User login
Microcystic Adnexal Carcinoma of the External Auditory Canal
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
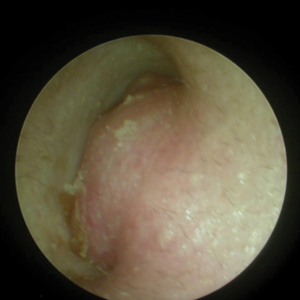
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.

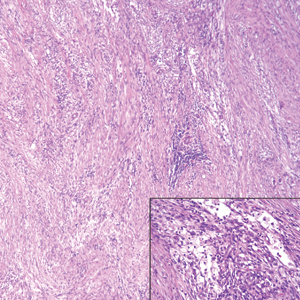
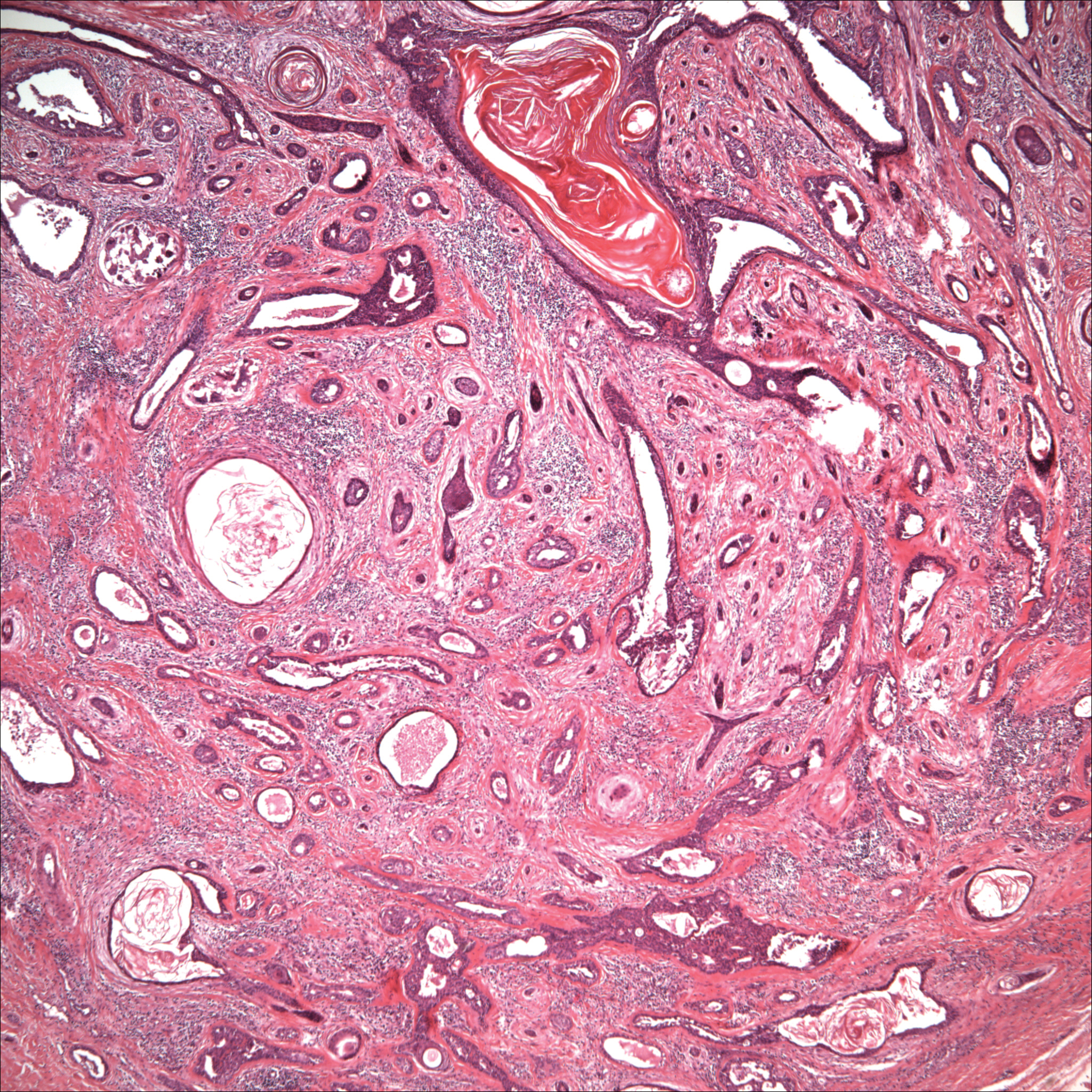
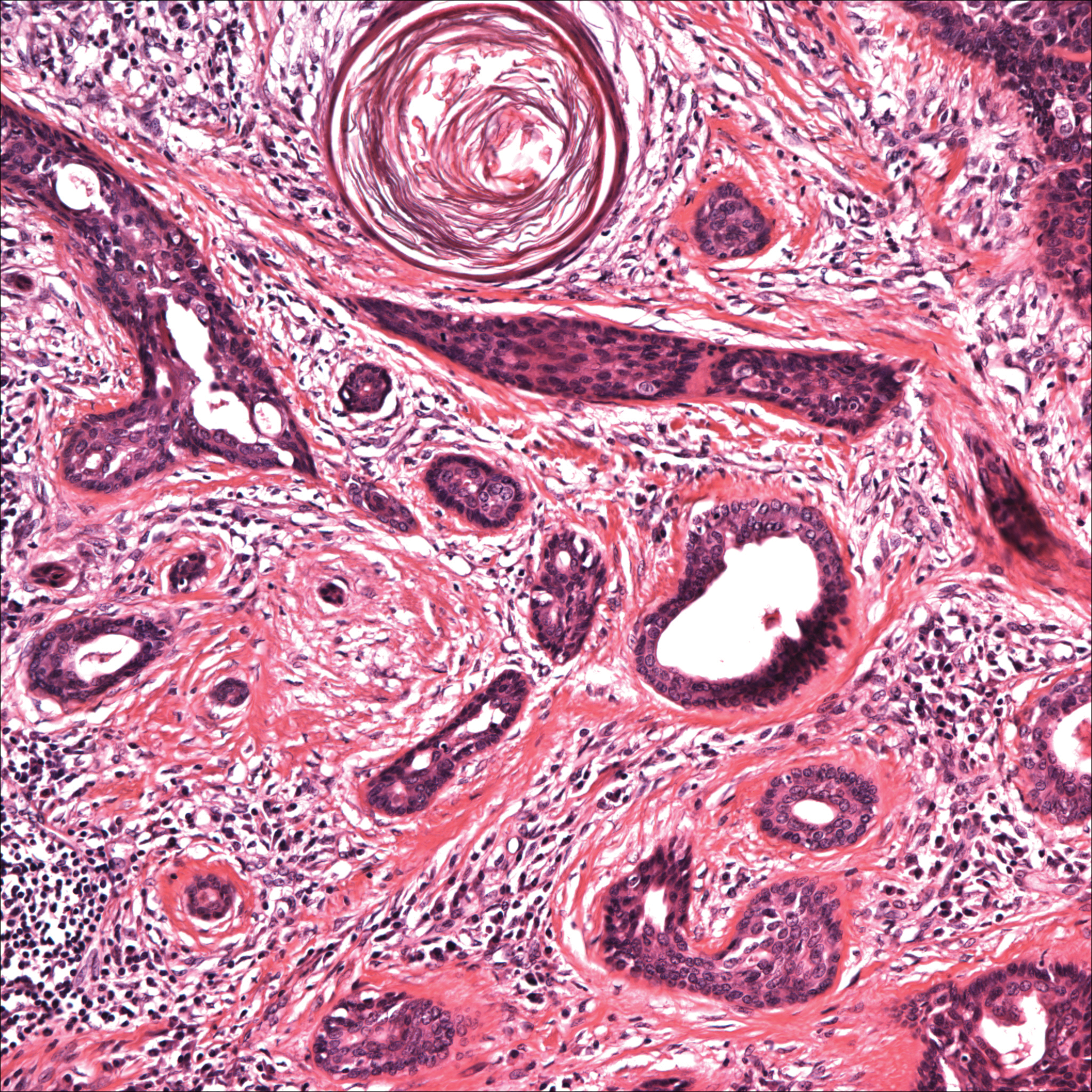
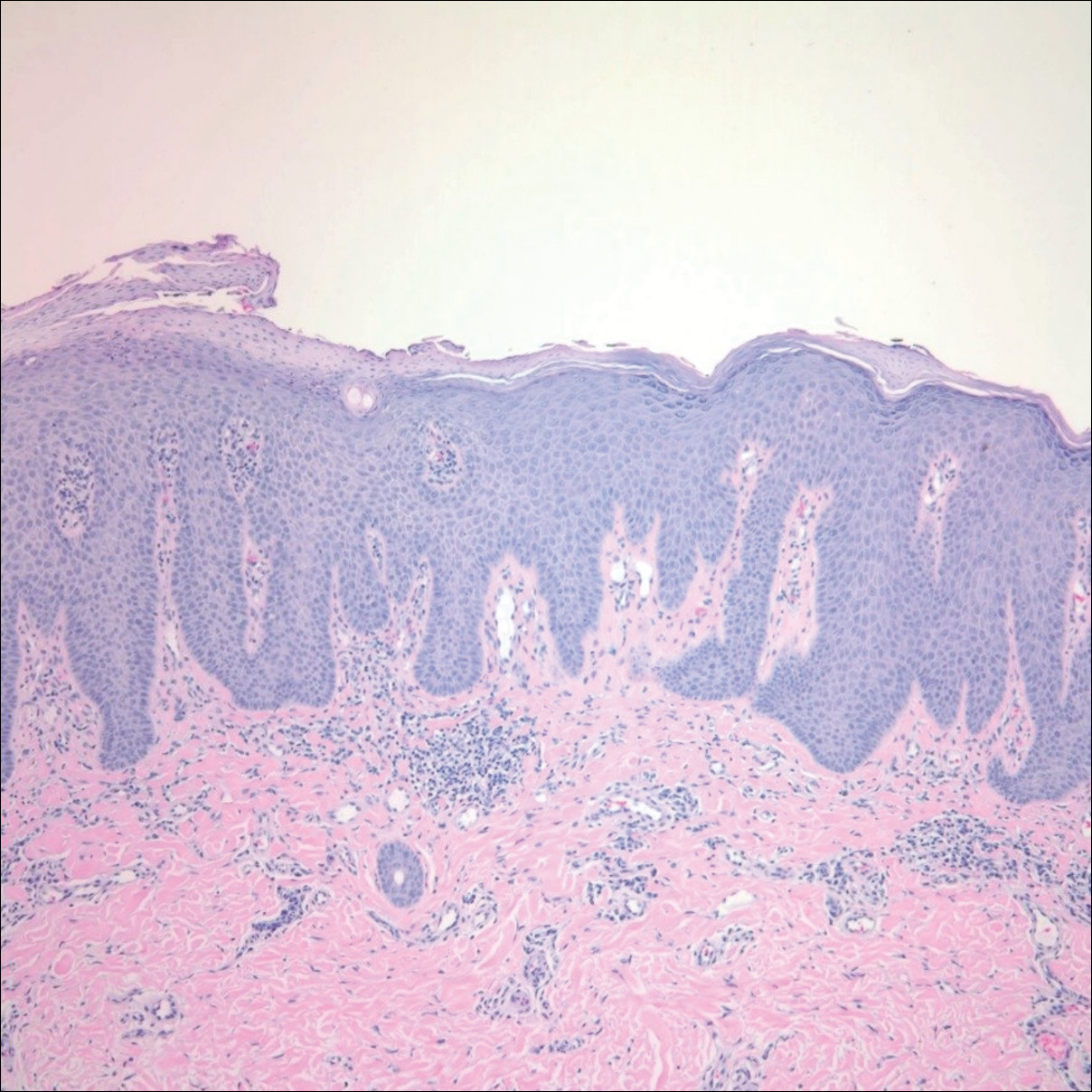
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
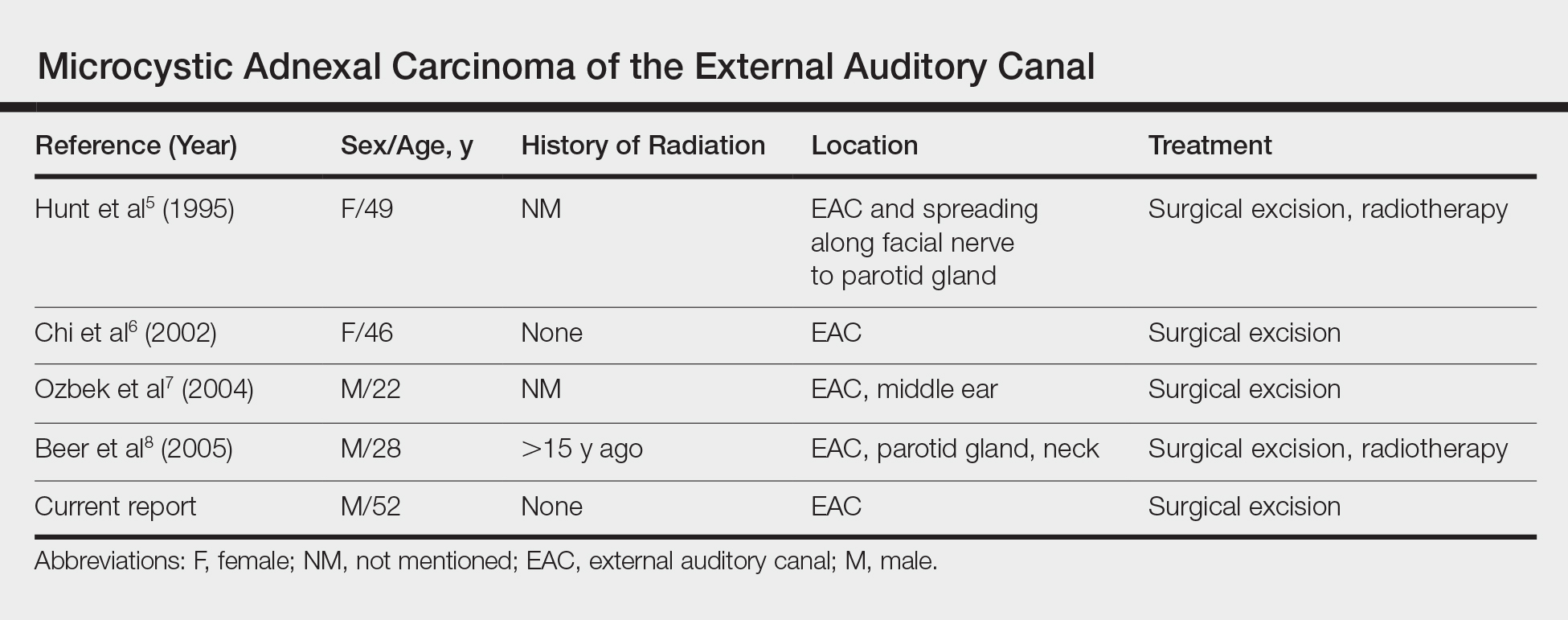
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
To the Editor:
Microcystic adnexal carcinoma (MAC), described by Goldstein et al1 in 1982, is a relatively uncommon cutaneous neoplasm. This locally aggressive malignant adnexal tumor has high potential for local recurrence. The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.2 Involvement of the external auditory canal (EAC) is relatively rare. We report a case of MAC of the EAC.
A 52-year-old man presented with 1 palpable nodule on the right EAC of approximately 1 year’s duration. The lesion was asymptomatic, and the patient had no history of radiation exposure. The patient was an airport employee required to wear an earplug in the right ear. Endoscopic examination identified a 1×1 cm2 erythematous nodule on the anterior inferior quadrant of the right external ear canal orifice (Figure 1). Axial and coronal computed tomography demonstrated a soft tissue mass in the right EAC without any bony erosion. No clinical signs of regional lymphadenopathy or distant metastasis were present. Excision was performed under microscopic visualization.
Histopathology of the nodule showed marked proliferation of multiple keratin-containing cysts, irregular ductal structures, and solid epithelial nests in the deep dermis (Figure 2). Irregular ductal structures with 2 cell layer walls and several epithelial strands or small nests of tumor cells within desmoplastic stroma were noted (Figure 3). No perineural infiltration or tumor infiltration existed at the margin. Based on the clinical and histopathologic findings, the final diagnosis was MAC. Complete resolution was noted after the excision. The patient returned for regular follow-up and no signs of recurrence were noted for 7 years postoperatively.
Microcystic adnexal carcinoma, also known as sclerosing sweat duct (syringomatous) carcinoma, malignant syringoma, and syringoid eccrine carcinoma, is characterized by slow and locally aggressive growth with high likelihood of perineural invasion and frequent recurrence.2 Regional lymph node metastasis is uncommon, and systemic metastasis is rare.2-4
Although the head most often is involved, a PubMed search of articles indexed for MEDLINE using the terms microcystic adnexal carcinoma and external auditory canal revealed 4 cases (Table).5-8 Our report adds another case of MAC arising solely in the EAC. Although the etiology of MAC is unknown, prior studies indicated that radiotherapy is a risk factor for MAC. Other possible risk factors include UV light exposure and immunodeficiency.2 Our patient had no history of these factors and experienced chronic friction caused by use of an occupational unilateral earplug, which may be a notable factor. Locations of MAC arising outside the head region include the axilla, vulva, breast, palm, toe, perianal skin, buttock, chest, and an ovarian cystic teratoma.3,9 Friction commonly occurs in many of these areas. Therefore, we propose that friction may be a risk factor for MAC.
Microcystic adnexal carcinoma should be included in the differential diagnosis of any slowly growing cutaneous tumor, even in the EAC. Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence signs is essential.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Brenn T, Mckee PH. Tumors of the sweat glands. In: McKee PH, Calonje E, Granter SR, eds. Pathology of the Skin With Clinical Correlations. 3rd ed. Philadelphia, PA: Elsevier Mosby; 2005:1647-1651.
- Ohtsuka H, Nagamatsu S. Microcystic adnexal carcinoma: review of 51 Japanese patients. Dermatology. 2002;204:190-193.
- Yu JB, Blitzblau RC, Patel SC, et al. Surveillance, Epidemiology, and End Results (SEER) database analysis of microcystic adnexal carcinoma (sclerosing sweat duct carcinoma) of the skin. Am J Clin Oncol. 2010;33:125-127.
- Hunt JT, Stack BC Jr, Futran ND, et al. Pathologic quiz case 1. microcystic adnexal carcinoma (MAC). Arch Otolaryngol Head Neck Surg. 1995;121:1430-1433.
- Chi J, Jung YG, Rho YS, et al. Microcystic adnexal carcinoma of external auditory canal: report of a case. Otolaryngol Head Neck Surg. 2002;127:241-242.
- Ozbek C, Celikkanat S, Beriat K, et al. Microcystic adnexal carcinoma of the external ear canal. Otolaryngol Head Neck Surg. 2004;130:148-150.
- Beer KT, Bühler SS, Mullis P, et al. A microcystic adnexal carcinoma in the auditory canal 15 years after radiotherapy of a 12-year-old boy with nasopharynx carcinoma. Strahlenther Onkol. 2005;181:405-410.
- Nadiminti H, Nadiminti U, Washington C. Microcystic adnexal carcinoma in African Americans. Dermatol Surg. 2007;33:1384-1387.
Practice Points
- Microcystic adnexal carcinoma is a locally aggressive malignant adnexal tumor with a high potential for local recurrence.
- The skin of the head, particularly in the nasolabial and periorbital regions, most often is involved.
- Once diagnosed, the tumor should be surgically excised. Because local recurrence is common and may occur several decades after excision, lifetime follow-up for recurrence is essential.
Inflammatory Linear Verrucous Epidermal Nevus Responsive to 308-nm Excimer Laser Treatment
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare entity that presents with linear and pruritic psoriasiform plaques and most commonly occurs during childhood. It represents a dysregulation of keratinocytes exhibiting genetic mosaicism.1,2 Epidermal nevi may derive from keratinocytic, follicular, sebaceous, apocrine, or eccrine origin. Inflammatory linear verrucous epidermal nevus is classified under the keratinocytic type of epidermal nevus and represents approximately 6% of all epidermal nevi.3 The condition presents as erythematous and verrucous plaques along the lines of Blaschko.2,4 There is a predilection for the legs, and girls are 4 times more commonly affected than boys.1 Cases of ILVEN are predominantly sporadic, though rare familial cases have been reported.4
Inflammatory linear verrucous epidermal nevus is notoriously refractory to treatment. First-line therapies include topical agents such as corticosteroids, calcipotriol, retinoids, and 5-fluorouracil.3,4 Other treatments include intralesional corticosteroids, cryotherapy, electrodesiccation and curettage, and surgical excision.3 Several case reports have shown promising results using the pulsed dye and ablative CO2 lasers.5-8
Case Report
An otherwise healthy 20-year-old woman presented with dry, pruritic, red lesions on the right leg that had been present and stable since she was an infant (2 weeks of age). Her medical history included acne vulgaris, but she denied any personal or family history of psoriasis as well as any arthralgia or arthritis. Physical examination revealed discrete, oval, hyperkeratotic, scaly, red plaques on the lateral right leg with a larger hyperkeratotic, linear, red plaque extending from the right popliteal fossa to the posterior thigh (Figure 1A). The nails, scalp, buttocks, and upper extremities were unaffected. Bacterial culture of the right leg demonstrated Staphylococcus aureus colonization. Biopsy of the right popliteal fossa showed psoriasiform dermatitis with psoriasiform hyperplasia, a slightly verruciform surface, broad zones of superficial pallor, and parakeratosis with conspicuous colonies of bacteria (Figure 2).
 and after 18 treatment sessions with the 308-nm excimer laser (B)...")
Following the positive bacterial culture, the patient was treated with a short course of oral doxycycline, which did not alter the clinical appearance of the lesions or improve symptoms of pruritus. Pruritus improved moderately with topical corticosteroid treatment, but clinically the lesions appeared unchanged. The plaque on the superior right leg was treated with a superpulsed CO2 laser and the plaque on the inferior right leg was treated with a fractional CO2 laser, both with minimal improvement.
Because of the clinical and histopathologic similarities of the patient's lesions to psoriasis, a trial of the UV 308-nm excimer laser was initiated. Following initial test spots, she completed a total of 18 treatments to all lesions with noticeable clinical improvement (Figure 1B). Initially, the patient returned for treatment biweekly for approximately 5 weeks with 2 small spots being targeted at each session, with an average surface area of approximately 16 cm2. She was started at 225 mJ/cm2 with 25% increases at each session and ultimately reached up to 1676 mJ/cm2 at the end of the 10 sessions. She tolerated the procedure well with some minor blistering. Treatment was deferred for 3 months due to the patient's schedule, then biweekly treatments resumed for 4 weeks, totaling 8 more sessions. At that time, all lesions on the right leg were targeted, with an average surface area of approximately 100 cm2. The laser settings were initiated at 225 mJ/cm2 with 20% increases at each session and ultimately reached 560 mJ/cm2. The treatment was well tolerated throughout; however, the patient initially reported residual pruritus. The plaques continued to improve, and most notably, there was thinning of the hyperkeratotic scale of the plaques in addition to decreased erythema and complete resolution of pruritus. Ultimately, treatment was discontinued because of lack of insurance coverage and financial burden. The patient was lost to follow-up.
