User login
Incidence, outcomes, and management of bleeding in non-ST-elevation acute coronary syndromes
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?

Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13

BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
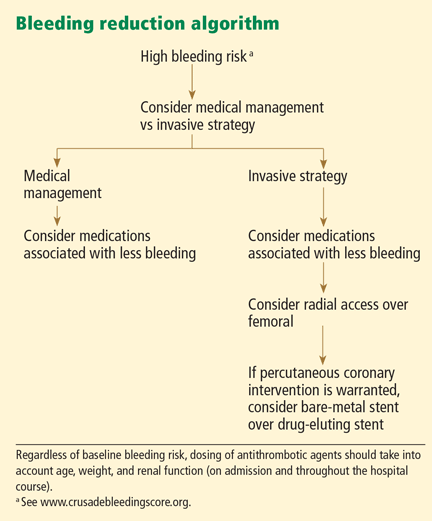
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
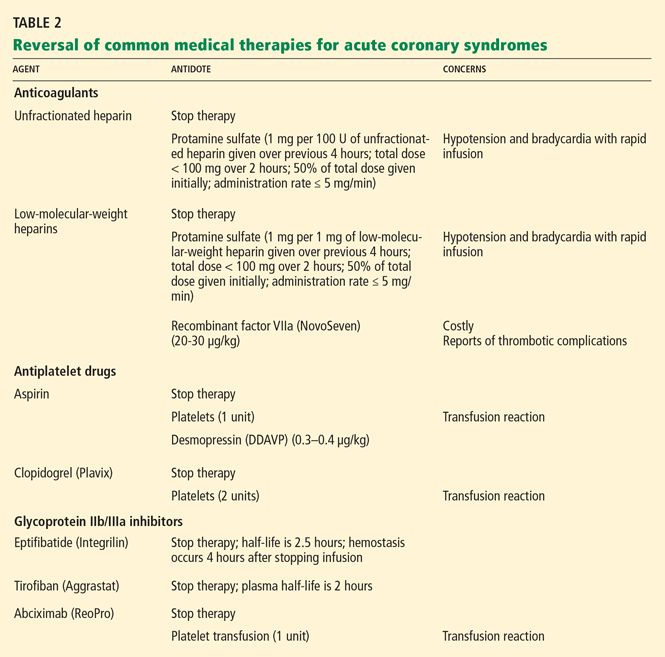
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?
Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13
BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
The medical management of non-ST-elevation acute coronary syndromes focuses on blocking the coagulation cascade and inhibiting platelets. This—plus diagnostic angiography followed, if needed, by revascularization—has reduced the rates of death and recurrent ischemic events.1 However, the combination of potent antithrombotic drugs and invasive procedures also increases the risk of bleeding.
This review discusses the incidence and complications associated with bleeding during the treatment of acute coronary syndromes and summarizes recommendations for preventing and managing bleeding in this setting.
THE TRUE INCIDENCE OF BLEEDING IS HARD TO DETERMINE
The optimal way to detect and analyze bleeding events in clinical trials and registries is highly debated. The reported incidences of bleeding during antithrombotic and antiplatelet therapy for non-ST-elevation acute coronary syndromes depend on how bleeding was defined, how the acute coronary syndromes were treated, and on other factors such as how the study was designed.
How was bleeding defined?
Since these classification schemes are based on different types of data, they yield different numbers when applied to the same study population. For instance, Rao et al4 pooled the data from the PURSUIT and PARAGON B trials (15,454 patients in all) and found that the incidence of severe bleeding (by the GUSTO criteria) was 1.2%, while the rate of major bleeding (by the TIMI criteria) was 8.2%.
What was the treatment strategy?
Another reason that the true incidence of bleeding is hard to determine is that different studies used treatment strategies that differed in the type, timing, and dose of antithrombotic agents and whether invasive procedures were used early. For example, if unfractionated heparin is used aggressively in regimens that are not adjusted for weight and with a higher target for the activated clotting time, the risk of bleeding is higher than with conservative dosing.5–7
Subherwal et al8 evaluated the effect of treatment strategy on the incidence of bleeding in patients with non-ST-elevation acute coronary syndromes who received two or more antithrombotic drugs in the CRUSADE Quality Improvement Initiative. The risk of bleeding was higher with an invasive approach (catheterization) than with a conservative approach (no catheterization), regardless of baseline bleeding risk.
What type of study was it?
Another source of variation is the design of the study. Registries differ from clinical trials in patient characteristics and in the way data are gathered (prospectively vs retrospectively).
In registries, data are often collected retrospectively, whereas in clinical trials the data are prospectively collected. For this reason, the definition of bleeding in registries is often based on events that are easily identified through chart review, such as transfusion. This may lead to a lower reported rate of bleeding, since other, less serious bleeding events such as access-site hematomas and epistaxis may not be documented in the medical record.
On the other hand, registries often include older and sicker patients, who may be more prone to bleeding and who are often excluded from clinical trials. This may lead to a higher rate of reported bleeding.9
Where the study was conducted makes a difference as well, owing to regional practice differences. For example, Moscucci et al10 reported that the incidence of major bleeding in 24,045 patients with non-ST-elevation acute coronary syndromes in the GRACE registry (in 14 countries worldwide) was 3.9%. In contrast, Yang et al11 reported that the rate of bleeding in the CRUSADE registry (in the United States) was 10.3%.
This difference was partly influenced by different definitions of bleeding. The GRACE registry defined major bleeding as life-threatening events requiring transfusion of two or more units of packed red blood cells, or resulting in an absolute decrease in the hematocrit of 10% or more or death, or hemorrhagic subdural hematoma. In contrast, the CRUSADE data reflect bleeding requiring transfusion. However, practice patterns such as greater use of invasive procedures in the United States may also be responsible.
Rao and colleagues12 examined international variation in blood transfusion rates among patients with acute coronary syndromes. Patients outside the United States were significantly less likely to receive transfusions, even after adjusting for patient and practice differences.
Taking these confounders into account, it is reasonable to estimate that the frequency of bleeding in patients with non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%.13
BLEEDING IS ASSOCIATED WITH POOR OUTCOMES
Regardless of the definition or the data source, hemorrhagic complications are associated with a higher risk of death and nonfatal adverse events, both in the short term and in the long term.
Short-term outcomes
A higher risk of death. In the GRACE registry study by Moscucci et al10 discussed above, patients who had major bleeding were significantly more likely to die during their hospitalization than those who did not (odds ratio [OR] 1.64, 95% confidence interval [CI] 1.18–2.28).
Rao et al14 evaluated pooled data from the multicenter international GUSTO IIb, PURSUIT, and PARAGON A and B trials and found that the effects of bleeding in non-ST-elevation acute coronary syndromes extended beyond the hospital stay. The more severe the bleeding (by the GUSTO criteria), the greater the adjusted hazard ratio (HR) for death within 30 days:
- With mild bleeding—HR 1.6, 95% CI 1.3–1.9
- With moderate bleeding—HR 2.7, 95% CI 2.3–3.4
- With severe bleeding—HR 10.6, 95% CI 8.3–13.6.
The pattern was the same for death within 6 months:
- With mild bleeding—HR 1.4, 95% CI 1.2–1.6
- With moderate bleeding—HR 2.1, 95% CI 1.8–2.4
- With severe bleeding, HR 7.5, 95% CI 6.1–9.3.
These findings were confirmed by Eikelboom et al15 in 34,146 patients with acute coronary syndromes in the OASIS registry, the OASIS-2 trial, and the CURE randomized trial. In the first 30 days, five times as many patients died (12.8% vs 2.5%; P < .0009) among those who developed major bleeding compared with those who did not. These investigators defined major bleeding as bleeding that was life-threatening or significantly disabling or that required transfusion of two or more units of packed red blood cells.
A higher risk of nonfatal adverse events. Bleeding after antithrombotic therapy for non-ST-elevation acute coronary syndromes has also been associated with nonfatal adverse events such as stroke and stent thrombosis.
For example, in the study by Eikelboom et al,15 major bleeding was associated with a higher risk of recurrent ischemic events. Approximately 1 in 5 patients in the OASIS trials who developed major bleeding during the first 30 days died or had a myocardial infarction or stroke by 30 days, compared with 1 in 20 of those who did not develop major bleeding during the first 30 days. However, after events that occurred during the first 30 days were excluded, the association between major bleeding and both myocardial infarction and stroke was no longer evident between 30 days and 6 months.
Manoukian et al16 evaluated the impact of major bleeding in 13,819 patients with highrisk acute coronary syndromes undergoing treatment with an early invasive strategy in the ACUITY trial. At 30 days, patients with major bleeding had higher rates of the composite end point of death, myocardial infarction, or unplanned revascularization for ischemia (23.1% vs 6.8%, P < .0001) and of stent thrombosis (3.4% vs 0.6%, P < .0001).
Long-term outcomes
The association between bleeding and adverse outcomes persists in the long term as well, although the mechanisms underlying this association are not well studied.
Kinnaird et al17 examined the data from 10,974 unselected patients who underwent percutaneous coronary intervention. At 1 year, the following percentages of patients had died:
- After TIMI major bleeding—17.2%
- After TIMI minor bleeding—9.1%
- After no bleeding—5.5%.
However, after adjustment for potential confounders, only transfusion remained a significant predictor of 1-year mortality.
Mehran et al18 evaluated 1-year mortality data from the ACUITY trial. Compared with the rate in patients who had no major bleeding and no myocardial infarction, the hazard ratios for death were:
- After major bleeding—HR 3.5, 95% CI 2.7–4.4
- After myocardial infarction—HR 3.1, 95% CI 2.4–3.9.
Interestingly, the risk of death associated with myocardial infarction abated after 7 days, while the risk associated with bleeding persisted beyond 30 days and remained constant throughout the first year following the bleeding event.
Similarly, Ndrepepa and colleagues19 examined pooled data from four ISAR trials using the TIMI bleeding scale and found that myocardial infarction, target vessel revascularization, and major bleeding all had similar discriminatory ability at predicting 1-year mortality.
In patients undergoing elective or urgent percutaneous coronary intervention in the REPLACE-2 trial,20 independent predictors of death by 1 year were21:
- Major hemorrhage (OR 2.66, 95% CI 1.44–4.92)
- Periprocedural myocardial infarction (OR 2.46, 95% CI 1.44–4.20).
THEORIES OF HOW BLEEDING MAY CAUSE ADVERSE OUTCOMES
Several mechanisms have been proposed to explain the association between bleeding during treatment for acute coronary syndromes and adverse clinical outcomes.13,22
The immediate effects of bleeding are thought to be hypotension and a reflex hyperadrenergic state to compensate for the loss of intravascular volume.23 This physiologic response is believed to contribute to myocardial ischemia by further decreasing myocardial oxygen supply in obstructive coronary disease.
Trying to minimize blood loss, physicians may withhold anticoagulation and antiplatelet therapy, which in turn may lead to further ischemia.24 To compensate for blood loss, physicians may also resort to blood transfusion. However, depletion of 2,3-diphosphoglycerate and nitric oxide in stored donor red blood cells is postulated to reduce oxygen delivery by increasing hemoglobin’s affinity for oxygen, leading to induced microvascular obstruction and adverse inflammatory reactions.15,25
Recent data have also begun to elucidate the long-term effects of bleeding during acute coronary syndrome management. Patients with anemia during the acute phase of infarction have greater neurohormonal activation.26 These adaptive responses to anemia may lead to eccentric left ventricular remodeling that may lead to higher oxygen consumption, increased diastolic wall stress, interstitial fibrosis, and accelerated myocyte loss.27–30
Nevertheless, we must point out that although strong associations between bleeding and adverse outcomes have been established, direct causality has not.
TO PREVENT BLEEDING, START BY ASSESSING RISK
The CRUSADE bleeding risk score
The CRUSADE bleeding score (calculator available at http://www.crusadebleedingscore.org/) was developed and validated in more than 89,000 community-treated patients with non-ST-elevation acute coronary syndromes.8 It is based on eight variables:
- Sex (higher risk in women)
- History of diabetes (higher risk)
- Prior vascular disease (higher risk)
- Heart rate (the higher the rate, the higher the risk)
- Systolic blood pressure (higher risk with pressures above or below the 121–180 mm Hg range)
- Signs of congestive heart failure (higher risk)
- Baseline hematocrit (the lower the hematocrit, the higher the risk)
- Creatinine clearance (by the Cockcroft-Gault formula; the lower the creatinine clearance, the higher the risk).
Patients who are found to have bleeding scores suggesting a moderate or higher risk of bleeding should be considered for medications associated with a favorable bleeding profile, and for radial access at the time of coronary angiography. Scores are graded as follows8:
- < 21: Very low risk
- 21–30: Low risk
- 31–40: Moderate risk
- 41–50: High risk
- > 50: Very high risk.
The CRUSADE bleeding score is unique in that, unlike earlier risk stratification tools, it was developed in a “real world” population, not in subgroups or in a clinical trial. It can be calculated at baseline to help guide the selection of treatment.8
Adjusting the heparin regimen in patients at risk of bleeding
Both the joint American College of Cardiology/American Heart Association1 and the European Society of Cardiology guidelines31 for the treatment of non-ST-elevation acute coronary syndromes recommend taking steps to prevent bleeding, such as adjusting the dosage of unfractionated heparin, using safer drugs, reducing the duration of antithrombotic treatment, and using combinations of antithrombotic and antiplatelet agents according to proven indications.31
In the CRUSADE registry, 42% of patients with non-ST-elevation acute coronary syndromes received at least one initial dose of antithrombotic drug outside the recommended range, resulting in an estimated 15% excess of bleeding events.32 Thus, proper dosing is a target for prevention.
Appropriate antithrombotic dosing takes into account the patient’s age, weight, and renal function. However, heparin dosage in the current guidelines1 is based on weight only: a loading dose of 60 U/kg (maximum 4,000 U) by intravenous bolus, then 12 U/kg/hour (maximum 1,000 U/hour) to maintain an activated partial thromboplastin time of 50 to 70 seconds.1
Renal dysfunction is particularly worrisome in patients with non-ST-elevation acute coronary syndromes because it is associated with higher rates of major bleeding and death. In the OASIS-5 trial,33 the overall risk of death was approximately five times higher in patients in the lowest quartile of renal function (glomerular filtration rate < 58 mL/min/1.73 m2) than in the highest quartile (glomerular filtration rate ≥ 86 mL/min/1.73 m2).
Renal function must be evaluated not only on admission but also throughout the hospital stay. Patients presenting with acute coronary syndromes often experience fluctuations in renal function that would call for adjustment of heparin dosing, either increasing the dose to maximize the drug’s efficacy if renal function is recovering or decreasing the dose to prevent bleeding if renal function is deteriorating.
Clopidogrel vs prasugrel
Certain medications should be avoided when the risk of bleeding outweighs any potential benefit in terms of ischemia.
For example, in a randomized trial,34 prasugrel (Effient), a potent thienopyridine, was associated with a significantly lower rate of the composite end point of stroke, myocardial infarction, or death than clopidogrel (Plavix) in patients with acute coronary syndromes undergoing percutaneous coronary interventions. However, it did not seem to offer any advantage in patients 75 years old and older, those with prior stroke or transient ischemic attack, or those weighing less than 60 kg, and it posed a substantially higher risk of bleeding.
With clopidogrel, the risk of acute bleeding is primarily in patients who undergo coronary artery bypass grafting within 5 days of receiving a dose.35,36 Therefore, clopidogrel should be stopped 5 to 7 days before bypass surgery.1 Importantly, there is no increased risk of recurrent ischemic events during this 5-day waiting period in patients who receive clopidogrel early. Therefore, the recommendation to stop clopidogrel before surgery does not negate the benefits of early treatment.36
Lower-risk drugs: Fondaparinux and bivalirudin
At this time, only two agents have been studied in clinical trials that have specifically focused on reducing bleeding risk: fondaparinux (Arixtra) and bivalirudin (Angiomax).20,37–39
Fondaparinux
OASIS-5 was a randomized, double-blind trial that compared fondaparinux and enoxaparin (Lovenox) in patients with acute coronary syndromes.38 Fondaparinux was similar to enoxaparin in terms of the combined end point of death, myocardial infarction, or refractory ischemia at 9 days, and fewer patients on fondaparinux developed bleeding (2.2% vs 4.1%, HR 0.52; 95% CI 0.44–0.61). This difference persisted during long-term follow-up.
Importantly, fewer patients died in the fondaparinux group. At 180 days, 638 (6.5%) of the patients in the enoxaparin group had died, compared with 574 (5.8%) in the fondaparinux group, a difference of 64 deaths (P = .05). The authors found that 41 fewer patients in the fondaparinux group than in the enoxaparin group died after major bleeding, and 20 fewer patients in the fondaparinux group died after minor bleeding.38 Thus, most of the difference in mortality rates between the two groups was attributed to a lower incidence of bleeding with fondaparinux.
Unfortunately, despite its safe bleeding profile, fondaparinux has fallen out of favor for use in acute coronary syndromes, owing to a higher risk of catheter thrombosis in the fondaparinux group (0.9%) than in those undergoing percutaneous coronary interventions with enoxaparin alone (0.4%) in the OASIS-5 trial.40
Bivalirudin
The direct thrombin inhibitor bivalirudin has been studied in three large randomized trials in patients undergoing percutaneous coronary interventions.20,37,41
The ACUITY trial37 was a prospective, open-label, randomized, multicenter trial that compared three regimens in patients with moderate or high-risk non-ST-elevation acute coronary syndromes:
- Heparin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin plus a glycoprotein IIb/IIIa inhibitor
- Bivalirudin alone.
Bivalirudin alone was as effective as heparin plus a glycoprotein IIb/IIIa inhibitor with respect to the composite ischemia end point, which at 30 days had occurred in 7.8% vs 7.3% of the patients in these treatment groups (P = .32, RR 1.08; 95% CI 0.93–1.24), and it was superior with respect to major bleeding (3.0% vs 5.7%, P < .001, RR 0.53; 95% CI 0.43–0.65).
The HORIZONS-AMI study41 was a prospective, open-label, randomized, multicenter trial that compared bivalirudin alone vs heparin plus a glycoprotein IIb/IIIa inhibitor in patients with ST-elevation acute coronary syndromes who were undergoing primary percutaneous coronary interventions. The two primary end points were major bleeding and net adverse events.
At 1 year, patients assigned to bivalirudin had a lower rate of major bleeding than did controls (5.8% vs 9.2%, HR 0.61, 95% CI 0.48–0.78, P < .0001), with similar rates of major adverse cardiac events in both groups (11.9% vs 11.9%, HR 1.00, 95% CI 0.82– 1.21, P = .98).41
Both OASIS 5 and HORIZONS-AMI are examples of clinical trials in which strategies that reduced bleeding risk were also associated with improved survival.
For cardiac catheterization, inserting the catheter in the wrist poses less risk
Bleeding is currently the most common noncardiac complication in patients undergoing percutaneous coronary interventions, and it most often occurs at the vascular access site.17
Rao et al12 evaluated data from 593,094 procedures in the National Cardiovascular Data Registry and found that, compared with the femoral approach, the use of transradial percutaneous coronary intervention was associated with a similar rate of procedural success (OR 1.02, 95% CI 0.93–1.12) but a significantly lower risk of bleeding complications (OR 0.42, 95% CI 0.31–0.56) after multivariable adjustment.
The use of smaller sheath sizes (4F–6F) and preferential use of bivalirudin over unfractionated heparin and glycoprotein IIb/IIIa inhibitor therapy are other methods described to decrease the risk of bleeding after percutaneous coronary interventions.20,41–49
IF BLEEDING OCCURS
Once a bleeding complication occurs, cessation of therapy is a potential option. Stopping or reversing antithrombotic and antiplatelet therapy is warranted in the event of major bleeding (eg, gastrointestinal, retroperitoneal, intracranial).31
Stopping antithrombotic and antiplatelet therapy
Whether bleeding is minor or major, the risk of a recurrent thrombotic event must be considered, especially in patients who have undergone revascularization, stent implantation, or both. The risk of acute thrombotic events after interrupting antithrombotic or antiplatelet agents is considered greatest 4 to 5 days following revascularization or percutaneous coronary intervention.15 If bleeding can be controlled with local treatment such as pressure, packing, or dressing, antithrombotic and antiplatelet therapy need not be interrupted.50
Current guidelines recommend strict control of hemorrhage for at least 24 hours before reintroducing antiplatelet or antithrombotic agents.
It is also important to remember that in the setting of gastrointestinal bleeding due to peptic ulcer disease, adjunctive proton pump inhibitors are recommended after restarting antiplatelet or antithrombotic therapy or both.
Importantly, evidence-based antithrombotic medications (especially dual antiplatelet therapy) should be restarted once the acute bleeding event has resolved.31
Reversal of anticoagulant and antiplatelet therapies
Unfractionated heparin is reversed with infusion of protamine sulfate at a dose of 1 mg per 100 U of unfractionated heparin given over the previous 4 hours.51,52 The rate of protamine sulfate infusion should be less than 100 mg over 2 hours, with 50% of the dose given initially and subsequent doses titrated according to bleeding response.52,53 Protamine sulfate is associated with a risk of hypotension and bradycardia, and for this reason it should be given no faster than 5 mg/min.
Low-molecular-weight heparin (LMWH) can be inhibited by 1 mg of protamine sulfate for each 1 mg of LMWH given over the previous 4 hours.51,52
However, protamine sulfate only partially neutralizes the anticoagulant effect of LMWH. In cases in which protamine sulfate is unsuccessful in abating bleeding associated with LMWH use, guidelines allow for the use of recombinant factor VIIa (NovoSeven).31 In healthy volunteers given fondaparinux, recombinant factor VIIa normalized coagulation times and thrombin generation within 1.5 hours, with a sustained effect for 6 hours.52
It is important to note that the use of this agent has not been fully studied, it is very costly (a single dose of 40 μg/kg costs from $3,000 to $4,000), and it is linked to reports of increased risk of thrombotic complications.54,55
Antiplatelet agents are more complex to reverse. The antiplatelet actions of aspirin and clopidogrel wear off as new platelets are produced. Approximately 10% of a patient’s platelet count is produced daily; thus, the antiplatelet effects of aspirin and clopidogrel can persist for 5 to 10 days.31,56
If these agents need to be reversed quickly to stop bleeding, according to expert consensus the aspirin effect can be reversed by transfusion of one unit of platelets. The antiplatelet effect of clopidogrel is more significant than that of aspirin; thus, two units of platelets are recommended.56
Glycoprotein IIb/IIIa inhibitors. If a major bleeding event requires the reversal of glycoprotein IIb/IIIa inhibitor therapy, the treatment must take into consideration the pharmacodynamics of the target drug. Both eptifibatide (Integrilin) and tirofiban (Aggrastat) competitively inhibit glycoprotein IIb/IIIa receptors; thus, their effects depend on dosing, elimination, and time. Due to the stoichiometry of both drugs, transfusion of platelets is ineffective. Both eptifibatide and tirofiban are eliminated by the kidney; thus, normal renal function is key to the amount of time it takes for platelet function to return to baseline.57 Evidence suggests that fibrinogen-rich plasma can be administered to restore platelet function.31,58,59
Abciximab (ReoPro). Whereas reversal of eptifibatide and tirofiban focuses on overcoming competitive inhibition, neutralization of abciximab involves overcoming its high receptor affinity. At 24 hours after abciximab infusion is stopped, platelet aggregation may still be inhibited by up to 50%. Fortunately, owing to abciximab’s short plasma half-life and its dilution in serum, platelet transfusion is effective in reversing its antiplatelet effects.31,57
Blood transfusion
Long considered beneficial to critically ill patients, blood transfusion to maintain hematocrit levels during acute coronary syndromes has come under intense scrutiny. Randomized trials have shown that transfusion should not be given aggressively to critically ill patients.60 In acute coronary syndromes, there are only observational data.
Rao et al61 used detailed clinical data from 24,112 patients with acute coronary syndromes in the GUSTO IIb, PURSUIT, and PARAGON B trials to determine the association between blood transfusion and outcomes in patients who developed moderate to severe bleeding, anemia, or both during their hospitalization. The rates of death in the hospital and at 30 days were significantly higher in patients who received a transfusion (30-day mortality HR 3.94; 95% CI 3.36–4.75). However, there was no significant association between transfusion and the 30-day mortality rate if the nadir hematocrit was 25% or less.
Of note: no randomized clinical trial has evaluated transfusion strategies in acute coronary syndromes at this time. Until such data are available, it is reasonable to follow published guidelines and to avoid transfusion in stable patients with ischemic heart disease unless the hematocrit is 25% or less.31 The risks and benefits of blood transfusion should be carefully weighed. Routine use of transfusion to maintain predefined hemoglobin levels is not recommended in stable patients.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. J Am Coll Cardiol 2007; 50:e1–e157.
- The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329:673–682.
- Chesebro JH, Knatterud G, Roberts R, et al. Thrombolysis in Myocardial Infarction (TIMI) Trial, Phase I: a comparison between intravenous tissue plasminogen activator and intravenous streptokinase. Clinical findings through hospital discharge. Circulation 1987; 76:142–154.
- Rao SV, O’Grady K, Pieper KS, et al. A comparison of the clinical impact of bleeding measured by two different classifications among patients with acute coronary syndromes. J Am Coll Cardiol 2006; 47:809–816.
- Granger CB, Hirsch J, Califf RM, et al. Activated partial thromboplastin time and outcome after thrombolytic therapy for acute myocardial infarction: results from the GUSTO-I trial. Circulation 1996; 93:870–878.
- Gilchrist IC, Berkowitz SD, Thompson TD, Califf RM, Granger CB. Heparin dosing and outcome in acute coronary syndromes: the GUSTO-IIb experience. Global Use of Strategies to Open Occluded Coronary Arteries. Am Heart J 2002; 144:73–80.
- Tolleson TR, O’Shea JC, Bittl JA, et al. Relationship between heparin anticoagulation and clinical outcomes in coronary stent intervention: observations from the ESPRIT trial. J Am Coll Cardiol 2003; 41:386–393.
- Subherwal S, Bach RG, Chen AY, et al. Baseline risk of major bleeding in non-ST-segment-elevation myocardial infarction: the CRUSADE (Can Rapid risk stratification of Unstable angina patients Suppress ADverse outcomes with Early implementation of the ACC/AHA Guidelines) Bleeding Score. Circulation 2009; 119:1873–1882.
- Bassand JP. Bleeding and transfusion in acute coronary syndromes: a shift in the paradigm. Heart 2008; 94:661–666.
- Moscucci M, Fox KA, Cannon CP, et al. Predictors of major bleeding in acute coronary syndromes: the Global Registry of Acute Coronary Events (GRACE). Eur Heart J 2003; 24:1815–1823.
- Yang X, Alexander KP, Chen AY, et al; CRUSADE Investigators. The implications of blood transfusions for patients with non-ST-segment elevation acute coronary syndromes: results from the CRUSADE National Quality Improvement Initiative. J Am Coll Cardiol 2005; 46:1490–1495.
- Rao SV, Ou FS, Wang TY, et al. Trends in the prevalence and outcomes of radial and femoral approaches to percutaneous coronary intervention: a report from the National Cardiovascular Data Registry. JACC Cardiovasc Interv 2008; 1:379–386.
- Rao SV, Eikelboom JA, Granger CB, Harrington RA, Califf RM, Bassand JP. Bleeding and blood transfusion issues in patients with non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1193–1204.
- Rao SV, O’Grady K, Pieper KS, et al. Impact of bleeding severity on clinical outcomes among patients with acute coronary syndromes. Am J Cardiol 2005; 96:1200–1206.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Kinnaird TD, Stabile E, Mintz GS, et al. Incidence, predictors, and prognostic implications of bleeding and blood transfusion following percutaneous coronary interventions. Am J Cardiol 2003; 92:930–935.
- Mehran R, Pocock SJ, Stone GW, et al. Associations of major bleeding and myocardial infarction with the incidence and timing of mortality in patients presenting with non-ST-elevation acute coronary syndromes: a risk model from the ACUITY trial. Eur Heart J 2009; 30:1457–1466.
- Ndrepepa G, Berger PB, Mehilli J, et al. Periprocedural bleeding and 1-year outcome after percutaneous coronary interventions: appropriateness of including bleeding as a component of a quadruple end point. J Am Coll Cardiol 2008; 51:690–697.
- Lincoff AM, Bittl JA, Harrington RA, et al; REPLACE-2 Investigators. Bivalirudin and provisional glycoprotein IIb/IIIa blockade compared with heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary intervention: REPLACE-2 randomized trial. JAMA 2003; 289:853–863.
- Feit F, Voeltz MD, Attubato MJ, et al. Predictors and impact of major hemorrhage on mortality following percutaneous coronary intervention from the REPLACE-2 Trial. Am J Cardiol 2007; 100:1364–1369.
- Fitchett D. The impact of bleeding in patients with acute coronary syndromes: how to optimize the benefits of treatment and minimize the risk. Can J Cardiol 2007; 23:663–671.
- Bassand JP. Impact of anaemia, bleeding, and transfusions in acute coronary syndromes: a shift in the paradigm. Eur Heart J 2007; 28:1273–1274.
- Yan AT, Yan RT, Huynh T, et al; INTERACT Investigators. Bleeding and outcome in acute coronary syndrome: insights from continuous electrocardiogram monitoring in the Integrilin and Enoxaparin Randomized Assessment of Acute Coronary Syndrome Treatment (INTERACT) Trial. Am Heart J 2008; 156:769–775.
- Jolicoeur EM, O’Neill WW, Hellkamp A, et al; APEX-AMI Investigators. Transfusion and mortality in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur Heart J 2009; 30:2575–2583.
- Gehi A, Ix J, Shlipak M, Pipkin SS, Whooley MA. Relation of anemia to low heart rate variability in patients with coronary heart disease (from the Heart and Soul study). Am J Cardiol 2005; 95:1474–1477.
- Anand I, McMurray JJ, Whitmore J, et al. Anemia and its relationship to clinical outcome in heart failure. Circulation 2004; 110:149–154.
- O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant 2000; 15(suppl 3):19–22.
- Olivetti G, Quaini F, Lagrasta C, et al. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am J Pathol 1992; 141:227–239.
- Aronson D, Suleiman M, Agmon Y, et al. Changes in haemoglobin levels during hospital course and long-term outcome after acute myocardial infarction. Eur Heart J 2007; 28:1289–1296.
- Task Force for Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of European Society of Cardiology; Bassand JP, Hamm CW, Ardissino D, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. Eur Heart J 2007; 28:1598–1660.
- Alexander KP, Chen AY, Roe MT, et al; CRUSADE Investigators. Excess dosing of antiplatelet and antithrombin agents in the treatment of non-ST-segment elevation acute coronary syndromes. JAMA 2005; 294:3108–3116.
- Fox KA, Bassand JP, Mehta SR, et al; OASIS 5 Investigators. Influence of renal function on the efficacy and safety of fondaparinux relative to enoxaparin in non ST-segment elevation acute coronary syndromes. Ann Intern Med 2007; 147:304–310.
- Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med 2007; 357:2001–2015.
- Berger JS, Frye CB, Harshaw Q, Edwards FH, Steinhubl SR, Becker RC. Impact of clopidogrel in patients with acute coronary syndromes requiring coronary artery bypass surgery: a multicenter analysis. J Am Coll Cardiol 2008; 52:1693–1701.
- Fox KA, Mehta SR, Peters R, et al; Clopidogrel in Unstable angina to prevent Recurrent ischemic Events Trial. Benefits and risks of the combination of clopidogrel and aspirin in patients undergoing surgical revascularization for non-ST-elevation acute coronary syndrome: the Clopidogrel in Unstable angina to prevent Recurrent ischemic Events (CURE) Trial. Circulation 2004; 110:1202–1208.
- Stone GW, McLaurin BT, Cox DA, et al; ACUITY Investigators. Bivalirudin for patients with acute coronary syndromes. N Engl J Med 2006; 355:2203–2216.
- Fifth Organization to Assess Strategies in Acute Ischemic Syndromes Investigators; Yusuf S, Mehta SR, Chrolavicius S, et al. Comparison of fondaparinux and enoxaparin in acute coronary syndromes. N Engl J Med 2006; 354:1464–1476.
- Potsis TZ, Katsouras C, Goudevenos JA. Avoiding and managing bleeding complications in patients with non-ST-segment elevation acute coronary syndromes. Angiology 2009; 60:148–158.
- Mehta SR, Granger CB, Eikelboom JW, et al. Efficacy and safety of fondaparinux versus enoxaparin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: results from the OASIS-5 trial. J Am Coll Cardiol 2007; 50:1742–1751.
- Mehran R, Lansky AJ, Witzenbichler B, et al; HORIZONS-AMI Trial Investigators. Bivalirudin in patients undergoing primary angioplasty for acute myocardial infarction (HORIZONS-AMI): 1-year results of a randomised controlled trial. Lancet 2009; 374:1149–1159.
- Stone GW, Ware JH, Bertrand ME, et al; ACUITY Investigators. Antithrombotic strategies in patients with acute coronary syndromes undergoing early invasive management: one-year results from the ACUITY trial. JAMA 2007; 298:2497–2506.
- Cantor WJ, Mahaffey KW, Huang Z, et al. Bleeding complications in patients with acute coronary syndrome undergoing early invasive management can be reduced with radial access, smaller sheath sizes, and timely sheath removal. Catheter Cardiovasc Interv 2007; 69:73–83.
- Büchler JR, Ribeiro EE, Falcão JL, et al. A randomized trial of 5 versus 7 French guiding catheters for transfemoral percutaneous coronary stent implantation. J Interv Cardiol 2008; 21:50–55.
- Shammas NW, Allie D, Hall P, et al; APPROVE Investigators. Predictors of in-hospital and 30-day complications of peripheral vascular interventions using bivalirudin as the primary anticoagulant: results from the APPROVE Registry. J Invasive Cardiol 2005; 17:356–359.
- Doyle BJ, Ting HH, Bell MR, et al. Major femoral bleeding complications after percutaneous coronary intervention: incidence, predictors, and impact on long-term survival among 17,901 patients treated at the Mayo Clinic from 1994 to 2005. JACC Cardiovasc Interv 2008; 1:202–209.
- Stone GW, White HD, Ohman EM, et al; Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet 2007; 369:907–919.
- Stone GW, Bertrand ME, Moses JW, et al; ACUITY Investigators. Routine upstream initiation vs deferred selective use of glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: the ACUITY Timing trial. JAMA 2007; 297:591–602.
- Lincoff AM, Bittl JA, Kleiman NS, et al; REPLACE-1 Investigators. Comparison of bivalirudin versus heparin during percutaneous coronary intervention (the Randomized Evaluation of PCI Linking Angiomax to Reduced Clinical Events [REPLACE]-1 trial). Am J Cardiol 2004; 93:1092–1096.
- Barkun A, Bardou M, Marshall JK; Nonvariceal Upper GI Bleeding Consensus Conference Group. Consensus recommendations for managing patients with nonvariceal upper gastrointestinal bleeding. Ann Intern Med 2003; 139:843–857.
- Warkentin TE, Crowther MA. Reversing anticoagulants both old and new. Can J Anaesth 2002; 49:S11–S25.
- Crowther MA, Warkentin TE. Bleeding risk and the management of bleeding complications in patients undergoing anticoagulant therapy: focus on new anticoagulant agents. Blood 2008; 111:4871–4879.
- Kessler CM. Current and future challenges of antithrombotic agents and anticoagulants: strategies for reversal of hemorrhagic complications. Semin Hematol 2004; 41(suppl 1):44–50.
- Ganguly S, Spengel K, Tilzer LL, O’Neal B, Simpson SQ. Recombinant factor VIIa: unregulated continuous use in patients with bleeding and coagulopathy does not alter mortality and outcome. Clin Lab Haematol 2006; 28:309–312.
- O’Connell KA, Wood JJ, Wise RP, Lozier JN, Braun MM. Thromboembolic adverse events after use of recombinant human coagulation factor VIIa. JAMA 2006; 295:293–298.
- Beshay JE, Morgan H, Madden C, Yu W, Sarode R. Emergency reversal of anticoagulation and antiplatelet therapies in neurosurgical patients. J Neurosurg 2010; 112:307–318.
- Tcheng JE. Clinical challenges of platelet glycoprotein IIb/IIIa receptor inhibitor therapy: bleeding, reversal, thrombocytopenia, and retreatment. Am Heart J 2000; 139:S38–S45.
- Li YF, Spencer FA, Becker RC. Comparative efficacy of fibrinogen and platelet supplementation on the in vitro reversibility of competitive glycoprotein IIb/IIIa receptor-directed platelet inhibition. Am Heart J 2002; 143:725–732.
- Schroeder WS, Gandhi PJ. Emergency management of hemorrhagic complications in the era of glycoprotein IIb/IIIa receptor antagonists, clopidogrel, low molecular weight heparin, and third-generation fibrinolytic agents. Curr Cardiol Rep 2003; 5:310–317.
- Hébert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999; 340:409–417.
- Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA 2004; 292:1555–1562.
KEY POINTS
- The reported incidence of bleeding after treatment for non-ST-elevation acute coronary syndromes ranges from less than 1% to 10%, depending on a number of factors.
- Bleeding is strongly associated with adverse outcomes, although a causal relationship has not been established.
- Patients should be assessed for risk of bleeding so that the antithrombotic and antiplatelet regimen can be adjusted, safer alternatives can be considered, and percutaneous interventions can be used less aggressively for those at high risk.
- If bleeding develops and the risk of continued bleeding outweighs the risk of recurrent ischemia, antithrombotic and antiplatelet drug therapy can be interrupted and other agents given to reverse the effects of these drugs.
