acanthosis nigricans and alopecia can occur due to insulin resistance and hyperandrogenism, but concurrent clitoromegaly and hirsutism are common.6 Sudden onset of extensive acanthosis nigricans also is among the paraneoplastic dermatoses; it has been associated with multiple malignancies, but in these cases, hypotrichosis and hypohidrosis are not observed. Adenocarcinomas are the most common neoplasms associated with paraneoplastic acanthosis nigricans, which occurs through growth factor secretion by tumor cells stimulating hyperkeratosis and papillomatosis.6
Lelis syndrome is rare, and our case is unique because the patient had severe manifestations of acanthosis nigricans and hypotrichosis. Because the inheritance pattern and specific genetics of the condition have not been fully elucidated, the diagnosis primarily is clinical.1,8 Diagnosis may be complicated by the variety of other signs that can accompany acanthosis nigricans, hypohidrosis, and hypotrichosis.1,2 The condition also may alter or obscure presentation of other dermatologic conditions, as in our case.
Although there is no cure for Lelis syndrome, one case report described treatment with acitretin that resulted in marked improvement of the patient’s hyperkeratosis and acanthosis nigricans.9 Due to lack of health insurance coverage of acitretin, our patient was started on tazarotene cream 0.1% for acanthosis nigricans. General treatment of ectodermal dysplasia primarily consists of multidisciplinary symptom management, including careful monitoring of temperature and heat intolerance as well as provision of dental prosthetics.4,10 For ectodermal dysplasias caused by identified genetic mutations, prenatal interventions targeting gene pathways offer potentially curative treatment.10 However, for Lelis syndrome, along with many other disorders of ectodermal dysplasia, mitigation of signs and symptoms remains the primary treatment objective. Despite its rarity, increased awareness of Lelis syndrome is important to increase knowledge of ectodermal dysplasia syndromes and allow for the investigation of potential treatment options.
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
- Steiner CE, Cintra ML, Marques-de-Faria AP. Ectodermal dysplasia with acanthosis nigricans (Lelis syndrome). Am J Med Genet. 2002;113:381-384. doi:10.1002/ajmg.b.10787
- Lelis J. Autosomal recessive ectodermal dysplasia. Cutis. 1992; 49:435-437.
- Itin PH, Fistarol SK. Ectodermal dysplasias. Am J Med Genet C Semin Med Genet. 2004;131C:45-51. doi:10.1002/ajmg.c.30033
- Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-399. doi:10.1016/j .earlhumdev.2010.04.008
- Le C, Bedocs PM. Confluent and reticulated papillomatosis. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK459130/
- Das A, Datta D, Kassir M, et al. Acanthosis nigricans: a review. J Cosmet Dermatol. 2020;19:1857-1865. doi:10.1111/jocd.13544
- Torley D, Bellus GA, Munro CS. Genes, growth factors and acanthosis nigricans. Br J Dermatol. 2002;147:1096-1101. doi:10 .1046/j.1365-2133.2002.05150.x
- van Steensel MAM, van der Hout AH. Lelis syndrome may be a manifestation of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:1612-1613. doi:10.1002/ajmg.a.32945
- Yoshimura AM, Neves Ferreira Velho PE, Ferreira Magalhães R, et al. Lelis’ syndrome: treatment with acitretin. Int J Dermatol. 2008;47: 1330-1331. doi:10.1111/j.1365-4632.2008.03874.x
- Schneider H. Ectodermal dysplasias: new perspectives on the treatment of so far immedicable genetic disorders. Front Genet. 2022;13:1000744. doi:10.3389/fgene.2022.1000744
A 61-year-old woman with a history of hypohidrosis and deafness presented with a pruritic rash on the neck and antecubital fossae of several years’ duration. Prior treatment with topical corticosteroids failed to resolve the rash. Physical examination revealed thick, velvety, hyperpigmented plaques on the inframammary folds, axillae, groin, posterior neck, and antecubital fossae with lichenification of the latter 2 areas. Many pedunculated papules were seen on the face, chest, shoulders, and trunk, as well as diffuse hair thinning, particularly of the frontal and vertex scalp. Eyebrows, eyelashes, and axillary hair were absent. Two 5-mm punch biopsies of the antecubital fossa and inframammary fold were obtained for histopathologic analysis.