Comment
Presentation
Inflammatory linear verrucous epidermal nevus is a rare type of keratinocytic epidermal nevus4 that clinically presents as small, discrete, pruritic, scaly plaques coalescing into a linear plaque along the lines of Blaschko.9 Considerable pruritus and resistance to treatment are hallmarks of the disease.10 Histopathologically, ILVEN is characterized by alternating orthokeratosis and parakeratosis with a lack of neutrophils in an acanthotic epidermis.11-13 Inflammatory linear verrucous epidermal nevus presents at birth or in early childhood. Adult onset is rare.9,14 Approximately 75% of lesions present by 5 years of age, with a majority occurring within the first 6 months of life.15 The differential diagnosis includes linear psoriasis, epidermal nevi, linear lichen planus, linear verrucae, linear lichen simplex chronicus, and mycosis fungoides.4,11
Differentiation From Psoriasis
Despite the histopathologic overlap with psoriasis, ILVEN exhibits fewer Ki-67-positive keratinocyte nuclei (proliferative marker) and more cytokeratin 10-positive cells (epidermal differentiation marker) than psoriasis.16 Furthermore, ILVEN has demonstrated fewer CD4−, CD8−, CD45RO−, CD2−, CD25−, CD94−, and CD161+ cells within the dermis and epidermis than psoriasis.16
The clinical presentations of ILVEN and psoriasis may be similar, as some patients with linear psoriasis also present with psoriatic plaques along the lines of Blaschko.17 Additionally, ILVEN may be a precursor to psoriasis. Altman and Mehregan1 found that ILVEN patients who developed psoriasis did so in areas previously affected by ILVEN; however, they continued to distinguish the 2 pathologies as distinct entities. Another early report also hypothesized that the dermoepidermal defect caused by epidermal nevi provided a site for the development of psoriatic lesions because of the Koebner phenomenon.18
Patients with ILVEN also have been found to have extracutaneous manifestations and symptoms commonly seen in psoriasis patients. A 2012 retrospective review revealed that 37% (7/19) of patients with ILVEN also had psoriatic arthritis, cutaneous psoriatic lesions, and/or nail pitting. The authors concluded that ILVEN may lead to the onset of psoriasis later in life and may indicate an underlying psoriatic predisposition.19 Genetic theories also have been proposed, stating that ILVEN may be a mosaic of psoriasis2 or that a postzygotic mutation leads to the predisposition for developing psoriasis.20
Treatment
Inflammatory linear verrucous epidermal nevus frequently is refractory to treatment; however, the associated pruritus and distressing cosmesis make treatment attempts worthwhile.11 No single therapy has been found to be successful in all patients. A widely used first-line treatment is topical or intralesional corticosteroids, with the former typically used with occlusion.13 Other treatments include adalimumab, calcipotriol,22,23 tretinoin,24 and 5-fluorouracil.24 Physical modalities such as cryotherapy, electrodesiccation, and dermabrasion have been reported with varying success.15,24 Surgical treatments include tangential25 and full-thickness excisions.26
The CO2 laser also has demonstrated success. One study showed considerable improvement of pruritus and partial resolution of lesions only 5 weeks following a single CO2 laser treatment.5 Another study showed promising results when combining CO2 pulsed laser therapy with fractional CO2 laser treatment.6 Other laser therapies including the argon27 and flashlamp-pumped pulsed dye lasers8 have been used with limited success. The use of light therapy and lasers in psoriasis have now increased the treatment options for ILVEN based on the rationale of their shared histopathologic characteristics. Photodynamic therapy also has been attempted because of its successful use in psoriasis patients. It has been found to be successful in diminishing ILVEN lesions and associated pruritus after a few weeks of therapy; however, treatment is limited by the associated pain and requirement for local anesthesia.28
The excimer laser is a form of targeted phototherapy that emits monochromatic light at 308 nm.29 It is ideal for inflammatory skin lesions because the UVB light induces apoptosis.30 Psoriasis lesions treated with the excimer laser show a decrease in keratinocyte proliferation, which in turn reverses epidermal acanthosis and causes T-cell depletion due to upregulation of p53.29,31 This mechanism of action addresses the overproliferation of keratinocytes mediated by T cells in psoriasis and contributes to the success of excimer laser treatment.31 A considerable advantage is its localized treatment, resulting in lower cumulative doses of UVB and reducing the possible carcinogenic and phototoxic risks of whole-body phototherapy.32
One study examined the antipruritic effects of the excimer laser following the treatment of epidermal hyperinnervation leading to intractable pruritus in patients with atopic dermatitis. The researchers suggested that a potential explanation for the antipruritic effect of the excimer laser may be secondary to nerve degeneration.33 Additionally, low doses of UVB light also may inhibit mast cell degranulation and prevent histamine release, further supporting the antipruritic properties of excimer laser.34
In our patient, failed treatment with other modalities led to trial of excimer laser therapy because of the overlapping clinical and histopathologic findings with psoriasis. Excimer laser improved the clinical appearance and overall texture of the ILVEN lesions and decreased pruritus. The reasons for treatment success may be two-fold. By decreasing the number of keratinocytes and mast cells, the excimer laser may have improved the epidermal hyperplasia and pruritus in the ILVEN lesions. Alternatively, because the patient had ILVEN lesions since infancy, psoriasis may have developed in the location of the ILVEN lesions due to koebnerization, resulting in the clinical response to excimer therapy; however, she had no other clinical evidence of psoriasis.
Because of the recalcitrance of ILVEN lesions to conventional therapies, it is important to investigate therapies that may be of possible benefit. Our novel case documents successful use of the excimer laser in the treatment of ILVEN.
Conclusion
Our case of ILVEN in a woman that had been present since infancy highlights the disease pathology as well as a potential new treatment modality. The patient was refractory to first-line treatments and was concerned about the cosmetic appearance of the lesions. The patient was subsequently treated with a trial of a 308-nm excimer laser with clinical improvement of the lesions. It is possible that the similarity of ILVEN and psoriasis may have contributed to the clinical improvement in our patient, but the mechanism of action remains unknown. Due to the paucity of evidence regarding optimal treatment of ILVEN, the current case offers dermatologists an option for patients who are refractory to other treatments.
- Altman J, Mehregan AH. Inflammatory linear verrucose epidermal nevus. Arch Dermatol. 1971;104:385-389.
- Hofer T. Does inflammatory linear verrucous epidermal nevus represent a segmental type 1/type 2 mosaic of psoriasis? Dermatology. 2006;212:103-107.
- Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome: a review of 131 cases. J Am Acad Dermatol. 1989;20:476-488.
- Khachemoune A, Janjua S, Guldbakke K. Inflammatory linear verrucous epidermal nevus: a case report and short review of the literature. Cutis. 2006;78:261-267.
- Ulkur E, Celikoz B, Yuksel F, et al. Carbon dioxide laser therapy for an inflammatory linear verrucous epidermal nevus: a case report. Aesthetic Plast Surg. 2004;28:428-430.
- Conti R, Bruscino N, Campolmi P, et al. Inflammatory linear verrucous epidermal nevus: why a combined laser therapy. J Cosmet Laser Ther. 2013;15:242-245.
- Alonso-Castro L, Boixeda P, Reig I, et al. Carbon dioxide laser treatment of epidermal nevi: response and long-term follow-up. Actas Dermosifiliogr. 2012;103:910-918.
- Alster TS. Inflammatory linear verrucous epidermal nevus: successful treatment with the 585 nm flashlamp-pumped dye laser. J Am Acad Dermatol. 1994;31:513-514.
- Kruse LL. Differential diagnosis of linear eruptions in children. Pediatr Ann. 2015;44:194-198.
- Renner R, Colsman A, Sticherling M. ILVEN: is it psoriasis? debate based on successful treatment with etanercept. Acta Derm Venereol. 2008;88:631-632.
- Lee SH, Rogers M. Inflammatory linear verrucous epidermal naevi: a review of 23 cases. Australas J Dermatol. 2001;42:252-256.
- Ito M, Shimizu N, Fujiwara H, et al. Histopathogenesis of inflammatory linear verrucose epidermal nevus: histochemistry, immunohistochemistry and ultrastructure. Arch Dermatol Res. 1991;283:491-499.
- Cerio R, Jones EW, Eady RA. ILVEN responding to occlusive potent topical steroid therapy. Clin Exp Dermatol. 1992;17:279-281.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus. J Dermatol. 1999;26:599-602.
- Behera B, Devi B, Nayak BB, et al. Giant inflammatory linear verrucous epidermal nevus: successfully treated with full thickness excision and skin grafting. Indian J Dermatol. 2013;58:461-463.
- Vissers WH, Muys L, Erp PE, et al. Immunohistochemical differentiation between ILVEN and psoriasis. Eur J Dermatol. 2004;14:216-220.
- Agarwal US, Besarwal RK, Gupta R, et a. Inflammatory linear verrucous epidermal nevus with psoriasiform histology. Indian J Dermatol. 2014;59:211.
- Bennett RG, Burns L, Wood MG. Systematized epidermal nevus: a determinant for the localization of psoriasis. Arch Dermatol. 1973;108:705-757.
- Tran K, Jao-Tan C, Ho N. ILVEN and psoriasis: a retrospective study among pediatric patients. J Am Acad Dermatol. 2012;66(suppl 1):AB163.
- Happle R. Superimposed linear psoriasis: a historical case revisited. J Dtsch Dermatol Ges. 2011;9:1027-1028; discussion 1029.
- Özdemir M, Balevi A, Esen H. An inflammatory verrucous epidermal nevus concomitant with psoriasis: treatment with adalimumab. Dermatol Online J. 2012;18:11.
- Zvulunov A, Grunwald MH, Halvy S. Topical calcipotriol for treatment of inflammatory linear verrucous epidermal nevus. Arch Dermatol. 1997;133:567-568.
- Gatti S, Carrozzo AM, Orlandi A, et al. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132:837-839.
- Fox BJ, Lapins NA. Comparison of treatment modalities for epidermal nevus: a case report and review. J Dermatol Surg Oncol. 1983;9:879-885.
- Pilanci O, Tas B, Ceran F, et al. A novel technique used in the treatment of inflammatory linear verrucous epidermal nevus: tangential excision. Aesthetic Plast Surg. 2014;38:1066-1067.
- Lee BJ, Mancini AJ, Renucci J, et al. Full-thickness surgical excision for the treatment of inflammatory linear verrucous epidermal nevus. Ann Plast Surg. 2001;47:285-292.
- Hohenleutner U, Landthaler M. Laser therapy of verrucous epidermal naevi. Clin Exp Dermatol. 1993;18:124-127.
- Parera E, Gallardo F, Toll A, et al. Inflammatory linear verrucous epidermal nevus successfully treated with methyl-aminolevulinate photodynamic therapy. Dermatol Surg. 2010;36:253-256.
- Situm M, Bulat V, Majcen K, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253.
- Beggs S, Short J, Rengifo-Pardo M, et al. Applications of the excimer laser: a review. Dermatol Surg. 2015;41:1201-1211.
- Bianchi B, Campolmi P, Mavilia L, et al. Monochromatic excimer light (308 nm): an immunohistochemical study of cutaneous T cells and apoptosis-related molecules in psoriasis. J Eur Acad Dermatol Venereol. 2003;17:408-413.
- Mudigonda T, Dabade TS, Feldman SR. A review of targeted ultraviolet B phototherapy for psoriasis. J Am Acad Dermatol. 2012;66:664-672.
- Kamo A, Tominaga M, Kamata Y, et al. The excimer lamp induces cutaneous nerve degeneration and reduces scratching in a dry-skin mouse model. J Invest Dermatol. 2014;134:2977-2984.
- Bulat V, Majcen K, Dzapo A, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare entity that presents with linear and pruritic psoriasiform plaques and most commonly occurs during childhood. It represents a dysregulation of keratinocytes exhibiting genetic mosaicism.1,2 Epidermal nevi may derive from keratinocytic, follicular, sebaceous, apocrine, or eccrine origin. Inflammatory linear verrucous epidermal nevus is classified under the keratinocytic type of epidermal nevus and represents approximately 6% of all epidermal nevi.3 The condition presents as erythematous and verrucous plaques along the lines of Blaschko.2,4 There is a predilection for the legs, and girls are 4 times more commonly affected than boys.1 Cases of ILVEN are predominantly sporadic, though rare familial cases have been reported.4
Inflammatory linear verrucous epidermal nevus is notoriously refractory to treatment. First-line therapies include topical agents such as corticosteroids, calcipotriol, retinoids, and 5-fluorouracil.3,4 Other treatments include intralesional corticosteroids, cryotherapy, electrodesiccation and curettage, and surgical excision.3 Several case reports have shown promising results using the pulsed dye and ablative CO2 lasers.5-8
Case Report
An otherwise healthy 20-year-old woman presented with dry, pruritic, red lesions on the right leg that had been present and stable since she was an infant (2 weeks of age). Her medical history included acne vulgaris, but she denied any personal or family history of psoriasis as well as any arthralgia or arthritis. Physical examination revealed discrete, oval, hyperkeratotic, scaly, red plaques on the lateral right leg with a larger hyperkeratotic, linear, red plaque extending from the right popliteal fossa to the posterior thigh (Figure 1A). The nails, scalp, buttocks, and upper extremities were unaffected. Bacterial culture of the right leg demonstrated Staphylococcus aureus colonization. Biopsy of the right popliteal fossa showed psoriasiform dermatitis with psoriasiform hyperplasia, a slightly verruciform surface, broad zones of superficial pallor, and parakeratosis with conspicuous colonies of bacteria (Figure 2).
Following the positive bacterial culture, the patient was treated with a short course of oral doxycycline, which did not alter the clinical appearance of the lesions or improve symptoms of pruritus. Pruritus improved moderately with topical corticosteroid treatment, but clinically the lesions appeared unchanged. The plaque on the superior right leg was treated with a superpulsed CO2 laser and the plaque on the inferior right leg was treated with a fractional CO2 laser, both with minimal improvement.
Because of the clinical and histopathologic similarities of the patient's lesions to psoriasis, a trial of the UV 308-nm excimer laser was initiated. Following initial test spots, she completed a total of 18 treatments to all lesions with noticeable clinical improvement (Figure 1B). Initially, the patient returned for treatment biweekly for approximately 5 weeks with 2 small spots being targeted at each session, with an average surface area of approximately 16 cm2. She was started at 225 mJ/cm2 with 25% increases at each session and ultimately reached up to 1676 mJ/cm2 at the end of the 10 sessions. She tolerated the procedure well with some minor blistering. Treatment was deferred for 3 months due to the patient's schedule, then biweekly treatments resumed for 4 weeks, totaling 8 more sessions. At that time, all lesions on the right leg were targeted, with an average surface area of approximately 100 cm2. The laser settings were initiated at 225 mJ/cm2 with 20% increases at each session and ultimately reached 560 mJ/cm2. The treatment was well tolerated throughout; however, the patient initially reported residual pruritus. The plaques continued to improve, and most notably, there was thinning of the hyperkeratotic scale of the plaques in addition to decreased erythema and complete resolution of pruritus. Ultimately, treatment was discontinued because of lack of insurance coverage and financial burden. The patient was lost to follow-up.
Comment
Presentation
Inflammatory linear verrucous epidermal nevus is a rare type of keratinocytic epidermal nevus4 that clinically presents as small, discrete, pruritic, scaly plaques coalescing into a linear plaque along the lines of Blaschko.9 Considerable pruritus and resistance to treatment are hallmarks of the disease.10 Histopathologically, ILVEN is characterized by alternating orthokeratosis and parakeratosis with a lack of neutrophils in an acanthotic epidermis.11-13 Inflammatory linear verrucous epidermal nevus presents at birth or in early childhood. Adult onset is rare.9,14 Approximately 75% of lesions present by 5 years of age, with a majority occurring within the first 6 months of life.15 The differential diagnosis includes linear psoriasis, epidermal nevi, linear lichen planus, linear verrucae, linear lichen simplex chronicus, and mycosis fungoides.4,11
Differentiation From Psoriasis
Despite the histopathologic overlap with psoriasis, ILVEN exhibits fewer Ki-67-positive keratinocyte nuclei (proliferative marker) and more cytokeratin 10-positive cells (epidermal differentiation marker) than psoriasis.16 Furthermore, ILVEN has demonstrated fewer CD4−, CD8−, CD45RO−, CD2−, CD25−, CD94−, and CD161+ cells within the dermis and epidermis than psoriasis.16
The clinical presentations of ILVEN and psoriasis may be similar, as some patients with linear psoriasis also present with psoriatic plaques along the lines of Blaschko.17 Additionally, ILVEN may be a precursor to psoriasis. Altman and Mehregan1 found that ILVEN patients who developed psoriasis did so in areas previously affected by ILVEN; however, they continued to distinguish the 2 pathologies as distinct entities. Another early report also hypothesized that the dermoepidermal defect caused by epidermal nevi provided a site for the development of psoriatic lesions because of the Koebner phenomenon.18
Patients with ILVEN also have been found to have extracutaneous manifestations and symptoms commonly seen in psoriasis patients. A 2012 retrospective review revealed that 37% (7/19) of patients with ILVEN also had psoriatic arthritis, cutaneous psoriatic lesions, and/or nail pitting. The authors concluded that ILVEN may lead to the onset of psoriasis later in life and may indicate an underlying psoriatic predisposition.19 Genetic theories also have been proposed, stating that ILVEN may be a mosaic of psoriasis2 or that a postzygotic mutation leads to the predisposition for developing psoriasis.20
Treatment
Inflammatory linear verrucous epidermal nevus frequently is refractory to treatment; however, the associated pruritus and distressing cosmesis make treatment attempts worthwhile.11 No single therapy has been found to be successful in all patients. A widely used first-line treatment is topical or intralesional corticosteroids, with the former typically used with occlusion.13 Other treatments include adalimumab, calcipotriol,22,23 tretinoin,24 and 5-fluorouracil.24 Physical modalities such as cryotherapy, electrodesiccation, and dermabrasion have been reported with varying success.15,24 Surgical treatments include tangential25 and full-thickness excisions.26
The CO2 laser also has demonstrated success. One study showed considerable improvement of pruritus and partial resolution of lesions only 5 weeks following a single CO2 laser treatment.5 Another study showed promising results when combining CO2 pulsed laser therapy with fractional CO2 laser treatment.6 Other laser therapies including the argon27 and flashlamp-pumped pulsed dye lasers8 have been used with limited success. The use of light therapy and lasers in psoriasis have now increased the treatment options for ILVEN based on the rationale of their shared histopathologic characteristics. Photodynamic therapy also has been attempted because of its successful use in psoriasis patients. It has been found to be successful in diminishing ILVEN lesions and associated pruritus after a few weeks of therapy; however, treatment is limited by the associated pain and requirement for local anesthesia.28
The excimer laser is a form of targeted phototherapy that emits monochromatic light at 308 nm.29 It is ideal for inflammatory skin lesions because the UVB light induces apoptosis.30 Psoriasis lesions treated with the excimer laser show a decrease in keratinocyte proliferation, which in turn reverses epidermal acanthosis and causes T-cell depletion due to upregulation of p53.29,31 This mechanism of action addresses the overproliferation of keratinocytes mediated by T cells in psoriasis and contributes to the success of excimer laser treatment.31 A considerable advantage is its localized treatment, resulting in lower cumulative doses of UVB and reducing the possible carcinogenic and phototoxic risks of whole-body phototherapy.32
One study examined the antipruritic effects of the excimer laser following the treatment of epidermal hyperinnervation leading to intractable pruritus in patients with atopic dermatitis. The researchers suggested that a potential explanation for the antipruritic effect of the excimer laser may be secondary to nerve degeneration.33 Additionally, low doses of UVB light also may inhibit mast cell degranulation and prevent histamine release, further supporting the antipruritic properties of excimer laser.34
In our patient, failed treatment with other modalities led to trial of excimer laser therapy because of the overlapping clinical and histopathologic findings with psoriasis. Excimer laser improved the clinical appearance and overall texture of the ILVEN lesions and decreased pruritus. The reasons for treatment success may be two-fold. By decreasing the number of keratinocytes and mast cells, the excimer laser may have improved the epidermal hyperplasia and pruritus in the ILVEN lesions. Alternatively, because the patient had ILVEN lesions since infancy, psoriasis may have developed in the location of the ILVEN lesions due to koebnerization, resulting in the clinical response to excimer therapy; however, she had no other clinical evidence of psoriasis.
Because of the recalcitrance of ILVEN lesions to conventional therapies, it is important to investigate therapies that may be of possible benefit. Our novel case documents successful use of the excimer laser in the treatment of ILVEN.
Conclusion
Our case of ILVEN in a woman that had been present since infancy highlights the disease pathology as well as a potential new treatment modality. The patient was refractory to first-line treatments and was concerned about the cosmetic appearance of the lesions. The patient was subsequently treated with a trial of a 308-nm excimer laser with clinical improvement of the lesions. It is possible that the similarity of ILVEN and psoriasis may have contributed to the clinical improvement in our patient, but the mechanism of action remains unknown. Due to the paucity of evidence regarding optimal treatment of ILVEN, the current case offers dermatologists an option for patients who are refractory to other treatments.
Inflammatory linear verrucous epidermal nevus (ILVEN) is a rare entity that presents with linear and pruritic psoriasiform plaques and most commonly occurs during childhood. It represents a dysregulation of keratinocytes exhibiting genetic mosaicism.1,2 Epidermal nevi may derive from keratinocytic, follicular, sebaceous, apocrine, or eccrine origin. Inflammatory linear verrucous epidermal nevus is classified under the keratinocytic type of epidermal nevus and represents approximately 6% of all epidermal nevi.3 The condition presents as erythematous and verrucous plaques along the lines of Blaschko.2,4 There is a predilection for the legs, and girls are 4 times more commonly affected than boys.1 Cases of ILVEN are predominantly sporadic, though rare familial cases have been reported.4
Inflammatory linear verrucous epidermal nevus is notoriously refractory to treatment. First-line therapies include topical agents such as corticosteroids, calcipotriol, retinoids, and 5-fluorouracil.3,4 Other treatments include intralesional corticosteroids, cryotherapy, electrodesiccation and curettage, and surgical excision.3 Several case reports have shown promising results using the pulsed dye and ablative CO2 lasers.5-8
Case Report
An otherwise healthy 20-year-old woman presented with dry, pruritic, red lesions on the right leg that had been present and stable since she was an infant (2 weeks of age). Her medical history included acne vulgaris, but she denied any personal or family history of psoriasis as well as any arthralgia or arthritis. Physical examination revealed discrete, oval, hyperkeratotic, scaly, red plaques on the lateral right leg with a larger hyperkeratotic, linear, red plaque extending from the right popliteal fossa to the posterior thigh (Figure 1A). The nails, scalp, buttocks, and upper extremities were unaffected. Bacterial culture of the right leg demonstrated Staphylococcus aureus colonization. Biopsy of the right popliteal fossa showed psoriasiform dermatitis with psoriasiform hyperplasia, a slightly verruciform surface, broad zones of superficial pallor, and parakeratosis with conspicuous colonies of bacteria (Figure 2).
Following the positive bacterial culture, the patient was treated with a short course of oral doxycycline, which did not alter the clinical appearance of the lesions or improve symptoms of pruritus. Pruritus improved moderately with topical corticosteroid treatment, but clinically the lesions appeared unchanged. The plaque on the superior right leg was treated with a superpulsed CO2 laser and the plaque on the inferior right leg was treated with a fractional CO2 laser, both with minimal improvement.
Because of the clinical and histopathologic similarities of the patient's lesions to psoriasis, a trial of the UV 308-nm excimer laser was initiated. Following initial test spots, she completed a total of 18 treatments to all lesions with noticeable clinical improvement (Figure 1B). Initially, the patient returned for treatment biweekly for approximately 5 weeks with 2 small spots being targeted at each session, with an average surface area of approximately 16 cm2. She was started at 225 mJ/cm2 with 25% increases at each session and ultimately reached up to 1676 mJ/cm2 at the end of the 10 sessions. She tolerated the procedure well with some minor blistering. Treatment was deferred for 3 months due to the patient's schedule, then biweekly treatments resumed for 4 weeks, totaling 8 more sessions. At that time, all lesions on the right leg were targeted, with an average surface area of approximately 100 cm2. The laser settings were initiated at 225 mJ/cm2 with 20% increases at each session and ultimately reached 560 mJ/cm2. The treatment was well tolerated throughout; however, the patient initially reported residual pruritus. The plaques continued to improve, and most notably, there was thinning of the hyperkeratotic scale of the plaques in addition to decreased erythema and complete resolution of pruritus. Ultimately, treatment was discontinued because of lack of insurance coverage and financial burden. The patient was lost to follow-up.
