User login
Proceedings of the 4th Annual Perioperative Medicine Summit

Supplement Editor:
Amir K. Jaffer, MD, FHM
Associate Editors:
David L. Hepner, MD, and Franklin A. Michota, MD, FHM
Contents
Public reporting and pay-for-performance programs in perioperative medicine
Peter Lindenauer, MD MSc
Cardiac risk stratification for noncardiac surgery: Update from the American College of Cardiology/American Heart Association 2007 guidelines
Lee A. Fleisher, MD
Perioperative care of the elderly patient: An update
Robert M. Palmer, MD, MPH
The role of testing in the preoperative evaluation
David L. Hepner, MD
Perioperative fluid management: Progress despite lingering controversies
Mark A. Hamilton, MBBS, MRCP, FRCA
Giving anesthesiologists what they want: How to write a useful preoperative consult
David Lubarsky, MD, MBA, and Keith Candiotti, MD
Perioperative management of warfarin and antiplatelet therapy
Amir K. Jaffer, MD, FHM
Prevention of venous thromboembolism after surgery
Franklin A. Michota, MD, FHM
Perioperative management of diabetes: Translating evidence into practice
Luigi F. Meneghini, MD, MBA
Postoperative pulmonary complications: An update on risk assessment and reduction
Gerald W. Smetana, MD
Postoperative gastrointestinal tract dysfunction: An overview of causes and management strategies
Michael G. (Monty) Mythen, MD
Case studies in perioperative management: Challenges, controversies, and common ground
Steven L. Cohn, MD, and BobbieJean Sweitzer, MD
Statins and noncardiac surgery: Current evidence and practical considerations
Don Poldermans, MD, PhD
The experts debate: perioperative beta-blockade for noncardiac surgery patients—proven safe or not?
Don Poldermans, MD, PhD, and P.J. Devereaux, MD, PhD
Perioperative considerations for patients with liver disease
Paul Martin, MD
Perioperative management of obstructive sleep apnea: Ready for prime time?
Shirin Shafazand, MD, MS
Nuts and bolts of preoperative clinics: The view from three institutions
Angela M. Bader, MD, MPH; BobbieJean Sweitzer, MD; and Ajay Kumar, MD
Perioperative management of anemia: Limits of blood transfusion and alternatives to it
Ajay Kumar, MD
Medicolegal issues in perioperative medicine: Lessons from real cases
Franklin A. Michota, MD, FHM, and Matthew J. Donnelly, Esq
Perioperative medication management: General principles and practical applications
Christopher Whinney, MD
Supplement Editor:
Amir K. Jaffer, MD, FHM
Associate Editors:
David L. Hepner, MD, and Franklin A. Michota, MD, FHM
Contents
Public reporting and pay-for-performance programs in perioperative medicine
Peter Lindenauer, MD MSc
Cardiac risk stratification for noncardiac surgery: Update from the American College of Cardiology/American Heart Association 2007 guidelines
Lee A. Fleisher, MD
Perioperative care of the elderly patient: An update
Robert M. Palmer, MD, MPH
The role of testing in the preoperative evaluation
David L. Hepner, MD
Perioperative fluid management: Progress despite lingering controversies
Mark A. Hamilton, MBBS, MRCP, FRCA
Giving anesthesiologists what they want: How to write a useful preoperative consult
David Lubarsky, MD, MBA, and Keith Candiotti, MD
Perioperative management of warfarin and antiplatelet therapy
Amir K. Jaffer, MD, FHM
Prevention of venous thromboembolism after surgery
Franklin A. Michota, MD, FHM
Perioperative management of diabetes: Translating evidence into practice
Luigi F. Meneghini, MD, MBA
Postoperative pulmonary complications: An update on risk assessment and reduction
Gerald W. Smetana, MD
Postoperative gastrointestinal tract dysfunction: An overview of causes and management strategies
Michael G. (Monty) Mythen, MD
Case studies in perioperative management: Challenges, controversies, and common ground
Steven L. Cohn, MD, and BobbieJean Sweitzer, MD
Statins and noncardiac surgery: Current evidence and practical considerations
Don Poldermans, MD, PhD
The experts debate: perioperative beta-blockade for noncardiac surgery patients—proven safe or not?
Don Poldermans, MD, PhD, and P.J. Devereaux, MD, PhD
Perioperative considerations for patients with liver disease
Paul Martin, MD
Perioperative management of obstructive sleep apnea: Ready for prime time?
Shirin Shafazand, MD, MS
Nuts and bolts of preoperative clinics: The view from three institutions
Angela M. Bader, MD, MPH; BobbieJean Sweitzer, MD; and Ajay Kumar, MD
Perioperative management of anemia: Limits of blood transfusion and alternatives to it
Ajay Kumar, MD
Medicolegal issues in perioperative medicine: Lessons from real cases
Franklin A. Michota, MD, FHM, and Matthew J. Donnelly, Esq
Perioperative medication management: General principles and practical applications
Christopher Whinney, MD
Supplement Editor:
Amir K. Jaffer, MD, FHM
Associate Editors:
David L. Hepner, MD, and Franklin A. Michota, MD, FHM
Contents
Public reporting and pay-for-performance programs in perioperative medicine
Peter Lindenauer, MD MSc
Cardiac risk stratification for noncardiac surgery: Update from the American College of Cardiology/American Heart Association 2007 guidelines
Lee A. Fleisher, MD
Perioperative care of the elderly patient: An update
Robert M. Palmer, MD, MPH
The role of testing in the preoperative evaluation
David L. Hepner, MD
Perioperative fluid management: Progress despite lingering controversies
Mark A. Hamilton, MBBS, MRCP, FRCA
Giving anesthesiologists what they want: How to write a useful preoperative consult
David Lubarsky, MD, MBA, and Keith Candiotti, MD
Perioperative management of warfarin and antiplatelet therapy
Amir K. Jaffer, MD, FHM
Prevention of venous thromboembolism after surgery
Franklin A. Michota, MD, FHM
Perioperative management of diabetes: Translating evidence into practice
Luigi F. Meneghini, MD, MBA
Postoperative pulmonary complications: An update on risk assessment and reduction
Gerald W. Smetana, MD
Postoperative gastrointestinal tract dysfunction: An overview of causes and management strategies
Michael G. (Monty) Mythen, MD
Case studies in perioperative management: Challenges, controversies, and common ground
Steven L. Cohn, MD, and BobbieJean Sweitzer, MD
Statins and noncardiac surgery: Current evidence and practical considerations
Don Poldermans, MD, PhD
The experts debate: perioperative beta-blockade for noncardiac surgery patients—proven safe or not?
Don Poldermans, MD, PhD, and P.J. Devereaux, MD, PhD
Perioperative considerations for patients with liver disease
Paul Martin, MD
Perioperative management of obstructive sleep apnea: Ready for prime time?
Shirin Shafazand, MD, MS
Nuts and bolts of preoperative clinics: The view from three institutions
Angela M. Bader, MD, MPH; BobbieJean Sweitzer, MD; and Ajay Kumar, MD
Perioperative management of anemia: Limits of blood transfusion and alternatives to it
Ajay Kumar, MD
Medicolegal issues in perioperative medicine: Lessons from real cases
Franklin A. Michota, MD, FHM, and Matthew J. Donnelly, Esq
Perioperative medication management: General principles and practical applications
Christopher Whinney, MD

Managing diabetes in hemodialysis patients: Observations and recommendations
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
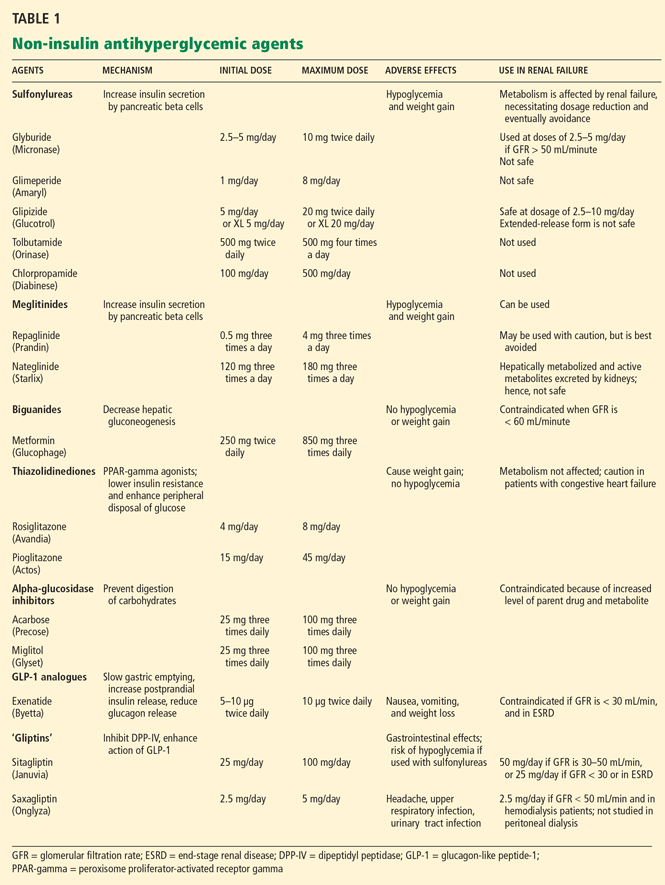
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
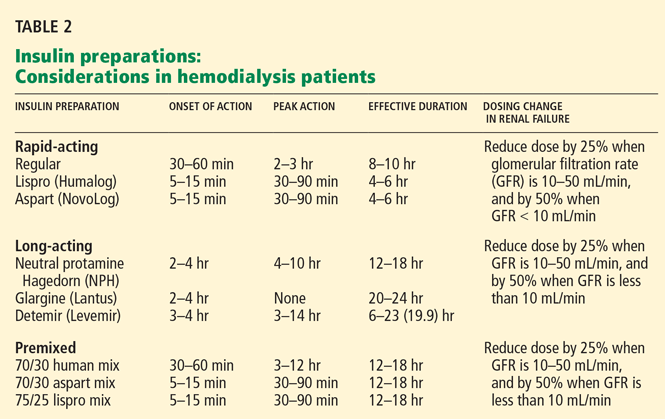
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
Although diabetes is the most common cause of end-stage renal disease (ESRD) worldwide, accounting for 44.2% of ESRD patients in the US Renal Data System in 2005,1 data are scarce on how diabetes should best be treated in patients in ESRD.
We do know that blood glucose levels need to be well controlled in these patients. Several observational studies and one nonrandomized interventional study2–10 showed that higher levels of hemoglobin A1c were associated with higher death rates in patients with diabetes and chronic kidney disease after adjusting for markers of inflammation and malnutrition.
However, ESRD significantly alters glycemic control, the results of hemoglobin A1c testing, and the excretion of antidiabetic medications. The various and opposing effects of ESRD and dialysis can make blood glucose levels fluctuate widely, placing patients at risk of hypoglycemia—and presenting a challenge for nephrologists and internists.
In this review, we summarize the available evidence and make practical recommendations for managing diabetes in patients on hemodialysis.
GLUCOSE LEVELS MAY FLUCTUATE WIDELY
In ESRD, both uremia and dialysis can complicate glycemic control by affecting the secretion, clearance, and peripheral tissue sensitivity of insulin.
Several factors, including uremic toxins, may increase insulin resistance in ESRD, leading to a blunted ability to suppress hepatic gluconeogenesis and regulate peripheral glucose utilization. In type 2 diabetes without kidney disease, insulin resistance leads to increased insulin secretion. This does not occur in ESRD because of concomitant metabolic acidosis, deficiency of 1,25 dihydroxyvitamin D, and secondary hyperparathyroidism.11,12 Hemodialysis further alters insulin secretion, clearance, and resistance as the result of periodic improvement in uremia, acidosis, and phosphate handling.
The dextrose concentration in the dialysate can also affect glucose control. In general, dialysates with lower dextrose concentrations are used and may be associated with hypoglycemia. Conversely, dialysates with higher dextrose concentrations are occasionally used in peritoneal dialysis to increase ultrafiltration, but this can lead to hyperglycemia.10,13
Thus, ESRD and hemodialysis exert opposing forces on insulin secretion, action, and metabolism, often creating unpredictable serum glucose values. For example, one would think that a patient who has insulin resistance would need more supplemental insulin; however, the reduced renal gluconeogenesis and insulin clearance seen in ESRD may result in variable net effects in different patients. In addition, ESRD and hemodialysis alter the pharmacokinetics of diabetic medications. Together, all of these factors contribute to wide fluctuations in glucose levels and increase the risk of hypoglycemic events.
HEMOGLOBIN A1c MAY BE FALSELY HIGH
Self-monitoring of blood glucose plus serial hemoglobin A1c measurements are the standard of care in diabetic patients without renal failure.
However, in diabetic patients with ESRD, elevated blood urea nitrogen causes formation of carbamylated hemoglobin, which is indistinguishable from glycosylated hemoglobin by electrical-charge-based assays and can cause the hemoglobin A1c measurement to be falsely elevated. Other factors such as the shorter red life span of red blood cells, iron deficiency, recent transfusion, and use of erythropoietin-stimulating agents may also cause underestimation of glucose control.14
Despite these limitations, the hemoglobin A1c level is considered a reasonable measure of glycemic control in ESRD. Glycated fructosamine and albumin are other measures of glycemic control with some advantages over hemoglobin A1c in dialysis patients. However, they are not readily available and can be affected by conditions that alter protein metabolism, including ESRD.15–18
Self-monitoring of blood glucose and continuous glucose monitoring systems provide real-time assessments of glycemic control, but both have limitations. Self-monitoring is subject to errors from poor technique, problems with the meters and strips, and lower sensitivity in measuring low blood glucose levels. Continuous monitoring is expensive and is less reliable at lower glucose concentrations, and thus it needs to be used in conjunction with other measures of glucose control. For these reasons, continuous glucose monitoring is not yet widely used.
The guidelines of the 2005 National Kidney Foundation Kidney Disease Outcomes Quality Initiative did not clearly establish a target hemoglobin A1c level for patients with diabetes and ESRD, but levels of 6% to 7% appear to be safe. The target fasting plasma glucose level should be lower than 140 mg/dL, and the target postprandial glucose level should be lower than 200 mg/dL.19
MOST ORAL DIABETES DRUGS ARE CONTRAINDICATED IN ESRD
Sulfonylureas
Sulfonylureas reduce blood glucose by stimulating the pancreatic beta cells to increase insulin secretion.
Sulfonylureas have a wide volume of distribution and are highly protein-bound,20 but only the unbound drug exerts a clinical effect. Because of protein binding, dialysis cannot effectively clear elevated levels of sulfonylurea drugs. Furthermore, many ESRD patients take drugs such as salicylates, sulfonamides, vitamin K antagonists, beta-blockers, and fibric acid derivatives, which may displace sulfonylureas from albumin, thus increasing the risk of severe hypoglycemia.
The first-generation sulfonylureas—chlorpropamide (Diabinese), acetohexamide (Dymelor), tolbutamide (Orinase), and tolazamide (Tolinase)—are almost exclusively excreted by the kidney and are therefore contraindicated in ESRD.21 Second-generation agents include glipizide (Glucotrol), glimepiride (Amaryl), glyburide (Micronase), and gliclazide (not available in the United States). Although these drugs are metabolized in the liver, their active metabolites are excreted in the urine, and so they should be avoided in ESRD.22
The only sulfonylurea recommended in ESRD is glipizide, which is also metabolized in the liver but has inactive or weakly active metabolites excreted in the urine. The suggested dose of glipizide is 2.5 to 10 mg/day. In ESRD, sustained-release forms should be avoided because of concerns of hypoglycemia.23
Meglitinides
The meglitinides repaglinide (Prandin) and nateglinide (Starlix) are insulin secretagogues that stimulate pancreatic beta cells. Like the sulfonylureas, nateglinide is hepatically metabolized, with renal excretion of active metabolites. Repaglinide, in contrast, is almost completely converted to inactive metabolites in the liver, and less than 10% is excreted by the kidneys.24,25 The meglitinides still pose a risk of hypoglycemia, especially in ESRD, and hence are not recommended for patients on hemodialysis.24,25
Biguanides
Metformin (Glucophage) is a biguanide that reduces hepatic gluconeogenesis and glucose output. It is excreted essentially unchanged in the urine and is therefore contraindicated in patients with renal disease due to the risks of bioaccumulation and lactic acidosis.22
Thiazolidinediones
The thiazolidinediones rosiglitazone (Avandia) and pioglitazone (Actos) are highly potent, selective agonists that work by binding to and activating a nuclear transcription factor, specifically, peroxisome proliferator-activated receptor gamma (PPAR-gamma). These drugs do not bioaccumulate in renal failure and so do not need dosing adjustments.26
The main adverse effect of these agents is edema, especially when they are combined with insulin therapy. Because of this effect, a joint statement of the American Diabetes Association and the American Heart Association recommends avoiding thiazolidinediones in patients in New York Heart Association (NYHA) class III or IV heart failure.27 Furthermore, caution is required in patients in compensated heart failure (NYHA class I or II) or in those at risk of heart failure, such as patients with previous myocardial infarction or angina, hypertension, left ventricular hypertrophy, significant aortic or mitral valve disease, age greater than 70 years, or diabetes for more than 10 years.27
In summary, although ESRD and dialysis do not affect the metabolism of thiazolidinediones, these agents are not recommended in ESRD because of the associated risk of fluid accumulation and precipitation of heart failure.
Alpha-glucosidase inhibitors
The alpha-glucosidase inhibitors acarbose (Precose) and miglitol (Glyset) slow carbohydrate absorption from the intestine. The levels of these drugs and their active metabolites are higher in renal failure,22 and since data are scarce on the use of these drugs in ESRD, they are contraindicated in ESRD.
GLP-1 ANALOGUES AND ‘GLIPTINS,’ NEW CLASSES OF DRUGS
Glucagon-like peptide-1 (GLP-1) stimulates glucose-dependent insulin release from pancreatic beta cells and inhibits inappropriate postprandial glucagon release. It also slows gastric emptying and reduces food intake. Dipeptidyl peptidase IV (DPP-IV) is an active ubiquitous enzyme that deactivates a variety of bioactive peptides, including GLP-1.
Exenatide (Byetta) is a naturally occurring GLP-1 analogue that is resistant to degradation by DPP-IV and has a longer half-life. Given subcutaneously, exenatide undergoes minimal systemic metabolism and is excreted in the urine.
No dose adjustment is required if the glomerular filtration rate (GFR) is greater than 30 mL/min, but exenatide is contraindicated in patients undergoing hemodialysis or in patients who have a GFR less than 30 mL/min (Table 1).
Sitagliptin (Januvia) is a DPP-IV inhibitor, or “gliptin,” that can be used as initial pharmacologic therapy for type 2 diabetes, as a second agent in those who do not respond to a single agent such as a sulfonylurea,28 metformin,29–31 or a thiazolidinedione,32 and as an additional agent when dual therapy with metformin and a sulfonylurea does not provide adequate glycemic control.28 Sitagliptin is not extensively metabolized and is mainly excreted in the urine.
The usual dose of sitagliptin is 100 mg orally once daily, with reduction to 50 mg for patients with a GFR of 30 to 50 mL/min, and 25 mg for patients with a GFR less than 30 mL/min.33 Sitagliptin may be used at doses of 25 mg daily in ESRD, irrespective of dialysis timing (Table 1).
Other drugs of this class are being developed. Saxagliptin (Onglyza) was recently approved by the US Food and Drug Administration and can be used at a dosage of 2.5 mg daily after dialysis.
Sitagliptin has been associated with gastrointestinal adverse effects. Anaphylaxis, angioedema, and Steven-Johnson syndrome have been reported. The risk of hypoglycemia increases when sitagliptin is used with sulfonylureas.
ESRD REDUCES INSULIN CLEARANCE
In healthy nondiabetic people, the pancreatic beta cells secrete half of the daily insulin requirement (about 0.5 units/kg/day) at a steady basal rate independent of glucose levels. The other half is secreted in response to prandial glucose stimulation.
Secreted into the portal system, insulin passes through the liver, where about 75% is metabolized, with the remaining 25% metabolized by the kidneys. About 60% of the insulin in the arterial bed is filtered by the glomerulus, and 40% is actively secreted into the nephric tubules.34 Most of the insulin in the tubules is metabolized into amino acids, and only 1% of insulin is secreted intact.
For diabetic patients receiving exogenous insulin, renal metabolism plays a more significant role since there is no first-pass metabolism in the liver. As renal function starts to decline, insulin clearance does not change appreciably, due to compensatory peritubular insulin uptake.35 But once the GFR drops below 20 mL/min, the kidneys clear markedly less insulin, an effect compounded by a decrease in the hepatic metabolism of insulin that occurs in uremia.36 Thus, despite the increase in insulin resistance caused by renal failure, the net effect is a reduced requirement for exogenous insulin in ESRD.37
Aisenpreis et al38 showed that the pharmacokinetic profile of insulin lispro (Humalog), which has a short onset of action and a short duration of action, may not only facilitate the correction of hyperglycemia but may also decrease the risk of late hypoglycemic episodes, which is of increased relevance in hemodialysis patients.
On the basis of the available evidence,39,40 we recommend a long-acting insulin such as insulin glargine (Lantus) or NPH insulin for basal requirements, along with a rapid-acting insulin analogue such as lispro or insulin aspart (NovoLog) before meals two or three times daily.
When the GFR drops to between 10 and 50 mL/min, the total insulin dose should be reduced by 25%; once the filtration rate is below 10 mL/min, as in ESRD, the insulin dose should be decreased by 50% from the previous amount.41,42
The newer insulins such as glargine and lispro are more favorable than NPH and regular insulin, but they cost more, which can be an obstacle for some patients.
OBSERVATIONS AND RECOMMENDATIONS
After reviewing the available evidence for the use of diabetic therapy in ESRD, we offer the following observations and recommendations:
- Glycemic control and monitoring in ESRD are complex.
- Patients with ESRD are especially susceptible to hypoglycemia, so diabetic drug therapy requires special caution.
- ESRD patients need ongoing diabetes education, with an emphasis on how to recognize and treat hypoglycemia.
- Diabetic pharmacotherapy in ESRD should be individualized. The targets of therapy are a hemoglobin A1c value between 6% and 7%, a fasting blood glucose level less than 140 mg/dL, and a postprandial glucose level less than 200 mg/dL.
- Of the oral antidiabetic drugs available, glipizide, sitagliptin, and saxagliptin may be used in ESRD. Glipizide, starting with 2.5 mg daily, should be reserved for ESRD patients with a hemoglobin A1c value less than 8.5%.
- Thiazolidinediones may cause fluid overload and thus should be avoided in ESRD.
- We recommend a long-acting insulin (glargine or NPH) for basal requirements, along with rapid-acting insulin before meals two or three times daily.
- The newer basal insulin (glargine) and rapid-acting insulin analogues (lispro or aspart insulin) are more favorable than NPH and regular insulin, but their higher cost could be an issue.
- Some patients may prefer a premixed insulin combination for convenience of dosing. In that case, NPH plus lispro insulin may be better than NPH plus regular insulin.
- For ESRD patients with type 1 diabetes, insulin therapy should be started at 0.5 IU/kg, which is half the calculated dose in patients without renal failure.
- For ESRD patients with type 2 diabetes, insulin therapy should be started at a total daily dose of 0.25 IU/kg.
- Further adjustments to the regimen should be individualized based on self-monitored blood glucose patterns.
- We recommend consulting an endocrinologist with expertise in managing diabetes in ESRD.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
- National Institute of Diabetes and Digestive and Kidney Diseases: United States Renal Data System: USRDS 2005 Annual Data Report. Bethesda, MD: National Institutes of Health, 2005.
- Wu MS, Yu CC, Yang CW, et al. Poor pre-dialysis glycaemic control is a predictor of mortality in type II diabetic patients on maintenance haemodialysis. Nephrol Dial Transplant 1997; 12:2105–2110.
- Morioka T, Emoto M, Tabata T, et al. Glycemic control is a predictor of survival for diabetic patients on hemodialysis. Diabetes Care 2001; 24:909–913.
- McMurray SD, Johnson G, Davis S, McDougall K. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis 2002; 40:566–575.
- Oomichi T, Emoto M, Tabata T, et al. Impact of glycemic control on survival of diabetic patients on chronic regular hemodialysis: a 7-year observational study. Diabetes Care 2006; 29:1496–1500.
- Williams ME, Lacson E, Teng M, Ofsthun N, Lazarus JM. Hemodialyzed type I and type II diabetic patients in the US: characteristics, glycemic control, and survival. Kidney Int 2006; 70:1503–1509.
- Tzamaloukas AH, Yuan ZY, Murata GH, Avasthi PS, Oreopoulos DG. Clinical associations of glycemic control in diabetics on CAPD. Adv Perit Dial 1993; 9:291–294.
- Tzamaloukas AH, Murata GH, Zager PG, Eisenberg B, Avasthi PS. The relationship between glycemic control and morbidity and mortality for diabetics on dialysis. ASAIO J 1993; 39:880–885.
- Kalantar-Zadeh K, Kopple JD, Regidor DL, et al. A1C and survival in maintenance hemodialysis patients. Diabetes Care 2007; 30:1049–1055.
- Kovesdy C, Sharma K, Kalantar-Zadeh. Glycemic control in diabetic CKD patients: where do we stand? Am J Kidney Dis 2008; 52:766–777.
- Mak RH. Intravenous 1,25-dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int 1992; 41:1049–1054.
- Hajjar SM, Fadda GZ, Thanakitcharu P, Smogorzewski M, Massry SG. Reduced activity of Na(+)-K+ ATPase of pancreatic islet cells in chronic renal failure: role of secondary hyperparathyroidism. J Am Soc Nephrol 1992; 2:1355–1359.
- Grodstein GP, Blumenkrantz MJ, Kopple JD, Moran JK, Coburn JW. Glucose absorption during continuous ambulatory peritoneal dialysis. Kidney Int 1981; 19:564–567.
- Joy MS, Cefali WT, Hogan SL, Nachman PH. Long-term glycemic control measurements in diabetic patients receiving hemodialysis. Am J Kidney Dis 2002; 39:297–307.
- Lamb E, Venton TR, Cattell WR, Dawnay A. Serum glycated albumin and fructosamine in renal dialysis patients. Nephron 1993; 64:82–88.
- Inaba M, Okuno S, Kumeda Y, et al; Osaka CKD Expert Research Group. Glycated albumin is a better glycemic indicator than glycated hemoglobin values in hemodialysis patients with diabetes: effect of anemia and erythropoietin injection. J Am Soc Nephrol 2007; 18:896–903.
- Constanti C, Simo JM, Joven J, Camps J. Serum fructosamine concentration in patients with nephrotic syndrome and with cirrhosis of the liver: the influence of hypoalbuminaemia and hypergammaglobulinaemia. Ann Clin Biochem 1992; 29:437–442.
- Ford HC, Lim WC, Crooke MJ. Hemoglobin A1 and serum fructosamine levels in hyperthyroidism. Clin Chim Acta 1987; 166:317–321.
- Mak RH. Impact of end-stage renal disease and dialysis on glycemic control. Semin Dial 2000; 13:4–8.
- Skillman TG, Feldman JM. The pharmacology of sulfonylureas. Am J Med 1981; 70:361–372.
- Krepinsky J, Ingram AJ, Clase CM. Prolonged sulfonylurea-induced hypoglycemia in diabetic patients with end-stage renal disease. Am J Kidney Dis 2000; 35:500–505.
- Snyder RW, Berns JS. Use of insulin and oral hypoglycemic medications in patients with diabetes mellitus and advanced kidney disease. Semin Dial 2004; 17:365–370.
- United Kingdom Prospective Diabetes Study (UKPDS) 13. Relative efficacy of randomly allocated diet, sulphonylureas, insulin, or metformin in patients with newly diagnosed non-insulin dependent diabetes followed for three years. BMJ 1995; 310:83–88.
- Inoue T, Shibahara N, Miyagawa K, et al. Pharmacokinetics of nateglinide and its metabolites in subjects with type 2 diabetes mellitus and renal failure. Clin Nephrol 2003; 60:90–95.
- Nagai T, Imamura M, Iizuka K, Mori M. Hypoglycemia due to nateglinide administration in diabetic patient with chronic renal failure. Diabetes Res Clin Pract 2003; 59:191–194.
- Thompson-Culkin K, Zussman B, Miller AK, Freed MI. Pharmacokinetics of rosiglitazone in patients with end-stage renal disease. J Int Med Res 2002; 30:391–399.
- Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004; 27:256–263.
- Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P; Sitagliptin Study 035 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab 2007; 9:733–745.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G, et al; Sitagliptin Study 020 Group Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Goldstein BJ, Feinglos MN, Lunceford JK, Johnson J, Williams-Herman DE; Sitagliptin 036 Study Group. Effect of initial combination therapy with sitagliptin, a dipeptidyl peptidase-4 inhibitor, and metformin on glycemic control in patients with type 2 diabetes. Diabetes Care 2007; 30:1979–1987.
- Nauck MA, Meininger G, Sheng D, Terranella L, Stein PP; Sitagliptin Study 024 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, compared with the sulfonylurea, glipizide, in patients with type 2 diabetes inadequately controlled on metformin alone: a randomized, double-blind, non-inferiority trial. Diabetes Obes Metab 2007; 9:194–205.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- Bergman AJ, Cote J, Yi B, et al. Effect of renal insufficiency on the pharmacokinetics of sitagliptin, a dipeptidyl peptidase-4 inhibitor. Diabetes Care 2007; 30:1862–1864.
- Carone FA, Peterson DR. Hydrolysis and transport of small peptides by the proximal tubule. Am J Physiol 1980; 238:F151–F158.
- Rabkin R, Simon NM, Steiner S, Colwell JA. Effects of renal disease on renal uptake and excretion of insulin in man. N Engl J Med 1970; 282:182–187.
- Mak RH, DeFronzo RA. Glucose and insulin metabolism in uremia. Nephron 1992; 61:377–382.
- Biesenbach G, Raml A, Schmekal B, Eichbauer-Sturm G. Decreased insulin requirement in relation to GFR in nephropathic type 1 and insulin-treated type 2 diabetic patients. Diabet Med 2003; 20:642–645.
- Aisenpreis U, Pfützner A, Giehl M, Keller F, Jehle PM. Pharmacokinetics and pharmacodynamics of insulin Lispro compared with regular insulin in hemodialysis patients with diabetes mellitus. Nephrol Dial Transplant 1999; 14( suppl 4):5–6.
- Tunbridge FK, Newens A, Home PD, et al. A comparison of human ultralente- and lente-based twice-daily injection regimens. Diabet Med 1989; 6:496–501.
- Freeman SL, O’Brien PC, Rizza RA. Use of human ultralente as the basal insulin component in treatment of patients with IDDM. Diabetes Res Clin Pract 1991; 12:187–192.
- Charpentier G, Riveline JP, Varroud-Vial M. Management of drugs affecting blood glucose in diabetic patients with renal failure. Diabetes Metab 2000; 26( suppl 4):73–85.
- Aronoff GR, Berns JS, Brier ME, et al, eds. Drug Prescribing in Renal Failure: Dosing Guidelines for Adults, 4th ed. Philadelphia, PA: American College of Physicians, 1999.
KEY POINTS
- Blood glucose levels can fluctuate widely due to various and opposing effects of ESRD and dialysis.
- The hemoglobin A1c level can be falsely high in ESRD, but it is still a reasonable measure of glycemic control in this population.
- Most diabetes drugs are excreted at least in part by the kidney, so that patients in ESRD are at greater risk of hypoglycemia.
- Insulin is the cornerstone of treatment, since most oral diabetes drugs are contraindicated or not recommended in this population. Insulin doses should be lowered in those with low glomerular filtration rates.
Alternative modes of mechanical ventilation: A review for the hospitalist
Technologic advances and computerized control of mechanical ventilators have made it possible to deliver ventilatory assistance in new modes. Driving these innovations is the desire to prevent ventilator-induced lung injury, improve patient comfort, and liberate the patient from mechanical ventilation as soon as possible.
We call these innovations “alternative” modes to differentiate them from the plain volume-control and pressure-control modes. Some clinicians rarely use these new modes, but in some medical centers they have become the most common ones used, or are being used unknowingly (the operator misunderstands the mode name). The information we provide on these modes of ventilation is by no means an endorsement of their use, but rather a tool to help the clinician understand their physiologic, theoretical, and clinical effects.
We focused on two goals:
- Explain what the mode does
- Briefly review the theoretical benefits and the actual evidence supporting these alternative modes of ventilation.