Conditional recommendations rule in new SARD-associated interstitial lung disease guidelines
SAN DIEGO – In the spring of 2024, the American College of Rheumatology is expected to release guidelines to help inform the screening, monitoring, and treatment of interstitial lung disease (ILD) in people with systemic autoimmune rheumatic diseases (SARDs).
The guidelines, which were previewed during a session at the ACR’s annual meeting, will include 50 recommendations, 3 of which met criteria for a strong rating:
- For people with SARDs at increased risk of developing ILD, the authors strongly recommend against screening with surgical lung biopsy.
- For people with systemic sclerosis (SSc)-related ILD, the authors strongly recommend against glucocorticoids as a first-line ILD treatment.
- For people with SSc-related ILD progression despite an initial ILD treatment, the authors strongly recommend against using long-term glucocorticoids.
Elana J. Bernstein, MD, MSc, a rheumatologist who directs the Columbia/New York-Presbyterian Scleroderma Center, and Sindhu R. Johnson, MD, a rheumatologist who directs the Toronto Scleroderma Program at the University of Toronto, provided a sneak peek of the recommendations to attendees before anticipated publication in Arthritis & Rheumatology and Arthritis Care & Research. For now, guideline summaries for screening and monitoring and treatment are currently available, and three manuscripts are under peer review: one about screening and monitoring, one about treatment, and one about the patient panel that participated in the effort.

“ILD is a significant cause of morbidity and mortality in people with SARDs,” said Dr. Bernstein, who is co-first author of the guidelines. “People with systemic sclerosis, rheumatoid arthritis, idiopathic inflammatory myopathies, mixed connective tissue disease, and Sjögren’s disease are at greatest risk of developing ILD.”
Pediatric patients with SARDs excluded
The guidelines’ population of interest was people 17 years of age and older who were diagnosed with SARDs with a high risk of ILD. Pediatric patients with SARDs were excluded from the endeavor, as were those with systemic lupus erythematosus, antineutrophil cytoplasmic antibody–associated vasculitis, sarcoidosis, ankylosing spondylitis, undifferentiated connective tissue disease, interstitial pneumonia with autoimmune features, and those with unclassifiable ILD.
In the realm of screening, the guideline authors conditionally recommend two screening tests for patients considered at increased risk of ILD: pulmonary function tests and high-resolution chest CT (HRCT). Pulmonary function tests should include spirometry, lung volumes, and diffusion capacity. “Office spirometry alone is insufficient,” said Dr. Johnson, who served as lead author of the guidelines. And while a HRCT scan is recommended, “some patients may present to the emergency room with acute onset shortness of breath, and they may receive a CT angiogram to screen for pulmonary embolism,” she said. “It’s important to note that CT angiograms are performed in incomplete inspiration to maximize pulmonary artery enhancement. This may produce atelectasis that may obscure or mimic ILD. As a result, CTA studies are often inadequate to screen for ILD.”
Once a patient is diagnosed with ILD, three tests are recommended for monitoring: pulmonary function testing (every 3-6 months the first year in patients with IIM and SSc, then less frequently once stable, and every 3-12 months in the first year in patients with RA, SjD, and MCTD, then less frequently once stable); ambulatory desaturation testing every 3-12 months; and HRCT as needed. Dr. Johnson noted that while that the screening of ILD lies within the realm of rheumatologists, “once a patient is diagnosed, we are encouraged to comanage these patients with pulmonologists,” she said. “Ambulatory desaturation testing is not an infrequent test in the hands of pulmonologists. This is where co-management can be helpful.” She characterized a 6-minute walk test with continuous oximetry as “insufficient and is not synonymous with ambulatory desaturation testing. Ambulatory desaturation testing includes up titration of oxygen if a patient desaturates.”