Comment
Presentation
Inflammatory linear verrucous epidermal nevus is a rare type of keratinocytic epidermal nevus4 that clinically presents as small, discrete, pruritic, scaly plaques coalescing into a linear plaque along the lines of Blaschko.9 Considerable pruritus and resistance to treatment are hallmarks of the disease.10 Histopathologically, ILVEN is characterized by alternating orthokeratosis and parakeratosis with a lack of neutrophils in an acanthotic epidermis.11-13 Inflammatory linear verrucous epidermal nevus presents at birth or in early childhood. Adult onset is rare.9,14 Approximately 75% of lesions present by 5 years of age, with a majority occurring within the first 6 months of life.15 The differential diagnosis includes linear psoriasis, epidermal nevi, linear lichen planus, linear verrucae, linear lichen simplex chronicus, and mycosis fungoides.4,11
Differentiation From Psoriasis
Despite the histopathologic overlap with psoriasis, ILVEN exhibits fewer Ki-67-positive keratinocyte nuclei (proliferative marker) and more cytokeratin 10-positive cells (epidermal differentiation marker) than psoriasis.16 Furthermore, ILVEN has demonstrated fewer CD4−, CD8−, CD45RO−, CD2−, CD25−, CD94−, and CD161+ cells within the dermis and epidermis than psoriasis.16
The clinical presentations of ILVEN and psoriasis may be similar, as some patients with linear psoriasis also present with psoriatic plaques along the lines of Blaschko.17 Additionally, ILVEN may be a precursor to psoriasis. Altman and Mehregan1 found that ILVEN patients who developed psoriasis did so in areas previously affected by ILVEN; however, they continued to distinguish the 2 pathologies as distinct entities. Another early report also hypothesized that the dermoepidermal defect caused by epidermal nevi provided a site for the development of psoriatic lesions because of the Koebner phenomenon.18
Patients with ILVEN also have been found to have extracutaneous manifestations and symptoms commonly seen in psoriasis patients. A 2012 retrospective review revealed that 37% (7/19) of patients with ILVEN also had psoriatic arthritis, cutaneous psoriatic lesions, and/or nail pitting. The authors concluded that ILVEN may lead to the onset of psoriasis later in life and may indicate an underlying psoriatic predisposition.19 Genetic theories also have been proposed, stating that ILVEN may be a mosaic of psoriasis2 or that a postzygotic mutation leads to the predisposition for developing psoriasis.20
Treatment
Inflammatory linear verrucous epidermal nevus frequently is refractory to treatment; however, the associated pruritus and distressing cosmesis make treatment attempts worthwhile.11 No single therapy has been found to be successful in all patients. A widely used first-line treatment is topical or intralesional corticosteroids, with the former typically used with occlusion.13 Other treatments include adalimumab, calcipotriol,22,23 tretinoin,24 and 5-fluorouracil.24 Physical modalities such as cryotherapy, electrodesiccation, and dermabrasion have been reported with varying success.15,24 Surgical treatments include tangential25 and full-thickness excisions.26
The CO2 laser also has demonstrated success. One study showed considerable improvement of pruritus and partial resolution of lesions only 5 weeks following a single CO2 laser treatment.5 Another study showed promising results when combining CO2 pulsed laser therapy with fractional CO2 laser treatment.6 Other laser therapies including the argon27 and flashlamp-pumped pulsed dye lasers8 have been used with limited success. The use of light therapy and lasers in psoriasis have now increased the treatment options for ILVEN based on the rationale of their shared histopathologic characteristics. Photodynamic therapy also has been attempted because of its successful use in psoriasis patients. It has been found to be successful in diminishing ILVEN lesions and associated pruritus after a few weeks of therapy; however, treatment is limited by the associated pain and requirement for local anesthesia.28
The excimer laser is a form of targeted phototherapy that emits monochromatic light at 308 nm.29 It is ideal for inflammatory skin lesions because the UVB light induces apoptosis.30 Psoriasis lesions treated with the excimer laser show a decrease in keratinocyte proliferation, which in turn reverses epidermal acanthosis and causes T-cell depletion due to upregulation of p53.29,31 This mechanism of action addresses the overproliferation of keratinocytes mediated by T cells in psoriasis and contributes to the success of excimer laser treatment.31 A considerable advantage is its localized treatment, resulting in lower cumulative doses of UVB and reducing the possible carcinogenic and phototoxic risks of whole-body phototherapy.32
One study examined the antipruritic effects of the excimer laser following the treatment of epidermal hyperinnervation leading to intractable pruritus in patients with atopic dermatitis. The researchers suggested that a potential explanation for the antipruritic effect of the excimer laser may be secondary to nerve degeneration.33 Additionally, low doses of UVB light also may inhibit mast cell degranulation and prevent histamine release, further supporting the antipruritic properties of excimer laser.34
In our patient, failed treatment with other modalities led to trial of excimer laser therapy because of the overlapping clinical and histopathologic findings with psoriasis. Excimer laser improved the clinical appearance and overall texture of the ILVEN lesions and decreased pruritus. The reasons for treatment success may be two-fold. By decreasing the number of keratinocytes and mast cells, the excimer laser may have improved the epidermal hyperplasia and pruritus in the ILVEN lesions. Alternatively, because the patient had ILVEN lesions since infancy, psoriasis may have developed in the location of the ILVEN lesions due to koebnerization, resulting in the clinical response to excimer therapy; however, she had no other clinical evidence of psoriasis.
Because of the recalcitrance of ILVEN lesions to conventional therapies, it is important to investigate therapies that may be of possible benefit. Our novel case documents successful use of the excimer laser in the treatment of ILVEN.
Conclusion
Our case of ILVEN in a woman that had been present since infancy highlights the disease pathology as well as a potential new treatment modality. The patient was refractory to first-line treatments and was concerned about the cosmetic appearance of the lesions. The patient was subsequently treated with a trial of a 308-nm excimer laser with clinical improvement of the lesions. It is possible that the similarity of ILVEN and psoriasis may have contributed to the clinical improvement in our patient, but the mechanism of action remains unknown. Due to the paucity of evidence regarding optimal treatment of ILVEN, the current case offers dermatologists an option for patients who are refractory to other treatments.
- Altman J, Mehregan AH. Inflammatory linear verrucose epidermal nevus. Arch Dermatol. 1971;104:385-389.
- Hofer T. Does inflammatory linear verrucous epidermal nevus represent a segmental type 1/type 2 mosaic of psoriasis? Dermatology. 2006;212:103-107.
- Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome: a review of 131 cases. J Am Acad Dermatol. 1989;20:476-488.
- Khachemoune A, Janjua S, Guldbakke K. Inflammatory linear verrucous epidermal nevus: a case report and short review of the literature. Cutis. 2006;78:261-267.
- Ulkur E, Celikoz B, Yuksel F, et al. Carbon dioxide laser therapy for an inflammatory linear verrucous epidermal nevus: a case report. Aesthetic Plast Surg. 2004;28:428-430.
- Conti R, Bruscino N, Campolmi P, et al. Inflammatory linear verrucous epidermal nevus: why a combined laser therapy. J Cosmet Laser Ther. 2013;15:242-245.
- Alonso-Castro L, Boixeda P, Reig I, et al. Carbon dioxide laser treatment of epidermal nevi: response and long-term follow-up. Actas Dermosifiliogr. 2012;103:910-918.
- Alster TS. Inflammatory linear verrucous epidermal nevus: successful treatment with the 585 nm flashlamp-pumped dye laser. J Am Acad Dermatol. 1994;31:513-514.
- Kruse LL. Differential diagnosis of linear eruptions in children. Pediatr Ann. 2015;44:194-198.
- Renner R, Colsman A, Sticherling M. ILVEN: is it psoriasis? debate based on successful treatment with etanercept. Acta Derm Venereol. 2008;88:631-632.
- Lee SH, Rogers M. Inflammatory linear verrucous epidermal naevi: a review of 23 cases. Australas J Dermatol. 2001;42:252-256.
- Ito M, Shimizu N, Fujiwara H, et al. Histopathogenesis of inflammatory linear verrucose epidermal nevus: histochemistry, immunohistochemistry and ultrastructure. Arch Dermatol Res. 1991;283:491-499.
- Cerio R, Jones EW, Eady RA. ILVEN responding to occlusive potent topical steroid therapy. Clin Exp Dermatol. 1992;17:279-281.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus. J Dermatol. 1999;26:599-602.
- Behera B, Devi B, Nayak BB, et al. Giant inflammatory linear verrucous epidermal nevus: successfully treated with full thickness excision and skin grafting. Indian J Dermatol. 2013;58:461-463.
- Vissers WH, Muys L, Erp PE, et al. Immunohistochemical differentiation between ILVEN and psoriasis. Eur J Dermatol. 2004;14:216-220.
- Agarwal US, Besarwal RK, Gupta R, et a. Inflammatory linear verrucous epidermal nevus with psoriasiform histology. Indian J Dermatol. 2014;59:211.
- Bennett RG, Burns L, Wood MG. Systematized epidermal nevus: a determinant for the localization of psoriasis. Arch Dermatol. 1973;108:705-757.
- Tran K, Jao-Tan C, Ho N. ILVEN and psoriasis: a retrospective study among pediatric patients. J Am Acad Dermatol. 2012;66(suppl 1):AB163.
- Happle R. Superimposed linear psoriasis: a historical case revisited. J Dtsch Dermatol Ges. 2011;9:1027-1028; discussion 1029.
- Özdemir M, Balevi A, Esen H. An inflammatory verrucous epidermal nevus concomitant with psoriasis: treatment with adalimumab. Dermatol Online J. 2012;18:11.
- Zvulunov A, Grunwald MH, Halvy S. Topical calcipotriol for treatment of inflammatory linear verrucous epidermal nevus. Arch Dermatol. 1997;133:567-568.
- Gatti S, Carrozzo AM, Orlandi A, et al. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132:837-839.
- Fox BJ, Lapins NA. Comparison of treatment modalities for epidermal nevus: a case report and review. J Dermatol Surg Oncol. 1983;9:879-885.
- Pilanci O, Tas B, Ceran F, et al. A novel technique used in the treatment of inflammatory linear verrucous epidermal nevus: tangential excision. Aesthetic Plast Surg. 2014;38:1066-1067.
- Lee BJ, Mancini AJ, Renucci J, et al. Full-thickness surgical excision for the treatment of inflammatory linear verrucous epidermal nevus. Ann Plast Surg. 2001;47:285-292.
- Hohenleutner U, Landthaler M. Laser therapy of verrucous epidermal naevi. Clin Exp Dermatol. 1993;18:124-127.
- Parera E, Gallardo F, Toll A, et al. Inflammatory linear verrucous epidermal nevus successfully treated with methyl-aminolevulinate photodynamic therapy. Dermatol Surg. 2010;36:253-256.
- Situm M, Bulat V, Majcen K, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253.
- Beggs S, Short J, Rengifo-Pardo M, et al. Applications of the excimer laser: a review. Dermatol Surg. 2015;41:1201-1211.
- Bianchi B, Campolmi P, Mavilia L, et al. Monochromatic excimer light (308 nm): an immunohistochemical study of cutaneous T cells and apoptosis-related molecules in psoriasis. J Eur Acad Dermatol Venereol. 2003;17:408-413.
- Mudigonda T, Dabade TS, Feldman SR. A review of targeted ultraviolet B phototherapy for psoriasis. J Am Acad Dermatol. 2012;66:664-672.
- Kamo A, Tominaga M, Kamata Y, et al. The excimer lamp induces cutaneous nerve degeneration and reduces scratching in a dry-skin mouse model. J Invest Dermatol. 2014;134:2977-2984.
- Bulat V, Majcen K, Dzapo A, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253
- Altman J, Mehregan AH. Inflammatory linear verrucose epidermal nevus. Arch Dermatol. 1971;104:385-389.
- Hofer T. Does inflammatory linear verrucous epidermal nevus represent a segmental type 1/type 2 mosaic of psoriasis? Dermatology. 2006;212:103-107.
- Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome: a review of 131 cases. J Am Acad Dermatol. 1989;20:476-488.
- Khachemoune A, Janjua S, Guldbakke K. Inflammatory linear verrucous epidermal nevus: a case report and short review of the literature. Cutis. 2006;78:261-267.
- Ulkur E, Celikoz B, Yuksel F, et al. Carbon dioxide laser therapy for an inflammatory linear verrucous epidermal nevus: a case report. Aesthetic Plast Surg. 2004;28:428-430.
- Conti R, Bruscino N, Campolmi P, et al. Inflammatory linear verrucous epidermal nevus: why a combined laser therapy. J Cosmet Laser Ther. 2013;15:242-245.
- Alonso-Castro L, Boixeda P, Reig I, et al. Carbon dioxide laser treatment of epidermal nevi: response and long-term follow-up. Actas Dermosifiliogr. 2012;103:910-918.
- Alster TS. Inflammatory linear verrucous epidermal nevus: successful treatment with the 585 nm flashlamp-pumped dye laser. J Am Acad Dermatol. 1994;31:513-514.
- Kruse LL. Differential diagnosis of linear eruptions in children. Pediatr Ann. 2015;44:194-198.
- Renner R, Colsman A, Sticherling M. ILVEN: is it psoriasis? debate based on successful treatment with etanercept. Acta Derm Venereol. 2008;88:631-632.
- Lee SH, Rogers M. Inflammatory linear verrucous epidermal naevi: a review of 23 cases. Australas J Dermatol. 2001;42:252-256.
- Ito M, Shimizu N, Fujiwara H, et al. Histopathogenesis of inflammatory linear verrucose epidermal nevus: histochemistry, immunohistochemistry and ultrastructure. Arch Dermatol Res. 1991;283:491-499.
- Cerio R, Jones EW, Eady RA. ILVEN responding to occlusive potent topical steroid therapy. Clin Exp Dermatol. 1992;17:279-281.
- Kawaguchi H, Takeuchi M, Ono H, et al. Adult onset of inflammatory linear verrucous epidermal nevus. J Dermatol. 1999;26:599-602.
- Behera B, Devi B, Nayak BB, et al. Giant inflammatory linear verrucous epidermal nevus: successfully treated with full thickness excision and skin grafting. Indian J Dermatol. 2013;58:461-463.
- Vissers WH, Muys L, Erp PE, et al. Immunohistochemical differentiation between ILVEN and psoriasis. Eur J Dermatol. 2004;14:216-220.
- Agarwal US, Besarwal RK, Gupta R, et a. Inflammatory linear verrucous epidermal nevus with psoriasiform histology. Indian J Dermatol. 2014;59:211.
- Bennett RG, Burns L, Wood MG. Systematized epidermal nevus: a determinant for the localization of psoriasis. Arch Dermatol. 1973;108:705-757.
- Tran K, Jao-Tan C, Ho N. ILVEN and psoriasis: a retrospective study among pediatric patients. J Am Acad Dermatol. 2012;66(suppl 1):AB163.
- Happle R. Superimposed linear psoriasis: a historical case revisited. J Dtsch Dermatol Ges. 2011;9:1027-1028; discussion 1029.
- Özdemir M, Balevi A, Esen H. An inflammatory verrucous epidermal nevus concomitant with psoriasis: treatment with adalimumab. Dermatol Online J. 2012;18:11.
- Zvulunov A, Grunwald MH, Halvy S. Topical calcipotriol for treatment of inflammatory linear verrucous epidermal nevus. Arch Dermatol. 1997;133:567-568.
- Gatti S, Carrozzo AM, Orlandi A, et al. Treatment of inflammatory linear verrucous epidermal naevus with calcipotriol. Br J Dermatol. 1995;132:837-839.
- Fox BJ, Lapins NA. Comparison of treatment modalities for epidermal nevus: a case report and review. J Dermatol Surg Oncol. 1983;9:879-885.
- Pilanci O, Tas B, Ceran F, et al. A novel technique used in the treatment of inflammatory linear verrucous epidermal nevus: tangential excision. Aesthetic Plast Surg. 2014;38:1066-1067.
- Lee BJ, Mancini AJ, Renucci J, et al. Full-thickness surgical excision for the treatment of inflammatory linear verrucous epidermal nevus. Ann Plast Surg. 2001;47:285-292.
- Hohenleutner U, Landthaler M. Laser therapy of verrucous epidermal naevi. Clin Exp Dermatol. 1993;18:124-127.
- Parera E, Gallardo F, Toll A, et al. Inflammatory linear verrucous epidermal nevus successfully treated with methyl-aminolevulinate photodynamic therapy. Dermatol Surg. 2010;36:253-256.
- Situm M, Bulat V, Majcen K, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253.
- Beggs S, Short J, Rengifo-Pardo M, et al. Applications of the excimer laser: a review. Dermatol Surg. 2015;41:1201-1211.
- Bianchi B, Campolmi P, Mavilia L, et al. Monochromatic excimer light (308 nm): an immunohistochemical study of cutaneous T cells and apoptosis-related molecules in psoriasis. J Eur Acad Dermatol Venereol. 2003;17:408-413.
- Mudigonda T, Dabade TS, Feldman SR. A review of targeted ultraviolet B phototherapy for psoriasis. J Am Acad Dermatol. 2012;66:664-672.
- Kamo A, Tominaga M, Kamata Y, et al. The excimer lamp induces cutaneous nerve degeneration and reduces scratching in a dry-skin mouse model. J Invest Dermatol. 2014;134:2977-2984.
- Bulat V, Majcen K, Dzapo A, et al. Benefits of controlled ultraviolet radiation in the treatment of dermatological diseases. Coll Antropol. 2014;38:1249-1253
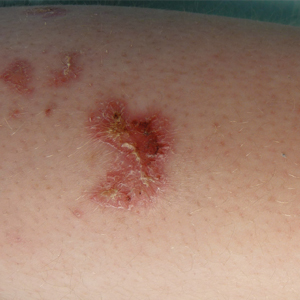
Pigmented Lesion on the Forearm
The Diagnosis: Monsel Solution Reaction
Exogenous substances can cause interesting incongruities in cutaneous biopsies of which pathologists and dermatologists should be cognizant. Exogenous lesions are caused by externally introduced foreign bodies, substances, or materials, such as sterile compressed sponges, aluminum chloride hexahydrate and anhydrous ethyl alcohol, silica, paraffin, and Monsel solution. Monsel solution reaction is a florid fibrohistiocytic proliferation stimulated by the application of Monsel solution. Monsel solution is a ferric subsulfate that often is used to achieve hemostasis after shave biopsies. Hemostasis is thought to result from the ability of ferric ions to denature and agglutinate proteins such as fibrinogen.1,2 Application of Monsel solution likely causes ferrugination of fibrin, dermal collagen, and striated muscle fibers. Some ferruginated collagen fibers are eliminated through the epidermis as the epidermis regenerates, while some fibers become calcified. Siderophages (iron-containing macrophages) are present in these areas. The ferrugination of collagen fibers becomes less pronounced as the biopsy sites heal and the iron pigment subsequently is absorbed by macrophages. Ferruginated skeletal muscles can act as foreign bodies and may elicit granulomatous reactions.2
It is currently unclear why fibrohistiocytic responses occur in some instances but not others. Iron stains (eg, Perls Prussian blue stain) make interpretation clear, provided the pathologist is familiar with Monsel solution. The primary differential diagnosis of these lesions centers on heavily pigmented melanocytic proliferations. It is critical to review prior biopsy sections or to have definite knowledge of the prior biopsy diagnosis. Histologically, the epidermis may demonstrate nonspecific reactive changes such as hyperkeratosis with foci of irregular acanthosis. The prominent features are present in the dermis where there is a proliferation of spindle- and polyhedral-shaped cells that may show cytologic atypia and occasional mitotic figures. The cells contain refractile brown pigment scattered in the dermis and deposited on collagen fibers (quiz images). Occasional large black or brown encrustations may be identified. Monsel-containing cells may indiscernibly blend with foci of more blatantly fibrohistiocytic differentiation, in which case iron stains are strongly positive (Figure 1). If the clinician uses Monsel solution for hemostasis during the removal of a nevomelanocytic neoplasm, it might be necessary to use melanin stains or immunohistochemistry on the reexcision specimen to distinguish between residual nevomelanocytic and fibrohistiocytic cells.3
.")
Common blue nevus is a benign, typically intradermal melanocytic lesion. It most frequently occurs in young adults and has a predilection for females. Clinically, it can be found anywhere on the body as a single, asymptomatic, well-circumscribed, blue-black, dome-shaped papule measuring less than 1 cm in diameter. Histologically, it is characterized by pigmented, dendritic, spindle-shaped melanocytes that typically are separated by thick collagen bundles (Figure 2). The melanocytes typically have small nuclei with occasional basophilic nucleolus. Melanocytes typically are diffusely positive for melanocytic markers including human melanoma black (HMB) 45, S-100, Melan-A, and microphthalmia transcription factor 1. In contrast to most other benign melanocytic nevi, HMB-45 strongly stains the entire lesion in blue nevi.4
.")
Desmoplastic melanoma accounts for 1% to 4% of all melanomas. The median age at diagnosis is 62 years and, as in other types of melanoma, men are more commonly affected.5 Clinically, desmoplastic melanoma typically presents on the head and neck as a painless indurated plaque, though it can present as a small papule or nodule. Nearly half of desmoplastic melanomas lack obvious pigmentation, which may lead to the misdiagnosis of basal cell carcinoma or a scar. Histologically, desmoplastic melanomas are composed of spindled melanocytes separated by collagen fibers or fibrous stroma (Figure 3). Histology displays variable cytologic atypia and stromal fibrosis. Characteristically there are small islands of lymphocytes and plasma cells within or at the edge of the tumor. The spindle cells stain positive with S-100 and Sry-related HMg-box gene 10, SOX10. Type IV collagen and laminin often are expressed in desmoplastic melanoma. In contrast to many other subtypes of melanoma, HMB-45 and Melan-A usually are negative.6
.")
Animal-type melanoma is a rare neoplasm that differs from other subtypes of melanoma both clinically and histologically. Most frequently, animal-type melanoma affects younger adults (median age, 27 years) and arises on the arms and legs, head and neck, or trunk; men and women are affected equally.7 It most commonly presents with a blue or blue-black nodule with a blue-white veil or irregular white areas. Histologically, animal-type melanoma is a predominantly dermal-based melanocytic proliferation with heavily pigmented epithelioid and spindled melanocytes (Figure 4). The pigmentation pattern ranges widely from fine, granular, light brown deposits to coarse dark brown deposits with malignant cells often arranged in fascicles or sheets. Frequently, there is periadnexal and perieccrine spread. Often, there is epidermal hyperplasia above the dermis. As with conventional melanoma, the immunohistochemistry of animal-type melanoma is positive for S-100 protein, HMB-45, SOX10, and Melan-A.7
.")
Recurrent nevi typically arise within 6 months of a previously biopsied melanocytic nevus. Most recurrent nevi originate from common banal nevi (most often a compound nevus). Recurrent nevi also may arise from congenital, atypical/dysplastic, and Spitz nevi. Most often they are found on the back of women aged 20 to 30 years.8 Clinically, they manifest as a macular area of scar with variegated hyperpigmentation and hypopigmentation as well as linear streaking. They may demonstrate variable diffuse, stippled, and halo pigmentation patterns. Classically, recurrent nevi present with a trizonal histologic pattern. Within the epidermis there is a proliferation of melanocytes along the dermoepidermal junction, which may show varying degrees of atypia and pagetoid migration. The melanocytes often are described as epithelioid with round nuclei and even chromatin (Figure 5). The atypical features should be confined to the epidermis overlying the prior biopsy site. Within the dermis there is dense dermal collagen and fibrosis with vertically oriented blood vessels. Finally, features of the original nevus may be seen at the base of the lesion. Although immunohistochemistry may be helpful in some cases, an appropriate clinical history and comparison to the prior biopsy can be invaluable.8
.")