Difficulty swallowing solid foods; food ‘getting stuck in the chest’
A 61-year-old woman presents to her primary care physician because for the last 4 weeks she has had difficulty swallowing solid food and a feeling of food “getting stuck in the chest.” She also reports having nausea, mild epigastric pain, and heartburn. She denies having fevers, chills, night sweats, weight loss, vomiting, diarrhea, hematochezia, or melena.
Medical history
For the past 20 years, she has had gastroesophageal reflux disease (GERD), intermittently treated with a proton pump inhibitor. She also has arthritis, hyperlipidemia, hypertension, and asthma, and she has undergone right hip replacement for a hip fracture. She has no known allergies.
She lives in the Midwest region of the United States and is on disability due to her arthritis. She is divorced and has three children.
She quit smoking 3 years ago after smoking half a pack per day for 30 years. She drinks socially; she has never used recreational drugs.
She recalls that an uncle had cancer, but she does not know the specific type.
Physical examination
The patient’s temperature is 96.7°F (35.9°C), heart rate 86 per minute, blood pressure 150/92 mm Hg, respiratory rate 16 per minute, and oxygen saturation 100% on room air.
Differential diagnosis
Although the differential diagnosis at this stage is broad, a few conditions that commonly present like this are:
- Esophageal cancer
- Esophageal stricture
- Esophageal webs
- Esophagitis (infectious, inflammatory)
- Peptic ulcer disease.
WHICH TEST SHOULD BE ORDERED?
1. Which test will you order now for this patient?
- Endoscopy (esophagogastroduodenoscopy)
- Serum Helicobacter pylori antibody testing
- Wireless pH monitoring
- Barium swallow
Endoscopy would be the best test to order. Esophageal cancer and esophageal stricture must be ruled out, in view of her long history of GERD, gastritis, and smoking and her alarming symptoms of difficulty swallowing and food sticking. In this situation, endoscopy is the first test recommended. In addition to its diagnostic value, it offers an opportunity to obtain tissue samples and to perform a therapeutic intervention, if necessary.1,2
H pyloriantibody testing is used in the “test-and-treat approach” for H pylori infection, an established management strategy for patients who have uninvestigated dyspepsia and who are younger than 55 years and have no “alarm features,” ie, red flags for cancer. The alarm features commonly described are anemia, early satiety, unexplained weight loss, bleeding, odynophagia, progressive dysphagia, unexplained vomiting, and a family history or prior history of gastrointestinal malignancy.3
Our patient’s symptoms raise the possibility of cancer, so that H pylori testing would not be the best test to order at this point.
Ambulatory wireless pH monitoring with a wireless pH capsule is useful for confirming GERD in those with persistent symptoms (whether typical or atypical) who do not have evidence of mucosal damage on initial endoscopy, particularly if a trial of acid suppression has failed.4–6
Barium swallow is an x-ray examination of the esophagus with contrast. It can show both the anatomy and the function of the esophagus, and it would be the initial diagnostic procedure of choice for patients with dysphagia who have no alarm symptoms.7 However, our patient does have alarm symptoms.
First highlight point
- Endoscopy is the first test in patients with dysphagia with alarm symptoms.
CASE CONTINUES: ENDOSCOPY
Multiple biopsies of the nodules show atypical lymphoid infiltrates with small cleaved lymphocytes that are mostly positive for CD5, CD20, and CD43 and negative for CD10 and CD23 by flow cytometry. In addition, a staining test for H pylori is positive.
Comment. Our patient has had GERD for the past 20 years, intermittently treated with a proton pump inhibitor. Acid suppressive therapy with a proton pump inhibitor is the standard of care of patients with erosive esophagitis. In standard doses, these drugs control symptoms in most cases and heal esophagitis in almost 90% of cases within 4 to 8 weeks.9 Proton pump inhibitors are also effective for maintaining healing of esophagitis and controlling symptoms in patients who respond to an acute course of therapy for a period of 6 to 12 months.10
WHAT IS THE DIAGNOSIS?
2. Which is the most likely diagnosis for our patient?
- Fundic gland polyps
- Gastric hyperplastic polyps
- Gastric adenomas
- Mucosa-associated lymphoid tissue (MALT) lymphoma
Fundic gland polyps are small (0.1–0.8 cm), hyperemic, sessile, flat, nodular lesions that have a smooth surface. They occur exclusively in the gastric corpus and are composed of normal gastric corpus-type epithelium arranged in a disorderly or microcystic configuration. 11 This pattern does not match our patient’s findings.
Gastric hyperplastic polyps are elongated, cystic, and distorted foveolar epithelium with marked regeneration. Other histologic findings are stromal inflammation, edema, and smooth muscle hyperplasia.12 This does not match our patient’s findings.
Adenomas can be flat or polypoid and range in size from a few millimeters to several centimeters. Endoscopically, adenomatous polyps have a velvety, lobulated appearance. Most are solitary (82% of cases), located in the antrum, and less than 2 cm in diameter.13 This does not match our patient’s findings.
MALT lymphoma, the correct answer, is characterized by small cleaved lymphocytes positive for CD4, CD20, and CD43. Although CD5 positivity is not characteristic, rare cases of MALT lymphoma may be CD5-positive and may be more aggressive.14
Other common features of MALT lymphoma are erosions, small nodules, thickening of gastric folds—generally suggesting a benign condition—or hyperemic or even normal gastric mucosa.15 Our patient’s complaint of food sticking in her chest and difficulty swallowing was most likely related to the erosive esophagitis found on endoscopy.
A TYPE OF NON-HODGKIN LYMPHOMA
Normal gastric mucosa contains no lymphoid tissue.16,17 Primary gastric lymphoma, of which MALT lymphoma is a subtype, accounts for around 5% of gastric malignancies, with an annual incidence rate of 0.5 per 100,000 people. 18–20 Although rare, it accounts for 60% to 70% of cases of non-Hodgkin lymphoma of the gastrointestinal tract and can involve the perigastric or abdominal lymph nodes or both.21–23 Although earlier studies suggested that its incidence was increasing, recent data indicate the incidence may be decreasing, thanks to active H pylori treatment.24–26
Two subtypes of primary gastric non-Hodgkin lymphoma commonly described are MALT lymphoma and diffuse large B-cell (DLBC) lymphoma. In the Revised European-American Lymphoma Classification, high-grade MALT lymphoma is comparable to DLBC lymphoma and may have transformed from low-grade MALT lymphoma.27,28 Another reported subtype, mantle cell lymphoma with MALT lymphoma features, should be considered in the differential diagnosis, although it is rare.29
MALT lymphoma is linked to H pylori
H pylori infection is a factor in the development of MALT lymphoma,30 as multiple lines of evidence show:
- H pylori infection has been reported in more than 90% of patients with MALT lymphoma.31–35
- H pylori antibodies have been found in stored serum drawn from patients who subsequently developed MALT lymphoma.35
- In response to H pylori antigens, T cells from MALT lymphoma proliferate and cause an increase in tumor immunoglobulin production.36
- In animals experimentally infected with H pylori, around one-third develop lymphoid follicles and lymphoepithelial lesions including B cells, which are similar to human MALT lymphoma.37
However, only a minority of patients with H pylori develop lymphoma, owing to a host immune response that is not well defined.
Second highlight point
- Gastric MALT lymphoma is associated with H pylori.
Associated genetic translocations
Three translocations, t(11;18), t(1;14), and t(14;18), are specifically associated with MALT lymphoma, and the genes involved have been characterized.
The t(11;18) translocation, seen in gastric and nongastric MALT lymphoma, is not seen in H pylori gastritis.38 This translocation is usually associated with extension of the disease outside the stomach (ie, to regional lymph nodes or distal sites).27 Most cases that do not respond to H pylori eradication involve the t(11;18) and t(1;14) translocations.28
Clinical presentation of gastric MALT lymphoma
The average age at presentation with gastric MALT lymphoma is 54 to 58 years.
The most common complaint is nonspecific abdominal pain in the epigastric region, sometimes accompanied by weight loss, nausea, vomiting, and, in a quarter of cases, acute or chronic bleeding.39–41 Weight loss is common, and its extent is associated with the location and the grade of the disease.
Most cases of MALT lymphoma are found serendipitously during endoscopy, on which the appearance of the lesions ranges from small ulcerations to polypoid masses with infiltrated, thickened folds involving predominantly the antrum or prepyloric region.15,41
MANAGING MALT LYMPHOMA
Our patient undergoes endoscopic ultrasonography, which reveals she has stage I disease, ie, it is limited to the stomach without involving the lymph nodes (stage II), adjacent organs (stage III), or distant organs (stage IV).
3. How will you treat this patient, given the present information?
- Chemotherapy
- Radiation therapy
- Surgery
- Antibiotics with a proton pump inhibitor
Antibiotics with a proton pump inhibitor would be best. According to the Maastricht III Consensus Report,42H pylori eradication is the treatment of first choice for H pylori infection in patients with stage I low-grade gastric MALT lymphoma. This therapy can induce complete histologic remission in 80% to 100% of patients with MALT lymphoma. 43 Several studies have shown regression44 or complete remission of low-grade gastric MALT lymphoma after eradication of H pylori with antibiotics, making it a reasonable initial treatment.45–49
Several regimens are used. The first choice in populations in which the prevalence of resistance to clarithromycin (Biaxin) is less than 15% to 20% is a proton pump inhibitor, clarithromycin, and either amoxicillin or metronidazole (Flagyl). (Metronidazole is preferable to amoxicillin if the prevalence of resistance to metronidazole is less than 40%.)
Sequential treatment—ie, 5 days of a proton pump inhibitor plus amoxicillin followed by 5 additional days of a proton pump inhibitor plus clarithromycin plus tinidazole (Tindamax)— may be better than a 7-day course of the combination of a proton pump inhibitor, amoxicillin, and clarithromycin.50,51
Treatment with a proton pump inhibitor, clarithromycin (500 mg twice a day), and either amoxicillin (1,000 mg twice a day) or metronidazole (400 or 500 mg twice a day) for 14 days is more effective than treatment for 7 days.52
H pylori reinfection in the general population is quite rare, with an estimated yearly rate as low as 2%.53 Recurrence of the infection is a risk factor for lymphoma relapse.17,54
Several predictors of the response of MALT lymphoma to eradication therapy have been recognized: H pylori positivity, stage I, lymphoma confined to the stomach; gastric wall invasion confined to mucosa and submucosa, and the absence of the t(11;18) translocation.
The time between H pylori eradication and complete remission of primary gastric lymphoma varies and can be longer than 12 months.55
Chemotherapy. In a single study,56 complete remission was achieved with oral cyclophosphamide (Cytoxan) in cases of antibiotic-refractory gastric MALT lymphoma. Comparable results were achieved after radiation therapy (see below); hence, oral monotherapy with cyclophosphamide might also be a suitable second-line therapy.57
The regimen of cyclophosphamide, hydroxydaunomycin, vincristine, and prednisone (CHOP) has been recommended for patients with stage III and IV disease.41,58
Rituximab (Rituxan) has been proven effective in treating t(11;18)-positive MALT lymphoma.59
Radiation therapy. Two studies have shown a 100% complete response rate after radiation therapy with a median dose of 30 Gy.57,60 Tsang et al61 reported complete remission in up to 90% of patients receiving radiation therapy alone, with excellent 5-year progression-free and overall survival rates of 98% and 77%, respectively.
Although surgery, radiotherapy, and chemotherapy have been used in cases in which eradication therapy failed and in more advanced stages of MALT lymphoma, there is no consensus about their use, so therapy must be individualized.
Fourth highlight point
- Antibiotic treatment for eradication of H pylori infection is the recommended treatment only for stage I low-grade MALT lymphoma.
FOLOW-UP
4. How should you follow patients with MALT lymphoma?
- Endoscopy
- H pylori testing
- Computed tomography and magnetic resonance imaging
- No surveillance required after treatment
Endoscopy is the correct answer. As initial diagnostic biopsies do not exclude aggressive lymphoma, careful endoscopic follow-up is recommended. A recommended schedule is a breath test for H pylori every 2 months in conjunction with repeat endoscopy with biopsies every 3 to 6 months for the first 2 years, and then annually.62
Although H pylori may be eradicated within 1 month of drug therapy, lymphoma may take several months to disappear histologically. In patients with stage I disease with residual lymphoma after H pylori eradication therapy, a simple wait-and-watch strategy is a suitable alternative to oncologic therapy.63,64
Local relapse may occur after many years of complete remission; thus, patients should be followed closely long-term with endoscopy and possibly endoscopic ultrasonography. 47–49,63
Patients who fail to attain a complete remission within 12 months should undergo radiation therapy, with or without chemotherapy. The same therapy should be started as soon as possible in patients with progressive disease after antibiotic therapy. Patients negative for H pylori, patients with stage II disease, and patients with t(11;18) translocation should receive antibiotic treatment with endoscopic surveillance every 3 months.
Fifth highlight point
- Surveillance endoscopy is recommended for follow-up of MALT lymphoma.
CASE CONTINUES: HER CONDITION IMPROVES, THEN WORSENS
Computed tomography of the chest, abdomen, and pelvis reveals no evidence of additional sites of tumor. Positron emission tomography reveals increased uptake in the left tonsillar region, for which she has undergoes an ear, nose, and throat evaluation, and no pathology is found.
Due to recurrence of her marginal zone Bcell lymphoma of MALT type of the stomach, the patient is referred to an oncology service. She is treated with radiation, receiving 15 sessions of 30 Gy localized to the stomach. Three months after radiation therapy, she undergoes endoscopy again, which shows no evidence of the previously described nodules. Repeat biopsies are negative for H pylori and MALT lymphoma.
Annual surveillance endoscopy and computed tomography for the past 3 years have been negative for any tumor recurrence.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808–1825.
- Pandolfino JE, Richter JE, Ours T, Guardino JM, Chapman J, Kahrilas PJ. Ambulatory esophageal pH monitoring using a wireless system. Am J Gastroenterol 2003; 98:740–749.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Furlow B. Barium swallow. Radiol Technol 2004; 76:49–58.
- Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999; 45:172–180.
- Havelund T, Laursen LS, Skoubo-Kristensen E, et al. Omeprazole and ranitidine in treatment of reflux oesophagitis: double blind comparative trial. Br Med J (Clin Res Ed) 1988; 296:89–92.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Odze RD, Marcial MA, Antonioli D. Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996; 27:896–903.
- Snover DC. Benign epithelial polyps of the stomach. Pathol Annu 1985; 20:303–329.
- Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009; 6:331–341.
- Wenzel C, Dieckmann K, Fiebiger W, Mannhalter C, Chott A, Raderer M. CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001; 42:823–829.
- Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98:975–986.
- Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med 1999; 131:88–95.
- Stolte M, Bayerdörffer E, Morgner A, et al. Helicobacter and gastric MALT lymphoma. Gut 2002; 50(suppl 3):III19–III24.
- Ducreux M, Boutron MC, Piard F, Carli PM, Faivre J. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: a population-based study. Br J Cancer 1998; 77:511–514.
- Gurney KA, Cartwright RA, Gilman EA. Descriptive epidemiology of gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer 1999; 79:1929–1934.
- d’Amore F, Brincker H, Grønbaek K, et al. Non-Hodgkin’s lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 1994; 12:1673–1684.
- Koch P, del Valle F, Berdel WE, et al; German Multicenter Study Group. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001; 19:3861–3873.
- Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma 2006; 47:2140–2146.
- Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992; 20:29–34.
- Wotherspoon AC. Choosing the right MALT. Gut 1996; 39:617–618.
- Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer 2003; 97:2462–2473.
- Luminari S, Cesaretti M, Marcheselli L, et al. Decreasing incidence of gastric MALT lymphomas in the era of anti-Helicobacter pylori interventions: results from a population-based study on extranodal marginal zone lymphomas. Ann Oncol 2009; epub ahead of print.
- Liu H, Ye H, Dogan A, et al. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001; 98:1182–1187.
- Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001; 357:39–40.
- Shibata K, Shimamoto Y, Nakano S, Miyahara M, Nakano H, Yamaguchi M. Mantle cell lymphoma with the features of mucosa-associated lymphoid tissue (MALT) lymphoma in an HTLV-I-seropositive patient. Ann Hematol 1995; 70:47–51.
- Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol 2005; 23:6370–6378.
- de Jong D, Boot H, van Heerde P, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology 1997; 112:1466–1474.
- Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338:1175–1176.
- Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol 1994; 47:436–439.
- Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet 1992; 339:834–835.
- Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994; 330:1267–1271.
- Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 342:571–574.
- Lee A, O’Rourke J, Enno A. Gastric mucosa-associated lymphoid tissue lymphoma: implications of animal models on pathogenic and therapeutic considerations—mouse models of gastric lymphoma. Recent Results Cancer Res 2000; 156:42–51.
- Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997; 8:979–985.
- Morgner A, Bayerdörffer E, Neubauer A, Stolte M. Malignant tumors of the stomach. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori. Gastroenterol Clin North Am 2000; 29:593–607.
- Ruskoné-Fourmestraux A, Aegerter P, Delmer A, Brousse N, Galian A, Rambaud JC. Primary digestive tract lymphoma: a prospective multicentric study of 91 patients. Groupe d’Etude des Lymphomes Digestifs. Gastroenterology 1993; 105:1662–1671.
- Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991; 101:1159–1170.
- Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 2007; 56:772–781.
- Boot H, de Jong D. Gastric lymphoma: the revolution of the past decade. Scand J Gastroenterol Suppl 2002; 236:27–36.
- Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993; 342:575–577.
- Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 1995; 345:1591–1594.
- Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med 1995; 122:767–769.
- Ruskoné-Fourmestraux A. Gastrointestinal lymphomas: the French experience of the Groupe d’Étude des Lymphomes Digestifs (GELD). Recent Results Cancer Res 2000; 156:99–103.
- Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005; 23:8018–8024.
- Wündisch T, Mösch C, Neubauer A, Stolte M. Helicobacter pylori eradication in gastric mucosa-associated lymphoid tissue lymphoma: results of a 196-patient series. Leuk Lymphoma 2006; 47:2110–2114.
- De Francesco V, Zullo A, Margiotta M, et al. Sequential treatment for Helicobacter pylori does not share the risk factors of triple therapy failure. Aliment Pharmacol Ther 2004; 19:407–414.
- Zullo A, Vaira D, Vakil N, et al. High eradication rates of Helicobacter pylori with a new sequential treatment. Aliment Pharmacol Ther 2003; 17:719–726.
- Paoluzi P, Iacopini F, Crispino P, et al. 2-week triple therapy for Helicobacter pylori infection is better than 1-week in clinical practice: a large prospective single-center randomized study. Helicobacter 2006; 11:562–568.
- Gisbert JP, Olivares D, Jimenez I, Pajares JM. Long-term follow-up of 13C-urea breath test results after Helicobacter pylori eradication: frequency and significance of borderline delta13CO2 values. Aliment Pharmacol Ther 2006; 23:275–280.
- Bayerdörffer E, Morgner A. Gastric marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type: management of the disease. Dig Liver Dis 2000; 32:192–194.
- Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: true relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res 2000; 156:116–124.
- Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer 2005; 104:532–540.
- Schechter NR, Portlock CS, Yahalom J. Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone. J Clin Oncol 1998; 16:1916–1921.
- Solidoro A, Payet C, Sanchez-Lihon J, Montalbetti JA. Gastric lymphomas: chemotherapy as a primary treatment. Semin Surg Oncol 1990; 6:218–225.
- Lévy M, Copie-Bergman C, Molinier-Frenkel V, et al. Treatment of t(11;18)-positive gastric mucosa-associated lymphoid tissue lymphoma with rituximab and chlorambucil: clinical, histological, and molecular follow-up. Leuk Lymphoma 2010; 51:284–290.
- Yahalom J. MALT lymphomas: a radiation oncology viewpoint. Ann Hematol 2001; 80(suppl 3):B100–B105.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosaassociated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 2003; 21:4157–4164.
- Hung PD, Schubert ML, Mihas AA. Marginal zone B-cell lymphoma (MALT lymphoma). Curr Treat Options Gastroenterol 2004; 7:133–138.
- Zucca E, Cavalli F. Are antibiotics the treatment of choice for gastric lymphoma? Curr Hematol Rep 2004; 3:11–16.
- Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, et al; EGI LS (European Gastro-Intestinal Lymphoma Study) Group. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007; 56:1685–1687.
A 61-year-old woman presents to her primary care physician because for the last 4 weeks she has had difficulty swallowing solid food and a feeling of food “getting stuck in the chest.” She also reports having nausea, mild epigastric pain, and heartburn. She denies having fevers, chills, night sweats, weight loss, vomiting, diarrhea, hematochezia, or melena.
Medical history
For the past 20 years, she has had gastroesophageal reflux disease (GERD), intermittently treated with a proton pump inhibitor. She also has arthritis, hyperlipidemia, hypertension, and asthma, and she has undergone right hip replacement for a hip fracture. She has no known allergies.
She lives in the Midwest region of the United States and is on disability due to her arthritis. She is divorced and has three children.
She quit smoking 3 years ago after smoking half a pack per day for 30 years. She drinks socially; she has never used recreational drugs.
She recalls that an uncle had cancer, but she does not know the specific type.
Physical examination
The patient’s temperature is 96.7°F (35.9°C), heart rate 86 per minute, blood pressure 150/92 mm Hg, respiratory rate 16 per minute, and oxygen saturation 100% on room air.
Differential diagnosis
Although the differential diagnosis at this stage is broad, a few conditions that commonly present like this are:
- Esophageal cancer
- Esophageal stricture
- Esophageal webs
- Esophagitis (infectious, inflammatory)
- Peptic ulcer disease.
WHICH TEST SHOULD BE ORDERED?
1. Which test will you order now for this patient?
- Endoscopy (esophagogastroduodenoscopy)
- Serum Helicobacter pylori antibody testing
- Wireless pH monitoring
- Barium swallow
Endoscopy would be the best test to order. Esophageal cancer and esophageal stricture must be ruled out, in view of her long history of GERD, gastritis, and smoking and her alarming symptoms of difficulty swallowing and food sticking. In this situation, endoscopy is the first test recommended. In addition to its diagnostic value, it offers an opportunity to obtain tissue samples and to perform a therapeutic intervention, if necessary.1,2
H pyloriantibody testing is used in the “test-and-treat approach” for H pylori infection, an established management strategy for patients who have uninvestigated dyspepsia and who are younger than 55 years and have no “alarm features,” ie, red flags for cancer. The alarm features commonly described are anemia, early satiety, unexplained weight loss, bleeding, odynophagia, progressive dysphagia, unexplained vomiting, and a family history or prior history of gastrointestinal malignancy.3
Our patient’s symptoms raise the possibility of cancer, so that H pylori testing would not be the best test to order at this point.
Ambulatory wireless pH monitoring with a wireless pH capsule is useful for confirming GERD in those with persistent symptoms (whether typical or atypical) who do not have evidence of mucosal damage on initial endoscopy, particularly if a trial of acid suppression has failed.4–6
Barium swallow is an x-ray examination of the esophagus with contrast. It can show both the anatomy and the function of the esophagus, and it would be the initial diagnostic procedure of choice for patients with dysphagia who have no alarm symptoms.7 However, our patient does have alarm symptoms.
First highlight point
- Endoscopy is the first test in patients with dysphagia with alarm symptoms.
CASE CONTINUES: ENDOSCOPY
Multiple biopsies of the nodules show atypical lymphoid infiltrates with small cleaved lymphocytes that are mostly positive for CD5, CD20, and CD43 and negative for CD10 and CD23 by flow cytometry. In addition, a staining test for H pylori is positive.
Comment. Our patient has had GERD for the past 20 years, intermittently treated with a proton pump inhibitor. Acid suppressive therapy with a proton pump inhibitor is the standard of care of patients with erosive esophagitis. In standard doses, these drugs control symptoms in most cases and heal esophagitis in almost 90% of cases within 4 to 8 weeks.9 Proton pump inhibitors are also effective for maintaining healing of esophagitis and controlling symptoms in patients who respond to an acute course of therapy for a period of 6 to 12 months.10
WHAT IS THE DIAGNOSIS?
2. Which is the most likely diagnosis for our patient?
- Fundic gland polyps
- Gastric hyperplastic polyps
- Gastric adenomas
- Mucosa-associated lymphoid tissue (MALT) lymphoma
Fundic gland polyps are small (0.1–0.8 cm), hyperemic, sessile, flat, nodular lesions that have a smooth surface. They occur exclusively in the gastric corpus and are composed of normal gastric corpus-type epithelium arranged in a disorderly or microcystic configuration. 11 This pattern does not match our patient’s findings.
Gastric hyperplastic polyps are elongated, cystic, and distorted foveolar epithelium with marked regeneration. Other histologic findings are stromal inflammation, edema, and smooth muscle hyperplasia.12 This does not match our patient’s findings.
Adenomas can be flat or polypoid and range in size from a few millimeters to several centimeters. Endoscopically, adenomatous polyps have a velvety, lobulated appearance. Most are solitary (82% of cases), located in the antrum, and less than 2 cm in diameter.13 This does not match our patient’s findings.
MALT lymphoma, the correct answer, is characterized by small cleaved lymphocytes positive for CD4, CD20, and CD43. Although CD5 positivity is not characteristic, rare cases of MALT lymphoma may be CD5-positive and may be more aggressive.14
Other common features of MALT lymphoma are erosions, small nodules, thickening of gastric folds—generally suggesting a benign condition—or hyperemic or even normal gastric mucosa.15 Our patient’s complaint of food sticking in her chest and difficulty swallowing was most likely related to the erosive esophagitis found on endoscopy.
A TYPE OF NON-HODGKIN LYMPHOMA
Normal gastric mucosa contains no lymphoid tissue.16,17 Primary gastric lymphoma, of which MALT lymphoma is a subtype, accounts for around 5% of gastric malignancies, with an annual incidence rate of 0.5 per 100,000 people. 18–20 Although rare, it accounts for 60% to 70% of cases of non-Hodgkin lymphoma of the gastrointestinal tract and can involve the perigastric or abdominal lymph nodes or both.21–23 Although earlier studies suggested that its incidence was increasing, recent data indicate the incidence may be decreasing, thanks to active H pylori treatment.24–26
Two subtypes of primary gastric non-Hodgkin lymphoma commonly described are MALT lymphoma and diffuse large B-cell (DLBC) lymphoma. In the Revised European-American Lymphoma Classification, high-grade MALT lymphoma is comparable to DLBC lymphoma and may have transformed from low-grade MALT lymphoma.27,28 Another reported subtype, mantle cell lymphoma with MALT lymphoma features, should be considered in the differential diagnosis, although it is rare.29
MALT lymphoma is linked to H pylori
H pylori infection is a factor in the development of MALT lymphoma,30 as multiple lines of evidence show:
- H pylori infection has been reported in more than 90% of patients with MALT lymphoma.31–35
- H pylori antibodies have been found in stored serum drawn from patients who subsequently developed MALT lymphoma.35
- In response to H pylori antigens, T cells from MALT lymphoma proliferate and cause an increase in tumor immunoglobulin production.36
- In animals experimentally infected with H pylori, around one-third develop lymphoid follicles and lymphoepithelial lesions including B cells, which are similar to human MALT lymphoma.37
However, only a minority of patients with H pylori develop lymphoma, owing to a host immune response that is not well defined.
Second highlight point
- Gastric MALT lymphoma is associated with H pylori.
Associated genetic translocations
Three translocations, t(11;18), t(1;14), and t(14;18), are specifically associated with MALT lymphoma, and the genes involved have been characterized.
The t(11;18) translocation, seen in gastric and nongastric MALT lymphoma, is not seen in H pylori gastritis.38 This translocation is usually associated with extension of the disease outside the stomach (ie, to regional lymph nodes or distal sites).27 Most cases that do not respond to H pylori eradication involve the t(11;18) and t(1;14) translocations.28
Clinical presentation of gastric MALT lymphoma
The average age at presentation with gastric MALT lymphoma is 54 to 58 years.
The most common complaint is nonspecific abdominal pain in the epigastric region, sometimes accompanied by weight loss, nausea, vomiting, and, in a quarter of cases, acute or chronic bleeding.39–41 Weight loss is common, and its extent is associated with the location and the grade of the disease.
Most cases of MALT lymphoma are found serendipitously during endoscopy, on which the appearance of the lesions ranges from small ulcerations to polypoid masses with infiltrated, thickened folds involving predominantly the antrum or prepyloric region.15,41
MANAGING MALT LYMPHOMA
Our patient undergoes endoscopic ultrasonography, which reveals she has stage I disease, ie, it is limited to the stomach without involving the lymph nodes (stage II), adjacent organs (stage III), or distant organs (stage IV).
3. How will you treat this patient, given the present information?
- Chemotherapy
- Radiation therapy
- Surgery
- Antibiotics with a proton pump inhibitor
Antibiotics with a proton pump inhibitor would be best. According to the Maastricht III Consensus Report,42H pylori eradication is the treatment of first choice for H pylori infection in patients with stage I low-grade gastric MALT lymphoma. This therapy can induce complete histologic remission in 80% to 100% of patients with MALT lymphoma. 43 Several studies have shown regression44 or complete remission of low-grade gastric MALT lymphoma after eradication of H pylori with antibiotics, making it a reasonable initial treatment.45–49
Several regimens are used. The first choice in populations in which the prevalence of resistance to clarithromycin (Biaxin) is less than 15% to 20% is a proton pump inhibitor, clarithromycin, and either amoxicillin or metronidazole (Flagyl). (Metronidazole is preferable to amoxicillin if the prevalence of resistance to metronidazole is less than 40%.)
Sequential treatment—ie, 5 days of a proton pump inhibitor plus amoxicillin followed by 5 additional days of a proton pump inhibitor plus clarithromycin plus tinidazole (Tindamax)— may be better than a 7-day course of the combination of a proton pump inhibitor, amoxicillin, and clarithromycin.50,51
Treatment with a proton pump inhibitor, clarithromycin (500 mg twice a day), and either amoxicillin (1,000 mg twice a day) or metronidazole (400 or 500 mg twice a day) for 14 days is more effective than treatment for 7 days.52
H pylori reinfection in the general population is quite rare, with an estimated yearly rate as low as 2%.53 Recurrence of the infection is a risk factor for lymphoma relapse.17,54
Several predictors of the response of MALT lymphoma to eradication therapy have been recognized: H pylori positivity, stage I, lymphoma confined to the stomach; gastric wall invasion confined to mucosa and submucosa, and the absence of the t(11;18) translocation.
The time between H pylori eradication and complete remission of primary gastric lymphoma varies and can be longer than 12 months.55
Chemotherapy. In a single study,56 complete remission was achieved with oral cyclophosphamide (Cytoxan) in cases of antibiotic-refractory gastric MALT lymphoma. Comparable results were achieved after radiation therapy (see below); hence, oral monotherapy with cyclophosphamide might also be a suitable second-line therapy.57
The regimen of cyclophosphamide, hydroxydaunomycin, vincristine, and prednisone (CHOP) has been recommended for patients with stage III and IV disease.41,58
Rituximab (Rituxan) has been proven effective in treating t(11;18)-positive MALT lymphoma.59
Radiation therapy. Two studies have shown a 100% complete response rate after radiation therapy with a median dose of 30 Gy.57,60 Tsang et al61 reported complete remission in up to 90% of patients receiving radiation therapy alone, with excellent 5-year progression-free and overall survival rates of 98% and 77%, respectively.
Although surgery, radiotherapy, and chemotherapy have been used in cases in which eradication therapy failed and in more advanced stages of MALT lymphoma, there is no consensus about their use, so therapy must be individualized.
Fourth highlight point
- Antibiotic treatment for eradication of H pylori infection is the recommended treatment only for stage I low-grade MALT lymphoma.
FOLOW-UP
4. How should you follow patients with MALT lymphoma?
- Endoscopy
- H pylori testing
- Computed tomography and magnetic resonance imaging
- No surveillance required after treatment
Endoscopy is the correct answer. As initial diagnostic biopsies do not exclude aggressive lymphoma, careful endoscopic follow-up is recommended. A recommended schedule is a breath test for H pylori every 2 months in conjunction with repeat endoscopy with biopsies every 3 to 6 months for the first 2 years, and then annually.62
Although H pylori may be eradicated within 1 month of drug therapy, lymphoma may take several months to disappear histologically. In patients with stage I disease with residual lymphoma after H pylori eradication therapy, a simple wait-and-watch strategy is a suitable alternative to oncologic therapy.63,64
Local relapse may occur after many years of complete remission; thus, patients should be followed closely long-term with endoscopy and possibly endoscopic ultrasonography. 47–49,63
Patients who fail to attain a complete remission within 12 months should undergo radiation therapy, with or without chemotherapy. The same therapy should be started as soon as possible in patients with progressive disease after antibiotic therapy. Patients negative for H pylori, patients with stage II disease, and patients with t(11;18) translocation should receive antibiotic treatment with endoscopic surveillance every 3 months.
Fifth highlight point
- Surveillance endoscopy is recommended for follow-up of MALT lymphoma.
CASE CONTINUES: HER CONDITION IMPROVES, THEN WORSENS
Computed tomography of the chest, abdomen, and pelvis reveals no evidence of additional sites of tumor. Positron emission tomography reveals increased uptake in the left tonsillar region, for which she has undergoes an ear, nose, and throat evaluation, and no pathology is found.
Due to recurrence of her marginal zone Bcell lymphoma of MALT type of the stomach, the patient is referred to an oncology service. She is treated with radiation, receiving 15 sessions of 30 Gy localized to the stomach. Three months after radiation therapy, she undergoes endoscopy again, which shows no evidence of the previously described nodules. Repeat biopsies are negative for H pylori and MALT lymphoma.
Annual surveillance endoscopy and computed tomography for the past 3 years have been negative for any tumor recurrence.
A 61-year-old woman presents to her primary care physician because for the last 4 weeks she has had difficulty swallowing solid food and a feeling of food “getting stuck in the chest.” She also reports having nausea, mild epigastric pain, and heartburn. She denies having fevers, chills, night sweats, weight loss, vomiting, diarrhea, hematochezia, or melena.
Medical history
For the past 20 years, she has had gastroesophageal reflux disease (GERD), intermittently treated with a proton pump inhibitor. She also has arthritis, hyperlipidemia, hypertension, and asthma, and she has undergone right hip replacement for a hip fracture. She has no known allergies.
She lives in the Midwest region of the United States and is on disability due to her arthritis. She is divorced and has three children.
She quit smoking 3 years ago after smoking half a pack per day for 30 years. She drinks socially; she has never used recreational drugs.
She recalls that an uncle had cancer, but she does not know the specific type.
Physical examination
The patient’s temperature is 96.7°F (35.9°C), heart rate 86 per minute, blood pressure 150/92 mm Hg, respiratory rate 16 per minute, and oxygen saturation 100% on room air.
Differential diagnosis
Although the differential diagnosis at this stage is broad, a few conditions that commonly present like this are:
- Esophageal cancer
- Esophageal stricture
- Esophageal webs
- Esophagitis (infectious, inflammatory)
- Peptic ulcer disease.
WHICH TEST SHOULD BE ORDERED?
1. Which test will you order now for this patient?
- Endoscopy (esophagogastroduodenoscopy)
- Serum Helicobacter pylori antibody testing
- Wireless pH monitoring
- Barium swallow
Endoscopy would be the best test to order. Esophageal cancer and esophageal stricture must be ruled out, in view of her long history of GERD, gastritis, and smoking and her alarming symptoms of difficulty swallowing and food sticking. In this situation, endoscopy is the first test recommended. In addition to its diagnostic value, it offers an opportunity to obtain tissue samples and to perform a therapeutic intervention, if necessary.1,2
H pyloriantibody testing is used in the “test-and-treat approach” for H pylori infection, an established management strategy for patients who have uninvestigated dyspepsia and who are younger than 55 years and have no “alarm features,” ie, red flags for cancer. The alarm features commonly described are anemia, early satiety, unexplained weight loss, bleeding, odynophagia, progressive dysphagia, unexplained vomiting, and a family history or prior history of gastrointestinal malignancy.3
Our patient’s symptoms raise the possibility of cancer, so that H pylori testing would not be the best test to order at this point.
Ambulatory wireless pH monitoring with a wireless pH capsule is useful for confirming GERD in those with persistent symptoms (whether typical or atypical) who do not have evidence of mucosal damage on initial endoscopy, particularly if a trial of acid suppression has failed.4–6
Barium swallow is an x-ray examination of the esophagus with contrast. It can show both the anatomy and the function of the esophagus, and it would be the initial diagnostic procedure of choice for patients with dysphagia who have no alarm symptoms.7 However, our patient does have alarm symptoms.
First highlight point
- Endoscopy is the first test in patients with dysphagia with alarm symptoms.
CASE CONTINUES: ENDOSCOPY
Multiple biopsies of the nodules show atypical lymphoid infiltrates with small cleaved lymphocytes that are mostly positive for CD5, CD20, and CD43 and negative for CD10 and CD23 by flow cytometry. In addition, a staining test for H pylori is positive.
Comment. Our patient has had GERD for the past 20 years, intermittently treated with a proton pump inhibitor. Acid suppressive therapy with a proton pump inhibitor is the standard of care of patients with erosive esophagitis. In standard doses, these drugs control symptoms in most cases and heal esophagitis in almost 90% of cases within 4 to 8 weeks.9 Proton pump inhibitors are also effective for maintaining healing of esophagitis and controlling symptoms in patients who respond to an acute course of therapy for a period of 6 to 12 months.10
WHAT IS THE DIAGNOSIS?