The guidelines conditionally recommend against using chest radiography, 6-minute walk test distance, ambulatory desaturation testing, and bronchoscopy for ILD screening, and there is a strong recommendation against surgical lung biopsy. “However, there are unique circumstances where these tests may be considered,” Dr. Johnson said. “For example, ambulatory desaturation testing may be helpful if a patient is unable to perform a pulmonary function test. Bronchoscopy may be used to rule out infection, sarcoidosis, lymphoma, or alveolar hemorrhage, and surgical lung biopsy may be considered if you’re trying to rule out a malignancy.”
Similarly, several tests are conditionally recommended against for the monitoring of ILD, including chest radiography, the 6-minute walk test distance, and bronchoscopy. “But there are unique circumstances where they may be considered,” she said. “The 6-minute walk test may be used if a patient is unable to perform a pulmonary function test or if they’re being assessed for lung transplantation. Bronchoscopy may be used to rule out infection or alveolar hemorrhage.”
Preferred treatment options described
First-line treatment recommendations for ILD were based on the best available published evidence, voting panel expertise, and patient preferences. For SSc, the preferred treatment options include mycophenolate (CellCept), tocilizumab (Actemra), or rituximab (Rituxan and biosimilars), while additional options include cyclophosphamide, nintedanib (Ofev), and azathioprine. For myositis, the preferred treatment options include mycophenolate, azathioprine, rituximab, or calcineurin inhibitors, while additional options include a Janus kinase (JAK) inhibitor or cyclophosphamide. For MCTD, the preferred treatment options include mycophenolate, azathioprine, or rituximab, while additional options include tocilizumab or cyclophosphamide. For RA and Sjögren’s, the preferred treatment options include mycophenolate, azathioprine, or rituximab, while additional options include cyclophosphamide. Dr. Johnson emphasized that there was low certainty evidence to recommend one treatment over another. “Many situations might lead a provider to choose a different option for ILD treatment, such as the presence of comorbidities or extra-pulmonary disease,” she said. “So, while our guidelines were focused on effectiveness for ILD, providers may choose therapies that will help ILD and other disease manifestations.”
The guidelines conditionally recommend a short course of glucocorticoids as a bridging therapy or for treatment of a flare of ILD in patients with myositis, MCTD, RA, and Sjögren’s. The panel strongly recommends against the use of glucocorticoids in patients with SSc due to the concern for inducing a scleroderma renal crisis. “While this may be common knowledge for rheumatologists, it may not be common knowledge for pulmonologists,” she said. “So here is an opportunity to educate our pulmonology colleagues in our consultation notes.”
The guidelines also include recommendations for progression of ILD, which was defined using the INBUILD trial criteria. Mycophenolate is conditionally recommended to be the first ILD treatment for all SARDs when progression occurs, if it wasn’t the first ILD treatment used. “If it was, then other medications that rheumatologists are used to can be considered as the next ILD treatment in the face of progression: rituximab, nintedanib, tocilizumab, and cyclophosphamide,” she said. The guidelines include a conditional recommendation against long-term glucocorticoid use in myositis, MCTD, RA, and Sjögren’s, plus a strong recommendation against long-term glucocorticoid use in SSc. Finally, there is a conditional recommendation of referral for lung transplant evaluation at the appropriate time at experienced centers.

Another group of recommendations has to do with cases of rapidly progressive ILD, which is characterized by rapid progression from no oxygen or a patient’s baseline oxygen requirement to a high oxygen requirement or intubation usually within days to weeks without a documented cause, such as infection or heart failure. “In cases of rapidly progressive ILD, which typically occurs in the setting of anti-MDA5 antibodies, there is a conditional recommendation for IV glucocorticoids plus two additional therapies: traditionally rituximab and mycophenolate,” Dr. Johnson said. “However, what may be new to some clinicians is combination IVIG [intravenous immunoglobulin] and a calcineurin inhibitor, notably tacrolimus,” she said. “This is the situation where experience at expert centers is influencing our guidelines in advance of data.”