Host tissue reactions resulting in artefactual changes caused by foreign bodies or substances may confound the untrained eye. Monsel solution reaction may be confused for a blue nevus, desmoplastic melanoma, animal-type melanoma, and a residual/recurrent nevus. This confusion could lead to serious diagnostic errors that could cause an unfavorable outcome for the patient. It is critical to know the salient points in the patient's clinical history. Knowledge of the Monsel solution reaction and other exogenous lesions as well as the subsequent unique tissue reaction patterns can aid in facilitating an accurate and prompt pathologic diagnosis.
- Olmstead PM, Lund HZ, Leonard DD. Monsel's solution: a histologic nuisance. J Am Acad Dermatol. 1980;3:492-498.
- Amazon K, Robinson MJ, Rywlin AM. Ferrugination caused by Monsel's solution. clinical observations and experimentations. Am J Dermatopathol. 1980;2:197-205.
- Del Rosario RN, Barr RJ, Graham BS, et al. Exogenous and endogenous cutaneous anomalies and curiosities. Am J Dermatopathol. 2005;27:259-267.
- Calonje E, Blessing K, Glusac E, et al. Blue naevi. In: LeBoit PE, Burg G, Weedon D, et al, eds. World Health Organization Classification of Tumours, Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006:95-99.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- McCarthy SW, Crotty KA, Scolyer RA. Desmoplastic melanoma and desmoplastic neurotropic melanoma. In: LeBoit PE, Burg G, Weedon D, et al, eds. World Health Organization Classification of Tumours, Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006:76-78.
- Vyas R, Keller JJ, Honda K, et al. A systematic review and meta-analysis of animal-type melanoma. J Am Acad Dermatol. 2015;73:1031-1039.
- Fox JC, Reed JA, Shea CR. The recurrent nevus phenomenon: a history of challenge, controversy, and discovery. Arch Pathol Lab Med. 2011;135:842-846.
The Diagnosis: Monsel Solution Reaction
Exogenous substances can cause interesting incongruities in cutaneous biopsies of which pathologists and dermatologists should be cognizant. Exogenous lesions are caused by externally introduced foreign bodies, substances, or materials, such as sterile compressed sponges, aluminum chloride hexahydrate and anhydrous ethyl alcohol, silica, paraffin, and Monsel solution. Monsel solution reaction is a florid fibrohistiocytic proliferation stimulated by the application of Monsel solution. Monsel solution is a ferric subsulfate that often is used to achieve hemostasis after shave biopsies. Hemostasis is thought to result from the ability of ferric ions to denature and agglutinate proteins such as fibrinogen.1,2 Application of Monsel solution likely causes ferrugination of fibrin, dermal collagen, and striated muscle fibers. Some ferruginated collagen fibers are eliminated through the epidermis as the epidermis regenerates, while some fibers become calcified. Siderophages (iron-containing macrophages) are present in these areas. The ferrugination of collagen fibers becomes less pronounced as the biopsy sites heal and the iron pigment subsequently is absorbed by macrophages. Ferruginated skeletal muscles can act as foreign bodies and may elicit granulomatous reactions.2
It is currently unclear why fibrohistiocytic responses occur in some instances but not others. Iron stains (eg, Perls Prussian blue stain) make interpretation clear, provided the pathologist is familiar with Monsel solution. The primary differential diagnosis of these lesions centers on heavily pigmented melanocytic proliferations. It is critical to review prior biopsy sections or to have definite knowledge of the prior biopsy diagnosis. Histologically, the epidermis may demonstrate nonspecific reactive changes such as hyperkeratosis with foci of irregular acanthosis. The prominent features are present in the dermis where there is a proliferation of spindle- and polyhedral-shaped cells that may show cytologic atypia and occasional mitotic figures. The cells contain refractile brown pigment scattered in the dermis and deposited on collagen fibers (quiz images). Occasional large black or brown encrustations may be identified. Monsel-containing cells may indiscernibly blend with foci of more blatantly fibrohistiocytic differentiation, in which case iron stains are strongly positive (Figure 1). If the clinician uses Monsel solution for hemostasis during the removal of a nevomelanocytic neoplasm, it might be necessary to use melanin stains or immunohistochemistry on the reexcision specimen to distinguish between residual nevomelanocytic and fibrohistiocytic cells.3
Common blue nevus is a benign, typically intradermal melanocytic lesion. It most frequently occurs in young adults and has a predilection for females. Clinically, it can be found anywhere on the body as a single, asymptomatic, well-circumscribed, blue-black, dome-shaped papule measuring less than 1 cm in diameter. Histologically, it is characterized by pigmented, dendritic, spindle-shaped melanocytes that typically are separated by thick collagen bundles (Figure 2). The melanocytes typically have small nuclei with occasional basophilic nucleolus. Melanocytes typically are diffusely positive for melanocytic markers including human melanoma black (HMB) 45, S-100, Melan-A, and microphthalmia transcription factor 1. In contrast to most other benign melanocytic nevi, HMB-45 strongly stains the entire lesion in blue nevi.4
Desmoplastic melanoma accounts for 1% to 4% of all melanomas. The median age at diagnosis is 62 years and, as in other types of melanoma, men are more commonly affected.5 Clinically, desmoplastic melanoma typically presents on the head and neck as a painless indurated plaque, though it can present as a small papule or nodule. Nearly half of desmoplastic melanomas lack obvious pigmentation, which may lead to the misdiagnosis of basal cell carcinoma or a scar. Histologically, desmoplastic melanomas are composed of spindled melanocytes separated by collagen fibers or fibrous stroma (Figure 3). Histology displays variable cytologic atypia and stromal fibrosis. Characteristically there are small islands of lymphocytes and plasma cells within or at the edge of the tumor. The spindle cells stain positive with S-100 and Sry-related HMg-box gene 10, SOX10. Type IV collagen and laminin often are expressed in desmoplastic melanoma. In contrast to many other subtypes of melanoma, HMB-45 and Melan-A usually are negative.6
Animal-type melanoma is a rare neoplasm that differs from other subtypes of melanoma both clinically and histologically. Most frequently, animal-type melanoma affects younger adults (median age, 27 years) and arises on the arms and legs, head and neck, or trunk; men and women are affected equally.7 It most commonly presents with a blue or blue-black nodule with a blue-white veil or irregular white areas. Histologically, animal-type melanoma is a predominantly dermal-based melanocytic proliferation with heavily pigmented epithelioid and spindled melanocytes (Figure 4). The pigmentation pattern ranges widely from fine, granular, light brown deposits to coarse dark brown deposits with malignant cells often arranged in fascicles or sheets. Frequently, there is periadnexal and perieccrine spread. Often, there is epidermal hyperplasia above the dermis. As with conventional melanoma, the immunohistochemistry of animal-type melanoma is positive for S-100 protein, HMB-45, SOX10, and Melan-A.7
Recurrent nevi typically arise within 6 months of a previously biopsied melanocytic nevus. Most recurrent nevi originate from common banal nevi (most often a compound nevus). Recurrent nevi also may arise from congenital, atypical/dysplastic, and Spitz nevi. Most often they are found on the back of women aged 20 to 30 years.8 Clinically, they manifest as a macular area of scar with variegated hyperpigmentation and hypopigmentation as well as linear streaking. They may demonstrate variable diffuse, stippled, and halo pigmentation patterns. Classically, recurrent nevi present with a trizonal histologic pattern. Within the epidermis there is a proliferation of melanocytes along the dermoepidermal junction, which may show varying degrees of atypia and pagetoid migration. The melanocytes often are described as epithelioid with round nuclei and even chromatin (Figure 5). The atypical features should be confined to the epidermis overlying the prior biopsy site. Within the dermis there is dense dermal collagen and fibrosis with vertically oriented blood vessels. Finally, features of the original nevus may be seen at the base of the lesion. Although immunohistochemistry may be helpful in some cases, an appropriate clinical history and comparison to the prior biopsy can be invaluable.8
Host tissue reactions resulting in artefactual changes caused by foreign bodies or substances may confound the untrained eye. Monsel solution reaction may be confused for a blue nevus, desmoplastic melanoma, animal-type melanoma, and a residual/recurrent nevus. This confusion could lead to serious diagnostic errors that could cause an unfavorable outcome for the patient. It is critical to know the salient points in the patient's clinical history. Knowledge of the Monsel solution reaction and other exogenous lesions as well as the subsequent unique tissue reaction patterns can aid in facilitating an accurate and prompt pathologic diagnosis.
The Diagnosis: Monsel Solution Reaction
Exogenous substances can cause interesting incongruities in cutaneous biopsies of which pathologists and dermatologists should be cognizant. Exogenous lesions are caused by externally introduced foreign bodies, substances, or materials, such as sterile compressed sponges, aluminum chloride hexahydrate and anhydrous ethyl alcohol, silica, paraffin, and Monsel solution. Monsel solution reaction is a florid fibrohistiocytic proliferation stimulated by the application of Monsel solution. Monsel solution is a ferric subsulfate that often is used to achieve hemostasis after shave biopsies. Hemostasis is thought to result from the ability of ferric ions to denature and agglutinate proteins such as fibrinogen.1,2 Application of Monsel solution likely causes ferrugination of fibrin, dermal collagen, and striated muscle fibers. Some ferruginated collagen fibers are eliminated through the epidermis as the epidermis regenerates, while some fibers become calcified. Siderophages (iron-containing macrophages) are present in these areas. The ferrugination of collagen fibers becomes less pronounced as the biopsy sites heal and the iron pigment subsequently is absorbed by macrophages. Ferruginated skeletal muscles can act as foreign bodies and may elicit granulomatous reactions.2
It is currently unclear why fibrohistiocytic responses occur in some instances but not others. Iron stains (eg, Perls Prussian blue stain) make interpretation clear, provided the pathologist is familiar with Monsel solution. The primary differential diagnosis of these lesions centers on heavily pigmented melanocytic proliferations. It is critical to review prior biopsy sections or to have definite knowledge of the prior biopsy diagnosis. Histologically, the epidermis may demonstrate nonspecific reactive changes such as hyperkeratosis with foci of irregular acanthosis. The prominent features are present in the dermis where there is a proliferation of spindle- and polyhedral-shaped cells that may show cytologic atypia and occasional mitotic figures. The cells contain refractile brown pigment scattered in the dermis and deposited on collagen fibers (quiz images). Occasional large black or brown encrustations may be identified. Monsel-containing cells may indiscernibly blend with foci of more blatantly fibrohistiocytic differentiation, in which case iron stains are strongly positive (Figure 1). If the clinician uses Monsel solution for hemostasis during the removal of a nevomelanocytic neoplasm, it might be necessary to use melanin stains or immunohistochemistry on the reexcision specimen to distinguish between residual nevomelanocytic and fibrohistiocytic cells.3
Common blue nevus is a benign, typically intradermal melanocytic lesion. It most frequently occurs in young adults and has a predilection for females. Clinically, it can be found anywhere on the body as a single, asymptomatic, well-circumscribed, blue-black, dome-shaped papule measuring less than 1 cm in diameter. Histologically, it is characterized by pigmented, dendritic, spindle-shaped melanocytes that typically are separated by thick collagen bundles (Figure 2). The melanocytes typically have small nuclei with occasional basophilic nucleolus. Melanocytes typically are diffusely positive for melanocytic markers including human melanoma black (HMB) 45, S-100, Melan-A, and microphthalmia transcription factor 1. In contrast to most other benign melanocytic nevi, HMB-45 strongly stains the entire lesion in blue nevi.4
Desmoplastic melanoma accounts for 1% to 4% of all melanomas. The median age at diagnosis is 62 years and, as in other types of melanoma, men are more commonly affected.5 Clinically, desmoplastic melanoma typically presents on the head and neck as a painless indurated plaque, though it can present as a small papule or nodule. Nearly half of desmoplastic melanomas lack obvious pigmentation, which may lead to the misdiagnosis of basal cell carcinoma or a scar. Histologically, desmoplastic melanomas are composed of spindled melanocytes separated by collagen fibers or fibrous stroma (Figure 3). Histology displays variable cytologic atypia and stromal fibrosis. Characteristically there are small islands of lymphocytes and plasma cells within or at the edge of the tumor. The spindle cells stain positive with S-100 and Sry-related HMg-box gene 10, SOX10. Type IV collagen and laminin often are expressed in desmoplastic melanoma. In contrast to many other subtypes of melanoma, HMB-45 and Melan-A usually are negative.6
Animal-type melanoma is a rare neoplasm that differs from other subtypes of melanoma both clinically and histologically. Most frequently, animal-type melanoma affects younger adults (median age, 27 years) and arises on the arms and legs, head and neck, or trunk; men and women are affected equally.7 It most commonly presents with a blue or blue-black nodule with a blue-white veil or irregular white areas. Histologically, animal-type melanoma is a predominantly dermal-based melanocytic proliferation with heavily pigmented epithelioid and spindled melanocytes (Figure 4). The pigmentation pattern ranges widely from fine, granular, light brown deposits to coarse dark brown deposits with malignant cells often arranged in fascicles or sheets. Frequently, there is periadnexal and perieccrine spread. Often, there is epidermal hyperplasia above the dermis. As with conventional melanoma, the immunohistochemistry of animal-type melanoma is positive for S-100 protein, HMB-45, SOX10, and Melan-A.7
Recurrent nevi typically arise within 6 months of a previously biopsied melanocytic nevus. Most recurrent nevi originate from common banal nevi (most often a compound nevus). Recurrent nevi also may arise from congenital, atypical/dysplastic, and Spitz nevi. Most often they are found on the back of women aged 20 to 30 years.8 Clinically, they manifest as a macular area of scar with variegated hyperpigmentation and hypopigmentation as well as linear streaking. They may demonstrate variable diffuse, stippled, and halo pigmentation patterns. Classically, recurrent nevi present with a trizonal histologic pattern. Within the epidermis there is a proliferation of melanocytes along the dermoepidermal junction, which may show varying degrees of atypia and pagetoid migration. The melanocytes often are described as epithelioid with round nuclei and even chromatin (Figure 5). The atypical features should be confined to the epidermis overlying the prior biopsy site. Within the dermis there is dense dermal collagen and fibrosis with vertically oriented blood vessels. Finally, features of the original nevus may be seen at the base of the lesion. Although immunohistochemistry may be helpful in some cases, an appropriate clinical history and comparison to the prior biopsy can be invaluable.8
Host tissue reactions resulting in artefactual changes caused by foreign bodies or substances may confound the untrained eye. Monsel solution reaction may be confused for a blue nevus, desmoplastic melanoma, animal-type melanoma, and a residual/recurrent nevus. This confusion could lead to serious diagnostic errors that could cause an unfavorable outcome for the patient. It is critical to know the salient points in the patient's clinical history. Knowledge of the Monsel solution reaction and other exogenous lesions as well as the subsequent unique tissue reaction patterns can aid in facilitating an accurate and prompt pathologic diagnosis.
- Olmstead PM, Lund HZ, Leonard DD. Monsel's solution: a histologic nuisance. J Am Acad Dermatol. 1980;3:492-498.
- Amazon K, Robinson MJ, Rywlin AM. Ferrugination caused by Monsel's solution. clinical observations and experimentations. Am J Dermatopathol. 1980;2:197-205.
- Del Rosario RN, Barr RJ, Graham BS, et al. Exogenous and endogenous cutaneous anomalies and curiosities. Am J Dermatopathol. 2005;27:259-267.
- Calonje E, Blessing K, Glusac E, et al. Blue naevi. In: LeBoit PE, Burg G, Weedon D, et al, eds. World Health Organization Classification of Tumours, Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006:95-99.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- McCarthy SW, Crotty KA, Scolyer RA. Desmoplastic melanoma and desmoplastic neurotropic melanoma. In: LeBoit PE, Burg G, Weedon D, et al, eds. World Health Organization Classification of Tumours, Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006:76-78.
- Vyas R, Keller JJ, Honda K, et al. A systematic review and meta-analysis of animal-type melanoma. J Am Acad Dermatol. 2015;73:1031-1039.
- Fox JC, Reed JA, Shea CR. The recurrent nevus phenomenon: a history of challenge, controversy, and discovery. Arch Pathol Lab Med. 2011;135:842-846.
- Olmstead PM, Lund HZ, Leonard DD. Monsel's solution: a histologic nuisance. J Am Acad Dermatol. 1980;3:492-498.
- Amazon K, Robinson MJ, Rywlin AM. Ferrugination caused by Monsel's solution. clinical observations and experimentations. Am J Dermatopathol. 1980;2:197-205.
- Del Rosario RN, Barr RJ, Graham BS, et al. Exogenous and endogenous cutaneous anomalies and curiosities. Am J Dermatopathol. 2005;27:259-267.
- Calonje E, Blessing K, Glusac E, et al. Blue naevi. In: LeBoit PE, Burg G, Weedon D, et al, eds. World Health Organization Classification of Tumours, Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006:95-99.
- Jain S, Allen PW. Desmoplastic malignant melanoma and its variants. a study of 45 cases. Am J Surg Pathol. 1989;13:358-373.
- McCarthy SW, Crotty KA, Scolyer RA. Desmoplastic melanoma and desmoplastic neurotropic melanoma. In: LeBoit PE, Burg G, Weedon D, et al, eds. World Health Organization Classification of Tumours, Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006:76-78.
- Vyas R, Keller JJ, Honda K, et al. A systematic review and meta-analysis of animal-type melanoma. J Am Acad Dermatol. 2015;73:1031-1039.
- Fox JC, Reed JA, Shea CR. The recurrent nevus phenomenon: a history of challenge, controversy, and discovery. Arch Pathol Lab Med. 2011;135:842-846.


A 67-year-old man presented to the dermatology clinic with a 2-cm pigmented lesion on the forearm. An excisional biopsy was obtained.
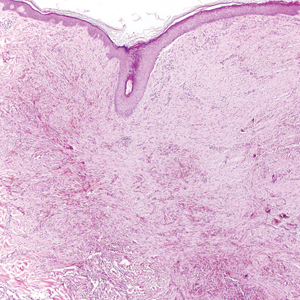
Solitary Nodule on the Proximal Nail Fold
The Diagnosis: Superficial Acral Fibromyxoma
A shave biopsy revealed an uninvolved grenz zone and mildly cellular spindle cell dermal proliferation in a collagenous and myxoid background (Figure 1). Spindle cells were seen in a myxoid background among dense coarse collagen (Figure 2A). Spindle cells also were seen in a myxoid background with mast cells and capillary network (Figure 2B). Histopathologic examination of the biopsy specimen revealed spindle cells that were diffusely positive for CD34 (Figure 3); focally positive for epithelial membrane antigen; and negative for melanocytic markers, smooth muscle markers, and cytokeratin. A diagnosis of superficial acral fibromyxoma (SAFM) was made based on clinical, histopathologic, and immunohistochemical findings.
.")
 as well as with mast cells and capillary network (B)(H&E, original magnifications ×200 and ×400).")
.")
Superficial acral fibromyxomas, also known as digital fibromyxomas, are soft, slow-growing tumors that have a predilection for subungual or periungual regions of the hands and feet. Superficial acral fibromyxomas most frequently occur on the hallux and rarely occur on the ankle or leg. They can present as nodular, dome-shaped, polyploid, or verrucous masses. They can be soft to firm, gelatinous or solid, off-white to gray-white and can have fasciculate cut surfaces. Superficial acral fibromyxomas can be either painful or painless and present with a deformed nail in 9% of cases. Superficial acral fibromyxoma is a superficial lesion with frequent infiltration of the dermal collagen and subcutaneous tissue and may even erode or infiltrate into the underlying bone in rare cases.1-4 Although SAFMs are rare tumors, documented cases of SAFM have been reported at an increasing rate since the first published report by Fetsch et al2 in 2001.
Patients often delay seeking medical treatment and present with a solitary mass that has been slowly growing for months to years. In a study of 124 patients, Hollmann et al1 found that symptoms exist for a mean of 35 months and present with a small mass with a mean tumor size of 1.7 cm before biopsy or excision. Although the age range is broad, SAFM mostly affects middle-aged adults (median age, 49 years).1 Hollmann et al1 also reported a male predominance (1.3:1 ratio), and preexisting local trauma is reported in 25% of cases.2-4
The differential for SAFM should include dermatofibroma, keloid, dermatofibrosarcoma protuberans, acquired digital fibrokeratoma, infantile digital fibromatosis, neurolemmoma, sclerosing perineurioma, superficial angiomyxoma, low-grade fibromyxoid sarcoma, and acral myxoinflammatory fibroblastic sarcoma.1-4
Superficial acral fibromyxomas are composed of CD34+ spindle or stellate-shaped cells that are embedded in a myxoid and/or dense hyalinized collagenous stroma in a random or loosely fascicular growth pattern. The spindle or stellate-shaped cells in SAFMs also have been found to be focally positive for epithelial membrane antigen and CD99. Lesions have accentuated microvasculature and increased mast cells.5-8
Conservative management is reasonable, but patients presenting with persistent pain and/or local deformity should be definitively treated with complete excision and follow-up. Hollmann et al1 found that 24% of tumors recurred locally upon incomplete excision after a mean interval of 27 months. All recurrent tumors had positive margins at excision or initial biopsy.1 To date, no reports of tumors metastasizing have been documented.1-4
- Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
- Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
- Ashby-Richardson H, Rogers GS, Stadecker MJ. Superficial acral fibromyxoma: an overview. Arch Pathol Lab Med. 2011;135:1064-1066.
- Quaba O, Evans A, Al-Nafussi AA, et al. Superficial acral fibromyxoma. Br J Plast Surg. 2005;58:561-564.
- Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx: a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
- Meyerle J, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
- Kazakov DV, Mentzel T, Buro G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
The Diagnosis: Superficial Acral Fibromyxoma
A shave biopsy revealed an uninvolved grenz zone and mildly cellular spindle cell dermal proliferation in a collagenous and myxoid background (Figure 1). Spindle cells were seen in a myxoid background among dense coarse collagen (Figure 2A). Spindle cells also were seen in a myxoid background with mast cells and capillary network (Figure 2B). Histopathologic examination of the biopsy specimen revealed spindle cells that were diffusely positive for CD34 (Figure 3); focally positive for epithelial membrane antigen; and negative for melanocytic markers, smooth muscle markers, and cytokeratin. A diagnosis of superficial acral fibromyxoma (SAFM) was made based on clinical, histopathologic, and immunohistochemical findings.
Superficial acral fibromyxomas, also known as digital fibromyxomas, are soft, slow-growing tumors that have a predilection for subungual or periungual regions of the hands and feet. Superficial acral fibromyxomas most frequently occur on the hallux and rarely occur on the ankle or leg. They can present as nodular, dome-shaped, polyploid, or verrucous masses. They can be soft to firm, gelatinous or solid, off-white to gray-white and can have fasciculate cut surfaces. Superficial acral fibromyxomas can be either painful or painless and present with a deformed nail in 9% of cases. Superficial acral fibromyxoma is a superficial lesion with frequent infiltration of the dermal collagen and subcutaneous tissue and may even erode or infiltrate into the underlying bone in rare cases.1-4 Although SAFMs are rare tumors, documented cases of SAFM have been reported at an increasing rate since the first published report by Fetsch et al2 in 2001.
Patients often delay seeking medical treatment and present with a solitary mass that has been slowly growing for months to years. In a study of 124 patients, Hollmann et al1 found that symptoms exist for a mean of 35 months and present with a small mass with a mean tumor size of 1.7 cm before biopsy or excision. Although the age range is broad, SAFM mostly affects middle-aged adults (median age, 49 years).1 Hollmann et al1 also reported a male predominance (1.3:1 ratio), and preexisting local trauma is reported in 25% of cases.2-4
The differential for SAFM should include dermatofibroma, keloid, dermatofibrosarcoma protuberans, acquired digital fibrokeratoma, infantile digital fibromatosis, neurolemmoma, sclerosing perineurioma, superficial angiomyxoma, low-grade fibromyxoid sarcoma, and acral myxoinflammatory fibroblastic sarcoma.1-4
Superficial acral fibromyxomas are composed of CD34+ spindle or stellate-shaped cells that are embedded in a myxoid and/or dense hyalinized collagenous stroma in a random or loosely fascicular growth pattern. The spindle or stellate-shaped cells in SAFMs also have been found to be focally positive for epithelial membrane antigen and CD99. Lesions have accentuated microvasculature and increased mast cells.5-8
Conservative management is reasonable, but patients presenting with persistent pain and/or local deformity should be definitively treated with complete excision and follow-up. Hollmann et al1 found that 24% of tumors recurred locally upon incomplete excision after a mean interval of 27 months. All recurrent tumors had positive margins at excision or initial biopsy.1 To date, no reports of tumors metastasizing have been documented.1-4
The Diagnosis: Superficial Acral Fibromyxoma
A shave biopsy revealed an uninvolved grenz zone and mildly cellular spindle cell dermal proliferation in a collagenous and myxoid background (Figure 1). Spindle cells were seen in a myxoid background among dense coarse collagen (Figure 2A). Spindle cells also were seen in a myxoid background with mast cells and capillary network (Figure 2B). Histopathologic examination of the biopsy specimen revealed spindle cells that were diffusely positive for CD34 (Figure 3); focally positive for epithelial membrane antigen; and negative for melanocytic markers, smooth muscle markers, and cytokeratin. A diagnosis of superficial acral fibromyxoma (SAFM) was made based on clinical, histopathologic, and immunohistochemical findings.