STANDARD NOMENCLATURE NEEDED
Since its invention, mechanical ventilation has been plagued by multiple names being used to describe the same things. For example, volume-control ventilation is also called volume-cycled ventilation, assist-control ventilation, volume-limited ventilation, and controlled mechanical ventilation. Similarly, multiple abbreviations are used, each depending on the brand of ventilator, and new acronyms have been added in recent years as new modes have been developed. The vast number of names and modes can confuse even the most seasoned critical care physician.
Efforts to establish a common nomenclature are under way.1
WHAT IS A MODE?
A mode of mechanical ventilation has three essential components:
- The control variable
- The breath sequence
- The targeting scheme.
Similar modes may require more detailed descriptions to distinguish them, but the basic function can be explained by these three components.
The control variable
In general, inspiration is an active process, driven by the patient’s effort, the ventilator, or both, while expiration is passive. For simplicity, in this article a mechanical breath means the inspiratory phase of the breath.
The machine can only control the volume (and flow) or the pressure given. The breaths can be further described on the basis of what triggers the breath, what limits it (the maximum value of a control variable), and what ends (cycles) it.
The breath sequence
There are three possible breath sequences:

- Intermittent mandatory ventilation, in which the patient can take spontaneous breaths between mandatory breaths
- Continuous spontaneous ventilation, in which all breaths are spontaneous (Table 1).

