User login
To the Editor:
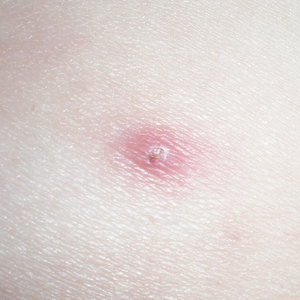
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.


Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
To the Editor:
A 38-year-old woman was diagnosed with common variable immune deficiency (CVID) by an immunologist at an outside institution 1 year prior to the current presentation. The diagnosis was based on history of severe recurrent sinopulmonary tract, inner ear, Clostridium difficile, urinary tract, and herpes zoster infections of approximately 6 years’ duration, as well as persistently low IgG, IgA, and IgM levels of 530 mg/dL (reference range, 690–1400 mg/dL), 29 mg/dL (reference range, 88–410 mg/dL), and 30 mg/dL (reference range, 34–210 mg/dL), respectively, with failure to respond to vaccinations (ie, Haemophilus influenzae type B, Streptococcus pneumoniae, diphtheria IgG antibody, tetanus antibody). She was started on replacement intravenous immunoglobulin (IVIG) 40 g monthly (400 mg/kg) for CVID. She had a family history of CVID diagnosed in her son and sister.
One year after the CVID diagnosis, she was diagnosed with Sweet syndrome (SS) by a physician at our institution via biopsy of a lesion on the left arm (Figure 1) that showed dense dermal infiltrate of neutrophils with scattered background apoptotic nuclear debris without evidence of vasculitis (Figure 2). Gram stain and microbial biopsy cultures were negative for mycobacterial, fungal, and bacterial organisms. Cutaneous lesions failed to respond to courses of intravenous antibiotics. Sarcoidosis workup was unremarkable and was pursued to exclude the association with SS. Other negative testing included antinuclear antibody, human immunodeficiency virus, rheumatoid factor, thyroid-stimulating hormone, Ro and La autoantibodies, cytoplasmic antineutrophil cytoplasmic antibody, perinuclear antineutrophil cytoplasmic antibody, antimitochondrial antibody, and urinalysis. Occult malignancy was excluded with negative bone marrow biopsy; cerebrospinal fluid analysis; esophagogastroduodenoscopy; colonoscopy; and computed tomography of the chest, abdomen, and pelvis.
Sweet syndrome flares in this patient began with a prodromal syndrome of fever, chills, fatigue, diarrhea, and severe local neuropathic pain. Cutaneous lesions erupted 2 days later, most frequently on the arms and fingers. Preemptive treatment with prednisone 30 to 40 mg when the prodrome was present did not arrest cutaneous lesion development. Flares initially occurred every 3 to 5 weeks.
She initially was successfully treated with high-dose prednisone 100 mg daily during SS flares. Prolonged low-dose prednisone maintenance (10–20 mg) and hydroxychloroquine failed to control her frequent exacerbations. Dapsone was intolerable secondary to an adverse reaction. She continued to have frequent exacerbations of the SS requiring hospitalizations.
During SS flares, CVID was stable with infrequent systemic infections. Although a causal relationship between CVID and SS was unclear, an empiric increase in IVIG dose was made by her immunologist to test if it would decrease the frequency of the cutaneous flares. Subsequently, the IVIG dose was increased to 60 g monthly followed by 200 g monthly after approximately 4 months with a partial initial response in the beginning of therapy for the first 6 months. However, episodes resumed with increasing frequency with cutaneous lesion flares every 2 to 3 weeks. In a 3-month period, the patient had at least 4 hospitalizations for SS flares. Finally, 18 months after the diagnosis of SS was made, she was started on metronomic cyclophosphamide at a daily oral dose of 100 mg, later reduced to 50 mg daily after she developed mild neutropenia. She was continued on monthly IVIG replacement at a higher dose of 200 g divided over 2 days for CVID throughout the course of the disease and to the present time. Since then, the frequency of SS flares has notably reduced. She required 1 hospitalization after cyclophosphamide was initiated. She uses short-pulse prednisone (1 mg/kg) for 3 to 5 days when new skin lesions appear in addition to cyclophosphamide.
Common variable immune deficiency, the most common primary immunodeficiency, initially can present in adulthood.1,2 Its hallmarks include low levels of serum immunoglobulin, most notably IgG with most patients having concurrent deficiencies of IgA and IgM, and impaired antibody responses with recurrent or atypical infections. It has been associated with autoimmune diseases, granulomatous disease, and inflammatory disorders.2 Failure to mount protective levels of antibody titer after vaccination demonstrates the deficiency of antibody production.1 Lack of recognition of this clinical spectrum may lead to delayed diagnosis and more importantly stalls the initiation of immunoglobulin replacement therapy.1 The customary dose of immunoglobulin replacement is 400 mg/kg given in a single monthly infusion2; however, doses should be individualized and based on clinical response.1
Sweet syndrome is characterized by the constellation of pyrexia; leukocytosis; and eruption of painful, edematous, dermal, and neutrophil-dense plaques that occur in the setting of infection or malignancy or are drug induced.3,4 Although not fully elucidated, the pathogenesis is thought to involve the effects of cytokines that precipitate neutrophil activation and infiltration inducing a hypersensitivity reaction and escalation of the immunologic cascade.3 Because SS can represent a paraneoplastic phenomenon or a dermal manifestation of a solid neoplasm or hematologic dyscrasia, it is important to rule out occult malignancy.3 The mainstay of treatment is systemic corticosteroids to which classical SS lesions readily respond. Alternatively, topical or intralesional corticosteroids may be used as adjuvant therapy. Alternate first-line treatments include potassium iodide and colchicine. Second-line therapies include indomethacin, cyclosporine, dapsone, and other immunosuppressive agents.5 The lesions may become superinfected with bacterial pathogens requiring antimicrobials.3 Spontaneous resolution seldom occurs. The risk for relapse is lifelong following spontaneous or therapy-induced clinical remission.3 There is a growing body of literature of SS-associated conditions.