Superficial acral fibromyxomas, also known as digital fibromyxomas, are soft, slow-growing tumors that have a predilection for subungual or periungual regions of the hands and feet. Superficial acral fibromyxomas most frequently occur on the hallux and rarely occur on the ankle or leg. They can present as nodular, dome-shaped, polyploid, or verrucous masses. They can be soft to firm, gelatinous or solid, off-white to gray-white and can have fasciculate cut surfaces. Superficial acral fibromyxomas can be either painful or painless and present with a deformed nail in 9% of cases. Superficial acral fibromyxoma is a superficial lesion with frequent infiltration of the dermal collagen and subcutaneous tissue and may even erode or infiltrate into the underlying bone in rare cases.1-4 Although SAFMs are rare tumors, documented cases of SAFM have been reported at an increasing rate since the first published report by Fetsch et al2 in 2001.
Patients often delay seeking medical treatment and present with a solitary mass that has been slowly growing for months to years. In a study of 124 patients, Hollmann et al1 found that symptoms exist for a mean of 35 months and present with a small mass with a mean tumor size of 1.7 cm before biopsy or excision. Although the age range is broad, SAFM mostly affects middle-aged adults (median age, 49 years).1 Hollmann et al1 also reported a male predominance (1.3:1 ratio), and preexisting local trauma is reported in 25% of cases.2-4
The differential for SAFM should include dermatofibroma, keloid, dermatofibrosarcoma protuberans, acquired digital fibrokeratoma, infantile digital fibromatosis, neurolemmoma, sclerosing perineurioma, superficial angiomyxoma, low-grade fibromyxoid sarcoma, and acral myxoinflammatory fibroblastic sarcoma.1-4
Superficial acral fibromyxomas are composed of CD34+ spindle or stellate-shaped cells that are embedded in a myxoid and/or dense hyalinized collagenous stroma in a random or loosely fascicular growth pattern. The spindle or stellate-shaped cells in SAFMs also have been found to be focally positive for epithelial membrane antigen and CD99. Lesions have accentuated microvasculature and increased mast cells.5-8
Conservative management is reasonable, but patients presenting with persistent pain and/or local deformity should be definitively treated with complete excision and follow-up. Hollmann et al1 found that 24% of tumors recurred locally upon incomplete excision after a mean interval of 27 months. All recurrent tumors had positive margins at excision or initial biopsy.1 To date, no reports of tumors metastasizing have been documented.1-4
- Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
- Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
- Ashby-Richardson H, Rogers GS, Stadecker MJ. Superficial acral fibromyxoma: an overview. Arch Pathol Lab Med. 2011;135:1064-1066.
- Quaba O, Evans A, Al-Nafussi AA, et al. Superficial acral fibromyxoma. Br J Plast Surg. 2005;58:561-564.
- Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx: a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
- Meyerle J, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
- Kazakov DV, Mentzel T, Buro G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
- Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
- Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
- Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
- Ashby-Richardson H, Rogers GS, Stadecker MJ. Superficial acral fibromyxoma: an overview. Arch Pathol Lab Med. 2011;135:1064-1066.
- Quaba O, Evans A, Al-Nafussi AA, et al. Superficial acral fibromyxoma. Br J Plast Surg. 2005;58:561-564.
- Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx: a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
- Meyerle J, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
- Kazakov DV, Mentzel T, Buro G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
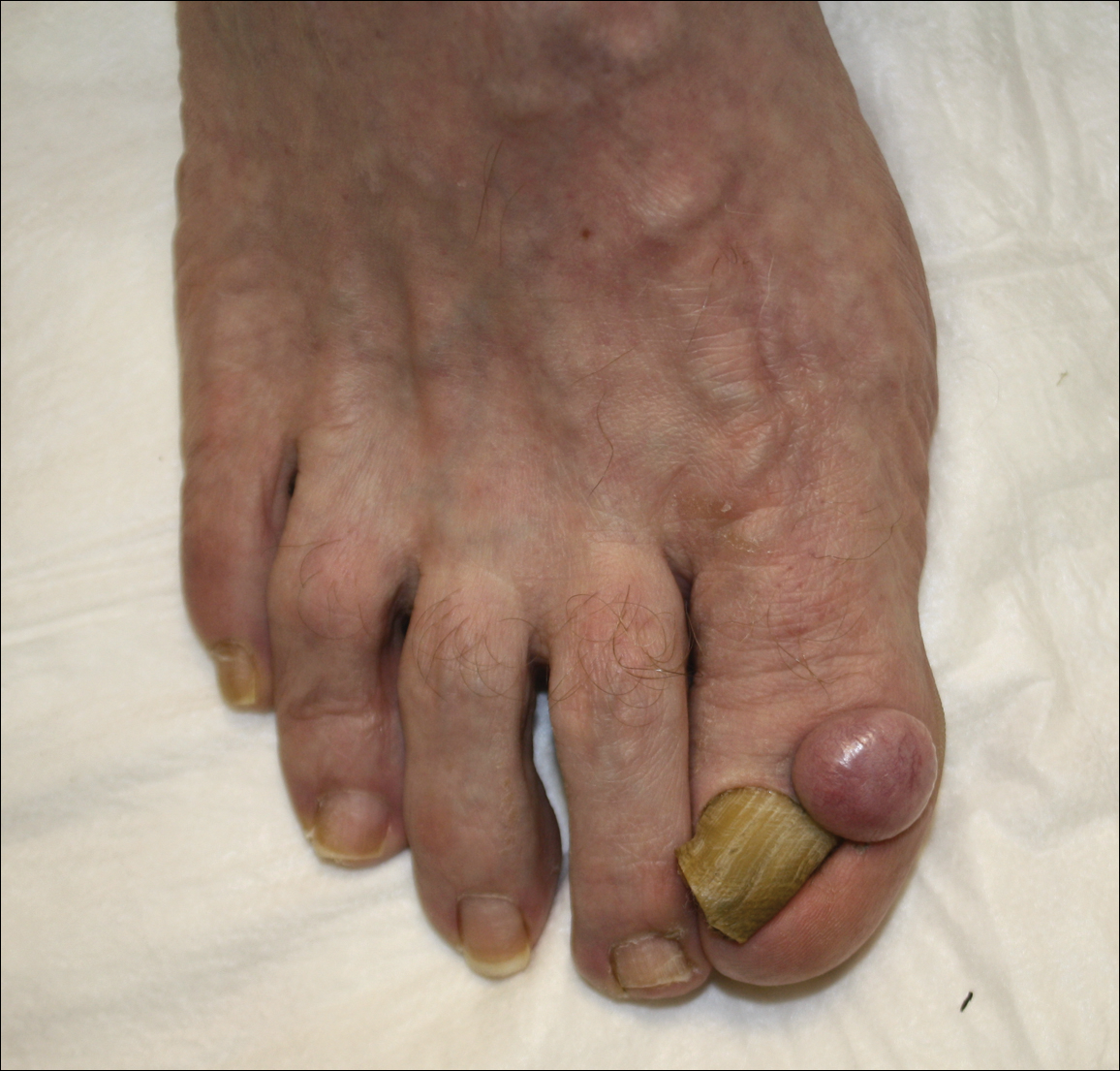
A 62-year-old man presented for evaluation of a slowly growing, nonpainful nodule on the first proximal toenail fold of the right foot of 6 years' duration. He reported that the nail plate of the affected toe was thickened and malaligned. He denied a history of trauma. Physical examination revealed a 2.0×1.6-cm, flesh-colored, nontender, well-defined, rubbery nodule with prominent overlying tortuous telangiectases on the medial aspect of the first proximal toenail fold of the right foot. The associated nail plate was yellow, thickened, and angled laterally into the second toe. Radiograph of the right hallux identified a soft tissue density contiguous with the dorsal aspect of the distal portion of the phalanx. There was no evidence of bony involvement. A shave saucerization biopsy specimen was obtained and sent for hematoxylin and eosin and immunohistochemical staining. The spindle cells were diffusely positive for CD34.
How to watch – and when to biopsy – atypical nevi
CHICAGO – Among the many difficult decisions dermatologists have to make, some of the more challenging involve caring for patients with atypical melanocytic lesions. A session at the summer meeting of the American Academy of Dermatology provided some guidance for surveillance of these patients.

Caroline C. Kim, MD, directs the pigmented lesion clinic at Beth Israel Deaconess Medical Center, Boston, and shared the evidence base for her management schema, along with some clinical pearls. No dermatologist ever wants to miss a melanoma, she acknowledged. “We want to avoid those scenarios but not make people feel like Swiss cheese” from multiple biopsies, she said during her presentation.
One key concept that can help physicians find the balance, she said, is that although the presence of atypical or dysplastic nevi increases the risk for melanoma in a given patient, the actual transformation rate of dysplastic nevi to melanomas is not known. In fact, she said, between 50% and 75% of melanomas may arise de novo.
From a dermatopathologic perspective, nevi exist along a continuum of mild to moderate to severe dysplasia, and some lesions are melanomas. But mildly dysplastic nevi are not fated to continue a transformation to increasingly severely dysplastic ones, or to melanomas.
Bringing these ideas to the patient discussion means that one should avoid ever calling a dysplastic nevus “precancerous,” said Dr. Kim; not only is this inaccurate, but it is unnecessarily anxiety provoking, she said.
Within this framework, . Each patient will have a pattern, or several patterns, that typify their nevi. Though the markings may be “atypical,” they’ll have some consistency; if the nevus has several neighbors that look just like it, it’s much less likely to be melanoma. “If they are matching partners, it’s more likely that it’s your typical nevus pattern,” said Dr. Kim, also an assistant professor of dermatology at Harvard Medical School, Boston.
By contrast, some lesions stand out from the patient’s other atypical nevi. They may be larger, darker, more elevated, but sometimes, “Even from the doorway, they just stand out,” Dr. Kim said. And these dual concepts of signature patterns and ugly ducklings are useful to talk over with patients, she said. “It’s so easy for patients to grab on to – they totally get it.”
“Use dermoscopy” when you get to the detailed skin exam, she said. “Data have shown that as clinicians, we are pretty good at picking up melanomas ... But with dermoscopy, our detection rate goes up to 70%-95%,” Dr. Kim said. The caveat is that dermoscopy without proper training is a dangerous tool: Several studies have shown that melanoma detection rates drop compared to the naked eye when dermoscopy is performed by untrained users, she said. “Training matters.”
A further tool to help train the eye and mind to recognize benign and malignant patterns when performing dermoscopy of atypical nevi is a now-classic paper that maps these patterns out, she said (Dermatol Surg. 2007;33[11]:1388-91).
“Beware of de novo and changing lesions,” Dr. Kim said. “A picture truly is worth a thousand words” for tracking these, she said.
Total body digital photography, if it’s available, is the best way to track subtle changes, and to spot new lesions as they crop up, said Dr. Kim. In head-to-head studies with dermoscopy and visual exam alone, digital photography can reduce the number of lesions excised, detect early melanoma, and reduce patient anxiety. One study found a 3.8-fold reduction in the mean rate of nevus biopsies when total body digital photography was used, she said (J Am Acad Dermatol. 2016 Mar. doi: 10.1016/j.jaad.2016.02.1152).
A patient care pearl Dr. Kim shared is that she’ll ask patients for their smartphones and take a photograph of the patients’ backs with those phones. This lets them have a handy reference image for monitoring their own skin in the intervals between visits. But make sure, she said, that patients know that “all change is not bad change – you can get new nevi through your 50s.
“Consider sharing care with a local pigmented lesion clinic” if digital photography is not available at your site, said Dr. Kim. She does this for several of her patients, alternating visits with the primary dermatologist.
When should you perform a biopsy?
“You don’t need to biopsy an atypical nevus to call it atypical. You biopsy lesions if you’re suspicious for calling it melanoma,” Dr. Kim said. Removal also can be considered if, for example, a patient lives alone and the nevi of concern are on her back so home monitoring is a challenge, she said.
Once you’ve decided to biopsy, a narrow excisional biopsy with saucerization and 1- to 3-mm margins is preferred when there’s a high suspicion for melanoma, said Dr. Kim, citing a study that found that 2-mm margins using this method yielded an 87% rate of clear pathologic specimen margins in dysplastic nevi (J Am Acad Dermatol. 2017 Dec;77[6]:1096-9). There is some leeway in the guidelines, but “the preferred technique is a narrow excisional biopsy when you are worried,” she said.
There may be times when a partial or incisional biopsy is a rational choice, as when lesions are very large, located on the face or acral areas, or when suspicion for melanoma is low. “If you do partial biopsies, you really have to be aware of the limitations” of the technique since it may miss the nidus of melanoma within an otherwise bland lesion, Dr. Kim pointed out.
And don’t forget to plan your closure with future follow-up in mind: Dr. Kim related that she’d seen a patient for melanoma who’d had the large excisional biopsy performed elsewhere; the patient’s site was closed with an advancement flap, which made sentinel node biopsy impossible.
When the results come back, then what?
Studies have found that atypical nevi are characterized differently at different sites and that management strategies vary geographically, Dr. Kim said. “There’s a need for large-scale data to further investigate the role of observation versus re-excision of dysplastic nevi,” and a multicenter study is underway to do just that, she said, under the auspices of the Pigmented Lesion Subcommittee of the Melanoma Prevention Working Group (ECOG/SWOG).
That same subcommittee has issued a consensus statement for dealing with histologically positive excisional biopsy margins. For mildly dysplastic lesions without clinically observable residual pigment, observation is preferred. Severely dysplastic lesions with unpigmented margins should be re-excised, says the statement (JAMA Dermatol. 2015;151[2]:212-18).
For the intermediate lesions, the group recommended that a reasonable option is to observe a moderately dysplastic nevus site that’s been excisionally biopsied with a finding of positive margins, while acknowledging that more data are needed.
All biopsy sites should be followed for regrowth, though recurrence of pigment alone doesn’t necessarily mean another excision is in the patient’s future, Dr. Kim said.
She reported no conflicts of interest.
SOURCE: Kim C. Summer AAD 2018, Presentation F014.
CHICAGO – Among the many difficult decisions dermatologists have to make, some of the more challenging involve caring for patients with atypical melanocytic lesions. A session at the summer meeting of the American Academy of Dermatology provided some guidance for surveillance of these patients.
Caroline C. Kim, MD, directs the pigmented lesion clinic at Beth Israel Deaconess Medical Center, Boston, and shared the evidence base for her management schema, along with some clinical pearls. No dermatologist ever wants to miss a melanoma, she acknowledged. “We want to avoid those scenarios but not make people feel like Swiss cheese” from multiple biopsies, she said during her presentation.
One key concept that can help physicians find the balance, she said, is that although the presence of atypical or dysplastic nevi increases the risk for melanoma in a given patient, the actual transformation rate of dysplastic nevi to melanomas is not known. In fact, she said, between 50% and 75% of melanomas may arise de novo.
From a dermatopathologic perspective, nevi exist along a continuum of mild to moderate to severe dysplasia, and some lesions are melanomas. But mildly dysplastic nevi are not fated to continue a transformation to increasingly severely dysplastic ones, or to melanomas.
Bringing these ideas to the patient discussion means that one should avoid ever calling a dysplastic nevus “precancerous,” said Dr. Kim; not only is this inaccurate, but it is unnecessarily anxiety provoking, she said.
Within this framework, . Each patient will have a pattern, or several patterns, that typify their nevi. Though the markings may be “atypical,” they’ll have some consistency; if the nevus has several neighbors that look just like it, it’s much less likely to be melanoma. “If they are matching partners, it’s more likely that it’s your typical nevus pattern,” said Dr. Kim, also an assistant professor of dermatology at Harvard Medical School, Boston.
By contrast, some lesions stand out from the patient’s other atypical nevi. They may be larger, darker, more elevated, but sometimes, “Even from the doorway, they just stand out,” Dr. Kim said. And these dual concepts of signature patterns and ugly ducklings are useful to talk over with patients, she said. “It’s so easy for patients to grab on to – they totally get it.”
“Use dermoscopy” when you get to the detailed skin exam, she said. “Data have shown that as clinicians, we are pretty good at picking up melanomas ... But with dermoscopy, our detection rate goes up to 70%-95%,” Dr. Kim said. The caveat is that dermoscopy without proper training is a dangerous tool: Several studies have shown that melanoma detection rates drop compared to the naked eye when dermoscopy is performed by untrained users, she said. “Training matters.”
A further tool to help train the eye and mind to recognize benign and malignant patterns when performing dermoscopy of atypical nevi is a now-classic paper that maps these patterns out, she said (Dermatol Surg. 2007;33[11]:1388-91).
“Beware of de novo and changing lesions,” Dr. Kim said. “A picture truly is worth a thousand words” for tracking these, she said.
Total body digital photography, if it’s available, is the best way to track subtle changes, and to spot new lesions as they crop up, said Dr. Kim. In head-to-head studies with dermoscopy and visual exam alone, digital photography can reduce the number of lesions excised, detect early melanoma, and reduce patient anxiety. One study found a 3.8-fold reduction in the mean rate of nevus biopsies when total body digital photography was used, she said (J Am Acad Dermatol. 2016 Mar. doi: 10.1016/j.jaad.2016.02.1152).
A patient care pearl Dr. Kim shared is that she’ll ask patients for their smartphones and take a photograph of the patients’ backs with those phones. This lets them have a handy reference image for monitoring their own skin in the intervals between visits. But make sure, she said, that patients know that “all change is not bad change – you can get new nevi through your 50s.
“Consider sharing care with a local pigmented lesion clinic” if digital photography is not available at your site, said Dr. Kim. She does this for several of her patients, alternating visits with the primary dermatologist.
When should you perform a biopsy?
“You don’t need to biopsy an atypical nevus to call it atypical. You biopsy lesions if you’re suspicious for calling it melanoma,” Dr. Kim said. Removal also can be considered if, for example, a patient lives alone and the nevi of concern are on her back so home monitoring is a challenge, she said.
Once you’ve decided to biopsy, a narrow excisional biopsy with saucerization and 1- to 3-mm margins is preferred when there’s a high suspicion for melanoma, said Dr. Kim, citing a study that found that 2-mm margins using this method yielded an 87% rate of clear pathologic specimen margins in dysplastic nevi (J Am Acad Dermatol. 2017 Dec;77[6]:1096-9). There is some leeway in the guidelines, but “the preferred technique is a narrow excisional biopsy when you are worried,” she said.
There may be times when a partial or incisional biopsy is a rational choice, as when lesions are very large, located on the face or acral areas, or when suspicion for melanoma is low. “If you do partial biopsies, you really have to be aware of the limitations” of the technique since it may miss the nidus of melanoma within an otherwise bland lesion, Dr. Kim pointed out.
And don’t forget to plan your closure with future follow-up in mind: Dr. Kim related that she’d seen a patient for melanoma who’d had the large excisional biopsy performed elsewhere; the patient’s site was closed with an advancement flap, which made sentinel node biopsy impossible.
When the results come back, then what?
Studies have found that atypical nevi are characterized differently at different sites and that management strategies vary geographically, Dr. Kim said. “There’s a need for large-scale data to further investigate the role of observation versus re-excision of dysplastic nevi,” and a multicenter study is underway to do just that, she said, under the auspices of the Pigmented Lesion Subcommittee of the Melanoma Prevention Working Group (ECOG/SWOG).
That same subcommittee has issued a consensus statement for dealing with histologically positive excisional biopsy margins. For mildly dysplastic lesions without clinically observable residual pigment, observation is preferred. Severely dysplastic lesions with unpigmented margins should be re-excised, says the statement (JAMA Dermatol. 2015;151[2]:212-18).
For the intermediate lesions, the group recommended that a reasonable option is to observe a moderately dysplastic nevus site that’s been excisionally biopsied with a finding of positive margins, while acknowledging that more data are needed.
All biopsy sites should be followed for regrowth, though recurrence of pigment alone doesn’t necessarily mean another excision is in the patient’s future, Dr. Kim said.
She reported no conflicts of interest.
SOURCE: Kim C. Summer AAD 2018, Presentation F014.
CHICAGO – Among the many difficult decisions dermatologists have to make, some of the more challenging involve caring for patients with atypical melanocytic lesions. A session at the summer meeting of the American Academy of Dermatology provided some guidance for surveillance of these patients.
Caroline C. Kim, MD, directs the pigmented lesion clinic at Beth Israel Deaconess Medical Center, Boston, and shared the evidence base for her management schema, along with some clinical pearls. No dermatologist ever wants to miss a melanoma, she acknowledged. “We want to avoid those scenarios but not make people feel like Swiss cheese” from multiple biopsies, she said during her presentation.
One key concept that can help physicians find the balance, she said, is that although the presence of atypical or dysplastic nevi increases the risk for melanoma in a given patient, the actual transformation rate of dysplastic nevi to melanomas is not known. In fact, she said, between 50% and 75% of melanomas may arise de novo.
From a dermatopathologic perspective, nevi exist along a continuum of mild to moderate to severe dysplasia, and some lesions are melanomas. But mildly dysplastic nevi are not fated to continue a transformation to increasingly severely dysplastic ones, or to melanomas.
Bringing these ideas to the patient discussion means that one should avoid ever calling a dysplastic nevus “precancerous,” said Dr. Kim; not only is this inaccurate, but it is unnecessarily anxiety provoking, she said.
Within this framework, . Each patient will have a pattern, or several patterns, that typify their nevi. Though the markings may be “atypical,” they’ll have some consistency; if the nevus has several neighbors that look just like it, it’s much less likely to be melanoma. “If they are matching partners, it’s more likely that it’s your typical nevus pattern,” said Dr. Kim, also an assistant professor of dermatology at Harvard Medical School, Boston.
By contrast, some lesions stand out from the patient’s other atypical nevi. They may be larger, darker, more elevated, but sometimes, “Even from the doorway, they just stand out,” Dr. Kim said. And these dual concepts of signature patterns and ugly ducklings are useful to talk over with patients, she said. “It’s so easy for patients to grab on to – they totally get it.”
“Use dermoscopy” when you get to the detailed skin exam, she said. “Data have shown that as clinicians, we are pretty good at picking up melanomas ... But with dermoscopy, our detection rate goes up to 70%-95%,” Dr. Kim said. The caveat is that dermoscopy without proper training is a dangerous tool: Several studies have shown that melanoma detection rates drop compared to the naked eye when dermoscopy is performed by untrained users, she said. “Training matters.”
A further tool to help train the eye and mind to recognize benign and malignant patterns when performing dermoscopy of atypical nevi is a now-classic paper that maps these patterns out, she said (Dermatol Surg. 2007;33[11]:1388-91).
“Beware of de novo and changing lesions,” Dr. Kim said. “A picture truly is worth a thousand words” for tracking these, she said.
Total body digital photography, if it’s available, is the best way to track subtle changes, and to spot new lesions as they crop up, said Dr. Kim. In head-to-head studies with dermoscopy and visual exam alone, digital photography can reduce the number of lesions excised, detect early melanoma, and reduce patient anxiety. One study found a 3.8-fold reduction in the mean rate of nevus biopsies when total body digital photography was used, she said (J Am Acad Dermatol. 2016 Mar. doi: 10.1016/j.jaad.2016.02.1152).