A patient panel provided input
For the undertaking, a core team that included six rheumatologists; one pulmonologist; one thoracic radiologist; one expert on the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology; and two literature review experts developed clinically relevant population, intervention, comparator, and outcomes (PICO) questions. The literature review team included 13 rheumatologists, 8 pulmonologists, and 3 methodologists. Finally, a 21-member patient panel was convened to share their values and preferences regarding screening, monitoring, and treatment of SARD-related ILD. Of these, Dr. Bernstein said that 4 were at risk for ILD and 17 had been diagnosed with ILD. Next, the literature review team conducted a systematic review and used the GRADE methodology to rate the available evidence as high, moderate, low, or very low. Then, a voting panel comprising 13 rheumatologists, 10 pulmonologists, 1 radiologist, and 3 patients from the patient panel cast votes for each PICO question and made final recommendations.
The review of evidence left the guidelines authors with 241 PICO questions, “which is a lot,” Dr. Bernstein said. “To put this in perspective, some guidelines address only 10 or 15 PICO questions. Fortunately, we had a dedicated group of experts who were up to the challenge.” Dr. Johnson emphasized that the forthcoming guidelines should not be used by insurers to mandate a specific order of prescribing. “Clinicians must retain the latitude to prescribe medications based on individual patient factors and preferences,” she said.
Dr. Bernstein disclosed that she is an adviser to, a consultant for, and has received grant or research support from Boehringer Ingelheim and has also received grant or research support from Kadmon and Pfizer. Dr. Johnson disclosed that she has received research support from the American College of Rheumatology to develop these guidelines. She has also been an investigator for trials sponsored by Bristol-Myers Squibb, Roche, and Boehringer Ingelheim and has mitigated these relevant conflicts of interest 1 year prior to the development of these guidelines, and will continue to do so for the foreseeable future.
SAN DIEGO – In the spring of 2024, the American College of Rheumatology is expected to release guidelines to help inform the screening, monitoring, and treatment of interstitial lung disease (ILD) in people with systemic autoimmune rheumatic diseases (SARDs).
The guidelines, which were previewed during a session at the ACR’s annual meeting, will include 50 recommendations, 3 of which met criteria for a strong rating:
- For people with SARDs at increased risk of developing ILD, the authors strongly recommend against screening with surgical lung biopsy.
- For people with systemic sclerosis (SSc)-related ILD, the authors strongly recommend against glucocorticoids as a first-line ILD treatment.
- For people with SSc-related ILD progression despite an initial ILD treatment, the authors strongly recommend against using long-term glucocorticoids.
Elana J. Bernstein, MD, MSc, a rheumatologist who directs the Columbia/New York-Presbyterian Scleroderma Center, and Sindhu R. Johnson, MD, a rheumatologist who directs the Toronto Scleroderma Program at the University of Toronto, provided a sneak peek of the recommendations to attendees before anticipated publication in Arthritis & Rheumatology and Arthritis Care & Research. For now, guideline summaries for screening and monitoring and treatment are currently available, and three manuscripts are under peer review: one about screening and monitoring, one about treatment, and one about the patient panel that participated in the effort.
“ILD is a significant cause of morbidity and mortality in people with SARDs,” said Dr. Bernstein, who is co-first author of the guidelines. “People with systemic sclerosis, rheumatoid arthritis, idiopathic inflammatory myopathies, mixed connective tissue disease, and Sjögren’s disease are at greatest risk of developing ILD.”
Pediatric patients with SARDs excluded
The guidelines’ population of interest was people 17 years of age and older who were diagnosed with SARDs with a high risk of ILD. Pediatric patients with SARDs were excluded from the endeavor, as were those with systemic lupus erythematosus, antineutrophil cytoplasmic antibody–associated vasculitis, sarcoidosis, ankylosing spondylitis, undifferentiated connective tissue disease, interstitial pneumonia with autoimmune features, and those with unclassifiable ILD.
In the realm of screening, the guideline authors conditionally recommend two screening tests for patients considered at increased risk of ILD: pulmonary function tests and high-resolution chest CT (HRCT). Pulmonary function tests should include spirometry, lung volumes, and diffusion capacity. “Office spirometry alone is insufficient,” said Dr. Johnson, who served as lead author of the guidelines. And while a HRCT scan is recommended, “some patients may present to the emergency room with acute onset shortness of breath, and they may receive a CT angiogram to screen for pulmonary embolism,” she said. “It’s important to note that CT angiograms are performed in incomplete inspiration to maximize pulmonary artery enhancement. This may produce atelectasis that may obscure or mimic ILD. As a result, CTA studies are often inadequate to screen for ILD.”