2. Which is the most likely diagnosis for our patient?
- Fundic gland polyps
- Gastric hyperplastic polyps
- Gastric adenomas
- Mucosa-associated lymphoid tissue (MALT) lymphoma
Fundic gland polyps are small (0.1–0.8 cm), hyperemic, sessile, flat, nodular lesions that have a smooth surface. They occur exclusively in the gastric corpus and are composed of normal gastric corpus-type epithelium arranged in a disorderly or microcystic configuration. 11 This pattern does not match our patient’s findings.
Gastric hyperplastic polyps are elongated, cystic, and distorted foveolar epithelium with marked regeneration. Other histologic findings are stromal inflammation, edema, and smooth muscle hyperplasia.12 This does not match our patient’s findings.
Adenomas can be flat or polypoid and range in size from a few millimeters to several centimeters. Endoscopically, adenomatous polyps have a velvety, lobulated appearance. Most are solitary (82% of cases), located in the antrum, and less than 2 cm in diameter.13 This does not match our patient’s findings.
MALT lymphoma, the correct answer, is characterized by small cleaved lymphocytes positive for CD4, CD20, and CD43. Although CD5 positivity is not characteristic, rare cases of MALT lymphoma may be CD5-positive and may be more aggressive.14
Other common features of MALT lymphoma are erosions, small nodules, thickening of gastric folds—generally suggesting a benign condition—or hyperemic or even normal gastric mucosa.15 Our patient’s complaint of food sticking in her chest and difficulty swallowing was most likely related to the erosive esophagitis found on endoscopy.
A TYPE OF NON-HODGKIN LYMPHOMA
Normal gastric mucosa contains no lymphoid tissue.16,17 Primary gastric lymphoma, of which MALT lymphoma is a subtype, accounts for around 5% of gastric malignancies, with an annual incidence rate of 0.5 per 100,000 people. 18–20 Although rare, it accounts for 60% to 70% of cases of non-Hodgkin lymphoma of the gastrointestinal tract and can involve the perigastric or abdominal lymph nodes or both.21–23 Although earlier studies suggested that its incidence was increasing, recent data indicate the incidence may be decreasing, thanks to active H pylori treatment.24–26
Two subtypes of primary gastric non-Hodgkin lymphoma commonly described are MALT lymphoma and diffuse large B-cell (DLBC) lymphoma. In the Revised European-American Lymphoma Classification, high-grade MALT lymphoma is comparable to DLBC lymphoma and may have transformed from low-grade MALT lymphoma.27,28 Another reported subtype, mantle cell lymphoma with MALT lymphoma features, should be considered in the differential diagnosis, although it is rare.29
MALT lymphoma is linked to H pylori
H pylori infection is a factor in the development of MALT lymphoma,30 as multiple lines of evidence show:
- H pylori infection has been reported in more than 90% of patients with MALT lymphoma.31–35
- H pylori antibodies have been found in stored serum drawn from patients who subsequently developed MALT lymphoma.35
- In response to H pylori antigens, T cells from MALT lymphoma proliferate and cause an increase in tumor immunoglobulin production.36
- In animals experimentally infected with H pylori, around one-third develop lymphoid follicles and lymphoepithelial lesions including B cells, which are similar to human MALT lymphoma.37
However, only a minority of patients with H pylori develop lymphoma, owing to a host immune response that is not well defined.
Second highlight point
- Gastric MALT lymphoma is associated with H pylori.
Associated genetic translocations
Three translocations, t(11;18), t(1;14), and t(14;18), are specifically associated with MALT lymphoma, and the genes involved have been characterized.
The t(11;18) translocation, seen in gastric and nongastric MALT lymphoma, is not seen in H pylori gastritis.38 This translocation is usually associated with extension of the disease outside the stomach (ie, to regional lymph nodes or distal sites).27 Most cases that do not respond to H pylori eradication involve the t(11;18) and t(1;14) translocations.28
Clinical presentation of gastric MALT lymphoma
The average age at presentation with gastric MALT lymphoma is 54 to 58 years.
The most common complaint is nonspecific abdominal pain in the epigastric region, sometimes accompanied by weight loss, nausea, vomiting, and, in a quarter of cases, acute or chronic bleeding.39–41 Weight loss is common, and its extent is associated with the location and the grade of the disease.
Most cases of MALT lymphoma are found serendipitously during endoscopy, on which the appearance of the lesions ranges from small ulcerations to polypoid masses with infiltrated, thickened folds involving predominantly the antrum or prepyloric region.15,41
MANAGING MALT LYMPHOMA
Our patient undergoes endoscopic ultrasonography, which reveals she has stage I disease, ie, it is limited to the stomach without involving the lymph nodes (stage II), adjacent organs (stage III), or distant organs (stage IV).
3. How will you treat this patient, given the present information?
- Chemotherapy
- Radiation therapy
- Surgery
- Antibiotics with a proton pump inhibitor
Antibiotics with a proton pump inhibitor would be best. According to the Maastricht III Consensus Report,42H pylori eradication is the treatment of first choice for H pylori infection in patients with stage I low-grade gastric MALT lymphoma. This therapy can induce complete histologic remission in 80% to 100% of patients with MALT lymphoma. 43 Several studies have shown regression44 or complete remission of low-grade gastric MALT lymphoma after eradication of H pylori with antibiotics, making it a reasonable initial treatment.45–49
Several regimens are used. The first choice in populations in which the prevalence of resistance to clarithromycin (Biaxin) is less than 15% to 20% is a proton pump inhibitor, clarithromycin, and either amoxicillin or metronidazole (Flagyl). (Metronidazole is preferable to amoxicillin if the prevalence of resistance to metronidazole is less than 40%.)
Sequential treatment—ie, 5 days of a proton pump inhibitor plus amoxicillin followed by 5 additional days of a proton pump inhibitor plus clarithromycin plus tinidazole (Tindamax)— may be better than a 7-day course of the combination of a proton pump inhibitor, amoxicillin, and clarithromycin.50,51
Treatment with a proton pump inhibitor, clarithromycin (500 mg twice a day), and either amoxicillin (1,000 mg twice a day) or metronidazole (400 or 500 mg twice a day) for 14 days is more effective than treatment for 7 days.52
H pylori reinfection in the general population is quite rare, with an estimated yearly rate as low as 2%.53 Recurrence of the infection is a risk factor for lymphoma relapse.17,54
Several predictors of the response of MALT lymphoma to eradication therapy have been recognized: H pylori positivity, stage I, lymphoma confined to the stomach; gastric wall invasion confined to mucosa and submucosa, and the absence of the t(11;18) translocation.
The time between H pylori eradication and complete remission of primary gastric lymphoma varies and can be longer than 12 months.55
Chemotherapy. In a single study,56 complete remission was achieved with oral cyclophosphamide (Cytoxan) in cases of antibiotic-refractory gastric MALT lymphoma. Comparable results were achieved after radiation therapy (see below); hence, oral monotherapy with cyclophosphamide might also be a suitable second-line therapy.57
The regimen of cyclophosphamide, hydroxydaunomycin, vincristine, and prednisone (CHOP) has been recommended for patients with stage III and IV disease.41,58
Rituximab (Rituxan) has been proven effective in treating t(11;18)-positive MALT lymphoma.59
Radiation therapy. Two studies have shown a 100% complete response rate after radiation therapy with a median dose of 30 Gy.57,60 Tsang et al61 reported complete remission in up to 90% of patients receiving radiation therapy alone, with excellent 5-year progression-free and overall survival rates of 98% and 77%, respectively.
Although surgery, radiotherapy, and chemotherapy have been used in cases in which eradication therapy failed and in more advanced stages of MALT lymphoma, there is no consensus about their use, so therapy must be individualized.
Fourth highlight point
- Antibiotic treatment for eradication of H pylori infection is the recommended treatment only for stage I low-grade MALT lymphoma.
FOLOW-UP
4. How should you follow patients with MALT lymphoma?
- Endoscopy
- H pylori testing
- Computed tomography and magnetic resonance imaging
- No surveillance required after treatment
Endoscopy is the correct answer. As initial diagnostic biopsies do not exclude aggressive lymphoma, careful endoscopic follow-up is recommended. A recommended schedule is a breath test for H pylori every 2 months in conjunction with repeat endoscopy with biopsies every 3 to 6 months for the first 2 years, and then annually.62
Although H pylori may be eradicated within 1 month of drug therapy, lymphoma may take several months to disappear histologically. In patients with stage I disease with residual lymphoma after H pylori eradication therapy, a simple wait-and-watch strategy is a suitable alternative to oncologic therapy.63,64
Local relapse may occur after many years of complete remission; thus, patients should be followed closely long-term with endoscopy and possibly endoscopic ultrasonography. 47–49,63
Patients who fail to attain a complete remission within 12 months should undergo radiation therapy, with or without chemotherapy. The same therapy should be started as soon as possible in patients with progressive disease after antibiotic therapy. Patients negative for H pylori, patients with stage II disease, and patients with t(11;18) translocation should receive antibiotic treatment with endoscopic surveillance every 3 months.
Fifth highlight point
- Surveillance endoscopy is recommended for follow-up of MALT lymphoma.
CASE CONTINUES: HER CONDITION IMPROVES, THEN WORSENS
Computed tomography of the chest, abdomen, and pelvis reveals no evidence of additional sites of tumor. Positron emission tomography reveals increased uptake in the left tonsillar region, for which she has undergoes an ear, nose, and throat evaluation, and no pathology is found.
Due to recurrence of her marginal zone Bcell lymphoma of MALT type of the stomach, the patient is referred to an oncology service. She is treated with radiation, receiving 15 sessions of 30 Gy localized to the stomach. Three months after radiation therapy, she undergoes endoscopy again, which shows no evidence of the previously described nodules. Repeat biopsies are negative for H pylori and MALT lymphoma.
Annual surveillance endoscopy and computed tomography for the past 3 years have been negative for any tumor recurrence.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808–1825.
- Pandolfino JE, Richter JE, Ours T, Guardino JM, Chapman J, Kahrilas PJ. Ambulatory esophageal pH monitoring using a wireless system. Am J Gastroenterol 2003; 98:740–749.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Furlow B. Barium swallow. Radiol Technol 2004; 76:49–58.
- Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999; 45:172–180.
- Havelund T, Laursen LS, Skoubo-Kristensen E, et al. Omeprazole and ranitidine in treatment of reflux oesophagitis: double blind comparative trial. Br Med J (Clin Res Ed) 1988; 296:89–92.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Odze RD, Marcial MA, Antonioli D. Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996; 27:896–903.
- Snover DC. Benign epithelial polyps of the stomach. Pathol Annu 1985; 20:303–329.
- Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009; 6:331–341.
- Wenzel C, Dieckmann K, Fiebiger W, Mannhalter C, Chott A, Raderer M. CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001; 42:823–829.
- Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98:975–986.
- Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med 1999; 131:88–95.
- Stolte M, Bayerdörffer E, Morgner A, et al. Helicobacter and gastric MALT lymphoma. Gut 2002; 50(suppl 3):III19–III24.
- Ducreux M, Boutron MC, Piard F, Carli PM, Faivre J. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: a population-based study. Br J Cancer 1998; 77:511–514.
- Gurney KA, Cartwright RA, Gilman EA. Descriptive epidemiology of gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer 1999; 79:1929–1934.
- d’Amore F, Brincker H, Grønbaek K, et al. Non-Hodgkin’s lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 1994; 12:1673–1684.
- Koch P, del Valle F, Berdel WE, et al; German Multicenter Study Group. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001; 19:3861–3873.
- Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma 2006; 47:2140–2146.
- Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992; 20:29–34.
- Wotherspoon AC. Choosing the right MALT. Gut 1996; 39:617–618.
- Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer 2003; 97:2462–2473.
- Luminari S, Cesaretti M, Marcheselli L, et al. Decreasing incidence of gastric MALT lymphomas in the era of anti-Helicobacter pylori interventions: results from a population-based study on extranodal marginal zone lymphomas. Ann Oncol 2009; epub ahead of print.
- Liu H, Ye H, Dogan A, et al. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001; 98:1182–1187.
- Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001; 357:39–40.
- Shibata K, Shimamoto Y, Nakano S, Miyahara M, Nakano H, Yamaguchi M. Mantle cell lymphoma with the features of mucosa-associated lymphoid tissue (MALT) lymphoma in an HTLV-I-seropositive patient. Ann Hematol 1995; 70:47–51.
- Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol 2005; 23:6370–6378.
- de Jong D, Boot H, van Heerde P, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology 1997; 112:1466–1474.
- Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338:1175–1176.
- Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol 1994; 47:436–439.
- Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet 1992; 339:834–835.
- Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994; 330:1267–1271.
- Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 342:571–574.
- Lee A, O’Rourke J, Enno A. Gastric mucosa-associated lymphoid tissue lymphoma: implications of animal models on pathogenic and therapeutic considerations—mouse models of gastric lymphoma. Recent Results Cancer Res 2000; 156:42–51.
- Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997; 8:979–985.
- Morgner A, Bayerdörffer E, Neubauer A, Stolte M. Malignant tumors of the stomach. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori. Gastroenterol Clin North Am 2000; 29:593–607.
- Ruskoné-Fourmestraux A, Aegerter P, Delmer A, Brousse N, Galian A, Rambaud JC. Primary digestive tract lymphoma: a prospective multicentric study of 91 patients. Groupe d’Etude des Lymphomes Digestifs. Gastroenterology 1993; 105:1662–1671.
- Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991; 101:1159–1170.
- Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 2007; 56:772–781.
- Boot H, de Jong D. Gastric lymphoma: the revolution of the past decade. Scand J Gastroenterol Suppl 2002; 236:27–36.
- Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993; 342:575–577.
- Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 1995; 345:1591–1594.
- Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med 1995; 122:767–769.
- Ruskoné-Fourmestraux A. Gastrointestinal lymphomas: the French experience of the Groupe d’Étude des Lymphomes Digestifs (GELD). Recent Results Cancer Res 2000; 156:99–103.
- Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005; 23:8018–8024.
- Wündisch T, Mösch C, Neubauer A, Stolte M. Helicobacter pylori eradication in gastric mucosa-associated lymphoid tissue lymphoma: results of a 196-patient series. Leuk Lymphoma 2006; 47:2110–2114.
- De Francesco V, Zullo A, Margiotta M, et al. Sequential treatment for Helicobacter pylori does not share the risk factors of triple therapy failure. Aliment Pharmacol Ther 2004; 19:407–414.
- Zullo A, Vaira D, Vakil N, et al. High eradication rates of Helicobacter pylori with a new sequential treatment. Aliment Pharmacol Ther 2003; 17:719–726.
- Paoluzi P, Iacopini F, Crispino P, et al. 2-week triple therapy for Helicobacter pylori infection is better than 1-week in clinical practice: a large prospective single-center randomized study. Helicobacter 2006; 11:562–568.
- Gisbert JP, Olivares D, Jimenez I, Pajares JM. Long-term follow-up of 13C-urea breath test results after Helicobacter pylori eradication: frequency and significance of borderline delta13CO2 values. Aliment Pharmacol Ther 2006; 23:275–280.
- Bayerdörffer E, Morgner A. Gastric marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type: management of the disease. Dig Liver Dis 2000; 32:192–194.
- Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: true relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res 2000; 156:116–124.
- Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer 2005; 104:532–540.
- Schechter NR, Portlock CS, Yahalom J. Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone. J Clin Oncol 1998; 16:1916–1921.
- Solidoro A, Payet C, Sanchez-Lihon J, Montalbetti JA. Gastric lymphomas: chemotherapy as a primary treatment. Semin Surg Oncol 1990; 6:218–225.
- Lévy M, Copie-Bergman C, Molinier-Frenkel V, et al. Treatment of t(11;18)-positive gastric mucosa-associated lymphoid tissue lymphoma with rituximab and chlorambucil: clinical, histological, and molecular follow-up. Leuk Lymphoma 2010; 51:284–290.
- Yahalom J. MALT lymphomas: a radiation oncology viewpoint. Ann Hematol 2001; 80(suppl 3):B100–B105.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosaassociated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 2003; 21:4157–4164.
- Hung PD, Schubert ML, Mihas AA. Marginal zone B-cell lymphoma (MALT lymphoma). Curr Treat Options Gastroenterol 2004; 7:133–138.
- Zucca E, Cavalli F. Are antibiotics the treatment of choice for gastric lymphoma? Curr Hematol Rep 2004; 3:11–16.
- Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, et al; EGI LS (European Gastro-Intestinal Lymphoma Study) Group. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007; 56:1685–1687.
- Esfandyari T, Potter JW, Vaezi MF. Dysphagia: a cost analysis of the diagnostic approach. Am J Gastroenterol 2002; 97:2733–2737.
- Varadarajulu S, Eloubeidi MA, Patel RS, et al. The yield and the predictors of esophageal pathology when upper endoscopy is used for the initial evaluation of dysphagia. Gastrointest Endosc 2005; 61:804–808.
- Chey WD, Wong BC; Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808–1825.
- Pandolfino JE, Richter JE, Ours T, Guardino JM, Chapman J, Kahrilas PJ. Ambulatory esophageal pH monitoring using a wireless system. Am J Gastroenterol 2003; 98:740–749.
- DeVault KR, Castell DO; American College of Gastroenterology. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. Am J Gastroenterol 2005; 100:190–200.
- Vakil N, van Zanten SV, Kahrilas P, Dent J, Jones R; Global Consensus Group. The Montreal definition and classification of gastroesophageal reflux disease: a global evidence-based consensus. Am J Gastroenterol 2006; 101:1900–1920.
- Furlow B. Barium swallow. Radiol Technol 2004; 76:49–58.
- Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oesophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999; 45:172–180.
- Havelund T, Laursen LS, Skoubo-Kristensen E, et al. Omeprazole and ranitidine in treatment of reflux oesophagitis: double blind comparative trial. Br Med J (Clin Res Ed) 1988; 296:89–92.
- Kahrilas PJ, Shaheen NJ, Vaezi MF; American Gastroenterological Association Institute. American Gastroenterological Association Institute technical review on the management of gastroesophageal reflux disease. Gastroenterology 2008; 135:1392–1413.
- Odze RD, Marcial MA, Antonioli D. Gastric fundic gland polyps: a morphological study including mucin histochemistry, stereometry, and MIB-1 immunohistochemistry. Hum Pathol 1996; 27:896–903.
- Snover DC. Benign epithelial polyps of the stomach. Pathol Annu 1985; 20:303–329.
- Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009; 6:331–341.
- Wenzel C, Dieckmann K, Fiebiger W, Mannhalter C, Chott A, Raderer M. CD5 expression in a lymphoma of the mucosa-associated lymphoid tissue (MALT)-type as a marker for early dissemination and aggressive clinical behaviour. Leuk Lymphoma 2001; 42:823–829.
- Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98:975–986.
- Steinbach G, Ford R, Glober G, et al. Antibiotic treatment of gastric lymphoma of mucosa-associated lymphoid tissue. An uncontrolled trial. Ann Intern Med 1999; 131:88–95.
- Stolte M, Bayerdörffer E, Morgner A, et al. Helicobacter and gastric MALT lymphoma. Gut 2002; 50(suppl 3):III19–III24.
- Ducreux M, Boutron MC, Piard F, Carli PM, Faivre J. A 15-year series of gastrointestinal non-Hodgkin’s lymphomas: a population-based study. Br J Cancer 1998; 77:511–514.
- Gurney KA, Cartwright RA, Gilman EA. Descriptive epidemiology of gastrointestinal non-Hodgkin’s lymphoma in a population-based registry. Br J Cancer 1999; 79:1929–1934.
- d’Amore F, Brincker H, Grønbaek K, et al. Non-Hodgkin’s lymphoma of the gastrointestinal tract: a population-based analysis of incidence, geographic distribution, clinicopathologic presentation features, and prognosis. Danish Lymphoma Study Group. J Clin Oncol 1994; 12:1673–1684.
- Koch P, del Valle F, Berdel WE, et al; German Multicenter Study Group. Primary gastrointestinal non-Hodgkin’s lymphoma: I. Anatomic and histologic distribution, clinical features, and survival data of 371 patients registered in the German Multicenter Study GIT NHL 01/92. J Clin Oncol 2001; 19:3861–3873.
- Papaxoinis G, Papageorgiou S, Rontogianni D, et al. Primary gastrointestinal non-Hodgkin’s lymphoma: a clinicopathologic study of 128 cases in Greece. A Hellenic Cooperative Oncology Group study (HeCOG). Leuk Lymphoma 2006; 47:2140–2146.
- Wotherspoon AC, Doglioni C, Isaacson PG. Low-grade gastric B-cell lymphoma of mucosa-associated lymphoid tissue (MALT): a multifocal disease. Histopathology 1992; 20:29–34.
- Wotherspoon AC. Choosing the right MALT. Gut 1996; 39:617–618.
- Nakamura S, Matsumoto T, Iida M, Yao T, Tsuneyoshi M. Primary gastrointestinal lymphoma in Japan: a clinicopathologic analysis of 455 patients with special reference to its time trends. Cancer 2003; 97:2462–2473.
- Luminari S, Cesaretti M, Marcheselli L, et al. Decreasing incidence of gastric MALT lymphomas in the era of anti-Helicobacter pylori interventions: results from a population-based study on extranodal marginal zone lymphomas. Ann Oncol 2009; epub ahead of print.
- Liu H, Ye H, Dogan A, et al. T(11;18)(q21;q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood 2001; 98:1182–1187.
- Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, et al. Resistance of t(11;18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet 2001; 357:39–40.
- Shibata K, Shimamoto Y, Nakano S, Miyahara M, Nakano H, Yamaguchi M. Mantle cell lymphoma with the features of mucosa-associated lymphoid tissue (MALT) lymphoma in an HTLV-I-seropositive patient. Ann Hematol 1995; 70:47–51.
- Farinha P, Gascoyne RD. Molecular pathogenesis of mucosa-associated lymphoid tissue lymphoma. J Clin Oncol 2005; 23:6370–6378.
- de Jong D, Boot H, van Heerde P, Hart GA, Taal BG. Histological grading in gastric lymphoma: pretreatment criteria and clinical relevance. Gastroenterology 1997; 112:1466–1474.
- Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338:1175–1176.
- Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol 1994; 47:436–439.
- Doglioni C, Wotherspoon AC, Moschini A, de Boni M, Isaacson PG. High incidence of primary gastric lymphoma in northeastern Italy. Lancet 1992; 339:834–835.
- Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994; 330:1267–1271.
- Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993; 342:571–574.
- Lee A, O’Rourke J, Enno A. Gastric mucosa-associated lymphoid tissue lymphoma: implications of animal models on pathogenic and therapeutic considerations—mouse models of gastric lymphoma. Recent Results Cancer Res 2000; 156:42–51.
- Auer IA, Gascoyne RD, Connors JM, et al. t(11;18)(q21;q21) is the most common translocation in MALT lymphomas. Ann Oncol 1997; 8:979–985.
- Morgner A, Bayerdörffer E, Neubauer A, Stolte M. Malignant tumors of the stomach. Gastric mucosa-associated lymphoid tissue lymphoma and Helicobacter pylori. Gastroenterol Clin North Am 2000; 29:593–607.
- Ruskoné-Fourmestraux A, Aegerter P, Delmer A, Brousse N, Galian A, Rambaud JC. Primary digestive tract lymphoma: a prospective multicentric study of 91 patients. Groupe d’Etude des Lymphomes Digestifs. Gastroenterology 1993; 105:1662–1671.
- Cogliatti SB, Schmid U, Schumacher U, et al. Primary B-cell gastric lymphoma: a clinicopathological study of 145 patients. Gastroenterology 1991; 101:1159–1170.
- Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 2007; 56:772–781.
- Boot H, de Jong D. Gastric lymphoma: the revolution of the past decade. Scand J Gastroenterol Suppl 2002; 236:27–36.
- Wotherspoon AC, Doglioni C, Diss TC, et al. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet. 1993; 342:575–577.
- Bayerdörffer E, Neubauer A, Rudolph B, et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. MALT Lymphoma Study Group. Lancet 1995; 345:1591–1594.
- Roggero E, Zucca E, Pinotti G, et al. Eradication of Helicobacter pylori infection in primary low-grade gastric lymphoma of mucosa-associated lymphoid tissue. Ann Intern Med 1995; 122:767–769.
- Ruskoné-Fourmestraux A. Gastrointestinal lymphomas: the French experience of the Groupe d’Étude des Lymphomes Digestifs (GELD). Recent Results Cancer Res 2000; 156:99–103.
- Wündisch T, Thiede C, Morgner A, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol 2005; 23:8018–8024.
- Wündisch T, Mösch C, Neubauer A, Stolte M. Helicobacter pylori eradication in gastric mucosa-associated lymphoid tissue lymphoma: results of a 196-patient series. Leuk Lymphoma 2006; 47:2110–2114.
- De Francesco V, Zullo A, Margiotta M, et al. Sequential treatment for Helicobacter pylori does not share the risk factors of triple therapy failure. Aliment Pharmacol Ther 2004; 19:407–414.
- Zullo A, Vaira D, Vakil N, et al. High eradication rates of Helicobacter pylori with a new sequential treatment. Aliment Pharmacol Ther 2003; 17:719–726.
- Paoluzi P, Iacopini F, Crispino P, et al. 2-week triple therapy for Helicobacter pylori infection is better than 1-week in clinical practice: a large prospective single-center randomized study. Helicobacter 2006; 11:562–568.
- Gisbert JP, Olivares D, Jimenez I, Pajares JM. Long-term follow-up of 13C-urea breath test results after Helicobacter pylori eradication: frequency and significance of borderline delta13CO2 values. Aliment Pharmacol Ther 2006; 23:275–280.
- Bayerdörffer E, Morgner A. Gastric marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue type: management of the disease. Dig Liver Dis 2000; 32:192–194.
- Savio A, Zamboni G, Capelli P, et al. Relapse of low-grade gastric MALT lymphoma after Helicobacter pylori eradication: true relapse or persistence? Long-term post-treatment follow-up of a multicenter trial in the north-east of Italy and evaluation of the diagnostic protocol’s adequacy. Recent Results Cancer Res 2000; 156:116–124.
- Nakamura S, Matsumoto T, Suekane H, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer 2005; 104:532–540.
- Schechter NR, Portlock CS, Yahalom J. Treatment of mucosa-associated lymphoid tissue lymphoma of the stomach with radiation alone. J Clin Oncol 1998; 16:1916–1921.
- Solidoro A, Payet C, Sanchez-Lihon J, Montalbetti JA. Gastric lymphomas: chemotherapy as a primary treatment. Semin Surg Oncol 1990; 6:218–225.
- Lévy M, Copie-Bergman C, Molinier-Frenkel V, et al. Treatment of t(11;18)-positive gastric mucosa-associated lymphoid tissue lymphoma with rituximab and chlorambucil: clinical, histological, and molecular follow-up. Leuk Lymphoma 2010; 51:284–290.
- Yahalom J. MALT lymphomas: a radiation oncology viewpoint. Ann Hematol 2001; 80(suppl 3):B100–B105.
- Tsang RW, Gospodarowicz MK, Pintilie M, et al. Localized mucosaassociated lymphoid tissue lymphoma treated with radiation therapy has excellent clinical outcome. J Clin Oncol 2003; 21:4157–4164.
- Hung PD, Schubert ML, Mihas AA. Marginal zone B-cell lymphoma (MALT lymphoma). Curr Treat Options Gastroenterol 2004; 7:133–138.
- Zucca E, Cavalli F. Are antibiotics the treatment of choice for gastric lymphoma? Curr Hematol Rep 2004; 3:11–16.
- Fischbach W, Goebeler ME, Ruskone-Fourmestraux A, et al; EGI LS (European Gastro-Intestinal Lymphoma Study) Group. Most patients with minimal histological residuals of gastric MALT lymphoma after successful eradication of Helicobacter pylori can be managed safely by a watch and wait strategy: experience from a large international series. Gut 2007; 56:1685–1687.
Does vitamin D deficiency play a role in the pathogenesis of chronic heart failure? Do supplements improve survival?
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
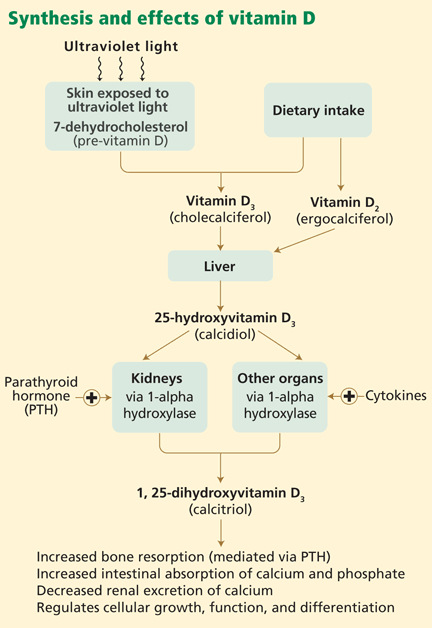
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE

The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
Vitamin D deficiency may play a role in the pathogenesis of chronic heart failure, but whether giving patients supplements to raise their vitamin D levels into the normal range improves their survival is not clear.
ASSOCIATION BETWEEN VITAMIN D DEFICIENCY AND OTHER DISORDERS
In the mid-17th century, Whistler and Glisson independently described rickets as a severe bone-deforming disease characterized by growth retardation, bending of the spine, deformities of the legs, and weak and toneless muscles. Histologically, rickets is characterized by impaired mineralization of the cartilage in the epiphyseal growth plates in children. In 1919, Sir Edward Mellanby identified vitamin D deficiency as the cause.
Osteomalacia, another disease caused by vitamin D deficiency, is a disorder of mineralization of newly formed bone matrix in adults. Vitamin D, therefore, has well-known roles in maintaining bone health and calcium and phosphorus homeostasis.
In addition, vitamin D deficiency has been shown in recent years to be associated with myocardial dysfunction.1,2
VITAMIN D METABOLISM IS COMPLEX
In skin exposed to ultraviolet B light, the provitamin 7-dehydrocholesterol is converted to vitamin D3 (cholecalciferol). Vitamin D3 is also obtained from dietary sources. However, many scientists consider vitamin D more a hormone than a classic vitamin, as adequate exposure to sunlight may negate the need for dietary supplements.
The active form of vitamin D is synthesized by hydroxylation in the liver and kidney. In the liver, hepatic enzymes add a hydroxyl (OH) group to vitamin D3, changing it to 25-hydroxyvitamin D3. In the kidney, 25-hydroxyvitamin D3 receives another hydroxyl group, converting it to the biologically active metabolite 1,25-dihydroxyvitamin D3 (calcitriol). This renal hydroxylation is via 1-alpha-hydroxylase activity and is directly under control of parathyroid hormone (PTH), and indirectly under control of the serum concentrations of calcium.
Interestingly, a number of different organ cells, including cardiomyocytes, also express 1-alpha-hydroxylase and therefore also convert 25-hydroxyvitamin D3 to 1,25-dihydroxyvitamin D3. Unlike the renal hydroxylation, this extrarenal process depends on cytokine activation and on serum levels of 25-hydroxyvitamin D3.3 Low levels of 25-hydroxyvitamin D3 lead to alterations in cellular control over growth, differentiation, and function.
The active form of vitamin D is transported protein-bound in the blood to various target organs, where it is delivered in free form to cells. Specific nuclear receptor proteins are found in many organs not classically considered target organs for vitamin D, including the skin, brain, skeletal muscles, cardiomyocytes, vascular endothelial cells, circulating monocytes, and activated B and T lymphocytes. Vitamin D plays a significant role in the autocrine and paracrine regulation of cellular function, growth, and differentiation in various organs.3
MOST HEART FAILURE PATIENTS HAVE LOW VITAMIN D LEVELS
More than 40% of men and 50% of women in the United States have low vitamin D levels (< 30 ng/mL [75 nmol/L])—and low levels in adults are associated with both coronary artery disease and heart failure.4 Most patients with heart failure have low levels.5,6 Therefore, screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
Low vitamin D levels carry a poor prognosis. Pilz et al5 measured baseline 25-hydroxyvitamin D3 levels in 3,299 patients referred for elective coronary angiography and followed them prospectively for a median of 7.7 years. Even after adjustment for cardiac risk factors, patients who had low 25-hydroxyvitamin D3 levels were more likely to die of heart failure or sudden cardiac death than patients with normal levels.
Boxer et al7 found an association between low 25-hydroxyvitamin D3 levels and low exercise capacity and frailty in patients with systolic heart failure.
LOW VITAMIN D CONTRIBUTES TO THE PATHOGENESIS OF HEART FAILURE
In recent years, ideas about the pathophysiology of heart failure have expanded from a purely hemodynamic view to a more complex concept involving inflammatory cytokines and neurohormonal overactivation.8
Animal studies first showed vitamin D to inhibit the renin-angiotensin-aldosterone system, activation of which contributes to the salt and water retention seen in heart failure.4,9
In addition, vitamin D has a number of effects that should help prevent hypertension, an important risk factor for heart failure. It protects the kidney by suppressing the reninangiotensin-aldosterone system, prevents secondary hyperparathyroidism and its effects on vascular stiffness, prevents insulin resistance, and suppresses inflammation, which protects vascular endothelial cells.10
The first studies to show a connection between cardiovascular homeostasis and vitamin D status were in animal models more than 20 years ago. These studies showed that 1,25-dihydroxyvitamin D3 acts directly on cardiomyocyte vitamin D receptors, which are widely distributed throughout the body in several tissue types.11
Excess PTH levels associated with low vitamin D levels may play a role in cardiovascular disease by leading to cardiomyocyte hypertrophy and interstitial fibrosis of the heart.12 Animal studies have found that vitamin D suppresses cardiac hypertrophy.13 Vitamin D also plays a role in cardiomyocyte relaxation and may abrogate the hypercontractility associated with diastolic heart failure.2,14
Currently, it is unclear whether vitamin D deficiency is a causative risk factor for heart failure or simply a reflection of the poor functional status of patients with heart failure that leads to decreased exposure to sunlight. This debate will continue until further randomized clinical trials address this association.
VITAMIN D AND HEART TRANSPLANTATION
One would expect that patients with endstage organ failure would be at high risk of vitamin D deficiency because of limited sunlight exposure. However, few studies have evaluated the role of this vitamin in heart transplant recipients.
Stein and colleagues15 measured serum 25-hydroxyvitamin D3 immediately after transplantation in 46 heart and 23 liver transplant recipients. Levels were low in both types of transplant recipients, but liver transplant recipients had significantly lower levels than heart transplant patients. This could be explained by malabsorption and impaired synthesis of 25-hydroxyvitamin D3 in end-stage liver disease.
Also, an important point is that osteoporosis is prevalent in postcardiac transplant patients and likely related to the immunosuppressive agents these patients must take.16 In theory, increasing the body’s stores of vitamin D during the pretransplant period could lower the rate of bone loss and osteoporosis after cardiac transplantation.
Further investigation is needed to determine whether restoring adequate levels of vitamin D at the time of or after transplantation prevents graft rejection or improves survival.
VITAMIN D SUPPLEMENTATION AND SURVIVAL IN HEART FAILURE
The best laboratory test to assess vitamin D levels is the serum 25-hydroxyvitamin D3 concentration. A level between 20 and 30 ng/mL (50–75 nmol/L) is considered insufficient, and a level below 20 ng/mL (50 nmol/L) represents vitamin D deficiency.4,5,11
Vitamin D insufficiency is typically treated with 800 to 1,000 IU of vitamin D3 daily, whereas deficiency requires 50,000 IU of vitamin D3 weekly for 6 to 8 weeks, followed by 800 to 1,000 IU daily.19 The goal of therapy is to increase the serum 25-hydroxyvitamin D3 level above 30 ng/mL.19
Currently, it is unknown if vitamin D supplementation improves survival in heart failure. We recommend testing for vitamin D deficiency in all patients with heart failure and treating them as described above. For heart failure patients that are not deficient, daily intake of 800 to 1,000 IU of vitamin D is reasonable. Our review underscores the need for more studies to evaluate the efficacy of vitamin D replacement in improving survival in patients with heart failure.
KEY POINTS
- Screening for vitamin D deficiency in patients with heart failure is appropriate and encouraged.
- Vitamin D deficiency is common in patients with heart failure and in heart transplant recipients.
- In theory, achieving adequate levels of vitamin D would have a beneficial effect on patients with heart failure.
- Randomized controlled trials are needed to determine if vitamin D supplementation confers a survival benefit in patients with heart failure who have deficient vitamin D levels.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
- Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol 2007; 103:533–537.
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 2008; 149:558–564.
- Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest 2005; 35:290–304.
- Kim DH, Sabour S, Sagar UN, Adams S, Whellan DJ. Prevalence of hypovitaminosis D in cardiovascular diseases (from the National Health and Nutrition Examination Survey 2001 to 2004). Am J Cardiol 2008; 102:1540–1544.
- Pilz S, März W, Wellnitz B, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 2008; 93:3927–3935.
- Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev 2006; 11:25–33.
- Boxer RS, Dauser DA, Walsh SJ, Hager WD, Kenny AM. The association between vitamin D and inflammation with the 6-minute walk and frailty in patients with heart failure. J Am Geriatr Soc 2008; 56:454–461.
- Schleithoff SS, Zittermann A, Tenderich G, Berthold HK, Stehle P, Koerfer R. Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 2006; 83:754–759.
- Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 2002; 110:229–238.
- Pilz S, Tomaschitz A, Ritz E, Pieber TR; Medscape. Vitamin D status and arterial hypertension: a systematic review. Nat Rev Cardiol 2009; 6:621–630.
- Nemerovski CW, Dorsch MP, Simpson RU, Bone HG, Aaronson KD, Bleske BE. Vitamin D and cardiovascular disease. Pharmacotherapy 2009; 29:691–708.