A patient care pearl Dr. Kim shared is that she’ll ask patients for their smartphones and take a photograph of the patients’ backs with those phones. This lets them have a handy reference image for monitoring their own skin in the intervals between visits. But make sure, she said, that patients know that “all change is not bad change – you can get new nevi through your 50s.
“Consider sharing care with a local pigmented lesion clinic” if digital photography is not available at your site, said Dr. Kim. She does this for several of her patients, alternating visits with the primary dermatologist.
When should you perform a biopsy?
“You don’t need to biopsy an atypical nevus to call it atypical. You biopsy lesions if you’re suspicious for calling it melanoma,” Dr. Kim said. Removal also can be considered if, for example, a patient lives alone and the nevi of concern are on her back so home monitoring is a challenge, she said.
Once you’ve decided to biopsy, a narrow excisional biopsy with saucerization and 1- to 3-mm margins is preferred when there’s a high suspicion for melanoma, said Dr. Kim, citing a study that found that 2-mm margins using this method yielded an 87% rate of clear pathologic specimen margins in dysplastic nevi (J Am Acad Dermatol. 2017 Dec;77[6]:1096-9). There is some leeway in the guidelines, but “the preferred technique is a narrow excisional biopsy when you are worried,” she said.
There may be times when a partial or incisional biopsy is a rational choice, as when lesions are very large, located on the face or acral areas, or when suspicion for melanoma is low. “If you do partial biopsies, you really have to be aware of the limitations” of the technique since it may miss the nidus of melanoma within an otherwise bland lesion, Dr. Kim pointed out.
And don’t forget to plan your closure with future follow-up in mind: Dr. Kim related that she’d seen a patient for melanoma who’d had the large excisional biopsy performed elsewhere; the patient’s site was closed with an advancement flap, which made sentinel node biopsy impossible.
When the results come back, then what?
Studies have found that atypical nevi are characterized differently at different sites and that management strategies vary geographically, Dr. Kim said. “There’s a need for large-scale data to further investigate the role of observation versus re-excision of dysplastic nevi,” and a multicenter study is underway to do just that, she said, under the auspices of the Pigmented Lesion Subcommittee of the Melanoma Prevention Working Group (ECOG/SWOG).
That same subcommittee has issued a consensus statement for dealing with histologically positive excisional biopsy margins. For mildly dysplastic lesions without clinically observable residual pigment, observation is preferred. Severely dysplastic lesions with unpigmented margins should be re-excised, says the statement (JAMA Dermatol. 2015;151[2]:212-18).
For the intermediate lesions, the group recommended that a reasonable option is to observe a moderately dysplastic nevus site that’s been excisionally biopsied with a finding of positive margins, while acknowledging that more data are needed.
All biopsy sites should be followed for regrowth, though recurrence of pigment alone doesn’t necessarily mean another excision is in the patient’s future, Dr. Kim said.
She reported no conflicts of interest.
SOURCE: Kim C. Summer AAD 2018, Presentation F014.
EXPERT ANALYSIS FROM SUMMER AAD 2018
Gottron Papules Mimicking Dermatomyositis: An Unusual Manifestation of Systemic Lupus Erythematosus
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
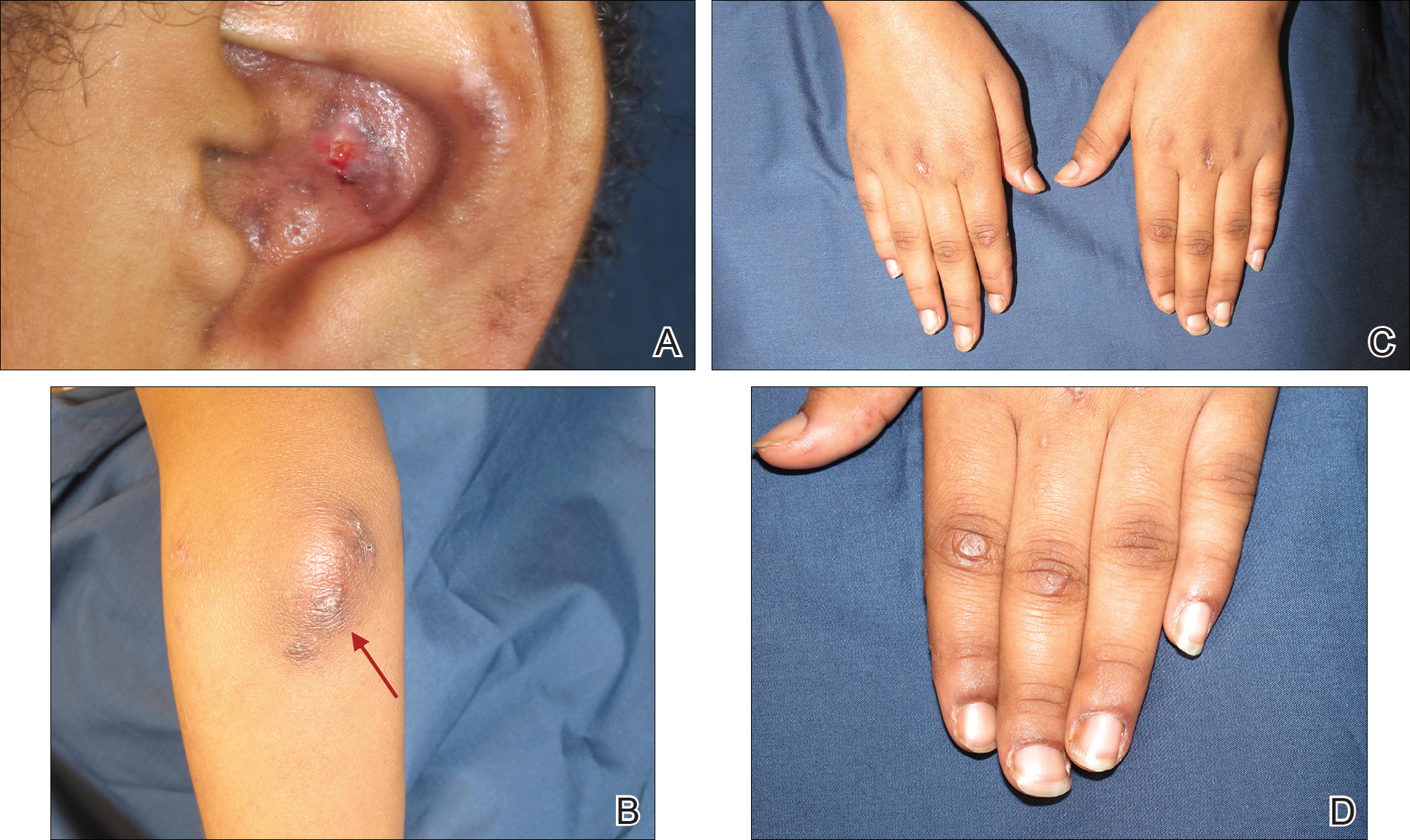
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
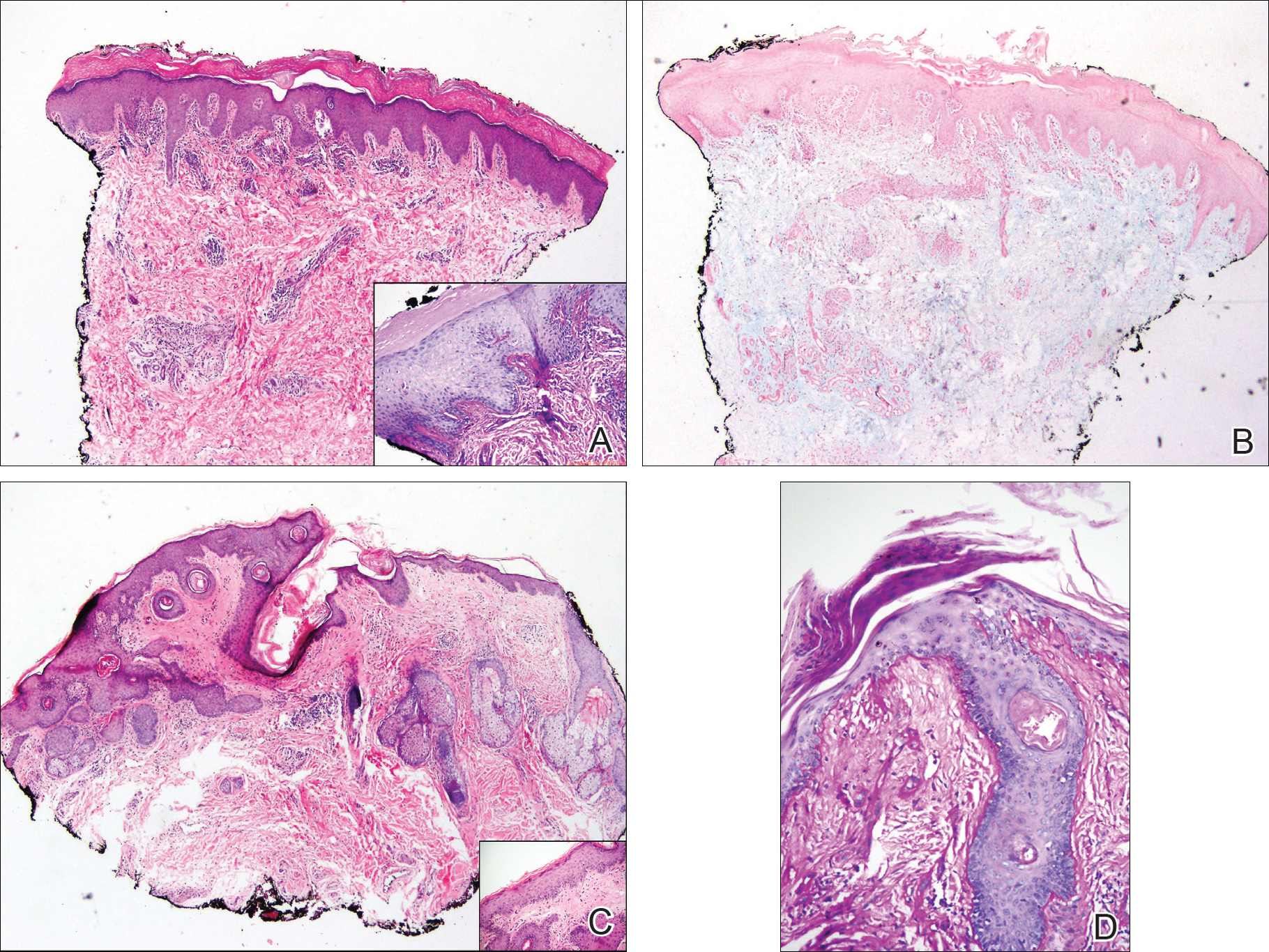
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
To the Editor:
An 11-year-old girl presented to the dermatology clinic with an asymptomatic rash on the bilateral forearms, dorsal hands, and ears of 1 month’s duration. Recent history was notable for persistent low-grade fevers, dizziness, headaches, arthralgia, and swelling of multiple joints, as well as difficulty ambulating due to the joint pain. A thorough review of systems revealed no photosensitivity, oral sores, weight loss, pulmonary symptoms, Raynaud phenomenon, or dysphagia.
Medical history was notable for presumed viral pancreatitis and transaminitis requiring inpatient hospitalization 1 year prior to presentation. The patient underwent extensive workup at that time, which was notable for a positive antinuclear antibody level of 1:2560, an elevated erythrocyte sedimentation rate level of 75 mm/h (reference range, 0–22 mm/h), hemolytic anemia with a hemoglobin of 10.9 g/dL (14.0–17.5 g/dL), and leukopenia with a white blood cell count of 3700/µL (4500–11,000/µL). Additional laboratory tests were performed and were found to be within reference range, including creatine kinase, aldolase, complete metabolic panel, extractable nuclear antigen, complement levels, C-reactive protein level, antiphospholipid antibodies,partial thromboplastin time, prothrombin time, anti–double-stranded DNA, rheumatoid factor, β2-glycoprotein, and antineutrophil cytoplasmic antibody tests. Skin purified protein derivative (tuberculin) test and chest radiograph also were unremarkable. The patient also was evaluated and found negative for Wilson disease, hemochromatosis, α1-antitrypsin disease, and autoimmune hepatitis.
Physical examination revealed erythematous plaques with crusted hyperpigmented erosions and central hypopigmentation on the bilateral conchal bowls and antihelices, findings characteristic of discoid lupus erythematosus (Figure 1A). On the bilateral elbows, metacarpophalangeal (MCP) joints, and proximal interphalangeal (PIP) joints, there were firm, erythematous to violaceous, keratotic papules that were clinically suggestive of Gottron-like papules (Figures 1B and 1C). However, there were no lesions on the skin between the MCP, PIP, and distal interphalangeal joints. The MCP joints were associated with swelling and were tender to palpation. Examination of the fingernails showed dilated telangiectasia of the proximal nail folds and ragged hyperkeratotic cuticles of all 10 digits (Figure 1D). On the extensor aspects of the bilateral forearms, there were erythematous excoriated papules and papulovesicular lesions with central hemorrhagic crusting. The patient showed no shawl sign, heliotrope rash, calcinosis, malar rash, oral lesions, or hair loss.
Additional physical examinations performed by the neufigrology and rheumatology departments revealed no impairment of muscle strength, soreness of muscles, and muscular atrophy. Joint examination was notable for restriction in range of motion of the hands, hips, and ankles due to swelling and pain of the joints. Radiographs and ultrasound of the feet showed fluid accumulation and synovial thickening of the metatarsal phalangeal joints and one of the PIP joints of the right hand without erosion.
The patient did not undergo magnetic resonance imaging of muscles due to the lack of muscular symptoms and normal myositis laboratory markers. Dermatomyositis-specific antibody testing, such as anti–Jo-1 and anti–Mi-2, also was not performed.
After reviewing the biopsy results, laboratory findings, and clinical presentation, the patient was diagnosed with systemic lupus erythematosus (SLE), as she met American College of Rheumatology criteria1 with the following: discoid rash, hemolytic anemia, positive antinuclear antibodies, and nonerosive arthritis. Due to her abnormal constellation of laboratory values and symptoms, she was evaluated by 2 pediatric rheumatologists at 2 different medical centers who agreed with a primary diagnosis of SLE rather than dermatomyositis sine myositis. The hemolytic anemia was attributed to underlying connective tissue disease, as the hemoglobin levels were found to be persistently low for 1 year prior to the diagnosis of systemic lupus, and there was no alternative cause of the hematologic disorder.
A punch biopsy obtained from a Gottron-like papule on the dorsal aspect of the left hand revealed lymphocytic interface dermatitis and slight thickening of the basement membrane zone (Figure 2A). There was a dense superficial and deep periadnexal and perivascular lymphocytic inflammation as well as increased dermal mucin, which can be seen in both lupus erythematosus and dermatomyositis (Figure 2B). Perniosis also was considered from histologic findings but was excluded based on clinical history and physical findings. A second biopsy of the left conchal bowl showed hyperkeratosis, epidermal atrophy, interface changes, follicular plugging, and basement membrane thickening. These findings can be seen in dermatomyositis, but when considered together with the clinical appearance of the patient’s eruption on the ears, they were more consistent with discoid lupus erythematosus (Figures 2C and 2D).
Finally, although ragged cuticles and proximal nail fold telangiectasia typically are seen in dermatomyositis, nail fold hyperkeratosis, ragged cuticles, and nail bed telangiectasia also have been reported in lupus erythematosus.2,3 Therefore, the findings overlying our patient’s knuckles and elbows can be considered Gottron-like papules in the setting of SLE.
Dermatomyositis has several characteristic dermatologic manifestations, including Gottron papules, shawl sign, facial heliotrope rash, periungual telangiectasia, and mechanic’s hands. Of them, Gottron papules have been the most pathognomonic, while the other skin findings are less specific and can be seen in other disease entities.4,5
The pathogenesis of Gottron papules in dermatomyositis remains largely unknown. Prior molecular studies have proposed that stretch CD44 variant 7 and abnormal osteopontin levels may contribute to the pathogenesis of Gottron papules by increasing local inflammation.6 Studies also have linked abnormal osteopontin levels and CD44 variant 7 expression with other diseases of autoimmunity, including lupus erythematosus.7 Because lupus erythematosus can have a large variety of cutaneous findings, Gottron-like papules may be considered a rare dermatologic presentation of lupus erythematosus.
We present a case of Gottron-like papules as an unusual dermatologic manifestation of SLE, challenging the concept of Gottron papules as a pathognomonic finding of dermatomyositis.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
- Singal A, Arora R. Nail as a window of systemic diseases. Indian Dermatol Online J. 2015;6:67-74.
- Trüeb RM. Hair and nail involvement in lupus erythematosus. Clin Dermatol. 2004;22:139-147.
- Koler RA, Montemarano A. Dermatomyositis. Am Fam Physician. 2001;64:1565-1572.
- Muro Y, Sugiura K, Akiyama M. Cutaneous manifestations in dermatomyositis: key clinical and serological features—a comprehensive review. Clin Rev Allergy Immunol. 2016;51:293-302.
- Kim JS, Bashir MM, Werth VP. Gottron’s papules exhibit dermal accumulation of CD44 variant 7 (CD44v7) and its binding partner osteopontin: a unique molecular signature. J Invest Dermatol. 2012;132:1825-1832.
- Kim JS, Werth VP. Identification of specific chondroitin sulfate species in cutaneous autoimmune disease. J Histochem Cytochem. 2011;59:780-790.
Practice Points
- Gottron-like papules can be a dermatologic presentation of lupus erythematosus.
- When present along with other findings of lupus erythematosus without any clinical manifestations of dermatomyositis, Gottron-like papules can be thought of as a manifestation of lupus erythematosus rather than dermatomyositis.

Ecthyma Gangrenosum Due to Pseudomonas fluorescens
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
...")
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
(H&E, original magnification ×100); biopsy from the periphery of the lesion showed leukocytoclastic vasculitis (B)...")
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
Practice Points
- Immunocompromised patients with a high exposure to agricultural products may be at increased risk for systemic infection by Pseudomonas fluorescens.
- Pseudomonas fluorescens is an opportunistic pathogen that can cause ecthyma gangrenosum, which necessitates rapid diagnosis and treatment to prevent mortality.
Copresentation of Common Variable Immune Deficiency and Sweet Syndrome
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.


Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
Practice Points
- Suggested workup for Sweet syndrome includes ruling out connective tissue disorders and malignancies.
- Familial common variable immune deficiency is rare and can first manifest in adulthood.
Reflectance Confocal Microscopy as a First-Line Diagnostic Technique for Mycosis Fungoides
Case Report
A 60-year-old man with a history of Hodgkin lymphoma that had been treated with chemotherapy 6 years prior presented to our dermatology clinic with a persistent pruritic rash on the back, abdomen, and bilateral arms and legs. The eruption initially began as localized discrete lesions on the lower back 1 year prior to the current presentation; at that time a diagnosis of psoriasis was made at an outside dermatology clinic, and treatment with mometasone furoate cream was initiated. Despite the patient’s compliance with this treatment, the lesions did not resolve and began spreading to the arms, legs, chest, and abdomen. His current medications included lisinopril, escitalopram, aspirin, and omeprazole.
On presentation to our clinic, physical examination revealed round, scaly, pink plaques and tumors of variable sizes (3–10 cm) distributed asymmetrically on the chest, back, abdomen, arms, and legs (Figure 1). The lesions were grouped in well-defined areas encompassing approximately 30% of the body surface area. No lymphadenopathy was appreciated. In vivo reflectance confocal microscopy (RCM) performed on one of the lesions revealed disarray of the epidermis with small, weakly refractile, round to oval cells scattered within the spinous layer and dermoepidermal junction (Figure 2). Additionally, these weakly refractile, round to oval cells also were seen in vesiclelike dark spaces, and hyporefractile basal cells were appreciated surrounding the dermal papillae. Mycosis fungoides (MF) was diagnosed following correlation of the RCM findings with the clinical picture.
.")
 scattered among keratinocytes in vesiclelike dark spaces (A)...")
A biopsy was performed, with pathologic examination confirming the diagnosis of tumor-stage MF. Parakeratosis with epidermotropism of lymphocytes was noted along the basal layer and into the spinous layer of the epidermis (Figure 3). Underlying the epidermis there was a dense mononuclear infiltrate and conspicuous eosinophils extending to the deeper reticular dermis. The infiltrating cells had cerebriform nuclei and large pale cytoplasm. On immunostaining, the lymphocytes were positive for CD3 and CD4, and negative for CD5, CD7, and CD8. The patient was referred to the oncology department for disease management. Staging workup including computed tomography, flow cytometry, and T-cell receptor gene rearrangement were consistent with tumor-stage MF (T3N0M0B0).
 as well as a dense infiltrate in the dermis (A)(H&E, original magnifications ×10 and ×50 [inset])...")
Comment
Clinical Presentation of MF
Mycosis fungoides, a non-Hodgkin lymphoma of T-cell origin, is the most commonly diagnosed cutaneous lymphoma worldwide.1 It has an annual incidence of approximately 0.36 per 100,000 persons, and this number continues to rise.2,3 The median age of diagnosis is 55 to 60 years, and MF occurs twice as often in men versus women.4
The clinical presentation of MF varies and is classified by stages including patches, plaques, tumors, and erythroderma.5 Classically, MF is slowly progressive and begins as pruritic erythematous patches that have a predilection for non–sun-exposed areas of the skin. Over time, these patches may evolve into plaques and tumors. Early or patch-stage MF often presents as well-demarcated lesions of various sizes and shapes that tend to enlarge.6 These lesions may resemble eczema or psoriasis if there is scaling, such as in our patient. At the tumor stage, flat or dome-shaped nodules that may vary in color and are deeper than plaques begin to appear. Ulcerations, which were absent in our case, may often be seen.
Because of the diverse clinical manifestations of MF, which can mimic other common dermatoses, diagnosis often is challenging for clinicians. Furthermore, histology can yield nonspecific diagnostic results and may even resemble chronic inflammatory dermatoses.7 As a result, patients frequently are subjected to multiple skin biopsies to establish the diagnosis,8 and diagnosis may be delayed, with the median time from onset of skin symptoms to diagnosis being approximately 6 years.9
Reflectance Confocal Microscopy
In vivo RCM is a noninvasive technique that allows visualization of the skin at a cellular level and recently has been evaluated as a diagnostic tool for many skin conditions.10,11 Reflectance confocal microscopy findings have been well established for many cutaneous malignancies as well as inflammatory conditions such as psoriasis and atopic dermatitis.12,13 Specifically, 2 preliminary descriptive studies utilized RCM to visualize the characteristic features of MF in vivo.14,15 These studies reported the histopathologic correlation of RCM findings in biopsy-proven MF lesions. Consistent in all stages of MF is the presence of small, weakly refractile, round to oval cells within the spinous layer that correlate with atypical lymphocytes, in addition to hyporefractile basal cells surrounding the dermal papillae. Patch-stage MF lesions have more subtle epidermal findings compared to plaque-stage lesions, which tend to have more prominent vesiclelike dark spaces filled with collections of monomorphous, weakly refractile, round to oval cells corresponding with Pautrier microabscesses and evidence of spongiosis.14,15 The first descriptive study of RCM in the diagnosis of MF failed to identify features of tumor-stage MF that would distinguish it from patch- or plaque-stage disease. The investigators also stated that deep nodular collections of atypical lymphocytes seen on histopathology in tumor-stage MF were missed on RCM evaluation.14 Furthermore, the second descriptive study of RCM and MF, which included 2 patients with tumor-stage disease, also failed to differentiate tumor-stage MF from the patch or plaque stages.15
Because of these 2 descriptive studies, a pilot study was conducted to determine the applicability and reproducibility of RCM findings for MF diagnosis.16 Two blinded confocalists were asked to diagnose RCM images as MF when compared to either normal skin or a variety of lymphoproliferative disorders. Of 15 patients, the confocalists correctly diagnosed MF in 84% and 90% of cases, respectively. Additionally, they reported the specificity and sensitivity of the following RCM features in the diagnosis of MF: spongiosis, 88.9% and 94.7%; loss of demarcation, 88.9% and 94.7%; disarray of the epidermis, 77.8% and 89.5%; hyporefractile rings, 88.9% and 78.9%; junctional atypical lymphocytes, 100% and 73.7%; and vesiclelike structures (Pautrier microabscesses), 100% and 73.7%. Importantly, this study did not evaluate the specificity and sensitivity of MF diagnosis compared to other eczematous or inflammatory conditions that may share similar RCM findings; therefore, these results are not generalizable, and many of the RCM findings characteristically seen in MF are not specific to its diagnosis.16
One study assessed the diagnostic accuracy of RCM in evaluating erythematosquamous diseases including MF, psoriasis, contact dermatitis, discoid lupus, and subacute cutaneous lupus.17 In this study, 3 blinded confocalists achieved a 95.41% and 92.89% specificity and 89.13% and 63.33% sensitivity for psoriasis and MF, respectively. Typical features of psoriasis on RCM included parakeratosis, reduction or absence of the granular layer, papillomatosis, acanthosis with normal honeycomb pattern of the epidermis, and dilated vessels in the upper dermis. Features that were more specific to MF included epidermotropic atypical lymphocytes, interface dermatitis, pleomorphic tumor cells, and dendritic cells.17 However, atypical lymphocytes and interface dermatitis also may be seen in cutaneous lupus; therefore, additional studies are still needed to validate RCM’s utility in differentiating between erythematosquamous skin diseases, including psoriasis, cutaneous lupus, and MF. Currently, RCM findings must be interpreted in conjunction with the clinical and histologic picture.