The targeting scheme
The targeting or feedback scheme refers to the ventilator settings and programming that dictate its response to the patient’s lung compliance, lung resistance, and respiratory effort. The regulation can be as simple as controlling the pressure in pressure-control mode, or it can be based on a complicated algorithm.
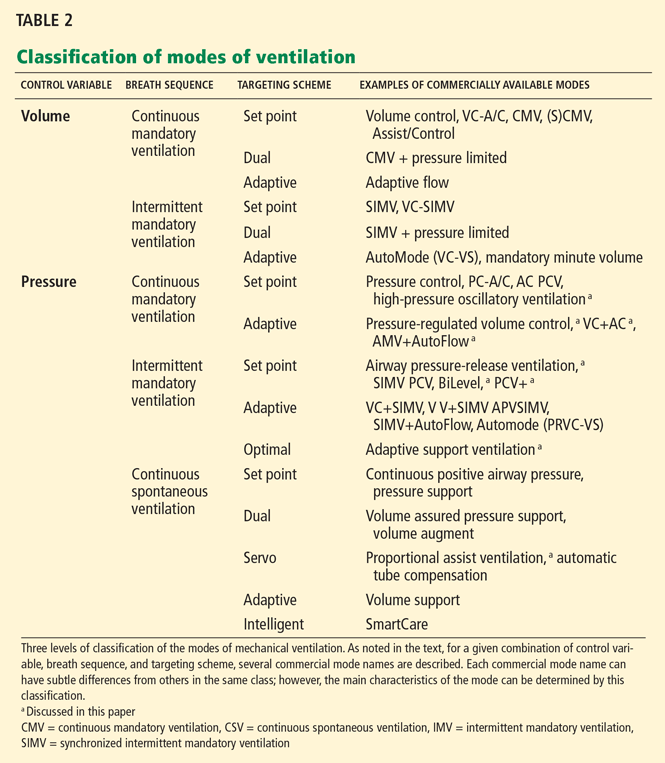
ADAPTIVE PRESSURE CONTROL
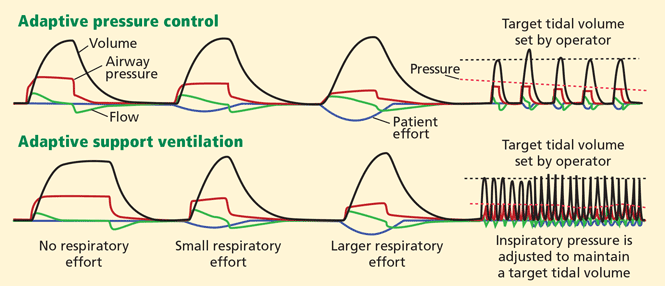
Other names for adaptive pressure control
- Pressure Regulated Volume Control (Maquet Servo-i, Rastatt, Germany)
- AutoFlow (Dräger Medical AG, Lübeck, Germany)
- Adaptive Pressure Ventilation (Hamilton Galileo, Hamilton Medical AG, Bonaduz, Switzerland)
- Volume Control+ (Puritan Bennett, Tyco Healthcare; Mansfield, MA)
- Volume Targeted Pressure Control, Pressure Controlled Volume Guaranteed (Engström, General Electric, Madison, WI).
What does adaptive pressure control do?
The APC mode delivers pressure-controlled breaths with an adaptive targeting scheme (Table 2).
In pressure-control ventilation, tidal volumes depend on the lung’s physiologic mechanics (compliance and resistance) and patient effort (Figure 1). Therefore, the tidal volume varies with changes in lung physiology (ie, larger or smaller tidal volumes than targeted).
To overcome this effect, a machine in APC mode adjusts the inspiratory pressure to deliver the set minimal target tidal volume. If tidal volume increases, the machine decreases the inspiratory pressure, and if tidal volume decreases, the machine increases the inspiratory pressure. However, if the patient effort is large enough, the tidal volume will increase in spite of decreasing the inspiratory pressure (Figure 2). The adjustments to the inspiratory pressure occur after the tidal volume is off-target in a number of breaths.
Common sources of confusion with adaptive pressure control
First, APC is not a volume-control mode. In volume control, the tidal volume does not change; in APC the tidal volume can increase or decrease, and the ventilator will adjust the inflation pressure to achieve the target volume. Thus, APC guarantees an average minimum tidal volume but not a maximum tidal volume.
Second, a characteristic of pressure control (and hence, APC) is that the flow of gas varies to maintain constant airway pressure (ie, maintain the set inspiratory pressure). This characteristic allows a patient who generates an inspiratory effort to receive flow as demanded, which is likely more comfortable. This is essentially different from volume control, in which flow is set by the operator and hence is fixed. Thus, if the patient effort is strong enough (Figure 1), this leads to what is called flow asynchrony, in which the patient does not get the flow asked for in a breath.
Ventilator settings in adaptive pressure control
Ventilator settings in APC are:
- Tidal volume
- Time spent in inspiration (inspiratory time)
- Frequency
- Fraction of inspired oxygen (Fio2)
- Positive end-expiratory pressure (PEEP).
Some ventilators also require setting the speed to reach the peak pressure (also known as slope percent or inspiratory rise time).
Clinical applications of adaptive pressure control
This mode is designed to maintain a consistent tidal volume during pressure-control ventilation and to promote inspiratory flow synchrony. It is a means of automatically reducing ventilatory support (ie, weaning) as the patient’s inspiratory effort becomes stronger, as in awakening from anesthesia.
APC may not be ideal for patients who have an inappropriately increased respiratory drive (eg, in severe metabolic acidosis), since the inspiratory pressure will decrease to maintain the targeted average tidal volume, inappropriately shifting the work of breathing onto the patient.
Theoretical benefits of adaptive pressure control
APC guarantees a minimum average tidal volume (unless the pressure alarm threshold is set too low, so that the target tidal volume is not delivered). Other theoretical benefits are flow synchrony, less ventilator manipulation by the operator, and automatic weaning of ventilator support.
Evidence of benefit of adaptive pressure control
Physiologic benefits. This mode has lower peak inspiratory pressures than does volume-control ventilation,3,4 which is often reported as a positive finding. However, in volume-control mode (the usual comparator), the peak inspiratory pressure is a manifestation of both resistance and compliance. Hence, peak inspiratory pressure is expected to be higher but does not reflect actual lung-distending pressures. It is the plateau pressure, a manifestation of lung compliance, that is related to lung injury.
Patient comfort. APC may increase the work of breathing when using low tidal volume ventilation and when there is increased respiratory effort (drive).5 Interestingly, APC was less comfortable than pressure support ventilation in a small trial.6
Outcomes have not been studied.7
Adaptive pressure control: Bottom line
APC is widely available and widely used, sometimes unknowingly (eg, if the operator thinks it is volume control). It is relatively easy to use and to set; however, evidence of its benefit is scant.
ADAPTIVE SUPPORT VENTILATION
Adaptive support ventilation (ASV) evolved as a form of mandatory minute ventilation implemented with adaptive pressure control. Mandatory minute ventilation is a mode that allows the operator to preset a target minute ventilation, and the ventilator then supplies mandatory breaths, either volume- or pressure-controlled, if the patient’s spontaneous breaths generate a lower minute ventilation.
ASV automatically selects the appropriate tidal volume and frequency for mandatory breaths and the appropriate tidal volume for spontaneous breaths on the basis of the respiratory system mechanics and target minute alveolar ventilation.
Described in 1994 by Laubscher et al,8,9 ASV became commercially available in 1998 in Europe and in 2007 in the United States (Hamilton Galileo ventilator, Hamilton Medical AG). This is the first commercially available ventilator that uses an “optimal” targeting scheme (see below).
What does adaptive support ventilation do?
ASV delivers pressure-controlled breaths using an adaptive (optimal) scheme (Table 2). “Optimal,” in this context, means minimizing the mechanical work of breathing: the machine selects a tidal volume and frequency that the patient’s brain would presumably select if the patient were not connected to a ventilator. This pattern is assumed to encourage the patient to generate spontaneous breaths.
The ventilator calculates the normal required minute ventilation based on the patient’s ideal weight and estimated dead space volume (ie, 2.2 mL/kg). This calculation represents 100% of minute ventilation. The clinician at the bedside sets a target percent of minute ventilation that the ventilator will support—higher than 100% if the patient has increased requirements due, eg, to sepsis or increased dead space, or less than 100% during weaning.
The ventilator initially delivers test breaths, in which it measures the expiratory time constant for the respiratory system and then uses this along with the estimated dead space and normal minute ventilation to calculate an optimal breathing frequency in terms of mechanical work.
The optimal or target tidal volume is calculated as the normal minute ventilation divided by the optimal frequency. The target tidal volume is achieved by the use of APC (see above) (Figure 2). This means that the pressure limit is automatically adjusted to achieve an average delivered tidal volume equal to the target. The ventilator continuously monitors the respiratory system mechanics and adjusts its settings accordingly.
The ventilator adjusts its breaths to avoid air trapping by allowing enough time to exhale, to avoid hypoventilation by delivering tidal volume greater than the dead space, and to avoid volutrauma by avoiding large tidal volumes.
Ventilator settings in adaptive support ventilation
Ventilator settings in ASV are:
- Patient height (to calculate the ideal body weight)
- Sex
- Percent of normal predicted minute ventilation goal
- Fio2
- PEEP.
Clinical applications of adaptive support ventilation
ASV is intended as a sole mode of ventilation, from initial support to weaning.
Theoretical benefits of adaptive support ventilation
In theory, ASV offers automatic selection of ventilator settings, automatic adaptation to changing patient lung mechanics, less need for human manipulation of the machine, improved synchrony, and automatic weaning.
Evidence of benefit of adaptive support ventilation
Physiologic benefits. Ventilator settings are adjusted automatically. ASV selects different tidal volume-respiratory rate combinations based on respiratory mechanics in passive and paralyzed patients.10–12 In actively breathing patients, there was no difference in the ventilator settings chosen by ASV for different clinical scenarios (and lung physiology).10 Compared with pressure-controlled intermittent mandatory ventilation, with ASV, the inspiratory load is less and patient-ventilator interaction is better.13
Patient-ventilator synchrony and comfort have not been studied.
Outcomes. Two trials suggest that ASV may decrease time on mechanical ventilation.14,15 However, in another trial,16 compared with a standard protocol, ASV led to fewer ventilator adjustments but achieved similar postsurgical weaning outcomes. The effect of this mode on the death rate has not been examined.17,18
Adaptive support ventilation: Bottom line
ASV is the first commercially available mode that automatically selects all the ventilator settings except PEEP and Fio2. These seem appropriate for different clinical scenarios in patients with poor respiratory effort or in paralyzed patients. Evidence of the effect in actively breathing patients and on outcomes such as length of stay or death is still lacking.
PROPORTIONAL ASSIST VENTILATION
Patients who have normal respiratory drive but who have difficulty sustaining adequate spontaneous ventilation are often subjected to pressure support ventilation (PSV), in which the ventilator generates a constant pressure throughout inspiration regardless of the intensity of the patient’s effort.
In 1992, Younes and colleagues19,20 developed proportional assist ventilation (PAV) as an alternative in which the ventilator generates pressure in proportion to the patient’s effort. PAV became commercially available in Europe in 1999 and was approved in the United States in 2006, available on the Puritan Bennett 840 ventilator (Puritan Bennett Co, Boulder, CO). PAV has also been used for noninvasive ventilation, but this is not available in the United States.
Other names for proportional assist ventilation
Proportional Pressure Support (Dräger Medical; not yet available in the United States).
What does proportional assist ventilation do?
This mode delivers pressure-controlled breaths with a servo control scheme (Table 2).
To better understand PAV, we can compare it with PSV. With PSV, the pressure applied by the ventilator rises to a preset level that is held constant (a set-point scheme) until a cycling criterion (a percent of the maximum inspiratory flow value) is reached. The inspiratory flow and tidal volume are the result of the patient’s inspiratory effort, the level of pressure applied, and the respiratory system mechanics.
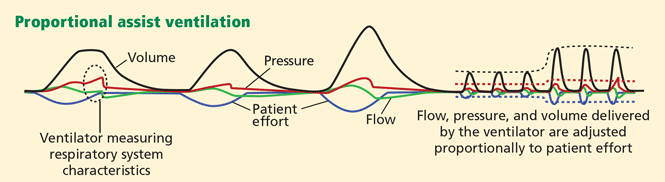
In PAV, as in PSV, all breaths are spontaneous (Table 1). The patient controls the timing and size of the breath. There are no preset pressure, flow, or volume goals, but safety limits on the volume and pressure delivered can be set.
Ventilator settings in proportional assist ventilation
Ventilator settings in PAV are:
- Airway type (endotracheal tube, tracheostomy)
- Airway size (inner diameter)
- Percentage of work supported (assist range 5%–95%)
- Tidal volume limit
- Pressure limit
- Expiratory sensitivity (normally, as inspiration ends, flow should stop; this parameter tells the ventilator at what flow to end inspiration).
Caution when assessing the literature. Earlier ventilator versions, ie, Dräger and Manitoba (University of Manitoba, Winnipeg, MB, Canada), which are not available in the United States, required the repeated calculation of the respiratory system mechanics and the manual setting of flow and volume assists (amplification factors) independently. To overcome this limitation, new software automatically adjusts the flow and volume amplification to support the loads imposed by the automatically measured values of resistance and elastance (inverse of compliance) of the respiratory system.21 This software is included in the model (Puritan Bennett) available in the United States.
Clinical applications of proportional assist ventilation
The PAV mode is indicated for maximizing ventilator patient synchrony for assisted spontaneous ventilation.
PAV is contraindicated in patients with respiratory depression (bradypnea) or large air leaks (eg, bronchopleural fistulas). It should be used with caution in patients with severe hyperinflation, in which the patient may still be exhaling but the ventilator doesn’t recognize it. Another group in which PAV should be used with caution is those with high ventilatory drives, in which the ventilator overestimates respiratory system mechanics. This situation can lead to overassistance due to the “runaway phenomenon,” in which the ventilator continues to provide support even if the patient has stopped inspiration.22
Theoretical benefits of proportional assist ventilation
In theory, PAV should reduce the work of breathing, improve synchrony, automatically adapt to changing patient lung mechanics and effort, decrease the need for ventilator intervention and manipulation, decrease the need for sedation, and improve sleep.
Evidence of benefit of proportional assist ventilation
Physiologic benefits. PAV reduces the work of breathing better than PSV,21 even in the face of changing respiratory mechanics or increased respiratory demand (hypercapnia).23–25 The hemodynamic profile is similar to that in PSV. Tidal volumes are variable; however, in recent reports the tidal volumes were within the lung-protective range (6–8 mL/kg, plateau pressure < 30 cm H20).26,27
Comfort. PAV entails less patient effort and discomfort that PSV does.23,25 PAV significantly reduces asynchrony,27 which in turn may favorably affect sleep in critically ill patients. 28
Outcomes. The probability of spontaneous breathing without assistance was significantly better in critically ill patients ventilated with PAV than with PSV. No trial has reported the effect of PAV on deaths.27,29
Proportional assist ventilation: Bottom line
Extensive basic research has been done with PAV in different forms of respiratory failure, such as obstructive lung disease, acute respiratory distress syndrome (ARDS), and chronic respiratory failure. It fulfills its main goal, which is to improve patient-ventilator synchrony. Clinical experience with PAV in the United States is limited, as it was only recently approved.
AIRWAY PRESSURE-RELEASE VENTILATION AND BIPHASIC POSITIVE AIRWAY PRESSURE
Airway pressure-release ventilation (APRV) was described in 1987 by Stock et al30 as a mode for delivering ventilation in acute lung injury while avoiding high airway pressures. APRV combines high constant positive airway pressure (improving oxygenation and promoting alveolar recruitment) with intermittent releases (causing exhalation).
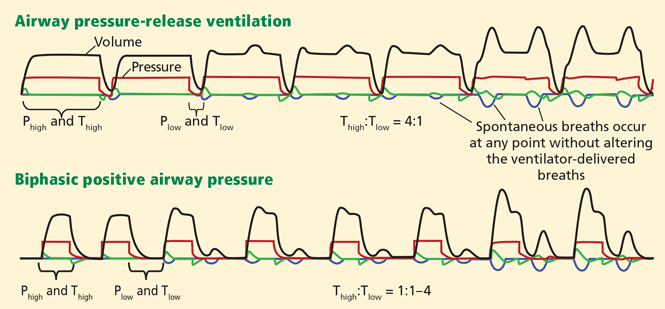
Other names for biphasic positive airway pressure
Other names for biphasic positive airway pressure are:
- BiLevel (Puritan Bennett)
- BIPAP (Dräger Europe)
- Bi Vent (Siemens)
- BiPhasic (Avea, Cardinal Health, Inc, Dublin, OH)
- PCV+ (Dräger Medical)
- DuoPAP (Hamilton).
Caution—name confusion. In North America, BiPAP (Respironics, Murrysville, PA) and BiLevel are used to refer to noninvasive modes of ventilation.
APRV has no other name.
What do these modes do?
These modes deliver pressure-controlled, time-triggered, and time-cycled breaths using a set-point targeting scheme (Table 2). This means that the ventilator maintains a constant pressure (set point) even in the face of spontaneous breaths.
Caution—source of confusion. The term continuous positive airway pressure (CPAP) is often used to describe this mode. However, CPAP is pressure that is applied continuously at the same level; the patient generates all the work to maintain ventilation (“pressure-controlled continuous spontaneous ventilation” in the current nomenclature). In APRV, the airway pressure is intermittently released and reapplied, generating a tidal volume that supports ventilation. In other words, this is a pressure-controlled breath with a very prolonged inspiratory time and a short expiratory time in which spontaneous ventilation is possible at any point (“pressure-controlled intermittent mandatory ventilation” in the current nomenclature).
How these modes are set in the ventilator may also be a source of confusion. To describe the time spent in high and low airway pressures, we use the terms Thigh and Tlow, respectively. By convention, the difference between APRV and biphasic mode is the duration of Tlow (< 1.5 sec for APRV).
Similarly, Phigh and Plow are used to describe the high and low airway pressure. To better understand this concept, you can create the same mode in conventional pressure-control ventilation by thinking of the Thigh as the inspiratory time, the Tlow as the expiratory time, the Phigh as inspiratory pressure, and the Plow as PEEP.
Hence, APRV is an extreme form of inverse ratio ventilation, with an inspiration-to-expiration ratio of 4:1. This means a patient spends most of the time in Phigh and Thigh, and exhalations are short (Tlow and Plow). In contrast, the biphasic mode uses conventional inspiration-expiration ratios (Figure 4).
As with any form of pressure control, the tidal volume is generated by airway pressure rising above baseline (ie, the end-expiratory value). Hence, to ensure an increase in minute ventilation, the mandatory breath rate must be increased (ie, decreasing Thigh, Tlow, or both) or the tidal volume must be increased (ie, increasing the difference between Phigh and Plow). This means that in APRV the Tlow has to happen more often (by increasing the number of breaths) or be more prolonged (allowing more air to exhale). Because unrestricted spontaneous breaths are permitted at any point of the cycle, the patient contributes to the total minute ventilation (usually 10%–40%).
In APRV and biphasic mode, the operator’s set time and pressure in inspiration and expiration will be delivered regardless of the patient’s breathing efforts—the patient’s spontaneous breath does not trigger a mechanical breath. Some ventilators have automatic adjustments to improve the trigger synchrony.
Ventilator settings in APRV and biphasic mode
These modes require the setting of two pressure levels (Phigh and Plow) and two time durations (Thigh and Tlow). One can add pressure support or automatic tube compensation to assist spontaneous breaths. The difference in Tlow generates differences in the Thigh:Tlow ratio: APRV has a short Tlow (an inspiration-expiration ratio of 4:1). Biphasic mode has a conventional inspiration-expiration ratio of 1:1 to 1:4.
Clinical applications
APRV is used in acute lung injury and ARDS. This mode should be used with caution or not at all in patients with obstructive lung disease or inappropriately increased respiratory drive.32–35
Biphasic mode is intended for both ventilation and weaning. In a patient who has poor respiratory effort or who is paralyzed, biphasic is identical to pressure-control/continuous mandatory ventilation.
Theoretical benefits of APRV and biphasic mode
Multiple benefits have been ascribed to these modes. In theory, APRV will maximize and maintain alveolar recruitment, improve oxygenation, lower inflation pressures, and decrease overinflation. Both APRV and biphasic, by preserving spontaneous breathing, will improve ventilation-perfusion matching and gas diffusion, improve the hemodynamic profile (less need for vasopressors, higher cardiac output, reduced ventricular workload, improved organ perfusion), and improve synchrony (decrease the work of breathing and the need for sedation).
Evidence of benefit of APRV and biphasic mode
APRV and biphasic are different modes. However studies evaluating their effects are combined. This is in part the result of the nomenclature confusion and different practice in different countries.36
Physiologic benefits. In studies, spontaneous breaths contributed to 10% to 40% of minute ventilation,37,38 improved ventilation of dependent areas of the lung, improved ventilation-perfusion match and recruitment,39 and improved hemodynamic profile.40
Patient comfort. These modes are thought to decrease the need for analgesia and sedation,38 but a recent trial showed no difference with pressure-controlled intermittent mandatory ventilation.41 Patient ventilator synchrony and comfort have not been studied.32,42
Outcomes. In small trials, these modes made no difference in terms of deaths, but they may decrease the length of mechanical ventilation.38,41,43,44
APRV and biphasic mode: Bottom line
Maintaining spontaneous breathing while on mechanical ventilation has hemodynamic and ventilatory benefits.
APRV and biphasic mode are not the same thing. APRV’s main goal is to maximize mean airway pressure and, hence, lung recruitment, whereas the main goal of the biphasic mode is synchrony.
There is a plethora of ventilator settings and questions related to physiologic effects.33,34,36
Although these modes are widely used in some centers, there is no evidence yet that they are superior to conventional volume- or pressure-control ventilation with low tidal volume for ARDS and acute lung injury. There is no conclusive evidence that these modes improve synchrony, time to weaning, or patient comfort.
HIGH-FREQUENCY OSCILLATORY VENTILATION
High-frequency oscillatory ventilation (HFOV) was first described and patented in 1952 by Emerson and was clinically developed in the early 1970s by Lunkenheimer.45
The goal of HFOV is to minimize lung injury; its characteristics (discussed below) make it useful in patients with severe ARDS. The US Food and Drug Administration approved it for infants in 1991 and for children in 1995. The adult model has been available since 1993, but it was not approved until 2001 (SensorMedics 3100B, Cardinal Health, Inc).
Other names for high-frequency oscillatory ventilation
While HFOV has no alternative names, the following acronyms describe similar modes:
- HFPPV (high-frequency positive pressure ventilation)
- HFJV (high-frequency jet ventilation)
- HFFI (high-frequency flow interruption)
- HFPV (high-frequency percussive ventilation)
- HFCWO (high-frequency chest wall oscillation).
All of these modes require different specialized ventilators.
What does high-frequency oscillatory ventilation do?
Conceptually, HFOV is a form of pressure-controlled intermittent mandatory ventilation with a set-point control scheme. In contrast to conventional pressure-controlled intermittent mandatory ventilation, in which relatively small spontaneous breaths may be superimposed on relatively large mandatory breaths, HFOV superimposes very small mandatory breaths (oscillations) on top of spontaneous breaths.
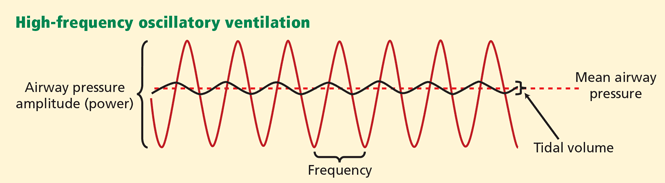
Adult patients are usually paralyzed or deeply sedated, since deep spontaneous breathing will trigger alarms and affect ventilator performance.
To manage ventilation (CO2 clearance), one or several of the following maneuvers can be done: decrease the oscillation frequency, increase the amplitude of the oscillations, increase the inspiratory time, or increase bias flow (while allowing an endotracheal tube cuff leak). Oxygenation adjustments are controlled by manipulating the mean airway pressure and the Fio2.
Ventilator settings in high-frequency oscillatory ventilation
Ventilator settings in HFOV are46:
- Airway pressure amplitude (delta P or power)
- Mean airway pressure
- Percent inspiration
- Inspiratory bias flow
- Fio2.
Clinical applications of high-frequency oscillatory ventilation
This mode is usually reserved for ARDS patients for whom conventional ventilation is failing. A recently published protocol46 suggests considering HFOV when there is oxygenation failure (Fio2 ≥ 0.7 and PEEP ≥ 14 cm H2O) or ventilation failure (pH < 7.25 with tidal volume ≥ 6 mL/kg predicted body weight and plateau airway pressure ≥ 30 cm H2O).
This mode is contraindicated when there is known severe airflow obstruction or intracranial hypertension.
Theoretical benefits of high-frequency oscillatory ventilation
Conceptually, HFOV can provide the highest mean airway pressure paired with the lowest tidal volume of any mode. These benefits might make HFOV the ideal lung-protective ventilation strategy.
Evidence of benefit of high-frequency oscillatory ventilation
Physiologic benefits. Animal models have shown less histologic damage and lung inflammation with HFOV than with high-tidal-volume conventional ventilation47,48 and low-tidal-volume conventional ventilation.49
Patient comfort has not been studied. However, current technology does impose undue work of breathing in spontaneously breathing patients.50
Outcomes. Several retrospective case series have described better oxygenation with HFOV as rescue therapy for severe ARDS than with conventional mechanical ventilation. Two randomized controlled trials have studied HFOV vs high-tidal-volume conventional mechanical ventilation for early severe ARDS; HFOV was safe but made no difference in terms of deaths.42,51–54
High-frequency oscillatory ventilation: Bottom line
In theory, HFOV provides all the benefits of an ideal lung-protective strategy, at least for paralyzed or deeply sedated patients. Animal studies support these concepts. In human adults, HFOV has been shown to be safe and to provide better oxygenation but no improvement in death rates compared with conventional mechanical ventilation. Currently, HFOV is better reserved for patients with severe ARDS for whom conventional mechanical ventilation is failing.
- Chatburn RL. Classification of ventilator modes: update and proposal for implementation. Respir Care 2007; 52:301–323.
- Chatburn RL. Computer control of mechanical ventilation. Respir Care 2004; 49:507–517.
- Alvarez A, Subirana M, Benito S. Decelerating flow ventilation effects in acute respiratory failure. J Crit Care 1998; 13:21–25.
- Guldager H, Nielsen SL, Carl P, Soerensen MB. A comparison of volume control and pressure regulated volume control ventilation in acute respiratory failure. Crit Care 1997; 1:75–77.
- Kallet RH, Campbell AR, Dicker RA, Katz JA, Mackersie RC. Work of breathing during lung protective ventilation in patients with acute lung injury and acute respiratory distress syndrome: a comparison between volume and pressure regulated breathing modes. Respir Care 2005; 50:1623–1631.
- Betensley AD, Khalid I, Crawford J, Pensler RA, DiGiovine B. Patient comfort during pressure support and volume controlled continuous mandatory ventilation. Respir Care 2008; 53:897–902.
- Branson RD, Chatburn RL. Controversies in the critical care setting. Should adaptive pressure control modes be utilized for virtually all patients receiving mechanical ventilation? Respir Care 2007; 52:478–485.
- Laubscher TP, Frutiger A, Fanconi S, Jutzi H, Brunner JX. Automatic selection of tidal volume, respiratory frequency and minute ventilation in intubated ICU patients as start up procedure for closed-loop controlled ventilation. Int J Clin Monit Comput 1994; 11:19–30.
- Laubscher TP, Heinrichs W, Weiler N, Hartmann G, Brunner JX. An adaptive lung ventilation controller. IEEE Trans Biomed Eng 1994; 41:51–59.
- Arnal JM, Wysocki M, Nafati C, et al. Automatic selection of breathing pattern using adaptive support ventilation. Intensive Care Med 2008; 34:75–81.
- Campbell RS, Sinamban RP, Johannigman JA, et al. Clinical evaluation of a new closed loop ventilation mode: adaptive supportive ventilation (ASV). Crit Care 1999; 3( suppl 1):083.
- Belliato M, Palo A, Pasero D, Iotti GA, Mojoli F, Braschi A. Evaluation of adaptive support ventilation in paralysed patients and in a physical lung model. Int J Artif Organs 2004; 27:709–716.
- Tassaux D, Dalmas E, Gratadour P, Jolliet P. Patient ventilator interactions during partial ventilatory support: a preliminary study comparing the effects of adaptive support ventilation with synchronized intermittent mandatory ventilation plus inspiratory pressure support. Crit Care Med 2002; 30:801–807.
- Gruber PC, Gomersall CD, Leung P, et al. Randomized controlled trial comparing adaptive-support ventilation with pressure-regulated volume-controlled ventilation with automode in weaning patients after cardiac surgery. Anesthesiology 2008; 109:81–87.
- Sulzer CF, Chiolero R, Chassot PG, et al. Adaptive support ventilation for fast tracheal extubation after cardiac surgery: a randomized controlled study. Anesthesiology 2001; 95:1339–1345.
- Petter AH, Chiolèro RL, Cassina T, Chassot PG, Müller XM, Revelly JP. Automatic “respirator/weaning” with adaptive support ventilation: the effect on duration of endotracheal intubation and patient management. Anesth Analg 2003; 97:1743–1750.
- Brunner JX, Iotti GA. Adaptive support ventilation (ASV). Minerva Anestesiol 2002; 68:365–368.
- Campbell RS, Branson RD, Johannigman JA. Adaptive support ventilation. Respir Care Clin North Am 2001; 7:425–440.
- Younes M. Proportional assist ventilation, a new approach to ventilatory support. Theory. Am Rev Respir Dis 1992; 145:114–120.
- Younes M, Puddy A, Roberts D, et al. Proportional assist ventilation. Results of an initial clinical trial. Am Rev Respir Dis 1992; 145:121–129.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Respiratory load compensation during mechanical ventilatio—proportional assist ventilation with load-adjustable gain factors versus pressure support. Intensive Care Med 2006; 32:692–699.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Effect of different levels of pressure support and proportional assist ventilation on breathing pattern, work of breathing and gas exchange in mechanically ventilated hypercapnic COPD patients with acute respiratory failure. Respiration 2003; 70:355–361.
- Grasso S, Puntillo F, Mascia L, et al. Compensation for increase in respiratory workload during mechanical ventilation. Pressure support versus proportional assist ventilation. Am J Respir Crit Care Med 2000; 161:819–826.
- Wrigge H, Golisch W, Zinserling J, Sydow M, Almeling G, Burchardi H. Proportional assist versus pressure support ventilation: effects on breathing pattern and respiratory work of patients with chronic obstructive pulmonary disease. Intensive Care Med 1999; 25:790–798.
- Ranieri VM, Giuliani R, Mascia L, et al. Patient ventilator interaction during acute hypercapnia: pressure support vs. proportional assist ventilation. J Appl Physiol 1996; 81:426–436.
- Kondili E, Xirouchaki N, Vaporidi K, Klimathianaki M, Georgopoulos D. Short-term cardiorespiratory effects of proportional assist and pressure support ventilation in patients with acute lung injury/acute respiratory distress syndrome. Anesthesiology 2006; 105:703–708.
- Xirouchaki N, Kondili E, Vaporidi K, et al. Proportional assist ventilation with load-adjustable gain factors in critically ill patients: comparison with pressure support. Intensive Care Med 2008; 34:2026–2034.
- Bosma K, Ferreyra G, Ambrogio C, et al. Patient ventilator interaction and sleep in mechanically ventilated patients: pressure support versus proportional assist ventilation. Crit Care Med 2007; 35:1048–1054.
- Sinderby C, Beck J. Proportional assist ventilation and neurally adjusted ventilatory assist—better approaches to patient ventilator synchrony? Clin Chest Med 2008; 29:329–342.
- Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med 1987; 15:462–466.
- Baum M, Benzer H, Putensen C, Koller W, Putz G. [Biphasic positive airway pressure (BIPAP)—a new form of augmented ventilation]. Anaesthesist 1989; 38:452–458.
- Seymour CW, Frazer M, Reilly PM, Fuchs BD. Airway pressure release and biphasic intermittent positive airway pressure ventilation: are they ready for prime time? J Trauma 2007; 62:1298–1308.
- Myers TR, MacIntyre NR. Respiratory controversies in the critical care setting. Does airway pressure release ventilation offer important new advantages in mechanical ventilator support? Respir Care 2007; 52:452–458.
- Neumann P, Golisch W, Strohmeyer A, Buscher H, Burchardi H, Sydow M. Influence of different release times on spontaneous breathing pattern during airway pressure release ventilation. Intensive Care Med 2002; 28:1742–1749.
- Calzia E, Lindner KH, Witt S, et al. Pressure-time product and work of breathing during biphasic continuous positive airway pressure and assisted spontaneous breathing. Am J Respir Crit Care Med 1994; 150:904–910.
- Rose L, Hawkins M. Airway pressure release ventilation and biphasic positive airway pressure: a systematic review of definitional criteria. Intensive Care Med 2008; 34:1766–1773.
- Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Longterm effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med 1994; 149:1550–1556.
- Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001; 164:43–49.
- Davis K, Johnson DJ, Branson RD, Campbell RS, Johannigman JA, Porembka D. Airway pressure release ventilation. Arch Surg 1993; 128:1348–1352.
- Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001; 5:221–226.
- Varpula T, Valta P, Niemi R, Takkunen O, Hynynen M, Pettilä VV. Airway pressure release ventilation as a primary ventilatory mode in acute respiratory distress syndrome. Acta Anaesthesiol Scand 2004; 48:722–731.
- Siau C, Stewart TE. Current role of high frequency oscillatory ventilation and airway pressure release ventilation in acute lung injury and acute respiratory distress syndrome. Clin Chest Med 2008; 29:265–275.
- Rathgeber J, Schorn B, Falk V, Kazmaier S, Spiegel T, Burchardi H. The influence of controlled mandatory ventilation (CMV), intermittent mandatory ventilation (IMV) and biphasic intermittent positive airway pressure (BIPAP) on duration of intubation and consumption of analgesics and sedatives. A prospective analysis in 596 patients following adult cardiac surgery. Eur J Anaesthesiol 1997; 14:576–582.
- Habashi NM. Other approaches to open lung ventilation: airway pressure release ventilation. Crit Care Med 2005; 33 suppl 3:S228–S240.
- Hess D, Mason S, Branson R. High-frequency ventilation design and equipment issues. Respir Care Clin North Am 2001; 7:577–598.
- Fessler HE, Derdak S, Ferguson ND, et al. A protocol for high frequency oscillatory ventilation in adults: results from a roundtable discussion. Crit Care Med 2007; 35:1649–1654.
- Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol 1983; 55:131–138.
- Sedeek KA, Takeuchi M, Suchodolski K, et al. Open-lung protective ventilation with pressure control ventilation, high-frequency oscillation, and intratracheal pulmonary ventilation results in similar gas exchange, hemodynamics, and lung mechanics. Anesthesiology 2003; 99:1102–1111.
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol 2001; 91:1836–1844.
- van Heerde M, Roubik K, Kopelent V, Plötz FB, Markhorst DG. Unloading work of breathing during high-frequency oscillatory ventilation: a bench study. Crit Care 2006; 10:R103.
- Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med 2002; 166:801–808.
- Bollen CW, van Well GT, Sherry T, et al. High-frequency oscillatory ventilation compared with conventional mechanical ventilation in adult respiratory distress syndrome: a randomized controlled trial [ISRCTN24242669]. Crit Care 2005; 9:R430–R439.
- Mehta S, Granton J, MacDonald RJ, et al. High frequency oscillatory ventilation in adults: the Toronto experience. Chest 2004; 126:518–527.
- Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest 2007; 131:1907–1916.
Technologic advances and computerized control of mechanical ventilators have made it possible to deliver ventilatory assistance in new modes. Driving these innovations is the desire to prevent ventilator-induced lung injury, improve patient comfort, and liberate the patient from mechanical ventilation as soon as possible.
We call these innovations “alternative” modes to differentiate them from the plain volume-control and pressure-control modes. Some clinicians rarely use these new modes, but in some medical centers they have become the most common ones used, or are being used unknowingly (the operator misunderstands the mode name). The information we provide on these modes of ventilation is by no means an endorsement of their use, but rather a tool to help the clinician understand their physiologic, theoretical, and clinical effects.
We focused on two goals:
- Explain what the mode does
- Briefly review the theoretical benefits and the actual evidence supporting these alternative modes of ventilation.
STANDARD NOMENCLATURE NEEDED
Since its invention, mechanical ventilation has been plagued by multiple names being used to describe the same things. For example, volume-control ventilation is also called volume-cycled ventilation, assist-control ventilation, volume-limited ventilation, and controlled mechanical ventilation. Similarly, multiple abbreviations are used, each depending on the brand of ventilator, and new acronyms have been added in recent years as new modes have been developed. The vast number of names and modes can confuse even the most seasoned critical care physician.
Efforts to establish a common nomenclature are under way.1
WHAT IS A MODE?
A mode of mechanical ventilation has three essential components:
- The control variable
- The breath sequence
- The targeting scheme.
Similar modes may require more detailed descriptions to distinguish them, but the basic function can be explained by these three components.
The control variable
In general, inspiration is an active process, driven by the patient’s effort, the ventilator, or both, while expiration is passive. For simplicity, in this article a mechanical breath means the inspiratory phase of the breath.
The machine can only control the volume (and flow) or the pressure given. The breaths can be further described on the basis of what triggers the breath, what limits it (the maximum value of a control variable), and what ends (cycles) it.
The breath sequence
There are three possible breath sequences:
- Intermittent mandatory ventilation, in which the patient can take spontaneous breaths between mandatory breaths
- Continuous spontaneous ventilation, in which all breaths are spontaneous (Table 1).
The targeting scheme
The targeting or feedback scheme refers to the ventilator settings and programming that dictate its response to the patient’s lung compliance, lung resistance, and respiratory effort. The regulation can be as simple as controlling the pressure in pressure-control mode, or it can be based on a complicated algorithm.
ADAPTIVE PRESSURE CONTROL
Other names for adaptive pressure control
- Pressure Regulated Volume Control (Maquet Servo-i, Rastatt, Germany)
- AutoFlow (Dräger Medical AG, Lübeck, Germany)
- Adaptive Pressure Ventilation (Hamilton Galileo, Hamilton Medical AG, Bonaduz, Switzerland)
- Volume Control+ (Puritan Bennett, Tyco Healthcare; Mansfield, MA)
- Volume Targeted Pressure Control, Pressure Controlled Volume Guaranteed (Engström, General Electric, Madison, WI).
What does adaptive pressure control do?
The APC mode delivers pressure-controlled breaths with an adaptive targeting scheme (Table 2).
In pressure-control ventilation, tidal volumes depend on the lung’s physiologic mechanics (compliance and resistance) and patient effort (Figure 1). Therefore, the tidal volume varies with changes in lung physiology (ie, larger or smaller tidal volumes than targeted).
To overcome this effect, a machine in APC mode adjusts the inspiratory pressure to deliver the set minimal target tidal volume. If tidal volume increases, the machine decreases the inspiratory pressure, and if tidal volume decreases, the machine increases the inspiratory pressure. However, if the patient effort is large enough, the tidal volume will increase in spite of decreasing the inspiratory pressure (Figure 2). The adjustments to the inspiratory pressure occur after the tidal volume is off-target in a number of breaths.
Common sources of confusion with adaptive pressure control
First, APC is not a volume-control mode. In volume control, the tidal volume does not change; in APC the tidal volume can increase or decrease, and the ventilator will adjust the inflation pressure to achieve the target volume. Thus, APC guarantees an average minimum tidal volume but not a maximum tidal volume.
Second, a characteristic of pressure control (and hence, APC) is that the flow of gas varies to maintain constant airway pressure (ie, maintain the set inspiratory pressure). This characteristic allows a patient who generates an inspiratory effort to receive flow as demanded, which is likely more comfortable. This is essentially different from volume control, in which flow is set by the operator and hence is fixed. Thus, if the patient effort is strong enough (Figure 1), this leads to what is called flow asynchrony, in which the patient does not get the flow asked for in a breath.
Ventilator settings in adaptive pressure control
Ventilator settings in APC are:
- Tidal volume
- Time spent in inspiration (inspiratory time)
- Frequency
- Fraction of inspired oxygen (Fio2)
- Positive end-expiratory pressure (PEEP).
Some ventilators also require setting the speed to reach the peak pressure (also known as slope percent or inspiratory rise time).
Clinical applications of adaptive pressure control
This mode is designed to maintain a consistent tidal volume during pressure-control ventilation and to promote inspiratory flow synchrony. It is a means of automatically reducing ventilatory support (ie, weaning) as the patient’s inspiratory effort becomes stronger, as in awakening from anesthesia.
APC may not be ideal for patients who have an inappropriately increased respiratory drive (eg, in severe metabolic acidosis), since the inspiratory pressure will decrease to maintain the targeted average tidal volume, inappropriately shifting the work of breathing onto the patient.
Theoretical benefits of adaptive pressure control
APC guarantees a minimum average tidal volume (unless the pressure alarm threshold is set too low, so that the target tidal volume is not delivered). Other theoretical benefits are flow synchrony, less ventilator manipulation by the operator, and automatic weaning of ventilator support.
Evidence of benefit of adaptive pressure control
Physiologic benefits. This mode has lower peak inspiratory pressures than does volume-control ventilation,3,4 which is often reported as a positive finding. However, in volume-control mode (the usual comparator), the peak inspiratory pressure is a manifestation of both resistance and compliance. Hence, peak inspiratory pressure is expected to be higher but does not reflect actual lung-distending pressures. It is the plateau pressure, a manifestation of lung compliance, that is related to lung injury.
Patient comfort. APC may increase the work of breathing when using low tidal volume ventilation and when there is increased respiratory effort (drive).5 Interestingly, APC was less comfortable than pressure support ventilation in a small trial.6
Outcomes have not been studied.7
Adaptive pressure control: Bottom line
APC is widely available and widely used, sometimes unknowingly (eg, if the operator thinks it is volume control). It is relatively easy to use and to set; however, evidence of its benefit is scant.
ADAPTIVE SUPPORT VENTILATION
Adaptive support ventilation (ASV) evolved as a form of mandatory minute ventilation implemented with adaptive pressure control. Mandatory minute ventilation is a mode that allows the operator to preset a target minute ventilation, and the ventilator then supplies mandatory breaths, either volume- or pressure-controlled, if the patient’s spontaneous breaths generate a lower minute ventilation.
ASV automatically selects the appropriate tidal volume and frequency for mandatory breaths and the appropriate tidal volume for spontaneous breaths on the basis of the respiratory system mechanics and target minute alveolar ventilation.
Described in 1994 by Laubscher et al,8,9 ASV became commercially available in 1998 in Europe and in 2007 in the United States (Hamilton Galileo ventilator, Hamilton Medical AG). This is the first commercially available ventilator that uses an “optimal” targeting scheme (see below).
What does adaptive support ventilation do?
ASV delivers pressure-controlled breaths using an adaptive (optimal) scheme (Table 2). “Optimal,” in this context, means minimizing the mechanical work of breathing: the machine selects a tidal volume and frequency that the patient’s brain would presumably select if the patient were not connected to a ventilator. This pattern is assumed to encourage the patient to generate spontaneous breaths.
The ventilator calculates the normal required minute ventilation based on the patient’s ideal weight and estimated dead space volume (ie, 2.2 mL/kg). This calculation represents 100% of minute ventilation. The clinician at the bedside sets a target percent of minute ventilation that the ventilator will support—higher than 100% if the patient has increased requirements due, eg, to sepsis or increased dead space, or less than 100% during weaning.
The ventilator initially delivers test breaths, in which it measures the expiratory time constant for the respiratory system and then uses this along with the estimated dead space and normal minute ventilation to calculate an optimal breathing frequency in terms of mechanical work.
The optimal or target tidal volume is calculated as the normal minute ventilation divided by the optimal frequency. The target tidal volume is achieved by the use of APC (see above) (Figure 2). This means that the pressure limit is automatically adjusted to achieve an average delivered tidal volume equal to the target. The ventilator continuously monitors the respiratory system mechanics and adjusts its settings accordingly.
The ventilator adjusts its breaths to avoid air trapping by allowing enough time to exhale, to avoid hypoventilation by delivering tidal volume greater than the dead space, and to avoid volutrauma by avoiding large tidal volumes.
Ventilator settings in adaptive support ventilation
Ventilator settings in ASV are:
- Patient height (to calculate the ideal body weight)
- Sex
- Percent of normal predicted minute ventilation goal
- Fio2
- PEEP.
Clinical applications of adaptive support ventilation
ASV is intended as a sole mode of ventilation, from initial support to weaning.
Theoretical benefits of adaptive support ventilation
In theory, ASV offers automatic selection of ventilator settings, automatic adaptation to changing patient lung mechanics, less need for human manipulation of the machine, improved synchrony, and automatic weaning.
Evidence of benefit of adaptive support ventilation
Physiologic benefits. Ventilator settings are adjusted automatically. ASV selects different tidal volume-respiratory rate combinations based on respiratory mechanics in passive and paralyzed patients.10–12 In actively breathing patients, there was no difference in the ventilator settings chosen by ASV for different clinical scenarios (and lung physiology).10 Compared with pressure-controlled intermittent mandatory ventilation, with ASV, the inspiratory load is less and patient-ventilator interaction is better.13
Patient-ventilator synchrony and comfort have not been studied.
Outcomes. Two trials suggest that ASV may decrease time on mechanical ventilation.14,15 However, in another trial,16 compared with a standard protocol, ASV led to fewer ventilator adjustments but achieved similar postsurgical weaning outcomes. The effect of this mode on the death rate has not been examined.17,18
Adaptive support ventilation: Bottom line
ASV is the first commercially available mode that automatically selects all the ventilator settings except PEEP and Fio2. These seem appropriate for different clinical scenarios in patients with poor respiratory effort or in paralyzed patients. Evidence of the effect in actively breathing patients and on outcomes such as length of stay or death is still lacking.
PROPORTIONAL ASSIST VENTILATION
Patients who have normal respiratory drive but who have difficulty sustaining adequate spontaneous ventilation are often subjected to pressure support ventilation (PSV), in which the ventilator generates a constant pressure throughout inspiration regardless of the intensity of the patient’s effort.
In 1992, Younes and colleagues19,20 developed proportional assist ventilation (PAV) as an alternative in which the ventilator generates pressure in proportion to the patient’s effort. PAV became commercially available in Europe in 1999 and was approved in the United States in 2006, available on the Puritan Bennett 840 ventilator (Puritan Bennett Co, Boulder, CO). PAV has also been used for noninvasive ventilation, but this is not available in the United States.
Other names for proportional assist ventilation
Proportional Pressure Support (Dräger Medical; not yet available in the United States).
What does proportional assist ventilation do?
This mode delivers pressure-controlled breaths with a servo control scheme (Table 2).
To better understand PAV, we can compare it with PSV. With PSV, the pressure applied by the ventilator rises to a preset level that is held constant (a set-point scheme) until a cycling criterion (a percent of the maximum inspiratory flow value) is reached. The inspiratory flow and tidal volume are the result of the patient’s inspiratory effort, the level of pressure applied, and the respiratory system mechanics.
In PAV, as in PSV, all breaths are spontaneous (Table 1). The patient controls the timing and size of the breath. There are no preset pressure, flow, or volume goals, but safety limits on the volume and pressure delivered can be set.
Ventilator settings in proportional assist ventilation
Ventilator settings in PAV are:
- Airway type (endotracheal tube, tracheostomy)
- Airway size (inner diameter)
- Percentage of work supported (assist range 5%–95%)
- Tidal volume limit
- Pressure limit
- Expiratory sensitivity (normally, as inspiration ends, flow should stop; this parameter tells the ventilator at what flow to end inspiration).
Caution when assessing the literature. Earlier ventilator versions, ie, Dräger and Manitoba (University of Manitoba, Winnipeg, MB, Canada), which are not available in the United States, required the repeated calculation of the respiratory system mechanics and the manual setting of flow and volume assists (amplification factors) independently. To overcome this limitation, new software automatically adjusts the flow and volume amplification to support the loads imposed by the automatically measured values of resistance and elastance (inverse of compliance) of the respiratory system.21 This software is included in the model (Puritan Bennett) available in the United States.
Clinical applications of proportional assist ventilation
The PAV mode is indicated for maximizing ventilator patient synchrony for assisted spontaneous ventilation.
PAV is contraindicated in patients with respiratory depression (bradypnea) or large air leaks (eg, bronchopleural fistulas). It should be used with caution in patients with severe hyperinflation, in which the patient may still be exhaling but the ventilator doesn’t recognize it. Another group in which PAV should be used with caution is those with high ventilatory drives, in which the ventilator overestimates respiratory system mechanics. This situation can lead to overassistance due to the “runaway phenomenon,” in which the ventilator continues to provide support even if the patient has stopped inspiration.22
Theoretical benefits of proportional assist ventilation
In theory, PAV should reduce the work of breathing, improve synchrony, automatically adapt to changing patient lung mechanics and effort, decrease the need for ventilator intervention and manipulation, decrease the need for sedation, and improve sleep.
Evidence of benefit of proportional assist ventilation
Physiologic benefits. PAV reduces the work of breathing better than PSV,21 even in the face of changing respiratory mechanics or increased respiratory demand (hypercapnia).23–25 The hemodynamic profile is similar to that in PSV. Tidal volumes are variable; however, in recent reports the tidal volumes were within the lung-protective range (6–8 mL/kg, plateau pressure < 30 cm H20).26,27
Comfort. PAV entails less patient effort and discomfort that PSV does.23,25 PAV significantly reduces asynchrony,27 which in turn may favorably affect sleep in critically ill patients. 28
Outcomes. The probability of spontaneous breathing without assistance was significantly better in critically ill patients ventilated with PAV than with PSV. No trial has reported the effect of PAV on deaths.27,29
Proportional assist ventilation: Bottom line
Extensive basic research has been done with PAV in different forms of respiratory failure, such as obstructive lung disease, acute respiratory distress syndrome (ARDS), and chronic respiratory failure. It fulfills its main goal, which is to improve patient-ventilator synchrony. Clinical experience with PAV in the United States is limited, as it was only recently approved.
AIRWAY PRESSURE-RELEASE VENTILATION AND BIPHASIC POSITIVE AIRWAY PRESSURE
Airway pressure-release ventilation (APRV) was described in 1987 by Stock et al30 as a mode for delivering ventilation in acute lung injury while avoiding high airway pressures. APRV combines high constant positive airway pressure (improving oxygenation and promoting alveolar recruitment) with intermittent releases (causing exhalation).
Other names for biphasic positive airway pressure
Other names for biphasic positive airway pressure are:
- BiLevel (Puritan Bennett)
- BIPAP (Dräger Europe)
- Bi Vent (Siemens)
- BiPhasic (Avea, Cardinal Health, Inc, Dublin, OH)
- PCV+ (Dräger Medical)
- DuoPAP (Hamilton).
Caution—name confusion. In North America, BiPAP (Respironics, Murrysville, PA) and BiLevel are used to refer to noninvasive modes of ventilation.
APRV has no other name.
What do these modes do?
These modes deliver pressure-controlled, time-triggered, and time-cycled breaths using a set-point targeting scheme (Table 2). This means that the ventilator maintains a constant pressure (set point) even in the face of spontaneous breaths.
Caution—source of confusion. The term continuous positive airway pressure (CPAP) is often used to describe this mode. However, CPAP is pressure that is applied continuously at the same level; the patient generates all the work to maintain ventilation (“pressure-controlled continuous spontaneous ventilation” in the current nomenclature). In APRV, the airway pressure is intermittently released and reapplied, generating a tidal volume that supports ventilation. In other words, this is a pressure-controlled breath with a very prolonged inspiratory time and a short expiratory time in which spontaneous ventilation is possible at any point (“pressure-controlled intermittent mandatory ventilation” in the current nomenclature).
How these modes are set in the ventilator may also be a source of confusion. To describe the time spent in high and low airway pressures, we use the terms Thigh and Tlow, respectively. By convention, the difference between APRV and biphasic mode is the duration of Tlow (< 1.5 sec for APRV).
Similarly, Phigh and Plow are used to describe the high and low airway pressure. To better understand this concept, you can create the same mode in conventional pressure-control ventilation by thinking of the Thigh as the inspiratory time, the Tlow as the expiratory time, the Phigh as inspiratory pressure, and the Plow as PEEP.
Hence, APRV is an extreme form of inverse ratio ventilation, with an inspiration-to-expiration ratio of 4:1. This means a patient spends most of the time in Phigh and Thigh, and exhalations are short (Tlow and Plow). In contrast, the biphasic mode uses conventional inspiration-expiration ratios (Figure 4).
As with any form of pressure control, the tidal volume is generated by airway pressure rising above baseline (ie, the end-expiratory value). Hence, to ensure an increase in minute ventilation, the mandatory breath rate must be increased (ie, decreasing Thigh, Tlow, or both) or the tidal volume must be increased (ie, increasing the difference between Phigh and Plow). This means that in APRV the Tlow has to happen more often (by increasing the number of breaths) or be more prolonged (allowing more air to exhale). Because unrestricted spontaneous breaths are permitted at any point of the cycle, the patient contributes to the total minute ventilation (usually 10%–40%).
In APRV and biphasic mode, the operator’s set time and pressure in inspiration and expiration will be delivered regardless of the patient’s breathing efforts—the patient’s spontaneous breath does not trigger a mechanical breath. Some ventilators have automatic adjustments to improve the trigger synchrony.
Ventilator settings in APRV and biphasic mode
These modes require the setting of two pressure levels (Phigh and Plow) and two time durations (Thigh and Tlow). One can add pressure support or automatic tube compensation to assist spontaneous breaths. The difference in Tlow generates differences in the Thigh:Tlow ratio: APRV has a short Tlow (an inspiration-expiration ratio of 4:1). Biphasic mode has a conventional inspiration-expiration ratio of 1:1 to 1:4.
Clinical applications