- Rostand SG, Drüeke TB. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney Int 1999; 56:383–392.
- Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest 1996; 97:1577–1588.
- Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV. Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 2006; 41:350–359.
- Stein EM, Cohen A, Freeby M, et al. Severe vitamin D deficiency among heart and liver transplant recipients. Clin Transplant 2009; (Epub ahead of print)
- Shane E, Rivas M, McMahon DJ, et al. Bone loss and turnover after cardiac transplantation. J Clin Endocrinol Metab 1997; 82:1497–1506.
- Norman AW, Bouillon R, Whiting SJ, Vieth R, Lips P. 13th Workshop consensus for vitamin D nutritional guidelines. J Steroid Biochem Mol Biol 2007; 103:204–205.
- Vieth R, Bischoff-Ferrari H, Boucher BJ, et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr 2007; 85:649–650.
- Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporos Int 2005; 16:713–716.
Perioperative Medicine Summit 2010
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Venous thromboembolism after total hip and knee replacement in older adults with single and co-occurring comorbidities
Alok Kapoor, MD, MSc; A. Labonte; M. Winter; J.B. Segal; R.A. Silliman; J.N. Katz; E. Losina; and D.R. Berlowitz
Abstract 2: Are there consequences of discontinuing angiotensin system inhibitors preoperatively in ambulatory and same-day admission patients?
Vasudha Goel, MBBS; David Rahmani, BS; Roy Braid, BS; Dmitry Rozin, BS; and Rebecca Twersky, MD, MPH
Abstract 3: Residents’ knowledge of ACC/AHA guidelines for preoperative cardiac evaluation is limited
BobbieJean Sweitzer, MD; Michael Vigoda, MD, MBA; Nikola Milokjic; Ben Boedeker, DVM, MD, PhD, MBA; Kip D. Robinson, MD, FACP; Michael A. Pilla, MD; Robert Gaiser, MD; Angela F. Edwards, MD; Ronald P. Olson, MD; Matthew D. Caldwell, MD; Shawn T. Beaman, MD; Jeffrey A. Green, MD; Jesse M. Ehrenfeld, MD, MPH; Marsha L. Wakefield, MD; Praveen Kalra, MD; David M. Feinstein, MD; Deborah C. Richman, MBChB, FFA(SA); Gail Van Norman; Gary E. Loyd, MD, MMM; Paul W. Kranner, MD; Stevin Dubin, MD; Sunil Eappen, MD; Sergio D. Bergese, MD; Suzanne Karan, MD; James R. Rowbottom, MD, FCCP; and Keith Candiotti, MD
Abstract 4: Descriptive perioperative BNP and CRP in vascular surgery patients
Thomas Barrett, MD, MCR, and Rebecca Duby, BS
Abstract 5: Selective serotonin reuptake inhibitors and risk of intraoperative bleeding
Adriana Oprea, MD, and Paula Zimbrean, MD
Abstract 6: Incidence and nature of postoperative complications in patients with obstructive sleep apnea undergoing noncardiac surgery
Roop Kaw, MD; Vinay Pasupuleti, MBBS, PhD; Esteban Walker, PhD; Anuradha Ramaswamy, MD; Thadeo Catacutan, MD; and Nancy Foldvary, DO
Abstract 7: HMG-CoA reductase inhibitor therapy and the risk of venous thromboembolism in joint replacement surgery
William Ho, MBBS; Brendan Flaim, MBBS, FRACP; and Andrea Chan, MBBS, FRACP
Abstract 8: Risk prediction models for cardiac morbidity and mortality in noncardiac surgery: A systematic review of the literature
Ramani Moonesinghe, MBBS, MRCP, FRCA; Kathy Rowan, PhD; Judith Hulf, CBE, FRCA; Michael G. Mythen, MD, FRCA; and Michael P.W. Grocott, MD, FRCA
Abstract 9: Economic aspects of preoperative testing
Gerhard Fritsch, MD; Maria Flamm, MD; Josef Seer, MD; and Andreas Soennichsen, MD
Abstract 10: Postoperative myocardial infarction and in-hospital mortality predictors in patients undergoing rlective noncardiac surgery
Anitha Rajamanickam, MD; Ali Usmani, MD; Jelica Janicijevic, MD; Preethi Patel, MD; Eric Hixson; Omeed Zardkoohi, MD; Michael Pecic; Changhong Yu; Michael Kattan, PhD; Sagar Kalahasti, MD; and Mina K. Chung, MD
Abstract 11: Incidence and predictors of postoperative heart failure in patients undergoing elective noncardiac surgery
Anitha Rajamanickam, MD; Ali Usmani, MD; Jelica Janicijevic, MD; Preethi Patel, MD; Eric Hixson; Omeed Zardkoohi, MD; Michael Pecic; Changhong Yu; Michael Kattan, PhD; Sagar Kalahasti, MD; and Mina K. Chung, MD
Abstract 12: Predictors of length of stay in patients undergoing total knee replacement surgery
Vishal Sehgal, MD; Pardeep Bansal, MD; Praveen Reddy, MD; Vishal Sharma, MD; Rajendra Palepu, MD; Linda Thomas, MD; and Jeremiah Eagan, MD
Abstract 13: Analysis of surgeon utilization of the Preoperative Assessment Communication Education (PACE) center in the pediatric population
Lisa Price Stevens, MD, and Ezinne Akamiro, BA, MD/MHA
Abstract 14: Use of the BATHE method to increase satisfaction amongst patients undergoing cardiac and major vascular operations
Samuel DeMaria, MD; Anthony P. DeMaria, MA; Menachem Weiner, MD; and George Silvay, MD
Abstract 15: Indication for surgery predicts long-term but not in-hospital mortality in patients undergoing lower extremity bypass vascular surgery
Brigid C. Flynn, MD; Michael Mazzeffi , MD; Carol Bodian, PhD; and Vivek Moitra, MD
Abstract 16: Research and outcomes on analgesia and nociception during surgery
Jinu Kim, MD; Tehila Adams, MD; Deepak Sreedharan, MD; Shanti Raju, MD; and Henry Bennett, PhD
Abstract 17: A snapshot survey of fluid prescribing
Helen Grote, MD; Luke Evans, MRCS; Abdel Omer, MD, PhD, FRCS; and Rob Lewis, MD, FRCA
Abstract 18: Predictors of difficult intubation with the video laryngoscope
Dario Galante, MD
Abstract 19: Use of technology to improve operational efficiency
Lucy Duffy, RN, MA, and Rita Lanaras, RN, BS, CNOR
Abstract 20: The ASA physical status score for the nonanesthesiologist
Adriana Oprea, MD, and David Silverman, MD
Abstract 21: Development of a shared multidisciplinary electronic preanesthetic record
Meghan Tadel, MD; R. Boyer, DO, MS; N. Smith; and P. Kallas, MD
Abstract 22: Development of a patient selection protocol prior to robotic radical prostatectomy (RRP) in the Preoperative Assessment Unit (PAU)
James Dyer, MD
Abstract 23: Protocol-driven preoperative testing in the Preoperative Assessment Unit (PAU): Which patients should receive a resting transthoracic echocardiogram (TTE) prior to elective noncardiac surgery?
James Dyer, MD
Abstract 24: High-risk preoperative assessment for elective orthopedic surgery patients
Terrence Adam, MD, PhD; Connie Parenti, MD; Terence Gioe, MD; Karen Ringsred, MD; and Joseph Wels, MD
Abstract 25: A novel use of web-based software to efficiently triage presurgical patients based on perioperative risk: A pilot
Alicia Kalamas, MD
Abstract 26: Value of a specialized clinic for day admission surgery for cardiac and major vascular operations
George Silvay, MD, PhD; Samuel DeMaria, MD; Marietta dePerio, NP, CCRN; Ellen Hughes, MA, RN; Samantha Silvay; Marina Krol, PhD; Brigid C. Flynn, MD; and David L. Reich, MD
Abstract 27: Preoperative evaluation for parathyroidectomy—rule out pheochromocytoma
Rubin Bahuva, MD; Sudhir Manda, MD; and Saurabh Kandpal, MD
Abstract 28: Should we stop the oral selective estrogen receptor modulator raloxifene prior to surgery?
Vesselin Dimov, MD; Tarek Hamieh, MD; and Ajay Kumar, MD
Abstract 29: Should mesalamine be stopped prior to noncardiac surgery to avoid bleeding complications?
Vesselin Dimov, MD; Tarek Hamieh, MD; and Ajay Kumar, MD
Abstract 30: Thyroidectomy: Perioperative management of acute thyroid storm
Stephen VanHaerents, MD, and Aashish A. Shah, MD
Abstract 31: Core competencies: Not just for the ACGME—but for successful and ethical perioperative management of a young respiratory cripple
Deborah Richman, MBChB, FFA(SA); Misako P. Sakamaki, MD; and Slawomir P. Oleszak, MD
Abstract 32: ‘If I have to be transfused I only want my wwn blood, or blood from family members’—what is best-practice advice to be given in the preoperative clinic?
Deborah Richman, MBChB, FFA(SA), and Joseph L. Conrad, MD
Abstract 33: Prolonged QTc and hypokalemia: A bad combination before surgery
Chadi Alraies, MD, and Abdul Hamid Alraiyes, MD
Abstract 34: Perioperative management of a parturient with neuromyelitis optica
Neeti Sadana, MD; Michael Orosco, MD; Michaela Farber, MD; and Scott Segal, MD
Abstract 35: ‘High’-pertension
Anuradha Ramaswamy, MD, and Franklin A. Michota, Jr., MD
Abstract 36: Perioperative care in neuromuscular scoliosis
Saurabh Basu Kandpal, MD, and Priya Baronia, MD
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Venous thromboembolism after total hip and knee replacement in older adults with single and co-occurring comorbidities
Alok Kapoor, MD, MSc; A. Labonte; M. Winter; J.B. Segal; R.A. Silliman; J.N. Katz; E. Losina; and D.R. Berlowitz
Abstract 2: Are there consequences of discontinuing angiotensin system inhibitors preoperatively in ambulatory and same-day admission patients?
Vasudha Goel, MBBS; David Rahmani, BS; Roy Braid, BS; Dmitry Rozin, BS; and Rebecca Twersky, MD, MPH
Abstract 3: Residents’ knowledge of ACC/AHA guidelines for preoperative cardiac evaluation is limited
BobbieJean Sweitzer, MD; Michael Vigoda, MD, MBA; Nikola Milokjic; Ben Boedeker, DVM, MD, PhD, MBA; Kip D. Robinson, MD, FACP; Michael A. Pilla, MD; Robert Gaiser, MD; Angela F. Edwards, MD; Ronald P. Olson, MD; Matthew D. Caldwell, MD; Shawn T. Beaman, MD; Jeffrey A. Green, MD; Jesse M. Ehrenfeld, MD, MPH; Marsha L. Wakefield, MD; Praveen Kalra, MD; David M. Feinstein, MD; Deborah C. Richman, MBChB, FFA(SA); Gail Van Norman; Gary E. Loyd, MD, MMM; Paul W. Kranner, MD; Stevin Dubin, MD; Sunil Eappen, MD; Sergio D. Bergese, MD; Suzanne Karan, MD; James R. Rowbottom, MD, FCCP; and Keith Candiotti, MD
Abstract 4: Descriptive perioperative BNP and CRP in vascular surgery patients
Thomas Barrett, MD, MCR, and Rebecca Duby, BS
Abstract 5: Selective serotonin reuptake inhibitors and risk of intraoperative bleeding
Adriana Oprea, MD, and Paula Zimbrean, MD
Abstract 6: Incidence and nature of postoperative complications in patients with obstructive sleep apnea undergoing noncardiac surgery
Roop Kaw, MD; Vinay Pasupuleti, MBBS, PhD; Esteban Walker, PhD; Anuradha Ramaswamy, MD; Thadeo Catacutan, MD; and Nancy Foldvary, DO
Abstract 7: HMG-CoA reductase inhibitor therapy and the risk of venous thromboembolism in joint replacement surgery
William Ho, MBBS; Brendan Flaim, MBBS, FRACP; and Andrea Chan, MBBS, FRACP
Abstract 8: Risk prediction models for cardiac morbidity and mortality in noncardiac surgery: A systematic review of the literature
Ramani Moonesinghe, MBBS, MRCP, FRCA; Kathy Rowan, PhD; Judith Hulf, CBE, FRCA; Michael G. Mythen, MD, FRCA; and Michael P.W. Grocott, MD, FRCA
Abstract 9: Economic aspects of preoperative testing
Gerhard Fritsch, MD; Maria Flamm, MD; Josef Seer, MD; and Andreas Soennichsen, MD
Abstract 10: Postoperative myocardial infarction and in-hospital mortality predictors in patients undergoing rlective noncardiac surgery
Anitha Rajamanickam, MD; Ali Usmani, MD; Jelica Janicijevic, MD; Preethi Patel, MD; Eric Hixson; Omeed Zardkoohi, MD; Michael Pecic; Changhong Yu; Michael Kattan, PhD; Sagar Kalahasti, MD; and Mina K. Chung, MD
Abstract 11: Incidence and predictors of postoperative heart failure in patients undergoing elective noncardiac surgery
Anitha Rajamanickam, MD; Ali Usmani, MD; Jelica Janicijevic, MD; Preethi Patel, MD; Eric Hixson; Omeed Zardkoohi, MD; Michael Pecic; Changhong Yu; Michael Kattan, PhD; Sagar Kalahasti, MD; and Mina K. Chung, MD
Abstract 12: Predictors of length of stay in patients undergoing total knee replacement surgery
Vishal Sehgal, MD; Pardeep Bansal, MD; Praveen Reddy, MD; Vishal Sharma, MD; Rajendra Palepu, MD; Linda Thomas, MD; and Jeremiah Eagan, MD
Abstract 13: Analysis of surgeon utilization of the Preoperative Assessment Communication Education (PACE) center in the pediatric population
Lisa Price Stevens, MD, and Ezinne Akamiro, BA, MD/MHA
Abstract 14: Use of the BATHE method to increase satisfaction amongst patients undergoing cardiac and major vascular operations
Samuel DeMaria, MD; Anthony P. DeMaria, MA; Menachem Weiner, MD; and George Silvay, MD
Abstract 15: Indication for surgery predicts long-term but not in-hospital mortality in patients undergoing lower extremity bypass vascular surgery
Brigid C. Flynn, MD; Michael Mazzeffi , MD; Carol Bodian, PhD; and Vivek Moitra, MD
Abstract 16: Research and outcomes on analgesia and nociception during surgery
Jinu Kim, MD; Tehila Adams, MD; Deepak Sreedharan, MD; Shanti Raju, MD; and Henry Bennett, PhD
Abstract 17: A snapshot survey of fluid prescribing
Helen Grote, MD; Luke Evans, MRCS; Abdel Omer, MD, PhD, FRCS; and Rob Lewis, MD, FRCA
Abstract 18: Predictors of difficult intubation with the video laryngoscope
Dario Galante, MD
Abstract 19: Use of technology to improve operational efficiency
Lucy Duffy, RN, MA, and Rita Lanaras, RN, BS, CNOR
Abstract 20: The ASA physical status score for the nonanesthesiologist
Adriana Oprea, MD, and David Silverman, MD
Abstract 21: Development of a shared multidisciplinary electronic preanesthetic record
Meghan Tadel, MD; R. Boyer, DO, MS; N. Smith; and P. Kallas, MD
Abstract 22: Development of a patient selection protocol prior to robotic radical prostatectomy (RRP) in the Preoperative Assessment Unit (PAU)
James Dyer, MD
Abstract 23: Protocol-driven preoperative testing in the Preoperative Assessment Unit (PAU): Which patients should receive a resting transthoracic echocardiogram (TTE) prior to elective noncardiac surgery?
James Dyer, MD
Abstract 24: High-risk preoperative assessment for elective orthopedic surgery patients
Terrence Adam, MD, PhD; Connie Parenti, MD; Terence Gioe, MD; Karen Ringsred, MD; and Joseph Wels, MD
Abstract 25: A novel use of web-based software to efficiently triage presurgical patients based on perioperative risk: A pilot
Alicia Kalamas, MD
Abstract 26: Value of a specialized clinic for day admission surgery for cardiac and major vascular operations
George Silvay, MD, PhD; Samuel DeMaria, MD; Marietta dePerio, NP, CCRN; Ellen Hughes, MA, RN; Samantha Silvay; Marina Krol, PhD; Brigid C. Flynn, MD; and David L. Reich, MD
Abstract 27: Preoperative evaluation for parathyroidectomy—rule out pheochromocytoma
Rubin Bahuva, MD; Sudhir Manda, MD; and Saurabh Kandpal, MD
Abstract 28: Should we stop the oral selective estrogen receptor modulator raloxifene prior to surgery?
Vesselin Dimov, MD; Tarek Hamieh, MD; and Ajay Kumar, MD
Abstract 29: Should mesalamine be stopped prior to noncardiac surgery to avoid bleeding complications?
Vesselin Dimov, MD; Tarek Hamieh, MD; and Ajay Kumar, MD
Abstract 30: Thyroidectomy: Perioperative management of acute thyroid storm
Stephen VanHaerents, MD, and Aashish A. Shah, MD
Abstract 31: Core competencies: Not just for the ACGME—but for successful and ethical perioperative management of a young respiratory cripple
Deborah Richman, MBChB, FFA(SA); Misako P. Sakamaki, MD; and Slawomir P. Oleszak, MD
Abstract 32: ‘If I have to be transfused I only want my wwn blood, or blood from family members’—what is best-practice advice to be given in the preoperative clinic?
Deborah Richman, MBChB, FFA(SA), and Joseph L. Conrad, MD
Abstract 33: Prolonged QTc and hypokalemia: A bad combination before surgery
Chadi Alraies, MD, and Abdul Hamid Alraiyes, MD
Abstract 34: Perioperative management of a parturient with neuromyelitis optica
Neeti Sadana, MD; Michael Orosco, MD; Michaela Farber, MD; and Scott Segal, MD
Abstract 35: ‘High’-pertension
Anuradha Ramaswamy, MD, and Franklin A. Michota, Jr., MD
Abstract 36: Perioperative care in neuromuscular scoliosis
Saurabh Basu Kandpal, MD, and Priya Baronia, MD
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Venous thromboembolism after total hip and knee replacement in older adults with single and co-occurring comorbidities
Alok Kapoor, MD, MSc; A. Labonte; M. Winter; J.B. Segal; R.A. Silliman; J.N. Katz; E. Losina; and D.R. Berlowitz
Abstract 2: Are there consequences of discontinuing angiotensin system inhibitors preoperatively in ambulatory and same-day admission patients?
Vasudha Goel, MBBS; David Rahmani, BS; Roy Braid, BS; Dmitry Rozin, BS; and Rebecca Twersky, MD, MPH
Abstract 3: Residents’ knowledge of ACC/AHA guidelines for preoperative cardiac evaluation is limited
BobbieJean Sweitzer, MD; Michael Vigoda, MD, MBA; Nikola Milokjic; Ben Boedeker, DVM, MD, PhD, MBA; Kip D. Robinson, MD, FACP; Michael A. Pilla, MD; Robert Gaiser, MD; Angela F. Edwards, MD; Ronald P. Olson, MD; Matthew D. Caldwell, MD; Shawn T. Beaman, MD; Jeffrey A. Green, MD; Jesse M. Ehrenfeld, MD, MPH; Marsha L. Wakefield, MD; Praveen Kalra, MD; David M. Feinstein, MD; Deborah C. Richman, MBChB, FFA(SA); Gail Van Norman; Gary E. Loyd, MD, MMM; Paul W. Kranner, MD; Stevin Dubin, MD; Sunil Eappen, MD; Sergio D. Bergese, MD; Suzanne Karan, MD; James R. Rowbottom, MD, FCCP; and Keith Candiotti, MD
Abstract 4: Descriptive perioperative BNP and CRP in vascular surgery patients
Thomas Barrett, MD, MCR, and Rebecca Duby, BS
Abstract 5: Selective serotonin reuptake inhibitors and risk of intraoperative bleeding
Adriana Oprea, MD, and Paula Zimbrean, MD
Abstract 6: Incidence and nature of postoperative complications in patients with obstructive sleep apnea undergoing noncardiac surgery
Roop Kaw, MD; Vinay Pasupuleti, MBBS, PhD; Esteban Walker, PhD; Anuradha Ramaswamy, MD; Thadeo Catacutan, MD; and Nancy Foldvary, DO
Abstract 7: HMG-CoA reductase inhibitor therapy and the risk of venous thromboembolism in joint replacement surgery
William Ho, MBBS; Brendan Flaim, MBBS, FRACP; and Andrea Chan, MBBS, FRACP
Abstract 8: Risk prediction models for cardiac morbidity and mortality in noncardiac surgery: A systematic review of the literature
Ramani Moonesinghe, MBBS, MRCP, FRCA; Kathy Rowan, PhD; Judith Hulf, CBE, FRCA; Michael G. Mythen, MD, FRCA; and Michael P.W. Grocott, MD, FRCA
Abstract 9: Economic aspects of preoperative testing
Gerhard Fritsch, MD; Maria Flamm, MD; Josef Seer, MD; and Andreas Soennichsen, MD
Abstract 10: Postoperative myocardial infarction and in-hospital mortality predictors in patients undergoing rlective noncardiac surgery
Anitha Rajamanickam, MD; Ali Usmani, MD; Jelica Janicijevic, MD; Preethi Patel, MD; Eric Hixson; Omeed Zardkoohi, MD; Michael Pecic; Changhong Yu; Michael Kattan, PhD; Sagar Kalahasti, MD; and Mina K. Chung, MD
Abstract 11: Incidence and predictors of postoperative heart failure in patients undergoing elective noncardiac surgery
Anitha Rajamanickam, MD; Ali Usmani, MD; Jelica Janicijevic, MD; Preethi Patel, MD; Eric Hixson; Omeed Zardkoohi, MD; Michael Pecic; Changhong Yu; Michael Kattan, PhD; Sagar Kalahasti, MD; and Mina K. Chung, MD
Abstract 12: Predictors of length of stay in patients undergoing total knee replacement surgery
Vishal Sehgal, MD; Pardeep Bansal, MD; Praveen Reddy, MD; Vishal Sharma, MD; Rajendra Palepu, MD; Linda Thomas, MD; and Jeremiah Eagan, MD
Abstract 13: Analysis of surgeon utilization of the Preoperative Assessment Communication Education (PACE) center in the pediatric population
Lisa Price Stevens, MD, and Ezinne Akamiro, BA, MD/MHA
Abstract 14: Use of the BATHE method to increase satisfaction amongst patients undergoing cardiac and major vascular operations
Samuel DeMaria, MD; Anthony P. DeMaria, MA; Menachem Weiner, MD; and George Silvay, MD
Abstract 15: Indication for surgery predicts long-term but not in-hospital mortality in patients undergoing lower extremity bypass vascular surgery
Brigid C. Flynn, MD; Michael Mazzeffi , MD; Carol Bodian, PhD; and Vivek Moitra, MD
Abstract 16: Research and outcomes on analgesia and nociception during surgery
Jinu Kim, MD; Tehila Adams, MD; Deepak Sreedharan, MD; Shanti Raju, MD; and Henry Bennett, PhD
Abstract 17: A snapshot survey of fluid prescribing
Helen Grote, MD; Luke Evans, MRCS; Abdel Omer, MD, PhD, FRCS; and Rob Lewis, MD, FRCA
Abstract 18: Predictors of difficult intubation with the video laryngoscope
Dario Galante, MD
Abstract 19: Use of technology to improve operational efficiency
Lucy Duffy, RN, MA, and Rita Lanaras, RN, BS, CNOR
Abstract 20: The ASA physical status score for the nonanesthesiologist
Adriana Oprea, MD, and David Silverman, MD
Abstract 21: Development of a shared multidisciplinary electronic preanesthetic record
Meghan Tadel, MD; R. Boyer, DO, MS; N. Smith; and P. Kallas, MD
Abstract 22: Development of a patient selection protocol prior to robotic radical prostatectomy (RRP) in the Preoperative Assessment Unit (PAU)
James Dyer, MD
Abstract 23: Protocol-driven preoperative testing in the Preoperative Assessment Unit (PAU): Which patients should receive a resting transthoracic echocardiogram (TTE) prior to elective noncardiac surgery?
James Dyer, MD
Abstract 24: High-risk preoperative assessment for elective orthopedic surgery patients
Terrence Adam, MD, PhD; Connie Parenti, MD; Terence Gioe, MD; Karen Ringsred, MD; and Joseph Wels, MD
Abstract 25: A novel use of web-based software to efficiently triage presurgical patients based on perioperative risk: A pilot
Alicia Kalamas, MD
Abstract 26: Value of a specialized clinic for day admission surgery for cardiac and major vascular operations
George Silvay, MD, PhD; Samuel DeMaria, MD; Marietta dePerio, NP, CCRN; Ellen Hughes, MA, RN; Samantha Silvay; Marina Krol, PhD; Brigid C. Flynn, MD; and David L. Reich, MD
Abstract 27: Preoperative evaluation for parathyroidectomy—rule out pheochromocytoma
Rubin Bahuva, MD; Sudhir Manda, MD; and Saurabh Kandpal, MD
Abstract 28: Should we stop the oral selective estrogen receptor modulator raloxifene prior to surgery?
Vesselin Dimov, MD; Tarek Hamieh, MD; and Ajay Kumar, MD
Abstract 29: Should mesalamine be stopped prior to noncardiac surgery to avoid bleeding complications?
Vesselin Dimov, MD; Tarek Hamieh, MD; and Ajay Kumar, MD
Abstract 30: Thyroidectomy: Perioperative management of acute thyroid storm
Stephen VanHaerents, MD, and Aashish A. Shah, MD
Abstract 31: Core competencies: Not just for the ACGME—but for successful and ethical perioperative management of a young respiratory cripple
Deborah Richman, MBChB, FFA(SA); Misako P. Sakamaki, MD; and Slawomir P. Oleszak, MD
Abstract 32: ‘If I have to be transfused I only want my wwn blood, or blood from family members’—what is best-practice advice to be given in the preoperative clinic?
Deborah Richman, MBChB, FFA(SA), and Joseph L. Conrad, MD
Abstract 33: Prolonged QTc and hypokalemia: A bad combination before surgery
Chadi Alraies, MD, and Abdul Hamid Alraiyes, MD
Abstract 34: Perioperative management of a parturient with neuromyelitis optica
Neeti Sadana, MD; Michael Orosco, MD; Michaela Farber, MD; and Scott Segal, MD
Abstract 35: ‘High’-pertension
Anuradha Ramaswamy, MD, and Franklin A. Michota, Jr., MD
Abstract 36: Perioperative care in neuromuscular scoliosis
Saurabh Basu Kandpal, MD, and Priya Baronia, MD

A 40-year-old man with spells of generalized weakness and paresthesias
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
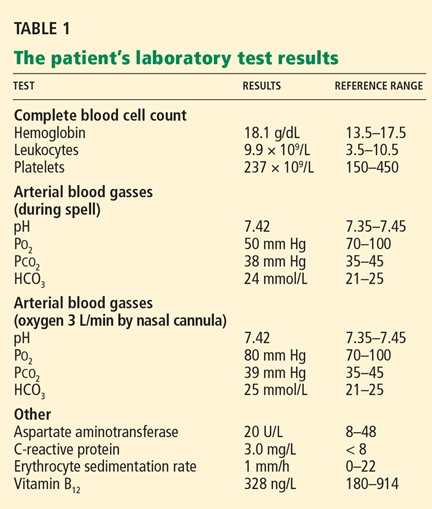
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
Abdominal pain in a 20-year-old woman
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
A 20-year-old woman presents to the emergency department with postprandial epigastric and right-upper-quadrant pain, sometimes associated with nausea. She has been having six to eight loose bowel movements every day, with no blood or mucus, and she has lost about 20 lb despite a good appetite. The diarrhea did not improve when she tried omitting milk products and carbohydrates.
Her symptoms began several months ago, but she says that 3 days ago the pain worsened steadily, radiating to the middle of her back, with associated episodes of nonbloody, nonbilious emesis. She cannot keep down liquids or solids. She says she has never had such episodes in the past.
She reports no oral ulcers, urinary symptoms, skin rashes, musculoskeletal pain, or neurologic symptoms, and she denies being anxious or depressed.
She has no history of serious illness, surgery, or hospitalization. She smokes a half pack of cigarettes a day, drinks alcohol occasionally, and smokes marijuana occasionally. She is employed as a certified nursing assistant.
She is taking ethinyl estradiol-levonorgestrel pills for birth control and takes calcium carbonate as needed for abdominal discomfort. She is taking no other medications, including nonsteroidal anti-inflammatory drugs (NSAIDs).
Her maternal uncle died of colon cancer at age 32, and her mother had colon polyps on colonoscopy. There is no family history of inflammatory bowel disease or celiac sprue. Her father committed suicide.
Her laboratory values
- White blood cell count 10.2 × 109/L (normal range 4–11)
- Red blood cell count 4.71 × 1012/L (3.9–5.5)
- Hemoglobin 14.4 g/dL (12–16)
- Hematocrit 42.4% (37%–47%)
- Mean corpuscular volume 90 fL (83–99)
- Mean corpuscular hemoglobin 30.6 pg (27–33)
- Platelet count 230 × 109/L (150–400)
- Red cell distribution width 13.3% (11.5%–14.5%)
- Sodium 140 mmol/L (132–148)
- Potassium 3.3 mmol/L (3.5–5.0)
- Chloride 104 mmol/L (98–111)
- Bicarbonate 28 mmol/L (23–32)
- Blood urea nitrogen 9 mg/dL (8–25)
- Creatinine 0.8 mg/dL (0.7–1.4)
- Glucose 87 mg/dL (65–100)
- Alanine aminotransferase 26 U/L (0–45)
- Aspartate aminotransferase 21 U/L (7–40)
- Alkaline phosphatase 101 U/L (40–150)
- Total bilirubin 0.8 mg/dL (0–1.5)
- Albumin 3.5 g/dL (3.5–5)
- Pregnancy screen negative
- Urine toxicology screen negative.
Physical examination
The patient is very thin and appears quite uncomfortable. Her temperature is 99.7°F (37.6°C), pulse rate 101, respiratory rate 18, blood pressure 111/67 mm Hg, and oxygen saturation 96% on room air. Her skin is warm and dry. Her height is 66 inches, weight 116 lb, and body mass index 18.7.
Examination of the head and neck shows normal dentition, dry mucus membranes, and no oral exudates. The thyroid is normal, and no masses or lymphadenopathy are noted.
Heart sounds and rhythm are normal, and the lungs are clear with no crackles or rubs. The abdomen is scaphoid and soft, with no distention. She has epigastric tenderness but no rebound, guarding, rigidity, palpable mass, or costovertebral angle tenderness. Bowel sounds are normal. The neurologic examination is normal.
NARROWING THE DIAGNOSIS
1. Given the history and findings so far, which is the least likely cause of her symptoms?
- Lactose intolerance
- Celiac disease
- Crohn disease
- Duodenal ulcer
- Eating disorder
This young woman’s presentation has some features found in all of these conditions. However, the least likely is lactose intolerance.
Lactose intolerance results from a shortage of the enzyme lactase, which is normally produced by the cells that line the small intestine. Close to 50 million American adults have lactose intolerance. Common symptoms include nausea, cramps, bloating, gas, and diarrhea, which begin about 30 minutes to 2 hours after eating or drinking foods containing lactose.
Since the patient’s symptoms did not improve when she tried omitting milk products, and since lactose intolerance is rarely associated with pain radiating to the back and with severe vomiting, this is the least likely cause of her symptoms.
Celiac disease presents with a myriad of symptoms—sometimes without gastrointestinal (GI) symptoms. Anemia is the most common laboratory finding, due most often to iron deficiency, but also due to deficiencies of vitamin B12 and folate as a result of malabsorption.1
Our patient’s laboratory values—especially her red cell indices—do not confirm this finding. One must also remember, however, that hemoglobin tends to be falsely elevated in patients who are dehydrated.
Crohn disease often presents with occult blood loss, low-grade fever, weight loss, and anemia. Though the condition is most often ileocolic, it can affect any part of the gastrointestinal tract. Nevertheless, most patients with gastroduodenal involvement have previously been diagnosed with ileocolic disease, and gastroduodenal involvement manifests later. Nonradiating epigastric pain is very common. Obstructive symptoms due to gastroduodenal strictures (eg, postprandial vomiting, epigastric pain, weight loss, bloating) are also common. 2
Duodenal ulcer. The most important factors responsible for duodenal ulcers are NSAID use and Helicobacter pylori infection.3 Duodenal ulcers have a variety of clinical presentations, ranging from no symptoms to severe pain. Epigastric pain can be sharp, dull, burning, or penetrating. Many patients complain of a feeling of hunger and weight gain—as opposed to gastric ulcer, in which patients experience anorexia and weight loss. Abdominal pain generally occurs several hours after meals and often awakens the patient at night. Pain is often relieved by food, but this phenomenon is present in only 20% to 60% of patients and probably is not specific for duodenal ulcer.
Our patient does not use NSAIDs, but some of her symptoms, such as postprandial pain, epigastric pain radiating to the back, and nausea and vomiting are seen with duodenal ulcer.
Eating disorders. The two main types of eating disorders—anorexia nervosa and bulimia nervosa—have a significant diagnostic overlap,4 and a third type, binge-eating disorder, is currently being investigated and defined. Girls and women are 10 times as likely as boys and men to develop an eating disorder.
People with anorexia have a distorted view of their bodies. Even when they are extremely thin, they see themselves as too fat.
Bulimia is characterized by binge-eating, purging, and overexercising to compensate for the excess calories. Patients are often close to normal weight.
Binge-eating disorder involves the consumption of very large amounts of food in a short period of time. About 2% of all young adults in the United States struggle with bingeeating. They are either overweight or obese.
These disorders tend to be associated with other psychiatric disorders such as depression or obsessive-compulsive disorder. Our patient sought medical attention and was appropriately concerned about her weight loss, which make an eating disorder unlikely.
CASE CONTINUED: SHE UNDERGOES CT
2. Which of the following is the most likely diagnosis at this point?
- SMA syndrome
- Chronic mesenteric ischemia involving the SMA
- Megaduodenum due to a connective tissue disorder
SMA syndrome is the most likely diagnosis. Despite its name, this syndrome is not a vascular condition. It is an uncommon cause of proximal intestinal obstruction in which the duodenum is compressed between the SMA and the aorta. First described in 1861, it has also been known as cast syndrome, Wilkie syndrome, and arteriomesenteric duodenal obstruction.5
To date, more than 400 cases of this syndrome have been reported, twice as many in women as in men. Most patients are between 20 and 40 years of age at the time of diagnosis. Common presenting symptoms include postprandial abdominal pain, nausea, vomiting, and weight loss, which may further reduce the angle between the SMA and the aorta. Diarrhea is not generally associated with this syndrome, and in our patient’s case the diarrhea was thought to be unrelated to the SMA syndrome, since it subsided spontaneously.
Conditions and events that cause, contribute to, or worsen SMA syndrome include:
- Rapid weight loss (as in cancer or burns) or lean body habitus
- Prolonged bed rest
- Use of a body cast
- Malabsorption
- Spinal disease, deformity, or trauma
- Scoliosis surgery
- Rapid linear growth without compensatory weight gain
- Abnormally high and fixed position of the ligament of Treitz
- Abdominal surgery
- Cardiac cachexia
- Unusually low origin of the SMA.7
More common causes of mechanical smallbowel obstruction are adhesions, hernias, and tumors.8 Hyperactive, high-pitched peristalsis with rushes coinciding with cramps is typical. Abdominal cramps are centered around the umbilicus or in the epigastrium and are associated with vomiting; obstipation develops in patients with complete obstruction. Patients with partial obstruction may develop diarrhea. Paralytic ileus secondary to hypokalemia is an important consideration in partial obstruction. However, abdominal radiography and CT did not confirm an obstruction, and her symptoms persisted despite correction of the potassium level.
Chronic mesenteric ischemia can be caused by vasculitis, nonocclusive conditions that cause prolonged vasoconstriction (eg, cocaine ingestion), or reduced cardiac output.9 Symptoms are due to the gradual reduction in blood flow to the intestine that occurs during eating. Our patient’s toxicology report did not suggest cocaine abuse, and her history and the workup thus far do not suggest heart failure. A workup for vasculitis was negative.
Megaduodenum, SMA-like syndrome. In rare cases, dilation of the duodenum at the level of the SMA may be part of a generalized duodenal dilation caused by something other than obstruction due to mechanical compression. There are conditions, as described below, that cause an SMA-like syndrome.
A compression defect of the duodenum at the site where the SMA crossed the duodenum was found in a series of 11 cases of systemic sclerosis.10 These patients had definite dilation of the duodenum, but it was a result of atrophy of the muscle layers and replacement by collagenous tissue, changes that result in diminished peristalsis, loss of muscle tone, and dilation. The duodenum yields to pressure in its third portion under the SMA.
Several pathologic conditions, particularly connective tissue disorders, may predispose to the development of a megaduodenum that may result in an imprint on the duodenum at the level of the SMA. The most noteworthy of these conditions is scleroderma. Other conditions that can cause reduced duodenal peristalsis include diabetes, pancreatitis, dermatomyositis, lupus erythematosus, myxedema, and amyloidosis.11
It is important to distinguish SMA syndrome from SMA-like syndromes for several reasons.12 SMA-like syndromes result in loss of normal peristalsis. Further, the conditions have different outcomes, even though they are managed similarly initially, ie, with rehydration and parenteral nutrition. Surgery is to be avoided if possible in conditions that affect widespread areas of the intestine, such as scleroderma or diabetic neuropathy.