Once a patient is diagnosed with ILD, three tests are recommended for monitoring: pulmonary function testing (every 3-6 months the first year in patients with IIM and SSc, then less frequently once stable, and every 3-12 months in the first year in patients with RA, SjD, and MCTD, then less frequently once stable); ambulatory desaturation testing every 3-12 months; and HRCT as needed. Dr. Johnson noted that while that the screening of ILD lies within the realm of rheumatologists, “once a patient is diagnosed, we are encouraged to comanage these patients with pulmonologists,” she said. “Ambulatory desaturation testing is not an infrequent test in the hands of pulmonologists. This is where co-management can be helpful.” She characterized a 6-minute walk test with continuous oximetry as “insufficient and is not synonymous with ambulatory desaturation testing. Ambulatory desaturation testing includes up titration of oxygen if a patient desaturates.”
The guidelines conditionally recommend against using chest radiography, 6-minute walk test distance, ambulatory desaturation testing, and bronchoscopy for ILD screening, and there is a strong recommendation against surgical lung biopsy. “However, there are unique circumstances where these tests may be considered,” Dr. Johnson said. “For example, ambulatory desaturation testing may be helpful if a patient is unable to perform a pulmonary function test. Bronchoscopy may be used to rule out infection, sarcoidosis, lymphoma, or alveolar hemorrhage, and surgical lung biopsy may be considered if you’re trying to rule out a malignancy.”
Similarly, several tests are conditionally recommended against for the monitoring of ILD, including chest radiography, the 6-minute walk test distance, and bronchoscopy. “But there are unique circumstances where they may be considered,” she said. “The 6-minute walk test may be used if a patient is unable to perform a pulmonary function test or if they’re being assessed for lung transplantation. Bronchoscopy may be used to rule out infection or alveolar hemorrhage.”
Preferred treatment options described
First-line treatment recommendations for ILD were based on the best available published evidence, voting panel expertise, and patient preferences. For SSc, the preferred treatment options include mycophenolate (CellCept), tocilizumab (Actemra), or rituximab (Rituxan and biosimilars), while additional options include cyclophosphamide, nintedanib (Ofev), and azathioprine. For myositis, the preferred treatment options include mycophenolate, azathioprine, rituximab, or calcineurin inhibitors, while additional options include a Janus kinase (JAK) inhibitor or cyclophosphamide. For MCTD, the preferred treatment options include mycophenolate, azathioprine, or rituximab, while additional options include tocilizumab or cyclophosphamide. For RA and Sjögren’s, the preferred treatment options include mycophenolate, azathioprine, or rituximab, while additional options include cyclophosphamide. Dr. Johnson emphasized that there was low certainty evidence to recommend one treatment over another. “Many situations might lead a provider to choose a different option for ILD treatment, such as the presence of comorbidities or extra-pulmonary disease,” she said. “So, while our guidelines were focused on effectiveness for ILD, providers may choose therapies that will help ILD and other disease manifestations.”
The guidelines conditionally recommend a short course of glucocorticoids as a bridging therapy or for treatment of a flare of ILD in patients with myositis, MCTD, RA, and Sjögren’s. The panel strongly recommends against the use of glucocorticoids in patients with SSc due to the concern for inducing a scleroderma renal crisis. “While this may be common knowledge for rheumatologists, it may not be common knowledge for pulmonologists,” she said. “So here is an opportunity to educate our pulmonology colleagues in our consultation notes.”
The guidelines also include recommendations for progression of ILD, which was defined using the INBUILD trial criteria. Mycophenolate is conditionally recommended to be the first ILD treatment for all SARDs when progression occurs, if it wasn’t the first ILD treatment used. “If it was, then other medications that rheumatologists are used to can be considered as the next ILD treatment in the face of progression: rituximab, nintedanib, tocilizumab, and cyclophosphamide,” she said. The guidelines include a conditional recommendation against long-term glucocorticoid use in myositis, MCTD, RA, and Sjögren’s, plus a strong recommendation against long-term glucocorticoid use in SSc. Finally, there is a conditional recommendation of referral for lung transplant evaluation at the appropriate time at experienced centers.