Common variable immune deficiency is a collection of disorders resulting in antibody deficiency and recurrent infections.6 Despite the humeral defects in CVID, patients paradoxically may develop various autoimmune, hematologic, and inflammatory disorders.7 Sweet syndrome, first described in 1964, is a constellation of fever, neutrophilia, and neutrophilic dermatosis of unknown pathogenesis.8 Copresentation of CVID and SS has not been commonly reported. O’Regan et al8 described a 17-year-old adolescent boy with both SS and CVID but SS preceded the diagnosis of CVID. In our case, the patient presented with CVID first and then manifested SS 1 year later.
Common variable immune deficiency is the most frequent symptomatic primary immunodeficiency in adults. Because adults with CVID have varied manifestations, CVID is thought to be late-onset antibody failure. The genetic basis of these disorders has not been identified in the majority of individuals. More than 100 genetic defects have been ascribed to primary immunodeficiencies,9 though none are consistently found to be associated with CVID. The majority of CVID cases are sporadic, but the positive family history in our patient suggests a familial form. Approximately 10% to 20% of patients have an identified heritable cause of CVID.10 Our patient’s diagnosis of CVID was confirmed by meeting the diagnostic triad set by the European Society for Immunodeficiencies11 of marked reduction of IgG and IgA or IgM plus onset after 2 years of age, recurrent infections, and defective vaccination response. Additional complications including autoimmunity, malignancy, and granulomatous inflammation were extensively ruled out.
The etiology of SS is unknown and its pathogenesis not fully elucidated, though it is presumed to be a hypersensitivity reaction.12 Sweet syndrome can be classified into 3 major subtypes: classical or idiopathic, malignancy associated, or drug induced.3 Our patient’s presentation is consistent with the classical variant, as malignancy was ruled out and the patient was not on any medication other than IVIG at the time of diagnosis. The treatment of SS consists of systemic steroids, initially high dose followed by a prolonged taper over 4 to 6 weeks.3 This treatment causes a pronounced and sustained decrease in serum IgG due to increased catabolism during drug administration and decreased synthesis during and for a variable time after drug administration.13 In refractory cases, intravenous pulse administration of methylprednisolone sodium succinate for 3 to 5 days may enhance the response to standard therapies.5
The concurrent development of neutrophilic dermatoses/SS in an individual with CVID has not been fully described. There is a credible association of SS with infections, inflammatory bowel disease, pregnancy, malignancy, and medications, as well as a possible association with Behçet disease, erythema nodosum, relapsing polychondritis, rheumatoid arthritis, sarcoidosis, and thyroid disease.5 The association between immunoglobulin deficiencies and SS is markedly unusual. Despite regular IVIG replacement, adequate treatment of CVID did not seem to modulate SS flares in our patient. A case report in a pediatric patient does not provide specific guidance regarding treatment options.8
A particularly challenging aspect of our case was tailoring a treatment regimen to suppress SS flares. We have attained partial response to the refractory cutaneous lesions (decreased frequency and amplitude of outbreaks) with IVIG replacement 200 g every 4 weeks in combination with metronomic cyclophosphamide 50 mg daily (use of a repetitive, low-dose daily chemotherapy regimen to minimize side effects). Intermittent SS flares were managed acutely with pulse high-dose steroids. We report a case of SS with CVID, raising the plausibility of correlated pathogenesis. However, the exact mechanisms remain undefined.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
- Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-1426.
- Sicherer SH, Winkelstein JA. Primary immunodeficiency diseases in adults. JAMA. 1998;279:58-61.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Sweet RD. Acute febrile neutrophilic dermatosis. Br J Dermatol. 1979;100:93-99.
- Cohen PR. Neutrophilic dermatoses a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is not CVID: common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:48-77.
- Giannouli S, Anagnostou D, Soliotis F, et al. Autoimmune manifestations in common variable immunodeficiency. Clin Rheumatol. 2004;23:449-452.
- O’Regan GM, Ho WL, Limaye S, et al. Sweet’s syndrome in association with common variable immunodeficiency. Clin Exp Dermatol. 2008;34:192-194.
- Bergbreiter A, Salzer U. Common variable immunodeficiency: a multifaceted and puzzling disorder. Expert Rev Clin Immunol. 2009;5:167-180.
- Ameratunga R, Woon S-T, Gillis D, et al. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-211.
- Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol. 1999;93:190-197.
- Yi S, Bhate C, Schwartz RA. Sweet’s syndrome: an update and review. G Ital Dermatol Venereol. 2009;144:603-612.
- Butler WT, Rossen RD. Effects of corticosteroids on immunity in man. I. decreased serum IgG concentration caused by 3 or 5 days of high doses of methylprednisone. J Clin Invest. 1973;52:2629-2640.
Practice Points
- Suggested workup for Sweet syndrome includes ruling out connective tissue disorders and malignancies.
- Familial common variable immune deficiency is rare and can first manifest in adulthood.