3. Which of the following is helpful in confirming SMA syndrome?
- CT of the abdomen
- Upper GI radiography series
- Upper GI endoscopy
All three can help confirm the diagnosis.
CT of the abdomen is a convenient, safe, rapid, readily available, and relatively noninvasive way to evaluate the aortomesenteric angle and to view retroperitoneal and mesenteric fat.13 Rehydration before injecting intravenous dye is important to avoid precipitating renal failure. In this patient, CT findings that helped make the diagnosis included a narrow aortomesenteric angle, compression of the duodenum, and a paucity of fat around the SMA.
An upper GI series can reveal dilation of the first and second portions of the duodenum and abrupt compression of the duodenal mucosal folds. Other findings can include a delay of 4 to 6 hours in gastroduodenal transit and relief of the obstruction when the patient is in the left lateral decubitus position. The Hayes maneuver refers to the disappearance of these radiologic features in the knee-chest position on cinefluoroscopy.14 The findings mentioned above are best noted in the supine position on both radiography and CT.
Endoscopy is necessary to rule out mechanical causes of duodenal obstruction. A pulsatile extrinsic compression suggests this condition but is found only occasionally.
Other imaging studies, such as ultrasonography, arteriography, and hypotonic duodenography, are used less often.
4. At this time, which of the following would be the most appropriate initial treatment in this patient?
- Conservative treatment
- Narcotics
- Duodenojejunostomy
Conservative treatment is indicated initially in all cases of SMA syndrome.15 This involves reversing precipitating factors and replacing fluid, electrolytes, and nutrition via total parenteral nutrition and nasogastric decompression.
To avoid keeping the patient on intravenous therapy for a prolonged time, it is important to start enteral feeding once the pain has subsided and the patient can tolerate it. A double-lumen nasojejunal tube is passed distal to the obstruction under fluoroscopic guidance. During feedings, the patient should be in the modified knee-chest, prone, or leftside-down position, all of which increase the aortomesenteric angle.
Delaying the treatment of SMA syndrome can increase the risk of morbidity and mortality by progressive malnutrition, dehydration, oliguria, electrolyte abnormalities (eg, hypokalemia), or intestinal perforation from prolonged ischemia.16,17
Narcotics and other drugs known to slow gut motility should be avoided.
Symptoms typically improve after restoration of normal body weight. If conservative treatment fails, or if the case is severe or chronic, surgery is required.18 Fortunately, this is not required often.
Duodenojejunostomy is the most common surgical treatment and involves creation of an alternate route between the duodenum and the jejunum, bypassing the compression between the aorta and the SMA. Other procedures include gastrojejunostomy, laparoscopic duodenojejunostomy, 19 a Roux-en-Y procedure, robotically assisted duodenojejunostomy, and anterior transposition of the third portion of the duodenum. Cleavage of the ligament of Treitz is another option, enabling the duodenum to drop away from the apex of the sharpened aortomesenteric angle.
WHEN TO CONSIDER SMA SYNDROME
The SMA syndrome is an uncommon cause of a very common presenting symptom, ie, abdominal pain. Nevertheless, it should be considered in the differential diagnosis of abdominal pain, especially in patients who have conditions that cause significant weight loss, such as anorexia nervosa, malabsorption, or hypercatabolic states such as burns, major surgery, severe injuries, or malignancies. The diagnosis is based on a thorough history and on supportive findings from CT and endoscopy.
In our patient, weight loss began with nonspecific diarrhea but perpetuated itself as SMA syndrome occurred.
Appropriate management consists of interrupting the cycle of weight loss and secondary upper gut obstruction. For patients in whom more definitive therapy is not feasible, a gastrostomy tube for decompression with a jejunal extension available for feeding appears to be a reasonable and safe treatment option. Duodenojejunostomy is considered the procedure of choice in severe cases.
CASE CONCLUDED
Fortunately, our patient responded well to conservative management. She was treated with intravenous hydration and correction of electrolytes and 10 days later was able to tolerate a soft diet. She was discharged in stable condition. At a follow-up visit 2 weeks later, she reported minimal abdominal discomfort, was able to tolerate meals, and had gained a few pounds. She continues to do well.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
- Iovino P, Ciacci C, Sabbatini F, Acioli DM, D'Argenio G, Mazzacca G. Esophageal impairment in adult celiac disease with steatorrhea. Am J Gastroenterol 1998; 93:1243–1249.
- Loftus EV. Upper gastrointestinal tract Crohn’s disease. Clin Perspect Gastroenterol 2002; 5:188–191.
- Zapata-Colindres JC, Zepeda-Gómez S, Montaño-Loza A, Vázquez-Ballesteros E, de Jesús Villalobos J, Valdovinos-Andraca F. The association of Helicobacter pylori infection and nonsteroidal antiinflammatory drugs in peptic ulcer disease. Can J Gastroenterol 2006; 20:277–280.
- Milos G, Spindler A, Schnyder U, Fairburn CG. Instability of eating disorder diagnoses: prospective study. Br J Psychiatry 2005; 187:573–578.
- Wilkie DP. Chronic duodenal ileus. Br J Surg 1921; 9:204–214.
- Ozkurt H, Cenker MM, Bas N, Erturk SM, Basak M. Measurement of the distance and angle between the aorta and superior mesenteric artery: normal values in different BMI categories. Surg Radiol Anat 2007; 29:595–599.
- Lippl F, Hannig C, Weiss W, Allescher HD, Classen M, Kurjak M. Superior mesenteric artery syndrome: diagnosis and treatment from the gastroenterologist's view. J Gastroenterol 2002; 37:640–643.
- Balthazar EJ. George W. Holmes Lecture. CT of small-bowel obstruction. AJR Am J Roentgenol 1994; 162:255–261.
- Chang JB, Stein TA. Mesenteric ischemia: acute and chronic. Ann Vasc Surg 2003; 17:323–328.
- Gondos B. Duodenal compression defect and the “superior mesenteric artery syndrome” 1. Radiology 1977; 123:575–580.
- Cohen LB, Field SP, Sachar DB. The superior mesenteric artery syndrome. The disease that isn't, or is it? J Clin Gastroenterol 1985; 7:113–716.
- Ahmed AR, Taylor I. Superior mesenteric artery syndrome. Postgrad Med J 1997; 73:776–778.
- Santer R, Young C, Rossi T, Riddlesberger MM. Computed tomography in superior mesenteric artery syndrome. Pediatr Radiol 1991; 21:154–155.
- Lukes PJ, Rolny P, Nilson AE, Gamklou R, Darle N, Dotevall G. Diagnostic value of hypotonic duodenography in superior mesenteric artery syndrome. Acta Chir Scand 1978; 144:39–43.
- Dietz UA, Debus ES, Heuko-Valiati L, et al. Aorto-mesenteric artery compression syndrome. Chirurg 2000; 71:1345–1351.
- Lim JE, Duke GL, Eachempati SR. Superior mesenteric artery syndrome presenting with acute massive gastric dilatation, gastric wall pneumatosis, and portal venous gas. Surgery 2003; 134:840–843.
- Fuhrman MA, Felig DM, Tanchel ME. Superior mesenteric artery syndrome with obstructing duodenal bezoar. Gastrointest Endosc 2003; 57:387.
- Hines JR, Gore RM, Ballantyne GH. Superior mesenteric artery syndrome. Diagnostic criteria and therapeutic approaches. Am J Surg 1984; 148:630–632.
- Gersin KS, Heniford BT. Laparoscopic duodenojejunostomy for treatment of superior mesenteric artery syndrome. JSLS 1998; 2:281–284.
Proceedings of the 4th Annual Perioperative Medicine Summit

Supplement Editor:
Amir K. Jaffer, MD, FHM
Associate Editors:
David L. Hepner, MD, and Franklin A. Michota, MD, FHM
Contents
Public reporting and pay-for-performance programs in perioperative medicine
Peter Lindenauer, MD MSc
Cardiac risk stratification for noncardiac surgery: Update from the American College of Cardiology/American Heart Association 2007 guidelines
Lee A. Fleisher, MD
Perioperative care of the elderly patient: An update
Robert M. Palmer, MD, MPH
The role of testing in the preoperative evaluation
David L. Hepner, MD
Perioperative fluid management: Progress despite lingering controversies
Mark A. Hamilton, MBBS, MRCP, FRCA
Giving anesthesiologists what they want: How to write a useful preoperative consult
David Lubarsky, MD, MBA, and Keith Candiotti, MD
Perioperative management of warfarin and antiplatelet therapy
Amir K. Jaffer, MD, FHM
Prevention of venous thromboembolism after surgery
Franklin A. Michota, MD, FHM
Perioperative management of diabetes: Translating evidence into practice
Luigi F. Meneghini, MD, MBA
Postoperative pulmonary complications: An update on risk assessment and reduction
Gerald W. Smetana, MD
Postoperative gastrointestinal tract dysfunction: An overview of causes and management strategies
Michael G. (Monty) Mythen, MD
Case studies in perioperative management: Challenges, controversies, and common ground
Steven L. Cohn, MD, and BobbieJean Sweitzer, MD
Statins and noncardiac surgery: Current evidence and practical considerations
Don Poldermans, MD, PhD
The experts debate: perioperative beta-blockade for noncardiac surgery patients—proven safe or not?
Don Poldermans, MD, PhD, and P.J. Devereaux, MD, PhD
Perioperative considerations for patients with liver disease
Paul Martin, MD
Perioperative management of obstructive sleep apnea: Ready for prime time?
Shirin Shafazand, MD, MS
Nuts and bolts of preoperative clinics: The view from three institutions
Angela M. Bader, MD, MPH; BobbieJean Sweitzer, MD; and Ajay Kumar, MD
Perioperative management of anemia: Limits of blood transfusion and alternatives to it
Ajay Kumar, MD
Medicolegal issues in perioperative medicine: Lessons from real cases
Franklin A. Michota, MD, FHM, and Matthew J. Donnelly, Esq
Perioperative medication management: General principles and practical applications
Christopher Whinney, MD
Supplement Editor:
Amir K. Jaffer, MD, FHM
Associate Editors:
David L. Hepner, MD, and Franklin A. Michota, MD, FHM
Contents
Public reporting and pay-for-performance programs in perioperative medicine
Peter Lindenauer, MD MSc
Cardiac risk stratification for noncardiac surgery: Update from the American College of Cardiology/American Heart Association 2007 guidelines
Lee A. Fleisher, MD
Perioperative care of the elderly patient: An update
Robert M. Palmer, MD, MPH
The role of testing in the preoperative evaluation
David L. Hepner, MD
Perioperative fluid management: Progress despite lingering controversies
Mark A. Hamilton, MBBS, MRCP, FRCA
Giving anesthesiologists what they want: How to write a useful preoperative consult
David Lubarsky, MD, MBA, and Keith Candiotti, MD
Perioperative management of warfarin and antiplatelet therapy
Amir K. Jaffer, MD, FHM
Prevention of venous thromboembolism after surgery
Franklin A. Michota, MD, FHM
Perioperative management of diabetes: Translating evidence into practice
Luigi F. Meneghini, MD, MBA
Postoperative pulmonary complications: An update on risk assessment and reduction
Gerald W. Smetana, MD
Postoperative gastrointestinal tract dysfunction: An overview of causes and management strategies
Michael G. (Monty) Mythen, MD
Case studies in perioperative management: Challenges, controversies, and common ground
Steven L. Cohn, MD, and BobbieJean Sweitzer, MD
Statins and noncardiac surgery: Current evidence and practical considerations
Don Poldermans, MD, PhD
The experts debate: perioperative beta-blockade for noncardiac surgery patients—proven safe or not?
Don Poldermans, MD, PhD, and P.J. Devereaux, MD, PhD
Perioperative considerations for patients with liver disease
Paul Martin, MD
Perioperative management of obstructive sleep apnea: Ready for prime time?
Shirin Shafazand, MD, MS
Nuts and bolts of preoperative clinics: The view from three institutions
Angela M. Bader, MD, MPH; BobbieJean Sweitzer, MD; and Ajay Kumar, MD
Perioperative management of anemia: Limits of blood transfusion and alternatives to it
Ajay Kumar, MD
Medicolegal issues in perioperative medicine: Lessons from real cases
Franklin A. Michota, MD, FHM, and Matthew J. Donnelly, Esq
Perioperative medication management: General principles and practical applications
Christopher Whinney, MD
Supplement Editor:
Amir K. Jaffer, MD, FHM
Associate Editors:
David L. Hepner, MD, and Franklin A. Michota, MD, FHM
Contents
Public reporting and pay-for-performance programs in perioperative medicine
Peter Lindenauer, MD MSc
Cardiac risk stratification for noncardiac surgery: Update from the American College of Cardiology/American Heart Association 2007 guidelines
Lee A. Fleisher, MD
Perioperative care of the elderly patient: An update
Robert M. Palmer, MD, MPH
The role of testing in the preoperative evaluation
David L. Hepner, MD
Perioperative fluid management: Progress despite lingering controversies
Mark A. Hamilton, MBBS, MRCP, FRCA
Giving anesthesiologists what they want: How to write a useful preoperative consult
David Lubarsky, MD, MBA, and Keith Candiotti, MD
Perioperative management of warfarin and antiplatelet therapy
Amir K. Jaffer, MD, FHM
Prevention of venous thromboembolism after surgery
Franklin A. Michota, MD, FHM
Perioperative management of diabetes: Translating evidence into practice
Luigi F. Meneghini, MD, MBA
Postoperative pulmonary complications: An update on risk assessment and reduction
Gerald W. Smetana, MD
Postoperative gastrointestinal tract dysfunction: An overview of causes and management strategies
Michael G. (Monty) Mythen, MD
Case studies in perioperative management: Challenges, controversies, and common ground
Steven L. Cohn, MD, and BobbieJean Sweitzer, MD
Statins and noncardiac surgery: Current evidence and practical considerations
Don Poldermans, MD, PhD
The experts debate: perioperative beta-blockade for noncardiac surgery patients—proven safe or not?
Don Poldermans, MD, PhD, and P.J. Devereaux, MD, PhD
Perioperative considerations for patients with liver disease
Paul Martin, MD
Perioperative management of obstructive sleep apnea: Ready for prime time?
Shirin Shafazand, MD, MS
Nuts and bolts of preoperative clinics: The view from three institutions
Angela M. Bader, MD, MPH; BobbieJean Sweitzer, MD; and Ajay Kumar, MD
Perioperative management of anemia: Limits of blood transfusion and alternatives to it
Ajay Kumar, MD
Medicolegal issues in perioperative medicine: Lessons from real cases
Franklin A. Michota, MD, FHM, and Matthew J. Donnelly, Esq
Perioperative medication management: General principles and practical applications
Christopher Whinney, MD

Managing diabetes in hemodialysis patients: Observations and recommendations
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
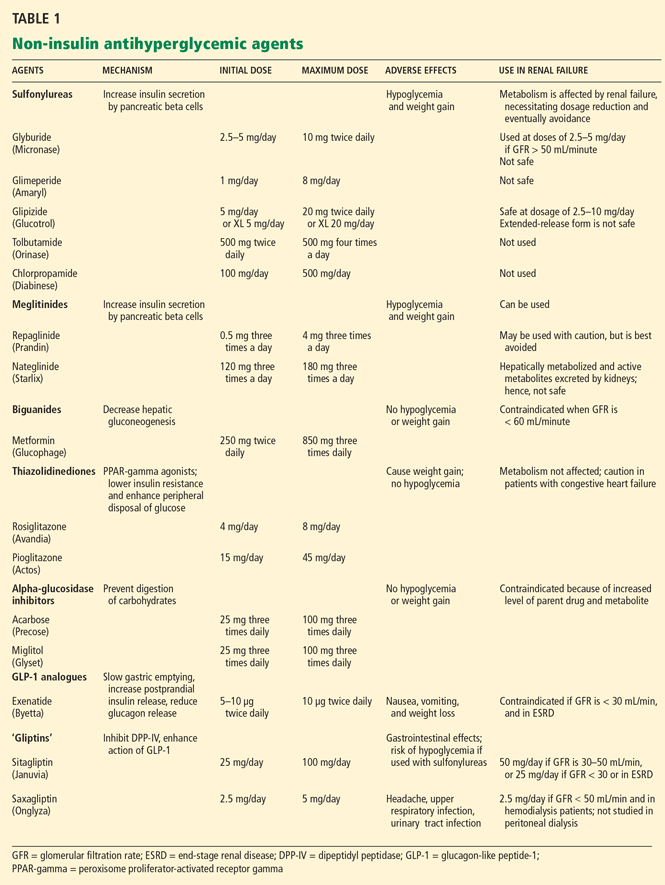
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
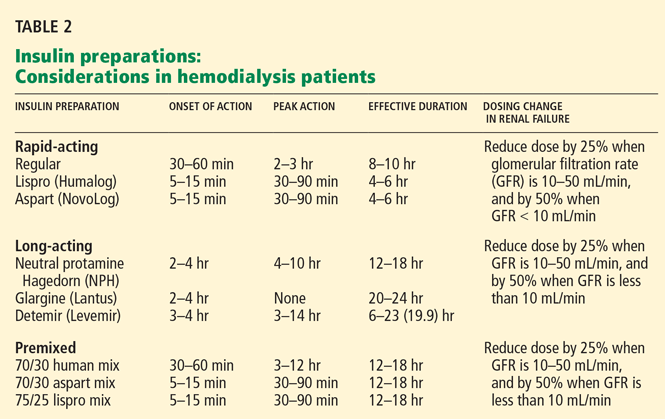
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
KEY POINTS
- Blood glucose levels can fluctuate widely due to various and opposing effects of ESRD and dialysis.
- The hemoglobin A1c level can be falsely high in ESRD, but it is still a reasonable measure of glycemic control in this population.
- Most diabetes drugs are excreted at least in part by the kidney, so that patients in ESRD are at greater risk of hypoglycemia.
- Insulin is the cornerstone of treatment, since most oral diabetes drugs are contraindicated or not recommended in this population. Insulin doses should be lowered in those with low glomerular filtration rates.
Alternative modes of mechanical ventilation: A review for the hospitalist
Technologic advances and computerized control of mechanical ventilators have made it possible to deliver ventilatory assistance in new modes. Driving these innovations is the desire to prevent ventilator-induced lung injury, improve patient comfort, and liberate the patient from mechanical ventilation as soon as possible.
We call these innovations “alternative” modes to differentiate them from the plain volume-control and pressure-control modes. Some clinicians rarely use these new modes, but in some medical centers they have become the most common ones used, or are being used unknowingly (the operator misunderstands the mode name). The information we provide on these modes of ventilation is by no means an endorsement of their use, but rather a tool to help the clinician understand their physiologic, theoretical, and clinical effects.
We focused on two goals:
- Explain what the mode does
- Briefly review the theoretical benefits and the actual evidence supporting these alternative modes of ventilation.

STANDARD NOMENCLATURE NEEDED
Since its invention, mechanical ventilation has been plagued by multiple names being used to describe the same things. For example, volume-control ventilation is also called volume-cycled ventilation, assist-control ventilation, volume-limited ventilation, and controlled mechanical ventilation. Similarly, multiple abbreviations are used, each depending on the brand of ventilator, and new acronyms have been added in recent years as new modes have been developed. The vast number of names and modes can confuse even the most seasoned critical care physician.
Efforts to establish a common nomenclature are under way.1
WHAT IS A MODE?
A mode of mechanical ventilation has three essential components:
- The control variable
- The breath sequence
- The targeting scheme.
Similar modes may require more detailed descriptions to distinguish them, but the basic function can be explained by these three components.
The control variable
In general, inspiration is an active process, driven by the patient’s effort, the ventilator, or both, while expiration is passive. For simplicity, in this article a mechanical breath means the inspiratory phase of the breath.
The machine can only control the volume (and flow) or the pressure given. The breaths can be further described on the basis of what triggers the breath, what limits it (the maximum value of a control variable), and what ends (cycles) it.
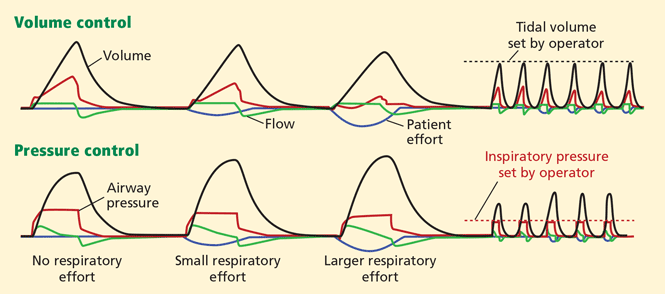
The breath sequence
There are three possible breath sequences:

- Intermittent mandatory ventilation, in which the patient can take spontaneous breaths between mandatory breaths
- Continuous spontaneous ventilation, in which all breaths are spontaneous (Table 1).

The targeting scheme
The targeting or feedback scheme refers to the ventilator settings and programming that dictate its response to the patient’s lung compliance, lung resistance, and respiratory effort. The regulation can be as simple as controlling the pressure in pressure-control mode, or it can be based on a complicated algorithm.
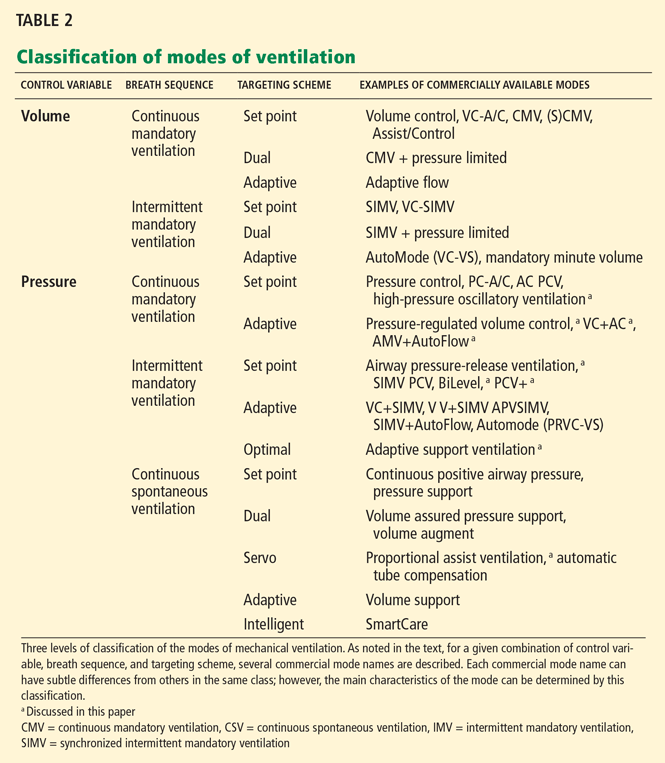
ADAPTIVE PRESSURE CONTROL
Other names for adaptive pressure control
- Pressure Regulated Volume Control (Maquet Servo-i, Rastatt, Germany)
- AutoFlow (Dräger Medical AG, Lübeck, Germany)
- Adaptive Pressure Ventilation (Hamilton Galileo, Hamilton Medical AG, Bonaduz, Switzerland)
- Volume Control+ (Puritan Bennett, Tyco Healthcare; Mansfield, MA)
- Volume Targeted Pressure Control, Pressure Controlled Volume Guaranteed (Engström, General Electric, Madison, WI).
What does adaptive pressure control do?
The APC mode delivers pressure-controlled breaths with an adaptive targeting scheme (Table 2).
In pressure-control ventilation, tidal volumes depend on the lung’s physiologic mechanics (compliance and resistance) and patient effort (Figure 1). Therefore, the tidal volume varies with changes in lung physiology (ie, larger or smaller tidal volumes than targeted).
To overcome this effect, a machine in APC mode adjusts the inspiratory pressure to deliver the set minimal target tidal volume. If tidal volume increases, the machine decreases the inspiratory pressure, and if tidal volume decreases, the machine increases the inspiratory pressure. However, if the patient effort is large enough, the tidal volume will increase in spite of decreasing the inspiratory pressure (Figure 2). The adjustments to the inspiratory pressure occur after the tidal volume is off-target in a number of breaths.
Common sources of confusion with adaptive pressure control
First, APC is not a volume-control mode. In volume control, the tidal volume does not change; in APC the tidal volume can increase or decrease, and the ventilator will adjust the inflation pressure to achieve the target volume. Thus, APC guarantees an average minimum tidal volume but not a maximum tidal volume.
Second, a characteristic of pressure control (and hence, APC) is that the flow of gas varies to maintain constant airway pressure (ie, maintain the set inspiratory pressure). This characteristic allows a patient who generates an inspiratory effort to receive flow as demanded, which is likely more comfortable. This is essentially different from volume control, in which flow is set by the operator and hence is fixed. Thus, if the patient effort is strong enough (Figure 1), this leads to what is called flow asynchrony, in which the patient does not get the flow asked for in a breath.
Ventilator settings in adaptive pressure control
Ventilator settings in APC are:
- Tidal volume
- Time spent in inspiration (inspiratory time)
- Frequency
- Fraction of inspired oxygen (Fio2)
- Positive end-expiratory pressure (PEEP).
Some ventilators also require setting the speed to reach the peak pressure (also known as slope percent or inspiratory rise time).
Clinical applications of adaptive pressure control
This mode is designed to maintain a consistent tidal volume during pressure-control ventilation and to promote inspiratory flow synchrony. It is a means of automatically reducing ventilatory support (ie, weaning) as the patient’s inspiratory effort becomes stronger, as in awakening from anesthesia.
APC may not be ideal for patients who have an inappropriately increased respiratory drive (eg, in severe metabolic acidosis), since the inspiratory pressure will decrease to maintain the targeted average tidal volume, inappropriately shifting the work of breathing onto the patient.
Theoretical benefits of adaptive pressure control
APC guarantees a minimum average tidal volume (unless the pressure alarm threshold is set too low, so that the target tidal volume is not delivered). Other theoretical benefits are flow synchrony, less ventilator manipulation by the operator, and automatic weaning of ventilator support.
Evidence of benefit of adaptive pressure control
Physiologic benefits. This mode has lower peak inspiratory pressures than does volume-control ventilation,3,4 which is often reported as a positive finding. However, in volume-control mode (the usual comparator), the peak inspiratory pressure is a manifestation of both resistance and compliance. Hence, peak inspiratory pressure is expected to be higher but does not reflect actual lung-distending pressures. It is the plateau pressure, a manifestation of lung compliance, that is related to lung injury.
Patient comfort. APC may increase the work of breathing when using low tidal volume ventilation and when there is increased respiratory effort (drive).5 Interestingly, APC was less comfortable than pressure support ventilation in a small trial.6
Outcomes have not been studied.7
Adaptive pressure control: Bottom line
APC is widely available and widely used, sometimes unknowingly (eg, if the operator thinks it is volume control). It is relatively easy to use and to set; however, evidence of its benefit is scant.
ADAPTIVE SUPPORT VENTILATION
Adaptive support ventilation (ASV) evolved as a form of mandatory minute ventilation implemented with adaptive pressure control. Mandatory minute ventilation is a mode that allows the operator to preset a target minute ventilation, and the ventilator then supplies mandatory breaths, either volume- or pressure-controlled, if the patient’s spontaneous breaths generate a lower minute ventilation.
ASV automatically selects the appropriate tidal volume and frequency for mandatory breaths and the appropriate tidal volume for spontaneous breaths on the basis of the respiratory system mechanics and target minute alveolar ventilation.
Described in 1994 by Laubscher et al,8,9 ASV became commercially available in 1998 in Europe and in 2007 in the United States (Hamilton Galileo ventilator, Hamilton Medical AG). This is the first commercially available ventilator that uses an “optimal” targeting scheme (see below).
What does adaptive support ventilation do?
ASV delivers pressure-controlled breaths using an adaptive (optimal) scheme (Table 2). “Optimal,” in this context, means minimizing the mechanical work of breathing: the machine selects a tidal volume and frequency that the patient’s brain would presumably select if the patient were not connected to a ventilator. This pattern is assumed to encourage the patient to generate spontaneous breaths.
The ventilator calculates the normal required minute ventilation based on the patient’s ideal weight and estimated dead space volume (ie, 2.2 mL/kg). This calculation represents 100% of minute ventilation. The clinician at the bedside sets a target percent of minute ventilation that the ventilator will support—higher than 100% if the patient has increased requirements due, eg, to sepsis or increased dead space, or less than 100% during weaning.
The ventilator initially delivers test breaths, in which it measures the expiratory time constant for the respiratory system and then uses this along with the estimated dead space and normal minute ventilation to calculate an optimal breathing frequency in terms of mechanical work.
The optimal or target tidal volume is calculated as the normal minute ventilation divided by the optimal frequency. The target tidal volume is achieved by the use of APC (see above) (Figure 2). This means that the pressure limit is automatically adjusted to achieve an average delivered tidal volume equal to the target. The ventilator continuously monitors the respiratory system mechanics and adjusts its settings accordingly.
The ventilator adjusts its breaths to avoid air trapping by allowing enough time to exhale, to avoid hypoventilation by delivering tidal volume greater than the dead space, and to avoid volutrauma by avoiding large tidal volumes.
Ventilator settings in adaptive support ventilation
Ventilator settings in ASV are:
- Patient height (to calculate the ideal body weight)
- Sex
- Percent of normal predicted minute ventilation goal
- Fio2
- PEEP.
Clinical applications of adaptive support ventilation
ASV is intended as a sole mode of ventilation, from initial support to weaning.
Theoretical benefits of adaptive support ventilation
In theory, ASV offers automatic selection of ventilator settings, automatic adaptation to changing patient lung mechanics, less need for human manipulation of the machine, improved synchrony, and automatic weaning.
Evidence of benefit of adaptive support ventilation
Physiologic benefits. Ventilator settings are adjusted automatically. ASV selects different tidal volume-respiratory rate combinations based on respiratory mechanics in passive and paralyzed patients.10–12 In actively breathing patients, there was no difference in the ventilator settings chosen by ASV for different clinical scenarios (and lung physiology).10 Compared with pressure-controlled intermittent mandatory ventilation, with ASV, the inspiratory load is less and patient-ventilator interaction is better.13
Patient-ventilator synchrony and comfort have not been studied.
Outcomes. Two trials suggest that ASV may decrease time on mechanical ventilation.14,15 However, in another trial,16 compared with a standard protocol, ASV led to fewer ventilator adjustments but achieved similar postsurgical weaning outcomes. The effect of this mode on the death rate has not been examined.17,18
Adaptive support ventilation: Bottom line
ASV is the first commercially available mode that automatically selects all the ventilator settings except PEEP and Fio2. These seem appropriate for different clinical scenarios in patients with poor respiratory effort or in paralyzed patients. Evidence of the effect in actively breathing patients and on outcomes such as length of stay or death is still lacking.
PROPORTIONAL ASSIST VENTILATION
Patients who have normal respiratory drive but who have difficulty sustaining adequate spontaneous ventilation are often subjected to pressure support ventilation (PSV), in which the ventilator generates a constant pressure throughout inspiration regardless of the intensity of the patient’s effort.
In 1992, Younes and colleagues19,20 developed proportional assist ventilation (PAV) as an alternative in which the ventilator generates pressure in proportion to the patient’s effort. PAV became commercially available in Europe in 1999 and was approved in the United States in 2006, available on the Puritan Bennett 840 ventilator (Puritan Bennett Co, Boulder, CO). PAV has also been used for noninvasive ventilation, but this is not available in the United States.
Other names for proportional assist ventilation
Proportional Pressure Support (Dräger Medical; not yet available in the United States).
What does proportional assist ventilation do?
This mode delivers pressure-controlled breaths with a servo control scheme (Table 2).
To better understand PAV, we can compare it with PSV. With PSV, the pressure applied by the ventilator rises to a preset level that is held constant (a set-point scheme) until a cycling criterion (a percent of the maximum inspiratory flow value) is reached. The inspiratory flow and tidal volume are the result of the patient’s inspiratory effort, the level of pressure applied, and the respiratory system mechanics.
In PAV, as in PSV, all breaths are spontaneous (Table 1). The patient controls the timing and size of the breath. There are no preset pressure, flow, or volume goals, but safety limits on the volume and pressure delivered can be set.
Ventilator settings in proportional assist ventilation
Ventilator settings in PAV are:
- Airway type (endotracheal tube, tracheostomy)
- Airway size (inner diameter)
- Percentage of work supported (assist range 5%–95%)
- Tidal volume limit
- Pressure limit
- Expiratory sensitivity (normally, as inspiration ends, flow should stop; this parameter tells the ventilator at what flow to end inspiration).
Caution when assessing the literature. Earlier ventilator versions, ie, Dräger and Manitoba (University of Manitoba, Winnipeg, MB, Canada), which are not available in the United States, required the repeated calculation of the respiratory system mechanics and the manual setting of flow and volume assists (amplification factors) independently. To overcome this limitation, new software automatically adjusts the flow and volume amplification to support the loads imposed by the automatically measured values of resistance and elastance (inverse of compliance) of the respiratory system.21 This software is included in the model (Puritan Bennett) available in the United States.
Clinical applications of proportional assist ventilation
The PAV mode is indicated for maximizing ventilator patient synchrony for assisted spontaneous ventilation.
PAV is contraindicated in patients with respiratory depression (bradypnea) or large air leaks (eg, bronchopleural fistulas). It should be used with caution in patients with severe hyperinflation, in which the patient may still be exhaling but the ventilator doesn’t recognize it. Another group in which PAV should be used with caution is those with high ventilatory drives, in which the ventilator overestimates respiratory system mechanics. This situation can lead to overassistance due to the “runaway phenomenon,” in which the ventilator continues to provide support even if the patient has stopped inspiration.22
Theoretical benefits of proportional assist ventilation
In theory, PAV should reduce the work of breathing, improve synchrony, automatically adapt to changing patient lung mechanics and effort, decrease the need for ventilator intervention and manipulation, decrease the need for sedation, and improve sleep.
Evidence of benefit of proportional assist ventilation
Physiologic benefits. PAV reduces the work of breathing better than PSV,21 even in the face of changing respiratory mechanics or increased respiratory demand (hypercapnia).23–25 The hemodynamic profile is similar to that in PSV. Tidal volumes are variable; however, in recent reports the tidal volumes were within the lung-protective range (6–8 mL/kg, plateau pressure < 30 cm H20).26,27
Comfort. PAV entails less patient effort and discomfort that PSV does.23,25 PAV significantly reduces asynchrony,27 which in turn may favorably affect sleep in critically ill patients. 28
Outcomes. The probability of spontaneous breathing without assistance was significantly better in critically ill patients ventilated with PAV than with PSV. No trial has reported the effect of PAV on deaths.27,29
Proportional assist ventilation: Bottom line
Extensive basic research has been done with PAV in different forms of respiratory failure, such as obstructive lung disease, acute respiratory distress syndrome (ARDS), and chronic respiratory failure. It fulfills its main goal, which is to improve patient-ventilator synchrony. Clinical experience with PAV in the United States is limited, as it was only recently approved.
AIRWAY PRESSURE-RELEASE VENTILATION AND BIPHASIC POSITIVE AIRWAY PRESSURE
Airway pressure-release ventilation (APRV) was described in 1987 by Stock et al30 as a mode for delivering ventilation in acute lung injury while avoiding high airway pressures. APRV combines high constant positive airway pressure (improving oxygenation and promoting alveolar recruitment) with intermittent releases (causing exhalation).
Other names for biphasic positive airway pressure
Other names for biphasic positive airway pressure are:
- BiLevel (Puritan Bennett)
- BIPAP (Dräger Europe)
- Bi Vent (Siemens)
- BiPhasic (Avea, Cardinal Health, Inc, Dublin, OH)
- PCV+ (Dräger Medical)
- DuoPAP (Hamilton).
Caution—name confusion. In North America, BiPAP (Respironics, Murrysville, PA) and BiLevel are used to refer to noninvasive modes of ventilation.
APRV has no other name.
What do these modes do?
These modes deliver pressure-controlled, time-triggered, and time-cycled breaths using a set-point targeting scheme (Table 2). This means that the ventilator maintains a constant pressure (set point) even in the face of spontaneous breaths.
Caution—source of confusion. The term continuous positive airway pressure (CPAP) is often used to describe this mode. However, CPAP is pressure that is applied continuously at the same level; the patient generates all the work to maintain ventilation (“pressure-controlled continuous spontaneous ventilation” in the current nomenclature). In APRV, the airway pressure is intermittently released and reapplied, generating a tidal volume that supports ventilation. In other words, this is a pressure-controlled breath with a very prolonged inspiratory time and a short expiratory time in which spontaneous ventilation is possible at any point (“pressure-controlled intermittent mandatory ventilation” in the current nomenclature).
How these modes are set in the ventilator may also be a source of confusion. To describe the time spent in high and low airway pressures, we use the terms Thigh and Tlow, respectively. By convention, the difference between APRV and biphasic mode is the duration of Tlow (< 1.5 sec for APRV).
Similarly, Phigh and Plow are used to describe the high and low airway pressure. To better understand this concept, you can create the same mode in conventional pressure-control ventilation by thinking of the Thigh as the inspiratory time, the Tlow as the expiratory time, the Phigh as inspiratory pressure, and the Plow as PEEP.
Hence, APRV is an extreme form of inverse ratio ventilation, with an inspiration-to-expiration ratio of 4:1. This means a patient spends most of the time in Phigh and Thigh, and exhalations are short (Tlow and Plow). In contrast, the biphasic mode uses conventional inspiration-expiration ratios (Figure 4).