APRV is used in acute lung injury and ARDS. This mode should be used with caution or not at all in patients with obstructive lung disease or inappropriately increased respiratory drive.32–35
Biphasic mode is intended for both ventilation and weaning. In a patient who has poor respiratory effort or who is paralyzed, biphasic is identical to pressure-control/continuous mandatory ventilation.
Theoretical benefits of APRV and biphasic mode
Multiple benefits have been ascribed to these modes. In theory, APRV will maximize and maintain alveolar recruitment, improve oxygenation, lower inflation pressures, and decrease overinflation. Both APRV and biphasic, by preserving spontaneous breathing, will improve ventilation-perfusion matching and gas diffusion, improve the hemodynamic profile (less need for vasopressors, higher cardiac output, reduced ventricular workload, improved organ perfusion), and improve synchrony (decrease the work of breathing and the need for sedation).
Evidence of benefit of APRV and biphasic mode
APRV and biphasic are different modes. However studies evaluating their effects are combined. This is in part the result of the nomenclature confusion and different practice in different countries.36
Physiologic benefits. In studies, spontaneous breaths contributed to 10% to 40% of minute ventilation,37,38 improved ventilation of dependent areas of the lung, improved ventilation-perfusion match and recruitment,39 and improved hemodynamic profile.40
Patient comfort. These modes are thought to decrease the need for analgesia and sedation,38 but a recent trial showed no difference with pressure-controlled intermittent mandatory ventilation.41 Patient ventilator synchrony and comfort have not been studied.32,42
Outcomes. In small trials, these modes made no difference in terms of deaths, but they may decrease the length of mechanical ventilation.38,41,43,44
APRV and biphasic mode: Bottom line
Maintaining spontaneous breathing while on mechanical ventilation has hemodynamic and ventilatory benefits.
APRV and biphasic mode are not the same thing. APRV’s main goal is to maximize mean airway pressure and, hence, lung recruitment, whereas the main goal of the biphasic mode is synchrony.
There is a plethora of ventilator settings and questions related to physiologic effects.33,34,36
Although these modes are widely used in some centers, there is no evidence yet that they are superior to conventional volume- or pressure-control ventilation with low tidal volume for ARDS and acute lung injury. There is no conclusive evidence that these modes improve synchrony, time to weaning, or patient comfort.
HIGH-FREQUENCY OSCILLATORY VENTILATION
High-frequency oscillatory ventilation (HFOV) was first described and patented in 1952 by Emerson and was clinically developed in the early 1970s by Lunkenheimer.45
The goal of HFOV is to minimize lung injury; its characteristics (discussed below) make it useful in patients with severe ARDS. The US Food and Drug Administration approved it for infants in 1991 and for children in 1995. The adult model has been available since 1993, but it was not approved until 2001 (SensorMedics 3100B, Cardinal Health, Inc).
Other names for high-frequency oscillatory ventilation
While HFOV has no alternative names, the following acronyms describe similar modes:
- HFPPV (high-frequency positive pressure ventilation)
- HFJV (high-frequency jet ventilation)
- HFFI (high-frequency flow interruption)
- HFPV (high-frequency percussive ventilation)
- HFCWO (high-frequency chest wall oscillation).
All of these modes require different specialized ventilators.
What does high-frequency oscillatory ventilation do?
Conceptually, HFOV is a form of pressure-controlled intermittent mandatory ventilation with a set-point control scheme. In contrast to conventional pressure-controlled intermittent mandatory ventilation, in which relatively small spontaneous breaths may be superimposed on relatively large mandatory breaths, HFOV superimposes very small mandatory breaths (oscillations) on top of spontaneous breaths.
Adult patients are usually paralyzed or deeply sedated, since deep spontaneous breathing will trigger alarms and affect ventilator performance.
To manage ventilation (CO2 clearance), one or several of the following maneuvers can be done: decrease the oscillation frequency, increase the amplitude of the oscillations, increase the inspiratory time, or increase bias flow (while allowing an endotracheal tube cuff leak). Oxygenation adjustments are controlled by manipulating the mean airway pressure and the Fio2.
Ventilator settings in high-frequency oscillatory ventilation
Ventilator settings in HFOV are46:
- Airway pressure amplitude (delta P or power)
- Mean airway pressure
- Percent inspiration
- Inspiratory bias flow
- Fio2.
Clinical applications of high-frequency oscillatory ventilation
This mode is usually reserved for ARDS patients for whom conventional ventilation is failing. A recently published protocol46 suggests considering HFOV when there is oxygenation failure (Fio2 ≥ 0.7 and PEEP ≥ 14 cm H2O) or ventilation failure (pH < 7.25 with tidal volume ≥ 6 mL/kg predicted body weight and plateau airway pressure ≥ 30 cm H2O).
This mode is contraindicated when there is known severe airflow obstruction or intracranial hypertension.
Theoretical benefits of high-frequency oscillatory ventilation
Conceptually, HFOV can provide the highest mean airway pressure paired with the lowest tidal volume of any mode. These benefits might make HFOV the ideal lung-protective ventilation strategy.
Evidence of benefit of high-frequency oscillatory ventilation
Physiologic benefits. Animal models have shown less histologic damage and lung inflammation with HFOV than with high-tidal-volume conventional ventilation47,48 and low-tidal-volume conventional ventilation.49
Patient comfort has not been studied. However, current technology does impose undue work of breathing in spontaneously breathing patients.50
Outcomes. Several retrospective case series have described better oxygenation with HFOV as rescue therapy for severe ARDS than with conventional mechanical ventilation. Two randomized controlled trials have studied HFOV vs high-tidal-volume conventional mechanical ventilation for early severe ARDS; HFOV was safe but made no difference in terms of deaths.42,51–54
High-frequency oscillatory ventilation: Bottom line
In theory, HFOV provides all the benefits of an ideal lung-protective strategy, at least for paralyzed or deeply sedated patients. Animal studies support these concepts. In human adults, HFOV has been shown to be safe and to provide better oxygenation but no improvement in death rates compared with conventional mechanical ventilation. Currently, HFOV is better reserved for patients with severe ARDS for whom conventional mechanical ventilation is failing.
Technologic advances and computerized control of mechanical ventilators have made it possible to deliver ventilatory assistance in new modes. Driving these innovations is the desire to prevent ventilator-induced lung injury, improve patient comfort, and liberate the patient from mechanical ventilation as soon as possible.
We call these innovations “alternative” modes to differentiate them from the plain volume-control and pressure-control modes. Some clinicians rarely use these new modes, but in some medical centers they have become the most common ones used, or are being used unknowingly (the operator misunderstands the mode name). The information we provide on these modes of ventilation is by no means an endorsement of their use, but rather a tool to help the clinician understand their physiologic, theoretical, and clinical effects.
We focused on two goals:
- Explain what the mode does
- Briefly review the theoretical benefits and the actual evidence supporting these alternative modes of ventilation.
STANDARD NOMENCLATURE NEEDED
Since its invention, mechanical ventilation has been plagued by multiple names being used to describe the same things. For example, volume-control ventilation is also called volume-cycled ventilation, assist-control ventilation, volume-limited ventilation, and controlled mechanical ventilation. Similarly, multiple abbreviations are used, each depending on the brand of ventilator, and new acronyms have been added in recent years as new modes have been developed. The vast number of names and modes can confuse even the most seasoned critical care physician.
Efforts to establish a common nomenclature are under way.1
WHAT IS A MODE?
A mode of mechanical ventilation has three essential components:
- The control variable
- The breath sequence
- The targeting scheme.
Similar modes may require more detailed descriptions to distinguish them, but the basic function can be explained by these three components.
The control variable
In general, inspiration is an active process, driven by the patient’s effort, the ventilator, or both, while expiration is passive. For simplicity, in this article a mechanical breath means the inspiratory phase of the breath.
The machine can only control the volume (and flow) or the pressure given. The breaths can be further described on the basis of what triggers the breath, what limits it (the maximum value of a control variable), and what ends (cycles) it.
The breath sequence
There are three possible breath sequences:
- Intermittent mandatory ventilation, in which the patient can take spontaneous breaths between mandatory breaths
- Continuous spontaneous ventilation, in which all breaths are spontaneous (Table 1).
The targeting scheme
The targeting or feedback scheme refers to the ventilator settings and programming that dictate its response to the patient’s lung compliance, lung resistance, and respiratory effort. The regulation can be as simple as controlling the pressure in pressure-control mode, or it can be based on a complicated algorithm.
ADAPTIVE PRESSURE CONTROL
Other names for adaptive pressure control
- Pressure Regulated Volume Control (Maquet Servo-i, Rastatt, Germany)
- AutoFlow (Dräger Medical AG, Lübeck, Germany)
- Adaptive Pressure Ventilation (Hamilton Galileo, Hamilton Medical AG, Bonaduz, Switzerland)
- Volume Control+ (Puritan Bennett, Tyco Healthcare; Mansfield, MA)
- Volume Targeted Pressure Control, Pressure Controlled Volume Guaranteed (Engström, General Electric, Madison, WI).
What does adaptive pressure control do?
The APC mode delivers pressure-controlled breaths with an adaptive targeting scheme (Table 2).
In pressure-control ventilation, tidal volumes depend on the lung’s physiologic mechanics (compliance and resistance) and patient effort (Figure 1). Therefore, the tidal volume varies with changes in lung physiology (ie, larger or smaller tidal volumes than targeted).
To overcome this effect, a machine in APC mode adjusts the inspiratory pressure to deliver the set minimal target tidal volume. If tidal volume increases, the machine decreases the inspiratory pressure, and if tidal volume decreases, the machine increases the inspiratory pressure. However, if the patient effort is large enough, the tidal volume will increase in spite of decreasing the inspiratory pressure (Figure 2). The adjustments to the inspiratory pressure occur after the tidal volume is off-target in a number of breaths.
Common sources of confusion with adaptive pressure control
First, APC is not a volume-control mode. In volume control, the tidal volume does not change; in APC the tidal volume can increase or decrease, and the ventilator will adjust the inflation pressure to achieve the target volume. Thus, APC guarantees an average minimum tidal volume but not a maximum tidal volume.
Second, a characteristic of pressure control (and hence, APC) is that the flow of gas varies to maintain constant airway pressure (ie, maintain the set inspiratory pressure). This characteristic allows a patient who generates an inspiratory effort to receive flow as demanded, which is likely more comfortable. This is essentially different from volume control, in which flow is set by the operator and hence is fixed. Thus, if the patient effort is strong enough (Figure 1), this leads to what is called flow asynchrony, in which the patient does not get the flow asked for in a breath.
Ventilator settings in adaptive pressure control
Ventilator settings in APC are:
- Tidal volume
- Time spent in inspiration (inspiratory time)
- Frequency
- Fraction of inspired oxygen (Fio2)
- Positive end-expiratory pressure (PEEP).
Some ventilators also require setting the speed to reach the peak pressure (also known as slope percent or inspiratory rise time).
Clinical applications of adaptive pressure control
This mode is designed to maintain a consistent tidal volume during pressure-control ventilation and to promote inspiratory flow synchrony. It is a means of automatically reducing ventilatory support (ie, weaning) as the patient’s inspiratory effort becomes stronger, as in awakening from anesthesia.
APC may not be ideal for patients who have an inappropriately increased respiratory drive (eg, in severe metabolic acidosis), since the inspiratory pressure will decrease to maintain the targeted average tidal volume, inappropriately shifting the work of breathing onto the patient.
Theoretical benefits of adaptive pressure control
APC guarantees a minimum average tidal volume (unless the pressure alarm threshold is set too low, so that the target tidal volume is not delivered). Other theoretical benefits are flow synchrony, less ventilator manipulation by the operator, and automatic weaning of ventilator support.
Evidence of benefit of adaptive pressure control
Physiologic benefits. This mode has lower peak inspiratory pressures than does volume-control ventilation,3,4 which is often reported as a positive finding. However, in volume-control mode (the usual comparator), the peak inspiratory pressure is a manifestation of both resistance and compliance. Hence, peak inspiratory pressure is expected to be higher but does not reflect actual lung-distending pressures. It is the plateau pressure, a manifestation of lung compliance, that is related to lung injury.
Patient comfort. APC may increase the work of breathing when using low tidal volume ventilation and when there is increased respiratory effort (drive).5 Interestingly, APC was less comfortable than pressure support ventilation in a small trial.6
Outcomes have not been studied.7
Adaptive pressure control: Bottom line
APC is widely available and widely used, sometimes unknowingly (eg, if the operator thinks it is volume control). It is relatively easy to use and to set; however, evidence of its benefit is scant.
ADAPTIVE SUPPORT VENTILATION
Adaptive support ventilation (ASV) evolved as a form of mandatory minute ventilation implemented with adaptive pressure control. Mandatory minute ventilation is a mode that allows the operator to preset a target minute ventilation, and the ventilator then supplies mandatory breaths, either volume- or pressure-controlled, if the patient’s spontaneous breaths generate a lower minute ventilation.
ASV automatically selects the appropriate tidal volume and frequency for mandatory breaths and the appropriate tidal volume for spontaneous breaths on the basis of the respiratory system mechanics and target minute alveolar ventilation.
Described in 1994 by Laubscher et al,8,9 ASV became commercially available in 1998 in Europe and in 2007 in the United States (Hamilton Galileo ventilator, Hamilton Medical AG). This is the first commercially available ventilator that uses an “optimal” targeting scheme (see below).
What does adaptive support ventilation do?
ASV delivers pressure-controlled breaths using an adaptive (optimal) scheme (Table 2). “Optimal,” in this context, means minimizing the mechanical work of breathing: the machine selects a tidal volume and frequency that the patient’s brain would presumably select if the patient were not connected to a ventilator. This pattern is assumed to encourage the patient to generate spontaneous breaths.
The ventilator calculates the normal required minute ventilation based on the patient’s ideal weight and estimated dead space volume (ie, 2.2 mL/kg). This calculation represents 100% of minute ventilation. The clinician at the bedside sets a target percent of minute ventilation that the ventilator will support—higher than 100% if the patient has increased requirements due, eg, to sepsis or increased dead space, or less than 100% during weaning.
The ventilator initially delivers test breaths, in which it measures the expiratory time constant for the respiratory system and then uses this along with the estimated dead space and normal minute ventilation to calculate an optimal breathing frequency in terms of mechanical work.
The optimal or target tidal volume is calculated as the normal minute ventilation divided by the optimal frequency. The target tidal volume is achieved by the use of APC (see above) (Figure 2). This means that the pressure limit is automatically adjusted to achieve an average delivered tidal volume equal to the target. The ventilator continuously monitors the respiratory system mechanics and adjusts its settings accordingly.
The ventilator adjusts its breaths to avoid air trapping by allowing enough time to exhale, to avoid hypoventilation by delivering tidal volume greater than the dead space, and to avoid volutrauma by avoiding large tidal volumes.
Ventilator settings in adaptive support ventilation
Ventilator settings in ASV are:
- Patient height (to calculate the ideal body weight)
- Sex
- Percent of normal predicted minute ventilation goal
- Fio2
- PEEP.
Clinical applications of adaptive support ventilation
ASV is intended as a sole mode of ventilation, from initial support to weaning.
Theoretical benefits of adaptive support ventilation
In theory, ASV offers automatic selection of ventilator settings, automatic adaptation to changing patient lung mechanics, less need for human manipulation of the machine, improved synchrony, and automatic weaning.
Evidence of benefit of adaptive support ventilation
Physiologic benefits. Ventilator settings are adjusted automatically. ASV selects different tidal volume-respiratory rate combinations based on respiratory mechanics in passive and paralyzed patients.10–12 In actively breathing patients, there was no difference in the ventilator settings chosen by ASV for different clinical scenarios (and lung physiology).10 Compared with pressure-controlled intermittent mandatory ventilation, with ASV, the inspiratory load is less and patient-ventilator interaction is better.13
Patient-ventilator synchrony and comfort have not been studied.
Outcomes. Two trials suggest that ASV may decrease time on mechanical ventilation.14,15 However, in another trial,16 compared with a standard protocol, ASV led to fewer ventilator adjustments but achieved similar postsurgical weaning outcomes. The effect of this mode on the death rate has not been examined.17,18
Adaptive support ventilation: Bottom line
ASV is the first commercially available mode that automatically selects all the ventilator settings except PEEP and Fio2. These seem appropriate for different clinical scenarios in patients with poor respiratory effort or in paralyzed patients. Evidence of the effect in actively breathing patients and on outcomes such as length of stay or death is still lacking.
PROPORTIONAL ASSIST VENTILATION
Patients who have normal respiratory drive but who have difficulty sustaining adequate spontaneous ventilation are often subjected to pressure support ventilation (PSV), in which the ventilator generates a constant pressure throughout inspiration regardless of the intensity of the patient’s effort.
In 1992, Younes and colleagues19,20 developed proportional assist ventilation (PAV) as an alternative in which the ventilator generates pressure in proportion to the patient’s effort. PAV became commercially available in Europe in 1999 and was approved in the United States in 2006, available on the Puritan Bennett 840 ventilator (Puritan Bennett Co, Boulder, CO). PAV has also been used for noninvasive ventilation, but this is not available in the United States.
Other names for proportional assist ventilation
Proportional Pressure Support (Dräger Medical; not yet available in the United States).
What does proportional assist ventilation do?
This mode delivers pressure-controlled breaths with a servo control scheme (Table 2).
To better understand PAV, we can compare it with PSV. With PSV, the pressure applied by the ventilator rises to a preset level that is held constant (a set-point scheme) until a cycling criterion (a percent of the maximum inspiratory flow value) is reached. The inspiratory flow and tidal volume are the result of the patient’s inspiratory effort, the level of pressure applied, and the respiratory system mechanics.
In PAV, as in PSV, all breaths are spontaneous (Table 1). The patient controls the timing and size of the breath. There are no preset pressure, flow, or volume goals, but safety limits on the volume and pressure delivered can be set.
Ventilator settings in proportional assist ventilation
Ventilator settings in PAV are:
- Airway type (endotracheal tube, tracheostomy)
- Airway size (inner diameter)
- Percentage of work supported (assist range 5%–95%)
- Tidal volume limit
- Pressure limit
- Expiratory sensitivity (normally, as inspiration ends, flow should stop; this parameter tells the ventilator at what flow to end inspiration).
Caution when assessing the literature. Earlier ventilator versions, ie, Dräger and Manitoba (University of Manitoba, Winnipeg, MB, Canada), which are not available in the United States, required the repeated calculation of the respiratory system mechanics and the manual setting of flow and volume assists (amplification factors) independently. To overcome this limitation, new software automatically adjusts the flow and volume amplification to support the loads imposed by the automatically measured values of resistance and elastance (inverse of compliance) of the respiratory system.21 This software is included in the model (Puritan Bennett) available in the United States.
Clinical applications of proportional assist ventilation
The PAV mode is indicated for maximizing ventilator patient synchrony for assisted spontaneous ventilation.
PAV is contraindicated in patients with respiratory depression (bradypnea) or large air leaks (eg, bronchopleural fistulas). It should be used with caution in patients with severe hyperinflation, in which the patient may still be exhaling but the ventilator doesn’t recognize it. Another group in which PAV should be used with caution is those with high ventilatory drives, in which the ventilator overestimates respiratory system mechanics. This situation can lead to overassistance due to the “runaway phenomenon,” in which the ventilator continues to provide support even if the patient has stopped inspiration.22
Theoretical benefits of proportional assist ventilation
In theory, PAV should reduce the work of breathing, improve synchrony, automatically adapt to changing patient lung mechanics and effort, decrease the need for ventilator intervention and manipulation, decrease the need for sedation, and improve sleep.
Evidence of benefit of proportional assist ventilation
Physiologic benefits. PAV reduces the work of breathing better than PSV,21 even in the face of changing respiratory mechanics or increased respiratory demand (hypercapnia).23–25 The hemodynamic profile is similar to that in PSV. Tidal volumes are variable; however, in recent reports the tidal volumes were within the lung-protective range (6–8 mL/kg, plateau pressure < 30 cm H20).26,27
Comfort. PAV entails less patient effort and discomfort that PSV does.23,25 PAV significantly reduces asynchrony,27 which in turn may favorably affect sleep in critically ill patients. 28
Outcomes. The probability of spontaneous breathing without assistance was significantly better in critically ill patients ventilated with PAV than with PSV. No trial has reported the effect of PAV on deaths.27,29
Proportional assist ventilation: Bottom line
Extensive basic research has been done with PAV in different forms of respiratory failure, such as obstructive lung disease, acute respiratory distress syndrome (ARDS), and chronic respiratory failure. It fulfills its main goal, which is to improve patient-ventilator synchrony. Clinical experience with PAV in the United States is limited, as it was only recently approved.
AIRWAY PRESSURE-RELEASE VENTILATION AND BIPHASIC POSITIVE AIRWAY PRESSURE
Airway pressure-release ventilation (APRV) was described in 1987 by Stock et al30 as a mode for delivering ventilation in acute lung injury while avoiding high airway pressures. APRV combines high constant positive airway pressure (improving oxygenation and promoting alveolar recruitment) with intermittent releases (causing exhalation).
Other names for biphasic positive airway pressure
Other names for biphasic positive airway pressure are:
- BiLevel (Puritan Bennett)
- BIPAP (Dräger Europe)
- Bi Vent (Siemens)
- BiPhasic (Avea, Cardinal Health, Inc, Dublin, OH)
- PCV+ (Dräger Medical)
- DuoPAP (Hamilton).
Caution—name confusion. In North America, BiPAP (Respironics, Murrysville, PA) and BiLevel are used to refer to noninvasive modes of ventilation.
APRV has no other name.
What do these modes do?
These modes deliver pressure-controlled, time-triggered, and time-cycled breaths using a set-point targeting scheme (Table 2). This means that the ventilator maintains a constant pressure (set point) even in the face of spontaneous breaths.
Caution—source of confusion. The term continuous positive airway pressure (CPAP) is often used to describe this mode. However, CPAP is pressure that is applied continuously at the same level; the patient generates all the work to maintain ventilation (“pressure-controlled continuous spontaneous ventilation” in the current nomenclature). In APRV, the airway pressure is intermittently released and reapplied, generating a tidal volume that supports ventilation. In other words, this is a pressure-controlled breath with a very prolonged inspiratory time and a short expiratory time in which spontaneous ventilation is possible at any point (“pressure-controlled intermittent mandatory ventilation” in the current nomenclature).
How these modes are set in the ventilator may also be a source of confusion. To describe the time spent in high and low airway pressures, we use the terms Thigh and Tlow, respectively. By convention, the difference between APRV and biphasic mode is the duration of Tlow (< 1.5 sec for APRV).
Similarly, Phigh and Plow are used to describe the high and low airway pressure. To better understand this concept, you can create the same mode in conventional pressure-control ventilation by thinking of the Thigh as the inspiratory time, the Tlow as the expiratory time, the Phigh as inspiratory pressure, and the Plow as PEEP.
Hence, APRV is an extreme form of inverse ratio ventilation, with an inspiration-to-expiration ratio of 4:1. This means a patient spends most of the time in Phigh and Thigh, and exhalations are short (Tlow and Plow). In contrast, the biphasic mode uses conventional inspiration-expiration ratios (Figure 4).
As with any form of pressure control, the tidal volume is generated by airway pressure rising above baseline (ie, the end-expiratory value). Hence, to ensure an increase in minute ventilation, the mandatory breath rate must be increased (ie, decreasing Thigh, Tlow, or both) or the tidal volume must be increased (ie, increasing the difference between Phigh and Plow). This means that in APRV the Tlow has to happen more often (by increasing the number of breaths) or be more prolonged (allowing more air to exhale). Because unrestricted spontaneous breaths are permitted at any point of the cycle, the patient contributes to the total minute ventilation (usually 10%–40%).
In APRV and biphasic mode, the operator’s set time and pressure in inspiration and expiration will be delivered regardless of the patient’s breathing efforts—the patient’s spontaneous breath does not trigger a mechanical breath. Some ventilators have automatic adjustments to improve the trigger synchrony.
Ventilator settings in APRV and biphasic mode
These modes require the setting of two pressure levels (Phigh and Plow) and two time durations (Thigh and Tlow). One can add pressure support or automatic tube compensation to assist spontaneous breaths. The difference in Tlow generates differences in the Thigh:Tlow ratio: APRV has a short Tlow (an inspiration-expiration ratio of 4:1). Biphasic mode has a conventional inspiration-expiration ratio of 1:1 to 1:4.
Clinical applications
APRV is used in acute lung injury and ARDS. This mode should be used with caution or not at all in patients with obstructive lung disease or inappropriately increased respiratory drive.32–35
Biphasic mode is intended for both ventilation and weaning. In a patient who has poor respiratory effort or who is paralyzed, biphasic is identical to pressure-control/continuous mandatory ventilation.
Theoretical benefits of APRV and biphasic mode
Multiple benefits have been ascribed to these modes. In theory, APRV will maximize and maintain alveolar recruitment, improve oxygenation, lower inflation pressures, and decrease overinflation. Both APRV and biphasic, by preserving spontaneous breathing, will improve ventilation-perfusion matching and gas diffusion, improve the hemodynamic profile (less need for vasopressors, higher cardiac output, reduced ventricular workload, improved organ perfusion), and improve synchrony (decrease the work of breathing and the need for sedation).
Evidence of benefit of APRV and biphasic mode
APRV and biphasic are different modes. However studies evaluating their effects are combined. This is in part the result of the nomenclature confusion and different practice in different countries.36
Physiologic benefits. In studies, spontaneous breaths contributed to 10% to 40% of minute ventilation,37,38 improved ventilation of dependent areas of the lung, improved ventilation-perfusion match and recruitment,39 and improved hemodynamic profile.40
Patient comfort. These modes are thought to decrease the need for analgesia and sedation,38 but a recent trial showed no difference with pressure-controlled intermittent mandatory ventilation.41 Patient ventilator synchrony and comfort have not been studied.32,42
Outcomes. In small trials, these modes made no difference in terms of deaths, but they may decrease the length of mechanical ventilation.38,41,43,44
APRV and biphasic mode: Bottom line
Maintaining spontaneous breathing while on mechanical ventilation has hemodynamic and ventilatory benefits.
APRV and biphasic mode are not the same thing. APRV’s main goal is to maximize mean airway pressure and, hence, lung recruitment, whereas the main goal of the biphasic mode is synchrony.
There is a plethora of ventilator settings and questions related to physiologic effects.33,34,36
Although these modes are widely used in some centers, there is no evidence yet that they are superior to conventional volume- or pressure-control ventilation with low tidal volume for ARDS and acute lung injury. There is no conclusive evidence that these modes improve synchrony, time to weaning, or patient comfort.
HIGH-FREQUENCY OSCILLATORY VENTILATION
High-frequency oscillatory ventilation (HFOV) was first described and patented in 1952 by Emerson and was clinically developed in the early 1970s by Lunkenheimer.45
The goal of HFOV is to minimize lung injury; its characteristics (discussed below) make it useful in patients with severe ARDS. The US Food and Drug Administration approved it for infants in 1991 and for children in 1995. The adult model has been available since 1993, but it was not approved until 2001 (SensorMedics 3100B, Cardinal Health, Inc).
Other names for high-frequency oscillatory ventilation
While HFOV has no alternative names, the following acronyms describe similar modes:
- HFPPV (high-frequency positive pressure ventilation)
- HFJV (high-frequency jet ventilation)
- HFFI (high-frequency flow interruption)
- HFPV (high-frequency percussive ventilation)
- HFCWO (high-frequency chest wall oscillation).
All of these modes require different specialized ventilators.
What does high-frequency oscillatory ventilation do?
Conceptually, HFOV is a form of pressure-controlled intermittent mandatory ventilation with a set-point control scheme. In contrast to conventional pressure-controlled intermittent mandatory ventilation, in which relatively small spontaneous breaths may be superimposed on relatively large mandatory breaths, HFOV superimposes very small mandatory breaths (oscillations) on top of spontaneous breaths.
Adult patients are usually paralyzed or deeply sedated, since deep spontaneous breathing will trigger alarms and affect ventilator performance.
To manage ventilation (CO2 clearance), one or several of the following maneuvers can be done: decrease the oscillation frequency, increase the amplitude of the oscillations, increase the inspiratory time, or increase bias flow (while allowing an endotracheal tube cuff leak). Oxygenation adjustments are controlled by manipulating the mean airway pressure and the Fio2.
Ventilator settings in high-frequency oscillatory ventilation
Ventilator settings in HFOV are46:
- Airway pressure amplitude (delta P or power)
- Mean airway pressure
- Percent inspiration
- Inspiratory bias flow
- Fio2.
Clinical applications of high-frequency oscillatory ventilation
This mode is usually reserved for ARDS patients for whom conventional ventilation is failing. A recently published protocol46 suggests considering HFOV when there is oxygenation failure (Fio2 ≥ 0.7 and PEEP ≥ 14 cm H2O) or ventilation failure (pH < 7.25 with tidal volume ≥ 6 mL/kg predicted body weight and plateau airway pressure ≥ 30 cm H2O).
This mode is contraindicated when there is known severe airflow obstruction or intracranial hypertension.
Theoretical benefits of high-frequency oscillatory ventilation
Conceptually, HFOV can provide the highest mean airway pressure paired with the lowest tidal volume of any mode. These benefits might make HFOV the ideal lung-protective ventilation strategy.
Evidence of benefit of high-frequency oscillatory ventilation
Physiologic benefits. Animal models have shown less histologic damage and lung inflammation with HFOV than with high-tidal-volume conventional ventilation47,48 and low-tidal-volume conventional ventilation.49
Patient comfort has not been studied. However, current technology does impose undue work of breathing in spontaneously breathing patients.50
Outcomes. Several retrospective case series have described better oxygenation with HFOV as rescue therapy for severe ARDS than with conventional mechanical ventilation. Two randomized controlled trials have studied HFOV vs high-tidal-volume conventional mechanical ventilation for early severe ARDS; HFOV was safe but made no difference in terms of deaths.42,51–54
High-frequency oscillatory ventilation: Bottom line
In theory, HFOV provides all the benefits of an ideal lung-protective strategy, at least for paralyzed or deeply sedated patients. Animal studies support these concepts. In human adults, HFOV has been shown to be safe and to provide better oxygenation but no improvement in death rates compared with conventional mechanical ventilation. Currently, HFOV is better reserved for patients with severe ARDS for whom conventional mechanical ventilation is failing.
- Chatburn RL. Classification of ventilator modes: update and proposal for implementation. Respir Care 2007; 52:301–323.
- Chatburn RL. Computer control of mechanical ventilation. Respir Care 2004; 49:507–517.
- Alvarez A, Subirana M, Benito S. Decelerating flow ventilation effects in acute respiratory failure. J Crit Care 1998; 13:21–25.
- Guldager H, Nielsen SL, Carl P, Soerensen MB. A comparison of volume control and pressure regulated volume control ventilation in acute respiratory failure. Crit Care 1997; 1:75–77.
- Kallet RH, Campbell AR, Dicker RA, Katz JA, Mackersie RC. Work of breathing during lung protective ventilation in patients with acute lung injury and acute respiratory distress syndrome: a comparison between volume and pressure regulated breathing modes. Respir Care 2005; 50:1623–1631.
- Betensley AD, Khalid I, Crawford J, Pensler RA, DiGiovine B. Patient comfort during pressure support and volume controlled continuous mandatory ventilation. Respir Care 2008; 53:897–902.
- Branson RD, Chatburn RL. Controversies in the critical care setting. Should adaptive pressure control modes be utilized for virtually all patients receiving mechanical ventilation? Respir Care 2007; 52:478–485.
- Laubscher TP, Frutiger A, Fanconi S, Jutzi H, Brunner JX. Automatic selection of tidal volume, respiratory frequency and minute ventilation in intubated ICU patients as start up procedure for closed-loop controlled ventilation. Int J Clin Monit Comput 1994; 11:19–30.
- Laubscher TP, Heinrichs W, Weiler N, Hartmann G, Brunner JX. An adaptive lung ventilation controller. IEEE Trans Biomed Eng 1994; 41:51–59.
- Arnal JM, Wysocki M, Nafati C, et al. Automatic selection of breathing pattern using adaptive support ventilation. Intensive Care Med 2008; 34:75–81.
- Campbell RS, Sinamban RP, Johannigman JA, et al. Clinical evaluation of a new closed loop ventilation mode: adaptive supportive ventilation (ASV). Crit Care 1999; 3( suppl 1):083.
- Belliato M, Palo A, Pasero D, Iotti GA, Mojoli F, Braschi A. Evaluation of adaptive support ventilation in paralysed patients and in a physical lung model. Int J Artif Organs 2004; 27:709–716.
- Tassaux D, Dalmas E, Gratadour P, Jolliet P. Patient ventilator interactions during partial ventilatory support: a preliminary study comparing the effects of adaptive support ventilation with synchronized intermittent mandatory ventilation plus inspiratory pressure support. Crit Care Med 2002; 30:801–807.
- Gruber PC, Gomersall CD, Leung P, et al. Randomized controlled trial comparing adaptive-support ventilation with pressure-regulated volume-controlled ventilation with automode in weaning patients after cardiac surgery. Anesthesiology 2008; 109:81–87.
- Sulzer CF, Chiolero R, Chassot PG, et al. Adaptive support ventilation for fast tracheal extubation after cardiac surgery: a randomized controlled study. Anesthesiology 2001; 95:1339–1345.
- Petter AH, Chiolèro RL, Cassina T, Chassot PG, Müller XM, Revelly JP. Automatic “respirator/weaning” with adaptive support ventilation: the effect on duration of endotracheal intubation and patient management. Anesth Analg 2003; 97:1743–1750.
- Brunner JX, Iotti GA. Adaptive support ventilation (ASV). Minerva Anestesiol 2002; 68:365–368.
- Campbell RS, Branson RD, Johannigman JA. Adaptive support ventilation. Respir Care Clin North Am 2001; 7:425–440.
- Younes M. Proportional assist ventilation, a new approach to ventilatory support. Theory. Am Rev Respir Dis 1992; 145:114–120.
- Younes M, Puddy A, Roberts D, et al. Proportional assist ventilation. Results of an initial clinical trial. Am Rev Respir Dis 1992; 145:121–129.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Respiratory load compensation during mechanical ventilatio—proportional assist ventilation with load-adjustable gain factors versus pressure support. Intensive Care Med 2006; 32:692–699.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Effect of different levels of pressure support and proportional assist ventilation on breathing pattern, work of breathing and gas exchange in mechanically ventilated hypercapnic COPD patients with acute respiratory failure. Respiration 2003; 70:355–361.
- Grasso S, Puntillo F, Mascia L, et al. Compensation for increase in respiratory workload during mechanical ventilation. Pressure support versus proportional assist ventilation. Am J Respir Crit Care Med 2000; 161:819–826.
- Wrigge H, Golisch W, Zinserling J, Sydow M, Almeling G, Burchardi H. Proportional assist versus pressure support ventilation: effects on breathing pattern and respiratory work of patients with chronic obstructive pulmonary disease. Intensive Care Med 1999; 25:790–798.
- Ranieri VM, Giuliani R, Mascia L, et al. Patient ventilator interaction during acute hypercapnia: pressure support vs. proportional assist ventilation. J Appl Physiol 1996; 81:426–436.
- Kondili E, Xirouchaki N, Vaporidi K, Klimathianaki M, Georgopoulos D. Short-term cardiorespiratory effects of proportional assist and pressure support ventilation in patients with acute lung injury/acute respiratory distress syndrome. Anesthesiology 2006; 105:703–708.
- Xirouchaki N, Kondili E, Vaporidi K, et al. Proportional assist ventilation with load-adjustable gain factors in critically ill patients: comparison with pressure support. Intensive Care Med 2008; 34:2026–2034.
- Bosma K, Ferreyra G, Ambrogio C, et al. Patient ventilator interaction and sleep in mechanically ventilated patients: pressure support versus proportional assist ventilation. Crit Care Med 2007; 35:1048–1054.
- Sinderby C, Beck J. Proportional assist ventilation and neurally adjusted ventilatory assist—better approaches to patient ventilator synchrony? Clin Chest Med 2008; 29:329–342.
- Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med 1987; 15:462–466.
- Baum M, Benzer H, Putensen C, Koller W, Putz G. [Biphasic positive airway pressure (BIPAP)—a new form of augmented ventilation]. Anaesthesist 1989; 38:452–458.
- Seymour CW, Frazer M, Reilly PM, Fuchs BD. Airway pressure release and biphasic intermittent positive airway pressure ventilation: are they ready for prime time? J Trauma 2007; 62:1298–1308.
- Myers TR, MacIntyre NR. Respiratory controversies in the critical care setting. Does airway pressure release ventilation offer important new advantages in mechanical ventilator support? Respir Care 2007; 52:452–458.
- Neumann P, Golisch W, Strohmeyer A, Buscher H, Burchardi H, Sydow M. Influence of different release times on spontaneous breathing pattern during airway pressure release ventilation. Intensive Care Med 2002; 28:1742–1749.
- Calzia E, Lindner KH, Witt S, et al. Pressure-time product and work of breathing during biphasic continuous positive airway pressure and assisted spontaneous breathing. Am J Respir Crit Care Med 1994; 150:904–910.
- Rose L, Hawkins M. Airway pressure release ventilation and biphasic positive airway pressure: a systematic review of definitional criteria. Intensive Care Med 2008; 34:1766–1773.
- Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Longterm effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med 1994; 149:1550–1556.
- Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001; 164:43–49.
- Davis K, Johnson DJ, Branson RD, Campbell RS, Johannigman JA, Porembka D. Airway pressure release ventilation. Arch Surg 1993; 128:1348–1352.
- Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001; 5:221–226.
- Varpula T, Valta P, Niemi R, Takkunen O, Hynynen M, Pettilä VV. Airway pressure release ventilation as a primary ventilatory mode in acute respiratory distress syndrome. Acta Anaesthesiol Scand 2004; 48:722–731.
- Siau C, Stewart TE. Current role of high frequency oscillatory ventilation and airway pressure release ventilation in acute lung injury and acute respiratory distress syndrome. Clin Chest Med 2008; 29:265–275.
- Rathgeber J, Schorn B, Falk V, Kazmaier S, Spiegel T, Burchardi H. The influence of controlled mandatory ventilation (CMV), intermittent mandatory ventilation (IMV) and biphasic intermittent positive airway pressure (BIPAP) on duration of intubation and consumption of analgesics and sedatives. A prospective analysis in 596 patients following adult cardiac surgery. Eur J Anaesthesiol 1997; 14:576–582.
- Habashi NM. Other approaches to open lung ventilation: airway pressure release ventilation. Crit Care Med 2005; 33 suppl 3:S228–S240.
- Hess D, Mason S, Branson R. High-frequency ventilation design and equipment issues. Respir Care Clin North Am 2001; 7:577–598.
- Fessler HE, Derdak S, Ferguson ND, et al. A protocol for high frequency oscillatory ventilation in adults: results from a roundtable discussion. Crit Care Med 2007; 35:1649–1654.
- Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol 1983; 55:131–138.
- Sedeek KA, Takeuchi M, Suchodolski K, et al. Open-lung protective ventilation with pressure control ventilation, high-frequency oscillation, and intratracheal pulmonary ventilation results in similar gas exchange, hemodynamics, and lung mechanics. Anesthesiology 2003; 99:1102–1111.
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol 2001; 91:1836–1844.
- van Heerde M, Roubik K, Kopelent V, Plötz FB, Markhorst DG. Unloading work of breathing during high-frequency oscillatory ventilation: a bench study. Crit Care 2006; 10:R103.
- Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med 2002; 166:801–808.
- Bollen CW, van Well GT, Sherry T, et al. High-frequency oscillatory ventilation compared with conventional mechanical ventilation in adult respiratory distress syndrome: a randomized controlled trial [ISRCTN24242669]. Crit Care 2005; 9:R430–R439.
- Mehta S, Granton J, MacDonald RJ, et al. High frequency oscillatory ventilation in adults: the Toronto experience. Chest 2004; 126:518–527.
- Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest 2007; 131:1907–1916.
- Chatburn RL. Classification of ventilator modes: update and proposal for implementation. Respir Care 2007; 52:301–323.
- Chatburn RL. Computer control of mechanical ventilation. Respir Care 2004; 49:507–517.
- Alvarez A, Subirana M, Benito S. Decelerating flow ventilation effects in acute respiratory failure. J Crit Care 1998; 13:21–25.
- Guldager H, Nielsen SL, Carl P, Soerensen MB. A comparison of volume control and pressure regulated volume control ventilation in acute respiratory failure. Crit Care 1997; 1:75–77.
- Kallet RH, Campbell AR, Dicker RA, Katz JA, Mackersie RC. Work of breathing during lung protective ventilation in patients with acute lung injury and acute respiratory distress syndrome: a comparison between volume and pressure regulated breathing modes. Respir Care 2005; 50:1623–1631.
- Betensley AD, Khalid I, Crawford J, Pensler RA, DiGiovine B. Patient comfort during pressure support and volume controlled continuous mandatory ventilation. Respir Care 2008; 53:897–902.
- Branson RD, Chatburn RL. Controversies in the critical care setting. Should adaptive pressure control modes be utilized for virtually all patients receiving mechanical ventilation? Respir Care 2007; 52:478–485.
- Laubscher TP, Frutiger A, Fanconi S, Jutzi H, Brunner JX. Automatic selection of tidal volume, respiratory frequency and minute ventilation in intubated ICU patients as start up procedure for closed-loop controlled ventilation. Int J Clin Monit Comput 1994; 11:19–30.
- Laubscher TP, Heinrichs W, Weiler N, Hartmann G, Brunner JX. An adaptive lung ventilation controller. IEEE Trans Biomed Eng 1994; 41:51–59.
- Arnal JM, Wysocki M, Nafati C, et al. Automatic selection of breathing pattern using adaptive support ventilation. Intensive Care Med 2008; 34:75–81.
- Campbell RS, Sinamban RP, Johannigman JA, et al. Clinical evaluation of a new closed loop ventilation mode: adaptive supportive ventilation (ASV). Crit Care 1999; 3( suppl 1):083.
- Belliato M, Palo A, Pasero D, Iotti GA, Mojoli F, Braschi A. Evaluation of adaptive support ventilation in paralysed patients and in a physical lung model. Int J Artif Organs 2004; 27:709–716.
- Tassaux D, Dalmas E, Gratadour P, Jolliet P. Patient ventilator interactions during partial ventilatory support: a preliminary study comparing the effects of adaptive support ventilation with synchronized intermittent mandatory ventilation plus inspiratory pressure support. Crit Care Med 2002; 30:801–807.
- Gruber PC, Gomersall CD, Leung P, et al. Randomized controlled trial comparing adaptive-support ventilation with pressure-regulated volume-controlled ventilation with automode in weaning patients after cardiac surgery. Anesthesiology 2008; 109:81–87.
- Sulzer CF, Chiolero R, Chassot PG, et al. Adaptive support ventilation for fast tracheal extubation after cardiac surgery: a randomized controlled study. Anesthesiology 2001; 95:1339–1345.
- Petter AH, Chiolèro RL, Cassina T, Chassot PG, Müller XM, Revelly JP. Automatic “respirator/weaning” with adaptive support ventilation: the effect on duration of endotracheal intubation and patient management. Anesth Analg 2003; 97:1743–1750.
- Brunner JX, Iotti GA. Adaptive support ventilation (ASV). Minerva Anestesiol 2002; 68:365–368.
- Campbell RS, Branson RD, Johannigman JA. Adaptive support ventilation. Respir Care Clin North Am 2001; 7:425–440.
- Younes M. Proportional assist ventilation, a new approach to ventilatory support. Theory. Am Rev Respir Dis 1992; 145:114–120.
- Younes M, Puddy A, Roberts D, et al. Proportional assist ventilation. Results of an initial clinical trial. Am Rev Respir Dis 1992; 145:121–129.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Respiratory load compensation during mechanical ventilatio—proportional assist ventilation with load-adjustable gain factors versus pressure support. Intensive Care Med 2006; 32:692–699.
- Kondili E, Prinianakis G, Alexopoulou C, Vakouti E, Klimathianaki M, Georgopoulos D. Effect of different levels of pressure support and proportional assist ventilation on breathing pattern, work of breathing and gas exchange in mechanically ventilated hypercapnic COPD patients with acute respiratory failure. Respiration 2003; 70:355–361.
- Grasso S, Puntillo F, Mascia L, et al. Compensation for increase in respiratory workload during mechanical ventilation. Pressure support versus proportional assist ventilation. Am J Respir Crit Care Med 2000; 161:819–826.
- Wrigge H, Golisch W, Zinserling J, Sydow M, Almeling G, Burchardi H. Proportional assist versus pressure support ventilation: effects on breathing pattern and respiratory work of patients with chronic obstructive pulmonary disease. Intensive Care Med 1999; 25:790–798.
- Ranieri VM, Giuliani R, Mascia L, et al. Patient ventilator interaction during acute hypercapnia: pressure support vs. proportional assist ventilation. J Appl Physiol 1996; 81:426–436.
- Kondili E, Xirouchaki N, Vaporidi K, Klimathianaki M, Georgopoulos D. Short-term cardiorespiratory effects of proportional assist and pressure support ventilation in patients with acute lung injury/acute respiratory distress syndrome. Anesthesiology 2006; 105:703–708.
- Xirouchaki N, Kondili E, Vaporidi K, et al. Proportional assist ventilation with load-adjustable gain factors in critically ill patients: comparison with pressure support. Intensive Care Med 2008; 34:2026–2034.
- Bosma K, Ferreyra G, Ambrogio C, et al. Patient ventilator interaction and sleep in mechanically ventilated patients: pressure support versus proportional assist ventilation. Crit Care Med 2007; 35:1048–1054.
- Sinderby C, Beck J. Proportional assist ventilation and neurally adjusted ventilatory assist—better approaches to patient ventilator synchrony? Clin Chest Med 2008; 29:329–342.
- Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med 1987; 15:462–466.
- Baum M, Benzer H, Putensen C, Koller W, Putz G. [Biphasic positive airway pressure (BIPAP)—a new form of augmented ventilation]. Anaesthesist 1989; 38:452–458.
- Seymour CW, Frazer M, Reilly PM, Fuchs BD. Airway pressure release and biphasic intermittent positive airway pressure ventilation: are they ready for prime time? J Trauma 2007; 62:1298–1308.
- Myers TR, MacIntyre NR. Respiratory controversies in the critical care setting. Does airway pressure release ventilation offer important new advantages in mechanical ventilator support? Respir Care 2007; 52:452–458.
- Neumann P, Golisch W, Strohmeyer A, Buscher H, Burchardi H, Sydow M. Influence of different release times on spontaneous breathing pattern during airway pressure release ventilation. Intensive Care Med 2002; 28:1742–1749.
- Calzia E, Lindner KH, Witt S, et al. Pressure-time product and work of breathing during biphasic continuous positive airway pressure and assisted spontaneous breathing. Am J Respir Crit Care Med 1994; 150:904–910.
- Rose L, Hawkins M. Airway pressure release ventilation and biphasic positive airway pressure: a systematic review of definitional criteria. Intensive Care Med 2008; 34:1766–1773.
- Sydow M, Burchardi H, Ephraim E, Zielmann S, Crozier TA. Longterm effects of two different ventilatory modes on oxygenation in acute lung injury. Comparison of airway pressure release ventilation and volume-controlled inverse ratio ventilation. Am J Respir Crit Care Med 1994; 149:1550–1556.
- Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001; 164:43–49.
- Davis K, Johnson DJ, Branson RD, Campbell RS, Johannigman JA, Porembka D. Airway pressure release ventilation. Arch Surg 1993; 128:1348–1352.
- Kaplan LJ, Bailey H, Formosa V. Airway pressure release ventilation increases cardiac performance in patients with acute lung injury/adult respiratory distress syndrome. Crit Care 2001; 5:221–226.
- Varpula T, Valta P, Niemi R, Takkunen O, Hynynen M, Pettilä VV. Airway pressure release ventilation as a primary ventilatory mode in acute respiratory distress syndrome. Acta Anaesthesiol Scand 2004; 48:722–731.
- Siau C, Stewart TE. Current role of high frequency oscillatory ventilation and airway pressure release ventilation in acute lung injury and acute respiratory distress syndrome. Clin Chest Med 2008; 29:265–275.
- Rathgeber J, Schorn B, Falk V, Kazmaier S, Spiegel T, Burchardi H. The influence of controlled mandatory ventilation (CMV), intermittent mandatory ventilation (IMV) and biphasic intermittent positive airway pressure (BIPAP) on duration of intubation and consumption of analgesics and sedatives. A prospective analysis in 596 patients following adult cardiac surgery. Eur J Anaesthesiol 1997; 14:576–582.
- Habashi NM. Other approaches to open lung ventilation: airway pressure release ventilation. Crit Care Med 2005; 33 suppl 3:S228–S240.
- Hess D, Mason S, Branson R. High-frequency ventilation design and equipment issues. Respir Care Clin North Am 2001; 7:577–598.
- Fessler HE, Derdak S, Ferguson ND, et al. A protocol for high frequency oscillatory ventilation in adults: results from a roundtable discussion. Crit Care Med 2007; 35:1649–1654.
- Hamilton PP, Onayemi A, Smyth JA, et al. Comparison of conventional and high-frequency ventilation: oxygenation and lung pathology. J Appl Physiol 1983; 55:131–138.
- Sedeek KA, Takeuchi M, Suchodolski K, et al. Open-lung protective ventilation with pressure control ventilation, high-frequency oscillation, and intratracheal pulmonary ventilation results in similar gas exchange, hemodynamics, and lung mechanics. Anesthesiology 2003; 99:1102–1111.
- Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol 2001; 91:1836–1844.
- van Heerde M, Roubik K, Kopelent V, Plötz FB, Markhorst DG. Unloading work of breathing during high-frequency oscillatory ventilation: a bench study. Crit Care 2006; 10:R103.
- Derdak S, Mehta S, Stewart TE, et al., Multicenter Oscillatory Ventilation For Acute Respiratory Distress Syndrome Trial (MOAT) Study Investigators. High-frequency oscillatory ventilation for acute respiratory distress syndrome in adults: a randomized, controlled trial. Am J Respir Crit Care Med 2002; 166:801–808.
- Bollen CW, van Well GT, Sherry T, et al. High-frequency oscillatory ventilation compared with conventional mechanical ventilation in adult respiratory distress syndrome: a randomized controlled trial [ISRCTN24242669]. Crit Care 2005; 9:R430–R439.
- Mehta S, Granton J, MacDonald RJ, et al. High frequency oscillatory ventilation in adults: the Toronto experience. Chest 2004; 126:518–527.
- Chan KP, Stewart TE, Mehta S. High-frequency oscillatory ventilation for adult patients with ARDS. Chest 2007; 131:1907–1916.
KEY POINTS
- The alternative modes of ventilation were developed to prevent lung injury and asynchrony, promote better oxygenation and faster weaning, and be easier to use. However, evidence of their benefit is scant.
- Until now, we have lacked a standard nomenclature for mechanical ventilation, leading to confusion.
- Regardless of the mode used, the goals are to avoid lung injury, keep the patient comfortable, and wean the patient from mechanical ventilation as soon as possible.
Is telemetry overused? Is it as helpful as thought?
Telemetry—from the Greek words tele (remote) and metron (measure)—for cardiac monitoring was developed in the mid-1960s by Spacelabs Medical for use in spaceflight.1 The system was later adopted in hospitals to detect life-threatening arrhythmias.
Guidelines for the use of telemetry were published in 1991 by the American College of Cardiology (ACC)2 in response to concerns raised by its increasing use in noncritical care settings during the 30 years after its introduction to clinical medicine. The latest revision of the guidelines was published in 2004 by the American Heart Association (AHA).3
However, the guidelines are based largely on expert opinion and on research data in electrocardiography. Few clinical trials of telemetry have been published, and they were either retrospective or nonrandomized. In fact, there were no published randomized trials at the time the 2004 guidelines were written. Moreover, very few of these studies evaluated the impact of cardiac telemetry monitoring on physician management decisions.
We reviewed the literature to find out how cardiac telemetry is being used in clinical practice and how it might be used more selectively. The literature search was performed using Ovid MEDLINE (1996 to present) and PubMed Central using the key search terms “cardiac monitoring,” “telemetry monitoring,” “telemetry,” and “inpatient.” References from articles identified using Ovid MEDLINE (1996 to present) and PubMed Central that were relevant to our review were also included.
THREE CLASSES OF RISK