Another group of recommendations has to do with cases of rapidly progressive ILD, which is characterized by rapid progression from no oxygen or a patient’s baseline oxygen requirement to a high oxygen requirement or intubation usually within days to weeks without a documented cause, such as infection or heart failure. “In cases of rapidly progressive ILD, which typically occurs in the setting of anti-MDA5 antibodies, there is a conditional recommendation for IV glucocorticoids plus two additional therapies: traditionally rituximab and mycophenolate,” Dr. Johnson said. “However, what may be new to some clinicians is combination IVIG [intravenous immunoglobulin] and a calcineurin inhibitor, notably tacrolimus,” she said. “This is the situation where experience at expert centers is influencing our guidelines in advance of data.”
A patient panel provided input
For the undertaking, a core team that included six rheumatologists; one pulmonologist; one thoracic radiologist; one expert on the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology; and two literature review experts developed clinically relevant population, intervention, comparator, and outcomes (PICO) questions. The literature review team included 13 rheumatologists, 8 pulmonologists, and 3 methodologists. Finally, a 21-member patient panel was convened to share their values and preferences regarding screening, monitoring, and treatment of SARD-related ILD. Of these, Dr. Bernstein said that 4 were at risk for ILD and 17 had been diagnosed with ILD. Next, the literature review team conducted a systematic review and used the GRADE methodology to rate the available evidence as high, moderate, low, or very low. Then, a voting panel comprising 13 rheumatologists, 10 pulmonologists, 1 radiologist, and 3 patients from the patient panel cast votes for each PICO question and made final recommendations.
The review of evidence left the guidelines authors with 241 PICO questions, “which is a lot,” Dr. Bernstein said. “To put this in perspective, some guidelines address only 10 or 15 PICO questions. Fortunately, we had a dedicated group of experts who were up to the challenge.” Dr. Johnson emphasized that the forthcoming guidelines should not be used by insurers to mandate a specific order of prescribing. “Clinicians must retain the latitude to prescribe medications based on individual patient factors and preferences,” she said.
Dr. Bernstein disclosed that she is an adviser to, a consultant for, and has received grant or research support from Boehringer Ingelheim and has also received grant or research support from Kadmon and Pfizer. Dr. Johnson disclosed that she has received research support from the American College of Rheumatology to develop these guidelines. She has also been an investigator for trials sponsored by Bristol-Myers Squibb, Roche, and Boehringer Ingelheim and has mitigated these relevant conflicts of interest 1 year prior to the development of these guidelines, and will continue to do so for the foreseeable future.
SAN DIEGO – In the spring of 2024, the American College of Rheumatology is expected to release guidelines to help inform the screening, monitoring, and treatment of interstitial lung disease (ILD) in people with systemic autoimmune rheumatic diseases (SARDs).
The guidelines, which were previewed during a session at the ACR’s annual meeting, will include 50 recommendations, 3 of which met criteria for a strong rating:
- For people with SARDs at increased risk of developing ILD, the authors strongly recommend against screening with surgical lung biopsy.
- For people with systemic sclerosis (SSc)-related ILD, the authors strongly recommend against glucocorticoids as a first-line ILD treatment.
- For people with SSc-related ILD progression despite an initial ILD treatment, the authors strongly recommend against using long-term glucocorticoids.
Elana J. Bernstein, MD, MSc, a rheumatologist who directs the Columbia/New York-Presbyterian Scleroderma Center, and Sindhu R. Johnson, MD, a rheumatologist who directs the Toronto Scleroderma Program at the University of Toronto, provided a sneak peek of the recommendations to attendees before anticipated publication in Arthritis & Rheumatology and Arthritis Care & Research. For now, guideline summaries for screening and monitoring and treatment are currently available, and three manuscripts are under peer review: one about screening and monitoring, one about treatment, and one about the patient panel that participated in the effort.