As with any form of pressure control, the tidal volume is generated by airway pressure rising above baseline (ie, the end-expiratory value). Hence, to ensure an increase in minute ventilation, the mandatory breath rate must be increased (ie, decreasing Thigh, Tlow, or both) or the tidal volume must be increased (ie, increasing the difference between Phigh and Plow). This means that in APRV the Tlow has to happen more often (by increasing the number of breaths) or be more prolonged (allowing more air to exhale). Because unrestricted spontaneous breaths are permitted at any point of the cycle, the patient contributes to the total minute ventilation (usually 10%–40%).
In APRV and biphasic mode, the operator’s set time and pressure in inspiration and expiration will be delivered regardless of the patient’s breathing efforts—the patient’s spontaneous breath does not trigger a mechanical breath. Some ventilators have automatic adjustments to improve the trigger synchrony.
Ventilator settings in APRV and biphasic mode
These modes require the setting of two pressure levels (Phigh and Plow) and two time durations (Thigh and Tlow). One can add pressure support or automatic tube compensation to assist spontaneous breaths. The difference in Tlow generates differences in the Thigh:Tlow ratio: APRV has a short Tlow (an inspiration-expiration ratio of 4:1). Biphasic mode has a conventional inspiration-expiration ratio of 1:1 to 1:4.
Clinical applications
APRV is used in acute lung injury and ARDS. This mode should be used with caution or not at all in patients with obstructive lung disease or inappropriately increased respiratory drive.32–35
Biphasic mode is intended for both ventilation and weaning. In a patient who has poor respiratory effort or who is paralyzed, biphasic is identical to pressure-control/continuous mandatory ventilation.
Theoretical benefits of APRV and biphasic mode
Multiple benefits have been ascribed to these modes. In theory, APRV will maximize and maintain alveolar recruitment, improve oxygenation, lower inflation pressures, and decrease overinflation. Both APRV and biphasic, by preserving spontaneous breathing, will improve ventilation-perfusion matching and gas diffusion, improve the hemodynamic profile (less need for vasopressors, higher cardiac output, reduced ventricular workload, improved organ perfusion), and improve synchrony (decrease the work of breathing and the need for sedation).
Evidence of benefit of APRV and biphasic mode
APRV and biphasic are different modes. However studies evaluating their effects are combined. This is in part the result of the nomenclature confusion and different practice in different countries.36
Physiologic benefits. In studies, spontaneous breaths contributed to 10% to 40% of minute ventilation,37,38 improved ventilation of dependent areas of the lung, improved ventilation-perfusion match and recruitment,39 and improved hemodynamic profile.40
Patient comfort. These modes are thought to decrease the need for analgesia and sedation,38 but a recent trial showed no difference with pressure-controlled intermittent mandatory ventilation.41 Patient ventilator synchrony and comfort have not been studied.32,42
Outcomes. In small trials, these modes made no difference in terms of deaths, but they may decrease the length of mechanical ventilation.38,41,43,44
APRV and biphasic mode: Bottom line
Maintaining spontaneous breathing while on mechanical ventilation has hemodynamic and ventilatory benefits.
APRV and biphasic mode are not the same thing. APRV’s main goal is to maximize mean airway pressure and, hence, lung recruitment, whereas the main goal of the biphasic mode is synchrony.
There is a plethora of ventilator settings and questions related to physiologic effects.33,34,36
Although these modes are widely used in some centers, there is no evidence yet that they are superior to conventional volume- or pressure-control ventilation with low tidal volume for ARDS and acute lung injury. There is no conclusive evidence that these modes improve synchrony, time to weaning, or patient comfort.
HIGH-FREQUENCY OSCILLATORY VENTILATION
High-frequency oscillatory ventilation (HFOV) was first described and patented in 1952 by Emerson and was clinically developed in the early 1970s by Lunkenheimer.45
The goal of HFOV is to minimize lung injury; its characteristics (discussed below) make it useful in patients with severe ARDS. The US Food and Drug Administration approved it for infants in 1991 and for children in 1995. The adult model has been available since 1993, but it was not approved until 2001 (SensorMedics 3100B, Cardinal Health, Inc).
Other names for high-frequency oscillatory ventilation
While HFOV has no alternative names, the following acronyms describe similar modes:
- HFPPV (high-frequency positive pressure ventilation)
- HFJV (high-frequency jet ventilation)
- HFFI (high-frequency flow interruption)
- HFPV (high-frequency percussive ventilation)
- HFCWO (high-frequency chest wall oscillation).
All of these modes require different specialized ventilators.
What does high-frequency oscillatory ventilation do?
Conceptually, HFOV is a form of pressure-controlled intermittent mandatory ventilation with a set-point control scheme. In contrast to conventional pressure-controlled intermittent mandatory ventilation, in which relatively small spontaneous breaths may be superimposed on relatively large mandatory breaths, HFOV superimposes very small mandatory breaths (oscillations) on top of spontaneous breaths.
Adult patients are usually paralyzed or deeply sedated, since deep spontaneous breathing will trigger alarms and affect ventilator performance.
To manage ventilation (CO2 clearance), one or several of the following maneuvers can be done: decrease the oscillation frequency, increase the amplitude of the oscillations, increase the inspiratory time, or increase bias flow (while allowing an endotracheal tube cuff leak). Oxygenation adjustments are controlled by manipulating the mean airway pressure and the Fio2.
Ventilator settings in high-frequency oscillatory ventilation
Ventilator settings in HFOV are46:
- Airway pressure amplitude (delta P or power)
- Mean airway pressure
- Percent inspiration
- Inspiratory bias flow
- Fio2.
Clinical applications of high-frequency oscillatory ventilation
This mode is usually reserved for ARDS patients for whom conventional ventilation is failing. A recently published protocol46 suggests considering HFOV when there is oxygenation failure (Fio2 ≥ 0.7 and PEEP ≥ 14 cm H2O) or ventilation failure (pH < 7.25 with tidal volume ≥ 6 mL/kg predicted body weight and plateau airway pressure ≥ 30 cm H2O).
This mode is contraindicated when there is known severe airflow obstruction or intracranial hypertension.
Theoretical benefits of high-frequency oscillatory ventilation
Conceptually, HFOV can provide the highest mean airway pressure paired with the lowest tidal volume of any mode. These benefits might make HFOV the ideal lung-protective ventilation strategy.
Evidence of benefit of high-frequency oscillatory ventilation
Physiologic benefits. Animal models have shown less histologic damage and lung inflammation with HFOV than with high-tidal-volume conventional ventilation47,48 and low-tidal-volume conventional ventilation.49
Patient comfort has not been studied. However, current technology does impose undue work of breathing in spontaneously breathing patients.50
Outcomes. Several retrospective case series have described better oxygenation with HFOV as rescue therapy for severe ARDS than with conventional mechanical ventilation. Two randomized controlled trials have studied HFOV vs high-tidal-volume conventional mechanical ventilation for early severe ARDS; HFOV was safe but made no difference in terms of deaths.42,51–54
High-frequency oscillatory ventilation: Bottom line
In theory, HFOV provides all the benefits of an ideal lung-protective strategy, at least for paralyzed or deeply sedated patients. Animal studies support these concepts. In human adults, HFOV has been shown to be safe and to provide better oxygenation but no improvement in death rates compared with conventional mechanical ventilation. Currently, HFOV is better reserved for patients with severe ARDS for whom conventional mechanical ventilation is failing.
- Chatburn RL. Classification of ventilator modes: update and proposal for implementation. Respir Care 2007; 52:301–323.
- Chatburn RL. Computer control of mechanical ventilation. Respir Care 2004; 49:507–517.
- Alvarez A, Subirana M, Benito S. Decelerating flow ventilation effects in acute respiratory failure. J Crit Care 1998; 13:21–25.
- Guldager H, Nielsen SL, Carl P, Soerensen MB. A comparison of volume control and pressure regulated volume control ventilation in acute respiratory failure. Crit Care 1997; 1:75–77.
- Kallet RH, Campbell AR, Dicker RA, Katz JA, Mackersie RC. Work of breathing during lung protective ventilation in patients with acute lung injury and acute respiratory distress syndrome: a comparison between volume and pressure regulated breathing modes. Respir Care 2005; 50:1623–1631.
- Betensley AD, Khalid I, Crawford J, Pensler RA, DiGiovine B. Patient comfort during pressure support and volume controlled continuous mandatory ventilation. Respir Care 2008; 53:897–902.
- Branson RD, Chatburn RL. Controversies in the critical care setting. Should adaptive pressure control modes be utilized for virtually all patients receiving mechanical ventilation? Respir Care 2007; 52:478–485.
- Laubscher TP, Frutiger A, Fanconi S, Jutzi H, Brunner JX. Automatic selection of tidal volume, respiratory frequency and minute ventilation in intubated ICU patients as start up procedure for closed-loop controlled ventilation. Int J Clin Monit Comput 1994; 11:19–30.
- Laubscher TP, Heinrichs W, Weiler N, Hartmann G, Brunner JX. An adaptive lung ventilation controller. IEEE Trans Biomed Eng 1994; 41:51–59.
- Arnal JM, Wysocki M, Nafati C, et al. Automatic selection of breathing pattern using adaptive support ventilation. Intensive Care Med 2008; 34:75–81.
- Campbell RS, Sinamban RP, Johannigman JA, et al. Clinical evaluation of a new closed loop ventilation mode: adaptive supportive ventilation (ASV). Crit Care 1999; 3( suppl 1):083.
- Belliato M, Palo A, Pasero D, Iotti GA, Mojoli F, Braschi A. Evaluation of adaptive support ventilation in paralysed patients and in a physical lung model. Int J Artif Organs 2004; 27:709–716.
- Tassaux D, Dalmas E, Gratadour P, Jolliet P. Patient ventilator interactions during partial ventilatory support: a preliminary study comparing the effects of adaptive support ventilation with synchronized intermittent mandatory ventilation plus inspiratory pressure support. Crit Care Med 2002; 30:801–807.
- Gruber PC, Gomersall CD, Leung P, et al. Randomized controlled trial comparing adaptive-support ventilation with pressure-regulated volume-controlled ventilation with automode in weaning patients after cardiac surgery. Anesthesiology 2008; 109:81–87.
- Sulzer CF, Chiolero R, Chassot PG, et al. Adaptive support ventilation for fast tracheal extubation after cardiac surgery: a randomized controlled study. Anesthesiology 2001; 95:1339–1345.
- Petter AH, Chiolèro RL, Cassina T, Chassot PG, Müller XM, Revelly JP. Automatic “respirator/weaning” with adaptive support ventilation: the effect on duration of endotracheal intubation and patient management. Anesth Analg 2003; 97:1743–1750.
- Brunner JX, Iotti GA. Adaptive support ventilation (ASV). Minerva Anestesiol 2002; 68:365–368.
- Campbell RS, Branson RD, Johannigman JA. Adaptive support ventilation. Respir Care Clin North Am 2001; 7:425–440.
- Younes M. Proportional assist ventilation, a new approach to ventilatory support. Theory. Am Rev Respir Dis 1992; 145:114–120.
- Younes M, Puddy A, Roberts D, et al. Proportional assist ventilation. Results of an initial clinical trial. Am Rev Respir Dis 1992; 145:121–129.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Respiratory load compensation during mechanical ventilatio—proportional assist ventilation with load-adjustable gain factors versus pressure support. Intensive Care Med 2006; 32:692–699.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Effect of different levels of pressure support and proportional assist ventilation on breathing pattern, work of breathing and gas exchange in mechanically ventilated hypercapnic COPD patients with acute respiratory failure. Respiration 2003; 70:355–361.
- Grasso S, Puntillo F, Mascia L, et al. Compensation for increase in respiratory workload during mechanical ventilation. Pressure support versus proportional assist ventilation. Am J Respir Crit Care Med 2000; 161:819–826.
- Wrigge H, Golisch W, Zinserling J, Sydow M, Almeling G, Burchardi H. Proportional assist versus pressure support ventilation: effects on breathing pattern and respiratory work of patients with chronic obstructive pulmonary disease. Intensive Care Med 1999; 25:790–798.
- Ranieri VM, Giuliani R, Mascia L, et al. Patient ventilator interaction during acute hypercapnia: pressure support vs. proportional assist ventilation. J Appl Physiol 1996; 81:426–436.
- Kondili E, Xirouchaki N, Vaporidi K, Klimathianaki M, Georgopoulos D. Short-term cardiorespiratory effects of proportional assist and pressure support ventilation in patients with acute lung injury/acute respiratory distress syndrome. Anesthesiology 2006; 105:703–708.
- Xirouchaki N, Kondili E, Vaporidi K, et al. Proportional assist ventilation with load-adjustable gain factors in critically ill patients: comparison with pressure support. Intensive Care Med 2008; 34:2026–2034.
- Bosma K, Ferreyra G, Ambrogio C, et al. Patient ventilator interaction and sleep in mechanically ventilated patients: pressure support versus proportional assist ventilation. Crit Care Med 2007; 35:1048–1054.
- Sinderby C, Beck J. Proportional assist ventilation and neurally adjusted ventilatory assist—better approaches to patient ventilator synchrony? Clin Chest Med 2008; 29:329–342.
- Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med 1987; 15:462–466.
- Baum M, Benzer H, Putensen C, Koller W, Putz G. [Biphasic positive airway pressure (BIPAP)—a new form of augmented ventilation]. Anaesthesist 1989; 38:452–458.
- Seymour CW, Frazer M, Reilly PM, Fuchs BD. Airway pressure release and biphasic intermittent positive airway pressure ventilation: are they ready for prime time? J Trauma 2007; 62:1298–1308.
- Myers TR, MacIntyre NR. Respiratory controversies in the critical care setting. Does airway pressure release ventilation offer important new advantages in mechanical ventilator support? Respir Care 2007; 52:452–458.
- Neumann P, Golisch W, Strohmeyer A, Buscher H, Burchardi H, Sydow M. Influence of different release times on spontaneous breathing pattern during airway pressure release ventilation. Intensive Care Med 2002; 28:1742–1749.
- Calzia E, Lindner KH, Witt S, et al. Pressure-time product and work of breathing during biphasic continuous positive airway pressure and assisted spontaneous breathing. Am J Respir Crit Care Med 1994; 150:904–910.
- Rose L, Hawkins M. Airway pressure release ventilation and biphasic positive airway pressure: a systematic review of definitional criteria. Intensive Care Med 2008; 34:1766–1773.
- Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Longterm effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med 1994; 149:1550–1556.
- Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001; 164:43–49.
- Davis K, Johnson DJ, Branson RD, Campbell RS, Johannigman JA, Porembka D. Airway pressure release ventilation. Arch Surg 1993; 128:1348–1352.
- Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001; 5:221–226.
- Varpula T, Valta P, Niemi R, Takkunen O, Hynynen M, Pettilä VV. Airway pressure release ventilation as a primary ventilatory mode in acute respiratory distress syndrome. Acta Anaesthesiol Scand 2004; 48:722–731.
- Siau C, Stewart TE. Current role of high frequency oscillatory ventilation and airway pressure release ventilation in acute lung injury and acute respiratory distress syndrome. Clin Chest Med 2008; 29:265–275.
- Rathgeber J, Schorn B, Falk V, Kazmaier S, Spiegel T, Burchardi H. The influence of controlled mandatory ventilation (CMV), intermittent mandatory ventilation (IMV) and biphasic intermittent positive airway pressure (BIPAP) on duration of intubation and consumption of analgesics and sedatives. A prospective analysis in 596 patients following adult cardiac surgery. Eur J Anaesthesiol 1997; 14:576–582.
- Habashi NM. Other approaches to open lung ventilation: airway pressure release ventilation. Crit Care Med 2005; 33 suppl 3:S228–S240.
- Hess D, Mason S, Branson R. High-frequency ventilation design and equipment issues. Respir Care Clin North Am 2001; 7:577–598.
- Fessler HE, Derdak S, Ferguson ND, et al. A protocol for high frequency oscillatory ventilation in adults: results from a roundtable discussion. Crit Care Med 2007; 35:1649–1654.
- Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol 1983; 55:131–138.
- Sedeek KA, Takeuchi M, Suchodolski K, et al. Open-lung protective ventilation with pressure control ventilation, high-frequency oscillation, and intratracheal pulmonary ventilation results in similar gas exchange, hemodynamics, and lung mechanics. Anesthesiology 2003; 99:1102–1111.
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol 2001; 91:1836–1844.
- van Heerde M, Roubik K, Kopelent V, Plötz FB, Markhorst DG. Unloading work of breathing during high-frequency oscillatory ventilation: a bench study. Crit Care 2006; 10:R103.
- Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med 2002; 166:801–808.
- Bollen CW, van Well GT, Sherry T, et al. High-frequency oscillatory ventilation compared with conventional mechanical ventilation in adult respiratory distress syndrome: a randomized controlled trial [ISRCTN24242669]. Crit Care 2005; 9:R430–R439.
- Mehta S, Granton J, MacDonald RJ, et al. High frequency oscillatory ventilation in adults: the Toronto experience. Chest 2004; 126:518–527.
- Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest 2007; 131:1907–1916.
Technologic advances and computerized control of mechanical ventilators have made it possible to deliver ventilatory assistance in new modes. Driving these innovations is the desire to prevent ventilator-induced lung injury, improve patient comfort, and liberate the patient from mechanical ventilation as soon as possible.
We call these innovations “alternative” modes to differentiate them from the plain volume-control and pressure-control modes. Some clinicians rarely use these new modes, but in some medical centers they have become the most common ones used, or are being used unknowingly (the operator misunderstands the mode name). The information we provide on these modes of ventilation is by no means an endorsement of their use, but rather a tool to help the clinician understand their physiologic, theoretical, and clinical effects.
We focused on two goals:
- Explain what the mode does
- Briefly review the theoretical benefits and the actual evidence supporting these alternative modes of ventilation.
STANDARD NOMENCLATURE NEEDED
Since its invention, mechanical ventilation has been plagued by multiple names being used to describe the same things. For example, volume-control ventilation is also called volume-cycled ventilation, assist-control ventilation, volume-limited ventilation, and controlled mechanical ventilation. Similarly, multiple abbreviations are used, each depending on the brand of ventilator, and new acronyms have been added in recent years as new modes have been developed. The vast number of names and modes can confuse even the most seasoned critical care physician.
Efforts to establish a common nomenclature are under way.1
WHAT IS A MODE?
A mode of mechanical ventilation has three essential components:
- The control variable
- The breath sequence
- The targeting scheme.
Similar modes may require more detailed descriptions to distinguish them, but the basic function can be explained by these three components.
The control variable
In general, inspiration is an active process, driven by the patient’s effort, the ventilator, or both, while expiration is passive. For simplicity, in this article a mechanical breath means the inspiratory phase of the breath.
The machine can only control the volume (and flow) or the pressure given. The breaths can be further described on the basis of what triggers the breath, what limits it (the maximum value of a control variable), and what ends (cycles) it.
The breath sequence
There are three possible breath sequences:
- Intermittent mandatory ventilation, in which the patient can take spontaneous breaths between mandatory breaths
- Continuous spontaneous ventilation, in which all breaths are spontaneous (Table 1).
The targeting scheme
The targeting or feedback scheme refers to the ventilator settings and programming that dictate its response to the patient’s lung compliance, lung resistance, and respiratory effort. The regulation can be as simple as controlling the pressure in pressure-control mode, or it can be based on a complicated algorithm.
ADAPTIVE PRESSURE CONTROL
Other names for adaptive pressure control
- Pressure Regulated Volume Control (Maquet Servo-i, Rastatt, Germany)
- AutoFlow (Dräger Medical AG, Lübeck, Germany)
- Adaptive Pressure Ventilation (Hamilton Galileo, Hamilton Medical AG, Bonaduz, Switzerland)
- Volume Control+ (Puritan Bennett, Tyco Healthcare; Mansfield, MA)
- Volume Targeted Pressure Control, Pressure Controlled Volume Guaranteed (Engström, General Electric, Madison, WI).
What does adaptive pressure control do?
The APC mode delivers pressure-controlled breaths with an adaptive targeting scheme (Table 2).
In pressure-control ventilation, tidal volumes depend on the lung’s physiologic mechanics (compliance and resistance) and patient effort (Figure 1). Therefore, the tidal volume varies with changes in lung physiology (ie, larger or smaller tidal volumes than targeted).
To overcome this effect, a machine in APC mode adjusts the inspiratory pressure to deliver the set minimal target tidal volume. If tidal volume increases, the machine decreases the inspiratory pressure, and if tidal volume decreases, the machine increases the inspiratory pressure. However, if the patient effort is large enough, the tidal volume will increase in spite of decreasing the inspiratory pressure (Figure 2). The adjustments to the inspiratory pressure occur after the tidal volume is off-target in a number of breaths.
Common sources of confusion with adaptive pressure control
First, APC is not a volume-control mode. In volume control, the tidal volume does not change; in APC the tidal volume can increase or decrease, and the ventilator will adjust the inflation pressure to achieve the target volume. Thus, APC guarantees an average minimum tidal volume but not a maximum tidal volume.
Second, a characteristic of pressure control (and hence, APC) is that the flow of gas varies to maintain constant airway pressure (ie, maintain the set inspiratory pressure). This characteristic allows a patient who generates an inspiratory effort to receive flow as demanded, which is likely more comfortable. This is essentially different from volume control, in which flow is set by the operator and hence is fixed. Thus, if the patient effort is strong enough (Figure 1), this leads to what is called flow asynchrony, in which the patient does not get the flow asked for in a breath.
Ventilator settings in adaptive pressure control
Ventilator settings in APC are:
- Tidal volume
- Time spent in inspiration (inspiratory time)
- Frequency
- Fraction of inspired oxygen (Fio2)
- Positive end-expiratory pressure (PEEP).
Some ventilators also require setting the speed to reach the peak pressure (also known as slope percent or inspiratory rise time).
Clinical applications of adaptive pressure control
This mode is designed to maintain a consistent tidal volume during pressure-control ventilation and to promote inspiratory flow synchrony. It is a means of automatically reducing ventilatory support (ie, weaning) as the patient’s inspiratory effort becomes stronger, as in awakening from anesthesia.
APC may not be ideal for patients who have an inappropriately increased respiratory drive (eg, in severe metabolic acidosis), since the inspiratory pressure will decrease to maintain the targeted average tidal volume, inappropriately shifting the work of breathing onto the patient.
Theoretical benefits of adaptive pressure control
APC guarantees a minimum average tidal volume (unless the pressure alarm threshold is set too low, so that the target tidal volume is not delivered). Other theoretical benefits are flow synchrony, less ventilator manipulation by the operator, and automatic weaning of ventilator support.
Evidence of benefit of adaptive pressure control
Physiologic benefits. This mode has lower peak inspiratory pressures than does volume-control ventilation,3,4 which is often reported as a positive finding. However, in volume-control mode (the usual comparator), the peak inspiratory pressure is a manifestation of both resistance and compliance. Hence, peak inspiratory pressure is expected to be higher but does not reflect actual lung-distending pressures. It is the plateau pressure, a manifestation of lung compliance, that is related to lung injury.
Patient comfort. APC may increase the work of breathing when using low tidal volume ventilation and when there is increased respiratory effort (drive).5 Interestingly, APC was less comfortable than pressure support ventilation in a small trial.6
Outcomes have not been studied.7
Adaptive pressure control: Bottom line
APC is widely available and widely used, sometimes unknowingly (eg, if the operator thinks it is volume control). It is relatively easy to use and to set; however, evidence of its benefit is scant.
ADAPTIVE SUPPORT VENTILATION
Adaptive support ventilation (ASV) evolved as a form of mandatory minute ventilation implemented with adaptive pressure control. Mandatory minute ventilation is a mode that allows the operator to preset a target minute ventilation, and the ventilator then supplies mandatory breaths, either volume- or pressure-controlled, if the patient’s spontaneous breaths generate a lower minute ventilation.
ASV automatically selects the appropriate tidal volume and frequency for mandatory breaths and the appropriate tidal volume for spontaneous breaths on the basis of the respiratory system mechanics and target minute alveolar ventilation.
Described in 1994 by Laubscher et al,8,9 ASV became commercially available in 1998 in Europe and in 2007 in the United States (Hamilton Galileo ventilator, Hamilton Medical AG). This is the first commercially available ventilator that uses an “optimal” targeting scheme (see below).
What does adaptive support ventilation do?
ASV delivers pressure-controlled breaths using an adaptive (optimal) scheme (Table 2). “Optimal,” in this context, means minimizing the mechanical work of breathing: the machine selects a tidal volume and frequency that the patient’s brain would presumably select if the patient were not connected to a ventilator. This pattern is assumed to encourage the patient to generate spontaneous breaths.
The ventilator calculates the normal required minute ventilation based on the patient’s ideal weight and estimated dead space volume (ie, 2.2 mL/kg). This calculation represents 100% of minute ventilation. The clinician at the bedside sets a target percent of minute ventilation that the ventilator will support—higher than 100% if the patient has increased requirements due, eg, to sepsis or increased dead space, or less than 100% during weaning.
The ventilator initially delivers test breaths, in which it measures the expiratory time constant for the respiratory system and then uses this along with the estimated dead space and normal minute ventilation to calculate an optimal breathing frequency in terms of mechanical work.
The optimal or target tidal volume is calculated as the normal minute ventilation divided by the optimal frequency. The target tidal volume is achieved by the use of APC (see above) (Figure 2). This means that the pressure limit is automatically adjusted to achieve an average delivered tidal volume equal to the target. The ventilator continuously monitors the respiratory system mechanics and adjusts its settings accordingly.
The ventilator adjusts its breaths to avoid air trapping by allowing enough time to exhale, to avoid hypoventilation by delivering tidal volume greater than the dead space, and to avoid volutrauma by avoiding large tidal volumes.
Ventilator settings in adaptive support ventilation
Ventilator settings in ASV are:
- Patient height (to calculate the ideal body weight)
- Sex
- Percent of normal predicted minute ventilation goal
- Fio2
- PEEP.
Clinical applications of adaptive support ventilation
ASV is intended as a sole mode of ventilation, from initial support to weaning.
Theoretical benefits of adaptive support ventilation
In theory, ASV offers automatic selection of ventilator settings, automatic adaptation to changing patient lung mechanics, less need for human manipulation of the machine, improved synchrony, and automatic weaning.
Evidence of benefit of adaptive support ventilation
Physiologic benefits. Ventilator settings are adjusted automatically. ASV selects different tidal volume-respiratory rate combinations based on respiratory mechanics in passive and paralyzed patients.10–12 In actively breathing patients, there was no difference in the ventilator settings chosen by ASV for different clinical scenarios (and lung physiology).10 Compared with pressure-controlled intermittent mandatory ventilation, with ASV, the inspiratory load is less and patient-ventilator interaction is better.13
Patient-ventilator synchrony and comfort have not been studied.
Outcomes. Two trials suggest that ASV may decrease time on mechanical ventilation.14,15 However, in another trial,16 compared with a standard protocol, ASV led to fewer ventilator adjustments but achieved similar postsurgical weaning outcomes. The effect of this mode on the death rate has not been examined.17,18
Adaptive support ventilation: Bottom line
ASV is the first commercially available mode that automatically selects all the ventilator settings except PEEP and Fio2. These seem appropriate for different clinical scenarios in patients with poor respiratory effort or in paralyzed patients. Evidence of the effect in actively breathing patients and on outcomes such as length of stay or death is still lacking.
PROPORTIONAL ASSIST VENTILATION
Patients who have normal respiratory drive but who have difficulty sustaining adequate spontaneous ventilation are often subjected to pressure support ventilation (PSV), in which the ventilator generates a constant pressure throughout inspiration regardless of the intensity of the patient’s effort.
In 1992, Younes and colleagues19,20 developed proportional assist ventilation (PAV) as an alternative in which the ventilator generates pressure in proportion to the patient’s effort. PAV became commercially available in Europe in 1999 and was approved in the United States in 2006, available on the Puritan Bennett 840 ventilator (Puritan Bennett Co, Boulder, CO). PAV has also been used for noninvasive ventilation, but this is not available in the United States.
Other names for proportional assist ventilation
Proportional Pressure Support (Dräger Medical; not yet available in the United States).
What does proportional assist ventilation do?
This mode delivers pressure-controlled breaths with a servo control scheme (Table 2).
To better understand PAV, we can compare it with PSV. With PSV, the pressure applied by the ventilator rises to a preset level that is held constant (a set-point scheme) until a cycling criterion (a percent of the maximum inspiratory flow value) is reached. The inspiratory flow and tidal volume are the result of the patient’s inspiratory effort, the level of pressure applied, and the respiratory system mechanics.
In PAV, as in PSV, all breaths are spontaneous (Table 1). The patient controls the timing and size of the breath. There are no preset pressure, flow, or volume goals, but safety limits on the volume and pressure delivered can be set.
Ventilator settings in proportional assist ventilation
Ventilator settings in PAV are:
- Airway type (endotracheal tube, tracheostomy)
- Airway size (inner diameter)
- Percentage of work supported (assist range 5%–95%)
- Tidal volume limit
- Pressure limit
- Expiratory sensitivity (normally, as inspiration ends, flow should stop; this parameter tells the ventilator at what flow to end inspiration).
Caution when assessing the literature. Earlier ventilator versions, ie, Dräger and Manitoba (University of Manitoba, Winnipeg, MB, Canada), which are not available in the United States, required the repeated calculation of the respiratory system mechanics and the manual setting of flow and volume assists (amplification factors) independently. To overcome this limitation, new software automatically adjusts the flow and volume amplification to support the loads imposed by the automatically measured values of resistance and elastance (inverse of compliance) of the respiratory system.21 This software is included in the model (Puritan Bennett) available in the United States.
Clinical applications of proportional assist ventilation
The PAV mode is indicated for maximizing ventilator patient synchrony for assisted spontaneous ventilation.
PAV is contraindicated in patients with respiratory depression (bradypnea) or large air leaks (eg, bronchopleural fistulas). It should be used with caution in patients with severe hyperinflation, in which the patient may still be exhaling but the ventilator doesn’t recognize it. Another group in which PAV should be used with caution is those with high ventilatory drives, in which the ventilator overestimates respiratory system mechanics. This situation can lead to overassistance due to the “runaway phenomenon,” in which the ventilator continues to provide support even if the patient has stopped inspiration.22
Theoretical benefits of proportional assist ventilation
In theory, PAV should reduce the work of breathing, improve synchrony, automatically adapt to changing patient lung mechanics and effort, decrease the need for ventilator intervention and manipulation, decrease the need for sedation, and improve sleep.
Evidence of benefit of proportional assist ventilation
Physiologic benefits. PAV reduces the work of breathing better than PSV,21 even in the face of changing respiratory mechanics or increased respiratory demand (hypercapnia).23–25 The hemodynamic profile is similar to that in PSV. Tidal volumes are variable; however, in recent reports the tidal volumes were within the lung-protective range (6–8 mL/kg, plateau pressure < 30 cm H20).26,27
Comfort. PAV entails less patient effort and discomfort that PSV does.23,25 PAV significantly reduces asynchrony,27 which in turn may favorably affect sleep in critically ill patients. 28
Outcomes. The probability of spontaneous breathing without assistance was significantly better in critically ill patients ventilated with PAV than with PSV. No trial has reported the effect of PAV on deaths.27,29
Proportional assist ventilation: Bottom line
Extensive basic research has been done with PAV in different forms of respiratory failure, such as obstructive lung disease, acute respiratory distress syndrome (ARDS), and chronic respiratory failure. It fulfills its main goal, which is to improve patient-ventilator synchrony. Clinical experience with PAV in the United States is limited, as it was only recently approved.
AIRWAY PRESSURE-RELEASE VENTILATION AND BIPHASIC POSITIVE AIRWAY PRESSURE
Airway pressure-release ventilation (APRV) was described in 1987 by Stock et al30 as a mode for delivering ventilation in acute lung injury while avoiding high airway pressures. APRV combines high constant positive airway pressure (improving oxygenation and promoting alveolar recruitment) with intermittent releases (causing exhalation).
Other names for biphasic positive airway pressure
Other names for biphasic positive airway pressure are:
- BiLevel (Puritan Bennett)
- BIPAP (Dräger Europe)
- Bi Vent (Siemens)
- BiPhasic (Avea, Cardinal Health, Inc, Dublin, OH)
- PCV+ (Dräger Medical)
- DuoPAP (Hamilton).
Caution—name confusion. In North America, BiPAP (Respironics, Murrysville, PA) and BiLevel are used to refer to noninvasive modes of ventilation.
APRV has no other name.
What do these modes do?
These modes deliver pressure-controlled, time-triggered, and time-cycled breaths using a set-point targeting scheme (Table 2). This means that the ventilator maintains a constant pressure (set point) even in the face of spontaneous breaths.
Caution—source of confusion. The term continuous positive airway pressure (CPAP) is often used to describe this mode. However, CPAP is pressure that is applied continuously at the same level; the patient generates all the work to maintain ventilation (“pressure-controlled continuous spontaneous ventilation” in the current nomenclature). In APRV, the airway pressure is intermittently released and reapplied, generating a tidal volume that supports ventilation. In other words, this is a pressure-controlled breath with a very prolonged inspiratory time and a short expiratory time in which spontaneous ventilation is possible at any point (“pressure-controlled intermittent mandatory ventilation” in the current nomenclature).
How these modes are set in the ventilator may also be a source of confusion. To describe the time spent in high and low airway pressures, we use the terms Thigh and Tlow, respectively. By convention, the difference between APRV and biphasic mode is the duration of Tlow (< 1.5 sec for APRV).
Similarly, Phigh and Plow are used to describe the high and low airway pressure. To better understand this concept, you can create the same mode in conventional pressure-control ventilation by thinking of the Thigh as the inspiratory time, the Tlow as the expiratory time, the Phigh as inspiratory pressure, and the Plow as PEEP.
Hence, APRV is an extreme form of inverse ratio ventilation, with an inspiration-to-expiration ratio of 4:1. This means a patient spends most of the time in Phigh and Thigh, and exhalations are short (Tlow and Plow). In contrast, the biphasic mode uses conventional inspiration-expiration ratios (Figure 4).
As with any form of pressure control, the tidal volume is generated by airway pressure rising above baseline (ie, the end-expiratory value). Hence, to ensure an increase in minute ventilation, the mandatory breath rate must be increased (ie, decreasing Thigh, Tlow, or both) or the tidal volume must be increased (ie, increasing the difference between Phigh and Plow). This means that in APRV the Tlow has to happen more often (by increasing the number of breaths) or be more prolonged (allowing more air to exhale). Because unrestricted spontaneous breaths are permitted at any point of the cycle, the patient contributes to the total minute ventilation (usually 10%–40%).
In APRV and biphasic mode, the operator’s set time and pressure in inspiration and expiration will be delivered regardless of the patient’s breathing efforts—the patient’s spontaneous breath does not trigger a mechanical breath. Some ventilators have automatic adjustments to improve the trigger synchrony.
Ventilator settings in APRV and biphasic mode
These modes require the setting of two pressure levels (Phigh and Plow) and two time durations (Thigh and Tlow). One can add pressure support or automatic tube compensation to assist spontaneous breaths. The difference in Tlow generates differences in the Thigh:Tlow ratio: APRV has a short Tlow (an inspiration-expiration ratio of 4:1). Biphasic mode has a conventional inspiration-expiration ratio of 1:1 to 1:4.
Clinical applications
APRV is used in acute lung injury and ARDS. This mode should be used with caution or not at all in patients with obstructive lung disease or inappropriately increased respiratory drive.32–35
Biphasic mode is intended for both ventilation and weaning. In a patient who has poor respiratory effort or who is paralyzed, biphasic is identical to pressure-control/continuous mandatory ventilation.
Theoretical benefits of APRV and biphasic mode
Multiple benefits have been ascribed to these modes. In theory, APRV will maximize and maintain alveolar recruitment, improve oxygenation, lower inflation pressures, and decrease overinflation. Both APRV and biphasic, by preserving spontaneous breathing, will improve ventilation-perfusion matching and gas diffusion, improve the hemodynamic profile (less need for vasopressors, higher cardiac output, reduced ventricular workload, improved organ perfusion), and improve synchrony (decrease the work of breathing and the need for sedation).
Evidence of benefit of APRV and biphasic mode
APRV and biphasic are different modes. However studies evaluating their effects are combined. This is in part the result of the nomenclature confusion and different practice in different countries.36
Physiologic benefits. In studies, spontaneous breaths contributed to 10% to 40% of minute ventilation,37,38 improved ventilation of dependent areas of the lung, improved ventilation-perfusion match and recruitment,39 and improved hemodynamic profile.40
Patient comfort. These modes are thought to decrease the need for analgesia and sedation,38 but a recent trial showed no difference with pressure-controlled intermittent mandatory ventilation.41 Patient ventilator synchrony and comfort have not been studied.32,42
Outcomes. In small trials, these modes made no difference in terms of deaths, but they may decrease the length of mechanical ventilation.38,41,43,44
APRV and biphasic mode: Bottom line
Maintaining spontaneous breathing while on mechanical ventilation has hemodynamic and ventilatory benefits.
APRV and biphasic mode are not the same thing. APRV’s main goal is to maximize mean airway pressure and, hence, lung recruitment, whereas the main goal of the biphasic mode is synchrony.
There is a plethora of ventilator settings and questions related to physiologic effects.33,34,36
Although these modes are widely used in some centers, there is no evidence yet that they are superior to conventional volume- or pressure-control ventilation with low tidal volume for ARDS and acute lung injury. There is no conclusive evidence that these modes improve synchrony, time to weaning, or patient comfort.