- Class II consists of patients for whom cardiac monitoring may be of benefit in some cases but is not essential for all (Table 2).



PATIENTS AT LOW RISK DO NOT BENEFIT
Telemetry monitoring has become an essential and commonly used clinical tool in most hospital systems. However, physicians do not seem to be using the risk stratification guidelines routinely or appropriately. The result is that many patients are being monitored needlessly, because telemetric monitoring neither affects how patients at low risk are managed nor improves their clinical outcomes.
Saleem et al4 reported that, of 105 patients at low risk who presented with chest pain and were admitted to a telemetry unit, none experienced a cardiac event or arrhythmia warranting changes in management while in the hospital.
Durairaj et al5 conducted a prospective cohort study of 1,033 patients admitted consecutively from an emergency department to an inpatient telemetry unit from July 1998 to January 1999. Patients were initially stratified according to a prediction model proposed by Goldman et al6 into groups at high, moderate, low, and very low risk. The risk groups were substratified according to the presence or absence of chest pain. The outcomes measured were transfer to an intensive care unit and a major cardiac complication, which included acute myocardial infarction, cardiac arrest, ventricular fibrillation, temporary pacemaker implantation, cardiogenic shock, emergency cardioversion, use of an intraaortic balloon assist device, intubation, and recurrent ischemic pain requiring coronary revascularization within 72 hours after admission or requiring cardiac catheterization followed by coronary revascularization before discharge from the hospital. The subgroup of patients who were classified as being at very low risk and who did not have chest pain (n = 318) did not experience any major cardiac complication.
Sivaram and colleagues7 studied the role of telemetric monitoring in the management of patients with class I, II, and III indications for telemetric monitoring outside of critical care units. The class was assigned at the time of discharge for the purpose of the study. A total of 297 telemetry events were noted during the study, but only 12 (4%) of the events led to changes in patient management: a change in medication in 8 patients, cardioversion for unstable atrial flutter in 1 patient, insertion of a pacemaker for sinus pause in 1, and electrophysiology studies in 2 patients.
Estrada et al8 examined the clinical outcomes of 2,240 patients admitted to a non-intensive care unit. The physicians perceived telemetric monitoring as helpful in 283 (12.6%) of the patients. However, data obtained from telemetry monitoring directly affected management decisions in only 156 patients (7% of the original study population). The researchers concluded that physicians may overestimate the role of telemetry in guiding patient management.
Hollander et al9 examined the outcomes of 261 patients admitted because of chest pain who had normal or nonspecific findings on electrocardiography on presentation. Only 4 patients (1.5%) experienced arrhythmias. The authors concluded that the policy of admitting patients at low risk to monitored beds should be reevaluated.
Snider et al10 showed that patients presenting with atypical chest pain and normal electrocardiographic findings were at low risk of arrhythmias and did not benefit from telemetric monitoring.
Schull and Redelmeier11 performed a 5-year observational study in which they reviewed all telemetry admissions (N = 8,932) to a tertiary care facility. Twenty patients experienced cardiac arrest during the study period, but telemetric monitoring was in use at the time in only 16 of the 20. Furthermore, the telemetry monitors signalled the onset of cardiac arrest in only 9 of these 16 patients. Three of the patients whose hearts stopped beating survived until discharge: two in whom telemetry actually signalled the onset of cardiac arrest and one in whom it did not.
TELEMETRY CAN GIVE FALSE-POSITIVE ALARMS
Inappropriate use of telemetric monitoring increases the chance of artifacts or false-positive rhythms being misinterpreted as dysrhythmias and can potentially lead to errors in management.
Cases have been reported of patients undergoing invasive procedures because of artifacts seen during telemetric monitoring. Knight et al12 described 12 patients who underwent unnecessary diagnostic or therapeutic interventions as a result of misdiagnosis of artifacts as ventricular tachycardia.
We did not discover in our review any data correlating the frequency of false-positive telemetric monitoring findings to management errors. On the other hand, it is also not possible to discern from these studies how often cardiac telemetric monitoring reaffirmed the clinical impression and facilitated ongoing therapy.
TELEMETRY IS EXPENSIVE
Telemetry requires specialized equipment and trained personnel, making it both costly and labor-intensive. The additional costs and cost-effectiveness of telemetry remain uncertain. Studies of its medical costs have found wide variations across different hospital systems. Sivaram et al,7 in an observational study published in 1998, estimated the cost per patient at $683. At our hospital, the current cost of telemetric monitoring is at least $1,400 per patient per 24 hours.
Whatever the true cost, inappropriate use of telemetry creates a financial burden on the health care system and adds to unnecessary costs incurred by patients.
POTENTIAL BARRIERS TO APPROPRIATE USE OF TELEMETRY
A number of factors contribute to the inappropriate use of telemetry. Possible causes for its overuse may be a lack of awareness of the ACC and AHA guidelines, nonadherence to the guidelines, or a combination of factors.
Even when physicians are aware of these guidelines, adherence may be suboptimal for a variety of reasons (reviewed by Mehta13). Adams et al14 revealed that most studies evaluating adherence were biased by overreporting, since the levels of adherence were self-reported.
OUR RECOMMENDATIONS
To improve on the appropriate use of telemetry, we recommend that several strategies be implemented.
Current guidelines for in-hospital cardiac monitoring need to be updated, particularly since the recommendations were based on evidence that is several decades old. Also, medical care has improved since the publication of the last guidelines, justifying an update in the guidelines.
Guidelines for cardiac monitoring should be incorporated into the curriculum for physician education to increase awareness of the guidelines. Hospitals should ensure that the emergency medicine staff is educated with regard to ensuring appropriateness of admissions to telemetry units.
Finally, the implementation of predictive models similar to that developed by Goldman et al6 and implemented in the study by Durairaj5 could help to ensure that cardiac telemetry is reserved for patients who will benefit from it the most.
- NASA Scientific and Technical Information (STI). Space-proven medical monitor: the total patient care package. Health and medicine. Originating technology/NASA contribution. Spinoff 2006. www.sti.nasa.gov/Textonly/tto/Spinoff2006/hm_2.html. Accessed 1/2009.
- Jaffe AS, Atkins JM, Field JM, et al. Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. J Am Coll Cardiol 1991; 18:1431–1433.
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation 2004; 110:2721–2746.
- Saleem MA, McClung JA, Aronow WS, Kannam H. Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in-hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci 2005; 60:605–606.
- Durairaj L, Reilly B, Das K, et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med 2001; 110:7–11.
- Goldman A, Cook EF, Johnson PA, et al. Prediction of the need for intensive care in patients who come to emergency departments with acute chest pain. N Engl J Med 1996; 334:1498–1504.
- Sivaram CA, Summers JH, Ahmed N. Telemetry outside critical care units: patterns of utilization and influence on management decisions. Clin Cardiol 1998; 21:503–505.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol 1995; 76:960–965.
- Hollander JE, Valentine SM, McCuskey CF, Brogan GX. Are monitored telemetry beds necessary for patients with nontraumatic chest pain and normal or nonspecific electrocardiograms? Am J Cardiol 1997; 79:1110–1111.
- Snider A, Papaleo M, Beldner S, et al. Is telemetry monitoring necessary in low-risk suspected acute chest pain syndromes? Chest 2002; 122:517–523.
- Schull MJ, Redelmeier DA. Continuous electrocardiographic monitoring and cardiac arrest outcomes in 8,932 telemetry ward patients. Acad Emerg Med 2000; 7:647–652.
- Knight BP, Pelosi F, Michaud GF, Strickberger SA, Morady F. Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med 1999; 341:1270–1274.
- Mehta NB. The doctors’ challenge: How can we follow guidelines better? Cleve Clin J Med 2004; 71:81–85.
- Adams AS, Soumerai SB, Lomas J, Ross-Degnan D. Evidence of self-reporting bias in assessing adherence to guidelines. Int J Quality Health Care 1999; 11:187–192.
Telemetry—from the Greek words tele (remote) and metron (measure)—for cardiac monitoring was developed in the mid-1960s by Spacelabs Medical for use in spaceflight.1 The system was later adopted in hospitals to detect life-threatening arrhythmias.
Guidelines for the use of telemetry were published in 1991 by the American College of Cardiology (ACC)2 in response to concerns raised by its increasing use in noncritical care settings during the 30 years after its introduction to clinical medicine. The latest revision of the guidelines was published in 2004 by the American Heart Association (AHA).3
However, the guidelines are based largely on expert opinion and on research data in electrocardiography. Few clinical trials of telemetry have been published, and they were either retrospective or nonrandomized. In fact, there were no published randomized trials at the time the 2004 guidelines were written. Moreover, very few of these studies evaluated the impact of cardiac telemetry monitoring on physician management decisions.
We reviewed the literature to find out how cardiac telemetry is being used in clinical practice and how it might be used more selectively. The literature search was performed using Ovid MEDLINE (1996 to present) and PubMed Central using the key search terms “cardiac monitoring,” “telemetry monitoring,” “telemetry,” and “inpatient.” References from articles identified using Ovid MEDLINE (1996 to present) and PubMed Central that were relevant to our review were also included.
THREE CLASSES OF RISK
- Class II consists of patients for whom cardiac monitoring may be of benefit in some cases but is not essential for all (Table 2).
PATIENTS AT LOW RISK DO NOT BENEFIT
Telemetry monitoring has become an essential and commonly used clinical tool in most hospital systems. However, physicians do not seem to be using the risk stratification guidelines routinely or appropriately. The result is that many patients are being monitored needlessly, because telemetric monitoring neither affects how patients at low risk are managed nor improves their clinical outcomes.
Saleem et al4 reported that, of 105 patients at low risk who presented with chest pain and were admitted to a telemetry unit, none experienced a cardiac event or arrhythmia warranting changes in management while in the hospital.
Durairaj et al5 conducted a prospective cohort study of 1,033 patients admitted consecutively from an emergency department to an inpatient telemetry unit from July 1998 to January 1999. Patients were initially stratified according to a prediction model proposed by Goldman et al6 into groups at high, moderate, low, and very low risk. The risk groups were substratified according to the presence or absence of chest pain. The outcomes measured were transfer to an intensive care unit and a major cardiac complication, which included acute myocardial infarction, cardiac arrest, ventricular fibrillation, temporary pacemaker implantation, cardiogenic shock, emergency cardioversion, use of an intraaortic balloon assist device, intubation, and recurrent ischemic pain requiring coronary revascularization within 72 hours after admission or requiring cardiac catheterization followed by coronary revascularization before discharge from the hospital. The subgroup of patients who were classified as being at very low risk and who did not have chest pain (n = 318) did not experience any major cardiac complication.
Sivaram and colleagues7 studied the role of telemetric monitoring in the management of patients with class I, II, and III indications for telemetric monitoring outside of critical care units. The class was assigned at the time of discharge for the purpose of the study. A total of 297 telemetry events were noted during the study, but only 12 (4%) of the events led to changes in patient management: a change in medication in 8 patients, cardioversion for unstable atrial flutter in 1 patient, insertion of a pacemaker for sinus pause in 1, and electrophysiology studies in 2 patients.
Estrada et al8 examined the clinical outcomes of 2,240 patients admitted to a non-intensive care unit. The physicians perceived telemetric monitoring as helpful in 283 (12.6%) of the patients. However, data obtained from telemetry monitoring directly affected management decisions in only 156 patients (7% of the original study population). The researchers concluded that physicians may overestimate the role of telemetry in guiding patient management.
Hollander et al9 examined the outcomes of 261 patients admitted because of chest pain who had normal or nonspecific findings on electrocardiography on presentation. Only 4 patients (1.5%) experienced arrhythmias. The authors concluded that the policy of admitting patients at low risk to monitored beds should be reevaluated.
Snider et al10 showed that patients presenting with atypical chest pain and normal electrocardiographic findings were at low risk of arrhythmias and did not benefit from telemetric monitoring.
Schull and Redelmeier11 performed a 5-year observational study in which they reviewed all telemetry admissions (N = 8,932) to a tertiary care facility. Twenty patients experienced cardiac arrest during the study period, but telemetric monitoring was in use at the time in only 16 of the 20. Furthermore, the telemetry monitors signalled the onset of cardiac arrest in only 9 of these 16 patients. Three of the patients whose hearts stopped beating survived until discharge: two in whom telemetry actually signalled the onset of cardiac arrest and one in whom it did not.
TELEMETRY CAN GIVE FALSE-POSITIVE ALARMS
Inappropriate use of telemetric monitoring increases the chance of artifacts or false-positive rhythms being misinterpreted as dysrhythmias and can potentially lead to errors in management.
Cases have been reported of patients undergoing invasive procedures because of artifacts seen during telemetric monitoring. Knight et al12 described 12 patients who underwent unnecessary diagnostic or therapeutic interventions as a result of misdiagnosis of artifacts as ventricular tachycardia.
We did not discover in our review any data correlating the frequency of false-positive telemetric monitoring findings to management errors. On the other hand, it is also not possible to discern from these studies how often cardiac telemetric monitoring reaffirmed the clinical impression and facilitated ongoing therapy.
TELEMETRY IS EXPENSIVE
Telemetry requires specialized equipment and trained personnel, making it both costly and labor-intensive. The additional costs and cost-effectiveness of telemetry remain uncertain. Studies of its medical costs have found wide variations across different hospital systems. Sivaram et al,7 in an observational study published in 1998, estimated the cost per patient at $683. At our hospital, the current cost of telemetric monitoring is at least $1,400 per patient per 24 hours.
Whatever the true cost, inappropriate use of telemetry creates a financial burden on the health care system and adds to unnecessary costs incurred by patients.
POTENTIAL BARRIERS TO APPROPRIATE USE OF TELEMETRY
A number of factors contribute to the inappropriate use of telemetry. Possible causes for its overuse may be a lack of awareness of the ACC and AHA guidelines, nonadherence to the guidelines, or a combination of factors.
Even when physicians are aware of these guidelines, adherence may be suboptimal for a variety of reasons (reviewed by Mehta13). Adams et al14 revealed that most studies evaluating adherence were biased by overreporting, since the levels of adherence were self-reported.
OUR RECOMMENDATIONS
To improve on the appropriate use of telemetry, we recommend that several strategies be implemented.
Current guidelines for in-hospital cardiac monitoring need to be updated, particularly since the recommendations were based on evidence that is several decades old. Also, medical care has improved since the publication of the last guidelines, justifying an update in the guidelines.
Guidelines for cardiac monitoring should be incorporated into the curriculum for physician education to increase awareness of the guidelines. Hospitals should ensure that the emergency medicine staff is educated with regard to ensuring appropriateness of admissions to telemetry units.
Finally, the implementation of predictive models similar to that developed by Goldman et al6 and implemented in the study by Durairaj5 could help to ensure that cardiac telemetry is reserved for patients who will benefit from it the most.
Telemetry—from the Greek words tele (remote) and metron (measure)—for cardiac monitoring was developed in the mid-1960s by Spacelabs Medical for use in spaceflight.1 The system was later adopted in hospitals to detect life-threatening arrhythmias.
Guidelines for the use of telemetry were published in 1991 by the American College of Cardiology (ACC)2 in response to concerns raised by its increasing use in noncritical care settings during the 30 years after its introduction to clinical medicine. The latest revision of the guidelines was published in 2004 by the American Heart Association (AHA).3
However, the guidelines are based largely on expert opinion and on research data in electrocardiography. Few clinical trials of telemetry have been published, and they were either retrospective or nonrandomized. In fact, there were no published randomized trials at the time the 2004 guidelines were written. Moreover, very few of these studies evaluated the impact of cardiac telemetry monitoring on physician management decisions.
We reviewed the literature to find out how cardiac telemetry is being used in clinical practice and how it might be used more selectively. The literature search was performed using Ovid MEDLINE (1996 to present) and PubMed Central using the key search terms “cardiac monitoring,” “telemetry monitoring,” “telemetry,” and “inpatient.” References from articles identified using Ovid MEDLINE (1996 to present) and PubMed Central that were relevant to our review were also included.
THREE CLASSES OF RISK
- Class II consists of patients for whom cardiac monitoring may be of benefit in some cases but is not essential for all (Table 2).
PATIENTS AT LOW RISK DO NOT BENEFIT
Telemetry monitoring has become an essential and commonly used clinical tool in most hospital systems. However, physicians do not seem to be using the risk stratification guidelines routinely or appropriately. The result is that many patients are being monitored needlessly, because telemetric monitoring neither affects how patients at low risk are managed nor improves their clinical outcomes.
Saleem et al4 reported that, of 105 patients at low risk who presented with chest pain and were admitted to a telemetry unit, none experienced a cardiac event or arrhythmia warranting changes in management while in the hospital.
Durairaj et al5 conducted a prospective cohort study of 1,033 patients admitted consecutively from an emergency department to an inpatient telemetry unit from July 1998 to January 1999. Patients were initially stratified according to a prediction model proposed by Goldman et al6 into groups at high, moderate, low, and very low risk. The risk groups were substratified according to the presence or absence of chest pain. The outcomes measured were transfer to an intensive care unit and a major cardiac complication, which included acute myocardial infarction, cardiac arrest, ventricular fibrillation, temporary pacemaker implantation, cardiogenic shock, emergency cardioversion, use of an intraaortic balloon assist device, intubation, and recurrent ischemic pain requiring coronary revascularization within 72 hours after admission or requiring cardiac catheterization followed by coronary revascularization before discharge from the hospital. The subgroup of patients who were classified as being at very low risk and who did not have chest pain (n = 318) did not experience any major cardiac complication.
Sivaram and colleagues7 studied the role of telemetric monitoring in the management of patients with class I, II, and III indications for telemetric monitoring outside of critical care units. The class was assigned at the time of discharge for the purpose of the study. A total of 297 telemetry events were noted during the study, but only 12 (4%) of the events led to changes in patient management: a change in medication in 8 patients, cardioversion for unstable atrial flutter in 1 patient, insertion of a pacemaker for sinus pause in 1, and electrophysiology studies in 2 patients.
Estrada et al8 examined the clinical outcomes of 2,240 patients admitted to a non-intensive care unit. The physicians perceived telemetric monitoring as helpful in 283 (12.6%) of the patients. However, data obtained from telemetry monitoring directly affected management decisions in only 156 patients (7% of the original study population). The researchers concluded that physicians may overestimate the role of telemetry in guiding patient management.
Hollander et al9 examined the outcomes of 261 patients admitted because of chest pain who had normal or nonspecific findings on electrocardiography on presentation. Only 4 patients (1.5%) experienced arrhythmias. The authors concluded that the policy of admitting patients at low risk to monitored beds should be reevaluated.
Snider et al10 showed that patients presenting with atypical chest pain and normal electrocardiographic findings were at low risk of arrhythmias and did not benefit from telemetric monitoring.
Schull and Redelmeier11 performed a 5-year observational study in which they reviewed all telemetry admissions (N = 8,932) to a tertiary care facility. Twenty patients experienced cardiac arrest during the study period, but telemetric monitoring was in use at the time in only 16 of the 20. Furthermore, the telemetry monitors signalled the onset of cardiac arrest in only 9 of these 16 patients. Three of the patients whose hearts stopped beating survived until discharge: two in whom telemetry actually signalled the onset of cardiac arrest and one in whom it did not.
TELEMETRY CAN GIVE FALSE-POSITIVE ALARMS
Inappropriate use of telemetric monitoring increases the chance of artifacts or false-positive rhythms being misinterpreted as dysrhythmias and can potentially lead to errors in management.
Cases have been reported of patients undergoing invasive procedures because of artifacts seen during telemetric monitoring. Knight et al12 described 12 patients who underwent unnecessary diagnostic or therapeutic interventions as a result of misdiagnosis of artifacts as ventricular tachycardia.
We did not discover in our review any data correlating the frequency of false-positive telemetric monitoring findings to management errors. On the other hand, it is also not possible to discern from these studies how often cardiac telemetric monitoring reaffirmed the clinical impression and facilitated ongoing therapy.
TELEMETRY IS EXPENSIVE
Telemetry requires specialized equipment and trained personnel, making it both costly and labor-intensive. The additional costs and cost-effectiveness of telemetry remain uncertain. Studies of its medical costs have found wide variations across different hospital systems. Sivaram et al,7 in an observational study published in 1998, estimated the cost per patient at $683. At our hospital, the current cost of telemetric monitoring is at least $1,400 per patient per 24 hours.
Whatever the true cost, inappropriate use of telemetry creates a financial burden on the health care system and adds to unnecessary costs incurred by patients.
POTENTIAL BARRIERS TO APPROPRIATE USE OF TELEMETRY
A number of factors contribute to the inappropriate use of telemetry. Possible causes for its overuse may be a lack of awareness of the ACC and AHA guidelines, nonadherence to the guidelines, or a combination of factors.
Even when physicians are aware of these guidelines, adherence may be suboptimal for a variety of reasons (reviewed by Mehta13). Adams et al14 revealed that most studies evaluating adherence were biased by overreporting, since the levels of adherence were self-reported.
OUR RECOMMENDATIONS
To improve on the appropriate use of telemetry, we recommend that several strategies be implemented.
Current guidelines for in-hospital cardiac monitoring need to be updated, particularly since the recommendations were based on evidence that is several decades old. Also, medical care has improved since the publication of the last guidelines, justifying an update in the guidelines.
Guidelines for cardiac monitoring should be incorporated into the curriculum for physician education to increase awareness of the guidelines. Hospitals should ensure that the emergency medicine staff is educated with regard to ensuring appropriateness of admissions to telemetry units.
Finally, the implementation of predictive models similar to that developed by Goldman et al6 and implemented in the study by Durairaj5 could help to ensure that cardiac telemetry is reserved for patients who will benefit from it the most.
- NASA Scientific and Technical Information (STI). Space-proven medical monitor: the total patient care package. Health and medicine. Originating technology/NASA contribution. Spinoff 2006. www.sti.nasa.gov/Textonly/tto/Spinoff2006/hm_2.html. Accessed 1/2009.
- Jaffe AS, Atkins JM, Field JM, et al. Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. J Am Coll Cardiol 1991; 18:1431–1433.
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation 2004; 110:2721–2746.
- Saleem MA, McClung JA, Aronow WS, Kannam H. Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in-hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci 2005; 60:605–606.
- Durairaj L, Reilly B, Das K, et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med 2001; 110:7–11.
- Goldman A, Cook EF, Johnson PA, et al. Prediction of the need for intensive care in patients who come to emergency departments with acute chest pain. N Engl J Med 1996; 334:1498–1504.
- Sivaram CA, Summers JH, Ahmed N. Telemetry outside critical care units: patterns of utilization and influence on management decisions. Clin Cardiol 1998; 21:503–505.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol 1995; 76:960–965.
- Hollander JE, Valentine SM, McCuskey CF, Brogan GX. Are monitored telemetry beds necessary for patients with nontraumatic chest pain and normal or nonspecific electrocardiograms? Am J Cardiol 1997; 79:1110–1111.
- Snider A, Papaleo M, Beldner S, et al. Is telemetry monitoring necessary in low-risk suspected acute chest pain syndromes? Chest 2002; 122:517–523.
- Schull MJ, Redelmeier DA. Continuous electrocardiographic monitoring and cardiac arrest outcomes in 8,932 telemetry ward patients. Acad Emerg Med 2000; 7:647–652.
- Knight BP, Pelosi F, Michaud GF, Strickberger SA, Morady F. Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med 1999; 341:1270–1274.
- Mehta NB. The doctors’ challenge: How can we follow guidelines better? Cleve Clin J Med 2004; 71:81–85.
- Adams AS, Soumerai SB, Lomas J, Ross-Degnan D. Evidence of self-reporting bias in assessing adherence to guidelines. Int J Quality Health Care 1999; 11:187–192.
- NASA Scientific and Technical Information (STI). Space-proven medical monitor: the total patient care package. Health and medicine. Originating technology/NASA contribution. Spinoff 2006. www.sti.nasa.gov/Textonly/tto/Spinoff2006/hm_2.html. Accessed 1/2009.
- Jaffe AS, Atkins JM, Field JM, et al. Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. J Am Coll Cardiol 1991; 18:1431–1433.
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation 2004; 110:2721–2746.
- Saleem MA, McClung JA, Aronow WS, Kannam H. Inpatient telemetry does not need to be used in the management of older patients hospitalized with chest pain at low risk for in-hospital coronary events and mortality. J Gerontol A Biol Sci Med Sci 2005; 60:605–606.
- Durairaj L, Reilly B, Das K, et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med 2001; 110:7–11.
- Goldman A, Cook EF, Johnson PA, et al. Prediction of the need for intensive care in patients who come to emergency departments with acute chest pain. N Engl J Med 1996; 334:1498–1504.
- Sivaram CA, Summers JH, Ahmed N. Telemetry outside critical care units: patterns of utilization and influence on management decisions. Clin Cardiol 1998; 21:503–505.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol 1995; 76:960–965.
- Hollander JE, Valentine SM, McCuskey CF, Brogan GX. Are monitored telemetry beds necessary for patients with nontraumatic chest pain and normal or nonspecific electrocardiograms? Am J Cardiol 1997; 79:1110–1111.
- Snider A, Papaleo M, Beldner S, et al. Is telemetry monitoring necessary in low-risk suspected acute chest pain syndromes? Chest 2002; 122:517–523.
- Schull MJ, Redelmeier DA. Continuous electrocardiographic monitoring and cardiac arrest outcomes in 8,932 telemetry ward patients. Acad Emerg Med 2000; 7:647–652.
- Knight BP, Pelosi F, Michaud GF, Strickberger SA, Morady F. Clinical consequences of electrocardiographic artifact mimicking ventricular tachycardia. N Engl J Med 1999; 341:1270–1274.
- Mehta NB. The doctors’ challenge: How can we follow guidelines better? Cleve Clin J Med 2004; 71:81–85.
- Adams AS, Soumerai SB, Lomas J, Ross-Degnan D. Evidence of self-reporting bias in assessing adherence to guidelines. Int J Quality Health Care 1999; 11:187–192.
KEY POINTS
- Guidelines from the American College of Cardiology (1991) and American Heart Association (2004) divide patients into three risk classes for whom telemetry is, may be, or is not indicated.
- Few studies have addressed whether telemetry is beneficial in clinical practice.
- The available evidence suggests that telemetry infrequently influences physician management decisions for patients at low risk, although it may in a relatively small subset at high risk.
- Inappropriate use of telemetry is associated with unnecessary testing and treatment and higher cost of care.
- Better risk-assessment and selection strategies are needed to identify patients for whom telemetry monitoring will be most beneficial.
Platelet Response in Acute Coronary Syndromes