Importantly, RCM also is limited when evaluating MF due to its limited depth of visualization, as it allows imaging only to the superficial papillary dermis. Furthermore, any infiltrative process such as epidermal hyperplasia, spongiosis, or scaling, which can be seen in MF, may further impair the imaging quality of the deeper dermis.
Conclusion
Despite its limitations, RCM has the potential to be advantageous in evaluating skin lesions suspicious for MF in real time and is a promising technology for a quick noninvasive bedside adjunct tool. Its utility in selecting the optimal site for biopsy for better yield of histopathologic results in suspected MF cases has been demonstrated.16 However, large-scale studies still are needed to evaluate RCM in the diagnosis of the wide diversity of MF lesions as well as its efficacy in selecting optimal biopsy sites.
- Lutzner M, Edelson R, Schein P, et al. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83:534-552.
- Akinbami AA, Osikomaiya BI, John-Olabode SO, et al. Mycosis fungoides: case report and literature review. Clin Med Insights Case Rep. 2014;7:95-98.
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-959.
- Bradford PT, Devesa SS, Anderson WF, et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113:5064-5073.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nashan D, Faulhaber D, Stander S. Mycosis fungoides: a dermatological masquerader. Br J Dermatol. 2007;157:1-10.
- Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
- Glass LF, Keller KL, Messina JL, et al. Cutaneous T-cell lymphoma. Cancer Control. 1998;5:11-18.
- Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14:293-371.
- Tannous ZS, Mihm MC, Flotte TJ, et al. In vivo examination of lentigo maligna and malignant melanoma in situ, lentigo maligna type by near-infrared reflectance confocal microscopy: comparison of in vivo confocal images with histologic sections. J Am Acad Dermatol. 2002;46:260-263.
- Gerger A, Koller S, Weger W, et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer. 2006;107:193-200.
- Branzan AL, Landthaler M, Szeimies RM. In vivo confocal scanning laser microscopy in dermatology [published online November 18, 2006]. Lasers Med Sci. 2007;22:73-82.
- González S. Confocal reflectance microscopy in dermatology: promise and reality of non-invasive diagnosis and monitoring. Actas Dermosifiliogr. 2009;100(suppl 2):59-69.
- Agero AL, Gill M, Ardigo M, et al. In vivo reflectance confocal microscopy of mycosis fungoides: a preliminary study [published online April 16, 2007]. J Am Acad Dermatol. 2007;57:435-441.
- Wi L, Dai H, Li Z, et al. Reflectance confocal microscopy for the characteristics of mycosis fungoides and correlation with histology: a pilot study [published online April 18, 2013]. Skin Res Technol. 2013;19:352-355.
- Lange-Asschenfeldt S, Babilli J, Beyer M, et al. Consistency and distribution of reflectance confocal microscopy features for diagnosis of cutaneous T cell lymphoma. J Biomed Opt. 2012;17:016001.
- Koller S, Gerger A, Ahlgrimm-Siess V. In vivo reflectance confocal microscopy of erythematosquamous skin diseases [published online March 6, 2009]. Exp Dermatol. 2009;18:536-540.
Case Report
A 60-year-old man with a history of Hodgkin lymphoma that had been treated with chemotherapy 6 years prior presented to our dermatology clinic with a persistent pruritic rash on the back, abdomen, and bilateral arms and legs. The eruption initially began as localized discrete lesions on the lower back 1 year prior to the current presentation; at that time a diagnosis of psoriasis was made at an outside dermatology clinic, and treatment with mometasone furoate cream was initiated. Despite the patient’s compliance with this treatment, the lesions did not resolve and began spreading to the arms, legs, chest, and abdomen. His current medications included lisinopril, escitalopram, aspirin, and omeprazole.
On presentation to our clinic, physical examination revealed round, scaly, pink plaques and tumors of variable sizes (3–10 cm) distributed asymmetrically on the chest, back, abdomen, arms, and legs (Figure 1). The lesions were grouped in well-defined areas encompassing approximately 30% of the body surface area. No lymphadenopathy was appreciated. In vivo reflectance confocal microscopy (RCM) performed on one of the lesions revealed disarray of the epidermis with small, weakly refractile, round to oval cells scattered within the spinous layer and dermoepidermal junction (Figure 2). Additionally, these weakly refractile, round to oval cells also were seen in vesiclelike dark spaces, and hyporefractile basal cells were appreciated surrounding the dermal papillae. Mycosis fungoides (MF) was diagnosed following correlation of the RCM findings with the clinical picture.
A biopsy was performed, with pathologic examination confirming the diagnosis of tumor-stage MF. Parakeratosis with epidermotropism of lymphocytes was noted along the basal layer and into the spinous layer of the epidermis (Figure 3). Underlying the epidermis there was a dense mononuclear infiltrate and conspicuous eosinophils extending to the deeper reticular dermis. The infiltrating cells had cerebriform nuclei and large pale cytoplasm. On immunostaining, the lymphocytes were positive for CD3 and CD4, and negative for CD5, CD7, and CD8. The patient was referred to the oncology department for disease management. Staging workup including computed tomography, flow cytometry, and T-cell receptor gene rearrangement were consistent with tumor-stage MF (T3N0M0B0).
Comment
Clinical Presentation of MF
Mycosis fungoides, a non-Hodgkin lymphoma of T-cell origin, is the most commonly diagnosed cutaneous lymphoma worldwide.1 It has an annual incidence of approximately 0.36 per 100,000 persons, and this number continues to rise.2,3 The median age of diagnosis is 55 to 60 years, and MF occurs twice as often in men versus women.4
The clinical presentation of MF varies and is classified by stages including patches, plaques, tumors, and erythroderma.5 Classically, MF is slowly progressive and begins as pruritic erythematous patches that have a predilection for non–sun-exposed areas of the skin. Over time, these patches may evolve into plaques and tumors. Early or patch-stage MF often presents as well-demarcated lesions of various sizes and shapes that tend to enlarge.6 These lesions may resemble eczema or psoriasis if there is scaling, such as in our patient. At the tumor stage, flat or dome-shaped nodules that may vary in color and are deeper than plaques begin to appear. Ulcerations, which were absent in our case, may often be seen.
Because of the diverse clinical manifestations of MF, which can mimic other common dermatoses, diagnosis often is challenging for clinicians. Furthermore, histology can yield nonspecific diagnostic results and may even resemble chronic inflammatory dermatoses.7 As a result, patients frequently are subjected to multiple skin biopsies to establish the diagnosis,8 and diagnosis may be delayed, with the median time from onset of skin symptoms to diagnosis being approximately 6 years.9
Reflectance Confocal Microscopy
In vivo RCM is a noninvasive technique that allows visualization of the skin at a cellular level and recently has been evaluated as a diagnostic tool for many skin conditions.10,11 Reflectance confocal microscopy findings have been well established for many cutaneous malignancies as well as inflammatory conditions such as psoriasis and atopic dermatitis.12,13 Specifically, 2 preliminary descriptive studies utilized RCM to visualize the characteristic features of MF in vivo.14,15 These studies reported the histopathologic correlation of RCM findings in biopsy-proven MF lesions. Consistent in all stages of MF is the presence of small, weakly refractile, round to oval cells within the spinous layer that correlate with atypical lymphocytes, in addition to hyporefractile basal cells surrounding the dermal papillae. Patch-stage MF lesions have more subtle epidermal findings compared to plaque-stage lesions, which tend to have more prominent vesiclelike dark spaces filled with collections of monomorphous, weakly refractile, round to oval cells corresponding with Pautrier microabscesses and evidence of spongiosis.14,15 The first descriptive study of RCM in the diagnosis of MF failed to identify features of tumor-stage MF that would distinguish it from patch- or plaque-stage disease. The investigators also stated that deep nodular collections of atypical lymphocytes seen on histopathology in tumor-stage MF were missed on RCM evaluation.14 Furthermore, the second descriptive study of RCM and MF, which included 2 patients with tumor-stage disease, also failed to differentiate tumor-stage MF from the patch or plaque stages.15
Because of these 2 descriptive studies, a pilot study was conducted to determine the applicability and reproducibility of RCM findings for MF diagnosis.16 Two blinded confocalists were asked to diagnose RCM images as MF when compared to either normal skin or a variety of lymphoproliferative disorders. Of 15 patients, the confocalists correctly diagnosed MF in 84% and 90% of cases, respectively. Additionally, they reported the specificity and sensitivity of the following RCM features in the diagnosis of MF: spongiosis, 88.9% and 94.7%; loss of demarcation, 88.9% and 94.7%; disarray of the epidermis, 77.8% and 89.5%; hyporefractile rings, 88.9% and 78.9%; junctional atypical lymphocytes, 100% and 73.7%; and vesiclelike structures (Pautrier microabscesses), 100% and 73.7%. Importantly, this study did not evaluate the specificity and sensitivity of MF diagnosis compared to other eczematous or inflammatory conditions that may share similar RCM findings; therefore, these results are not generalizable, and many of the RCM findings characteristically seen in MF are not specific to its diagnosis.16
One study assessed the diagnostic accuracy of RCM in evaluating erythematosquamous diseases including MF, psoriasis, contact dermatitis, discoid lupus, and subacute cutaneous lupus.17 In this study, 3 blinded confocalists achieved a 95.41% and 92.89% specificity and 89.13% and 63.33% sensitivity for psoriasis and MF, respectively. Typical features of psoriasis on RCM included parakeratosis, reduction or absence of the granular layer, papillomatosis, acanthosis with normal honeycomb pattern of the epidermis, and dilated vessels in the upper dermis. Features that were more specific to MF included epidermotropic atypical lymphocytes, interface dermatitis, pleomorphic tumor cells, and dendritic cells.17 However, atypical lymphocytes and interface dermatitis also may be seen in cutaneous lupus; therefore, additional studies are still needed to validate RCM’s utility in differentiating between erythematosquamous skin diseases, including psoriasis, cutaneous lupus, and MF. Currently, RCM findings must be interpreted in conjunction with the clinical and histologic picture.
Importantly, RCM also is limited when evaluating MF due to its limited depth of visualization, as it allows imaging only to the superficial papillary dermis. Furthermore, any infiltrative process such as epidermal hyperplasia, spongiosis, or scaling, which can be seen in MF, may further impair the imaging quality of the deeper dermis.
Conclusion
Despite its limitations, RCM has the potential to be advantageous in evaluating skin lesions suspicious for MF in real time and is a promising technology for a quick noninvasive bedside adjunct tool. Its utility in selecting the optimal site for biopsy for better yield of histopathologic results in suspected MF cases has been demonstrated.16 However, large-scale studies still are needed to evaluate RCM in the diagnosis of the wide diversity of MF lesions as well as its efficacy in selecting optimal biopsy sites.
Case Report
A 60-year-old man with a history of Hodgkin lymphoma that had been treated with chemotherapy 6 years prior presented to our dermatology clinic with a persistent pruritic rash on the back, abdomen, and bilateral arms and legs. The eruption initially began as localized discrete lesions on the lower back 1 year prior to the current presentation; at that time a diagnosis of psoriasis was made at an outside dermatology clinic, and treatment with mometasone furoate cream was initiated. Despite the patient’s compliance with this treatment, the lesions did not resolve and began spreading to the arms, legs, chest, and abdomen. His current medications included lisinopril, escitalopram, aspirin, and omeprazole.
On presentation to our clinic, physical examination revealed round, scaly, pink plaques and tumors of variable sizes (3–10 cm) distributed asymmetrically on the chest, back, abdomen, arms, and legs (Figure 1). The lesions were grouped in well-defined areas encompassing approximately 30% of the body surface area. No lymphadenopathy was appreciated. In vivo reflectance confocal microscopy (RCM) performed on one of the lesions revealed disarray of the epidermis with small, weakly refractile, round to oval cells scattered within the spinous layer and dermoepidermal junction (Figure 2). Additionally, these weakly refractile, round to oval cells also were seen in vesiclelike dark spaces, and hyporefractile basal cells were appreciated surrounding the dermal papillae. Mycosis fungoides (MF) was diagnosed following correlation of the RCM findings with the clinical picture.
A biopsy was performed, with pathologic examination confirming the diagnosis of tumor-stage MF. Parakeratosis with epidermotropism of lymphocytes was noted along the basal layer and into the spinous layer of the epidermis (Figure 3). Underlying the epidermis there was a dense mononuclear infiltrate and conspicuous eosinophils extending to the deeper reticular dermis. The infiltrating cells had cerebriform nuclei and large pale cytoplasm. On immunostaining, the lymphocytes were positive for CD3 and CD4, and negative for CD5, CD7, and CD8. The patient was referred to the oncology department for disease management. Staging workup including computed tomography, flow cytometry, and T-cell receptor gene rearrangement were consistent with tumor-stage MF (T3N0M0B0).
Comment
Clinical Presentation of MF
Mycosis fungoides, a non-Hodgkin lymphoma of T-cell origin, is the most commonly diagnosed cutaneous lymphoma worldwide.1 It has an annual incidence of approximately 0.36 per 100,000 persons, and this number continues to rise.2,3 The median age of diagnosis is 55 to 60 years, and MF occurs twice as often in men versus women.4
The clinical presentation of MF varies and is classified by stages including patches, plaques, tumors, and erythroderma.5 Classically, MF is slowly progressive and begins as pruritic erythematous patches that have a predilection for non–sun-exposed areas of the skin. Over time, these patches may evolve into plaques and tumors. Early or patch-stage MF often presents as well-demarcated lesions of various sizes and shapes that tend to enlarge.6 These lesions may resemble eczema or psoriasis if there is scaling, such as in our patient. At the tumor stage, flat or dome-shaped nodules that may vary in color and are deeper than plaques begin to appear. Ulcerations, which were absent in our case, may often be seen.
Because of the diverse clinical manifestations of MF, which can mimic other common dermatoses, diagnosis often is challenging for clinicians. Furthermore, histology can yield nonspecific diagnostic results and may even resemble chronic inflammatory dermatoses.7 As a result, patients frequently are subjected to multiple skin biopsies to establish the diagnosis,8 and diagnosis may be delayed, with the median time from onset of skin symptoms to diagnosis being approximately 6 years.9
Reflectance Confocal Microscopy
In vivo RCM is a noninvasive technique that allows visualization of the skin at a cellular level and recently has been evaluated as a diagnostic tool for many skin conditions.10,11 Reflectance confocal microscopy findings have been well established for many cutaneous malignancies as well as inflammatory conditions such as psoriasis and atopic dermatitis.12,13 Specifically, 2 preliminary descriptive studies utilized RCM to visualize the characteristic features of MF in vivo.14,15 These studies reported the histopathologic correlation of RCM findings in biopsy-proven MF lesions. Consistent in all stages of MF is the presence of small, weakly refractile, round to oval cells within the spinous layer that correlate with atypical lymphocytes, in addition to hyporefractile basal cells surrounding the dermal papillae. Patch-stage MF lesions have more subtle epidermal findings compared to plaque-stage lesions, which tend to have more prominent vesiclelike dark spaces filled with collections of monomorphous, weakly refractile, round to oval cells corresponding with Pautrier microabscesses and evidence of spongiosis.14,15 The first descriptive study of RCM in the diagnosis of MF failed to identify features of tumor-stage MF that would distinguish it from patch- or plaque-stage disease. The investigators also stated that deep nodular collections of atypical lymphocytes seen on histopathology in tumor-stage MF were missed on RCM evaluation.14 Furthermore, the second descriptive study of RCM and MF, which included 2 patients with tumor-stage disease, also failed to differentiate tumor-stage MF from the patch or plaque stages.15
Because of these 2 descriptive studies, a pilot study was conducted to determine the applicability and reproducibility of RCM findings for MF diagnosis.16 Two blinded confocalists were asked to diagnose RCM images as MF when compared to either normal skin or a variety of lymphoproliferative disorders. Of 15 patients, the confocalists correctly diagnosed MF in 84% and 90% of cases, respectively. Additionally, they reported the specificity and sensitivity of the following RCM features in the diagnosis of MF: spongiosis, 88.9% and 94.7%; loss of demarcation, 88.9% and 94.7%; disarray of the epidermis, 77.8% and 89.5%; hyporefractile rings, 88.9% and 78.9%; junctional atypical lymphocytes, 100% and 73.7%; and vesiclelike structures (Pautrier microabscesses), 100% and 73.7%. Importantly, this study did not evaluate the specificity and sensitivity of MF diagnosis compared to other eczematous or inflammatory conditions that may share similar RCM findings; therefore, these results are not generalizable, and many of the RCM findings characteristically seen in MF are not specific to its diagnosis.16
One study assessed the diagnostic accuracy of RCM in evaluating erythematosquamous diseases including MF, psoriasis, contact dermatitis, discoid lupus, and subacute cutaneous lupus.17 In this study, 3 blinded confocalists achieved a 95.41% and 92.89% specificity and 89.13% and 63.33% sensitivity for psoriasis and MF, respectively. Typical features of psoriasis on RCM included parakeratosis, reduction or absence of the granular layer, papillomatosis, acanthosis with normal honeycomb pattern of the epidermis, and dilated vessels in the upper dermis. Features that were more specific to MF included epidermotropic atypical lymphocytes, interface dermatitis, pleomorphic tumor cells, and dendritic cells.17 However, atypical lymphocytes and interface dermatitis also may be seen in cutaneous lupus; therefore, additional studies are still needed to validate RCM’s utility in differentiating between erythematosquamous skin diseases, including psoriasis, cutaneous lupus, and MF. Currently, RCM findings must be interpreted in conjunction with the clinical and histologic picture.
Importantly, RCM also is limited when evaluating MF due to its limited depth of visualization, as it allows imaging only to the superficial papillary dermis. Furthermore, any infiltrative process such as epidermal hyperplasia, spongiosis, or scaling, which can be seen in MF, may further impair the imaging quality of the deeper dermis.
Conclusion
Despite its limitations, RCM has the potential to be advantageous in evaluating skin lesions suspicious for MF in real time and is a promising technology for a quick noninvasive bedside adjunct tool. Its utility in selecting the optimal site for biopsy for better yield of histopathologic results in suspected MF cases has been demonstrated.16 However, large-scale studies still are needed to evaluate RCM in the diagnosis of the wide diversity of MF lesions as well as its efficacy in selecting optimal biopsy sites.
- Lutzner M, Edelson R, Schein P, et al. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83:534-552.
- Akinbami AA, Osikomaiya BI, John-Olabode SO, et al. Mycosis fungoides: case report and literature review. Clin Med Insights Case Rep. 2014;7:95-98.
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-959.
- Bradford PT, Devesa SS, Anderson WF, et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113:5064-5073.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nashan D, Faulhaber D, Stander S. Mycosis fungoides: a dermatological masquerader. Br J Dermatol. 2007;157:1-10.
- Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
- Glass LF, Keller KL, Messina JL, et al. Cutaneous T-cell lymphoma. Cancer Control. 1998;5:11-18.
- Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14:293-371.
- Tannous ZS, Mihm MC, Flotte TJ, et al. In vivo examination of lentigo maligna and malignant melanoma in situ, lentigo maligna type by near-infrared reflectance confocal microscopy: comparison of in vivo confocal images with histologic sections. J Am Acad Dermatol. 2002;46:260-263.
- Gerger A, Koller S, Weger W, et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer. 2006;107:193-200.
- Branzan AL, Landthaler M, Szeimies RM. In vivo confocal scanning laser microscopy in dermatology [published online November 18, 2006]. Lasers Med Sci. 2007;22:73-82.
- González S. Confocal reflectance microscopy in dermatology: promise and reality of non-invasive diagnosis and monitoring. Actas Dermosifiliogr. 2009;100(suppl 2):59-69.
- Agero AL, Gill M, Ardigo M, et al. In vivo reflectance confocal microscopy of mycosis fungoides: a preliminary study [published online April 16, 2007]. J Am Acad Dermatol. 2007;57:435-441.
- Wi L, Dai H, Li Z, et al. Reflectance confocal microscopy for the characteristics of mycosis fungoides and correlation with histology: a pilot study [published online April 18, 2013]. Skin Res Technol. 2013;19:352-355.
- Lange-Asschenfeldt S, Babilli J, Beyer M, et al. Consistency and distribution of reflectance confocal microscopy features for diagnosis of cutaneous T cell lymphoma. J Biomed Opt. 2012;17:016001.
- Koller S, Gerger A, Ahlgrimm-Siess V. In vivo reflectance confocal microscopy of erythematosquamous skin diseases [published online March 6, 2009]. Exp Dermatol. 2009;18:536-540.
- Lutzner M, Edelson R, Schein P, et al. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83:534-552.
- Akinbami AA, Osikomaiya BI, John-Olabode SO, et al. Mycosis fungoides: case report and literature review. Clin Med Insights Case Rep. 2014;7:95-98.
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-959.
- Bradford PT, Devesa SS, Anderson WF, et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113:5064-5073.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nashan D, Faulhaber D, Stander S. Mycosis fungoides: a dermatological masquerader. Br J Dermatol. 2007;157:1-10.
- Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
- Glass LF, Keller KL, Messina JL, et al. Cutaneous T-cell lymphoma. Cancer Control. 1998;5:11-18.
- Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14:293-371.
- Tannous ZS, Mihm MC, Flotte TJ, et al. In vivo examination of lentigo maligna and malignant melanoma in situ, lentigo maligna type by near-infrared reflectance confocal microscopy: comparison of in vivo confocal images with histologic sections. J Am Acad Dermatol. 2002;46:260-263.
- Gerger A, Koller S, Weger W, et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer. 2006;107:193-200.
- Branzan AL, Landthaler M, Szeimies RM. In vivo confocal scanning laser microscopy in dermatology [published online November 18, 2006]. Lasers Med Sci. 2007;22:73-82.
- González S. Confocal reflectance microscopy in dermatology: promise and reality of non-invasive diagnosis and monitoring. Actas Dermosifiliogr. 2009;100(suppl 2):59-69.
- Agero AL, Gill M, Ardigo M, et al. In vivo reflectance confocal microscopy of mycosis fungoides: a preliminary study [published online April 16, 2007]. J Am Acad Dermatol. 2007;57:435-441.
- Wi L, Dai H, Li Z, et al. Reflectance confocal microscopy for the characteristics of mycosis fungoides and correlation with histology: a pilot study [published online April 18, 2013]. Skin Res Technol. 2013;19:352-355.