HIGH-FREQUENCY OSCILLATORY VENTILATION
High-frequency oscillatory ventilation (HFOV) was first described and patented in 1952 by Emerson and was clinically developed in the early 1970s by Lunkenheimer.45
The goal of HFOV is to minimize lung injury; its characteristics (discussed below) make it useful in patients with severe ARDS. The US Food and Drug Administration approved it for infants in 1991 and for children in 1995. The adult model has been available since 1993, but it was not approved until 2001 (SensorMedics 3100B, Cardinal Health, Inc).
Other names for high-frequency oscillatory ventilation
While HFOV has no alternative names, the following acronyms describe similar modes:
- HFPPV (high-frequency positive pressure ventilation)
- HFJV (high-frequency jet ventilation)
- HFFI (high-frequency flow interruption)
- HFPV (high-frequency percussive ventilation)
- HFCWO (high-frequency chest wall oscillation).
All of these modes require different specialized ventilators.
What does high-frequency oscillatory ventilation do?
Conceptually, HFOV is a form of pressure-controlled intermittent mandatory ventilation with a set-point control scheme. In contrast to conventional pressure-controlled intermittent mandatory ventilation, in which relatively small spontaneous breaths may be superimposed on relatively large mandatory breaths, HFOV superimposes very small mandatory breaths (oscillations) on top of spontaneous breaths.
Adult patients are usually paralyzed or deeply sedated, since deep spontaneous breathing will trigger alarms and affect ventilator performance.
To manage ventilation (CO2 clearance), one or several of the following maneuvers can be done: decrease the oscillation frequency, increase the amplitude of the oscillations, increase the inspiratory time, or increase bias flow (while allowing an endotracheal tube cuff leak). Oxygenation adjustments are controlled by manipulating the mean airway pressure and the Fio2.
Ventilator settings in high-frequency oscillatory ventilation
Ventilator settings in HFOV are46:
- Airway pressure amplitude (delta P or power)
- Mean airway pressure
- Percent inspiration
- Inspiratory bias flow
- Fio2.
Clinical applications of high-frequency oscillatory ventilation
This mode is usually reserved for ARDS patients for whom conventional ventilation is failing. A recently published protocol46 suggests considering HFOV when there is oxygenation failure (Fio2 ≥ 0.7 and PEEP ≥ 14 cm H2O) or ventilation failure (pH < 7.25 with tidal volume ≥ 6 mL/kg predicted body weight and plateau airway pressure ≥ 30 cm H2O).
This mode is contraindicated when there is known severe airflow obstruction or intracranial hypertension.
Theoretical benefits of high-frequency oscillatory ventilation
Conceptually, HFOV can provide the highest mean airway pressure paired with the lowest tidal volume of any mode. These benefits might make HFOV the ideal lung-protective ventilation strategy.
Evidence of benefit of high-frequency oscillatory ventilation
Physiologic benefits. Animal models have shown less histologic damage and lung inflammation with HFOV than with high-tidal-volume conventional ventilation47,48 and low-tidal-volume conventional ventilation.49
Patient comfort has not been studied. However, current technology does impose undue work of breathing in spontaneously breathing patients.50
Outcomes. Several retrospective case series have described better oxygenation with HFOV as rescue therapy for severe ARDS than with conventional mechanical ventilation. Two randomized controlled trials have studied HFOV vs high-tidal-volume conventional mechanical ventilation for early severe ARDS; HFOV was safe but made no difference in terms of deaths.42,51–54
High-frequency oscillatory ventilation: Bottom line
In theory, HFOV provides all the benefits of an ideal lung-protective strategy, at least for paralyzed or deeply sedated patients. Animal studies support these concepts. In human adults, HFOV has been shown to be safe and to provide better oxygenation but no improvement in death rates compared with conventional mechanical ventilation. Currently, HFOV is better reserved for patients with severe ARDS for whom conventional mechanical ventilation is failing.
Technologic advances and computerized control of mechanical ventilators have made it possible to deliver ventilatory assistance in new modes. Driving these innovations is the desire to prevent ventilator-induced lung injury, improve patient comfort, and liberate the patient from mechanical ventilation as soon as possible.
We call these innovations “alternative” modes to differentiate them from the plain volume-control and pressure-control modes. Some clinicians rarely use these new modes, but in some medical centers they have become the most common ones used, or are being used unknowingly (the operator misunderstands the mode name). The information we provide on these modes of ventilation is by no means an endorsement of their use, but rather a tool to help the clinician understand their physiologic, theoretical, and clinical effects.
We focused on two goals:
- Explain what the mode does
- Briefly review the theoretical benefits and the actual evidence supporting these alternative modes of ventilation.
STANDARD NOMENCLATURE NEEDED
Since its invention, mechanical ventilation has been plagued by multiple names being used to describe the same things. For example, volume-control ventilation is also called volume-cycled ventilation, assist-control ventilation, volume-limited ventilation, and controlled mechanical ventilation. Similarly, multiple abbreviations are used, each depending on the brand of ventilator, and new acronyms have been added in recent years as new modes have been developed. The vast number of names and modes can confuse even the most seasoned critical care physician.
Efforts to establish a common nomenclature are under way.1
WHAT IS A MODE?
A mode of mechanical ventilation has three essential components:
- The control variable
- The breath sequence
- The targeting scheme.
Similar modes may require more detailed descriptions to distinguish them, but the basic function can be explained by these three components.
The control variable
In general, inspiration is an active process, driven by the patient’s effort, the ventilator, or both, while expiration is passive. For simplicity, in this article a mechanical breath means the inspiratory phase of the breath.
The machine can only control the volume (and flow) or the pressure given. The breaths can be further described on the basis of what triggers the breath, what limits it (the maximum value of a control variable), and what ends (cycles) it.
The breath sequence
There are three possible breath sequences:
- Intermittent mandatory ventilation, in which the patient can take spontaneous breaths between mandatory breaths
- Continuous spontaneous ventilation, in which all breaths are spontaneous (Table 1).
The targeting scheme
The targeting or feedback scheme refers to the ventilator settings and programming that dictate its response to the patient’s lung compliance, lung resistance, and respiratory effort. The regulation can be as simple as controlling the pressure in pressure-control mode, or it can be based on a complicated algorithm.
ADAPTIVE PRESSURE CONTROL
Other names for adaptive pressure control
- Pressure Regulated Volume Control (Maquet Servo-i, Rastatt, Germany)
- AutoFlow (Dräger Medical AG, Lübeck, Germany)
- Adaptive Pressure Ventilation (Hamilton Galileo, Hamilton Medical AG, Bonaduz, Switzerland)
- Volume Control+ (Puritan Bennett, Tyco Healthcare; Mansfield, MA)
- Volume Targeted Pressure Control, Pressure Controlled Volume Guaranteed (Engström, General Electric, Madison, WI).
What does adaptive pressure control do?
The APC mode delivers pressure-controlled breaths with an adaptive targeting scheme (Table 2).
In pressure-control ventilation, tidal volumes depend on the lung’s physiologic mechanics (compliance and resistance) and patient effort (Figure 1). Therefore, the tidal volume varies with changes in lung physiology (ie, larger or smaller tidal volumes than targeted).
To overcome this effect, a machine in APC mode adjusts the inspiratory pressure to deliver the set minimal target tidal volume. If tidal volume increases, the machine decreases the inspiratory pressure, and if tidal volume decreases, the machine increases the inspiratory pressure. However, if the patient effort is large enough, the tidal volume will increase in spite of decreasing the inspiratory pressure (Figure 2). The adjustments to the inspiratory pressure occur after the tidal volume is off-target in a number of breaths.
Common sources of confusion with adaptive pressure control
First, APC is not a volume-control mode. In volume control, the tidal volume does not change; in APC the tidal volume can increase or decrease, and the ventilator will adjust the inflation pressure to achieve the target volume. Thus, APC guarantees an average minimum tidal volume but not a maximum tidal volume.
Second, a characteristic of pressure control (and hence, APC) is that the flow of gas varies to maintain constant airway pressure (ie, maintain the set inspiratory pressure). This characteristic allows a patient who generates an inspiratory effort to receive flow as demanded, which is likely more comfortable. This is essentially different from volume control, in which flow is set by the operator and hence is fixed. Thus, if the patient effort is strong enough (Figure 1), this leads to what is called flow asynchrony, in which the patient does not get the flow asked for in a breath.
Ventilator settings in adaptive pressure control
Ventilator settings in APC are:
- Tidal volume
- Time spent in inspiration (inspiratory time)
- Frequency
- Fraction of inspired oxygen (Fio2)
- Positive end-expiratory pressure (PEEP).
Some ventilators also require setting the speed to reach the peak pressure (also known as slope percent or inspiratory rise time).
Clinical applications of adaptive pressure control
This mode is designed to maintain a consistent tidal volume during pressure-control ventilation and to promote inspiratory flow synchrony. It is a means of automatically reducing ventilatory support (ie, weaning) as the patient’s inspiratory effort becomes stronger, as in awakening from anesthesia.
APC may not be ideal for patients who have an inappropriately increased respiratory drive (eg, in severe metabolic acidosis), since the inspiratory pressure will decrease to maintain the targeted average tidal volume, inappropriately shifting the work of breathing onto the patient.
Theoretical benefits of adaptive pressure control
APC guarantees a minimum average tidal volume (unless the pressure alarm threshold is set too low, so that the target tidal volume is not delivered). Other theoretical benefits are flow synchrony, less ventilator manipulation by the operator, and automatic weaning of ventilator support.
Evidence of benefit of adaptive pressure control
Physiologic benefits. This mode has lower peak inspiratory pressures than does volume-control ventilation,3,4 which is often reported as a positive finding. However, in volume-control mode (the usual comparator), the peak inspiratory pressure is a manifestation of both resistance and compliance. Hence, peak inspiratory pressure is expected to be higher but does not reflect actual lung-distending pressures. It is the plateau pressure, a manifestation of lung compliance, that is related to lung injury.
Patient comfort. APC may increase the work of breathing when using low tidal volume ventilation and when there is increased respiratory effort (drive).5 Interestingly, APC was less comfortable than pressure support ventilation in a small trial.6
Outcomes have not been studied.7
Adaptive pressure control: Bottom line
APC is widely available and widely used, sometimes unknowingly (eg, if the operator thinks it is volume control). It is relatively easy to use and to set; however, evidence of its benefit is scant.
ADAPTIVE SUPPORT VENTILATION
Adaptive support ventilation (ASV) evolved as a form of mandatory minute ventilation implemented with adaptive pressure control. Mandatory minute ventilation is a mode that allows the operator to preset a target minute ventilation, and the ventilator then supplies mandatory breaths, either volume- or pressure-controlled, if the patient’s spontaneous breaths generate a lower minute ventilation.
ASV automatically selects the appropriate tidal volume and frequency for mandatory breaths and the appropriate tidal volume for spontaneous breaths on the basis of the respiratory system mechanics and target minute alveolar ventilation.
Described in 1994 by Laubscher et al,8,9 ASV became commercially available in 1998 in Europe and in 2007 in the United States (Hamilton Galileo ventilator, Hamilton Medical AG). This is the first commercially available ventilator that uses an “optimal” targeting scheme (see below).
What does adaptive support ventilation do?
ASV delivers pressure-controlled breaths using an adaptive (optimal) scheme (Table 2). “Optimal,” in this context, means minimizing the mechanical work of breathing: the machine selects a tidal volume and frequency that the patient’s brain would presumably select if the patient were not connected to a ventilator. This pattern is assumed to encourage the patient to generate spontaneous breaths.
The ventilator calculates the normal required minute ventilation based on the patient’s ideal weight and estimated dead space volume (ie, 2.2 mL/kg). This calculation represents 100% of minute ventilation. The clinician at the bedside sets a target percent of minute ventilation that the ventilator will support—higher than 100% if the patient has increased requirements due, eg, to sepsis or increased dead space, or less than 100% during weaning.
The ventilator initially delivers test breaths, in which it measures the expiratory time constant for the respiratory system and then uses this along with the estimated dead space and normal minute ventilation to calculate an optimal breathing frequency in terms of mechanical work.
The optimal or target tidal volume is calculated as the normal minute ventilation divided by the optimal frequency. The target tidal volume is achieved by the use of APC (see above) (Figure 2). This means that the pressure limit is automatically adjusted to achieve an average delivered tidal volume equal to the target. The ventilator continuously monitors the respiratory system mechanics and adjusts its settings accordingly.
The ventilator adjusts its breaths to avoid air trapping by allowing enough time to exhale, to avoid hypoventilation by delivering tidal volume greater than the dead space, and to avoid volutrauma by avoiding large tidal volumes.
Ventilator settings in adaptive support ventilation
Ventilator settings in ASV are:
- Patient height (to calculate the ideal body weight)
- Sex
- Percent of normal predicted minute ventilation goal
- Fio2
- PEEP.
Clinical applications of adaptive support ventilation
ASV is intended as a sole mode of ventilation, from initial support to weaning.
Theoretical benefits of adaptive support ventilation
In theory, ASV offers automatic selection of ventilator settings, automatic adaptation to changing patient lung mechanics, less need for human manipulation of the machine, improved synchrony, and automatic weaning.
Evidence of benefit of adaptive support ventilation
Physiologic benefits. Ventilator settings are adjusted automatically. ASV selects different tidal volume-respiratory rate combinations based on respiratory mechanics in passive and paralyzed patients.10–12 In actively breathing patients, there was no difference in the ventilator settings chosen by ASV for different clinical scenarios (and lung physiology).10 Compared with pressure-controlled intermittent mandatory ventilation, with ASV, the inspiratory load is less and patient-ventilator interaction is better.13
Patient-ventilator synchrony and comfort have not been studied.
Outcomes. Two trials suggest that ASV may decrease time on mechanical ventilation.14,15 However, in another trial,16 compared with a standard protocol, ASV led to fewer ventilator adjustments but achieved similar postsurgical weaning outcomes. The effect of this mode on the death rate has not been examined.17,18
Adaptive support ventilation: Bottom line
ASV is the first commercially available mode that automatically selects all the ventilator settings except PEEP and Fio2. These seem appropriate for different clinical scenarios in patients with poor respiratory effort or in paralyzed patients. Evidence of the effect in actively breathing patients and on outcomes such as length of stay or death is still lacking.
PROPORTIONAL ASSIST VENTILATION
Patients who have normal respiratory drive but who have difficulty sustaining adequate spontaneous ventilation are often subjected to pressure support ventilation (PSV), in which the ventilator generates a constant pressure throughout inspiration regardless of the intensity of the patient’s effort.
In 1992, Younes and colleagues19,20 developed proportional assist ventilation (PAV) as an alternative in which the ventilator generates pressure in proportion to the patient’s effort. PAV became commercially available in Europe in 1999 and was approved in the United States in 2006, available on the Puritan Bennett 840 ventilator (Puritan Bennett Co, Boulder, CO). PAV has also been used for noninvasive ventilation, but this is not available in the United States.
Other names for proportional assist ventilation
Proportional Pressure Support (Dräger Medical; not yet available in the United States).
What does proportional assist ventilation do?
This mode delivers pressure-controlled breaths with a servo control scheme (Table 2).
To better understand PAV, we can compare it with PSV. With PSV, the pressure applied by the ventilator rises to a preset level that is held constant (a set-point scheme) until a cycling criterion (a percent of the maximum inspiratory flow value) is reached. The inspiratory flow and tidal volume are the result of the patient’s inspiratory effort, the level of pressure applied, and the respiratory system mechanics.
In PAV, as in PSV, all breaths are spontaneous (Table 1). The patient controls the timing and size of the breath. There are no preset pressure, flow, or volume goals, but safety limits on the volume and pressure delivered can be set.
Ventilator settings in proportional assist ventilation
Ventilator settings in PAV are:
- Airway type (endotracheal tube, tracheostomy)
- Airway size (inner diameter)
- Percentage of work supported (assist range 5%–95%)
- Tidal volume limit
- Pressure limit
- Expiratory sensitivity (normally, as inspiration ends, flow should stop; this parameter tells the ventilator at what flow to end inspiration).
Caution when assessing the literature. Earlier ventilator versions, ie, Dräger and Manitoba (University of Manitoba, Winnipeg, MB, Canada), which are not available in the United States, required the repeated calculation of the respiratory system mechanics and the manual setting of flow and volume assists (amplification factors) independently. To overcome this limitation, new software automatically adjusts the flow and volume amplification to support the loads imposed by the automatically measured values of resistance and elastance (inverse of compliance) of the respiratory system.21 This software is included in the model (Puritan Bennett) available in the United States.
Clinical applications of proportional assist ventilation
The PAV mode is indicated for maximizing ventilator patient synchrony for assisted spontaneous ventilation.
PAV is contraindicated in patients with respiratory depression (bradypnea) or large air leaks (eg, bronchopleural fistulas). It should be used with caution in patients with severe hyperinflation, in which the patient may still be exhaling but the ventilator doesn’t recognize it. Another group in which PAV should be used with caution is those with high ventilatory drives, in which the ventilator overestimates respiratory system mechanics. This situation can lead to overassistance due to the “runaway phenomenon,” in which the ventilator continues to provide support even if the patient has stopped inspiration.22
Theoretical benefits of proportional assist ventilation
In theory, PAV should reduce the work of breathing, improve synchrony, automatically adapt to changing patient lung mechanics and effort, decrease the need for ventilator intervention and manipulation, decrease the need for sedation, and improve sleep.
Evidence of benefit of proportional assist ventilation
Physiologic benefits. PAV reduces the work of breathing better than PSV,21 even in the face of changing respiratory mechanics or increased respiratory demand (hypercapnia).23–25 The hemodynamic profile is similar to that in PSV. Tidal volumes are variable; however, in recent reports the tidal volumes were within the lung-protective range (6–8 mL/kg, plateau pressure < 30 cm H20).26,27
Comfort. PAV entails less patient effort and discomfort that PSV does.23,25 PAV significantly reduces asynchrony,27 which in turn may favorably affect sleep in critically ill patients. 28
Outcomes. The probability of spontaneous breathing without assistance was significantly better in critically ill patients ventilated with PAV than with PSV. No trial has reported the effect of PAV on deaths.27,29
Proportional assist ventilation: Bottom line
Extensive basic research has been done with PAV in different forms of respiratory failure, such as obstructive lung disease, acute respiratory distress syndrome (ARDS), and chronic respiratory failure. It fulfills its main goal, which is to improve patient-ventilator synchrony. Clinical experience with PAV in the United States is limited, as it was only recently approved.
AIRWAY PRESSURE-RELEASE VENTILATION AND BIPHASIC POSITIVE AIRWAY PRESSURE
Airway pressure-release ventilation (APRV) was described in 1987 by Stock et al30 as a mode for delivering ventilation in acute lung injury while avoiding high airway pressures. APRV combines high constant positive airway pressure (improving oxygenation and promoting alveolar recruitment) with intermittent releases (causing exhalation).
Other names for biphasic positive airway pressure
Other names for biphasic positive airway pressure are:
- BiLevel (Puritan Bennett)
- BIPAP (Dräger Europe)
- Bi Vent (Siemens)
- BiPhasic (Avea, Cardinal Health, Inc, Dublin, OH)
- PCV+ (Dräger Medical)
- DuoPAP (Hamilton).
Caution—name confusion. In North America, BiPAP (Respironics, Murrysville, PA) and BiLevel are used to refer to noninvasive modes of ventilation.
APRV has no other name.
What do these modes do?
These modes deliver pressure-controlled, time-triggered, and time-cycled breaths using a set-point targeting scheme (Table 2). This means that the ventilator maintains a constant pressure (set point) even in the face of spontaneous breaths.
Caution—source of confusion. The term continuous positive airway pressure (CPAP) is often used to describe this mode. However, CPAP is pressure that is applied continuously at the same level; the patient generates all the work to maintain ventilation (“pressure-controlled continuous spontaneous ventilation” in the current nomenclature). In APRV, the airway pressure is intermittently released and reapplied, generating a tidal volume that supports ventilation. In other words, this is a pressure-controlled breath with a very prolonged inspiratory time and a short expiratory time in which spontaneous ventilation is possible at any point (“pressure-controlled intermittent mandatory ventilation” in the current nomenclature).
How these modes are set in the ventilator may also be a source of confusion. To describe the time spent in high and low airway pressures, we use the terms Thigh and Tlow, respectively. By convention, the difference between APRV and biphasic mode is the duration of Tlow (< 1.5 sec for APRV).
Similarly, Phigh and Plow are used to describe the high and low airway pressure. To better understand this concept, you can create the same mode in conventional pressure-control ventilation by thinking of the Thigh as the inspiratory time, the Tlow as the expiratory time, the Phigh as inspiratory pressure, and the Plow as PEEP.
Hence, APRV is an extreme form of inverse ratio ventilation, with an inspiration-to-expiration ratio of 4:1. This means a patient spends most of the time in Phigh and Thigh, and exhalations are short (Tlow and Plow). In contrast, the biphasic mode uses conventional inspiration-expiration ratios (Figure 4).
As with any form of pressure control, the tidal volume is generated by airway pressure rising above baseline (ie, the end-expiratory value). Hence, to ensure an increase in minute ventilation, the mandatory breath rate must be increased (ie, decreasing Thigh, Tlow, or both) or the tidal volume must be increased (ie, increasing the difference between Phigh and Plow). This means that in APRV the Tlow has to happen more often (by increasing the number of breaths) or be more prolonged (allowing more air to exhale). Because unrestricted spontaneous breaths are permitted at any point of the cycle, the patient contributes to the total minute ventilation (usually 10%–40%).
In APRV and biphasic mode, the operator’s set time and pressure in inspiration and expiration will be delivered regardless of the patient’s breathing efforts—the patient’s spontaneous breath does not trigger a mechanical breath. Some ventilators have automatic adjustments to improve the trigger synchrony.
Ventilator settings in APRV and biphasic mode
These modes require the setting of two pressure levels (Phigh and Plow) and two time durations (Thigh and Tlow). One can add pressure support or automatic tube compensation to assist spontaneous breaths. The difference in Tlow generates differences in the Thigh:Tlow ratio: APRV has a short Tlow (an inspiration-expiration ratio of 4:1). Biphasic mode has a conventional inspiration-expiration ratio of 1:1 to 1:4.
Clinical applications
APRV is used in acute lung injury and ARDS. This mode should be used with caution or not at all in patients with obstructive lung disease or inappropriately increased respiratory drive.32–35
Biphasic mode is intended for both ventilation and weaning. In a patient who has poor respiratory effort or who is paralyzed, biphasic is identical to pressure-control/continuous mandatory ventilation.
Theoretical benefits of APRV and biphasic mode
Multiple benefits have been ascribed to these modes. In theory, APRV will maximize and maintain alveolar recruitment, improve oxygenation, lower inflation pressures, and decrease overinflation. Both APRV and biphasic, by preserving spontaneous breathing, will improve ventilation-perfusion matching and gas diffusion, improve the hemodynamic profile (less need for vasopressors, higher cardiac output, reduced ventricular workload, improved organ perfusion), and improve synchrony (decrease the work of breathing and the need for sedation).
Evidence of benefit of APRV and biphasic mode
APRV and biphasic are different modes. However studies evaluating their effects are combined. This is in part the result of the nomenclature confusion and different practice in different countries.36
Physiologic benefits. In studies, spontaneous breaths contributed to 10% to 40% of minute ventilation,37,38 improved ventilation of dependent areas of the lung, improved ventilation-perfusion match and recruitment,39 and improved hemodynamic profile.40
Patient comfort. These modes are thought to decrease the need for analgesia and sedation,38 but a recent trial showed no difference with pressure-controlled intermittent mandatory ventilation.41 Patient ventilator synchrony and comfort have not been studied.32,42
Outcomes. In small trials, these modes made no difference in terms of deaths, but they may decrease the length of mechanical ventilation.38,41,43,44
APRV and biphasic mode: Bottom line
Maintaining spontaneous breathing while on mechanical ventilation has hemodynamic and ventilatory benefits.
APRV and biphasic mode are not the same thing. APRV’s main goal is to maximize mean airway pressure and, hence, lung recruitment, whereas the main goal of the biphasic mode is synchrony.
There is a plethora of ventilator settings and questions related to physiologic effects.33,34,36
Although these modes are widely used in some centers, there is no evidence yet that they are superior to conventional volume- or pressure-control ventilation with low tidal volume for ARDS and acute lung injury. There is no conclusive evidence that these modes improve synchrony, time to weaning, or patient comfort.
HIGH-FREQUENCY OSCILLATORY VENTILATION
High-frequency oscillatory ventilation (HFOV) was first described and patented in 1952 by Emerson and was clinically developed in the early 1970s by Lunkenheimer.45
The goal of HFOV is to minimize lung injury; its characteristics (discussed below) make it useful in patients with severe ARDS. The US Food and Drug Administration approved it for infants in 1991 and for children in 1995. The adult model has been available since 1993, but it was not approved until 2001 (SensorMedics 3100B, Cardinal Health, Inc).
Other names for high-frequency oscillatory ventilation
While HFOV has no alternative names, the following acronyms describe similar modes:
- HFPPV (high-frequency positive pressure ventilation)
- HFJV (high-frequency jet ventilation)
- HFFI (high-frequency flow interruption)
- HFPV (high-frequency percussive ventilation)
- HFCWO (high-frequency chest wall oscillation).
All of these modes require different specialized ventilators.
What does high-frequency oscillatory ventilation do?
Conceptually, HFOV is a form of pressure-controlled intermittent mandatory ventilation with a set-point control scheme. In contrast to conventional pressure-controlled intermittent mandatory ventilation, in which relatively small spontaneous breaths may be superimposed on relatively large mandatory breaths, HFOV superimposes very small mandatory breaths (oscillations) on top of spontaneous breaths.
Adult patients are usually paralyzed or deeply sedated, since deep spontaneous breathing will trigger alarms and affect ventilator performance.
To manage ventilation (CO2 clearance), one or several of the following maneuvers can be done: decrease the oscillation frequency, increase the amplitude of the oscillations, increase the inspiratory time, or increase bias flow (while allowing an endotracheal tube cuff leak). Oxygenation adjustments are controlled by manipulating the mean airway pressure and the Fio2.
Ventilator settings in high-frequency oscillatory ventilation
Ventilator settings in HFOV are46:
- Airway pressure amplitude (delta P or power)
- Mean airway pressure
- Percent inspiration
- Inspiratory bias flow
- Fio2.
Clinical applications of high-frequency oscillatory ventilation
This mode is usually reserved for ARDS patients for whom conventional ventilation is failing. A recently published protocol46 suggests considering HFOV when there is oxygenation failure (Fio2 ≥ 0.7 and PEEP ≥ 14 cm H2O) or ventilation failure (pH < 7.25 with tidal volume ≥ 6 mL/kg predicted body weight and plateau airway pressure ≥ 30 cm H2O).
This mode is contraindicated when there is known severe airflow obstruction or intracranial hypertension.
Theoretical benefits of high-frequency oscillatory ventilation
Conceptually, HFOV can provide the highest mean airway pressure paired with the lowest tidal volume of any mode. These benefits might make HFOV the ideal lung-protective ventilation strategy.
Evidence of benefit of high-frequency oscillatory ventilation
Physiologic benefits. Animal models have shown less histologic damage and lung inflammation with HFOV than with high-tidal-volume conventional ventilation47,48 and low-tidal-volume conventional ventilation.49
Patient comfort has not been studied. However, current technology does impose undue work of breathing in spontaneously breathing patients.50
Outcomes. Several retrospective case series have described better oxygenation with HFOV as rescue therapy for severe ARDS than with conventional mechanical ventilation. Two randomized controlled trials have studied HFOV vs high-tidal-volume conventional mechanical ventilation for early severe ARDS; HFOV was safe but made no difference in terms of deaths.42,51–54
High-frequency oscillatory ventilation: Bottom line
In theory, HFOV provides all the benefits of an ideal lung-protective strategy, at least for paralyzed or deeply sedated patients. Animal studies support these concepts. In human adults, HFOV has been shown to be safe and to provide better oxygenation but no improvement in death rates compared with conventional mechanical ventilation. Currently, HFOV is better reserved for patients with severe ARDS for whom conventional mechanical ventilation is failing.
- Chatburn RL. Classification of ventilator modes: update and proposal for implementation. Respir Care 2007; 52:301–323.
- Chatburn RL. Computer control of mechanical ventilation. Respir Care 2004; 49:507–517.
- Alvarez A, Subirana M, Benito S. Decelerating flow ventilation effects in acute respiratory failure. J Crit Care 1998; 13:21–25.
- Guldager H, Nielsen SL, Carl P, Soerensen MB. A comparison of volume control and pressure regulated volume control ventilation in acute respiratory failure. Crit Care 1997; 1:75–77.
- Kallet RH, Campbell AR, Dicker RA, Katz JA, Mackersie RC. Work of breathing during lung protective ventilation in patients with acute lung injury and acute respiratory distress syndrome: a comparison between volume and pressure regulated breathing modes. Respir Care 2005; 50:1623–1631.
- Betensley AD, Khalid I, Crawford J, Pensler RA, DiGiovine B. Patient comfort during pressure support and volume controlled continuous mandatory ventilation. Respir Care 2008; 53:897–902.
- Branson RD, Chatburn RL. Controversies in the critical care setting. Should adaptive pressure control modes be utilized for virtually all patients receiving mechanical ventilation? Respir Care 2007; 52:478–485.
- Laubscher TP, Frutiger A, Fanconi S, Jutzi H, Brunner JX. Automatic selection of tidal volume, respiratory frequency and minute ventilation in intubated ICU patients as start up procedure for closed-loop controlled ventilation. Int J Clin Monit Comput 1994; 11:19–30.
- Laubscher TP, Heinrichs W, Weiler N, Hartmann G, Brunner JX. An adaptive lung ventilation controller. IEEE Trans Biomed Eng 1994; 41:51–59.
- Arnal JM, Wysocki M, Nafati C, et al. Automatic selection of breathing pattern using adaptive support ventilation. Intensive Care Med 2008; 34:75–81.
- Campbell RS, Sinamban RP, Johannigman JA, et al. Clinical evaluation of a new closed loop ventilation mode: adaptive supportive ventilation (ASV). Crit Care 1999; 3( suppl 1):083.
- Belliato M, Palo A, Pasero D, Iotti GA, Mojoli F, Braschi A. Evaluation of adaptive support ventilation in paralysed patients and in a physical lung model. Int J Artif Organs 2004; 27:709–716.
- Tassaux D, Dalmas E, Gratadour P, Jolliet P. Patient ventilator interactions during partial ventilatory support: a preliminary study comparing the effects of adaptive support ventilation with synchronized intermittent mandatory ventilation plus inspiratory pressure support. Crit Care Med 2002; 30:801–807.
- Gruber PC, Gomersall CD, Leung P, et al. Randomized controlled trial comparing adaptive-support ventilation with pressure-regulated volume-controlled ventilation with automode in weaning patients after cardiac surgery. Anesthesiology 2008; 109:81–87.
- Sulzer CF, Chiolero R, Chassot PG, et al. Adaptive support ventilation for fast tracheal extubation after cardiac surgery: a randomized controlled study. Anesthesiology 2001; 95:1339–1345.
- Petter AH, Chiolèro RL, Cassina T, Chassot PG, Müller XM, Revelly JP. Automatic “respirator/weaning” with adaptive support ventilation: the effect on duration of endotracheal intubation and patient management. Anesth Analg 2003; 97:1743–1750.
- Brunner JX, Iotti GA. Adaptive support ventilation (ASV). Minerva Anestesiol 2002; 68:365–368.
- Campbell RS, Branson RD, Johannigman JA. Adaptive support ventilation. Respir Care Clin North Am 2001; 7:425–440.
- Younes M. Proportional assist ventilation, a new approach to ventilatory support. Theory. Am Rev Respir Dis 1992; 145:114–120.
- Younes M, Puddy A, Roberts D, et al. Proportional assist ventilation. Results of an initial clinical trial. Am Rev Respir Dis 1992; 145:121–129.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Respiratory load compensation during mechanical ventilatio—proportional assist ventilation with load-adjustable gain factors versus pressure support. Intensive Care Med 2006; 32:692–699.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Effect of different levels of pressure support and proportional assist ventilation on breathing pattern, work of breathing and gas exchange in mechanically ventilated hypercapnic COPD patients with acute respiratory failure. Respiration 2003; 70:355–361.
- Grasso S, Puntillo F, Mascia L, et al. Compensation for increase in respiratory workload during mechanical ventilation. Pressure support versus proportional assist ventilation. Am J Respir Crit Care Med 2000; 161:819–826.
- Wrigge H, Golisch W, Zinserling J, Sydow M, Almeling G, Burchardi H. Proportional assist versus pressure support ventilation: effects on breathing pattern and respiratory work of patients with chronic obstructive pulmonary disease. Intensive Care Med 1999; 25:790–798.
- Ranieri VM, Giuliani R, Mascia L, et al. Patient ventilator interaction during acute hypercapnia: pressure support vs. proportional assist ventilation. J Appl Physiol 1996; 81:426–436.
- Kondili E, Xirouchaki N, Vaporidi K, Klimathianaki M, Georgopoulos D. Short-term cardiorespiratory effects of proportional assist and pressure support ventilation in patients with acute lung injury/acute respiratory distress syndrome. Anesthesiology 2006; 105:703–708.
- Xirouchaki N, Kondili E, Vaporidi K, et al. Proportional assist ventilation with load-adjustable gain factors in critically ill patients: comparison with pressure support. Intensive Care Med 2008; 34:2026–2034.
- Bosma K, Ferreyra G, Ambrogio C, et al. Patient ventilator interaction and sleep in mechanically ventilated patients: pressure support versus proportional assist ventilation. Crit Care Med 2007; 35:1048–1054.
- Sinderby C, Beck J. Proportional assist ventilation and neurally adjusted ventilatory assist—better approaches to patient ventilator synchrony? Clin Chest Med 2008; 29:329–342.
- Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med 1987; 15:462–466.
- Baum M, Benzer H, Putensen C, Koller W, Putz G. [Biphasic positive airway pressure (BIPAP)—a new form of augmented ventilation]. Anaesthesist 1989; 38:452–458.
- Seymour CW, Frazer M, Reilly PM, Fuchs BD. Airway pressure release and biphasic intermittent positive airway pressure ventilation: are they ready for prime time? J Trauma 2007; 62:1298–1308.
- Myers TR, MacIntyre NR. Respiratory controversies in the critical care setting. Does airway pressure release ventilation offer important new advantages in mechanical ventilator support? Respir Care 2007; 52:452–458.
- Neumann P, Golisch W, Strohmeyer A, Buscher H, Burchardi H, Sydow M. Influence of different release times on spontaneous breathing pattern during airway pressure release ventilation. Intensive Care Med 2002; 28:1742–1749.
- Calzia E, Lindner KH, Witt S, et al. Pressure-time product and work of breathing during biphasic continuous positive airway pressure and assisted spontaneous breathing. Am J Respir Crit Care Med 1994; 150:904–910.
- Rose L, Hawkins M. Airway pressure release ventilation and biphasic positive airway pressure: a systematic review of definitional criteria. Intensive Care Med 2008; 34:1766–1773.
- Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Longterm effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med 1994; 149:1550–1556.
- Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001; 164:43–49.
- Davis K, Johnson DJ, Branson RD, Campbell RS, Johannigman JA, Porembka D. Airway pressure release ventilation. Arch Surg 1993; 128:1348–1352.
- Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001; 5:221–226.
- Varpula T, Valta P, Niemi R, Takkunen O, Hynynen M, Pettilä VV. Airway pressure release ventilation as a primary ventilatory mode in acute respiratory distress syndrome. Acta Anaesthesiol Scand 2004; 48:722–731.
- Siau C, Stewart TE. Current role of high frequency oscillatory ventilation and airway pressure release ventilation in acute lung injury and acute respiratory distress syndrome. Clin Chest Med 2008; 29:265–275.
- Rathgeber J, Schorn B, Falk V, Kazmaier S, Spiegel T, Burchardi H. The influence of controlled mandatory ventilation (CMV), intermittent mandatory ventilation (IMV) and biphasic intermittent positive airway pressure (BIPAP) on duration of intubation and consumption of analgesics and sedatives. A prospective analysis in 596 patients following adult cardiac surgery. Eur J Anaesthesiol 1997; 14:576–582.
- Habashi NM. Other approaches to open lung ventilation: airway pressure release ventilation. Crit Care Med 2005; 33 suppl 3:S228–S240.
- Hess D, Mason S, Branson R. High-frequency ventilation design and equipment issues. Respir Care Clin North Am 2001; 7:577–598.
- Fessler HE, Derdak S, Ferguson ND, et al. A protocol for high frequency oscillatory ventilation in adults: results from a roundtable discussion. Crit Care Med 2007; 35:1649–1654.
- Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol 1983; 55:131–138.
- Sedeek KA, Takeuchi M, Suchodolski K, et al. Open-lung protective ventilation with pressure control ventilation, high-frequency oscillation, and intratracheal pulmonary ventilation results in similar gas exchange, hemodynamics, and lung mechanics. Anesthesiology 2003; 99:1102–1111.
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol 2001; 91:1836–1844.
- van Heerde M, Roubik K, Kopelent V, Plötz FB, Markhorst DG. Unloading work of breathing during high-frequency oscillatory ventilation: a bench study. Crit Care 2006; 10:R103.
- Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med 2002; 166:801–808.
- Bollen CW, van Well GT, Sherry T, et al. High-frequency oscillatory ventilation compared with conventional mechanical ventilation in adult respiratory distress syndrome: a randomized controlled trial [ISRCTN24242669]. Crit Care 2005; 9:R430–R439.
- Mehta S, Granton J, MacDonald RJ, et al. High frequency oscillatory ventilation in adults: the Toronto experience. Chest 2004; 126:518–527.
- Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest 2007; 131:1907–1916.
- Chatburn RL. Classification of ventilator modes: update and proposal for implementation. Respir Care 2007; 52:301–323.
- Chatburn RL. Computer control of mechanical ventilation. Respir Care 2004; 49:507–517.
- Alvarez A, Subirana M, Benito S. Decelerating flow ventilation effects in acute respiratory failure. J Crit Care 1998; 13:21–25.
- Guldager H, Nielsen SL, Carl P, Soerensen MB. A comparison of volume control and pressure regulated volume control ventilation in acute respiratory failure. Crit Care 1997; 1:75–77.
- Kallet RH, Campbell AR, Dicker RA, Katz JA, Mackersie RC. Work of breathing during lung protective ventilation in patients with acute lung injury and acute respiratory distress syndrome: a comparison between volume and pressure regulated breathing modes. Respir Care 2005; 50:1623–1631.
- Betensley AD, Khalid I, Crawford J, Pensler RA, DiGiovine B. Patient comfort during pressure support and volume controlled continuous mandatory ventilation. Respir Care 2008; 53:897–902.
- Branson RD, Chatburn RL. Controversies in the critical care setting. Should adaptive pressure control modes be utilized for virtually all patients receiving mechanical ventilation? Respir Care 2007; 52:478–485.
- Laubscher TP, Frutiger A, Fanconi S, Jutzi H, Brunner JX. Automatic selection of tidal volume, respiratory frequency and minute ventilation in intubated ICU patients as start up procedure for closed-loop controlled ventilation. Int J Clin Monit Comput 1994; 11:19–30.
- Laubscher TP, Heinrichs W, Weiler N, Hartmann G, Brunner JX. An adaptive lung ventilation controller. IEEE Trans Biomed Eng 1994; 41:51–59.
- Arnal JM, Wysocki M, Nafati C, et al. Automatic selection of breathing pattern using adaptive support ventilation. Intensive Care Med 2008; 34:75–81.
- Campbell RS, Sinamban RP, Johannigman JA, et al. Clinical evaluation of a new closed loop ventilation mode: adaptive supportive ventilation (ASV). Crit Care 1999; 3( suppl 1):083.
- Belliato M, Palo A, Pasero D, Iotti GA, Mojoli F, Braschi A. Evaluation of adaptive support ventilation in paralysed patients and in a physical lung model. Int J Artif Organs 2004; 27:709–716.
- Tassaux D, Dalmas E, Gratadour P, Jolliet P. Patient ventilator interactions during partial ventilatory support: a preliminary study comparing the effects of adaptive support ventilation with synchronized intermittent mandatory ventilation plus inspiratory pressure support. Crit Care Med 2002; 30:801–807.
- Gruber PC, Gomersall CD, Leung P, et al. Randomized controlled trial comparing adaptive-support ventilation with pressure-regulated volume-controlled ventilation with automode in weaning patients after cardiac surgery. Anesthesiology 2008; 109:81–87.
- Sulzer CF, Chiolero R, Chassot PG, et al. Adaptive support ventilation for fast tracheal extubation after cardiac surgery: a randomized controlled study. Anesthesiology 2001; 95:1339–1345.
- Petter AH, Chiolèro RL, Cassina T, Chassot PG, Müller XM, Revelly JP. Automatic “respirator/weaning” with adaptive support ventilation: the effect on duration of endotracheal intubation and patient management. Anesth Analg 2003; 97:1743–1750.
- Brunner JX, Iotti GA. Adaptive support ventilation (ASV). Minerva Anestesiol 2002; 68:365–368.
- Campbell RS, Branson RD, Johannigman JA. Adaptive support ventilation. Respir Care Clin North Am 2001; 7:425–440.
- Younes M. Proportional assist ventilation, a new approach to ventilatory support. Theory. Am Rev Respir Dis 1992; 145:114–120.
- Younes M, Puddy A, Roberts D, et al. Proportional assist ventilation. Results of an initial clinical trial. Am Rev Respir Dis 1992; 145:121–129.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Respiratory load compensation during mechanical ventilatio—proportional assist ventilation with load-adjustable gain factors versus pressure support. Intensive Care Med 2006; 32:692–699.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Effect of different levels of pressure support and proportional assist ventilation on breathing pattern, work of breathing and gas exchange in mechanically ventilated hypercapnic COPD patients with acute respiratory failure. Respiration 2003; 70:355–361.
- Grasso S, Puntillo F, Mascia L, et al. Compensation for increase in respiratory workload during mechanical ventilation. Pressure support versus proportional assist ventilation. Am J Respir Crit Care Med 2000; 161:819–826.
- Wrigge H, Golisch W, Zinserling J, Sydow M, Almeling G, Burchardi H. Proportional assist versus pressure support ventilation: effects on breathing pattern and respiratory work of patients with chronic obstructive pulmonary disease. Intensive Care Med 1999; 25:790–798.
- Ranieri VM, Giuliani R, Mascia L, et al. Patient ventilator interaction during acute hypercapnia: pressure support vs. proportional assist ventilation. J Appl Physiol 1996; 81:426–436.
- Kondili E, Xirouchaki N, Vaporidi K, Klimathianaki M, Georgopoulos D. Short-term cardiorespiratory effects of proportional assist and pressure support ventilation in patients with acute lung injury/acute respiratory distress syndrome. Anesthesiology 2006; 105:703–708.
- Xirouchaki N, Kondili E, Vaporidi K, et al. Proportional assist ventilation with load-adjustable gain factors in critically ill patients: comparison with pressure support. Intensive Care Med 2008; 34:2026–2034.
- Bosma K, Ferreyra G, Ambrogio C, et al. Patient ventilator interaction and sleep in mechanically ventilated patients: pressure support versus proportional assist ventilation. Crit Care Med 2007; 35:1048–1054.
- Sinderby C, Beck J. Proportional assist ventilation and neurally adjusted ventilatory assist—better approaches to patient ventilator synchrony? Clin Chest Med 2008; 29:329–342.
- Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med 1987; 15:462–466.
- Baum M, Benzer H, Putensen C, Koller W, Putz G. [Biphasic positive airway pressure (BIPAP)—a new form of augmented ventilation]. Anaesthesist 1989; 38:452–458.
- Seymour CW, Frazer M, Reilly PM, Fuchs BD. Airway pressure release and biphasic intermittent positive airway pressure ventilation: are they ready for prime time? J Trauma 2007; 62:1298–1308.
- Myers TR, MacIntyre NR. Respiratory controversies in the critical care setting. Does airway pressure release ventilation offer important new advantages in mechanical ventilator support? Respir Care 2007; 52:452–458.
- Neumann P, Golisch W, Strohmeyer A, Buscher H, Burchardi H, Sydow M. Influence of different release times on spontaneous breathing pattern during airway pressure release ventilation. Intensive Care Med 2002; 28:1742–1749.
- Calzia E, Lindner KH, Witt S, et al. Pressure-time product and work of breathing during biphasic continuous positive airway pressure and assisted spontaneous breathing. Am J Respir Crit Care Med 1994; 150:904–910.
- Rose L, Hawkins M. Airway pressure release ventilation and biphasic positive airway pressure: a systematic review of definitional criteria. Intensive Care Med 2008; 34:1766–1773.
- Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Longterm effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med 1994; 149:1550–1556.
- Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001; 164:43–49.
- Davis K, Johnson DJ, Branson RD, Campbell RS, Johannigman JA, Porembka D. Airway pressure release ventilation. Arch Surg 1993; 128:1348–1352.
- Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001; 5:221–226.
- Varpula T, Valta P, Niemi R, Takkunen O, Hynynen M, Pettilä VV. Airway pressure release ventilation as a primary ventilatory mode in acute respiratory distress syndrome. Acta Anaesthesiol Scand 2004; 48:722–731.
- Siau C, Stewart TE. Current role of high frequency oscillatory ventilation and airway pressure release ventilation in acute lung injury and acute respiratory distress syndrome. Clin Chest Med 2008; 29:265–275.
- Rathgeber J, Schorn B, Falk V, Kazmaier S, Spiegel T, Burchardi H. The influence of controlled mandatory ventilation (CMV), intermittent mandatory ventilation (IMV) and biphasic intermittent positive airway pressure (BIPAP) on duration of intubation and consumption of analgesics and sedatives. A prospective analysis in 596 patients following adult cardiac surgery. Eur J Anaesthesiol 1997; 14:576–582.
- Habashi NM. Other approaches to open lung ventilation: airway pressure release ventilation. Crit Care Med 2005; 33 suppl 3:S228–S240.
- Hess D, Mason S, Branson R. High-frequency ventilation design and equipment issues. Respir Care Clin North Am 2001; 7:577–598.
- Fessler HE, Derdak S, Ferguson ND, et al. A protocol for high frequency oscillatory ventilation in adults: results from a roundtable discussion. Crit Care Med 2007; 35:1649–1654.
- Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol 1983; 55:131–138.
- Sedeek KA, Takeuchi M, Suchodolski K, et al. Open-lung protective ventilation with pressure control ventilation, high-frequency oscillation, and intratracheal pulmonary ventilation results in similar gas exchange, hemodynamics, and lung mechanics. Anesthesiology 2003; 99:1102–1111.
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol 2001; 91:1836–1844.
- van Heerde M, Roubik K, Kopelent V, Plötz FB, Markhorst DG. Unloading work of breathing during high-frequency oscillatory ventilation: a bench study. Crit Care 2006; 10:R103.
- Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med 2002; 166:801–808.
- Bollen CW, van Well GT, Sherry T, et al. High-frequency oscillatory ventilation compared with conventional mechanical ventilation in adult respiratory distress syndrome: a randomized controlled trial [ISRCTN24242669]. Crit Care 2005; 9:R430–R439.
- Mehta S, Granton J, MacDonald RJ, et al. High frequency oscillatory ventilation in adults: the Toronto experience. Chest 2004; 126:518–527.
- Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest 2007; 131:1907–1916.
KEY POINTS
- The alternative modes of ventilation were developed to prevent lung injury and asynchrony, promote better oxygenation and faster weaning, and be easier to use. However, evidence of their benefit is scant.
- Until now, we have lacked a standard nomenclature for mechanical ventilation, leading to confusion.
- Regardless of the mode used, the goals are to avoid lung injury, keep the patient comfortable, and wean the patient from mechanical ventilation as soon as possible.
Is telemetry overused? Is it as helpful as thought?
Telemetry—from the Greek words tele (remote) and metron (measure)—for cardiac monitoring was developed in the mid-1960s by Spacelabs Medical for use in spaceflight.1 The system was later adopted in hospitals to detect life-threatening arrhythmias.
Guidelines for the use of telemetry were published in 1991 by the American College of Cardiology (ACC)2 in response to concerns raised by its increasing use in noncritical care settings during the 30 years after its introduction to clinical medicine. The latest revision of the guidelines was published in 2004 by the American Heart Association (AHA).3
However, the guidelines are based largely on expert opinion and on research data in electrocardiography. Few clinical trials of telemetry have been published, and they were either retrospective or nonrandomized. In fact, there were no published randomized trials at the time the 2004 guidelines were written. Moreover, very few of these studies evaluated the impact of cardiac telemetry monitoring on physician management decisions.
We reviewed the literature to find out how cardiac telemetry is being used in clinical practice and how it might be used more selectively. The literature search was performed using Ovid MEDLINE (1996 to present) and PubMed Central using the key search terms “cardiac monitoring,” “telemetry monitoring,” “telemetry,” and “inpatient.” References from articles identified using Ovid MEDLINE (1996 to present) and PubMed Central that were relevant to our review were also included.
THREE CLASSES OF RISK


- Class II consists of patients for whom cardiac monitoring may be of benefit in some cases but is not essential for all (Table 2).



PATIENTS AT LOW RISK DO NOT BENEFIT
Telemetry monitoring has become an essential and commonly used clinical tool in most hospital systems. However, physicians do not seem to be using the risk stratification guidelines routinely or appropriately. The result is that many patients are being monitored needlessly, because telemetric monitoring neither affects how patients at low risk are managed nor improves their clinical outcomes.
Saleem et al4 reported that, of 105 patients at low risk who presented with chest pain and were admitted to a telemetry unit, none experienced a cardiac event or arrhythmia warranting changes in management while in the hospital.
Durairaj et al5 conducted a prospective cohort study of 1,033 patients admitted consecutively from an emergency department to an inpatient telemetry unit from July 1998 to January 1999. Patients were initially stratified according to a prediction model proposed by Goldman et al6 into groups at high, moderate, low, and very low risk. The risk groups were substratified according to the presence or absence of chest pain. The outcomes measured were transfer to an intensive care unit and a major cardiac complication, which included acute myocardial infarction, cardiac arrest, ventricular fibrillation, temporary pacemaker implantation, cardiogenic shock, emergency cardioversion, use of an intraaortic balloon assist device, intubation, and recurrent ischemic pain requiring coronary revascularization within 72 hours after admission or requiring cardiac catheterization followed by coronary revascularization before discharge from the hospital. The subgroup of patients who were classified as being at very low risk and who did not have chest pain (n = 318) did not experience any major cardiac complication.
Sivaram and colleagues7 studied the role of telemetric monitoring in the management of patients with class I, II, and III indications for telemetric monitoring outside of critical care units. The class was assigned at the time of discharge for the purpose of the study. A total of 297 telemetry events were noted during the study, but only 12 (4%) of the events led to changes in patient management: a change in medication in 8 patients, cardioversion for unstable atrial flutter in 1 patient, insertion of a pacemaker for sinus pause in 1, and electrophysiology studies in 2 patients.
Estrada et al8 examined the clinical outcomes of 2,240 patients admitted to a non-intensive care unit. The physicians perceived telemetric monitoring as helpful in 283 (12.6%) of the patients. However, data obtained from telemetry monitoring directly affected management decisions in only 156 patients (7% of the original study population). The researchers concluded that physicians may overestimate the role of telemetry in guiding patient management.
Hollander et al9 examined the outcomes of 261 patients admitted because of chest pain who had normal or nonspecific findings on electrocardiography on presentation. Only 4 patients (1.5%) experienced arrhythmias. The authors concluded that the policy of admitting patients at low risk to monitored beds should be reevaluated.
Snider et al10 showed that patients presenting with atypical chest pain and normal electrocardiographic findings were at low risk of arrhythmias and did not benefit from telemetric monitoring.
Schull and Redelmeier11 performed a 5-year observational study in which they reviewed all telemetry admissions (N = 8,932) to a tertiary care facility. Twenty patients experienced cardiac arrest during the study period, but telemetric monitoring was in use at the time in only 16 of the 20. Furthermore, the telemetry monitors signalled the onset of cardiac arrest in only 9 of these 16 patients. Three of the patients whose hearts stopped beating survived until discharge: two in whom telemetry actually signalled the onset of cardiac arrest and one in whom it did not.
TELEMETRY CAN GIVE FALSE-POSITIVE ALARMS
Inappropriate use of telemetric monitoring increases the chance of artifacts or false-positive rhythms being misinterpreted as dysrhythmias and can potentially lead to errors in management.
Cases have been reported of patients undergoing invasive procedures because of artifacts seen during telemetric monitoring. Knight et al12 described 12 patients who underwent unnecessary diagnostic or therapeutic interventions as a result of misdiagnosis of artifacts as ventricular tachycardia.
We did not discover in our review any data correlating the frequency of false-positive telemetric monitoring findings to management errors. On the other hand, it is also not possible to discern from these studies how often cardiac telemetric monitoring reaffirmed the clinical impression and facilitated ongoing therapy.
TELEMETRY IS EXPENSIVE
Telemetry requires specialized equipment and trained personnel, making it both costly and labor-intensive. The additional costs and cost-effectiveness of telemetry remain uncertain. Studies of its medical costs have found wide variations across different hospital systems. Sivaram et al,7 in an observational study published in 1998, estimated the cost per patient at $683. At our hospital, the current cost of telemetric monitoring is at least $1,400 per patient per 24 hours.
Whatever the true cost, inappropriate use of telemetry creates a financial burden on the health care system and adds to unnecessary costs incurred by patients.
POTENTIAL BARRIERS TO APPROPRIATE USE OF TELEMETRY
A number of factors contribute to the inappropriate use of telemetry. Possible causes for its overuse may be a lack of awareness of the ACC and AHA guidelines, nonadherence to the guidelines, or a combination of factors.
Even when physicians are aware of these guidelines, adherence may be suboptimal for a variety of reasons (reviewed by Mehta13). Adams et al14 revealed that most studies evaluating adherence were biased by overreporting, since the levels of adherence were self-reported.
OUR RECOMMENDATIONS
To improve on the appropriate use of telemetry, we recommend that several strategies be implemented.
Current guidelines for in-hospital cardiac monitoring need to be updated, particularly since the recommendations were based on evidence that is several decades old. Also, medical care has improved since the publication of the last guidelines, justifying an update in the guidelines.
Guidelines for cardiac monitoring should be incorporated into the curriculum for physician education to increase awareness of the guidelines. Hospitals should ensure that the emergency medicine staff is educated with regard to ensuring appropriateness of admissions to telemetry units.
Finally, the implementation of predictive models similar to that developed by Goldman et al6 and implemented in the study by Durairaj5 could help to ensure that cardiac telemetry is reserved for patients who will benefit from it the most.
- NASA Scientific and Technical Information (STI). Space-proven medical monitor: the total patient care package. Health and medicine. Originating technology/NASA contribution. Spinoff 2006. www.sti.nasa.gov/Textonly/tto/Spinoff2006/hm_2.html. Accessed 1/2009.
- Jaffe AS, Atkins JM, Field JM, et al. Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. J Am Coll Cardiol 1991; 18:1431–1433.
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation 2004; 110:2721–2746.
- Saleem MA, McClung JA, Aronow WS, Kannam H. Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in-hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci 2005; 60:605–606.
- Durairaj L, Reilly B, Das K, et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med 2001; 110:7–11.
- Goldman A, Cook EF, Johnson PA, et al. Prediction of the need for intensive care in patients who come to emergency departments with acute chest pain. N Engl J Med 1996; 334:1498–1504.
- Sivaram CA, Summers JH, Ahmed N. Telemetry outside critical care units: patterns of utilization and influence on management decisions. Clin Cardiol 1998; 21:503–505.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol 1995; 76:960–965.
- Hollander JE, Valentine SM, McCuskey CF, Brogan GX. Are monitored telemetry beds necessary for patients with nontraumatic chest pain and normal or nonspecific electrocardiograms? Am J Cardiol 1997; 79:1110–1111.
- Snider A, Papaleo M, Beldner S, et al. Is telemetry monitoring necessary in low-risk suspected acute chest pain syndromes? Chest 2002; 122:517–523.
- Schull MJ, Redelmeier DA. Continuous electrocardiographic monitoring and cardiac arrest outcomes in 8,932 telemetry ward patients. Acad Emerg Med 2000; 7:647–652.
- Knight BP, Pelosi F, Michaud GF, Strickberger SA, Morady F. Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med 1999; 341:1270–1274.
- Mehta NB. The doctors’ challenge: How can we follow guidelines better? Cleve Clin J Med 2004; 71:81–85.
- Adams AS, Soumerai SB, Lomas J, Ross-Degnan D. Evidence of self-reporting bias in assessing adherence to guidelines. Int J Quality Health Care 1999; 11:187–192.
Telemetry—from the Greek words tele (remote) and metron (measure)—for cardiac monitoring was developed in the mid-1960s by Spacelabs Medical for use in spaceflight.1 The system was later adopted in hospitals to detect life-threatening arrhythmias.
Guidelines for the use of telemetry were published in 1991 by the American College of Cardiology (ACC)2 in response to concerns raised by its increasing use in noncritical care settings during the 30 years after its introduction to clinical medicine. The latest revision of the guidelines was published in 2004 by the American Heart Association (AHA).3
However, the guidelines are based largely on expert opinion and on research data in electrocardiography. Few clinical trials of telemetry have been published, and they were either retrospective or nonrandomized. In fact, there were no published randomized trials at the time the 2004 guidelines were written. Moreover, very few of these studies evaluated the impact of cardiac telemetry monitoring on physician management decisions.
We reviewed the literature to find out how cardiac telemetry is being used in clinical practice and how it might be used more selectively. The literature search was performed using Ovid MEDLINE (1996 to present) and PubMed Central using the key search terms “cardiac monitoring,” “telemetry monitoring,” “telemetry,” and “inpatient.” References from articles identified using Ovid MEDLINE (1996 to present) and PubMed Central that were relevant to our review were also included.
THREE CLASSES OF RISK
- Class II consists of patients for whom cardiac monitoring may be of benefit in some cases but is not essential for all (Table 2).
PATIENTS AT LOW RISK DO NOT BENEFIT
Telemetry monitoring has become an essential and commonly used clinical tool in most hospital systems. However, physicians do not seem to be using the risk stratification guidelines routinely or appropriately. The result is that many patients are being monitored needlessly, because telemetric monitoring neither affects how patients at low risk are managed nor improves their clinical outcomes.
Saleem et al4 reported that, of 105 patients at low risk who presented with chest pain and were admitted to a telemetry unit, none experienced a cardiac event or arrhythmia warranting changes in management while in the hospital.
Durairaj et al5 conducted a prospective cohort study of 1,033 patients admitted consecutively from an emergency department to an inpatient telemetry unit from July 1998 to January 1999. Patients were initially stratified according to a prediction model proposed by Goldman et al6 into groups at high, moderate, low, and very low risk. The risk groups were substratified according to the presence or absence of chest pain. The outcomes measured were transfer to an intensive care unit and a major cardiac complication, which included acute myocardial infarction, cardiac arrest, ventricular fibrillation, temporary pacemaker implantation, cardiogenic shock, emergency cardioversion, use of an intraaortic balloon assist device, intubation, and recurrent ischemic pain requiring coronary revascularization within 72 hours after admission or requiring cardiac catheterization followed by coronary revascularization before discharge from the hospital. The subgroup of patients who were classified as being at very low risk and who did not have chest pain (n = 318) did not experience any major cardiac complication.
Sivaram and colleagues7 studied the role of telemetric monitoring in the management of patients with class I, II, and III indications for telemetric monitoring outside of critical care units. The class was assigned at the time of discharge for the purpose of the study. A total of 297 telemetry events were noted during the study, but only 12 (4%) of the events led to changes in patient management: a change in medication in 8 patients, cardioversion for unstable atrial flutter in 1 patient, insertion of a pacemaker for sinus pause in 1, and electrophysiology studies in 2 patients.
Estrada et al8 examined the clinical outcomes of 2,240 patients admitted to a non-intensive care unit. The physicians perceived telemetric monitoring as helpful in 283 (12.6%) of the patients. However, data obtained from telemetry monitoring directly affected management decisions in only 156 patients (7% of the original study population). The researchers concluded that physicians may overestimate the role of telemetry in guiding patient management.
Hollander et al9 examined the outcomes of 261 patients admitted because of chest pain who had normal or nonspecific findings on electrocardiography on presentation. Only 4 patients (1.5%) experienced arrhythmias. The authors concluded that the policy of admitting patients at low risk to monitored beds should be reevaluated.
Snider et al10 showed that patients presenting with atypical chest pain and normal electrocardiographic findings were at low risk of arrhythmias and did not benefit from telemetric monitoring.
Schull and Redelmeier11 performed a 5-year observational study in which they reviewed all telemetry admissions (N = 8,932) to a tertiary care facility. Twenty patients experienced cardiac arrest during the study period, but telemetric monitoring was in use at the time in only 16 of the 20. Furthermore, the telemetry monitors signalled the onset of cardiac arrest in only 9 of these 16 patients. Three of the patients whose hearts stopped beating survived until discharge: two in whom telemetry actually signalled the onset of cardiac arrest and one in whom it did not.
TELEMETRY CAN GIVE FALSE-POSITIVE ALARMS
Inappropriate use of telemetric monitoring increases the chance of artifacts or false-positive rhythms being misinterpreted as dysrhythmias and can potentially lead to errors in management.
Cases have been reported of patients undergoing invasive procedures because of artifacts seen during telemetric monitoring. Knight et al12 described 12 patients who underwent unnecessary diagnostic or therapeutic interventions as a result of misdiagnosis of artifacts as ventricular tachycardia.
We did not discover in our review any data correlating the frequency of false-positive telemetric monitoring findings to management errors. On the other hand, it is also not possible to discern from these studies how often cardiac telemetric monitoring reaffirmed the clinical impression and facilitated ongoing therapy.
TELEMETRY IS EXPENSIVE
Telemetry requires specialized equipment and trained personnel, making it both costly and labor-intensive. The additional costs and cost-effectiveness of telemetry remain uncertain. Studies of its medical costs have found wide variations across different hospital systems. Sivaram et al,7 in an observational study published in 1998, estimated the cost per patient at $683. At our hospital, the current cost of telemetric monitoring is at least $1,400 per patient per 24 hours.
Whatever the true cost, inappropriate use of telemetry creates a financial burden on the health care system and adds to unnecessary costs incurred by patients.
POTENTIAL BARRIERS TO APPROPRIATE USE OF TELEMETRY
A number of factors contribute to the inappropriate use of telemetry. Possible causes for its overuse may be a lack of awareness of the ACC and AHA guidelines, nonadherence to the guidelines, or a combination of factors.
Even when physicians are aware of these guidelines, adherence may be suboptimal for a variety of reasons (reviewed by Mehta13). Adams et al14 revealed that most studies evaluating adherence were biased by overreporting, since the levels of adherence were self-reported.
OUR RECOMMENDATIONS
To improve on the appropriate use of telemetry, we recommend that several strategies be implemented.
Current guidelines for in-hospital cardiac monitoring need to be updated, particularly since the recommendations were based on evidence that is several decades old. Also, medical care has improved since the publication of the last guidelines, justifying an update in the guidelines.
Guidelines for cardiac monitoring should be incorporated into the curriculum for physician education to increase awareness of the guidelines. Hospitals should ensure that the emergency medicine staff is educated with regard to ensuring appropriateness of admissions to telemetry units.
Finally, the implementation of predictive models similar to that developed by Goldman et al6 and implemented in the study by Durairaj5 could help to ensure that cardiac telemetry is reserved for patients who will benefit from it the most.
Telemetry—from the Greek words tele (remote) and metron (measure)—for cardiac monitoring was developed in the mid-1960s by Spacelabs Medical for use in spaceflight.1 The system was later adopted in hospitals to detect life-threatening arrhythmias.
Guidelines for the use of telemetry were published in 1991 by the American College of Cardiology (ACC)2 in response to concerns raised by its increasing use in noncritical care settings during the 30 years after its introduction to clinical medicine. The latest revision of the guidelines was published in 2004 by the American Heart Association (AHA).3
However, the guidelines are based largely on expert opinion and on research data in electrocardiography. Few clinical trials of telemetry have been published, and they were either retrospective or nonrandomized. In fact, there were no published randomized trials at the time the 2004 guidelines were written. Moreover, very few of these studies evaluated the impact of cardiac telemetry monitoring on physician management decisions.
We reviewed the literature to find out how cardiac telemetry is being used in clinical practice and how it might be used more selectively. The literature search was performed using Ovid MEDLINE (1996 to present) and PubMed Central using the key search terms “cardiac monitoring,” “telemetry monitoring,” “telemetry,” and “inpatient.” References from articles identified using Ovid MEDLINE (1996 to present) and PubMed Central that were relevant to our review were also included.
THREE CLASSES OF RISK
- Class II consists of patients for whom cardiac monitoring may be of benefit in some cases but is not essential for all (Table 2).
PATIENTS AT LOW RISK DO NOT BENEFIT
Telemetry monitoring has become an essential and commonly used clinical tool in most hospital systems. However, physicians do not seem to be using the risk stratification guidelines routinely or appropriately. The result is that many patients are being monitored needlessly, because telemetric monitoring neither affects how patients at low risk are managed nor improves their clinical outcomes.
Saleem et al4 reported that, of 105 patients at low risk who presented with chest pain and were admitted to a telemetry unit, none experienced a cardiac event or arrhythmia warranting changes in management while in the hospital.
Durairaj et al5 conducted a prospective cohort study of 1,033 patients admitted consecutively from an emergency department to an inpatient telemetry unit from July 1998 to January 1999. Patients were initially stratified according to a prediction model proposed by Goldman et al6 into groups at high, moderate, low, and very low risk. The risk groups were substratified according to the presence or absence of chest pain. The outcomes measured were transfer to an intensive care unit and a major cardiac complication, which included acute myocardial infarction, cardiac arrest, ventricular fibrillation, temporary pacemaker implantation, cardiogenic shock, emergency cardioversion, use of an intraaortic balloon assist device, intubation, and recurrent ischemic pain requiring coronary revascularization within 72 hours after admission or requiring cardiac catheterization followed by coronary revascularization before discharge from the hospital. The subgroup of patients who were classified as being at very low risk and who did not have chest pain (n = 318) did not experience any major cardiac complication.
Sivaram and colleagues7 studied the role of telemetric monitoring in the management of patients with class I, II, and III indications for telemetric monitoring outside of critical care units. The class was assigned at the time of discharge for the purpose of the study. A total of 297 telemetry events were noted during the study, but only 12 (4%) of the events led to changes in patient management: a change in medication in 8 patients, cardioversion for unstable atrial flutter in 1 patient, insertion of a pacemaker for sinus pause in 1, and electrophysiology studies in 2 patients.
Estrada et al8 examined the clinical outcomes of 2,240 patients admitted to a non-intensive care unit. The physicians perceived telemetric monitoring as helpful in 283 (12.6%) of the patients. However, data obtained from telemetry monitoring directly affected management decisions in only 156 patients (7% of the original study population). The researchers concluded that physicians may overestimate the role of telemetry in guiding patient management.
Hollander et al9 examined the outcomes of 261 patients admitted because of chest pain who had normal or nonspecific findings on electrocardiography on presentation. Only 4 patients (1.5%) experienced arrhythmias. The authors concluded that the policy of admitting patients at low risk to monitored beds should be reevaluated.
Snider et al10 showed that patients presenting with atypical chest pain and normal electrocardiographic findings were at low risk of arrhythmias and did not benefit from telemetric monitoring.
Schull and Redelmeier11 performed a 5-year observational study in which they reviewed all telemetry admissions (N = 8,932) to a tertiary care facility. Twenty patients experienced cardiac arrest during the study period, but telemetric monitoring was in use at the time in only 16 of the 20. Furthermore, the telemetry monitors signalled the onset of cardiac arrest in only 9 of these 16 patients. Three of the patients whose hearts stopped beating survived until discharge: two in whom telemetry actually signalled the onset of cardiac arrest and one in whom it did not.
TELEMETRY CAN GIVE FALSE-POSITIVE ALARMS
Inappropriate use of telemetric monitoring increases the chance of artifacts or false-positive rhythms being misinterpreted as dysrhythmias and can potentially lead to errors in management.
Cases have been reported of patients undergoing invasive procedures because of artifacts seen during telemetric monitoring. Knight et al12 described 12 patients who underwent unnecessary diagnostic or therapeutic interventions as a result of misdiagnosis of artifacts as ventricular tachycardia.
We did not discover in our review any data correlating the frequency of false-positive telemetric monitoring findings to management errors. On the other hand, it is also not possible to discern from these studies how often cardiac telemetric monitoring reaffirmed the clinical impression and facilitated ongoing therapy.
TELEMETRY IS EXPENSIVE
Telemetry requires specialized equipment and trained personnel, making it both costly and labor-intensive. The additional costs and cost-effectiveness of telemetry remain uncertain. Studies of its medical costs have found wide variations across different hospital systems. Sivaram et al,7 in an observational study published in 1998, estimated the cost per patient at $683. At our hospital, the current cost of telemetric monitoring is at least $1,400 per patient per 24 hours.
Whatever the true cost, inappropriate use of telemetry creates a financial burden on the health care system and adds to unnecessary costs incurred by patients.
POTENTIAL BARRIERS TO APPROPRIATE USE OF TELEMETRY
A number of factors contribute to the inappropriate use of telemetry. Possible causes for its overuse may be a lack of awareness of the ACC and AHA guidelines, nonadherence to the guidelines, or a combination of factors.
Even when physicians are aware of these guidelines, adherence may be suboptimal for a variety of reasons (reviewed by Mehta13). Adams et al14 revealed that most studies evaluating adherence were biased by overreporting, since the levels of adherence were self-reported.
OUR RECOMMENDATIONS
To improve on the appropriate use of telemetry, we recommend that several strategies be implemented.
Current guidelines for in-hospital cardiac monitoring need to be updated, particularly since the recommendations were based on evidence that is several decades old. Also, medical care has improved since the publication of the last guidelines, justifying an update in the guidelines.
Guidelines for cardiac monitoring should be incorporated into the curriculum for physician education to increase awareness of the guidelines. Hospitals should ensure that the emergency medicine staff is educated with regard to ensuring appropriateness of admissions to telemetry units.
Finally, the implementation of predictive models similar to that developed by Goldman et al6 and implemented in the study by Durairaj5 could help to ensure that cardiac telemetry is reserved for patients who will benefit from it the most.
- NASA Scientific and Technical Information (STI). Space-proven medical monitor: the total patient care package. Health and medicine. Originating technology/NASA contribution. Spinoff 2006. www.sti.nasa.gov/Textonly/tto/Spinoff2006/hm_2.html. Accessed 1/2009.
- Jaffe AS, Atkins JM, Field JM, et al. Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. J Am Coll Cardiol 1991; 18:1431–1433.
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation 2004; 110:2721–2746.
- Saleem MA, McClung JA, Aronow WS, Kannam H. Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in-hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci 2005; 60:605–606.
- Durairaj L, Reilly B, Das K, et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med 2001; 110:7–11.
- Goldman A, Cook EF, Johnson PA, et al. Prediction of the need for intensive care in patients who come to emergency departments with acute chest pain. N Engl J Med 1996; 334:1498–1504.
- Sivaram CA, Summers JH, Ahmed N. Telemetry outside critical care units: patterns of utilization and influence on management decisions. Clin Cardiol 1998; 21:503–505.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol 1995; 76:960–965.
- Hollander JE, Valentine SM, McCuskey CF, Brogan GX. Are monitored telemetry beds necessary for patients with nontraumatic chest pain and normal or nonspecific electrocardiograms? Am J Cardiol 1997; 79:1110–1111.
- Snider A, Papaleo M, Beldner S, et al. Is telemetry monitoring necessary in low-risk suspected acute chest pain syndromes? Chest 2002; 122:517–523.
- Schull MJ, Redelmeier DA. Continuous electrocardiographic monitoring and cardiac arrest outcomes in 8,932 telemetry ward patients. Acad Emerg Med 2000; 7:647–652.
- Knight BP, Pelosi F, Michaud GF, Strickberger SA, Morady F. Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med 1999; 341:1270–1274.
- Mehta NB. The doctors’ challenge: How can we follow guidelines better? Cleve Clin J Med 2004; 71:81–85.
- Adams AS, Soumerai SB, Lomas J, Ross-Degnan D. Evidence of self-reporting bias in assessing adherence to guidelines. Int J Quality Health Care 1999; 11:187–192.
- NASA Scientific and Technical Information (STI). Space-proven medical monitor: the total patient care package. Health and medicine. Originating technology/NASA contribution. Spinoff 2006. www.sti.nasa.gov/Textonly/tto/Spinoff2006/hm_2.html. Accessed 1/2009.
- Jaffe AS, Atkins JM, Field JM, et al. Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. J Am Coll Cardiol 1991; 18:1431–1433.
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation 2004; 110:2721–2746.
- Saleem MA, McClung JA, Aronow WS, Kannam H. Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in-hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci 2005; 60:605–606.
- Durairaj L, Reilly B, Das K, et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med 2001; 110:7–11.
- Goldman A, Cook EF, Johnson PA, et al. Prediction of the need for intensive care in patients who come to emergency departments with acute chest pain. N Engl J Med 1996; 334:1498–1504.
- Sivaram CA, Summers JH, Ahmed N. Telemetry outside critical care units: patterns of utilization and influence on management decisions. Clin Cardiol 1998; 21:503–505.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol 1995; 76:960–965.
- Hollander JE, Valentine SM, McCuskey CF, Brogan GX. Are monitored telemetry beds necessary for patients with nontraumatic chest pain and normal or nonspecific electrocardiograms? Am J Cardiol 1997; 79:1110–1111.
- Snider A, Papaleo M, Beldner S, et al. Is telemetry monitoring necessary in low-risk suspected acute chest pain syndromes? Chest 2002; 122:517–523.
- Schull MJ, Redelmeier DA. Continuous electrocardiographic monitoring and cardiac arrest outcomes in 8,932 telemetry ward patients. Acad Emerg Med 2000; 7:647–652.
- Knight BP, Pelosi F, Michaud GF, Strickberger SA, Morady F. Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med 1999; 341:1270–1274.
- Mehta NB. The doctors’ challenge: How can we follow guidelines better? Cleve Clin J Med 2004; 71:81–85.
- Adams AS, Soumerai SB, Lomas J, Ross-Degnan D. Evidence of self-reporting bias in assessing adherence to guidelines. Int J Quality Health Care 1999; 11:187–192.
KEY POINTS
- Guidelines from the American College of Cardiology (1991) and American Heart Association (2004) divide patients into three risk classes for whom telemetry is, may be, or is not indicated.
- Few studies have addressed whether telemetry is beneficial in clinical practice.
- The available evidence suggests that telemetry infrequently influences physician management decisions for patients at low risk, although it may in a relatively small subset at high risk.
- Inappropriate use of telemetry is associated with unnecessary testing and treatment and higher cost of care.
- Better risk-assessment and selection strategies are needed to identify patients for whom telemetry monitoring will be most beneficial.