Supplement Editors:
Deepak L. Bhatt, MD, MPH, and W. Frank Peacock, MD
Contents
Importance of platelets and platelet response in acute coronary syndromes
Kandice Kottke-Marchant, MD, PhD
Novel antiplatelet strategies in acute coronary syndromes
Marc S. Sabatine, MD, MPH
The current state of antiplatelet therapy in acute coronary syndromes: The data and the real word
John H. Alexander, MD, MHSc
Platelet response in practice: Applying new insights and tools for testing and treatment
Deepak L. Bhatt, MD, MPH; Kandice Kottke-Marchant, MD, PhD; John H. Alexander, MD, MHSc; W. Frank Peacock, MD; and Marc S. Sabatine, MD, MPH
Supplement Editors:
Deepak L. Bhatt, MD, MPH, and W. Frank Peacock, MD
Contents
Importance of platelets and platelet response in acute coronary syndromes
Kandice Kottke-Marchant, MD, PhD
Novel antiplatelet strategies in acute coronary syndromes
Marc S. Sabatine, MD, MPH
The current state of antiplatelet therapy in acute coronary syndromes: The data and the real word
John H. Alexander, MD, MHSc
Platelet response in practice: Applying new insights and tools for testing and treatment
Deepak L. Bhatt, MD, MPH; Kandice Kottke-Marchant, MD, PhD; John H. Alexander, MD, MHSc; W. Frank Peacock, MD; and Marc S. Sabatine, MD, MPH
Supplement Editors:
Deepak L. Bhatt, MD, MPH, and W. Frank Peacock, MD
Contents
Importance of platelets and platelet response in acute coronary syndromes
Kandice Kottke-Marchant, MD, PhD
Novel antiplatelet strategies in acute coronary syndromes
Marc S. Sabatine, MD, MPH
The current state of antiplatelet therapy in acute coronary syndromes: The data and the real word
John H. Alexander, MD, MHSc
Platelet response in practice: Applying new insights and tools for testing and treatment
Deepak L. Bhatt, MD, MPH; Kandice Kottke-Marchant, MD, PhD; John H. Alexander, MD, MHSc; W. Frank Peacock, MD; and Marc S. Sabatine, MD, MPH

Endoscopic therapy of recurrent acute pancreatitis
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
- Autoimmune disorders
- Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
- Chronic pancreatitis
- Drugs
- Heredity
- Metabolic abnormalities (hypertriglyceridemia, hypercalcemia)
- Sphincter of Oddi dysfunction
- Structural or congenital abnormalities (pancreas divisum)
- Trauma.
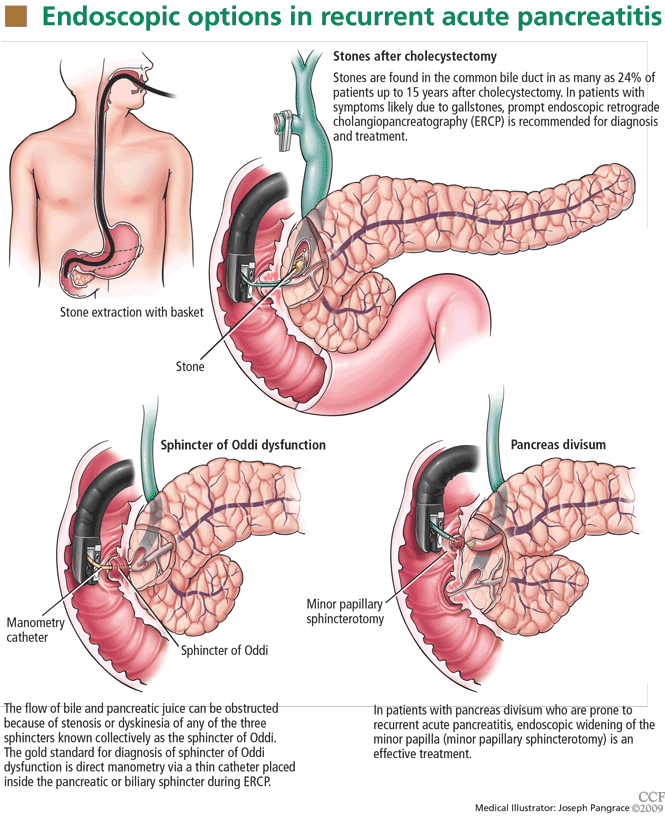
- Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
- Sphincter of Oddi dysfunction
- Pancreas divisum
- Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis (Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
- Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol 2001; 96:2540–2555.
- Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol 2002; 97:1959–1962.
- Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol 2006; 41:681–685.
- Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology 2001; 120:708–717.
- Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol 2007; 102:1781–1788.
- Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc 2007; 66:213–218.
- Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology 1994; 20:523–525.
- Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 1901; 12:182–188.
- Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373.
- Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology 1991; 101:1701–1709.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med 1992; 326:589–593.
- Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts? Gastrointest Endosc 2002; 55:286–293.
- Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology 1986; 6:118–122.
- Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology 1986; 90:677–686.
- Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol 2000; 95:1702–1707.
- Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther 2006; 24:1059–1066.
- Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas 2003; 27:103–117.
- Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am 2003; 13:695–716.
- Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc 2005; 62:1–8.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology 2005; 128:756–763.
- Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy 2003; 35:92.
- Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg 1977; 185:598–604.
- Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol 2008; 14:1023–1026.
- Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335:909–918.
- Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology 1996; 110:894–899.
- Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology 1990; 98:1008–1012.
- Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up? Gastrointest Endosc 2002; 55:523–526.
- Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc 2001; 54:42–48.
- Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy 2002; 34:273–279.
- Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc 2006; 64:60–65.
- Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc 2002; 55:157–162.
- Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc 1999; 49:599–604.
- Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc 2008; 67:AB102.
- Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil 2005; 17 suppl 1:31–40.
- McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol 2007; 13:6333–6343.
- Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci 2007; 52:1211–1218.
- Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep 2002; 4:153–159.
- Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas 2005; 30:359–362.
- Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP 2004; 5:322–327.
- Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc 2004; 59:499–505.
- Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility? Rev Gastroenterol Disord 2004; 4:211–213.
- Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy? Curr Gastroenterol Rep 2002; 4:172–176.
- Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol 2007; 41:94–102.
- Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther 2006; 24:237–246.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989; 34:56–60.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy 2002; 34:280–285.
- Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol 2007; 5:1354–1365.
- Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford) 2003; 5:142–145.
- Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP 2001; 2:382–400.
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004; 60:419–425.
- Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas 2007; 34:21–45.
- Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol 2003; 17:381–383.
- Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am 1995; 5:145–170.
- Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis? Pancreas 1988; 3:108–110.
- Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas 2008; 37:151–153.
- Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg 1990; 159:59–64.
- Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg 1996; 183:65–70.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc 2002; 55:376–381.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc 1992; 38:430–434.
- Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol 2008; 14:1027–1033.
- Finnie IA, Ghosh P, Garvey C, Poston GJ, Rhodes JM. Intraluminal duodenal diverticulum causing recurrent pancreatitis: treatment by endoscopic incision. Gut 1994; 35:557–559.
- Guzzardo G, Kleinman MS, Krackov JH, Schwartz SI. Recurrent acute pancreatitis caused by ampullary villous adenoma. J Clin Gastroenterol 1990; 12:200–202.
- Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg 1999; 134:311–315.
- Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg 2004; 67:309–310.
- Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol 2007; 13:2892–2894.
- Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc 1990; 36:63–65.
- Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J 2000; 41:528–532.
- Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol 2000; 31:172–174.
- Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol 2001; 16:1070,1075.
- Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery 2004; 136:909–916.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol 2007; 5:75–79.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc 2004; 60:673–678.
- Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 2007; 356:676–684.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
- Autoimmune disorders
- Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
- Chronic pancreatitis
- Drugs
- Heredity
- Metabolic abnormalities (hypertriglyceridemia, hypercalcemia)
- Sphincter of Oddi dysfunction
- Structural or congenital abnormalities (pancreas divisum)
- Trauma.
- Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
- Sphincter of Oddi dysfunction
- Pancreas divisum
- Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis (Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
Endoscopic therapy has become an alternative to surgery for some patients with acute recurrent pancreatitis, ie, those whose disease is caused by gallstones or other mechanical processes that can obstruct the outflow from the pancreas.
In this paper, we review the specific situations in which endoscopic therapy might be useful in patients with acute recurrent pancreatitis.
ACUTE PANCREATITIS IS MANAGED DIFFERENTLY IF IT RECURS
Recurrent acute pancreatitis is defined as more than one episode of acute pancreatitis.1 In clinical practice, it is important to distinguish between the first and recurrent episodes of acute pancreatitis.
Most patients who have one episode of acute pancreatitis never have another one.2,3 Therefore, for patients having an initial attack, we recommend a limited workup that includes a detailed history, laboratory evaluation, and a noninvasive imaging study such as transcutaneous ultrasonography or computed tomography.
On the other hand, people who have a second attack are at higher risk of more recurrences. Therefore, patients having recurrent attacks need a more extensive workup to determine the underlying cause. We recommend referring them to a gastroenterologist for further evaluation.
WHICH CAUSES CAN BE MANAGED ENDOSCOPICALLY?
In the Western world, 70% to 80% of cases of recurrent pancreatitis are due to either alcohol abuse or gallstone disease.2,4 The rest are related to:
- Autoimmune disorders
- Cancer, including occult malignancies and premalignant conditions such as intraductal papillary mucinous neoplasm
- Chronic pancreatitis
- Drugs
- Heredity
- Metabolic abnormalities (hypertriglyceridemia, hypercalcemia)
- Sphincter of Oddi dysfunction
- Structural or congenital abnormalities (pancreas divisum)
- Trauma.
- Gallstone disease, including biliary microlithiasis and sludge (in patients with or without a gallbladder)
- Sphincter of Oddi dysfunction
- Pancreas divisum
- Obstruction to flow of pancreatic juice.
Endoscopy is not completely benign
Although endoscopic procedures are less invasive than surgery, they are not completely benign. They can cause anxiety and are associated with risks such as bleeding, perforation, and pancreatitis.5 The risks, benefits, and alternatives to these procedures should be discussed with the patient, and informed consent should be obtained before any endoscopic procedure.6
STONES (LARGE OR SMALL) OR SLUDGE IN PATIENTS WITH A GALLBLADDER
Gallstones can be large, but small stones (microlithiasis) and sludge are more common and therefore account for more cases of pancreatitis.
Strictly defined, microlithiasis refers to stones smaller than 2 mm in diameter in the biliary tract, whereas sludge is a suspension of biliary crystals, mucin, and cellular debris in the gallbladder or bile ducts.7 The terms are often used interchangeably, since the conditions often coexist and their treatment is similar.
Theories differ as to how microlithiasis or sludge can cause recurrent pancreatitis. According to one theory, the debris blocks the common channel, increasing the pancreatic intraductal pressure and leading to pancreatitis.8 A second theory is that small stones or biliary crystals passing through the sphincter of Oddi cause inflammation, and that repeated inflammation eventually leads to stenosis or dyskinesia of the sphincter, both of which have been associated with pancreatitis.9
Studies suggest that microlithiasis and sludge are common causes of recurrent pancreatitis, accounting for about two-thirds of cases according to estimates by Ros et al10 and Lee et al.11
Detecting small stones and sludge
The diagnosis of microlithiasis and biliary sludge in patients with a gallbladder is based on imaging studies and bile microscopy.12
Transabdominal ultrasonography is the imaging study most often used for diagnosing microlithiasis. The technology and expertise for this test are widely available, and it is relatively inexpensive.
Endoscopic ultrasonography is more sensitive for detecting microlithiasis and can examine the distal common bile duct.
Bile microscopy involves obtaining bile from the second portion of the duodenum (via an endoscope or a duodenal tube) or from the bile ducts (by cannulating the common bile duct and stimulating the gallbladder with cholecystokinin). The bile sample is centrifuged and inspected microscopically under plain light and polarized light (which aids the visualization of biliary crystals). The crystals can be cholesterol monohydrate, calcium bilirubinate, or calcium carbonate.7,13,14
Removing the gallbladder is the treatment of choice for small stones and sludge
Treatments to prevent recurrent attacks of acute pancreatitis due to microlithiasis and sludge include cholecystectomy, biliary sphincterotomy, and ursodioxycholic acid.10,11,15
In prospective observational studies by Ros et al10 and Lee et al,11 about half of the patients with recurrent pancreatitis were treated with cholecystectomy, endoscopic sphincterotomy, or ursodioxycholic acid in a nonrandomized fashion. The choice of therapy was based on the patient’s medical status and the preferences of the patient and the physician. Half the patients received no treatment. In both studies the median follow-up was 4 years. Treated patients had a significantly lower rate of recurrent attacks of pancreatitis during follow-up: less than 20% with therapy compared with more than 60% without therapy. Unfortunately, no published study has compared these three treatments head to head.
Cholecystectomy, however, is the most definitive therapy and is generally considered the treatment of choice.
Biliary sphincterotomy is an endoscopic procedure that involves cutting the sphincter of Oddi to allow the stones and sludge to pass more freely. It is as effective as cholecystectomy in preventing recurrent attacks but does not eliminate the risk of cholecystitis and cholangitis (Figure 1). For this reason, it is usually reserved for patients who cannot tolerate surgery due to comorbidities, those who refuse surgery, or those who are pregnant.16
Ursodeoxycholic acid is a reasonable alternative in patients who cannot tolerate surgical or endoscopic biliary sphincterotomy.1,17–20 The dosage is 10 mg/kg/day, which can be in two or three divided doses. The optimal duration of treatment is not known; however, since this drug works slowly, it may need to be taken for 2 years or more. Ursodeoxycholic acid is more effective in patients with cholesterol-based stones and crystals. It is not effective for large stones (> 1 cm in diameter) or calcified stones.
STONES AFTER CHOLECYSTECTOMY
Bile duct stones can be classified as primary or secondary. A primary stone is one that remains where it was formed, whereas a secondary stone is one that has migrated from its site of formation.21
Some suggest that bile duct stones that are detected within 2 years of cholecystectomy originated in the gallbladder and were missed when the gallbladder was removed (and therefore are considered secondary stones), and that stones that present more than 2 years after cholecystectomy are de novo (ie, primary) stones.22,23
In any event, stones have been found in the common bile duct in 4% to 24% of patients up to 15 years after cholecystectomy.24–26 A fair number of these patients have no symptoms.27 Risk factors for stone recurrence are lithogenic bile (ie, high concentration of cholesterol, low concentration of bile salts), biliary stasis, strictures, dilated bile ducts, and advanced age.28–30
No role for crystal analysis after cholecystectomy
Biliary crystal analysis does not seem to have diagnostic value in patients with recurrent acute pancreatitis after cholecystectomy,31 because removing the gallbladder eliminates the crystals and sludge. Imaging studies are therefore the cornerstone of diagnosis.
Transabdominal ultrasonography is the most commonly used initial imaging test. However, abdominal fat and gas in the duodenum can obscure the distal common bile duct and decrease the sensitivity of this test.32
Endoscopic ultrasonography involves positioning the transducer in the second part of the duodenum, where it can show the adjacent biliary tree without interference from digestive gas or abdominal fat.
Magnetic resonance cholangiopancreatography (MRCP) and endoscopic ultrasonography are both highly sensitive for detecting common bile duct stones and are recommended if they can be done without delay.
Endoscopic retrograde cholangiopancreatography (ERCP). As a rule, patients who are very likely to have gallstones are best served by proceeding directly to ERCP, a procedure that enables both imaging and treatment. However, ERCP exposes the patient to radiation and the risk of pancreatitis, so in some patients (eg, pregnant women, people who recently had acute pancreatitis), one may want to do ultrasonography first.
ERCP is the treatment of choice after cholecystectomy
The treatment of choice in patients with choledocholithiasis is ERCP with biliary sphincterotomy and stone extraction. Success at clearing the biliary tree of all stones depends on the size, number, and location of the stones, the anatomy of the digestive tract and the bile duct, and the experience of the endoscopist. At specialized centers, the rate of successful clearance with subsequent procedures is close to 100%. Large stones may require fragmentation inside the bile duct to aid their removal.33
SPHINCTER OF ODDI DYSFUNCTION
The sphincter of Oddi, located where the bile and pancreatic ducts penetrate the wall of the duodenum, actually consists of three sphincters: the common, the biliary, and the pancreatic. Its physiologic role is to regulate the flow of bile and pancreatic juice into the duodenum and to prevent reflux into the ducts from the duodenum.34 Its basal pressure is the main regulating mechanism for pancreatic and biliary secretions into the intestine, and its phasic contractile activity is closely associated with duodenal motility.
Sphincter dysfunction: Stenosis, dyskinesia
The sphincter of Oddi can obstruct the flow of bile and pancreatic juice owing either to stenosis or to dyskinesia.35,36 Stenosis refers to structural alteration of the sphincter, probably from inflammation and subsequent fibrosis. In contrast, dyskinesia refers to a motor abnormality of the sphincter that makes it hypertonic.
Stenosis or dyskinesia can occur in the biliary sphincter, the pancreatic sphincter, the common sphincter, or any combination of the three. For example, dysfunction of the biliary sphincter can cause abnormalities in liver-associated enzyme levels and biliary-type pain, whereas pancreatic sphincter dysfunction can cause recurrent attacks of pancreatitis and pancreatic-type pain.37 Elevated pancreatic sphincter pressure has been shown to correlate with increased pancreatic ductal pressure, suggesting that the sphincter plays a role in the pathogenesis of acute pancreatitis.23,38
Sphincter pressure can be measured during ERCP, but ERCP is risky
The gold standard for the diagnosis of sphincter of Oddi dysfunction is manometry,23,35 ie, direct measurement of sphincter pressure via a thin catheter placed inside the pancreatic or biliary sphincter during ERCP (Figure 1).
However, in patients with suspected sphincter of Oddi dysfunction, ERCP with or without manometry is associated with a high rate of complications, with pancreatitis occurring in up to 25% of cases.39–41 Therefore, several noninvasive and provocative tests have been designed in an attempt to identify patients with this disorder. Unfortunately, none of them seems to be as sensitive and specific as manometry for diagnosing sphincter of Oddi dysfunction, and so they have not gained widespread use.
Opening the sphincter of Oddi with drugs, endoscopy, or surgery
Drug treatment of sphincter of Oddi dysfunction is based on drugs that relax smooth muscle, such as calcium channel blockers and nitrates. The treatment must be lifelong. Also, it does not improve sphincter stenosis, and only half of patients with sphincter dyskinesia respond to it. For these reasons, drug treatment of sphincter of Oddi dysfunction has not gained widespread acceptance.36,42
Endoscopic sphincterotomy is the current standard endoscopic therapy for sphincter of Oddi dysfunction. This procedure is performed during ERCP and involves cutting the sphincter with electrocautery.
Endoscopic pancreatic sphincterotomy prevents recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction in more than 60% of cases.23,43–46 A potential complication is pancreatitis, which occurs more often in patients with pancreatic sphincter dyskinesia. Placing a stent in the pancreatic duct after pancreatic sphincterotomy reduces the risk of pancreatitis after ERCP.37,47,48
Surgery. Pancreatic sphincterotomy can also be done surgically, most commonly via transduodenal pancreatic sphincteroplasty. Surgical sphincteroplasty is as effective as endoscopic sphincterotomy for preventing recurrent attacks of pancreatitis in patients with pancreatic sphincter dysfunction.49 However, endoscopic therapy is much less invasive and remains the preferred treatment for sphincter of Oddi dysfunction in most centers with experience in this technique.50
PANCREAS DIVISUM
Pancreas divisum is the most common congenital anomaly of the pancreatic duct. Autopsy studies show it occurs in 5% to 10% of the population.51–53
At approximately the 5th week of gestation, there are two pancreatic buds: a ventral and a dorsal bud. The ventral bud eventually gives rise to part of the pancreatic head and uncinate process of the pancreas in the adult. The dorsal bud eventually gives rise to the rest of the pancreatic head, the pancreatic body, and the pancreatic tail. At 6 to 7 weeks of gestation, the ventral bud rotates clockwise and lies posterior to the dorsal bud. At this stage, both the dorsal and ventral pancreata have their own ducts, which do not communicate with each other. Normally, the ventral and dorsal pancreas and their ducts fuse together at 8 weeks of gestation; in people with pancreas divisum, this ductal fusion does not occur.51
The pancreas secretes 1.5 L of fluid per day. Normally, 90% to 95% of this volume drains through the major papilla. In people with pancreas divisum, 90% to 95% of the fluid drains through the minor papilla.
People with pancreas divisum are a heterogeneous group. Most have no symptoms, and their ductal anatomy is diagnosed only incidentally. However, a subgroup is prone to develop acute pancreatitis. The cause is thought to be the small diameter of the minor papilla, which poses a relative obstruction to the flow of pancreatic juice.54 Direct support for this theory comes from a study in which investigators measured pancreatic ductal pressures in eight people with normal anatomy and six people with pancreas divisum. The pressure in the main pancreatic duct in those with pancreas divisum was significantly higher than in those with normal anatomy.55 Additional evidence in favor of this theory is the effectiveness of treatment, which involves widening the minor papillary opening (minor papillary sphincterotomy).
Diagnosis of pancreas divisum
The diagnosis of pancreas divisum is based on imaging studies, and ERCP remains the gold standard for patients with equivocal results on noninvasive imaging. However, MRCP, especially secretin-enhanced MRCP, is as accurate as ERCP. In most cases, MRCP has replaced ERCP for the diagnosis of this condition, although a recent study suggests that MRCP is inferior to ERCP in the diagnosis of pancreas divisum.56 We recommend secretin-enhanced MRCP for this purpose.
Computed tomography and endoscopic ultrasonography can also diagnose pancreas divisum, but their diagnostic accuracy is lower than that of ERCP and MRCP.
Minor papillary sphincterotomy
Treating recurrent pancreatitis due to pancreas divisum involves relieving the relative obstruction of the minor papilla by minor papillary sphincterotomy. This can be done surgically or endoscopically (Figure 1).
Surgery. No randomized, controlled study has yet assessed the efficacy of surgical sphincteroplasty for recurrent pancreatitis in patients with pancreas divisum. However, retrospective studies and one prospective study have been published.57,58
In the retrospective study with the largest number of patients, Warshaw et al57 reported their experience in 49 patients who had recurrent pancreatitis due to pancreas divisum. After surgical sphincteroplasty, the patients were followed for a mean of 53 months; 40 (82%) of the 49 patients had no further episodes of acute pancreatitis during this time.
Bradley and Stephan58 studied 37 patients with pancreas divisum and recurrent pancreatitis.58 After surgical sphincteroplasty, the patients were followed for a mean of 60 months; 31 of the 37 patients had no further attacks, a success rate of 84%.
Endoscopic therapy. As with surgical therapy trials, most trials of endoscopic therapy of recurrent pancreatitis in patients with pancreas divisum are small case series. In a retrospective study with one of the largest number of patients, Heyries et al59 reported their experience with 24 patients with pancreas divisum and recurrent pancreatitis. After undergoing endoscopic minor papillary sphincterotomy, all patients were followed for a mean of 39 months, during which 22 (92%) did not have further episodes of acute pancreatitis.
In the only randomized controlled trial available, 19 patients with recurrent pancreatitis and pancreas divisum underwent either no treatment or endoscopic minor papillary sphincterotomy.60 In the treatment group, 9 of 10 patients had no further episodes of acute pancreatitis during the 3 years of follow-up, while 6 of 9 patients who were randomized to no treatment had at least one episode.60
Although surgical and endoscopic minor papillary sphincterotomy are equally effective, endoscopic therapy is preferred since it is less invasive, is associated with less morbidity, and costs less. It is also more convenient for patients, since it is an outpatient procedure. Surgical treatment is usually reserved for those in whom endoscopic treatment has failed or is not technically possible.
OTHER PROCESSES OBSTRUCTING THE FLOW OF PANCREATIC JUICE
Any process preventing free flow of pancreatic juice can lead to acute pancreatitis. The cause of the blockage can be around the ampulla, in the ampulla, or in the duct.61
Periampullary lesions, tumors, or polyps can press on the ampulla and cause complete or relative obstruction of the pancreatic duct with a subsequent increase in intraductal pressure and, thus, acute pancreatitis.62 Tumors or polyps of the ampulla, such as ampullary adenoma or carcinoma, can cause pancreatitis by directly obstructing the pancreatic duct where it opens into the duodenum.63–66 Intraductal processes such as ductal adenocarcinoma, intraductal papillary mucinous tumor, pancreatic duct stone, and intraductal stricture due to cancer, chronic pancreatitis, or trauma can also cause pancreatitis by preventing free flow of pancreatic juice.67–71
Although it is well known that sequelae of severe chronic pancreatitis such as ductal strictures or intraductal stones can lead to recurrent attacks of acute pancreatitis by preventing the free flow of pancreatic juice, a relationship also seems to exist between early chronic pancreatitis and recurrent acute pancreatitis.72 Several studies have shown that up to 50% of patients with idiopathic recurrent pancreatitis have evidence of chronic pancreatitis.72–74 However, it is still unclear whether early chronic pancreatitis is the underlying cause of the recurrent attacks of acute pancreatitis or whether recurrent attacks of acute pancreatitis might have led to the development of chronic pancreatitis.
Diagnosis
Ampullary and periampullary neoplasms can be diagnosed endoscopically. Intraductal lesions such as strictures can be diagnosed by MRCP, especially secretin-enhanced MRCP, or by ERCP. ERCP has the additional advantage of being able to deliver treatment, ie, balloon dilation and stenting. In the case of ductal strictures, upsizing of the stents or placement of multiple stents during subsequent procedures is usually needed. Pancreatic ductal calcifications associated with chronic pancreatitis are usually radiopaque and are easily visible on plain films or computed tomography of the abdomen. Parenchymal and ductal changes of chronic pancreatitis can be diagnosed by endoscopic ultrasonography.
Treatment
The treatment is to relieve the obstruction and re-establish the free flow of pancreatic juice.
Periampullary tumors or polyps can be resected surgically or, if they involve only the mucosa, by endoscopic mucosal resection. Ampullary adenomas can be resected endoscopically. Ampullary carcinomas usually require surgical resection.
Small, nonobstructive stones in the pancreatic duct can be removed during ERCP.75 Larger stones may need to be fragmented by extracorporeal shock wave lithotripsy to facilitate removal by ERCP.75
Intraductal strictures should raise the suspicion of pancreatic adenocarcinoma, especially in older patients.61 In these cases, relief of the obstruction by placement of a pancreatic stent can prevent further attacks of pancreatitis until a diagnosis can be established and a more definitive treatment can be offered.
- Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol 2001; 96:2540–2555.
- Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol 2002; 97:1959–1962.
- Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol 2006; 41:681–685.
- Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology 2001; 120:708–717.
- Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol 2007; 102:1781–1788.
- Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc 2007; 66:213–218.
- Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology 1994; 20:523–525.
- Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 1901; 12:182–188.
- Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373.
- Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology 1991; 101:1701–1709.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med 1992; 326:589–593.
- Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts? Gastrointest Endosc 2002; 55:286–293.
- Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology 1986; 6:118–122.
- Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology 1986; 90:677–686.
- Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol 2000; 95:1702–1707.
- Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther 2006; 24:1059–1066.
- Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas 2003; 27:103–117.
- Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am 2003; 13:695–716.
- Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc 2005; 62:1–8.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology 2005; 128:756–763.
- Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy 2003; 35:92.
- Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg 1977; 185:598–604.
- Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol 2008; 14:1023–1026.
- Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335:909–918.
- Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology 1996; 110:894–899.
- Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology 1990; 98:1008–1012.
- Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up? Gastrointest Endosc 2002; 55:523–526.
- Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc 2001; 54:42–48.
- Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy 2002; 34:273–279.
- Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc 2006; 64:60–65.
- Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc 2002; 55:157–162.
- Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc 1999; 49:599–604.
- Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc 2008; 67:AB102.
- Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil 2005; 17 suppl 1:31–40.
- McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol 2007; 13:6333–6343.
- Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci 2007; 52:1211–1218.
- Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep 2002; 4:153–159.
- Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas 2005; 30:359–362.
- Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP 2004; 5:322–327.
- Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc 2004; 59:499–505.
- Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility? Rev Gastroenterol Disord 2004; 4:211–213.
- Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy? Curr Gastroenterol Rep 2002; 4:172–176.
- Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol 2007; 41:94–102.
- Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther 2006; 24:237–246.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989; 34:56–60.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy 2002; 34:280–285.
- Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol 2007; 5:1354–1365.
- Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford) 2003; 5:142–145.
- Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP 2001; 2:382–400.
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004; 60:419–425.
- Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas 2007; 34:21–45.
- Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol 2003; 17:381–383.
- Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am 1995; 5:145–170.
- Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis? Pancreas 1988; 3:108–110.
- Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas 2008; 37:151–153.
- Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg 1990; 159:59–64.
- Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg 1996; 183:65–70.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc 2002; 55:376–381.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc 1992; 38:430–434.
- Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol 2008; 14:1027–1033.
- Finnie IA, Ghosh P, Garvey C, Poston GJ, Rhodes JM. Intraluminal duodenal diverticulum causing recurrent pancreatitis: treatment by endoscopic incision. Gut 1994; 35:557–559.
- Guzzardo G, Kleinman MS, Krackov JH, Schwartz SI. Recurrent acute pancreatitis caused by ampullary villous adenoma. J Clin Gastroenterol 1990; 12:200–202.
- Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg 1999; 134:311–315.
- Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg 2004; 67:309–310.
- Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol 2007; 13:2892–2894.
- Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc 1990; 36:63–65.
- Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J 2000; 41:528–532.
- Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol 2000; 31:172–174.
- Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol 2001; 16:1070,1075.
- Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery 2004; 136:909–916.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol 2007; 5:75–79.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc 2004; 60:673–678.
- Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 2007; 356:676–684.
- Levy MJ, Geenen JE. Idiopathic acute recurrent pancreatitis. Am J Gastroenterol 2001; 96:2540–2555.
- Gullo L, Migliori M, Pezzilli R, et al. An update on recurrent acute pancreatitis: data from five European countries. Am J Gastroenterol 2002; 97:1959–1962.
- Gao YJ, Li YQ, Wang Q, et al. Analysis of the clinical features of recurrent acute pancreatitis in China. J Gastroenterol 2006; 41:681–685.
- Somogyi L, Martin SP, Venkatesan T, Ulrich CD. Recurrent acute pancreatitis: an algorithmic approach to identification and elimination of inciting factors. Gastroenterology 2001; 120:708–717.
- Andriulli A, Loperfido S, Napolitano G, et al. Incidence rates of post-ERCP complications: a systematic survey of prospective studies. Am J Gastroenterol 2007; 102:1781–1788.
- Standards of Practice Committee,Zuckerman MJ, Shen B, Harrison ME, et al. Informed consent for GI endoscopy. Gastrointest Endosc 2007; 66:213–218.
- Lee SP, Hayashi A, Kim YS. Biliary sludge: curiosity or culprit? Hepatology 1994; 20:523–525.
- Opie E. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp 1901; 12:182–188.
- Hernandez CA, Lerch MM. Sphincter stenosis and gallstone migration through the biliary tract. Lancet 1993; 341:1371–1373.
- Ros E, Navarro S, Bru C, Garcia-Puges A, Valderrama R. Occult microlithiasis in 'idiopathic' acute pancreatitis: prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology 1991; 101:1701–1709.
- Lee SP, Nicholls JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med 1992; 326:589–593.
- Levy MJ. The hunt for microlithiasis in idiopathic acute recurrent pancreatitis: should we abandon the search or intensify our efforts? Gastrointest Endosc 2002; 55:286–293.
- Delchier JC, Benfredj P, Preaux AM, Metreau JM, Dhumeaux D. The usefulness of microscopic bile examination in patients with suspected microlithiasis: a prospective evaluation. Hepatology 1986; 6:118–122.
- Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology 1986; 90:677–686.
- Testoni PA, Caporuscio S, Bagnolo F, Lella F. Idiopathic recurrent pancreatitis: long-term results after ERCP, endoscopic sphincterotomy, or ursodeoxycholic acid treatment. Am J Gastroenterol 2000; 95:1702–1707.
- Siddiqui AA, Mitroo P, Kowalski T, Loren D. Endoscopic sphincterotomy with or without cholecystectomy for choledocholithiasis in high-risk surgical patients: a decision analysis. Aliment Pharmacol Ther 2006; 24:1059–1066.
- Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: management of acute idiopathic recurrent pancreatitis. Pancreas 2003; 27:103–117.
- Khalid A, Slivka A. Approach to idiopathic recurrent pancreatitis. Gastrointest Endosc Clin North Am 2003; 13:695–716.
- Adler DG, Baron TH, Davila RE, et al; Standards of Practice Committee of American Society for Gastrointestinal Endoscopy. ASGE guideline: the role of ERCP in diseases of the biliary tract and the pancreas. Gastrointest Endosc 2005; 62:1–8.
- Draganov P, Forsmark CE. “Idiopathic” pancreatitis. Gastroenterology 2005; 128:756–763.
- Chung EJ, Kim MH, Lee SS, Lee SK. Primary vs. secondary common bile duct stones: apples and oranges. Endoscopy 2003; 35:92.
- Saharia PC, Zuidema GD, Cameron JL. Primary common duct stones. Ann Surg 1977; 185:598–604.
- Elta GH. Sphincter of Oddi dysfunction and bile duct microlithiasis in acute idiopathic pancreatitis. World J Gastroenterol 2008; 14:1023–1026.
- Freeman ML, Nelson DB, Sherman S, et al. Complications of endoscopic biliary sphincterotomy. N Engl J Med 1996; 335:909–918.
- Prat F, Malak NA, Pelletier G, et al. Biliary symptoms and complications more than 8 years after endoscopic sphincterotomy for choledocholithiasis. Gastroenterology 1996; 110:894–899.
- Hawes RH, Cotton PB, Vallon AG. Follow-up 6 to 11 years after duo-denoscopic sphincterotomy for stones in patients with prior cholecystectomy. Gastroenterology 1990; 98:1008–1012.
- Lai KH, Lo GH, Lin CK, et al. Do patients with recurrent choledocholithiasis after endoscopic sphincterotomy benefit from regular follow-up? Gastrointest Endosc 2002; 55:523–526.
- Kim DI, Kim MH, Lee SK, et al. Risk factors for recurrence of primary bile duct stones after endoscopic biliary sphincterotomy. Gastrointest Endosc 2001; 54:42–48.
- Costamagna G, Tringali A, Shah SK, Mutignani M, Zuccala G, Perri V. Long-term follow-up of patients after endoscopic sphincterotomy for choledocholithiasis, and risk factors for recurrence. Endoscopy 2002; 34:273–279.
- Keizman D, Ish Shalom M, Konikoff FM. Recurrent symptomatic common bile duct stones after endoscopic stone extraction in elderly patients. Gastrointest Endosc 2006; 64:60–65.
- Kaw M, Brodmerkel GJ. ERCP, biliary crystal analysis, and sphincter of Oddi manometry in idiopathic recurrent pancreatitis. Gastrointest Endosc 2002; 55:157–162.
- Chak A, Hawes RH, Cooper GS, et al. Prospective assessment of the utility of EUS in the evaluation of gallstone pancreatitis. Gastrointest Endosc 1999; 49:599–604.
- Parsi MA, Neuhaus H, Pleskow D, et al. Peroral cholangioscopy guided stone therapy—report of an international multicenter registry [abstract]. Gastrointest Endosc 2008; 67:AB102.
- Woods CM, Mawe GM, Toouli J, Saccone GT. The sphincter of Oddi: understanding its control and function. Neurogastroenterol Motil 2005; 17 suppl 1:31–40.
- McLoughlin MT, Mitchell RM. Sphincter of Oddi dysfunction and pancreatitis. World J Gastroenterol 2007; 13:6333–6343.
- Bosch A, Pena LR. The sphincter of Oddi. Dig Dis Sci 2007; 52:1211–1218.
- Devereaux BM, Sherman S, Lehman GA. Sphincter of Oddi (pancreatic) hypertension and recurrent pancreatitis. Curr Gastroenterol Rep 2002; 4:153–159.
- Fazel A, Geenen JE, MoezArdalan K, Catalano MF. Intrapancreatic ductal pressure in sphincter of Oddi dysfunction. Pancreas 2005; 30:359–362.
- Freeman ML. Role of pancreatic stents in prevention of post-ERCP pancreatitis. JOP 2004; 5:322–327.
- Singh P, Gurudu SR, Davidoff S, et al. Sphincter of Oddi manometry does not predispose to post-ERCP acute pancreatitis. Gastrointest Endosc 2004; 59:499–505.
- Guda NM, Freeman ML. True culprit or guilt by association? Is sphincter of Oddi manometry the cause of post-ERCP pancreatitis in patients with suspected sphincter of Oddi dysfunction, or is it the patients' susceptibility? Rev Gastroenterol Disord 2004; 4:211–213.
- Craig A, Toouli J. Sphincter of Oddi dysfunction: is there a role for medical therapy? Curr Gastroenterol Rep 2002; 4:172–176.
- Freeman ML, Gill M, Overby C, Cen YY. Predictors of outcomes after biliary and pancreatic sphincterotomy for sphincter of Oddi dysfunction. J Clin Gastroenterol 2007; 41:94–102.
- Sgouros SN, Pereira SP. Systematic review: sphincter of Oddi dysfunction—non-invasive diagnostic methods and long-term outcome after endoscopic sphincterotomy. Aliment Pharmacol Ther 2006; 24:237–246.
- Venu RP, Geenen JE, Hogan W, Stone J, Johnson GK, Soergel K. Idiopathic recurrent pancreatitis. An approach to diagnosis and treatment. Dig Dis Sci 1989; 34:56–60.
- Geenen JE, Hogan WJ, Dodds WJ, Toouli J, Venu RP. The efficacy of endoscopic sphincterotomy after cholecystectomy in patients with sphincter-of-Oddi dysfunction. N Engl J Med 1989; 320:82–87.
- Fogel EL, Eversman D, Jamidar P, Sherman S, Lehman GA. Sphincter of Oddi dysfunction: pancreaticobiliary sphincterotomy with pancreatic stent placement has a lower rate of pancreatitis than biliary sphincterotomy alone. Endoscopy 2002; 34:280–285.
- Freeman ML. Pancreatic stents for prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis. Clin Gastroenterol Hepatol 2007; 5:1354–1365.
- Toouli J. The sphincter of Oddi and acute pancreatitis - revisited. HPB (Oxford) 2003; 5:142–145.
- Sherman S, Lehman GA. Sphincter of Oddi dysfunction: diagnosis and treatment. JOP 2001; 2:382–400.
- Klein SD, Affronti JP. Pancreas divisum, an evidence-based review: part I, pathophysiology. Gastrointest Endosc 2004; 60:419–425.
- Fogel EL, Toth TG, Lehman GA, DiMagno MJ, DiMagno EP. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas 2007; 34:21–45.
- Lehman GA. Acute recurrent pancreatitis. Can J Gastroenterol 2003; 17:381–383.
- Lehman GA, Sherman S. Pancreas divisum. Diagnosis, clinical significance, and management alternatives. Gastrointest Endosc Clin N Am 1995; 5:145–170.
- Staritz M, Meyer zum Buschenfelde KH. Elevated pressure in the dorsal part of pancreas divisum: the cause of chronic pancreatitis? Pancreas 1988; 3:108–110.
- Carnes M, Romagnuolo J, Cotton P. Miss rate of pancreas divisum by magnetic resonance cholangiopancreatography in clinical practice. Pancreas 2008; 37:151–153.
- Warshaw AL, Simeone JF, Schapiro RH, Flavin-Warshaw B. Evaluation and treatment of the dominant dorsal duct syndrome (pancreas divisum redefined). Am J Surg 1990; 159:59–64.
- Bradley EL, Stephan RN. Accessory duct sphincteroplasty is preferred for long-term prevention of recurrent acute pancreatitis in patients with pancreas divisum. J Am Coll Surg 1996; 183:65–70.
- Heyries L, Barthet M, Delvasto C, Zamora C, Bernard JP, Sahel J. Long-term results of endoscopic management of pancreas divisum with recurrent acute pancreatitis. Gastrointest Endosc 2002; 55:376–381.
- Lans JI, Geenen JE, Johanson JF, Hogan WJ. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis: a prospective, randomized, controlled clinical trial. Gastrointest Endosc 1992; 38:430–434.
- Delhaye M, Matos C, Arvanitakis M, Deviere J. Pancreatic ductal system obstruction and acute recurrent pancreatitis. World J Gastroenterol 2008; 14:1027–1033.
- Finnie IA, Ghosh P, Garvey C, Poston GJ, Rhodes JM. Intraluminal duodenal diverticulum causing recurrent pancreatitis: treatment by endoscopic incision. Gut 1994; 35:557–559.
- Guzzardo G, Kleinman MS, Krackov JH, Schwartz SI. Recurrent acute pancreatitis caused by ampullary villous adenoma. J Clin Gastroenterol 1990; 12:200–202.
- Wright BE, Kozarek RA, Traverso LW, Wechter D, Thirlby R, Raltz SL. Recurrent pancreatitis in Gardner variant familial polyposis: etiology, diagnostic approach, and interventional results. Arch Surg 1999; 134:311–315.
- Tanasijtchouk T, Vaisbein E, Lachter J, Nassar F. Carcinoma of Papilla Vateri presenting as recurrent acute pancreatitis. Acta Gastroenterol Belg 2004; 67:309–310.
- Kwon TH, Park do H, Shim KY, et al. Ampullary adenomyoma presenting as acute recurrent pancreatitis. World J Gastroenterol 2007; 13:2892–2894.
- Lorente JA, Ruiz del Arbol L, Moreira VF, Garcia-Plaza A. Recurrent pancreatitis in a young patient associated with a solitary nonopaque concretion in the main pancreatic duct. Gastrointest Endosc 1990; 36:63–65.
- Chung JP, Chi SW, Park YN, et al. A case of minute intraductal papillary mucinous tumor of the pancreas presenting with recurrent acute pancreatitis. Yonsei Med J 2000; 41:528–532.
- Tikhomirov V, Tikhomirova S, Sieber S, Schiffman MK. A pancreatic intraductal papillary mucinous tumor causing recurrent acute pancreatitis at the onset of menstrual periods. J Clin Gastroenterol 2000; 31:172–174.
- Mosca S, Bottino V, Molino C. Hepatobiliary and pancreatic: a woman with recurrent idiopathic acute pancreatitis. Intraductal papillary mucinous tumor of the pancreas. J Gastroenterol Hepatol 2001; 16:1070,1075.
- Howard TJ, Moore SA, Saxena R, Matthews DE, Schmidt CM, Wiebke EA. Pancreatic duct strictures are a common cause of recurrent pancreatitis after successful management of pancreatic necrosis. Surgery 2004; 136:909–916.
- Garg PK, Tandon RK, Madan K. Is biliary microlithiasis a significant cause of idiopathic recurrent acute pancreatitis? A long-term follow-up study. Clin Gastroenterol Hepatol 2007; 5:75–79.
- Tandon M, Topazian M. Endoscopic ultrasound in idiopathic acute pancreatitis. Am J Gastroenterol 2001; 96:705–709.
- Yusoff IF, Raymond G, Sahai AV. A prospective comparison of the yield of EUS in primary vs. recurrent idiopathic acute pancreatitis. Gastrointest Endosc 2004; 60:673–678.
- Cahen DL, Gouma DJ, Nio Y, et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N Engl J Med 2007; 356:676–684.
KEY POINTS
- Recurrent attacks of acute pancreatitis can be prevented only by determining and treating the underlying cause.
- Endoscopic procedures can cause anxiety and carry a risk of bleeding, perforation, and pancreatitis. The risks, benefits, and other treatment options should be discussed with the patient.
- Endoscopic therapy is now the preferred treatment of sphincter of Oddi dysfunction at centers that have experience with this technique.
- In patients with pancreas divisum and recurrent acute pancreatitis, surgical and endoscopic minor sphincterotomy are equally effective.
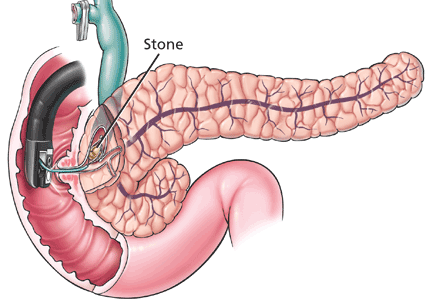
Multiple huge bullae after renal transplant
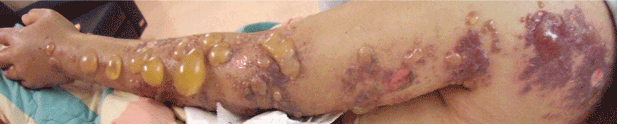
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
Q: What is the most likely diagnosis?
- Contact dermatitis
- Herpes zoster
- Herpes simplex
- Pemphigus
- Bullous pemphigoid
- Graft-vs-host disease
A: The correct answer is herpes zoster (shingles), which represents reactivation of varicella-zoster virus.
The diagnosis of herpes zoster is usually based solely on the clinical presentation. It is typically characterized in immunocompetent patients by a unilateral vesicular eruption with a well-defined dermatomal distribution. But occasionally, as in this patient on immunosuppressant drugs, it presents with atypical skin lesions such as multiple huge bullae involving multiple dermatomes.1,2
Patients treated with immunosuppressive agents after organ transplantation are at high risk of herpes zoster. A recent published retrospective study of adult kidney transplant recipients showed an average incidence of approximately 28 per 1,000 person-years.3
Treatment involves analgesics and sometimes antiviral drugs, and the decisions should take into account the patient’s age and immune status.1
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
- Nagel MA, Gilden DH. The protean neurologic manifestations of varicella-zoster virus infection. Cleve Clin J Med 2007; 74:489–504.
- Albrecht MA. Clinical manifestations of varicella-zoster virus infection: Herpes zoster. InRose BD, editor: UpToDate. Waltham, MA: UpToDate, 2008.
- Arness T, Pedersen R, Dierkhising R, Kremers W, Patel R. Varicella zoster virus-associated disease in adult kidney transplant recipients: incidence and risk-factor analysis. Transpl Infect Dis 2008; 10:260–268.
A 43-year-old woman with chest pressure
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma

The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4

At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.

DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
A 43-year-old woman presents to the emergency department with substernal chest pressure of moderate intensity that started approximately 6 hours ago. The pressure radiates to both arms and is accompanied by nausea. She says she has had no emesis, diaphoresis, fevers, chills, shortness of breath, abdominal pain, melena, dysuria, weight loss, headaches, change in vision, seizures, joint pain, or skin rashes. She also says she has had no prior similar episodes and has no history of myocardial infarction (MI) or stroke.
The patient has a history of gastroesophageal reflux disease and uterine fibroids. She has had three pregnancies, one ending in spontaneous abortion at 12 weeks and two ending with healthy children delivered by cesarean section. She does not take any daily medications. She has smoked one pack per day over the last 25 years. She denies using alcohol or illicit drugs.
The patient’s mother had idiopathic deep vein thrombosis (DVT) at age 46, her father had an MI at age 65, and her sister had an MI at age 43.
On examination, she is in mild distress but is alert and oriented. Her temperature is 99.0°F (37.2°C), blood pressure 98/66 mm Hg, heart rate 65 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation 99% on room air. Her body mass index is 19.5 (normal range 18.5–24.9). Her skin appears normal. Her head and neck show no obvious abnormalities, lymphadenopathy, thyromegaly, or bruits. Her heart, lungs, and abdomen are normal, as are her strength, sensation, reflexes, and gait.
Laboratory values at the time of admission:
- White blood cell count 12.58 × 109/L (reference range 4.0–11.0)
- Hemoglobin 15.4 g/dL (12.0–16.0)
- Platelet count 122 × 109/L (150–400)
- International normalized ratio (INR) 1.1 (0.9–1.1)
- Activated partial thromboplastin time 29.1 seconds (24.6–34).
A heart attack, and then a stroke
An initial electrocardiogram shows normal sinus rhythm, left anterior hemiblock, and nonspecific T-wave abnormalities. Cardiac enzymes are measured at intervals: her troponin T level is less than 0.01 ng/mL at the time of admission but rises to 0.75 ng/mL 3 hours later (normal range 0.0–0.1 ng/mL). Similarly, her creatine kinase-MB level is 3.3 ng/mL at admission but rises to 71.9 ng/mL 3 hours later (normal range 0.0–8.0 ng/mL).
The patient is diagnosed with non-ST-elevation MI. An intravenous heparin drip is started, and she is sent for urgent cardiac catheterization, which shows a total occlusion in a lateral obtuse marginal branch of the left circumflex artery due to a thrombus in the vessel. Otherwise, her coronary arteries are angiographically free of disease. The heparin drip is continued, and treatment is started with abciximab (ReoPro) and tissue plasminogen activator (Alteplase). She is sent to the cardiac intensive care unit for recovery, where she is placed on continuous cardiac monitoring, with no evidence of arrhythmia.
One day later, the left side of her face is drooping, her left arm is weak, and her speech is slurred. Magnetic resonance imaging of the brain shows an acute ischemic infarct in the right temporoparietal area and multiple areas of subacute to chronic ischemia. Magnetic resonance angiography of the brain indicates patent vessels. Both transthoracic and transesophageal echocardiography are performed and indicate normal left ventricular size, ejection fraction of 55%, valves without thrombus or vegetations, aorta with mild atheroma, and no patent foramen ovale by Doppler flow or agitated saline contrast study. Carotid artery Doppler ultrasonography shows 40% to 59% stenosis bilaterally.
ARTERIAL THROMBOSIS
1. Which of the following is a risk factor for arterial thrombosis?
- Atherosclerosis
- Protein C deficiency
- Use of oral contraceptive pills
- The factor V Leiden mutation
Protein C deficiency, the use of oral contraceptives, and the factor V Leiden mutation are typically associated with venous thrombosis1; they have been documented as a cause of arterial thrombosis only in some case reports. In contrast, atherosclerosis is a well-established risk factor for arterial thrombosis.
Arterial occlusion can be due to thrombosis, embolism, or trauma
The causes of arterial occlusion can be categorized as thrombotic, embolic, or traumatic (Table 1).
Atherosclerosis is a risk factor for thrombosis and can be a source of emboli. Atherosclerotic plaque rupture may release inflammatory mediators, which also predispose to thrombosis.2 This patient’s coronary arteries are essentially free of atherosclerotic disease per angiography. However, studies of intravascular ultrasonography have shown that coronary angiography may not detect all atherosclerotic plaques, as angiography can show only the lumen of the artery and not the plaque itself.3 For that reason, atherosclerosis has not been ruled out completely, and further workup is needed to evaluate other possible causes of her thrombotic events.
Embolism is the most likely cause of her stroke, however. Cases of arterial embolism can be classified on the basis of the origin of the thrombus, ie, the heart, an artery, or the venous system via a patent foramen ovale (paradoxical embolism). This patient’s echocardiogram reveals mild aortic atheroma, which can be a source of emboli, especially soon after intervention.
Case continues: Acute and recurrent DVT
While recovering from her MI and stroke, the patient develops edema and pain in both legs. Doppler ultrasonography is performed, which reveals acute DVT in the right gastrocnemius and posterior tibial veins and left soleal vein, despite her continued heparin therapy.
Her platelet count is 189 × 109/L, so heparin-induced thrombocytopenia is not suspected; the new DVT is thought to be due to her hospitalization. Several days later, oral warfarin (Coumadin) is started and titrated to an INR of 2.0 to 3.0, the heparin is phased out, and the patient is sent home.
In the first few months after discharge, the patient presents to the emergency department three times with severe leg pain, and each time she is found to have extensive DVT in various leg veins even though she is complying with her warfarin therapy. At each visit, her INR is in the range of 2.5 to 3.1.
Comment. Her recurrent DVT warrants further evaluation for risk factors for venous thrombosis, which can be divided into hereditary and acquired factors.
Hereditary risk factors include the factor V Leiden mutation, the prothrombin gene mutation, hyperhomocysteinemia, dysfibrinogenemia, and deficiencies of protein C, protein S, and antithrombin.
Acquired risk factors include the antiphospholipid antibody syndrome, cancer, immobilization, surgery, congestive heart failure, pregnancy, use of hormonal contraceptives, hormone replacement therapy, nephrotic syndrome, trauma, and infection.1,4
TESTING FOR HYPERCOAGULABLE STATES
2. In view of our patient’s recurrent thrombotic episodes, should she be tested for hypercoagulable states?
- Yes
- No
Testing for hypercoagulable conditions is warranted if it will affect the patient’s management or outcome. Some authorities recommend testing patients who are clinically characterized as “strongly” thrombophilic,5 ie, those who present with DVT and are younger than age 50, have recurrent thrombotic episodes, have a first-degree relative with documented thromboembolism before age 50, or have thrombotic episodes despite warfarin therapy.
This patient should be tested for hypercoagulable conditions because her initial DVT occurred before age 50 (at age 43), she has had recurrent, apparently idiopathic thrombotic episodes, she has a family history of thromboembolism, and she had clots while on therapeutic warfarin therapy, all of which suggest a hypercoagulable state. Furthermore, the confirmation of her diagnosis may affect her medical management, as it may determine if further testing and therapies are needed.
Case continues: Tests are negative
Laboratory tests for hypercoagulable conditions are performed and are negative for the factor V Leiden mutation, the prothrombin gene mutation, antithrombin deficiency, and protein C and S deficiencies. A screen for antiphospholipid antibodies is indeterminate.
TREATMENT AFFECTS TEST RESULTS
3. If a patient is on warfarin therapy, which test results may be affected?
- Antithrombin levels
- Protein C and S levels
- Factor V Leiden mutation
Warfarin decreases the levels of proteins C and S; therefore, the levels of these substances cannot be accurately interpreted in a patient taking warfarin.
All anticoagulants prolong the clotting time and may affect the results of assays based on the clotting time, such as the prothrombin time, the partial thromboplastin time, the dilute Russell’s viper venom time (DRVVT), the hexagonal phase phospholipid neutralization assay, the thrombin time, and clottable protein C and protein S. Heparin reduces the level of antithrombin; however, laboratories now have heparin-binding agents that reduce the effect of heparin in clotting studies.
Acute thrombotic states lower the levels of antithrombin and proteins C and S.
Assays not based on the clotting time (immunogenic or genetic tests such as those for anticardiolipin antibodies and the factor V Leiden and prothrombin gene mutations) are not affected by anticoagulant use.5
However, the presence or absence of a hypercoagulable state should not affect the treatment of acute DVT, and a full 6- to 12-month course of anticoagulation should be completed.6,7 If possible, lupus anticoagulant testing should be repeated 2 weeks after anticoagulation is stopped.8
This patient needs lifelong anticoagulation because of her repeated thrombotic episodes. Stopping the medication for 2 weeks for testing would increase the risk of rethrombosis in this patient, and most experts would not advise it.
In summary, testing for hypercoagulable conditions is not recommended during an acute thrombotic episode and is preferably performed while the patient is not on anticoagulation therapy. If the patient is already on anticoagulation, the results of tests for hypercoagulable conditions should be interpreted with caution.
Case continues: Another stroke
During the subsequent year, the patient’s primary care physician monitors her warfarin use and sends her for age-appropriate cancer screening, including a breast examination, Papanicolaou smear, and mammography. Also, given her history of smoking, a chest radiograph is ordered. All of these studies are normal. In addition, evaluations for hematologic disorders such as myelodysplastic syndrome, polycythemia vera, and Waldenström macroglobulinema reveal normal complete blood counts and normal results on serum and urine protein electrophoresis.
Later that year, she returns to the emergency department with complete aphasia and total right-sided paralysis. Magnetic resonance imaging shows an acute infarct in the left frontal operculum, a subacute infarct in the right cerebellum, and multiple chronic cortical and subcortical infarcts throughout the brain. Ultrasonography shows an extensive new DVT in her right leg. Her INR at this time is 3.1.
WHAT CONDITIONS CAUSE BOTH ARTERIAL AND VENOUS THROMBOSIS?
4. Given that the patient has evidence of both recurrent arterial and venous thromboses, which of the following conditions is likely?
- Antiphospholipid antibody syndrome
- Heparin-induced thrombocytopenia
- Malignancy
- All of the above
Conditions associated with both arterial and venous thrombosis include antiphospholipid antibody syndrome, heparin-induced thrombocytopenia, malignancy, paradoxical embolism, hyperhomocysteinemia, myeloproliferative disorders, myelodysplastic disorder, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.1,4
At present, the exact mechanism that causes Trousseau syndrome is unknown. Some hypotheses implicate mucin (produced by the cancer),10 tissue factor,11 tumor-associated cysteine proteinase,12 tumor hypoxia,13 and oncogene activation as plausible triggers for this syndrome.
DOES SHE HAVE ANTIPHOSPHOLIPID ANTIBODY SYNDROME?
5. The patient is positive for lupus anticoagulant. Does she have antiphospholipid antibody syndrome?
- Yes
- No
- Repeat testing is needed to meet the diagnostic criteria
The Sapporo criteria15 indicate that antiphospholipid antibody syndrome is present if at least one clinical criterion and one laboratory criterion are met. The clinical criteria are one or more episodes of arterial or venous thrombosis or pregnancy-related morbidity, ie:
- Unexplained intrauterine fetal death at 10 weeks gestation or later with no apparent fetal abnormality
- Premature births of a morphologically normal fetus at less than 34 weeks of gestation due to preeclampsia, eclampsia, or placental insufficiency
- Three or more spontaneous abortions at 10 weeks of gestation or earlier, with no known paternal chromosomal abnormalities or maternal hormonal abnormalities and normal maternal anatomy.
The laboratory criteria are:
- Lupus anticoagulant present
- Anticardiolipin antibody (IgG or IgM) titer greater than 40 IgG antiphospholipid units (GPL) or IgM antiphospholipid units (MPL) or higher than the 99th percentile of the testing laboratory normal reference range
- Anti-beta-2 glycoprotein-I antibody (IgG or IgM) titer greater than 20 GPL or MPL or higher than the 99th percentile of the testing laboratory normal reference range.
The patient likely has antiphospholipid antibody syndrome because her lupus anticoagulant screen is positive and she meets the clinical criteria of thrombosis, and she should continue to be treated accordingly. However, to officially meet the revised Sapporo criteria, she would need to have laboratory tests that are positive on two or more occasions at least 12 weeks apart.
Case continues: Lung cancer is found
The patient reports that she has lost 10 pounds in 4 months. Since age-appropriate cancer testing was previously performed, a more extensive evaluation for weight loss is undertaken, with computed tomography of the chest, abdomen, and pelvis. These tests reveal a nodule in the right upper lobe of the lung, scarring in the right middle and left lower lung lobes, and hilar lymphadenopathy. Bronchoscopy with transbronchial biopsy confirms that she has adenocarcinoma of the lung.
6. What is suggested as a sufficient workup for malignancy in patients with idiopathic venous thromboembolism?
- Computed tomography of the chest, abdomen, and pelvis for every patient with idiopathic venous thromboembolism
- Positron emission tomography and tumor marker levels
- A comprehensive history and physical examination, routine laboratory tests, chest radiography, age- and sex-specific cancer screening, and patient-specific testing as indicated clinically
To date, there is no evidence to support a cancer evaluation beyond a comprehensive medical history and physical examination, routine laboratory testing, chest radiography, and age- and sex-specific cancer screening unless it is dictated by the patient’s clinical presentation. A study by Cornuz et al16 suggested that this approach is appropriate for detecting cancer in patients with idiopathic venous thromboembolism.
A 2004 study17 attempted to answer the question of what to do about patients who have idiopathic venous thromboembolism but no other signs or symptoms that raise any clinical suspicion of cancer. This study randomized patients with idiopathic venous thromboembolism to undergo either routine medical management or an extensive malignancy evaluation. The evaluation included ultrasonography of the abdomen and pelvis, computed tomography of the abdomen and pelvis, gastroscopy or a double-contrast barium swallow study, colonoscopy or sigmoidoscopy followed by a barium enema, stool occult blood testing, and sputum cytology. Women were also tested for the tumor markers carcinoembryonic antigen, alpha-fetoprotein, and CA-125, and they underwent mammography and Papanicolaou testing; men were tested for prostate-specific antigen and underwent ultrasonography of the prostate. The results of the study did not reveal a statistically significant survival benefit in the group that underwent extensive cancer evaluation.
These studies indicate that the decision to test for cancer should be guided by clinical suspicion. Our patient lost 10 pounds in 4 months, smokes, and has had recurrent venous thromboembolism, so testing was appropriate.
After her diagnosis with adenocarcinoma of the lung, the patient has yet another DVT despite an INR of 3.1 and treatment with warfarin and aspirin.
LOW-MOLECULAR-WEIGHT HEPARIN FOR PATIENTS WITH CANCER?
7. True or false? Low-molecular-weight heparin is more effective than warfarin in preventing DVT in cancer patients without increasing the bleeding risk.
- True
- False
This statement is true. The American College of Chest Physicians (ACCP) recommends immediate treatment of DVT with low-molecular-weight heparin for 6 to 12 months after a thrombotic event in a patient with malignancy.6,18
Two major studies provide evidence for these recommendations: the Comparison of Low-Molecular-Weight Heparin Versus Oral Anticoagulant Therapy for the Prevention of Recurrent Venous Thromboembolism in Patients With Cancer (CLOT)19 and the Trial of the Effect of Low-Molecular-Weight Heparin Versus Warfarin on Mortality in the Long-Term Treatment of Proximal Deep Vein Thrombosis (LITE)20 studies.
The CLOT19 study showed that dalteparin (Fragmin) 200 IU/kg subcutaneously once daily for l month and then 150 IU/kg once daily was more effective than oral warfarin titrated to an INR of 2.5 and did not increase the risk of bleeding.
The LITE trial20 showed the efficacy of tinzaparin (Innohep) 175 IU/kg subcutaneously daily, which can be used as an alternative.
Enoxaparin sodium (Lovenox) 1.5 mg/kg once daily has also been used. However, if low-molecular-weight heparin is not available, warfarin titrated to an INR of 2 to 3 is also acceptable.18
The ACCP consensus panel recommends giving anticoagulation for an initial 6 to 12 months and continuing it as long as there is evidence of active malignancy.6 The American Society for Clinical Oncology also recommends placement of an inferior vena cava filter for patients who have contraindications to anticoagulation or for whom low-molecular-weight heparin fails.18
Case continues: Summing up
In conclusion, our patient had an underlying malignancy, causing Trousseau syndrome. Before her cancer was diagnosed, she also had test results that suggested antiphospholipid antibody syndrome. Both of these conditions likely contributed to her hypercoagulable state, increasing her propensity for clotting and causing her recurrent thrombosis. The patient is currently on low-molecular-weight heparin and is undergoing palliative chemotherapy for metastatic adenocarcinoma of the lung. To this date, she has not had any new thrombotic events.
TAKE-HOME POINTS
- Risk factors for arterial occlusion can be divided into thrombotic, embolic, and traumatic categories.
- Risk factors for venous thrombosis can be divided into hereditary and acquired categories.
- Evaluation for hypercoagulable conditions is recommended if it will affect patient management or outcome. Patients to be considered for testing include those with idiopathic DVT and who are under age 50, those with a history of recurrent thrombosis, and those with a first-degree relative with documented venous thromboembolism before age 50.
- Evaluation for hypercoagulable conditions should ideally be performed either before starting anticoagulation therapy or 2 weeks after completing it.
- Potential causes of both arterial and venous thrombosis include antiphospholipid antibody syndrome, cancer, hyperhomocysteinemia, heparin-induced thrombocytopenia, paradoxical emboli, myeloproliferative disorders, myelodysplastic syndrome, paraproteinemia, vasculitis, and paroxysmal nocturnal hemoglobinuria.
- Current evidence does not support an extensive cancer evaluation in patients with idiopathic venous thromboembolism, unless dictated by the patient’s clinical condition.
- In patients with venous thromboembolism and active malignancy, anticoagulation is recommended for at least 6 to 12 months and as long as there is evidence of active malignancy.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
- Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med 2002; 346:752–763.
- Lee KW, Lip GY. Acute coronary syndromes: Virchow’s triad revisited. Blood Coagul Fibrinolysis 2003; 14:605–625.
- Yamashita T, Colombo A, Tobis JM. Limitations of coronary angiography compared with intravascular ultrasound: implications for coronary interventions. Prog Cardiovasc Dis 1999; 42:91–138.
- Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe’s Clinical Hematology. 11th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2004.
- Bauer KA. The thrombophilias: well-defined risk factors with uncertain therapeutic implications. Ann Intern Med 2001; 135:367–373.
- Buller HR, Agnelli G, Hull RD, Hyers TM, Prins MH, Raskob GE. Antithrombotic therapy for venous thromboembolic disease: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 2004; 126 suppl 3:401S–428S.
- Locke CF, Evans NC. Evaluating idiopathic venous thromboembolism: what is necessary, what is not. J Fam Pract 2003; 52:770–777.
- Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Investigation and management of heritable thrombophilia. Br J Haematol 2001; 114:512–528.
- Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood 2007; 110:1723–1729.
- Pineo GF, Brain MC, Gallus AS, Hirsh J, Hatton MW, Regoeczi E. Tumors, mucus production, and hypercoagulability. Ann N Y Acad Sci 1974; 230:262–270.
- Zacharski LR, Schned AR, Sorenson GD. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res 1983; 43:3963–3968.
- Falanga A, Gordon SG. Isolation and characterization of cancer pro-coagulant: a cysteine proteinase from malignant tissue. Biochemistry 1985; 24:5558–5567.
- Denko NC, Giaccia AJ. Tumor hypoxia, the physiological link between Trousseau’s syndrome (carcinoma-induced coagulopathy) and metastasis. Cancer Res 2001; 61:795–798.
- Brandt JT, Barna LK, Triplett DA. Laboratory identification of lupus anticoagulants: results of the Second International Workshop for Identification of Lupus Anticoagulants. On behalf of the Subcommittee on Lupus Anticoagulants/Antiphospholipid Antibodies of the ISTH. Thromb Haemost 1995; 74:1597–1603.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4:295–306.
- Cornuz J, Pearson SD, Creager MA, Cook EF, Goldman L. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann Intern Med 1996; 125:785–793.
- Piccioli A, Lensing AW, Prins MH, et al. Extensive screening for occult malignant disease in idiopathic venous thromboembolism: a prospective randomized clinical trial. J Thromb Haemost 2004; 2:884–889.
- Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol 2007; 25:5490–5505.
- Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med 2003; 349:146–153.
- Hull RD, Pineo GF, Brant RF, et al. Long-term low-molecular-weight heparin versus usual care in proximal-vein thrombosis patients with cancer. Am J Med 2006; 119:1062–1072.
A new, precise definition of acute myocardial infarction
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.

Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT

ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
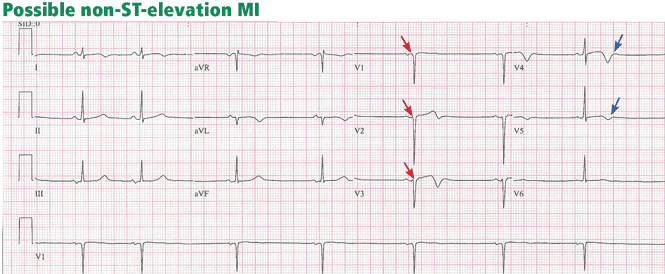
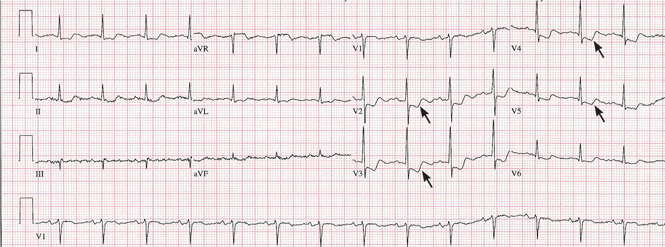
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.

Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
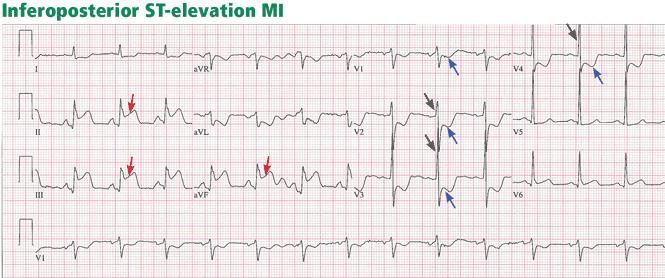
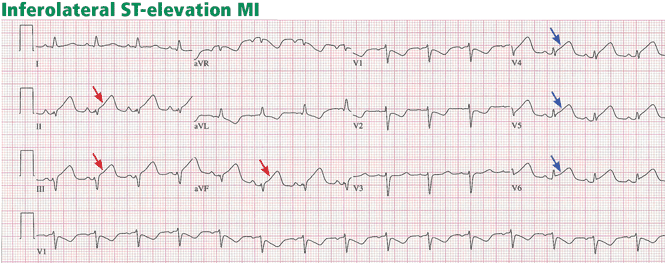
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI

The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
KEY POINTS
- The clinical presentation of acute MI varies considerably from patient to patient. Therefore, one must consider the symptoms, serial electrocardiographic findings, and serial biomarker results in concert.
- Troponin I or T is now the preferred biomarker of myocardial necrosis. Still, troponin can be elevated in many conditions other than ischemic heart disease.
- Electrocardiographic signs of acute ischemia have been precisely defined, but electrocardiography can give false-positive or false-negative results in a number of conditions.
- MI is now categorized into five types depending on cause.
Perioperative Medicine Summit 2009
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Pulmonary hypertension is an important predictor of perioperative outcomes in patients undergoing noncardiac surgery
Roop Kaw, MD; Esteban Walker, PhD; Vinay Pasupuleti, MD, PhD; Abhishek Deshpande, MD, PhD; Tarek Hamieh, MD; and Omar A. Minai, MD
Abstract 2: Analysis of administrative practices and residency training curricula in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 3: Is percent body fat a better predictor of surgical site infection risk than body mass index?
Emily Waisbren, BS; Angela M. Bader, MD, MPH; Heather Rosen, MD, MPH; Selwyn O. Rogers, Jr., MD, MPH; and Elof Eriksson, MD, PhD
Abstract 4: A nomogram for prediction of survival for patients undergoing elective major noncardiac surgery
Y. Olivia Xu-Cai, MD; and Michael W. Kattan, PhD
Abstract 5: Sustainability of an osteoporosis pathway
Catherine Gibb, MBBS, FRACP; Christopher Butcher, FRACS; Lesley Thomas, BNsg; and Jennifer Pink, BPharm
Abstract 6: Length of hospital stay is predicted by comorbidities
Catherine Gibb, MBBS, FRACP; and Professor Villis Marshall, FRACS
Abstract 7: Generalization of the POISE and Mangano studies on beta-blocker use in the perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; and Luc Lanthier, MD, MSc
Abstract 8: Impact of antihypertensive medication on perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; Danielle Pilon, MD, MSc; and Luc Lanthier, MD, MSc
Abstract 9: An analysis of preoperative testing protocols in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 10: Preoperative biomarkers of inflammation, ischemia, and heart failure and outcomes of vascular surgery
Matthew Griffee, MD; Ansgar Brambrink, MD, PhD; and Thomas Barrett, MD
Abstract 11: Alcohol-related predictors of postoperative delirium in major head and neck cancer surgery
Harrison Weed, MD; Summit Shah, BS; Xin He, PhD; Amit Agrawal, MD; Enver Ozer, MD; and David E. Schuller, MD
Abstract 12: Intraoperative coagulopathy: A low-volume treatment protocol that completely replaces fresh frozen plasma
Peter Kallas, MD; Mary Lou Green, MHS; and Anjali Desai, MD
Abstract 13: Is the Berlin Questionnaire an effective screening tool for obstructive sleep apnea in the preoperative total joint replacement population?
Peter Kallas, MD; Mark Schumacher; Mona Lazar, DO; and Anjali Desai, MD
Abstract 14: The impact of preoperative medical optimization on head and neck cancer surgery
Christopher Tan, MBBS; Catherine Gibb, MBBS, FRACP; and Suren Krishnan, MBBS, FRACS
Abstract 15: Reconceptualizing the preoperative process
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 16: Development of an electronic medical record smart set form to increase standardization, consistency, and compliance with ACC/AHA perioperative guidelines
Anitha Rajamanickam, MD; Ali Usmani, MD; Ajay Kumar, MD; and Brian Harte, MD
Abstract 17: Development of a perioperative electronic medical record research and quality improvement database
Anitha Rajamanickam, MD; Ali Usmani, MD; Feza Remzi, MD; Brian Harte, MD; and Ajay Kumar, MD
Abstract 18: An innovative perioperative/consultative curriculum for third-year internal medicine residents
Alex Rico, MD; Joshua Lenchus, DO; and Amir Jaffer, MD
Abstract 19: Preoperative medicine infobutton
Terrence J. Adam, MD, PhD
Abstract 20: Nurse practitioners: Bridging the gap in perioperative care
Sally Morgan, RN, MS, ANP-BC, ACNS-BC; and Angela Wright, RN, MSN, APRN, BC
Abstract 21: Intubation training of deploying far forward combat medical personnel with the video laryngoscope
Ben Boedeker, MD; Mary Barak-Bernhagen, BS; Kirsten Boedeker; and W. Bosseau Murray, MD
Abstract 22: The establishment of a perioperative skin integrity committee
Jeanne Lanchester, RN, MEd; Ann Leary, BSN, RNC; and Susan Vargas, AD, RN
Abstract 23: Development and implementation of a perianesthesia integrative care committee
Jeanne Lanchester, RN, MEd; Jeanette Cote, BWN, RN; Terri Jamros, RN; Charla Delillo, RN; Sherie Lavoie, BSN, RN; Jennifer Therminos, SN; Joan Compagnone, RN; and Nicole Engel, MSN, RN
Abstract 24: Development of a screening system to identify patients preoperatively who may benefit from a postoperative hospitalist consult
Elizabeth Marlow, MD, MA; and Chad Whelan, MD
Abstract 25: An algorithm for preoperative screening and management of sleep apnea: Have we created a monster?
Deborah C. Richman, MBChB, FFA(SA); Jorge M. Mallea, MD; Paul S. Richman, MD; and Pater S.A. Glass, MBChB
Abstract 26: Constructing a collaborative neuroscience hospitalist program
Rachel Thompson, MD; Christy Gilmore, MD; Kamal Ajam, MD; and Jennifer Thompson, MD
Abstract 27: The development of algorithms for preoperative management of antiplatelet and anticoagulation therapy in patients undergoing surgical or invasive procedures
Catherine McGowan, MSN, and Patricia Kidik, MSN
Abstract 28: Surgeon-initiated preoperative screening: A new approach
Christina Johnson, RN, PA-C; and Edward J. denBraven, CRNA
Abstract 29: A new process for ensuring the safety of patients having anesthesia outside of the operating room
Ellen Leary, MSN; Catherine McGowan, MSN; Kathleen McGrath, MSN; Sheila McCabe Hassan, MSN; and Theresa Kennedy, MSN
Abstract 30: Establishing a virtual preoperative evaluation clinic
Corey Zetterman, MD; Bobbie J. Sweitzer, MD; and Ben H. Boedeker, MD
Abstract 31: Perioperative hypoxemia and rhabdomyolysis in a medically complicated patient
Sarah Bodin, MD
Abstract 32: How soon is too soon? General anesthesia after coronary intervention with bare metal stents
Meghan Tadel, MD
Abstract 33: Can patients with critical aortic stenosis undergo noncardiac surgery without intervening aortic valve replacement?
M. Chadi Alraies, MD; Abdul Alraiyes, MD; Anitha Rajamanickam, MD; and Frank Michota, MD
Abstract 34: Is it safe to operate on cocaine-positive patients?
M. Chadi Alraies, MD; Abdul Hamid Alraiyes, MD; and Brian Harte, MD
Abstract 35: To intensive care or not?
Mona Lazar, DO; and Peter Kallas, MD
Abstract 36: Predicting surgical complications from liver disease
Mona Lazar, DO, and Peter Kallas, MD
Abstract 37: Preoperative coronary angiography: Friend or foe?
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 38: Heparin-induced thrombocytopenia with low molecular weight heparin after total knee replacement
Steven Cohn, MD
Abstract 39: Patient with Parkinson’s disease treated with implanted deep brain stimulators for laparotomy
Deborah C. Richman, MBChB, FFA(SA); Daryn H. Moller, MD; and Khoa N. Nguyen, MD
Abstract 40: Ethical dilemma in the preoperative assessment clinic: Can a patient refuse an indicated cardiac workup? Can we refuse to anesthetize?
Deborah C. Richman, MBChB, FFA(SA)
Abstract 41: Coronary artery bypass grafting as a precipitatin factor in diabetic ketoacidosis in type 2 diabetes
Vishal Sehgral, MD, and Abbas Kitabchi, MD
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Pulmonary hypertension is an important predictor of perioperative outcomes in patients undergoing noncardiac surgery
Roop Kaw, MD; Esteban Walker, PhD; Vinay Pasupuleti, MD, PhD; Abhishek Deshpande, MD, PhD; Tarek Hamieh, MD; and Omar A. Minai, MD
Abstract 2: Analysis of administrative practices and residency training curricula in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 3: Is percent body fat a better predictor of surgical site infection risk than body mass index?
Emily Waisbren, BS; Angela M. Bader, MD, MPH; Heather Rosen, MD, MPH; Selwyn O. Rogers, Jr., MD, MPH; and Elof Eriksson, MD, PhD
Abstract 4: A nomogram for prediction of survival for patients undergoing elective major noncardiac surgery
Y. Olivia Xu-Cai, MD; and Michael W. Kattan, PhD
Abstract 5: Sustainability of an osteoporosis pathway
Catherine Gibb, MBBS, FRACP; Christopher Butcher, FRACS; Lesley Thomas, BNsg; and Jennifer Pink, BPharm
Abstract 6: Length of hospital stay is predicted by comorbidities
Catherine Gibb, MBBS, FRACP; and Professor Villis Marshall, FRACS
Abstract 7: Generalization of the POISE and Mangano studies on beta-blocker use in the perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; and Luc Lanthier, MD, MSc
Abstract 8: Impact of antihypertensive medication on perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; Danielle Pilon, MD, MSc; and Luc Lanthier, MD, MSc
Abstract 9: An analysis of preoperative testing protocols in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 10: Preoperative biomarkers of inflammation, ischemia, and heart failure and outcomes of vascular surgery
Matthew Griffee, MD; Ansgar Brambrink, MD, PhD; and Thomas Barrett, MD
Abstract 11: Alcohol-related predictors of postoperative delirium in major head and neck cancer surgery
Harrison Weed, MD; Summit Shah, BS; Xin He, PhD; Amit Agrawal, MD; Enver Ozer, MD; and David E. Schuller, MD
Abstract 12: Intraoperative coagulopathy: A low-volume treatment protocol that completely replaces fresh frozen plasma
Peter Kallas, MD; Mary Lou Green, MHS; and Anjali Desai, MD
Abstract 13: Is the Berlin Questionnaire an effective screening tool for obstructive sleep apnea in the preoperative total joint replacement population?
Peter Kallas, MD; Mark Schumacher; Mona Lazar, DO; and Anjali Desai, MD
Abstract 14: The impact of preoperative medical optimization on head and neck cancer surgery
Christopher Tan, MBBS; Catherine Gibb, MBBS, FRACP; and Suren Krishnan, MBBS, FRACS
Abstract 15: Reconceptualizing the preoperative process
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 16: Development of an electronic medical record smart set form to increase standardization, consistency, and compliance with ACC/AHA perioperative guidelines
Anitha Rajamanickam, MD; Ali Usmani, MD; Ajay Kumar, MD; and Brian Harte, MD
Abstract 17: Development of a perioperative electronic medical record research and quality improvement database
Anitha Rajamanickam, MD; Ali Usmani, MD; Feza Remzi, MD; Brian Harte, MD; and Ajay Kumar, MD
Abstract 18: An innovative perioperative/consultative curriculum for third-year internal medicine residents
Alex Rico, MD; Joshua Lenchus, DO; and Amir Jaffer, MD
Abstract 19: Preoperative medicine infobutton
Terrence J. Adam, MD, PhD
Abstract 20: Nurse practitioners: Bridging the gap in perioperative care
Sally Morgan, RN, MS, ANP-BC, ACNS-BC; and Angela Wright, RN, MSN, APRN, BC
Abstract 21: Intubation training of deploying far forward combat medical personnel with the video laryngoscope
Ben Boedeker, MD; Mary Barak-Bernhagen, BS; Kirsten Boedeker; and W. Bosseau Murray, MD
Abstract 22: The establishment of a perioperative skin integrity committee
Jeanne Lanchester, RN, MEd; Ann Leary, BSN, RNC; and Susan Vargas, AD, RN
Abstract 23: Development and implementation of a perianesthesia integrative care committee
Jeanne Lanchester, RN, MEd; Jeanette Cote, BWN, RN; Terri Jamros, RN; Charla Delillo, RN; Sherie Lavoie, BSN, RN; Jennifer Therminos, SN; Joan Compagnone, RN; and Nicole Engel, MSN, RN
Abstract 24: Development of a screening system to identify patients preoperatively who may benefit from a postoperative hospitalist consult
Elizabeth Marlow, MD, MA; and Chad Whelan, MD
Abstract 25: An algorithm for preoperative screening and management of sleep apnea: Have we created a monster?
Deborah C. Richman, MBChB, FFA(SA); Jorge M. Mallea, MD; Paul S. Richman, MD; and Pater S.A. Glass, MBChB
Abstract 26: Constructing a collaborative neuroscience hospitalist program
Rachel Thompson, MD; Christy Gilmore, MD; Kamal Ajam, MD; and Jennifer Thompson, MD
Abstract 27: The development of algorithms for preoperative management of antiplatelet and anticoagulation therapy in patients undergoing surgical or invasive procedures
Catherine McGowan, MSN, and Patricia Kidik, MSN
Abstract 28: Surgeon-initiated preoperative screening: A new approach
Christina Johnson, RN, PA-C; and Edward J. denBraven, CRNA
Abstract 29: A new process for ensuring the safety of patients having anesthesia outside of the operating room
Ellen Leary, MSN; Catherine McGowan, MSN; Kathleen McGrath, MSN; Sheila McCabe Hassan, MSN; and Theresa Kennedy, MSN
Abstract 30: Establishing a virtual preoperative evaluation clinic
Corey Zetterman, MD; Bobbie J. Sweitzer, MD; and Ben H. Boedeker, MD
Abstract 31: Perioperative hypoxemia and rhabdomyolysis in a medically complicated patient
Sarah Bodin, MD
Abstract 32: How soon is too soon? General anesthesia after coronary intervention with bare metal stents
Meghan Tadel, MD
Abstract 33: Can patients with critical aortic stenosis undergo noncardiac surgery without intervening aortic valve replacement?
M. Chadi Alraies, MD; Abdul Alraiyes, MD; Anitha Rajamanickam, MD; and Frank Michota, MD
Abstract 34: Is it safe to operate on cocaine-positive patients?
M. Chadi Alraies, MD; Abdul Hamid Alraiyes, MD; and Brian Harte, MD
Abstract 35: To intensive care or not?
Mona Lazar, DO; and Peter Kallas, MD
Abstract 36: Predicting surgical complications from liver disease
Mona Lazar, DO, and Peter Kallas, MD
Abstract 37: Preoperative coronary angiography: Friend or foe?
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 38: Heparin-induced thrombocytopenia with low molecular weight heparin after total knee replacement
Steven Cohn, MD
Abstract 39: Patient with Parkinson’s disease treated with implanted deep brain stimulators for laparotomy
Deborah C. Richman, MBChB, FFA(SA); Daryn H. Moller, MD; and Khoa N. Nguyen, MD
Abstract 40: Ethical dilemma in the preoperative assessment clinic: Can a patient refuse an indicated cardiac workup? Can we refuse to anesthetize?
Deborah C. Richman, MBChB, FFA(SA)
Abstract 41: Coronary artery bypass grafting as a precipitatin factor in diabetic ketoacidosis in type 2 diabetes
Vishal Sehgral, MD, and Abbas Kitabchi, MD
Summit Director:
Amir K. Jaffer, MD
Contents
Abstract 1: Pulmonary hypertension is an important predictor of perioperative outcomes in patients undergoing noncardiac surgery
Roop Kaw, MD; Esteban Walker, PhD; Vinay Pasupuleti, MD, PhD; Abhishek Deshpande, MD, PhD; Tarek Hamieh, MD; and Omar A. Minai, MD
Abstract 2: Analysis of administrative practices and residency training curricula in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 3: Is percent body fat a better predictor of surgical site infection risk than body mass index?
Emily Waisbren, BS; Angela M. Bader, MD, MPH; Heather Rosen, MD, MPH; Selwyn O. Rogers, Jr., MD, MPH; and Elof Eriksson, MD, PhD
Abstract 4: A nomogram for prediction of survival for patients undergoing elective major noncardiac surgery
Y. Olivia Xu-Cai, MD; and Michael W. Kattan, PhD
Abstract 5: Sustainability of an osteoporosis pathway
Catherine Gibb, MBBS, FRACP; Christopher Butcher, FRACS; Lesley Thomas, BNsg; and Jennifer Pink, BPharm
Abstract 6: Length of hospital stay is predicted by comorbidities
Catherine Gibb, MBBS, FRACP; and Professor Villis Marshall, FRACS
Abstract 7: Generalization of the POISE and Mangano studies on beta-blocker use in the perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; and Luc Lanthier, MD, MSc
Abstract 8: Impact of antihypertensive medication on perioperative period
Matthieu Touchette, MD; Odile Paquette, MD; Catherine St-Georges, MD; Danielle Pilon, MD, MSc; and Luc Lanthier, MD, MSc
Abstract 9: An analysis of preoperative testing protocols in academic anesthesiology programs
David Hepner, A.R. Bader, D. Correll, L.C. Tsen, B.S. Segal, and A.M. Bader
Abstract 10: Preoperative biomarkers of inflammation, ischemia, and heart failure and outcomes of vascular surgery
Matthew Griffee, MD; Ansgar Brambrink, MD, PhD; and Thomas Barrett, MD
Abstract 11: Alcohol-related predictors of postoperative delirium in major head and neck cancer surgery
Harrison Weed, MD; Summit Shah, BS; Xin He, PhD; Amit Agrawal, MD; Enver Ozer, MD; and David E. Schuller, MD
Abstract 12: Intraoperative coagulopathy: A low-volume treatment protocol that completely replaces fresh frozen plasma
Peter Kallas, MD; Mary Lou Green, MHS; and Anjali Desai, MD
Abstract 13: Is the Berlin Questionnaire an effective screening tool for obstructive sleep apnea in the preoperative total joint replacement population?
Peter Kallas, MD; Mark Schumacher; Mona Lazar, DO; and Anjali Desai, MD
Abstract 14: The impact of preoperative medical optimization on head and neck cancer surgery
Christopher Tan, MBBS; Catherine Gibb, MBBS, FRACP; and Suren Krishnan, MBBS, FRACS
Abstract 15: Reconceptualizing the preoperative process
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 16: Development of an electronic medical record smart set form to increase standardization, consistency, and compliance with ACC/AHA perioperative guidelines
Anitha Rajamanickam, MD; Ali Usmani, MD; Ajay Kumar, MD; and Brian Harte, MD
Abstract 17: Development of a perioperative electronic medical record research and quality improvement database
Anitha Rajamanickam, MD; Ali Usmani, MD; Feza Remzi, MD; Brian Harte, MD; and Ajay Kumar, MD
Abstract 18: An innovative perioperative/consultative curriculum for third-year internal medicine residents
Alex Rico, MD; Joshua Lenchus, DO; and Amir Jaffer, MD
Abstract 19: Preoperative medicine infobutton
Terrence J. Adam, MD, PhD
Abstract 20: Nurse practitioners: Bridging the gap in perioperative care
Sally Morgan, RN, MS, ANP-BC, ACNS-BC; and Angela Wright, RN, MSN, APRN, BC
Abstract 21: Intubation training of deploying far forward combat medical personnel with the video laryngoscope
Ben Boedeker, MD; Mary Barak-Bernhagen, BS; Kirsten Boedeker; and W. Bosseau Murray, MD
Abstract 22: The establishment of a perioperative skin integrity committee
Jeanne Lanchester, RN, MEd; Ann Leary, BSN, RNC; and Susan Vargas, AD, RN
Abstract 23: Development and implementation of a perianesthesia integrative care committee
Jeanne Lanchester, RN, MEd; Jeanette Cote, BWN, RN; Terri Jamros, RN; Charla Delillo, RN; Sherie Lavoie, BSN, RN; Jennifer Therminos, SN; Joan Compagnone, RN; and Nicole Engel, MSN, RN
Abstract 24: Development of a screening system to identify patients preoperatively who may benefit from a postoperative hospitalist consult
Elizabeth Marlow, MD, MA; and Chad Whelan, MD
Abstract 25: An algorithm for preoperative screening and management of sleep apnea: Have we created a monster?
Deborah C. Richman, MBChB, FFA(SA); Jorge M. Mallea, MD; Paul S. Richman, MD; and Pater S.A. Glass, MBChB
Abstract 26: Constructing a collaborative neuroscience hospitalist program
Rachel Thompson, MD; Christy Gilmore, MD; Kamal Ajam, MD; and Jennifer Thompson, MD
Abstract 27: The development of algorithms for preoperative management of antiplatelet and anticoagulation therapy in patients undergoing surgical or invasive procedures
Catherine McGowan, MSN, and Patricia Kidik, MSN
Abstract 28: Surgeon-initiated preoperative screening: A new approach
Christina Johnson, RN, PA-C; and Edward J. denBraven, CRNA
Abstract 29: A new process for ensuring the safety of patients having anesthesia outside of the operating room
Ellen Leary, MSN; Catherine McGowan, MSN; Kathleen McGrath, MSN; Sheila McCabe Hassan, MSN; and Theresa Kennedy, MSN
Abstract 30: Establishing a virtual preoperative evaluation clinic
Corey Zetterman, MD; Bobbie J. Sweitzer, MD; and Ben H. Boedeker, MD
Abstract 31: Perioperative hypoxemia and rhabdomyolysis in a medically complicated patient
Sarah Bodin, MD
Abstract 32: How soon is too soon? General anesthesia after coronary intervention with bare metal stents
Meghan Tadel, MD
Abstract 33: Can patients with critical aortic stenosis undergo noncardiac surgery without intervening aortic valve replacement?
M. Chadi Alraies, MD; Abdul Alraiyes, MD; Anitha Rajamanickam, MD; and Frank Michota, MD
Abstract 34: Is it safe to operate on cocaine-positive patients?
M. Chadi Alraies, MD; Abdul Hamid Alraiyes, MD; and Brian Harte, MD
Abstract 35: To intensive care or not?
Mona Lazar, DO; and Peter Kallas, MD
Abstract 36: Predicting surgical complications from liver disease
Mona Lazar, DO, and Peter Kallas, MD
Abstract 37: Preoperative coronary angiography: Friend or foe?
Ross Kerridge, MBBS, FRCA, FANZCA
Abstract 38: Heparin-induced thrombocytopenia with low molecular weight heparin after total knee replacement
Steven Cohn, MD
Abstract 39: Patient with Parkinson’s disease treated with implanted deep brain stimulators for laparotomy
Deborah C. Richman, MBChB, FFA(SA); Daryn H. Moller, MD; and Khoa N. Nguyen, MD
Abstract 40: Ethical dilemma in the preoperative assessment clinic: Can a patient refuse an indicated cardiac workup? Can we refuse to anesthetize?
Deborah C. Richman, MBChB, FFA(SA)
Abstract 41: Coronary artery bypass grafting as a precipitatin factor in diabetic ketoacidosis in type 2 diabetes
Vishal Sehgral, MD, and Abbas Kitabchi, MD