“ILD is a significant cause of morbidity and mortality in people with SARDs,” said Dr. Bernstein, who is co-first author of the guidelines. “People with systemic sclerosis, rheumatoid arthritis, idiopathic inflammatory myopathies, mixed connective tissue disease, and Sjögren’s disease are at greatest risk of developing ILD.”
Pediatric patients with SARDs excluded
The guidelines’ population of interest was people 17 years of age and older who were diagnosed with SARDs with a high risk of ILD. Pediatric patients with SARDs were excluded from the endeavor, as were those with systemic lupus erythematosus, antineutrophil cytoplasmic antibody–associated vasculitis, sarcoidosis, ankylosing spondylitis, undifferentiated connective tissue disease, interstitial pneumonia with autoimmune features, and those with unclassifiable ILD.
In the realm of screening, the guideline authors conditionally recommend two screening tests for patients considered at increased risk of ILD: pulmonary function tests and high-resolution chest CT (HRCT). Pulmonary function tests should include spirometry, lung volumes, and diffusion capacity. “Office spirometry alone is insufficient,” said Dr. Johnson, who served as lead author of the guidelines. And while a HRCT scan is recommended, “some patients may present to the emergency room with acute onset shortness of breath, and they may receive a CT angiogram to screen for pulmonary embolism,” she said. “It’s important to note that CT angiograms are performed in incomplete inspiration to maximize pulmonary artery enhancement. This may produce atelectasis that may obscure or mimic ILD. As a result, CTA studies are often inadequate to screen for ILD.”
Once a patient is diagnosed with ILD, three tests are recommended for monitoring: pulmonary function testing (every 3-6 months the first year in patients with IIM and SSc, then less frequently once stable, and every 3-12 months in the first year in patients with RA, SjD, and MCTD, then less frequently once stable); ambulatory desaturation testing every 3-12 months; and HRCT as needed. Dr. Johnson noted that while that the screening of ILD lies within the realm of rheumatologists, “once a patient is diagnosed, we are encouraged to comanage these patients with pulmonologists,” she said. “Ambulatory desaturation testing is not an infrequent test in the hands of pulmonologists. This is where co-management can be helpful.” She characterized a 6-minute walk test with continuous oximetry as “insufficient and is not synonymous with ambulatory desaturation testing. Ambulatory desaturation testing includes up titration of oxygen if a patient desaturates.”
The guidelines conditionally recommend against using chest radiography, 6-minute walk test distance, ambulatory desaturation testing, and bronchoscopy for ILD screening, and there is a strong recommendation against surgical lung biopsy. “However, there are unique circumstances where these tests may be considered,” Dr. Johnson said. “For example, ambulatory desaturation testing may be helpful if a patient is unable to perform a pulmonary function test. Bronchoscopy may be used to rule out infection, sarcoidosis, lymphoma, or alveolar hemorrhage, and surgical lung biopsy may be considered if you’re trying to rule out a malignancy.”
Similarly, several tests are conditionally recommended against for the monitoring of ILD, including chest radiography, the 6-minute walk test distance, and bronchoscopy. “But there are unique circumstances where they may be considered,” she said. “The 6-minute walk test may be used if a patient is unable to perform a pulmonary function test or if they’re being assessed for lung transplantation. Bronchoscopy may be used to rule out infection or alveolar hemorrhage.”
Preferred treatment options described
First-line treatment recommendations for ILD were based on the best available published evidence, voting panel expertise, and patient preferences. For SSc, the preferred treatment options include mycophenolate (CellCept), tocilizumab (Actemra), or rituximab (Rituxan and biosimilars), while additional options include cyclophosphamide, nintedanib (Ofev), and azathioprine. For myositis, the preferred treatment options include mycophenolate, azathioprine, rituximab, or calcineurin inhibitors, while additional options include a Janus kinase (JAK) inhibitor or cyclophosphamide. For MCTD, the preferred treatment options include mycophenolate, azathioprine, or rituximab, while additional options include tocilizumab or cyclophosphamide. For RA and Sjögren’s, the preferred treatment options include mycophenolate, azathioprine, or rituximab, while additional options include cyclophosphamide. Dr. Johnson emphasized that there was low certainty evidence to recommend one treatment over another. “Many situations might lead a provider to choose a different option for ILD treatment, such as the presence of comorbidities or extra-pulmonary disease,” she said. “So, while our guidelines were focused on effectiveness for ILD, providers may choose therapies that will help ILD and other disease manifestations.”