- Lange-Asschenfeldt S, Babilli J, Beyer M, et al. Consistency and distribution of reflectance confocal microscopy features for diagnosis of cutaneous T cell lymphoma. J Biomed Opt. 2012;17:016001.
- Koller S, Gerger A, Ahlgrimm-Siess V. In vivo reflectance confocal microscopy of erythematosquamous skin diseases [published online March 6, 2009]. Exp Dermatol. 2009;18:536-540.
Practice Points
- Mycosis fungoides (MF) can be a challenging diagnosis to establish and often requires multiple biopsies.
- Reflectance confocal microscopy (RCM) may be helpful as a bedside noninvasive diagnostic technique.
- In suspected MF cases, RCM may assist in selecting the optimal biopsy site for better yield of histopathologic results.
Deep Soft Tissue Mass of the Knee
The Diagnosis: Nodular Fasciitis
The diagnosis of spindle cell tumors can be challenging; however, by using a variety of immunoperoxidase stains and fluorescent in situ hybridization (FISH) testing in conjunction with histology, it often is possible to arrive at a definitive diagnosis. For this case, the histologic features in conjunction with the immunoperoxidase stains and FISH were consistent with a diagnosis of nodular fasciitis.
Nodular fasciitis is a benign, self-limiting, myofibroblastic, soft-tissue proliferation typically found in the subcutaneous tissue.1 It can be found anywhere on the body but most commonly on the upper arms and trunk. It most often is seen in young adults, and many cases have been reported in association with a history of trauma to the area.1,2 It typically measures less than 2 cm in diameter.3 The diagnosis of nodular fasciitis is particularly challenging because it mimics sarcoma, both in presentation and in histologic findings with rapid growth, high mitotic activity, and increased cellularity.1,4-7 In contrast to malignancy, nodular fasciitis has no atypical mitoses and little cytologic atypia.8,9 Rather, it contains plump myofibroblasts loosely arranged in a myxoid or fibrous stroma that also may contain lymphocytes, extravasated erythrocytes, and osteoclastlike giant cells distributed throughout.5,10,11 In this case, lymphocytes, extravasated red blood cells, and myxoid change are present, suggesting the diagnosis of nodular fasciitis. In other cases, however, these features may be much more limited, making the diagnosis more challenging. The spindle cells are arranged in poorly defined short fascicles. The tumor cells do not infiltrate between individual adipocytes. There is no notable cytologic atypia.
Because of the difficulty in making the diagnosis, overtreatment of this benign condition can be a problem, causing increased morbidity.1 Erickson-Johnson et al12 identified the role of an ubiquitin-specific peptidase 6, USP6, gene rearrangement on chromosome 17p13 in 92% (44/48) of cases of nodular fasciitis. The USP6 gene most often is rearranged with the myosin heavy chain 9 gene, MYH9, on chromosome 22q12.3. With this rearrangement, the MYH9 promoter leads to the overexpression of USP6, causing tumor formation.2,13 The use of multiple immunoperoxidase stains can be important in the identification of nodular fasciitis. Nodular fasciitis stains negative for S-100, epithelial membrane antigen, CD34, β-catenin, and cytokeratin, but typically stains positive for smooth muscle actin.9
Although dermatofibrosarcoma protuberans (DFSP) was in the differential diagnosis, these tumors tend to have greater cellularity than nodular fasciitis. In addition, the spindle cells of DFSP typically are arranged in a storiform pattern. Another characteristic feature of DFSP is that the tumor cells will infiltrate between adipose cells creating a lacelike or honeycomblike appearance within the subcutaneous tissue (Figure 1). Immunohistochemistry staining and FISH testing may be useful in making a diagnosis of DFSP. These tumors typically are positive for CD34 by immunoperoxidase staining and demonstrate a translocation t(17;22)(q21;q13) between platelet-derived growth factor subunit B gene, PDGFB, and collagen type I alpha 1 chain gene, COL1A1, by FISH.
.")
The distinction between the fibrous phase of nodular fasciitis and fibromatosis can be challenging. The size of the lesion may be helpful, with most lesions of nodular fasciitis being less than 3 cm, while lesions of fibromatosis have a mean diameter of 7 cm.5,14 Microscopically, both tumors demonstrate a fascicular growth pattern; however, the fascicles in nodular fasciitis tend to be short and irregular compared to the longer fascicles seen in fibromatosis (Figure 2). Immunohistochemistry staining has limited utility with only 56% (14/25) of superficial fibromatoses having positive nuclear staining for β-catenin.15
.")
Low-grade fibromyxoid sarcoma (LGFMS) would be unusual in this clinical scenario. Only 13% to 19% of cases present in patients younger than 18 years (mean age, 33 years).16 In LGFMS there are cytologically bland spindle cells that are typically arranged in a patternless or whorled pattern (Figure 3), though fascicular architecture may be seen. There are alternating areas of fibrous and myxoid stroma. A curvilinear vasculature network and lack of lymphocytes and extravasated red blood cells are histologic features favoring LGFMS over nodular fasciitis. Immunohistochemistry staining and FISH testing can be useful in making the diagnosis of LGFMS. These tumors are characterized by a translocation t(7;16)(q34;p11) involving the fusion in sarcoma, FUS, and cAMP responsive element binding protein 3 like 2, CREB3L2, genes.16 Positive immunohistochemistry staining for MUC4 can be seen in up to 100% of LGFMS and is absent in many other spindle cell tumors.16
.")
Plexiform fibrohistiocytic tumor (PFT) is least likely to be confused with nodular fasciitis. Histologically these tumors are characterized by multiple small nodules arranged in a plexiform pattern (Figure 4). Within the nodules, 3 cell types may be noted: spindle fibroblast-like cells, mononuclear histiocyte-like cells, and osteoclastlike cells.17 Either the spindle cells or the mononuclear cells may predominate in cases of PFT. Immunohistochemistry staining of PFT is nonspecific and there are no molecular/FISH studies that can be used to help confirm the diagnosis.
.")
- Shin C, Low I, Ng D, et al. USP6 gene rearrangement in nodular fasciitis and histological mimics. Histopathology. 2016;69:784-791.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Nishio J. Updates on the cytogenetics and molecular cytogenetics of benign and intermediate soft tissue tumors. Oncol Lett. 2013;5:12-18.
- Lin X, Wang L, Zhang Y, et al. Variable Ki67 proliferative index in 65 cases of nodular fasciitis, compared with fibrosarcoma and fibromatosis. Diagn Pathol. 2013;8:50.
- Goldstein J, Cates J. Differential diagnostic considerations of desmoid-type fibromatosis. Adv Anat Pathol. 2015;22:260-266.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al, eds. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyons, France: IARC Press; 2013.
- Bridge JA, Cushman-Vokoun AM. Molecular diagnostics of soft tissue tumors. Arch Pathol Lab Med. 2011;135:588-601.
- Anzeljc AJ, Oliveira AM, Grossniklaus HE, et al. Nodular fasciitis of the orbit: a case report confirmed by molecular cytogenetic analysis. Ophthalmic Plast Reconstr Surg. 2017;33(3S suppl 1):S152-S155.
- de Paula SA, Cruz AA, de Alencar VM, et al. Nodular fasciitis presenting as a large mass in the upper eyelid. Ophthalmic Plast Reconstr Surg. 2006;22:494-495.
- Bernstein KE, Lattes R. Nodular (pseudosarcomatous) fasciitis, a nonrecurrent lesion: clinicopathologic study of 134 cases. Cancer. 1982;49:1668-1678.
- Shimizu S, Hashimoto H, Enjoji M. Nodular fasciitis: an analysis of 250 patients. Pathology. 1984;16:161-166.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Amary MF, Ye H, Berisha F, et al. Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch. 2013;463:97-98.
- Wirth L, Klein A, Baur-Melnyk A. Desmoid tumors of the extremity and trunk. a retrospective study of 44 patients. BMC Musculoskelet Disord. 2018;19:2.
- Carlson JW, Fletcher CD. Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cells lesions: analysis of a series and review of the literature. Histopathology. 2007;51:509-514.
- Mohamed M, Fisher C, Thway K. Low-grade fibromyxoid sarcoma: clinical, morphologic and genetic features. Ann Diagn Pathol. 2017;28:60-67.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med. 2007;131:1135-1138.
The Diagnosis: Nodular Fasciitis
The diagnosis of spindle cell tumors can be challenging; however, by using a variety of immunoperoxidase stains and fluorescent in situ hybridization (FISH) testing in conjunction with histology, it often is possible to arrive at a definitive diagnosis. For this case, the histologic features in conjunction with the immunoperoxidase stains and FISH were consistent with a diagnosis of nodular fasciitis.
Nodular fasciitis is a benign, self-limiting, myofibroblastic, soft-tissue proliferation typically found in the subcutaneous tissue.1 It can be found anywhere on the body but most commonly on the upper arms and trunk. It most often is seen in young adults, and many cases have been reported in association with a history of trauma to the area.1,2 It typically measures less than 2 cm in diameter.3 The diagnosis of nodular fasciitis is particularly challenging because it mimics sarcoma, both in presentation and in histologic findings with rapid growth, high mitotic activity, and increased cellularity.1,4-7 In contrast to malignancy, nodular fasciitis has no atypical mitoses and little cytologic atypia.8,9 Rather, it contains plump myofibroblasts loosely arranged in a myxoid or fibrous stroma that also may contain lymphocytes, extravasated erythrocytes, and osteoclastlike giant cells distributed throughout.5,10,11 In this case, lymphocytes, extravasated red blood cells, and myxoid change are present, suggesting the diagnosis of nodular fasciitis. In other cases, however, these features may be much more limited, making the diagnosis more challenging. The spindle cells are arranged in poorly defined short fascicles. The tumor cells do not infiltrate between individual adipocytes. There is no notable cytologic atypia.
Because of the difficulty in making the diagnosis, overtreatment of this benign condition can be a problem, causing increased morbidity.1 Erickson-Johnson et al12 identified the role of an ubiquitin-specific peptidase 6, USP6, gene rearrangement on chromosome 17p13 in 92% (44/48) of cases of nodular fasciitis. The USP6 gene most often is rearranged with the myosin heavy chain 9 gene, MYH9, on chromosome 22q12.3. With this rearrangement, the MYH9 promoter leads to the overexpression of USP6, causing tumor formation.2,13 The use of multiple immunoperoxidase stains can be important in the identification of nodular fasciitis. Nodular fasciitis stains negative for S-100, epithelial membrane antigen, CD34, β-catenin, and cytokeratin, but typically stains positive for smooth muscle actin.9
Although dermatofibrosarcoma protuberans (DFSP) was in the differential diagnosis, these tumors tend to have greater cellularity than nodular fasciitis. In addition, the spindle cells of DFSP typically are arranged in a storiform pattern. Another characteristic feature of DFSP is that the tumor cells will infiltrate between adipose cells creating a lacelike or honeycomblike appearance within the subcutaneous tissue (Figure 1). Immunohistochemistry staining and FISH testing may be useful in making a diagnosis of DFSP. These tumors typically are positive for CD34 by immunoperoxidase staining and demonstrate a translocation t(17;22)(q21;q13) between platelet-derived growth factor subunit B gene, PDGFB, and collagen type I alpha 1 chain gene, COL1A1, by FISH.
The distinction between the fibrous phase of nodular fasciitis and fibromatosis can be challenging. The size of the lesion may be helpful, with most lesions of nodular fasciitis being less than 3 cm, while lesions of fibromatosis have a mean diameter of 7 cm.5,14 Microscopically, both tumors demonstrate a fascicular growth pattern; however, the fascicles in nodular fasciitis tend to be short and irregular compared to the longer fascicles seen in fibromatosis (Figure 2). Immunohistochemistry staining has limited utility with only 56% (14/25) of superficial fibromatoses having positive nuclear staining for β-catenin.15
Low-grade fibromyxoid sarcoma (LGFMS) would be unusual in this clinical scenario. Only 13% to 19% of cases present in patients younger than 18 years (mean age, 33 years).16 In LGFMS there are cytologically bland spindle cells that are typically arranged in a patternless or whorled pattern (Figure 3), though fascicular architecture may be seen. There are alternating areas of fibrous and myxoid stroma. A curvilinear vasculature network and lack of lymphocytes and extravasated red blood cells are histologic features favoring LGFMS over nodular fasciitis. Immunohistochemistry staining and FISH testing can be useful in making the diagnosis of LGFMS. These tumors are characterized by a translocation t(7;16)(q34;p11) involving the fusion in sarcoma, FUS, and cAMP responsive element binding protein 3 like 2, CREB3L2, genes.16 Positive immunohistochemistry staining for MUC4 can be seen in up to 100% of LGFMS and is absent in many other spindle cell tumors.16
Plexiform fibrohistiocytic tumor (PFT) is least likely to be confused with nodular fasciitis. Histologically these tumors are characterized by multiple small nodules arranged in a plexiform pattern (Figure 4). Within the nodules, 3 cell types may be noted: spindle fibroblast-like cells, mononuclear histiocyte-like cells, and osteoclastlike cells.17 Either the spindle cells or the mononuclear cells may predominate in cases of PFT. Immunohistochemistry staining of PFT is nonspecific and there are no molecular/FISH studies that can be used to help confirm the diagnosis.
The Diagnosis: Nodular Fasciitis
The diagnosis of spindle cell tumors can be challenging; however, by using a variety of immunoperoxidase stains and fluorescent in situ hybridization (FISH) testing in conjunction with histology, it often is possible to arrive at a definitive diagnosis. For this case, the histologic features in conjunction with the immunoperoxidase stains and FISH were consistent with a diagnosis of nodular fasciitis.
Nodular fasciitis is a benign, self-limiting, myofibroblastic, soft-tissue proliferation typically found in the subcutaneous tissue.1 It can be found anywhere on the body but most commonly on the upper arms and trunk. It most often is seen in young adults, and many cases have been reported in association with a history of trauma to the area.1,2 It typically measures less than 2 cm in diameter.3 The diagnosis of nodular fasciitis is particularly challenging because it mimics sarcoma, both in presentation and in histologic findings with rapid growth, high mitotic activity, and increased cellularity.1,4-7 In contrast to malignancy, nodular fasciitis has no atypical mitoses and little cytologic atypia.8,9 Rather, it contains plump myofibroblasts loosely arranged in a myxoid or fibrous stroma that also may contain lymphocytes, extravasated erythrocytes, and osteoclastlike giant cells distributed throughout.5,10,11 In this case, lymphocytes, extravasated red blood cells, and myxoid change are present, suggesting the diagnosis of nodular fasciitis. In other cases, however, these features may be much more limited, making the diagnosis more challenging. The spindle cells are arranged in poorly defined short fascicles. The tumor cells do not infiltrate between individual adipocytes. There is no notable cytologic atypia.
Because of the difficulty in making the diagnosis, overtreatment of this benign condition can be a problem, causing increased morbidity.1 Erickson-Johnson et al12 identified the role of an ubiquitin-specific peptidase 6, USP6, gene rearrangement on chromosome 17p13 in 92% (44/48) of cases of nodular fasciitis. The USP6 gene most often is rearranged with the myosin heavy chain 9 gene, MYH9, on chromosome 22q12.3. With this rearrangement, the MYH9 promoter leads to the overexpression of USP6, causing tumor formation.2,13 The use of multiple immunoperoxidase stains can be important in the identification of nodular fasciitis. Nodular fasciitis stains negative for S-100, epithelial membrane antigen, CD34, β-catenin, and cytokeratin, but typically stains positive for smooth muscle actin.9
Although dermatofibrosarcoma protuberans (DFSP) was in the differential diagnosis, these tumors tend to have greater cellularity than nodular fasciitis. In addition, the spindle cells of DFSP typically are arranged in a storiform pattern. Another characteristic feature of DFSP is that the tumor cells will infiltrate between adipose cells creating a lacelike or honeycomblike appearance within the subcutaneous tissue (Figure 1). Immunohistochemistry staining and FISH testing may be useful in making a diagnosis of DFSP. These tumors typically are positive for CD34 by immunoperoxidase staining and demonstrate a translocation t(17;22)(q21;q13) between platelet-derived growth factor subunit B gene, PDGFB, and collagen type I alpha 1 chain gene, COL1A1, by FISH.
The distinction between the fibrous phase of nodular fasciitis and fibromatosis can be challenging. The size of the lesion may be helpful, with most lesions of nodular fasciitis being less than 3 cm, while lesions of fibromatosis have a mean diameter of 7 cm.5,14 Microscopically, both tumors demonstrate a fascicular growth pattern; however, the fascicles in nodular fasciitis tend to be short and irregular compared to the longer fascicles seen in fibromatosis (Figure 2). Immunohistochemistry staining has limited utility with only 56% (14/25) of superficial fibromatoses having positive nuclear staining for β-catenin.15
Low-grade fibromyxoid sarcoma (LGFMS) would be unusual in this clinical scenario. Only 13% to 19% of cases present in patients younger than 18 years (mean age, 33 years).16 In LGFMS there are cytologically bland spindle cells that are typically arranged in a patternless or whorled pattern (Figure 3), though fascicular architecture may be seen. There are alternating areas of fibrous and myxoid stroma. A curvilinear vasculature network and lack of lymphocytes and extravasated red blood cells are histologic features favoring LGFMS over nodular fasciitis. Immunohistochemistry staining and FISH testing can be useful in making the diagnosis of LGFMS. These tumors are characterized by a translocation t(7;16)(q34;p11) involving the fusion in sarcoma, FUS, and cAMP responsive element binding protein 3 like 2, CREB3L2, genes.16 Positive immunohistochemistry staining for MUC4 can be seen in up to 100% of LGFMS and is absent in many other spindle cell tumors.16
Plexiform fibrohistiocytic tumor (PFT) is least likely to be confused with nodular fasciitis. Histologically these tumors are characterized by multiple small nodules arranged in a plexiform pattern (Figure 4). Within the nodules, 3 cell types may be noted: spindle fibroblast-like cells, mononuclear histiocyte-like cells, and osteoclastlike cells.17 Either the spindle cells or the mononuclear cells may predominate in cases of PFT. Immunohistochemistry staining of PFT is nonspecific and there are no molecular/FISH studies that can be used to help confirm the diagnosis.
- Shin C, Low I, Ng D, et al. USP6 gene rearrangement in nodular fasciitis and histological mimics. Histopathology. 2016;69:784-791.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Nishio J. Updates on the cytogenetics and molecular cytogenetics of benign and intermediate soft tissue tumors. Oncol Lett. 2013;5:12-18.
- Lin X, Wang L, Zhang Y, et al. Variable Ki67 proliferative index in 65 cases of nodular fasciitis, compared with fibrosarcoma and fibromatosis. Diagn Pathol. 2013;8:50.
- Goldstein J, Cates J. Differential diagnostic considerations of desmoid-type fibromatosis. Adv Anat Pathol. 2015;22:260-266.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al, eds. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyons, France: IARC Press; 2013.
- Bridge JA, Cushman-Vokoun AM. Molecular diagnostics of soft tissue tumors. Arch Pathol Lab Med. 2011;135:588-601.
- Anzeljc AJ, Oliveira AM, Grossniklaus HE, et al. Nodular fasciitis of the orbit: a case report confirmed by molecular cytogenetic analysis. Ophthalmic Plast Reconstr Surg. 2017;33(3S suppl 1):S152-S155.
- de Paula SA, Cruz AA, de Alencar VM, et al. Nodular fasciitis presenting as a large mass in the upper eyelid. Ophthalmic Plast Reconstr Surg. 2006;22:494-495.
- Bernstein KE, Lattes R. Nodular (pseudosarcomatous) fasciitis, a nonrecurrent lesion: clinicopathologic study of 134 cases. Cancer. 1982;49:1668-1678.
- Shimizu S, Hashimoto H, Enjoji M. Nodular fasciitis: an analysis of 250 patients. Pathology. 1984;16:161-166.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Amary MF, Ye H, Berisha F, et al. Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch. 2013;463:97-98.
- Wirth L, Klein A, Baur-Melnyk A. Desmoid tumors of the extremity and trunk. a retrospective study of 44 patients. BMC Musculoskelet Disord. 2018;19:2.
- Carlson JW, Fletcher CD. Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cells lesions: analysis of a series and review of the literature. Histopathology. 2007;51:509-514.
- Mohamed M, Fisher C, Thway K. Low-grade fibromyxoid sarcoma: clinical, morphologic and genetic features. Ann Diagn Pathol. 2017;28:60-67.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med. 2007;131:1135-1138.
- Shin C, Low I, Ng D, et al. USP6 gene rearrangement in nodular fasciitis and histological mimics. Histopathology. 2016;69:784-791.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Nishio J. Updates on the cytogenetics and molecular cytogenetics of benign and intermediate soft tissue tumors. Oncol Lett. 2013;5:12-18.
- Lin X, Wang L, Zhang Y, et al. Variable Ki67 proliferative index in 65 cases of nodular fasciitis, compared with fibrosarcoma and fibromatosis. Diagn Pathol. 2013;8:50.
- Goldstein J, Cates J. Differential diagnostic considerations of desmoid-type fibromatosis. Adv Anat Pathol. 2015;22:260-266.
- Fletcher CDM, Bridge JA, Hogendoorn PCW, et al, eds. WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. Lyons, France: IARC Press; 2013.
- Bridge JA, Cushman-Vokoun AM. Molecular diagnostics of soft tissue tumors. Arch Pathol Lab Med. 2011;135:588-601.
- Anzeljc AJ, Oliveira AM, Grossniklaus HE, et al. Nodular fasciitis of the orbit: a case report confirmed by molecular cytogenetic analysis. Ophthalmic Plast Reconstr Surg. 2017;33(3S suppl 1):S152-S155.
- de Paula SA, Cruz AA, de Alencar VM, et al. Nodular fasciitis presenting as a large mass in the upper eyelid. Ophthalmic Plast Reconstr Surg. 2006;22:494-495.
- Bernstein KE, Lattes R. Nodular (pseudosarcomatous) fasciitis, a nonrecurrent lesion: clinicopathologic study of 134 cases. Cancer. 1982;49:1668-1678.
- Shimizu S, Hashimoto H, Enjoji M. Nodular fasciitis: an analysis of 250 patients. Pathology. 1984;16:161-166.
- Erickson-Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9-USP6 gene fusion. Lab Invest. 2011;91:1427-1433.
- Amary MF, Ye H, Berisha F, et al. Detection of USP6 gene rearrangement in nodular fasciitis: an important diagnostic tool. Virchows Arch. 2013;463:97-98.
- Wirth L, Klein A, Baur-Melnyk A. Desmoid tumors of the extremity and trunk. a retrospective study of 44 patients. BMC Musculoskelet Disord. 2018;19:2.
- Carlson JW, Fletcher CD. Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cells lesions: analysis of a series and review of the literature. Histopathology. 2007;51:509-514.
- Mohamed M, Fisher C, Thway K. Low-grade fibromyxoid sarcoma: clinical, morphologic and genetic features. Ann Diagn Pathol. 2017;28:60-67.
- Taher A, Pushpanathan C. Plexiform fibrohistiocytic tumor: a brief review. Arch Pathol Lab Med. 2007;131:1135-1138.
.")
A 16-year-old adolescent girl presented with a bump over the left posterior knee of 1 month's duration. Her medical history was unremarkable. She denied recent trauma or injury to the area. On physical examination there was a visible and palpable tense nontender mass the size of an egg over the left posterior knee. Magnetic resonance imaging showed a lobulated mass-like focus of T2 hyperintensity centered at the subcutaneous tissues and superficial myofascial plane of the gastrocnemius on the posterior knee. Complete excision of the lesion was performed and demonstrated a 2.6.2 ×2.9.2 ×2.1-cm mass within subcutaneous adipose tissue. There was no microscopic involvement of skeletal muscle. Immunohistochemistry staining of the tumor was performed that was positive for smooth muscle actin and negative for desmin, S-100, CD34, pan-cytokeratin, and β-catenin. Fluorescent in situ hybridization testing demonstrated rearrangement of the ubiquitin-specific peptidase 6 gene, USP6, locus (17p13).