The guidelines conditionally recommend a short course of glucocorticoids as a bridging therapy or for treatment of a flare of ILD in patients with myositis, MCTD, RA, and Sjögren’s. The panel strongly recommends against the use of glucocorticoids in patients with SSc due to the concern for inducing a scleroderma renal crisis. “While this may be common knowledge for rheumatologists, it may not be common knowledge for pulmonologists,” she said. “So here is an opportunity to educate our pulmonology colleagues in our consultation notes.”
The guidelines also include recommendations for progression of ILD, which was defined using the INBUILD trial criteria. Mycophenolate is conditionally recommended to be the first ILD treatment for all SARDs when progression occurs, if it wasn’t the first ILD treatment used. “If it was, then other medications that rheumatologists are used to can be considered as the next ILD treatment in the face of progression: rituximab, nintedanib, tocilizumab, and cyclophosphamide,” she said. The guidelines include a conditional recommendation against long-term glucocorticoid use in myositis, MCTD, RA, and Sjögren’s, plus a strong recommendation against long-term glucocorticoid use in SSc. Finally, there is a conditional recommendation of referral for lung transplant evaluation at the appropriate time at experienced centers.
Another group of recommendations has to do with cases of rapidly progressive ILD, which is characterized by rapid progression from no oxygen or a patient’s baseline oxygen requirement to a high oxygen requirement or intubation usually within days to weeks without a documented cause, such as infection or heart failure. “In cases of rapidly progressive ILD, which typically occurs in the setting of anti-MDA5 antibodies, there is a conditional recommendation for IV glucocorticoids plus two additional therapies: traditionally rituximab and mycophenolate,” Dr. Johnson said. “However, what may be new to some clinicians is combination IVIG [intravenous immunoglobulin] and a calcineurin inhibitor, notably tacrolimus,” she said. “This is the situation where experience at expert centers is influencing our guidelines in advance of data.”
A patient panel provided input
For the undertaking, a core team that included six rheumatologists; one pulmonologist; one thoracic radiologist; one expert on the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology; and two literature review experts developed clinically relevant population, intervention, comparator, and outcomes (PICO) questions. The literature review team included 13 rheumatologists, 8 pulmonologists, and 3 methodologists. Finally, a 21-member patient panel was convened to share their values and preferences regarding screening, monitoring, and treatment of SARD-related ILD. Of these, Dr. Bernstein said that 4 were at risk for ILD and 17 had been diagnosed with ILD. Next, the literature review team conducted a systematic review and used the GRADE methodology to rate the available evidence as high, moderate, low, or very low. Then, a voting panel comprising 13 rheumatologists, 10 pulmonologists, 1 radiologist, and 3 patients from the patient panel cast votes for each PICO question and made final recommendations.
The review of evidence left the guidelines authors with 241 PICO questions, “which is a lot,” Dr. Bernstein said. “To put this in perspective, some guidelines address only 10 or 15 PICO questions. Fortunately, we had a dedicated group of experts who were up to the challenge.” Dr. Johnson emphasized that the forthcoming guidelines should not be used by insurers to mandate a specific order of prescribing. “Clinicians must retain the latitude to prescribe medications based on individual patient factors and preferences,” she said.
Dr. Bernstein disclosed that she is an adviser to, a consultant for, and has received grant or research support from Boehringer Ingelheim and has also received grant or research support from Kadmon and Pfizer. Dr. Johnson disclosed that she has received research support from the American College of Rheumatology to develop these guidelines. She has also been an investigator for trials sponsored by Bristol-Myers Squibb, Roche, and Boehringer Ingelheim and has mitigated these relevant conflicts of interest 1 year prior to the development of these guidelines, and will continue to do so for the foreseeable future.
AT ACR 2023